detección del receptor trka como un posible marcador ... · tesis para optar al grado de magíster...
TRANSCRIPT

Detección del Receptor trkA como un posible marcador pronóstico y de
progresión en Cáncer Ovárico Epitelial (COE)
MARCELA FRANCISCA MUÑOZ CARVACHO
Tesis para optar al grado de Magíster en Bioquímica
Área de especialización: Bioquímica Clínica
Directora de Tesis: Dra. Carmen Romero Osses
2008
Universidad de Chile Facultad de Ciencias Químicas y Farmacéuticas

2
UNIVERSIDAD DE CHILE
Facultad de Ciencias Químicas y Farmacéuticas
Informe de Aprobación
TESIS DE MAGISTER Se informa a la comisión de Postgrado de la Facultad de Ciencias Químicas y Farmacéuticas que la Tesis de Magíster presentada por la candidata:
MARCELA FRANCISCA MUÑOZ CARVACHO Ha sido aprobada por la comisión informante de Tesis como requisito de tesis para optar al Grado de Magíster en Bioquímica, en el examen de defensa de tesis rendido el………………………………………………………………….
Directora de Tesis: Dra. Carmen Romero Osses…………………………………………………….
Comisión informante de Tesis: Dr. Alfonso Paredes V…………………………………………………………. Dr. Javier Puente P……………………………………………………………... Dra. Margarita Vega B………………………………………………………….

3
A mis queridos padres: Yolanda y Raúl
Y por supuesto al amor de mi vida Jaime

4
AGRADECIMIENTOS Durante todo este camino me han acompañado y ayudado muchas personas, de
quienes estoy muy agradecida, por lo que espero nombrarlos a todos. A mi madre, porque
gracias a su inmenso esfuerzo puedo estar hoy en esta situación y por ello siempre estaré
inmensamente agradecida. Además le agradezco por todo el apoyo, preocupación, cariño y
ayuda que siempre me ha brindado. A mi padre, del cual también he recibido mucho apoyo,
cariño, ayuda, preocupación y consejos. Muchas gracias a los dos y recuerden que los
quiero muchísimo, los admiro y estoy muy orgullosa de ustedes. A mis hermanos y
sobrinos, quienes han estado presentes de una u otra forma durante este proceso y me han
hecho sentir muy apoyada y querida. Muchas gracias y recuerden que también yo los quiero
mucho ustedes. A mi novio Jaime el cual ha sido mi pilar durante todos estos años, me ha
brindado siempre su ayuda, su apoyo, cariño y protección. Me ayudó a salir adelante en los
momentos difíciles y a solucionar los problemas que se me presentaron. Estoy muy
agradecida y muy contenta de tenerte a mi lado, porque contigo soy muy feliz, Te amo con
todo mi corazón y muchísimas gracias por todo.
A mis compañeros del laboratorio: Ale, Maca, Vero, Pauli, Fran, Carlos, María Paz,
Rodrigo, y a unos excompañeros: Luis, Enrique y Ketty. Porque siempre conté con su
ayuda, me dieron ánimo en los momentos difíciles e hicieron que este tiempo en el
laboratorio fuese muy agradable. Muchas gracias a todos, los quiero mucho.
A mi directora de tesis, Carmen Romero, a quien le agradezco mucho por haberme
recibido, por todo lo que me enseñó y por haber estado conmigo en los buenos y malos
momentos de este proceso, muchas gracias. A la Dra. Margarita Vega, por todo el apoyo y
ayuda que he recibido de su parte. Estoy muy agradecida y feliz de haberla conocido
porque es una gran persona. Al Dr. Alfonso Paredes por su colaboración en esta tesis, y su
buena voluntad y también al Dr. Javier Puente por su participación en este proceso, muchas
gracias a ambos. A Fernando Gabler, le agradezco por toda la ayuda y buena voluntad que
siempre ha tenido con todos nosotros. Muchas gracias.
Y finalmente a todo el resto del personal del laboratorio de Endocrinología y
Biología de la Reproducción, con quienes compartí muchos momentos agradables y les
tengo mucho aprecio. En fin muchas gracias a todos quienes de alguna forma me ayudaron
a cumplir esta etapa de la mejor manera.

5
INDICE
Resumen 7
Summary 9
Financiamiento 11
I. Introducción 12
I.1 El ovario y cáncer ovárico epitelial (COE) 12
I.2 Neurotrofinas y sus receptores 15
I.3 La importancia de NGF y su receptor trkA en la fisiología ovárica y en COE 17
I.4 Angiogénesis en la progresión del COE 19
II Hipótesis 21
III Objetivos 21
III.1 Objetivo General 21
III.2 Objetivos Específicos 21
IV Metodología 22
IV.1. Obtención de las Muestras 22
IV.1.2 Diseño Experimental 23
IV.2 Análisis de Muestras 24
IV.2.1 Estudio Inmunohistoquimico 24
IV.2.1.1 Estudio de RT-PCR 26
IV.3. Análisis Estadístico 27
V Resultados 28
V.1 Niveles de mRNA de VEGF durante la progresión carcinogénica ovárica 28
V.1.1 Localización y semicuantificación de la proteína VEGF durante la progresión
carcinogénica 32
V.2 Niveles de mRNA de NGF durante la progresión carcinogénica ovárica 34
V.2.1 Localización y semicuantificación de la proteína NGF durante la progresión
carcinogénica 35
V.3 Niveles de mRNA de trkA durante la progresión carcinogénica ovárica 37
V.3.1 Localización y semicuantificación de la proteína trkA durante la progresión
carcinogénica 38

6
V.4 Localización y semicuantificación de la proteína p-trkA durante la progresión
carcinogénica 40
VI Discusión 45
VII Conclusiones 51
VIII Proyecciones 52
IX Referencias 53
X Anexo 1 59

7
RESUMEN El cáncer ovárico representa la segunda causa de muerte de origen ginecológico, y
el 80-90% corresponde a carcinoma ovárico epitelial (COE), del cual el 40% es de tipo
seroso. Esta patología se caracteriza por presentar un transcurso silente, una detección
tardía, responder pobremente a terapias, ser altamente angiogénico y de sobrevida muy
baja. La etiología del COE no está clara y se postulan varias hipótesis al respecto. En
carcinoma de páncreas, colon, tiroide papilar y medular, próstata y neuroblastoma, se ha
reportado una sobreexpresión del receptor tirosina kinasa (trkA). En cuanto a su ligando, el
factor de crecimiento nervioso (NGF) se ha reportado su participación directa e indirecta a
través de su receptor trkA, en la angiogénesis, evento crucial para el desarrollo y progresión
del tumor. El factor angiogénico más estudiado es el factor de crecimiento de endotelio
vascular (VEGF) y se ha encontrado una sobreexpresión de las isoformas 121, 165 y 189,
las cuales aumentan por efecto de NGF en explantes de cáncer ovárico epitelial. Si bien, ya
se sabe que el receptor trkA y su ligando NGF participan en la angiogénesis del ovario, no
hay antecedentes claros que indiquen si el receptor trkA y su ligando NGF están
involucrados en la génesis y progresión del COE. Por lo que nos planteamos la siguiente
Hipótesis: trkA modifica su expresión durante la carcinogénesis ovárica constituyendo un
nuevo marcador tumoral. Para desarrollar este estudio nos propusimos el siguiente
Objetivo General: Evaluar los niveles del receptor trkA y de NGF, durante las distintas
etapas de transición del epitelio ovárico (desde el tejido ovárico normal al cáncer ovárico
epitelial) y si esta expresión se relaciona con la expresión de VEGF.
Metodología: En 30 muestras de ovarios: ovarios inactivos (OV-I), tumores
benignos (T.Ben), borderline (T.Bord) y COE grados I, II y III (n=5 para cada grupo), se
evaluó la localización celular y niveles proteicos del receptor trkA total, trkA-p, NGF y
VEGF mediante inmunohistoquímica (IHQ). Para la semicuantificación se sumaron los
porcentajes de las intensidades 2 y 3 y se expresó como el porcentaje de células con tinción
positiva en el epitelio. Además, en tejidos congelados se analizaron los niveles de mRNA
del receptor trkA, NGF y VEGF por RT-PCR, los resultados por PCR fueron normalizados
con el gen constituivo β – actina.
Resultados: Se encontró un aumento discreto en los niveles de mRNA de NGF
durante la progresión carcinogénica, y estos aumentos fueron significativos entre OV-I vs

8
COE I, COE II COE III (p<0,05). En cuanto a la expresión proteica de NGF, también se
encontró un aumento significativo entre OV-I vs COE II y COE III (p<0,01). Los productos
de splicing alternativos que codifican para las isoformas de VEGF 121, 165 y 189
aumentaron significativamente durante la progresión carcinogénica. VEGF 121 aumentó
significativamente entre OV-I vs COE I, II y III vs (p<0,001). VEGF 165 aumentó
significativamente entre OV-I vs COE I, II y III (p<0,001), y para VEGF 189 entre OV-I vs
COE I, COE II y III (p<0,01). En cuanto a la expresión proteica de VEGF, se encontró un
aumento durante la progresión, el cual fue significativo entre OV-I vs COE I, II y III (p<
0,001). Se encontró un aumento gradual y significativo en los niveles de mRNA de trkA, a
medida que se va perdiendo la diferenciación celular. Este aumento fue significativo entre
T.Bord vs COE I, COE II y III (p<0,01), COE I vs COE III (p<0,001) y COE II vs COE III
(p<0,01). En las muestras de ovario normal inactivo y tumores benignos no fue posible
encontrar la expresión génica de trkA, en parte por la poca cantidad de epitelio que
encontramos en estos grupos. En cuanto a la expresión proteica del receptor trkA total, se
observó un aumento gradual y significativo durante la progresión; principalmente entre
OV-I vs COE I, II y III (p<0,001). De igual forma, los niveles proteicos del receptor activo
p-trkA, aumentaron significativamente durante la progresión del COE, entre COE I vs COE
III (p<0,01). En los grupos de OV-I, T.Ben y T.Bord, no se encontró tinción positiva para
p-trkA.
Discusión: Tanto la expresión proteica del receptor trkA total y p-trkA, como los
niveles de mRNA de trkA, aumentan durante la progresión carcinogénica, principalmente
entre los COE, y dichos aumentos se elevan significativamente con la indiferenciación del
tejido, lo que confirmaría su posible uso como marcador de progresión del COE y también
como marcador de mal pronóstico, ya que la proteína p-trkA se detectó desde COE I en
adelante y a nivel de mRNA no fue detectado en los grupos de ovario inactivo y tumor
benigno, sólo a partir de los tumores borderline comienza a detectarse pero muy
débilmente, a diferencia de lo que ocurre en COE donde su expresión génica como proteica
es muy alta. Este aumento en la expresión proteica de trkA durante la progresión se
correlaciona positiva y significativamente con la expresión proteica de NGF y VEGF. Estos
últimos corresponden a factores proangiogénicos, importantes para el desarrollo, progresión
y metástasis tumoral.

