universidad catÓlica de cuenca - …dspace.ucacue.edu.ec/bitstream/reducacue/6486/1/estudio de...
TRANSCRIPT

1
UNIVERSIDAD CATÓLICA DE CUENCA
Unidad Académica de Ingeniería Química, Biofarmacia,
Industrias y Producción.
FACULTAD DE BIOFARMACIA
ESTUDIO DE LOS PRODUCTOS FINALES DEL
METABOLISMO DE COMPUESTOS
NITROGENADOS
Investigadora:
María Enidt Cuenca Saraguro.
Director:
Ing. César Juela.
CUENCA - ECUADOR
2013
Monografía previa a la
obtención del título de
Químico Farmaceuta.

2
DEDICATORIA
A mi esposo Juan Carlos, y a mi hijo Juan Pablo,
por su constante apoyo, amor y comprensión,
incentivándome así a alcanzar el éxito profesional.
A mis queridos padres, Luis y Edita,
por su amor, cariño y concejos;
para Magali, Fanny, Ana, Jeanelly, Marvin, Liliana, Luis y Ronald,
mis hermanos, gracias por estar siempre conmigo.

3
AGRADECIMIENTO
Mi reconocimiento y gratitud:
A la Universidad Católica de Cuenca, de manera especial a la
Unidad Académica de Ingeniería Química, Biofarmacia, Industrias
y Producción, en la persona de sus Autoridades y Maestros por
haberme recibido en sus aulas y haber hecho de mí un profesional en
Quimica Farmacéutica.
A mi Director de monografía, Ingeniero Cesar Juela, quien con su
valiosa orientación me permitió culminar exitosamente mi
investigación.
A mi esposo, mi hijo, y a mis queridos padrespor su constante apoyo,
amor y comprensión, que me supieron prodigar a cada instante.

4
INDICE
CONTENIDOS
PRELIMINARES:
páginas
CARÁTULA……………………………………………………………………...……..
I
DEDICATORIA……....………..………………………………………………………
II
AGRADECIMIENTO……………………..…..….………………………….……….
III
INDICE………..…….…………………..….……………………………….………….
IV
INTRODUCCIÓN…………………………….…………………………….………..
VII
OBJETIVOS……………………………………………………………………………
VIII
CONTENIDOS:
CAPITULO I
METABOLISMO CELULAR Y COMPUESTOS ORGÁNICOS
NITROGENADOS
1.1 Aspectos generales del Metabolismo Celular...…………..........
10
1.2 Catabolismo……………………………........................................
11
1.3 Anabolismo……………………………………………………………..
13
1.4 Fases del metabolismo.………............………………………….….
14
1.5Generalidades de los Compuestos Orgánicos Nitrogenados….…
14
1.6Clases de compuestos nitrogenados…………………………….....
15
1.7 Fijación Biológica del Nitrógeno…………………………………….
20
CAPITULO II
AMINOÁCIDOS
2.1 Los Aminoácidos son las subunidades de las proteínas…..…….
21
2.2 Funciones………………………………………………………………
22
2.3 Estructura química………………….………………………………
22
2.4Clasificación de los Aminoácidos………..…….……………………
23

5
2.5 Propiedades físico-quimicas delos aminoácidos………...………. 26
2.6 Clases de Amoniácidos………………..………………………………
29
2.7 Funciones de los Aminoácidos…………………………………
30
2.8 Enlace peptídico……….…………………………………………
33
CAPITULO III
PÉPTIDOS Y PROTEÍNAS
3.1Generalidades de los péptidos…………………………………
34
3.2Nomenclatura…………………………………………………….
35
3.3 Funciones de los péptidos………………………………………..…..
36
3.4 Proteínas……………………………………………………..........................
40
3.5 Estructura de las proteínas…………………..…………………
40
3.6 Propiedades de las proteínas…..…………..………………………
45
3.7 Clasificación de las proteínas…..………...…………………………...
46
3.8Funciones de las proteínas………..…………………………………….
48
3.9 Alimentación, Digestión y Absorción de Proteínas….……..
49
CAPITULO IV
ÁCIDOS NUCLÉICOS
4.1 Características Generales..….………………………………….
52
4.2 Estructura de los ácidos nucleicos…………………..…………
52
4.4 El ARN. Ácido ribonucleico……………………………………..
57
4.5 El ADN. Ácido desoxirribonucleico………………..…………..
61
4.6 Funciones Biológicas….…………………….……………..…….
66
CAPITULO V
METABOLISMO DE LOS COMPUESTOS NITROGENADOS
5.1 Metabolismo de Proteínas……………..………………………….
67

6
5.2 Metabolismo de Aminoácidos…..………….…………………….
70
5.3 Metabolismo de los Nucléotidos…………………………………
90
CAPITULO VI
ESTUDIO DE LOS PRODUCTOS FINALES DEL METABOLISMO
DE LOS COMPUESTOS NITROGENADOS
6.1 Amoníaco………...……………………………………………..…...
101
6.2 Urea…………………………………………………………….…….
104
6.3 Ácido Úrico………..………………………………………………..
109
6.4 Creatina….………………………………………………………….
111
6.5 Creatinina…………………………………………….………….....
114
CAPITULO VII
ENFERMEDADES CAUSADAS POR EL DESEQUILIBRIO DEL
BALANCE DEL NITRÓGENO.
7.1 Enfermedades causadas por el aumento de productos finales
del metabolismo…………………………………………………….
117
7.2 Enfermedades asociadas a deficiencias de proteínas y
aminoácidos………………………………………………………….
122
CAPITULO VII
DETERMINACIÓN DEL ANALISIS CLÍNICO DE LOS
PRODUCTOS FINALES DE COMPUESTOSNITROGENADOS.
8.1 Análisis clínico de los productos finales de compuestos
nitrogenados y su significado clínico en muestra de orina y
sangre…………………………………………………..…………….
125
CONCLUSIONES……………………………………………………
130
BILBIOGRAFÍA………………………………………………………
132

7
INTRODUCCIÓN
El Nitrógeno junto a otros elementos, como Carbono, Oxígeno e Hidrógeno
participan en la constitución de las moléculas orgánicas fundamentales de
la materia viva. Este compuesto orgánico de gran jerarquía biológicale
están asignadas funciones muy importantes, como lo son las proteínas y
los nucleótidos. Este elemento constituye por sí solo el 3% del peso
corporal.
En el ser humano, la principal fuente de sustancias nitrogenadas son las
proteínas de la dieta. Como estos compuestos, a diferencia de
carbohidratos y grasas, no se almacenan como reserva, los niveles en las
células se regulan por el equilibrio entre anabolismo y catabolismo, es
decir un balance entre biosíntesis y degradación de proteínas, a lo que
también se conoce como recambio normal de proteínas. Por lo tanto, un
adulto sano que ingiere una dieta variada y completa se encuentra
generalmente en situación de “equilibrio nitrogenado”, un estado en el
que la cantidad de nitrógeno ingerida cada día es equilibrada por la
cantidad excretada por heces, orina y sudor, sin que se produzca ningún
cambio neto en la cantidad de nitrógeno del organismo. Sin embargo, en
ciertas condiciones, el organismo se halla en equilibrio nitrogenado
negativo o positivo.
En la situación de equilibrio nitrogenado negativo se excreta mayor
cantidad de nitrógeno del que se ingiere. Esto tiene lugar en la inanición,
la desnutrición proteica y en ciertas enfermedades que cursan con
catabolismo aumentado. Durante la inanición prolongada las cadenas
carbonadas de los aminoácidos son necesarias para la gluconeogénesis; el
amoníaco (nitrógeno) liberado de los aminoácidos es excretado
principalmente en forma de urea y no se reincorpora a las proteínas.
También puede darse un equilibrio negativo durante la vejez, la fiebre
severa, diabetes no controlada y, de gran importancia médica, neoplasias,

8
donde el catabolismo se encuentra exacerbado. En el otro extremo, puede
hallarse equilibrio nitrogenado positivo cuando lo ingerido supera a lo
excretado, tal caso se da en niños en edad de crecimiento, puesto que están
aumentando su peso corporal e incorporando más aminoácidos en las
proteínas somáticas. Puede darse equilibrio nitrogenado positivo durante
el embarazo.
El estudio del metabolismo de los compuestos nitrogenados dentro del
organismo comprende uno de los más grandes temas de la Bioquímica. En
esta recopilación bibliográfica me ocupare, por un lado de los procesos que
implican su metabolismo y de igual forma la importancia de la
determinación de los productos finales formadas de dicho proceso,
debido a que repercuten en la fisiología normal del cuerpo
humano,por lo tantola determinación del balance de nitrógeno en un ser
humano es un parámetro eficaz en la control de deficiencias o excesos de
proteínas en su dieta y junto a otros indicadores, su estado nutricional y de
salud.
La importancia más grande que tiene su determinación es que cualquier
elevación en suero puede ser señal de un trastorno renal.

9
OBJETIVOS
1.1 Objetivo General:
1.1.1 Explicar el fundamento, procesos químicos de producción y
alteraciones fisiológicas que producen en el organismo los
productos finales de compuestos nitrogenados, al mismo tiempo
revisar las técnicas para su determinación analítica en el
laboratorio clínico.
1.2 Objetivo Específicos:
1.2.1 Describir las generalidades de los compuestos orgánicos
nitrogenados.
1.2.2 Analizar la estructura, propiedades y función de los
aminoácidos.
1.2.3 Examinar la estructura, clasificación y desnaturalización de las
proteínas.
1.2.4 Establecer la estructura, clases y formación de los ácidos
nucleicos.
1.2.5 Detallar el metabolismo de los aminoácidos, proteínas y
nucléotidos.
1.2.6 Caracterizar cada uno de los productos finales del metabolismo
de los compuestos nitrogenados.
1.2.7 Tipificar las enfermedades causadas por el desequilibrio de los
productos finales del metabolismo de compuestos nitrogenados.
1.2.8 Determinar las técnicas para el análisis clínico de los productos
finales de compuestos nitrogenados.

10
CAPITULO I
METABOLISMO CELULAR Y COMPUESTOS ORGÁNICOS
NITROGENADOS
ASPECTOS GENERALES
El metabolismo celularcomprende una serie de transformaciones químicas y
procesos energéticos que ocurren en el ser vivo.
El conjunto de reacciones químicas y enzimáticas se denomina ruta o vía
metabólica.
LUGARES ESPECÍFICOSDE LAS CÉLULAS DONDE SE
DESARROLLAN LAS RUTAS METABÓLICAS
Fig.1. Núcleo celular (Replicación de DNA, Síntesis de tRNA y
mRNA).Nucléolo(Síntesis de rRNA). Ribosomas (Síntesis de proteínas). Aparato de
Golgi (Maduración de glicoproteínas y otros componentes de las membranas). Retículo
endoplasmático liso (Síntesis de lípidos, Transporte intracelular). Mitocondria
(Oxidación del piruvato, Ciclo de Krebs, Fosfoliración oxidativa intracelular).
Citoplasma (glucólisis).
Funciones del metabolismo:
a) Obtener energía química (ATP) degradando nutrientes ricos en energía.
b) Convertir moléculas nutrientes en moléculas celulares.
c) Polimerizar precursores monoméricos a proteínas, ácidos nucleicos,
polisacáridos, etc.
d) Sintetizar y degradar biomoléculas requeridas en funciones celulares
especializadas.
El metabolismo va a descomponerse en dos series de reacciones, que aunque
son dos procesos contrarios, los dos funcionan coordinada y armónicamente, y
constituyen una unidad difícil de separar.

11
CATABOLISMO.- Es un proceso destructivo generador de energía, las
moléculas complejas son degradadas formándose moléculas más simples; como
por ejemplo: la glucólisis.
VÍAS DEL CATABOLISMO
Los organismos autótrofos fijan la
energía solarenforma de energía
químicacontenida en loscompuestos
orgánicos, glucosa, en particular.Esta
energía, convenientemente liberada,
seráutilizada posteriormente por las
partes de la plantaque no tienen
cloroplastos, como suele ser elcaso de las
raíces y tallos no verdes, o por toda
laplanta cuando falta la energía solar. Es
tambiénesta energía la que permite la
vida de losorganismos heterótrofos.
Existen dos tipos de catabolismos según
sea el aceptor final de electrones:
La respiración celular ylas
fermentaciones son las vías catabólicas máscorrientes para la obtención de la
energía contenidaen las sustancias orgánicas. Ambas vías, noobstante, tienen
una primera fase común: laglucolisis.
1. Fermentación. Si se degrada a un compuesto todavía orgánico pero
mássencillo. En ella tanto el aceptor final de electrones es un
compuestos orgánicos.
2. Respiración celular. Si la degradación del compuesto orgánico es
hasta CO₂y H₂O. El aceptor final de electrones es una sustancia
inorgánica. La respiración celular puede ser:
a. Respiración aerobia. Se necesita oxígeno.
b. Respiración anaerobia. No necesita oxígeno

12
FASES DEL CATABOLISMO
a. Fase I, inicial o
preparatoria.-las
grandes moléculas de los
elementos nutritivos se
degradan hasta liberar sus
principales componentes (los
polisacáridos se degradan en
monosacáridos; los lípidos a
ácidos grasos y glicerina, y
las proteínas liberan sus
aminoácidos).
b. Fase II o fase
intermedia.-en ella los
diversos productos formados
en la faseI, son convertidos
en una misma moléculas,
más sencillas el Acetil-
coenzima A (acetil CoA).
c. Fase III o fase final, en
la que el acetil-CoA (se
incorpora al ciclo de Krebs)
da lugar a moléculas
elementales CO₂ y H₂O.
De estas tres fases, la
intermedia y la final son
comunes para todos los
principios inmediatos orgánicos,
glúcidos, lípidos y proteínas.
El catabolismo de cada uno deellos
difiere en la fase inicial, losglúcidos
(glucólisis) y las
proteínas(desaminaciónytransaminac
ión),ocurre en el hialoplasma, mientras
quepara los lípidos (β-oxidación),
ocurreen la matriz mitocondrial.

13
ANABOLISMO:Es la parte constructiva del metabolismo, consiste en la
síntesis de moléculas complejas a partir de otras más sencillas, con gasto de
energía, tomada de los ATP producidos durante las fases catabólicas: por
ejemplo, la fotosíntesis, la síntesis de proteínas o la replicación del ADN.
Estas moléculas sintetizadas pueden:
Formar parte de la propia estructura de la célula.
Ser almacenadas para su posterior utilización como fuente de energía.
Ser exportadas al exterior de la célula.
Se puede distinguir dos tipos de anabolismo en los distintos tipos de células:
1. Anabolismo autótrofo. Las células autótrofas son las células
vegetales y algunos tipos de bacterias. Todas ellas son capaces de
aprovechar distintas fuentes de energía localizadas en el exterior de la
célula, fabrican moléculas orgánicas a partir de materia inorgánica (agua,
dióxido de carbono y sales minerales), y una fuente de energía. A su vez
presenta dos tipos:
Quimiosíntesis.-utiliza como fuente de energía ciertas reacciones de
óxido-reducción de materia inorgánica. La realizan algunos grupos de
bacterias (bacterias del Fe, del H, etc.)
Fotosíntesis.-utiliza la luz solar como fuente de energía. También
presenta distintos tipos, laanoxigénica, que no desprende O2 (la que
realizan las bacterias púrpuras fotosintéticas, en la que el H2S cede los
electronesy se desprende S elemental), y la oxigénica(que realizan las
cianobacterias y los vegetales, en la que el H2O cede los electronesy se
desprende O2).
2. Anabolismo heterótrofo.- Las células de los animales, de los hongos y
de muchas bacterias son heterótrofas porque solo pueden utilizar en su
anabolismo energía química que procede de la destrucción de compuestos
orgánicos que previamente han sido tomados del exterior. En este caso, por
tanto, la fuente de energía procede del interior de la propia célula. En el
anabolismo heterótrofo, se parte de sustancias orgánicas sencillas y con
ellas se elaboran otras más complejas.

14
FASES DEL METABOLISMO: El mantenimiento de la vida requiere de
un cambio continuo de sustancias y una constante transformación de la
energía, para que ocurran estos cambios se deben cumplir tres fases que son las
siguientes:
1. Absorción.- Es la fase donde penetran en el protoplasma las sustancias
químicas y la energía que procede del medio ambiente, a través de la
membrana plasmática. Esto implica que todo lo que absorbe el
protoplasma debe hallarse en solución sean, sólidas, líquidas o gaseosas.
2. Transformación.- El protoplasma transforma las especies químicas y
la energía absorbidas. Comprende especialmente:
2.1 Secreción.- Consiste en que el protoplasma produzca compuestos
(enzimas o fermentos) que intervienen en las transformaciones.
2.2 Digestión.- Consiste en hacer solubles las sustancias absorbidas que
las pone en condiciones de entrar en reacción con formación de otras
sustancias químicas.
2.2.1 Asimilación.- Consiste en que el protoplasma se transforme en
algunos de sus componentes propios.
2.2.2 Desasimilación.- Consiste en que en el protoplasma se
desintegra parte de sus componentes o de sus reservas, de los
que resultan los compuestos y la energía que interviene en la
asimilación.
3. Excreción.-Consiste en la eliminación de las especies químicas que no sé
incorporaron al protoplasma o se dispersa energía (calor, luz).
La absorción, transformación y excreción que constantemente se produce en los
organismos vivos dan un crecimiento de la materia y de la energía (anabolismo)
o de un decrecimiento o pérdida de materia y energía (catabolismo).
GENERALIDADES DE LOS COMPUESTOS ORGÁNICOS
NITROGENADOS
El Nitrógeno (N) junto a otros elementos, como Carbono, Oxígeno e Hidrógeno
participan en la constitución de las moléculas orgánicas fundamentales de la
materia viva. Entre los compuestos constituyentes del organismo, el Nitrógeno
forma parte de un grupo de compuestos orgánicos de gran jerarquía biológica a
los cuales están asignadas funciones muy importantes, como lo son las
proteínas y los nucleótidos. Este elemento constituye por sí solo el 3% del peso
corporal.

15
En la atmósfera, el Nitrógeno molecular (N2), es muy abundante. Esta
molécula es casi no reactiva o inerte debido a su triple enlace que la estabiliza.
Antes de poder ser utilizado por los animales, el nitrógeno atmosférico debe ser
“fijado” mediante una cadena de reacciones.
Las legumbres son capaces de absorber el nitrógeno directamente del aire,
siendo éste transformado en amoníaco (amonificación) y luego en nitrato por
bacterias que viven en simbiosis con la planta en sus raíces. El nitrato es
posteriormente utilizado por la planta para formar el grupo
amino(nitrificación), este grupo amino es el componente esencial de los
aminoácidos y los ácidos nucleicos, vitales para la vida y los seres vivos.
CLASES DE COMPUESTOS NITROGENADOS
AMINAS
ESTRUCTURA Y NOMENCLATURA
Son compuestos químicos orgánicos que se consideran como derivados del
amoníaco (NH₃) y resultan de la sustitución de los hidrógenos de la molécula
por los radicales alquilo.

16
Según se sustituyan uno, dos o tres hidrógenos, las aminas serán primarios,
secundarios o terciarios, respectivamente.
Fig.6 Tipos de aminas.
Se los nombra deacuerdo al sistema IUC, el o los radicales con la terminación il
seguido de la palabra amina.
Fig. 7 Ejemplos de aminas.
PROPIEDADES FÍSICAS
Las de peso molecular bajo son líquidas y de olor desagradable.
Son muy solubles en disolventes orgánicos pero no en disolventes iónicos o
polares como el agua.
Su principal característica es que tienen un marcado carácter básico (como
el amoniaco del que provienen). Por ejemplo:

17
AMINAS DE IMPORTANCIA BILÓGICA
Histamina
Ácido γ–aminobutírico (GABA)
Catecolaminas. (Dopamina, Adrenalina y Noradrenalina).
Hormona Tiroidea.
Melatonina.
Serotonina.
Creatina.
AMIDAS
ESTRUCTURA Y NOMENCLATURA.
Son compuestos cuaternarios nitrogenados y oxigenados; R` Y R`` pueden ser
radicales alquilo o hidrógenos.
PROPIEDADES FÍSICAS
Las amidas sencillas a excepción de (formamida), son todas sólidas y
solubles en agua, sus puntos de ebullición son bastante más altos que los
de los ácidos correspondientes.
Casi todas las amidas son incoloras e inodoras.
Son neutras frente a los indicadores.
Fig. 8 Estructura de
las amidas.
Fig. 9 Se nombra según el ácido del que
proviene seguido con la terminación –amida.

18
Los puntos de fusión y ebullición de las amidas secundarias son bastante
menores.
Las amidas terciarias no pueden asociarse, por lo que son líquidos
normales, con puntos de fusión y ebullición de acuerdo con su peso
molecular.
AMIDAS DE ESPECIAL IMPORTANCIA
La Ureaes uno de los compuestos más importantes derivado del ácido
carbónico. Es el producto final del metabolismo de las proteínas y el
primer compuesto orgánico que se sintetizó en un laboratorio.
El Nylon es una fibra poliamídica que se produce en la condensación de
una diamida y un ácido dicarboxílico. La poliamida, con una masa
molecular entre 10000 y 25000, funde a unos 250ºC y el material fundido
puede ser estirado en finos hilos. Estos hilos sometidos, a temperatura
ambiente, a una tensión que los alarga hasta una longitud 4 veces
superior, a diferencia de los materiales elásticos, no se contrae cuando
cesa la tensión, transformándose en este proceso de estiramiento en un
material fibroso que posee mayor resistencia y brillo, y es parecido a la
seda natural. Se utiliza para fabricar medias, paracaídas, alfombras y
muchos otros artículos.
NITRILOS
ESTRUCTURA Y NOMENCLATURA
Son compuesto químicos en cuya molécula existe el
grupo funcional cianuro o ciano.
Se los nombran añadiendo el sufijo nitrilo,alnombre de la cadena principal;
también los nombres de cianuros, pero aquí se disminuye un átomo de carbono
y la terminación en ilo. Ejemplo:
PROPIEDADES FÍSICAS
Su característica principal es la polaridad del triple enlace.
La mayoría de los nitrilos son líquidos muy tóxicos.

19
NITRILOS DE ESPECIAL IMPORTANCIA
a. Cianuro de hidrógeno: también llamado ácido cianhídrico o
metanonitrilo es unlíquido incoloro muy venenoso, pues la dosis mortal
para una persona es de 0'05mgr. También se emplea como fumigante.
b. Cianuro de vinilo: se utiliza en la fabricación del caucho sintético Buna-
N, que esun copolimetil de este compuesto y butadieno.También se utiliza
en la fabricaciónde fibras poliacrílicas como el orlón.
NITROCOMPUESTOS
ESTRUCTURA Y NOMENCLATURA:
La estructura de los nitrocompuestos es la siguiente:
Se debe distinguir entre los nitrocompuestos, en los que el grupo nitro (-NO2)
estádirectamente unido a un átomo de carbono y los esteres nitrosos o nítricos,
donde el gruponitro está unido a un oxígeno:
Se nombran colocando el prefijo-nitro indicando la posición que ocupa dentro
de lacadena principal cuando sea necesario, delante del nombre del alcano
correspondiente.
APLICACIONES DE LOS NITROCOMPUESTOS
Los nitrocompuestos son muy utilizados como explosivos.
Se llama explosivo a cualquier sustancia que por la acción de una causa
externa (roce, percusión, temperatura) se transforma en un tiempo muy breve
en gases con un gran contenido térmico. Este calor, al no poderse disipar,
provoca un violento estallido, transformándose en energía mecánica. La
explosión de los nitroderivados es una combustión rápida en la que el oxígeno
procede de la misma molécula. Puestoque los grupos nitro son los
suministradores de oxígeno, la fuerza explosiva aumenta con el número de los
mismos.
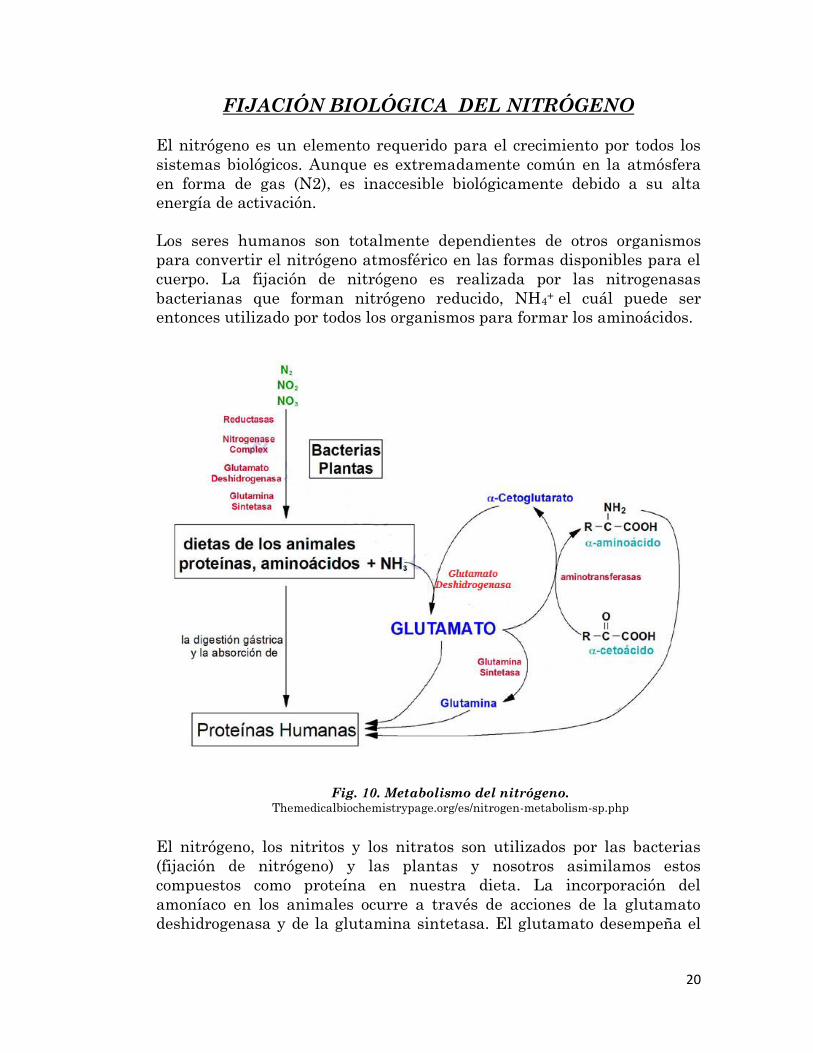
20
FIJACIÓN BIOLÓGICA DEL NITRÓGENO
El nitrógeno es un elemento requerido para el crecimiento por todos los
sistemas biológicos. Aunque es extremadamente común en la atmósfera
en forma de gas (N2), es inaccesible biológicamente debido a su alta
energía de activación.
Los seres humanos son totalmente dependientes de otros organismos
para convertir el nitrógeno atmosférico en las formas disponibles para el
cuerpo. La fijación de nitrógeno es realizada por las nitrogenasas
bacterianas que forman nitrógeno reducido, NH4+ el cuál puede ser
entonces utilizado por todos los organismos para formar los aminoácidos.
El nitrógeno, los nitritos y los nitratos son utilizados por las bacterias
(fijación de nitrógeno) y las plantas y nosotros asimilamos estos
compuestos como proteína en nuestra dieta. La incorporación del
amoníaco en los animales ocurre a través de acciones de la glutamato
deshidrogenasa y de la glutamina sintetasa. El glutamato desempeña el
Fig. 10. Metabolismo del nitrógeno. Themedicalbiochemistrypage.org/es/nitrogen-metabolism-sp.php

