guia de esteroides corregida 13 09 10±o... · objetivos al finalizar el estudio de esta clase ud....
TRANSCRIPT

BIOQUIMICA HUMANA
Biosíntesis y metabolismo de
hormonas esteroides y vitamina D

OBJETIVOS
Al finalizar el estudio de esta clase Ud. deberá ser capaz de:
• Reconocer las estructuras de las diferentes hormonas esteroideas, su origen biosintético,
órganos blancos y efectos fisiológicos
• Describir la biosíntesis de las principales hormonas esteroideas: mineralocorticoides,
glucocorticoides y hormonas sexuales
• Describir la histología del testículo, la regulación del eje hipotálamo-hipófiso-testicular y la
biosíntesis de andrógenos, el transporte y catabolismo de dichos esteroides
• Describir la histología del ovario, la regulación del eje hipotálamo-hipófiso-ovárico, y la
biosíntesis de estrógenos y progesterona, el transporte y catabolismo de dichos
esteroides y sus variaciones a lo largo del ciclo menstrual
• Describir la histología de la corteza adrenal, la regulación del eje hipotálamo-hipófiso-
adrenal y la síntesis de glucocorticoides, la regulación de la biosíntesis de
mineralocorticoides (sistema renina-angiotensina), y el transporte y catabolismo de
dichas hormonas corticales.
• Describir las patologías asociadas con la hipo/hiper función de la corteza adrenal en
relación con la producción de glucocorticoides.
• Explicar el camino biosintético y las funciones asociadas a la vitamina D.

HORMONAS ESTEROIDEAS Las hormonas esteroideas son derivados del colesterol y por lo tanto formadas por el
ciclopentanoperhidrofenantreno, compuesto por tres anillos de 6 carbonos (Ilamados A, B
y C) y uno de 5 carbonos (D). A este ciclo se le pueden agregar dos metilos en las
posiciones C10 (metilo C19) y C13 (metilo C18) o una cadena lateral en C17. (Figura 1). Los grupos metilos C19 y C18 se proyectan sobre el plano de los anillos y se usan como
punto de referencia en la nomenclatura. Los sustituyentes que se orientan en el mismo
plano se denominan cis o beta y los que se orientan en el otro sentido se nombran como
trans o alpha. Las hormonas esteroideas se agrupan en 3 familias de acuerdo al número de
metilos y si llevan o no cadena lateral (Figura 1): A) C21 grupo pregnano: poseen metilos
C19,C18 y cadena lateral C21, comprenden los glucocorticoides, mineralocorticoides y
progesterona. B) C19 grupo androstano: poseen metilos C19 y C18, entre ellos figuran los
andrógenos. C) C18 grupo estrano: solo posee metilo C18, derivan de este grupo los
estrógenos, los cuales además poseen un anillo A aromático.
Figura 1: Clasificación de esteroides y estructura del colesterol. Las hormonas esteroideas tienen funciones muy diversas y son sintetizadas en diferentes
glándulas endócrinas como se muestra en el cuadro 1.

ESTRUCTURA Y FUNCIÓN DE LAS PRINCIPALES HORMONAS ESTEROIDEAS
BIOSÍNTESIS DE HORMONAS ESTEROIDEAS El colesterol es el precursor obligado en la síntesis de hormonas esteroideas. Las fuentes
celulares de colesterol son:
• síntesis celular de novo,
• lipoproteínas plasmáticas transportadoras de colesterol,
• gotas citoplasmáticas de ésteres de colesterol que actúan a modo de reservas
intracelulares.
El paso limitante en el camino biosintético del colesterol corresponde a la producción
irreversible de mevalonato catalizada por la enzima β-hidroxi-metil-glutaril coenzima A
(CoA) reductasa (HMG CoA reductasa). La actividad de esta enzima se encuentra
altamente regulada por el aporte de colesterol dietario, por un mecanismo de inhibición

alostérico vía mevalonato, y por la acción conjunta de quinasas y fosfatasas. En las
células esteroidogénicas existe un equilibrio entre el colesterol libre y los ésteres de
colesterol. Dicho equilibrio es regulado por la enzima acilCoA colesterol aciltransferasa
que esterifica al colesterol (ACAT), y por la enzima colesterol éster hidrolasa o esterasa
que cataliza la reacción inversa. (Se sugiere el repaso de la Unidad de colesterol que se
dictó previamente.)
El primer paso en la biosíntesis de hormonas esteroideas es el transporte del colesterol a
la mitocondria, mediado primeramente por la proteína StAR (steroidogenic acute
regulatory protein). Las células esteroidogénicas, en respuesta al estímulo hormonal
trófico, sintetizan una proteína citoplasmática precursora de StAR de 37KDa. Este
precursor se asociaría a proteínas chaperonas que impedirían el plegado y
consecuentemente, su transporte hacia la mitocondria. La proteína de 37KDa sería
entonces incorporada a esta organela subcelular a través de su interacción con un
receptor específico presente en la membrana externa mitocondrial. Posteriormente, se
formarían puntos de contacto entre las membranas externa e interna de la mitocondria, y
ocurriría el primer evento de clivaje, obteniéndose una forma intermediaria de la proteína
StAR de 32KDa. Esta fusión de membranas, facilitaría el transporte del colesterol hacia la
membrana interna mitocondrial. Finalmente, las membranas se fusionarían, y la proteína
de 32KDa sufriría un segundo clivaje originándose un producto de 30KDa incapaz de
participar en el transporte de colesterol. Así, el ingreso de colesterol en la mitocondria,
estaría controlado a través de la continúa síntesis y procesamiento de StAR, y la
formación de sitios de contacto entre las dos membranas de la organela (Figura 2).
Figura 2: Transporte de colesterol hacia la matriz mitocondrial. Mecanismo de acción de
StAR.

Además de la proteína StAR, existe otro importante grupo de proteínas involucradas en
la regulación de la producción de hormonas esteroideas, entre las cuales podemos citar a
un transportador proteico de colesterol (SCP2), que moviliza dicho compuesto desde las
gotas lipídicas citoplasmáticas a la mitocondria, un polipéptido activador de la
esteroidogénesis (SAP), una proteína inductora de la esteroidogénesis (SIP) y la proteína
traslocadora (TSPO) presente en altas concentraciones en la membrana mitocondrial
externa de tejidos esteroidogénicos.
Las enzimas e intermediarios involucrados en el camino biosintético de las hormonas
esteroideas se encuentran esquematizados en la Figura 3.
Figura 3: Camino biosintético de las hormonas esteroideas. La pregnenolona (21 átomos de C) da origen a la progesterona, que no sólo tiene acción por sí misma como hormona sexual femenina, sino que también permite sintetizar todas las hormonas esteroideas: 1) las hormonas suprarrenales con 21 átomos de C (aldosterona y cortisol); 2) las hormonas sexuales masculinas (andrógenos) con 19 átomos de C a nivel suprarrenal testicular y ovárico (dehidroepiandrosterona-DHEA, androstenediona, testosterona y dihidrotestosterona- cuadro celeste), y 3) hormonas sexuales femeninas (estradiol, estrona y estriol, en rosa) con 18 átomos de C a nivel ovárico.

Como se explicó más arriba, el primer paso de la biosíntesis de hormonas esteroides
sucede en la membrana interna mitocondrial. Dicha biosíntesis involucra la conversión
del colesterol a pregnenolona, catalizada por un complejo multienzimático dependiente
del citocromo P-450 que escinde la cadena lateral del colesterol (desmolasa o P450scc)
entre los carbonos 20 y 22.
En el retículo endoplasmático liso, la pregnenolona puede seguir dos caminos diferentes:
Δ4 o Δ5. Esta terminología designa la posición que el enlace insaturado mantiene durante
la transformación de los intermediarios. Así, la pregnenolona puede ser sustrato de la
enzima 3β-hidroxiesteroide deshidrogenasa Δ5-Δ4 isomerasa (3β-HSD) dando origen a la
progesterona a través del camino Δ4. Pero también, la pregnenolona puede seguir la vía
Δ5 obteniéndose primeramente 17-Hidroxipregnenolona por acción de la enzima 17α-
hidroxilasa.
Las transformaciones químicas en las cuales participarán la progesterona y la 17-
hidroxipregnenolona dependerán de cada glándula esteroidogénica en particular. Dado
que las distintas enzimas esteroidogénicas se encuentran algunas en la mitocondria y
otras en el retículo endoplásmico liso (Figura 3), habrá un movimiento de lanzadera
repetido de sustratos, dentro y fuera de la mitocondria.
Hidroxilaciones mediadas por el citocromo p450
Las reacciones de hidroxilación juegan un rol muy importante en la síntesis de colesterol,
hormonas esteroideas y sales biliares y requieren NADPH y O2. Uno de los átomos de O2
queda unido al esteroide formando el grupo hidroxilo y el otro forma agua como se
muestra en el esquema:
Las enzimas que catalizan este tipo de reacciones se llaman monooxigenasas, y en
particular en la síntesis de esteroides se las denomina citocromo P-450 y poseen un
grupo prostético hemo. La hidroxilación requiere la activación del O2. A pesar de ser
reacciones de oxidación, este proceso involucra al NADPH. El NADPH transfiere los
electrones a una flavoproteína que a su vez los transfiere a una proteína que no posee
grupo prostético hemo, llamada adrenodoxina. La adrenodoxina transfiere los electrones
de a uno para reducir el hierro férrico (Fe+++) del P450 a ferroso (Fe++) (Figura 4). Al
incorporar este electrón, el citocromo P450 puede unir la molécula de O2. Cuando se
pega el O2, la adrenodoxina cede el otro electrón, el cual conduce al clivaje de la
molécula de O2. Uno de los átomos de O2 es protonado para formar agua y el otro

permanece como un intermediario altamente reactivo unido al hierro. Este intermediario
reacciona con el esteroide el cual se hidroxila y el hierro retorna a su estado férrico
(Fe+++)
Figura 4: Mecanismo de acción del citocromo P450 en la biosíntesis de esteroides
Transporte plasmático de hormonas esteroides En cada sección se detallará el transporte específico de cada hormona esteroide. Sin
embargo, es necesario recordar que dada su característica lipofílica, gran porcentaje de
estas hormonas circula unida a proteínas, mientras que la fracción libre de la hormona es
activa.
Catabolismo El catabolismo es principalmente hepático. El objetivo es aumentar la solubilidad en agua
del esteroide a través de su conjugación entre otros con el ácido glucurónico. De esta
manera, el esteroide conjugado puede ser eliminado por orina.

