universidad de buenos aires facultad de … · y hormonales, y a nivel central, como factores de...
TRANSCRIPT

UNIVERSIDAD DE BUENOS AIRES
FACULTAD DE FARMACIA Y BIOQUIMICA
Estudio molecular de alteraciones del eje Hipotálamo-
hipófiso-IGF-1 en población pediátrica Argentina con
déficit de crecimiento.
TESISTA: Juanes Matías Hernán.
DIRECTORA: Dra. Belgorosky Alicia.
CONSEJERO DE ESTUDIOS: Dr. Dominici Fernando.
Tesis presentada para optar por el título de Doctor de la Universidad de
Buenos Aires
-2015-
Servicio de Endocrinología, Laboratorio de Biología Molecular,
Hospital de Pediatría J.P. Garrahan.
Buenos Aires, Argentina

AGRADECIMIENTOS
A mi directora de tesis, Dra. Alicia Belgorosky, en lo académico por compartir
su amplio conocimiento en la rama de la endocrinología y de la investigación y
por darme la oportunidad de conocer y discutir resultados con profesionales de
la salud de gran reconocimiento, tanto a nivel local como internacional, que me
permitieron crecer y profundizar mis conocimientos. En lo personal, por estar
siempre durante estos cinco años para escucharme y aconsejarme, ayudándome
en momentos difíciles y compartiendo los momentos felices. Estoy orgulloso y
enormemente agradecido de que la Dra. Belgorosky haya sido mi directora de
tesis.
Al Dr. Marco Aurelio Rivarola por su humildad, consejos y por tener siempre las
palabras justas en los momentos indicados. Por compartir su vasta experiencia
en la endocrinología pediátrica.
A la Dra. Roxana Marino, por recibirme e incorporarme a su grupo de trabajo
como uno más y hacerme sentir como en mi casa cuando recién llegaba a
Buenos Aires. Por enseñarme todo lo que logre aprender del laboratorio de
Biología Molecular, por su humildad, dedicación y compromiso con los
pacientes que me inculcó desde el primer día. Por lograr un ambiente laboral en
donde todos los integrantes del laboratorio nos sentimos cómodos y podemos
compartir el trabajo, no con compañeros, sino con amigos. Por último y lo más
valiosos de estos 5 años, por su amistad.
A mis compañeros y amigos del laboratorio, Pablo, Nati PG, Sol, Esperanza,
Nati P y Jesi por su ayuda y participación en el desarrollo de la tesis, por tantas
charlas entre mates compartidas, por estar siempre, tanto en los momentos
difíciles como felices, por la buena onda de todos los días, por hacer que el
trabajo sea algo que se disfruta y principalmente por sus amistades.
A todos los integrantes del servicio de Endocrinología del Hospital Garrahan por
su excelencia en el manejo y en el compromiso con los pacientes, tanto en el
consultorio como en el laboratorio. Por sus enseñanzas diarias en el estudio de
los pacientes. En especial quiero agradecer a las Dras. Marta Ciaccio, Gabriela

Guercio e Isabel Di Palma por su ayuda y aportes durante el desarrollo de la
tesis en los temas relacionados con los aspectos clínicos de los pacientes
estudiados.
A la dirección asociada de Docencia e Investigación del Hospital Garrahan por
permitirme desarrollar mi tesis doctoral en la institución.
A mi consejero de estudios el Dr. Fernando Dominici por su buena
predisposición y recomendaciones en la escritura de la tesis y en los aspectos
administrativos de la facultad de Farmacia y Bioquímica de la UBA.
A mi mujer Mariana, por su amor incondicional, por estar siempre, por
escucharme, por acompañarme en todos los momentos de mi vida, por ser mi
sostén y mi cable a tierra, por su bondad, por creer en mí, por volver a elegirme
cada día, por cambiarme la vida y hacerme feliz.
A mis padres, Roberto y Ana María, por ser mis ejemplos a seguir y mi orgullo,
por inculcarme los valores para ser la persona que soy, por esforzarse al máximo
por darme la educación que tuve que me permitió lograr los objetivos que me
propuse. Por estar siempre y aconsejarme en las decisiones de mi vida. Por su
amor incondicional.
A mi hermano Ignacio, por mostrarme la fortaleza que se necesita para afrontar
las distintas etapas de la vida, por enseñarme que hay que mirar siempre para
adelante, por su cariño, por su confianza, por tantas risas y momentos felices que
pasamos y seguiremos pasando.
A mi hermano Martín, por enseñarme el valor de la familia y de la vida.
A mi cuñada Marisa por su amistad, por su enorme fortaleza, dedicación y por
hacer feliz a mi hermano.
A Joaquín por ser estar siempre conmigo, a mi lado, por enseñarme a pelear por
lo que quiero y no bajar nunca los brazos, por hacernos tan feliz a todos y
cuidarnos siempre.

A mi hermano del alma y amigo de toda la vida, Esteban, por estos 23 años de
amistad, por su generosidad y su confianza. Por permitir que crezcamos a la par,
por estar siempre para lo que necesite, por las charlas, las comidas, las anécdotas
de cuando éramos más chicos, por tantos momentos de felicidad que pasamos,
por tantas risas y alegrías, que seguro van a ser muchas más.
A mis amigos, Sebastián y Pablo, por soportarme en la convivencia en los
primeros años en Buenos Aires, por las charlas, las comidas, las salidas, las
anécdotas, los consejos y también por estar siempre en las buenas y en las malas.
A mi abuela que tanto la quiero, por enseñarme que hay que tener perseverancia
para lograr las cosas y llevar siempre una sonrisa a todos lados, por su cariño.
A mis Tías Mirta y Mercedes por su preocupación y por estar siempre pendiente
de mí y de mi familia.
A Adriana por ser parte de mi crianza, por ayudarme a crecer, por permitirme
compartir mañanas, tardes, noches y veranos durante 23 años junto a su familia.
Por estar en las buenas y en las malas.
A mis amigos de la Universidad Guillermo, Pablo y Federico por sus amistades,
por estar cuando se los necesita, por tantos asados y risas compartidas.
A mi ciudad Bahía Blanca que siempre me recuerda de dónde vengo y la llevo
en el corazón.
A la Universidad Nacional del Sur por formarme en las ciencias de la salud y
haberme garantizado las herramientas necesarias para ingresar al ambiente
profesional.

Esta tesis pudo llevarse a cabo gracias al aporte económico de:
Hospital de Pediatría Dr. Juan P. Garrahan.
Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
Agencia Nacional de Promoción Científica y Tecnológica, Ministerio de Ciencia
y Técnica de la Nación.
Ministerio de Salud de la Nación.

INTRODUCCIÓN
1. Embriogénesis Hipofisaria 1
1.1 Generalidades
1.2 Factores de transcripción de relevancia clínica 5
1.2.1 Factores de acción temprana 6
1.2.2 Factores de acción tardía 13
2. Crecimiento Fetal
2.1 Generalidades 16
2.2 El gen GH2 y la PGH 16
2.3 Propiedades biológicas de la PGH 17
2.4 Rol fisiológico de la PGH 18
2.5 Rol del sistema de los IGFs durante el desarrollo fetal 18
2.6 Regulación de la secreción de PGH 19
2.7 PGH en casos patológicos 20
3. Crecimiento postnatal
3.1 Eje de crecimiento GHRH-GH-IGF1 21
3.2 Defectos moleculares que afectan el eje de crecimiento GHRH-GH-IGF1 24
3.2.1 Déficit aislado de GH-1 (IGHD) 24
3.2.1.1 IGHD tipo IA 25
3.2.1.2 IGHD tipo IB 26
3.2.1.3 IGHD tipo II 26
3.2.1.4 IGHD tipo III 27
3.2.2 Síndrome de insensibilidad a la GH 28
3.2.2.1 Alteración del receptor de GH-1 28
3.2.2.2 Alteración en el traductor de señal STAT5b 30
3.2.2.3 Deficiencia de ALS 33
3.2.3 Resistencia IGF-1: Alteración del receptor de IGFs tipo 1 34
3.2.3.1 Receptor de IGFs tipo 1 34
3.2.3.2 Mutaciones en el gen IGF1R 36
4. OBJETIVOS
4.1 Objetivos Generales 38
4.2 Objetivos Específicos 38
5. MATERIALES Y METODOS
5.1 Selección de pacientes 39
5.1.1 Grupo déficit de hormona de crecimiento 39
5.1.2 Grupo síndrome de insensibilidad a la GH-1 40
5.1.3 Grupo con alteraciones en la cascada señalización del GHR 40
5.1.4 Grupo resistencia a IGF-1 40
5.2 Determinaciones clínicas y bioquímicas 41

5.3 Análisis Molecular
5.3.1 Extracción de ADN 41
5.3.2 Extracción de ARN 42
5.3.3 Amplificación por PCR y secuenciación 42
5.3.4 RFLP 45
5.3.5 MLPA 45
5.3.6 Ensayos in silico 46
5.3.7 Cultivo primario de fibroblastos 46
5.3.8 Ensayo funcionales in vitro
5.3.8.1 Ensayo de proliferación celular 46
5.3.8.2 Ensayo de fosforilación de AKT 47
5.3.8.3 Análisis estadístico 47
6. RESULTADOS 48
6.1 Alteraciones moleculares en pacientes con IGHD 48
6.2 Alteraciones moleculares en pacientes con síndrome de insensibilidad a la GH 52
6.3 Alteraciones moleculares en pacientes con resistencia a IGF-1 54
6.3.1 Evaluación Clínica de los pacientes afectados 55
6.3.2 Análisis molecular 57
6.3.3 Ensayos funcionales 58
6.3.4 MLPA 60
6.4 Alteraciones moleculares en pacientes con MPHD
6.4.1 Evaluación clínica de pacientes afectados por alteraciones en el gen GLI2
6.4.2 Análisis Molecular 64
7. DISCUSION 66
8. CONCLUSION 74
RESUMEN 75
BIBLIOGRAFIA 77

1
1. Embriogénesis Hipofisaria
1.1 Generalidades
El crecimiento es una propiedad inherente de la vida. El crecimiento somático normal requiere
de la integración de múltiples factores que actúan a nivel periférico, como factores metabólicos
y hormonales, y a nivel central, como factores de trascripción involucrados en el eje
hipotálamo-hipofisario-IGF-1.
El desarrollo de la glándula hipofisaria está determinado por la expresión secuencial
coordinada, de manera temporo-espacial, de distintos factores de transcripción de expresión
temprana en el desarrollo embrionario (Takuma N et al 1998; Treier M et al 1998). Estos factores
generar la activación, o inhibición, de distintas vías de señalización celular que van a dar lugar
a tres etapas específicas del desarrollo hipofisario, 1) formación de la hipófisis, 2) expansión y
proliferación de las células progenitoras y 3) diferenciación celular. Este proceso permite el
establecimiento de los distintos linajes celulares hipofisarios que ejercen las funciones vitales
de la glándula regulando el crecimiento (eje somatotropo), la reproducción (eje gonadotropo),
el metabolismo (eje tirotropo) y la respuesta al stress (eje corticotropo). Alteraciones en
distintas etapas de la embriogénesis resultan en la pérdida, o reducción, de las células
secretoras de hormonas hipofisarias y se manifiesta en una disminución funcional de la
glándula hipófisis conocida como hipopituitarismo congénito (Shannon W. Davis et al 2013). El
hipopituitarismo congénito puede variar desde déficit múltiple de hormonas hipofisarias
(MPHD), donde más de un linaje celular se ve afectado, hasta déficit aislado, por alteración de
un único linaje celular. Dentro de este último, el déficit más frecuente es el que afecta al linaje
somatotropo, que da lugar a déficit aislado de hormona de crecimiento (IGHD) (Shannon W.
Davis et al 2013).
El desarrollo hipofisario se inicia con la formación de la glándula hipófisis propiamente dicha. La
hipófisis deriva de dos estructuras ectodérmicas, el ectodermo ventral (o diencéfalo ventral), el
cual da lugar al lóbulo posterior, y la superficie del ectodermo, que da lugar a lo que se
denomina como bolsa de Rathke, el precursor primordial del lóbulo anterior e intermedio de la
hipófisis. El patrón de desarrollo del diencéfalo ventral es crítico no solo para el establecimiento
de la neurohipófisis (lóbulo posterior) sino también para la formación de un centro organizador
que delimita el tamaño y la forma apropiada de la bolsa de Rathke. Sobre este centro
organizador comienzan a expresarse e interactuar una serie de factores de transcripción que
dan lugar a la formación de las estructuras primitivas de la hipófisis, por un lado el infundíbulo,
que se genera por una invaginación del diéncefalo ventral, y por otro lado la bolsa de Rathke, a
partir del engrosamiento da la superficie del ectodermo (Shannon W. Davis et al 2013). Existen
una gran cantidad de factores de trascripción que se expresan y ejercen sus funciones en esta
etapa del desarrollo (Treier M et al 1998), de los cuales cabe destacar a las proteínas
morfogenéticas óseas, BMP2 y BMP4 (Davis SW et al 2007), y a los factores de crecimiento de
fibroblastos, FGF8 y FGF10 (Ericson J et al 1998; Ohuchi H et al 2000).

2
Las proteínas BMP, mayoritariamente la BMP4, son una señal inductiva esencial para la
formación de la bolsa de Rathke mientras que la señalización FGF es necesaria para la
proliferación celular una vez formada la bolsa (Takuma N et al 1998; De Moerlooze L et al 2000;
Ohuchi H et al 2000; McCabe MJ). Estas vías de señalización no son las únicas que se expresan
en el diencéfalo ventral, sino que existe un intrincado solapamiento entre distintas vías de
señalización. Un ejemplo de este solapamiento es la expresión de los genes de la familia Wnt.
Esta familia está compuesta por numerosos genes que se expresan durante el desarrollo
embrionario, de los cuales los más relevantes son el Wnt5a y el Wnt4 (Cha KB et al 2004; Potok
MA et al 2008; Ho HY et al 2012). Modelos de ratones Knock-out para Wnt5a (Wnt5a-/-), han
demostrado el rol de este factor en establecer la forma de la hipófisis, aunque no se conoce
exactamente el mecanismo por el cual interviene en esta función (se sospecha que es a partir
de la regulación de la adhesión celular). A su vez estos ratones tienen un aumento en la
actividad de las vías BMP y FGF, lo que demuestra la importancia de la regulación negativa de
estas vías durante la embriogénesis (Potok MA et al 2008; Ho HY et al 2012). Por otro lado,
ratones knock-out para Wnt4 muestran una clara reducción de las células de la hipófisis
anterior, con drástica disminución en los fenotipos ventrales (principalmente gonadotropos), lo
que demuestra la importancia de este factor en la diferenciación y proliferación celular en
etapas más avanzados del desarrollo hipofisario (Olson LE et al 2006; Gaston-Massuet C et al
2011).
Dentro de la gran variedad de caminos de señalización que se activan durante la
embriogénesis hipofisaria, uno de los más importantes es el del Sonic Hegdehog (SHH), el cual
tiene como principal función restringir el crecimiento de la glándula (Zhao L et al 2012). La
expresión del SHH está regulada por la acción de los factores SOX2 y SOX3, los cuales se
unen a regiones regulatorias del ADN y promueven su activación. Las acciones del SHH están
mediadas por la familia de factores de transcripción Gli, los cuales tienen efectos opuestos
sobre los genes blancos río abajo en la cascada de señalización (Wang Y et al 2010). El Gli2
tiene acciones estimulantes mientras que el Gli3 tiene acciones inhibitorias (Hui CC et al 2011).
Debido al efecto dual de las proteínas Gli aún no se ha determinado exactamente el
mecanismo por el cual el SHH ejerce sus funciones. Se ha demostrado por medio de ratones
KO que ambas proteínas son necesarias para el desarrollo normal de la hipófisis y la
regulación de BMPs y FGFs (Wang Y et al 2010).
Como se mencionó previamente, la interacción de los distintos caminos de señalización es muy
importante para la formación de las estructuras primordiales de la hipófisis. A su vez es
necesario que todas estas señales se integren dentro de la bolsa de Ratkhe para permitir la
proliferación y diferenciación de los distintos linajes celulares de la hipófisis. A partir de
modelos animales, con pérdida de función, existe evidencia de que los BMPs y FGFs tienen un
rol importante en el crecimiento y establecimiento de la estructura de la glándula hipofisaria
pero no tendrían injerencia en la especificación celular (Shannon W. Davis et al 2013). Mientras
que por otro lado, modelos animales con ganancia de función sugieren que un exceso en la
señalización de estas vías puede influir en la especificación celular o afectar el tamaño de una

3
población celular específica (Mullis P. 2005; Shannon W. Davis et al 2013). En base a estas
evidencias bibliográficas se originó lo que se denomina como “Teoría de Gradientes de
expresión”. Esta teoría se fundamenta en base a las regiones, dentro de la bolsa de Rathke, en
donde se expresan los FGFs y los BMPs. Se ha reportado que el FGF8 y FGF10 se expresan
en cercanías del infundíbulo, dorsal a la bolsa de Rathke, mientras que BMP2 se expresa del
lado ventral y adyacente al mesodermo, por lo que se cree que existen gradientes de
señalización opuestos de FGF y BMP (Mullis P. 2005; Shannon W. Davis et al 2013). Estos
gradientes activan distintos factores de transcripción, que son dorsales (Nkx 3, SIX-3, Prop1) o
ventrales (Brn4, Isl 1, P-Frk) a la bolsa de Rathke, y dependiendo de la ubicación relativa de las
células progenitoras con respecto a estos gradientes, varía el impacto de la señalización que
reciban de estos factores permitiendo la diferenciación en los distintos linajes hipofisarios
(Ericson J et al 1998; Treier M et al 1998). Es decir las células progenitoras ubicadas del lado
ventral de la bolsa de Rathke, adyacentes al mesodermo, recibirán mayor estímulo por parte
del gradiente de BMP (factores ventrales), diferenciándose a gonadotropos-corticotropos,
mientras que las células cercanas a la fuente de FGF (factores dorsales) se diferencian a
somatotropos-lactotropos y en última instancia a tirotropos (Figura 1).
Genetic control of growth, Mullis P, EJE 2005
Figura 1. Diferenciación celular en base a la Teoría del gradiente de expresión. Gradientes opuestos de señalización
de FGFs y BMPs. FGF8 y FGF10 se expresan en cercanías del infundíbulo, dorsal a la bolsa de Rathke, mientras que
BMP2 se expresa del lado ventral y adyacente al mesodermo. Estos gradientes activan distintos factores de
transcripción, que son dorsales (Nkx 3, SIX-3, PROP1) o ventrales (Brn4, Isl 1, P-Frk) a la bolsa de Rathke, y
dependiendo de la ubicación relativa de las células progenitoras con respecto a estos gradientes se diferencian a los
distintos linajes hipofisarios. Región ventral: diferenciación a gonadotropo-corticotropo, región dorsal: diferenciación a
somatotropo-lactotropo-tirotropo.

4
El descubrimiento de redes específicas de un tipo celular dentro del lóbulo anterior, y el
movimiento de células secretoras de hormonas para formar estas redes, sugieren que la
posición final de las células en el lóbulo anterior no puede estar correlacionada directamente
con la posición inicial de la célula progenitora en la bolsa de Rathke, de tal manera que la
teoría de gradientes de expresión no puede explicar por sí sola todo el proceso de
especificación y diferenciación celular (Shannon W. Davis et al 2013). Además, cabe destacar
que no existe evidencia científica que permita analizar el seguimiento de una célula progenitora
y su descendencia desde su posición inicial, en la luz de la bolsa de Rathke, hasta su
localización y diferenciación en un determinado tipo celular en el lóbulo anterior, por lo que se
han propuesto otros modelos que puedan explicar el proceso de especificación y diferenciación
celular (Shannon W. Davis et al 2013). Dentro de estos modelos, el de mayor sustento propone
que todos los eventos de diferenciación celular se inician a partir de dos precursores comunes;
el pre-somato-lacto-tirotropo (PSLT) y el pre-gonado-corticotropo (Ericson J et al 1998; Dasen JS
et al 1999; Pulichino AM et al 2003; Mullis P. 2005). Este modelo se basa en la evidencia de que
la acción del factor de transcripción Prop1, en la región dorsal del lóbulo anterior, da lugar al
precursor PSLT que, posteriormente, se diferencia a los distintos linajes celulares a partir de la
expresión de factores de transcripción más tardíos, como Pit-1 y Gata2, entre otros (Dasen JS
et al 1999). El precursor pre-gonado-corticotropo tiene un comportamiento similar, de tal modo
que la expresión de los factores T-pit y NeuroD1 dan lugar al precursor cortico-melanotropo
mientras que la acción de SF-1 y Gata2 influyen en la diferenciación a gonadotropo (Dasen JS
et al 1999; Pulichino AM 2003).
Estos hallazgos junto con la gran cantidad de interacciones entre distintos ligandos y
receptores y las distintas vías de señalización demuestran el alto grado de complejidad del
proceso de diferenciación que no permite ser explicado por una única teoría. A su vez, la
dificultad de estudiar estas vías de señalización in vivo, junto con la incapacidad de
relacionarlas entre ellas, impide que el proceso de diferenciación sea revelado por completo.
En el siguiente esquema se muestra un resumen de las distintas vías de señalización y cómo
interactúan sus factores durante la embriogénesis hipofisaria.

5
Christopher J Romero, et al, Journal of Molecular Endocrinology (2011)
Figura 2. Resumen del desarrollo hipofisario murino que muestra las secuencia de eventos que determinan la
proliferación y diferenciación de los distintos tipos celulares. En el período embriológico e9.5, la expresión de BMP4,
Nkx2.1 y Shh inician el desarrollo del la BR. La expresión de los factores Gli1, Gli2, Lhx3 y Lhx4 permite el desarrollo
de las células progenitoras. Hesx1, Isl1, Pax6, Six3 y Six6 participan en los procesos de desarrollo, proliferación y
migración celular. La disminución en la expresión de Hesx1 es requerida para la expresión de Prop1. Entre los días
e12.5 y e17.5, la diferenciación de los distintos linajes celulares hipofisarios se completa (Dasen & Rosendfeld 2001,
Scully & Rosenfeld 2002, Kelberman & Dattani 2006, Zhu et al 2007).
1.2 Factores de trascripción de relevancia clínica
Se ha demostrado que una gran cantidad de factores de trascripción juegan un importante rol
en la especificación y expansión de las células productoras de hormonas hipofisarias y, a su
vez, deficiencias de los mismos se han relacionados con patologías en humanos. Estos
factores se clasifican en dos grandes grupos, aquellos que actúan en etapas muy tempranas
del desarrollo (antes del período e11 del desarrollo embrionario murino) y aquellos que actúan
en etapas más tardías (posterior al período e11) (Dasen & Rosendfeld 2001, Scully & Rosenfeld
2002, Kelberman & Dattani 2006, Zhu et al 2007). Nos centraremos específicamente en aquellos
factores que estén implicados con patologías en humanos relacionadas con déficit de
crecimiento, ya sea por déficit múltiple de hormonas hipofisarias (MPHD) o por deficiencia
aislada de hormona de crecimiento (IGHD). Dentro de los factores de acción temprana
Señales BMP4 Nkx2.1
Otx2
Shh, Gli1,2
Lhx3, Lhx4
Señales FGF8/10 Wnt5a
Hesx1 Prop1
e9.5 e11
Isl1 Pax6 Six3,6
e12
GATA 2
somatotropo
lactotropo
Caudal Tirotropo Rostral
gonadotropo
T pit Neuro D1
corticotropo
Factores de desarrollo temprano Factores de desarrollo tardíos
LINAJE CELULAR
HIPOFISIS ANTERIOR
e17.5
Pit1
SF-1

6
relacionados con este tipo de patología se describen el Otx2, Hesx1, Lhx3, Lhx4 y Gli2
mientras que, con respecto a los factores de acción más tardía, el Prop 1 y Pit-1.
1.2.1 Factores de acción temprana
Otx2
El gen Orthodenticle Homebox 2 (NG_008204.1) está localizado en la posición cromosómica
14q23.1, es el gen ortólogo en los vertebrados al gen Otd de Drosophila, el cual es requerido
para la formación del cerebro anterior, los ojos y las antenas (Finkelstein R, Boncinelli E. 1994) y
para la regulación del desarrollo de los foto receptores y la rodopsina (Tahayato A., et al 2003).
Los mamíferos poseen 3 genes OTX: OTX1, OTX2 y OTX5 (o Crx). Los tres genes se expresan
en el ojo en desarrollo. En ratones, el OTX 1 y 2 se expresan en estructuras neuronales y
sensoriales en desarrollo que incluyen el cerebro, las orejas, la nariz y el ojo (Simeone A, et al
1993; Martinez-Morales JR, et al 2001). Alteraciones en el gen OTX2 han sido reportadas en
humanos asociadas a malformaciones del ojo y defectos estructurales y funcionales de la
hipófisis (Ragge NK et al 2005; Chatelain G et al 2006). El gen OTX2 está formado por 5 exones,
de los cuales los exones 1 y 2 son no codificantes mientras que del 3 al 5 codifican para la
proteína Otx2 (Figura 3). La proteína consta de un homeodominio de unión al ADN y un
dominio transactivador que permite la regulación de distintos genes blancos relacionados con
el desarrollo cerebral y ocular.
Ratones KO para el gen OTX2 mueren durante la gestación debido a anomalías cerebrales
severas mientras que ratones heterocigotas para OTX2 han revelado un fenotipo muy variable
que abarca desde anencefalia, micrognatia, anoftalmia y microftalmia (Matsuo I et al 1995;
Acampora D et al 1995; Ang SL
et al 1996; Kurokawa D
et al 2004).
En humanos, las mutaciones en el gen OTX2 son raras (menor al 1%) y poseen un patrón de
herencia autosómica dominante. Se han reportado en pacientes con malformaciones oculares
severas y anomalías cerebrales junto con convulsiones y retardo en el desarrollo (Ragge NK et
al 2005; Chatelain G et al 2006). Aunque la mayoría de las mutaciones se asocian a
malformaciones del ojo, el 30% de los pacientes con mutaciones en el gen OTX2 poseen
defectos estructurales y funcionales de la hipófisis, lo que indica su participación durante la
embriogénesis hipofisaria (Schilter et al 2010). Se han reportado mutaciones heterocigotas, sin
sentido y con cambio en el marco de lectura, relacionadas con MPHD junto con anomalías
oculares. Por otro lado, también se han reportado microdeleciones en la región donde mapea el
gen OTX2, relacionadas con anoftalmia y alteraciones hipotálamo-hipofisarias (Dateki et al
2009). Estudios in vitro demostraron que mutantes para el gen OTX2 pierden la capacidad de
activar los promotores de los genes HESX1 y POU1F1 (gen que codifica para el factor de
transcripción Pit-1), lo cual demuestra el impacto directo que posee este factor en la regulación
del proceso de proliferación y diferenciación de los distintos linajes celulares hipofisarios
(Tajima et al 2009).

7
Modificado de Christopher J Romero, et al 2011
Figura 3. Mutaciones reportadas en el gen OTX2 asociadas a MPHD en humanos. El gen se organiza en 5 exones de
los cuales solo tres codifican para los distintos dominios de la proteína. HD, homeodominio. TF, dominio transactivador
(factor de transcripción). Dateki et al. 2008 y 2009, Tajima et al. 2009, Diaczok et al. 2008, Henderson et al.2009.
Hesx1
También denominado Rpx (Rathke´s Pouch Homeobox Gene), es un factor de trascripción con
un dominio represor, localizado en la región amino terminal (N), y un homeodominio de unión al
ADN (Hermesz E et al 1996; Dasen JS et al 2001). El gen HESX1 se localiza en la posición
cromosómica 3p14.3 (NG_008242.1), está conformado por 4 exones los cuales codifican para
una proteína de 185 aminoácidos que actúa como un represor transcripcional promotor
específico que se esquematiza en la figura 4 (Dattani MT et al 1998; Brickman JM et al 2001). La
expresión de Hesx1 se inicia muy temprano en el desarrollo del ratón (gastrulación) y luego se
restringe completamente a la bolsa de Rathke (Hermesz E et al 1996). Su rol como represor se
ha demostrado a partir de la disminución de su expresión en el día e13 del desarrollo
embrionario, haciéndose indetectable en el período e14-15.5 (Sornson MW et al 1996), lo que se
correlaciona con el tiempo exacto en donde comienza la diferenciación celular de los distintos
linajes hipofisarios. Hesx1 inhibe la expresión de otro factor de transcripción de la misma
familia, denominado Prop1, el cual posee funciones activantes durante el desarrollo y la
diferenciación celular. Ambos factores de transcripción se unen a los mismos elementos de
respuesta en el ADN, pero con acciones opuestas, de tal forma que la regulación temporo-
espacial de estos factores es vital para el desarrollo normal de la hipófisis (Shannon W. Davis et
al 2013). Hesx1, se expresa en etapas tempranas del desarrollo, regulando negativamente
estas regiones e impidiendo la acción de Prop1, permitiendo el crecimiento y la proliferación de
la hipófisis primitiva. La disminución en la expresión de Hesx1 sigue una dirección ventro-
dorsal, en espejo con la diferenciación celular, y coincide temporo-espacialmente con el
1 2 3 4 5
HD PolyQ Otx1 TF region
A72fsX86
K74fsX103
L135fsX136
S138X
W152X
S186fsX187
G188X
N233S
S136fsX178
1 2 3 4 5
HD PolyQ Otx1 TF region
A72fsX86
K74fsX103
L135fsX136
S138X
W152X
S186fsX187
G188X
N233S
S136fsX178

8
aumento en la expresión de Prop1. Este aumento genera una regulación positiva sobre genes
implicados en la diferenciación celular, como POU1F1 y GATA2, permitiendo la formación de la
hipófisis definitiva (Mullis P. 2005; Shannon W. Davis et al 2013).
Mutaciones en el gen HESX1 están relacionadas a displasia septo óptica (DSO) e
hipopituitarismo, con una frecuencia del 1% (Dattani MT et al 1998; Brickman JM et al 2001;
Thomas PQ et al 2001). La DSO es un síndrome que se caracteriza por hipoplasia del nervio
óptico, disfunción de la glándula hipófisis y anormalidades de la línea media que involucran al
cuerpo calloso y al septum pellucidum (Dattani MT et al 1998; Cohen LE et al 2002). Todos los
casos reportados de DSO, causados por mutaciones en el gen HESX1, resultan en
alteraciones en la región de unión al ADN de la proteína (homeodominio) o en el dominio
represor, impidiendo la regulación negativa necesaria para el correcto desarrollo hipofisario. El
modo de herencia de esta patología no es claro, se han reportado casos de herencia
autosómica recesiva y dominante con penetrancia incompleta (Dattani MT et al 1998; Brickman
JM et al 2001; Thomas PQ et al 2001).
Modificado de Christopher J Romero, et al 2011
Figura 4. Mutaciones en el gen Hesx1 asociadas con MPHD. El gen se esquematiza en la región superior mientras que
la proteína se encuentra en la región inferior de la figura. RD, dominio represor; HD, homeodominio. (Carvalho et al.
2003, Brickman et al. 2001, Sobrier et al. 2006, Dattani et al. 1998, Thomas et al 2001)
LHX3
El factor Lhx3 es un miembro de la familia de factores de transcripción con homeodominio LIM.
El gen LHX3 (NG_008097.1) consta de 7 exones, que codifican para una proteína de 397
aminoácidos, ubicado en la región subtelomérica del cromosoma 9, en la banda 9q34.3. La
proteína posee dos dominios LIM involucrados en interacciones proteína-proteína, un dominio
de unión al ADN (homeodominio) y un dominio de transactivación carboxilo terminal que se
esquematiza en la figura 5 (Sloop KW et al 2001). En humanos se han identificado y estudiado
1 2 3 4
HD
Q6H I26T Q117P
E149K
E150delX167
S170L
R160C
Lfs110X K176T
T181A
RD

9
dos isoformas, Lhx3a y Lhx3b, siendo la isoforma “a” la de mayor actividad (West BE et al 2004;
McGillivray SM et al 2005). Ambas isoformas comparten todos sus dominios y se diferencian en
la secuencia amino terminal. Estas diferencias les imparte la capacidad de activar distintos
blancos transcripcionales, atribuyéndoles distintas funciones durante el desarrollo hipofisario
(Sloop KW et al 2001; West BE et al 2004; McGillivray SM et al 2005).
La expresión de Lhx3 durante el período de interacción entre la superficie del ectodermo y el
diencefalo ventral, para formar la bolsa de Rathke y el lóbulo posterior de la hipófisis, indica
que es indispensable para el normal desarrollo hipofisario (Seidah NG et al 1995; Bach I
et al
1995; Zhadanov AB et al 1995). Ratones KO para el gen LHX3 detienen el desarrollo de la bolsa
de Rathke y no producen los distintos linajes celulares de la adenohipófisis, demostrando que
la proteína Lhx3 es necesaria tanto para el desarrollo estructural temprano como para la
diferenciación celular tardía de la hipófisis (Sheng HZ et al 1996).
En humanos, alteraciones en el gen LHX3 se relacionan a déficit múltiple de hormonas
hipofisarias con una frecuencia del 1-2%. En la mayoría de los casos el MPHD se acompaña
de escoliosis y rigidez de la columna cervical con limitación en la rotación de la cabeza. A su
vez, reportes recientes indican que la presentación del fenotipo clínico puede ser variable,
incluyendo deficiencia aislada de hormona de crecimiento, hipoplasia hipofisaria, retardo
mental, alteraciones en la audición y anormalidades esqueléticas (Pfaeffle RW et al 2007; Rajab
A et al 2008; Kristrom B et al 2009). Todas las mutaciones reportadas en este gen tienen un
modo de herencia autosómica recesiva y, como se mencionó previamente, afectan las tres
etapas del desarrollo embrionario de la hipófisis (Romero CJ et al 2011).
Modificado de Christopher J Romero, et al 2011
Figura 5. Mutaciones en el gen LHX3 asociadas a MPHD. Se ilustra el gen del LHX3 con la expresión de la isoforma A
y los dominios LIM y el homeodominio (HD). Kristrom et al. 2009, Rajab et al.2008, Pfaeffle et al. 2007, Bhangoo et
al. 2006.
c.150 A>G splice site
1 2 3 4 5 6
HDLIM 1 LIM2
1a b
3088 bp del intragenica
g.159 delT
K50X
Y116C
E173X
23bp del
A210V
W224X
c.150 A>G splice site
1 2 3 4 5 6
HDLIM 1 LIM2
1a b
1 2 3 4 5 6
HDLIM 1 LIM2
1a b
3088 bp del intragenica
g.159 delT
K50X
Y116C
E173X
23bp del
A210V
W224X

10
LHX4
Lhx4 es otro miembro de la familia de factores de transcripción con homeodominio LIM. El gen
se localiza en la posición cromosómica 1q25 (NG_008081.1) y consta de 6 exones que
codifican para una proteína de 390 aminoácidos (Figura 6). Al igual que el Lhx3, este factor
contiene dos dominios LIM en la región amino terminal y un homeodominio de unión al ADN
(Yamashita T et al 1997; Howard PW, Maurer RA
2000). Modelos animales demostraron que el gen
LHX4 se expresa en el tubo neuronal en desarrollo, en la bolsa de Rathke y en la hipófisis
definitiva. Ratones KO para LHX4 demuestran que este factor es importante para la
proliferación y diferenciación celular. Se observa que en ausencia de Lhx4, el desarrollo
estructural temprano hipofisario no se ve afectado, mientras que la proliferación y
diferenciación celular se ve muy disminuida (Sheng HZ et al 1997). Se determinó que este efecto
es causado por un aumento en la apoptosis de las células progenitoras, que se traduce en un
fenotipo de hipoplasia adenohipofisaria con déficit aislado o múltiple de hormonas hipofisarias
(Sheng HZ et al 1997; Raetzman LT et al 2002).
La frecuencia de aparición de variantes en el gen LHX4 es de alrededor del 2% y las
manifestaciones clínicas son muy variables, presentándose pacientes con IGHD y otros con
MPHD, que incluye déficit de GH, TSH, ACTH y gonadotrofinas (Machinis K et al 2001 y 2005;
Pfaeffe RW et al 2008; Castinetti F et al 2008). En la mayoría de los casos las malformaciones
hipofisarias comprometen la hipófisis anterior, dando lugar a hipoplasia adenohipofisaria, sin
embargo en algunos pacientes se ha reportado alargamiento y/o tamaño normal de la hipófisis
anterior. En aproximadamente la mitad de los casos descriptos se demuestra un pobre
desarrollo de la silla turca, lo cual pareciera ser una característica distintiva de mutaciones en el
gen LHX4, que permitiría distinguir a los pacientes con MPHD con defectos en LHX4 de
aquellos que tengan alteraciones en otros genes (Tajima T et al 2007 y 2010; Dateki S et al 2010;
Filges I et al 2012; Takagi M et al 2012).
Todas las mutaciones y deleciones reportadas en el gen LHX4 son heterocigotas y tienen una
frecuencia muy baja. El fenotipo clínico se manifiesta a causa de haploinsuficiencia de Lhx4, a
pesar de que en un primer momento se generaron confusiones con respecto al tipo de herencia
de la patología. Se creía que la herencia era del tipo dominante, aunque se observaron casos
en donde más de un integrante de la familia poseía la misma mutación pero no manifestaba el
fenotipo clínico (penetrancia incompleta). La herencia dominante se descartó a partir de
distintos estudios in vitro donde se demuestra que mutaciones heterocigotas no poseen un
efecto dominante negativo sobre el alelo no afectado (Pfaeffe RW et al 2008; Castinetti F et al
2008; Dateki S et al 2010; Tajima T et al 2007 y 2010; Takagi M et al 2012).

