5. talasemia
TRANSCRIPT

HEMATOLOGÍA IM.C. ADRIANA GONZÁLEZ FLORES
TALASEMIA
DÁVILA GARRIDO BRENDADE ITA MÁRQUEZ ITZEL PAOLA
HERNÁNDEZ BRINDIS JULIOJUÁREZ MEJÍA BETSAIDA
ORDAZ REYES ERENIA NÍNIVEVILLEGAS AGUIRRE AMERICA
BENEMÉRITA UNIVERSIDAD AUTÓNOMA DE PUEBLAFACULTAD DE CIENCIAS QUÍMICAS
LICENCIATURA QUÍMICO FARMACOBIÓLOGO

Las talasemias son trastornos hereditarios,
caracterizados por la producción anormal de
hemoglobina, que ocasionan disminución en
su producción y destrucción excesiva de los glóbulos
rojos.
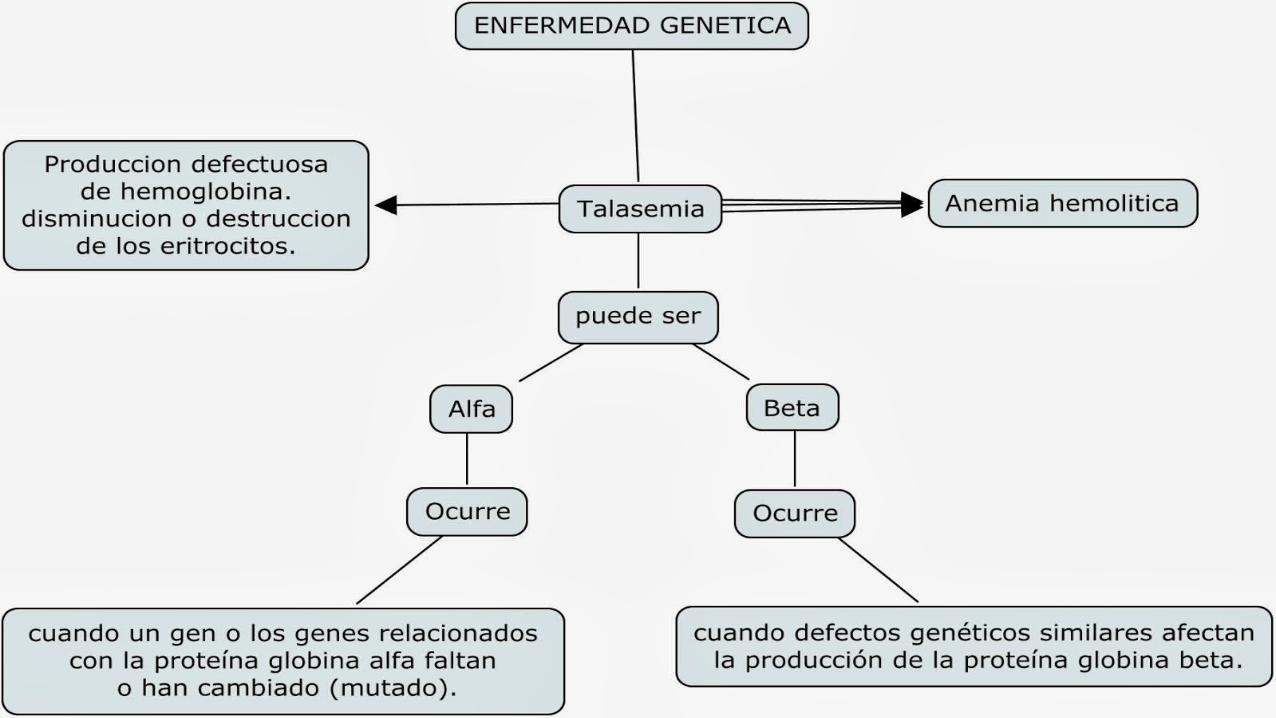
Las talasemias beta son causadas por una mutación en la cadena de la globina beta. Para adquirir la forma mayor de esta enfermedad, los genes mutados se deben heredar de
ambos padres. Si se hereda un solo gen mutado, la persona será portadora de la enfermedad, pero no experimenta los
síntomas, lo cual corresponde a la forma menor de la enfermedad.
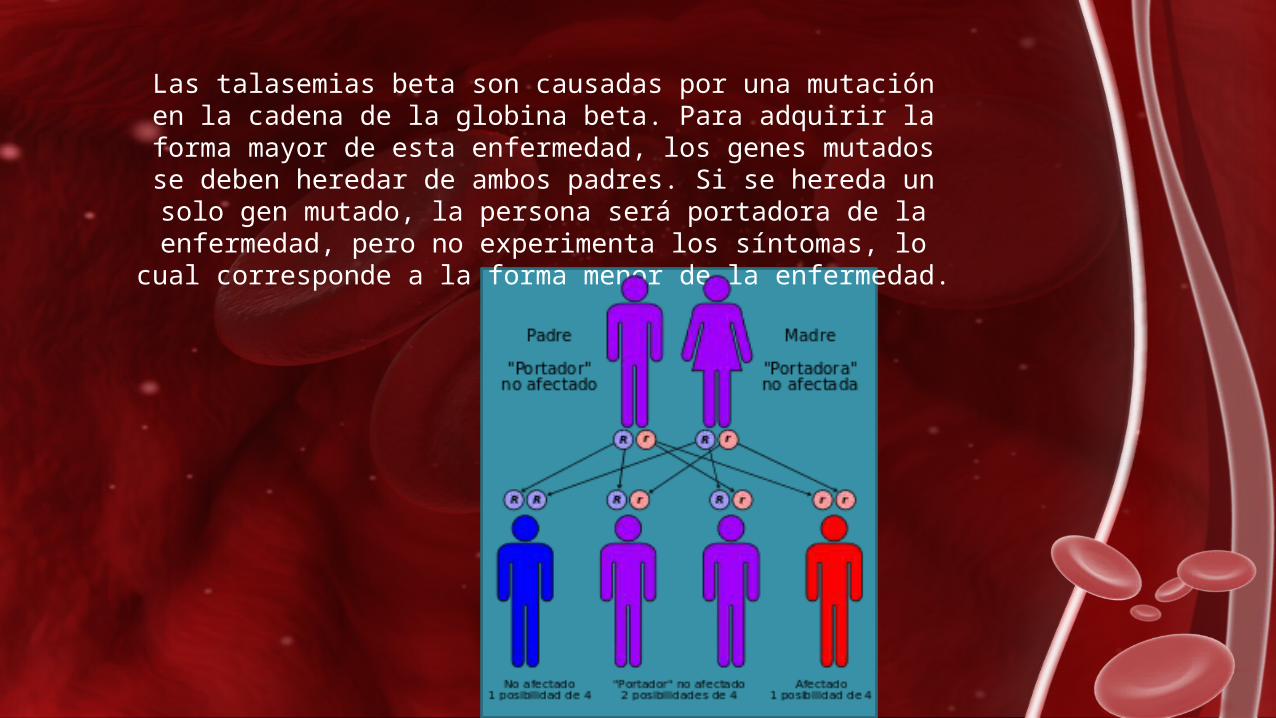
En la forma mayor, los niños son normales al nacer, pero desarrollan anemia durante el primer año de vida.
