enfermedades mitocondriales
TRANSCRIPT

SEDE - PISCO
ESCUELA ACADEMICA PROFESIONAL DE OBSTETRICIA
CICLO: III
CURSO: Bioquímica y nutrición.
ALUMNA: Huamani Villa, Josselin Adriana.
DOCENTE: Marisol Quispe Nuñez
2015
Enfermedades mitocondriales

Las enfermedades mitocondriales son desordenes resultantes de la deficiencia de una o mas proteínas localizadas en el mitocondrias e involucradas en el metabolismo celular.
DEFINICIÓN :

Son orgánelos celulares ubicados en el citoplasma, su función es la apoptosis y TRIFOSFATO DE ADENOSINA que es la energía utilizada por las células para realizar sus actividades.
MITOCONDRIA :
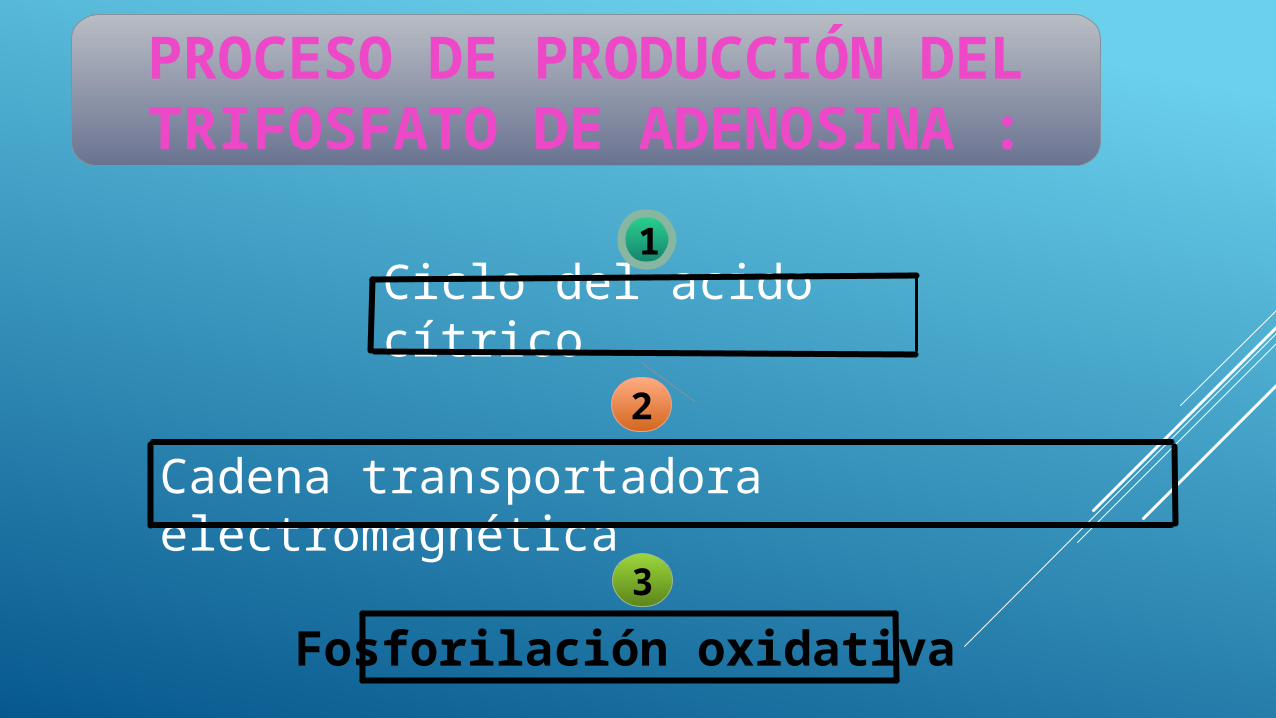
Ciclo del acido cítrico
PROCESO DE PRODUCCIÓN DEL TRIFOSFATO DE
ADENOSINA :1
2
Cadena transportadora electromagnética
3
Fosforilación oxidativa
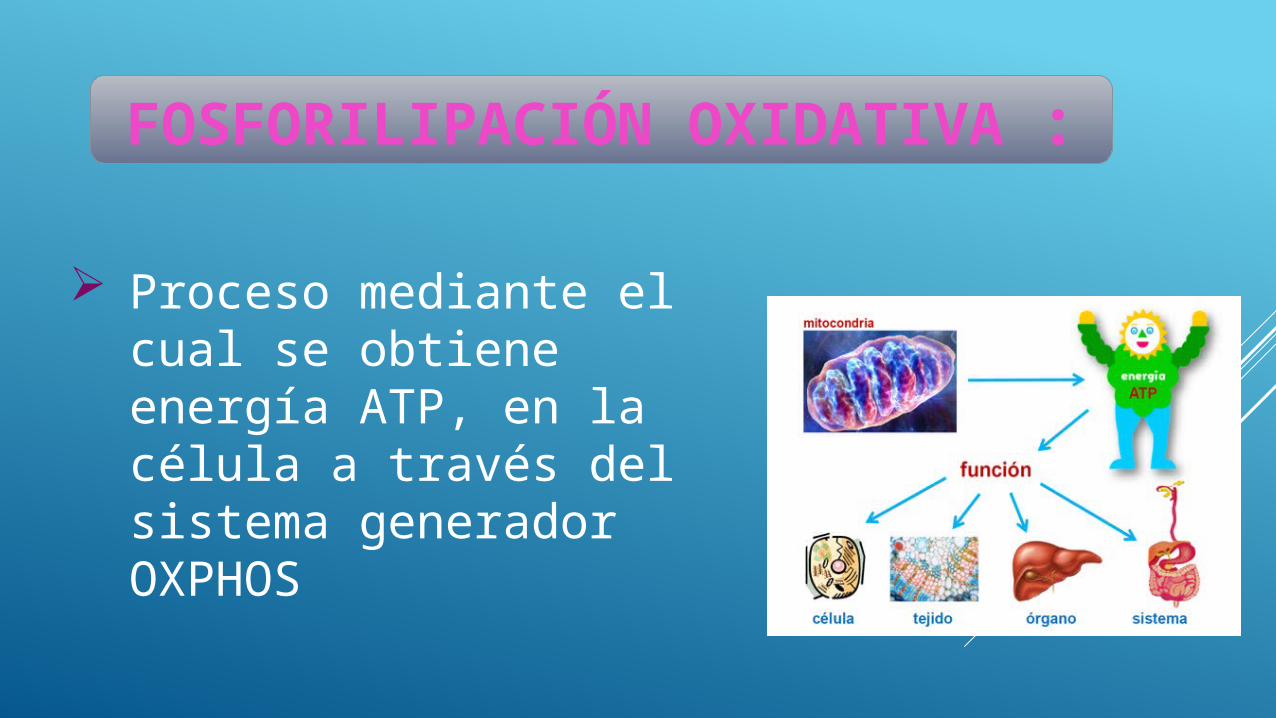
Proceso mediante el cual se obtiene energía ATP, en la célula a través del sistema generador OXPHOS
FOSFORILIPACIÓN OXIDATIVA :