9
SUMMARY Ovarian cancer is the second cause of death from gynecological origin, of which 80-
90% correspond to epithelial ovarian carcinoma (EOC). This pathology is characterized by
presenting a silent course, a late detection, poor response to therapy, highly angiogenic and
has a very low survival rate. The EOC etiology is unclear, and several hypotheses put
forward in this regard. Carcinoma of pancreas, colon, thyroid papillary and marrow,
prostate, neuroblastoma and miloide acute leukemia have been reported to overexpress
tyrosine kinase receptor (trkA). Its ligand nerve growth factor (NGF), a direct and indirect
action in angiogenesis through its receptor trkA, a crucial event in the development and
progression of tumors. The most studied angiogenic factor correspond to vascular
endothelial growth factor (VEGF) and it has been reported that isoforms 121, 165 and 189
are overexpressed, wich are increased due to NGF in explants of ovarian cancer. The trkA
receptor and its ligand NGF are involved in angiogenesis, but is no clear if the trkA
receptor and its ligand NGF are involved in the genesis and progression of the EOC. Then
our hypothesis is that: trkA modifies its expression during carcinogenesis and as a new
ovarian tumor marker. To develop this study, our general objectives was: To evaluate the
expression of trkA receptor and NGF, during the various stages of epithelial ovarian
transition (from normal ovarian tissue to epithelial ovarian cancer) and to evaluate if this
expression is related to the VEGF expression.
Methodology: 30 samples of ovaries: inactive ovaries (OV-I), benign tumors (T.
Ben), borderline (T. Bord) and EOC grades I, II and III (n = 5 for each group) were
evaluated for trkA, p-trkA, NGF and VEGF by immunohistochemistry (IHQ).
Immunohistochemical evaluation for each protein was performed by a semicuantitative
analysis, where intensity of the staining is scored on a scale of 1- 3 (1= low intensity; 2=
mild intensity, and 3= higher intensity). Results are expressed as percentage of positive
stained epitelials cells wich correspond to the sum of the percentage of cells stained in the
scale 2 and 3. In addition, frozen tissues we used to analyzed mRNA levels of trkA
receptor, NGF and VEGF by RT-PCR, results were normalizated relative to β – actin gen
product.
Results: We found an increase for the mRNA levels of NGF, during the
carcinogenic progression, and these increases were significant between OV-I vs EOC I

10
(p<0,05), OV-I vs EOC II (p<0,01) and OV-I vs EOC III (p<0,001). Protein expression of
NGF during the carcinogenic progression increased between OV-I vs EOC II y EOC III
(p<0,01). We also found that mRNA levels of VEGF 121, 165 and 189 significantly
increased during carcinogenic progression. For VEGF 121, the increase was significant
between OV-I vs EOC I, II and III vs (p<0,001), for VEGF 165 the difference was between
OV-I vs EOC I, II y III vs (p<0,001), and for VEGF 189 between OV-I vs EOC I (p<0,05),
OV-I vs EOC II y III (p<0,001). We further found that protein expression of VEGF had a
moderate increase during carcinogenic progression, and there was a significant increase
between OV-I vs EOC I, II y III (p< 0,001). Our results show a gradual increase in mRNA
levels of trkA during the carcinogenic progression, and this increase was also significative
when as the cells lose its differentiation, obtaining differences between the T.Bord vs EOC
I (p<0,01), T.Bord vs EOC II and III (p<0,001), between EOC I vs EOC III (p<0,001) and
EOC II vs EOC III (p<0,01). In benign tumors and inactive ovaries we did not find the
expression of trkA, probably due to the low number of epithelium in the samples of these
groups. Protein expression of trkA receptor increased gradually and significantly during the
carcinogenic progression: mainly between OV-I vs EOC I, II and III (p<0,001). Likewise,
protein levels of p-trkA increased significantly during EOC progression between EOC I vs
EOC III (p<0,01). In the other groups studied (OV-I, and Ben T. T. Bord) no positive
staining was found for p-trkA.
Discussion: Both, total trkA receptor protein expression and p-trkA, as well as
levels of mRNA of trkA increase during carcinogenic progression, mainly among the
EOCs, and this increase was significant with higher tissue indifferentiation (tumor
progression), which would confirm its possible use as a progression marker for EOC and
also as a potential marker of poor prognosis because it was not detected either at the mRNA
level or biologically active protein (p-trkA) in inactive ovaries and benign tumor, but from
the borderline tumors it begins to be detected but very weakly, unlike what occurs in EOC,
where its protein expression is very high.
The increase in protein and trkA mRNA levels during carcinogenic progression
correlated positively and significantly with both gene and protein expression of NGF and
VEGF, both of which are proangiogenic factors related with the development, progression
and metastasis of tumors.

11
FINANCIAMIENTO
Esta tesis fue realizada en el Laboratorio de Endocrinología y Biología de la
Reproducción, Hospital Clínico Universidad de Chile.
Esta Investigación se desarrolló en el marco del proyecto FONDECYT 1030661 y
1071036.

12
I INTRODUCCIÓN
I.1 El ovario y Cáncer Ovárico Epitelial (COE):
El ovario es un órgano que presenta múltiples compartimientos con diferentes y
variadas propiedades biológicas. Las funciones del ovario son de dos tipos: una
gametogénica (formación del gameto femenino) y la otra endocrina (secreción de hormonas
esteroidales, peptídicas y factores de crecimiento). Morfológicamente, está organizado en
una zona central llamada médula (nervios y vasos sanguíneos), rodeada de una zona
periférica llamada corteza (crecimiento y desarrollo folicular). Rodeando a este órgano, se
encuentra una monocapa de células epiteliales, también llamada epitelio de la superficie
ovárica (ESO). Esta monocapa de células planas a cuboidales, deriva del epitelio celómico,
secreta hormonas, factores de crecimiento y citoquinas, además presenta receptores para
GnRH, FSH, LH, estrógeno, progesterona, andrógeno y EGF. Su función es el transporte de
materiales hacia y desde la cavidad peritoneal y participa en la ruptura ovulatoria cíclica
(capacidad proteolítica, remodelamiento) y en su reparación (proliferación). Su
funcionamiento es dependiente de hormonas y factores de crecimiento.1, 2
El cáncer constituye la segunda causa de muerte en la población chilena, después de
las enfermedades cardiovasculares. El cáncer de ovario representa la segunda causa de
muerte de origen ginecológico, después del cáncer cervicouterino. De todas las neoplasias
malignas ováricas humanas, aproximadamente entre el 80-90%, corresponden a carcinoma
ovárico epitelial (COE), el resto es originado en células de la granulosa, o raramente en
células del estroma o germinales. En el progreso a la malignidad, el ESO, pierde las
características estromales y adquiere las características del epitelio derivado del conducto
Mulleriano. De esta forma hablamos de adenocarcinoma seroso si adquiere las
características del epitelio de la trompa de Falopio, endometrioide si es del endometrio y
mucinoso si es endocervical.1 El carcinoma seroso corresponde al 40% de todos los COE;
además, se presenta en mayor porcentaje de forma invasiva, se caracteriza por responder
pobremente a terapia y ser detectados tardíamente, por lo cual, la sobrevida es pobre.3, 4
Existen algunos factores que predisponen a desarrollar más tempranamente COE y
otros que lo previenen, hablamos entonces de factores de riesgo: como la ovulación, altos
niveles de gonadotrofinas, historia familiar de cáncer ovárico (5-10%), drogas para

13
fertilidad, terapia hormonal de reemplazo post-menopáusica con estrógenos, síndrome de
ovario poliquístico (SOP)1,4 y factores protectores: como progesterona, anticonceptivos
orales, multiparidad, lactancia, salpingoligadura e histerectomía.1,4
La patogénesis del carcinoma ovárico aún no está del todo clara y numerosos
mecanismos han sido propuestos para explicar la etiología de esta patología. En este
contexto se mencionan las hipótesis que se postulan para la etiología del COE:
Hipótesis de la ovulación incesante:
La continua ruptura y reparación del ESO, provoca daño al DNA, originando
mutaciones en genes supresores de tumores, lo que llevaría a la transformación maligna de
las células, provocando la estimulación del crecimiento, expansión clonal, proteólisis y
angiogénesis, para finalmente llevar a metástasis.5
Hipótesis de las gonadotrofinas:
La excesiva exposición a gonadotrofinas incrementa la estimulación estrogénica del
ESO, conduciendo posiblemente a la transformación maligna. Las gonadotrofinas podrían
actuar directamente sobre el ESO, aumentando la transformación, o indirectamente
mediante la estimulación de la producción de estrógenos locales. Los niveles de
gonadotrofinas aumentan con la edad y son particularmente altos durante la menopausia,
período en que se registran las mayores tasas de COE.6
Hipótesis hormonal:
La estimulación excesiva de andrógenos sobre el ESO conduce a un riesgo
aumentado de cáncer. Los andrógenos estimulan la proliferación celular en quistes de
inclusión y ESO. Una de las características claves del SOP, son los niveles elevados de
andrógenos, y estos podrían contribuir al riesgo aumentado de COE en esta condición.6
Hipótesis de la inflamación:
El proceso ovulatorio consiste en una reacción inflamatoria, con producción de
mediadores inflamatorios. Este proceso involucra la liberación de colagenasas y proteasas
características de reacciones inflamatorias en respuesta al tejido. La persistencia del daño
genético causado por factores inflamatorios puede ser un factor importante en la
transformación de células del ESO.6
Hipótesis conversión epitelio-mesénquima de células del ESO:

14
Dos caminos por los cuales el ESO traspasa hacia la corteza ovárica e invade el
estroma:
1) Fragmentos del ESO producto de la ruptura folicular en la ovulación pasan
al estroma.
2) Invaginaciones de la superficie, las cuales se forman con el transcurso de los
años.
Ambas situaciones llevan a la formación de quistes de inclusión, donde ocurren
cambios displásticos y metaplásticos, procesos pre-malignos que pueden llevar a
tumorigénesis.1
La carcinogénesis ovárica presenta cambios en la diferenciación celular que
acompañan a la progresión neoplásica. Se ha postulado un modelo dualista para la
progresión del COE, que postula la existencia de dos tipos de tumores:
• Tumores tipo I o de bajo grado: Seroso de bajo grado, mucinoso,
endometrioide, células claras, tumor de Brenner maligno.
Todos estos tumores progresarían desde un quiste de inclusión de ESO, a un tumor
benigno, luego a un tumor borderline, posteriormente a un carcinoma seroso de bajo grado,
y finalmente a un carcinoma seroso de alto grado.
• Tumores tipo II o de alto grado: Seroso de alto grado, tumor mesodermal
maligno mixto (carcinosarcomas).
Estos se desarrollarían directamente desde el ESO, o bien desde quistes de inclusión de
ESO para dar origen a un carcinoma seroso de alto grado.7
Modelo dualista basado en evidencias clínico-patológicas y moleculares:
• Tumores tipo I: Presentan algunos cambios genéticos comunes como
mutaciones en genes supresores de tumores BRAF y KRAS (30% tumores serosos
de bajo potencial maligno) resulta en activación constitutiva de la vía RAS-MAPK
y otros cambios fenotípicos como una baja proliferación celular.
• Tumores tipo II: Presentan algunos cambios genéticos comunes como
mutaciones en el gen supresor de tumores p53 (50% carcinoma seroso invasivo), y
otros cambios fenotípicos como una alta proliferación celular.7
Además, existe la clasificación del COE de acuerdo a su diferenciación histológica
en:

15
COE I o bien diferenciado: donde se observan estructuras papilares irregulares con
malignidad celular, epitelio pseudoestratificado y algunos focos de invasión.
COE II o moderadamente diferenciado: en los cuales existe una disminución de las
estructuras papilares por presencia de zonas indiferenciadas, con mucha atipia nuclear y
mayores focos de invasión.
COE III o pobremente diferenciado: los que se presentan mayoritariamente como
una masa sólida indiferenciada, con evidente pleomorfismo celular y nuclear, y con
muchos focos de invasión. 8
Las células cancerosas se caracterizan por presentar un crecimiento celular alterado,
en parte por el uso de señales de crecimiento generadas por una variedad de receptores
de factores de crecimiento, tales como los receptores tirosina kinasa. En la mayoría de
las células, la sobreexpresión de estos receptores o vías de transducción de señales río
abajo, están involucradas en la transformación maligna de la célula48. Por lo que, a
continuación se mencionan algunos antecedentes de estos receptores y sus ligandos,
como las neurotrofinas, y su relación con el COE.
I.2 Neurotrofinas y sus receptores:
Las neurotrofinas representan una familia de proteínas homodiméricas estructural y
funcionalmente relacionadas que incluye: el factor de crecimiento nervioso (NGF), factor
neurotrófico derivado de cerebro (BDNF), neurotrofina 3 (NT-3), neurotrofina 4/5 (NT 4/5)
y neurotrofina 6 (NT-6). Las neurotrofinas median su señal a través de la unión a dos clases
distintas de receptores de superficie celular: el receptor de alta afinidad trk y el de baja
afinidad p75.9, 10
El receptor p75 une todas las neurotrofinas, la activación del receptor inicia
apoptosis en el contexto de neuronas trk negativas, pero promueve sobrevida incluso a
bajas concentraciones de NGF si trkA es coexpresado en la superficie celular.11
Se conocen tres receptores trk diferentes, los cuales presentan una alta afinidad por
sus ligandos. Los receptores trkA, trkB y trkC que unen NGF, BDNF y NT-3 y NT-4/5,
respectivamente.10
La función biológica de NGF es el mantenimiento y sobrevida del sistema nervioso
central y periférico. A su vez, su receptor trkA, es crítico para el desarrollo y maduración