Algunos problemas que se pueden presentar son: insuficiencia en el crecimiento deformidades de los huesos agrandamiento del hígado del bazo.
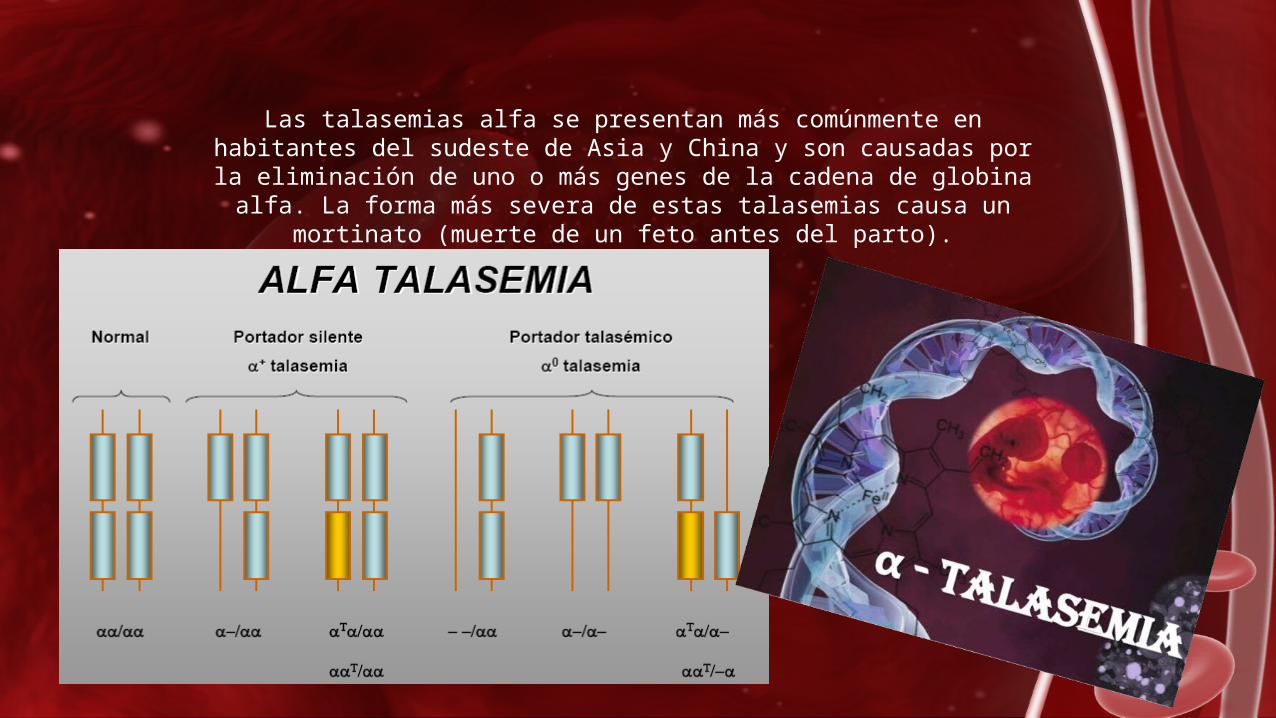
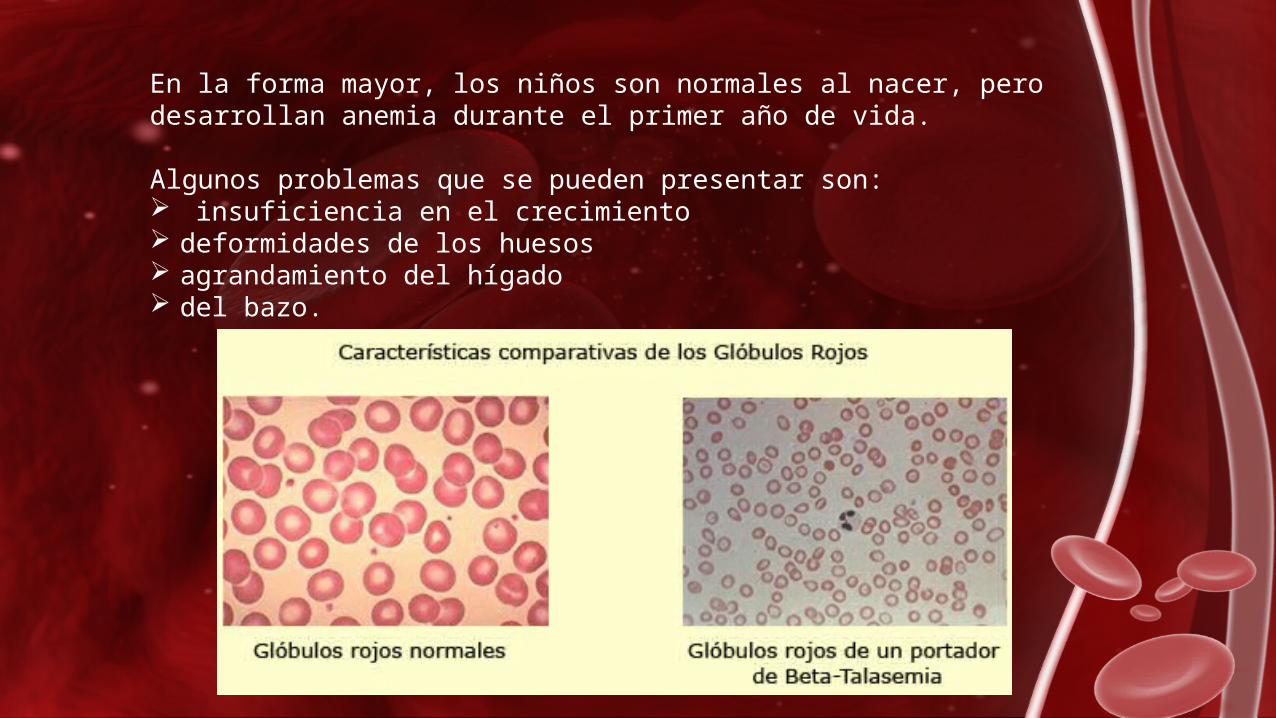
Las talasemias alfa se presentan más comúnmente en habitantes del sudeste de Asia y China y son causadas por la eliminación de uno o
más genes de la cadena de globina alfa. La forma más severa de estas talasemias causa un mortinato (muerte de un feto antes del
parto).
FISIOPATOLOGÍA
- Talasemia
o
0 +
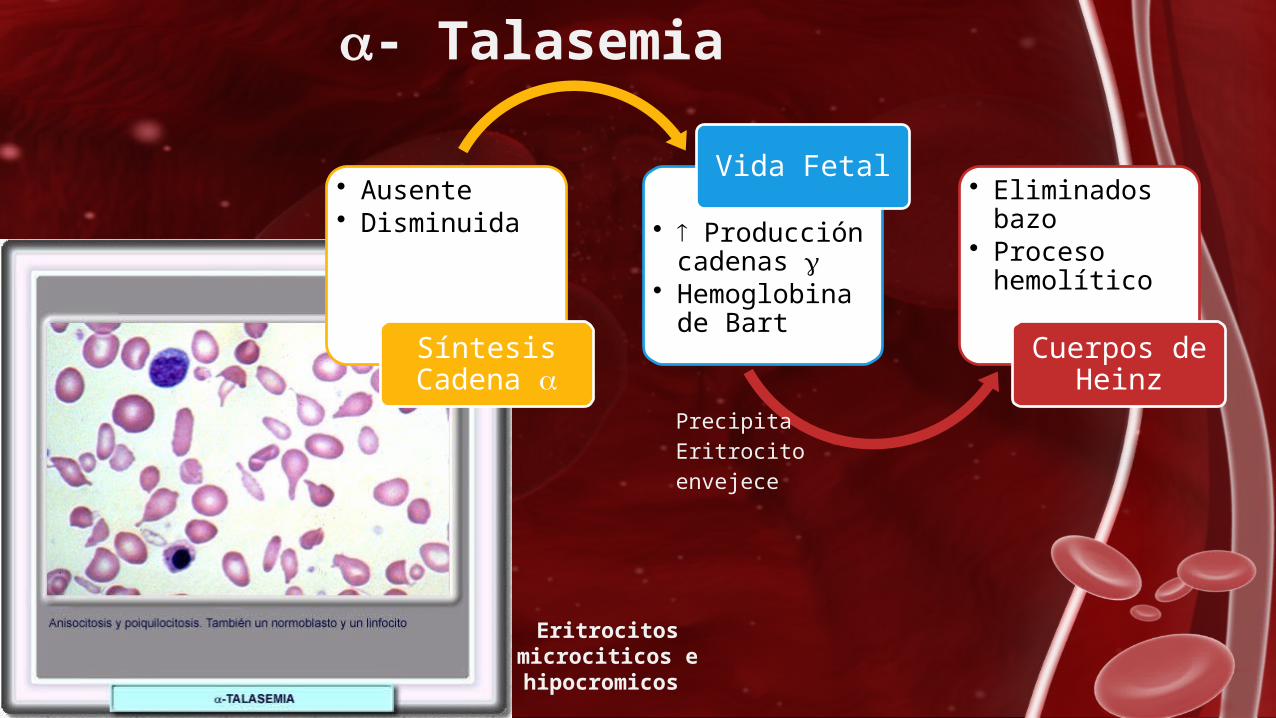
- Talasemia
• Ausente • Disminuida
Síntesis Cadena
• Producción cadenas
• Hemoglobina de Bart
Vida Fetal • Eliminados bazo
• Proceso hemolítico
Cuerpos de Heinz
Precipita Eritrocito envejece
Eritrocitos microciticos e hipocromicos
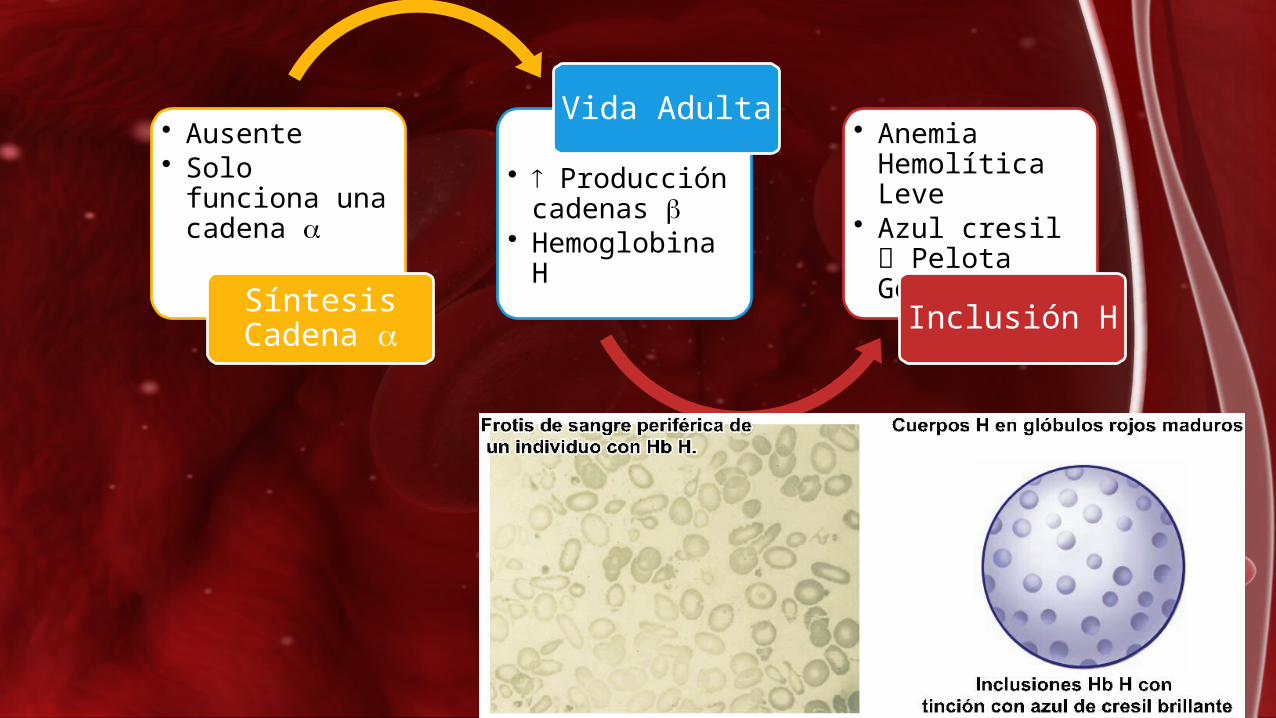
• Ausente • Solo funciona
una cadena
Síntesis Cadena
• Producción cadenas
• Hemoglobina H
Vida Adulta • Anemia Hemolítica Leve
• Azul cresil Pelota Golf
Inclusión H

