universidad central del ecuador facultad de … · derechos morales y patrimoniales del trabajo de...
TRANSCRIPT

UNIVERSIDAD CENTRAL DEL ECUADOR
FACULTAD DE INGENIERÍA QUÍMICA
CARRERA DE INGENIERÍA QUÍMICA
Validación de los métodos de ensayo para la determinación de sulfatos, sulfuros y
fenoles en el Laboratorio Ambiental Environovalab
Trabajo de titulación, modalidad proyecto técnico para la obtención del título de
Ingeniera Química
Autor: Bertha Estefanía Mena Flores
Tutor: Ing. Washington Ruíz
QUITO
2018

iii
DERECHOS DE AUTOR
Yo, BERTHA ESTEFANÍA MENA FLORES, en calidad de autor y titular de los
derechos morales y patrimoniales del trabajo de titulación, VALIDACIÓN DE LOS
MÉTODOS DE ENSAYO PARA LA DETERMINACIÓN DE SULFATOS,
SULFUROS Y FENOLES EN EL LABORATORIO AMBIENTAL
ENVIRONOVALAB, modalidad Proyecto Técnico, de conformidad con el Art. 114 del
CÓDIGO ORGÁNICO DE LA ECONOMÍA SOCIAL DE LOS CONOCIMIENTOS,
CREATIVIDAD E INNOVACIÓN, concedo a favor de la Universidad Central del
Ecuador una licencia gratuita, intransferible y no exclusiva para el uso no comercial de la
obra, con fines estrictamente académicos. Conservo a mi favor todos los derechos de autor
sobre la obra, establecidos en la normativa citada.
Asimismo, autorizo a la Universidad Central del Ecuador para que realice la digitalización
y publicación de este trabajo de titulación en el repositorio virtual, de conformidad a lo
dispuesto en el Art. 144 de la Ley Orgánica de Educación Superior.
El autor declara que la obra objeto de la presente autorización es original en su forma de
expresión y no infringe el derecho de autor de terceros, asumiendo la responsabilidad por
cualquier reclamación que pudiera presentarse por esta causa y liberando a la Universidad
de toda responsabilidad.
Firma:
________________________
Bertha Estefanía Mena Flores
CC. 1724802556

iv
APROBACIÓN DEL TUTOR
Yo, WASHINGTON POLIVIO RUÍZ LÓPEZ en calidad de tutor del trabajo de
titulación, modalidad proyecto técnico, cuyo título es VALIDACIÓN DE LOS
MÉTODOS DE ENSAYO PARA LA DETERMINACIÓN DE SULFATOS,
SULFUROS Y FENOLES EN EL LABORATORIO AMBIENTAL
ENVIRONOVALAB, elaborado por la estudiante MENA FLORES BERTHA
ESTEFANÍA, para la obtención del título de Ingeniera Química, considero que el mismo
reúne los requisitos y méritos necesarios en el campo metodológico y en el campo
epistemológico, para ser sometido a la evaluación por parte del jurado examinador que se
designe, por lo que lo APRUEBO, a fin de que el trabajo sea habilitado para continuar
con el proceso de titulación determinado por la Universidad Central del Ecuador.
En la ciudad de Quito, a los 23 días del mes de Abril de 2018.
_________________________
Firma del Tutor
ING. WASHINGTON POLIVIO RUÍZ LÓPEZ
CC.1711337939

v
A Dios, a mis
padres y a mi
hermana por
haberme
brindado su
apoyo
incondicional
en esta etapa.

vi
AGRADECIMIENTOS
La Autora expresa sus agradecimientos a:
A mis padres, por estar presentes en los momentos más hermosos y también en los más
difíciles de mi vida, en especial a mi madre por ser mi motor, por sus consejos y sus
palabras de aliento que me permitieron llenarme de valor y de fuerzas para concluir con
esta etapa de mi vida. Gracias a los dos por nunca dejarme sola y hacerme saber que
puedo confiar en ustedes sin importar las circunstancias.
A mi hermana Dayana, por ser mi ejemplo a seguir y mi cómplice durante toda mi vida,
apoyándome en mis decisiones y brindándome toda su comprensión y cariño cuando se
me presentaron dificultades. Gracias por nunca haber soltado mi mano desde que
nacimos. Siempre juntas ya lo sabes. No hay mejor amiga que una hermana y no hay
mejor hermana que tú.
A mi gran amiga Priscila con quien compartí demasiadas experiencias dentro y fuera de
las aulas, sobre todo por estar junto a mí cuando nadie más estuvo, estaré siempre
eternamente agradecida con la vida y con Dios por haber cruzado nuestros caminos.
Ingeniero Washington Ruíz, tutor de mi tesis de grado, por su valioso tiempo y
orientación que me sirvieron de gran ayuda para la realización de este trabajo.
A la Universidad Central del Ecuador, por abrirme sus puertas y haber permitido mi
formación profesional.
Al Laboratorio Ambiental Environovalab por permitirme realizar la parte experimental
del presente trabajo de titulación.

vii
CONTENIDO
pág.
LISTA DE TABLAS ...................................................................................................... xii
LISTA DE FIGURAS ....................................................................................................xiv
LISTA DE GRÁFICOS ................................................................................................... xv
LISTA DE ANEXOS .....................................................................................................xvi
RESUMEN ................................................................................................................... xvii
ABSTRACT ................................................................................................................ xviii
INTRODUCCIÓN ............................................................................................................. 1
1. MARCO TEÓRICO ..................................................................................................... 3
1.1. Agua. ......................................................................................................................... 3
1.1.1. Composición del agua. ........................................................................................... 3
1.2. Propiedades del agua. ................................................................................................ 4
1.2.1. Propiedades físicas. ................................................................................................ 4
1.2.2. Propiedades químicas. ............................................................................................ 4
1.3. Sulfatos. ..................................................................................................................... 5
1.3.1. Características generales y origen. ......................................................................... 5
1.3.2. Aplicaciones. .......................................................................................................... 5
1.4. Sulfuros. ..................................................................................................................... 5
1.4.1. Características generales y origen. ......................................................................... 5
1.4.2. Aplicaciones. .......................................................................................................... 6
1.5. Fenoles. ...................................................................................................................... 7

viii
1.5.1. Características generales y origen. ......................................................................... 7
1.5.2. Propiedades físicas. ................................................................................................ 7
1.5.3. Propiedades químicas. ............................................................................................ 7
1.6. Espectro electromagnético. ........................................................................................ 7
1.6.1. Luz ultravioleta. ...................................................................................................... 8
1.7. Espectrofotómetro. .................................................................................................... 8
1.8. Estadística básica. ...................................................................................................... 9
1.8.1. Población. ............................................................................................................... 9
1.8.2. Muestra. .................................................................................................................. 9
1.8.3. Media aritmética. .................................................................................................... 9
1.8.4. Desviación estándar. ............................................................................................... 9
1.8.5. ANOVA. ............................................................................................................... 10
1.9. Validación e incertidumbre. .................................................................................... 10
1.9.1. Principio de la validación. .................................................................................... 10
1.9.2. Norma Internacional ISO/IEC 17025:2017. ......................................................... 10
1.9.3. Establecimiento del alcance y objetivos de la validación..................................... 11
1.9.4. Exactitud. .............................................................................................................. 11
1.9.5. Veracidad. ............................................................................................................. 11
1.9.6. Precisión. .............................................................................................................. 11
1.9.7. Reproducibilidad. ................................................................................................. 12
1.9.8. Repetibilidad. ........................................................................................................ 12
1.9.9. Límite de detección. ............................................................................................. 12
1.9.10. Límite de cuantificación. .................................................................................... 12
1.9.11. Intervalo de trabajo. ............................................................................................ 12
1.9.12. Incertidumbre...................................................................................................... 12
1.9.12.1. Tipos de evaluación de incertidumbre. ........................................................... 12
1.9.12.2. Incertidumbre combinada. ............................................................................... 13

ix
1.9.12.3. Incertidumbre expandida. ................................................................................ 13
1.9.12.4. Factor de cobertura. ......................................................................................... 13
2. DISEÑO EXPERIMENTAL Y ESTADÍSTICO ....................................................... 14
2.1. Fijación de objetivos para los diferentes analitos y selección de los parámetros
de validación. ................................................................................................................... 14
2.2. Diseño experimental. ............................................................................................... 18
2.3. Diseño experimental por tipo de ensayo. ................................................................ 19
2.4. Tratamiento estadístico. ........................................................................................... 23
2.5. Puesta a punto. ......................................................................................................... 25
2.5.1. Determinación cuantitativa de sulfatos. ............................................................... 25
2.5.1.1. Limitaciones de la determinación. ..................................................................... 26
2.5.1.2. Interferencias. .................................................................................................... 26
2.5.2. Determinación cuantitativa de sulfuros. ............................................................... 26
2.5.2.1. Limitaciones de la determinación. ..................................................................... 27
2.5.2.2. Interferencias. .................................................................................................... 27
2.5.3. Determinación cuantitativa de fenoles. ................................................................ 27
2.5.3.1. Limitaciones de la determinación. ..................................................................... 28
2.5.3.2. Interferencias. .................................................................................................... 28
2.6. Verificación de la balanza analítica. ........................................................................ 28
2.7. Calibración del espectrofotómetro........................................................................... 29
2.8. Preparación de las muestras fortificadas utilizadas en la validación. ...................... 29
2.9. Procedimientos de ensayo. ...................................................................................... 30
2.9.1. Procedimiento de ensayo para sulfatos. ................................................................ 30
2.9.2. Procedimiento de ensayo para sulfuros. ............................................................... 30
2.9.3. Procedimiento de ensayo para fenoles. ................................................................ 31
2.10. Datos experimentales. ............................................................................................ 32
2.10.1. Datos obtenidos de la función respuesta durante la puesta a punto.................... 32

x
3. VALIDACIÓN ........................................................................................................... 35
3.1. Planteamiento de la validación. ............................................................................... 35
3.2. Datos para la validación. ......................................................................................... 36
4. CÁLCULOS ............................................................................................................... 39
4.1. Cálculo para la preparación de las muestras fortificadas. ....................................... 39
4.1.1. Cálculo modelo para la muestra fortificada de 7 mg/L en sulfatos. ..................... 39
4.2. Determinación de la linealidad de la función de respuesta. .................................... 40
4.3. Cálculo de los parámetros de validación. ................................................................ 45
4.3.1. Límite de detección (LD). .................................................................................... 45
4.3.2. Límite de cuantificación (LC). ............................................................................. 45
4.3.3. Veracidad. ............................................................................................................. 45
4.3.3.1. Veracidad para muestras fortificadas. ............................................................... 45
4.3.3.2. Veracidad para material de referencia certificado. ............................................ 46
4.3.4. Precisión. .............................................................................................................. 47
4.3.4.1. Cálculo modelo para la precisión para determinar sulfatos en aguas para la
concentración de 7 mg/L. ................................................................................................ 47
4.3.5. Cálculo de la incertidumbre. ................................................................................. 52
4.3.5.1. Cálculo de la incertidumbre estándar por reproducibilidad............................... 53
4.3.5.2. Cálculo de la incertidumbre estándar del equipo............................................... 54
4.3.5.3. Cálculo de la incertidumbre debida al cronómetro. ........................................... 65
4.3.5.4. Cálculo de la incertidumbre debida a la probeta. .............................................. 67
4.3.5.5. Correcciones no aplicadas ................................................................................. 70
4.3.6. Cálculo de la veracidad. ....................................................................................... 70
5. RESULTADOS .......................................................................................................... 72
5.1. Resultados de validación de sulfatos en aguas. ............................................. 72
5.2. Resultados de validación de sulfuros en aguas. ............................................ 73

xi
5.3. Resultados de validación de fenoles en aguas. ........................................................ 74
6. DISCUSIÓN ............................................................................................................... 75
6.1. Discusión general. ................................................................................................... 75
6.2. Sulfatos en aguas. .................................................................................................... 76
6.3. Sulfuros en aguas. .................................................................................................... 77
6.4. Fenoles en aguas. ..................................................................................................... 78
7. CONCLUSIONES ...................................................................................................... 80
7.1. Conclusión general. ................................................................................................. 80
7.2. Sulfatos en aguas. .................................................................................................... 80
7.3. Sulfuros en aguas. .................................................................................................... 81
7.4. Fenoles en aguas. ..................................................................................................... 82
8. RECOMENDACIONES ............................................................................................. 83
CITAS BIBLIOGRÁFICAS ........................................................................................... 84
BIBLIOGRAFÍA ............................................................................................................. 88
ANEXOS ....................................................................................................................... 90

xii
LISTA DE TABLAS
pág.
Tabla 1. Datos físicos del agua a presión normal=1 atm. ................................................. 4
Tabla 2. Límites permisibles según los organismos nacionales. .................................... 14
Tabla 3. Fijación de objetivos para el método de sulfatos.............................................. 15
Tabla 4. Fijación de objetivos para el método de sulfuros. ............................................ 16
Tabla 5. Fijación de objetivos para el método de fenoles. ............................................. 17
Tabla 6. Diseño experimental para el ensayo de sulfatos. .............................................. 19
Tabla 7. Diseño experimental para el ensayo de sulfuros. ............................................. 20
Tabla 8. Diseño experimental para el ensayo de fenoles. ............................................... 22
Tabla 9. Tabla de datos para el ANOVA de un factor. .................................................. 24
Tabla 10. Ecuaciones para el ANOVA de un factor....................................................... 25
Tabla 11. Datos obtenidos de las rectas de calibración construidas durante la puesta
a punto del método de sulfatos. ...................................................................................... 32
Tabla 12. Datos obtenidos de las rectas de calibración construidas durante la puesta
a punto del método de sulfuros. ...................................................................................... 33
Tabla 13. Datos obtenidos de las rectas de calibración construidas durante la puesta
a punto del método de fenoles. ....................................................................................... 34
Tabla 14. Datos primarios para validación de sulfatos en aguas, concentraciones
obtenidas a diferentes niveles. ........................................................................................ 36
Tabla 15. Datos primarios para validación de sulfuros en aguas, concentraciones
obtenidas a diferentes niveles. ........................................................................................ 37
Tabla 16. Datos primarios para validación de fenoles en aguas, concentraciones
obtenidas a diferentes niveles. ........................................................................................ 37
Tabla 17. Concentración inicial de la muestra para sulfatos. ......................................... 39
Tabla 18. Concentración a añadir a la muestra para sulfatos. ........................................ 40
Tabla 19. Fórmulas para el cálculo de datos estadísticos. .............................................. 41

xiii
Tabla 20. Cálculos estadísticos de la puesta a punto. ..................................................... 42
Tabla 21. Intervalo de trabajo para pendiente e intercepto............................................. 43
Tabla 22. Rectas de calibración para la validación. ....................................................... 43
Tabla 23. Datos estadísticos de las curvas de calibración utilizadas para la
validación. ...................................................................................................................... 44
Tabla 24. Análisis para fijar la recta de calibración. ...................................................... 46
Tabla 25. Datos reportados por día. ................................................................................ 47
Tabla 26. Valor medio de las determinaciones............................................................... 48
Tabla 27. Incertidumbre estándar por reproducibilidad. ................................................ 54
Tabla 28. Incertidumbre estándar del MRI. .................................................................... 64
Tabla 29. Incertidumbre estándar debida a la lectura de las concentraciones en el
espectrofotómetro. .......................................................................................................... 65
Tabla 30. Incertidumbre estándar del equipo por niveles. .............................................. 65
Tabla 31. Cálculo de incertidumbre por niveles para sulfatos en aguas. ........................ 70
Tabla 32. Veracidad con MRC para sulfatos. ................................................................. 71
Tabla 33. Veracidad con MRC de sulfatos en aguas. ..................................................... 72
Tabla 34. Precisión, veracidad e incertidumbre de sulfatos en aguas. ........................... 72
Tabla 35. Veracidad con MRC de sulfuros en aguas. .................................................... 73
Tabla 36. Precisión, veracidad e incertidumbre de sulfuros en aguas. ........................... 73
Tabla 37. Veracidad con MRC de fenoles en aguas. ...................................................... 74
Tabla 38. Precisión, veracidad e incertidumbre de fenoles en aguas. ............................ 74

xiv
LISTA DE FIGURAS
pág.
Figura 1. Modelo de la molécula del agua. ...................................................................... 3
Figura 2. Síntesis de fenol a escala industrial por el método de Dow Chemical.............. 7
Figura 3. Espectro electromagnético. ............................................................................... 8
Figura 4. Partes básicas de un espectrofotómetro de absorción. ...................................... 9
Figura 5. Diseño experimental para la validación de métodos de ensayo. ..................... 18
Figura 6. Reacción de dimetil- para- fenilendiamina con el cloruro férrico y sulfuro
de hidrógeno. .................................................................................................................. 27
Figura 7. Reacción de 4-aminoantipirina con fenol o compuesto fenólico. ................... 28
Figura 8. Diagrama de flujo del planteamiento de la validación. ................................... 35
Figura 9. Diagrama de Ishikawa para incertidumbre del método................................... 52
Figura 10. Diagrama de Ishikawa para incertidumbre del material de referencia
interno. ............................................................................................................................ 52

xv
LISTA DE GRÁFICOS
pág.
Gráfico 1. Absorbancia en función de la concentración de la puesta a punto ............... 41
Gráfico 2. Absorbancia en función de la concentración para la validación .................. 44

xvi
LISTA DE ANEXOS
pág.
ANEXO A. Declaración del método validado para sulfatos en aguas .......................... 91
ANEXO B. Declaración del método validado para sulfuros en aguas .......................... 94
ANEXO C. Declaración del método validado para fenoles en aguas ............................ 97
ANEXO D. Cálculos de Excel para el método de sulfatos .......................................... 100
ANEXO E. Cálculos de Excel para el método de sulfuros .......................................... 107
ANEXO F. Cálculos de Excel para el método de fenoles ........................................... 113
ANEXO G. Reporte fotográfico del equipo ................................................................. 119

xvii
Validación de los métodos de ensayo para la determinación de sulfatos, sulfuros y
fenoles en el Laboratorio Ambiental Environovalab
RESUMEN
Se validaron los métodos de ensayo por espectrofotometría UV-VIS para la
determinación de sulfatos en aguas residuales y de consumo, sulfuros en aguas residuales
y fenoles en aguas residuales, en el Laboratorio Ambiental Environovalab de acuerdo a
la aplicación de la norma ISO/IEC 17025:2017 y a los procedimientos definidos por los
Métodos Estándar.
En primer lugar se fijaron los objetivos, se realizó la puesta a punto para cada analito y
se determinó la función respuesta de los mismos. Se realizó el diseño experimental bajo
condiciones de repetibilidad y reproducibilidad para diferentes concentraciones y se
obtuvieron los resultados mediante el tratamiento estadístico de los datos (ANOVA) y el
cálculo de la incertidumbre (k=2).
Los intervalos validados son: para sulfatos en aguas residuales y de consumo de 7 a 48
mg/L; sulfuros en aguas residuales de 0,28 a 1,00 mg/L y fenoles en aguas residuales de
0,10 a 0,20 mg/L, rangos que se encuentran dentro de los criterios establecidos en la
normativa nacional vigente. Por lo cual, estos métodos son aptos para ser sometidos al
proceso de acreditación conforme lo establece la norma ISO/IEC 17025:2017.
PALABRAS CLAVES: / VALIDACIÓN/ MÉTODOS DE ENSAYO/ SULFATOS/
SULFUROS/ FENOLES/ AGUA POTABLE/ AGUAS RESIDUALES/

xviii
Validation of the test methods for the determination of sulfates, sulfides and
phenols in Environovalab Environmental Laboratory
ABSTRACT
The test methods were validated by UV-VIS spectrophotometry for the determination of
sulfates in wastewater and water for consumption, sulfides in wastewater and phenols in
wastewater in Environovalab Environmental Laboratory according to the application of
ISO/IEC 17025:2017 and to the procedures defined by the Standard Methods.
In the first place, the objectives were set, the set-up was carried out for each analyte and
the response function was determined. The experimental design was implemented under
conditions of repeatability and reproducibility for different concentrations and the results
were obtained by means of the statistical treatment of the data (ANOVA) and the
calculation of the uncertainty (k = 2).
The validated intervals are: for sulfates in wastewater and water for consumption from 7
to 48 mg/L; sulfides in wastewater from 0,28 to 1,00 mg/L and phenols in wastewater
from 0,10 to 0,20 mg/L, these ranges are within the criteria established in the current
national regulation. Therefore, these methods are suitable to be submitted to the
accreditation process as established by ISO/IEC 17025:2017.
KEY WORDS: / VALIDATION/ TEST METHODS/ SULFATES/ SULFIDES/
PHENOLS/ WATER FOR CONSUMPTION/ WASTEWATER/

1
INTRODUCCIÓN
Una de las principales preocupaciones para las distintas entidades de regulación ha sido
la calidad de agua potable que llega al consumidor final, así como también el agua
residual procedente de las actividades de empresas públicas y privadas. Por tal causa,
la Norma ISO/IEC 17025 fue diseñada para que se utilice por los laboratorios de ensayo
y calibración cuando se desarrollen los sistemas de gestión para sus actividades de
calidad, administrativas y técnicas. Al trabajar bajo los estándares de esta norma se
reconoce su competencia técnica y la validez de los resultados, se responde a las
exigencias de los organismos nacionales e internacionales y se obtiene credibilidad ante
los clientes.
Actualmente, en el Ecuador existe un gran número de laboratorios que buscan
implementar un sistema de gestión de calidad, con el fin de que sus resultados sean
técnicamente confiables y se pueda mejorar la prestación de servicios al público,
cumpliendo con los requerimientos de los parámetros para la acreditación que solicita el
Servicio de Acreditación Ecuatoriana (SAE).
El propósito inicial del Laboratorio Ambiental Environovalab al ser una empresa que está
incursionando en el mercado, es incluir la prestación de servicios en la matriz agua, por
lo cual ha visto la necesidad de implementar la validación de métodos de ensayo para la
determinación de sulfatos, en aguas residuales y de consumo, sulfuros en aguas residuales
y fenoles en aguas residuales, a su vez el laboratorio busca la validación de los atributos
básicos de los métodos estadísticos que aseguren que los análisis realizados son veraces
y que demuestren que los mismos están estandarizados acorde con las políticas de calidad.
La validación de métodos analíticos se basa en la reglamentación actual para el control
de calidad del agua, además se utiliza una técnica ya establecida por los organismos
nacionales tales como el SAE e internacionales como la ISO.

2
El presente trabajo tiene por objetivos específicos:
Validar métodos analíticos por espectrofotometría UV-VIS para la determinación de
varios parámetros en la matriz agua, basándose en las reglamentaciones vigentes.
Documentar la información necesaria de los distintos métodos de análisis que permita
llevar a cabo la determinación en la matriz agua.
Determinar los criterios de aceptación y rechazo a través del tratamiento estadístico de
los datos obtenidos.
Para lo cual, inicialmente es importante realizar una selección de los procedimientos
estándar más adecuados para cada uno de los métodos de ensayo antes mencionados,
entonces de acuerdo a los resultados que se obtienen se hacen las respectivas
modificaciones pertinentes para asegurar la conformidad con la normativa.
Se fijan entonces los objetivos y se seleccionan los parámetros de la validación. El cálculo
adecuado de los parámetros de rendimiento para cada método requiere de un diseño
experimental acorde con las necesidades del laboratorio y del alcance de la validación.
Estableciendo un estimado de la incertidumbre de medida y teniendo pleno conocimiento
de los fenómenos que tienen lugar durante la validación de cada método de ensayo, se
fijará el grado de confianza en los resultados aportados, con lo que se puede determinar
su factibilidad y verificar que los mismos son aptos para su aplicación en aguas limpias
(en el caso de sulfatos) y residuales bajo condiciones específicas con fines de
cumplimiento de las normas ambientales nacionales vigentes.
Los métodos analíticos que se validaron fueron: sulfatos en aguas residuales y de
consumo, sulfuros en aguas residuales y fenoles en aguas residuales. A partir del diseño
experimental basado en la normativa vigente, en cada uno de los ensayos se cumplió con
los objetivos planteados inicialmente para cada analito y su matriz, tales como coeficiente
de variación de repetibilidad y reproducibilidad (≤ 10% ó ≤ 15%); veracidad (80 < % R
< 120) e incertidumbre (U ≤ 30 %), mismos que le permitirán al laboratorio garantizar la
calidad de los procedimientos.

3
1. MARCO TEÓRICO
1.1. Agua.
El agua es uno de los constituyentes principales de la naturaleza viva o inanimada que
nos rodea y el cuerpo humano la contiene en un 60 a 75%.
El agua natural presente en el medio ambiente siempre ha experimentado un ciclo
hidrológico por lo que puede contener una gran variedad de impurezas, ésta puede llegar
a ser empleada para procesos industriales, así como también para el consumo humano.
(Rigola Lapeña, 1990)
“Cuando las impurezas representan elementos nocivos para el uso a que va destinada el
agua las denominamos contaminantes.” (Rigola Lapeña, 1990, pág. 11)
1.1.1. Composición del agua. “El agua es una combinación de 88,9% en masa de
oxígeno (𝑂2) y 11,1% en masa de hidrógeno (𝐻2), se forma por combustión del
hidrógeno o de combinaciones hidrogenadas en atmósfera de oxígeno.” (Hopp, 2005, pág.
34)
𝐻2 + 1
2𝑂2 → 𝐻2𝑂 (1)
ℎ𝑖𝑑𝑟ó𝑔𝑒𝑛𝑜 𝑜𝑥í𝑔𝑒𝑛𝑜 𝑎𝑔𝑢𝑎
La molécula del agua vista como un tetraedro irregular con el átomo de oxígeno en su
centro se muestra en la figura (1).
Figura 1. Modelo de la molécula del agua (Bolaños Chombo, 2003).

4
1.2. Propiedades del agua.
El agua como la biomolécula inorgánica más importante que existe posee como cualquier
compuesto químico propiedades físicas y químicas que se detallan a continuación.
1.2.1. Propiedades físicas. El agua pura es un líquido sin olor, incoloro, insípido y
transparente que en espesores grandes tiende a tomar un color azul. (Hopp, 2005)
El agua es considerada como el disolvente universal por su abundancia y la polaridad de
su molécula, la facilidad de utilización y su amplio intervalo de temperaturas en la cual
permanece líquida.
Tabla 1. Datos físicos del agua a presión normal=1 atm (Hopp, 2005).
Punto de congelación 0ºC
Punto de ebullición 100ºC
Calor de fusión 334,96 kJ/kg (80 kcal/kg)
Calor de vaporización a 100ºC 2256,8 kJ/kg (539 kcal/kg)
Densidad máxima a 4ºC 1 kg/L
Calor de formación -68,3 kcal/mol
Peso molecular 18,015 g/mol
Temperatura crítica 374,2ºC
Presión crítica 218,4 atm
Calor específico 1 kcal/kgºC
1.2.2. Propiedades químicas. “El agua es una combinación pobre en energía y, por ello,
químicamente estable. Inversamente, se debe aportar energía para descomponer el agua
en sus componentes elementales hidrógeno y oxígeno. A causa de su alto contenido
energético, el hidrógeno y el oxígeno son sustancias muy reactivas.” (Hopp, 2005, pág.
34)
“El agua se ioniza ligeramente formando 𝐻+y 𝑂𝐻−. Las concentraciones de estos iones
se encuentran relacionadas por el producto iónico del agua.” (Teijón, 2006, pág. 34)
𝐾𝑤 = [1,0 𝑥 10−7][1,0 𝑥 10−7] = [1,0 𝑥 10−14] 25 ºC (2)

5
1.3. Sulfatos.
1.3.1. Características generales y origen. Los sulfatos (𝑆𝑂42−) se encuentran en las
aguas naturales y su contenido obedece a un intervalo de concentraciones bastante
variable. La mayoría de los sulfatos son altamente solubles en agua y otros disolventes
polares debido a que son sustancias iónicas, a excepción de unos pocos como es el caso
del sulfato de calcio. (Rice, Baird, Eaton, & Clesceri, 2012)
Para aguas residuales, el límite máximo permisible por cuerpo receptor debe ser de 1000
mg/L según la tabla Nº 1 de la norma técnica para el control de descargas líquidas
(NT002) perteneciente a la Ordenanza Metropolitana Nº 138, a mayor concentración los
sulfatos se vuelven altamente contaminantes.
Los sulfatos son sales que por lo general se originan de la reacción del ácido
sulfúrico (𝐻2𝑆𝑂4) con bases. También son producto de la reacción de dicho ácido con
metales.
𝐻2𝑆𝑂4 + 𝐶𝑢𝐶𝑂3 → 𝐶𝑢𝑆𝑂4+ 𝐶𝑂2 + 𝐻2𝑂 (3)
3𝐻2𝑆𝑂4 + 2𝐴𝑙(𝑂𝐻)3 → 𝐴𝑙2(𝑆𝑂4)3 + 6𝐻2𝑂 (4)
1.3.2. Aplicaciones. Los sulfatos tienen aplicaciones importantes en múltiples campos.
Algunos de los más importantes son:
Sulfato de Sodio (Na2SO4). Es empleado para fabricar vidrio, es usado también como
desecante debido a sus propiedades higroscópicas.
Sulfato de Calcio (CaSO4). Es un material ampliamente utilizado en la construcción y
también en esculturas y es usado como coagulante.
Sulfato de Cobre (CuSO4). Es utilizado para prevenir las plagas en horticultura
especialmente en las vides. (Químicas.net, 2015)
1.4. Sulfuros.
1.4.1. Características generales y origen. En cuanto se refiere a los sulfuros, se
encuentran localizados comúnmente en aguas subterráneas, en su mayoría en manantiales

6
con temperaturas cálidas e inclusive en sedimentos. Muchas veces se puede localizarlos
en las aguas residuales, esto debido en su mayor parte a la descomposición de la materia
orgánica. Una vez que la materia orgánica empieza su descomposición, esta produce
olores desagradables, esto es el resultado de la liberación de sulfuro de hidrógeno. Cabe
añadir que la zona abarcada por este olor, puede llegar a ser muy nociva para los
trabajadores de los alcantarillados. Incluso se han registrado varias muertes por la
excesiva inhalación de este gas. (Rice, Baird, Eaton, & Clesceri, 2012)
Al momento de categorizar al sulfuro se tienen tres tipos:
Sulfuro total. Esta categoría, añade 𝐻2𝑆 y 𝐻𝑆 en la calidad de disuelto, además de
sulfuros metálicos, los cuales son solubles en acido. Cabe acotar que no aplica para los
sulfuros de cobre y plata, ya que presentan un alto grado de insolubilidad.
Sulfuro disuelto. Es aquel que aún está presente incluso después de haber erradicado
toda presencia de sólidos suspendidos por floculación y depósito al igual que las
interferencias.
Sulfuro de hidrógeno no ionizado. Valorado a partir de la concentración de sulfuro
disuelto, el pH de la muestra y la constante de ionización práctica de 𝐻2𝑆.
“La concentración del umbral de detección de sulfuro de hidrógeno en aguas limpias está
entre 0.025µg/L y 0.25µg/L. Disuelto el 𝐻2𝑆 es tóxico para peces y otros organismos
acuáticos.” (Rickard & Morse, 2005, pág. 182)
1.4.2. Aplicaciones. Los sulfuros al estar aliados con algún metal, tienen aplicaciones
importantes. Algunos de los sulfuros más comunes y difundidos son empleados como
menas metálicas.
La separación entre sulfuros, una vez separados de las gangas, también se realiza
mediante flotación selectiva, empleando reactivos que deprimen y activan las superficies
de los sulfuros que se desea separar. Las tostaciones se realizan en hornos especiales,
generalmente de rastrillos, y en corriente de oxígeno, para favorecer el proceso, que en el
caso de la blenda ocurre según la reacción siguiente:
2𝑍𝑛𝑆 + 3𝑂2 → 2𝑍𝑛𝑂 + 2𝑆𝑂2 (5)

7
1.5. Fenoles.
1.5.1. Características generales y origen. Los fenoles son compuestos hidroxilados
aromáticos en los que el grupo hidroxilo está directamente unido al anillo aromático.
“Los fenoles tienen una semejanza formal, y, de hecho, experimentan algunas reacciones
comunes a los alcoholes; pero exigen un tratamiento especial debido a la profunda
influencia por el anillo aromático sobre las propiedades del anillo.
El fenol es una sustancia muy tóxica y ataca a los tejidos vivos en virtud de su acción
sobre las proteínas.” (Geissman, 1974, pág. 733)
Figura 2. Síntesis de fenol a escala industrial por el método de Dow Chemical
(Acuña Arias, 2006).
1.5.2. Propiedades físicas. Los fenoles sencillos son líquidos o sólidos de bajo punto de
fusión, tienden a formar puentes de hidrógeno intermolecular y es por ello que presentan
puntos de ebullición demasiado elevados. (Acuña Arias, 2006)
1.5.3. Propiedades químicas. Los alcoholes y los fenoles presentan el mismo grupo
funcional, OH, y algunas de sus propiedades son similares, se comportan también como
ácidos pero más fuertes que los alcoholes, el enlace C-OH es muy difícil de romper, por
lo tanto no se comportan como bases, esto se debe a que el grupo OH está directamente
unido al anillo aromático. (Acuña Arias, 2006)
1.6. Espectro electromagnético.
Se llama espectro electromagnético al conjunto de todas las radiaciones
electromagnéticas ordenadas en función de su frecuencia o de su longitud de onda.
(Cabrerizo, Antón, & Barrio, 2010)

8
Figura 3. Espectro electromagnético (Esopo, 2016).
1.6.1. Luz ultravioleta. La radiación ultravioleta (UV) pertenece a la franja del espectro
electromagnético con longitudes de onda entre 400 y 100 nm aproximadamente. Se
extiende desde la parte violeta del espectro visible hasta la zona de rayos X blandos,
aunque ambos límites son arbitrarios. (Portero, 2007)
1.7. Espectrofotómetro.
El espectrofotómetro es un instrumento diseñado para cuantificar la absorción de
radiación ultravioleta o visible por sustancias químicas y sus componentes básicos son:
Fuente. Emite la radiación que posteriormente interactúa con la muestra.
Sistema monocromador. Está constituido por lentes, prismas de refracción, rendijas,
etc; y permiten separar bandas de luz estrechas, antes o después de que la luz interaccionó
con la muestra.
Compartimentos para celdas o cubetas. Están colocados de una forma adecuada para
que el haz de luz de la fuente atraviese de manera perpendicular la muestra y dependiendo
de la región del espectro utilizada y del método a analizar son de diferentes formas y
tamaños.
Sistema detector. Detecta la radiación que ha atravesado la muestra.
Sistemas electrónicos. De amplificación, transformación y comparación de señal.