16
de sistema nervioso central y periférico, regulando proliferación, diferenciación y
apoptosis.10, 11
El receptor trkA es una proteína glicosilada transmembrana de 140 kDa, que posee
un dominio extracelular, transmembrana e intracelular12. Su mapa genético es de 25 Kb,
ubicado en el cromosoma 1q21-q22, y está organizado en 17 exones para la isoforma I
(trkAI), las isoformas II y III (trkAII y trkAIII) son originadas por splicing alternativo del
exón 9 y de los exones 6, 7 y 9, respectivamente. El dominio extracelular contiene dos
regiones ricas en cisteína interrumpida por un dominio rico en leucina y dos regiones
semejantes a inmunoglobulina (IGC1 y IGC2).13 El dominio transmembrana y
yuxtamembrana son críticos para la internalización y transducción de señales. El dominio
intracelular exhibe actividad tirosina kinasa bajo dimerización, e inicia la transducción de
señales intracelular por autofosforilación de residuos de tirosinas. Estas interacciones
inducen transducciones de señales vía RAS/MAPK, PI3K, y/o PKC, las cuales median los
efectos de NGF en proliferación, diferenciación y sobrevida.11, 13
En tejidos normales, la activación del receptor está altamente regulada. La
activación promueve su internalización dentro de la vía de tránsito endocítica, y
posteriormente, son regulados negativamente mediante ubiquitinización y degradación
lisosomal o proteosómica. La ubiquitinización ocurre en una región conservada del dominio
N-terminal del receptor, esta interacción es esencial para su regulación14.
Su inapropiada activación (activación oncogénica) por mutación puntual, deleción o
formación quimérica debido a recombinación cromosómica, resulta en dimerización y
activación espontánea del receptor independiente del ligando, con la consiguiente
transducción de señal constante y transformación maligna, asociada con el desarrollo y
progresión de varios tipos de cáncer 13, 14.
Los oncogenes trk quiméricos son generados por una recombinación genética, en la
cual se reemplaza el dominio extracelular por otro gen, conservando el dominio
transmembrana y el dominio tirosina kinasa del receptor trkA. Se han descrito a la fecha
tres oncogenes quiméricos. Uno de ellos corresponde a TRK, el cual reemplaza el dominio
extracelular (dominio de unión del ligando putativo) por el gen TPM3 que codifica para una
tropomiosina no muscular, que le confiere un cambio conformacional al dominio kinasa
que lo mantiene activo constitutivamente e independiente del ligando.15, 16, 17 La activación

17
de este receptor ha sido implicada en el desarrollo de ciertos cánceres humanos, que
incluyen carcinoma de tiroide papilar y colon18, 19. El segundo corresponde a TRK-T1 y
TRK-T2 (dependiendo del largo de la fusión), en el cual se reemplaza el dominio
extracelular por el gen TPR, que codifica para una proteína del complejo de poro nuclear, y
ha sido detectado en hepatocarcinoma de rata, y el tercero corresponde a TRK-T3, en el
cual se reemplaza el dominio extracelular por el gen TFG que codifica una proteína de
función desconocida, y ha sido reportado en condrosarcoma mixoide extraesqueletal.12 La
activación constitutiva del receptor por una deleción de 75 aminoácidos en el dominio
extracelular ha sido detectada en pacientes con leucemia mieloide aguda. En carcinoma
prostático un loop autocrino que afecta a NGF y trkA es responsable de la progresión y
crecimiento del tumor, y recientemente se ha visto lo mismo en cáncer de mama20.
El oncogen trkA (trkAIII), originado por splicing alternativo, carece de los exones
6,7 y 9 del dominio extracelular, es inducido por hipoxia y posee actividad tirosina kinasa
constitutiva (vía PI3K/AKT/NF-kB) promoviendo el crecimiento tumoral12, y ha sido
asociado con distintos tipos de cáncer, que incluyen: neuroblastoma, carcinomas de colon,
tiroide medular, próstata, y leucemia mieloide aguda. 21.
Existen evidencias de que los oncogenes escapan de los mecanismos de regulación
negativa, debido a los cambios que presentan en su dominio extracelular, lo que impide su
ubiquitinización y posterior degradación por la vía lisosomal, permitiendo la dimerización y
activación constitutiva del receptor14
I.3 La importancia de NGF y su receptor trkA en la fisiología ovárica y en COE:
Se ha visto que el ovario de rata fetal y en etapa neonatal expresa 4 de las cinco
neurotrofinas conocidas (NGF, BDNF, NT-3 y NT-4/5), y sus receptores de alta afinidad
(trkA, trkB y trkC) que están presentes en células no neuronales del ovario. El incremento
preovulatorio de la expresión de trkA es dramático (> 100 veces), y es seguido por un
aumento sustancial de los niveles de mRNA de NGF. Estos cambios en la expresión génica
de trkA y NGF ocurren previos a la primera ovulación.22, 23, 24
Estudios en ovario de rata han demostrado la presencia del receptor trkA y NGF en
ovocitos de folículos primordiales. Utilizando ratones con deleción del gen para NGF y
trkA se demostró que NGF participa en la regulación del desarrollo folicular temprano a

18
través de su receptor trkA25, promoviendo la proliferación de células mesenquimales en el
folículo primordial, la diferenciación y el crecimiento de folículos primarios e inducción
del receptor de FSH en folículos secundarios 25,26, es por esto que, NGF puede representar
una señal facilitadora para la ruptura folicular en la ovulación, ya que junto al aumento de
NGF y trkA previo a la ovulación, se produce un aumento en la secreción de progesterona,
producción de COX 2 y proliferación celular.27 Por todo lo anterior, se demuestra que NGF
juega un rol importante en la regulación del desarrollo de la función ovárica.28
También se demostró la presencia de NGF y trkA en folículos preantrales y antrales
de ovarios humanos normales y células de la granulosa aisladas. En células de la granulosa
humana, NGF induce la secreción de estrógeno e inhibe la secreción de progesterona
(previniendo luteinización temprana) y aumenta la expresión de receptores para FSH, 29al
igual que en ovario de rata,30, 31 donde además se encontró que participan en la formación
de quistes ováricos.32, 33 En las células de la teca, NGF, contribuye en el proceso ovulatorio
mediante la estimulación de la secreción de progesterona, estimulación de la producción de
prostaglandina 2 (PG2) y proliferación celular antes de la ruptura ovulatoria34. Por todo lo
anterior, podemos decir que NGF tiene una participación tanto en la foliculogénesis como
en la esteroidogénesis ovárica.
Además, se ha postulado un rol indirecto y directo de NGF en la angiogénesis. En
cuanto a su rol indirecto se ha reportado que NGF al unirse a su receptor trkA, aumenta la
inmunorreactividad del factor de crecimiento de endotelio vascular (VEGF), en neuronas de
ganglio cervical superior de ratas recién nacidas, tratadas con NGF por 8 días.35 A su vez,
en ovario de rata NGF aumenta dos principales parámetros angiogénicos: la expresión de
VEGF y el área de vasos sanguíneos.36 En explantes de COE se encontró un aumento en la
expresión de VEGF por acción de NGF mediante la activación del receptor trkA37. Con
respecto a su rol directo se ha visto que en células endoteliales de la vena umbilical humana
(HUVEC), NGF promueve la proliferación y migración, a través, de trkA por la vía
MAPK38, pero en menor grado comparado con VEGF, sin embargo, estimula
significativamente la invasión y formación de vasos en matrigel, a través del aumento de la
expresión de la metaloproteinasa de la matriz extracelular 2 (MMP-2). Estos efectos son
regulados a través de su receptor trkA, activando la via PI3K/AkT39.

19
Resultados preliminares de nuestro laboratorio indican que durante la progresión del
cáncer ovárico (desde OV-I hasta COE III), se observa un aumento en el grosor de la pared
de los vasos sanguineos, existiendo una relación directa con la expresión de trkA en células
endoteliales y epiteliales. La expresión de trkA en células endoteliales del ovario, tendría
directa relación con la vasculogénesis, tanto en el ovario normal como en cáncer ovárico
epitelial. Congreso Reproducción y Desarrollo, Chillán, 2008.
Por otra parte, trkA ha sido asociado generalmente con progresión y mal pronóstico
en varios cánceres. Se ha visto que la expresión de p-trkA está aumentada en estados
avanzados de carcinoma ovárico.40 La coexpresión de NGF con moléculas involucradas en
la angiogénesis y expresión de p-trkA en células endoteliales, sugiere que el rol
proangiogénico atribuido a NGF in vitro e in vivo puede ser relevante en cáncer.41
I.4 Angiogénesis en la progresión del COE:
La angiogénesis es el proceso en el cual se desarrollan nuevos capilares desde vasos
preexistentes, inducido por inflamación, heridas, reacciones inmunes y neoplasia. Además,
se requiere para el crecimiento del tumor y progresión.42 La vascularización del tumor es un
proceso vital para la progresión de una neoplasia, ya que de un tumor pequeño y localizado
se transforma rápidamente en un tumor de gran tamaño con la capacidad de metastizar. Un
gran número de factores proangiogénicos y sus receptores han sido identificados. El factor
de crecimiento de endotelio vascular (VEGF) ha sido el mejor caracterizado.43 El gen de
VEGF presenta splicing alternativo, originando cinco mRNA que codifican para cinco
isoformas proteicas distintas VEGF 121, 145, 165, 189 y 206. La isoforma VEGF121 es
secretada desde la célula, a diferencia de la isoforma VEGF165 que es parcialmente retenida
en la superficie celular, y las otras isoformas son retenidas.37 VEGF tiene tres receptores
tirosina kinasa que están localizados en las células endoteliales (Flt-1, KDR/Flk-1, y Flt-4),
es un potente mitógeno, morfógeno, y quimioatractante para células endoteliales y es
estimulado por hipoxia, citoquinas y hormonas.42
Una elevada expresión del mRNA de VEGF fue encontrada en todos los tumores
primarios y metastásicos de ovario, por lo que se cree que existiría una correlación entre
sobreexpresión de VEGF y pronóstico en pacientes con carcinoma ovárico.42 Es por esto,

20
que actualmente existen proyectos multicéntricos donde se está utilizando terapias
antiangiogénicas en conjunto con las quimioterapias.
Los factores proangiogénicos y las metaloproteinasas de la matriz promueven la
angiogénesis. Estas últimas, lo hacen mediante la remodelación de la matriz extracelular
permitiendo la liberación de factores angiogénicos44,45,46.
Recientemente, nuestro laboratorio ha encontrado una baja expresión de NGF y
muy baja de trkA en células del ESO en ovarios normales, a diferencia de lo encontrado en
cáncer ovárico, donde existe una alta inmunodetección de ambos. Además, en tejido
ovárico normal se expresan las isoformas de VEGF 121,165 y 189 y en COE se
sobreexpresan las isoformas de VEGF 165 y 18937. Al analizar la expresión de VEGF por
efecto de NGF en cultivos de explantes de COE, existe un aumento de la expresión génica
y proteica de las tres isoformas de VEGF (121,165 y 189). Este efecto es inhibido por
inmunobloqueo de NGF y por K252a, un inhibidor de receptores tirosina kinasa.37 Lo que
indica que el aumento de VEGF por NGF es mediado por la activación del receptor trkA.
La expresión de NGF y trkA en el ESO no sería solamente importante para los
estados iniciales de la angiogénesis, sino que, también podría jugar un rol en la progresión
carcinogénica del COE, aumentando los niveles de expresión de VEGF, y si bien, ya se
sabe que el receptor trkA y su ligando NGF participan en la angiogénesis, no hay
antecedentes claros que indiquen si el receptor trkA y su ligando NGF están involucrados
en la génesis y progresión del COE, y si estos podrían ser usados como marcadores de
pronóstico y/o progresión en este tipo de cáncer. Por lo que nos planteamos la siguiente
hipótesis:

21
II HIPOTESIS trkA modifica su expresión durante la carcinogénesis ovárica
constituyendo un nuevo marcador tumoral
III. OBJETIVOS III.1 OBJETIVO GENERAL
Evaluar la expresión del receptor trkA y de NGF, durante las distintas etapas
de transición del epitelio ovárico (desde el tejido ovárico normal al cáncer ovárico
epitelial) y si esta expresión se relaciona con la expresión de VEGF.
III 2 OBJETIVO ESPECÍFICOS
1) Caracterizar los niveles de mRNA de trkA, NGF y VEGF en ovarios inactivos,
tumores benignos, borderline y en los distintos grados de diferenciación del
COE.
2) Determinar la localización y semicuantificar la expresión proteíca de trkA,
NGF y VEGF durante la progresión carcinogénica.
3) Evaluar la localización y semicuantificar la expresión proteíca del receptor
activo p-trkA durante la progresión carcinogénica.
OV-I T.Bord T.Ben
COE I COE II COE III
COE II COE I Tumor Benigno
Quiste de Inclusión
COE III Tumor Borderline
PROGRESIÓN CARCINOGÉNICA OVÁRICA

