estructura y función de la hemoglobina
DESCRIPTION
Se estudia la estrcutura y funciòn de la hemoglobina, como un estrutura importante en la supervivencia del ser humano.TRANSCRIPT

[Escriba texto] Página 1
HEMOGLOBINA 1.1.- DEFINICIÓN
La hemoglobina (HB) es una proteína globular, que está presente en altas concentraciones en lo glóbulos rojos y se encarga del transporte de O del aparato respiratorio hacia los tejidos periféricos; y del transporte de CO y protones (H) de los tejidos periféricos hasta los pulmones para ser excretados. Los valores normales en sangre son de 13 – 18 g/ dl en el hombre y 12 – 16 g/ dl en la mujer 4. 1.2.- ORGANIZACIÓN ESTRUCTURAL
Todas la hemoglobinas evolucionaron de una cadena polipeptídica primitiva , la
protoglobina , similar a la mioglobina y predecesora de las cadenas 𝝰, 𝝱, Ƴ, 𝞭 ,𝝴
y 𝞯 , tal evolución explica el alto grado de hemologia entre las cadenas 𝝰 y no –
𝝰 (𝝱, 𝞭 y Ƴ) , así como las extraordinarias similitudes entre las globinas no-à.
La evolución se realizó por mutaciones cromosómicas simples, como lo
demuestra la existencia de más de un centenar de Hb humanas anormales, no
difieren de la Hb A más que por un solo aminoácido, lo que indica la variación
de una única base nitrogenada en un triplete de código genético. Estas
mutaciones consistieron en duplicaciones y translocaciones sucesivas, al ritmo
de alrededor de 104 en 500 millones de años. La hemoglobina de la lamprea
es un verdadero fósil molecular , que existe solo en la forma de monómero ; los
dímeros aparecieron en los peces y más tarde surgieron los tetrámeros, Así ,
las cadenas polipeptidicas 𝝰 y no – 𝝰 probablemente se originaron por
duplicación genética y posteriormente los genes para estas proteínas
individuales genética y posteriormente los genes para estas proteínas
individuales evolucionaron independientemente , de modo que las cadenas 𝝱,
Ƴ y 𝞭 también se habrían originado por duplicación genética . 1
Fig. 1 ESTRUCTURA DE LA HEMOGLOBINA (primaria –secundaria-terciaria-
cuaternaria)

[Escriba texto] Página 2
Las cadenas de globinas humanas han sido denominadas con letras del
alfabeto griego: Alfa (α), Beta (β), Gamma(γ), Delta (δ), Epsilon (ε) y Zeta (δ).
De las combinaciones dos a dos de las diferentes cadenas de globinas se van
a formar las distintas hemoglobinas, en los períodos embrionario, fetal,
neonatal y adulto.
La hemoglobina es una proteína con estructura cuaternaria, es decir, está
constituida por cuatro cadenas polipeptídicas dos 𝝰 y dos b (hemoglobina
adulta- HbA); dos α y dos δ (forma minoritaria de hemoglobina adulta- HbA2-
normal 2%); dos α y dos γ (hemoglobina fetal- HbF). En el feto humano, en un
principio, no se sintetizan cadenas alfa ni beta, sino zeta (δ ) y epsilon (ε) (Hb
Gower I). Al final del primer trimestre la subunidades a han reemplazado a las
subunidades δ (Hb Gower II) y las subunidades γ a los péptidos ε. Por esto, la
HbF tiene la composición 𝝰2 γ 2. Las subunidades β comienzan su síntesis en
el tercer trimestre y no reemplazan a γ en su totalidad hasta algunas semanas
después del nacimiento.
La estructura primaria de globina se refiere a la secuencia de aminoácidos de
los diversos tipos de cadena. Numeración desde el extremo N-terminal
identifica la posición de los aminoácidos individuales. La identidad y la posición
de estos aminoácidos no se pueden cambiar sin causar deterioro bruto a la
función molecular. La cadena alfa normal contiene 142 residuos de
aminoácidos en secuencia lineal .La no – 𝝰 (𝝱, 𝞭 y Ƴ) cadenas son todos los
146aminoacidos de longitud, la cadena beta comienza con vallina e histidina .
Los residuos C- terminal son Tyr B145 y B146 Su .La cadena delta (de la Hb
A2) difiera de la cadena beta ( de la Hb A) en solo 10 residuos .Los ocho
primeros residuos y los residuos C- terminal (127-146) son los mismo en delta y
Fig. 2 Combinación de las cadenas de globinas

[Escriba texto] Página 3
beta cadenas .Tetrámeros de cadena beta (Hb H) se pueden encontrar en una
talasemia la cadena gamma de la Hb F difiera de la cadena beta por 39
residuos. Los residuos N-terminal de la cadena gamma y de la cadena beta son
glicina y valina , respectivamente . mientras de los residuos C-terminales ,
Tyr145 y His 146, son los mismos que en gamma y beta cadenas .Cantidades
apreciables de cadenas gamma libres se encuentran en los glóbulos rojos de
algunos bebes con una talasemia ; cadenas gammas gratuitas , como cadenas
beta , pueden formar homotetrameros conocidos como la hemoglobina de Bart.
Los genes gamma se duplican, uno para los códigos de glicina (Gg) y el otro de
alanina (Ag) 7 en el residuo 176, dando lugar a dos tipos de cadenas gamma.
La estructura secundaria tiene alrededor del 75% de los aminoácidos alfa y
beta cadenas en una disposición helicoidal .Todas las hemoglobinas
estudiadas tienen un contenido helicoidal similar .ocho zonas helicoidales, con
letras A a H , se producen en las cadenas beta .La nomenclatura especifica de
la hemoglobina de los aminoácidos dentro de las hélices son designadas por el
número de aminoácidos y la letra hélice , mientras que los aminoácidos entre
las hélices llevan el número del aminoácido y las letras de las dos hélices .Por
lo tanto EF3 residuo es el tercer residuo del segmento que conecta el E y F
hélices , mientras residuo F8 es el octavo residuo de la hélice F .Alineamiento
de acuerdo de la designación helicoidal hace homología evidente ,F8 residuo
es el histidina hemo enlace proximal y la histidina en el lado distal de la hemo
es E7.
La estructura terciaria de alfa y beta cadenas, el plegamiento terciario de
cada cadena de globina forma una esfera aproximada, el plegamiento terciario
da lugar a al menos 3 características funcionales importantes del a molécula de
hemoglobina.
Las cadenas laterales polares o cargadas tienden a ser dirigido a la
superficie exterior de la subunidad y a la inversa, las estructuras no
polares tienden a ser dirigidos hacia el interior .El efecto de esto es que
la superficie de la molécula hidrófila y la hidrófoba al interior.
Una hendidura abierta en la superficie de la subunidad conocida como
bolsillo hemo se crea .Esta hendidura hidrofobia protege el ion ferroso
de la oxidación.
Los aminoácidos que forman os lazos entre la subunidad responsable
del mantenimiento de la estructura cuaternaria , y polo tanto la función
del a molécula de hemoglobina se ponen en la orientación correcta para
permitir que estos enlaces se puedan formar .
El grupo hemo se encuentra en una grieta entre las hélices E y F en cada
cadena .Las cadenas laterales sumamente polares proporcionado por el hem
están sobre la superficie de la molécula y son ionizados por el pH fisiológico. El
resto del hemo está dentro de la molécula, excepto por dos histonas .E l átomo

[Escriba texto] Página 4
de hierro está ligado por un enlace coordinado con el nitrógeno del imidazol (N)
de F8 histidina ; la histidina distal E7 , en el lado del plano de hemo , no está
unido al átomo de hierro , pero es muy cerca del sitio de unión al ligando.2
La estructura cuaternaria se caracteriza por la existencia de zonas de unión
entre las 4 cadenas, las uniones entre alfa 1 y beta 2 y alfa 2 y beta 2, estos son
móviles y desempeñan un papel activo en los cambios de conformación
molecular , por lo contrario , los enlaces entre las cadenas alfa 1 y beta2 , y alfa
2 y beta 2 . Son fijos y pasivos en el curso de las transformaciones de la
molécula. Alrededor de estas zonas fijas bisagras, las cadenas tienen cierta
movilidad por eso se puede decir que es de suerte que existe bajo dos formas
de equilibrio, desoxigenada o tensa T u oxigenada o relajada R cuyas
proporciones varían con la concentración de los ligando en el medio.1
1.3.- GRUPO PROSTETICO DE LA HEMOGLOBINA
Las cuatro cadenas polipeptídicas de la Hb contienen cada una un grupo
prostético, el Hem, un tetrapirrol cíclico, que les proporciona el color rojo a los
hematíes. Un grupo prostético es una porción no polipeptídica que forma parte
de una proteína en su estado funcional. El átomo de hierro se encuentra en
estado de oxidación ferroso (+2) y puede formar 5 o 6 enlaces de coordinación
dependiendo de la unión del oxígeno a la Hb (oxiHb, desoxiHb). Cuatro de estos
enlaces se producen con los nitrógenos pirrólicos de la porfirina en un plano
horizontal. El quinto enlace de coordinación se realiza con el nitrógeno del
imidazol de una histidina denominada histidina proximal. Finalmente, el sexto
enlace del átomo ferroso es con el O2, que además está unido a un segundo
imidazol de una histidina denominada histidina distal.
Tanto el quinto como el sexto enlace se encuentran en un plano perpendicular al
plano del anillo de porfirina. La parte porfirínica del Hem se sitúa dentro de una
bolsa hidrofóbica que se forma en cada una de las cadenas polipeptídicas.
Cuando una proteína esta con su grupo prostético se denomina holoproteina, y
cuando esta sin este, se lo denomina apoproteina. Además por poseer un grupo
prostético se dice que la Hb es una proteína conjugada, es una hemoproteina.4
Fig. 3 Estructura de la
hemoglobina
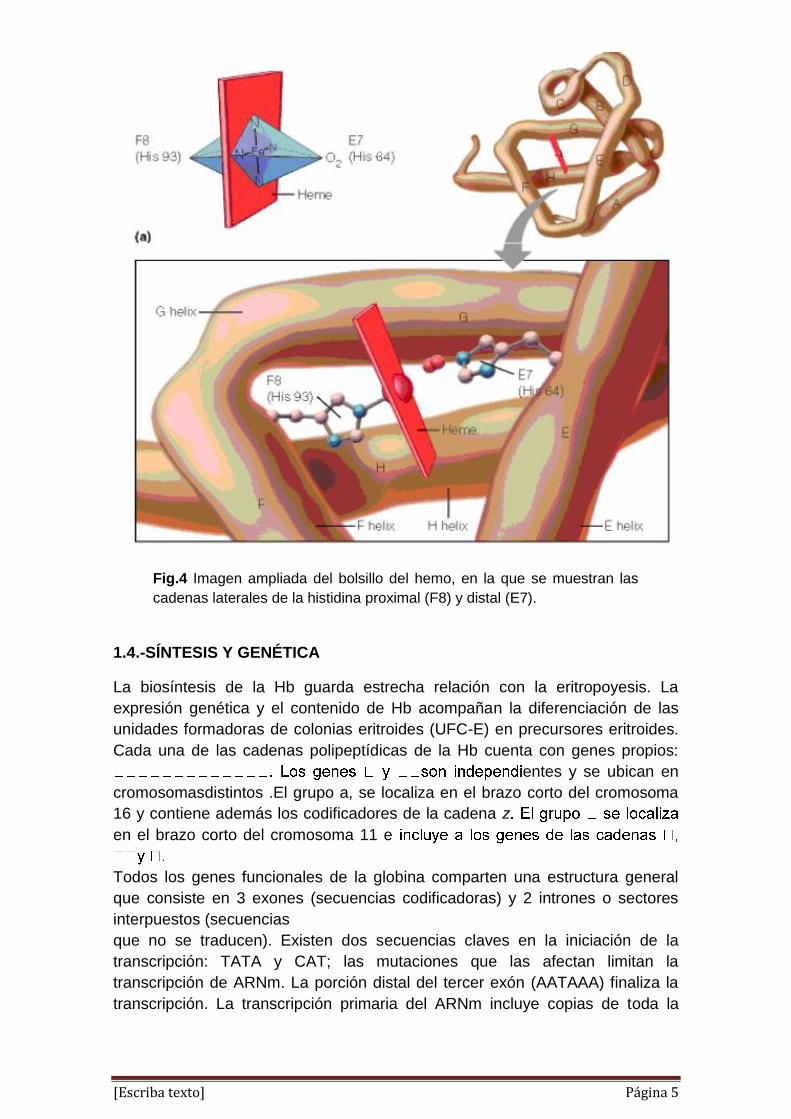
[Escriba texto] Página 5
1.4.-SÍNTESIS Y GENÉTICA
La biosíntesis de la Hb guarda estrecha relación con la eritropoyesis. La
expresión genética y el contenido de Hb acompañan la diferenciación de las
unidades formadoras de colonias eritroides (UFC-E) en precursores eritroides.
Cada una de las cadenas polipeptídicas de la Hb cuenta con genes propios:
entes y se ubican en
cromosomasdistintos .El grupo a, se localiza en el brazo corto del cromosoma
16 y contiene además los codificadores de la cadena z
en el brazo corto del cromosoma 11 e
Todos los genes funcionales de la globina comparten una estructura general
que consiste en 3 exones (secuencias codificadoras) y 2 intrones o sectores
interpuestos (secuencias
que no se traducen). Existen dos secuencias claves en la iniciación de la
transcripción: TATA y CAT; las mutaciones que las afectan limitan la
transcripción de ARNm. La porción distal del tercer exón (AATAAA) finaliza la
transcripción. La transcripción primaria del ARNm incluye copias de toda la
Fig.4 Imagen ampliada del bolsillo del hemo, en la que se muestran las
cadenas laterales de la histidina proximal (F8) y distal (E7).

