metodolog ía sint ética aplicada a la s íntesis de f …€¦ · tema i. introducción 1...
TRANSCRIPT

MetodologMetodolog íía Sinta Sint éética Aplicada tica Aplicada a la Sa la Sííntesis de Fntesis de F áármacosrmacos
Miguel CardaMiguel CardaMMááster en Quster en Qu íímica Aplicada y Farmacolmica Aplicada y Farmacol óógicagica
Universidad Jaume IUniversidad Jaume I
NH2
HOOCMeO
HN
N
NN
NPh3C
(PriO)2B
Br
Br
Síntesis de losartan


Tema 1
Introducción a la síntesis de fármacos
Máster en Química Aplicada y Farmacológica
Universidad Jaume I


Tema I. Introducción 1
Introducción a la síntesis de fármacos
El diseño de una síntesis de un compuesto orgánico se aborda de forma racional mediante el denominado análisis retrosintético. Esta metodología permite la propuesta de rutas sintéticas para una molécula objetivo mediante la desconexión de enlaces. La molécula objetivo se desconecta a moléculas más simples, que a su vez se desconectan a otras moléculas y así sucesivamente hasta llegar a compuestos comerciales o fácilmente accesibles.
1.1. Desconexión de los compuestos orgánicos: sintón y equivalente sintético
Cada etapa de desconexión se basa en una reacción química y formalmente supone la ruptura de un enlace para dar lugar a dos fragmentos denominados sintones. Por tanto, el sintón se define como el fragmento que surge de una desconexión. Por ejemplo, el cloruro de t-butilo se puede desconectar en el enlace C-Cl para generar un sintón catiónico (el carbocatión t-butilo) y un sintón aniónico (el anión cloruro).
En la desconexión del cloruro de t-butilo surgen dos sintones que tienen existencia real: el carbocatión t-butilo y el anión cloruro.
Los sintones que surgen de una desconexión pueden tener existencia real, o pueden ser fragmentos idealizados para los cuales hay que encontrar un equivalente sintético. Por tanto, una vez desconectada la estructura y analizados los sintones se tienen que proponer los correspondientes equivalentes sintéticos, es decir los reactivos que harán el papel de los sintones en la reacción.
El carbocatión t-butilo es un sintón que tiene una existencia real, pero no se encuentran accesibles sales que contengan este catión porque es muy inestable. Todo lo contrario ocurre con el sintón cloruro. Su gran estabilidad hace que existan un gran número de sales que lo contienen.
El reactivo para el sintón cloruro puede ser el HCl, que liberará en disolución acuosa Cl-, o
NaCl, que se disolverá para dar iones Na+ y Cl-.
El reactivo para el carbocatión t-butilo no es tan evidente y dependerá de la reacción que se elija para la obtención del cloruro de t-butilo.
Por ejemplo, basándonos en la desconexión propuesta anteriormente se podría proponer una síntesis del cloruro de t-butilo mediante la reacción de tipo SN1 entre el t-butanol y el HCl:
ClOH + HCl + H2O
La síntesis del cloruro de t-butilo se habría podido efectuar también empleando como material de partida 2-metilpropeno, como se indica a continuación:

Metodología síntetica 2
En este caso, la reacción que permite la obtención del cloruro de t-butilo es una adición electrofílica al doble enlace.
Así, un compuesto de estructura tan simple como el cloruro de t-butilo se puede obtener mediante dos síntesis diferentes, tanto por lo que hace al sustrato carbonado (t-butanol o 2-metilpropeno) como al tipo de mecanismo que interviene en el proceso (SN1 o adición electrofílica a doble enlace).
La molécula objetivo, por ejemplo el cloruro de t-butilo, marca los reactivos y por tanto el tipo de mecanismo que participará en el proceso de síntesis. La síntesis del cloruro de t-butilo a partir del t-butanol es un proceso de sustitución formal de OH por Cl.
2.1. Selectividad en las reacciones orgánicas
La selectividad de las reacciones orgánicas se puede clasificar de la siguiente forma:
2.1.1. Reacciones quimioselectivas
Las reacciones quimioselectivas aquéllas en las que un grupo funcional reacciona con preferencia a otros presentes en el sustrato orgánico.
Es el caso de la síntesis del paracetamol, que se puede llevar a cabo de forma quimioselectiva empleando un equivalente de anhídrido acético en presencia de piridina como base. La quimoselectividad de esta reacción se explica por la mayor nucleofília del grupo amino en relación con el grupo hidroxilo, lo que provoca que la acetilación se produzca sobre el grupo amino.
2.1.1.a. Grupos protectores
Cuando no es posible conseguir la quimioselectividad en una reacción orgánica se recurre al empleo de los grupos protectores. Por ejemplo, no es posible conseguir la adición quimioselectiva del bromuro de finilmagnesio al cetoéster que se indica a continuación, porque el grupo carbonilo cetónico es más reactivo que el grupo carbonilo del éster
Para conseguir la transformación anterior se recurre a la protección provisional del carbonilo cetónico, por ejemplo en forma de acetal cíclico, tal y como se indica en la siguiente secuencia sintética. Una vez transformado el carbonilo cetnico en acetal, se puede llevar a cabo la reacción de adición del PhMgBr sobre el carbonilo del éster, porque la función acetálica es