BIOSÍNTESIS DE ESTEROIDES EN EL TESTÍCULO Y SU REGULACIÓN HISTOLOGÍA Y FUNCIÓN Los testículos (gr. orchis o didymis) son glándulas endócrinas productoras de
andrógenos y factores, que proporcionan el medio adecuado para el desarrollo de la
espermatogénesis.
En los mamíferos, los testículos se encuentran de a pares ubicados fuera del abdomen,
en el escroto, y rodeados por una cápsula de tejido conectivo, la túnica albugínea. Desde
la túnica albugínea se extiende un engrosamiento hacia el interior del órgano, el
mediastino testicular o cuerpo de Highmore, del cual emergen tabiques de tejido
conectivo que separan el tejido glandular en 200-300 lobulillos testiculares. Cada lobulillo
contiene varios túbulos seminíferos, representantes del compartimiento productor de
espermatozoides del testículo. Los túbulos seminíferos poseen un diámetro aproximado
de 200-250µm con longitudes que varían según la especie. La pared de los túbulos
seminíferos está compuesta por cuatro capas: 1) la membrana basal, 2) células mioides
responsables de los movimientos peristálticos del túbulo, 3) fibras de colágeno, y la zona
más externa está integrada por células endoteliales. Por dentro de la membrana basal,
los túbulos se encuentran revestidos por un epitelio estratificado que contiene dos tipos
de células: las denominadas células de sostén, nodrizas o de Sertoli, y las células espermatogénicas que comprenden espermatogonias, espermatocitos primarios y
secundarios, espermátides y espermatozoides. Los túbulos seminíferos se continúan en
túbulos rectos que desembocan en la rete testis, un sistema de conductillos ubicado en el
mediastino. El intersticio, que rodea los túbulos seminíferos, contiene a las células intersticiales o de Leydig, que presentan función endócrina y, por ende, producen
andrógenos como testosterona (Figura 5).
Figura 5: Corte histológico del testículo. ST: túbulos seminíferos

Células de Leydig: Las células de Leydig se encuentran generalmente agrupadas en cúmulos relacionados
con capilares o bien cercanos a la pared de los túbulos seminíferos. Son células
poligonales o fusiformes, con un diámetro de 8-12 µm, que presentan a menudo
proyecciones que fueron descriptas por algunos autores como filapodios o
microvellosidades. Su núcleo es grande y redondo, y presenta uno o dos nucleolos
prominentes. El citoplasma acidófilo, contiene lisosomas, peroxisomas, vesículas
pinocíticas, cuerpos multivesiculares, un retículo endoplasmático liso y un aparato de
Golgi muy desarrollados, y múltiples mitocondrias. Las células de Leydig se desarrollan,
bajo el estímulo de la LH, a partir de precursores que han sido definidos en la literatura
como células mesenquimáticas, células mioides peritubulares y macrófagos. El 95% de la
testosterona circulante es producida por las células de Leydig. Se cree que la forma
activa de la testosterona es la 5α-dihidrotestosterona (DHT), y que la especial
sensibilidad a la testosterona de ciertos tejidos, se debe a que sus células son capaces
de reducirla a DHT. En la actualidad es reconocido que las células de Leydig sintetizan
también otras sustancias bioactivas involucradas en la regulación parácrina y autócrina
de la función testicular. Así, proveen numerosas hormonas y factores potencialmente
activos tales como péptidos derivados de la pro-opiomelanocortina (POMC),
prostaglandinas, activina, inhibina, neurotransmisores (GABA, glutamato, serotonina) y
neuropéptidos (NPY, VIP).
EJE HIPOTÁLAMO-HIPÓFISO-TESTICULAR Las hormonas LH y FSH son sintetizadas y secretadas por la adenohipófisis en
respuesta al estímulo hipotalámico generado por la hormona liberadora de
gonadotrofinas (GnRH o LHRH). GnRH es un decapéptido producido en las neuronas
peptidérgicas del área preóptica del hipotálamo, liberado en forma pulsátil al sistema
porta hipofisario desde las terminales nerviosas de la eminencia media. Así, esta
hormona llega a la hipófisis en donde interactúa con receptores de membrana presentes
en los gonadotropos.
Las gonadotrofinas hipofisarias actúan sobre los testículos promoviendo y estimulando la
actividad reproductiva. Mientras que FSH ejerce su acción sobre las células de Sertoli
regulando la espermatogénesis; LH actúa sobre las células de Leydig induciendo la
síntesis de andrógenos (Figura 6). Sin embargo, ambos procesos gonadales son
interactivos, requiriendo la integración y coordinación entre las distintas poblaciones
celulares del testículo.
Los andrógenos estimulan el crecimiento y desarrollo de los órganos reproductivos,
participan en la expresión de los caracteres sexuales secundarios, inducen y mantienen
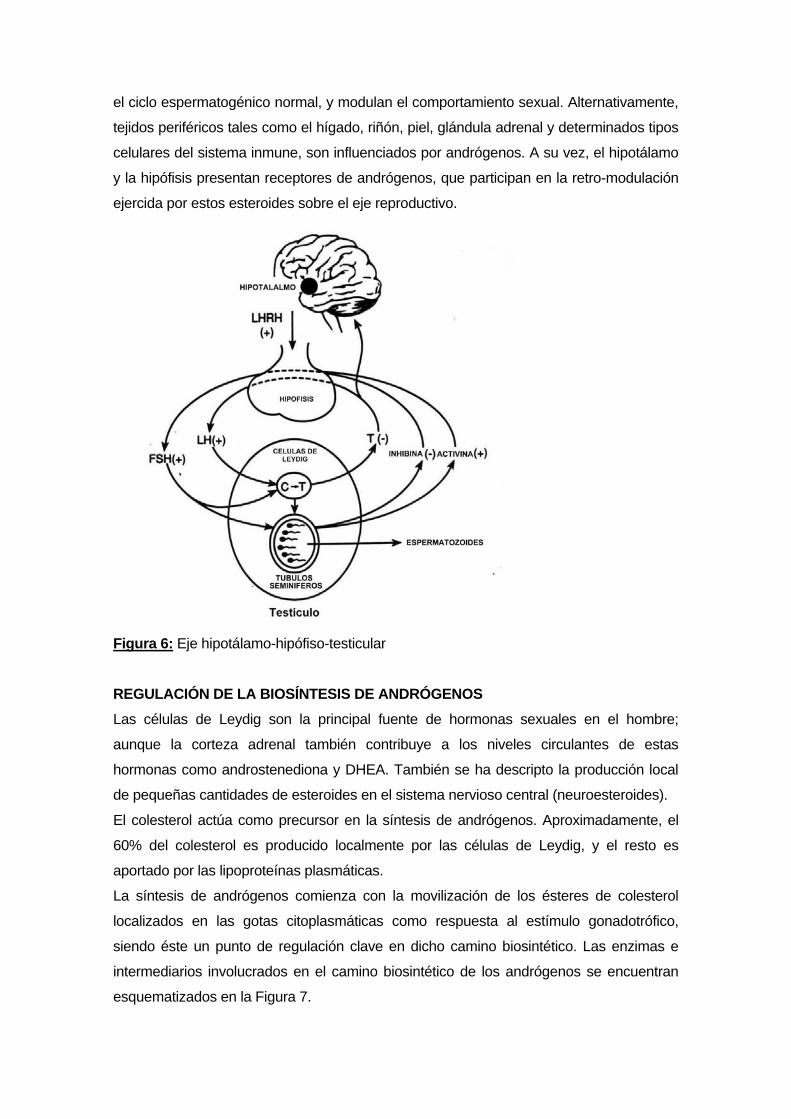
el ciclo espermatogénico normal, y modulan el comportamiento sexual. Alternativamente,
tejidos periféricos tales como el hígado, riñón, piel, glándula adrenal y determinados tipos
celulares del sistema inmune, son influenciados por andrógenos. A su vez, el hipotálamo
y la hipófisis presentan receptores de andrógenos, que participan en la retro-modulación
ejercida por estos esteroides sobre el eje reproductivo.
Figura 6: Eje hipotálamo-hipófiso-testicular
REGULACIÓN DE LA BIOSÍNTESIS DE ANDRÓGENOS Las células de Leydig son la principal fuente de hormonas sexuales en el hombre;
aunque la corteza adrenal también contribuye a los niveles circulantes de estas
hormonas como androstenediona y DHEA. También se ha descripto la producción local
de pequeñas cantidades de esteroides en el sistema nervioso central (neuroesteroides).
El colesterol actúa como precursor en la síntesis de andrógenos. Aproximadamente, el
60% del colesterol es producido localmente por las células de Leydig, y el resto es
aportado por las lipoproteínas plasmáticas.
La síntesis de andrógenos comienza con la movilización de los ésteres de colesterol
localizados en las gotas citoplasmáticas como respuesta al estímulo gonadotrófico,
siendo éste un punto de regulación clave en dicho camino biosintético. Las enzimas e
intermediarios involucrados en el camino biosintético de los andrógenos se encuentran
esquematizados en la Figura 7.

