ciencia ultrarrÁpida: desvelar y … · roturas de enlace, dinámica del estado de transición) y...
TRANSCRIPT

Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp)Vol. 101, Nº. 2, pp 329-346, 2007VIII Programa de Promoción de la Cultura Científica y Tecnológica
CIENCIA ULTRARRÁPIDA: DESVELAR Y CONTROLAR LA DANZADE LOS ÁTOMOS
JESÚS SANTAMARÍA ANTONIO *
* Real Academia de Ciencias Exactas, Físicas y Naturales. Valverde 22. 28004 Madrid. Ftad. de Ciencias Químicas. Universidad Complutense. 28040 Madrid. [email protected]
RESUMEN
Se describe el carácter, significado y alcance de losestudios en tiempo real, mediante pulsos láser, de ladinámica y el control de los movimientos atómicos enlas moléculas, cuando se rompe o modifica un enlacequímico. Tras recordar los conceptos que fundamentanesta nueva disciplina, Femtoquímica y Control Cuán-tico, se describen los experimentos seminales, y losmodelos teóricos y técnicas de simulación que seemplean. Finalmente se ponen en contexto estosavances dentro de la ciencia atómico-molecular.
I. INTRODUCCIÓN (1)
Para desvelar y controlar las dinámicas reactiva yestructural de los diversos agregados atómicos (molé-culas, cristales, clusters, líquidos, etc.) se necesitanpulsos ultracortos de luz láser y Rayos X, que operenen la escala de tiempo en que los átomos se mueven.Esta escala viene dada por la velocidad del sonido enun medio denso, es decir aproximadamente 1000 m/s,o sea 0.01 Ångstroms/ femtosegundo. Un femtose-gundo (fs), segundos, es a un segundo lo que unsegundo es a 32 millones de años, mientras que 0.01 Åes la centésima parte de una distancia interatómicatípica.
El problema para seguir el movimiento atómico noes tan simple como en el uso del estroboscopio paraobjetos macroscópicos, donde el flash de luz no modi-fica los estados dinámicos y la resolución ∆x, viene
dada por ∆x v∆t, donde v es la velocidad del objeto y∆t el intervalo entre disparos. En las partículasmicroscópicas, su comportamiento dinámico vienedescrito por un paquete de ondas coherente localizadoen el espacio ∆x, que se mueve con una velocidad degrupo en un rango de impulsos (k vector deondas).
El paquete cumple la relación (prin-cipio de incertidumbre). Por otra parte, para unapartícula libre con Energía , la incer-tidumbre energética es , que debe ser delorden de la anchura del pulso láser en función delintervalo de frecuencias del mismo, es decir,
, con lo cual se cumple (otra relación, que no principio, de incertidumbre).Esto supone una incertidumbre grande en la energíapara la escala de tiempos del fs. Así a un pulso de50-100 fs le corresponde . Sinembargo, combinando ambas relaciones de incer-tidumbre, se llega a la expresión , similara la resolución clásica, pero con distinto significado ydonde queda obviado el principio de incertidumbre encuanto que un pequeño valor de ∆t conduce a unpequeño valor de ∆x, es decir hemos logrado la locali-zación en el espacio del paquete de ondas, aunque sudispersión en energía sea grande.
En un experimento típico, con el primer pulso láser(pulso de bombeo) se excitan instantánea y coherente-mente un conjunto de niveles vibracionales de unenlace químico (oscilador anarmónico) formando unpaquete de ondas no estacionario (estado coherente)
=
1510−
p k∆ = ∆h
2x p∆ ∆ ≥ h
2 2E p m=
E tω∆ = ∆ ≈ ∆h h E t∆ ∆ ≈ h
1100 50 cmE −∆ = −

Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio330
que evoluciona como un objeto clásico en el estadoexcitado. A continuación, el segundo pulso (pulso desonda) con un retardo del orden de los picosegundos,baja el sistema al estado fundamental en una posicióndistinta a la inicial o lo sube a otro estado excitado(primer punto estroboscópico). Repitiendo el proceso,pero cambiando el retardo del segundo pulso (cambiodel camino óptico) resulta otra distancia y así sucesi-vamente de forma que se obtiene la visión estro-boscópica del movimiento relativo de los átomos en elestado excitado. Estos experimentos no son de molé-cula única (como aquellos que hoy son posibles sobresuperficies mediante microscopios de efecto túnel)sino que se realizan sobre millones de moléculas sin-cronizadas en el instante inicial dentro de una posiciónrelativa inicial de átomos ∆x(0).
De esta manera se han estudiado procesos elemen-tales unimoleculares (pulsos cuánticos vibracionales,roturas de enlace, dinámica del estado de transición) ybimoleculares (rotura y formación simultánea deenlaces) y reacciones complejas de Q. Orgánica eInorgánica, así como procesos de transferencia pro-tónica y electrónica en clusters y disoluciones, etc.
Entender los procesos atómicos ha sido el propó-sito de muchos científicos durante siglos. Al principiocambiando parámetros macroscópicos como p, T, pH,concentración, etc. se pudo controlar los rendimientosde productos químicos deseados. Sin embargo, esosmétodos, incluyendo la catálisis, estaban basados encolisiones incoherentes entre las moléculas y no per-mitían un control activo de los procesos microscó-picos. El control activo implica el uso de camposexternos, cuya intensidad, fase y distribución de fre-cuencias puede variar en el tiempo, conduciendo a lamodificación de la dinámica molecular (los procesosdinámicos cuánticos son procesos de ondas sujetos ainterferencias constructivas y destructivas) cambiandola evolución de los reactivos y generando un rendi-miento más alto de los productos deseados.
Se han propuesto métodos muy diversos, desde eldescrito de bombeo y sonda y otros tales como elcontrol de amplitud y fase, el camino adiábático deRaman estimulado, etc., hasta esquemas generalesvariacionales (control óptimo) y algoritmos genera-tivos automáticos, siendo hoy día este tema del controlcuántico un campo muy activo de investigación.
El trabajo se estructura como sigue. Se repasan, enprimer lugar, los conceptos en que se apoya la disci-plina Femtoquímica y Control Cuántico, para describira continuación los experimentos originarios y luegolos fundamentos de los modelos teóricos y las técnicasde simulación. Finalmente se delimita el campo deesta disciplina en relación con otros recientes avancesen la ciencia atómico-molecular.
II. CONCEPTOS FUNDAMENTALES (2,3)
Los descubrimientos más notables del siglo xx enciencia atomico-molecular fueron el establecimientodefinitivo de la existencia de los átomos (J.B.Perrin,1908), la formulación de la Mecánica Cuántica(W. Heisenberg, E. Schrödinger, 1925-26) y el des-cubrimiento del láser (T. H. Maiman,1960).
Según la teoría atómica, la velocidad de los áto-mos, en un modelo balístico o elástico, viene dada porla velocidad del sonido en el medio en que se muevany es del orden de 1000 m/s, es decir 0.01 A/fs. Losmovimientos de los átomos en las moléculas (vibra-ciones moleculares) ocurren en el rango 1000-3000 , o sea 33-10 fs, mientras que las rotacionesse realizan en tiempos de aproximadamente 300 fs.Por otra parte, la velocidad de la luz es de 3000 A/fs,es decir los fotones se transmiten a una velocidad3.105 veces rápida que los átomos y puede conside-rarse instantánea a escala molecular. Además eltamaño de los átomos, establecido en medidas dedifracción electrónica y rayos X es del orden delängstrom. El establecimiento de la existencia de lasmoléculas (T. Svedberg, 1906) y su estructura espacialatómica (van´t Hoff, 1874) ayudó a crear la idea deSuperficie de energía potencial (Marcelin 1915;Eyring y Polanyi 1931) como campo de fuerzas con-servativo interatómico, a cuya determinación dedicasus esfuerzos la Química Cuántica.
La ecuación de Schrödinger dependiente deltiempo es la ecuación fundamental de la MecánicaCuántica no relativista, en la que se describen todoslos fenómenos microscópicos. El tiempo es la mag-nitud más importante en la aplicación de la MecánicaCuántica a dos de los procesos más importantes enQuímica Física: La Dinámica Reactiva y la Fotoquí-
1cm−