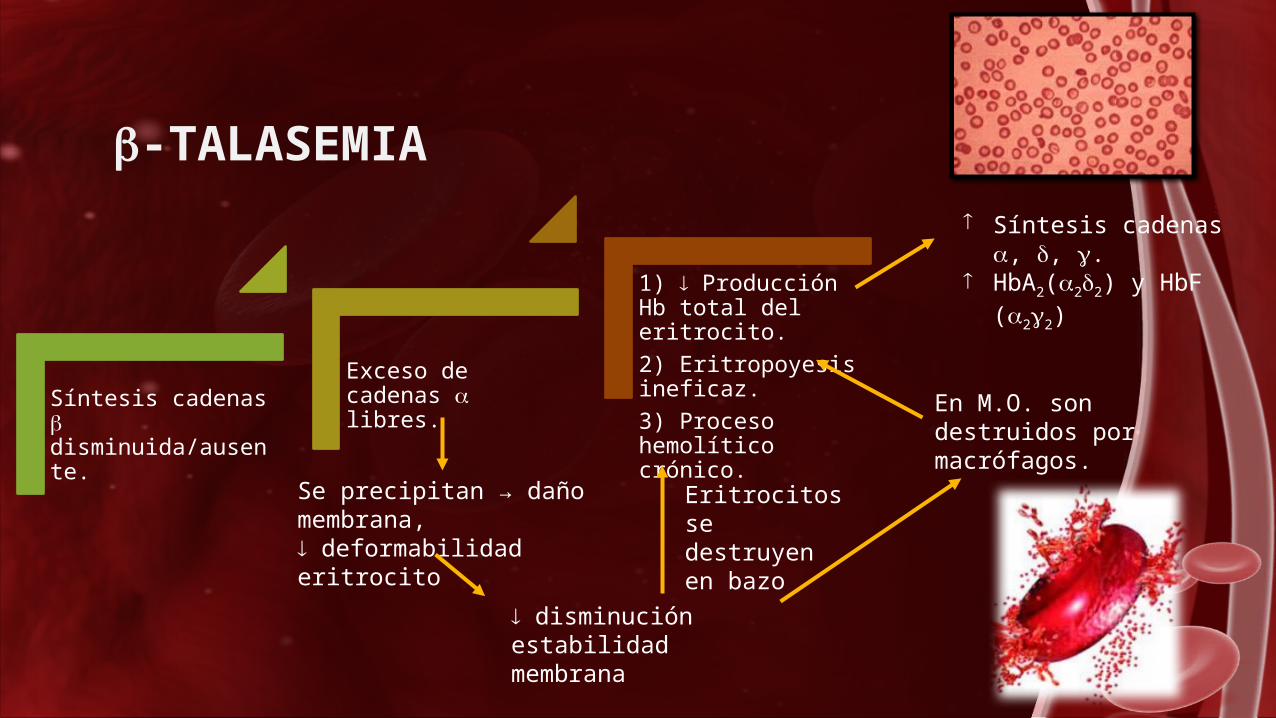
-TALASEMIA
Síntesis cadenas disminuida/ausente.
Exceso de cadenas libres.
1) Producción Hb total del eritrocito.2) Eritropoyesis ineficaz. 3) Proceso hemolítico crónico.
Síntesis cadenas , , .
HbA2(22) y HbF (22)
Se precipitan → daño membrana, deformabilidad eritrocito
disminución estabilidad membrana
En M.O. son destruidos por macrófagos.
Eritrocitos se destruyen en bazo

