insuficiencia de la médula Ósea
DESCRIPTION
Presentacion Insuficiencia de la Medula OseaTRANSCRIPT

Insuficiencia de la Médula ósea. Agranulocitosis.
Dra. Beatriz Molinas
CORONEL OVIEDO – PARAGUAY2015
UNIVERSIDAD NACIONAL DE CAAGUAZU Sede Coronel Oviedo
Creada por Ley Nº 3.198 del 4 de Mayo de 2.007
FACULTAD DE MEDICINA
Patología Medica I

Integrantes
• Claudia Helena Escobar Reiniak.
• Luis Enrique Martínez Espinola
• Marcos Antonio Irala Burgos
• Perla Romina González Gutierrez

Insuficiencia de la médula ósea
Aplasia Medular
Insuficiencia medular selectiva
Hemoglobinuria Paroxística Nocturna

Aplasia Medular

Aplasia Medular
•Engloba un grupo heterogéneo de enfermedades que se caracterizan por pancitopenia, sin evidencia de infiltración neoplásica ni de síndrome mieloproliferativo.
Etiológicamente la AM se divide en dos:
•Aplasia Medular Adquirida.
•Aplasia Medular Constitucional o congénita
Farreras - Rozman

Aplasia Medular Adquirida• La aplasia medular adquirida (AM) es una
insuficiencia medular cuantitativa adquirida que afecta, en mayor o menor medida, a las tres series hematopoyéticas.
Epidemiología : •Muy variable
•Mayor en el oriente
• De 2 a 6 casos por cada millón de personas
•Afecta a ambos sexos
•Tiene dos picos de incidencia a los 30 y 60 añosFarreras - Rozman

Etiología:
Radiaciones ionizantes
Medicamentos
Agentes químicos
Virus
Farreras - Rozman

Cuadro Clínico
• Relacionado a las citopenias y el grado de estas.
• No se palpan esplenomegalias ni adenopatias.
• Combinación de anemia, leucopenia y trombocitopenia.
Farreras - Rozman

Exploraciones Complementarias:
• Pancitopenia, linfocitos normales, se presenta una anemia normocítica y normocrómica.
• Hb inferior a 80g/L.
• Plaquetas inferiores a 50000 mm3
• Depósitos de hierro en abundancia.
• Escases de las tres series hematopoyéticas.
Farreras - Rozman

PronósticoLa Aplasia Medular Adquirida se subdivide en:
•Granulocitos inferior a 500/mm3•Plaquetas menor a 20.000 /mm3•Reticulocitos inferior al 1% en presencia de anemia
Grave
•Las características de la AMG mas una disminución de granulocitos a 200/mm3
Muy grave
•Incluyen menos de dos criterios de la aplasia graveModerada
Farreras - Rozman

Tratamiento
• Pacientes con trombocitopenia intensa asociada a hemorragias, deben de recibir transfusiones de plaquetas de preferencia de un donante único.
• Se deben de tratar las infecciones por bacterias con antibióticos de amplio espectro.
• Aplicar concentrados de hematíes.
Farreras - Rozman

Transporte de progenitores hematopoyeticos (TPH)
• Para aplasias graves y muy graves.
• Edad inferior a 40 años.
• Disponer de un donante histocompatible (generalmente un hermano).
• Depende del numero de transfusiones previas al TPH.
Farreras - Rozman

Tratamiento Inmunodepresor
• Fármacos como la globulina antilinfocitica/antitimocitica, la ciclosporina A y los glucocorticoides. Generalmente se combinan dos.
• Raramente es completa ya que con frecuencia queda alguna citopenia residual y alteraciones displásicas de la serie roja.
• Recomendado en pacientes mayores de 40 años o con granulocitos superiores a 500/mm3 sin donante.
Farreras - Rozman

Aplasias congénitas o constitucionalesAnemia de Fanconi
• Aplasia medular asociada a malformaciones congénitas.
• Defecto en la reparación del ADN y la mayor sensibilidad a los radicales toxico del oxigeno determinan entrecruzamientos y roturas del ADN.
• El patrón de herencia es autosomica recesiva.
• El diagnostico se realiza entre los 7 y 9 años.
Farreras - Rozman

Las anomalías congénitas asociadas son:
• Baja estatura
• Pulgares anormales
• Microcefalea
• Manchas cutáneas
• Base nasal ancha
Farreras - Rozman