La progesterona, originada a través del camino Δ4 es transformada en androstenediona
por el complejo enzimático 17α hidroxilasa C17-20 liasa, dependiente del citocromo P-
450 (P-450c17). Pero también, la pregnenolona puede seguir la vía Δ5 obteniéndose
primeramente dehidroepiandrosterona por acción del complejo P-450c17 y luego, por
isomerización, androstenediona.
El paso final en la síntesis de testosterona está regulado por la enzima microsomal
17β-hidroxiesteroide deshidrogenasa (17βHSD), que cataliza la conversión de
androstenediona a testosterona. Dicha reacción es reversible siendo su dirección
regulada por la concentración de sustrato y producto. Esta enzima posee dos sitios
activos y presenta múltiples isoformas.
Figura 7: BIOSÍNTESIS DE ANDRÓGENOS TESTICULARES. (1) complejo enzimático que escinde la cadena lateral del colesterol, (2) 3β-hidroxiesteroide deshidrogenasa/4-5 isomerasa, (3) 17α-hidroxilasa. 4: 17,20 liasa, (5) 17β-hidroxiesteroide deshidrogenasa.

La principal regulación fisiológica de la síntesis testicular de andrógenos es ejercida por la
adenohipófisis a través de la secreción pulsátil de LH; aunque otras hormonas y factores
producidos localmente, también estarían involucrados en este proceso mediante un
control autócrino y/ó parácrino entre las células de Leydig y los restantes componentes
testiculares.
La acción estimulatoria de la LH sobre la producción de andrógenos se ejerce a través de
su unión a receptores específicos ubicados en la membrana de las células de Leydig.
Dicha interacción ligando-receptor induce un cambio conformacional en la proteína G,
que activa una adenilato ciclasa, dependiente de GTP y Mg+2, ocasionando un aumento
en la síntesis de AMPc. El AMPc obtenido se une a la subunidad regulatoria de una PKA
con la subsiguiente liberación de la subunidad catalítica de la misma. Esta subunidad
catalítica activa, fosforila proteínas intracelulares, en sus residuos de treonina y serina,
que intervienen en la síntesis de esteroides regulando la expresión génica y traducción
de enzimas claves del proceso. Si bien la función del AMPc como segundo mensajero en
la acción de LH se encuentra bien establecida, recientemente se han descripto
mensajeros alternativos y respuestas rápidas, que involucran cambios en el flujo iónico
transmembrana, depósitos intracelulares de calcio, actividad de la proteína calmodulina, y
alteraciones en el metabolismo y recambio de fosfolípidos.
La FSH ejerce también un efecto modulatorio sobre la actividad de las células de Leydig
Trabajos recientes sugieren que FSH estimula la síntesis y/o secreción de un factor en
las células de Sertoli. Este factor estimula el efecto de LH sobre la actividad de las
enzimas 3β-HSD y citocromo P450 17α-hidroxilasa, y consecuentemente, sobre la
síntesis de andrógenos.
Por otra parte, otras hormonas hipofisarias como la prolactina y adrenocorticotrofina
(ACTH) serían capaces de influenciar el estado funcional de estas células
esteroidogénicas.
Además, la producción endógena de esteroides en la célula de Leydig regula la
expresión de enzimas involucradas en la biosíntesis de testosterona.
TRANSPORTE DE LOS ANDRÓGENOS Como consecuencia de su hidrofobicidad, la testosterona es transportada en sangre
unida, en su mayor parte, a proteínas. El principal transportador, es una β-globulina
denominda β-globulina fijadora de hormona sexuales (SHBG). La albúmina sérica actúa
también como un transportador inespecífico de testosterona.

Las células de Sértoli producen una proteína transportadora de andrógenos conocida
como ABP (androgen binding protein), que sirve para transportar la testosterona en el
fluído luminal de los túbulos seminíferos del testículo.
Sólo una pequeña porción de la testosterona en sangre permanece libre. Esta porción
puede atravesar rápidamente las membranas celulares y por ello es la forma activa
desde el punto de vista biológico.
CATABOLISMO DE LOS ANDRÓGENOS Los niveles séricos de testosterona resultan de un balance entre la producción de esta
hormona y su desaparición de la circulación vía excreción y transformación metabólica.
Algunos esteroides se excretan libres. Sin embargo, el catabolismo y excreción de la
mayoría de los andrógenos, involucra la inicial transformación de los mismos en
compuestos de mayor solubilidad en agua a través de su conjugación con glucurónidos y
sulfatos en la posición 3 ó 17. Con excepción de los esteroides fenólicos, las reacciones
catabólicas son reductivas y ocurren mayoritariamente en el hígado; aunque la próstata y
la piel también contribuyen significativamente al metabolismo degradativo de los
andrógenos. Los 3α-hidroxiesteroides aparecen principalmente conjugados como
glucurónidos; mientras que, la mayoría de los 3β-hidroxiesteroides son excretados en
forma de sulfatos. Una gran parte de los metabolitos de los andrógenos son eliminados a
través de la orina. Los compuestos 3α-hidroxi-5α-androstano-17-ona (androsterona) y
3α-hidroxi-5β-androstano-17-ona (etiocolanolona) son los metabolitos urinarios más
abundantes excretados mayoritariamente como glucurónidos.
Sin embargo, las transformaciones metabólicas no siempre están asociadas con una
disminución en la actividad biológica. La metabolización de la testosterona a través de la
5α-reducción irreversible origina DHT, y una notoria amplificación de sus acciones
biológicas. A su vez, la DHT puede ser metabolizada a 5α-androstano 3α/β, 17β-diol
(Diol) por acción de las enzimas 3α/β hidroxi-esteroide óxido reductasa, que requieren
nicotinamida-adenina-dinucleótido fosfato reducido (NADPH) como cofactor (Figura 8).
En testículos inmaduros y púberes, se describe una importante actividad de las enzimas
5α-reductasa y 3α-hidroxiesteroide óxido-reductasa, sintetizándose metabolitos 5α-
reducidos tales como Diol. En el caso de especies fotoperiódicas, a través de 5β-
reducción se obtienen concentraciones importantes de compuestos 5β-reducidos de
función desconocida. Por otro lado, la testosterona y la androstenediona son
interconvertibles vía 17β-HSD. Testosterona y androstenediona pueden estar sujetas a
hidroxilaciones, pero dichas reacciones no contribuyen significativamente al metabolismo
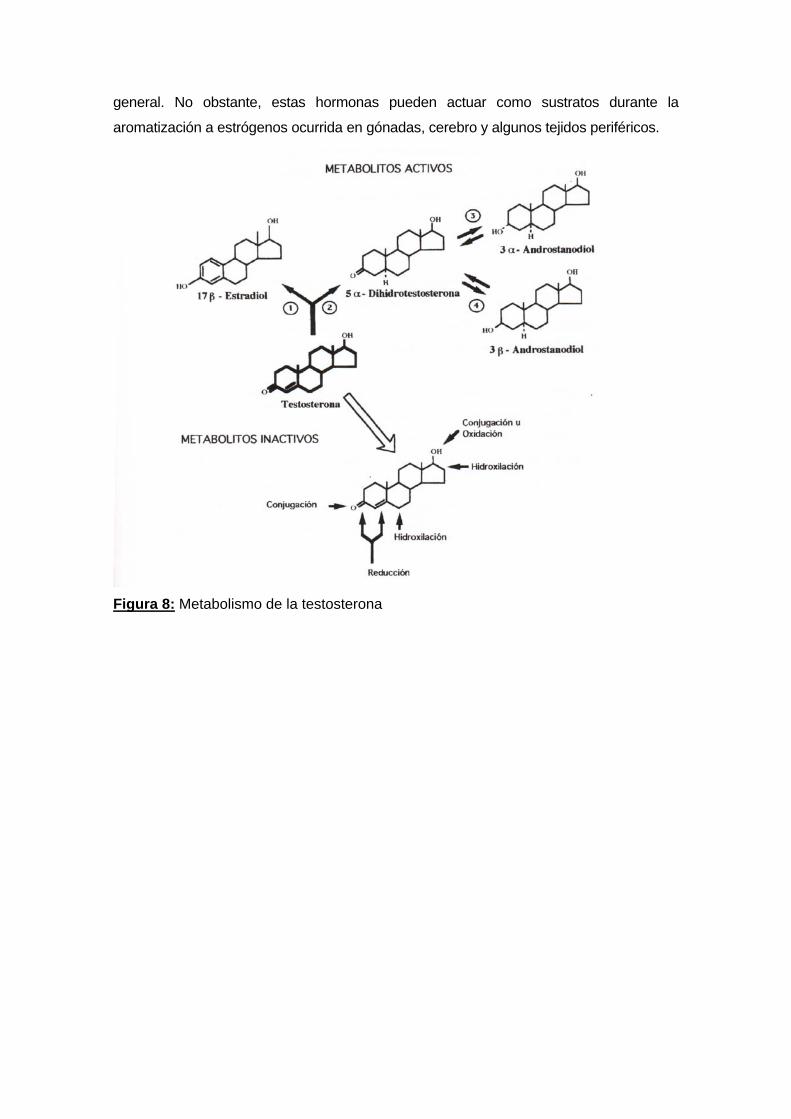
general. No obstante, estas hormonas pueden actuar como sustratos durante la
aromatización a estrógenos ocurrida en gónadas, cerebro y algunos tejidos periféricos.
Figura 8: Metabolismo de la testosterona

