filogenÉtica y genÉtica de poblaciones de potos …
TRANSCRIPT

1
FILOGENÉTICA Y GENÉTICA DE POBLACIONES DE Potos flavus (CARNIVORA:
PROCYONIDAE) CON SECUENCIAS DE ADN MITOCONDRIAL
MARIA FERNANDA JARAMILLO TRUJILLO
TRABAJO DE GRADO
Presentado como requisito parcial para optar al título
MAGISTER EN CIENCIAS BIOLÓGICAS
PONTIFICIA UNIVERSIDAD JAVERIANA
FACULTAD DE CIENCIAS
MAESTRIA EN CIENCIAS BIOLÓGICAS
Bogotá, D. C.
2014

2
FILOGENÉTICA Y GENÉTICA DE POBLACIONES DE Potos flavus (CARNIVORA:
PROCYONIDAE) CON SECUENCIAS DE ADN MITOCONDRIAL
MARIA FERNANDA JARAMILLO TRUJILLO
APROBÓ
______________________________ ______________________________
Ingrid Schuler Ph.D Manuel Antonio Franco Cortes MD PhD
Decana Académica Director Posgrado en Ciencias Biológicas

3
FILOGENÉTICA Y GENÉTICA DE POBLACIONES DE Potos flavus (CARNIVORA:
PROCYONIDAE) CON SECUENCIAS DE ADN MITOCONDRIAL
MARIA FERNANDA JARAMILLO TRUJILLO
APROBÓ
Manuel Ruiz García Ph.D María Ignacia Castillo Amezquita PhD
Director Jurado 1
______________________________
Ignacio Manuel Zarante Montoya MD Ph.D Joao Víctor Muñoz Duran PhD
Jurado 2 Jurado 3

4
NOTA DE ADVERTENCIA
“La Universidad no se hace responsable de los conceptos emitidos por sus alumnos en sus
trabajos de tesis. Solo velará por qué no se publique nada contrario al dogma y a la moral católica
y porque la tesis no contenga ataques personales contra persona alguna, antes bien se vea en ellas
el anhelo de buscar la verdad y la justicia”.
Artículo 23 de la Resolución N° 13 de julio de 1946.

5
AGRADECIMIENTOS
A Manuel Ruiz García que me permitió hacer parte de este proyecto y que me confió las muestras
de una especie tan maravillosa y poco estudiada, por su confianza y paciencia gracias.
A Myrera Pinedo Castro por toda su disposición y enseñanzas, gracias.
A todos mis compañeros del laboratorio por sus aportes y colaboración, gracias.
A mi familia por todo su apoyo, gracias.
A mi hijo que me acompañó durante todo este proceso y que me enseño que los grandes sueños
merecen un esfuerzo adicional pero que siempre será bien recompensado.

6
TABLA DE CONTENIDO 1. RESUMEN ......................................................................................................................................... 7
2. INTRODUCCIÓN ................................................................................................................................ 7
3. JUSTIFICACION Y PLANTEAMIENTO DEL PROBLEMA ........................................................................ 9
4. MARCO TEÓRICO .............................................................................................................................10
5. OBJETIVO .........................................................................................................................................14
5.1 Objetivo general ............................................................................................................................14
5.2 Objetivos específicos .....................................................................................................................14
6. METODOLOGÍA ...............................................................................................................................15
6.1 Obtención de muestras y localidades ......................................................................................15
6.2 Extracción, amplificación y secuenciación de ADN ...................................................................18
6.3 Alineamiento de secuencias de ADN mitocondrial ..................................................................21
6.4 Análisis filogenéticos ................................................................................................................21
6.5 Análisis de diversidad, heterogeneidad y flujo génico ............................................................24
6.6 Detección de cambios demográficos .......................................................................................24
6.7 Tiempos de divergencia ...........................................................................................................25
7. RESULTADOS ...................................................................................................................................26
7.1 Análisis de filogenia ..................................................................................................................26
7.2 Diversidad genética ..................................................................................................................39
7.3 Heterogeneidad y flujo génico entre las poblaciones ..............................................................40
7.4 Cambios demográficos .............................................................................................................43
7.5 Tiempos de divergencia ...........................................................................................................49
8. DISCUSIÓN /CONCLUSIONES ..............................................................................................................
8.1 Filogenia y correspondencia de supuestas especies definidas morfológicamente .................59
8.2 Diversidad genética, heterogeneidad genética y flujo génico .................................................63
8.3 Cambios demográficos y tiempos de divergencia ....................................................................64
9. BIBLIOGRAFIA ................................................................................................................................68

7
1. RESUMEN
Este es el primer estudio desarrollado sobre sistemática molecular, genética de poblaciones y
filogeografía dentro de Potos flavus (Procyonidae) con un numero representativo de muestras
(n=100) y que abarca una parte importante de la distribución de este carnívoro neotropical.
Para llevar a cabo este trabajo se analizaron secuencias del gen mitocondrial NADH – 5 (280
pares de bases; pb), de 99 individuos de P. flavus, representando cuatro poblaciones diferentes
(megalotus (15), chapadensis (17), chiriquensis (3) y Amazonas Occidental (64)) provenientes de
5 países (Colombia, Perú, Brasil, Ecuador y Bolivia). Adicionalmente 24 muestras fueron
secuenciadas también para el gen mitocondrial Citocromo Oxidasa-b, Cyt-b (426 pb),
representando las poblaciones anteriormente mencionadas con excepción de chiriquensis.
A partir de las secuencias obtenidas se realizó un análisis de las relaciones filogenéticas. Se
indagó si los datos moleculares obtenidos permiten diferenciar las subespecies propuestas
morfológicamente, se evaluó el estado genético de las poblaciones (diversidad genética,
heterogeneidad genética y flujo génico), y sobre su historia evolutiva (se calcularon tiempos de
divergencia y cambios demográficos de la especie y se relacionaron con posibles causas
climatológicas y geológicas).
Los principales resultados obtenidos fueron los siguientes: 1- La especie muestra altos niveles de
diversidad genética mitocondrial; 2- Las muestras que representaron las poblaciones de
megalotus, chiriquensis y amazonas occidental, pertenecen a una única población, mientras
chapadensis en Bolivia y unos individuos peruanos provenientes de ciertos departamentos de ese
país formaron dos poblaciones diferentes. 3- El proceso de diversificación parece haberse dado
en el mioceno medio.
2. INTRODUCCIÓN
El orden Carnivora incluye 11 familias y clásicamente han sido divididos en dos superfamilias
monofileticas: Caniformia y Feliformia (Eisenberg 1989, Wozencraft, 1989, Wyss y Flynn 1993).
Caniformia ha sido usualmente organizada en las familias de Canidae, Ursidae, Procyonidae,

8
Mustelidae, Otariidae, Odobenidae, y Phocidae mientras Feliformia fue dividida en las familias
Viverridae, Felidae, Herpestidae y Hyaenidae ((Eisenberg 1989, Flynn & Nedbal 1998).
Los prociónidos son una de las 11 familias tradicionales dentro del orden carnívora. Esta familia
está compuesta por 14 especies incluidas en 6 géneros (Wozencraft 2005) que están distribuidas
geográficamente a través de las américas e incluye frugívoros arborícolas (olingos [Bassaricyon
spp.] y los del cola prensil Kinkajou [Potos flavus]) y omnívoros terrestres y arbóreos
encontrados a través de diversos hábitats (cola anillada y cacomistle [Bassariscus spp.]; coaties
[Nasua spp. and Nasuella olivácea]; y mapaches [Procyon spp.]) (Zeveloff 2002).
Procyonidae muestra un importante éxito de colonización en Sur América. La diversificación de
los actuales prociónidos neotropicales parece ser antes de la formación del puente de panamá
(alrededor de 3 millones de años atrás) (Koepfli et al. 2007). Así la radiación de especies de
Procyonidae precede el gran intercambio biótico americano en desacuerdo con muchas
explicaciones tradicionales paleontológicas. Los primeros prociónidos aparecen en el registro
fósil de Norte América a principios del Mioceno (Hunt 1996, Baskin 1998, 2004).
Potos flavus es la especie más basal de los prociónidos y de acuerdo con Koepfli et al. (2007), su
diversificación se dió alrededor de hace 21,6 a 24 MA.
Se obtuvieron secuencias de los genes mitocondriales Cyt-b y NADH5 para 24 y 99 individuos
respectivamente. Mediante la construcción de árboles filogenéticos de máxima verosimilitud,
Neighbour-Joining, y bayesianos se exploraron las relaciones de parentesco entre las mismas.
Con el mismo conjunto de datos, se calcularon diferentes medidas de diversidad genética para la
población en general y para 4 subpoblaciones divididas geográficamente y con base en algunos
morfotipos determinados previamente por algunos autores.
Se evaluaron adicionalmente estadísticos de heterogeneidad y flujo génico entre las
subpoblaciones asignadas, se aplicaron diferentes test para evaluar posibles cambios
demográficos y finalmente se establecieron los tiempos de divergencia mediante el análisis de los
árboles bayesianos, y la construcción de una red de haplotipos y “Bayesian skyline plot”
3. JUSTIFICACION Y PLANTEAMIENTO DEL PROBLEMA

9
El Neotrópico es rico en biodiversidad y podría decirse que contiene algunos de los menos
comprendidos y más estudiados depredadores del mundo. Los carnívoros representan los más
altos niveles tróficos dentro de las áreas neotropicales siendo especies claves que pueden alterar
comunidades de mamíferos omnívoros y herbívoros e indirectamente a comunidades de plantas.
Desafortunadamente debido a las presiones de las poblaciones humanas, muchas áreas tropicales
y los mamíferos dentro de ellas están aumentando su riesgo. Este problema se ve agravado por la
carencia de datos genéticos, de la biología evolutiva y conservación de estas especies. (Ruiz-
García & Shostell 2013).
La mayor parte de trabajos filogenéticos en carnívoros se han centrado en aclarar las relaciones
existentes entre las familias como por ejemplo Li Yu et al. (2004), Agnarsson et al. (2010),
Fulton & Strobeck (2006). Las relaciones entre prociónidos por su parte también han sido
abordadas desde la perspectiva molecular, Koepfli et al. (2007) realiza un análisis de la filogenia
al interior de la familia a partir de secuencias nucleares y estima los posibles tiempos de
divergencia entre cada uno de los géneros de Procyonidae, adicionalmente realiza un análisis de
los posibles fenómenos que afectaron la colonización de esta familia en centro y Sur América.
Sin embargo son pocos los estudios que se han realizado a nivel de genero para la familia
Procyonidae, específicamente para Potos flavus solo existe un estudio preliminar de filogenia y
posible existencia molecular de subespecies definidas morfológicamente (Ruiz – García et al.
2012 a), en este estudio preliminar se calculan unos tiempos de divergencia para Potos mucho
menores de los calculados por Koepfli et al. (2007) y se da una idea original de la existencia de
algunas subpoblaciones aisladas.
Este es el primer estudio con un número de muestras representativas de todo el rango de
distribución en Sur américa, que permitirá determinar la correspondencia entre las subespecies
definidas y su relación molecular, adicionalmente se realizarán las estimaciones de divergencia
temporal para ver que tanto se ajustan con lo descrito previamente por otros autores y como se
relacionan con los grandes periodos históricos que han influido en la distribución de la biota que
conocemos actualmente.
Por último, el uso de las herramientas moleculares, como por ejemplo el estudio del ADN
mitocondrial permite determinar estrategias de conservación efectivas basadas en la clasificación

10
sistemática de las especies y de sus poblaciones diferenciadas por debajo del nivel de especies
(subespecies). La incertidumbre sobre las unidades de conservación guía tanto a la confusión en
el establecimiento de planes de manejo como a errores en el establecimiento de prioridades
(O´Brien 1994). Precisamente por eso, se hace enormemente importante describir la estructura
genética, la filogeografía y la determinación de linajes evolutivamente significativos (ESU’s;
Moritz 1994) y su posible correspondencia con subespecies definidas morfológicamente para las
poblaciones muestreadas de P. flavus.
4. MARCO TEORICO
P. flavus también conocido como Kinkajou, perro de monte o martucha, es la única especie
perteneciente al género Potos.
De acuerdo con Ford & Hoffmann (1988) se han identificado las siguientes subespecies, con la
localidad tipo.
1. P. f. prehensilis, Atoyac River in Veracruz, México
2. P. f. chiriquensis, Chiriqui Panama
3. P. f. megalotus, Santa Marta Magdalena
4. P. f. meridensis, Merida Venezuela
5. P. f. modestus, Balzar Ecuador
6. P. f. flavus, Surinam
7. P. f. nocturnus, Alagoas State in Brazil
8. P. f. chapadensis, Matto Grosso State in Brazil
El mapa de distribución para estas subespecies de acuerdo con los mismos autores es el siguiente:

11
Figura 1. Mapa tomado de Ford & Hoffmann (1988). 1. P.f. chapadendis; 2 P.f.chiriquensis.; 3,
P.f. flavus; 4, P.f megalotus; 5, P.f. meridensis; 6, P. f. modestus; 7, P.f. nocturnos; 8, P.f.
prehensilis. El símbolo de interrogación cuestiona la distribución pata Trinidad.
A continuación se presenta la descripción detallada para la especie de acuerdo con lo compilado
en un artículo por estos autores.
El género Potos difiere de otros géneros de procyonidos en tener orejas pequeñas y redondeadas,
la presencia de dos glándulas cutáneas ventrales (esternal y medio abdominal), una cola prensil,
una larga y estrecha lengua extensible y un báculo único que termina en 4 cortos brazos de punta
redondeada.
Distribución
El Kinkajou probablemente se encuentra a los largo del trópico de Centro y Sur América. El
rango conocido es desde Mato Grosso en el centro de Brasil, oriente de Perú, norte de Ecuador, e
incluye las Guyanas, Surinam, Venezuela y Colombia; hacia el norte a lo largo de Centro
América a San Luis Potosi y al sur Tamaulipas en el este de México y Guerrero en el occidente

12
de México. Un avistamiento cerca a Acuna, Tamaulipas, México por Leopold (1959) es el
registro más al norte.
Algunas áreas dentro del rango de distribución en las cuales no se encuentre Kinkajú son altas
montañas andinas, Catinga y Chaco de Brasil y aparentemente los estados secos en Venezuela.
El Kinkajou fue reportado en la isla de trinidad; pero Allen & Cahpman (1897) considera esta
distribución incorrecta o dudosa porque está basada en pruebas insuficientes. Los Kinkajous se
encuentra en altitudes hasta los 2500 msnm; en Venezuela, 162 animales fueron colectados entre
los 24 y los 1.750 msnm, pero el 97% fueron encontrados alrededor de los 500 m.
Registro fósil
Los prociónidos se encuentran entre los más raros fósiles conocidos de carnívoros, y no hay
registro fósil de Potos. El pobre registro fósil es atribuido a los hábitos arbóreos de la especie y a
la carencia de datos fósiles de los bosques lluviosos que parecen ser sus centros evolutivos. Potos
ha sido un linaje independiente desde aproximadamente el Mioceno (Simpson 1945). El género
parece ser originario de Centro o Suramerica (Hershkovitz 1972), o de Norte América con
posterior migración a Sur América durante el Pleistoceno (Patterson & Pacual, 1972).
Dieta
Kinkajou parece ser principalmente frugívoro, pero suplementa su dieta. Reportes de
observaciones alimenticias y análisis de contenido estomacal incluyen una variedad de pulpa de
frutas y semillas, flores, miel, pequeños escarabajos, algunas larvas de insectos, hojas y brotes
jóvenes.
Algunos Kinkajous fueron encontrados alimentandose del nectar de Quararibea cordata en Perú
ya que ellos visitan más de un árbol por noche, los Kinkajous contribuyen potencialmente a la
polinización cruzada. Su larga y delgada lengua sugiere una adaptación para una dieta frugívora,
para romper el cono y remover la miel de las colmenas y para capturar insectos y abejas sin
aguijón de la colmena. El Kinkajou tiene un intestino simple, sin ciego, por lo que al comer
grandes cantidades de fruta presenta una limitada absorción.

13
Oroaetus isidori es un predador de Kinkajous en Colombia . El aguila Arpia (Harpia harpyja)
fue observada trayendo un Kinkajou a su polluelo en dos ocasiones durante 4 meses de estudio.
Algunas veces se han reportado ser comidos por jaguares, el mayor predador de la especie son los
humanos, son vendidos como mascotas y su pelaje es comercializado. Mas de 100 animales
vivos por año, mas cientos de pieles (216 en 1966) fueron exportadas desde Perú solamente.
La destrucción de sus hábitats ha reducido el rango y número de estos individuos. Los Kinkajous
parecen preferir hábitats boscosos imperturbados, solo el 3% de 162 animales colectados en
Venezuela fueron encontrados en áreas abiertas como huertos o cultivos.
Los Kinkajous son estrictamente nocturnos y usualmente duermen en sitios oscuros y retirados
durante el dia.
De acuerdo con la Unión Internacional para la Conservación de la Naturaleza, esta especie se
encuentra clasificada en la categoría LC preocupación menor, el mapa de distribución para la
especie se observa a continuación:

14
Figura 2. Distribución geográfica de Potos flavus. Tomado de IUCN (International Union for
Conservation of Nature) 2008. Potos flavus. In: IUCN 2013. IUCN Red List of Threatened
Species. Version 2013.2 <www.iucnredlist.org
5. OBJETIVO
5.1 Objetivo general
Determinar la estructura genética y la filogenia molecular de P.flavus a través de su rango
de distribución en Colombia y en Suramérica, mediante marcadores mitocondriales.
5.2 Objetivos específicos
Reconstruir la filogeografía y las relaciones filogenéticas internas, de las subespecies
consideradas a partir de las secuencias de dos genes mitocondriales y determinar si existe
correspondencia entre los diferentes acervos genéticos encontrados y las subespecies
morfológicas definidas previamente por otros autores.
Determinar los niveles de variabilidad del gen mitocondrial NADH5 como un todo y en
cada una de los subpoblaciones analizadas de P. flavus.
Detectar los posibles cambios demográficos a través de la historia natural de P. flavus
(Expansiones demográficas, flujo génico, cuellos de botella entre otros).
Estimar posibles brechas de divergencia temporal para la especie y entre las subespecies
estudiadas y encontrar relaciones entre esas brechas temporales con los cambios
climáticos y geológicos.
Buscar en cuantos linajes evolutivamente significativos (ESU’s) quedan asignados los
individuos analizados en el seno de cada una de las subespecies estudiadas.