[Escriba texto] Página 6
secuencia del ADN genómico (intrones y exones). Antes de su transporte al
citoplasma se procesa por clivaje del extremo 5’, hay separación de las
secuencias transcriptas de los intrones y poliadenilación del extremo 3’. Los
puntos de consenso son secuencias de nucleótidos adyacentes que
perfeccionan la síntesis del ARNm. Las mutaciones que involucran tanto los
puntos de unión, así como los de consenso, alteran la separación y crean
ARNm anormales.
La causa más común de las hemoglobinopatías es la mutación puntual, es
decir, la sustitución de un nucleótido de ADN por otro, lo que modifica el código
genético y puede inducir un cambio en un aminoácido de la globina resultante.
La traducción es un proceso ribosómico, en donde se sintetiza una cadena
polipeptídica de acuerdo al patrón de codones del ARNm. La terminación se
produce cuando se llega a un codón de finalización UAA, la cadena
polipeptídica se completa y se separa del ribosoma. Los polipéptidos libres
El grupo Hem se sintetiza en virtualmente todos los tejidos, pero su síntesis es
más pronunciada en la médula ósea y el hígado, debido a la necesidad de
incorporarlo en la Hb y los citocromos, respectivamente. Es una molécula plana
que consta de un hierro ferroso y un anillo tetrapirrólico, la protoporfirina III o IX.
El Hem es un factor fundamental en la regulación de la tasa de síntesis de la
globina. Su principal efecto se ejerce en la iniciación de la traducción, donde
bloquea la acción de un inhibidor de la producción de globina. También
participa en la transcripción y el procesamiento del ARNm.
Normalmente los eritrocitos envejecidos se degradan hacia el día 120 de vida
en la médula ósea, el hígado y el bazo. En algunas circunstancias sin embargo,
los eritrocitos sufren lisis intravascular, liberando Hb, que puede ser tóxica para
los tejidos a menos que se remueva rápidamente. La haptoglobina (Hp) es una
proteína plasmática que une Hb libre, a través de la formación de un complejo
Hp-Hb. Este complejo es reconocido a través de una proteína situada en la
superficie de los macrófagos y monocitos denominada CD163, permitiendo su
digestión y la seguida liberación de hierro y bilirrubina.4
Fig.5 Los genes y son independientes y se ubican en cromosomas distintos.

[Escriba texto] Página 7
Síntesis
El primer paso en la síntesis del hemo es la combinación de succinil CoA
y glicina para producir ácido aminolevulínico δ (δ ALA). Esta reacción es
dependiente de la energía y así se produce en la mitocondria
Se catalizada por la enzima δ ALA sintetasa.
Este paso es una primera etapa limitante para todo el proceso de la
síntesis de hemo.
Se estimulada por la presencia de cadenas de globina e inhibida por la
presencia de grupos hemo libres
Esto representa un importante mecanismo de control de la tasa de
síntesis de hemo y es la coordinación con la síntesis de globina.
Se requieren varios factores para este paso, incluyendo la vitamina B6,
iones ferrosos y de cobre libres.
Síntesis de la enzima δ ALA sintetasa es inhibida por la presencia del
grupo hemo libre.
Esto representa un mecanismo de retroalimentación adicional para la
síntesis de hemo
Ácido aminolevulínico δ mitocondrial (ALA) es transportado al
citoplasma, donde ALA deshidratasa (también llamada porfobilinógeno
sintasa) dimeriza 2 moléculas de ALA para producir el compuesto de
anillo pirrol porfobilinógeno (PBG).
El siguiente paso requiere la síntesis de anillo de porfirina.
Las reacciones implicadas son extremadamente complejos, pero pueden
resumirse como la condensación de cuatro moléculas de PBG para
formar el uroporfirinógeno cíclico asimétrico III (UPGIII).
Síntesis de UPGIII requiere la presencia de dos enzimas
(uroporfirinógeno me sintetasa y uroporfirinógeno III cosynthetase) y
consiste en la formación de varios intermedios de vida corta.
UPG III se convierte en coproporfirinógeno III (CPGIII) por
descarboxilación de las cadenas laterales de etilo bajo la influencia de la
enzima uroporfirinógeno descarboxilasa.
CPGIII entra en las mitocondrias, donde se convirtieron al
protoporfirinógeno IX (PPG IX) por un mecanismo desconocido. Esta
reacción es catalizada por la enzima oxidasa coproporhyrinogen.
PPG IX se convierte además en la mitocondria para protoporphrin IX.
Sólo queda para el ión ferroso central para ser insertado para completar
la síntesis del grupo hemo. Esta reacción es catalizada por la enzima
ferroquelatasa y requiere la presencia de agentes reductores.5
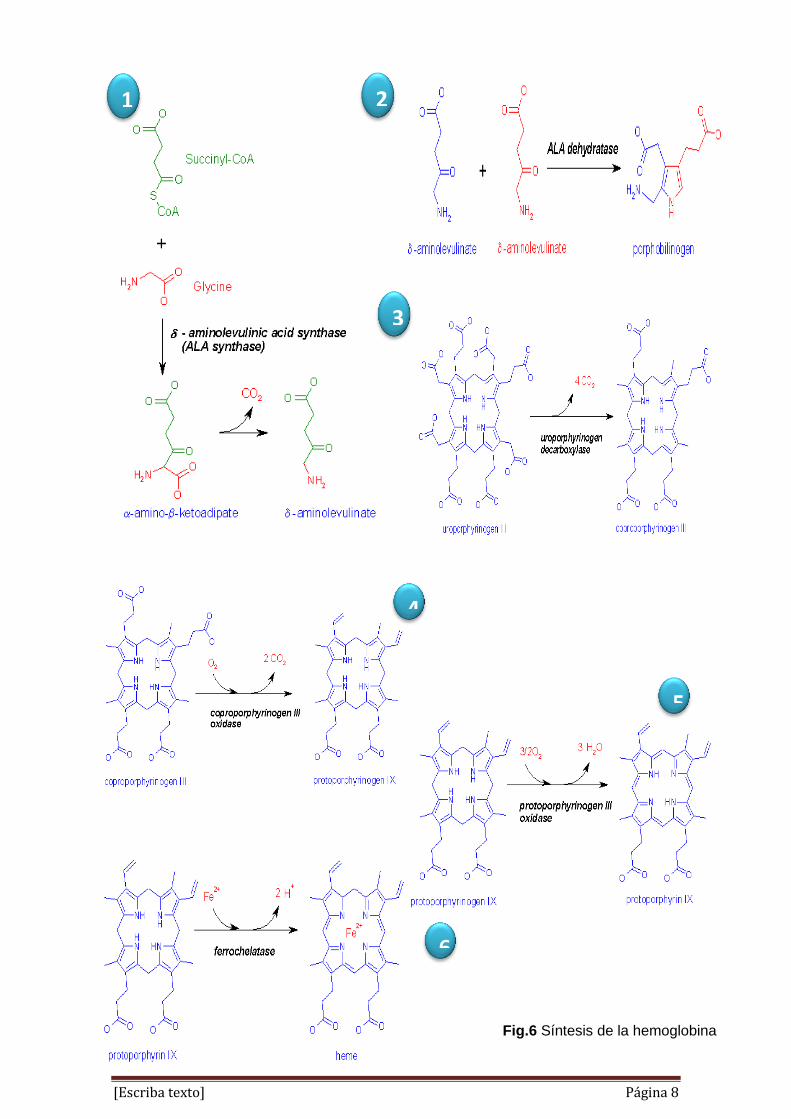
[Escriba texto] Página 8
1 2
3
4
5
6
Fig.6 Síntesis de la hemoglobina
[Escriba texto] Página 9
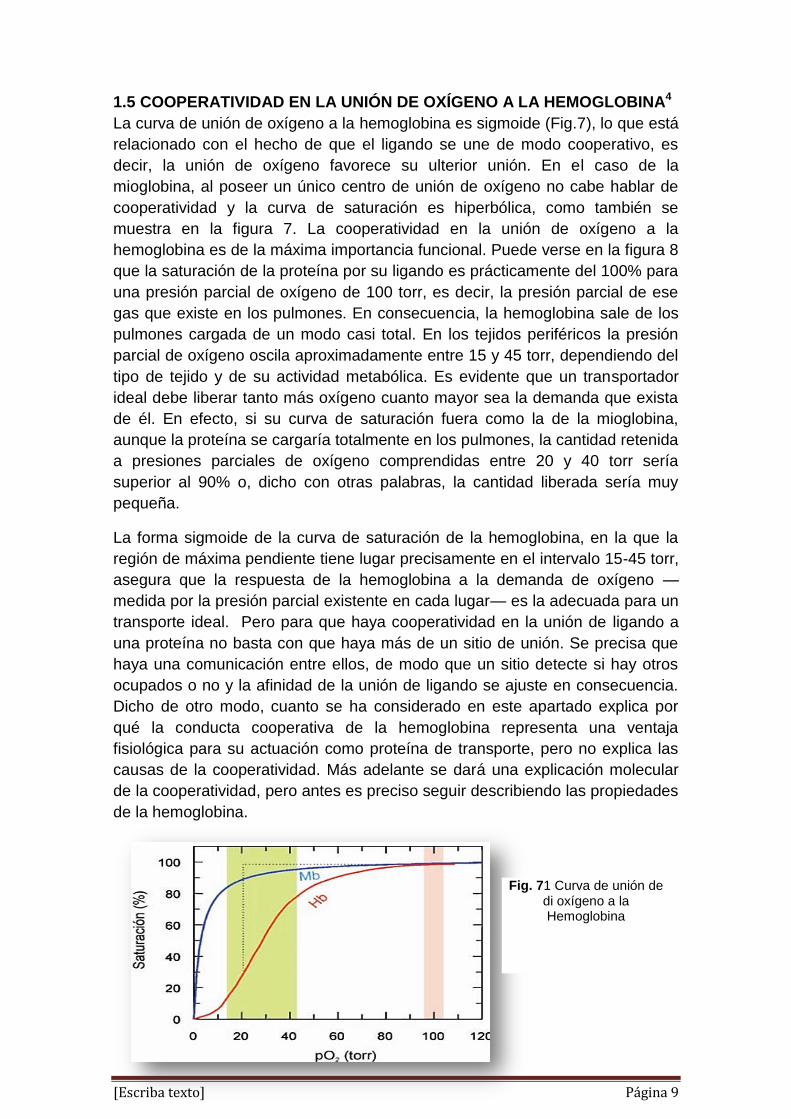
1.5 COOPERATIVIDAD EN LA UNIÓN DE OXÍGENO A LA HEMOGLOBINA4
La curva de unión de oxígeno a la hemoglobina es sigmoide (Fig.7), lo que está
relacionado con el hecho de que el ligando se une de modo cooperativo, es
decir, la unión de oxígeno favorece su ulterior unión. En el caso de la
mioglobina, al poseer un único centro de unión de oxígeno no cabe hablar de
cooperatividad y la curva de saturación es hiperbólica, como también se
muestra en la figura 7. La cooperatividad en la unión de oxígeno a la
hemoglobina es de la máxima importancia funcional. Puede verse en la figura 8
que la saturación de la proteína por su ligando es prácticamente del 100% para
una presión parcial de oxígeno de 100 torr, es decir, la presión parcial de ese
gas que existe en los pulmones. En consecuencia, la hemoglobina sale de los
pulmones cargada de un modo casi total. En los tejidos periféricos la presión
parcial de oxígeno oscila aproximadamente entre 15 y 45 torr, dependiendo del
tipo de tejido y de su actividad metabólica. Es evidente que un transportador
ideal debe liberar tanto más oxígeno cuanto mayor sea la demanda que exista
de él. En efecto, si su curva de saturación fuera como la de la mioglobina,
aunque la proteína se cargaría totalmente en los pulmones, la cantidad retenida
a presiones parciales de oxígeno comprendidas entre 20 y 40 torr sería
superior al 90% o, dicho con otras palabras, la cantidad liberada sería muy
pequeña.
La forma sigmoide de la curva de saturación de la hemoglobina, en la que la
región de máxima pendiente tiene lugar precisamente en el intervalo 15-45 torr,
asegura que la respuesta de la hemoglobina a la demanda de oxígeno —
medida por la presión parcial existente en cada lugar— es la adecuada para un
transporte ideal. Pero para que haya cooperatividad en la unión de ligando a
una proteína no basta con que haya más de un sitio de unión. Se precisa que
haya una comunicación entre ellos, de modo que un sitio detecte si hay otros
ocupados o no y la afinidad de la unión de ligando se ajuste en consecuencia.
Dicho de otro modo, cuanto se ha considerado en este apartado explica por
qué la conducta cooperativa de la hemoglobina representa una ventaja
fisiológica para su actuación como proteína de transporte, pero no explica las
causas de la cooperatividad. Más adelante se dará una explicación molecular
de la cooperatividad, pero antes es preciso seguir describiendo las propiedades
de la hemoglobina.
Fig. 71 Curva de unión de di oxígeno a la Hemoglobina