mica. Los átomos no se pueden ver, en primer lugarporque la longitud de onda de la luz visible (~5000A)es muy superior al tamaño del átomo, pero además yfundamentalmente porque éstos caen dentro delmundo microscópico mecanocuántico, que está some-tido al principio de incertidumbre. La descripcióndinámica de los átomos debe hacerse mediantepaquetes de ondas, que son los objetos centrales de lamecánica cuántica dependiente del tiempo, los cualespor otra parte permiten una descripción muy cercana ala clásica. Según la mecánica cuántica, los paquetes noson observables mecanocuánticos, sino que lo son úni-camente sus cuadrados. Veremos además que, en losexperimentos de Femtoquímica, lo que es observable(debido a limitaciones de las señales de medida) es elcomportamiento coherente del colectivo de varios mi-llones de moléculas en procesos de preparación yprueba con pulsos ultracortos (generalmente en elrango 10-100 fs) del láser. En efecto, la sincronizaciónde los pulsos de luz con las oscilaciones molecularesda una imagen estroboscópica de las vibraciones mole-culares de los átomos.
1. Paquetes de ondas
Los procesos dinámicos al nivel más detallado(nivel microscópico cuántico) son fenómenos de ondasy manifiestan por lo tanto interferencias cuánticas. Ladescripción dinámica de los átomos debe hacersemediante paquetes de ondas, que son los objetos cen-trales de la mecánica cuántica dependiente del tiempo,los cuales por otra parte permiten una descripción muycercana a la clásica. En efecto, el centro del paquete,tanto en posición como en momento se describe me-diante una trayectoria clásica (teoría de Ehrenfest) a lavez que su anchura espacial es una manifestacióndirecta del principio de incertidumbre de Heisenberg.En concreto, el movimiento de los átomos dentro deuna molécula se describe por una superposición cohe-rente de estados vibracionales (paquete de ondas). Delas relaciones de incertidumbre y ,resulta , que indica que la corta inter-acción entre la radiación del pulso láser da un ensan-chamiento en la energía que es compatible con unalocalización en la posición. Esta idea de la formaciónde un paquete de ondas en la superficie de potencial deun estado excitado, supone la plasmación teórica de loque se realiza en un laboratorio experimental con pul-
sos láser ultracortos, donde el proceso de excitaciónFranck-Condon desde el estado fundamental, se puedeconsiderar instantáneo y la evolución temporal estáasegurada al no ser el paquete una solución estacio-naria del estado excitado.
2. Pulsos láser ultracortos
En Espectroscopia tradicional, las moléculas sonirradiadas por un láser de onda continua más o menosmonocromático o con una lámpara, obteniéndose unespectro con líneas definidas. Si el proceso es incohe-rente (caso más corriente) se produce una saturación ysolo el 50% se transfiere desde el estado inicial al final,alcanzándose, en ausencia de emisión espontánea, unestado estacionario. En cambio si el proceso es cohe-rente, las amplitudes de probabilidad de cada transi-ción dependen de la frecuencia de Rabi, ,donde µE es el momento de transición, y la absorcióny emisión estimulada se suceden alternativamente enel tiempo. Las frecuencias ópticas están en el intervalo1013-1015 , mientras que la frecuencia de Rabi estípicamente del orden de 108 para campos no exce-sivamente fuertes (107-109 ). En el caso inco-herente, la evolución del sistema viene determinadapor el valor de los coeficientes de Einstein y se puedepredecir integrando las ecuaciones de balance de flujode poblaciones. En el caso coherente, hay que resolverla ecuación de Schrödinger dependiente del tiempo delsistema, donde las probabilidades de cada procesodependen de la amplitud de probabilidad de los estadosy el comportamiento varía según las fases del sistemay de la interacción.
Con pulsos láser ultracortos, la interacción con lamolécula es más eficiente, dado que en la anchuraespectral del pulso láser existe una distribución conti-nua y coherente de componentes Fourier de diferentelongitud de onda. Por tanto, cuando una molécula esexcitada por un pulso láser, se crea un paquete deondas que da cuenta del movimiento de la molécula enun nuevo estado superposición coherente. Si esepaquete de ondas en un estado excitado ha sidopreparado por un pulso láser de débil intensidad, suevolución puede seguirse por teoría de perturbacionesdependiente del tiempo, mientras que si la intensidades grande (>1012 ) no se puede emplear la teoríade perturbaciones.
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 331
x p∆ ∆ ≈ h E t∆ ∆ ≈ hx p m t∆ ≈ ∆
R Eω µ= h
1s−
1s−
2Wcm−
2Wcm−

