análisis de la solidificación en metales puros por el
TRANSCRIPT

Análisis de la solidificación en metales puros por el
Método de Campo de Fases
T E S I S Que para obtener el título de:
Ingeniero en Metalurgia y Materiales
P R E S E N T A Cristóbal Ricardo Escamilla Illescas
Director de tesis Dr. Víctor Manuel López Hirata
Ciudad de México Agosto de 2016
Instituto Politécnico Nacional
Escuela Superior de Ingeniería Química e Industrias Extractivas
Departamento de Ingeniería en Metalurgia y Materiales

Dedicatoria

Dedicatoria

Dedicatoria
A mis padres y abuelos; que con su vida, hicieron posible la mía.

Agradecimientos
Agradecimientos
Llegar al final de una etapa no es simple, y es estando aquí, al mirar atrás, en donde me doy
cuenta de todo lo que necesité y se me fue dado para construir un camino que llevara hasta
este lugar. Quiero agradecer:
A Dios, por todas las fortuitas casualidades que nos permiten existir y dejar una
evidencia de nuestra presencia. Por la vida, el tiempo, el amor y por este fascinante mundo
en el que vivimos.
Al Instituto Politécnico Nacional, a la Escuela Superior de Ingeniería Química e
Industrias Extractivas y al Departamento de Ingeniería en Metalurgia y Materiales, por ser
el pilar de mi formación. Por darme una identidad como Politécnico y Metalúrgico, la cual
honrar y de la cual estar orgulloso.
A mis padres, María Victoria Illescas Faustino y Aurelio Escamilla Téllez, a quienes
debo más que el amor recibido, el techo bajo el que he vivido o todo lo que he aprendido.
Es por ustedes que me encuentro aquí, que soy este Ricardo. Han formado mi ser cada día
de mi vida. Lo bueno en mí se los debo a ustedes.
A mis hermanos, Juan Pablo, David y Esmeralda, que son la fuerza cohesiva de la
familia Escamilla Illescas, por mostrarme el camino con su ejemplo.
A mi asesor, el Dr. Víctor Manuel López Hirata, quien con su incansable guía hizo
posible este trabajo. Por llenar mis dudas con entendimiento y siempre encontrar tiempo
para escucharlas, por la interminable paciencia e interés, por compartir sus conocimientos
conmigo y por, incluso, sentarse conmigo a hacer cálculo vectorial.
A la Dra. Maribel Leticia Saucedo Muñoz, por hacer este trabajo cada vez más completo
con cada observación y por hacerme sentir parte de este fabuloso grupo de investigación.
A la Dra. Lucía Graciela Díaz Barriga Arceo, al Dr. Héctor Javier Dorantes Rosales y al
M. en C. Sergio Javier García Núñez, por tomarse el tiempo para leer y revisar este trabajo,
a fin de mejorarlo.
A Valeria Miranda López, quien ha caminado a mi lado cada día de estos últimos cinco
años, quien me ha visto y hecho crecer. Has iluminado mi vida con recuerdos rebosantes de
amor y felicidad. Eres verdaderamente la mejor compañera de viaje. Tengamos el viaje más
largo, ¿Sí?

Agradecimientos
Al Pbro. Felipe Galicia Reyes, quien me formó musical y humanamente, y me dio un
segundo hogar en Donceles número 49.
A la Ing. Tania Soriano Cruz, a Karen Ledezma Gutiérrez y Mauricio Trejo Cristerna,
que me brindaron su apoyo, amistad y compañía durante muchos días de trabajo.
Al Consejo Nacional de Ciencia y Tecnología, en particular a los proyectos 220929 y
220984, así como al programa de Becas de Estímulo Institucional de Formación de
Investigadores del Instituto Politécnico Nacional por el apoyo brindado para la realización
de este trabajo.
A todas las personas que han sido parte de mi formación personal y profesional, y que en
una clara muestra de ingratitud que sabrán perdonar, he olvidado mencionar.
Muchas gracias.
C. R. E. I.

Contenido
Contenido
Resumen ..................................................................................................................................I
Lista de figuras ..................................................................................................................... II
Lista de tablas ..................................................................................................................... VI
Introducción .......................................................................................................................... 1
1. Consideraciones teóricas ............................................................................................... 3
1.1 Solidificación en metales puros ............................................................................. 3
1.1.1 Nucleación en metales puros ............................................................................ 3
1.1.1.1 Nucleación homogénea ..................................................................................... 4
1.1.1.2 Nucleación heterogénea ................................................................................... 8
1.1.2 Crecimiento y microestructura dendrítica ...................................................... 12
1.1.2.1 Anisotropía en crecimiento dendrítico ........................................................... 17
1.2 Método de Campo de Fases ................................................................................. 19
1.3 Formulación de Kobayashi ................................................................................. 21
1.4 Método de Diferencias Finitas ............................................................................ 24
2. Metodología numérica ................................................................................................ 29
3. Resultados .................................................................................................................... 33
3.1 Simulaciones de simetría de anisotropía perfecta ............................................. 33
3.2 Simulaciones de magnitud de anisotropía ......................................................... 36
3.3 Simulaciones de calor latente adimensional ...................................................... 43
4. Discusión ....................................................................................................................... 48
4.1 Comparación de dendritas en metales puros con dendritas simuladas .......... 48
4.2 Efecto de la anisotropía en la solidificación ....................................................... 52
4.3 Efecto del calor latente adimensional en la solidificación ................................ 57
Conclusiones ........................................................................................................................ 62
Referencias .......................................................................................................................... 63
Apéndices ............................................................................................................................. 66
Apéndice I. Código usado para la simulación .............................................................. 66

Resumen
Resumen
Actualmente el método de campo de fases es una de las técnicas más efectivas para
modelar la evolución microestructural. La principal característica de los modelos de campo
de fases es el carácter difuso y finito de la intercara entre dos fases, que se describe por una
transición continua, en espacio x, de la variable de campo de fases ϕ(x,t) entre dos estados:
sólido y líquido en el caso estudiado.
En el presente trabajo se aplicó el método de campo de fases a la solidificación de
metales puros, haciendo énfasis en el crecimiento de cristales dendríticos usando la
formulación propuesta por Kobayashi para analizar los diferentes parámetros que afectan el
proceso de solidificación.
Para simular la evolución de la microestructura dendrítica, se usó un código en
FORTRAN, en el cual, las ecuaciones diferenciales parciales del modelo se resolvieron
numéricamente por el Método Explícito de Diferencias Finitas para generar archivos de
datos, que posteriormente se graficaron y se usaron para crear secuencias de video.
Los resultados de la simulación mostraron la evolución de cristales dendríticos en
metales con simetría cúbica y hexagonal, el efecto del calor latente de solidificación y la
magnitud de anisotropía sobre la formación de dendritas.
Al incrementar el valor de calor latente adimensional se observó una reducción en la
velocidad de crecimiento de los cristales. Además, a valores bajos de calor latente de
fusión, este mostró ser el mecanismo que determina la geometría de los cristales en
crecimiento, permitiendo la formación de geometrías simples. En contraste, a valores altos
de calor latente de fusión, la magnitud de anisotropía es el mecanismo determinante de la
geometría de los cristales, permitiendo la formación de geometrías complejas.
Por otra parte, en condiciones de isotropía cristalina o de baja anisotropía, se observó la
formación de cristales de puntas separadas. Además, al aumentar la magnitud de
anisotropía, se observó un aumento en la velocidad de crecimiento de los cristales, con la
presencia de un máximo, tras el cual la velocidad de crecimiento de los cristales disminuye.
I

Lista de figuras
Lista de figuras
Figura Descripción Página
1 Diferencia en energía libre entre líquido y sólido cerca del punto de fusión. (7) 3
2 Nucleación homogénea. (7) 5
3 Cambio de energía libre asociado con la nucleación homogénea de una esfera
de radio r. (7) 6
4 Nucleación heterogénea de una capa esférica en una pared de molde plana. (7) 8
5 Exceso de energía libre de embriones para nucleación homogénea y
heterogénea. (7) 10
6 Variación de ΔG* con respecto al sobreenfriamiento, ΔT, para nucleación
homogénea y heterogénea. (7) 11
7
(a) Distribución de temperatura para solidificación cuando el calor es extraído
a través del sólido. Isotermas (b) para una intercara plana S/L y (c) para una
protuberancia. (7)
12
8 Lo expuesto en la figura 7, con conducción de calor hacia el líquido (7). 13
9
Desarrollo de dendritas térmicas: (a) núcleo esférico; (b) comienzo de
inestabilidad en la intercara; (c) desarrollo de brazos primarios en direcciones
cristalográficas; (d) desarrollo de brazos secundarios y terciarios. (7)
14
10
Cambio secuencial de la morfología de la intercara a medida que la velocidad
de solidificación aumenta: (a) Célula creciendo en la dirección de la extracción
de calor; (b) Célula creciendo en la dirección <100> (c) Transición
célula/dendrita (d) Dendrita. (11)
15
11 Distribución de temperaturas en la punta de una dendrita térmica en
crecimiento. (7) 16
II

Lista de figuras
12 Patrón de evolución espacial para una secuencia de separación de puntas
obtenido en las simulaciones de Kessler, Koplik y Levine. (13) 17
13 Crecimiento de un cristal en el cual se observa separación de puntas obtenido
en las simulaciones de Brush y Sekerka. (14) 18
14 Patrón de separación de puntas formado en la experimentación de Ben-Jacob y
colaboradores. (15) 19
15
Representación gráfica del Método Explícito de Diferencias Finitas:
(a) Aproximación para una ecuación diferencial parcial de primer orden
(b) Aproximación para una ecuación diferencial parcial de segundo orden. (21)
28
16 Diagrama de flujo correspondiente a la simulación de la solidificación en
metales puros por el método de campo de fases. 30
17
Evolución de la solidificación de cristales bajo condiciones de j = 0.0 y
κ = 1.0. (a) t = 0.08; (b) t = 0.2; (c) t = 0.32; con δ = 0.000. (d) t = 0.08;
(e) t = 0.2; (f) t = 0.32; con δ = 0.020.
34
18
Evolución de la solidificación de cristales bajo condiciones de j = 0.0 y
κ = 1.8. (a) t = 0.2; (b) t = 0.4; (c) t = 0.6; con δ = 0.000. (d) t = 0.2;
(e) t = 0.4; (f) t = 0.6; con δ = 0.020.
35
19
Evolución de la solidificación de cristales bajo condiciones de κ = 1.8 y
δ = 0.000. (a) t = 0.2; (b) t = 0.4; (c) t = 0.6; con j = 4.0. (d) t = 0.2;
(e) t = 0.4; (f) t = 0.6; con j = 6.0.
37
20
Evolución de la solidificación de cristales bajo condiciones de j = 4.0 y
κ = 1.8. (a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.005. (d) t = 0.2;
(e) t = 0.32; (f) t = 0.4; con δ = 0.010. (g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con
δ = 0.020.
38
21
Evolución de la solidificación de cristales bajo condiciones de j = 4.0 y
κ = 1.8. (a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.050. (d) t = 0.2;
(e) t = 0.32; (f) t = 0.4; con δ = 0.080. (g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con
δ = 0.120.
39
III

Lista de figuras
22
Evolución de la solidificación de cristales bajo condiciones de j = 6.0 y
κ = 1.8. (a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.005. (d) t = 0.2;
(e) t = 0.32; (f) t = 0.4; con δ = 0.010. (g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con
δ = 0.020.
41
23
Evolución de la solidificación de cristales bajo condiciones de j = 6.0 y
κ = 1.8. (a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.050. (d) t = 0.2;
(e) t = 0.32; (f) t = 0.4; con δ = 0.080.
42
24
Evolución de la solidificación de cristales bajo condiciones de κ = 1.0 y
δ = 0.020. (a) t = 0.08; (b) t = 0.2; (c) t = 0.32; con j = 4.0. (d) t = 0.08;
(e) t = 0.2; (f) t = 0.32; con j = 6.0.
44
25
Evolución de la solidificación de cristales bajo condiciones de j = 4.0 y
δ = 0.020. (a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con κ = 1.5. (d) t = 0.2;
(e) t = 0.32; (f) t = 0.4; con κ = 1.8. (g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con
κ = 2.2.
45
26
Evolución de la solidificación de cristales bajo condiciones de j = 6.0 y
δ = 0.020. (a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con κ = 1.5. (d) t = 0.2;
(e) t = 0.32; (f) t = 0.4; con κ = 1.8. (g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con
κ = 2.2.
46
27
Comparación entre (a) Imagen de Microscopio Electrónico de Barrido (MEB)
de una dendrita de austenita; (b) Cristal equiaxial calculado con un modelo de
Campo de Fases correspondiente a dicha dendrita; (22) (c) Simulación obtenida
con las condiciones de t = 0.2, j = 4.0, κ = 1.8 y δ = 0.020.
48
28
Comparación entre (a) Micrografía de una dendrita equiaxial de Succinonitrilo
(SCN); (b) Dendrita de Succinonitrilo con brazos secundarios casi
periódicos; (3) (c) Simulación obtenida con las condiciones de t = 0.2, j = 4.0,
κ = 1.5 y δ = 0.020.
49
IV

Lista de figuras
29
Comparación entre (a) Dendrita de hielo solidificada en agua pura
sobreenfriada a - 0.4 °C; (23) (b) Simulación obtenida con las condiciones de
t = 0.2, j = 6.0, κ = 1.5 y δ = 0.020; (c) Dendrita de hielo solidificada en una
solución de agua y 5% de etilenglicol sobreenfriada a -2.0 °C; (23)
(d) Simulación obtenida con las condiciones de t = 0.4, j = 6.0, κ = 1.8 y
δ = 0.005.
50
30
Comparación entre (a) Micrografía de cristales con puntas separadas en una
aleación Al-4%Cu; (25) (b) Micrografía de cristales con puntas separadas en una
aleación Al-4.5%Cu; (26) (c) Simulación obtenida con las condiciones de
t = 0.6, j = 4.0, κ = 1.8 y δ = 0.000.
51
31
Simulaciones obtenidas con las condiciones fijas de t = 0.4, j = 4.0, κ = 1.8 y
magnitud de anisotropía variable: (a) δ = 0.000; (b) δ = 0.005; (c) δ = 0.010;
(d) δ = 0.20; (e) δ = 0.050; (f) δ = 0.080; (g) δ = 0.120.
53
32
Simulaciones obtenidas con las condiciones fijas de t = 0.4, j = 6.0, κ = 1.8 y
magnitud de anisotropía variable: (a) δ = 0.000; (b) δ = 0.005; (c) δ = 0.010;
(d) δ = 0.20; (e) δ = 0.050; (f) δ = 0.080.
54
33
Simulaciones obtenidas con las condiciones fijas de t = 0.32, j = 4.0, δ = 0.020
y calor latente adimensional variable: (a) κ = 1.0; (b) κ = 1.5; (c) κ = 1.8;
(d) κ = 2.2.
58
34
Simulaciones obtenidas con las condiciones fijas de t = 0.32, j = 6.0, δ = 0.020
y calor latente adimensional variable: (a) κ = 1.0; (b) κ = 1.5; (c) κ = 1.8;
(d) κ = 2.2.
59
35
Simulaciones obtenidas con las condiciones fijas de t = 0.2, j = 0.0 con
condiciones de calor latente adimensional y magnitud de anisotropía variable:
(a) κ = 1.0, δ = 0.000; (b) κ = 1.0, δ = 0.020; (c) κ = 1.8, δ = 0.000;
(d) κ = 1.8, δ = 0.020.
60
V

Lista de tablas
Lista de tablas
Tabla Descripción Página
1 Parámetros de la formulación de Kobayashi usados durante las simulaciones 29
2 Condiciones usadas para las simulaciones de simetría de anisotropía perfecta 33
3 Condiciones usadas para las simulaciones de anisotropía 36
4 Condiciones usadas para las simulaciones de calor latente adimensional 43
VI

Introducción
Introducción
Se estima que las piezas fundidas se usan en el 90% de los productos fabricados, tales como
motores de vehículos, maquinaria para construcción y agricultura, válvulas, bombas,
compresores, electrodomésticos, computadoras, herramientas y en toda la maquinaria
empleada en la manufactura de dichos productos.
Las principales razones para utilizar la fundición son el amplio rango de propiedades
físicas y mecánicas cubierto por aleaciones fundidas, la obtención de un producto con poca
necesidad de acabado superficial, la versatilidad del proceso con respecto al peso y la forma
del componente, que puede ser desde gramos hasta toneladas y formas elaboradas que no
pueden ser obtenidas por otros procesos, así como un costo competitivo de los productos
obtenidos.
El proceso de solidificación es una parte inherente de los procesos de fundición, y
durante esta se genera la estructura de colada de la pieza fundida. Puesto que diversas
piezas se usan sin tratamientos térmicos o conformado posterior, las propiedades mecánicas
de estas, que son consecuencia directa de su microestructura, deben ser controladas durante
el proceso de solidificación. (1) (2)
Los cristales dendríticos o dendritas son estructuras de no-equilibrio que persisten a lo
largo del proceso de solidificación y que están presentes en todas las piezas fundidas. Estas
consisten en cristales individuales ramificados, que presentan morfologías con
direccionalidad cristalográfica, como brazos o ramas primarias, secundarias o terciarias que
guardan relaciones angulares especiales entre ellas: los brazos primarios de una dendrita en
un sistema cúbico se desarrollan en la dirección < 100 >, con los brazos secundarios y
terciarios desarrollándose en dirección ortogonal al brazo anterior.
Las dendritas son de gran importancia en materiales fundidos debido a que establecen la
textura de solidificación y tamaño de grano en la pieza fundida, factores de influencia en las
propiedades mecánicas y procesamiento posteriores al vaciado. (3) Por ello es de gran
importancia modelar la formación y evolución de esta microestructura.
Actualmente el Método de Campo de Fases es una de las técnicas más efectivas para
modelar la evolución microestructural. La principal característica de los modelos de campo
de fases es el carácter difuso de la intercara entre dos fases, razón por la cual se llaman
1

