enfermedad de los hematies y trastornos hemorragicos
DESCRIPTION
Enfermedad de los Hematies y Trastornos Hemorragicos. Los glóbulos rojos. contienen en su interior la hemoglobina. Son los principales portadores de oxígeno a las células y tejidos del cuerpo. - PowerPoint PPT PresentationTRANSCRIPT

Enfermedad de los Hematies y Trastornos
Hemorragicos

Los glóbulos rojos contienen en su interior la hemoglobina. Son los principales portadores de oxígeno a las
células y tejidos del cuerpo. Tienen una forma bicóncava para adaptarse a
una mayor superficie de intercambio de oxígeno por dióxido de carbono en los tejidos.
Además su membrana es flexible lo que permite a los glóbulos rojos atravesar los más estrechos capilares.

PRODUCCIÓN
Se producen en la médula ósea, a partir de células madre que se multiplican a gran velocidad.
La producción de glóbulos rojos esta regulada por la eritropoyetina, que es una hormona producida por el riñón.

Una disminución de la oxígenación de los tejidos aumenta la producción de
eritropoyetina, que actúa en la médula ósea estimulando la producción de
glóbulos rojos.

Patología Anemias
Reducción por debajo de los limites normales de la masa eritrocitaria total circulante medido por el hematocrito o una reducción de la hemoglobina.
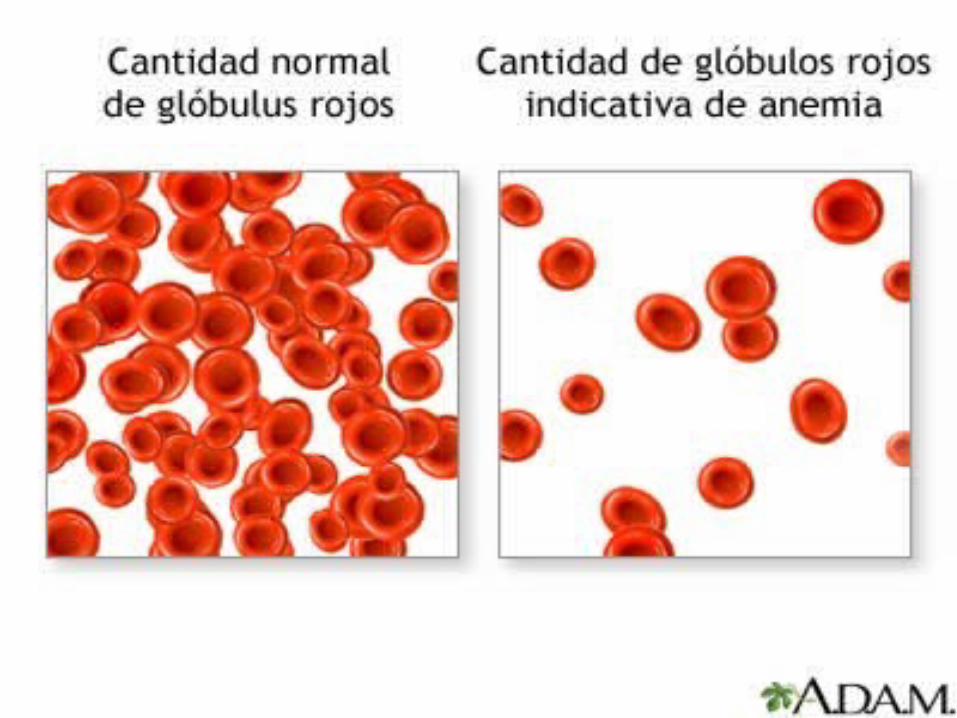

Clasificacion Etiologica de las Anemias
Anemias por Perdida de Sangre Anemias Hemolíticas Anemias Por Deficiente Producción de
Hematíes

Anemias por Pérdida de SangreHemorragia Aguda
Los efectos de la perdida de sangre aguda se deben principalmente a la perdida de volumen intravascular, lo que puede conducir a colapso cardiovascular, shock y muerte

Hemorragia Cronica La perdida de sangre solo induce anemia
cuando la rapidez de la perdida supera la capacidad regeneradora de la medula, o cuando las reservas de hierro estan agotadas.