22
IV. METODOLOGÍA IV.1. Obtención de las Muestras
Las muestras de tejido ovárico humano fueron obtenidas de mujeres que acudieron
al Servicio de Obstetricia y Ginecología del Hospital Clínico de la Universidad de Chile y
Clínica Las Condes. Previamente estas mujeres debieron firmar un Consentimiento
Escrito, aprobado por el Comité de Ética del Hospital Clínico Universidad de Chile y
Clínica Las Condes (ANEXO 1).
Para este estudio se consideraron 6 tipos de muestras:
1) Ovario Inactivo (OV-I): Ovario de una mujer menopáusica o postmenopáusica,
con uno o más quistes de inclusión rodeados por una monocapa de células epiteliales.
2) Tumor Benigno (T.BEN): Quiste de inclusión con un diámetro mayor a 1 cm, y
rodeado por una monocapa de células epiteliales.
3) Tumor Borderline (T.BORD): Monocapa de células epiteliales que ha proliferado
y se observa como un epitelio pseudoestratificado con atipia celular leve a moderada.
4) Cáncer ovárico epitelial (COE I) o Bien Diferenciado: Estructuras papilares
irregulares con malignidad celular, epitelio pseudoestratificado y algunos focos de invasión
de más de 3 mm.
5) Cáncer ovárico epitelial (COE II) o Moderadamente Diferenciado: Disminución
de las estructuras papilares por presencia de zonas indiferenciadas, con mucha atipia
nuclear y mayores focos de invasión.
6) Cáncer ovárico epitelial (COE III) o Pobremente Diferenciado: Masa sólida
indiferenciada, con evidente pleomorfismo celular y nuclear, y con muchos focos de
invasión.
Las muestras de ovarios inactivos correspondieron a tejidos provenientes de ovarios
de mujeres voluntarias que acudieron al Servicio de Obstetricia y Ginecología del Hospital

23
Clínico Universidad de Chile y Clínica Las Condes, para ser sometidas a histerectomía con
ooforectomía por patología no ovárica.
Los otros grupos del estudio se obtuvieron mediante histerectomía con ooforectomía
o por histerectomía radical, en mujeres que acudieron al Servicio de Obstetricia y
Ginecología del Hospital Clínico Universidad de Chile y Clínica Las Condes a quienes se
les diagnosticó un tumor ovárico o cáncer ovárico.
Como se muestra en el siguiente recuadro, todas las pacientes consideradas dentro
del estudio son menopáusicas o postmenopáusicas:
Grupos de Estudios Edades Promedio + Error Estándar
Ovario Inactivo 46 + 1,25 años Tumor Benigno 46 + 3,08 años
Tumor Borderline 46 + 5,67 años COE I (Bien Diferenciado) 53 + 4,07 años
COE II (Moderadamente Diferenciado) 53 + 4,54 años COE III (Pobremente Diferenciado) 68 + 2,24 años
IV.1.2 Diseño Experimental
Una vez obtenida la muestra, un trozo de tejido se congeló en N2 líquido y fue
guardado a –80° C, para posteriormente realizar estudios de expresión génica mediante RT-
PCR convencional. El otro trozo fue incluido en parafina, del cual se obtuvieron cortes de 5
µm de espesor, a un corte se le realizó una tinción de hematoxilina–eosina para el análisis
de morfología. El resto de los cortes se utilizaron para la evaluación de la expresión
proteica por inmunohistoquímica, tal como se muestra en el esquema 1.

24
Esquema 1: Diseño experimental: OV-I: Ovario Inactivo, T.Ben: Tumor Benigno, T.Bord:
Tumor Borderline, COE I, II, III: Cáncer Ovárico Epitelial grado I, II, III.
IV.2 Análisis de muestras
IV.2.1 Estudio Inmunohistoquimico
Este estudio se realizó con el objetivo de identificar la localización y
semicuantificación de las proteínas: trkA, p-trkA, NGF y VEGF, durante la progresión
carcinogénica del ovario. La técnica se realizó en cortes de tejidos previamente incluidos en
parafina. Dichos cortes tenían un espesor de 4 a 6 μm, los cuales fueron desparafinados en
xilol y luego hidratados en concentraciones seriadas de alcohol. Para la recuperación
antigénica, las muestras se incubaron en buffer citrato 10 mM a pH 6.0, a 96-98° C por 20
min. Posteriormente, con el objetivo de bloquear la acción de peroxidasas endógenas, los
tejidos fueron tratados con peróxido de hidrógeno al 3% en agua destilada. Para evitar las
uniones inespecíficas, las muestras se incubaron por 10 min con el bloqueador específico
del kit HistostainRSP (Zymed Laboratorios Inc, San Francisco, California, USA), para
Muestras de Ovario OV-I, T.Ben, T.Bord., COE I, II y III
Tejido Congelado N2 liquido / -80°C
Estudio de expresión
génica (RT-PCR)
Tejido Incluido en
parafina
Estudio de Expresión Proteica
(Inmunohistoquimica)
trkA, NGF y VEGF trkA, p-trkA, NGF y VEGF
Evaluación Morfológica
25
posteriormente incubar los cortes con los anticuerpos primarios, diluidos en PBS-BSA al
2%, correspondiente a cada ensayo. Dicha incubación se realizó a 4°C por 20 h con las
diluciones de anticuerpos señaladas en la Tabla 1. Luego, se incubó con el segundo
anticuerpo biotinilado por 20 min y, posteriormente, con estreptavidina marcada con
peroxidasa por 20 min a temperatura ambiente. Se utilizó como sustrato final de la reacción
el cromógeno diaminobencidina, DAB, (DakoCytomation, Inc., CA, USA), dando una
tinción positiva de color café. Los tejidos fueron contrateñidos con hematoxilina para
evidenciar los núcleos de color azul claro. Cada placa contó con su control negativo
incubado sólo con PBS-BSA 2% (no se le agregó el anticuerpo primario). La evaluación de
las placas se llevó a cabo por tres observadores independientes y ciegos a la categoría de la
muestra. Cada sección del tejido analizado se dividió en cuatro cuadrantes. En cada uno de
ellos se contaron 250 células epiteliales, dando un total de 1000 células epiteliales por
muestra, asignándoles distintas intensidades de tinción (i):0- sin tinción; 1- tinción leve; 2-
tinción moderada; 3- tinción intensa. Para calcular el porcentaje de células con tinción
positiva se sumaron los porcentajes de las intensidades 2 + 3. Para el cálculo de la
intensidad de tinción de cada compartimiento se aplicó la siguiente fórmula de HScore
(HS), validada en nuestro laboratorio: HS= S % células teñidas x (i+1)/100. Las fotografías
se hicieron con el programa computacional Image-ProPlus 6.2.
Tabla 1: Anticuerpos utilizados en el estudio inmunohistoquimico.
Anticuerpo 1° Dilución Procedencia Tipo de Anticuerpo
Anti NGF 1:750
Donación del Dr. HF Urbansky Oregon Regional Primate Research Center
Policlonal
Anti trkA 1:300 Santa Cruz H-190: sc-14024 Policlonal
Anti VEGF 1:1000 Abcam: ab 1316 Monoclonal
Anti p-trkA 1:100 Abcam: ab 51185 Policlonal

26
IV.2.1.1 Estudio de RT-PCR
Este estudio se realizó con el objetivo de caracterizar los niveles de mRNA de NGF,
VEGF y trkA, durante la progresión carcinogénica. La técnica de extracción de RNA, se
realizó a partir de tejidos congelados, utilizando apróximadamente 100 mg de tejido. Se les
agregó TRIZOL (Invitrogen, Life Technologies, Carlsbad, CA) para homogeneizarlos,
cloroformo para eliminar las trazas de fenol, y se incubó durante 15 min, y luego se
centrifugó a 16.000 g por 15 min a 4°C. A continuación se extrajo 300-500 μl de la fase
acuosa, donde se encuentra el RNA, se le agregó el mismo volumen de isopropanol, y se
incubó toda la noche a -20°C, para precipitar el RNA. Luego se centrifugó a 16.000 g por
15 min a 4°C. El pellet se lavó con etanol al 75% y se resuspendió en H2ODEPC. La
concentración y pureza del RNA, fue medida en un espectrofotómetro a 260 y 280 nm. El
DNA complementario (cDNA) fue sintetizado en una mezcla de reacción de 20 μl,
utilizando 5 μg de RNA total, dNTPs 10 mM, Random Primers (500 μg/ml, Madison, WI,
USA) y un volumen de H20 necesario para completar 10,5 μl. Esta mezcla se calentó por 5
min a 65°C y se dejó enfriar otros 5 min en hielo. Posteriormente, se le agregó Buffer 5X
first strand, DTT 0,1 M, RNAsa OUT 40 U/ul, y la enzima Superscript II (Invitrogen,
200U/μl), se incubó 10 min a 25 °C y luego 50 min a 42°C. El cDNA fue amplificado en
una mezcla de reacción de 25 µL, utilizando Buffer 10X, MgCl2 50mM, dNTPs 10mM,
DNA polimerasa (Biotools, 5U/μl), partidores sense y antisense, un volumen de H2O
necesario para llegar a 23 μl y 2 µL de cDNA. Los partidores específicos para cada gen y
su respectivo programa de PCR se resumen en las Tablas 2 y 3 respectivamente. Como
control interno (gen constitutivo) se utilizó β-actina, que en el caso de NGF y VEGF se
incorporó en la mezcla de reacción, estandarizado en nuestro laboratorio, a diferencia de
trkA que se realizó por separado. Los productos de PCR fueron separados en un gel de
agarosa al 2%, para validar el tamaño predicho de las bandas del amplificado, se utilizó un
estándar de peso molecular. Los geles fueron visualizados bajo luz ultravioleta,
fotografiados y analizados con un programa de densitometría UV Transilluminator UVP
with Doc-it Software Image Acquisition and 1 D Analysis (UVP, Inc. Laboratory Products,
Upland, CA, USA)

27
Como control negativo del PCR, se utilizó agua estéril (ausencia de cDNA), y como
control positivo del PCR, se utilizó cDNA de células de la granulosa humana, en las cuales
se había descrito anteriormente la presencia de NGF, VEGF y trkA.
Tabla 2: Partidores específicos para la amplificación por PCR Partidor Producto (pb) Sense Antisense
NGF 167 5’ TAA AAA GCG GCG ACT CCG TT 3’
5’ ATT CGC CCC TGT GGA AGA TG 3’
trkA 900 5′-AAC CTC ACC ATC GTG AAG AGT GGT-3′
5′-GGT TGA ACT CGA AAG GGT TGT CCA-3′
VEGF 307 (VEGF189),
236(VEGF165) y 104 (VEGF121)
5’ AGG CCA GCA CAT AGG AGA GA 3’
5’ ACC GCC TCG GCT TGT CAC AT 3’
β-actina 661 5’ –TGA CCG GGT CAC CCA CAC TGT GCC CAT CTA 3’
5’ –CTA GAA GCA TTG CGG TGG ACG ATG GAG GG 3’
Tabla 3: Programa de PCR para cada gen estudiado.
IV.3. Análisis Estadístico
El cálculo del n muestral se realizó mediante la fórmula de comparación de medias,
estableciéndose en 10 para cada grupo estudiado. Los resultados fueron expresados en
todos los ensayos como promedio ± error estándar del promedio (EEM) y todos los grupos
estudiados se compararon con el grupo de ovario inactivo, ya que este último es un control
de ovario normal, aunque se pueden gestar cambios displásticos en las células epiteliales y
posteriormente originar un carcinoma, por ello está dentro de la progresión carcinogénica.
Para el análisis estadístico se utilizó el programa Graph Pad 5.0. En cada análisis, se realizó
un test de normalidad (Kolmogorov–Smirnov) con el objetivo de conocer la distribución de
los datos. Dado que los datos obtenidos en el presente estudio fueron paramétricos, se
realizó el test ANOVA, para conocer la existencia de diferencias significativas, y
finalmente se utilizó el post test Bonferroni para identificar entre cuales grupos están las
diferencias significativas. Como criterio de significancia se consideró un p < 0,05.
Programa NGF TrkA VEGF Denaturación 94°C 30’’ 94°C 1’ 94°C 30’’ Alineamiento 62°C 1’ 55°C 1’ 62°C 1’
Extensión 72°C 1’ 72°C 2’ 72°C 1’ N° de Ciclos 32 35 27

28
V RESULTADOS
V 1 Niveles de mRNA de VEGF durante la progresión carcinogénica ovárica.
Se encontró un aumento en los niveles de mRNA de VEGF durante la progresión
carcinogénica, en sus tres productos de splicing alternativo que codifican para las isoformas
proteicas de VEGF 121, 165 y 189
El producto que codifica la isoforma de VEGF 121, aumentó significativamente a
medida que se va perdiendo la diferenciación celular, obteniéndose diferencias
significativas entre, COE I vs OV-I, COE II vs OV-I y COE III vs OV-I (p<0,001). Entre
los distintos grados del COE se encontraron diferencias significativas entre COE I vs COE
III (p<0,001) y COE II vs COE III (p<0,01). Como se observa en la figura 1. Los niveles de
mRNA de VEGF fueron normalizados con los niveles de mRNA de β-actina.
661 pb
104 pb
236 pb 307 pb
VEGF 121
β-actina
VEGF 165 VEGF 189
C(+) C(-) OV-I T.Ben T.Bor COEI COEII COEIII