El desarrollo de pulsos láser ultracortos se haperseguido desde la aparición del láser. Así, medianteel método de conmutación rápida del factor Q (Q-switching), factor que da la relación entre la energíaalmacenada en el medio y la energía perdida en unciclo óptico, se consiguen pulsos de nanosegundos(Hellwarth,1961). Posteriormente, mediante la técnicade acoplamiento de pulsos (mode locking), es decirsintonizando las fases de los modos de la cavidad láser,se consiguen pulsos del orden de picosegundos (deMaria, 1966), menores que picosegundos con láseresde colorante (Shank,1974), y finalmente de femtose-gundos (Shank, 1987). Actualmente la técnica másusada consiste en el empleo de láseres de estado sólidode Ti- Zafiro que da pulsos de unos pocos femtosegun-dos de forma muy estable (Sibbet, 1991).
3. Coherencia y coherencia en fase
La coherencia viene ligada a conceptos mecano-cuánticos de superposición, estados coherentes, pa-quetes de onda, etc., e indica que interviene la natu-raleza compleja de las amplitudes de probabilidad, y esla base de la Mecánica Cuántica. Si se habla de lacoherencia en fase de la interacción entre el campo lá-ser y las moléculas, nos referimos a que las amplitudesde probabilidad de cada transición espectral dependede las amplitudes de probabilidad del estado del sis-tema y del campo láser.
En relación con la óptica y las ondas, la coherenciaindica que la fase de la onda de radiación varía de for-ma suave, continua y no al azar. Por tanto, la coheren-cia en fase de la onda de radiación indica una distribu-ción muy estrecha de frecuencias en la vecindad deuna resonancia, dando una forma lorentziana en casode ensanchamiento homogéneo. La coherencia de unaradiación puede ser espacial (en dirección perpendicu-lar a la propagación) o temporal (en la dirección depropagación), definiéndose en cada caso la longitud detiempo de coherencia.
4. Espectroscopia dinámica
En resumen, los conceptos anteriores modifican lamanera tradicional de proceder en Espectroscopiamolecular. Hasta hace poco, el problema se reducía a
analizar los elementos de matriz del “operador de tran-sición” en distintos órdenes de perturbación y asídeterminar a priori las reglas de selección por aspectosde simetría de las moléculas, donde la dinámica nojuega ningún papel y el campo externo sólo hace acti-var el operador de transición. Ahora, con la nueva tec-nología de láseres pulsados, se debe tener en cuenta lacoherencia del láser (los fotones deben estar en fase),la coherencia de la interacción (el láser genera super-posiciones coherentes en el sistema), el tiempo deinteracción y la intensidad del campo láser.
En esta situación, el espectro de absorción seobtiene por transformada de Fourier del tren de pulsosal dominio de frecuencias. En el dominio temporal, elpaquete de ondas es la convolución de la secuencia depulsos con la dinámica del estado excitado, mientrasque en el dominio de frecuencias, el paquete en el esta-do excitado es simplemente el producto de la envol-vente de frecuencias con el espectro Franck-Condon(esto es el solapamiento entre el estado inicial vibra-cional y el excitado). Como veremos, con un pulso tipoδ-Dirac, la transformada de Fourier es una excitaciónconstante afectando por igual a todo el espectroFranck-Condon (FC). Con dos pulsos δ-Dirac y unretardo t entre ellos, la transformada de Fourier es unespectro de picos espaciados 2π/t. Si la frecuencia esresonante y el desfase nulo entre pulsos, el espectro defrecuencias estará justamente sobre el espectro FCreforzándolo. Si, por el contrario, el desfase es de π(oposición de fase), la frecuencia del espectro de pul-sos estará sobre el espectro FC anulándolo y dandoabsorción nula. En el caso de un pulso de duraciónfinita, habrá una envolvente menor que el espectro FCtotal y el proceso de absorción dependerá del sola-pamiento del espectro de frecuencias con el FC y portanto dependerá de la forma del pulso y de la frecuen-cia del láser.
Conviene recordar que existe una dificultad inhe-rente en la relación de la Dinámica con la Espectros-copia: Para modificar espacialmente las propiedadesde una molécula, solamente puede manipularse elcampo externo en el dominio temporal o de frecuen-cias, mientras que el acoplamiento entre las dosdimensiones de la interacción (espacial y temporal) através del momento de transición no admite interven-ción por parte del experimentador (excepto en el casode campos muy fuertes).
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio332
5. Femtoquímica y Control cuántico deprocesos moleculares
La Femtoquímica trata de la visión dinámica de losátomos en la molécula al vibrar, rotar y reaccionarquímicamente; movimientos cuya escala natural detiempos es del orden del femtosegundo ( 10s).Esta dinámica es consecuencia de la excitación de lasmoléculas por medio de pulsos láser ultracortos, loscuales localizan dicha excitación en un espacio muyreducido bajo la forma de un paquete de ondas cohe-rente que evoluciona en el tiempo, generandomecanocuánticamente el movimiento real de los áto-mos en el proceso que se siga. Transcurridas dosdécadas desde que se produjeran las primeras aplica-ciones de los láseres de pulso ultracorto (láseres defemtosegundo) al estudio de los procesos químicos y
biológicos en tiempo real, hoy en día la Femtoquímicay Femtobiología constituyen nuevos campos en plenoauge dentro de la Química y la Biología. De hecho, nopuede dejar de mencionarse que el Premio Nobel deQuímica 1999 fuera otorgado al Prof. Ahmed H.Zewail del Instituto Tecnológico de California enPasedena (USA) por sus pioneros trabajos enFemtoquímica, concretamente en la espectroscopiadinámica del estado de transición.
Por otra parte, el campo no se refiere únicamente ala observación de la dinámica intramolecular, sino queincluye también el control cuántico de esos movimien-tos. Desde un punto de vista general, sabemos que lasinteracciones entre electrones y núcleos están media-das por los fotones. El objetivo sería ahora, al contrarioque en la mera observación, el usar los fotones para
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 333
1510−
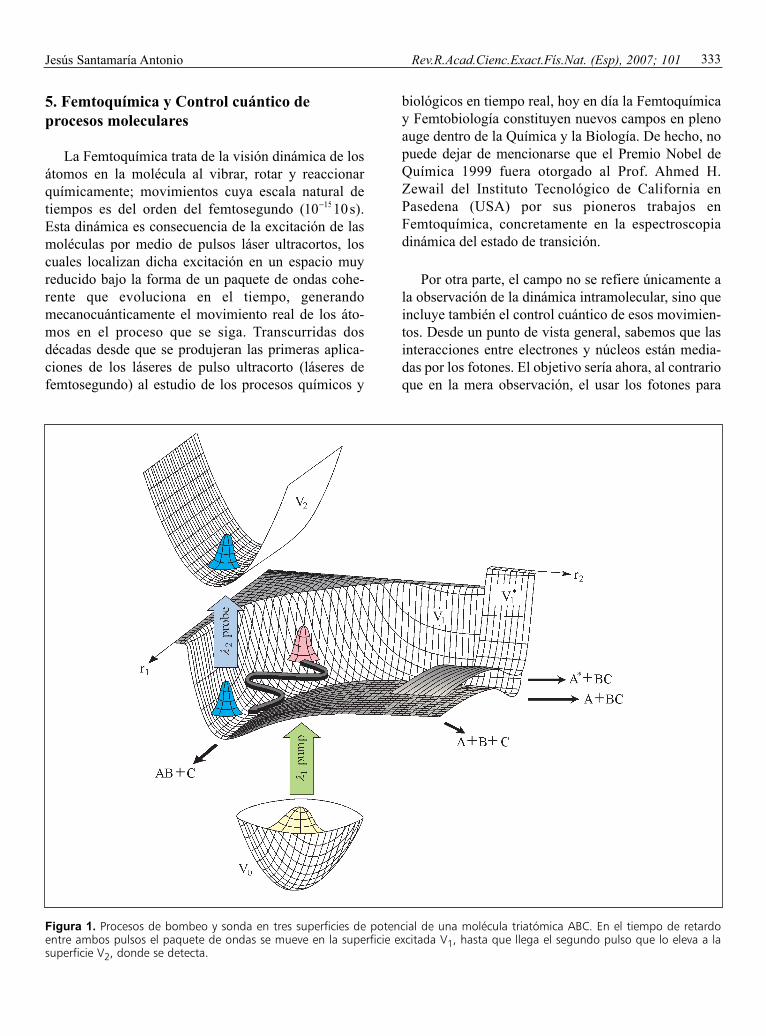
Figura 1. Procesos de bombeo y sonda en tres superficies de potencial de una molécula triatómica ABC. En el tiempo de retardoentre ambos pulsos el paquete de ondas se mueve en la superficie excitada V1, hasta que llega el segundo pulso que lo eleva a lasuperficie V2, donde se detecta.

controlar el movimiento de electrones y núcleos, sa-biendo que esos procesos son fenómenos de ondas,que manifiestan interferencias cuánticas. El objetivode un proceso de control cuántico consiste en alterarlos paquetes de ondas (superposición coherente deestados) de forma tal que, por ejemplo, las interferen-cias constructivas aparezcan a lo largo de un ciertocamino deseado de reacción, mientras que las destruc-tivas lo hagan en el resto de los posibles caminos.
III. RESULTADOS EXPERIMENTALES (4,5)
Los primeros experimentos en Femtoquímicafueron realizados por A. Zewail y colaboradores(1987,1988) utilizando láseres ultrarrápidos que pro-ducían pulsos de fotones de unas decenas de femtose-gundos, lo que era imprescindible si se quería seguir ladinámica cuántica de un proceso químico reactivo con
resolución de 0.1 A. En efecto, si la velocidad de losátomos (centro del paquete de ondas de Ehrenfest) esdel orden de 105cm/s, se requiere a partir de
, un intervalo ∆t del orden de decenas defs.
Para conseguir la película estroboscópica de unareacción química en el campo de fuerzas interatómicas(superficie electrónica de energía potencial mecano-cuántica), un primer pulso láser de bombeo excita lasmoléculas de la superficie fundamental a un punto deuna superficie excitada previsiblemente disociativa,iniciándose (origen de tiempos) el movimiento delpaquete de ondas en ella. Con un retardo variable delorden de un centenar de fs se dispara un segundopulso de sonda, que detecta el estado en tránsito(estado de transición) del sistema molecular en esetiempo de desfase entre ambos pulsos. Repitiendonumerosas veces el proceso con diferentes desfases
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio334
Figura 2. Fotodisociación del ICN sobre la superficie excitada a t=100, 200 fs (superior izda). Máximos de detección del fragmentoCN a diferentes tiempos (superior decha). Simulación cuántica de la evolución del paquete (inferior izda) y probabilidad asintóticaclásica frente a tiempo de desfase (inferior decha). (tomado de A. H. Zewail, 1988).
x p m t∆ = ∆

