implementación de un método de orbitales moleculares de ... · pdf filepropuesto...
TRANSCRIPT

Introduccion Metodologıa Teorıa Resultados Conclusiones
Implementacion de un metodo de orbitalesmoleculares de fragmentos para cualquier partıcula
en el programa LOWDIN
Ronald GonzalezDirector: Andres Reyes
Grupo de quımica cuantica y computacional
Universidad Nacional de Colombia
18 de septiembre de 2014

Introduccion Metodologıa Teorıa Resultados Conclusiones
Contenido
• Introduccion
Quımica cuanticaEfectos cuanticos nuclearesDiferentes especies cuanticas (e−, H+, µ−, µ+, e+)Metodos FMO y APMO
• Metodologıa
Union de los metodos APMO y FMOImplementacion en el programa LOWDIN
• Resultados
Esquema FMO-APMO
• Conclusiones
1
Introduccion Metodologıa Teorıa Resultados Conclusiones
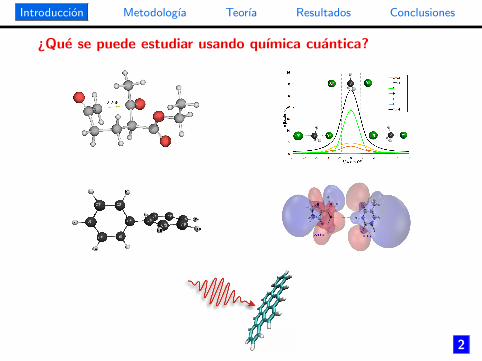
¿Que se puede estudiar usando quımica cuantica?
2
Introduccion Metodologıa Teorıa Resultados Conclusiones
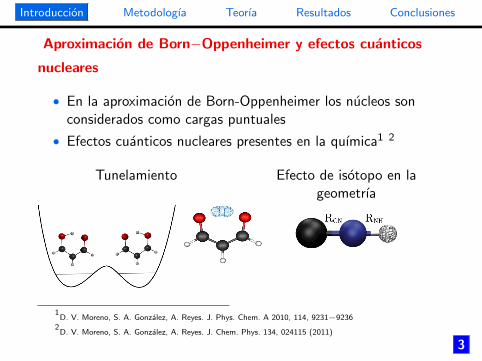
Aproximacion de Born−Oppenheimer y efectos cuanticos
nucleares
• En la aproximacion de Born-Oppenheimer los nucleos sonconsiderados como cargas puntuales
• Efectos cuanticos nucleares presentes en la quımica1 2
Tunelamiento Efecto de isotopo en lageometrıa
1D. V. Moreno, S. A. Gonzalez, A. Reyes. J. Phys. Chem. A 2010, 114, 9231−9236
2D. V. Moreno, S. A. Gonzalez, A. Reyes. J. Chem. Phys. 134, 024115 (2011)
3

Introduccion Metodologıa Teorıa Resultados Conclusiones
Orbital molecular para cualquier partıcula (APMO)
• La aproximacion: orbital molecular para cualquier partıcula(APMO), es una metodologıa desarrollada en el grupo dequımica cuantica y computacional (QCC) de la universidadNacional de Colombia.
• Diferentes especies cuanticas (e−, H+, µ−, µ+, e+)
4

Introduccion Metodologıa Teorıa Resultados Conclusiones
Positrones e+, muones µ− µ+ y cualquier partıcula cuantica
• Aplicaciones de los positrones en la quımica3 4
3J. Charry, J. Romero, M. Varella, A. Reyes. ”Positron binding energies of amino acids with the generalized
any-particle propagator method”, Phys. Rev. A., 89, 052709 (2014)4
Katsuhiko Koyanagi, Yukiumi Kita, Yasuteru Shigeta, Masanori Tachikawa. ”Binding of a Positron to Nucleic
Base Molecules and Their Pairs”, ChemPhysChem, Communication, 2013, 14, 3458−3462
5

Introduccion Metodologıa Teorıa Resultados Conclusiones
Positrones e+, muones µ− µ+ y cualquier partıcula cuantica
• Aplicaciones de los muones en la quımica5 6
−0.02 −0.01 0.00 0.01 0.02
density/a.u.−3
distance/a.u.
H2µ+
H2µ2
5F. Moncada, D. Cruz, A. Reyes. ”Muonic alchemy: Transmuting elements with the inclusion of negative
muons”, Chemical Physics Letters 539−540 (2012) 209−2136
F. Moncada, D. Cruz, A. Reyes. .Electronic properties of atoms and molecules containing one and two
negative muons”, Chemical Physics Letters 570 (2013) 16−21
6