21
papel central en el flujo de nitrógeno en los mamíferos, sirviendo como
donante y receptor de nitrógeno.
El nitrógeno reducido ingresa al cuerpo humano como aminoácidos libres
dietéticos, proteína, y amoníaco producido por las bacterias del tracto
intestinal. Un par de enzimas principales, la glutamato deshidrogenasa y
la glutamina sintasa, se encuentran en todos los organismos y efectúan la
conversión de amoníaco en los aminoácidos glutamato y glutamina,
respectivamente. Los grupos amino y amido de estas dos substancias son
libremente transferidos a otros esqueletos de carbono por reacciones de
transaminación y transamidación.
CAPITULO II
AMINOÁCIDOS
LOS AMINOÁCIDOS SON LAS SUBUNIDADES DE LAS
PROTEÍNAS
Los aminoácidos, son los constituyentes principales de las proteínas. Hay 20
aminoácidos comunmente presentes en las proteínas, cada uno identificado de
manera específica por el grupo variable (grupo R) unido al carbono alfa.
Los aminoácidos en solución con pH neutro son sobre todo ionesdipolares, que
es la forma en que por lo general están presentes los aminoácidos en el pH
celular. Cada grupo carboxilo (COOH) dona un protón y se disocia (-COO-),
mientras que cada grupo amino (-NH2) acepta un protón y seconvierte en(-
NH3⁺). Debido a sus grupos amino y carboxilo, los aminoácidos en solución
resisten los cambios de acidez y alcalinidad, de tal suerte que son
amortiguadores biológicos importantes.
Fig. 11. Biosíntesis de aminoácidos. Themedicalbiochemistrypage.org/es/nitrogen-metabolism-sp.php

22
FUNCIONES:
Son precursores de los peptidos y las proteínas.
Biosintesis de purinas, pirimidinas y porfirinas.
Forman parte de vitaminas.
Son precursores de la síntesis de algunas hormonas.
Algunos amminoácidos son antibióticos (cloramfenicol).
Algunos son metabolitos intermediarios de importantes vías metabolicas.
ESTRUCTURA QUÍMICA.- se caracterizan por poseer un grupo carboxilo (-
COOH) y otro amino (-NH₂), un átomo de hidrógeno y un grupo distintivo, el
radical (-R), todos enlazados a un mismo átomo de carbono asimétrico, el
carbonop alfa.
Fig. 12 Estructura química de un aminoácido.
El carbono α corresponde al C2 y –R representa al radical, diferente para cada
aminoácido. En todos los aminoácidos ecepto la glicina el Cα tiene cuatro
sustituyentes diferentes; es un centro quiral.
Los 20 aminoácidos se diferencian en: tamaño, forma, carga, capacidad de
formar puentes de hidrógeno o reactividad química del –R. para la

23
denominación de los aminoácidos se usan las tres primeras letras de su nombre
común, por ejemplo: glicina Gly; alanina Ala, etc.
CLASIFICACIÓN DE LOS AMINOÁCIDOS
Los aminoácidos se agrupan según las propiedades de sus cadenas laterales.
Estas categorías generales en realidad incluyen aminoácidos con un intervalo
bastante amplio de propiedades. Los aminoácidos clasificados como poseedores
de cadenas laterales no polares tienden a presentar propiedades hidrófobas,
en tanto que los clasificados como polares, son más hidrófilos. Los aminoácidos
tienen cadenas laterales con un grupo carboxilo. A pH celular este grupo se
disocia, de manera que el grupo –R queda con carga negativa. Los aminoácidos
básicos adquieren carga positiva cuando el grupo amino en su cadena lateral
acepta un ion hidrógeno. Las cadenas laterales ácidas y básicas son iónicas al
pH celular y por tanto hidrófilas.
C
L
A
S
I
F
I
C
A
C
I
Ó
N
DE
LOS
A
M
I
N
O
Á
C
I
D
O
S
1.1 Sin carga
eléctrica o
carganeutra
1.2 Con carga
eléctrica.
1.2.1 Aminoácidos
básicos
Ácido aspártico.
Ácido glutámico.
2.1 Alanina
2.2 Valina.
2.3 Leucina.
2.4 Isoleucina.
2.5 Metionina.
2.6 Prolina.
2.7 Fenilalanina.
2.8 Triptófano.
1.1.1 Glicina.
1.1.2 Serina.
1.1.3 Treonina.
1.1.4 Cisteína.
1.1.5 Tirosina.
1.1.6 Glutamina.
1.1.7 Asparagina.
1. Aminoácidos
Polares
2. Aminoácidos
Apolares
1.2.2 Aminoácidos
ácidos.
Arginina.
Lisina.
Histidina
.

24
Aminoácidos Polares:aquellos con tendencia ainteractuar con el medio
acuoso, característica clave para el ordenamiento en el espacio delas proteínas.
Polares sin carga.- Siete son los aminoácidos cuyo grupo –R es polar pero
sin carga. La glicinaposee la cadena más simple, un átomo de hidrógeno. La
serinay treoninason portadores de un grupo hidroxilo (-OH). La
asparaginay glutamina, poseen cadenas laterales portadoras de un grupo
amida, y por hidrólisis dan lugar, respectivamente, al aspartato y glutamato,
dos aminoácidos con carga negativa. La tirosinaposee un grupo fenólico y la
cisteína debe su polaridad a la presencia de un grupo tiólico(-SH).
Fig. 13 Aminoácidos Polares Neutros.
Polares con carga.-presentan un grupo adicional ácido o base en el grupo-
R.
1. Aminoácidos Ácidos o Polares con carga negativa.- Existen
dos aminoácidos cuyo resto polar posee carga negativa a pH fisiológico,
debida a la presencia de un grupo carboxilo (-COOH) en el radical, el
ácido glutámicoy el ácido aspártico.
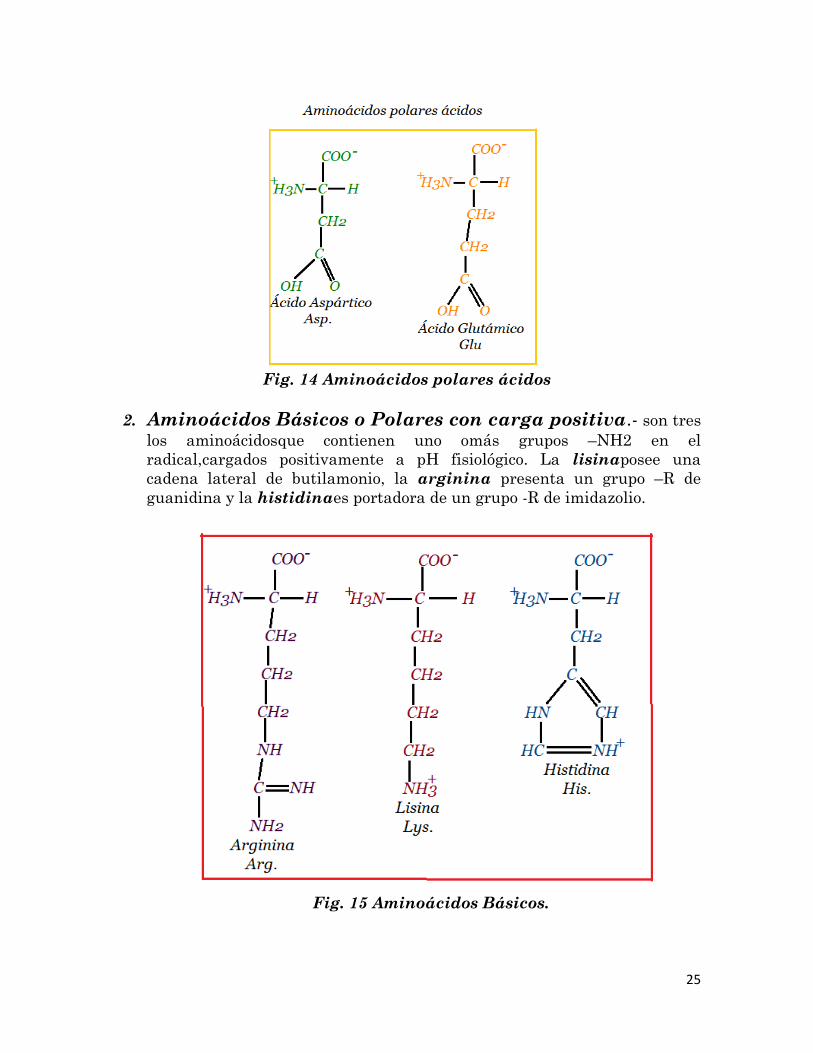
25
Fig. 14 Aminoácidos polares ácidos
2. Aminoácidos Básicos o Polares con carga positiva.- son tres
los aminoácidosque contienen uno omás grupos –NH2 en el
radical,cargados positivamente a pH fisiológico. La lisinaposee una
cadena lateral de butilamonio, la arginina presenta un grupo –R de
guanidina y la histidinaes portadora de un grupo -R de imidazolio.
Fig. 15 Aminoácidos Básicos.

26
Aminoácidos Apolares.-se caracterizan porque su –R es una cadena
alifática. Debido a ello no se generan zonas electronegativas o electropositivas.
Cuando formanparte de las proteínaslos – R tienden a alejarse del medio
acuoso e interaccionar entre sí en zonasinternas de la molécula. Son 8 los
aminoácidos que pertenecen a este grupo;alanina, valina, leucina e
isoleucina, poseen cadenas laterales de hidrocarburos alifáticos. La
metioninatiene una cadena lateral de éter tiólico (C-S-C). La prolina es el
único aminoácido cíclico, pues el grupo -R se cierra sobre el N del grupoamino
(realmente es un amina secundaria). Por su parte, la fenilalaninay el
triptófanocontienen grupos aromáticos.
Fig. 16 Aminoácidos apolares.
PROPIEDADES FÍSICO-QUÍMICAS DE LOS AMINOÁCIDOS
Son anfóteros (tienen grupo amino y otro carboxílico)
La mayoría presenta un punto de fusión cercano a 300˚C.
Son compuestos iónicos, por lo tanto son más solubles en solventespolares.
Muy solubles en agua.
Mayoría insolubles en solventes orgánicos.
Polaridad.
Aromaticidad.

27
Tamaño, flexibilidad de conformación.
Capacidadde formar enlaces cruzados
Reactividad química. Capacidad de unión a hidrógeno.
CARÁCTERANFÓTERO: Una molécula se denomina anfótera cuando
puede comportarse como un ácido o como una base dependiendo del pH del
medio donde se encuentre. Éste es el caso de los aminoácidos: al tener un grupo
carboxilo pueden desprender protones (H+) por lo que tienen carácter ácido; por
otra parte, al poseer un grupo amino, son capaces de aceptar protones (H+) y,
por tanto, también tienen carácter básico.
Al pH existente habitualmente en los medios biológicos, cercano a la
neutralidad (pH=7) ambos grupos suelen estar ionizados y los aminoácidos
aparecen como iones dobles o dipolar, conocido como zwitterión.
Fig. 17 Carácter anfótero de una molécula.
A un pH más ácido, los aminoácidos tendrán carga neta positiva, pues los H+
del medio son captados por el grupo carboxilo ionizado, neutralizando éste y
quedando únicamente la carga del grupo amino.Por el contrario, en un pH más
básico, el grupo amino cederá un H+ al medio y el aminoácido quedará con
carga negativa.
La cadena lateral (R) puede tener grupos ionizables que participan en la carga
eléctrica del aminoácido.
El valorde pH para el cual un aminoácido tiene carga neta 0, es decir, posee
tantas cargas positivas como negativas, se denomina punto isoeléctrico.
Como cada aminoácido presenta un punto isoeléctrico diferente, ya que posee

28
cadenas laterales distintas, se puede utilizar un método de separación de
aminoácidos, denominado electroforesis, que se basa en este concepto. Dicho
método consiste en situar una disolución de los aminoácidos que se quieren
separar en un campo eléctrico. Los aminoácidos con carga neta negativa se
desplazarán hacia el ánodo, mientras que los que tengan carga positiva lo
harán hacia el cátodo y los que se encuentren en su punto isoeléctrico no se mo-
verán. Al modificar el pH de la disolución, las cargas de los aminoácidos irán
variando y se podrán separar en el campo eléctrico.
ESTEREOISOMERÍA:es decir presenta isómeros que difieren en su
orientación espacial.
Por ser el carbono α un carbonoasimétrico o quiral, los aminoácidos pueden
presentar dos posiciones espaciales, formando imágenes especulares. Excepto
la Gly, que el R es H.
Para diferenciar ambos isómeros en una fórmula plana, se escribe la cadena
lateral R hacia arriba y los grupos amino y carboxilo a ambos lados del carbono:
el grupo amino se sitúa a la derecha para representar el estereoisómero
D[dextrorrotatorio (+)] y a la izquierda para representar el estereoisómero L
[Levorrotatorio (-)].
Fig. 18 Estereoisomería.

29
Todos los aminoácidos proteinogénicos son isómeros L, aunque es posible
encontrar D-aminoácidos en determinados compuestos biológicos, en la pared
bacteriana o en ciertos antibióticos.
Los estereoisómeros tienen idénticas propiedades físicas y químicas, con
excepción claro está de la polarización de la luz.
Pero los estereoisómeros presentan además diferentes propiedades
bioquímicas:
Todos los aminoácidos que forman las proteínas están en su forma L, con
excepción claro de la glicina.
Los D-aminoácidos los encontramos en los antibióticos o en algunas paredes
bacterianas, o incluso en algunas plantas.
Algunos aminoácidos presentan más estereoisómeros debido a que poseen
más carbonos quirales, y por lo tanto poseen 2n estereoisómeros.
CLASES DE AMINOÁCIDOS
Con algunas ecepciones, las bacterias y las plantas pueden sintetizar todos los
aminoácidos que necesitan a partir de sustancias más sencillas. Cuando
disponen de la materia prima necesaria, las células de los seres humanos y
otros animales pueden producir casi todos los aminoácidos de importancia
biológica. Los que no pueden y por ello deben provenir de los alimentos, se
denominan aminoácidos esenciales y los que el organismo humano produce
por sí solo se denominan,aminoácidos no esenciales.
1. AMONIÁCIDOS ESENCIALES:son aquellos que el cuerpo humano
no puede generar por sí solo. Esto implica que la única fuente de estos
aminoácidos en esos organismos es la ingesta directa a través de la dieta.
Las rutas para la obtención de estos aminoácidos esenciales suelen ser
largas y energéticamente costosas. Cuando un alimento contiene proteínas
con todos los aminoácidos esenciales, se dice que son de alta o de buena
calidad. Algunos de estos alimentos son: la carne, los huevos, los lácteos y
algunos vegetales como la espelta, la soja y la quínoa.
Los animales diferien en su capacidad de biosintesis; lo que es un
amonoácido esencial en una especie no siempre lo es en la otra. Entre los
aminoácidos esenciales para el ser humano se incluyen: valina, leucina,
isoleucina, fenilalanina, triptófano, metionina, treonina, lisina, histidina y
en ninos, arginina.

30
FUNCIONES DE LOS AMINOÁCIDOS ESENCIALES
1.1 L-Valina:Es un aminoácido hidrofóbico de cadena alifática. Estimula el
crecimiento y reparación de los tejidos, el mantenimiento de diversos
sistemas y balance de nitrógeno.
1.2 L-Leucina: Junto con la isoleucina y la hormona del crecimiento (HGH)
interviene con la formación y reparación del tejido muscular.
1.3 L-Isoleucina: Junto con la leucina y la hormona del crecimiento
intervienen en la formación y reparación del tejido muscular.
1.4 L-Fenilalanina:es un aminoácido aromático. Interviene en la producción
del colágeno, fundamentalmente en la estructura de la piel y el tejido
conectivo, y también en la formación de diversas neurohormonas. Es el
precursor de las catecolaminas en nuestro cuerpo y constituyente
importante de neuropéptidos cerebrales (somastostatina, vasopresina,
melanotropina). Muchas drogas psicotrópicas, contienen fenilalanina.
La fuente más importante de fenilalanina son los alimentos ricos en
proteínas, como es la carne y productos lácteos. En la industria de la
alimentación se utiliza para elaborar edulcorantes artificiales.
1.5 L-Triptófano: Es un aminoácido aromático. Está implicado en el
crecimiento y en la producción hormonal, especialmente en la función de las
glándulas de secreción adrenal. También interviene en la síntesis de la
serotonina, neurohormona involucrada en la relajación y el sueño.
1.6 L-Metionina: Colabora en la síntesis de proteínas y constituye el principal
limitante en las proteínas de la dieta. El aminoácido limitante determina el
porcentaje de alimento que va a utilizarse a nivel celular.
Aminoácidos
Esenciales
Apolares
1.1 Valina.
1.2 Luecina.
1.3 Isoleucina.
1.4 Fenilalanina.
1.5 Triptófano.
1.6 Metionina.
1.7 Treonina.
1.8 Lisina.
1.9 Arginina
1.10 Histidina.
Polar neutro
Polares Básicos

31
1.7 L-Treonina: Junto con la con la metionina y el ácido aspártico ayuda al
hígado en sus funciones generales de desintoxicación.
1.8 L-Lisina: Es uno de los más importantes aminoácidos porque, en asociación
con varios aminoácidos más, interviene en diversas funciones, incluyendo el
crecimiento, reparación de tejidos, anticuerpos del sistema inmunológico y
síntesis de hormonas.
1.9 L-Arginina:implicada en la conservación del equilibrio de nitrógeno y de
dióxido de carbono. Importante en la producción de la hormona del
crecimiento, involucrada directamente en el crecimiento de los tejidos y
músculos y en el mantenimiento y reparación del sistema inmunológico.
1.10 L-Histidina: En combinación con la hormona de crecimiento (HGH) y
algunos aminoácidos asociados, contribuyen al crecimiento y reparación de
los tejidos del sistema cardiovascular.
2. AMINOÁCIDOS NO ESENCIALES: son aquellos que nuestro
organismo los pueden sintetizar por sí solo, a partir de otros aminoácidos,
sustancias o proteínas. Encontramos los siguientes tipos:
FUNCIONES DE LOS AMINOÁCIDOS NO ESENCIALES
2.1 L-Glicina: se forma a partir de la serina. Su función e generar tejidos,
participa en la neurotransmisión donde cumple una función inhibitoria.
2.2 L-Serina: Junto con algunos aminoácidos mencionados, interviene en la
desintoxicación del organismo, crecimiento muscular, y metabolismo de
grasas y ácidos grasos.
Aminoácidos
No Esenciales
Apolares
2.1 Glicina.
2.2 Serina.
2.3 Cisteína.
2.4 Tirosina.
2.5 Asparagina.
2.6 Glutamina.
2.7 Ácido aspártico.
2.8 Ácido Glutámico
2.9 Alanina.
2.10 Prolina.
Polar neutro
Polares ácidos

32
2.3 L-Cisteína: es un aminoácido no esencial azufrado, se sintetiza a través
de la metionina. Interviene en procesos metabólicos del sistema nervioso
central. Va a formar parte de proteínas de gran importancia biológica
como son la taurina y el glutatión. Junto con lacistina, realiza una
función de desintoxicación, principalmente como antagonista de los
radicales libres. Por su elevado contenido de azufre, se considera
apropiada para el cabello.
2.4 L-Tirosina:se sintetiza por medio de la degradación de la fenilalanina, a
través de la acción de la fenilalanina-hidroxilasa. Es un neurotransmisor
directo y puede ser muy eficaz en el tratamiento de la depresión, en
combinación con otros aminoácidos necesarios. Está relacionado con la
síntesis de catecolaminas en el cerebro. A partir de la tirosina, la enzima
tirosina-hidroxilasa (TH) actúa y lo transforma en DOPA
(dihidroxifenilalanina) y sobre esta actúa la DOPA descarboxilasa
formando dopamina, está a su vez puede transformarse, en aquellas
células que contengan la enzima dopamina-b-hidroxilasa (DBH), en
noradrenalina. La noradrenalina puede transformarse en adrenalina por
otra transferencia de metilos.
2.5 L-Asparagina: Interviene en los procesos metabólicos del Sistema
Nervioso Central (SNC).
2.6 L-Glutamina:se forma a partir del glutamato, por acción de la enzima
glutamina sintetasa. Entre sus funciones tenemos:
Es un nutriente cerebral, que interviene específicamente en la
utilización de la glucosa por el cerebro.
Sirve de sustrato energético para células que se dividen rápidamente
como linfocitos, enterocitos, reticulocitos, fibroblastos y células
tumorales. Cuando la glutamina se oxida en el enterocito, se produce
alanina, citrulina y prolina, además de amoníaco y CO₂.
Es el precursor en la síntesis de las bases purínicas y pirimidínicas.
Es un sistema de transporte de aminoácidos y de nitrógeno desde
tejidos periféricos hacia el hígado.
2.7 Ácido L-Aspártico: Está relacionado con el correcto funcionamiento del
hígado ya que colabora en su desintoxicación. Se combina con otros
aminoácidos formando moléculas capaces de absorber toxinas del
torrente sanguíneo.

33
2.8 Ácido L-Glutamínico:o glutamato, tiene gran importancia en el
funcionamiento del Sistema Nervioso Central y actúa como estimulante
del sistema inmunológico.
2.9 L-Prolina: Está involucrada también en la producción de colágeno y
tiene gran importancia en la reparación y mantenimiento del músculo y
huesos.
2.10 L-Alanina: Interviene en el metabolismo de la glucosa. La
glucosa es un carbohidrato simple que el organismo utiliza como fuente
de energía.
ENLACE PEPTÍDICO
Los aminoácidos se combinan entre sí mediante una relación de condensación
que une al carbono del grupo carboxilo (-COOH) de una molécula con el
nitrógeno del grupo amino (-NH₂) de otra molécula, con la perdida de una
molécula agua.
Fig. 19 Enlace peptídico.
Este enlace covalente carbono-nitrógeno que une dos aminoácidos se denomina
enlace peptídico. Los péptidos y las proteínas están formados por la unión de
aminoácidos mediante enlaces peptídicos.Dependiendo del número de
aminoácidos que intervengan en la formación, la molécula resultante recibirá el
nombre(di-, tri-, tetra-, etc.) y péptido.

34
CAPITULO III
PÉPTIDOS Y PROTEÍNAS
GENERALIDADES DE LOS PÉPTIDOS
Los péptidos son moléculas formadas por la unión de varios aminoácidos
mediante enlaces peptídicos.
La unión de un bajo número de aminoácidos da lugar a un péptido, y si el
número es alto a una proteína.
Oligopéptido: de 2 a 9 aminoácidos.
Polipéptido: entre 10 y 100 aminoácidos.
Proteína: más de 100 aminoácidos.
Los péptidos se diferencian de las proteínas en que son más pequeños (tienen
menos de diez mil o doce mil Daltons de masa) y que las proteínas pueden estar
formadas por la unión de varios polipéptidos y a veces grupos prostéticos.Un
ejemplo de polipéptido es la insulina, compuesta por 51 aminoácidos y conocida
como una hormona de acuerdo a la función que tiene en el organismo de los
seres humanos.
Fig. 20 Estructura primaria de un polipéptido.- la insulina es una
proteína muy pequeña constituida por dos polipéptidos, cada uno con su
propia estructura primaria. La secuencia lineal de aminoácidos se
indica por medio de óvalos que contienen los nombres abreviados.

35
La larga secuencia repetida de enlaces peptídicos forman una cadena, la
estructura primaria o esqueleto de las proteínas. Por convención siempre se
escriben los péptidos con el aminoácido N-terminal a la izquierda y el
aminoácido C-terminal a la derecha. Lo que varía de unos péptidos a otros, y
por extensión, de unas proteínas a otras, es el número, la naturaleza y el orden
o secuencia de sus aminoácidos.
Fig. 21 Estructura de un péptido.
NOMENCLATURA DE LOS PÉPTIDOS
Para nombrar un péptido se empieza por el aminoácido que porta el grupo –
NH2 terminal, y se termina por el aminoácido que porta el grupo -COOH. En el
sistema clásico cada aminoácido se representa por tres letras, y en el moderno,
impuesto por la genética molecular, por una letra. Si el primer aminoácido de
nuestro péptido fuera serina y el segundo alanina tendríamos el péptido serinil
alanina;Ser-Ala; o SA.Los nombre de los distintos residuos se van acabando en
-il, con excepción del último, donde se lo nombra totalmente.
Fig. 22 Nomenclatura de los péptidos.

36
FUNCIONES DE LOS PÉPTIDOS
Entre las funciones biológicas más importantesque realizan los péptidos
podemos destacar las siguientes:
1. Agentes vasoactivos.
2. Hormonas.
3. Antibióticos.
4. Antioxidantes.
AGENTES VASOACTIVOS
Angiotensina II.-octapéptido, hipertensor se origina mediante la
hidrólisis de una proteína precursora que se llama angiotensinógeno, y
que no tiene actividad vasopresora.
Fig. 23 Estructura de la Angiotensina II.
Bradiquinina.- nonapéptido, hipotensor y potente vasodilatador, se
origina mediante la hidrólisis de una proteína precursora que se llama
quininógeno.
Fig. 24 Estructura de la Bradiquinina.

