pediatria utm: hipotiroidismo y-cetoacidosis
TRANSCRIPT

UNIVERSIDAD TECNICA DE MANABIFACULTAD DE CIENCIAS DE LA SALUD
CARRERA DE MEDICINAPEDIATRIA CLINICA
DRA: BETZABHE PICO
TEMA: hipotiroidismo y cetoacidosis diabética
RESPONSABLES:
o BAZURTO BASURTO GALOo GILER MARCO STEBANo FLORES MORENO RUTH
o MECIAS MANZABA HOLGERo MEJIA MARQUEZ MARIA G.
o MENENDEZ SALTOS JONATHANo MIELES ANDRADE ANGI
OCTAVO “C”
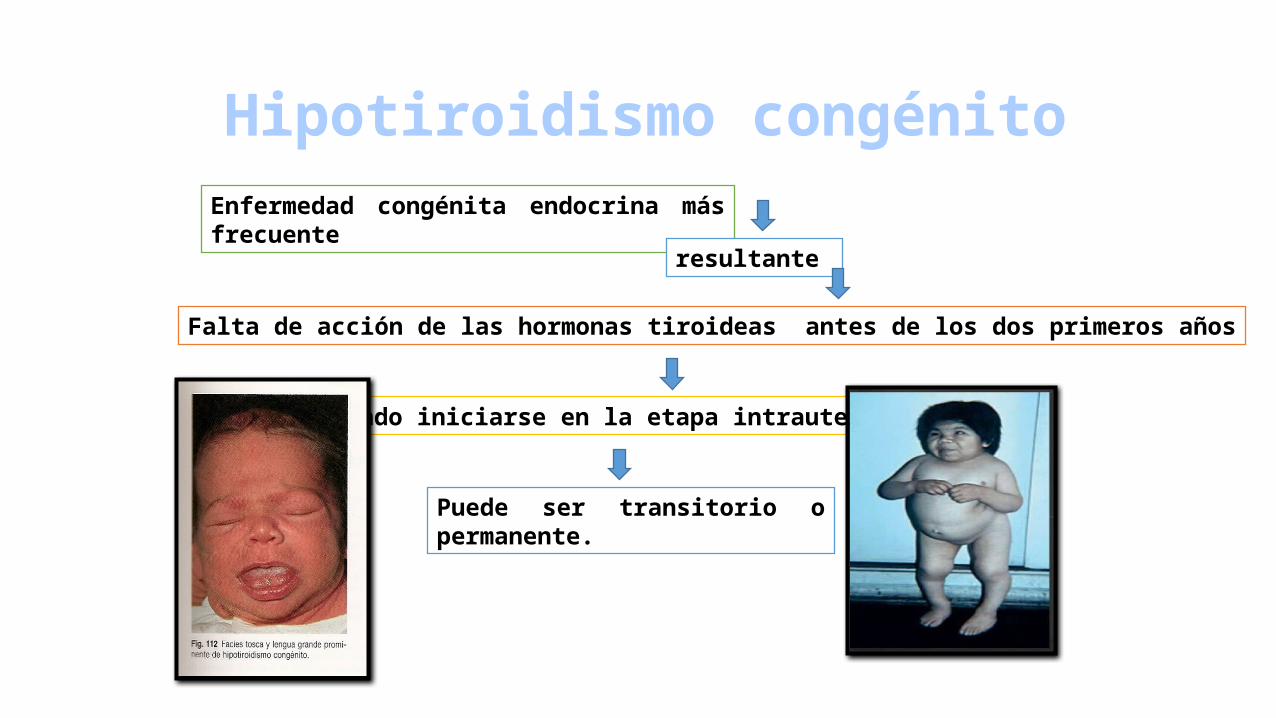
Hipotiroidismo congénitoEnfermedad congénita endocrina más frecuente
resultante
Falta de acción de las hormonas tiroideas antes de los dos primeros años
Pudiendo iniciarse en la etapa intrauterina
Puede ser transitorio o permanente.

El límite de edad se ha fijado en forma artificial basado en:
La importancia de las hormonas tiroideas
El desarrollo del sistema nervioso central (SNC)
El período crítico que abarca los 2 primeros años de vida,
especialmente, los primeros 6 meses
La ausencia de dichas hormonas se traduce en
Retardo de la mielinización
Retardo de la arborización dendrítica
Retardo de la formación de sinapsis
Retardo de la migración neuronal
Disminución de la vascularización
Lesiones irreversibles del SNC
lo que lleva a
Múltiples alteraciones neurológicas.
Retardo mental
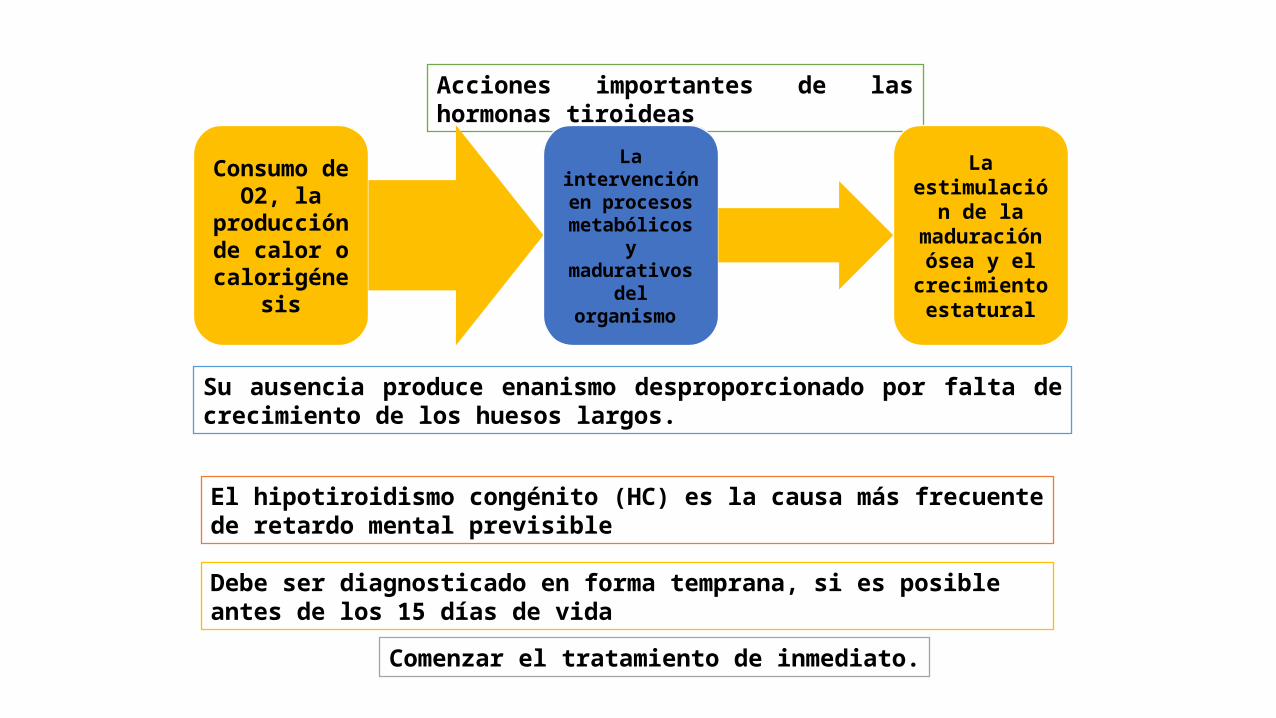
Acciones importantes de las hormonas tiroideas
Consumo de O2, la
producción de calor o
calorigénesis
La intervención en procesos
metabólicos y madurativos
del organismo
La estimulación
de la maduración
ósea y el crecimiento
estatural
El hipotiroidismo congénito (HC) es la causa más frecuente de retardo mental previsible
Su ausencia produce enanismo desproporcionado por falta de crecimiento de los huesos largos.
Debe ser diagnosticado en forma temprana, si es posible antes de los 15 días de vida
Comenzar el tratamiento de inmediato.