BIOSINTESIS DE ESTEROIDES EN EL OVARIO Y SU REGULACION. HISTOLOGÍA Y FUNCIÓN El ovario humano es una glándula endócrina cuyo peso y tamaño varía
durante el ciclo menstrual. Está formado por varias estructuras, que están ligadas a la
síntesis de distintas hormonas sexuales. Por ello, resulta conveniente efectuar un
breve repaso histológico. Durante la edad reproductiva, la composición estructural del
ovario y su actividad hormonal están en continuo cambio. Normalmente, el ovario está
cubierto por un epitelio germinal (ver Figura 9) del que nacen los folículos
primordiales. Estos están compuestos por una membrana basal, células granulosas y
un pequeño oocito. A partir de los folículos primordiales se forman los folículos
preantrales, debido a la proliferación de las células de la granulosa. Cuando el folículo
preantral alcanza a tener 6-7 capas de células granulosas, la teca interna aumenta de
tamaño y comienza la formación de la cavidad antral. En ausencia de estimulación
gonadotrófica apropiada, los folículos no pasan del estado antral temprano y se
vuelven atrésicos. Los folículos preantrales crecen a antrales, compuestos en su
pared por la teca externa, teca interna y granulosa, y ya contienen el fluido folicular.
Antes de la ovulación se llaman “foliculos de Graaf” midiendo 2-2.5 cm. Los folículos
son el sitio de mayor producción de estrógenos. Producida la ovulación, se forma
primero un cuerpo hemorrágico y luego el cuerpo lúteo, que se caracteriza por
producir progesterona. Si no se produce la fecundación y embarazo, el cuerpo
amarillo dura unos 14 días y se convierte en tejido fibroso, llamándoselo corpus
albicans. Este mecanismo se denomina luteólisis.
Figura 9: Etapas del desarrollo folicular
El ovario cumple dos funciones esenciales para la reproducción: 1) produce y libera
un óvulo cada 28-30 días, y 2) sintetiza y secreta a la sangre las hormonas sexuales
femeninas: estrógenos y progesterona, además de cierta cantidad de andrógenos, y
de otras sustancias no esteroides (inhibina, activina). Cerca de 400 óvulos maduros

son producidos durante la vida de una mujer, aunque muchísimos más son los
folículos que nunca llegan al estadio maduro.
EJE SISTEMA NERVIOSO CENTRAL (SNC)-HIPÓFISO-GONADAL
El control del crecimiento folicular es debido a varios factores. Al principio, la
estimulación del crecimiento de los folículos primordiales hasta la etapa de folículos
preantrales parece no depender de las gonadotrofinas hipofisarias: hormona folículo-
estimulante (FSH) y hormona luteinizante (LH). En cambio, las células granulosas
desarrolladas poseen receptores para FSH. La unión de FSH a su receptor estimula
la síntesis de la enzima aromatasa que convierte a los precursores androgénicos en
estradiol. Así, la acción concertada de FSH y estradiol sobre los folículos pequeños
estimula su crecimiento hasta el grado de folículo antral. Asimismo, la acción
concertada de FSH y estradiol induce la síntesis de receptores de hormona
luteinizante (LH) en las células granulosas. Existen también receptores para LH en las
células de la teca, intersticiales y obviamente, en las del cuerpo lúteo. Por
consiguiente, la acción de LH sobre: 1- las células de la teca estimula la síntesis y
secreción de andrógenos, 2- las células de la granulosa, la secreción de estrógenos y
3- las células del cuerpo lúteo, la secreción de progesterona.
Biosíntesis de estrógenos:
Las hormonas esteroides ováricas se originan a partir del colesterol, el cual está
presente en la glándula como colesterol libre o esterificado con ácidos grasos. El
colesterol a su vez proviene de la captación por el ovario de las lipoproteínas
circulantes ricas en colesterol (LDL, HDL) o bien puede ser sintetizado in situ a partir
del acetato. El primer paso en la biosíntesis es común con las otras glándulas
endócrinas, descripto previamente. Está controlado por la hormona LH hipofisaria por
un paso mediado por la unión de la LH a su receptor, síntesis de AMP cíclico y
activación por el AMP cíclico de la proteína quinasa A. Esta última promueve la
fosforilación de una proteína que transporta el colesterol desde sus depósitos
intracelulares hacia la mitocondria, donde es convertido en pregnenolona por la
enzima dependiente del citocromo P450 llamada desmolasa o P450scc. La
pregnenolona formada en esta vía puede convertirse en progesterona o 17 α-
hidroxipregnenolona (Figura 3 y Figura 9). La conversión de pregnenolona a
progesterona ocurre por intermedio de 2 enzimas: la 3β-ol deshidrogenasa Δ4-Δ5
isomerasa (abreviada como 3β-HSD en la Figura). La pregnenolona puede, además,
convertirse en 17α-hidroxipregnenolona al actuar una 17α-hidroxilasa o P45017αOH

(enzima microsomal que requiere NADPH, como todas la hidroxilasas de los tejidos
esteroidogénicos). La progesterona no puede convertirse como tal en andrógenos (los
precursores de los estrógenos) sino que también debe 17 hidroxilarse a 17α-
hidroxiprogesterona por la P45017αOH, en forma similar a la pregnenolona. Ahora,
tenemos los dos productos: la 17α-hidroxiprogesterona y la 17α-hidroxipregenolona.
La 17 hidroxilación es un mecanismo que provoca el corte la cadena lateral, y que
convierte a los compuestos de 21 átomos de carbono en andrógenos de 19 carbonos.
Así, a partir de la 17α-hidroxiprogesterona se forma androstenediona, mientras que a
partir de la 17α-hidroxipregnenolona se origina la dehidroepiandrosterona (DHEA).
Ambos son andrógenos débiles, y se pueden convertir en testosterona, en forma
directa la androstenediona por acción de la 17β-hidroxiesteroide
deshidrogenasa/reductasa (17 beta-HSO en la Figura 9). La DHEA necesita
convertirse primero en androstenediona para formar testosterona. La síntesis de
andrógenos en el ovario ocurre en la teca, siendo la misma estimulada por la LH
hipofisaria.
La conversión de andrógenos a estrógenos es catalizada por la enzima
aromatasa. La estrona se sintetiza a partir de la androstenediona, y el estradiol a
partir de la testosterona. La aromatasa o P450arom, de las células granulosas está
estimulada por la FSH, y el estradiol así formado puede en parte ingresar al fluido
folicular, o en su mayoría pasar a sangre y ser secretado a la periferia por medio de la
vena ovárica. La teca también secreta andrógenos a la sangre, preferentemente la
androstenediona que es un andrógeno débil.
Figura 10: Rutas biosintéticas en el ovario
Se ha postulado la “teoría de las 2 células“para explicar la formación del estradiol en
el folículo. De acuerdo a esta hipótesis, la LH actúa sobre las células de la teca

interna estimulando la síntesis de andrógenos (Figura 11). Esta síntesis ocurre porque
las células tecales poseen receptores que unen LH, activándose la adenilato ciclasa y
la síntesis de AMP cíclico, el cual por activación de proteína quinasa A y
fosforilaciones estimulan la conversión de colesterol en pregnenolona. La
pregnenolona, por los pasos detallados anteriormente, se transforma en
androstenediona y en menor grado testosterona. Estas hormonas difunden a través
de la membrana basal que separa las células de la granulosa de las tecales, y aunque
una pequeña cantidad de las mismas entra al fluido antral, la mayoría se aromatiza a
estradiol en las células de la granulosa. Las células de la granulosa, a su vez, poseen
receptores para FSH, estimulándose allí la síntesis de AMP cíclico, el cual estimula la
aromatasa responsable de la conversión de andrógenos en estrógenos, como se
observa en la Figura 11. Como se dijo anteriormente, la FSH es responsable de la
diferenciación y desarrollo de los folículos. De esta manera, las acciones de la LH y
FSH se complementan actuando sobre dos tipos celulares diferentes (teca y
granulosa), conducentes a la síntesis final del estradiol.
Figura 11: TEORÍA DE LAS 2 CÉLULAS