15
6. METODOLOGIA
6.1 Obtención de muestras y localidades
Las muestras fueron obtenidas por el profesor Manuel Ruíz García en una porción representativa
del rango de distribución de la especie. Las muestras consistían en pelos con bulbo, pieles,
dientes y huesos.
Se extrajeron, amplificaron y secuenciaron muestras de un total de 100 individuos identificados
morfológicamente y con un origen geográfico determinado. 99 de estas muestras fueron
analizadas para el gen mitocondrial NADH5 con una longitud de 280 pares de bases;
adicionalmente se obtuvieron las secuencias de 23 de estas muestras, más 1 individuo para el
también gen mitocondrial Citocromo Oxidasa-b (Cyt-b) de 426 pares de bases.
Adicionalmente se extrajo, amplificó y secuenció para NADH5 (280 pb) una muestra de la
especie Bassaricyon alleni, proveniente de Ecuador la cual fue utilizada como grupo externo en
los análisis de filogenia.
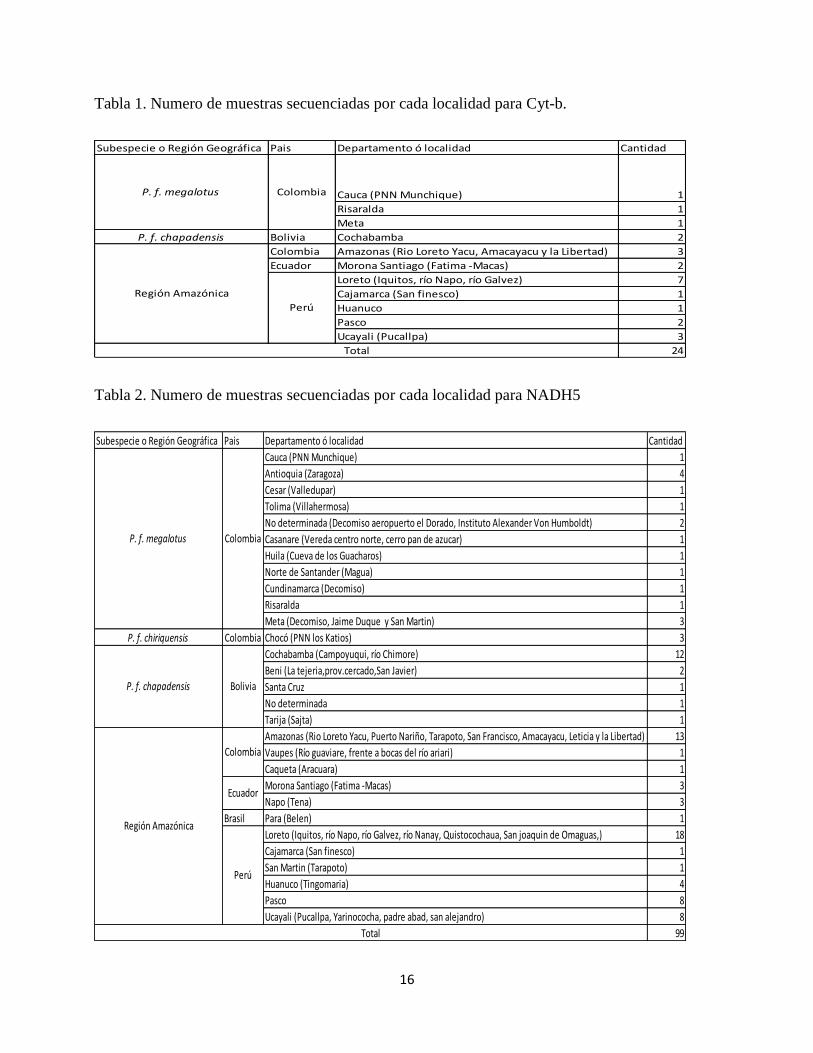
En la tabla 1 y 2 se muestra el origen geográfico de las muestras amplificadas para cada
marcador; de acuerdo con este origen se asignaron las posibles subespecies morfológicas
encontrando representación de tres (P. f. chiriquensis, P. f. megalotus y P. f. chapadensis) de las
ocho propuestas por Ford & Hoffmann (1988), los individuos muestreados en la Amazonia
Occidental que incluye localidades de cuatro países (Colombia, Ecuador, Perú y Brasil), no
fueron asignados a ninguna subespecie ya que no existe un morfotipo claro en la descripción
realizada por los precitados autores en el que puedan agruparse las muestras obtenidas de esta
región Amazónica. En la figura 3 pueden observarse las localidades muestreadas para los 100
individuos de Potos flavus.
16
Tabla 1. Numero de muestras secuenciadas por cada localidad para Cyt-b.
Tabla 2. Numero de muestras secuenciadas por cada localidad para NADH5
Subespecie o Región Geográfica Pais Departamento ó localidad Cantidad
Cauca (PNN Munchique) 1
Risaralda 1
Meta 1
P. f. chapadensis Bolivia Cochabamba 2
Colombia Amazonas (Rio Loreto Yacu, Amacayacu y la Libertad) 3
Ecuador Morona Santiago (Fatima -Macas) 2
Loreto (Iquitos, río Napo, río Galvez) 7
Cajamarca (San finesco) 1
Huanuco 1
Pasco 2
Ucayali (Pucallpa) 3
24Total
P. f. megalotus Colombia
Región Amazónica
Perú
Subespecie o Región Geográfica Pais Departamento ó localidad Cantidad
Cauca (PNN Munchique) 1
Antioquia (Zaragoza) 4
Cesar (Valledupar) 1
Tolima (Villahermosa) 1
No determinada (Decomiso aeropuerto el Dorado, Instituto Alexander Von Humboldt) 2
Casanare (Vereda centro norte, cerro pan de azucar) 1
Huila (Cueva de los Guacharos) 1
Norte de Santander (Magua) 1
Cundinamarca (Decomiso) 1
Risaralda 1
Meta (Decomiso, Jaime Duque y San Martin) 3
P. f. chiriquensis Colombia Chocó (PNN los Katios) 3
Cochabamba (Campoyuqui, río Chimore) 12
Beni (La tejeria,prov.cercado,San Javier) 2
Santa Cruz 1
No determinada 1
Tarija (Sajta) 1
Amazonas (Rio Loreto Yacu, Puerto Nariño, Tarapoto, San Francisco, Amacayacu, Leticia y la Libertad) 13
Vaupes (Río guaviare, frente a bocas del río ariari) 1
Caqueta (Aracuara) 1
Morona Santiago (Fatima -Macas) 3
Napo (Tena) 3
Brasil Para (Belen) 1
Loreto (Iquitos, río Napo, río Galvez, río Nanay, Quistocochaua, San joaquin de Omaguas,) 18
Cajamarca (San finesco) 1
San Martin (Tarapoto) 1
Huanuco (Tingomaria) 4
Pasco 8
Ucayali (Pucallpa, Yarinococha, padre abad, san alejandro) 8
99
P. f. megalotus Colombia
Región Amazónica
Perú
Total
P. f. chapadensis Bolivia
Colombia
Ecuador
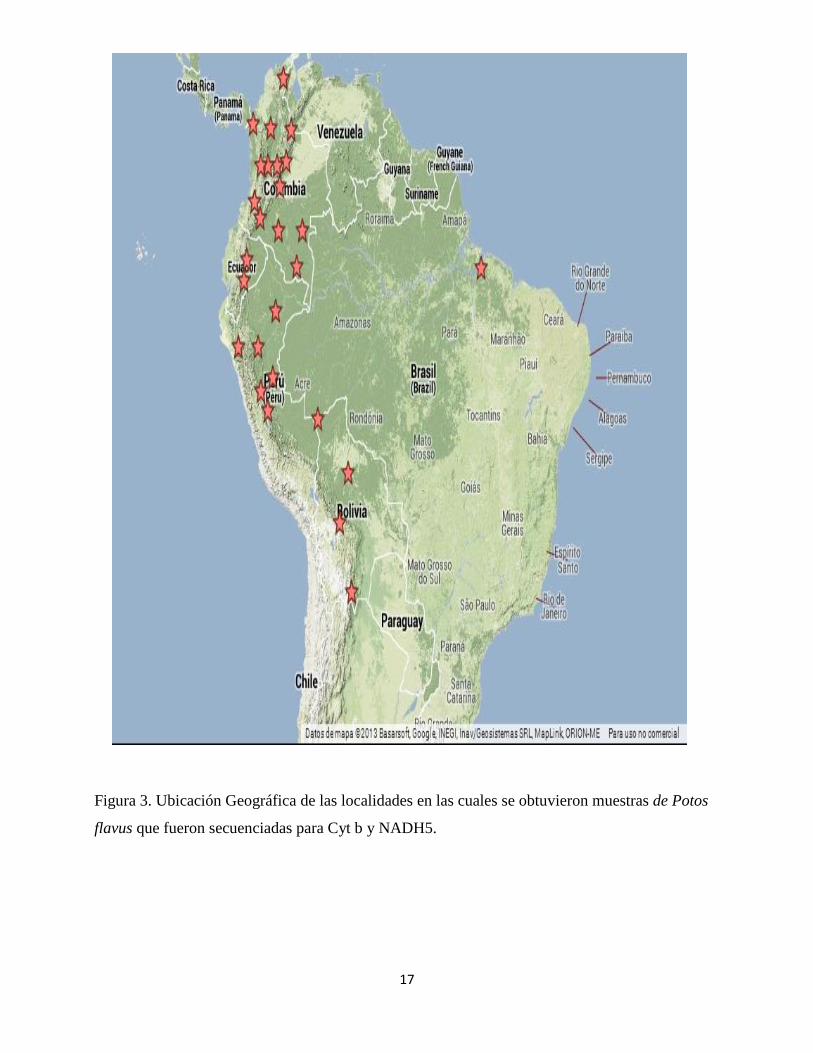
17
Figura 3. Ubicación Geográfica de las localidades en las cuales se obtuvieron muestras de Potos
flavus que fueron secuenciadas para Cyt b y NADH5.

18
6.2 Extracción, amplificación y secuenciación de ADN
El ADN fue extraído de trozos de piel, pulpa dentaria y médula de hueso cuando las muestras
provenían de animales muertos y de bulbo de pelo cuando los individuos se muestrearon vivos.
La preparación de los tejidos se realizó de la siguiente manera:
Piel: se cortó una muestra de aproximadamente 1cm², ésta fue picada y macerada usando 300μl
de buffer de lisis de piel (solución de 2.5 ml de Tris-HCL, 0,1 ml de EDTA, 1 ml de NaCl y 2ml
de SDS 10% en 44,4 ml de H2O), este macerado fue transferido a un tubo al que se agregaron
300μl adicionales de buffer, 10 μl de proteinasa-k [20 mg/ml] y 20 μl de ditiotreitol (DTT) y
finalmente fue llevado a incubación en baño de maría (56°C) durante dos noches.
Pulpa dentaria: se tomaron uno o dos dientes de cada individuo dependiendo de la cantidad de
pulpa dentaria que presentaran, los dientes se lavaron con hipoclorito y agua destilada y se
rompieron para extraer la pulpa interior, este tejido fue transferido a un tubo eppendorf.
Pelo: se tomaron pelos en los que se distinguiera fácilmente el bulbo y se lavaron con detergente
dodecilsulfato sódico (SDS) al 20% y 1 ml de agua destilada, de la siguiente manera:
Se incluyeron los pelos con el bulbo hacia abajo en un tubo eppendorf de 1.5 μl con 1 ml de agua
destilada, se agregaron 40 μl de SDS al 20%, se dió vortex a la muestra, se retiró el sobrenadante
y se agregó nuevamente agua destilada llevando al vórtex nuevamente. Este procedimiento fue
repetido hasta que se eliminara la espuma de la muestra, por último se realizó un lavado con 500
μl de Etanol absoluto, se dió vórtex, se descartó el sobrenadante y se dejó secar en horno por 1
minuto a 50 °C.
Medula de hueso: se realizó una limpieza previa del hueso con hipoclorito y agua destilada,
después, se rompieron los huesos para extraer la medula, se tomó una pequeña cantidad de este
tejido y se llevó a un tubo eppendorf.
La extracción del ADN se realizó para las muestras de piel con algunas variaciones de los
protocolos descritos en Sambrook et al. (1989).

19
Una vez la muestra de piel se encontraba preparada y digerida como se describió anteriormente,
se procedió a realizar la extracción utilizando fenol – cloroformo:
Se colocó en un tubo eppendorf la mayor cantidad de muestra digerida posible y se añadieron
500 μl de la solución fenol – cloroformo – alcohol - isoamílico (25:24:1), en el mismo volumen
de la muestra. Se realizó una mezcla suave por 20 segundos mediante inversión, se centrifugó a
13000 rpm por 15 minutos, se extrajó la mayor cantidad de sobrenadante y se añadió en un nuevo
tubo descartando el precipitado.
A ese sobrenadante se adicionó el mismo volumen de cloroformo – alcohol isoamilico (24:1) y se
mezcló suavemente por inversión. Se centrifugó a 13000 rpm por 15 minutos, en el caso en el
que algunas muestras después de este lavado presentaron en la interfase una capa blanquecina se
repitió el lavado con la solución cloroformo – alcohol isoamilico (24:1).
Sobre el volumen final obtenido se agregó dos veces el volumen de etanol al 100% y se dejó en
nevera a 20° C durante una noche. Al día siguiente la muestra fue centrifugada por 15 minutos a
13000 rpm, posteriormente se descartó el sobrenadante y se dejó secar el alcohol en el horno a
una temperatura de 40° C.
Finalmente el pellet se resuspende en agua Mili – Q, esteril y se guarda en nevera.
Para las muestras de pelo, medula de hueso y pulpa dentaria se añadieron 200 µl de una solución
de resina Chelex® al 20%, 20 µl de DTT y 10 µl de Proteinasa – K [20 mg/ml], a cada una de
las muestras de los tejidos antes mencionados. Posteriormente las muestras fueron dejadas por
una noche en baño maría (56°C), el día siguiente se llevaron a temperatura de ebullición durante
8 minutos. Finalmente fueron centrifugadas y el sobrenadante fue utilizado para protocolo de
PCR.
Para la amplificación del segmento de los dos genes mitocondriales se utilizó la Reacción en
Cadena de la Polimerasa, PCR. Para cada muestra se preparó un mix que contenía 2 μL de DNA
(diluido o no según el caso), 13,5μL de agua Mili-Q, 1 μL de genTaq® polimerasa, 1 μL de
DNTP’s, 3,0 μL de MgCl2, 1 μL de cada cebador, (para Citocromo-b se utilizaron los cebadores
de Irwin et al. (1991) con algunas modificaciones, H15149 y L14724, 5’-

20
CAGAATGATATTTGTCCTCA-3’ y 5´-GATATGAAAAACCATCGTTG-3’, y para NADH5
los cebadores utilizados fueron L12673 y H12977, 5’ GGTGCAACTCCAAATAAAAGTA -3’ y
5´- AGAATTCTATGATGGATCATGT 3’ propuestos por Waits et al. (1999), 2,5 μL de buffer
10X; el producto siguió un protocolo de PCR de denaturación a 94°C durante 5 minutos, 35
ciclos a 94°C por 1 minuto, 35 ciclos a 50°C por 30 segundos, 35 ciclos a 72°C por 1 minuto, y
una extensión final de 72°C por 5 minutos.
Todas las amplificaciones incluyendo controles positivos y negativos fueron chequeados en gel
de agarosa al 2%. Las muestras que presentaron una banda nítida con la longitud esperada,
fueron enviadas para secuenciación automática de la cadena “L” a MACROGEN U.S.A.
Este tipo de secuenciación ha sido realizada de esta manera en diversas especies estudiadas en el
laboratorio de Genética de poblaciones de la Pontificia Universidad Javeriana para los dos genes
objetos de análisis en este estudio. Las secuencias obtenidas para otros carnívoros tales como
Eira barbara, Tremarctos ornatus, Puma concolor, Panthera onca, varias especies del genero
Pseudalopex entre otros, han sido secuencias confiables una vez verificada la calidad de las
mismas comparando ambas cadenas, lo cual permite confiar en los análisis obtenidos con este
tipo de secuencias.
Por otra parte las muestras fueron alineadas manualmente y chequedas con los cromatogramas
enviados por MACROGEN U.S.A (pueden verse esas figuras en el Anexo 1), de esta manera se
verifica una vez más la calidad de las secuencias, tomando para el análisis solo las muestras que
presentaran picos estables y una secuencia coherente.
De igual manera para tener certeza adicional sobre la secuencia obtenida se enviaron las
amplificaciones del mismo individuo a partir de varios tejidos (ejemplo piel y diente).
Las secuencias que no cumplieron con la longitud o coherencia esperada para las secuencias
fueron excluidas del análisis.
6.3 Alineamiento de secuencias de ADN mitocondrial
Las secuencias fueron alineadas manualmente y con el Software DNA Alignment (Fluxus
Technology Ltd).

21
6.4 Análisis filogenéticos
Los análisis filogenéticos fueron realizados usando los programas MEGA versión 5.2.2 (Tamura
et al. 2011) y BEAST v. 1.6.2 (Drummond & Rambaut, 2007), en este último programa además
de las relaciones filogenéticas se determinaron los tiempos de divergencia entre las diferentes
poblaciones analizadas y entre las secuencias obtenidas para Potos y las especies utilizadas como
grupos externos, los carnívoros: Bassaricyon alleni (Procyonidae) para las secuencias de NADH5
y Pseudalopex culpaeus (Canidae) para Citocromo b (Cyt b).
Inicialmente se escogió el mejor modelo de substitución nucleotídica para las secuencias
obtenidas entre 24 modelos diferentes (Nei & Kumar, 2000). Se eligió el modelo que presentara
los valores más bajos para el parámetro “Akaike” Criterio de Información, corregido, AICc.
Se construyeron arboles utilizando tres metodologías para cada gen mitocondrial, por métodos
basados en distancias (Neighbour-Joinig), métodos de máxima verosimilitud (Maximum-
Likelihood) y por métodos bayesianos.
Primero, mediante el software MEGA 5.2.2, se construyeron árboles basados en el método
Neighbour-Joining (Saitou & Nei M. 1987). Los valores de bootstrap se determinaron con 500
réplicas, las distancias evolutivas fueron computarizadas usando el método parámetro-3 de
Tamura (Tamura K. 1992).
El segundo grupo de árboles fue obtenido utilizando el mismo software, siguiendo el método de
Maximum-Likelihood y el modelo GTR (Nei & Kumar, 2000) para NADH5 y el modelo HKY
(Hasegawa et al. 1985) para Cyt b. Los valores de bootstrap se determinaron con 500 réplicas.
Finalmente se construyeron arboles con procedimientos bayesianos llevados a cabo con el
programa BEAST v. 1.6.2 (Drummond & Rambaut 2007), estos análisis fueron realizados usando
el Hasegawa – Kishino – Yano (HKY), como modelo de substitución nucleotídica, con base en
las frecuencias estimadas con un modelo de heterogeneidad de sitios gamma y tasas que varían
entre los sitios. Los análisis fueron corridos asumiendo un reloj molecular relajado con una tasa
log-normal no-correlacionada (Drummond et al. 2006). El tamaño efectivo de la muestra (ESS)
para las estimaciones de los parámetros y la convergencia se chequeo utilizando el programa

22
Tracer v 1.5 (Rambaut & Drummond, 2007). Las estimaciones de las divergencias temporales y
los intervalos superiores e inferiores al 95 % de las densidades posteriores mayores (HPD) de
estos parámetros al igual que las medias, las medias geométricas, medianas, densidades
marginales y trazas fueron también estimados con el programa Tracer v 1.5. Para determinar los
valores reales de estos parámetros, se obtuvó el árbol de autocorrelación (ACT) y ESS para las
estimaciones de los parámetros. El árbol final se estimó en TreeAnnotator v1.6.2 y visualizado en
FigTree v. 1.3.1. Adicionalmente, este programa fue corrido para estimar el tiempo del más
reciente ancestro común (TMRCA) para los diferentes linajes de haplotipos encontrados. Este
valor se tomó como un prior para el “treeModel.rootHeight” en el análisis Bayesiano con una
distribución normal y una desviación estándar. El prior utilizado en el análisis realizado para las
secuencias de NADH5 fue de 21 MA + 0.5 MA, teniendo en cuenta que Koepfli et al. (2007),
estimó que la división entre Potos y los demás prociónidos ocurrió hace 21.6 – 24 MA (95%
densidades posteriores mayores (HPD): 12.1–36 MA). Por otra parte para el conjunto de datos
obtenidos de Cyt b se utilizó un prior de 41.2 MA que es el tiempo estimado para la separación
de Canidae y Procyonidae, esto teniendo en cuenta que la especie utilizada como grupo externo
es un zorro (Pseudalopex culpaeus) (Bininda - Emonds et al. 1999).
6.5 Análisis de diversidad, heterogeneidad y flujo génico
Estos análisis se realizaron para las secuencias obtenidas para el gen mitocondrial NADH5, con
el software DNAsp 5.10 (Rozas et al. 2010), se determinaron el número de sitios polimórficos
(S), el número de haplotipos (h), la diversidad Haplotípica (Hd), la diversidad nucleotídica (π), el
número promedio de diferencias nucleotídicas (k), y el coeficiente de coancestralidad (θ) por
secuencia para determinar la diversidad genética en P. flavus. Inicialmente se tomaron estas
estimaciones para la totalidad de las muestras (n=99), después se dividieron en cuatro grupos
teniendo en cuenta la distribución geográfica y las posibles subespecies asignadas (P. f.
megalotus (n=15), P. f. chiriquensis (n=3), P. f. chapadensis (17), Región Amazónica (n=64)) y
se realizó el mismo análisis para cada población por separado.
Se utilizaron diversas pruebas para medir la heterogeneidad genética y determinar estimas de
posible flujo génico entre las supuestas subespecies: HST, HST, KST, KST*, Z and Z* (Hudson et
al. 1992a, b), Snn (Hudson 2000), X2 (en las frecuencias haplotipicas con pruebas de permutación

23
de 10.000 réplicas), GST (a partir de las frecuencias haplotipicas) y, γST, NST, FST (a partir de las
secuencias de los nucleótidos; Hudson et al. 1992a).
6.6 Detección de cambios demográficos
Para detectar la existencia de cambios demográficos se utilizaron diversos procedimientos.
1. Distribución de desemparejamientos (Mismatch distribution), fue obtenida siguiendo el
método de Rogers & Harpending (1992) and Rogers et al., (1996). Se compararon las curvas
obtenidas asumiendo tamaños constantes y no constantes para la distribución empírica observada.
Se usó el estadístico “raggedness” rg (Harpending et al. 1993, Harpending 1994) y el estadístico
R2 de Ramos-Onsins & Rozas (2002) para determinar la similitud entre las curvas observadas y
las teóricas. Estas gráficas fueron obtenidas analizando el conjunto de secuencias en general y
posteriormente para tres de los grupos construidos a partir de la ubicación geográfica y posibles
subespecies, en esta ocasión los individuos provenientes del Parque Nacional los Katios en
Colombia (3) que fueron clasificados en los análisis anteriores como P. f. chiriquensis, se
incluyeron dentro del grupo colombiano de P. f. megalotus, de esta manera se evaluaron posibles
fenómenos de expansión o contracción poblacional para las siguientes subpoblaciones: P. f.
megalotus (n=18), P. f. chapadensis (n= 17) y región Amazónica Occidental (n= 64).
2. Tests Fu and Li D* and F* (Fu & Li 1993), el estadístico Fu FS (Fu, 1997) y el test Tajima D
(Tajima 1989) para determinar posibles cambios en los tamaños poblacionales (Simonsen et al.
1995, Ramos-Onsins & Rozas 2002). Estos estadísticos se calcularon para las secuencias
obtenidas de NADH5.
3. “Bayesian skyline plot” (BSP) fue obtenido para las secuencias del Cyt b y NADH5 por medio
de los software BEAST v. 1.6.2 y Tracer v1.5. Los priores de los árboles fueron seleccionado con
cinco pasos y un modelo skyline por tramos constante con 10.000.000 generaciones (el primer
1,000,000 fue descartado como “burn-in”). Se seleccionó como el tiempo máximo que el 95%
superior de HPD (densidades posteriores mayores) y la traza de la altura de la raíz como la
treeModel.rootHeigh. Estos análisis se llevaron a cabo para los últimos 1,15 MA para NADH5, y
1,3 MA para Cyt b.