Introducción
también ‘modelos de intercara difusa’. La intercara se describe por una transición continua,
en espacio x, de la variable de campo de fases ϕ(x,t) entre dos estados. (4)
El presente trabajo tiene el propósito de aplicar el Método de Campo de Fases al
crecimiento de cristales dendríticos en un metal puro, usando la formulación propuesta por
Kobayashi (5) para analizar los diferentes parámetros que afectan el proceso de
solidificación.
2
Consideraciones teóricas
1. Consideraciones teóricas
La forma y el tamaño de los granos en un metal puro policristalino se determinan
inicialmente por la nucleación y crecimiento que tiene lugar durante la solidificación, (6) por
lo que es necesario comprender los mecanismos de estos fenómenos.
1.1 Solidificación en metales puros La solidificación y la fusión son transformaciones entre estados cristalográficos y no-
cristalográficos de un metal o aleación. Estas trasformaciones son básicas para aplicaciones
tecnológicas como vaciado de lingotes y piezas, colada continua, crecimiento de
monocristales para semiconductores, solidificación direccionada de aleaciones compuestas,
solidificación rápida de aleaciones y vidrios, así como soldadura por fusión.
Al entenderse el mecanismo de solidificación y cómo lo afectan parámetros tales como la
distribución de temperaturas o la velocidad de enfriamiento, se puede tener control en las
propiedades mecánicas de metales vaciados y uniones soldadas. (7)
1.1.1 Nucleación en metales puros
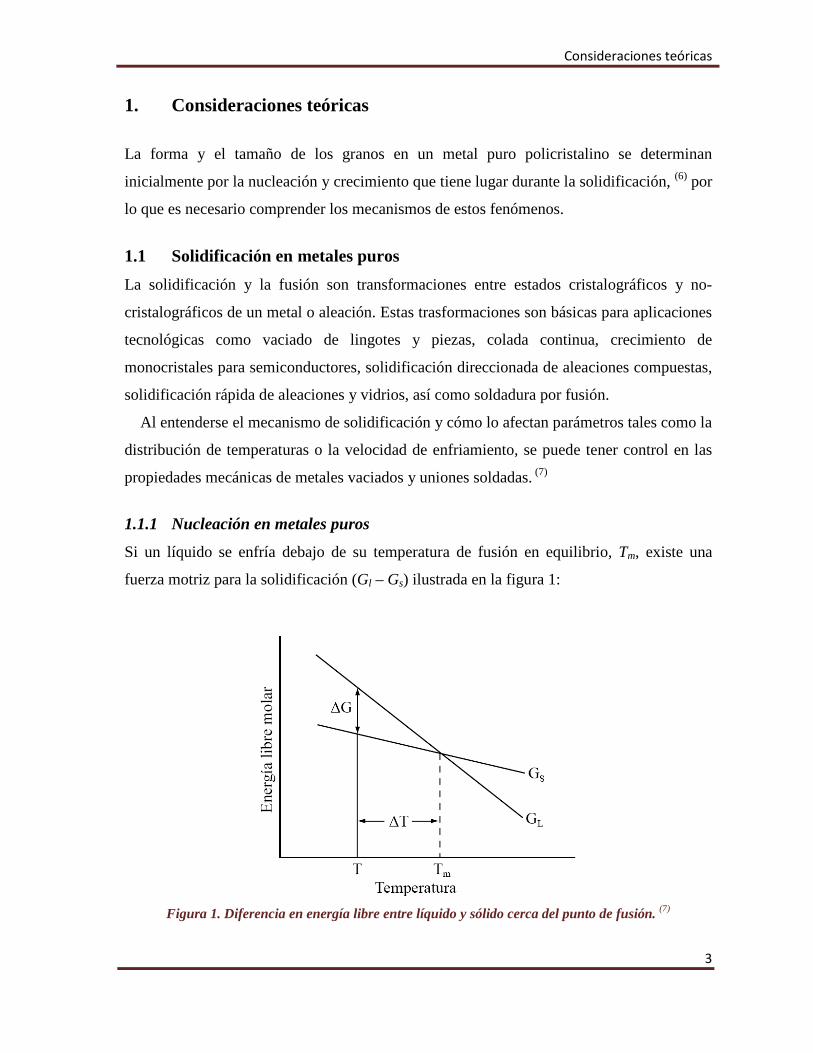
Si un líquido se enfría debajo de su temperatura de fusión en equilibrio, Tm, existe una
fuerza motriz para la solidificación (Gl – Gs) ilustrada en la figura 1:
Figura 1. Diferencia en energía libre entre líquido y sólido cerca del punto de fusión. (7)
3

Consideraciones teóricas
Por su presencia se esperaría que la fase líquida solidifique espontáneamente, sin
embargo este no es siempre el caso.
Por ejemplo, bajo condiciones apropiadas, el níquel puede ser sobreenfriado 250 K
debajo de Tm, 1453°C para Ni, y mantenido indefinidamente sin que ocurra transformación
alguna. La razón de este comportamiento es que la transformación comienza con la
formación de pequeñas partículas sólidas o núcleos.
Normalmente no se observan sobreenfriamientos tan grandes como 250 K, ya que en la
práctica las paredes del recipiente que contiene al líquido o las partículas sólidas de
impurezas en el líquido catalizan la nucleación del sólido a sobreenfriamientos tan bajos
como 1 K. Esto se conoce como nucleación heterogénea.
Los sobreenfriamientos grandes, mencionados antes, se obtienen cuando no hay sitios
disponibles para nucleación heterogénea, es decir, cuando los núcleos sólidos deben
formarse de forma homogénea a partir del líquido. (7)
1.1.1.1 Nucleación homogénea
La figura 2(a) muestra un volumen de líquido determinado a una temperatura ΔT debajo de
Tm, con una energía libre G1. Si algunos átomos del líquido se agrupan para formar una
pequeña esfera de sólido, figura 2 (b), la energía libre del sistema cambiará a G2, expresado
por:
𝐺𝐺2 = 𝑉𝑉𝑆𝑆𝐺𝐺𝑣𝑣𝑆𝑆 + 𝑉𝑉𝐿𝐿𝐺𝐺𝑣𝑣𝐿𝐿 + 𝐴𝐴𝑆𝑆𝐿𝐿𝛾𝛾𝑆𝑆𝐿𝐿 ( 1 )
donde VS es el volumen de la esfera sólida, VL el volumen del líquido, ASL es el área
interfacial sólido/líquido, GvS y Gv
L con las energías libres por unidad de volumen de sólido
y líquido, respectivamente, y γSL la energía libre interfacial sólido/líquido. La energía libre
del sistema sin sólido presente se describe por:
𝐺𝐺1 = (𝑉𝑉𝑆𝑆 + 𝑉𝑉𝐿𝐿) 𝐺𝐺𝑣𝑣𝐿𝐿 ( 2 )
4

Consideraciones teóricas
Figura 2. Nucleación homogénea. (7)
La formación de sólido consiste en un cambio de energía libre ΔG = G2 – G1, donde:
𝛥𝛥𝐺𝐺 = − 𝑉𝑉𝑆𝑆 𝛥𝛥𝐺𝐺𝑣𝑣 + 𝐴𝐴𝑆𝑆𝐿𝐿𝛾𝛾𝑆𝑆𝐿𝐿 ( 3 )
𝛥𝛥𝐺𝐺𝑣𝑣 = 𝐺𝐺𝑣𝑣𝐿𝐿 − 𝐺𝐺𝑣𝑣𝑆𝑆 ( 4 )
Para un sobreenfriamiento ΔT, ΔGv se expresa como:
𝛥𝛥𝐺𝐺𝑣𝑣 =𝐿𝐿𝑣𝑣∆𝑇𝑇𝑇𝑇𝑚𝑚
( 5 )
donde Lv es el calor latente de fusión por unidad de volumen.
Debajo de Tm, ΔGv es positivo. El cambio de energía asociado con la formación de un
pequeño volumen de sólido tiene una contribución negativa debida a la transformación del
sistema de un estado de líquido, con mayor energía libre, a un estado sólido, con menor
energía libre; así como una contribución positiva debida a la creación de una intercara
sólido/líquido.
5
Consideraciones teóricas
El exceso de energía libre puede minimizarse con la elección correcta de la forma de
partícula. Si γSL es isotrópica, ésta es una esfera de radio r y la Ecuación (3) se reescribe
como: (7) (8)
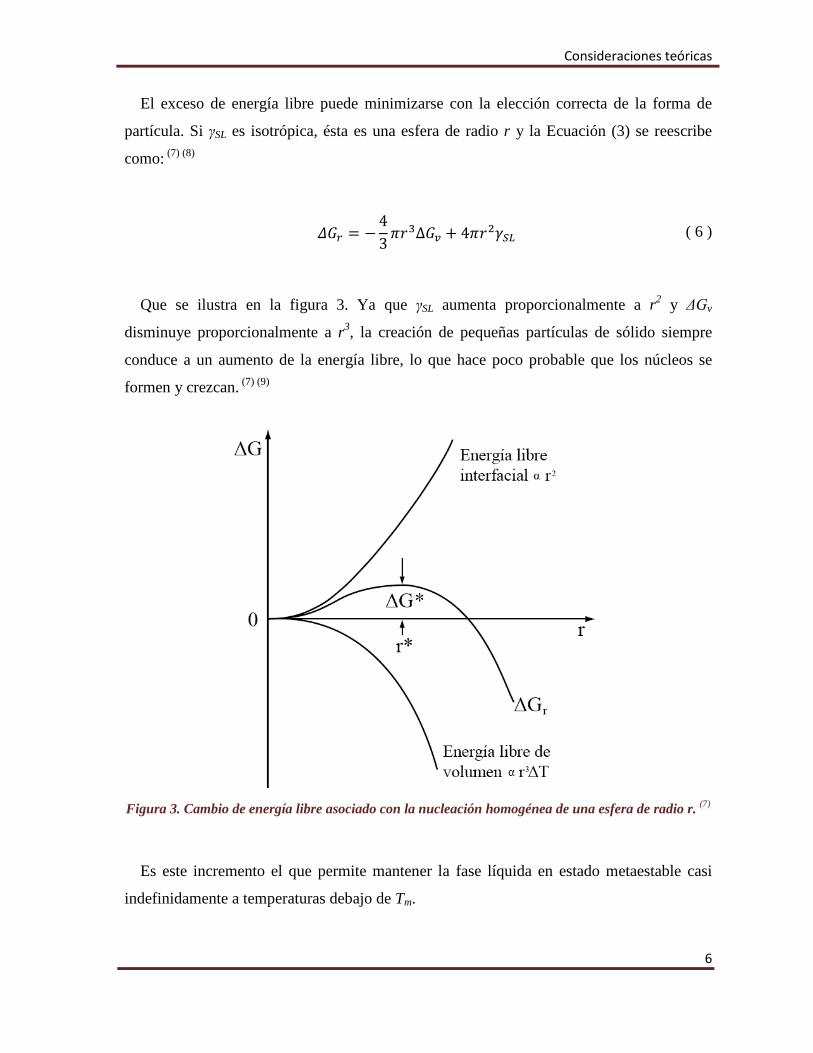
𝛥𝛥𝐺𝐺𝑟𝑟 = −43𝜋𝜋𝑟𝑟3∆𝐺𝐺𝑣𝑣 + 4𝜋𝜋𝑟𝑟2𝛾𝛾𝑆𝑆𝐿𝐿 ( 6 )
Que se ilustra en la figura 3. Ya que γSL aumenta proporcionalmente a r2 y ΔGv
disminuye proporcionalmente a r3, la creación de pequeñas partículas de sólido siempre
conduce a un aumento de la energía libre, lo que hace poco probable que los núcleos se
formen y crezcan. (7) (9)
Figura 3. Cambio de energía libre asociado con la nucleación homogénea de una esfera de radio r. (7)
Es este incremento el que permite mantener la fase líquida en estado metaestable casi
indefinidamente a temperaturas debajo de Tm.
6

Consideraciones teóricas
Dado que la contribución debida a la trasformación de un volumen, proporcional a r3,
difiere de la contribución correspondiente a la formación de la intercara, proporcional a r2,
la función ΔGr presenta un máximo en ΔG*: para un sobreenfriamiento dado hay un cierto
radio, r*, asociado al máximo exceso de energía libre que forma una barrera de energía de
activación que se opone a la cristalización, por lo que es crucial para el comportamiento de
sobreenfriamiento del metal fundido.
Si r < r* el sistema puede disminuir su energía libre con la disolución del sólido,
mientras que cuando r > r* la energía libre del sistema disminuye si el sólido crece.
Las partículas sólidas inestables con r < r* reciben el nombre de embriones, mientras que
las partículas estables con r > r* se llaman núcleos. r* es el tamaño crítico de núcleo, ya que
dG = 0 cuando r = r*, por lo que el núcleo se encuentra en equilibrio con el líquido que lo
rodea. (7) (8)
La Ecuación (6) puede diferenciarse para obtener:
𝑟𝑟∗ =2𝛾𝛾𝑆𝑆𝐿𝐿∆𝐺𝐺𝑣𝑣
( 7 )
Al sustituir la Ecuación (7) en la Ecuación (6) se obtiene:
∆𝐺𝐺ℎ𝑜𝑜𝑚𝑚∗ =16𝜋𝜋𝛾𝛾𝑆𝑆𝐿𝐿3
3∆𝐺𝐺𝑣𝑣2 ( 8 )
Al sustituir ΔGv de la Ecuación (7) en la Ecuación (5) se obtiene:
𝑟𝑟∗ = �2𝛾𝛾𝑆𝑆𝐿𝐿𝑇𝑇𝑚𝑚𝐿𝐿𝑣𝑣
�1𝛥𝛥𝑇𝑇
( 9 )
Y al sustituir la Ecuación (5) en la Ecuación (8) se obtiene:
7

Consideraciones teóricas
∆𝐺𝐺ℎ𝑜𝑜𝑚𝑚∗ = �16𝜋𝜋𝛾𝛾𝑆𝑆𝐿𝐿3 𝑇𝑇𝑚𝑚2
3𝐿𝐿𝑣𝑣2�
1𝛥𝛥𝑇𝑇2
( 10 )
Así, r* y ΔGhom* disminuyen al aumentar el sobreenfriamiento, ΔT. (7) (9)
1.1.1.2 Nucleación heterogénea
De acuerdo a la Ecuación (9), para facilitar la nucleación a sobreenfriamientos bajos el
término de energía interfacial, γSL, debe reducirse. Dos maneras simples de lograr esto es
que el núcleo se forme en contacto con la pared del molde o con la superficie de partículas
inoculantes externas.
La figura 4 muestra un embrión sólido formándose en contacto con una pared de molde
perfectamente plana:
Figura 4. Nucleación heterogénea de una capa esférica en una pared de molde plana. (7)
Asumiendo que γSL es isotrópica se puede mostrar que para un volumen de sólido
determinado, la energía interfacial total del sistema se minimiza si el embrión tiene la
forma de una capa esférica con un ángulo de mojado θ, expresado por el balance de
tensiones interfaciales γML, γSM y γSL en la pared del molde: (7) (9)
𝛾𝛾𝑀𝑀𝐿𝐿 = 𝛾𝛾𝑆𝑆𝑀𝑀 + 𝛾𝛾𝑆𝑆𝐿𝐿𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐 ( 11 )
8