[Escriba texto] Página 10
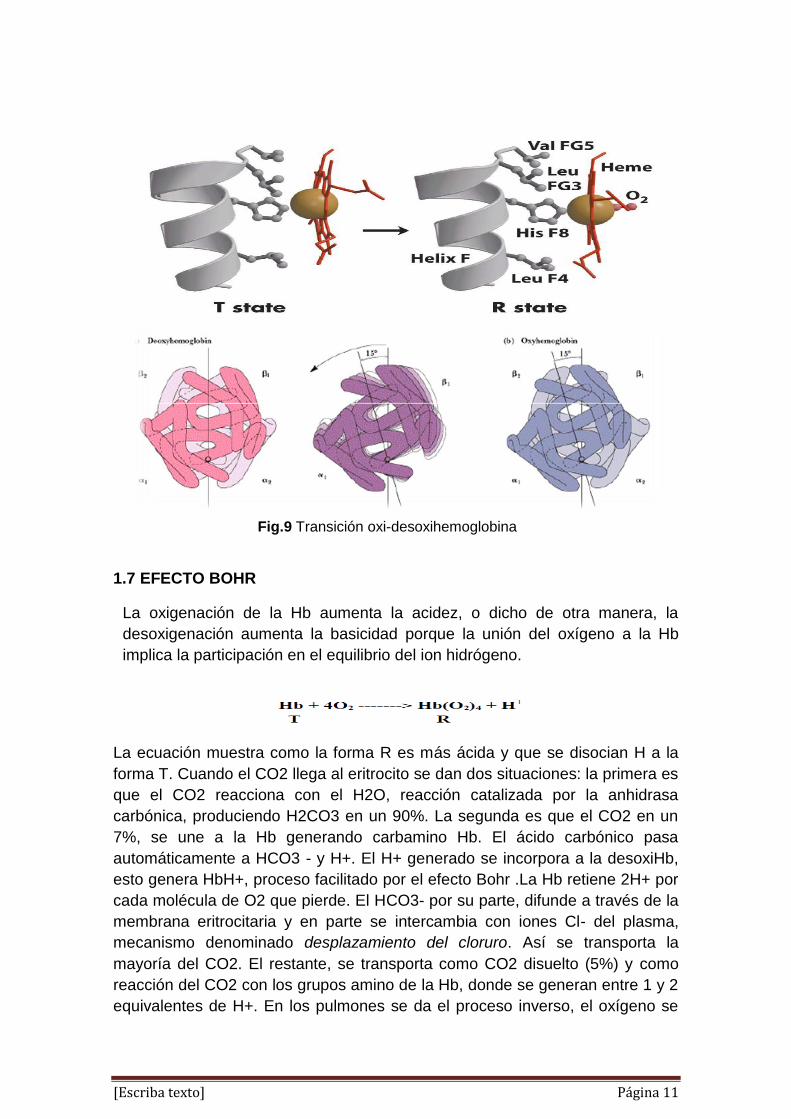
1.6 MODIFICACIONES CONFORMACIONALES DURANTE LA
OXIGENACIÓN
Estudios estructurales han demostrado que existe hemoglobina en una de las
dos conformaciones, conocidos como T (tenso) y R (relajado). Hemoglobina
desoxigenada (azul) se encuentra en el estado T, y de unión a oxígeno (rojo)
provoca la transición al estado R.
En el estado desoxigenado (T), el átomo de hierro no es plana con el resto del
grupo hemo debido a su asociación con una cadena lateral de histidina. Unión
de oxígeno hace que el átomo de hierro en el hemo se mueva de tal manera
que se convierte en planar con el resto del grupo hemo, que luego se tira de la
histidina, causando un cambio estructural mayor escala en la proteína.
El cambio conformacional que se produce durante la T a R transición tiene
lugar principalmente en las posiciones de estos dos dímeros uno con relación
al otro (en vez de entre las subunidades alfa y beta dentro de la misma
dímero).
El T a R transición requiere que al menos dos de las subunidades de la
hemoglobina en obligarse por el oxígeno. Dado que la hemoglobina en el
estado T solamente tiene una baja afinidad por el oxígeno, el cambio
conformacional sólo puede ocurrir bajo las concentraciones de oxígeno
relativamente altas (como en los capilares pulmonares).En el estado R, la
hemoglobina se une al oxígeno con afinidad mucho mayor, dando lugar a
ningún resto de subunidades de unión desoxigenadas rápidamente a oxígeno.
El cambio de colores. Eso es debido a que el oxígeno en el aire "oxida" los
metales, por lo tanto cuando el oxígeno se combina con la hemoglobina, la
molécula se vuelve un poco más rojo. 3
Fig.8 Modificaciones conformacionales durante la oxigenación
[Escriba texto] Página 11
1.7 EFECTO BOHR
La oxigenación de la Hb aumenta la acidez, o dicho de otra manera, la
desoxigenación aumenta la basicidad porque la unión del oxígeno a la Hb
implica la participación en el equilibrio del ion hidrógeno.
La ecuación muestra como la forma R es más ácida y que se disocian H a la
forma T. Cuando el CO2 llega al eritrocito se dan dos situaciones: la primera es
que el CO2 reacciona con el H2O, reacción catalizada por la anhidrasa
carbónica, produciendo H2CO3 en un 90%. La segunda es que el CO2 en un
7%, se une a la Hb generando carbamino Hb. El ácido carbónico pasa
automáticamente a HCO3 - y H+. El H+ generado se incorpora a la desoxiHb,
esto genera HbH+, proceso facilitado por el efecto Bohr .La Hb retiene 2H+ por
cada molécula de O2 que pierde. El HCO3- por su parte, difunde a través de la
membrana eritrocitaria y en parte se intercambia con iones Cl- del plasma,
mecanismo denominado desplazamiento del cloruro. Así se transporta la
mayoría del CO2. El restante, se transporta como CO2 disuelto (5%) y como
reacción del CO2 con los grupos amino de la Hb, donde se generan entre 1 y 2
equivalentes de H+. En los pulmones se da el proceso inverso, el oxígeno se
Fig.9 Transición oxi-desoxihemoglobina
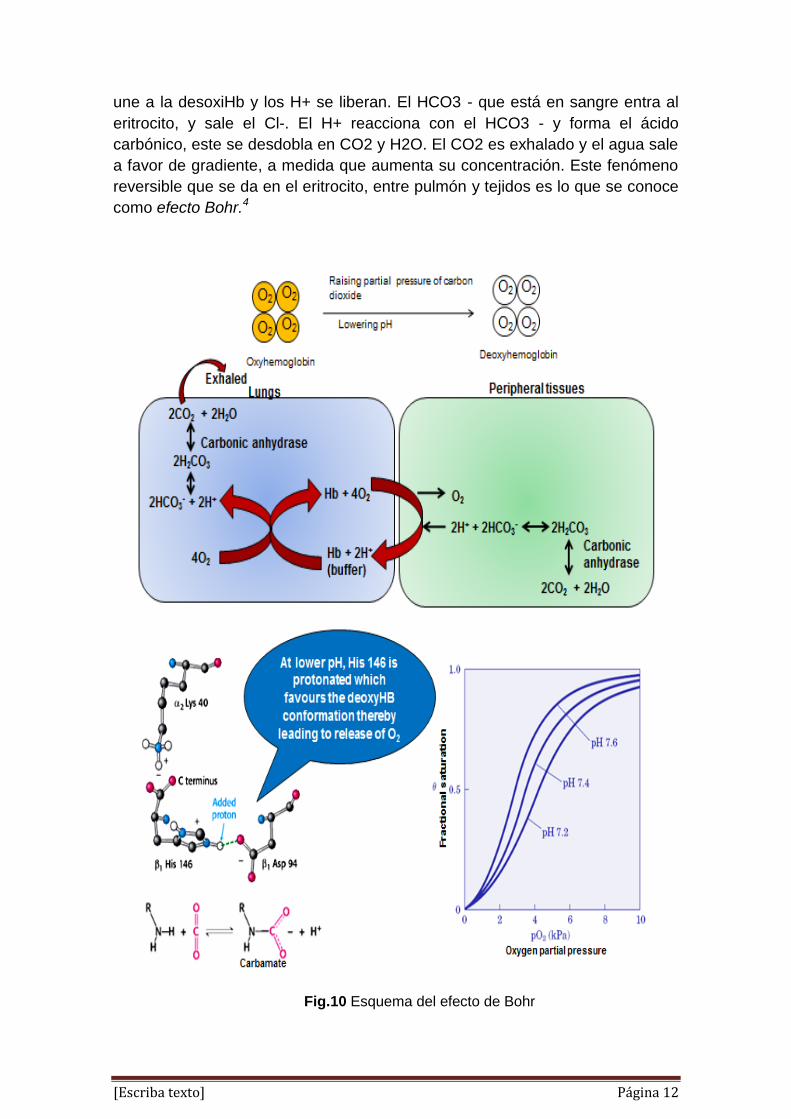
[Escriba texto] Página 12
une a la desoxiHb y los H+ se liberan. El HCO3 - que está en sangre entra al
eritrocito, y sale el Cl-. El H+ reacciona con el HCO3 - y forma el ácido
carbónico, este se desdobla en CO2 y H2O. El CO2 es exhalado y el agua sale
a favor de gradiente, a medida que aumenta su concentración. Este fenómeno
reversible que se da en el eritrocito, entre pulmón y tejidos es lo que se conoce
como efecto Bohr.4
Fig.10 Esquema del efecto de Bohr
[Escriba texto] Página 13
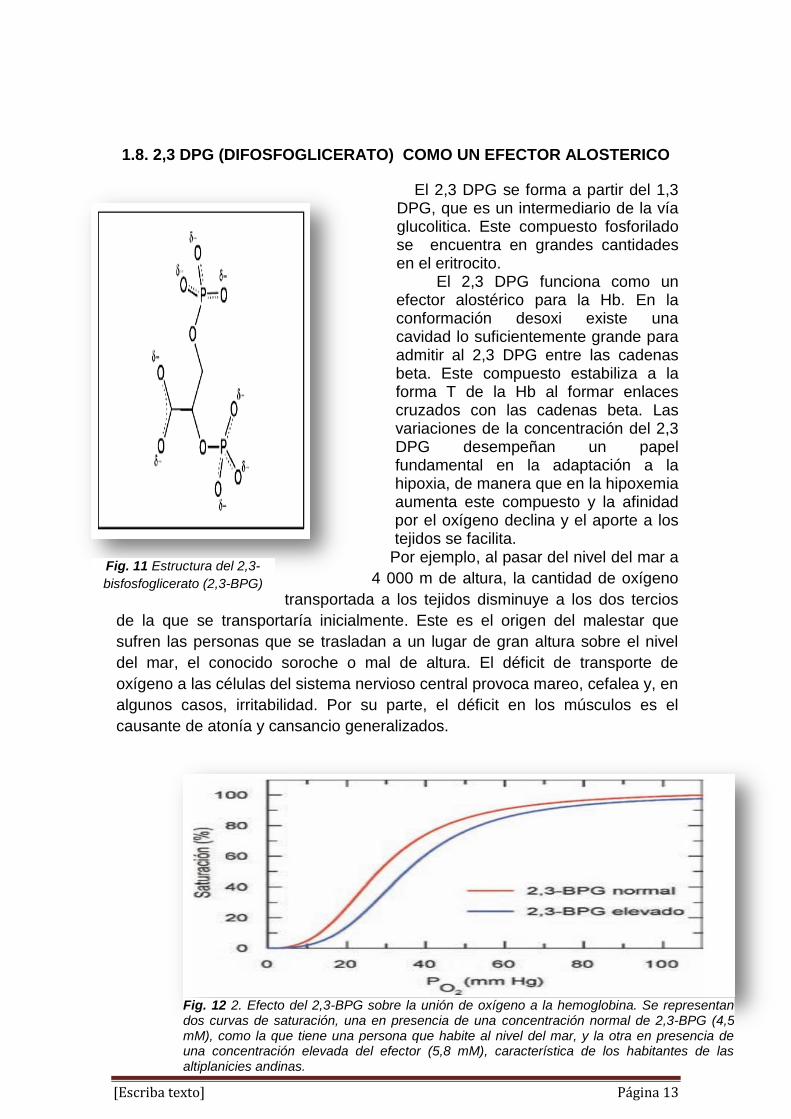
1.8. 2,3 DPG (DIFOSFOGLICERATO) COMO UN EFECTOR ALOSTERICO
El 2,3 DPG se forma a partir del 1,3 DPG, que es un intermediario de la vía glucolitica. Este compuesto fosforilado se encuentra en grandes cantidades en el eritrocito. El 2,3 DPG funciona como un efector alostérico para la Hb. En la conformación desoxi existe una cavidad lo suficientemente grande para admitir al 2,3 DPG entre las cadenas beta. Este compuesto estabiliza a la forma T de la Hb al formar enlaces cruzados con las cadenas beta. Las variaciones de la concentración del 2,3 DPG desempeñan un papel fundamental en la adaptación a la hipoxia, de manera que en la hipoxemia aumenta este compuesto y la afinidad por el oxígeno declina y el aporte a los tejidos se facilita.
Por ejemplo, al pasar del nivel del mar a
4 000 m de altura, la cantidad de oxígeno
transportada a los tejidos disminuye a los dos tercios
de la que se transportaría inicialmente. Este es el origen del malestar que
sufren las personas que se trasladan a un lugar de gran altura sobre el nivel
del mar, el conocido soroche o mal de altura. El déficit de transporte de
oxígeno a las células del sistema nervioso central provoca mareo, cefalea y, en
algunos casos, irritabilidad. Por su parte, el déficit en los músculos es el
causante de atonía y cansancio generalizados.
Fig. 11 Estructura del 2,3-
bisfosfoglicerato (2,3-BPG)
Fig. 12 2. Efecto del 2,3-BPG sobre la unión de oxígeno a la hemoglobina. Se representan dos curvas de saturación, una en presencia de una concentración normal de 2,3-BPG (4,5 mM), como la que tiene una persona que habite al nivel del mar, y la otra en presencia de una concentración elevada del efector (5,8 mM), característica de los habitantes de las altiplanicies andinas.
[Escriba texto] Página 14
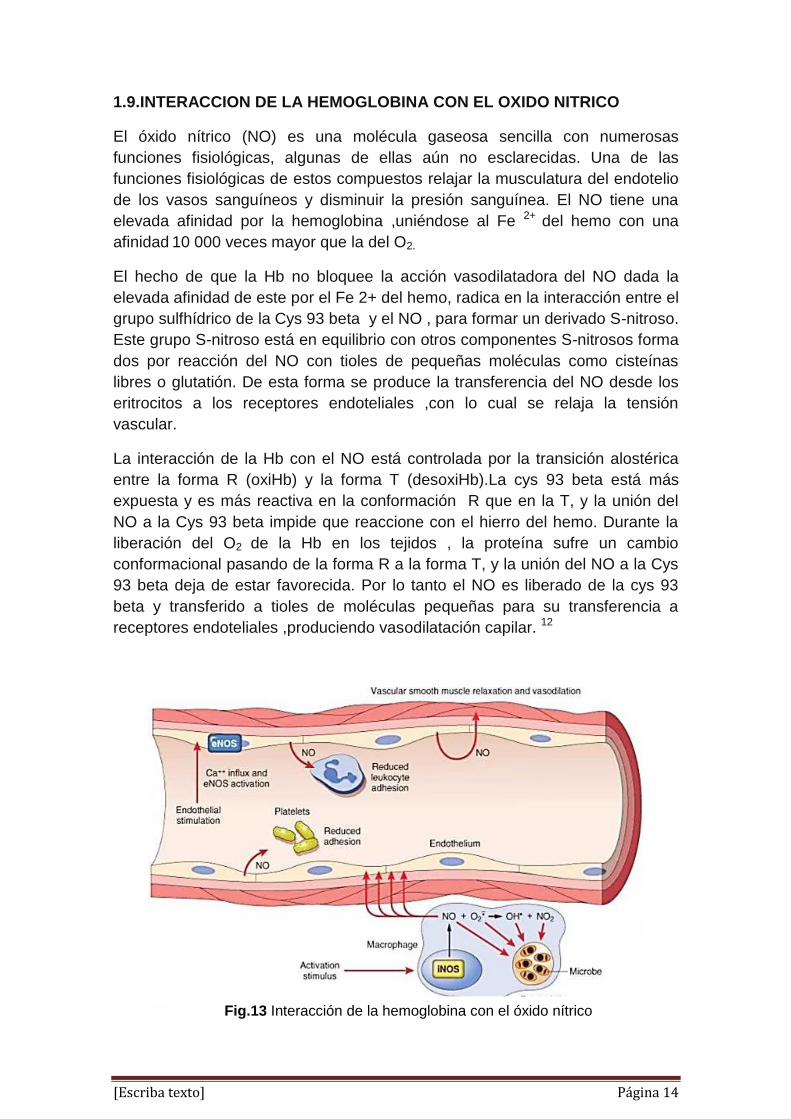
1.9.INTERACCION DE LA HEMOGLOBINA CON EL OXIDO NITRICO
El óxido nítrico (NO) es una molécula gaseosa sencilla con numerosas
funciones fisiológicas, algunas de ellas aún no esclarecidas. Una de las
funciones fisiológicas de estos compuestos relajar la musculatura del endotelio
de los vasos sanguíneos y disminuir la presión sanguínea. El NO tiene una
elevada afinidad por la hemoglobina ,uniéndose al Fe 2+ del hemo con una
afinidad 10 000 veces mayor que la del O2.
El hecho de que la Hb no bloquee la acción vasodilatadora del NO dada la
elevada afinidad de este por el Fe 2+ del hemo, radica en la interacción entre el
grupo sulfhídrico de la Cys 93 beta y el NO , para formar un derivado S-nitroso.
Este grupo S-nitroso está en equilibrio con otros componentes S-nitrosos forma
dos por reacción del NO con tioles de pequeñas moléculas como cisteínas
libres o glutatión. De esta forma se produce la transferencia del NO desde los
eritrocitos a los receptores endoteliales ,con lo cual se relaja la tensión
vascular.
La interacción de la Hb con el NO está controlada por la transición alostérica
entre la forma R (oxiHb) y la forma T (desoxiHb).La cys 93 beta está más
expuesta y es más reactiva en la conformación R que en la T, y la unión del
NO a la Cys 93 beta impide que reaccione con el hierro del hemo. Durante la
liberación del O2 de la Hb en los tejidos , la proteína sufre un cambio
conformacional pasando de la forma R a la forma T, y la unión del NO a la Cys
93 beta deja de estar favorecida. Por lo tanto el NO es liberado de la cys 93
beta y transferido a tioles de moléculas pequeñas para su transferencia a
receptores endoteliales ,produciendo vasodilatación capilar. 12
Fig.13 Interacción de la hemoglobina con el óxido nítrico
[Escriba texto] Página 15
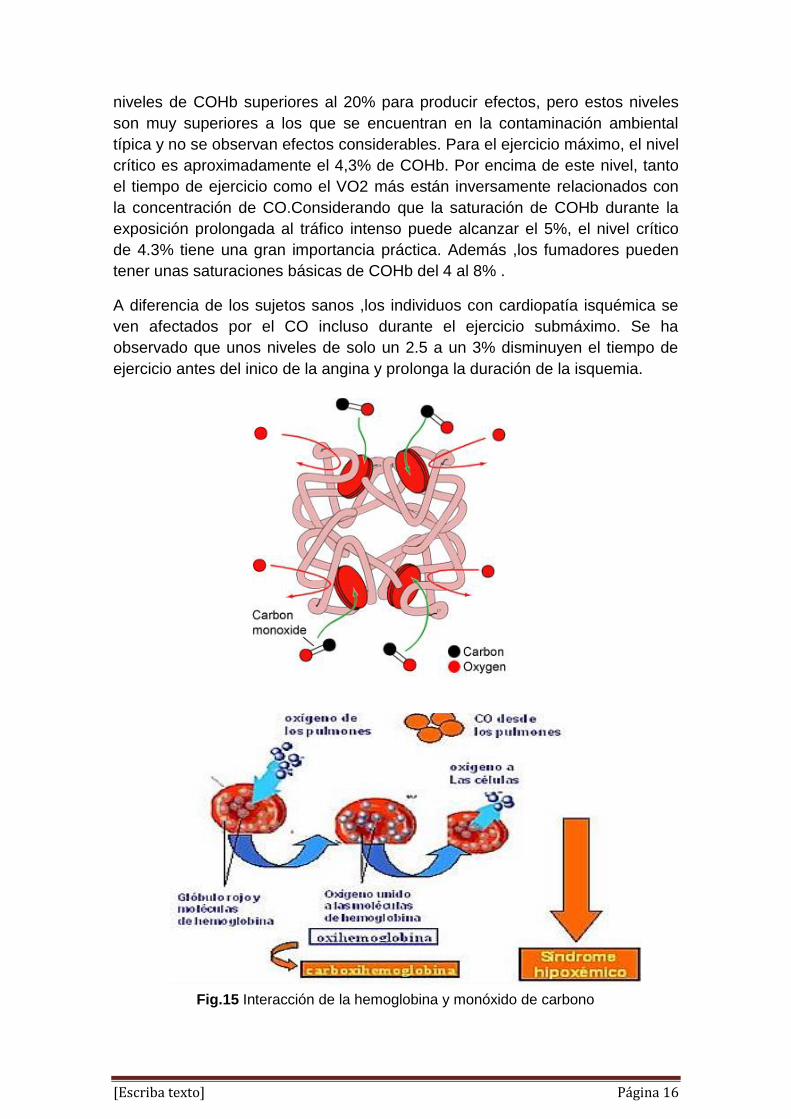
2.- INTERACCION DE LA HEMOGLOBINA CON EL MONOXIDO DE
CARBONO
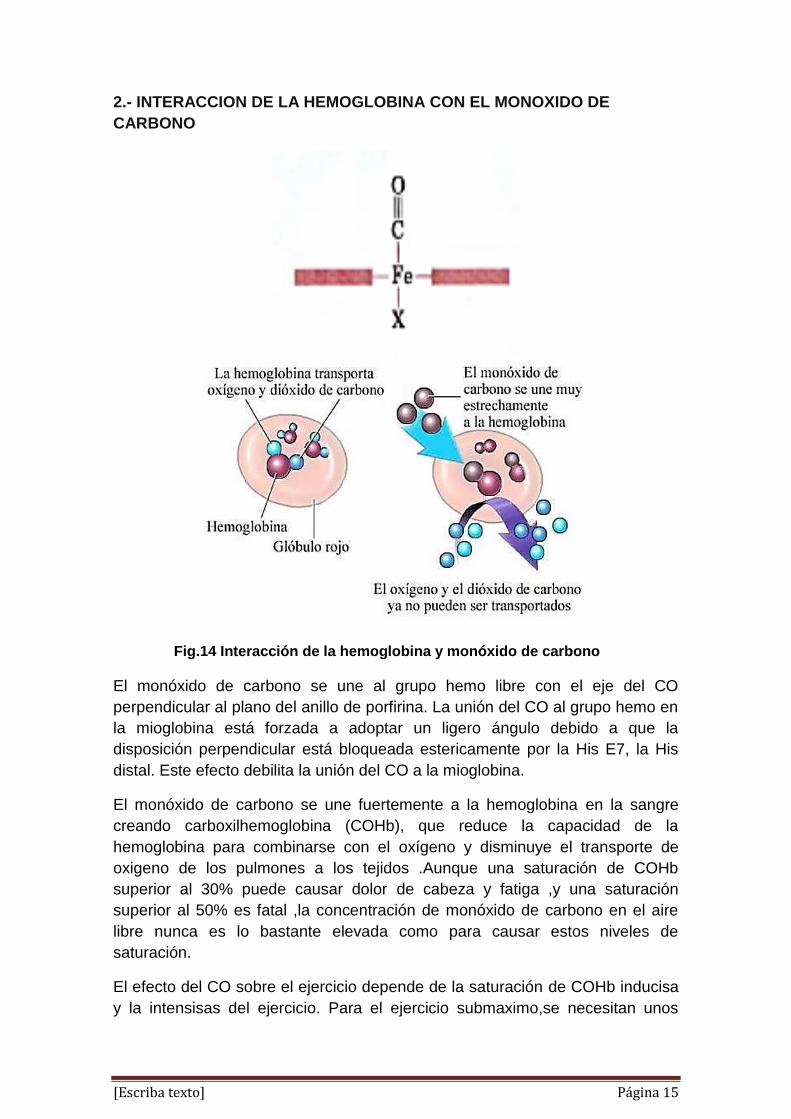
El monóxido de carbono se une al grupo hemo libre con el eje del CO
perpendicular al plano del anillo de porfirina. La unión del CO al grupo hemo en
la mioglobina está forzada a adoptar un ligero ángulo debido a que la
disposición perpendicular está bloqueada estericamente por la His E7, la His
distal. Este efecto debilita la unión del CO a la mioglobina.
El monóxido de carbono se une fuertemente a la hemoglobina en la sangre
creando carboxilhemoglobina (COHb), que reduce la capacidad de la
hemoglobina para combinarse con el oxígeno y disminuye el transporte de
oxigeno de los pulmones a los tejidos .Aunque una saturación de COHb
superior al 30% puede causar dolor de cabeza y fatiga ,y una saturación
superior al 50% es fatal ,la concentración de monóxido de carbono en el aire
libre nunca es lo bastante elevada como para causar estos niveles de
saturación.
El efecto del CO sobre el ejercicio depende de la saturación de COHb inducisa
y la intensisas del ejercicio. Para el ejercicio submaximo,se necesitan unos
Fig.14 Interacción de la hemoglobina y monóxido de carbono
[Escriba texto] Página 16
niveles de COHb superiores al 20% para producir efectos, pero estos niveles
son muy superiores a los que se encuentran en la contaminación ambiental
típica y no se observan efectos considerables. Para el ejercicio máximo, el nivel
crítico es aproximadamente el 4,3% de COHb. Por encima de este nivel, tanto
el tiempo de ejercicio como el VO2 más están inversamente relacionados con
la concentración de CO.Considerando que la saturación de COHb durante la
exposición prolongada al tráfico intenso puede alcanzar el 5%, el nivel crítico
de 4.3% tiene una gran importancia práctica. Además ,los fumadores pueden
tener unas saturaciones básicas de COHb del 4 al 8% .
A diferencia de los sujetos sanos ,los individuos con cardiopatía isquémica se
ven afectados por el CO incluso durante el ejercicio submáximo. Se ha
observado que unos niveles de solo un 2.5 a un 3% disminuyen el tiempo de
ejercicio antes del inico de la angina y prolonga la duración de la isquemia.
Fig.15 Interacción de la hemoglobina y monóxido de carbono

