introducciÓn a la experimentaciÓn...
TRANSCRIPT

INTRODUCCIÓN A LA EXPERIMENTACIÓN QUÍMICA Y A LAS TÉCNICAS INSTRUMENTALES
Ciclo Superior de Laboratorio de Análisis y Control de Calidad
(MÓDULO DE TÉCNICAS INSTRUMENTALES)
Curso 2010-2011

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 1
VALORACIONES POTENCIOMÉTRICAS al acabar las prácticas el alumno TIENE QUE SER CAPAZ DE:
• Escribir y ajustar las reacciones de la valoración. • Definir qué es un patrón primario. Conocer sus características y los diferentes tipos,
así como saber nombrar algunos ejemplos. • Utilizar correctamente los diferentes tipos de balanzas. • Identificar los diferentes tipos de electrodos indicadores (ácido-base y redox) y de
referencia. • Definir qué son las curvas de valoración y determinar el punto final de una valoración
mediante su uso. • Identificar los errores que se pueden cometer en el análisis cuantitativo. • Determinar la concentración de las especies indicadas a las prácticas P1-P8 mediante
valoración potenciométrica.
FUNDAMENTO TEÓRICO
Son los métodos analíticos basados en las medidas de potencial Ya hemos comentado que el potencial de un solo electrodo, es decir, el potencial de una semirreacción aislada, resulta imposible de medir; es decir que solamente es posible obtener experimentalmente los potenciales de celdas electroquímicas. Esta celda se puede representar como:
El electrodo de referencia es un electrodo con un potencial perfectamente conocido (εref), independiente de la concentración de analito o de la concentración de cualquier otro ión de la disolución. Por convenio se toma siempre como ánodo en las medidas potenciométricas. El electrodo indicador al sumergirse en la disolución de analito genera un potencial (εind) que depende de la actividad del analito. El puente salino impide que los componentes de la disolución del analito se mezclen con los del electrodo de referencia. En las interfases entre el puente salino y las disoluciones aparece un potencial de unión líquida (εj).

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 2
El potencial de la celda electroquímica que acabamos de describir será igual a la suma de todos los potenciales que aparecen en el sistema.
Potencial de unión líquida. Siempre que dos disoluciones electrolíticas están en contacto, se establece una diferencia de potencial eléctrico en la interfase o zona de separación de ambas. El potencial de unión es por tanto un potencial de interfase. Para explicar por qué ocurre esto consideremos una
disolución que contiene NaCl en contacto con agua destilada (Figura. 3). Los iones Cl y Na+ empiezan a difundirse a partir de la disolución de la sal hacia el agua. Sin embargo el ión cloruro tiene mayor movilidad que el ión sodio, es decir, se difunde con mayor velocidad. De ello resulta un frente de difusión rico en iones cloruro. Este frente contiene un exceso de cargas negativas. Detrás de él se encuentra una región empobrecida en cargas negativas y que, por tanto, contiene un exceso de cargas positivas. El resultado es una diferencia de potencial eléctrico en la unión entre las fases, disolución de NaCl y agua pura. Este potencial de unión se opone al movimiento de los iones Cl - y acelera el de los iones Na + . La diferencia de potencial que resulta de esta separación de cargas puede ser de varias centésimas de voltio. La conexión interna entre el electrodo indicador y el de referencia en algunos tipos de celdas utilizadas en potenciometría se lleva a cabo generalmente mediante una unión líquida, que permite el paso de iones (flujo de corriente) sin mezcla física de las disoluciones. Si las movilidades de los iones positivos y negativos que difunden a través de la unión de ambas semiceldas son diferentes, aparece un potencial de unión liquida. La velocidad de difusión de un ión depende de su movilidad iónica. Los potenciales de unión liquida que se crean de esta forma se minimizan cuando los iones del electrolito que la constituye poseen movilidades parecidas. Así, las movilidades del ión potasio (73.5x10 -5 cm/s) y del ión nitrato (71.6x10-5 cm/s) son parecidas a la del ión cloruro. Por regla general, cuando resulte deseable mantener el potencial de unión líquida en un mínimo, se utiliza un puente salino cuya concentración de KCl es varias veces superior a la concentración del electrolito de la disolución con la que va a establecer contacto. Los potenciales de unión son entonces pequeños y del orden de algunos milivoltios. Mediante selección adecuada de mezcla de sales pueden obtenerse puentes salinos con valores del potencial de unión menores incluso que el de cloruro de potasio.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 3
Electrodos de referencia Como se ha comentado antes, las medidas prácticas en potenciometría se llevan a cabo con un sistema que incluye el electrodo de media (electrodo indicador) y un electrodo de referencia. La semicelda de referencia o electrodo de referencia debe ser fácil de preparar y utilizar, y su potencial debe ser exactamente conocido, constante y completamente insensible a la composición de la disolución del analito, y además, debe permanecer invariable con el paso de corrientes de baja intensidad a través del sistema del que forma parte. El electrodo normal de hidrógeno es el electrodo de referencia universal, pero debido a lo peligroso de su manejo (utiliza una corriente de H2) y a lo dificultoso de su preparación y manejo se emplean otros diferentes electrodos de referencia secundarios más cómodos y fácil de manejar. Los electrodos de referencias más comunes son el de calomelanos y el electrodo de plata-cloruro de plata. El electrodo de calomelanos (Figura 4) puede representarse esquemáticamente como sigue:
Hg | Hg2Cl2 (saturada), KCl (x M) |
Donde x representa la concentración molar de KCl en la disolución. Tres son las concentraciones más utilizadas, 0,1 M, 1 M y saturada (~ 4,6 M), la más corriente de las cuales es la saturada también denominada electrodo de calomelanos saturado (ECS) con un potencial de 0,244V más positivo que el del electrodo normal de hidrógeno. La reacción que tiene lugar en la semicelda de calomelanos es:
valor del potencial del electrodo de calomelanos (en cualquiera de sus versiones) depende de la actividad del ión cloruro, como puede deducirse de la expresión de la ecuación de Nernst para el par Hg (I)/Hg, si se sustituye la actividad del ión mercurio (I) por la correspondiente obtenida del producto de solubilidad de la sal poco soluble Hg2Cl2 (consultar el concepto de potencial condicional).
El electrodo de referencia Ag/AgCl (Figura 5) está constituido por un hilo de plata recubierto de cloruro de plata -sal poco soluble al igual que el cloruro de Hg (I)-sumergido en una disolución de NaCl o KCl de concentración 0,l M, 1 M o saturada. La semirreacción que tiene lugar es:
AgCl (s) + e Ag (s) + Cl -

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 4
Y la expresión de la semicelda:
Ag | AgCl (saturada), KCl (x M) | Al igual que antes, el potencial del electrodo viene determinado por la actividad del ión Ag (I), que a su vez viene condicionada por el producto de solubilidad del cloruro de plata y por la actividad del ión cloruro.
Celdas utilizadas en potenciometría Se utilizan tres tipos diferentes de celdas para potenciometría directa y en valoraciones potenciométricas. La primera contiene un electrodo indicador de metal activo, la segunda un electrodo indicador inerte y la tercera dos electrodos de referencia unidos a cada lado de una membrana. El tercero y más importante tipo de celda está constituido por dos semiceldas de referencia separadas por un material de unión. Por ejemplo ECS// disolución problema/material de unión/ECS Los potenciales de dos ECS tienen nominalmente el mismo valor y no varían de forma significativa con el tiempo. El voltaje de la celda sería cero si no fuese por la existencia del potencial de unión para cuya medida se ha diseñado precisamente el dispositivo electroquímico. Su aplicación más importante es en el empleo de electrodos selectivos de iones, de los cuales el más conocido es el electrodo de vidrio para medidas potenciométricas del pH. El electrodo de vidrio El electrodo selectivo más antiguo y mejor conocido es el electrodo de vidrio para medidas de la concentración del ión hidrógeno. Un sistema de medida mediante electrodo selectivo está formado por dos semiceldas de referencia, una a cada lado de una membrana liquida o sólida que responde más o menos selectivamente a una especie individual. Un lado de la membrana está en contacto con una disolución que contiene el ión (su actividad) a determinar, y el otro lado con una disolución de actividad constante de dicho ión

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 5
Las semiceldas de referencia hacen contacto eléctrico con los lados de la membrana en la que se establecen los potenciales de unión. Aparecen dos potenciales diferentes como resultado del equilibrio entre las disoluciones y las superficies de la membranas. Una unión es la de contacto de un lado de la membrana con la disolución interna o de referencia cuyo potencial de unión permanece virtualmente invariable. La otra es la de contacto del otro lado de la membrana con la disolución problema. Como las dos semiceldas de referencia, así como el lado de referencia de la membrana, poseen valores de potencial constantes, la variación del potencial global de la celda tiene como origen el cambio del potencial de unión de la interfase membrana-disolución problema. Este cambio se produce al modificar la actividad del ión de interés de la disolución problema o las condiciones de la misma. Probablemente en la actualidad se realizan más medidas de pH que de cualquier otro parámetro químico. La mayor parte de ellas se hacen potenciométricamente utilizando un electrodo indicador de vidrio. Después de varias décadas de estudio y utilización, el funcionamiento de este tipo de electrodo se conoce razonablemente bien a pesar de que incluso hoy las mejoras que se realizan en las membranas de vidrio son con mucho de tipo empírico. Un electrodo típico de pH (Figura 6) consiste en una membrana delgada de vidrio, generalmente en forma de bulbo, sellada en el extremo de un tubo de vidrio. En el interior se mantiene un pH constante mediante un relleno con una disolución generalmente de HCl 0,l M. El contacto entre esta disolución y el aparato de media (voltímetro) se lleva a cabo mediante un electrodo de referencia, calomelanos o Ag-cloruro de plata en el caso de la figura al que se denomina electrodo de referencia interno. por motivos de estabilidad y facilidad de manejo, la disolución de pH constante y el electrodo de referencia interno se fabrican en un conjunto cerrado. El conjunto se completa con un segundo electrodo de referencia (externo) que en este caso es un ECS. El vidrio de la membrana debe tener una composición apropiada, por ejemplo, 72 % de SiO2, 22 % de Na2O y 6 % de CaO (Esta composición, propuesta en 1930, ha sido comercializada durante muchos años por la firma Corning Glass Works con el nombre de Corning 015; presenta el inconveniente de responder ligeramente al ión sodio, lo que lleva al llamado "error del sodio" o "error alcalino" en medios de pH superior a 10. Los electrodos modernos sustituyen el óxido de sodio por el de litio, entre otras modificaciones. Estos electrodos se pueden utilizar desde pH 0 hasta 14 con pequeño error de respuesta a otros iones La membrana de vidrio puede visualizarse de la forma siguiente

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 6
Las dos uniones de interés son la unión entre la disolución problema y la capa hidratada (Ej (1)) y la unión entre la otra capa hidratada y la disolución interna (Ej (2)). En estas dos uniones se establece rápidamente un equilibrio en la superficie, apareciendo los potenciales correspondientes de unión debido a la transferencia de protones a un lado a otro dependiendo del valor de la actividad del ión hidrógeno en la disolución y en las capas hidratadas. El equilibrio es
H+ vidrio H+ dón
No existen métodos conocidos para determinar los valores absolutos de estos potenciales; pero cualitativamente hablando, si la actividad del ión hidrógeno de la disolución aumenta, los protones se transfieren a la capa de vidrio y el potencial en la superficie de dicha capa se hace más positivo. Es importante darse cuenta de que no hay transferencia de electrones, es decir, no hay procesos redox en términos generales. El potencial en la interfase de interés crece simplemente como resultado del equilibrio establecido en ambas fases. Cuantitativamente el potencial de interfase vine dado por:
En la segunda capa hidratada (interna) aparece un potencial similar cuyo valor viene dado por una expresión semejante. Como la actividad del ión hidrógeno en la disolución interna se mantiene constante, el potencial de interfase de la capa interna no varia. El potencial neto a través de toda la membrana es la diferencia entre los anteriores:
Desde un punto de vista práctico, las dos superficies de la membrana de vidrio no se comportan exactamente de la misma forma, hay una pequeña diferencia en respuesta relacionada con la historia de cada superficie individual. Estas variaciones dan lugar a una ligera diferencia de potencial a través de la membrana que resulta observable aún cuando las disoluciones a cada lado sean exactamente iguales. Esta diferencia, denominada potencial de asimetría, es generalmente del orden de algunos milivoltios. Su magnitud varía de un día a otro y para cada membrana. Se corrige en el momento en que el electrodo se calibra con una disolución de referencia. La velocidad de variación del potencial de asimetría es lo suficientemente lenta como para no necesitar recalibrado durante la medida. Medida práctica del pH Por comodidad los electrodos comerciales para medida del pH se construyen en una sola unidad y se conoce como electrodo combinado de vidrio (Figura 8). Tienen la ventaja de ocupar menos espacio y la posibilidad por tanto de su utilización con volúmenes pequeños de