9
Sistemas de registro. De señal o almacenamiento de datos. (Olsen, 1990)
Figura 4. Partes básicas de un espectrofotómetro de absorción (Olsen, 1990).
1.8. Estadística básica.
1.8.1. Población. Es un conjunto de individuos o cosas que poseen la información de
aquello que se encuentra en estudio, es decir que son objeto del análisis. (Jurado, 2008)
1.8.2. Muestra. Subconjunto de una población determinada, que es representativo de la
misma y que tiene la misma probabilidad de ser escogido que cualquiera de los elementos.
(Jurado, 2008)
1.8.3. Media aritmética. Es el valor promedio de las muestras y es independiente de las
amplitudes de los intervalos. Se simboliza como �̅� y se encuentra sólo para variables
cuantitativas. Es un número significativo del conjunto de datos. (Jurado, 2008)
�̅� =∑ 𝒙𝒊
𝒏𝒊=𝟏
𝒏 (6)
Donde:
𝑥𝑖 = cada uno de los datos
𝑛 = número de datos totales
1.8.4. Desviación estándar. Es una medida de dispersión básica, que indica qué tan
dispersos están los datos con respecto a la media. Mientras mayor sea la desviación
estándar, mayor será la dispersión de los datos. También se puede utilizar para establecer
un valor de referencia para estimar la variación general de un proceso. (Jurado, 2008)

10
𝑺 = √∑ (𝒙−�̅�)𝟐𝒏
𝒊−𝟏
𝒏−𝟏 (7)
1.8.5. ANOVA. “El análisis de la varianza (ANOVA) es una potente herramienta
estadística, de gran utilidad tanto en la industria, para el control de procesos, como en el
laboratorio de análisis, para el control de métodos analíticos. Los ejemplos de aplicación
son múltiples, pudiéndose agrupar, según el objetivo que persiguen, en dos
principalmente: la comparación de múltiples columnas de datos y la estimación de los
componentes de variación de un proceso.” (Boqué & Maroto, 2011, pág. 1)
1.9. Validación e incertidumbre.
1.9.1. Principio de la validación. “Cada validación de un procedimiento consiste en
cuatro pasos principales:
a) Establecimientos de las condiciones por cumplir, es decir los objetivos de validación
por ejemplo: límite de detección < 1 mg/L, intervalo lineal, incertidumbre de los
resultados.
b) Determinación de los parámetros estadísticos del método.
c) Valoración de los resultados de la validación por comparación de los parámetros
estadísticos obtenidos con las condiciones (objetivos) y decisión sobre la validez del
procedimiento para el propósito establecido.
d) Declaración de la conformidad del método” (ISO & Calidad-Universidad CEU San
Pablo Madrid, 2013, págs. 10, 11)
1.9.2. Norma Internacional ISO/IEC 17025:2017. Esta norma establece los requisitos
a cumplir para acreditar la competencia de los laboratorios de ensayo y calibración. Esta
norma introduce cambios en la traducción para mejorar su comprensión y para armonizar
dicha traducción con los diferentes países de lengua española.
Por otra parte se incorpora algún nuevo matiz sobre las funciones de la alta dirección,
servicio al cliente, personal y aseguramiento de la calidad de los resultados de ensayo y
calibraciones. (García Bermejo, 2006)

11
1.9.3. Establecimiento del alcance y objetivos de la validación. “Se diferencian tres
casos, en los que la dificultad de la validación aumenta:
a) Método de ensayo estandarizado y normalizado, que se aplica exactamente como está
descrito en la norma. El objetivo de la validación es comprobar que el laboratorio domina
el ensayo y lo utiliza correctamente.
b) Modificación a un método de ensayo normalizado, se hicieron modificaciones a los
métodos descritos en la norma que pueden tener una repercusión sobre la calidad de los
resultados. El objetivo de la validación es comprobar que la repetitividad, la
reproducibilidad, la precisión intermedia y la exactitud del método original no dependen
de la modificación introducida y que el laboratorio domina el ensayo y lo utiliza
correctamente.
c) Método de ensayo interno, elaborado en el laboratorio y que no se encuentra en normas
u otras colecciones de métodos. El objetivo de la validación es comprobar que el método
tiene la repetitividad, la reproducibilidad, la precisión intermedia y la exactitud
suficientes para el objetivo de aplicación y que el laboratorio domina el ensayo y lo realiza
correctamente.” (ISO & CALIDAD-Universidad CEU San Pablo Madrid, 2013, pág. 13)
1.9.4. Exactitud. Grado de concordancia que existe entre el resultado del ensayo y un
valor verdadero que se ha tomado como referencia. Se estudia en dos componentes:
veracidad y precisión. (ISO, 1994)
1.9.5. Veracidad. Grado de concordancia que existe entre el valor medio obtenido de
una gran serie de resultados y un valor real que se ha tomado como referencia. Se expresa
en términos de sesgo. (ISO, 1994)
1.9.6. Precisión. Cercanía que existe entre los resultados independientes de un ensayo,
obtenidos en condiciones previstas. Se expresa en términos de desviación estándar u otra
variable de dispersión. (ISO, 1994)

12
1.9.7. Reproducibilidad. Los resultados de un ensayo se obtienen mediante la
aplicación de un mismo método pero a medidas hechas a distintas condiciones (operarios,
aparatos, laboratorios o épocas distintas), para que esta expresión sea válida se deben
aplicar las condiciones que pueden cambiar de una medida a otra. (Griful Ponsati &
Canela Campos, 2002)
1.9.8. Repetibilidad. Los resultados de un ensayo se obtienen mediante la aplicación de
un mismo método con las medidas realizadas a condiciones lo más estables posible, por
un mismo operario, con un mismo equipo y a breves intervalos de tiempo. (Griful Ponsati
& Canela Campos, 2002)
1.9.9. Límite de detección. Describe la concentración más baja de un analito en una
muestra la cual puede ser detectada pero no necesariamente cuantificada con un valor
exacto. Se expresa en unidades de concentración. (Eurachem, 2016)
1.9.10. Límite de cuantificación. Es la concentración mínima del analito que puede
determinarse con un nivel aceptable de exactitud y precisión. (Eurachem, 2016)
1.9.11. Intervalo de trabajo. Después de demostrar que el método analítico tiene un
nivel adecuado de precisión, veracidad y linealidad, se procede a fijar el intervalo de
trabajo, el mismo que se encuentra entre la más alta y la más baja concentración del
analito de la muestra. (Ich Harmonised Tripartite Guideline, 2005)
1.9.12. Incertidumbre. Un parámetro asociado con el resultado de una medición, que
caracteriza al intervalo de los valores dentro de los cuales se afirma que esta el valor
verdadero. (Eurachem, 2016)
1.9.12.1. Tipos de evaluación de incertidumbre. El laboratorio debe realizar una
evaluación de las incertidumbres tipo A y B que se encuentren presentes en el método:
Evaluación de incertidumbre tipo A. Evaluación de un componente por un análisis
estadístico de los valores de mediciones obtenidos en condiciones de medición definidas.
Ejemplo: realizar varias mediciones en condiciones de repetibilidad.
Evaluación de incertidumbre tipo B. Evaluación de un componente incertidumbre
de la medición realizada por otros medios distinto a los del tipo A. Ejemplos: La

13
evaluación basada en la información, obtenidos a partir de un certificado de calibración,
obtenidos a partir de los límites deducirse a través de personal, la experiencia, etc.
(Eurachem, 2000)
1.9.12.2. Incertidumbre combinada. “Incertidumbre típica del resultado de una
medición, cuando el resultado se obtiene a partir de los valores de otras magnitudes, e
igual a la raíz cuadrada positiva de la suma de las varianzas o covarianzas de esas otras
magnitudes, ponderadas según el factor de sensibilidad del resultado de medición
respecto a la variación de dichas magnitudes.” (Centro Español de Metrología, 2011, pág.
7)
1.9.12.3. Incertidumbre expandida. “Cantidad que define un intervalo en torno al
resultado de una medición, en el que puede esperarse encontrar una fracción amplia de la
distribución de valores que pueden ser razonablemente atribuidos al mensurando.
La fracción puede entenderse como la probabilidad o el nivel de confianza del
intervalo.
Para asociar un nivel específico de confianza a un intervalo definido por la
incertidumbre expandida, se requieren hipótesis explícitas o implícitas sobre la
distribución de probabilidad caracterizada por el resultado de medida y su incertidumbre
típica combinada. El nivel de confianza que puede atribuirse a este intervalo posee la
misma validez que las hipótesis realizadas.” (Centro Español de Metrología, 2011, pág.
7)
1.9.12.4. Factor de cobertura. “Factor numérico utilizado como multiplicador de la
incertidumbre típica combinada, para obtener una incertidumbre expandida. Un factor de
cobertura típico, toma valores comprendidos entre 2 y 3.” (Centro Español de Metrología,
2011, pág. 6)
14
2. DISEÑO EXPERIMENTAL Y ESTADÍSTICO
2.1. Fijación de objetivos para los diferentes analitos y selección de los parámetros
de validación.
Los objetivos de validación se fijaron de acuerdo con la tabla Nº 1 de la norma técnica
para el control de descargas líquidas (NT002) de la Ordenanza Metropolitana Nº 138, y
con las tablas Nº 1, 8 y 9 del acuerdo 097-A del Anexo 1 del libro VI del Texto Unificado
de Legislación Ambiental Secundaria del Ministerio de Ambiente (TULSMA), porque
estos cuerpos legales presentan los límites máximos permisibles para cada analito por
cuerpo recetor, mismos que fueron definidos acorde a lo que dicta la Constitución de la
República del Ecuador. Además, de acuerdo con el laboratorio ISO & Calidad (2013),
para el establecimiento de la validación, el tipo de caso que se presenta para sulfatos,
sulfuros y fenoles es el de un método de ensayo estandarizado y normalizado, por ello se
debe aplicar exactamente lo que describen las normas ya que el objetivo de ésta validación
es comprobar que el laboratorio domina el ensayo y lo utiliza correctamente.
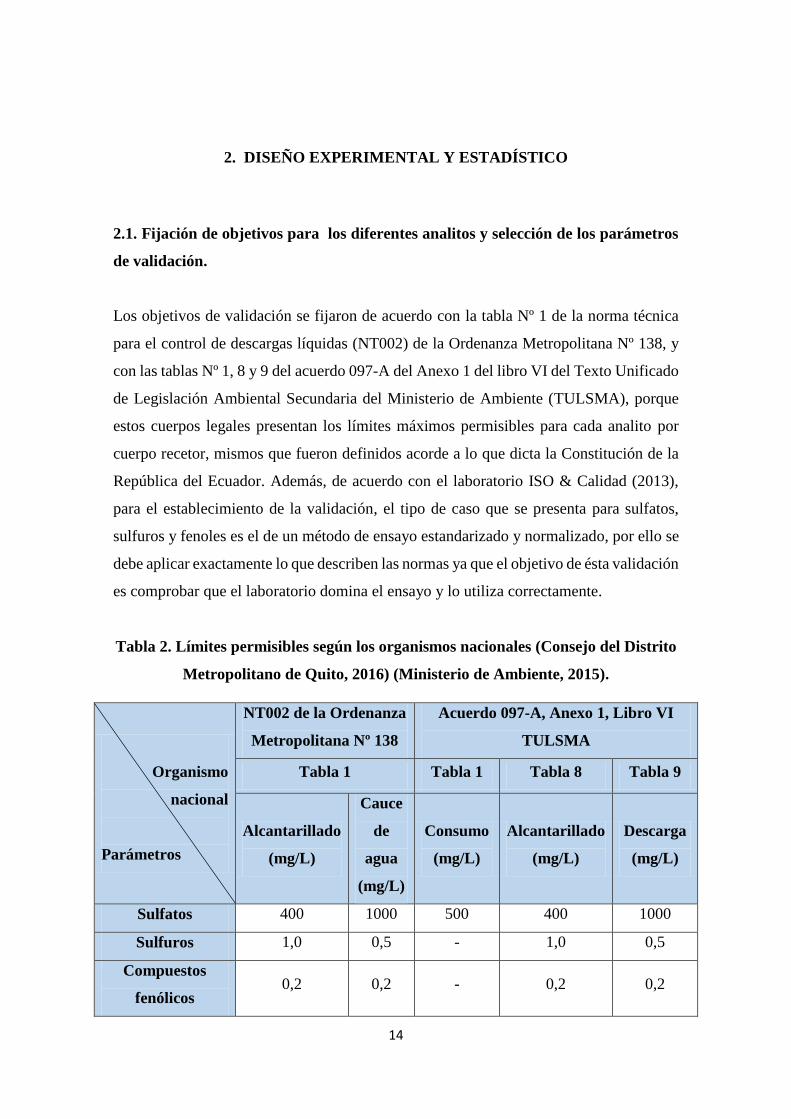
Tabla 2. Límites permisibles según los organismos nacionales (Consejo del Distrito
Metropolitano de Quito, 2016) (Ministerio de Ambiente, 2015).
Organismo
nacional
Parámetros
NT002 de la Ordenanza
Metropolitana Nº 138
Acuerdo 097-A, Anexo 1, Libro VI
TULSMA
Tabla 1 Tabla 1 Tabla 8 Tabla 9
Alcantarillado
(mg/L)
Cauce
de
agua
(mg/L)
Consumo
(mg/L)
Alcantarillado
(mg/L)
Descarga
(mg/L)
Sulfatos 400 1000 500 400 1000
Sulfuros 1,0 0,5 - 1,0 0,5
Compuestos
fenólicos 0,2 0,2 - 0,2 0,2
15
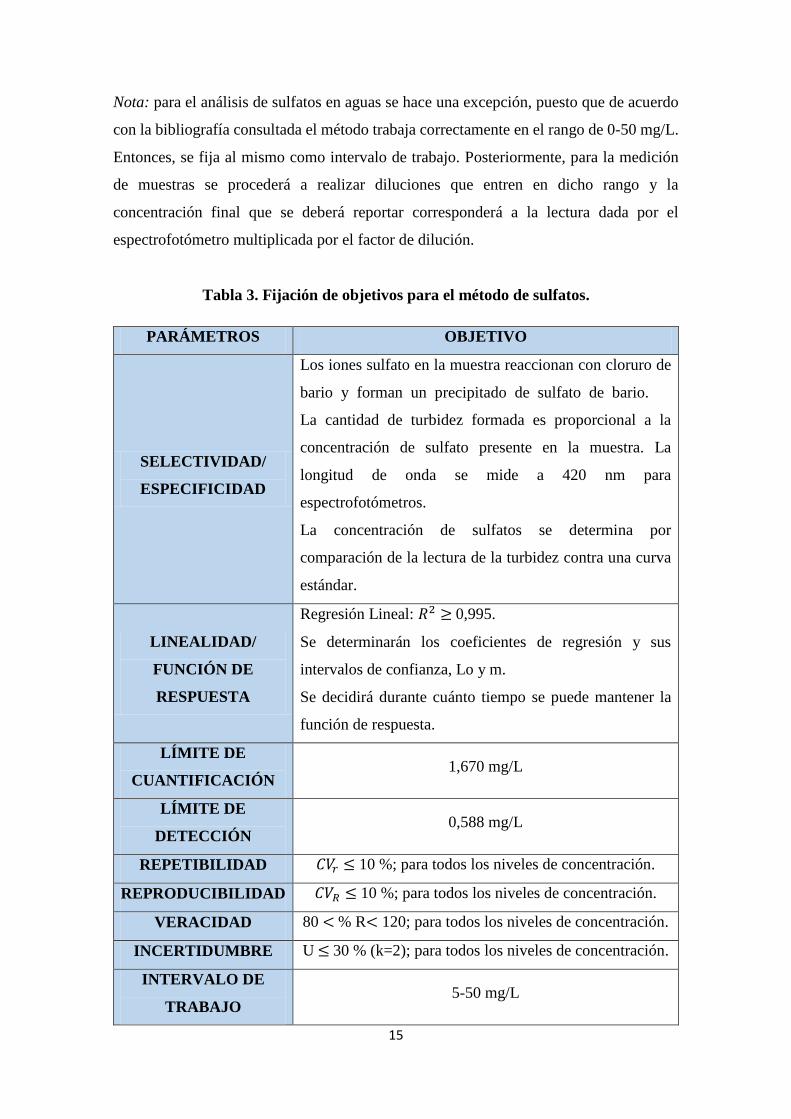
Nota: para el análisis de sulfatos en aguas se hace una excepción, puesto que de acuerdo
con la bibliografía consultada el método trabaja correctamente en el rango de 0-50 mg/L.
Entonces, se fija al mismo como intervalo de trabajo. Posteriormente, para la medición
de muestras se procederá a realizar diluciones que entren en dicho rango y la
concentración final que se deberá reportar corresponderá a la lectura dada por el
espectrofotómetro multiplicada por el factor de dilución.
Tabla 3. Fijación de objetivos para el método de sulfatos.
PARÁMETROS OBJETIVO
SELECTIVIDAD/
ESPECIFICIDAD
Los iones sulfato en la muestra reaccionan con cloruro de
bario y forman un precipitado de sulfato de bario.
La cantidad de turbidez formada es proporcional a la
concentración de sulfato presente en la muestra. La
longitud de onda se mide a 420 nm para
espectrofotómetros.
La concentración de sulfatos se determina por
comparación de la lectura de la turbidez contra una curva
estándar.
LINEALIDAD/
FUNCIÓN DE
RESPUESTA
Regresión Lineal: 𝑅2 ≥ 0,995.
Se determinarán los coeficientes de regresión y sus
intervalos de confianza, Lo y m.
Se decidirá durante cuánto tiempo se puede mantener la
función de respuesta.
LÍMITE DE
CUANTIFICACIÓN 1,670 mg/L
LÍMITE DE
DETECCIÓN 0,588 mg/L
REPETIBILIDAD 𝐶𝑉𝑟 ≤ 10 %; para todos los niveles de concentración.
REPRODUCIBILIDAD 𝐶𝑉𝑅 ≤ 10 %; para todos los niveles de concentración.
VERACIDAD 80 < % R< 120; para todos los niveles de concentración.
INCERTIDUMBRE U ≤ 30 % (k=2); para todos los niveles de concentración.
INTERVALO DE
TRABAJO 5-50 mg/L

16
Tabla 4. Fijación de objetivos para el método de sulfuros.
PARÁMETROS OBJETIVO
SELECTIVIDAD/
ESPECIFICIDAD
El sulfuro de hidrógeno y los sulfuros metálicos solubles
en ácido reaccionan con N, N-dimetil-fenilendiamina
sulfato para formar azul de metileno. La intensidad del
color azul es proporcional a la concentración de sulfuro.
La longitud de onda de medición es 665 nm para
espectrofotómetros.
La concentración de sulfuros se determina por
comparación de la lectura del color azul contra una curva
estándar.
LINEALIDAD/
FUNCIÓN DE
RESPUESTA
Regresión Lineal: 𝑅2 ≥ 0,995.
Se determinarán los coeficientes de regresión y sus
intervalos de confianza, Lo y m.
Se decidirá durante cuánto tiempo se puede mantener la
función de respuesta.
LÍMITE DE
CUANTIFICACIÓN 0,029 mg/L
LÍMITE DE
DETECCIÓN 0,011 mg/L
REPETIBILIDAD 𝐶𝑉𝑟 ≤ 10 %; para todos los niveles de concentración.
REPRODUCIBILIDAD 𝐶𝑉𝑅 ≤ 10 %; para todos los niveles de concentración.
VERACIDAD 80 < % R< 120; para todos los niveles de concentración.
INCERTIDUMBRE U ≤ 30 % (k=2); para todos los niveles de concentración.
INTERVALO DE
TRABAJO 0,25-1,1 mg/L

17
Tabla 5. Fijación de objetivos para el método de fenoles.
PARÁMETROS OBJETIVO
SELECTIVIDAD/
ESPECIFICIDAD
El método de 4-aminoantipirina mide todos los fenoles
orto-y meta-sustituidos. Estos fenoles reaccionan con 4-
aminoantipirina en presencia de ferricianuro de potasio
para formar un colorante de antipirina. La sensibilidad del
método cambia con el tipo de compuesto fenólico.
Debido a que las muestras de agua pueden contener varios
tipos de compuestos fenólicos, los resultados se muestran
como la concentración equivalente de fenol. La medida de
la longitud de onda es 460 nm.
La concentración de fenoles se determina por comparación
de la lectura de color contra una curva estándar.
LINEALIDAD/
FUNCIÓN DE
RESPUESTA
Regresión Lineal: 𝑅2 ≥ 0,995.
Se determinarán los coeficientes de regresión y sus
intervalos de confianza, Lo y m.
Se decidirá durante cuánto tiempo se puede mantener la
función de respuesta.
LÍMITE DE
CUANTIFICACIÓN 0,007 mg/L
LÍMITE DE
DETECCIÓN 0,003 mg/L
REPETIBILIDAD 𝐶𝑉𝑟 ≤ 15 %; para todos los niveles de concentración.
REPRODUCIBILIDAD 𝐶𝑉𝑅 ≤ 15 %; para todos los niveles de concentración.
VERACIDAD 80 < % R < 120; para todos los niveles de concentración.
INCERTIDUMBRE U ≤ 30 % (k=2); para todos los niveles de concentración.
INTERVALO DE
TRABAJO 0,05-0,25 mg/L

18
2.2. Diseño experimental.
Se eligió el esquema de la figura 5 para representar el diseño experimental, porque de
acuerdo con Fernanda Toasa (2012), el mismo detalla las principales etapas a seguir
durante la validación de un método instrumental. El número de niveles se fijó de acuerdo
al tipo de agua que se analizaría, es decir, para sulfatos se estableció trabajar con aguas
residuales y de consumo, es por ello que se escogió cinco niveles de concentración, en
cambio para sulfuros y fenoles fue necesario definir tres niveles de concentración, puesto
que se resolvió trabajar sólo en aguas residuales. Se realizaron las lecturas de cada nivel
por quintuplicado para valorar el grado de repetibilidad de los análisis, ya que de acuerdo
con Antonio Creus Solé (2011), la repetibilidad es sinónimo de precisión y una buena
manera de aumentarla es realizando varias mediciones del mismo carácter (mínimo dos).
Además este procedimiento se llevó a cabo durante tres días distintos para determinar el
grado de reproducibilidad de los análisis y debido al tiempo que se demora cada ensayo.
Muestra o
blanco
Submuestra 1
Submuestra 2
Submuestra 3
Submuestra 4
Submuestra 5
Procesado 1
Procesado 2
Procesado 3
Procesado 4
Procesado 5
Resultado 1
Resultado 2
Resultado 3
Resultado 4
Resultado 5
Resultado
final sobre la
muestra
Procesamiento en
condiciones de
repetibilidad y
reproducibilidad:
Pesada
Extracción
Disolución
Lectura
instrumental e
interpolación contra
recta de calibrado
Cálculos para
llegar al resultado
final sobre la
muestra
Figura 5. Diseño experimental para la validación de métodos de ensayo (Toasa,
2012).

19
2.3. Diseño experimental por tipo de ensayo.
Tabla 6. Diseño experimental para el ensayo de sulfatos.
DISEÑO
EXPERIMENTAL
Condiciones de repetibilidad. Determinación de sulfatos
en cinco niveles de concentración por quintuplicado en cada
nivel.
Condiciones de reproducibilidad. Determinación de
sulfatos en cinco niveles de concentración por quintuplicado
en cada nivel en tres días distintos.
Veracidad. Determinación del porcentaje de recuperación
utilizando material de referencia certificado, diariamente por
quintuplicado; tomando como valor verdadero el
proporcionado en el certificado de análisis y como valor
experimental, la media de todos los valores obtenidos, en
condiciones de repetibilidad y reproducibilidad.
MUESTRAS
Se eligen cinco muestras diferentes a las cuales se les añade
material de referencia para que cumplan con los niveles de
concentración requeridos.
Muestras fortificadas. Tres muestras de aguas residuales de
diferentes empresas, fortificadas a tres niveles de
concentración; 1 a nivel bajo, 1 a nivel medio del intervalo de
trabajo y 1 a nivel alto del intervalo de trabajo.
Dos muestras de agua de consumo (llave) diferentes,
fortificadas a dos niveles de concentración; 1 a un valor bajo
y 1 a un valor medio del intervalo de trabajo.
2 bajos, cercanos al menor valor del intervalo de trabajo.
2 medios, cercanos al valor medio del intervalo de trabajo.
1 alto, cercano al mayor valor del intervalo de trabajo.

20
Tabla 6. (Continuación)
SUBMUESTRAS
De cada muestra disponible para la validación se obtienen
cinco submuestras, que se analizarán por tres días diferentes
cambiando diariamente de analista. Obteniéndose un total de
75 datos. Estas submuestras siguen el procedimiento
completo.
PROCESAMIENTO
Se procesará todo el conjunto de datos obtenidos, tanto en
condiciones de repetibilidad y reproducibilidad, para cada
nivel de concentración.
LECTURA Concentración de la muestra en mg/L.
FUNCIÓN DE
RESPUESTA
Se realiza diariamente una recta de calibración, la cual por
regresión lineal debe estar dentro de los límites del grado de
ajuste (𝑅2), esto permitirá hacer la corrección de los datos
leídos en el equipo conjuntamente con el material de
referencia.
TRATAMIENTO
ESTADÍSTICO
Análisis de varianza simple de los resultados obtenidos para
obtención de la precisión por niveles de concentración.
Obtención del intervalo de trabajo y de la incertidumbre
asociada a cada nivel.
Tabla 7. Diseño experimental para el ensayo de sulfuros.
DISEÑO
EXPERIMENTAL
Condiciones de repetibilidad. Determinación de sulfuros
en cinco niveles de concentración por quintuplicado en cada
nivel.
Condiciones de reproducibilidad. Determinación de
sulfuros en cinco niveles de concentración por quintuplicado
en cada nivel en tres días distintos.

21
Tabla 7. (Continuación)
DISEÑO
EXPERIMENTAL
Veracidad. Determinación del porcentaje de recuperación
utilizando material de referencia certificado, diariamente
por quintuplicado; tomando como valor verdadero el
proporcionado en el certificado de análisis y como valor
experimental, la media de todos los valores obtenidos, en
condiciones de repetibilidad y reproducibilidad.
MUESTRAS
Se eligen tres muestras diferentes a las cuales se les añade
material de referencia para que cumplan con los niveles de
concentración requeridos.
Muestras fortificadas. Tres muestras de aguas residuales de
diferentes empresas fortificadas a tres niveles de
concentración.
1 bajo, cercano al menor valor del intervalo de trabajo.
1 medio, cercano al valor medio del intervalo de trabajo.
1 alto, cercano al mayor valor del intervalo de trabajo.
SUBMUESTRAS
De cada muestra disponible para la validación se obtienen
cinco submuestras, que se analizarán por tres días diferentes
cambiando diariamente de analista. Obteniéndose un total de
45 datos. Estas submuestras siguen el procedimiento
completo.
PROCESAMIENTO
Se procesará todo el conjunto de datos obtenidos, tanto en
condiciones de repetibilidad y reproducibilidad, para cada
nivel de concentración.
LECTURA Concentración de la muestra en mg/L.
FUNCIÓN DE
RESPUESTA
Se realiza diariamente una recta de calibración, la cual por
regresión lineal debe estar dentro de los límites del grado de
ajuste (𝑅2), esto permitirá hacer la corrección de los datos
leídos en el equipo conjuntamente con el material de
referencia.
22
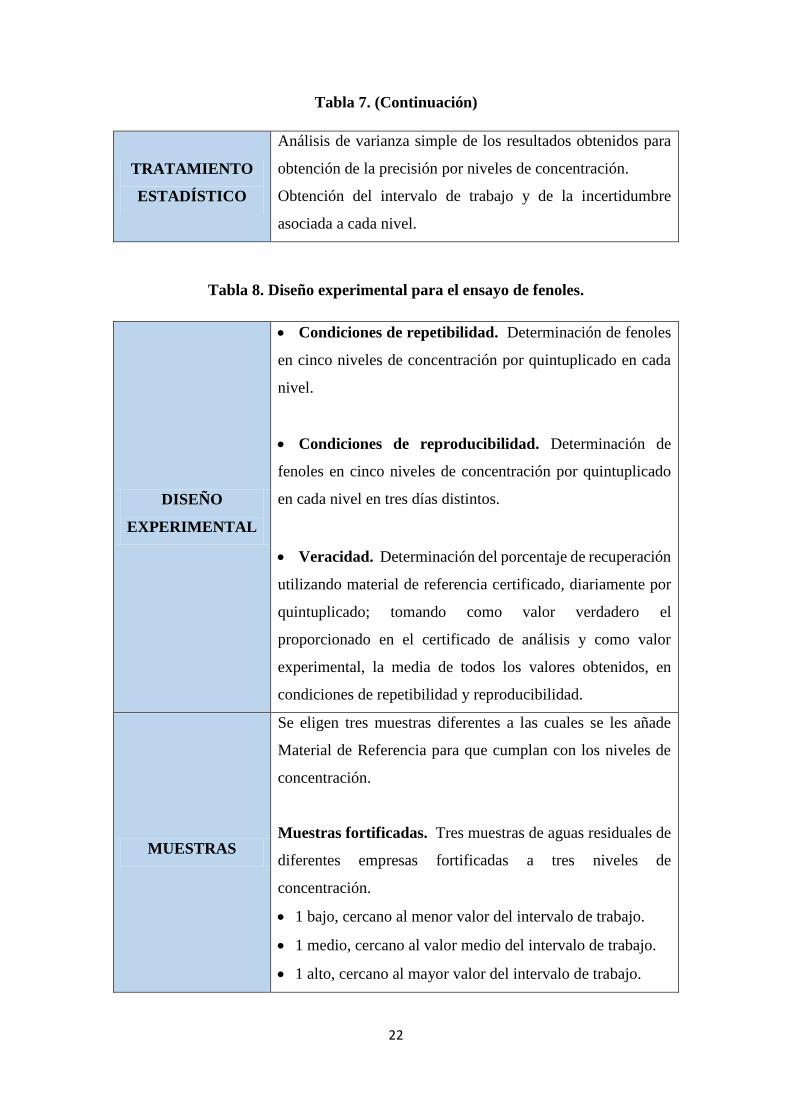
Tabla 7. (Continuación)
TRATAMIENTO
ESTADÍSTICO
Análisis de varianza simple de los resultados obtenidos para
obtención de la precisión por niveles de concentración.
Obtención del intervalo de trabajo y de la incertidumbre
asociada a cada nivel.
Tabla 8. Diseño experimental para el ensayo de fenoles.
DISEÑO
EXPERIMENTAL
Condiciones de repetibilidad. Determinación de fenoles
en cinco niveles de concentración por quintuplicado en cada
nivel.
Condiciones de reproducibilidad. Determinación de
fenoles en cinco niveles de concentración por quintuplicado
en cada nivel en tres días distintos.
Veracidad. Determinación del porcentaje de recuperación
utilizando material de referencia certificado, diariamente por
quintuplicado; tomando como valor verdadero el
proporcionado en el certificado de análisis y como valor
experimental, la media de todos los valores obtenidos, en
condiciones de repetibilidad y reproducibilidad.
MUESTRAS
Se eligen tres muestras diferentes a las cuales se les añade
Material de Referencia para que cumplan con los niveles de
concentración.
Muestras fortificadas. Tres muestras de aguas residuales de
diferentes empresas fortificadas a tres niveles de
concentración.
1 bajo, cercano al menor valor del intervalo de trabajo.
1 medio, cercano al valor medio del intervalo de trabajo.
1 alto, cercano al mayor valor del intervalo de trabajo.