29
OV-I
T.Ben
.
T. Bord
.COE I
COE II
COE III0.0
0.5
1.0
1.5
2.0
****
**
Progresión Carcinogénica
mR
NA
VEG
F/m
RN
A b
-act
ina
Figura 1: Niveles de mRNA de VEGF 121 durante la progresión carcinogénica mediante RT-PCR. Los resultados de este experimento representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado en unidades arbitrarias normalizados con los niveles de β-actina y representado por la razón mRNA VEGF/mRNA β-actina. ** p<0,001 OV-I vs COE I, II III. ** p<0,001 COE I vs COE III y * p<0,01 COE II vs COE III. El producto que codifica la isoforma de VEGF 165, aumentó significativamente desde
COE I y a medida que se va perdiendo la diferenciación celular, obteniéndose diferencias
significativas entre, COE I vs OV-I, COE II vs OV-I y COE III vs OV-I (p<0,001). Entre
los distintos grados del COE se encuentran diferencias significativas entre COE I vs COE
III (p<0,001) y COE II vs COE III (p<0,01). Los niveles de mRNA de VEGF fueron
normalizados con los niveles de mRNA de β-actina. Como se observa en la figura 2.
*
**

30
OV-I
T.Ben
.
T.Bord
.COE I
COE II
COE III0.0
0.5
1.0
1.5
2.0
2.5
** **
**
Progresión Carcinogénica
mR
NA
VEG
F/m
RN
A b
-act
ina
Figura 2: Niveles de mRNA de VEGF 165 durante la progresión carcinogénica mediante RT-PCR. Los resultados de este experimento representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado en unidades arbitrarias normalizados con los niveles de β-actina y representado por la razón mRNA VEGF/mRNA β-actina. ** p<0,001 OV-I vs COE I, II y III. ** p<0,001 COE I vs COE III y * p<0,01 COE II vs COE III. El producto que codifica la isoforma de VEGF 189, aumentó gradual y
significativamente durante la progresión carcinogénica, como se observa en la figura 3. Se
encontraron diferencias significativas entre COE I vs OV-I (p<0,05), COE II vs OV-I y
COE III vs OV-I (p<0,001). Entre los distintos grados del COE hubo diferencia
significativa entre COE I y COE III (p<0,001) y COE II y COE III (p<0,05). Los niveles de
mRNA de VEGF fueron normalizados con los niveles de mRNA de β-actina.
*
**

31
OV-I
T.Ben
.
T.Bor.
COE I
COE II
COE III0.0
0.5
1.0
1.5
***
**
Progresión Carcinogénica
mR
NA
VEG
F/m
RN
A b
-act
ina
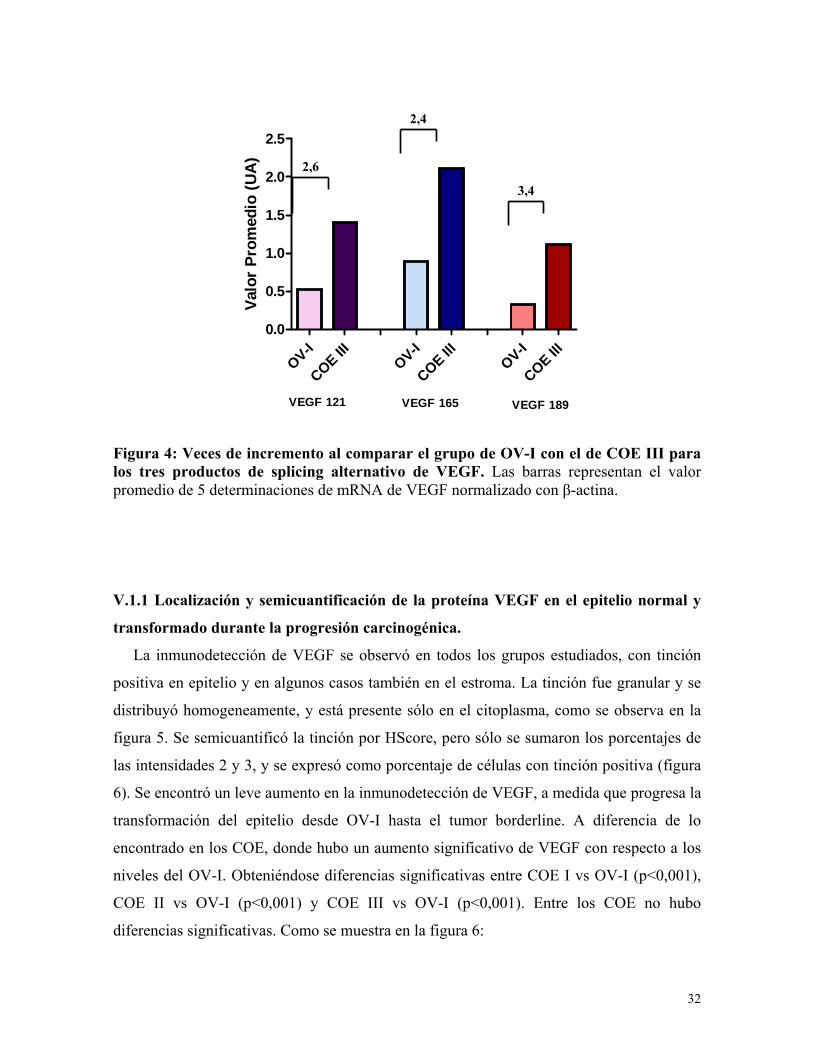
Figura 3: Niveles de mRNA de VEGF 189 durante la progresión carcinogénica mediante RT-PCR. Los resultados de este experimento representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado en unidades arbitrarias normalizados con los niveles de β-actina y representado por la razón mRNA VEGF/mRNA β-actina. * p<0,05 COE I vs OV-I, ** p<0,001 OV-I vs COE II y III ** p<0,001 COE I vs COE III y * p<0,05 COE II vs COE III. Al analizar los niveles de mRNA de VEGF, se pudo ratificar que las isoformas 121 y
165 son las que se expresan mayoritariamente, sin embargo, el aumento en los niveles de la
isoforma 189 entre OV-I vs COE III fue el mayor, aumentando 3,4 veces su valor, a
diferencia de los otros que aumentaron 2,6 y 2,4 respectivamente. Como se muestra en la
figura 4.
*
***
32
OV-I
COE III OV-I
COE III OV-I
COE III0.0
0.5
1.0
1.5
2.0
2.5
VEGF 121 VEGF 165 VEGF 189
Valo
r Pr
omed
io (U
A)
Figura 4: Veces de incremento al comparar el grupo de OV-I con el de COE III para los tres productos de splicing alternativo de VEGF. Las barras representan el valor promedio de 5 determinaciones de mRNA de VEGF normalizado con β-actina. V.1.1 Localización y semicuantificación de la proteína VEGF en el epitelio normal y
transformado durante la progresión carcinogénica.
La inmunodetección de VEGF se observó en todos los grupos estudiados, con tinción
positiva en epitelio y en algunos casos también en el estroma. La tinción fue granular y se
distribuyó homogeneamente, y está presente sólo en el citoplasma, como se observa en la
figura 5. Se semicuantificó la tinción por HScore, pero sólo se sumaron los porcentajes de
las intensidades 2 y 3, y se expresó como porcentaje de células con tinción positiva (figura
6). Se encontró un leve aumento en la inmunodetección de VEGF, a medida que progresa la
transformación del epitelio desde OV-I hasta el tumor borderline. A diferencia de lo
encontrado en los COE, donde hubo un aumento significativo de VEGF con respecto a los
niveles del OV-I. Obteniéndose diferencias significativas entre COE I vs OV-I (p<0,001),
COE II vs OV-I (p<0,001) y COE III vs OV-I (p<0,001). Entre los COE no hubo
diferencias significativas. Como se muestra en la figura 6:
3,4
2,4
2,6

33
Figura 5: Microfotografías representativas de la inmunodetección de la proteína VEGF durante la progresión carcinogénica. A. Ovario Inactivo (OV-I), B. Tumor Benigno (T.Ben), C. Tumor Borderline (T.Bord) D. E. y F. Cáncer Ovárico Epitelial (COE I, II, III respectivamente). El recuadro pequeño en cada microfotografía corresponde al control negativo. La barra en cada recuadro representa 50 um, Aumento 1000X. La flecha indica la tinción positiva en el epitelio.
A B
D
E F
C

34
OV-I
T.BEN
T.BORD
COE I
COE II
COE III0
20
40
60
80
100* * *
Progresión Carcinogénica
%C
élul
as P
ositi
vas
Figura 6: Semicuantificación de la proteína VEGF por inmunohistoquimica durante la progresión carcinogénica. Las barras representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado como porcentaje de células con tinción positiva. * p<0,001 OV-I vs COE I, II, III.
V.2 Niveles de mRNA de NGF durante la progresión carcinogénica ovárica.
En las muestras de ovario inactivo, tumor benigno, borderline, COE I, II y III, se
encontró un leve aumento al evaluar los niveles de mRNA para NGF, durante la progresión
carcinogénica. Este aumento fue significativo a medida que se va perdiendo la
diferenciación celular, obteniéndose diferencias significativas entre COE I vs OV-I
(p<0,05), COE II vs OV-I (p<0,01) y COE III vs OV-I (p<0,001). Como se observa en la
figura 7. Entre los distintos grados de los COE no hubo diferencias significativas. Los
niveles de mRNA de NGF fueron normalizados con los niveles de mRNA de β-actina.
NGF
β-actina
167 pb
661 pb
C(+) C(-) OV-I T.Ben T.Bor COEI COEII COEIII

35
OV-I
T.Ben
.
T. Bord
.COE I
COE II
COE III0.0
0.2
0.4
0.6
* *****
Progresión Carcinogénica
mR
NA
NG
F/m
RN
A b
-act
ina
(UA
)
Figura 7: Niveles de mRNA de NGF durante la progresión carcinogénica mediante RT-PCR. Los resultados de este experimento representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado en unidades arbitrarias normalizados con los niveles de β-actina y representado por la razón mRNA NGF/mRNA β-actina.*p<0,05, **p<0,01, ***p<0,001 OV-I. vs COE I, II y III.
V.2.1 Localización y semicuantificación de la proteína NGF durante la progresión
carcinogénica ovárica.
La inmunodetección de NGF se observó en todos los grupos estudiados, con tinción
positiva en epitelio y en algunos casos también en el estroma. La tinción fue homogénea y
granular, y está presente sólo en el citoplasma, como se observa en la figura 8. Se
semicuantificó la tinción por HScore, pero sólo se sumaron los porcentajes de las
intensidades 2 y 3, y se expresó como porcentaje de células con tinción positiva. Se
encontró un aumento en la inmunodetección de NGF, el cual fue discreto a medida que
progresa la transformación del epitelio desde OV-I hasta el tumor borderline. A diferencia
de los COE donde se encontró un aumento significativo de NGF, con respecto a los niveles
del OV-I. Obteniéndose así diferencias significativas entre COE II vs OV-I (p<0,01) y COE
III vs OV-I (p<0,001). Entre los distintos grados del COE no hubo diferencias
significativas. Como se muestra en la figura 9:

36
Figura 8: Microfotografías representativas de la inmunodetección de la proteína NGF durante la progresión carcinogénica. A. Ovario Inactivo (OV-I), B. Tumor Benigno (T.Ben), C. Tumor Borderline (T.Bord) D. E. y F. Cáncer Ovárico Epitelial (COE I, II, III respectivamente). El recuadro pequeño en cada microfotografía corresponde al control negativo. La barra en cada recuadro representa 50 um, Aumento 1000X. La flecha indica la tinción positiva en el epitelio.
A B
C D
E F