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 7
muestra. Para medir el pH de una disolución es preciso calibrar previamente el aparato. Para ello se utilizan las llamadas disoluciones tampones o reguladoras de pH. Cada casa comercial suministra algunas de ellas con el instrumento. Además, el aparato posee una función de calibrado. El calibrado se realiza introduciendo el electrodo en la disolución apropiada de calibrado y utilizando la citada función de calibración, de acuerdo con las instrucciones del manual. Suele llevarse a cabo un calibrado en la zona ácida y otro en la zona básica. Una vez realizado esto, el pH-metro está en condiciones de medir la actividad del protón de la disolución problema. Aunque el electrodo de vidrio es con mucho el más ampliamente utilizado, otros electrodos son también sensibles al pH y algunos soportan entornos más adversos que el electrodo de vidrio. Un electrodo de Ir recubierto de IrO2 se comporta como detector de pH [S. Bordi, M. Carlá y G. Papeschi, Anal. Chem., 56, 317 (1984)]. Para medir el pH de disoluciones hasta 300ºC suele usarse un electrodo basado en ZrO2 [L.W. Niedrach, Angew. Chem., 26, 161, (1987)]. Las superficies de platino y carbón también son sensibles al pH, pero se les debe recubrir para hacerlas selectivas al ión hidrógeno [G. Cheek, C.P. Wales y R.J. Novak, Anal.Chem., 58, 38 (1983)]. También se han descrito ingeniosos electrodos de alambre con la superficie modificada par a la medición del pH [Rubinstein, Anal. Chem., 56, 1135 (1984)].
EJEMPLO A LEER CUIDADOSAMENTE MATERIALES Y REACTIVOS Materiales Electrodo de pH Sonda CAT pH-metro Agitador magnético y barra magnética. Pipeta 25 ml. Bureta. Matraz aforado de 100 ml. Vasos de precipitado, probeta de 25 ml, etc... Reactivos NaOH de concentración exacta. Disolución problema de ac. acético y ac. clorhídrico.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 8
MÉTODO EXPERIMENTAL Descripción general La práctica consiste en la valoración potenciométrica de una mezcla de dos ácidos. Dicha valoración se realiza midiendo el pH de la disolución problema después de cada adición del valorante (disolución de NaOH de molaridad perfectamente conocida). Se obtiene de esta forma una tabla de datos pH-ml y se representa gráficamente la variación del pH frente a los mililitros añadidos. Se determinan a continuación los puntos de equivalencia (puntos de inflexión o de máxima pendiente de la curva) expresados en ml. Se transforman los ml anteriores en milimoles de OH- o, lo que es lo mismo, en milimoles de protones y por tanto del ácido valorado. Se da finalmente la molaridad de la disolución inicial en cada uno de los ácidos que constituyen la mezcla. La recogida de datos durante el curso de la valoración debe ser cuidadosa ya que de ella depende, si las demás operaciones se han realizado bien, la exactitud de los resultados. Para la representación gráfica es muy útil un programa de ordenador que dé además la gráfica de la primera derivada y los datos numéricos de los puntos de inflexión. Modo de operación. Se toma el problema en un matraz de 100 ml que contiene la una mezcla de HCl y de ácido acético y se enrasa en primer lugar con agua destilada hasta la marca exactamente (ver Figura 9). Si el volumen queda por encima de la marca de aforo el problema queda invalidado. Para que esto no ocurra, los últimos mililitros anteriores a la señal de aforo deben añadirse con cuidado (por ejemplo, utilizando una pipeta limpia). Se pone el tapón al matraz y se agita para homogeneizar. Se toman 25 ml de la disolución anterior, exactamente medidos (con pipeta) y se transfieren al vaso de valoración, dejando caer libremente el contenido de la pipeta (Figura 10).
Se añaden a continuación unos 20-25 ml de agua destilada (aproximadamente, no hace falta medirlos) al vaso de valoración y se coloca en su interior la barra magnética. Se enrasa la bureta a cero con la disolución patrón de NaOH (anotar su molaridad exacta) con el mismo criterio de enrase que el de la figura 9. Se coloca el vaso de valoración en el centro de la placa de agitación y se introduce en el mismo el electrodo de pH y la sonda de control automático de
temperatura (CAT) procurando que queden a una altura tal que al girar el imán no los golpee. El dispositivo debe quedar como aparece en la figura 11.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 9
El pH-metro no es necesario que esté exactamente calibrado, pero sí debe funcionar correctamente. Se mide el pH de la disolución inicial y se anota. A continuación se empieza la valoración añadiendo la disolución de NaOH de la bureta de 1 ml en 1 ml y agitación moderada, anotando las lecturas de pH estables correspondientes. Entre pH 2 y 3,5 deberá hacerse la adición de 0,2 en 0,2 ml para situar con exactitud el primer punto de equivalencia que pasa desapercibido (no se nota para el mismo una variación brusca del pH). Pasado este valor de pH se continua la adición de 1 ml en 1 ml hasta un pH de aproximadamente 6. A partir de aquí la adición se hace de 0,1 en 0,1 ml, anotando siempre la lectura de pH. Una vez conseguido el pH de 11 se vuelve la adición de 1 ml en 1 ml hasta un valor de alrededor de 12, en el que se da por finalizada la valoración. Apagar el agitador. Se retiran a continuación los electrodos con cuidado, se enjuagan con agua destilada y se introducen en el vaso de electrodos. Se limpia el vaso de valoración y se repite la toma de muestra, enrase de bureta con NaOH y valoración una vez más para obtener otra serie de datos. Una vez finalizada esta segunda valoración y si no se va a repetir más la experiencia, dejar el material (bureta, vaso y pipeta) limpios y los electrodos lavados y sumergidos en el vaso “electrodos”. Mediante un programa de ordenador se representan los dados de pH frente a ml añadidos, así como la gráfica de la primera derivada (pH/vol. frente a ml) para cada experiencia. En caso de no disponer de ordenador, se representará en papel milimetrado. En la Tabla de datos de la primera derivada se determinan los puntos de equivalencia (puntos de máxima pendiente, máximo valor de la ordenada) y se transforman en milimoles de cada uno de los ácidos de la mezcla. El resultado final se da en concentración molar de HCl y de ácido acético en los 100 ml del problema original. Hacer para ello los cálculos correspondientes. CÁLCULOS Y RESULTADOS 1º) Representar en papel milimetrado (o gráfica de ordenador) los dados de pH frente a volumen de NaOH (en ml) añadido, 2º) Obtener también la gráfica de la primera derivada (pH/vol. frente a vol. en ml) para cada experiencia. Identificar en la misma los puntos de equivalencia. 3º) Concentración molar de HCl y de ácido acético del problema original.
BIBLIOGRAFIA
1. A.I. Vogel, Química Analítica Cuantitativa, vol. II, Ed. Kapelusz, Buenos Aires, (1960).
2. I.M. Kolthoff, E.B. Sandell, Análisis Químico Cuantitativo, Ed. Nigar, Buenos Aires, (1972).
3. J. Basset, R.C. Denney, G.H. Geffery, J. Mendham, Vogel´s Textbook of Quantitative Inorganic Analysis, Ed. Longman, New York, 4ª edición (1972).
4. D.A. Skoog, D.M. West, F.J. Holler, Fundamentos de Química Analítica, vol. II, Ed. Reverté, Barcelona, 4ª edición (1996).
Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 10
RECOMENDACIONES PRÁCTICAS Para cualquier valoración potenciométrica/conductimétrica es conveniente seguir los siguientes pasos :
• Calibrar el pHmetro o conductivímetro, respectivamente. • Llenar la bureta con disolución de la sustancia a valorar o valorante.. • Pipetear a un vaso de precipitados, de 100 ml, 25 ml de valorante (o sustancia a
valorar) de concentración conocida. Introducir en el mismo un imán, colocarlo sobre un agitador magnético e introducir el electrodo bien lavado.
• Poner a funcionar el agitador y anotar a medida do aparato. • Desde la bureta se va adicionando lentamente y sin salpicaduras volúmenes iguales de
reactivo, p. ej. 2-3 ml , y anotar la medida del aparato. Cuando nos aproximamos al punto de equivalencia es conveniente añadir el reactivo en incrementos de 0.1 ml. NOTA: A veces la agitación puede causar errores de medida, por tanto para realizar la lectura es conveniente parar momentáneamente el motor.
• Representar gráficamente para obtener el resultado.
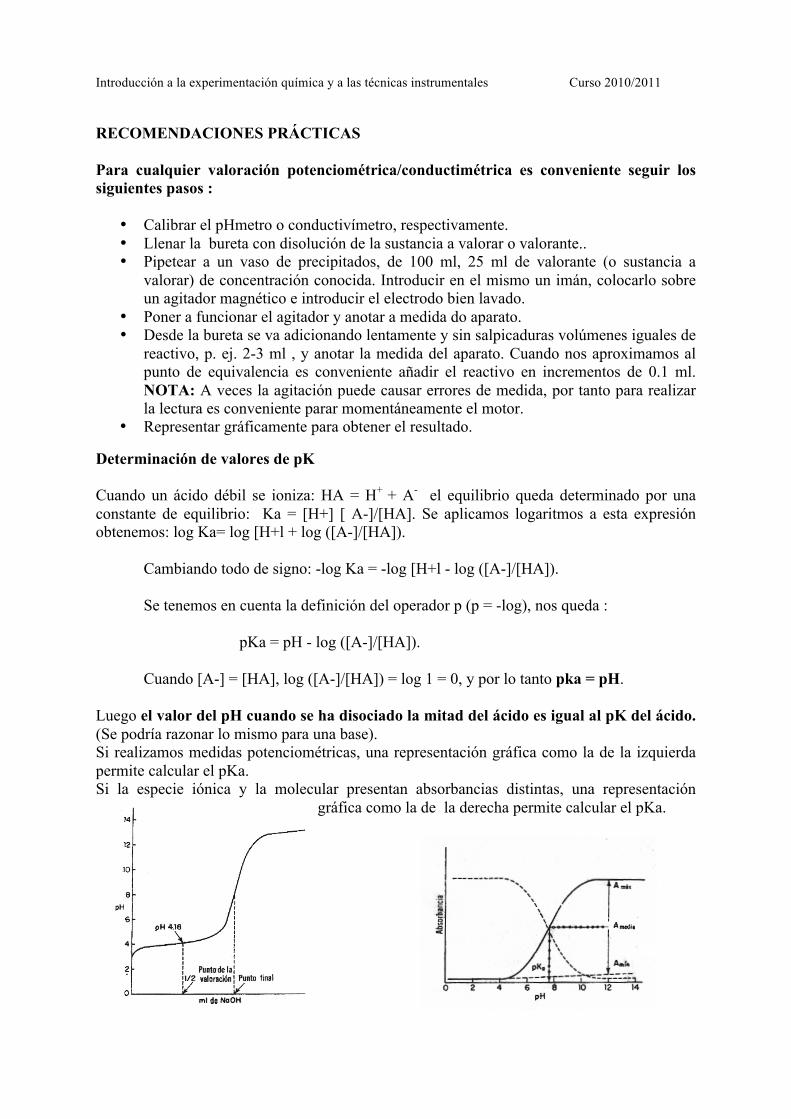
Determinación de valores de pK Cuando un ácido débil se ioniza: HA = H+ + A- el equilibrio queda determinado por una constante de equilibrio: Ka = [H+] [ A-]/[HA]. Se aplicamos logaritmos a esta expresión obtenemos: log Ka= log [H+l + log ([A-]/[HA]).
Cambiando todo de signo: -log Ka = -log [H+l - log ([A-]/[HA]). Se tenemos en cuenta la definición del operador p (p = -log), nos queda :
pKa = pH - log ([A-]/[HA]). Cuando [A-] = [HA], log ([A-]/[HA]) = log 1 = 0, y por lo tanto pka = pH.
Luego el valor del pH cuando se ha disociado la mitad del ácido es igual al pK del ácido. (Se podría razonar lo mismo para una base). Si realizamos medidas potenciométricas, una representación gráfica como la de la izquierda permite calcular el pKa. Si la especie iónica y la molecular presentan absorbancias distintas, una representación
gráfica como la de la derecha permite calcular el pKa.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 11
VALORACIONES POTENCIOMÉTRICAS ACIDO-BASE
PRÁCTICA 1
• Preparar 250 ml de disolución 0, 1 M de NaOH.
• Preparar 250 ml de disolución 0,1 M de ácido acético
• Preparar 100 ml HCl 0.1 M
• Valoración clásica del NaOH, HCl y ácido acético (volumétricamente)
• Valorar potenciométricamente el HCl con el carbonato.
• Valorar el hidróxido sódico potenciométricamente con el ftalato ácido de potasio
(patrón primario)
• Valorar el ácido acético con el hidróxido sódico potenciométricamente
• Comparar los resultados clásicos con los instrumentales.
PRÁCTICA 2
DETERMINACIÓN DE LA ACIDEZ TOTAL EN UN VINO TINTO
OBJETIVO
Calcular la acidez total en una muestra de vino tinto expresada como porcentaje (p/v) de ácido tartárico, mediante una valoración potenciométrica ácido-base.
FUNDAMENTO
La acidez total en un vino viene dada por la suma de los ácidos que contiene, siendo el ácido tartárico el componente mayoritario, de tal modo que la acidez total queda expresada como porcentaje de este ácido. El ácido tartárico (PM=150) es un ácido diprótico que se valora con NaOH 0.1 M.
MATERIAL
Potenciómetro, electrodo combinado de vidrio, 1 bureta de 25 ml, erlenmeyers, vidrios de reloj, matraces aforados, embudo, kitasato.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 12
REACTIVOS
Biftalato potásico, NaOH lentejas
PROCEDIMIENTO
Preparación de la solución valorante.
Preparar una solución de NaOH 0.1 M.
Estandarización de la solución valorante.
Con biftalato potásico. Se debe realizar por triplicado.
Tratamiento de la muestra.
Antes de llevar a término la práctica es necesario eliminar el CO2 de la muestra. Para hacerlo, se coloca la muestra en un matraz kitasato y se realiza el vacío con trompa de agua. (Repetir el proceso tres veces).
Valoración de la muestra.
A partir de la concentración aproximada de la acidez total que contiene la muestra (dato que proporciona el profesor), se calcula la cantidad de muestra necesaria por llevar a término la valoración, teniendo en cuenta que se tienen que gastar aproximadamente 2/3 del volumen de la bureta. Se pone en funcionamiento el instrumento seleccionando la opción pH para medir
potenciales y se procede a la valoración. Tener en cuenta que cada vez que se añade la disolución valorante se debe agitar el erlenmeyer antes de hacer la lectura. Para saber dónde está aproximadamente el punto de equivalencia, se aconseja hacer una primera valoración rápida. Finalmente, la muestra se valora por triplicado.
RESULTADOS
a) Escribe las reacciones que tienen sitio en la valoración potenciométrica. b) Dibuja la gráfica de valoración así como la primera y segunda derivada (ΔpH/Δ volumen respecto volumen valorante, ΔpH2/Δvolumen2 respecto volumen valorante). c) Encuentra el porcentaje (p/v) de ácido tartárico a la muestra inicial con la correspondiente incertidumbre.
TRATAMIENTO DE RESIDUOS
Los residuos se deben tirar por el fregadero bien diluidos.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 13
PRÁCTICA 3
DETERMINACIÓN DE UNA MEZCLA DE HIDRÓGENOFOSFATO DE SODIO - DIHIDRÓGENOFOSFATO DE SODIO EN UNA MUESTRA LÍQUIDA MEDIANTE UNA VALORACIÓN POTENCIOMÉTRICA
OBJETIVO
Determinar las concentraciones de hidrógenofosfato y dihidrógenofosfato sódico en una muestra líquida preparada, mediante una valoración potenciométrica ácido-base.
FUNDAMENTO
La determinación volumétrica de monohidrógeno y dihidrógeno fosfato sódico se fundamenta en dos reacciones de neutralización, una entre la muestra y una base fuerte y otra entre la muestra y un ácido fuerte. Así, el dihidrógeno fosfato actúa como un ácido ante una base fuerte como el NaOH y el monohidrógeno fosfato actúa como una base ante un ácido fuerte como el HCl. Por lo tanto, para poder cuantificar cada una de las especies el procedimiento que habitualmente se utiliza consiste, mediante la utilización de una potenciometría, en valorar una alícuota de la muestra con NaOH para determinar el dihidrógeno fosfato y otra alícuota se valora con HCl para determinar el monohidrógeno fosfato.
MATERIAL
Potenciómetro, electrodo combinado de vidrio, 1 bureta, erlenmeyers de boca ancha, matraces aforados, embudo, vidrios de reloj.
REACTIVOS NaOH lentejas, HCl 0.1 M, biftalato potásico, Na2CO3.
PROCEDIMIENTO
Preparación de las soluciones valorantes.
Preparar soluciones de NaOH y HCl 0.1 M.
Estandarización de las soluciones valoranets.
Con biftalato potásico y carbonato sódico, respectivamente. Se debe realizar por triplicado.
Valoración de la muestra.
A partir de la concentración aproximada de hidrógenofosfato y dihidrógenofosfato sódico que contiene la muestra (dato que proporciona el profesor), se calcula la cantidad de muestra necesaria por llevar a término la valoración, teniendo en cuenta que se tienen que gastar aproximadamente 2/3 del volumen de la bureta. . Se pone en funcionamiento el instrumento