Regulación de la aromatasa La enzima aromatasa es clave en la síntesis de estrógenos ováricos y extra-
ováricos. En el caso del ovario, la FSH induce la transcripción del gen de la
aromatasa. El mecanismo es genómico, como se detalla en la Figura 12. La FSH se
une a su receptor de membrana, lo cual resulta en la activación de la adenilato ciclasa
vía la activación del sistema de la proteína G. La estimulación de la síntesis del AMP
cíclico activa la PKA, y aunque los eventos posteriores están en discusión, el CREB
(cAMP response element binding protein) parece estar involucrado. El factor
esteroidogénico 1 (SF-1) y el CREB se unen a un sitio de reconocimiento específico
del gen de la aromatasa (llamado “CYP19” ) y estimulan la transcripción del ARNm.
Figura 12: Regulación de la expresión de aromatasa por FSH
Biosíntesis de estrógenos extraováricos:
En la mujer postmenopáusica y en hombres ancianos, el principal sitio de biosíntesis
de estrógenos es extraovárico, ocurriendo principalmente en tejido adiposo
glúteofemoral y mamario. Los estrógenos en este tejido derivan de los andrógenos
suprarrenales, ováricos o testiculares que actúan como precursores. La aromatasa
encargada de convertirlos en estrógenos, tiene una regulación totalmente distinta a la
del ovario ya que aquí la FSH no juega ningún papel.
Relevancia en Cáncer de Mama: en el tejido estromal que rodea al tumor mamario la
prostaglandina PGE2 producida por el epitelio tumoral, fibroblastos del tumor y/o
macrófagos infiltrantes estimulan a la enzima aromatasa con la consiguiente

producción local de estrógenos. Este mecanismo conduce al crecimiento continuo y
desarrollo del tumor mamario.
Biosíntesis de progesterona: La progesterona es producida y secretada por el
cuerpo lúteo porque sus células no contienen enzimas que la metabolicen a otras
hormonas esteroides.
Transporte plasmático de esteroides ováricos: Luego de ser liberados a la
circulación, los esteroides ováricos se unen a proteínas plasmáticas. El estradiol se
liga ávidamente a la SHBG en un 37 % y con mucha menor afinidad pero en mayor
proporción (61%) a la albúmina. El 2% restante circula libre. La SHBG se sintetiza en
el hígado, y su síntesis se estimula por los estrógenos e inhibe por los andrógenos, de
manera que sus niveles son el doble en mujeres que en hombres.
La progesterona se une fuertemente a la CBG o transcortina (la proteína que también
une cortisol) y débilmente a la albúmina. Los porcentajes de unión correspondientes a
la progesterona son: 20% a CBG, 2.5 % libre y el resto a albúmina.
Catabolismo de los estrógenos y progesterona: El estradiol circulante es captado
por el hígado donde se convierte en estrona por la acción de la 17-β-hidroxiesteroide
deshidrogenasa (17betaHSO). En pequeña proporción la estrona se escapa a la
circulación donde puede actuar en sus células blanco ya que es un estrógeno débil.
Sin embargo, la mayoría de la estrona se metaboliza a estriol (un metabolito urinario
muy importante) (Figura 13).
Figura 13: Catabolismo de los estrógenos

En orina se excretan los derivados conjugados de los estrógenos, a saber: estrona
sulfato, estriol-3-sulfato y estriol-16-glucurónido. Las conjugaciones con acido sulfúrico
y glucurónico se producen en el hígado y convierten a los estrógenos en formas
solubles aptas para su excreción urinaria.
La progesterona sintetizada en el ovario desaparece rápidamente de la circulación, ya
que su vida media es de solamente 5 minutos. Es captada en el hígado, donde se
reduce a pregnandiol (pregnano-3 α, 20 α-diol) por la acción de las enzimas 5
reductasa y 20 reductasa. El pregnandiol se conjuga con ácido glucurónico y el
pregnandiol glucurónido es el principal metabolito de la progesterona hallado en
orina y cuya determinación tiene importante significado clínico para diagnóstico de la
ovulación y de embarazo. (Figura 14). Otro metabolito importante de la progesterona
es la 20 α -hidroxiprogesterona, pero que posee solamente un quinto de la actividad
biológica de su precursor.
PROGESTERONA PREGNANDIOL (3 β-pregnano-3 α :20 α-diol)
Figura 14: El pregnandiol como catabolito de la progesterona.
Variaciones hormonales durante el ciclo menstrual
El ovario secreta estrógenos, progesterona y andrógenos. Su síntesis y secreción está
gobernada por las gonadotrofinas hipofisarias (Figura 15). En el ciclo menstrual
normal, ambas FSH y LH comienzan a aumentar en plasma en los días previos a la
menstruación. Las concentraciones de FSH son mayores que las de LH y son
máximas durante la primera mitad de la fase folicular, para mostrar un pico durante la
ovulación y luego disminuir durante la fase luteal o secretoria. Las concentraciones de
LH aumentan gradualmente durante la fase folicular y presentan un gran pico que
dura 1-3 días en la mitad del ciclo, con posterior declinación en la fase luteal. Las

determinaciones frecuentes de LH sérica muestran que su secreción es pulsátil,
debida a la secreción pulsátil del factor liberador hipotalámico GnRH.
Con relación a las variaciones del estrógeno biológicamente activo, el estradiol (Figura
15), sus niveles plasmáticos en la primera parte de la fase folicular son bajos,
menores que 50 pg/ml de plasma para subir rápidamente a 200-300 pg/ml el día del
pico de LH o precediéndolo. Este aumento se correlaciona con el aumento de tamaño
del foliculo preovulatorio. Luego de la ovulación, los niveles bajan, aunque se produce
un segundo pico en la mitad de la fase luteal debida a la secreción estrogénica por el
cuerpo amarillo. Los niveles de estradiol caen abruptamente antes de la
menstruación.
Durante toda la fase folicular, los niveles de progesterona son bajos, menores a 1
ng/ml (Figura 15), subiendo luego de la ovulación debido a la gran secreción de la
misma por el cuerpo amarillo. Alcanza un plateau de 20 ng/ml en la mitad de la fase
luteal, para descender abruptamente antes de la menstruación. La determinación de
la concentración plasmática de progesterona es un valioso signo de ovulación cuando
su concentración supera los 5 ng/ml de plasma.
Figura 15: Variaciones hormonales en el ciclo menstrual femenino

BIOSINTESIS DE ESTEROIDES EN LA CORTEZA SUPRARRENAL Y SU REGULACIÓN
En 1929 ya existían evidencias sugiriendo que las suprarrenales producían factores con
importancia fundamental para mantener la vida. La corteza suprarrenal, por medio de sus
hormonas, regula el metabolismo intermedio y el balance hidroelectrolítico, mientras que la
médula, que se encuentra en el centro del órgano, es parte del sistema nervioso y sintetiza
catecolaminas. Morfológicamente, en la corteza suprarrenal se distinguen tres zonas. La
zona glomerulosa o externa, es la zona donde se sintetizan los mineralocorticoides, entre
los que se encuentra la aldosterona. La zona fascicular o media constituye la mayor parte
de la corteza; sus células se ordenan en columnas y en ella se sintetizan los
glucocorticoides siendo el principal en el hombre, el cortisol. Por último, la zona reticular o interna donde las células se disponen irregularmente produciendo principalmente
esteroides sexuales.
Figura 16: Esquema histológico de la glándula suprarrenal y compartimentalización de la síntesis de esteroides

BIOSÍNTESIS DE ESTEROIDES EN LA SUPRARRENAL:
Zona fasciculada-Glucocorticoides (grupo prengano C21)
Los glucocorticoides son el cortisol en el hombre, primates y algunas especies animales
(perro, gato, cobayo, etc.) y la corticosterona, secretada principalmente por los roedores. El
hombre secreta entre 15 y 30 mg diarios de cortisol. El cortisol proviene del colesterol
(Figura 3). En los pasos de la biosíntesis intervienen enzimas dependientes del citocromo
P450. El primer paso consiste en el clivaje de la cadena lateral del colesterol para formar
pregnenolona mediante la colesterol desmolasa o P450scc. Luego ocurre una hidroxilación
en C17 en la cual interviene la 17α-hidroxilasa (P45017OH) y una 3 β-deshidrogenación e
isomerización para formar 17-hidroxiprogesterona. Posteriormente suceden dos
hidroxilaciones sucesivas (C21 y C11) llevadas a cabo por la 21 hidroxilasa (P450 21OH) y
11β-hidroxilasa (P450 11OH, modernamente llamada CYP11B). En la rata no ocurre la 17
hidroxilación y el producto final es la corticosterona. La 3 β HSD, la 17 α hidroxilasa y la 21
hidroxilasa son enzimas del reticulo endoplasmático mientras que la 11 β-hidroxilasa es una
enzima mitocondrial, lo que supone un movimiento de lanzadera repetido de sustratos,
dentro y fuera de la mitocondria.
Zona glomerulosa- Mineralocorticoides (grupo prengano C21)
De los mineralocorticoides, el más activo es la aldosterona, de la cual el hombre secreta
0,07-0,2 mg por 24 horas. Los mineralocorticoides se forman a partir de la progesterona, la
cual proviene de la pregnenolona (Figura 3). En la zona glomerulosa no se expresa la 17 α-
hidroxilasa del retículo endoplasmático liso, por lo que la progesterona es hidroxilada en
C21 para formar 11-Desoxicorticosterona (DOC) que es un mineralocorticoide activo de
acción similar a la aldosterona pero 20 veces menos potente. La hidroxilación siguiente en
C11 produce corticosterona que tiene actividad de glucocorticoide y es un
mineralocorticoide débil. La DOC y la corticosterona pueden hidroxilarse en posición 18
formando la 18-hidroxi-DOC y la 18-hidroxicorticosterona. La síntesis de la aldosterona
proviene de la corticosterona por 18-hidroxilación seguida de deshidrogenación. En el
organismo, la aldosterona se encuentra como un hemiacetal, ya que el aldehído en C18 no
esta libre, sino que reacciona con el grupo alcohol del C11.
Existe otra isoenzima mitocondrial con actividad de 11 β-hidroxilasa en las células de la
glomerulosa, responsable de la biosíntesis de aldosterona. Como hemos visto, la
nomenclatura de la 11 β-hidroxilasa que sintetiza cortisol a partir del 11-desoxicortisol es
CYP11B1, mientras que la isoenzima que sintetiza aldosterona a partir de 11-