[Escriba texto] Página 17
2.1.
2.1.1 HEMOGLOBINOPATÍAS ESTRUCTURALES
Hb con alteraciones de la secuencia de aminoácidos que causan alteraciones de la función o de las propiedades físicas o químicas. A) Polimerización anómala de la Hb: HbS. B) Afinidad por el O2 alterada: - Alta afinidad: Policitemia - Baja afinidad: Cianosis, pseudoanemia. C) Hb que se oxidan fácilmente: - Hb inestables: Anemia Hemolítica, Ictericia. - Hb M: metahemoglobinemia, Cianosis. Hb S: también llamada anemia Falciforme o Drepanocítica. En 1949, Pauling
descubrió que en la anemia falciforme había alteración en la molécula de Hb.
Es una enfermedad hereditaria, autosómica recesiva, ya que es necesario que
el individuo sea homocigoto para tener la enfermedad. Esta enfermedad que se
encuentra con frecuencia en personas de raza negra y su mestizaje ,debido a
que son portadoras de la hemoglobina S en su forma homocigoto (HbSHbS).
Sin embargo, también puede presentarse como heterocigoto, es decir HbA y
HbS produciendo tan sólo el rasgo falciforme y una resistencia a la malaria. En
esta patología se produce un cambio de aminoácido en la posición 6 de beta
globina normal, cambiando ácido glutámico por valina, lo que disminuye la
solubilidad de la proteína, de tal manera que la hemoglobina S forma polímeros
produciendo un glóbulo rojo en Estos glóbulos rojos falciformes no son flexibles
y forman tapones en los vasos sanguíneos pequeños, produciendo una
interrupción de la circulación de la sangre que puede dañar los órganos de
cualquier parte del cuerpo. En un estudio realizado por Robert Hebbel y sus
colaboradores, demostraron que el componente hemo de la hemoglobina
tiende a liberarse de la proteína debido a episodios repetidos de la
polimerización de la hemoglobina S. Algunos de estos grupos hemo libres
tienden a alojarse en la membrana de los hematíes, el hierro de este grupo
promueve la formación de componentes muy peligrosos llamados especies
reactivas de oxígeno. Estas moléculas dañan los componentes lipídicos y
proteicos de la membrana de los glóbulos rojos, produciendo su destrucción
(hemólisis). Por lo tanto, en la anemia falciforme se incrementa la hemólisis y
desciende el valor de la hemoglobina y el hematocrito, es decir que se presenta
como una anemia hemolítica crónica, donde las manifestaciones clínicas se
inician a los seis meses de edad. Hay un mayor número de reticulocitos en
sangre a causa de la hemólisis. El balance entre vasoconstrictores y
vasodilatadores se altera a favor de los primeros y el flujo de la sangre se hace
lento. También se observan trastornos en el crecimiento y desarrollo del niño.
Generalmente se observan retardos en la maduración sexual. La vasooclusión
posee manifestaciones diversas: isquemia dolorosa, microinfartos, crisis de

