m. zandio, m. ferrín, m.j. cuesta · en la neurobiología molecular de los tras-tornos afectivos....
TRANSCRIPT

43ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
RESUMEN
El modelo neurobiológico en el que durantemuchos años se han basado la etiología, y por lotanto el tratamiento de la depresión, comprendíabásicamente alteraciones en el funcionamiento de losneurotransmisores, o en los receptores de los mis-mos. Sin embargo, investigaciones recientes hantransformado el escenario de la patofisiología de ladepresión, implicándose distintos niveles y sistemas,tanto nerviosos como endocrinos e inmunes, e inclu-so celulares, moleculares y genéticas. Desde estanueva perspectiva se pueden entender mejor los sín-tomas de la depresión y muchas de sus alteracionesneurobiológicas.
El presente trabajo pretende integrar de una mane-ra global los distintos mecanismos biológicos que sehan relacionado con la etiología de la depresión, parapermitir un nuevo abordaje conceptual de los trastor-nos depresivos y abrir nuevas posibilidades terapeúti-cas en el futuro.
Palabras clave: Depresión. Aspectos neurobioló-gicos. Trastornos afectivos. Afectividad. Patofisiolo-gía. Neuroquímica. Neurotransmisores. Serotonina.Noradrenalina. Neuropatología. Transductores de laseñal plasticidad neuronal.
ABSTRACT
During decades, both aetiology and treatment inDepressive Disorders relied on neurotransmisors’physiopathology or on abnomalities in their receptorsfunction. However, recently evidences from researchon neurobiological grounds suggest that there aremultiple and complex systems involved in thepathophysiology of Depressive Disorders. Severalneurobiological structures, such as the neural, inmuneand endocrine systems seems to interact amongthemselves and to influence on clinical manifestationsof illness. Moreover, disregulations on lower levels,such as intracellular and genetic systems might causeanomalities in protein expression, and in consequencemight modullate receptors’ disfunction anddisturbances at the intramolecular level of signaltransmission. The above distrubances at differentlevels of complexity are finally integrated within theframe of most recent theoretical approaches toDepressive Disorders. Specifically, recent theoriesimplicating neuronal plasticity and survival-death cellmechanisms are described.
The aim of this review is to integrate recentevidence on pathophysiological mechanisms ofDepressive Disorders. New lines of treatment basedupon these ‘new pathophysiology’ of depression willbe wellcome.
Keywords: Depression. Neurobiology. Mood disor-ders. Neurotransmitters. Neuronal plasticity.
Neurobiología de la depresión
Neurobiology of depression
M. Zandio, M. Ferrín, M.J. Cuesta
Correspondencia:Dr. Manuel J. CuestaUnidad de PsiquiatríaHospital Virgen del CaminoIrunlarrea, 431008 PamplonaE-mail: [email protected]
Unidad de Psiquiatría. Hospital Virgen del Camino
ANALES Sis San Navarra 2002; 25 (Supl. 3): 43-62.

M. Zandio et al.
44 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
INTRODUCCIÓN
Para comprender de forma adecuada lafisiopatología de los trastornos del humory no conformarnos con las interpretacio-nes reduccionistas previas, debemos teneruna visión global que abarque los distintosniveles fisiopatológicos de la enfermedad.Los niveles implicados van desde las alte-raciones moleculares, pasando por la dis-regulación de la neurotransmisión neuro-endocrinológica y neuroinmune, hasta lasmanifestaciones afectivas, cognitivas yconductuales de la enfermedad. Estos dife-rentes niveles neurofisiológicos interactú-an y conforman un complejo sistema quepermitiría explicar de una forma integral lafisiopatología de los Trastornos Afectivos1.Se cree que alteraciones en la expresióngénica, todavía sin identificar2, intervienenen la neurobiología molecular de los tras-tornos afectivos. A este nivel sí se están
empezando a describir alteraciones en elfuncionamiento de algunas proteínas, quedeterminan los mecanismos celulares yque producen alteraciones en el neurotro-fismo y la neuroplasticidad de determina-das poblaciones celulares, tanto neurona-les como gliales3,4. Con las nuevas técnicasde neuroimagen se han localizado algunasde estas poblaciones, postulándose que laalteración de determinados circuitos neu-ronales podría causar la sintomatología delos trastornos afectivos5. El esquema de lafigura 1 refleja de manera gráfica uno delos modelos fisiopatológicos de los tras-tornos afectivos, el propuesto por Manji ycol1 y modificado por los autores de estetrabajo. Representa los distintos nivelesfisiopatológicos implicados y nos sirve deesquema para ir desarrollando los diferen-tes niveles de disfuncionalidad neurobioló-gica presentes en la depresión.
CONDUCTA
SISTEMASNEURONALES
CELULAR
MOLECULAR
CogniciónAfectividadPsicomotricidad
Sistema NeuroendocrinoNeurotrasmisiónNeurotrofismoNeuroplasticidadRemodelación del citoesqueleto
CREBFactores de transcripciónmRNAImportación/exportación nuclear
Proteínas -GPKC & MARCKSGSK-3β & sustratosMAP KinasasCalcioFamilia de proteínas Bcl-2BDNF
CREB: cAMP response element binding protein (elemento unido a proteína en la respuesta AMPcícli-ca), mRNA: RNA mensajero, PKC: proteinkinasa C, GSK3ß: enzima glicogeno sintasa kinasa 3ß, BDNF:brain-derived neurotrophic factor (factor neurotrófico derivado del cerebro)
Figura 1. Modelo fisiopatológico (modificado de Manji y col, 2000).

45ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
RELACIONES MORFOFUNCIONALESDE LA SINTOMATOLOGÍA AFECTIVA
Los síntomas de la enfermedad depresi-va son la última expresión de las alteracio-nes que se producen en los distintos nive-les fisiopatológicos causantes de ladepresión. Expresan la alteración de variasfunciones psicológicas como, la afectividad(tristeza), la cognición (desesperanza), lapsicomotricidad (inhibición) y la conducta(hipoactividad). Durante años, neurólogos,psiquiatras y neuropsicólogos se han dedi-cado a buscar cuáles eran los sustratosneuroanatómicos de las funciones psicoló-gicas y dónde se localizaban las áreas y loscentros que intervenían en la elaboraciónde las funciones psicológicas del hombre.Tanto las descripciones de casos depacientes con lesiones cerebrales en áreasespecíficas presentes en la literatura médi-
ca desde inicios del siglo XX, como las nue-vas técnicas de neuroimagen (Tomografíade emisión de positrones-PET-), que permi-ten visualizar la activación de determina-das áreas cerebrales cuando al sujeto se lesomete a tareas o estímulos estipulados,han aportado gran información sobre estetema.
En la figura 2 se enumeran algunas delas áreas involucradas en la afectividadhumana. Entre ellas, las relacionadas conlos trastornos depresivos son el córtexprefrontal, el núcleo estriado, la amígdalay el hipotálamo. Se cree que el neocórtex yel hipocampo están involucrados en losaspectos cognitivos de la depresión, esdecir, en las ideas o sentimientos de culpa,de falta de autoestima, de desesperanza yautolíticas, y en las alteraciones de memo-ria. Lo cual se relaciona con el aumento
Figura 2. Neuroanatomía de la afectividad.

M. Zandio et al.
46 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
significativo del riesgo de depresiones enpacientes con antecedentes de lesionestumorales o vasculares a nivel del lóbulofrontal. Algunos autores defienden la exis-tencia de una especificidad regional, argu-mentando la existencia de un mayor riesgode depresión en aquellos pacientes coninfartos corticales pequeños y/o localiza-dos en la región frontal izquierda6. En losúltimos años, como se puntualizará másadelante, se han realizado grandes esfuer-zos para demostrar la posible relaciónetiológica entre la depresión de inicio tar-dío y la patología vascular cerebral, deno-minándose a este posible subtipo dedepresión “depresión vascular”7. Se definecomo aquella depresión que es debida amúltiples infartos cerebrales, con frecuen-cia “silentes”8, es decir sin síntomas neuro-lógicos. Parece ser que este nuevo concep-to tiene implicaciones etiológicas9, clínicas(criterios diagnósticos de Steffens y col10),terapéuticas (tratamiento de los factoresde riesgo vascular) y pronósticas (cursocrónico y recurrente) que difieren delresto de depresiones.
Dentro del córtex prefrontal se encuen-tra en el cíngulo anterior el área subge-nual. Los pacientes con lesiones en esteárea cerebral presentan alteraciones en larespuesta autonómica a estímulos emocio-nales, además de incapacidad para expre-sar emoción ante situaciones normalmen-te emotivas e imposibilidad para utilizarinformación sobre la probabilidad de cas-tigo o recompensa como guía en el com-portamiento social11. Hallazgos similaresse obtuvieron del estudio de ratas conlesiones bilaterales o derechas en áreasprelímbicas. Se objetivaba una atenuaciónde la respuesta autonómica, de la respues-ta corticoesteroidea y de la respuesta gás-trica a estresantes12-14. En cambio, las lesio-nes en el lado izquierdo producían locontrario. Ante estos hallazgos se postulóla hipótesis de que la región subgenualderecha facilitaba la expresión de la res-puesta visceral al estrés, y en cambio laregión izquierda modulaba esta respuesta.Apoyan esta hipótesis las numerosas cone-xiones recíprocas del área subgenual conotras estructuras, como el córtex orbital,el área tegmental ventral, la sustancianegra, los núcleos del rafe, el locus coeru-
leus, la sustancia gris periacueductal y elnúcleo del tracto solitario15,16. También enratas se ha estudiado la posible interven-ción de este área en el comportamientocondicionado por las percepciones emoti-vas17.
En el cíngulo anterior se encuentratambién el área pregenual, que si se esti-mula con electricidad produce miedo,pánico y presentimientos18. Estudios reali-zados del córtex orbital prefrontal sugie-ren su relación con la modulación del com-portamiento, más específicamente en larespuesta cognitiva de defensa al miedo yel comportamiento dirigido a la recompen-sa. Se ha observado un aumento del flujocerebral en la zona posterior del córtexorbital cuando se induce a individuossanos tristeza, pensamiento obsesivo yansiedad. Pacientes con lesiones en el cór-tex orbital presentan un deterioro en laplanificación de las tareas que requiereninformación relacionada con la recompen-sa y el castigo, y muestran dificultades enconfigurar estrategias ante circunstanciascambiantes, perseverando en actitudesinadecuadas19,20. Se cree que en la depre-sión este área pueda estar relacionada conla respuesta emocional excesiva a estre-santes y la ideación obsesiva.
Con respecto a las áreas dorsomedial ydorsoanterolateral del córtex prefrontal sepostula que cuando se activan, modulan laexpresión emocional, disminuyendo laansiedad y la frecuencia cardíaca. Ratascon lesiones en el área homóloga presen-tan una respuesta cardíaca exagerada a unestímulo condicionado negativo.
También en los Trastornos Afectivos sehallan lesiones en áreas que no estándirectamente relacionadas con el procesa-miento emocional, ya que debemos teneren cuenta que en la depresión existen sín-tomas de tipo cognitivo, psicomotor y neu-rovegetativo. Se ha visto que las áreas dor-solateral del córtex prefrontal y el áreadorsal del cíngulo anterior están relaciona-das con el lenguaje, la atención, la memo-ria, la función visoespacial y la memoria.
El núcleo estriado, en especial el estria-do ventral o núcleo accumbens, quedurante años se había involucrado en losmecanismos de recompensa del consumo

47ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
de tóxicos, se ha visto que tiene un papelimportante en la regulación del humor. Enél se encuentran neuronas dopaminérgicasdel sistema dopaminérgico mesolímbico yse ha objetivado un aumento de la trans-cripción mediada por CREB (cAMP respon-se element binding protein o proteína ligadaa la respuesta AMPcíclica), en respuesta alestrés agudo o crónico, que como másadelante veremos es un factor de trascrip-ción que se ha involucrado en los meca-nismos moleculares de la depresión.
La amígdala condiciona de forma fun-damental la respuesta a estímulos temero-sos y a estímulos agradables, es decir ela-bora la memoria emocional.
Por último, parece ser que es el hipotá-lamo quien media los síntomas neurovege-tativos, como son el sueño, el apetito, laenergía y la líbido. Una de sus característi-cas fundamentales es su papel de coordi-nador de las distintas funciones neuroen-docrinas y neurovegetativas que se alteranen la depresión (hiperactividad del ejeHipotálamo-Hipofiso-Adrenal [HHA], hipo-tiroidismo subclínico, alteraciones en lafunción de la hormona del crecimiento yde la prolactina)21.
AVANCES EN LA NEUROIMAGEN YEN LA NEUROANATOMÍA EN LOSTRASTORNOS AFECTIVOS
Aunque se sospechaba que algunas deestas áreas estaban involucradas en lafisiopatología de la depresión, hasta hacemenos de una década desconocíamos siexistía un sustrato anatomopatológico quejustificase la disfunción de las distintasáreas cerebrales implicadas en los trastor-nos del humor. Los avances en el área de laneuroimagen han permitido hallar altera-ciones morfológicas que sugieren ser posi-bles correlatos neuroanatómicos de algu-nas enfermedades mentales como laesquizofrenia, el trastorno obsesivo-com-pulsivo, la depresión, etc. Además hanorientado en los últimos años los estudiosneuroanatómicos hacia la búsqueda de lossustratos neurohistológicos en las áreascerebrales que se mostraban alteradas enlas técnicas de neuroimagen. Fundamen-talmente dos han sido las técnicas implica-das en la búsqueda: el PET y la RMF (Reso-
nancia magnética funcional). El PET cere-bral es una técnica que estudia el flujo vas-cular y el metabolismo de la glucosa en lasdiferentes regiones cerebrales mediante elmarcaje con radioisótopos. La RMF permi-te objetivar alteraciones de la neuromorfo-logía y neuromorfometría cerebral. No obs-tante, la interpretación de estos hallazgoses muy compleja, ya que las alteracionesde los parámetros que miden (Flujo Cere-bral Vascular FCV, cambios en la morfolo-gía, etc.), pueden tener como correlato laafectación de uno o varios niveles de laneurofisiología cerebral. Es decir, cambiosen la neurotransmisión (cambios en lasconcentraciones de neurotransmisores,alteraciones en la función de los recepto-res), alteraciones a nivel neuropatológico(disminución del número de células, desinapsis) o alteraciones a nivel vascular22-25.
A diferencia de otras patologías consustrato neuroanatómico, como son laEnfermedad de Parkinson o la Corea deHuntington, donde las alteraciones neuro-anatómicas se localizan en una regiónespecífica del cerebro, en la depresión secree que están involucradas múltiplesáreas que interrelacionan entre sí produ-ciendo los síntomas de la enfermedad.Aunque en los últimos años se han locali-zado varias áreas que pueden estar impli-cadas en la fisiopatología de la depresión,todavía desconocemos cómo funcionan ycómo se interrelacionan formando diferen-tes circuitos.
Las técnicas de neuroimagen han seña-lado al córtex cingulado anterior del cór-tex prefrontal, que tiene la capacidad demodular la respuesta emocional, como unade las áreas cerebrales probablementealteradas en la depresión, además de laamígdala, el núcleo estriado y el tálamo.
Los hallazgos más representativos enel córtex prefrontal a nivel del córtex cin-gulado, se localizan fundamentalmente encuatro áreas: la región subgenual del cín-gulo anterior, el área pregenual, tambiénsituada en el cíngulo anterior, las regionesorbital y ventrolateral del córtex prefron-tal, y por último, las áreas dorsolateral delcórtex prefrontal y dorsal del cíngulo ante-rior.

M. Zandio et al.
48 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
Se ha descrito una disminución signifi-cativa del tamaño de la región subgenualsg24 del cíngulo anterior en pacientes conTrastorno Depresivo Familiar (TDF) conrespecto a los controles26,27. También se hademostrado en unipolares y bipolares unadisminución del flujo vascular y del meta-bolismo de la glucosa en esta misma área yen el área pregenual de pacientes diagnos-ticados de Trastorno Depresivo Mayor(TDM)28. Se pensó que este último paráme-tro podría ser un factor predictivo de res-puesta al tratamiento29,30, pero los hallaz-gos han sido contradictorios.
En cuanto a las regiones orbital y ven-trolateral del córtex prefrontal se objetivóun aumento del metabolismo y del flujosanguíneo cerebral en pacientes no medi-cados con Trastorno Afectivo Primario(TAP)31-35. Se ha demostrado que esteaumento es reversible con tratamientoantidepresivo eficaz28,29,34,36,37.
A pesar de que el área dorsolateral delcórtex prefrontal y el área dorsal del cín-gulo anterior son áreas que no se hanimplicado en el procesamiento emocional,presentan una disminución de metabolis-mo y de flujo vascular reversible en lospacientes con trastornos del humor28,36,38.Parece ser que esta disminución de activi-dad pueda estar relacionado con en el défi-cit de atención, deterioro de la memoria yde la función visuoespacial en los pacien-tes con trastornos del ánimo38.
Otra área de gran interés en la patofi-siología de la emoción es la amígdala,donde se ha objetivado un incrementoanormal del flujo sanguíneo vascular y delmetabolismo de la glucosa en pacientesdiagnosticados de TDF y de TrastornosBipolares (TB) tipo I y tipo II sin síntomaspsicóticos34,39,40. Con el tratamiento conantidepresivos el metabolismo de la amíg-dala disminuye a niveles normales28. Portodo ello se postula que la actividad anó-mala de la amígdala pueda correlacionar-se con la severidad de los EpisodiosDepresivos Mayores (EDM) y con una sus-ceptibilidad hacia la recurrencia del tras-torno34,39,41.
Respecto a las alteraciones morfomé-tricas del hipotálamo los resultados soncontradictorios, ya que la disminución de
volumen que se objetivó en el TDM en losprimeros estudios, no se ha corroboradoposteriormente23,28.
Price y col18 describieron la existenciade un gran número de conexiones de laamígdala y las áreas orbital, ventrolateral ysubgenual del córtex prefrontal con elnúcleo mediodorsal del tálamo y el núcleoventral del estriado. Mediante el PET se haobjetivado en el núcleo mediodorsal detálamo y en el núcleo ventral del estriadoun incremento anormal del flujo sanguíneocerebral y del metabolismo en individuosdiagnosticados de TDM y TB34,39. En cam-bio, se ha observado una disminución delos dos parámetros en el caudado depacientes con TDM31. Tanto el caudadocomo el estriado ventral se encuentrandisminuidos de volumen tanto en los estu-dios con RMF como en los estudios post-mortem de pacientes con TDM42,43.
Por último, uno de los hallazgos de lastécnicas de neuroimagen que más ha apor-tado al conocimiento de los mecanismosfisiopatológicos de la depresión, ha sido elestudio de las alteraciones neurofisiológi-cas de los pacientes con Depresión Mayorde inicio tardía (primer episodio despuésde los 55 años). Se han descrito con la RMFun mayor número y un mayor tamaño deseñales focales hiperintensas en T2 en lasustancia blanca periventricular, lacunar yprofunda del córtex y en el núcleo estria-do43. Además en estudios anatomopatoló-gicos post-mortem se han encontrado alte-raciones propias de una isquemia en lasmismas áreas22,44 y de una disminución delflujo en el PET24,44. Todo esto sugiere queeste subgrupo de trastorno afectivos,como se ha señalado anteriormente,pueda ser secundario a la enfermedadcerebrovascular45. Algunos autores postu-lan que el notable enlentecimiento cogniti-vo que se observa en sujetos depresivosancianos pueda ser consecuencia de lainterrupción de las conexiones axonalesfronto-temporales46,47. Además con RMF seha objetivado una estrecha relación entretrastornos del humor familiares y el núme-ro y severidad de señales focales hiperin-tensas en T2 en la sustancia blanca pro-funda48,49, es decir lesiones similares, encuanto a localización y morfología a lasdescritas en depresiones tardías. La dife-

49ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
rencia es la naturaleza no vascular deestas lesiones en los trastornos afectivosde inicio temprano. Se cree que la presen-cia de estos hallazgos puede conferir unavulnerabilidad para los trastornos de tipoafectivo.
Los hallazgos proporcionados por lastécnicas de neuroimagen en los trastornosafectivos han impulsado de forma notablela realización de investigaciones en elámbito de la neuroanatomía con el fin deencontrar los correlatos histológicos delas alteraciones neurorradiológicas de lasdiferentes áreas cerebrales afectadas.
En la tabla 1 se resumen los hallazgosneurohistológicos de los estudios másrelevantes que se han llevado a cabo en elámbito de la neuroanatomía en los últimosaños50. A diferencia que en las enfermeda-des neurodegenerativas en las que seobserva el fenómeno de la gliosis (prolife-ración e hipertorfia de la glía), en los tras-tornos del humor no se produce un proce-so degenerativo convencional, sino una
disminución de la glía, en especial de losastrocitos, y una disminución del tamañoy/o número de algunas poblaciones neuro-nales. Se conoce que los astrocitos estánimplicados en la migración neuronal, en lasinaptogénesis, en la neurotransmisión, enla plasticidad sináptica y en el manteni-miento de la estructura neuronal51-57. Porotra parte, los astrocitos contribuyen deforma importante en la señal del PET y laRMF58 y poseen también receptores ytransportadores en su membrana59,60. Loanteriormente expuesto nos puede hacerpensar como postularon Rajkowska y col61
que el déficit de glía sea el evento patoló-gico central de la enfermedad, causante delas alteraciones neuronales, sinápticas ydendríticas, pero como Cotter y col62 pos-teriormente expusieron pueden existirotras posibles explicaciones y secuenciasde eventos. Tanto la glía como la morfolo-gía y el número de neuronas están influen-ciadas por factores ambientales y por laexpresión génica, pero hasta la fecha des-conocemos si las alteraciones neuroanató-
Tabla 1. Hallazgos neuroanatómicos de los trastornos del humor.
DM: Depresión mayor. TB: Trastorno Bipolar
AREA CEREBRAL HALLAZGOS NEUROANATÓMICOS
CÓRTEX PREFRONTAL
SUBGENUAL Disminución del número y la densidad de las células gliales en la DM y el TB. Ongür y col (1998)109
PREGENUAL Disminución de la densidad de las neuronas no piramidales en lámina II. Nueronas piramidales aumentadas de tamaño enlámina II. No alteraciones en la glia. Benes y col (2001) 110
SUPRAGENUAL En DM disminución de la densidad de la glia y reducción deltamaño de las neuronas.No cambios en la densidad de lasneuronas. No alteraciones en el TB. Cotter y col (2001) 62
DORSOLATERAL/ORBITAL Disminución de la densidad de la glía y reducción del tamaño de las neuronas. Rajkowska y col en DM (1999)111 y en TB (2001)61
Disminución del número de astrocitos en los DM menores de 45 años. Miguel- Hidalgo y col (2000) 112
HIPOCAMPO Disminución de la densidad de la arborización y de las espinasde las dendritas apicales. Rosoklija y col (2000) 113
No diferencias significativas entre los dos grupos. Lucassen ycol (2001) 107
No alteraciones en las proteínas de la sinapsis. Müller y col (2001) 108
HIPOTÁLAMO Aumento del número de neuronas en el núcleo paraventricular.Zhou y col (2001) 114
LOCUS COERULEUS Y NÚCLEOS DEL RAFE No se han encontrado alteraciones consistentes. Baumann ycol (1999)115
SUSTANCIA BLANCA SUBCORTICAL No existen estudios consistentes

M. Zandio et al.
50 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
micas descritas son un epifenómeno de laenfermedad o son causantes de la enfer-medad. Tampoco se han establecido alte-raciones neuroanatómicas comunes y/odiferenciadoras entre el TDM y el TB, y porúltimo se desconoce la evolución en eltiempo de las alteraciones histológicasdescritas, si son estables, reversibles oprogresivas.
Cabe señalar que una de las necesida-des importantes en el estudio de la neuro-biología de la depresión es conocer mejorlos circuitos cerebrales que regulan elhumor y sus interconexiones. Dos circui-tos se han involucrado, el límbico-talámi-co-cortical, que comprende la amígdala, eltálamo medial y las áreas orbital y medialdel córtex prefrontal, y el sistema límbico-cortical-estriatal-pálido-talámico, que com-prende áreas del circuito límbico-talámico-cortical que se interrelacionan con elnúcleo estriado y el pallidum.
ALTERACIONES EXTRACELULARESDE LOS TRASTORNOS AFECTIVOS
Los avances tecnológicos y en la meto-dología de investigación han permitidoestudiar con más precisión los supuestosmodelos fisiopatológicos y descubrir deforma simultánea multitud de nuevosmecanismos también relacionados con laenfermedad. Se han dedicado importantesesfuerzos en dilucidar si los diferentesmecanismos descritos podían ser factorescausantes de la enfermedad, pero en losúltimos años, como proponen Manji y col1,se ha desarrollado una nueva manera deorientar las investigaciones que consisteen identificar la contribución de estoshallazgos en el complejo sistema que con-forma la neurobiología de los TrastornosAfectivos. Esta nueva tendencia implicauna ardua tarea de estudiar y delimitar lasrelaciones entre los diferentes sistemas yniveles que conforme un sistema capaz deexplicar la fisiopatología de los TrastornosAfectivos.
En las décadas de los 60 y 70 se dedicóun gran esfuerzo al estudio de los aspectosextracelulares de la transmisión sináptica,ya que se creía que éste era el sustratoprincipal de la fisiopatología de los Tras-tornos Afectivos. Se describieron altera-
ciones en los sistemas de neurotransmi-sión, principalmente de noradrenalina(NA), serotonina (5HT) y dopamina (DA), ala par que se desarrollaban agentes tera-péuticos que incidían a este nivel. Pero enestos últimos años se ha visto que la causaúltima de los Trastornos Afectivos esmucho más compleja que el simple hechode una alteración en la actividad de los sis-temas de neurotransmisión. Se propusoque las monoaminas no tenían un efectodirecto sobre la regulación del humor, sinoque tenían un papel fundamental en lamodulación de otros sistemas neurobioló-gicos implicados en la recuperación de ladepresión.
Se cree que la disregulación del siste-ma noradrenérgico media algunos de lossíntomas fundamentales de la depresión,como déficit atencional, dificultades deconcentración y de memoria, aislamientosocial y estados de excitación. Su funciónes coordinar la respuesta central y perifé-rica precoz al estrés63. Se han relacionadocon la disfunción del sistema noradrenér-gico tanto un aumento de la frecuencia dedescarga como una disminución de la des-carga noradrenérgica. Esto produce unaalteración en la sensibilidad de los recep-tores y en las interacciones con otros sis-temas moduladores, resultando una modu-lación noradrenérgica postsinápticainefectiva64. En la modulación del sistemanoradrenérgico están involucrados otrasáreas cerebrales además del locus coeru-leus (córtex, tálamo, núcleo del rafe, hipo-campo) y otras sistemas como el GABAér-gico, glutamatérgico, de las encefalinas,neuropeptídicos, etc. Está descrita unarelación recíproca entre el sistema seroto-ninérgico y noradrenérgico65,66 y su relacióncon el eje HHA, como mediadores de la res-puesta sistémica al estrés67.
Se han descrito que predisponen apadecer una Depresión Mayor alteracionesde la actividad presináptica serotoninérgi-ca y alteraciones en los receptores postsi-nápticos 5-HT2 y 5-HT1A, y también se haobjetivado una relación recíproca entre lasalteraciones del sistema serotoninérgico yel eje HHA68. Existen multitud de hallazgosque apoyan la influencia del sistema sero-toninérgico en la fisiopatología de la depre-

51ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
sión, como por ejemplo, la posibilidad deinducir sintomatología depresiva con téc-nicas que depleccionan el L-triptófano, elprecursor de 5-HT69,70, las bajas concentra-ciones de L-triptófano, 5-HT y un metaboli-to de la 5-HT, 5-HIAA halladas en estudioshistológicos post-mortem de pacientes sui-cidas y el aumento de la concentración deL-triptófano con tratamiento antidepresi-vo. Además en la Depresión Mayor se obje-tiva un incremento del número y de la afi-nidad de los receptores postsinápticos5-HT271,72 y una regulación a la baja y desen-sibilización de los receptores 5-HT1A post-sinápticos. En cuanto a la relación con eleje HHA, se ha demostrado que la 5-HTestimula este eje a través de la activaciónde los receptores 5-HT1A y 5-HT2 que ac-túan sobre la Hormona estimulante corti-cotropínica.
Existen evidencias de que alteracionesen los mecanismos de adaptación al estrésestán involucrados en el desarrollo, trata-miento y prevención de los trastornos delhumor. El eje HHA es el sistema endocrinofundamental en la respuesta al estrés. Poruna parte, se ha objetivado en estudiosepidemiológicos recientes que la mayoríade los cuadros afectivos se asocian a unevento estresante vital y por otra, estábien establecido que en pacientes conDepresión Mayor, especialmente aquelloscon características melancólicas, presen-tan alteraciones significativas en el ejeHHA73. Estos pacientes exhiben un incre-mento de la actividad del eje HHA, unaumento del número de pulsos secretoresde hormona adrenocortico-trópica y unaumento de la magnitud de los pulsos decortisol. Se conoce que el efecto de losglucocorticoides consiste en la regulacióndel metabolismo general y del comporta-miento afectivo por medio de su accióndirecta en numerosas regiones cerebrales.La actividad del eje HHA está controladapor determinados circuitos cerebrales,que incluyen el hipocampo (que ejerce unainfluencia inhibidora sobre la síntesis deCRF, el factor estimulador corticoideo, enel núcleo paraventricular del hipotálamo)y la amígdala. Los glucocorticoidesmediante su efecto directo en el hipocam-po e hipotálamo realizan una retroalimen-tación negativa en el eje HHA. Se ha
demostrado que las concentraciones ele-vadas de corticoides mantenidas en eltiempo pueden dañar las neuronas delhipocampo, en particular las neuronaspiramidales CA3, produciendo una reduc-ción de la arborización dendrítica74,75 y unadisminución de la proliferación de neuro-nas granulares en el giro dentado, como seexplicará de forma más detallada poste-riormente. Este daño a nivel del hipocam-po conlleva una reducción de la retroali-mentación negativa sobre el eje HHA,manteniendo la elevada actividad gluco-corticoidea y causando un mayor dañohipocampal. Se cree que este deterioro anivel del hipocampo contribuye a las alte-raciones cognitivas de la depresión. Lahiperactividad del eje HHA no sólo contri-buye a la depresión por medio de la hiper-cortisolemia, sino que también influye elaumento de la transmisión CRH. Pero toda-vía se desconoce si la alteración del ejeHHA es una causa primaria de la depre-sión, o por el contrario es secundaria aotra causa inicial76. También otros siste-mas hormonales como el tiroideo y elsomatotrópico se han involucrado en lafisiopatología de la depresión.
ALTERACIONES CELULARES YMOLECULARES EN LOSTRASTORNOS AFECTIVOS
En los últimos años se ha realizado unestudio exhaustivo de los mecanismos detransducción de la señal implicados en larespuesta neuronal. En el SNC los mecanis-mos de transducción de señal intracelularson los responsables de coordinar la infor-mación y la respuesta celular, de tal mane-ra que dichas alteraciones molecularespueden conducir a un desequilibrio enmúltiples mecanismos neurotransmiso-res77. Esto podría explicar la variedad clíni-ca observada en los trastornos depresi-vos, así como el tiempo que los fármacosnecesitan para ejercer su acción antide-presiva.
Los procesos de transmisión sinápticaimplican mecanismos complejos de acti-vación celular, los cuales se han intentadorepresentar en la figura 3. Los neurotrans-misores se unen a una molécula de recep-tor transmembrana, que interactúa con
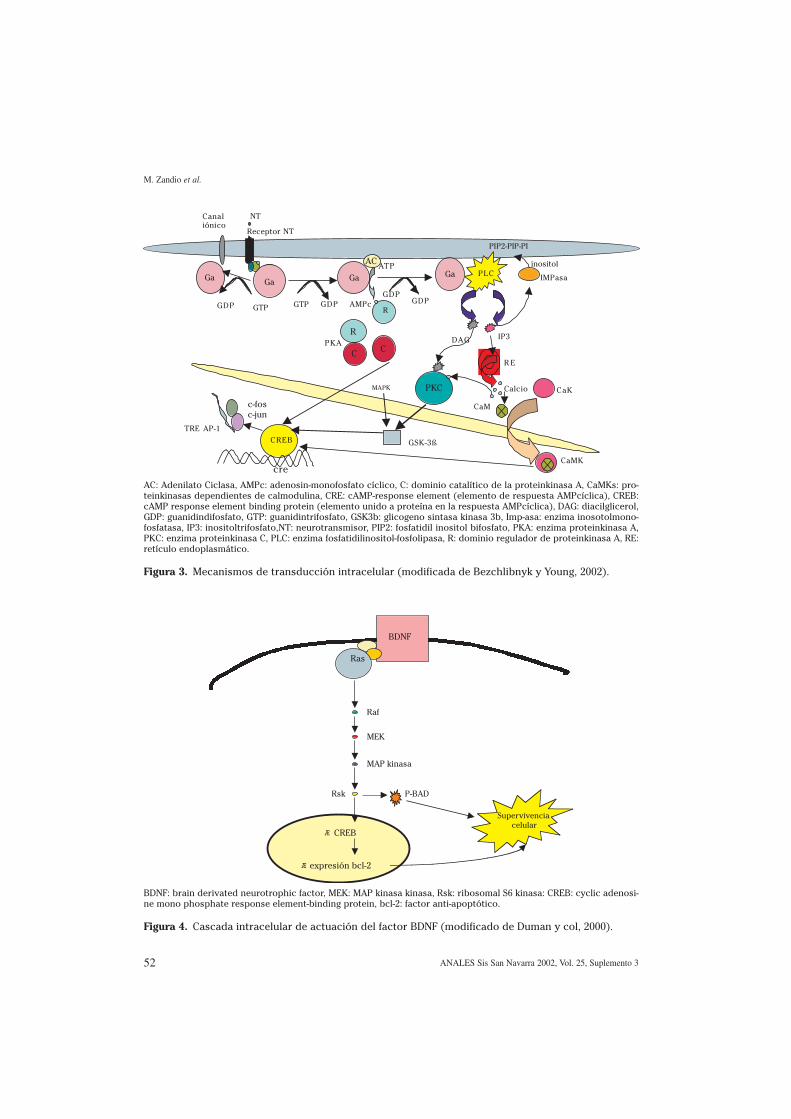
M. Zandio et al.
52 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
GaGaGa
Canaliónico
NT
AC
Ga
PKAC
GDPGTPGTPGDP
GDPGDP
R
C
AMPc
PLC
DAG IP3
IMPasa
inositol
PIP2-PIP-PI
R E
CalcioPKC
ATP
Receptor NT
CaK
CaM
CaMK
CREB GSK-3ß
MAPK
cre
R
c-fos
c-jun
TRE AP-1
AC: Adenilato Ciclasa, AMPc: adenosin-monofosfato cíclico, C: dominio catalítico de la proteinkinasa A, CaMKs: pro-teinkinasas dependientes de calmodulina, CRE: cAMP-response element (elemento de respuesta AMPcíclica), CREB:cAMP response element binding protein (elemento unido a proteína en la respuesta AMPcíclica), DAG: diacilglicerol,GDP: guanidindifosfato, GTP: guanidintrifosfato, GSK3b: glicogeno sintasa kinasa 3b, Imp-asa: enzima inosotolmono-fosfatasa, IP3: inositoltrifosfato,NT: neurotransmisor, PIP2: fosfatidil inositol bifosfato, PKA: enzima proteinkinasa A,PKC: enzima proteinkinasa C, PLC: enzima fosfatidilinositol-fosfolipasa, R: dominio regulador de proteinkinasa A, RE:retículo endoplasmático.
Figura 3. Mecanismos de transducción intracelular (modificada de Bezchlibnyk y Young, 2002).
BDNF
Ras
MEK
Raf
MAP kinasa
Rsk P-BAD
CREB
expresión bcl-2
Supervivencia
celularÆ
Æ
BDNF: brain derivated neurotrophic factor, MEK: MAP kinasa kinasa, Rsk: ribosomal S6 kinasa: CREB: cyclic adenosi-ne mono phosphate response element-binding protein, bcl-2: factor anti-apoptótico.
Figura 4. Cascada intracelular de actuación del factor BDNF (modificado de Duman y col, 2000).

53ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
proteínas G de membrana. Estas proteí-nas están compuestas por subunidades α,β y γ. La activación del receptor induce uncambio conformacional en el receptorasociado a la proteína G, como resultadode un intercambio de GDP (guanidindifos-fato) por GTP (guanidintrifosfato) en lasubunidad a. La subunidad α a su vez con-duce a la activación de canales iónicos oa la producción de segundos mensaje-ros78. Los niveles del subtipo estimuladorde la subunidad α (Gαs) parecen tenerrelevancia en los trastornos afectivos.Varios estudios realizados en células san-guíneas han encontrado una relaciónentre los niveles y las alteraciones funcio-nales de proteínas G con la presentaciónde síntomas depresivos79,80. La compleji-dad generada por las interacciones de losreceptores acoplados a proteínas G puedeser un mecanismo mediante el cual lasneuronas adquieren la capacidad de gene-rar la amplia gama de respuestas que seobservan en el sistema nervioso. De estamanera, las vías de transducción neuro-nal acopladas a proteínas G de membranaestarían implicadas en la regulación defunciones como el apetito, la debilidad oel estado de ánimo en las áreas cerebralescorrespondientes81.
La señal extracelular es posteriormen-te integrada, amplificada y transmitida aenzimas intracelulares específicas, llama-das efectoras. Los subtipos estimulador einhibidor de la proteína G se asocian a laenzima Adenilato Ciclasa, la cual catalizala formación de AMPcíclico, uno de lossegundos mensajeros más importantes,desde una molécula de ATP. La producciónde AMP cíclico es regulada por fosfodies-terasas de diversos tipos. El AMPc regulafunciones celulares, como el metabolismoy la transcripción génica. La principaldiana del AMPc es otra enzima, llamadaproteinkinasa A (PKA), formada por dossubunidades. La unión al dominio regula-dor (R) induce la disociación del dominiocatalítico (C). Esta enzima es crucial paraasociar los distintos cambios de señaliza-ción en neurotransmisores con los cam-bios neurobiológicos que se suceden82. Laactividad de esta enzima parece estar alte-rada en el córtex temporal de pacientescon trastornos afectivos83,84.
Otros neurotransmisores se asocian amecanismos de señalización que implicanla fosfatidilinositol-fosfolipasa (PLC), acti-vadas mediante la unión específica a iso-formas Gαq. La subunidad Gαq disocia yactiva la PLC, que cataliza la hidrólisis delfosfatidilinositolbifosfato (PIP2) a dosmensajeros: diacilglicerol (DAG) e inositol-trifosfato (IP3). El IP3 libera calcio al cito-sol desde el retículo endoplasmático. Porotro lado, la molécula de DAG es capaz deactivar la proteinkinasa C (PKC). Las molé-culas de inositol atraviesan con dificultadla barrera hematoencefálica, por lo que lascélulas deben mantener una cantidad sufi-ciente de mio-inositol para resintetizar elPIP2. Éste se obtiene de la desfosforilaciónde inositol fosfatos, donde la enzima inosi-tolmonofosfatasa (IMPasa) juega un impor-tante papel. Existen suficientes evidenciasacerca de la alteración de Gαq y PLC, tantoen tejidos periféricos como en estudiospost-mortem del córtex occipital enpacientes con alteraciones afectivas85,86.También la alteración de la PKC se ha vistoimplicada en estos procesos.
El calcio juega un papel también princi-pal en la mediación de distintos eventosintracelulares, que incluyen la plasticidadneuronal, la supervivencia celular y lamuerte celular87,88. Se ha observado cómomecanismos que implican cambios en losniveles de calcio intracelular conllevandiferentes cambios bioquímicos. El aumen-to de calcio procede tanto del exterior dela célula como de su retículo endoplasmá-tico, y es nuevamente regulado a la bajagracias a la hidrólisis del IP3. El gradientede calcio se mantiene gracias a bombas deCa/ATPasa o intercambiadoras de Na/Casituadas en la membrana celular. El calciose une a la calmodulina, y el complejoregula otras enzimas (como las proteinki-nasas dependientes de calmodulina, oCaMKs). Estudios que se refieren a las con-centraciones de calcio en plaquetas o lin-focitos de pacientes depresivos sugierenuna estrecha relación de éstos tanto con laclínica, como con los niveles de otras enzi-mas (por ejemplo, la PKC y los receptoresGα?). De esta manera, se ha sugerido queson los niveles alterados de calcio los quesecundariamente alteran los niveles deotras enzimas. También se han encontrado

