los aá no se almacenan en ninguna proteína … · ciclo de la urea sistema nervioso: principal...
TRANSCRIPT

Metabolismo de aminoácidos
Marijose Artolozaga Sustacha, MSc

Energía metabólica generada en los tejidos:
90% proviene de la oxidación de carbohidratos y triglicéridos
Sólo 10% de la oxidación de las proteínas
1-2% de las proteínas del cuerpo sufren recambio = degradación normal a sus aá
75-80% de esos aá se utilizan para síntesis de
proteínas nuevas
otros compuestos nitrogenados
Sólo 20-25% de los aá se degradan:
Energía
CO2
Urea
Intermediarios>>Glucosa y/o cuerpos cetónicos

Los aá no se almacenan en ninguna proteína con función especial de reserva:
Aminoácidos se deben conseguir por
La digestión de proteínas de la dieta
La degradación normal de proteínas del organismo
La síntesis de novo
Los aá que rebasen las necesidades son degradados rápidamente

Equilibrio de nitrógeno
Nitrógeno del organismo < aá < proteínas
Diariamente:
Equilibrio:
Síntesis-Degradación
Anabolismo-Catabolismo
Proteínas endógenas
aminoácidosUrea
(orina)
Proteínas de la dieta
Excreción
Degradación
Síntesis
300g/día
100g/día
100g/día
Digestión
300g de proteínas se “recambian”
+ 100g de proteína de la dieta
100g se elimina en la orina: urea

Equilibrio de nitrógeno
Equilibrio = balance nitrogenado, normal
Desequilibrios:
Equilibrio negativo: se excreta más de lo que se ingiere
Ej. Falta de aá esenciales
No se puede sintetizar proteína normales >> se degradan >> aumenta excreción
Equilibrio positivo: se excreta menos de los que se ingiere
Ej. Embarazadas y niños

Anabolismo y catabolismo de aá y proteínas
Anabolismo:
Si la producción de aá por la degradación de proteínas
endógenas o de la dieta es insuficiente:
Síntesis de aá
Síntesis de proteínas
Síntesis de otros compuestos nitrogenados
Catabolismo
Degradación de proteínas >> aá
Degradación de aá:
Nitrógeno se convierte en urea > excreción
Degradación del esqueleto de Carbono de los aá

Temas: metabolismo de aá
1.Degradación de aá y proteínas:
Degradación de proteínas
Síntesis de urea > excreción
Degradación del esqueleto de Carbono de los aá
2. Síntesis de aá
3. Síntesis de productos especializados a partir de aá
Degradación de aminoácidos
y proteínas
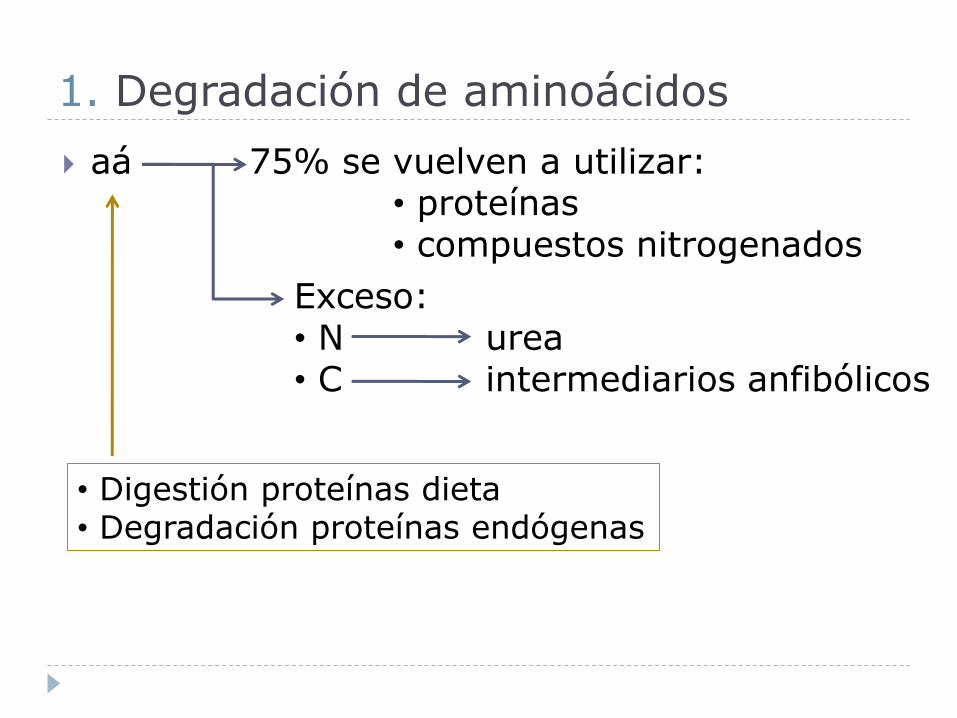
1. Degradación de aminoácidos
aá 75% se vuelven a utilizar:• proteínas• compuestos nitrogenados
Exceso:• N urea• C intermediarios anfibólicos
• Digestión proteínas dieta• Degradación proteínas endógenas