11
Modificado de Christopher J Romero, et al 2011
Figura 6. Mutaciones en el gen LHX4, reportadas desde el 2001, asociadas con MPHD. Se observan las regiones
codificantes del gen y los dominios LIM y HD de la proteína (Machinis K et al 2001 y 2005; Pfaeffe RW et al 2008;
Castinetti F et al 2008; Tajima T et al 2007 y 2010; Dateki S et al 2010; Filges I et al 2012; Takagi M et al 2012)
SHH
Como se mencionó previamente, el camino de señalización Sonic Hedgehog (SHH) es una de
las principales vías de señalización que participan en el desarrollo hipofisario. El SHH se
expresa fuertemente en la superficie del ectodermo y en el diencéfalo ventral, inmediatamente
adyacente y por debajo de la Bolsa de Rathke, y tiene como principal función delimitar las
dimensiones de la bolsa de Rathke, de tal forma de establecer el tamaño y la estructura de la
hipófisis (Zhu X. et al 2007; Kelberman, D et al 2009). La sobreexpresión de SHH genera
hiperplasia hipofisaria mientras que el bloqueo de la señal se traduce en hipoplasia hipofisaria,
debido a la incapacidad de SHH de regular las dimensiones de la bolsa de Rathke primitiva.
La expresión del SHH está regulada por la acción de los factores SOX2 y SOX3, los cuales se
unen a regiones regulatorias del ADN y promueven su activación. A su vez, SHH media sus
efectos a través de tres factores de transcripción zinc finger de la familia Gli (Gli 1, Gli2 y Gli3).
Gli1 y Gli2 tienen efectos activantes mientras que Gli3 posee efectos inhibitorios sobre los
genes blancos involucrados en el camino de señalización SHH (Wang Y et al 2010). Debido a
que se demostró que la inactivación de Gli1 y Gli3, en ratones KO, no genera anormalidades
en el desarrollo hipofisario nos centraremos específicamente en las acciones del Gli2.
Gli2
El factor de transcripción Gli2 esta codificado por el gen GLI2 (NG_009030.1), ubicado en la
posición cromosómica 2q14. Este gen, formado por 13 exones, codifica para una proteína de
1586 aminoácidos que posee 3 dominios funcionales; uno central de unión al ADN, que posee
1
c.607-1 G>C
1
HD LIM 1 LIM2
R84C
T99fs53X
V101A L190R
A210P
P366T
1 2 3 4 5 6

12
5 dedos de Zinc, uno represor amino terminal, y uno transactivador carboxilo Terminal (Figura
7) (Sasaki H et al 1999).
Gli2 se expresa tanto en el diencéfalo ventral como en la superficie del ectodermo, regulando la
expresión de BMP4 y FGF8 y la proliferación de las células progenitoras, respectivamente.
Modelos animales demuestran que la inactivación de Gli2 genera una disminución en el
número de células progenitoras y reducción en la expresión de los BMPs y FGFs, lo que da
lugar a un lóbulo anterior hipoplásico y un lóbulo posterior ausente (Wang Y et al 2010).
En humanos, defectos en la señalización SHH, en etapas tempranas del desarrollo, son
considerados como la principal causa de holoprosencefalia (HPE), una anomalía anatómica
que resulta del clivaje incompleto del prosencéfalo en desarrollo (Roessler E, et al 2005 y 2010).
Mutaciones en el gen GLI2 han sido descriptas asociadas a una gran variedad de fenotipos,
los cuales incluyen HPE en los casos más severos (mutaciones heterocigotas que dan lugar a
proteínas truncadas) (Roessler E, et al 2003 y 2005). Por otro lado, se han reportado mutaciones
heterocigotas en pacientes con fenotipos más leves, de los cuales los más frecuentes son el
déficit aislado o múltiple de hormonas hipofisarias asociados, en la mayoría de los casos, con
neurohipófisis ectópica y polidactilia. Estas mutaciones son del tipo de variantes que afectan
aminoácidos específicos, de tal manera que un cambio nucleotídico en la secuencia de ADN se
traduce como un cambio de aminoácido a nivel proteico que afecta la funcionalidad de Gli2
(Bear KA et al 2014). La frecuencia de aparición de este tipo de variantes es de alrededor del
10% en pacientes con IGHD o MPHD, acompañadas de alteraciones de la neurohipófisis
(França MM et al. 2013).
Nuevamente, como en el caso de LHX4, el patrón de herencia dominante se caracteriza por
una penetrancia incompleta y una alta variabilidad en la expresividad fenotípica. La pérdida del
modelo clásico de herencia Mendeliana puede atribuirse, en parte, a la co-ocurrencia de
mutaciones en genes que interactúan entre sí, aunque es necesario mayor evidencia para
corroborar esta hipótesis.
Modificado de França MM et al 2013 Figura 7. Representación esquemática de la proteína GLI2 con mutaciones asociadas a MPHD. Rahimov F et al.2006, França MM et al 2013, Roessler et al. 2003 y 2005, Flemming GM et al. 2013, Bertolacinia CD. et al 2012, Bear KA et al. 2014.
Dominio represor Union al ADN
Dominio activador
NH2 COOH
p.A203T
p.P253S
p.A268V
p.V432M
p.R473H
p.A780V
p.F830L
p.R933H
p.G947D
p.V1117L
p.G1197D
p.(M1241I;
P1485A)
p.P1315S
p.(M1444I;
L1445F)
p.(M1352V; D1520N)

13
1.2.2 Factores de acción tardía Prop1
También conocido como el profeta de Pit-1, el gen se localiza en la posición cromosómica 5q35
(NG_015889.1). Está formado por tres exones que codifican para una proteína de 226
aminoácidos con un homeodominio de unión al ADN y un dominio activador transcripcional
carboxilo Terminal (Figura 8) (Duquesnoy P et al 1998). Prop1 es un factor de transcripción con
acciones activantes e inhibitorias. Se postula que ejerce sus funciones a través del camino de
señalización Wnt/β-catenina, en donde la unión de Prop1 con β-catenina reprime la expresión
del gen HESX1 y promueve, simultáneamente, el desarrollo celular dependiente de Pit-1 (Olson
LE
et al 2006). La expresión de PROP1 mantiene el linaje celular Pit-1 y permite la
diferenciación de las células progenitoras a lactotropos, somatotropos y tirotropos. El normal
desarrollo y proliferación de estos linajes celulares permite, a su vez, la correcta diferenciación
del linaje adrenal y gonadal a partir de la interacción de distintos factores que son secretados y
que ejercen sus funciones a nivel hipofisario. Se ha demostrado que este proceso de
diferenciación es dependiente del camino de señalización Notch, el cual es indispensable para
la diferenciación tardía de los tres tipos celulares (Zhu X et al 2006).
Para analizar las funciones de Prop1 en un modelo de enfermedad se desarrolló un ratón,
denominado Ames, con una mutación puntual, homocigota, en el dominio de unión al ADN de
Prop1. Este ratón presenta un retardo de crecimiento severo, hipotiroidismo e infertilidad junto
con hipoplasia adenohipofisaria. Cuando se vuelve a restablecer la actividad de Prop1, a partir
de un transgen artificial, se revierte el fenotipo observado en estos ratones (Andersen et al.
1995, Sornson et al. 1996). Estos resultados demuestran la importancia y la participación de
Prop1 en el proceso de diferenciación de las células progenitoras a somatotropos, lactotropos y
tirotropos (Sornson et al.1996).
Las alteraciones en el gen PROP1 son la principal causa de MPHD, alcanzando una frecuencia
del 30-50 % en casos familiares (Kelberman D et al 2009). En casos esporádicos la frecuencia se
mantiene baja, similar a lo mencionado previamente cuando se afectan otros genes
relacionados con el desarrollo embrionario. Más de 20 mutaciones han sido reportadas con un
modo de herencia autosómica recesiva, que incluyen mutaciones homocigotas o heterocigotas
compuesto (Kelberman D & Dattani 2007). La mayoría de las mutaciones afectan el dominio de
unión al ADN y en parte el dominio transcripcional. El fenotipo clínico observado en los
pacientes afectados es variable y dinámico. Típicamente, los pacientes presentan déficit de
GH, PRL, y TSH, aunque en algunos casos, con la edad, aumenta el riesgo de que se afecten
los ejes adrenal y gonadal a causa de una disminución en la interacción de los distintos linajes
celulares hipofisarios necesarios para la correcta proliferación. A nivel estructural, la
neurohipófisis es normal mientras que la afectación de la adenohipófisis es más variables,
pudiéndose observar una hipófisis anterior hipoplásica, normal o alargada (Turton et al. 2005,
Kelberman & Dattani 2007).

14
Modificado de Christopher J Romero, et al 2011
Figura 8. Mutaciones en el gen PROP1 asociadas a MPHD. El gen PROP1 se muestra en la región superior mientras
que, en la región inferior, se observa la proteína con el homeodominio (HD) y el dominio transactivador (TD). Agarwal
et al. 2000, Turton et al. 2005, Kelberman & Dattani 2006 y 2007, Lemos et al. 2006, Abrao et al. 2006, Kelberman
et al. 2008
Pit-1 (POU1F1)
El factor de transcripción específico hipofisario (Pit-1) es un miembro de la familia de
homeoproteínas POU. Esta codificado por 6 exones en el gen POU1F1 (NG_008225.2),
localizado en la posición cromosómica 3p11, que da lugar a una proteína de 291 aminoácidos.
Posee un dominio transactivador carboxilo terminal y dos dominios, altamentes conservados,
de unión al ADN; el homeodominio POU y el dominio específico POU (Figura 9) (Rosenfeld MG
1991). Se expresa en el lóbulo anterior de la hipófisis y regula la expresión de distintos genes
mediante la unión a múltiples regiones promotoras, entre los cuales se encuentran los genes
que codifican para GH-1 y PRL, así como también genes que participan en la regulación de la
producción de TSHβ, AMPc y PRL (Xu et al. 1998, Cohen et al. 1999).
Se han desarrollado distintos ratones con mutaciones puntuales en el gen POU1F1 para
determinar sus efectos en un modelo de enfermedad. Uno de los ratones más usados es el
llamado Snell, el cual presenta un fenotipo asociado con hipoplasia hipofisaria y deficiencia de
GH-1, PRL y TSH, y bajos niveles de expresión de Pit-1(Camper et al.1990).
Las manifestaciones clínicas, en pacientes afectados por mutaciones en el gen POU1F1,
incluyen deficiencia de GH-1, TSH y PRL, con inconsistentes hallazgos de hipoplasia
adenohipofisaria en la resonancia magnética nuclear (RMN). El déficit de GH-1 y PRL son de
aparición temprana mientras que el déficit de TSH es más variable, pudiendo aparecer más
tarde en la infancia (Cohen LE, Radovick S 2002).
Se han reportado más de 25 mutaciones distintas, con herencia autosómica recesiva y
dominante (Kelberman D. et al 2009; Christopher J Romero, et al 2011). Se ha postulado que las
mutaciones que afectan los dominios de unión al ADN, POU específico y homeodominio POU,
causan un MPHD con herencia autosómica recesiva mientras que las mutaciones que afectan
11
HD
c.112_124delFs37X
c.150delA,fsX164
p.R71C
p.R71H
p.F99X
p.F99Q
c.310delC
p.W194X
1 2 3
TD
c.157delA,fsX164
p.R73H
p.Q83X
p.F88Sp.R125W
c.467insT
c.629delC
11
HD
c.112_124delFs37X
c.150delA,fsX164
p.R71C
p.R71H
p.F99X
p.F99Q
c.310delC
p.W194X
1 2 3
TD
c.157delA,fsX164
p.R73H
p.Q83X
p.F88Sp.R125W
c.467insT
c.629delC

15
otras regiones de la proteína causan un MPHD con un patrón de herencia autosómica
dominante (Blankenstein O et al 2001; Salemi S et al 2003, Mullis P 2005; Christopher J Romero, et
al 2011).
Modificado de Christopher J Romero, et al 2011
Figura 9. Mutaciones en el gen POU1F1 asociadas a MPHD. Se ilustra el gen y la proteína Pit-1 con el homeodominio (HD) y el dominio especifico POU (SD). Turton et al. 2005b, Miyata et al. 2006, Snabboon et al. 2008, Carlomagno et al. 2009, Kelberman et al. 2009
1
IVS2-3insA ter82
1
HDSD
p.Q4X p.R143L
p.K145X
p.R172Q
p.S179R
p.F262L
1 2 3 4 5 6
c.682+1G>A
p.W193R
p.W193X
p.L194Q
p.E230K
p.F233L
p.Q242R
c.747delA,fs250X
p.V272X
c778insA,
fs284x
1
IVS2-3insA ter82
1
HDSD
p.Q4X p.R143L
p.K145X
p.R172Q
p.S179R
p.F262L
1 2 3 4 5 6
c.682+1G>A
p.W193R
p.W193X
p.L194Q
p.E230K
p.F233L
p.Q242R
c.747delA,fs250X
p.V272X
c778insA,
fs284x

16
2. Crecimiento Fetal
2.1 Generalidades
El crecimiento fetal es un proceso complejo y dinámico regulado por factores de origen
materno, placentario y fetal. En las primeras etapas de la vida fetal el genoma fetal es el mayor
determinante del crecimiento mientras que con el avance del embarazo aumenta la importancia
de los factores nutricionales, ambientales y hormonales. La regulación hormonal del
crecimiento fetal está diseñada de tal forma de asegurar la disponibilidad de nutrientes para el
feto. A su vez, la disponibilidad de nutrientes depende de las funciones placentarias y
endocrinas que permiten el intercambio de nutrientes entre la madre y el feto. Durante el
embarazo, la placenta se desarrolla de manera conjunta con el feto, situándose como una
barrera entre la madre y el feto que selecciona específicamente los componentes a transferir
entre ambos. La placenta secreta una gran variedad de sustancias, de las cuales algunas son
secretadas dentro del compartimiento materno, otras dentro compartimiento fetal y otras dentro
de ambos. Estas sustancias pueden actuar localmente en la placenta o tener efectos
sistémicos. La circulación fetal esta estrictamente separada de la circulación materna de tal
manera que, para que los nutrientes lleguen al feto, es necesario que atraviesen la barrera
placentaria (Lacroix M., et al. 2002; Jens Fuglsang, Per Ovesen 2006).
Nosotros nos centraremos, principalmente, en las funciones endocrinas que regulan el
crecimiento fetal y en la participación de la hormona de crecimiento placentaria (PGH) en la
adaptación del metabolismo materno al embarazo.
2.2 El gen GH2 y la PGH
En humanos, las hormonas de crecimientos están codificadas por cinco genes organizados en
tandem y con alta homología entre sí, que incluyen a los genes de la GH hipofisaria (GH1), la
GH placentaria (GH2), y tres somatotrofinas coriónicas (CSH-1, CSH-2 y CSH-P), también
llamadas lactógenos placentarias. Estos genes se localizan en un cluster de 65Kb, en el brazo
largo del cromosoma 17 (q22-q24) que se muestra en la figura 9 (Barsh et al., 1983; Hirt et
al.,1987; Chen et al.,1989). Se cree que estos genes han evolucionado por duplicación génica
debido a la alta homología estructural (91-99% de secuencias conservadas), en el patrón de
organización (5 exones separados por 4 intrones), en la orientación transcripcional y en las
secuencias de ADN flanqueantes a cada uno de ellos (DeNoto FM et al 1981; Chen et al.,1989).
Cada gen codifica para una proteína madura de 217 aminoácidos precedida por un péptido
señal de 26 aminoácidos. De los cinco genes que conforman el cluster, cuatro son de
expresión placentaria; CSH 1, 2 y P, GH2 (Barrera-Saldana et al., 1983), mientras que el gen
GH1 se expresa en la hipófisis (Martial et al., 1979; Nachtigal et al., 1993). La expresión eficiente
de GH1, en las células somatotropas, depende de la unión del factor de transcripción Pit-1 a las
regiones promotoras del gen. A pesar de la presencia de sitios de unión para Pit-1 en las

17
regiones regulatorias del locus GH/CSH y la elevada expresión de este factor en la hipófisis, el
gen GH2 no se expresa en la hipófisis. (Jin et al., 1999; Lefevre et al., 1987; Ingraham et al., 1988;
Castrillo et al., 1989). A su vez, existe evidencia de un miembro de la familia de factores
nucleares tipo 1 involucrado en la inhibición de la expresión de los genes placentarios en el
tejido hipofisario (Norquay LD et al 2001).
El gen GH2 se expresa en la placenta y codifica para una proteína de 191 aminoácidos (PGH,
22kDa), que difiere en 13 aminoácidos de la GH hipofisaria (GH-1, 22kDa). Posee un punto
isoeléctrico más básico y un sitio de N-glicolisación en la posición 140-142. Ensayos de hibridización
in situ, con una sonda específica para GH2, han demostrado que el sitio exacto de producción de
PGH es el sincitiotrofoblasto. (Cooke N., et al. 1988; Níkel B., et al. 1990; Frankenne F., et al. 1990)
Genetic control of growth, Mullis P. 2005
Figura 9. Cluster de GH.
2.3 Propiedades biológicas de la PGH
La PGH se une a los receptores específicos de GH, en la superficie celular, con similar afinidad
que la GH hipofisaria, sin embargo tiene una afinidad significativamente menor por los
receptores lactogénicos (Hizuka et al., 1982; Ray et al., 1990). Esto se correlaciona directamente
con la alta actividad somatogénica y la baja actividad lactogénica que posee la PGH. Circula
unida a la proteína trasportadora de GH (GHBP), de tal forma que en la circulación materna
puede estar de manera libre o unida a la GHBP (McIntyre et al., 2000).
La actividad somatogénica se ha demostrado a partir de estudios con ratones transgénicos,
donde aquellos que sobreexpresan el gen GH2 poseen un tamaño entre 40-90% mayor
comparado con los ratones salvajes (Selden et al., 1988). A su vez, se ha reportado que la PGH
induce la ganancia de peso en ratas hipofisosectomizadas de manera comparable a la GH
hipofisaria (MacLeod et al., 1991).
Cabe destacar que todos los experimentos realizados para determinar las propiedades
biológicas de la PGH se han realizado in vitro, en líneas celulares y en roedores, de tal manera
que los efectos observados no son del todo extrapolables al humano.
GH-1 CSHP CSH-1 GH-2 CSH-2
HHIIPPOOFFIISSAARRIIAA PPLLAACCEENNTTAARRIIAA PPLLAACCEENNTTAARRIIAA PPLLAACCEENNTTAARRIIAA PPLLAACCEENNTTAARRIIAA
5´ 3´
17q22-24

18
2.4 Rol fisiológico de la PGH
La PGH regula los niveles de IGF-1 en la circulación materna. Se ha estudiado la presencia de
PGH en sangre de cordón umbilical, sin embargo sólo fue detectada en la sangre materna,
consistente con el sitio de síntesis de la PGH en la interfaz materno-placentaria (Frankenne F.,
et al 1990; Heinrichs C., 1994; Linnemann K., 2000). Estudios transversales y longitudinales de
embarazos normales y patológicos, han demostrado una correlación positiva entre los niveles
de PGH y los niveles de IGF-1 en la circulación materna (Caufriez F., et al 1993; Chellakooty K.,
et al. 2004; Mcintyre H.D., et al 2000). En mujeres con acromegalia, a pesar de los altos niveles
de GH-1 y las altas concentraciones basales de IGF-1, los niveles de IGF-1 materno aumentan
gradualmente durante el embarazo siguiendo el patrón de secreción de la PGH. Por otro lado,
se ha observado un aumento progresivo de IGF-1 circulante en mujeres embarazas con
deficiencia de Pit-1, lo que indica que la PGH es el principal regulador de los niveles séricos de
IGF-1 maternos durante el embarazo (Verhaeghe J et al 2001).
La presencia de receptores para PGH en las vellosidades trofoblásticas así como la expresión
de PGH en las vellosidades extra trofoblásticas sugieren que esta hormona tiene acciones
paracrinas y autocrinas en el desarrollo placentario (Frankenne et al., 1992; Hill et al., 1992;
Mertani et al., 1995).
Con respecto a sus funciones metabólicas la PGH estimula fuertemente la gluconeogénesis, la
lipólisis y el anabolismo en distintos órganos, además de aumentar la disponibilidad de
nutrientes en la unidad feto-placenta (Selden R.F., et al. 1988; Barbour L.A., et al. 2002).
2.5 Rol del sistema de los IGFs durante el desarrollo fetal
Tanto el IGF-1 como el IGF-2 se expresan en el tejido fetal desde etapas muy tempranas hasta
la fase final de maduración del tejido, justo antes del nacimiento. El IGF-2 es el principal factor
de crecimiento durante el desarrollo embrionario mientras que el IGF-1 toma mayor relevancia
en las etapas más avanzadas de la gestación. Todos los tejidos fetales humanos expresan
ARNm de IGF-1 e IGF-2 y esta expresión varía entre los distintos tejidos fetales y según el
período gestacional (Jones JI., et al. 1995). Ambos IGFs están presentes en la circulación fetal y
actúan mayoritariamente de manera autocrina, aunque acciones endocrinas-sistémicas pueden
ocurrir durante la segunda mitad de la gestación. Las concentraciones séricas de IGF-2 son
mucho más altas que las concentraciones de IGF-1 en las etapas más avanzadas del
embarazo, sin embargo la mayoría de los reportes no encuentran asociación entre los niveles
séricos de IGF-2 y el peso al nacer. Por otro lado, niveles séricos de IGF-1 del recién nacido y
de cordón umbilical se correlacionan directamente con el peso al nacer. A su vez, se ha
observado que los niveles de IGF-1, en el útero y al nacer, en recién nacidos con retardo de
crecimiento intrauterino (RCIU) se encuentran disminuidos mientras que están aumentados en
neonatos que nacen grandes para la edad gestacional (LGA) (Fowden AL., et al. 2003 y 2004;
Kayak NR., et al 2003, Gluckman PD., et al. 2003).

19
Los efectos del sistema de los IGFs en el crecimiento y desarrollo fetal se han demostrado a
partir de modelos de ratones KO que invalidan los genes que codifican para varias de sus
proteínas. El bloqueo aislado de IGF1, IGF2 y IGF1R da lugar a retardo de crecimiento
intrauterino, el cual aumenta su gravedad si se combinan con alguno del los otros déficit. El
déficit de IGF-1 o IGF-2 da lugar a un retardo de crecimiento similar (disminución del 40% del
peso corporal comparado con el normal) mientras que la pérdida del IGF1R lleva a un retardo
de crecimiento más severo (55% de pérdida del peso corporal con respecto al normal),
sugiriendo que ambos IGFs actúan a través del IGF1R. El fenotipo de retardo de crecimiento
más severo (70% de disminución del peso corporal) es observado cuando se combina la
pérdida del IGF1R o el IGF-1 con la pérdida de IGF-2, lo que indica de que existe otro receptor
por el cual el IGF-2 ejerce sus efectos en el crecimiento fetal (Butler AA., et al. 2001).
Se ha observado en modelos experimentales que el gen IGF1 posee gran sensibilidad a los
cambios en el estatus nutricional, de tal manera que una restricción de nutrientes lleva a una
menor expresión del gen IGF1 que se traduce en bajos niveles séricos de IGF-1 (Fowden AL., et
al. 2003; Gluckman PD., et al. 2003). La insulina regula positivamente los niveles de IGF-1,
probablemente por un aumento en la disponibilidad celular de glucosa (Fowden AL., et al. 2003).
A su vez, este mismo grupo en el 2004 demostró que los glucocorticoides también regulan la
expresión de ambos IGFs de manera tejido específica.
Luego del nacimiento, la producción de IGF-1 se hace dependiente de GH-1, reseteando el
mecanismo de regulación de crecimiento para asegurar el correcto desarrollo postnatal en un
nuevo ambiente nutricional (Gicquel C., Le Bouc Y., 2006).
2.6 Regulación de la secreción de PGH
La regulación de la secreción de la PGH es muy diferente a la regulación de la GH hipofisaria.
La hormona, hipotalámica, liberadora de la hormona de crecimiento (GHRH) no modula la
secreción de PGH in vivo o in vitro (de Zegher et al.1990; Evain-Brion et al., 1990). La secreción
de PGH es inhibida, in vitro, por la glucosa tanto en células placentarias como en células
trofoblásticas en cultivo (Patel et al., 1995). En mujeres con diabetes gestacional, las
concentraciones de PGH disminuyen durante el test de tolerancia a la glucosa, mientras que no
se observan cambios en la secreción de lactógeno placentaria, leptina o gonadotrofina
coriónica (Alsat et al., 1997; Chaignaud et al., 2000). Bjorklund et al en 1998 reportaron un
aumento del 27% en la PGH durante los episodios hipoglucémicos hiperinsulinémicos en
mujeres embarazas con diabetes insulino dependiente. Estos resultados sugieren que el
sincitiotrofoblástico, el cual está en contacto directo con la sangre materna y expresa la mayor
cantidad de trasportadores de glucosa (Glut1), responde de manera muy rápida a las
variaciones en los niveles de glucosa en la sangre materna modificando la secreción de PGH.

20
2.7 PGH en casos patológicos En los casos de anencefalia fetal los niveles de PGH en la circulación materna no sufren
modificaciones, lo que demuestra la independencia de la regulación de la PGH del eje
hipofisario (Evain-Brion et al., 1994). Se han reportado embarazos sin complicaciones, con
recién nacidos normales, en mujeres que portan una deleción completa del locus CSH P-1-2-
GH2 y que no tienen niveles circulantes de lactógeno placentaria. Sin embargo, en estos
casos, se observa una inesperada expresión del gen GH1 (hipofisaria) en la placenta, lo que
sugiere que existe un mecanismo compensatorio que permite que la placenta secrete una
hormona con el mismo efecto metabólico que la PGH (Alsat et al., 1997).
Existen casos donde pacientes portadores de deleciones nacen sin inconvenientes y solo se
ven afectadas las proporciones corporales. Estos casos de deleciones completas del cluster
GH evidencian que la viabilidad de los fetos no se ve afectada por la incapacidad de la placenta
de sintetizar GH (Akinci et al., 1992; Ghizzoni et al., 1994). La pérdida de PGH genera un déficit
en la síntesis de IGF-1, que se manifiesta en una menor disponibilidad de los tejidos a los
factores de crecimiento necesarios para un normal desarrollo, dando lugar a un retardo de
crecimiento intrauterino (RCIU).
Existen varias causas de RCIU, además del déficit de PGH, entre las cuales podemos
mencionar el déficit de IGF-1, propiamente dicho, y la resistencia a IGF-1. Aunque el sistema
de los IGFs tiene un rol crucial en el crecimiento fetal, las mutaciones son raras. Solo seis
familias han sido reportadas con alteraciones en el gen IGF1, los cuales se presentan con
retardo de crecimiento pre y postnatal muy severo. Por otro lado, la resistencia a IGF-1 se
presenta en pacientes con haploinsuficiencia del receptor de IGFs tipo 1 (IGF1R), causado por
mutaciones heterocigotas en el gen IGF1R. En los capítulos siguientes nos centraremos en
esta patología.

21
3. Crecimiento postnatal
3.1 Eje de crecimiento GHRH-GH-IGF1
El crecimiento somático normal requiere de la integración funcional de distintos factores
hormonales, metabólicos y de crecimiento que conforman el eje hipotálamo-hipofisario de
crecimiento, o lo que también se conoce como eje GHRH-GH-IGF1.
La secreción de GH-1 es regulada por la acción de dos péptidos hipotalámicos, la hormona
liberadora de GH (GHRH) y el factor inhibitorio de GH (GHIF). Estos factores a su vez,
dependen de la acción de neurotransmisores pertenecientes a vías de señalización del sistema
nervioso central. Las células somatotróficas de la adenohipófisis poseen receptores de
membrana que responden a estos factores, de tal forma que cuando la GHRH se une a su
receptor específico de membrana (GHRHR) desencadena la secreción de GH-1 (Mullis P. 2005).
Las acciones de la GH-1 son mediadas por una combinación de componentes del sistema de
los IGFs, que incluyen al IGF-1, proteínas de unión a los IGFs (IGFBP), el receptor de IGFs tipo
1 (IGF1R), y por efectos independientes de IGF-1, a través de las acciones directas de la GH-1
en distintos tejidos (David A. et al., 2011). La hipótesis original de la somatomedina, propuesta
por Salmon y Daughaday en 1957, afirmaba que la GH-1 se unía a su receptor específico y
estimulaba la producción de IGF-1, el cual controlaba todos los procesos relacionados con el
crecimiento de los tejidos de manera independiente (acción endocrina). Sin embargo, el
descubrimiento de que los IGFs se expresan en la mayoría de los tejidos y órganos
cuestionaba esta hipótesis original. Posteriormente Green et al, en 1985 propusieron una teoría
de efecto dual, donde la GH-1 regula la expresión de la producción local de IGF-1, el cual actúa
de manera autócrina/parácrina, indicando que la GH-1 tiene efectos locales independientes de
aquellos que son mediados por los niveles de IGF-1 circulantes por acción de GH-1 de manera
endocrina.
La GH-1 ejerce sus efectos sobre el crecimiento a través de la regulación de la expresión del
IGF-1 en tejido hepático y tejidos periféricos. En tejidos no hepáticos, además de la GH-1, otros
factores de crecimiento, así como también algunas citoquinas, pueden ejercer efectos locales
mediante la regulación de la síntesis de IGF-1 y/o mediante mecanismos independientes de la
expresión de IGF-1 (David A. et al., 2011). La vía GH/IGF-1 se inicia con la unión de la GH-1 a
su receptor específico (GHR). Este receptor se encuentra presente en la mayoría de los tejidos,
siendo el hígado el órgano con mayor expresión (Ballesteros M., et al. 2000). Es una proteína
transmembrana homodimérica perteneciente a la superfamilia de receptores citoquina tipo 1.
Esta formado por un dominio extracelular que posee dos sitios de unión para la GH-1, un
dominio transmembrana que ancla el receptor a la membrana plasmática y un dominio
intracelular involucrado en la traducción de señal. El GHR posee una región soluble,
denominada GHBP, que se genera por clivaje del dominio extracelular del receptor y cumple la
función de trasportar la GH-1 en circulación (Leung DW., et al. 1987). La unión de una molécula
de GH-1 al GHR homodimérico genera un cambio conformacional que permite la asociación del

22
receptor con una quinasa intracelular denominada JAK2 (Rowland JE., et al 2005). La proteína
JAK2 es una quinasa que fosforila residuos de tirosina en el dominio intracelular del receptor,
los cuales sirven como puntos de anclaje para las proteínas STATs, en particular STAT5a y
STAT5b. Las proteínas STATs fosforiladas se dimerizan y se traslocan al núcleo donde se
unen a elementos de respuesta específicos en el ADN y regulan la expresión de distintos
genes (Derr MA., et al. 2011). Modelos animales junto con casos clínicos de sujetos afectados,
han demostrado que el principal factor involucrado en la regulación de la síntesis de IGF-1 es la
STAT5b (Udy GB., et al 1997; Teglund S., et al 1998; Rosendfeld RG., et al 2007), de tal manera
que la unión de la GH-1 a su receptor genera la formación de dímeros de STAT5b que se
traslocan al núcleo y se unen a los elementos de respuesta a la GH (GHRE), regulando la
transcripción de los genes IGF1, IGFBP3 e IGFALS, los cuales codifican para las proteínas
IGF-1, IGFBP-3 y la subunidad ácido lábil (ALS), respectivamente (Wang Y., et al. 2005;
Rowlinson SW et al 2008; Chia DJ., et al. 2010).
Por mucho tiempo se creyó que solo este mecanismo era utilizado por el GHR para traducir la
señal de la GH-1, sin embargo distintos reportes han evidenciado la existencia de otros
caminos de señalización, entre los cuales cabe mencionar las vías Src/Ras/ERK y PI3K/AKT
(Milward A. et al. 2004). Barclay et al en 2010 caracterizaron un ratón con la vía JAK2-STAT5
suprimida y mostraron que el sistema Src/Ras/ERK se activaba de manera dependiente de GH-
1. El camino de señalización que involucra a la familia de proteínas tirosina quinasa Src es
independiente de JAK2 y activa a las proteínas ERK 1 y 2 (p44/42 MAPK) (Rowlinson SW et al
2008; Barclay et al. 2010). La quinasa Src Lyn se une directamente al GHR en la parte proximal
de la membrana plasmática, independientemente de la unión de la GH-1 al receptor (a
diferencia de JAK2). Cuando un agonista de la GH-1 se une al GHR, ocurre un cambio
conformacional en un loop específico del receptor que da lugar a la activación de Src, que inicia
todo el proceso de señalización vía Ras/ERK (Barclay et al. 2010). Se han reportado mutaciones
que afectan genes que codifican para proteínas implicadas en el camino de señalización
Ras/ERK que causan una serie de enfermedades denominadas RASopatías, entre las cuales
se encuentra el síndrome de Noonan (Tartaglia M. et al 2002). Pacientes con este síndrome
poseen retardo de crecimiento asociado a niveles bajos de IGF-1, lo que demuestra la
implicancia de este camino de señalización en la traducción de señal del GHR (Binder G., et al
2005; Limal JM., et al 2006).
Los factores de crecimiento símil a la insulina tipo 1 y 2 (IGF-1, IGF-2) fueron reportados por
primera vez en 1950 como factores de crecimiento producidos en el hígado en respuesta a la
GH-1 y tienen la función de regular el crecimiento somático de todo el organismo (Daughaday
WH. et al 1999). Tanto el IGF-1 como el IGF-2 tienen múltiples funciones sobre las células, que
son mediadas a través de un mismo receptor, el receptor de IGFs tipo 1 (IGF1R). De manera
similar a sus ligandos, el IGF1R tiene una alta homología con el receptor de insulina (INSR),
principalmente en el dominio tirosina quinasa, y comparten los mismos caminos de
señalización intracelular cuando son activados (Kim JJ., et al 2002). Aunque los IGFs, sus
receptores y consecuentemente los caminos de señalización intracelulares son muy similares a
los de la insulina, han evolucionado funcionalmente de manera muy diferente. La síntesis de

23
insulina se restringe al páncreas, donde se almacena en gránulos secretores en las células β, y
es liberada en repuesta a distintos estímulos, como por ejemplo cambios en los niveles de
glucosa. Por otro lado, los IGFs se expresan a lo largo de la mayoría de los tejidos y órganos y,
como la mayoría de los factores de crecimiento y citoquinas, no se almacenan en gránulos
secretorios dentro de las células sino que son secretados a medida que se sintetizan (Holly J.,
et al 2006). A su vez existen diferencias en el modo en el que circulan en el torrente sanguíneo.
Cuando la insulina es secretada desde el páncreas entra en la circulación y atraviesa el
organismo hasta encontrar su receptor específico en los tejidos blancos mientras que los IGFs
se asocian con alta afinidad a proteínas de unión (IGFBP) específicas, que los estabilizan y
aumentan su vida media (Hintz RL., et al 1977, Holly J., et al 2006).
Existen 6 proteínas de unión a los IGFs (IGFBP-1 a IGFBP-6) que, a diferencia de la GHBP, no
están relacionadas con los receptores de membrana. Las 6 proteínas transportadoras están
íntimamente relacionadas desde el punto de vista estructural aunque provienen de distintos
genes y tienen distintas propiedades funcionales (Firth SM., et al 2002). En términos fisiológicos,
aún es limitado el entendimiento del rol exacto de la mayoría de los IGFBPs. Se sabe que la
formación de complejos entre los IGFBPs y los IGFs reduce considerablemente el clearance de
estos últimos. En circulación, el IGFBP3 e IGFBP5 se unen a una glicoproteína (ALS) que se
encuentra en exceso y forman un complejo ternario que disminuye aún más el clearence de los
IGFs. El mayor transportador de IGFs en circulación es la IGFBP3, que a su vez es la proteína
trasportadora más abundante. El 75-80% de los IGFs que circulan en el torrente sanguíneo
forman parte de un complejo de 150 kDa que contiene a uno de los IGFs, la IGFBP-3 y la ALS.
El restante 20-25% está asociado a alguna otra IGFBP en forma de complejo binario mientras
que menos del 1% circula de manera libre (Jones JI., et al. 1995; Hwa V., et al. 1999; Firth SM., et
al 2002).
Como se menciono previamente, los efectos metabólicos y mitogénicos de los IGFs son
mediados a través del receptor de IGFs tipo 1 (IGF1R), un receptor de superficie celular
perteneciente a la familia de los receptores tirosina quinasa. La unión del IGF-1 a su receptor
promueve la autofosforilación del dominio intracelular del receptor resultando en la activación
de los caminos de señalización PI3K/Akt y Ras/ERK, los cuales median los efectos
proliferativos, metabólicos y de supervivencia celular del IGF-1 (Adams TE., et al. 2000). En
capítulos posteriores nos centraremos en la estructura y funciones del IGF1R. La regulación del
eje GH-IGF1 se puede observar en la figura 10.