Anemias Hemoliticas
Sus características principales son las siguientes: Acortamiento de la vida de los hematíes (normal = 120dias) Concentraciones elevadas de eritropoyetina y
eritropoyesis. Acumulación de los productos de desechos.

La anemia hemolítica es una afección en la cual hay un número insuficiente de glóbulos rojos en la sangre y es ocasionada por la destrucción prematura de éstos
se presenta cuando la médula ósea es incapaz de compensar la destrucción prematura de los glóbulos rojos por medio del aumento en su producción

Tipos de Anemia Hemolítica Anemia drepanocitica Hemoglobinuria paroxistica nocturna Anemia hemolitica por deficiencia de G-6-PD Esferocitosis hereditaria Anemia hemolitica autoinmunitaria Talasemia

Esferocitosis Hereditaria (EH) Este trastorno hereditario esta causado por
defectos intrínsecos en la membrana de los hematíes, que convierten a las células rojas en esferoides, deformables y vulnerables al secuestro y la destrucción en el bazo.

MANIFESTACIONES CLINICAS
ANEMIA ESPLENOMEGALIA ICTERICIA


ANEMIA HEMOLÍTICA DEFICIENCIA DE G-6-PD Es un defecto hereditario enzimatico, ligado
al sexo, que produce la descomposición de los glóbulos rojos.
La deficiencia de G6PD causa hemolisis episodica intravascular y extravascular.
El daño menos grave de la membrana conduce a disminución de la capacidad de deformación de los hematíes

MEDICAMENTOS QUE PUEDEN PRECIPITAR LA REACCIÓN
Medicamentos antipalúdicos Aspirina Medicamentos antiinflamatorios no esteroides
(AINES) Quinidina Quinina Sulfamidas (antibióticos) Nitrofurantoína

SIGNOS Y SINTOMAS Orina oscura Esplenomegalia Fatiga Palidez Frecuencia cardiaca rapida Dificultad para respirar Ictericia

ANEMIA DREPANOCÍTICA Es un tipo de enfermedad caracterizada
por producción de hemoglobinas defectuosas.
Esta causada por una mutación puntual en la sexta porsicion de la cadena de
globina B que conduce a sustitución de un residuo de valina por un residuo de acido glutámico

Anemia Drepanocitica Potencialmente mortal. Puede presentar “crisis” o episodios
dolorosos y agudos causados por vasos sanguíneos bloqueados y órganos dañados
Crisis repetitivas pueden ocasionar daño a Riñones, Pulmones, Huesos, Hígado y Sistema Nervioso Central

ANEMIA DREPANOCITICA

TALASEMIA
Los síndromes talasemicos forman un grupo heterogéneo de trastornos hereditarios causados por lesiones geneticas, que conducen a síntesis disminuida de las cadenas de globina A o B.

B- Talasemia La gravedad clínica de la anemia varia,
dado el carácter heterogeneo de las mutaciones causales.
La síntesis alterada de la globina B provoca anemia por dos mecanismos

B TALASEMIA El defecto de la síntesis de la HbB produce
hematíes microciticos hipocromicos subhemolizados, con capacidad de transporte de oxigeno por debajo de lo normal.
Un factor mas importante es la disminución de la supervivencia de los hematíes.

Muchos de los pacientes llegan a requerir repetidas transfusiones para evitar la gran debilidad y la descompensación cardíaca.
Frecuentemente requieren extirpación del bazo


Las principales alteraciones morfológicas afectan a la medula ósea y al bazo.
En el paciente no tranfundido existe una expansión notable de la medula hematopoyetica activa, sobre los huesos faciales.

los huesos de la cara y el cráneo se deforman dando lugar a una cara peculiar (prominencia de los dientes incisivos superiores, separación de las órbitas)
aumenta el tamaño del hígado y el bazo.