1. Degradación de proteínas endógenas
Vida media de una proteína: t1/2
= tiempo requerido para disminuir la concentración a la mitad del valor inicial
Proteínas hepáticas: 30’ a 150h
Hemoglobina: 120 días !
Colágeno es estable: meses o años
Proteínas con anomalías: vida muy corta
Prot. reguladoras clave: 30’ a 2h
En enfermedad o ayuno se degrada la proteína del músculo para suplir al cuerpo de aá esenciales y energía

1. Degradación de proteínas endógenas
Señales químicas para degradación de proteínas:
aá en extremo terminal:
Ser: vida larga
Asp, Arg: vida corta
Abundancia de ciertos aá:
Secuencia PEST: Pro, Glu, Ser, Thr: vida corta
Las proteínas con anormalidades se ubiquitinan> degradación rápida

1. Degradación de proteínas endógenas
En lisosomas> proteasas lisosomales = catepsinas
Prot extracelulares, ej plasmáticas
Prot de membrana
Prot intracelulares de vida larga
En el citosol> endoproteasas y exoproteasas; Vía proteolítica de la ubitiquitina-proteosoma (ATP)
Proteínas intracelulares de vida corta
Proteínas anormales

1. Degradación de proteínas endógenas
Proteasas lisosomales
Lisosomas contienen alta concentración de proteasas
Degradan primariamente:
Proteínas plasmáticas que entran en la célula por endocitosis
Proteínas de la superficie de la membrana que se utilizan en la endocitosis mediada por receptores
Algunas hormonas y señales se degradan después de haber producido la respuesta celular
Algunos receptores se degradan y otros se reciclan
Lisosomas

1. Degradación de proteínas endógenas
Vía proteolítica de la ubitiquitina-proteasoma
Ubiquitina= proteína globular pequeña
Proteínas (lys) se unen a la ubiquitina (gly)= “ubiquitinación” es selectiva *
Proteína-Cadena de poliubiquitina
Proteasoma= completo proteolítico en forma de tonel *
Proteasoma reconoce las proteínas marcadas y las parte en fragmentos: *
Actividad enzimática: desnaturalizante + endopeptidasa
Exopeptidasas citosólicas >> aá
* Gasto de ATP en 3 de los pasos
En citosol

1. Degradación de proteínas endógenas
Vía proteolítica de la ubitiquitina-proteasoma
Proteasoma
Conjugación de la Ubiquitina
Ubiquitina
Proteína
Complejo 19 S
Proteasoma 20 S
Degradación de proteínas
Amino-
ácidos
Péptidos
Presentación
de
Antígenos

1. Degradación de aminoácidos
Ocurre en 2 etapas generales:
N: Desaminación: eliminación del N del amino a
Síntesis de urea
C: Degradación de los esqueletos carbonados:
intermediarios anfibólicos
Energía, CO2…
Utilización para síntesis

1. Síntesis de urea
Para eliminación del N del amino a
4 etapas:
1. Transaminación
2. Desaminación oxidativa de Glu
3. Transporte del NH3 al hígado
4. Reacciones del ciclo de la urea

1. Síntesis de urea: 4 etapas
1. Transaminación
2. Desaminaciónoxidativa
1
4
3
2
3
4. Ciclo de la urea
3. Transporte al hígado
4. Ciclo de la urea
3. Transporte al hígado

1. Síntesis de urea
Para eliminación del N del amino a
4 etapas:
1. Transaminación
2. Desaminación oxidativa de Glu
3. Transporte del NH3 al hígado
4. Reacciones del ciclo de la urea

