perfil oncogÉnico del carcinoma adenoide …seorl.net/pdf/tesis/2007 perfil oncogénico del...
TRANSCRIPT

Universidad de Oviedo Departamento de Cirugía y Especialidades Médico-Quirúrgicas
“PERFIL ONCOGÉNICO
DEL CARCINOMA ADENOIDE QUÍSTICO
DE GLÁNDULAS SALIVALES”
Guadalupe Sequeiros Santiago Tesis Doctoral (que opta al grado de Doctor por la Universidad
de Oviedo)

Universidad de Oviedo Departamento de Cirugía y Especialidades Médico-Quirúrgicas
“PERFIL ONCOGÉNICO
DEL CARCINOMA ADENOIDE QUÍSTICO
DE GLÁNDULAS SALIVALES”
Tesis Doctoral (que opta al grado de Doctor por la Universidad
de Oviedo)
Autor Guadalupe Sequeiros Santiago
Directores Maria Victoria González Meana Juan Pablo Rodrigo Tapia

“Lo que sabemos es una gota de agua;
lo que ignoramos es el océano”
Isaac Newton
ii

a mi hermana
que forma parte de mi existencia
por los sueños no realizados
a mis padres
iii

AGRADECIMIENTOS
Quiero expresar mi agradecimiento más sincero a todas
aquellas personas que, con su colaboración, han contribuido a
la realidad de este trabajo:
A la Dra. Mª Victoria González Meana, directora de la Tesis,
por su dedicación, contribuyendo con su experiencia y la gran
calidad de su trabajo a elaborar esta Tesis.
Al Dr. Juan Pablo Rodrigo Tapia, codirector de la Tesis, que
encauzó mi trabajo de investigación hacia el carcinoma
adenoide quístico, por brindarme su dilatada y valiosa
experiencia en el campo de la investigación y la práctica
clínica para forjar y dar forma a esta Tesis.
Al Dr. Carlos Suárez Nieto, por su apoyo permanente y
estímulo constante fomentando la investigación en el cáncer
de cabeza y cuello, por la seriedad y rigor con la que lleva a
cabo su trabajo, siendo una referencia a seguir; y por darme
la oportunidad de realizar la Tesis en su servicio.
ii

Al Servicio de Anatomía Patológica del Hospital Central de
Asturias, por su inestimable colaboración en la obtención de
muestras tumorales de sus archivos; especialmente al Dr. D.
Manuel Florentino Fresno Forcelledo, Jefe del Servicio, por su
importantísima participación en la realización e interpretación
de las técnicas inmunohistoquímicas.
Al Servicio de Cirugía Máxilofacial del Hospital Central de
Asturias, que me permitió utilizar los pacientes intervenidos
por ellos para realizar este trabajo; especialmente al Dr. D.
Juan Sebastián López-Arranz, Jefe del Servicio, por su buen
asesoramiento y por brindarme todo tipo de facilidades, de
manera desinteresada, para la obtención del material a
estudio; al Dr. D. Santiago Llorente Pendás, por su generosa
colaboración y disponibilidad, proporcionándome información
clínica de sus pacientes; y al Dr. Jacobo Sánchez Mayoral, que
colaboró en la búsqueda inicial de los pacientes intervenidos
en su servicio.
A los compañeros del Laboratorio de Oncología Quirúrgica del
IUOPA, Doctores y doctorandos, Darío, Juani, Cristina, Emma,
iii

Pablo, Marta y Mariló, por su ayuda y comprensión siempre
que tuve que recurrir a ellos.
A todos mis antiguos compañeros otorrinolaringólogos del
Hospital Central de Asturias, adjuntos y residentes, por
proporcionarme información sobre la evolución de los
pacientes, en especial al Dr. José Luís Llorente Pendás, que,
con su buen hacer, me transmitió sus valiosos conocimientos.
A quienes me quieren y compartieron mi esfuerzo: mis
amigos y mi familia.
iv

Índice
v

1. INTRODUCCIÓN........................................................... 1
1.1. GENERALIDADES DEL CARCINOMA ADENOIDE
QUÍSTICO DE GLÁNDULAS SALIVALES ......................... 2
1.1.1. Epidemiología ................................................ 2
1.1.2. Histología ...................................................... 2
1.1.3. Clínica........................................................... 6
1.1.4. Tratamiento................................................... 8
1.1.5. Pronóstico ..................................................... 9
1.2. ASPECTOS MOLECULARES DE LA CARCINOGÉNESIS ..10
1.2.1. Generalidades ...............................................10
1.2.2. Tipos de genes implicados en cáncer ................14
1.2.2.1. Genes supresores de tumores......................14
1.2.2.2. Genes de reparación ..................................14
1.2.2.3. Oncogenes................................................14
1.2.3. Oncogenes estudiados....................................16
1.2.3.1. ERBB1....................................................16
1.2.3.2. c-kit.......................................................18
1.2.3.3. PIK3CA...................................................21
1.2.3.4. CCND1/PRAD1.........................................24
1.2.3.5. c-myc ....................................................26
1.2.3.6. MDM2 ....................................................28
2. HIPOTESIS DE TRABAJO Y OBJETIVOS...........................32
3. MATERIAL Y METODOS ................................................35
3.1. CARACTERÍSTICAS CLINICO-PATOLÓGICAS DE LOS
PACIENTES Y SUS TUMORES......................................36
3.2. ESTUDIO DE AMPLIFICACIÓN GÉNICA......................40
3.2.1. Procesamiento de muestras ............................40
3.2.1.1. Desparafinado .......................................40
3.2.1.2. Lisis .....................................................40
ii

3.2.1.3. Extracción de ADN .................................40
3.2.2. Análisis de amplificación génica. PCR múltiplex ..41
3.2.2.1. Validación del método.............................45
3.3. ESTUDIO DE EXPRESIÓN PROTEICA ........................46
3.3.1. Técnica inmunohistoquímica empleada .............46
3.3.2. Análisis de expresión de EGFR, c-kit y ciclina D1 47
3.4. ANÁLISIS ESTADÍSTICO.........................................49
4. RESULTADOS .............................................................51
4.1. AMPLIFICACIÓN ONCOGÉNICA EN LOS TUMORES
PRIMARIOS .............................................................52
4.2. AMPLIFICACIÓN ONCOGÉNICA EN LAS RECIDIVAS
TUMORALES ............................................................55
4.3. NIVELES DE EXPRESIÓN PROTEICA .........................56
4.3.1. Expresión de c-kit..........................................56
4.3.2. Expresión de EGFR.........................................59
4.3.3. Expresión de ciclina D1...................................59
4.4. RELACIÓN ENTRE LA AMPLIFICACIÓN GÉNICA DE
DIVERSAS DIANAS...................................................62
4.5. RELACIÓN ENTRE LA AMPLIFICACIÓN GÉNICA Y LA
EXPRESIÓN PROTEICA ..............................................64
4.5.1. ERBB1- EGFR................................................64
4.5.2. c-kit – c-kit...................................................64
4.5.3. CCND1-ciclina D1 ..........................................64
4.6. RELACIÓN ENTRE LAS ALTERACIONES MOLECULARES
Y LOS PARÁMETROS CLINICO-PATOLÓGICOS...............67
4.7. ANÁLISIS DE SUPERVIVENCIA ................................73
5. DISCUSIÓN................................................................85
6. CONCLUSIONES .......................................................102
7. BIBLIOGRAFÍA..........................................................105
iii

ABREVIATURAS
CAQ, Carcinoma Adenoide
Quístico
CDK, del inglés, Cyclin
Dependent Kinase
CEA, del inglés,
CarcinoEmbrionic Antigen
CGH, del inglés, Comparative
Genomic Hybridization
CKI, del inglés, Cyclin-
dependent Kinase Inhibitor
DAB, diaminobencidina
dNTP, desoxinucleótido
trifosfato.
EDTA, ácido
etilendiaminotetraacético
EGF, del inglés, Epidermal
Growth Factor
EGFR, del inglés, Epidermal
Growth Factor Receptor
EMA, del inglés, Epithelial
Membrane Antigen
FISH, del inglés, Fluorescence
In Situ Hybridization
GF, del inglés, Growth Factor
GIST, del inglés,
GastroIntestinal Stromal Tumor
HNSCC, del inglés Head and
Neck Squamous Cell Carcinoma
IHQ, inmunohistoquímica
ILK, del inglés, Integrin Linked
Kinase
MDM2, del inglés, Murine
Double Minute
PCR, del inglés, Polymerase
Chain Reaction
PDK1, 2 del inglés,
Phosphoinositide-Dependent
protein Kinase 1
PH, del inglés, Plekstrin
Homology
PIP2, del inglés,
Phosphoinositide 3,4
bisphosphate
PIP3, del inglés,
Phosphoinositide 3,4,5-
trisphosphate
Rb, retinoblastoma
SCF, del inglés, Stem Cell
Factor
TAE, Tris-acético-EDTA.
TGFα, del inglés, Transforming
Growth Factor α
TNM, del inglés, Tumor-Node-
Metastasis.
TSG, del inglés, Tumor
Supresor Gene

NOTA
El trabajo realizado durante esta Tesis Doctoral, ha dado lugar a la siguiente publicación: Sequeiros Santiago G, Rodrigo Tapia JP, Garcia-Carracedo D, Garcia Pedrero J, Suarez Nieto C, Gonzalez Meana MV. CCND1 gene amplification in the adenoid cystic carcinoma of the minor salivary glands. Acta Otorrinolaringol Esp. 2004 Feb;55(2):88-92.

1
Introducción

Introducción
1.1. GENERALIDADES DEL CARCINOMA ADENOIDE
QUÍSTICO DE GLÁNDULAS SALIVALES
1.1.1. Epidemiología
El carcinoma adenoide quístico (CAQ) es un tumor maligno
de estirpe epitelial, descrito por primera vez por Billroth en
1856 (1), quien lo denominó inicialmente “cilindroma” por su
apariencia histológica cilíndrica. Representa aproximadamente
el 1% de los tumores de cabeza y cuello y el 10% de los
tumores de glándulas salivales (2, 3). Es el tumor más
frecuente de las glándulas salivales menores, donde
representa el 60%, siendo el paladar la localización más
frecuente en este tipo de glándulas. Globalmente, a nivel de
cabeza y cuello, la glándula parótida es la localización más
frecuente del CAQ (25%) (4). Se desconocen los factores
etiológicos ligados a su desarrollo.
1.1.2. Histología
De forma característica, este tumor presenta
diferenciación mioepitelial y produce gran cantidad de
proteínas que forman parte de membranas basales y de la
2

Introducción
matriz extracelular. Las tinciones inmunohistoquímicas
confirman la presencia de, al menos, dos tipos de células:
• Células ductales que expresan el antígeno
carcinoembrionario (CEA) y el antígeno de membrana
epitelial (EMA).
• Células no ductales que expresan la actina muscular
específica, característica del mioepitelio.
El CAQ presenta tres patrones de crecimiento o subtipos
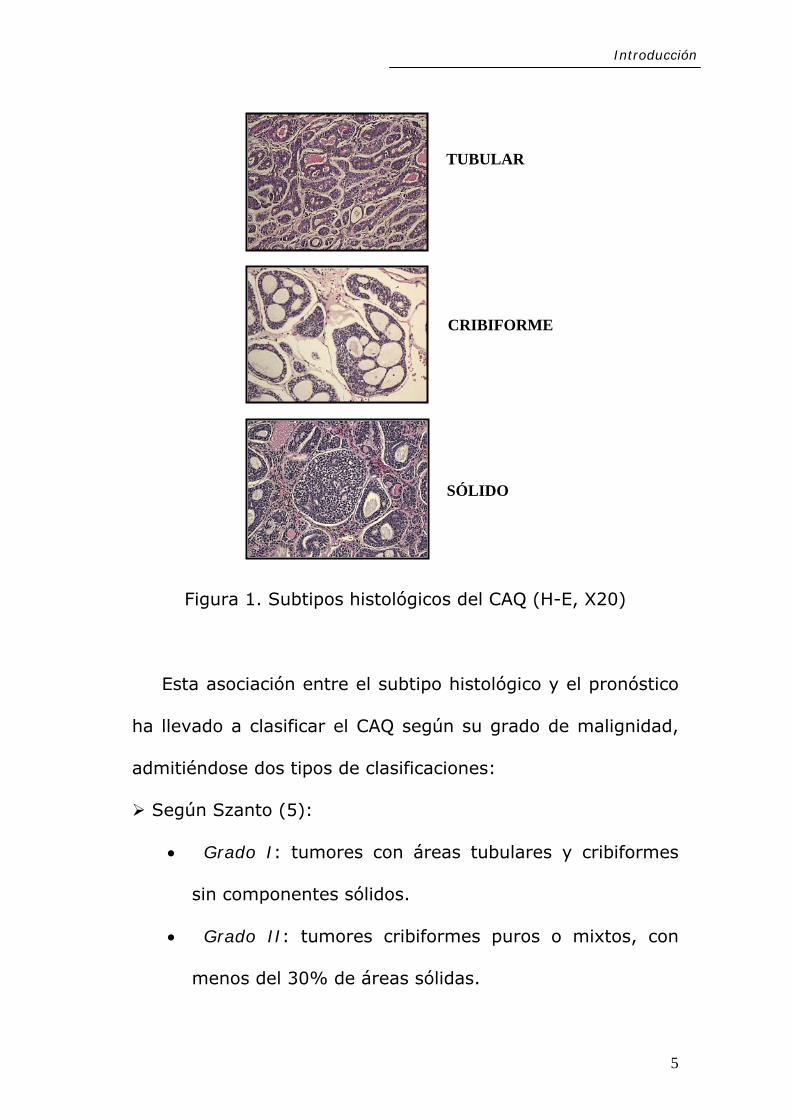
histológicos (Figura 1) (4):
• Tubular, presenta conductos bien formados y túbulos
con luces centrales que están delimitadas por una doble capa
de células epiteliales internas y mioepiteliales externas.
• Cribiforme, que es el más frecuente, se caracteriza por
nidos de células con espacios microquísticos cilindromatosos.
Estos espacios están llenos de material hialino o mucoide
basófilo.
• Sólido o basaloide, único en el que se observan focos de
necrosis, pleomorfismo celular y mitosis. Formado por
sábanas de células basaloides uniformes que pierden la
formación tubular o microquística. Es el menos frecuente.
3

Introducción
Ciertos tumores del subtipo sólido contienen estructuras
trabeculares, por lo que algunos autores han reconocido un
cuarto subtipo, el trabecular.
En la mayoría de los tumores se observan los cuatro
patrones en proporciones variables, siendo el de mayor
representación el que condiciona el diagnóstico patológico.
Numerosos estudios han descrito una asociación entre el
subtipo histológico y el pronóstico de la enfermedad (5); así,
el subtipo tubular tiene el mejor pronóstico con respecto a la
recurrencia o metástasis, el cribiforme tiene un pronóstico
intermedio, y el subtipo sólido tiene el peor pronóstico, ya que
se asocia con recurrencia precoz de la enfermedad, desarrollo
temprano de metástasis a distancia (2, 6) y elevada tasa de
mortalidad. El patrón trabecular presenta un comportamiento
intermedio entre los subtipos cribiforme y sólido (7).
4
Introducción
TUBULAR
CRIBIFORME
SÓLIDO
Figura 1. Subtipos histológicos del CAQ (H-E, X20)
Esta asociación entre el subtipo histológico y el pronóstico
ha llevado a clasificar el CAQ según su grado de malignidad,
admitiéndose dos tipos de clasificaciones:
Según Szanto (5):
• Grado I: tumores con áreas tubulares y cribiformes
sin componentes sólidos.
• Grado II: tumores cribiformes puros o mixtos, con
menos del 30% de áreas sólidas.
5

Introducción
• Grado III: tumores consistentes en más del 30% de
patrón sólido.
Según Yamamoto et al. (7):
- Tumores de bajo grado (subtipos tubular y cribiforme)
- Tumores de alto grado (subtipos trabecular y sólido).
A nivel histológico un fenómeno único en el CAQ es la
propensión a la invasión perineural, tanto microscópica como
macroscópica, a través de nervios mayores y menores (3, 4,
8, 9). No se conoce la causa de este patrón de crecimiento,
presente por igual en los tumores de alto y bajo grado de
malignidad, y también en estadios tumorales precoces. La
existencia de invasión perineural se relaciona con un peor
pronóstico de la enfermedad cuando afecta a nervios mayores
(10, 11).
1.1.3. Clínica
El CAQ se caracteriza por presentar un curso clínico
prolongado, con un crecimiento lento y progresivo, y una
infiltración insidiosa de los tejidos adyacentes. Parece existir
una correlación entre el sitio de origen y el pronóstico; los
CAQs que asientan en glándulas salivales menores, por lo
general, están avanzados en el momento del diagnóstico, ya
6

Introducción
que, debido a este lento crecimiento, el diagnóstico suele ser
tardío. A diferencia de éstos, los tumores de glándulas
salivales mayores presentan un mejor pronóstico por su
diagnóstico más precoz y su más fácil acceso. Así, se ha
descrito que los CAQs de la cavidad nasal y senos paranasales
presentan peor pronóstico que los de cualquier otra
localización en el área de cabeza y cuello (12, 13).
Debido a su crecimiento y progresión perineural, a través
de nervios mayores y menores (3, 4, 8, 9), no es infrecuente
que los carcinomas localizados en las fosas nasales pasen a la
cavidad intracraneal por los agujeros oval o redondo mayor, o
bien aparezcan recidivas intracraneales tiempo después de la
cirugía.
Otra característica de este tumor es su tendencia a las
recidivas locales múltiples; así, a pesar de realizar un
tratamiento local agresivo, la mayoría de los pacientes (60%)
desarrollan recurrencias. Aproximadamente el 2% de las
recurrencias son clínicamente evidentes en los dos primeros
años después del tratamiento.
Las metástasis ganglionares no son frecuentes en este
tumor (5%), ni al diagnóstico ni durante el curso de la
enfermedad. Las metástasis a distancia ocurren en un 33-
7

Introducción
40% de los casos, y pueden aparecer incluso décadas después
del tratamiento del tumor primario (14) y tras haber logrado
un adecuado control locorregional; suelen asentar en pulmón,
seguido del hueso y, con menor frecuencia, en el hígado. A
diferencia de otros tumores malignos de cabeza y cuello, los
pacientes con CAQ a menudo sobreviven durante largos
períodos de tiempo con metástasis a distancia (15).
1.1.4. Tratamiento
El tratamiento de elección es la cirugía. La extensión de la
resección quirúrgica (completa vs residual microscópica) tiene
un impacto significativo en la supervivencia libre de
enfermedad y en el control local, pero carece de impacto
sobre la supervivencia global debido al lento crecimiento de
este tipo de tumores (16).
El CAQ es un tumor radiosensible pero no radiocurable.
Varios estudios señalan que la combinación de cirugía y
radioterapia ofrece las mejores tasas de control de la
enfermedad (15). Así, la radioterapia estaría indicada en los
siguientes casos: localización tumoral próxima a base de
cráneo, márgenes quirúrgicos positivos, estadio tumoral
8

Introducción
avanzado, subtipo histológico sólido, presencia de metástasis
linfáticas al diagnóstico y en caso de recurrencias (15, 17).
La quimioterapia tiene un papel paliativo, y se utiliza en el
caso de enfermedad localmente avanzada o metástasis a
distancia, aunque la respuesta del tumor es limitada.
1.1.5. Pronóstico
No es fácil predecir el curso clínico de los CAQs de
glándulas salivales. Se han establecido una serie de
parámetros clínico-patológicos con valor pronóstico, entre los
que se encuentran la presencia de metástasis linfáticas
cervicales al diagnóstico, el estadio tumoral avanzado, la
presencia de márgenes quirúrgicos afectados, el tipo
histológico sólido, y la existencia de invasión perineural
macroscópica (8, 10, 11, 14, 17). La invasión macroscópica
perineural, los márgenes positivos, y el patrón histológico
sólido predicen el control locoregional, mientras que las
metástasis linfáticas, la invasión perineural de nervios
mayores, el subtipo histológico sólido y la presencia de
múltiples síntomas al diagnóstico influyen de manera negativa
en la supervivencia (14).
9

Introducción
Asimismo, se han estudiado otros marcadores de carácter
molecular, como son la ploidía del ADN (18), las regiones de
organización nucleolar (19), la expresión del antígeno Ki-67
(20), y el porcentaje de células en fase S (21). Sin embargo,
las alteraciones moleculares responsables de la progresión
tumoral del CAQ no se conocen aún.
Como se mencionó, una característica de estos tumores
son las recidivas múltiples, que pueden ocurrir más de diez
años después del tratamiento inicial, por lo que es difícil
hablar estrictamente de curación de la enfermedad; por lo
tanto, los resultados del tratamiento y el pronóstico final
deben ser evaluados al término de diez a quince años.
Diversos estudios demuestran cómo la tasa de supervivencia
libre de enfermedad disminuye del 90 al 40% a los 5 y 15
años, respectivamente (14).
1.2. ASPECTOS MOLECULARES DE LA
CARCINOGÉNESIS
1.2.1. Generalidades
El cáncer es una enfermedad genética a nivel celular, en la
que la acumulación de alteraciones genéticas dota a la célula
10