24
6.7 Tiempos de divergencia
Para calcular el tiempo de divergencia entre las diferentes subpoblaciones de Potos y los grupos
externos se utilizó el método bayesiano descrito anteriormente en los análisis filogenéticos.
Adicionalmente se calculó el tiempo de divergencia entre los haplotipos encontrados para los dos
genes mitocondriales, por medio de Median Joining Network (MJ) (Bandelt et al. 1999). Esta red
fue construida por medio del software Network 4.6 (Fluxus Technology Ltd). Adicionalmente, el
estadístico ρ fue estimado y transformado en años (Morral et al. 1994). La desviación estándar de
ρ también fue calculada (Saillard et al. 2000). El estadístico ρ es imparcial y altamente
independiente de los acontecimientos demográficos pasados. Las tasas de mutación utilizadas
fueron para Cyt b una divergencia de nucleótidos del 1,55% por cada millón de años (Wayne et
al. 1997), lo cual representa una mutación cada 153.976 años y para NADH5, una divergencia
nucleotidica de 1.22% por cada millón de años (Culver et al. 2000), que representa una mutación
cada 309.310 años. Estas estimaciones fueron obtenidas para Canidae y Felidae, respectivamente.
Se asume entonces en este trabajo que estas tasas de mutación pueden ser similares para
Procyonidae.
7. RESULTADOS
De todas las amplificaciones obtenidas y enviadas a secuenciar, 99 muestras de 280 pares de
bases del gen mitocondrial NADH5 y 24 secuencias para el caso del gen mitocondrial Cyt b, con
una longitud de 426 pares de bases, mostraron unas secuencias confiables para ser incluidas en el
análisis del presente trabajo de investigación.
Para evaluar la calidad de las secuencias se tuvieron en cuenta los cromatogramas remitidos para
cada secuencia por MACRO GEN USA, el envío de más de un amplificado para un mismo
individuo provenientes de diferentes tejidos que al final resultaron en secuencias idénticas y la
alineación manual de las mismas que permitió detectar posibles inconsistencias.
7.1 Análisis de filogenia
Se determinó que el mejor modelo mutacional entre 24 modelos evolutivos probados para las
secuencias de Cyt B incluyendo la secuencia del grupo externo, fue el modelo de substitución

25
nucleotídica Hasegawa-Kishino-Yano (HKY), con un valor de “Akaike” Criterio de Información,
corregido, AICc, de 2706.703, mientras para NADH5 y el grupo externo el mejor modelo fue el
de General Time Reversible (GTR), con un valor de AICc de 10074.127.
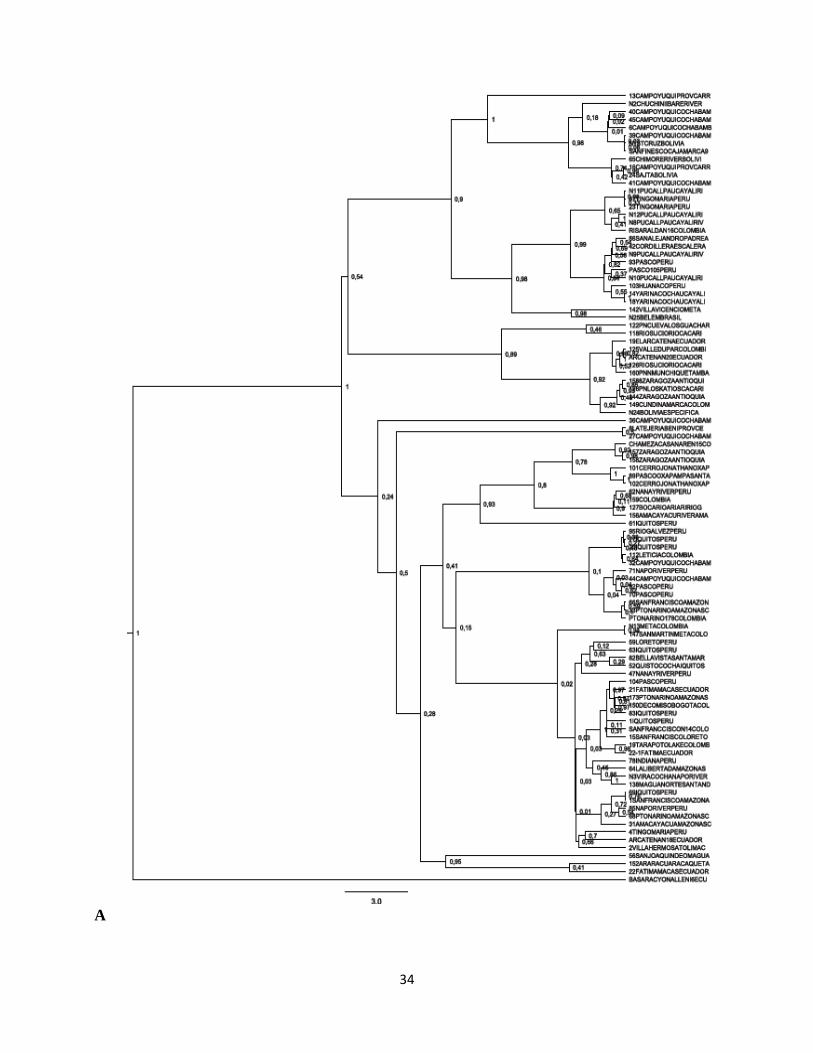
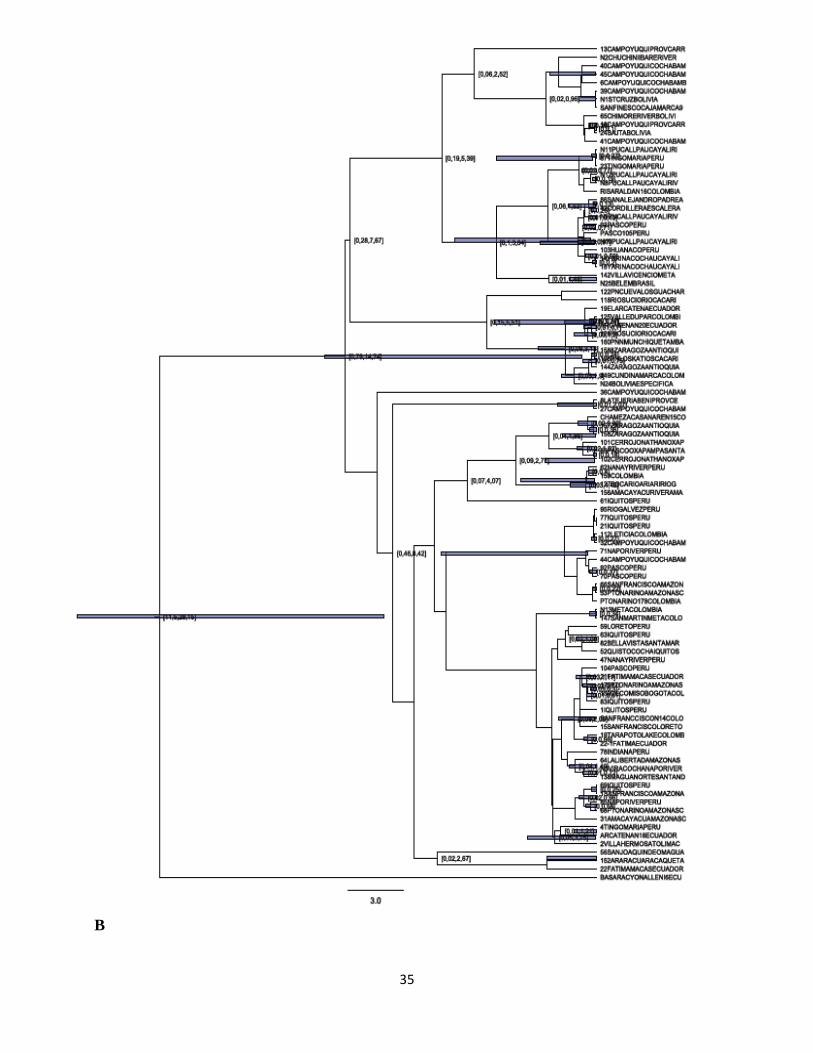
De acuerdo con los árboles obtenidos se observa que el género Potos es monofiletico. Las
gráficas construidas a partir de los métodos de máxima verosimilitud y algoritmo Neighbour-
Joining, para el caso de las secuencias analizadas para el gen mitocondrial Cyt-b (n=24),
muestran una topología muy similar figura 4 (A y B) tal como se describe a continuación; se
forman 2 grupos principales soportados por diferentes valores de boostrap para cada árbol, pero
incluyendo a los mismos individuos para cada uno de los grupos conformados; el primer clado
está constituido por individuos de Perú, Colombia y Ecuador todos muestreados en la región
amazónica con excepción de un individuo (identificado N13 Jaime Duque) a quien se le señaló
para efectos del presente estudio como origen geográfico la región oriental de Colombia. Sin
embargo, por la manera como se agrupa en estos análisis preliminares y como se agrupa en la red
de haplotipos tal como se mencionará más adelante, de acuerdo con sus características genéticas
este parece ser un individuo proveniente de la región amazónica. Los trece individuos reunidos
dentro de este primer grupo, de acuerdo con los valores obtenidos de bootstrap, no parecen estar
relacionados de una manera específica. Sin embargo, en los dos árboles si se observa que dentro
de este grupo hay una muestra que se diferencia más que las demás.
Esta secuencia se obtuvo a partir de un individuo muestreado en el departamento de Cajamarca
en Perú. En el segundo grupo aparecen 11 individuos provenientes de Colombia, Perú y Bolivia;
dentro de este grupo, para los dos árboles se presenta una asociación soportada con unos valores
de boostrap altos (97 y 92) entre dos muestras provenientes de la región de Cochabamba en
Bolivia. Las otras muestras se encuentran mezcladas sin asociación aparente para estos dos
árboles, entre estas se encuentran individuos provenientes de la región amazónica de Colombia y
Perú y muestras de la zona andina colombiana (Cauca y Risaralda).
Se obtuvieron dos árboles más, construidos con las mismas metodologías (máxima verosimilitud
y Neighbour-Joining), para el gen NADH5 con un número mucho mayor de muestras. Estos
pueden observarse en la figura 5 (A y B) y de manera general presentan las siguientes
características: en los dos árboles se observa que la mayoría de las muestras no tiene una

26
conformación en grupos fuertemente soportados. Se encuentran mezclados individuos de todas
las localidades para todos los países muestreados. Sin embargo los dos casos muestran 2
agrupaciones, uno de individuos peruanos y otro de bolivianos que se resaltan entre la mezcla de
las muestras restantes. Estos grupos son, en el árbol construido con la metodología de máxima
verosimilitud, un conjunto de 12 individuos más dos muestras que no se encuentran dentro del
mismo grupo pero si están muy cercanas a estos, 13 individuos provienen de Bolivia
(Cochabamba, Beni, Tarija y Santa Cruz) y un individuo proveniente de Cajamarca, Perú. El
segundo grupo incluye 14 muestras peruanas de los departamentos de Pasco, Ucayali, Huanuco y
San Martin y una muestra colombiana proveniente del departamento de Risaralda, para un total
de 15. El árbol construido con el algoritmo Neighbour-Joining también realiza estas dos
agrupaciones. Para el caso del grupo boliviano se encuentra representado por las mismas 12
muestras del árbol anterior incluyendo un individuo peruano, pero en este caso las 2 muestras
también de origen boliviano que se encontraron muy próximas en el árbol anterior quedaron muy
distantes en este árbol. Por su parte el grupo peruano también se encontró conformado por las 15
muestras representadas en el árbol anterior incluyendo la muestra colombiana.
Con respecto a la muestra (N13 Jaime Duque) mencionada en la primer parte de los resultados,
para los arboles construidos con las secuencias de NADH5, en ambos casos se encontró
estrechamente relacionada con una muestra proveniente de San Martin Meta. Sin embargo esta
relación no está soportada por valores significativos de boostrap.
Por otra parte dos individuos secuenciados para NADH5 de los cuales no se contaba con
localidad geográfica exacta, (POTOSFLAVUS150DECOMISOBOGOTACOLOMBIA,
POTOSFLAVUS159COLOMBIA), se mostraron relacionados en los dos árboles con individuos
provenientes de la región amazónica. Sin embargo, estas relaciones no se encuentran soportadas
con valores altos de boostrap.
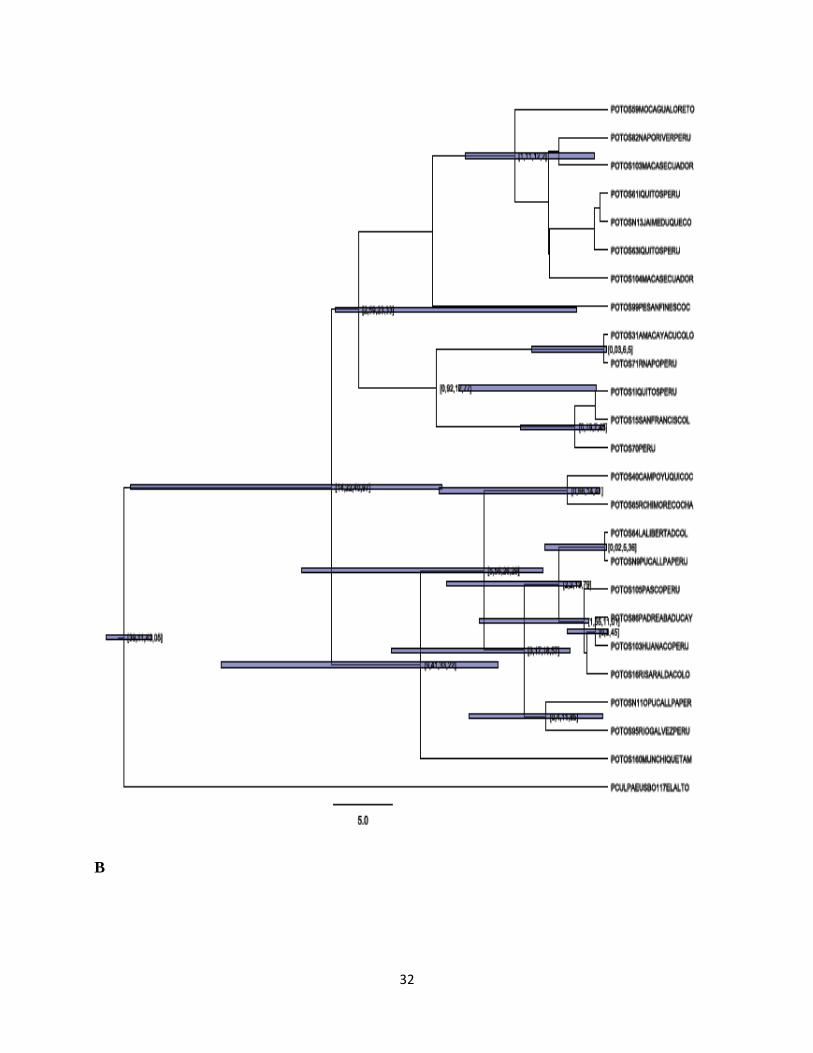
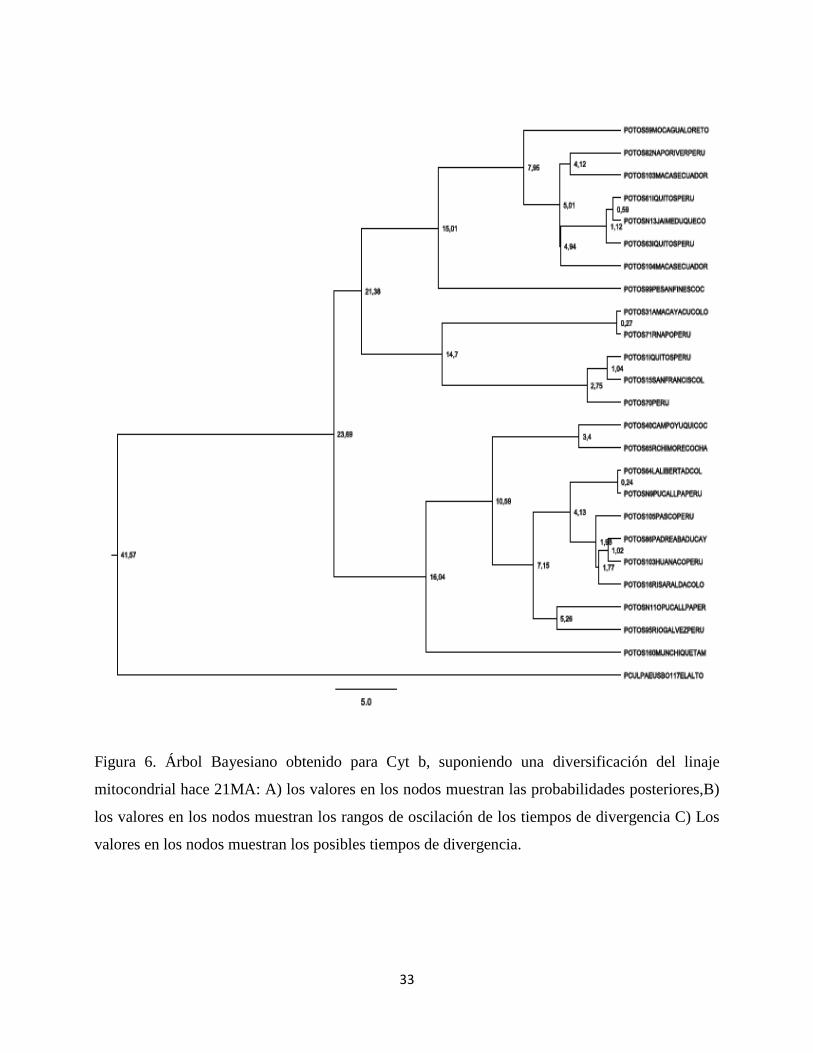
Para los arboles construidos con métodos bayesianos se obtuvieron 3 gráficas para cada árbol:
una muestra las probabilidades posteriores, otra muestra los tiempos de divergencia y otra los
intervalos entre estos tiempos.
Para el primer grupo de árboles con secuencias de Cyt b (Figura 6, A, B Y C) se observa una
tipología muy similar a la encontrada en los arboles construidos con métodos de distancias

27
genéticas. En general, se evidencia una división en dos grandes grupos, soportados fuertemente
con una probalidad igual a 1. A su vez, la organización de los individuos dentro de cada grupo se
soporta con probabilidades del 0,98 y 0,56; dentro del primer grupo se encuentran mezcladas
entre sí muestras de Ecuador, Perú y Colombia incluyendo la muestra (N13 Jaime Duque). El
segundo grupo se caracteriza por tener muestras peruanas y colombianas entremezcladas. Así
mismo, se encontró nuevamente una estrecha relación soportada por una por una probabilidad de
1 entre las dos muestras bolivianas.
Con respecto a los arboles construidos con NADH5, se encuentran que los dos grupos
observados anteriormente en los arboles basados en distancias genéticas, conformados por
individuos de una parte de las subpoblaciones peruanas y bolivianas están soportados con valores
altos de probabilidades posteriores. El primer grupo reúne con una probabilidad igual a 1, 11
individuos bolivianos y 1 peruano; el grupo peruano por su parte con una probabilidad del 0,99
agrupa los 14 individuos peruanos más 1 muestra colombiana de manera idéntica a lo descrito
para los arboles basados en distancias genéticas. En este caso la muestra (N13 Jaime Duque), se
encontró relacionada a un individuo proveniente de San Martin, Meta en Colombia con una
probabilidad de 0,94.
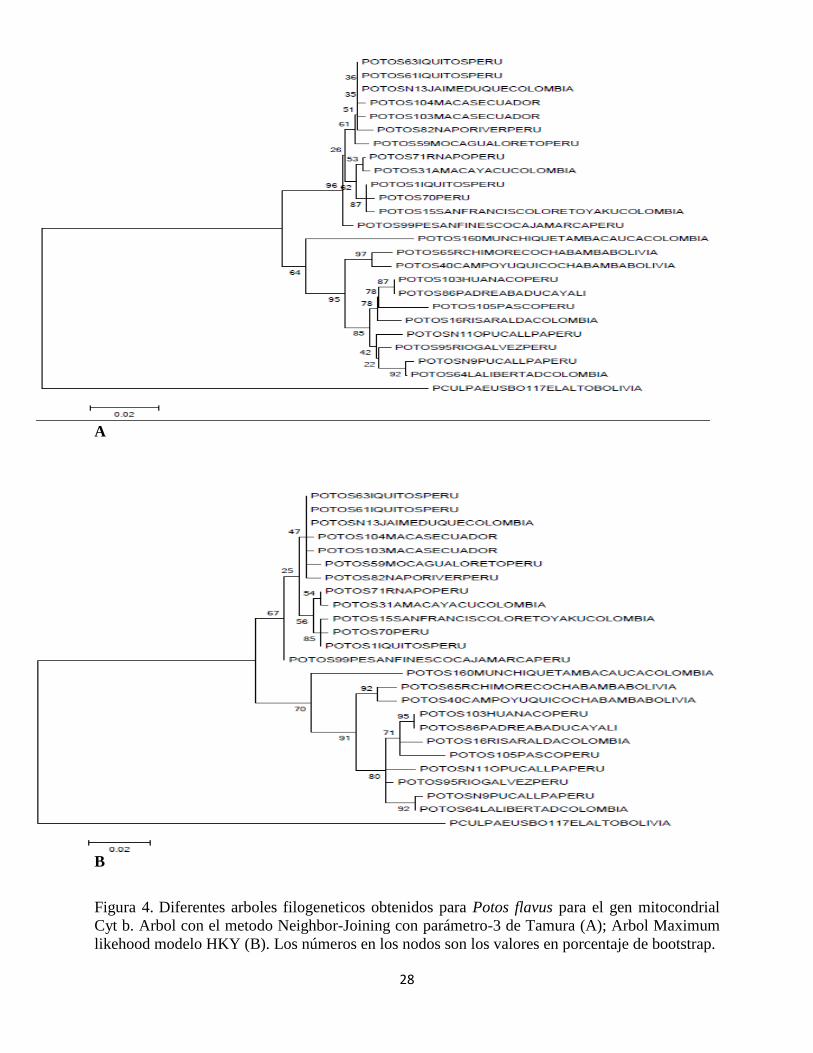
28
A
B
Figura 4. Diferentes arboles filogeneticos obtenidos para Potos flavus para el gen mitocondrial
Cyt b. Arbol con el metodo Neighbor-Joining con parámetro-3 de Tamura (A); Arbol Maximum
likehood modelo HKY (B). Los números en los nodos son los valores en porcentaje de bootstrap.
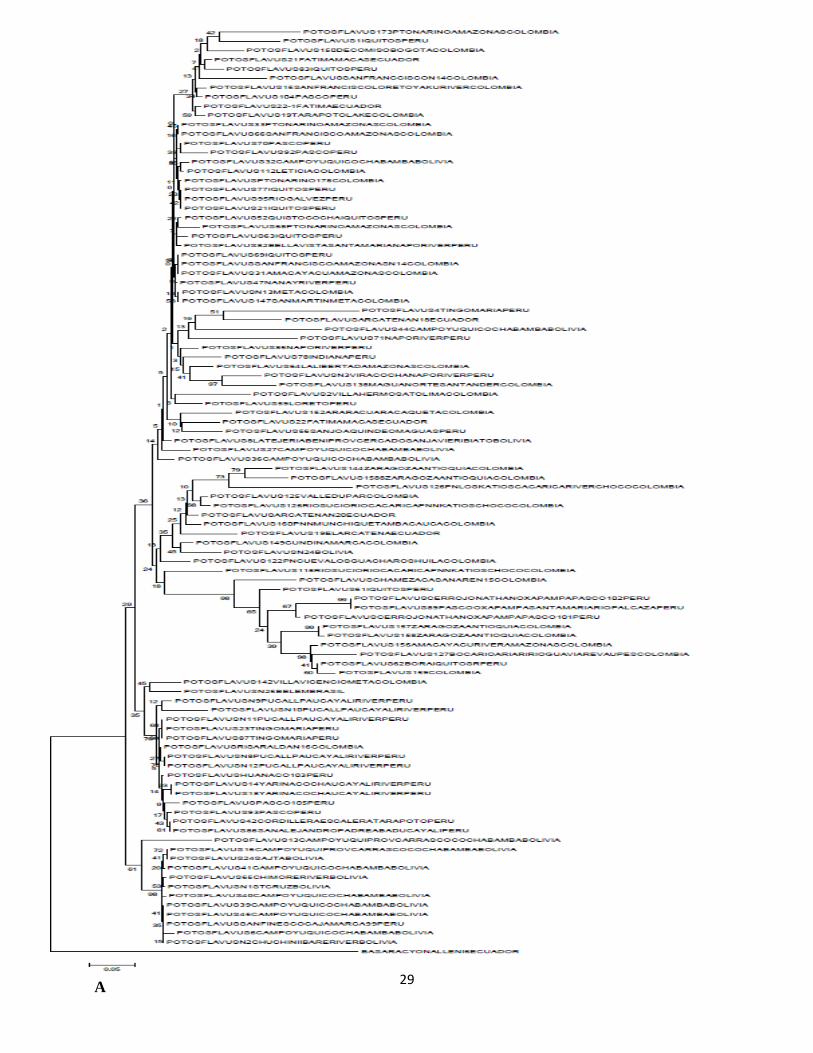
29 A