No hay una característica única para identificar una enfermedad mitocondrial. Los pacientes presentan varios problemas que pueden surgir desde el nacimiento hasta en la edad adulta madura. Piense en las mitocondrias cuando:
Una “enfermedad común” presente características atípicas que la distingan del resto.
Haya tres o más órganos involucrados Se presenten recaídas recurrentes, o cuando
ocurran brotes de infección en una enfermedad crónica.
CUÁNDO SOSPECHAR QUE HAY UNA DIFUCIÓN MITOCONDRIAL

Encefalopatía Convulsiones Retraso en el Desarrollo o Regresión
(incluyendo demencia temprana o episodios tardíos)
Desórdenes de Movimiento (distonia, disquinesias, corea, etc.)
Migraña Complicada Infartos
SINTOMAS:

Neuropatía Defectos en los Conductos Cardiacos
Cardiomiopatías
Deficiencias en la Audición Estatura corta Desórdenes de Músculos Extraoculares
tosis estrabismo adquirido Oftalmoplegia
Diabetes Enfermedad Renal Tubular Pérdida de la Visión
retinitis pigmentosa atrofia óptica
Acidosis láctica (puede ser leve)

DIAGNOSTICO:

Deficiencia del Complejo I Nombre completo: Deficiencia NADH dehidrogenasa (NADH-Co!
Reductasa) Síntomas: Tres formas principales: Desorden fatal infantil multisistémico, caracterizado por un retraso en
el desarrollo, debilidad muscular, enfermedad cardiaca, acidosis láctica congénita y paro respiratorio.
Miopatía que inicia en la infancia o en la vida adulta, manifestándose como intolerancia al ejercicio o debilidad. Es común la elevación del ácido láctico.
Encefalomiopatía mitocondrial (incluyendo MELAS), que puede comenzar en la infancia o en la edad adulta y que consiste en combinaciones variables de síntomas y signos, incluyendo oftalmoplegia, convulsiones, demencia, ataxia, pérdida auditiva, retinopatía pigmentosa, neuropatía sensorial y movimientos incontrolables. Puede provocar el Síndrome de Leigh.

