tema 3. equilibrio de fases en sistemas heterogéneos · pdf filediferentes clases de...
TRANSCRIPT

Apuntes de TF-1122 - Susana Curbelo 3.1
Tema 3. Equilibrio de Fases en Sistemas Heterogéneos Unarios Diagramas P-T y representaciones alternativas de las superficies termodinámicas. Potencial químico y energías de Gibbs como coordenadas de diagramas y superficies. Ecuación de Clausius-Clapeyron; curvas de vaporización y sublimación. Puntos triples. Cálculo de diagramas P-T de sistemas Unarios.
El estudio de los cambios en composición es necesario no solo para sistemas con reacciones químicas, sino para un número importante de operaciones industriales de transferencia de masa. En gran cantidad de experiencias cotidianas experimentamos transferencia de una sustancia de una fase a otra: al respirar (tomamos oxígeno del aire y lo disolvemos en la sangre, mientras que CO2 abandona la sangre y se incorpora al aire), en la cafetera (los ingredientes solubles del café pasan al agua), líquido quitamanchas disuelve la mancha de salsa o grasas de la ropa). De igual modo la composición es una variable importante en procesos de separación como destilación, adsorción, absorción y extracción, que implican la transferencia de masa entre fases de diferente composición cuando no están en equilibrio. De esta forma para el tratamiento cuantitativo de la transferencia de masa se verá que será necesario estimar el estado termodinámico en el equilibrio. La termodinámica del equilibrio de fases trata de establecer las relaciones entre las distintas propiedades cuando dos o más fases alcanzan su estado de equilibrio.
3.1 Equilibrio Equilibrio es una palabra que denota condición estática, ausencia de un cambio. En termodinámica significa no solo ausencia de un cambio sino de cualquier tendencia hacia el cambio en una escala macroscópica. Luego, no puede existir un cambio de estado en un sistema al equilibrio (Smith y Van Ness, 2003). La idea de estado de equilibrio tiene dos componentes:
• Es un estado de reposo: es decir, la condición del sistema es independiente del tiempo, es estacionaria. No hay cambio en las propiedades macroscópicas del sistema.
• Es un estado de balance: si el sistema es perturbado de su condición de equilibrio
por alguna influencia externa, el retornará a la misma condición de equilibrio.
Estado de equilibrio es aquel que no tienen tendencia alguna de ser abandonado espontáneamente, respecto a ciertos cambios o procesos permitidos, por ejemplo, transferencia de calor, trabajo de expansión y para sistemas abiertos, transferencia de masa. En un estado de equilibrio las propiedades son independientes del tiempo y de la historia previa del sistema; además son estables, es decir, no sufren cambios drásticos cuando condiciones externas varían ligeramente.

Apuntes de TF-1122 - Susana Curbelo 3.2
Lo anterior implica la ausencia de cualquier fuerza impulsora que induzca un cambio, es decir, dichas fuerzas deben estar balanceadas. Diferentes clases de fuerzas impulsoras tienden a producir tipos de cambios diferentes:
• Las fuerzas mecánicas no balanceadas como la presión en un pistón llegan a ocasionar transferencia de energía en forma de trabajo.
• Las diferencias de temperatura pueden producir transferencia de calor • Los gradientes de potencial químico tienden a originar transferencia de masa de
una fase a otra.
3.2 Criterios Generales de Equilibrio Termodinámico La termodinámica se ocupa, normalmente, de cambios finitos en el estado de equilibrio de un sistema, o de la variación de un estado de equilibrio sujeto a determinadas condiciones. Un cambio entre estados de equilibrio es lo que llamamos proceso. Y si el proceso es reversible el sistema mantiene en un estado virtual durante todo el proceso, pasado por sucesivos estados de equilibrio, de forma que la dirección del proceso puede ser invertida sin afectar a sus alrededores. Este principio termodinámico puede ser formulado de forma matemática según un grupo de ecuaciones denominadas condiciones de equilibrio, que describe como deben ser las relaciones entre las propiedades internas de un sistema aislado cuando este alcanza su estado de equilibrio. Se verá que para esto es requisito que:
La temperatura debe ser uniforme a través del sistema, si esta condición no se cumple existirá transferencia de calor entre una u otra locación del sistema mientras el sistema esté aislado.
Además no debe haber desbalance de fuerzas entre las partes del sistema, esto garantiza que el sistema está térmica y mecánicamente en equilibrio.
Finalmente, se debe considerar si en el sistema se puede dar una reacción química, una transferencia de masa entre las fases o ambas.

Apuntes de TF-1122 - Susana Curbelo 3.3
3.3 Equilibrio de fases: Sistemas Heterogéneos Se entiende por equilibrio de fases, aquella situación donde se encuentran dos o mas fases o estados de agregación de la materia coexistiendo en condiciones de equilibrio termodinámico.
Esto ocurre por que cuando dos fases se ponen en contacto, tienden a intercambiar sus componentes hasta que la composición en las dos fases alcanza un valor constante, cuando se alcanza este estado se dice que las fases están en equilibrio. La coexistencia de fases más encontrada a nivel industrial es el líquido-vapor, sin embargo, se consiguen también sistemas en equilibrio líquido-líquido, sólido-vapor, líquido-sólido, líquido-líquido-vapor, líquido-sólido-vapor y sólido-sólido-líquido. Entendiendo por Fase toda región homogénea de materia.
Un gas o una mezcla de gases, un líquido o una solución líquida y un sólido cristalino, son algunos ejemplos de fases.
Una fase no necesita ser continua, por ejemplo, gases dispersos como burbujas en
un líquido, un líquido disperso como gotas en otro líquido inmiscible, y un sólido cristalino disperso, ya sea en un gas o en un líquido. En cada caso la fase dispersa está distribuida a lo largo de la fase continua.
Las composiciones en el equilibrio de las dos fases frecuentemente son distintas entre sí, esa diferencia es la que permite separar mezclas mediante destilación, extracción, absorción, adsorción, lixiviación y otras operaciones similares. Las composiciones finales o en el equilibrio dependen de diversas variables, como T y P, la naturaleza química de la sustancia y las concentraciones de las sustancias en la mezcla.
3.4 Condiciones de Equilibrio Termodinámico Los criterios que se deben aplicar para decidir cuando un sistema está o no en equilibrio pueden ser desarrollados a partir de las 1era y 2da Ley de la termodinámica:
3.4.1 Sistemas Cerrados Homogéneos Un sistema monofásico u homogéneo, es aquel que posee propiedades uniformes en todo el sistema. Una fase es un sistema homogéneo. Consideremos un sistema homogéneo compresible de masa fija (cerrado) en el cual la temperatura y la presión son uniformes con la posición (aunque no necesariamente constantes). 1era ley:

Apuntes de TF-1122 - Susana Curbelo 3.4
WQdU δδ −= (3.1)
Si el cambio de volumen es la única posibilidad de hacer trabajo y la presión es uniforme en el sistema, entonces:
PdVWREV =δ (3.2) Por tanto:
PdVdUQ +=δ (3.3)
Si la temperatura es uniforme en el sistema la 2da Ley en forma diferencia queda:
δσδ+=
TQdS
(3.4)
Eliminado el calor de ambas ecuaciones:
δσTPdVdUTdS =−− (3.5) Recordemos que se produce entropía en todo proceso real y se conserva solo en ausencia de irreversibilidades, por tanto,
0≥−− PdVdUTdS (3.6) Puesto que tal relación implica solo propiedades, debe ser satisfecha para cambios de estado de cualquier sistema cerrado de T y P uniformes, sin restricción a las condiciones de reversibilidad mecánica supuesta en su deducción.
La desigualdad se aplica a cada cambio diferencial del sistema entre los estados de no equilibrio y dicta la dirección del cambio que conduce al equilibrio.
El empleo de esta expresión general suele ser poco aplicable a sistemas prácticos, por ello se deducen versiones restringidas. Por ejemplo, un sistema aislado está restringido a energía interna y volumen constante: dV=0 y dU=0, entonces el cambio de entropía:
0, ≥VUdS (3.7)
Esto sugiere que los cambios de entropía de un sistema cerrado de volumen y energía interna constantes solo pueden seguir la dirección del incremento de entropía. Lo anterior implica que la entropía alcanza un máximo cuando el sistema alcanza el equilibrio.
“En un sistema Aislado, en equilibrio la entropía es máxima”

Apuntes de TF-1122 - Susana Curbelo 3.5
Si se restringe a un proceso que ocurre a temperatura y presión constante. En este caso es conveniente trabajar con la energía libre de Gibbs:
TSHTSPVUG −=−+≡ (3.8)
SdTTdSVdPPdVdUdG −−++≡ (3.9)
( )PdVdUTdSSdTVdPdG −−−=+− (3.10)
El lado derecho de esta expresión lo tenemos arriba, por tanto podemos concluir que:
0≤+− SdTVdPdG (3.11) Para un proceso a dT=0 y dP=0 queda que: 0, ≤PTdG
(3.12) Esto indica que la energía libre de Gibbs de un sistema a T y P fijas decrece durante procesos irreversibles.
Por tanto, el estado de equilibrio involucra un mínimo en la energía libre de Gibbs. Cuando 0, =PTdG , el sistema está en equilibrio.
En general, en un sistema mantenido a temperatura y presión constante, el cambio de Energía Libre de Gibbs decrece espontáneamente. G es mínima en el equilibrio.
La manera en el cual el equilibrio es alcanzado no tiene importancia, una vez obtenido el estado de equilibrio, el sistema existe a una T y P particular y no toman lugar más cambios. Puede demostrarse que las relaciones obtenidas para un sistema aislado, son válidas para cualquier tipo de sistema en el equilibrio aunque no esté aislado durante el proceso.
De forma similar obtenemos para el resto de las funciones de estados H, A, U:
En un sistema mantenido a entropía y volumen constante, el cambio de energía interna decrece espontáneamente. U es mínima en el equilibrio.
0, ≤VSdU (3.13)
En un sistema mantenido a entropía y presión constante, el cambio de entalpía decrece espontáneamente. H es mínima en el equilibrio.
0, ≤PSdH (3.14)