30
B

31
Figura 5. Diferentes arboles filogeneticos obtenidos para Potos flavus para el gen mitocondrial
NADH5. Arbol con el metodo Neighbor-Joining con parámetro-3 de Tamura (A); Árbol
Maximum likehood modelo GTR (B). Los numeros en los nodos son los valores en porcentaje de
bootstrap.
A
32
B
33
Figura 6. Árbol Bayesiano obtenido para Cyt b, suponiendo una diversificación del linaje
mitocondrial hace 21MA: A) los valores en los nodos muestran las probabilidades posteriores,B)
los valores en los nodos muestran los rangos de oscilación de los tiempos de divergencia C) Los
valores en los nodos muestran los posibles tiempos de divergencia.
34
A
35
B

36
C
37
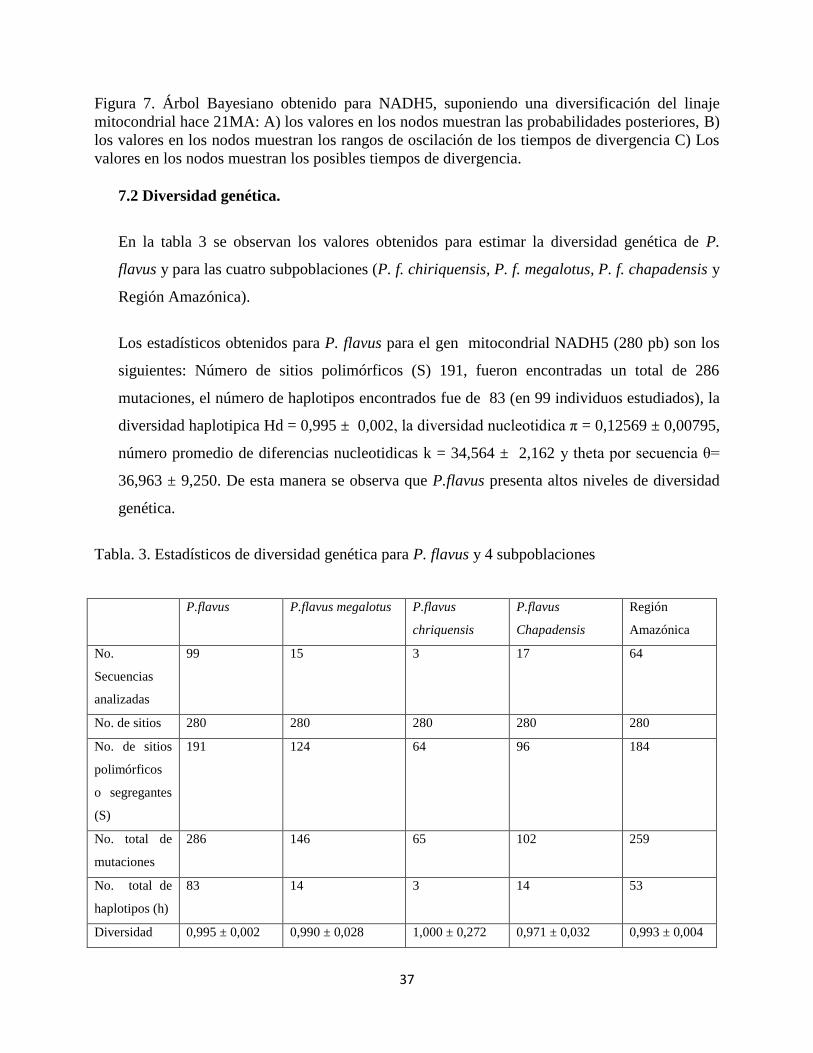
Figura 7. Árbol Bayesiano obtenido para NADH5, suponiendo una diversificación del linaje
mitocondrial hace 21MA: A) los valores en los nodos muestran las probabilidades posteriores, B)
los valores en los nodos muestran los rangos de oscilación de los tiempos de divergencia C) Los
valores en los nodos muestran los posibles tiempos de divergencia.
7.2 Diversidad genética.
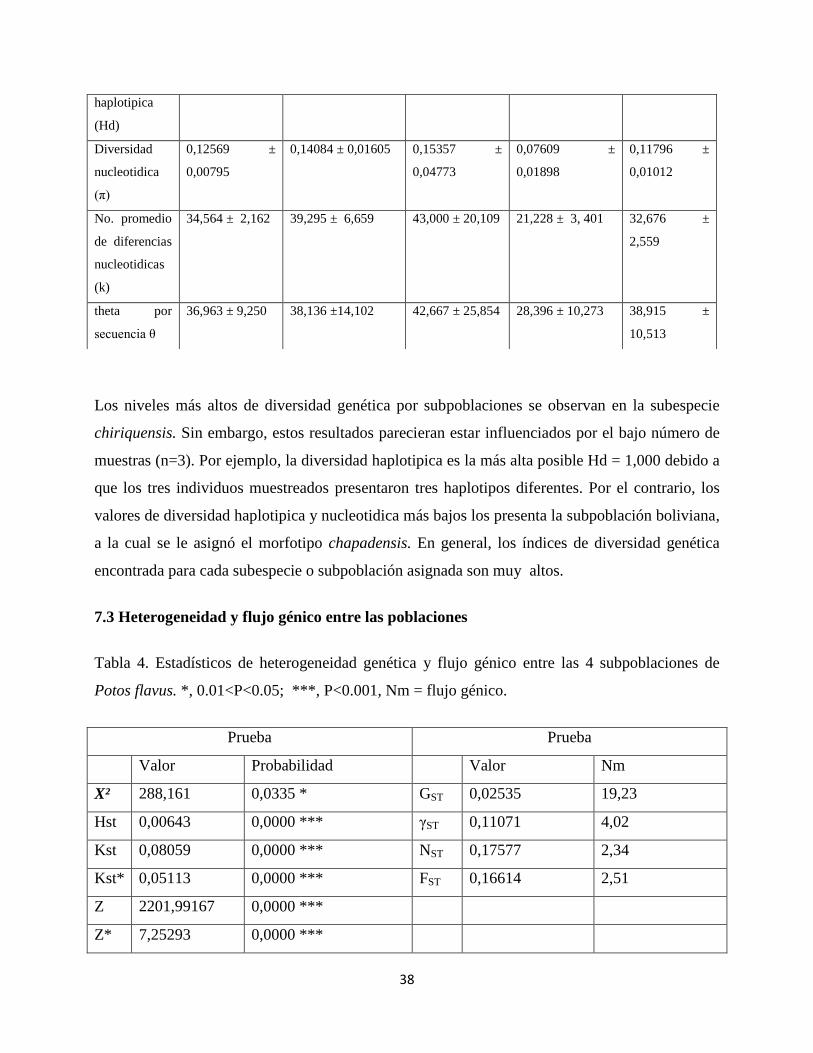
En la tabla 3 se observan los valores obtenidos para estimar la diversidad genética de P.
flavus y para las cuatro subpoblaciones (P. f. chiriquensis, P. f. megalotus, P. f. chapadensis y
Región Amazónica).
Los estadísticos obtenidos para P. flavus para el gen mitocondrial NADH5 (280 pb) son los
siguientes: Número de sitios polimórficos (S) 191, fueron encontradas un total de 286
mutaciones, el número de haplotipos encontrados fue de 83 (en 99 individuos estudiados), la
diversidad haplotipica Hd = 0,995 ± 0,002, la diversidad nucleotidica π = 0,12569 ± 0,00795,
número promedio de diferencias nucleotidicas k = 34,564 ± 2,162 y theta por secuencia θ=
36,963 ± 9,250. De esta manera se observa que P.flavus presenta altos niveles de diversidad
genética.
Tabla. 3. Estadísticos de diversidad genética para P. flavus y 4 subpoblaciones
P.flavus P.flavus megalotus P.flavus
chriquensis
P.flavus
Chapadensis
Región
Amazónica
No.
Secuencias
analizadas
99 15 3 17 64
No. de sitios 280 280 280 280 280
No. de sitios
polimórficos
o segregantes
(S)
191 124 64 96 184
No. total de
mutaciones
286 146 65 102 259
No. total de
haplotipos (h)
83 14 3 14 53
Diversidad 0,995 ± 0,002 0,990 ± 0,028 1,000 ± 0,272 0,971 ± 0,032 0,993 ± 0,004
38
Los niveles más altos de diversidad genética por subpoblaciones se observan en la subespecie
chiriquensis. Sin embargo, estos resultados parecieran estar influenciados por el bajo número de
muestras (n=3). Por ejemplo, la diversidad haplotipica es la más alta posible Hd = 1,000 debido a
que los tres individuos muestreados presentaron tres haplotipos diferentes. Por el contrario, los
valores de diversidad haplotipica y nucleotidica más bajos los presenta la subpoblación boliviana,
a la cual se le asignó el morfotipo chapadensis. En general, los índices de diversidad genética
encontrada para cada subespecie o subpoblación asignada son muy altos.
7.3 Heterogeneidad y flujo génico entre las poblaciones
Tabla 4. Estadísticos de heterogeneidad genética y flujo génico entre las 4 subpoblaciones de
Potos flavus. *, 0.01<P<0.05; ***, P<0.001, Nm = flujo génico.
Prueba Prueba
Valor Probabilidad Valor Nm
X² 288,161 0,0335 * GST 0,02535 19,23
Hst 0,00643 0,0000 *** γST 0,11071 4,02
Kst 0,08059 0,0000 *** NST 0,17577 2,34
Kst* 0,05113 0,0000 *** FST 0,16614 2,51
Z 2201,99167 0,0000 ***
Z* 7,25293 0,0000 ***
haplotipica
(Hd)
Diversidad
nucleotidica
(π)
0,12569 ±
0,00795
0,14084 ± 0,01605 0,15357 ±
0,04773
0,07609 ±
0,01898
0,11796 ±
0,01012
No. promedio
de diferencias
nucleotidicas
(k)
34,564 ± 2,162 39,295 ± 6,659 43,000 ± 20,109 21,228 ± 3, 401 32,676 ±
2,559
theta por
secuencia θ
36,963 ± 9,250 38,136 ±14,102 42,667 ± 25,854 28,396 ± 10,273 38,915 ±
10,513