[Escriba texto] Página 18
secuestro esplénico (que puede ser causa de muerte súbita en niños),
neovascularización, necrosis de órganos isquémicos afectados, entre otras.
2.1.2. TALASEMIAS
Los dos tipos principales de talasemia se denominan talasemia alfa y talasemia beta. Los individuos afectados por el primer tipo no producen suficiente cantidad de globina alfa y los afectados por el segundo, de globina beta. A su vez, cada uno de estos tipos de talasemia puede adoptar formas diferentes, con síntomas que van de leves a severos. Los términos ―mayor, menor, intermedia y mínima‖, utilizadas para indicar la gravedad de las manifestaciones clínicas, no necesariamente indican Heterocigota u Homocigota. Las talasemias más importantes se heredan por genes autosómicos recesivos. Tanto la alfa como la beta talasemia ocasionan disminución de la Hb dentro del eritrocito, lo que da lugar a una disminución del color (hipocromia) y del tamaño (microcitica) del hematíe. Otras talasemias descriptas son la delta y la gamma, de escasa frecuencia.
ALFA TALASEMIAS
Son cuatro los genes que controlan la producción de la globina alfa y la cantidad de genes faltantes o anormales determina la severidad de la enfermedad. El principal mecanismo por el que se producen las alfa talasemias es la deleción o pérdida total de un gen. Las formas no delecionales son menos frecuentes y obedecen a mutaciones, alteraciones en la transcripción del ARN o producción de ARN anómalo.
Perdida en un solo gen alfa: En este caso no existe manifestación clínica. Solo se diagnostica mediante técnicas complejas de análisis de ADN.
Perdida de dos genes alfa: produce un cuadro denominado talasemia menor o rasgo talasemico; no suele provocar problemas de salud importantes pero los individuos afectados pueden padecer una ligera anemia y transmitir la enfermedad a sus descendientes.
Perdida de tres genes alfa: constituye la denominada enfermedad de Hb H, esto produce anormalidades en los glóbulos rojos que derivan en su destrucción rápida. En esta enfermedad la producción de Hb A va de 25 a 30%.en el adulto, la cadenas beta sin pareja se acumulan y forman tetrámeros b4, denominadas Hb H. Esfrecuente en China e Indonesia y se han descrito también algunos casos en Italia y Sudamérica y en España. Cursan con un cuadro clínico de anemia hemolítica de intensidad moderada exacerbada por infecciones o por la ingesta de algunos medicamentos oxidantes, y moderada esplenomegalia. Es frecuente la supervivencia hasta la etapa media de la edad adulta, sin transfusiones.

[Escriba texto] Página 19
Perdida de cuatro genes alfa: es la denominada talasemia grave o mayor en la cual se produce la muerte del niño durante la gestación o en el periodo que sigue al parto. Esta enfermedad es incompatible con la vida del niño. Como la síntesis de la cadena alfa falla la HbA y la HbF disminuyen y en su lugar aumentan la Hb de Bart (cuatro cadenas gamma), que tiene una extraordinaria afinidad por el O2, y casi no lo suministra a los tejidos, causando asfixia y muerte; o la Hb H (cuatro cadenas beta).
BETA TALASEMIAS
Las beta talasemias son el resultados de la falta de síntesis de las cadenas beta de globina. Beta talasemia heterocigota o menor (rasgo talasemico): aparece cuando sólo está afectada una de las copias del gen que codifica la cadena. Es la mutación del gen beta, caracterizada por una hematíes elevada, con concentración de hemoglobina normal o disminuida y generalmente presenta unaumento de la Hb A2. Las personas portadoras de talasemia menor, no presentan manifestaciones clínicas, aunque en ocasiones pueden tener una ligera anemia que se pone de manifiesto al realizar un análisis. Los glóbulos rojos de los portadores del rasgo talasémico son más pequeños de lo normal. La talasemia menor está presente desde el nacimiento, permanece durante toda la vida y puede transmitirse de los padres a los hijos. Beta Talasemia Homocigota O Mayor (Anemia De Cooley): Es la forma más grave anemia congénita. La talasemia homocigótica, es en la que las dos copias del gen para una cadena de la hemoglobina son defectuosas, ocurre cuando no se sintetizan cadenas. Dependiendo de las mutaciones genéticas beta, se producirá una cantidad nula o muy escasa de cadenas beta, y un menor o mayor número de cadenas alfa. La talasemia mayor es una anemia hereditaria grave. Los pacientes afectados con esta anomalía no pueden fabricar suficientes glóbulos rojos y requieren frecuentes transfusiones de sangre. La enfermedad se manifiesta durante los primeros meses de vida, habitualmente entre el tercer y octavo mes. Estos pacientes presentan palidez, alteraciones del sueño, rechazo de los alimentos y vómitos. Desarrollan hemosiderosis (depósito en todos los tejidos del hierro liberado tras la hemólisis). Es frecuente la presencia de cálculos biliares por la hemólisis crónica. Adquieren un color pardo-verdoso por la anemia, la ictericia (la hemólisis libera bilirrubina que produce un color amarillo en la piel y mucosas) y la hemosiderosis. Se detiene el crecimiento, se retrasa la pubertad. Y finalmente se produce un fallo cardíaco. Actualmente algunos pacientes pueden también ser tratados, e incluso curados, mediante un trasplante de médula ósea. Beta Talasemia Intermedia: Se designa así al síndrome talasémico de moderada Hb E: (a2b2 26 Glu--->Lys) es extremadamente común en Camboya, Tailandia y Vietnam. Es ligeramente inestable pero no lo suficiente como para acortar