Tema I. Introducción 3
inerte al reactivo de Grignard. Después de la adición se procede a la desprotección del carbonilo cetónico, tal y como se representa en el siguiente esquema:
En la siguiente tabla se indican algunos de los grupos protectores más empleados en síntesis orgánica.
Grupos Protectores
Grupo Grupo
Protector
Síntesis Eliminación El GP resiste: El GP reacciona
con:
Aldehido
Cetona
Acetal
RCH(OR´)2
R´OH, H+ H2O, H+ Nucleófilos, bases,
reductores
Electrófilos, acidos
Acidos
RCOOH
Esteres:
RCOOMe
RCOOEt
RCOOBn
RCOOt-Bu
Anion:
RCOO-
CH2N2
EtOH, H+
BnOH, H+
t-BuOH, H+
base
H2O, OH-
H2, o HBr
H+
ácido
bases débiles,
electrófilos
Nucleófilos
bases fuertes
nucleófilos, agentes
reductores
“
“
Electrófilos
Alcohol
ROH
Acetales:THP
Eteres:
ROBn
ROTr
Sililéteres:
TES
TBDMS
TBDPS
Esteres:
R´COOR
DHP, H+
BnBr, NaH
TrCl, base
TESCl,
TBDMSCl,
TBDPSCl,
R´COCl,
piridina
H2O, H+
H2, o HBr
H2O, H+
F-, o H2O, H+
“
“
“
H2O, H+ o
con
H2O, OH-
Nucleófilos, bases,
reductores
Nucleófilos, bases, y
oxidantes
Bases, nucleófilos, y
oxidantes
oxidantes,nucleófilos
“
“
“
Electrófilos,
oxidantes
Electrófilos, ácidos
HX (X=nucleófilo)
Acidos
Acidos
“
“
“
Acidos, bases y
nucleófilos
Amina
RNH2
Amida:
R´CONHR
R´COCl, base H2O, OH- o
H2O, H+
Electrófilos
Uretanos:
R´OCONHR
R´OCOCl, base
si R´=Bn:
H2, cat., o
HBr
si R´=t-Bu:
Electrófilos,
oxidantes
Electrófilos,
oxidantes
Bases, nucleófilos
Bases, nucleófilos,
H2O, H+

Metodología síntetica 4
Los grupos protectores deben cumplir una serie de requisitos:
1) Las condiciones de reacción para introducir el grupo protector debe reaccionar selectivamente y con buen rendimiento para dar el sustrato protegido.
2) Los grupos protectores deben poder eliminarse selectivamente y con buen rendimiento.
3) Los grupos protectores no deben presentar ninguna funcionalidad adicional que pudiera dar lugar a reacciones secundarias.
Conviene indicar que la utilización de grupos protectores en una secuencia sintética va en detrimento de la eficiencia de ésta, ya que aumenta el número de pasos, puesto que cada protección implica un paso de instalación y otro de desinstalación. Como muy difícilmente se consigue un rendimiento del 100% en las etapas de protección/desprotección, el rendimiento global de la secuencia sintética disminuye en relación a la misma secuencia llevada a cabo sin empleo de grupos protectores. Además de diminuir el rendimiento químico de la secuencia sintética, el empleo de los grupos protectores encarece la síntesis, ya que hay que emplear instalación, y la desinstalación del grupo protector, requiere necesariamente del empleo de reactivos. Por todo lo anteriormente comentado, una síntesis ideal es la que no emplea grupos protectores. La química orgánica moderna pone a disposición de aquéllos dedicados a la síntesis orgánica de un vasto arsenal de reactivos, algunos de los cuales son altamente quimioselectivos. No obstante, en la síntesis de moléculas altamente funcionarizadas se hace imprescindible recurrir a la estrategia de protección de grupos funcionales.
2.1.2. Reacciones regioselectivas
Las reacciones regioselectivas son aquéllas en las reacciona preferentemente una zona o región del sustrato orgánico.
Un ejemplo de reacción regioselectiva es la que se produce en la reacción de bromación de la acetanilida. Así, cuando la reacción se lleva a cabo con bromo molecular en ácido acético se obtiene muy preferentemente la p-bromoacetanilida, debido a que el grupo acetamido es electrón-donante, y por tanto activa las posiciones orto/para, y a que el impedimento estérico de las posiciones orto evita la bromación en estas regiones del anillo aromático.
2.1.3. Reacciones estereoselectivas
Las reacciones estereoselectivas aquéllas en las que se forma preferentemente un estereoisómero de entre varios posibles.
Un ejemplo de reacción estereoselectiva es la reacción del fenil-litio con la 1,3-dimetilpiperidin-4-ona. La adición del reactivo organometálico se produce de forma axial, obteniéndose muy mayoritariamente el estereoisómero que se indica a continuación:

Tema I. Introducción 5
N
O
CH3
CH3
N
O
Me
H
Me
PhLi
PhLiN
Ph
Me
H
Me
OH
N
OHCH3
CH3
Ph
2.1.4. Reacciones estereoespecíficas
Un caso extremo de reacciones estereoselectivas son las denominadas reacciones estereoespecíficas, que se pueden definir como aquéllas en las únicamente se forma un estereoisómero porque que el mecanismo de la reacción no ofrece alternativas a la formación de otros estereoisómeros.
Un ejemplo de reacciones estereoespecíficas son las reacciones SN2, ya que siempre tienen lugar con inversión de la configuración. Por ejemplo, la reacción del tosilato derivado del (S)-1-fenilpropan-1-ol con acetato de tetrabutilamonio, en DMF, proporciona exclusivamente el acetato del (R)-1-fenilpropan-1-ol. La reacción transcurre mediante el mecanismo SN2, que por su propia naturaleza impide la formación del estereoisómero S.
Ph (S)
OTsBu4N AcO
DMF Ph (R)
OAc
Reacción SN2
Las reacciones E2 también son estereoespecíficas ya que transcurren a través de un estado de transición antiperiplanar. Por ejemplo, en la reacción de eliminación del bromuro que se indica a continuación se forma únicamente la olefina de configuración Z:
2.2. Análisis retrosintético
Para llevar a cabo el proceso de desconexión de un compuesto orgánico es aconsejable seguir los siguientes puntos:
1) Las desconexiones se deben basar en reacciones conocidas.
2) En el caso de compuestos que presenten dos partes unidas por un heteroátomo, conviene desconectar dicho heteroátomo del carbono al cual está enlazado.
3) Conviene considerar desconexiones alternativas y elegir aquellas rutas que no conlleven problemas de quimioselectividad, lo que implica desconectar el grupo funcional más reactivo.
4) Para evitar problemas de quimioselectividad muchas veces se puede recurrir a la interconversión de grupos funcionales. Para simbolizarlo en el análisis retrosintético se escriben las siglas IGF encima de la flecha retrosintética. Las interconversiones de grupos funcionales suponen normalmente reacciones de sustitución nucleofílica, reducciones y oxidaciones.

Metodología síntetica 6
A continuación se muestra la estructura de la ofornina, un fármaco antihipertensivo. La retrosíntesis se inicia con la desconexión del enlace amida (véase el esquema que se indica a continución:
La operación de desconexión genera el sintón catiónico 1 y el sintón aniónico 2, cuyo equivalente sintético es la piperidina 3. El sintón catiónico 1 puede generarse a partir del ácido 4, o de sus derivados reactivos, como el correspondiente cloruro de ácido o anhidrido. Por último, la desconexión del enlace C-N en el ácido 4 forma el sintón aniónico 5 y el sintón catiónico 6.
En el siguiente esquema se muestra la síntesis de la ofornina con los reactivos reales.
O
N
NH
N
Ofornina
HN
O
NH
N
Cl
O
NH
N
OHO
NH2
OHN
Cl
SOCl2
Síntesis de ofornina
En la siguiente tabla se muestran algunas de las desconexiones más usuales empleadas en los análisis retrosintéticos:

Tema I. Introducción 7
R OH1,2 C-C
R OH
Molécula objetivo Sintones
Nombre de ladesconexión Equivalentes sintéticos Reacción
R MgBr O
H
H adición 1,2
R OH1,3 C-C
R OH R MgBr adición 1,3O
RX
R'1,2-diX R X adición 1,2
RX
O1,2-diX
O
R XC-X
R X R Y SN2
OH
R X1,3-diX adición 1,4
O
OR'
OH
R'R X
R X OCl
R X SN2
R R'1,3-diO adición
aldólica
O OH
R X
O
R X
O
R R'
O OH
R R'
O O
RO R'1,3-diCO adición
Claisen
O O
RO R'
O O
RO R'
O O
R'O
1,5-diCO adición 1,4O O
RO
OO O O
CO2RWittigC=C
CO2RO
PPh3
CO2R
2.3. Umpolung o inversión de la polaridad
Se define umpolung, o inversión de la polaridad, a la modificación química de un grupo funcional con el objetivo de revertir su polaridad.
El átomo de oxígeno del grupo carbonilo es más electronegativo que el átomo de carbono y, en consecuencia, la polarización natural del grupo carbonilo es la que coloca la carga parcial positiva sobre el átomo de carbono y la carga parcial negativa sobre el átomo de oxígeno. La desconexión de un grupo carbonilo origina un sintón catiónico, denominado catión acilo, y un sintón aniónico, como se indica en el siguiente esquema:

Metodología síntetica 8
La desconexión umpolung del grupo carbonilo origina un anión acilo y el equivalente sintético de este sintón. Los aniones derivados de ditioacetales y los aniones derivados de nitroalcanos son equivalentes sintéticos de los aniones acilos, como se indica en el siguiente esquema:
Cuando el ditiano deriva de un aldehído, el protón acílico puede ser abstraído por n-butil-litio en THF a temperaturas bajas. El 2-litio-1,3-ditiano generado reacciona como un nucleófilo con halogenuros de alquilo, como el bromuro de bencilo, con compuestos carbonílicos y con oxiranos. Después de la eliminación del grupo ditiano los productos finales de la reacción son cetonas, β-hidroxicetonas o α-hidroxicetonas. Un reactivo común para la hidrólisis del ditiano es el (bis(trifluoroacetoxi)yodo)benceno.
En el análisis retrosintético de una 1,4-dicetona se forma un sintón catiónico, cuya polaridad es lógica, o natural, y un anión acilo, con polaridad no natural, como se observa en el siguiente esquema:

Tema I. Introducción 9
El equivalente sintético del sintón catiónico es el correspondiente compuesto carbonílico α,β-insaturado. El equivalente sintético del anión acilo puede ser el correspondiente anión de nitroalcano. La síntesis de la 1,4-dicetona se diseñaría del siguiente modo:
2.4. Obtención de compuestos ópticamente puros
La mayor parte de los receptores biológicos son quirales y por tanto pueden distinguir entres dos compuestos enantioméricos, de ahí la importancia de conseguir la preparación de fármacos en su versión ópticamente activa, no racémica. .
2.4.1. Obtención de compuestos quirales de la Naturaleza
El método más tradicional empleado en la obtención de compuestos homoquirales es recurrir a los productos obtenidos directamente de la Naturaleza, ya que muchas moléculas quirales empleadas como medicamentos son productos naturales, o derivados de ellos obtenidos mediante manipulación sintética. Es el caso del taxol y del docetaxel, cuyas estructuras se indican a continuación:
El taxol se emplea en el tratamiento del cáncer de pulmón, ovario, mama y formas avanzadas del Sarcoma de Kaposi. El taxol fue aislado en 1968 por M. E. Wall y M. C. Wani de la corteza del Taxus brevifolia (tejo del pacífico).
El docetaxel es un derivado semisintético del taxol y se emplea en el tratamiento del cáncer de mama, cáncer de ovario, cáncer de pulmón no microcítico, cáncer de próstata y cáncer gástrico.
2.4.2. Obtención de compuestos quirales mediante la resolución de mezclas racémicas
El otro método tradicional para la obtención de compuestos ópticamente puros es el de la resolución de mezclas racémicas. La separación de los enantiómeros de una mezcla racémica se denomina resolución y se lleva a cabo convirtiendo la mezcla racémica en una mezcla