Diagnóstico
• Se efectúa mediante estudios citogenéticos mediante estimulación con diepoxibutano, mitomicina C, o cisplatino.
• La supervivencia es de 2 a 6 años.
• El TPH cambia la historia natural de la enfermedad ya que corrige las alteraciones hematológicas.
Farreras - Rozman

Insuficiencia Medular Selectiva

Insuficiencia Medular Selectiva Clasificación
Insuficiencia Medular Selectiva
Hipoplasia de la Serie Roja
Trombocitopenias Hipomegacariocíticas
Farreras - Rozman

Hipoplasia de la Serie Roja

• Anemia normocítica
• Anemia normocrómica
• Reticulocitopenia
• Eritroblastopenia
Hipoplasia de la Serie RojaCaracterísticas
Farreras - Rozman

Hipoplasia de la Serie RojaClasificación
Hipoplasia de la Serie RojaEritroblastopenia congénita de
Blackfan-Diamond
Hipoplasia adquirida de la Serie Roja
Farreras - Rozman

Eritroblastopenia congénita de Blackfan – Diamond
Comprende un grupo heterogéneo de pacientes:
• Algunos con herencia autosómica dominante o recesiva.
• La mayoría sin historia familiar.
Farreras - Rozman

Eritroblastopenia congénita de Blackfan – Diamond. Tratamiento
• Glucocorticoides (2mg/kg de peso x día).
• TPH (Transplante de Progenitores Hemopoyéticos).
Farreras - Rozman

Hipoplasia Adquirida de la Serie Roja Clasificación
Hipoplasia Adquirida de la Serie Roja
Formas Agudas Formas Crónicas
Farreras - Rozman

Hipoplasia Adquirida de la Serie RojaFormas Agudas
Causas más frecuentes:
• Fármacos
• Infecciones
Farreras - Rozman

Hipoplasia Adquirida de la Serie RojaFormas Agudas
Tratamiento:
• Recuperación espontánea.
• En pacientes con infección por PV-B19 (Parvovirus B19) puede ser útil el uso de IgG Humana
Farreras - Rozman

Hipoplasia Adquirida de la Serie RojaFormas Crónicas
• Ocurren en mayores de 50 años con timoma y miastenia grave.
Farreras - Rozman

Hipoplasia Adquirida de la Serie RojaFormas Crónicas
Tratamiento:
• Timectomía.
• Inmunodepresores (IgG, ciclofosfamida, azatioprina).
Farreras - Rozman

Trombocitopenia Hipomegacariocítica

Trombocitopenia HipomegacariocíticaClasificación
Trombocitopenia HipomegacariocíticaTrombocitopenia amegacariocítica
congénita
Trombocitopenia Hipomegacariocítica adquirida
Farreras - Rozman

Trombocitopenia amegacariocítica congénita. Características
• Se asocia a alteraciones esqueléticas (TAR).
• Herencia autosómica recesiva.
• Se presentan complicaciones hemorrágicas.
Farreras - Rozman

Trombocitopenia amegacariocítica congénita. Tratamiento
• TPH (Transplante de progenitores hemopoyéticos).
Farreras - Rozman

Trombocitopenia Hipomegacariocítica adquirida.
Causas más frecuentes:
• Exposición a fármacos o tóxicos.
• Infecciones víricas.
• Lupus eritematoso sistémico.
Farreras - Rozman

Trombocitopenia Hipomegacariocítica adquirida. Tratamiento
• Inmunodepresores.
Farreras - Rozman

Hemoglobinuria Paroxística Nocturna

Hemoglobinuria Paroxística NocturnaConcepto
• Enfermedad adquirida y clonal
• PIGA glucosilfosfatidilinositol
Farreras - Rozman

Hemoglobinuria Paroxística NocturnaCuadro Clínico
• Anemia hemolítica crónica
• Trombosis en territorio venoso
• Defecto hemopoyético
• Infecciones repetidas
Farreras - Rozman

Hemoglobinuria Paroxística NocturnaDiagnóstico
• Anemia
• Leucopenia
• Trombocitopenia
• Signos de hemolisis
• FerropeniaFarreras - Rozman

• Hemolisis eritrocitaria provocada en un medio acificado
• Prueba de la sacarosa
Farreras - Rozman

Hemoglobinuria Paroxística NocturnaPronostico
• Esperanza de vida
• Causas de muertes
Farreras - Rozman