Introducción
de una ventaja selectiva de crecimiento, y posterior capacidad
transformante (22).
Las células normales son capaces de mantener un balance
finamente controlado entre las señales promotoras e
inhibidoras del crecimiento, de manera que la proliferación y
la diferenciación ocurren de una manera ordenada. Sin
embargo, en las células tumorales este equilibrio desaparece,
favoreciéndose una proliferación continuada frente a la
diferenciación. Además, éstas son capaces de sobrevivir en
situaciones en las que una célula normal entraría en la muerte
celular programada o apoptosis. Estas alteraciones se
manifiestan como consecuencia de la acumulación de diversos
daños genéticos, que proporcionan a las células una ventaja
de crecimiento selectivo frente al resto, escapando al control
normal de la proliferación. Esta acumulación tiene lugar de
forma escalonada, de manera que se pueden distinguir
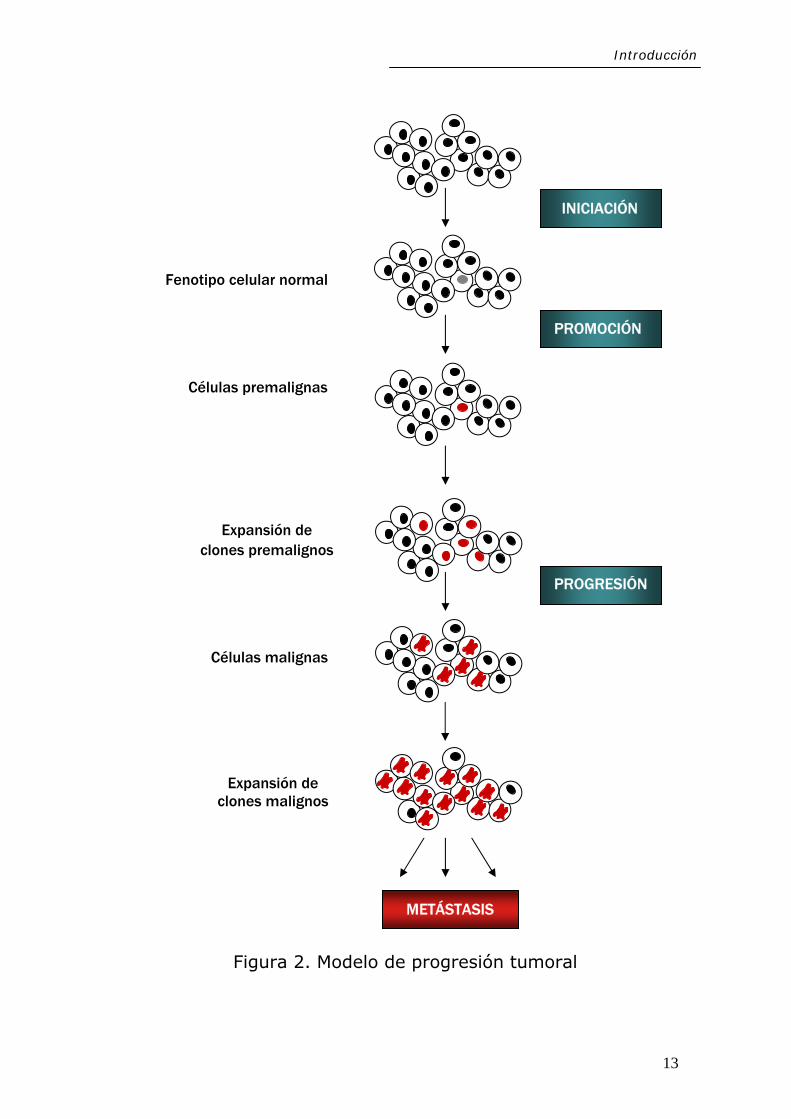
básicamente tres fases: iniciación, promoción y progresión
(Figura 2). La célula mutada que adquiere ventaja selectiva
genera un clon homogéneo, en el que se van acumulando
mutaciones adicionales, surgiendo diversos clones celulares,
motivo por el que los tumores presentan un alto grado de
heterogeneidad. Algunas de las mutaciones acumuladas
11

Introducción
potencian el carácter maligno de los clones celulares
afectados. Adicionalmente, los tumores malignos invaden los
tejidos adyacentes y se diseminan por el organismo dando
lugar a las metástasis, siendo este proceso el más
amenazante para la vida. Para ello, las células tumorales
adquieren capacidades determinadas genéticamente. Múltiples
estudios apuntan a que es la acumulación de eventos lo que
es importante, más que el orden en el cual ocurren.
Hanahan y Weinberg (23) describieron seis alteraciones
esenciales en la fisiología celular que son requeridas para la
transformación maligna:
• Independencia de factores de crecimiento
• Insensibilidad a las señales de inhibición del
crecimiento
• Escape de la muerte celular programada
(apoptosis)
• Capacidad replicativa ilimitada
• Angiogénesis sostenida
• Invasividad y metástasis
12
Introducción
INICIACIÓN
Figura 2. Modelo de progresión tumoral
PROGRESIÓN
Fenotipo celular normal
PROMOCIÓN
Células premalignas
Expansión de clones premalignos
Células malignas
Expansión de clones malignos
METÁSTASIS
13

Introducción
1.2.2. Tipos de genes implicados en cáncer
Las mutaciones causantes de la aparición del cáncer tienen
lugar en tres tipos de genes: los genes supresores de tumores
(TSGs), los implicados en los procesos de reparación del ADN
o genes de estabilidad y los proto-oncogenes.
1.2.2.1. Genes supresores de tumores
Son aquellos que codifican proteínas cuya función
contribuye al control de la proliferación, por lo que las
mutaciones en estos genes, que provoquen la pérdida de
función de la proteína, privan a las células de un mecanismo
de control esencial en el mantenimiento del equilibrio
proliferación-diferenciación.
1.2.2.2. Genes de reparación
Son aquellos que codifican proteínas implicadas en corregir
los errores que se producen tanto en la incorporación de
nucleótidos durante el proceso de replicación del ADN como
por agentes físicos y químicos.
1.2.2.3. Oncogenes
Los protooncogenes son genes que, en condiciones
normales, están involucrados en las funciones básicas
esenciales de las células, como la proliferación. Ante estímulos
extracelulares, como factores de crecimiento, las células
14

Introducción
ponen en marcha rutas de señalización intracelular,
comenzando por la activación de receptores de superficie y
finalizando en el núcleo, donde se produce la expresión
selectiva de genes en respuesta a los estímulos externos
recibidos.
Los oncogenes son formas alteradas de los proto-
oncogenes. Esta alteración implica una ganancia de función,
reflejada en una proliferación incontrolada (exceso de
proliferación o defecto de muerte celular).
Algunos de los principales mecanismos de activación
oncogénica son:
amplificación génica (ganancia del número de copias
del gen).
aumento de la expresión provocada por traslocaciones
cromosómicas.
mutaciones intragénicas, que afectan a residuos
cruciales que regulan la actividad de la proteína.
15

Introducción
1.2.3. Oncogenes estudiados
1.2.3.1. ERBB1
ERBB1 (localizado en 7p12-14) es el gen que codifica el
receptor del factor de crecimiento epidérmico (EGFR),
perteneciente a la familia de los receptores tirosin quinasa.
Entre sus ligandos se encuentran el factor de crecimiento
epidérmico (EGF), el factor de crecimiento transformante
(TGFα), anfiregulina, betacelulina y epiregulina, de los cuales
TGFα ha sido el ligando más frecuentemente implicado en
cáncer.
Una vez que el ligando se une al receptor tirosin quinasa
se produce la dimerización, u oligomerización, resultando en
un cambio conformacional en el receptor, el cual lleva a la
activación de su actividad tirosina quinasa intrínseca.
Las principales vías de señalización iniciadas mediante las
interacciones con EGFR son las vías de la Ras-Raf-MAP
quinasa y la ruta del metabolismo fosfolipídico (Figura 3).
16

Introducción
Ligando
PI3-K
PKD1
AKT/PKB
RAS
RAF
MEK
JAK
STAT
Oncogenes nucleares
TRANSCRIPCIÓN DE GENES
ERK
EGFR
Progresión Supervivencia Proliferación ciclo celular celular
Figura 3. Vías de señalización intracelular activadas por EGFR
17

Introducción
El mecanismo más frecuente mediante el cual EGFR
contribuye al cáncer es a través su sobreexpresión, a menudo
junto con uno de sus ligandos, TGFα o EGF.
Esta alteración ha sido descrita para varios tipos de
tumores humanos, entre ellos, el carcinoma epidermoide de
cabeza y cuello (HNSCC, 50-100% de los casos), alteración
asociada a enfermedad avanzada, desarrollo de un fenotipo
metastático y un pronóstico pobre (24, 25).
El hecho de tratarse de un receptor de membrana, cuya
sobreexpresión es específica de las células tumorales,
convierte a EGFR en una atractiva diana para el desarrollo de
fármacos dirigidos a bloquear su sobreactivación, como
anticuerpos monoclonales (cetuximab – Erbitux®) y pequeñas
moléculas inhibidoras (gefitinib - Iressa®, erlotinib -
Tarceva®).
1.2.3.2. c-kit
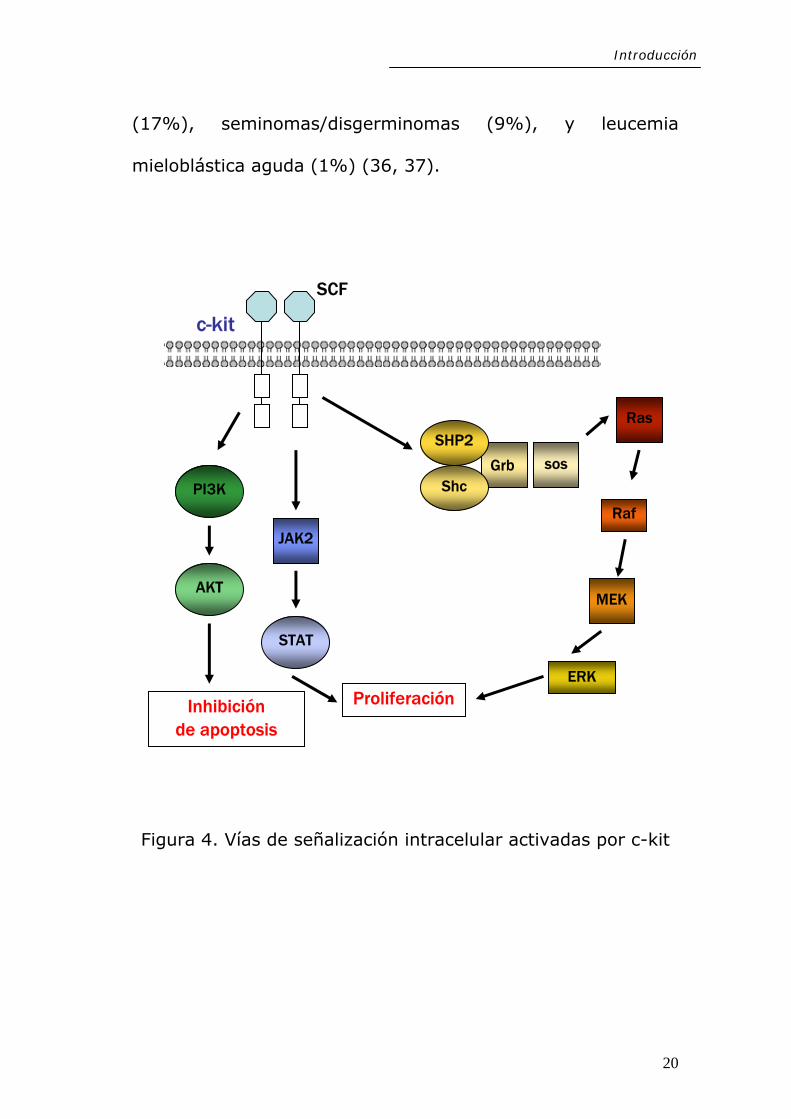
El proto-oncogén c-kit (localizado en 4q11-12), codifica un
receptor transmembrana tirosin quinasa tipo III de 145-160
kDa, llamado también CD117. La interacción de c-kit con su
ligando, SCF (del inglés, Stem Cell Factor), promueve la
fosforilación y la activación de rutas de señalización
18

Introducción
intracelular – dianas como Akt, la familia de quinasas Src, PI
3-K, Fosfolipasa Cγ, y Ras/MAPK- esenciales en procesos
como embriogénesis, hematopoyesis, desarrollo, proliferación
y migración (26-28) (Figura 4).
Son varios los tipos de neoplasias malignas que muestran
una sobreexpresión de c-kit, entre ellos, los tumores
estromales gastrointestinales (GIST), para los que se ha
demostrado una relación entre la activación de c-kit y la
patogénesis de la enfermedad (29, 30); tumores de células
germinales testiculares (31), carcinomas endometriales (32),
cáncer de pulmón de células pequeñas y no pequeñas – para
los que se ha establecido como indicador de pronóstico
adverso - (33), carcinoma seroso de ovario avanzado, para el
que la expresión de c-kit ha sido asociada con resistencia a la
quimioterapia (34), y diversos tipos de sarcoma (35).
Asimismo, se han descrito mutaciones puntuales en c-kit,
localizadas en el dominio quinasa, que provocan un aumento
de función que genera una progresión del ciclo celular y
supresión de la apoptosis. Estas han sido descritas en un
porcentaje de neoplasias humanas, incluyendo mastocitomas
(> 90%), GIST (> 70%), linfomas nasosinusales de células T
19
Introducción
(17%), seminomas/disgerminomas (9%), y leucemia
mieloblástica aguda (1%) (36, 37).
JAK2
STAT
PI3K
AKT
Ras
Raf
MEK
ERK
Grb
SHP2
Shc sos
SCF
c-kit
Proliferación Inhibición de apoptosis
Figura 4. Vías de señalización intracelular activadas por c-kit
20

Introducción
1.2.3.3. PIK3CA
El gen PIK3CA se localiza en el locus 3q26.3, incluido en
una región cromosómica frecuentemente afectada por una
ganancia de material genético en varios tipos de tumores
humanos, como pulmón, cervix, y cabeza y cuello (38-40).
Varios estudios de hibridación genomica comparativa (CGH)
han sugerido el valor de dicha alteración como marcador de
t
escamosas. La detección de la amplificación de 3q en el 3%
de la mucosa normal, 25% de tejido premaligno, y 56% de
HNSCC invasivo sugiere que podría ser un marcador de
transición al cáncer (41). Se ha propuesto al oncogén PIK3CA
como posible diana de tales ganancias, encontrándose
am
ransición en la progresión al carcinoma invasivo de células
plificado en el 37% de los HNSCC, en el 80% de los
cánceres de ovario (42, 43), y cérvix (44). También se han
descrito mutaciones puntuales en PIK3CA en el cáncer
humano, tales como tumores colorectales, cáncer de mama,
cánceres gástricos, de pulmón, de ovario y en tumores
cerebrales malignos como oligodendrogliomas anaplásicos,
astrocitomas anaplásicos, glioblastoma multiforme, y
meduloblastomas (45-47).
21

Introducción
En el CAQ de glándulas salivales también se ha descrito
polisomía/ganancia del número de copias del cromosoma 3
mediante análisis de hibridación in situ fluorescentes (FISH),
sugiriendo que tal alteración ocurre durante la tumorigénesis
(48). Sin embargo, en este tipo de tumor, no se conoce aún
cuál es el gen implicado.
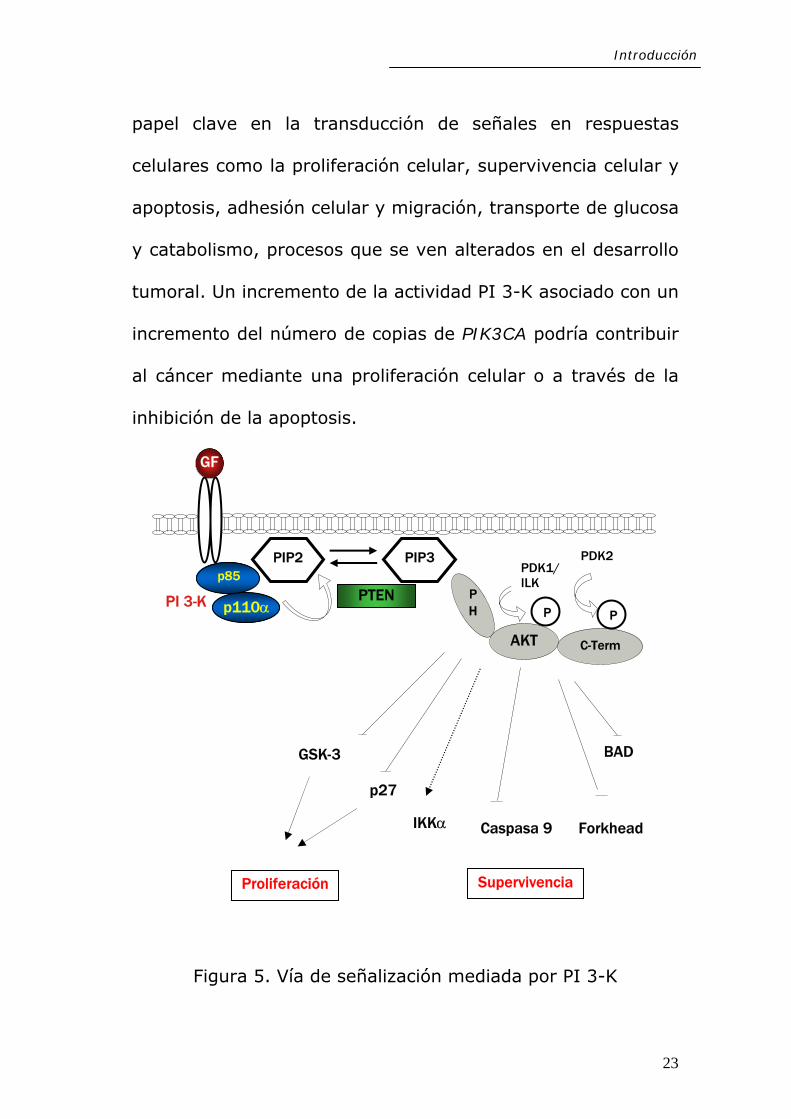
PIK3CA codifica la subunidad catalítica p110α que, junto
con la subunidad reguladora, p85, constituyen PI 3-K
(fosfatidil inositol 3 quinasa Ia). Tras la unión de p85 al
receptor fosforilado, p110α cataliza la fosforilación de
fosfatidilinositol 3,4-bisfosfato (PIP2) en fosfatidilinositol
3,4
su activación (PDK1/ILK y PDK2)
(49-51). Una vez fosforilada, Akt activa una variedad de
,5-trisfosfato (PIP3). Estos mensajeros lipídicos unen
proteínas (como la serín-treonín quinasa Akt/PKB) que poseen
un dominio homólogo de pleckstrina (PH), permitiendo la
modulación de su actividad a través de su localización en la
cara interna de la membrana citoplasmática junto con las
quinasas responsables de
sustratos, de los que se han reconocido al menos trece, los
cuales se reúnen en dos grupos principales: (a) reguladores
de la apoptosis y (b) reguladores de la proliferación celular
(Figura 5). Así, la vía de señalización PI 3-K/Akt juega un
22
Introducción
papel clave en la transducción de señales en respuestas
celulares como la proliferación celular, supervivencia celular y
apoptosis, adhesión celular y migración, transporte de glucosa
y catabolismo, procesos que se ven alterados en el desarrollo
tumoral. Un incremento de la actividad PI 3-K asociado con un
incremento del número de copias de PIK3CA podría contribuir
al cáncer mediante una proliferación celular o a través de la
inhibición de la apoptosis.
Figura 5. Vía de señalización mediada por PI 3-K
GF
PTEN PH
C-Term AKT
P
GSK-3
p27
Proliferación
IKKα Forkhead
Supervivencia
Caspasa 9
BAD
PI 3-K
PIP2
p110α
PIP3PDK1/ILK
PDK2
P
p85
23

Introducción
1.2.3.4. CCND1/PRAD1
El ciclo celular es una secuencia de procesos altamente
ordenados que dan lugar a la división celular. Cada una de
sus etapas (G0, G1, S, G2 y M) está cuidadosamente
controlada mediante la activación o inactivación de las
quinasas dependientes de ciclinas (CDKs), mediante la
formación de complejos heterodiméricos con las ciclinas,
proteínas reguladoras. Su expresión/degradación en cada
punto de control, junto con la interacción de los inhibidores de
quinasas dependientes de ciclina (CKIs: familias INK4,
Cip/Kip) permite a la célula entrar en te fase del
ciclo de forma controlada.
La transición en ) es
un punto crítico en el control de
superar este punto, la
irreversiblemente a dividirse. Durante la fase G1, la ciclina D1
Esta ruta se encuentra alterada en alguno de sus
componentes en más del 80% de las neoplasias humanas, ya
la siguien
tre las fases G1-S (punto de restricción
l ciclo celular, ya que, tras
célula se compromete
activa CDK4 y 6, resultando en la fosforilación de la proteína
del retinoblastoma (Rb), liberándose los factores de
transcripción E2Fs unidos, para iniciar la transcripción de
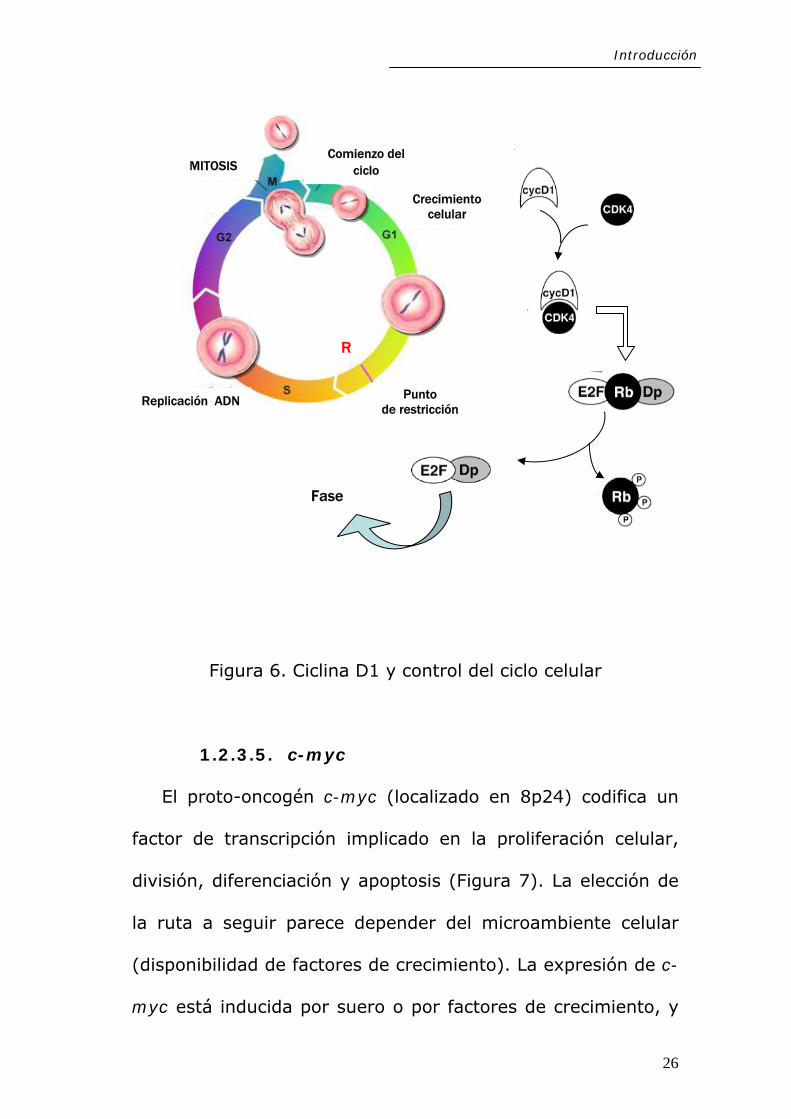
genes requeridos para la progresión hacia la fase S (Figura 6).
24