M. Zandio et al.
54 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
en glóbulos rojos de pacientes deprimidosun aumento en la actividad de la Ca/ATPa-sa y un descenso de actividad en la bombaNa/K/ATPasa, que regula el intercambio deNa/Ca89.
La consecuencia final de la activaciónlas distintas vías antes descritas es la regu-lación de factores de transcripción. Uno delos factores más estudiados es el factorCREB. La activación de este factor se damediante la fosforilación de un aminoáci-do (Ser-133) por medio de distintas kina-sas (PKA, PKC, CaMK y elementos de lacascada MAPK). El factor fosforilado(pCREB) se une a un sitio específico de laregión promotora llamada CRE (cAMP-res-ponse element), que conduce a la expre-sión de RNA mensajero que regula la pro-ducción de la consiguiente proteína89. Lasfluctuaciones rápidas en los niveles deneurotransmisor y receptor asociadas a laalteración en la expresión de proteínaspueden alterar permanentemente la fun-ción o estructura de determinadas regio-nes cerebrales.
La glicógeno sintasa kinasa 3β (GSK3β)es otra proteína moduladora del factorCREB, cuya función es crucial en la regula-ción de los acontecimientos que se suce-den en el núcleo. Además de producir unajuste fino de este factor, está implicada enla regulación de microtúbulos, microfila-mentos, proteína básica de la mielina, fac-tor de crecimiento nervioso y proteína tauen el cerebro. Se han encontrado alteracio-nes en los niveles de esta enzima, así comode la proteína tau con relación a pacientescontrol.
En la regulación del factor CREB inter-vienen distintas neurotrofinas localizadasen la región del hipocampo. Una de las másinvestigadas es el denominado BDNF(brain-derived neurotrophic factor o factorneurotrófico cerebral). El BDNF juega unpapel principal en mecanismos de apren-dizaje y memoria. Es transportado desdelas neuronas granulares de la circunvolu-ción dentada a las neuronas piramidalesCA3. En ausencia de este factor, las célulasentran en procesos de muerte celular pro-gramada, o apoptosis. Las proteínas nece-sarias para la muerte celular son las cas-pasas, una familia de cisteín-proteasas que
se activa durante el proceso proteolítico.Por el contrario, otros factores como lasproteínas Bcl-2 inhiben la activación de lascaspasas, aumentando por tanto la super-vivencia celular. Finalmente el factor BADbloquea la acción del Bcl-2, conduciendo ala muerte celular.
Una de las vías intramoleculares acti-vadas por el BDNF es la cascada de la MAP-kinasa, gracias a la cual se fosforila y blo-quea el factor pro-apoptótico BAD (Fig. 4).El producto final de esta vía es la activa-ción la proteína CREB, encargada en laexpresión del factor antiapoptótico Bcl-2.Se cree que la expresión de este factorpuede conducir a la diferenciación neuro-nal, dependiendo de si las células expresanlos receptores de NA o 5HT apropiados. Deesta manera, la pérdida neuronal observa-da en la depresión puede deberse a altera-ciones de los factores que controlan lamuerte celular programada (por ejemplo,un descenso de Bcl-2 o un aumento de fac-tor BAD). Es en la regulación del CREB yBcl-2 donde se están centrando actualmen-te ciertas líneas de investigación de nue-vos tratamientos90.
¿INFLUYE LA PLASTICIDADNEURONAL EN LA PATOFISIOLOGÍADE LOS TRASTORNOS AFECTIVOS?
Se están realizando importantes esfuer-zos para dilucidar los mecanismos impli-cados en la remodelación de la citoarqui-tectura cerebral en la depresión. Por loque gran parte de las hipótesis actuales dela depresión se basan en lo que denominan“plasticidad neuronal”, que representaríala capacidad que tienen las células cere-brales para adaptarse a las distintas situa-ciones. De esta manera, la plasticidad nosólo quedaría determinada en el momentodel nacimiento, sino que persistiría nece-sariamente en el cerebro del adulto, refle-jando la interacción del sujeto con elmedio ambiente como fenómenos de géne-sis y muerte neuronal. Constituye unmecanismo necesario para la superviven-cia neuronal e incluye mecanismos deadaptación de señales intracelulares asícomo la expresión de determinadosgenes91.

55ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
La posibilidad de que los fallos en laplasticidad neuronal puedan contribuir aenfermedades neurodegenerativas es unplanteamiento relativamente reciente. Laszonas cerebrales que se han implicado ycoinciden con las detectadas mediantetécnicas de imagen serían las estructuraslímbicas, y más específicamente el hipo-campo, en la circunvolución dentada (neu-ronas piramidales CA3)76. Mediante meca-nismos intramoleculares, diversassituaciones (o sustancias actuando comonoxas), originarían una pérdida de plastici-dad neuronal, y secundariamente un des-censo en las funciones neurotróficas. Estoa su vez produciría un descenso en lasupervivencia neuronal, que conduciríanuevamente a la alteración de la plastici-dad neuronal. De esta manera el círculo ini-ciado por una noxa tendería a perpetuarsedentro de un círculo vicioso, en el que elresultado final sería la pérdida celular en lazona del hipocampo, neuronas piramida-les CA3 y neuronas granulares91.
En estos mecanismos parecen estarimplicadas distintas sustancias. Las másestudiadas han sido los glucocorticoides,como se ha expuesto anteriormente, y losaminoácidos excitadores, liberados antesituaciones de estrés vital. También se hanimplicado otras sustancias como la seroto-nina y los receptores GABA. Estas sustan-cias serían las responsables de modular laexcitabilidad de las neuronas del hipocam-po. Por ejemplo, diversos autores propo-nen que serían capaces de originar unainhibición de la neurogénesis92,93. Otromecanismo que se plantea sería una inte-rrupción de conexiones entre la circunvo-lución y la amígdala (alterando de estamanera procesos cognitivos como elaprendizaje y la memoria). También podrí-an ser los responsables de producir una“remodelación dendrítica” de las neuronasde esta zona, que consiste en un descensoen la longitud y arborización de las dendri-tas94,95, o de una pérdida de neuronas en elhipocampo en la depresión prolongada75.En la vida adulta, las neuronas granularesvan siendo reemplazadas, reguladas demanera continua por mecanismos de neu-rogénesis y apoptosis celular.
Los aminoácidos excitadores (AAE)actúan a través del receptor NMDA (N-
metil-D-aspartato) inhibiendo la neurogé-nesis en la circunvolución dentada. Entreestos AAE, el más potente descrito es elglutamato, y está implicado entre otros, enlos fenómenos de aprendizaje. De manerafisiológica, el glutamato se uniría al recep-tor NMDA para producir su activación. Através de este receptor entraría calcio enla célula, que también se liberaría de losorgánulos intracelulares. Esta movilizaciónde calcio citosólico sería responsable delos cambios en la excitabilidad sináptica,cambios que probablemente conforman la“memoria de aprendizaje”. Sin embargo,un exceso de glutamato origina que en lasinapsis éste se comporte de manera exci-totóxica, de tal manera que se originaríauna “hiperactivación” de las enzimasdependientes de calcio. Esto conduciría auna degradación del citoesqueleto de lasdendritas, a la malformación proteica y a laliberación de radicales libres, con la con-secuente muerte neuronal en esa zona. Enlos últimos años se observó que el excesode glutamato era capaz de inducir apopto-sis en determinados grupos neuronales, encondiciones de exceso de calcio y radica-les libres74-76.
El transporte de glutamato en la sinap-sis precisa de un cotransporte activo consodio, y por lo tanto de un exceso de ésteen el proceso antes descrito. De esta mane-ra, fenómenos que alteren la producciónde energía en esta zona (como la isquemia,hipoxia, hipoglucemia o epilepsia) altera-rán a capacidad del transportadorNa/K/ATPasa, y modificarán las concentra-ciones de sodio extracelulares. Se hademostrado que si bien el glutamato es unrequisito indispensable para que se inicienlos mecanismos de neurotoxicidad, luegono es necesario para que otros actúen.Esta convicción se basa en lo que se deno-mina como “fragilidad excitotóxica”, y serefiere al periodo durante el cual el equili-brio energético neuronal está tan compro-metido que niveles normales de glutamatopueden originar muerte celular por losmecanismos explicados96,97.
Los glucocorticoides intervienen demanera fisiológica en los mecanismos deconcentración y memoria. Sin embargo,como hemos señalado anteriormente, elexceso de éstos se ha comprobado neuro-