En el 95% de los casos la
falla es de la glándula tiroides o
hipotiroidismo primario.
El defecto reside en
estructuras supratiroideas
; hipotálamohipofisarias=hipo
tiroidismo secundario
Insensibilidad de los
receptores esperiféricos a
la triyodotironin
a (T3) y a la tiroxina (T4).
ETIOPATOGENIA
INCIDENCIAEntre 1 x 3.000 a 1 x 3.500 recién nacidos vivos.
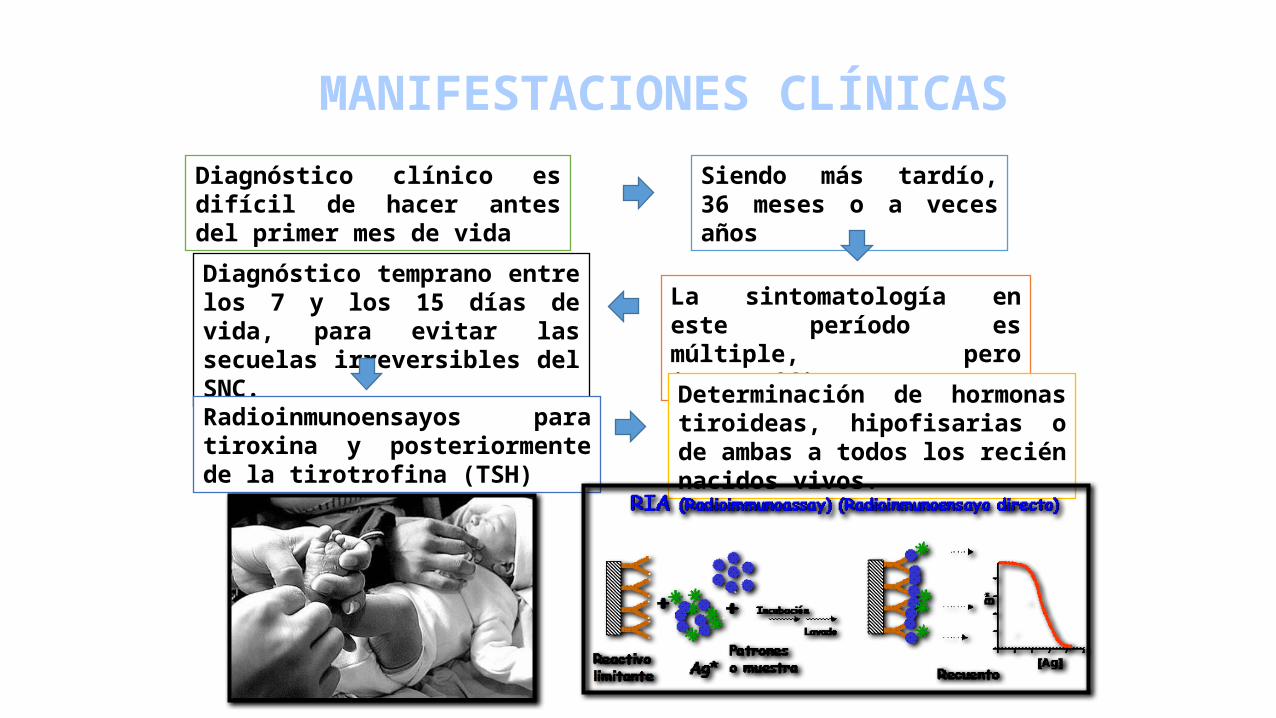
MANIFESTACIONES CLÍNICAS
Diagnóstico clínico es difícil de hacer antes del primer mes de vida
La sintomatología en este período es múltiple, pero inespecífica.
Siendo más tardío, 36 meses o a veces años
Diagnóstico temprano entre los 7 y los 15 días de vida, para evitar las secuelas irreversibles del SNC.
Radioinmunoensayos para tiroxina y posteriormente de la tirotrofina (TSH)
Determinación de hormonas tiroideas, hipofisarias o de ambas a todos los recién nacidos vivos.

Se obtiene una gota de sangre por punción en el talón, a las 48 o 72 horas en el RN de término y al 5 día en los prematuros
Se la deposita en una tarjeta de papel filtro adecuado diseñada con círculos para recibir una gota en cada uno
La sangre así obtenida se deja secar a temperatura ambiente y luego se envía al laboratorio especializado
IRMA (Inmuno Radiometric with Monoclonal Antibodies )
La detección puede efectuarse con mediciones de T4
Confirmaciones de los casos positivos con TSH
Confirmación con medición en suero de TSH por IRMA y T4 por radioinmunoensayo (RIA).
Este método no diagnostica el hipotiroidismo secundario hipotalámo-hipofisario, que presenta TSH bajo
El ideal sería medir TSH y T4

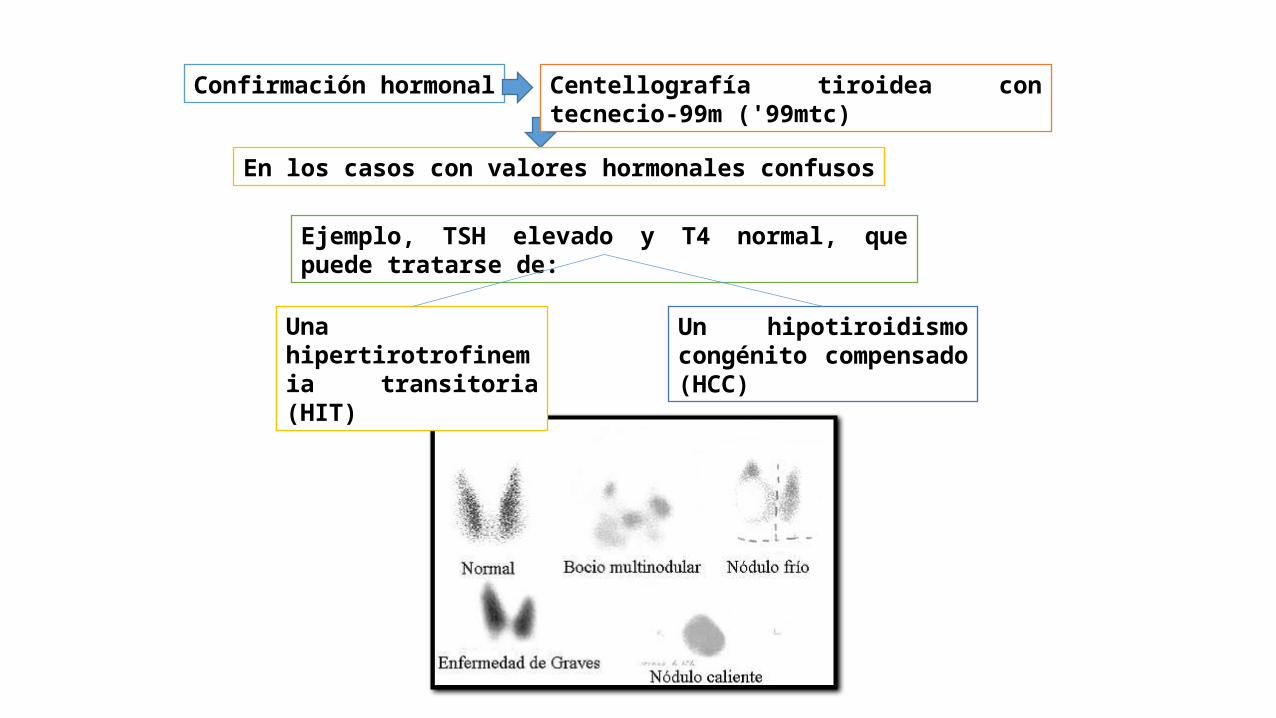
Confirmación hormonal Centellografía tiroidea con tecnecio-99m ('99mtc)
En los casos con valores hormonales confusos
Ejemplo, TSH elevado y T4 normal, que puede tratarse de:
Una hipertirotrofinemia transitoria (HIT)
Un hipotiroidismo congénito compensado (HCC)
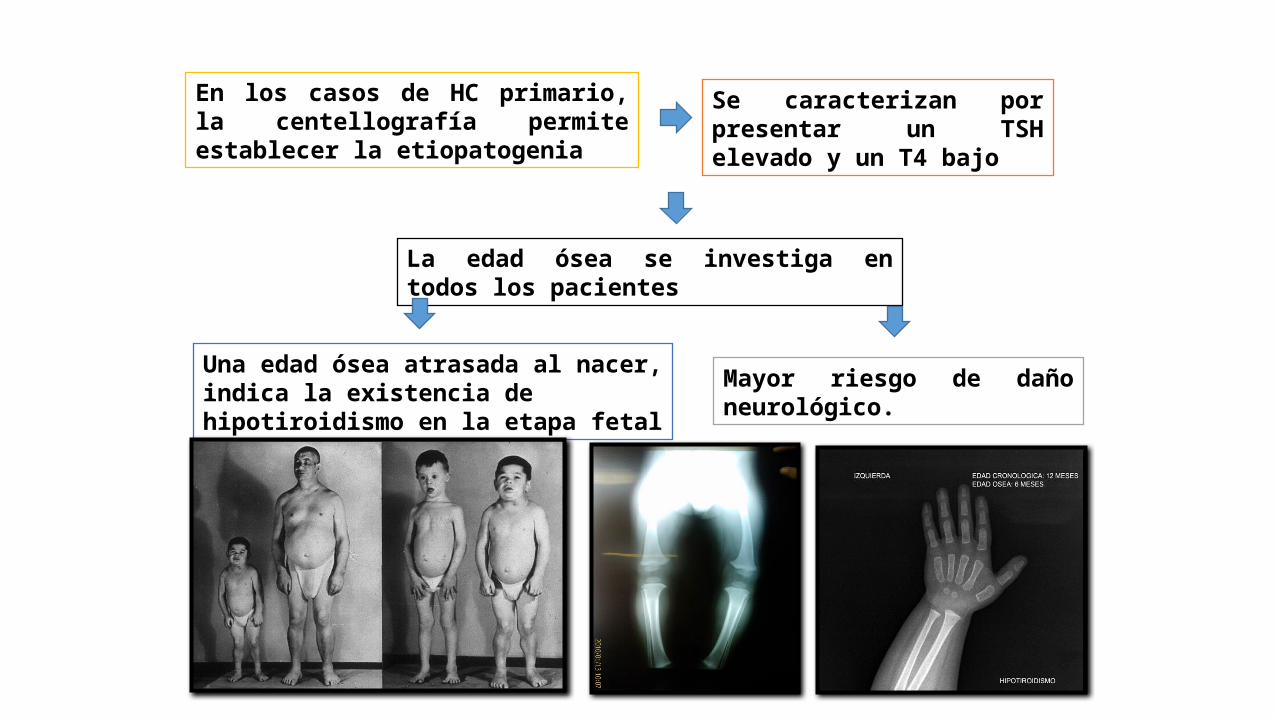
En los casos de HC primario, la centellografía permite establecer la etiopatogenia
Se caracterizan por presentar un TSH elevado y un T4 bajo
La edad ósea se investiga en todos los pacientes
Una edad ósea atrasada al nacer, indica la existencia de hipotiroidismo en la etapa fetal
Mayor riesgo de daño neurológico.
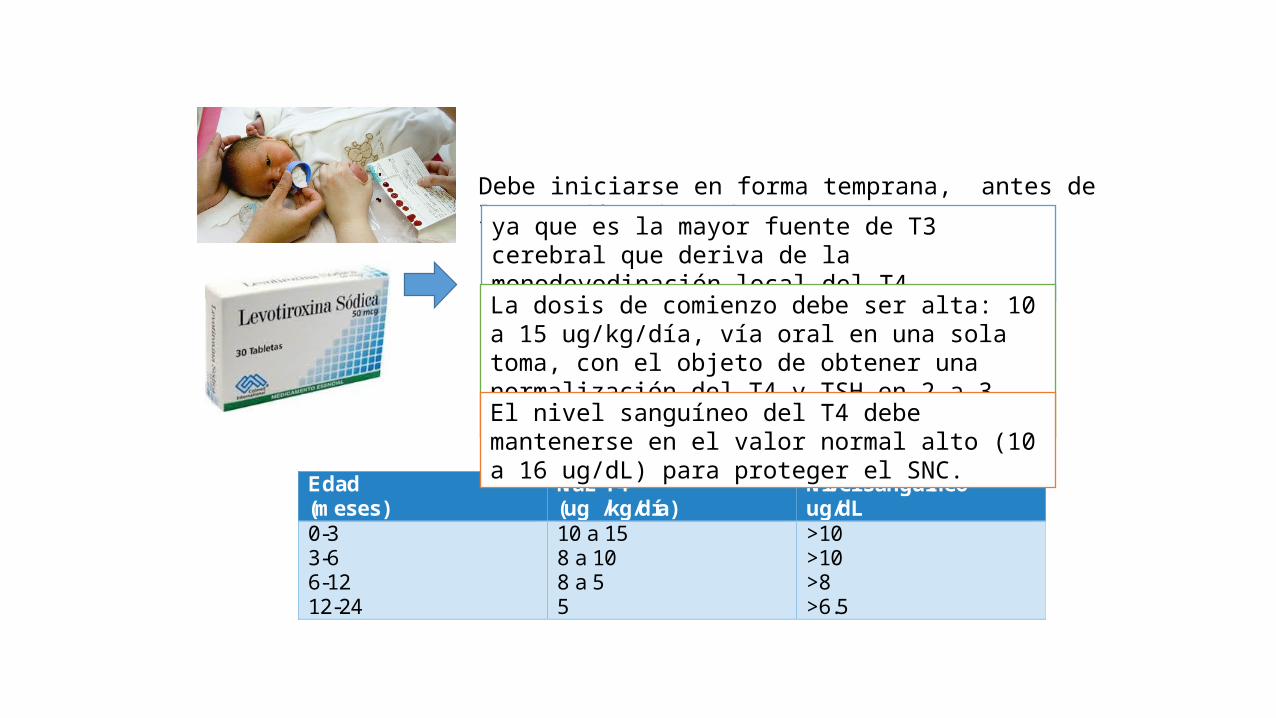
TRATAMIENTO
Edad (meses)
NaL-T4 (ug /kg/día)
Nivel sanguineo ug/dL
0-3 3-6 6-12 12-24
10 a 15 8 a 10 8 a 5 5
>10 >10 >8 >6.5
Debe iniciarse en forma temprana, antes de los 15 días de vida
ya que es la mayor fuente de T3 cerebral que deriva de la monodeyodinación local del T4
La dosis de comienzo debe ser alta: 10 a 15 ug/kg/día, vía oral en una sola toma, con el objeto de obtener una normalización del T4 y TSH en 2 a 3 semanas
El nivel sanguíneo del T4 debe mantenerse en el valor normal alto (10 a 16 ug/dL) para proteger el SNC.