Introducción
sea por mutaciones en los genes que codifican estas proteínas
o en sus reguladores. La ciclina D1 está codificada por el
proto-oncogén CCND1/PRAD1/BCL1 (localizado en 11q13). Su
sobreexpresión es un suceso común en muchos tipos
diferentes de neoplasias tales como tumores de mama,
esófago, carcinomas colorectales, pulmón y cabeza y cuello,
para el que se ha demostrado su valor pronóstico (52-56).
Se ha propuesto que la amplificación oncogénica podría
justificar los elevados niveles de proteína observados en
algunos tipos de tumores humanos, como es el caso de
HNSCC. Sin embargo, en otros tipos de tumores la
sobreexpresión se justifica mediante translocaciones
cromosómicas, como las observadas en los linfomas de
células del manto (54) y en adenomas de paratiroides (57).
25
Introducción
Replicación ADN
MITOSIS
Crecimiento celular
Comienzo del ciclo
Figura 6. Ciclina D1 y control del ciclo celular
1.2.3.5. c-myc
El proto-oncogén c-myc
Punto de restricción
R
Fase
(localizado en 8p24) codifica un
factor de transcripción implicado en la proliferación celular,
división, diferenciación y apoptosis (Figura 7). La elección de
la ruta a seguir parece depender del microambiente celular
(disponibilidad de factores de crecimiento). La expresión de c-
myc está inducida por suero o por factores de crecimiento, y
26

Introducción
es mín ciadas
terminalmente. En células en proliferación, acelera el ciclo
celular acortando la fase G1, y reduce o elimina el
requerimiento de factores de crecimiento. Además, causa
(65).
ima en células en reposo o diferen
inmortalización celular e inhibe la diferenciación de varios
tipos de células y, en ciertas condiciones restringidas, puede
inducir apoptosis.
c-myc se encuentra sobreexpresado en varios tipos de
tumores humanos, como consecuencia de su amplificación
génica: carcinomas de mama, estómago, pulmón y colon,
neuroblastomas y glioblastomas, y cervix (58-61). En el
carcinoma epidermoide de cabeza y cuello se han descrito
tasas muy variables de amplificación (5-25%), encontrándose
asociado con estadios tumorales avanzados (62-64). Su
activación por translocación, se presenta en el 100% de los
linfomas de Burkitt
27

Introducción
P
P P
P P P
Ras
Raf
MEK
MAPK
GrbSHP
Shc
sos
PI3K
Figura 7. Rutas de señalización intracelular implicadas en la activaci
AKT
TCF4 c-jun
c-myc
c-fos
Síntesis ADN
PDGFR
ón de c-myc
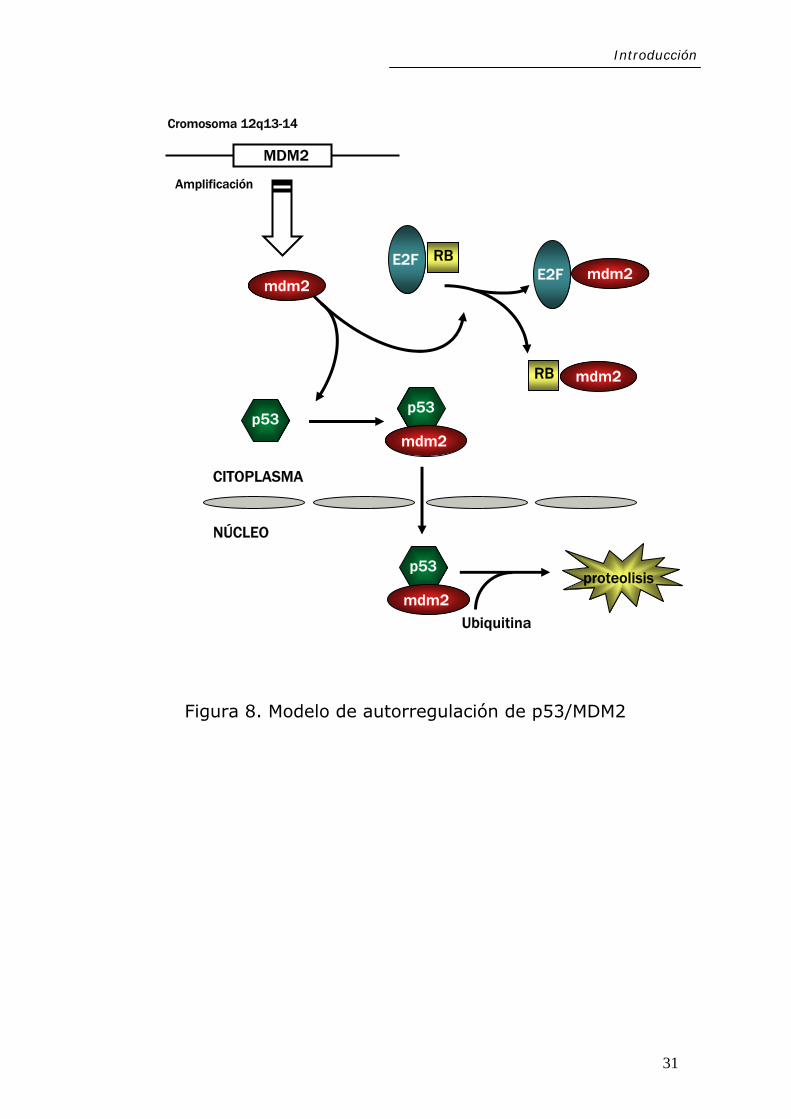
1.2.3.6. MDM2
MDM2 (Murine Double Minute-2, también llamado HDM2
en humanos) (localizado en 12q ca una proteína
c
13-14) codifi
uya función principal es la unión e inactivación de dos
28

Introducción
importantes proteínas codificadas por genes supresores de
tumores, como son p53 y Rb.
celular motivado, entre otras
cau
En condiciones normales, MDM2 y p53, cuyas vidas medias
son cortas, establecen un ciclo de retroalimentación (Figura
8), que permite mantener bajos los niveles de ambas
proteínas en el núcleo, lo cual favorece la proliferación celular.
Frente a situaciones de stress
sas, por daño genético, p53 se sobreexpresa y transactiva
genes implicados en la parada del ciclo celular, lo cual
proporciona a la célula un tiempo para reparar el daño
genético antes de que el ciclo prosiga o bien para entrar en
apoptosis en caso de que el daño sea irreparable. A su vez,
estimula la expresión de MDM2, que se une a p53
inactivándola, bien mediante cambios estructurales, o bien
mediante la activación de la degradación de p53 por el
proteasoma vía ubiquitina (MDM2 funciona como una
ubiquitina E3 ligasa que promueve la unión de p53 a la
ubiquitina). Así, se consigue un fino control de los niveles de
p53 y MDM2 en las células. Si la función de MDM2 se
encontrase sobreactivada, este equilibrio se vería afectado,
resultando en la pérdida de función de p53 y en la privación
de uno de los mecanismos de control más eficaces.
29

Introducción
Por otro lado, MDM2 también inactiva a la proteína Rb, la
cual tiene un papel clave en la regulación de genes esenciales
para la proliferación celular a través de su interacción con los
factores de transcripción heterodiméricos E2F-DP. La
inactivación de Rb tiene como consecuencia la liberación del
fac
mores humanos es del 7%, con la mayor
frec
tor de transcripción E2F y la progresión descontrolada del
ciclo celular.
La proteína MDM2 se encuentra sobreexpresada en
tumores humanos y líneas tumorales como consecuencia de
amplificación génica o de niveles de transcripción
incrementados. La frecuencia global de la amplificación de
MDM2 en tu
uencia observada en los sarcomas de tejidos blandos (20-
30%), osteosarcomas (16%) y carcinomas esofágicos (13%)
(66). La mutación simultánea de p53 y la amplificación de
MDM2 no ocurre generalmente en el mismo tumor, sugiriendo
que la amplificación de MDM2 es un medio eficaz para la
inactivacion de la función de p53 (67).
30
Introducción
Cromosoma 12q13-14
MDM2
RB E2F
mdm2
Figura 8. Modelo de autorregulación de p53/M
E2F mdm2
p53
mdm2
mdm2
NÚCLEO
CITOPLASMA
Ubiquitina
Amplificación
RB
p53
p53
mdm2 proteolisis
DM2
31

2
Hipótesis de trabajo
y objetivos

Hipótesis de trabajo y objetivos
El espectacular avance en el conocimiento del cáncer de
las pasadas dos décadas ha proporcionado una visión de la
complejidad de esta enfermedad a nivel molecular. Los
cambios genéticos moleculares involucrados en la
tumorigénesis y la transformación maligna de los tumores
humanos han permitido disponer de nuevas dianas para el
diagnóstico del cáncer y para su tratamiento.
Dado el carácter incierto de la progresión clínica del CAQ,
basado en los parámetros considerados actualmente, se hace
necesaria la búsqueda de marcadores moleculares con valor
pronóstico, que ayuden en la toma de decisiones en el manejo
de los CAQs.
Por todo lo anteriormente expuesto y con el fin de
contribuir al conocimiento de la biología molecular del
carcinoma adenoide quístico, nos propusimos los siguientes
objetivos:
1. Determinar la frecuencia de amplificación de
los oncogenes ERBB1, c-kit, PIK3CA, MDM2, CCND1
y c-myc en los CAQs.
2. Determinar el nivel de expresión de las
proteínas EGFR, c-kit y ciclina D1 en los CAQs.
33

Hipótesis de trabajo y objetivos
3. Determinar las correlaciones entre las
alteraciones moleculares encontradas y los
parámetros clínico-patológicos.
4. Determinar la influencia de las alteraciones
moleculares encontradas en el pronóstico de la
enfermedad.
34

3
Material y métodos

Material y métodos
3.1. CARACTERÍSTICAS CLINICO-PATOLÓGICAS
DE LOS PACIENTES Y SUS TUMORES
Se llevó a cabo un estudio retrospectivo de 26 pacientes
intervenidos de carcinoma adenoide quístico (CAQ) de
glándulas salivales mayores y menores entre los años 1984 y
2004 por los Servicios de Otorrinolaringología y Cirugía
Maxilofacial del Hospital Central de Asturias (HUCA). De ellos,
se recogieron muestras de 24 tumores primarios y 5 recidivas
– para tres de los cuales se disponía del tumor primario-
conservadas en parafina en el archivo del Servicio de
Anatomía Patológica del Hospital Central de Asturias. Todos
los tumores fueron revisados histológicamente por dos
observadores, confirmando el diagnóstico. Para algunos de los
pacientes fue posible obtener, asimismo, muestra de tejido
normal parafinado, que tomamos como control. Los datos de
las variables clínico-patológicas de interés se obtuvieron de
las historias clínicas y se recogen en la Tabla 1. Los tumores
se estadificaron siguiendo la clasificación TNM (Tumor, Node,
Metastasis) de la UICC (Unión Internacional contra el Cáncer),
6ª Edición (68).
Atendiendo al origen tumoral, el 70% de los casos eran
tumores de glándulas salivales menores (17 pacientes), y el
36

Material y métodos
30% restante corresponden a glándulas salivales mayores (7
pacientes). La localización tumoral primaria más frecuente fue
el seno maxilar (5 casos, 21%), seguido de la parótida (4
casos, 16%), el paladar y glándula submaxilar (3 casos cada
uno, 12%), el etmoides, las fosas nasales y glándula
sublingual con dos casos en cada localización (8%) y,
finalmente, un caso en la nasofaringe, uno en la fosa
pterigopalatina y uno en la infratemporal. En los tumores de
glándulas salivales menores lo más habitual era la afectación
de más de una estructura al diagnóstico, aunque
predominando la extensión en la estructura de origen.
Ninguno de los pacientes presentó metástasis a distancia
en el momento del diagnóstico. Sólo en uno de los casos
(tumor de origen en la glándula sublingual) se objetivaron
metástasis regionales. Todos los tumores fueron considerados
resecables, por lo que recibieron tratamiento quirúrgico,
realizándose en todos los casos resección macroscópicamente
completa del tumor. En cinco pacientes éste fue el único
tratamiento realizado (15%); en los 21 pacientes restantes se
complementó la cirugía con radioterapia (18 casos) y/o
quimioterapia (3 casos) coincidiendo con tumores localmente
avanzados o con invasión de los márgenes de resección. En
37

Material y métodos
dos casos, de tumores con progresión a base de cráneo, se
administró radiocirugía tras la cirugía para completar el
tratamiento. El seguimiento de los pacientes se llevó a cabo
durante un máximo de 23 años, siendo el seguimiento mínimo
de 3 años. Se recogió la información referente a la aparición
durante este tiempo de recidivas locales (6 casos, 3 de los
cuales fue posible estudiar ya que disponíamos de muestra) y
de metástasis a distancia (7 casos).
38

Material y métodos
Parámetro Nº casos (%)
Edad media (años) 54 (rango 14-79) Sexo
Mujer 16 (66,7) Hombre 8 (33,3)
Clasificación T 1 1 (4,2) 2 6 (25) 3 6 (25) 4 11 (45,8)
Invasión perineural no 7 (29,17) si 17 (70,83)
Subtipo histológico tubular 3 (12,5) cribiforme 18 (75) sólido 3 (12,5)
Márgenes de resección libres 7 (29,17) afectados 17 (70,84)
Estadio Tumoral I 1 (4,17) II 6 (25) III 5 (20,83) IV 12 (50)
Hábitos tóxicos Ninguno 15 (62,5) Tabaco 6 (25) Tabaco y alcohol 3 (12,5)
Tabla 1. Características clínico-
patológicas de los pacientes y sus tumores primarios
(N= 24).
39

Material y métodos
3.2. ESTUDIO DE AMPLIFICACIÓN GÉNICA
3.2.1. Procesamiento de muestras
3.2.1.1. Desparafinado
Para cada muestra, se procesaron 4 cortes de 4 µm de
grosor, recogidos en tubos de polipropileno estériles. Se
incubaron en 1 ml de xileno a temperatura ambiente durante
20 min. Tras la centrifugación (5 min a 13.000 rpm) se retiró
el sobrenadante y se repitió la operación, obteniéndose un
sedimento correspondiente al tejido desparafinado. Se
añadieron 500 µl de etanol absoluto, y tras centrifugación y
eliminación del sobrenadante, se dejó secar el tejido en
bloque térmico.
3.2.1.2. Lisis
Una vez bien seco, se lisó el tejido a 55ºC, durante una
noche, en cuatro volúmenes de tampón de digestión (50 mM
Tris pH 8.5, 10 mM EDTA, 0.5% Tween 20, 1 mg/ml
proteinasa K).
3.2.1.3. Extracción del ADN
El tejido digerido se sometió a dos rondas sucesivas de
extracción con fenol-cloroformo-alcohol isoamílico y el ADN se
40

Material y métodos
precipitó con acetato sódico 3 M (1/10 volumen) y etanol
absoluto frío (2 volúmenes). Tras un lavado con etanol al 70%
se secó el ADN al aire y se resuspendió en agua bidestilada
estéril. Las muestras se conservaron a –20ºC hasta su uso. La
concentración del ADN se determinó midiendo la absorbancia
a 260 nm. Se trabajó con concentraciones de ADN de 0,2
µg/µl.
3.2.2. Análisis de amplificación génica: PCR
múltiplex
La reacción en cadena de la polimerasa (PCR), permite la
amplificación in vitro de un segmento específico de ADN de
secuencia conocida. Dos oligonucleótidos de secuencia
complementaria a los extremos del fragmento actúan como
cebadores para iniciar la reacción de síntesis de ADN por la
ADN polimerasa, en cuya repetición cíclica se basa la técnica
de la PCR. Cuando el objetivo es amplificar simultáneamente
más de una diana en una sola reacción (en el mismo tubo),
nos encontramos ante una PCR múltiplex. Esta técnica fue
utilizada en nuestro estudio como herramienta
semicuantitativa, ya que permite de forma rápida y sencilla
detectar la presencia de más de una copia de un gen, siempre
41

Material y métodos
que tomemos como referencia la señal generada a partir de
un gen control de copia única (no descrito como diana
alterada en cáncer).
Para la puesta a punto del método, se establecen las
condiciones experimentales que permitan obtener, en una
muestra de ADN de un tejido sano (en nuestro estudio se
utilizó el tejido sano disponible de los pacientes), la misma
intensidad de banda para el gen diana (ej. CCND1) y el gen
control (ej. TH, Tirosin-hidroxilasa) (lo cual correspondería a
una sola copia). Así, al mantener estas condiciones para el
estudio del ADN tumoral podremos detectar la presencia de
más de una copia del gen diana por el aumento en la
intensidad de la banda correspondiente. El gen control elegido
se sitúa en el mismo cromosoma que el diana, lo cual permite
excluir falsos positivos procedentes de una posible polisomía
del cromosoma en cuestión.
Los oncogenes estudiados fueron: ERBB1, c-kit, PIK3CA,
MDM2, CCND1, c-myc.
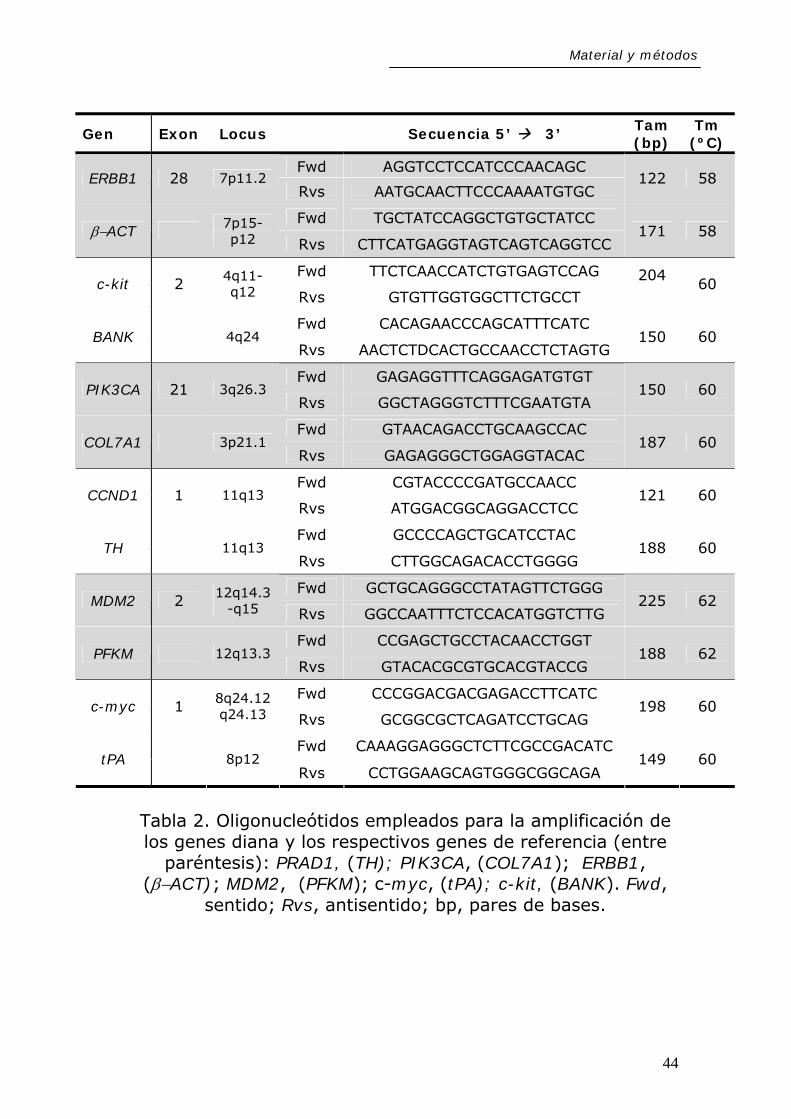
La Tabla 2 recoge las secuencias de cada uno de los
oligonucleótidos cebadores utilizados para la amplificación de
fragmentos de los citados genes (exones indicados) y sus
correspondientes controles, el tamaño del fragmento
42

Material y métodos
amplificado, y las temperaturas de cebamiento específicas
para cada gen.
Las reacciones se realizaron en tubos de propileno de
pared fina de 0,2 ml en un volumen final de 20 µl,
conteniendo 200 µM de deoxinucleótidos trifosfato (dNTPs), 2
mM de MgCl2, 200 ng de ADN, 0,5 µM de cada
oligonucleótido, 1X tampón de AmpliTaq GOLD y 0,5 U
AmpliTaq GOLD (Applied Biosystems). Tras un paso previo de
10 min a 94ºC, requerido para la activación del enzima
empleado, las muestras se sometieron a 32 ciclos de
amplificación con una temperatura de anillamiento de 60ºC
para los genes CCND1, c-kit, c-myc y PIK3CA, 58ºC para
ERBB1, y 62ºC para el gen MDM2. Los productos de las
reacciones (9 µl) se sometieron a electroforesis en gel de
agarosa al 2% (conteniendo 0,5 mg/ml de bromuro de etidio)
en tampón TAE. Las bandas correspondientes se visualizaron
en un transiluminador de luz ultravioleta, y se fotografiaron
con una cámara digital (Canon digital Ixus 400). La
amplificación se estimó contrastando siempre las opiniones de
dos observadores. Todos los análisis se llevaron a cabo al
menos dos veces en experimentos independientes, para
confirmar los resultados.
43
Material y métodos
Gen Exon Locus Secuencia 5’ 3’ Tam(bp)
Tm (ºC)
Fwd AGGTCCTCCATCCCAACAGC ERBB1 28 7p11.2
Rvs AATGCAACTTCCCAAAATGTGC 122 58
Fwd TGCTATCCAGGCTGTGCTATCC β−ACT 7p15-
p12 Rvs CTTCATGAGGTAGTCAGTCAGGTCC 171 58
Fwd TTCTCAACCATCTGTGAGTCCAG c-kit 2 4q11-
q12 Rvs GTGTTGGTGGCTTCTGCCT
204
60
Fwd CACAGAACCCAGCATTTCATC BANK 4q24
Rvs AACTCTDCACTGCCAACCTCTAGTG 150 60
Fwd GAGAGGTTTCAGGAGATGTGT PIK3CA 21 3q26.3
Rvs GGCTAGGGTCTTTCGAATGTA 150 60
Fwd GTAACAGACCTGCAAGCCAC COL7A1 3p21.1
Rvs GAGAGGGCTGGAGGTACAC 187 60
Fwd CGTACCCCGATGCCAACC CCND1 1 11q13
Rvs ATGGACGGCAGGACCTCC 121 60
Fwd GCCCCAGCTGCATCCTAC TH 11q13
Rvs CTTGGCAGACACCTGGGG 188 60
Fwd GCTGCAGGGCCTATAGTTCTGGG MDM2 2 12q14.3
-q15 Rvs GGCCAATTTCTCCACATGGTCTTG 225 62
Fwd CCGAGCTGCCTACAACCTGGT PFKM 12q13.3
Rvs GTACACGCGTGCACGTACCG 188 62
Fwd CCCGGACGACGAGACCTTCATC c-myc 1 8q24.12
q24.13 Rvs GCGGCGCTCAGATCCTGCAG 198 60
Fwd CAAAGGAGGGCTCTTCGCCGACATC tPA 8p12
Rvs CCTGGAAGCAGTGGGCGGCAGA 149 60
Tabla 2. Oligonucleótidos empleados para la amplificación de los genes diana y los respectivos genes de referencia (entre
paréntesis): PRAD1, (TH); PIK3CA, (COL7A1); ERBB1, (β−ACT); MDM2, (PFKM); c-myc, (tPA); c-kit, (BANK). Fwd,
sentido; Rvs, antisentido; bp, pares de bases.
44