Introduccion Metodologıa Teorıa Resultados Conclusiones
Orbitales moleculares de fragmentos (FMO)
• El metodo (FMO) ha sido aplicado a una gran variedad de sistemas moleculares
con cientos y miles de atomos, en diversos campos de investigacion7 y fue
propuesto por Kazuo Kitaura8 en 1999.
0
5
10
15
20
10 20 30 40 50 60 70 80 90 100
Tim
e / h
ours
Atoms
O(N3)
O(N4)
O(N!)
7T. Sawada, D.G. Fedorov and K. Kitaura. Role of the Key Mutation in the Selective Binding of Avian and
Human Influenza Hemagglutinin to Sialosides Revealed by Quantum−Mechanical Calculations”, J. Am. Chem.
Soc. 2010, 132, 16862−168728
K. Kitaura, E. Ikeo, T. Asada, T. Nakano and M. Uebayasi, Chem. Phys. Letters. 313,701 19997
Introduccion Metodologıa Teorıa Resultados Conclusiones
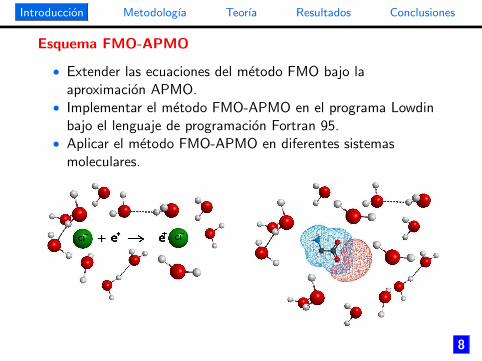
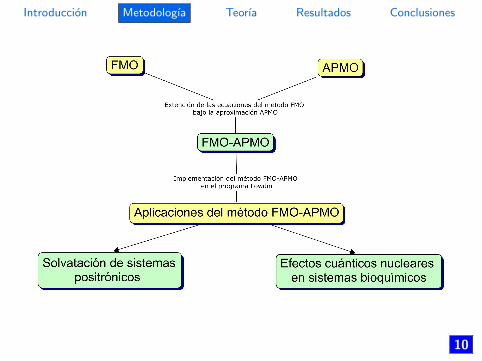
Esquema FMO-APMO
• Extender las ecuaciones del metodo FMO bajo laaproximacion APMO.
• Implementar el metodo FMO-APMO en el programa Lowdinbajo el lenguaje de programacion Fortran 95.
• Aplicar el metodo FMO-APMO en diferentes sistemasmoleculares.
8
Introduccion Metodologıa Teorıa Resultados Conclusiones
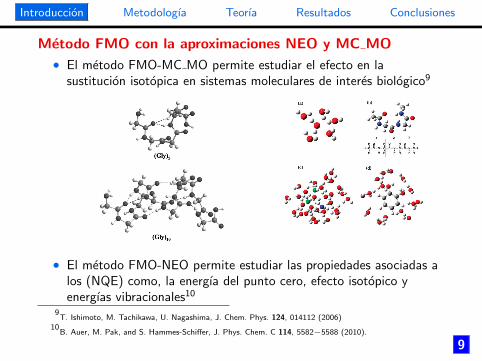
Metodo FMO con la aproximaciones NEO y MC MO
• El metodo FMO-MC MO permite estudiar el efecto en lasustitucion isotopica en sistemas moleculares de interes biologico9
• El metodo FMO-NEO permite estudiar las propiedades asociadas alos (NQE) como, la energıa del punto cero, efecto isotopico yenergıas vibracionales10
9T. Ishimoto, M. Tachikawa, U. Nagashima, J. Chem. Phys. 124, 014112 (2006)
10B. Auer, M. Pak, and S. Hammes-Schiffer, J. Phys. Chem. C 114, 5582−5588 (2010).
9
Introduccion Metodologıa Teorıa Resultados Conclusiones
10

Introduccion Metodologıa Teorıa Resultados Conclusiones
¿En que consiste el metodo FMO?
11

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa total en el esquema FMO
12

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa total en el esquema FMO
13

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa total en el esquema FMO
14

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa total en el esquema FMO
15