PRONÓSTICO Es fundamental que los padres estén informados de las
características de la enfermedad, de la importancia del tratamiento permanente y constante para conseguir la adhesión al tratamiento
Estudios a largo plazo han demostrado que aun con iniciación temprana del tratamiento, el tener uno o varios valores elevados de TSH en el año coincide con secuelas en la esfera intelectual y motora.
Así se ha comprobado en los niños con signos claros de hipotiroidismo fetal y valores hormonales muy alterado al nacer. Generalmente coincide con atireosis y con aquellos que presentan retraso de la edad ósea.
Se aconseja que de haber una sospecha de que el niño es portador un HC, es preferible iniciar el tratamiento en forma temprana, mantenerlo durante toda la etapa crítica del desarrollo del SNC y suspenderlo alrededor de los 3 años de vida.

HIPOTIROIDISMO ADQUIRIDO
DEL NIÑO Y DEL ADOLESCENTE

HIPOTIROIDISMO
enfermedad endocrinológica y metabólica
se produce como resultado de una alteración
que conduce a una disminución en la producción de hormonas tiroideas.
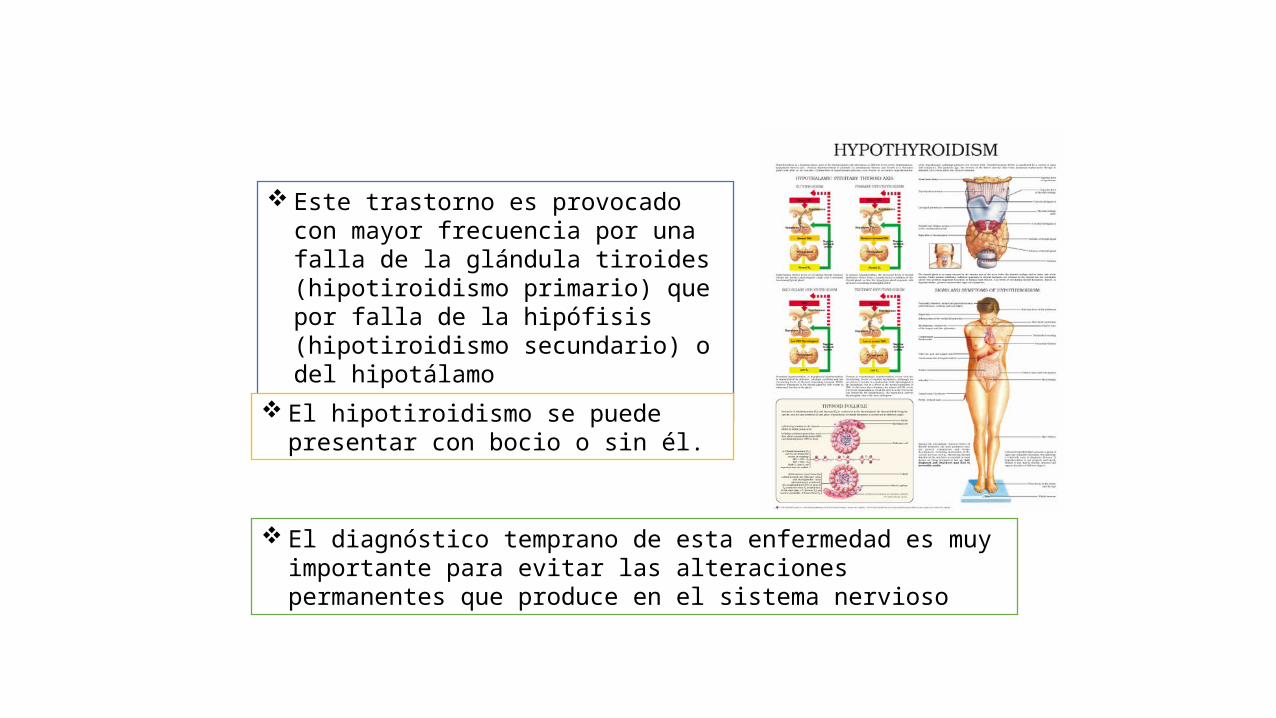
Este trastorno es provocado con mayor frecuencia por una falla de la glándula tiroides (hipotiroidismo primario) que por falla de la hipófisis (hipotiroidismo secundario) o del hipotálamo (hipotiroidismo terciario).
El hipotiroidismo se puede presentar con bocio o sin él.
El diagnóstico temprano de esta enfermedad es muy importante para evitar las alteraciones permanentes que produce en el sistema nervioso