Material y métodos
3.2.2.1. Validación del método
Para comprobar la sensibilidad del método empleado en la
detección de más de una copia del gen diana, presente en la
mezcla de reacción, se llevó a cabo la amplificación de un
fragmento de 121 bp del gen CCND1. El producto de la
reacción se purificó a partir de un gel de agarosa, utilizando
un kit comercial (QUIAGEN). Volúmenes iguales de diluciones
sucesivas de este molde, se añadieron a varios tubos de
reacción en los que estaba también presente el ADN genómico
de una muestra de mucosa sana. Como cabía esperar, la
imagen obtenida (Figura 9) muestra la capacidad de la PCR
múltiple (amplificación del gen diana, CCND1 y un gen
control, TH) utilizada para detectar la presencia de cantidades
crecientes de CCND1 en el tubo de reacción, estableciéndose
una competición de los reactivos por ambas dianas (CCND1 y
TH) hasta el punto de ser CCND1 la única amplificada.
Diluciones CCND1 -3- -6 -5 -471 1 1 1 1 1
Figura 9. PCR muestra de la validación del método
CCND1TH
45

Material y métodos
3.3. ESTUDIO DE EXPRESIÓN PROTEICA
3.3.1. Técnica inmunohistoquímica empleada
La inmunohistoquímica permite determinar la expresión de
proteínas en un tejido mediante la utilización de anticuerpos
específicos. La reacción específica entre anticuerpo y antígeno
se puede detectar de maneras muy diversas. En nuestro
estudio, se ha utilizado el sistema de polímeros de dextrano
EnVisionTM plus (PDE) Dako, desarrollada recientemente, que
presenta alta sensibilidad y una menor señal de fondo. Se
trata de una técnica de tinción en dos pasos en la que, tras el
anticuerpo primario, se utiliza dextrano, polímero de alto peso
molecular, conjugado covalentemente con un gran número de
moléculas de enzima (por ejemplo peroxidasa de rábano,
hasta 100 moléculas por cadena) y de anticuerpo secundario
(hasta 20 moléculas por cadena). Con ello se aumenta mucho
la sensibilidad, lo que permite incrementar las diluciones de la
mayoría de los anticuerpos primarios. Los resultados son
altamente reproducibles y comparables.
46

Material y métodos
3.3.2. Análisis de expresión de EGFR, c-kit y
ciclina D1
Se llevó a cabo el estudio de expresión proteica por
inmunohistoquímica (IHQ) de EGFR, c-kit y ciclina D1, para
los que disponíamos de anticuerpos primarios que
proporcionaban una tinción específica esperada. Previamente
se realizó una tinción con hematoxilina de secciones de 4 µm
de tejido tumoral de cada caso incluido en la serie, con el fin
de verificar la presencia en la muestra de CAQ representativo
y de comprobar una calidad de fijación adecuada para un
análisis IHQ. Las piezas tumorales fijadas en formol e
incluidas en parafina fueron cortadas previo control
microscópico, asegurando la presencia de tejido tumoral, en
secciones de 4 µm sobre portas tratados (Dako/ChemMate,
Santa Barbara, CA, USA). Las secciones se incubaron durante
una noche en estufa a 54 - 56ºC, y posteriormente fueron
desparafinadas con xilol y rehidratadas utilizando soluciones
de etanol en concentración de etanol decreciente. La
recuperación antigénica se realizó calentando las secciones en
tampón citrato a pH 6.4 durante 4 minutos en una olla a
presión para analizar la expresión de c-kit y ciclina D1; para
analizar EGFR, tras los lavados con el tampón suministrado,
47

Material y métodos
se llevó a cabo la recuperación antigénica, con solución de
proteinasa K durante 5 minutos (kit EGFR PharmDx™ K 1494).
Las tinciones se realizaron a temperatura ambiente de forma
automática de una sola vez para cada marcador en una
estación de trabajo automatizada (TechMate 500,
DakoCytomation). Las muestras se mantuvieron durante tres
periodos de 2,5 minutos en un medio bloqueante de la
actividad peroxidasa endógena consistente en peroxido de
hidrógeno al 3%, y posteriormente se incubaron con el
anticuerpo primario correspondiente: anti-EGFR 2-18C9
(monoclonal, pharmDx-Ab - Kit EGFR PharmDx™ K 1494,
DakoCytomation, Carpinteria, CA), anti-c-kit (CD117) A4502
(dilución 1:200, policlonal, DAKO, Carpintería, CA), o anti-
ciclina D1 DSC-6 (dilución 1:20, monoclonal, Novocastra
Laboratorios Co., Ltd., Newcastle-upon-Tyne, UK). La
incubación tuvo una duración de 30 minutos en el caso de los
anticuerpos anti-EGFR y anti-c-kit, y de 24 horas en el caso
del anti-ciclina D1. La inmunodetección se realizó con el
sistema EnVision Plus (Dako, Carpinteria, CA, USA) de ratón
(EGFR y ciclina D1) o de conejo (c-kit), añadiendo la solución
PDE (polímeros de dextrano Envision) que se incuba a
temperatura ambiente durante media hora. Como cromógeno
48

Material y métodos
se empleó la diaminobenzidina (DAB) en un substrato
tamponado (Dako, Carpinteria, CA, USA). Por último, se
contrastó la preparación mediante tinción con hematoxilina
durante un minuto. Las muestras fueron deshidratadas en
diferentes grados de alcohol según el método convencional y
montadas empleando un medio estándar.
Como controles positivos se utilizaron muestras tumorales
obtenidas de cirugía oncológica de carcinoma de colon en el
caso de EGFR y de melanoma en el caso de c-kit; como
control positivo para la ciclina D1 se empleó tejido amigdalar
normal. Como control negativo, se llevó a cabo, en cada caso,
todo el proceso en paralelo, sobre un porta que fue incubado
con suero en sustitución del anticuerpo primario.
3.4. ANÁLISIS ESTADÍSTICO
El análisis estadístico se realizó empleando el método de
χ2 con la corrección de Yates en caso necesario, con la ayuda
del programa informático SPSS 14.0. Se calcularon las curvas
de supervivencia mediante el método de Kaplan-Meier,
considerándose censuradas las muertes por motivos
diferentes al CAQ. Las diferencias entre las curvas de
supervivencia se compararon mediante el método log-rank.
49

Material y métodos
Los valores de p < 0,05 fueron considerados estadísticamente
significativos.
50

4
Resultados

Resultados
4.1 AMPLIFICACIÓN ONCOGÉNICA EN LOS
TUMORES PRIMARIOS
La Tabla 3 recoge los resultados de amplificaciones
oncogénicas estudiados para cada muestra. El oncogén cuya
amplificación se encontró con mayor frecuencia fue ERBB1
(66,67%), seguido de CCND1 (45,8%), PIK3CA (37,9%), c-
myc (8,7%), c-kit (4,8%) y MDM2, para el que ningún caso
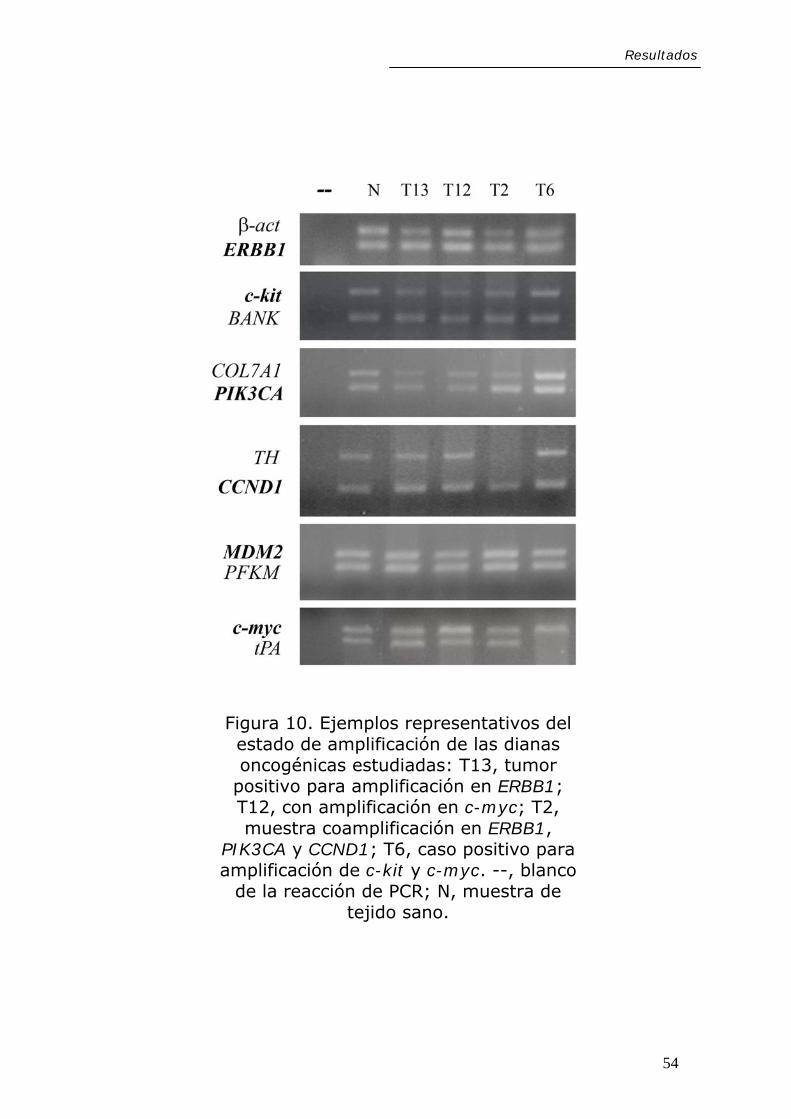
resultó positivo. La Figura 10 muestra ejemplos
representativos de los resultados obtenidos en las reacciones
de PCR para cada una de las dianas estudiadas.
52

Resultados
Caso Nº ERBB1 c-kit PIK3CA CCND1 MDM2 c-myc T2 + -- + + -- -- T3 + SR + -- -- -- T4 -- -- -- -- -- -- T6 -- + -- -- -- + T7 + -- + + -- -- T8 + -- -- -- -- -- T9 + -- + + -- --
T11 + -- -- + -- -- T12 -- -- -- -- -- + T13 + -- -- -- -- -- T14 -- -- -- -- -- -- T15 + -- -- -- -- -- T19 + -- -- -- -- -- T21 + -- + + -- -- T22 + -- + + -- -- T23 -- -- -- + -- -- T24 + -- + + -- -- T25 + -- + + -- -- T26 + SR -- + -- -- T27 -- -- -- -- -- -- T28 -- SR -- -- SR SR T29 + -- -- + -- -- T30 + -- -- -- -- -- T31 -- -- + -- -- --
TOTAL (%)
16/24 (66,67)
1/21 (4,8)
9/24 (37,9)
11/24 (45,8)
0/23 (0)
2/23 (8,7)
Tabla 3. Resultados de amplificación génica de los
tumores primarios; SR, sin resultado
53
Resultados
Figura 10. Ejemplos representativos del estado de amplificación de las dianas oncogénicas estudiadas: T13, tumor
positivo para amplificación en ERBB1; T12, con amplificación en c-myc; T2, muestra coamplificación en ERBB1,
PIK3CA y CCND1; T6, caso positivo para amplificación de c-kit y c-myc. --, blanco
de la reacción de PCR; N, muestra de tejido sano.
54

Resultados
La Tabla 4 recoge el número de casos según el diferente
número de genes amplificados, así como la frecuencia de los
patrones predominantes.
Número de genes
amplificados
Número de casos Patrón más frecuente
0 4 -- 1 8 ERBB1 - 5/8 (62,5%) 2 5 ERBB1 y CCND1 - 3/5 (60%)
3 7 ERBB1, CCND1 y PIK3CA - 7/7 (100%)
Tabla 4. Distribución de los casos según el número de dianas amplificadas, especificando el patrón encontrado con mayor
frecuencia para cada caso.
4.2. AMPLIFICACIÓN ONCOGÉNICA EN LAS
RECIDIVAS TUMORALES
ERBB1 se encontró amplificado en 3 de las 5 recidivas
estudiadas. En uno de los casos el tumor primario
correspondiente no presentaba tal alteración. Una de las
recidivas presentaba amplificación de CCND1, alteración que
estaba ya presente en el tumor primario. Una recidiva, para la
que no disponíamos del tumor primario correspondiente, fue
positiva para amplificación de c-myc.
Ninguna de las recidivas presentó amplificación de los
oncogenes c-kit, PIK3CA y MDM2.
55
Resultados
Los resultados hallados se recogen en la Tabla 5.
Caso nº ERBB1 c-kit PIK3CA CCND1 MDM2 c-myc R5 + - - - - - R10 - - - - - + T8 + - - - - - R8 + - - - - - T23 - - - + - - R23 + - - + - - T27 - - - - - - R27 - - - - - -
Tabla 5. Resultados de amplificación génica de las recidivas
(R) y sus tumores primarios (T) correspondientes, en caso de estar disponibles.
4.3. NIVELES DE EXPRESIÓN PROTEICA
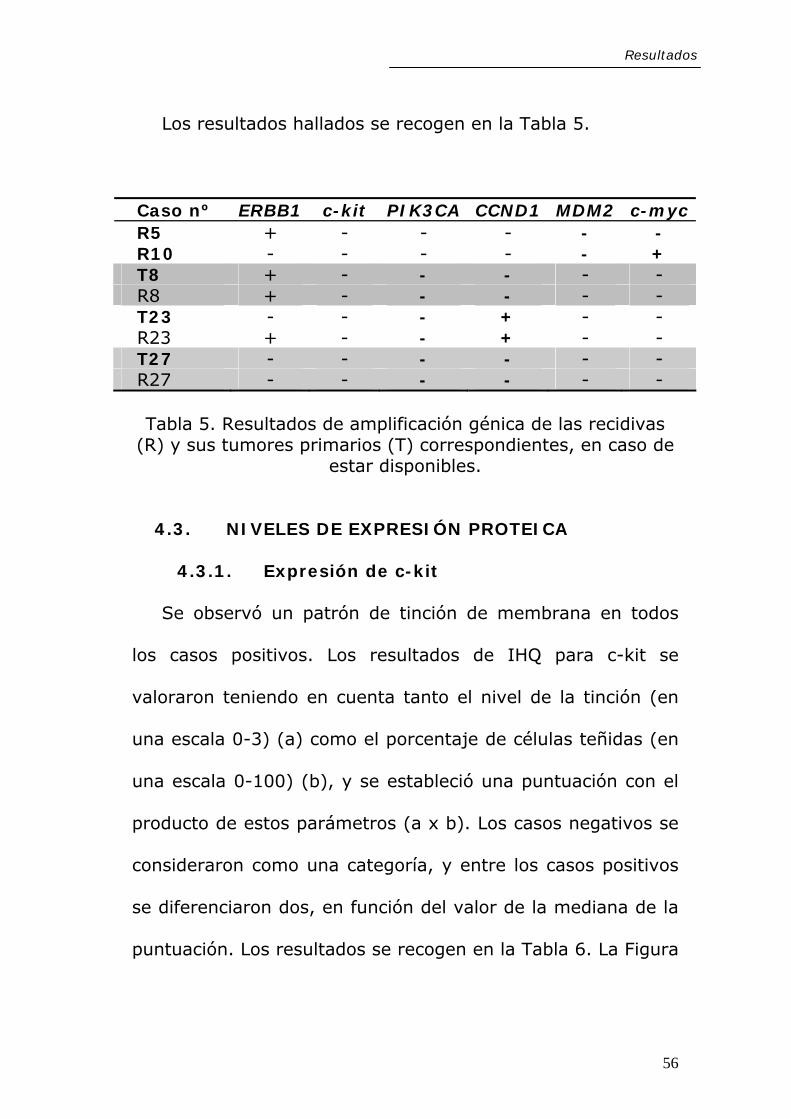
4.3.1. Expresión de c-kit
Se observó un patrón de tinción de membrana en todos
los casos positivos. Los resultados de IHQ para c-kit se
valoraron teniendo en cuenta tanto el nivel de la tinción (en
una escala 0-3) (a) como el porcentaje de células teñidas (en
una escala 0-100) (b), y se estableció una puntuación con el
producto de estos parámetros (a x b). Los casos negativos se
consideraron como una categoría, y entre los casos positivos
se diferenciaron dos, en función del valor de la mediana de la
puntuación. Los resultados se recogen en la Tabla 6. La Figura
56

Resultados
11 muestra imágenes representativas de los distintos niveles
de tinción observados.
IHQ c-kit Nº casos % Negativo Moderado Intenso
9 7 5
42,86 33,33 23,81
TOTAL 21 100
Tabla 6. Distribución de los casos según el nivel de
expresión de c-kit
57
Resultados
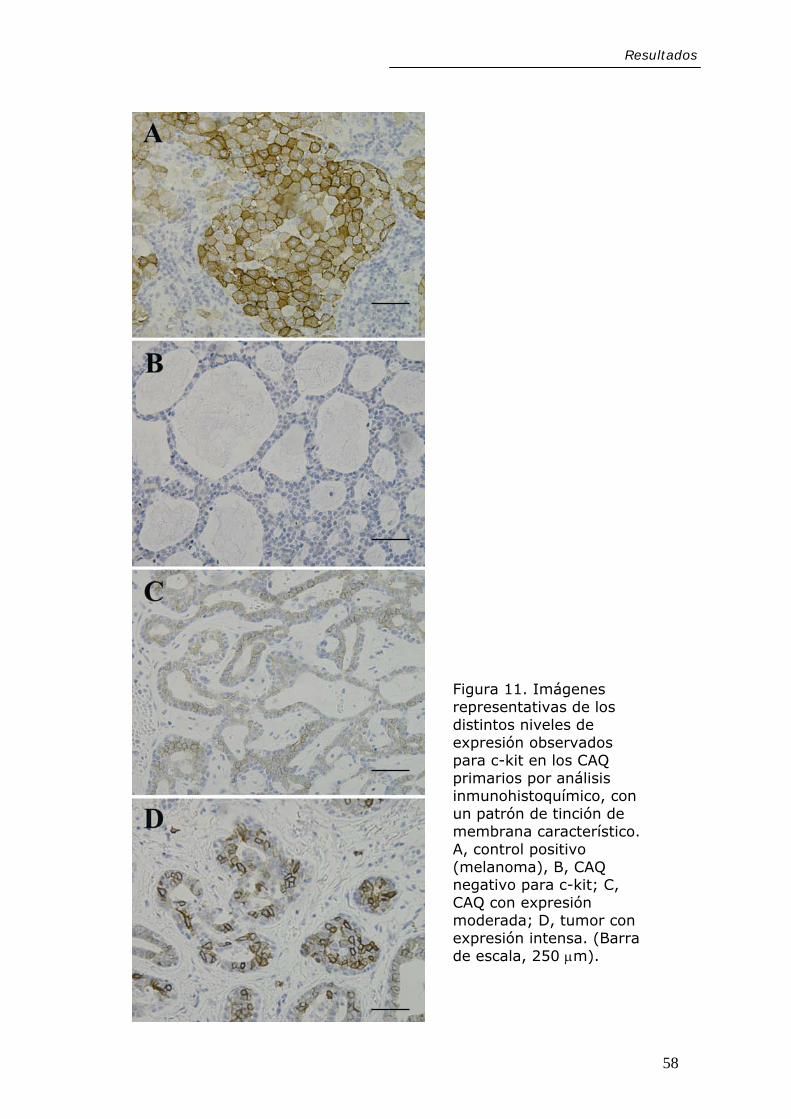
Figura 11. Imágenes representativas de los distintos niveles de expresión observados para c-kit en los CAQ primarios por análisis inmunohistoquímico, con un patrón de tinción de membrana característico.A, control positivo (melanoma), B, CAQ negativo para c-kit; C, CAQ con expresión moderada; D, tumor con expresión intensa. (Barra de escala, 250 µm).
58

Resultados
4.3.2. Expresión de EGFR
La expresión de EGFR fue negativa en todos los casos
analizados (Figura 12).
Figura 12. Imagen representativa de los resultados obtenidos mediante IHQ para la detección de EGFR. En
ningún caso se detectó señal. El control positivo (muestra de carcinoma de colon) presenta el patrón de membrana esperado con una intensidad elevada (Barra
de escala, 250 µm).
4.3.3. Expresión de ciclina D1
La tinción IHQ de ciclina D1 presentaba un patrón nuclear.
Únicamente resultaron positivas las células epiteliales. Estas
células forman ductos que contienen mucopolisacáridos, a
diferencia de las células mioepiteliales que las rodean
(negativas para ciclina D1), que secretan componentes de la
membrana basal. Dado que el 100% de las células epiteliales
59