entre ambos pulsos, se obtiene cada vez una imagencongelada, y en conjunto una visión estroboscópica dela molécula evolucionando en un estado excitado. Elpulso de sonda permite detectar una propiedad delestado de transición (fluorescencia, etc.) íntimamenteligada a la estructura instantánea geométrico-espacialdel mismo (ver Figura 1).
Para ver el movimiento en sistemas reales, se debeformar, en primer lugar, un paquete de ondas en cadamolécula a través de la coherencia en fase entre la con-figuración inicial de equilibrio de la molécula (confi-namiento espacial de típicamente 0.05 A) y la interac-ción instantánea del pulso láser que crea el paquete enestado excitado, con un confinamiento también muypequeño. Por otra parte, debe existir un ensan-chamiento limitado (del orden de 0.1 A) en las posi-ciones del colectivo formado por los varios millonesde moléculas sobre los que se hace la medida experi-mental. Estos experimentos no son, por tanto, demolécula única, pero las coherencias molecular y delcolectivo garantizan que el movimiento detectado seaen la práctica el de la trayectoria de una molécula.
1. Fotodisociación del ICN (Zewail y col., 1987)
Se trata de investigar la ruptura de la molécula decianuro de yodo (ICN) en sus fragmentos yodo (I) yradical ciano (CN). Para ello, un primer pulso láserexcita electrónicamente las moléculas de ICN desde elestado fundamental al primer estado excitado, que esrepulsivo (ver Figura 2). El paquete de ondas, genera-do en la superficie repulsiva, evoluciona hacia la rup-tura del enlace I-C dando lugar a la formación de losfragmentos I y CN. A continuación se dispara el pulsoláser de sonda, cuya longitud de onda corresponde a latransición electrónica entre el estado fundamental y elprimer estado excitado del fragmento CN (B2Σ+). Elláser de sonda induce entonces la fluorescencia de esefragmento, que se mide en función del tiempo de retra-so entre el pulso de bombeo y el de sonda. De estemodo es posible medir el tiempo total de la reacción,que resulta ser de aproximadamente 200 fs. Cuando sesintoniza el láser de sonda a longitudes de onda próxi-mas, pero fuera de la resonancia del fragmento CN, esposible detectar configuraciones moleculares interme-dias entre el estado inicial y el final, es decir, estadosen tránsito desde los reactivos a los productos. De esta
forma es posible detectar y seguir la evolución de loscomplejos activados a lo largo de la coordenada dereacción.
2. Fotodisociación del NaI (Zewail y col., 1988)
En este segundo experimento, considerado comoparadigma de los libros de texto de Química Física, elgrupo de Zewail estudió la fotodisociación del yodurode sodio (NaI) en una superficie excitada provenientedel cruce prohibido a 6.9 A de dos superficies, unaiónica (muy profunda y estrecha, que a energías bajases la fundamental) y otra covalente (poco profunda ymás ancha) (ver Figura 3). A distancias menores de 6.9A, el paquete se mueve en la superficie covalente, peroen torno a ese punto existe una probabilidad de cruce ala otra superficie iónica. Se observa que si los átomosde Na y I se aproximan en la superficie covalente, serepelen entre sí, en especial a distancias cortas, mien-tras que sus iones y se atraen. En este experi-mento el láser de bombeo excita la molécula de NaI,propagando un paquete de ondas desde una distanciade 2.8 A en la superficie covalente. La evolución tem-poral y espacial del paquete de ondas es sondeada adistintos tiempos por un segundo pulso láser, detectán-dose la fluorescencia del fragmento Na o de los com-plejos activados . No todos los complejos activa-dos se disocian en Na y I, sino que existe una probabi-lidad finita de que, cuando se alcanza la zona de cruceentre ambas curvas, pasen a la curva iónica. De hecholos complejos sobreviven aproximadamente unas diezoscilaciones antes de disociarse completamente en susfragmentos atómicos.
3. Otras reacciones
Durante la década de los 90, el grupo de Zewail haaplicado la espectroscopia de femtosegundo al estudiode una gran variedad de reacciones químicas, tanto enfase gas como en fase condensada, unimoleculares(disociaciones, isomerizaciones, transferencias deproptón y electrón, reacciones retro Diles-Alder) ybimoleculares (como , y
), así como a procesos más comple-jos, que son importantes en los mecanismos involucra-dos en el mecanismo de la visión. Estudios de otros
Na +I−
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 335
NaI±
2H CO OH CO+ → +2Br I BrI I+ → +

investigadores abarcan muchas otras reacciones y tam-bién a procesos de la Biología, como la fotosíntesis einteracciones con el ADN, dando origen a la Femtobi-ología.
IV. TRATAMIENTOS TEÓRICOS (2,6,7)
El acoplamiento de la molécula con el láser generasuperposiciones coherentes entre los estados de lamolécula, cuya dinámica se sigue resolviendo laecuación de Schrödinger dependiente del tiempo. Parasimplificar el problema conviene trabajar en las sigu-ientes aproximaciones:
a) Separación de la dinámica molecular de ladinámica del campo externo. Consideramos que el
láser es suficientemente intenso y la muestra suficien-temente fina, desde el punto de vista óptico, como parapoder despreciar los efectos dispersivos del medio enla propagación del pulso. El hecho de asumir que elnúmero de fotones a la frecuencia deseada es mayorque el número de moléculas absorbentes, nos permiteutilizar la aproximación semiclásica para describir lainteracción del sistema con el láser, pero nos impide,en general, el utilizar aproximaciones perturbativaspara solucionar la ecuación de Schrödinger, exceptopara campos débiles.
b) Sistema molecular aislado. Asumimos que elestado inicial del sistema es el fundamental y despre-ciamos los procesos físicos que provocan pérdida decoherencia del proceso, bien en el láser (fluctuacionesde fase, etc.), bien en el sistema (fluorescencia, coli-siones). Esto permite prescindir de la formulación más
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio336
Figura 3. Femtoquímica de la reacción de NaI. Evolución del paquete entre las superficies iónica y covalente (superior izda). Evolucióntemporal frente a la separación internuclear (superior decha). Detección creciente de los fragmentos y detección menguante del com-plejo (inferior). (Tomado de A. H. Zewail, 1988).

general en función de la matriz densidad (ecuación deLiouville-von Neumann) que es más costosa de tratarnuméricamente.
c) Validez de la aproximación Born-Oppenheimer,es decir separación de los movimientos nuclear y elec-trónicos, , donde función de ondaelectrónica, depende paramétricamente de la coor-denada nuclear R; es decir existe un potencial elec-trónico promedio que interacciona con los núcleos encada instante del tiempo.
En general, los mecanismos para iniciar la excita-ción y obtener resolución temporal de los movimientosmoleculares (vibracionales y reactivos) son coherentesen fase y de tipo impulsivo, al contrario que la absor-ción resonante y el scattering Raman estimulado. Laexcitación está presente durante una fracción delperiodo vibracional del estado fundamental y creauna superposición de estados electrónicos (funda-mental y excitado ) y vibracionales ( y ):
donde Vg y Ve son los potenciales adiabáticos y µE esla interacción dipolar. Puede hallarse una solución entérminos de la frecuencia de Rabi, pero el problemapuede simplificarse introduciendo potenciales adia-báticos vestidos y transformaciones unitarias, queanulan los términos no diagonales del Hamiltoniano.
En la práctica, sin embargo, se supone que eldesfase electrónico es instantáneo y se define así unestado ficticio promocionado (paquete de ondas) concoherencia vibracional en fase que evoluciona porcierto periodo de tiempo. Por tanto, cuando se usanpulsos cortos, se obtienen varias ventajas: Localiza-ción de la excitación Franck-Condon en la coordenada,corta evolución durante el proceso de excitación yfacilidad de resonancia. La corta duración del pulsosimplifica los cálculos porque se puede ignorar elproceso de excitación y el estado fundamental. Elestado inicial en este caso es el estado excitado en lamisma posición de coordenada que el estado funda-mental original.
Un tratamiento general para el caso de dos estadospuede hacerse en el formalismo del operador de
evolución temporal y de la teoría de perturbaciones,suponiendo que ésta sea válida, lo que ocurre cuando
. Haciendo
y
con
se obtiene para el primer orden de perturbación:
La interpretación física de la anterior ecuación es lasiguiente: La función de onda inicial , evolucionaentre y bajo el hamiltoniano del estado fun-damental Hg. Al tiempo , el campo eléctrico interac-ciona con el momento dipolar de la transición pro-moviendo la amplitud al estado excitado, la cualevoluciona bajo influencia de He desde el tiempo altiempo t.
Cuando no es válida la teoría de perturbaciones y elhamiltoniano es función del tiempo, la representaciónintegral formalmente correcta de la función de onda es
donde T es el operador de orden temporal que pordefinición ordena cronológicamente los operadores enla serie de Taylor del llamado desarrollo de Magnus.Este desarrollo es una alternativa elegante a la teoríade perturbaciones y se puede truncar a cualquier ordendando expresiones unitarias para el propagador
donde , con
t′
t′t t′=0t =
V H
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 337
eψ
gψ
geψ eeψ gNψ eNψ
0 1H H Hλ= +
gψ