37
OV-I
T.BEN
T.BORD
COE I
COE II
COE III0
20
40
60
80
100
***
Progresión Carcinogénica
%C
élul
as P
ositi
vas
Figura 9: Semicuantificación de la proteína NGF en el epitelio normal y transformado por inmunohistoquimica durante la progresión carcinogénica. Las barras representan el promedio ± el error estándar del promedio, de 5 muestras en cada grupo de estudio, expresado como porcentaje de células con tinción positiva. *p<0,01 OV-I vs COE II, **p<0,001 OV-I vs COE III.
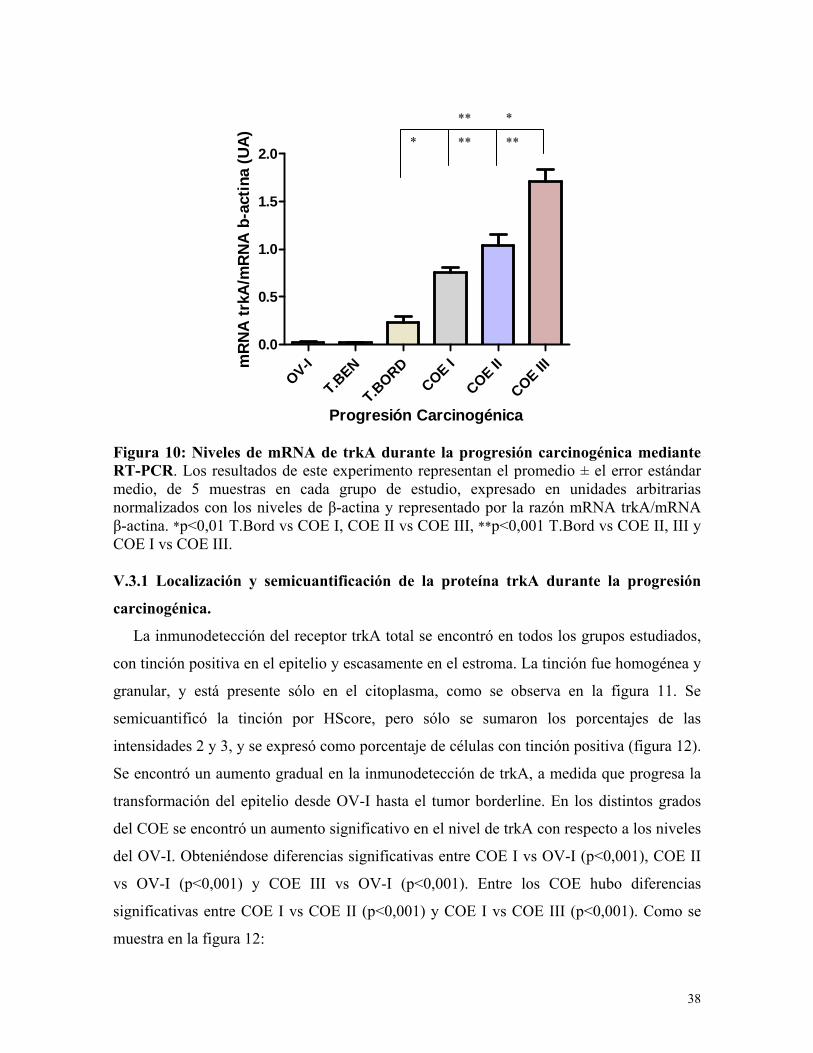
V.3 Niveles de mRNA de trkA durante la progresión carcinogénica ovárica.
En las muestras de ovario inactivo y tumor benigno, no se detectó el mRNA de trkA,
mediante la técnica utilizada. En los grupos borderline, COE I, II y III, se encontró un
aumento gradual en los niveles de mRNA para trkA, como se observa en la figura 10. Este
aumento fue significativo a medida que se va perdiendo la diferenciación celular,
obteniéndose diferencias significativas entre T.Bord vs COE I, COE II y COE III
(p<0,001). Además hubo diferencias significativas entre COE I vs COE III (p<0,01) y COE
II vs COE III (p<0,01). Los niveles de mRNA de trkA fueron normalizados con los niveles
de mRNA de β-actina.
C(+) C(-) TBord COEI COEII COEIII
trkA
β-actina
900pb
661p
38
Figura 10: Niveles de mRNA de trkA durante la progresión carcinogénica mediante RT-PCR. Los resultados de este experimento representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado en unidades arbitrarias normalizados con los niveles de β-actina y representado por la razón mRNA trkA/mRNA β-actina. *p<0,01 T.Bord vs COE I, COE II vs COE III, **p<0,001 T.Bord vs COE II, III y COE I vs COE III. V.3.1 Localización y semicuantificación de la proteína trkA durante la progresión
carcinogénica.
La inmunodetección del receptor trkA total se encontró en todos los grupos estudiados,
con tinción positiva en el epitelio y escasamente en el estroma. La tinción fue homogénea y
granular, y está presente sólo en el citoplasma, como se observa en la figura 11. Se
semicuantificó la tinción por HScore, pero sólo se sumaron los porcentajes de las
intensidades 2 y 3, y se expresó como porcentaje de células con tinción positiva (figura 12).
Se encontró un aumento gradual en la inmunodetección de trkA, a medida que progresa la
transformación del epitelio desde OV-I hasta el tumor borderline. En los distintos grados
del COE se encontró un aumento significativo en el nivel de trkA con respecto a los niveles
del OV-I. Obteniéndose diferencias significativas entre COE I vs OV-I (p<0,001), COE II
vs OV-I (p<0,001) y COE III vs OV-I (p<0,001). Entre los COE hubo diferencias
significativas entre COE I vs COE II (p<0,001) y COE I vs COE III (p<0,001). Como se
muestra en la figura 12:
OV-I
T.BEN
T.BORD
COE I
COE II
COE III0.0
0.5
1.0
1.5
2.0
Progresión Carcinogénica
mR
NA
trkA
/mR
NA
b-a
ctin
a (U
A) * ** **
***

39
Figura 11: Microfotografías representativas de la inmunodetección de la proteína trkA durante la progresión carcinogénica. A. Ovario Inactivo (OV-I), B. Tumor Benigno (T.Ben), C. Tumor Borderline (T.Bord) D. E. y F. Cáncer Ovárico Epitelial (COE I, II, III respectivamente). El recuadro pequeño en cada microfotografía corresponde al control negativo. La barra en cada recuadro representa 50 um, Aumento 1000X. La flecha indica la tinción positiva en el epitelio.
A B
C D
E F

40
OV-I
T.BEN
T.BORD
COE I
COE II
COE III0
20
40
60
80
100*
*
*
Progresión Carcinogénica
%C
élul
as P
ositi
vas
Figura 12: Semicuantificación de la proteína trkA por inmunohistoquimica durante la progresión carcinogénica. Las barras representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado como porcentaje de células con tinción positiva. *p<0,001 OV-I vs COE I, II y III, *p<0,001 T.Ben vs COE I, II y III, T.Bord vs COE I, II, III, COE I vs COE II y COE III.
V.4 Localización y semicuantificación del receptor p-trkA en epitelio normal y
transformado durante la progresión carcinogénica.
La inmunodetección del receptor p-trkA se encontró principalmente en el epitelio
transformado de las muestras de COE, con tinción positiva en epitelio. La tinción fue
homogénea y granular, y está presente sólo en el citoplasma, como se observa en la figura
13. Se semicuantificó la tinción por HScore, pero sólo se sumaron los porcentajes de las
intensidades 2 y 3, y se expresó como porcentaje de células con tinción positiva. Se
encontró un aumento gradual y significativo en la inmunodetección de p-trkA, a medida
que progresa la transformación del epitelio en los distintos grados del COE. Este aumento
fue significativo entre COE I vs COE III (p<0,01). Como se muestra en la figura 14. Para
evaluar la razón p-trkA / trkA total, figura 15, se normalizaron los valores del trkA total con
respecto a su control (OV-I), para así poder realizar la razón con el p-trkA, ya que cada
anticuerpo reconoce distintos dominios del receptor, por lo que no son comparables ambas
mediciones sin esta normalización.
*

41
Figura 13: Microfotografías representativas de la inmunodetección de la proteína trkA-p durante la progresión carcinogénica. D. E. y F. Cáncer Ovárico Epitelial (COE I, II, III respectivamente). El recuadro pequeño en cada microfotografía corresponde al control negativo. La barra en cada recuadro representa 50 um, Aumento 1000X. La flecha indica la tinción positiva en el epitelio.
D
E
F

42
OV-I
T.BEN
T.BORD
COE I
COE II
COE III0
20
40
60
80
Progresión Carcinogénica
%C
élul
as P
ositi
vas
Figura 14: Semicuantificación del receptor p-trkA por inmunohistoquimica durante la progresión carcinogénica. Las barras representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, expresado como porcentaje de células con tinción positiva. *p<0,01 COE I vs COE III.
Razón p-trkA / trkA total
En cuanto a la razón entre el receptor biológicamente activado, p-trkA, y el receptor
trkA total, se encontró un aumento significativo durante la progresión del COE. Este
aumento fue significativo entre COE I vs COE III (*p<0,05). Como se observa en la figura
15:
OV-I
T.BEN
T.BORD
COE I
COE II
COE III0
2
4
6
8
10
Progresión Carcinogénica
Raz
ón p
-trk
A/tr
kA to
tal (
UA
)
Figura 15: Razón entre el receptor p-trkA y trkA total por inmunohistoquimica durante la progresión carcinogénica. Las barras representan el promedio ± el error estándar medio, de 5 muestras en cada grupo de estudio, tanto para el p-trkA como para trkA total. *p<0,05 COE II vs COE III, **p<0,01 COE I vs COE III.
*
*

43
En las muestras de ovario inactivo, tumor benigno y borderline, no se encontró una
tinción positiva para p-trkA, como se observa en la figura 16. Sin embargo, encontramos en
algunas zonas una muy leve presencia del mismo, (designadas con intensidad 1), y otras
totalmente negativas (designadas con intensidad 0). Por ello, no se incluyeron en el gráfico
anterior, donde se muestra el % de células con tinción positiva.
Figura 16: Microfotografía representativa de la inmunodetección de la proteína p-trkA durante la progresión carcinogénica. A. Ovario Inactivo (OV-I), B. Tumor Benigno (T.Ben), C. Tumor Borderline (T.Bord) El recuadro pequeño en cada microfotografía corresponde al control negativo. La barra en cada recuadro representa 50 um, Aumento 1000X. La flecha indica la tinción positiva en el epitelio.
C B
B C
A
A

44
Conjuntamente se evaluó la correlación proteica entre trkA / NGF y trkA / VEGF
durante la progresión carcinogénica ovárica, como se observa en la figura 17. En ambos
casos se obtuvo una correlación positiva y significativa. En el caso de trkA / NGF se
obtuvo un r = 0.8 y en el caso de trkA/VEGF un r = 0.7, ambos con un ***p<0.001.
2.0 2.5 3.0 3.50
1
2
3
4
5OV-IT.BEN.T.BORD.COE ICOE IICOE III
NGF
trkA
2.0 2.5 3.0 3.5 4.0 4.50
1
2
3
4
5OV-IT.BEN.T.BORD.COE ICOE IICOE III
VEGF
trkA
Figura 17: Correlación entre los niveles proteicos de trkA y NGF (parte superior) y
entre los niveles proteico de trkA y VEGF (parte inferior) durante la progresión
carcinogénica ovárica.

45
VI DISCUSIÓN
En estudios previos realizados en roedores se determinó la presencia y función de
NGF y de trkA en el estroma ovárico, estableciéndose su participación en la
esteroidogénesis ovárica, mediante el aumento de la secreción de progesterona en las
células de la teca previo a la ruptura folicular durante la ovulación, y también en la
foliculogénesis ovárica participando en la regulación del desarrollo folicular temprano y en
su diferenciación. Además, se encontró la presencia de NGF y trkA en folículos preantrales
y antrales de ovarios humanos.24,26,28,30,33. Por otra parte VEGF también participa en el
desarrollo folicular, induciendo el desarrollo hasta folículo antral, mediante la formación de
la vasculatura tecal en ratas inmaduras 50,51 y también en monos. De esta forma tanto NGF
como trkA y VEGF comparten un rol fisiológico en el ovario normal funcional.
Cuando se produce el cese de la función ovárica, deja de ciclar, etapa conocida
como menopausia, el ovario es un ovario inactivo, donde se observan con mayor frecuencia
invaginaciones de la pared ovárica hacia el estroma, las cuales se cierran dando origen a los
quistes de inclusión, rodeados por una monocapa de células epiteliales de la superficie
ovárica. En esta etapa se pueden gestar cambios displásticos en estas células epiteliales,1,2
que pueden originar un carcinoma, principal hipótesis para la etiología del COE, cuyas
mayores tasas de incidencia se registran precisamente en mujeres postmenopáusicas. Es por
ello, que este estudio ha considerado como grupo control o normal al ovario inactivo, para
entender y conocer los cambios que acompañan a la progresión carcinogénica ovárica, y
que participación tendrían en esta patología; NGF, trkA y VEGF, de los cuales poco se
conoce a la fecha.
La expresión génica y proteica de NGF en el epitelio, presentó un leve aumento
durante toda la progresión carcinogénica, pero este aumento fue significativo a partir del
COE I en adelante, aumentando a medida que el tejido se va desdiferenciando, lo cual nos
da indicios de su importancia en la progresión de esta patología. Además, recordemos que
NGF participa en la angiogénesis, tanto de forma directa en las células endoteliales como
indirecta en las células epiteliales, descrito en otros estudios35,36,37,39. La angiogénesis es un
evento muy importante para el crecimiento y posterior metástasis tumoral, y el COE seroso
se caracteriza por ser altamente angiogénico, lo que le permite alcanzar grandes tamaños,
una alta agresividad y una baja sobrevida.4,5