39
Snn 0,80325 0,0000 ***
Antes de describir los resultados encontrados, se aclara que los valores de flujo génico que se
tomaron en cuenta fueron los suministrados por los estadísticos (γST, NST y FST), ya que el valor
de flujo génico estimado con el coeficiente GST está basado en diferencias entre haplotipos y no
entre secuencias por lo cual los valores calculados por los otros métodos son más precisos.
Tal como se observa en la tabla anterior, todas las pruebas de heterogeneidad presentaron valores
significativos. Sin embargo, cuando se observan los valores de flujo génico (Nm) que resultaron
ser valores altos y teniendo en cuenta que Nm > 1 indica homogeneidad genética, se puede
deducir que las diferencias significativas que muestran las diversas pruebas realizadas, se deben a
que el tamaño de la muestra (n=99) es grande por lo tanto los estadísticos utilizados se hacen más
sensibles para detectar cualquier cambio que ocurra por más pequeño que este sea. Esta hipótesis
que indica que existen diferencias significativas pero que estas diferencias solo las están
aportando una pequeña parte de la muestra ya que el flujo génico entre las poblaciones es alta, se
corrobora cuando se excluye la subpoblación Boliviana tal como se explica mas adelante.
Por otra parte cuando se compara la diferenciación genética por pares de subespecies los
resultados son los siguientes:
Tabla 5. Estadísticos de heterogeneidad genética (KXY, GST, ΔST, γST, NST, FST y Da) por pares
de subespecies.
Población 1 Población 2 Kxy GST ΔST γST NST FST Da
Megalotus Chiriquensis 41,24445 0,04377 0,00957 0,07097 0,03637 0,03029 0,00454
Megalotus Chapadensis 37,43137 0,00992 0,01731 0,14810 0,20780 0,20452 0,02784
Megalotus REGIÓN_AMAZÓNICA 37,69167 0,01036 0,00422 0,03418 0,06362 0,05938 0,00814
Chiriquensis Chapadensis 45,54902 0,04933 0,02009 0,20716 0,32868 0,30959 0,05128
Chiriquensis REGIÓN_AMAZÓNICA 45,39063 0,06770 0,00481 0,03984 0,20022 0,18210 0,03006
Chapadensis REGIÓN_AMAZÓNICA 33,38511 0,01131 0,00887 0,07632 0,19388 0,19408 0,02356
Cuando se observan los valores de FST y se tiene en cuenta lo siguiente: valores FST de 0,05 a
0,15 indican poca diferenciación genética, de 0,15 a 0,25, diferenciación genética grande, > 0,25
diferenciación genética muy grande, se concluye que se presenta heterogeneidad genética grande
(0,204) entre las poblaciones de megalotus y chapadensis, entre las poblaciones de chiriquensis y
Región Amazónica (0,182) y entre chapadensis y Región Amazónica Por su parte, la
diferenciación genética entre chiriquensis y chapadensis es muy grande (0,309). No existe
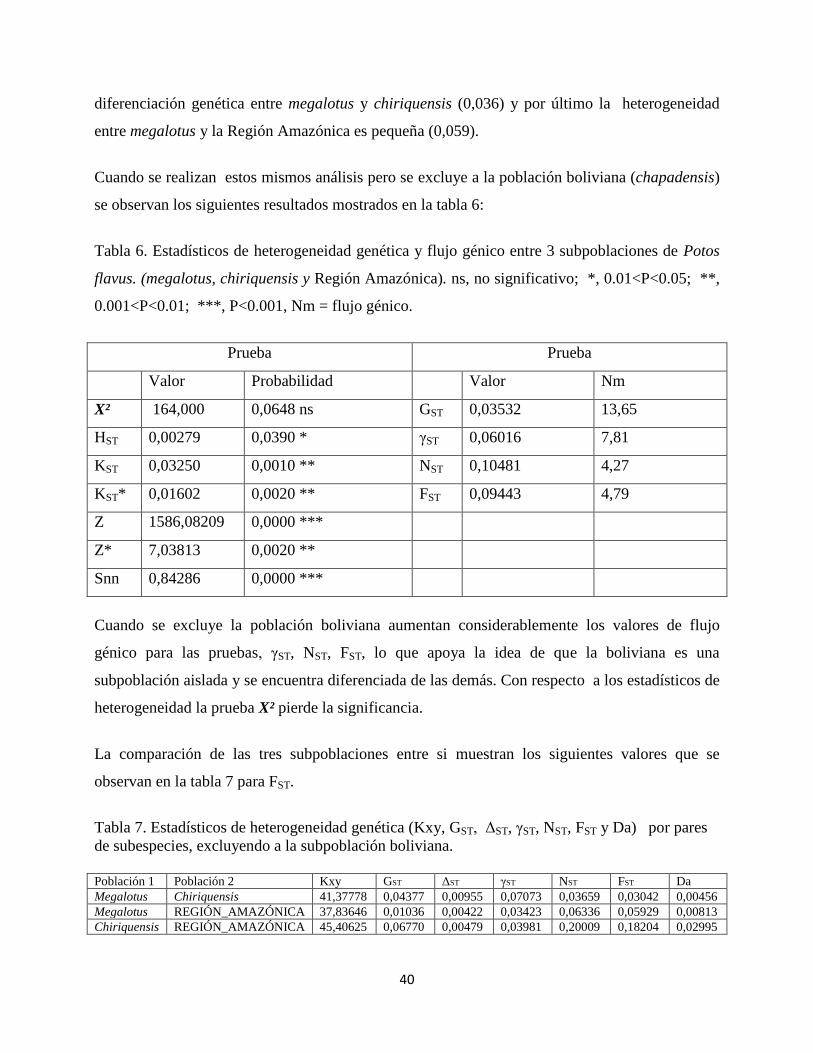
40
diferenciación genética entre megalotus y chiriquensis (0,036) y por último la heterogeneidad
entre megalotus y la Región Amazónica es pequeña (0,059).
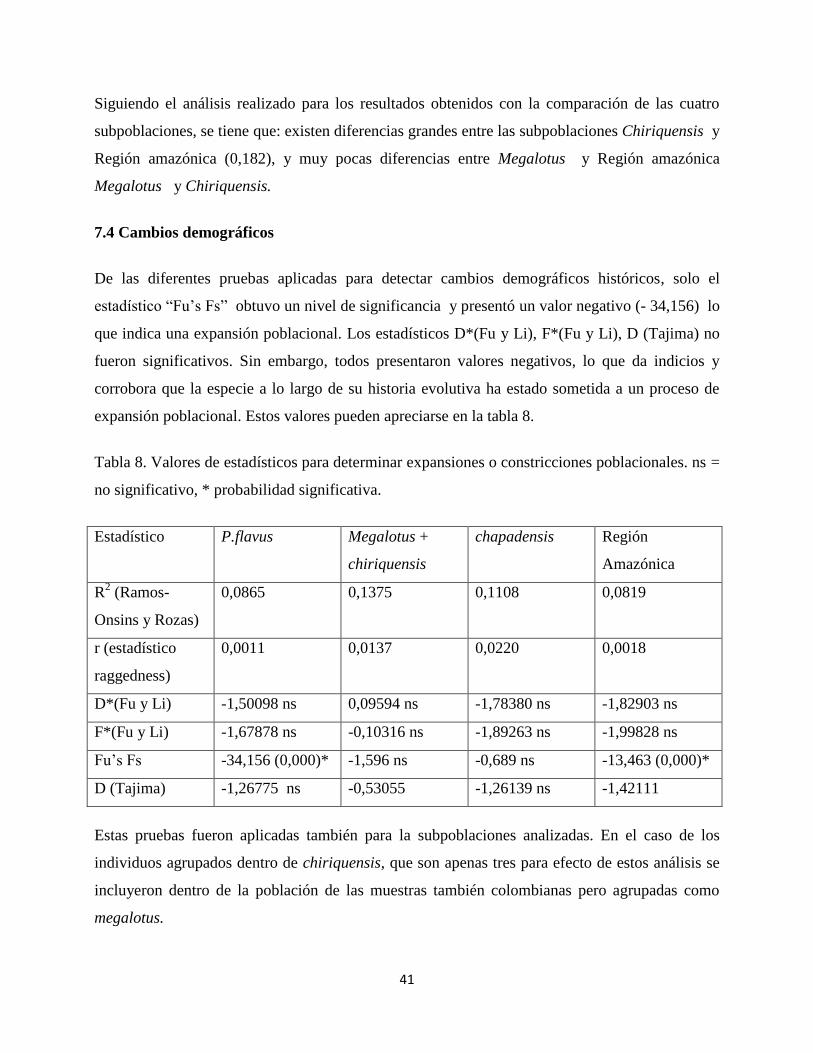
Cuando se realizan estos mismos análisis pero se excluye a la población boliviana (chapadensis)
se observan los siguientes resultados mostrados en la tabla 6:
Tabla 6. Estadísticos de heterogeneidad genética y flujo génico entre 3 subpoblaciones de Potos
flavus. (megalotus, chiriquensis y Región Amazónica). ns, no significativo; *, 0.01<P<0.05; **,
0.001<P<0.01; ***, P<0.001, Nm = flujo génico.
Prueba Prueba
Valor Probabilidad Valor Nm
X² 164,000 0,0648 ns GST 0,03532 13,65
HST 0,00279 0,0390 * γST 0,06016 7,81
KST 0,03250 0,0010 ** NST 0,10481 4,27
KST* 0,01602 0,0020 ** FST 0,09443 4,79
Z 1586,08209 0,0000 ***
Z* 7,03813 0,0020 **
Snn 0,84286 0,0000 ***
Cuando se excluye la población boliviana aumentan considerablemente los valores de flujo
génico para las pruebas, γST, NST, FST, lo que apoya la idea de que la boliviana es una
subpoblación aislada y se encuentra diferenciada de las demás. Con respecto a los estadísticos de
heterogeneidad la prueba X² pierde la significancia.
La comparación de las tres subpoblaciones entre si muestran los siguientes valores que se
observan en la tabla 7 para FST.
Tabla 7. Estadísticos de heterogeneidad genética (Kxy, GST, ΔST, γST, NST, FST y Da) por pares
de subespecies, excluyendo a la subpoblación boliviana.
Población 1 Población 2 Kxy GST ΔST γST NST FST Da
Megalotus Chiriquensis 41,37778 0,04377 0,00955 0,07073 0,03659 0,03042 0,00456
Megalotus REGIÓN_AMAZÓNICA 37,83646 0,01036 0,00422 0,03423 0,06336 0,05929 0,00813
Chiriquensis REGIÓN_AMAZÓNICA 45,40625 0,06770 0,00479 0,03981 0,20009 0,18204 0,02995
41
Siguiendo el análisis realizado para los resultados obtenidos con la comparación de las cuatro
subpoblaciones, se tiene que: existen diferencias grandes entre las subpoblaciones Chiriquensis y
Región amazónica (0,182), y muy pocas diferencias entre Megalotus y Región amazónica
Megalotus y Chiriquensis.
7.4 Cambios demográficos
De las diferentes pruebas aplicadas para detectar cambios demográficos históricos, solo el
estadístico “Fu’s Fs” obtuvo un nivel de significancia y presentó un valor negativo (- 34,156) lo
que indica una expansión poblacional. Los estadísticos D*(Fu y Li), F*(Fu y Li), D (Tajima) no
fueron significativos. Sin embargo, todos presentaron valores negativos, lo que da indicios y
corrobora que la especie a lo largo de su historia evolutiva ha estado sometida a un proceso de
expansión poblacional. Estos valores pueden apreciarse en la tabla 8.
Tabla 8. Valores de estadísticos para determinar expansiones o constricciones poblacionales. ns =
no significativo, * probabilidad significativa.
Estadístico P.flavus Megalotus +
chiriquensis
chapadensis Región
Amazónica
R2 (Ramos-
Onsins y Rozas)
0,0865 0,1375 0,1108 0,0819
r (estadístico
raggedness)
0,0011 0,0137 0,0220 0,0018
D*(Fu y Li) -1,50098 ns 0,09594 ns -1,78380 ns -1,82903 ns
F*(Fu y Li) -1,67878 ns -0,10316 ns -1,89263 ns -1,99828 ns
Fu’s Fs -34,156 (0,000)* -1,596 ns -0,689 ns -13,463 (0,000)*
D (Tajima) -1,26775 ns -0,53055 -1,26139 ns -1,42111
Estas pruebas fueron aplicadas también para la subpoblaciones analizadas. En el caso de los
individuos agrupados dentro de chiriquensis, que son apenas tres para efecto de estos análisis se
incluyeron dentro de la población de las muestras también colombianas pero agrupadas como
megalotus.
42
En general, todos los estadísticos presentan valores negativos con excepción de D*(Fu y Li) para
la subpoblación Megalotus + chiriquensis (0,09594). Solamente el estadístico Fu’s Fs fue
significativo para la Región amazónica, lo que también sugiere indicios de expansión cuando el
análisis se realiza por subpoblaciones.
Con respecto al análisis de distribución Mismatch fueron calculados los estadísticos R2 y r que
pueden ser observados en la tabla 8. Se aplicó la prueba a la población en general y a cada una de
las subpoblaciones que se definieron para las demás pruebas de cambios demográficos. Las
figuras 8 a la 15 muestran lo encontrado para cada caso.
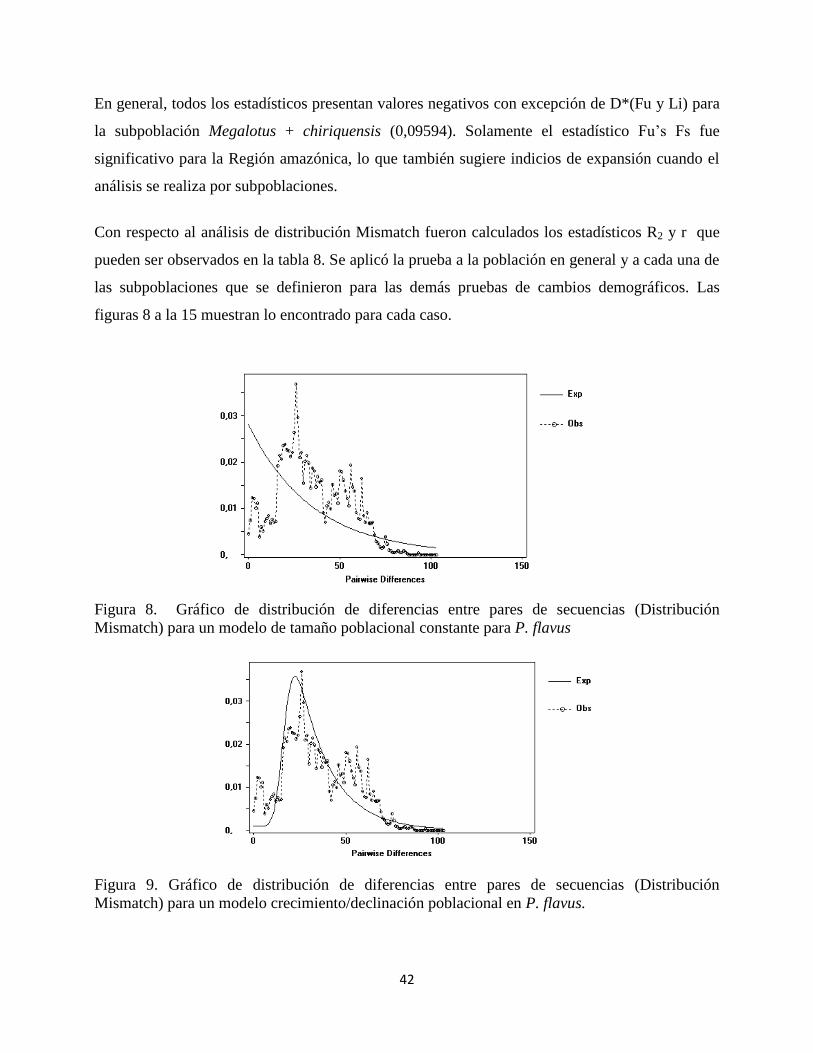
Figura 8. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo de tamaño poblacional constante para P. flavus
Figura 9. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo crecimiento/declinación poblacional en P. flavus.
43
En el caso de la población en general, se observa claramente que la distribución más ajustada se
da cuando hay un modelo de expansión poblacional, lo cual es validado por el pequeño valor del
estadístico r.
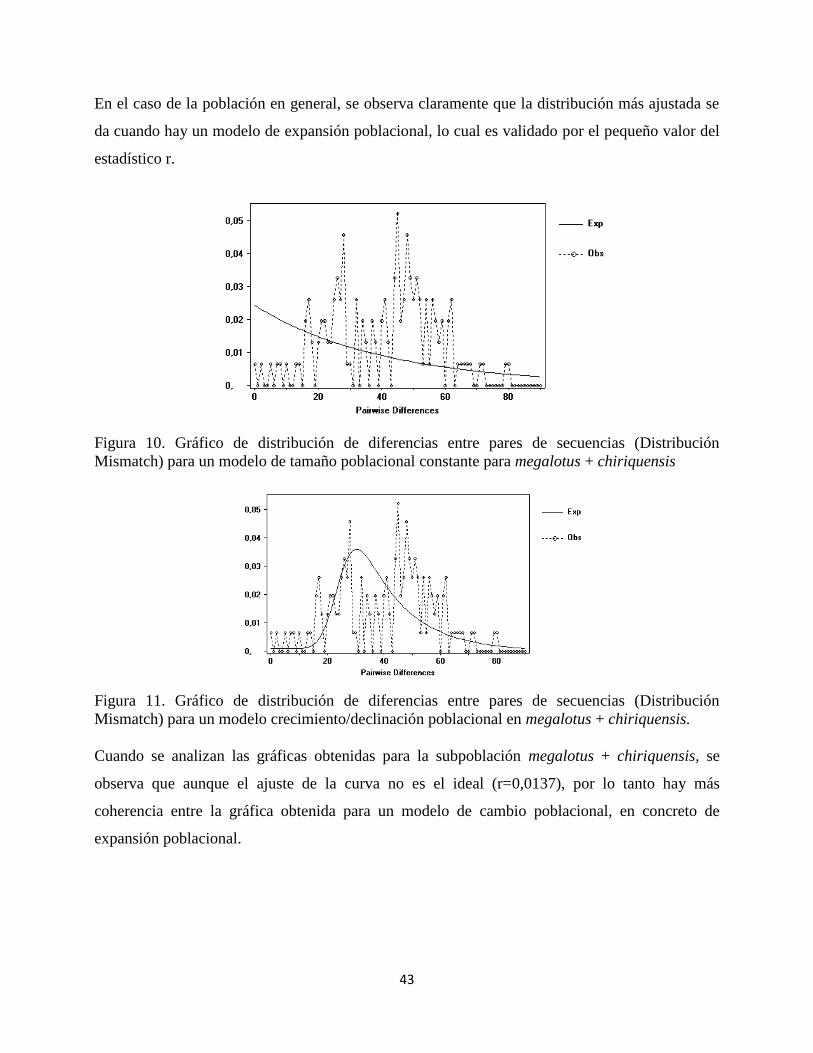
Figura 10. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo de tamaño poblacional constante para megalotus + chiriquensis
Figura 11. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo crecimiento/declinación poblacional en megalotus + chiriquensis.
Cuando se analizan las gráficas obtenidas para la subpoblación megalotus + chiriquensis, se
observa que aunque el ajuste de la curva no es el ideal (r=0,0137), por lo tanto hay más
coherencia entre la gráfica obtenida para un modelo de cambio poblacional, en concreto de
expansión poblacional.
44
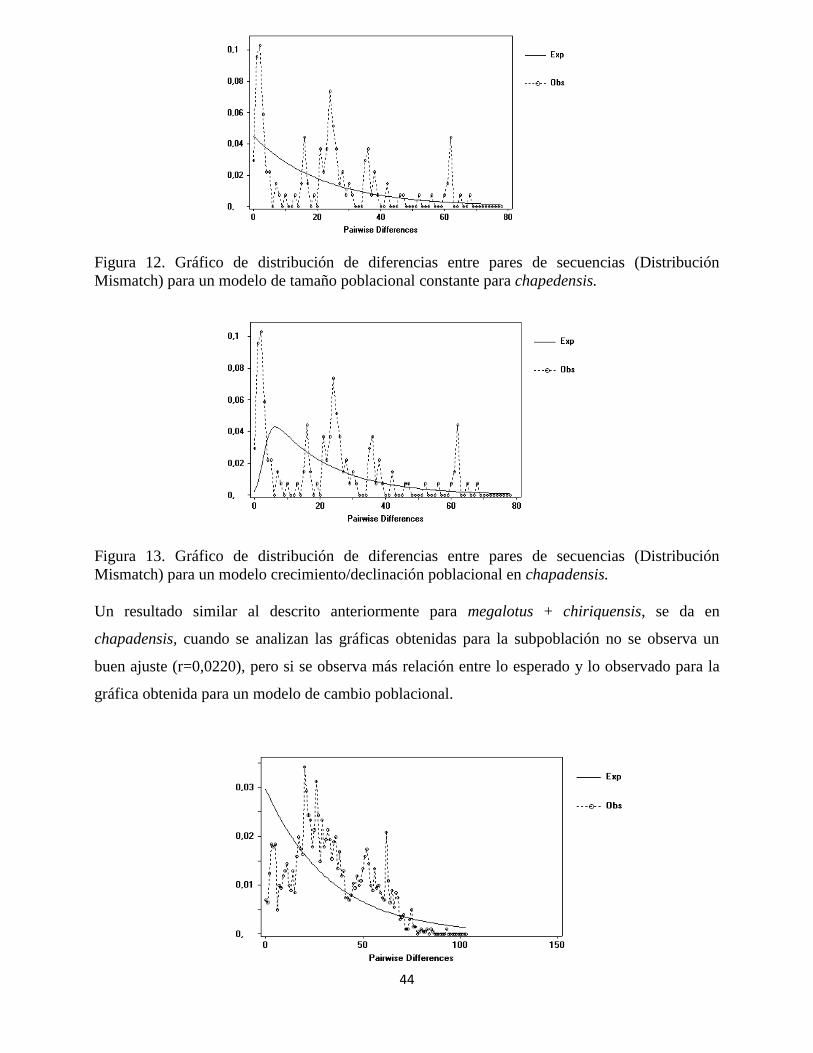
Figura 12. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo de tamaño poblacional constante para chapedensis.
Figura 13. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo crecimiento/declinación poblacional en chapadensis.
Un resultado similar al descrito anteriormente para megalotus + chiriquensis, se da en
chapadensis, cuando se analizan las gráficas obtenidas para la subpoblación no se observa un
buen ajuste (r=0,0220), pero si se observa más relación entre lo esperado y lo observado para la
gráfica obtenida para un modelo de cambio poblacional.
45
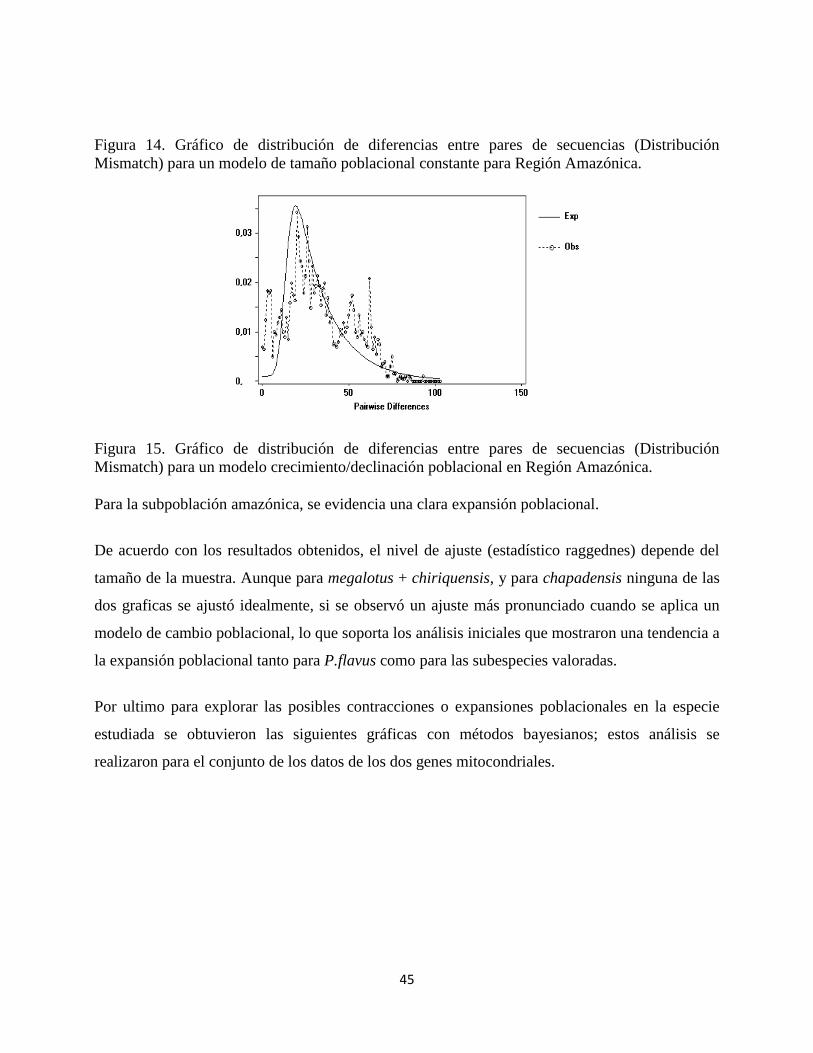
Figura 14. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo de tamaño poblacional constante para Región Amazónica.
Figura 15. Gráfico de distribución de diferencias entre pares de secuencias (Distribución
Mismatch) para un modelo crecimiento/declinación poblacional en Región Amazónica.
Para la subpoblación amazónica, se evidencia una clara expansión poblacional.
De acuerdo con los resultados obtenidos, el nivel de ajuste (estadístico raggednes) depende del
tamaño de la muestra. Aunque para megalotus + chiriquensis, y para chapadensis ninguna de las
dos graficas se ajustó idealmente, si se observó un ajuste más pronunciado cuando se aplica un
modelo de cambio poblacional, lo que soporta los análisis iniciales que mostraron una tendencia a
la expansión poblacional tanto para P.flavus como para las subespecies valoradas.
Por ultimo para explorar las posibles contracciones o expansiones poblacionales en la especie
estudiada se obtuvieron las siguientes gráficas con métodos bayesianos; estos análisis se
realizaron para el conjunto de los datos de los dos genes mitocondriales.
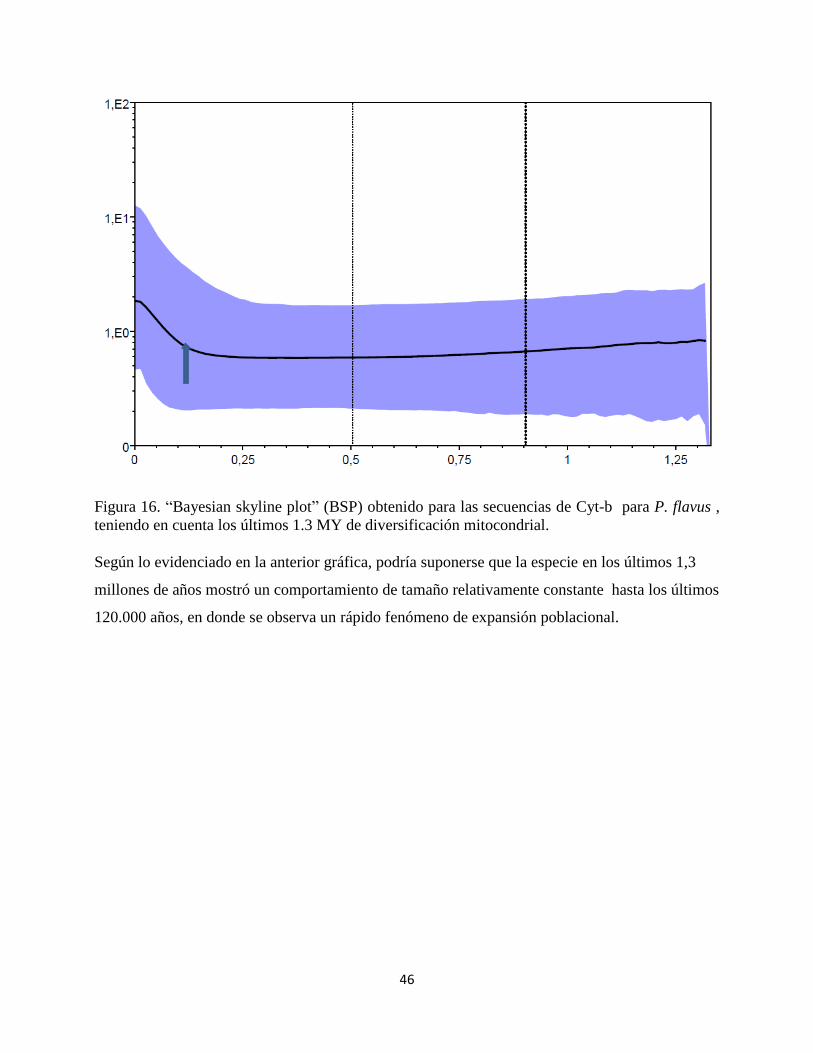
46
Figura 16. “Bayesian skyline plot” (BSP) obtenido para las secuencias de Cyt-b para P. flavus ,
teniendo en cuenta los últimos 1.3 MY de diversificación mitocondrial.
Según lo evidenciado en la anterior gráfica, podría suponerse que la especie en los últimos 1,3
millones de años mostró un comportamiento de tamaño relativamente constante hasta los últimos
120.000 años, en donde se observa un rápido fenómeno de expansión poblacional.
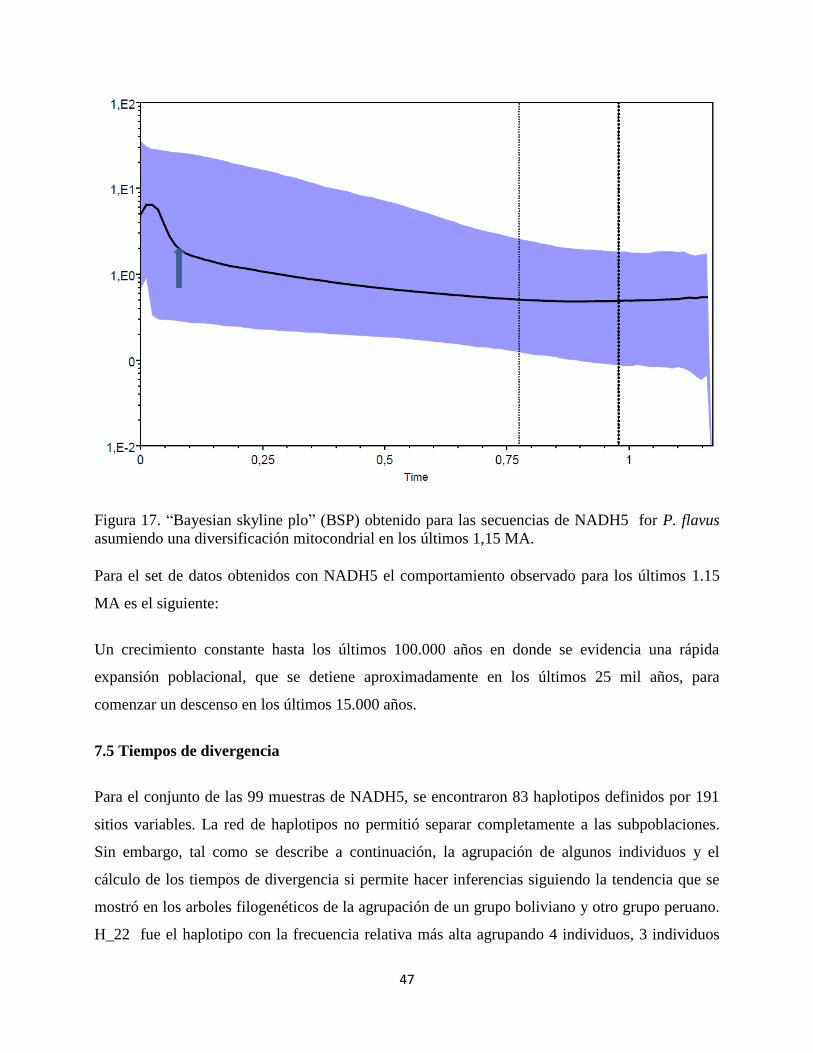
47
Figura 17. “Bayesian skyline plo” (BSP) obtenido para las secuencias de NADH5 for P. flavus
asumiendo una diversificación mitocondrial en los últimos 1,15 MA.
Para el set de datos obtenidos con NADH5 el comportamiento observado para los últimos 1.15
MA es el siguiente:
Un crecimiento constante hasta los últimos 100.000 años en donde se evidencia una rápida
expansión poblacional, que se detiene aproximadamente en los últimos 25 mil años, para
comenzar un descenso en los últimos 15.000 años.
7.5 Tiempos de divergencia
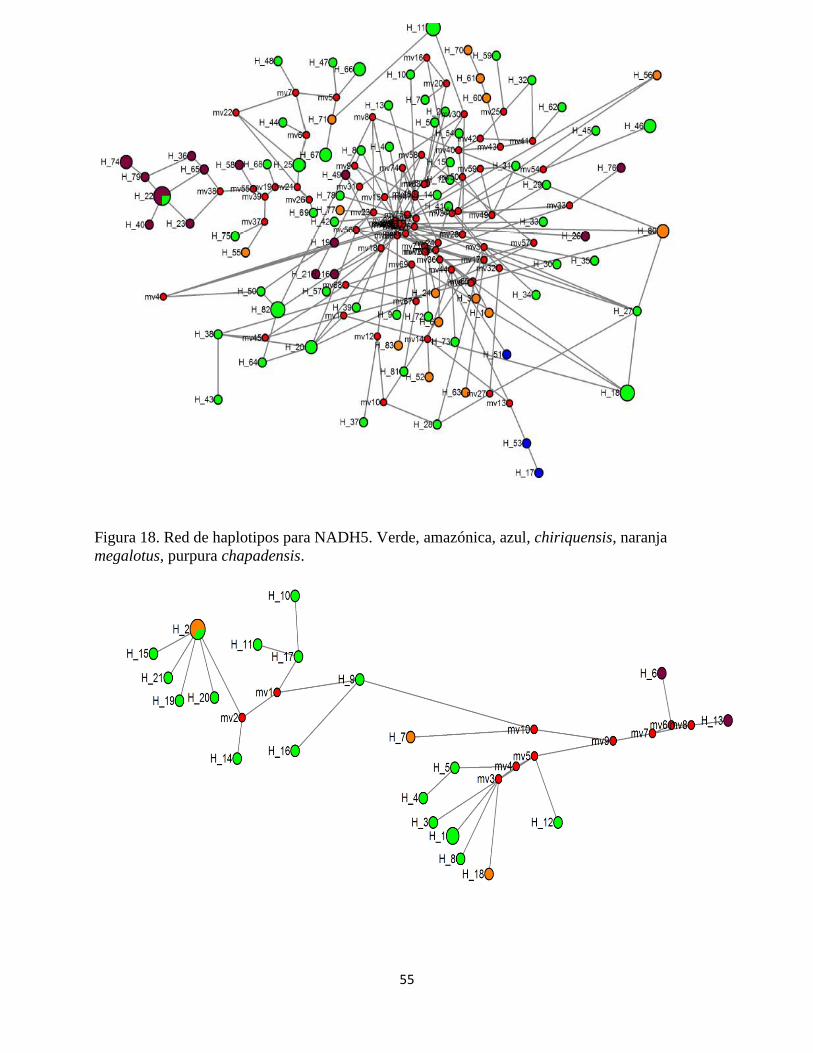
Para el conjunto de las 99 muestras de NADH5, se encontraron 83 haplotipos definidos por 191
sitios variables. La red de haplotipos no permitió separar completamente a las subpoblaciones.
Sin embargo, tal como se describe a continuación, la agrupación de algunos individuos y el
cálculo de los tiempos de divergencia si permite hacer inferencias siguiendo la tendencia que se
mostró en los arboles filogenéticos de la agrupación de un grupo boliviano y otro grupo peruano.
H_22 fue el haplotipo con la frecuencia relativa más alta agrupando 4 individuos, 3 individuos