23
Tabla 8. (Continuación)
SUBMUESTRAS
De cada muestra disponible para la validación se obtienen
cinco submuestras, que se analizarán por tres días diferentes
cambiando diariamente de analista. Obteniéndose un total de
45 datos. Estas submuestras siguen el procedimiento
completo.
PROCESAMIENTO
Se procesaran todo el conjunto de datos obtenidos tanto en
condiciones de repetibilidad y reproducibilidad, para cada
nivel de concentración.
LECTURA Concentración de la muestra en mg/L.
FUNCIÓN DE
RESPUESTA
Se realiza diariamente una recta de calibración, la cual por
regresión lineal debe estar dentro de los límites del grado de
ajuste (𝑅2), esto permitirá hacer la corrección de los datos
leídos en el equipo conjuntamente con el material de
referencia.
TRATAMIENTO
ESTADÍSTICO
Análisis de varianza simple de los resultados obtenidos para
obtención de la precisión por niveles de concentración.
Obtención del intervalo de trabajo y de la incertidumbre
asociada a cada nivel.
2.4. Tratamiento estadístico.
Para el tratamiento estadístico de los métodos de ensayo se utiliza ANOVA.
“Los resultados de un experimento de un factor se acostumbra presentarlos en una tabla
con a renglones y b columnas, como la tabla 9. Aquí, 𝑋𝑖𝑗 denota la medición del renglón
i y columna j, donde i = 1, 2,. . ., a y donde j = 1, 2,. . ., b. Por ejemplo, 𝑋35 significa la
quinta medición del tercer tratamiento.” (Spiegel & Stephens, 2009, pág. 403)

24
Tabla 9. Tabla de datos para el ANOVA de un factor.
Réplicas
Tratamientos
Réplica 1 Réplica 2 … Réplica b
Tratamiento 1 𝑋11 𝑋12 … 𝑋1𝑏 𝑋1̅̅ ̅
Tratamiento 2 𝑋21 𝑋22 … 𝑋2𝑏 𝑋2̅̅ ̅
… … … … … …
Tratamiento a 𝑋𝑎1 𝑋𝑎2 … 𝑋𝑎𝑏 𝑋𝑎̅̅̅̅
“La medida de las mediciones en el reglón i se denota:
𝑋𝑖.̅̅ ̅ =
1
𝑏∑ 𝑋𝑖𝑗
𝑏𝑘=1 (8)
Donde i=1, 2,..., a
El punto que aparece en 𝑋𝑖. sirve para indicar que se suma sobre el índice j. A los valores
𝑋𝑖. se les llama medias de grupo, medias de tratamiento o medias de reglón. La gran
media o media general es la media de todas las mediciones de todos los grupos y se denota
�̅�:
�̅�=1
𝑎𝑏∑ ∑ 𝑋𝑖𝑗
𝑏𝑗=1
𝑎𝑖=1 ” (9)
(Spiegel & Stephens, 2009, pág. 404)
25
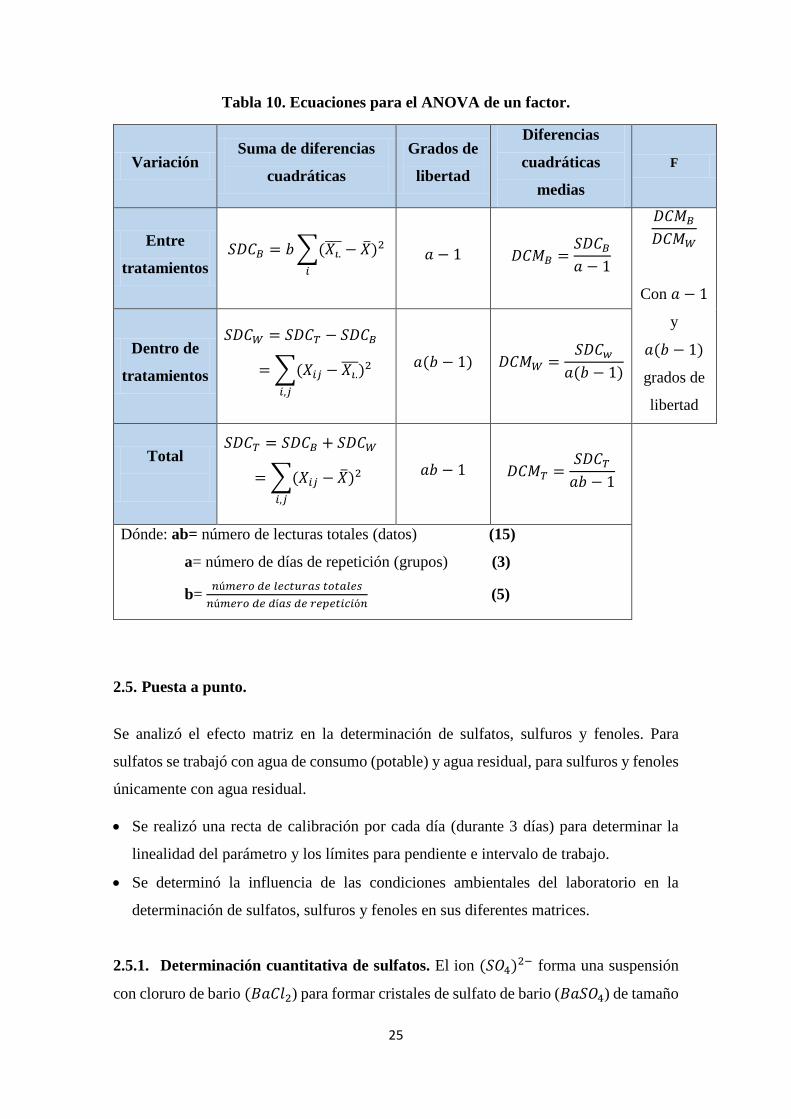
Tabla 10. Ecuaciones para el ANOVA de un factor.
Variación Suma de diferencias
cuadráticas
Grados de
libertad
Diferencias
cuadráticas
medias
F
Entre
tratamientos
𝑆𝐷𝐶𝐵 = 𝑏 ∑(𝑋𝑖.̅̅ ̅ − �̅�)2
𝑖
𝑎 − 1
𝐷𝐶𝑀𝐵 =𝑆𝐷𝐶𝐵
𝑎 − 1
𝐷𝐶𝑀𝐵
𝐷𝐶𝑀𝑊
Con 𝑎 − 1
y
𝑎(𝑏 − 1)
grados de
libertad
Dentro de
tratamientos
𝑆𝐷𝐶𝑊 = 𝑆𝐷𝐶𝑇 − 𝑆𝐷𝐶𝐵
= ∑(𝑋𝑖𝑗 − 𝑋𝑖.̅̅ ̅)2
𝑖,𝑗
𝑎(𝑏 − 1) 𝐷𝐶𝑀𝑊 =
𝑆𝐷𝐶𝑤
𝑎(𝑏 − 1)
Total
𝑆𝐷𝐶𝑇 = 𝑆𝐷𝐶𝐵 + 𝑆𝐷𝐶𝑊
= ∑(𝑋𝑖𝑗 − �̅�)2
𝑖,𝑗
𝑎𝑏 − 1
𝐷𝐶𝑀𝑇 =𝑆𝐷𝐶𝑇
𝑎𝑏 − 1
Dónde: ab= número de lecturas totales (datos) (15)
a= número de días de repetición (grupos) (3)
b= 𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑙𝑒𝑐𝑡𝑢𝑟𝑎𝑠 𝑡𝑜𝑡𝑎𝑙𝑒𝑠
𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑑í𝑎𝑠 𝑑𝑒 𝑟𝑒𝑝𝑒𝑡𝑖𝑐𝑖ó𝑛 (5)
2.5. Puesta a punto.
Se analizó el efecto matriz en la determinación de sulfatos, sulfuros y fenoles. Para
sulfatos se trabajó con agua de consumo (potable) y agua residual, para sulfuros y fenoles
únicamente con agua residual.
Se realizó una recta de calibración por cada día (durante 3 días) para determinar la
linealidad del parámetro y los límites para pendiente e intervalo de trabajo.
Se determinó la influencia de las condiciones ambientales del laboratorio en la
determinación de sulfatos, sulfuros y fenoles en sus diferentes matrices.
2.5.1. Determinación cuantitativa de sulfatos. El ion (𝑆𝑂4)2− forma una suspensión
con cloruro de bario (𝐵𝑎𝐶𝑙2) para formar cristales de sulfato de bario (𝐵𝑎𝑆𝑂4) de tamaño

26
uniforme. Se mide la dispersión de la luz de la suspensión de 𝐵𝑎𝑆𝑂4 con un
espectrofotómetro o turbidímetro, y la concentración de (𝑆𝑂4)2− se determina por
comparación de la lectura contra una curva estándar.
Este método es aplicable a aguas potables, lluvia, superficiales, y efluentes domésticos e
industriales (aguas residuales).
(𝑆𝑂4)2− + 2𝐻𝐶𝑙 → 𝐻2𝑆𝑂4 + 2𝐶𝑙− (10)
𝐻2𝑆𝑂4 + 𝐵𝑎𝐶𝑙2 → 𝐵𝑎𝑆𝑂4 + 2𝐻𝐶𝑙 (11)
2.5.1.1. Limitaciones de la determinación. El intervalo de aplicación de este método
va de 0 a 50 mg (𝑆𝑂4)2−/L. Para muestras con concentraciones superiores, debe usarse
una alícuota que contenga menos de 50 mg (𝑆𝑂4)2−/L.
2.5.1.2. Interferencias. El color o la materia en suspensión en grandes cantidades
pueden interferir con la determinación; ésta última se puede remover por filtración. Si
ambas son pequeñas en comparación con la concentración de (𝑆𝑂4)2−, se corrige la
interferencia mediante un blanco de muestra en el que se omite la adición de 𝐵𝑎𝑆𝑂4.
La sílice en cantidad superior a 500 mg/L interfiere. Se debe realizar una dilución o
muestra adicionada para observar la interferencia por matriz.
En presencia de materia orgánica algunas bacterias reducen el (𝑆𝑂4)2−a (𝑆)2−, lo que se
evita por refrigeración de la muestra. (Rice, Baird, Eaton, & Clesceri, 2012)
2.5.2. Determinación cuantitativa de sulfuros. El método más preciso para la
determinación de sulfuros es el método de azul de metileno, que se basa en la reacción
del sulfuro, el cloruro férrico y la dimetil-para-fenilendiamina para producir el azul de
metileno.

27
Figura 6. Reacción de dimetil-para-fenilendiamina con el cloruro férrico y sulfuro
de hidrógeno (Brender, 2016).
2.5.2.1. Limitaciones de la determinación. El método del azul de metileno se aplica
para aguas que contengan hasta 20 mg/L de sulfuros. Para concentraciones mayores,
deberán hacerse las diluciones correspondientes.
2.5.2.2. Interferencias. En el método del azul de metileno interfieren los fuertes agentes
oxidantes enmascarando la formación del color azul. Cuando la concentración de
tiosulfatos es mayor de 10 mg/L, la formación del color se retarda.
La misma concentración de sulfuros, si es de varios cientos de miligramos por cada litro,
representa una interferencia. Las interferencias debido a sulfitos, tiosulfatos, yoduros y
muchas otras sustancias solubles, exceptuando ferrocianuros, se eliminan adicionando
sulfuro de zinc, removiendo el sobrenadante y reemplazándolo por agua. Este mismo
procedimiento se usa para concentrar los sulfuros, incluso si no hay necesidad de remover
las interferencias. (Rice, Baird, Eaton, & Clesceri, 2012)
2.5.3. Determinación cuantitativa de fenoles. “Los procedimientos analíticos casi
siempre utilizan el método colorimétrico de la 4-aminoantipirina que determina el fenol,
los fenoles sustituidos en orto y meta y bajo condiciones apropiadas de pH, los fenoles
sustituidos en para en los que los sustituyentes son grupos carboxilo, halógeno, metoxilo
o ácido sulfónico.
El método de extracción con cloroformo, es el de la máxima sensibilidad, es adaptable a
muestras de agua que contengan menos de 1 mg fenol/l. Concentra el color en una
solución no acuosa.” (Rice, Baird, Eaton, & Clesceri, 2012, págs. 5-56)
Los compuestos fenólicos destilados reaccionan con 4-aminoantipirina a pH 10.0 ± 0,1
en presencia de ferricianuro de potasio, formando un compuesto coloreado de antipirina.

28
Este tinte es extraído de la solución acuosa con Cloroformo 𝐶𝐻𝐶𝑙3 y la absorbancia es
medida a 460 nm.
Figura 7. Reacción de 4-aminoantipirina con fenol o compuesto fenólico (IDEAM,
2007).
2.5.3.1. Limitaciones de la determinación. El método de la 4-aminoantipirina no
determina los fenoles para-sustituidos donde la sustitución es un grupo alquilo, arilo,
nitro, benzoilo, nitroso o aldehído, esto se debe a que está desactivada la posición del
anillo bencénico tanto para las reacciones de copulación, como para las de condensación.
Un ejemplo típico de estos últimos grupos es el paracresol, que puede estar presente en
ciertas aguas residuales industriales y en las aguas de superficie contaminadas.
2.5.3.2. Interferencias. La mayoría de sustancias interferentes son eliminadas
destilando los fenoles de la muestra.
Las interferencias tales como bacterias descomponedoras de fenoles, sustancias oxidantes
o reductoras, y valores de pH alcalinos se eliminan por acidificación con ácido fosfórico.
(Rice, Baird, Eaton, & Clesceri, 2012)
2.6. Verificación de la balanza analítica.
a) Conectar la balanza, esperar 10 minutos para que se estabilice.
b) Si el centro de masas no se encuentra en el centro, se debe ajustar el mismo utilizando
las perillas que se encuentran al pie de la balanza analítica.
c) Empezar con las mediciones correspondientes de las pesas de 0,5 g; 1 g; 10 g; 50 g y
100 g.

29
d) Anotar los valores correspondientes en el registro de verificación de la balanza con la
fecha correspondiente.
e) Tener en cuenta que los valores mostrados por la balanza analítica no pueden estar
fuera de los límites mostrados en el registro de verificación.
f) Anotar también los valores correspondientes a humedad relativa y temperatura que
indica el higrómetro.
2.7. Calibración del espectrofotómetro.
a) Conectar el espectrofotómetro y encender con <ON/OFF>.
b) Quitar todas las cubetas y cerrar la tapa del compartimiento de cubetas.
c) Con <STAR ENTER> iniciar el Auto-Prueba (en el display aparecerá prueba del
sistema, prueba de filtro, prueba de lámpara, calibración de longitud de onda).
d) Seleccionar calibración de longitud de onda.
e) Para realizar las mediciones el espectrofotómetro requiere un tiempo de calentamiento
de 15 minutos después de haberlo encendido, no se debe llevar a cabo mediciones durante
el tiempo de calentamiento, cuando haya finalizado el calentamiento, en la parte superior
derecha del display a lado de la fecha aparece la indicación con un llenado, es decir que
se puede proceder a realizar mediciones.
f) Se introducen los cristales patrón y se comprueban a las longitudes de onda
establecidas para dichos cristales que la transmitancia es la correcta.
g) Se anotan los valores en el cuaderno de trabajo destinado para la calibración del
espectrofotómetro.
h) Se procede a utilizar el espectrofotómetro para cualquiera de los modos concentración,
absorción/% transmisión, múltiples longitud de onda, espectro, cinética.
2.8. Preparación de las muestras fortificadas utilizadas en la validación.
Tanto para sulfatos, sulfuros y fenoles, para preparar las muestras fortificadas a utilizarse,
se debe:
a) Medir la concentración inicial de la muestra a fortificar.

30
b) Definir la concentración de la muestra fortificada que se desea preparar.
c) Calcular el volumen que se debe añadir del patrón, solución de trabajo o del material
de referencia certificado a la muestra que se desea fortificar.
d) Aforar en el material volumétrico correspondiente con la muestra inicial.
e) Seguir el mismo procedimiento para preparar las muestras fortificadas a los demás
niveles.
2.9. Procedimientos de ensayo.
2.9.1. Procedimiento de ensayo para sulfatos.
a) Tomar un mínimo de 200 mL de muestra en frascos de plástico o vidrio y mantener a
temperatura de refrigeración por un máximo de 28 días.
b) Homogenizar la muestra (previamente estabilizada al ambiente).
c) Transferir una alícuota de 25 mL de las soluciones de calibración o muestra a un
matraz Erlenmeyer con una pipeta volumétrica.
d) Añadir el contenido de un sachet de SULFAVER 4, para la formación de turbidez del
sulfato de bario.
e) Agitar durante 60 segundos, hasta que el contenido se disuelva totalmente.
f) Se requiere de 5 minutos para el desarrollo completo de la turbidez.
g) Medir la absorbancia en el espectrofotómetro a 420 nm en un tiempo máximo de 10
minutos desde que se disolvió el contenido del sachet, previamente encerar el equipo con
el blanco (muestra sin reactivo).
h) Anotar los valores en el registro.
2.9.2. Procedimiento de ensayo para sulfuros.
a) Recoger las muestras evitando la aireación de las mismas y mantenerlas a
temperatura de refrigeración por un máximo de 28 días.
b) Preparar un blanco llenando un tubo de ensayo con 10 mL de agua destilada (blanco
agua destilada con reactivos).
c) Con una pipeta volumétrica tomar 10 mL de las soluciones de calibración o muestra
y no agitarla más de lo necesario para evitar la pérdida de sulfuro.
d) Añadir 0,5 mL del reactivo Sulfuro 1, mover el tubo de ensayo suavemente en forma
circular (máximo dos veces) y tapar.

31
e) Añadir 0,5 mL del reactivo Sulfuro 2 a los tubos de ensayo, tapar e invertir de forma
suave y corta para mezclar. Un color rosado se desarrolla inicialmente, si el sulfuro está
presente la solución se vuelve azul.
f) Dejar reaccionar por 5 minutos.
g) Cuando el tiempo termine inmediatamente encerar el equipo con el blanco y proceder
a medir las muestras a 665 nm.
h) Anotar los valores en el registro.
2.9.3. Procedimiento de ensayo para fenoles.
a) Mantener las muestras que se van a analizar a temperatura de refrigeración hasta su
análisis por un máximo de 28 días.
b) Tomar 100 mL de la muestra, ajustar el pH a un valor aproximado de 4 con una
solución de ácido fosfórico diluido 1:10, utilizando para ello un indicador de naranja de
metilo (hasta cloración rosa) o el potenciómetro y colocar en el aparato de destilación.
c) Destilar por completo (una destilación debe ser suficiente para purificar la muestra
de una forma adecuada, pero si el destilado es turbio se debe acidificar con solución de
ácido fosfórico y destilar de nuevo según las especificaciones anteriores).
d) Para el blanco, medir 100 mL de agua destilada en una probeta y colocarla en un
embudo de separación.
e) Colocar 100 mL de las soluciones de calibración o muestra destilada en embudos de
separación.
f) Agregar 5 mL de la solución buffer Hardness en los embudos de separación y agitar.
g) Añadir 1 sachet del reactivo Phenol 1, agitar hasta que el reactivo se haya disuelto
por completo.
h) Añadir 1 sachet del reactivo Phenol 2, agitar hasta que el reactivo se haya disuelto
por completo.
i) Añadir 25 mL de cloroformo a cada embudo de separación, agitar vigorosamente
durante 30 segundos. Se debe abrir la válvula de los embudos de vez en cuando para
liberar la presión.
j) Esperar a que las dos fases se separen.
k) Colocar algodón en la punta de cada vástago y proceder a recoger la fase de
cloroformo en vasos de precipitación limpios y secos.
l) Proceder a leer la absorbancia a 460 nm.
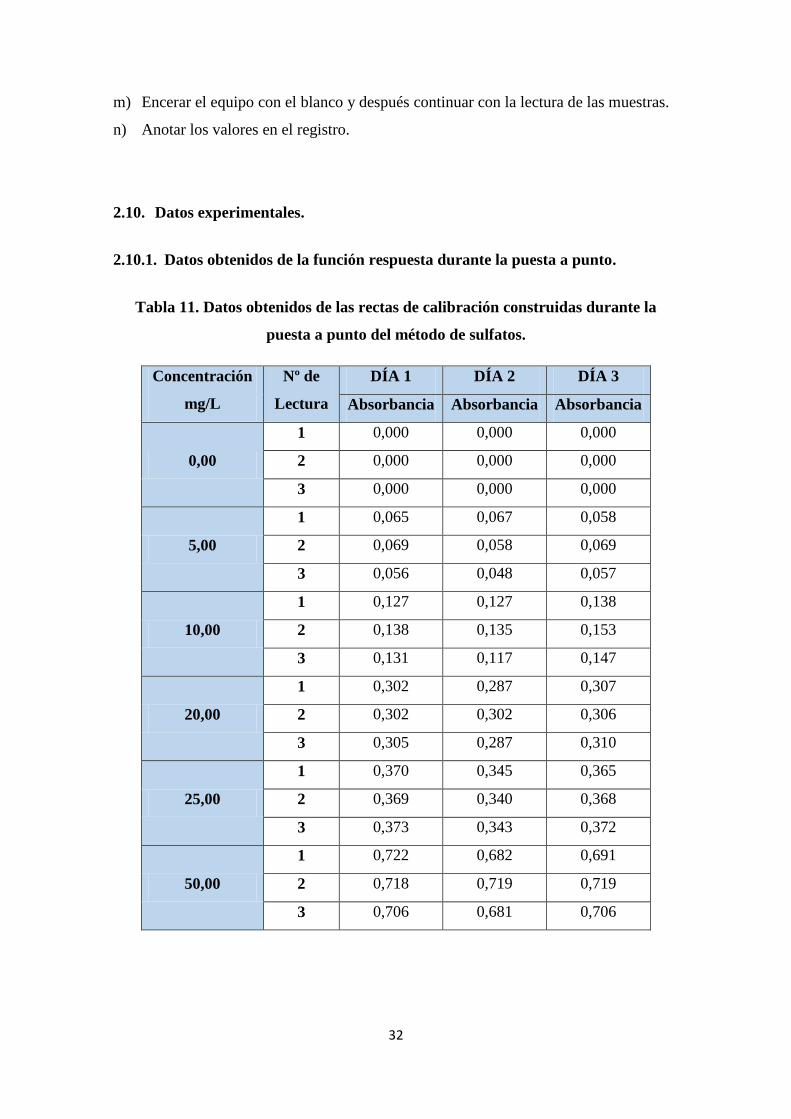
32
m) Encerar el equipo con el blanco y después continuar con la lectura de las muestras.
n) Anotar los valores en el registro.
2.10. Datos experimentales.
2.10.1. Datos obtenidos de la función respuesta durante la puesta a punto.
Tabla 11. Datos obtenidos de las rectas de calibración construidas durante la
puesta a punto del método de sulfatos.
Concentración
mg/L
Nº de
Lectura
DÍA 1 DÍA 2 DÍA 3
Absorbancia Absorbancia Absorbancia
0,00
1 0,000 0,000 0,000
2 0,000 0,000 0,000
3 0,000 0,000 0,000
5,00
1 0,065 0,067 0,058
2 0,069 0,058 0,069
3 0,056 0,048 0,057
10,00
1 0,127 0,127 0,138
2 0,138 0,135 0,153
3 0,131 0,117 0,147
20,00
1 0,302 0,287 0,307
2 0,302 0,302 0,306
3 0,305 0,287 0,310
25,00
1 0,370 0,345 0,365
2 0,369 0,340 0,368
3 0,373 0,343 0,372
50,00
1 0,722 0,682 0,691
2 0,718 0,719 0,719
3 0,706 0,681 0,706
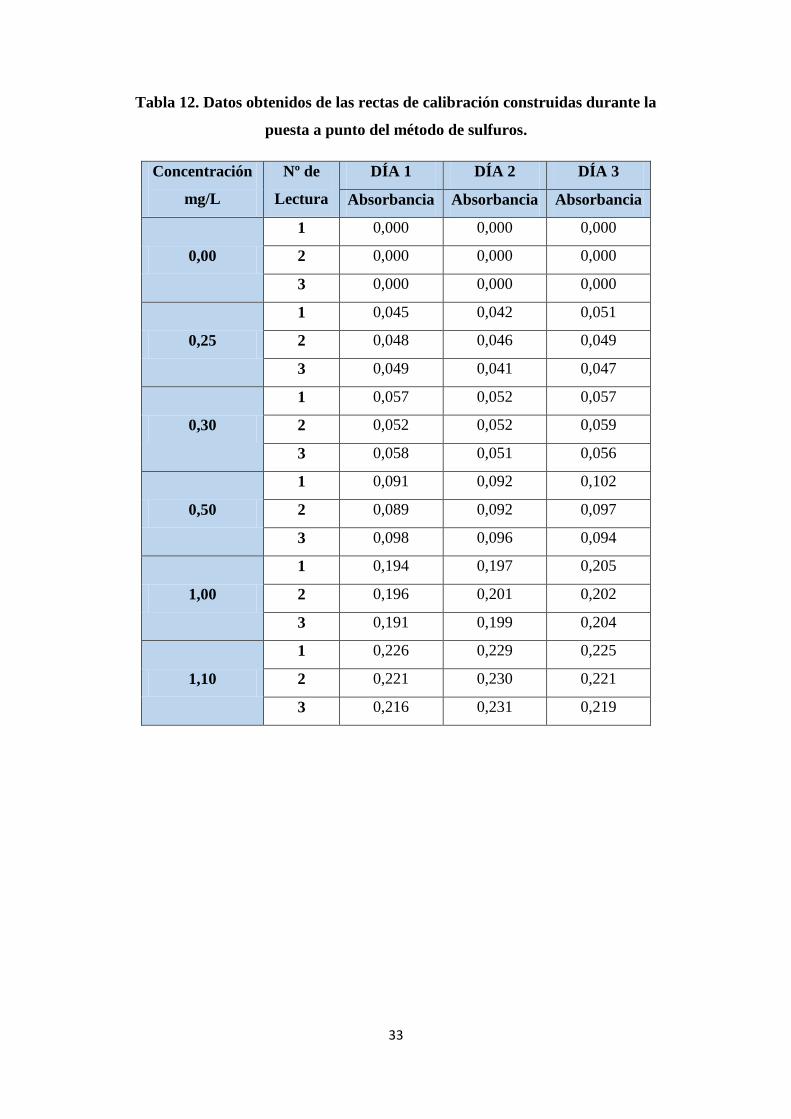
33
Tabla 12. Datos obtenidos de las rectas de calibración construidas durante la
puesta a punto del método de sulfuros.
Concentración
mg/L
Nº de
Lectura
DÍA 1 DÍA 2 DÍA 3
Absorbancia Absorbancia Absorbancia
0,00
1 0,000 0,000 0,000
2 0,000 0,000 0,000
3 0,000 0,000 0,000
0,25
1 0,045 0,042 0,051
2 0,048 0,046 0,049
3 0,049 0,041 0,047
0,30
1 0,057 0,052 0,057
2 0,052 0,052 0,059
3 0,058 0,051 0,056
0,50
1 0,091 0,092 0,102
2 0,089 0,092 0,097
3 0,098 0,096 0,094
1,00
1 0,194 0,197 0,205
2 0,196 0,201 0,202
3 0,191 0,199 0,204
1,10
1 0,226 0,229 0,225
2 0,221 0,230 0,221
3 0,216 0,231 0,219
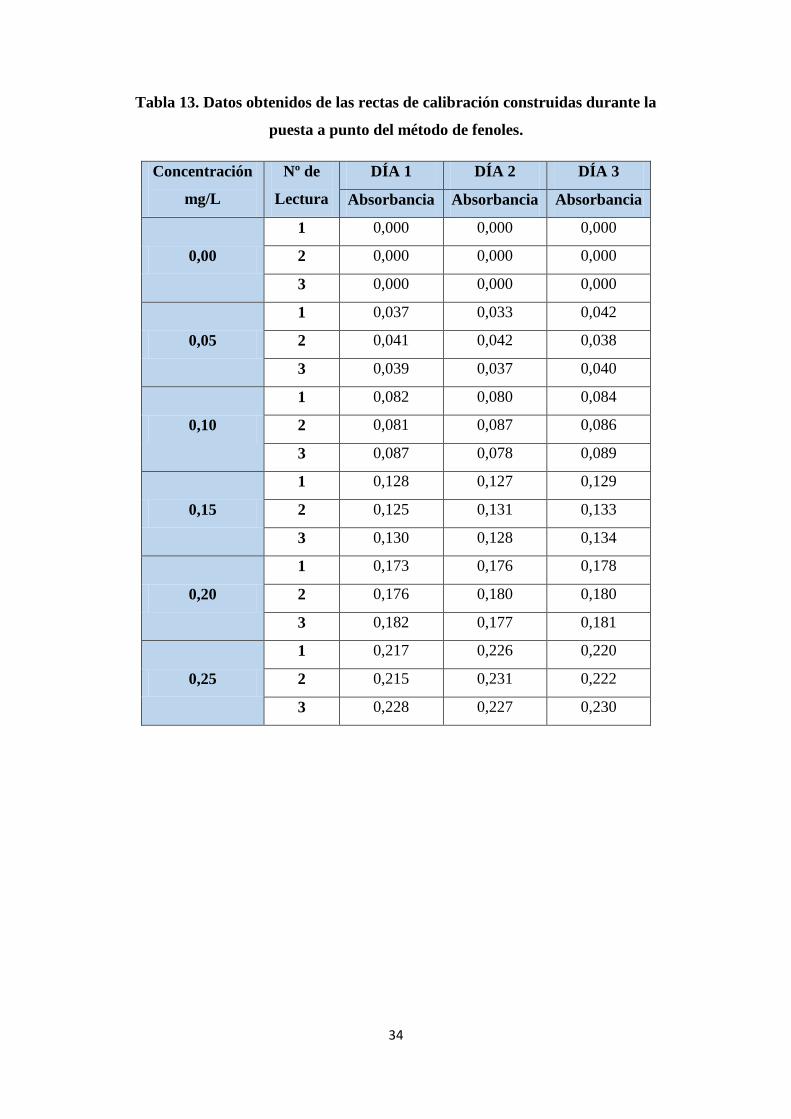
34
Tabla 13. Datos obtenidos de las rectas de calibración construidas durante la
puesta a punto del método de fenoles.
Concentración
mg/L
Nº de
Lectura
DÍA 1 DÍA 2 DÍA 3
Absorbancia Absorbancia Absorbancia
0,00
1 0,000 0,000 0,000
2 0,000 0,000 0,000
3 0,000 0,000 0,000
0,05
1 0,037 0,033 0,042
2 0,041 0,042 0,038
3 0,039 0,037 0,040
0,10
1 0,082 0,080 0,084
2 0,081 0,087 0,086
3 0,087 0,078 0,089
0,15
1 0,128 0,127 0,129
2 0,125 0,131 0,133
3 0,130 0,128 0,134
0,20
1 0,173 0,176 0,178
2 0,176 0,180 0,180
3 0,182 0,177 0,181
0,25
1 0,217 0,226 0,220
2 0,215 0,231 0,222
3 0,228 0,227 0,230
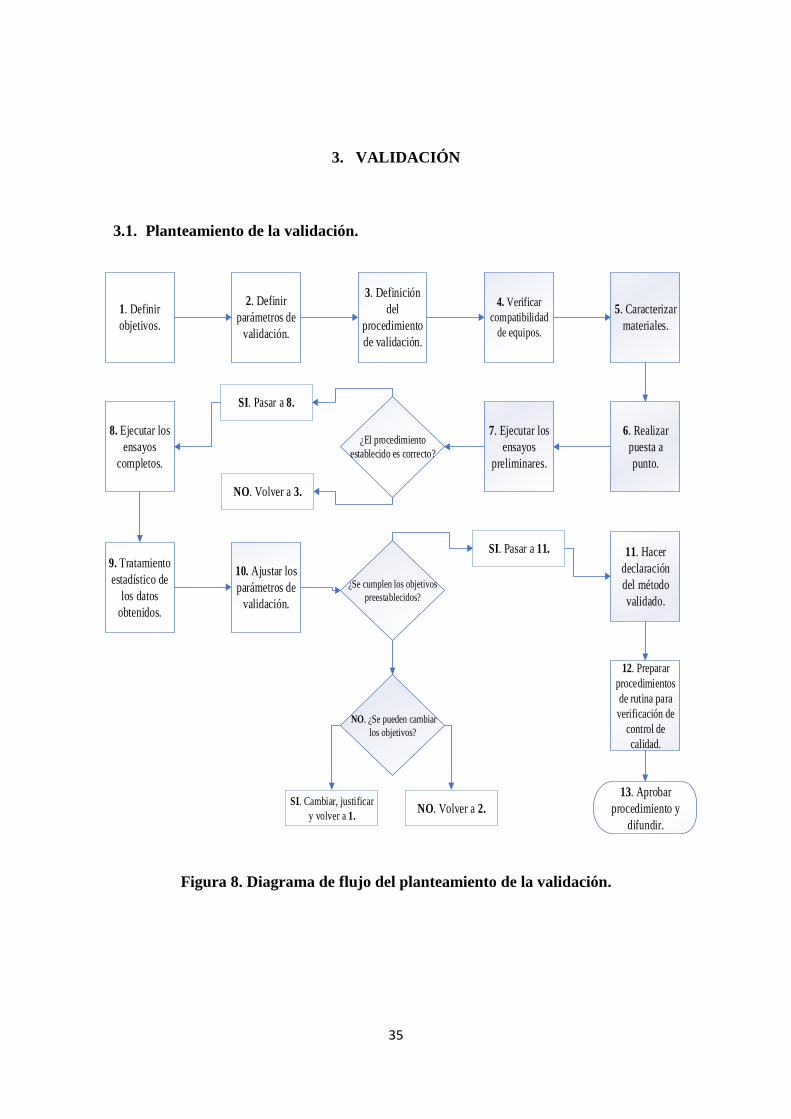
35
3. VALIDACIÓN
3.1. Planteamiento de la validación.
1. Definir
objetivos.
2. Definir
parámetros de
validación.
3. Definición
del
procedimiento
de validación.
5. Caracterizar
materiales.
6. Realizar
puesta a
punto.
7. Ejecutar los
ensayos
preliminares.
9. Tratamiento
estadístico de
los datos
obtenidos.
10. Ajustar los
parámetros de
validación.
8. Ejecutar los
ensayos
completos.
4. Verificar
compatibilidad
de equipos.
SI. Pasar a 11. 11. Hacer
declaración
del método
validado.
12. Preparar
procedimientos
de rutina para
verificación de
control de
calidad.
¿El procedimiento
establecido es correcto?
SI. Pasar a 8.
NO. Volver a 3.
¿Se cumplen los objetivos
preestablecidos?
NO. ¿Se pueden cambiar
los objetivos?
SI. Cambiar, justificar
y volver a 1.NO. Volver a 2.
13. Aprobar
procedimiento y
difundir.
Figura 8. Diagrama de flujo del planteamiento de la validación.