Así el primer coeficiente del desarrollo de Magnuses muy preciso para perturbaciones súbitas, mientrasque el segundo lo es para hamiltonianos que cambianadiabáticamente.
Volviendo a la fórmula del primer orden de pertur-bación, podemos definir un estado inicial efectivo
, como un estado que evoluciona enel potencial del estado excitado, dado que esúnicamente función propia en el potencial del estadofundamental. Dado que µ se puede considerar comouna función suave de x, ese estado efectivo es unaréplica del estado vibracional inicial, lo se conocecomo la aproximación Condon en la región Franck-Condon.
Las aplicaciones a diferentes secuencias de pulsosson muy interesantes. Así cuando tenemos un pulsotipo función delta, es decir , la pertur-bación de primer orden queda
es decir, la amplitud de primer orden es simplementeuna constante multiplicada por el estado efectivoinicial que se propaga en la superficie delestado excitado. Cualquier secuencia complicada depulsos se puede suponer como una superposición depaquetes de onda que pueden interferir. Así, si tenemosuna superposición de dos pulsos delta de la misma fre-cuencia, el primer pulso sube el estado vibracionalinicial al estado excitado donde evoluciona. A contin-uación, jugando con el retardo entre pulsos (t2-t1)podemos conseguir que el segundo pulso actúe en fase,en oposición de fase o en otras situaciones, pro-duciendo interferencias constructivas o destructivas.
La teoría de perturbaciones de segundo orden esnecesaria para explicar procesos de segundo orden, esdecir aquellos en que ocurre una segunda interaccióncon el campo eléctrico y se origina una evoluciónsobre una tercera superficie con Hamiltoniano Hc. Laamplitud de segundo orden viene dada como
con y . Esta fórmulasirve para interpretar los experimentos de tempo-
rización de reacciones químicas llevadas a cabo porZewail y que constituyeron el nacimiento de laFemtoquímica que hemos descrito en el apartadoanterior. Disponemos de tres intervalos de tiempo: Enel primero el sistema está en la superficie fun-damental con un Hamiltoniano Ha, en el segundo
en la superficie de hamiltoniano Hb y en eltercero en la superficie disociativa con Hc. Deesta forma se puede seguir la dinámica del paquete deondas. Anotando la frecuencia de la luz absorbida porel segundo pulso, se calcula la diferencia de energíasentre ambos estados excitados, lo que se puede co-rrelacionar con el retardo entre ambos pulsos parainferir el movimiento del paquete en el primer estadoexcitado y también el tiempo que tarda en alcanzar laregión asintótica del potencial, describiendo así laespectroscopia del estado de transición en la escala delos femtosegundos.
V. TÉCNICAS DE SIMULACIÓNNUMÉRICA (2,9-13)
Mientras que, en los métodos teóricos de aproxi-mación, basta con un pequeño número de términos deldesarrollo para obtener una representación valiosa dela solución, en el caso de los métodos numéricos seprecisan cientos de funciones de base o de iteracionespara aproximarnos al valor numérico, careciendo desentido cuando se usan pocas funciones o inter-acciones.
Existen dos tipos de métodos numéricos pararesolver la ecuación de Schrödinger, los basados en larepresentación de autofunciones y los de propa-gadores. Los primeros responden normalmente alenfoque tradicional de desarrollo en autofunciones debase del Hamiltoniano en ausencia de perturbación.Estas bases son de dos tipos: Representación en basefinita (FBR) y en variable discreta (DVR). Separandoel Hamiltoniano en dos partes , y eva-luando los elementos de matriz talque los elementos de matriz con respecto a H0 sondiagonales, mientras que la integral con respecto a Vpuede resolverse mediante cuadratura gussiana, seprocede a diagonalizar mediante algún algoritmoestándar (Jacobi, Lanczos, etc.) obteniéndose los auto-valores y autofunciones correspondientes.
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio338
jk j kH dx Hφ φ= ∫

Existe, además, otro método basado en autofun-ciones de la posición (tipo ondas planas) y no de laenergía, llamado de Hamiltoniano en malla de Fourier(Fourier grid Hamiltonian, FGH), que hace un usomuy inteligente del hecho que el operador de energíacinética es diagonal en la representación de momentos.De esta forma, los términos cinético y potencial delHamiltoniano, se evalúan en la representación en queson diagonales, realizándose a continuación el cambiode representación por transformadas de Fourier. En lamalla discreta de puntos equiespaciados, en que sedivide el espacio, se cumple que el producto delintervalo por la función delta de Dirac es igual alíndice Kronecker, es decir, . Los ele-mentos de la matriz hamiltoniana en una malla discretade puntos equiespaciados son
donde
Los elementos de matriz cinética vienen en funciónde los momentos , por lo cual hay que pasar a larepresentación de coordenadas, mediante una trans-formada de Fourier rápida, para obtener la función deondas en cada punto de la malla. Lo resultadosobtenidos por este método son similares a losobtenidos por el método DVR, utilizando en éste casocomo polinomios ortogonales al conjunto de ondasplanas.
Los métodos de propagadores se basan en lasolución formal de la ecuación de Schrödinger
, donde el factor expo-nencial es el operador de evolución temporal o propa-gador. Dos son los más usados. El primero es elmétodo de Feit-Fleck o del operador desdoblado (splitoperador) en parte cinética o potencial, que al tener encuenta la no conmutatividad de ambas, puede aproxi-
marse de la forma: . Este tipo de propagadores
pertenecen al grupo de los llamados locales, dado quetruncan el desarrollo en los primeros términos o bien,como en este caso, factorizan la exponencial. Para elcaso de dos estados moleculares acoplados con unláser, el propagador se define como
donde , es el acoplamiento vía momentobipolar y , es el hamiltoniano para cadaestado electrónico. A continuación, el operador expo-nencial se descompone en tres factores al aplicar laaproximación de desdoblar la parte potencial.
El otro método es el de propagadores de tipo globaldado que se desarrollan en polinomios ortogonales, enconcreto los de Chebyshev, que minimizan el errormáximo en el intervalo . En este método seemplea también la representación de momentos paradiagonalizar el operador de energía cinética. Porejemplo, dado un estado en un espacio bidi-mensional (molécula con dos grados de libertad) suevolución en una malla viene dada por
donde Tv son los polinomios de Chebyshev, αv son loscoeficientes del desarrollo y los elementos de matrizdel Hamiltoniano vienen definidoscomo
donde K es el operador cinético en la representación demomentos y V el operador potencial en la repre-sentación de coordenadas. Este método es muy precisoy se ha utilizado de forma rutinaria, habiendo servidopara popularizar los métodos numéricos de solución dela ecuación de Schrödinger dependiente del tiempo.
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 339
i ijx x Ixj δ∆ < >=
kαh
i iH T V= +
i j m nx y H x y