Apuntes de TF-1122 - Susana Curbelo 3.6
En un sistema mantenido a temperatura y volumen constante, el cambio de Energía Libre de Helmholtz decrece espontáneamente. A es mínima en el equilibrio.
0, ≤VTdA (3.15)
3.4.2 Potencial químico: Sistemas Abiertos Homogéneos Un sistema cerrado heterogéneo puede describirse como la suma de dos sistemas abiertos homogéneos que intercambian materia y energía entre sí. Recordemos que cualquier propiedad extensiva de un sistema unario, homogéneo es función de dos propiedades intensivas independientes entre sí e independientes del tamaño del sistema. Esto se conoce como el Postulado de Estado.
( ) 0, , =⇒= PTdGPTGG (3.16)
Sin embargo si el sistema es abierto, o la composición y tamaño del sistema cambian (si hay reacción química) hay que especificar más variables para determinar el estado. Esta puede ser la cantidad de sustancia de cada componente presente, ni. Tomando T y P como propiedades independientes y el número de moles n como una medida del tamaño del sistema. La Función de Gibbs puede ser expresada para un sistema multicomponente homogéneo como:
( )jnnnPTGG ,...,,,, 21= (3.17)
Ahora,
( ) 0,...,, , ≠⇒= PTi dGnPTGG (3.18)
Y la derivada total de la función como:
knPk
inPinTnP
dnnGdn
nGdP
PGdT
TGdG
ikikkk ≠≠
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
++⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=,,,,
K
(3.19) Donde sabemos que:
∑= ≠
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
++−=k
ii
nPidn
nGVdPSdTdG
ik1 , (3.20)
Este último térmico lleva el nombre de Potencial Químico de la especie i en una fase homogénea y se denota:
,...,,, jnPTii n
G⎟⎟⎠
⎞⎜⎜⎝
⎛
∂∂
=μ o ∑=
=k
iiinG
1
μ (3.21)

Apuntes de TF-1122 - Susana Curbelo 3.7
E indica como cambia la función de Gibbs con nj a T, P y nk constantes. Es una propiedad intensiva que puede ser aplicada tanto a sistemas abiertos como cerrados con cambios en la composición. El cambio de energía puede ser escrito ahora como:
ii
idnVdPSdTdG ∑=
++−=1
μ (3.22)
Y un criterio de equilibrio más general puede ser escrito, ahora en función del potencial químico:
∑∑==
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=k
iii
k
ii
nPTiPT dndn
nGdG
j 1.
1 ,...,,,, μ (3.23)
Por tanto cuando el estado de equilibrio toma lugar 0, =PTdG se puede emplear la forma:
00 22111
=+++⇒=∑=
kki
k
ii dndndndn μμμμ K (3.24)
El resto de las propiedades cambia similarmente, por ejemplo:
ii
idnPdVTdSdU ∑=
+−=1
μ (3.25)
Donde para un sistema cerrado a n ctte:
WQdU δδ −= (3.26)
TdSQ =δ (3.27)
∑=
+=k
iii dnPdVW
1μδ (3.28)
Se puede interpretar como un trabajo químico
Para las otras funciones de energía:
∑=
++=k
iii dnVdPTdSdH
1μ (3.29)
∑=
+−−=k
iii dnPdVSdTdA
1μ (3.30)
Por lo tanto, hay cuatro expresiones para el potencial cada una derivada de una propiedad extensiva con respecto a la cantidad del componente que se considere:

Apuntes de TF-1122 - Susana Curbelo 3.8
,...,,,,...,,,,...,,,,...,,, jjjj nVTinPSinVSinPTii n
AnH
nU
nG
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
≡μ (3.31)
Mas adelante veremos que la magnitud del potencial químico es la energía de Gibbs molar parcial, pero no es la energía interna, entalpía o energía de de Helmholtz, molar parcial. Eso de debe a que las propiedades elegidas para definir las propiedades molares parciales son T y P, las mimas variables independientes fundamentales de G.
3.4.3 Determinación de Condiciones de Equilibrio de Fases
Ya se dispone de un grupo de condiciones de equilibrio para sistema cerrado expresada en función de U, H, A y G. Estas condiciones pueden ser expresadas también en términos de variables intensivas como T, P y iμ .
Dado un sistema unario, no reactivo bifásico (con dos fases βα , en equilibrio termodinámico). Cada una de las fases puede ser tratada como un sistema de una fase con su propio grupo de propiedades intensivas y extensivas. La descripción de un sistema de dos fases usa el hecho que, para todas las propiedades extensivas definidas, el valor para el sistema es la suma de los valores para cada una de sus partes. Entonces, cada una de las propiedades S, V, U, H, A, y G es la suma de los valores de las correspondientes cantidades para cada fases separada. Sin embargo, es necesario tratar cada fase como un sistema abierto, ya que el número de moles en cada fase puede cambiar durante el proceso. Paso 1. Expresión para el cambio de entropía del sistema
( ) βαβα dSdSSSddSsistema +=+= (3.32)
Entonces será necesario calcular el cambio de entropía de cada fase durante el proceso como U=U(S,V,n) que está dado por la ecuación (3.25)
ii
idnPdVTdSdU ∑=
+−=1
μ
(3.33)
αα
αα
α
α
αα μ dn
TdV
TP
TdUdS −⎟
⎠
⎞⎜⎝
⎛+= (3.34a)
ββ
ββ
β
β
ββ μ dn
TdV
TP
TdUdS −⎟
⎠
⎞⎜⎝
⎛+= (3.34b)
ββ
ββ
β
β
βα
α
αα
α
α
α μμ dnT
dVTP
TdUdn
TdV
TP
TdUdSsistema −⎟
⎠
⎞⎜⎝
⎛++−⎟⎠
⎞⎜⎝
⎛+= (3.35)
Paso 2. Evaluación de las condiciones impuestas por el sistema aislado

Apuntes de TF-1122 - Susana Curbelo 3.9
Si el sistema es aislado no se ve influenciado por sus alrededores entonces es rígido (no hace ni le aplican trabajo), es térmicamente aislado (no hay intercambio de calor), y es impermeable (no hay transferencia de masa).
( ) 0=+=+= βαβα dUdUUUddUsistema (3.36)
( ) 0=+=+= βαβα dVdVVVddVsistema (3.37)
( ) 0=+=+= βαβα dndnnnddnsistema (3.38)
Por lo cual resulta:
βα dUdU −= (3.39)
βα dVdV −= (3.40)
βα dndn −= (3.41)
Estas propiedades pueden ser intercambiadas entre las fases pero conservadas en el sistema aislado.
αβ
β
α
αα
βαα
βαμμ dnTT
dVTP
TPdU
TTdSsistema ⎟
⎟⎠
⎞⎜⎜⎝
⎛−−
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠
⎞⎜⎝
⎛−⎟⎠
⎞⎜⎝
⎛+⎟⎠
⎞⎜⎝
⎛−=
11 (3.42)
Paso 3. Obtención de Condiciones de Equilibrio Las condiciones que describen el máximo en la entropía para un sistema aislado son obtenidas haciendo los coeficientes de cada diferencial igual a cero:
0,, ≥nVUdS (3.43)
βα
βαTT
TT=⇒=⎟
⎠
⎞⎜⎝
⎛− 011 (Condición de Equilibrio Térmico) (3.44)
βαβα
PPTP
TP
=⇒=⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠
⎞⎜⎝
⎛−⎟⎠
⎞⎜⎝
⎛ 0 (Condición de Equilibrio Mecánico) (3.45)
βαβ
β
α
αμμμμ
=⇒=⎟⎟⎠
⎞⎜⎜⎝
⎛− 0
TT (Condición de Equilibrio Químico) (3.46)
Conclusión: Para una sustancia pura con dos fases en el equilibrio en un sistema aislado o no:
1. La Temperatura de las dos fases debe ser la misma 2. La presión de las dos fases debe ser la misma 3. Los potenciales químicos de ambas fases deben ser iguales

Apuntes de TF-1122 - Susana Curbelo 3.10
Para que un sistema esté en equilibrio mecánico o térmico es de esperar que la temperatura y la presión sean uniformes en toda la masa del sistema heterogéneo. Además se puede esperar que iμ tenga un valor uniforme en todo el sistema heterogéneo en equilibrio
El resultado general para un sistema heterogéneo compuesto por π fases y m componentes, es que, en el equilibrio se cumple:
πβα TTT === ... (Condición de Equilibrio Térmico)
πβα PPP === ... (Condición de Equilibrio Mecánico)
πβα μμμ 111 ... === (Condición de Equilibrio Químico)
πβα μμμ 222 ... ===
πβα μμμ mmm === ...
Este juego de ecuaciones representa el criterio básico del equilibrio entre fases.
Consideremos el sistema constituido por dos fases de una sustancia pura en el equilibrio. Desde el momento en el cual el sistema llegó al equilibrio, cada fase está a la misma T y P, por tal razón:
( ) ( )PTGnPTGnG ,,' ββαα += (3.47) Donde βα, son las fases en cuestión. La forma diferencia de lo anterior queda:
ββαα dnGdnGdG PT +=,' (3.48)
Así, si βα dndn −= para masa constante, luego:
( ) ββα dnGGdG PT −=,' (3.49)
En el equilibrio como 0, =PTdG , también se cumple que:
βα GG = (3.50)
“En el equilibrio la Energía de Gibbs molar de ambas fases son iguales”

Apuntes de TF-1122 - Susana Curbelo 3.11
3.4.4 Regla de Fases de Gibbs El postulado de estado establece que el estado termodinámico de un fluido homogéneo puro es determinado al conocer dos propiedades termodinámicas intensivas independientes. En contraste cuando dos fases están en equilibrio, el estado del sistema se establece al especificar solo una propiedad. Por ejemplo, una mezcla de vapor y agua líquida a 101,325 kPa solo puede existir a 100 ◦C. Es imposible cambiar la temperatura sin alterar la presión, si se requiere que el vapor y el líquido continúen en equilibrio. Esto indica una dependencia entre la temperatura y la presión en régimen de transición de fases. En general, pueden coexistir varias fases, pero deben estar en equilibrio para aplicar la regla de fases. Para cualquier sistema en equilibrio, el número de variables independientes que deben fijarse en forma arbitraria a fin de establecer su estado intensivo se determina por la célebre regla de fases de J. Willard Gibbs (1839-1903), quien la dedujo en 1875 por medio de razonamiento teórico. En ausencia de reacción química:
π−+= mGL 2 (3.51) Donde π es el número de fases, m el número de especies químicas y GL son los grados de libertad del sistema.
El número de grados de libertad, es el número de variables intensivas utilizadas para caracterizar el sistema menos el número de restricciones o relaciones entre ellas.
Cuando se considera el número de grados de libertad de un sistema heterogéneo, es preciso tener en cuenta lo anterior. Si el sistema no se encuentra en un estado de equilibrio interno, pero cada fase si lo está, el número de variables independientes es
( )1+mπ , por que cada fase tiene ( )1+m grados de libertad. Ahora si el sistema total se encuentra en un estado de equilibrio interno, entonces hay que tener en cuenta que de las ( )1+mπ variables, hay ( )( )21 +− mπ relaciones de equilibrio representadas por:
πβα TTT === ... (Condición de Equilibrio Térmico)
πβα PPP === ... (Condición de Equilibrio Mecánico)
πβα μμμ 111 ... === (Condición de Equilibrio Químico)
πβα μμμ 222 ... ===
πβα μμμ mmm === ...
Por tal razón los grados de libertad del sistema se calculan como:

Apuntes de TF-1122 - Susana Curbelo 3.12
( ) ( )( ) πππ −+=+−−+= 2211 mmmGL (3.52)
El número mínimo de grados de libertad para cualquier sistema son cero, cuando 0=GL el sistema es invariante, es decir está correctamente especificado. En este caso la ecuación (3.1) se transforma en: 2+= mπ (3.53) Este es el número de fases máximo que puede coexistir en equilibrio para un sistema que contiene m especies. Cuando 1=m el número máximo de fases en equilibrio es 3=π , resultado característico de un punto triple, donde coexiste una fase líquida, una gaseosa y una sólida, por ejemplo. Por ejemplo, dada una solución salina acuosa saturada en su punto de ebullición con un exceso de sal cristalina. Las tres fases son: sales cristalinas, solución acuosa y el vapor generado en el punto de ebullición ( )3=π . Las especies químicas son dos ( )2=m , el agua y la sal. Por tanto, los grados de libertad de este sistema son 1=GL .
Pero ¿Cual es el mayor número de fases que pueden coexistir en el equilibrio?
Si, ( )2=m , para que 0=GL , entonces: π =4, es decir, cuatro fases pueden coexistir en equilibrio en un punto dado, en soluciones binarias.
Esto se puede deducir además ya que hay π +2 variables (P, T, nI, nII,...) y 2π potenciales químicos, es decir, hay 2(π-1) ecuaciones. Esto tiene una única solución, si π+2=2(π-1), o π=4.
Figura 3.1. Diagrama P-T y Teorema de Gibbs
Apuntes de TF-1122 - Susana Curbelo 3.13
3.5 Estabilidad de fases y Cálculo de Superficies de Potencial Químico Como ya se discutió, en el equilibrio termodinámico:
πβα TTT === ... πβα PPP === ...
πβα μμμ 111 ... === Para un sistema en ELV a T y P:
VLV
PT
L
PT
VL
GGn
nGn
nG=⇒⎟
⎠
⎞⎜⎝
⎛∂
∂=⎟
⎠
⎞⎜⎝
⎛∂
∂
=
,,
11 μμ
( ) ( )⎪⎪⎩
⎪⎪⎨
⎧
⇒=⇒≠⇒=
−⇒>⇒>⇒≤
−⇒<⇒<⇒<
−=−=
ELVdndGc
vaporfasedndGb
líquidafasedndGa
dndnGGdGVLV
VLV
VLV
VVLVVLPT
LV
μμ
μμ
μμ
μμ
00)
00)
00)
,
De acuerdo con el criterio de mínima energía, G tenderá a decrecer hasta alcanzar el cero en el equilibrio, por tanto, esto determinará la dirección del flujo de materia. La materia debe ir desde la fase de mayor energía a la fase de menor energía, por tanto, desde la fase de mayor potencial a la fase de menor potencial.
El potencial químico es la fuerza impulsora que induce la transferencia de masa entre las fases en el equilibrio termodinámico.
Luego, en el equilibrio de fases, a presión constante la temperatura a la cual ocurre la transición de fases, TTR, es fija y los potenciales entre las fases son iguales.
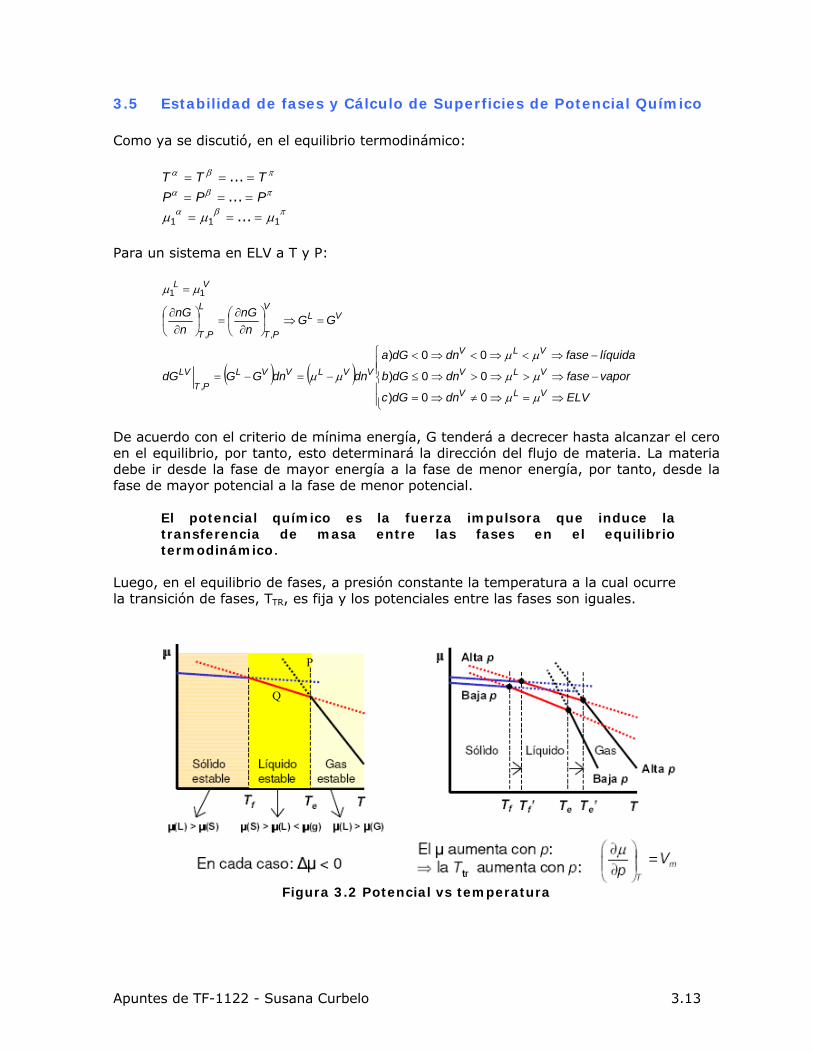
Figura 3.2 Potencial vs temperatura
P
Q

Apuntes de TF-1122 - Susana Curbelo 3.14
• A una temperatura dada la distancia vertical entre las curvas, reporta la diferencia
en la energía libre de Gibbs o potencial químico entre las fases, y es una medida cuantitativa de la estabilidad relativa entre las fases.
• La intersección de la curva de potencial del sólido con la del gas (P), indica el
equilibrio sólido-gas. Pero a la misma temperatura el potencial químico de la fase líquida es menor, por lo que la fase líquida (Q) es la más estable en dicha condición y el pto P. representa un punto de metaestabilidad.
• Todos los puntos de la curva deben estar localizados con respecto a una misma referencia (p.ej. 298 K).
• 298<T<Tfusión: el sólido es la fase estable, posee el menor potencial químico.
• Tfusión<T<Tebullicion: el líquido es la fase estable, posee menor potencial químico.
• T>Tebullición: el gas es la fase estable, posee menor potencial químico.
3.5.1 Cálculo de Superficies de Potencial Químico Ya se vió que, una superficie de potencial químico está representada por la función
( )αααα μμ PT ,= , y puede ser calculada por integración de la ecuación:
αααααμ dPVdTSd +−= donde si: dP=0
αααμ dTSd −= (3.59)
Esta integración puede ser llevada a cabo fijando un valor de P e integrado el término: –SdT sobre la temperatura, de forma de construir una sección isobárica a través de la superficie. Repitiendo este proceso para una serie de valores consecutivos de P se forma la superficie para la fase en cuestión. Para ello se requiere la información de la dependencia de la entropía con la temperatura de la fase, para lo cual se emplea:
dTT
CdPVdT
TC
dS PPαα
α =−= ya que dP=0 (3.60)
o
( ) ( ) ( )∫+=T
P dTT
TCSTS
298
298α
αα (3.61)
Sustituyendo la entropía en la expresión del potencial químico:
Apuntes de TF-1122 - Susana Curbelo 3.15
( ) ( )dTdT
TTC
SdT
P
⎥⎥⎦
⎤
⎢⎢⎣
⎡+−= ∫
298
298α
ααμ (3.62)
e integrando,
( ) ( )dTdT
TTC
SdT T
PT
∫ ∫∫ ⎥⎥⎦
⎤
⎢⎢⎣
⎡+−=
298 298298
298α
ααμ (3.63)
( ) ( ) ( ) ( ) ( ) ( )dTdT
TTC
SGTGTT T
P∫ ∫ ⎥⎥⎦
⎤
⎢⎢⎣
⎡+−=−=−
298 298
298298298α
ααααα μμ (3.64)
La evaluación de esta integral requiere del conocimiento del valor de la entropía de la fase alfa a 298K y del calor específico de la fase alfa como una función de la temperatura.
3.6 Estados termodinámicos de Metaestabilidad El equilibrio metaestable es importante para el conocimiento de las transformaciones de fase. Si la cinética de transformación de fase es lenta, el sistema puede encontarse en un estado termodinámico de equilibrio aparente o metaestable, y puede quedarse en este de forma indefinida.
La secuencia de estados termodinámicos que sufre un sistema mientras alcanza el verdadero estado termodinámico de equilibrio son denominados estados termodinámicos metaestables.

Apuntes de TF-1122 - Susana Curbelo 3.16
La metaestabilidad es la propiedad que un sistema con varios estados de equilibrio, tiene de exhibir, durante un considerable espacio de tiempo, un estado de equilibrio débilmente estable. Sin embargo, bajo la acción de perturbaciones externas (a veces no fácilmente detectables) dichos sistemas exhiben una evolución temporal hacia un estado de equilibrio fuertemente estable. Si representamos un sistema físico-químico por su energía potencial, un estado metaestable estará caracterizado por un estado que corresponde a un pseudo-mínimo de energía. Para que el sistema pueda alcanzar el estado de energía mínima que corresponde al estado de equilibrio termodinámico, es necesario suministrarle una cantidad de energía llamada energía de activación.