36
3.2. Datos para la validación.
Tabla 14. Datos primarios para validación de sulfatos en aguas, concentraciones
obtenidas a diferentes niveles.
Nivel Concentración
mg/L
Nº de
Lectura DÍA 1 DÍA 2 DÍA 3
1 7,00
1 7,00 6,00 7,00
2 7,00 7,00 8,00
3 7,00 7,00 6,00
4 7,00 7,00 7,00
5 8,00 6,00 7,00
2 14,00
1 13,00 13,00 14,00
2 13,00 13,00 13,00
3 13,00 14,00 14,00
4 13,00 13,00 14,00
5 14,00 14,00 13,00
3 20,00
1 19,00 21,00 21,00
2 21,00 21,00 20,00
3 21,00 20,00 21,00
4 20,00 21,00 21,00
5 21,00 20,00 20,00
4 25,00
1 24,00 23,00 25,00
2 25,00 24,00 24,00
3 25,00 24,00 24,00
4 25,00 25,00 24,00
5 25,00 26,00 25,00
5 48,00
1 48,00 47,00 46,00
2 49,00 46,00 47,00
3 49,00 46,00 49,00
4 47,00 49,00 47,00
5 48,00 47,00 46,00

37
Tabla 15. Datos primarios para validación de sulfuros en aguas, concentraciones
obtenidas a diferentes niveles.
Nivel Concentración
mg/L
Nº de
Lectura DÍA 1 DÍA 2 DÍA 3
1 0,28
1 0,27 0,28 0,27
2 0,27 0,28 0,28
3 0,28 0,28 0,28
4 0,28 0,26 0,26
5 0,28 0,27 0,28
2 0,45
1 0,46 0,45 0,45
2 0,44 0,46 0,46
3 0,46 0,45 0,44
4 0,46 0,45 0,45
5 0,45 0,44 0,46
3 1,00
1 1,00 0,98 0,97
2 1,02 1,00 0,99
3 1,00 1,04 1,04
4 1,00 1,00 1,02
5 1,00 1,00 1,00
Tabla 16. Datos primarios para validación de fenoles en aguas, concentraciones
obtenidas a diferentes niveles.
Nivel Concentración
mg/L
Nº de
Lectura DÍA 1 DÍA 2 DÍA 3
1 0,10
1 0,09 0,09 0,10
2 0,10 0,08 0,11
3 0,09 0,09 0,11
4 0,08 0,08 0,10
5 0,10 0,11 0,10
2 0,14
1 0,13 0,14 0,13
2 0,14 0,12 0,12
3 0,16 0,13 0,13
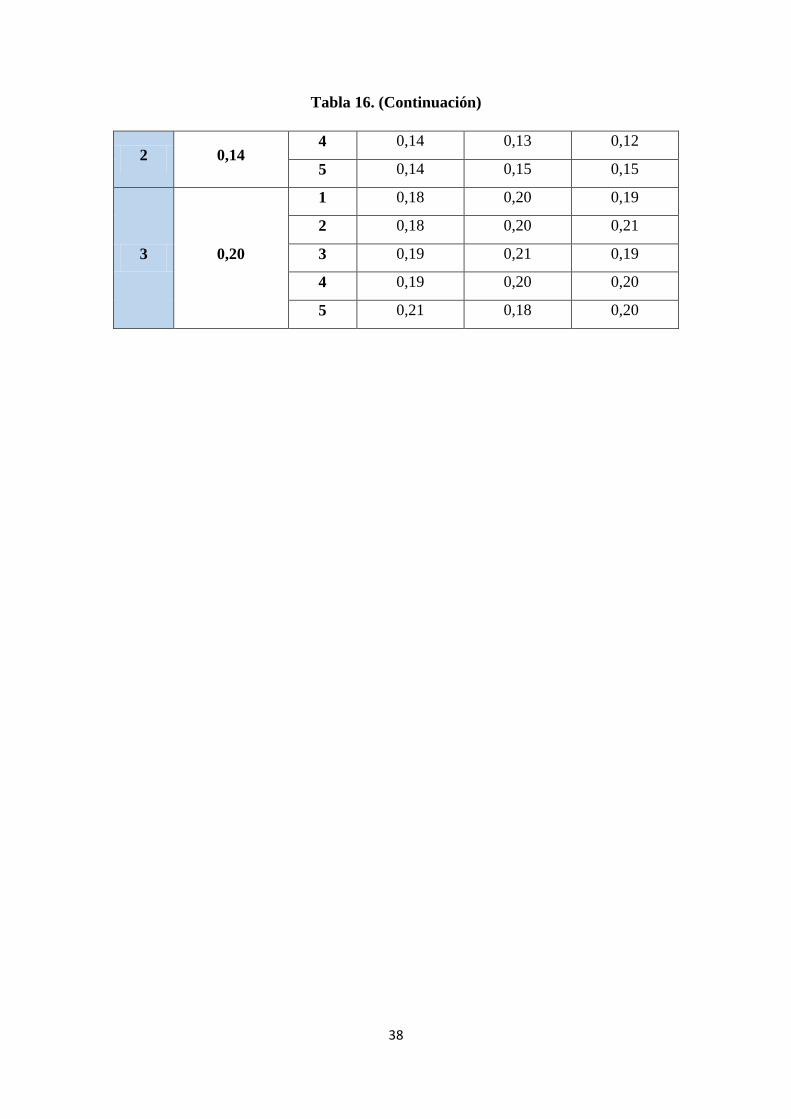
38
Tabla 16. (Continuación)
2 0,14 4 0,14 0,13 0,12
5 0,14 0,15 0,15
3 0,20
1 0,18 0,20 0,19
2 0,18 0,20 0,21
3 0,19 0,21 0,19
4 0,19 0,20 0,20
5 0,21 0,18 0,20

39
4. CÁLCULOS
4.1. Cálculo para la preparación de las muestras fortificadas.
a) Realizar la lectura de concentración inicial de la muestra a fortificar (cinco veces) y
sacar el promedio, mg/L.
b) Calcular con las siguientes ecuaciones el volumen del material de referencia
certificado (MRC) a añadir a la muestra inicial para tener la concentración final requerida.
𝐶𝐴𝑀 = 𝐶𝐹𝑅 − 𝑃𝑀𝐼 (12)
𝑉𝑀𝑅𝐶 =𝐶𝐴𝑀∗𝑉𝐴
𝐶𝑀𝑅𝐶 (13)
Donde:
𝐶𝐴𝑀 = concentración a añadir para obtener la concentración final requerida, mg/L.
𝐶𝐹𝑅 = concentración final requerida, mg/L.
𝑃𝑀𝐼 = promedio de concentraciones de la muestra inicial, mg/L.
𝑉𝑀𝑅𝐶 = volumen del material de referencia certificado a añadir a la muestra inicial, L.
𝐶𝑀𝑅𝐶 = concentración del material de referencia certificado, mg/L.
𝑉𝐴 = volumen de aforo, L.
c) Preparar la muestra fortificada.
4.1.1. Cálculo modelo para la muestra fortificada de 7 mg/L en sulfatos.
Tabla 17. Concentración inicial de la muestra para sulfatos.
Concentración 1, mg/L 3,00
Concentración 2, mg/L 2,00
Concentración 3, mg/L 3,00
Concentración 4, mg/L 3,00
Concentración 5, mg/L 2,00
Promedio, mg/L 2,60

40
Tabla 18. Concentración a añadir a la muestra para sulfatos.
Concentración MRC, mg/L 500
Concentración inicial, mg/L 2,60
Concentración a añadir, mg/L 4,40
Se aplica la ecuación 13:
𝑉𝑀𝑅 =4,40
𝑚𝑔𝐿 ∗ 0,2𝐿
500𝑚𝑔
𝐿
𝑉𝑀𝑅 = 0,0018 𝐿
𝑉𝑀𝑅 = 1,80 𝑚𝐿
Se debe añadir 1,80 mL del material de referencia a la muestra de concentración inicial
de 2,60 mg/L, para obtener una concentración final requerida de 7 mg/L.
Nota: se prepara las muestras fortificadas de sulfatos en balones de 200 mL.
4.2. Determinación de la linealidad de la función de respuesta.
Se determina utilizando el método de mínimos cuadrados. Los cálculos modelo que a
continuación se describen hacen referencia al método para la determinación de sulfatos
en aguas.
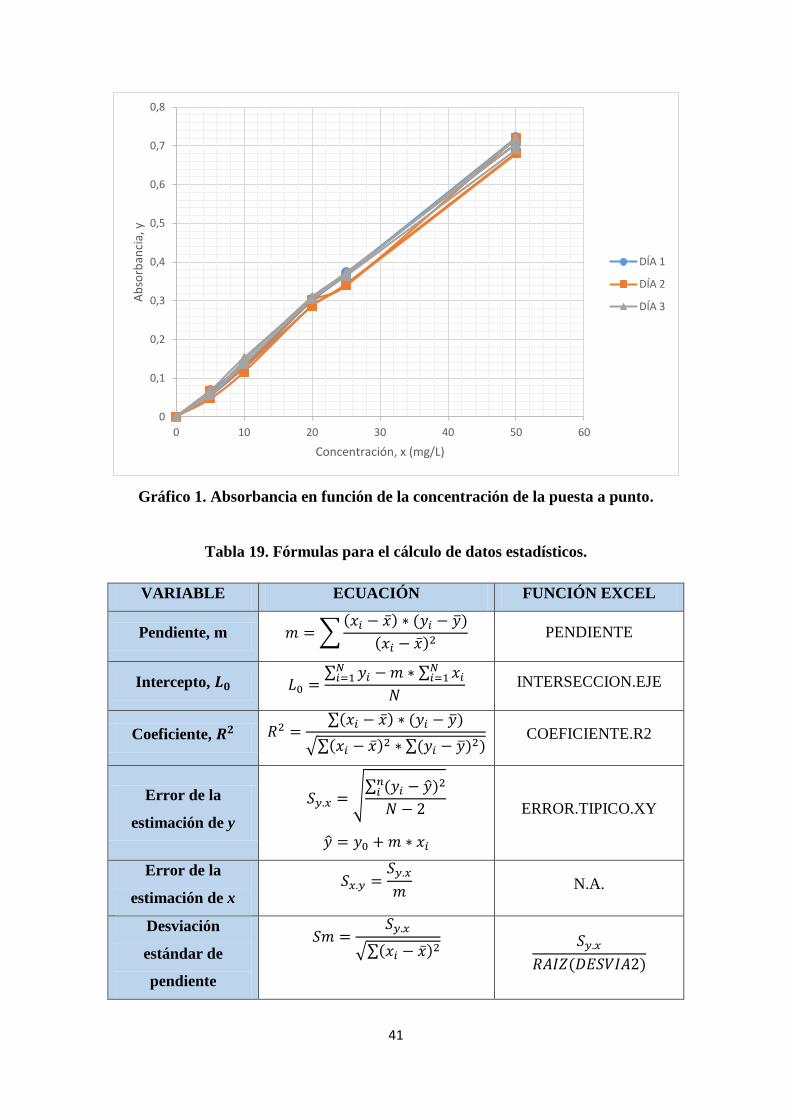
41
Gráfico 1. Absorbancia en función de la concentración de la puesta a punto.
Tabla 19. Fórmulas para el cálculo de datos estadísticos.
VARIABLE ECUACIÓN FUNCIÓN EXCEL
Pendiente, m 𝑚 = ∑(𝑥𝑖 − �̅�) ∗ (𝑦𝑖 − �̅�)
(𝑥𝑖 − �̅�)2 PENDIENTE
Intercepto, 𝑳𝟎 𝐿0 =∑ 𝑦𝑖 − 𝑚 ∗ ∑ 𝑥𝑖
𝑁𝑖=1
𝑁𝑖=1
𝑁 INTERSECCION.EJE
Coeficiente, 𝑹𝟐 𝑅2 =∑(𝑥𝑖 − �̅�) ∗ (𝑦𝑖 − �̅�)
√∑(𝑥𝑖 − �̅�)2 ∗ ∑(𝑦𝑖 − �̅�)2) COEFICIENTE.R2
Error de la
estimación de y
𝑆𝑦.𝑥 = √∑ (𝑦𝑖 − �̂�)2𝑛
𝑖
𝑁 − 2
�̂� = 𝑦0 + 𝑚 ∗ 𝑥𝑖
ERROR.TIPICO.XY
Error de la
estimación de x 𝑆𝑥.𝑦 =
𝑆𝑦.𝑥
𝑚 N.A.
Desviación
estándar de
pendiente
𝑆𝑚 =𝑆𝑦.𝑥
√∑(𝑥𝑖 − �̅�)2 𝑆𝑦.𝑥
𝑅𝐴𝐼𝑍(𝐷𝐸𝑆𝑉𝐼𝐴2)
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0 10 20 30 40 50 60
Ab
sorb
anci
a, y
Concentración, x (mg/L)
DÍA 1
DÍA 2
DÍA 3
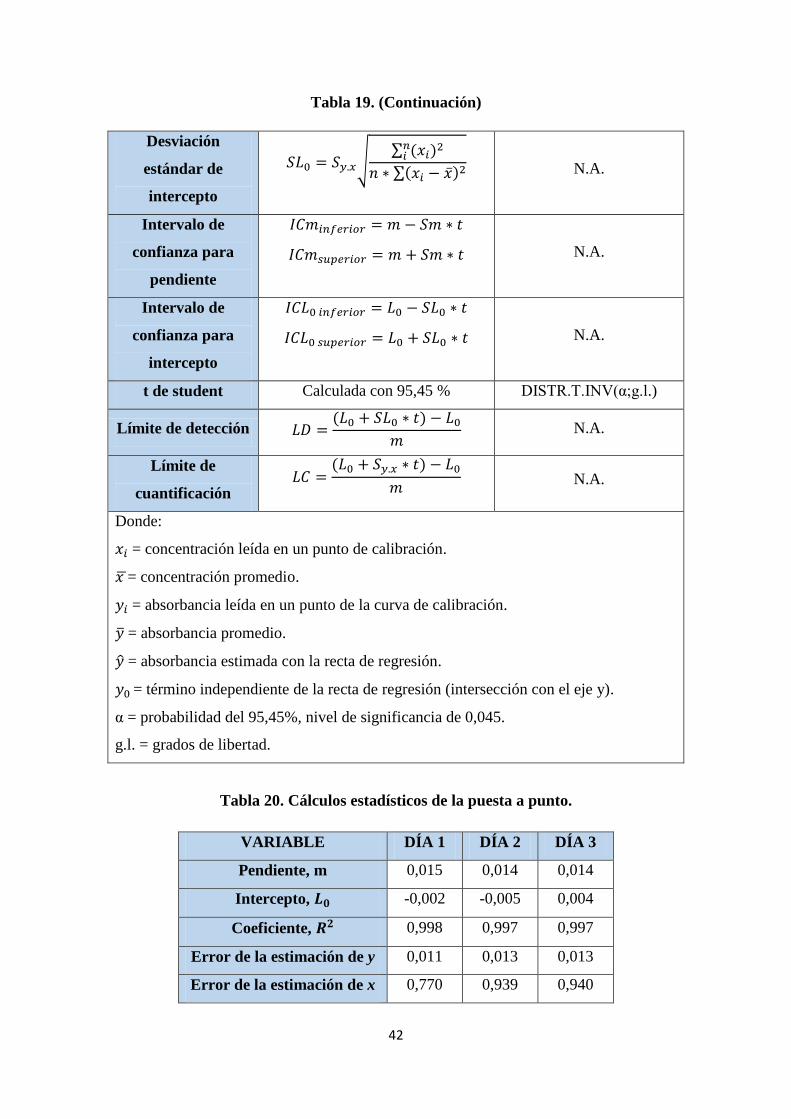
42
Tabla 19. (Continuación)
Desviación
estándar de
intercepto
𝑆𝐿0 = 𝑆𝑦.𝑥√∑ (𝑥𝑖)2𝑛
𝑖
𝑛 ∗ ∑(𝑥𝑖 − �̅�)2 N.A.
Intervalo de
confianza para
pendiente
𝐼𝐶𝑚𝑖𝑛𝑓𝑒𝑟𝑖𝑜𝑟 = 𝑚 − 𝑆𝑚 ∗ 𝑡
𝐼𝐶𝑚𝑠𝑢𝑝𝑒𝑟𝑖𝑜𝑟 = 𝑚 + 𝑆𝑚 ∗ 𝑡 N.A.
Intervalo de
confianza para
intercepto
𝐼𝐶𝐿0 𝑖𝑛𝑓𝑒𝑟𝑖𝑜𝑟 = 𝐿0 − 𝑆𝐿0 ∗ 𝑡
𝐼𝐶𝐿0 𝑠𝑢𝑝𝑒𝑟𝑖𝑜𝑟 = 𝐿0 + 𝑆𝐿0 ∗ 𝑡 N.A.
t de student Calculada con 95,45 % DISTR.T.INV(α;g.l.)
Límite de detección 𝐿𝐷 =(𝐿0 + 𝑆𝐿0 ∗ 𝑡) − 𝐿0
𝑚 N.A.
Límite de
cuantificación 𝐿𝐶 =
(𝐿0 + 𝑆𝑦.𝑥 ∗ 𝑡) − 𝐿0
𝑚 N.A.
Donde:
𝑥𝑖 = concentración leída en un punto de calibración.
𝑥 ̅= concentración promedio.
𝑦𝑖 = absorbancia leída en un punto de la curva de calibración.
�̅� = absorbancia promedio.
�̂� = absorbancia estimada con la recta de regresión.
𝑦0 = término independiente de la recta de regresión (intersección con el eje y).
α = probabilidad del 95,45%, nivel de significancia de 0,045.
g.l. = grados de libertad.
Tabla 20. Cálculos estadísticos de la puesta a punto.
VARIABLE DÍA 1 DÍA 2 DÍA 3
Pendiente, m 0,015 0,014 0,014
Intercepto, 𝑳𝟎 -0,002 -0,005 0,004
Coeficiente, 𝑹𝟐 0,998 0,997 0,997
Error de la estimación de y 0,011 0,013 0,013
Error de la estimación de x 0,770 0,939 0,940

43
Tabla 20. (Continuación)
Desviación estándar de
pendiente 0,0002 0,0002 0,0002
Desviación estándar de
intercepto 0,0039 0,0046 0,0047
Intervalo de confianza para
pendiente
0,014
0,015
0,014
0,014
0,014
0,015
Intervalo de confianza para
intercepto
-0,010
0,007
-0,015
0,005
-0,007
0,014
t de student 2,169 2,169 2,169
Límite de detección 0,588 0,718 0,718
Límite de cuantificación 1,670 2,038 2,039
Con los estadísticos obtenidos de la puesta a punto se establecen los rangos para pendiente
e intercepto, que deberán cumplir las curvas de validación diarias.
Tabla 21. Intervalo de trabajo para pendiente e intercepto.
m m min m máx Lo Lo min Lo máx
DÍA 1 0,015 0,014 0,015 -0,002 -0,010 0,007
DÍA 2 0,014 0,014 0,014 -0,005 -0,015 0,005
DÍA 3 0,014 0,014 0,015 0,004 -0,007 0,014
CONJUNTO 0,014 0,014 0,015 -0,001 -0,015 0,014
Se procede a construir las rectas de calibración para la validación de los métodos de
ensayo que deben cumplir con los requisitos antes impuestos y después se fijará la mejor
curva para los diferentes parámetros.
Tabla 22. Rectas de calibración para la validación.
Concentración (x),
mg/L
DÍA 1
Absorbancia (y)
DÍA 2
Absorbancia (y)
DÍA 3
Absorbancia (y)
0 0,000 0,000 0,000

44
Tabla 22. (Continuación)
5 0,052 0,049 0,066
10 0,134 0,135 0,126
20 0,284 0,298 0,295
25 0,358 0,353 0,364
50 0,687 0,701 0,696
Gráfico 2. Absorbancia en función de la concentración para la validación.
Tabla 23. Datos estadísticos de las curvas de calibración utilizadas para la
validación.
PARÁMETRO DÍA 1
Absorbancia (y)
DÍA 2
Absorbancia (y)
DÍA 3
Absorbancia (y)
m 0,014 0,014 0,014
𝑳𝟎 -0,003 -0,005 -0,001
𝑹𝟐 0,998 0,998 0,998
𝑆𝑦.𝑥 0,011 0,013 0,012
𝑆𝑥.𝑦 0,803 0,927 0,886
y = 0,014x - 0,0035R² = 0,9984
y = 0,0142x - 0,0052R² = 0,9979
y = 0,0141x - 0,0006R² = 0,9981
-0,1
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0 10 20 30 40 50 60
Ab
sorb
anci
a, y
Concentración, x (mg/L)
DÍA 1
DÍA 2
DÍA 3
Lineal (DÍA 1)
Lineal (DÍA 2)
Lineal (DÍA 3)

45
Tabla 23. (Continuación)
g.l. 4 4 4
t (95,45%) 2,869 2,869 2,869
4.3. Cálculo de los parámetros de validación.
Los resultados que se obtiene a continuación corresponden a la curva de calibración
utilizada el primer día de la puesta a punto.
4.3.1. Límite de detección (LD). El límite de detección se calcula a partir de la recta de
calibración (recta del día 1) obtenida durante la puesta a punto de los métodos. Se utiliza
la fórmula descrita en la tabla 19 para el límite de detección:
𝐿𝐷 =(𝐿0+𝑆𝐿0∗𝑡)−𝐿0
𝑚 (14)
𝐿𝐷 =(−0,002 + (0,0039 ∗ 2,169)) − (−0,002)
0,015
𝐿𝐷 =0,588 mg/L
4.3.2. Límite de cuantificación (LC). El límite de cuantificación de igual manera se
calcula a partir de la recta de calibración (recta del día 1) obtenida durante la puesta a
punto de los respectivos métodos. Se utiliza la fórmula descrita en la tabla 19 para el
límite de cuantificación:
𝐿𝐶 =(𝐿0+𝑆𝑦.𝑥∗𝑡)−𝐿0
𝑚 (15)
𝐿𝐶 =(−0,002 + (0,011) ∗ (2,169)) − (−0,002)
0,015
𝐿𝐷 =1,670 mg/L
4.3.3. Veracidad.
4.3.3.1. Veracidad para muestras fortificadas.
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜−𝑥𝑖𝑛𝑖𝑐𝑖𝑎𝑙
𝑥𝑎𝑑𝑖𝑐𝑖𝑜𝑛𝑎𝑑𝑜∗ 100 (16)
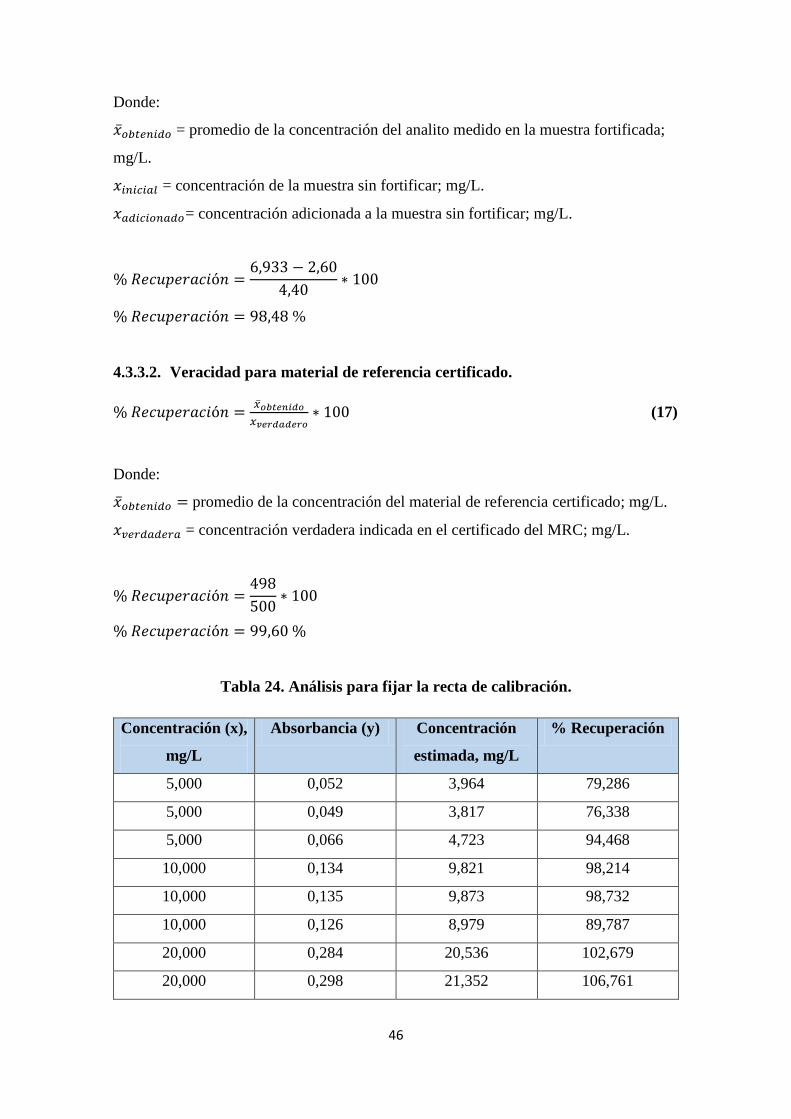
46
Donde:
�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜 = promedio de la concentración del analito medido en la muestra fortificada;
mg/L.
𝑥𝑖𝑛𝑖𝑐𝑖𝑎𝑙 = concentración de la muestra sin fortificar; mg/L.
𝑥𝑎𝑑𝑖𝑐𝑖𝑜𝑛𝑎𝑑𝑜= concentración adicionada a la muestra sin fortificar; mg/L.
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =6,933 − 2,60
4,40∗ 100
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 = 98,48 %
4.3.3.2. Veracidad para material de referencia certificado.
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜
𝑥𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑜∗ 100 (17)
Donde:
�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜 = promedio de la concentración del material de referencia certificado; mg/L.
𝑥𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑎 = concentración verdadera indicada en el certificado del MRC; mg/L.
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =498
500∗ 100
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 = 99,60 %
Tabla 24. Análisis para fijar la recta de calibración.
Concentración (x),
mg/L
Absorbancia (y) Concentración
estimada, mg/L
% Recuperación
5,000 0,052 3,964 79,286
5,000 0,049 3,817 76,338
5,000 0,066 4,723 94,468
10,000 0,134 9,821 98,214
10,000 0,135 9,873 98,732
10,000 0,126 8,979 89,787
20,000 0,284 20,536 102,679
20,000 0,298 21,352 106,761