[Escriba texto] Página 20
significativamente la vida de los glóbulos rojos. Los heterocigotos son similares a los que tienen un rasgo talasemico b leve. Los homocigotos presentan anomalías algo más intensas, pero son asintomático intensidad, que condiciona la aparición de una anemia leve y alteraciones óseas. Presentan sintomatología clínica y requieren transfusiones de sangre durante alguna época de su vida, pueden desarrollar hemosiderosis. Sus manifestaciones no son tan graves como en los pacientes afectados de la forma mayor de la enfermedad.
VARIANTES ESTRUCTURALES TALASEMICAS
Hb Lepore: (a2 (db)2) surge de un entrecruzamiento y recombinación desigual
que fusiona el extremo proximal del gen beta con el extremo distal del gen delta
estrechamente ligado a él. El cromosoma resultante contiene el gen db
fusionado.
Persistencia Hereditaria de la Hb Fetal: se caracteriza por la síntesis
ininterrumpida de concentraciones altas de Hb F en la edad adulta. No son
detectables efectos nocivos ni anemia, incluso cuando toda la Hb producidaes
HbF.
3.- MIOGLOBINA
3.1 DEFINICIÓN
La mioglobina es una proteína globular que contiene una sola cadena
polipeptídica, constituida por ocho segmentos de hélice-α . La mioglobina se
halla, principalmente en las células de los músculos esqueléticos y es
especialmente abundante en los mamíferos buceadores, en los que no sólo
actúa almacenando oxígeno, sino también contribuyendo al aumento de la
velocidad de difusión del oxígeno.
Fig. 16.-Estructura de la Mioglobina donde se aprecia hemo (en rojo) y 8
segmentos de alfa hélice
[Escriba texto] Página 21
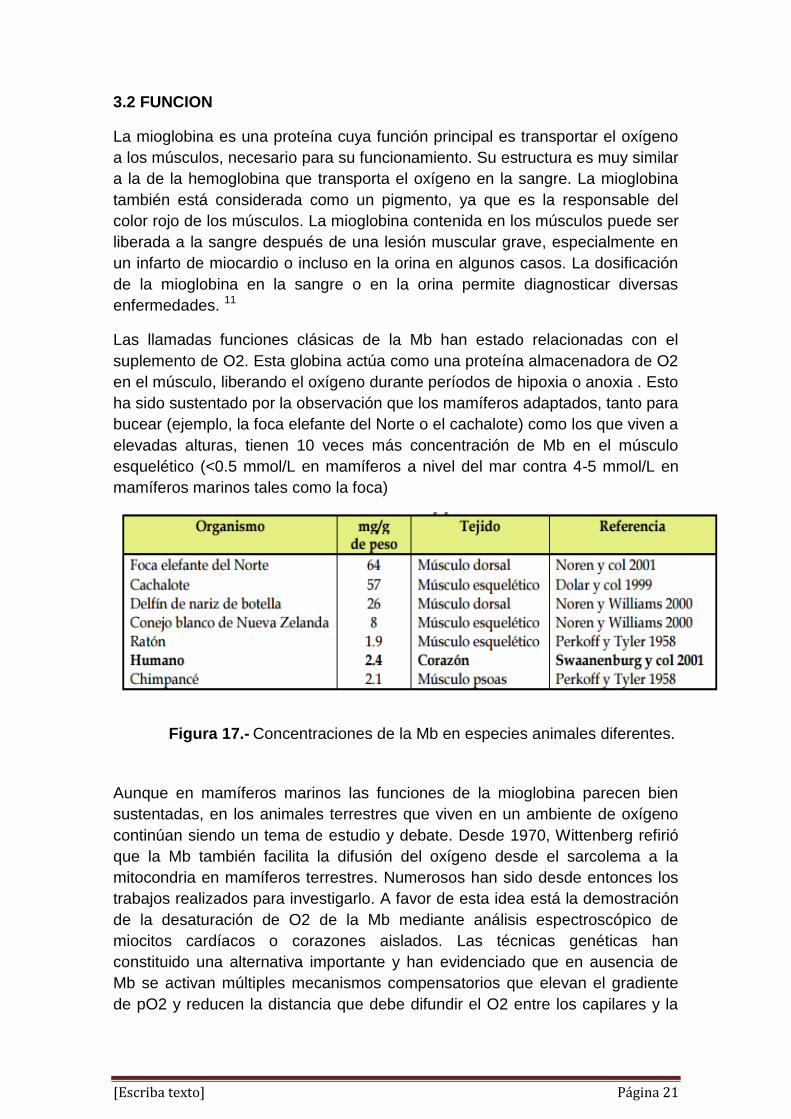
Figura 17.- Concentraciones de la Mb en especies animales diferentes.
3.2 FUNCION
La mioglobina es una proteína cuya función principal es transportar el oxígeno
a los músculos, necesario para su funcionamiento. Su estructura es muy similar
a la de la hemoglobina que transporta el oxígeno en la sangre. La mioglobina
también está considerada como un pigmento, ya que es la responsable del
color rojo de los músculos. La mioglobina contenida en los músculos puede ser
liberada a la sangre después de una lesión muscular grave, especialmente en
un infarto de miocardio o incluso en la orina en algunos casos. La dosificación
de la mioglobina en la sangre o en la orina permite diagnosticar diversas
enfermedades. 11
Las llamadas funciones clásicas de la Mb han estado relacionadas con el
suplemento de O2. Esta globina actúa como una proteína almacenadora de O2
en el músculo, liberando el oxígeno durante períodos de hipoxia o anoxia . Esto
ha sido sustentado por la observación que los mamíferos adaptados, tanto para
bucear (ejemplo, la foca elefante del Norte o el cachalote) como los que viven a
elevadas alturas, tienen 10 veces más concentración de Mb en el músculo
esquelético (<0.5 mmol/L en mamíferos a nivel del mar contra 4-5 mmol/L en
mamíferos marinos tales como la foca)
Aunque en mamíferos marinos las funciones de la mioglobina parecen bien
sustentadas, en los animales terrestres que viven en un ambiente de oxígeno
continúan siendo un tema de estudio y debate. Desde 1970, Wittenberg refirió
que la Mb también facilita la difusión del oxígeno desde el sarcolema a la
mitocondria en mamíferos terrestres. Numerosos han sido desde entonces los
trabajos realizados para investigarlo. A favor de esta idea está la demostración
de la desaturación de O2 de la Mb mediante análisis espectroscópico de
miocitos cardíacos o corazones aislados. Las técnicas genéticas han
constituido una alternativa importante y han evidenciado que en ausencia de
Mb se activan múltiples mecanismos compensatorios que elevan el gradiente
de pO2 y reducen la distancia que debe difundir el O2 entre los capilares y la
[Escriba texto] Página 22
mitocondria. Se ha demostrado que la Mb funciona facilitando la difusión del
oxígeno en el cardiomiocito y típicamente permanece saturada al 40- 70 % con
O2 (Mb + O2 ↔ MbO2). Estudios muy recientes apoyan este criterio. La
actividad peroxidasa de esta proteína fue informada por primera vez por
George e Irving, en 1955.
3.3.- ORGANIZACIÓN ESTRUCTURAL
La mioglobina es el ejemplo clásico de una proteína globular, se compone de
una cadena de polipéptido (en la que la mayoría de los aminoácidos hidrófobos
se encuentran en el interior y muchos de los residuos polares están expuestos
en la superficie) e incluye un grupo prostético, grupo hemo, que también
aparece en la hemoglobina (dentro de una cavidad hidrófoba de la proteína).
Esta unidad no polipeptídica se encuentra unida de manera no covalente a la
mioglobina y es esencial para la actividad biológica de unión de O2 de la
proteína. Se pliega en forma compacta que mide 4.5x3.5x2.5 nm 9
ESTRUCTURA PRIMARIA Y SECUNDARIA
La mioglobina es una cadena polipeptídica de 153 residuos aminoaciso (PM
17.000 daltons). Es una proteína globular con la superficie polar (rojo) y el
interior apolar (amarrillo), patrón característico de las proteínas globulares.
Sin contar los dos residuos de His que intervienen en el proceso de fijación del
oxígeno, el interior de la mioglobina sólo contiene residuos no polares (por
ejemplo, Leu, Val, Phe y Met). 3
Fig. 18.-Distribución espacial de los residuos de aminoácidos polares y apolares.
Los aminos hidrofóbicos están en el interior de la globina: hidrofóbicos (en rojo) ,
hidrofilicos (en verde)

[Escriba texto] Página 23
Fig. 19.-Estructura de la Mioglobina
ESTRUCTURA TERCIARIA
La estructura terciaria depende lógicamente de su estructura primaria, así las
cadenas laterales de los aminoácidos en las proteínas globulares se hallan
distribuidas espacialmente de acuerdo con sus polaridades, de tal forma que:
Los restos no polares aparecen, casi siempre, en el interior de la proteína,
para no entrar en contacto con el disolvente acuoso que la envuelve, creando
un ambiente hidrofóbico.
Los residuos polares con carga se hallan situados, normalmente en la zona
externa, interaccionando con el medio acuoso. A veces, se requiere de estos
centros en la parte interna de la proteína y en estos casos también ocurre que
están directamente implicados en alguna funcionalidad de la proteína, bien a
nivel estructural o bien a nivel catalítico.
La estructura terciaria de la mioglobina es la de una proteína típica globular
soluble en agua. Su estructura secundaria es inusual ya que contiene una alta
proporción (75%) de la estructura secundaria α-helicoidal. 7
El color de la cadena polipeptídica está graduado a lo largo del espectro visible
desde azul (N terminal) hasta marrón claro (c terminal). El grupo prostético hem
se muestra en color rojo. las regiones helicoidales α están designadas de la a a
la H. los residuos histidina distal (E7) y proximal (F8) se resaltan en azul y
naranja, respectivamente. Note de qué modo los sustituyentes propionato
polares (pr) se proyectan hacia afuera del hem hacia el solvente.