Resultados
mostraban tinción de ciclina D1, se estableció la puntuación
para este marcador atendiendo únicamente al nivel de
expresión nuclear, en las siguientes categorías: bajo,
moderado e intenso (Figura 13). Los resultados se recogen en
la Tabla 7.
Por otro lado, se recogió la información referente al
porcentaje de células epiteliales observadas en cada caso.
Tras una división de la población en función del valor de la
mediana, se clasificaron los tumores en dos grupos:
proporción reducida (11/22 casos) vs proporción elevada de
componente epitelial (11/22 casos).
IHQ ciclina D1 Nº casos % Bajo
Moderado Intenso
4 7 11
18,18 31,82
50 TOTAL 22 100
Tabla 7. Distribución de los casos
según el nivel de expresión de ciclina D1
60
Resultados
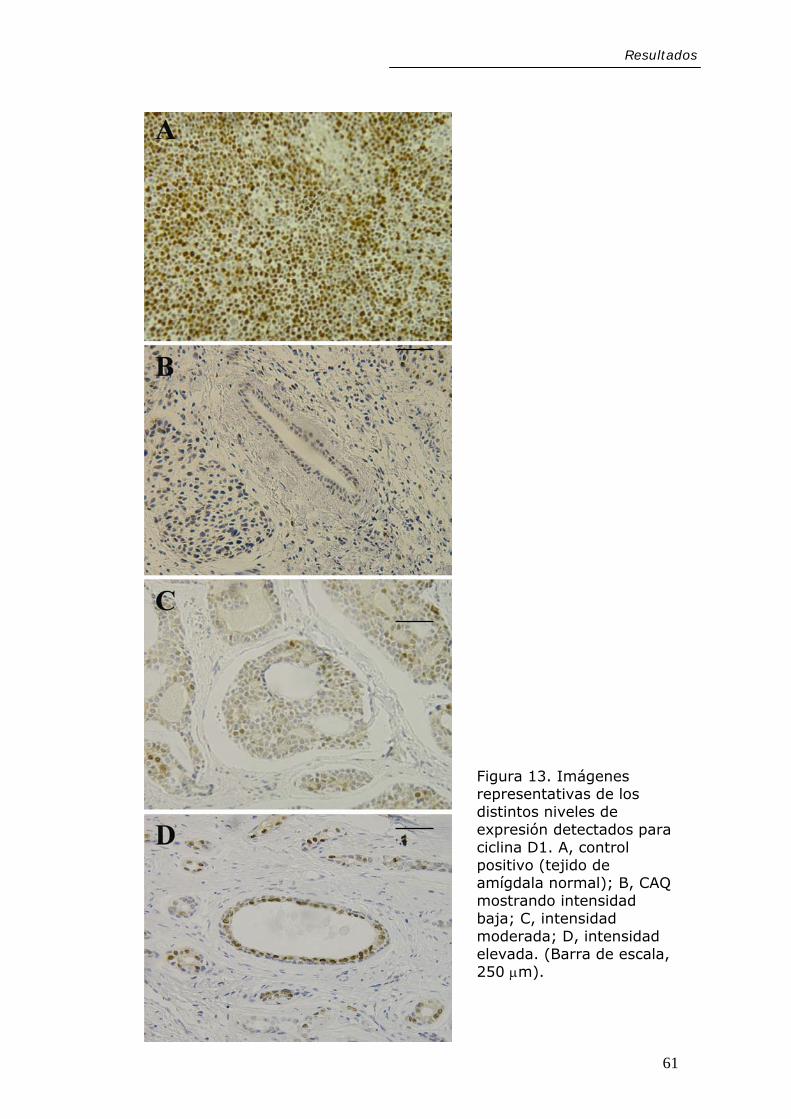
Figura 13. Imágenes representativas de los distintos niveles de expresión detectados para ciclina D1. A, control positivo (tejido de amígdala normal); B, CAQ mostrando intensidad baja; C, intensidad moderada; D, intensidad elevada. (Barra de escala, 250 µm).
61

Resultados
4.4. RELACIÓN ENTRE LA AMPLIFICACIÓN GÉNICA
DE DIVERSAS DIANAS
• Se encontró una correlación estadísticamente
significativa entre las amplificaciones génicas de PIK3CA
y CCND1 (χ2 = 5,919, p = 0,015) (Tabla 8).
CCND1 PIK3CA No amplificado Amplificado Total
No amplificado 11 4 15 Amplificado 2 7 9
Total 13 11 24
Tabla 8. Distribución de los CAQs según el estado de amplificación de PIK3CA y CCND1.
• De los 16 casos con amplificación de ERBB1, 8
mostraban esta alteración para PIK3CA (χ2 = 3,2, p =
0,074) (Tabla 9), y en 10/16 se hallaba su
coamplificación con CCND1 (χ2 = 5,371, p = 0,02)
(Tabla 10).
62

Resultados
PIK3CA ERBB1 No amplificado Amplificado Total
No amplificado 7 1 8 Amplificado 8 8 16
Total 15 9 24
Tabla 9. Distribución de los CAQs según el estado de amplificación de ERBB1 y PIK3CA.
CCND1 ERBB1 No amplificado Amplificado Total
No amplificado Amplificado
7 6
1 10
8 16
Total 13 11 24
Tabla 10. Distribución de los CAQs según el estado de amplificación de ERBB1 y CCND1.
• 7/24 (29,1%) casos contaban con la amplificación
de los genes ERBB1, CCND1 y PIK3CA, lo cual convierte
a este patrón de amplificación oncogénica en el más
frecuentemente observado en nuestra serie de CAQs.
63

Resultados
4.5. RELACIÓN ENTRE LA AMPLIFICACIÓN GÉNICA
Y LA EXPRESIÓN PROTEICA
4.5.1. ERBB1 – EGFR
La amplificación génica de ERBB1 no se refleja en un
aumento de la expresión protéica de EGFR, ya que ningún
caso presentaba expresión de EGFR.
4.5.2. c-kit - c-kit
El único caso positivo para amplificación génica mostraba
un nivel moderado de expresión de c-kit.
4.5.3. CCND1 – ciclina D1
La variable que recoge los resultados de expresión
proteica de ciclina D1 se recodificó, con fines estadísticos, en
dos categorías: bajo vs moderado-intenso. Se observó una
tendencia en la correlación de esta variable con el nivel de
amplificación de CCND1, aunque no se alcanzó significación
estadística (χ2 = 0,306, p = 0,08). La distribución de los casos
en función de estas variables se recoge en la Tabla 11. Como
se puede apreciar, la expresión de la ciclina D1 fue
moderada/intensa en todos los casos, excepto uno, con
64

Resultados
amplificación del gen. Sin embargo, 8 de los 11 casos sin
amplificación también mostraron expresión moderada/intensa,
lo que sugiere que otros mecanismos, además de la
amplificación, pueden ser responsables de la sobreexpresión
de la ciclina D1.
Ciclina D1 CCND1 Bajo Moderado/intenso Total
No amplificado 3 8 11 Amplificado 1 10 11
Total 4 18 22
Tabla 11. Distribución de los CAQs según el nivel de expresión de ciclina D1 y el estado de
amplificación génica para CCND1.
La Tabla 12 muestra el detalle de los resultados obtenidos
para los estudios de amplificación génica y la correspondiente
expresión proteica de los marcadores estudiados para cada
caso.
65

Resultados
Caso nº
ERBB1 EGFR c-kit C-KIT CCND1 Ciclina
D1 T2 Positivo Negativo Negativo Negativo Positivo Intenso T3 Positivo Negativo SR Negativo Negativo Bajo T4 Negativo Negativo Negativo Negativo Negativo Intenso T6 Negativo Negativo Positivo Moderado Negativo Intenso T7 Positivo Negativo Negativo Negativo Positivo Intenso T9 Positivo Negativo Negativo Negativo Positivo Moderado T11 Positivo Negativo Negativo Intenso Positivo Intenso T12 Negativo Negativo Negativo Moderado Negativo Moderado T13 Positivo Negativo Negativo Negativo Negativo Bajo T14 Negativo Negativo Negativo Moderado Negativo Moderado T19 Positivo Negativo Negativo Negativo Negativo Moderado T21 Positivo Negativo Negativo Negativo Positivo Moderado T22 Positivo Negativo Negativo Negativo Positivo Intenso T23 Negativo Negativo Negativo Intenso Positivo Moderado T24 Positivo Negativo Negativo Moderado Positivo Intenso T25 Positivo Negativo Negativo Moderado Positivo Bajo T26 Positivo Negativo SR Moderado Positivo Intenso T27 Negativo Negativo Negativo Moderado Negativo Intenso T28 Negativo Negativo SR Intenso Negativo Intenso T29 Positivo Negativo Negativo Intenso Positivo Intenso T30 Positivo Negativo Negativo SR Negativo Moderado T31 Negativo Negativo Negativo Intenso Negativo Bajo
Tabla 12. Resultados obtenidos del estudio de amplificación génica y su correspondiente proteína. SR, sin resultado. Los casos T8 y T15 no pudieron ser estudiados por IHQ por no
disponer de tejido parafinado.
66

Resultados
67
4.6. RELACIÓN ENTRE LAS ALTERACIONES
MOLECULARES Y LOS PARÁMETROS CLÍNICO-
PATOLÓGICOS
Una vez dicotomizada la variable “categoría T” en los
grupos 1-2 y 3-4, se observó una asociación entre los
tumores avanzados (T3-T4) y la presencia de amplificación en
CCND1 (χ2 = 3,962, p = 0,047).
Asimismo, se encontró una asociación entre la
amplificación de PIK3CA y el subtipo histológico, siendo más
frecuente en los subtipos tubular y sólido (100% y 66%,
respectivamente) (χ2 = 5,04, p = 0,019). Las Tablas 13 y 14
muestran la distribución de los casos en función de los
estudios llevados a cabo en los genes y proteínas,
respectivamente, y las variables clínico-patológicas
consideradas.
ERBB1 c-kit PIK3CA CCND1 c-mycPatrón más frecuente
Amp TOTAL Amp TOTAL Amp TOTAL Amp TOTAL Amp TOTAL SI TOTAL Menor 11 17 1 16 7 17 8 17 2 17 5 17 Mayor 5 7 0 5 2 7 3 7 0 6 2 7 TOTAL 16 24 1 21 9 24 11 24 2 23 7 24
Glándula
p 0,751 0,567 0,562 0,851 0,379 0,967 1-2 3 7 1 7 2 7 1 7 2 7 1 73-4 13 17 0 14 7 17 10 17 0 16 6 17
TOTAL 16 24 1 21 9 24 11 24 2 23 7 24Categoría
T p 0,112 0,147 0,562 0,047 0,025 0,303
NO 3 7 0 7 3 7 4 7 1 7 2 7 SI 13 17 1 14 6 17 7 17 1 16 5 17
TOTAL 16 24 1 21 9 24 11 24 2 23 7 24 Invasión
perineural p 0,112 0,467 0,727 0,475 0,529 0,9671
Tubular 2 3 0 2 3 3 2 3 0 2 2 3Cribiforme 12 18 1 16 4 18 8 18 2 18 4 18
Sólido 2 3 0 3 2 3 1 3 0 3 1 3TOTAL 16 24 1 21 9 24 11 24 2 23 7 24
Subtipo histológico
p 0,619 0,849 0,019 0,695 0,738 0,288
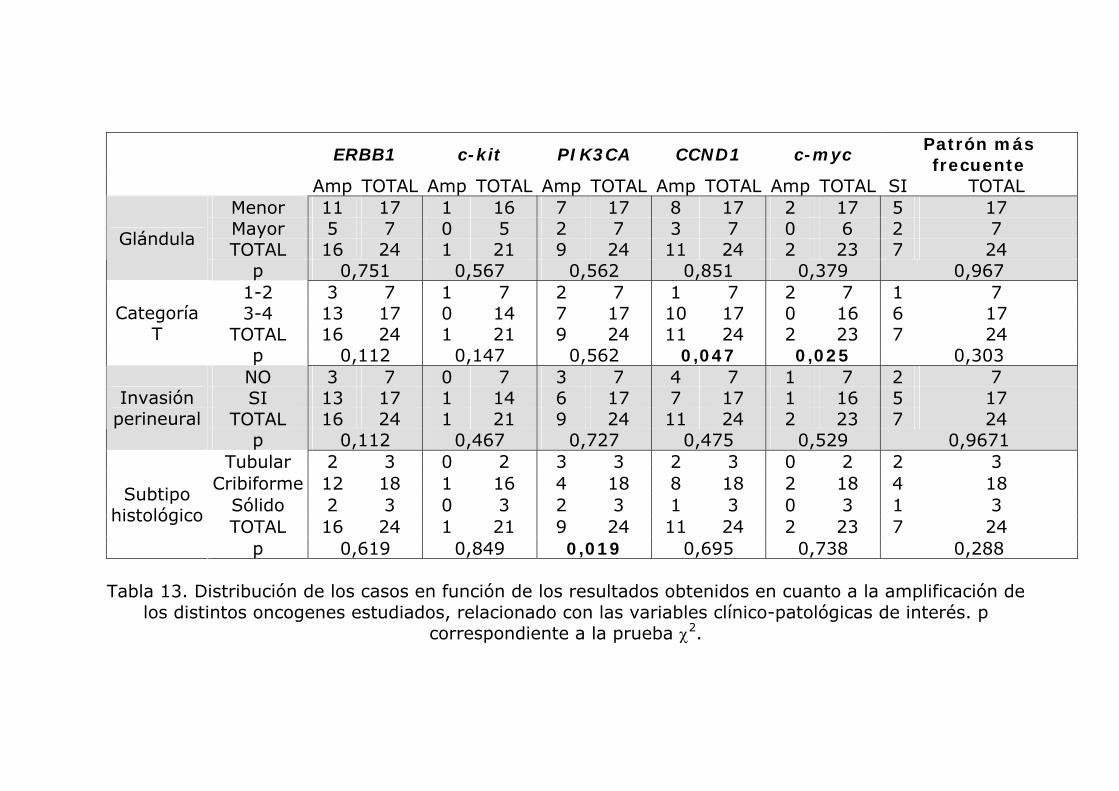
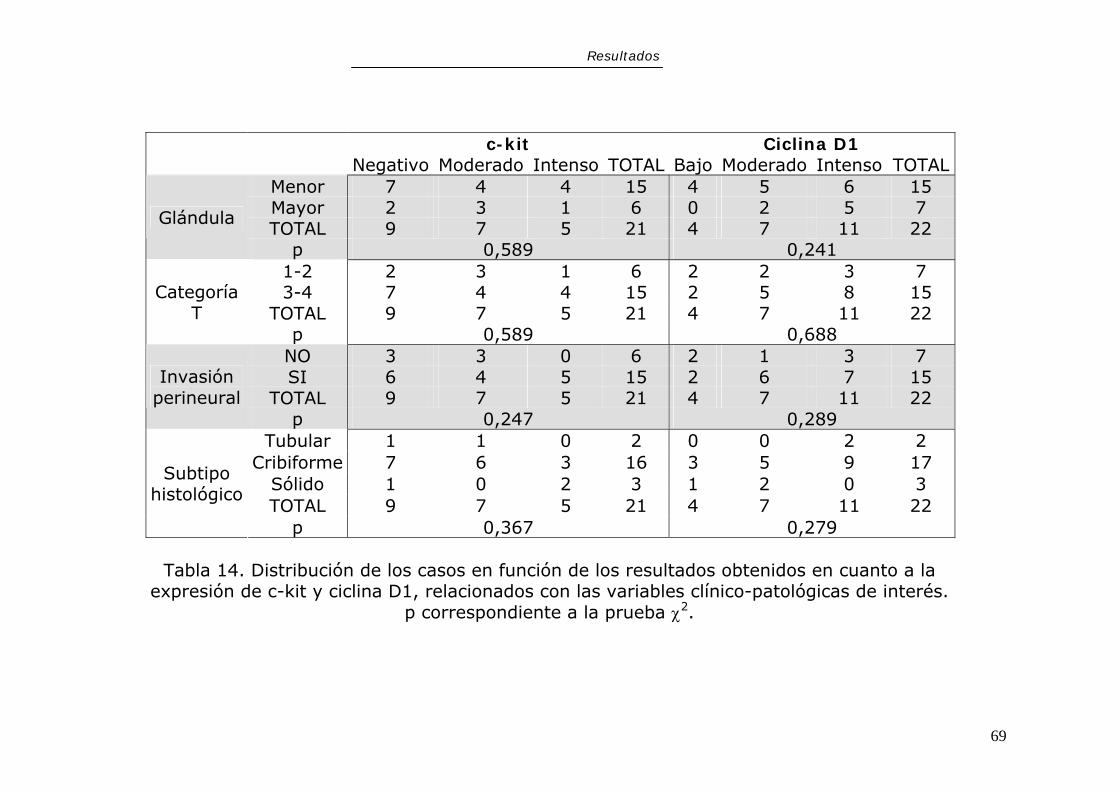
Tabla 13. Distribución de los casos en función de los resultados obtenidos en cuanto a la amplificación de los distintos oncogenes estudiados, relacionado con las variables clínico-patológicas de interés. p
correspondiente a la prueba χ2.
Resultados
69
c-kit Ciclina D1 Negativo Moderado Intenso TOTAL Bajo Moderado Intenso TOTAL
Menor 7 4 4 15 4 5 6 15 Mayor 2 3 1 6 0 2 5 7 TOTAL 9 7 5 21 4 7 11 22
Glándula
p 0,589 0,241 1-2 2 3 1 6 2 2 3 73-4 7 4 4 15 2 5 8 15
TOTAL 9 7 5 21 4 7 11 22Categoría
T p 0,589 0,688
NO 3 3 0 6 2 1 3 7 SI 6 4 5 15 2 6 7 15
TOTAL 9 7 5 21 4 7 11 22 Invasión
perineural p 0,247 0,289
Tubular 1 1 0 2 0 0 2 2Cribiforme 7 6 3 16 3 5 9 17
Sólido 1 0 2 3 1 2 0 3TOTAL 9 7 5 21 4 7 11 22
Subtipo histológico
p 0,367 0,279
Tabla 14. Distribución de los casos en función de los resultados obtenidos en cuanto a la expresión de c-kit y ciclina D1, relacionados con las variables clínico-patológicas de interés.
p correspondiente a la prueba χ2.

Resultados
Conviene apuntar que, dado que la práctica totalidad de
los casos no mostraban afectación ganglionar ni metástasis a
distancia en el momento del diagnóstico, su distribución
según la categoría T y el estadio tumoral son coincidentes, por
lo que en las tablas sólo se muestra el primero.
Cabe destacar que los casos en los que se observaba un
mayor porcentaje de células epiteliales, la expresión de ciclina
D1 presentaba mayor intensidad (χ2 = 4,961, p = 0,084)
(Tabla 15). La proporción de componente epitelial no se
correlacionaba con ninguna otra variable clínico-patológica.
Nivel expresión ciclina D1 % células epiteliales Bajo Moderado Intenso Total Reducido 4 3 4 11 Elevado 0 4 7 11
Total 4 7 11 22
Tabla 15. Distribución de los tumores en función del nivel de expresión de ciclina D1 y el porcentaje de
células epiteliales observado.
Teniendo en cuenta el número de oncogenes amplificado
en cada caso (ninguno-uno vs dos o más), se observó una
tendencia a encontrar mayor frecuencia de invasión perineural
en los casos con amplificación en dos o más oncogenes frente
70

Resultados
a los que tenían uno o ningún oncogén amplificado (p > 0,05)
(Tabla 16).
Nº oncogenes amplificados Invasión
perineural Ninguno o
uno Dos a cuatro
Total
NO 4 2 6 SI 5 8 13
Total 9 10 19
Tabla 16. Distribución de los CAQs según el número de oncongenes amplificados en relación con la invasión
perineural.
La aparición de recidivas era independiente de cualquier
variable molecular. Sin embargo, se encontró una asociación
entre la amplificación de ERBB1 y la aparición de metástasis a
distancia (χ2 = 4,941, p = 0,026). Así, mientras el 44% de los
casos (7/16) con amplificación en ERBB1 desarrollaron
metástasis a distancia, éstas no se presentaron en ninguno de
los casos que carecían de esta alteración génica (Tabla 17).
71

Resultados
ERBB1 Metástasis No amplificado Amplificado Total
NO 8 9 17 SI 0 7 7
Total 8 16 24
Tabla 17. Distribución de los CAQs según el estado de amplificación de ERBB1 en relación con la
aparición de metástasis.
Análogamente, la aparición de metástasis a distancia
tendía a ser más frecuente (χ2 = 3,601, p = 0,058) en los
casos que presentaban coamplificación de las dianas ERBB1 y
CCND1 (50% vs 14% de los casos sin coamplificación) (Tabla
18).
Coamplificación ERBB1/CCND1
Metástasis Negativo Positivo Total NO SI
12 2
5 5
17 7
Total 14 10 24
Tabla 18. Distribución de los CAQs según el estado de coamplificación de ERBB1 y CCND1
en relación con la aparición de metástasis.
72

Resultados
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Función de supervivencia Censurados
4.7. ANÁLISIS DE SUPERVIVENCIA
La media de supervivencia para la población global fue de
6 años y 1,5 meses (73,54 meses, rango 1-276). La media de
supervivencia para los pacientes que murieron por CAQ fue de
3 años y 11 meses (47 meses, rango 1-159). Los resultados
del análisis univariante de supervivencia (Kaplan-Meier)
mostraron una supervivencia global del 81% a los 5 años y
del 58% a los 15 años (Figura 14).
Figura 14. Supervivencia global de los pacientes estudiados.
73

Resultados
En cuanto a la influencia de las distintas variables clínico-
patológicas consideradas, cabe destacar el impacto del
subtipo histológico sólido (supervivencia media 28 meses
(S.E. 14)), asociado de manera estadísticamente significativa
con una supervivencia reducida (log rank p = 0.0206), frente
al subtipo histológico cribiforme (supervivencia media 193
meses (S.E. 40)). La Figura 15 muestra las curvas de
supervivencia para los tres subtipos histológicos
considerados: tubular, cribiforme y sólido.
Subtipo histológico Cribiforme Tubular Sólido Censurado
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
peen
cia
acum
ulad
aSu
rviv
Shis
T
Figura 15 . Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según el subtipo
histológico.
74