47
Tabla 24. (Continuación)
20,000 0,295 20,965 104,823
25,000 0,358 25,821 103,286
25,000 0,353 25,225 100,901
25,000 0,364 25,858 103,433
50,000 0,687 49,321 98,643
50,000 0,701 49,732 99,465
50,000 0,696 49,404 98,809
De acuerdo con los resultados de la tabla 24, la curva que cumple con los parámetros de
porcentaje de recuperación para todos los niveles es la curva 3, puesto que 79,286 %
(curva 1) y 76,338 % (curva 2) son menores al 80 % de veracidad fijado en los objetivos.
La ecuación lineal ajustada a la curva de calibración 3 (ver gráfico 2) es la siguiente:
𝑦 = 0,0141𝑥 − 0,0006 (18)
Donde:
𝑦 = absorbancia ajustada a la ecuación lineal, abs.
𝑥 = concentración ajustada a la ecuación lineal, mg/L.
4.3.4. Precisión.
4.3.4.1. Cálculo modelo para la precisión para determinar sulfatos en aguas para
la concentración de 7 mg/L.
Tabla 25. Datos reportados por día.
Concentración
mg/L
DÍA 1 DÍA 2 DÍA 3
7
7 6 7
7 7 8
7 7 6
7 7 7
8 6 7

48
Tabla 26. Valor medio de las determinaciones.
Concentración
mg/L
DÍA 1 DÍA 2 DÍA 3
7
7 6 7
7 7 8
7 7 6
7 7 7
8 6 7
Suma 36,000 33,000 35,000
𝑿𝒎 7,200 6,600 7,000
∑(𝑿 − 𝑿𝒎)𝟐 0,800 1,200 2,000
Cálculo de la suma de diferencias dentro de grupos “𝑺𝑫𝑪𝑾”.
𝑆𝐷𝐶𝑊 = ∑ ∑ (𝑥𝑖𝑗 − 𝑥�̅�)25
𝑗=15𝑖=1 (19)
𝑆𝐷𝐶𝑊 = ∑(0,800 + 1,200 + 2,000)
5
𝑖=1
𝑆𝐷𝐶𝑊 = 4,000
Cálculo de la determinación del valor medio de las sumas de las diferencias al
cuadrado dentro de grupos “𝑫𝑪𝑴𝑾”.
𝐷𝐶𝑀𝑊 =𝑆𝐷𝐶𝑊
𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑑𝑎𝑡𝑜𝑠−𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑔𝑟𝑢𝑝𝑜𝑠 (20)
𝐷𝐶𝑀𝑊 =4,000
12
𝐷𝐶𝑀𝑊 = 0,333
Cálculo de la media de las medias.
�̅� =𝑥𝑚1+𝑥𝑚2+𝑥𝑚3
𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑚𝑒𝑑𝑖𝑎𝑠 (21)
�̅� =20,800
3

49
�̅� = 6,933 𝑚𝑔/𝐿
Cálculo de la desviación estándar global de la muestra.
𝑆 = √∑ (𝑥−�̅�)2𝑛
𝑖−1
𝑛−1 (22)
𝑆 = √4,933
15 − 1
𝑆 = 0,594
Cálculo de la suma de diferencias entre grupos “𝑺𝑫𝑪𝑩”.
𝑆𝐷𝐶𝐵 = ∑ 5(𝑥�̅� − �̅�)25𝑖=1 (23)
𝑆𝐷𝐶𝐵=5*(7,200 − 6,933)2+5*(6,600 − 6,933)2+5*(7,000 − 6,933)2
𝑆𝐷𝐶𝐵 = 0,933
Cálculo de la determinación del valor medio de las sumas de las diferencias al
cuadrado entre grupos “𝑫𝑪𝑴𝑩”.
𝐷𝐶𝑀𝐵 =𝑆𝐷𝐶𝐵
𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑔𝑟𝑢𝑝𝑜𝑠−1 (24)
𝐷𝐶𝑀𝐵 =0,933
2
𝐷𝐶𝑀𝐵 = 0,467
Cálculo de desviación estándar por repetibilidad “𝑺𝒓”.
𝑆𝑟 = √𝐷𝐶𝑀𝑊 (25)
𝑆𝑟 = √0,333
𝑆𝑟 = 0,577

50
Cálculo de precisión intermedia “𝑺𝑳𝟐”.
𝑆𝐿2 =
𝐷𝐶𝑀𝐵−𝐷𝐶𝑀𝑊
3 (26)
𝑆𝐿2 =
0,467 − 0,333
3
𝑆𝐿2 = 0,044
Cálculo de la desviación estándar por reproducibilidad “𝑺𝑹”.
𝑆𝑅 = √𝑆𝑟2+𝑆𝐿
2 (27)
𝑆𝑅 = √0,333 + 0,044
𝑆𝑅 = 0,615
Cálculo del coeficiente de variación de repetibilidad “𝑪𝑽𝒓”.
%𝐶𝑉𝑟 =𝑆𝑟
�̅�∗ 100 (28)
%𝐶𝑉𝑟 =0,577
6,933∗ 100
%𝐶𝑉𝑟 = 8,327 %
Cálculo del coeficiente de variación de reproducibilidad “𝑪𝑽𝑹”.
%𝐶𝑉𝑅 =𝑆𝑅
�̅�∗ 100 (29)
%𝐶𝑉𝑅 =0,615
6,933∗ 100
%𝐶𝑉𝑅 = 8,865 %
Prueba de Hipótesis.
Se fijan las hipótesis:
Hipótesis nula (𝐻0) = no existe diferencia significativa entre datos.
Hipótesis alternativa (𝐻1) = existe diferencia significativa entre datos.

51
Aplicando la prueba F de Fisher:
𝐹𝑐𝑎𝑙 =𝑆𝐷𝐶𝐵𝑎−1
𝑆𝐷𝐶𝑊𝑎(𝑏−1)
(30)
𝐹𝑐𝑎𝑙 =
0,9333 − 14,00015 − 3
𝐹𝑐𝑎𝑙 = 1,400
Se obtiene el valor crítico de F de las tablas de Fisher tomado grados de libertad para el
numerador y denominador:
G.L para el numerador= 𝑎 − 1 = 3 − 1 = 2
G.L para el denominador= 𝑎(𝑏 − 1) = 3(5 − 1) = 12
Los niveles de significancia de 0,01 y 0,05 son los más utilizados de acuerdo con Stigler
y Stephen (1986). En este caso se escogió el nivel de significancia de 0,05 por ser próximo
al valor utilizado en la distribución t de student (0,045) en cálculos anteriores (ver tabla
19).
𝐹𝑡𝑎𝑏= 3,885
Entonces:
Si 𝐹𝑐𝑎𝑙<𝐹𝑡𝑎𝑏→No existe diferencia significativa entre datos.
Utilizando el valor P: se compara el valor P con el nivel de significancia escogido.
Del ANOVA calculado por Excel (ver anexos) se obtiene el valor de P= 0,284; lo cual
significa que existe una probabilidad de cometer una equivocación de 0,284; que es mayor
que el máximo de error que se permite con el 95% de confianza.
Entonces:
Si 𝑃>α→ Se ratifica que la hipótesis nula es válida, es decir, no existe diferencia
significativa entre datos.
Se sigue el mismo algoritmo de cálculo para el análisis de varianza de todos los métodos.
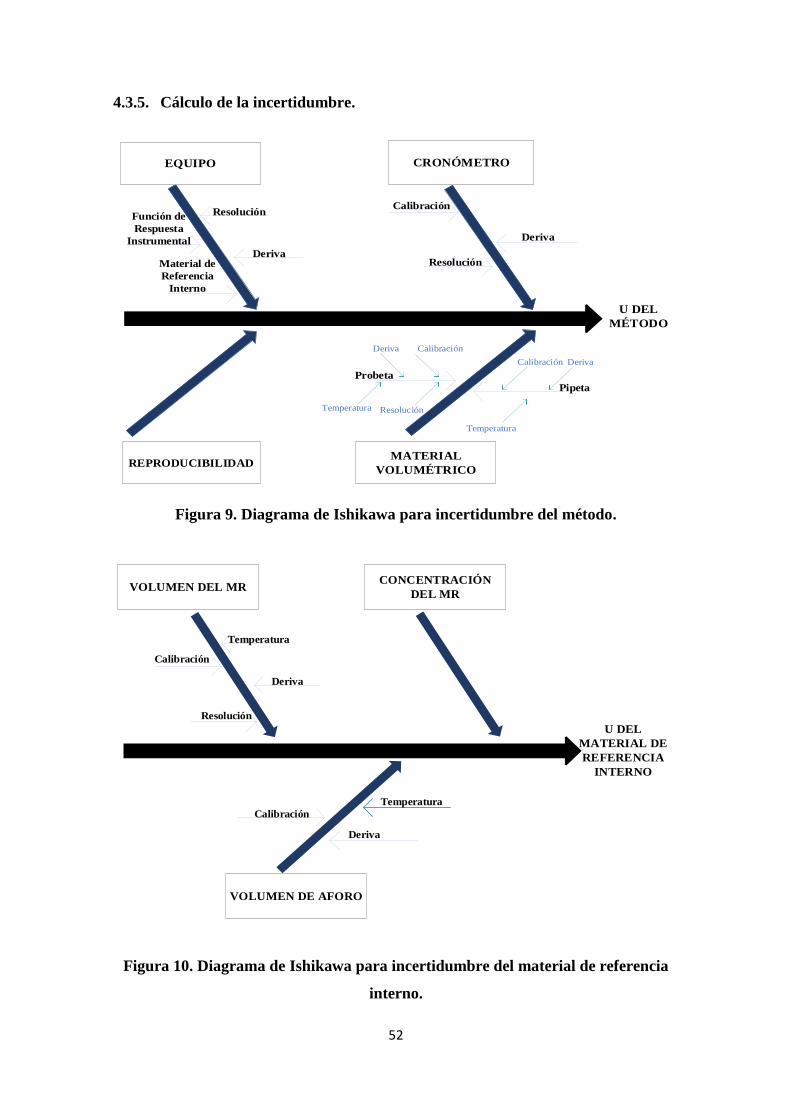
52
4.3.5. Cálculo de la incertidumbre.
Resolución
Deriva
Función de
Respuesta
Instrumental
Material de
Referencia
Interno
Calibración
Resolución
Deriva
Probeta
Pipeta
EQUIPO CRONÓMETRO
Calibración
Temperatura
CalibraciónDeriva
ResoluciónTemperatura
U DEL
MÉTODO
Deriva
MATERIAL
VOLUMÉTRICOREPRODUCIBILIDAD
Figura 9. Diagrama de Ishikawa para incertidumbre del método.
Temperatura
Deriva
Calibración
Resolución
Calibración
Deriva
VOLUMEN DEL MRCONCENTRACIÓN
DEL MR
VOLUMEN DE AFORO
Temperatura
U DEL
MATERIAL DE
REFERENCIA
INTERNO
Figura 10. Diagrama de Ishikawa para incertidumbre del material de referencia
interno.

53
Con el fin de establecer todas las contribuciones a la incertidumbre de medida, a
continuación se detalla el procedimiento modelo el cual es aplicado para su determinación
en el método de sulfatos.
La siguiente ecuación define la forma de determinar la incertidumbre estándar, aplicando
incertidumbres relativas, debido a que las variables que contribuyen a la incertidumbre
tienen diferentes unidades:
𝑢𝑆𝑂4−2
𝑆𝑂4−2 = √(
𝑢𝑅𝑒𝑝𝑟𝑜𝑑𝑢𝑐𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑
𝑆𝑂4−2 )
2
+ (𝑢𝐸𝑞𝑢𝑖𝑝𝑜
𝑆𝑂4−2 )
2
+ (𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜
𝑇𝑖𝑒𝑚𝑝𝑜)
2
+ (𝑢𝑀𝑉
𝑉𝑀)
2
(31)
𝑢𝑆𝑂4−2 = 𝑆𝑂4
−2√(𝑢𝑅𝑒𝑝𝑟𝑜𝑑𝑢𝑐𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑
𝑆𝑂4−2 )
2
+ (𝑢𝐸𝑞𝑢𝑖𝑝𝑜
𝑆𝑂4−2 )
2
+ (𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜
𝑇𝑖𝑒𝑚𝑝𝑜)
2
+ (𝑢𝑀𝑉
𝑉𝑀)
2
(32)
Donde:
𝑢𝑆𝑂4−2 = incertidumbre estándar del método de determinación de sulfatos, mg/L.
𝑆𝑂4−2= concentración de sulfatos, mg/L.
𝑢𝑅𝑒𝑝𝑟𝑜𝑑𝑢𝑐𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑= incertidumbre estándar de reproducibilidad, mg/L.
𝑢𝐸𝑞𝑢𝑖𝑝𝑜= incertidumbre estándar del equipo, mg/L.
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜= incertidumbre estándar del cronómetro, s.
𝑢𝑀𝑉= incertidumbre estándar del material volumétrico, mL.
VM= volumen del material volumétrico, mL.
La siguiente ecuación define la forma de determinar la incertidumbre total expandida de
la concentración de sulfatos:
𝑈𝑆𝑂4−2 = 𝑘𝑢𝑆𝑂4
−2 + ∑|𝐶𝑜𝑟𝑟𝑒𝑐𝑐𝑖𝑜𝑛𝑒𝑠 𝑛𝑜 𝐴𝑝𝑙𝑖𝑐𝑎𝑑𝑎𝑠| (33)
𝑘=2 para un nivel de confianza de 95,45 %; valores obtenidos de las especificaciones de
los fabricantes de equipos, materiales y reactivos del laboratorio que se emplearon.
4.3.5.1. Cálculo de la incertidumbre estándar por reproducibilidad.
De acuerdo con el Laboratorio Metrycal (2016), la fuente de incertidumbre por
reproducibilidad de la medición se obtiene mediante:

54
𝑢𝑅𝑒𝑝𝑟𝑜𝑑𝑢𝑐𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑 =𝑆𝑅
√𝑛 (34)
Donde:
𝑆𝑅= desviación estándar por reproducibilidad, mg/L.
n= número de repeticiones.
𝑢𝑅𝑒𝑝𝑟𝑜𝑑𝑢𝑐𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑 =0,615
√15
𝑢𝑅𝑒𝑝𝑟𝑜𝑑𝑢𝑐𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑=0,159 mg/L
Tabla 27. Incertidumbre estándar por reproducibilidad.
Concentración
mg/L
𝑺𝑹
mg/L
𝒖𝑹𝒆𝒑𝒓𝒐𝒅𝒖𝒄𝒊𝒃𝒊𝒍𝒊𝒅𝒂𝒅
mg/L
7,000 0,615 0,159
14,000 0,494 0,128
20,000 0,577 0,149
25,000 0,699 0,181
48,000 1,274 0,329
4.3.5.2. Cálculo de la incertidumbre estándar del equipo.
𝑢𝐸𝑞𝑢𝑖𝑝𝑜 = √(𝑢𝐹𝑅 )2 + (𝑢𝑅𝑒𝑠𝑜𝑙𝑐𝑖ó𝑛)2 + (𝑢𝐷𝑒𝑟𝑖𝑣𝑎)2 + (𝑢𝑀𝑅𝐼)2 (35)
Donde:
𝑢𝐹𝑅 = incertidumbre de la función respuesta.
𝑢𝑅𝑒𝑠𝑜𝑙𝑐𝑖ó𝑛= incertidumbre de resolución.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎= incertidumbre de deriva.
𝑢𝑀𝑅𝐼= incertidumbre del material de referencia interno.
Cálculo de la incertidumbre de la función respuesta.
De acuerdo con Fernanda Toasa (2012), la incertidumbre de la función de respuesta se
obtiene mediante:

55
𝑢𝐹𝑅 =|𝐶𝑒𝑠𝑡𝑖𝑚𝑎𝑑𝑎−𝐶𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑎|𝑚𝑎𝑥
√3 (36)
Donde:
𝐶𝑒𝑠𝑡𝑖𝑚𝑎𝑑𝑎 = concentración estimada de cada nivel de la curva de calibración, mg/L.
𝐶𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑎 = concentración verdadera de cada nivel de la curva de calibración, mg/L.
Distribución rectangular √3 = la especificación de incertidumbre no es explícita sino
que se da un límite máximo para el error del instrumento y de acuerdo con Carlos
Echeverri (2014), esto implica que el comportamiento del instrumento tiene
características de una distribución tipo rectangular o uniforme dentro de límites
establecidos.
𝑢𝐹𝑅 =|6,496−7|𝑚𝑎𝑥
√3
𝑢𝐹𝑅 =|−0,504|
√3
𝑢𝐹𝑅 = 0,291 𝑚𝑔/𝐿
𝑢𝐹𝑅 =0,291 𝑚𝑔/𝐿
7 𝑚𝑔/𝐿
𝑢𝐹𝑅 = 0,042
Cálculo de la incertidumbre de resolución.
De acuerdo con Fernanda Toasa (2012), la incertidumbre de resolución del
espectrofotómetro se obtiene mediante:
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒
√3 (37)
Donde:
Especificación del fabricante= se considera la resolución del espectrofotómetro, abs.
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =0,001 𝑎𝑏𝑠
√3
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 = 0,0006 𝑎𝑏𝑠
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =0,0006 𝑎𝑏𝑠
0,091 𝑎𝑏𝑠

56
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 = 0,006
Cálculo de la incertidumbre de deriva.
De acuerdo con Fernanda Toasa (2012), la incertidumbre de deriva del espectrofotómetro
se obtiene mediante:
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =𝑈𝐶𝑎𝑙
√3 (38)
Donde:
𝑈𝐶𝑎𝑙= incertidumbre expandida del espectrofotómetro Spectroquant Pharo 300, abs.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =0,009 𝑎𝑏𝑠
√3
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 = 0,0052 𝑎𝑏𝑠
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =0,0052 𝑎𝑏𝑠
0,091 𝑎𝑏𝑠
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 = 0,057
Cálculo de la incertidumbre del material de referencia interno.
𝑄𝑀𝑅𝐼 =𝑄𝑀𝑅𝐶∗𝑉𝑀𝑅𝐶
𝑉𝑎𝑓 (39)
Donde:
𝑄𝑀𝑅𝐼= concentración del material de referencia interno, (MRI) mg/L.
𝑄𝑀𝑅𝐶= concentración del material de referencia certificado (MRC), mg/L.
𝑉𝑀𝑅𝐶= volumen del material de referencia certificado empleado, mL.
𝑉𝑎𝑓= volumen de aforo, mL.
Se establece la ecuación de propagación de incertidumbres:
𝑢𝐶2
2 = ∑ 𝐶𝑖2𝑢2(𝑥𝑖)
𝑛𝑖=1 (40)
𝑢𝑄𝑀𝑅𝐼= √(𝐶𝑄𝑀𝑅𝐶
∗ 𝑢𝑄𝑀𝑅𝐶)2
+ (𝐶𝑉𝑀𝑅𝐶∗ 𝑢𝑉𝑀𝑅𝐶)
2+ (𝐶𝑉𝑎𝑓
∗ 𝑢𝑉𝑎𝑓)2
(41)

57
𝐶𝑄𝑀𝑅𝐶, 𝐶𝑉𝑀𝑅𝐶
y 𝐶𝑉𝑎𝑓= coeficientes de sensibilidad.
Coeficientes de sensibilidad (𝑪𝒊):
(𝜕𝑄𝑀𝑅𝐼
𝜕𝑄𝑀𝑅𝐶) 𝑉𝑀𝑅𝐶,𝑉𝑎𝑓
=𝑉𝑀𝑅𝐶
𝑉𝑎𝑓= 𝐶𝑄𝑀𝑅𝐶
(42)
(𝜕𝑄𝑀𝑅𝐼
𝜕𝑉𝑀𝑅𝐶) 𝑄𝑀𝑅𝐶,𝑉𝑎𝑓
=𝑄𝑀𝑅𝐶
𝑉𝑎𝑓= 𝐶𝑉𝑀𝑅𝐶
(43)
(𝜕𝑄𝑀𝑅𝐼
𝜕𝑉𝑎𝑓) 𝑄𝑀𝑅𝐶,𝑉𝑀𝑅𝐶
=−𝑄𝑀𝑅𝐶∗𝑉𝑀𝑅𝐶
𝑉𝑎𝑓2 = 𝐶𝑉𝑎𝑓
(44)
Donde:
(𝜕𝑄𝑀𝑅𝐼
𝜕𝑄𝑀𝑅𝐶) 𝑉𝑀𝑅𝐶,𝑉𝑎𝑓
= coeficiente de sensibilidad de la concentración del material de
referencia interno con respecto a 𝑄𝑀𝑅𝐶.
(𝜕𝑄𝑀𝑅𝐼
𝜕𝑉𝑀𝑅𝐶) 𝑄𝑀𝑅𝐶,𝑉𝑎𝑓
= coeficiente de sensibilidad de la concentración del material de
referencia interno con respecto a 𝑉𝑀𝑅𝐶.
(𝜕𝑄𝑀𝑅𝐼
𝜕𝑉𝑎𝑓) 𝑄𝑀𝑅𝐶,𝑉𝑀𝑅𝐶
= coeficiente de sensibilidad de la concentración del material de
referencia interno con respecto a 𝑉𝑎𝑓.
Reemplazando los valores de los coeficientes de sensibilidad 𝐶𝑄𝑀𝑅𝐶, 𝐶𝑉𝑀𝑅𝐶
y 𝐶𝑉𝑎𝑓, en la
ecuación (41) se tiene lo siguiente:
𝑢𝑄𝑀𝑅𝐼= √(
𝑉𝑀𝑅𝐶
𝑉𝑎𝑓∗ 𝑢𝑄𝑀𝑅𝐶)
2
+ (𝑄𝑀𝑅𝐶
𝑉𝑎𝑓∗ 𝑢𝑉𝑀𝑅𝐶)
2
+ (−𝑄𝑀𝑅𝐶∗𝑉𝑀𝑅𝐶
𝑉𝑎𝑓2 ∗ 𝑢𝑉𝑎𝑓)
2
(45)
a) Cálculo de incertidumbre de concentración del MRC.
De acuerdo con Fernanda Toasa (2012), la incertidumbre de concentración del MRC se
obtiene mediante:

58
𝑢𝑄𝑀𝑅𝐶 =𝑈𝑀𝑅𝐶
𝑘 (46)
Donde:
𝑈𝑀𝑅𝐶 = incertidumbre expandida del material de referencia obtenida del certificado,
mg/L.
𝑘 = factor de cobertura (generalmente es igual a 2, que corresponde a un nivel de
confianza de 95,45 %; valores obtenidos de las especificaciones del fabricante del MRC
que se empleó).
𝑢𝑄𝑀𝑅𝐶 =1,92
2
𝑢𝑄𝑀𝑅𝐶 = 0,96 𝑚𝑔/𝐿
b) Cálculo de incertidumbre de volumen del material de referencia certificado.
𝑢𝑉𝑀𝑅𝐶 = √(𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ó𝑛 )2 + (𝑢𝑅𝑒𝑠𝑜𝑙𝑐𝑖ó𝑛)2 + (𝑢𝐷𝑒𝑟𝑖𝑣𝑎)2 + (𝑢𝑡𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎)2 (47)
Donde:
Incertidumbre de calibración.
De acuerdo con el Laboratorio Metrycal (2016), la incertidumbre de calibración de
volumen del material de referencia certificado empleado se obtiene mediante:
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =𝑈
𝑘 (48)
Donde:
U = incertidumbre expandida que se obtiene del certificado de calibración del material
volumétrico (probeta o pipeta) mL.
𝑘 = factor de cobertura (generalmente es igual a 2, que corresponde a un nivel de
confianza de 95,45 %; valores obtenidos de las especificaciones de los fabricantes del
material volumétrico que se empleó).

59
Probeta:
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =0,033 𝑚𝐿
2= 0,017 𝑚𝐿
Pipeta:
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =0,033 𝑚𝐿
2= 0,017 𝑚𝐿
Nota: se utilizó una probeta calibrada para medir volúmenes que contengan decimales y
que no se puedan medir con pipeta volumétrica.
Incertidumbre de resolución.
De acuerdo con Fernanda Toasa (2012), la incertidumbre de resolución de volumen del
material de referencia certificado empleado se obtiene mediante:
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒
2√3 (49)
Donde:
𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒 = se considera la resolución del material volumétrico,
mL.
Distribución rectangular 2√3 = incertidumbre asociada a la resolución de la indicación
de un instrumento de medición, de acuerdo con Carlos Echeverri (2014), la incertidumbre
básica asociada a este problema se obtiene considerando que la información que se puede
contener en la porción menos significativa de la indicación de un instrumento, tiene una
función de distribución tipo rectangular, para este caso, la incertidumbre básica
corresponde a la sensibilidad del dígito menos significativo, dividido entre dos y entre la
raíz cuadrada de tres.
Probeta:
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =0,1 𝑚𝐿
2√3= 0,029𝑚𝐿

60
Incertidumbre de deriva.
De acuerdo con Fernanda Toasa (2012), la incertidumbre de deriva de volumen del
material de referencia certificado empleado se obtiene mediante:
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|𝐶𝑛−𝐶𝑛−1|𝑚𝑎𝑥
√3 (50)
Donde:
𝐶𝑛 =corrección del certificado de calibración n, mL.
𝐶𝑛−1 = corrección del certificado de calibración n-1, mL.
Probeta:
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|−0,050 − (−0,076)| 𝑚𝐿
√3= 0,015 𝑚𝐿
Pipeta:
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|−0,0013 − (−0,0020)|𝑚𝐿
√3= 0,0004 𝑚𝐿
Incertidumbre por temperatura.
De acuerdo con Gladys Ventura y Ángela Lluque (2008), la incertidumbre de volumen
del material de referencia certificado empleado se obtiene mediante:
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(𝑇−𝑇20)∗𝑉∗𝛼
√3 (51)
Donde:
T = temperatura ambiental, ºC.
𝑇20 = temperatura de calibración del material volumétrico, ºC.
V = volumen medido, mL.
𝛼 = coeficiente de dilatación del agua (0,000207 º𝐶−1) (physics.stackexchange.com,
2011).

61
Probeta:
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(16,6 − 20) ∗ 1,800 ∗ 0,0002
√3
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 = −0,001 𝑚𝐿
Pipeta:
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(16,6 − 20) ∗ 1,000 ∗ 0,0002
√3
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 = −0,0004 𝑚𝐿
Al aplicar la ecuación (47), se tiene:
Probeta:
𝑢𝑉𝑀𝑅𝐶= √(0,017)2 + (0,029)2 + (0,015)2 + (−0,001)2
𝑢𝑉𝑀𝑅𝐶= 0,036 𝑚𝐿
Pipeta:
𝑢𝑉𝑀𝑅𝐶= √(0,017)2 + (0,0004)2 + (−0,0004)2
𝑢𝑉𝑀𝑅𝐶= 0,017 𝑚𝐿
𝑈𝑉𝑀𝑅𝐶= 2𝑢𝑉𝑀𝑅𝐶
+ |𝐶𝑜𝑟𝑟𝑒𝑐𝑖ó𝑛 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎|𝑚𝑎𝑡𝑒𝑟𝑖𝑎𝑙 𝑣𝑜𝑙𝑢𝑚é𝑡𝑟𝑖𝑐𝑜 (52)
Probeta:
𝑈𝑉𝑀𝑅𝐶= 2(0,037) + |−0,050|𝑝𝑟𝑜𝑏𝑒𝑡𝑎
𝑈𝑉𝑀𝑅𝐶= 0,123 𝑚𝐿
Pipeta:
𝑈𝑉𝑀𝑅𝐶= 2(0,017) + |−0,0013|𝑝𝑖𝑝𝑒𝑡𝑎
𝑈𝑉𝑀𝑅𝐶= 0,034 𝑚𝐿
𝑢𝑉𝑀𝑅𝐶=
𝑈𝑉𝑀𝑅𝐶
2 (53)

62
Probeta:
𝑢𝑉𝑀𝑅=
0,123
2
𝑢𝑉𝑀𝑅= 0,061 𝑚𝐿
Pipeta:
𝑢𝑉𝑀𝑅=
0,034
2
𝑢𝑉𝑀𝑅= 0,017 𝑚𝐿
c) Cálculo de incertidumbre de volumen de aforo.
𝑢𝑉𝑎𝑓= √(𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ó𝑛 )2 + (𝑢𝐷𝑒𝑟𝑖𝑣𝑎)2 + (𝑢𝑡𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎)
2 (54)
Donde:
Incertidumbre de calibración.
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =𝑈
𝑘 (55)
Donde:
U = se obtiene del certificado de calibración del balón aforado, mL.
𝑘 = factor de cobertura (generalmente es igual a 2, corresponde a un nivel de confianza
de 95,45 %, valores obtenidos de las especificaciones del fabricante del balón de aforo
que se empleó).
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =0,033
2
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 = 0,017 𝑚𝐿
Incertidumbre de deriva.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|𝐶𝑛−𝐶𝑛−1|𝑚𝑎𝑥
√3 (56)
Donde:

63
𝐶𝑛 = corrección del certificado de calibración n, mL.
𝐶𝑛−1 = corrección del certificado de calibración n-1, mL.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|−0,268 − (−0,271)| 𝑚𝐿
√3
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 = 0,002 𝑚𝐿
Incertidumbre por temperatura.
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(𝑇−𝑇20)∗𝑉∗𝛼
√3 (57)
Donde:
T = temperatura ambiental, ºC.
𝑇20 = temperatura de calibración del balón aforado, ºC.
V = volumen de aforo, mL.
𝛼 = coeficiente de dilatación del agua (0,000207 º𝐶−1).
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(16,6 − 20) ∗ 200 ∗ 0,0002
√3
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 = −0,081 𝑚𝐿
Al aplicar la ecuación (54), se tiene:
𝑢𝑉𝑎𝑓= √(0,017)2 + (0,0002)2 + (−0,039)2
𝑢𝑉𝑎𝑓= 0,083 𝑚𝐿
𝑈𝑉𝑎𝑓= 2𝑢𝑉𝑎𝑓
+ |𝐶𝑜𝑟𝑟𝑒𝑐𝑖ó𝑛 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎|𝑏𝑎𝑙ó𝑛 𝑎𝑓𝑜𝑟𝑎𝑑𝑜 (58)
𝑈𝑉𝑎𝑓= 2(0,083) + |−0,268|𝑏𝑎𝑙ó𝑛 𝑎𝑓𝑜𝑟𝑎𝑑𝑜
𝑈𝑉𝑎𝑓= 0,434 mL