Consideraciones teóricas
Reescribiendo:
𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐 =𝛾𝛾𝑀𝑀𝐿𝐿 + 𝛾𝛾𝑆𝑆𝑀𝑀
𝛾𝛾𝑆𝑆𝐿𝐿 ( 12 )
Debe notarse que la componente vertical de γSL permanece sin balancear. Tras un tiempo,
esta fuerza tiraría de la superficie del molde hasta que las fuerzas de tensión superficial se
balancearan en todas las direcciones. Por ello, la Ecuación (12) sólo muestra la forma
óptima del embrión.
La formación de dicho embrión se asocia a un exceso de energía libre expresado por:
∆𝐺𝐺ℎ𝑒𝑒𝑒𝑒 = −𝑉𝑉𝑠𝑠∆𝐺𝐺𝑣𝑣 + 𝐴𝐴𝑆𝑆𝐿𝐿𝛾𝛾𝑆𝑆𝐿𝐿 + 𝐴𝐴𝑆𝑆𝑀𝑀𝛾𝛾𝑆𝑆𝑀𝑀 − 𝐴𝐴𝑆𝑆𝑀𝑀𝛾𝛾𝑀𝑀𝐿𝐿 ( 13 )
donde Vs es el volumen de la capa esférica, ASL y ASM son las áreas de las intercaras
sólido/líquido y sólido/molde, y γSL, γSM y γML son las energías libres de las intercaras
sólido/líquido, sólido/molde y molde/líquido.
De esta manera existen tres contribuciones de energía interfacial. Las primeras dos son
positivas ya que surgen de intercaras creadas durante el proceso de nucleación. La tercera,
sin embargo, se debe a la destrucción de la intercara molde/líquido bajo la capa esférica y
resulta en una contribución negativa a la energía.
La Ecuación (13) puede escribirse en términos del ángulo de mojado, θ, y del radio de la
capa esférica, r.
∆𝐺𝐺ℎ𝑒𝑒𝑒𝑒 = �−43𝜋𝜋𝑟𝑟3∆𝐺𝐺𝑣𝑣 + 4𝜋𝜋𝑟𝑟2𝛾𝛾𝑆𝑆𝐿𝐿� �
(2 + 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐)(1 − 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐)2
4� ( 14 )
Debe hacerse énfasis en que la Ecuación (14) sólo es distinta de la Ecuación (6), que
describe el cambio de energía libre necesario para que ocurra nucleación homogénea, en la
9
Consideraciones teóricas
existencia del segundo término. Este tiene un valor numérico ≤ 1, dependiente sólo del
ángulo de mojado θ, esto es, la forma del núcleo. Por ello se denomina factor de forma.
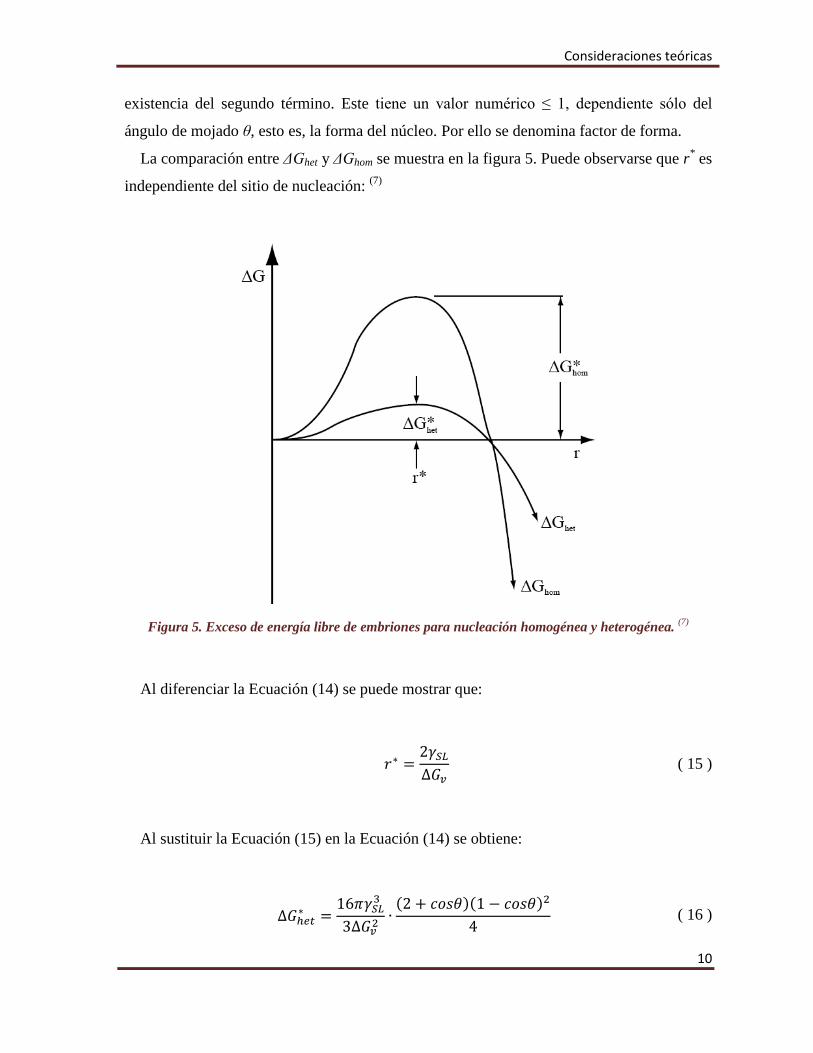
La comparación entre ΔGhet y ΔGhom se muestra en la figura 5. Puede observarse que r* es
independiente del sitio de nucleación: (7)
Figura 5. Exceso de energía libre de embriones para nucleación homogénea y heterogénea. (7)
Al diferenciar la Ecuación (14) se puede mostrar que:
𝑟𝑟∗ =2𝛾𝛾𝑆𝑆𝐿𝐿∆𝐺𝐺𝑣𝑣
( 15 )
Al sustituir la Ecuación (15) en la Ecuación (14) se obtiene:
∆𝐺𝐺ℎ𝑒𝑒𝑒𝑒∗ =16𝜋𝜋𝛾𝛾𝑆𝑆𝐿𝐿3
3∆𝐺𝐺𝑣𝑣2∙
(2 + 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐)(1 − 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐)2
4 ( 16 )
10
Consideraciones teóricas
Por lo que la barrera de energía de activación opuesta a la nucleación heterogénea ΔG*het
es menor que aquella para nucleación homogénea, ΔG*hom, debido al factor de forma. El
radio crítico de núcleo, r*, no se afecta por la pared del molde y sólo depende del
sobreenfriamiento. Un núcleo que crece se vuelve estable cuando alcanza un radio r0 =
1.5r*. Una vez que este ha crecido más allá de r0, la cristalización comienza.
Al combinar las Ecuaciones (16) y (8), se obtiene: (7) (8) (9)
∆𝐺𝐺ℎ𝑒𝑒𝑒𝑒∗ = �(2 + 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐)(1 − 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐)2
4� ∆𝐺𝐺ℎ𝑜𝑜𝑚𝑚∗ ( 17 )
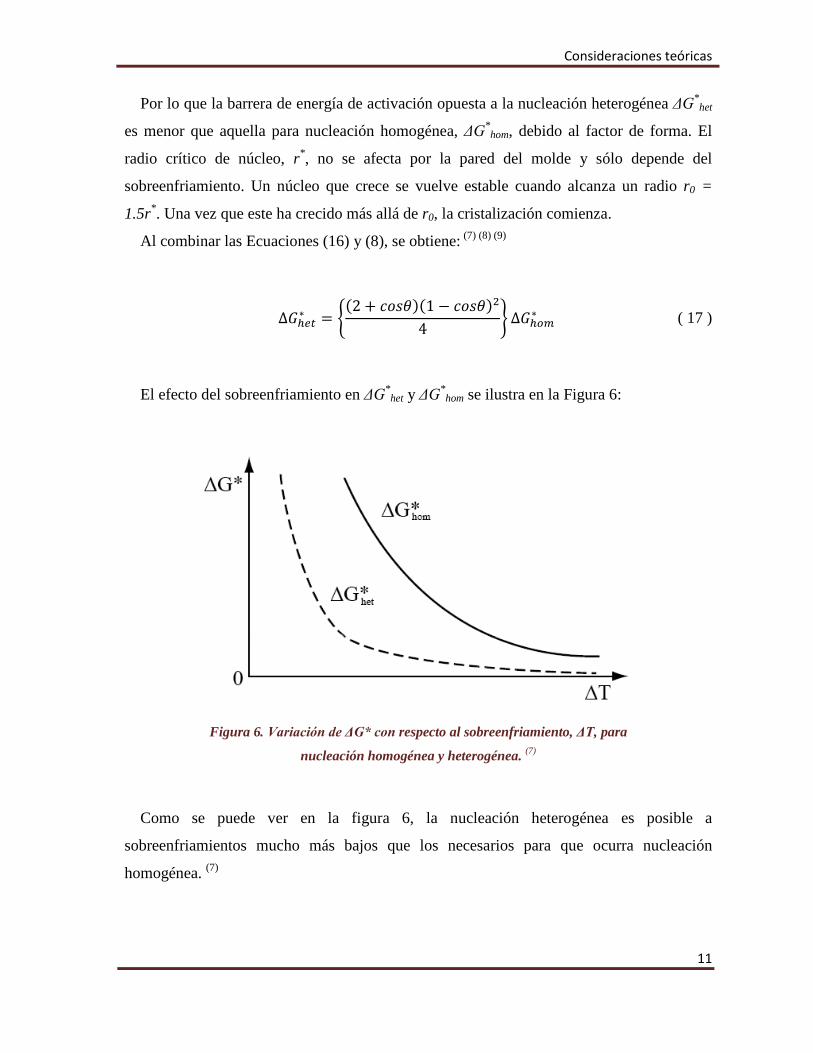
El efecto del sobreenfriamiento en ΔG*het y ΔG*
hom se ilustra en la Figura 6:
Figura 6. Variación de ΔG* con respecto al sobreenfriamiento, ΔT, para
nucleación homogénea y heterogénea. (7)
Como se puede ver en la figura 6, la nucleación heterogénea es posible a
sobreenfriamientos mucho más bajos que los necesarios para que ocurra nucleación
homogénea. (7)
11

Consideraciones teóricas
1.1.2 Crecimiento y microestructura dendrítica
La solidificación de metales es un proceso controlado por difusión. En metales puros, la
velocidad de solidificación y la distribución de temperaturas en la zona circundante al
núcleo están controladas por la velocidad a la cual el calor latente de solidificación puede
conducirse lejos de la intercara sólido/líquido. (7) (9)
La conducción se puede dar ya sea a través del sólido o del líquido, dependiendo de los
gradientes de temperatura en la intercara.
La figura 7 muestra un sólido con una intercara plana creciendo a una velocidad v hacia
un líquido sobrecalentado. El flujo de calor hacia el exterior de la intercara a través del
sólido debe balancear el flujo desde el líquido además del calor latente generado en la
intercara:
𝐾𝐾𝑆𝑆𝑇𝑇𝑆𝑆′ = 𝐾𝐾𝐿𝐿𝑇𝑇𝐿𝐿′ + 𝑣𝑣𝐿𝐿𝑣𝑣 ( 18 )
donde K es la conductividad térmica, T’ es el gradiente de temperatura, dT/dx, los
subíndices S y L significan sólido y líquido, v es la velocidad de crecimiento del sólido y Lv
es el calor latente de fusión por unidad de volumen.
Cuando un sólido crece hacia un líquido sobrecalentado, una intercara plana
sólido/líquido es estable, como se muestra en la figura 7:
Figura 7. (a) Distribución de temperatura para solidificación cuando el calor se extrae a través del
sólido. Isotermas (b) para una intercara plana S/L y (c) para una protuberancia. (7) 12

Consideraciones teóricas
Si como resultado de un incremento local en v, se forma una pequeña protuberancia en la
intercara, figura 7 (c), y la intercara de ésta se mantiene a Tm, el gradiente de temperatura en
el líquido frente al nódulo incrementará, mientras que el gradiente del sólido disminuye.
Como consecuencia, se conducirá más calor hacia la protuberancia y menos hacia el
exterior de ésta, por lo que la velocidad de crecimiento de la protuberancia será menor que
la velocidad en las zonas planas, y la protuberancia desaparecerá.
El resultado es distinto para un sólido creciendo hacia un líquido sobreenfriado debajo de
Tm, figura 8. Si se forma una protuberancia en el sólido, el gradiente de temperatura
negativo en el líquido se vuelve aún más negativo. Por ello, el calor se extrae con mayor
eficiencia desde la punta de la protuberancia que desde las regiones circundantes,
permitiendo que ésta crezca preferentemente. Una intercara sólido/líquido avanzando
dentro de un líquido sobreenfriado es inherentemente inestable.
Figura 8. Lo expuesto en la figura 7, con conducción de calor hacia el líquido. (7)
Esta situación se puede presentar al comienzo de la solidificación si ocurre nucleación en
las partículas de impurezas en el seno del líquido y se forma una protuberancia: ya que se
necesita un determinado sobreenfriamiento antes de que la nucleación pueda ocurrir en el
resto del líquido, las primeras partículas sólidas crecerán hacia el líquido sobreenfriado y el
calor latente de solidificación será conducido hacia el líquido. (7)
El líquido, al conducir el calor latente de las protuberancias, elevará su temperatura, con
lo que se formará una estructura de protuberancias y líquido entre ellas. Este último sólo
13

Consideraciones teóricas
podrá solidificarse por medio de la conducción de calor a través del sólido, como se
muestra en la figura 7, cuando las paredes del molde y las protuberancias sólidas tengan
menor temperatura que el líquido.
Si la nucleación ocurre en las paredes del molde y el líquido no está subenfriado, se
presentará una zona pequeña de estructuras de protuberancias cerca de la pared, donde el
líquido está sobreenfriado. (6) (7)
Cuando r alcanza el radio crítico necesario para que suceda la nucleación, r*, cualquier
inestabilidad morfológica provoca que la intercara esférica se vuelva inestable a
alteraciones espaciales continuas, favoreciendo el crecimiento de protuberancias en la
intercara. Debido a que el crecimiento de protuberancias se favorece si el calor latente se
conduce hacia el líquido, una partícula originalmente esférica desarrollará brazos en varias
direcciones como se muestra en la figura 9: (7) (10)
Figura 9. Desarrollo de dendritas térmicas: (a) núcleo esférico; (b) comienzo de inestabilidad en la
intercara; (c) desarrollo de brazos primarios en direcciones cristalográficas;
(d) desarrollo de brazos secundarios y terciarios. (7) 14

Consideraciones teóricas
El crecimiento tras la nucleación se favorece en la dirección de la extracción de calor,
perpendicular a la intercara sólido/líquido, formando una estructura de brazos primarios,
también llamada estructura celular. Posteriormente se da una transición de estructura
celular a estructura dendrítica, figura 10, acompañada de un cambio en la dirección de
crecimiento a las direcciones preferenciales de cada sistema cristalino, debido a que al
incrementarse la velocidad de solidificación, el requerimiento para el transporte atómico es
más alto y este es alcanzado con mayor facilidad en las direcciones preferenciales de cada
estructura, por ejemplo: < 100 > en metales cúbicos y < 11�00 > en metales hexagonales
compactos. (2) (7)
A la vez que los brazos primarios se alargan, su sección transversal cambia de forma
circular a la de una cruz. Posteriormente sus superficies se vuelven inestables y se
descomponen en brazos secundarios e incluso terciarios. Esta estructura se conoce como
dendrita, que se deriva de la palabra griega para árbol, δενδρων. (2) (3) (7)
Figura 10. Cambio secuencial de la morfología de la intercara a medida que la velocidad de
solidificación aumenta: (a) Célula creciendo en la dirección de la extracción de calor;
(b) Célula creciendo en la dirección <100> (c) Transición célula/dendrita
(d) Dendrita. (11)
15

Consideraciones teóricas
En la punta de una dendrita en crecimiento la situación es distinta de aquella presentada
para una intercara plana, ya que el calor puede disiparse de la punta de la dendrita en tres
dimensiones. Si se asume que el sólido es isotérmico, T’S = 0, y que el gradiente negativo
de temperatura T’L es aproximadamente igual a ΔTc / r, donde es la diferencia entre la
temperatura de la intercara, Ti, y la temperatura del líquido sobreenfriado lejano a la
dendrita, T∞, como se muestra en la figura 11, y se sustituyen ambas condiciones en la
Ecuación (18) se obtiene:
𝑣𝑣 =−𝐾𝐾𝐿𝐿𝑇𝑇𝐿𝐿′
𝐿𝐿𝑣𝑣≈𝐾𝐾𝐿𝐿𝐿𝐿𝑣𝑣
∙∆𝑇𝑇𝑟𝑟
( 19 )
Por lo que, para un determinado sobreenfriamiento, el crecimiento será rápido para
valores pequeños de r debido a la mejora de la conductividad térmica a medida que r
disminuye. (7)
Figura 11. Distribución de temperaturas en la punta de una dendrita térmica en crecimiento. (7)
16

Consideraciones teóricas
La microestructura dendrítica es posiblemente la microestructura más común, estando
presente en todas las piezas fundidas macroscópicas así como en las transformaciones de
estado sólido. De hecho, sólo se puede evitar si se usan limitantes de velocidad o
sobreenfriamiento.
Si un material que presenta una microestructura dendrítica gruesa es re-procesado, se
agrietará o presentará defectos. Por ello es deseable obtener una microestructura lo
suficientemente fina para permitir re-procesamiento sin defectos. En cualquier caso, habrá
remanentes de la estructura dendrítica en las piezas re-procesadas. (10) (12)
1.1.2.1 Anisotropía en crecimiento dendrítico
La anisotropía de la cinética de crecimiento de cristales se origina en la tensión superficial,
en el transporte de calor y en los procesos microscópicos en la intercara, y puede causar que
la morfología de un cristal en crecimiento cambie.
Diversos experimentos y cálculos en sistemas con elementos puros y propiedades
superficiales isotrópicas muestran que en una orientación correspondiente a la punta de una
perturbación en la forma inicial del cristal, un punto de máxima curvatura en la forma
inicial, la curvatura de la intercara puede disminuir e incluso volverse negativa a medida
que la intercara crece, como se muestra en la figura 12:
Figura 12. Patrón de evolución espacial para una secuencia de separación de puntas
obtenido en las simulaciones de Kessler, Koplik y Levine. (13) 17

Consideraciones teóricas
Este fenómeno se conoce como separación de puntas, y se presenta a valores mínimos de
anisotropía. Se requiere un valor crítico de anisotropía cristalina para que las puntas de los
cristales dendríticos sean estables y no se presente separación de las mismas.
La figura 12 muestra una de las simulaciones de crecimiento dendrítico obtenidas por
Kessler, Koplik y Levine con un modelo geométrico, considerando condiciones isotrópicas.
Cada contorno representa una etapa de tiempo, y se puede observar que a medida que el
tiempo transcurre, las puntas de la dendrita tienden a hacerse cóncavas y a formar dos
puntas redondeadas, es decir, a separarse.
Al aumentar el valor de la anisotropía, aumenta el número de brazos emitidos antes de
que ocurra la separación de puntas. Si el valor de anisotropía es igual al valor crítico, la
formación de brazos se estabiliza y el sistema es capaz de formar brazos secundarios. Así,
el valor crítico de tensión superficial anisotrópica puede evitar que la punta del cristal
dendrítico se vuelva negativa o cóncava en la orientación donde se presenta la separación
de puntas. (14) (13)
La figura 13 muestra una de las simulaciones obtenidas por Brush y Sekerka (14) con un
modelo de crecimiento de cristales, considerando tensión superficial isotrópica. Es evidente
la similitud entre esta figura y la presentada por Kessler, Koplik y Levine. (13)
Figura 13. Crecimiento de un cristal en el cual se observa separación de puntas
obtenido en las simulaciones de Brush y Sekerka. (14)
18