desoxicorticosterona se denomina CYP11B2 o aldosterona sintetasa. Esta enzima además
de actuar como 11 β-hidroxilasa, también actúa como 18-hidroxilasa y luego como 18-
oxidasa. Por ello, se considera que una vez que la 11-desoxicorticosterona se une a la
CYP11B2, este esteroide sustrato queda ligado a la enzima que cataliza las tres
conversiones sin que se liberen los intermediarios (es decir, 11-desoxicorticosterona →
corticosterona → 18-hidroxicorticosterona → aldosterona)
Zona reticular- Andrógenos (grupo androstano C19)
De los andrógenos suprarrenales, el más importante es la dehidroepiandrosterona, que
como compuesto libre y como sulfato, alcanza una tasa de secreción similar a la del
cortisol. Es un andrógeno débil, pero tendría importancia como producto de la suprarrenal
fetal y porque puede originar andrógenos activos. La corteza suprarrenal secreta pequeñas
cantidades de progesterona (0,4-0,8 mg por día) y trazas de estrógenos.
Los andrógenos suprarrenales se forman por ruptura de la cadena lateral de la 17α-
hidroxipregnenoIona y la 17α-hidroxiprogesterona, que dan lugar a la
dehidroepiandrosterona y la androstenediona, respectivamente. Esta última puede
convertirse en testosterona y 11 β-hidroxiandrostenediona. La síntesis de estrógenos por la
suprarrenal normal es muy limitada.
REGULACION DE LA FUNCIÓN SUPRARRENAL
La ACTH (hormona proteica sintetizada en los adenocorticotropos de la hipófisis) regula la
función y el metabolismo de la glándula suprarrenal, sin embargo hay evidencia
experimental de que la suprarrenal posee cierto grado, muy limitado, de autonomía. En la
rata, la hipofisectomía provoca la atrofia de las zonas fascicular y reticular, mientras que la
zona glomerulosa, que es regulada principalmente por la angiotensina II y por cationes, se
ve menos afectada.
Regulación de la secreción de los glucocorticoides: Está gobernada por la hipófisis y el
sistema nervioso central (Figura 17).

Figura 17: Esquema de la regulación del Eje Hipotálamo-hipofisario-adrenal.
En el hipotálamo, en su núcleo paraventricular, se secretan dos factores liberadores: el
“factor liberador de corticotrofina” o CRF, y la vasopresina. Ambos se cosintetizan en la
porción del nucleo paraventricular llamada “parvocelular” porque contiene celulas
pequeñas, en contraposición con la porción magnocelular de células grandes que sintetizan
vasopresina únicamente. La vasopresina del magnocelular no actúa como secretagogo de
ACTH sino como hormona antidiurética liberada por el lóbulo posterior de la hipófisis.
El CRF es un péptido de 41 aminoácidos, que libera ACTH de la hipófisis anterior y
estimula su síntesis. CRF y la vasopresina se estimulan por estrés y factores que actúan
sobre el sistema nervioso central, incluyendo agentes químicos (toxinas bacterianas,
metabolitos), factores nerviosos (traumatismos, reflejos), condiciones ambientales (calor,
frio, sonido, luz), fenómenos psicológicos (miedo, dolor, ansiedad, frustración, fatiga),
hemorragia, anoxia, hipoglucemia, etc. Estos estímulos inducen cambios en
neurotrasmisores (serotonina, acetilcolina, catecolaminas), los que a su vez estimulan a las
neuronas hipotalámicas productoras de factores liberadores. Los estímulos mencionados
activan al eje sistema nervioso-hipófisis-suprarrenal (Figura 17), con el consiguiente
aumento de ACTH, la cual, actuando sobre la corteza, aumenta la secreción de corticoides.
Los corticoides a su vez, poseen un efecto de regulación negativa sobre la secreción de
ACTH. Para ello, actúan sobre el hipotálamo, inhibiendo la síntesis y secreción de CRF y
vasopresina, sobre la hipófisis, inhibiendo la síntesis y secreción de ACTH.

Los niveles de CRF del hipotálamo no son constantes sino que presentan variaciones
diarias, que a su vez producen variaciones paralelas en los niveles de la ACTH plasmática,
los cuales preceden a cambios diurnos en los corticoides circulantes. En sujetos normales,
el cortisol plasmático presenta un ritmo circadiano, observándose un nivel máximo antes de
despertarse a la mañana (5-7 horas); luego los valores decaen hacia la noche,
alcanzándose cifras inferiores a los 5 pg por 100 ml a las 22-24 horas. Recientemente se
ha comprobado que los ascensos o descensos no son continuos, sino que el cortisol
presenta breves pulsos secretorios de 20 a 100 minutos de duración; estos picos están
precedidos por pulsos secretorios de ACTH, por lo cual se ha denominado a este
mecanismo “secreción episódica”.
Regulación de la esteroidogénesis por ACTH
La acción del ACTH sobre la corteza suprarrenal produce rápidos aumentos de la síntesis y
secreción de esteroides: los niveles plasmáticos de corticoides aumentan pocos minutos
luego del estrés o administración de ACTH. El ACTH aumenta el ARN, ADN y la síntesis
proteica y actuando crónicamente produce hipertrofia e hiperplasia suprarrenales. Para
estimular la síntesis de esteroides, el ACTH se une a receptores de membrana de alta
afinidad, activándose la adenilato ciclasa y la producción de AMP cíclico, el cual a su vez
activa a la PKA. La PKA activada por un lado estimula la enzima colesterol ester hidrolasa,
que hidroliza colesterol esterificado a libre, el cual es apto para ingresar a la mitocondria,
por un proceso dirigido por la proteína transportadora de colesterol. La PKA también activa
la síntesis “de novo” de la proteína StAR (steroidogenic acute regulatory protein). ACTH
también estimula la captación del colesterol de las lipoproteínas plasmáticas como fuente
esteroidogénica.
Regulación de la secreción de mineralocorticoides.
La aldosterona, que se produce exclusivamente en la zona glomerulosa, es estimulada por
la ACTH en forma aguda y pasajera. Existen otros mecanismos más importantes que
estimulan la secreción de aldosterona. Por ejemplo, el exceso de sodio en la dieta atrofia a
la zona glomerulosa; en cambio, la disminución de sodio estimula la secreción de
aldosterona, efecto que también puede observarse si se aumenta el potasio. El riñón
cumple un papel fundamental a través de las células yuxtaglomerulares que elaboran
renina. Esta enzima convierte al angiotensinógeno, producido en el hígado, en
angiotensina I, que es un decapéptido, el cual, a su vez, en el pulmón, es trasformado en

angiotensina II un octapéptido activo en la estimulación de la producción de aldosterona
(Figura 18).
Figura 18: Regulación de la síntesis de aldosterona por el sistema Renina-angiotensina.
El incremento del potasio, la ACTH y la angiotensina II estimulan la síntesis de aldosterona,
actuando en la conversión de colesterol a pregnenolona en la zona glomerulosa. Las
alteraciones en el balance del sodio actuarían en parte a través del simpático, activando la
producción de renina (y de esta manera, favoreciendo la síntesis de angiotensina II), pero
también estimulan directamente los últimos pasos de la ruta biosintética de la aldosterona.
Las modificaciones circulatorias que incrementan la secreción de aldosterona (hemorragia,
constricción de la vena cava inferior, cambios vasculares de la hipertensión maligna,
insuficiencia cardiaca, etc.) activarían el sistema renina-angiotensina.
Es de considerable importancia la acción del sistema nervioso simpático como mediador de
ciertos factores en la regulación de la secreción de renina. Los cambios posturales (de la
posición horizontal a la vertical), la depleción de sodio, las hemorragias, etc., que conducen
a una disminución del volumen plasmático, se acompañan de hiperactividad simpática con
liberación de catecolaminas. Estas últimas producen constricción de la arteriola renal
aferente, lo cual lleva a aumentos en la secreción de renina, responsables a su vez de
aumentos en la producción de aldosterona.
TRANSPORTE, CATABOLISMO Y EXCRECIÓN DE ESTEROIDES
El cortisol y la corticosterona circulan en el plasma unidos a una α1-globulina llamada
transcortina; en condiciones normales esta proteína liga el 77% de estas hormonas,
mientras que la albúmina une el 15 %, y el 8 % restante viaja libre. Cuando los niveles de
cortisol plasmático, que normalmente son de 10 a 20 pg/ 100 ml, superan los 30-40 pg/ 100

ml, el exceso es trasportado por la albúmina. En cambio, la aldosterona, por su condición
de hemiacetal, se une poco a la transcortina, ligándose principalmente a la albúmina (45
%), y un 25 % viaja libre.
La transcortina es producida en el hígado, proceso estimulado por los estrógenos. En el
embarazo, los estrógenos aumentan los valores de cortisol ligado a la transcortina; sin
embargo, no hay síntomas de hipercortisolismo puesto que la hormona activa es siempre la
libre y no la ligada a proteínas. Las hormonas sintéticas (dexametasona, triamcinolona) no
son ligadas por las proteínas plasmáticas y deben en parte su gran actividad al hecho de
circular libres.
La vida media del cortisol en el plasma es de 90 minutos. En el hígado el cortisol es
inactivado: se reduce en el anillo A para formar dihidrocortisol (Figura 19), y luego el grupo
cetona en posición 3 es convertido a un hidroxilo; el compuesto resultante es el
tetrahidrocortisol, que se conjuga con ácido glucurónico. El tetrahidrocortisol-glucurónido
circula en el plasma sin unirse a proteínas, y a causa de su solubilidad en agua, se excreta
por orina. El mismo proceso de degradación le ocurre a la aldosterona, la cual se excreta
como tetrahidroaldosterona-glucurónido. La excreción urinaria de tetrahidroglucurónidos es
de 5-8 mg en el hombre y de 3 mg en la mujer; en cambio, la cantidad de cortisol libre
excretada es de solo 30 ug diarios. Una pequeña fracción del cortisol -no mayor del 10 %-
es excretada como 17 cetosteroides. En un adulto con ingestión salina normal, se excretan
de 30 a 60 μg diarios de los derivados conjugados de la aldosterona
(tetrahidroaldosterona).
Figura 19: Mecanismo de inactivación del cortisol