VI. CONTROL CUÁNTICO DE PROCESOSQUÍMICOS (14-20)
Uno de los sueños más largamente esperados porlos químicos es conseguir romper y formar selectiva-mente enlaces en una molécula, mediante manipu-lación de fotones provenientes de un láser. Desde suaparición, los láseres han sido considerados la herra-mienta idónea para ello, es decir para controlar lasreacciones químicas. La ruptura selectiva de enlacesen moléculas poliatómicas fue una búsqueda pro-longada y fallida durante más de 30 años. El esquemabásico era simple: Elegir la frecuencia de un modolocal asociado a un enlace y bombear intensamentecon un láser sintonizado a dicha frecuencia, hasta queel enlace se rompa. La dificultad del problema estribaen que la energía depositada localmente se puede dis-tribuir muy rápidamente en toda la molécula, a travésde acoplamientos con otros modos, lo que se llamarelajación vibracional intramolecular (IVR), destru-yendo la selectividad del proceso. Por otra parte, laexcitación multifotónica hasta disociación con camposláser intensos en el infrarrojo (IR), que implica laabsorción de muchos fotones, se logró experimental-mente en la década de los 70. La explicación de esteproceso, en moléculas de cuatro y más átomos, implicaun bombeo coherente de varios fotones (tres o cuatro)simultáneos (proceso no lineal) seguido de un bombeoincoherente de muchos más (varias decenas), peroahora consecutivos, hasta el continuo de disociación.Este proceso es muy poco específico en la energía delos productos o fragmentos por la dificultad de con-trolar el número de fotones absorbidos. En el caso de laabsorción multifotónica, con fotones del visible o UV,se obtiene la ionización multifotónica que puede serresonante o no resonante.
Como se ha dicho, los procesos dinámicos cuán-ticos son fenómenos de ondas, que estarán sometidos ainterferencias constructivas y destructivas. La selec-tividad de una actuación debe venir asegurada por lacoherencia de las ondas que describen el compor-tamiento de los átomos y las moléculas (Warren,1993).En la materia ordinaria, las interacciones entre núcleosy electrones están mediadas por fotones y, en lamayoría de las aplicaciones químicas, es una buenaaproximación suponer que la ecuación de Schrödingerse puede separar paramétricamente en un producto de
las partes nuclear y electrónica (aproximación deBorn-Oppenheimer), es decir, se puede separar elmovimiento de los núcleos del de los electrones. Laexcitación de un láser de onda continua de frecuenciafija produce, en condiciones de resonancia, un tránsitoentre niveles definidos, bien sean nucleares (excitaciónen IR) o electrónico-nucleares (excitación en el visibleo ultravioleta). Por el contrario, cuando e trabaja conpulsos láser cortos (por ejemplo de duración del ordendel fs.) existe una distribución continua y coherente decomponentes Fourier de diferente longitud de onda,dentro de la anchura espectral del pulso. Cuando lamolécula se excita por un pulso láser, se crea unpaquete de ondas que da cuenta del movimiento de lamolécula en un estado coherente, que es una super-posición de estados excitados.
El objetivo de un proceso de control cuántico con-siste en alterar esa superposición de estados, de formaque la interferencia constructiva máxima ocurra a lolargo del camino deseado, mientras que las interfe-rencias destructivas lo hagan en el resto de caminos,consiguiéndose de ese modo el control selectivo.
En general, lo que se busca son esquemas para opti-mizar el rendimiento de la transferencia de poblaciónentre niveles vibracionales de moléculas, a través de lainteracción coherente de esos movimientos vibra-cionales y las características de modulación deamplitud, frecuencia y fase del campo láser, es decir, através de efectos de interferencias cuánticas. El controlcuántico se puede aplicar a reacciones unimolecularesmediante esquemas de bombeo y sonda (métodopump-probe de Tannor y Rice,1985), también lla-mados de control o transferencia de amplitud, dondeesta transferencia no es sensible a la fase de laamplitud preexistente. Estas contribuciones teóricastuvieron especial relevancia en el nacimiento de laFemtoquímica. También puede emplearse el métodode control de fase y amplitud de Scherer (1990), con elque la excitación producida en una molécula (moléculade I2) oscila dramáticamente en función del retardoentre pulsos. Brumer y Shapiro (1986) propusieron elmétodo de control coherente a través del uso de doscaminos con frecuencias muy distintas, donde la pro-babilidad de cada camino es proporcional al cuadradode la suma de amplitudes de transición desde el estadoinicial al final (este método es equivalente a ladifracción de la luz visible en doble rendija).
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio340

Para preparar moléculas en estados cuánticosespecíficos, se pueden usar métodos tipo Raman esti-mulado (Bergmann, 1989) llamados STIRAP (stimu-lated Raman via adiabatic passage) que, en sistemas detres niveles tipo (lambda) consiguen una gran efi-cacia, lanzando primero el láser de sonda paradespoblar el estado intermedio antes del láser debombeo (método antiintuitivo). El método APLIP (adi-abatic passage via light induced potential) fue pro-puesto por Garraway y Suominen en la década de los80, como extensión del método STIRAP para camposintensos y procesos de absorción de dos fotones. Otrosmétodos también adiabáticos, con control de amplitudy frecuencia de barrido, son los denominados ARP(adiabatic rapid passage), como el esquema de fre-cuencia de barrido CARP (chirped ARP) que va va-riando la frecuencia del pulso láser, etc. Chelkowski,Bandrauk y Corkum (1990) emplearon una variante delos métodos ARP que podemos llamar subida enescalera vibracional (vibrational ladder climbing)para pasar a niveles altos con campos fuertes.
Se ha ideado además un esquema general varia-cional especialmente útil que trata de optimizar elrendimiento teniendo en cuenta una serie de restric-ciones, llamado algoritmo de control óptimo (Rabitz,1988). Finalmente se han desarrollado algoritmos evo-lutivos que van adaptando cíclicamente las caracterís-ticas del pulso al efecto deseado (Gerber, 1998).
El fundamento físico del control con láser de latransferencia de población entre niveles radica en losesquemas de transferencia coherente de población. Enefecto, en la descripción incoherente, basada en loscoeficientes de Einstein, la probabilidad de transiciónviene dada por la fórmula ,donde B es el coeficiente de emisión o absorciónestimulada e I(t) es la intensidad de radiación, con locual el valor de saturación es 0.5, que es la mejor efi-ciencia de transferencia que se puede lograr. Es nece-sario trabajar en condiciones de excitación coherentepróximo a la resonancia para lograr una transferenciatotal en fase de población, según la fórmula
, donde Ω es la conocida frecuenciade Rabi.
Puede hacerse una clasificación de todos losesquemas de control en impulsivos, adiabáticos yvariacionales, antes de pasar a describir brevementelos más usados.
A. Mecanismos impulsivos (o súbitos a tiemposgeneralmente cortos)
a. Mecanismo de control de fase.
Este esquema de Brumer y Shapiro (1986) estábasado en el papel de la fase del pulso para el controlde la dinámica entre un estado inicial acoplado a dosestados finales degenerados mediante sendos láseres,lo cual exige igual paridad en el número de fotones enlas dos rutas elegidas. El objetivo consiste en maxi-mizar el flujo de población hacia uno de los estadosfinales y minimizar la excitación del otro estado. Lacombinación lineal del campo externo da la probabili-dad de excitar un estado final, que depende de un tér-mino de interferencia entre ambas rutas, el cual es fun-ción del coseno de la fase relativa entre ambos pulsos,la cual es controlable. El método se ha aplicado a tran-siciones entre estados discretos de moléculas diatómi-cas (HCl, CO) y poliatómicas (NH3, CH3I) así como ala fotodisociación de IBr y Na2.
b. Mecanismo de control local.
En el límite impulsivo, la función de ondas inicialse traslada a la ventana Franck-Condon en el potencialexcitado con el pulso de bombeo y comienza a evolu-cionar como un paquete de ondas hasta que, con unretardo temporal controlado, un segundo pulso de son-da baja el paquete al potencial inicial o lo sube a unsegundo estado excitado.
Repitiendo el experimento de par de pulsos condiferentes retardos, se logra detectar y seguir en eltiempo el solapamiento entre el paquete de ondas conel canal de reacción deseado. Tannor y Rice (1985),basándose en ideas de Heller sobre la espectroscopiaRaman, desarrollaron este esquema de bombeo y son-da como un tratamiento de teoría de perturbaciones desegundo orden dependiente del tiempo. Las posibili-dades de simular la dinámica controlada del sistema,sobretodo cuando se pretende aprovechar laspropiedades coherentes que exhiben los paquetes deonda, pasaban por desarrollar algoritmos numéricosmuy eficaces para solucionar la ecuación deSchrödinger dependiente del tiempo. La aparición devarios algoritmos numéricos, en particular los desa-rrollados por Kosloff (1988) provocaron la aceleraciónde estudios en Dinámica Molecular Cuántica. La reali-zación de experimentos de bombeo y sonda con láseres
∧
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 341