ALFA-TALASEMIA Se caracteriza por la síntesis reducida o
ausente de cadenas de Globina Alfa. Normalmente existen cuatro genes de
globina alfa. Su gravedad varia dependiendo del
numero de genes de globina alfa que se encuentren afectados.

Alfa Talasemia Estado de portador: este estado ocurre si
se elimina un solo gen de globina alfa. Se asocia con reduccion apenas detectable de la síntesis de cadenas.
Estos permanecen asintomaticos

Alfa Talasemia Rasgo de las alfa-talasemia: se debe a
deleccion de dos genes de alfa-globina. El cuadro clinico de la alfa talasemia es: MICROCITOSIS ANEMIA MINIMA O INEXISTENTE AUSENCIA DE SIGNOS FISICOS
ANORMALES.

Enfermedad de la hemoglobina H: causada por delección de tres genes de la globina alfa.
Hidropesía fetal: es la forma mas grave de la talasemia alfa, causada por delección de cuatro genes de globina alfa. En el feto el exceso de cadenas de globina alfa forma tetrameros con una afinidad tan alta por el oxigeno que no suministran casi ningún oxigeno a los tejidos.

ANEMIA MEGALOBLASTICA Las anemias megaloblasticas constituyen
un grupo de entidades diversas, que tienen en común el trastorno en la síntesis de DNA y cambios morfológicos distintivos en la sangre y la medula ósea.

El examen de sangre periférica suele revelar pancitopenia.
La hiperplasia medular que se suele observar en las anemias megaloblasticas es una respuesta a los niveles aumentados de factores de crecimiento, como la eritropoyetina.

ANEMIA PERNICIOSA Existe una marcada variación en el tamaño y la
forma de los hematíes, los neutrófilos son mas grandes de lo normal e hipersegmentados.
Los principales cambios específicos de la anemia perniciosa se encuentran en Medula Osea, Tubo Digestivo y el SNC

ANEMIA POR DEFICIENCIA DE FOLATO Existen tres causas principales de
deficiencia de acido folico: 1. Ingesta aumentada 2. Aumento de los requisitos 3. Alteraciones del uso.

Anemia Ferropénica Causada por una deficiencia de hierro debido a:1. Bajo aporte en la dieta 2. Transtorno de la absorción 3. Aumento de las necesidades4. Perdida cronica de sangre

Microciticos, Hipocromicos

ANEMIA APLÁSICA Síndrome de insuficiencia medular que
cursa con: Pancitopenia (anemia, neutropenia y
trombocitopenia. La mayoría de los casos de anemia
aplásica con etiología conocida guardan relación con la exposición a sustancia químicas y fármacos.

Anemia Aplasica El comienzo suele ser insidioso. Las manifestaciones clinicas varian
dependiendo de la linea celular predominantemente afectada.
La anemia puede causar debilidad progresiva , palidez y disnea.

POLICITEMIA Denota una concentración anormalmente
alta de hematíes, en general con un aumento correspondiente a la concentración de hemoglobina.
La policitemia relativa se origina por deshidratación: deprivacion de agua, vomitos prolongados, diarrea o abuso de diureticos.

La policitemia absoluta es primaria cuando se debe a una anomalia intrinseca de las celulas madre mieloides,
Secundaria cuando los progenitores eritroides estan respondiendo a niveles aumentados de eritropoyetina


Transtornos hemorragicos: Diatesis hemorragicas La hemorragia excesiva se puede deber
a: 1. fragilidad aumentada de los vasos 2. deficiencia o disfuncion de las plaquetas
alteracion de la coagulacion 3. combinaciones de los factores anteriores

TRASTORNOS HEMORRÁGICOS CAUSADOS POR ANOMALÍAS DE LAS PAREDES VASCULARES. Llamados a veces púrpuras no
trombocitopenicas, son relativamente frecuentes, pero no suelen causar problemas hemorrágicos graves.
La mayoría de las veces inducen a hemorragias pequeñas (petequias y purpura) en la piel o en la mucosa, sobre todo en las encías.