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 14
seleccionando la opción pH para medir potenciales y se procede a la valoración. Tener en cuenta que cada vez que se añade la disolución valorante se debe agitar el erlenmeyer antes de hacer la lectura. Para saber dónde está aproximadamente el punto de equivalencia, se aconseja hacer una primera valoración rápida. Finalmente, la muestra se valora por triplicado.
RESULTADOS
a. Escribe las reacciones que tienen sitio en la valoración potenciométrica. b. Dibuja la gráfica de valoración así como la primera y segunda derivadas
(ΔpH/Δ volumen respecto volumen valorante, ΔpH2/Δ volumen2 respecto volumen valorante).
c. Encuentra las concentraciones de monohidrógenofosfato y dihidrógenofosfato a la muestra líquida inicial expresadas en normalidades y su incertidumbre.
TRATAMIENTO DE RESIDUOS
Los residuos se deben tirar por el fregadero bien diluidos.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 15
VALORACIONES POTENCIOMÉTRICAS REDOX
PRÁCTICA 4
DETERMINACIÓN DE HIERRO EN PÍLDORAS ANTIANÉMICAS MEDIANTE UNA VALORACIÓN POTENCIOMÉTRICA
OBJETIVO
Determinar la cantidad de hierro (II) en una píldora antianémica, mediante una valoración potenciométrica redox.
FUNDAMENTO
Existen diferentes métodos para la determinación del ión Fe(II) mediante una valoración redox, como la volumetría con dicromato como valorante, que es patrón primario y, por lo tanto, no es necesaria su estandarización, haciendo la determinación más rápida. Pero en este caso, la solución adquiere una coloración que hace difícil la determinación del punto final de la valoración mediante la adición de indicadores. Por esto, se realiza una valoración potenciométrica, que permite la determinación del punto final de la valoración todo y tener esta coloración.
MATERIAL
Potenciómetro, electrodo combinado de platino, 1 bureta, erlenmeyers de boca ancha, vidrios de reloj, matraces aforados, bunsen, embudo.
REACTIVOS
K2Cr2O7 sólido, HCl 6 M y 1 M.
PROCEDIMIENTO
Preparación de la solución valorante. Preparar una solución de K2Cr2O7 0.02 M.
Tratamiento de la muestra. La píldora se disuelve calentándola durante 20 minutos en 25 ml de una solución de HCl 6 M. A continuación se filtra y se lleva hasta un volumen de 100 ml con HCl 1M.
Valoración de la muestra. A partir de la concentración aproximada de sulfato de hierro que contiene la píldora (dato que proporciona el profesor) se calcula el volumen de muestra

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 16
necesario por realizar la valoración teniendo en cuenta que se tienen que gastar aproximadamente 2/3 del volumen de la bureta en cada valoración. Si es necesario aumentar el volumen adicionado al erlenmeyer para que los electrodos queden sumergidos, utilizar HCl 1 M. Se pone en funcionamiento el instrumento, seleccionando la opción mV, para medir potenciales y se procede a la valoración. Es necesario comprobar que tanto la solución con la muestra como la solución con el dicromato tengan pH ácido (menor o igual que 2). De lo contrario, añadir HCl. Para saber dónde está aproximadamente el punto de equivalencia, se aconseja hacer una primera valoración rápida. Finalmente, la muestra se valora por triplicado.
RESULTADOS
a) Escribe las reacciones que tienen lugar en la valoración potenciométrica. b) Dibuja la gráfica de valoración así como la primera y segunda derivada
(Δpotencial/Δ volumen respecto volumen valorante, Δ2potencial/Δ volumen2 respecto volumen valorante).
c) Encuentra la cantidad de sulfato de Fe (II) a la píldora, expresada en mg de sulfato ferroso, y la correspondiente incertidumbre.
TRATAMIENTO DE RESIDUOS
Los residuos que contengan cromo se tienen que depositar en bidón de residuos correspondiente.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 17
PRÁCTICA 5
DETERMINACIÓN DE Fe(II) EN UN PREPARADO FARMACÉUTICO MEDIANTE VALORACIÓN POTENCIOMÉTRICA CON KMnO4
Fundamento
En una valoración potenciométrica el punto final se detecta determinando el volumen
en el cual ocurre un cambio de potencial relativamente grande cuando se adiciona el agente valorante.
Este método instrumental se puede utilizar para todas las reacciones adecuadas para
los propósitos volumétricos: ácido-base, rédox, precipitación y formación de complejos. Se selecciona un electrodo indicador adecuado y con un electrodo de referencia (de calomelanos o de Ag/AgCl) se completa la celda electroquímica, siendo su potencial:
Ecelda = EInd. - ERef.
El EInd. contiene la información que se requiere sobre la concentración de analito. Para determinar el punto final se pueden utilizar varios métodos. El más directo es el
de la gráfica E vs. V, en la cual el punto medio de la porción ascendente de la curva se considera como el punto final. Otros métodos que pueden utilizarse son los que usan la representación de la primera derivada (ΔE/ ΔV) vs. Vmedio y en este caso el máximo de la curva corresponde al punto final; y el que utiliza la representación de la segunda derivada (Δ2E/ ΔV2) vs. Vmedio, en la que el corte con el eje de abcisas determina el punto final.
Disoluciones necesarias
- Disolución de KMnO4 0.02 N de factor conocido (PM = 158 g/mol).
- Disolución de H2SO4 1:4. - Disolución de H3PO4 al 85%
Material e instrumentación necesarios
• Vaso de precipitados de 250 ml. • Bureta de 50 ml. • Matraz aforado de 100 ml • Pipeta de 10 ml. • Potenciométro. • Electrodo de Pt . • Electrodo de Ag/ClAg. • Agitador magnético.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 18
Preparación de la muestra Se toma una pastilla de FERO-GRADUMET y se pulveriza en un mortero de vidrio. A continuación, el polvo obtenido se trasvasa a un matraz aforado de 100 ml arrastrandolo con un poco de agua destilada, se añaden 5 ml de de la disolución de H2SO4 1:4 y se enrasa con agua destilada. [Según indica el fabricante cada pastilla contiene 525 mg de FeSO4, lo que equivale a 105 mg de Fe(II)].
Procedimiento
En un vaso de precipitados de 250 ml, introducir 10 ml de la disolución de muestra
problema preparada anteriormente, añadir 5 ml de la disolución de H2SO4 1:4, 2 ml de H3PO4 al 85% y diluir posteriormente con agua hasta 200 ml. Llenar la bureta con la disolución de KMnO4 0.02 M e instalarla de modo que el pico toque la disolución a valorar. Introducir los electrodos de Pt y Ag/ClAg en el vaso de modo que no se rocen con la varilla agitadora. Añadir desde la bureta sucesivas porciones de 1 o 0.5 ml (según corresponda) y tras un breve tiempo de agitación leer el valor de E (mV). En las proximidades del punto de equivalencia añadir porciones de 0.2 ml y continuar la adición hasta rebasar el punto de equivalencia y llegar a un volumen final de agente valorante de 20 ml. 1.- Representar gráficamente sobre papel milimetrado a) E(mV) vs. V (ml) b) Primera derivada: (ΔE/ ΔV) vs. Vmedio (ml) 2.- Deducir de las gráficas el contenido de Fe(II) en la pastilla de FERO-GRADUMET y comparar con el valor especificado por el fabricante.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 19
PRÁCTICA 6
DETERMINACIÓN DE COBRE EN UNA MUESTRA LÍQUIDA MEDIANTE UNA VALORACIÓN POTENCIOMÉTRICA
OBJETIVO
Determinar la concentración de una solución de cobre preparada, mediante una valoración potenciométrica redox.
FUNDAMENTO
Existen diferentes métodos para la determinación del Cu(II). Uno de ellos es una determinación iodométrica, que consiste al valorar el yodo liberado en la reacción redox que tiene sitio entre el Cu(II) y el yoduro, dado que la cantidad de yodo liberada es equivalente a la del cobre que hay en la muestra. Puesto que la solución resultante presenta turbidez y coloración, es difícil la determinación del punto final de la valoración mediante la adición de indicadores, por esto, es más adecuado hacer una valoración potenciométrica.
MATERIAL
Potenciómetro, electrodo combinado de platino, 1 bureta, erlenmeyers de boca ancha, vidrios de reloj y matraces aforados.
REACTIVOS
KI sólido, Na2S2O3 sólido, H2SO4 1M, KIO3 sólido.
PROCEDIMIENTO
Preparación de la solución valorante.
Preparar una solución de tiosulfato sódico 0.05 M.
Estandarización de la solución valorante.
Con iodato potásico. Se debe añadir yoduro potásico en exceso (1.5 - 2 g) y acidificar el medio con 5 ml de ácido sulfúrico. La estandarización se tiene que hacer por triplicado.
Valoración de la muestra.
A partir de la concentración aproximada de cobre que contiene la muestra (dato que proporciona el profesor) se calcula el volumen de muestra necesario por realizar la valoración teniendo en cuenta que se tienen que gastar aproximadamente 2/3 del volumen de la bureta en cada valoración. Se debe añadir la cantidad necesaria de yoduro potásico más un ligero