1. Síntesis de urea:
1. Transaminación
Casi todos los aminoácidos
Mismas enzimas transaminasas participan en síntesis y degradación< muy reversibles
Coenzima PLP= Piridoxal Fosfato, porta el -NH3+
Un aminoácido transfiere su amino-a al a-
cetoglutarato Glutamato
Los -NH3+ quedan “acumulados” en el Glutamato
Desaminación Oxidativa
a-cetoglutarato
Glu
aá
ceto-á
Transaminaciónsíntesis de aá

1. Síntesis de urea:
1. Transaminación
Aminotransferasas
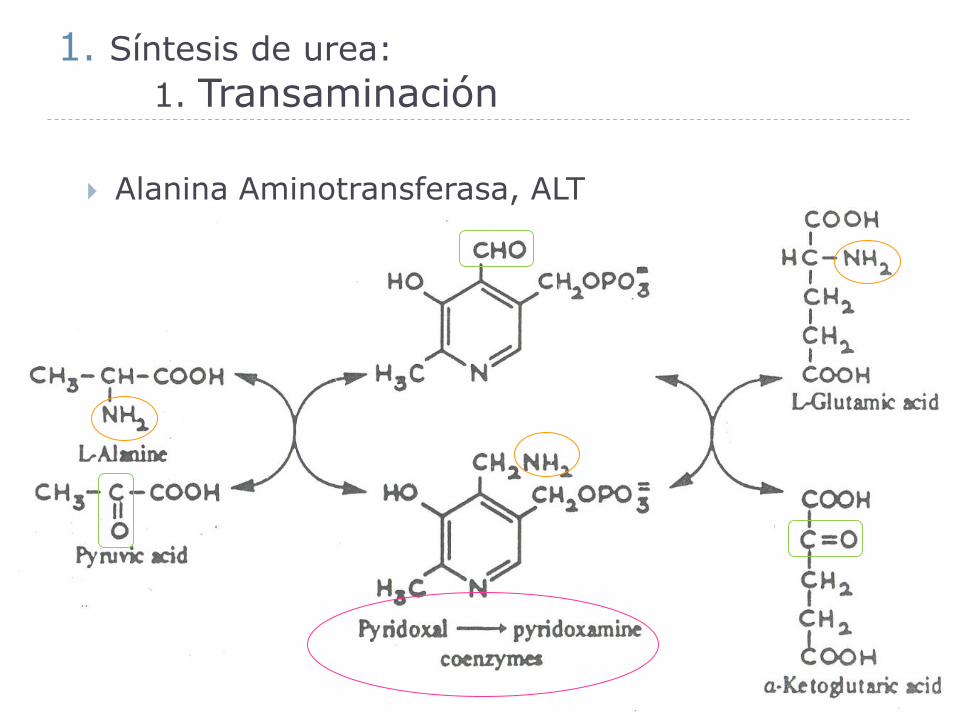
1. Síntesis de urea:
1. Transaminación
Coenzima de las transaminasas:
Piridoxal-P
Piridoxamina-P
Alanina Aminotransferasa, ALT

1. Síntesis de urea:
1. Transaminación
Coenzima:
Piridoxal-P (PLP)
Piridoxamina-P
=Vitamina B6

1. Síntesis de urea:
1. Transaminación
Transaminasas en el citosol de casi todas las células del cuerpo
En especial en hígado, riñón, intestino y músculo
Transaminasas en plasma:
Aspartato Aminotransferasa, AST
Alanina Aminotransferasa, ALT
Se elevan en
enfermedades
del hígado.También trastornos
musculares e
infarto
Antes se llamaban:
ALT: Transaminasa Glutámico-pirúvica, GPT
AST: Transaminasa Glutámico –oxalacética, GOT
La AST es una excepción: se utiliza más en la dirección de formar Asp ciclo de la urea

1. Síntesis de urea
Para eliminación del N del amino a
4 etapas:
1. Transaminación
2. Desaminación oxidativa del Glu
3. Transporte del NH3 al hígado
4. Reacciones del ciclo de la urea

1. Síntesis de urea:
2. Desaminación oxidativa del Glu
Enzima GluDH= Glutamato Deshidrogenasa
Principalmente en hígado y riñón
En la mitocondria