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa FMO2
• Mezcla orbital y potencial electrostatico11
11R. Zalesny, M. Papadopoulos, P. Mezey, J. Leszczynski, Linear−Scaling Techniques in Computational
Chemistry and Physics, Springer. 2011
16

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
Las energıas en el metodo FMO se obtienen resolviendo lasecuaciones de Hartree-Fock-Roothaan, para el caso FMO-RHF
FXCX = SXCXεX
Donde X es el ındice del fragmento o pares de fragmentos(monomeros o dımeros). En la expresion anterior el operador deFock se define como
FX = HX + GX
Donde HX es el operador mono-electronico modificado
17

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
HXµν = HX
µν + V Xµν +B
∑i∈X
P iµν
En la expresion anterior el termino V Xµν corresponde al potencial
electrostatico externo debido a la presencia de los otros fragmentosy se define como
V Xµν =
N∑K(K 6=X)
{∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
}
18

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
19

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
20

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
21

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
22

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
23

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
24

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩︸ ︷︷ ︸Potencial atractivo
+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉
25

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉︸ ︷︷ ︸
Potencial repulsivo
26
Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉︸ ︷︷ ︸
Potencial repulsivo
27

Introduccion Metodologıa Teorıa Resultados Conclusiones
Descripcion matematica del metodo FMO
V Xµν =
N∑K(K 6=X)
∑A∈K
⟨φµ
∣∣∣∣ −Za|r −RA|
∣∣∣∣φν⟩+∑σλ∈K
DKσλ 〈φµφν |φσφλ〉︸ ︷︷ ︸
Potencial repulsivo
28

Introduccion Metodologıa Teorıa Resultados Conclusiones
Fragmentacion de enlaces covalentes
29

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa de monomeros y dımeros
EHFX =1
2Tr[DHFX
(HX + FX
)]+ ERNX
Aquı, se introduce una nueva energıa que se obtiene mediante laexclusion de la contribucion del potencial electrostatico de EHFX ,es decir,
E′HFX = EHFX − Tr
(DHFX VX
)La energıa total de la aproximacion FMO1 en el metodo FMO esdefinida como,
EHFFMO1 =∑I
E′HFI
30

Introduccion Metodologıa Teorıa Resultados Conclusiones
Energıa de monomeros y dımeros
Ahora se introduce una nueva matriz DIJ , esta matriz recibe elnombre de matriz de densidad de dımero IJ , empleando estamatriz, la energıa FMO2-RHF se escribe como,
EHFFMO2 =∑I
E′HFI +
∑I>J
(E′HFIJ − E′HFI − E′HFj
)+∑I>J
Tr(∆DHF
IJ VHFIJ
)
31

Calculo de energıa total del esquema (FMO)

Introduccion Metodologıa Teorıa Resultados Conclusiones
¿En que consiste el metodo APMO?
En un sistema molecular que contiene Nq partıculas cuanticas y Nc
partıculas clasicas, el Hamiltoniano puede ser expresado enterminos de energıa cinetica y potencial12
Htot = T + V = −Nq∑i
1
2mi∇2i +
Nq∑i
Nc∑j
qiqjRij
+
Nq∑i
Nq∑j>i
qiqjrij
Al nivel de teorıa APMO Hartree-Fock (APMO-HF) la funcion deonda del estado basal Ψ0 es construida como un producto defunciones de onda, φα de cada especie cuantica α
Ψ0 =
Nespecies∏α
φα
12S.A. Gonzalez, N.F. Aguirre and A. Reyes, Int. J. Quant. Chem. 108, 1742 (2008).
33

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo de fragmentos para cualquier partıcula FMO-APMO
Las ecuaciones resultantes Hartree-Fock-Roothaan en una base deorbitales atomicos (AO) establecidos para las especies cuanticas αcorresponden a
Fα,XCα,X = Sα,XCα,Xεα,X
Donde S,F, C y ε son las matrices de solapamiento, Fock,coeficientes y valores propios, respectivamente.El ındice α indica especies cuanticas y el ındice X denotamonomeros I o dımeros IJ .
Fα,X = Hα,X + Gα,X + Cα,XAcoplamiento
34