ETIO
LOGÍ
A DE
L HIP
OTIR
OID
ISM
O
ETIOLOGÍA DEL HIPOTIROIDISMO A) CONGENITO
B) ADQUIRIDO
l. Aplasia tiroidea 2. Hipoplasia tiroidea 3. Ecropia tiroidea 4. Dishormonogénesis
a) Defecto del arrapamienro del yodo b) Defecto de la peroxidasa c) Defecto del acoplamiento de tirosinas d) Defecto de la halogenasa (deyodación) e) Producción de proteína yodada anormal f) Defecto de utilización periférica de T4
5. Bociógenos o yodo radiactivo durante el embarazo 6. Deficiencia de yodo (bocio y cretinismo endémico) 7. Tiroiditis auroinmune 8. Hipopituitarlsmo
Hipotiroidisrno Primario - Tiroiditis crónica autoinrnune (Hashimoto): • Con bocio • Atrófica -Inducido por drogas (yodo, litio, PTU, metimazol, carbimazol) - Bocio endémico - Irradiación del tiroides • Irradiación externa de tumores no tiroideos • Tratamiento con yodo radiactivo - Tiroidecromfa Hipotiroidismo familiar - Dishormonogénesis - Resistencia generalizada a las hormonas tiroideas Hipotiroidismo central (secundario y terciario) - Panhipopituitarismo: - ldiopático -Familiar • Asociado con defectos de la línea media - Tratamiento de tumores cerebrales: - Quirúrgico -Radiación - Déficit aislado de TRH - Deficit aislado de TSH
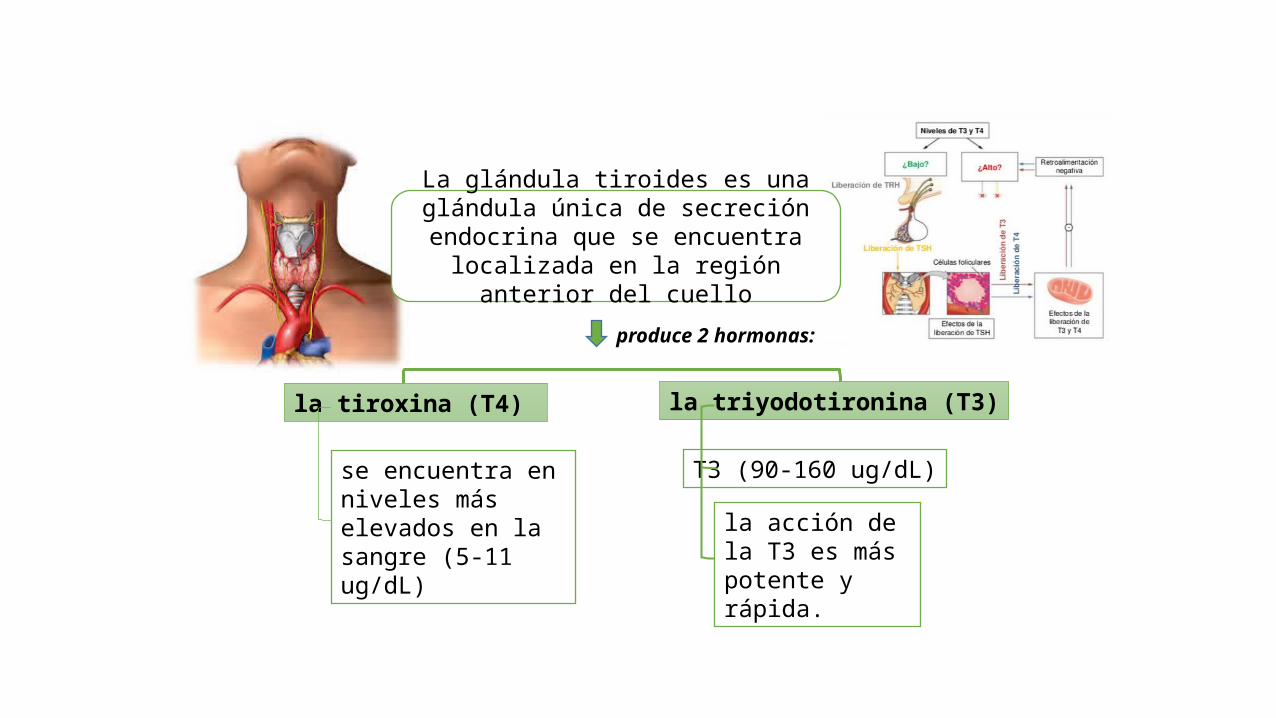
FISIOPATOLOGÍALa glándula tiroides es una glándula única de secreción endocrina que se encuentra localizada en la región anterior del cuello
produce 2 hormonas:
la tiroxina (T4) la triyodotironina (T3)
se encuentra en niveles más elevados en la sangre (5-11 ug/dL)
T3 (90-160 ug/dL)
la acción de la T3 es más potente y rápida.
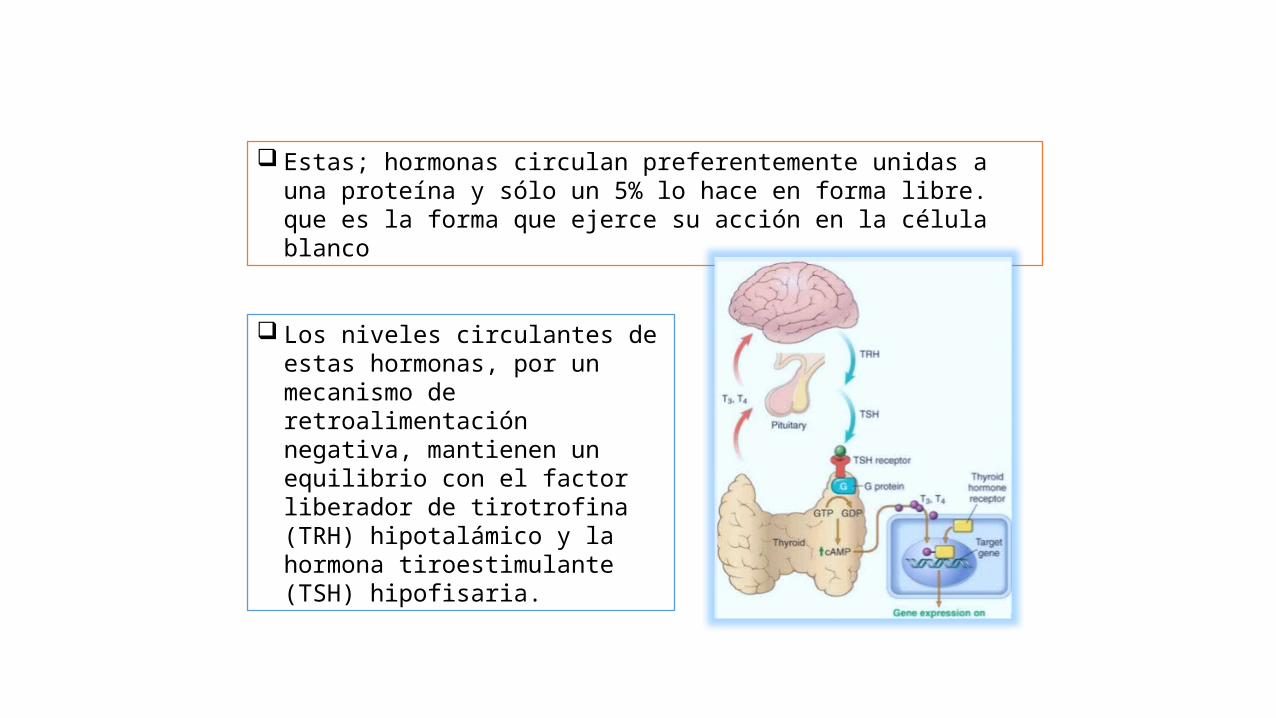
Estas; hormonas circulan preferentemente unidas a una proteína y sólo un 5% lo hace en forma libre. que es la forma que ejerce su acción en la célula blanco
Los niveles circulantes de estas hormonas, por un mecanismo de retroalimentación negativa, mantienen un equilibrio con el factor liberador de tirotrofina (TRH) hipotalámico y la hormona tiroestimulante (TSH) hipofisaria.

Desde el punto de
vista histológico
• este déficit produce una disminución severa del número de células, de las sinapsis y de las prolongaciones nerviosas.
Desde una perspectiva bioquímica
• disminuye la mielina, los neurotransmisores y la compartimentación de los aminoácidos y funcionalmente lleva a un retardo mental.

MANIFESTACIONES CLÍNICAS Y ETIOLOGÍASINTOMAS Y SIGNOS DEL HIPOTIROIDISMO ADQUIRIDO
Iniciación entre los 6 meses y 3 años
•Velocidad de crecimiento disminuida•Rasgos faciales toscos•Piel seca•Macroglosia•Llanto ronco•Hernia Umbilical•Seudohipertrofia muscular

SINTOMAS Y SIGNOS DEL HIPOTIROIDISMO ADQUIRIDO
Iniciación durante la etapa escolar
•Retraso de crecimiento •Retraso de la edad osea•Retraso de la maduracion dental•Seudohipertrofia muscular y miopatía•Constipación•Piel seca•Mixedema•Desarrollo puberal precoz

SINTOMAS Y SIGNOS DEL HIPOTIROIDISMO ADQUIRIDO
Iniciación durante la adolescencia
•Pubertad retrasada•Retraso del crecimiento y de la maduracion osea•Constipación•Piel seca•Mixedema•Retraso de la maduracion dental•Galactorrea

La sintomatología que produce el déficit de hormonas tiroideas es consecuencia de las alteraciones del metabolismo intermedio, del consumo de oxígeno por disminución de la síntesis proteica y del
metabolismo celular.
más marcada en el hipotiroidismo primario que en el hipotiroidismo central.
BOCEO ENDEMICO TCA
presencia de un bocio de superficie irregular, y de consistencia aumentada, aveces asimétrico y generalmente no doloroso: se palpan pequeños nódulos similares a semillas; a veces pueden
encontrarse adenopatías cervicales indoJoras
presentación más frecuente el bocio simple eutiroideo
EL LABORATORIO: centellograma irregular y un aumento de los anticuerpos
antitiroideos. Estos últimos se pueden elevar 4 a 6 meses después de iniciarse la enfermedad.
EL LABORATORIO: Por técnicas de hemaglutinación se detectan anIicuerpos
antitiruglobulina y antimicrosomicos en el 50 al 90% de los casos de TCA confirmada por biopsia

HIPOTIROIDISMO CENTRAL
Mas frecuente en niños mayores que en lactantes, se sospecha en pacientes que con deficit de otras trofinas hipofisiarias o de hormona
del crecimiento.

ANTES DE LOS 2 AÑOS DESPUES DE LOS 2 AÑOS
•Seudohipertrofia muscular•Alteración neuropsicológica
•No compromete SN•Bocio, retraso mental, fatigabilidad,disminución de rendimiento•Acumulación de liquido en el SN – HIC ( Pseudotumor cerebral)•Retraso de la maduracion osea y dental•Disgenesia Epifisiaria •Pubertad Precoz•Hiperprolactinemia
hipotiroidismo congénito, los síntomas pueden observarse ya al primer mes de vida, en los lactantes con
hipotiroidismo adquirido la sintomatología comienza después del sexto mes.