Apuntes de TF-1122 - Susana Curbelo 3.17
3.7 Comportamiento Cualitativo del Equilibrio de Fases en Sistemas Cerrados Heterogéneos
Las condiciones antes desarrolladas pueden emplearse para describir el equilibrio entre dos o más fases de un sistema. Comenzaremos con el caso elemental de equilibrio entre dos fases de una sustancia pura para finalmente llegar al caso general de varios componentes presentes en varias fases.
3.7.1 Procesos de Cambios de Fases Transformaciones Alotrópicas A 1 atm y hasta 910 ◦C, el hierro puro en equilibrio, existe como una estructura cristalina (tipo Body Centered Cubic, BCC). Entre 910 ◦C y 1455 ◦C, el estado de equilibrio del Hierro es del tipo cristal FCC (Face Centered Cubic). Entre 1455 ◦C y su punto de fusión (1537 ◦C) la forma más estable es otra vez como BCC.
Figura 3.2 Estructuras cristalinas más comunes a) cúbicas centradas en el cuerpo BCC b) cúbicas centradas en las caras FCC, c) Hexagonal compacta HC
BCC
P=1 atm T<910 C
Δ
P=1 atm T=910 C
FCC
Δ
P=1 atm T>910 C
Sistema Heterogéneo
FCC +
BCC
P=1 atm T=1455 C
Δ BCC
P=1 atm T<1537 C
Δ BCC +
FCC
Sistema Heterogéneo
Apuntes de TF-1122 - Susana Curbelo 3.18
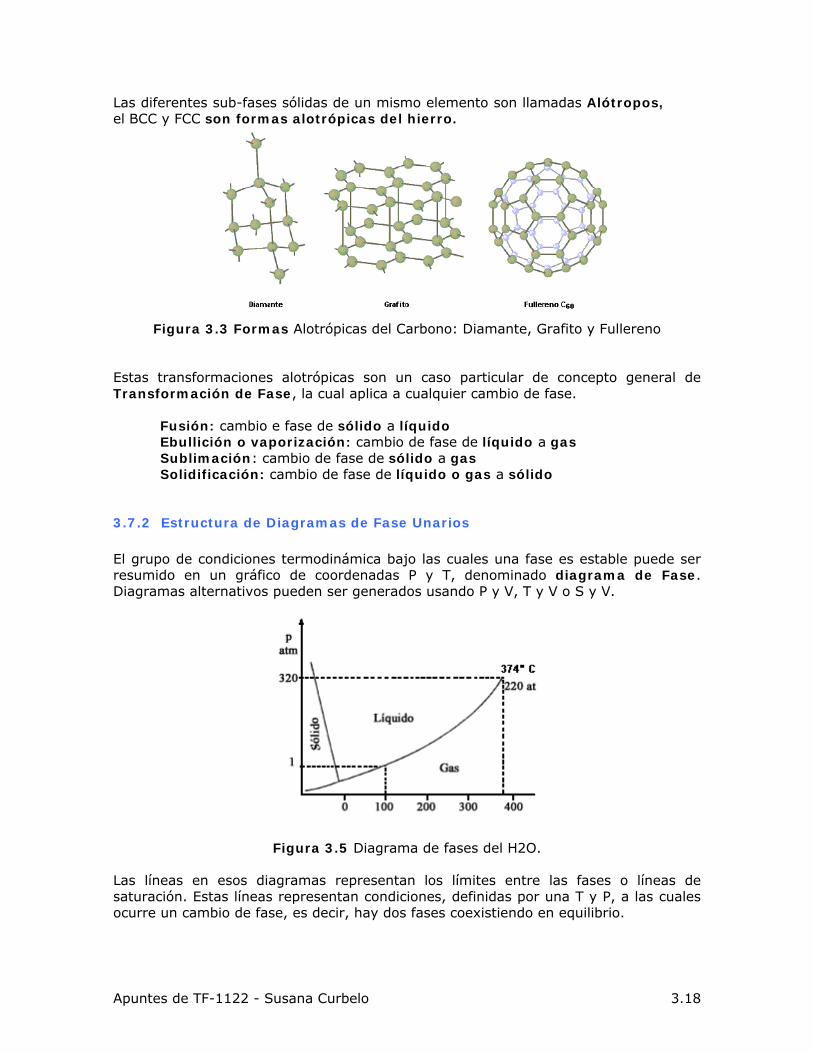
Las diferentes sub-fases sólidas de un mismo elemento son llamadas Alótropos, el BCC y FCC son formas alotrópicas del hierro.
Figura 3.3 Formas Alotrópicas del Carbono: Diamante, Grafito y Fullereno
Estas transformaciones alotrópicas son un caso particular de concepto general de Transformación de Fase, la cual aplica a cualquier cambio de fase.
Fusión: cambio e fase de sólido a líquido Ebullición o vaporización: cambio de fase de líquido a gas Sublimación: cambio de fase de sólido a gas Solidificación: cambio de fase de líquido o gas a sólido
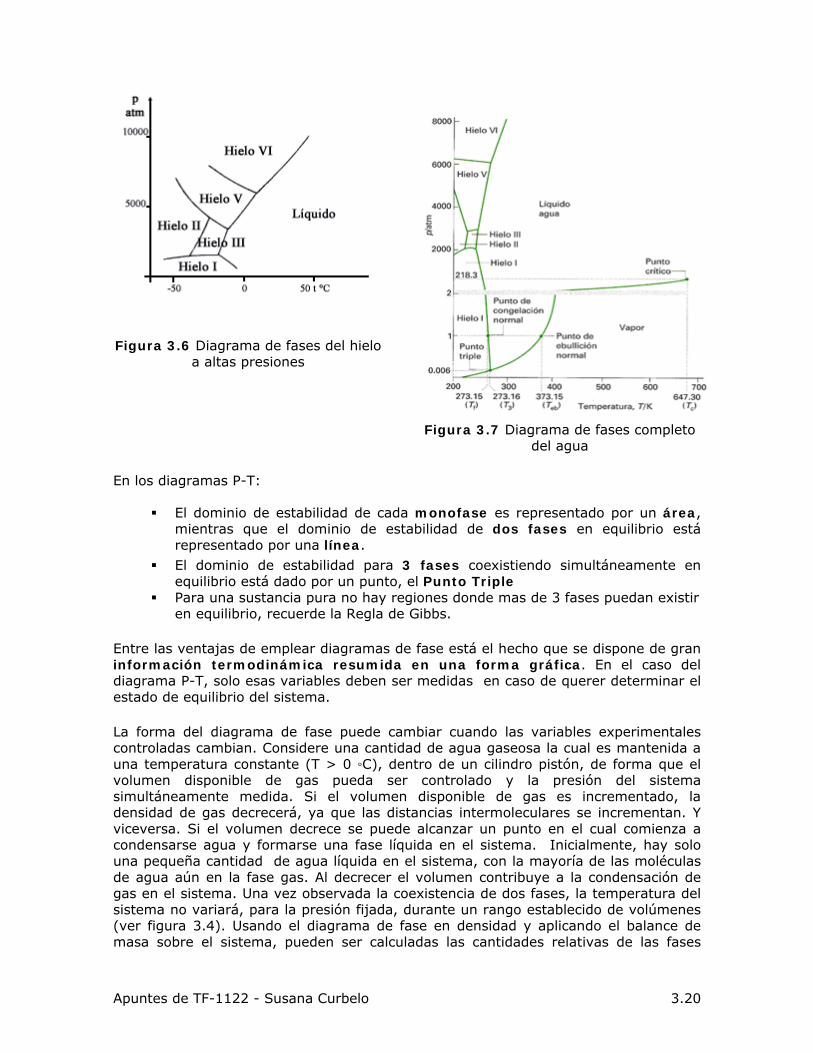
3.7.2 Estructura de Diagramas de Fase Unarios El grupo de condiciones termodinámica bajo las cuales una fase es estable puede ser resumido en un gráfico de coordenadas P y T, denominado diagrama de Fase. Diagramas alternativos pueden ser generados usando P y V, T y V o S y V.
Figura 3.5 Diagrama de fases del H2O.
Las líneas en esos diagramas representan los límites entre las fases o líneas de saturación. Estas líneas representan condiciones, definidas por una T y P, a las cuales ocurre un cambio de fase, es decir, hay dos fases coexistiendo en equilibrio.