Consideraciones teóricas
El fenómeno de separación de puntas se demostró experimentalmente por Ben-Jacob y
colaboradores (15) mediante un experimento en el cual se observa la formación de patrones
en la intercara entre dos líquidos inmiscibles, y donde el efecto de la anisotropía se incluye
en el experimento al llevarse a cabo sobre una superficie ranurada que forma una retícula.
La figura 14 muestra el patrón de separación de puntas obtenido por Ben-Jacob y
colaboradores (15) con bajos valores de anisotropía.
Figura 14. Patrón de separación de puntas formado en la experimentación
de Ben-Jacob y colaboradores. (15)
1.2 Método de Campo de Fases La solidificación de un metal puro se modela tradicionalmente usando una serie de
ecuaciones que describen el avance del frente de solidificación, que se limita por la difusión
de calor latente al exterior de la intercara sólido/líquido y por la habilidad de la intercara de
mantener dos condiciones límite:
• El flujo de calor hacia un lado de la intercara se balancea con un flujo de calor
equivalente lejos del lado contrario.
• La temperatura en la intercara experimenta una corrección debida a la curvatura,
conocida como condición de Gibbs-Thomson.
19

Consideraciones teóricas
Estas condiciones se expresan matemáticamente por el modelo de intercara definida
conocido como Problema de Stefan:
𝜕𝜕𝑇𝑇𝜕𝜕𝜕𝜕
= ∇ ∙ �𝐾𝐾𝜌𝜌𝑐𝑐𝜌𝜌
∇𝑇𝑇� ≡ ∇ ∙ (𝐷𝐷∇𝑇𝑇) ( 20 )
𝜌𝜌𝐿𝐿𝑓𝑓𝑉𝑉𝑛𝑛 = 𝐾𝐾𝑆𝑆∇𝑇𝑇 ∙ 𝑛𝑛�⃑ |𝑖𝑖𝑛𝑛𝑒𝑒𝑆𝑆 − 𝐾𝐾𝐿𝐿∇𝑇𝑇 ∙ 𝑛𝑛�⃑ |𝑖𝑖𝑛𝑛𝑒𝑒𝐿𝐿 ( 21 )
𝑇𝑇𝑖𝑖𝑛𝑛𝑒𝑒 = 𝑇𝑇𝑚𝑚 − �𝛶𝛶𝑇𝑇𝑀𝑀𝜌𝜌𝐿𝐿𝑓𝑓
� 𝜅𝜅𝑆𝑆𝑆𝑆 −𝑉𝑉𝑛𝑛𝜇𝜇
( 22 )
donde T ≡ T(x,t) denota la temperatura, K la conductividad térmica, con subíndices S y L
para el sólido y el líquido, respectivamente, ρ la densidad del sólido y del líquido, cp el
calor específico a presión constante, D el coeficiente de difusión térmica, Lf el calor latente
de fusión por masa de solidificación, ϒ la energía de la superficie sólido/líquido, Tm la
temperatura de fusión, κSL la curvatura local de la intercara sólido/líquido, Vn la velocidad
normal local de la intercara y μ la movilidad atómica local de la intercara. El subíndice int
se refiere a la intercara y los superíndices S y L se refieren a la evaluación en la intercara en
el lado del sólido y del líquido, respectivamente. (12)
El Problema de Stefan es un problema de frontera libre, en el cual se busca una solución
u a una ecuación diferencial parcial en un dominio, con una parte desconocida Γ de su
frontera. Así, se deben determinar u y Γ. Adicionalmente a las condiciones frontera de un
dominio desconocido, se debe indicar una condición en la frontera libre. Si se considera la
fusión de hielo, la temperatura satisface la ecuación de calor en la región de líquido, y sin
embargo esta región, o la intercara sólido/líquido, se desconoce y debe determinarse a la
vez que la temperatura dentro de la región del líquido. (16)
La manera clásica de resolver un problema de frontera libre es por medio de un modelo
con una intercara de espesor igual a cero, es decir, una intercara definida. En él, una
discontinuidad clara en propiedades o una variación en los flujos y funciones
termodinámicas ocurre a través de la intercara. Las propiedades interfaciales de la
transformación de solidificación (como de otras transformaciones limitadas por difusión)
20

Consideraciones teóricas
pueden describirse por ecuaciones de intercara definida análogas al Problema de Stefan, sin
embargo, existen varias limitantes al modelar problemas de frontera libre mediante modelos
de intercara definida:
• El modelo de intercara definida apropiado no se conoce para muchos fenómenos.
• La simulación numérica es de extrema dificultad debido a las interacciones
complejas entre intercaras complicadas, que se someten a unión y constricción
durante una transformación de fase.
• Los códigos numéricos son largos y complejos, especialmente en tres dimensiones.
Como alternativa, surgió el método de campo de fases, una técnica relativamente nueva
de modelado de evolución microestructural caracterizada por considerar un espesor de
intercara finito, cuyo uso ha ido en incremento debido a sus orígenes fundamentales y a que
evita algunos de los problemas asociados con los modelos de intercara definida. (4) (8)
El método de campo de fases incluye además del campo de temperaturas usual, un
campo continuo denominado campo de fases o parámetro de orden. Este campo asume
valores constantes en el interior de cada fase, interpolando continuamente entre sus valores
en cada fase a través de una capa límite delgada, usada para describir la intercara entre
fases; es decir, permite bosquejar la intercara en una región difusa. Por ello, a los modelos
usados en el método de campo de fases se les conoce también como modelos de intercara
difusa. (4) (12)
Un parámetro de orden es una cantidad que parametriza el cambio de simetría de una
fase madre, desordenada en el caso del líquido a solidificar, a una fase hija, ordenada en el
caso del sólido formado, que aparece tras la transformación. Por ello, el campo de fases
puede verse como un parámetro que describe el grado de cristalinidad u orden o desorden
atómico dentro de una fase. Puede ser visto también como un parámetro que proporciona
una descripción fundamental de una intercara atómica difusa. (12)
1.3 Formulación de Kobayashi La formulación propuesta por Kobayashi (4) (5) (17) consiste en un modelo de campo de fases
para simular la formación de estructuras dendríticas en metales puros, formadas según el
balance de la tensión superficial y la fuerza motriz termodinámica, sobreenfriamiento,
21

Consideraciones teóricas
tomando en cuenta la anisotropía de energía interfacial, que afecta drásticamente la forma
de los cristales. (5) (17)
Al simular la solidificación de metales puros, se considera que la velocidad de
crecimiento de los cristales está limitada sólo por el proceso de extracción de calor de la
intercara, que mantiene la temperatura interfacial debajo de la temperatura de equilibrio.
El modelo incluye dos variables: un campo de fases ϕ(x,t) y un campo de temperaturas
T(x,t). La variable de campo de fases ϕ(x,t) es un parámetro de orden. En la posición x y el
tiempo t, ϕ = 0 indica la existencia de fase líquida y ϕ = 1 indica la existencia de fase
sólida. La intercara sólido/líquido se expresa por una transición suave de los valores entre 0
y 1. (4) (5)
Se considera que la energía libre es del tipo Ginzburg-Landau:
𝐺𝐺(𝜙𝜙,𝑚𝑚) = �12𝜀𝜀2|∇𝜙𝜙|2 + 𝐹𝐹(𝜙𝜙,𝑚𝑚)𝑑𝑑𝒙𝒙 ( 23 )
donde ε es un parámetro que determina el espesor de la capa y la movilidad de la intercara.
La anisotropía se incluye en el modelo al asumir que ε depende de la dirección del vector
normal externo en la intercara (representado por -∇ϕ). Así, la Ecuación (23) se modifica:
𝐺𝐺(𝜙𝜙,𝑚𝑚) = � 12𝜀𝜀(−∇𝜙𝜙)2|∇𝜙𝜙|2 + 𝐹𝐹(𝜙𝜙,𝑚𝑚)𝑑𝑑𝒙𝒙 ( 24 )
El valor de ε se puede determinar mediante la relación:
𝜀𝜀 = 𝜀𝜀�̅�𝜎(𝑐𝑐) ( 25 )
donde 𝜀𝜀 ̅representa el valor promedio de ε y σ(θ) la anisotropía, que a su vez se expresa por:
𝜎𝜎(𝑐𝑐) = 1 + 𝛿𝛿𝑐𝑐𝑐𝑐𝑐𝑐𝛿𝛿(𝑐𝑐 − 𝑐𝑐𝑜𝑜) ( 26 )
22

Consideraciones teóricas
donde el parámetro δ se refiere a la magnitud de anisotropía, j al número de simetría de
anisotropía y θ es un ángulo entre el vector normal externo en la intercara y una dirección
determinada. El valor de (θ – θ0) se asigna a cada valor de ∇ϕ.
Se considera que la energía libre F(ϕ,m) reproduce una función potencial de doble pozo,
donde un pozo representa la fase líquida y otro la fase sólida, que se expresa
matemáticamente como:
𝐹𝐹(𝜙𝜙,𝑚𝑚) =14𝜙𝜙4 − �
12−
13𝑚𝑚�𝜙𝜙3 + �
14−
12𝑚𝑚�𝜙𝜙2 ( 27 )
donde m representa la fuerza motriz para la solidificación y su valor absoluto es menor a
0.5. (5)
La ecuación diferencial parcial de Allen-Cahn (18) a resolver es:
𝜏𝜏𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕
= −𝜕𝜕𝐺𝐺𝜕𝜕𝜙𝜙
( 28 )
donde τ es una constante positiva que fija la escala temporal. Al sustituir la Ecuación (27)
en la Ecuación (24) y el resultado en la Ecuación (28), se obtiene la siguiente ecuación
diferencial, que representa la evolución de la intercara y es una de las ecuaciones a resolver
en el modelo:
𝜏𝜏𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕
= −𝜕𝜕𝜕𝜕𝜕𝜕
�𝜀𝜀𝜀𝜀′𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕� +
𝜕𝜕𝜕𝜕𝜕𝜕
�𝜀𝜀𝜀𝜀′𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕� + ∇ ∙ (𝜀𝜀2∇𝜙𝜙)
+ 𝜙𝜙(1 − 𝜙𝜙) �𝜙𝜙 −12
+ 𝑚𝑚� ( 29 )
donde ε’ es la primera derivada de ε con respecto a θ. La fuerza motriz m se define como:
23

Consideraciones teóricas
𝑚𝑚(𝑇𝑇) = �𝛼𝛼𝜋𝜋� atan{𝛾𝛾(𝑇𝑇𝑒𝑒 − 𝑇𝑇)} ( 30 )
donde α es una constante positiva menor a 1, requisito para que se cumpla la condición
previamente mencionada de |m| < 0.5 en todos los valores de T, γ es un parámetro
adimensional y Te y T son las temperaturas de equilibrio y enfriamiento, respectivamente.
La ecuación para la temperatura adimensional T como una función del tiempo
adimensional t, que es la segunda ecuación a resolver en el modelo, se deriva de la ley de la
conservación de la energía y se expresa como:
𝜕𝜕𝑇𝑇𝜕𝜕𝜕𝜕
= ∇2𝑇𝑇 + 𝜅𝜅𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕
( 31 )
donde κ es el calor latente adimensional. La temperatura T varía entre 0 y 1, que
corresponden a las temperaturas de enfriamiento y equilibrio, respectivamente. Las
soluciones a las Ecuaciones (29) y (31) producen resultados equivalentes. (4) (5)
1.4 Método de Diferencias Finitas El primer paso de toda simulación es proponer el problema física, química o
termodinámicamente en la forma de un formalismo matemático, que incluye
frecuentemente una serie de ecuaciones diferenciales parciales.
En la mayoría de los casos el problema es no lineal y no existe una solución analítica, por
lo que la resolución de dichas ecuaciones es complicada. Por ello, se recurre a un método
numérico para obtener la solución.
Entre los métodos numéricos desarrollados hasta ahora, el Método de Diferencias Finitas
es el más simple y de más fácil implementación en un lenguaje de programación. (19)
El principio que emplea el Método de Diferencias Finitas para resolver ecuaciones es la
sustitución de las derivadas en las ecuaciones diferenciales parciales, ya sean derivadas
temporales o espaciales, por una expresión de diferencia apropiada, encargada de
aproximarlas. (20)
Dicha expresión es calculada mediante diferencias entre los valores de una función en
puntos determinados, llamados nodos, de una malla. 24

Consideraciones teóricas
Si se considera una serie de nodos en una malla regular, el espaciado puede ser
expresado por:
ℎ =𝐿𝐿
𝑁𝑁 + 1 ( 32 )
donde (N + 1) es el número de intervalos en que está dividida la longitud L. En ella se
pretende calcular los valores de una función, c(xk), en los N puntos internos del dominio. La
función c(x) puede ser expandida en una serie de Taylor alrededor de los puntos xk de la
malla:
𝑐𝑐(𝜕𝜕) = 𝑐𝑐(𝜕𝜕𝑘𝑘) + �𝑑𝑑𝑐𝑐𝑑𝑑𝜕𝜕�𝑥𝑥𝑘𝑘
(𝜕𝜕 − 𝜕𝜕𝑘𝑘) + �𝑑𝑑2𝑐𝑐𝑑𝑑𝜕𝜕2
�𝑥𝑥𝑘𝑘
(𝜕𝜕 − 𝜕𝜕𝑘𝑘)2
2!
+ �𝑑𝑑3𝑐𝑐𝑑𝑑𝜕𝜕3
�𝑥𝑥𝑘𝑘
(𝜕𝜕 − 𝜕𝜕𝑘𝑘)3
3!+ ⋯
( 33 )
Si se adopta la siguiente nomenclatura:
𝑐𝑐𝑘𝑘 = 𝑐𝑐(𝜕𝜕𝑘𝑘) ( 34 )
𝑐𝑐𝑘𝑘±1 = 𝑐𝑐(𝜕𝜕𝑘𝑘±1) ( 35 )
𝑐𝑐𝑘𝑘′ = �𝑑𝑑𝑐𝑐𝑑𝑑𝜕𝜕�𝑥𝑥𝑘𝑘
( 36 )
𝑐𝑐𝑘𝑘′′ = �𝑑𝑑2𝑐𝑐𝑑𝑑𝜕𝜕2
�𝑥𝑥𝑘𝑘
( 37 )
𝑐𝑐𝑘𝑘′′′ = �𝑑𝑑3𝑐𝑐𝑑𝑑𝜕𝜕3
�𝑥𝑥𝑘𝑘
( 38 )
los valores de c(x) en los nodos a la derecha e izquierda del nodo k están dados por: 25

Consideraciones teóricas
𝑐𝑐𝑘𝑘+1 = 𝑐𝑐𝑘𝑘 + 𝑐𝑐𝑘𝑘′ ℎ + 𝑐𝑐𝑘𝑘′′ℎ2
2+ 𝑐𝑐𝑘𝑘′′′
ℎ3
6+ 𝑂𝑂(ℎ4) ( 39 )
𝑐𝑐𝑘𝑘−1 = 𝑐𝑐𝑘𝑘 − 𝑐𝑐𝑘𝑘′ ℎ + 𝑐𝑐𝑘𝑘′′ℎ2
2− 𝑐𝑐𝑘𝑘′′′
ℎ3
6+ 𝑂𝑂(ℎ4) ( 40 )
Al combinar linealmente estas dos ecuaciones, se obtienen las aproximaciones de la
primera y segunda derivada para todos los puntos k de la malla:
𝑐𝑐𝑘𝑘+′ =𝑐𝑐𝑘𝑘+1 − 𝑐𝑐𝑘𝑘
ℎ+ 𝑂𝑂(ℎ) ( 41 )
𝑐𝑐𝑘𝑘−′ =𝑐𝑐𝑘𝑘 − 𝑐𝑐𝑘𝑘−1
ℎ+ 𝑂𝑂(ℎ) ( 42 )
𝑐𝑐𝑘𝑘′ =𝑐𝑐𝑘𝑘+1 − 𝑐𝑐𝑘𝑘−1
2ℎ+ 𝑂𝑂(ℎ2) ( 43 )
𝑐𝑐𝑘𝑘′′ =𝑐𝑐𝑘𝑘+1 − 2𝑐𝑐𝑘𝑘 + 𝑐𝑐𝑘𝑘−1
ℎ2+ 𝑂𝑂(ℎ2) ( 44 )
Los subíndices + y – se usan para distinguir la derivada central de la derivada de la
derecha y de la derivada de la izquierda, respectivamente.
Las Ecuaciones (41) y (42) tienen precisión de orden h, es decir, al sustituir las derivadas
por alguna de las expresiones de diferencia, el error cometido al truncar la serie de Taylor
tiende a cero de forma proporcional a h cuando el número de nodos tiende al infinito. Así,
si el espaciamiento entre nodos h se divide entre dos, el error de truncamiento también se
divide entre dos, y se gana precisión en los cálculos.
En el caso de las Ecuaciones (43) y (44), que tienen precisión de orden h2, al dividir el
espaciamiento de nodos entre dos, el error de truncamiento se divide entre cuatro.
La primera derivada de una función con respecto al tiempo puede aproximarse al
modificar la Ecuación (41):
26