64
𝑢𝑉𝑎𝑓=
𝑈𝑉𝑎𝑓
2 (59)
𝑢𝑉𝑎𝑓=
0,434
2
𝑢𝑉𝑎𝑓= 0,217 𝑚𝐿
Al aplicar la ecuación (45), se tiene:
𝑢𝑄𝑀𝑅𝐼= √(
1,76 𝑚𝐿
200 𝑚𝐿∗ 0,96 𝑚𝑔/𝐿)
2
+ (500 𝑚𝑔/𝐿
200 𝑚𝐿∗ 0,061 𝑚𝐿)
2
+ (−500
𝑚𝑔
𝐿∗1,76 𝑚𝐿
200 𝑚𝐿2 ∗ 0,217 𝑚𝐿)2
𝑢𝑄𝑀𝑅𝐼= 0,154 𝑚𝑔/𝐿
Tabla 28. Incertidumbre estándar del MRI.
Concentración; 𝒎𝒈/𝑳 𝒖𝑸𝑴𝑹𝑰; 𝒎𝒈/𝑳 𝒖𝑸𝑴𝑹𝑰
𝑹𝒆𝒍𝒂𝒕𝒊𝒗𝒂
7,000 0,154 0,022
14,000 0,154 0,011
20,000 0,156 0,008
25,000 0,157 0,006
48,000 0,174 0,004
Al aplicar la ecuación (35), se tiene:
𝑢𝐸𝑞𝑢𝑖𝑝𝑜 = √(0,042)2 + (0,006)2 + (0,057)2 + (0,022)2
𝑢𝐸𝑞𝑢𝑖𝑝𝑜 = 0,074
𝑢𝐸𝑞𝑢𝑖𝑝𝑜 = 0,074 ∗ (7,000𝑚𝑔
𝐿)
𝑢𝐸𝑞𝑢𝑖𝑝𝑜 = 0,516 𝑚𝑔/𝐿

65
Tabla 29. Incertidumbre estándar debida a la lectura de las concentraciones en el
espectrofotómetro.
Nivel,
mg/L
𝒖𝑹𝒆𝒔𝒐𝒍𝒖𝒄𝒊ó𝒏 𝒖𝑫𝒆𝒓𝒊𝒗𝒂 𝒖𝑸𝑴𝑹𝑰 𝒖𝑭𝑹 𝒖𝑬𝒒𝒖𝒊𝒑𝒐
Relativa
𝒖𝑬𝒒𝒖𝒊𝒑𝒐
mg/L
5 0,012 0,106 0,017 0,137 0,174 0,871
10 0,005 0,041 0,009 0,059 0,073 0,726
20 0,002 0,017 0,009 0,039 0,044 0,875
25 0,002 0,014 0,004 0,020 0,025 0,622
50 0,001 0,008 0,003 0,008 0,011 0,568
Tabla 30. Incertidumbre estándar del equipo por niveles.
Nivel, mg/L 𝒖𝑹𝒆𝒔𝒐𝒍𝒖𝒄𝒊ó𝒏 𝒖𝑫𝒆𝒓𝒊𝒗𝒂 𝒖𝑸𝑴𝑹𝑰 𝒖𝑭𝑹 𝒖𝑬𝒒𝒖𝒊𝒑𝒐
Relativa
𝒖𝑬𝒒𝒖𝒊𝒑𝒐
mg/L
7 0,006 0,057 0,022 0,042 0,074 0,515
14 0,003 0,030 0,011 0,020 0,037 0,499
20 0,002 0,018 0,008 0,022 0,029 0,599
25 0,002 0,015 0,006 0,018 0,024 0,597
48 0,001 0,008 0,004 0,009 0,013 0,607
4.3.5.3. Cálculo de la incertidumbre debida al cronómetro.
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 = √(𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ó𝑛 )2 + (𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛)2 + (𝑢𝐷𝑒𝑟𝑖𝑣𝑎)2 (60)
Nota: se utilizan las mismas fórmulas para calcular calibración, resolución y deriva y se
sigue el mismo procedimiento que para el cálculo de la incertidumbre del volumen del
material de referencia (ecuaciones (47) a la (53)).
Cálculo de la incertidumbre de calibración.
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =𝑈
𝑘
Donde:

66
U = incertidumbre expandida que se obtiene del certificado de calibración del cronómetro
del laboratorio, s.
𝑘 = factor de cobertura (generalmente es igual a 2, corresponde a un nivel de confianza
de 95,45 %; valores obtenidos de las especificaciones del fabricante del cronómetro que
se empleó).
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =0,060 𝑚𝐿
2= 0,030 𝑠
Incertidumbre de resolución.
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒
2√3
Donde:
𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒 = se considera la resolución del cronómetro del
laboratorio, s.
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =0,01 𝑠
2√3= 0,003 𝑠
Incertidumbre de deriva.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|𝐶𝑛−𝐶𝑛−1|𝑚𝑎𝑥
√3
Donde:
𝐶𝑛 = corrección del certificado de calibración del cronómetro n, s.
𝐶𝑛−1 = corrección del certificado de calibración del cronómetro n-1, s.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|0,004 − 0,017| 𝑚𝐿
√3= 0,008 𝑚𝐿
Al aplicar la ecuación (60) se tiene:
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 = √(0,030)2 + (0,003)2 + (0,008)2
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 = 0,031 𝑠
𝑈cronómetro = 2𝑢𝑐𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 + |𝐶𝑜𝑟𝑟𝑒𝑐𝑖ó𝑛 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎|𝑐𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 (61)

67
𝑈cronómetro = 2(0,031) + |0,004|𝑐𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜
𝑈cronómetro = 0,066 𝑠
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 =𝑈𝑐𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜
2 (62)
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 =0,066
2
𝑢𝐶𝑟𝑜𝑛ó𝑚𝑒𝑡𝑟𝑜 = 0,033 𝑠
4.3.5.4. Cálculo de la incertidumbre debida a la probeta.
𝑢𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = √(𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ó𝑛 )2 + (𝑢𝑅𝑒𝑠𝑜𝑙𝑐𝑖ó𝑛)2 + (𝑢𝐷𝑒𝑟𝑖𝑣𝑎)2 + (𝑢𝑡𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎)2 (63)
Nota: se utilizan las mismas fórmulas para el cálculo de calibración, resolución, deriva y
temperatura y se sigue el mismo procedimiento que para el cálculo de la incertidumbre
del volumen del material de referencia (ecuaciones (47) a la (53)).
Donde:
Incertidumbre de calibración.
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =𝑈
𝑘
Donde:
U = incertidumbre expandida que se obtiene del certificado de calibración de la probeta,
mL.
𝑘 = factor de cobertura (generalmente es igual a 2, corresponde a un nivel de confianza
de 95,45 %; valores que se obtienen de las especificaciones del fabricante de la probeta
que se empleó).
𝑢𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑐𝑖ò𝑛 =0,033 𝑚𝐿
2= 0,017 𝑚𝐿

68
Incertidumbre de resolución.
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒
2√3
Donde:
𝐸𝑠𝑝𝑒𝑐𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑓𝑎𝑏𝑟𝑖𝑐𝑎𝑛𝑡𝑒= se considera la resolución del material volumétrico,
mL.
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =0,1 𝑚𝐿
2√3
𝑢𝑅𝑒𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 = 0,029𝑚𝐿
Incertidumbre de deriva.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|𝐶𝑛−𝐶𝑛−1|𝑚𝑎𝑥
√3
Donde:
𝐶𝑛 = corrección del certificado de calibración n, mL.
𝐶𝑛−1 = corrección del certificado de calibración n-1, mL.
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 =|−0,050 − (−0,076)| 𝑚𝐿
√3
𝑢𝐷𝑒𝑟𝑖𝑣𝑎 = 0,015 𝑚𝐿
Incertidumbre por temperatura.
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(𝑇−𝑇20)∗𝑉∗𝛼
√3
Donde:
T = temperatura ambiental, ºC.
𝑇20 = temperatura de calibración del material volumétrico, ºC.
V = volumen medido, mL.
𝛼 = coeficiente de dilatación del agua (0,000207 º𝐶−1).

69
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 =(16,6 − 20) ∗ 1,8 ∗ 0,0002
√3
𝑢𝑇𝑒𝑚𝑝𝑒𝑟𝑎𝑡𝑢𝑟𝑎 = −0,0007 𝑚𝐿
Al aplicar la ecuación (63), se tiene:
𝑢𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = √(0,017)2 + (0,029)2 + (0,015)2 + (−0,0007)2
𝑢𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = 0,037 𝑚𝐿
𝑈𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = 2𝑢probeta + |𝐶𝑜𝑟𝑟𝑒𝑐𝑖ó𝑛 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎|𝑚𝑎𝑡𝑒𝑟𝑖𝑎𝑙 𝑣𝑜𝑙𝑢𝑚é𝑡𝑟𝑖𝑐𝑜 (64)
𝑈𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = 2(0,037) + |−0,050|𝑝𝑟𝑜𝑏𝑒𝑡𝑎
𝑈𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = 0,123 𝑚𝐿
𝑢probeta =𝑈𝑝𝑟𝑜𝑏𝑒𝑡𝑎
2 (65)
𝑢𝑝𝑟𝑜𝑏𝑒𝑡𝑎 =0,123
2
𝑢𝑝𝑟𝑜𝑏𝑒𝑡𝑎 = 0,061 𝑚𝐿
Al aplicar la ecuación (31) se tiene:
𝑢𝑆𝑂4−2
𝑆𝑂4−2 = √(
0,159
7,000)
2
+ (0,516
7,000)
2
+ (0,033
300)
2
+ (0,061
25)
2
𝑢𝑆𝑂4−2
𝑆𝑂4−2 = 0,077
Al aplicar la ecuación (32) se tiene:
𝑢𝑆𝑂4−2 = 7,000
𝑚𝑔
𝐿∗ 0,077
𝑢𝑆𝑂4−2 = 0,539 𝑚𝑔/𝐿

70
4.3.5.5. Correcciones no aplicadas
Las correcciones fueron aplicadas al inicio de cada cálculo según el caso. De acuerdo con
Fernanda Toasa (2012), las correcciones no aplicadas están dadas por:
∑|𝑐𝑜𝑟𝑟𝑒𝑐𝑐𝑖𝑜𝑛𝑒𝑠 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎𝑠| = |𝐶�̅�𝑏𝑡𝑒𝑛𝑖𝑑𝑎 − 𝐶𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑎|𝑏𝑖𝑎𝑠 (66)
Donde:
𝐶�̅�𝑏𝑡𝑒𝑛𝑖𝑑𝑜 = promedio de la concentración obtenida, mg/L.
𝐶𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑜 = concentración verdadera, mg/L.
∑|𝑐𝑜𝑟𝑟𝑒𝑐𝑐𝑖𝑜𝑛𝑒𝑠 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎𝑠| = |6,933 − 7,000|𝑏𝑖𝑎𝑠
∑|𝑐𝑜𝑟𝑟𝑒𝑐𝑐𝑖𝑜𝑛𝑒𝑠 𝑛𝑜 𝑎𝑝𝑙𝑖𝑐𝑎𝑑𝑎𝑠| = 0,067 𝑚𝑔/𝐿
𝑈𝑆𝑂4−2 = (2 ∗ 0,539) + |0,067|
𝑈𝑆𝑂4−2 = 1,144 𝑚𝑔/𝐿
%𝑈 =𝑈
𝑆𝑂4−2
𝑆𝑂4−2 ∗ 100 (67)
%𝑈 =1,144
7,000∗ 100
%𝑈 = 16,348 %
Tabla 31. Cálculo de incertidumbre por niveles para sulfatos en aguas.
Nivel 𝑿𝒎 u
Reprod
u
Equipo
u
Cron
u
Prob
𝒖𝒙
𝑿𝒎
ux
mg/L CNR
U
mg/L %U
7,000 6,933 0,159 0,074 0,033 0,061 0,078 0,539 0,067 1,144 16,348
14,000 13,400 0,128 0,037 0,033 0,061 0,039 0,516 0,600 1,633 11,662
20,000 20,533 0,149 0,029 0,033 0,062 0,030 0,620 0,533 1,773 8,863
25,000 24,533 0,181 0,024 0,033 0,062 0,026 0,627 0,467 1,721 6,884
48,000 47,400 0,329 0,013 0,033 0,062 0,015 0,701 0,600 2,001 4,170
4.3.6. Cálculo de la veracidad.
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜
𝑥𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑜∗ 100 (68)

71
Donde:
�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜 = promedio de la concentración del analito medido en el MRC, mg/L.
𝑥𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑜 = concentración verdadera del analito en el MRC, mg/L.
Tabla 32. Veracidad con MRC para sulfatos.
NIVEL
FECHA
MATERIAL DE REFERENCIA CERTIFICADO
19/12/2017 20/12/2017 21/12/2017
500 500 498 500
500 496 499 500
500 498 500 498
500 496 495 496
500 499 498 495
�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜 498 mg/L
Al aplicar la ecuación (68) se tiene:
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =�̅�𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑜
𝑥𝑣𝑒𝑟𝑑𝑎𝑑𝑒𝑟𝑜*100
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 =498
500*100
% 𝑅𝑒𝑐𝑢𝑝𝑒𝑟𝑎𝑐𝑖ó𝑛 = 99,60 %

72
5. RESULTADOS
5.1. Resultados de validación de sulfatos en aguas.
Tabla 33. Veracidad con MRC de sulfatos en aguas.
NIVEL
FECHA
MATERIAL DE REFERENCIA CERTIFICADO
19/12/2017 20/12/2017 21/12/2017
500 500 498 500
500 496 499 500
500 498 500 498
500 496 495 496
500 499 498 495
Media General (�̅� ) 498 mg/L
% Recuperación 99,60
Tabla 34. Precisión, veracidad e incertidumbre de sulfatos en aguas.
PRECISIÓN, VERACIDAD E INCERTIDUMBRE
Nivel de
Concentración,
mg/L
Repetibilidad Reproducibilidad Veracidad U
expandida
𝑺𝒓 % 𝑪𝑽𝒓 𝑺𝑹 % 𝑪𝑽𝑹 %
Recuperación
% U
(k=2)
7,000 0,577 8,327 0,615 8,865 99,048 16,348
14,000 0,516 3,854 0,494 3,690 95,714 11,662
20,000 0,683 3,327 0,577 2,812 102,667 8,863
25,000 0,775 3,157 0,699 2,850 98,133 6,884
48,000 1,111 2,343 1,274 2,687 98,750 4,170

73
5.2. Resultados de validación de sulfuros en aguas.
Tabla 35. Veracidad con MRC de sulfuros en aguas.
NIVEL
FECHA
MATERIAL DE REFERENCIA CERTIFICADO
08/01/2018 09/01/2018 10/01/2018
50,00 48,25 50,00 50,00
50,00 48,30 48,60 49,65
50,00 49,52 50,00 49,87
50,00 48,80 48,32 50,00
50,00 48,12 48,87 49,15
Media General (�̅� ) 49,16 mg/L
% Recuperación 98,33
Tabla 36. Precisión, veracidad e incertidumbre de sulfuros en aguas.
PRECISIÓN, VERACIDAD E INCERTIDUMBRE
Nivel de
Concentración,
mg/L
Repetibilidad Reproducibilidad Veracidad U
expandida
𝑺𝒓 % 𝑪𝑽𝒓 𝑺𝑹 % 𝑪𝑽𝑹 %
Recuperación
% U
(k=2)
0,28 0,008 2,897 0,007 2,427 98,095 29,888
0,45 0,008 1,806 0,007 1,582 100,444 18,854
1,00 0,021 2,065 0,017 1,686 100,400 8,905

74
5.3. Resultados de validación de fenoles en aguas.
Tabla 37. Veracidad con MRC de fenoles en aguas.
NIVEL
FECHA
MATERIAL DE REFERENCIA CERTIFICADO
20/12/2017 21/12/2017 22/12/2017
10,00 10,02 9,91 9,75
10,00 10,05 10,00 9,82
10,00 10,00 9,85 9,97
10,00 9,67 9,39 9,34
10,00 9,83 9,61 9,62
Media General (�̅� ) 9,79 mg/L
% Recuperación 97,89
Tabla 38. Precisión, veracidad e incertidumbre de fenoles en aguas.
PRECISIÓN, VERACIDAD E INCERTIDUMBRE
Nivel de
Concentración,
mg/L
Repetibilidad Reproducibilidad Veracidad U
expandida
𝑺𝒓 % 𝑪𝑽𝒓 𝑺𝑹 % 𝑪𝑽𝑹 %
Recuperación
% U
(k=2)
0,100 0,009 9,576 0,012 12,895 95,333 29,411
0,140 0,012 7,945 0,012 8,458 96,667 27,010
0,200 0,011 5,450 0,011 5,396 97,667 23,452

75
6. DISCUSIÓN
6.1. Discusión general.
En el proceso de validación del laboratorio se planteó cumplir con los diferentes
criterios para cada analito y su matriz, tales como coeficiente de variación de repetibilidad
y reproducibilidad ≤ 10 % ó ≤ 15 %; veracidad 80 < % R < 120 e incertidumbre U ≤ 30
%, cantidades que se establecieron en función de la información bibliográfica (requisitos
normativos) y de los datos de la puesta a punto. El análisis de fenoles se realizó en niveles
de concentración más bajos que el resto de métodos (ver tablas 34, 36 y 38); por lo cual
para este ensayo es necesario elevar el valor de los coeficientes de variación al 15 %.
Los parámetros de la función respuesta (pendiente e intervalo, intercepto e intervalo y
coeficiente de determinación) fueron establecidos después de haber realizado las curvas
de calibración de la puesta a punto mediante condiciones de reproducibilidad y
repetibilidad, es decir durante tres días y con repeticiones de cada nivel por triplicado, lo
cual significa que de acuerdo a la Norma ISO/IEC 17025:2017 se realizó el seguimiento
adecuado de la validez de los ensayos llevados a cabo dentro del laboratorio.
En los días de validación, se establecieron las curvas de calibración y se fijó la que
obedeció el porcentaje de recuperación de los objetivos planteados. Durante la
experimentación, se observó que a niveles bajos de concentración se debe tener mayor
precaución al momento de fortificar las muestras, especialmente al tomar las alícuotas a
añadir, debido a que se presenta mayor dificultad al tratarse de volúmenes pequeños, por
lo que una inadecuada medición provocará una variación apreciable en los datos
obtenidos, misma que interferirá con el cumplimiento de los criterios de validación.
Además, para todos los métodos de análisis el nivel más bajo siempre es el que presenta
los porcentajes de coeficientes de variación y de incertidumbre más altos en comparación
con el resto de niveles (ver tablas 34, 36 y 38). Por estas razones se consideraron como
los más críticos.

76
6.2. Sulfatos en aguas.
Para la determinación de sulfatos se utilizó SulfaVer 4 que contiene cloruro de bario y
un medio ácido que en presencia de estos iones permite la formación de sulfato de bario,
lo que permite la cuantificación del sulfato. Durante la experimentación se comprobó que
las lecturas de la concentración de las muestras deben realizarse transcurrido un período
de 5 a 10 minutos, de lo contrario no serán correctas, puesto que pasado ese tiempo el
sulfato presente disminuye considerablemente (el método pierde estabilidad); así, la
lectura del primer nivel a los cinco minutos fue de 7 mg/L, a los ocho minutos fue de 6
mg/L y finalmente después de diez minutos fue de 3 mg/L.
La curva de validación fue realizada desde 5 mg/L, ya que se calculó la menor cantidad
de material de referencia certificado que se podía tomar fácilmente con el material
volumétrico disponible (en el caso del laboratorio, eran una pipeta calibrada de
apreciación 1 mL para volúmenes exactos y para volúmenes menores se utilizaba una
probeta calibrada de apreciación 0,1 mL), por lo cual el rango de validación empezó con
un valor cercano de 7 mg/L, que sí formó parte de los valores fijados en objetivos por
tener un % 𝐶𝑉𝑅 y % 𝐶𝑉𝑟 dentro del 10 %, una incertidumbre expandida dentro del 30 %,
así como una veracidad entre el 80 y el 120 %, a pesar de que el límite de cuantificación
fijado o definido de la recta de calibración de la puesta a punto con la ecuación (15) fue
de 1,670 mg/L. Es importante recordar que a niveles muy bajos no se cumpliría con el
criterio máximo de incertidumbre, porque al calcular la incertidumbre relativa del método
es necesario realizar la división para la concentración respectiva, entonces al tratarse de
un número pequeño (exponente negativo), el resultado será un valor alto, situación que
se debe evitar, ya que gracias a la experiencia se pudo evidenciar que dichos valores
ocasionan que el porcentaje de veracidad se incremente notablemente. Finalmente todos
los niveles obedecen a los rangos establecidos, por lo que quedan dentro del intervalo de
validación.
Una de las principales interferencias fue ocasionada por la materia en suspensión en
aguas residuales, lo que hizo necesario realizar filtración previa al procedimiento de
ensayo. Además, la linealidad del método sólo está presente en el rango de 0 a 50 mg/L.
De acuerdo con la ley de Lambert Beer conforme una solución se vuelve más concentrada,
las moléculas de soluto interactúan entre si debido a su proximidad, cambiando sus
propiedades (incluyendo la absorción de luz); por lo que para la lectura de muestras que

77
presentaron un mayor nivel de sulfatos, se realizó diluciones de las mismas y al reportar
los resultados reales se multiplicó por el factor de dilución.
La recta de calibración que se establece para el análisis de sulfatos dentro del
laboratorio es la del tercer día de validación, puesto que cumplió con los parámetros
definidos en objetivos. Entonces, se la fija durante el mes de evaluación.
6.3. Sulfuros en aguas.
Para la determinación de sulfuros se utilizó los reactivos Sulfuro 1 y Sulfuro 2 que
contienen dimetil-p-fenilendiamina y cloruro férrico respectivamente, que permiten la
formación de azul de metileno en presencia de sulfuro de hidrógeno y sulfuros metálicos
solubles en ácido. Durante la experimentación se comprobó que las lecturas de la
concentración de las muestras deben realizarse transcurrido un período máximo de 5
minutos, de lo contrario no serán correctas, puesto que pasado ese tiempo el sulfuro
presente disminuye de manera considerable o se pierde totalmente (el método es bastante
inestable); así, la lectura del primer nivel al minuto fue de 0,280 mg/L, a los cuatro
minutos fue de 0,260 mg/L y finalmente después de cinco minutos fue de 0,150 mg/L.
La curva de validación fue realizada desde 0,250 mg/L ya que se calculó la menor
cantidad de material de referencia certificado que se podía tomar fácilmente con el
material volumétrico disponible (en el caso del laboratorio eran una pipeta calibrada de
apreciación 1 mL para volúmenes exactos y para volúmenes menores se utilizaba una
probeta calibrada de apreciación 0,1 mL), por lo cual el rango de validación empezó con
un valor cercano de 0,280 mg/L, que sí formó parte de los valores fijados en objetivos
por tener un % 𝐶𝑉𝑅 y % 𝐶𝑉𝑟 dentro del 10 %, una incertidumbre expandida dentro del 30
%, así como una veracidad entre el 80 y el 120 %; a pesar de que el límite de
cuantificación fijado o definido de la recta de calibración al aplicar la ecuación (15) fue
de 0,029 mg/L. Es importante recordar que a concentraciones muy bajas no se cumpliría
con el criterio máximo de incertidumbre, porque al calcular la incertidumbre relativa del
método, es necesario realizar la división para la concentración respectiva, entonces al
tratarse de un número pequeño (exponente negativo), el resultado será un valor alto,
situación que se debe evitar ya que gracias a la experiencia se pudo evidenciar que dichos
valores ocasionan que el porcentaje de veracidad se incremente notablemente. Finalmente

78
todos los niveles obedecen a los rangos establecidos, por lo que quedan dentro del
intervalo de validación.
Uno de los principales inconvenientes fue ocasionado debido a que las muestras no se
pueden preservar y deben ser analizadas inmediatamente, puesto que como ya se
mencionó, con el tiempo el sulfuro presente en las mismas entra en contacto con el aire
(se volatiliza rápidamente), hasta perderse por completo, razón por la cual durante la
experimentación se identificó a este método como el más inestable en comparación con
los demás.
La recta de calibración que se establece para el análisis de sulfuros dentro del
laboratorio es la del segundo día de validación, puesto que cumplió con los parámetros
definidos en objetivos. Entonces, se la fija durante el mes de evaluación.
6.4. Fenoles en aguas.
Para la determinación de fenoles se utilizó los reactivos Fenol 1 y Fenol 2 que
contienen 4- aminoantipirina y ferricianuro de potasio respectivamente, para formar un
colorante antipirina en presencia de fenoles orto y meta sustituidos, este colorante se
elimina de la fase acuosa por extracción con cloroformo y al realizar las lecturas en el
espectrofotómetro no existen cambios significativos en la concentración de fenol presente
en los extractos con el paso del tiempo; así, la lectura del primer nivel al minuto fue de
0,10 mg/L, a los cinco minutos fue de 0,10 mg/L, a los ocho minutos fue de 0,10 mg/L y
finalmente después de diez minutos fue de 0,11 mg/L.
La curva de validación fue realizada desde 0,050 mg/L ya que se calculó la menor
cantidad de material de referencia certificado que se podía tomar fácilmente con el
material volumétrico disponible (en el caso del laboratorio eran una pipeta calibrada de
apreciación 1 mL para volúmenes exactos y para volúmenes menores se utilizaba una
probeta calibrada de apreciación 0,1 mL), por lo cual el rango de validación empezó con
un valor cercano de 0,100 mg/L, que sí formó parte de los valores fijados en objetivos
por tener un % 𝐶𝑉𝑅 y % 𝐶𝑉𝑟 dentro del 15 %, una incertidumbre expandida dentro del 30
%, así como una veracidad entre el 80 y el 120 %; a pesar de que el límite de
cuantificación fijado o definido de la recta de calibración al aplicar la ecuación (15) fue
de 0,007 mg/L. Es importante recordar que a concentraciones muy bajas no se cumpliría

79
con el criterio máximo de incertidumbre, porque al calcular la incertidumbre relativa del
método, es necesario realizar la división para la concentración respectiva, entonces al
tratarse de un número pequeño (exponente negativo), el resultado será un valor alto,
situación que se debe evitar ya que gracias a la experiencia se pudo evidenciar que dichos
valores ocasionan que el porcentaje de veracidad se incremente notablemente. Finalmente
todos los niveles obedecen a los rangos establecidos, por lo que quedan dentro del
intervalo de validación.
En fenoles se establecen los porcentajes de 𝐶𝑉𝑟 y 𝐶𝑉𝑅 ≤ 15 %, debido a que de todos
los métodos es el que requiere las determinaciones de los niveles más bajos (hasta 0,2
mg/L) de acuerdo con los límites máximos permisibles tabulados en la Ordenanza
Metropolitana y en el TULSMA (ver tabla 2) y por ello resulta más difícil realizar las
mediciones correspondientes durante la experimentación, por lo que conforme a la
información bibliográfica se necesita que los parámetros para coeficientes de variación
de repetibilidad y reproducibilidad sean de ≤ 15 %.
Se debe destilar las muestras previo al desarrollo del método, con ello todas las
interferencias se reducen al mínimo, pero su principal problema es que cuando se realiza
esta operación se debe obtener un destilado transparente, lo cual a veces no ocurre y el
resultado es turbio (como se comprobó durante la experimentación en muestras que
contenían detergentes), entonces se procede a realizar las destilaciones necesarias hasta
cumplir el objetivo, por lo cual el método de fenoles es el que requiere de más tiempo y
vigilancia con respecto a los demás, aunque durante la experimentación se lo identificó
como el más estable, puesto que al pasar el tiempo la concentración de fenol presente en
los extractos de cloroformo a analizar no presenta variaciones significativas como ya se
indicó anteriormente.
La recta de calibración que se establece para el análisis de fenoles dentro del
laboratorio es la del segundo día de validación, puesto que cumplió con los parámetros
definidos en objetivos. Entonces, se la mantiene fija durante el mes de evaluación.

80
7. CONCLUSIONES
7.1. Conclusión general.
Se cumplió con los objetivos planteados inicialmente para cada analito y su matriz,
tales como coeficiente de variación de repetibilidad y reproducibilidad ≤ 10% ó ≤ 15%;
veracidad 80 < % R < 120 e incertidumbre U ≤ 30 %.
7.2. Sulfatos en aguas.
El método analítico para la cuantificación de sulfatos en aguas mantiene la linealidad,
precisión y veracidad desde 0 a 50 mg/L, por ello se efectuó la validación en dicho rango,
pero debido a que las normas presentan valores superiores (ver tabla 2), se realizó
diluciones para aquellas muestras que superaron el intervalo, como ya se indicó en la
discusión de sulfatos.
Se realizó la validación en los siguientes niveles: 7, 14, 20, 25 y 48 mg/L, debido a
que en dichos niveles se facilitó la medición (con el material volumétrico disponible) de
las respectivas alícuotas del material de referencia certificado a añadir en las muestras a
fortificar.
Se realizaron los ensayos para valores de concentración que varían desde 7 hasta 48
mg/L, de esta manera se obtuvieron como resultados valores que se encuentran dentro de
los objetivos establecidos, por lo cual todos estos niveles pasan a formar parte del método
validado.
Como se puede observar en la tabla 3, se cumplió con los criterios establecidos de
precisión (% 𝐶𝑉𝑟 ≤ 10 % y % 𝐶𝑉𝑅 ≤ 10); veracidad (80 < % R < 120) e incertidumbre (≤
30 %). Así se puede observar en la tabla 34 que se obtuvo el mayor valor de % 𝐶𝑉𝑟 de
8,327 % y % 𝐶𝑉𝑅 de 8,865 %; el rango de veracidad fue de 95,714 % a 102,667 % y la
mayor incertidumbre de 16,348 %.

81
Los parámetros de la función respuesta son: pendiente 0,014; intervalo estudiado de
pendiente 0,014 a 0,015; intercepto -0,001; intervalo estudiado de intercepto -0,015 a -
0,014 y el coeficiente de determinación ≥ 0,995.
En función de los resultados anteriores y considerando los criterios de validación
establecidos en los objetivos, se concluye que el método de análisis queda validado en el
rango de 7 a 48 mg/L en aguas residuales y de consumo.
7.3. Sulfuros en aguas.
Se realizó la validación en el rango de 0,280 a 1,000 mg/L, debido a que las normas
presentan como límite máximo en cauce 0,5 mg/L y en alcantarillado 1 mg/L.
Se realizó la validación en los siguientes niveles: 0,280; 0,450 y 1,000 mg/L, debido a
que a dichos niveles se facilitó la medición (con el material volumétrico disponible) de
las respectivas alícuotas del material de referencia certificado a añadir en las muestras a
fortificar.
Se realizaron los ensayos para valores de concentración que varían desde 0,280 hasta
1,000 mg/L, de esta manera se obtuvieron como resultados valores que se encuentran
dentro de los objetivos establecidos, por lo cual todos estos niveles pasan a formar parte
del método validado.
Como se puede observar en la tabla 4, se cumplió con los criterios establecidos de
precisión (% 𝐶𝑉𝑟 ≤ 10 % y % 𝐶𝑉𝑅 ≤ 10); veracidad (80 < % R < 120) e incertidumbre (≤
30 %). Así se puede observar en la tabla 36 que se obtuvo el mayor valor de % 𝐶𝑉𝑟 de
2,897 % y % 𝐶𝑉𝑅 de 2,427 %; el rango de veracidad fue de 98,095 % a 100,444 % y la
mayor incertidumbre de 29,888 %.
Los parámetros de la función respuesta son: pendiente 0,204; intervalo estudiado de
pendiente 0,194 a 0,217; intercepto -0,004; intervalo estudiado de intercepto -0,012 a
0,001 y el coeficiente de determinación ≥ 0,995.
En función de los resultados anteriores y considerando los criterios de validación
establecidos en los objetivos, se concluye que el método de análisis queda validado en el
rango de 0,280 a 1,000 mg/L en aguas residuales.