1. Síntesis de urea:
2. Desaminación oxidativa del Glu
Enzima GluDH= Glutamato Deshidrogenasa
Reversible:
en la degradación: NAD+ NADH
en la biosíntesis: NADPH

1. Síntesis de urea:
2. Desaminación oxidativa del Glu
Enzima GluDH= Glutamato Deshidrogenasa
Productos:
a-cetoglutarato Ciclo de Krebs Energía
NH3 ¡tóxico!
Inhibidores alostéricos: ATP, GTP, NADH
Activadores alostéricos: ADP, GDP

NH3 es muy tóxico:
en el sistema nervioso central se une al a-cetoglutarato y forma Glutamato >>>
concentración de a-cetoglutarato
función del Ciclo de Krebs
>>> retardo mental, etc
>>> encefalopatía
>>> coma
Llevarlo al hígado
para eliminarlo!

1. Síntesis de urea
Para eliminación del N del amino a
4 etapas:
1. Transaminación
2. Desaminación oxidativa de Glu
3. Transporte del NH3 al hígado
4. Reacciones del ciclo de la urea

1. Síntesis de urea:
3. Transporte del NH3 al hígado
Hígado elimina rápidamente el amoniaco NH3
de la circulación sanguínea (10-20ml/dl en sangre)
El NH3 producido en otros tejidos se transporta en forma de Glutamina (Gln)
El NH3 producido en los músculos también en forma de Alanina (Ala)
También hay NH3 producido por bacterias intestinales

1. Síntesis de urea:
3. Transporte del NH3 al hígado
Como Glutamina:
Glutamina Sintetasa en tejidos
NH3 se une al Glu
Gasto de ATP
Gln se transporta al hígado
Glutaminasa en hígado (mitocondria)
Libera el NH3
Ciclo de la urea
Sistema nervioso:
Principal mecanismo para remoción
de amoniaco del cerebro

1. Síntesis de urea:
3. Transporte del NH3 al hígado
Como Alanina:
Piruvato producido en la glicólisis en músculo + Glu
Transaminasa en músculo
Alanina + a-cetoglutarato
Transaminasa en hígado
Piruvato + Glu
Gluconeogénesis
Glucosa sangre músculo
Ciclo de la
Alanina- Glucosa

1. Síntesis de urea
Para eliminación del N del amino a
4 etapas:
1. Transaminación
2. Desaminación oxidativa de Glu
3. Transporte del NH3 al hígado
4. Reacciones del ciclo de la urea

1. Síntesis de urea:
4. Ciclo de la urea
1 CO2
1 NH4+ ó NH3
1 Asp-NH3+
3 ATP
aá especiales participan en el ciclo, pero no se consumen:
ornitina
citrulina
argininosuccinato
arginina
1
En el hígado:
• reacciones 1-2 en matriz mitocondrial
• reacciones 3-5 en citosol

1. Síntesis de urea:
4. Ciclo de la urea1
2
3
4
5
Matriz mitocondrialEnzimas:
1. Carbamoil-P-Sintasa I
2. Ornitina Transcarbamoilasa
3. Argininosuccinato Sintasa
4. Argininosuccinasa
5. Arginasa
CO2
NH3
Citosol
NH3

2
3a
4
5
3b
CO2
NH3
1
Glu NH3
Ciclo de la urea
Glu

1. Síntesis de urea:
Regulación
GlutamatoDH
Es alostérica:
Activadores: ADP
Inhibidores: GTP, ATP, NADH
Arginasa:
Inhibidores competitivos: Lisina, ornitina (=producto)
Carbamoil-P Sintasa I:
Activador alostérico: N-acetil-Glu
Acetil CoA + Glu
ArgActivada por dieta
alta en proteínas

1. Trastornos metabólicos de la síntesis de urea
Son enfermedades metabólicas
Algunas se detectan en la prueba de tamizaje
Cualquier defecto en enzimas:
Intoxicación con amoniaco >>
vómito, aversión por proteínas, retraso mental severo
Se debe consumir proteínas con frecuencia pero en cantidades muy pequeñas

1. Otros trastornos relacionados
Patologías que reducen la función hepática
Cirrosis o hepatitis
Falla en el ciclo de la urea >> NH3
Encefalopatía hepática
Coma hepático