Resultados
Asimismo, el tipo de tratamiento elegido, considerando
únicamente a los pacientes tratados con cirugía vs cirugía y
radioterapia, mostró tener un efecto sobre la supervivencia.
Así, los pacientes sometidos a cirugía y radioterapia
presentaron una supervivencia media de 222,93 meses (S.E.
32,5), frente al otro grupo, cuya supervivencia media fue de
36 meses (S.E. 17,0) (log rank p = 0,008) (Figura 16).
Tratamiento Cirugía Cirugía &radio
Censurados
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Tra
Figura 16. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según el
tratamiento aplicado.
75
Resultados
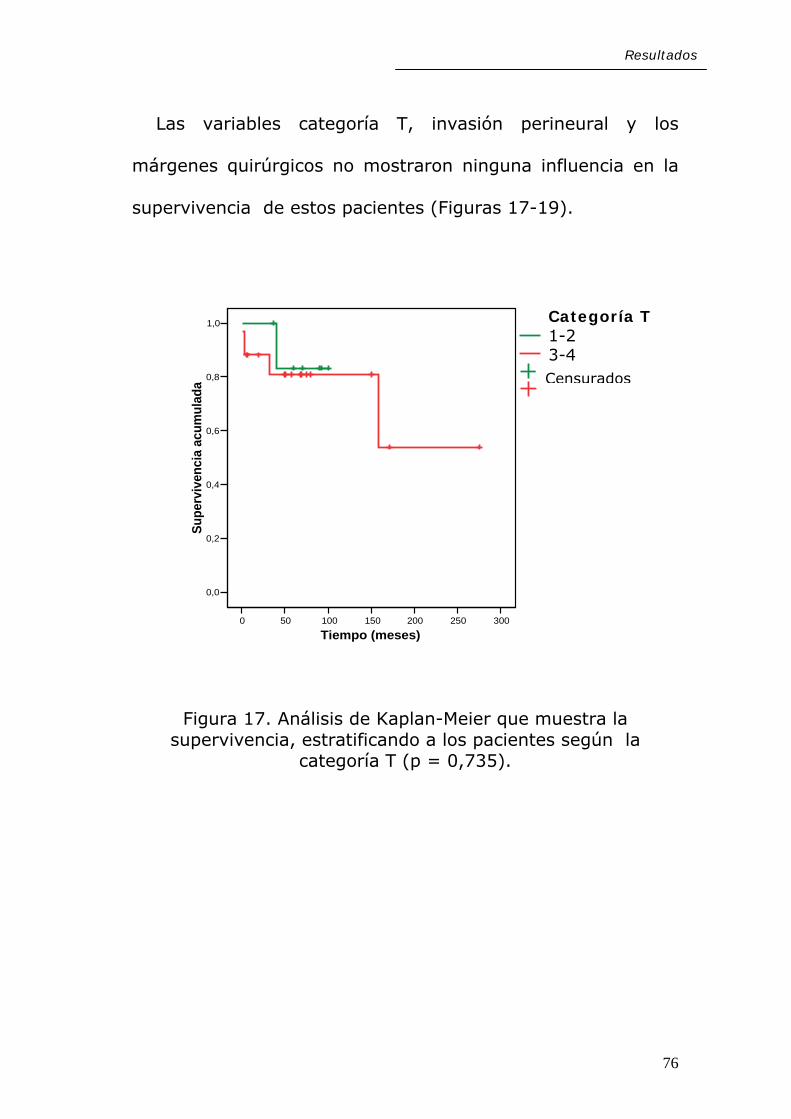
Las variables categoría T, invasión perineural y los
márgenes quirúrgicos no mostraron ninguna influencia en la
supervivencia de estos pacientes (Figuras 17-19).
Categoría T 1-2 3-4
0 50 100 150 00 250 300
Tiempo (meses)2
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Ca
Censurados
Figura 17. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según la
categoría T (p = 0,735).
76
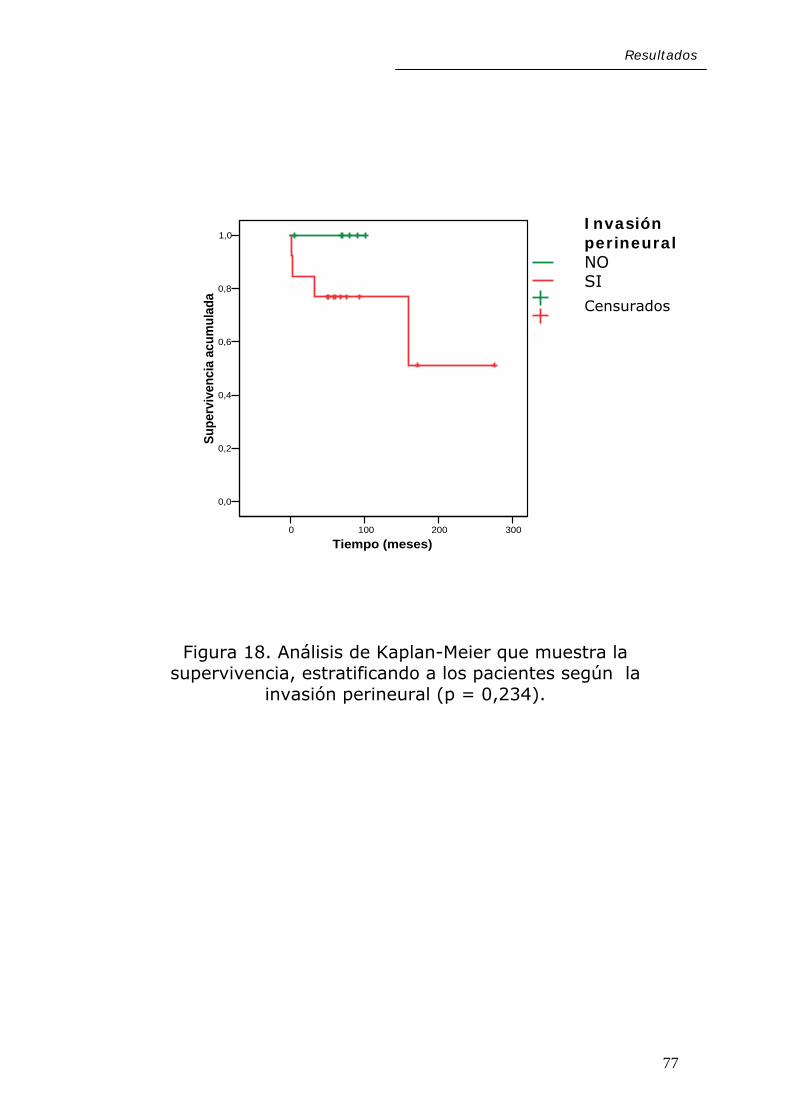
Resultados
Invasión perineural NO SI
0 100 200 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Inpe
Censurados
Figura 18. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según la
invasión perineural (p = 0,234).
77
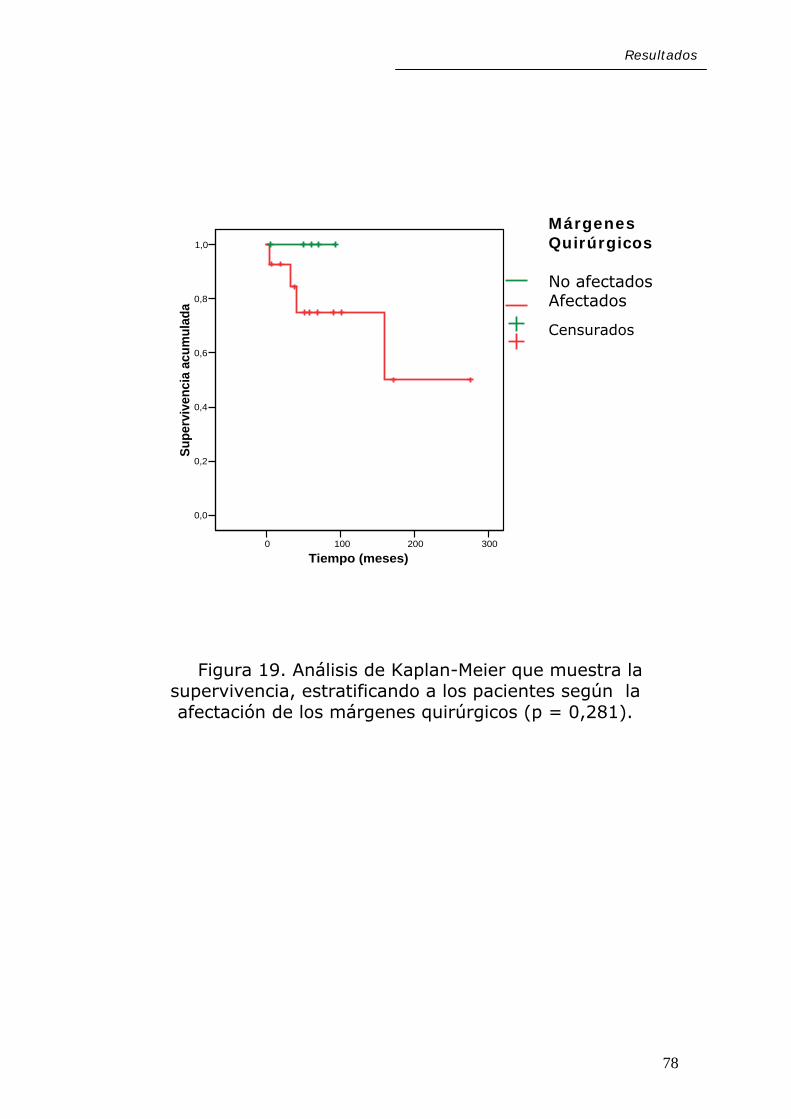
Resultados
Márgenes Quirúrgicos No afectados Afectados
Censurados
0 100 0 300
Tiempo (meses)
20
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Mqu
Figura 19. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según la afectación de los márgenes quirúrgicos (p = 0,281).
78
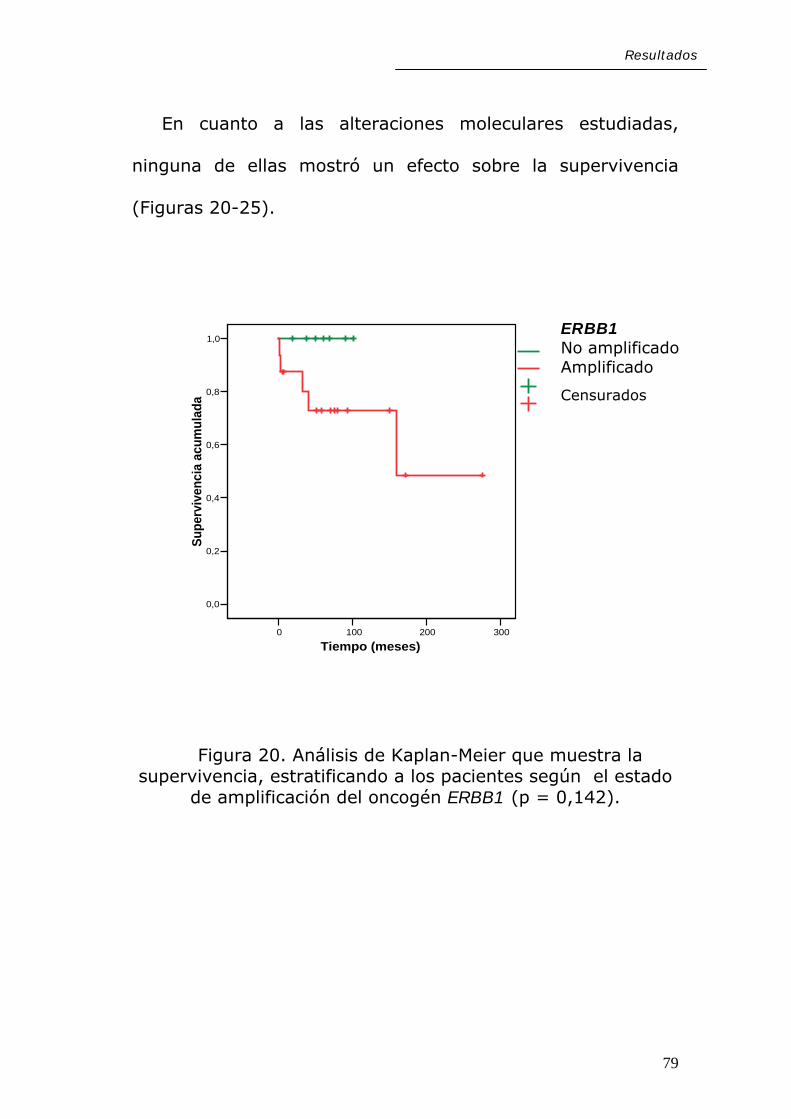
Resultados
En cuanto a las alteraciones moleculares estudiadas,
ninguna de ellas mostró un efecto sobre la supervivencia
(Figuras 20-25).
0 100 200 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
ERBB1 No amplificado Amplificado
Censurados
Figura 20. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según el estado
de amplificación del oncogén ERBB1 (p = 0,142).
79
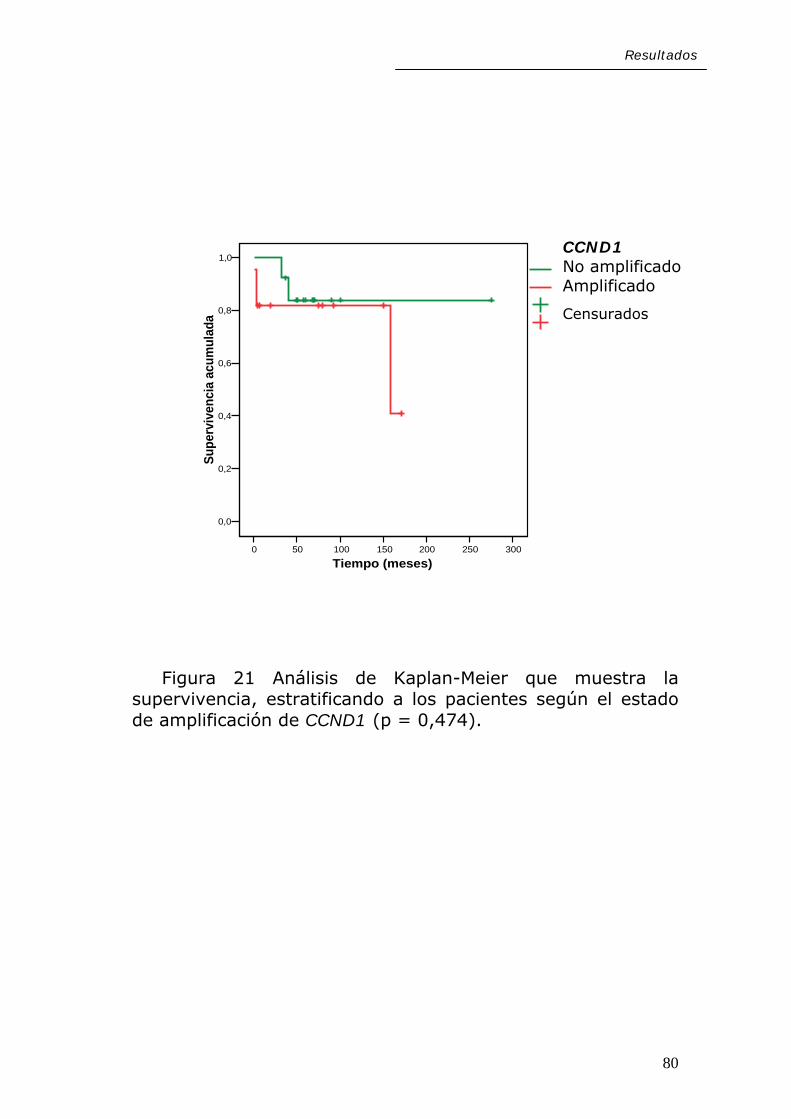
Resultados
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
CCND1 No amplificado
Amplificado
Censurados
Figura 21 Análisis de Kaplan-Meier que muestra la
supervivencia, estratificando a los pacientes según el estado de amplificación de CCND1 (p = 0,474).
80

Resultados
PIK3CA No amplificado Amplificado
Censurados
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
P
Figura 22. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según el estado
de amplificación de PIK3CA (p = 0,342).
81
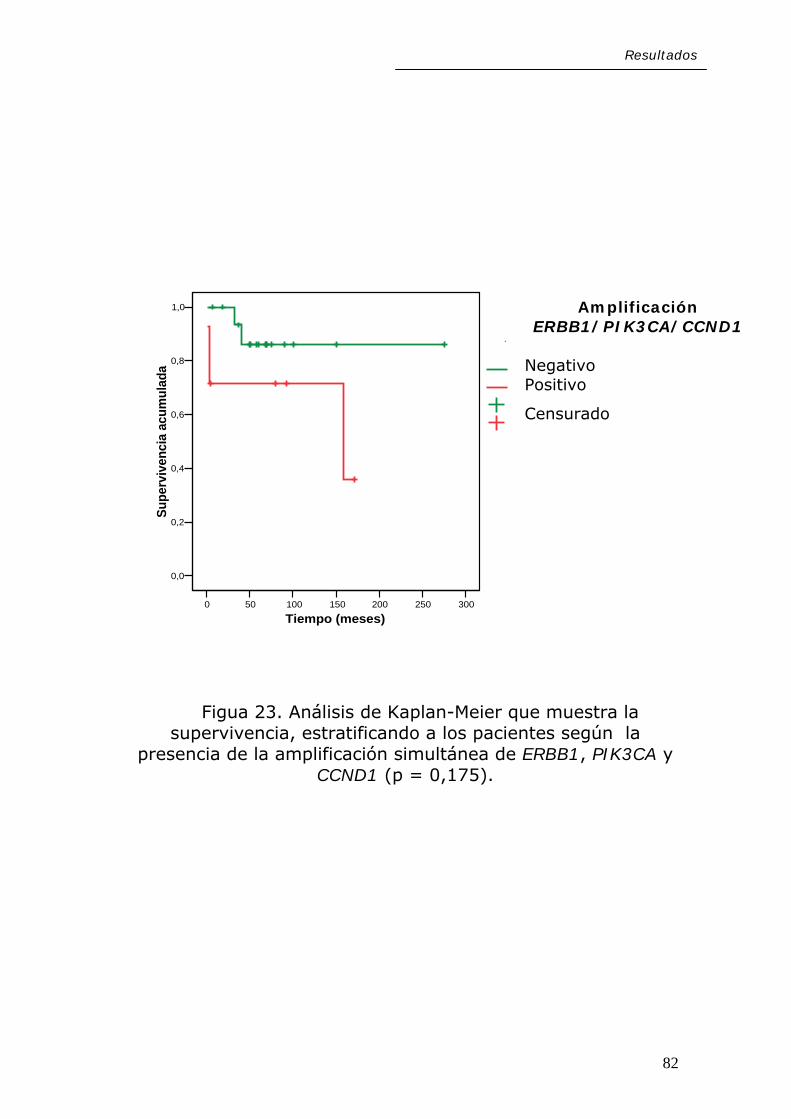
Resultados
Amplificación ERBB1/PIK3CA/CCND1
Negativo Positivo
Censurado
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
EP
Amp
Figua 23. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según la
presencia de la amplificación simultánea de ERBB1, PIK3CA y CCND1 (p = 0,175).
82

Resultados
Número oncogenes Amplificados
0-1 2 o más
Censurados
0 50 100 150 200 250 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Non
am
Figura 24. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según el número
de oncogenes con amplificación (p = 0,674).
83
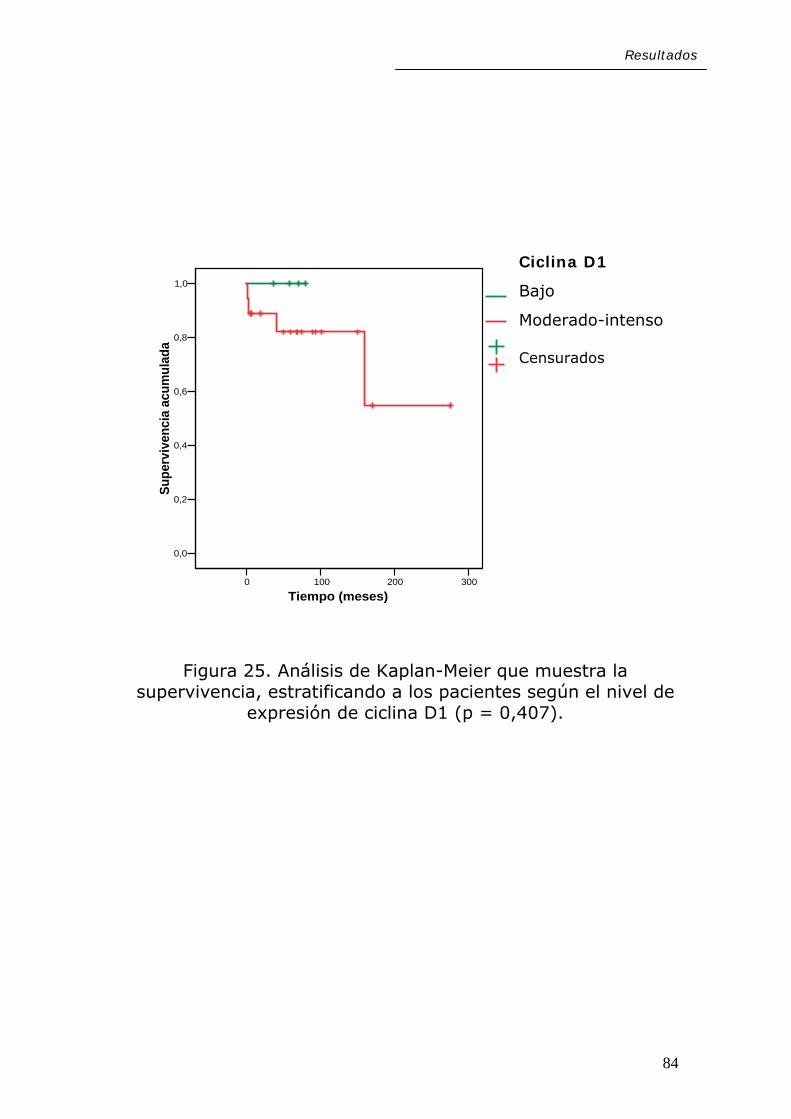
Resultados
Ciclina D1
Bajo
Moderado-intenso
Censurados
0 100 200 300
Tiempo (meses)
0,0
0,2
0,4
0,6
0,8
1,0
Supe
rviv
enci
a ac
umul
ada
Ci
Figura 25. Análisis de Kaplan-Meier que muestra la supervivencia, estratificando a los pacientes según el nivel de
expresión de ciclina D1 (p = 0,407).
84