82
7.4. Fenoles en aguas.
Se realizó la validación en el rango de 0,100 a 0,200 mg/L, debido a que las normas
presentan como límite máximo en cauce y alcantarillado 0,200 mg/L.
Se realizó la validación en los siguientes niveles: 0,100; 0,140 y 0,200 mg/L, debido a
que a dichos niveles se facilitó la medición (con el material volumétrico disponible) de
las respectivas alícuotas del material de referencia certificado a añadir en las muestras a
fortificar.
Se realizaron los ensayos para valores de concentración que varían desde 0,100 hasta
0,200 mg/L, de esta manera se obtuvieron como resultados valores que se encuentran
dentro de los objetivos establecidos, por lo cual todos estos niveles pasan a formar parte
del método validado.
Como se puede observar en la tabla 5, se cumplió con los criterios establecidos de
precisión (% 𝐶𝑉𝑟 ≤ 15 % y % 𝐶𝑉𝑅 ≤ 15); veracidad (80 < % R < 120) e incertidumbre (≤
30 %). Así se puede observar en la tabla 38 que se obtuvo el mayor valor de % 𝐶𝑉𝑟 de
9,576 % y % 𝐶𝑉𝑅 de 12,895 %; el rango de veracidad fue de 95,333 % a 97,667 % y la
mayor incertidumbre de 29,411 %.
Los parámetros de la función respuesta son: pendiente 0,905; intervalo estudiado de
pendiente 0,865 a 0,948; intercepto -0,004; intervalo estudiado de intercepto -0,010 a
0,000 y el coeficiente de determinación ≥ 0,995.
En función de los resultados anteriores y considerando los criterios de validación
establecidos en los objetivos, se concluye que el método de análisis queda validado en el
rango de 0,100 a 0,200 mg/L en aguas residuales.

83
8. RECOMENDACIONES
El Laboratorio Ambiental Environovalab contará con los parámetros validados, por lo
que se recomienda acreditarlos, debido a que con ello se asegura que los resultados
obtenidos sean confiables y competentes.
Al realizar curvas de calibración, se debe tener máximo cuidado en los puntos más
bajos ya que son los más críticos en la validación y en los cuales se presenta mayor
dificultad al momento de realizar las mediciones correspondientes.
Es estrictamente necesario que cada método de análisis tenga su propio material
volumétrico de trabajo, destinado únicamente para cada analito, de esta manera se
pretende evitar contaminación cruzada durante el análisis de las muestras.
Cumplir con los plazos establecidos para realizar los procesos de calibración de los
equipos, así como para el material volumétrico. Realizar la calibración externa del equipo
y calibración interna del método de acuerdo a las diferentes necesidades.
Se debe realizar una verificación interna y mensual de la recta de calibración fija. En
caso de que no cumpla con las especificaciones se deberá realizar una verificación y/o
mantenimiento externo del equipo, lo recomendable es una vez al año; recurriendo al
personal técnico especializado de confianza. Si no existiese problema con los equipos
indicados se extenderá la periodicidad de mantenimiento, a su vez se recomienda cambiar
las celdas de calibración si fuera necesario, así como asegurarse de la fecha de caducidad
de las mismas.
Para el cumplimiento de la veracidad de los métodos de análisis para los analitos
establecidos, efectuar la verificación de cada analito en su matriz tres veces al año, es
decir cada 4 meses, si no se dispone de muestras se debe proceder utilizando material de
referencia interno o material de referencia certificado.

84
CITAS BIBLIOGRÁFICAS
Acuña Arias, F. (2006). Química Orgánica. San José: EUNED.
Bolaños Chombo, V. (2003). Química Analítica Cuantitativa (Reacciones en Solución).
Ciudad de México : Universidad Autónoma del Estado de México.
Boqué, R., & Maroto, A. (2011). El análisis de la varianza- Comparación de múltiples
poblaciones. Tarragona: Grupo de Quimiometría y Cualimetría. Universitat Rovira i
Virgili.
Brender, J. (9 de Julio de 2016). Quora. Obtenido de How does one measure H2S in the
cell?: https://www.quora.com/How-does-one-measure-H2S-in-the-cell.
Centro Español de Metrología. (2011). Ministerio de Economía, Industria y
Competitividad- Gobierno de España. Obtenido de Glosario de términos:
http://www.cem.es/cem/metrologia/glosario_de_terminos?term_node_tid_depth_1=24.
Consejo del Distrio Metropolitano de Quito. (2016). Ordenanza Metropolitana Nº 138-
Norma técnica para el control de descargas líquidas 002. Quito: Consejo del Distrio
Metropolitano de Quito.
Creus Solé, A. (2011). Instrumentación Industrial. España: Marcombo.
Echeverri Molina, C. A. (2014). Laboratorios de Física- Universidad Tecnológica del
Perú. Obtenido de http://labs123.galeon.com:
http://labs123.galeon.com/metrologia3.htm.
Esopo. (2016). iie.fing.edu.uy. Obtenido de Espectro electromagnético:
https://iie.fing.edu.uy/proyectos/esopo/eem/.

85
Eurachem. (2000). depa.fquim.unam.mx. Obtenido de Cuantificación de la incertidumbre
en mediciones analíticas:
http://depa.fquim.unam.mx/amyd/archivero/Calculo_Incertidumbres_Mediciones_Anali
ticas_25390.pdf.
Eurachem. (2016). Eurachem. Obtenido de Una guía de laboratorio para validación de
métodos y temas relacionados:
https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_ES.pdf.
García Bermejo, M. J. (2006). Auxiliares de Laboratorio de la Xunta de Galicia . Sevilla:
MAD.
Geissman, T. (1974). Principios de Química Orgánica. Barcelona: Reverté.
Griful Ponsati, E., & Canela Campos, M. Á. (2002). Gestión de la Calidad. Catalunya:
EDICIONS UPC.
Hopp, V. (2005). Fundamentos de Tecnología Química para Formación Profesional.
Berlín: Verlag Chemie, Weinheim.
Ich Harmonised Tripartite Guideline. (Noviembre de 2005). www.ich.org. Obtenido de
Conferencia internacional sobre la armonización de los requisitos técnicos para el registro
de productos farmacéuticos para uso humano:
https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_
R1/Step4/Q2_R1__Guideline.pdf.
IDEAM. (28 de Diciembre de 2007). Instituto de Hidrología, Metrología y Estudios
Ambientales . Obtenido de Fenoles en agua por destilación, extracción con cloroformo y
determinación espectrofotométrica:
http://www.ideam.gov.co/documents/14691/38155/Fenoles+por+Destilaci%C3%B3n%
2C+Extracci%C3%B3n+con+CHCL3+y+Espectrofotometr%C3%ADa.pdf/3de90b8a-
b7aa-419d-b8b5-4f71892f696e.

86
ISO & Calidad-Universidad CEU San Pablo Madrid. (2 de mayo de 2013). Capítulo 3,
Validación de métodos analíticos. Obtenido de https://www.docsity.com/es/capitulo-3-
validacion-de-metodos-analiticos/568385/.
ISO. (1994). ISO Plataforma de navegación en línea. Obtenido de ISO 5725-1:1994
Exactitud (veracidad y precisión) de los métodos de medición y resultados - Parte 1:
Principios generales y definiciones:
https://www.aenor.es/aenor/descarga_extracto.asp?producto=N0038154
Jurado, M. J. (1 de abril de 2008). Aplicación de Microsoft Excel a la química analítica:
validación de métodos analíticos. Obtenido de
http://personal.us.es/jmjurado/docs/AQAEXCEL.pdf.
Laboratorio Metrycal. (2016). www.metrycal.com. Obtenido de Estimación de la
incertidumbre de medida:
http://www.metrycal.com/Main/Estimacion_de_la_incertidumbre_de_medida.pdf.
Ministerio de Ambiente. (2015). TULSMA-Norma de calidad ambiental y de descarga de
efluentes: Recurso Agua. Libro VI. Anexo 1. Quito: CEP. Corporación de estudios y
pulicaciones.
Olsen, E. D. (1990). Métodos Ópticos de Análisis. Barcelona: Reverté.
physics.stackexchange.com. (2011). physics.stackexchange.com. Obtenido de
https://physics.stackexchange.com/questions/225211/ what-is-the-coefficient-of
volumetric-expansion-of-water.
Portero, S. (2007). Portal de la Red de Salud Públiba de Cuba. Obtenido de Radiación
ultravioleta: http://www.sld.cu/galerias/pdf/sitios/rehabilitacion-fis/ultravioleta-
morrillo.pdf.
Químicas.net. (8 de septiembre de 2015). w.w.w.químicas.net. Obtenido de Los sulfatos:
http://www.quimicas.net/2015/09/los-sulfatos.html.

87
Rice, E., Baird, R., Eaton, A., & Clesceri, L. (2012). Standard Methods for the
Examination of Water and Wastewater. USA: APHA, AWWA, WPCF.
Rickard, D., & Morse, J. (2005). Acid Volatile Sulfide (AVS). Texas USA: ELSEVIER.
Rigola Lapeña, M. (1990). Tratamiento de Aguas Industriales: Agua de Proceso y
Residuales. Barcelona: Prodúctica.
Spiegel, M. R., & Stephens, L. J. (2009). Estadística Schaum. México: Mc Graw Hill.
Teijón, J. M. (2006). Fundamentos de Bioquímica Estructural. Madrid: Editorial Tebar.
Toasa, F. (2012). Validación de los métodos de ensayo para fenoles, tensoactivos, sólidos
suspendidos y total de sólidos disueltos (TDS). Quito: Tesis de Grado para la obtención
del Título de Ingeniera Química.
Ventura Egusquiza, G., & Lluque Aquino, A. (11 de junio de 2008). Ministerio de Salud
del Perú. Obtenido de Cálculo de la incertidumbre de la medición:
http://www.ins.gob.pe/insvirtual/images/normatividad/proctec/ PRT-CNSP-
008%20Ed%2001%20C%C3%A1lculo%20de%20la%20incertidumbre%20de%20la%2
0medici%C3%B3n_FINAL.pdf.

88
BIBLIOGRAFÍA
Atkins, P., & De Paula, J. (2006). Physical Chemistry. USA: Oxford University.
Ayus, J. C., & Caramelo, C. (2007). Agua, Electrolitos y Equilibrio Ácido-Base:
Aprendizaje Mediante Casos Clínicos. Buenos Aires: Médica Panamericana.
Cabrerizo, D., Antón, J., & Barrio, J. (2010). Física y Química . Madrid: Editex.
Camero Jiménez, J. (Octubre de 2007). Colegio de Ingenieros de Perú- Capítulo de
Ingeniería Química. Obtenido de Técnicas estadísticas aplicadas al sistema de gestión de
laboratorios bajo la ISO/IEC 17025:
http://inocua.org/site/images/colaboradores/Hoja_de_Vida_JOSE_CAMERO.pdf.
Campos Gómez, I. (2003). Saneamiento Ambiental. San José: EUNED.
Carmona Baez, M. (2014). Instituto de Salud Pública de Chile. Obtenido de Validación
de métodos de ensayo:
http://www.ispch.cl/sites/default/files/Presesntacion_Taller_Validacion_Analitica14_10
_2014%20Marco%20Carmona.pdf.
Chapman, D. (1996). Water Quality Assessment . Cambridge : UNESCO.
ECA Ente Costarricense de Acreditación. (18 de febrero de 2014). ECA. Obtenido de
Guía para la validación de métodos: www.eca.or.cr/docus/v2/4003.
Instituto de Salud Pública de Chile. (2010). Validación de Métodos y Determinación de
la Incertidumbre de la Medición. Santiago: Instituto de Salud Pública de Chile.

89
ISO. (1994). ISO Plataforma de navegación en línea. Obtenido de ISO 5725-1:1994
Exactitud (veracidad y precisión) de los métodos de medición y resultados - Parte 1:
Principios generales y definiciones:
https://www.aenor.es/aenor/descarga_extracto.asp?producto=N0038154
Laboratorio Metrycal. (2016). www.metrycal.com. Obtenido de Estimación de la
incertidumbre de medida:
http://www.metrycal.com/Main/Estimacion_de_la_incertidumbre_de_medida.pdf.
Ministerio de Salud de Panamá. (1986). Memorias del Seminario- Taller: Normas de
Caldad de Agua. Panamá: CATIE.
OSAKITEDZA- Servicio Vasco de Salud. (2006). Técnicos Especialistas de
Laboratorio. Madrid: MAD.
Pino Pérez, F., & Pérez Bendito, D. (1983). Análisis de Elementos-traza por
Espectrofotometría de Absorción Molecular Ultravioleta-visible. Sevilla: Universidad de
Sevilla, Imprenta San Pablo.
Rodríguez García, J., & Virgós, J. (1998). Fundamentos de Óptica Ondulatoria. Oviedo:
Universidad de Oviedo.
Skoog, D., Holler, J., & Timothy, N. (1992). Principios de Análisis Instrumental. Madrid:
Mc Graw Hill.
Spectroquant MERCK. (2011). Operating Manual Spectroquant Pharo 300. Darmstadt,
Alemania: MERK.
Vilanova, E., & Sogorb, M. Á. (2015). Técnicas Analíticas de Contaminantes Químicos:
Aplicaciones Toxicológicas, Medioambientales y Alimentarias. España: Díaz de Santos.
Walton, H., & Reyes, J. (2005). Análsis Químico e Intrumental Moderno. Barcelona:
Reverté.

90
ANEXOS
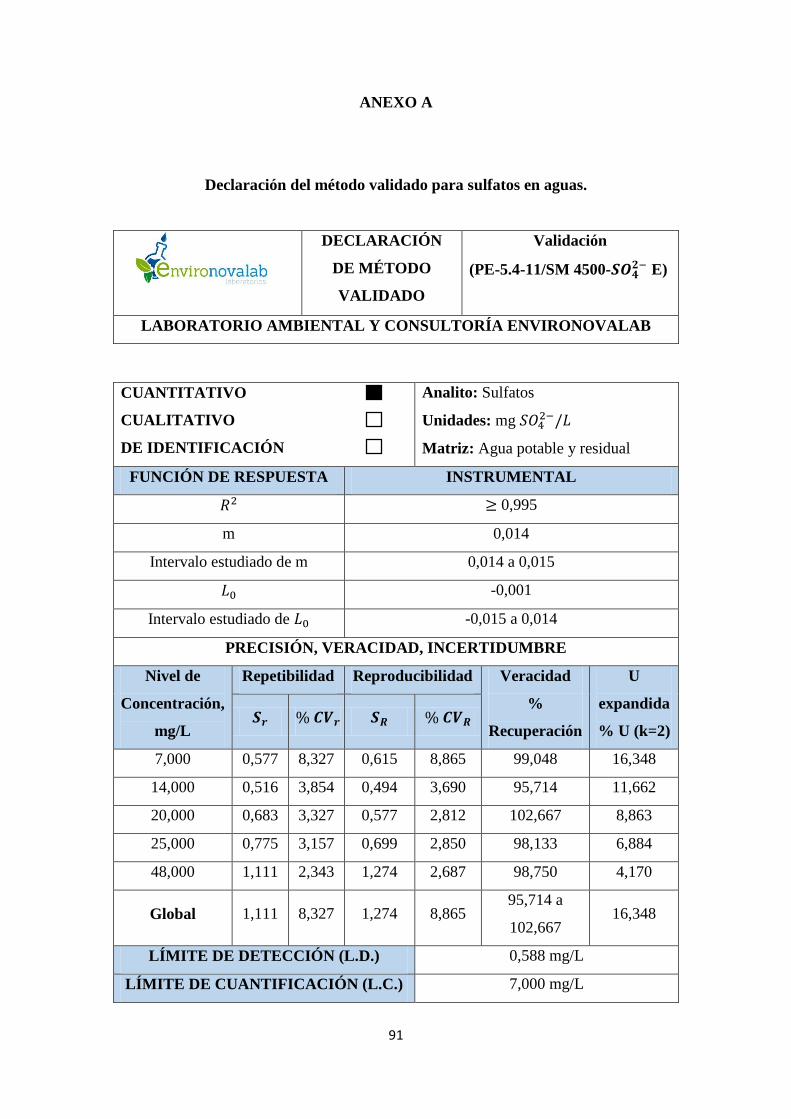
91
ANEXO A
Declaración del método validado para sulfatos en aguas.
DECLARACIÓN
DE MÉTODO
VALIDADO
Validación
(PE-5.4-11/SM 4500-𝑺𝑶𝟒𝟐− E)
LABORATORIO AMBIENTAL Y CONSULTORÍA ENVIRONOVALAB
CUANTITATIVO
CUALITATIVO
DE IDENTIFICACIÓN
Analito: Sulfatos
Unidades: mg 𝑆𝑂42−/𝐿
Matriz: Agua potable y residual
FUNCIÓN DE RESPUESTA INSTRUMENTAL
𝑅2 ≥ 0,995
m 0,014
Intervalo estudiado de m 0,014 a 0,015
𝐿0 -0,001
Intervalo estudiado de 𝐿0 -0,015 a 0,014
PRECISIÓN, VERACIDAD, INCERTIDUMBRE
Nivel de
Concentración,
mg/L
Repetibilidad Reproducibilidad Veracidad
%
Recuperación
U
expandida
% U (k=2) 𝑺𝒓 % 𝑪𝑽𝒓 𝑺𝑹 % 𝑪𝑽𝑹
7,000 0,577 8,327 0,615 8,865 99,048 16,348
14,000 0,516 3,854 0,494 3,690 95,714 11,662
20,000 0,683 3,327 0,577 2,812 102,667 8,863
25,000 0,775 3,157 0,699 2,850 98,133 6,884
48,000 1,111 2,343 1,274 2,687 98,750 4,170
Global 1,111 8,327 1,274 8,865 95,714 a
102,667 16,348
LÍMITE DE DETECCIÓN (L.D.) 0,588 mg/L
LÍMITE DE CUANTIFICACIÓN (L.C.) 7,000 mg/L

92
SELECTIVIDAD / ESPECIFICIDAD
INTERFERENCIAS CONOCIDAS:
Color o la materia en suspensión: En grandes cantidades pueden interferir con la
determinación.
Sílice: En cantidad superior a 500 mg/L interfiere.
Calcio: En cantidad superior a 20000 mg/L como 𝐶𝑎𝐶𝑂3.
Materia orgánica: En grandes cantidades puede no ser posible precipitar 𝐵𝑎𝑆𝑂4
satisfactoriamente.
CORRECCIÓN DE INTERFERENCIAS: Para el tratamiento de las interferencias,
se debe preparar las muestras previamente como se indica en el numeral 7 del
procedimiento de ensayo de sulfatos PE-5.4-11/SM 4500-𝑆𝑂42− E.
INTERVALO DE TRABAJO VALIDADO: 7-48 mg/L
CRITERIOS DE ACEPTACIÓN / RECHAZO
LINEALIDAD: 𝑅2 ≥ 0,995
VERACIDAD: 80 < % R < 120
El porcentaje de recuperación obtenido con las muestras fortificadas va desde 95,714
a 102,667 %.
El porcentaje de recuperación que caracteriza al método es el obtenido a partir del
material de referencia certificado y es del 99,60 %.
INCERTIDUMBRE ≤ 30 % (k=2) → Sulfatos ≥ 7,000 mg/L
Las incertidumbres fueron estimadas con un nivel de confianza (k=2), obteniéndose el
mayor porcentaje de incertidumbre (16,348 %) en el nivel de 7,000 mg/L.
Las incertidumbres que deberán reportarse en el informe de resultados serán:
Rango: 7,000 a 14,000 mg/L → U = 1,633 mg/L (k=2)
Rango: 15,000 a 20,000 mg/L → U = 1,773 mg/L (k=2)
Rango: 21,000 a 25,000 mg/L → U = 1,721 mg/L (k=2)
Rango: 26,000 a 48,000 mg/L → U = 2,001 mg/L (k=2)

93
PRECISIÓN:
Repetibilidad: 𝐶𝑉𝑟 ≤ 10% → Sulfatos ≥ 7,000 mg/L
El mayor coeficiente de variación obtenido es de 8,327 % que corresponde a la
concentración de 7,000 mg/L.
Reproducibilidad: 𝐶𝑉𝑅 ≤ 10% → Sulfatos ≥ 7,000 mg/L
El mayor coeficiente de variación obtenido es de 8,865 % que corresponde a la
concentración de 7,000 mg/L.
CONCLUSIÓN:
Al cumplirse todos los parámetros de desempeño, la validación queda aceptada.

94
ANEXO B
Declaración del método validado para sulfuros en aguas.
DECLARACIÓN DE
MÉTODO
VALIDADO
Validación
(PE-5.4-10/SM/4500-𝑺𝟐− D)
LABORATORIO AMBIENTAL Y CONSULTORÍA ENVIRONOVALAB
CUANTITATIVO
CUALITATIVO
DE IDENTIFICACIÓN
Analito: Sulfuros
Unidades: mg 𝑆2−/𝐿
Matriz: Agua residual
FUNCIÓN DE RESPUESTA INSTRUMENTAL
𝑅2 ≥ 0,995
m 0,204
Intervalo estudiado de m 0,194 a 0,217
𝐿0 -0,004
Intervalo estudiado de 𝐿0 -0,012 a 0,001
PRECISIÓN, VERACIDAD, INCERTIDUMBRE
Nivel de
Concentración,
mg/L
Repetibilidad Reproducibilidad Veracidad
%
Recuperación
U
expandida
% U (k=2) 𝑺𝒓 % 𝑪𝑽𝒓 𝑺𝑹 % 𝑪𝑽𝑹
0,280 0,008 2,897 0,007 2,427 98,095 29,888
0,450 0,008 1,806 0,007 1,582 100,444 18,854
1,000 0,021 2,065 0,017 1,686 100,400 8,905
Global 0,021 2,897 0,017 2,427 98,095 a
100,444 29,888
LÍMITE DE DETECCIÓN (L.D.) 0,011 mg/L
LÍMITE DE CUANTIFICACIÓN (L.C.) 0,280 mg/L

95
SELECTIVIDAD / ESPECIFICIDAD
INTERFERENCIAS CONOCIDAS:
Tiosulfatos: Cuando la concentración es mayor de 10 mg/L, la formación del color
se retarda.
La misma concentración de sulfuros: Cuando es de varios cientos de miligramos
por cada litro, representa una interferencia.
CORRECCIÓN DE INTERFERENCIAS: Para el tratamiento de las interferencias,
se debe preparar las muestras previamente como se indica en el numeral 7 del
procedimiento de ensayo de sulfuros PE-5.4-10/SM/4500-𝑆2− D.
INTERVALO DE TRABAJO VALIDADO: 0,28-1,00 mg/L
CRITERIOS DE ACEPTACIÓN / RECHAZO
LINEALIDAD: 𝑅2 ≥ 0,995
VERACIDAD: 80 < % R < 120
El porcentaje de recuperación obtenido con las muestras fortificadas va desde 98,095 a
100,444 %.
El porcentaje de recuperación que caracteriza al método es el obtenido a partir del
material de referencia certificado y es del 98,33 %.
INCERTIDUMBRE ≤ 30 % (k=2) → Sulfuros ≥ 0,280 mg/L
Las incertidumbres fueron estimadas con un nivel de confianza (k=2), obteniéndose el
mayor porcentaje de incertidumbre (29,888 %) en el nivel de 0,280 mg/L.
Las incertidumbres que deberán reportarse en el informe de resultados serán:
Rango: 0,280 a 0,450 mg/L → U = 0,085 mg/L (k=2)
Rango: 0,460 a 1,000 mg/L → U = 0,089 mg/L (k=2)

96
PRECISIÓN:
Repetibilidad: 𝐶𝑉𝑟 ≤ 10% → Sulfuros ≥ 0,280 mg/L
El mayor coeficiente de variación obtenido es de 2,897 % que corresponde a la
concentración de 0,280 mg/L.
Reproducibilidad: 𝐶𝑉𝑅 ≤ 10% → Sulfuros ≥ 0,280 mg/L
El mayor coeficiente de variación obtenido es de 2,427 % que corresponde a la
concentración de 0,280 mg/L.
CONCLUSIÓN:
Al cumplirse todos los parámetros de desempeño, la validación queda aceptada.
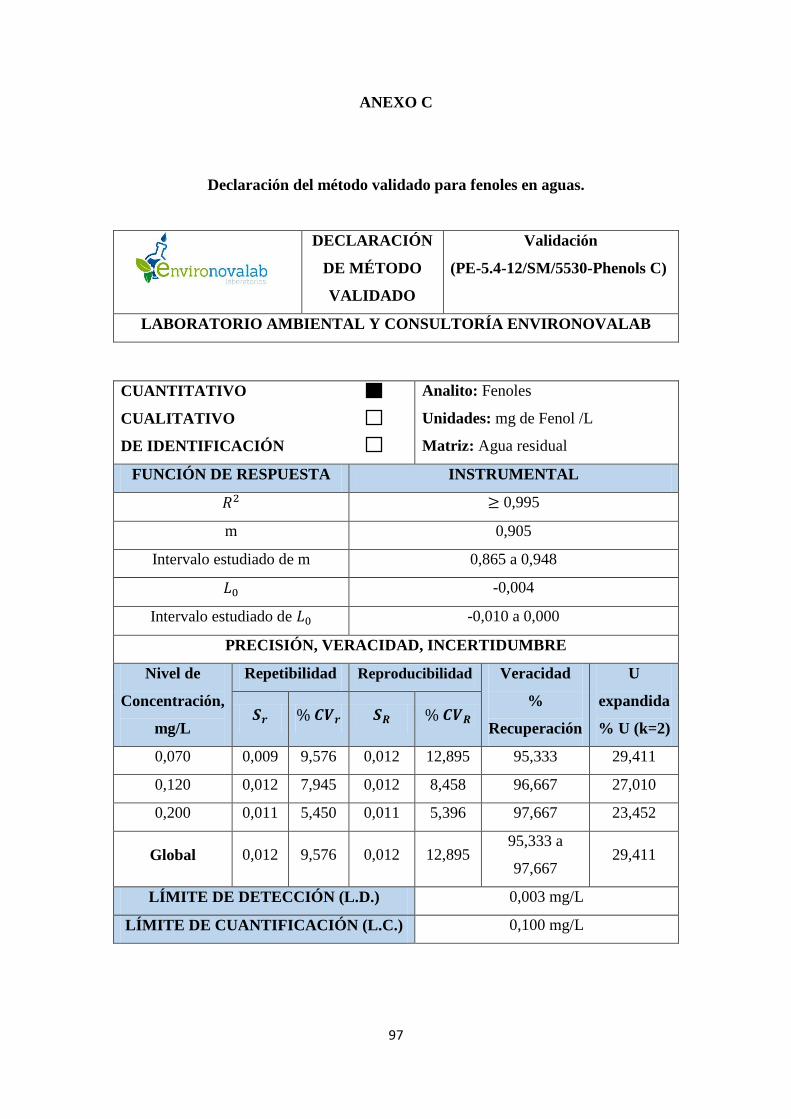
97
ANEXO C
Declaración del método validado para fenoles en aguas.
DECLARACIÓN
DE MÉTODO
VALIDADO
Validación
(PE-5.4-12/SM/5530-Phenols C)
LABORATORIO AMBIENTAL Y CONSULTORÍA ENVIRONOVALAB
CUANTITATIVO
CUALITATIVO
DE IDENTIFICACIÓN
Analito: Fenoles
Unidades: mg de Fenol /L
Matriz: Agua residual
FUNCIÓN DE RESPUESTA INSTRUMENTAL
𝑅2 ≥ 0,995
m 0,905
Intervalo estudiado de m 0,865 a 0,948
𝐿0 -0,004
Intervalo estudiado de 𝐿0 -0,010 a 0,000
PRECISIÓN, VERACIDAD, INCERTIDUMBRE
Nivel de
Concentración,
mg/L
Repetibilidad Reproducibilidad Veracidad
%
Recuperación
U
expandida
% U (k=2) 𝑺𝒓 % 𝑪𝑽𝒓 𝑺𝑹 % 𝑪𝑽𝑹
0,070 0,009 9,576 0,012 12,895 95,333 29,411
0,120 0,012 7,945 0,012 8,458 96,667 27,010
0,200 0,011 5,450 0,011 5,396 97,667 23,452
Global 0,012 9,576 0,012 12,895 95,333 a
97,667 29,411
LÍMITE DE DETECCIÓN (L.D.) 0,003 mg/L
LÍMITE DE CUANTIFICACIÓN (L.C.) 0,100 mg/L

98
SELECTIVIDAD / ESPECIFICIDAD
INTERFERENCIAS CONOCIDAS:
La mayoría de sustancias interferentes son eliminadas destilando los fenoles de la
muestra.
Bacterias descomponedoras de fenoles, sustancias oxidantes o reductoras, y valores
de pH alcalinos: Pueden interferir en el desarrollo del color.
CORRECCIÓN DE INTERFERENCIAS: Para el tratamiento de las interferencias,
se debe preparar las muestras previamente como se indica en el numeral 7 del
procedimiento de ensayo de fenoles PE-5.4-12/SM/5530-Phenols C.
INTERVALO DE TRABAJO VALIDADO: 0,100-0,200 mg/L
CRITERIOS DE ACEPTACIÓN / RECHAZO
LINEALIDAD: 𝑅2 ≥ 0,995
VERACIDAD: 80 < % R < 120
El porcentaje de recuperación obtenido con las muestras fortificadas va desde 95,333 a
97,667 %.
El porcentaje de recuperación que caracteriza al método es el obtenido a partir del
material de referencia certificado y es del 97,89 %.
INCERTIDUMBRE ≤ 30 % (k=2) → Fenoles ≥ 0,100 mg/L
Las incertidumbres fueron estimadas con un nivel de confianza (k=2), obteniéndose el
mayor porcentaje de incertidumbre (29,411 %) en el nivel de 0,100 mg/L.
Las incertidumbres que deberán reportarse en el informe de resultados serán:
Rango: 0,100 a 0,140 mg/L → U = 0,038 mg/L (k=2)
Rango: 0,150 a 0,200 mg/L → U = 0,047 mg/L (k=2)

99
PRECISIÓN:
Repetibilidad: 𝐶𝑉𝑟 ≤ 15% → Fenoles ≥ 0,100 mg/L
El mayor coeficiente de variación obtenido es de 9,576 % que corresponde a la
concentración de 0,100 mg/L.
Reproducibilidad: 𝐶𝑉𝑅 ≤ 15% → Fenoles ≥ 0,100 mg/L
El mayor coeficiente de variación obtenido es de 12,895 % que corresponde a la
concentración de 0,100 mg/L.
CONCLUSIÓN:
Al cumplirse todos los parámetros de desempeño, la validación queda aceptada.

100
ANEXO D
Cálculos de Excel para el método de sulfatos.
Figura D.1. Curva de calibración fijada para el método de sulfatos.