24
Modificado de David et al 2011 Endocrine Reviews
Figura 10. Eje de crecimiento GH-IGF1
3.2 Defectos moleculares que afectan el eje de crecimiento GHRH-GH-IGF1.
3.2.1 Déficit aislado de GH (IGHD)
La talla baja afecta al 3% de la población. Se la define como una talla menor a -2 score de
desvíos estándar (SDS) de la población de referencia en función de la edad y el sexo. Las
causas son múltiples, desde variantes fisiológicas hasta enfermedades crónicas y patología
endocrinológica. Dentro de estas últimas, la prevalencia del déficit de hormona de crecimiento
(GHD) es poco frecuente, hallándose en 1 de cada 4000-10000 niños (Mullis P. 2005; Wit J.M. et
al 2011). La mayoría de los casos son esporádicos, y se cree que resultan de anomalías del
desarrollo, sin embargo entre un 3-30 % de los casos reportados tienen un familiar directo
afectado, lo que sugiere una etiología genética de la patología (Lacey KA & Parkin JM 1974;
Rona RJ & Tanner JM 1977; Mullis P. 2005; Wit J.M. et al 2011).
El déficit aislado de hormona de crecimiento (IGHD) se asocia, al menos, con cuatro
desordenes Mendelianos (Tabla 1). Estos incluyen a dos formas de herencia autosómica
recesiva (tipo IA y IB), una autosómica dominante (tipo II) y una forma ligada al X (tipo III)
(Mullis P. 2005).
Como ya se mencionó en el capítulo de crecimiento fetal, el cluster GH consiste en 5 genes
con alta homología entre sí, ubicados en dirección 5’ a 3’ ordenados de la siguiente forma:
GH1, CSHP (pseudogen somatotrofina corionica), CSH1 (somatotrofina corionica), GH2 y
CSH2 (somatotrofina corionica). Con la excepción de CSHP, cada gen codifica para una
prehormona de 217 aminoácidos, la cual pierde el péptido señal para formar una proteína
IGF-1 IGFBP-3
ALS
IGF1R
Efectos metabólicos y mitogénicos
CRECIMIENTO
SHC
IRS-1
ERK1/2 AKT2
GHRE IGF-1
GH-GHBP
GHR
JAK2
ERK1/2
STAT5b PI3K

25
madura de 191 aminoácidos y 22kDa de peso molecular. (Barsh et al., 1983; Hirt et al.,1987;
Chen et al.,1989) (Figura 11)
Genetic control of growth, Mullis P. 2005
Figura 11. Cluster GH. Se observa la estructura del gen GH1, conformado por 5 exones y 4
intrones.
3.2.1.1 IGHD tipo IA
Este tipo de IGHD fue reportada por primera vez en 1970 en tres niños Suizos con alteración
severa del crecimiento (Illig R. 1970). Los individuos afectados, ocasionalmente, presentan talla
baja al nacer pero alrededor de los seis meses desarrollan un retardo de crecimiento severo
con episodios de hipoglucemias. La principal causa molecular del IGHD tipo IA son deleciones
que abarcan todo el gen GH1, mientras que en menor medida se han reportado mutaciones sin
sentido o mutaciones que generan un cambio en el marco de lectura que afectan la región que
codifica para el péptido señal en el gen GH1 (Mullis P. 2005; Wit J.M. et al 2011).
La frecuencia de deleciones en el gen GH1 como causa de IGHD difiere entre las distintas
poblaciones estudiadas en base, principalmente, al criterio con el que se define la talla baja
para la población en cuestión. El análisis de pacientes con severo retardo crecimiento (< -4
SDS) reveló que la prevalencia de deleciones es aproximadamente del 10-15% en las distintas
poblaciones (Parks JS. et al 1989; Kamijo T. et al 1991; Mullis P. et al 1992). Los tamaños de las
deleciones son heterogéneas, siendo la más frecuente (70-80%) la que abarca una distancia
de 6.7Kb en el clúster del gen GH1 (Mullis P. 2005). A su vez, se han reportado deleciones de
mayor tamaño, como las de 7, 7.6 y 45 kb, pero con una frecuencia de aparición menor
(Vnencak-Jones CL., et al. 1988 y 1990).
Los pacientes afectados por este tipo de alteraciones poseen un retardo de crecimiento severo
(menor a -4 SDS de talla) con niveles indetectables de GH-1 sérica. La principal característica
G H-1 C S HP C S H-1 G H-2 C S H-2
HIP OF IS AR IAHIP OF IS AR IA P L AC E NT AR IAP L AC E NT AR IA P L AC E NT AR IAP L AC E NT AR IA P L AC E NT AR IAP L AC E NT AR IAP L AC E NT AR IAP L AC E NT AR IA
5´ 3´
17q23
1 I 2 II 3 III 4 IV 55´ 3´
G H-1G H-1 C S HPC S HP C S H-1C S H-1 G H-2G H-2 C S H-2C S H-2
HIP OF IS AR IAHIP OF IS AR IA P L AC E NT AR IAP L AC E NT AR IA P L AC E NT AR IAP L AC E NT AR IA P L AC E NT AR IAP L AC E NT AR IAP L AC E NT AR IAP L AC E NT AR IA
5´ 3´
17q23
1 I 2 II 3 III 4 IV 55´ 3´
11 II 22 IIII 33 IIIIII 44 IVIV 555´ 3´

26
es que desarrollan anticuerpos contra la GH exógena, cuando es suministrada como
tratamiento de reemplazo hormonal. Esto hace que, en un primer momento, los pacientes
tengan una buena respuesta al tratamiento hasta el momento en que desarrollan Ac anti GH,
en donde el crecimiento se enlentece dramáticamente. El desarrollo de Ac por parte de los
pacientes se debe a que en ausencia del gen GH1 las células somatotróficas son incapaces de
sintetizar GH-1, de tal forma que cuando es administrada de manera exógena las células del
sistema inmunológico no la reconocen como propia y sintetizan anticuerpos para eliminarla (Illig
R. et al 1970).
3.2.1.2 IGHD tipo IB
El fenotipo clínico de este déficit es más variable que el tipo IA. Los pacientes con IGHD tipo IB
también presentan un retardo de crecimiento con niveles muy bajos pero detectables de GH-1
en suero, tanto en estado basal como bajo estímulo. Múltiples estudios clínicos en diferentes
cohortes describen que el tratamiento con GH recombinante humana mejora el pronóstico de la
talla adulta ya que no se observa que estos pacientes desarrollen anticuerpos anti GH (Mullis P.
2005; Wit J.M. et al 2011).
Este tipo de deficiencia se produce por mutaciones homocigotas que afectan regiones
regulatorias que participan en la normal transcripción del gen GH1 (mutaciones de splicing). A
su vez, se han reportado mutaciones que dan lugar a la aparición de un codón de terminación
prematuro (mutaciones nonsense y frameshifts), que generan una proteína truncada
(Alatzoglou K., et al. 2009). Por otro lado, en la población asiática, se han reportado alteraciones
en el gen que codifica para el receptor de la hormona liberadora de la GH (GHRHR) en
pacientes con retardo de crecimiento asociado a IGHD tipo IB
(Wajnrajch MP et al 1996;
Baumann G & Maheshwari H 1997).
El GHRHR es un miembro de la familia de receptores con siete pasos transmembrana
acoplados a proteína G. La unión de la GHRH hipotalámica a sus receptores específicos en la
superficie de las células somatotróficas, activa a la proteína Gs estimulatoria la cual induce un
aumento en la síntesis de AMPc que promueve la proliferación celular y la secreción de GH-1
(Mayo K., 1992; Lin SC., et al. 1993). Mutaciones con pérdida de sentido en el gen GHRHR han
sido asociadas a un síndrome caracterizado por IGHD tipo 1B e hipoplasia adenohipofisaria
(Wajnrajch MP et al 1996; Baumann G & Maheshwari H 1997).
3.2.1.3 IGHD tipo II
La forma autosómica dominante de IGHD se caracteriza por presentar, al igual que la tipo IB,
retardo de crecimiento junto con niveles muy bajos pero detectables de GH-1 sérica, tanto en
estado basal como bajo estímulo. Como principal característica, debido a su modo de herencia,
se observa la presencia de casos familiares. Esto permite una mayor facilidad para detectar
este tipo de patología y orientar al diagnóstico ya que, generalmente, hay un padre afectado del
cual el niño heredó el alelo alterado (Mullis P. et al 2002 y 2005; Wit J.M. et al 2011).

27
IGHD tipo II es causada principalmente por mutaciones entre los primeros 6 nucleótidos de la
secuencia consenso de splicing del intrón 3 del gen GH1 (Mullis P. et al. 2002). A su vez las
primeras 7 bases del exón 3 también son cruciales para el correcto splicing del ARNm del gen
GH1, de tal forma que mutaciones en estas regiones generan un procesamiento anómalo del
ARNm que se traduce en la pérdida del exón 3, lo que da lugar a la producción de una isoforma
de 17.5 kDa (Binder G & Ranke MB, 1995). Esta isoforma de la GH pierde los aminoácidos
comprendidos entre las posiciones 32-71, los cuales codifican para el loop que conecta la
hélice 1 y 2 de la estructura ternaria de la hormona (Cunningham BC., et al. 1991; deVos AM., et
al 1992). A nivel funcional, la isoforma de 17.5 kDa, posee un efecto dominante negativo sobre
la secreción de la isoforma de 22 kDa, tanto en cultivo celular como en animales transgénicos
(Lee MS., et al 2000). La isoforma de 17.5 kDa es retenida en el retículo endoplasmático de la
células somatotropas e impide el tráfico normal de las vesículas secretoras (que poseen
moléculas de ambas isoformas) hacia la membrana plasmática. Por otro lado, el aumento de
las vesículas secretoras en el citoplasma aumenta la toxicidad en las células, lo que genera
una disminución de las células somatotropas en el tejido adenohipofisario (Hayashi Y., et al.
1999; Graves TK., et al. 2001; McGuinness L., et al. 2003).
A su vez, se han reportado tres mutaciones con pérdida de sentido (p.Pro89Leu, p.Arg183Hys
y p.Val110Phe) en el gen GH1, que también dan lugar a IGHD tipo II. Se cree que estas
mutaciones poseen el mismo efecto funcional que las mutaciones de splicing previamente
mencionadas, aumentando el nivel de toxicidad celular y disminuyendo el número de
somatotropos en la hipófisis anterior (Deladoey J., et al 2001; Binder G., et al 2001).
3.2.1.4 IGHD tipo III
Este tipo de deficiencia de GH es un desorden de herencia recesiva ligada al cromosoma X. En las
familias afectadas, se observa que los hombres poseen déficit de GH-1 y de inmunoglobulinas. Se
ha reportado mutaciones y/o deleciones en el brazo largo del cromosoma X, en regiones que son
importantes para la normal producción de inmunoglobulinas y expresión de GH1, y que estarían
involucradas en este tipo de déficit (Fleisher TA et al 1980; Sitz KV et al 1990; Conley ME et al 1991).
A su vez, Duriez et al, en 1994, reportaron una mutación en el gen BTK en un paciente con IGHD y
agamaglobulinemia ligada al X. El gen BTK codifica para una tirosina quinasa denominada Bruton,
en honor a su descubridor (Bruton et al 1952), la cual está involucrada en la regulación del
desarrollo de las células B del sistema inmune (Rawlings and Witte 1994).
Tabla 1. Tipos de deficiencia aislada de GH.
Categoria Herencia Gen candidato Tipo de mutaciones
IGHD tipo IA Recesiva GH1 Deleciones 6.7, 7 , 7.6 y 45 Kb
Mutaciones Nonsense y frameshift
que afectan el péptido señal
IGHD tipo IB Recesiva GH1 Mutaciones de splicing, nonsense y
GHRHR frameshift
IGHD tipo II Dominante GH1 Mutaciones de splicing y missense
IGHD tipo III Ligada al X BTK
Cromosoma X
Modificado de Genetic control of growth, Mullis P. 2005

28
3.2.2 Síndrome de insensibilidad a la GH-1
3.2.2.1 Alteración del receptor de GH-1
Desde 1966 a la fecha se han reportado más de 250 pacientes con insensibilidad a la hormona
de crecimiento (GHI) debido a mutaciones en el gen que codifica para el receptor de GH (GHR)
(David A., et al. 2011). Esta patología también es conocida como síndrome de Laron, en honor al
Dr. Laron quien caracterizó el síndrome por primera vez en 1966 (Laron Z 2004). La mayoría de
los casos de GHI tienen una forma de herencia autosómica recesiva, y el defecto molecular
identificado involucra a mutaciones, homocigotas o heterocigotas compuestas, a lo largo del
gen GHR.
El GHR media los efectos de la GH sobre el crecimiento y el metabolismo. El gen GHR (RefSeq
NG_011688.1) fue caracterizado por Godowski et al en 1989. Está conformado por 9 exones,
numerados de 2 al 10, ubicados en la posición cromosómica 5p13-p12. El exón 2 codifica para
un péptido señal mientras que los exones 3-7 codifican para el dominio extracelular, el exón 8
para el dominio transmembrana y los exones 9-10 para el dominio citoplasmático. El GHR es
un receptor transmembrana homodimérico con dos sitios de unión para la GH-1 en el dominio
extracelular, lo que permite asegurar la unión de la GH-1 al receptor. A su vez este dominio
extracelular da lugar a la proteína transportadora de GH-1(GHBP), la cual se genera por un
proceso de clivaje luego de que la GH-1 se una al GHR y gatille el inicio de la cascada de
señalización intracelular (Leung DW., et al. 1987). En la figura 12 se muestra la organización del
gen GHR y la estructura del receptor.
Análisis genéticos han demostrado una alta heterogeneidad molecular del síndrome de GHI; a
la fecha más de 70 mutaciones en el gen GHR han sido identificadas, las cuales abarcan
desde deleciones a mutaciones puntuales, incluyendo mutaciones con pérdida de sentido, sin
sentido y cambio en el marco de lectura (David A., et al. 2011). Mutaciones que afectan el
splicing normal del ARNm representan aproximadamente el 20% de los defectos del gen GHR
y generalmente afectan regiones regulatorias importantes, como los sitios dadores y aceptores
de splicing (David A., et al. 2011). Dentro de los defectos que causan un splicing deletéreo del
gen GHR, se encuentra un cambio de base en el intrón 6, que lleva al reconocimiento e
inclusión de 108 pares de bases, entre el exón 6 y 7, correspondiente a la secuencia de un
pseudoexón, generando un cambio en el marco de lectura que da lugar a una proteína no
funcional (Metherell LA et al 2001; Maamra M et al 2006). El fenotipo observado en pacientes con
esta variante varían desde alteración de crecimiento severa ha moderada (David A et al 2007).
Mutaciones intrónicas que dan lugar a la activación de un pseudoexón son raras en
enfermedades genéticas (Akker SA et al 2007) por lo que es importante tener en cuenta este
fenómeno cuando se estudia el gen GHR.
La GHBP es un buen indicador para determinar qué dominio del receptor se encuentra
afectado en pacientes con GHIS. Como se mencionó previamente, la GHBP resulta del clivaje
de una porción del dominio extracelular del GHR y se ha observado que los niveles en suero

29
disminuyen en casos de GHIS causado por mutaciones que afectan los exones 2-7
(corresponde al 70% de las mutaciones descriptas) (Laron Z 2004). Por otro lado, en los casos
donde las mutaciones afectan los dominios transmembrana y citoplasmático, los niveles de
GHBP se mantienen normales o elevados (Leung DW., et al. 1987). Existen excepciones donde
mutaciones que involucran el dominio extracelular presentan niveles normales de GHBP y
alteraciones en los dominios transmembrana e intracelular poseen niveles bajos (Godowski PJ
1987; Laron Z 2004; David A et al 2011).
El fenotipo clínico observado, en pacientes con GHIS, es variable dependiendo del tipo de
mutaciones presentes y del dominio que se afecta en el receptor. Los pacientes con
mutaciones en el gen GHR se caracterizan por un retardo de crecimiento severo con niveles
normales, o elevados, de GH-1 y niveles muy bajos de IGF-1 en suero, tanto en estado basal
como luego de las pruebas de estímulo (Mullis P. 2005; David A et al 2011).
Cabe mencionar, que mutaciones heterocigotas en el gen GHR se han relacionado con casos
de insensibilidad parcial a la GH-1. En un primer momento se propuso el estudio del gen GHR
en casos de talla baja sin diagnóstico aparente (talla baja idiopática), con la hipótesis de que
mutaciones heterocigotas pueden dar lugar a fenotipos intermedios, no tan severos (Woods
KA., et al. 1997; Wit J.M. et al 2011). Sin embargo, se han reportado pacientes y familiares con
mutaciones heterocigotas que poseen talla normal, de tal manera que la implicancia de estas
variantes en pacientes con talla baja idiopática es incierta (Sanchez JE., et al. 1998). A pesar de
estos resultados, se recomienda que mutaciones en el gen GHR se consideren cuando otras
causas de talla baja se hayan descartado (Mullis P. 2005; David A et al 2011; Wit J.M. et al 2011).

30
Modificado de Godowski et al. 1989, PNAS
Figura 12. Esquema del gen GHR y de los dominios del hemireceptor.
3.2.2.2 Alteración en el traductor de señal STAT5b
Existe evidencia de que distintos caminos de señalización intracelular actúan río abajo en la
cascada de señalización del receptor de GH. Múltiples reportes han establecido que el factor
de transcripción STAT5b es el principal intermediario en el camino de señalización de la GH-1
regulando el normal crecimiento somático y muchos otras acciones biológicas de la GH-1
(Kofoed EM et al 2003; Rowland JE et al 2005; Hwa V et al 2011).
La primer proteína STAT fue caracterizada en 1990 como un factor de señalización en la vía de
los interferones α/β y γ. Estudios posteriores determinaron que esta familia de proteínas es
escencial para la regulación de múltiples procesos biológicos relacionados con el crecimiento y
la respuesta inmunológica (Levy DE et al 2002; Hennighausen L
et al 2008; Schindler C and
Plumlee C 2008).
Las proteínas STATs se encuentran en el citoplasma de células que responden a estímulos
hormonales y de citoquinas. Cuando estas células se activan, por unión de estos ligandos a
sus receptores específicos de membranas, las proteínas STATs son reclutadas hacia los
segmentos intracelulares de los receptores activados y se unen a residuos fosforilados de
tirosina a través de sus dominios SH2. Mientras se encuentran unidos al receptor, las proteínas
2 3 4 5 6 7 8 9 10
Dominio extracelular
Dominio transmembrana
Dominio intracelular
DOMINIO TRANSMEMBRANA ANCLAJE A LA MEMBRANA
DOMINIO INTRACELULAR
SEÑALIZACIÓN INTRACELULAR
DOMINIO EXTRACELULAR UNION A LA GH DIMERIZACION

31
STATs son fosforiladas por proteínas tirosina quinasas asociadas al receptor (proteína JAK2 en
el caso del GHR). Una vez fosforiladas se disocian del receptor y forman dímeros, a través de
interacciones recíprocas entre los dominios SH2, y se traslocan al núcleo donde se unen a
regiones especificas del ADN para inducir la transcripción de distintos genes relacionados con
múltiples procesos biológicos (Levy DE et al 2002; Hennighausen L
et al 2008; Schindler C and
Plumlee C 2008).
Existen 7 proteínas STATs en las células de los mamíferos, STAT1-4, 5a, 5b y 6 (Levy DE et al
2002; Hiu Kiu and Sandra Nicholson 2012). STAT1, 2, 3, 4 y 6 se expresan en la mayoría de los
tejidos (Zhong Z et al 2004). STAT1 y 4 tiene altos niveles de expresión en el timo y el bazo
(Herrada G et al 1997; Zhong Z et al 2004) mientras que STAT5a y 5b exhiben un patrón de
expresión diferencial en músculo, cerebro, glándula mamaria, vesícula seminal y glándulas
salivales (Liu X et al 1995). Ratones deficientes en el gen Stat1 y Stat2 poseen alteración de la
respuesta inmune mediada por INF α y γ y son altamente susceptibles a infecciones virales
(Durbin JE et al 1996; Park C et al 2000). A su vez, ratones KO para los genes Stat4 y Stat6
desarrollan una falla en la respuesta a IL4, IL12, IL13 e IL23, todas interleuquinas relacionadas
con la diferenciación de los linfocitos Th y las funciones de las células NK (Kaplan MH et al 1996;
Shimoda K et al 1996). Por otro lado, ratones deficientes en Stat3 mueren en el período
embrionario probablemente a causa de la incapacidad de formar el endodermo visceral (Takeda
K et al 1997).
Las proteínas STAT5a y STAT5b están involucradas en la respuesta biológica a la prolactina
(PRL) y GH-1, respectivamente (Udy GB et al 1997; Imada K et al 1998) y, a su vez, participan en
la respuesta inmune vía IL-3 y citoquinas γ (Teglund S et al 1998). Consistente con estas
funciones, los ratones doble deficientes desarrollan alteraciones en las glándulas mamarias, en
la respuesta inmune y en el eje de crecimiento GH-IGF-1(Cui Y et al 2004).
Con respecto al eje GH-IGF-1, la GH-1 promueve el crecimiento postnatal normal regulando la
expresión del gen IGF1 a través de la vía de señalización que incluye a STAT5b. Los sujetos
con mutaciones en el gen STAT5b presentan un retardo de crecimiento severo asociado con
resistencia a la GH-1 y deficiencia severa de IGF-1, lo que enfatiza la importancia de la vía de
señalización STAT5b en la regulación de la expresión de IGF1, IGFBP3 y IGFALS (Hwa V., et al
2011). A su vez, similar a los defectos en los genes STAT1 y STAT3, las alteraciones en el gen
STAT5b dan lugar a disfunción inmune que se correlaciona con la participación de este factor
en la regulación de la expresión de distintas citoquinas (Hennighausen L et al 2008).
El gen STAT5b tiene un tamaño de 77 Kb dentro de una región de 204 Kb, que incluye a los
genes STAT5a y STAT3, en la posición cromosómica 17q11.2. Los genes STAT5b y STAT5a
se encuentran en posiciones invertidas, distanciados por 11 Kb. Aunque el gen STAT5a (24,4
Kb) es considerablemente menor que el gen STAT5b, ambos comparten una homología del
93% en la secuencia codificante, lo que sugiere que se originaron por un evento evolutivo de
duplicación génica (Ambrosio R., et al 2002). El gen STAT5b consiste de 19 exones que
codifican para una proteína de 794 aminoácidos con 6 dominios funcionales: un dominio amino
Terminal (ND, 141 aminoácidos codificados desde el exon 2 al 5) y un dominio coiled coiled

32
(CCD, 172 aminoácidos, exones 5 a 8) que participan en las interacciones proteína-proteína;
un dominio de unión al ADN (DBD, 172 residuos, exones 8 a 12); una región de unión entre los
dominios DBD y SH(L, 103 residuos, exones 12 a 14) cuya función se desconoce; un dominio
SH2 (88 residuos, exones 14 a 16) que, como se mencionó previamente, le permite a las
proteínas STAT unirse a los residuos de tirosina fosforilados en la región intracelular de los
receptores activados; y un dominio de transactivación carboxilo Terminal (TAD, 110 residuos,
exones 16 a 19) crítico para la interacción con otros factores de transcripción (Ambrosio R., et al
2002).
En humanos, el primer defecto genético reportado en STAT5b fue identificado en un paciente
con severo retardo de crecimiento postnatal, disfunción inmune y enfermedad pulmonar severa
(Kofoed EM et al 2003). A la fecha, se han reportado 7 mutaciones en el gen STAT5b en 10
pacientes con déficit de crecimiento y alteración del sistema inmunológico (figura 13), a
excepción de un paciente adulto en donde el sistema inmune parece no afectarse (Vidarsdottir
S., et al 2006).
En base a la caracterización molecular, la mayoría de los individuos afectados no sintetizan la
proteína en cantidades adecuadas debido a mutaciones homocigotas (herencia recesiva) que
dan lugar a la aparición de un codón de terminación prematuro (mutaciones nonsense y
frameshift). Solo se han reportado dos individuos con mutaciones que generan una substitución
aminoacídica en el gen STAT5b y, en ambos casos, afectan el dominio SH2, provocando la
acumulación y agregación de la proteína en el citoplasma e impidiendo que ejerza sus
funciones a nivel nuclear (Hwa V., et al 2011 STAT5b deficiency: Lessons from STAT5b gene
mutations; David Alessia et al. Endocrine Reviews 2011).
Modificado de David Alessia et al. Endocrine Reviews 2011
Figura 13. Esquema de la estructura proteica de STAT5b codificada por sus correspondientes exones y las mutaciones
identificadas hasta la fecha. CCD. Coiled-Coiled domain; DBD, DNA binding domain; L, linker; ND, N-terminal domain;
TAD, transactivation domain.
2 193 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
ND CCD DBD L SH2 TAD
c.424_427del: Pugliese 2010
p.R152X: Bernasconi 2006
p.Q368fsX376: Vidarsottir 2006
p.N398fsX423: Hwa 2005
c.1680delG: Hwa 2007
p.A630P: Kofoed 2003
p.F646S: Martinez A 2007
2 193 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
ND CCD DBD L SH2 TADND CCD DBD L SH2 TAD
c.424_427del: Pugliese 2010
p.R152X: Bernasconi 2006
p.Q368fsX376: Vidarsottir 2006
p.N398fsX423: Hwa 2005
c.1680delG: Hwa 2007
p.A630P: Kofoed 2003
p.F646S: Martinez A 2007

33
3.2.2.3 Deficiencia de ALS
La subunidad acido lábil (ALS) es una proteína glicosilada sintetizada en el hígado en
respuesta al estimulo de la GH-1. Su principal función es extender la vida media de los IGFs a
partir de la formación de complejos ternarios con los IGFs y las IGFBPs (Leong SR., et al.1992;
Ooi GT., et al. 1997). En estado libre, los IGFs, poseen una vida media menor a 10 minutos
mientras que formando los complejos ternarios aumenta a más de 12 horas. La formación de
los complejos impide la proteólisis de los IGFs y las IGFBPs y, a su vez, actúan como
reservorios para prevenir efectos metabólicos, como puede ser las hipoglucemias (Baxter RC.,
1994)
La ALS es una proteína de 578 aminoácidos codificado por el gen IGFALS, ubicado en la
posición cromosómica 16p13.3. El 75% de la ALS se organiza en dominios ricos en leucina
(LRR) que participan en interacciones proteína-proteína (Domené H. et al. 2004).
El primer paciente con una mutación homocigoto en el gen IGFALS fue reportado por Domené
et al en el año 2004. La característica más llamativa de este tipo de insensibilidad a la GH-1 es
la escasa correlación que existe entre los niveles extremadamente bajos de IGF-1, IGFBP3 y
ALS circulantes con respecto al déficit moderado de crecimiento, que se normaliza en la vida
adulta en algunos pacientes (Domené H., et al. 2007 y 2009). A la fecha se han reportado 16
mutaciones, con herencia autosómica recesiva, en 21 pacientes afectados (Figura 14). Estas
mutaciones corresponden a variantes con pérdida de sentido y alteraciones que dan lugar a la
aparición de un codón de terminación prematuro. En todos los casos los niveles de ALS
circulantes son extremadamente bajos y no se forman los complejos ternarios, sin embargo la
producción local de IGF-1 en los tejidos periféricos mantiene, e incluso aumenta, la regulación
positiva de la secreción de GH-1. Este fenómeno podría explicar la escasa correlación entre los
niveles bajos de IGF-1 y el retardo moderado de crecimiento en la infancia.
Modificado de David Alessia et al. Endocrine Reviews 2011
Figura 14. Representación esquemática de la proteína ALS indicando la localización de las mutaciones identificadas en
humanos. El gen IGFALS está formado por dos exones, el exón 1 codifica para el péptido conformado por 5 residuos
aminoacídicos mientras que el resto de la proteína es codificada por el exón 2. David A et al 2011.
1
2
Repeticiones ricas en leucina Región rica en cisteina
Peptido señal
103delC
p.C60S
p.P73L
p.L134Q
p.L172F
546-547insA 546-548delGGCinsAG
p.L241P
p.L244F
p.N276S
p.Q320X
p.L437-L439dup
p.D440
N
1490dupT p.C540R
p.S195-R197dup

34
3.2.3 Resistencia IGF-1: Alteración del receptor de IGFs tipo 1
3.2.3.1 Receptor de IGFs tipo 1
El IGF1R pertenece a la familia del receptor de insulina (INSR), que poseen un dominio tirosina
quinasa. Los miembros de la familia de receptores INSR son homodímeros preensamblados
(α2β2) que se diferencian de otros miembros de la superfamilia de receptores tirosina quinasa
en que necesitan experimentar determinados rearreglos estructurales para su dimerización y
activación (Ullrich A et al 1986; Klammt J et al 2011). El INSR y IGF1R son altamente homólogos,
con la mayor identidad en el dominio tirosina quinasa citoplasmático. Debido a esta alta
homología se cree que la división funcional entre ellos se originó a partir de un evento de
duplicación de un gen ancestral, durante la evolución de los vertebrados, que generó la
adquisición de un exón 11 adicional en el gen del INSR (Hernandez sanchez C et al 2008).
El gen del receptor de IGFs tipo 1 (IGF1R, OMIM 147370) mapea en la región cromosómica
15q26.3 y abarca aproximadamente 315Kb. Está conformado por 21 exones que codifican para
un ARNm de 11.242pb (GenBank NM_000875), que incluyen regiones 5´ y 3´ no traducibles
extraordinariamente extensas (UTR). El marco abierto de lectura del ARNm consta de 4104
nucleótidos que codifican para una proteína de 1367 aminoácidos (Ullrich A et al 1986). Se
expresa constitutivamente en todas las células y tejidos pero su expresión es regulada
dependiendo del estado de desarrollo en el cual se encuentre la célula, las condiciones
nutricionales, los niveles hormonales y de factores intracelulares (Werner H et al 1995). En
embriones humanos, la expresión del gen IGF1R es detectable a partir del estadio de 8 células
y persiste a través del desarrollo embrionario, fetal y postnatal, iniciando su disminución hacia
la adultez (Hardy K & Spanos S 2002).
La síntesis del IGF1R se inicia con la traducción del ARNm, dando lugar a un preproreceptor
simple cadena nuclear que es transportado al retículo endoplasmático. Una vez eliminado el
péptido señal, el proreceptor se pliega y dimeriza a partir de puentes disulfuro. El proreceptor
dimerizado se transporta a la red trans-Golgi donde completa su maduración mediante un
proceso de glicosilación y clivaje que da lugar al receptor maduro, una glicoproteína
heterotetramérica con dos subunidades α extracelulares idénticas y dos subunidades β
transmembrana con actividad tirosina quinasa intrínseca (figura 15) (Adams TE et al 2000).
Las funciones de la insulina e IGF-1 son fisiológicamente distintas pero pueden solaparse. En
mamíferos, la insulina actúa principalmente regulando la homeostasis metabólica en tejido
hepático, adiposo y muscular, mientras que el IGF-1 promueve el crecimiento, la supervivencia
y diferenciación celular (Siddle K et al 2001). La insulina y el IGF-1 se unen con alta afinidad a
sus receptores específicos, sin embargo a elevadas concentraciones de insulina puede haber
una reacción cruzada con el IGF1R y viceversa (Laviola L. et al 2007). Ambos receptores se
expresan, a diferentes niveles, en la mayoría de los tejidos. En células que expresan ambos
receptores, los pro-receptores pueden sufrir un proceso de homodimerización, así como de
heterodimerización, dando lugar a la formación de receptores híbridos entre el IGF1R y el INSR

35
junto con receptores clásicos (Moxham CP et al 1989; Treadway JL et al 1989). Los heterodímeros
parecen ensamblarse con eficiencia comparable a los homodímeros y cuando un receptor se
encuentra en exceso, el receptor menos abundante se presenta enteramente como híbrido. Por
ejemplo, en el músculo esquelético el IGF1R se presenta principalmente como híbrido junto
con un exceso del receptor de insulina clásico, mientras que lo opuesto ocurre en fibroblastos
(Bailyes EM et al 1997). Estos receptores híbridos unen tanto insulina como IGF-1, aunque
tienen menor afinidad por la insulina que el INSR clásico (Soos MA et al 1994).
El IGF-1 se une a regiones asimétricas en el ectodominio del receptor (cadena α) y permite el
acercamiento de las regiones citoplasmáticas de ambos hemireceptores (αβ), de tal manera de
facilitar la trans-autofosforilación (DeMeyts P & Whittaker J et al 2002). Este proceso se inicia con
la activación del dominio tirosina quinasa intrínseca, el cual fosforila residuos de tirosina en el
dominio juxtamembranoso (JM), tirosina quinasa (TK) y en la región carboxilo Terminal (C-tail)
del otro hemireceptor. Estos sitios fosforilados representan puntos de anclajes para distintas
moléculas de señalización que activan dos vías de traducción de señal, la vía PI3K/AKT y
Src/Ras/ERK (figura 15) (Adams TE et al 2000).
A) B)
Figura 15. Panel A. Estructura del receptor de IGFs tipo 1, se observa el hemireceptor citoplasmático inmaduro y el
receptor maduro heterotetramerico anclado a la membrana.Panel B. Vías de señalización involucradas en el sistema
IGF1-IGF1R.
Alpha Chain Beta Chain
SignalSignal peptidepeptide
NH2 NH2
COOH COOH
COOHNH2
TK DomainTransmembrane
Domain
Alpha Chain
Beta Chain
IR SS HC
G rb2
mS oS
R as
p85
p110P I3KP I3K
P DK 1 (P DK 2)P DK 1 (P DK 2)
MAP K MAP K K inas eK inas e (ME K )(ME K )
MAP K (MAP K (E R K sE R K s )) AK TAK T
R af
Butler and Butler and LeRoithLeRoith Endocrinology Endocrinology 20012001
Alpha Chain Beta Chain
SignalSignal peptidepeptide
NH2 NH2
COOH COOH
COOHNH2
TK DomainTransmembrane
Domain
Alpha Chain
Beta Chain
Alpha Chain Beta Chain
SignalSignal peptidepeptide
NH2 NH2
COOH COOH
COOHNH2
TK DomainTransmembrane
Domain
Alpha Chain
Beta Chain
IR SS HC
G rb2
mS oS
R as
p85
p110P I3KP I3K
P DK 1 (P DK 2)P DK 1 (P DK 2)
MAP K MAP K K inas eK inas e (ME K )(ME K )
MAP K (MAP K (E R K sE R K s )) AK TAK T
R af
Butler and Butler and LeRoithLeRoith Endocrinology Endocrinology 20012001
IR SIR SS HCS HC
G rb2G rb2
mS oSmS oS
R asR as
p85p85
p110p110P I3KP I3K
P DK 1 (P DK 2)P DK 1 (P DK 2)
MAP K MAP K K inas eK inas e (ME K )(ME K )
MAP K (MAP K (E R K sE R K s )) AK TAK T
R afR af
Butler and Butler and LeRoithLeRoith Endocrinology Endocrinology 20012001

36
3.2.3.2 Mutaciones en el gen IGF1R
Abuzzahab y colaboradores en el año 2003, reportaron el primer caso de resistencia a IGF-1 a
causa de una mutación heterocigota en el gen IGF1R. Desde el año 2003 a la fecha se han
reportado 18 mutaciones, las cuales se ubican a lo largo de toda la secuencia codificante del
gen IGF1R (figura 16). Predominan las mutaciones con pérdida de sentido, donde un
aminoácido es sustituido por otro, aunque también se reportaron mutaciones que dan lugar a la
aparición de un codón de terminación prematuro y duplicaciones pequeñas. Todas las
mutaciones afectan aminoácidos que se encuentran altamente conservados filogenéticamente
entre las distintas especies de vertebrados y no vertebrados, lo que indica que son esenciales
para el normal funcionamiento del receptor (Klammt J et al 2011).
La relevancia patogénica de todas las variantes es demostrada a través de estudios
comparativos, genéticos, estructurales y de datos bioquímicos. Por ejemplo, ensayos in vitro
con fibroblastos de pacientes afectados se utilizan para demostrar una disminución en la
activación del receptor o en las moléculas de señalización río abajo en la cascada de
traducción de señal. Todas las mutaciones reportadas muestran una disminución significativa
en la autofosforilación de la subunidad β seguido de una reducción en la activación de, al
menos, uno de los caminos de señalización. A su vez, se ha demostrado una falta de respuesta
al estímulo con IGF-1, que se ve reflejado en una disminución en la proliferación celular in vitro
(Klammt J et al 2011).
Los pacientes con mutaciones en el gen IGF1R nacen pequeños para la edad gestacional y
pierden la capacidad de generar el catch-up de crecimiento en los años subsiguientes. Los
niveles séricos de GH-1 son normales, en estado basal y bajo estímulo, mientras que los
niveles de IGF-1 e IGFBP-3 se mantienen normales o elevados. El peso o la talla al nacer son
menores a -2 SDS mientras que la talla, después del año de vida, puede comprometerse aún
más alcanzando valores menores a -2.5 SDS. La microcefalia es reportada en aquellos casos
en donde el perímetro cefálico es tenido en cuenta para la selección de pacientes. Se estima
que la frecuencia de alteraciones en el gen IGF1R se encuentra entre 2-4%. Sin embargo, la
mayoría de los pacientes presentan una gran variabilidad en las manifestaciones bioquímicas y
clínicas, lo que dificulta el diagnóstico. A su vez, la baja expresividad o penetrancia de los
alelos mutados sugieren que la haploinsuficiencia del receptor sería el fenómeno que mejor
explica el origen de la patología (Klammt J et al 2011).