Anomalias de paredes de los vasos En ocasiones pueden dar lugar a
hemorragias mas importantes en las articulaciones, en los músculos y en localizaciones subperiosticas

Hemorragia Relacionada con Disminución del numero de plaquetas:
TROMBOCITOPENIA La reducción del número de plaquetas
constituye una causa importante de hemorragia generalizada.
Se considera que un recuento inferior a 100.000/ul constituye trombocitopenia.

Causas de Trombocitopenia Producción disminuida de plaquetas. Disminución de la vida media de las
Plaquetas Secuestro
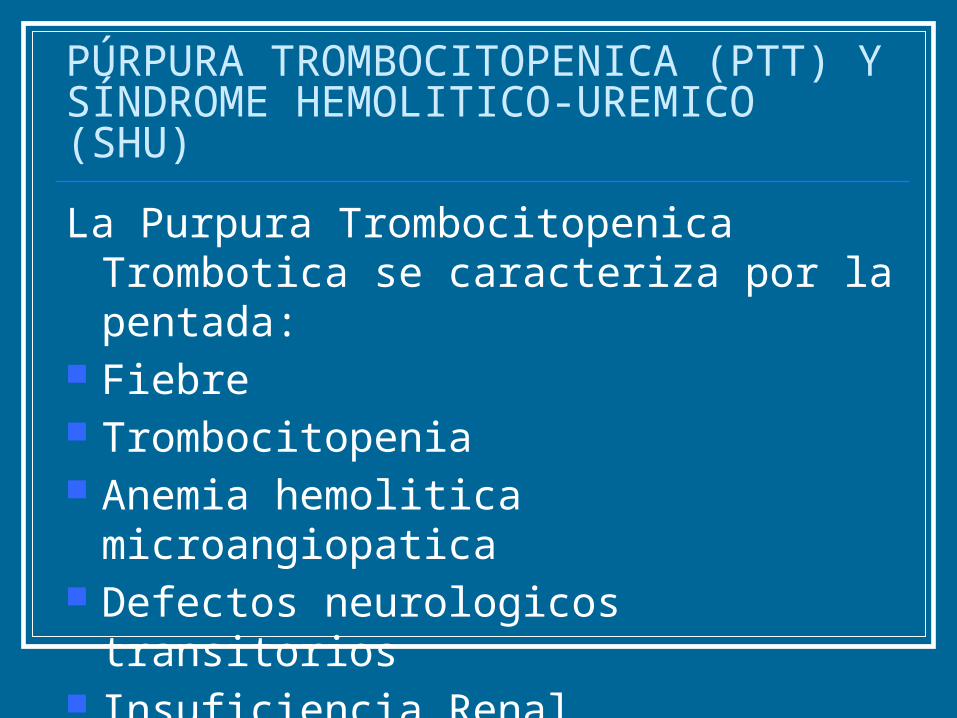
PÚRPURA TROMBOCITOPENICA (PTT) Y SÍNDROME HEMOLITICO-UREMICO (SHU)
La Purpura Trombocitopenica Trombotica se caracteriza por la pentada:
Fiebre Trombocitopenia Anemia hemolitica microangiopatica Defectos neurologicos transitorios Insuficiencia Renal.

SINDROME UREMICO HEMOLITICO El SHU tambien cursa con: Anemia Hemolitica Microangiopatica Trobocitopenia Se diferencia de la PTT por Ausencia de sintomas neurologicos Insuficiencia Renal Aguda Afección frecuente a niños.