37
HORMONAS
Las hormonas son señales químicas que ejercensu acción sobre órganos y
tejidos situados lejos del lugar donde se han sintetizado. Muchas hormonas
tienen estructura peptídica, como por ejemplo:
Oxitocina: nonapéptido segregado por la hipófisis. Provoca la
contracción uterina y la secreción de leche por la glándula mamaria,
facilitando el parto y la alimentación del recién nacido.
Fig. 25. Estructura lineal de la Oxitocina.
Fig. 26. Estructura química de la Oxitocina. Enlaces disulfuro son
enlaces covalentes entre los átomos de azufre de dos
cisteínas.http://commons.wikimedia.org/wiki/File:Oxytocin_with_la
bels.png

38
Vasopresina:nonapéptido, que induce la reabsorción de agua en el
riñón, llamado también hormona antidiurética.
Somatostatina: tetradecapéptido, inhibidor de la liberación de la
hormona del crecimiento.
Insulina: Hormona compuesta por 51 aminoácidos sintetizada en el
páncreas. Estimula la absorción de glucosa por parte de las células. La
insulina fue el primer péptido que se secuenció por métodos químicos.
Está formada por dos cadenas polipeptídicas unidas entre sí mediante tres
puentes disulfuro. Interviene en el aprovechamiento metabólico de los
nutrientes sobre todo con el anabolismo de los carbohidratos.Su déficit
provoca la diabetes mellitus y su exceso provoca hiperinsulinismo con
hipoglucemia.
Glucagón:Compuesta por 29aminoácidos liberada por el páncreas
cuando los niveles de azúcar en sangre son altos. Hace que en el hígado, el
glucógeno se hidrolice para generar glucosa. Sus efectos son los contrarios
a los de la insulina. NH2-His-Ser-Gln-Gly-Thr-Phe-Thr-Ser-Asp-Tyr-Ser-
Lys-Tyr-Leu-Asp-Ser-Arg-Arg-Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-Asn-
Thr-COOH.
NEUROTRANSMISORES
Unneurotransmisor es una biomolécula que transmite información de una
neurona a otra neurona consecutiva, unidas mediante una sinapsis.El
neurotransmisor se libera por las vesículas en la extremidad de la neurona
presináptica durante la propagación del impulso nervioso, atraviesa el espacio
sináptico y actúa cambiando el potencial de acción en la neurona siguiente
(denominada postsináptica) fijándose en puntos precisos de su membrana
plasmática.
Fig. 27. Estructura de la
Vasopresina.Enlaces disulfuro son enlaces
covalentes entre los átomos de azufre de dos
cisteínas.

39
Son neurotransmisores peptídicos las encefalinas (pentapéptidos), sustancia P
(undecapéptido):
Las endorfinas y encefalinas:son péptidos opioides endógenos que
funcionan como neurotransmisores. Producidas por la glándula
pituitaria yel hipotálamo envertebrados duranteel ejercicio, la excitación,
el dolor, el consumo de alimentos picantes o el consumo de chocolate,
actúan como moduladores del dolor, reproducción, temperatura corporal,
hambre y funciones reproductivas.
Son similares a los opiáceos en su efecto analgésico y de sensación de
bienestar.B-endorfina; (31 aminoácidos) Tyr-Gly-Gly-Phe-Met-Thr-Ser-
Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-Thr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-
Asn-Ala-Tyr-Lys-Lys-Gly-Glu.
ANTIBIÓTICOS
La valinomicinay la gramicidina son dos péptidos cíclicos con acción
antibiótica. Los dos contienen aminoácidos de la serie D, además de otros
aminoácidos no proteicos. La valinomicina es un ionóforo: es capaz de
transportar iones potasio a través de la membrana biológica.
ANTIOXIDANTES
El glutatión.- es un tripéptido que actúa como antioxidante celular. Reduce
las especiesreactivas del oxígeno (como el peróxido de H) gracias a la enzima
glutatión peroxidasa.
Fig. 28 Sinapsis. Acción de
neurotransmisores.
http://commons.wikimedia.org/
wiki/File:Sinapsis.png

40
PROTEÍNAS
Son polímeros formados por la unión de aminoácidos, mediante enlaces
peptídicos. Estas macromoléculas también pueden contener otras moléculas
orgánicas (lípidos, glúcidos, etc.) recibiendo el nombre de prótidos.El 50%
delpeso seco de la célula son proteínas. Están constituidas, fundamentalmente,
por C, H, O yN y casi todas tienen también azufre. Algunas tienen, además,
otros elementos químicos como: P, Fe, Zn o Cu.
El elemento más característico de las proteínas es elnitrógeno. Son los
compuestos nitrogenados por excelencia de los seres vivos.
Las proteínas son moléculas específicas que marcan la a individualidad de cada
ser vivo. Son además de una gran importancia porque a través de ellas se va a
expresar lainformación genética, de hecho el dogma central de la genética
molecular nos dice:
DNA RNA Proteína
ESTRUCTURA DE LAS PROTEÍNAS
La conformación de una proteína es la disposición espacial que adopta la
molécula proteica. Las cadenas peptídicas, en condiciones normales de pH y
temperatura, poseen solamente una conformación y ésta determina en gran
medida su función.Pueden distinguirse cuatro niveles de organización
principales en las proteínas: primario, secundario, terciario y cuaternario.
Fig. 29 Niveles de organización de las proteínas.Estructura
primaria, secundaria, terciaria y cuaternaria.

41
1. La Estructura primaria.-es la secuencia de aminoácidos de la
proteína, nos indica que aminoácidos componen la cadena polipeptídica y el
orden en que se encuentran, determinando la función de la proteína.
La alteración de la estructura primaria por eliminación, adición o
intercambio de los aminoácidos, cambia su configuración general, dando
lugar a una proteína diferente y por lo tanto no puede realizar su función.
2. La Estructura Secundaria.-es muy constante debido a que es
mantenida por enlaces de hidrógeno entre determinados átomos del
esqueleto de la cadena polipeptídica, imponiendo restricciones que obligan a
que las proteínas adopten una determinada estructura secundaria. Ésta
puede ser en hélice α, hélice de colágeno o en conformación ß.
Es de destacar que las tres son configuraciones en hélice diferenciandose en
el número de aminoácidos por vuelta (n) y en el diámetro de la hélice. En la
hélice αlfa, n=4; en la hélice de colágeno, n=3 y en la conformación ß, n=2.
A continuación estudiaremos solo la hélice α y la conformación ß por ser las
configuraciones más frecuentes.
2.1 Estructura en hélice αlfa.-la molécula adopta una disposición
helicoidal, determinada y mantenida por la formación de puentes de hidrógeno
entre los esqueletos de los aminoácidos en vueltas sucesivas de la espiral. Cada
enlace de hidrógeno se forma entre el oxígeno, con carga parcial negativa
residuo del grupo carboxilo y el hidrógeno, con carga parcial positiva residuo
Fig. 30.Estructura primaria de las proteínas.
http://proteinasnacha.blogspot.com/2011/04/estructura-de-las-proteinas.html

42
del grupo amino del cuarto aminoácido siguiente en la cadena polipeptídica.
En consecunecia cada vuelta completa en la hélice comprende 3,6 aminoácidos.
La hélice alfa es la unidad estructural básica de algunas proteínas fibrosas,
como las de la lana, pelo y uñas. La fibra es elástica por una combinación de
factores físicos (la forma helicoidal) y químicos (los puentes de hidrógeno).
Aunque los enlaces de hidrógeno mantienen la estructura helicoidal, pueden
romperse, lo que permite que las fibras se estiren al aplicar tensión. Cuando la
tensión se libera las fibras retroceden y los enlaces se forman de nuevo. A esto
se debe que el pelo humano se pueda estirar hasta cierto punto y luego regresar
a su longitud original.
2.2 Conformación β o Lámina plegada beta.- Los puentes de
hidrógeno se forman entre cadenas polipeptídicas distintas o entre
diferentes regiones de una cadena polipeptídica que se ha enrrollado en sí
misma. Cada cadena está extendida por completo, pero dado que cada una
tiene estructura en zigzag, la lámina resultante tiene conformación global
plegada (parecido a una hoja de papel que se ha plegado para formar un
abanico). Aunque está estructura es resistente y flexible, no es elástica. Esto
Fig. 31Estructura secundaria de la
proteína.Conformación hélice
alfa.http://payala.mayo.uson.mx/Programa/bi
omoleculas,proteinas.htm

43
se debe a que la distancia entre los plegamientos es fija, determinada por
los fuertes enlaces covalentes de los esqueletos del polipéptido.
No es raro que una misma cadena polipeptídica contenga tanto regiones con
conformaciones hélice alfa como regiones con lámina plegada beta. Además,
las propiedades de algunos materiales biológicos complejos se deben a tales
combinaciones. La seda de las arañas es en extremo fuerte, flexible y
elástica, se trata de una combinación de proteínas con conformaciones de
hélice alfa (que dan elasticidad) y otras conformaciones de lámina plegada
beta (que dan resistencia).
3. Estructura Terciaria.- de una molécula proteíca es la forma
global que asume cada cadena polipeptídica individual. Esta estructura
tridimencional depende de cuatro factores principales, que implican
interacciones entre los grupos R (cadenas laterales) pertenecientes a la
misma cadena polipeptídica.
Se forman enlaces de hidrógeno entre grupos R de determinados
aminoácidos.
Puede haber atracción iónica entre un grupo R con carga positiva
y otro con carga negativa unitaria.
Fig. 32 Estructura secundaria de la proteína. Lámina plegada
beta.http://my.opera.com/tutoriabiologiaUBAXXI/blog/blog-2

44
Resultan interacciones hidrófobas de la tendencia de los grupos R
no polares a ser rechazados por el agua circundante y de este
modo asociarse en el interior de la estructura globular.
Pueden formarse enlaces covalentes, conocidos como enlaces o
puentes disulfuro ( S S ), entre los átomos de azufre de dos
subunidades de cisteína pertenecientes a la misma cadena. Esto
ocurre cuando los grupos sulfhidrilo de dos cisteínas reaccionan;
los dos hidrógenos son eliminados, y los dos átomos de azufre que
quedan se unen de modo covalente.
4. Estructura Cuaternaria.- es la arquitectura tridimensional
resultante de la unión de dos o más cadenas polipeptídicas que forman
una molécula de proteína con actividad biológica (cada una con sus
propias estructuras primaria, secundaria y terciaria). Los mismos tipos
de interacciones que determinan las estructuras secundaria y terciaria
contribuyen a la estructura caternaria; entre ellas se incluye enlaces de
hidrógeno, enlaces iónicos, interacciones hidrofóbicas y puentes
disulfuro.
La hemoglobina proteína de los eritrocitos de la que depende el
transporte de oxígeno, es un claro ejemplo de proteína globular con
estructura cuaternaria. La hemoglobina consta de 574 aminoácidos
dispuestos en cuatro cadenas polipeptídicas: Dos cadenas alfa idénticas
entre sí y otras dos beta, tambien iguales entre sí.
Fig. 33 Estructura terciaria de la proteína.
http://proteinas-biologia.blogspot.com/2011/04/estructura-de-las-proteinas.html

45
PROPIEDADES DE LAS PROTEÍNAS
1. Especificidad.- se refiere a su función; cada una lleva a cabo una
determinada función y la realiza porque posee una determinada estructura
primaria y una conformación espacial propia; por lo que un cambio en la
estructura de la proteína puede significar una pérdida de la función.
Además, no todos los individuosposeen proteínas específicas suyas que se
ponen de manifiesto en los procesos de rechazo de órganos grado de
parentesco entre individuos, por lo que sirve para la construcción de
filogenéticos.
2. Desnaturalización.- Pérdida de la estructura tridimensional o
conformación, y por tanto también de la actividad biológica. Se produce por
cambios de la temperatura, variación de pH o por agentes químicos
desnaturalizante, etc. Esto provoca la rotura de los puentes de hidrógeno
que mantienen las estructuras secundaria y terciaria, y las proteínas se
convierten en fibras insolubles en agua. En algunos casos, si las condiciones
se restablecen, unaproteína desnaturalizada puede volver a su anterior
plegamiento o conformación,proceso que se denomina renaturalización.
3. Solubilidad.- Las proteínas globulares son solubles en agua, debido a
que sus radicales polares o hidrófilos se sitúan hacia el exterior, formando
puentes de hidrógeno con el agua, constituyendo una capa de solvatación.
Esta solubilidad varía dependiendo del tamaño, de la forma, de la
disposición de los radicales y del pH.
Fig. 34 Estructura cuaternaria de las
proteínas.http://biotecnologiageneral.blogspot.com/2011/10/proteina
s.html

46
CLASIFICACIÓN DE PROTEÍNAS
Atendiendo al criterio según su composición las proteínas se clasifican en:
1. HOLOPROTEÍNAS O PROTEÍNAS SIMPLES.- Son proteínas
formadas únicamente por aminoácidos. Pueden ser globulares o fibrosas.
1.1Proteínas globulares.-se caracterizan por doblar sus cadenas en una
forma esférica compacta dejando grupos hidrófobos hacia adentro de la
proteína y grupos hidrófilos hacia afuera, lo que hace que sean solubles en
disolventes polares como el agua. La mayoría de las enzimas, anticuerpos,
algunas hormonas y proteínas de transporte, son ejemplos de proteínas
globulares. Algunos tipos son:
Albúminas: son proteínas solubles en agua. Ejemplo: seroalbúmina
(sangre), ovoalbúmina (huevo), lactoalbúmina (leche).
Hormonas: insulina, hormona del crecimiento, prolactina, tirotropina.
Enzimas: hidrolasas, oxidasas, ligasas, liasas, transferasas. etc.
Prolaminas: ricas en prolina e insolubles en agua. Aparecen en las
semillas. Ejemplos: zeína (maíz), gliadina (trigo), hordeína (cebada).
Gluteninas: son proteínas insolubles en agua pero solubles en ácidos y
bases diluidas. Ejemplos: glutenina (trigo), orizanina (arroz).
1.2 Proteínas fibrosas.- presentan cadenas polipeptídicas largas y una
estructura secundaria atípica. Son insolubles en agua y en disoluciones
acuosas. Algunas proteínas fibrosas son:
Colágenos: proteína con función estructural y aparece en el tejido
conjuntivo, cartilaginoso y óseo.
Queratinas: son proteínas ricas en cisteína que tiene función
estructural aparecen en formaciones epidérmicas: pelos, uñas, plumas,
cuernos.
CLASIFICACIÓN
DE
PROTEÍNAS
1.1 Globulares
1.2 Fibrosas
2.1 Glicoproteínas.
2.2 Lipoproteínas.
2.3 Nucleoproteínas.
2.4 Cromoproteínas
1. Holoproteínas o
Proteínas simples
2. Heteroproteínas
o Proteínas
Conjugadas

47
Elastinas: su función es estructural, forma parte de tendones y vasos
sanguíneos.
Fibroínas: tiene función estructural o de resistencia mecánica, presente
en hilos de seda, (arañas, insectos).
2. HETEROPROTEÍNAS O PROTEÍNAS CONJUGADAS.- formadas por un número determinado de aminoácidos más una parte no
proteica, denominado grupo prostético. Dependiendo del grupo prostético
existen varios tipos:
2.1 Glicoproteínas.- Son moléculas compuestas por una proteína unida a
uno o varios glúcidos. Algunas de ellas son:
Ribonucleasa.
Mucoproteínas.
Anticuerpos.
Hormona luteinizante.
2.2. Lipoproteínas.- Son complejos macromoleculares esféricos formados
por un núcleo que contiene lípidos apolares (colesterol esterificado y
triglicéridos) y una capa externa polar formada por fosfolípidos, colesterol libre
y proteínas. Su función principal es el transporte de triglicéridos, colesterol y
otros lípidos entre los tejidos a través de la sangre.
Las lipoproteínas se clasifican según su densidad:
Lipoproteínas de alta densidad
Lipoproteínas de baja densidad
Lipoproteínas de muy baja densidad
Fig.35 Lipoproteínas.
http://www.ehu.es/biomoleculas/proteinas/prot45.htm

48
2.3.Nucleoproteínas.- Son proteínas estructuralmente asociadas con un
ácido nucleicos (que puede ser ARN o ADN). El ejemplo prototípico sería
cualquiera de las histonas, que son identificables en las hebras de cromatina.
Su característica fundamental es que forman complejos estables con los ácidos
nucleicos, a diferencia de otras proteínas que sólo se unen a éstos de manera
transitoria, como las que intervienen en la regulación, síntesis y degradación
del ADN.
2.4.Cromoproteínas.- Las cromoproteínas poseen como grupo prostético
una sustancia coloreada, por lo que reciben también el nombre de pigmentos.
Según la naturaleza del grupo prostético, pueden ser pigmentos porfirínicos
como la hemoglobina encargada de transportar el oxígeno en la sangre o no
porfirínicos como la hemocianina, un pigmento respiratorio que contiene cobre
y aparece en crustáceos y moluscos por ejemplo. También los citocromos, que
transportan electrones.
FUNCIONES Y EJEMPLOS DE PROTEÍNAS
Las proteínas están entre las sustancias que realizan las funciones más
importantes en los seres vivos. De entre todas pueden destacarse las
siguientes:
1.1 Función estructural:Las proteínas constituyen muchas estructuras
celulares.
1.1 Glucoproteínas: forman parte de las membranas celulares y actúan
como receptores o facilitan el transporte de sustancias.
Histonas: forman parte de los cromosomas que regulan la expresión de los
genes.
El Colágeno: confiere resistencia al tejido conjuntivo fibroso.
La Elastina: confiere elasticidad al tejido conjuntivo elástico.
1.2 Función enzimática: Las proteínas con función enzimática son las
más numerosas y especializadas. Actúan como biocatalizadores
ascelerando las reacciones químicas del metabolismo celular.
1.3 Función hormonal: Algunas hormonas son de naturaleza proteica,
como la insulina y el glucagón (que regulan los niveles de glucosa en sangre)
o las hormonas segregadas por la hipófisis como la del crecimiento o la
adrenocorticotrópica (que regula la síntesis de corticosteroides) o la
calcitonina (que regula el metabolismo del calcio).
1.4 Función defensiva: Lasproteínas crean anticuerpos y regulan factores
contra agentes extraños o infecciones.

49
Las inmunoglobulinas.- actúan como anticuerpos frente a posibles
antígenos.
La trombina y el fibrinógeno.- contribuyen a la formación de coágulos
sanguíneos para evitar hemorragias.
Las mucinas.- tienen efecto germicida y protegen a las mucosas.
Algunas toxinas bacterianas, como la del botulismo, o venenos de
serpientes, son proteínas fabricadas con funciones defensivas.
1.5 Función de transporte:
La hemoglobina.- transporta oxígeno en la sangre de los vertebrados.
La hemocianina.- transporta oxígeno en la sangre de los invertebrados.
La mioglobina.- transporta oxígeno en los músculos.
Las lipoproteínas.- transportan lípidos por la sangre.
Los citocromos.- transportan electrones.
1.6 Función de reserva:
La ovoalbúmina.- de la clara de huevo, La gliadina del grano de trigo y
La hordeína de la cebada, constituyen la reserva de aminoácidos para el
desarrollo del embrión.
La lactoalbúmina.- de la leche.
1.7 Función reguladora: Algunas proteínas como la ciclina regulan la
expresión de ciertos genes y otras regulan la división celular.
1.8 Función contráctil:La actina y la miosina constituyen las
miofibrillas responsables de la contracción muscular.
La dineína está relacionada con el movimiento de cilios y flagelos.
1.9 Función homeostática: Son las proteínas que mantienen el
equilibrio osmótico y actúan junto con otros sistemas amortiguadores para
mantener constante el pH del medio interno.
ALIMENTACIÓN, DIGESTIÓN Y ABSORCIÓN DE
PROTEÍNAS
Requerimiento de proteínas.-Las proteínas dietarias deben proveer los
aminoácidos necesarios para mantener el balance nitrogenado. Un adulto debe
incorporarse 0.8gr de proteínas por kg de peso corporal por día. En
embarazadas deben adicionarse al requerimiento para un adulto 30gr por día
durante toda la gestación. Durante la lactancia debe agregarse 20gr por día
para cubrir la necesidad de síntesis de proteínas de la leche. Lactantes menores
de 1 año deben recibir 2gr/kg/día, niños de 1 a 10 años 1.2gr/kg/día y

50
adolescentes 1gr/kg/día. En todos los grupos de edades el requerimiento
aumenta ante procesos que acrecienten el catabolismo.
Alimentos ricos en proteínas.- Entre estos tenemos a los de origen
animal: carnes, huevos y leche; y a los de origen vegetal, donde la soja ocupa el
primer lugar en contenido proteico, seguida por los cereales. Los alimentos de
origen animal son también llamados alimentos con proteínas de alto valor
biológico, debido a que contienen gran cantidad de aminoácidos que el cuerpo
requiere en forma indispensable por no poder sintetizarlos (esenciales); por el
contrario, las proteínas aportadas por la soja, por ejemplo, son de muy bajo
valor biológico por su bajo contenido en aminoácidos esenciales.
Una alimentación pobre en proteínas es la causa más frecuente de
desnutrición. Los cuadros más serios de malnutrición proteica son el
kwashiorkor, observado en niños con dietas pobres en proteínas de buen valor
biológico y dietas ricas en carbohidratos, caracterizado por retardo del
crecimiento, abdomen globoso, disminución de albúmina en plasma, anemia y
hepatomegalia; y el marasmo, producido por déficit crónico de proteínas y
calorías en la dieta, con perdida del tejido graso y gran parte de la masa
muscular en un proceso de consumición severo.
Digestión.- La hidrólisis de las proteínas de los alimentos se inicia en el
estómago. Aquí la pepsina, una endopeptidasa secretada como pepsinógeno por
las células parietales de la mucosa gástrica, escinde las proteínas en segmentos
de menor peso molecular. Estos pasan al duodeno donde se encuentran tres
endopeptidasas: tripsina, quimiotripsina y elastasa del jugo pancreático, que
los degradan en trozos menores, del tipo polipéptidos. Hasta aquí no se han
producido aminoácidos libres; estos comienzan a aparecer gracias a la acción
de dos exopeptidasas que van atacando los péptidos desde sus extremos. La
carboxipeptidasa, de origen pancreático, y la aminopeptidasa intestinal.
Finalmente quedan tri- y dipéptidos, cuya hidrólisis es catalizada por
tripeptidasas y dipeptidasas del borde en sepillo del intestino. De esta manera,
las proteínas de la dieta son degradadas hasta aminoácidos libres, di- y
tripéptidos.
Absorción.- Los productos finales de la digestión de proteínas son
incorporados a los enterocitos utilizando distintos mecanismos. Un grupo de
aminoácidos libres se incorporan por un cotransporte activo estereoespecífico.
El proceso es similar al de absorción de la glucosa. Se trata de un cotransporte
con Na+, dependiente del funcionamiento de la Bomba Na+/K+ ATPasa. Este

51
sistema es utilizado por los aminoácidos neutros, aromático, alifáticos,
fenilalanina, metionina, aminoácidos ácidos y prolina. Un grupo menor de
aminoácidos (básicos y neutros hidrófobos) ingresan a la célula por difusión
facilitada (Na+ independiente).
Por otro lado, los di- y tripeptidos son transportados por sistemas propios que
dependen del gradiente químico del Na+ y una vez dentro de la célula son
escindidos a aminoácidos libres por peptidasas intracelulares. Los aminoácidos
liberados en el citoplasma pasan luego al intersticio y a los capilares
sanguíneos por difusión facilitada. Una vez en el torrente sanguíneo portal, los
aminoácidos ramificados son deportados preferentemente al músculo mientras
que los no ramificados se dirigen al hígado.
En condición normal solo llegan a la sangre aminoácidos libres; sin embargo, se
dan algunas situaciones fisiológicas y patológicas en las cuales debe aceptarse
la posibilidad de la absorción de proteínas enteras o trozos moleculares de gran
tamaño. Esto explicaría el mecanismo de la enfermedad celíaca, en la cual
existe un defecto de la mucosa que posibilita la absorción de polipéptidos
(gliadina) resultantes de la digestión del gluten, la principal proteína del trigo,
avena, centeno y cebada. Se produce intolerancia a dicha proteína,
determinando un cuadro clínico muy severo.
Fig. 36Esquema de la absorción de aminoácidosen intestino. 1. Co-transporte estereoespecífico
(transporte activo secundario Na+dependiente). 2a. Difusión facilitada sistema y+ (aminoácidos
básicos). 2b. Difusión facilitada sistema L (aminoácidos neutros hidrófobos). 3. Transportadores
de di- y tripéptidos Na+ dependiente.
52
CAPITULO IV
ÁCIDOS NUCLEICOS
CARACTERÍSTICAS GENERALES
Todas las células contiene la información necesaria para realizar distintas
reacciones químicas mediante las cuales las células crecen, obtienen energía y
sintetizan sus componentes. Esta información esta almacenada en el material
genético, el cual debe copiarse con exactitud para transmitir dicha información
a las células hijas. Sin embargo estas instrucciones pueden ser modificadas
levemente, es por eso que un individuo no es exactamente igual a otro de la
misma especie. De este modo, podemos decir que el material genético es lo
suficientemente maleable como para hacer posible la evolución.
Los ácidos nucleicos son macromoléculas formadas por C, H, O, N y P, y que
están constituídas por unidades denominadas nucleótidos.
La información genética o genoma, está contenida en unas moléculas llamadas
ácidos nucleicos. Existen dos tipo de ácidos nucleicos: ADN y ARN.
En las células, la información genética, indispensable para la continuación de
la vida, se halla en el ADN o ácido desoxirribonucleico. Para que dicha
información pueda expresarse se requiere otra molécula, el ARN o ácido
ribonucleico. Ambos ácidos nucleicos están formados por la repetición de unas
unidades denominadas nucleótidos.
ESTRUCTURADE LOS ÁCIDOS NUCLEICOS
Ácidos
Nucleicos
Bases
Nitrogenadas
Pentosa Grupo
Fosfato
Localización Estructura Funciones
ADN
Adenina
Guanina
Citocina
Timina
2-
desoxi-
D-
ribosa
Cromatina
Bicatenaria
Portador
de la
herencia
ARN
Adenina
Guanina
Citocina
Uracilo
Ribosa
Nucleolo
y
Citoplasma
Monocatenaria
Síntesis
de
proteínas
Fig. 37 Componentes básicos, localización, estructura y función de los ácidos nucleicos.