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo de fragmentos para cualquier partıcula FMO-APMO
Hα,Xµν = Hα,X
µν + V α,Xµν +B
∑i∈X
P iµν
El primer termino Hα,Xµν es la matriz mono-partıcula estandar para
las especies cuanticas α en el fragmento X.La matriz de acoplamiento entre la especie alfa y todas las demasespecies del mismo fragmento (acoplamiento inter-especie).
Cα,XAcoplamiento =
N∑K
Nespecies∑β>α
Nα∑i
Nβ∑j
qαqβ
rij
35

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo de fragmentos para cualquier partıcula FMO-APMO
Potencial electrostatico para cualquier partıcula
V A,α,Xµν =N∑
K(K 6=X)
∑L∈K
⟨φαµ
∣∣∣∣ −Za|r −RL|
∣∣∣∣φαν⟩
V B,α,Xµν =N∑
K(K 6=X)
∑σλ∈K
Dα,Kσλ 〈φαµφ
ασ |φαν φαλ〉 −
Nespecies∑β 6=α
∑σλ∈K
Dβ,Kσλ 〈φαµφ
βσ |φαν φ
βλ〉
V α,Xµν = V A,α,Xµν + V B,α,Xµν
Donde el ındice K es sumado sobre todo los monomeros exceptoX, el ındice L es sumado sobre los nucleos clasicos NK
c en elmonomero K, los ındices µ ν son sumados sobre todas lasfunciones base de las especies cuanticas α en el monomero K.
36

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo de fragmentos para cualquier partıcula FMO-APMO
Energıa FMO-APMO-HF
EAPMO−HFX =
1
2Tr[DAPMO−HFX
(HX + FindepX
)]+EγX +ERNX
Aquı EγX se denomina energıa de acoplamiento inter-especie y dacuenta de la atraccion o repulsion entre las partıculas de especiesdiferentes.
Energıa FMO2-APMO-RHF
EAPMO−HFFMO2 =
∑I
E′APMO−HFI +
∑I>J
(E
′APMO−HFIJ − E
′APMO−HFI − E
′APMO−HFj
)+∑I>J
Tr(
∆DAPMO−HFIJ VAPMO−HF
IJ
)
37

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo FMO-APMO en el programa LOWDIN
• El programa Lowdincontiene mas de 80.000lıneas de codigo13
• Disenado bajo la filosofıade programacion orientadaa objetos
• Input Manager parafragmentos con especiescuanticas
• Programa SCFmulti-partıcula
• Programa de integrales
13Int. J. Quantum Chem., 114(1), 50-56 (2014)38

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo de fragmentos para cualquier partıcula FMO-APMO
39

Introduccion Metodologıa Teorıa Resultados Conclusiones
Metodo de fragmentos para cualquier partıcula FMO-APMO
40

Introduccion Metodologıa Teorıa Resultados Conclusiones
Aplicacion del Esquema FMO-APMO
41

Introduccion Metodologıa Teorıa Resultados Conclusiones
Aplicacion del Esquema FMO-APMO
Correccion de energıa a primer orden FMO1-RHF
Energıa total FMO1-RHF
Sistema molecular Conjunto base GAMESS LOWDIN
Energıa (Hartrees) Energıa (Hartrees) ∆E (kcal/mol)6-31G(d,p)
(H2O)3 -228,066491 -228,066490 0,0(H2O)4 -304,075594 -304,075594 0,0(H2O)5 -380,090681 -380,090681 0,0(H2O)6 -456,113002 -456,113001 0,0(H2O)8 -608,160068 -608,160068 0,0Etanol +(H2O)3 -382,151699 -382,151699 0,0Etanol +(H2O)6 -610,213126 -610,213232 0,0Fenol +(H2O)3 -533,634927 -533,634927 0,0Fenol +(H2O)6 -761,699424 -761,699349 0,0
42

Introduccion Metodologıa Teorıa Resultados Conclusiones
Aplicacion del Esquema FMO-APMO
Correccion de energıa a segundo orden FMO2-RHF
Energıa total FMO2-RHF
Sistema molecular Conjunto base GAMESS LOWDIN
Energıa (Hartrees) Energıa (Hartrees) ∆E (kcal/mol)6-31G(d,p)
(H2O)3 -228,098450 -228,098451 0,0(H2O)4 -304,137466 -304,137466 0,0(H2O)5 -380,173702 -380,173702 0,0(H2O)6 -456,214013 -456,214012 0,0(H2O)8 -608,313884 -608,313883 0,0Etanol +(H2O)3 -382,202826 -382,202827 0,0Etanol +(H2O)6 -610,322908 -610,322869 0,0Fenol +(H2O)3 -533,685758 -533,685760 0,0Fenol +(H2O)6 -761,783975 -761,783995 0,0
43