Características Clínicas

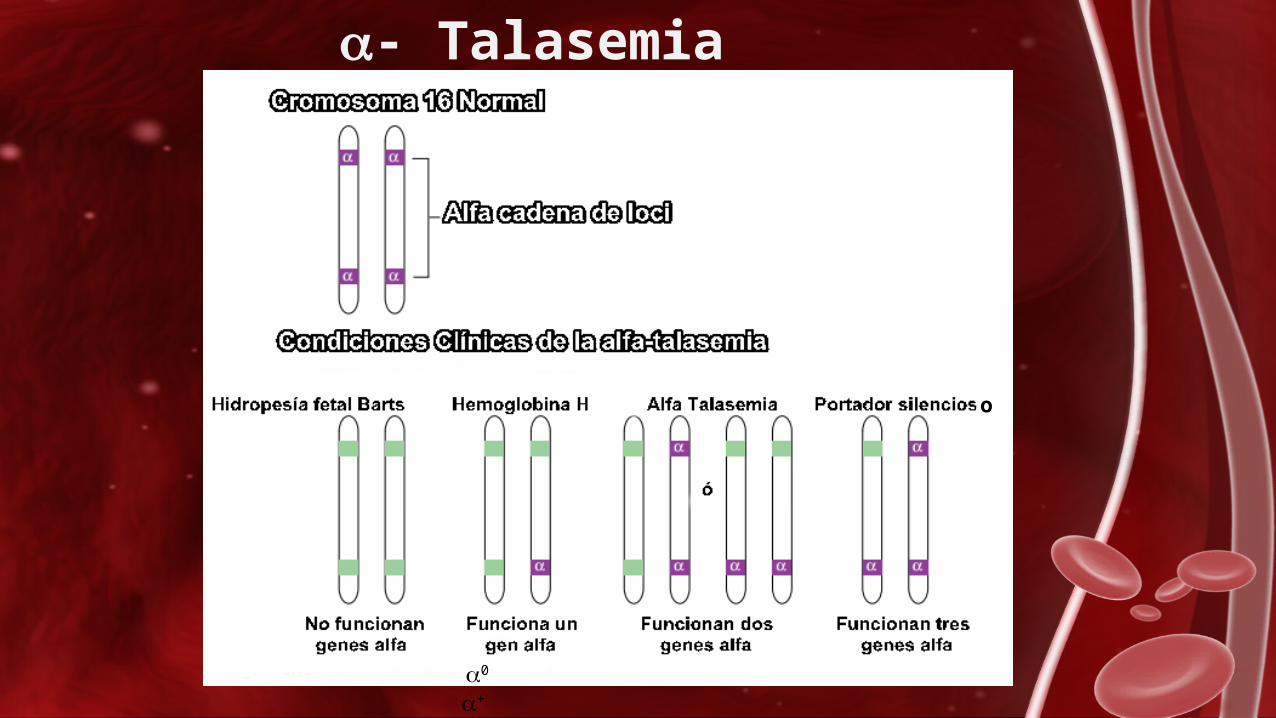
Alfa-TalasemiaLa alfa-talasemia es una hemoglobinopatía hereditaria caracterizada por un fallo en
la síntesis de las cadenas de globina-alfa que da lugar a un cuadro clínico variable dependiendo del número de alelos afectados.
La alfa talasemia no afecta el crecimiento, desarrollo, inteligencia, ó la habilidad de aprendizaje. No se puede distinguir si alguien tiene alfa talasemia por sólo mirarlo.
α Talasemia portador saliente
No son enfermedades y no le causará enfermedad en alguna forma. No se espera que los individuos con portador silente tengan fatiga ó poca energía (Asintomático)
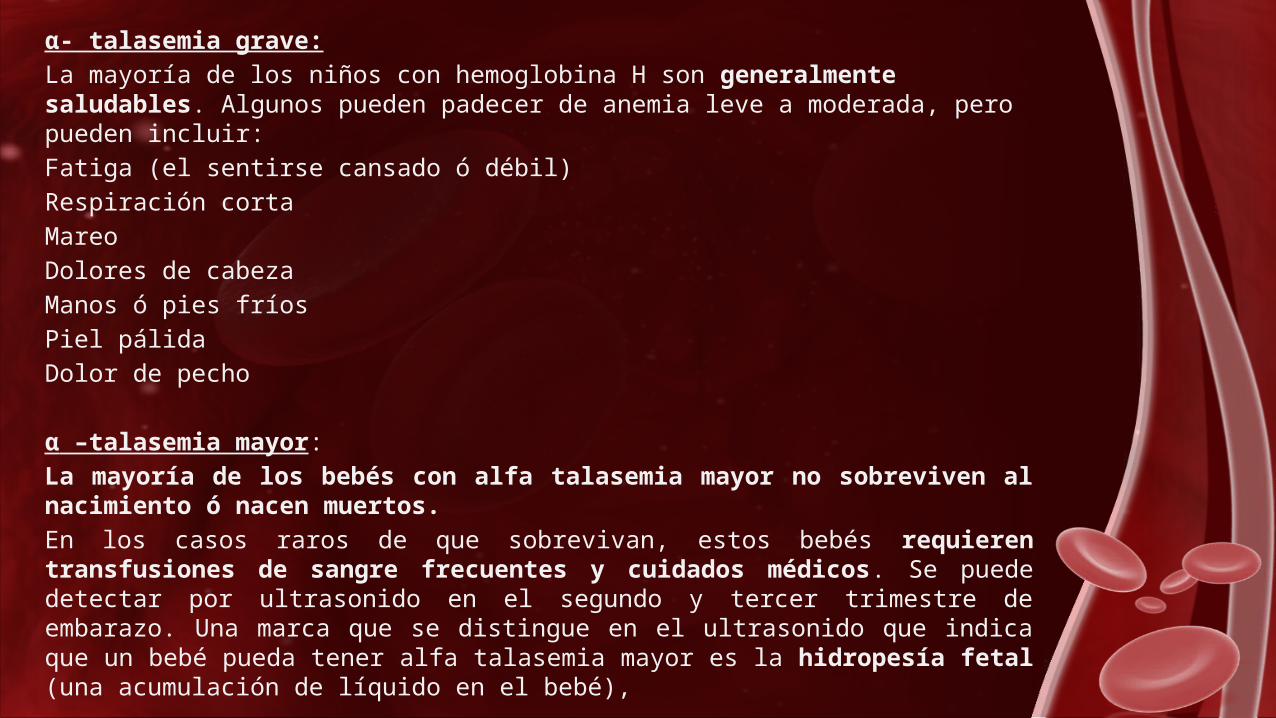
α- talasemia grave:
La mayoría de los niños con hemoglobina H son generalmente saludables. Algunos pueden padecer de anemia leve a moderada, pero pueden incluir:
Fatiga (el sentirse cansado ó débil)
Respiración corta
Mareo
Dolores de cabeza
Manos ó pies fríos
Piel pálida
Dolor de pecho
α –talasemia mayor:
La mayoría de los bebés con alfa talasemia mayor no sobreviven al nacimiento ó nacen muertos.
En los casos raros de que sobrevivan, estos bebés requieren transfusiones de sangre frecuentes y cuidados médicos. Se puede detectar por ultrasonido en el segundo y tercer trimestre de embarazo. Una marca que se distingue en el ultrasonido que indica que un bebé pueda tener alfa talasemia mayor es la hidropesía fetal (una acumulación de líquido en el bebé),