Consideraciones teóricas
𝑐𝑐𝑒𝑒+′ =𝑐𝑐𝑘𝑘𝑒𝑒+1 − 𝑐𝑐𝑘𝑘𝑒𝑒
∆𝜕𝜕+ 𝑂𝑂(∆𝜕𝜕) ( 45 )
No es posible aproximarla con la Ecuación (42), debido a que la condición previa a la
condición inicial de la función es desconocida.
La Ecuación (45), con precisión de orden Δt, permite calcular valores en las etapas de
tiempo t + 1, y es llamada una aproximación hacia adelante.
Si como ejemplo se pretende encontrar una solución numérica a una ecuación general de
flujo conservativo:
𝜕𝜕𝜕𝜕𝜕𝜕𝜕𝜕
= −𝑉𝑉𝜕𝜕𝜕𝜕𝜕𝜕𝜕𝜕
( 46 )
donde V es una constante, usando las aproximaciones correspondientes a las Ecuaciones
(43) y (45), la Ecuación (46) puede expresarse como:
𝑐𝑐𝑘𝑘𝑒𝑒+1 − 𝑐𝑐𝑘𝑘𝑒𝑒
∆𝜕𝜕= −𝑉𝑉 �
𝑐𝑐𝑘𝑘+1𝑒𝑒 − 𝑐𝑐𝑘𝑘−1𝑒𝑒
2ℎ� ( 47 )
que puede ser reescrita como:
𝑐𝑐𝑘𝑘𝑒𝑒+1 = 𝑐𝑐𝑘𝑘𝑒𝑒 −𝑉𝑉∆𝜕𝜕2ℎ
(𝑐𝑐𝑘𝑘+1𝑒𝑒 − 𝑐𝑐𝑘𝑘−1𝑒𝑒 ) ( 48 )
Si se pretende resolver numéricamente una ecuación de difusión:
𝜕𝜕𝜕𝜕𝜕𝜕𝜕𝜕
= −𝐷𝐷𝜕𝜕2𝜕𝜕𝜕𝜕𝜕𝜕2
( 49 )
la aproximación puede expresarse como:
27
Consideraciones teóricas
𝑐𝑐𝑘𝑘𝑒𝑒+1 = 𝑐𝑐𝑘𝑘𝑒𝑒 −𝐷𝐷∆𝜕𝜕ℎ2
(𝑐𝑐𝑘𝑘+1𝑒𝑒 − 2𝑐𝑐𝑘𝑘𝑒𝑒 + 𝑐𝑐𝑘𝑘−1𝑒𝑒 ) ( 50 )
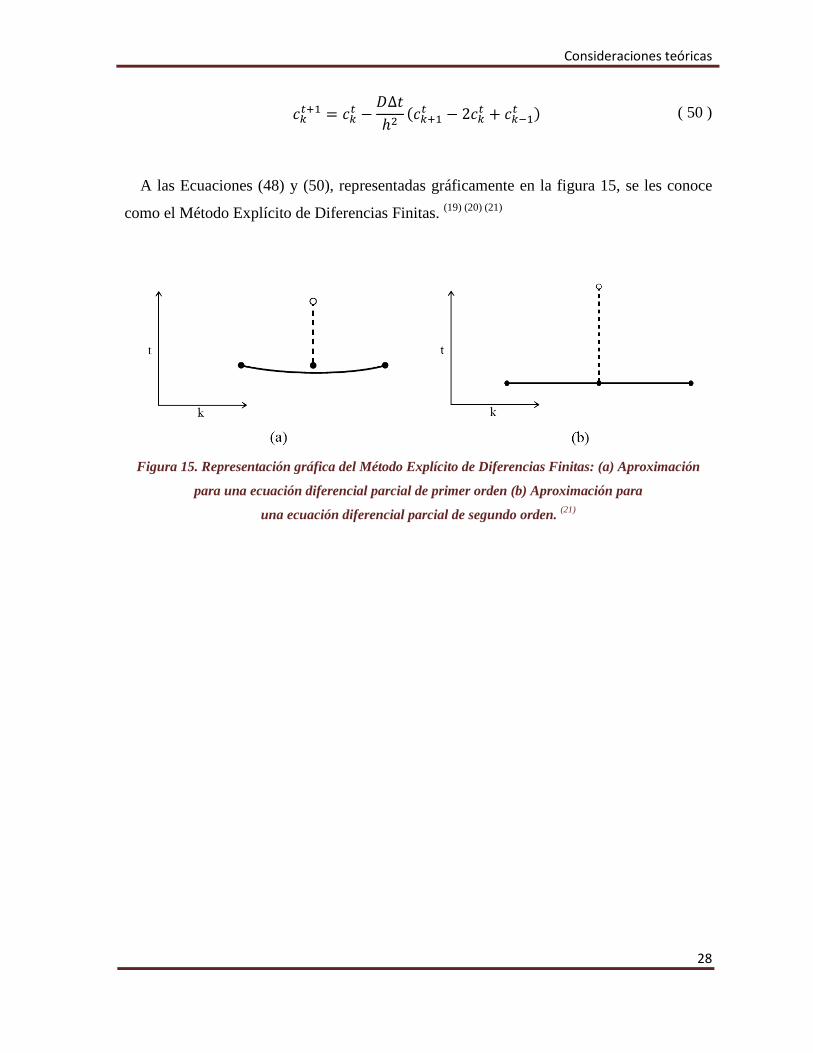
A las Ecuaciones (48) y (50), representadas gráficamente en la figura 15, se les conoce
como el Método Explícito de Diferencias Finitas. (19) (20) (21)
Figura 15. Representación gráfica del Método Explícito de Diferencias Finitas: (a) Aproximación
para una ecuación diferencial parcial de primer orden (b) Aproximación para
una ecuación diferencial parcial de segundo orden. (21)
28

Metodología numérica
2. Metodología numérica
Para simular la evolución de la microestructura dendrítica, se usó un código en
FORTRAN llamado “Dendrite.f95”, en el cual, las Ecuaciones (29) y (31) se resolvieron
simultáneamente en dos dimensiones usando el Método Explícito de Diferencias Finitas
utilizando una malla de 301 x 301 nodos.
Las etapas de distancia Δx y de tiempo Δt elegidas fueron 0.03 unidades de distancia y
2x10-4 unidades de tiempo, respectivamente. Los valores seleccionados para los parámetros
restantes en las distintas simulaciones se resumen en la tabla 1.
Tabla 1. Parámetros de la formulación de Kobayashi usados durante las simulaciones
Parámetro Valor
𝜺𝜺� 0.01
δ 0.00, 0.02, 0.05, 0.08
j 0.0, 4.0, 6.0
m 1.0
α 0.9
γ 10.0
τ 0.0003
κ 1.0, 1.5, 1.8, 2.2
Los archivos de datos generados por el programa Dendrite.f95 posteriormente se
graficaron y se usaron para crear secuencias de video.
En cada nueva simulación se modificaron las condiciones iniciales de magnitud de
anisotropía, δ, número de simetría de anisotropía, j, y calor latente adimensional, κ, a fin de
simular las condiciones indicadas en la tabla 1.
Se modificaron las condiciones de un parámetro a la vez, δ constante y κ variable o lo
opuesto, usando condiciones de δ = 0.02 y κ = 1.8 cuando éstos son el parámetro constante;
y se simularon dichas condiciones para dos estructuras cristalinas, cúbica y hexagonal, para
lo cual se usó un valor de j = 4.0 y j = 6.0, respectivamente. 29
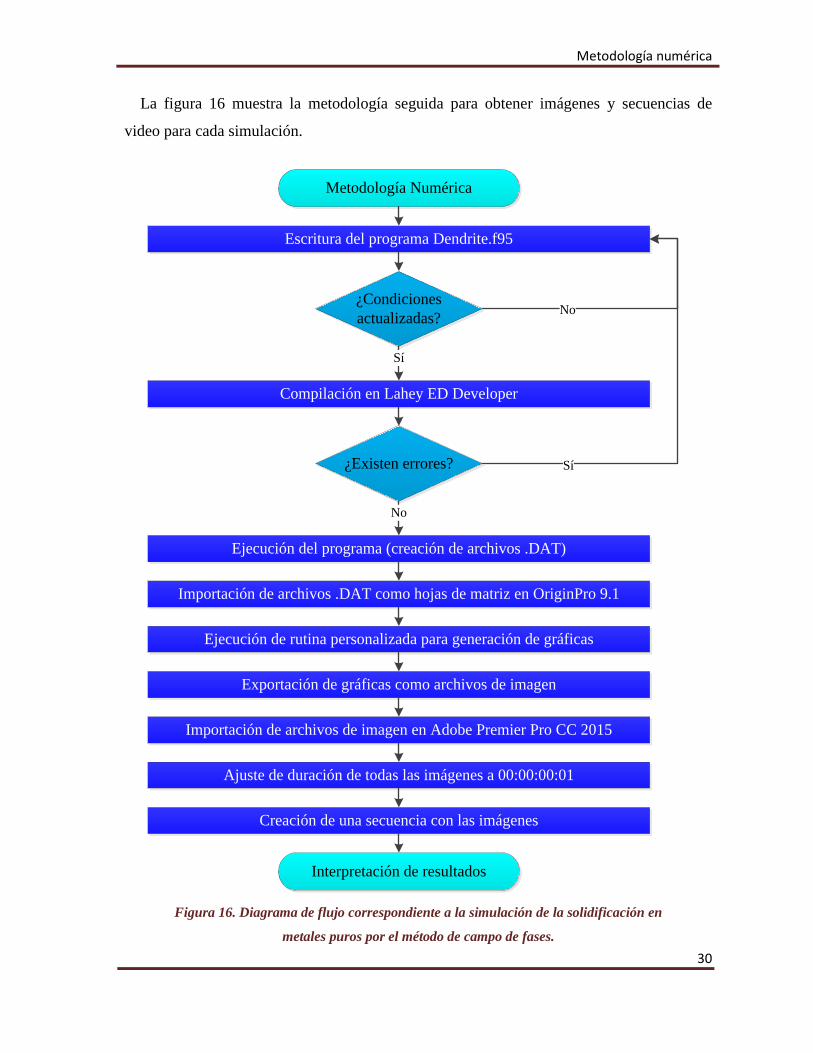
Metodología numérica
La figura 16 muestra la metodología seguida para obtener imágenes y secuencias de
video para cada simulación.
Metodología Numérica
Escritura del programa Dendrite.f95
Compilación en Lahey ED Developer
Ejecución del programa (creación de archivos .DAT)
¿Condiciones actualizadas?
Sí
No
Importación de archivos .DAT como hojas de matriz en OriginPro 9.1
¿Existen errores?
No
Ejecución de rutina personalizada para generación de gráficas
Exportación de gráficas como archivos de imagen
Importación de archivos de imagen en Adobe Premier Pro CC 2015
Ajuste de duración de todas las imágenes a 00:00:00:01
Creación de una secuencia con las imágenes
Interpretación de resultados
Sí
Figura 16. Diagrama de flujo correspondiente a la simulación de la solidificación en
metales puros por el método de campo de fases. 30

Metodología numérica
Para crear el código del programa Dendrite.f95 se usó FORTRAN 95 como lenguaje de
programación y el software Lahey ED Developer versión 3.80 como compilador. El código
se incluye como apéndice en este trabajo.
El programa consiste de los siguientes elementos:
• Un módulo “mdata” para almacenar la información sobre las variables a usar, a fin
de poder ser usada en las distintas subrutinas del programa.
• Una subrutina “initial” donde se indican las condiciones iniciales de la simulación:
valores de variables y posición de núcleos.
• Una subrutina “gradandlaplace” cuya función es calcular las variables y operadores
necesarios para la resolución de las ecuaciones.
• Una subrutina “evolution” usada para la resolución de las ecuaciones.
• Una subrutina “printdata” encargada de la generación de archivos de datos (con
formato .DAT) donde se almacena la información de la evolución cada vez que se
cumplen 20 ciclos del programa.
Tras cada ejecución del programa se obtuvieron 500 archivos de datos para cada campo,
ϕ y T, debido a que el programa ejecuta 10000 ciclos y se crea un archivo de datos
únicamente de los ciclos múltiplos de 20.
A fin de poder analizar la evolución microestructural, los archivos de datos generados
por el programa deben transformarse en gráficas y posteriormente en secuencias de video.
Para estos fines se usaron los softwares OriginPro 9.1 y Adobe Premiere Pro CC 2015,
respectivamente.
Para graficar la información guardada en los archivos de datos se empleó la siguiente
serie de pasos en OriginPro 9.1:
i. Abrir un nuevo libro de matrices dando clic el ícono “New Matrix” en la barra de
herramientas.
ii. Abrir el menú de importación de archivos ASCII dando clic en el ícono “Import
Multiple ASCII” en la barra de herramientas. Una vez abierto este menú,
seleccionar los archivos a importar y dar clic en “Add Files” y posteriormente en
“OK”.
iii. En el cuadro de diálogo que se abre al ejecutar la orden anterior, seleccionar la
opción “Start New Sheets” en el menú “Import Mode” y dar clic en “OK”, lo que 31

Metodología numérica
comenzará la traducción de los archivos ASCII a matrices, almacenando los datos
en hojas consecutivas de un mismo libro.
iv. Iniciar la rutina personalizada, que se encargará de graficar cada hoja de matriz en
el proyecto, dando clic en el ícono “Custom Routine” en la barra de herramientas.
La rutina programada consiste en el siguiente código, escrito en el lenguaje
LabTalk, desarrollado para crear rutinas en Origin:
doc -e LB {plotm im:=<active> plot:=PlotTypeID ogl:=<new
template:=TemplateName>;}
en ella, debe reemplazarse el texto “PlotTypeID” por un número de tres cifras
característico de cada tipo de gráfica en Origin, “226” para las gráficas usadas en
este trabajo, y el texto “Template Name” por el nombre de una plantilla creada
previamente, que contiene el formato de las gráficas a generar, “Dendrite” en este
trabajo.
Este código debe haber sido designado previamente como la rutina personalizada,
seleccionando la opción “Script Window” en el menú “Window”, y seleccionando
la opción “Open” en el menú “File(Text)” para posteriormente seleccionar el
archivo Custom.ogs y reemplazar el contenido de este por el código de la rutina
creada.
v. Seleccionar la opción “Export Graphs” en el menú “File”. En el cuadro de diálogo
que se abre, seleccionar el formato de salida y la resolución de la imagen: en este
trabajo se eligieron imágenes .png con 300 DPI. Al dar clic en “OK”, se exportan
los archivos de imagen.
32
Resultados
3. Resultados
Los resultados que se presentan a continuación representan el crecimiento simulado de
cristales individuales, cuya nucleación fue heterogénea sobre la superficie de una partícula
de impureza localizada en el centro de la malla. Se considera que el metal solidifica a partir
de líquido estático, es decir, en ausencia de convección.
En las simulaciones se presenta el efecto del cambio en tres variables: magnitud de
anisotropía cristalina, δ, que representa la simulación de metales solidificándose bajo
distintas condiciones de tensión superficial, transporte de calor y de interacción entre su
intercara y el entorno; calor latente adimensional, κ, que representa la simulación de
metales con distintos calores latentes de fusión; y número de simetría de anisotropía, j, que
representa la simulación de metales con diferentes simetrías de crecimiento, es decir,
distintas estructuras cristalinas.
3.1 Simulaciones de simetría de anisotropía perfecta En esta sección se incluyen las simulaciones que corresponden a los casos ideales de
cristales que se solidifican con simetría de anisotropía perfecta, es decir, sin direcciones
preferenciales de crecimiento características de alguna estructura cristalina. Las
condiciones usadas en las simulaciones correspondientes a esta sección se resumen en la
tabla 2.
Tabla 2. Condiciones usadas para las simulaciones de simetría de anisotropía perfecta
Número de Figura
Magnitud de
Anisotropía
Cristalina ( δ )
Calor Latente
Adimensional ( κ )
Número de
Simetría de
Anisotropía ( j )
17 0.000 1.0 0.0
17 0.020 1.0 0.0
18 0.000 1.8 0.0
18 0.020 1.8 0.0
La figura 17 muestra las simulaciones donde el calor latente adimensional, κ, es unitario. 33

Resultados
Las imágenes (a), (b) y (c) de la figura 17 muestran la evolución de un cristal poligonal
simulado bajo condiciones de simetría de anisotropía perfecta e isotropía cristalina, que
presenta geometría casi circular.
Las imágenes (d), (e) y (f) de la figura 17 muestran la evolución de un cristal poligonal
simulado bajo condiciones de simetría de anisotropía perfecta y baja anisotropía cristalina,
δ = 0.020. A pesar del valor de anisotropía distinto a cero con el que se simuló, no se
aprecian cambios en la geometría del cristal.
Es evidente que ambos cristales presentan altas velocidades de crecimiento, ya que a un
tiempo de 0.32, casi todo el líquido disponible ha solidificado.
Figura 17. Evolución de la solidificación de cristales bajo condiciones de j = 0.0 y κ = 1.0.
(a) t = 0.08; (b) t = 0.2; (c) t = 0.32; con δ = 0.000. (d) t = 0.08; (e) t = 0.2; (f) t = 0.32; con δ = 0.020.
La figura 18 muestra las simulaciones donde el calor latente adimensional tiene un valor
alto, κ = 1.8. 34