Discusión
5
Discusión
85

Discusión
Uno de los retos actuales de la Biología Molecular es llegar
a esclarecer las bases genéticas y moleculares que conducen
al desarrollo y progresión de una neoplasia. El carcinoma
adenoide quístico (CAQ) es un tipo de tumor caracterizado por
un lento crecimiento, pero con una elevada tendencia al
desarrollo de metástasis a distancia. Las alteraciones
moleculares y citogenéticas subyacentes a su desarrollo,
están aún por determinar, a diferencia de otros cánceres,
para los que se ha logrado establecer un patrón de
alteraciones genético-moleculares responsables de la
tumorigénesis y con implicaciones terapéuticas y pronósticas
(como es el caso del adenocarcinoma de colon o del
carcinoma epidermoide de cabeza y cuello, HNSCC). Por ello,
se hace necesaria la identificación de las alteraciones
genéticas y los consiguientes cambios de expresión proteica
responsables del crecimiento y la progresión de los CAQs.
En este estudio hemos analizado seis oncogenes cuya
ganancia de función se relaciona con el desarrollo tumoral y
hemos valorado su grado de implicación en el desarrollo del
CAQ de glándulas salivales.
La sobreexpresión de ciclina D1 es una característica de
muchos tumores y resulta con frecuencia de la amplificación
86

Discusión
de su gen, CCND1. Se cree que ambos fenómenos
contribuyen al crecimiento de las células tumorales y a la
progresión del cáncer (54).
En nuestro estudio, el nivel de expresión de ciclina D1 fue
intenso en la mitad de los casos y un 31,82% adicional
mostraba tinción moderada, resultados que concuerdan con
estudios previos que han mostrado una sobreexpresión entre
el 32 y el 90% de los casos (69-71). Estos resultados
sugieren que se trata de un fenómeno muy frecuente en este
tipo de tumor. Sin embargo, el hecho de que ciclina D1 se
encuentre sobreexpresada de forma independiente del subtipo
histológico, sugiere que este fenómeno podría jugar un papel
en la progresión del CAQ global, más que en la generación de
las características propias de un subtipo concreto. En la línea
de nuestros resultados, otros autores han demostrado que la
presencia de la sobreexpresión de ciclina D1 no es diferente
entre los tumores benignos o malignos de las glándulas
salivales (72), lo cual limita su valor pronóstico.
El impacto de la sobreexpresión de ciclina D1 en la
proliferación celular en el CAQ es una cuestión controvertida.
Estudios básicos previos demuestran que la inhibición de la
expresión de ciclina D1 en líneas celulares de CAQ, no influye
87

Discusión
en el crecimiento celular (73). En nuestro estudio se describe
por primera vez que la tinción IHQ de ciclina D1 se observaba
únicamente en el componente epitelial de cada tumor
estudiado. Estas células forman ductos que contienen
mucopolisacáridos, a diferencia de las células mioepiteliales,
que secretan componentes de la membrana basal. El 100%
de las células epiteliales mostraban tinción de ciclina D1. El
hecho de que la intensidad de expresión de ciclina D1
tendiese a ser superior en aquellos casos que mostraban una
proporción de células epiteliales más elevada sugeriría que la
expresión de ciclina D1 contribuiría a una mayor tasa de
proliferación, lo cual concuerda con el papel establecido para
esta proteína en el control del ciclo celular.
La distribución de los casos en función de la proporción de
componente epitelial era independiente del subtipo histológico
(tubular, cribiforme y sólido), lo cual sugiere una ausencia de
relación entre la abundancia de células epiteliales (asociada a
una expresión más intensa de ciclina D1) y el pronóstico de la
enfermedad derivado de la clasificación histológica.
Los mecanismos exactos responsables de la
sobreexpresión de ciclina D1 en CAQ están aún por
determinar. La amplificación génica podría ser uno de ellos y
88

Discusión
se encuentra presente en otros tipos de carcinomas humanos,
como es el caso de HNSCC, para el que se describe con una
frecuencia en torno al 30% (74, 75). La tasa de amplificación
génica de CCND1 descrita para CAQ en la literatura se
encuentra en un 5% (71). En nuestro estudio encontramos
esta alteración en el 45,8% de los casos (11/24). Los
diferentes abordajes técnicos podrían justificar la diferencia,
siendo la detección basada en PCR, como la del presente
estudio, de mayor sensibilidad frente a FISH. En nuestra
serie, aunque no observamos una asociación significativa
entre la amplificación de CCND1 y la sobreexpresión de la
proteína (p = 0,08), 10/11 casos con amplificación génica
muestran una expresión proteica moderada/intensa. Sin
embargo, 8/18 casos con expresión moderada/intensa no
tenían amplificado el gen, lo cual indica que la sobreexpresión
de ciclina D1 podría justificarse sólo en parte por amplificación
génica y sugeriría la presencia de otros mecanismos
responsables de tal sobreexpresión. Entre ellos, se podría
mencionar un aumento en la tasa de transcripción o de la
estabilidad del mRNA. Adicionalmente, los niveles de ciclina
D1 podrían estar modulados por la degradación en el
proteasoma mediada por ubiquitina (76).
89

Discusión
Cabe destacar la asociación encontrada en el presente
estudio entre la presencia de amplificación en CCND1 y una
categoría T/estadio tumoral más avanzado (p = 0,047),
observación que ha sido descrita en el HNSCC (74, 77).
La sobreexpresión de c-kit se encuentra en varios tipos de
neoplasias humanas, entre los que cabe destacar los tumores
estromales gastrointestinales (GIST) (29), para los que se ha
demostrado una relación entre la activación de c-kit y la
patogénesis de la enfermedad (30). El papel de la expresión
de c-kit en la historia natural del CAQ es desconocido. En
nuestro estudio, hemos utilizado el anticuerpo A4502 de
DAKO, que ha sido testado previamente junto con una batería
de diversos anticuerpos anti-c-kit, mostrando los resultados
más reproducibles y específicos frente a varios tipos de
tumores humanos (78). Mediante IHQ, hemos encontrado una
expresión moderada/intensa en el 57,14% (12/21) de los
CAQs. Un mecanismo que podría justificar esta
sobreexpresión de c-kit podría ser un incremento del número
de copias del gen, como fue demostrado para otros tipos de
tirosin quinasas transmembrana tipo III, como ABL y PDGFR,
donde una tasa de transcripción incrementada resulta de un
efecto de dosis del gen. Sin embargo, sólo uno de los CAQs
90

Discusión
estudiados contaba con amplificación génica, caso que
mostraba un nivel de expresión moderado. Estos resultados
coinciden con un estudio previo en el que, mediante el uso del
mismo anticuerpo y la misma técnica de detección, se
muestra sobreexpresión en 29/55 (52,7%) tumores, de los
que sólo 3 fueron positivos para amplificación génica,
detectada por FISH. La tinción era menos intensa en los CAQs
de subtipo sólido frente a los tubulares o cribiformes (79),
resultado que no se reproduce en nuestro estudio, en el que
el nivel de expresión de c-kit era independiente del subtipo
histológico. Otros estudios detectaron una sobreexpresión de
c-kit en el 78% y 71% de los CAQs, respectivamente (28,
80). Estos resultados sugieren que la amplificación génica
podría no contribuir a la frecuente sobreexpresión proteica,
cuyos mecanismos responsables están, por tanto, por
determinar.
Ninguna de las recidivas presentó amplificación de c-kit, y
tampoco recidivó el único caso que presentó amplificación del
gen. Las tasas de recurrencia permanecieron similares en los
grupos c-kit positivos y c-kit negativos. Estos resultados están
en concordancia con el estudio de Aslan (81) en el que no se
encontró relación entre la expresión de c-kit y la recurrencia
91

Discusión
de la enfermedad, lo cual sugeriría que c-kit podría no influir
en el curso natural del CAQ.
c-kit es una diana molecular frente a la que actúa el
mesilato de imatinib (Glivec®). El éxito de esta terapia en
GIST que expresan c-kit, hizo pensar en su posible utilidad en
el tratamiento de los CAQs, donde, como se ha demostrado,
la sobreexpresión de c-kit es también muy frecuente. Sin
embargo, los resultados del uso terapéutico de Glivec® en
CAQ son controvertidos. Varios estudios, entre ellos un
ensayo en fase II, han mostrado que Glivec® no tiene efecto
sobre el CAQ avanzado de cabeza y cuello (82, 83). No se
sabe con certeza qué factor determina el éxito del
tratamiento. Algunos autores sugieren que el nivel de
expresión de c-kit, podría establecer un subgrupo de
pacientes con mayor probabilidad de beneficiarse del mismo
(79). Sin embargo, la idea de que los tumores que expresan
c-kit serán sensibles a Glivec® es simplista, ya que la
expresión de c-kit por sí sola no predice la respuesta a este
fármaco. La presencia de mutaciones activantes puede ser
crítica (82), como las descritas en GIST, en el que se han
encontrado mutaciones somáticas en los exones 9 y 11, que
codifican el dominio juxtamembrana de c-kit (84) y que
92

Discusión
confieren a la proteína una ganancia de función. Por otro lado,
éstas no se han encontrado en tumores de mama (85), ni en
neoplasias de las glándulas salivales (86), hecho que podría
justificar el fracaso de Glivec® en el tratamiento del CAQ.
El gen ERBB1 se encuentra frecuentemente
sobreexpresado en varios tipos de tumores humanos. En
HNSCC esta alteración se produce con elevada frecuencia y se
asocia a enfermedad avanzada, desarrollo de un fenotipo
metastático y un pronóstico pobre (24, 25).
El hecho de que se trate de un receptor de membrana ha
permitido el desarrollo de anticuerpos monoclonales y
pequeñas moléculas inhibidoras dirigidos a disminuir su
actividad en aquellos casos que muestren sobreexpresión de
EGFR.
Hasta el momento, la prevalencia y el papel que pueda
jugar la sobreexpresión de EGFR en CAQ no están claros. Los
diferentes estudios realizados hasta la fecha para su detección
presentan datos contradictorios. Mientras un solo estudio
clasifica como positivos el 85% de los casos estudiados
(23/27) (87), la mayoría apuntan a una prevalencia mínima
de sobreexpresión de EGFR (0/16 y 1/43 casos) (88, 89). Así,
nuestro estudio se sumaría al argumento de la baja o nula
93

Discusión
prevalencia de sobreexpresión de EGFR en CAQ, ya que
ninguno de los casos estudiados resultó positivo. Las
discrepancias frente al estudio de Vered (87) se podrían deber
a cuestiones técnicas, dada la variedad de anticuerpos
disponibles y diversos protocolos utilizados en cada
laboratorio. En nuestro caso, el hecho de haber utilizado un
anticuerpo anti-EGFR con los debidos controles y
sobradamente validado en la práctica hospitalaria, nos
permite confiar en la validez de los resultados obtenidos.
Uno de los mecanismos mejor descritos para justificar la
sobreexpresión de EGFR es el de la amplificación oncogénica,
presente en los casos de HNSCC con niveles elevados de la
proteína. La frecuencia descrita en este tipo de tumor varía
entre el 15% y el 58%, habiéndose observado su asociación
con un pronóstico pobre, con tumores clínicamente más
avanzados, pobremente diferenciados, con presencia de
metástasis ganglionares y de localización hipofaríngea (24,
90-92).
Nuestro estudio es el primero en el que se evalúa el nivel
de amplificación génica de ERBB1 en CAQ. En nuestra serie,
ERBB1 fue el oncogén que presentaba esta alteración con
mayor frecuencia (66,67%, 16/24 tumores primarios), y este
94

Discusión
fenómeno se asociaba a la aparición de metástasis (p =
0,026), lo cual convierte a este oncogén en una diana de
potencial interés en la práctica clínica.
El hecho de que la amplificación génica no se refleje en un
aumento de la expresión proteica induce a pensar en la
posibilidad de que otros genes localizados en la región en la
que se localiza ERBB1 (7p11.2) sean las verdaderas dianas de
la amplificación, proceso que podría arrastrar a ERBB1. Uno
de estos genes candidatos localizado en 7p11.2 podría ser
Grb10, que codifica una proteína adaptadora que interacciona
con varias tirosin quinasa celulares, y se ha visto implicado en
la tumorigénesis humana (93).
Cabe destacar que 10 de los 11 tumores primarios que
mostraron amplificación para CCND1, también mostraban esta
alteración para ERBB1 (p = 0,02). La aparición de metástasis
tendía a ser más frecuente entre los casos que presentaban
coamplificación de estas dos dianas (p > 0,05).
La amplificación génica afectaba al gen PIK3CA en 9/24
casos (37,9%), frecuencia similar a la encontrada para
HNSCC en nuestro laboratorio (94), siendo éste el primer
trabajo en el que se describe esta alteración en CAQ.
Teniendo en cuenta la función de la proteína codificada por
95

Discusión
PIK3CA, p110α, y a falta de estudios moleculares básicos
llevados a cabo de forma específica sobre líneas de CAQ, se
podría especular con la posible activación constitutiva de la
vía de señalización de la que forma parte y que, a través de la
activación de una de las proteínas clave, la serin-treonín
quinasa Akt, conduciría a respuestas celulares de resistencia a
apoptosis y mayor tasa de proliferación celular.
Se encontró una asociación entre la amplificación de
PIK3CA y el subtipo histológico. Mientras el 100% y el 67% de
los tumores tubulares y sólidos, respectivamente, mostraban
esta alteración, tan sólo estaba presente en el 22,22% de los
cribiformes (p = 0,019). Sin embargo, esta observación
carece de valor pronóstico, dado que la asociación encontrada
relaciona la amplificación de PIK3CA, tanto con el subtipo
histológico de mejor evolución como con el de peor evolución.
Por otro lado, se encontró una correlación entre la
presencia de amplificación génica en CCND1 y en PIK3CA (p =
0,015), con una frecuencia del 21,17% (7/24). 7 de los 11
casos (63,64%) con amplificación en CCND1 contaban con
esta alteración en PIK3CA. Este fenómeno ha sido observado
también en una serie de HNSCC estudiada en nuestro
laboratorio, en la que aparecía con una frecuencia del 28%
96

Discusión
(32/111), y se encontraba asociado a estadio tumoral
avanzado. Algunos autores (95) muestran que el 50% de los
casos de HNSCC con amplificación en 11q13 mostraban una
recombinación con la parte distal de 3q, lo cual podría reflejar
un efecto sinérgico entre la amplificación de CCND1 y la
ganancia en 3q en la tumorigénesis. Se podría especular
acerca de la posibilidad de que este tipo de mecanismos
estuviera presente en los CAQs, para los que la
coamplificación se observa con una frecuencia similar.
Además, ERBB1 se encontró amplificado en 8/9 casos que
mostraban esta alteración para PIK3CA (p = 0,074), por lo
que se podría también especular con un efecto sinérgico entre
las ganancias en 3q y 7p, aunque esto no ha sido descrito
hasta la fecha.
La amplificación de MDM2 confiere potencial tumorigénico
a células de roedores, por lo que se ha clasificado como
oncogén (96). En tumores humanos, la amplificación se
produce típicamente en sarcomas (97), mientras que la
proteína se encuentra sobreexpresada, en ausencia de
amplificación génica, en tumores de mama (98) y en
leucemias (99), para los que se ha descrito una ganancia de
función durante la transcripción.
97

Discusión
En el CAQ, se ha descrito la sobreexpresión de MDM2 en el
76.9% de los casos (100), con valor pronóstico adverso,
particularmente cuando ocurre junto con mutaciones de p53
(101). En otro estudio llevado a cabo sobre diversos tumores
de las glándulas salivales no se detecta amplificación génica,
aunque sí se describe la acumulación de tinción nuclear (102).
En líneas celulares de CAQ se ha sugerido que la
sobreexpresión de MDM2 se produce a nivel de RNA (103). En
nuestra serie de CAQs ningún caso mostró amplificación del
oncogén MDM2, resultado que apoya los estudios previos en
los que se sugiere la posibilidad de que sean otros
mecanismos los responsables de la acumulación proteica
observada en CAQ. La imposibilidad de obtener RNA de buena
calidad a partir de tejido embebido en parafina nos impide
abordar la detección del nivel de RNA de MDM2 en nuestra
serie de CAQs.
La incidencia de la amplificación de c-myc no ha sido
previamente estudiada en CAQ. Tampoco se encuentran en la
literatura trabajos de investigación básica que hagan
referencia al posible papel de este oncogén en estos tumores.
En nuestra serie de CAQs la incidencia de la amplificación
génica afectando a c-myc fue reducida (8,7%), encontrándose
98

Discusión
asociada a estadios tumorales tempranos (p = 0,025). Los
resultados descritos para HNSCC son controvertidos, ya que
mientras unos autores muestran una baja incidencia de esta
alteración (17%) (104), asociada al estadio tumoral avanzado
(105), otros autores han observado un peor pronóstico para
los casos con una disminución en el nivel de mRNA de c-myc
en carcinoma epidermoide de lengua (106).
El estado de amplificación oncogénica en las recidivas
tumorales estudiadas era semejante a la encontrada en los
correspondientes tumores primarios disponibles. Tan sólo en
un caso la recidiva contaba con una amplificación génica de
ERBB1 que no estaba presente en el primario.
La mitad de los tumores primarios (12/24) contaban con
dos o más oncogenes amplificados. Cabe destacar que el 61%
(8/13) de los tumores con invasión perineural portaban
amplificación en dos o más oncogenes, frente al 33% (2/6)
entre los que no presentaban tal invasión (p > 0,05).
El patrón de amplificación génica que se repetía con mayor
frecuencia (7/24 casos, 29%) consistía en la acumulación de
esta alteración en los oncogenes ERBB1, CCND1 y PIK3CA.
Aunque este patrón no mostró utilidad como factor pronóstico
(con resultados estadísticamente significativos), se observó
99

Discusión
una tendencia de los casos positivos a presentar una
supervivencia reducida (p = 0,175). Este resultado merece
ser tenido en cuenta en el diseño de futuros trabajos
encaminados a validar el aquí presentado, ya que el reducido
número de muestras que componen la serie estudiada
compromete la posibilidad de extraer conclusiones definitivas.
Sin embargo, esta serie presenta un comportamiento
esperado, de acuerdo con lo descrito en la literatura. Así, los
resultados obtenidos en cuanto a la supervivencia global
(85% a los 5 años y 58% a los 15 años), así como el impacto
del subtipo histológico sobre la supervivencia (peor pronóstico
para los sólidos, p = 0,0206) y la supervivencia más larga
para los pacientes tratados con cirugía y radioterapia, frente a
los tratados únicamente con cirugía (p = 0,008) dan cuenta
del carácter representativo de la serie de CAQs considerada.
La dificultad de acumular un número elevado de estos
tumores es conocida en el ámbito de la práctica de
Otorrinolaringología y una constante en la mayoría de los
trabajos encontrados en la literatura. En el presente estudio
se han recogido los casos intervenidos en el Servicio de
Otorrinolaringología y en el Servicio de Cirugía Maxilofacial de
un centro de referencia como es el HUCA entre los años 1984
100

Discusión
y 2004, lo cual da muestra de la baja incidencia de este tipo
de tumor, y la dificultad de contar con series que permitan
hacer estudios estadísticos concluyentes.
101

Conclusiones
6
Conclusiones
102

Conclusiones
1. La amplificación oncogénica más frecuentemente
observada en nuestra serie de CAQs es la que afecta al
gen ERBB1. Su presencia en el tumor primario se asocia
con la aparición de las metástasis, observación que
sugiere el potencial interés del estudio de esta alteración
en la práctica clínica.
2. La frecuente sobreexpresión de la ciclina D1
(81,82%) se justifica sólo en parte mediante la
amplificación génica (45,8%), alteración más frecuente
entre los tumores en estadio avanzado.
3. c-kit se encuentra sobreexpresado en un 57,14%
de los casos, fenómeno no justificado por la
amplificación génica, observada en tan solo un caso.
4. Mientras que la mayoría de los tumores de los
subtipos tubular y sólido mostraban amplificación de
PIK3CA, esta alteración no era frecuente entre los del
subtipo cribiforme.
5. La mitad de los CAQs mostraban amplificación en
dos o más oncogenes. Esta alteración se presentaba rara
103

Conclusiones
vez en los oncogenes c-myc, c-kit y MDM2. La
coamplificación de ERBB1, CCND1 y PIK3CA es el patrón
oncogénico presente con mayor frecuencia en nuestra
serie de CAQs, apuntándose una tendencia hacia una
reducción en la supervivencia de los casos afectados.
104

Bibliografía
7
Bibliografía
105

Bibliografía
1. Billroth T: Die Cylindergeschwalst. Untersuchungen
ueber die Entwicklung der Blutgefefasse. Edited by Berlin, G
Reimer, 1856, 55-69.
2. Kim KH, Sung MW, Chung PS, Rhee CS, Park CI, Kim
WH: Adenoid cystic carcinoma of the head and neck. Arch
Otolaryngol Head Neck Surg 1994, 120:721-726.
3. Haddad A, Enepekides DJ, Manolidis S, Black M: Adenoid
cystic carcinoma of the head and neck: a clinicopathologic
study of 37 cases. J Otolaryngol 1995, 24:201-205.
4. Bradley PJ: Adenoid cystic carcinoma of the head and
neck: a review. Curr Opin Otolaryngol Head Neck Surg 2004,
12:127-132.
5. Szanto PA, Luna MA, Tortoledo ME, White RA: Histologic
grading of adenoid cystic carcinoma of the salivary glands.
Cancer 1984, 54:1062-1069.
6. Matsuba HM, Spector GJ, Thawley SE, Simpson JR,
Mauney M, Pikul FJ: Adenoid cystic salivary gland carcinoma.
A histopathologic review of treatment failure patterns. Cancer
1986, 57:519-524.
7. Yamamoto Y, Saka T, Makimoto K, Takahashi H:
Histological changes during progression of adenoid cystic
carcinoma. J Laryngol Otol 1992, 106:1016-1020.
106

Bibliografía
8. Spiro RH, Huvos AG, Strong EW: Adenoid cystic
carcinoma: factors influencing survival. Am J Surg 1979,
138:579-583.
9. Goepfert H, Luna MA, Lindberg RD, White AK: Malignant
salivary gland tumors of the paranasal sinuses and nasal
cavity. Arch Otolaryngol 1983, 109:662-668.
10. Huang M, Ma D, Sun K, Yu G, Guo C, Gao F: Factors
influencing survival rate in adenoid cystic carcinoma of the
salivary glands. Int J Oral Maxillofac Surg 1997, 26:435-439.
11. Garden AS, Weber RS, Morrison WH, Ang KK, Peters LJ:
The influence of positive margins and nerve invasion in
adenoid cystic carcinoma of the head and neck treated with
surgery and radiation. Int J Radiat Oncol Biol Phys 1995,
32:619-626.
12. Howard DJ, Lund VJ: Reflections on the management of
adenoid cystic carcinoma of the nasal cavity and paranasal
sinuses. Otolaryngol Head Neck Surg 1985, 93:338-341.
13. Prokopakis EP, Snyderman CH, Hanna EY, Carrau RL,
Johnson JT, D'Amico F: Risk factors for local recurrence of
adenoid cystic carcinoma: the role of postoperative radiation
therapy. Am J Otolaryngol 1999, 20:281-286.
107