Apuntes de TF-1122 - Susana Curbelo 3.19
Las ecuaciones que definen las condiciones de equilibrio en un sistema unario con 2 fases en equilibrio fueron desarrolladas en el Punto (3.5). La línea:
1-2, o curva de sublimación, separa las regiones de sólido y gas (ESV) 2-3, o curva de fusión, separa las regiones de sólido y líquido (ESL) 2-C, o curva de vaporización, separa las regiones de líquido y vapor (ELV) las tres líneas se encuentran en el llamado punto triple, donde las tres fases
coexisten en equilibrio. La curva de vaporización culmina en el punto crítico C, de coordenadas Pc y
Tc, que son la temperatura y presión más altas para las cuales una especie química puede existir en ELV.
La región por encima de Pc y Tc, se denomina supercrítica. Dependiendo de las condiciones de T y P alguna de las fases será más estable que la otra: sólido, líquido o gas. También puede darse que las condiciones de P y T sean las del equilibrio y coexistan ambas fases.
Cuando el hielo es calentado a presión atmosférica, el fundirá una vez que la temperatura suba a 0 ◦C. El hielo experimentará un cambio o transformación de fase en la cual la fase sólida es transformada a fase líquida.
Si el sistema es mantenido a 0 ◦C, luego el hielo puede coexistir con el agua. Si el sistema es mantenido a un temperatura levemente por debajo de 0 ◦C, solo puede ser observado hielo, mientras que a temperaturas por encima de 0 ◦C solo puede observase agua líquida.
Cuando el sistema como líquido es calentado otra transformación de fase ocurre a 100 ◦C, momento en el cual el líquido hierve y se transforma en gas. Estas transformaciones de fase toman lugar a una temperatura específica y se dice que ocurre isotérmicamente.
Las observaciones arriba graficadas fueron hechas a 1 atm. Si los experimentos son repetidos a presiones mas altas, se puede observar que el hielo funde a una temperatura mas baja mientras que el agua ebulle a una temperatura mas alta.
Cualquiera se la sustancia, una única temperatura de ebullición y fusión puede ser medida para cada presión examinada. Dichos valores pueden ser tabulados para cada presión.
De acuerdo a la regla de fases, el punto triple es invariante (GL=0), un sistema donde coexisten dos fases es univariante (GL=1) y una monofase es divariante (GL=2).
Apuntes de TF-1122 - Susana Curbelo 3.20
Figura 3.6 Diagrama de fases del hielo a altas presiones
Figura 3.7 Diagrama de fases completo del agua
En los diagramas P-T:
El dominio de estabilidad de cada monofase es representado por un área, mientras que el dominio de estabilidad de dos fases en equilibrio está representado por una línea.
El dominio de estabilidad para 3 fases coexistiendo simultáneamente en equilibrio está dado por un punto, el Punto Triple
Para una sustancia pura no hay regiones donde mas de 3 fases puedan existir en equilibrio, recuerde la Regla de Gibbs.
Entre las ventajas de emplear diagramas de fase está el hecho que se dispone de gran información termodinámica resumida en una forma gráfica. En el caso del diagrama P-T, solo esas variables deben ser medidas en caso de querer determinar el estado de equilibrio del sistema.
La forma del diagrama de fase puede cambiar cuando las variables experimentales controladas cambian. Considere una cantidad de agua gaseosa la cual es mantenida a una temperatura constante (T > 0 ◦C), dentro de un cilindro pistón, de forma que el volumen disponible de gas pueda ser controlado y la presión del sistema simultáneamente medida. Si el volumen disponible de gas es incrementado, la densidad de gas decrecerá, ya que las distancias intermoleculares se incrementan. Y viceversa. Si el volumen decrece se puede alcanzar un punto en el cual comienza a condensarse agua y formarse una fase líquida en el sistema. Inicialmente, hay solo una pequeña cantidad de agua líquida en el sistema, con la mayoría de las moléculas de agua aún en la fase gas. Al decrecer el volumen contribuye a la condensación de gas en el sistema. Una vez observada la coexistencia de dos fases, la temperatura del sistema no variará, para la presión fijada, durante un rango establecido de volúmenes (ver figura 3.4). Usando el diagrama de fase en densidad y aplicando el balance de masa sobre el sistema, pueden ser calculadas las cantidades relativas de las fases
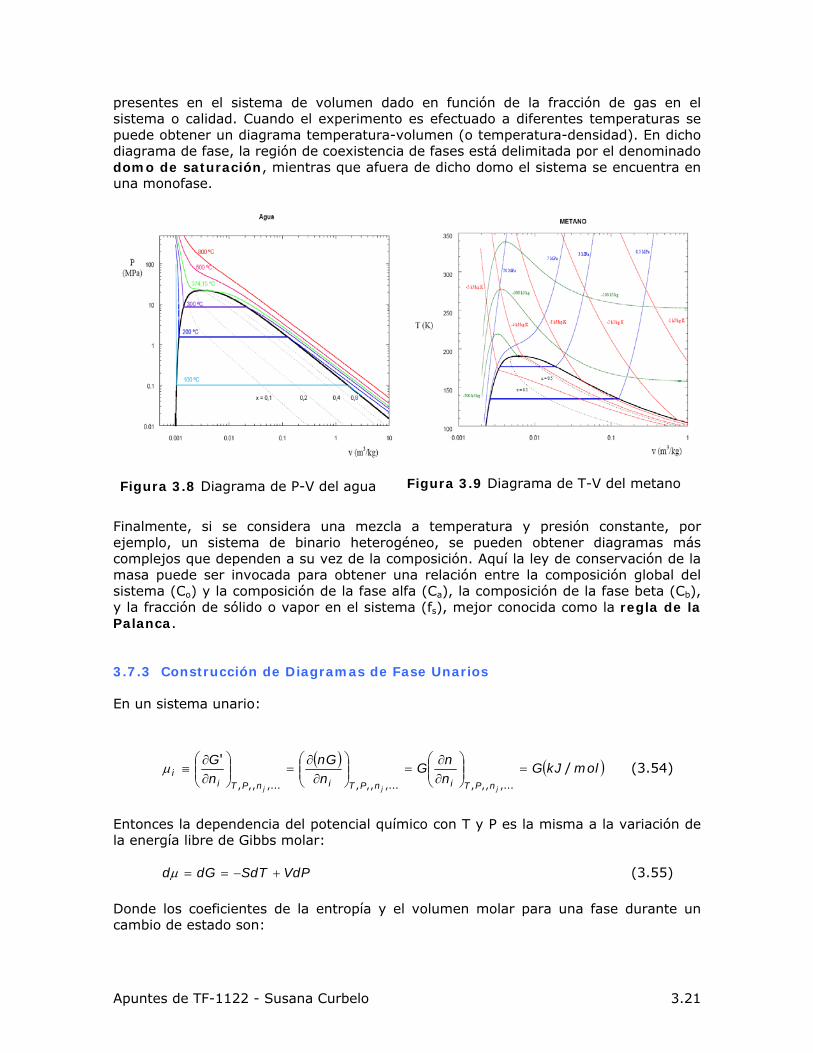
Apuntes de TF-1122 - Susana Curbelo 3.21
presentes en el sistema de volumen dado en función de la fracción de gas en el sistema o calidad. Cuando el experimento es efectuado a diferentes temperaturas se puede obtener un diagrama temperatura-volumen (o temperatura-densidad). En dicho diagrama de fase, la región de coexistencia de fases está delimitada por el denominado domo de saturación, mientras que afuera de dicho domo el sistema se encuentra en una monofase.
Figura 3.8 Diagrama de P-V del agua
Figura 3.9 Diagrama de T-V del metano
Finalmente, si se considera una mezcla a temperatura y presión constante, por ejemplo, un sistema de binario heterogéneo, se pueden obtener diagramas más complejos que dependen a su vez de la composición. Aquí la ley de conservación de la masa puede ser invocada para obtener una relación entre la composición global del sistema (Co) y la composición de la fase alfa (Ca), la composición de la fase beta (Cb), y la fracción de sólido o vapor en el sistema (fs), mejor conocida como la regla de la Palanca.
3.7.3 Construcción de Diagramas de Fase Unarios
En un sistema unario:
( ) ( )molkJGnnG
nnG
nG
jjj nPTinPTinPTii /'
,...,,,,...,,,,...,,,
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂
∂=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
≡μ (3.54)
Entonces la dependencia del potencial químico con T y P es la misma a la variación de la energía libre de Gibbs molar:
VdPSdTdGd +−==μ (3.55)
Donde los coeficientes de la entropía y el volumen molar para una fase durante un cambio de estado son:

Apuntes de TF-1122 - Susana Curbelo 3.22
αααααμ dPVdTSd +−= (3.56)
S y V pueden ser evaluados como una función de T y P, a partir de calor específico, coeficiente de expansión isotérmica y compresión isobárica, para luego ser integrada y obtener:
( )αααα μμ PT ,= (3.57) Un argumento similar puede ser aplicado a cada fase, pudiendo representar visualmente como una superficie (Dehoff Figura 7.3). Ojo: estos valores pueden ser comparados mientras el estado de referencia utilizado sea el mismo para cada fase (To, Po, fase L o S). La conexión entre la fase líquida y sólida puede ser hecha en el punto de fusión.
La intersección de cada superficie obtenida para cada fase, origina una curva, a la cual la T, P y μ, de las dos fases son idénticas
SLSLSL PPTT μμ === ,, (3.58)
Las cuales representan las condiciones a las cuales se encuentran esas fases L y S en equilibrio termodinámico. La proyección de esas líneas en el plano P-T representa la curva de saturación S+L (curva de fusión). Y los límites de estabilidad de las fases sólida y líquida. De igual forma se pueden obtener las respectivas curvas de vaporización y sublimación.
3.8 Ecuaciones de Clapeyron y de Clausius-Clapeyron
Las curvas que están en el dominio de dos fases βα, en equilibrio (por ejemplo, sólido de líquido, líquido de vapor, etc) en un espacio (P,T), para una sustancia pura, no son arbitrarias. Las mismas están determinadas por los principios de la termodinámica y los criterios de equilibrio, y pueden ser descritas matemáticamente como una función P=P(T).
La ecuación de Clausius-Clapeyron, es la forma diferencial de esta ecuación.
Esta expresa como para dos fases en el equilibrio, variaciones en la presión están únicamente relacionadas con variaciones en la temperatura: ( )TPP Sat=
Para cualquier par de fases en un sistema unario, la integración de la ecuación de Clausius-Clapeyron suministra una expresión para las curvas de saturación en un diagrama de fase. La intersección de dos curvas de saturación produce el punto triple, en el cual las tres fases coexisten.

Apuntes de TF-1122 - Susana Curbelo 3.23
3.8.1 Ecuación de Clapeyron
La transición de fases ocurre a temperatura y presión constante, correspondiente a cada punto de la línea. Como ya se demostró la transición de fase entre βα, , el cambio en la energía libre de Gibbs para la transición es cero en cualquier punto de la línea βα, .
0=ΔG (3.64)
Como el cambio de energía libre de Gibbs es constante a la largo de la línea y su valor es cero la pendiente de esa curva vendrá dada por:
GT
P
Δ
⎟⎠
⎞⎜⎝
⎛∂∂ (3.65)
Relación que puede ser resuelta en función de las anteriores a través de las relaciones de Maxwell y demás propiedades, hasta llegar a:
VS
TP
G ΔΔ
=⎟⎠
⎞⎜⎝
⎛∂∂
Δ
o
βα
βα
→
→
Δ
Δ=
VS
dTdP (3.66)
Además se sabe que para cualquier proceso a temperatura constante:
ST- G ΔΔ=Δ H (3.67)
Por lo que, para la transición, donde 0=ΔG , se cumple que:
THS Δ
=Δ (3.68)
Quedando una ecuación:
VTH
dTdP
ΔΔ
= (3.69)
La cual resulta de mayor utilidad, ya que el cambio de entropía no se puede medir directamente pero si los datos calorimétricos de los calores de transformación: entalpía de vaporización, fusión, sublimación. Hay que recordar que si el cambio de fase ocurre isobárica y reversiblemente, el calor transferido es igual al cambio de entalpía del proceso:
αββαβα HHHQ −=Δ= →→ (3.70) Esta es la denominada ecuación de Clapeyron, e indica que a cualquier punto en una curva de saturación que representa a dos fases en equilibrio en un diagrama de fases

Apuntes de TF-1122 - Susana Curbelo 3.24
unario, la pendiente es igual al radio entre el cambio de entropía y el cambio de volumen asociado a un cambio de fase. Donde en cada caso, los deltas representan los cambios en la propiedad por el cambio de fase y son:
αβ G- G G=Δ (3.71a)
αβ H- H =ΔH (3.71b)
αβ S- S =ΔS (3.71c)
Adicionalmente se define:
αββα VVV −=Δ → (3.71d) Otra forma de obtenerla es a partir del potencial químico, que para cada fase, viene dado por la ecuación (3.55):
αααααμ dPVdTSd +−= y βββββμ dPVdTSd +−=
Si durante este proceso infinitesimal, alfa y beta se mantienen en equilibrio, luego los cambio de P, T y μ, de cada fase vienen dado por las condiciones de equilibrio derivadas en la clase anterior:
dTdTdTTT ==⇒= βαβα dPdPdPPP ==⇒= βαβα
μμμμμ βαβα ddd ==⇒= 11 (3.72) Si están en equilibrio ambas fases experimentan el mismo cambio de temperatura, presión y potencial químico durante el proceso. De esta forma los superíndices pueden ser obviados, en las ecuaciones del potencial químico.
βββααα μμ ddPVdTSdPVdTSd ≡+−=+−≡ (3.73) Reagrupando:
( ) ( )dPVVdTSS αβαβ −=− (3.74)
dPVdTS βαβα →→ Δ=Δ (3.75) Lo cual representa el cambio de entropía y volumen por unidad de mol, asociado a la transformación de un mol de α a un mol de β a la T y P dadas. Arreglando la ecuación obtenemos nuevamente la ecuación 3.66:
βα
βα
→
→
Δ
Δ=
VS
dTdP (3.76)