48
correspondientes al morfotipo chapadensis y uno proveniente de Cajamarca en Perú. Los
haplotipos que siguen en frecuencia relativa fueron: Hap_11 con tres individuos peruanos, 2
provenientes de Tingomaria, (Huanaco) y uno proveniente de la región de Pucallpa en Ucayali;
Hap_18, representado por tres individuos, uno de Amacayacu, Amazonas colombiano, otro de
San Francisco también localidad amazónica colombiana y otro individuo proveniente de Iquitos,
amazonia peruana; Hap_82 agrupó 3 individuos todos provenientes del departamento de Loreto
en la amazonia peruana; el siguiente grupo de haplotipos estuvo representado por 2 individuos
cada uno: Hap_20 , individuos provenientes de Puerto Nariño y San Francisco, dos localidades
amazónicas colombianas; Hap_25, los individuos provienen del departamento de Ucayali en el
Perú; Hap_46, fueron muestreados en Pasco, Perú; Hap_66, de Yarinococha, departamento de
Ucayali (Perú); Hap_67, este también se encontró representando por dos muestras peruanas de
Ucayali; Hap_74, este haplotipo representó dos individuos bolivianos provenientes de
Cochabamba y Tarija; por último el Hap_80 agrupó dos muestras provenientes de la región
oriental de Colombia, Meta. Los 72 haplotipos restantes se encontraron en un individuo cada uno.
Debido al gran número de haplotipos teniendo en cuenta el número de muestras y que no
existieron uno o varios haplotipos con una frecuencia relativa alta, no fue posible considerar
haplotipos ancestrales o descendientes. Sin embargo, se calculó el estadístico rho para determinar
los posibles tiempos de divergencia entre los más frecuentes y los restantes, en la tabla 9 se
observa el valor estimado de divergencia calculado para algunos haplotipos.
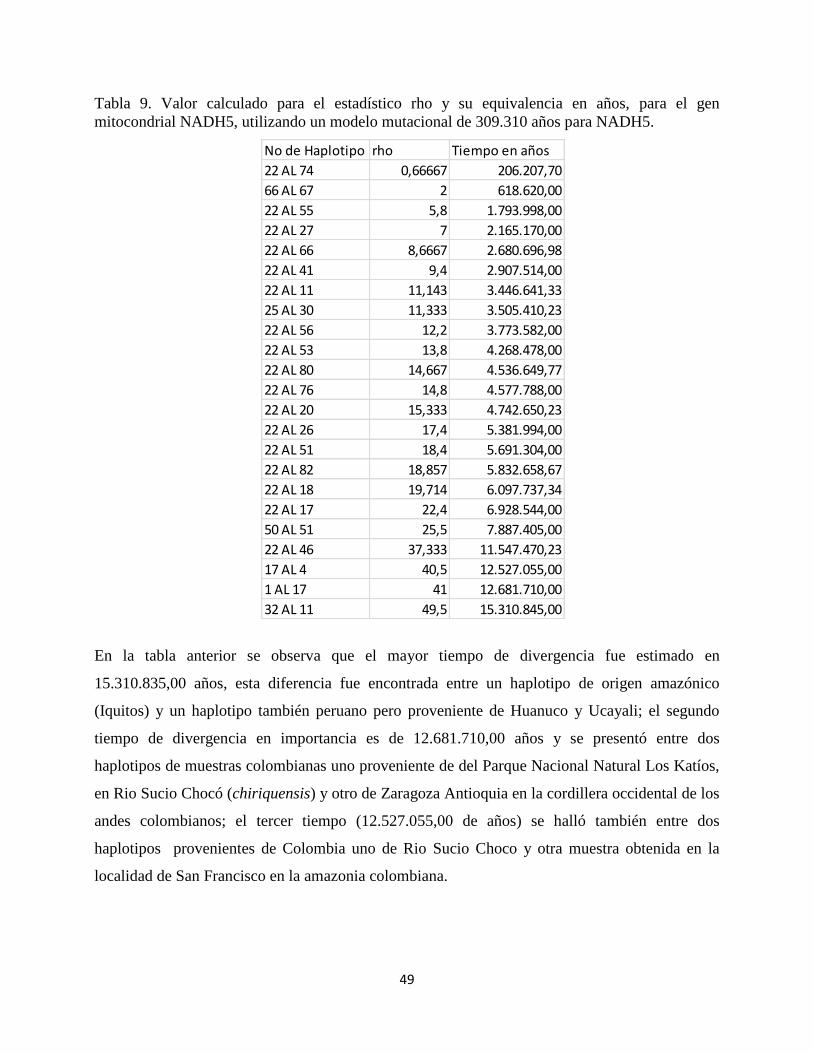
49
Tabla 9. Valor calculado para el estadístico rho y su equivalencia en años, para el gen
mitocondrial NADH5, utilizando un modelo mutacional de 309.310 años para NADH5.
En la tabla anterior se observa que el mayor tiempo de divergencia fue estimado en
15.310.835,00 años, esta diferencia fue encontrada entre un haplotipo de origen amazónico
(Iquitos) y un haplotipo también peruano pero proveniente de Huanuco y Ucayali; el segundo
tiempo de divergencia en importancia es de 12.681.710,00 años y se presentó entre dos
haplotipos de muestras colombianas uno proveniente de del Parque Nacional Natural Los Katíos,
en Rio Sucio Chocó (chiriquensis) y otro de Zaragoza Antioquia en la cordillera occidental de los
andes colombianos; el tercer tiempo (12.527.055,00 de años) se halló también entre dos
haplotipos provenientes de Colombia uno de Rio Sucio Choco y otra muestra obtenida en la
localidad de San Francisco en la amazonia colombiana.
No de Haplotipo rho Tiempo en años
22 AL 74 0,66667 206.207,70
66 AL 67 2 618.620,00
22 AL 55 5,8 1.793.998,00
22 AL 27 7 2.165.170,00
22 AL 66 8,6667 2.680.696,98
22 AL 41 9,4 2.907.514,00
22 AL 11 11,143 3.446.641,33
25 AL 30 11,333 3.505.410,23
22 AL 56 12,2 3.773.582,00
22 AL 53 13,8 4.268.478,00
22 AL 80 14,667 4.536.649,77
22 AL 76 14,8 4.577.788,00
22 AL 20 15,333 4.742.650,23
22 AL 26 17,4 5.381.994,00
22 AL 51 18,4 5.691.304,00
22 AL 82 18,857 5.832.658,67
22 AL 18 19,714 6.097.737,34
22 AL 17 22,4 6.928.544,00
50 AL 51 25,5 7.887.405,00
22 AL 46 37,333 11.547.470,23
17 AL 4 40,5 12.527.055,00
1 AL 17 41 12.681.710,00
32 AL 11 49,5 15.310.845,00

50
Otros tiempos de divergencia en orden descendente son mostrados a continuación; 11.547.470,23
años de divergencia fueron encontrados para un haplotipo representado por muestras de 3
individuos bolivianos más uno proveniente del departamento de Cajamarca (Perú) y otro
haplotipo que contiene muestras de 2 individuos muestreados en Pasco (Perú); 7.887.405,00
años, esta diferencia corresponde al tiempo de divergencia entre un haplotipo representado por un
individuo de Leticia (Colombia) contra un haplotipo representando por un individuo asignado al
morfotipo chiriquensis; por el contrario la divergencia más reciente ocurrió hace 206.207,70 años
y se dio entre un haplotipo boliviano y un haplotipo representado por dos muestras asignadas a la
misma subespecie chapadensis.
A continuación (tabla 10) se presenta la descripción de los haplotipos obtenidos para los
resultados descritos anteriormente y para el grupo de secuencias de cyt b (tabla 11).
Tabla 10. Descripción de haplotipos encontrados para el gen mitocondrial NADH5
Haplotipo Frecuencia Subespecie o
subpoblación
asignada
Muestra y localidad
Hap_1 1 megalotus POTOSFLAVUS144ZARAGOCOLOM
Hap_2 1 Región
Amazónica
POTOSFLAVUS150DECOMISOELDORADO
Hap_3 1 megalotus POTOSFLAVUS1588ZARAGOCOLOM
Hap_4 1 Región
Amazónica
POTOSFLAVUS15SANFRANCISCOLOM
Hap_5 1 Región
Amazónica
POTOSFLAVUS173PTONARCOLOM
Hap_6 1 Región
Amazónica
POTOSFLAVUS19ELARATENAECUA
Hap_7 1 Región
Amazónica
POTOSFLAVUS21FATIMAECUA
Hap_8 1 Región
Amazónica
POTOSFLAVUS22-1FATIMAECUA
Hap_9 1 Región
Amazónica
POTOSFLAVUS22FATIMAECUA
Hap_10 1 Región
Amazónica
POTOSFLAVUS104PASCOPERU
Hap_11 3 Región
Amazónica
POTOSFLAVUS87TINGOMAPERU
POTOSFLAVUSN11PUCALLPAPERU
POTOSFLAVUS23TINGOMA
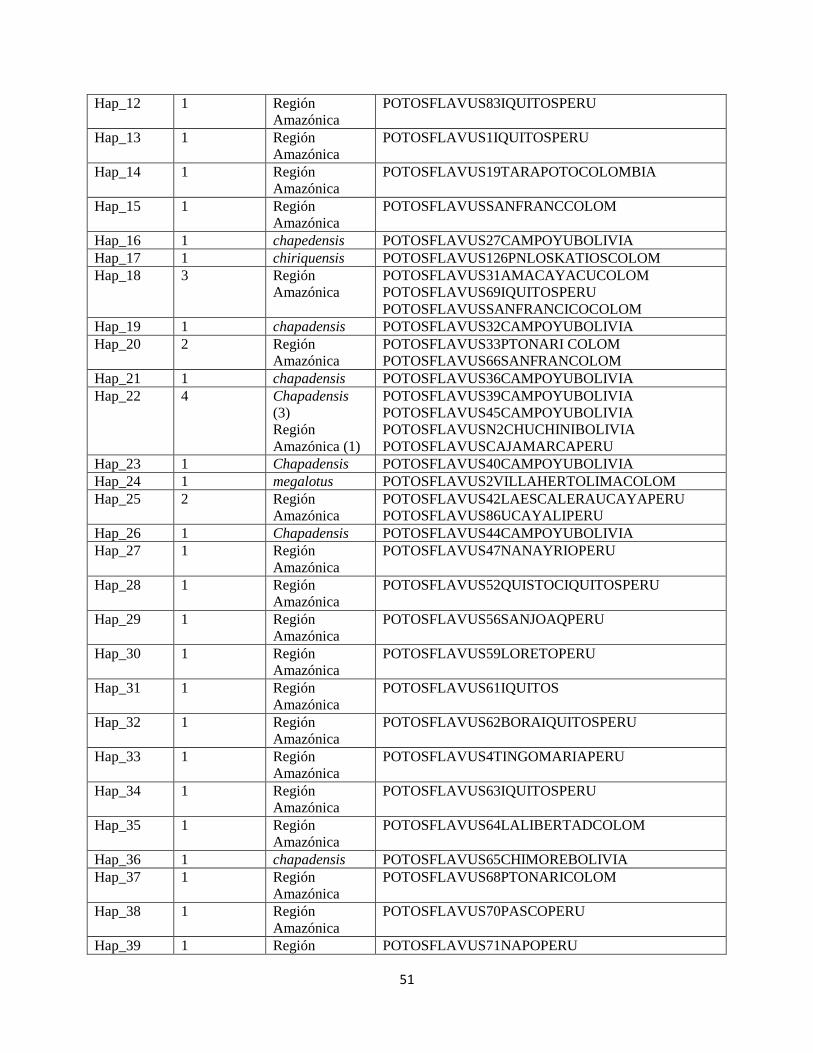
51
Hap_12 1 Región
Amazónica
POTOSFLAVUS83IQUITOSPERU
Hap_13 1 Región
Amazónica
POTOSFLAVUS1IQUITOSPERU
Hap_14 1 Región
Amazónica
POTOSFLAVUS19TARAPOTOCOLOMBIA
Hap_15 1 Región
Amazónica
POTOSFLAVUSSANFRANCCOLOM
Hap_16 1 chapedensis POTOSFLAVUS27CAMPOYUBOLIVIA
Hap_17 1 chiriquensis POTOSFLAVUS126PNLOSKATIOSCOLOM
Hap_18 3 Región
Amazónica
POTOSFLAVUS31AMACAYACUCOLOM
POTOSFLAVUS69IQUITOSPERU
POTOSFLAVUSSANFRANCICOCOLOM
Hap_19 1 chapadensis POTOSFLAVUS32CAMPOYUBOLIVIA
Hap_20 2 Región
Amazónica
POTOSFLAVUS33PTONARI COLOM
POTOSFLAVUS66SANFRANCOLOM
Hap_21 1 chapadensis POTOSFLAVUS36CAMPOYUBOLIVIA
Hap_22 4 Chapadensis
(3)
Región
Amazónica (1)
POTOSFLAVUS39CAMPOYUBOLIVIA
POTOSFLAVUS45CAMPOYUBOLIVIA
POTOSFLAVUSN2CHUCHINIBOLIVIA
POTOSFLAVUSCAJAMARCAPERU
Hap_23 1 Chapadensis POTOSFLAVUS40CAMPOYUBOLIVIA
Hap_24 1 megalotus POTOSFLAVUS2VILLAHERTOLIMACOLOM
Hap_25 2 Región
Amazónica
POTOSFLAVUS42LAESCALERAUCAYAPERU
POTOSFLAVUS86UCAYALIPERU
Hap_26 1 Chapadensis POTOSFLAVUS44CAMPOYUBOLIVIA
Hap_27 1 Región
Amazónica
POTOSFLAVUS47NANAYRIOPERU
Hap_28 1 Región
Amazónica
POTOSFLAVUS52QUISTOCIQUITOSPERU
Hap_29 1 Región
Amazónica
POTOSFLAVUS56SANJOAQPERU
Hap_30 1 Región
Amazónica
POTOSFLAVUS59LORETOPERU
Hap_31 1 Región
Amazónica
POTOSFLAVUS61IQUITOS
Hap_32 1 Región
Amazónica
POTOSFLAVUS62BORAIQUITOSPERU
Hap_33 1 Región
Amazónica
POTOSFLAVUS4TINGOMARIAPERU
Hap_34 1 Región
Amazónica
POTOSFLAVUS63IQUITOSPERU
Hap_35 1 Región
Amazónica
POTOSFLAVUS64LALIBERTADCOLOM
Hap_36 1 chapadensis POTOSFLAVUS65CHIMOREBOLIVIA
Hap_37 1 Región
Amazónica
POTOSFLAVUS68PTONARICOLOM
Hap_38 1 Región
Amazónica
POTOSFLAVUS70PASCOPERU
Hap_39 1 Región POTOSFLAVUS71NAPOPERU
52
Amazónica
Hap_40 1 chapadensis POTOSFLAVUS6CAMPOYUQBOLIVIA
Hap_41 1 Región
Amazónica
POTOSFLAVUS82BELLAVINAPOPERU
Hap_42 1 Región
Amazónica
POTOSFLAVUS85NAPORIOPERU
Hap_43 1 Región
Amazónica
POTOSFLAVUS92PASCOPERU
Hap_44 1 Región
Amazónica
POTOSFLAVUS93PASCOPERU
Hap_45 1 Región
Amazónica
POTOSFLAVUSCERROJONAPASCOPERU
Hap_46 2 Región
Amazónica
POTOSFLAVUSCERROJONAPASCOPERU
POTOSFLAVUS89PASCOPERU
Hap_47 1 Región
Amazónica
POTOSFLAVUSHUANACO10PERU
Hap_48 1 Región
Amazónica
POTOSFLAVUSPASCO105PERU
Hap_49 1 chapedensis POTOSFLAVUS8LATEJERIABOLIVIA
Hap_50 1 Región
Amazónica
POTOSFLAVUS112LETICIACOLOM
Hap_51 1 chiriquensis POTOSFLAVUS118RIOSUCIOCOLOM
Hap_52 1 megalotus POTOSFLAVUS125VALLEDCOLOM
Hap_53 1 chiriquensis POTOSFLAVUS126RIOSUCIOCCOLOM
Hap_54 1 megalotus POTOSFLAVUS127BOCARIARIVAUPESCOLOM
Hap_55 1 megalotus POTOSFLAVUS142VILLAVMETACOLOM
Hap_56 1 megalotus POTOSFLAVUS149CUNDINCOLOM
Hap_57 1 Región
Amazónica
POTOSFLAVUS152ARARACAQUETACOLOM
Hap_58 1 chapadensis POTOSFLAVUS13CAMPOYUBOLIVIA
Hap_59 1 Región
Amazónica
POTOSFLAVUS156AMACAYACUCOLOM
Hap_60 1 megalotus POTOSFLAVUS157ZARAGOZACOLOM
Hap_61 1 megalotus POTOSFLAVUS158ZARAGOZACOLOM
Hap_62 1 Región
Amazónica
POTOSFLAVUS159COLOMBIA
Hap_63 1 megalotus POTOSFLAVUS160PNNMUNCHIQUECOLOM
Hap_64 1 Región
Amazónica
POTOSFLAVUSPTONARINOCOLOM
Hap_65 1 chapadensis POTOSFLAVUSN1STCRUZBOLIVIA
Hap_66 2 Región
Amazónica
POTOSFLAVUS14YARINACPERU
POTOSFLAVUS18YARINACPERU
Hap_67 2 Región
Amazónica
POTOSFLAVUSN8PUCALLPPERU
POTOSFLAVUSN12PUCALLPERU
Hap_68 1 Región
Amazónica
POTOSFLAVUSN9PUCALLPPERU
Hap_69 1 Región
Amazónica
POTOSFLAVUSN10PUCALLPERU
Hap_70 1 megalotus POTOSFLAVUSCHAMEZACASANARECOLOMBIA
Hap_71 1 megalotus POTOSFLAVUSRISARALDACOLOMBIA
53
Hap_72 1 Región
Amazónica
POTOSFLAVUSARCATENANECUADOR
Hap_73 1 Región
Amazónica
POTOSFLAVUSARCATENANECUADOR
Hap_74 2 chapadensis POTOSFLAVUS16CAMPOYU BOLIVIA
POTOSFLAVUS24SAJTABOLIVIA
Hap_75 1 Región
Amazónica
POTOSFLAVUSN25BELEMBRASIL
Hap_76 1 chapadensis POTOSFLAVUSN24BOLIVIA
Hap_77 1 modestus POTOSFLAVUS122PNCUEVAGUACHAROSCOLO
M
Hap_78 1 Región
Amazónica
POTOSFLAVUS78INDIANAPERU
Hap_79 1 chapadensis POTOSFLAVUS41CAMPOYUBOLIVIA
Hap_80 2 modestus POTOSFLAVUSN13METACOLOMBIA
POTOSFLAVUS147SANMARTINMETACOLOMBIA
Hap_81 1 Región
Amazónica
POTOSFLAVUSN3VIRACOCPERU
Hap_82 3 Región
Amazónica
POTOSFLAVUS77IQUITOS PERU
POTOSFLAVUS95RIOGALV PERU
POTOSFLAVUS21IQUITOSPERU
Hap_83 1 modestus POTOSFLAVUS138MAGUANORSANTANDERCOL
OM
Tabla. 11 Descripción de haplotipos encontrados para el gen mitocondrial Cyt-b
Haplotipo Frecuencia Subespecie o
subpoblación asignada
Muestra y localidad
Hap_1 2 Región Amazónica POTOS103HUANACOPERU
POTOS86PADREABADUCAY
Hap_2 3 megalotus (1)
Región Amazónica (2)
POTOSN13JAIMEDUQUECOMETA
POTOS63IQUITOSPERU
POTOS61IQUITOSPERU
Hap_3 1 Región Amazónica POTOSN11OPUCALLPAPER
Hap_4 1 Región Amazónica POTOSN9PUCALLPAPERU
Hap_5 1 Región Amazónica POTOS64LALIBERTADCOLOMBIA
Hap_6 1 Chapedensis POTOS65RCHIMORECOCHABAMBA
Hap_7 1 Megalotus POTOS160MUNCHIQUETAMBOCOLOM
Hap_8 1 Región Amazónica POTOS105PASCOPERU
Hap_9 1 Región Amazónica POTOS71RNAPOPERU
Hap_10 1 Región Amazónica POTOS70PASCOPERU
54
Hap_11 1 Región Amazónica POTOS15SANFRANCISCOLOM
Hap_12: 1 Región Amazónica POTOS95RIOGALVEZPERU
Hap_13 1 Chapadensis POTOS40CAMPOYUQUIBOLIVIA
Hap_14 1 Región Amazónica POTOS99CAJAMARCAPERU
Hap_15 1 Región Amazónica POTOS59LORETOPERU
Hap_16 1 Región Amazónica POTOS31AMACAYACUCOLOM
Hap_17 1 Región Amazónica POTOS1IQUITOSPERU
Hap_18 1 Megalotus POTOS16RISARALDACOLOM
Hap_19 1 Región Amazónica POTOS103MACASECUADOR
Hap_20 1 Región Amazónica POTOS104MACASECUADOR
Hap_21 1 Región Amazónica POTOS82RIONAPOPERU
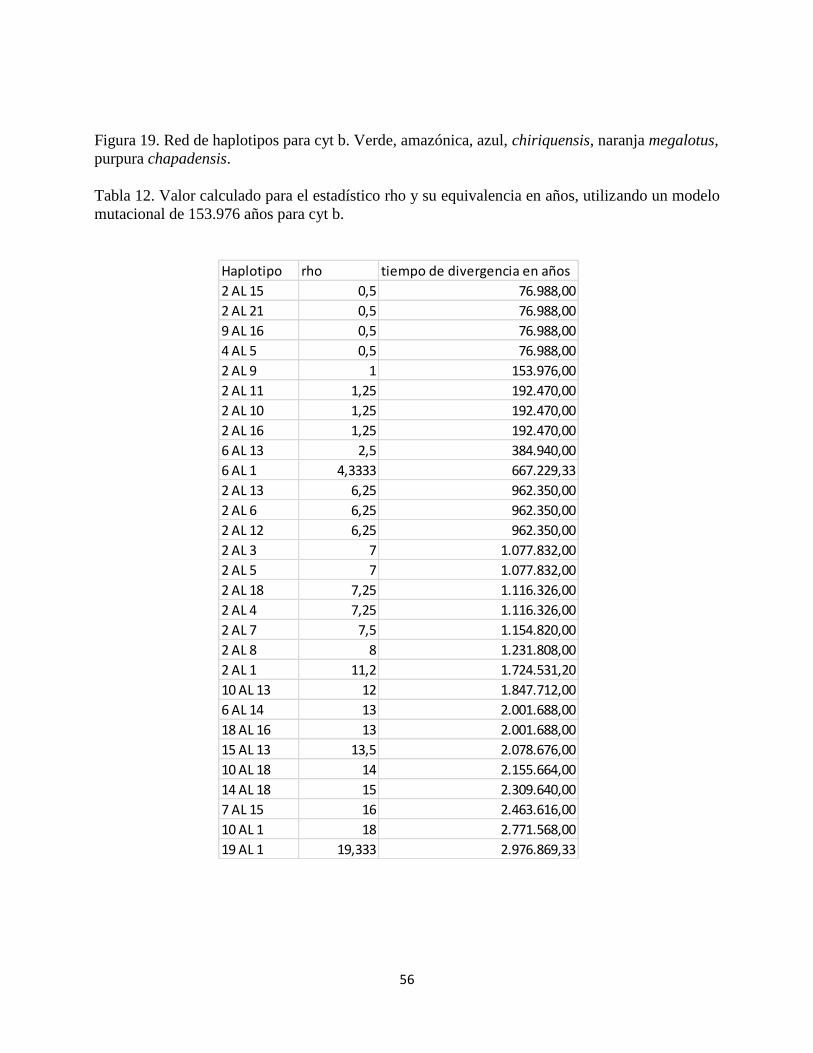
Con respecto a la red de haplotipos obtenida para el gen mitondrial Cyt b, se encontraron 21
haplotipos de 24 muestras y 59 sitios variables, el haplotipo más representado (Hap_2), mostró
una frecuencia relativa de tres individuos, seguido por el Hap_2 que incluyó dos individuos
provenientes de Ucayali (Perú9). Los demás 19 haplotipos están representados por un solo
individuos cada uno, como en el caso de los análisis realizados con NADH5 no es posible
determinar unos haplotipos ancestros o descendientes. Las figuras 18 y 19 muestras las redes de
haplotipos para cada gen, por otra parte en la tabla 12 se presentan algunos cálculos de tiempos
de divergencia entre los haplotipos obtenidos con cyt b.
55
Figura 18. Red de haplotipos para NADH5. Verde, amazónica, azul, chiriquensis, naranja
megalotus, purpura chapadensis.
56
Figura 19. Red de haplotipos para cyt b. Verde, amazónica, azul, chiriquensis, naranja megalotus,
purpura chapadensis.
Tabla 12. Valor calculado para el estadístico rho y su equivalencia en años, utilizando un modelo
mutacional de 153.976 años para cyt b.
Haplotipo rho tiempo de divergencia en años
2 AL 15 0,5 76.988,00
2 AL 21 0,5 76.988,00
9 AL 16 0,5 76.988,00
4 AL 5 0,5 76.988,00
2 AL 9 1 153.976,00
2 AL 11 1,25 192.470,00
2 AL 10 1,25 192.470,00
2 AL 16 1,25 192.470,00
6 AL 13 2,5 384.940,00
6 AL 1 4,3333 667.229,33
2 AL 13 6,25 962.350,00
2 AL 6 6,25 962.350,00
2 AL 12 6,25 962.350,00
2 AL 3 7 1.077.832,00
2 AL 5 7 1.077.832,00
2 AL 18 7,25 1.116.326,00
2 AL 4 7,25 1.116.326,00
2 AL 7 7,5 1.154.820,00
2 AL 8 8 1.231.808,00
2 AL 1 11,2 1.724.531,20
10 AL 13 12 1.847.712,00
6 AL 14 13 2.001.688,00
18 AL 16 13 2.001.688,00
15 AL 13 13,5 2.078.676,00
10 AL 18 14 2.155.664,00
14 AL 18 15 2.309.640,00
7 AL 15 16 2.463.616,00
10 AL 1 18 2.771.568,00
19 AL 1 19,333 2.976.869,33