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 20
exceso de aprox. 0.3 g a la muestra antes de valorarla. Se pone en funcionamiento el instrumento, seleccionando la opción mV para medir potenciales y se procede a la valoración. Sí no se cubre el electrodo, añadir agua hasta que quede totalmente sumergido. Para saber dónde está aproximadamente el punto de equivalencia, se aconseja hacer una primera valoración rápida. Finalmente, la muestra se valora por triplicado.
RESULTADOS
a. Escribe las reacciones que tienen sitio en la valoración potenciométrica. b. Dibuja la gráfica de valoración así como la primera y segunda derivada
(Δpotencial/Δvolumen respecto volumen valorante, Δ2potencial/Δvolumen2 respecto volumen valorante).
c. Encuentra la concentración de Cu(II) en la muestra expresada en gramos por litro y su incertidumbre.
TRATAMIENTO DE RESIDUOS
Los residuos se deben tirar por el fregadero bien diluidos.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 21
VALORACIONES POTENCIOMÉTRICAS ESI
Determinación de Amonio en una muestra acuosa
1.- Principio. Determinación por electrodo específico del tipo «Sensible a un gas». Se basa en medir la modificación que experimenta el pH de una solución interna del electrodo, provocada al difundirse amoniaco, a través de una membrana permeable al gas hecha de un material poroso hidrófobo. Como el electrodo mide concentración de NH3 es necesario que todo el ion NH4
+ se convierta en gas amoniaco, para lo cual es imprescindible que el pH de la solución se encuentre en un valor de 11 o superior. El método es de aplicación para concentraciones comprendidas entre 0,01 y 0,6 mg/litro, expresadas en ión amonio. 2.- Material y aparatos.
Medidor digital para electrodos selectivos.
Electrodo selectivo de amoníaco.
Soporte para mantener el electrodo formando un ángulo de 60.° con la vertical.
Agitador magnético.
Material de uso corriente en el laboratorio.
3.- Reactivos.
1. Solución patrón concentrada de cloruro amónico de 3.140 mg/litro de ión amonio. Pesar 0,314 g de cloruro amónico P.A. disolver en agua exenta de ión amonio y aforar a 100 ml.
2. Solución de cloruro amónico de 100 mg/litro de ión amonio, obtenida por
dilución de la anterior.
3. Solución de hidróxido sódico 10 M.
4. Solución tampón de pH = 4, con adición de cloruros, de tal manera que resulte una concentración de este ión 0,1 M.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 22
4.- Procedimiento. Conservación de la muestra.
Si ésta no va a ser analizada inmediatamente, se la debe acidificar a pH 6 aproximadamente con ácido clorhídrico 1 M.
El electrodo se conservará sumergido en una solución de cloruros al 1%.
Antes de su uso se introducirá unos diez minutos en disolución tampón de pH = 4, con adición de cloruros, de tal manera que resulte una concentración de este ión 0,1 M.
Comprobación del funcionamiento correcto del electrodo.
Se trata de determinar la pendiente de la ecuación de Nerst, definida como el cambio de potencial observado cuando la concentración cambia con un factor 10.
Poner 100 ml de agua exenta de amoniaco, añadir 1 ml de solución 10 M de
NaOH
Introducir el electrodo, agitando continuamente.
Añadir 1 ml de disolución 3.1. Dejar que se estabilice y medir el potencial.
Añadir 10 ml de la misma disolución 3.1
Dejar que se estabilice y medir el potencial.
El estado del electrodo es satisfactorio si la diferencia entre ambas lecturas es 57 ± 2 mV a 20. °C.
5.- Curva de calibrado.
1. Preparar las disoluciones patrón que se indican el la tabla a partir de la disolución “madre” de 1000 ppm en NH3
Volumen (ml) Conc. mg/litro de NH3
1 2.5 2 5 3 7.5 4 10 5 12.5
Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 23
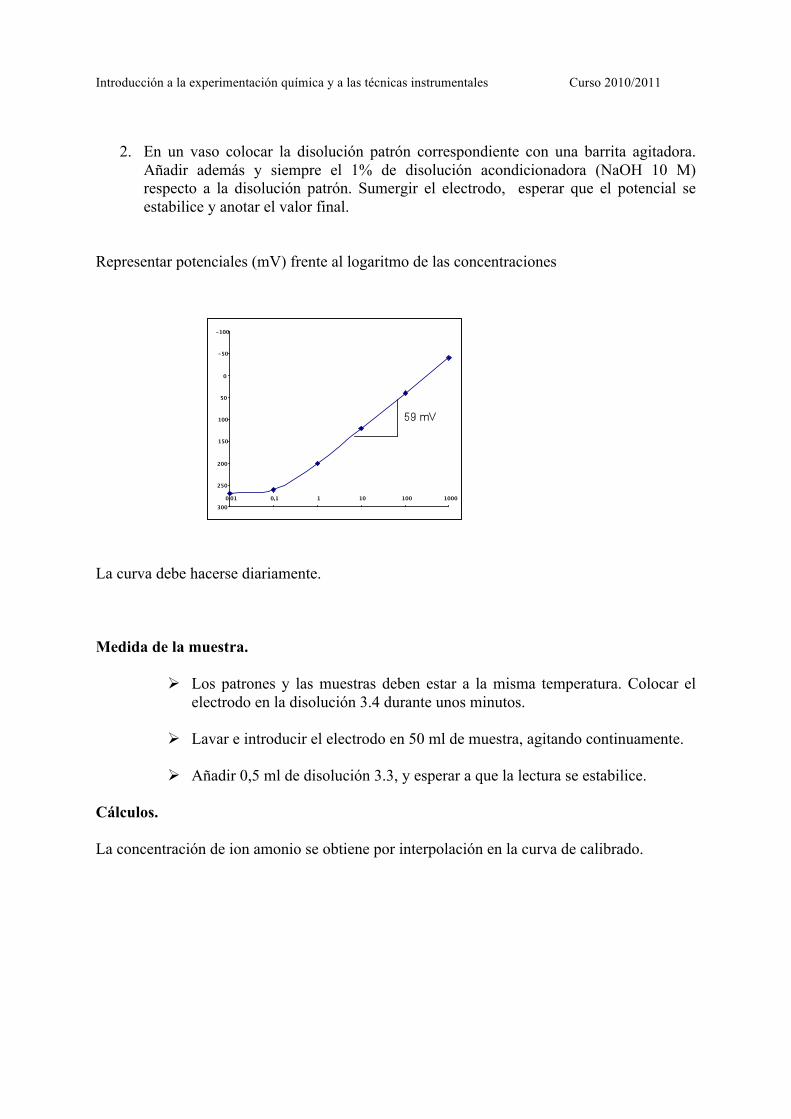
2. En un vaso colocar la disolución patrón correspondiente con una barrita agitadora.
Añadir además y siempre el 1% de disolución acondicionadora (NaOH 10 M) respecto a la disolución patrón. Sumergir el electrodo, esperar que el potencial se estabilice y anotar el valor final.
Representar potenciales (mV) frente al logaritmo de las concentraciones La curva debe hacerse diariamente. Medida de la muestra.
Los patrones y las muestras deben estar a la misma temperatura. Colocar el electrodo en la disolución 3.4 durante unos minutos.
Lavar e introducir el electrodo en 50 ml de muestra, agitando continuamente.
Añadir 0,5 ml de disolución 3.3, y esperar a que la lectura se estabilice.
Cálculos. La concentración de ion amonio se obtiene por interpolación en la curva de calibrado.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 24
VALORACIONES CONDUCTIMÉTRICAS
OBJETIVOS
• Manejo de parámetros conductimétricos. • Aplicación analítica de medidas de conductividad para determinar el punto final de
una valoración. • Determinación del valorante más apropiado para un determinado ión según sus curvas
de valoración. FUNDAMENTO TEÓRICO La conducción de corriente eléctrica a través de una disolución implica una migración hacia los electrodos de las especies cargadas positiva y negativamente. Todas las partículas cargadas contribuyen al proceso de conducción y la contribución de cualquier especie viene gobernada por su concentración y su movilidad. Cuando todos los demás factores se mantienen constantes, las variaciones en la concentración de un electrolito dan lugar a cambios en las conducción eléctrica. Por tanto, es posible relacionar la concentración con la conductividad eléctrica. El empleo de la conductividad para la determinación cuantitativa directa de un ión dado está muy limitado. Las aplicaciones de medida directa de conductividad incluyen el análisis de mezclas binarias agua-electrolito y la determinación de la concentración total de electrolitos de una disolución. Esta última aplicación es particularmente importante a la hora de emplearla como criterio de pureza del agua destilada. Una aplicación analítica más importante de las medidas de conductividad es su uso para señalar el punto final de una valoración. En este caso lo importante es el cambio de conductividad de la disolución. La principal ventaja del punto final conductimétrico es su aplicabilidad a disoluciones muy diluídas y a sistemas que implican reacciones relativamente incompletas. Valoraciones conductimétricas Todos los conceptos y parámetros utilizados en esta práctica son los mismos que los ya vistos en la práctica 4: el concepto de conductividad y todos los parámetros asociados a su cálculo. Es por ello que obviamos su definición y pasamos a dar una pequeña reseña de lo que son las valoraciones con indicador conductimétrico o valoraciones conductimétricas. Una valoración conductimétrica implica la medida de la conductividad de la muestra después de sucesivas adiciones de reactivo. El punto final es determinado a partir de la representación de la conductividad o de la conductividad específica como función del volumen de reactivo añadido. En general estas representaciones se caracterizan por segmentos rectilíneos con pendientes desiguales a ambos lados del punto de equivalencia. Cuanto más diferenciadas sean las dos pendientes, más exactamente se determinará el punto final. Las medidas de conductividad se realizan en una célula conductimétrica, que poseen dos electrodos de igual área y normalmente de platino.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 25
Ya se ha visto que la relación entre conductividad y conductividad específica viene dada por la ecuación Donde al cociente l/A lo denominábamos constante de la célula. Dicho cociente representa características físicas de los electrodos, y por tanto su valor es constante para cada célula conductimétrica. Un factor que puede provocar grandes variaciones en la conductividad es la temperatura de la muestra; por ello, si se producen grandes cambios en la conductividad es posible que sea necesario el control termostático de la misma. Durante la valoración se producen igualmente cambios en el volumen de la disolución, y si este efecto no se corrige, es posible que se obtengan relaciones no lineales en las curvas de valoración. La corrección se lleva a cabo multiplicando la conductividad medida por el factor (V + v) / V ; donde V es el volumen inicial de la muestra y v es el volumen total de reactivo añadido hasta ese momento. Con el fin de mantener v en un valor pequeño, el reactivo empleado en la valoración conductimétrica suele emplearse varias veces más concentrado que la disolución a valorar. Curvas de valoración Aunque la conductividad no es una propiedad física específica de las disoluciones de electrolitos, pues depende de todas las especies iónicas presentes, la sustitución de un ión por otro hará que la conductividad cambie al diferir las movilidades de estos dos iones. Este es el fundamento de las valoraciones con indicador conductimétrico (valoraciones conductimétricas) que tienen como objeto primordial la localización de puntos de equivalencia de valoraciones. Sólo tienen importancia los cambios de conductividad durante las valoraciones y no los valores absolutos de conductividad. En principio el método conductimétrico es aplicable a los cuatro tipos de volumetrías, si bien la mayoría de las valoraciones conductimétricas satisfactorias son las de los tipos ácido-base y precipitación. La representación de la conductividad frente al volumen (o la fracción) de valorante añadido, corresponde a dos líneas rectas que se cortan en el punto de equivalencia. El punto de equivalencia se calcula de modo gráfico, tomando tres o cuatro medidas antes del punto de equivalencia (punto final) y otras tantas después de él y con la misma separación; o bien resolviendo analíticamente el sistema de ecuaciones de las dos rectas de valoración, que responden a la expresión:
Λ= n + m·f
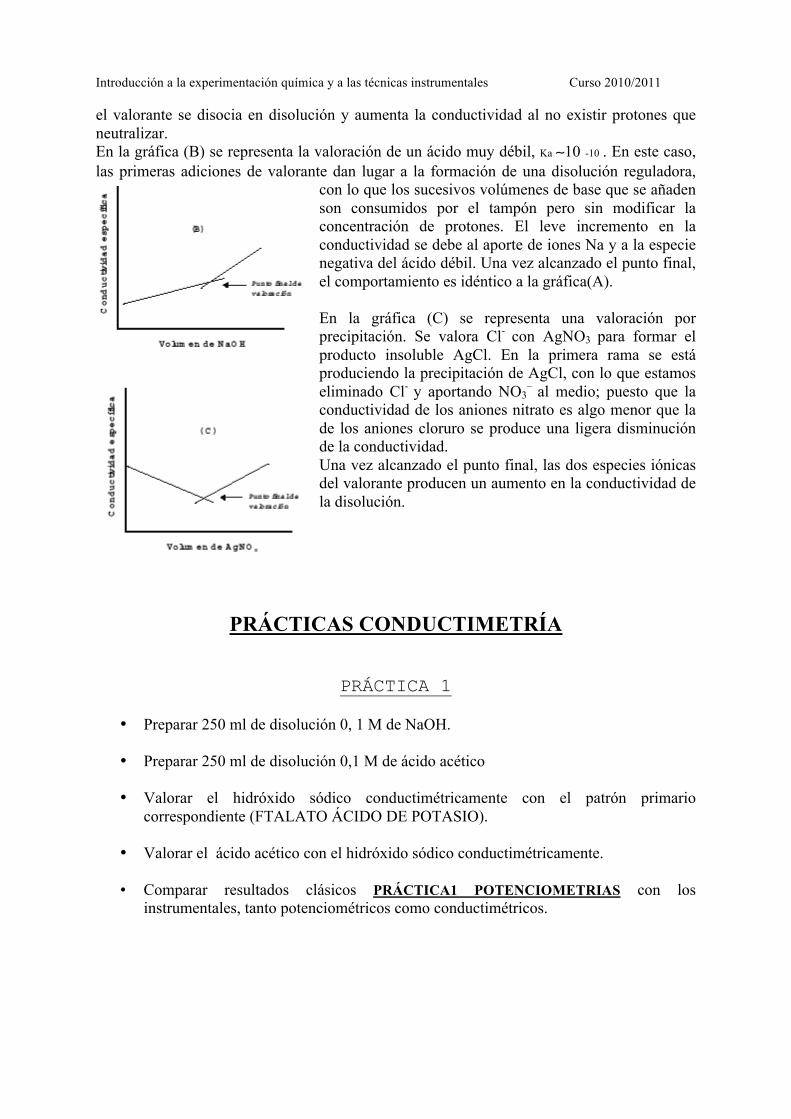
donde n y m son constantes (ordenada en el origen y pendiente), y f la fracción de valorante. Ejemplos de curvas de valoración Veamos algunos ejemplos de curvas de valoración conductimétrica: En la representación (A) se ha realizado la valoración de un ácido fuerte, HCl, con NaOH. La rama descendente se produce por la neutralización de los protones por parte de los OH - del valorante; se está sustituyendo una especie de gran conductividad, protones, por otra de mucha menor conductividad, Na+. Una vez que se alcanza el punto final,
Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 26
el valorante se disocia en disolución y aumenta la conductividad al no existir protones que neutralizar. En la gráfica (B) se representa la valoración de un ácido muy débil, Ka ∼10 -10 . En este caso, las primeras adiciones de valorante dan lugar a la formación de una disolución reguladora,
con lo que los sucesivos volúmenes de base que se añaden son consumidos por el tampón pero sin modificar la concentración de protones. El leve incremento en la conductividad se debe al aporte de iones Na y a la especie negativa del ácido débil. Una vez alcanzado el punto final, el comportamiento es idéntico a la gráfica(A). En la gráfica (C) se representa una valoración por precipitación. Se valora Cl-
con AgNO3 para formar el producto insoluble AgCl. En la primera rama se está produciendo la precipitación de AgCl, con lo que estamos eliminado Cl-
y aportando NO3–
al medio; puesto que la conductividad de los aniones nitrato es algo menor que la de los aniones cloruro se produce una ligera disminución de la conductividad. Una vez alcanzado el punto final, las dos especies iónicas del valorante producen un aumento en la conductividad de la disolución.
PRÁCTICAS CONDUCTIMETRÍA
PRÁCTICA 1
• Preparar 250 ml de disolución 0, 1 M de NaOH.
• Preparar 250 ml de disolución 0,1 M de ácido acético
• Valorar el hidróxido sódico conductimétricamente con el patrón primario correspondiente (FTALATO ÁCIDO DE POTASIO).
• Valorar el ácido acético con el hidróxido sódico conductimétricamente.
• Comparar resultados clásicos PRÁCTICA1 POTENCIOMETRIAS con los
instrumentales, tanto potenciométricos como conductimétricos.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 27
PRÁCTICA 2
DETERMINACIÓN CONDUCTIMÉTRICA DE LA COMPOSICIÓN DE UNA MUESTRA DE ÁCIDO CLORHÍDRICO E ÁCIDO ACÉTICO
• Calibrar el conductivímetro
• Pipetear a un vaso de precipitados de 250 ml, 25 ml de la mezcla problema, un imán y suficiente agua destilada para que queden cubiertos los electrodos. Medir la conductividad inicial.
• Llenar una bureta con NaOH 0,1 M, colocarla sobre o vaso de precipitados y dejar
caer NaOH de 0,5 en 0,5 ml, anotando la medida del conductivímetro después de cada adición. Se deben apreciar tres cambios en la variación de la conductividad, parándose la valoración cuando tengamos diez medidas del último tipo.
• Representar gráficamente la conductividad frente al volumen, determinar las
ecuaciones de las tres rectas y calcular los puntos de corte de las mismas, para conocer el volumen gastado por cada ácido.
• Calcular la composición de la muestra.
PRACTICAS 3 y 4 MATERIALES Y REACTIVOS Reactivos H2SO4 96% comercial. Na2SO4 comercial BaCl2 comercial Disolución problema Ba2+ Disolución de AEDT Materiales Célula conductimétrica Conductímetro Balanza Agitador magnético Bureta, Pipeta, Probeta Matraces aforados, vasos de precipitado, etc...