Metodología síntetica 10
diastereoisomérica mediante la reacción de aquélla con un compuesto ópticamente puro. Las diferencias en las propiedades físicas y/o químicas de los diastereoisómeros permiten su separación. Una vez conseguida ésta, los enantiómeros que constituían la mezcla racémica se obtienen puros mediante reversión de reacción empleada en la conversión de la mezcla racémcia en mezcla diastereoisomérica.
En la siguiente figura se esquematiza un proceso de separación (resolución) de una mezcla de tornillos de diferente helicidad. Una tuerca de una helicidad determinada sólo encajará eficazmente con los tornillos de helicidad complementaria. Se formarán dos conjuntos de tornillo-tuerca (diastereoisómeros) cuya estabilidad y propiedades serán muy diferentes. Esta diferencia de propiedades permite la separación de los tornillos según su helicidad.
A continuación se representa de forma esquemática el proceso de resolución de una mezcla racémica.
En el esquema que se da a continuación se representa el proceso de resolución de una mezcla racémica formada por (R)- y (S)-3-amino-1-butino. La reacción ácido-base con el ácido (+)-tartárico forma una mezcla de tartratos diastereoisóméricos. La cristalización fraccionada de la mezcla diastereoisomérica permite la separación de los diastereoisómeros, ya la sal de amonio derivada del (R)-3-amino-1-butino cristaliza mientras que la sal de amonio derivada del (S)-3-amino-1-butino permanece en disolución. Una vez conseguida la separación mediante filtración, cada sal de amonio se trata con una disolución acuosa de carbonato potásico, lo que permite la obtención del (R)-3-amino-1-butino y del (S)-3-amino-1-butino puros.
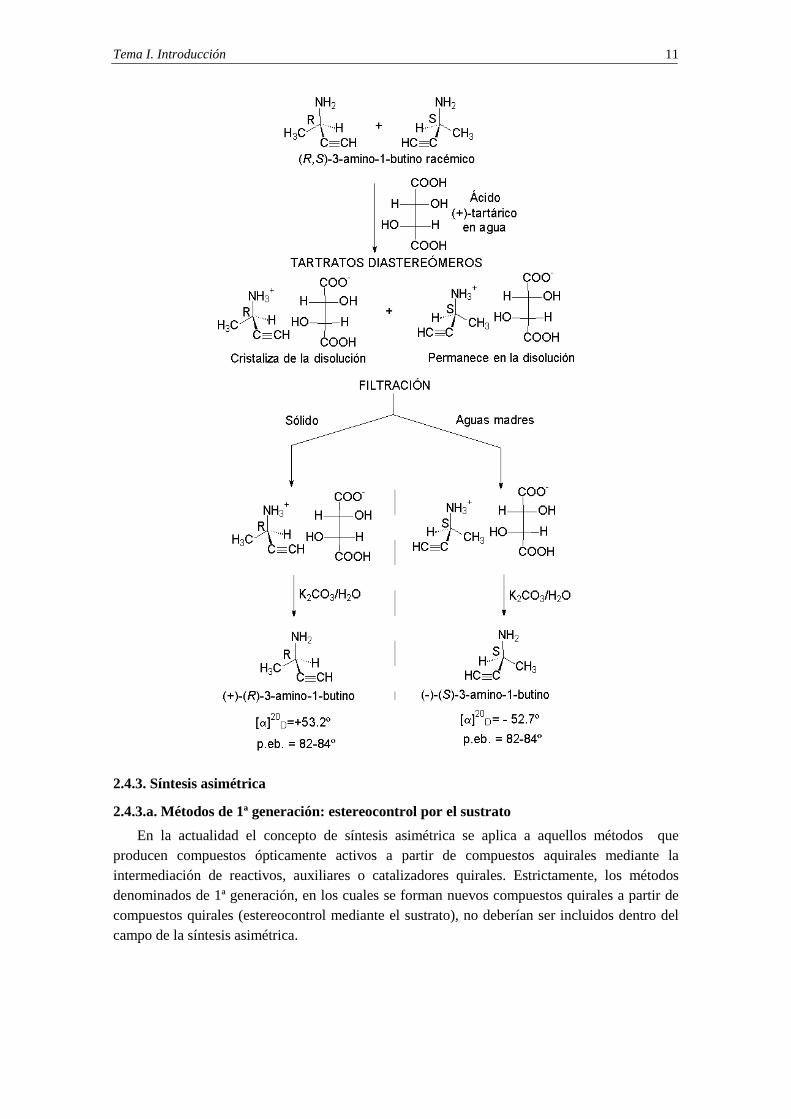
Tema I. Introducción 11
2.4.3. Síntesis asimétrica
2.4.3.a. Métodos de 1ª generación: estereocontrol por el sustrato
En la actualidad el concepto de síntesis asimétrica se aplica a aquellos métodos que producen compuestos ópticamente activos a partir de compuestos aquirales mediante la intermediación de reactivos, auxiliares o catalizadores quirales. Estrictamente, los métodos denominados de 1ª generación, en los cuales se forman nuevos compuestos quirales a partir de compuestos quirales (estereocontrol mediante el sustrato), no deberían ser incluidos dentro del campo de la síntesis asimétrica.

Metodología síntetica 12
Un ejemplo de método de síntesis asimétrica de 1ª generación lo constituye la adición del yoduro de metilmagnesio a la (S)-2-metilciclohexanona. En esta reacción se obtiene mayoritariamente el producto resultante del ataque nucleofílico a la cara Si del grupo carbonilo:
2.4.3.b. Métodos de 2ª generación: estereocontrol mediante el empleo de un auxiliar quiral
En estos métodos el estereocontrol lo ejerce también un centro estereogénico presente en un sustrato quiral, obtenido mediante una reacción previa entre un sustrato aquiral y un componente quiral. Después de la reacción asimétrica el componente quiral se libera del producto de la reacción.
Un ejemplo de esta clase de métodos es el que se emplea en la α-alquilación enantioselectiva de la ciclohexanona (sustrato aquiral) mediante el empleo del (S)-fenilalaninol (auxiliar quiral). Así, la reacción entre la ciclohexanona y el (S)-fenilalaninol forma una imina quiral, la cual se alquila estereoselectivamente mediante metalación y subsiguiente alquilación con yoduro de metilo. La eliminación del auxiliar quiral proporciona la (S)-2-metilciclohexanona ópticamente activa:
2.4.3.c. Métodos de 3ª generación: estereocontrol por medio del reactivo
Estos métodos se basan en la reacción de un sustrato aquiral con un reactivo quiral. La ventaja de estos métodos con respecto a los de segunda generación es que ahorran la etapa de instalación y desinstalación del auxiliar quiral.
Un ejemplo de esta clase de métodos es el que se utiliza en la reducción enantioselectiva de la acetofenona con el complejo quiral geenrado in situ al reaccionar el LiAlH4 con el aminoalcohol quiral denominado Darvon ((2S,3R)-dimetilamino-1,2-difenil-3-metil-2-butanol.
Cuando la acetofenona reacciona con el complejo reductor quiral, simbolizado en el siguiente esquema como Al(L*2)H2
-, se generan dos estados de transición diastereomórficos, y
por tanto de diferente energía. Como la reacción está sometida a control cinético, el producto mayoritario que se obtiene en la reacción es el que se forma a través del estado de transición de menor energía., que en este caso es el alcohol de configuración R.