Resultados
Las imágenes (a), (b) y (c) de la figura 18 muestran la evolución de un cristal simulado
bajo condiciones de simetría de anisotropía perfecta e isotropía cristalina, que presenta
separación de puntas.
Las imágenes (d), (e) y (f) de la figura 18 muestran la evolución de un cristal simulado
bajo condiciones de simetría de anisotropía perfecta y baja anisotropía cristalina, δ = 0.020.
Este cristal presenta separación de puntas y una velocidad de crecimiento mayor que la del
cristal correspondiente a las imágenes (a), (b) y (c).
Ambos cristales presentan bajas velocidades de crecimiento, ya que a un tiempo de 0.6,
aún falta por solidificar una importante porción de líquido.
Figura 18. Evolución de la solidificación de cristales bajo condiciones de j = 0.0 y κ = 1.8.
(a) t = 0.2; (b) t = 0.4; (c) t = 0.6; con δ = 0.000. (d) t = 0.2; (e) t = 0.4; (f) t = 0.6; con δ = 0.020.
35
Resultados
3.2 Simulaciones de magnitud de anisotropía En esta sección se presentan las evoluciones de cristales con distintos valores de anisotropía
cristalina. En todas las simulaciones presentadas en esta sección se usó un valor fijo de κ =
1.8. Los valores de anisotropía cristalina y número de simetría de anisotropía usados en esta
sección se resumen en la tabla 3.
Tabla 3. Condiciones usadas para las simulaciones de anisotropía
Número de Figura Magnitud de Anisotropía
Cristalina ( δ )
Número de Simetría de
Anisotropía ( j )
19 0.000 4.0
19 0.000 6.0
20 0.005 4.0
20 0.010 4.0
20 0.020 4.0
20 0.050 4.0
20 0.080 4.0
20 0.120 4.0
21 0.005 6.0
21 0.010 6.0
21 0.020 6.0
21 0.050 6.0
21 0.080 6.0
La figura 19 muestra las simulaciones en condiciones de isotropía cristalina, δ = 0.000,
para la estructura cúbica, imágenes (a), (b) y (c); y la estructura hexagonal, imágenes (d),
(e) y (f).
Las imágenes (a), (b) y (c) de la figura 19 muestran la evolución de un cristal de
estructura cúbica simulado bajo condiciones de isotropía cristalina, δ = 0.000, que aunque
inicialmente presenta una forma poligonal, con el tiempo desarrolla separación de puntas.
36

Resultados
Las imágenes (d), (e) y (f) de la figura 19 muestran la evolución de un cristal de
estructura hexagonal simulado bajo condiciones de isotropía cristalina, δ = 0.000. No se
aprecian rasgos característicos de dicha estructura. Este cristal presenta separación de
puntas y una velocidad de crecimiento menor que la del cristal correspondiente a las
imágenes (a), (b) y (c).
Ambos cristales presentan bajas velocidades de crecimiento.
Figura 19. Evolución de la solidificación de cristales bajo condiciones de κ = 1.8 y δ = 0.000.
(a) t = 0.2; (b) t = 0.4; (c) t = 0.6; con j = 4.0. (d) t = 0.2; (e) t = 0.4; (f) t = 0.6; con j = 6.0.
La figuras 20 y 21 muestran cristales con estructura cúbica, simulados con diferentes
condiciones de magnitud de anisotropía cristalina, usando valores de δ = 0.005, 0.01, 0.020
y δ = 0.050, 0.080, 0.120, respectivamente.
37
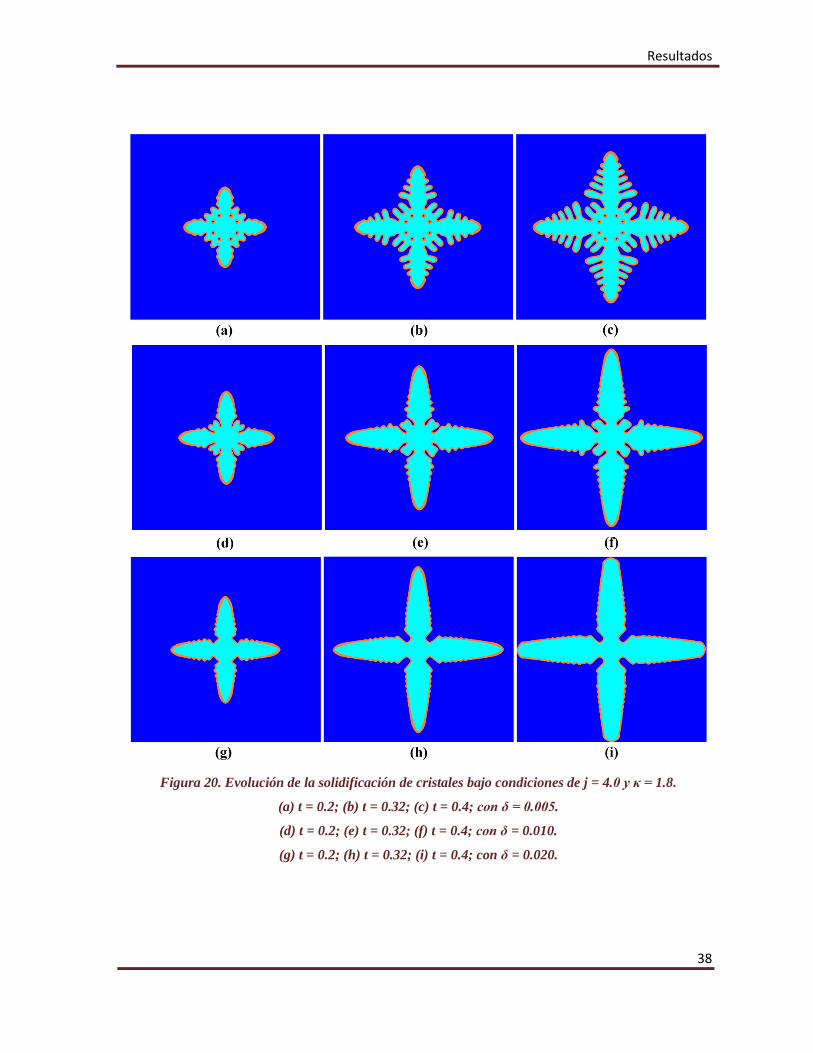
Resultados
Figura 20. Evolución de la solidificación de cristales bajo condiciones de j = 4.0 y κ = 1.8.
(a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.005.
(d) t = 0.2; (e) t = 0.32; (f) t = 0.4; con δ = 0.010.
(g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con δ = 0.020.
38

Resultados
Figura 21. Evolución de la solidificación de cristales bajo condiciones de j = 4.0 y κ = 1.8.
(a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.050.
(d) t = 0.2; (e) t = 0.32; (f) t = 0.4; con δ = 0.080.
(g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con δ = 0.120.
39

Resultados
Las imágenes (a), (b) y (c) de la figura 20 muestran la evolución de un cristal cuya
geometría es parcialmente dendrítica. Aunque se presentan brazos primarios y secundarios,
se observan remanentes de la estructura no dendrítica: puntas adicionales en direcciones
que no corresponden a las de crecimiento de acuerdo a la estructura cristalina simulada e
inestabilidades de la intercara en la zona central.
Las imágenes (d), (e) y (f) de la figura 20 muestran la evolución de un cristal cuya
geometría es casi totalmente dendrítica. Aunque aún existen remanentes de la estructura de
puntas separadas, la geometría del cristal es casi completamente dendrítica.
Las imágenes (g), (h) e (i) de la figura 20 muestran la evolución de un cristal dendrítico
típico de la estructura cúbica simulada, que presenta brazos primarios y brazos secundarios
pequeños creciendo con dirección ortogonal a estos.
Las imágenes (a), (b) y (c) de la figura 21 muestran la evolución de un cristal dendrítico
cúbico con brazos primarios gruesos y crecimiento diferente en cada brazo secundario,
obtenida al simular la solidificación del cristal con un valor moderadamente alto de
anisotropía cristalina.
Las imágenes (d), (e) y (f) de la figura 21 muestran la evolución de un cristal dendrítico
cúbico con brazos primarios gruesos y un crecimiento notable en los brazos secundarios.
Puede observarse la interferencia de los brazos secundarios con crecimiento rápido en el
desarrollo de los brazos secundarios con crecimiento lento.
Las imágenes (g), (h) e (i) de la figura 21 muestran la evolución de un cristal dendrítico
cúbico simulado bajo condiciones de alta anisotropía cristalina, δ = 0.120, que presenta
brazos secundarios asimétricos y que crece con una velocidad menor a la de los cristales
presentados en las imágenes (a), (b), (c) y (d), (e), (f).
Puede observarse un aumento gradual en la velocidad de crecimiento de los cristales de
las figuras 20 y 21, que no continúa en la evolución expuesta en las imágenes (g), (h) e (i)
de la figura 21.
La figuras 22 y 23 muestran cristales con estructura hexagonal, simulados con diferentes
condiciones de magnitud de anisotropía cristalina, usando valores de δ = 0.005, 0.01, 0.020
y δ = 0.050, 0.080, respectivamente.
40

Resultados
Figura 22. Evolución de la solidificación de cristales bajo condiciones de j = 6.0 y κ = 1.8.
(a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.005.
(d) t = 0.2; (e) t = 0.32; (f) t = 0.4; con δ = 0.010.
(g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con δ = 0.020.
41

Resultados
Figura 23. Evolución de la solidificación de cristales bajo condiciones de j = 6.0 y κ = 1.8.
(a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con δ = 0.050.
(d) t = 0.2; (e) t = 0.32; (f) t = 0.4; con δ = 0.080.
Las imágenes (a), (b) y (c) de la figura 22 muestran la evolución de un cristal dendrítico
típico de la estructura hexagonal simulada, que presenta brazos primarios y brazos
secundarios.
Las imágenes (d), (e) y (f) de la figura 22 muestran la evolución de un cristal dendrítico
hexagonal similar al presentado en las imágenes (a), (b) y (c), distinto de este en el
crecimiento ligeramente más rápido del cristal y en la geometría de los brazos secundarios.
Las imágenes (g), (h) e (i) de la figura 22 muestran la evolución de un cristal dendrítico
similar a los presentados en las imágenes (a), (b), (c) y (d), (e), (f), distinto de estos en el
menor número de brazos secundarios y la ligeramente mayor velocidad de crecimiento.
42
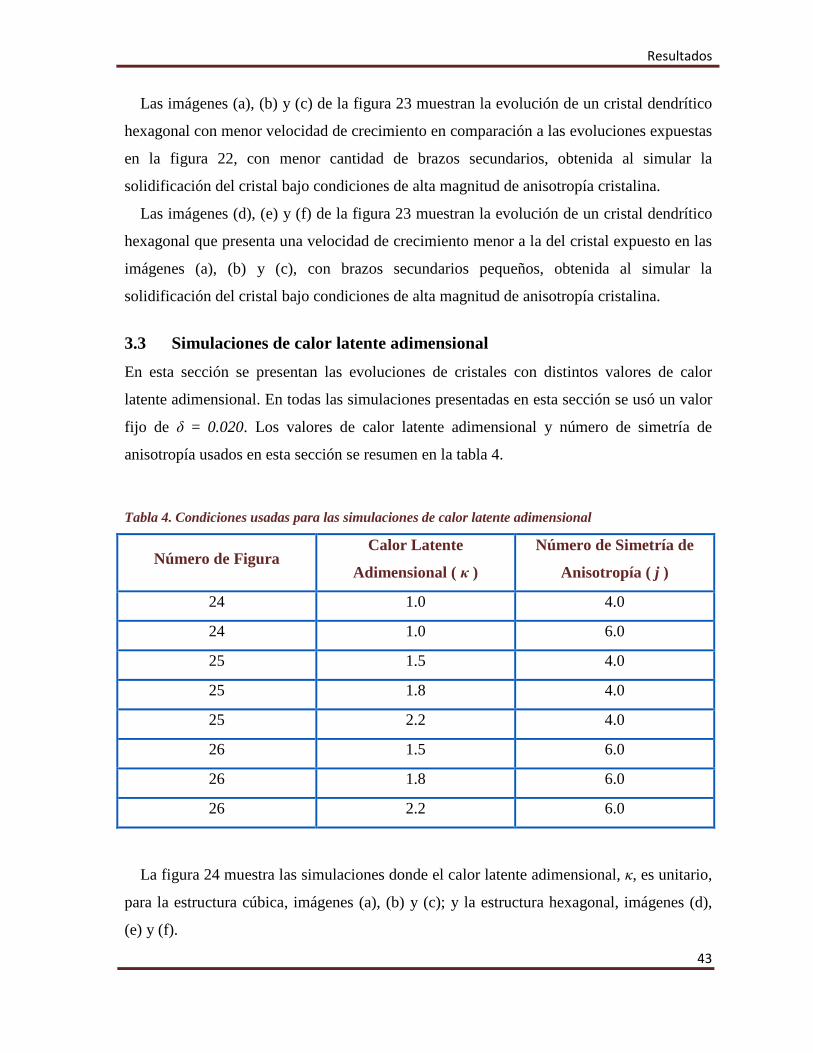
Resultados
Las imágenes (a), (b) y (c) de la figura 23 muestran la evolución de un cristal dendrítico
hexagonal con menor velocidad de crecimiento en comparación a las evoluciones expuestas
en la figura 22, con menor cantidad de brazos secundarios, obtenida al simular la
solidificación del cristal bajo condiciones de alta magnitud de anisotropía cristalina.
Las imágenes (d), (e) y (f) de la figura 23 muestran la evolución de un cristal dendrítico
hexagonal que presenta una velocidad de crecimiento menor a la del cristal expuesto en las
imágenes (a), (b) y (c), con brazos secundarios pequeños, obtenida al simular la
solidificación del cristal bajo condiciones de alta magnitud de anisotropía cristalina.
3.3 Simulaciones de calor latente adimensional En esta sección se presentan las evoluciones de cristales con distintos valores de calor
latente adimensional. En todas las simulaciones presentadas en esta sección se usó un valor
fijo de δ = 0.020. Los valores de calor latente adimensional y número de simetría de
anisotropía usados en esta sección se resumen en la tabla 4.
Tabla 4. Condiciones usadas para las simulaciones de calor latente adimensional
Número de Figura Calor Latente
Adimensional ( κ )
Número de Simetría de
Anisotropía ( j )
24 1.0 4.0
24 1.0 6.0
25 1.5 4.0
25 1.8 4.0
25 2.2 4.0
26 1.5 6.0
26 1.8 6.0
26 2.2 6.0
La figura 24 muestra las simulaciones donde el calor latente adimensional, κ, es unitario,
para la estructura cúbica, imágenes (a), (b) y (c); y la estructura hexagonal, imágenes (d),
(e) y (f).
43

Resultados
Las imágenes (a), (b) y (c) de la figura 24 muestran la evolución de un cristal poligonal,
con forma similar a un cuadrado, que se vuelve convexa con el tiempo hasta ser similar a
un círculo.
Las imágenes (d), (e) y (f) de la figura 24 muestran la evolución de un cristal poligonal,
con forma similar a un hexágono, que se vuelve cóncava con el tiempo.
Ambos cristales presentan altas velocidades de crecimiento.
Figura 24. Evolución de la solidificación de cristales bajo condiciones de κ = 1.0 y δ = 0.020.
(a) t = 0.08; (b) t = 0.2; (c) t = 0.32; con j = 4.0. (d) t = 0.08; (e) t = 0.2; (f) t = 0.32; con j = 6.0.
Las figuras 25 y 26 muestran cristales simulados con diferentes condiciones de calor
latente adimensional, usando valores de κ = 1.5, 1.8 y 2.2, con estructura cúbica y
hexagonal, respectivamente.
44

Resultados
Figura 25. Evolución de la solidificación de cristales bajo condiciones de j = 4.0 y δ = 0.020.
(a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con κ = 1.5.
(d) t = 0.2; (e) t = 0.32; (f) t = 0.4; con κ = 1.8.
(g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con κ = 2.2.
45

Resultados
Figura 26. Evolución de la solidificación de cristales bajo condiciones de j = 6.0 y δ = 0.020.
(a) t = 0.2; (b) t = 0.32; (c) t = 0.4; con κ = 1.5.
(d) t = 0.2; (e) t = 0.32; (f) t = 0.4; con κ = 1.8.
(g) t = 0.2; (h) t = 0.32; (i) t = 0.4; con κ = 2.2.
46

Resultados
Las imágenes (a), (b) y (c) de la figura 25 muestran la evolución de un cristal dendrítico
cúbico, que presenta brazos primarios gruesos y brazos secundarios pequeños.
Las imágenes (d), (e) y (f) de la figura 25 muestran la evolución de un cristal dendrítico
cúbico similar al presentado en las imágenes (a), (b) y (c), distinto de este en el crecimiento
más lento del cristal.
Las imágenes (g), (h) e (i) de la figura 25 muestran la evolución de un cristal dendrítico
similar a los presentados en las imágenes (a), (b), (c) y (d), (e), (f), distinto de estos en la
menor velocidad de crecimiento.
Las imágenes (a), (b) y (c) de la figura 26 muestran la evolución de un cristal dendrítico
hexagonal, que presenta brazos primarios gruesos y brazos secundarios poco desarrollados.
Puede observarse la interferencia de los brazos secundarios con crecimiento rápido en el
desarrollo de los brazos con crecimiento lento, lo que ocasiona una geometría asimétrica.
Las imágenes (d), (e) y (f) de la figura 26 muestran la evolución de un cristal dendrítico
hexagonal con brazos secundarios claramente desarrollados, con una velocidad de
crecimiento menor a la del cristal presentado en las imágenes (a), (b) y (c).
Las imágenes (g), (h) e (i) de la figura 26 muestran la evolución de un cristal dendrítico
hexagonal con una velocidad de crecimiento menor a la de los cristales expuestos en las
imágenes (a), (b), (c) y (d), (e), (f), que presenta brazos primarios delgados y brazos
secundarios poco desarrollados, con una geometría carente de simetría.
Puede observarse una disminución gradual en la velocidad de crecimiento de los cristales
de las figuras 25 y 26.
47