DIAGNOSTICO
T4 y T4 libre TSH
HIPOTIROIDISMO PRIMARIO
test de TRH sirve para diferenciar los distintos tipos de hipotiroidismo; así, en los terciarios la respuesta de la TSH a la TRH es lenta, de menor magnitud y con una caída también más lenta, mientras que en los primarios es más rápida y alta: en los secundarios, debido a la falla hipofisaria, no hay respuesta de la TSH a la TRH
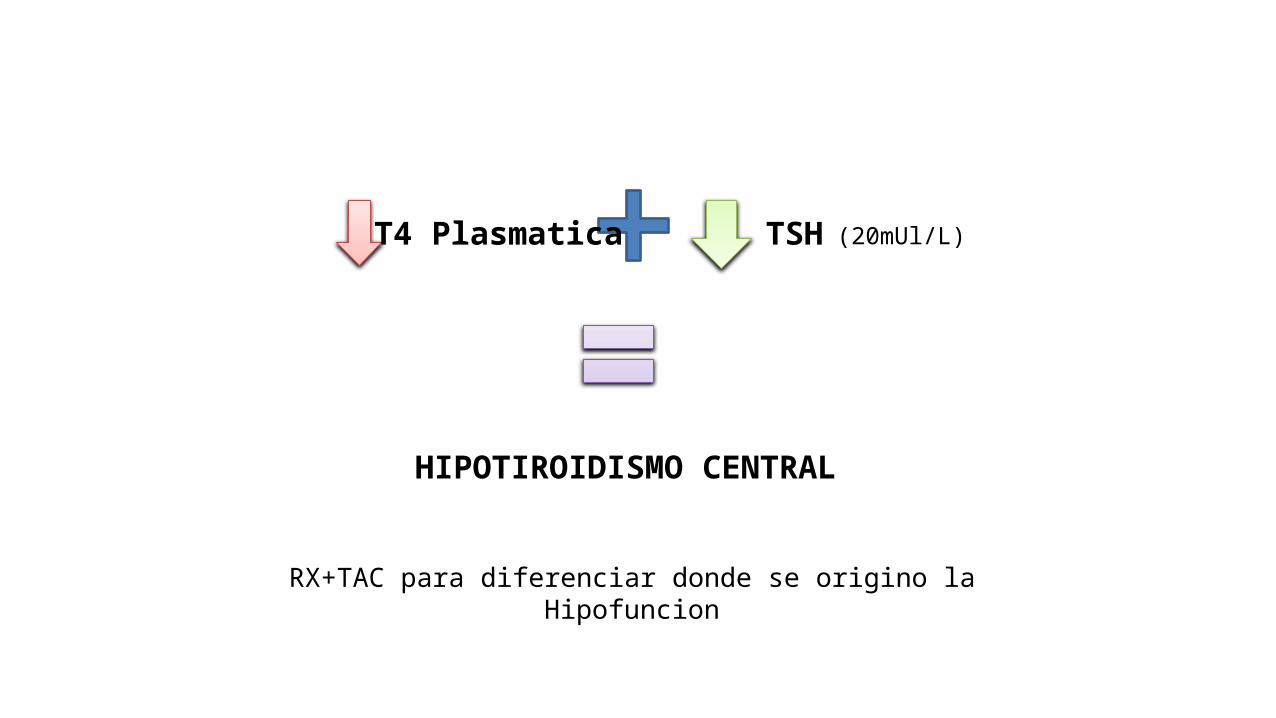
T4 Plasmatica TSH
HIPOTIROIDISMO CENTRAL
(20mUl/L)
RX+TAC para diferenciar donde se origino la Hipofuncion

TRATAMIENTO
El tratamiento del hipotiroidismo se efectúa con tiroxina en una única dosis diaria de 5-6 ug/kg durante el primer año de vida,
de 4 ug/kg después de los dos años y de 3-4 ug/kg en los preescolares y los adolescentes.
En el hipotiroidismo severo la dosis inicial debe ser el 25% de la dosis de mantenimiento, con aumentos paulatinos debido a que una reabsorción brusca del mixedema,
en caso de estar presente, puede provocar una insuficiencia cardíaca. La absorción del medicamento es mejor si se lo administra media hora antes de las comidas.
Se debe evitar el hipertiroidismo iatrogénico en estos pacientes ya que acelera la maduración ósea y el cierre de las suturas, disminuye el número de células cerebrales ,
Disminuye la mineralización ósea y altera el desarrollo neurológico y conductual.
En raras ocasiones puede haber hipertensión intracraneana al iniciar el tratamiento (manifiestada por cefaleas, vómitos y signos visuales).
TRATAMIENTO

el tratamiento produce un excelente resultado con resolución de la mayoría de los signos y síntomas del hipotiroidismo.
TRATAMIENTO
Una vez conseguida la dosis adecuada de tiroxina basta con controlar cada 6 meses a los niños mayores y cada 3 meses a los lactantes, por la posibilidad de tener que cambiar la dosis con los incrementos de peso.
El tratamiento temprano, por la detección precoz de laboratorio en el RN, es el más eficaz para evitar el retraso mental que puede producir un hipotiroidismo.

Es un estado catabólico agudo
que se caracteriza por la falta de utilización de glucosa a nivel celular, lo que ocasiona
un desequilibrio metabólico con predominio de las hormonas contrarreguladoras, con la finalidad de preservar la formación de energía a nivel mitocondrial que se acompaña de deshidratación hiperosmolar,
CETOACIDOSIS DIABETICAgenerado por la deficiencia de insulina, y la ausencia total o casi total de sus acciones,
alteraciones electrolíticas, exceso de formación de cuerpos cetónicos y acidosis.
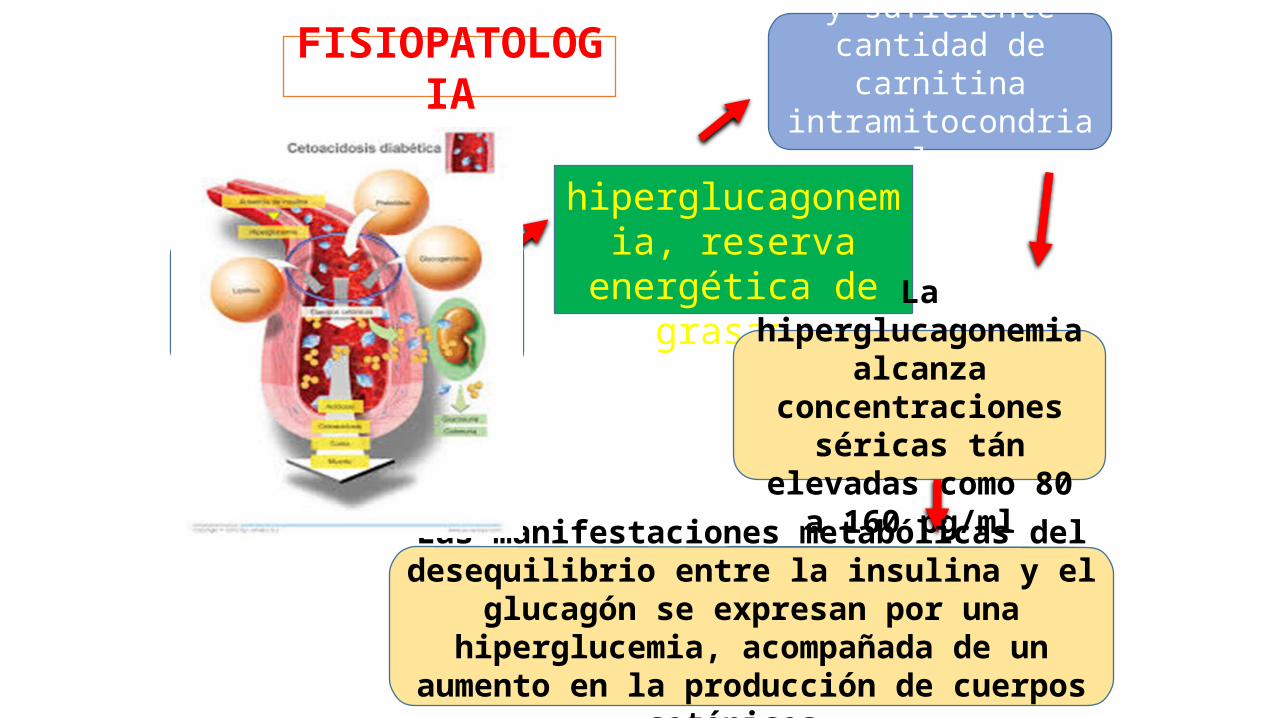
La C.D. sólo se desarrolla si coexisten
insulinopenia,
hiperglucagonemia, reserva energética de
grasas
y suficiente cantidad de carnitina
intramitocondrial.
La hiperglucagonemia alcanza concentraciones
séricas tán elevadas como 80 a 160 pg/ml
y se presenta 6 a 10 horas después de que se instala la
insulínopenia.
FISIOPATOLOGIA
Las manifestaciones metabólicas del desequilibrio entre la insulina y el glucagón se expresan por una hiperglucemia, acompañada de un aumento en la
producción de cuerpos cetónicos.

La deficiencia de insulina disminuye la cantidad de transportadores "Glu T -4", lo que dificulta el ingreso de la glucosa en las células
y disminuye la acción de las enzimas insulino-dependientes reguladoras de la glucolisis, la glucogénesis y el ciclo de Krebs.
CAUSAS DE LA HIPERGLUCEMIA
El exceso de glucagón y el hipercortisolismo,aumentan la gluconeogénesis y la proteinólisis con la finalidad de generar energía u partir de las proteínas.
Aumenta la cantidad de transportadores ''Glut-2" hepáticos, y permite la salida de la glucosa a una velocidad aproximada de 4 mg/kg/min con la finalidad de preservar el aporte de glúcidos al SNC.