Los 17-cetosteroides (compuestos de 19 carbonos con grupo cetona en posición 17)
urinarios tienen un doble origen; 70 % provienen de la suprarrenal y 30 % del testículo. La
excreción diaria de 17 cetosteroides es de 10 mg en la mujer y de 15 mg en el hombre. Los
17-cetosteroides de origen suprarrenal provienen principalmente de la
dehidroepiandrosterona, que se convierte en el hígado en androsterona y etiocolanolona;
los dos últimos y la dehidroepiandrosterona se excretan conjugados principalmente con
sulfato, pero también con ácido glucurónico. Un 10 % del cortisol metabolizado en el
hígado lo hace a 17-cetosteroides, que tienen la particularidad de estar oxigenados en
posición 11 (11-oxi- 17-cetosteroides), ya que provienen del cortisol, que es un esteroide
11-hidroxilado.
CONDICIONES PATOLÓGICAS
Desde un punto de vista funcional, los trastornos de la corteza adrenal se clasifican en
los que producen un aumento, una disminución o ausencia de la liberación de
esteroides. El hiperadrenalismo o hipercorticalismo se define como un proceso que
provoca el aumento de la liberación de hormonas adrenocorticales. El
hipoadrenalismo o hipocorticalismo es el que produce disminución de la liberación
de hormonas adrenocorticales.
DEFICIENCIA DE CORTISOL El paciente afectado por deficiencia de cortisol carece generalmente de vigor físico y
mental. Por lo común, experimenta anorexia y disminución de peso y a menudo
presenta náuseas, vómitos y dolor abdominal. No excreta agua libre con la rapidez
normal y puede presentar intoxicaciones hídricas con síntomas como confusión.
incoordinación y convulsiones en caso de hiponatremia severa. Presenta capacidad
limitada para acumular y movilizar hidratos carbono y grasas, puede presentar
hipogIucemias durante ayunos prolongados. Es vulnerable a tensiones físicas y
muchas veces no sobrevive a un traumatismo o infección. Se observa deficiencia de
cortisol en la insuficiencia adrenocortical y en la hiperplasia suprarrenal congénita
grave. La insuficiencia suprarrenal es consecuencia de la destrucción o disfunción
de la corteza (insuficiencia primara adrenocortical o enfermedad de Addison) o
secundaria a una deficiencia en la secreción de ACTH por la hipófisis. La insuficiencia
adrenocortical secundaria a un tratamiento con glucocorticoides es la causa más
frecuente.

1- HIPERPLASIA SUPRARRENAL CONGENITA La variedad más común es consecuencia de un defecto congénito de la enzima 21-hidroxilasa. En consecuencia está alterada la síntesis de cortisol, lo cual lleva a un
aumento de la secreción de ACTH. Por influencia del aumento de ACTH se produce
hiperplasia de la corteza suprarrenal, la que secreta exceso de precursores de cortisol
y productos secundarios, algunos de estos productos son androgénicos y por eso en
estos pacientes se presenta virilización en mujeres y desarrollo sexual precoz en
niños.. Dado que la aldosterona también es un 21-hidroxiesteroide, también está
alterada su secreción. En consecuencia los pacientes tienen una hiperplasia suprarrenal congénita virilizante perdedora de sal. Las consecuencias clínicas se
ponen de manifiesto al nacimiento, sobre todo en las niñas debido al desarrollo
irregular de sus genitales. En los niños puede pasar desapercibida al nacimiento y se
manifiesta con el desarrollo sexual precoz.
Otra variedad de hiperplasia suprarrenal congénita es consecuencia de una deficiencia
de la enzima 17-hidroxilasa. La biosíntesis de cortisol, andrógenos y estrógenos
depende de esta enzima. En consecuencia se produce deficiencia de cortisol y falta de
desarrollo sexual. En respuesta a la deficiencia de cortisol se produce un aumento de
la secreción de ACTH que, en esta enfermedad, lleva a la hipersecreción del
mineralocorticoide 11-desoxicorticosterona, lo cual conduce a hipertensión. Ambos
tipos de hiperplasia suprarrenal congénita responden favorablemente al tratamiento
con cortisol, que reduce la secreción de ACTH, y en consecuencia limita la
hiperproducción de metabolitos tales como andrógenos o 11desoxicorticosterona. En
la figura 20 se observan las consecuencias de deficiencia de 21-hidroxilasa (A) Y
deficiencia de 17 α-hidroxilasa (B).

Figura 20: Deficiencia de 21 y 17-hidroxilasas
2 INSUFICIENCIA ADRENOCORTICAL PRIMARIA O ENFERMEDAD DE ADDISON La principal causa es de carácter autoinmune (80% de los casos) en la que la
infiltración linfocítica de la corteza suprarrenal es la característica histológica
sobresaliente. Las suprarrenales son pequeñas y atróficas la cápsula está engrosada.
La tuberculosis de las glándulas suprarrenales es responsable de la mayor parte de
los otros casos. La tuberculosis suprarrenal se debe a una infección hematógena que
usualmente ocurre como complicación de la infección tuberculosa sistémica (pulmón,
aparato digestivo o riñón) produciendo destrucción gradual de la corteza y médula. En
casos como septicemia y hemorragias la destrucción se produce con rapidez llevando
a cuadros de insuficiencia aguda. La enfermedad de Addison usualmente se
diagnostica entre la tercera a quinta década de vida. Es más frecuente en la mujer que
en el varón, especialmente la de tipo autoinmune. En pacientes con SIDA, el examen
postmorten revela que la suprarrenal es la glándula endócrina más frecuentemente
afectada. Esto se puede relacionar con la presencia de citomegalovirus, lesiones
metastásicas (sarcoma de Kaposi), infecciones tuberculosas o micosis. Sin embargo
es poco frecuente la insuficiencia clínica. El ketoconazol, agente antimicótico, usado
en pacientes con SIDA, actúa sobre las enzimas que utilizan el citocromo P450 en
varios órganos disminuyendo la estereidogénesis.

Fisiopatología y características clínicas Con la destrucción gradual adrenocortical, la secreción basal de esteroides es normal,
pero la reserva adrenal está disminuída, es decir la secreción no aumenta en
respuesta al esfuerzo. Es así que se puede desarrollar una crisis suprarrenal aguda
por esfuerzo de cirugía, traumatismo o infección. Con una pérdida posterior de tejido
cortical aparecen las manifestaciones de insuficiencia adrenocortical crónica.
Signos y síntomas: La hiperpigmentación de la piel y mucosa es una de las
manifestaciones más tempranas de la enfermedad de Addison. Se incrementa en las
áreas expuestas a la luz y se acentúa en las áreas de presión como los nudillos, codos
y rodillas. Esto se debe a la elevación de ACTH, además de varios productos
secundarios estimulantes de los melanocitos. La deficiencia de cortisol provoca
debilidad, fatiga, anorexia, pérdida de peso, características que están siempre
presentes en este trastorno. En la mayoría de los pacientes se producen desórdenes
gastrointestinales, en especial náuseas y vómitos. La hipoglucemia es poco frecuente
en adultos pero puede estar provocada por ayuno, fiebre o infección. La deficiencia de
mineralocorticoides produce pérdida de sodio renal y retención de potasio y puede
llevar a una deshidratación grave, hipotensión, hiponatremia, hipercalemia y acidosis.
La amenorrea es frecuente y puede deberse a la pérdida de peso (o a una
insuficiencia ovárica primaria), la pérdida de vello axilar y púbico puede presentarse
como resultado de la disminución de andrógenos suprarrenales.
Tratamiento: la finalidad del tratamiento es producir concentraciones de
glucocorticoides y mineralocorticoides equivalentes a los normales en circunstancias
similares. El tratamiento de la crisis Addisoniana deberá administrarse tan pronto como
se sospeche el diagnóstico. El tratamiento incluye administración de glucocorticoides,
corrección de la deshidratación, hipovolemia y anomalías de electrolitos. Los pacientes
requieren una terapéutica de mantenimiento por toda la vida, habitualmente con
glucocorticoides y mineralocorticoides.
3 HIPERCORTISOLISMO CRÓNICO - SINDROME DE CUSHING Los excesos transitorios de glucocorticoides como los que podrían producirse como
parte de la respuesta fisiológica al stress o como consecuencia de la administración
durante breves períodos de cortisol no conducen a anomalías significativas. Sin
embargo, la elevación persistente, inapropiada y mantenida de los niveles circulantes
de glucocorticoides lleva a una constelación de signos y síntomas que se denominan

Síndrome de Cushing.
La causa más frecuente del síndrome de Cushing es la yatrogénica, por administración
de dosis suprafisiológicas de corticosteroides o ACTH en el tratamiento de
enfermedades sistémicas. El síndrome de Cushing espontáneo o endógeno se debe a
la secreción excesiva y mantenida de cortisol por las suprarrenales. Este tipo de
hipercortisolismo puede clasificarse en varios grupos (Figura 21).
1.- Dependientes de ACTH. Se caracterizan por hipersecreción çrónica de ACTH, que
da como resultado hiperplasia de las zonas reticular y fasciculada de las glándulas
suprarrenales y por lo tanto un aumento de la secreción de cortisol, andrógenos y
desoxicorticosterona. Se puede deber a:
a) adenoma hipofisario - enfermedad de Cushing. Es el tipo más frecuente de
síndrome de Cushing,
es la responsable de casi el 70% de los casos. Es mucho más frecuente en mujeres
que en varones y la edad del diagnóstico es generalmente entre los 20 y 40 años. En
la enfermedad de Cushing, la hipersecreción de ACTH es al azar y provoca
hipersecreción de cortisol en ausencia de un ritmo circadiano normal. No se produce
retroalimentación inhibidora de ACTH por concentraciones fisiológicas de
glucocorticoides, así persiste la hipersecreción de ACTH a pesar de la elevación del
cortisol y da como resultado exceso crónico de glucocorticoides.