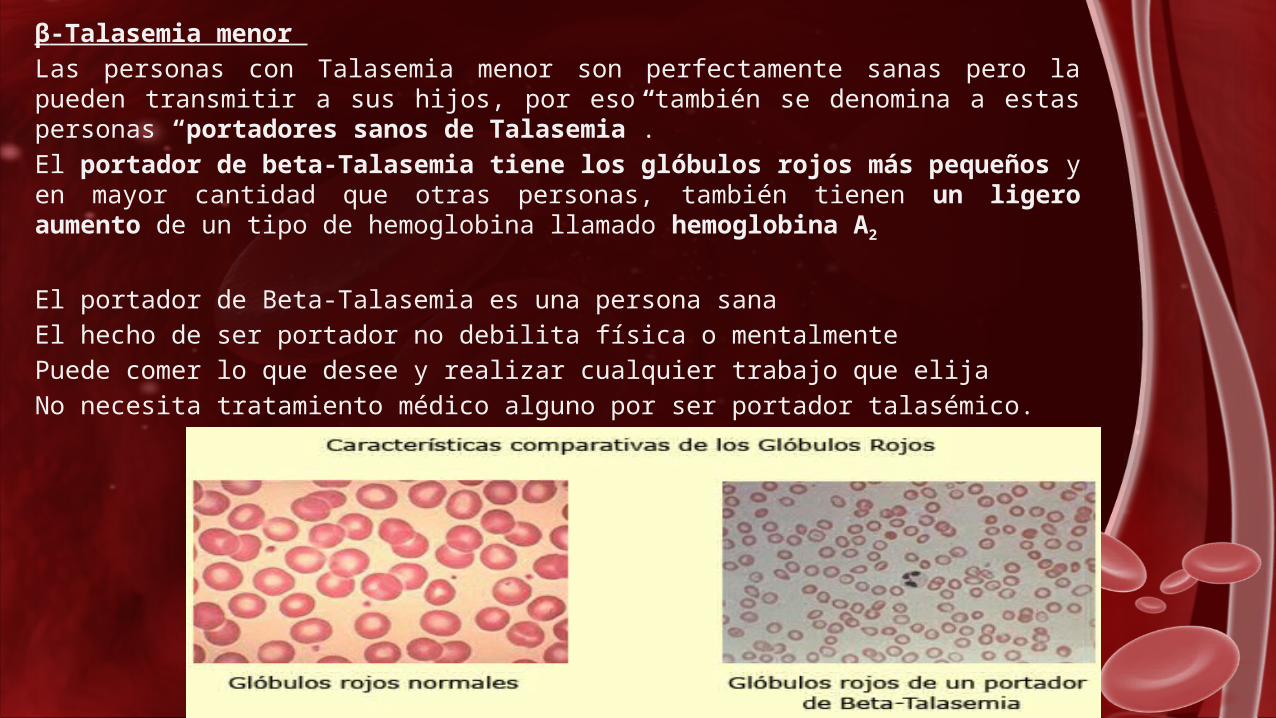
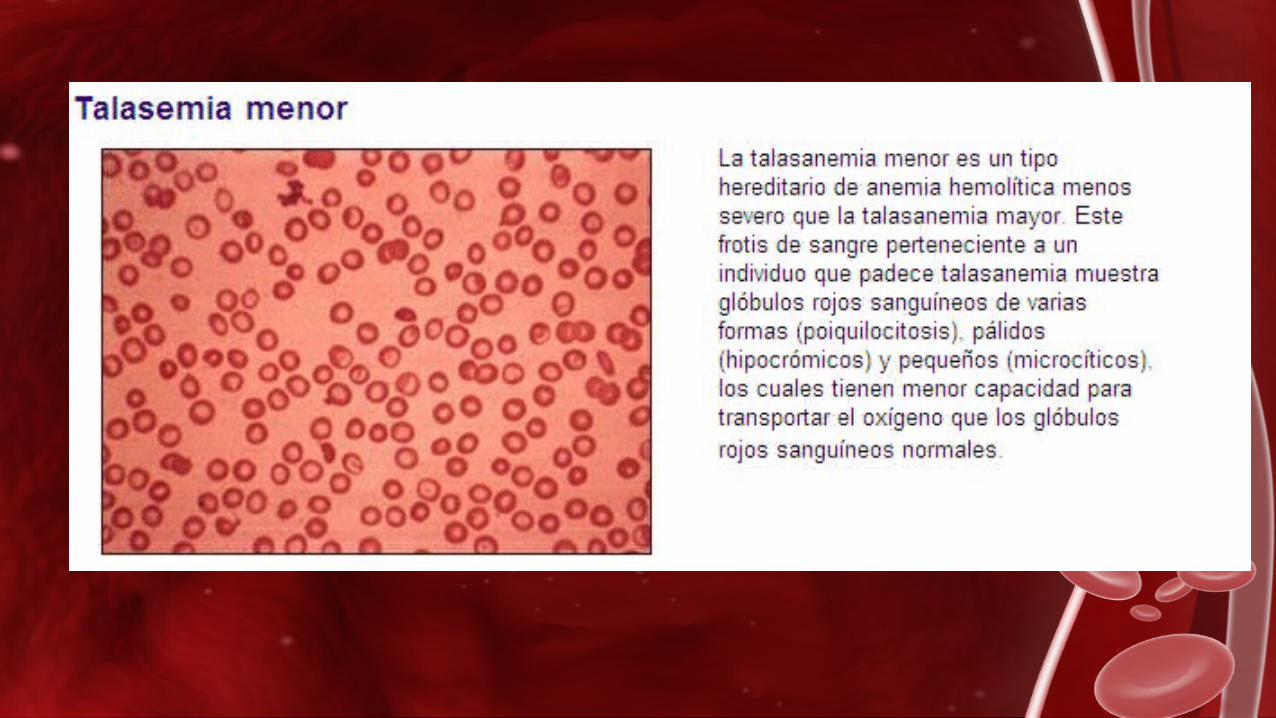
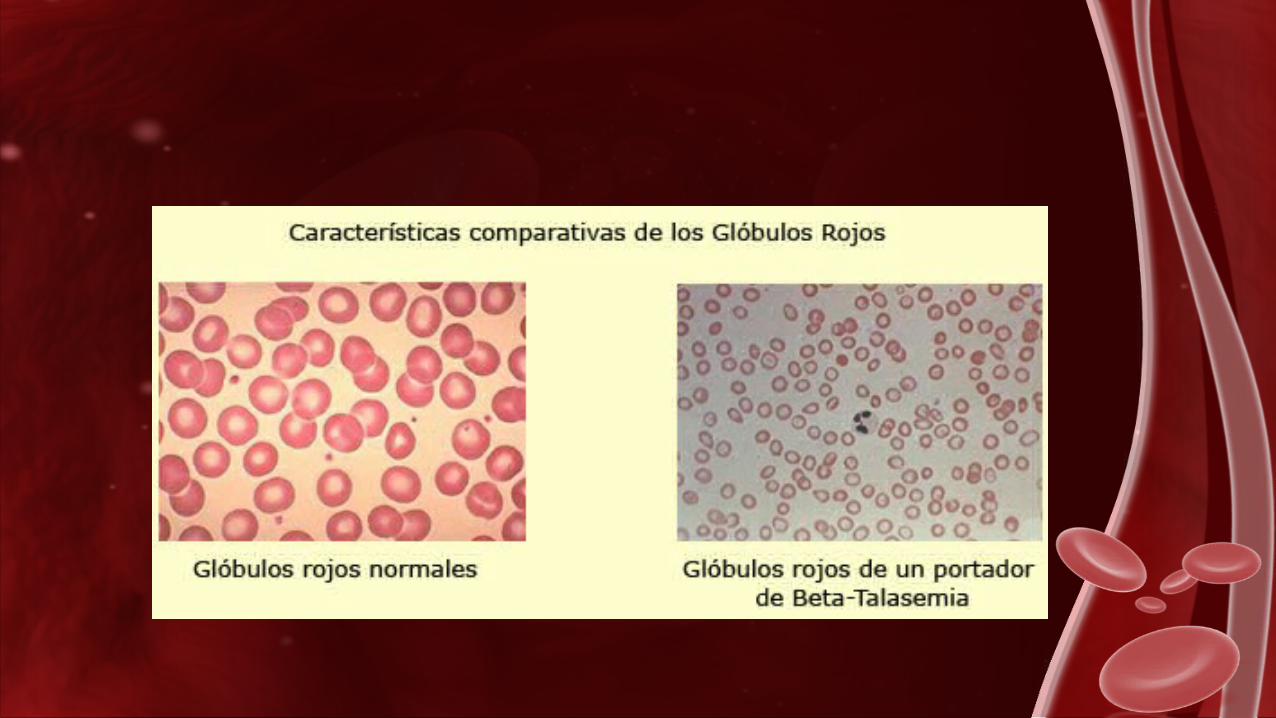
β-Talasemia menor
Las personas con Talasemia menor son perfectamente sanas pero la pueden transmitir a sus hijos, por eso también se denomina a estas personas “portadores sanos de Talasemia”.
El portador de beta-Talasemia tiene los glóbulos rojos más pequeños y en mayor cantidad que otras personas, también tienen un ligero aumento de un tipo de hemoglobina llamado hemoglobina A2
El portador de Beta-Talasemia es una persona sana
El hecho de ser portador no debilita física o mentalmente
Puede comer lo que desee y realizar cualquier trabajo que elija
No necesita tratamiento médico alguno por ser portador talasémico.
Enfermedad de la hemoglobina H
Los rasgos clínicos son muy variables y generalmente se desarrollan en los primeros años de vida, aunque pueden no hacerlo hasta la edad adulta en algunos pacientes.
Los pacientes tienen una anemia microcítica, hipocrómica y hemolítica variable, esplenomegalia (de 50 a 80%)
Y menos frecuentemente hepatomegalia (50%), ictericia leve (20-40%), y cambios esqueléticos