Discusión
4. Discusión
4.1 Comparación de dendritas en metales puros con dendritas simuladas Los cristales y estructuras dendríticas han sido observados de manera experimental en gran
cantidad de trabajos. A continuación se presentan fotografías de cristales dendríticos y se
comparan con las simulaciones obtenidas en este trabajo.
La figura 27 muestra (a) una dendrita de austenita, (b) el cristal correspondiente a dicha
dendrita modelado en tres dimensiones por medio del Método de Campo de Fases y (c) una
simulación obtenida en este trabajo.
Figura 27. Comparación entre (a) Imagen de Microscopio Electrónico de Barrido (MEB) de una
dendrita de austenita; (b) Cristal equiaxial calculado con un modelo de Campo de
Fases correspondiente a dicha dendrita; (22) (c) Simulación obtenida con
las condiciones de t = 0.2, j = 4.0, κ = 1.8 y δ = 0.020.
48

Discusión
Aunque en la figura 27 la dendrita de austenita, imagen (a), puede parecer distinta a la
obtenida en dos dimensiones en este trabajo, imagen (c), la imagen muestra un brazo
primario sobre el cual se desarrollan brazos secundarios, al igual que en la simulación de
este trabajo, que muestra una vista total del cristal, con cuatro brazos primarios y brazos
secundarios pequeños creciendo desde estos. Esto puede comprobarse con el cristal
modelado correspondiente a la dendrita de austenita, imagen (b), cuya vista superior
mostraría una geometría igual a la de la simulación del presente trabajo.
Las imágenes (a) y (b) de la figura 28 muestran dendritas de Succinonitrilo (SCN), un
compuesto orgánico usado como alternativa a los metales durante estudios experimentales
debido a que solidifica a temperatura ambiente en una estructura cúbica centrada en el
cuerpo (BCC) y es transparente, lo que permite un análisis a fondo de las estructuras
formadas. Al solidificar en una estructura BCC, los brazos primarios de la dendrita se
desarrollan en las seis direcciones < 100 >, manteniendo un ángulo ortogonal entre cada
brazo. (3) (12)
Figura 28. Comparación entre (a) Micrografía de una dendrita equiaxial de Succinonitrilo (SCN);
(b) Dendrita de Succinonitrilo con brazos secundarios casi periódicos; (3) (c) Simulación
obtenida con las condiciones de t = 0.2, j = 4.0, κ = 1.5 y δ = 0.020.
La imagen (a) de la figura 28 muestra la vista superior de un cristal equiaxial similar al
simulado en la imagen (b) de la figura 27. Al comparar la imagen (a) con la imagen (c) se
49

Discusión
prueba la similitud entre las dendritas observadas experimentalmente y las simuladas en
este trabajo, como se afirma anteriormente.
La figura 29 muestra la comparación entre dos dendritas de hielo, imágenes (a) y (c) y
dos simulaciones obtenidas en este trabajo, imágenes (b) y (d).
Figura 29. Comparación entre (a) Dendrita de hielo solidificada en agua pura sobreenfriada a
- 0.4 °C; (23) (b) Simulación obtenida con las condiciones de t = 0.2, j = 6.0, κ = 1.5 y δ = 0.020;
(c) Dendrita de hielo solidificada en una solución de agua y 5% de etilenglicol
sobreenfriada a -2.0 °C; (23) (d) Simulación obtenida con las
condiciones de t = 0.4, j = 6.0, κ = 1.8 y δ = 0.005.
50

Discusión
Aunque la geometría de las imágenes en la figura 29 es similar a la de un copo de nieve,
las dendritas mostradas no equivalen a estos, debido a que los copos de nieve se forman por
la transformación de vapor de agua en hielo.
Aunque tanto los copos de nieve, como el hielo solidificado en líquido sobreenfriado
presentan estructuras hexagonales, las dendritas expuestas se forman tras la nucleación y
crecimiento de un sólido en un líquido sobreenfriado. (5) (24)
La figura 30 muestra la comparación entre dos microestructuras donde se muestra el
fenómeno de separación de puntas, imágenes (a) y (b), y una simulación obtenida en este
trabajo, imagen (c).
Figura 30. Comparación entre (a) Micrografía de cristales con puntas separadas en una aleación
Al-4%Cu; (25) (b) Micrografía de cristales con puntas separadas en una aleación
Al-4.5%Cu; (26) (c) Simulación obtenida con las condiciones de
t = 0.6, j = 4.0, κ = 1.8 y δ = 0.000.
Además de haber similitud entre las imágenes presentadas en la figura 30, las imágenes
son comparables con las simulaciones de Kessler, Koplik y Levine (13) y de Brush y
Sekerka (14), presentadas en las figuras 12 y 13, respectivamente, y con las fotografías
obtenidas por Ben-Jacob y colaboradores (15) en su trabajo experimental, expuestas en la
figura 14.
Al comparar la geometría de las dendritas observadas experimentalmente de las figuras
27 y 28, con estructura FCC y BCC, respectivamente, con la geometría de las simulaciones 51

Discusión
presentadas en las respectivas figuras, se observa que las dendritas simuladas son
representativas de un metal con estructura cúbica.
Así mismo, al comparar la geometría de las dendritas hexagonales observadas
experimentalmente, de la figura 29, con las simulaciones expuestas en las mismas figuras,
se observa que las dendritas simuladas ofrecen una predicción realista de las dendritas en
metales con estructura hexagonal.
Lo anterior, en conjunto con la similitud entre las geometrías no dendríticas
experimentales, como los cristales con puntas separadas, y las simuladas, mostrada en la
figura 30, demuestra la capacidad del modelo usado para simular con precisión el
crecimiento de cristales dendríticos y de puntas separadas, y además, de simularlos en
diferentes estructuras cristalinas al variar el valor del número simetría de anisotropía, j.
4.2 Efecto de la anisotropía en la solidificación En la figura 31 se comparan las simulaciones de estructura cúbica obtenidas cuando la
magnitud de anisotropía se incrementa gradualmente desde cero y el resto de los
parámetros son constantes, mostrando el estado de los cristales en t = 0.4, a fin de exponer
el efecto de este parámetro sobre la geometría de los cristales formados.
La figura 31 (a), donde se simula una condición de isotropía, muestra un cristal con gran
número de protuberancias creciendo a partir de una zona con forma cuadrada de gran
tamaño y sin brazos característicos de un cristal dendrítico. Debido a la forma redondeada
de las protuberancias y a la evolución que presentan estas con el tiempo, como se presenta
en la figura 19 (c), se puede afirmar que se trata de un cristal que presenta separación de
puntas.
La imagen (b) de la figura 31, donde se muestra la simulación en condiciones de baja
magnitud de anisotropía, δ = 0.005, muestra un cristal con apariencia intermedia entre un
cristal dendrítico y uno con puntas separadas. En las cuatro direcciones principales para la
simetría cúbica se aprecian brazos primarios y secundarios, pero en las direcciones a 45° de
estas se presentan pares de protuberancias redondeadas. En la imagen (c) se observa un
caso similar, con un remanente de la estructura de puntas separadas y brazos primarios que
se desarrollan más a la vez que los brazos secundarios se vuelven menos notorios. En las
imágenes (d), (e) y (f) se muestran cristales completamente dendríticos, con brazos
52

Discusión
secundarios más notorios a mayores valores de anisotropía. Finalmente, la imagen (g)
muestra un cristal dendrítico con crecimiento limitado aún a valores mayores de
anisotropía.
Figura 31. Simulaciones obtenidas con las condiciones fijas de t = 0.4, j = 4.0, κ = 1.8 y magnitud de
anisotropía variable: (a) δ = 0.000; (b) δ = 0.005; (c) δ = 0.010; (d) δ = 0.20; (e) δ = 0.050;
(f) δ = 0.080; (g) δ = 0.120.
53

Discusión
En la figura 32 se comparan las simulaciones de la estructura hexagonal obtenidas
cuando la magnitud de anisotropía se incrementa gradualmente desde cero y el resto de los
parámetros son constantes, mostrando el estado de los cristales en t = 0.4.
Figura 32. Simulaciones obtenidas con las condiciones fijas de t = 0.4, j = 6.0, κ = 1.8 y magnitud de
anisotropía variable: (a) δ = 0.000; (b) δ = 0.005; (c) δ = 0.010; (d) δ = 0.20;
(e) δ = 0.050; (f) δ = 0.080.
La imagen (a) de la figura 32 muestra un cristal de puntas separadas carente de la
simetría hexagonal simulada. En las imágenes (b), (c) y (d), correspondientes a
simulaciones de baja anisotropía, se presentan cristales dendríticos altamente simétricos,
con brazos secundarios que crecen en la dirección de extracción de calor.
54

Discusión
Las imágenes (e) y (f) de la figura 32 muestran cristales dendríticos con crecimiento más
limitado a mayores valores de anisotropía, tanto en los brazos primarios como en los
secundarios.
En las simulaciones presentadas en las figuras 31 y 32, se pueden notar dos efectos:
a) La velocidad de crecimiento del cristal aumenta al aumentar la magnitud de
anisotropía, pero se presenta un máximo a valores altos de anisotropía, tras el cual,
la velocidad disminuye al aumentar δ.
Este efecto puede explicarse usando la Ecuación (26) en la Sección 1.3:
𝜎𝜎(𝑐𝑐) = 1 + 𝛿𝛿𝑐𝑐𝑐𝑐𝑐𝑐𝛿𝛿(𝑐𝑐 − 𝑐𝑐𝑜𝑜) ( 26 )
Debe recordarse que el valor de (θ – θ0) se asigna a cada valor de ∇ϕ. Cuando δ tiene
valor de 0.000, el segundo término se vuelve cero, con lo que σ(θ) tiene el mismo valor
para todos los valores de ∇ϕ, causando a su vez que ε tenga el mismo valor en todos los
puntos.
Así, la Ecuación (29) de la Sección 1.3,
𝜏𝜏𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕
= −𝜕𝜕𝜕𝜕𝜕𝜕
�𝜀𝜀𝜀𝜀′𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕� +
𝜕𝜕𝜕𝜕𝜕𝜕
�𝜀𝜀𝜀𝜀′𝜕𝜕𝜙𝜙𝜕𝜕𝜕𝜕� + ∇ ∙ (𝜀𝜀2∇𝜙𝜙)
+ 𝜙𝜙(1 − 𝜙𝜙) �𝜙𝜙 −12
+ 𝑚𝑚� ( 29 )
sólo dependerá del valor de ϕ, que al ser determinado por el Método de Diferencias Finitas,
estará determinado por los valores de ϕ en las posiciones anteriores y posteriores a un nodo.
Como consecuencia, todas las direcciones en que puede crecer el núcleo presentan la
misma facilidad de crecimiento, en contraste a cuando existe un valor de δ, donde el
segundo término de la Ecuación (26) es de importancia, y junto con este término, el valor
asignado a (θ – θ0), que determina las direcciones preferenciales de crecimiento.
Considerando esto, en el escenario en el que δ tiene valor de 0.000, el crecimiento ideal
de acuerdo a la Ecuación (26) es aquel en el que la intercara crece siempre en todas las
55

Discusión
direcciones, formando una figura casi circular. Sin embargo, deben considerarse las
fluctuaciones debidas a la Ecuación (30) de la Sección 1.3:
𝑚𝑚(𝑇𝑇) = �𝛼𝛼𝜋𝜋� atan{𝛾𝛾(𝑇𝑇𝑒𝑒 − 𝑇𝑇)} ( 30 )
que contiene una función trigonométrica, y que causan la inestabilidad de la intercara, lo
que a su vez permite la formación de protuberancias, cuyo crecimiento, como se explicó en
la Sección 1.1.2, es termodinámicamente estable en un líquido sobreenfriado, como es el
caso de las simulaciones de este trabajo.
Al aumentar δ, la influencia del segundo término de la Ecuación (26) es mayor, con lo
que el crecimiento del cristal se limita cada vez más a las direcciones preferenciales de cada
estructura.
Esto explica que la disminución en la cantidad de brazos secundarios que se presentan a
mayores magnitudes de anisotropía sea más marcada en la estructura hexagonal, donde las
direcciones preferenciales se presentan cada 60°, por lo que la dirección de crecimiento de
un brazo secundario no es preferencial, resultando en que el crecimiento del brazo
secundario se suprima o este crezca con dificultad en la dirección de extracción de calor, ya
que los brazos primarios siguen creciendo con preferencia.
En contraste, en la estructura cúbica, donde las direcciones preferenciales se presentan
cada 90°, la dirección de crecimiento de los brazos primarios y secundarios coincide, por lo
que los brazos pueden crecer con mayor facilidad que en la estructura hexagonal.
El máximo en la velocidad de crecimiento presente a altos valores de anisotropía
cristalina se puede explicar al considerarse que las direcciones de extracción de calor están
siendo restringidas a aquellas en las que se desarrollan los brazos primarios a medida que se
aumenta la magnitud de anisotropía. Así, los brazos primarios y los pocos brazos
secundarios presentes son los únicos capaces de disipar calor, al crecer o volverse gruesos,
limitando el crecimiento del cristal completo.
b) La separación de puntas se suprime a medida que la magnitud de anisotropía
aumenta, y en las etapas previas a la supresión completa de esta estructura, se forma
un cristal con geometría intermedia, con brazos primarios que se forman antes de 56

Discusión
que ocurra la separación de puntas, como se describe en el trabajo de Kessler,
Koplik y Levine (13). Además, la supresión de la estructura de puntas separadas
ocurre a valores más bajos de δ cuando la simetría de anisotropía es mayor.
La presencia de remanentes de una estructura de puntas separadas en las estructuras
intermedias una vez más se debe a la Ecuación (26). Un valor cercano a cero de δ se
traduce en que el término (θ – θ0) tiene menos impacto en el valor de σ(θ). Con ello, la
influencia de (θ – θ0) que como ya se mencionó, determina las direcciones preferenciales de
crecimiento, se ve reducida, y aunque el cristal se desarrolla preferentemente en las cuatro
direcciones preferenciales de la estructura, existen remanentes de la separación de puntas.
A valores mayores de magnitud de anisotropía la influencia de (θ – θ0) se ve
magnificada, lo que causa que el cristal se desarrolle solo en las direcciones preferenciales
y la estructura de puntas separadas no esté presente.
La separación de puntas a bajos valores de δ en la estructura cristalina se debe a que el
número de direcciones preferenciales de la estructura hexagonal es mayor que el de la
estructura cúbica. Los brazos primarios, al desarrollarse de forma estable y en gran número
antes que la estructura de separación de puntas, impiden el posible crecimiento de
protuberancias entre ellos.
4.3 Efecto del calor latente adimensional en la solidificación En la figura 33 se comparan las simulaciones de estructura cúbica obtenidas cuando el calor
latente adimensional se incrementa gradualmente desde uno y el resto de los parámetros
son constantes, mostrando el estado de los cristales en t = 0.32, a fin de hacer evidente el
efecto de este parámetro sobre la geometría de los cristales formados.
La imagen (a) de la figura 33, donde se simula una condición de κ = 1.0, muestra un
cristal regular de forma redondeada, que como se puede ver en su evolución, en las
imágenes (a), (b) y (c) de la figura 24, crece a partir de un cristal con forma casi cuadrada.
La imagen (b) de la figura 45 muestra un cristal dendrítico con remanentes de una
estructura de separación de puntas, mientras que las imágenes (c) y (d) muestran cristales
dendríticos con brazos secundarios pequeños.
57

Discusión
Figura 33. Simulaciones obtenidas con las condiciones fijas de t = 0.32, j = 4.0, δ = 0.020 y calor
latente adimensional variable: (a) κ = 1.0; (b) κ = 1.5; (c) κ = 1.8; (d) κ = 2.2.
En la figura 34 se comparan las simulaciones de estructura hexagonal obtenidas cuando
el calor latente adimensional se incrementa gradualmente desde uno y el resto de los
parámetros son constantes, mostrando el estado de los cristales en t = 0.32.
La imagen (a) de la figura 34, donde se simula una condición de κ = 1.0, muestra el
equivalente a la figura 34 (a): un cristal regular hexagonal que crece a partir de un cristal
hexagonal convexo, cuya evolución se presenta en las imágenes (d), (e) y (f) de la figura
24. Las imágenes (b), (c) y (d) muestran cristales dendríticos con brazos secundarios de
tamaño variable según el caso.
58

Discusión
Figura 34. Simulaciones obtenidas con las condiciones fijas de t = 0.32, j = 6.0, δ = 0.020 y calor
latente adimensional variable: (a) κ = 1.0; (b) κ = 1.5; (c) κ = 1.8; (d) κ = 2.2.
En las simulaciones presentadas en las figuras 33 y 34, se pueden notar dos efectos:
c) Cuando el calor latente adimensional tiene valor de 1.0, el cristal no presenta
estructura de separación de puntas o dendrítica, sino que se forma un cristal con
forma poligonal.
Ya que se debe extraer el calor latente de fusión desde el sólido para que la solidificación
ocurra, éste se vuelve una limitante al crecimiento: si el valor es alto, debe retirarse más
calor, lo que ralentiza el crecimiento del cristal. Esto último está relacionado con la
observación d).
Cuando el calor latente a extraer es menor, el efecto de la anisotropía, que dicta las
direcciones preferenciales de crecimiento del cristal, se ignora en favor de la necesidad de
extraer el calor para que el cristal crezca. Esto se demuestra en la figura 35:
59