Deficiencia del Complejo III Nombre completo: Deficiencia ubiquinona-citocroma c
óxidoreductasa Síntomas: Cuatro formas principales: Encefalomiopatía mortal infantil, acidosis láctica congénita,
hipotonia, postura distrófica, convulsiones y coma. Es común la fibrosis roja.
Encefalomiopatía de brotes posteriores (desde la infancia hasta la edad adulta): varias combinaciones de debilidad, estatura corta, ataxia, demencia, pérdida auditiva, neuropatía sensorial, retinopatía pigmentosa y síntomas piramidales. Es común la fibrosis roja. Posibilidad de acidosis láctica.
Miopatía, con intolerancia al ejercicio que deviene en debilidad continua. Es común la fibrosis roja. Posibilidad de acidosis láctica.
Cardiomiopatía histiocitoide infantil.
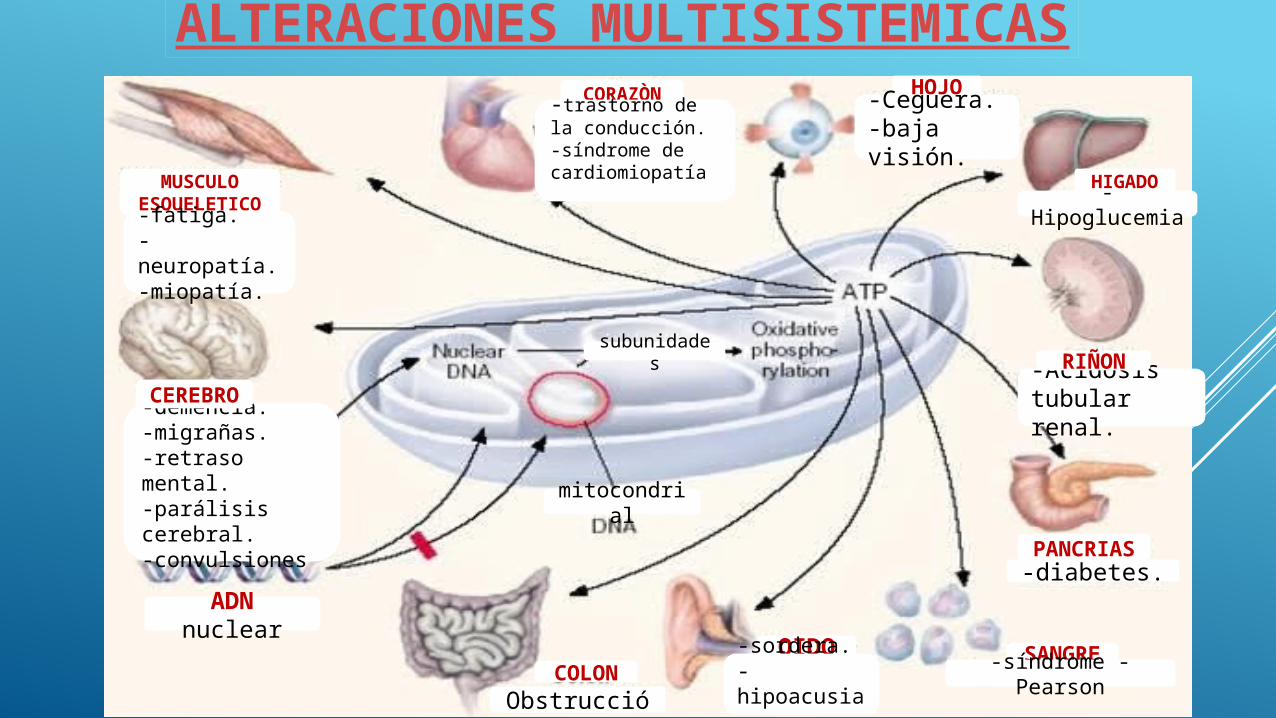
ALTERACIONES MULTISISTEMICAS
MUSCULO ESQUELETIC
O-fatiga.-neuropatía.-miopatía.
CORAZÒN-trastorno de la
conducción.-síndrome de
cardiomiopatía.
HOJO-Ceguera.-baja visión.
-demencia.-migrañas.-retraso mental.-parálisis cerebral.-convulsiones
-Hipoglucemia
CEREBRO
HIGADO
OIDO
-Acidosis tubular renal.
-diabetes.
PANCRIAS
RIÑON
-sordera.-hipoacusia.
SANGRE
subunidades
ADN nuclear
-síndrome - Pearson-
Obstrucción
COLON
mitocondrial