Apuntes de TF-1122 - Susana Curbelo 3.25
Las ecuaciones 3.66 y 3.69 son exactas y válidas para todo tipo de transición de fases, sólido → líquido, sólido → gas, líquido → gas.
La integración de esta ecuación requiere del conocimiento de cómo varia la entropía o entalpía, y el volumen con la temperatura, y origina la curva P=P(T) para el equilibrio entre α y β . El signo de la pendiente depende del signo del cambio de volumen ya que el numerador siempre es positivo. A excepción del paso de líquido a sólido en el agua, en el resto de los casos, este denominador también será positivo.
3.8.2 Integración de la Ecuación de Clapeyron
Sino es posible la consideración de cambio de entalpía y volumen constante, será necesario desarrollar expresiones para el cambio de entalpía HΔ y de volumen VΔ , durante el cambio de fase como función de T y P.
( )( ) ( ) αβαββα
αββαβα
dHdHHHdHd
HHPTHH
−=−=Δ
−=Δ=Δ→
→→ , (3.77a)
y ( )
( ) ( ) αβαββα
αββα
dVdVVVdVd
VVPTV
−=−=Δ
−=Δ→
→ , (3.77b)
En el tema 2 se desarrollaron las expresiones para su cálculo:
( )dPTVdTCdH P α−+= 1 y dPVdTVdV βα −=
Por tanto: ( ) ( )[ ]dPTVdTCHd P α−Δ+Δ=Δ 1 (3.78)
Donde:
αβPPP CCC −=Δ (3.79)
y ( )[ ] ( ) ( )ααββ ααα TVTVTV −−−=−Δ 111 (3.80)
Sin embargo, se ha demostrado que para valores típicos de las propiedades este último término es despreciable para cambios de presión hasta 100.000 atm.
Por lo que para propósitos prácticos el cambio de entalpía puede ser considerado como función de la temperatura
( ) dTCHd PΔ=Δ →αβ (3.81)
La capacidad calorífica es generalmente expresada como:

Apuntes de TF-1122 - Susana Curbelo 3.26
22 TcbTaC
TcbTaC PP
Δ+Δ+Δ=Δ⇒++= (3.82)
Donde αβ aaa −=Δ , αβ bbb −=Δ , etc.
Estos resultados pueden ser usados para integrar el cambio de entalpía desde una To de referencia a un límite superior variable T:
( ) ( )T
Too T
cTbaTTHTH ⎥⎦
⎤⎢⎣
⎡ Δ−
Δ+Δ=Δ−Δ 2
2
( ) ( ) ⎥⎦
⎤⎢⎣
⎡ Δ−
Δ+Δ−Δ+⎥
⎦
⎤⎢⎣
⎡ Δ−
Δ+Δ=Δ
oooo T
cTbaTTHTcTbaTTH 22
22 Quedando el calor de transformación como:
( ) oCTcTbaTTH +⎥
⎦
⎤⎢⎣
⎡ Δ−
Δ+Δ=Δ 2
2 (3.83)
Por otro lado, la evaluación del numerador de la ecuación de Clausius-Clapeyron requiere del cálculo del cambio de volumen en función de T y P:
( ) ( ) ( )PTVPTVPTV ,,, αββα −=Δ →
Los cuales pueden calcular como dPVdTVdV βα −= , pero requieren del conocimiento de los coeficientes de expansión térmica y compresión isobárica, y su dependencia con T y P. Lo cual suele ser difícil. Ahora, si se analiza un sistema constituido por una fase vapor β y una fase sólida o
líquida α , luego αβ VV >> en la mayoría de los casos prácticos (a temperatura presión estándar el volumen molar de gas ocupa 22.400 cc mientras que la fase condensada está típicamente alrededor de 10 cc). Lo que simplifica considerablemente la matemática. Sin embargo, para fases condensadas en equilibrio (sólido o líquido) si se requiere información acerca de los coeficientes de expansión y compresibilidad de las dos fases para calcular el cambio de volumen como una función de T y P. Si el rango de presión considerado es del orden de algunas decenas de atmósferas, luego se puede asumir cambio de volumen constante en la mayoría de los cálculos. Pero dicha suposición no es válida si el rango de presión al cual será calculado el diagrama es de cientos de atmósferas.
3.8.2.1 Curvas de vaporización y Sublimación A condiciones en las cuales el vapor es típicamente encontrado para la mayoría de las sustancias, la desviación del comportamiento de gas ideal es despreciable.

Apuntes de TF-1122 - Susana Curbelo 3.27
Por tanto, para las curvas de vaporización o sublimación en el diagrama de fase:
( )P
RTVVVPTV GGG =≅−=Δ → αα , (3.84)
Entonces para el cálculo de curvas de vaporización o sublimación se puede considerar que ( ) ( )PTVPTV GG ,, ≈Δ →α obteniendo entonces la ecuación de Clausius-Clapeyron:
PRTH
dTdP
2
βα →Δ=
o
dTRTH
PdP
2
βα →Δ= (3.85)
Esta ecuación no es exacta ya que involucra dos suposiciones a) volumen de fase condensada despreciable frente al del vapor y b) comportamiento de gas ideal.
La expresión anterior ahora puede ser integrada si se conoce una expresión como la 3.82 para el cambio de la entalpía de vaporización o sublimación con la temperatura, o en su defecto si se puede considerar constante. Por ejemplo:
( ) ( )[ ]( )PRT
CTcTbaT
VTH
dTdP o
2
2 12 +Δ−Δ+Δ
=Δ
Δ=
→
→
βα
βα (3.86)
La cual se integra por separación de variables como:
dTTC
Tcb
Ta
RPdP o
⎥⎦
⎤⎢⎣
⎡+
Δ−
Δ+
Δ=
2321 (3.87)
Integrando,
( ) ⎥⎦
⎤⎢⎣
⎡−−
Δ+
Δ+Δ=⎟⎟
⎠
⎞⎜⎜⎝
⎛1
02
0 2ln1ln C
TC
TcTbTa
RPP (3.88)
Donde la constante de integración esta dada por:
( )⎥⎥⎦
⎤
⎢⎢⎣
⎡−
Δ+
Δ+Δ=
0
02
0001 2
lnTC
TcTbTaC (3.89)
Los coeficientes cba ΔΔΔ ,, son obtenidos empíricamente de información de capacidad calorífica definida anteriormente, y donde T0 y P0 son coordenadas de un punto conocido en la curva de saturación.

Apuntes de TF-1122 - Susana Curbelo 3.28
Esta ecuación describe la curva de saturación en el rango que la descripción empírica de la capacidad calorífica y aproximación de gas ideal son válidas. Si por otro lado, la misma es integrada suponiendo un rango de entalpía constante, entre dos temperaturas y presiones conocidas, da:
⎟⎟⎠
⎞⎜⎜⎝
⎛−
Δ−=⎟⎟
⎠
⎞⎜⎜⎝
⎛
00
11lnTTR
HPP (3.90a)
o
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛−
Δ−=
00
11expTTR
HPP (3.90b)
La anterior es válida en rangos de interés restringidos a algunos cientos de grados Kelvin, rango en el cual el cambio de entalpía relacionado a la vaporización o sublimación es de 100 kJ. Por lo cual, el cambio de entalpía del proceso puede ser supuesto independiente de la temperatura: ( ) ( ) ( ) ( )oo THTHTHTH Δ=Δ⇒=Δ−Δ 0
A presiones por debajo de 1 atm es posible calcular, por tanto, el calor de vaporización (o sublimación) de la pendiente de la curva de presión de vapor, en un gráfico del logaritmo de la presión vs el inverso de la temperatura.
3.8.2.2 Curvas de saturación entre fases condensadas Como ya se comentó, la variación de SHV ΔΔΔ ,, con T y P está relacionada con la diferencia entre capacidades caloríficas y coeficientes de expansión y compresión de las dos fases involucradas. Un valor aproximado de esas curvas puede ser obtenido despreciando la dependencia de esos términos con T y P. De forma que quede:
VS
TP
ΔΔ
=ΔΔ (3.91a)
o
( )00 TTVSPP −
ΔΔ
=− (3.91b)
Donde T0 y P0 es un punto conocido de la curva, generalmente la temperatura de equilibrio a 1 atm. De esta suposición, la curva de saturación es una línea recta que
pasa por (T0, P0) y tiene pendiente VS ΔΔ . Alternativamente, se puede obtener una solución equivalente a la expresión 3.89 por integración de:
TdT
VHdP
VTH
dTdP
ΔΔ
=⇒Δ
Δ=
Quedando,

Apuntes de TF-1122 - Susana Curbelo 3.29
⎟⎟⎠
⎞⎜⎜⎝
⎛ΔΔ
=−0
0 lnTT
VHPP (3.92)
Resolviendo el logaritmo por expansión en serie:
...1311
211ln
3
0
2
000−⎟⎟
⎠
⎞⎜⎜⎝
⎛−+⎟⎟
⎠
⎞⎜⎜⎝
⎛−−⎟⎟
⎠
⎞⎜⎜⎝
⎛−=⎟⎟
⎠
⎞⎜⎜⎝
⎛TT
TT
TT
TT
(3.93)
y dado que el rango de temperatura considerado es pequeño comprado a T0, luego:
⎟⎟⎠
⎞⎜⎜⎝
⎛ −ΔΔ
=−0
00 T
TTVHPP (3.94)
La cual es idéntica a la ecuación 3.92, ya que: 0THS Δ=Δ
Para el cálculo preciso de la curva entre dos fases condensadas, se requieren valores experimentales de las capacidades caloríficas y coeficientes de expansión y compresión como función de T y P para ambas fases.