La adrenalina favorece la glucogenólisis al activar a la fosforilasa de glucógeno y la hormona del crecimiento disminuye la captación de glucosa a nivel periférico.
El determinante principal de la formación de cuerpos cetónicos es la lipólisis generada por la adrenalina, que aumenta la actividad de la lipasa del tejido adiposo a través de sus receptores β,
CAUSAS DE LA CETOGENESIS
Pero requiere la existencia de las modificaciones metabólicas debidas al desequilibrio entre la insulina y el glucagón.

A nivel mitocondrial; la de malonil-CoA permite un actividad de la palmitoiltransferasa- 1, con incremento
En la transferencia de acil-CoA del citoplasma hacia el espacio intermembranoso, en donde el en la concentración de carnitina los transforma en acil-carnitina, que por acción de la palmitoiltransferasa-II son convertidos en aciL-Coa
Al atravesar la membrana interna y, ya en la matriz, son degradados por la β-oxidación y forman cuerpos cetónicos.

Consecuencias de la hiperglucemia y de la cetogénesis
Al existir glucemias superiores a los 180 a 200 mg/dl se sobrepasa la capacidad de reabsorción tubular renal y se produce glucosuria, la cual se acompaña de pérdida de agua, situación conocida como diuresis osmótica
El aumento del volumen urinario se acompaña de pérdidas renales muy elevadas de sodio, potasio, cloro y fosfato
Debido al efecto osmolar de la hiperglucemia, se favorecen la salida de agua con aumento de la osmolaridad intracelular
Cuando esta condición dura más de 8 horas
las células aumentan la síntesis de proteínas no metabolizables en el corto plazo, lo que permite aumentar sus condiciones osmolares y evitar una mayor pérdida de líquidos
Estos osmoles idogenicos son particularmente importantes a nivel neuronal, tanto para preservar la actividad celular como para mantener la función tisular
Si bien inicialmente existen expansión y dilución extracelular, en el largo plazo las pérdidas de agua son mayores que las de sodio y otros compuestos, por lo que la osmolaridad sérica se normaliza y posteriormente aumenta hasta 360 mOsm/l o más
La pérdida de líquidos intravasculares comprometen el flujo visceral y a nivel renal disminuye la presión de filtración glomerular y por lo tanto la eliminación de sustratos lo que favorece la persistencia de la hiperglucemia, la hipercetonemia y la acidosis
La excesiva oxidación de las grasas y la disminución del ciclo de Krebs disminuyen la producción de CO2 y facilitan la acumulación sérica de acetato, la cual libera un hidrogenión de manera hipermolar,
por lo que el pH sérico disminuye una vez sobrepasada la capacidad buffer del bicarbonato, la hemoglobina y otros sistemas intraoculares y extraoculares
Para disminuir la producción excesiva de hidrogeniones,
dos moléculas de acetato se unen por una reacción no enzimática y forma acetato, con la que la velocidad de acidificación disminuye en un 50 %,
pero dado que este es inestable a pH bajo, se lleva a cabo una reacción enzimática que utiliza NADH+H para convertirlo en B- hidroxibutirato, un compuesto con menor capacidad reacción
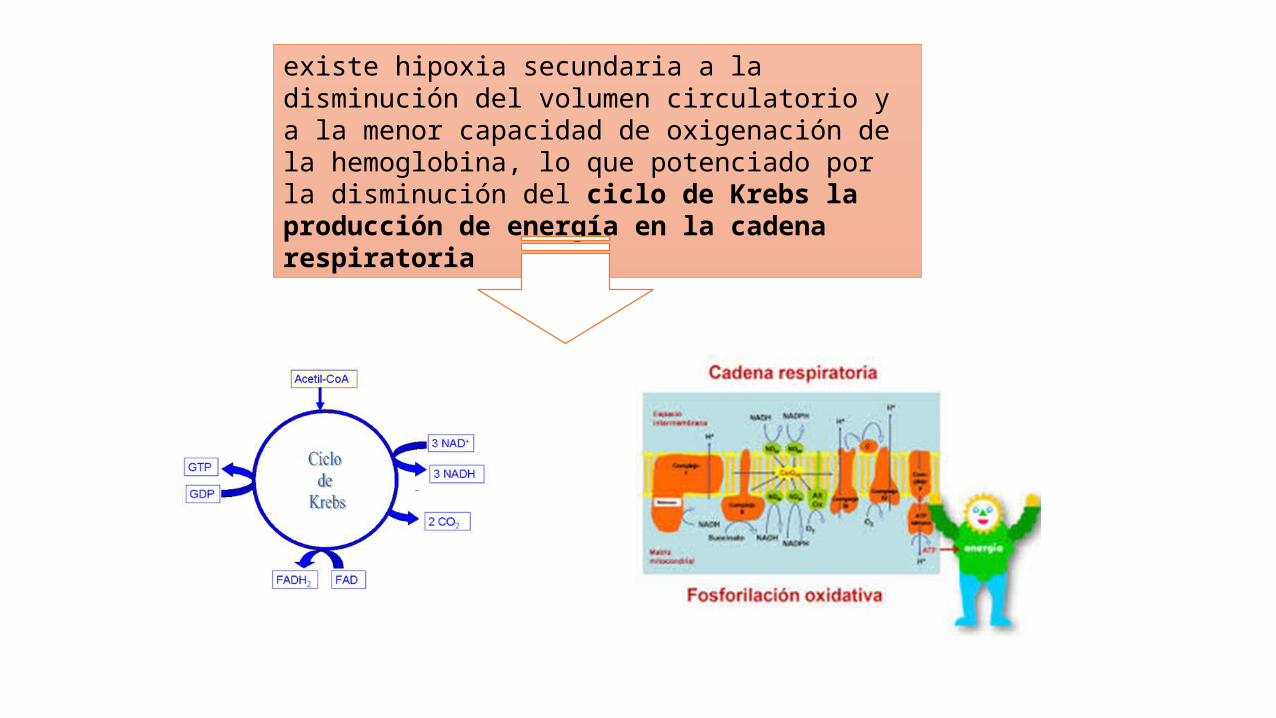
existe hipoxia secundaria a la disminución del volumen circulatorio y a la menor capacidad de oxigenación de la hemoglobina, lo que potenciado por la disminución del ciclo de Krebs la producción de energía en la cadena respiratoria

Diagnostico
• Glucemia superior a 250 mg/dl• Cetonemia y cetonuria que permanecen positivos a diluciones mayores de 1:2
• Acidosis metabólica con pH menor a 7,3 y bicarbonato menor a 15 mEq/l
Es preciso que existan las siguientes condiciones
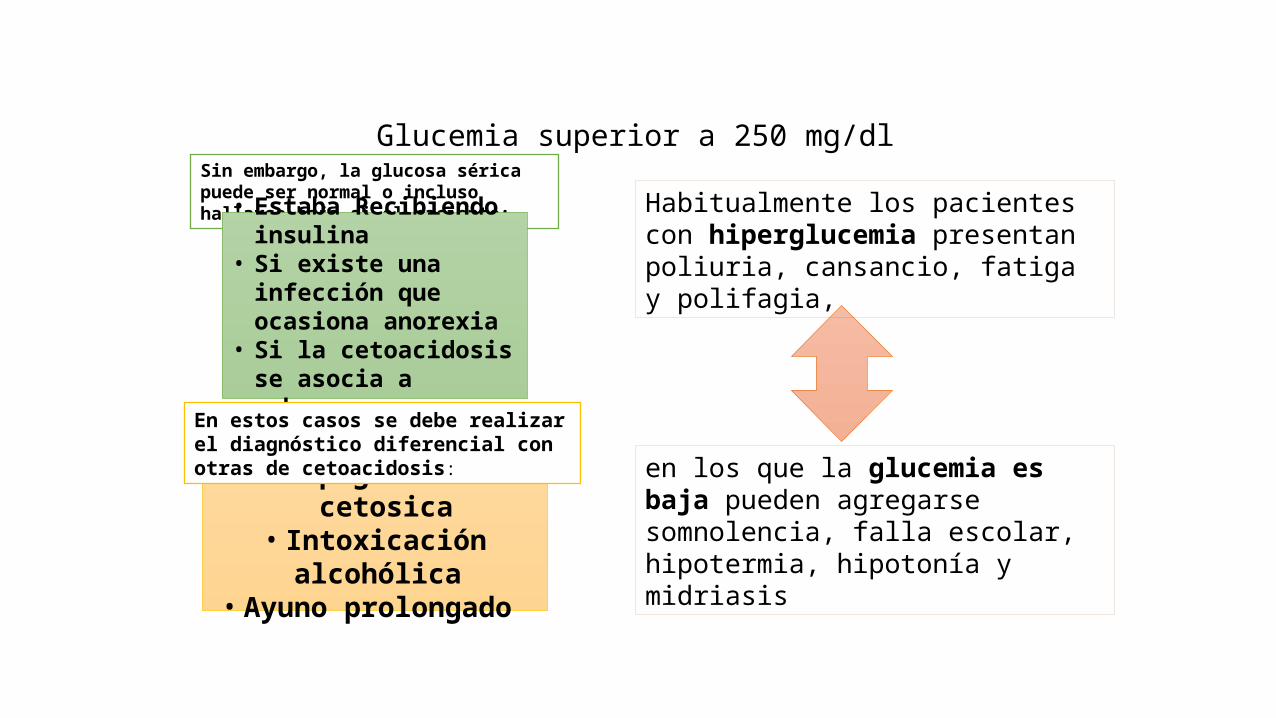
Glucemia superior a 250 mg/dlSin embargo, la glucosa sérica puede ser normal o incluso hallarse baja si el paciente:
• Estaba Recibiendo insulina
• Si existe una infección que ocasiona anorexia
• Si la cetoacidosis se asocia a embarazo
• Hipoglucemia cetosica• Intoxicación alcohólica
• Ayuno prolongado
En estos casos se debe realizar el diagnóstico diferencial con otras de cetoacidosis:
Habitualmente los pacientes con hiperglucemia presentan poliuria, cansancio, fatiga y polifagia,
en los que la glucemia es baja pueden agregarse somnolencia, falla escolar, hipotermia, hipotonía y midriasis