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 28
PROCEDIMIENTO EXPERIMENTAL
PRÁCTICA 3
En primer lugar, se va a proceder a valorar conductimétricamente una disolución de una sal de Ba2+ de concentración conocida (0,01 M) con dos valorantes: Na2SO4 y H2SO4. Se pretende determinar cual es el mejor valorante de los dos.
a) Valoración con Na2SO4: Se toman 50 ml de la disolución de Ba2+ 0,01 M y se introducen en el vaso de valoración. Se calibra el aparato según las instrucciones del profesor. Se introduce la célula conductimétrica de modo que los electrodos queden cubiertos por la disolución, evitando que la barra de agitación magnética golpee la célula. Se valora frente a la disolución 0,1 M de Na2SO4.
b) Valoración con H2SO4: Se toman otros 50 ml de disolución 0,01 M de Ba2+ y se
valoran de forma similar con H2SO4 0,l M. Los valores de conductividad específica leídos se multiplican por el factor de corrección de variación de volumen (V+ v)/V y se representan frente al volumen de valorante. Se trazan las rectas para cada una de las valoraciones, se calcula por el método de los mínimos cuadrados las ecuaciones de las dos rectas en cada valoración y se determina el punto de corte de las mismas, el cual será el punto final de la valoración. Indicar cual de los métodos resulta más recomendable para la valoración del ión bario.
PRÁCTICA 4
Una vez elegido cual es el valorante más adecuado, se realiza la valoración conductimétrica de una muestra problema de Ba2+ . Resolviendo de forma análoga el punto final de la valoración y calculando la concentración de Ba2+ en la muestra problema. BIBLIOGRAFÍA Problemas y experimentos en análisis instrumental, Clifton E. Meloan y Robert M. Kiser, Ed. Reverté, 1973. Instrumental methods of analysis, Hobart H. Willard, Lynne L. Merritt y John A. Dean, D. Van Nostrand Company, 1974. Fundamentos de química analítica, Douglas A. Skoog y Donald N. West, Ed. Reverté, 1976.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 29
ESPECTROSCOPÍA UV-VIS
al acabar las prácticas el alumno TIENE QUE SER CAPAZ DE:
• Enunciar la expresión de la ley de Beer y conocer sus limitaciones. • Relacionar absorbancia y porcentaje de transmitáncia. • Relacionar coeficiente de absortivitad molar y longitud del camino óptico con
sensibilidad y tope de detección. • Justificar la selección de la longitud de onda óptima del espectro. • Evaluar la importancia de las sustancias/substancias interferentes. • Diseñar y preparar una serie de patrones y construir una recta de calibración. • Justificar la necesidad de obtener medidas del blanco analítico. • Identificar las fuentes de error asociadas a una determinación colorimétrica. • Determinar colorimétricamente la concentración de Cu o Fe en diferentes tipos de
muestras.
FUNDAMENTO TEÓRICO
La espectrofotometría de absorción molecular UV-Visible se basa en la medida de la disminución de la intensidad de la radiación electromagnética como consecuencia de la absorción que se produce por la interacción con el analito. Esta disminución de la intensidad se puede relacionar con la concentración del analito mediante la ley de Beer :
A = log(Io/It) = ε b c
dónde A es la absorbancia a una longitud de ola determinada, Io es la intensidad del haz incidente, It es la intensidad del haz transmitido, ε es el absortividad molar, b es la longitud del camino óptico, o sea, la longitud de la sección de la cubeta por dónde pasa el haz de luz (en cm) y c es la concentración del analito (moles/l). La ley de Beer se cumple para radiación monocromática y para un determinado intervalo de concentración, conocido como intervalo de linealidad, que es necesario determinar por cada analito en las condiciones de trabajo.
BIBLIOGRAFÍA
1. A.I. Vogel, Vogel's Tetxbook ofoff Quantitative Inorganic Analysis, 4ª ed., Longman, London, 1978. 2. A.I. Vogel, Química Analítica Cuantitativa: Teoría y Práctica, 2ª ed., Ed. Kapelusz, Buenos Aires, 1969. 3. E.B. Sandell. Colorimetric Determination ofoff trazo ofoff metals. Interescience Publishers, New York, 1950. 4. L. Meites (ed), Handbook ofoff Analytical Chemistry. McGraw Hill, New York, 1982.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 30
PRÁCTICA 1-PRÁCTICA MODELO-LEER DETENIDAMENTE
1.- OBJETIVO DE LA PRÁCTICA. Determinación cuantitaviva de la concentración de MnO4-- en una muestra problema mediante espectroscopía de absorción molecular UV-VIS. 2.- MATERIAL Y REACTIVOS. 2.1.- MATERIAL.
• 3 matraces aforados de 25 mL. • 1 matraz aforado de 100 mL. • 2 vasos de precipitado de 100 mL. • 2 pipetas graduadas. • 2 cubetas para espectrofotómetro • 1 frasco de agua destilada • 1 pipeteado
3.- REACTIVOS. Disolución KMnO4 0,02 M Muestra problema 4.- PROCEDIMIENTO EXPERIMENTAL. 4.1.- Obtención del espectro de absorbancia Spectronic 20
• Con una pipeta graduada, tomar 1 mL de la disolución 0,02M de KMnO4 y verterlo en un vaso de precipitado de 100 mL. Añadir poco a poco, y agitando cuidadosamente, agua destilada hasta que la disolución resultante presente un color similar al de la muestra problema.
• Con la disolución preparada se obtendrá el espectro de absorción como sigue:
Debido al color púrpura de la disolución de MnO4- , podemos predecir la zona
del espectro en la que se obtendrá la máxima absorbancia. Después de poner a punto el espectrofotómetro, con la ayuda del profesor, seleccionar una longitud de onda 60 nm por debajo del límite inferior del intervalo correspondiente.
"Hacer el blanco" para dicha longitud de onda. Es decir, toma una de las cubetas y, tras llenarla con agua destilada (es el blanco para este caso particular), sitúala en el portacubetas del espectrofotómetro y ajusta la lectura al cero de Absorbancia.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 31
A continuación llena la otra cubeta con la disolución previamente preparada, sitúala en el portacubetas y espera unos segundos hasta que la lectura de absorbancia se estabilice. Anota el valor de absorbancia obtenido para esta longitud de onda.
Selecciona en el espectrofotómetro una longitud de onda 10 nm superior a la anterior y repite el proceso anteriormente descrito. Repite de nuevo todo el proceso hasta alcanzar una longitud de onda 40 nm por encima del límite superior del intervalo de longitudes de onda correspondiente al espectro de absorción del MnO4
-
• Introduce los datos en una hoja de cálculo Excel y representa el espectro de absorción del KMnO4 en la forma A vs. λ.
NOTA IMPORTANTE: Selecciona la longitud de onda correspondiente al máximo de absorbancia. Dicha longitud de onda será la utilizada en el análisis cuantitativo del MnO4
-
Helios γ
• Se coloca el blanco en la cubeta, agua destilada, y se hace un barrido entre longitudes de onda de 400 a 600 nm.
• Se coloca la disolución de permanganato en la cubeta y se hace el barrido entre las
mismas longitudes de onda, obteniéndose el espectro automáticamente. 4. 2.- Preparación de la recta de calibrado
A. A partir de esta disolución de KMnO4 0,02M, prepara, utilizando los matraces aforados de 25 ml, y siguiendo el mismo procedimiento experimental, 5 disoluciones patrón de KMnO4 de concentraciones entre 10-4 M y 10-5 M
4. 3.- Medición de los patrones y obtención de la curva de calibrado
• Siguiendo el procedimiento ya conocido, medir la absorbancia de cada una de las disoluciones patrón a la longitud de onda seleccionada. Recordar que antes de la medición de cada patrón debe hacerse el blanco. La medición de la absorbancia de los cinco patrones debe realizarse en orden creciente de concentraciones y utilizando una misma cubeta. Para ello, después de cada medida: - Desechar la disolución patrón ya medida - Limpiar la cubeta con agua destilada varias veces - Limpiar la cubeta con la nueva disolución patrón una vez - Llenar la cubeta con la nueva disolución patrón y medir
• Con las absorbancias anotadas para cada una de las disoluciones patrón, representar
gráficamente, en un papel milimetrado, la curva de calibrado en la forma: A vs. C

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 32
• Mediante un ajuste de regresión por mínimos cuadrados, calcular la ecuación de la
recta obtenida. 4. 4.- Medición de la muestra problema
A. Siguiendo el procedimiento conocido, medir la absorbancia de la muestra problema. B. Calcular la concentración de dicha muestra mediante dos métodos:
- Gráficamente, mediante interpolación del valor de absorbancia obtenido - Matemáticamente, mediante la utilización de la ecuación de la recta de calibrado5.- RESULTADOS EXPERIMENTALES - Longitud de onda seleccionada: nm - Ecuación de la recta de calibrado:
• Concentración de MnO4-- en la muestra problema:
a. Gráficamente: M b. Matemáticamente: M

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 33
PRÁCTICA 2
DETERMINACIÓN ESPECTROFOTOMETRICA DE HIERRO TOTAL EN VINOS o AGUAS
Fundamento
La espectroscopia de absorción molecular es uno de los métodos mas usados en el
análisis cuantitativo. Una gran variedad de especies inorgánicas y orgánicas absorben
radiación en las longitudes de onda del ultravioleta y visible y por tanto, es posible su
cuantificación mediante medida de la radiación absorbida, utilizando un espectrofotómetro.
La absorción se cuantificar por unidades arbitrarias llamadas "unidades de absorbancia", las
cuales están relacionadas linealmente con la concentración de analito existente.
En esta práctica, se aplica esta técnica para determinar el contenido total de hierro
(Fe(II) y Fe(III)) en vinos blancos y poco coloreados, mediante medida de la absorción del
complejo rojo formado entre el ion Fe(II) y la 1,10-Fenantrolina.
Disoluciones necesarias
- Disolución 2.10-4 M de sulfato ferroso amónico hexahidratado
- Disolución 2.10-3 M de 1,10-Fenantrolina
- Disolución de clorhidrato de hidroxilamina al 10% ¿qué función tiene esta disolución?
- Disolución 0.1 M de acetato sódico ¿qué función tiene esta disolución?
Material e instrumentación necesarios - Matraces aforados de 25 mL
- Pipetas de 2, 5 y 10 mL o micropipetas.
- Espectrofotómetro
Procedimiento
Construir una recta de calibrado, adicionando en matraces de 25 ml, 1, 2, 3, 4 y 5 mL
de disolución patrón de sulfato ferroso amónico hexahidratado (pipeta de 10 ml), 1 mL de
clorhidrato de hidroxilamina (pipeta de 2 ml), 5 ml de acetato sódico (pipeta de 10 mL),
2.5 mL de 1,10-Fenantrolina (pipeta de 5 ml) y enrasar con agua destilada. Esperar 10
minutos y medir la absorbancia a 510 nm de longitud de onda.