53
ÁCIDO ORTOFOSFÓRICO
La unión entre la base nitrogenada y el azúcar se
realiza por un enlace N-glicosídico entre un grupo
amino de la base y el C-1 de la pentosa, originándose
un nucleósido que se denominará: adenosina,
guanosina, citidina, timidina u uridina, en función de
la base nitrogenada presente.
Fig. 38 Bases nitrogenadas; Pirimidinas (Citosina, Timina, Uracilo);
Purinas (Adenina, Guanina). Pentosas; Ribosa y Desoxirribosa.
http://www.cede.es/n_temas_2012/t35_biologia.pdf
Fig. 39 Ácido
Ortofosfórico.

54
FORMACIÓN DE NUCLEÓSIDOS
Los nucleósidos están formados por una pentosa y una base nitrogenada. Dependiendo
de la pentosa constituída, los nucléosidos son ribonucléosidos o desoxi-ribonucléosidos.
En ambos casos la unión de la base nitrogenada siempre ocurre en la posición 1’ de la
pentosa.
FORMACIÓN DE NUCLEÓTIDOS
Éste nucleósido se une al ión fosfato por un enlace fosfo-ester entre un OH del C3’ o
5’ de la pentosa y el OH del grupo fosfato, originandose así el nucleótido, que se
denominará con el nombre del nucleósido seguido de la palabra 3’-fosfato o 5’-fosfato.
Ejemplo: Adenosín 5’-monofosfato.
Fig. 40 Formación de un nucleósido.
http://www.cede.es/n_temas_2012/t35_biologia.pdf
Fig. 41 Formación de un nucleótido.
http://www.cede.es/n_temas_2012/t35_biologia.pdf
55
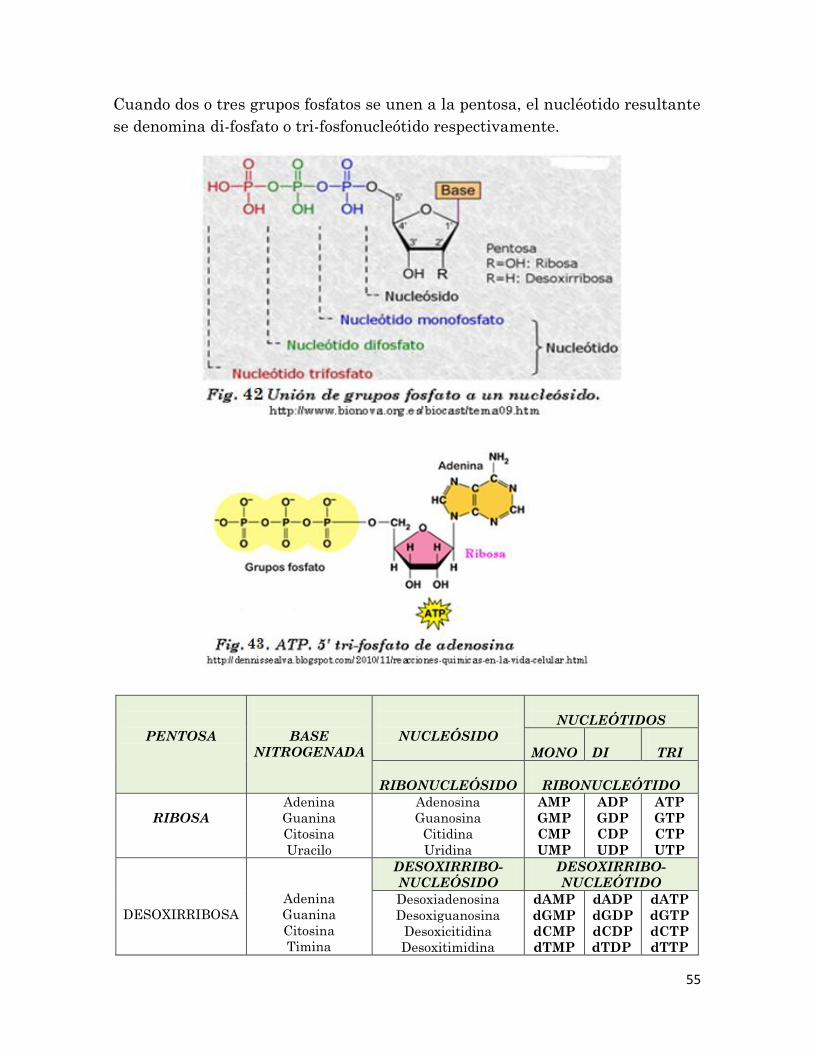
Cuando dos o tres grupos fosfatos se unen a la pentosa, el nucléotido resultante
se denomina di-fosfato o tri-fosfonucleótido respectivamente.
PENTOSA
BASE
NITROGENADA
NUCLEÓSIDO
NUCLEÓTIDOS
MONO
DI
TRI
RIBONUCLEÓSIDO
RIBONUCLEÓTIDO
RIBOSA
Adenina
Guanina
Citosina
Uracilo
Adenosina
Guanosina
Citidina
Uridina
AMP
GMP
CMP
UMP
ADP
GDP
CDP
UDP
ATP
GTP
CTP
UTP
DESOXIRRIBOSA
Adenina
Guanina
Citosina
Timina
DESOXIRRIBO-
NUCLEÓSIDO
DESOXIRRIBO-
NUCLEÓTIDO
Desoxiadenosina
Desoxiguanosina
Desoxicitidina
Desoxitimidina
dAMP
dGMP
dCMP
dTMP
dADP
dGDP
dCDP
dTDP
dATP
dGTP
dCTP
dTTP

56
FORMACIÓN DE POLINUCLEÓTIDOS
Dos nucleótidos van a poder unirse entre sí mediante un enlace fosfo-ester.
Este enlace se forma entre un OH del grupo fosfato y el OH (hidroxilo) del
carbono 3’ del azúcar del otro nucleótido con formación de una molécula de
agua. La unión de otros nucleótidos dará lugar a un polinucleótido. Es de
destacar que en la cadena de polinucleótidos el nucleótido de uno de los
extremos tendrá libre el OH del azúcar en posición 3’, este será el extremo 3’
de la cadena. El grupo fosfato del nucleótido que se encuentre en el extremo
opuesto también estará libre, este será el extremo 5’. Esto marca un sentido en
la cadena de policucleótidos.
Fig. 44 Formación de polinucleótidos.
http://www.biologiasur.org/apuntes/base-fisico-quimica/base/acidos-
nucleicos/nucleotidos.html
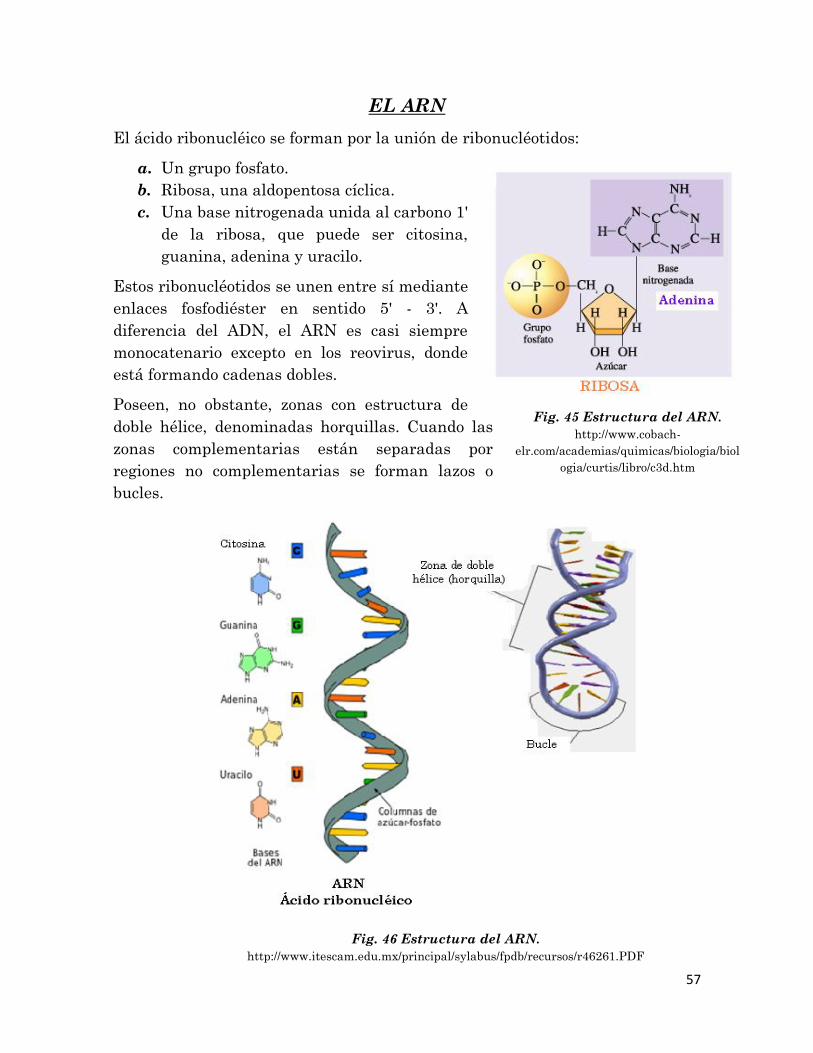
57
EL ARN
El ácido ribonucléico se forman por la unión de ribonucléotidos:
a. Un grupo fosfato.
b. Ribosa, una aldopentosa cíclica.
c. Una base nitrogenada unida al carbono 1'
de la ribosa, que puede ser citosina,
guanina, adenina y uracilo.
Estos ribonucléotidos se unen entre sí mediante
enlaces fosfodiéster en sentido 5' - 3'. A
diferencia del ADN, el ARN es casi siempre
monocatenario excepto en los reovirus, donde
está formando cadenas dobles.
Poseen, no obstante, zonas con estructura de
doble hélice, denominadas horquillas. Cuando las
zonas complementarias están separadas por
regiones no complementarias se forman lazos o
bucles.
Fig. 46 Estructura del ARN.
http://www.itescam.edu.mx/principal/sylabus/fpdb/recursos/r46261.PDF
Fig. 45 Estructura del ARN.
http://www.cobach-
elr.com/academias/quimicas/biologia/biol
ogia/curtis/libro/c3d.htm

58
TIPOS Y FUNCIÓN
Por su localización celular, su estructura y la función que desempeñan se
distinguen varios tipos de ARN:
1. ARN mensajero. (ARNm)
2. ARN ribosomal. (ARNr)
3. ARN de transferencia. (ARNt)
ARNm.- el ARN mensajero, forman cadenas cortas y
lineales que poseen unicamente estructura primaria,
pueden llegar a estar formadas hasta por 5.000
nucleótidos. Lleva la información sobre la secuencia de
aminoácidos de la proteína desde el ADN, lugar en que
está inscrita, hasta el ribosoma, lugar en que se sintetizan
las proteínas de la célula. En eucariotas, el ARNm se
sintetiza en el nucleoplasma del núcleo celular y de allí
accede al citosol, donde se hallan los ribosomas, a través
de los poros de la envoltura nuclear. También se pueden
distinguir:
Exones.- secuencias de bases que codifican proteínas.
Intrones.- secuencias sin información.
Fig. 48 Exones e intrones.
http://www.um.es/molecula/anucl03.htm
Fig. 47 Tipos de ARN.
http://www.itescam.edu.mx/principal/sylabus/fpdb/recursos/r46261.PDF

59
ARNt.-están formados por moléculas relativamente pequeñas que contienen
entre 70 y 90 nucleótidos y constituyen una única hebra o cadena. Esta cadena
presenta zonas con doble hélice, que dan lugar a la estructura secundaria en
“hoja de trébol”. Los distintos ARNt dispersos en elhialoplasma se encargan de
recoger los diferentes aminoácidos y de transportarloshasta los ribosomas.
En el ARNt podemos distinguirun sitio específico para la fijación del
aminoácido (extremo 3') y un anticodón formado por un triplete de nucleótidos
que se une al codón complementario del ARNm mediante puentes de
hidrógeno.
ARNr.- son los más abundantes (90 - 95 % de losARN), se halla combinado
con proteínas para formar los ribosomas. Los ribosomas son los orgánulos
encargados de la biosíntesis deproteínas;concretamente, “traducen” la
secuencia de bases del ARNm en lasecuencia correspondiente de
aminoácidos.
Fig. 49tARN.
http://temasbiologiamolecular.blogspot.com/2012/03/acidos-nucleicos-
estructura-y-funcion.html

60
Fig. 51ARNr. Síntesis de proteínas.
http://biologiamolecular-miguel.blogspot.com/2012/05/74-eucariotico.html
Fig. 50 Ribosoma. ARNr. Síntesis de proteínas.
http://estructurayfuncioncelularbacteriana.wikispaces.com/ribosomas

61
EL ADN
Es una macromolécula muy larga, de aspecto filamentoso formada por un gran
número de nucleótidos, cada uno de ellos compuesto por:
1. Un grupo fosfato.
2. Un azúcar, desoxirribosa.
3. Una base nitrogenada unida al carbono 1' de la desoxirribosa.
Fig. 52Estructura del ADN.
http://www.efn.uncor.edu/dep/biologia/intrbiol/adntema1.htm
http://www.itescam.edu.mx/principal/sylabus/fpdb/recursos/r46261.PDF

62
ESTRUCTURA PRIMARIA DEL ADN
Es la secuencia de nucleótidos unidos por enlaces fosfodiéster. En dicha
secuencia de bases reside la información genética o mensaje biológico
hereditario, en tanto que los grupos fosfatos y el azúcar tienen un papel
estructural. La cadena presenta dos extremos libres: el 5' unido al grupo fosfato
y el extremo 3' unido al grupo hidroxilo. Cada cadena se diferencia de otra por
su tamaño, su composición y la secuencia de sus bases nitrogenadas,
considerando que en un ADN existen 4 nucleótidos diferentes, el número de
moléculas distintas de ADN que puede haber es de 4ⁿ, siendo n el número de
nucleótidos que contiene dicho ADN.
.
Fig. 53Estructura primaria del ADN.
http://www.um.es/molecula/anucl02.htm

63
ESTRUCTURA SECUNDARIA
James Watson y Francis Crick en 1953 demostraron que la estructura del
ADN estáconstituida por dos cadenas o hebras de polinucleótidos enrolladas
helicoidalmente en sentido dextrógiro sobre un mismo eje formando una
doblehélice.Ambas cadenas son antiparalelas, una va en sentido 3'- 5' y la otra
en sentido inverso, 5' - 3'.Los grupos fosfato están hacia elexterior, de este modo
sus cargasnegativas interaccionan con los cationespresentes en el
nucleoplasma dando másestabilidad a la molécula.Las bases nitrogenadas
están hacia el interior de la hélice con sus planos paralelos entre sí y las bases
de cada una de las hélices están apareadas con las de la otra asociándose
mediante puentes de hidrógeno.El apareamiento se realiza únicamente entre
adenina-timina, mediante dos puentes de hidrógeno, y guanina-citosina,
mediante tres puentes de hidrógeno. Porlo tanto, la estructura primaria de
unacadena está determinada por la de la otra, ambas cadenas son
complementarias.La complementariedad de las cadenas sugiere el mecanismo
por el cual el ADN secopia se replica para ser trasferido a las células hijas.
Ambas cadenas o hebras sepueden separar parcialmente y servir de molde para
la síntesis de una nueva cadenacomplementaria.
Fig. 54 Estructura secundaria del ADN.
http://www.maristasgranada.net/webcole/documentos/Ciencias/Bach-
2%BA/Biologia/1_Bioquimica/04_Ac_Nucleicos/ADN_Estructura_1%AA,2%AA
,3%AAy4%AA.pdf

64
PROPIEDADES DE LA ESTRUCTURA SECUNDARIA DEL
ADN
DESNATURALIZACIÓN:
Si una disolución de ADN se calientasuficientemente ambas cadenas se
separan,pues se rompen los enlaces de hidrógenoque unen las bases, y el ADN
sedesnaturaliza. La temperatura dedesnaturalización depende de la
proporciónde bases. A mayor proporción de C-G,mayor temperatura de
desnaturalización,pues la citosina y la guanina establecen trespuentes de
hidrógeno, mientras que laadenina y la timina sólo dos y, por lo tanto,a mayor
proporción de C-G, más puentes dehidrógeno unirán ambas cadenas.
Ladesnaturalización se produce tambiénvariando el pH o a concentraciones
salinaselevadas. Si se restablecen las condiciones,el ADN se renaturaliza y
ambas cadenas seunen de nuevo.
ESTRUCTURA TERCIARIA DEL ADN
Modelo del “collar de perlas”
El ADN es una molécula muy larga en algunas especies y, sin embargo, en las
células eucariotas se encuentra alojado dentro del minúsculo núcleo. Cuando el
ADN se une a proteínas básicas, su estructura se compacta mucho. Dichas
proteínas son las Histonas.
Fig. 55 Estructura secundaria del ADN.
http://www.biologia.arizona.edu/molecular_bio/problem_sets/mol_genetics
_of_eukaryotes/05t.html

65
La unión con Histonas genera la estructura denominada NUCLEOSOMA.
Cada nucleosoma está compuesto por una estructura voluminosa, denominada
core, seguida por un eslabón o "Linker". El core está compuesto por un
octámero de Histonas, denominadas H2A, H2B, H3 y H4; cada tipo de histona
se presenta en número par.
Esta estructura está rodeada por un tramo de ADN que da una vuelta y 3/4 en
torno al octámero.El Linker está formado por un tramo de ADN que une un
nucleosoma con otro y, además, se une a una histona H1. (El llamado linker se
traduce como ADN espaciador o ligador).
El conjunto de la estructura se denomina fibra de cromatina de 10 nm (100Å) o
collar de perlas, debido a su aspecto repetitivo en el que las perlas serían los
nucleosomas, unidos por los linker.
ESTRUCTURA CUATERNARIA del ADN
Modelo del “solenoide”
La fibra de cromatina de 10nm (collar de perlas) se empaqueta formando una
fibra de cromatina de 30nm: el conjunto de nucleosomassufre un enrollamiento
helicoidal, que recibe el nombre de solenoide, con 6 nucleosomas por vuelta
unidos mediante las histonas H1que, a su vez, están unidas al ADN espaciador.
Fig. 56 Estructura terciaria del ADN.
http://recursos.cnice.mec.es/biosfera/alumno/2bachillerato/biomol/contenidos18.htm
http://quimica-biologia-12-13.wikispaces.com/Biomol%C3%A9culas+para+la+vida.+ADN.

66
Los solenoides se enrollan formando la cromatina del núcleo interfásico de la
célula eucariota. Cuando la célula entra en división,el ADN se compacta aún
más, formando los cromosomas.
FUNCIONES BIOLÓGICAS DE LOS ÁCIDOS NUCLEÍCOS
Las funciones biológicas de los ácidos nucleicos pueden resumirse en el
almacenamiento y transmisión de la información genética. El conocimiento de
estas funciones demuestra que la información genética está contenida en las
moléculas de ADN. La confluencia de estudios entre la Bioquímica yla
Genética, tuvo su máximo resultado cuando en el año 1953, Watson y Crick
postularon el modelo estructural de doble hélice y el proceso de duplicación del
ADN. Estos trabajos condujeron rápidamente a una notable confluencia de
ideas así como nuevos enfoques experimentales de la Genética y la Bioquímica,
que desembocaron en lo que Crick denomino Dogma Central de la Genética
Molecular y que en un principio se podía resumir de la siguiente forma:
ADN ----------- ARN ---------------- proteína
Replicación----- Transcripción-------Traducción
Fig. 57 Estructura cuaternaria del ADN.
http://www.maristasgranada.net/webcole/documentos/Ciencias/Bach-
2%BA/Biologia/1_Bioquimica/04_Ac_Nucleicos/ADN_Estructura_1%AA,2%AA,3%
AAy4%AA.pdf

67
CAPITULO V
METABOLISMO DE LOS COMPUESTOS NITROGENADOS
METABOLISMO DE PROTEÍNAS
La síntesis de proteínas se lleva a cabo en dos etapas: la primera
etapa(transcripción) ocurre dentro del núcleo de las células eucariotas, aquí
lasecuencia se transcribe en una molécula de ARN, el cual es denominado ARN
mensajero (ARNm) y la segunda etapa (traducción o síntesis de
proteínapropiamente dicha) el ARNm pasa del núcleo al citoplasma donde el
mensajees traducido por los ribosomas que arman una proteína.
TRANSCRIPCIÓN
Para formar la cadena de ARN a partir del ADN se debe tener en cuenta que
cada nucleótido del ADN se ensambla con un determinado nucleótido del ARN.
La molécula helicoidal de ADN se desenrolla y deja accesible la cadena a partir
de la cual se inicia la síntesis (armado) del ARN. La enzima (polimerasa del
ARN) que controla la reacción detecta una región de la secuencia del ADN,
llamada promotor, que marca el punto de inicio de la síntesis. Los nucleótidos
se añaden uno por uno en orden complementario, de esta manera la adenina
Fig. 58 Traducción y Transcripción de proteínas.
http://mipaginapersonal.movistar.es/web3/mantmedina/flujo.htm

68
del ADN se combina con el uracilo del ARN (A – U), en el mismo orden,
latimina se ensambla con la adenina (T – A), y la citosina se combina con
laguanina y viceversa (C – G, G – C). Hay por lo tanto complementariedad
entre el ARN y el ADN de donde se copia. Al conservar la información impresa
enesta parte del genoma (dotación genética), el ARN se constituye en portador
de las instrucciones que determinan la secuencia de aminoácidos de una
proteína.
Dichas instrucciones, en clave, se descifran leyendo los nucleótidos de tres
entres ("tripletes"), y cada triplete de nucleótido, que determina uno de los 20
aminoácidos existentes, recibe el nombre de codón. Durante la traducción,
amedida que se "leen" los codones, se van añadiendo los
aminoácidoscorrespondientes a la proteína que se está formando.
TRADUCCIÓN
Queda claro que el ARNm es el que lleva la información que se decodificará en
la síntesis (armado) de proteínas, determina el orden en que se unirán
losaminoácidos. La síntesis de proteínas o traducción tiene lugar en los
ribosomasdel citoplasma celular. Los aminoácidos son transportados por el
ARN de transferencia (ARNt) específico para cada uno de ellos, y son llevados
hastaARNm, dónde se aparean el codón de éste y el anticodón del ARNt, por
complementariedad de bases, y de ésta forma se sitúan en la posición que
lescorresponde. Una vez finalizada la síntesis de una proteína, el ARNm
quedalibre y puede ser leído de nuevo. De hecho, es muy frecuente que antes de
quefinalice una proteína ya está comenzando otra, con lo cual, una misma
molécula de ARNm, está siendo utilizada por varios ribosomas
simultáneamente, esta estructura se conoce con el nombre de polirribosoma.
El trabajo de los ARNt consiste en tomar del citosol a los aminoácidos
yconducirlos al ribosoma en el orden marcado por los nucleótidos del
ARNm,que son los moldes del sistema. La síntesis de las proteínas comienza
con launión entre sí de dos aminoácidos y continúa por el agregado de nuevos
aminoácidos de a uno por vez, en uno extremos de la cadena.
Como se ha explicado, la clave de la traducción reside en el código genético,
compuesto por combinaciones de tres nucleótidos consecutivos o tripletes enel
ARNm. Los distintos tripletes se relacionan específicamente con tipos
deaminoácidos usados en la síntesis de las proteínas. Cada triplete constituye
uncodón, existen en total 64 codones, 61 de los cuales sirven para cifrar
aminoácidos y 3 paramarcar el cese de la traducción.