Apuntes de TF-1122 - Susana Curbelo 3.30
Caso curioso: Curva de Fusión del Agua
Como se mencionó en los diagramas de fases unarios la pendiente de la curva de fusión del agua es negativa. El agua es una de las pocas sustancias que presenta este comportamiento, el otro es el bismuto, el resto de las sustancias tiene curvas de fusión de pendiente positiva. La consecuencia termodinámica de este comportamiento puede ser analizada a partir de la ecuación de Clapeyron:
VS
TP
G ΔΔ
=⎟⎠
⎞⎜⎝
⎛∂∂
Δ
Donde en este caso los deltas de S y V se refieren a los del proceso de fusión o congelación. Por ejemplo para el proceso:
H2O(s) → H2O(l)
Una pendiente negativa en el diagrama, implica que dicha derivada es negativa
también: 0<ΔΔ
=⎟⎠
⎞⎜⎝
⎛∂∂
Δ VS
TP
G
Como se sabe que el cambio de entropía para el proceso de fusión debe ser positivo, debido al que el sistema está yendo desde un estado sólido cristalino altamente ordenado a uno mas desordenado, estado líquido, calculado como pffusion THΔ . Luego solo existe una vía para que dicha derivada sea negativa y es que el cambio de volumen ∆V sea negativo. Esto significa que el volumen molar del líquido es menor que el volumen molar del sólido, en el punto de fusión. Por lo tanto, la densidad, que es su recíproco, es mayor para el agua líquida que para la fase sólida.
Lo cual es un resultado que va en contra de la intuición, ya que se esperaría que los materiales expandan al derretirse. Pero que resulta en una consecuencia esencial para la vida en el planeta ya que si sol no puede incidir sobre las capas de hielo depositadas, ahora, en el fondo, los lagos, ríos y mares se congelarían, así como toda vida vegetal y animal.
Figura 3.10 Foto de un iceberg

Apuntes de TF-1122 - Susana Curbelo 3.31
3.9 Estimación de Propiedades de Saturación
3.9.1 Presión de Vapor
La presión de vapor es una medida de la habilidad de las moléculas para escapar de la superficie de un sólido o líquido. Un sólido o un líquido mantienen unidas sus moléculas debido a la presencia de fuerzas intermoleculares atractivas. Sin embargo, dichas moléculas poseen cierta energía cinética, la cual es proporcional a la temperatura absoluta del sistema, por lo que existe una competencia entre ambos fenómenos. La presión de vapor es el resultado de dicha competencia, y aumenta proporcionalmente con la temperatura del sistema.
El comportamiento de la presión de vapor con la temperatura puede ser obtenido de la integración de la ecuación de Clausius-Clapeyron (3.87), pero la misma implica el conocimiento de los calores de vaporización o sublimación.
Como siempre existen ecuaciones de naturaleza empírica cuyos parámetros dependen del material en cuestión y de las condiciones a la que los mismos fueron ajustados. En general, la forma de dichas ecuaciones se basa en el hecho que el comportamiento del
satPln vs T1 entre el punto triple y el crítico, es prácticamente lineal, por tanto, tienen la forma de:
TBAP Sat −=ln (3.95)
Donde A y B son constantes que dependen de la especie. Sin embargo, esta es una aproximación burda, por lo cual se han desarrollado otras ecuaciones que dan mejores resultados, tal como la Ecuación de Antoine:
CTBAP Sat
+−=ln (3.96)
De la cual sus constantes están disponibles en la literatura para una amplia variedad de constantes, que son válidas para un intervalo dado de temperatura.
Expresiones para el cálculo exacto de la misma suelen ser de mayor complejidad. Una de las más reconocidas al respecto es la ecuación de Wagner la cual se expresa como una función de la temperatura reducida:
τττττ
−+++
=1
ln635.1 DCBAP Sat (3.97)
Con rT−= 1τ y A, B, C y D constantes

Apuntes de TF-1122 - Susana Curbelo 3.32
Figura 3.3 Presión de vapor para varias sustancias
3.9.2 Calor de Latente de Vaporización y Fusión Cuando una sustancia pura en estado sólido se licua, o vaporiza a partir de un estado líquido a presión constante, no hay cambio observable de temperatura, sin embargo, el proceso requiere de la transferencia de una cantidad finita de calor a la sustancia. Estos efectos térmicos son llamados calor latente de fusión y calor latente de vaporización. De forma similar existen calores latentes de transición que acompañan el cambio de una fase sólida a otra, por ejemplo, el calor adsorbido cuando los cristales rómbicos de azufre cambian a una estructura monoclínica a 95 C y 1 bar, es 360 J/g. El calor latente que acompaña a un cambio de fase es función únicamente de la temperatura y está relacionada con otras propiedades termodinámicas a través de la ecuación exacta de Clapeyron:
dT
dPVTHsat
Δ=Δ (3.98)
Con ella se pueden calcular los valores de los calores latentes a partir de la presión de vapor y los datos volumétricos de la sustancia. Experimentalmente esto se hace a través de mediciones calorimétricas.

Apuntes de TF-1122 - Susana Curbelo 3.33
3.9.3 Punto Triple
En un diagrama P-T tres fases pueden coexistir en equilibrio solo en un punto aislado, representado por la intersección de las tres superficies, (para las fases gas, líquido y sólidos) de potencial químico. El punto triple, además es la intersección de las curvas S+L, S+G y L+G.
Si el sistema posee una cuarta fase con una superficie de potencial químico bajo este punto triple, luego el S+L+G punto triple es metaestable y no aparece en el diagrama de fase. Si no es el caso, el punto triple es estable y si aparece.
Este punto satisface la ecuación de Clausius-Clapeyron simultáneamente para todas las fases.
Además, es característico del punto triple que el cambio de las propiedades de estado para las transiciones estén relacionadas. Por ejemplo, para la transición sólidos–gas es la suma del cambio de sólido a líquido más el cambio de líquido a gas. Ya que el cambio es independiente del camino. Por tanto:
( ) ( ) LT
GLT
LLGGT
LLGGGT
VVVVVVV
VVVVVVV→→→
→
Δ+Δ=−+−=Δ
−+−=−=Δααα
ααα (3.99a)
GLT
LST
GST HHH →→→ Δ+Δ=Δ (3.99b)
GLT
LST
GST SSS →→→ Δ+Δ=Δ (3.99c)
Esto indica que el cambio de V, S o H, para la formación directa de vapor a partir de sólido, es idéntica a la obtenida para un proceso en el cual el sólido primero funde y luego se vaporiza.
Al considerar tres fases en equilibrio, por ejemplo S+L+G, como este punto es la intersección de las curvas de sublimación, fusión y vaporización, el valor de la presión y temperatura de este punto puede ser determinado, para equilibrios que involucran la fase vapor, a partir de la ecuación de Clausius-Clapeyron integrada a cambio de entalpía constante (en las cercanías del punto triple esta suposición es válida):
⎟⎟⎠
⎞⎜⎜⎝
⎛−
Δ=⎟⎟
⎠
⎞⎜⎜⎝
⎛
00
11lnTTR
HPP
Por tanto, para el equilibrio sólido-vapor, la presión de vapor se puede despejar de:
⎟⎠⎞⎜
⎝⎛ Δ−=
→
RTHAP
VSSs exp (3.100a)
y para el equilibrio líquido-vapor, de:
⎟⎠⎞⎜
⎝⎛ Δ−=
→
RTHAP
VLVV exp (3.100b)

Apuntes de TF-1122 - Susana Curbelo 3.34
Donde SV AA , son constantes que dependen del punto conocido o de referencia a partir del cual se integró la ecuación de Clausius-Clapeyron.
Como en el punto triple (TT, PT) se unen ambas curvas, se puede resolver la intercepción de las ecuaciones anteriores para despejar el valor de la temperatura de ese punto:
( )VS
VS
TAARHHT
lnΔ−Δ
= (3.101)
⎟⎟⎠
⎞⎜⎜⎝
⎛
Δ−Δ
Δ=
SV
SV
THH
HAP exp (3.102)
La forma mas fácil de calcular estas constantes y determinar luego el punto triple, es darse cuenta que las curvas de vaporización y sublimación son rectas en un gráfico del ln(P) vs (1/T) en el rango de bajas presiones.
3.9.4 Punto de Fusión y Ebullición
El punto de fusión es la temperatura del momento en el cual una sustancia pura pasa del estado sólido al estado líquido a la condición de 1 atm de presión. El proceso inverso se denomina punto de congelación de un líquido es la temperatura a la que dicho líquido se solidifica debido a una reducción de temperatura.
El material con el más alto punto de fusión es el grafito, con un punto de fusión de 3,948 K. En el caso del agua, el punto de fusión y de congelación es el mismo: 0 ºC (32º F), ya que hay presencia de histéresis. Este valor es en presencia de núcleos de cristalización en el líquido ya que sin éstos el agua líquida puede enfriarse hasta −42 °C (−43,6 °F) sin que se produzca la congelación en un proceso llamado superenfriamiento.
De forma similar el punto de ebullición de un compuesto puro es la temperatura que debe alcanzar este para pasar del estado líquido al estado gaseoso a la condición de 1 atm de presión. El proceso inverso se denomina punto de condensación. Para el agua esta temperatura es 100 °C.
Una definición alternativa para ambos Puntos (de Fusión y de Ebullición), sería las temperaturas a la cual la presión de vapor en equilibrio es 1 atm.
Caso curioso: Superenfiamiento y Supercalentamiento
Como se comentó, el punto de congelación del agua es de 273 K (0 °C), sin embargo, el agua se puede superenfriar a presión ambiental, si se enfría a un ritmo del orden de 1 millón de grados K por segundo. En este caso, se puede evitar la nucleación cristalina hasta lograr su nucleación homogénea, a casi 231 K (−42 °C), aquí el agua se convierte en un vidrio. Su temperatura de transición vítrea es muy inferior y difícil de determinar, pero hay estudios que la estiman en unos 165 K (−108 °C). El agua vítrea se puede calentar hasta aproximadamente 150 K (−123 °C). En el rango de

Apuntes de TF-1122 - Susana Curbelo 3.35
temperaturas entre 231 K (−42 °C) y 150 K (−123 °C), los experimentos sólo han logrado hielo cristalino [1,2].
Por ejemplo, en los estratos y los cúmulos, a menudo existen gotas de agua superenfriada. Cuando son golpeadas por el viento de un avión que pasa, cristalizan abruptamente formando hielo. Esto causa problemas en el despegue, por lo que los aviones que van a viajar en esas condiciones deben tener un sistema antihielo.
De forma similar, es posible supercalentar un líquido por encima de su punto de ebullición sin que se haga gaseoso. En física, supercalentar (a veces llamado retardación de la ebullición o defervescencia), es el fenómeno por el que un líquido se calienta a una temperatura superior a su punto de ebullición normal sin que se produzca ebullición. Esto se puede conseguir calentando rápidamente una sustancia homogénea sin perturbarla (para evitar introducir burbujas de agua en los puntos de nucleación). Como un fluido supercalentado es resultado de circunstancias artificiales, está en un estado metaestable, y puede decaer en cuanto desaparezcan las circunstancias, dando lugar a que el líquido ebullicione súbita y violentamente, una situación muy peligrosa.
A veces el supercalentamiento es una preocupación relacionada con los hornos microondas. Un recipiente lleno de agua supercalentada en un microondas podría quemar fácilmente a una persona, al hacer contacto con el vaso o al verter sustancias como café instantáneo o azúcar. Que pueden provocar que el agua hirviendo salga proyectada hacia arriba. La probabilidad de que se produzca supercalentamiento aumenta si el recipiente es muy liso, como un vaso de vidrio nuevo que no tenga arañazos (los arañazos pueden alojar pequeñas bolsas de aire que sirven de punto de nucleación; esto no equivale a decir que un recipiente antiguo es automáticamente seguro). Para evitar esto, se puede meter algo como un palo de helado de madera antes de calentar; los platos giratorios de los microondas modernos también pueden proporcionar la suficiente perturbación y evitar el supercalentamiento.
Figura 3.11 Efectos del Supercalentamiento de agua en un microondas