de fs. en el grupo de Zewail significaron la apariciónde la Femtoquímica. Por otra parte, la simulación delos experimentos de Zewail pudo hacerse en tiempopor Metiu, Imre, Engel, etc., debido a disponer de lospropagadores numéricos indicados. Un siguiente pasohacia el control fue logrado por Scherer (1990) medi-ante la técnica, ya indicada de interferometría depaquetes de onda. Finalmente Kosloff y Tannor (1992)resaltaron el papel de la fase relativa entre el campoláser y el estado del sistema, en lo que se ha llamado elcontrol local.
B. Mecanismos adiabáticos (no-impulsivos;tiempos generalmente largos)
a. Modelo de pulsos π.Para un sistema de dos niveles 1 y 2, sometido a un
campo láser , el hamiltoniano es, donde µ12 es el momento dipolar de
la transición y la probabilidad de encontrar al sistemaen el estado 2 viene dada por , donde
es la frecuencia de Rabi. Siempre que elproducto sea un número impar de veces π el sis-tema se encontrará en el estado final deseado.Independientemente de la forma del pulso, cuando elárea es igual a un múltiplo impar de π, laprobabilidad final de encontrar el sistema en un estadoexcitado es máxima y vale la unidad (teorema delárea). Los pulsos de área π son los más económicospara transiciones entre dos niveles, siendo el tiempomínimo para la transición igual a π multiplicado por elinverso de la frecuencia de Rabi. Este esquema se haaplicado en el grupo de Manz y Paramonov al estudiode inversión de poblaciones en el radical OH y enmoléculas como HOD, H2O y SH2 y también en proce-sos de control secuencial para dirigir reacciones uni-moleculares de isomerización en moléculas organo-metálicas y en moléculas como el NH3.
b. Modelo de barrido de frecuencias.Se puede estudiar la transferencia de población
entre dos niveles acoplados mediante un pulsos de fre-cuencia variable, , con
, donde χ es la velocidad de barridode frecuencias, también llamada trino o gorjeo (chirp).Un pulso de frecuencia variable se logra, descom-poniendo espectralmente un pulso de frecuencia fija eincertidumbre mínima en un prisma, e introduciendo a
continuación retardos entre los distintos componentes(por ejemplo, variando el camino óptico) y volviendo arecomponer el pulso en otro prisma. La ecuación deSchrödinger, gobernada por el Hamiltoniano de esteproceso, tiene también solución analítica y la probabil-idad de transición asintótica (a tiempo infinito) vienedada por la conocida fórmula de Landau-Zener,
, con Ω la frecuencia deRabi.
c. Método adiabático STIRAP (ver figura 4)
Cuando la perturbación del láser es grande, con-viene usar una descripción donde los estados son fun-ciones propias del hamiltoniano completo, incluido elimpulso láser. Dichos estados, llamados estados vesti-dos o adiabáticos, son representaciones rotadas de losestados sin perturbar, caracterizadas por un ángulo demezcla que, en el caso de dos niveles, está definidopor la relación , donde es lades-sintonización entre la frecuencia del láser y laespectroscópica. Los valores propios de los estadosadiabáticos son:
En la evolución adiabática no hay transición entrelos estados adiabáticos, lo cual ocurre cuando el pulso
θ
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio342
0tΩ0 12 0 2AµΩ =
Figura 4. Transferencia de población en un sistema de tresniveles en mediante STIRAP. Probabilidades frente a retardode pulsos de bombeo y Stokes. En el límite de la izquierda seobtiene el comportamiento alternante de los pulsos π.(Tomado de referencia 14).
∧

varía suavemente, el tiempo de interacción es largo ytambién lo es . Cuando hay tres estados, la config-uración puede ser en o en escalera, necesitándosedos pulsos que pueden ser consecutivos o simultáneos.En la configuración , los pulsos se llaman de bombeoΩp y Stokes Ωs, el ángulo de mezcla viene definido porla relación, y los valores propios son:
Si mantenemos el estado vestido de energía cero(que viene definido por una combinación de los esta-dos diabáticos 1 y 3) sin poblar en todo instante, elloimplica no poblar nunca el estado intermedio 2, para locual se exige que el pulso Stokes preceda en el tiempoal pulso de bombeo, en contra de la idea intuitiva. Estose ha conseguido experimentalmente en moléculasdiatómicas como Na2 y NO, así como en poliatómicas(SO2, etc.). El método STIRAP puede usarse en com-binación con el barrido de frecuencias (Chelkowski yBandrauk) con resultados muy satisfactorios.
d. Método de potenciales inducido por la luz (LIP)con campos intensos (fig. 5)
En el límite de grandes energías, la representaciónadiabática se aplica sólo al potencial, diagonizando
solamente la matriz potencial incluyendo elacoplamiento con el láser, con lo que se obtienen lospotenciales adiabáticos siguientes
con como potencial promedio de y , yla diferencia entre ambos. Los potenciales
inducidos por la luz tienen forma distinta a amboslados de la resonancia y su estructura depende de lafrecuencia de Rabi, originándose un cruce evitado alsepararse ambos potenciales en una cantidad igual a Ω.La realidad física de estos potenciales se pone de man-ifiesto en fenómenos tan curiosos como la estabi-lización y desestabilización de enlaces.
C. Métodos de control óptimo
Puede también controlarse la dinámica del paquetede ondas en cada potencial utilizando la técnica delcontrol óptimo desarrollada por Rabitz (1988). Ladinámica del sistema se estudia como un problemavariacional , donde la incognita es el pulso óptimo quese quiere obtener, mientras el sistema está sometido ala ecuación de Schrödinger. Las ecuaciones de controlse resuelven mediante diversos algoritmos (gradiente,Krotov, etc.), incluyendo también algoritmos evolu-tivos. Esto ha permitido ampliar el campo de sistemas
∧
∧
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 343
Figura 5. Transferencia de población mediante formación de potenciales inducidos por la luz. Secuencia de pulsos antiintuitiva.(Tomado de Ref. 20).

físicos estudiados a vibraciones en diatómicas, diso-ciaciones, reacciones de isomerización, etc.
VII. APLICACIONES Y EXTENSIONES(5,21)
Los campos relacionados con la Femtoquímica yControl Cuántico son los siguientes:
1. Información y Computación cuánticas.
La interacción entre electrones y núcleos a travésde fotones permite preparar estados superposición quedefinen los paquetes de ondas. Dentro de la teoría deinformación y computación cuántica, se han estudiadodiversos sistemas (rotaxanos, catenanos, dendrímeros,pentacianoferratos, etc.). Por ejemplo, se han estudia-do los isómeros ópticos con diferente quiralidad paradefinir un espacio binario apto para operaciones lógi-cas. En concreto se pueden estudiar diferentes secuen-cias de pulsos para implementar puertas lógicas deestados entrelazados, donde la fase de entrelazamientojuega un papel importante. Se pueden diseñar secuen-cias estables de pulsos para generalizarlo al caso de nqubits. Las secuencias fuera de resonancia juegan unpapel importante en moléculas con velocidad baja deefecto túnel.
2. Nuevo paradigma “Estructura-Dinámica-Función” en Biología.
Los movimientos de una proteína están determina-dos por su estructura y generalmente sus acciones sediscuten en términos de estructuras estáticas, pero esobvio que la función requiere movimiento, por lo cualel paradigma “Estructura-Función” sería incompletosin la Dinámica. Los movimientos de las proteinas vandesde la escala de femtosegundos en los movimientoselementales a la de milisegundos en movimientos con-formacionales de grupos, pero son los movimientos,entre grupos muy próximos, los que determinan latopología del estado plegado nativo, ya que elplegamiento es más rápido cuando abundan los con-tactos locales. En este sentido la femtodinámica esmuy útil en Biología.
3. Nueva visión de los paradigmas (orbitales,enlaces) de la Química.
El campo fuerte de un pulso láser de femtosegun-dos arranca los electrones de valencia de una molécu-la, los cuales se vuelven contra el ión molecular cuan-do cambia la dirección del campo en el siguientemedio período óptico. Este pulso de electrones en laescala de attosegundos choca con la nube electrónicadel ión molecular en diferente localización cada vez.Repitiendo el proceso, se obtiene por tomografía laforma del orbital electrónico más externo. Así se halogrado ver el orbital más externo de la molécula deN2. Se espera poder seguir en el tiempo cómo se reaco-moda la nube de carga electrónica (enlace) a medidaque avanza la reacción química, lo que supondrá cono-cer los procesos químicos con el máximo detalle en laescala de attosegundos.
4. Attofísica.
La Attofísica está establecida sobre los siguienteshechos:
a) El movimiento de los electrones en la escala deattosegundos se puede controlar con pulsos láserde femtosegundos.
b) Se puede obtener con esos pulsos electrónicos,al recolisionar con los iones moleculares, la evo-lución de los orbitales de valencia, así como losmovimientos rápidos de los núcleos y la dinámi-ca de relajación electrónica.
c) La recombinación electrón-ión produce pulsosláser en el UV extremo (XUV) de varios cientosde attosegundos, lo que supone incrementar elcontrol sobre los pulsos láser, llegándose inten-sidades de .
Las posibilidades que se abren para la Química sonimportantes pudiéndose estudiar los límites de validezde la aproximación de Born-Oppenheimer y, experi-mentalmente, los procesos que involucran movimien-tos electrónicos y nucleares, como la dinámicamolecular en las intersecciones cónicas de superficiesde potencial, proceso vital para entender la Fotoquí-mica moderna.
VIII. DISCUSIÓN FINAL
Es preciso concluir con unas reflexiones sobre elcontexto y alcance de los avances logrados en el tema
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio344
18 210 Wcm−