69
Fig. 59 Fases de la traducción.
http://mariielfashionn.blogspot.com/2012/05/7_19.html
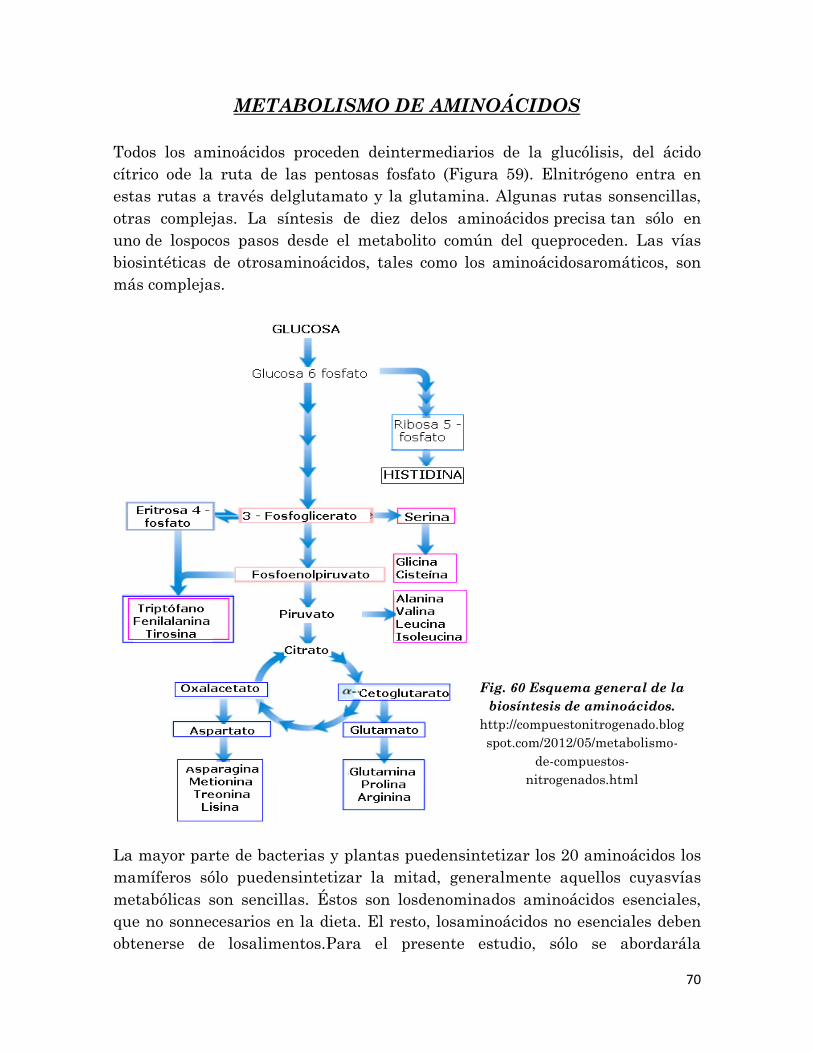
70
METABOLISMO DE AMINOÁCIDOS
Todos los aminoácidos proceden deintermediarios de la glucólisis, del ácido
cítrico ode la ruta de las pentosas fosfato (Figura 59). Elnitrógeno entra en
estas rutas a través delglutamato y la glutamina. Algunas rutas sonsencillas,
otras complejas. La síntesis de diez delos aminoácidos precisa tan sólo en
uno de lospocos pasos desde el metabolito común del queproceden. Las vías
biosintéticas de otrosaminoácidos, tales como los aminoácidosaromáticos, son
más complejas.
La mayor parte de bacterias y plantas puedensintetizar los 20 aminoácidos los
mamíferos sólo puedensintetizar la mitad, generalmente aquellos cuyasvías
metabólicas son sencillas. Éstos son losdenominados aminoácidos esenciales,
que no sonnecesarios en la dieta. El resto, losaminoácidos no esenciales deben
obtenerse de losalimentos.Para el presente estudio, sólo se abordarála
Fig. 60 Esquema general de la
biosíntesis de aminoácidos.
http://compuestonitrogenado.blog
spot.com/2012/05/metabolismo-
de-compuestos-
nitrogenados.html

71
biosíntesis de los aminoácidos no esenciales, larazón es que las rutas son
más cortas y el interésse enfoca a aquellos aminoácidos que sonsintetizados por
el ser humano.
BIOSÍNTESIS A PARTIR DEL α-CETOGLUTARATO
SÍNTESIS DELA GLUTAMINA A PARTIR DEL GLUTAMATO
Y DEL α- CETOGLUTARATO
La aminación reductiva del α-cetoglutarato
secataliza por la enzima glutamato
deshidrogenas, reacción reversible que
actuara en un sentido o en otro en
dependencias de las necesidades celulares y el
organismo. Con la colaboración del poder
reductor del NADPH, fija un grupo amino al α-
cetoácido.
La glutamina sintetasa cataliza la entrada
de un grupo amino sobre el carboxilo gamma,
formándose un grupo amida. La catálisis
necesita de la energía que aporta una molécula
de ATP. La glutamina sintetasa es un punto de
regulación importante en el metabolismo de
compuestos nitrogenados.
α- Cetoglutarato
Glutamato
Glutamina
Prolina
Arginina
Fig. 61 Reacción de la glutaminasintetasa.
http://es.scribd.com/doc/19096451/BIOSINTES-DE-
AMINOACIDOS

72
SÍNTESIS DE LA PROLINA A PARTIR DEL GLUTAMATO
El glutamato es el precursor de prolina y ornitina, siendo el glutamato
semialdehído un intermediario de ramificación llevando a uno o al otro de estos
2 productos. Mientras que la ornitina no es uno de los 20 aminoácidos usados
en síntesis de proteínas, juega un papel significativo como receptor del
carbamoil fosfato en el ciclo de la urea. La ornitina tiene un importante papel
adicional como precursor para la síntesis de poliaminas. La producción de
ornitina a partir del glutamato es importante cuando la arginina dietética, la
otra fuente principal de ornitina, es limitada.
Fig. 62 Biosíntesis de la prolina a partir del glutamato.
http://themedicalbiochemistrypage.org/es/amino-acid-metabolism-sp.php

73
SÍNTESIS DE LA SERINA Y GLICINA A PARTIR DEL
3-FOSFOGLICERATO
La principal vía para la biosíntesis de serina comienza con el intermediario
glicolítico 3-fosfoglicerato. Una deshidrogenasa ligada a NADH convierte el
3-fosfoglicerato en un cetoácido, 3-fosfopiruvato, adecuado para la
transaminación subsecuente. La actividad de la aminotransferasa con el
glutamato como donante produce 3-fosfoserina, que es convertido a serina
por la fosfoserina fosfatasa.
La serina puede ser derivada de la glicina (y viceversa) por una reacción de
un solo paso que envuelve la serina hidroximetiltransferasa y el
tetrahidrofolato (THF).
3-Fosfoglicerato
Serina
Glicina
Cisteína
Fig. 63 Biosíntesis de la serina a partir del 3-fosfoglicerato.
http://themedicalbiochemistrypage.org/es/amino-acid-metabolism-sp.php#top
Fig. 64 Biosíntesis de la glicina a partir de serina.
http://es.scribd.com/doc/19096451/BIOSINTES-DE-AMINOACIDOS
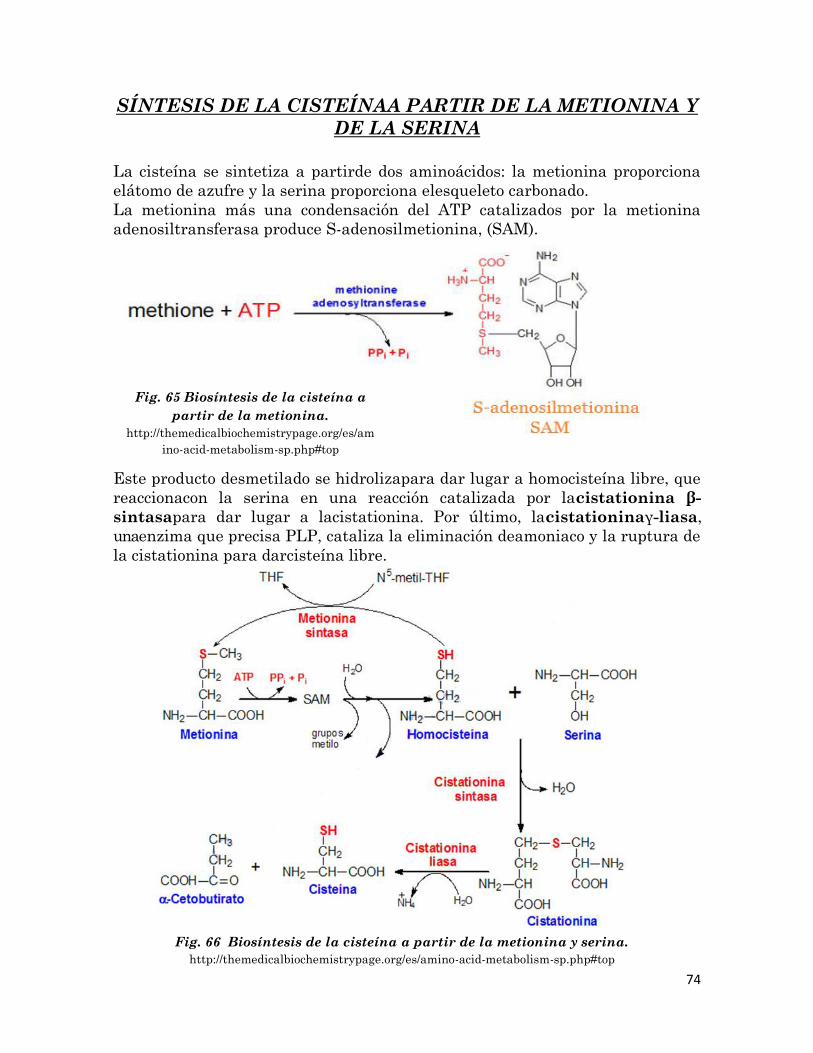
74
SÍNTESIS DE LA CISTEÍNAA PARTIR DE LA METIONINA Y
DE LA SERINA
La cisteína se sintetiza a partirde dos aminoácidos: la metionina proporciona
elátomo de azufre y la serina proporciona elesqueleto carbonado.
La metionina más una condensación del ATP catalizados por la metionina
adenosiltransferasa produce S-adenosilmetionina, (SAM).
Este producto desmetilado se hidrolizapara dar lugar a homocisteína libre, que
reaccionacon la serina en una reacción catalizada por lacistationina β-
sintasapara dar lugar a lacistationina. Por último, lacistationinaγ-liasa,
unaenzima que precisa PLP, cataliza la eliminación deamoniaco y la ruptura de
la cistationina para darcisteína libre.
Fig. 65 Biosíntesis de la cisteína a
partir de la metionina.
http://themedicalbiochemistrypage.org/es/am
ino-acid-metabolism-sp.php#top
Fig. 66 Biosíntesis de la cisteína a partir de la metionina y serina.
http://themedicalbiochemistrypage.org/es/amino-acid-metabolism-sp.php#top
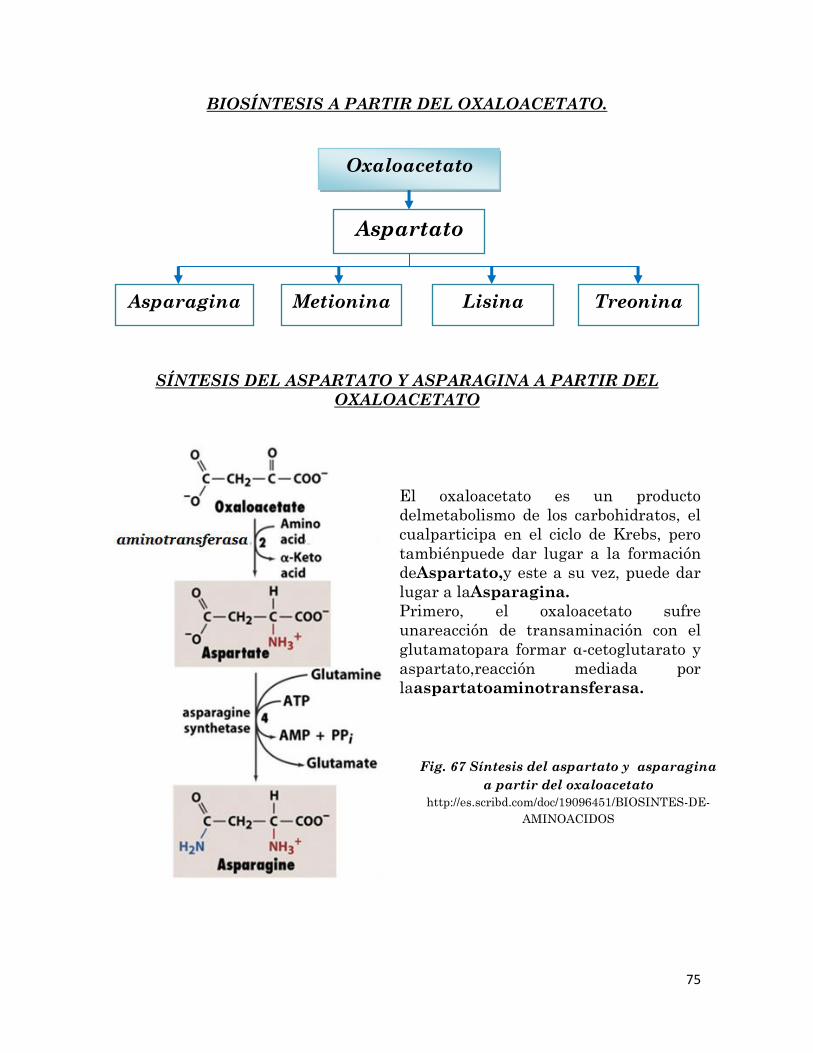
75
BIOSÍNTESIS A PARTIR DEL OXALOACETATO.
SÍNTESIS DEL ASPARTATO Y ASPARAGINA A PARTIR DEL
OXALOACETATO
El oxaloacetato es un producto
delmetabolismo de los carbohidratos, el
cualparticipa en el ciclo de Krebs, pero
tambiénpuede dar lugar a la formación
deAspartato,y este a su vez, puede dar
lugar a laAsparagina.
Primero, el oxaloacetato sufre
unareacción de transaminación con el
glutamatopara formar α-cetoglutarato y
aspartato,reacción mediada por
laaspartatoaminotransferasa.
Oxaloacetato
Aspartato
Asparagina
Lisina
Metionina
Treonina
Fig. 67 Síntesis del aspartato y asparagina
a partir del oxaloacetato
http://es.scribd.com/doc/19096451/BIOSINTES-DE-
AMINOACIDOS

76
BIOSÍNTESIS A PARTIR DEL PIRUVATO
SÍNTESIS DE LA ALANINA
La alanina se sintetiza a partir de piruvato portransaminación con el
glutamato o el aspartato.
SÍNTESIS DE TIROSINA A PARTIR DE LA FENILALANINA
Piruvato
Alanina
Leucina Valina
Isoleucina
Fig. 68 Síntesis de la alanina por transaminación del piruvato.
http://www2.uah.es/tejedor_bio/BBM-II_2F/tema15.htm
Fosfoenolpiruvato
Eritrosa 4 fosfato
Fenilalanina
Tirosina
Tirosina
Triptófano
a

77
SÍNTESIS DE LA TIROSINA
La tirosina es producida en las células por hidroxilación del aminoácido
esencial fenilalanina. Esta relación es como la que se da entre la cisteína y la
metionina. La mitad de la fenilalanina requerida va a la producción de
tirosina; si la dieta es rica en tirosina por sí misma, los requerimientos para la
fenilalanina se reducen en un 50%.
La fenilalanina hidroxilasa es una oxigenasa de funciones mixtas: un átomo
de oxígeno es incorporado en el agua y otro en el hidroxilo de la tirosina. El
agente reductor es el cofactor tetrahidrofolato relacionado con
latetrahidrobiopterina, que es mantenido en estado reducido por la enzima
dihidropteridina reductasa (DHPR) dependiente de NADH.
Fig. 69 Síntesis de la tirosina a partir de la fenilalanina.
http://es.scribd.com/doc/19096451/BIOSINTES-DE-AMINOACIDOS

78
CATABOLISMO DE AMINOÁCIDOS
El catabolismo de los aminoácidos se inicia con la eliminación del grupo amino,
después su α-cetoácido se incorpora al ciclo del ácido cítrico o del ácido
tricarboxílico (CAT) para la oxidación completa a CO₂ y H₂O.
Los 20 aminoácidos estándar tienen esqueletos de carbono muy diferentes, por
lo que sus conversiones a intermediarios del ciclo del ácido cítrico siguen vías
muy diversas, cada uno de ellos es degradado a siete intermediarios
metabólicos:
1. Acetil-CoA.
2. Acetoacetil-CoA.
3. Piruvato.
4. Alfa-cetoglutarato.
5. Succinil-CoA.
6. Fumarato.
7. Oxaloacetato.
Fig. 70 Catabolismo de los aminoácidos.
http://www2.uah.es/tejedor_bio/bioquimica_Farmacia/tema29.htm

79
Por lo tanto los aminoácidos pueden dividirse en dos grupos, según su ruta
catabólica:
1. Aminoácidos Glucogénicos.- cuyos esqueletos de carbono se degradan a
piruvato, α-cetoglutarato, succinil-CoA, fumarato u oxaloacetato, y son por
lo tanto, precursores de la glucosa.
2. Aminoácidos cetogénicos.- cuyos esqueletos de carbono son degradados a
acetil-CoA o acetato y pueden ser convertidos a ácidos grasos o cuerpos
cetónicos.
Por ejemplo la alanina es glucogénica porque su producto de transformación, el
piruvato, puede convertirse en glucosa por gluconeogénesis. La leucina, junto
con la lisina son cetogénicos, sus esqueletos de carbono se convierten en acetil-
CoA y en acetoacetato.
Fig. 71 Incorporación de esqueletos de carbono de los aminoácidos estándar al
ciclo del ácido cítrico. La leucina y triptófano se degradan y forman acetil-CoA.
http://www2.uah.es/tejedor_bio/bioquimica_Farmacia/tema31.htm

80
La Alanina, Cisteína, Glicina, Serina y Treonina se
degradan a piruvato.
La alanina es importante en el transporte del nitrógeno entre tejidos como
parte del ciclo glucosa-alanina.La vía catabólica de la alanina sufre una
reacción de transaminación con el α-cetoglutarato para formar piruvato y
glutamato. Generalmente el piruvato producido por esta vía resultará en la
formación del oxaloacetato, aunque cuando la carga de energía de una célula es
baja el piruvato será oxidado a CO2 y H2O por vía del complejo piruvato
deshidrogenasa (PDH) y del Ciclo del ácido tricarboxílico (TCA). Esto hace que
la alanina sea un aminoácido glucogénico.La transaminación de la alanina
para derivar en piruvato es la siguiente:
La serina puede ser convertida en piruvato con la serina deshidratasa, que
remueve el grupo hidroxilo y el grupo amino.
Fig. 72 La alanina se convierte en piruvato.
http://iqb.fcien.edu.uy/pdf/aminoacidos%20II%202004.pdf
Fig. 73 La serina se convierte
en piruvato.
http://iqb.fcien.edu.uy/pdf/aminoa
cidos%20II%202004.pdf

81
La conversión de serina a glicina y luego la oxidación de glicina a CO2 y NH3,
con la producción de dos equivalentes de N5,N10-metileno THF. La serina puede
ser catabolizada de nuevo al intermediario glicolítico, 3-fosfoglicerato, por una
vía que es esencialmente un reverso de la biosíntesis de serina. Sin embargo,
las enzimas son diferentes.
La Asparagina se degrada a aspartato y éste a su vez a
oxaloacetato
La asparaginasa también se distribuye extensamente dentro del cuerpo,
donde convierte la asparagina a amoníaco y aspartato. El aspartato se
transamina a oxaloacetato, que sigue el camino gluconeogénico a glucosa.
El glutamato y el aspartato son importantes en recoger y eliminar el
nitrógeno amino vía la glutamina sintetasa y el ciclo de la urea,
respectivamente. La trayectoria catabólica de los esqueletos de carbono
implica reacciones de aminotransferasa simples de 1 paso que producen
directamente cantidades netas de un intermediario del Ciclo del TCA. La
reacción de la glutamato deshidrogenasa que funciona en la dirección de la
producción de α-cetoglutarato proporciona una segunda ruta que conduce del
glutamato a la gluconeogénesis.
Del catabolismo de la asparagina se obtiene aspartato y de éste a su vez se
obtiene oxaloacetato.
Fig. 74 Conversión de la serina en glicina y ésta en CO₂ y
NH⁺₄.http://iqb.fcien.edu.uy/pdf/aminoacidos%20II%202004.pdf

82
La Arginina, Glutamato, Glutamina, Histidina y Prolina se
degradan a α-Cetoglutarato
La arginina, glutamina, histidina y prolina se degradan por conversión a
glutamato, el que, a su vez, se oxida a α-cetoglutarato por la glutamato
deshidrogenasa.
Fig. 76 Degradación de la arginina, glutamina, histidina y prolina
en glutamato y la conversión de este a α-cetoglutarato.
http://iqb.fcien.edu.uy/pdf/aminoacidos%20II%202004.pdf
Fig. 75 De la asparagina y el
aspartato se obtiene oxaloacetato.
http://iqb.fcien.edu.uy/pdf/aminoacidos
%20II%202004.pdf

83
La arginina sufre una reacción de hidrólisis exergónicas cataliza por la
arginasa para formar ornitina y urea.
La glutaminasa es una importante enzima del túbulo renal implicada en la
conversión de glutamina (del hígado y de otros tejidos) a glutamato y NH3+,
siendo el NH3+ excretado en la orina. La actividad de la glutaminasa está
presente en muchos otros tejidos también, aunque su actividad no es tan
prominente como en el riñón. El glutamato producido de la glutamina es
convertido a α-cetoglutarato, haciendo que la glutamina sea un aminoácido
glucogénico.
Fig. 77 La arginina se convierte
α-cetoglutarato.
http://iqb.fcien.edu.uy/pdf/aminoacido
s%20II%202004.pdf
Fig. 78 La glutamina se convierte
en α-cetoglutarato.
http://iqb.fcien.edu.uy/pdf/aminoacido
s%20II%202004.pdf

84
El catabolismo de la histidina comienza con la liberación del grupo α-amino
catalizado por la histidasa, introduciendo un doble enlace en la molécula.
Consecuentemente, el producto desaminado, urocanato, no es el usual α-
cetoácido asociado con la pérdida de α-amino nitrógenos. El producto final del
catabolismo de la histidina es glutamato, haciendo a la histidina uno de los
aminoácidos glucogénicos.
Otra característica dominante del catabolismo de la histidina es que sirve
como fuente de nitrógeno para combinarse con el tetrahidrofolato (THF),
produciendo el intermediario 1-carbono THF conocido como N5-formimino
THF. La última reacción es una de las dos rutas a N5- formimino. THF.
La decarboxilación de la histidina en el intestino por las bacterias da
lugar histamina.
Fig. 79 Catabolismo de la histidina en
α-cetoglutarato.
http://iqb.fcien.edu.uy/pdf/aminoacidos%20I
I%202004.pdf

85
El catabolismo de la prolina es un reverso de su proceso de la síntesis.
Comienza con una oxidación dependiente de flavoproteína para formar ∆' –
pirrolina 5-carboxilato. Posteriormente se añade agua al derivado, y el anillo se
abre no enzimaticamente para formar glutamato semialdehído, que por accion
de la glutamato deshidrogenasa se transforma en α-cetoglutarato.
Fig. 80 La prolina se convierte en α-
cetoglutarato.
http://iqb.fcien.edu.uy/pdf/aminoacidos%20II
%202004.pdf

86
LA METIONINA, VALINA, TREONINA E ISOLEUCINA SE
DEGRADAN A PROPIONIL-CoA
La metionina, valina, treonina y la isoleucina rinden propionil-CoA. La valina,
leucina e isoleucina son catabolizados principalmente en el músculo y produce
NADH y FADH2 que puede ser utilizado para la generación de ATP.
El producto principal de la valina es la propionil-CoA, el precursor glucogénico
del succinil-CoA. El catabolismo de la isoleucina termina con la producción de
acetil-CoA y propionil-CoA; así la isoleucina es tanto glucogénica como
cetogénica. La leucina da lugar a acetil-CoA y a acetoacetil-CoA, y se clasifica
así como estrictamente cetogénico.
Los principales destinos del aminoácido esencial metionina son la incorporación
en las cadenas del polipéptido, y la utilización en la producción de α-
cetobutirato y cisteína por vía de SAM como se describe en la síntesis de la
cisteína a partir de la metionina. Las reacciones del transulfuración que
producen cisteína a partir de la homocisteína y de la serina también producen
α-cetobutirato, el último que es convertido a succinil-CoA.
Fig. 81 Metionina, Valina, treonina, e isoleucina se degradan a propionil-CoA.
http://iqb.fcien.edu.uy/pdf/aminoacidos%20II%202004.pdf

87
LA LEUCINA, LISINA, SE DEGRADAN A ACETIL-CoA
Las proteínas del suero son ricas en los aminoácidos de cadena ramificada,
leucina, isoleucina y valina, y, por tanto, excelentes fuentes de producción de
energía en músculo esquelético.
Fig. 82 Leucina se degradan a
acetil-CoA.
http://iqb.fcien.edu.uy/pdf/aminoa
cidos%20II%202004.pdf
Fig. 83 Lisina se degradan a
acetil-CoA.
http://iqb.fcien.edu.uy/pdf/aminoacid
os%20II%202004.pdf

88
LA FENILALANINA Y LA TIROSINA, SE DEGRADAN A
FUMARATO Y ACETIL-CoA
La fenilalanina tiene normalmente solo dos destinos: la incorporación en las
cadenas del polipéptido, y la producción de tirosina por la fenilalanina
hidroxilasa dependiente de tetrahidrobiopterina. Así, el catabolismo de la
fenilalanina sigue siempre el camino del catabolismo de la tirosina. La
degradación de tirosina implica la conversión a fumarato y a acetoacetato,
permitiendo que la fenilalanina y la tirosina sean clasificadas como
glucogénicos y cetogénicos.
Fig. 84 Fenilalanina y Tirosina se degradan a fumarato y acetil-CoA.
http://iqb.fcien.edu.uy/pdf/aminoacidos%20II%202004.pdf

89
DEGRADACIÓN DEL TRIPTÓFANO A ALANINA
El triptófano tiene un metabolismo que puede dar lugar a la alanina que es
glucogénica y a acetil-CoA, que es cetogénica.
Esta ruta es la más compleja de los aminoácidos en los tejidos animales, consta
de trece etapas. La enzima triptófano pirrolasa cataliza la ruptura del anillo
indólico con la incorporación de dos átomos de oxígeno molecular, formando N-
formilquinurenina. La triptófano oxigenasa es inhibida mediante retroacción de
los derivados del ácido nicotínico, incluyendo NADPH.
Una pequeña porción de triptófano (2%) se convierte en nicotinato, una
vitamina. La cantidad de triptófano que es metabolizada diariamente es
pequeña algunos de los intermediarios se necesitan como precursores para la
biosíntesis de moléculas importantes como la serotonina, una neurohormona.
Fig. 85 Catabolismo del Triptófano.
http://books.google.com.ec/books?id=KHec9weY8Y0C&pg=PA262&lpg=PA262&dq=catabolismo+del+triptofa
no&source=bl&ots=5c0z42tJ5B&sig=-egeKxHAxecbYE-
lg0csC2Pfy_c&hl=es&sa=X&ei=gCFeUf7CFYn29gT_nIHgBA&sqi=2&ved=0CDYQ6AEwAQ#v=onepage&q=
catabolismo%20del%20triptofano&f=false

90
METABOLISMO DE LOSNUCLEÓTIDOS
Los nucleótidos son las unidades monómeras de las moléculas de ADN y ARN y
se encuentran en numerosas moléculas de importancia biológica, 5´-trifosfato
de adenosina (ATP), 5´-difosfato de adenosina (ADP), dinucleótido de
nicotinamida y adenina (NAD), dinucleótido fosfato de nicotinamida y adenina
(NADP), 5´-trifosfato de guanosina (GTP), y dinucléotido de flavina y adenina
(FAD); que actúan como mesajeros intracelulares e intervienen en reacciones
de transformación de energía y de fosforilación.
Las purinas y las pirimidinas forman parte de la estructura de los nucleótidos,
que estan formados por una base nitrogenada heterocíclica derivada de una
purina o de una pirimidina, una pentosa (ribosa en el ARN y desoxirribosa en
el ADN) y una molécula de ácido fosfórico.
BIOSÍNTESIS DE LAS PURINAS
Las purinas estan representadas en el organismo por la adneina, la guanina,
su precursor la inosina, y por algunos derivados menores que se encuentran en
los acidos nucleicos. El sitio principal de la síntesis de purinas está en el hígado
llevada a cabo por enzimas que sehallan en el citosol de la mayoría de las
células.
Estudios con isótopos han permitido establecer el origen de cada uno de los
átomos constituyentes del núcleo purina.
El N₁ surge del grupo
amino del aspartato; C₂ y
C₈ se originan de restos
formilos transportados
por tetrahidrofolatos
(THF); N₃ y N₉ son
aportados por el grupo
amida de la glutamina;
C₄, C₅ y N₇ derivan de la
glicina (lo que sugiere con
fireza que esta molecula
se incorpora totalmente
en el anillo de purina) y
el C₆ proviene del HCO₃ˉ. Fig.86 Precursores del anillo púrico.
http://www.paginasprodigy.com.mx/fiatlux/bioquimica/4.13
%20Metabolismo%20de%20purinas%20y%20pirimidinas.pdf