Discusión
Figura 35. Simulaciones obtenidas con las condiciones fijas de t = 0.2, j = 0.0 con condiciones de
calor latente adimensional y magnitud de anisotropía variable: (a) κ = 1.0, δ = 0.000;
(b) κ = 1.0, δ = 0.020; (c) κ = 1.8, δ = 0.000; (d) κ = 1.8, δ = 0.020.
La figura 35 muestra la comparación de las simulaciones de simetría de anisotropía (j)
perfecta. Las imágenes (a) y (b) muestran el crecimiento de cristales con bajo calor latente,
κ = 1.0, bajo condiciones de isotropía, imagen (a), y δ = 0.020, imagen (b). El efecto
mencionado anteriormente se comprueba, ya que ambas simulaciones muestran el mismo
resultado.
Por otra parte, las imágenes (c) y (d) de la figura 35 muestran el crecimiento de cristales
con mayor calor latente, κ = 1.8, bajo condiciones de isotropía, imagen (c), y δ = 0.020,
imagen (d), de forma análoga al caso anterior. Se observa que en estos cristales, la
magnitud de anisotropía se vuelve el mecanismo dominante y determina la estructura del
cristal, que presenta separación de puntas.
60

Discusión
d) Al aumentar el calor latente adimensional, la velocidad de crecimiento del cristal
disminuye, lo que causa que los brazos secundarios no se desarrollen a valores altos
de κ.
Este efecto es explicado por la Ecuación (19), que describe la velocidad de crecimiento
de la punta de una dendrita según la teoría de nucleación y crecimiento:
𝑣𝑣 =−𝐾𝐾𝐿𝐿𝑇𝑇𝐿𝐿′
𝐿𝐿𝑣𝑣≈𝐾𝐾𝐿𝐿𝐿𝐿𝑣𝑣
∙∆𝑇𝑇𝑟𝑟
( 19 )
La Ecuación (19) indica que la velocidad de crecimiento de la punta de la dendrita, v, es
inversamente proporcional al calor latente de fusión por unidad de volumen Lv, debido a
que una menor cantidad de calor latente por extraer se traduce en una mayor velocidad de
solidificación, mientras que una gran cantidad de calor latente por extraer limita la
velocidad de solidificación.
61

Conclusiones
Conclusiones
En este trabajo se utilizó el Método de Campo de Fases para simular la solidificación de
metales puros, considerando las variables de la formulación de Kobayashi, llegándose a las
siguientes conclusiones:
1. El modelo de Campo de Fases usado en este trabajo permite simular de forma
realista la solidificación de cristales dendríticos y no dendríticos, cristales con
puntas separadas y poligonales, en metales puros con diferentes estructuras
cristalinas.
2. La velocidad de solidificación de un cristal aumenta al incrementar la magnitud de
anisotropía, δ, y se presenta un máximo a altos valores de δ, tras el cual, la
velocidad disminuye al aumentar δ.
3. A medida que la magnitud de anisotropía, δ, aumenta, la estructura de puntas
separadas, existente a bajos valores de δ, se suprime para dar paso a una estructura
dendrítica, y en las etapas previas a la supresión completa de dicha estructura, se
forma un cristal con geometría intermedia.
4. A bajos valores de calor latente de fusión, la extracción de calor es el mecanismo de
crecimiento dominante durante la solidificación, impidiendo el desarrollo de
geometrías complejas. En contraste, a altos valores de calor latente de fusión, la
magnitud de anisotropía es el mecanismo de crecimiento dominante, provocando la
formación de geometrías complejas.
5. Al aumentar el calor latente de fusión, la velocidad de solidificación de un cristal
disminuye, por lo que un metal con calor latente de fusión bajo solidificará más
rápidamente.
62

Referencias
Referencias
1. ASM International Handbook Committee. ASM handbook. ASM International, 1992.
Vol. 15. ISBN: 0-87170-007-7.
2. Stefanescu, Doru M. Science and engineering of casting solidification. Springer, 2009.
pp. 1-2, 159. ISBN: 978-0-387-74609-8.
3. Glicksman, Martin E. Principles of solidification: An introduction to modern casting
and crystal growth concepts. Springer, 2011. p. 305. ISBN: 978-1-4419-7344-3.
4. Phase-field models in materials science. Steinbach, Ingo. 7, 2009, Modelling and
Simulation in Materials Science and Engineering, Vol. 17, pp. 1-31.
5. Modeling and numerical simulations of dendritic crystal growth. Kobayashi, Ryo. 3-4,
1993, Physica D, Vol. 63, pp. 410-423.
6. ASM International Handbook Committee. ASM handbook. ASM International, 1992.
Vol. 09. ISBN: 0-87170-007-7.
7. Porter, David A., Easterling, Kenneth E. and Sherif, Mohamed Y. Phase
transformations in metals and alloys. Taylor & Francis Group, 2009. pp. 189 - 209. ISBN:
978-1-4398-8357-0.
8. Herlach, Dieter M., Galenko, Peter and Holland-Moritz, Dirk. Metastable solids
from undercooled melts. Elsevier, 2007. pp. 133, 145-146. ISBN: 978-0-08-043638-8.
9. Fredriksson, Hasse and Akerlind, Ulla. Solidification and crystallization processing in
metals and alloys. John Wiley & Sons, Ltd., 2012. pp. 167-182, 362. ISBN: 978-1-119-
99305-6.
10. Davis, Stephen H. Theory of solidification. Cambridge University Press, 2004. p. 215.
ISBN: 0-511-03696-5.
11. The cell to dendrite transition. Morris, L. R. and Winegard, W. C. 1, 1969, Journal of
Crystal Growth, Vol. 6, pp. 61, 64-65.
12. Provatas, Nikolas and Elder, Ken. Phase-field methods in materials science and
engineering. Wiley-VCH, 2010. pp. 4-7. ISBN: 978-3-527-40747-7.
13. Geometric models of interface evolution. II: Numerical simulation. Kessler, David A.,
Koplik, Joel and Levine, Herbert. 6, 1984, Physical Review A, Vol. 30, pp. 3164-3167.
63

Referencias
14. A numerical study of two-dimensional crystal growth forms in the presence of
anisotropic growth kinetics. Brush, L. N. and Sekerka, R. F. 2, 1989, Journal of Crystal
Growth, Vol. 96, pp. 420, 427.
15. Experimental demonstration of the role of anisotropy in interfacial pattern formation.
Ben-Jacob, E., et al. 12, 1985, Physical Review Letters, Vol. 55, pp. 1316-1317.
16. Friedman, Avner and Miller, Willard. The IMA volumes in mathematics and its
aplications. Springer, 1993. p. xiii. Vol. 53. ISBN: 978-1-4613-8359-8.
17. A comparative study of dendritic growth using the extended Cahn-Hilliard model and
the conventional phase-field model. Choi, Jaeho, et al. 2015, Acta Materialia, Vol. 84, p.
55.
18. A microscopic theory for antiphase boundary motion and its application to antiphase
domain coarsening. Allen, Samuel M. and Cahn, John W. 6, 1979, Acta Metallurgica,
Vol. 27, pp. 1085-1095.
19. Rappaz, Michel, Bellet, Michel and Deville, Michel. Numerical modeling in
materials science and engineering. Springer, 2010. pp. 47, 52. ISBN: 978-3-642-11820-3.
20. Velten, Kai. Mathematical modeling and simulation: Introduction for scientists and
engineers. Wiley-VCH, 2009. p. 259. ISBN: 978-3-527-40758-3.
21. Press, William H., et al. Numerical recipes: the art of scientific computing. Cambridge
University Press, 2007. pp. 1032-1038. ISBN: 978-0-511-33555-6.
22. Solidification and modeling of cast iron - A short history of the defining moments.
Stefanescu, Doru M. 2005, Materials Science and Engineering A, Vol. 413-414, p. 327.
23. Ice crystal growth in supercooled solution. Teraoka, Yoshikazu, Saito, Akio and
Okawa, Seiji. 2, 2002, International Journal of Refrigeration, Vol. 25, p. 222.
24. De Graef, Marc and McHenry, Michael E. Structure of materials: An introduction to
crystallography, diffraction and symmetry. Cambridge University Press, 2007. p. 763.
ISBN: 978-521-65151-6.
25. Tip-splitting instability and transition to seaweed growth during alloy solidification in
anisotropically preferred growth direction. Chen, Yun, et al. 2014, Acta Materialia, Vol.
66, p. 221.
64

Referencias
26. Morphological instability of cell and dendrite during directional solidification under a
high magnetic field. Li, Xi, Fautrelle, Yves and Ren, Zhongming. 2008, Acta Materialia,
Vol. 56, p. 3148.
65

Apéndices
Apéndices
Apéndice I. Código usado para la simulación A continuación se presenta el código usado durante la simulación de las estructuras
dendríticas presentadas:
!El siguiente código permite la simulación de la solidificación en metales puros con base en
!la formulación de Kobayashi.
module mdata
!Creación del módulo “mdata” para guardar información sobre las variables a fin de ser
!usada en otras partes del programa (con el comando use mdata).
integer, parameter :: nx=300, ny=300
!Definición de dos constantes: "nx" y "ny", que corresponden al número de nodos a usar en
!x y y, respectivamente.
real, dimension(nx,ny) :: t,phi,lap_phi
real, dimension(nx,ny) :: partial_phi_x, partial_phi_y,lap_t,epsilon,epsilon_derivative
!Definición de variables reales dimensionadas, con dos columnas de nx y ny valores
!respectivamente.
real, save :: kappa, dx, dy, dt, pi
real, save :: anisotropy, tau, epsilonbar, delta, alpha, gamma, teq, angle
real, save :: phiold, m, term1, term2, term3
real, save:: partial_epsilon2_x, partial_epsilon2_y
!Definición de variables reales y orden para ser guardadas tras la ejecución del programa,
!evitando que queden indefinidas tras la ejecución.
end module mdata
!================================================================
program main
!Principio el programa principal.
use mdata
implicit none
integer :: itimes
!Definición de la variable “itimes”, que indica el número de ciclos a realizar. 66

Apéndices
print *, 'start'
call initial
!Uso de la subrutina "initial", encargada de establecer las condiciones iniciales.
do itimes=1,10000
!Recorrido de la variable itimes.
call gradandlaplace
call evolution
!Uso de las subrutinas "gradandlaplace" y "evolution", encargadas de los cálculos de
!gradientes y laplacianos y de los métodos numéricos usados para resolver las ecuaciones.
if ( mod(itimes,20).EQ.0 ) then
call printdata(itimes)
!Uso de la subrutina “printdata”, encargada de crear archivos de resultados, cada que se
!terminan 20 ciclos.
endif
end do
print *, 'stop'
stop
end program main
!================================================================
subroutine initial
use mdata
implicit none
integer :: i, j
!Definición de las variables “i” e “j”, que corresponden a los nodos en x y y,
!respectivamente.
!Valores de las constantes a usar en la simulación:
dx=0.03 !Distancia entre nodos en la dirección x.
dy=0.03 !Distancia entre nodos en la dirección y.
dt=2.e-4 !Etapa de tiempo entre cada ciclo.
tau=0.0003 !Constante positiva que fija la escala temporal.
epsilonbar= 0.01 !Valor promedio de épsilon.
67

Apéndices
kappa=1.8 !Calor latente adimensional.
delta=0.01 !Parámetro de magnitud de anisotropía.
anisotropy= 4.0 !Número de simetría de anisotropía (j).
alpha= 0.9 !Constante positiva menor a 1.
gamma= 10.0 !Parámetro adimensional.
t=0.0 !Temperatura de enfriamiento.
teq=1.0 !Temperatura de equilibrio.
pi=3.14
do i=1,nx
do j=1,ny
!Recorrido de las variables i e j.
phi(i,j)=0
!Primera condición inicial: en toda la malla existe líquido.
if ((i-nx/2)*(i-nx/2)+(j-ny/2)*(j-ny/2)<10) then
phi(i,j) = 1
endif
!Segunda condición inicial: en el centro de la malla existe un núcleo.
end do
end do
end subroutine initial
!================================================================
subroutine gradandlaplace
use mdata
implicit none
integer :: i, j, ip, im, jp, jm
!Definición de las posiciones de nodos a usar en los cálculos de gradientes, laplacianos y en
!las rutinas del método de diferencias finitas. La letra p indica el nodo inmediato superior y
!la letra m indica el nodo inmediato inferior.
do i=1,nx
do j=1,ny
jp = j+1
68

Apéndices
jm = j-1
ip = i+1
im = i-1
if (im.eq.(0)) im=nx
if (ip.eq.(nx+1)) ip=1
if (jm.eq.(0)) jm=ny
if (jp.eq.(ny+1)) jp=1
!Condición adicional para los nodos en los extremos de la malla: si el nodo inmediato
!inferior es 0, el nodo es sustituido por el que ocupa el borde opuesto (nx o ny); si el nodo
!inmediato superior es mayor a nx o ny, el nodo es sustituido por el nodo 1, en el extremo
!opuesto.
partial_phi_x(i,j) = (phi(ip,j) - phi(im,j))/dx
partial_phi_y(i,j) = (phi(i,jp) - phi(i,jm))/dy
!Cálculo del gradiente de phi.
lap_phi(i,j) = (2.0*(phi(ip,j)+phi(im,j)+phi(i,jp)+ phi(i,jm)) +
phi(ip,jp)+phi(im,jm)+phi(im,jp)+ phi(ip,jm) - 12.0*phi(i,j))/(3.0*dx*dx)
lap_t(i,j) = (2.0*(t(ip,j)+t(im,j)+t(i,jp)+t(i,jm)) + t(ip,jp)+t(im,jm)+t(im,jp)+t(ip,jm) -
12.0*t(i,j))/(3.0*dx*dx)
!Cálculo de los laplacianos de phi y t.
if (partial_phi_x(i,j).eq.0.0) then
if (partial_phi_y(i,j).le.0.0 ) angle =-0.5*pi
if (partial_phi_y(i,j).gt.0.0 ) angle = 0.5*pi
endif
if (partial_phi_x(i,j).gt.0.0) then
if (partial_phi_y(i,j).le.0.0) angle= 2.0*pi + atan(partial_phi_y(i,j)/partial_phi_x(i,j))
if (partial_phi_y(i,j).gt.0.0) angle= atan(partial_phi_y(i,j)/partial_phi_x(i,j))
end if
if (partial_phi_x(i,j).lt.0.0) angle = pi + atan(partial_phi_y(i,j)/partial_phi_x(i,j))
!Asignación de valores al ángulo (diferencia entre theta y theta0).
epsilon(i,j) = epsilonbar*(1.0 + delta*cos(anisotropy*(angle)))
!Cálculo de épsilon.
69

Apéndices
epsilon_derivative(i,j) = -epsilonbar*anisotropy*delta*sin(anisotropy*(angle))
!Derivada de la ecuación anterior.
end do
end do
end subroutine gradandlaplace
!================================================================
subroutine evolution
use mdata
implicit none
integer :: i, j, ip, im, jp, jm
do i=1,nx
do j=1,ny
jp = j+1
jm = j-1
ip = i+1
im = i-1
if (im.eq.(0)) im=nx
if (ip.eq.(nx+1)) ip=1
if (jm.eq.(0)) jm=ny
if (jp.eq.(ny+1)) jp=1
term1= (epsilon(i,jp)*epsilon_derivative(i,jp)*partial_phi_x(i,jp) -
epsilon(i,jm)*epsilon_derivative(i,jm)*partial_phi_x(i,jm))/dy
term2= -(epsilon(ip,j)*epsilon_derivative(ip,j)*partial_phi_y(ip,j) -
epsilon(im,j)*epsilon_derivative(im,j)*partial_phi_y(im,j))/dx
!Términos 1 y 2 de la Ecuación (29).
partial_epsilon2_x = (epsilon(ip,j)**2 - epsilon(im,j)**2)/dx
partial_epsilon2_y = (epsilon(i,jp)**2 - epsilon(i,jm)**2)/dy
!Cálculo del gradiente de épsilon al cuadrado.
term3= partial_epsilon2_x*partial_phi_x(i,j)+ partial_epsilon2_y*partial_phi_y(i,j)
!Parte del término 3 de la Ecuación (29).
phiold = phi(i,j)
70

Apéndices
!Uso de la variable phiold para almacenar información de pasos anteriores durante la
evolución.
m = alpha/pi * atan(gamma*(teq-t(i,j)))
!Cálculo de m.
phi(i,j) = phi(i,j) + (term1 + term2 + (epsilon(i,j)*epsilon(i,j)*lap_phi(i,j)) + term3 +
phiold*(1.0-phiold)*(phiold-0.5+m))*(dt/tau)
t(i,j) =t(i,j) + lap_t(i,j)*dt + kappa*(phi(i,j) - phiold)
!Cálculo de phi y t.
end do
end do
end subroutine evolution
!================================================================
subroutine printdata(p)
use mdata
implicit none
integer :: p,i,j
character(len=14) :: FileName1, FileName2
!Subrutina que se encarga de crear dos archivos .dat en los cuales se escriben los datos de
!phi y t calculados.
write(FileName1,FMT='(A4,I6.6,A4)') "phi_",p,".dat"
FileName1 = trim(FileName1)
open(unit=52,file=FileName1)
do j=1,ny
do i=1,nx
write(52,FMT='(F10.6)',ADVANCE='no') phi(i,j)
end do
write(52,FMT='(F10.6)',ADVANCE='yes')
end do
close(52)
write(FileName2,FMT='(A4,I6.6,A4)') "temp_",p,".dat"
FileName2 = trim(FileName2)
71

Apéndices
open(unit=53,file=FileName2)
do j=1,ny
do i=1,nx
write(53,FMT='(F10.6)',ADVANCE='no') t(i,j)
end do
write(53,FMT='(F10.6)',ADVANCE='yes')
end do
close(53)
print*,p
return
end subroutine printdata
!================================================================
72