Hemoglobinuria Paroxística NocturnaTratamiento
• Transfusiones
• Ciclosporina A
• Eculizumab
• TPH
Farreras - Rozman

Leucopenias neutropénicas

Clasificación
NEUTROPENIAS
ETIOLÓGICA
IINTENSIDAD
Farreras - Rozman

ETIOLOGÍA
CONGÉNITAS- PRIMARIAS
(IDIOPÁTICAS)- SECUNDARIAS
ADQUIRIDAS- PRIMARIAS
(IDIOPÁTICAS)- SECUNDARIAS
Farreras - Rozman
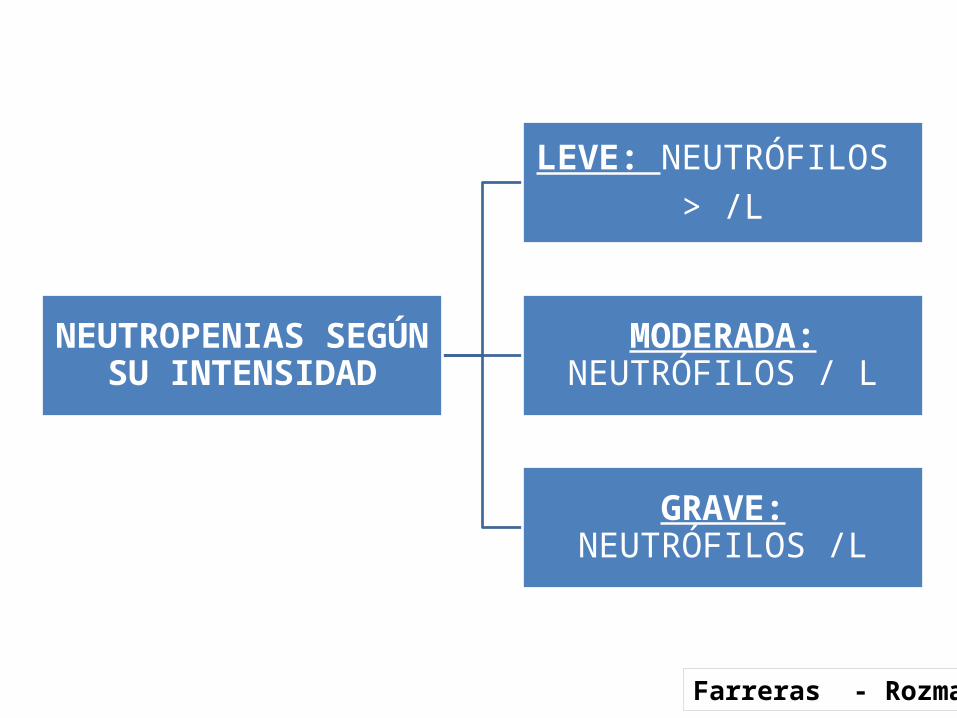
NEUTROPENIAS SEGÚN SU INTENSIDAD
LEVE: NEUTRÓFILOS > /L
MODERADA: NEUTRÓFILOS / L
GRAVE: NEUTRÓFILOS /L
Farreras - Rozman

Agranulocitosis inducida por fármacos

Epidemiología
• - INCIDENCIA: BAJA, (4 CASOS/ 1 000 000 HAB / AÑO)
• PREDOMINANCIA: MUJERES
• FRECUENCIA: PERSONAS ENTRE 40 – 60 AÑOS
Farreras - Rozman

Etiopatogenia
FÁRMACOS CAUSANTES DE AGRANULOCITOSIS:1. Aminopirinas2. Sulfanamidas3. Quinidinas4. Antitiroideos5. Fenotiazinas*
Farreras - Rozman

Cuadro clínico
• Fiebre
• Escalosfrios
• Postración
• Infecciones (Bacterias gramnegativas: E. Coli, Pseudomona Aeruginosa, Enterobacter, Klebsiella. Bacterias grammpositivas: S. aureus, S. epidermidis, Streptococcus spp.)
• Úlceras necróticas no purulentas en la mucosa orofaríngea
Farreras - Rozman

Laboratorio
• Recuento leucocitario: < 1 x / L (con menos de 0,2 x / L de neutrófilos, o ausencia virtual de los mismos)
• Linfocitopenia
• Anemia y/o trombocitopenia obliga a descartar otros diagnósticos
Farreras - Rozman

Muchas Gracias