Tema I. Introducción 13
Otro ejemplo de esta clase de métodos es la reacción de hidratación asimétrica del metilciclohexeno por hidroboración con el borano quiral derivado de (+)-α-pineno, que proporciona enantioselectivamente el (S,S)-2-metilciclohexan-1-ol.
BH2
CH3 CH31.
2. H2O2, NaOH OH
(S,S)-2-metilciclohexanol
2.4.3.d. Métodos de 4ª generación: estereocontrol mediante el catalizador
Estos métodos permiten la formación enantioselectiva del producto de la reacción mediante la combinación de un sustrato aquiral con un reactivo aquiral en presencia de un catalizador quiral.
Un ejemplo de esta clase de métodos es la adición conjugada de p-t-butiltiofenol a ciclohexenona en presencia de cantidades catalíticas del alcaloide cinconidina:

Metodología síntetica 14
Estos métodos son lo más eficientes en síntesis asimétrica puesto que en ellos se logra economía atómica, ya que el catalizador quiral no se consume en la reacción, y todos los átomos que forman parte del sustrato y del reactivo, ambos aquirales, entran a formar parte del producto de la reacción, que además se genera en forma quiral.
3. Síntesis de fármacos
Un fármaco, o principio activo, es una sustancia pura, químicamente definida, que se obtiene de fuentes naturales o mediante síntesis y que tiene una actividad biológica que puede ser utilizada con fines terapéuticos o de diagnóstico.
Un medicamento es un preparado que contiene uno o varios principios activos y uno o más excipientes.
Un receptor es una subestructura de un biopolímero, ya sea un enzima, un ácido nucleico, un canal iónico, etc, cuya interacción con una molécula endógena o exógena produce una respuesta biológica. La mayoría de los receptores se encuentran en las membranas celulares, pero también los hay intracelulares e incluso intranucleares.
3.1. Nomenclatura de los fármacos
La gran mayoría de los fármacos son compuestos orgánicos y sus nombres sistemáticos IUPAC son, en general, demasiado complejos para ser útiles a la hora de nombrarlos. En su lugar se emplea la nomenclatura DCI, Denominación Común Internacional.
Los nombres IUPAC y DCI de los fármacos son nombres sin propietarios y pueden ser utilizados libremente, en contraposición a los nombres registrados, que son propiedad de la compañía farmacéutica que los produce. Por ejemplo, el donezepilo es el nombre DCI de un fármaco inhibidor del enzima acetilcolinesterasa, que se emplea para mejorar las funciones cognitivas de los enfermos de Alzheimer.
N
OMeO
MeO
Nombre DCI = Donezepilo
Nombre IUPAC = 2-((1-bencilpiperidin-4-il)metil)-5,6-dimetoxi-2,3-dihidro-1H-inden-1-ona
La compañía farmacéutica Eisai le asignó el nombre de fabricante E-2020 y registró el medicamento que contiene donezepilo con el nombre de Aricept®.
Conviene destacar que el nombre registrado lo es del medicamento. Es muy usual que el mismo principio activo se comercialice bajo diversos nombres registrados, porque forma parte de distintos medicamentos en los cuales puede estar asociado con otros principios activos o con otros excipientes. Por ejemplo, el naproxeno es el nombre DCI de un principio activo con actividad antiinflamatoria, cuya sal sódica recibe los siguientes nombres registrados: Aleve®,

Tema I. Introducción 15
Anaprox®, Antalgin®, Apranax®, Axer Alfa®, Flanax®, Gynestrel®, Miranax®, Naprelan®, Primeral®, Synflex®.
3.3. La importancia de la quiralidad
Muchos de los fármacos actualmente comercializados lo son en forma de racematos, es decir mezclas de enantiómeros en igual proporción. Cuando cada uno de los enantiómeros que componen el racemato interacciona con el receptor, que es un compuesto quiral, se generan situaciones diastereomórficas, por tanto de diferente energía. Es muy usual que una de las interacciones sea más favorable que la otra y, en consecuencia, las respuestas biológicas pueden ser diferentes, tanto por lo que hace a su actividad como a su magnitud.
El enantiómero más activo se denomina eutómero. El menos activo de denomina distómero. El cociente eudísmico es el cociente entre la afinidad del eutómero y del distómero, y es una medida de la diastereoselectividad del compuesto.
Las fases de la acción biológica de un fármaco son la exposición, la farmacocinética y sobre todo la farmacodinámica, en la cual se produce la interacción del fármaco son su receptor:
La enantioselectividad es clave en la fase farmacodinámica, pero también en la fase farmacocinética ya que es posible que uno de los enantiómeros de un fármaco, administrado en forma racémica, pueda metabolizarse selectivamente dando lugar a metabolitos potencialmente tóxicos o con efectos secundarios no deseados, considerándose el distómero de una mezcla racémica como una impureza no deseable.