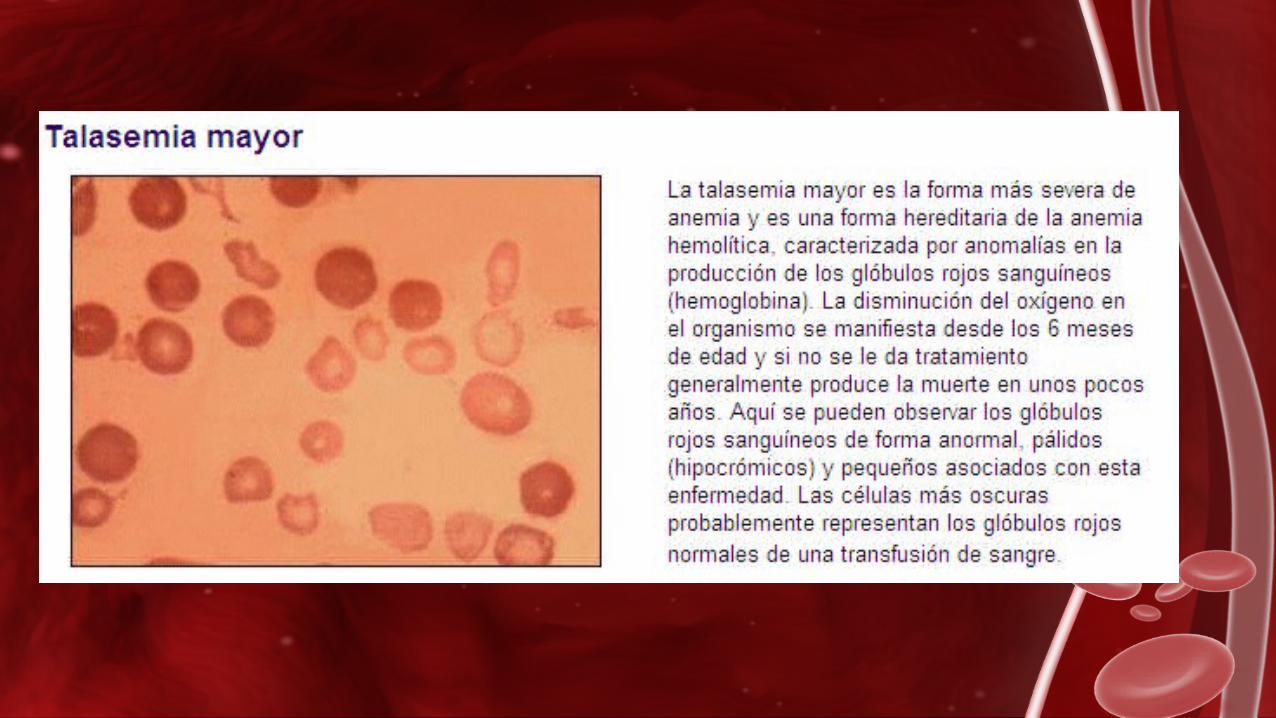
β. –Talasemia mayor:
La disminución del oxígeno en el organismo se manifiesta desde los 6 meses de edad y si no son tratados, llevan una mala calidad de vida. Generalmente, mueren entre el año y los 8 años de edad.
palidez
ictericia y manifestaciones acompañantes como:
diarrea
irritabilidad
episodios febriles recurrentes
distensión abdominal progresiva debida a la hepato/esplenenomegalia
Muestran un importante retraso del crecimiento con escasa musculatura
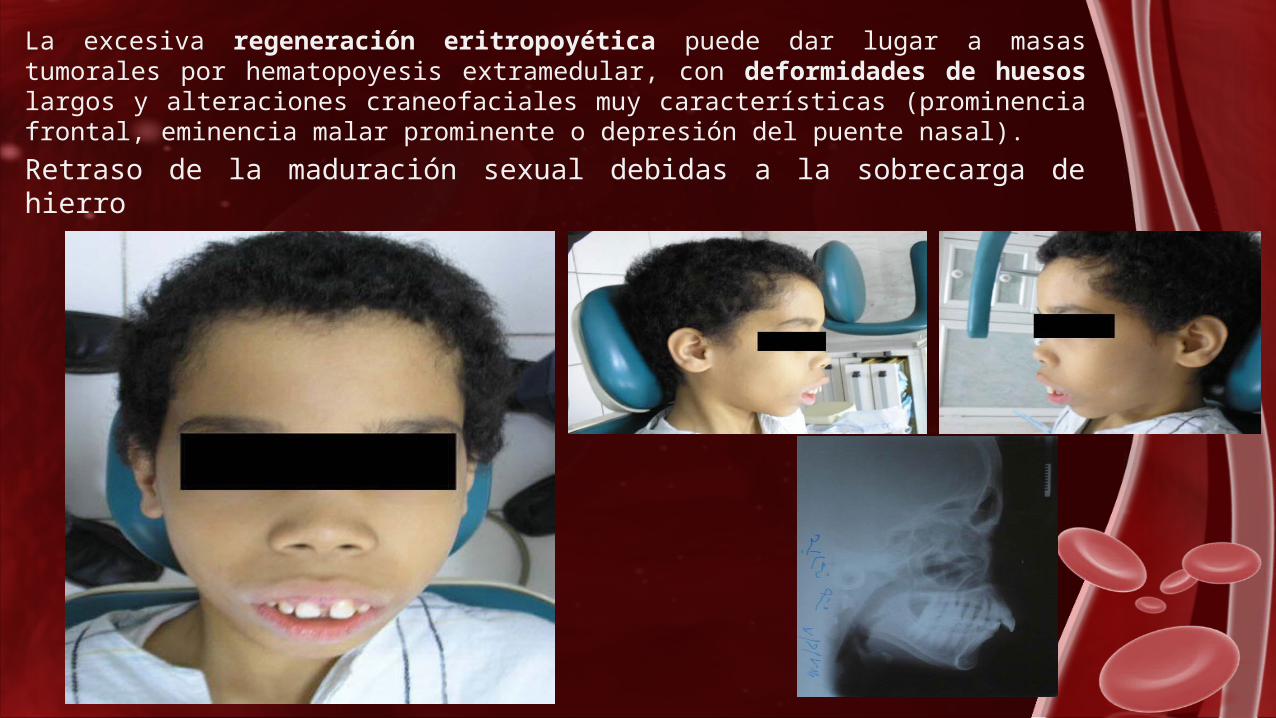
La excesiva regeneración eritropoyética puede dar lugar a masas tumorales por hematopoyesis extramedular, con deformidades de huesos largos y alteraciones craneofaciales muy características (prominencia frontal, eminencia malar prominente o depresión del puente nasal).
Retraso de la maduración sexual debidas a la sobrecarga de hierro

DIAGNÓSTICO HC, FROTIS, ELECTROFORESIS DE HEMOGLOBINA

HEMOGRAMA COMPLETOSe caracteriza por:Niveles bajos de hemoglobina (Hb) (< 7 g/dl)Volumen corpuscular medio (VCM) (50-70 fl)Hemoglobina corpuscular media (HCM) (12-20pg).En cambio, la amplitud de la distribución eritrocitaria (HEM) y el recuento de leucocitos suelen estar elevados. El recuento de plaquetas suele ser normal, excepto en casos de esplenomegalia.

