farmacodermia - smiba - sociedad de medicina … · cualquier efecto no deseado en la estructura de...
TRANSCRIPT
FARMACODERMIA

Introdución
� Las reacciones adversas son eventos no deseados e involuntarios que ocurren con dosis habituales del fármaco utilizadas para prevención, diagnóstico o tratamiento de una enfermedad, o para modificar cualquier función biológica.
� Se considera una reacción adversa cutánea causada por una droga, a cualquier efecto no deseado en la estructura de la piel, anexos o mucosas, e incluye todos los eventos adversos independientemente del mecanismo o la vía de administración del fármaco.
� La piel es el blanco más frecuente de las reacciones medicamentosas, es necesario que el personal de salud, y especialmente los médicos dermatólogos, conozcan las formas clínicas con las que se presentan, así como los fármacos más frecuentemente involucrados, su incidencia y prevalencia en la población general.
� La fisiopatogenia de las reacciones adversas a drogas es compleja.

� Se clasifican en dos grupos: tipo A y tipo B.
� Las primeras se consideran predecibles, dosis dependiente y explicables por la acción farmacológica de la droga.
� las segundas son impredecibles, menos frecuentes y dosis independiente. Estas últimas, a su vez, pueden ser reacciones idiosincráticas (debidas a alguna predisposición individual) o reacciones de hipersensibilidad.
� Las farmacodermias pertenecen a las reacciones de tipo B.

� Se mencionan diversos factores de riesgo para padecerlas, tales como sexo femenino, edad avanzada, polimedicación, infección por VIH y factores genéticos de predisposición, entre otros.
� Se caracterizan por poseer diversas formas clínicas. Pueden observarse desde erupciones autolimitadas a cuadros de necrosis extensa de la piel con riesgo de muerte; sin embargo, la incidencia de formas severas es baja.
� Los exantemas morbiliformes y la erupción fija por drogas se mencionan como las formas más frecuentes.

ERITEMA MULTIFORME

Historia
� Ferdinand Von Hebra en 1860 describió al eritema multiforme menor (EM menor) bajo la denominación de "eritema exudativo multiforme".
� Bazin en 1862 agregó la sintomatología sistémica y el compromiso mucoso, constituyéndose así el eritema multiforme mayor (EM mayor).
� Feissinger y Rendu en 1917 y Stevens y Johnson en 1922 reconocieron un síndrome con predominio óculo-mucoso, que hoy lleva el nombre de estos dos últimos autores (SSJ).
� Thomas en 1950 reunió a estas enfermedades en dos grupos: el EM menor por un lado y el EM mayor (con el SSJ como variedad) por el otro.
� 1956, se le agregó la "necrolisis epidérmica tóxica" (NET), descrita por Lyell.
� A partir de 1993:
� Reordenaron dermatosis preexistentes
� Creación de nuevas entidades como consecuencia de formas de superposición y pasajes entre las 4 variedades clásicas
� Denominación de diferentes términos de acuerdo a la nueva ubicación nosológica
� Progreso terapéuticos

Definición
� Sme. que abarca un grupo heterogéneo de enfermedades inflamatorias agudas de piel y mucosas, con o sin compromiso sistémico.
� Todas las razas
� Ambos sexos
� Todas las edades
� Multifactorial (infecciosas – farmacológicas)
� Mecanismo inmune
� Predisposición genética (HLA: B62 - B35 – A33 – DR53 – Dq 31)

Definición
� Este síndrome involucra a cuatro formas clínicas clásicas:
� EM menor
� EM mayor
� Síndrome de Stevens-Johnson
� Necrólisis epidérmica tóxica,
� En los últimos años se le han agregado nuevas variedades: inclasificado y de superposición.
� Es autorresolutivo, con tendencia a recurrir y de acuerdo con las características de las recaídas, se lo puede llegar a clasificar en:
� EM recurrente o EM persistente.
� El pronóstico es variable según la forma clínica de presentación.

Espectro del EM
� Existe un espectro de EM que desde el extremo mas leve (benigno) al mas severo (en ocasiones mortal) se muestra a continuación:
VHS
Fármacos
EBV
• Desencadenado por infecciones (HSV)
• Lesiones en escarapela• Tendencia a recidivar• Buen pronóstico
• Desencadenado por fármacos• Lesiones eritematopurpúricas,
ampollares extensas• Tendencia de epidermis a
desprenderse• Forma cutáneo mucosa grave de
mal pronóstico

Clasificación

EM(m/M)-Definición
� Sme. mucocutáneo agudo, autolimitado, habitualmente leve y, a menudo, recurrente
� Por lo general, la enfermedad está relacionada con una infección aguda, la mayoría de las veces una infección recurrente de HSV
� se define clínicamente por placas en forma de diana que predominan en cara y los miembros

Subtipos de EM
� EMm: lesiones cutáneas sin compromiso de mucosas
� EMM: lesiones cutáneas con compromiso de mucosas
� EM mucoso: (ectodermosis pluriorificial) lesiones mucosas sin compromiso cutáteneo.