57
Los tiempos de divergencia estimada mediante los arboles bayesianos tal como se observan en la
figura 7 C son los siguientes. De acuerdo con el gen mitocondrial NADH5 aparentemente Potos
divergió de Procyonidae, específicamente de Bassaricyon alleni, que fue la especie empleada
como grupo externo hace alrededor de 23,71 MA. La primera diversificación dentro del género se
dio hace 13,64 MA separándose en dos grandes grupos que a su vez se diversificaron hace 13,36
y 11,89 MA. Dentro del primer grupo el mayor número de linajes fueron diferenciados entre 6,61
MA y 1,2 MA, para el segundo de 8,79 MA a 1,73 MA.
Para el gen cyt b el árbol bayesiano muestra una separación inicial del 41,57 MA, tiempo en el
que Potos aparentemente divergió de los canidos teniendo en cuenta que la especie utilizada
como grupo externo es un zorro. El primer proceso de diversificación dentro del género ocurrió
hace 23,69 MA cuando se dividió en dos grandes grupos. Estos a su vez divergieron a los 21,38 y
16,04 MA. Dentro del primer grupo los principales procesos de diferenciación se han dado entre
los 15,01 MA y 2,75 MA; para el segundo grupo el mayor número de linajes fueron diferenciados
entre 10,59 y 1,77 MA. Estos resultados deben ser tomados con precaución ya que como la
especie utilizada como grupo externo es una especie de la cual Potos divergió hace mucho
tiempo, los valores obtenidos para el resto de las divergencias van a estar sesgados por ese valor
inicial que es muy distante.
8 DISCUSIÓN /CONCLUSIONES
8.1 Filogenia y correspondencia de supuestas especies definidas morfológicamente
La agrupación inicialmente evidenciada a partir de los arboles construidos por diversos métodos,
para los dos genes, que muestra un grupo boliviano parece ser soportado también por los análisis
de heterogeneidad, flujo genético y por la red de haplotipos.
Los doce individuos conformaron el mismo grupo en todos los árboles, incluyéndose también un
individuo de origen peruano proveniente de Cajamarca. Por otra parte en los análisis de
heterogeneidad genética se observó que cuando se comparan las poblaciones por pares, los
valores mayores de heterogeneidad resultan cuando se realiza la comparación contra chapensis,
encontrándose la mayor diferencia con la población más alejada geográficamente que en este
caso es chiriquensis. En ese mismo orden megalotus también diverge y se observa que la menor

58
diferenciación se da con la población amazónica que es también la más cercana. De esta manera
se sugiere que la población boliviana si está aislada de las demás poblaciones. Otra prueba de lo
anterior son los análisis de flujo génico cuando se excluye la población boliviana, estos valores
aumentan considerablemente, de la misma manera al excluir esta población del análisis de
heterogeneidad se muestra que el estadístico X² pierde su nivel de significancia estadística dando
una idea de que chapadensis aporta gran parte de las diferencias que fueron consideradas como
significativas.
Una vez se realizó el análisis de los haplotipos también se observó la agrupación de las 12
muestras que se presentaron en los arboles de filogenia. Por su parte, las muestras bolivianas que
se encuentran por fuera en estos árboles también se muestran distanciadas en la red de haplotipos.
Nuevamente, en este grupo se incluye la muestra proveniente de Cajarmarca Perú que comparte
haplotipo con dos muestras provenientes de Cochabamba (Bolivia). Estas dos regiones son
montañosas y se encuentran conectadas por la cordillera. Sin embargo, tampoco es claro porque
el haplotipo solamente se encuentra en Cajamarca y no se ha dispersado por otras zonas peruanas
cercanas. Sin embargo, como se verá más adelante existe también un aparente aislamiento
geográfico de algunas muestras peruanas cuando se calcula el tiempo de divergencia.
Por último los niveles más bajos de diversidad genética por subpoblación fueron presentados por
chapadensis.
Tal como lo indica preliminarmente Ruiz–García et al. (2012a) y de acuerdo con lo anterior se
sugiere que sí existe una correspondencia limitada entre las subespecies morfológicas tal como
las mencionan Ford & Hoffmann (1988), por lo menos para el morfotipo descrito como P. f.
chapadensis (Allen en 1904) con localidad tipo en Chapada, Matto Grosso en Brasil, región
continua a las localidades muestreadas en Bolivia, por lo que se esperaría que la subespecie
encontrada en esta región de Brasil sea igual a la que se tomó para efectos del presente estudio
como chapadensis.
Teniendo en cuenta estos resultados, se puede sugerir que este grupo, además de estar separado
geográficamente, pueden conformar un grupo históricamente aislado ameritando que se trate
como una Unidad Evolutiva Significativa (sensu Moritz 1994). Esto, sin embargo, se debe
verificar haciendo un análisis con un mayor número de muestras.

59
Por otra parte se observa que para los arboles construidos con las secuencias de NADH5 se forma
un grupo que fue llamado en la sección de resultados como peruano. Este incluye 15 individuos
provenientes de los departamentos de Huanuco, Ucayali y Pasco (zona centro y amazonía sur de
la geografía peruana) y una muestra que proviene del departamento de Risaralda en Colombia.
Con respecto a las 14 muestras peruanas, estas se encuentran alejadas de otras muestras peruanas
pero que provienen de los departamentos de Loreto y San Martin y de la muestra peruana
proveniente de Cajamarca de la que se habló en un análisis anterior (todas ellas más al norte en la
geografía peruana). La red de haplotipos mostró una conformación similar agrupándose de
manera cercana estas 15 muestras incluyendo la colombiana, como se puede observar en la tabla
8 el valor más alto de divergencia calculado entre la red de haplotipos, la presentaron dos
muestras peruanas, un haplotipo proveniente del departamento de Loreto y otro que incluía
individuos de los departamentos de Huanuco y Ucayali. De acuerdo con los datos moleculares, se
puede determinar que las muestras agrupadas inicialmente dentro de la población peruana que
provienen de los departamentos de Pasco, Huanuco y Ucayali, deben ser tratadas como una
subpoblación diferente, por lo menos, de acuerdo a los datos obtenidos en este caso. Sin
embargo, se observa cierto nivel de flujo génico ya que algunas muestras que provienen de esos
departamentos se encontraron relacionadas en los árboles y en la red de haplotipos con muestras
amazónicas. Una vez más existe un indicio de que hay ciertas barreras geográficas que podrían
haber aislado a ciertas poblaciones de Potos flavus. Posiblemente se trate de la cordillera andina,
ya que las poblaciones que se han mostrado con algún grado de aislamiento comparten como
característica estar sobre la cordillera en Bolivia y Perú o en el piedemonte de la misma. Sin
embargo, no existe un morfotipo definido para esa área geográfica que permita evaluar una
posible correspondencia entre una subespecie morfológica y los datos moleculares. No obstante,
de forma preliminar podría considerarse a esta población como una Unidad Evolutiva
Significativa.
Aparentemente para las muestras amazónicas de Colombia, Ecuador y norte de la Amazonía del
Perú y para las muestras de megalotus, no existe una diferenciación genética que permita
construir grupos determinados y bien soportados. Los análisis de flujo génico también
presentaron valores importantes que sugieren conexión entre esas subpoblaciones; sin embargo,
cuando se evalúan los datos de heterogeneidad por subpoblaciones, se observa que entre
chiriquensis y las muestras de la región amazónica (Perú, Colombia y Ecuador) si existen valores

60
que permiten determinar heterogeneidad genética entre esas dos subpoblaciones. Esto se observa
en los árboles y en la red de haplotipos para NADH5. Se evidencia que estas muestras
presentaron una distancia de separación importante. Sin embargo, este resultado está muy
seguramente influenciado por el bajo número de muestras con que se cuenta para chiriquensis.
Por lo tanto, se recomiendan estudios que incluyan más ejemplares que correspondan con el
morfotipo de chiriquensis y se incluyan poblaciones centro americanas que permitan determinar
si efectivamente corresponde a subespecies que deban ser tratadas como Unidades Evolutivas
Significativas.
Para el caso de las muestras denominadas (POTOSFLAVUS159COLOMB,
POTOSFLAVUS150COLOMB y POTOSFLAVUSN13METACO), para las que se conocía el
país de origen geográfico, pero no la localidad exacta, teniendo en cuenta los arboles
filogenéticos y la red de haplotipos se permitió determinar que posiblemente el individuo 159 y
150 provenientes de Colombia son individuos de origen amazónico dado su cercanía con
miembros de ese grupo en los árboles y ubicación en la red. Por su parte, para el individuo N13
se estableció que procede de la región oriental de Colombia ya que comparte el mismo haplotipo
con un individuo proveniente de San Martin Meta, individuo con el que se relacionó mediante
una alta probabilidad en el árbol bayesiano.
Se observa que la frecuencia relativa de los haplotipos se encuentran distribuidos por el rango de
distribución estudiado casi uniformemente con unas pequeñas excepciones que no agrupan más
de 4 individuos, por lo que se sugiere que esta especie no siguió un modelo de especiación que
permitiera establecer un haplotipo ancestral, al asentarse por un tiempo considerable en una zona
geográfica determinada, que le permita dejar una marca molecular y de allí dispersarse a otros
sitios convirtiéndolos en haplotipos descendientes. Al contrario, pareciera que la especie se
hubiera distribuido rápidamente a través de varios puntos geográficos sin tener un claro origen
geográfico, al menos, en Sudamérica. Esta hipótesis podría ser considerada si se tiene en cuenta
que Kays et al, (2000), mediante microsatélites, determinó la organización social del Potos
flavus encontrando que la hembra es la que se dispersa a otros lugares fuera de su grupo original.
Este modelo de dispersión es inusual en sistemas de mamíferos, y no se conoce en otras especies
de carnívoros (Greenwood 1980, Fuller et al. 1992, McNutt 1996, Waser 1996). Por lo tanto, los
kinkajous parecen ser inusuales entre los mamíferos por tener una dispersión sesgada hacia la

61
hembra (Greenwood 1980, Kays 1999). Teniendo en cuenta que los análisis son realizados con
secuencias de ADN mitocondrial, este comportamiento pudiera ser un indicio de la explicación
de los altos niveles de diversidad genética y de gran cantidad de haplotipos distribuidos por todo
el rango de distribución de la especie. Adicionalmente apoya la hipótesis de que una barrera
geográfica pudo interferir para la dispersión de las hembras de kinkajou que habitan en zonas de
la cordillera de los Andes peruanos y bolivianos.
8.2 Diversidad genética, heterogeneidad genética y flujo génico
La especie, en general, presenta niveles elevadísimos de diversidad genética (Hd=0,995 y
π=0,12569). Estos superan los niveles encontrados en otras especies tales como en lo mustélidos
Eyra barbara (Hd = 0.972 and π = 0.0661; Ruiz-García et al.2012 a) y Lontra felina (Hd = 0.93
and π = 0.005; Vianna et al. 2010), en 3 especies de zorros neotropicales, Pseudoalopex culpaeus
(Hd = 0.952 y π = 0.008), Pseudoalopex sechurae (Hd = 0.893 y π = 0.015; Ruiz-García et al.
2012b) y Cerdocyon thous (Hd = 0.830 y π = 0.019; Tchaika et al. 2006); también se muestra
niveles superiores a los encontrados para felinos tales como, Leopardus jacobita (Hd = 0.557 y π
= 0.0047; Ruiz-García et al. 2012 c) y Leopardus guigna (Hd = 0.94 y π = 0.00461; Napolitano et
al. 2012), incluso para felinos que han mostrado niveles de diversidad muy altos tales como el
jaguarundí, Puma yaguaroundi (Hd = 0.940 y π = 0.072; Ruiz-García and Pinedo-Castro 2012),
jaguar, Panthera onca, (Hd = 0.937 y π = 0.078; Ruiz-García et al. 2012d), Leopardus pajeros
(Hd = 0.932 and π = 0.0513; Ruiz-García et al. 2012c) y Leopardus pardalis (Hd = 0.962 and π =
0.068; Eizirik et al. 1998).
Estos altos niveles de diversidad genética pueden correlacionarse con las diferencias
morfológicas de los individuos encontrados a lo largo del rango de distribución de la especie tal
como lo describen autores como Cabrera (1958), Hall (1981) y Kortlucke (1973)
Con respecto a los resultados de heterogeneidad se observó significancia de los estadísticos
evaluados. Sin embargo, y teniendo en cuenta los valores elevados de flujo génico, esta
heterogeneidad es relativamente pequeña. Como el tamaño de la muestra es relativamente grande,
permite que los estadísticos detecten estas diferencias, aunque como se mencionó anteriormente,
la mayor parte de los estadísticos analizados fueron relativamente bajos. No obstante, el grupo

62
boliviano fue claramente detectable al igual que la población del centro y sur de la Amazonía
peruana, al menos, para el marcador NADH5.
8.3 Cambios demográficos y tiempos de divergencia.
Los test aplicados para evaluar posibles eventos de constricciones o expansiones poblacionales,
muestran una tendencia que sugiere que la especie ha expandido sus poblaciones en algún
momento de su historia evolutiva. Esto es corroborado por las gráficas obtenidas por métodos
bayesianos que sugieren que la especie aumentó sus poblaciones considerablemente en los
últimos 120.000 años en el caso de NADH5 y, para el cyt b, en los últimos 100.000 años.
De acuerdo con el árbol bayesiano para NADH5, la primera diversificación dentro del género se
dio hace 13,64 MA separándose en dos grandes grupos que a su vez se diversificaron hace 13,36
y 11,89 MA. Dentro del primer grupo, el mayor número de linajes fueron diferenciados entre
6,61 MA y 1,2 MA, mientras que para el segundo de 8,79 MA a 1,73 MA.
Para el gen cyt b, el árbol bayesiano mostró el primer proceso de diversificación dentro del
género hace 23,69 MA cuando se dividió en dos grandes grupos. Estos a su vez divergieron a los
21,38 y 16,04 MA. Dentro del primer grupo. Los principales procesos de diferenciación se dieron
entre los 15,01 MA y 2,75 MA; para el segundo grupo, el mayor número de linajes fueron
diferenciados entre 10,59 y 1,77 MA.
Las divergencias entre las redes de haplotipos para NADH5 no fueron iguales a los que se
calcularon para los análisis bayesianos, pero si resultan tener alguna similitud sobre todo con el
primer tiempo calculado para la divergencia dentro del género, los tiempos de divergencia entre
los haplotipos calculados oscilaron entre 206.207,70 y 15.310.845,00 años. Para el cyt b la
menor divergencia fue de 76.988,00 y la mayor de 2.976.869,33 años. La diferencia encontrada
para esta prueba entre los dos genes puede deberse a que el análisis para NADH5 se realizó con
muchas más muestras lo que permite encontrar haplotipos más o menos divergentes que si tengo
una población pequeña, adicionalmente debe tenerse en cuenta que las tasas de mutación para los
dos genes son muy diferentes.