La enfermedad de ALPERS se describió como una afectación cerebral difusa de presentación infantil
CAUSA:
El síndrome se debe a una alteración del metabolismo oxidativo en el que aparece implicado una depleción (menor cantidad de copias de ADN de las necesarias) del ADN y una mutación en el gen de la polimerasa.
ENFERMEDAD DE ALPERS

•Convulsiones.
•Demencia.
• Espasticidad.
•Ceguera
•Disfunción del hígado.
•Atrofia cerebral.
SINTOMAS :

Acrónimo de Neuropatía, Ataxia y Retinitis pigmentosa sin fibras rasgadas en la biopsia muscular.
CAUSA:Se asocia a una mutación del ATP; aunque este trastorno suele ser de inicio juvenil.
SINDROME DE NARP

SÍNDROME de epilepsia mioclònica asociada a fibras rojas rasgadas.
no siempre que se detecta esta mutación es un síndrome de NERRF, en ocasiones presenta que se manifiesta en otros cuadros como el síndrome de LEIGH (la degeneración espinocerebelar ).
SINDROME DE MERRF

•Epilepsia.
•Ataxia progresiva.
•Debilidad.
•Degeneración muscular.
•Sordera.
•Demencia.
SINTOMAS :

Se debe a una mutación del ADN que afecta a 3 genes, codificantes de una subunidad de complejo I lo que provoca el fallo de este.
Esta enfermedad se considera el paradigma de las neuropatías óptica de causa mitocondrial.
ENFERMEDAD DE LEBER

•CEGUERA .
•Demencia leve.
•Espasticidad.
•Neuropatía Periférica.
•Defectos en los conductos cardiacos.
SINTOMAS :

CAUSA: Se debe a una selección del ADN, en
los que siempre se ven afectados unos nucleótidos concretos, suele eliminar 5ARN mitocondriales y 7 proteínas implicadas en la fosforilaciòn oxidativa.
SINDROME DE KEARNS

• Bloqueo del corazón.
• Elevada proteína cerebroespinal.
• Alteraciones endocrinas.
• Sordera.
• Retinopatía pigmentaria.
SINTOMAS :

Es un desorden neurodegenerativo, caracterizado por miopatía mitocondrial(M), encefalopatía(E), acidosis láctica(LA), y episodios tipo stroke.
CAUSA:
Aparece un amplio espectro de mutaciones, tanto en la codificación proteica como en la síntesis genética, localizándose el fallo, en el 80% de los casos del nucleótido adenina en la posición 3243 por un nucleótido de guanina, lo que provoca una mutación ADN, que afecta concretamente al ARN.
SINDROME DE MELAS

Cuadro clínico reconocido o sugestivo
Neuroimagen sugestiva y otros exámenes complementarios, exploración oftalmológica, electromiograma.
Estudio bioquímico en sangre, orina y LCR (si precisa): elevación de lactato, piruvato, alanina, metabolitos del ciclo de Krebs.
Pruebas dinámicas: test del esfuerzo, según edad y estado clínico.
Biopsia muscular: anatomía patológica sugestiva (fibras ragged red, acúmulos subsarcolémnicos) Mitocondrias anómalas en microscopía electrónica.
Estudio enzimático en biopsia muscular o de tejido afecto: demostración de un defecto del sistema OXPHOS.
Estudio genético en diferentes tejidos: búsqueda de mutaciones del nDNA y mDNA.
DIAGNOSTICO :

Modificar la función de la fosforilación oxidativa, mejorando la síntesis de ATP. (ubiquinona, vitamina C, vitamina K3).
Reducir el acúmulo de metabolitos tóxicos: facilita la eliminación o impide la formación de metabolitos tóxicos (carnitina).
Reducir el estrés oxidativo causado por la mala función de la fosforilación oxidativa, administrando antioxidantes (vitaminas A,E,C y ubiquinona).
Terapia nutricional: Algunas enfermedades mitocondriales pueden beneficiarse de una terapia nutricional adecuada, que proporcione a los pacientes el adecuado aporte de macronutrientes (proteínas, carbohidratos y grasas) y micronutrientes (vitaminas y oligoelementos). Es importante el conocimiento del origen de la enfermedad para aplicar la terapia nutricional más adecuada a su trastorno.
TRATAMIENTO :