37
Figura 16. Gen del IGF1R y mutaciones reportadas en la literatura.
(1) Abuzzahab M et al. NEJM 2003
(2) Kawashima Y et al. JCEM 2005
(3) Walenkamp MJ et al. JCEM 2006
(4) Inagaki K et al. JCEM 2007
(5) Fang P et al. JCEM 2009
(6) Wallborn T et al. JCEM 2010
(7) Kruis T et al. JCEM 2010
5´ 3´
c.3348_3366 dup195
p.Gly1155Ala7
p.Arg89Ter1
p.Arg138Gln1
p.Arg511Gln4
p.Val629Glu6
p.Arg739Gln2
p.Glu1050Lys3
p.Lys145Asn1
p.Glu121Lys10 c.420del(p. Ala140fsX20)9
p.Glu234Lys10
p.Tyr387Ter8
p.Arg511Trp13
p.Arg461Leu11
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
Sin sentido
Pérdida de sentido
Duplicación
Deleciones pequeñas
p.Tyr487Phe12
(8) Mohn A et al. Horm Res Ped 2011
(9) Choi JH et al. JCEM 2011
(10) Fang P et al. JCEM 2012
(11) Kawashima Y et al. Clin Endocrinol 2012
(12) Labarta J.I et al. Clin Endocrinol 2013
(13) Leal AC et al. Clin Endocrinol 2013
(14) Gannage-Yared MH et al. Eur J Endocrinol 2013
p.Arg10Leu14
(1) Abuzzahab M et al. NEJM 2003
(2) Kawashima Y et al. JCEM 2005
(3) Walenkamp MJ et al. JCEM 2006
(4) Inagaki K et al. JCEM 2007
(5) Fang P et al. JCEM 2009
(6) Wallborn T et al. JCEM 2010
(7) Kruis T et al. JCEM 2010
5´ 3´
c.3348_3366 dup195
p.Gly1155Ala7
p.Arg89Ter1
p.Arg138Gln1
p.Arg511Gln4
p.Val629Glu6
p.Arg739Gln2
p.Glu1050Lys3
p.Lys145Asn1
p.Glu121Lys10 c.420del(p. Ala140fsX20)9
p.Glu234Lys10
p.Tyr387Ter8
p.Arg511Trp13
p.Arg461Leu11
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
Sin sentido
Pérdida de sentido
Duplicación
Deleciones pequeñas
p.Tyr487Phe12
(8) Mohn A et al. Horm Res Ped 2011
(9) Choi JH et al. JCEM 2011
(10) Fang P et al. JCEM 2012
(11) Kawashima Y et al. Clin Endocrinol 2012
(12) Labarta J.I et al. Clin Endocrinol 2013
(13) Leal AC et al. Clin Endocrinol 2013
(14) Gannage-Yared MH et al. Eur J Endocrinol 2013
p.Arg10Leu14

38
4. Objetivos
4.1 Objetivos Generales
La caracterización molecular de los genes relacionados con el eje GHRH/GH1/IGF-1 tiene
implicancias diagnósticas que impactan directamente en la estrategia terapéutica y el consejo
genético, el objetivo general de la presente tesis doctoral es la identificación molecular de
pacientes pediátricos con alteraciones en dicho eje. A su vez se analizará la frecuencia de
estas alteraciones en la población pediátrica argentina y se la comparará con los datos
disponibles reportados en otras poblaciones.
4.2 Objetivos Específicos
4.2.1 Estudio de alteraciones moleculares en pacientes con deficiencia aislada de hormona de
crecimiento (IGHD).
a) Análisis del gen GH1
b) Análisis del gen GHRHR
4.2.2 Estudio de alteraciones moleculares en pacientes con síndrome de insensibilidad a la
hormona de crecimiento (GHIS).
a) Análisis del gen GHR
b) Análisis del gen STAT5b
4.2.3 Estudio de alteraciones moleculares en pacientes con insensibilidad a IGF-1.
a) Análisis del gen IGF1R
4.2.4 Estudio de alteraciones moleculares en pacientes con déficit múltiple de hormonas
hipofisarias (MPHD).
Análisis de los genes OTX2, HESX1, LHX3, LHX4, GLI2, PROP1 y POU1F1.
4.2.5 Caracterización funcional de variantes que no se encuentren previamente descriptas en la
literatura.
Análisis de predicción in silico para determinar si las variantes nuevas
afectan la funcionalidad de la proteína en cuestión.
Ensayos in vitro para determinar los efectos funcionales de la variantes
presentes en el gen IGF1R.
a) Ensayo de proliferación celular a partir de cultivo primario de
fibroblastos de pacientes afectados y sujetos controles.
b) Ensayo de fosforilación del traductor de señal Akt en cultivo
primario de fibroblastos de pacientes afectados y sujetos controles.

39
5. Materiales y Métodos
5.1 Selección de pacientes Los pacientes de los distintos grupos de estudio fueron seleccionados en base a datos
auxológicos, de laboratorio y evaluación funcional del eje hipotálamo-hipófiso-IGF-1. Los datos
auxológicos fueron evaluados mediante tres mediciones de talla y peso tomados en forma
estandarizada en el último año, en intervalos adecuados, o tres mediciones separadas por un
año, o más, incluyendo la última medición. Todos los pacientes debían haber nacido luego de
un embarazo con parto normal, o cesárea sin complicaciones, y presentar, al momento del
diagnóstico, un examen clínico, de laboratorio (rutina) y cariotipo normal así como también
proporciones corporales, nutrición e ingesta calórica dentro de los valores establecidos.
Se descartaron aquellos pacientes con anomalías óseas así como también aquellos con
sospecha de enfermedad pediátrica crónica renal, pulmonar, cardiaca, gastrointestinal,
nutricional y Diabetes mellitus tipo 1.
El screening de los genes a estudiar en los pacientes afectados se realizó en base al análisis
de los distintos fenotipos clínicos y datos de laboratorio observados. Los pacientes fueron
subdivididos en distintos grupos siguiendo los algoritmos diagnósticos que se detallan a
continuación:
5.1.1 Grupo déficit de hormona de crecimiento Pacientes con déficit aislado de hormona de crecimiento fueron seleccionados a partir de los
siguientes criterios de inclusión.
1. Peso al nacer mayor al percentilo 10 para la edad gestacional. Examen perinatológico
normal.
2. Maduración psicomotriz y escolaridad normal.
3. Talla menor a -2.5 SDS para la edad y el sexo.
4. Velocidad de crecimiento menor a 1.3 SDS en el último año.
5. Respuesta máxima de GH sérica por debajo de 4.7 ng/ml, en al menos uno de dos
ensayos de estimulación farmacológicos (Chaler EA., et al 2013).
6. Niveles séricos basales de IGF-1 por debajo de -2 SDS (Chaler EA. 2009).
7. Niveles séricos de ACTH, PRL, TSH y hormonas tiroides dentro de los límites normales
de acuerdo a los valores de referencia para la edad.
8. Los niveles séricos de LH-FSH solo fueron considerados en la edad puberal.
Los valores de laboratorio y mediciones antropométricas fueron calculados para la población de
referencia Argentina (Lejarraga, H. et al). Se excluyeron los pacientes con déficit de GH-1 de
causa orgánica (tumores en SNC), por cirugía cerebral, radio terapia o síndromes conocidos.
Los pacientes que presentaron déficit de GH-1 asociada a deficiencias de otras hormonas
hipofisarias (TSH, ACTH, FSH, LH, PRL) de causa no orgánica, se seleccionaron para el
estudio de genes implicados en la embriogénesis hipofisaria. Dentro de este subgrupo se

40
incluyeron a aquellos pacientes con displasia septo óptica y alteraciones de la hipófisis
posterior, definidos por resonancia magnética nuclear (RMN).
5.1.2 Grupo síndrome de insensibilidad a la GH Dentro de este grupo de estudio, los pacientes con sospecha de insensibilidad a la GH, a
causa de alteraciones en el gen GHR, fueron seleccionados en base a los siguientes criterios:
1. Peso y longitud corporal mayor a -2 SDS al nacer.
2. Retardo de crecimiento postnatal muy severo, talla menor a -4 SDS para la edad y el
sexo.
3. Respuesta máxima de GH-1 sérica por encima de 4.7 ng/ml luego de ensayos de
estimulación farmacológicos. (Chaler EA., et al 2013)
4. Niveles séricos de IGF-1 menores a -2 SDS. (Chaler EA. 2009)
5. Niveles séricos de IGFBP-3 y ALS menores a los valores de referencia (Domené 2011 et
al).
Los valores de laboratorio y mediciones antropométricas fueron calculados para la población de
referencia Argentina (Lejarraga, H. et al).
5.1.3 Grupo con alteraciones en la cascada señalización del GHR
Los pacientes fueron seleccionados en función del fenotipo clínico de insensibilidad a la GH-1
asociados a alteraciones de la inmunidad celular con el objetivo de buscar mutaciones en el
gen STAT5b.
5.1.4 Grupo resistencia a IGF-1
Los pacientes con sospecha de insensibilidad a IGF-1, a causa de mutaciones en el gen
IGF1R, fueron seleccionados utilizando los siguientes criterios de inclusión:
1. Pacientes nacidos pequeños para la edad gestacional (SGA), definido como peso
y/o talla al nacer por debajo de -2 SDS para la edad gestacional.
2. Retardo de crecimiento postnatal, definido como peso o talla menor a -2 SDS para
la media normal establecida para la edad y el sexo.
3. Presencia de microcefalia, establecida como perímetro cefálico por debajo de -2
SDS para la edad y el sexo.
Los valores de laboratorio y mediciones antropométricas fueron calculados para la población de
referencia Argentina (Lejarraga, H. et al).

41
5.2 Determinaciones clínicas y bioquímicas
La talla, el peso y el perímetro cefálico fueron medidos con equipamiento estándar. Los valores
(SDS) antropométricos fueron calculados en base a las referencias para la población Argentina
(Lejarraga, H. et al). La edad ósea fue determinada por el método de Greulich and Pyle
(Greulich, W.W. & Pyle, S.J. 1959).
Los parámetros bioquímicos de rutina (hemograma completo, proteínas séricas, ionograma,
hepatograma, función renal y screening de enfermedad celíaca) fueron determinados a partir
de técnicas estándares.
Los valores séricos de GH-1 fueron determinados a partir del ensayo Immulite, utilizando el
estandar IRP IS 98/574 (valor de corte 4.7 ng/ml) (Chaler EA., et al 2013). Los valores de IGF-1 e
IGFBP-3 fueron analizados utilizando anticuerpos monoclonales anti-IGF-1 y anti-IGFBP-3 y el
sistema de quimioluminiscencia automatizado Immulite (ImmuliteR, Diagnostic Products Corp,
Los Angeles, CA, USA). Los valores fueron convertidos a SDS en base a los datos
normalizados de nuestro laboratorio (Chaler EA., et al 2009).
Los niveles de prolactina, TSH, T3, T4 y T4 libres fueron analizados utilizando el equipo
Architect 2000 de Abbott mientras que los valores de ACTH, cortisol, estradiol y testosterona
fueron medidos mediante el equipo de quimioluminisciencia Immulite de Siemens. Por otro
lado, los niveles de gonadotrofinas fueron determinados por medio del sistema MEIA Axdym de
Abbott.
Las resonancias magnéticas fueron realizadas con el equipamiento 1.5 T en diferentes centros.
5.3 Análisis Molecular
5.3.1 Extracción de ADN
Se utilizó el método, previamente reportado por Miller SA., et al en 1988 (Nucleic Acids
Research), de precipitación salina a partir de sangre entera anticoagulada con EDTA
(aproximadamente 5 ml), el cual se describe brevemente. La muestra se centrifuga a 1500 rpm
durante 10 minutos para separar el plasma. El paquete globular se mezcla con buffer de lisis de
glóbulos rojos (RCLB) y se vuelve a centrifugar durante 10 minutos. Este proceso se repite tres
veces hasta aislar completamente los glóbulos blancos. El siguiente paso consiste en lisar los
glóbulos blancos para extraer el ADN a partir de un buffer de lisis específico (WCLB). En esta
etapa también se agregan 15 ul de proteinasa K (PK), que inactivan las enzimas que degradan
el ADN y ARN (DNAsas y RNAsas), y se deja incubando toda la noche a 56 ºC. Luego de la
incubación el volumen es volcado en un tubo de 1,5 ml y se agregan 380 ul de NaCl 5M. Se
vortexea durante 5 minutos y se centrífuga a máxima velocidad (13000 rpm) durante 30
minutos. El sobrenadante obtenido se pasa a un tubo falcon de 15 ml y se agrega etanol
absoluto (aproximadamente el doble del volumen del sobrenadante). Una vez agregado el
etanol se forma el ovillo de ADN, el cual se transfiere a otro tubo de 1,5 ml donde se realizan 2

42
lavados con etanol al 70% y se deja secar (en estufa a 37 ºC 1 hora o toda la noche a
temperatura ambiente). Por último se resuspende el ADN en buffer TE y se mide la densidad
óptica (DO) a 260 nonómetros. La muestras de ADN son aceptadas si cumplen con el filtro de
calidad calculado a partir de la relación ácidos nucleicos-proteínas. La relación se considera
aceptable cuando el valor estimado se encuentra entre 1,8 y 2 al medir la DO a 260 nm y 280
nm.
5.3.2 Extracción de ARN
El primer paso en el proceso de extracción de ARN consiste en separar las células
mononucleares sanguíneas a partir de la técnica de Ficoll-Hypaque plus (GE healthcare Bio
Science AB). Se utiliza una muestra de sangre anticoagulada con EDTA (aproximadamente
10ml) la cual se debe diluir al medio con solución fisiológica. La muestra diluida se agrega a un
volumen de 10 ml de Ficoll-Hypaque y se centrifuga a 1500 rpm durante 10 minutos. Una vez
centrifugada la muestra, se forma una monocapa de células mononucleares entre la capa de
ficoll y plasma, la cual se extrae cuidadosamente y se lava con solución fisiológica.
El proceso de extracción de ARN, a partir de las células mononucleares, se realizó utilizando la
técnica de Trizol (TRI reagent, SIGMA life Science), la cual se describe brevemente. El Trizol
permite separar ADN, ARN y proteínas de una misma muestra biológica. En el caso particular
de la extracción del ARN, debido a la inestabilidad del mismo, todos los reactivos deben
utilizarse en frío a -4 ºC. A las células mononucleares tratadas con trizol (aproximadamente 1
ml) se le agregan 200 ul de cloroformo y se agita enérgicamente para que se formen dos fases,
una acuosa superior y una orgánica inferior. Se incuba durante 15 minutos, en frío a -4ºC, y se
centrifuga a 13000 rpm durante 10 minutos. Se transfiere cuidadosamente la fase acuosa a
otro tubo y se extrae el ARN con 500 ul de isopropanol. La muestra se centrifuga nuevamente
para precipitar el ARN y se realizan 2 lavados con etanol al 70% para eliminar impurezas. Por
último se resuspende el ARN en agua libre de RNAsas y se mide la DO a 260 nm. La muestras
de ARN son aceptadas si cumplen con el filtro de calidad calculado a partir de la relación
ácidos nucleicos-proteínas. La relación debe encontrase entre 1,8 y 2 cuando se mide la DO a
260 nm y 280 nm.
El ARN obtenido se retrotranscribe a ADN copia a partir de una RT-PCR utilizando random
hexámeros y la enzima SuperScrip 3 (Invitrogen).
5.3.3 Amplificación por PCR y secuenciación
Las secuencias codificantes y las regiones intronicas flanqueantes de cada exon fueron
amplificadas por la técnica de PCR a partir de ADN genómico y ADNc extraído de las muestras
de sangre de los pacientes, siguiendo procedimientos estándares. Se utilizaron primers
específicos para los distintos genes estudiados que se detallan en la tabla 2. Aquellos exones
que poseen un tamaño superior a las 800 pb se amplificaron en varios fragmentos con primers

43
internos solapados, de tal manera de no perder ninguna región en la lectura de secuencia. Los
fragmentos amplificados tienen un tamaño, en promedio, de aproximadamente 300 pb. Cabe
destacar que el gen GH1 se amplifica en un único fragmento de 2155pb y se secuencia a partir
de primers internos.
Las condiciones de la reacción de PCR se determinaron a partir de las indicaciones para la
enzima GoTaq DNA polimerasa de la empresa Promega. Los productos de PCR obtenidos se
sembraron en un gel de agaraso al 1% y se seleccionó la banda que corresponde al fragmento
de interés. Estos fragmentos fueron purificados a partir del Kit de purificación Qia Quick
(Qiagen, Buenos Aires, Argentina) y se secuenciaron por la técnica de Sanger, utilizando el kit
de secuenciación BigDye Terminador version 3.1 (Applied Biosystems, Buenos Aires,
Argentina) y el secuenciador automático 3130 Genetic Analyzer (Applied Biosystem).
Las secuencias nucleotídicas obtenidas fueron comparadas con las secuencias de referencias
disponibles en la base de datos de GeneBank (www.nbci.nlm.nih.gov/genebank).
Tabla 2. Primers específicos para los genes estudiados
GEN EXON PRIMERS FORWARS PRIMERS REVERSE
GHRHR 1 AGCCAAGGCTTACTGAGGCT CAGAGGCCAGAGGGTCTCAG Salvatori,R. & Levine,M.A. 1999 2-3 GACACCCAAATGGCTTGGCTCAT GCCACTTCCAGATGAAAGCACCTCCC
4 AGAGGGAAGGAGTTGTGGCTAGAG GTCCATGGCAGTCCTTCCTCCAA
5 TCCAAAGGCCCAGAAGCTGA TGGGAAATAAGGAGCCAA
6 CAGAATCCCCTCTCCCTGCTT CTGCCCAGCCCCTTCACCCATGG
7 CAGTGGGTGTCCCAGCTCTGAAGCACC TTTACCTCCCCATGGTGCCCA
8 AGATCTCAGAGTCAAGGATGCAGA TCCACTCCACACCCCATGTAGGA
9 AGGT GCCTGCGTCACCACAGTGA CAGTAGTTCCAGGGAAGTTGACT
10 GCCACCCAAGGCTACCCCCTGAC TGGATGAGACAGACACATTGAAT
11 TGAAGTGCACACGACAGTTTCTA GCAACAGCACCTCCCTCCAGCAC
12 AGGCCAAAGGGTTCTGATGGG TTAGGTCTGGTGGGAGGGGGA
13 GACCTTCCTAACGTCCTCTTC CAGCTGGGGTGGGGATGTGGC
GH1 1-5 GTGGGGGCAACAGTGGGAGAGAAGGGGCCAGGTAT TGTCCCACCGGTTGGGCATGGCAGGTAGCC
Chen EY et al 1989 SeqPrimers CCCGCTGGGAAATAAGAGGAGGAGACT AGTCTCCTCCTCTTATTTCCCAGCGGG
CTGACTTTGAGAGCTGTGTTAG CTAACACAGCTCTCAAAGTCAG
GGCACTGGAGTGGCAACTTCCAGGG
GHR 2 TGCTGGGCTTTACCTTACC GTTTCAAAACACTGAGGGTG
Pilotta P et al 2006 3 TACACAGGGTCATATCAGATT CTATTCCAGTTACTACCATCCC
4 ATATGACTCACCTGATTTATG TAGGTACATCCATGGAGAGGAA
5 ACTTAAGCTACAACATGATT GCTTCCCCATTTATTATTTAG
6 ATTGTGTCTGTCTGTGTACTAATG ATAGAAAGAAAAGTCAAAGTGTAAG
7 TTGAGTTGTTGACTCTTTGGCC AACTGTTATATTGACAAAAGC
8 GAAACTGTGCTTCAACTAGTC GGTCTAACACAACTGGTACA
9 GCTATAATTGAGAATATGTAG CATATGACAGGAGTCTTCAGGTG
10 A GAGTTTCTTTTCATAGATCTTC GGCATTGAAATCAGTCTCCAG
10 B GGACGTACCAGCTGTTGTGA CGATGTTTGACAGTGAACTTGG
10 C CACCAAGCTGCCCATATTCAG GGTGATGTAAATGTCCTCTTG
10 D GGTTGAATCACACATACAGCC TGCCCCAGTCAATTCTTTGCT
IN 6 GGCCACTCATCAGGTGATGC TTGAGAACCGCCACCTCAGG
44
STAT5B 2 CGCCCAAAGCACTGATTGTTTGT CCAAGAGTTCCATCCCCTCAATCTAAGATG Kofoed EM., et al 2003 3 GTTGGGCTGTTTCTTAAAATTTGCAGTGA GAAGACCCCCAAGGGAAGGTAATTAA
4 CATAGTGTGGAGTGGCACAAAGTGAT GTGCCCCTTAGGATGAAGCTCTCTTTA
5 CCATGATCTGTCCGCTCACCTGT GCCCAATCCTCAAGTTGCACAAT
6-8 CCGACTTGCCCTTAATGCAGTATCT GGGCCACAGCAACTGGAGAT
9 CGCCACCATCACGGACATTATCTC GGACATGAGACAAGTAGCAGGGAAT
10-12 GGGTTTGGAGCTGGTCTCTCT GAGATTCATGACACCGGCATTGATTTT
13 AATCATGCAACTGTTTTCTCATCAGATTCTG AGGGAGACTCCTGAGAGCATCT
14,15 GCAGTTTGGTATTTTCCCCTTCTTCCCTCTTTTCCTCTCCATG GAACTAACATCTTGGGTAGAAATGGTTGATGGCAATAT
16 GCAAGCTTGAATTTATAGTATGTTGGGGTTTTAA GCTCAAACTGCTCTCACTGAATGGTAA
17-19 GTGGTTGTGTTCTGTCCTAAGAAAAGAT CCAACTTCCAGGCTTCACGAAA
IGF1R ADNc 1-10 AAAAGGAATGAAGTCTGGCTCC ACCTCAGTCTTGGGGTTCTC Kawashima Y. et al 2005 11-21 GCAGCCTCAGGACGGCTACC GGGCAGCAAGGGCAGTTCTG
IGF1R ADNg 1 AGAAAGGGGAATTTCATCCCAA ACAGCTGCGACGGTGGCAA Kawashima Y. et al 2005 2 CTGAGACGTTTACCCTCTTGT GGCAGAGAGAAAGCAGCAGTC
3 GTGAATGACCCAGAAGGATG GTCTACCTTTGTGTGCTAGG
4 CC ATGTGACCACATGAACACTT C TTTTAGTGGTGACTCATTTACG
5 C TTACACCAAGTGAGCACACAG CACAGTTTCATAAGCACGCTGC
6 TGCGCTAACATCGATTTCTGT CGGTGTTTTGGATGCTGTCA
7 TGCTTTTCAGAGACACATG CCCTGTCAACAGAATGGCAT
8 CCCGAACTTTCTCTGAACTT GCTTGCGAAGAAGTGTGGATG
9 GCCAGAGTATCTGATAGCCTGAC TCCGTCCAGGCCGCTGCT
10 CGGCTTTCATTCCCACTCTT TGCCAACTGAAATTTCACAT
11 AAAGCCACATTTCTCTCCTCCTT AATGGGGCCAGCTGGATCAT
12 GTCACCTGTTTAAATTGTACA ATGGAAGTACATGCAGGCA
13 TGACATGTATGTTTTATTTCC AGTTTTACCACGTTTCTTCTC
14 AATGTGAAGAAATGAAATGA CAAAAATATCACCTTCACTC
15 TGTTGTAGCGAAGATGAAAGTA CAAAATAATGCAAACCTCCTT
16 TGCTTTAATTACGGTTTCTTC GCTTTTCAGGAACTTTCTCTT
17 TGACCCTCTGAGTCTTTCT CCGGTGGAAATGAAAACTG
18 AAATCCAAAATTCTCATGTGAAT GCCCAGACCTGGAAAGCTA
19 GTGCCTGCTCCAGCGTGT CTAGAGACTGAGCTGGTGGAA
20 TTCAGTCCATCCCTTTCCAA CCTAATCTCCTGTGACCCAA
21 CTCCCGTGTGTCT TGGCTGC TGTGAATGGATTGTTAAAACT
OTX2 1 TTTAAAAGCCTCTGCCTCG GAACAGGGTGTTGCATCC
Ragge NK et al 2005 2 GAGAGCATTGGTAGGCTCC TCTCCACAGTCCCATACTCG
3 GAGCCATTCTTGTCCTTAAGG AATGCCTGGCTAAAACTGG
HESX1 1 AGCTGTTGCTCTGTGCAGACCACGAGA ACAAAGAATTGAAACAATTAAGCTGTGGCA
Dattani M et al 1998 2 TGGAACATAAGATTGACCAT CTAAGACA AGCCTTTATATTATCATTATTGGGTGAA
3 AGCTCATTTTTGGAGACATACTGAATA ATAGGTCTCAGAAACATTATTTTATTATTC
4 AGTTGTTATTCATGATGTTGAAATGTTA TCATGCTCTGCAATTAGAAGATAATTTCAC
LHX3 1 TGACCTAGGAGGAGCGCGTCT CAACCGCTGTCCCGCACTCTT Kyle W. Sloop et al 2000 2 TACGAGGTGACCCAGAACTT CCTGGCCTTGGTGATTGTGA
3 CGAAATGAGCCTCGCGCTTC TTTCAGACCAGGAAAGGTGG
4-5 GCTGCCGCGCCTCACCGCT AGGAGTCCACTAACTCCATG
6 CGCTGACTGAGCCTCTGCTT CCTCGTGTGAGGTGCAGGGT
LHX4 1 TAGAGCGAGAGAGCGAGAGA CAGCACTCTCCCCAGCGTTC Machinas K et al 2001 2 TCTGCGGTTTGGGCCATGTCT GCTGCAAACTTCCCCCTCACT
3 GAAGCTAGATGGAGGCAGAGA CAGAGGTGGCGGTGCATGAGT
4-5 GGAAGGGAGGGTGTGGGAGG GAGTGCCAGGGATTACAGAT
6 TTGGGCATCTTTCCTGGTCAT CCAGGGGGGCAGGTAGGGTG

45
POU1F1 1 AACTCTGATTTGGGGAGCAG GGGTAAAATGAAAGATGCAAAGA Hiroshi Inoue et al 2012 2 TGAGAAAAACATATTGGCATGT CCATCTAAGTGTCCCCAAATTC
3 GGGCTAAGTCAGGCAAAACC TGCAAACCAAGTTCTTTTTCC
4 AGATTTGTGTGACAATGAACCAA TTTTAGGTTAAAACACAGCACAGC
5 GCATTTTTATTTTGCATTGTTTT GCCTCCCAATTCACCTTACA
6 TTGAGCTGCCAAAAACTTCA GAAAGGAATGAAACGGGAGA
PROP1 1 ATCTCAAGTCAGAGATTCAGGGACA TTTCTCCTCCTAACCTTCTTCATGGA Kyle W. Sloop et al 2000 2 ACAGGCACATGTGGTCCAGCACCGA ATTTCTTTCCTGAGAGAGGAGGATCCT
3 TGTCACCACCTATGTCAAGTGT ACCCCATTTTCTTGTCTTTTCACGA
GLI2 1 TGGGTTTGGGCTCAGTGT AATTAAATCCTAACGGGCTAATG França MM et al 2013 2 TGGCTGCTCTTGCTATGAAA GCAGGAGATGTGGCTGAGG
3 CATGTTGGTTTTGGGGTCTT GACCAAGGCTGAGGAGTTGA
4 TGTGCATTTCTCTCTGCCTTT CCTTGTCCCCAAAAGAAACA
5 CCTTGCAGGCTCTTCCTATC TCTTTCTCCTCGGGTCAAAA
6 TGGGCAAGGTTCTCTCTGTC CTTAGCATGAGCTGGCAGTG
7 TGTGCGGAGAGATCCTAGAG TTCACCACCAAGGGTACAGC
8 CACACACACTTGCATCCACA TCCAGCCCCTTCTGTCTAGT
9 GACAGCAGGGGGTGGTCT CCACCTCCAAACATGATCC
10 GGTTGGAGCAGAGCAGAGAA GGCACCTGGCTATCTACTGG
11 GCCCCTTCAGAGTTGATCCT GGGCACGGCTATGGAGTC
12 GCCTGTGCAGGCCTAGAG GTGGGTGCCAGCCTAGTTG
13.1 ATGACTGAGCACGGTCAAAG AGTGGCTGCCGCGTACTT
13.2 CTTCCACAGCACCCACAAC ACGTTCTCGCTGATGCTAGG
13.3 GACAGCACGCAGCCACAC GACCCCATGTTGCCCAT
13.4 GTGGGGTACGTGCTGTGC GGAAAAAGACAAGACAGCTGGA
5.3.4 RFLP
Deleciones en el gen GH1 fueron estudiadas mediante la técnica de RFLP (restriction fragment
lenght polymorphism) utilizando las enzimas de restricción Sma I, Bgl I y Hae II (Takashi K,
John Phillips III. 1992). Previamente al análisis por RFLP se amplificó, con un único par de
primers, dos fragmentos homólogos de 1900 y 1919 pb que flanquean al gen GH1. Estos
fragmentos son indistinguibles en tamaño cuando se los siembra en un gel de agarosa, pero
poseen un patrón de restricción diferente cuando se los digiere con las enzimas mencionadas,
permitiendo diferenciar entre las tres deleciones mas frecuentes de 6.7, 7.0 y 7.6 Kb.
5.3.5 MLPA
Variantes en el número de copias, en el gen IGF1R, fueron analizadas mediante la técnica de
MLPA (Multiplex ligation-dependent probe ampliflication). El estudio fue realizado utilizando el
kit comercial SALSA MLPA P217 IGF1R (MCR Holland, Amsterdam, Holanda) y el
secuenciador automático 3130 Genetic Analyzer (Applied Biosystem), siguiendo las
instrucciones del proveedor. Los resultados fueron analizados utilizando el software
GeneMarker v1.97 (SoftGenetics).

46
5.3.6 Ensayos In silico
Herramientas informáticas fueron aplicadas para identificar el potencial impacto funcional de las
nuevas variantes encontradas. Se utilizaron tres ensayos online:
1. SIFT (Sorting Intolerant from Tolerant, hyyp://sift.jcvi.org/) versión 2.06, basado en el
análisis de estructura, el cual determina si la mutación introducida genera un cambio
estructural en la proteína que repercute en su función.
2. PolyPhen2 (Polymorphism Phenotyping, http://genetics.bwh.harvard.edu/pph2), basado
en homología de secuencia, el cual analiza la conservación evolutiva de los
aminoácidos afectados en las distintas especies.
3. Mutation Taster (http://www.mutationtaster.org/) el cual combina los análisis anteriores.
5.3.7 Cultivo primario de fibroblastos
Se utilizó, como modelo biológico, el cultivo de fibroblastos en aquellos pacientes con
variantes, no reportadas previamente, en el gen IGF1R. Los cultivos de fibroblastos se
establecieron a partir de biopsias de piel, obtenidas de la zona abdominal, de los pacientes
afectados y de sujetos controles (misma edad, sexo y estadio de desarrollo puberal de Tanner
de los pacientes). Los explantos de piel se cultivaron en medio Dulbecco’s Eagle modificado y
F12 (DMEM/F12, SIGMA, Bueno Aires, Argentina) con suero fetal bovino (FBS) al 10 o 20%,
junto con el agregado de penistreptomicina, gentamicina y amfotericina B. Las placas de
cultivo se mantuvieron en estufa a 37ºC, en una atmósfera húmeda, con 5% de dióxido de
carbono y se realizaron tres subcultivos. Las células fueron almacenas en nitrógeno líquido
hasta el momento de realizar los ensayos funcionales. Finalmente, los fibroblastos fueron
subcultivados una vez más para obtener la cantidad de células necesarias para llevar a cabo
los estudios (25.000 cel/well en placas de 24 wells).
Los fibroblastos fueron estimulados con distintas concentraciones de IGF-1 (25, 50 y 100
ng/ml) y se observó que la mayor respuesta se obtiene con la dosis de 50 ng/ml.
5.3.8 Ensayo funcionales in vitro
5.3.8.1 Ensayo de proliferación celular
Los fibroblastos de los pacientes afectados y los sujetos controles se incubaron en placas de
24 wells con DMEM/F12 al 10% de FBS. Cuando las células llegaron a subconfluencia, se
lavaron con solución fisiológica y se incubaron 24 horas en DMEM/F12 con BSA 0.1%,
eliminado el suero fetal del medio de cultivo. La placa de cultivo de 24 wells fue dividida en 4
condiciones distintas para cada paciente y sujeto control:
1) Estado basal.
2) Estímulo de 16 hs con IGF-1 50 ng/ml.

47
3) Estímulo de 20 hs con IGF-1 50 ng/ml.
4) Estímulo de 24 hs con IGF-1 50 ng/ml.
Cuatro horas antes de que se cumplan los tiempos estipulados para cada condición se agregó
una solución de 3[H]-Timidina (1mCi/ml) para alcanzar una concentración final de 1 uCi/ml.
Posteriormente al tiempo establecido, las células fueron lavadas con PBS y fijadas con ácido
tricloroacético (TCA) al 10% durante 2 horas a 4ºC. Por último los fibroblastos fueron lisados
con una solución de NaOH 0.1 M y SDS 0.2 % y la radioactividad soluble se midió en un
contador de centelleo de emisión beta.
Cada estadio fue realizado por sextuplicado en simultáneo y en una placa de 24 wells para
cada paciente y sujeto control y los resultados se expresaron como veces de incremento con
respecto a la condición basal.
5.3.8.2 Ensayo de fosforilación de AKT
Nuevamente, los fibroblastos de los pacientes afectados y los sujetos controles se incubaron
en placas de 24 wells con DMEM/F12 al 10% de FBS. Cuando las células llegaron a
subconfluencia, se lavaron con solución fisiológica y se incubaron 24 horas en DMEM/F12 con
BSA 0.1%, eliminado el suero fetal del medio de cultivo. En este punto los cultivos de
fibroblastos se estimularon con 50 ng/ml de IGF-1 durante 10 minutos y, posteriormente, se
lavaron dos veces con una solución de PBS a -4ºC para eliminar el remanente de IGF-1
presente en el medio. En el siguiente paso las células fueron sometidas a un proceso de lisis
por medio del agregado de un buffer a base de Deoxicolato de sodio y triton a
concentraciones finales 0,5% y 1%, respectivamente, y se extrajeron las proteínas mediante
procedimientos estándares.
La fosforilación de AKT fue determinada por la técnica de ELISA utilizando el kit phospo-Akt
(Ser473) STAR ELISA (Merk Millipore, Tecnolab, Bs As, Argentina) siguiendo las instrucciones
del mismo. Por otro lado, la concentración proteica de cada well fue determinada por la
técnica de Bradford y los resultados del ensayo fueron normalizados por miligramo de
proteína. El ensayo fue realizado por duplicado para cada paciente y sujetos controles.
5.3.8.3 Análisis estadístico
Se aplicaron los test estadísticos de análisis de la varianza (ANOVA) y Bonferroni para analizar
si existen cambios estadísticamente significativos (p< 0.05), entre la condición basal y la
condición estimulada con IGF-1, en cada paciente y sujetos controles en el ensayo de
fosforilación de Akt y entre los distintos tiempos de estímulo para el ensayo de proliferación
celular.