91
Biosíntesis de novo de nucleótidos
de purinas.
1. El paso limitante en la síntesis
del novo nucléotido de purinas es la
sintesis del PRAapartir del PRPP y
glutamina.
La glutamina reacciona con el PRPP,
adquiriendo el N₉, que procede de la
cadena lateral de la glutamina,
desplaza al grupo pirofosfato unido al
C₁ del PRPP.
2. La glicina + ATP, reacciona con
PRA para obterner GAR, en este paso
se adquiere los átomos de C₄, C₅ y N₇
del anillo púrico.
3. La adquisición del C₈ del anillo
se da al reaccionar el GAR + N⁻¹⁰
formil-tetrahidrofólico(THF),
transfiriendo el grupo formil para
formar FGAR y eliminando THF.
4. FGAR + Glutamina + ATP
forman el N₃ del anillo, es decir:
FGAM + Glutamato + ADP + P .i
Fig.87 Biosíntesis de purinas.
http://www.paginasprodigy.com.mx/fiatlux/bioquimica/4.13%20
Metabolismo%20de%20purinas%20y%20pirimidinas.pdf

92
5. FGAM + ATP producen el
cierre del anillo imidazol, es decir
AIR + ADP + P .i
6. AIR + ATP + HCO⁻₃
producen N⁵-CAIR + ADP + P i.
7. Junto con el paso amterior se
adquiere el C₆ del anillo púrico.
8. CAIR + Aspartato + ATP y
por acción de la enzima SACAIR
sintetasa dan lugar a la adquisición
del N₁ es decir forman SACAIR +
ADP + P .i
9. SAICAR por acción de la
enzima adenilsuccinato liasa
elimina el fumarato formando
AICAR.
Fig.88 Biosíntesis de purinas.
http://www.paginasprodigy.com.mx/fiatlux/bioquimica/4.13
%20Metabolismo%20de%20purinas%20y%20pirimidinas.pdf
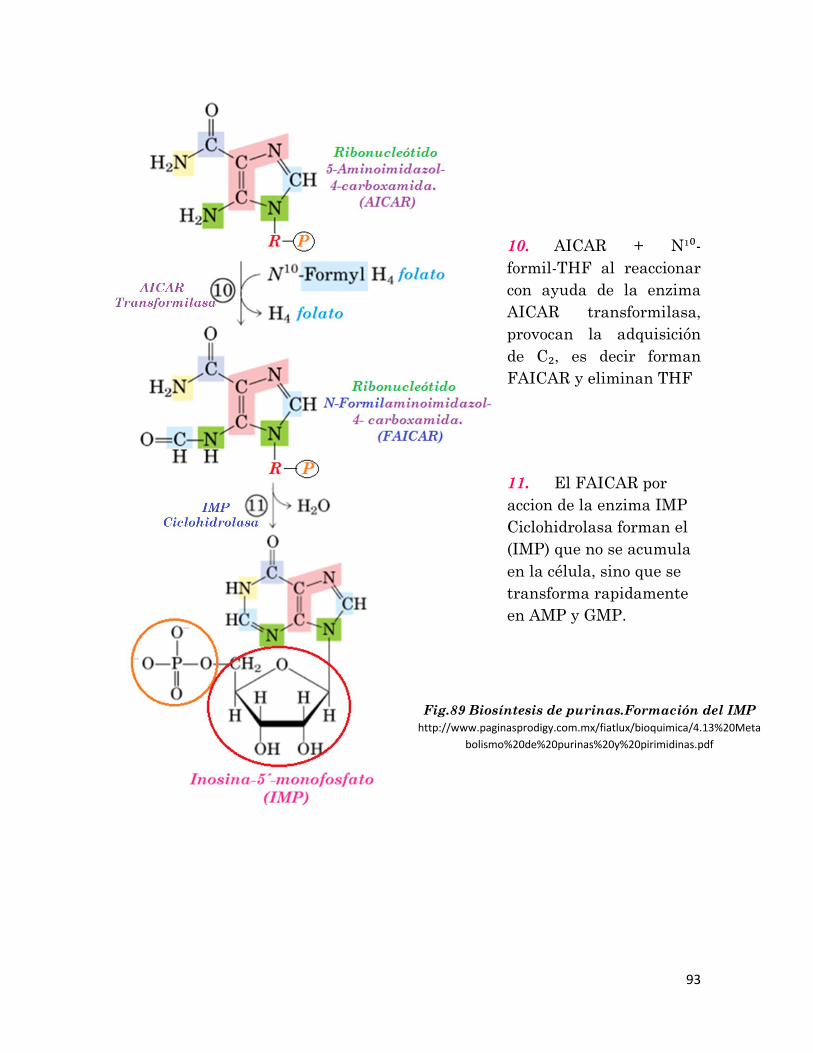
93
10. AICAR + N¹⁰-
formil-THF al reaccionar
con ayuda de la enzima
AICAR transformilasa,
provocan la adquisición
de C₂, es decir forman
FAICAR y eliminan THF
11. El FAICAR por
accion de la enzima IMP
Ciclohidrolasa forman el
(IMP) que no se acumula
en la célula, sino que se
transforma rapidamente
en AMP y GMP.
Fig.89 Biosíntesis de purinas.Formación del IMP
http://www.paginasprodigy.com.mx/fiatlux/bioquimica/4.13%20Meta
bolismo%20de%20purinas%20y%20pirimidinas.pdf

94
El IMP es el compuesto central de la síntesis de purinas (AMP)5´monofosfato
de adenosina y (GMP) 5´monofosfato de guanosina. Para evitar la
acumulación de uno de estos dos productos producida por una disminución de
la concentración del otro, estas vías están reguladas de forma que el GTP
actúa como cofactor en la síntesis de AMP, y el ATP como cofactor en la
GMP. Sin embargo, ello no evita la acumulación simultánea de AMP y GMP.
IMP + Aspartato + GTP Adenil-succinato + GDP +
P i
Adenil-succinato Fumarato + AMP
IMP + NAD i+ H₂O XMP + NADH + H i
XMP + Glutamina + ATP + H₂O GMP + Glutamato + AMP +
PP i
Fig. 90Biosíntesis de purinas. Formación del AMP y GMP a partir del IMP.
http://www.paginasprodigy.com.mx/fiatlux/bioquimica/4.13%20Metabolismo%20de%20purinas%20y%2
0pirimidinas.pdf

95
CATABOLISMO DE LAS PURINAS
El catabolismo de los nucleótidos de purina conduce en última instancia a la
producción de ácido úrico que es insoluble y es excretado en la orina como
cristales de urato de sodio.
Los ácidos nucleicos ya existentes en el organismo son hidrolizadospor endo y
exonucleasas que dan mononucleótidos que a su vez son degradados a
nucleósidos por la Fosfomonoesterasa, esta enzima libera guanosina y
adenosina. Estos 2 nucleósidos no pueden seguir exactamente la misma vía.La
adenosina debe ser desaminada previamente por la AdenosinaDesaminasa
para formar inosina.
Sobre la inosina actúa la Nucleósido Fosforilasa que la despoja de su ribosa y
da hipoxantina. La Nucleósido Fosforillasa actúa directamente sobre la
guanosina liberando guanina.Desde la hipoxantina y la guanina, como bases
púricas, se forma un compuesto llamadoxantina, que da origen al ácido úrico.
Estos últimos 2 pasos son catalizados por la Xantina Oxidasa (esta enzima
contiene FAD, molibdeno y hierro no hemo). La actividad de la Xantino
Oxidasa da lugar a laformación de ácidoúrico y luego al uratomonosódico.
Fig. 91Catabolismo de
purinas. Formación de
Ácido úrico.
http://themedicalbiochemistrypage
.org/es/nucleotide-metabolism-
sp.php

96
BIOSÍNTESIS DE NUCLEÓTIDOS DE PIRIMIDINA
Los precursores del anillo de pirimidina son: el Aspartato que otorga el
mayor aporte los C₄, C₅, C₆ y N₁; el HCO₃⁻ corresponde al C₂: Glutamina
aporta el N₃.
La biosíntesis de pirimidina se inicia con la formación del
carbamilfosfato(carbamoilfosfato), intermediario bioquímico que también
actúa comoprecursor para la síntesis de la urea (carbamida).
La primera base terminada se deriva a partir de un mol de glutamina, un
mol de ATP y un mol de CO2.
En los organismos eucariotas, la síntesis del dador de grupos carbonilos se
lleva a cabo en dos compartimentos celulares distintos: el carbamilfosfato que
se consume en la síntesis de pirimidinas se forma en el citosol (fracción soluble
del citoplasma); mientras que el carbamilfosfato que se utiliza en la síntesis de
la urea se forma en la mitocondria, proceso que es catalizado por una
carbamilfosfato-sintetasa diferente.La reacción del ciclo de la urea es catalizada
por la carbamoil fosfato sintetasa I (CPS-I) mientras que el precursor del
nucleótido de pirimidina es sintetizado por la CPS-II.
Otra diferencia es que el dador de nitrógeno para la síntesis
de carbamilfosfato citosólico es la glutamina, en lugar del amoniaco (NH4+).
Fig. 93Síntesis de carbamilfosfato derivado de la glutamina.
http://themedicalbiochemistrypage.org/es/nucleotide-metabolism-sp.php
Fig. 92 Precursores del anillo de
pirimidina
http://med.unne.edu.ar/catedras/bioquimica
/pdf/nitro.pdf
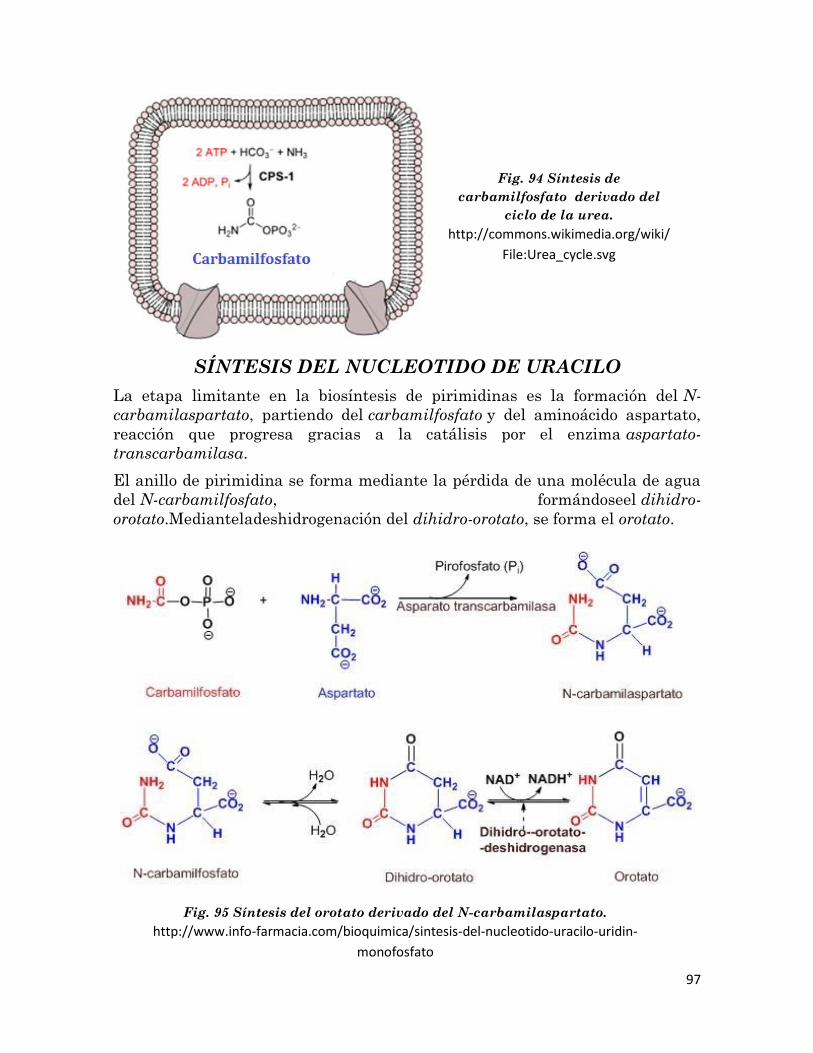
97
SÍNTESIS DEL NUCLEOTIDO DE URACILO
La etapa limitante en la biosíntesis de pirimidinas es la formación del N-
carbamilaspartato, partiendo del carbamilfosfato y del aminoácido aspartato,
reacción que progresa gracias a la catálisis por el enzima aspartato-
transcarbamilasa.
El anillo de pirimidina se forma mediante la pérdida de una molécula de agua
del N-carbamilfosfato, formándoseel dihidro-
orotato.Medianteladeshidrogenación del dihidro-orotato, se forma el orotato.
Fig. 94 Síntesis de
carbamilfosfato derivado del
ciclo de la urea.
http://commons.wikimedia.org/wiki/
File:Urea_cycle.svg
Fig. 95 Síntesis del orotato derivado del N-carbamilaspartato.
http://www.info-farmacia.com/bioquimica/sintesis-del-nucleotido-uracilo-uridin-
monofosfato

98
El orotato incorpora una ribosa fosfato, siendo el grupo
dadorFosforibosilpirofosfato (PPRP).
El orotato (una pirimidinalibre)reacciona con
elPPRPparaformar orotidilato (un nucleótido de pirimidina). La energía que
tenga para que progrese esa reacción proviene de la energía liberada por la
hidrólisis del pirofosfato. Finalmente, el orotidilato se descarboxila hasta
uridilato (uridina-monofosfato o, abreviadamente, UMP).
SÍNTESIS DEL NUCLEOTIDO DE CITOCINA
El Citidin-trifosfato (CTP) deriva del Uridin-trifosfato (UTP). La reacción
consiste en la aminación de un grupo carboxilo (indicado en color azul en la
reacción). En los organismos superiores (eucariotas) el dador del grupo amino
es laglutamina, que al ceder el grupo amino se transforma englutamato;
Fig. 96 Síntesis del Uridilato (UMP) derivado del Orotato.
http://www.info-farmacia.com/bioquimica/sintesis-del-nucleotido-
uracilo-uridin-monofosfato

99
mientras que en los procariotas (E. coli), el dador es un grupo amino (NH4+). En
el proceso evolutivo, los mamíferos evitan tener concentraciones elevadas del
grupo amino en plasma (tóxico), de tal manera que el grupo amino aportado por
la glutamina se usa a la vez que se genera.
SÍNTESIS DEL NUCLEOTIDO DE TIMINA
El Timidina monofosfato (TMP)se sintetiza a partir del UMP por la acción de
la timidilato sintasa con N5,N10-metileno-THF, donador de los grupos
metilos. La única propiedad de la acción de la timidilato sintasa es que el
THF es convertido a dihidrofolato (DHF). Para que la reacción de la
timidilato sintasa continúe, el THF debe ser regenerado desde DHF. Esto se
logra a través de la acción de la dihidrofolato reductasa (DHFR). El THF
entonces es convertido a N5,N10-THF por la acción de la serina hidroximetil
transferasa.
Fig. 97 Síntesis del Citidilato (CMP) derivado del Uridilato.
http://www.info-farmacia.com/bioquimica/sintesis-del-nucleotido-uracilo-uridin-monofosfato
Fig. 98 Síntesis del Timidilato (TMP) derivado del Uridilato.
http://www.info-farmacia.com/bioquimica/sintesis-del-nucleotido-uracilo-uridin-
monofosfato

100
CATABOLISMO DE NUCLEÓTIDOS DE PIRIMIDINA
Las bases pirmídicas son degradadas hasta productos muy solubles y
fácilmente eliminados o utilizados
porlas células.
El uracilo recibe dos hidrógenos
donados por NADPH para
convertirse en dihidrouracilo.
Posteriormente se produce, por
hidrólisis, apertura y ruptura del
anillo pirimidínico para formar β-
alanina, CO2 y NH3 como producto
final. Ninguno de estos productos es
exclusivo de la degradación del
uracilo, por lo que no puede
estimarse el recambio de los
nucleótidos de citosina y de uracilo a
partir de los productos finales de
esta vía.
La timina es hidrogenada a dihidrotimina en una reacción en la cual
participa NADPH. Luego el ciclo se abre y se produce β-aminoisobutirato,
CO2 y NH3. Eventualmente, el β-
aminoisobutirato puede
convertirseen succinil-CoA, que
puede ingresar al ciclo del ácido
cítrico. Por otro lado, el ácido β-
aminoisobutírico es excretado por la
orina en el hombre, y se origina
exclusivamente a partir de la
degradación de timina. Se puede, por
tanto, estimar el recambio de DNA o
de los nucleótidos de timidina
midiendo la excreción de β-amino-
isobutirato. Pacientes con cáncer
sometidos a quimioterapia o
radioterapia, procesos en los que se
matan grandes cantidades de células
y se degrada DNA, excretan niveles
aumentados de ácido β-
aminoisobutirico.
Fig. 100 Catabolismo de Timina a
ácido β-aminoisobutírico.
http://www.info-farmacia.com/bioquimica/sintesis-
del-nucleotido-uracilo-uridin-monofosfato
Fig. 99 Catabolismo de Citosina y
Uracilo a β-alanina.
http://www.info-farmacia.com/bioquimica/sintesis-
del-nucleotido-uracilo-uridin-monofosfato

101
Fig.101Reacción de desaminación
oxidativa. Catalizada por Glutamato
deshidrogenasa. NAD+ y NADP+
participan como cofactores y tanto los
nucleótidos trifosfatos (ATP y GTP) como
los difosfatos (ADP y GDP) ejercen control
como moduladores alostéricos positivos
según el sentido de la reacción.
http://med.unne.edu.ar/catedras/bioqu
imica/pdf/nitro.pdf
CAPITULO VI
ESTUDIO DE LOS PRODUCTOS FINALES DEL
METABOLISMO DE LOS COMPUESTOS NITROGENADOS
AMONÍACO
ASPECTOS GENERALES DEL AMONÍACO
El amoníaco es uno de los productos finales del metabolismo nitrogenado,
producido por células que se encuentran en todo el cuerpo, especialmente en los
intestinos, el hígado y los riñones. La mayor parte del amoníaco producido en el
organismo es utilizado por el hígado para producir urea.el valor plasmático
normal del amoníaco oscila entre 10-47 mcg/dL.Está constituido por nitrógeno e
hidrógeno. (NH₃ )i.
Todos los grupos α-amino de los aminoácidos sonfinalmente transferidos al α-
cetoglutarato mediante transaminación, formando L-glutamato. A partir de
esteaminoácido el grupo nitrogenado puede ser separado por un proceso
denominado desaminación oxidativa, una reacción catalizada por la L-
glutamato deshidrogenasas, una enzima presente de los tejidos de mamíferos
que utiliza como coenzima NAD+ o NADP+como oxidante. En la reacción
directa, generalmente se utiliza NAD+ y se forma α-cetoglutarato y amoníaco:
NH₃; este último, al pH fisiológico delmedio se carga con un protón,
presentándose casi en su totalidad como ion amonio (NH₄ )i. La reacción es
reversible, por lo que el amonio puede unirse a una α-cetoglutarato para formar
glutamato, usando como coenzima NADPH+. Es probable que in vivo la
reacción tenga mayormente una dirección haciala formación de amoníaco. La
concentración de amoniaco que sería necesario para que la reacción sedesplace
hacia la producción de glutamato es tóxica y, en condiciones normales, sería
raramente alcanzada, exceptuando la región periportal del hígado, donde llega
el amoníaco absorbido en el intestino y transportadoal hígado.

102
El glutamato forma parte del par obligado de la transaminación de los
aminoácidos y por tanto es la “puerta de acceso” del amoníaco libre a los grupos
amino de la mayoría de los aminoácidos; y a lainversa, es la “puerta de salida”
del nitrógeno de estos compuestos.
El papel predominante de la L-glutamato deshidrogenasas en la eliminación
del amoniaco queda marcado por su localización preponderante en las
mitocondrias del hígado en donde, como veremos más adelante, tienen lugar las
reacciones iniciales del ciclo de formación de urea. La enzima se implica
también en la producción de amoníaco a partir de aquellos aminoácidos que
sonrequerido para la producción de glucosa o para dar energía cuando se
agotan las reservas de otras moléculas: azucares y lípidos. Basándose en esto,
la L-glutamato deshidrogenasas se regula alostéricamente por los nucleótidos
púricos. Cuando es necesaria la oxidación de aminoácidos para la producción de
energía, la actividad en la dirección de la degradación del glutamato es
incrementada por el ADP y GDP, que son indicadores de un estado de bajo
nivel de energía enla célula. El GTP y ATP, indicativos de un nivel de energía
alto, son activadores alostéricos en la dirección de la síntesis de glutamato.
Regulación alostérica de la L-glutamato deshidrogenasa
Estado
Energético
ATP/GTP
ADP/GDP
Producto
Favorable
Alta
Baja
Glutamato
Desfavorable
Baja
Alta
Α-cetoglutarato +
NH⁴
TRANSPORTE DEL AMONÍACO
El amoníaco libre es tóxico, por lo que en la sangre es transportado
preferentemente en forma de grupos amina o amida. El 50% de los aminoácidos
circulantes está constituido por glutamina, untransportador de amoníaco. El
grupo amida de la glutamina es importante como dador de nitrógeno
paravarias clases de moléculas entre las que se encuentran las bases purínicas
y el grupo amino de la citosina. El glutamato y el amoníaco son el sustrato de la
glutamina sintetasa, la cual requiere ATP para la catálisis. Por otro lado, la
eliminación del grupo amida es catalizada por la glutaminasa.

103
El hígado contiene ambas enzimas, situadas en las células del parénquima, en
diferentes segmentos de esteórgano. La región periportal está en contacto con
la sangre que proviene del músculo esquelético y contiene glutaminasa (y las
enzimas del ciclo de la urea). Las células del área perivenosa, 5% del
parénquima, contienen glutamina sintetasa; la sangre fluye de este lugar hacía
el riñón. Este ciclo intercelular de laglutamina puede ser considerado un
mecanismo para recoger y eliminar el amoníaco que no ha sidoincorporado al
ciclo de la urea. El ciclo de la glutamina permite controlar el flujo de amoníaco
bien hacia laurea, bien hacia la glutamina, y por tanto hacia la excreción de
amoníaco por el riñón en diferentescondiciones de pH.
Una reacción similar a la de la glutaminasa es catalizada por la asparaginasa,
que hidroliza la asparagina a aspartato y amoníaco.
Un transportador de amoniaco extra lo constituye la alanina, la cual, en su
ciclo intertisula (ciclo de laalanina), moviliza, no solo su esqueleto carbonado
hasta el hígado para que este realice gluconeogenesis,sino también su grupo
amino para que sea transaminado a glutamato, posteriormente desaminado de
este aminoácido y sea convertido en urea.
TOXICIDAD DEL AMONÍACO
El amoníaco es tóxico y afecta principalmente al sistema nervioso central. La
encefalopatía asociada a defectos severos del ciclo de la urea se debe a aumento
de amoníaco en sangre y tejidos. Como el hígado es el principal órgano
encargado de la eliminación del amoníaco, cuando hay una falla o insuficiencia
Fig. 102 Formación y degradación de glutamina. La formación deglutamina es
un proceso que demanda gasto deenergía a diferencia de su procesoinverso.
med.unne.edu.ar/catedras/bioquimicas/pdf/nitro.pdf

104
hepáticagrave, la amonemia asciende y se produce un cuadro de intoxicación,
que pude llevar al coma e incluso lamuerte.
Como se menciono anteriormente, al pH fisiológico de los fluidos del organismo,
la casi totalidad, alrededor del 99%, del amoníaco que es una molécula neutra
se convierte en ión amonio, el cual no puede atravesar la membranas celulares.
Se han postulados diferentes mecanismos que podrían contribuir a la notable
toxicidad del amoníaco.
1. Acumulación de glutamina.- Los niveles de esta sustancia se
incrementan notablemente en las hiperamonemias. La acumulación de
glutamina en el cerebro, especialmente en astrositos, produce efecto
osmótico, aumentando la PIC (presión intracraneana) y dando hipoxia
cerebral.
2. Inhibición de la lanzadera malato-aspartato.- La síntesis exagerada
de glutamina reduce los niveles deglutamato, esto inhibe la lanzadera. Se
produce aumento de lactato y disminución del pH cerebral.
3. Actividad de la glucólisis.- El amoníaco estimula la fosfofructoquinasa y
con ello la actividad glucolítica. Aumenta el lactato y el valor de la relación
NADH/NAD+.
4. Inhibición del ciclo de Krebs. El aumento de amoníaco en la mitocondria
desvía la reacción de laglutamato deshidrogenasa hacia la aminación de α-
cetoglutarato para formar glutamato. Este “drenaje” de uno de los
intermediarios del ciclo del ácido cítrico deprime la marcha de esta vía de
oxidación, de la cual depende exclusivamente el cerebro para proveerse de
energía. El descenso de la concentración de ATP en las neuronas ocasiona
graves trastornos en su actividad, lo que conlleva en última instancia a su
muerte.
UREA La urea constituye el principal compuesto de excreción del amoníaco, que se
forma en el transcurso del catabolismo de los aminoácidos y las proteínas. Es
sintetizada en el hígado a partir de los aminoácidos y desde allí pasa a la
sangre. Finalmente es eliminada a nivel renal.
Fig. 103 Molécula de urea.
Compuesto rico en nitrógeno sintetizado en el hígado.