Clasificación desde el punto de vista evolutivo
� EM recurrente:
Brotes de 14 días de evolución que en general se presentan clínicamente como EMM o EMm. Adultos jóvenes ambos sexos por igual. Promedio 6 brotes anuales y evolución crónica (aprox. 10 años). 90% HSV.
� EM persistente o continua:
raro. Brotes similares a EM inclasificado o SSJ + neutropenia, hipocomplementemia y complejos inmunes circulantes.
EBV causa + frecuente y siempre investigar neoplasias asociadas (gástricas y renales)

Epidemiología
� Afecta a ptes. de todas las edades pero, en mayor medida, a adolescentes y adultos jóvenes
� Ligero predominio masculino 3:2
� Tasa de recurrencia mayor a 30%
� HIV y trastornos autoinmunitarios no aumentan el riesgo de EM a diferencia del SSJ
� Gen DQB1 66% de ptes con EM y 30% en controles

Etiología
� INFECCIONES:
HSV: 50% EM presentan clínica de herpes.
10-40% EM relación con HSV sin manifestación clínica
La erupción de EM comienza, en promedio a los 7 días (3-14) después de una recurrencia herpética.
no toda recurrencia sintomática es seguida de EM, y las asintomáticas pueden provocar EM

Etiología
� M. pneumoniae:
� 2° causa, puede ser la primera en ptes. pediátricos
La presentación clínica suele ser menos típica y más grave
DX: hisopado de fauces
serologías
� Otras infecciones: varicela zoster, parbovirus B19, HBV, HCV, EB
� Fármacos : causa rara

Manifestaciones clínicas
� Antecedentes:
Episodio o episodios previos en más del 30%
Repasar eventos de 3 semanas previas en busca de agentes precipitantes con especial interés en VHS

Lesiones cutáneas
� Erupción aguda (72 Hs)
� Mayormente simétricas
� Distribución acral en la superficie extensora de los miembros (manos, pies, codos y rodillas), cara y cuello
� Menos frecuentes muslos, nalgas y tronco
� Localización periférica con propagación centrípeta
� Generalmente asintomáticas

� Lesión típica: Comienza como una pápula o placa eritematosa, de limites netos, redondeada, que se torna violácea y oscura que a menudo se trasforma en una vesícula o ampolla tensa
� la zona siguiente es más pálida e infiltrada
� tercer componente es un halo eritematoso, resultando en la típica lesión en diana o blanco de tiro
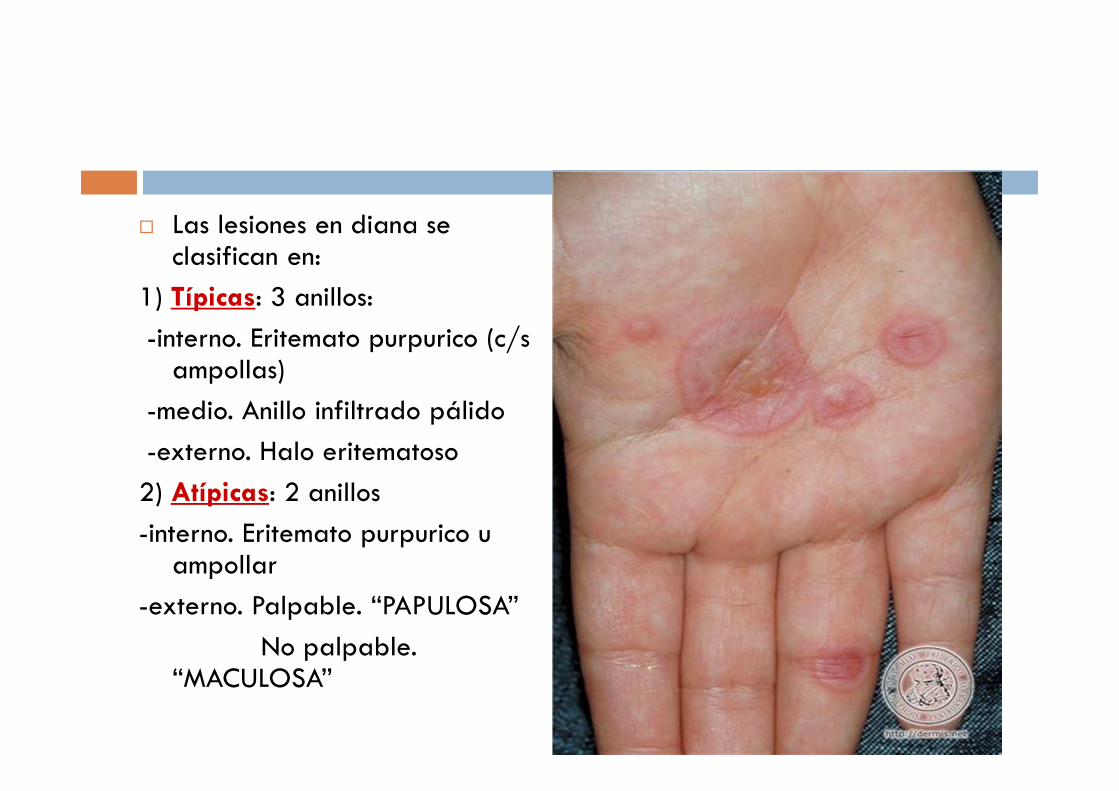
� Las lesiones en diana se clasifican en:
1) Típicas: 3 anillos:
-interno. Eritemato purpurico (c/s ampollas)
-medio. Anillo infiltrado pálido
-externo. Halo eritematoso
2) Atípicas: 2 anillos
-interno. Eritemato purpurico u ampollar
-externo. Palpable. “PAPULOSA”
No palpable. “MACULOSA”