46
Esta elevada angiogénesis, se ve reflejada en los altos niveles tanto de mRNA como
de proteína encontrados para VEGF, el principal factor angiogénico42. Este aumento en
cuanto a la proteína, fue significativo desde COE I en adelante relacionándose con lo
encontrado para NGF, por lo que podemos atribuirlo a su participación como factores
angiogénicos durante la progresión del tumor para permitir su desarrollo y posterior
metástasis. A su vez, los niveles de mRNA de VEGF variaron de acuerdo a las isoformas
estudiadas, en cuanto a la isoforma 121, aumentó significativamente en COE II y III, la
isoforma 165 desde COE I en delante, las cuales son secretadas (VEGF 121 totalmente y
VEGF 165 parcialmente) por tanto, estas dos isoformas actuarían en forma paracrina en las
células endoteliales y entregarían la señal de proliferación y migración. Esta función se
complementaría con la isoforma 189, la que experimenta mayores cambios, y cuyo
aumento ocurre en etapas más tempranas desde el tumor borderline en adelante. Se ha
postulado que esta isoforma permanece retenida en la matriz y actuaría como señal
quimioatractante, para guiar el viaje de las células endoteliales y así originar la formación
de nuevos vasos a partir de vasos existentes, lo que conocemos como angiogénesis.
En cuanto al receptor trkA, ha sido estudiado en varios tipos de cáncer, tales como
cáncer de páncreas, próstata, colon, tiroides, entre otros, en donde se ha encontrado una alta
presencia de trkA20, 47, 48. En el presente estudio se encontró muy bajos niveles de la
proteína trkA en el epitelio de ovarios inactivos, pero ésta fue aumentando gradual y
significativamente durante la progresión carcinogénica. En cuanto al receptor activo p-trkA,
no se encontró en ovarios inactivos, ni en tumores benignos y borderline; sólo se encontró
en algunas zonas, con una muy leve presencia, a diferencia de lo observado en COE donde
se encontró una alta presencia, la cual fue aumentando significativamente a medida que el
tejido va perdiendo su diferenciación, así como también fue aumentando la razón p-trkA /
trkA total durante la progresión del COE.
Con respecto a los niveles de mRNA para trkA, sólo fue detectado en tumores
borderline y COE. Este hecho se explicaría principalmente por la naturaleza de la muestra,
ya que, en muchas ocasiones puede ocurrir que el pequeño trozo extraído corresponda sólo
a estroma ovárico, o a tejido fibroso ovárico, en vez de ESO y/o tejido tumoral con alto
porcentaje de epitelio. En los grupos de tumores benignos y ovarios inactivos no se
encontró la expresión de trkA, probablemente debido a la poca cantidad de epitelio que

47
encontramos en estos grupos, además, del hecho de que los receptores de membrana, como
es el caso de trkA, son difíciles de detectar, por sus bajos niveles en condiciones
fisiológicas.
Estos resultados dan cuenta de la transformación y malignidad que presentan las
células epiteliales durante este proceso carcinogénico, considerando los cambios de
expresión encontrados tanto en el receptor trkA, como también en los factores angiogénicos
VEGF y NGF. En el grupo de estudio COE I, las células epiteliales están transformadas, ya
que presentan la capacidad de invadir, y es precisamente donde encontramos los mayores
cambios tanto a nivel de proteína como de mRNA de trkA, lo cual apoya nuestra hipótesis
de proponer al receptor trkA como un marcador tumoral de valor pronóstico y de
progresión del COE.
El motivo de proponer al receptor trkA como un marcador pronóstico, se debe
principalmente a los resultados obtenidos con la inmunodetección del receptor p-trkA,
donde sólo encontramos tinción positiva cuando las células epiteliales se han transformado
(COE I, II y III), así su presencia establecería un mal pronóstico, y podría ser útil en cuanto
al manejo y monitoreo del tratamiento de la paciente. Además, estos resultados se
relacionan con el aumento significativo en los grados del COE encontrado en la
inmunodetección del receptor trkA total, y también de los niveles de mRNA de trkA que se
detectan claramente a partir del COE.
Este aumento gradual y significativo del receptor trkA encontrado durante la
progresión del COE, lo postula como un marcador de progresión, ya que su expresión
génica y proteica aumenta a medida que el tejido va perdiendo su diferenciación. Además,
se correlaciona positiva y significativamente con la expresión de los factores angiogénicos
NGF y VEGF, que tienen gran importancia en la progresión tumoral.
El uso de trkA como marcador pronóstico y de progresión tumoral permitiría tomar
medidas en cuanto al manejo, monitoreo y tratamiento de esta patología, que se caracteriza
por tener un transcurso silente, de rápida progresión y responder pobremente a las terapias
que actualmente se utilizan, como la quimioterapia, por la alta resistencia a las drogas
utilizadas, lo que conlleva a una alta mortalidad4,5.
Actualmente no existen marcadores tumorales diagnóstico de esta patología, por lo
que, sólo se puede recurrir a la ecografía pélvica para confirmar el diagnóstico, a pesar de

48
que presenta falsos positivos, ya que no todos los tumores con diámetro mayor a 5 cm son
malignos, y además no está establecido como examen de rutina, por una parte por su costo
y por otra porque el cáncer ovárico no presenta altos porcentajes de incidencia, como si lo
es el cáncer uterino, para el cual si existen exámenes de rutina para su diagnóstico52.
La mayoría de las mujeres pre y post-menopáusicas, no se controlan periódicamente
por esta patología, sino que recurren cuando presentan los síntomas como molestias
abdominales, hinchazón, plenitud abdominal y saciedad precoz, dolor en la parte baja de la
espalda y sangrado vaginal. En este contexto el tumor se encuentra generalmente en
estadios avanzados, por lo que la medida que se toma en esos casos es la cirugía,
histerectomía con ooforectomía y/o histerectomía radical, dependiendo de la complejidad
del tumor, acompañado de quimioterapia en pacientes con enfermedad residual. Para el
monitoreo pre y post-operatorio se utiliza el marcador tumoral CA-125, que es altamente
inespecífico, por lo que no se utiliza para diagnosticar52.
Por todo lo anterior proponemos utilizar la inmunodetección del receptor p-trkA y
trkA total, en las muestras de biopsias de pacientes que han sido sometidas a cirugía, para
así conocer el pronóstico y progresión del tumor, y de esta manera tomar medidas en cuanto
al manejo del tratamiento a seguir con la paciente. También sería útil en mujeres
menopáusicas cuya ecografía pélvica no es normal, para conocer el pronóstico de ese tumor
y tomar medidas al respecto.
Por lo mismo sería muy interesante hacer estudios posteriores para determinar la
factibilidad de pesquisar trkA en líquidos peritoneales, para conocer el posible
comportamiento metastásico del tumor, y tomar medidas al respecto, para tener mejores
resultados en el tratamiento y en la sobrevida de las pacientes.
Efectivamente comprobamos que tanto trkA como NGF y VEGF están asociados
con el comportamiento celular maligno, ya sea en el desarrollo, como en la progresión del
COE seroso, al igual que en otros cánceres humanos.
De esta forma podemos concluir que trkA NGF y VEGF tendrían una participación
importante en esta patología, pero sería de vital importancia entender por qué ocurre este
cambio, es decir, desde el rol fisiológico en el ovario normal al rol patológico en cáncer
ovárico, para así llegar a conocer y entender mejor estos complejos eventos moleculares
que ocurren desde un rol funcional.

49
Posteriormente, sería necesario hacer estudios de cuantificación proteica mediante
western blot y de expresión génica mediante PCR en tiempo real durante la progresión
carcinogénica ovárica, y de esta manera corroborar los datos obtenidos con las técnicas de
semicuantificación utilizadas en este estudio.
Por otra parte, se ha descrito la presencia de oncogenes del receptor trkA en
diversos cánceres. Estos oncogenes pueden ser de dos tipos: los originados por splicing
alternativo como es el caso del oncogen trkA III descrito en carcinoma de próstata, tiroide
medular y neuroblastoma, 13 y los originados por recombinación génica como el oncogen
trK unido a tropomiosina, descrito en carcinoma de colon y tiroide papilar.17 Sin embargo,
en cáncer de ovario no ha sido descrito ningún oncogen hasta la fecha.
Este oncogen del receptor trkA participaría cooperando en el desarrollo y progresión
tumoral, promoviendo el crecimiento tumoral y aumentando la vascularidad tumoral13. Por
lo que, una vez encontrado en cáncer de ovario, podría ser promovido como un posible
candidato como blanco terapéutico, inhibiendo su expresión proteica mediante el uso de
RNA antisentido o combinados de vías de señalización trkA/PI3K/AKT49, para actuar sólo
en las células tumorales, y no en las normales, como ocurre con los actuales métodos
disponibles como las quimioterapia, que cabe decir, no tienen buenos resultados debido a la
alta resistencia a las drogas utilizadas. Por este motivo se han probado otras alternativas
terapéuticas como la terapia antiangiogénica en conjunto con las quimioterapias
(bloqueando el VEGF circulante mediante anticuerpos antiVEGF)43 la cual tampoco ha
tenido los resultados esperados, debido a que no solamente el VEGF sería responsable del
alto poder angiogénico, sino que, también lo sería NGF, ya que se ha encontrado que trkA
también se expresa en las células endoteliales del ovario41. Es por este motivo que es de
vital importancia el desarrollo de nuevas estrategias terapéuticas, las cuales podrían
aumentar la efectividad de agentes quimioterapéuticos en el tratamiento del cáncer ovárico,
mejorando la efectividad de la quimioterapia49 y aumentando con ello la sobrevida de las
pacientes con cáncer ovárico, que actualmente es muy baja.

50
Esquema 2: Los resultados de esta tesis junto con antecedentes previos nos permiten
postular este modelo. En las células epiteliales transformadas en COE, NGF actúa en forma
autocrina activando a su receptor trkA, incrementando la expresión de VEGF, el cual actúa
en las células endoteliales del tejido ovárico, aumentando la angiogénesis tumoral.
Demostrando de esta forma, la participación indirecta de NGF en la angiogénesis. Además,
NGF secretado por las células epiteliales transformadas, actuaría a nivel de las células
endoteliales del tejido ovárico, aumentado la proliferación, migración e invasión de estas
células y por tanto, NGF también puede ser considerado un factor angiogénico directo;
permitiendo el desarrollo y posterior metástasis tumoral.
Célula Epitelial Transformada
supresores
de tumores
mutados
mRNA
Oncogenes
Factores de crecimiento secretados
Ras
/Map
k
PI3K
/Akt
Transcripción
de genes
Células
Endoteliales
Proliferación
Invasión
Migración
Desarrollo y
Metástasis
Tumoral
COE IICOE I COE III
Proliferación
Invasión
Migración
Angiogénesis
invasiónOV-I T.BEN T.BORD
NGFNGF
VEGFVEGF
VEGFVEGF
NGFNGF
NGFNGF
flt-1trkA
trkA

51
VII CONCLUSIONES
En la progresión del COE existe un aumento en la expresión génica y proteica de los
factores angiogénicos VEGF y NGF, los cuales participan en la progresión y posterior
metástasis tumoral. Además, sus altos niveles explican por qué este cáncer es altamente
angiogénico.
Existe un aumento en la expresión génica y proteica del receptor trkA, en la
progresión carcinogénica. Sin embargo, el receptor activo p-trkA sólo se encuentra presente
en COE y aumenta a medida que se pierde la diferenciación, es decir, en los COE III o
pobremente diferenciado.
El receptor trkA activado por NGF, estaría mediando la alta proliferación celular y
la angiogénesis de este tipo de cáncer.
Por todo lo anterior proponemos al receptor trkA como marcador pronóstico y de
progresión del COE, para ser evaluado en muestras de tejido ovárico humano, de biopsias
de pacientes post-cirugía, ayudando en el manejo, monitoreo y tratamiento de esta
patología.

52
VIII PROYECCIONES
Proponemos el oncogen trkA como un nuevo blanco terapéutico, para realizar la
construcción de un RNA antisentido, que inhiba su expresión proteica, o combinados con
las vías de transducción de señales: oncogen trkA/PI3K/Akt, lo cual, sólo afectaría a las
células tumorales que expresan este oncogen y no a las normales como ocurre con la
quimioterapia. Esto aumentaría la sensibilidad a las drogas utilizadas en la quimioterapia,
ya que actualmente existe una alta resistencia a las drogas utilizadas.
Por otra parte, se podría mejorar las terapias combinadas de quimioterapia en
conjunto con la terapia antiangiogénica, las cuales no han entregado los resultados
esperados, ya que, hoy se sabe que no solamente VEGF es un importante factor
proangiogénico sino que también lo es NGF. Esta alternativa, sería de gran utilidad ya que
este cáncer es altamente angiogénico y por tanto es altamente agresivo y con una alta tasa
de mortalidad.