Metodología síntetica 16
3.1. Distómeros que dan lugar a metabolitos con efectos secundarios
La prilocaína es un anestésico local y su enantiómero R se metaboliza rápidamente por hidrólisis enzimática originando o-toluidina que produce metahemoglobinemia, una enfermedad que se caracteriza por el elevado contenido de metahemoglobina, la hemoglobina en sangre que contiene el Fe en estado de oxidación +3 en lugar de +2 y por tanto, no contribuye al transporte de oxígeno.
El enantiómero S, por el contrario, se hidroliza lentamente y no tiene efectos secundarios.
El deprenilo es un fármaco que se utiliza en el tratamiento de la depresión y de la enfermedad de Parkinson. Uno de los metabolitos del deprenilo es la metanfetamina, que es un estimulante del Sistema Nervioso Central y crea adicción. Si se administra deprenilo racémico el metabolismo humano lo transforma en metanfetamina racémica. Sin embargo, si se administra el (-)-deprenilo, enantiómero levorrotatorio, se genera (-)-metanfetamina que es mucho menos adictiva que la (+)-metanfetamina. En la actualidad el deprenilo se administra en su forma levorrotaria bajo el nombre de selegilina.
3.2. Distómeros sin efectos secundarios
Los bloqueantes β-adrenérgicos tienen estructura general de ariloxipropanoaminas y contienen un único centro estereogénico en sus estructuras. El efecto terapéutico se debe casi
Tema I. Introducción 17
exlusivamente al enantiómero S. El enantiómero R no tiene efectos secundarios y los β-bloqueantes se venden casi todos ellos en forma de racematos.
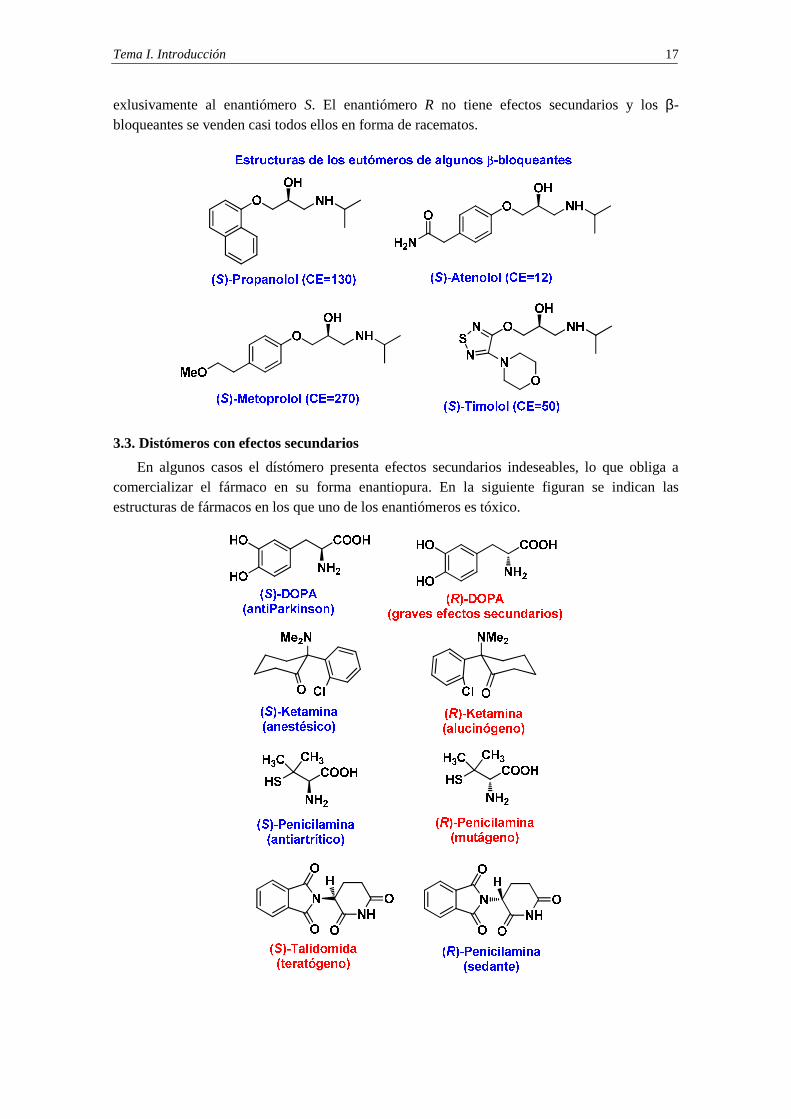
3.3. Distómeros con efectos secundarios
En algunos casos el dístómero presenta efectos secundarios indeseables, lo que obliga a comercializar el fármaco en su forma enantiopura. En la siguiente figuran se indican las estructuras de fármacos en los que uno de los enantiómeros es tóxico.