48
6. Resultados
6.1 Alteraciones moleculares en pacientes con IGHD
Alteraciones moleculares en el gen GH1 fueron estudiadas en una cohorte de pacientes
pediátricos pertenecientes a 41 pedigrees no consanguíneos (22 casos familiares) con
deficiencia aislada de hormona de crecimiento (IGHD). A su vez, en 12 pacientes con IGHD,
que no presentaron alteraciones en el gen GH1, se analizó la presencia de mutaciones en el
gen GHRHR.
Identificamos mutaciones en el gen GH1, previamente reportadas como causa de IGHD, en
9/41 pedigrees no consanguíneos que corresponde al 22% de nuestra cohorte (13 pacientes).
Analizando únicamente los casos familiares este porcentaje se elevó a 36.4%, que
corresponde a 8 familias con más de un miembro afectado, de un total de 22 familias
estudiadas. La información clínica, los datos de laboratorio y los estudios moleculares en los
pacientes con mutaciones en el gen GH1 se detallan en la tabla 3.
Tabla 3. Información clínica de los pacientes y familias afectadas con mutaciones en GH1
Paciente (A) (B1) (B2) (C1) (C2) (D) (E) (F1) (F2) (G) (H) (I1) (I2) X±SD
Sexo M M M M F F F M M M M M M
Tipo de IGHD IA
Del 6.7
Kb
IA
Del 6.7
Kb
IA
Del 6.7 Kb
II
p.R183H
II
p.R183H
II
p.R183H
II
p.R183H
II
p.R183H
II
p.R183H
II
p.R183H
II
p.R183H
II
p.V110I
II
p.V110I
Edad
cronológica
0.71 1.42 0.75 13.5 7 0.75 3.58 7.09 7.09 9.58 1.75 1.67 6.33
Edad ósea 0.5 0.5 ND 11 5.30 0.50 1.50 2.6 2.6 7 ND 0.75 2.50
Talla
SDS
-3.77 -5.97 -4.07 -2.46 -2.10 -3.65 -5.13 -5.86 -5.80 -3.39 -3.87 -4.40 -5.02 -4.26 ± 1.24
Peso
SDS
-1.51 -4.22 -1.83 -2.56 -3.38 -3.22 -4.70 -4.3 -4.13 -2.48 -3.00) -4.22) -3.97) -3.35 ± 1.02
Talla
Padre SDS
0.18 -3.35 -3.35 1.50 1.41 -4.34 -2.18 -1.88 -1.88 -3.54 -2.47 -1.67 -1.67 -1.97 ± 1.84
Talla
Madre SDS
-1.84 -2.90 -2.90 -2.57 -2.57 -2.10 -3.05 -2.16 -2.16 -2.90 0.67 -1.73 -1.73 -2.06 ± 1.13
Resp. Max
GH ng/ml
0.05 0.10 0.05 1.99 3.06 1.24 2.87 5.00 2.00 1.80 1.20 0.97 1.20 1.94 ± 1.32
IGF1SDS -3.02 -3.08 -2.77 -2.09 -2.89 -3.73 -2.91 -3.43 -3.39 -2.21 -2.31 -2.31 -4.73 -2.99 ± 0.73
IGFBP3 ND <-3.50 ND <-2.63 <-2.48 0.65 0.36 ND ND ND -2.01 -2.20 ND
Familiar
Afectado
M y P
HT
M y P
HT
M y P
HT
M M P M ND ND P P M M
Matias Juanes et al 2013 Clincal Endocrinology
Pedigrees son identificados con letras y los hermanos dentro del pedigree por numeros. M, masculino; F, femenino;
Resp. max, respuesta máxima de GH-1; X±SD, promedio ± desviación estandar; SDS, score de desvío estandar; P,
padre; M, madre; ND, no determinado; HT, heterocigota.

49
Análisis de los genes GH1 y GHRHR
1) Deleción del gen GH1: Identificamos la deleción de 6.7 kb, en estado homocigota, en 3
pacientes provenientes de 2 familias no consanguíneas (Paciente (p): pA, y pB1-B2). El análisis
molecular se muestra en la figura 17. La deleción del gen GH1 da lugar a la IGHD tipo 1A de
herencia recesiva y como es esperable los padres de los pacientes afectados son portadores
de la deleción.
A)
B)
Figura 17. Patrón de digestión de los fragmentos flanqueantes al gen GH1 con las enzimas Sma I, Bgl I y Hae II. En el panel A a la izquierda se observa
el gen GH1, normal, flanqueado por las regiones homólogas que poseen sitios de restricción para las enzimas. A la derecha se esquematiza el fragmento
resultante de la deleción de 6.7 kb con los sitios de restricción que se conservan. Las flechas indican la posición del par de primers utilizado para amplificar los
fragmentos. El panel B muestra el patrón de bandas obtenido, cuando se siembran los fragmentos digeridos en un gel de poliacrilamida. CTRL; sujeto control,
Pac; paciente afectado, P; padre del paciente afectado y M; madre del paciente afectado.
Sma I Bgl I Hae II
Pac P M Ctrl Pac P M Ctrl Pat P M ctrl

50
2) Mutaciones previamente reportadas en el gen GH1: Detectamos mutaciones puntuales en
10 pacientes, provenientes de 7 familias no consanguíneas (Tabla 3). Identificamos dos
mutaciones con pérdida de sentido en estado heterocigota previamente reportadas en la
literatura (Wajnrajch MP et al 2001; Millar, D. S. et al 2003). La mutación p.Arg183Hys fue
encontrada en 6 pedigrees (pacientes: pC1-C2, pD, pE, pF1-F2, pG y pH) mientras que la
variante p.Val110Ile fue hallada en una familia (pI1-I2).
3) Variantes nuevas en el gen GH1: Identificamos 3 nuevas variantes con pérdida de sentido
en tres pacientes no relacionados (pJ, pK y pL). El fenotipo clínico, los hallazgos de laboratorio
y los estudios moleculares se muestran en la tabla 4 y la figura 18.
El análisis de la secuencia nucleotídica de pJ reveló la presencia de una variante heterocigota
en el exón 2 del gen GH1, una substitución de citosina por timina en la posición 55 del ADNc.
Esta variante resulta en una substitución de Prolina por Serina en el codón -7 (p.Pro-7Ser) de
la proteína. Por otro lado, dado que el padre del paciente posee una talla de -2.84 SDS, se
sospechó que era portador de la variante pero la muestra de ADN no estuvo disponible para
confirmar el diagnóstico.
El estudio molecular del gen GH1 en pK reveló una substitución heterocigota de guanina por
adenina a nivel del exón dos, en la posición 162 del ADNc. Este cambio de base da lugar a un
cambio aminoacídico de Arginina por Histidina en la posición 19 de la proteína madura
(p.Arg19Hys). Cabe destacar, que en la madre del paciente K se detectó la variante en estado
heterocigota pero su talla se encontró dentro de los valores normales para la población de
referencia (-1.92 SDS).
Por último en pL se encontró una variante heterocigota a nivel del exón 4, una substitución de
timina por citosina en la posición nucleotídica 305 del ADNc. Esta variante genera un cambio a
nivel proteico de Leucina por Prolina en el codon 102 (p.Leu102Pro) del gen GH1. Esta
variante también fue encontrada en el padre del paciente, el cual posee un retardo de
crecimiento severo (talla: -5.12 SDS).
Para corroborar si las variantes halladas se encuentran presentes en la población general, se
analizó la presencia de las mismas en 60 sujetos controles (120 alelos) y no se encontró
ninguna de las variantes mencionadas. A su vez se analizó la presencia de las mismas en las
distintas bases de datos disponibles online (www.ncbi.nim.gov/SNP; www.ensembl.org;
www.exact.bradinstitute.org) y encontramos que la variante p.Arg19Hys, presente en el
paciente K, fue previamente reportada en el proyecto mil genomas con una frecuencia de 0.1%
(rs71640274).
Para analizar la conservación evolutiva de los aminoácidos afectados por las nuevas variantes
se observó el alineamiento de secuencias de la proteína GH-1 provenientes de distintas
especies. Se determinó que la substitución p.Pro-7Ser y p.Arg19Hys no afectan residuos
conservados en la proteína mientras que la variante p.Leu102Pro afecta un residuo altamente
conservado entre las distintas especies, sugiriendo que esta nueva variante posiblemente
tenga un efecto deletéreo sobre la función de la proteína. A su vez se utilizaron ensayos in

51
silico para predecir el efecto funcional de las variantes y las tres herramientas informáticas
concluyeron que únicamente la variante p.Leu102Pro tendría un efecto deletéreo sobre la
función de la proteína mientras que las variantes p.Pro-7Ser y p.Arg19Hys se tratarían de
polimorfismos (SNP) sin efecto sobre la funcionalidad de la proteína.
Tabla 4. Variantes nuevas encontradas en el gen GH1.
Paciente J K L SDS X±SD
Sexo M M M
Variante p.Pro-7Ser p.Arg19Hys p.Leu102Pro
IGHD Tipo II
PN(Kg) 3.600 1.200 3.350
LCN(cm) ND ND 49
EC
(años)
11.50 8.67 12.75
EO
(años)
5.50 5.00 9.50
Talla cm
(SDS)
124.3 (-2.58) 109.0 (-3.62) 126.2 (-2.79) -2.99±0.55
Peso kg
(SDS)
30.0 (-1.25) 19.5 (-3.38) 28.5 (-2.16) -2.26±1.07
Talla Padre cm
(SDS)
154.8 (-2.84) 166.0 (-1.00) 138.0 (-5.12) -2.92±2.07
Talla Madre
cm (SDS)
154.9 (-0.95) 149.0 (-1.92) 151.0 (-1.59) -1.48±0.49
Resp. Max
GH(ng/ml)
4.70 4.80 3.20 4.23±0.89
IGF-1 ng/ml
(SDS)
207 (-0.23) 31.85 (-2.61) 32.57 (-4.49) -2.44±2.13
Familiar
portador (HT)
ND Madre Padre
M, masculino; X±SD, promedio ± desvío estandar; PN, peso al nacer; LCN, longitud corporal al nacer; EC, edad
cronológica al diagnóstico; EO, Edad ósea al diagnóstico; HT, heterocigota; ND, no determinado.

52
Figura 18. El esquema muestra las variantes nuevas encontradas en el exón 2 y 4 del gen GH1. El electroferograma,
la posición de los aminoácidos afectados y la conservación evolutiva se muestran para cada variante identificada.
4) Mutaciones en el gen GHRHR: se analizó la presencia de mutaciones en el gen GHRHR en
12 casos índices con IGHD sin alteraciones en el gen GH1 y no se observó ninguna variante
relacionada con déficit de crecimiento postnatal.
6.2 Alteraciones moleculares en pacientes con síndrome de insensibilidad a la GH
Alteraciones moleculares en el gen GHR fueron estudiadas en una cohorte de 31 pacientes
pediátricos no consanguíneos con sospecha de insensibilidad a la hormona de crecimiento. A
su vez, 16 pacientes con resistencia a la GH e infecciones recurrentes, con alteración
inmunológica, fueron estudiados en busca de alteraciones en el gen STAT5B.
Identificamos 4 alteraciones moleculares en el gen GHR en 5 pacientes no relacionados, de las
cuales solo una no ha sido previamente reportada en la literatura. En dos pacientes
encontramos la variante p.Arg179Cys, previamente asociada en la literatura con Síndrome de
Laron (Amselum S et al 1993), insensibilidad parcial a la GH y talla baja idiopática. Cabe
destacar que esta variante fue hallada en estado heterocigota en los dos pacientes estudiados
mientras que la variante asociada a Síndrome de Laron fue reportada en estado homocigota. A
su vez esta variante fue hallada en el proyecto mil genomas y se encuentra reportada, en las
bases de datos disponibles, con una frecuencia mayor al 1% en la población Europea
(www.ncbi.nim.gov/SNP; www.ensembl.org; www.exact.bradinstitute.org; rs121909362).
La segunda variante identificada fue un cambio aminoacídico de Alanina por Valina en la
posición 488 del receptor de GH en estado heterocigota. Esta variante se encuentra reportada

53
en distintas bases de datos y en múltiples observaciones no se la ha relacionado con
alteraciones en la talla de tal manera que es considerada una variante polimórfica sin efecto
patológico sobre la funcionalidad del receptor (www.ncbi.nim.gov/SNP; www.ensembl.org;
www.exact.bradinstitute.org; rs200084031).
Identificamos una tercer variante heterocigota con pérdida de sentido, en un paciente y su
madre, donde se substituye una Tirosina por Histidina en la posición 240 del GHR. La variante
fue previamente reportada por Tauber et al en 1998 pero la información fenotípica del paciente
no se encuentra disponible, de tal manera que no es posible confirmar su relación directa con
la patología en cuestión. A su vez la variante se encuentra reportada en las distintas bases de
datos disponibles online indicando que no se trataría de una alteración poco frecuente en la
población general (www.ncbi.nim.gov/SNP;www.ensembl.org; www.exact.bradinstitute.org). Sin
embargo los ensayos de predicción in silico indican que la variante afectaría la funcionalidad
del GHR con un alto índice de score.
Por último, identificamos una nueva mutación, sin sentido, homocigota, a nivel del exón 4 del
gen GHR, en un paciente con severo retardo de crecimiento postnatal (talla baja extrema) y
niveles de IGF-1, basal y post-estímulo, muy por debajo de los valores de referencia para la
edad y el sexo. La variante es un cambio de citosina por adenina en la posición 198 del ADN
codificante, que da lugar a la aparición de un codón de terminación prematuro en la posición 66
de la proteína. Consecuentemente, la síntesis del receptor de GH se detiene a nivel del exón 4,
impidiendo la traducción de una región del dominio extracelular y los dominios intracelular y
transmembrana, perdiéndose la capacidad de unión y respuesta a la GH-1.
Por último, 16 pacientes con insensibilidad a la GH-1 y características clínicas de alteración del
sistema inmune fueron estudiados en busca de mutaciones en el gen STAT5b y no se identificó
ninguna variante con impacto funcional relacionada con la patología.
Figura 19. Mutaciones encontradas en el gen GHR. Se observan los electroferogramas de los distintos pacientes,
indicando las variantes a nivel del ADN codificante y de la proteína. La variante p.Cys66* fue hallada en estado
homocigota mientras que las otras tres variantes se encuentran en estado heterocigoto.
C N
Peptido señal
Dominio extracelular Domain
Dominio transmembrana
Dominio intracelular
p.Arg179Cys
6 3 2 10 4 5 7 8 9
c.535 C>T c.198 C>A
p.Cys66*
c.718 T>C
p.Tyr240Hys
c.1463 C>T
p.Ala488Val

54
6.3 Alteraciones moleculares en pacientes con resistencia a IGF-1
Estudiamos 28 pacientes con sospecha de insensibilidad a IGF-1 e identificamos cuatros
nuevas variantes heterocigotas en el gen IGF1R, en cuatro pacientes no relacionados. Las
alteraciones p.Arg1256Ser (P1) y p.Asp359Tyr (P2) fueron encontradas solo en los pacientes,
sin tener otro familiar afectado, por lo que se consideraron como variantes de novo. Por otro
lado, las variantes p.Tyr865Cys (P3) y p.Arg1337Cys (P4) fueron heredadas de la rama
materna y paterna, respectivamente. Las características fenotípicas de los pacientes y los
resultados obtenidos de los análisis moleculares y los ensayos funcionales, junto con la
segregación de las variantes con el fenotipo de alteración de crecimiento, sugieren que las
alteraciones encontradas en los pacientes P1, P2 y P3 afectan la función del receptor de IGFs
tipo 1 y fueron consideradas como nuevas mutaciones en el gen IGF1R. La variante presente
en el paciente P4 (p.Arg1337Cys) afecta un residuo conservado de la proteína y no fue
encontrada en 120 alelos estudiados en sujetos normales, sin embargo fue considerada como
una variante benigna rara ya que no cosegrega con el fenotipo de alteración de crecimiento
presente en la familia (hermana y padre portan la mutación y poseen talla normal). A su vez,
los ensayos funcionales aplicados indican que la función del receptor se encuentra conservada.
Las cuatro variantes se esquematizan en la siguiente figura.
Matias Juanes et al 2014 Clinical Endocrinology
Figura 20. A) Esquema del hemireceptor del IGF1R: L1, L2, dominios ricos en leucina; CR, dominios ricos en cisteína; FNIII, dominio de repeticiones de
fibronectina tipo 3; TM, dominio transmembrana; JM, región juxtamembranosa; TK, dominio tirosina quinasa; C-tail, región carboxilo Terminal. Las variantes
afectan los dominios L2, FNIII, TK y C-tail. B) Los electroferogramas muestran las substituciones a nivel nucleotídico en las posiciones 1075, 2594, 3768 y
4009 que se traducen en un cambio de aminoácido de p.Asn359Tyr, p.Tyr865Cys, p.Arg1256Ser y p.Arg1337Cys, respectivamente. C) Conservación evolutiva
de los aminoácidos afectados en las distintas especies. Se observa que los cuatro aminoácidos substituidos están altamente conservados en el receptor de
IGFs tipo 1.
L1L1 CRCR FNIII-1FNIII-1 FNIII-2aFNIII-2aL2L2
C adena alfa
JMJMTMTMFNIII-3FNIII-3FNIII
-2b
FNIII
-2b TKTK C-TAILC-TAIL
P P P P P P P P
C adena beta
A A A A G G GG G G C C A AA A T T T T T T
T T AA TT
c.1075 A>T
p.As n359T yr
S
S
p.T yr865C ys
c.2594 A>G
T T G G AA G G G G C C C C T T T T
AA G G TT
p.Arg 1256S er
c.3768 G>T
G G G G CC G G C C G G G G AA G GG G
G G C C AA
p.Arg 1337C ys
c .4009 C >T
T T A A A A T T G G T T AA TT G G
T T G G T T
S pec ies P atient 2 P atient 3 P atient 1 P atient 4
Human Q M L Q G C T I F K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
Mutated Q M L Q G C T I F K G Y L L I N I E P E N P N G L I L M C E I K Y G M R M C W Q Y N P K M S P S E N G P G P G V L V L C A S F D
C himp Q M L Q G C T I F K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
R hes us Q M L Q G C T I F K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
Mous e Q M L Q G C T I L K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
C hic ken Q M L Q G C T I L K G N L L I N I E P T N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N - - G P G V V V L R A S F E
Xenopus N L Q L N I E P K R P N G L I L M Y E I E Y K M R M C W Q Y N P K M R P S E N - - G P G V V V L R A S F E
Z ebrafis h V I K G N L Q I N I E P L H P NG L I L MY E I K Y R C W Q Y N P K M R P S A N - - G P - - V V L L G A S F E E
F ug u T V I DG N L D I N I E P I T P NG L I L MY E V K F R M R M C W Q Y N P K M R P S H V S A N G P V V VL R P N F E
S pec ies P atient 2 P atient 3 P atient 1 P atient 4
Human Q M L Q G C T I F K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
Mutated Q M L Q G C T I F K G Y L L I N I E P E N P N G L I L M C E I K Y G M R M C W Q Y N P K M S P S E N G P G P G V L V L C A S F D
C himp Q M L Q G C T I F K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
R hes us Q M L Q G C T I F K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
Mous e Q M L Q G C T I L K G N L L I N I E P E N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N G P G P G V L V L R A S F D
C hic ken Q M L Q G C T I L K G N L L I N I E P T N P N G L I L M Y E I K Y G M R M C W Q Y N P K M R P S E N - - G P G V V V L R A S F E
Xenopus N L Q L N I E P K R P N G L I L M Y E I E Y K M R M C W Q Y N P K M R P S E N - - G P G V V V L R A S F E
Z ebrafis h V I K G N L Q I N I E P L H P NG L I L MY E I K Y R C W Q Y N P K M R P S A N - - G P - - V V L L G A S F E E
F ug u T V I DG N L D I N I E P I T P NG L I L MY E V K F R M R M C W Q Y N P K M R P S H V S A N G P V V VL R P N F E
P E P TIDO S E ÑAL
A)
B)
C)

55
6.3.1 Evaluación Clínica de los pacientes afectados
La descripción clínica de los pacientes afectados se detalla en las tablas 5 y 6. Come se
observa en la tabla 5, todos los pacientes nacieron a término y presentaron un severo retardo
de crecimiento prenatal, ya sea por afectación de la talla o el peso al nacer. Por otro lado, se
observó la presencia de una marcada microcefalia en aquellos pacientes donde la medida del
perímetro cefálico estuvo disponible (P3 y P4).
La tabla 6 muestra el patrón de crecimiento corporal y el perímetro cefálico en los pacientes y
familiares afectados por las variantes encontradas. En la primera consulta al servicio de
endocrinología, los cuatro pacientes tenían 3.2 años o menos con tallas y pesos por debajo de
-2 SDSs. Se observó una pequeña recuperación del perímetro cefálico pero en todos los casos
siempre se mantuvo por debajo de -2 SDS. El desarrollo mental fue normal en P2 y P3
mientras que se observó un retardo moderado en los otros dos pacientes.
Los pacientes P1 y P2 fueron sometidos a tratamiento con hormona de crecimiento
recombinante humana (rhGH) durante dos años. Al inicio del tratamiento, P1 se encontraba en
un proceso de catch up de crecimiento espontáneo, el cual no se modificó con la
administración de rhGH mientras que el paciente P2 mostró una moderada respuesta al
tratamiento con respecto a la talla (Δ +0.9 SDS). Por otro lado, el perímetro cefálico no se
modificó en P1 mientras que se observó un leve incremento en P2.
Los pacientes P1 y P2 poseen variantes de novo en el gen IGF1R y no se registraron
antecedentes familiares de retardo de crecimiento prenatal, talla baja y/o microcefalia. La
madre de P3 y el padre y la hermana de P4 (P4s) compartían la misma variante encontrada en
los casos índices respectivos. La madre de P3 presentó microcefalia (PC -2.1 SDS) con talla
normal mientras que, tanto el padre como la hermana de P4 presentaron un fenotipo normal sin
alteración de crecimiento.
Los análisis de laboratorio de rutina junto con el estudio cromosómico fueron normales en
todos los pacientes, incluyendo el análisis del cromosoma 15 donde se ubica el gen IGF1R. Se
descartó el estudio del gen IGF1 debido a que todos los pacientes estudiados tuvieron niveles
de IGF-1 e IGFBP-3 séricos por encima de la media para la edad y el sexo según la población
de referencia y a que la frecuencia de aparición de mutaciones en la población es muy baja
(menor al 1%). A su vez el fenotipo observado en pacientes con mutaciones en el gen IGF1 es
muy marcado, presentándose con RCIU, retardo mental severo, sordera y microcefalia
(Walenkamp et al 2005; Netchine et al 2009).

56
Tabla 5. Características clínicas, al nacer, en pacientes portadores de variantes en el gen IGF1R.
Paciente, sexo
EG semanas
Peso Kg, (SDS)
Talla cm, (SDS)
PC cm, (SDS)
Cariotipo Alteración molecular
Fenotipos asociados
P1,
M
37
1.9, (-3.76)
42, (-4.39)
n.d.
46,XY
de novo p.Arg1256Ser
SGA, cara triangular, orejas pequeñas, clinodactilia, fhiltrum alargado.
P2,
M
38
2.37 (-2.33)
48, (-1.1)
n.d.
46,XY
de novo p.Asn359Tyr
SGA
P3,
F
38.5
2.23 (-2.49)
43, (-3.46)
31, (-2.6)
46,XX
Familial p.Tyr865Cys
SGA
P4,
F
38
2.65 (-1.32)
44, (-2,91)
30 (-3.0)
46,XX
Familial p.Arg1337Cys
SGA. Síndrome de Klippel Fell, braquicefalia, hipertelorimso, cuello corto
P4s,
F
38
3.3, (0,23)
49, (-0.16)
35, (0.5)
n.d.
Familial p.Arg1337Cys
AGA
EG: Edad gestacional, SDS: score de desvío standar, PC: perímetro cefálico, M: masculino, F: femenino, P4s, hermana
de P4, SGA: pequeño para la edad gestacional, AGA: adecuado para la edad gestacional, n.d: no disponible.
Tabla 6. Parámetros auxológicos, eje GH/IGF-1 y desarrollo mental en sujetos portadores de alteraciones
en el gen IGF1R
Sujeto, Seguimiento
EC a
EO a
Talla cm, (SDS)
Peso Kg, (SDS)
PC cm, (SDS)
GH basal ng/ml
IGF-1 ng/ml (SDS)
IGFBP3 mg/L (SDS)
Desarrollo Mental
P1 (M)
Admisión Exam. rhGH In. RhGH Res.
1.5 2.8 4.5 6.4
-
1.4 3.0 4.0
74 (-3.0) 86 (-2.1)
97 (-1.7) 109 (-1.5)
7.7 (-3.1) 9.8 (-3.1) 12.6(-2.7) 16.1(-2.2)
42.5 (-4.0) 44.8 (-3.5) 46.2 (-3.0) 47.0 (-3.0)
8.0
231(+2.9) 261(+1.7)
5.2 (+3.4) 5.2 (+2.3)
Retardo moderado
P2 (M)
Admisión rhGH Ons. RhGH Res.
2.8 5.0 7.0
-
3.0 6.0
81 (-3.6) 94 (-3.0) 109.5(-2.1)
10.5 (-2.5) 14.0 (-2.2)
17.8 (-1.87)
43.8 (-4.6) 47.0 (-2.5) 48.8 (-2.1)
0.27
58 (+0.5) 192(+0.8)
2.6(-0.2) 4.5(+0.7)
Normal
P3 (F) Adm
Ult Exam. 1.9 5.0
1.5 5.5
78 (-2.4)
99 (-1.6)
7.9 (-3.0) 12.3 (-3.4)
43.0 (-4.0) 45.6 (-3.0)
1.18 1.04
211(+1.7) 306(+1.9)
4.7 (+1.9) 4.8 (+1.6)
Normal
P3 madre 35 - 161(+0.0) 52.2 (-2.1) Normal
P4 (F)
Adm. Ult Exam.
3.2 4.8
1.6 n.d
84 (-2.9) 94.1(-2.2)
10.4 (-3.0) 13.8 (-2.4)
46.0 (-2.3) 46.8 (-2.6)
8.51
110(+0.4) 152(+0.7)
3.5 (+0.6) 3.2 (+0.3)
Retardo moderado
P4s 6.2 - 114(+0.0) 52.6 (+1.0) 0.45 123 (0.0) 3.1 (-0.1) Normal
P4p 34 - 177(+0.6) 56.0 (0) Normal
EC: Edad cronológica, EO: edad ósea, a: años, P: paciente, PC: perímetro cefálico, Exam.: examen clínico en el
seguimiento, rhGH In.: inicio del tratamiento con rhGH, Res.: respuesta a rhGH luego de dos años de trtamiento, Ult
exam: último examen clínico, M: masculino, F: femenino, P4s: hermana de P4, P4p: padre de P4, n.d: no disponible.

57
6.3.2 Análisis molecular
Mediante el análisis de secuenciación directa del ADN genómico y ADN codificante de los
pacientes con sospecha de resistencia a IGF-1, identificamos 4 nuevas variantes
heterociogotas en el gen IGF1R. Hallamos dos variantes de novo en los pacientes P1 y P2
mientras que las variantes encontradas en los pacientes P3 y P4 también estuvieron presentes
en al menos un familiar directo.
El estudio reveló en P1, una alteración en el exón 21 del gen IGF1R, una substitución de
guanina por timina en la posición 3768 del ADN codificante, que se traduce en un cambio de
Arginina por Serina en el codon 1256 (p.Arg1256Ser). La variante afecta el dominio tirosina
quinasa del receptor de IGFs tipo 1. En P2 identificamos una variante a nivel del exón 4, una
substitución de adenina por timina en la posición 1075 del ADN codificante que genera un
cambio aminoácidico en la cadena alfa del receptor, en el domino rico en leucina tipo 2 (L2). La
alteración resulta en un cambio de Asparagina por Tirosina en el codon 359 de la proteína,
p.Asn359Tyr. Por otro lado, en P3 detectamos una substitución de adenina por guanina en el
exon 12, en la posición 2594 del ADN codificante. La alteración genera un cambio de Tirosina
por Cisteína en el codón 865 de la proteína (p.Tyr865Cys) que afecta el dominio fibronectina
tipo III-3 del IGF1R. El análisis del ADN genómico de los padres del paciente P3 mostró la
presencia de la variante, en estado heterocigota, en la madre. Por último, en P4 hallamos una
alteración en la posición 4009 del ADN codificante, una substitución nucleotídica de citosina por
timina que se manifiesta como un cambio de Arginina por Cisteína en el codón 1337
(p.Arg1337Cys) de la proteína, en el dominio carboxilo terminal del IGF1R. El estudio de los
familiares de P4 (padres y 4 hermanos) reveló la presencia de la variante en estado
heterocigota tanto en el padre como en una hermana (P4s).
En base a que las variantes encontradas no se encuentran reportadas en la literatura nos
propusimos determinar si se encontraban presentes en la población general. Se estudiaron 60
sujetos controles (120 alelos) por secuenciación directa del ADN y no se encontró ninguna de
las variantes mencionadas, lo que sugiere que las alteraciones halladas en nuestros pacientes
no serían polimorfismos comunes. A su vez, ningunas de las variantes fueron halladas en las
bases de datos poblacionales disponibles online (www.ncbi.nim.gov/SNP; www.ensembl.org;
www.exact.bradinstitute.org).
El siguiente paso fue analizar si los aminoácidos afectados por la presencia de las variantes se
encuentran altamente conservados entre las distintas especies. Los ensayos in silico de
alineamiento de secuencia demostraron que los cuatro aminoácidos son residuos altamente
conservados en el receptor de IGFs tipo 1 entre las distintas especies (figura 20, panel C), de
tal manera que un cambio de los mismos afectaría de manera deletérea la actividad de la
proteína.
Se aplicaron ensayos in silico para predecir el posible efecto funcional de las variantes sobre la
actividad del IGF1R arrojando resultados variables entre cada uno de ellos. Polyphen2 y
Mutation Taster indican que las 4 variantes encontradas afectarían la función de la proteína y

58
serían causa de desarrollo de la patología, respectivamente. Por otro lado la herramienta SIFT
demostró que las alteraciones p.Arg1256Ser y p.Tyr865Cys también afectarían la normal
actividad de la proteína, con el mayor índice de score posible, mientras que los cambios
p.Asn359Tyr y p.Arg1337Cys serían tolerados y no afectarían las funciones del receptor.
6.3.3 Ensayos funcionales
Para determinar los efectos funcionales de las variantes encontradas sobre la actividad del
receptor de IGFs tipo 1, se realizaron dos ensayos in vitro a partir de cultivo primario de
fibroblastos obtenidos de muestras de piel abdominal de los pacientes afectados y de dos
sujetos controles. La hermana de P4 fue incluida en los análisis debido a que es portadora de
la variante y no posee retardo de crecimiento.
A) Síntesis de ADN en cultivo primario de fibroblastos estimulados con IGF-1.
El ensayo se basa en medir el incremento en la incorporación de timidina tritiada (3[H]-timidina)
al ADN de las células en cultivo, luego del estímulo con IGF-1, con respecto a la condición
basal. La proliferación de los fibroblastos en cultivo alcanzan un pico a las 20 hs de estímulo y
es proporcional a la incorporación de 3[H]-timidina al ADN en cada ciclo de división celular, de
tal manera que a partir de la medida de radioactividad de cada cultivo podemos inferir si las
variantes presentes en las células de los pacientes afectados alteran la respuesta del IGF1R a
su ligando (Kawashima, Y. et al 2005). Se determinó la síntesis de ADN en cultivo primario de
fibroblastos de P1, P2, P3, P4 y P4s y en dos sujetos controles (C1 y C2). La incorporación de
3[H]-timidina fue medida luego del tratamiento de los cultivos con IGF-1 (50 ng/ml) durante 16,
20 y 24hs. Se observó que luego de 20 horas de estímulo con IGF-1, la síntesis de ADN se
induce de manera significativa en C1 y C2 con respecto a la condición 16 y 24 hs (5.15 ± 0.67 y
6.37 ± 1.00 veces de incremento con respecto al basal, respectivamente, p<0.05, Test ANOVA
y Bonferroni). Sin embargo, no observamos diferencias significativas entre los tres tiempos de
estímulo analizados en P1, P2 y P3. A su vez, los fibroblastos de P4 y P4s tampoco mostraron
diferencias significativas en la síntesis de ADN luego de los tiempos de estímulo analizados, sin
embargo se observó que la respuesta al IGF-1 fue mayor que P1, P2 y P3, pero las diferencias
no son estadísticamente significativas.