La caracteristica fundamental comun en ambos procesos es la formación generalizada de trombos hialinos, compuestos principalmente por agregados de plaquetas, en la microcirculación

DIÁTESIS HEMORRÁGICA RELACIONADA CON ANOMALÍAS EN LOS FACTORES DE COAGULACIÓN
DEFICIENCIA DEL COMPLEJO DE FACTOR VIII-FVW
La hemofilia A y la enfermedad de von Willebrand, dos de los trastornos hemorrágicos hereditarios mas comunes, están causados por defectos culatitativos o cuantitativos que afectan el complejo factor VIII-FvW. El complejo plasmatico factor VIII-FvW dos proteinas separadas.

HEMOFILIA A La proteina procoagulante VIII es un
componente de la via intrinseca requerido para la activacion del factor X. la deficiencia de factor VIII da lugar a hemofilia A.
El factor VIII es fabricado en diversos tejidos: las celulas endoteliales sinusoidales y las celulas epiteliales tubulares y glomerulares del riñon.

ENFERMEDAD DE VON WILLEBRAND Se caracteriza por Hemorragias espontáneas en las mucosas Sangrado excesivo de las heridas Menorragia Tiempo de hemorragia prolongado, en
presencia de recuento de plaquetas normal.

COAGULACIÓN INTRAVASCULAR DISEMINADA (CID) Trastorno trombohemorrágico agudo, subagudo o
crónico, que ocurre como una complicación secundaria a diversas enfermedades.
Se caracteriza por activación de la secuencia de la coagulación, que conduce a la formación de microtrombos en la microcirculación del organismo, muchas veces con distribución por completo imprevisible.

La CID es activada por dos mecanismo:
1) Liberación de factor tisular o sustancias tromboplasticas hacia la circulación
2) Lesion generalizada de las celulas endoteliales.
Los trombos se encuentran en los siguientes sitios: encefalo, corazon, pulmones, riñones, suprarrenales, bazo e higado


ENFERMEDADES DE LOS
LEUCOCITOS, GANGLIOS
LINFATICOS, BAZO Y EL TIMO

Leucocitos y Ganglios Linfáticos Los ganglios linfáticos son glándulas que
juegan un papel importante en las defensas del cuerpo contra las infecciones. Estos producen linfa que viaja por todo el cuerpo en el sistema linfatico y filtran las impurezas del cuerpo.

Patologías Leucopenia
El número de leucocitos circulantes puede disminuir mucho en una variedad de trastornos. Un recuento de leucocitos anormalmente bajo (leucopenia) se suele deber a una disminución del numero de neutrofilos.

Neutropenia y Agranulocitosis La reducción del numero de granulocitos
de la sangre periférica (neutropenia) se puede ver en una amplia variedad de circunstancias. La reducción del numero de neutrofilos conocida como agranulocitosis, tiene consecuencias serias, puesto que convierte al individuo susceptible a infecciones.

Los signos y síntomas de las neutropenias guardan relacion con infecciones bacterianas o micoticas.
Incluyen malestar general, escalofrios y fibre, seguidos por marcada debilidad y fatiga facil.
En las agranulocitosis graves con ausencia virtual de neutrofilos, las infecciones pueden ser sobreagudas y causar la muerte en cuestion de pocos dias.


Linfadenitis inespecífica Aguda Desde el punto de vista clinico los ganglios
de la linfadenitis aguda están agrandados a causa de infiltración celular y el edema. Como consecuencia de la distencion de la capsula son dolorosos al tacto.

De forma característica los ganglios linfáticos no son dolorosos en las reacciones crónicas, puesto que sus capsulas no están sometidas a tensión aumentada.
La linfadenitis crónica es particularmente común en los ganglios inguinales y axilares, que drenan áreas relativamente grandes del cuerpo y son objeto de agresión frecuente.