53
IX. REFERENCIAS
1) Auersperg N, Wong A, Choi K-C, Kang S K, and Leung P, Ovarian Surface
Epithelium: Biology, Endocrinology and Pathology, Endocrine Reviews, 22:255-
288, 2001.
2) Dyck H, Hamilton T, Godwin A, Lynch H, Maines-Bandiera S and Ausperg N,
Autonomy of the epithelial phenotype in human ovarian surface epithelium:
changes with neoplastic progression and with a family history of ovarian cancer, Int
J Cancer, 69:429–436, 1996.
3) Auersperg N, Ota T, and Mitchell G W E, Early Events in Ovarian Epithelial
Carcinogenesis: Progress and Problems in Experimental Approaches, Int J Gynecol
Cancer, 12:691-703, 2002.
4) Jordan S, Green A and Webb P, Benign Epithelial Ovarian Tumours-Cancer
Precursors or Markers for Ovarian Cancer Risk?, Cancer Causes Control, 17:623-
632, 2006.
5) Murdoch W. and McDonnel A., Roles of the Ovarian Surface Epithelium in
Ovulation and Carcinogenesis, Reproducción, 123:743-750, 2002
6) Fleming JS, Beaugié CR, Haviv I, Chenevix-Trench G, and Tan OL, incessant
ovulation, inflammation and epithelial ovarian carcinogenesis: revisiting old
hypotheses, MCE, 247: 4-21, 2006.
7) Shih I. and Kurman R., Ovarian Tumorigenesis: A Proposed Model Based on
Morphological and Molecular Genetic Analysis, Am J Pathol, 164:1544-1518,
2004.
8) Scully R., Classification of human ovarian tumors, Environmental Health
Perspectives, 73: 15-24, 1987.
9) Pahlman S. and Hoehner J., Neurotrophin Receptors, Tumor Progression and Tumor
Maturation, Molecular Medicine Today, 432-438, 1996.
10) Wiesmann C. and De Vos A., Nerve Growth Factor: Structure and Function, Cell.
Mol. Life Sci., 58:748-759, 2001.
11) Nakagawara A, TrK Receptor Tyrosine Kinases: A Bridge Between Cancer and
Neural Development, Cancer Letters, 169:107-114, 2001.

54
12) Pierotti M and Greco A, Oncogenic rearrangements of the NTRK1/NGF receptor,
Cancer Letters, 232: 90-98, 2006.
13) Tacconelli A, Farina A, Cappabianca L., DeSantis G., Tessitore A., Vetuschi A.,
Sferra R., Rucci N., Argenti B., Screpanti I., Gulino A and Mackay A., TrKA
Alternative Splicing: A Regulated Tumor-Promoting Switch in Human
Neuroblastoma, Cancer Cell, 6:347-360, 2004.
14) Mak H., Peschard P, Lin T, Naujokas M, Zuo D and Park M, Oncogenic activation
of the met receptor tyrosine kinase fusion protein, Tpr-Met, involves exclusion from
the endocytic degradative pathway, Oncogene, 26:7213-7221, 2007.
15) Mitra G., Martin-Zanca D. and Barbacid M., Identification and biochemical
characterization of p70TRK, product of the human TRK oncogene, Proc. Natl. Acad.
Sci., 84:6707-6711, 1987
16) Coulier F., Martin-Zanca D., Ernst M., and Barbacid M., Mechanism of activation
of the human trk oncogene, Molecular and Celular Biology, 9: 15-23, 1989.
17) Martin-Zanca D., Oskam R., Mitra G., Copeland T. and Barbacid M., Molecular
and biochemical characterization of the human trk proto-oncogene, Molecular and
Celular Biology, 9:24-33, 1989.
18) Oskam R., Coulier F., Ernst M., Martin-Zanca D. and Barbacid M., Frequent
generation of oncogenes by in vitro recombination of TRK protooncogene
sequences, Proc. Natl. Acad. Sci., 85:2964-2968, 1988.
19) Coulier F., Kumar R., Ernst M., Klein R., Martin-Zanca D. and Barbacid M.,
Human trk oncogenes activated by point mutacion, in-frame deletion, and
duplication of the tyrosine kinase domain, Molecular and Cellular Biology,
10:4202-4210, 1990.
20) Descamps S, Toillon R, Adriaenssens E, Pawlowski V, Cool S M, Nurcombe V,
Bourhis X L, Boilly B, Peyrat J, and Hondermarck H, Expresión of nerve growth
factor receptors and their prognostic value in human breast cancer, cancer research,
61: 4337-4340, 2001.
21) Tacconelli A., Farina A, Cappabianca L., Gulino A and Mackay A., TrKAIII a
novel hypoxia-regulated alternative TrKA splice variant of potencial physiological
and pathological importance, Cell Cycle, 4: 8-9, 2005.

55
22) Lara H., Hill D., Katz K. and Ojeda S, The gene encoding nerve growth factor is
expressed in the immature rat ovary: effect of denervation and hormonal treatment,
Endocrinology, 126: 357-363, 1990.
23) Dissen G, Hirshfield N, Malamed S, and Ojeda S., Expression of neurotrophins and
their receptors in the mammalian ovary is developmentally regulated: changes at the
time of folliculogenesis, Endocrinology, 136:4681-4692, 1995.
24) Dissen G, Hill D, Costa M, Dees W, Lara H, and Ojeda S, A role for TrkA nerve
growth factor receptors in mammalian ovulation, Endocrinology, 137:198-209,
1996.
25) Abir R, Fisch B, Jin S, Barnnet M, Ben-Haroush A, Felz C, Kessler-Icekson G,
Feldberg D, Nitke S and Ao A, Presence of NGF and its receptors in ovaries from
human fetuses and adults, Molecular Human Reproduction, 11:229-236, 2005.
26) Dissen G, Romero C, Newman Hirshfield A, Ojeda S, and Dissen G., Nerve growth
factor is required for early follicular development in the mammalian ovary,
Endocrinology, 142: 2078-2086, 2001.
27) Dissen G., Romero C., Paredes A. and Ojeda S, Neurotrophic control of ovarian
development, Microsc. Res. Tech., 59: 509-515, 2002.
28) Anderson R, Robinson L, Brooks J, and Spears N, Neurotropins and their receptors
are expressed in the human fetal ovary, J Clin Endocrinol Metab., 87: 890-897,
2002.
29) Salas C, Julio−Pieper M, Valladares M, Pommer R, Vega M, Mastronardi C, Kerr
B, Ojeda S, Lara H, and Romero C, Nerve growth factor-dependent activation of
trkA receptors in the human ovary results in synthesis of follicle-stimulating
hormone recptors and estrogen secretion, J Clin Endocrinol Metab., 91:2396-2403,
2006.
30) Romero C, Paredes A, Dissen G, and Ojeda S, Nerve Growth Factor Induces the
Expression of Functional FSH Receptors in Newly Formed Follicles of the Rat
Ovary; Endocrinology; 143: 1485–1494; 2002.
31) Mayerhofer A, Dissen G, Costa M, and Ojeda S, A role for neurotransmitters in
early follicular development: induction of functional follicle-stimulating hormone

56
receptors in newly formed follicles of the rat ovary; Endocrinology; 138: 3320-
3329; 1997.
32) Lara H, Dissen G, Leyton V, Paredes A, Fuenzalida H, Fiedler J, and Ojeda S, An
increased intraovarian synthesis of nerve growth factor and its low affinity receptor
is a principal component of steroid-induced polycystic ovary in the rat;
Endocrinology; 141: 1059-1072; 2000.
33) Dissen G, Lara H, Paredes A, Hill D, Costa M, Martinez-Serrano A, and Ojeda S;
Direct effects of nerve growth factor on thecal cells from antral ovarian follicles;
Endocrinology; 141: 4736-4750; 2000.
34) Dissen G, Lara H, Paredes A, Hill D, Costa M, Martinez-Serrano A, and Ojeda S,
Intraovarian excess of nerve growth factor increases androgen secretion and disrupts
estrous cyclicity in the rat; Endocrinology; 141: 1073-1082, 2000.
35) Calzá L, Giardino L, Giuliani A, Aloe L, and Levi-Montalcini R; Nerve growth
factor control of neuronal expression of angiogenetic and vasoactive factors, PNAS,
98: 4160-4165, 2001.
36) Julio-Pieper M, Lara H, Bravo J and Romero C, Effects of nerve growth factor
(NGF) on blood vessels area and expression of the angiogénico factors VEGF and
TGFbeta I in the rat ovary, Reproductive biology and endocrinology, 4:57,2006.
37) Campos X, Muñoz Y, Selman A, Yazigi R, Moyano L, Weinstein-Oppenheimer C,
Lara H and Romero C, Nerve Growth Factor and its High-Affinity Receptor TrkA
Participate in the Control of Vascular Endothelial Growth Factor Expression in
Epithelial Ovarian Cancer, Gynecologic Oncology 104: 168-175, 2007.
38) Cantarella G, Lempereur L, Presta M, Ribatti D, Lombardo G, Lazarovici P,
Zappala G, Pafumi C, and Bernardini R, Nerve growth factor-endothelial cell
interaction leads to angiogenesis in vitro and in vivo, FASEB, 16:1307-1309, 2002.
39) Park M., Kwak H, Lee H., Yoo D., Park I., Kim M., Lee S., Rhee C. and Hong S.,
Nerve growth factor induces endothelial cell inacsion and cord formation by
promoting matrix metalloproteinase-2 expression through the phosphatidylinositol-3
kinase/akt signaling pathway and ap-2 transcription factor, the journal of biological
chemistry, 282:30485-30496, 2007.

57
40) Odegaard E, Staff A, Abeler V, Kopolovic J, Onsrud M, Lazarovici P and Davidson
B, The activated nerve growth factor receptor p-TrKA is selectively expressed in
advanced-stage ovarian carcinoma, Human Pathology, 38:140-146, 2007.
41) Davidson B, Reich R, Lazarivici P, Nesland J, Skrede M, Risberg B, Tropé C, and
Florenes V, Expression and activation of the nerve growth factor receptor TrkA in
serous ovarian carcinoma, Clinical Cancer Research, 9: 2248-2259, 2003.
42) Abulafia O. and Sherer D., Angiogenesis of the ovary, Am J Obstet Gynecol,
182:240-6, 2000.
43) McMahon G., VEGF receptor signaling in tumor angiogenesis, The Oncologist, 5:3-
10, 2000.
44) Schiffenbauer Y, Abramovitch R, Meir G, Nevo N, Holzinger M, Itin A, Keshet E,
and Neeman M, Loss of ovarian function promotes angiogenesis in human ovarian
carcinoma, PNAS, 94: 13203-13208, 1997.
45) Choi J, Choi K, Auesperg N, and Leung P, Gonadotropins activate proteolysis and
increase invasion through protein kinase A and phosphatidylinositol 3-kinase
pathways in human epithelial ovarian cancer cells, Cancer Res, 66: 3912-20, 2006.
46) Ito T, Ishii G, Chiba H and Ochiai A, The VEGF angiogenic switch of fibroblasts is
regulated by MMP-7 from cancer cells, Oncogene, 26:7194-7203, 2007.
47) Dang C, Zhang Y, Ma Q, and Shimahara Y, Expression of nerve growth factor
receptors is correlated with progression and prognosis of human pancreatic cancer,
21: 850-858, 2006.
48) Davidson B, Lazarivici P, Ezersky A, Nesland J, Berner A, Risberg B, Tropé C,
Kristensen G, Goscinski M, van de Putte G, and Reich R, Expression levels of the
nerve growth factor receptors trkA and p75 in effusions and solid tumors of serous
ovarian carcinoma patients, Clinical Cancer Research, 7: 3457-3464, 2001.
49) Liu D., Zhang Y, Dang C, Ma Q, Lee W and Chen W, siRNA directed against TrkA
sensitizes human pancreatic cancer cells to apoptosis induced by gemcitabine
through an inactivation of PI3K/Akt-dependent pathway, Oncology Reports, 18:
673-677, 2007.
50) Shimizu T., Jiang J., Iijima K., Miyabayashi K., Ogawa Y., Sasada H., and Sato E.,
Induction of Follicular Development by Direct Single Injection of Vascular

58
Endothelial Growth Factor Gene Fragments into the Ovary of Miniature Gilts,
Biology of Reproduction, 69:1388-1393, 2003.
51) Shimizu T., Iijima K., Miyabayashi K., Ogawa Y., Miyazaki H., Sasada H., and
Sato E., Effect of direct ovarian injection of vascular endothelial growth factor gene
fragments on follicular development in immature female rats, Reproduction, 134:
677-682, 2007.
52) Baker M., Leon-Casasola O., DiCioccio R., Foon K., Hempling R., Herman S.,
Hicks M., Ho A., Klippenstein D., Lurain J., Malfetano J., O´leary K., Orner J.,
Patsner B., Piver M., Rose P., Shin K., Shows T., Stomper P., Tsukada Y.,
Oncología Ginecológica, hospital manual, marban, segunda edición.

59
X. ANEXO I

60