59
Figura 21. Síntesis de ADN bajo estímulo con IGF-1 en cultivo primario de fibroblastos. La síntesis de ADN de los
pacientes afectados y dos controles normales fue determinada mediante el ensayo de incorporación de 3[H]-timidina. El
ensayo se realizó luego de 16, 20 y 24 horas de estímulo con IGF-1 (cada condición fue realizada por sextuplicado). Se
observa un aumento significativo en la incorporación de 3[H]-timidina en los sujetos controles luego de 20 horas de
estímulo con respecto a la condición 16 y 24 hs (5.15 ± 0.67 y 6.37 ± 1.00 veces de incremento con respecto al basal,
respectivamente, p<0.05, Tests ANOVA y Bonferroni). No se observa diferencias significativas en la incorporación de
3[H]-timidina en los pacientes afectados luego de los tres tiempos tiempos de estímulo.
B) Fosforilación de Akt bajo el estímulo de IGF-1.
El ensayo se basa en determinar el efecto del estímulo de IGF-1 sobre la fosforilación de Akt, el
cual es el último mediador en el camino de señalización PI3K/Akt relacionado con el IGF1R y el
principal efector en las funciones implicadas en el crecimiento y desarrollo (Labarta, J.I. et al
2013). El Akt fosforilado fue medido a partir del kit de Elisa phospo-Akt (Ser473) STAR ELISA y
la concentración proteica fue determinada por la técnica de Bradford. Un total de 80.000
células/well, de los pacientes afectados y dos sujetos controles, fueron incubadas en placas de
24 well y estimuladas con 50 ng/ml de IGF-1 durante 10 minutos y se determinó la
concentración proteica y la fosforilación de Akt en estado basal y luego del estímulo. Se
observó un aumento significativo en la fosforilación de Akt en los sujetos controles y en P4 y
P4s, luego del estímulo con IGF-1 con respecto a la condición basal (p<0.05 vs “0” dosis, Tests
de ANOVA y Bonferroni) mientras que no observamos un cambio significativo en ninguno de
los otros tres pacientes afectados. Estos resultados sugieren que la variante hallada en P4 no
sería deletérea para la actividad funcional del receptor de IGFs tipo 1.
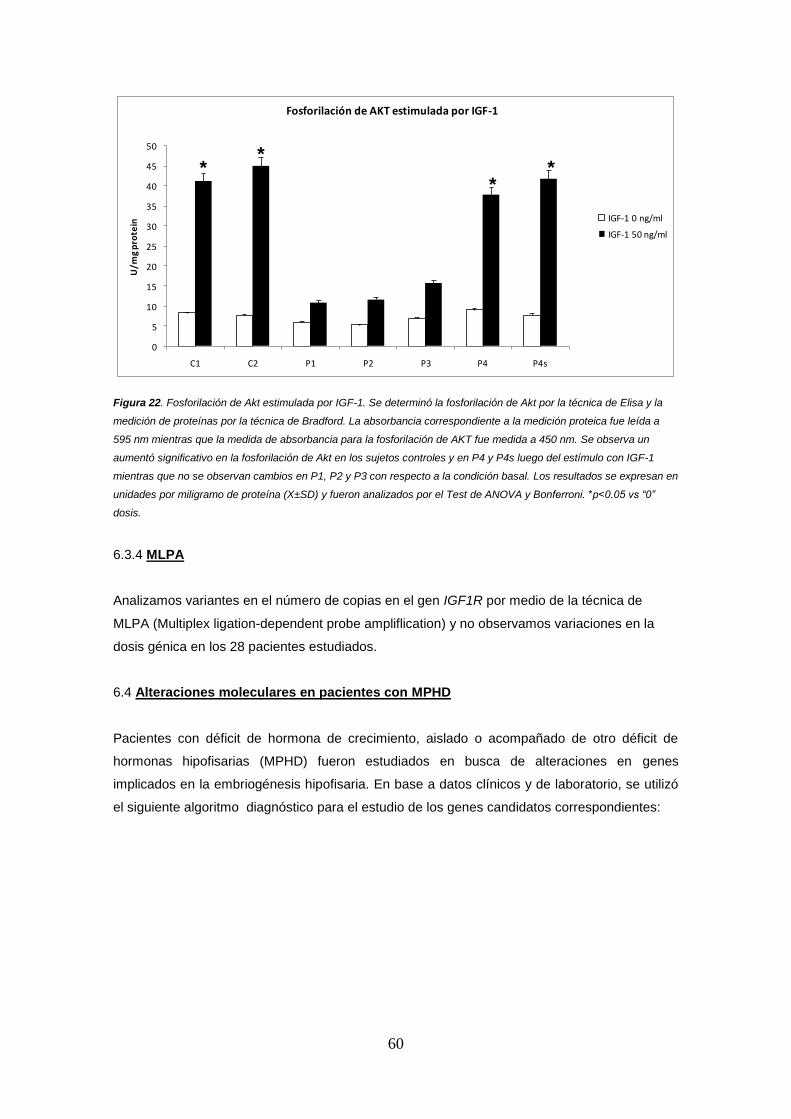
60
Figura 22. Fosforilación de Akt estimulada por IGF-1. Se determinó la fosforilación de Akt por la técnica de Elisa y la
medición de proteínas por la técnica de Bradford. La absorbancia correspondiente a la medición proteica fue leída a
595 nm mientras que la medida de absorbancia para la fosforilación de AKT fue medida a 450 nm. Se observa un
aumentó significativo en la fosforilación de Akt en los sujetos controles y en P4 y P4s luego del estímulo con IGF-1
mientras que no se observan cambios en P1, P2 y P3 con respecto a la condición basal. Los resultados se expresan en
unidades por miligramo de proteína (X±SD) y fueron analizados por el Test de ANOVA y Bonferroni. *p<0.05 vs “0”
dosis.
6.3.4 MLPA
Analizamos variantes en el número de copias en el gen IGF1R por medio de la técnica de
MLPA (Multiplex ligation-dependent probe ampliflication) y no observamos variaciones en la
dosis génica en los 28 pacientes estudiados.
6.4 Alteraciones moleculares en pacientes con MPHD
Pacientes con déficit de hormona de crecimiento, aislado o acompañado de otro déficit de
hormonas hipofisarias (MPHD) fueron estudiados en busca de alteraciones en genes
implicados en la embriogénesis hipofisaria. En base a datos clínicos y de laboratorio, se utilizó
el siguiente algoritmo diagnóstico para el estudio de los genes candidatos correspondientes:
0
5
10
15
20
25
30
35
40
45
50
C1 C2 P1 P2 P3 P4 P4s
U/m
g p
rote
in
Fosforilación de AKT estimulada por IGF-1
IGF-1 0 ng/ml
IGF-1 50 ng/ml
* *
* *

61
SW Davis et al, Molecular Cell Endocrinology, Julio 2010
Figura 23. Algoritmo diagnóstico para el estudio de los genes implicados en la embriogénesis hipofisaria.
Analizamos una cohorte de 47 pacientes, de los cuales se seleccionaron 18 para el estudio del
gen PROP1, 12 para el estudio del gen POU1F1, 11 para el estudio del gen HESX1, 2 para el
estudio de LHX3, LHX4 y OTX2. En ninguno de los casos analizados se encontraron
alteraciones implicadas en la patología que desarrollan los pacientes. Por otro lado, estudiamos
alteraciones en el gen GLI2, en 18 pacientes con IGHD o MHPD asociadas con alteraciones de
la neurohipófisis. Identificamos dos nuevas variantes heterocigotas y dos variantes
homocigotas en dos pacientes no relacionados. En el paciente 1 (P1) encontramos la variante
p.Arg231Gln en estado heterocigota mientras que en el paciente 2 (P2) detectamos la variante
p.Arg226Leu, en estado heterocigota, y las variantes p.Met1444Ile y p.Leu1445Phe, en estado
homocigota previamente reportadas en la literatura como variantes polimórficas sin efecto
funcional sobre Gli2 (Bear KA et al 2014).
Características Clínicas
Déficit Hormonal
Tamaño
Adenohipofisario
Neurohipófisis
Características
Sindrómicasasociadas
Gen candidato
GH, TSH, PRL
LH, FSH
± ACTH
GH, TSH, PRL
LH, FSH
ACTH
IGHD
O
GH, TSH, PRL
LH, FSH, ± ACTH
GH, TSH, PRL
± LH, FSH
ACTH
GH, TSH, PRL
LH, FSH
± ACTH
GH, TSH, PRL
Hipoplásica Hipoplásica Hipoplásica oNormal
Hipo/hiperplásicao normal
Hipo/hiperplásica
o normal
Hipo/hiperplásica
o normal
Hipoplásica o
Normal
Ectópica Normal oectópica
Normal o
ectópica
Normal o
ectópicaEctópica Ectópica Ectópica
Holoprosencefalia,
Cleft Lip,Incisivo central,
polidactilia
Malformaciones del
Cuerpo calloso,Hipocampo y el ojo.
Clinodactilia
DisplasiaSepto optica (DSO),
Coloboma,Polidactilia
Malformación
de Chiari
Rigidez del cuello,
Alteración de la audición
neurosensorial
Ninguna Ninguna
GLI2 OTX2 HESX1 LHX4 LHX3 PROP1 POU1F1
Características Clínicas
Déficit Hormonal
Tamaño
Adenohipofisario
Neurohipófisis
Características
Sindrómicasasociadas
Gen candidato
GH, TSH, PRL
LH, FSH
± ACTH
GH, TSH, PRL
LH, FSH
ACTH
IGHD
O
GH, TSH, PRL
LH, FSH, ± ACTH
GH, TSH, PRL
± LH, FSH
ACTH
GH, TSH, PRL
LH, FSH
± ACTH
GH, TSH, PRL
Hipoplásica Hipoplásica Hipoplásica oNormal
Hipo/hiperplásicao normal
Hipo/hiperplásica
o normal
Hipo/hiperplásica
o normal
Hipoplásica o
Normal
Ectópica Normal oectópica
Normal o
ectópica
Normal o
ectópicaEctópica Ectópica Ectópica
Holoprosencefalia,
Cleft Lip,Incisivo central,
polidactilia
Malformaciones del
Cuerpo calloso,Hipocampo y el ojo.
Clinodactilia
DisplasiaSepto optica (DSO),
Coloboma,Polidactilia
Malformación
de Chiari
Rigidez del cuello,
Alteración de la audición
neurosensorial
Ninguna Ninguna
GLI2 OTX2 HESX1 LHX4 LHX3 PROP1 POU1F1

62
6.4.1 Evaluación clínica de pacientes afectados por alteraciones en el gen GLI2
Estudiamos 18 pacientes con déficit de hormonas hipofisarias, de los cuales 16 presentaron
MPHD y 2 IGHD, acompañado de alteraciones de la neurohipófisis y se identificaron 2
pacientes con alteraciones en el gen GLI2. Las características clínicas y parámetros
auxológicos de los pacientes estudiados se detallan en la tabla 7 y 8.
P1 es el segundo hijo de una pareja no consanguínea. Nacido a término con un peso de 3.18
kg. Concurre al servicio de Endocrinología del Hospital Garrahan a causa de talla baja a los
dos años de edad. En la primer consulta se observó un retardo severo de crecimiento (talla -3.5
SDS), retardo en la edad ósea (1.24 años), microcefalia (perímetro cefálico -2.5 SDS), frente
amplia y prominente, puente nasal chato, fosas nasales hipoplásicas, hipotelorismo, asimetría
facial moderada, estrabismo del ojo izquierdo, retardo moderado en el desarrollo neuronal en el
área del lenguaje, cariotipo y parámetros bioquímicos de rutina normales. La resonancia
magnética nuclear (RMN) mostró una hipófisis anterior hipoplásica y neurohipófisis ectópica. El
estudio hormonal reveló niveles bajos de GH-1 sérica, basal y bajo estímulo, junto con niveles
séricos de IGF-1 e IGFBP-3 por debajo de los valores de referencia. Los niveles de cortisol,
prolactina y hormonas tiroideas se encontraron dentro de los parámetros normales para la edad
y el sexo para la población de referencia, confirmando el IGHD.
El P2 fue el quinto hijo de una pareja no consanguínea. Nacido a término con un peso de 3.48
kg. Concurre al servicio de Endocrinología del hospital a los 8 meses de edad. Durante el
período neonatal presentó alteraciones respiratorias que requirieron intubación, hipoglucemias
severas y convulsiones. A su vez las características clínicas de mayor relevancia fueron:
microcefalia (perímetro cefálico -2 SDS), micropene, testículos sin descender y retardo
moderado del desarrollo neuronal. La RMN reveló una silla turca pequeña con una
adenohipófisis hipoplásica y ausencia de neurohipófisis. Los estudios hormonales de
laboratorio revelaron un déficit múltiple de hormonas hipofisarias (GH, ACTH y TSH); niveles
bajos de hormonas tiroideas e inadecuados niveles de TSH sérica confirmaron el diagnóstico
de hipotiroidismo central. La respuesta inadecuada del cortisol sérico a la hipoglucemia
confirmó el diagnóstico de hipocortisolismo central mientras que la pérdida de respuesta a los
test de estímulos farmacológicos confirmo el diagnóstico de déficit de GH.
Cabe destacar que P1 y P2, a diferencia del resto de los pacientes, presentaron microcefalia.

63
TABLA 7. Características Clínicas y estudios hormonales de los pacientes analizados
Pacientes SexoEdad
(años)
Talla
(SDS)
Peso
(SDS)
IMC SDS
(kg/m2)
Resp.
Max.GH
(ng/ml)
Deficiencia
Hormonal
Hipófisis
posterior
RMN
Hipófisis
anterior
RMN
Tallo
hipofisarioInformación Clínica Variantes
P1 F 2 -3,5 -2,14 0,06 2,57 GH Ectópica Hipoplásica Ausente
microcefalia, frente amplia y
prominente, puente nasal chato, fosas
nasales hiploplásicas, hipotelorismo,
asimetría facial, estrabismo ojo
izquierdo, retardo neuromadurativo
moderado
p.Arg231Gln(ht)
P2 M 0,64 -1,99 -0,7 0,36 0,75 GH, TSH, ACTH Ausente Hipoplásica Ausente
microcefalia, micropene, testículo sin
descender, retardo neuromadurativo,
hipoglucemia.
p.Arg226Gln(ht);
p.Met1444Ile(hm)
p.Leu1445Phe(hm)
P3 F 2,16 -4,34 -1,38 - 0,89 GH, TSH, ACTH Ectópica Hipoplásica Ausentefrente amplia y prominente, puente
nasal chato.N
P4 F 2,64 -3,29 -0,19 0,26 0,1 GH, TSH, ACTH Ectópica Hipoplásica Ausente retardo neuromadurativo moderado N
P5 M 0,026 ND 0,84 ND 3,49GH, TSH, ACTH,
LH/FSHEctópica Hipoplásica Ausente micropene, hipoglucemia. N
P6 F 0,56 -2 -0,52 0,93 1,09 GH,TSH, ACTH Ectópica Hipoplásica Ausente Hipoglucemia. N
P7 M 1,24 -6,1 -4,53 -2,02 0,38 GH,TSH,LH/FSH* Ausente Hipoplásica Ausentemicropene, frente amplia y prominente,
puente nasal chato.N
P8 F 1,4 -4,44 -5,3 -1,16 0,15 GH,TSH Ectópica Hipoplásica - Escoliosis N
P9 F 6 -3,9 -1,17 2 0,55 GH,TSH, ACTH Ectópica Hipoplásica Ausentemicropene, frente amplia y prominente,
puente nasal chato,voz afinada.N
P10 F 0,16 -4,66 -3,42 -3,17 0,44 GH,TSH, ACTH Ausente Ausente Truncado Hipoglucemia N
P11 M 12,72 -3,92 -3,43 -1,87 0,33GH,TSH, ACTH,
LH/FSHEctópica Hipoplásica - Micropene N
P12 M 10,2 -2,74 -2,26 -0,32 1,4 GH,TSH Ectópica Hipoplásica Ausente N
P13 M 12,16 -3,12 -2,68 -0,97 2,42 GH,TSH Ectópica Hipoplásica - puente nasal chato N
P14 M 6,5 -4,27 -2,27 0,86 0,35 GH Ectópica Ausente - N
P15 F 5,4 -1,53 -1,98 -1,19 4,18 TSH,GH Ectópica Hipoplásica - N
P16 F 7,4 -6,54 -4,72 -1,98 O,70 GH,TSH Ectópica Hipoplásica Truncado
Malformación de Chiari, frente amplia y
prominente, puente nasal chato,
estrabismo convergente.
N
P17 M 0,16 -0,45 -1,13 -1,73 6 GH,TSH; ACTH Ectópica Hipoplásica AusenteHipoglucemia, micropene, retardo
neuromadurativo.N
P18 M 7 -2,72 -1,59 -0,73 3,19 GH,TSH Ectópica Hipoplásica - N
M, masculino; F, femenino; IMC, índice de masa corporal; RMN, resonancia magnética nuclear; N, normal; ND, no disponible available. * 15 años de edad con niveles indetectables de LH and FSH.
Tabla 8. Características clínicas y estudios hormonales en los pacientes afectados
Paciente P1 P2
Sexo F M
Edad (años) 2 0,64
Talla (SDS) -3,5 -1,99
Peso (SDS) -2,14 -0,7
Talla del Padre 0,76 -1.51
Talla de la Madre -1,75 -1.34
Talla objetivo genética -0,48 -1.33
Perímetro cefálico (SDS) -2.5 -2.0
Pico de GH (ng/ml) 2.57 0,75
IGF-1 (SDS) -4,2 -4
IGFBP3 (SDS) -2,09 ND
Deficiencias hormonales GH GH,TSH,ACTH
TSH µUI/ml (RV:0.84-4.31) 3,98 3,84
T4 µg/dl (RV:5.3-14.3) 8 4,6
FT4 ng/dl (RV: 0.86-1.9) 1,14 0,36
T3 ng/ml (VN 1.3-2.3) 1,39 1,88
Cortisol µg/dl RV:>18 bajo hipoglucemia ND 3,4
Cortisol Basal : 5-25µg/dl 10,8 2,3
Variantes p.Arg231Gln(ht) p.Arg226Gln(ht)
p.Met1444Ile(hm) p.Leu1445Phe(hm)
F: femenino, M: masculino, ND: no disponible; ht: heterocigota; hm: homocigota.

64
6.4.2 Análisis Molecular
El análisis de la secuencia nucleotídica, correspondiente al gen GLI2, en P1 reveló la presencia
de una variante heterocigota a nivel del exón 5, una substitución de guanina por adenina en la
posición 692, que genera un cambio de Arginina por Glutamina en el codon 231 (p.Arg231Gln)
en el dominio represor de la proteína. La variante también fue hallada en la madre de P1 en
estado heterocigota. Con respecto a P2, también encontramos una variante heterocigota a
nivel del exón 5 del gen GLI2. Esta alteración involucra un cambio de guanina por timina en la
posición nucleotídica 677, que se traduce en una substitución de Arginina por Leucina en el
codón 226 (p.Arg226Leu) del dominio represor de la molécula, que a su vez fue identificada en
el padre de P2. Además, detectamos dos variantes homocigotas en el dominio activante de la
proteína Gli2, p.Met1444Ile y p.Leu1445Phe, previamente reportadas en la literatura como
variantes polimórficas sin efecto funcional sobre la actividad de Gli2 (Bear KA et al 2014).
Para determinar si las variantes no reportadas en la literatura están presentes en la población
general, se estudiaron 60 sujetos controles (120 alelos) por secuenciación directa del ADN y no
se encontró ninguna de las variantes mencionadas, lo que sugiere que las alteraciones
halladas en nuestros pacientes no serían polimorfismos comunes. A su vez, ambas variantes
no se encuentran reportadas en las bases de datos poblacionales disponibles online
(www.ncbi.nim.gov/SNP; www.ensembl.org; www.exact.broadinstitute.org).
El siguiente paso fue analizar si los aminoácidos afectados se encuentran altamente
conservados entre las distintas especies. Los ensayos in silico de alineamiento de secuencia
demostraron que los dos aminoácidos afectados son residuos altamente conservados en la
proteína Gli2 (figura 24), sugiriendo que un cambio de los mismos podría ser deletéreo para la
actividad de la proteína.
Por último, se aplicaron ensayos in silico para predecir los posibles efectos funcionales de las
variantes sobre la actividad del factor de transcripción Gli2. Polyphen2, SIFT y Mutation Taster
indican que las variantes encontradas afectarían la funcionalidad de la proteína con el mayor
índice de score y serían la causa del fenotipo observado en los pacientes afectados.

65
Especies Paciente 2 Paciente 1
Humano V S R F S S P R V T P R L S R K R A L S I S P L S P R V T P R L S R K R A L S I S P L S D A S L
Mutado V S R F S S P R V T P L L S R K R R A L S I S S P R V T P R L S R K Q A L S I S P L S D
Ptroglodytes V S R F S S P R V T P R L S R K R A L S I S P S P R V T P R L S R K R A L S I S P L S
Mmulatta V S R F S S P R V T P R L S R K R A L S I S P S P R V T P R L S R K R A L S I S P L S
Mmusculus A S R F S S P R V T P R L S R K R A L S I S P S P R V T P R L S R K R A L S I S P L S
Trubripes T P R L S R K R A L S I S P T P R L S R K R A L S I S P L S
Drerio M S R F P S P R L T P R V S R K R A L S I S P S P R L T P R V S R KR A L S I S P V
Dmelanogaster I RA S I S R K R A L S S S P R K R A L S S S P Y S D
Figura 24. Panel (i) Esquema de la proteína Gli2: RD, dominio represor; ZFD, dominio zinc finger; AD, dominio
activante. (ii) Cromatogramas de las nuevas variantes encontradas en el gen GLI2. Los electroferogramas muestran las
substituciones de Arginina por Leucina en la posición 226 (c.667 G>T) y Arginina por Glutamina en la posición 231
(c.692 G>A) de la proteína en P2 y P1, respectivamente. (iii) Conservación evolutiva de los aminoácidos afectados. La
figura revela que ambos aminoácidos se encuentra altamente conservados en la secuencia proteíca de Gli2 entre las
distintas especies.
CN
473 594 1586
ZFDRD AD
C G C C T C
C G GC AG
i.
ii.
iii.
CN
473 594 1586
ZFDRD ADZFDZFDRDRD ADAD
C G C C T CC G C C T CC G C C T C
C G GC AG C G GC AG
i.
ii.
iii.

66
7. Discusión
La presente tesis doctoral fue diseñada con el fin de identificar alteraciones moleculares en el
eje Hipotálamo-Hipofiso-IGF-1, en pacientes pediátricos argentinos con déficit de crecimiento,
debido a la escasa información clínica y de laboratorio disponible en nuestra población de tal
manera de poder establecer una correcta y eficaz estrategia terapéutica y asesoramiento
genético.
Dentro de la pediatría, la estatura baja es una preocupación común, que cuando es severa
puede ser un indicador de una patología subyacente, además de ser invalidante. Se han
reportado una gran variedad de genes implicados con alteraciones del eje de crecimiento y aún
no se ha podido establecer la real prevalencia de estas alteraciones génicas en nuestra
población. Para resolver esta incógnita estudiamos cinco grupos de pacientes con alteraciones
en el eje GH-IGF-1 los cuales fueron seleccionados en base a datos auxológicos, de
laboratorio y a la evaluación funcional del eje hipotálamo-hipofisario. Siguiendo los algoritmos
diagnósticos detallados en materiales y métodos analizamos la presencia de alteraciones en el
gen GH1 en 46 pacientes con déficit aislado de hormona de crecimiento (41 familias), de los
cuales 12, que no presentaron anomalías, fueron estudiados en busca de mutaciones en el gen
GHRHR. A la fecha, al menos 60 mutaciones en el gen GH1 han sido reportadas asociadas a
IGHD, incluyendo mutaciones que alterar el splicing normal, mutaciones con cambio de sentido
y grandes deleciones (Mullis P et al 2005; Wit JM et al 2010). En nuestra cohorte identificamos
mutaciones en el gen GH1 (p.Arg183Hys, p.Val110Ile y la deleción de 6.7Kb), previamente
reportadas como causa de IGHD, en 9 de 41 familias estudiadas (22%), con una alta
prevalencia de la variante p.Arg183Hys (6 de 9 familias). De acuerdo a lo descripto por Wit et al,
este porcentaje se incrementa cuando solo se consideran los casos familiares, en donde más
de un individuo de la misma familia se ve afectado (8 de 22 familias, 36.4%). A su vez,
detectamos 3 nuevas variantes heterocigotas, en 3 pacientes no relacionados, que producen
un cambio aminoacídico en la secuencia codificante del gen GH1 (p.Pro-7Ser, p.Arh19Hys y
p.Leu102Pro). El impacto funcional de las variantes fue estimado de acuerdo a: 1) la presencia
de las variantes en las bases de datos poblacionales disponibles online, 2) el análisis de la
conservación evolutiva de los aminoácidos involucrados, 3) la aplicación de herramientas de
predicción in silico. En base a estos análisis, se puede concluir que únicamente la variante
p.Leu102Pro sería deletérea, ya que el aminoácido Leucina en la posición 102 está altamente
conservado entre las distintas especies y los ensayos de predicción in silico indican que la
variante afectaría la funcionaliadad de la proteína y sería causa de enfermedad.
Interesantemente, el padre del paciente afectado es portador de esta nueva variante y presenta
una talla extremadamente baja (-5.12 SDS), lo que refuerza la hipótesis de que la variante
p.Leu102Pro codificaría para una proteína inactiva, causando un severo retardo de crecimiento
postnatal tanto en el paciente como en el padre.
Al igual que lo reportado en otras poblaciones, el tipo de IGHD más frecuentemente hallado fue
el tipo II. Sin embargo, a diferencia de otras poblaciones donde en este tipo de déficit

67
prevalecen las variantes que producen un splicing anómalo del ARNm (Wit JM et al 2010), en
nuestra población predominan las mutaciones con cambio de sentido, principalmente la
variante p.Arg183Hys. A su vez cabe destacar que el tipo de herencia dominante del IGHD tipo
II facilita la detección de pacientes con alteraciones en el gen GH1 debido a que, en la mayoría
de los casos, existe un familiar directo afectado con retardo de crecimiento postnatal que
orienta a la búsqueda de un desorden genético. Con respecto a alteraciones hipofisarias en
este tipo de patología Alatzoglou et al en el año 2009 observaron la presencia de hipoplasia
hipofisaria únicamente en 2 pacientes pertenecientes a una misma familia con mutación en el
gen GH1, lo que indica que este tipo de anormalidades son claramente inusuales en pacientes
con IGHD.
El estudio del gen GHRHR, de manera similar a lo reportado en población caucásica, no reveló
alteraciones que se relacionen con la patología observada en los pacientes, de tal manera que
sólo es recomendable la búsqueda de mutaciones en este gen cuando existe evidencia de
descendencia étnica (Wajnrajch MP et al 1996; Salvatori R, Levine MA. 1999).
En conclusión, en el grupo de pacientes con IGHD reportamos que la mutación p.Arg183Hys,
asociada a la forma dominante tipo II, es relativamente común en nuestra población. A su vez,
de las tres nuevas variantes encontradas sólo la variante p.Leu102Pro es considerada como
una mutación que afecta la funcionalidad de GH-1, sugiriendo que otros factores pueden estar
implicados en la etiología del IGHD en estos pacientes.
Siguiendo con el análisis de los pacientes con alteraciones en el eje GH-IGF-1, estudiamos una
cohorte de 31 pacientes pediátricos, no consanguíneos, con sospecha de insensibilidad a la
hormona de crecimiento en búsqueda de alteraciones en el gen GHR. Identificamos 4 variantes
en 5 pacientes no relacionados, de las cuales sólo la variante p.Cys66* no se encuentra
reportada previamente en la literatura. Esta variante genera la aparición de un codón de
terminación prematuro durante la síntesis del GHR, produciendo la pérdida completa de los
dominios transmembrana e intracelular y de una porción del dominio extracelular del receptor.
Este fenómeno produce una incapacidad de respuesta a la GH-1 en las células que expresan
el gen GHR, de tal manera que los factores que median sus efectos (IGF1, IGFBP3 y IGFALS)
no se sintetizan. Esta variante, a diferencia de las otras 3, fue encontrada en estado
homocigota, lo cual concuerda con el modo de herencia de la patología y con la localización de
las mutaciones reportadas previamente, donde se observa que las alteraciones que afectan al
dominio extracelular del GHR son la principal causa de Síndrome de insensibilidad a la GH-1
(Laron Z 2004).
La segunda variante identificada fue el cambio p.Arg179Cys, en estado heterocigota, en dos
pacientes no relacionados. Esta variante fue previamente reportada por Amselum et al en 1993,
asociada a GHIS, insensibilidad parcial a la GH-1 y talla baja idiopática. Cabe mencionar, que
mutaciones heterocigotas en el gen GHR se han relacionado con casos de insensibilidad
parcial a la GH-1 (David A., et al 2011). En un primer momento se propuso el estudio del gen
GHR en casos de talla baja idiopática con la hipótesis de que mutaciones heterocigotas pueden
dar lugar a fenotipos intermedios no tan severos (Woods KA., et al. 1997; Wit J.M. et al 2011).
Este podría ser el caso de la variante p.Arg179Cys, la cual en estado homocigota se ha

68
demostrado que afecta la funcionalidad del GHR, dando lugar a GHIS. Es posible que, en
presencia de la variante en estado heterocigota, exista una disminución en el número de
moléculas normales del GHR en la membrana plasmática, de tal manera que la respuesta a la
GH-1 se encuentre parcialmente atenuada. Cabe destacar que esta variante se encuentra
reportada en el proyecto mil genomas y en las bases de datos poblacionales, con una
frecuencia mayor al 1% en la población Europea (rs121909362) por lo que es considerada
como una variante polimórfica con efecto deletéreo sobre el GHR confirmado por ensayos
funcionales (Amselum et al 1993, Meyer et al 2007).
La tercera variante identificada en el gen GHR fue el cambio p.Tyr240His, encontrada en una
familia (paciente y madre) en estado heterocigota. Esta variante fue previamente reportada por
Tauber et al en 1998, el cual describe a la variante pero no aporta información sobre el fenotipo
clínico de los pacientes afectados, de tal manera que no es posible confirmar su relación
directa con el GHIS. A su vez la variante se encuentra reportada en las bases de datos
poblacionales, con una frecuencia mayor al 1%, sin embargo los ensayos in silico de predicción
indican que la variante sería deletérea para la funcionalidad del receptor, no obstante son
necesarios ensayos funcionales que confirmen esta hipótesis.
Por último, identificamos una cuarta variante, p.Ala488Val, en estado heterocigota. Esta
variante se encuentra reportada con alta frecuencia en la población general y en múltiples
observaciones no se la ha relacionado con alteraciones que afecten la talla. Estos hallazgos
indican que la variante p.Ala488Val es altamente polimórfica, sin efecto funcional sobre el GHR
y no sería la causa del fenotipo de alteración de crecimiento observado en el paciente
estudiado.
Teniendo en cuenta que el GHIS es una patología caracterizada por la incapacidad de las
células de responder correctamente a la GH-1, defectos en la vía de señalización del GHR se
han relacionado con este tipo de patología. Kofoed et al en el 2003, describen por primera vez
la relación directa que existe entre alteraciones en los factores involucrados en el camino de
señalización del GHR y el GHIS, y observan que mutaciones en el gen que codifica para el
factor de transcripción STAT5b dan lugar a un fenotipo similar al GHIS que se acompaña, con
alta frecuencia, de alteración del sistema inmune. En base a estos resultados, nos propusimos
estudiar un grupo de pacientes con características clínicas de insensibilidad a la GH-1 y
alteración de la función inmunológica. Estudiamos 16 pacientes y no identificamos ninguna
variante con impacto funcional en el factor STAT5b. Estos resultados concuerdan con lo
descripto en la literatura donde, desde 2003 a la fecha, solo se han reportado 7 mutaciones en
el gen STAT5b (Hwa V et al 2011; David A et al 2011) asociadas a GHIS y alteración del sistema
inmune, lo que demuestra la baja frecuencia de aparición de este tipo de variantes. Las 7
mutaciones identificadas previamente tienen un modo de herencia autosómico recesivo, lo que
sugiere que la haploinsuficiencia del factor STAT5b tiene efectos mínimos sobre la expresión
de IGF-1 y el crecimiento. Sin embargo, a pesar de la baja frecuencia de aparición de
alteraciones en el gen STAT5b, es de vital importancia el estudio de este gen para la
identificación de pacientes con defectos en la cascada de señalización de GH-1, lo que

69
permitirá un mayor entendimiento de las bases moleculares implicadas en alteraciones de
crecimiento asociadas con deficiencia primaria de IGF-1.
Continuando río abajo en el estudio del eje GH-IGF-1, analizamos una cohorte de 28 pacientes
en busca de alteraciones en el gen IGF1R. Defectos en el gen IGF1R son considerados como
una causa infrecuente de retardo de crecimiento pre y postnatal, sin embargo no se conoce la
real prevalencia de alteraciones en este gen. Desde el primer paciente descripto por Abuzzahab
M et al en 2003, se han reportado varios individuos con alteraciones en el gen IGF1R, los
cuales muestran una amplia variabilidad bioquímica y fenotípica. Debido a esta amplia
variabilidad es necesario establecer un criterio de selección adecuado en base a las
características clínicas y de laboratorio más relevantes, de tal forma de incorporar al grupo de
estudio a los pacientes con mayor probabilidad de detectar un defecto molecular en el gen
IGF1R.
Se estima que la frecuencia de mutaciones en el gen IGF1R, en pacientes nacidos pequeños
para la edad gestacional, se encuentra entre el 2-4% (Leal AC. Et al 2013; Labarta JI et al 2013).
Sin embargo, los grupos de pacientes estudiados fueron seleccionados de acuerdo a diferentes
criterios de inclusión, que involucran diferentes grados de alteración de crecimiento y
parámetros bioquímicos (Klammt J et al 2011). En base a la información proveniente de distintos
reportes, donde se observa que existe una alta prevalencia de microcefalia en pacientes con
mutaciones en el gen IGF1R (Klammt J et al 2011), se ha incluido la medición del perímetro
cefálico (menor a -2 SDS) como un criterio de selección adicional al retardo de crecimiento pre
y postnatal que presentan los pacientes con sospecha de insensibilidad a IGF-1. Utilizando
este criterio identificamos 3 nuevas mutaciones como causa de la patología, en tres pacientes
no relacionados, de un total de 28 pacientes estudiados (10.7%). El aumento en la frecuencia
de aparición de alteraciones en el gen IGF1R en nuestra población refuerza la importancia de
determinar el perímetro cefálico en la evaluación de pacientes nacidos SGA con ausencia de
recuperación postnatal del crecimiento longitudinal.
Detectamos 4 variantes heterocigotas en el gen IGF1R que producen un cambio aminoacídico
a nivel proteico (p.Arg1256Ser, p.Asn359Tyr, p.Tyr865Cys y p.Arg1337Cys). Las variantes
p.Arg1256Ser (P1) y p.Asn359Tyr (P2) fueron variantes de novo mientras que las variantes
p.Tyr865Cys y p.Arg1337Cys (P4) fueron identificadas en al menos un familiar directo. Los
modelos de predicción in silico indican que los aminoácidos substituidos se encuentran en
regiones altamente conservadas de la proteína, de tal manera que un cambio de los mismos
probablemente afecte la función del receptor. Los ensayos funcionales realizados a partir de
cultivo primario de fibroblastos provenientes de biopsias de piel, muestran una disminución
significativa en la proliferación celular y en la fosforilación del traductor de señal Akt en P1, P2 y
P3, mientras que no se observan diferencias significativas en P4 luego de la estimulación con
IGF-1.
Las características fenotípicas de los pacientes y los resultados obtenidos de los análisis
moleculares y los ensayos funcionales, junto con la segregación de las variantes con el
fenotipo de alteración de crecimiento, sugieren que las alteraciones encontradas en los

70
pacientes P1, P2 y P3 afectan la función del receptor de IGFs tipo 1 y causan el fenotipo de
retardo de crecimiento pre y postnatal y microcefalia observado en los pacientes. Por otro lado,
a pesar de que la variante presente en el paciente P4 (p.Arg1337Cys) afecta un residuo
altamente conservado de la proteína, y no se encuentra reportada en las bases de datos
poblacionales, fue considerada como una variante benigna rara ya que no cosegrega con el
fenotipo de alteración de crecimiento presente en la familia (hermana y padre portan la
mutación y poseen talla normal). A su vez, los ensayos in vitro demuestran que la funcionalidad
del receptor se encuentra conservada.
La variante de novo p.Arg1256Ser afecta el dominio tirosina quinasa del receptor, de manera
similar a la mutación p.Gly1155Ala previamente reportada por Kruis et al. La unión del IGF-1 al
receptor lleva a la autofosforilación de residuos de tirosina en la región intracelular y a la
activación del dominio tirosina quinasa del IGF1R, que subsecuentemente fosforila distintos
componentes citoplasmáticos pertenecientes a los caminos de señalización PI3K/AKT y
MAPK/ERK (Le Roith et al 1995). En la presente tesis doctoral proponemos que la variante
p.Arg1256Cys afecta este camino de señalización llevando a la inhibición de la proliferación
celular. Por otro lado, la variante p.Asn359Tyr afecta el dominio L2 del IGF1R, que forma parte
de la subunidad α extracelular, involucrado en la unión del IGF-1 y la internalización del
receptor. En el año 2012 Kawashima et al describieron a un paciente Japonés con una
mutación en el dominio L2 del receptor (p.Arg413Leu) y demostraron que la variante afecta la
señalización y disminuye la internalización del IGF1R, lo que da lugar a la inhibición de la
proliferación celular inducida por IGF-1. De manera similar, la variante p.Asn359Tyr afecta el
camino de señalización vía Akt y, posiblemente, la internalización del IGF1R, resultando en la
inhibición de la proliferación celular que se traduce en el fenotipo clínico observado en nuestro
paciente. Por último, la variante p.Tyr865Cys, identificada en el P3 y su madre, se localiza en el
dominio extracelular fibronectina tipo III-3 de la subunidad β del receptor. Labarta et al, en el
2013, describieron una familia con una variante que afecta el dominio fibronectina tipo III-2a,
que se ubica en cercanías al primer puente disulfuro (Cys514) entre las dos subunidades α del
IGF1R maduro. Se demostró que esta variante afecta el proceso de dimerización del receptor y
produce una haploinsuficiencia del IGF1R. La variante p.Tyr865Cys afecta la funcionalidad del
receptor, ya que se observa que la vía de señalización PI3K/Akt se encuentra alterada y existe
una disminución en la proliferación celular, sin embargo no fue posible determinar el efecto
específico de la variante sobre el IGF1R.
La alta variabilidad fenotípica observada en los pacientes con alteraciones en el gen IGF1R
puede ser explicada a partir de los distintos efectos que causan las mutaciones sobre la
funcionalidad del receptor, dependiendo del dominio que se encuentre alterado. Cabe destacar
que se han observado distintas características fenotípicas dentro de una misma familia
afectada, por lo que es importante considerar el bagaje genético de cada paciente
individualmente. Por último, el seguimiento a largo plazo, así como el estudio de individuos
seleccionados con un criterio más laxo, es necesario para la evaluación del real impacto de las
mutaciones en el gen IGF1R en humanos.