Proliferación neoplasicas de los leucocitos El termino leucemia se usa para las
neoplasias linfoides que se presentan con afectación generalizada de la medula ósea, usualmente acompañada por la presencia de un gran numero de células tumorales en la sangre periférica.
Linfoma por otra parte se usa para escribir proliferaciones originadas como masas titulares discretas

Principales tipos de neoplasias linfoides:
Linfoma de Burkitt Los tejidos afectados aparecen borrados por un
infiltrado difuso de células linfoides de tamaño intermedio, que contiene nucleos redondos u ovales con cromatina.
Dentro de esta categoria se incluyen:1) linfoma de burkitt endemico 2)linfoma de burkitt esporadico, 3) un subconjunto que ocurren en afectados por VIH

Caracteristicas Clinicas: Adolescentes o adultos jóvenes con masas
mandibulares o abdominales extra ganglionares, rara vez se presenta como agresivo.


Linfoma B difuso de células grandes Existe un ligero predominio masculino, con
edad mediana alrededor de 60 años Presenta un tamaño relativamente grande
de las células y un patrón difuso de crecimiento.
Los pacientes se presentan con una masa de crecimiento rápido, frecuentemente sintomática, en un solo sitio ganglionar o extraganglionar

Pueden nacer en cualquier lugar.
La enfermedad extraganglionar puede comenzar dentro del tracto gastrointestinal, la piel, el hueso, el encéfalo y otros lugares.
La afección de la medula suele ocurrir en fases avanzadas de la enfermedad

Linfoma de células del manto Se suele presentar en las décadas quinta y
sexta de la vida y predomina en los hombres.
La proliferación consiste en una población homogénea de linfocitos pequeños con contornos nucleares entre redondos e irregulares y en ocasiones con hendiduras profundas.

La mayoria de los pacientes presentan linfadenopatia generalizada en el momento del diagnostico.
Los lugares frecuentes de afectacion extraganglionar incluyen la medula osea, ciertas zonas de la pulpa blanca esplenica, las areas periportales hepaticas y el intestino.
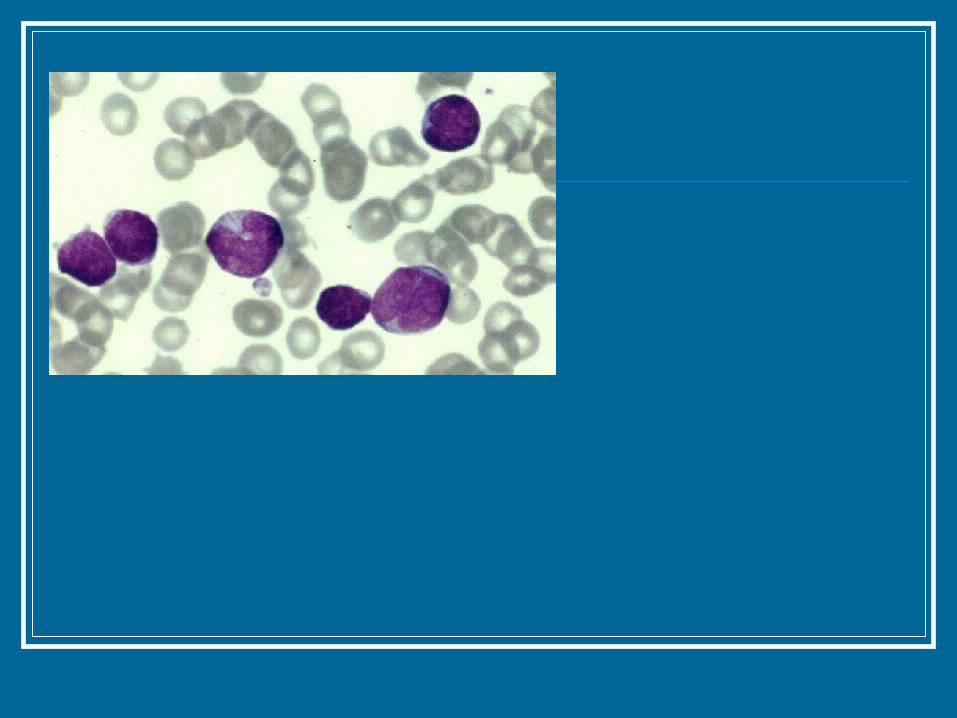

Tricoleucemia Se encuentra sobre todo en hombres de mediana
edad y raza blanca. Los tricocitos tienen núcleos redondos y
cantidades modestas de citoplasma azul palido. La pulpa roja del bazo se infiltra preferentemente. Las triadas portales hepaticas tanbien esta
afectadas con frecuencia.