Cetonemia y cetonuria que permanecen positivos a diluciones mayores de 1:2
Es posible que no haya formación de cuerpos cetónicos o que esta sea muy baja si existe hipoxia severa acompañada de sepsis, hipotensión arterial o shock o ambas, pero en estos casos si existe acidosis metabólica
diagnóstico diferencial
acidosis láctica acidosis hipercloremica salicilismo uremia intoxicación por medicamentos
La confirmación de la acidosis hipercloremica se realiza por una relación mayor de 4 entre delta de anión gap y delta de bicarbonato
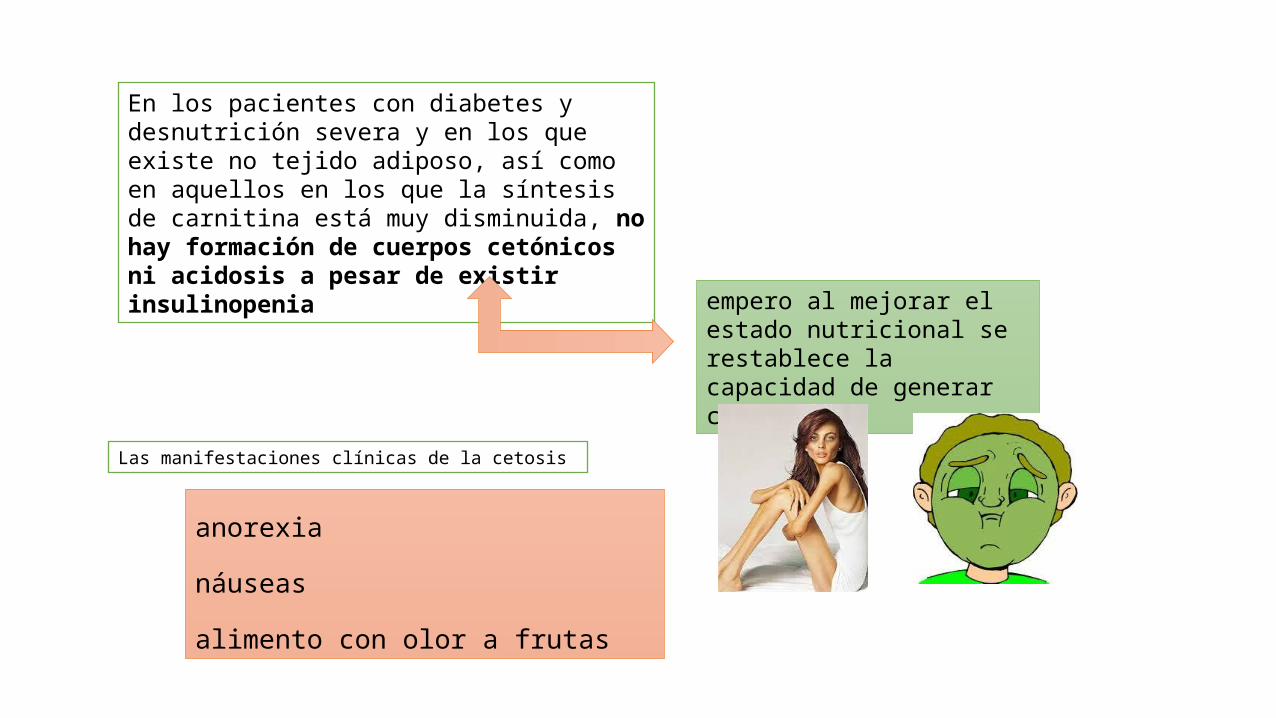
En los pacientes con diabetes y desnutrición severa y en los que existe no tejido adiposo, así como en aquellos en los que la síntesis de carnitina está muy disminuida, no hay formación de cuerpos cetónicos ni acidosis a pesar de existir insulinopenia
empero al mejorar el estado nutricional se restablece la capacidad de generar cetonas.
anorexia
náuseas
alimento con olor a frutas
Las manifestaciones clínicas de la cetosis

Acidosis metabólica con pH menor a 7,3 y bicarbonato menor a 15 mEq/l
La acidosis puede ser menos intensa que la esperada para el grado de cetosis, o incluso no existir, en aquellos pacientes con antecedentes de vómitos intensos y repetitivos, ingesta de álcalis, uso de diuréticos y síndrome de Cushing.
respiración de Kussmaul
enlentecimiento del relleno capilar
cianosis periférica
taquicardia sin hipotensión arterial
vómitos y dolor abdominal
La acidosis metabólica se manifiesta por : En estos casos la pCO2 es menor que la esperada para el pH

Estudios complementarios Potasio sérico
• acidosis metabólica• catabolismo de proteínas
intracelulares• disminución de la entrada de
glucosa en las células y degradación de glucógeno
• potasuria• falta de aporte por anorexia y
vómitos • hiperaldosteronismo secundario
Habitualmente existe depleción de potasio debido a la asociación de:
Recuérdese que por casa 0,1 que disminuye el pH el potasio sérico aumenta 0,4 a 1mEq
La disminución en el contenido total de potasio corporal se manifiesta por:
Constipación
hiporreflexia
dilatación gástrica y vómitos
incluso íleo paralitico.
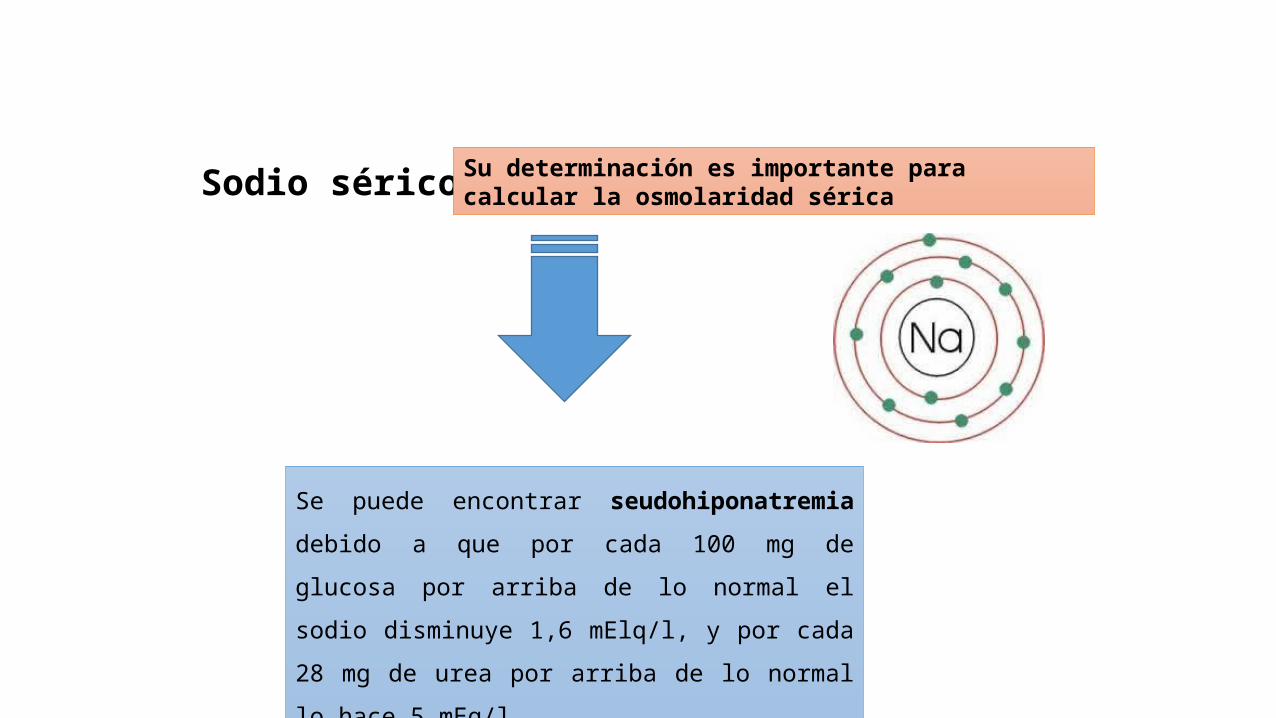
Sodio sérico Su determinación es importante para calcular la osmolaridad sérica
Se puede encontrar seudohiponatremia debido a que por
cada 100 mg de glucosa por arriba de lo normal el sodio
disminuye 1,6 mElq/l, y por cada 28 mg de urea por
arriba de lo normal lo hace 5 mEq/l.

UREA Y CREATININA SERICASSu determinación es útil para evaluar el grado de deshidratación y de hipovolemia. Habitualmente en una deshidratación del 10% los valores de estas aumentan 2 a 4 veces arriba de lo normal. Además el aumento en la producción de urea es un índice indirecto del grado de metabolismo proteico y un elemento que contribuye junto con la salida celular de potasio, fosforo, magnesio al aumento de la osmolaridad sérica y urinaria, favoreciendo la existencia de diuresis osmótica.
FosforoLas concentraciones séricas pueden ser normales a pesar de la presencia de una disminución en la cantidad corporal total y debe considerarse que existe una hipofosfatemia que amerita tratamiento si los valores son inferiores a 1,5mg/dl

Biometría hemáticaEs útil para descartar infecciones concomitantes, pero debe considerarse que durante la cetoacidosis es frecuente encontrar leucocitosis con cifras de 15.000 a 20.000 y neutrofilia asociada con hipertermia. Por ello la sospecha de infección es más firme en aquellos con 25.000 leucocitos que presentan bandemia y sobre todo si existe hipotermia. En ocasiones es particularmente difícil establecer el diagnóstico diferencial entre apendicitis y cetoacidosis diabética y debe recordarse que la identificación de un foco neumónico por radiografía no será fácil si el paciente esta deshidratado.