Metodología síntetica 18
La talidomida constituye el ejemplo más dramático de fármaco cuya administración y consumo en forma de mezcla racémica ha causado un gravísimo problema de salud. La talidomida se comercializó, en forma de racemato, entre los años 1958 y 1963 como sedante y como calmante de las náuseas durante los tres primeros meses de embarazo (hiperémesis gravídica).
Como sedante tuvo un gran éxito popular ya que no causaba casi ningún efecto secundario y en caso de ingestión masiva no era letal. Este medicamento, producido por Chemie Grünenthal, en Alemania, provocó miles de nacimientos de bebés afectados de focomelia, anomalía congénita caracterizada por la carencia o excesiva cortedad de las extremidades.
La talidomida afectaba a los fetos cuyas madres habían tomado el medicamento durante el embarazo, pero también afectaba a los fetos cuyos padres habían consumido talidomida durante el periodo de gestación, ya que la talidomida también afecta al esperma y transmite los efectos nocivos ya en el momento de la concepción. Cuando se comprobaron los efectos teratogénicos (malformaciones congénitas), se prohibió la venta de la talidomida.
3.4. Eutómero y distómero con funciones terapéuticas diferentes
En algunos casos los dos enantiómeros presentan actividades farmacológicas de interés, como el (2R,3S)-(+)-dextropropoxifeno, que es analgésico, y su enantiómero el (2S,3R)-(-)-levopropoxifeno, que es antitusivo.
3.5. Combinación del eutómero con el distómero con ventajas terapéuticas
El enantiómero R de la indacrinona es diurético, y como todos los diuréticos provoca retención de ácido úrico. Sin embargo, el enantiómero S actúa como uricosúrico, promoviendo la excreción de ácido úrico y antagonizando el efecto del enantiómero R.
La combinación ideal es una mezcla R/S en relación 9:1, mas que la combinación 1:1 de ambos enantiómeros, ya que es muy poco usual que ambos enantiómeros tengan la misma actividad cuantitativa.

Tema I. Introducción 19
3.6. Inversión de quiralidad provocada durante el metabolismo del fármaco
Algunos fármacos experimentan inversión de quiralidad in vivo, convirtiéndose el eutómero en el distómero, o viceversa. Es el caso de los ácidos α-arilpropiónicos, entre los que se encuentra el popular analgésico ibuprofeno. El proceso de inversión quiral. permite la transformación, en general unidireccional, del enantiómero R(-), inactivo, en la forma enantiomérica S(+), responsable de los efectos terapéuticos de los ácidos α-arilpropiónicos.1 Esta inversión de quiralidad tiene considerable implicación terapéutica ya que la eficacia anti-inflamatoria recae principalmente en el enantiómero de configuración S.2
En el caso del ibuprofeno el proceso de inversión de la configuración está controlado por el enzima acilcoenzima A sintetasa y convierte el (R)-ibuprofeno (distómero) en el (S)-ibuprofeno (eutómero). Así, el (R)-ibuprofeno reacciona con acilcoenzima A sintetasa y se convierte en el tioester del coenzima A. La racemización del tioéster, seguida de hidrólisis, forma el (S)-ibuprofeno.3
La hidrólisis del tioéster compite con un proceso de transesterificación con triacilgliceroles endógenos, que provoca acumulación de restos de (R)-ibuprofeno en los tejidos grasos. Hasta el momento no se sabe qué efectos puede tener en el organismo la acumulación de (R)-ibuprofeno en el organismo, aunque no pueden descartarse efectos tóxicos.
Por tanto, incluso en casos como el acabado de comentar, es muy recomendable la administración única del enantiómero activo.
1 Caldwell, J., Hutt, A. J.; Fournel-Gigleux, S. Biochem. Pharmacol. 1988, 37, 105-114. 2 (a) Mayer J. M.; Roy-De Vos M.; Audergon C.; Testa B.; Etter J. C. Int. J. Tissue React. 1994; 16, 59-72. (b) Hutt A.; Caldwell J. J. Pharm. Pharmacol. 1983; 35, 693-704. Ambos enantiómeros del ibuprofeno son igualmente efectivos, como analgésicos y antiinflamatorios, en modelos animales, a pesar de que el enantiómero-(S) es alrededor de 160 veces más activo que la forma (R) en la inhibición de la síntesis de prostaglandinas in vitro: Igarza L.; Soraci A.; Auza N.; Zeballos H. Vet. Res. Commun. 2002; 20, 29-37. 3 Nakamura Y.; Yamaguchi T.; Takahashi, S. J. Pharmacobio-Dynam. 1981; 4:S-1.