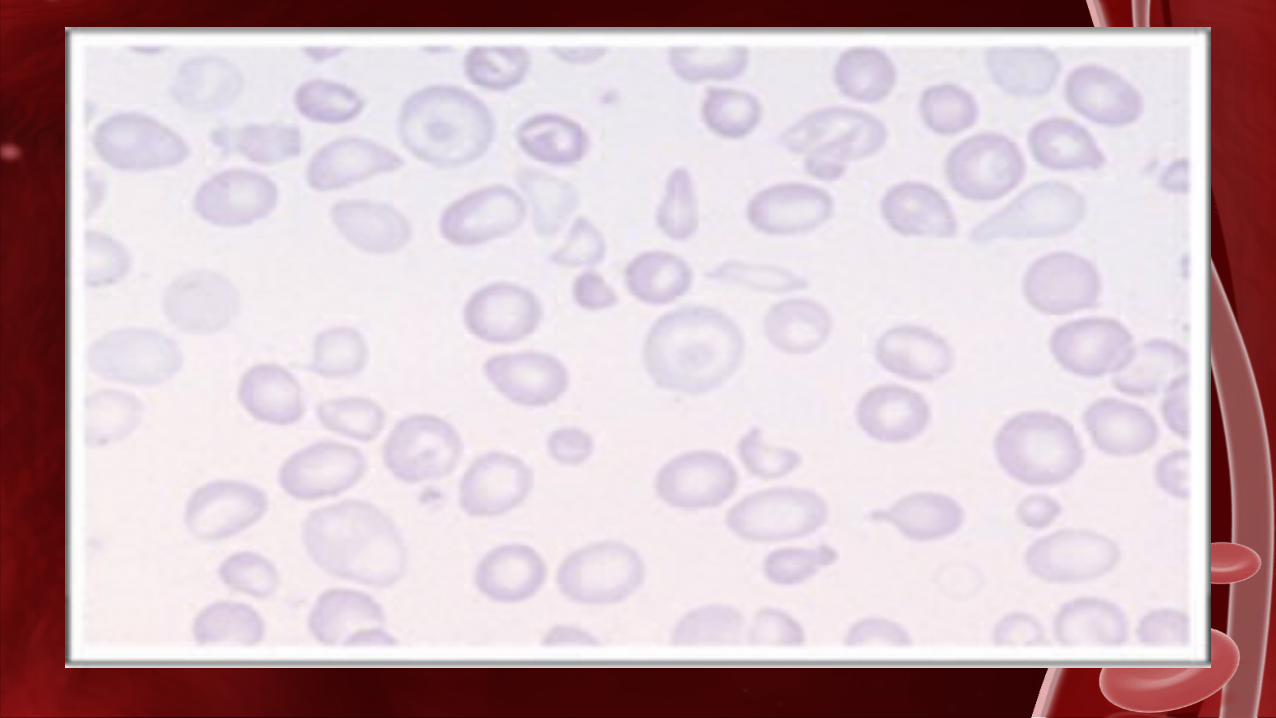
FROTIS•Hipocromía marcada (tinción menos intensa)•Microcitosis (presencia de hematíes anormalmente pequeños)•Macrocitos hipocrómicos (tinción menos intensa, presencia de hematíes anormalmente grandes)•Anisocitosis (variación de tamaño)•Poiquilocitosis (HEM especulados con proyecciones de tamaño y distribución superficial variable)•HEM nucleados•Punteado basófilo•Leucocitos inmaduros ocasionales
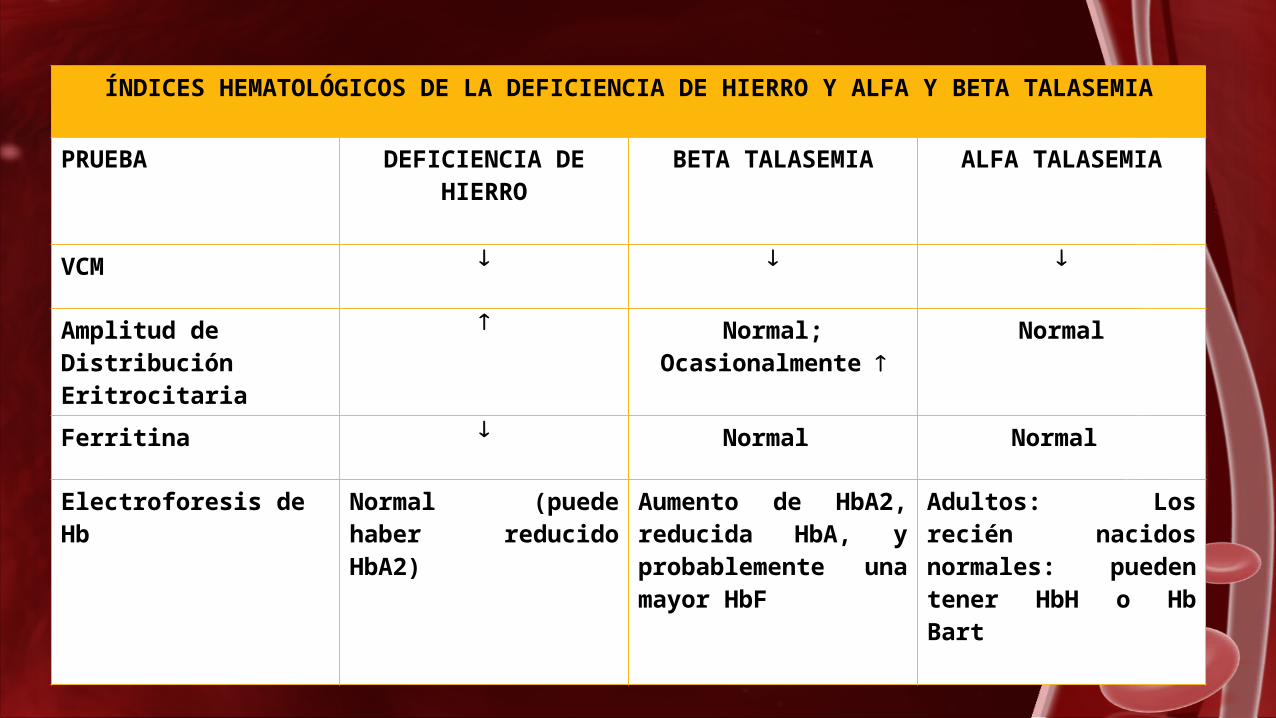
ÍNDICES HEMATOLÓGICOS DE LA DEFICIENCIA DE HIERRO Y ALFA Y BETA TALASEMIA
PRUEBA DEFICIENCIA DE HIERRO
BETA TALASEMIA ALFA TALASEMIA
VCM
Amplitud de Distribución Eritrocitaria
Normal; Ocasionalmente
Normal
Ferritina Normal Normal
Electroforesis de Hb
Normal (puede haber reducido HbA2)
Aumento de HbA2, reducida HbA, y probablemente una mayor HbF
Adultos: Los recién nacidos normales: pueden tener HbH o Hb Bart

ELECTROFORESIS DE HEMOGLOBINAUn médico suele solicitar una electroforesis de hemoglobina para poder diagnosticar enfermedades (llamadas "hemoglobinopatías") que implican la producción anormal de hemoglobina, como la anemia falciforme y la talasemia.

Electroforesis de Hb
Hemolisado50 ul de eritrocitos lavados450 ul de solución lisante
pH8.4-8.6
Acetato de celulosaGel de Agarosa
Cámara de electroforesis20 min. -200 Voltios
Hb normales corren por sus cargas

Talasemias. Este grupo de enfermedades hematológicas de tipo genético afecta la cantidad y el tipo de hemoglobina producida (por ejemplo, demasiada hemoglobina F y poca hemoglobina A en un niño).