Bibliografía
14. Fordice J, Kershaw C, El-Naggar A, Goepfert H: Adenoid
cystic carcinoma of the head and neck: predictors of morbidity
and mortality. Arch Otolaryngol Head Neck Surg 1999,
125:149-152.
15. Wiseman SM, Popat SR, Rigual NR, Hicks WL, Jr., Orner
JB, Wein RO, McGary CT, Loree TR: Adenoid cystic carcinoma
of the paranasal sinuses or nasal cavity: a 40-year review of
35 cases. Ear Nose Throat J 2002, 81:510-517.
16. Sur RK, Donde B, Levin V, Pacella J, Kotzen J, Cooper K,
Hale M: Adenoid cystic carcinoma of the salivary glands: a
review of 10 years. Laryngoscope 1997, 107:1276-1280.
17. Silverman DA, Carlson TP, Khuntia D, Bergstrom RT,
Saxton J, Esclamado RM: Role for postoperative radiation
therapy in adenoid cystic carcinoma of the head and neck.
Laryngoscope 2004, 114:1194-1199.
18. Franzen G, Klausen OG, Grenko RT, Carstensen J,
Nordenskjold B: Adenoid cystic carcinoma: DNA as a
prognostic indicator. Laryngoscope 1991, 101:669-673.
19. Xie X, Nordgard S, Halvorsen TB, Franzen G, Boysen M:
Prognostic significance of nucleolar organizer regions in
adenoid cystic carcinomas of the head and neck. Arch
Otolaryngol Head Neck Surg 1997, 123:615-620.
108

Bibliografía
20. Nordgard S, Franzen G, Boysen M, Halvorsen TB: Ki-67
as a prognostic marker in adenoid cystic carcinoma assessed
with the monoclonal antibody MIB1 in paraffin sections.
Laryngoscope 1997, 107:531-536.
21. Bang G, Donath K, Thoresen S, Clausen OP: DNA flow
cytometry of reclassified subtypes of malignant salivary gland
tumors. J Oral Pathol Med 1994, 23:291-297.
22. Nowell PC: The clonal evolution of tumor cell
populations. Science 1976, 194:23-28.
23. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell
2000, 100:57-70.
24. Irish JC, Bernstein A: Oncogenes in head and neck
cancer. Laryngoscope 1993, 103:42-52.
25. Di Marco E, Albanese E, Benso S, Beatrice F, Cancedda
R, Toma S: Expression of epidermal growth factor receptor
and transforming growth factor alpha in human larynx
carcinoma. Cancer Lett 1992, 65:189-199.
26. Ashman LK: The biology of stem cell factor and its
receptor C-kit. Int J Biochem Cell Biol 1999, 31:1037-1051.
109

Bibliografía
27. Huang E, Nocka K, Beier DR, Chu TY, Buck J, Lahm HW,
Wellner D, Leder P, Besmer P: The hematopoietic growth
factor KL is encoded by the Sl locus and is the ligand of the c-
kit receptor, the gene product of the W locus. Cell 1990,
63:225-233.
28. Andreadis D, Epivatianos A, Poulopoulos A, Nomikos A,
Papazoglou G, Antoniades D, Barbatis C: Detection of C-KIT
(CD117) molecule in benign and malignant salivary gland
tumours. Oral Oncol 2006, 42:57-65.
29. Graadt van Roggen JF, van Velthuysen ML, Hogendoorn
PC: The histopathological differential diagnosis of
gastrointestinal stromal tumours. J Clin Pathol 2001, 54:96-
102.
30. Fletcher JA: Role of KIT and platelet-derived growth
factor receptors as oncoproteins. Semin Oncol 2004, 31:4-11.
31. Leroy X, Augusto D, Leteurtre E, Gosselin B: CD30 and
CD117 (c-kit) used in combination are useful for
distinguishing embryonal carcinoma from seminoma. J
Histochem Cytochem 2002, 50:283-285.
110

Bibliografía
32. Scobie JV, Acs G, Bandera CA, Blank SV, Wheeler JE,
Pasha TL, Salscheider M, Zhang PJ: C-kit immunoreactivity in
endometrial adenocarcinomas and its clinicopathologic
significance. Int J Gynecol Pathol 2003, 22:149-155.
33. Pelosi G, Barisella M, Pasini F, Leon ME, Veronesi G,
Spaggiari L, Fraggetta F, Iannucci A, Masullo M, Sonzogni A,
Maffini F, Viale G: CD117 immunoreactivity in stage I
adenocarcinoma and squamous cell carcinoma of the lung:
relevance to prognosis in a subset of adenocarcinoma
patients. Mod Pathol 2004, 17:711-721.
34. Raspollini MR, Amunni G, Villanucci A, Baroni G, Taddei
A, Taddei GL: c-KIT expression and correlation with
chemotherapy resistance in ovarian carcinoma: an
immunocytochemical study. Ann Oncol 2004, 15:594-597.
35. Miettinen M: From morphological to molecular diagnosis
of soft tissue tumors. Adv Exp Med Biol 2006, 587:99-113.
36. Roskoski R, Jr.: Structure and regulation of Kit protein-
tyrosine kinase−−the stem cell factor receptor. Biochem
Biophys Res Commun 2005, 338:1307-1315.
111

Bibliografía
37. Heinrich MC, Blanke CD, Druker BJ, Corless CL:
Inhibition of KIT tyrosine kinase activity: a novel molecular
approach to the treatment of KIT-positive malignancies. J Clin
Oncol 2002, 20:1692-1703.
38. Balsara BR, Sonoda G, du Manoir S, Siegfried JM,
Gabrielson E, Testa JR: Comparative genomic hybridization
analysis detects frequent, often high-level, overrepresentation
of DNA sequences at 3q, 5p, 7p, and 8q in human non-small
cell lung carcinomas. Cancer Res 1997, 57:2116-2120.
39. Heselmeyer K, Macville M, Schrock E, Blegen H,
Hellstrom AC, Shah K, Auer G, Ried T: Advanced-stage
cervical carcinomas are defined by a recurrent pattern of
chromosomal aberrations revealing high genetic instability
and a consistent gain of chromosome arm 3q. Genes
Chromosomes Cancer 1997, 19:233-240.
40. Liehr T, Ries J, Wolff E, Fiedler W, Dahse R, Ernst G,
Steininger H, Koscielny S, Girod S, Gebhart E: Gain of DNA
copy number on chromosomes 3q26-qter and 5p14-pter is a
frequent finding in head and neck squamous cell carcinomas.
Int J Mol Med 1998, 2:173-179.
112

Bibliografía
41. Singh B, Stoffel A, Gogineni S, Poluri A, Pfister DG,
Shaha AR, Pathak A, Bosl G, Cordon-Cardo C, Shah JP, Rao
PH: Amplification of the 3q26.3 locus is associated with
progression to invasive cancer and is a negative prognostic
factor in head and neck squamous cell carcinomas. Am J
Pathol 2002, 161:365-371.
42. Volinia S, Hiles I, Ormondroyd E, Nizetic D, Antonacci R,
Rocchi M, Waterfield MD: Molecular cloning, cDNA sequence,
and chromosomal localization of the human
phosphatidylinositol 3-kinase p110 alpha (PIK3CA) gene.
Genomics 1994, 24:472-477.
43. Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T,
Collins C, Pinkel D, Powell B, Mills GB, Gray JW: PIK3CA is
implicated as an oncogene in ovarian cancer. Nat Genet 1999,
21:99-102.
44. Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng
J, Liu JM, Yang DM, Yang WK, Shen CY: PIK3CA as an
oncogene in cervical cancer. Oncogene 2000, 19:2739-2744.
113

Bibliografía
45. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo
S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK,
Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE: High
frequency of mutations of the PIK3CA gene in human cancers.
Science 2004, 304:554.
46. Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J,
Szabo S, Konishi H, Karakas B, Blair BG, Lin C, Peters BA,
Velculescu VE, Park BH: The PIK3CA gene is mutated with
high frequency in human breast cancers. Cancer Biol Ther
2004, 3:772-775.
47. Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins
JM, McLendon RE, Fults DW, Velculescu VE, Bigner DD, Yan H:
Mutations of PIK3CA in anaplastic oligodendrogliomas, high-
grade astrocytomas, and medulloblastomas. Cancer Res 2004,
64:5048-5050.
48. Li X, Tsuji T, Wen S, Mimura Y, Wang Z, Sasaki K,
Shinozaki F: A fluorescence in situ hybridization (FISH)
analysis with centromere-specific DNA probes of
chromosomes 3 and 17 in pleomorphic adenomas and adenoid
cystic carcinomas. J Oral Pathol Med 1995, 24:398-401.
114

Bibliografía
49. Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo
MA, Williams LT: Membrane localization of
phosphatidylinositol 3-kinase is sufficient to activate multiple
signal-transducing kinase pathways. Mol Cell Biol 1996,
16:4117-4127.
50. Marte BM, Rodriguez-Viciana P, Wennstrom S, Warne
PH, Downward J: R-Ras can activate the phosphoinositide 3-
kinase but not the MAP kinase arm of the Ras effector
pathways. Curr Biol 1997, 7:63-70.
51. Franke TF, Kaplan DR, Cantley LC, Toker A: Direct
regulation of the Akt proto-oncogene product by
phosphatidylinositol-3,4-bisphosphate. Science 1997,
275:665-668.
52. Weinberg RA: The retinoblastoma protein and cell cycle
control. Cell 1995, 81:323-330.
53. Sherr CJ: Cancer cell cycles. Science 1996, 274:1672-
1677.
54. Donnellan R, Chetty R: Cyclin D1 and human neoplasia.
Mol Pathol 1998, 51:1-7.
115

Bibliografía
55. Capaccio P, Pruneri G, Carboni N, Pagliari AV, Quatela M,
Cesana BM, Pignataro L: Cyclin D1 expression is predictive of
occult metastases in head and neck cancer patients with
clinically negative cervical lymph nodes. Head Neck 2000,
22:234-240.
56. Fracchiolla NS, Pruneri G, Pignataro L, Carboni N,
Capaccio P, Boletini A, Buffa R, Neri A: Molecular and
immunohistochemical analysis of the bcl-1/cyclin D1 gene in
laryngeal squamous cell carcinomas: correlation of protein
expression with lymph node metastases and advanced clinical
stage. Cancer 1997, 79:1114-1121.
57. Mallya SM, Arnold A: Cyclin D1 in parathyroid disease.
Front Biosci 2000, 5:D367-371.
58. Corzo C, Corominas JM, Tusquets I, Salido M, Bellet M,
Fabregat X, Serrano S, Sole F: The MYC oncogene in breast
cancer progression: from benign epithelium to invasive
carcinoma. Cancer Genet Cytogenet 2006, 165:151-156.
59. Aulmann S, Adler N, Rom J, Helmchen B, Schirmacher P,
Sinn HP: c-myc amplifications in primary breast carcinomas
and their local recurrences. J Clin Pathol 2006, 59:424-428.
116

Bibliografía
60. Prins J, De Vries EG, Mulder NH: The myc family of
oncogenes and their presence and importance in small-cell
lung carcinoma and other tumour types. Anticancer Res 1993,
13:1373-1385.
61. Hirvonen HE, Salonen R, Sandberg MM, Vuorio E, Vastrik
I, Kotilainen E, Kalimo H: Differential expression of myc, max
and RB1 genes in human gliomas and glioma cell lines. Br J
Cancer 1994, 69:16-25.
62. Leonard JH, Kearsley JH, Chenevix-Trench G, Hayward
NK: Analysis of gene amplification in head-and-neck
squamous-cell carcinoma. Int J Cancer 1991, 48:511-515.
63. Haughey BH, von Hoff DD, Windle BE, Wahl GM, Mock
PM: c-myc oncogene copy number in squamous carcinoma of
the head and neck. Am J Otolaryngol 1992, 13:168-171.
64. Rodrigo Tapia JP, Lazo PS, Ramos S, Alvarez Alvarez I,
Suarez Nieto C: [Analysis of rearrangements in the first exon
of c-myc oncogene in squamous cell carcinoma of the head
and neck]. Acta Otorrinolaringol Esp 1995, 46:287-292.
65. Rowley JD: The critical role of chromosome
translocations in human leukemias. Annu Rev Genet 1998,
32:495-519.
117

Bibliografía
66. Momand J, Jung D, Wilczynski S, Niland J: The MDM2
gene amplification database. Nucleic Acids Res 1998,
26:3453-3459.
67. Klein C, Vassilev LT: Targeting the p53-MDM2 interaction
to treat cancer. Br J Cancer 2004, 91:1415-1419.
68. Sobin LH, Wittekind, C: TNM classification of malignant
tumors. Edited by UICC. New York, Wiley-Liss, 2002.
69. Daa T, Kaku N, Kashima K, Nakayama I, Yokoyama S:
Expression of beta-catenin, E-cadherin and cyclin D1 in
adenoid cystic carcinoma of the salivary gland. J Exp Clin
Cancer Res 2005, 24:83-87.
70. Zhou CX, Gao Y: Aberrant expression of beta-catenin,
Pin1 and cylin D1 in salivary adenoid cystic carcinoma:
relation to tumor proliferation and metastasis. Oncol Rep
2006, 16:505-511.
71. Greer RO, Jr., Said S, Shroyer KR, Marileila VG, Weed
SA: Overexpression of cyclin D1 and cortactin is primarily
independent of gene amplification in salivary gland adenoid
cystic carcinoma. Oral Oncol 2007, 43:735-741.
118

Bibliografía
72. Etges A, Nunes FD, Ribeiro KC, Araujo VC:
Immunohistochemical expression of retinoblastoma pathway
proteins in normal salivary glands and in salivary gland
tumours. Oral Oncol 2004, 40:326-331.
73. Yasumatsu R, Kuratomi Y, Nakashima T, Masuda M,
Yamamoto T: Cyclin D1 expression does not effect cell
proliferation in adenoid cystic carcinoma of the salivary gland.
Eur Arch Otorhinolaryngol 2004, 261:526-530.
74. Callender T, el-Naggar AK, Lee MS, Frankenthaler R,
Luna MA, Batsakis JG: PRAD-1 (CCND1)/cyclin D1 oncogene
amplification in primary head and neck squamous cell
carcinoma. Cancer 1994, 74:152-158.
75. el-Naggar AK, Steck K, Batsakis JG: Heterogeneity of the
proliferative fraction and cyclin D1/CCND1 gene amplification
in head and neck squamous cell carcinoma. Cytometry 1995,
21:47-51.
76. Guo Y, Yang K, Harwalkar J, Nye JM, Mason DR, Garrett
MD, Hitomi M, Stacey DW: Phosphorylation of cyclin D1 at Thr
286 during S phase leads to its proteasomal degradation and
allows efficient DNA synthesis. Oncogene 2005, 24:2599-
2612.
119

Bibliografía
77. Rodrigo JP, Garcia LA, Ramos S, Lazo PS, Suarez C:
EMS1 gene amplification correlates with poor prognosis in
squamous cell carcinomas of the head and neck. Clin Cancer
Res 2000, 6:3177-3182.
78. Went PT, Dirnhofer S, Bundi M, Mirlacher M, Schraml P,
Mangialaio S, Dimitrijevic S, Kononen J, Lugli A, Simon R,
Sauter G: Prevalence of KIT expression in human tumors. J
Clin Oncol 2004, 22:4514-4522.
79. Freier K, Flechtenmacher C, Walch A, Devens F, Muhling
J, Lichter P, Joos S, Hofele C: Differential KIT expression in
histological subtypes of adenoid cystic carcinoma (ACC) of the
salivary gland. Oral Oncol 2005, 41:934-939.
80. Mino M, Pilch BZ, Faquin WC: Expression of KIT (CD117)
in neoplasms of the head and neck: an ancillary marker for
adenoid cystic carcinoma. Mod Pathol 2003, 16:1224-1231.
81. Aslan DL, Oprea GM, Jagush SM, Gulbahce HE, Adams
GL, Gaffney PM, Savik K, Pambuccian SE: c-kit expression in
adenoid cystic carcinoma does not have an impact on local or
distant tumor recurrence. Head Neck 2005, 27:1028-1034.
120

Bibliografía
82. Pfeffer MR, Talmi Y, Catane R, Symon Z, Yosepovitch A,
Levitt M: A phase II study of Imatinib for advanced adenoid
cystic carcinoma of head and neck salivary glands. Oral Oncol
2006, 43:33-36.
83. Hotte SJ, Winquist EW, Lamont E, MacKenzie M, Vokes
E, Chen EX, Brown S, Pond GR, Murgo A, Siu LL: Imatinib
mesylate in patients with adenoid cystic cancers of the
salivary glands expressing c-kit: a Princess Margaret Hospital
phase II consortium study. J Clin Oncol 2005, 23:585-590.
84. de Silva CM, Reid R: Gastrointestinal stromal tumors
(GIST): C-kit mutations, CD117 expression, differential
diagnosis and targeted cancer therapy with Imatinib. Pathol
Oncol Res 2003, 9:13-19.
85. Ulivi P, Zoli W, Medri L, Amadori D, Saragoni L, Barbanti
F, Calistri D, Silvestrini R: c-kit and SCF expression in normal
and tumor breast tissue. Breast Cancer Res Treat 2004,
83:33-42.
86. Holst VA, Marshall CE, Moskaluk CA, Frierson HF, Jr.:
KIT protein expression and analysis of c-kit gene mutation in
adenoid cystic carcinoma. Mod Pathol 1999, 12:956-960.
121

Bibliografía
87. Vered M, Braunstein E, Buchner A:
Immunohistochemical study of epidermal growth factor
receptor in adenoid cystic carcinoma of salivary gland origin.
Head Neck 2002, 24:632-636.
88. Shintani S, Funayama T, Yoshihama Y, Alcalde RE,
Ootsuki K, Terakado N, Matsumura T: Expression of c-erbB
family gene products in adenoid cystic carcinoma of salivary
glands: an immunohistochemical study. Anticancer Res 1995,
15:2623-2626.
89. Kusafuka K: [Expressions of oncogene products in
adenoid cystic carcinomas of salivary glands:
immunohistochemical study]. Kokubyo Gakkai Zasshi 1991,
58:696-717.
90. Rodrigo JP, Ramos S, Lazo PS, Alvarez I, Suarez C:
Amplification of ERBB oncogenes in squamous cell carcinomas
of the head and neck. Eur J Cancer 1996, 32A:2004-2010.
122

Bibliografía
91. Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker
J, Parker N, Jarrett C, Carter J, Murphy BA, Netterville J,
Burkey BB, Sinard R, Cmelak A, Levy S, Yarbrough WG,
Slebos RJ, Hirsch FR: Increased epidermal growth factor
receptor gene copy number is associated with poor prognosis
in head and neck squamous cell carcinomas. J Clin Oncol
2006, 24:4170-4176.
92. Rodrigo Tapia JP, Martinez Sancer JA, Sanchez Lazo P,
Ramos, S, Sampedro Nuno, A, Suarez Nieto, C.: Amplification
of oncogene c-erb-B1 and cellular DNA content in squamous
cell carcinoma of the head and neck. Acta Otorrinolaringol Esp
1996, 42:97-103.
93. Okino K, Konishi H, Doi D, Yoneyama K, Ota Y, Jin E,
Kawanami O, Takeshita T: Up-regulation of growth factor
receptor-bound protein 10 in cervical squamous cell
carcinoma. Oncol Rep 2005, 13:1069-1074.
94. Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH,
Rodrigo JP, Nieto CS, Gonzalez MV: Frequent genetic and
biochemical alterations of the PI 3-K/AKT/PTEN pathway in
head and neck squamous cell carcinoma. Int J Cancer 2005,
114:242-248.
123

Bibliografía
95. Jin Y, Jin C, Wennerberg J, Hoglund M, Mertens F: Cyclin
D1 amplification in chromosomal band 11q13 is associated
with overrepresentation of 3q21-q29 in head and neck
carcinomas. Int J Cancer 2002, 98:475-479.
96. Fakharzadeh SS, Trusko SP, George DL: Tumorigenic
potential associated with enhanced expression of a gene that
is amplified in a mouse tumor cell line. Embo J 1991,
10:1565-1569.
97. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein
B: Amplification of a gene encoding a p53-associated protein
in human sarcomas. Nature 1992, 358:80-83.
98. McCann AH, Kirley A, Carney DN, Corbally N, Magee HM,
Keating G, Dervan PA: Amplification of the MDM2 gene in
human breast cancer and its association with MDM2 and p53
protein status. Br J Cancer 1995, 71:981-985.
99. Bueso-Ramos CE, Yang Y, deLeon E, McCown P, Stass
SA, Albitar M: The human MDM-2 oncogene is overexpressed
in leukemias. Blood 1993, 82:2617-2623.
100. Jia L, Esguerra RL, Tang X, Yin H, Sakamoto K, Okada N,
Takagi M: Prognostic value of apoptosis and apoptosis-
associated proteins in salivary gland adenoid cystic
carcinoma. Pathol Int 2004, 54:217-223.
124

Bibliografía
101. de Araujo VC, Martins MT, Leite KR, Gomez RS, de
Araujo NS: Immunohistochemical Mdm2 expression in minor
salivary gland tumours and its relationship to p53 gene
status. Oral Oncol 2000, 36:67-69.
102. Schlott T, Nagel H, Laskawi R, Eiffert H, Droese M:
Genetic analysis of the human oncoprotein MDM2 in benign
and malignant tumors of the salivary gland. Pathobiology
2001, 69:67-76.
103. Mantesso A, Loducca SV, Bendit I, Garicochea B, Nunes
FD, de Araujo VC: Mdm2 mRNA expression in salivary gland
tumour cell lines. J Oral Pathol Med 2004, 33:96-101.
104. de la Guardia C, Casiano CA, Trinidad-Pinedo J, Baez A:
CENP-F gene amplification and overexpression in head and
neck squamous cell carcinomas. Head Neck 2001, 23:104-
112.
105. Nguyen DC, Parsa B, Close A, Magnusson B, Crowe DL,
Sinha UK: Overexpression of cell cycle regulatory proteins
correlates with advanced tumor stage in head and neck
squamous cell carcinomas. Int J Oncol 2003, 22:1285-1290.
106. Vora HH, Shah NG, Trivedi TI, Goswami JV, Shukla SN,
Shah PM: Expression of C-Myc mRNA in squamous cell
carcinoma of the tongue. J Surg Oncol 2007, 95:70-78.
125