Figura 21 : Etiopatología del sindrome de Cushing

b) síndrome de ACTH ectópico. Se presenta cuando los tumores no hipofisarios
sintetizan y secretan ACTH biológicamente activa. Se ha demostrado, además, la
producción de CRF en tumores ectópicos. Pueden dar origen a este síndrome
carcinomas de células pequeñas y tumores carcinoides del pulmón, timo, intestino,
páncreas, ovario e islotes pancreáticos. El síndrome de ACTH ectópica es más común
en hombres que en mujeres, y la mayor incidencia se da entre los 40 y 60 años. La
hipersecreción de ACTH y cortisol es mayor en pacientes con síndrome de ACTH
ectópico que en aquellos con enfermedad de Cushing, sin embargo las características
típicas del síndrome de Cushing están ausentes, quizá debido al inicio rápido del
hipercortisolismo y a que la semiología corresponde más a la expresión del tumor que
lo origina.
2.- Independientes de ACTH - hipercortisolismo suprarrenal primario. La glándula
suprarrenal produce hipercortisolismo de forma autónoma por un adenoma o
carcinoma. En estos casos el exceso de cortisol suprime la secreción hipofisaria de
ACTH y anula la actividad de la glándula contralateral no afectada por el tumor.
3.- De etiología no completamente determinada. a) hiperpiasia adrenal
micronodular bilateral. En esta forma de síndrome de Cushing se cree que los
pacientes tienen células adrenocorticales hipersensibles a ACTH, lo que hace que
niveles normales de ACTH puedan causar hipercortisolismo y transformación nodular
de la glándula la que llega a adquirir un comportamiento autónomo.
b) asociado a depresión endógena. Se presenta en algunos pacientes con niveles de
ACTH elevados, con amplitud de su ritmicidad e hipercortisolismo consecuente.
c) asociado a alcoholismo. En estos casos el cortisol está elevado y no mantiene el
ritmo circadiano y la ACTH está disminuída. El proceso revierte durante la abstinencia
alcohólica.

Manifestaciones clínicas del síndrome de Cushing
TEJIDOS NO TRADICIONALES DE SÍNTESIS DE ESTEROIDES
NEUROESTEROIDES
El sistema nervioso central y periférico se comporta como una glándula endócrina, ya
que posee la propiedad de sintetizar neuropéptidos y hormonas proteicas, tales como
ACTH, prolactina, insulina, glucagon, etc. Hace ya varios años, sin embargo, se
demostró que las células del sistema nervioso también poseían las enzimas
necesarias para sintetizar esteroides. Estos últimos fueron denominados por el Dr.
Baulieu (Hospital de Bicetre, Paris, Francia) como “neuroesteroides” .
Clásicamente, se sabía que las hormonas esteroides producidas por las glándulas
endocrinas tales como ovario, testículo, suprarrenal, y placenta, podían cruzar la
barrera hematoencefálica y actuar sobre las neuronas y células de la glia, las que
poseen receptores específicos para su reconocimiento. Estos esteroides, producidos
periféricamente y que actúan en el sistema nervioso, se denominan actualmente
“esteroides neuroactivos”, para distinguirlos de los neuroesteroides que se producen
en el sistema nervioso sin intervención periférica.

VITAMINA D
BIOSINTESIS DE VITAMINA D
Los animales requieren vitamina D para el metabolismo del calcio y del fósforo, necesarios
para el desarrollo normal de huesos y dientes. La vitamina D posee una interesante
historia, desde el tiempo que fue reconocida por McCollum como el componente del aceite
de hígado de bacalao que curaba el raquitismo. De Luca encontró 50 años mas tarde que
la forma activa de la vitamina requería el riñón para su formación. Los precursores de la
vitamina D están presentes en tejidos animales y vegetales; en los primeros el precursor
es el 7-dehidrocolesterol, mientras que en vegetales lo es el ergosterol.
Debido a varias particularidades, podríamos decir que más que una vitamina es una
hormona, ya que:
(1) No necesita ingerirse con la dieta por cuanto se puede sintetizar en el organismo a
partir del colesterol.
(2) Sus receptores pertenecen a la Superamilia de Receptores Nucleares, junto a los
receptores de glucocorticoides, mineralocorticoides, andrógenos, estrógenos, ácido
retinoico y hormonas tiroideas.
(3) Se sintetiza en tejidos (piel, hígado riñón), y luego de viajar por vía sanguínea alcanza
sus receptores intracelulares en los órganos blanco tales como intestino y hueso,
procesos habituales para las hormonas.
Síntesis de Vitamina D
En los animales se forma a partir del colesterol en la capa epidérmica de la piel, el cual
por vía enzimática se convierte en 7-deshidrocolesterol. Por medio de la radiación solar
ultravioleta, se rompe el anillo B del ciclopentanoperhidrofenantreno, formándose la
provitamina llamada colecalciferol o vitamina D3.
El colecalciferol llega a la sangre, y allí se une y es transportado por una proteína
plasmática conocida como DBP (vitamin D plasma binding protein) hacia el hígado, donde
continúa la biosíntesis de su metabolito activo. En el hígado existe una enzima
dependiente del citocromo P450 llamada 25-hidroxilasa, la que en presencia de NADPH
transforma al colecalciferol en 25-hidroxicolecalciferol (25OHD3). Este metabolito es 5
veces más potente que la vitamina D3 (Figura 22).
En cambio, normalmente el 25OHD3 se libera por el hígado, y transportado por la DBP
viaja al riñón, donde por la acción de una 1 hidroxilasa se forma el metabolito activo

llamado 1,25-dihidroxicolecalciferol (1,25OH2D3). Este compuesto, por poseer 3
hidroxilos, se llama también calcitriol, y es 10 veces más potente que la vitamina D3.
La síntesis de calcitriol se regula por los niveles de la hormona paratiroidea (PTH). La PTH
se libera por las glándulas paratiroideas en respuesta a la disminución sanguínea de
calcio y fósforo. De esta manera, la baja en la ingesta de calcio se refleja en una
disminución en la calcemia, la cual induce la liberación de PTH y el aumento subsiguiente
de la síntesis renal de calcitriol, ya que la PTH es un regulador positivo de la enzima 1
hidroxilasa renal. En el riñón existe también una 24-hidroxilasa que convierte al
colecalciferol en 24-25 dihidroxicolecalciferol o 1, 24, 25 trihidroxicolecalciferol
inactivándolo (Figura 22).
Figura 22: Síntesis de Vitamina D. El 1, 25dihidroxicolecalciferol es la forma activa
Funciones: El calcitriol promueve la absorción activa que requiere de energía, del
calcio intestinal ingerido con la dieta, estimulando la síntesis de una proteína que actúa
como transportador del calcio en las células de ribete en cepillo de la mucosa
intestinal. También aumenta la absorción intestinal de fósforo. Este mecanismo se
debe a la unión del calcitriol a su receptor, que actúa en el núcleo induciendo la
transcripción del gen del transportador intestinal de calcio (Figura 23).

Figura 23: El colecalciferol se metaboliza a sus formas activas 25OHD3 y 1,25OH2D3, las que actuando en intestino aumentan la reabsorción de calcio y fósforo, y también en el hueso para aumentar la resorción de calcio y fósforo. El resultado final es el aumento de calcemia y fosfatemia. La paratiroides monitorea el calcio sanguíneo, ya que su disminución provoca la descarga de PTH, la que a su vez induce la síntesis de calcitriol en el riñón
Necesidades y deficiencia de vitamina D : El adulto normal obtiene suficiente vitamina
D por exposición de su piel a la energía solar además de la ingestión de una pequeña
cantidad con los alimentos en forma de colecalciferol. Son fuentes de vitamina D el
aceite del hígado de pescado, y en menor escala manteca, huevos, crema. La leche
se adiciona de vitamina D.
La deficiencia de vitamina D se manifiesta como raquitismo en los niños y
osteomalacia en los adultos. Los niños raquíticos muestran huesos deformados, corta
estatura y rosario raquítico en el tórax, etc. Los adultos osteomalácicos presentan
deformidades de los huesos de los miembros, espina dorsal, tórax y pelvis; los dolores
óseos y la debilidad general son frecuentes.