Para el análisis de la muestra, pipetear 10 ml de vino a un matraz aforado de 50 ml y añadir
las mismas cantidades de los reactivos anteriormente citados. Esperar al menos 10 minutos
para que se desarrolle el color completamente y por último, medir la absorbancia en el
espectrofotómetro a 510 nm de longitud de onda.
Resultados 1. Obtención de la Recta de calibrado
Matraz nº
mL de Fe(II)
ppm de Fe(II)
A (510 nm)
1
1
2
2
3
3
4
4
5
5
2. Medida de las Muestras
Muestra nº
A (510 nm)
1
2
3
Tratamiento de datos
1.- Construir la recta de calibrado, representando en el eje de abcisas las concentraciones de
las disoluciones patrón y en el eje de ordenadas los correspondientes valores de absorbancia.
2.- Interpolar en la recta de calibrado los valores de la absorbancia encontrados para las
muestras problema y deducir el valor de la concentración de las mismas.

PRÁCTICA 3
DETERMINACIÓN ESPECTROFOTOMÉTRICA DE FE(III) POR FORMACIÓN DE
UN COMPLEJO COLOREADO CON SULFOCIANURO MATERIALES. Matraces de distintos volúmenes (25, 100, 250 mL) Pipetas de 1 a 25 ml Vasos de precipitado. Baño termostático. Espectrofotómetro con medida a 470- 490 nm. REACTIVOS Disolución patrón de hierro III (preparada). Se obtiene disolviendo una cantidad (pesada exactamente hasta la cuarta cifra decimal) tal de nitrato de hierro químicamente puro que la disolución resultante contenga alrededor de 500 mg de Fe(III) por litro. La disolución ya preparada aparece etiquetada con su contenido en hierro en partes por millón. HNO3 0.2 M (preparada) KSCN al 10 % (preparada) HCl comercial HNO3 comercial Muestra problema de Fe MÉTODO EXPERIMENTAL. Los iones SCN - reaccionan, en un medio moderadamente ácido con los iones Fe(III) dando lugar a un complejo de color rojo. En función de las cantidades relativas SCN - / Fe(III), pueden ser detectadas diversas formas complejas con índices de coordinación en el rango 1-6 [Fe(SCN)+2 ; Fe(SCN)2+ ; ...;Fe(SCN)6-3 ] (log 1 = 2,1; log 2 = 3,4; log 3 = 3,9; ...; log 6 = 3,7). La intensidad del color de estos complejos aumenta con el índice de coordinación, pudiéndose, pues, conseguir una mayor sensibilidad en la determinación trabajando en un exceso razonable de reactivo. Esto trae como consecuencia la necesidad de controlar de forma precisa el que tanto patrones como muestras posean la misma concentración de SCN –La solución acuosa debe de ser suficientemente ácida para evitar la hidrólisis del ion Fe(III) (pH = 3), pero no tan ácida que la concentración de SCN - se vea disminuida por polimerización. Por otra parte los complejos no son muy estables (30 min) y pueden existir durante cierto tiempo solamente en exceso de SCN-. Una concentración 0,05-0,2 N de ácido (HCl, H2SO4, HNO3 ó HClO4) es el punto óptimo que permite obviar todos los inconvenientes mencionados. La sensibilidad del procedimiento puede ser considerablemente aumentada por la presencia de disolventes orgánicos tales como la acetona o dioxano ( = 8,5 x 103
en solución acuosa; ε = 1,8 x 104
en solución al 50% acetona) o por extracción en disolventes oxigenados (éteres, ésteres y cetonas) ( = 2,4 x 104 en MIBK).

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 33
Selección de las condiciones experimentales de medida. Antes de realizar las medidas cuantitativas es necesario determinar la posición en el espectro del máximo o máximos de absorción del complejo, para lo cual se procede al trazado del espectro correspondiente a una concentración apropiada en la región 350-700 nm de forma automática o punto a punto. (Este apartado no se realizará en la Práctica). Trazado de la curva de calibrado. Se toman 5 ml (con pipeta) de la disolución patrón de 500 ppm de Fe y se transfieren a un matraz aforado de 100 ml, diluyéndose hasta la marca de enrase con HNO3 0.2 M. Con una pipeta se toman cuatro volúmenes exactos, comprendidos entre 0.5 y 5 ml de la disolución anterior, que se colocan en cuatro matraces aforados de 25 ml (disoluciones patrón de Fe). A un quinto matraz se agregan 12.5 ml (con pipeta) de KSCN y se enrasa hasta la marca con HNO3 0.2 M (disolución de referencia para ajustar a cero el espectrofotómetro antes de realizar las medidas). Tratar separadamente cada disolución patrón de Fe como sigue:
- Ajustar a cero el espectrofotómetro con la disolución de referencia. Agregar 12.5 ml de KSCN (con pipeta) a una de las cuatro disoluciones patrón de Fe (no agregar KSCN hasta justamente antes de hacer la medición espectrofotométrica) y enrasar con HNO3 0.2 M hasta 25 ml. Mezclar cuidadosamente. Se ha obtenido el complejo Fe(SCN)6
-3 . Pasar a la cubeta del espectrofotómetro la
disolución del complejo formado y leer la absorbancia inmediatamente a 480 nm. Anotar el resultado. Se realiza la misma operación para las tres restantes disoluciones patrón de Fe.
- Representar gráficamente la recta de calibrado (absorbancias frente concentraciones de Fe).
Determinación de la cantidad de Fe en una muestra problema. Se pesan exactamente entre 0.5 – 1 g de la muestra problema (anotar la referencia de la misma para entregarla con los resultados del análisis) en un vaso de 100 ml perfectamente seco. En campana de extracción se adicionan 5 ml de HCl 5M (que los alumnos deben preparar previamente) y 2 ml de HNO3 concentrado. Una vez disuelta totalmente la muestra, se transfiere a un matraz aforado de 250 ml (procurando evitar cualquier pérdida de la disolución). Se lava varias veces el vaso con agua destilada, pasando el líquido de lavado al matraz aforado y finalmente se enrasa el matraz hasta la marca. Agitar para homogeneizar. Esta es la disolución de trabajo (A) donde se va analizar el contenido de Fe de la muestra. Como la concentración de Fe es desconocida, es necesario inicialmente averiguar si el complejo presenta una absorbancia que quede dentro del rango de calibrado establecido anteriormente con los patrones de Fe. Para ello se toma un volumen cualquiera de la disolución A, por ejemplo 2 ml, y se trata de la misma manera que los patrones utilizados en el trazado de la curva de calibrado. Es decir, se ajusta el espectrofotómetro a cero, se transfieren los 2 ml de la disolución de trabajo A a un matraz aforado de 25 ml. Adicionar 12.5 ml de KSCN (pipeta) y enrasar con ácido HNO3 0.2 M a 25 ml. Mezclar cuidadosamente.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 34
Se ha obtenido el complejo Fe(SCN)6-3 . Pasar a la cubeta del espectrofotómetro la disolución
del complejo formado y leer la absorbancia inmediatamente a 480 nm. Anotar el resultado. a) Si la Absorbancia obtenida queda dentro del rango lineal de la recta de calibrado, el análisis es válido. En este caso se hacen los análisis de la disolución de trabajo A con el volumen elegido (2 ml) y se anotan los resultados de absorbancia. b) Si la Absorbancia fuese mayor que la correspondiente al patrón mas concentrado, es necesario tomar un volumen menor de muestra (1 ml, por ejemplo) con la finalidad de que la absorbancia quede dentro del rango lineal de la recta de calibrado. c) Si la absorbancia es menor que la del patrón menos concentrado, debe tomarse en este caso un volumen mayor por ejemplo 3 o 4 ml. Expresión del resultado. Se hace la media de las 6 determinaciones de absorbancia y se transforman en concentración de Fe a través de la recta de calibrado. Dar el resultado en mg de Fe de la muestra sólida original y el tanto por ciento en peso de la misma. Calcular el coeficiente de variación (CV) o RSD (Relative Standard Deviation) de los resultados del análisis mediante la expresión:
CV = s/x 100 El coeficiente de variación debe ser inferior al 5%.
PRÁCTICA 4
DETERMINACIÓN DE MANGANESO Y CROMO EN UNA MUESTRA
• Preparar una disolución estándar de dicromato potásico 0,001 M: disolver 0,075 g de dicromato potásico en unos 125 ml de agua, agregar 3 ml de ácido sulfúrico concentrado y diluir hasta 250 ml. • Preparar una disolución estándar de permanganato potásico 0,003 M: disolver 0,125 g de permanganato potásico en unos 125 ml de agua, agregar 3 ml de ácido sulfúrico concentrado y diluir hasta 250 ml. • A partir de las disoluciones estándar, preparar, por dilución, tres disoluciones más de cada reactivo, y medir la absorbancia de todas las disoluciones en las longitudes de onda en que absorben más el dicromato potásico (440 nm) y el permanganato potásico (525 nm). • Calcular la absortividad molar del permanganato y del dicromato a esas longitudes de onda. • Medir la absorbancia de la mezcla desconocida a las mismas longitudes de onda. • Calcular la concentración molar de cada especie en el problema. Expresar los resultados en mg de Cr/ml e mg de Mn/ml.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 35
PRÁCTICA 5
DETERMINACIÓN ESPECTROFOTOMÉTRICA DE COBRE EN UNA MUESTRA ACUOSA
OBJETIVO
Determinación de la concentración de cobre de una muestra acuosa por medida espectrofotométrica del complejo amoniacal.
MATERIAL
Pipetas, matraces aforados, vasos de precipitados.
REACTIVOS
Solución madre de Cu(II) de 2000 ppm, ácido clorhídrico 12 M, hidróxido amónico 15 M.
PROCEDIMIENTO
Preparación de las soluciones:
- Solución de Cu+2 a partir de la solución madre de 2000 ppm. - Solución de HCl 2 M a partir de HCl concentrado (12 M).
Preparación de los patrones de la recta de calibración y la muestra.
Añadir a cada matraz aforado (en el supuesto de que sean de 50 ml) 10 ml de HCl 2 M, 20 ml de NH3 concentrado (15 M), diferentes volúmenes de la solución de cobre hasta una concentración final de 0.4 mg/ml y enrasar con agua. Es necesario preparar también un blanco añadiendo todos los reactivos menos el analito a determinar. Se prepara también la muestra añadiendo la misma cantidad de reactivos a un volumen idéntico de muestra, dependiendo de la concentración aproximada (dato que proporciona el profesor). El análisis de la muestra es necesario hacerla por triplicado (preparar tres muestras en diferentes matraces aforados).
Medida espectrofotométrica.
Se mide la absorbancia de cada disolución a 580 nm (patrones y muestras a la vez).
RESULTADOS
a. Indica los resultados de absorbancia de los diferentes patrones y las muestras y representa gráficamente la recta de calibración obtenida.
b. Calcula la concentración de cobre a la muestra, expresada en ppm, y su incertidumbre.
TRATAMIENTO DE RESIDUOS
Los residuos que contengan cobre deben ir al bidón de residuos acuosos.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 36
PRÁCTICA 6
DETERMINACIÓN ESPECTROFOTOMÉTRICA DEL CONTENIDO DE HIERRO DE UNA PÍLDORA ANTIANÉMICA
OBJETIVO Y FUNDAMENTO
En esta práctica se determina colorimétricamente el Fe(III) de una muestra acuosa a partir de la formación del complejo del hierro con el tiocianato:
El tipo de complejo que se forma depende de la concentración de tiocianato . El color de estos complejos es pardo-rojizo , y la intensidad de color depende de diferentes factores, como el exceso de tiocianato, tipo de ácido presente y tiempo (hay una descomposición de los complejos con el tiempo).
MATERIAL
Pipetas, matraces aforados, vasos de precipitados, embudo, papel de filtro.
REACTIVOS
Solución madre de Fe(III) de 1000 ppm, tiocianato de potasio, ácido clorhídrico concentrado (12 M), H2O2 4 %.
PROCEDIMIENTO
Preparación de las soluciones:
- Solución patrón de Fe+3 a partir de la solución madre de 1000 ppm. - Solución de tiocianato potásico 3 M. - Solución de ácido clorhídrico 1 M. - Solución de ácido clorhídrico 6 M.
Tratamiento de la muestra.
Hacer hervir la píldora con 25 ml de HCl 6 M hasta completa disolución (aproximada-mente unos 20 minutos). Dejar enfriar la disolución, filtrarla y trasvasar el contenido a un matraz aforado de 100 ml.
Preparación de los patrones y la muestra.
Se preparan los diferentes patrones teniendo en cuenta que todos ellos tengan la misma concentración de todos los reactivos (es decir , 0.3 M de tiocianato potásico y 0.2 M de ácido clorhídrico) y diferentes cantidades del patrón de hierro (teniendo presente que el método es lineal hasta 10 ppm), según nos describe la bibliografía. Es necesario preparar también un