71
Para finalizar, en la presente tesis doctoral se estudio la presencia de alteraciones moleculares
en distintos genes que codifican para factores de transcripción involucrados en la
embriogénesis hipofisaria y que dan lugar a déficit múltiple de hormonas hipofisarias. La
etiología del MPHD es desconocida en la mayoría de los casos estudiados hasta la fecha.
Mutaciones en el gen PROP1 son la causa genética más frecuente de MHPD pero con una
incidencia variable dependiendo de la población en estudio (Romero CJ et al 2011). A su vez se
han reportado alteraciones en otros genes (OTX2, HESX1, POU1F1, LHX3 y LHX4) que dan
lugar a MHPD acompañado de una amplia variedad de características fenotípicas,
dependiendo del gen afectado, las cuales fueron descriptas en la introducción. Siguiendo el
algoritmo diagnóstico esquematizado en la figura 23, seleccionamos 46 pacientes para el
estudio de los genes mencionados (18 pacientes PROP1; de estos 18 se seleccionaron 12
para POU1F1; 11 pacientes HESX1; 2 pacientes LHX3; 2 pacientes LHX4; 2 pacientes OTX2).
No identificamos ninguna variante relacionada con la patología de base presente en los
pacientes. Estos resultados concuerdan con la baja frecuencia de mutaciones reportadas en
los genes que codifican para los factores que participan en la embriogénesis hipofisaria (Davis
SW et al 2010; Romero CJ et al 2011). Se estima que la frecuencia de alteraciones en estos
genes es menor al 1% en pacientes con MHPD y aumenta cuando se analizan casos
familiares. En los últimos años distintos estudios han reportado variantes en el gen GLI2 como
una causa frecuente de MPHD (França MM et al 2013; Flemming GM et al 2013; Bear KA et al
2014). França et al han reportado mutaciones sin sentido y alteraciones no sinónimas en el gen
GLI2 en 27/207 (13%) pacientes estudiados con MPHD. De los 207 pacientes estudiados 125
presentaban neurohipófisis ectópica y 18 fueron identificados con mutaciones en el gen GLI2
(14%). A su vez alteraciones en este gen se han reportado en casos de IGHD (Flemming GM et
al 2013; Bear KA et al 2014). En base a estos estudios, seleccionamos 18 pacientes con déficit
aislado de hormona de crecimiento, o MPHD acompañado de alteraciones de la neurohipófisis
para el análisis molecular del gen GLI2, e identificamos dos nuevas variantes, en dos pacientes
no relacionados. Estos resultados corresponden a una frecuencia de 11.1% (2/18) en nuestro
pequeño grupo de estudio, de manera similar a lo previamente reportado en otras poblaciones.
Bear K et al en el 2014 reportaron un gran número de individuos con variantes en el gen GLI2 y
observaron que todas las mutaciones con pérdida de función fueron heterocigotas, siguiendo
un patrón de herencia dominante con penetrancia incompleta. Consideraron a las variantes
como deletéreas si generaban una proteína truncada, o si afectaban la función de la proteína a
través de los ensayos de predicción in silico, siempre y cuando no se encontraran en las bases
de datos poblacionales. A su vez, compararon el fenotipo observado en los pacientes con
mutaciones con pérdida de función (mutaciones sin sentido, cambio en el marco de lectura o
grandes deleciones) con respecto a aquellas variantes de significado incierto (variantes con
pérdida de sentido) y mostraron que, en el primer caso, existe una asociación entre
anormalidades hipofisarias y polidactilia. Por otro lado, concluyeron que alteraciones en el gen
GLI2 no son una causa común de holoprocencefalia, observándose este fenotipo solo en
algunos casos donde los pacientes presentan mutaciones severas, a diferencia de lo que se

72
creía previamente en base a su relación con el camino de señalización de SHH. Estos
resultados concuerdan con lo observado en nuestros pacientes, donde prevalece el modo de
herencia autosómico dominante con penetrancia incompleta, ya que se identificó en ambos
casos, la presencia de las variantes en un padre no afectado. Ninguno de los dos pacientes
presentó polidactilia, consistente con la incidencia del 3% reportada en pacientes con
mutaciones con pérdida de sentido mientras que ambos presentaron alteraciones hipofisarias,
de manera similar al 58% de individuos descriptos con este tipo de alteraciones. Un dato
interesante es la presencia de microcefalia en los dos pacientes afectados, a diferencia de lo
observado en otras poblaciones. Estos resultados sugieren que el camino de señalización que
involucra al factor de transcripción Gli2 podría estar implicado en el desarrollo temprano del
sistema nervioso central, lo que concuerda con el hecho de que, en modelos animales, SHH
regula la proliferación de las células progenitoras durante el desarrollo cerebral (Balordi F et al
2007; Xu Q et al 2010).
Con respecto al efecto de las variantes sobre la funcionalidad de Gli2, Bear K et al identificaron
mutaciones en los tres dominios de la proteína y no observaron que exista alguna correlación
entre la localización de las variantes y el fenotipo de los pacientes afectados, lo que demuestra
que todos los dominios son esenciales para el normal funcionamiento de la proteína y que la
alteración de cualquiera de ellos genera un efecto deletéreo sobre el camino de señalización
SHH. Sin embargo el mecanismo molecular por el cual sucede este fenómeno aún se
desconoce.
En conclusión, mutaciones en los genes que codifican para los factores de transcripción
involucrados en la embriogénesis hipofisaria (HESX1, OTX2, LHX3, LH4, PROP1, POU1F1,
GLI2) en pacientes con MPHD y alteraciones hipofisarias son extremadamente raras y se
recomienda su estudio en casos donde las características clínicas y de laboratorio se
relacionen directamente con un gen candidato. A su vez, la presente tesis doctoral propone que
la triple asociación entre MPHD, neurohipofisis ectópica o ausente, junto con la presencia de
microcefalia sería un buen marcador clínico para la detección de variantes deletéreas en el gen
GLI2.
Con la aparición de las nuevas tecnologías de secuenciación automatizadas (NGS) es posible
analizar rápidamente un amplio número de genes al mismo tiempo y con un costo
significativamente menor. La evaluación selectiva de un único gen o un panel de genes por
NGS se recomienda para los subgrupos de pacientes que cumplen con los criterios de
selección de las distintas categorías diagnósticas mencionadas a lo largo de la presente tesis
doctoral. Aquellos pacientes cuyas características clínicas y de laboratorio no encajan dentro
de los distintos subgrupos de estudio, o para aquellos en donde el análisis genético inicial no
es determinante para establecer el diagnóstico sería importante considerar la posibilidad de
realizar un análisis a nivel genómico a través de la secuenciación del exoma y arrays
cromosómicos que permitan detectar alteraciones en la secuencia nucleotídica así como
variantes en el número de copias (CNV)(Andrew Dauber et al 2014). Posiblemente en el futuro,
con los avances tecnológicos, se podrán determinar ambas variantes con un único ensayo a

73
escala genómica. Cabe destacar que la secuenciación del exoma no permite identificar
modificaciones epigenéticas y variantes intrónicas o regulatorias que pueden tener efectos
patogénicos mientras que, por otro lado tiene la capacidad de detectar nuevas variantes de
significado incierto así como también hallazgos incidentales con potenciales implicancias sobre
la salud.
A la fecha el estudio de pacientes con sospecha de alguna patología de origen genético sigue
realizándose a partir del estudio del gen candidato teniendo en cuenta un análisis exhaustivo
de las características clínicas y de laboratorio y siguiendo los distintos algoritmos diagnósticos
mencionados previamente en la tesis. Esto se debe principalmente a que aún no existen
reportes de que las nuevas técnicas de secuenciación aumenten el porcentaje de detección de
alteraciones en genes previamente implicados como causa de talla baja o la aparición de
genes nuevos (Andrew Dauber et al 2014). A su vez, estas nuevas estrategias diagnósticas no
han sido validadas a partir de ensayos clínicos por lo que en todos los casos es necesario
confirmar los resultados por medio de las técnicas tradicionales. Dadas las múltiples
dificultades con la interpretación de los resultados de las nuevas variantes de significado
incierto como la posibilidad de identificar hallazgos incidentales, es que se recomienda que las
técnicas de NGS se apliquen en un contexto de investigación con un protocolo aprobado para
el estudio de pacientes con talla baja lo cual abrirá nuevos horizontes en el diagnóstico de
niños con alteración de crecimiento.

74
8. Conclusión
La significancia y relevancia de la presente tesis doctoral se basa en desarrollar las
herramientas diagnósticas adecuadas para la caracterización molecular de pacientes con
alteraciones en el eje de crecimiento GH-IGF-1. Estas herramientas permitirán establecer una
respuesta eficiente y veloz del personal médico frente a estas patologías, dando lugar a una
rápida detección, la selección de la estrategia terapéutica más eficaz y el asesoramiento
genético adecuado.
Los resultados de este estudio aportan nueva información sobre la frecuencia de las patologías
que afectan la talla en nuestra población y como impactan las alteraciones de los distintos
factores que forman parte del eje Hipotálamo-Hipófiso-IGF-1 en el crecimiento pre y postnatal,
aportando datos relevantes, tanto a nivel molecular como fisiológico, que permiten profundizar
el entendimiento de estas patologías endócrinas sobre la salud humana.

75
RESUMEN
El crecimiento somático normal requiere de la integración de múltiples factores que actúan a
nivel periférico, como factores metabólicos y hormonales, y a nivel central, como factores de
trascripción involucrados en el eje hipotálamo-hipofisario-IGF-1.
El desarrollo de la glándula hipofisaria está determinado por la expresión secuencial
coordinada, de manera temporo-espacial, de distintos factores de transcripción de expresión
temprana en el desarrollo embrionario (PROP1, POU1F1, LHX3, LHX4, HESX1, OTX2, GLI2).
Estos factores generar la activación, o inhibición, de distintas vías de señalización celular que
van a dar lugar a la formación de la glándula hipófisis. Este proceso permite el establecimiento
de los distintos linajes celulares hipofisarios que ejercen las funciones vitales de la glándula
regulando el crecimiento (eje somatotropo), la reproducción (eje gonadotropo), el metabolismo
(eje tirotropo) y la respuesta al stress (eje corticotropo). Alteraciones en distintas etapas de la
embriogénesis resultan en la pérdida, o reducción, de las células secretoras de hormonas
hipofisarias y se manifiesta en una disminución funcional de la glándula hipófisis conocida
como hipopituitarismo congénito. El hipopituitarismo congénito puede variar desde déficit
múltiple de hormonas hipofisarias (MPHD), donde más de un linaje celular se ve afectado,
hasta déficit aislado, por alteración de un único linaje celular. Dentro de este último, el déficit
más frecuente es el que afecta al linaje somatotropo, que da lugar a déficit aislado de hormona
de crecimiento (IGHD).
En la vida postnatal, la secreción de GH-1 es regulada por la acción de dos péptidos
hipotalámicos, la hormona liberadora de GH-1 (GHRH) y el factor inhibitorio de GH-1 (GHIF).
Estos factores a su vez, dependen de la acción de neurotransmisores pertenecientes a vías de
señalización del sistema nervioso central. Las células somatotróficas de la adenohipófisis
poseen receptores de membrana que responden a estos factores, de tal forma que cuando la
GHRH se une a su receptor específico de membrana (GHRHR) desencadena la secreción de
GH-1. La unión de una molécula de GH-1 a su receptor de membrana específico (GHR) genera
un cambio conformacional que permite la asociación del receptor con una quinasa intracelular
denominada JAK2. La proteína JAK2 es una quinasa que fosforila residuos de tirosina en el
dominio intracelular del receptor, los cuales sirven como puntos de anclaje para las proteínas
STATs, en particular STAT5a y STAT5b. Las proteínas STATs fosforiladas se dimerizan y se
traslocan al núcleo donde se unen a elementos de respuesta específicos en el ADN y regulan
la expresión de distintos genes. Modelos animales junto con casos clínicos de sujetos
afectados, han demostrado que el principal factor involucrado en la regulación de la síntesis de
IGF-1 es la STAT5b, de tal manera que la unión de la GH-1 a su receptor genera la formación
de dímeros de STAT5b que se traslocan al núcleo y se unen a los elementos de respuesta a la
GH-1 (GHRE), regulando la transcripción de los genes IGF1, IGFBP3 e IGFALS, los cuales
codifican para las proteínas IGF-1, IGFBP-3 y la subunidad ácido lábil (ALS), respectivamente,
los cuales median los efectos proliferativos y de crecimiento de la GH-1.

76
Dado que la caracterización molecular de los genes relacionados con el eje Hipotálamo-
Hipófiso-IGF-1 tiene implicancias diagnósticas que impactan directamente en la estrategia
terapéutica y el consejo genético, el objetivo general de la presente tesis doctoral es la
identificación molecular de pacientes pediátricos con alteración de dicho eje. A su vez se
analizará la frecuencia de estas alteraciones en la población pediátrica argentina y se la
comparará con los datos disponibles descriptos en otras poblaciones. Con el fin de cumplir con
los objetivos propuestos clasificamos las patologías en cuatro grupos de estudio: 1) Deficiencia
aislada de GH-1, por alteración del gen GH1 o GHRHR, 2) Síndrome de Insensibilidad a la GH-
1, por alteración de los genes GHR y STAT5b, 3) Resistencia a IGF-1, por alteración del gen
IGF1R y 4) Deficiencia múltiple de hormonas hipofisarias por alteraciones en distintos genes
que codifican para factores de transcripción que participan en la embriogénesis y diferenciación
de la hipófisis (PROP1, POU1F1, LHX3, LHX4, HESX1, OTX2, GLI2). Los genes mencionados
se estudiaron mediante la técnica de secuenciación directa, a partir de ADN y ARN extraído de
muestras de sangre periférica de los pacientes, incluidos en los distintos grupos, que acuden al
servicio de Endocrinología del Hospital Garrahan. En los casos en donde identificamos
alteraciones no reportadas previamente en la literatura se realizaron distintos ensayos in silico
e in vitro (dependiendo del tipo de alteración) para caracterizar funcionalmente las variantes
halladas y de esta forma completar el diagnóstico clínico y molecular de los pacientes.

77
BIBLIOGRAFIA
A. Caufriez, F. Frankenne, G. Hennen, G. Copinschi, Regulation of maternal IGF-I by
placental GH in normal and abnormal human pregnancies, Am.J.Physiol.265 (1993)
E572–E577.
Abrao MG, Leite MV, Carvalho LR, Billerbeck AE, Nishi MY, Barbosa AS, Martin RM,
Arnhold IJ & Mendonca BB. 2006 Combined pituitary hormone deficiency due to
complete PROP1 deletion. Clinical Endocrinology 65 294-300.
Acampora D, Mazan S, Lallemand Y, Avantaggiato V, Maury M, SimeoneA, Brulet P
1995 Forebrain and midbrain regions are deletedin Otx2 -/- mutants due to a defective
anterior neuroectoderm specification during gastrulation. Development 121:3279–3290
Adams TE, Epa VC, Garrett TP et al. Structure and function of the type 1 insulin-like
growth factor receptor. Cellular and Molecular Life Science 2000; 57: 1050–1093.
Adams TE, Epa VC, Garrett TP et al. Structure and function of the type 1 insulin-like
growth factor receptor. Cellular and Molecular Life Science 2000; 57: 1050–1093.
Agarwal G, Bhatia V, Cook S & Thomas PQ. 2000 Adrenocorticotropin deficiency in
combined pituitary hormone deficiency patients homozygous for a novel PROP1
deletion. Journal Clinical Endocrinology and Metabolism 85 4556-4561.
Akinci A, Kanaka C, Eble A, Akar N, Vidinlisan S & Mullis PE (1992) Isolated growth
(GH) deficiency type IA associated with a 45-kilobase gene deletion within the human
GH gene cluster. J Clin Endocrinol Metab, 75, 437–441.
Akker SA, Misra S, Aslam S, Morgan EL, Smith PJ, Khoo B, Chew SL 2007
Prespliceosomal binding of U1 small nuclear ribonucleo protein(RNP) and
heterogenous nuclear RNPE1 is associated with suppression of a growth hormone
receptor pseudoexon. Mol Endocrinol 21:2529–2540
Alatzoglou KS, Turton JP, Kelberman D et al. Expanding the spectrum of mutations in
GH1 and GHRHR: genetic screening in a large cohort of patients with congenital
isolated growth hormone deficiency. The Journal of Clinical Endocrinology and
Metabolism 2009; 94: 3191–3199.
Alsat E, Guibourdenche J, Luton D, Frankenne F & Evain-Brion D (1997) Human
placental growth hormone. Am J Obstet Gynecol, 177,1526–1534.
Ambrosio R, Fimiani G, Monfregola J et al. The structure of human STAT5A and B
genes reveals two regions of nearly identical sequence and an alternative tissue
specific STAT5B promoter. Gene 2002; 285: 311–318.
Amselum S, et al. Spectrum of growth hormone receptor mutation and associated
haplotypes in Laron syndrome. (1993) Human Mol Geneti 2, 355.
Andrew Dauber, Ron G. Rosenfeld, Joel N. Hirschhorn. Genetic Evaluation of Short
Stature. Journal Clinical Endocrinology and Metabolism, 2014 Sep;99(9):3080-92.

78
Ang SL, Jin O, Rhinn M, Daigle N, Stevenson L, Rossant J 1996 A targeted mouse
Otx2 mutation leads to severe defects in gastrulation and formation of axial mesoderm
and to deletion of rostral brain. Development122:243–252
B.E. Nickel, E. Kardami, P.A. Cattini, Differential expression of human placental growth-
hormone variant and chorionic somatomammotropin in culture, Biochem.J.
267(1990)653–658.
Bach I, Rhodes SJ, Pearse Jr RV, et al. 1995 P-Lim, a LIM homeodomain factor, is
expressed during pituitary organ and cell commitment and synergizes with Pit-1. Proc
Natl Acad Sci USA.92:2720–2724.
Bailyes EM, Navé BT, Soos MA, Orr SR, Hayward AC, Siddle K, et al. Insulin
receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues:
quantification of individual receptor species by selective immunoprecipitation and
immunoblotting. Biochem J 1997; 327: 209-15.
Ballesteros M, Leung KC, Ross RJ, Iismaa TP, Ho KK 2000 Distribution and abundance
of messenger ribonucleic acid for growth hormone receptor isoforms in human tissues.J
Clin Endocrinol Metab85:2865–2871.
Balordi F, Fishell G. Hedgehog signaling in the subventricular zone is required for both
the maintenance of stem cells and the migration of newborn neurons. J Neurosci.
2007;27:5936–5947.
Barclay JL, Kerr LM, Arthur L, Rowland JE, Nelson CN, Ishikawa M, d’Aniello EM,
White M, Noakes PG, Waters MJ 2010 In vivo targeting of the growth hormone receptor
(GHR) box1 sequence demonstrates that the GHR does not signal exclusively through
JAK2. Mol Endocrinol24:204–217.
Barrera-Saldana HA, Seeburg PH & Sauders GF (1983) Two structurally different
genes produce the same secreted human placental lactogen hormone. J Biol Chem,
258,3787–3793.
Barsh GS, Seeburg PH & Gelinas RE (1983) The human growth hormone gene family:
structure and evolution of the chromosomal locus. Nucleic Acids Res, 11,3939–3958.
Baumann G & Maheshwari H. The Dwarfs of Sindh: severe growth hormone (GH)
deficiency caused by a mutation in the GH releasing hormone receptor gene. Acta
Paediatrica Supplementum 1997 423 33–38.
Baxter RC 1994 Insulin-like growth factor binding proteins in the human circulation: a
review. Horm Res42:140–144.
Bear KA, Solomon BD, Antonini S, Arnhold IJ, França MM, Gerkes EH, Grange DK,
Hadley DW, Jääskeläinen J, Paulo SS, Rump P, Stratakis CA, Thompson EM, Willis M,
Winder TL, Jorge AA, Roessler E, Muenke M. Pathogenic mutations in GLI2 cause a
specific phenotype that is distinct from holoprosencephaly. J Med Genet. 2014
Jun;51(6):413-8. doi: 10.1136/jmedgenet-2013-102249. Epub 2014 Apr 17.
Bhangoo AP, Hunter CS, Savage JJ, Anhalt H, Pavlakis S, Walvoord EC, Ten S &
Rhodes SJ 2006 Clinical case seminar: a novel LHX3 mutation presenting as combined

79
pituitary hormonal deficiency. Journal of Clinical Endocrinology and Metabolism 91
747–753.
Binder G & Ranke MB. Screening for growth hormone (GH) gene splice site mutations
in sporadic cases with severe isolated GH deficiency using ectopic transcript analysis.
Journal of Clinical Endocrinology and Metabolism 1995 80 1247–1252.
Binder G, Keller E, Mix M, Massa GG, Stokvis-BrantsmaWH, Wit JM & Ranke MB.
Isolated GH deficiency with dominant inheritance: new mutations, new insights. Journal
of Clinical Endocrinology and Metabolism 2001 86 3877–3881.
Binder G, Neuer K, Ranke MB, Wittekindt NE 2005 PTPN11 mutations are associated
with mild growth hormone resistance in individuals with Noonan syndrome. J Clin
Endocrinol Metab 90: 5377–5381.
Blankenstein O, Muhleberg R, Kim C, Wuller S, Pfaffle R & Heimann G. A new C-
terminal located mutation (V272ter) in thePit-1 gene manifestating with severe
congenital hypothyroidism. Hormone Research 2001 56 81–86.
Brickman JM, Clements M, Tyrell R, McNay D, Woods K, Warner J, Stewart A,
Beddington RS & Dattani M 2001 Molecular effects of novel mutations in Hesx1/HESX1
associated with human pituitary disorders. Development 128 5189–5199.
Butler AA, LeRoith D: Minireview: tissue-specific versus generalized gene targeting of
the igf1 and igf1r genes and their roles in insulin like growth factor physiology.
Endocrinology 2001;142:1685-1688.
C. Heinrichs, F. de Zegher, F. Vansnick, A.Vokaer, C. Christophe, F. Frankenne, Fetal
hypopituitarism: perinatal endocrine and morphological studies in two cases, Acta
Paediatr.83(1994)448–451.
Camper SA, Saunders TL, Katz RW, Reeves RH. 1990 The pit-1 transcription factor
gene is a candidate for the murine Snell dwarf mutation. Genomics 8 586-590.
Carlomagno Y, Salerno M, Vivenza D, Capalbo D, Godi M, Mellone S, Tiradani L,
Corneli G, Momigliano-Richiardi et al. 2009 A novel recessive splicing mutation in the
POU1F1 gene causing combined pituitary hormone deficiency. Journal of
Endocrinological Investigation 32 653-658.
Carvalho LR, Woods KS, Mendonca BB, Marcal N, Zamparini AL, Stifani S, Brickman
JM, Arnhold IJ & Dattani MT. 2003 A homozygous mutation in HESX1 is associated
with evolving hypopituitarism due to impaired repressor-corepressor interaction. Journal
of Clinical Investigation 112 1192-1201.
Castinetti F, Saveanu A, Reynaud R, Quentien MH, Buffin A, Brauner R, et al. A novel
dysfunctional LHX4 mutation with high phenotypical variability in patients with
hypopituitarism. J Clin Endocrinol Metab 2008;93:2790–9.
Castrillo JL, Bodner M & Karin M (1989) Purification of growth hormone-specific
transcription factor GHF-1 containing homeobox. Science, 243,814–817.
Cha KB, Douglas KR, Potok MA, Liang H, Jones SN, Camper SA. WNT5A signaling
affects pituitary gland shape. Mech Dev. 2004; 121(2):183–94.

80
Chaignaud C, Guibourdenche J, Sibony O, Evain-Brion D, Luton D, Chevenne D &
Porquet D (2000) Modulation de la secretion des hormones placentaires au cours du
test de O’Sullivan. In Actualites en Pharmacie Biologique et Clinique (Ed.) Garnier JP,
LeMoe ¨lG,Nivet-AntoineV&BeaudeuxJL,pp.153–156.Paris.
Chaler E.A., Meazza, C., Guercio, G. et al. (2009) Serum IGF-I and IGFBP-3 reference
values from a chemiluminescent assay in normal children and adolescents of hispanic
and Italian origin: presence of sexual dimorphism in IGF-I values. Journal of Pediatrics
Endocrinology and Metabolism, 22, 1127–1135.
Chaler EA, Ballerini Ga, Lazzati JM, Maceiras M, Frusti M, Bergada I, Rivarola MA,
Belgorosky A, Ropelato G. Cut-off values of serum growth hormone (GH) in
pharmacological stimulation tests (PhT) evaluated in short-statured children using a
chemiluminescent immunometric assay (ICMA) calibrated with the International
Recombinant Human GH Standard 98/574. Clin Chem Lab Med. 2013 May;51(5):e95-7.
Chatelain G, Fossat N, Brun G, Lamonerie T. 2006 Molecular dissection reveals
decreased activity and not dominant negative effect in human OTX2
mutants.JMolMed84:604–615.
Chen EY, Liao YC, Smith DH, Barrera-Saldana HA, Gelinas RE & Seeburg PH (1989)
The human growth hormone locus: nucleotide sequence, biology and evolution.
Genomics, 4,479–497.
Chia DJ, Varco-Merth B& Rotwein P. Dispersed chromosomal STAT5B binding
elements mediate growth hormone activated insulin-like growth factor-I gene
transcription. Journal of Biological Chemistry 2010; 285: 17636–17646.
Christopher J Romero, Elyse Pine-Twaddell, Sally Radovick. (2011). Novel mutations
associated with combined pituitary hormone deficiency. Journal of Molecular
Endocrinology 46, R93–R102.
Claudia Danielli Pereira Bertolacinia, Lucilene Arilho Ribeiro-Bicudoa, Aline da Silva
Petrinb, Antonio Richieri-Costaa, and Jeffrey C. Murray. Hospital of Rehabilitation of
Craniofacial Anomalies, USP, Bauru SP, Brazil. Department of Pediatrics, University of
Iowa, Iowa City, IA, USA. Clinical findings in patients with GLI2 mutations – phenotypic
variability. Clin Genet. 2012 January ; 81(1): 70–75.
Cohen LE, Radovick S & Wondisford FE. 1999 Transcription factors and
hypopituitarism. Trends in Endocrinology & Metabolism 10 326-332.
Cohen LE, Radovick S 2002 Molecular basis of combined pituitary hormone
deficiencies. Endocr Rev 23:431–442.
Conley ME, Burks AW, Herrod HG & Puck JM. Molecular analysis of X-linked
agammaglobulinemia and isolated growth hormone deficiency. Journal of Pediatrics
1991 119 392–397.
Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen
L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct

81
functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004; 24:8037–
8047.
Cunningham BC, Ultsch M, DeVos AM, Mulkerrin MG, Clauser KR & Wells JA.
Dimerization of the extracellular domain of the human growth hormone receptor by a
single hormone molecule. Science 1991 25 821–825.
D.J.Hill, S.C. Riley, N.S. Bassett, M.J. Waters, Localization of the growth hormone
receptor, identified by immunocytochemistry, in second trimester human fetal tissues
and in placenta throughout gestation, J. Clin. Endocrinol. Metab.75 (1992) 646–650.
D.W. Leung, S.A. Spencer, G. Cachianes, R. G. Hammonds, C. Collins, W.J. Henzel,
R. Barnard, M.J. Waters, W.I.Wood, Growth hormone receptor and serum binding
protein: purification, cloning and expression, Nature 330 (1987) 537–543.
Dasen JS, Barbera JP, Herman TS, Connell SO, Olson L, Ju B, Tollkuhn J, Baek SH,
Rose DW, Rosenfeld MG 2001. Temporal regulation of a paired-like homeodomain
repressor/TLE corepressor complex and a related activator is required for pituitary
organogenesis. Genes Dev15:3193–207.
Dasen JS, O’Connell SM, Flynn SE, Treier M, Gleiberman AS, Szeto DP, Hooshmand
F, Aggarwal AK & Rosenfeld MG. Reciprocal interactions of Pit1 and GATA2 mediate
signaling gradient-induced determination of pituitary cell types. Cell 1999 97587–598.
Dateki S, Fukami M, Sato N, Muroya K, Adachi M & Ogata T. 2008 OTX2 mutation in a
patient with anophthalmia, short stature, and partial growth hormone deficiency:
functional studies using the IRBP, HESX1 and POU1F1 promoters. Journal of Clinical
Endocrinology and Metabolism 93 3697-3702.
Dateki S, Fukami M, Uematsu A, Kaji M, Iso M, Ono M, et al. Mutation and gene copy
number analyses of six pituitary transcription factor genes in 71 patients with combined
pituitary hormone deficiency: identifcation of a single patient with LHX4 deletion. J Clin
Endocrinol Metab 2010;95:4043–7.
Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, Muroya K, Adachi
M, Tajima T, Motumura K et al. 2009 Heterozygous orthodenticle homeobox 2
mutations are associated with variable pituitary phenotype. Journal of Clinical
Endocrinology Metabolism 95 756-764.
Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL,
Toresson H, Fox M, Wales JK, Hindmarsh PC et al. 1998 Mutations in the homeobox
gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse.
NatureGenetics 19 125–133
Daughaday WH, Salmon WD Jr: The Origins and Development of the Somatomedin
Hypothesis. Totowa, Humana Press, 1999.
David A, Camacho Hubner C, Bhangoo A, Rose SJ, Miraki Moud F, Akker SA, Butler
GE, Ten S, Clayton PE, Clark AJ, Savage MO, Metherell LA 2007 An intronic growth
hormone receptor mutation causing activation of a pseudoexon is associated with a

82
broad spectrum of growth hormone insensitivity phenotypes. J Clin Endocrinol
Metab92:655–659.
David A, Hwa V, Metherell LA, Netchine I, Camacho-Hubner C, Clark A, Rosenfeld RG,
Savage MO. 2011 Evidence for a continuum of genetic, phenotypic and biochemical
abnormalities in children with growth hormone insensitivity. Endocrine Reviews 32(4).
Davis SW, Camper SA. Noggin regulates Bmp4 activity during pituitary induction. Dev
Biol. 2007; 305(1):145–60.
De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C.
Animportant role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in
mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;
127(3):483–92.
De Vos AM, Ultsch M & Kossiakoff AA. Human growth hormone and extracellular
domain of its receptor: crystal structure of the complex. Science 1992 255 306–312.
De Zegher F, Vanderschueren-Lodeweyckx M, Spitz B, Faijerson Y, Blomberg F,
Beckers A, Hennen G & Frankenne F (1990) Perinatal growth hormone (GH)
physiology: effectof GH-releasing factor on maternal and fetal secretion of pituitary and
placental GH. J Clin Endocrinol Metab, 71,520–522.
Deladoey J, Stocker P & Mullis PE. Autosomal dominant GH deficiency due to an
Arg183His GH-1 gene mutation: clinical and molecular evidence of impaired regulated
GH secretion. Journal of Clinical Endocrinology and Metabolism 2001 86 3941–3947.
DeMeyts P & Whittaker J. Structural biology of insulin and IGF1 receptors: implications
for drug design. Nature Reviews. Drug Discovery 2002; 1: 769–783.
DeNoto FM, Moore DD & Goodman HM (1981) Human growth hormone DNA sequence
and mRNA structure: possible alternative splicing. Nucleic Acids Res,9,3719–3730.
Derr MA, Fang P, Sinha SK, Ten S, Hwa V, Rosenfeld RG 2011 A novel Y332C
missense mutation in the intracellular domain of the human growth hormone receptor
(GHR) does not alter STAT5b signaling: redundancy of GHR intracellular tyrosines
involved in STAT5b signaling. Horm Res Paediatr 75:187–199.
Diaczok D, Romero C, Zunich J, Marshall I & Radovick S. 2008 A novel dominant
negative mutation of OTX2 associated with combined pituitary hormone deficiency.
Journal of Clinical Endocrinology and Metabolism 93 4351-4359.
Domene HM, Bengolea SV, Martínez AS, Ropelato MG, Pennisi P, Scaglia P, Heinrich
JJ, Jasper HG 2004 Deficiency of the circulating insulin-like growth factor system
associated within activation of the acid-labile subunit gene. N Engl J Med350:570–577.
Domene HM, Hwa V, Argente J, Wit JM, Camacho-Hubner C, Jasper HG, Pozo J, van
Duyvenvoorde HA, Yakar S, Fofanova-Gambetti OV, Rosenfeld RG 2009 Human acid-
labile subunit deficiency: clinical, endocrine and metabolic consequences. Horm
Res72:129–141.
Domene HM, Scaglia PA, Lteif A, Mahmud FH, Kirmani S, Frystyk J, Bedecarrás P,
Gutiérrez M, Jasper HG 2007 Phenotypic effects of null and haploinsufficiency of acid

83
labile subunit in a family with two novel IGFALS gene mutations. J Clin Endocrinol
Metab 92:4444–4450.
Duquesnoy P, Roy A, Dastot F, Ghali I, Teinturier C, Netchine I, Cacheux V, Hafez M,
Salah N, Chaussain JL et al. 1998 Human prop1: cloning, mapping, genomic structure.
Mutations in familial combined pituitary hormone deficiency. FEBS Letters 437 216–
220.
Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse
Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996; 84:443–
450.
Duriez B, Duquesnoy P, Dastot F, Bougneres P, Amselem S & Goossens M. An exon
skipping mutation in the btk gene of a patient with x-linked agammaglobulinemia and
isolated growth hormone deficiency. FEBS Letters 1994 346 165–170
Ericson J, Norlin S, Jessell TM, Edlund T. Integrated FGF and BMP signaling controls
the progression of progenitor cell differentiation and the emergence of pattern in the
embryonic anterior pituitary. Development. 1998; 125(6):1005–15.
Evain-Brion D, Alsat E, Igout A, Frankenne F & Hennen G (1994) Placental growth
hormone variant: assay and clinical aspects. Acta Paediatr,399,49–51.
Evain-Brion D, Alsat E, Mirlesse V, Dodeur M, Scippo ML, Hennen G & Frankenne F
(1990) Regulation of growth hormone secretion in human trophoblastic cells in culture.
Horm Res, 33,256–259.
F. Frankenne, E. Alsat, M.L. Scippo, A. Igout, G. Hennen, D. Evain Brion, Evidence for
the expression of growth hormone receptors in human placenta, Biochem. Biophys.
Res.Commun. 182 (1992) 481–486.
F. Frankenne, M.L.Scippo, J. VanBeeumen, A. Igout, G. Hennen, Identification of
placental human growth hormone as the growth hormone-V gene expression product,
J.Clin.Endocrinol.Metab. 71(1990)15–18.
Filges I, Bischof-Renner A, Röthlisberger B, Potthoff C, Glanzmann R, Günthard J, et
al. Panhypopituitarism presenting as life-threatening heart failure caused by an inherited
microdeletion in 1q25 including LHX4. Pediatrics 2012;129:e529–34.
Finkelstein R, Boncinelli E (1994) From fly head to mammalian forebrain: the story of
otd and Otx. Trends Genet 10:310-315
Firth S, Baxter R. 2002 Cellular Actions of the Insulin-Like Growth Factor Binding
Proteins. Endocrine Reviews 23(6):824–854.
Fleisher TA, White RM, Broder S, Nissley SP, Blaese RM, Mulvihill JJ, Olive G &
Waldmann TA. X-linked hypogamma-globulinemia and isolated growth hormone
deficiency. New England Journal of Medicine 1980 302 1429–1439.
Flemming GM, Klammt J, Ambler G, Bao Y, Blum WF, Cowell C, Donaghue K, Howard
N, Kumar A, Sanchez J, Stobbe H, Pfäffle RW. Functional characterization of a
heterozygous GLI2 missense mutation in patients with multiple pituitary hormone

84
deficiency. J Clin Endocrinol Metab. 2013 Mar;98(3):E567-75. doi: 10.1210/jc.2012-
3224. Epub 2013 Feb 13.
Fowden Al, Forhead AJ: Endocrine mechanism of intrauterine programming.
Reproduction 2004; 127:515-526.
Fowden AL: The insulin like growth factors and feto-placental growth. Placenta 2003;
24: 803-812.
França MM, Jorge AA, Carvalho LR, Costalonga EF, Otto AP, Correa FA, Mendonca
BB, Arnhold IJ.Relatively high frequency of non-synonymous GLI2 variants in patients
with congenital hypopituitarism without holoprosencephaly. Clin Endocrinol (Oxf). 2013
Apr;78(4):551-7.
Gaston-Massuet C, Andoniadou CL, Signore M, et al. Increased Wingless (Wnt)
signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and
humans. Proc Natl Acad Sci U S A. 2011; 108(28):11482–7.
Ghizzoni L, Duquesnoy P, Torresani T, Vottero A, Goossens M & Bernasconi S (1994)
Isolated growth hormone deficiency typeIA associated with a 45-kilobase gene deletion
within the human growth hormone gene cluster in an Italian family. Pediatr Res,
36,654–659.
Gicquel C., Le Blouc Y. 2006 Hormonal regulation of fetal growth. Hormone Research
65(suppl 3) 28-33.
Gluckman PD, Pinal CS: Regulation of fetal growth by the somatotrophic axis. J Nutr
2003;133: 1741S-1746S.
Godowski PJ, Leung DW, Meacham LR, Galgani JP, Hellmiss R, Keret R, Rotwein PS,
Parks JS, Laron Z & Wood WI. Characterization of the human growth hormone receptor
gene and demonstration of a partial gene deletion in two patients with Laron-type
dwarfism. PNAS 1989 86 8083–8087.
Graves TK, Patel S, Dannies PS & Hinkle PM. Misfolded growth hormone causes
fragmentation of the Golgi apparatus and disrupts endoplasmic reticulum to Golgi traffic.
Journal of Cell Science 2001 114 3685–3694.
Green H, Morikawa M, Nixon T 1985 A dual effector theory of growth-hormone action.
Differentiation 29:195–198.
Greulich, W.W. & Pyle, S.J. (1959) Radiographic Atlas of Skeletal Development of the
hand and wrist. Stanfor University Press, Stanford, CA.
Hardy K & Spanos S. Growth factor expression and function in the human and mouse
pre implantation embryo. The Journal of Endocrinology 2002; 172: 221–236.
Hayashi Y, Yamamoto M, Ohmori S, Kamijo T, Ogawa M & Seo H. Inhibition of growth
hormone (GH) secretion by a mutant GH-I gene product in neuroendocrine cells
containing secretory granules: an implication for isolated GH deficiency inherited in an
autosomal dominant manner. Journal of Clinical Endocrinology and Metabolism 1999
84 2134–2139.