M. Zandio et al.
56 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
tóxico para las células del hipocampo, yalteraría estas funciones cognitivas. No seconoce exactamente si la neurotoxicidadvendría dada por la propia acción del cor-tisol en sí, o símplemente aumenta la capa-cidad neurotóxica de otras sustancias. Seha observado una interacción recíprocacon serotonina y con los AAE, en este últi-mo caso mediante múltiples vías (aumen-tando la expresión de NMDA en el hipo-campo, aumentando la concentraciónextracelular de glutamato en hipocampo,prolongando el tiempo de acción deestos)93,98. El cortisol sería capaz de aumen-tar los niveles de calcio citosólico, biendirectamente o secundariamente alaumento del glutamato. Sobre las propiasneuronas y células de la microglía, el corti-sol actúa inhibiendo moderadamente eltransporte de glucosa, de tal manera queese ligero descenso compromete la super-vivencia celular durante los eventos isqué-micos. Además el exceso de glucocorticoi-des originaría un descenso de la actividadde los antioxidantes, bloquearía la activi-dad de las proteínas neuroprotectoras ylimitaría la disponibilidad de NADPH pararesponder en los fenómenos de isquemia.
La serotonina también interaccionaríacon el glutamato, bien potenciando launión al receptor NMDA, o bien inducien-do la propia actividad de éste. Medianteneurotrazadores se ha podido investigar lafunción de los distintos receptores deserotonina, en concreto los más estudia-dos han sido el 5HT1A y 5HT2A. Asímismo, se ha sugerido que una actividaddisminuida en el receptor GABA-benzodia-cepínico, inhibidor de la sinapsis entrecélulas piramidales, puede influir en todoeste mecanismo. La gran variabilidad dehallazgos encontrados hasta el momentoindican que existe una gran heterogenei-dad de acción en lo referente a las basesfisiológicas de la depresión.
Se conoce que la glía tiene la función desoporte de las neuronas y de la transmi-sión sináptica99, por lo que se cree que ladisminución de la glía, que se ha objetiva-do en determinadas áreas cerebrales enlos Trastornos Afectivos, conlleva unareducción de la sinapsis asociada a unaretracción del neurópilo. El neurópilo es lared compleja y ordenada de prolongacio-
nes dendríticas, axónicas y gliales, cuyaestructura y relaciones están adaptadascon el fin de proporcionar un esqueletopara una actividad organizada. Se ha estu-diado que su volumen puede estar influen-ciado por varias circunstancias, como elaumento de concentración de aminoáci-dos excitadores y cortisol, y también porla disminución de la función de las neuro-trofinas, los receptores 5HT-1A, los recep-tores estrogénicos y otros factores quemantienen el citoesqueleto100,101. Tambiénse cree que la neurotransmisión glutama-érgica pueda influir en la inducción de alte-raciones en el neurópilo, ya que en Depre-siones Mayores existe un aumento de laactividad en la vía límbico-talámico-corti-cal, que está formada predominantementepor proyecciones glutamatérgicas34. Sonlos astrocitos quienes recaptan el glutama-to del líquido extracelular, por lo que lareducción de la astroglía descrita en lostrastornos del humor102, puede producir undeterioro en el transporte de glutamato.Además se cree que el efecto excitotóxicode las altas concentraciones de glutamatopueda estar facilitado por el aumento decortisol, que también se objetiva en lostrastornos del humor. Un hallazgo queapoya este posible mecanismo fisiopatoló-gico es que algunos agentes anticonvulsio-nantes efectivos en el Trastorno Bipolarreducen la transmisión glutamatérgica.
Con respecto a los receptores 5 HT-1A,se ha objetivado que la administración dedepleccionantes serotoninérgicos, antago-nistas de los receptores 5HT-1A producenuna pérdida de dendritas, espinas y sinap-sis en animales, efecto que se interrumpecon la administración de InhibidoresSelectivos de la Recaptación de Serotonina(ISRS) y agonistas de los receptores 5-HT-1A100. Se conoce que el tratamiento crónicocon antidepresivos aumenta la transmi-sión serotoninérgica y activa los recepto-res postsinápticos 5HT-1A. Se postula quemediante este mecanismo protejan de lareducción del neurópilo3,101,103-105.
El modelo de “plasticidad neuronal”que se acaba de exponer, une la vía de losfactores de crecimiento y de los segundosmensajeros con los efectos atróficos de losglucocorticoides y el estrés en las neuro-nas piramidales y sus dendritas3,76,106. Se

57ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
basa en el papel del hipocampo en la regu-lación del eje hipotálamo-hipofisario-adre-nal, con la hipercolesterolemia y la disfun-ción del eje HHA que ocurre en lostrastornos del humor. Toma como base elestrés como generador de patología psi-quiátrica mediante la alteración de losmecanismos fisiológicos de respuesta.Pero en contra de lo esperado no se hademostrado empíricamente a nivel neuro-patológico. Lucassen y col107 realizaron elprimer estudio neuropatológico sobre lahipótesis del estrés y la atrofia hipocam-pal. El modelo predecía una neurotoxici-dad mediada por la acción de los gluco-corticoides, asociado al estrés celular. Seutilizó una batería de marcadores inmuno-citoquímicos de estos procesos para com-parar un grupo de pacientes con trastornoafectivo, un grupo control normal y ungrupo de pacientes tratados con esteroi-des. En el grupo de los trastornos afectivossólo se observó un pequeño aumento delnúmero de células apoptóticas y los mar-cadores de estrés fueron negativos. Ade-más los resultados positivos no se locali-zaban en el hipocampo. Posteriormenteotros autores tampoco encontraron hallaz-gos significativos al estudiar de forma másdetallada los mismos cerebros108.
Respecto a la vulnerabilidad genética,ya Kraepelin constató la idea de que aque-llos individuos que presentaban predispo-sión genética para padecer un TrastornoAfectivo y se exponían a un estresantevital, tenían más riesgo de desarrollar unTrastorno Afectivo que aquellos que no.Además postuló que una vez iniciada laenfermedad el proceso era independientedel estresor. Estudios epidemiológios refle-jan que el 40-50% del riesgo de padecer untrastorno depresivo es de origen genético,por lo que se debe considerar al TrastornoDepresivo una enfermedad con alto com-ponente hereditario. Pero hasta la fecha labúsqueda de los genes específicos queconforman esta vulnerabilidad ha sidofrustrante, debido a varios factores, comola heterogeneidad clínica de la enferme-dad, su complejidad y la posibilidad deque varios genes estén implicados. Tampo-co debemos olvidar el papel etiológico delos factores no-genéticos, como el estrés,los traumas emocionales o las infecciones
virales, entre otros. Está bien evidenciadocómo los episodios depresivos suelen ocu-rrir habitualmente en el contexto de unestresante, pero debemos tener en cuentaque el estrés per se no es suficiente paracausar depresión76, ya que ante un estrésde igual intensidad no todo el mundo desa-rrolla un episodio depresivo. Todo estorecalca que la depresión es un trastornocausado por la interacción entre la predis-posición genética y algunos factoresambientales.
CONCLUSIÓN
Se ha tratado de realizar en esta revi-sión una integración de los diferentesmecanismos patofisiológicos implicadosen los Trastronos Afectivos sin ceñirseexclusivamente a las alteraciones de lossistemas monoaminérgicos. La interrela-ción entre los diferentes niveles de com-plejidad está por determinar, pero cadanivel permite explicar, al menos parcial-mente, los diferentes aspectos de la enfer-medad. Finalmente, se describen las hipó-tesis patofisiológicas más recientes de losTrastornos Afectivos que se fundamentanen alteraciones relacionadas con losmecanismos implicados en la plasticidadneuronal.
BIBLIOGRAFÍA
1. MANJI HK, LENOX HR. Signaling: Cellularinsights into pathophysiology of bipolardisorder. Biol Psychiatry 2000; 48: 518-527.
2. BURMEISTER, M. Basic concepts in the studyof diseases with complex genetics. BiolPsychiatry 1999; 45: 522-532.
3. DUMAN RS, HENINGER GR, NESTLER EJ. Amolecular and cellular theory of depression.Arch Gen Psychiatry 1997; 54: 597-606.
4. ALTAR CA. Neurotrophins and depression.Trends Pharmacol Sci 1999; 20: 59-61.
5. DREVETS WC. Neuroimaging Studies of MoodDisorders. Biol Psychiatry 2000; 48: 813-829.
6. STARKSTEIN SE, ROBINSON RG. Depression incerebrovascular disease. Depression inneurologic disease. The Johns HopkinsUniversity Press. 1993
7. THOMAS AJ, FERRIER IN, KALARIA NH, PERRY RH,BROWN A, O’BRIEN JT. A neuropathologicalstudy of vascular factors in late-lifedepression. J Neurol Neurosurg Psychiatry2001; 70: 83-87.

M. Zandio et al.
58 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
8. FUJIKAMA T, YANAI I, YAMAWAKI S. Psychosocialstressors in patients with mayor depressionand silent cerebral infarction. Stroke 1997;28: 1123-1125.
9. STARKSTEIN SE, ROBINSON RG, BERTHIER ML,PRICE TR. Depressive disorders followingposterior circulation compared with middlecerebral artery infarcts. Brain 1988;111: 375-387.
10. STEFFENS DC, KRISHNAN KR. Structuralneuroimaging and mood disorders: recentfindings, implications for classification, andfuture directions. Biol Psychiatry 1998; 43:705-712.
11. DAMASIO AR, TRANEL D, DAMASIO H. Individualswith sociopathic behavior caused by frontaldamage fail to respond autonomically tosocial stimuli. Behav Brain Res 1990; 41: 81-94.
12. FRYSTAZTAK RJ, NEAFSEY EJ. The effect ofmedial frontal cortex lessions oncardiovascular conditioned emotionalreponses in the rat. Brain Res 1994; 43:181-193.
13. MORGAN MA, LEDOUX JE. Differentialcontribution of dorsal and ventral medialprefrontal cortex to acquisition andextinction of conditioned fear in rats. BehavNeurosci 1995; 109: 681-688.
14. SULLIVAN RM, GRATTON A. Lateralized effectsof medial prefrontal cortex lesions onneuroendocrine and autonomic stressreponses in rats. J Neurosci 1999;19: 2834-2840.
15. CARMICHAEL ST, PRICE JL. Limbic connectionsof the orbital and medial prefrontal cortex inMacaque monkeys. J Comp Neurol 1995;363: 615-641.
16. LEUCHNETZ GR, ASTRUC J. The efferentprojections of the medial prefrontal cortexin the squirrel monkey (Saimiri sciureus).Brain Res 1976; 109: 455-472.
17. SCHULTZ W. Dopamine neurons and the rolein reward mechanisms. Curr Opin Neurobiol1997; 7: 191-197.
18. PRICE JL, CARMICHAEL ST, DREVETS WC.Networks related to the orbital and medialprefrontal cortex: A substrate for emotionalbehavior? En: Holstege G; Bandler R, SaperCB, editors. Progress in the Brain Research:The Emotional Motor System 1996; 107: 523-536.
19. BECHARA A, DAMASIO H, TRANEL D, ANDERSON
SW. Dissociation of working memory fromdecision-making within the humanprefrontal cortex. J Neurosci 1998;18: 428-437.
20. ROLLS ET. A theory emotion andconsciousness, and its application tounderstanding the neuronal basis ofemotion. En: Gazzaniga MS, editor. TheCognitive Neurosciences. Cambridge, MA:MIT Press, 1995; 1091-1106.
21. DINAN TG. Psyconeuroendocrinology ofmood disorders. Curr Opin Psychiatry 2001;14: 51-55.
22. CHIMOWITZ MI, ESTES ML, FURLAN AJ, AWAD IA.Further observations on the pathology ofsubcortical lesions identified on magneticresonance imaging. Arch Neurol 1992; 49:747-752.
23. DREVETS WC, GADDE K, KRISHNAN R.Neuroimaging studies of depression. En:Charney DS, Nestler EJ, Bunney BJ, Edit.Neurobiology of Mental Illness. New York:Oxford University Press, 1999b; 394-418.
24. FAZEKAS F. Magnetic resonance signalabnormalities in asymptomatic individuals:Their incidence and functional correlates.Eur Neurol 1989; 29: 164-168.
25. WOOTEN GF, COLLINS RC. Metabolic effects ofunilateral lesion of the substantia nigra. JNeurosci 1981; 1: 285-291.
26. DREVETS WC, PRICE JL, SIMPSON JR, TODD RD,REICH T, VANNIER M, RAICHLE ME. Subgenualprefrontal cortex abnormalities in mooddisorders. Nature 1997; 386: 824-827.
27. HIRAYASU Y, SHENTON ME, SALISBURY DF, KWON
JS, WIBLE CG, FISCHER IA et al. Subgenualcingulate cortex volume in first-episodepsychosis. Am J Psychiatry 1999; 156 :1091-1093.
28. DREVETS WC. Prefrontal cortical-amygdalarmetabolism in major depression. Ann N YAcad Sci 1999; 877: 614-637.
29. BRODY AL, SAXENA S, SILVERMAN DHS, ALBORZIAN
S, FAIRBANKS LA, PHELPS ME et al. Brainmetabolic changes in major depressivedisorder from pre- to post-treatment witnparoxetine. Psychiatry Res Neuroimaging1999; 91: 127-139.
30. KETTER T, KIMBRELL TA, LITTLE JT, GEORGE MS,SACHS N, WINSBERG ME et al. Differences andcommonalties in cerebral function in bipolarcompared to unipolar depression. Presentadoen el: 38th Annual Meeting of the AmericanCollege of Neuropsycopharmacology,Acapulco, Mexico.1999 Diciembre.
31. BAXTER LR, PHELPS ME, MAZZIOTTA JC, GUZE BH,SCHWARTZ JM, SELIN CE. Local cerebralglucose metabolic rates in obsessive-compulsive disorder-a comparison withrates in the unipolar depression and in

59ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
normal controls. Arch Gen Psychiatry 1987;44: 211-218.
32. BIVER F, GOLDMAN S, DELVENNE V, LUXEN A,DEMAERTELAER V, HUBAIN P et al. Frontal andparietal metabolic disturbances in unipolardepression. Biol Psychiatry 1994; 36: 381-388.
33. CHOEN RM, GROSS M, NORDAHL TE, SEMPLE WE,OREN DA, ROSENTHAL N. Preliminary data onthe metabolic brain pattern of patients withwinter seasonal affective disorder. Arch GenPsychiatry 1992; 49: 545-552.
34. DREVETS WC, VIDEEN TO, PRICE JL, PRESKORN
SH, CARMICHAEL ST, RAICHLE ME. A functionalanatomical study of unipolar depression. JNeurosci 1992; 12: 3628-3641.
35. EBERT D, FEISTEL H, BAROCKA A. Effects ofsleep deprivation on the limbic system andthe frontal lobes in affective disorders: Astudy with Tc-99m-HMPAO SPECT.Psychiatry Res Neuroimaging 1991; 40: 247-251.
36. MAYBERG HS, LIOTTI M, BRANNAN SK, MCGINNIS
BS, MAHURIN RK, JERABEK PA et al. Reciprocallimbic-cortical function and negative mood.Converging PET findings in depression andnormal sadness. Am J Psychiatry 1999; 156:675-682.
37. NOBLER MS, SACKEIM HA, PROHOVNIK I, MOELLER
JR, MUKHERJEE S, SCHNUR DB et al. Regionalcerebral BF in mood disorders, III.Treatment and clinical reponse. Arch GenPsychiatry 1994; 51: 884-897.
38. BENCH CJ, FRACKOWIAK RSJ, DOLAN RJ. Changesin the regional cerebral blood flow onrecovery from depression. Psycol Med 1995;25: 247-251.
39. DREVETS WC, SIMPSON JR, RAICHLE ME.Reciprocal suppession of regional cerebralblood flow during emotional versus highercognitive processes: Implications forinteractions between emotion andcognition. Cong Emotion 1995; 12: 353-385.
40. WU J, GILLIN C, BUCHSBAUM MS, HERSHEY T,JOHNSON JC, BUNNEY WE. Effect of sleepdeprivation on brain metabolism ofdepressed patients. Am J Psychiatry 1992;149: 538-543
41. ABERCROMBIE HC, LARSON CL, WARD RT,SCHAEFER SM, HOLDEN JE, PERLMAN SB et al.Metabolic rate in amygdala predictsnegative affect and depression severity indepressed patients: An FDG-PET study.Neuroimage 1996; 3: S217.
42. BAUMANN B, DANOS P, KRELL D, DIEKMANN S,LESCHINGER A, STAUCH R et al. Reduced volumeof limbic system-affiliated basal ganglia in
mood disorders: Preliminary data from apost mortem study. J Neuropsychiatry ClinNeurosci 1999; 11: 71-78
43. KRISHNAN KRR, MCDONALD WM, ESCOLANA PR,DORAISWAMY PM, NA C, HUSAIN MM et al.Magnetic resonance imaging of the caudatenuclei in depression: Preliminaryobservations. Arch Gen Psychiatry 1992; 49:553-337.
44. AWAD IA, JOHNSON PC, SPETZLER RJ, AWAD CA,CAREY R. Incidental subcortical lesionsidentified on magnetic resonance imaging inthe elderly, II: Postmortem pathologicalcorrelations. Stroke 1986; 17: 1090-1097.
45. FAZEKAS F. Magnetic resonance signalabnormalities in asymptomatic individuals:Their incidence and functional correlates.Eur Neurol 1989; 29: 164-168.
46. GRENNWALD BS, KRAMER-GINSBERG E, KRISHNAN
KR, ASHTARI M, AUERBACH C, PATEL M.Neuroanatomic localization of magneticresonance imaging signal hyperintensities ingeritatric depression. Stroke 1998; 29: 613-617.
47. MACFALL JR, PAYNE ME, PROVENZALE JE,KRISHNAN KR. Medial orbital frontal lesions inlate-onset depression. Biol Psychiatry 2001;49: 803-806.
48. VIDEBECH P. MRI findings in patients withaffective disorder: a meta-analysis. ActaPsychiatr Scand 1997; 97: 157-168.
49. BEARDEN CE, HOFFMAN KM, CANON TD. Theneuropsychology and neuroanatomy ofbipolar affective disorder: a critical review.[Review]. Bipolar Disord 2001; 3: 106-150.
50. HARRISON PJ. The neuropathology of primarymood disorder. [Review]. Brain 2002; 125:1428-1449.
51. AREQUE A, PARPURA V, SANZGIRI RP, HAYDON PG.Tripartite synapses: glia, theuncknowledged partner. [Review]. TrendsNeurosc 1999; 22: 208-215.
52. BARRES BA. A new role for glia: generation ofneurons [Review]. Cell 1999; 97: 667-670.
53. COYLE JT, SCHWARCZ R. Mind glue:implications of glial cell biology forpsychiatry. Arch Gen Psychiatry 2000; 57:90-93.
54. BEZZI P, VOLTERRA A. A neuron-glia signallingnetwork in the active brain. [Review]. CurrOpin Neurobiol 2001;11: 387-394.
55. OLIET SHR, PIET R, POULAIN DA. Control ofglutamate clearence and synaptic efficacyby glial coverage of neurons. Science 2001;292: 923-926.