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 37
blanco. La muestra se prepara añadiendo a un volumen de muestra determinado, dependiendo de la concentración aproximada (dato que proporciona el profesor), 5 gotas de H2O2 y la misma cantidad de ácido clorhídrico que a los patrones. Se agita bien y se añade el tiocianato potásico. El análisis de la muestra es necesario hacerlo por triplicado (tres muestras en matraces aforados diferentes).
Medida espectrofotométrica.
Se mide la absorbancia de los patrones y las muestras a la longitud de onda de 480 nm.
RESULTADOS
a. Indica los resultados de absorbancia de los diferentes patrones y las muestras y representa gráficamente la recta de calibración obtenida.
b. Calcula los miligramos de sulfato de hierro (II) que contiene la píldora, así como la incertidumbre.
TRATAMIENTO DE RESIDUOS
Las soluciones que contengan tiocianato se deben verter en el bidón de residuos acuosos. El resto pueden ir al fregadero bien diluidas.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 38
PRÁCTICA 7
DETERMINACIÓN ESPECTROFOTOMÉTRICA DE FÓSFORO
Fundamento
La determinación de fósforo en fluidos biológicos, fertilizantes, moléculas orgánicas y productos industriales tiene una gran importancia. En esta práctica se determina fósforo mediante un procedimiento espectrofotométrico que, con ligeras modificaciones, se aplica rutinariamente en los laboratorios clínicos al análisis de fosfatos en fluidos biológicos. El método se basa en la reacción del ión fosfato con molibdato(MoO4
2-) que da lugar a fosfomolibdato([PO4]2MoO3]3-). Este último por reducción origina un compuesto cuya estructura exacta se desconoce, denominado “azul de molibdeno”. Como reductores se pueden utilizar muchos compuestos, de los cuales el sulfohidrato de hidrazina, el cloruro estannoso, el ácido 1,2,4-aminonaftosulfónico y el ácido ascórbico son los más empleados.
Disoluciones necesarias
- Disolución de molibdato: Se prepara disolviendo 12.5 g de molibdato amónico en unos 100 ml de agua destilada, añadiendo 150 ml de H2SO4 5M y diluyendo a 500 ml con agua. La disolución es estable si se mantiene en frasco cerrado.
- Disolución reductora: Se prepara disolviendo 1 g de ácido ascórbico en 100 ml de agua.
- Disolución de 50 ppm de P: Se prepara disolviendo la cantidad adecuada de sal (NaH2PO4, Na2HPO4, KH2PO4, ó K2HPO4) en agua destilada, añadiendo 2 ml de HCl concentrado y diluyendo a 1 l con agua destilada.
Material e instrumentación necesarios
- Matraces aforados de 50, 100, 500 y 1000 ml. - 2 pipetas de 5 ml graduada. - Espectrofotómetro Spectronic 20.
Procedimiento
Establecimiento de la recta de calibrado En matraces de 25 ml se pipetea la cantidad adecuada de disolución patrón de fosfato. A continuación, con cuidado y sin dejar de agitar se añaden 2.5 ml de disolución de molibdato y 1.5 ml de disolución reductora. Se enrasa con agua y se agita la mezcla para homogeneizarla, dejándola reposar a continuación unos 6 minutos –es conveniente que el tiempo de reposo sea lo mas parecido posible para todos los matraces- . Seguidamente se miden las absorbancias a 660 nm, empleando como blanco una disolución preparada de igual manera pero sin analito.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 39
Determinación de fósforo en cascarón de huevo Se toman 5 ml de la disolución de muestra, procedente del tratamiento de cascarones de huevo, y se someten al procedimiento anterior. Se determina el contenido en fósforo en la muestra problema a partir de la recta de calibrado.
Resultados
Recta de calibrado
P(50 ppm) ml
[P] ppm
A λ=660 nm
1 1 2 2 3 3 4 4 5 5
Muestra
Alícuota A λ=660 nm
1 2 3
Dibujar la correspondiente recta de calibrado de A=f([P]), determinar su ecuación, y a
partir de la misma calcular el contenido de Fósforo en la muestra problema.
- Recta de calibrado y cálculos

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 40
FOTOMETRÍA DE LLAMA DETERMINACIÓN DEL CONTENIDO EN SODIO EN AGUAS NATURALES Y DE
POTASIO EN VINOS POR FOTOMETRÍA DE LLAMA Fundamento
Cuando se aspira una disolución acuosa de sales inorgánicas en una llama adecuada de un quemador, los iones presentes emiten una radiación característica, cuya intensidad de emisión es función lineal de su concentración, lo que permite su determinación cuantitativa.
La espectroscopía de llama, por tanto, mide la potencia radiante de una línea emitida por un analito aspirado dentro de una llama caliente. Para separar dicha línea de la radiación producida por la llama o por los demás componentes de la muestra se emplea un monocromador o filtro. Disoluciones necesarias
- Disoluciones de 1000 mg/l de Na+ y K+
Material e instrumentación
- Matraces aforados de 50 ml. - Pipetas graduadas de 5 ml. - Fotómetro de llama con filtros para sodio y potasio.
Procedimiento
1.- Puesta a punto del instrumento Conectar el aparato y dejar que transcurra el tiempo necesario para conseguir la
estabilización de la señal. Colocar el filtro de sodio o de potasio, según corresponda, y aspirar una disolución de los respectivos metales que contenga 150 mg/l de uno de los mismos y ajustar con ella el 100 de la escala. El cero de la escala se ajusta aspirando agua desionizada.
2.- Recta de calibrado
Se preparan dos series de matraces de 100 ml adicionando con una pipeta 2, 4, 6, 8 y
10 ml de disolución de 1000 mg/l de Na+ y 2, 4, 6, 8 y 10 ml de disolución de 1000 mg/l de K+ respectivamente y se completa hasta el enrase con agua destilada. Se mide la señal de estas disoluciones patrón empleando los filtros correspondientes. Hacer la representación gráfica de la variación de la señal medida en función de la concentración del respectivo analito.
3.- Medida de las muestras
Aspirar la disolución de la muestra y leer en la escala del instrumento la señal correspondiente a la emisión producida. Deducir el valor de la concentración de analito por

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 41
interpolación en la correspondiente recta de calibrado. (En el caso del vino diluir la muestra 200 veces, 5 ml llevados a 1000).
Resultados
* Recta de calibrado Matraz
ppm Na+
lectura
ppm K+
lectura
1
2
3
4
5
* Muestras
Muestras
lectura Na+
lectura K+
AGUA
----------
VINO
----------
Tratamiento de datos 1.- Construir las líneas de calibrado, representando la señal leída en el instrumento en el eje de ordenadas y la concentración de Na+ y de K+, según corresponda, en el eje de abcisas. 2.- Calcular las ecuaciones de las rectas de regresión correspondientes. 3.- A partir de las ecuaciones de las dichas rectas determinar las concentraciones de sodio y potasio en las muestras. Resultados
Muestras
Na+ (ppm)
K+ (ppm)
AGUA
----------
VINO
----------
Observaciones

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 42
ESPECTROSCOPIA ABSORCIÓN ATÓMICA
DETERMINACIÓN DE DE HIERRO Y COBRE EN VINO La presencia de algunos metales a nivel de traza en los vinos se debe a la propia naturaleza del suelo de cultivo de la vid y también al proceso de elaboración. El contenido de cobre es, por lo general muy bajo (< 0.3 ppm), mientras que el de hierro debe ser inferior a 10 ppm, ya que concentraciones superiores de este elemento pueden originar coprecipitación con polifenoles, proteínas o fosfatos, lo cual se manifiesta en la aparición turbidez, aspecto no deseable, en los procesos denominados quiebras del vino
• Determinación de cobre
El método es lineal entre 0.8 y 5 ppm.
En primer lugar preparamos tres disoluciones intermedias de 10, 15 y 20 ppm de cobre. Para esto se pipetean 1.0,1.5 y 2.0 ml, respectivamente, del patrón de 1000 ppm , se introducen en matraces aforados de 100 ml y se enrasan con ácido nítrico al 2 % .
NOTA: Para preparar una disolución patrón de cobre de 1000 ppm el procedimiento a seguir es el siguiente:
1. Pesar en una balanza analítica 0.5 g de Cu metal previamente desengrasado en alcohol etílico y secado en la estufa a 110 ºC.
2. Introducirlos en un vaso de precipitados de 200 ml y añadir 10 ml de ácido nítrico concentrado , dentro de una campana de gases, hasta su completa disolución.
3. Transferir la disolución a un matraz aforado de 500 ml y enrasar con ácido nítrico al 2% .
A continuación se pipetean los volúmenes que se indican para obtener los patrones también indicados.
V (ml) Disolución intermedia Patrones ppm Cu+2
Absorbancia
10 10 1.00 10 15 1.50 25 10 2.50 15 20 3.00 25 15 3.75 25 20 5.00
Seguidamente y utilizando el método de medida ya establecido se ajusta el cero mediante el blanco, que como sabemos tiene que se ácido nítrico al 2% , y se miden las absorbancias de los patrones y representando gráficamente estos valores frente a las concentraciones obtenemos la recta de calibrado.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 43
Para finalizar se aspira directamente la muestra y se mide su absorbancia. Por interpolación determinamos la concentración.
• Determinación de hierro
El método es lineal entre 0.5 y 5 ppm.
En primer lugar preparamos dos disoluciones intermedias de 25 y 50 ppm de hierro. Para esto se pipetean 2.5 y 5 ml, respectivamente, del patrón de 1000 ppm , se introducen en matraces aforados de 100 ml y se enrasan con ácido nítrico al 2 % .
NOTA: Para preparar una disolución patrón de hierro de 1000 ppm el procedimiento a seguir es el siguiente:
4. Pesar en una balanza analítica 0.5 g de Fe metal previamente desengrasado en alcohol etílico y secado en la estufa a 110 ºC.
5. Introducirlos en un vaso de precipitados de 200 ml y añadir 10 ml de ácido clorhídrico concentrado , dentro de una campana de gases, hasta su completa disolución. A continuación se añaden 10 ml de ácido nítrico concentrado para asegurar la oxidación a hierro (III), se calienta para eliminar el exceso de oxidante y se deja enfriar
6. Transferir la disolución a un matraz aforado de 500 ml y enrasar con ácido nítrico al 2% .
A continuación se pipetean los volúmenes que se indican para obtener los patrones también indicados.
V (ml) Disolución intermedia Patrones ppm Fe+2 Absorbancia 1 25 0.50 1 50 1.00 5 25 2.50
7.5 25 3.75 5 50 5.00
Seguidamente y utilizando el método de medida ya establecido se ajusta el cero mediante el blanco, que como sabemos tiene que se ácido nítrico al 2% , y se miden las absorbancias de los patrones y representando gráficamente estos valores frente a las concentraciones obtenemos la recta de calibrado.
Para finalizar se aspira directamente la muestra y se mide su absorbancia. Por interpolación determinamos la concentración.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 44
CROMATOGRAFÍA DE GASES
ANALISIS CUALITATIVO Y CUANTITATIVO DE UNA MUESTRA DE ALCOHOLES
• OBJETIVO:
1. Familiarización con el cromatógrafo de gases y el software que controla todo el proceso.
2. Identificar cuales son los alcoholes contenidos en una muestra de pentano que contiene una determinada cantidad de los mismos.
3. Cuantificar alguno de estos alcoholes identificados previamente mediante el método de patrón interno.
• EXPERIMENTO
Encender el cromatógrafo y el ordenador y ajustar todos los parámetros necesarios para que el ordenador y el CG se pongan en comunicación de un modo correcto. Manejo del software. Inyectar una muestra de alcoholes y analizar distintos parámetros que afectan a las separaciones cromatográficas (cantidad de muestra, temperatura del horno,...) Establecimiento de un método para separar los componentes de la muestra de alcoholes. Identificación de los componentes de la muestra mediante inyección individual de varios alcoholes para comprobar en que tR nos aparecen.
La muestra puede contener tres de los siguientes alcoholes: 1-propanol, 1-Butanol, 3-Metil-1-Butanol, 2-Butanol, 1-Pentanol. Para realizar la identificación se procederá en primer lugar a la separación cromatográfica mediante el desarrollo de un método bien en condiciones isotérmicas o en programación de temperatura. Una vez que tengamos desarrollado el método y realizada la separación, pasaremos a inyectar los diversos alcoholes que pueden estar presentes para comprobar cuales hay realmente en nuestra muestra. Para esto disolveremos 20 µl de cada alcohol en pentano en un matraz de 10 ml, y los inyectaremos en el cromatógrafo y según los tiempos de retención determinaremos cuales están presentes.
Cuantificar uno de los alcoholes mediante patrón interno Nuestra muestra problema tendrá dos alcoholes 1-Butanol y 3-Metil-1-Butanol y cuantificaremos el 1-Butanol mediante el uso de 1-Pentanol como patrón interno.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 45
Para ello se prepararán las siguientes disoluciones patrón en matraces de 10 ml y como disolvente pentano.
Patrones V (µ l) 1-Butanol V (µ l) 1-Pentanol 1 180 250 2 220 250 3 240 250 4 280 250 5 320 250
Una vez que estén preparados y guardados en sus viales respectivos, se procederá a su inyección con el método que hemos establecido para la separación. En cada inyección determinaremos el área de cada pico, es decir, del analito (1-Butanol) y del patrón interno (1-Pentanol). Mediante la representación gráfica de la relación de áreas Aanalito/ Apatrón interno frente a la relación de volúmenes VA /VPI podremos determinar que volumen de analito contiene nuestra muestra.