105
En forma práctica, la biosíntesis de este metabolito final encierra cuatro
etapas, incluyendo las reacciones recién tratadas:
1. Transaminación
2. Desaminación oxidativa
3. Transporte de amoníaco
4. Ciclo de la urea.
Estas etapas constituyen el flujo global del nitrógeno en el anfibolismo de los α-
aminoácidos.
CICLO DE LA UREA
La síntesis de la urea se realiza siguiendo un ciclo metabólico de reacciones
alimentado por el carbamil-fosfato. Algunas recciones son mitocondriales y
otras son citoplasmáticas. El conjunto de ellas se desarrolla exclusivamente en
el hígado, aunque algunas de las etapas de este ciclo metabólico se pueden dar
en otros tejidos. Concretamente, en las células de la mucosa intestinal se
desarrollan la mayor parte de estas etapas, aunque el producto final no es la
urea, sino los aminoácidos orinitina, prolina, citrulina y arginina.
Los dos nitrógenos de cada molécula de urea provienen de dos fuentes, el
amoníaco libre y el grupo amino del aspartato. El ciclo se inicia y finaliza en el
aminoácido ornitina. A diferencia del ciclo TCA, en donde los carbonos del
oxalacetato al principio son diferentes de los del final, los carbonos de la
ornitina final son los mismos que poseía la molécula inicialmente.
Fig. 104 Flujo global del nitrógeno en el catabolismo de los
aminoácidos.Las transaminaciones confluyen el nitrógeno amídico a glutamato, desde el cual
se lo extrae por desaminación para ser transportado en sangre (amoniaco libre, glutamina,
asparagina y alanina) hasta el hígado para sintetizar urea y ser excretada luego por el riñón. med.unne.edu.ar/catedras/bioquimicas/pdf/nitro.pdf

106
Cinco enzimas catalizan las reacciones de éste ciclo.De los seis aminoácidos que
participan, solo el N-acetilglutamato funcionan como activador enzimático; los
otros actúan como transportadores de los átomos que finalmente se convertirán
en urea. En los mamíferos, la principal función de la ornitina, citrulina y
argininosuccinato es la síntesis de la urea. Las reacciones del ciclo están
bicompartimentalizadas, algunas reacciones se llevan a cabo en la matriz
mitocondrial, en tanto que otras ocurren en el citosol. En forma esquemática
podemos enumerar las etapas de la biosíntesis de urea de la siguiente manera:
1. Inicio de la biosíntesis: Carbomoil fosfato sintetasa I.- La
biosíntesis de urea comienza con la combinación de bióxido de carbono,
amoníaco y 2 ATP, para formar carbamoil fosfato, reacción catalizada
por la carbamoil fosfato sintetasa I (CPSI). En los tejidos humanos
existen dos formas de CPS. La carbamoil fosfato sintetasa I, del la
síntesis de la urea, es una enzima mitocondrial hepática. La carbamoil
fosfato sintetasa II (CPSII), una enzima citisólica que emplea glutamina
en vez de amoníaco como donador de nitrógeno, participa en la
biosíntesis de pirimidinas.
Fig. 105 Reacciones e intermediarios en la biosíntesis de urea; relación con el
ciclo de Krebs. Los compuestos sombreados con gris corresponden a los que
contribuyen con nitrógeno para la formación de urea. A.Desaminación
oxidativa, B.Transaminación, C.Gluconeogénesis; 1.Carbamoil fosfato
sintetasa I, 2.Ornitina transcarbamoilasa, 3.Argininosuccinato sintetasa,
4.Argininosuccinasa, 5.Arginasa, 6.Fumarasa, 7.Malato deshidrogenasa. med.unne.edu.ar/catedras/bioquimicas/pdf/nitro.pdf

107
La CPSI es la enzima limitante de la velocidad, o marcapaso, del ciclo de
la urea. Esta enzima reguladora es activa sólo en presencia del activador
alostérico N-acetilglutamato, cuya unión induce un cambio
conformacional que aumenta la afinidad de la sintetasa por el ATP.
2. Formación de citrulina.- La L-ornitina transcarbamoilasa cataliza la
transferencia de la porción carbamoil del carbamoil fosfato a un
aminoácido ornitna, formando citrulina y ortofosfato. Esta reacción se
lleva a cabo en la matriz mitocondrial; la formación del sustrato ornitina
y la metabolización subsecuente del producto, citrulina, se lleva a cabo
en el citosol. Por tanto la entrada como la salida de ornitina y citrulina
de la mitocondria implica la participación de un sistema de transporte
situado en la membrana interna de esta organela, formado por un
contratransportador citrulina/ornitina.
3. Formación de argininosuccinato.- La reacción de la
argininosuccinato sintetasaune aspartato y citrulina a través del grupo
amino del aspartato, y suministra el segundo nitrógeno de la urea.
La reacción requiere ATP para formar un intermediario citrulina-AMP y
luego, desplazando el AMP por aspartato forma citrulina.
4. Formación de arginina y fumarato.- La escisión del
argininosuccinato, catalizado por la argininosuccinasa o arginino
succinato liasa, retiene nitrógeno en el producto arginina y libera el
esqueleto del aspartato como fumarato. La adición de agua al fumarato
genera malato, y la oxidación de éste, dependiente de NAD+, forma
oxalacetato. Estas dos reacciones, correspondientes a ciclo TCA, se
catalizan por la fumarasa y la malato deshidrogenasa citosólicas. La
transaminación del oxalacetato con el glutamato forma de nuevo
aspartato. El esqueletocarbonado del aspartato/fumarato, actúa como un
transportador para el paso del nitrógeno del glutamato a un precursor de
la urea.
5. Formación de ornitina y urea.- La reacción final del ciclo de la urea,
la ruptura hidrolítica de la arginina caltalizada por la arginasahepática,
libera urea. El otro producto, ornitina, reingresa a la mitocondria
hepática para ser utilizada nuevamente en el ciclo de la urea.
Cantidades menores de arginasa también se encuentran en los tejidos
renal, cerebral, mamario, testicular y en la piel. La ornitina y la lisina
son inhibidores potentes de la arginasa, y por tanto, compiten con la
arginina.
REGULACIÓN DEL CICLO DE LA UREA La regulación de la formación de urea se realiza en dos niveles, en la carbamoil
fosfato sintetasa I y por inducción enzimática.
La CPSI necesita de forma obligada el activador alostérico N-acetilglutamato.
Este compuesto es sintetizado a partir de glutamato y acetil-CoA por la N-
acetilglutamato sintetasa, que es activada por laarginina. El acetil-CoA, el

108
glutamato y la arginina son necesarios para suministrar intermediarios o
energía (ATP desde el ciclo TCA) al ciclo de la urea, y la presencia de N-
acetilglutamato indica que todos ellos están disponibles y en abundancia. Es
comprensible que una ruta que controla el nivel de amoníaco en plasma,
potencialmente tóxico, y que es además altamente dependiente de energía, esté
finamente regulado.
La inducción enzimática del ciclo de la urea (de 10 a 20 veces) tiene lugar
cuando aumenta el suministro de amoníaco o aminoácidos al hígado. La
concentración de los intermediarios del ciclo también desempeña un papel en
su regulación a través de la ley de acción de masa. Una dieta rica en proteínas
(exceso de aminoácidos) o la inanición (exceso de amoníaco por utilización de
cadenas carbonadas de aminoácidos para obtener energía), tienen como
resultado la inducción de las enzimas del ciclo de la urea.
RELACIÓN DEL CICLO DE LA UREA CON EL CICLO TCA
Los dos ciclos descriptos por Krebs se relacionan por medio del fumarato,
producto de la argininosuccinasa en el ciclo de la urea, éste metabolito puede
ingresar a la mitocondria y seguir, como vimos, el ciclo TCA, llegando a la
formación de oxalacetato, el cual puede seguir tres vías:
1. Continuar el TCA para dar energía.
2. Dar glucosa, vía fosfoenolpiruvato, en la gluconeogénesis.
3. Transaminarse con glutamato y dar α-cetoglutarato y aspartato; este
último es sustrato en el citosol de la argininosuccinato sintetasa,
aportando uno de los dos grupos nitrogenados para la formación de
urea.
Fig. 106 Relación entre el ciclo TCA y de la urea.
Destino del fumarato. 1. Producción de energía, 2.
Formación de glucosa y 3. Formación de aspartato. med.unne.edu.ar/catedras/bioquimicas/pdf/nitro.pdf

109
DESTINO DE LA UREA
El producto final del metabolismo de los aminoácidos es la urea, esta es
transportada por la circulación hasta el riñón para su excreción. Un adulto
normal, con una dieta equilibrada elimina alrededor de 25 a 35g de urea diarios
por orina, lo cual corresponde al 90% del nitrógeno total excretado por esta vía.
La urea es una sustancia soluble, fácilmente difusible a través de las
membranas celulares. Además, es completamente atoxica. Se encuentra en
sangre circulante en una concentración de 20 a 40mg/dL (mg por100ml) o
0,4mM. Este nivel aumenta en caso de insuficiencia renal. Comúnmente se
habla de uremia en las situaciones en las cuales la falla de la función renal
impide la excreción del metabolito.
ÁCIDO ÚRICO
Elácido úrico es el producto final del catabolismo de las purinas en el ser
humano. Su concentracion plasmática normal en varones es entre 3,4 y
7,0mg/dL y en las mujeres entre 2,4 y 6,0mg/dL,el execeso de ácido úrico se
deposita en los tejidos, principalmente en las articulaciones, es insoluble
yexcretado en la orina como cristales de urato de sodio.
Fig. 107 Catabolismo de purinas. Formación de Ácido úrico.
http://themedicalbiochemistrypage.org/es/nucleotide-metabolism-sp.php

110
METABOLISMO DEL ÁCIDO ÚRICO
El ácido úrico es creado cuando el cuerpo descompone sustancias llamadas
purinas, las cuales se encuentran en algunos alimentos y bebidas, como el
hígado, las anchoas, la caballa, las judías y arvejas secas, la cerveza y el vino.
Es el producto del desecho terminal del metabolismo purínico. Las dos purinas,
adeninay guanina, se encuentran en el organismo principalmente como
componentes de los ácidosnucleicos, ácido ribonucleico (ARN) y ácido
desoxirribonucleico (ADN). Normalmente existendos fuentes de purinas, las
que se obtienen por la hidrólisis de los ácidos nucleicos ingeridoso por los
endógenos. El ácido úrico o 2-6-8-trioxipurina, se forman por la oxidación
enzimáticade la adenina y guanina.
La mayor parte del ácido úrico se disuelve en la sangre y viaja a los riñones,
donde sale a través de la orina.Eliminamos 750 mg. diarios de ácido úrico, de
los cuales 500 mg. son eliminados por vía renal y 250 mg. por las heces. Todo
exceso de esta cantidad permite su acumulación. Los humanos no disponemos
de uricasa, única enzima que destruye el ácido úrico, sin embargo se ha
descrito una uricolisis a nivel intestinal.
FACTORES QUE AUMENTAN EL ÁCIDO ÚRICO
El ácido úrico aumenta por varios mecanismos:
Defectos enzimáticos
Destrucción celular (casos cáncer) traumas
Metabolismo de proteínas(purinas).
Muy poco por alimentación.
El alcohol en cualquiera de sus tipos impide su eliminación, por ello la
gota esfrecuente en alcoholizados.
Tomar café en exceso (mayor de 2 tazas).
Consumir muchas calorías.
Exceso de grasas saturadas y purinas.
Someterse a periodos de ayuno.
Falta de ingesta de líquidos.
Consumo crónico de aspirina y diuréticos tiazidicos.

111
CREATINA
La creatina es un ácido orgánico nitrogenado elaborado por el propio organismo
con la función de almacenar energía. Se encuentra en cada célula humana y
gracias a ella es posible el rendimiento físico y mental.
Nuestro organismo elabora alrededor de 1 g/día a partir de los aminoácidos
glicina, arginina y metionina en el hígado, riñones y en elpáncreas
principalmente.
De allí es transportada al torrente sanguíneo y posteriormente a todas las
células del cuerpo. Las células musculares, cerebrales y nerviosas ontienen
grandes cantidades de creatina yaque participan enprocesos que requieren
energía.
En menor proporción está presente en el cerebro, espermatozoides,
fotorreceptores de la retina, tejido adiposo marrón, intestino y vesículas
seminales.
Aproximadamente el 95% de la creatina está almacenada en la musculatura
estriada, desempeñando un papel esencial en la contracción muscular.
Fig. 108 Síntesis de creatina en el organismo y su metabolismo.
http://www.creapure.es/es/creapure-en-el-deporte-de-fuerza/por-que-es-tan-eficaz-creapure/la-creatina-en-el-cuerpo-humano

112
Alrededor del 60% de toda la creatina muscular se encuentra en forma de
fosfocreatina a (Pcr) constituyendo energía a corto plazo, mientras que el 40%
restante aparece en forma de creatina libre.
Todas las células del cuerpo emplean ATP (adenosín trifosfato) como fuente de
energía. Aparte de esta molécula, la creatina es la fuente energética más
importante del cuerpo.
Dado que los depósitos de ATP en el cuerpo están limitados (carbohidratos y
grasas), el metabolismo debe formar ATP de formacontinua. Cuando la célula
consume energía (ATP), se convierte en adenosín difosfato (ADP) “pobre en
energía”. Si los carbohidratos y las grasas se agotan en función de la intensidad
del ejercicio, la fuente energética que el organismo empleará será la
fosfocreatina.
Los músculos contienen entre 3 y 4 veces más fosfocreatina que ATP. Esto es
decisivo en ejercicios que requieren un gran esfuerzo ya que permite liberar y
proporcionar más energía.
La creatina se encuentra presente de forma natural en los alimentoscomo la
carne roja, el pescado, los productos lácteos y el huevo, aunque se tendría que
consumir grandes cantidades para rozar el gramo de creatina ingestada. En
Fig. 109 La creatina participa en la regeneración del ATP, la fuente
de energía de nuestro organismo.
https://s3-eu-west-1.amazonaws.com/nutritiendamagazine-images/n_2/guia-de-la-creatina/guia-de-la-creatina-n-02.pdf

113
vegetarianos, la concentración de creatina en la sangre es significativamente
baja (40 – 50 %) al suprimir estos alimentos de su dieta.
METABOLISMO DE LA CREATINA EN EL CUERPO
HUMANO
La cantidad de creatina total (PCr + creatina libre) en el músculo esquelético
para un individuo de 70 kg es de 120 gramos, sin embargo una persona puede
llegar a almacenar hasta160 gramos.
A diario, el organismo transforma aproximadamente 1-2 g de la reserva de
creatina en el músculo esquelético en creatinina y posteriormente se excreta
con la orina. Estas pérdidas se compensan enun 50% mediante la dieta y la
síntesis endógena.
TRANSPORTE Y CAPTACIÓN DE CREATINA POR LOS
TEJIDOS
Los órganos captan la creatina mediante la variación de concentración dentro y
fuera de la célula y gracias a la actividad de los trasportadores CREA T. Estos
transportadores, presentes en la membranacitoplasmática de las células
musculares, regulan el contenido de creatina dentro de la célula.
Existen dos aspectos críticos que pueden afectar a la captación y absorción de
creatina:
Fig. 110 Biodisponibilidad de creatina.
http://www.creapure.es/es/creapure-en-el-deporte-de-fuerza/por-que-es-tan-eficaz-creapure/la-creatina-en-el-cuerpo-humano

114
La concentración de creatina dentro de la célula, que determina la
necesidad de elevar sus niveles plasmáticos.
Las elevaciones reiteradas de los niveles plasmáticos de creatina que
afectan a los transportadores CREA T perjudicando la captación y
absorción celular.
La técnica nutricional y la suplementación con monohidrato decreatina pueden
incrementar el transporte y la captación de creatina por los tejidos al estimular
la actividad de los transportadores. Este efecto también puede potenciarse por
aminoácidos como la taurina o por la ingesta de hidratos de carbono que elevan
la glucemiay estimulan la secreción de insulina. Este hecho es
especialmenteefectivo durante las primeras 24 horas.
La concentración de CREA T es un factor fundamental que determina la
capacidad de captación y almacenamiento de creatina en el músculo. Estos
transportadores se saturan con una concentraciónplasmática de 100
moles/litro, por lo tanto es recomendable ingerir un suplemento nutricional que
aporte una concentración elevada demonohidrato de creatina de alta calidad
para que se asimile rápidamente en sangre, cause una elevación brusca de los
niveles plasmáticos y que este nivel se mantenga durante el tiempo o suficiente
para inducir su captación por las células musculares.
CREATININA
La creatinina es un compuesto orgánico generado a partir de la degradación de
la creatina. Es un producto de desecho del metabolismo normal de los músculos
que usualmente es producida por el cuerpo en una tasa muy constante, y
normalmente filtrada por los riñones y excretada en la orina. La medición de la
creatinina es la manera más simple de monitorizar la correcta función de los
riñones.
La creatinina es una molécula de desecho que se genera a partir del
metabolismo muscular. La creatinina proviene de la creatina, una molécula
muy importante para la producción de energía muscular. Aproximadamente el
2% de la creatina del cuerpo se convierte en creatinina cada día. La creatinina
Fig. 111Estructura química de
la creatinina.
http://es.wikipedia.org/wiki/Creatinina

115
se transporta desde los músculos por medio de la sangre hacia el riñón. Los
riñones filtran la mayoría de la creatinina y la eliminan en la orina.
Aunque es una sustancia de deshecho, la creatinina es una prueba diagnóstica
esencial, ya que se ha observado que su concentración en sangre indica con
bastante fiabilidad el estado de la función renal. Si los riñones no funcionan
bien, no eliminan bien la creatinina y por lo tanto ésta se acumula en la sangre.
Por esto la creatinina puede avisar de una posible disfunción o insuficiencia
renal, incluso antes de que se presenten síntomas. Por eso la creatinina suele
figurar en los análisis de sangre que se realizan comúnmente.
Los valores normales de creatinina en la sangre son aproximadamente0,6 a
1,2mg/dL en los varones adultos y 0,5 a 1,1 mg/dL en las mujeres adultas. Los
adultos con mucha masa muscular pueden tener más creatinina en la sangre
que la población normal. Las personas ancianas, por otro lado, pueden tener
menos creatinina en la sangre de lo normal.
Algunos fármacos pueden producir una elevación anormal de las
concentraciones de creatinina en sangre. Una concentración muy elevada de
creatinina en la sangre puede indicar la necesidad de someterse a diálisis para
eliminar las sustancias de deshecho de la sangre.
Fig. 112Síntesis de la creatinina.
http://creatininametabolismo.blogspot.com

116
La creatina es sintetizada en el hígado por metilación del guanidoacetato
usando SAM como donante metilo.El guanidoacetato se forma en el riñón a
partir de los aminoácidos arginina y glicina.
La creatina es utilizada como forma de almacenamiento del fosfato de alta
energía. El fosfato del ATP es transferido a la creatina, generando fosfato de
creatina, a través de la acción de la creatin-fosfocinasa. La reacción es
reversible cuando la demanda energética es altala creatinfosfato dona su
fosfato al ADP para producir ATP.
La creatina y la creatinfosfato se encuentran en músculo, cerebro y sangre. La
creatinina es formada en músculo a partir de la creatinfosfato por una
deshidratación no enzimática y perdida del fosfato. La cantidad de creatinina
producida se relaciona con la masa muscular y se mantiene constante día a día.
La creatinina es excretada por los riñones y el nivel de excreción (porcentaje de
clearance de creatinina) es una medida de la función renal.
Fig. 113Síntesis de la creatina y de creatinina.
http://creatininametabolismo.blogspot.com

117
CAPITULO VII
ENFERMEDADES CAUSADAS POR EL DESEQUILIBRIO
DEL BALANCE DELNITRÓGENO
Enfermedades causadas por el aumento de productos finales
del metabolismo humano
Los diferentes productos finales del metabolismo del nitrógeno son producidos
constantemente por el organismo y eliminados por los riñones, es por ello que
las pruebas de laboratorio se las solicita para evaluar la función renal, función
hepática.
Para la evaluación de los riñones, se solita la urea y la creatinina
conjuntamente. Sin embargo la creatinina es un mejor indicador, ya que la
urea puede ser alterada en casos de deshidratación, uso de diuréticos, sangrado
digestivo, alimentación rica en proteínas, enfermedad del hígado, etc.Si los
riñones y el hígado no funcionan bien, dichas sustancias comienzan a
acumularse en la sangre. Por lo tanto, cuanto peor sea la función renal, más
elevados serán los valores de urea, creatinina y ácido úrico.
HIPERAMONEMIA
La hiperamonemia tiene como característica principal, mostrar concentraciones
elevadas de amonio en la sangre, lo cual sucede cuando el amonio no es
eliminado del organismo a través de la orina. Si no se detecta rápido, puede
causar daños irreversibles en el sistema nervioso central, debido a que es
tóxico. Esta condición debe tratarse de forma inmediata y adecuada, pues de lo
contrario produce secuelas neurológicas graves y definitivas, que pueden
conducir a discapacidad intelectual y motora.
Un paciente puede presentar.
Letargo
Temblores
Habla balbuciente
Vómitos
Coma
Muerte

118
La hiperamonemia puede ser de dos tipos:
1. Hiperamonemia adquirida.- esta puede ser causada por: consumo
excesivo de etanol (bebidas alcohólicas); hepatitis C; exposición
prolongada a tetracloruro de carbono; amebiasis.
Todas estas razones causan cirrosis es decir pérdida de la función
hepática, ésta es devastadora debido a la extensa inflamación y necrosis
de los hepatocitos, lo que causa que estas células no puedan cumplir su
funciónalterando el ciclo de la urea, provocando acumulación de amonio.
2. Hiperamonemia congénita.- es un trastorno genético hereditario en
el cual una o varias enzimas del ciclo de la urea falta o es defectuosa, la
ausencia completa de una enzima del ciclo de la urea es mortal tras el
nacimiento ya que produce daño cerebral. Se la diagnostica al momento
del nacimiento.
Los desórdenes en el ciclo de la urea, causan:
HIPERAMONEMIA I.- Es el defecto en la falta de la enzima carbamoil
fosfato sintetasa I, la cual transforma el amoniaco a carbamoilfosfato. Se
caracteriza por:
• Amonio (NH₄) elevado en Sangre, Hígado y Orina.
• Vómitos
• Síntomas neurológicos
• Retraso mental
HIPERAMONEMIA II.- Es el defecto en la falta de la enzima ornitina
transcarbamilasa, la cual transforma el carbamoilfosfato a citrulina. Es
una enfermedad de herencia ligada al cromosoma X. Se caracteriza por:
• Glutamina elevada en Sangre, Líquido cefalorraquídeo (LCR) y
Orina.
• Vómitos
• Convulsiones
• Retraso mental. Letal en varones en período neonatal.
DIAGNÓSTICO
La evaluación inicial de los pacientes en quienes se sospecha hiperamonemia
consiste en la determinación sérica de amonio.
Una vez confirmado que existe hiperamonemia, deberán realizarse estudios
para determinar su etiología, entre los que se encuentran:
1. Pruebas de funcionamiento hepático

119
2. Pruebas de coagulación
3. Ultrasonido hepático
4. Tomografía axial computada de abdomen
Si con estos estudios no fuese suficiente para determinar la etiología, deberán
sospecharse errores innatos del metabolismo y para ello deberán realizarse las
siguientes determinaciones en suero y orina:
1. Citrulina
2. Glutamina
3. Ácido arginosuccínico
TRATAMIENTO 1. Administración de fenilacetato y benzoato de sodio para bajar los niveles
de amonio.
2. Llevar una dieta baja en proteínas.
HIPERURICEMIAO GOTA
La hiperuricemia o gota es un desorden orgánico del metabolismo de las
proteínas en el cual durante procesamiento de las mismas se puede producir
una cantidad excesiva de ácido úrico. Es posible también que el organismo no
pueda o exceda su capacidad de eliminarlo por el riñón y el sudor.
Cuando los niveles de ácido úrico en la sangre se elevan, comienzan a
depositarse principalmente en las articulaciones en forma de cristales de ácido
úrico.
Estos cristales ocasionan una inflamación intensa de la articulación con
enrojecimiento y dolor importante acompañado de impotencia
funcionalllamados“tofos”.
Aunque afecta con mayor frecuencia las pequeñas articulaciones del pie,
también puede afectar las manos o articulaciones mayores como rodilla, tobillo,
pie, codo y hombro.
Fig. 114Los cristales de ácido úrico se deposita en las articulaciones.
http://drsantamaria.blogspot.com/2011/01/que-es-la-gota.html

120
Este depósito es consecuencia directa de la hiperuricemia, y es reversible, ya
que cuando la uricemia se normaliza los cristales lentamente se disuelven y
acaban por desaparecer, por ello se ha considerado a la gota comouna
enfermedad curable. El depósito de cristales esasintomático pero los cristales
dentro de las articulaciones tienen capacidad de desencadenar episodios de
inflamación, súbitos y frecuentemente intensos que son los ataques de gota.
Además de los inconvenientes que representa para el paciente, los ataques
alertan al médico sobre la existencia del depósito subyacente.
ETIOLOGÍA
Las tres causas más frecuentes de la gota son:
1. La alimentación basada en carnes rojas o vegetales productores de ácido
úrico.
2. El consumo frecuente y excesivo de bebidas alcohólicas.
3. La obesidad ya que esta ocasiona un aumento en la producción.
Otras causas:
Hipertensión arterial
Diabetes
Anemia
Antecedentes familiares
Cáncer
Psoriasis
DIAGNOSTICO La gota se identifica mediante el examen clínico del paciente y por pruebas de
laboratorio:
Ácidoúrico en sangre.
Ácido úrico en la orina.
Análisis del líquido sinovial (muestra cristales de ácido úrico)
Radiografía de la articulación (puede ser normal)
Biopsia sinovial
Fig. 115 Articulaciones afectadas por cristales de ácido úrico.
http://drsantamaria.blogspot.com/2011/01/que-es-la-gota.html

121
TRATAMIENTO
Los antinflamatorios no esteroides (AINES), como ibuprofeno, naproxeno
o indometacina alivian los síntomastan pronto como empiezan. Hable
con el médico acerca de la dosis correcta. Usted necesitará dosis más
fuertes durante unos días.
El médico puede prescribir analgésicos fuertes de vez en cuando como
codeína, hidrocodona y oxicodona.
Un medicamento de venta con receta llamado colchicina ayuda a reducir
el dolor, la hinchazón y la inflamación.
Los corticosteroides también pueden ser muy eficaces. El médico puede
infiltrar la articulación inflamada con esteroides para aliviar el dolor.
El dolor con frecuencia desaparece al cabo de 12 horas de empezar el
tratamiento y se alivia completamente en 48 horas.
El uso diario de alopurinol o probenecida disminuye los niveles del ácido
úrico en la sangre. El médico puede recetar estos medicamentos si el
paciente presenta:
Varios ataques durante el mismo año o sus ataques son muy
intensos
Daño en las articulaciones
Tofos
Cálculos renales de ácido úrico
Algunos cambios en la dieta y en el estilo de vida pueden ayudar a
prevenir los ataques de gota:
Evite el alcohol.
Reduzca los alimentos ricos en purina que consume, especialmente
las anchoas, las sardinas, los aceites, el arenque, las vísceras
(hígado, riñón y mollejas), legumbres (frijoles y guisantes secos),
salsas, champiñones, espinaca, espárrago, coliflor, consomé y
levadura de cerveza o de panadería.
Reduzca la cantidad de carne que usted consume en cada comida.
Evite alimentos grasos como aderezos, helado y alimentos fritos.
Coma suficientes carbohidratos.
Si está perdiendo peso, hágalo lentamente. La pérdida rápida de
peso puede provocar la formación de cálculos renales de ácido úrico.
Incrementar la ingesta de líquidos (agua).