Micosis fungoides/ sindrome de Sezary Son manifestaciones de un tumos de
células T facilitadoras. Caracterizado por una tendencia marcada
de afectación de la piel. La progresión de la micosis se caracteriza
por diseminación extracutanea, con mas frecuencia en los ganglios linfáticos y la medula ósea.

El Sindrome de Sezary la afectación cutánea se manifiesta como una eritrodermia exfoliativa generaliza.
Rara vez progresan a tumefacción.


Linfoma de Hodgkin Nace en un solo ganglio o en una cadena
de ganglios. Se caracteriza por la presencia de células
gigante neoplásicas, que inducen a la acumulación de linfocitos reactivos, histiocitos y granulocitos.

Se reconocen 5 subtipos de LH Esclerosis nodular Celularidad mixta Rico en linfocitos Con deplecion de linfocitos Predominio linfocitico


Leucemia mieloide aguda Afectan primariamente a los adultos, con
una máxima incidencia entre los 15 y 39 años de edad.
Se asocia con alteraciones genéticas adquiridas que inhiben la diferenciación mieloide terminal.

La mayoría de los pacientes se presentan en las primeras semanas o pocos meses despues del comienzo de sintomas relacionados con anemia, neutropenia y trombocitopenia, sobre todo cansancio, fiebre y hemorragias mucosas y cutaneas espontaneas.


Bazo Forma parte del sistema linfocitico, que
combate las infecciones y mantiene el equilibrio de los líquidos del cuerpo. Contiene los glóbulos blancos que luchan contra los gérmenes.

Patología Esplenomegalia
El agrandamiento esplénico puede constituir un indicio diagnostico importante sobre la existencia de un trastorno subyacente, pero también puede causar problemas por si mismo.


El hiperesplenismo se caracteriza por: 1. esplenomegalia2. anemia, leucopenia, trombocitopenia o
cualquiera de ellas, 3. corrección de las citopenias sanguíneas

Esplenitis aguda Inespecífica El bazo puede aumentar de tamaño en
cualquier infección transportada por la sangre.
El bazo aparece agrandado y blando. En ocasiones existe necrosis aguda de los
centros de los folículos esplénicos, sobre todo cuando el germen causal es un estreptococo hemolítico.

Neoplasias En el bazo se pueden formar los tipos siguientes
de tumores benignos: fibromas osteomas Condroma linfangiomas hemangiomas. Los dos ultimos son los mas comunes y muchas
veces pertenecen al tipo cavernoso

Rotura La rotura del bazo suele ser precipitada por
aplastamiento o un golpe fuerte. La rotura puede seguirse de hemorragia
intraperitoneal extensa, en ocaciones masiva, que debe tratarse con esplenectomia pronta para evitar la muerte la muerte por hipovolemia y shock
Infarto esplenico

Timo Es uno de los controles centrales del
sistema inmunitario del organismo. Generalmente consta de dos lóbulos y se localiza en el mediastino, detrás del esternon.

Hiperplasia timica Los foliculos linfoides son similares a
centros germinales reactivos y contiene predominantemente linfocitos B de los que solo existe un numero pequeño en el timo normal.
Se encuentra con mas frecuencia en pacientes con miatenia gravis

Timomas pueden dividirse en las siguientes
categorías: Timoma benigno o encapsulado: citologica
y biológicamente benigno Timoma maligno: Tipo I, también llamado timoma invasivo Tipo II, también llamado carcinoma timico

Gracias por todo Dra.!!!!