de desvelar y controlar la danza de lo átomos, es decirsobre la Femtoquímica y el Control Cuántico.
1. Los átomos y moléculas no pueden verse direc-tamente no solamente porque su tamaño esmucho menor que la longitud de onda de la luzvisible (0.3-07 µm) sino principalmente porqueno son cosas, son únicamente partes de situacio-nes de observación, donde según el principio deincertidumbre conservan a nivel individual unacoherencia cuántica, que los aleja del comporta-miento clásico decoherente del mundo macros-cópico.
2. Los paquetes de ondas en evolución temporal,que describen átomos y moléculas, son funcio-nes matemáticas complejas (con parte real eimaginaria) que no pueden ser “observables”según la Mecánica Cuántica.
3. Lo que sí es observable es su cuadrado, es decirel comportamiento coherente de las moléculas yátomos en procesos de preparación y prueba(absorción, fluorescencia, etc.).
4. Los experimentos descritos no se refieren amoléculas individuales, sino a colectivos de lasmismas (típicamente 109-1011 moléculas) quemuestran un comportamiento coherente en fase.Además cada par de medidas (bombeo y sonda)se realizan se realizan sobre un colectivo demoléculas que es distinto para cada repetición, adiferente retardo, del par de pulsos.
5. Los experimentos de Femtoquímica y Control,aunque no dan la observación directa de losmovimientos atómicos en una molécula indivi-dual, sin embargo, sí suministran el camino dereacción sobre el campo de fuerzas conservativo(superficie de potencial) que define la entidadmolecular, lo que equivale a obtener la máximainformación posible sobre interacciones entreátomos en la ciencia atómico-molecular.
6. Existen por otra parte medidas sobre átomoslocalizados (en red cristalina) y moléculas indi-viduales (por ejemplo, biopolímeros anclados)realizados con microscopios de fuerza atómica,pinzas ópticas, etc. que permiten seguir proce-sos en esas entidades individuales.
7. Existen también medidas de difracción electró-nica ultrarrápida y por Rayos X (de láseres de
electrones libres, FEL) que dan la dinámicaestructural de sistemas complejos (clusters,fases condensadas, etc.), es decir, el movimien-to real de un sistema de muchos cuerpos en elespacio real cartesiano tridimensional.
REFERENCIAS
1. A. H. Zewail, Laser Chemistry, Science 242,1645(1988); A. H. Zewail, The birth of molecules,Scientific Amarican, dic.1990, p. 76; A. H. Zewail,Femtochemistry: Atomic- scale dynamics of the che-mical bond using ultrafast lasers (Nobel Lecture),Angew. Chem. Int. Ed. 39, 392586 (2000).
2. D. J. Tannor, Introduction to Quantum Dynamics: Atime dependent perspective (University ScienceBooks, Sausalito, CA, USA, 2007).
3. E. J. Heller, The semiclassical way to molecularSpectroscopy, Acc. Chem. Res. 14, 368 (1981); E. J.Heller y S. Tomsovic; Postmodern QuantumMechanics, Phys. Today 46, 38 ( Julio 1993).
4. Proceedings de las Conferencias bianuales enFemtochemistry desde 1993.
5. A. Douhal y J. Santamaría (eds.) Femtochemistryand Femtobiology ( World Scientific, Singapore,2002).
6. K. C. Kulander (ed.) The time dependent methods forQuantum Dynamics, Comput. Phys. Commun. 63(1991).
7. J. Broeckhove Y L. Lathouwers (eds.) Time depen-dent Quantum Molecular Dynamics ( Plenum, N.Y.1992).
8. J. C. Light y otros, Generalized discrete variableapproximation in Quantum Mechanics, J. Chem.Phys. 82, 1400 (1985).
9. C.C. Marston y G. C. Balint-Kurti, The Fourier gridHamiltonian method for bound state eigenvalues andeigenfunctions, J. Chem.Phys. 91, 3571 (1989).
10. M. D. Feit, J. A. Fleck y A Steiger, Solution of theSchrödinger equation by a spectral method, J.Comput. Phys. 47, 412 (1982).
11. R. Kosloff, The time dependent methods in molecu-lar dynamics, J. Phys. Chem. 92, 2087 (1988).
12. V. Engel, H. Metiu y otros, Chem.. Phys. Lett. 152,1(1988).
13. S. O. Williams y D. G. Imre, J. Chem. Phys. 92, 6648(1988).
14. I. R. Solá, Tesis Doctoral (UCM; Madrid 2000); B. Y.Chang, Tesis Doctoral (UCM Madrid, 2001).
15. W. S. Warren, H. Rabitz y M. Dahleh, Science 259,
Jesús Santamaría Antonio Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 345

1581 (1993).16. D. J. Tannor y S. A. Rice, J. Chem. Phys. 83, 5013
(1985) ; P. Brumer y M. Shapiro, J. Chem. Phys. 84,4013 (1986) ; N. F. Scherer y otros, J. Chem. Phys.93, 856 (1990).
17. K. Bergmann y otros, Phys. Rev. A 40, 6741 (1989);K. Bergmann y otros, Ann. Rev. Phys. Chem. 52, 763(2001); B. M. Garraway y K. A. Suominen, Rep.Prog. Phys. 58,365 (1995); S. Chelkowsky, A. D.Bandrauk y P. B. Corkum, Phys. Rev. Lett. 65, 2355(1990).
18. J. Manz y G. K. Paramonov, Chem. Phys. Lett 193,
429 81992);R. Kosloff y D. J: Tannor, Phys. Rev.Lett. 69, 2172 (1992).
19. S. A. Rice y M. Zhao, Optimal Control of moleculardynamics (J. Wiley & Sons, N.Y. 2000); H. Rabitz yotros, Phys. Rev.A 37, 4959 (1988); G. Gerber yotros, Science 282, 919 (1998).
20. B. Y. Chang, I. R. Solá, J. Santamaría, V. S.Malinovsky y J. L. Krause, J. Chem Phys. 114, 8820( 2001).
21. A. Scrinzi, M. Y. Ivanov, R. Kienberger y D. M.Villeneuve, Attosecond Physics, J. Phys. B: At.Mol.Opt. Phys. 39, D1 (2006).
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101Jesús Santamaría Antonio346