122
Enfermedades asociadas a deficiencias de proteínas y
aminoácidos
El déficit de aminoácidos proteínas causa: síndrome nefrótico, anemias graves
afecciones hepáticas crónicas
KWASHIORKOR
Es una forma de desnutrición que ocurre cuando no hay suficiente proteína en
la dieta.El kwashiorkor es más común en áreas donde hay: hambre y
suministro limitado de alimentos.
SÍNTOMAS
Cambios en la pigmentación de la piel.
Disminución de la masa muscular.
Diarrea.
Deficiencia en el aumento de peso y en el crecimiento.
Fatiga.
Cambios en el cabello (cambios en el color o la textura).
Aumento en el número y gravedad de las infecciones debido a daño en el
sistema inmunitario.
Irritabilidad.
Abdomen grande que sobresale.
Letargo o apatía.
Pérdida de la masa muscular
Salpullido.
Shock
Hinchazón
SIGNOS Y EXÁMENES
El examen físico puede mostrar un hígado agrandado(hepatomegalia) e
hinchazón generalizada.Los exámenes pueden abarcar:
Gasometría arterial
Nitrógeno ureico en la sangre(BUN)
Conteo sanguíneo completo (CSC)
Depuración de la creatinina
Creatinina en suero
Potasio sérico
Niveles de proteína total
Análisis de orina

123
TRATAMIENTO
El hecho de obtener más calorías y proteínas corregirá el kwashiorkor, si el
tratamiento se comienza a tiempo. No obstante, los niños que han padecido
esta afección nunca alcanzarán su potencial total con respecto a la estatura y el
crecimiento.El tratamiento depende de la gravedad de la afección. Las personas
que están en shock requieren tratamiento inmediato para restaurar la volemia
y mantener la presión arterial.
Primero se administran calorías en forma de carbohidratos, azúcares simples y
grasas. Las proteínas se administran después de que otras fuentes calóricas ya
han suministrado energía. Los suplementos de vitaminas y minerales son
esenciales.Debido a que la persona ha estado sin mucho alimento durante un
período largo de tiempo, el hecho de comer le puede ocasionar problemas,
especialmente si las calorías son demasiado altas al principio. Por lo tanto, los
alimentos deben introducirse gradualmente, comenzando por los carbohidratos
para proporcionar energía, seguidos por alimentos proteicos.
Muchos niños desnutridos desarrollarán intolerancia al azúcar de la leche y
será necesario suministrarles suplementos con la enzima lactasa para que
puedan tolerar productos lácteos.
SÍNDROME DE FANCONI
Es un trastorno de los túbulos renales en el cual ciertas sustancias
normalmente absorbidas en el torrente sanguíneo por los riñones son liberadas
en su lugar en la orina.
Causas, incidencia y factores de riesgo
El síndrome de Fanconi puede ser causado por genes defectuosos o puede
aparecer posteriormente en la vida debido a daño renal.
En los niños, las causas comunes de este síndrome son defectos genéticos que
afectan la capacidad del cuerpo para descomponer ciertos compuestos, como:
Cistina (cistinosis), es la causa más común.
Fructosa (intolerancia a la fructosa)
Galactosa (galactosemia)
Glucógeno (enfermedades por almacenamiento de glucógeno)
Otras causas en niños comprenden:
Exposición a metales pesados como el plomo, el mercurio y el cadmio
Enfermedad de Lowe, un raro trastorno genético de los ojos, el cerebro y
los riñones
Enfermedad de Wilson

124
En los adultos, el síndrome de Fanconi puede ser causado por diversas cosas
que provocan daño a los riñones, como:
Ciertos medicamentos como azatioprina, cidofovir, gentamicina y
tetraciclina
Trasplante de riñón
Enfermedad por precipitación de las cadenas ligeras
Mieloma múltiple
Amiloidosis primaria
SÍNTOMAS
La alteración en los túbulos proximales renales hace que su función se
deteriore y se eliminan por la orina cantidades altas de aminoácidos,
bicarbonato, fosfatos y otras sustancias.
Los niños que presentan la forma congénita tienen síntomas desde muy
pequeños, como hipofosfatemia, acidosis por eliminación excesiva de
bicarbonato en la orina, glucosuria (eliminación de glucosa por la orina),
hipocaliemia (disminución de potasio en sangre), retraso del
crecimiento, raquitismo, polidipsia (ingesta excesiva de agua) y poliuria. La
función renal se deteriora progresivamente y la enfermedad puede terminar
en insuficiencia renal que precise hemodialisis.
En la forma adquirida o del adulto, los trastornos metabólicos son similares y
las manifestaciones óseas suelen ser deosteomalacia.
SIGNOS Y EXÁMENES
Los exámenes de laboratorio pueden mostrar que cantidades excesivas de las
siguientes sustancias se pueden perder en la orina:
Aminoácidos
Bicarbonato
Glucosa
Magnesio
Fosfato
Potasio
Sodio
Ácido úrico
TRATAMIENTO
Muchas enfermedades diferentes pueden causar el síndrome de Fanconi. La
causa subyacente y sus síntomas se deben tratar en forma apropiada.

125
CAPITULO VIII
DETERMINACIÓN DEL ANALISIS CLÍNICO DE LOS
PRODUCTOS FINALES DE COMPUESTOS NITROGENADOS
Y SU SIGNIFICADO CLÍNICO
ANALISIS CLÍNICODE LOS PRODUCTOS FINALES DE
COMPUESTOS NITROGENADOS Y SU SIGNIFICADO CLÍNICO EN
MUESTRA DE ORINA Y SANGRE
AMONÍACO
FUNDAMENTO.-La orina se trata con formaldehído; éste reacciona con el
amoníaco formando hexametilentetramina e iones hidrógeno. Los iones
hidrógenos se valoran con un álcali.
MATERIALES REACTIVOS
• Puntas para pipetas ● Formaldehído 30-40%
• Matraz de erlenmeyer ● Disolución de fenolftaleína.
• Pipetas ● Hidróxido sódico 0,1N.
• Bureta
• Soporte universal
• Pinzas
PROCEDIMIENTO:
1. Pasar con una pipeta, a un matraz de valoración, 10,0ml de orina,
tratarlos con 1 gota de fenolftaleína y añadir desde un bureta, gota a
gota, hidróxido sódico 0,1N hasta el primer tono rojizo.
2. En un segundo matraz, añadir 25ml de disolución de formaldehído y 1
gota de fenolftaleína y llevar esta disolución también a débil color rosa
con el hidróxido sódico 0,1N.
3. Verter la disolución de formaldehído sobre la orina neutralizada y valora
la mezcla con hidróxido sódico hasta el primer tono rojo.
VALORES DE REFERENCIA:
15-45 mcg/dL
4NH₄ i+ 6CH₂O = 4H i+ (CH₂)₆N₄ +
6OH₂

126
CÁLCULOS:
OBSERVACIONES:
1. La orina debe estar absolutamente exenta de albúmina, pues las
proteínas también reaccionan con el formaldehído. Lo mejor es
eliminarlas por breve calentamiento de la orina débilmente ácida,
enfriar rápidamente y filtrar. El reconocimiento del primer tono rojizo
en la valoración está sujeto a variaciones individuales. Debido a la
acción reguladora de la orina el color rojo aparece lentamente y
aumenta de intensidad con mucha lentitud. El volumen de hidróxido
sódico consumido en esta valoración de la orina, frente a fenolftaleína,
se empleó previamente para calcular la acidez valorable de la orina.
2. El formaldehído es siempre ácido, por lo que es absolutamente necesario
neutralizarlo de antemano frente a fenolftaleína.En los cálculos se
introduce ahora solo el volumen de álcali que es necesario para valorar
los iones hidrógeno liberados al mezclar el formaldehído con la orina.
SIGNIFICADO CLÍNICO:
Los resultados anormales pueden significar que usted tiene un aumento en los
niveles de amoníaco en la sangre. Esto puede deberse a:
Insuficiencia cardíaca congestiva
Sangrado gastrointestinal, por lo general en las vías digestivas altas
Enfermedades genéticas del ciclo de la urea
Temperatura corporal alta (hipertermia)
Leucemia
Insuficiencia hepática
Nivel bajo de potasio en la sangre (hipocaliemia)
Alcalosis metabólica
Esfuerzo muscular intenso
UREA
FUNDAMENTO:Mide la cantidad de urea o nitrógeno ureico presente en la
sangre. La urea es el resultado final del metabolismo de las proteínas. Se forma
en el hígado a partir de la destrucción de las proteínas.
MATERIALES REACTIVOS
• Puntas para pipetas ●Suero
• Espectrofotómetro ●Reactivo 1 para urea.
• Pipetas serológicas ● Reactivo 2 para urea
• Pipetas automáticas
• Tubos de ensayo
ml de hidróxido sódico ƝOHNa x 170 = mg% de amoníaco
ml de hidróxido sódico x ҒOHNa x 17 = mg% de amoníaco

127
• Baño María
• Cubetas para espectrofotómetro
TÉCNICA:
BLANCO STANDARD MUESTRA
Suero 25 µl
Standard 25 µl
Reactivo 1 1ml 1ml 1 ml
Mezclar y añadir
Reactivo 2 1ml 1ml 1 ml
Mezclar por agitación suave e incubar 5 minutos a 37º. Luego leer la
absorbancia del St y de la muestra frente al blanco de reactivo a 510nm.
VALORES NORMALES: 15-45 mg/dl
Disminución de Urea Aumento de Urea
Insuficiencia hepática Insuficiencia renal
ACIDO ÚRICO
FUNDAMENTO:Esel resultado final del metabolismo de las purinas,
presentes en los ácidos nucleicos. Existen dos fuentes de ácido úrico:
1. Endógena.- originado por la destrucción de los tejidos orgánicos.
2. Exógena.- proveniente de la alimentación.
Este examen se realiza sobre todo para hacer un diagnóstico de gota.
MATERIALES: REACTIVOS:
• Tubos de ensayo ● Suero
• Pipetas serológicas ● Reactivo de trabajo
• Pipetas automáticas
• Puntas para pipetas
• Espectrofotómetro
• Cubetas para espectrofotómetro

128
TÉCNICA:
MUESTRA STANDARD
Suero 20 µl
Standard 20 µl
Reactivo de trabajo 1 ml 1 ml
Mezclar e incubar 5 nucleósido purínicos (adenosina y guanosina)
originarios de la vía
• metabólica minutos a 37°C
• Leer en espectrofotómetro a 520nm., frente a un blanco de agua
destilada.
VALORES NORMALES
Hombres: 3.4 - 7 mg/dl
Mujeres: 2.4 - 6 mg/dl
SIGNIFICADO CLÍNICO:
El ácido úrico es el mayor producto del catabolismo de los de las purinas. Estas
pueden sintetizarse endógenamente por descomposición de los ácidos nucleicos
o incorporarse externamente a través de dietas en que los ácidos nucleicos
estén presentes. El aumento anormal de ácido úrico circulante por encima de
7,0 mg/dL (0,42 mmol/L) se conoce como hiperuricemia siendo la gotala
expresión mayor de la dolencia que cursa con una saturación de ácido úrico en
los fluidos corporales y la consiguiente deposición de uratos en los tejidos
blandos, especialmente en las articulaciones. Aumentos elevados se hallan
también asociados a leucemias, toxemia del embarazo y fallo renal severo.
Menos comunes son los casos de hipouricemia, con concentraciones de ácido
úrico inferiores a 2,0 mg/dL (0,12mmol/L), por lo general secundarios a casos de
enfermedad hepatocelular, defectos de reabsorción renal o a sobredosis de
drogas uricosúricas empleadas en el tratamiento de la hiperuricemia.
CREATININA
Creatinina sérica o creatinina en suero es un producto de degradación de la
creatina, una parte importante del músculo.
FUNDAMENTO:Una gran parte de los métodos analíticos utilizados para la
determinación de creatina y creatinina están basados en la reacción química de
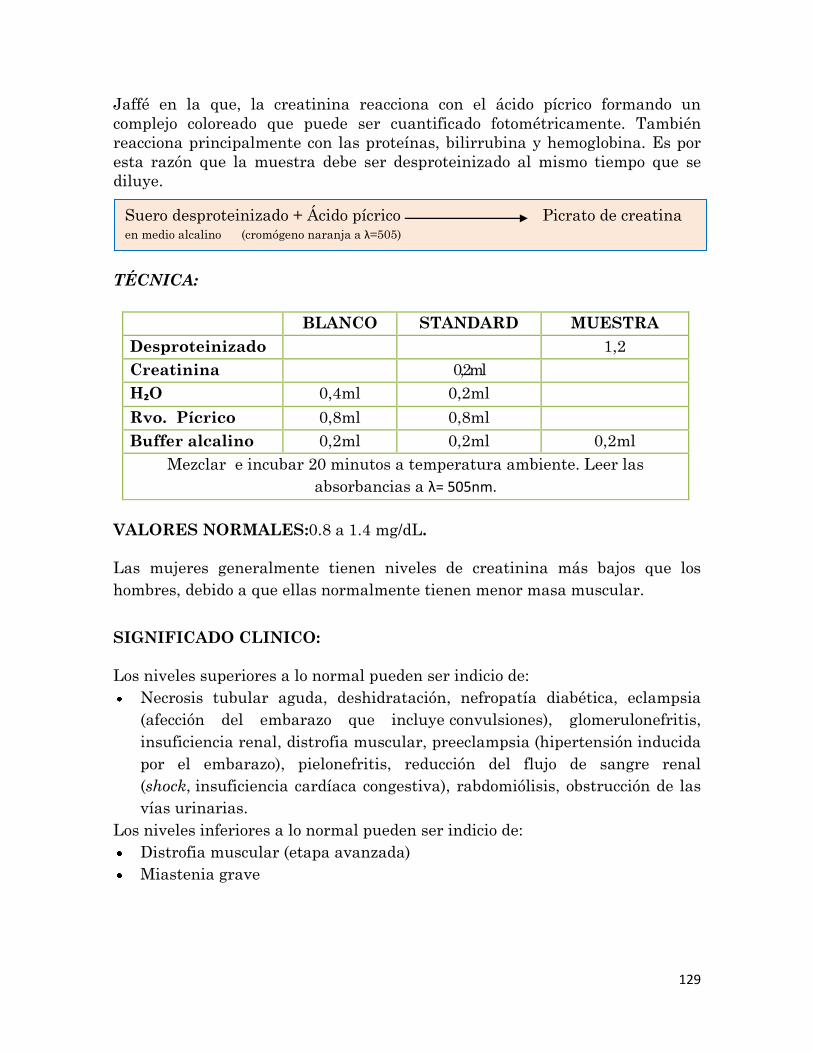
129
Jaffé en la que, la creatinina reacciona con el ácido pícrico formando un
complejo coloreado que puede ser cuantificado fotométricamente. También
reacciona principalmente con las proteínas, bilirrubina y hemoglobina. Es por
esta razón que la muestra debe ser desproteinizado al mismo tiempo que se
diluye.
TÉCNICA:
BLANCO STANDARD MUESTRA
Desproteinizado 1,2
Creatinina 0,2ml
H₂O 0,4ml 0,2ml
Rvo. Pícrico 0,8ml 0,8ml
Buffer alcalino 0,2ml 0,2ml 0,2ml
Mezclar e incubar 20 minutos a temperatura ambiente. Leer las
absorbancias a λ= 505nm.
VALORES NORMALES:0.8 a 1.4 mg/dL.
Las mujeres generalmente tienen niveles de creatinina más bajos que los
hombres, debido a que ellas normalmente tienen menor masa muscular.
SIGNIFICADO CLINICO:
Los niveles superiores a lo normal pueden ser indicio de:
Necrosis tubular aguda, deshidratación, nefropatía diabética, eclampsia
(afección del embarazo que incluye convulsiones), glomerulonefritis,
insuficiencia renal, distrofia muscular, preeclampsia (hipertensión inducida
por el embarazo), pielonefritis, reducción del flujo de sangre renal
(shock, insuficiencia cardíaca congestiva), rabdomiólisis, obstrucción de las
vías urinarias.
Los niveles inferiores a lo normal pueden ser indicio de:
Distrofia muscular (etapa avanzada)
Miastenia grave
Suero desproteinizado + Ácido pícrico Picrato de creatina en medio alcalino (cromógeno naranja a λ=505)

130
CONCLUSIONES
Expliqué el fundamento, procesos químicos de producción y alteraciones
fisiológicas que producen en el organismo los productos finales de compuestos
nitrogenados, para demostrar que el nitrógeno está presente en muchas
sustancias orgánicas en los seres vivos, en especial enforma de aminoácidos y
proteínas, en nucleótidos y ácidos nucléicos, el cual proviene delNitrógeno
molecular, sintetizado por nuestro propio organismo a partir de proteínas o
ingerido en la dieta, gracias a su constante biosíntesis y degradación, es decir
su metabolismo desencadenas multiples reacciones que con llevan a la
formación de amonio, urea, ácido urico, creatina y creatinina siendo éstos los
productos finales de dichos compuestos nitrogenados, indicadores primordiales
del buen funcionamiento del hígado y riñón, órganos encargados de la sístesis y
eliminación de éstos compuestos.
1. Describí las generalidades de los compuestos orgánicos nitrogenados
para comprender como se forman dichos compuestos y como el nitrógeno
molecular por medio de las plantas y algunos microorganismos pueden
sintetizar aminoácidos y así ingresar a nuestro organismo a través de la
dieta.
2. Analicé la estructura, propiedades y función de los aminoácidos, para
entender la importancia de estas unidades en los procesos físicos que
afectan el cuerpo, entre ellos se encuentran el crecimiento muscular y
recuperación, la producción de energía, la producción de hormonas,
síntesis de proteínas enzimáticas, de transporte, cuya existencia es
condición previa para la vida ya que si llegara a faltar un aminoácido
éstas no podrían sintetizarse.
3. Examiné la estructura, clasificación y desnaturalización de las proteínas,
por su elevado número de funciones biológicas que desempeñan en la
materia viva y sobre todo en la particular relación que las une con los
ácidos nucleicos, ya que constituyen el vehículo habitual de expresión de
la información genética contenida en ellos.
4. Establecí la estructura, clases y formación de los ácidos nucleicos porque
éstas moléulas dirigen y controlan la síntesis de proteína proporcionando
la informacion que determina su especificidad y características biológicas
siendo los responsables de todas las funciones básicas en los seres vivos
es decir el almacena y transmite de la información genética contituyendo
la base molecular de la herencia.

131
5. Detallé el metabolismo de los aminoácidos, proteínas y nucléotidos, para
comprender las reacciones químicas que conducen a la síntesis y
degradación de cada compuesto, transformandola en energía que
consumen las células del organismo.
6. Caractericé cada uno de los productos finales del metabolismo de los
compuestos nitrogenados, para valorar como los productos nitrogenados
a través de su catabolismo generan, amoníaco, urea, ácido úrico, creatina
y creatinina, productos de desecho que son indicadores indispensables en
la función del hígado y riñon, órganos encargados de su síntetisis y
eliminación.
7. Tipifiqué las enfermedades causadas por el desequilibrio de los
productos finales del metabolismo de compuestos nitrogenados, para
diagnosticarlas y prevenirlas a tiempo, su adecuado abordaje y
tratamiento son fundamentales para reducir la morbimortalidad,
concientizando a la sociedad sobre la gravedad del desorden metabólico
producido por daño hepático o renal o también por la alimentación
basada en carnes rojas, el consumo frecuente y excesivo de bebidas
alcohólicas y la falta de ejercicio que provoca obesidad ocasionando un
aumento en la producción de ácido úrico o urea.
8. Determiné las técnicas para el análisis clínico de los productos finales de
compuestos nitrogenados, porque a través de éstas se determina
alteraciones en sus niveles normales dentro del organismo, permitiendo
instituir rápidamente el tratamiento evitando su alta morbimortalidad.
Además, el diagnóstico permite el asesoramiento genético a la familia.

132
BIBLIOGRAFÍA
Brusilow SW, Horwich AL. Urea cycle enzymes. In: Scriver Ch R, Baudet
AL, Sly WS, Valle MD, eds. Eight edition. The Metabolic and Molecular
Bases of Inherited Disease. New York: McGraw Hill, 2001: 1187-232.
Sanjurjo P, Rubio V. Diagnóstico y tratamiento de las enfermedades del
ciclo de la urea. In: Sanjurjo P, Baldellou A, eds. Diagnóstico y tratamiento
de las enfermedades metabólicas hereditarias. Madrid: Ergon, 2001: 324-32.
Carrillo Esper R, et al. Amonio e hiperamonemia. Su significado clínico.
http://www.medigraphic.com/pdfs/medsur/ms-2008/ms083f.pdf 2008
Pastrana J, García JP, Civeira MP, Prieto JMª. Bioquímica hemática. En:
Prieto JMª, coordinador. La clínica y el laboratorio. 20ª ed. Barcelona:
Masson; 2006. p. 106.2009
Rezvani I. Defects in Metabolism of Aminoacids. In: Behrman RE, Kliegman
RM, Jenson HB, eds. Nelson Textbook ofPediatrics. 16th Edition.
Philadelphia: WB Sanders, 2000:344–76.
Tuchman M.The clinical, biochemical, and molecular spectrum of onithine
transcarbamylase deficiency. J Lab Clin Med 1992; 120(6): 836-50.
Tuchman M, McCullough B, Yodkoff M. The molecular basis of ornitine
transcarbamilase deficiency. Eur J Pediatr 2000; 159: S196-S198.
H. MALFATTI: Z, Métodos quelométricos y otros métodos volumétricos de
anállisis clínico. Editorial Reverte, S.A.
Berk PD, Korenblat KM. Approach to the patient with jaundice or abnormal
liver test results. In: Goldman L, Ausiello D, eds. Cecil Medicine. 24th ed.
Philadelphia, Pa: Saunders Elsevier; 2011:chap 149.
Recomendaciones para la utilización de la determinación de amonio en
plasma en el Laboratorio Clínico. Sociedad Española de Bioquímica Clínica
y Patología Molecular. Química Clínica 2007.
Bazari H. Approach to the patient with renal disease. En: Goldman L,
Ausiello D, eds. Cecil Medicina. 23rd ed. Philadelphia, Pa: Saunders
Elsevier; 2007:chap 115.
Páginas Web:
http://www.didactika.com/quimica/descargas/15_compuestos_nitrogen
ados_carbono.pdf
Propiedades de los aminoácidos. | La guía de
Biologíahttp://biologia.laguia2000.com/bioquimica/propiedades-de-los-
aminoacidos#ixzz2DMpC7WrC
Peptidos http://es.scribd.com/doc/2975077/7/Tipos-de-peptidos
http://www.ehu.es/biomoleculas/peptidos/pep4.htm

133
http://www.iespando.com/web/departamentos/biogeo/web/departamen
to/2BCH/PDFs/05Proteinas.pdf
http://www.uv.es/tunon/pdf_doc/proteinas_09.pdf
http://avdiaz.files.wordpress.com/2008/11/proteinas.pdf
http://siladin.cch-riente.unam.mx/coord_area_cienc_exp/biologia/Guia
BioI/ ANEXO_6SINTESIS.pdf
http://med.unne.edu.ar/catedras/bioquimica/pdf/nitro.pdf
http://med.unne.edu.ar/catedras/bioquimica/pdf/nitro.pdf
http://ocw.unican.es/ciencias-de-la-salud/fisiologia-
general/materiales-de-clase-1/tema-1.-introduccion-al-estudio-de-la-
fisiologia/Tema%204B-Bloque%20I-
Vias%20Degradacion%20Lipidos.pdf
http://www.unac.edu.pe/documentos/organizacion/vri/cdcitra/Informes
_Finales_Investigacion/Octubre_2011/IF_DECHECO%20EGUSQUIZ
A_FIPA/CAPITULO%20N%BA%2006.pdf
http://themedicalbiochemistrypage.org/es/amino-acid-metabolism-
sp.php
http://www.fmv-
uba.org.ar/grado/medicina/ciclo_biomedico/segundo_a%C3%B1o/bioqu
imica/Seminario12/sem12file3.pdf
http://www.bionova.org.es/biocast/documentos/tema09.pdf
http://www.cede.es/n_temas_2012/t35_biologia.pdf
http://www.biorom.uma.es/contenido/av_biomo/FigT8/tema8.pdf
http://ntorres.webs.ull.es/temas/tema23.pdf
http://es.scribd.com/doc/22027634/8-METABOLISMO-DE-
AMINOACIDOS
http://bioquimica6ce.wikispaces.com/Proteinas
http://es.scribd.com/doc/7067686/Metabolismo-de-Aminoacidos
http://www.medigraphic.com/pdfs/medsur/ms-2008/ms083f.pdf
http://www.biosalud.saber.ula.ve/db/ssalud/edocs/articulos/Acurico.pd
f
http://www.paginasprodigy.com.mx/fiatlux/bioquimica/4.13%20Metab
olismo%20de%20purinas%20y%20pirimidinas.pdf
http://docencia.izt.uam.mx/japg/RedVirtualJAP/CursoDRosado/4_Met
abolismo/4-%20PurinasyPirimidinas/1-Metabolismodepurinas.pdf
http://themedicalbiochemistrypage.org/es/nucleotide-metabolism-
sp.php
http://www.youtube.com/watch?v=OOMTdzOwohk