Introduccion Metodologıa Teorıa Resultados Conclusiones
Precision del Esquema FMO-APMO
Energıa total APMO-FMO1-RHF, APMO-FMO2-RHF y APMO-RHF
Sistema molecular APMO-FMO1-RHF APMO-FMO2-RHF APMO-RHF ∆E (Hartrees)
(H2O)3 -228,024385 -228,056442 -228,056121 0,000321(H2O)4 -304,075594 -304,095805 -304,094889 0,000916(H2O)5 -380,047920 -380,134556 -380,133776 0,000780(H2O)6 -456,070905 -456,173768 -456,170387 0,003381(H2O)8 -608,117991 -608,272132 -608,267625 0,004507Etanol +(H2O)3 -382,109601 -382,160834 -382,160207 0,000627Fenol +(H2O)3 -533,592882 -533,643894 -533,643300 0,000594
44

Introduccion Metodologıa Teorıa Resultados Conclusiones
Quımica del positron solvatado
La afinidad positronica (AP ) se define como la diferencia deenergıa entre el sistema molecular X y el correspondiente complejopositronico e+X,
AP (X) = E[X]− E[e+X]
Baeses positronicas: E+O7SPD-AUG-CC-PVDZ y E+F7SPD-AUG-CC-PVDZ
45

Introduccion Metodologıa Teorıa Resultados Conclusiones
Quımica del positron solvatado
Afinidad positronica al nivel de teorıa APMO-RHF
APMO-RHFSistema molecular E(X) E(e+X) AP(meV)
(H2O)3 -228,098129 -228,095162 -80,7(H2O)4 -304,136549 -304,132999 -96,6(H2O)5 -380,172922 -380,169543 -91,9(H2O)6 -456,210629 -456,209648 -26,7(H2O)8 -608,309374 -608,305599 -102,7
Afinidad positronica al nivel de teorıa APMO-FMO2
APMO-FMO2Sistema molecular E(X) E(e+X) AP(meV)
(H2O)3 -228,098451 -228,095503 -80,2(H2O)4 -304,137466 -304,133980 -94,8(H2O)5 -380,173702 -380,170381 -90,4(H2O)6 -456,214012 -456,213012 -27,2(H2O)8 -608,313883 -608,310169 -101,1
46

Introduccion Metodologıa Teorıa Resultados Conclusiones
Quımica del positron solvatado
Estudios experimentales14 indican que el ion F− y el e+ no seunen en solucion acuosa, mientras que estudios teoricos presentanvalores positivos de AP para el ion F− y el e+ en fase gaseosa,
Afinidad positronica al nivel de teorıa APMO-RHF
APMO-RHFSistema molecular E(X) E(e+X) AP(meV) µ (debye)
F− + (H2O)3 -327,611440 -327,714060 2792,4 2,27F− + (H2O)6 -555,763864 -555,843436 2165,3 3,17F− + (H2O)16 -1316,242620 -1316,302734 1635,8 6,09
Afinidad positronica al nivel de teorıa APMO-FMO2
APMO-FMO2Sistema molecular E(X) E(e+X ) AP(meV) µ (debye)
F− + (H2O)3 -327,612262 -327,714817 2790,7 2,27F− + (H2O)6 -555,765203 -555,844530 2158,6 3,17F− + (H2O)16 -1316,245417 -1316,305224 1627,4 6,09
14J. R. Andersen, N. J. Pedersen, O. E. Mogensen, Chem. Phys Lett. 1979, 63, 171-173
47

Introduccion Metodologıa Teorıa Resultados Conclusiones
Conclusiones
• Se implemento el metodo de orbitales moleculares defragmentos bajo la aproximacion del orbital molecular paracualquier partıcula (FMO-APMO) en el programa de quımicacuantica LOWDIN.
• Se comprobo la correcta implementacion comparando losresultados con el paquete de estructura electronica GAMESS.
• El metodo FMO-APMO se convierte en el primer esquemapara cualquier partıcula con un escalamiento de N2
• Se comprobo la precision del metodo FMO-APMO por mediodel calculo de afinidades positronicas.
48

Introduccion Metodologıa Teorıa Resultados Conclusiones
Gracias
• Preguntas...
49