TRATAMIENTO

Dosis diarias de ácido fólico
Transfusiones de sangre (según sea necesario)
Extirpación quirúrgica del bazo (si fuera necesario)
Alfa- Talasemia

Beta- Talasemia mayor
El tratamiento está compuesto por transfusiones de sangre regulares, cada 2 a 4 semanas, durante toda la vida. Pero este tratamiento tiene como consecuencia la acumulación de hierro en el organismo, que de no ser eliminado, causa la muerte antes de los 20 años.El hierro puede eliminarse mediante el tratamiento quelante, utilizando alguno medicamentos.
Deferoxamina
Deferiprona
Trasplantes de medula ósea

Enfermedad de la hemoglobina H
En las formas leves más comunes, los pacientes pueden requerir ocasionalmente una terapia de transfusiones de sangre.
En los casos más graves, se requieren transfusiones regulares.
La esplenectomía debe realizarse solo si existe un hiperesplenismo.
Pueden necesitarse un tratamiento con quelantes de hierro.
Monitorizarse los niveles de ferritina sérica.
Imágenes por resonancia magnética (RM) del hígado.
CASO CLÍNICO

Caso Clínico
Un paciente varón de 7 años de edad acude a la consulta de pediatría para un estudio de hipercolesterolemia familiar.
En el estudio realizado destaca una anemia moderada con un valor de volumen corpuscular
medio (VCM) disminuido.
Ante los hallazgos encontrados se pauta tratamiento de la anemia con hierro oral
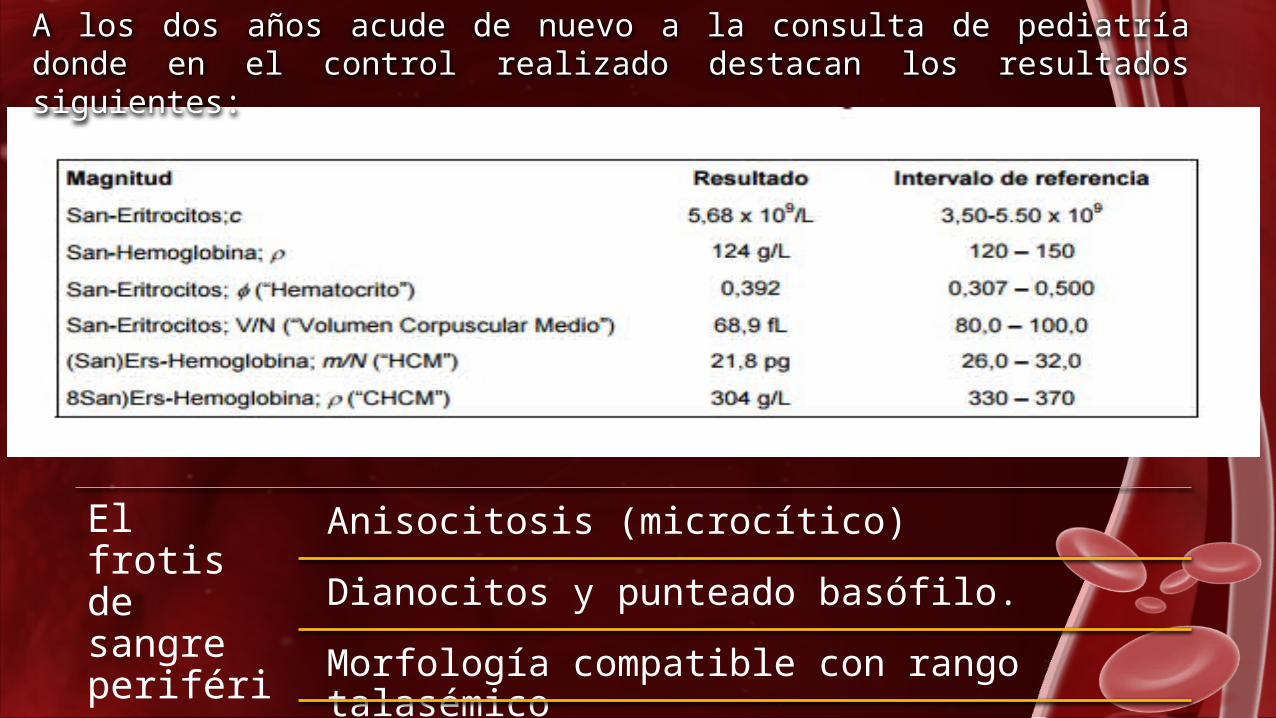
A los dos años acude de nuevo a la consulta de pediatría donde en el control realizado destacan los resultados siguientes:
El frotis de sangre periférica se observa
Anisocitosis (microcítico)
Dianocitos y punteado basófilo.
Morfología compatible con rango talasémico
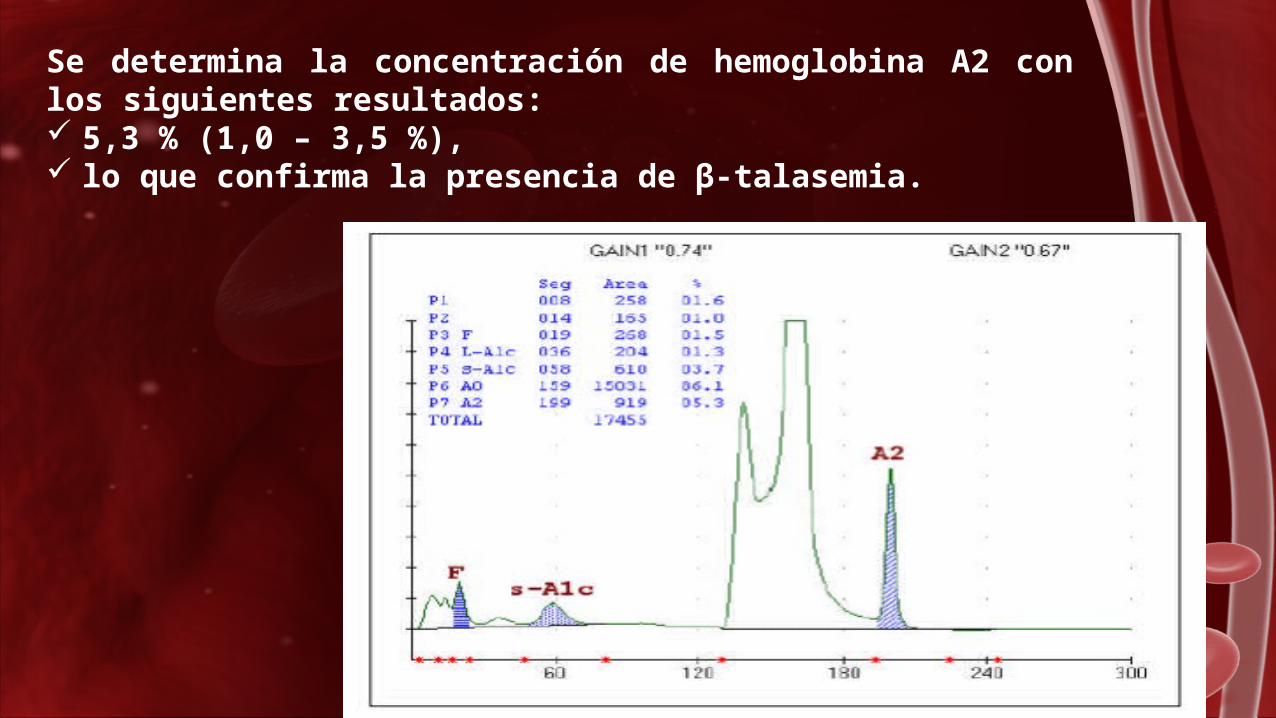
Se determina la concentración de hemoglobina A2 con los siguientes resultados: 5,3 % (1,0 – 3,5 %), lo que confirma la presencia de β-talasemia.

¡GRACIAS!
Bibliografía RODAK. Hematología y Aplicaciones
Clínicas. Segunda Edición. Editorial Panamericana
JAIME, José Carlos. Hematología La Sangre y Sus Enfermedades. Tercera
Edición. Editorial Mc Graw Hill.