M. Zandio et al.
60 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
56. PARRI HR, GOULD TM, CRUNELLI V. Spontaneusastrocytic Ca2+ oscillations in situ driveNMDAR-mediated neuronal excitation. NatNeurosci 2001; 4: 803-812.
57. ULLIAN EM, SAPPERSTEIN S, CHRISTOPHERSON K,BARRES BA. Control of synapse number byglia. Science 2001; 291: 657-661.
58. MAGISTRETTI PJ. Cellular bases of functionalbrain imaging: insights from neuron-gliametabolic coupling. [Review]. Brain Res2000; 886: 108-112.
59. PORTER JT, MCCARTHY KD. Astrocyticneurotransmitter receptors in situ and invivo. [Review]. Prog Neurobiol 1997; 51: 439-455.
60. GALLO V, GHIANI CA. Glutamate receptors inglia: new cells, new inputs and newfunctions. [Review]. Trends Pharmacol Sci2000; 21: 252-258.
61. RAJKOWSKA G, HALARIS A, SELEMON LD.Reductions in neuronal and glial densitycharacterize the dorsolateral prefrontalcortex in bipolar disorder. Biol Psychiatry2001; 49: 741-752.
62. COTTER DR, PARIANTE CM, EVERALL IP. Glial cellabnormalities in major psychiatricdisorders: the evidence and implications.[Review]. Brain Res Bull 2001; 55: 585-595.
63. ROBBINS T, EVERITT B. Central norepinephrineneurons and behavior. En: Bloom F, KupferD, Edit. Psychopharmacology: The FourthGeneration of Progress. New York: RavenPress Ltd., 1995; 363-372.
64. RESSLER KJ, NEMEROFF CB. Role ofnorepinephrine in the pathophysiology andtreatment of mood disorders. BiolPsychiatry 1999; 46: 1219-1233.
65. CLEARE A, MURRAY R, O’KEANE V. Donoradrenergic reuptake inhibitors affectserotonergic function in depression?Psychopharmacology 1997; 134: 406-410.
66. MONGEAU R, BLIER P, DE MONTIGNY C. Theserotonergic and noradrenergic systems ofthe hippocampus: Their interactions andthe effects of antidepressant treatments.Brain Res Rev 1997; 23: 145-195.
67. VALENTINO R, FOOTE S, PAGE M. The locuscoeruleus as a site for integratingcorticotropin-releasing factor andnoradrenergic mediation of stress reponses.Ann N Y Sci 1993; 697: 171-187.
68. MAES M, MELTZER HI. The serotoninehypothesys of Major Depression.www.acnp.org.
69. DELGADO PL, CHARNEY DS, PRICE LH, LANDIS H,HENINGER GR. Neuroendocrine and
behavioral effects of dietary tryptophanrestriction in healthy subjects. Life Sci 1990;45: 2323-2332.
70. HENINGER GR, DELGADO PL, CHARNEY DS, PRICE
LH, AGHAJANIAN GK. Tryptophan-deficientdiet and amino acid drink deplete plasmaTryptophan and induce a relapse ofdepression in susceptible patients. J ChemNeuroanatomy 1992; 5: 347-348.
71. ARORA RC, MELTZER HY. Increased serotonin2(5HT-2) recpetor binding as measured by3H-lysergic acid diethylamide (3H-LSD) inthe blood platelets of depressed patients.Life Sci 1989; 44: 725-734.
72. MIKUNI M, KAGAYA A, TAKAHASHI K, MELTZER HY.Serotonin but not norepinephrine-inducedcalcium mobilization of platelets isenhanced in affective disorders.Psychopharmacology 1992; 106: 311-314.
73. CARPENTER W, BUNNEY W. Adrenal corticalactivity in depressive ilness. Am JPsychiatry 1971: 128: 31-36.
74. MCEWEN BS. Effects of adverse experiencesfor brain structure and function. BiolPsychiatry 2000; 48: 721-731.
75. SAPOLSKY RM. The possibility ofneurotoxicity in the hippocampus in MajorDepresión: a primer on neuron death. BiolPsychiatry 2000; 48: 755-765.
76. NESTLER EJ, BARROT M, DILEONE RJ, AMELIA JE,GOLD S, MONTAGGIA LM. Neurobiology ofdepression. Neuron 2002; 34: 13-25.
77. ROSS EM. Signal sorting and amplificationthrough G protein-coupled receptors.Neuron 1989; 3: 141-52.
78. DUMAN RS, NESTLER EJ. Signal transductionpathways for catecholamine receptors.www.acnp.org/G4/GN401000028/CH.html.
79. SCHREIBER G, AVISSAR S, DANON A, BELMAKER RH.Hyperfunctional G proteins in mononuclearleukocytes of patines with mania. BiolPsychiatry 1991; 29: 273-80.
80. MANJI HK, CHEN G, SHIMON H, HSIAO JK, POTTER
WZ, BELMAKER RH. Guanine nucleotide-binding proteins in bipolar affectivedisorder. Effects of long-term lithiumtreatment. Arch Gen Psychiatry 1995; 52:135-144.
81. MANJI HK, MOORE GJ, CHEN G. BipolarDisorder: leads from the molecular andcellular mechanims of action of moodstabilisers. Br J Psychiatry 2001,178(Suppl.41): 107-119.
82. SCOTT JD. Cyclic nucleotide-dependentprotein kinases. Pharmacol Ther 1991; 50:123-145.

61ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
NEUROBIOLOGÍA DE LA DEPRESIÓN
83. SPAULDING SW. The ways in which hormoneschange cyclic adenosine 3’,5’-monophosphate-dependent proteinkinasessubunits, and how such changes affect cellbehavior. Endocr Rev 1993; 14: 632-650.
84. FIELDS A, LI PP, KISH SJ, WARSH JJ. Increasedcyclic AMP-dependent protein kinaseactivity in postmortem brain from patientswith Bipolar Affective Disorder. JNeurochem 1999; 73: 1704-1710.
85. MATHEWS R, LI PP, YOUNG LT, KISH SJ, WARSH JJ.Increased G alpha q/11 inmunoreactivity inpostmortem occipital cortex from patientswith Bipolar Affective Disorder. BiolPsychiatry 1997; 41: 649-656.
86. SOARES JC et al. Increased platelet membranephosphatidylinositol-4,5-biphosfate in drugfree depressed bipolar patients. NeurosciLett 2001; 299: 150-152.
87. RASMUSSEN H. The calcium messengersystem. N Engl J Med 1986; 314: 1094-1101.
88. BEBCHUCK JN, ARFKEN CL, DOLAN-MANJI S,MURPHY J, HASANAT K, MANJI HK. A preliminaryinvestigation of a proteinkinase C inhibitorin thetretment of actue mania. Arch GenPsychiatry 2000; 57: 95-97.
89. BEZCHLIBNYK Y, YOUNG T. The neurobiology ofbipolar disorder: focus on signaltransduction pathways and the regulation ofgene expresión. W Can J Psychiatry 2002; 47:2.
90. DUMAN RS, MALBERG J, NAKAGAWA S, D’SA C.Neuronal plasticity and survival in mooddisorders. Biol Psychiatry 2000; 48: 732-739.
91. GAGE FH. Structural Plasticity: cause, result,or correlate of depresión. Biol Psychiatry2000; 48: 713-714.
92. ERIKSSON P, PERFILIEVA E, BJORK-ERIKSSON T,ALBORN A, NORDBORG C, PETERSON D, GAGE F.Neurogenesis in the adult humanhippocampus. Nat Med 1998; 4: 1313-1317.
93. GOULD E, REEVES A, GRAZIANO M, GROSS C.Neurogenesis in the neocortex of addultprimates. Science 1999; 286: 548-552.
94. MCEWEN BS, SAPOLSKY RM. Stress andcognitive function. Curr Opin Neurobiol1995; 5: 205-216.
95. CONRAD CD, LEDOUX JE, MAGARINOS AM,MCEWEN BS. Repeat restraint stressfacilitates fear conditioning independentlyof causing hippocampal CA3 dendriticatrophy. Behav Neurosci 1999; 113: 902-913.
96. TURSKI L, TURSKI W. Towards an understandingof the role of glutamate in neurodegenerativedisorders: energy metabolism and
neuropathology. Experientia 1993; 49: 1064-1072.
97. TOMBAUGH G, SAPOLSKY R. Evolving conceptsabout the role of acidosis in ischemicneuropathology. J Neurochem 1993; 61: 793-803.
98. SAPOLSKY R. Stress, glucocorticoids and theiradverse neurological efects: revelance toaging. Exp Geront 1999; 34: 721-732.
99. MAGISTRETTI PJ, PELLERIN L, MARTIN JL. Brainenergy metabolism: An integrated cellularperspective. En: Bloom FE, Kupfer DJ, Edit.Psychopharmacology: The FourthGeneratiom of Progress. New York: Raven,1995; 921-932.
100.AZMITIA EC. Serotonin neurons,neuroplasticity, and homeostasis of neuronaltissue. Neuropsychopharmacology 1999;21(Suppl 2): 33S-45S.
101.MCEWEN BS. Stress and hippocampalplasticity. Annu Rev Neurosci 1999; 22: 105-122.
102.RAJKOWSKA G. Histopathology of the frontalcortex in major depression. What does it tellus about dysfunctional monoaminergiccircuits? [Review]. Prog Brain Res 2000; 126:397-412.
103.CHAPUT Y, DE MONTIGNY C, BLIER P. Presynapticand postsynaptic modifications of theserotonin system by long-termadministration of antidepressant treatments.An in vivo electrophysiologic study in rat.Neuropsychopharmacology 1991; 5: 219-229.
104.HADDJERI N, BLIER P, DE MONTIGNY C. Long-termantidepressant treatments result in tonicactivation of forebrain 5-HT1A receptors. JNeurosci 1998; 18: 10150-10156.
105.MAGARINOS AM, DESLANDES A, MCEWEN BS.Effects of antidepressant and benzodiazepinetreatments on the dendritic structure of CA3pyramidal neurons after chronic stress. Eur JPharmacol 1999; 371: 113-122.
106.BROWN ES, RUSH AJ, MCEWEN BS.Hippocampal remodeling and damage bycorticosteroids: implications for mooddisorders. Neuropsychopharmacology 1999;21: 474-484.
107.LUCASSEN PJ, MÜLLER MB, HOLSBOER F, BAUER J,HOLTROP A, WOUDA J et al. Hippocampalapoptosis in major depression is a minorevent and absent fron subareas at risk forglucocorticoid overexposure. Am J Pathol2001; 49: 803-806.
108.MÜLLER MB, LUCASSEN PJ, YASSOURIDIS A,HOOGENDIJK WJG, HOLSBOER F, SWAAB DF.Neither major depression nor

M. Zandio et al.
62 ANALES Sis San Navarra 2002, Vol. 25, Suplemento 3
glucocorticoid treatment affects the cellularintegrity of the human hippocampus. Eur JNeurosci 2001; 1063-1612.
109.ONGÜR D, DREVETS WC, PRICE JL. Glialreduction in the subgenual prefrontal cortexin mood disorders. Proc Natl Acad Sci USA1998; 95: 13290-13295.
110.BENES FM, VINCENT SL, TODTENKOPF M. Thedensity of pyramidal and nonpyramidalneurons in anterior cingulate cortex ofschizophrenic and bipolar subjetcs. BiolPsiquiatry 2001; 50: 395-404.
111.RAJKOWSKA G, MIGUEL-HIDALGO JJ, WEI J, DILLEY
G, PITTMAN SD, MELTZER HY et al.Morphometric evidence for neuronal andglial prefrontal cell pathology in majordepression. Biol Psychiatry 1999; 45: 1085-1098.
112.MIGUEL-HIDALGO JJ, BAUCOM C, DILLEY G,OVERHOLSER JC, MELTZRE HY, STOCKMEIER CA et
al. Glial fibrillary acidic proteininmunoreactivity in the prefrontal cortexdistinguises younger from older adults inmajor depressive disorder. Biol Psychiatry2000; 48: 861-873.
113.ROSOKLIJA G, TOOMAYAU G, ELLIS SP, KEILP A,MANN JJ, LATOV N. Structural abnormalities ofsubicular dendrites in subjects withschizophrenia and mood disorders:Preliminary findings. Arch Gen Psychiatry2000; 57: 349-356.
114.ZHOU JN, RIEMESMA RF, UNMEHOPA VA,HOODENDIJK WJ, VAN HEERIKHUIZE JJ. Alterationsin arginine vasopressin neurons in thesuprachiasmatic nucleus in depression.Arch Gen Psychiatry 2001; 58: 655-662.
115.BAUMANN B, DANOS P, DIEKMANN S, KRELL D,BIELAU H, GERETSEGGER C. Tyrosine hidroxilaseinmunoreactivity in the locus coeruleus isreduced in depression. Eur Arch PsychiatryClin Neurosci 1999; 249: 212-219.