Apuntes de TF-1122 - Susana Curbelo 3.36
Ejercicios Resueltos
E1. Determine una expresión para la presión de vapor del agua líquida si se conoce:
( )KJTTCVP /10.33.010.7.1030 253 −− ++= entre 298-2500K
( )KJCLP /44.75= entre 273-373 K
( )molJHvapK /41090373 =Δ a 1 atm
Solución:
Sabemos que: dTRT
HPdP
2Δ
= o ( )2
lnRT
HdT
Pd Δ=
Donde: ( ) CpdTHdCpdTdH Δ=Δ⇒=
Por tal razón: ( )KJTxTCCC LP
VP
VLP /1033,010.7,1044,45 253 −−→ ++−=−=Δ
∫∫ →→ ΔΔ=Δ=ΔT
VLP
vapT
VLP
vapT dTCHdTCH
373373
0
( ) ( )
TTT
TTTHvap
T
523
5223
10.33,010.35,544,4558872
3731110.33,037310.35,537344,4541090
−+−=
⎟⎠
⎞⎜⎝
⎛ −−−+−−+=Δ
−
−
Luego podemos integrar la ecuación de Clausius-Clapeyron: ( )2
lnRT
HdT
Pd Δ=
CT
TTTR
P +⎟⎟⎠
⎞⎜⎜⎝
⎛−+−= −
2
53
210.33.010.35.5ln44.45588721ln
La constante de integración puede ser evaluada a un punto de presión conocida como el punto de ebullición normal del agua:
092,51373,1 =⇒== CKTatmP

Apuntes de TF-1122 - Susana Curbelo 3.37
E2. La presión de vapor del Zinc sólido varía con la temperatura según:
25,19ln755,015775)(ln +−−= TT
atmPsub
y la presión de vapor del Zinc líquido según:
79,21ln255,115246)(ln +−−= TT
atmPvap
Calcule:
a. La temperatura de ebullición normal del Zinc líquido b. La temperatura y presión del punto triple c. El calor de vaporización del zinc a la temperatura de ebullición normal d. El calor de fusión del zinc a la temperatura del punto triple e. La diferencia entre las capacidades caloríficas del Zinc en la fase líquida y sólida
Solución:
a. La temperatura de ebullición normal del Zinc se define como la temperatura a la
cual la presión de vapor del zinc líquido es igual a 1 atm. Este valor se halla evaluado la ecuación de equilibrio líquido-vapor Ec (2) a 1 atm:
KTTT
atm B 1181079212551152461 =⇒=+−−= ,ln,)ln(
b. El punto triple representa la intersección de la curva de saturación líquido-vapor con la curva de saturación sólido-vapor. En dicho punto el sólido, líquido y vapor del zinc están en equilibrio a la misma temperatura y presión. Para ello basta con interceptar ambas ecuaciones:
2519755015775921255115246 ,ln,.,ln, +−−=+−− TT
TT
T
Tln,, 50542529
=−
Despejando: KTPT 708= para determinar la presión basta con sustituir en cualquiera de las dos ecuaciones: atmP 00034230,= Recordar que el calor de vaporización del zinc está relacionado con la presión de vapor según la ecuación de Clausius-Clapeyron:
dTRT
HPdP
2Δ
= o ( )2
lnRT
HdT
Pd Δ=
Donde la presión de vapor del líquido (vaporización) es:

Apuntes de TF-1122 - Susana Curbelo 3.38
7921255115246 ,ln,)ln( +−−= TT
Pvap y su derivada con respecto a T:
TTdTPd 255.115246)ln(
2 −=
Entonces: 22255115246
RTH
TTdTPd Δ
=−=,)ln(
( ) TTHvap 43.10126760255.1152463144.8 −=−=Δ
Evaluando a la temperatura de ebullición normal tenemos que:
molJHvapK /,4401141181 =Δ
c. Como no disponemos de información directa sobre el equilibrio sólido-líquido
(fusión), el calor de fusión del zinc a una temperatura dada puede ser estimado partiendo del conocimiento que:
VLT
VST
LST
VST
VLT
LST HHHHHH →→→→→→ Δ−Δ=Δ⇒Δ=Δ+Δ
Para ello necesitamos los valores de los calores de vaporización y sublimación del zinc:
Similarmente, a partir de la presión de vapor del sólido (fusión):
25.19ln755.015775)(ln +−−= TT
atmP
Entonces: 22755.015775)ln(
RTH
TTdTPd Δ
=−=
( ) )/(277.6131160755.0157753144.8 molJTTHvap −=−=Δ
( ) TTTHHH VLT
VST
LST 153.4440043.10126760277.6131160 +=−−−=Δ−Δ=Δ →→→
( ) )/(7340708153.44400708 molJH LSK =+=Δ →
d. La diferencia entre las capacidades caloríficas del Zinc en la fase líquida y sólida
( ) CpdTHdCpdTdh Δ=Δ⇒=
( ) ( ) ( )molJdT
TddTHdCCCp
LSSP
LPLS /,, 153415344400
=+
=Δ
=−=Δ→
→

Apuntes de TF-1122 - Susana Curbelo 3.39
E3. El carbón posee dos alótropos, grafito y diamante. A 25 oC y 1 atm el grafito es la forma más estable. Calcule la presión que debe ser aplicada al grafito a 25 oC de forma de llevarlo a diamante, si se conoce:
)/(1900298298 molJHH DK
GK −=− ,
)/(,
)/(,
molKJS
molKJSD
K
GK
432
735
298
298
=
=,
)/(,
)/(,
35153
3222
298
298
cmgmol
cmgmolD
K
GK
=
=
ρ
ρ
Solución:
Se necesita calcular la presión a la cual el grafito cambia a diamante a 298 K, para ello debemos relacionar P con V, S y H. Es más cómodo relacionar P y T con G, y G con H y S. Ya que en saturación, el cambio de la energía libre de Gibbs molar es cero.
Para la transformación de fase de grafito a diamante a 298 K, se requiere:
( ) )/(,, molJsThgDG
288373543229819000 =−−=Δ−Δ=Δ→
Para el mismo cambio de fase a 298 K el cambio volumétrico viene dado por:
GDDG
T
DGvvv
Pg
−=Δ=⎟⎟⎠
⎞⎜⎜⎝
⎛
∂Δ∂ →
→
Donde: ρρρ
1211===
PM
v de aquí obtenemos: )/(,
)/(,
molcmv
molcmvD
G
3
3
4153
4055
=
= por tanto:
)/(, molcmv DG 3991−=Δ →
Como en el equilibrio de fase el cambio de la energía libre de Gibbs molar es cero, debemos buscar cual presión satisface esta condición a 298 K:
Integrando:
( ) ( ) ∫∫ Δ+==Δ=Δ==Δ →PP
DG vdPKTatmPgvdPKTPg1
0
0
2981298 ,,
Si consideramos modelo incompresible para el carbón el volumen no es función de la presión y:
( ) ( ) 0879
1991288328832983
≡⎟⎠⎞
⎜⎝⎛
−−+=ΔΔ+==Δ →
atmcm
PPvKTPg DG
,
,,
atmxP 1430099.1
87.928831 =−
−=−

Apuntes de TF-1122 - Susana Curbelo 3.40
E4. Usando las relaciones entre temperatura y presión de vapor para CaF2(α), CaF2(β), CaF2(L), calcule:
a. La temperaturas y presiones de los ptos triples para los equilibrios: CaF2(α) - CaF2(β) - CaF2(v) CaF2(β)- CaF2(l)- CaF2(v)
b. La temperatura normal de ebullición del CaF2 c. El calor latente de transformación CaF2(α) CaF2(β) d. El calor latente de fusión de CaF2(β)
De los apéndices: CTBTAatmP ++−= ln)(ln donde:
Sustancia A B C Rango CaF2(α) 54.35 -4.525 56.57 298-1430 CaF2(β) 53.78 -4.525 56.08 1430-1691 (Tm) CaF2(l) 50.20 -4.525 53.96 1691-2783 (Tb)
Solución:
a. Ptos. Triples:
Para el equilibrio (α−β-v):
)(ln08.56ln525.478.5357.56ln525.435.54)(ln atmPTT
TT
atmP βα =+−−=+−−=
y despejo T=1163 K y P=2.52x10-10 atm Para el equilibrio (α-L-v):
)(ln96.53ln525.42.5057.56ln525.435.54)(ln atmPTT
TT
atmP L=+−−=+−−=α
y despejo T=1689 K y P=8.35 x10-5 atm
b. Para el pto de ebullición normal:
KTTT
atmP BL 277609653525420501 =⇒=+−−= .ln..)(ln
c. Calor latente molar de transformación (α−β):
⎥⎦
⎤⎢⎣
⎡−=Δ
+−−=
→
TTRTH
TT
atmP
v 525.435.54
57.56ln525.435.54)(ln
22α
α

Apuntes de TF-1122 - Susana Curbelo 3.41
⎥⎦
⎤⎢⎣
⎡−=Δ
+−−=
→
TTRTH
TT
atmP
v 52547853
085652547853
22 ..
.ln..)(ln
β
β
Por tanto: RH
HHH vv
57.0=Δ
Δ−Δ=Δ→
→→→
βα
βαβα
e. Calor latente molar de fusión del CaF2(β):
Ya se calculó: ⎥⎦
⎤⎢⎣
⎡−=Δ →
TTRTH v 52547853
22 ..β se necesita vLH →Δ
⎥⎦
⎤⎢⎣
⎡−=Δ
+−−=
→
TTRTH
TT
atmP
vL
L
52542050
965352542050
22 .,
.ln..)(ln
Por tanto: RH
HHHL
vLvL
583,=Δ
Δ−Δ=Δ→
→→→
β
ββ

Apuntes de TF-1122 - Susana Curbelo 3.42
REFERENCIAS BIBLIOGRÁFICAS 1 P. G. Debenedetti, P. G., and Stanley, H. E.; "Supercooled and Glassy Water", Physics Today 56 (6), 40–46 (2003) 2 Giovambattista, N., Angell, C. A., Sciortino, F. and Stanley, H. E.; "Glass-Transition Temperature of Water: A Simulation Study", Physical Review Letters 93 (4) 047801, (2004).