85
Henderson RH, Williamson KA, Kennedy JS, Wester AR, Holder GE, Robson AG,
FitzPatrick DR, van Heyningen V & Moore AT. 2009 A rare de novo nonsense mutation
in OTX2 causes early onset retinal dystrophy and pituitary dysfunction. Molecular vision
15 2442-2447.
Hennighausen L, Robinson GW 2008 Interpretation of cytokine signaling through the
transcription factors STAT5A and STAT5B. Genes Dev 22:711–721.
Hermesz E, Mackem S, Mahon KA 1996 Rpx: a novel anterior-restricted homeobox
gene progressively activated in the pre chordal plate, anterior neural plate and Rathke’s
pouch of the mouse embryo. Development 122:41–52
Hernandez sanchez C, Mansilla A, de Pablo F et al. Evolution of the insulin receptor
family and receptor isoform expression in vertebrates. Molecular Biology and Evolution
2008; 25: 1043–1053.
Herrada G, Wolgemuth DJ. The mouse transcription factor Stat4 is expressed in haploid
male germ cells and is present in the perinuclear theca of spermatozoa. J Cell Sci.
1997; 110 (Pt 14):1543–1553.
Hintz RL, Liu F: Demonstration of specific plasma protein binding sites for
somatomedin. J Clin Endocrinol Metab 1977; 45: 988–995.
Hiroshi Inoue, Tokuo Mukai, Yukiko Sakamoto, Chizuko Kimura, Natsumi Kangawa,
Mitsuo Itakura, Tsutomu Ogata, Yoshiya Ito and Kenji Fujieda. Identification of a novel
mutation in the exon 2 splice donor site of the POU1F1/PIT-1 gene in Japanese
identical twins with mild combined pituitary hormone deficiency. Clinical Endocrinology
(2012) 76,78–87
Hirt H, Kimelman J, Birnbaum MJ, Chen EY, Seeburg PH, Eberhardt NL & Barta A
(1987) The human growth hormone gene locus: structure, evolution, and allelic
variations. DNA, 6,59–70.
Hiu Kiu and Sandra E Nicholson. Biology and significance of the JAK/STAT signalling
pathways. Growth Factors. 2012 April ; 30(2): 88–106.
Hizuka N, Hendricks CM, Pavlakis GN, Hamer DH & Gorden P (1982) Properties of
human growth hormone polypeptides: purified from pituitary extracts and synthesized in
monkey kidney cells and bacteria. J Clin Endocrinol Metab, 55,545–550.
Ho HY, Susman MW, Bikoff JB, et al. Wnt5a-Ror-Dishevelled signaling constitutes a
core developmental pathway that controls tissue morphogenesis. Proc Natl Acad Sci U
S A. 2012;109(11):4044–51.
Holly J, Perks C. 2006 The role of insulin like growth factor binding proteins.
Neuroendocrinology 2006;83:154–160.
Howard PW, Maurer RA. Identifcation of a conserved protein that interacts with specifc
LIM homeodomain transcription factors. J Biol Chem 2000;275:13336–42.
Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol.
2011; 27:513–37.

86
Hwa V, Nadeau K, Wit JM, Rosenfeld RG 2011 STAT5b deficiency: lessons from
STAT5b gene mutations. Best Pract Res Clin EndocrinolMetab25:61–75.
HwavV,Oh Y, Rosenfeld RG 1999 The insulin-like growth factor binding protein (IGFBP)
superfamily. Endocr Rev 20:761–787.
Illig R. Growth hormone antibodies in patients treated with different preparations of
human growth hormone (hGH). Journal of Clinical Endocrinology and Metabolism 1970
31 679–688.
Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW,
Leonard WJ. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic
activity. J Exp Med. 1998; 188:2067–2074.
Ingraham HA, Chen RP, Mangalam HJ, Elsholtz HP, Flynn SE, Lin CR, Simmons DM,
Swanson L & Rosenfeld MG (1988) A tissue-specific transcription factor containing a
homeodomain specifies a pituitary phenotype. Cell, 55,519–529.
Jens Fuglsang, Per Ovesen. Aspects of placental growth hormone physiology. 2006
Growth Hormone & IGF Research 16 (2006) 67–85.
Jin Y, Surabhi RM, Fresnoza A, Lytras A & Cattini PA (1999) A role for A/T-rich
sequences and Pit-1/GHF-1 in a distal enhancer located in the human growth hormone
locus control region with preferential pituitary activity in culture and transgenic mice. Mol
Endocrinol, 13,1249–1266.
Jones JI and Clemmons D. 1995 Insulin-Like Growth Factors and Their Binding
Proteins: Biological Actions. Endocrine Reviews 16.
K. Linnemann, A. Malek, R. Sager, W.F. Blum, H. Schneider, C. Fusch, Leptin
production and release in the dually in vitro perfused human placenta, J.
Clin.Endocrinol.Metab. 85 (2000) 4298–4301.
Kamijo T, Phillips JA III, Ogawa M, Yuan L, Shi Y & Bao XL. Screening for growth
hormone gene deletions in patients with isolated growth hormone deficiency. Journal of
Pediatrics 1991 118 245–248.
Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced
development of Th2 cells in Stat4-deficient mice. Nature. 1996; 382:174–177.
Kawashima, Y., Kanzaki, S., Yang, F. et al. (2005) Mutation at cleavage site of insulin
like growth factor receptor in a short stature child born with intrauterine growth
retardation. Journal of Clinical Endocrinology and Metabolism, 90, 4679–4687.
Kelberman & Dattani MT. 2006 The role of transcription factors implicated in anterior
pituitary development in the aetiology of congenital hipopituitarism. Annals of Medicine
39 560-577.
Kelberman & Dattani. 2007 Hypopituitarism oddities: congenital causes. Hormon
Research 68 138-144.
Kelberman D., Rizzoti, K., Lovell-Badge, R. et al. (2009) Genetic regulation of pituitary
gland development in human and mouse. Endocrine Reviews, ,790–829.

87
Kim JJ, Accili D: Signalling through IGF-I and insulin receptors: where is the specificity?
Growth Horm IGF Res 2002; 2: 84-90.
Klammt, J., Kiess, W. & Pfaffle, R. (2011) IGF1R mutations as cause of SGA. Best
Practice and Research Clinical Endocrinology and Metabolism, 191–206.
Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L,
Jasper H, Tepper A, Heinrich JJ, Rosenfeld RG 2003 Growth hormone insensitivity
associated with a STAT5b mutation. N Engl J Med 349:1139–1147.
Kristrom B, Zdunek AM, Rydh A, Jonsson H, Sehlin P & Escher SA 2009 A novel
mutation in the LHX3 gene is responsible for combined pituitary hormone deficiency,
hearing impairment, and vertebral malformations. Journal of Clinical Endocrinology and
Metabolism 94 1154–1161.
Kurokawa D,Kiyonari H,Nakayama R,Kimura-Yoshida C,Matsuo I, Aizawa S 2004
Regulation of Otx2 expression and its functions in mouse forebrain and midbrain.
Development131:3319–3331
L.A. Barbour, J. Shao, L. Qiao, L.K. Pulawa, D.R. Jensen, A. Bartke, M. Garrity, B.
Draznin, J.E. Friedman, Human placental growth hormone causes severe insulin
resistance in transgenic mice, Am.J.Obstet.Gynecol. 186 (2002) 512–517.
Lacey KA & Parkin JM. Causes of short stature: a community study of children in
Newcastle upon Tyne. Lancet 1974 I 42–45.
Laron Z 2004 Laron syndrome (primary growth hormone resistance or insensitivity): the
personal experience 1958–2003. J Clin Endocrinol Metab 89:1031–1044.
Le Roith D, Werner H, Beitner-Johnson D, et al (1995) Molecular and cellular aspects of
insulin-like growth factor I receptor. Endocrine Reviews 16:143-163.
Lee MS, Wajnrajch MP, Kim SS, Plotnick LP, Wang J, Gertner JM, Leibel RL & Dannies
PS.Autosomal dominant growth hormone (GH) deficiency type II: the Del 32-71 GH
deletion mutants suppresses secretion of wild-type GH. Endocrinology 2000 141 883–
890
Lefevre C, Imagawa M, Dana S, Grindlay J, Bodner M & Karin M (1987) Tissue-specific
expression of the human growth hormone gene is conferred in part by the binding of a
specific trans-acting factor. EMBOJ,6,971–981.
Lejarraga, H., del Pino, M., Fano, V. et al. Referencias de peso y estatura desde el
nacimiento hasta la madurez para niñas y niños argentinos: Incorporación de datos de
la OMS de 0 a 2 añnos, recálculo de percentilos para obtención de valores LMS.
Archivos argentinos de pediatria, 107, 126–133.
Lemos MC, Gomes L, Bastos M, Leite V, Limbert E, Carvalho D, Bacelar C, Monteiro
M, Fonseca F, Agapito A et al. 2006 PROP1 gene análisis in Portuguese patients with
combined pituitary hormone deficiency. Clinical Endocrinology 65 479-485.
Leong SR, Baxter RC, Camerato T, Dai J, Wood WI 1992 Structure and functional
expression of the acid labile subunit of the insulin-like growth factor binding protein
complex. Mol Endocrinol 6:870–876.

88
Levy DE, Darnell Jr JE 2002 Stats: transcriptional control and biological impact. Nat
Rev Mol Cell Biol 3:651–662.
Limal JM, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S, Vidaud M, LeBouc Y
2006 Noonan syndrome: relationships between genotype, growth, and growth factors. J
Clin Endocrinol Metab 91: 300–306.
Lin SC, Lin CR & Gukovsky I. Molecular basis of the little mouse phenotype and
implications for cell type-specific growth. Nature 1993 364 208–213.
Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression
of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction
in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995; 92:8831–8835.
Luigi Laviola, Annalisa Natalicchio and Francesco Giorgino. The IGF-I Signaling
Pathway. Current Pharmaceutical Design, 2007, 13, 663-669.
M. Chellakooty, K.Vangsgaard, T. Larsen, T. Scheike, J. Falck Larsen,J. Legarth,A.M.
Andersson, K.M. Main, N. E. Skakkebaek, A. Juul, A longitudinal study of intrauterine
growth and the placental growth hormone(GH)-insulin-like growth factor I axis in
maternal circulation: association between placental GH and fetal growth,
J.Clin.Endocrinol.Metab.89 (2004) 384–391.
M.C.Lacroix, J.Guibourdenche, J.L. Frendo, F. Muller and D., Evain-Brion. Human
Placental Growth Hormone. Placenta (2002), 23, Supplement A, Trophoblast Research,
16,S87–S94.
Maamra M, Milward A, Esfahani HZ, Abbott LP, Metherell LA, Savage MO, Clark AJ,
Ross RJ 2006 A 36 residues insertion in the dimerization domain of the growth
hormone receptor results in defective trafficking rather than impaired signalling. J
Endocrinol188:251–261
Machinis K, Amselem S. Functional relationship between LHX4 and POU1F1 in light of
the LHX4 mutation identifed in patients with pituitary defects. J Clin Endocrinol Metab
2005;90:5456–62.
Machinis K, Pantel J, Netchine I, Léger J, Camand OJ, Sobrier ML, et al. Syndromic
short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J
Hum Genet 2001;69:961–8.
MacLeod JN, Worsley I, Ray J, Friesen HG, Liebhaber SA & Cooke NE (1991b) Human
growth hormone-variant is a biologically active somatogen and lactogen. Endocrinology,
128,1298–1302.
Martial JA, Hallewell RA, Baxter JD & Goodman HM (1979) Human growth hormone:
complementary DNA cloning and expression in bacteria. Science, 205,602–607.
Martinez-Morales JR,Signore M,Acampora D,Simeone A,Bovolenta P (2001) Otx genes
are required for tissue specification in the developing eye. Development1 28:2019–
2030.
Matias Juanes, Gabriela Guercio, Roxana Marino, Esperanza Berensztein, Diana
Monica Warman, Marta Ciaccio, Silvia Gil, Marcela Bailez, Marco A. Rivarola and

89
AliciaBelgorosky. Three novel IGF1R mutations in microcephalic patients with prenatal
and postnatal growth impairment. Clinical Endocrinology (2014), 0 ,1–8.
Matias Juanes, Roxana Marino, Marta Ciaccio, Isabel Di Palma, Pablo Ramirez, Diana
M. Warman, Valeria De Dona, Eduardo Chaler, Mercedes Maceiras, Marco A. Rivarola
and Alicia Belgorosky. Presence of GH1 and absence of GHRHR gene mutations in a
large cohort of Argentinian patients with severe short stature and isolated GH
deficiency. Clinical Endocrinology (2013), 0,1–2.
Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S 1995 Mouse Otx2 functions in the
formation and patterning of rostral head. Genes Dev 21:2646–2658
Mayo K. Molecular cloning and expression of a pituitary-specific receptor for growth
hormone releasing hormone. Molecular Endocrinology 1992 6 1734–1744.
McCabe MJ, Gaston-Massuet C, Tziaferi V, et al. Novel FGF8 mutations associated
with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary
dysfunction. J Clin Endocrinol metab. 2011; 96(10):E1709–18.
McGillivray SM, Bailey JS, Ramezani R, Kirkwood BJ &Mellon PL 2005 Mouse GnRH
receptor gene expression is mediated by the LHX3 homeodomain protein.
Endocrinology 146 2180–2185.
McGuinness L, Magoulas C, Sesay AK, Mathers K, Carmignac D, Manneville JB,
Christian H, Phillips JA 3rd
& Robinson IC. Autosomal dominant growth hormone
deficiency disrupts secretory vesicles in vitro and in vivo in transgenic mice.
Endocrinology 2003 144 720–731.
McIntyre HD, Serek R, Crane DI, Veveris-Lowe T, Parry A, Johnson S, Leung KC, Ho
KK, Bougassa M, Hennen G, Igout A, Chan FY, Cowley D, Cotterill A & Barnard R
(2000) Placental growth hormone (GH),GH-binding protein, and insulin-like growth
factor axis in normal, growth-retarded, and diabetic pregnancies: correlations with fetal
growth. J Clin Endocrinol Metab, 85,1143–1150.
Mertani HC, Delehaye-Zervas MC, Martini JF, Postel-Vinay MC & Morel G (1995)
Localization of growth hormone receptor messenger RNA in human tissues. Endocrine,
3,135–142.
Metherell LA, Akker SA, Munroe PB, Rose SJ, Caulfield M, Savage MO, Chew SL,
Clark AJ 2001 Pseudoexon activation as a novel mechanism for disease resulting in a
typical growth hormone insensitivity. Am J Hum Genet 69: 641–646.
Milward A, Metherell L, Maamra Metal. Growth hormone (GH) insensitivity syndrome
due to a GH receptor truncated after Box1, resulting in isolated failure of STAT5 signal
transduction. Journal of Clinical Endocrinology & Metabolism 2004;89: 1259–1266.
Miyata I, Vallette-Kasic S, Saveanu A, Takeuchi M, Yoshikawa H, Tajima A, Tojo K et
al. 2006 Identification and functional analysis of novel S179R POU1F1 mutation
associated with combined pituitary hormone deficiency. Journal of Clinical
Endocrinology and Metabolism 91 4981-4987.

90
Moxham CP, Duronio V, Jacobs S. Insulin-like growth factor I receptor beta-subunit
heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I
and insulin receptor heterodimers. J Biol Chem 1989; 264: 13238-44.
Mullis P. Genetic control of growth. European Journal of Endocrinology (2005) 152 11–
31.
Mullis PE, Akinci A, Kanaka C, Eble A & Brook CG. Prevalence of human growth
hormone 1 gene deletions among patients with isolated growth hormone deficiency
from different populations. Pediatric Research 1992 31 532–534.
Mullis PE, Deladoey J & Dannies PS. Molecular and cellular basis of isolated dominant
negative growth hormone deficiency, IGHD typeII: Insights on the secretory pathway of
peptide hormones. Hormone Research 2002 58 53–66.
N.E.Cooke, J. Ray, J.G. Emery, S.A. Liebhaber, Two distinct species of human growth
hormone-variant mRNA in the human placenta predict the expression of novel growth
hormone proteins, J. Biol. Chem. 263 (1988)9001–9006.
Nachtigal MW, Nickel BE & Cattini PA (1993) Pituitary-specific repression of placental
members of the human growth hormone gene family. A possible mechanism for locus
regulation. J Biol Chem, 268,8473–8479.
Nayak NR, Giudice LC: Comparative biology of the IGF system in te endometrium,
decidua, and placenta, and clinical implications for fetal growth and implantation
disorders. Placenta 2003; 24:281-296.
Netchine, I., Azzi, S., Le Bouc, Y. et al. (2011) IGF1 molecular anomalies demonstrate
its critical role in fetal, postnatal growth and brain development. Best Practice and
Research Clinical Endocrinology and Metabolism, 25,181–190.
Norquay LD, Jin Y, Surabbi RM, Gietz RD, Tanese N & Cattini P (2001) A member of
the nuclear-factor-1 family is involved in the pituitary repression of the human placental
growth hormone genes. Biochem J, 35 387–395.
Ohuchi H, Hori Y, Yamasaki M, et al. FGF10 acts as a major ligand for FGF receptor 2
IIIb in ouse multi-organ development. Biochem Biophys Res Commun. 2000;
277(3):643–9.
Olson LE, Tollkuhn J, Scafoglio C, et al. Homeodomain-mediated beta-catenin-
dependent switching events dictate cell-lineage determination. Cell. 2006; 125(3):593–
605.
Ooi GT, Cohen FJ, Tseng LY, Rechler MM, Boisclair YR 1997 Growth hormone
stimulates transcription of the gene encoding the acid-labile subunit(ALS) of the
circulating insulin-like growth factor-binding protein complex and ALS promoter activity
in rat liver. Mol Endocrinol 11:997–1007.
Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity.
2000;13:795–804.

91
Parks JS, Meacham LR & Mc Kean MC. Growth hormone (GH) gene deletion is the
most common cause of severe GH deficiency among Oriental Jewish children. Pediatric
Research 1989 25 90.
Patel N, Alsat E, Igout A, Baron F, Hennen G, Porquet D & Evain-Brion D (1995)
Glucose inhibits human placental GH secretion, in vitro. J Clin Endocinol Metab,
80,1743–1746.
Pfaeffe RW, Hunter CS, Savage JJ, Duran-Prado M, Mullen RD, Neeb ZP, et al. Three
novel missense mutations within the LHX4 gene are associated with variable pituitary
hormone deficiencies. J Clin Endocrinol Metab 2008;93:1062–71.
Pfaeffle RW, Savage JJ, Hunter CS, Palme C, Ahlmann M, Kumar P, Bellone J,
Schoenau E, Korsch E, Bramswig JH et al. 2007 Four novel mutations of the LHX3
gene cause combined pituitary hormone deficiencies with or with out limited neck
rotation. Journal of Clinical Endocrinology and Metabolism 92 1909–1919.
Pilotta P., et al. 2006 Common polymorphisms of the growth hormone receptor do not
correlate with the growth response to exogenous recombinant human GH in GH-
deficient children. Journal Clinical Endocrinology and metabolism 91 (3) 1178-1180.
Potok MA, Cha KB, Hunt A, et al. WNT signaling affects gene expression in the ventral
diencephalon and pituitary gland growth. Dev Dyn. 2008; 237(4):1006–20.
Pulichino AM,Vallette-Kasic S,Tsai JP,Couture C,Gauthier Y& Drouin J.T pit determines
alternate fates during pituitary cell differentiation. Genes & Development 2003 17 738–
747.
Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and
expansion of the pituitary primordia. Development 2002;129:4229–39.
Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-
Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, Ruddle P, Hurst J, Collin JR, Salt A,
Cooper ST, Thompson PJ, Sisodiya SM, Williamson KA, Fitzpatrick DR, vanHeyningen
V, Hanson IM 2005 Heterozygous mutations of OTX2 cause severe ocular
malformations. Am J HumGenet 76:1008–1022.
Rahimov F, Ribeiro LA, de Miranda E, Richieri-Costa A, Murray JC. GLI2 mutations in
four Brazilian patients: how wide is the phenotypic spectrum? Am J Med Genet A
2006;140:2571–6.
Rajab A, Kelberman D, deCastro SC, Biebermann H, Shaikh H, Pearce K, Hall CM,
Shaikh G, Gerrelli D, Grueters A et al. 2008 Novel mutations in LHX3 are associated
with hypopituitarism and sensorineural hearing loss. Human Molecular Genetics 17
2150–2159.(doi:10.1093/hmg/ddn114)
Ray J, Okamara H, Kelly PA, Cooke NE & Liebhaber SA (1990) Human growth
hormone-variant demonstrates a receptor binding profile distinct from that of normal
pituitary growth hormone. J Biol Chem, 265,7939–7944.

92
Roessler E, Ermilov AN, Grange DK, et al. A previously unidentified amino-terminal
domain regulates transcriptional activity of wild type and disease associated human
GLI2. Hum Mol Genet.2005; 14:2181–2188.
Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet
C Semin Med Genet.2010;154C:52–61.
Roessler, E., Du, Y.Z., Mullor, J.L. et al. (2003) Loss-of-function mutations in the human
GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features.
Proceedings of the National Academy of Sciences of the United States of America, 100,
13424–13429.
Rona RJ & Tanner JM. A etiology of idiopathic growth hormone deficiency in England
and Wales. Archives of Disease in Childhood 1977 52 197–208.
Rosenfeld MG 1991 POU-domain transcription factors: pou-er-ful developmental
regulators. Genes and Development 5 897–907.
Rosenfeld RG, Belgorosky A, Camacho-Hubner C et al. Defects in growth hormone
receptor signaling. Trends in Endocrinology & Metabolism 2007; 18: 134–141.
Rowland JE, Lichanska AM, Kerr LM, White M, d’Aniello EM, Maher SL, Brown R,
Teasdale RD, Noakes PG, Waters MJ 2005 In vivo analysis of growth hormone
receptor signaling domains and their associated transcripts. Mol Cell Biol 25:66–77
Rowlinson SW, Yoshizato H, Barclay JL, Brooks AJ, Behncken SN, Kerr LM, Millard K,
Palethorpe K, Nielsen K, Clyde-Smith J, Hancock JF, Waters MJ 2008 An agonist
induced conformational change in the growth hormone receptor determines the choice
of signaling pathway. Nat Cell Biol 10:740–747.
S. Meyer, M. Ipek, A. Keth, T. Minnemann, M.A. von Mach, A. Weise, J.R. Ittner, P.P.
Nawroth, U. Plockinger, G.K. Stalla, U. Tuschy, M.M. Weber, P.H. Kann. Short stature
and decreased insulin-like growth factor I (IGF-I)/growth hormone (GH)-ratio in an adult
GH-deficient patient pointing to additional partial GH insensitivity due to a R179C
mutation of the growth hormone receptor. Growth Hormone & IGF Research 17 (2007)
307–314.
Salemi S,Besson A, Eble A, Gallati S, Pfaffle RW & Mullis PE. New N-terminal located
mutation (Q4ter) within the POU1F1-gene (PIT-1) causes recessive combined pituitary
hormone deficiency and variable phenotype. Growth Hormone & IGF Research 2003 13
264–268.
Salvatori,R. & Levine,M.A.(1999) Familial dwarfism due to a novel mutation of growth
hormone-releasing hormone receptor gene. Journal of Clinical Endocrinology and
Metabolism, 84,917–923.
Sanchez JE, Perera E, Baumbach L & Cleveland WW. Growth hormone receptor
mutations in children with idiopathic short stature. Journal of Clinical Endocrinology and
Metabolism 1998 83 4079–4083.

93
Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3
activities by an amino terminal repression domain: implication of Gli2 and Gli3as
primary mediators of Shh signaling. Development.1999;126:3915–3924.
Schilter KF, Schneider A, Bardakjian T, Soucy JF, Tyler RC, Reis LM & Semina EV.
2010 OTX2 microphthalmia syndrome: four novel mutations and delineation of
phenotype. Clinical Genetics 79 158-168.
Schindler C, Plumlee C 2008 Inteferons pen the JAK-STAT pathway. Semin Cell Dev
Biol 19:311–318.
Scully KM & Rosenfeld MG. 2002 Pituitary development: regulatory codes in
mammalian organogenesis. Science 295 2231-2235.
Seidah NG, Barale JC, Marcinkiewicz M, Mattei MG, Day R, Chretien M. 1994 The
mouse homeoprotein LIM-3 is expressed early in cells derived from the neuro
epithelium and persists in adult pituitary. DNA Cell Biol. 13:1163–1180.
Selden RF, Wagner TE, Blethen S, Yun JS, Rowe ME & Goodman HM (1988)
Expression of the human growth hormone variant gene in cultured fibroblasts and
transgenic mice. Proc Natl Acad Sci USA, 85,8241–8245.
Shannon W. Davis, Buffy S. Ellsworth, Maria Peréz Millan, Peter Gergics, Vanessa
Schade, Nastaran Foyouzi, Michelle L. Brinkmeier, Amanda H. Mortensen, Sally A.
Camper. Pituitary Gland Development and Disease: From Stem Cell to Hormone
Production. Curr Top Dev Biol. 2013; 106: 1–47.
Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, et al. Multi step
control of pituitary organogenesis. Science.1997;278:1809–12.
Sheng HZ, Zhadanov AB, Mosinger Jr B, et al. 1996 Specification of pituitary cell line
ages by the LIM homeobox gene Lhx3.Science.272:1004–1007.
Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C,
Quelle FW, Nosaka T, Vignali DA, Doherty PC, Grosveld G, Paul WE, Ihle JN. Lack of
IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene.
Nature. 1996; 380:630–633.
Siddle K, Urso B, Niesler CA, Cope DL, Molina L, Surinya KH, et al. Specificity in ligand
binding and intracellular signalling by insulin and insulin-like growth factor receptors.
Biochem Soc Trans 2001; 29: 513-25.
Simeone A, Acampora D,Mallamaci A,Stornaiuolo A,D’Apice MR,Nigro V,Boncinelli E
(1993) A vertebrate gene related to orthodenticle contains a homeodomain of the bicoid
class and demarcates anterior neuroectodermin the gastrulating mouse
embryo.EMBOJ12:2735–2747
Sitz KV, Burks AW, Williams LW, Kemp SF & Steele RW.Confirmation of X linked
hypogammaglobuliemia with isolated growth hormone deficiency as a disease entity.
Journal of Pediatrics 1990 116 292–294.

94
Sloop KW, Dwyer CJ & Rhodes SJ 2001 An isoform specific inhibitory domain regulates
the LHX3 LIM homeodomain factor holoprotein and the production of a functional
alternate translation form. Journal of Biological Chemistry 276 36311–36319.
Snabboon T, Plengpanich W, Buranasupkajorn P et al. 2008 A novel germline
mutation, IVS4+1 G>A, of the POU1F1 gene underlying combined pituitary hormone
deficiency. Hormone Research 69 60-64.
Sobrier ML, Maghnie M, Vie-Luton MP, Secco A, di Iorgi N, Lorini R & Amselem S.
2006 Novel HESX1 mutations associated with a life threatening neonatal phenotype,
pituitary aplasia, but normally located posterior pituitary and no optic nerve
abnormalities. Journal of Clinical Endocrinology and Metabolism 91 4528-4536.
Soos MA, Field CE, Siddle K. Purified hybrid insulin/insulin-like growth factor-I
receptors bind insulin-like growth factor-I, but not insulin, with high affinity. Biochem J
1993; 290: 419-26.
Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, O’Connell SM Gukovsky I,
Carriere C, Ryan AK, Miller AP, Zuo L, Gleiberman AS, Andersen B, Beamer WG,
Rosenfeld MG 1996 Pituitary lineage determination by the Prophet of Pit-1
homeodomain factor defective in Ames dwarfism. Nature 384:327–333.
SW Davis, F Castinetti, LR Carvalho, et al (2010). Molecular mechanisms of pituitary
organogenesis: in search of novel regulatory genes. Mol Cell Endocrinol. 2010 July 8;
323(1): 4–19.
Tahayato A, Sonneville R, Pichaud F, Wernet MF, Papatsenko D, Beaufils P,Cook
T,Desplan C (2003) Otd/Crx,a dual regulator for the specification of ommatidia subtypes
in the Drosophila retina.DevCell5:391–402.
Tajima T, Hattori T, Nakajima T, Okuhara K, Tsubaki J, Fujieda K. A novel missense
mutation (P366T) of the LHX4 gene causes severe combined pituitary hormone
defciency with pituitary hypoplasia, ectopic posterior lobe and a poorly developed sella
turcica. Endocr J 2007;54:637–41.
Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K & Fujieda K. 2009
OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone
deficiency with small anterior and ectopic posterior pituitary, Journal of Clinical
Endocrinology and Metabolism 94 314-319.
Tajima T, Yorifuji T, Ishizu K, Fujieda K. A novel mutation (V101A) of the LHX4 gene in
a Japanese patient with combined pituitary hormone deficiency. Exp Clin Endocrinol
Diabetes 2010;118:405–9.
Takagi M, Ishii T, Inokuchi M, Amano N, Narumi S, Asakura Y, et al. Gradual loss of
ACTH due to a novel mutation in LHX4: Comprehensive mutation screening in
Japanese patients with congenital hypopituitarism. PLoS One. 2012;7:e46008.
Takashi K, John Phillips III. (1992) Detection of molecular heterogeneity in GH-1 gene
Deletions by analysis of PCR amplifications products. Journal of Clinical Endocrinology
and Metabolism, 74, 786-9.

95
Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira
S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc
Natl Acad Sci U S A. 1997; 94:3801–3804.
Takuma N, Sheng HZ, Furuta Y, et al. Formation of Rathke’s pouch requires dual
induction from the diencephalon. Development. 1998; 125(23):4835–40.
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt
I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD 2001
Mutations in PTPN11,encoding the protein tyrosine phosphatase SHP-2, cause Noonan
syndrome. Nat Genet 29:465–468.
Tauber MT, Porra V, Dastot F, Molinas C, Amselem S, Cholin S, Rochiccioli P, Bieth
E.Heterozygous mutation in the WSXWS equivalaent motif of the growth hormone
receptor in a child with poor response to growth hormone therapy. Growth Horm IGF
Res. 1998 Jun;8(3):211-6.
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M,
Bodner S, Grosveld G, Ihle JN 1998 Stat5a and Stat5b proteins have essential and non
essential, or redundant, roles in cytokine responses. Cell 93:841–850.
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M,
Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and
nonessential, or redundant, roles in cytokine responses. Cell. 1998; 93:841–850.
Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F,
Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS.
2001 Heterozygous HESX1 mutations associated with isolated congenital pituitary
hypoplasia and septo-optic dysplasia. Hum Mol Genet 10:39–45.
Treadway JL, Morrison BD, Goldfine ID, Pessin JE. Assembly of insulin/insulin-like
growth factor-1 hybrid receptors in vitro. J Biol Chem 1989; 264: 21450-3.
Treier M, Gleiberman AS, O’Connell SM, et al. Multistep signaling requirements for
pituitaryorganogenesis in vivo. Genes Dev. 1998; 12(11):1691–704.
Turton JP, Mehta A, Raza J, Woods KS, Tiulpakov A, Cassar J, Chong K, Thomas PQ,
Eunice M, Ammini AC et al. 2005 Mutations within the transcription factor PROP1 are
rare in a cohort of patients with sporadic combined pituitary hormone deficiency
(CPHD). Clinical Endocrinology 63 10-18.
Turton JP, Reynaud R, Mehta A, Torpiano J, Saveanu A, Woods KS, Tiulpakov A,
Zdravkovic V, Hamilton J et al. 2005 Novel mutations within the POU1F1 gene
associated with variable combined pituitary hormone deficiency. Journal of Clinical
Endocrinology and Metabolism 90 4762-4770.
Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW
1997 Requirement of STAT5b for sexual dimorphism of body growth rates and liver
gene expression. Proc Natl Acad Sci USA94:7239–7244.

96
Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey
HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver
gene expression. Proc Natl Acad Sci U S A. 1997; 94:7239–7244. [PubMed: 9207075]
Ullrich A, Gray A, Tam AW et al. Insulin-like growth factor I receptor primary structure:
comparison with insulin receptor suggests structural determinants that define functional
specificity. The EMBO Journal 1986; 5: 2503–2512.
Verhaeghe J, Bougassa M, VanHerck E, de Zegher F, Hennen G & Igout A (2001)
Placental growth hormone and IGF-I in a pregnant woman with Pit-1 deficiency. Clin
Endocrinol, 53,645–647.
Vidarsdottir S, Walenkamp MJE, Pereira AM et al. Clinical and biochemical
characteristics of a male patient with a novel homozygous STAT5b mutation. Journal of
Clinical Endocrinology & Metabolism 2006; 91: 3482–3485.
Vnencak-Jones CL, Phillips JA III, Chen EY & Seeburg PH. Molecular basis of human
growth hormone gene deletions. PNAS 1988 85 5615–5619.
Vnencak-Jones CL, Phillips JAIII & DeFen W. Use of polymerase chain reaction in
detection of growth hormone gene deletions. Journal of Clinical Endocrinology and
Metabolism 1990 70 1550–1553.
Wajnrajch MP, Gertner JM, Harbison MD, Chua SC Jr & Leibel RL. Nonsense mutation
in the human growth hormone receptor causes growth failure analogous to the little (lit)
mouse. Nature Genetics 1996 12 88–90.
Walenkamp, M.J., Karperien, M., Pereira, A.M. et al. (2005) Homozygous and
heterozygous expression of a novel insulin-like growth factor-I mutation. Journal of
Clinical Endocrinology and Metabolism, 90, 2855–2864.
Wang Y & Jiang H. Identification of a distal STAT-binding DNA region that may mediate
growth hormone regulation of insulin-like growth factor-I gene expression. Journal of
Biological Chemistry 2005; 280: 10955–10963.
Wang Y, Martin JF, Bai CB. Direct and indirect requirements of Shh/Gli signaling in
early pituitary development. Dev Biol. 2010; 348(2):199–209.
Werner H, Hernandez-Sanchez C, Karnieli E et al. The regulation of IGFI receptor gene
expression. The International Journal of Biochemistry & CellBiology 1995; 27: 987–994.
West BE, Parker GE, Savage JJ, Kiratipranon P, Toomey KS, Beach LR, Colvin SC,
Sloop KW & Rhodes SJ 2004 Regulation of the follicle stimulating hormone beta gene
by the LHX3 LIM-homeodomain transcription factor. Endocrinology 145 4866–4879.
Wit JM, Kiess W, Mullis P.2011 Genetic evaluation of short stature. Best Practice &
Research Clinical Endocrinology & Metabolism 25 1–17.
Woods KA, Dastot F, Preece MA, Clark AJ, Postel-Vinay MC, Chatelain PG, Ranke MB,
Rosenfeld RG, Amselam S & Savage MO. Phenotype: genotype relationships in growth
hormone insensitivity syndrome. Journal of Clinical Endocrinology and Metabolism 1997
82 3529–3535.

97
Xu L, Lavinsky RM, Dasen JS, Flynn SE, McInerney EM, Mullen TM, Heinzel T, Szeto
D, Korzus E, Kurokawa R et al. 1998 Signal-specific co-activator domain requirements
fot pit-1 activation. Nature 395 301-306.
Xu Q, Guo L, Moore H, Waclaw RR, Campbell K, Anderson SA. Sonic hedgehog
signaling confers ventral telencephalic progenitors with distinct cortical interneuron
fates. Neuron. 2010;65:328–340.
Yamashita T, Moriyama K, Sheng HZ, Westphal H. Lhx4, a LIM homeobox gene.
Genomics 1997;44:144–6.
Zhadanov AB, Bertuzzi S, Taira M, Dawid IB, Westphal H. 1995 Expression pattern of
the murine LIM class homeobox gene Lhx3 in subsets of neural and neuroendocrine
tissues. Dev Dyn.202:354–364.
Zhao L, Zevallos SE, Rizzoti K, Jeong Y, Lovell-Badge R, Epstein DJ. Disruption of
SoxB1-dependent Sonic hedgehog expression in the hypothalamus causes septo-optic
dysplasia. Dev Cell.2012; 22(3):585–96.
Zhong Z, Wen Z, Darnell JE Jr. Stat3 and Stat4: members of the family of signal
transducers and activators of transcription. Proc Natl Acad Sci U S A. 1994; 91:4806–
4810.
Zhu X, Zhang J, Tollkuhn J, Ohsawa R, Bresnick EH, Guillemot F, Kageyama R &
Rosenfeld MG 2006 Sustained notch signaling in progenitors is required for sequential
emergence of distinct cell lineages during organogenesis. Genes and Development 20
2739–2753.
Zhu X., Gleiberman, A.S. & Rosenfeld, M.G.(2007) Molecular physiology Of pituitary
development: signaling and transcriptional networks. Physiological Reviews, 933–963.