[Escriba texto] Página 24
3.4. LA MIOGLOBINA COMO MARCADOR CLINICO
La Mb es sintetizada y ejerce sus funciones principalmente en los músculos
esquelético y cardíaco, y está prácticamente ausente en el músculo liso. Se
encuentra además en el suero sanguíneo y la orina a muy bajas
concentraciones .Kitao determinó el contenido de Mb en músculos humanos y
encontraron que el contenido fue menor en el músculo cardíaco que en el
esquelético. Los niveles normales de Mb en el suero determinados por
radioinmunoensayo oscilan entre 6 y 85 ng/mL .Se han evidenciado diferencias
entre ambos sexos y se informaron como valores normales para las mujeres 3-
76 ng/mL y para los hombres, 12-78 ng/mL. Los niveles normales en la orina se
encuentran por debajo de 50 ng/mL Anesi y col (2000) encontraron diferencias
en los niveles séricos de Mb, no sólo relacionadas con el sexo, sino además
con la edad. 11
Fig. 20.-Cadenas Polipépticas de la mioglobina
Fig. 21.- Contenido de Mb de varios músculos esqueléticos humanos-Según Kitao

[Escriba texto] Página 25
3.5 LAS HISTIDINAS F8 Y E7 LLEVAN A CABO
FUNCIONES ÚNICAS EN LA FIJACIÓN DE OXÍGENO
El hem de la mioglobina, que se encuentra en un surco entre las hélices E y F (figura 22), se orienta con sus propionatos polares en la superficie. El resto se proyecta hacia el interior de la molécula de mioglobina en donde, con excepción de His E7 y de His FS, los residuos circundantes son no polares. La quinta posición de coordinación del átomo de hierro se une al nitrógeno de un anillo de la histidina proximal, His F8 (figura 2). Aunque no está enlazada a la sexta posición de coordinación del hierro, la histidina distal (His E7) permanece en el lado del anillo hemico al otro lado de His F8 (figura 23).
Figura 22. Modelo de mioglobina de baja resolución S610 se
muestran los Átomos de carbono alfa. Las regiones donde se
localizan las hélices alfa se designan con las letras que van de la A
hasta la H.
Figura 23. Adición del oxígeno al hierro hemico en
la oxigenación

[Escriba texto] Página 26
EL HIERRO SE MUEVE HACIA EL PLANO DEL HERN
CUANDO SE FIJA EL OXÍGENO En la rnioglobina no oxigenada, el hierro del hem se encuentra aproximadamente a
0.03 nm (0.3 A) fuera del plano del anillo en dirección a His F8. En la mioglobina
oxigenada, un átomo de oxigeno ocupa la sexta posición de coordinación del entorno
de hierro y entonces &te se desplaza a 0.01 nm (0.1 A) fuera del plano del hem. Por
tanto, la oxigenación de la rnioglobina va acornpañada por el movimiento del átomo de
hierro, y el consecuente desplazamiento de His F8 y los residuos aminoacidicos
enlazados por covalencia a CI, hacia el plano del anillo. Este movimiento produce una
nueva conformación de ciertas porciones de la proteína.
LA APOMIOGLOBINA PROPORCIONA UN AMBIENTE
ADVERSO PARA EL HIERRO HÉMICO Cuando el oxígeno se une a la rnioglobina, el enlace entre este y Fe2' es perpendicular al plano
del anillo hemico. Un segundo átomo de oxigeno se une en un ángulo de 121" con respecto al
plano del hem y orientado alejándose de la h histidina dista1 (figura 24).
El monóxido de carbono (CO) se une al hem aislado con una afinidad
aproximadamente de 25 000 veces mayor que la del oxígeno. Si la atmosfera
contiene restos de CO y el catabolismo normal del propio hem produce
cantidades pequeñas de este gas, ¿por qué entonces no es el CO (en lugar del
O2) es que ocupa la sexta posición de coordinación del hierro hemico de la
mioglobina? La respuesta se encuentra en el ambiente adverso del hem en la
mioglobina. La orientación preferida del CO para enlazarse al hierro hemico es
con los tres átomos (Fe, C, O) en posición perpendicular respecto al anillo
hemico (figura 3). Aunque esta orientación es posible para el hem aislado. En
la mioglobina la histidina distal obstaculiza estéricamente a los enlaces de CO
en este Angulo (figura 25). Esto obliga al CO a unirse en una configuración
Figura 24. Ángulos preferidos para el enlace del oxígeno y el rnonóxido de
carbono al átomo de hierro del hem libre

[Escriba texto] Página 27
menos favorable y reduce la fuerza de la unión hem-CO en más de dos
órdenes de magnitud, aproximadamente 200 veces la fuerza del enlace hem-
02. No obstante, por lo común, una pequeña cantidad (más o menos 1%) de la
mioglobina está presente en forma de mioglobina-CO.
3.6 LAS CURVAS DE DISOCIACIÓN DEL OXIGENO
PARA LA MIOGLOBINA EXPLICAN SUS FUNCIONES
FISIOLOGICAS
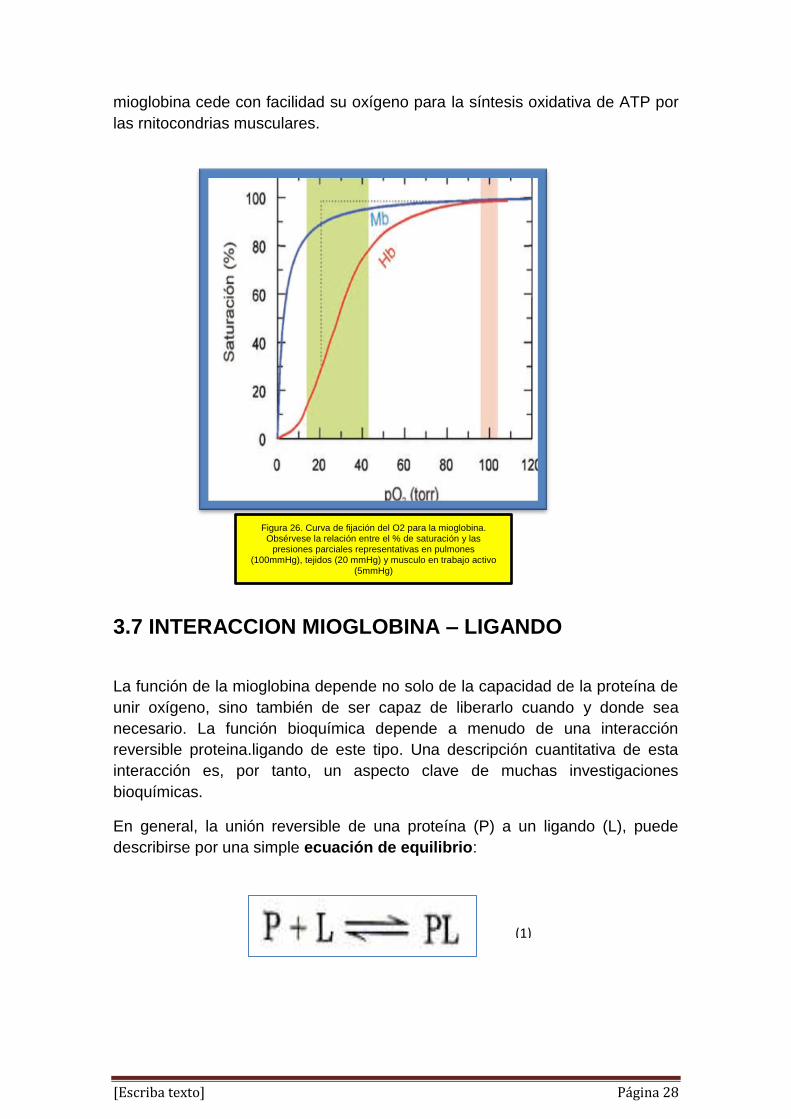
¿Por qué la mioglobina es inadecuada como proteína transportadora de
oxígeno, pero eficaz para almacenarlo?
La cantidad de oxigeno unido a la rnioglobina (expresada como "porcentaje de
saturación ") depende de la concentración de oxigeno (expresada como PO2 o
presión parcial de oxigeno) en el entorno inmediato del hierro hemico. En
relación entre PO2 y la cantidad de oxígeno unido puede graficarse como una
curva de saturación de oxigeno (disociación del oxígeno). Para la mioglobina, la
forma de la isoterma de absorción de oxígeno es hiperbólica (figura 26). Dado
que la PO2 en el lecho capilar pulmonar es de 100 mm Hg, la mioglobina
podría retener oxigeno de manera eficaz en los pulmones. Sin embargo, la PO2
de la sangre venosa es de 40 mm Hg y la del músculo activo aproximadamente
de 20 mm Hg. Puesto que la mioglobina no puede liberar una fracción grande
de su oxigeno unido aún a 20 mm Hg, no sirve como un vehículo eficaz para el
transporte del oxígeno de los pulmones a los tejidos periféricos. No obstante,
en la privación de oxigeno que acompaña al ejercicio físico extenuante, la PO2
del tejido muscular puede disminuir hasta 5 mm Hg. A esta presión, la
Figura 25. Ángulos de enlace del oxígeno y el rnonóxido de carbono
al hierro del hemico de la mioglobina.
[Escriba texto] Página 28
mioglobina cede con facilidad su oxígeno para la síntesis oxidativa de ATP por
las rnitocondrias musculares.
3.7 INTERACCION MIOGLOBINA – LIGANDO
La función de la mioglobina depende no solo de la capacidad de la proteína de
unir oxígeno, sino también de ser capaz de liberarlo cuando y donde sea
necesario. La función bioquímica depende a menudo de una interacción
reversible proteina.ligando de este tipo. Una descripción cuantitativa de esta
interacción es, por tanto, un aspecto clave de muchas investigaciones
bioquímicas.
En general, la unión reversible de una proteína (P) a un ligando (L), puede
describirse por una simple ecuación de equilibrio:
Figura 26. Curva de fijación del O2 para la mioglobina. Obsérvese la relación entre el % de saturación y las
presiones parciales representativas en pulmones (100mmHg), tejidos (20 mmHg) y musculo en trabajo activo
(5mmHg)
(1)

[Escriba texto] Página 29
La reacción se caracteriza por una constante de equilibrio Ka de manera que:
Donde Ka es una constante de velocidad. El termino Ka es una constante de
asociación (no9 hay que confundirla con la Ka que representa la constante de
disociación de una acido) que describe el equilibrio existente entre el complejo
y sus componentes libres no unidos. La constante de asociación proporciona
una medida de afinidad del ligando L por la proteína. La Ka tiene unidades en
M-1; un valor más alto de Ka corresponde a una mayor afinidad del ligando
para la proteína.
La redistribución de la primera parte de la ecuación muestra que la relación
entre la proteína unida y la proteína libre es directamente proporcional a la
concentración del ligando libre.
Cuando la concentración de ligando es mucho mayor que la concentración de
sitios de fijación de ligando, la unión de ligando por parte de la proteína no
afecta de manera significativa a la concentración de ligando libre (no unido),
con lo que [L] permanece constante. Esta situación es aplicable en general a la
mayoría de ligando que se unen a las proteínas en células y simplifica nuestra
descripción del equilibrio de unión.
Podemos ahora considerar la constante de equilibrio en función de la fracción θ
(teta), de sitios de fijación de ligando de la proteína que están ocupados por el
ligando:
Sustituyendo Ka [L] [P] por [PL] y reordenando los términos tenemos:
(2)
(3)
(4)