63
De acuerdo con las divergencias encontradas entre los haplotipos para las secuencias de NADH5
se sugiere una diversificación entre 206.207,70 y 15.310.845,00 años, lo que es un tiempo mucho
mayor al calculado preliminarmente por Ruiz–García et al, (2012a), con 24 secuencias de Cyt b,
y los tiempos para el mismo gen calculados en el presente estudio, en donde se encontró una
posible diversificación del género en los últimos 3 millones de años e incluso los últimos 40.000,
sin embargo como se mencionó antes este resultado esta problablemente muy sesgado al número
de muestras utilizadas y a que el valor de mutación utilizado para el gen de Cyt b es menor al que
se utiliza para NADH5.
De acuerdo con el punto de vista paleontológico, al parecer Potos está altamente emparentado
con el género Bassaricyon, basados en su presunta relación con los géneros extintos
Bassaricyonoides y Parapotos que de acuerdo a lo descrito por Baskin (2003) parecen divergir en
el Mioceno Temprano (15-20 Millones de años), lo cual concuerda con Simpson’s (1945) quien
supone un ancestro del Kinkajou en el Mioceno sobre la base de su morfología altamente
derivada.
Sin embargo, teniendo en cuenta los datos moleculares (Fulton y Strobeck, 2006, Koepfli et al,
2007), Potos no sería un linaje hermano de Bassaricyon. Así entonces el registro fósil más
cercano, ya que Potos no cuenta con registro fósil, es Parapotos cuya diversificación habría
ocurrido en el Mioceno en Norteamérica (15 Millones de años). Por lo tanto de acuerdo con los
datos paleontológicos, no está claro cuando el género Potos apareció y donde comenzó su
diversificación. La revisión realizada por Soibelzon & Prevosti (2013), acerca de los fósiles
sudamericanos de Carnívoros, muestra que sólo dos géneros actuales de Prociónidos aparecen en
el registro fósil (Procyon y Nasua) más otros dos géneros extintos (Cyonasua y Chapadmalania)
y ninguno de ellos está relacionado con Potos.
Por otra parte, para calcular los tiempos de divergencia mediante procedimientos bayesianos, se
debe partir de un registro fósil, teniendo en cuenta la carencia del registro en este caso, se tomó
como base los estudios moleculares de Koepfli et al. (2007).
Resulta interesante encontrar que los tiempos de divergencia tan antiguos encontrados en este
estudio, parecen ser lógicos siguiendo diversos estudios moleculares que han demostrado que

64
Potos es el género más basal de Procyonidae (Fulton y Strobeck, 2006, Koepfli et al, 2007),
siendo la primera taxa a divergir de los prociónidos existentes.
Los resultados obtenidos en este estudio apoyan la hipótesis formulada por Koepfli et al. (2007),
que sugiere que la diversificación de los actuales prociónidos neotropicales, parece ser antes de la
formación del istmo de panamá (alrededor de 3 millones de años atrás). Así la radiación de
especies de Procyonidae, por lo menos para P. flavus precede el gran intercambio biótico
americano dado por la formación del ismo de Panamá.
De acuerdo con estas estimaciones se sugiere que la diversificación de Potos se inició en el
Mioceno medio, continuando en el Mioceno tardío, este período coincide con el proceso de
mayor enfriamiento global, junto con la formación de la capa de hielo de la Antártida y una
importante caída en el nivel global del mar (Haq et al. 1987, Zachos et al. 2001; Miller et al.
2005), así mismo los bosques tropicales se redujeron y se transformaron en ambientes de Sabana
(Potts & Behrensmeyer 1992). Estos eventos jugaron un papel destacado en la diferenciación de
otras especies de mamíferos (Delsuc et al. 2004, Johnson et al. 2006.).
Estos cambios en el clima y en el nivel del mar aumentaron en conjunto la aridez terrestre y la
estacionalidad, que promovió un cambio de los hábitats de vegetación de ambientes cerrados
(bosques tropicales y subtropicales) a hábitats de vegetación más abiertos (bosques y pastizales),
lo que probablemente promovió la diversificación del Kinkajou.
Este tipo de fenómenos pueden explicar la diferenciación encontrada entre las poblaciones
restringidas a las cordilleras con otras poblaciones de tierras bajas. Sin embargo, se recomienda
realizar estudios que incluyan todas las subespecies propuestas morfológicamente, especialmente
de las poblaciones centro americanas para determinar si el punto de diversificación de la especie
se dio en esa zona y los haplotipos ancestrales se encuentran allí, adicionalmente el estudio de
estos haplotipos podrían confirmar los valores de divergencia encontrados en este estudio.
También se recomienda ampliar la población muestreada de chapadensis incluyendo individuos
provenientes del Matto Groso en Brasil para poder determinar si estos también se diferencias de
las poblaciones amazónicas o este fenómeno se da por estar en el límite de la cordillera en el caso
de las muestras bolivianas.

65
Por último un estudio con genes nucleares a través de microsatélites, podrían permitir la
evaluación de la tendencia poblacional más reciente de la especie, ya que como se observa en la
figura 17, hay indicios de un descenso poblacional en los últimos 15.000 años.
En conclusión:
1. P.f.chapadensis y los individuos peruanos provenientes de los departamentos de Pasco,
Huanaco, y Ucayali parecen conformar subespecies (sensu Ford y Hoffmann ) o cuando menos,
unidades evolutivas significativas diferentes (sensu Moritz 1994) que ameritan ser conservadas
independientemente de la restante distribución analizada de P. flavus.
2. La especie muestra niveles muy altos de diversidad genética. Esta tendencia se conservó tanto
a nivel global, como cuando el análisis se realizó por subpoblaciones.
3. Aunque los estadísticos muestran valores significativos para los análisis de heterogeniedad
genética, los valores de flujo génico encontrados son altos entre las subpoblaciones evaluadas.
Las diferencias entre poblaciones la está aportando, principalmente, chapadensis, que es la que
difiere primordialmente de las demás. La población peruana ubicada en la zona andina central y
en la parte sur de la Amazonía de ese país, y las muestras de chiriquensis aportaron cierto grado
de heterogeneidad pero menor al encontrado en la población boliviana.
4. La especie, en general, muestra tendencia a determinar una expansión poblacional a través de
su historia evolutiva.
5. Los valores de divergencia entre Potos de acuerdo con la red de haplotipos construida para
NADH5 varían entre 206.207,70 y 15.310.845,00. Debe tenerse en cuenta que las redes de
haplotipos se encuentran sesgadas a que el modelo mutacional que se utilice tenga el mismo
comportamiento para la especie que se calculó y para la especie objeto de estudio.
6. Se sugiere un inicio de la diversificación del género en el Mioceno medio, antes de la
formación del istmo de Panamá.
7. Todos los cambios climatológicos y ambientales ocurridos durante el Mioceno, parecen haber
influido sobre los procesos de diferenciación de la especie entre sí.

66
9. BIBLIOGRAFIA
Allen, J.A., y F.M. Chapman. (1897). On a second collection of mammals from the island of
Trinidad, with desciptions of new species, and a note on some mammals from the island of
Dominica, W. I. Bull. Amer. Mus. Nat. Hist., 9:13-30
Agnarsson I, Matjaz K, May-Collado L.J. (2010). Dogs, cats, and kin: A molecular species-level
phylogeny of Carnivora. Molecular Phylogenetics and Evolution 54, 726–745
Bandelt, H.J, Forster, P., and Rohl, A. (1999) Median-joining networks for inferring intraspecific
phylogenies. Molecular Biology and Evolution, 16, 37-48.
Baskin, J.A. (1998). Mustelidae. In C. M. Janis, K. M. Scott, and L. L. Jacobs (Eds.), Evolution
of Tertiary Mammals of North America Volume 1 (pp. 152-173). Cambridge, England:
Cambridge University Press.
Baskin, J.A. (2003). New procyonines from the Hemingfordian and Barstovian of the gulf coast
and Nevada, including the first fossil record of the Potosini. Bulletin of the American Museum of
Natural History, 279, 125–146.
Baskin, J.A. (2004). Bassariscus and Probassariscus (Mammalia, Carnivora, Procyonidae) from
the early Barstovian (Middle Miocene). Journal of Vertebrate Paleontology, 24, 709–720.
Bininda-Emonds, O.R. P., Gittleman, J.L. y Purvis, A. (1999). Building large trees by combining
phylogenetic information: a complete phylogeny of the extant Carnivora (Mammalia). Biol. Rev.
74, pp. 143±175
Cabrera, A. (1958). Catálogo de los mamíferos de América del Sur. I (Metatheria, Unguiculata,
Carnívora). Revista del Museo Argentino de Ciencias Naturales “Bernardo Rivadavia,” Zoología
4:1-307.
Culver, M., Johnson, W.E., Pecon-Slattery, J. and O’Brien, S.J. (2000). Genomic ancestry of the
American puma (Puma concolor). Journal of Heredity, 91, 186-197.
Delsuc, F., Vizcaíno, S.F., and Douzery, E.J.P. (2004). Influence of Tertiary paleoenvironmental
changes on the diversification of South American mammals: a relaxed molecular clock study
with xenarthrans. BMC Evolutionary Biology, 4, 11–24.

67
Drummond, A.J., Ho, S.Y.W., Phillips, M.J., and Rambaut, A. (2006). Relaxed phylogenetics
and dating with confidence. PLOS Biology, 4(5), e88.
Drummond, A.J., Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling
trees. BMC Evolutionary Biology 7: 214.
Eisenberg, J.F., (1989). An introduction to the Carnivora. In: Gittleman,J.L. (Ed.), Carnivore
Behavior, Ecology, and Evolution. Cornell University Press, Ithaca, NY, pp. 1–9.
Eizirik, E., Bonatto, S. L., Johnson, W. E., Crawshaw Jr. P. G., Vié, J. C., Brousset, D. M.,
O’Brien, S. J. and Salzano, F. M. (1998). Phylogeographic patterns and evolution of the
mitochondrial DNA control region in two Neotropical cats (Mammalia, Felidae). Journal of
Molecular Evolution, 47, 613-624.
Flynn, J.J., Nedbal, M.A., (1998). Phylogeny of the Carnivora (Mammalia):congruence vs
incompatibility among multiple data sets. Mol. Phylogenet. Evol. 9, 414–426.
Ford, L. S., and Hoffmann, R. S. (1988). Potos flavus. Mammalian Species, 321, 1-9.
Fu, Y. X. (1997). Statistical tests of neutrality against population growth, hitchhiking and
background selection. Genetics, 147, 915-925.
Fu, Y. X., and Li, W. H., (1993). Statistical tests of neutrality of mutations. Genetics, 133, 693-
709.
Fuller T.K, Mills M.G.L, Borner M. (1992) Long distance dispersal by African wild dogs in East
and South Africa. Journal of African Zoology, 106, 535–537.
Fulton, T.L., and Strobeck, C. (2006). Molecular phylogeny of the Arctoidea (Carnivora): effect
of missing data on supertree and supermatrix analyses of multiple gene data sets. Molecular
Phylogenetics and Evolution, 41, 165-181.
Greenwood PJ (1980) Mating systems, philopatry, and dispersal in birds and mammals. Animal
Behavior, 28, 1140–1162.

68
Haq, B.U., Hardenbol, J., and Vail, P.R. (1987). Chronology of fluctuating sea levels since the
Triassic. Science, 235, 1156-1167.
Harpending, H., (1994). Signature of ancient population growth in a low resolution mitochondrial
DNA mismatch distribution. Human Biology, 66, 591-600.
Harpending, H. C., Sherry, S. T., Rogers, A. R., and Stoneking, M. (1993). Genetic structure of
ancient human populations. Current Antrophology, 34, 483-496.
Hasegawa M., Kishino H., and Yano T., 1985. Dating the human-ape split by a molecular clock
of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.
Hall, E. R. (1981). The mammals of North America. Second Ed. New York, New York: John
Wiley and Sons.
Hershkovitz, P. (1972). The recent mammals of the Neotropical region: a zoogeographical and
ecological review. In A. Keast, F. Erk, and B. Glass (Eds.). Evolution, mammals, and southern
continents (pp. 311-431). Albany, New York: State University of New York Press.
Hudson, R.R., Boss, D.D., and Kaplan, N.L. (1992a). A statistical test for detecting population
subdivision. Molecular Biology and Evolution, 9, 138-151.
Hudson, R.R., Slatkin, M. and Maddison, W.P. (1992b). Estimations of levels of gene flow from
DNA sequence data. Genetics, 132, 583-589.
Hudson, R.R. (2000). A new statistic for detecting genetic differentiation. Genetics, 155, 2011-
2014.
Hunt Jr., R.M. (1996). Biogeography of the Order Carnivora. In J. L. Gittleman (Ed.), Carnivore
Behavior, Ecology and Evolution, vol. 2. (pp. 485-541). Ithaca, New York: Cornell University
Press.
Irwin, D.M., Kocher, T.D., and Wilson, A.C. (1991). Evolution of the cytochrome b gene of
mammals. Journal of Molecular Evolution, 32, 128–144.

69
Johnson, W.E., Eizirik, E., Pecon-Slattery, J., Murphy, W.J., Antunes, A., Teeling, E., and
O’Brien, S.J. (2006). The Late Miocene radiation of the modern Felidae: a genetic assessment.
Science, 311, 73–77.
Kays R.W (1999). The solitary group life of a frugivorous carnivore: ecology, behavior, and
genetics of kinkajous (Potos flavus). PhD Dissertation. University of Tennessee, Knoxville,TN.
Kays, R. W., Gittleman, J. L. y Wayne R. K. (2000). Microsatellite analysis of kinkajou social
organization. Molecular Ecology 9, 743–751.
Koepfli, K-P., Gompper, M.E., Eizirik, E., Ho, C.C., Linden, L., Maldonado, J.E., and Wayne,
R.K. (2007). Phylogeny of the Procyonidae (Mammalia: Carnivora): molecules, morphology and
the Great American Interchange. Molecular Phylogenetics and Evolution, 43, 1076-1095.
Kortlucke, S. M. (1973). Morphological variation in the kinkajou, Potos flavus (Mammalia:
Procyonidae) in Middle America. Occasional Papers of the Museum of Natural History,
University of Kansas, 17, 1-36.
Leopold, A.S. (1959). Wildlife of Mexico. Univ. California Press, Berkeley, 568 pp.
Li Yu A, Qing-wei L, O.A. Ryder,y Ya-ping Zhang.(2004). Phylogenetic relationships within
mammalian order Carnivora indicated by sequences of two nuclear DNA genes. Molecular
Phylogenetics and Evolution 33, 694–705.
McNutt, J.W (1996). Sex-biased dispersal in African wild dogs, Lycaon pictus. Animal Behavior,
52, 1067–1077.
Miller, K.G., Kominz, M.A., Browning, J.V., Wright, J.D., Mountain, G.S., Katz, M.E.,
Sugarman, P.J., Cramer, B.S., Christie-Blick, N., and Pekar, S.F. (2005). The Phanerozoic record
of global sea-level change. Science, 310, 1293–1298.
Moritz C. (1994.) Defining “evolutionary significant units” for conservation. Trends in Ecology
and Evolution. 9, 373-375.

70
Morral, N., Bertrantpetit, J., and Estivill,. X (1994). The origin of the major cystic fibrosis
mutation (delta F508) in European populations. Nature Genetics, 7, 169-175.
Napolitano, C., Sanderson, J., Johnson, W. E., O’Brien, S. J., Rus Hoelzel, A., Freer, R.,
Dunstone, N., Ritland, K., and Poulin E. (2012). Population Genetics of the felid Leopardus
guigna in Southern South America: Identifying intraspecific units for conservation. In M. Ruiz-
García and J. Shostell, (Eds.), Molecular Population Genetics, Phylogenetics, Evolutionary
Biology and Conservation of the Neotropical Carnivores. New York, New York: Nova Science
Publishers.
Nei M. and Kumar S. (2000). Molecular Evolution and Phylogenetics. Oxford University Press,
New York.
O’Brien, S. J. (1994). A role for molecular genetics in biological conservation. Proc. Natl. Acad.
Science USA 91: 5748-5755.
Patterson, B., and Pascual, R. (1972). The fossil mammal fauna of South America. In A. Keast, F.
C. Erk, and B. Glass (Eds.). Evolution, mammals and southern continents. (pp. 1-543) Albany,
New York: State University.
Potts, R., and Behrensmeyer, A.K. (1992). Late cenozoic terrestrial ecosystems. In A. K.
Behrensmeyer, J. D. Damuth, W. A. DiMichele, R. Potts, H. D. Sues, and S. L. Wing (Eds.),
Terrestrial Ecosystems Through Time: Evolutionary Paleoecology of Terrestrial Plants and
Animals. (pp. 419-541). Chicago, Illinois: University of Chicago Press.
Rambaut, A., and Drummond, A. J. (2007). Tracer v1.4. Available from http: beast. bio.ed.ac.uk
Tracer.
Ramos-Onsins, S. E., and Rozas, J. (2002). Statistical properties of new neutrality tests against
population growth. Molecular Biology and Evolution, 19, 2092-2100.
Rogers, A. R., Fraley, A. E., Bamshad, M. J., Watkins, W. S., and Jorde, L. B. (1996).
Mitochondrial mismatch analysis is insensitive to the mutational process. Molecular Biology and
Evolution, 13, 895-902.

71
Rogers, A. R., y Harpending, H. C. (1992). Population growth makes waves in the distribution of
pairwise genetic differences. Molecular Biology and Evolution, 9, 552-569.
Rogers, A. R., Fraley, A. E., Bamshad, M. J., Watkins, W. S., y Jorde, L. B. (1996).
Mitochondrial mismatch analysis is insensitive to the mutational process. Molecular Biology and
Evolution, 13, 895-902.
Rozas J, Sánchez-DelBarrio J, Messenguer X & Rozas R. (2010). DnaSP, DNA polymorphism
analyses by the coalescent and other methods. Bioinformatics 19, 2496-2497.
Ruiz-garcia. M.. J.M. Shostell. ( 2012). An Introduction to the Neotropical Carnivores In M.
Ruiz-Garcia & J.M. Shostell. Molecular Population Genetics. Phylogenetics. Evolutionary
Biology and Conservation of the Neotropical Carnivores. Hauppauge. New York: Nova Science
Publishers. Pp 1.
Ruiz-Garcia. M.. N. Lichilin- Ortiz y M. Jaramillo. (2012a). Molecular phylogenetics of two
neotropical carnivores, Potos flavus (PROCYONIDAE) and Eira barbara (MUSTELIDAE): No
clear existence of putative morphological subespecies. In M. Ruiz Garcia & J.M. Shostell.
Molecular Population Genetics. Phylogenetics. Evolutionary Biology and Conservation of the
Neotropical Carnivores. Hauppauge. New York: Nova Science Publishers. Pp 37.
Ruiz-García, M., Rivas-Sánchez, D., and Lichilín-Ortiz, N. (2012b) Phylogenetics relationships
among four putative taxa of foxes of the Pseudoalopex genus (Canidae, Carnivora) and molecular
population genetics of Ps. culpaeus and Ps. sechurae. In Ruiz-García, M., Shostell, J. (Eds.),
Molecular Population Genetics, Phylogenetics Evolutionary Biology and Conservation of the
Neotropical Carnivores. New York, New York: Nova Science Publishers.
Ruiz-García, M., Cossíos, D., Lucherini, M., Yáñez, J., Pinedo, M., and Angers, B. (2012c).
Population genetics and spatial structure in two Andean cats (the Pampas cat, Leopardus pajeros
and the Andean mountain cat, L. jacobita) by means of nuclear and mitochondrial markers and
some notes on skull biometrics. In Ruiz-García, M., Shostell, J. (Eds.), Molecular Population
Genetics, Phylogenetics, Evolutionary Biology and Conservation of the Neotropical Carnivores.
New York, New York: Nova Science Publishers.

72
Ruiz-García, M., Vásquez, C., Murillo, M., Pinedo, M., and Álvarez, D. (2012d). Population
genetics and phylogeography of the largest wild cat in the Americas: An analysis of the jaguar by
means of microsatellites and mitochondrial gene sequences. In Ruiz-García, M., Shostell, J.
(Eds.), Molecular Population Genetics, Phylogenetics, Evolutionary Biology and Conservation of
the Neotropical Carnivores. New York, New York: Nova Science Publishers.
Ruiz-García, M., y Pinedo-Castro, M. (2012). Population genetics and phylogeographic analyses
of the jaguarundi (Puma yagouaroundi) by means of three mitochondrial markers: the first
molecular population study of this species. In Ruiz-García, M., Shostell, J. (Eds.), Molecular
Population Genetics, Phylogenetics, Evolutionary Biology and Conservation of the Neotropical
Carnivores. New York, New York: Nova Science Publishers.
Saillard, J., Forster, P., Lynnerup, N., Bandelt, H-J, and Norby, S. (2000). mtDNA variation
among Greenland Eskimos: the edge of the Beringian expansion. American Journal of Human
Genetics, 67, 718-726.
Saitou N. and Nei M. (1987). The neighbor-joining method: A new method for reconstructing
phylogenetic trees. Molecular Biology and Evolution 4:406-425.
Sambrook J, Fritsch EF, Maniatis T. (1989). Molecular cloning: A laboratory manual. 2nd ed.
New York: Cold Spring Harbor Laboratory Press.
Simonsen, K.L., Churchill, G.A., Aquadro, C.F. 1995. Properties of statistical tests of neutrality
for DNA polymorphism data. Genetics 141: 413-429.
Soibelzon, L., Prevosti, F. J. 2013. Fossils of South America land Carnivores (Carnivora,
Mammalia). In Ruiz-García, M., Shostell, J. (Eds.), Molecular Population Genetics,
Phylogenetics Evolutionary Biology and Conservation of the Neotropical Carnivores. New York,
New York: Nova Science Publishers. Pp. 509-527.
Simpson, G.G. (1945). The principles of classification and a classification of mammals. Bulletin
of the American Museum of Natural History, 85, 1-350.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA
polymorphism. Genetics, 123, 585-595.

73
Tamura K. (1992). Estimation of the number of nucleotide substitutions when there are strong
transition-transversion and G + C-content biases. Molecular Biology and Evolution 9:678-687.
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., and Kumar S. (2011). MEGA5:
Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance,
and Maximum Parsimony Methods. Molecular Biology and Evolution 28: 2731-2739
Tchaicka, L., Eizirik, E., De Oliveira, T., Cándido, J.F., and Freitas, T. (2006). Phylogeography
and population history of the crab-eating fox (Cerdocyon thous). Molecular Ecology, 16, 819-
838.
Vianna, J. A., Ayerdi, P., Medina-Vogel, G., Mangel, J. C., Zeballos, H., Apaza, M., and
Faugeron, S. (2010). Phylogeography of the marine otter (Lontra felina): Historical and
contemporary factors determining its distribution. Journal of Heredity, 101, 676–689.
Waits, L.P., Sullivan, J., O’Brien, S.J., Ward, R.H., (1999). Rapid radiation events in the Family
Ursidae indicated by likelihood phylogenetic estimation from multiple fragments of mtDNA.
Mol. Phylogenet. Evol. 13, 82–92.
Waser P.M. (1996) Patterns and consequences of dispersal in gregarious carnivores, pp. En:
Carnivore Behavior, Ecology, and Evolution, Vol. 2 (ed. Gittleman JL), pp. 267–295. Cornell
University Press, Ithaca, New York.
Wayne, R.K., Geffen, E., Girman, D., Koepfli, K., Lau, L., and Marshal, C. (1997). Molecular
Systematics of the Canidae. Systematic Biology, 46, 622-653.
Wozencraft, W.C., (1989). The phylogeny of the recent carnivora. In: Gittleman, J.L. (Ed.),
Carnivore Behavior, Ecology, and Evolution.Cornell University Press, Ithaca, NY, pp. 495–535.
Wozencraft, W.C. (2005). Order Carnivora. in D.E. Wilson, D.M. Reeder (Eds.), Mammal
Species of the World: A Taxonomic and Geographic Reference, The Johns Hopkins University
Press, Baltimore pp. 279–348
Wyss, A.R., Flynn, J.J., 1993. A phylogenetic anaysis and definition of the carnivore. In: Szalay,
F.S., Novacek, M., McKenna, M. (Eds.), Mammal Phylogeny. Springer, NY.

74
Zachos, J., Pagani, M., Sloan, L., Thomas, E., and Billups, K. (2001). Trends, rhythms, and
aberrations in global climate 65 Ma to present. Science, 292, 686-693.
Zeveloff, S.I., (2002). Raccoons: A Natural History. Smithsonian Institution Press, Washington,
DC.
75
ANEXOS
ANEXO 1. CROMATOGRAMAS