Amilasa séricaDado que hasta 79% de los pacientes pueden presentar daño pancreático por hiperosmolaridad o hipoperfusion visceral
Osmolaridad séricaEn su defecto, calcularla sobre la base de esta fórmula:
Osmolaridad = 1,86 Na + glucosa / BUN/2,8 (o urea/5,6) +9
Existe una relación directamente proporcional entre el aumento de la osmolaridad y las alteraciones neurológicas. Los valores inferiores a 320 mOsm/L no modifican el estado de conciencia, en tanto que las cifras superiores a 330 se asocian con somnolencia, las superiores a 140 con estupor, y por arriba de 350 se presenta coma. Si un paciente con osmolaridad menor de 320 se encuentra comatoso deberán descartarse otras causas de daño neurológico

TRATAMIENTO: MEDIDAS GENERALESes preciso consignar cada hora la
frecuencia cardiaca y respiratoria, la tensión arterial, la temperatura, la
coloración de la piel, el llenado capilar, la diuresis, el estado de hidratación, los reflejos osteotendinosos, el perímetro abdominal y el estado de conciencia.
Debe buscarse un foco infeccioso, si no se encuentra la causa de la descompensación
metabólica en niños previamente diagnosticados y tratados, o si la respuesta al tratamiento es inadecuada, debe evaluarse la utilidad de exámenes específicos de laboratorio y gabinete, si como cultivos de orina, heces, exudado faríngeo, exudado vagina, e incluso
líquido cefalorraquídeo
Es necesario realizar un electrocardiograma
con búsqueda específica de arritmias
Es indispensable determinar el peso del paciente. Si no se conoce el que tenía antes de la cetoacidosis recuérdese que, por la deshidratación, en el
momento de iniciar el tratamiento existe por lo menos un 10% de déficit
que debe agregarse al peso consignado.

TRATAMIENTO ESPECIFICOLa rehidratación del paciente como tratamiento único normaliza el volumen intravascular con mejoría del estado acido-base, disminuye la concentración de hormonas contrareguladoras y reduce la glicemia hasta en un 18%, pero no revierte la cetogenesis. La reposición de volumen con líquidos isotónicos permite restituir el volumen intravascular pero facilita la acumulación de líquidos en el espacio intersticial y mantiene deplecionado el espacio intracelular, en tanto que el uso de soluciones hipotónicas o la asociación de esas con coloides, normaliza los 3 espacios.

Reposición rápida del volumen intravascular mediante una carga calculada en 30ml/kg o 600ml/m2 para pasar en una hora, utilizando una solución de NaCl al 0,9% y sin potasio (por la posibilidad de producir arritmias cardiacas)
si el paciente presenta oliguria puede evaluarse una segunda carga. Se considera conveniente el uso de
bicarbonato en si el pH sérico es inferior a 7,0 en cuyo caso debe considerarse la aplicación de soluciones de
bicarbonato 1/6 molar en dosis de 10 ml/kg dentro de los líquidos calculados para la primera hora del manejo
Líquidos de mantenimiento que se recomienda 3000 ml/m2/día los cuales la mitad se pasa en las primeras 8 horas y el 50% restante en las próximas 16 horas. Debe considerarse que si la osmolaridad sérica se encuentra entre 320 y 340 mOsm/L la reposición de hídrica debe
calcularse para 36 horas, en tanto que si es superior a 340 debe plantearse por lo menos para 48 horas, con la
finalidad de no producir cambios osmolares rápidos que facilitan la aparición de complicaciones cardiovasculares y
neurológicas. La osmolaridad sérica debe medirse o calcularse cada 2 horas.

LIQUIDOS DE MANTENIMIENTO
En los líquidos de mantenimiento se
agregan 40 mEq/L de cloruro de potasio, pero si
las concentraciones reales de este catión, una vez determinado el efecto
de la acidosis, son inferiores a 2,6 mEq/L, se recomienda un aporte de 6mEq/L, c0on monitoreo de la actividad eléctrica
cardiaca
El uso de fosfato de potasio es controvertido y no es necesario al menos que el paciente tenga una desnutrición severa, mantenga un
ayuno mayor de 48 horas o presente hipoxia, hemoglobina inferior a 8g/L, bronconeumonía con insuficiencia respiratoria,
insuficiencia cardiaca congestiva o concentraciones séricas inferiores
a 1mg/dl. En este caso se recomienda aplicar 1/3 parte de los requerimientos de potasio en forma de fosfato (recordando que la concentración de la solución de la solución comercial de fosfato de potasio es de 2mEq/ml, en tanto que la de cloruro de potasio es de
4mEq/L
La utilización de soluciones glucosadas en los líquidos de mantenimiento permite
aportar una cantidad de glucosa suficiente para cubrir los requerimientos del sistema nervioso central, al mismo tiempo que
disminuye la osmolaridad de la solución de NaCl al 0,9%. En términos generales se recomienda comenzar con solución glucosada en el momento en que la glucemia es inferior a 250mg/dl y
calculando un aporte de 3 a 5 mg/kg/min.

Si el paciente no presenta contraindicaciones para la vía oral esta debe ser iniciada una vez terminadas las soluciones de la primera hora de manejo, los niños toleran adecuadamente los líquidos por vía oral incluso en presencia de acidosis metabólica, y la utilización conjunta de hidratación enteral y parenteral permite acortar el tiempo requerido para la rehidratación.
La glucemia debe evaluarse cada hora mediante determinación capilar, y sobre la base de ésta, decidir la cantidad de glucosa a infundir. No se recomienda la determinación de la glucosa venosa o sérica, a menos que los resultados estén disponibles en un lapso menor de 15 minutos, ya que de otra manera las decisiones terapéuticas se toman a tiempo pasado
CETOACIDOSIS DIABÉTICA
TRATAMIENTO ESPECÍFICO
CONTROL DE LA CETOGÉNESIS
INSULINA de acción rap. 0.1U/kg/h
• Evitar la hipopotasemia y la hipoglucemia
El descenso de la glucemia es similar si se
aplica IV. IM. SC. (50-70mg/h)
• Aunque la reversión de la cetogénesis es más rápida con infusión IV.
Determinar cetonemia y cetonuria cada 1 a 2 horas, y agregar Glu en soluciones si
se presenta hipoglucemia
CETONURIA NEGATIVA NO SIGNIFICA AUSENCIA DE
CETOSIS
CETOACIDOSIS DIABÉTICATRATAMIENTO ESPECÍFICO
CONTROL DE LA ACIDOSIS
La acidosis metabólica se corrige con rehidratación y reversión de la cetogénesis
El uso de bicarbonato está restringido a: pacientes que no muestran mejoría en concentraciones de HCO3
y PH en las primeras 2-3 horas de manejo.
Utilizar HCO3
en infusión 40mEq/m2
pasar en 2 h si el PH es <de7.0

CETOACIDOSIS DIABÉTICA
TRATAMIENTO ESPECÍFICO
CONTROL DE LA ACIDOSIS
RIESGOS DE UTILIZAR HCO3 DE FORMA INADECUADA
HIPOXIA TISULAR RIESGO DE HIPOPOTASEMIAAGRAVA EL ESTADO HIPEROSMOLAR
ACIDOSIS PARADÓJICA DEL SNCALCALOSIS METABÓLICA
CETOACIDOSIS DIABÉTICA
TRATAMIENTO ESPECÍFICO
DETERMINACIÓN DE LA CAUSA
FALTA DE APLICACIÓN DE INSULINA (25%)
INFECCIONES: Inf. Resp. Altas o bajas, IVU, Inf. Enterales,
Apendicitis, Enf. Exantemáticas y Meningoencefalitis. (25-37%)
USO DE MEDICAMENTOS O INGESTA DE ALCOHOL
SUSPENSIÓN DEL USO DE INSULINA O MALA TÉCNICA DE
APLICACIÓN. (20%)
CETOACIDOSIS DIABÉTICA
TRATAMIENTO ESPECÍFICO
COMPLICACIONES DEL TRATAMIENTO
TROMBOSIS MICROVASCULA
R
• Producida por disminución del flujo sanguíneo durante deshidratación
EDEMA CEREBRAL• Desde antes de iniciarse tto, lleva a daño severo y muerte
• Más frecuente <5años, con hiponatremia persistente o periodos prolongados de cetoacidosis.
• Tto manitol 1-2mg/kg pasar en 30min y Dexametazona 0,25-0.5mg/kg/dia cada 6h.
EDEMA PULMONAR
• Asociado con el uso prolongado de soluciones cristaloides.
HIPOGLUCEMIA
• Por aporte inadecuado de sol. Glucosadas durante manejo de cetogénesis
MUCORMICOSIS
• Es rara. Sospecharse si: dolor facial en senos paranasales y epistaxis.
• Tto con anfotericina.

BIBLIOGRAFIA
•MENEGELLO. (1998). HIPOTIROIDISMO CONGENITO-ADQUIRIDO. CETOACIDOSIS DIABETICA (5ta ed., Vol. 2). Obtenido de http://booksmedicos.org/pediatria-meneghello/•Gómez Gila, Ana. González Casado Isabel. Barrio Castellanos, Raquel. Sociedad Española de Endocrinología Pediátrica SEEP 2012. Tratamiento de la Cetoacidosis Diabética (en línea). Disponible en: http://www.seep.es/privado/gdiabetes/tratamiento_cad_seep.pdf

Gracias