[Escriba texto] Página 30
El valor de Ka se puede determinar a partir de una representación de θ frente a
la concentración de ligando libre, [L]. Cualquier ecuación de la forma x = y/(yaz)
describe una hipérbole, por lo que θ resulta ser una función hiperbólica de [L].
La fracción de sitios de fijación de ligando ocupados tiende a la saturación de
manera asintótica a medida que aumenta [L]. La [L] a la que la mitad de los
sitios de fijación de ligando disponibles están ocupados(es decir, θ = 0,5)
corresponde a 1/Ka.
Sin embargo, a veces es más frecuente considerar la constante de
disociación, Kd, que es el reciproco de Ka (Kd=1/Ka) y que se expresa en
unidades de concentración molar (M). Kd es la constante de equilibrio para la
liberación del ligando. Las ecuaciones cambian a:
Cuando [L] es igual al Kd la mitad de los sitios de fijación de ligando están
ocupados. Cuando [L] desciende por debajo de Kd, cada vez hay menos
proteína con ligando unido. Para que un 90% de los sitios de fijación de ligando
disponibles estén ocupados, [L] debe ser nueve veces mayor que Kd.
En la práctica Kd se utiliza mucho más a menudo que Ka, para expresar la
afinidad de una proteína por su ligando.
Observe que un valor más bajo de Kd corresponde a una mayor afinidad de
ligando por la proteína. Las operaciones matemáticas pueden reducirse a
frases simple: Kd equivale a la concentración molar de ligando a la cual la
mitad de los sitios de fijación del ligando disponibles están ocupados. En este
punto, se dice que la proteína ha alcanzado semisaturacion con respecto a la
unión de ligando. Cuanto más fuerte es la unión de una proteína a su
(5)
(6)
(7)
[Escriba texto] Página 31
ligando,menor será la concentración del ligando requerida para que estén
ocupados la mitad de los sitios de fijación y, por tanto, menor será el valor de
Kd. La unión de oxígeno a la mioglobina sigue los patrones descritos
anteriormente. Sin embargo, al ser el oxígeno un gas, debemos hacer algunos
ajustes mínimos en las ecuaciones para poder proceder con más facilidad a los
experimentos de laboratorio necesarios. En primer lugar sustituimos la
concentración de oxígeno disuelto por [L] en la ecuación:
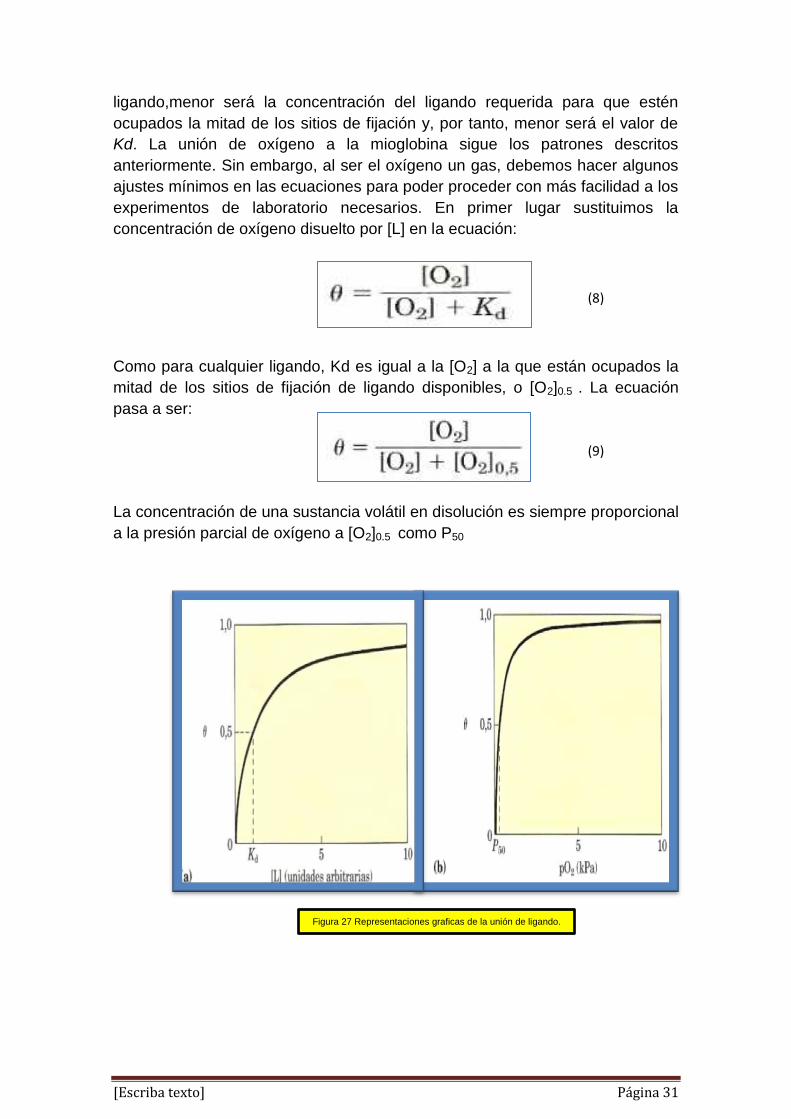
Como para cualquier ligando, Kd es igual a la [O2] a la que están ocupados la
mitad de los sitios de fijación de ligando disponibles, o [O2]0.5 . La ecuación
pasa a ser:
La concentración de una sustancia volátil en disolución es siempre proporcional
a la presión parcial de oxígeno a [O2]0.5 como P50
Figura 27 Representaciones graficas de la unión de ligando.
(9)
(8)
[Escriba texto] Página 32
3.8 ENFERMEDADES RELACIONADAS CON LA
MIOGLOBINA
RABDOMIÓLISIS
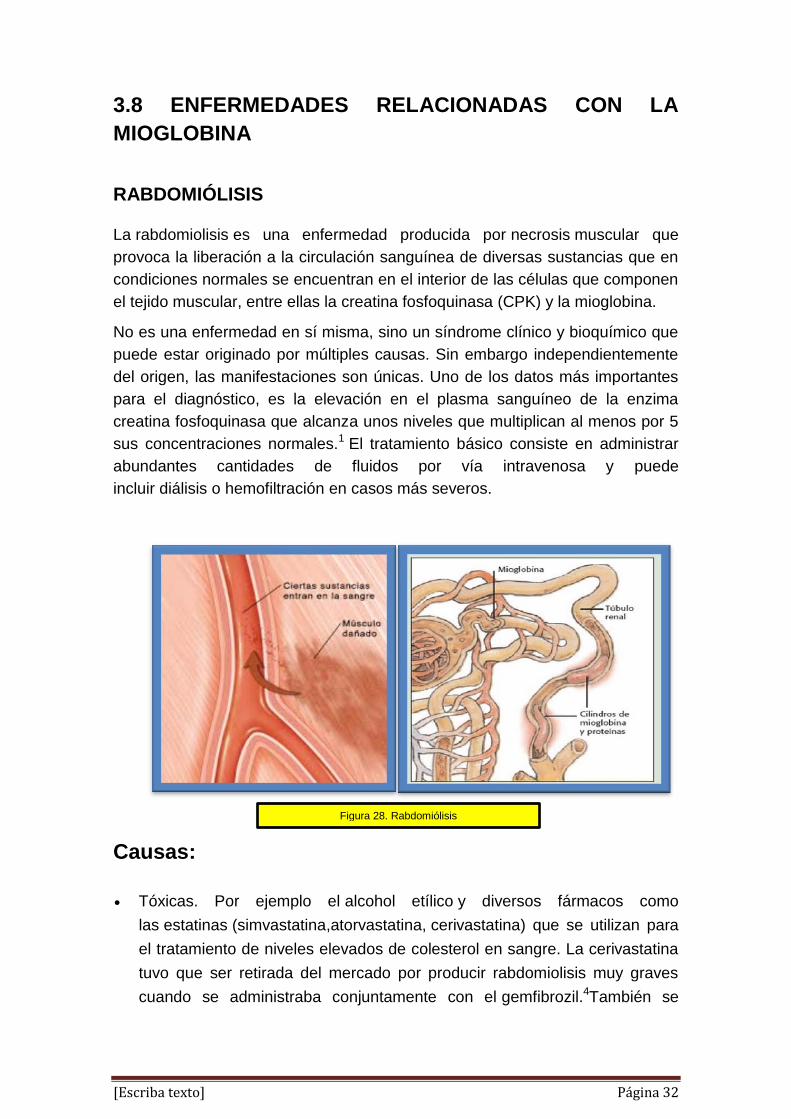
La rabdomiolisis es una enfermedad producida por necrosis muscular que
provoca la liberación a la circulación sanguínea de diversas sustancias que en
condiciones normales se encuentran en el interior de las células que componen
el tejido muscular, entre ellas la creatina fosfoquinasa (CPK) y la mioglobina.
No es una enfermedad en sí misma, sino un síndrome clínico y bioquímico que
puede estar originado por múltiples causas. Sin embargo independientemente
del origen, las manifestaciones son únicas. Uno de los datos más importantes
para el diagnóstico, es la elevación en el plasma sanguíneo de la enzima
creatina fosfoquinasa que alcanza unos niveles que multiplican al menos por 5
sus concentraciones normales.1 El tratamiento básico consiste en administrar
abundantes cantidades de fluidos por vía intravenosa y puede
incluir diálisis o hemofiltración en casos más severos.
Causas:
Tóxicas. Por ejemplo el alcohol etílico y diversos fármacos como
las estatinas (simvastatina,atorvastatina, cerivastatina) que se utilizan para
el tratamiento de niveles elevados de colesterol en sangre. La cerivastatina
tuvo que ser retirada del mercado por producir rabdomiolisis muy graves
cuando se administraba conjuntamente con el gemfibrozil.4También se
Figura 28. Rabdomiólisis

[Escriba texto] Página 33
asocia la rabdomiolisis al consumo de grandes cantidades en un periodo de
tiempo corto de la seta Tricholoma equestre, hasta hace poco tiempo
clasificada como comestible.
Daño muscular directo ocasionado por congelaciones, quemaduras
o isquemia por oclusión de los vasos sanguíneos que transportan el
oxígeno a los grupos musculares.
Ejercicio excesivo y entrenamientos en condiciones extremas (por ejemplo:
bailar, correr o hacer spinning crossfit hasta caer extenuado). También se
han documentado casos de rabdomiolisis en personas sometidas
a electroestimulación.5
Procesos infecciosos que afectan al tejido muscular.
Síntomas:
Los síntomas iniciales son fiebre, náuseas, vómitos y debilidad muscular. Los
músculos más afectados suelen ser los gemelos. Más adelante surgen
complicaciones que incluyen insuficiencia cardíaca y arritmias potenciadas por
los niveles altos de potasio en sangre. También coagulación intravascular
diseminada y fallo renal agudo desencadenado por los depósitos en el riñón de
la mioglobina liberada en la destrucción de las células musculares.
IMPLICACIONES BIOMEDICAS
MIOGLOBINURIA
Después de lesiones múltiples, la mioglobina
liberada de las fibras musculares rotas aparece
en la orina, coloreándola de rojo oscuro. Aunque
después de un infarto del miocardio es posible
detectar la mioglobina en el plasma, el análisis
de las enzimas séricas proporciona un índice
más sensible del daño miocárdico.
Figura 29. Mioglobinuria.

[Escriba texto] Página 34
4.- REFERENCIA BIBLIOGRAFICA
1. Velasquez J.,Gallardo D.. Fisiología de la sangre y del sistema
Inmunologico.1ra Ed. Lima -Perú :FACULTAD DE MEDICINA UNMSM-
FACULTAD DE MEDICINA UNIV. PARTICULAR SAN MARTN DE
PORRES;1999.pp 59-66.
2. Structure of hemoglobin – a overview [homepage on the Internet]:
BIOCHEMESTRY FOR MEDICS; c2015 [cited 2015 May 1]. Available
from: http://www.namrata.co/category/hemoglobin-and-
hemoglobinopathies/structure-of-normal-hemoglobin/
3. Iwasa J.. Hemoglobin: Studying the T to R Transition [homepage on the
Internet]:Visualization of molecular processes; [cited 2015 May 1].
Available from:
http://biochem.web.utah.edu/iwasa/projects/hemoglobin.html
4. Brandan N. , Aguirre M., Giménez C..HEMOGLOBINA Cátedra de
Bioquímica-Facultad de Medicina UNME;2008
5. Synthesis, Structure and functions of hemoglobin . [Homepage on the
Internet]:DOW UNIVERSITY OF HEALTH SCIENCES[cited 2015 May
1]. Available from: http://www.duhs.edu.pk/curriculum/downloads/lec4-
sem1-BMWK1-20140221.pdf
6. Vagara Fernández J. Bioquímica Biología Molecular .Universidad de
Huelva -Facultad de Ciencias Experimentales Campus de "El Carmen".
Capítulo 5 .España .2008
7. Fernández Sevilla J. Estructura y función de proteínas y péptidos:
mioglobina, hemoglobina, misiona, caseína, colágeno, gluten, lacto
albúmina y ovoalbúmina. [Monografía en internet]. Dpt. Ingeniería
Química. Ampliación de Tecnología de los Alimentos[Actualizado el 27
de Julio de 2005; consultado: 22 de Abril]. [9 p.]. Disponible en:
http://www.itescham.com/Syllabus/Doctos/r637.PDF
8. Maldonado V. Bioquímica de la nutrición 2 .Universidad Tolteca de
México A.C. Grupo Nutrición .2013
9. Harper. Bioquímica avanzada. 26 edición. Biblioteca Nacional de
México. México: Editorial El Manual Moderno. 2004.
10. Baynes.Bioquímica Médica.3ª ed. Edit. Elsevier, Caps 2,
5..España.2011.
11. Maisel AS, McCord J, Nowak RM, Hollander JE, Wu AH, Duc P, et al.
Bedside B-Type natriuretic peptide in the emergency diagnosis of heart
failure with reduced or preserved ejection fraction. Results from the
Breathing Not Properly Multinational Study. J Am Coll
12. Teijón J M. Fundamentos de bioquímica estructural. Segunda Edición. Editorial Tébar .Madrid. 2006

[Escriba texto] Página 35
13. Harper. Bioquímica avanzada. 26 edición. Biblioteca Nacional de
México. México: Editorial El Manual Moderno. 2004.
14. Lehninger. Principios de la Bioquímica. 6 edición. Barcelona: Ediciones
Omega. 2015
15.-Guillén Astete, CA; Zegarra Mondragón, S; Medina Quiñones, C (9 de
febrero de 2015).