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 46
HPLC-CROMATOGRAFÍA LIQUIDA ALTA RESOLUCIÓN
1.- ANALISIS CUALITATIVO Y CUANTITATIVO DE UNA MUESTRA DE PARACETAMOL (Acetoaminofén) Y ÁCIDO ACETILSALICILICO
Introducción:
Los analgésicos son fármacos que alivian el dolor sin producir pérdida de conciencia. Se dividen en opiáceos, derivados del opio, como la morfina y la codeína, y no opiáceos, como la aspirina, el paracetamol y el ibuprofeno.
Tanto el ácido salicílico como el paracetamol forman parte de muchos analgésicos, ya sea solos o mezclados con otros componentes como la cafeína.
En esta práctica se propone la determinación de una muestra que contiene uno de los componentes antes indicados mediante cromatografía de líquidos con detección UV. La cuantificación se lleva a cabo mediante una recta de calibrado externa.
Instrumentación:
• Cromatógrafo líquidos Varian Polaris 100 Isocrático, con detector UV. • Columna octadecilsilano (250 x 4.6 mm d.i). Fase reversa
Condiciones de trabajo:
• Fase móvil: disolución de ácido acético 0.01 M / metanol ( 60/40-V/V). • Flujo de la fase móvil: 1 ml/min • Volumen de inyección: 20 µl • Longitud de onda del detector UV: 254 nm.
Reactivos:
• Acetoaminofén (PM: 151.17 g/mol) • Ácido acetilsalicílico (PM: 168.084 g/mol) • Cafeína (PM:194.19 g/mol) • Ácido acético concentrado. • Metanol de calidad HPLC • Agua de calidad HPLC

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 47
Procedimiento:
Preparación de fase móvil: Como la fase móvil va a estar formada por una disolución de ácido acético/agua (1% / 59%) y metanol (40%) , lo que haremos será decidir qué cantidad de fase móvil vamos a preparar para nuestra práctica. Por tanto, si queremos preparar un litro de fase móvil, ¿qué tendremos que hacer?:
• Tomar luego 594 ml de esta agua ultrapura y 6 ml de ácetico glacial.
• Mezclar esos 600 ml de dón de acético con 400 ml de MeOH.
NOTA: Teóricamente tendríamos que filtrar ambos componentes de la fase móvil ena través de una membrana de nailon de 0.22 µm de poro y desgasificar el litro de fase móvil 15 minutos en un baño ultrasonidos.
Preparación de las disoluciones patrón de los distintos componentes que vamos a separar e identificar.
Disoluciones patrón originales: Matraces de 100 ml
• 100 ml de disolución patrón de 1000 ppm de acetoaminofén. • 100 ml de disolución patrón de 1000 ppm de acetilsalicílico. • 100 ml de disolución patrón de 1000 ppm de cafeína.
NOTA: Todas las disoluciones se preparan en metanol calidad HPLC.
Disoluciones patrón a inyectar: Matraces de 10 o 25 ml
Patrones Concentración (ppm) Acetoaminofén Cafeína Acetilsalicilico 1 10 10 10 2 15 15 15 3 20 20 20 4 25 25 25 5 30 30 30

Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 48
Análisis cualitativo:
• Inyectamos el patrón de 30 ppm de cada uno de los componentes y determinamos su orden de elución a través de los tiempos de retención.
Análisis cuantitativo:
• Inyectamos cada uno de los patrones de la tabla anterior y obtenemos mediante el software del aparato las áreas de los picos de cada componente del cromatograma.
• Según el componente o componentes que queramos cuantificar, representamos el área frente a la concentración obteniéndose una recta de calibrado para cada componente de la muestra.
• Inyectamos la muestra e identificamos (en caso de que no se nos diga) mediante los tiempos de retención el/los componentes presentes.
• Determinamos el área de el/los componente/s. • Si el componente es cafeína, ¿qué haríamos?:
1. Representación gráfica de las áreas de los patrones de
cafeína frente a sus concentraciones respectivas. Recta de calibrado
2. Comprobamos que el área cae dentro de los valores de nuestra recta de calibrado. Si es así ya está. Despejamos de la ecuación de nuestra recta y obtenemos la concentración.
3. Si el área cae fuera, tenemos que realizar alguna dilución para que nos entre dentro de los valores de nuestra recta y luego realizar el cálculo.
4. Si hubiese más componentes en nuestra muestra y los quisiéramos cuantificar tendríamos que repetir el procedimiento tantas veces como componentes.
NOTA: Antes de realizar cualquier inyección debemos acondicionar la columna y el aparato haciendo circular fase móvil por lo menos durante 15-30 minutos. Además, se deberían filtrar todos los patrones a través de filtros de membrana de nilón de 0.45 µm
Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 49
2.- ANALISIS CUALITATIVO Y CUANTITATIVO DE CAFEINA EN BEBIDAS CARBONATADAS. (TÉCNICA DE ADICIÓN ESTÁNDAR)
• Procedimiento
1. Preparar 1 L de fase móvil MeOH/Agua (35/65). 2. Filtrar la fase móvil con un filtro de 0,45 µm a vacio. 3. Desgasificar la bebida carbonatada mediante agitación. Filtrarla a través de un filtro
filtro de 0,45 µm a vacio. 4. Pesar 100 mg de cafeína y preparar 100 mL de disolución. 5. Preparar las diluciones de la siguiente tabla en matraces de 25 mL
Dilución Volumen de
muestra Volumen Patrón
de cafeína Volumen total
1 15,00 0,00 25,00 2 15,00 1,00 25,00 3 15,00 2,00 25,00 4 15,00 3,00 25,00 5 15,00 4,00 25,00 6 15,00 5,00 25,00
6. Obtenga los cromatogramas de las disoluciones por triplicado.
• Condiciones de trabajo
Las condiciones que vamos a poner son orientativas. Se tienen que determinar experimentalmente.
Instrumentación:
• Cromatógrafo líquidos Varian Polaris 100 Isocrático, con detector UV. • Columna octadecilsilano (250 x 4.6 mm d.i). Fase reversa
Condiciones de trabajo:
• Fase móvil: disolución agua / metanol ( 65/35-V/V). • Flujo de la fase móvil: 1 ml/min ó 1,5 ml/min • Volumen de inyección: 20 µl • Longitud de onda del detector UV: 254 nm.
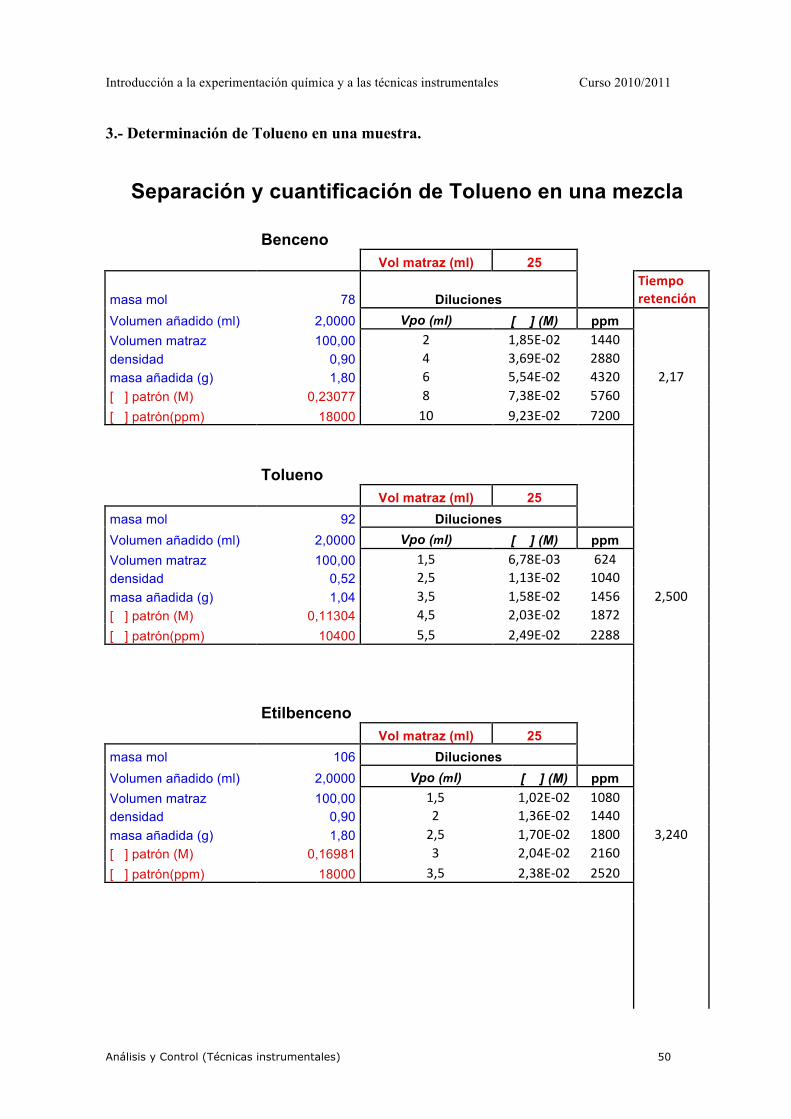
Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 50
3.- Determinación de Tolueno en una muestra.
Separación y cuantificación de Tolueno en una mezcla
Benceno
Vol matraz (ml) 25
masa mol 78 Diluciones Tiempo retención
Volumen añadido (ml) 2,0000 Vpo (ml) [ ] (M) ppm
Volumen matraz 100,00 2 1,85E-‐02 1440 densidad 0,90 4 3,69E-‐02 2880 masa añadida (g) 1,80 6 5,54E-‐02 4320 2,17 [ ] patrón (M) 0,23077 8 7,38E-‐02 5760
[ ] patrón(ppm) 18000 10 9,23E-‐02 7200
Tolueno
Vol matraz (ml) 25
masa mol 92 Diluciones
Volumen añadido (ml) 2,0000 Vpo (ml) [ ] (M) ppm
Volumen matraz 100,00 1,5 6,78E-‐03 624 densidad 0,52 2,5 1,13E-‐02 1040 masa añadida (g) 1,04 3,5 1,58E-‐02 1456 2,500 [ ] patrón (M) 0,11304 4,5 2,03E-‐02 1872
[ ] patrón(ppm) 10400 5,5 2,49E-‐02 2288
Etilbenceno
Vol matraz (ml) 25
masa mol 106 Diluciones
Volumen añadido (ml) 2,0000 Vpo (ml) [ ] (M) ppm
Volumen matraz 100,00 1,5 1,02E-‐02 1080 densidad 0,90 2 1,36E-‐02 1440 masa añadida (g) 1,80 2,5 1,70E-‐02 1800 3,240 [ ] patrón (M) 0,16981 3 2,04E-‐02 2160
[ ] patrón(ppm) 18000 3,5 2,38E-‐02 2520
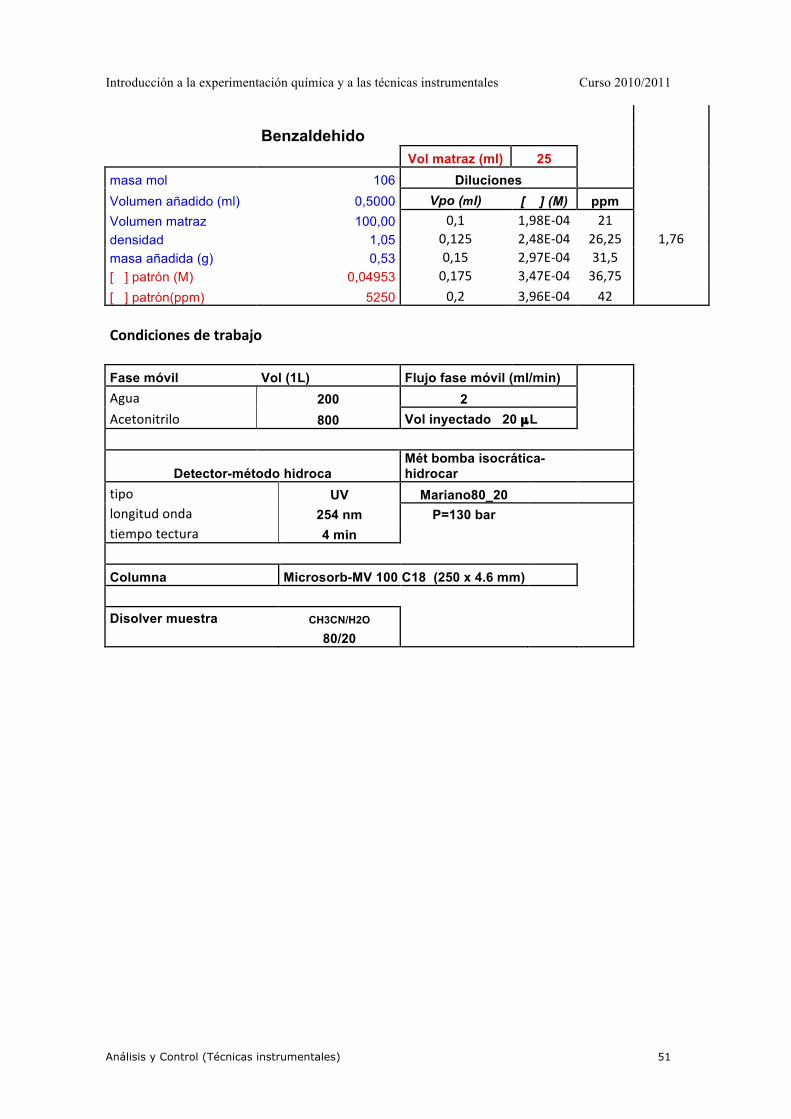
Introducción a la experimentación química y a las técnicas instrumentales Curso 2010/2011
Análisis y Control (Técnicas instrumentales) 51
Benzaldehido
Vol matraz (ml) 25
masa mol 106 Diluciones
Volumen añadido (ml) 0,5000 Vpo (ml) [ ] (M) ppm
Volumen matraz 100,00 0,1 1,98E-‐04 21 densidad 1,05 0,125 2,48E-‐04 26,25 1,76 masa añadida (g) 0,53 0,15 2,97E-‐04 31,5 [ ] patrón (M) 0,04953 0,175 3,47E-‐04 36,75
[ ] patrón(ppm) 5250 0,2 3,96E-‐04 42 Condiciones de trabajo
Fase móvil Vol (1L) Flujo fase móvil (ml/min)
Agua 200 2
Acetonitrilo 800 Vol inyectado 20 µL
Detector-método hidroca Mét bomba isocrática-hidrocar
tipo UV Mariano80_20 longitud onda 254 nm P=130 bar tiempo tectura 4 min
Columna Microsorb-MV 100 C18 (250 x 4.6 mm)
Disolver muestra CH3CN/H2O
80/20