101
Figura D.2. ANOVA detallado para el método de sulfatos.

102
Figura D.3. ANOVA de sulfatos calculado por Excel.
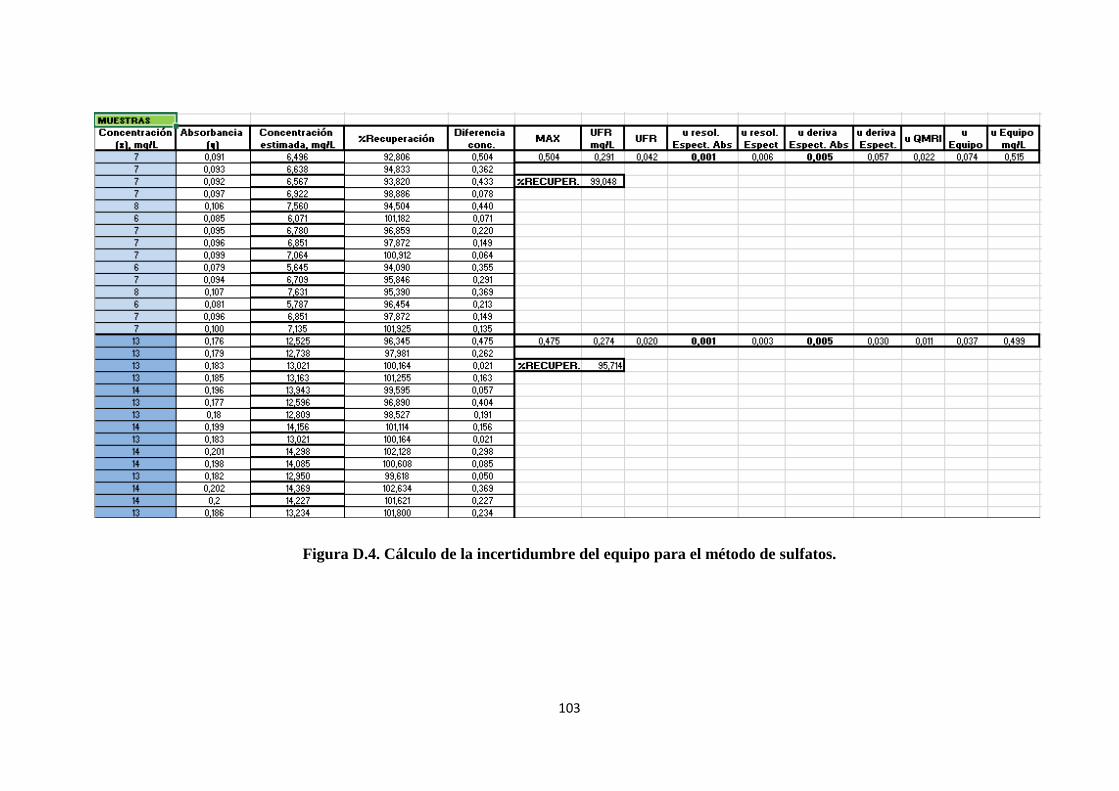
103
Figura D.4. Cálculo de la incertidumbre del equipo para el método de sulfatos.

104
Figura D.4. (Continuación)
Figura D.4. Cálculo de la incertidumbre del equipo para el método de sulfatos.
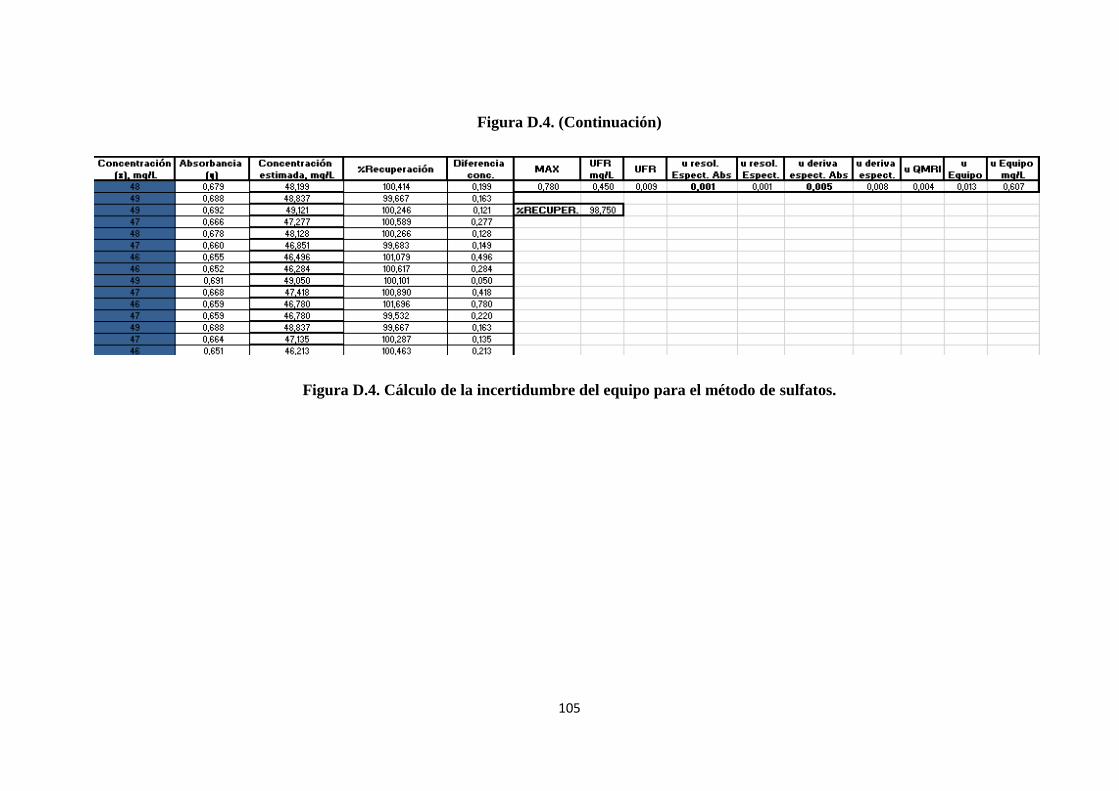
105
Figura D.4. (Continuación)
Figura D.4. Cálculo de la incertidumbre del equipo para el método de sulfatos.
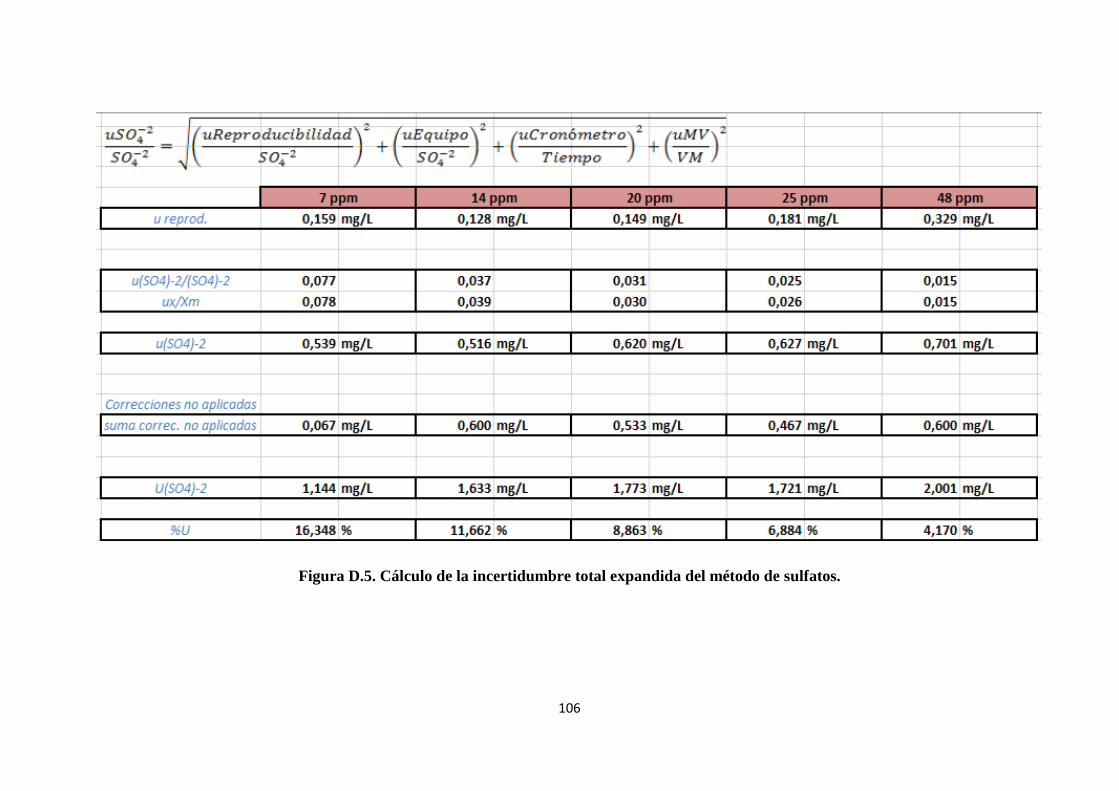
106
Figura D.5. Cálculo de la incertidumbre total expandida del método de sulfatos.

107
ANEXO E
Cálculos de Excel para el método de sulfuros.
Figura E.1. Curva de calibración fijada para el método de sulfuros.
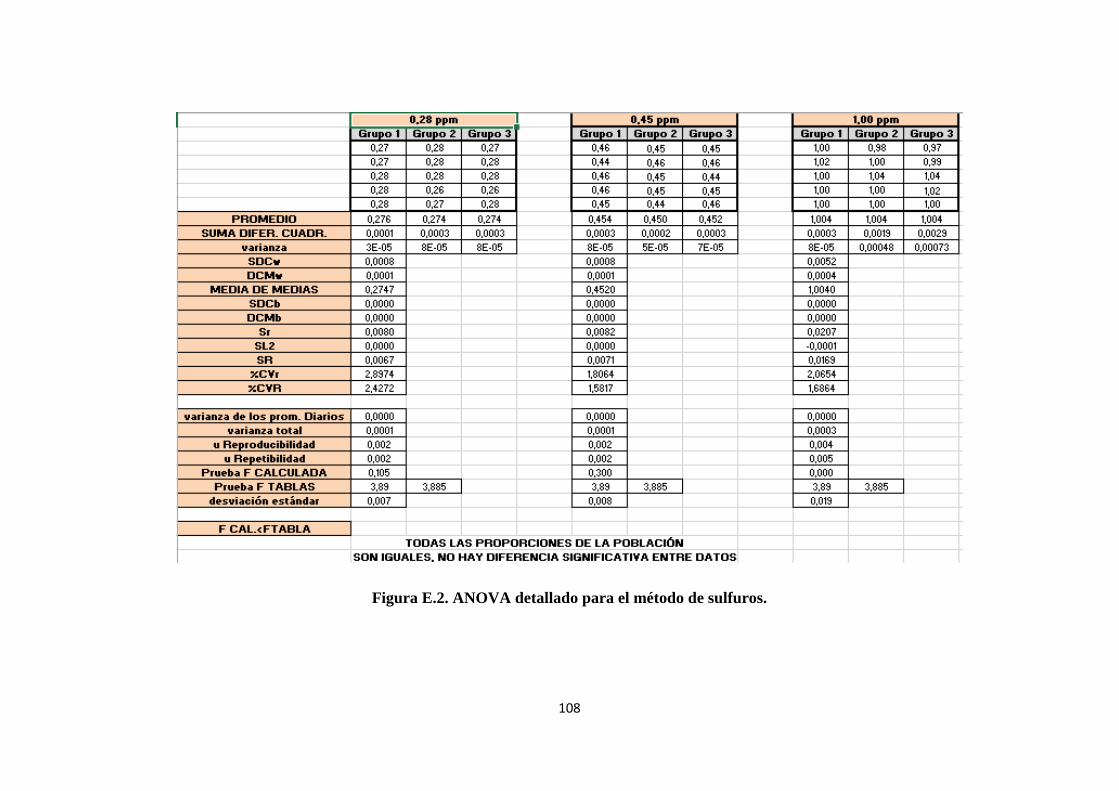
108
Figura E.2. ANOVA detallado para el método de sulfuros.
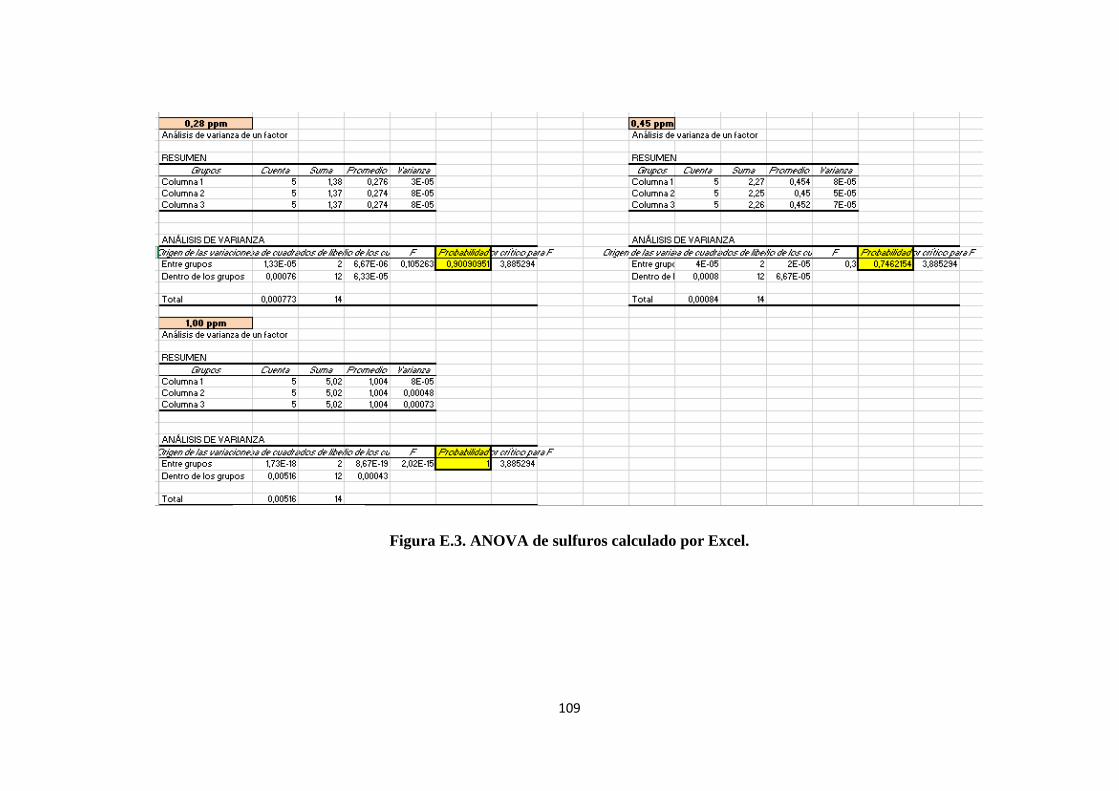
109
Figura E.3. ANOVA de sulfuros calculado por Excel.
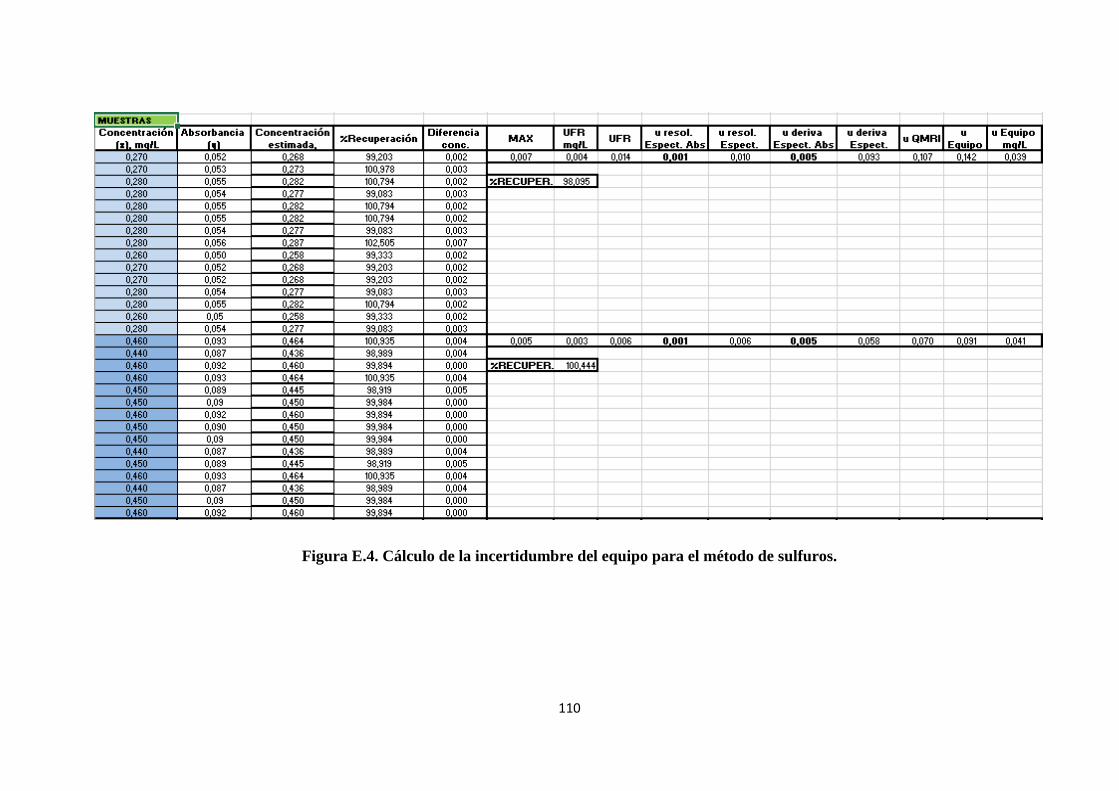
110
Figura E.4. Cálculo de la incertidumbre del equipo para el método de sulfuros.
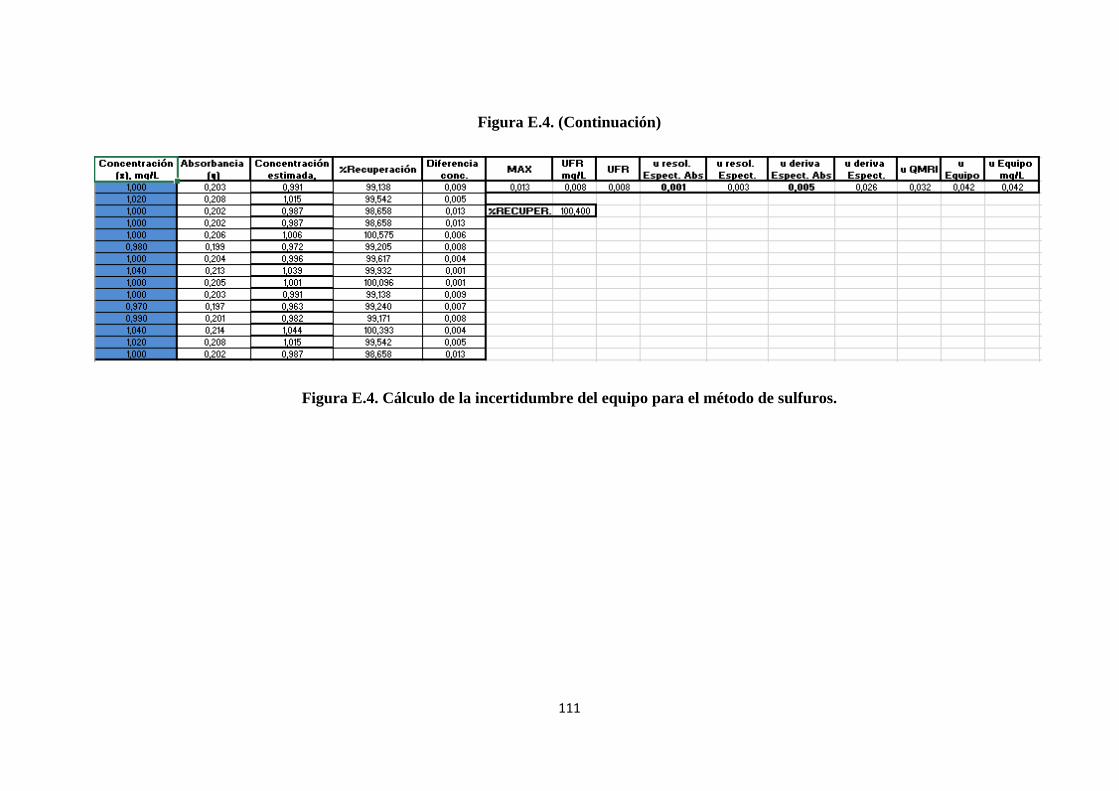
111
Figura E.4. (Continuación)
Figura E.4. Cálculo de la incertidumbre del equipo para el método de sulfuros.

112
Figura E.5. Cálculo de la incertidumbre total expandida del método de sulfuros.

113
ANEXO F
Cálculos de Excel para el método de fenoles.
Figura F.1. Curva de calibración fijada para el método de fenoles.

114
Figura F.2. ANOVA detallado para el método de fenoles.
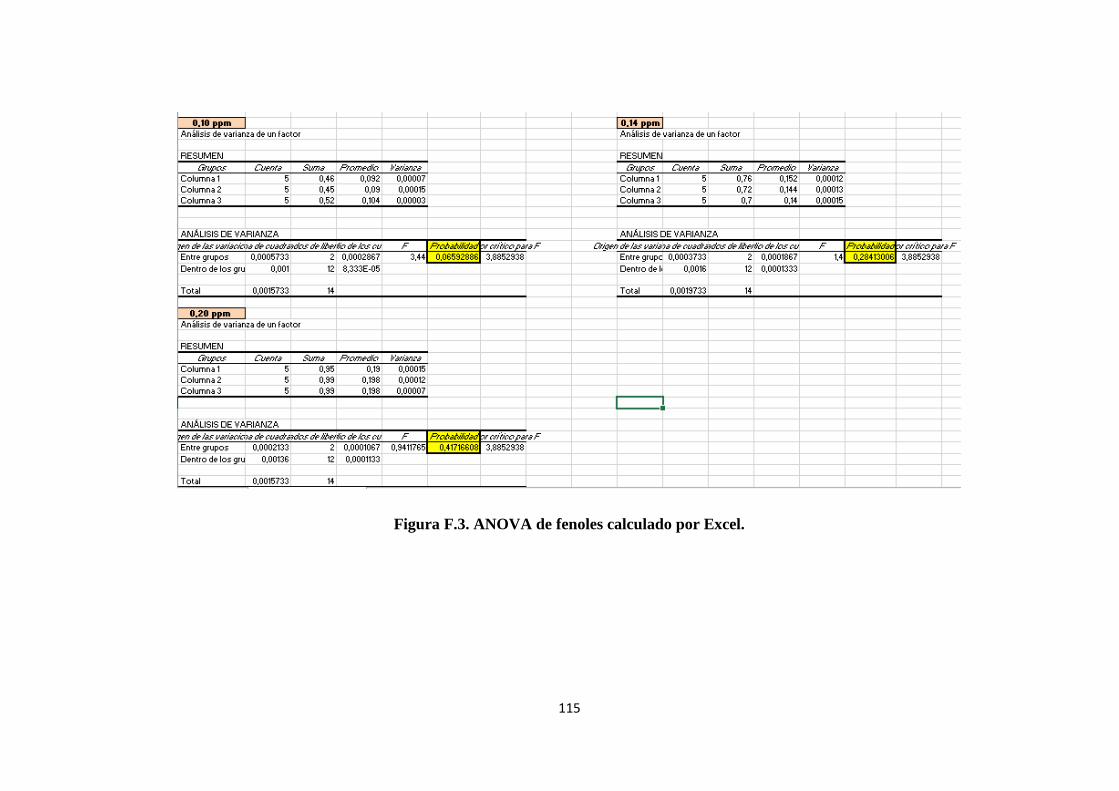
115
Figura F.3. ANOVA de fenoles calculado por Excel.
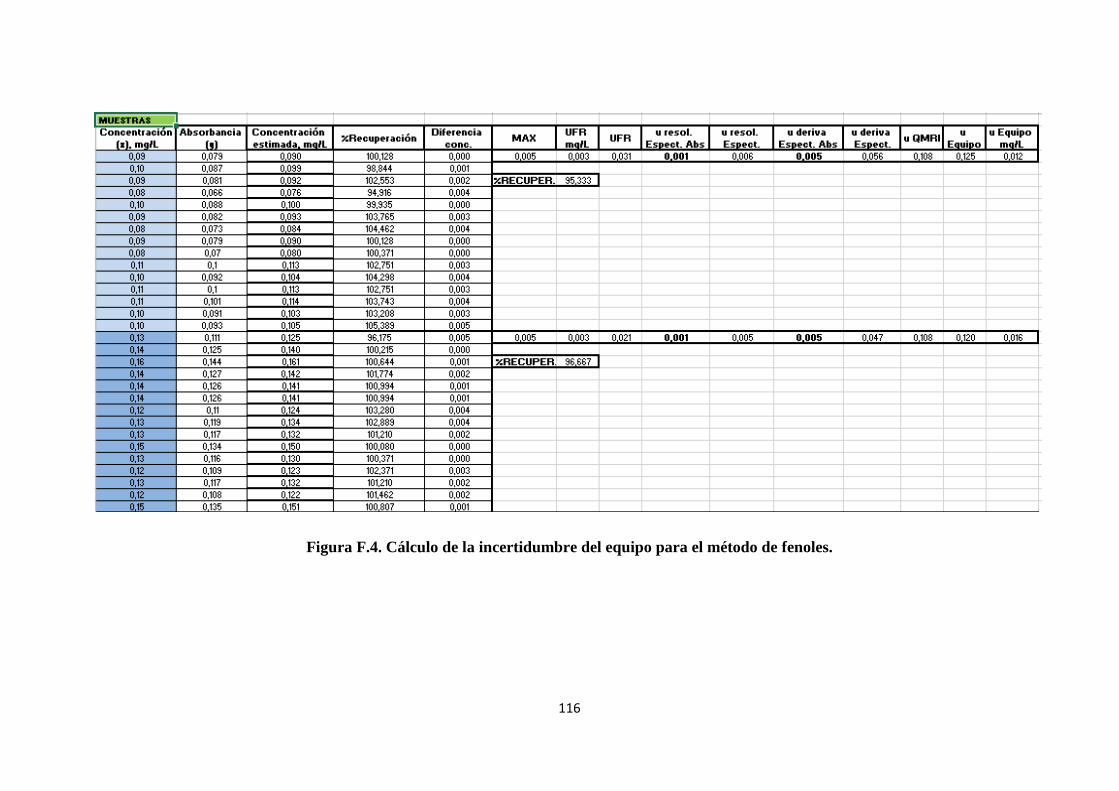
116
Figura F.4. Cálculo de la incertidumbre del equipo para el método de fenoles.
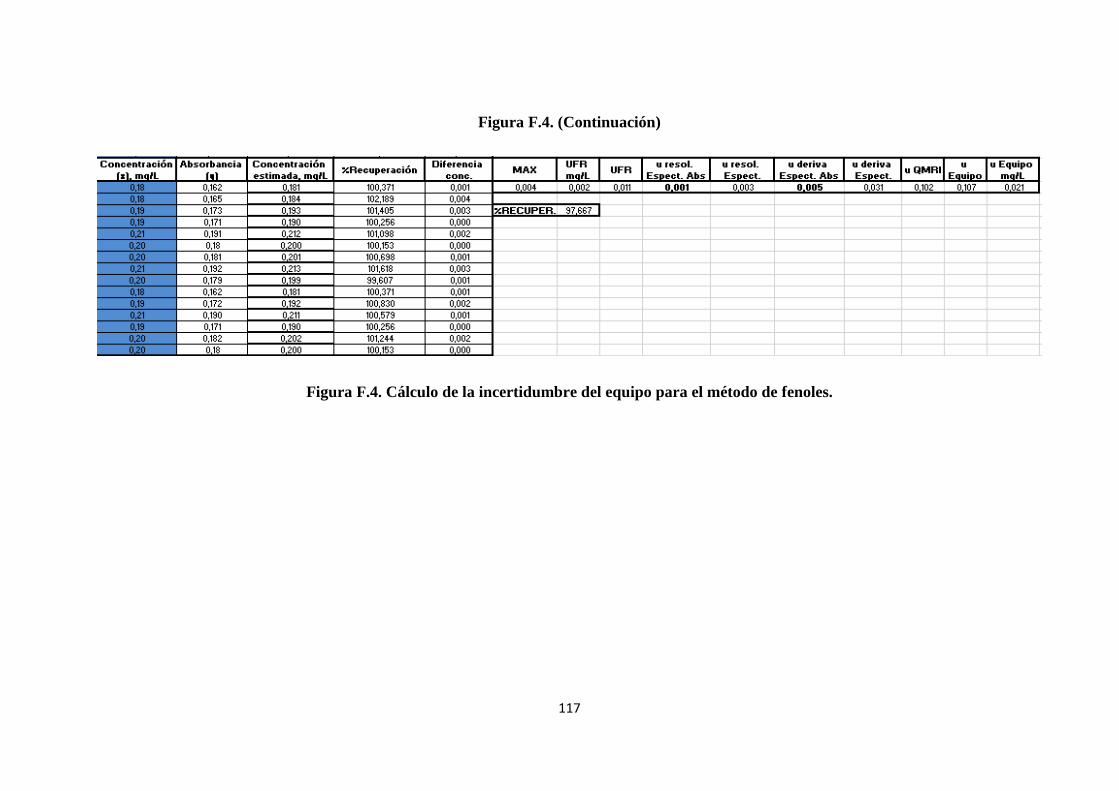
117
Figura F.4. (Continuación)
Figura F.4. Cálculo de la incertidumbre del equipo para el método de fenoles.
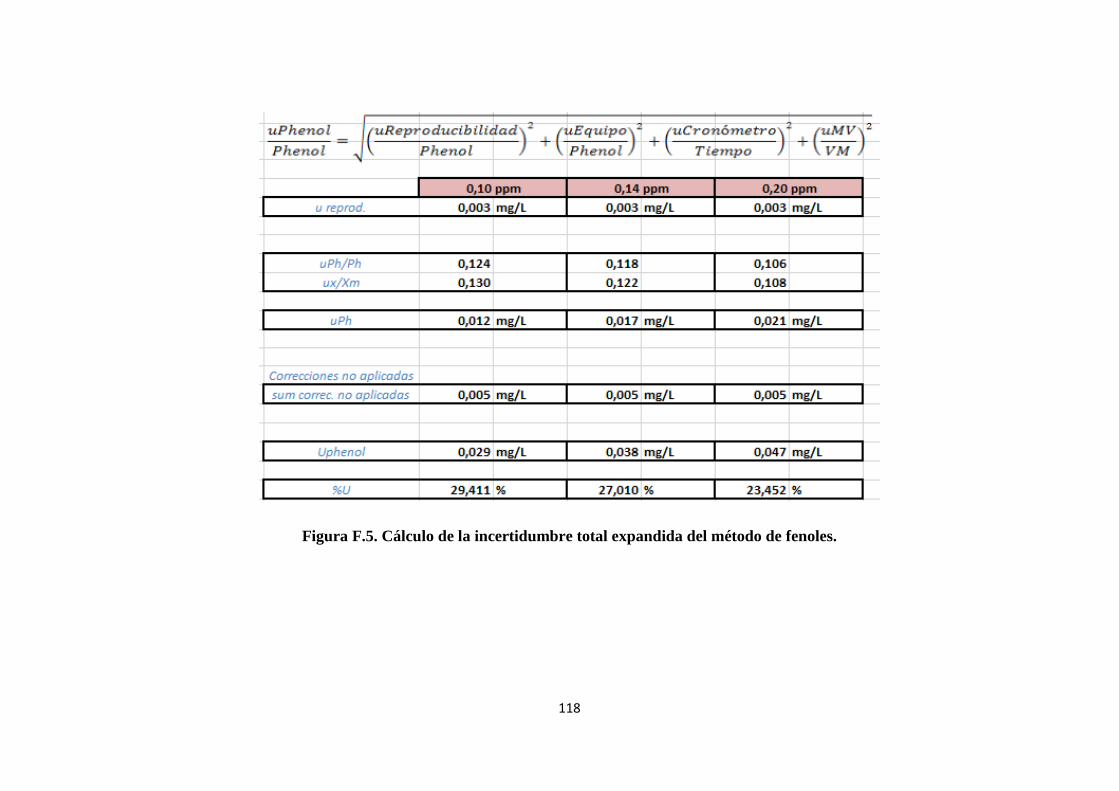
118
Figura F.5. Cálculo de la incertidumbre total expandida del método de fenoles.

119
ANEXO G
Reporte fotográfico del equipo.
Figura G.1. Equipo Espectrofotómetro Spectroquant Pharo 300