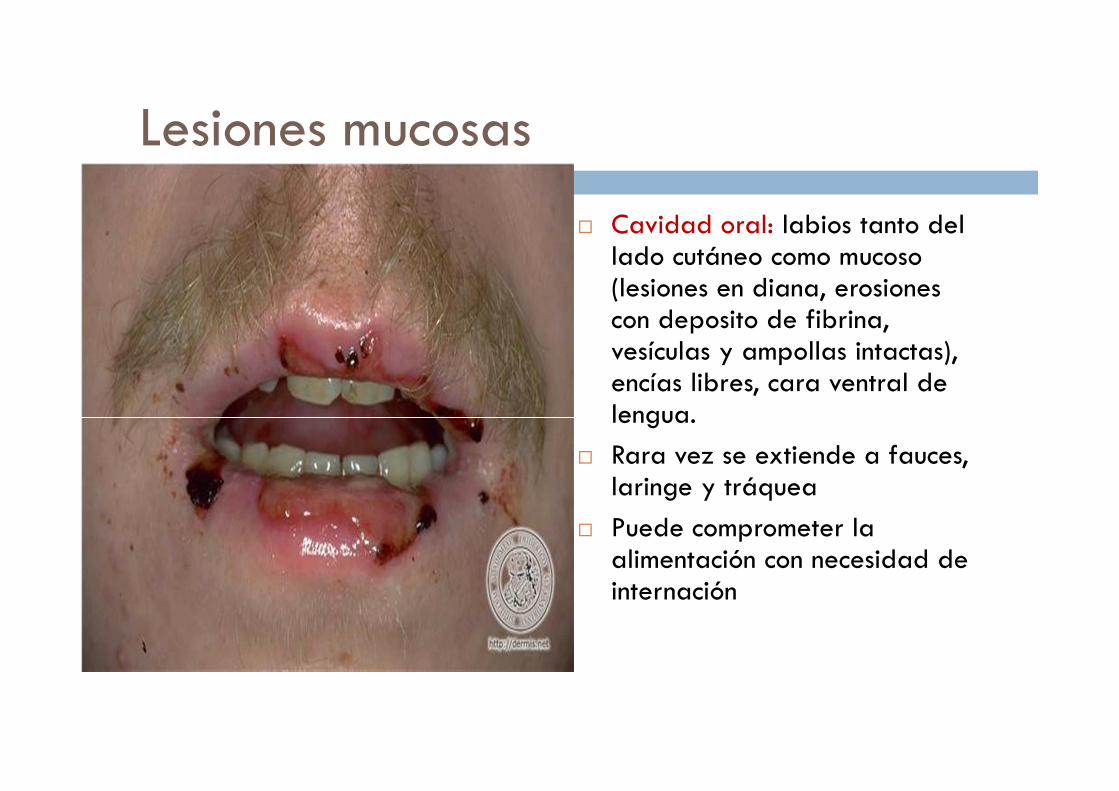
Lesiones mucosas
� Cavidad oral: labios tanto del lado cutáneo como mucoso (lesiones en diana, erosiones con deposito de fibrina, vesículas y ampollas intactas), encías libres, cara ventral de lengua.
� Rara vez se extiende a fauces, laringe y tráquea
� Puede comprometer la alimentación con necesidad de internación


� Compromiso ocular:
Comienza con dolor y conjuntivitis bilateral con posible aparición de vesículas y erosiones
� También puede haber inflamación y erosión de mucosa nasal uretral y anal

EMm EMM
tipo Escarapela típica, limites netos
(raro ampolla)
Escarapela típica (ampollas) y atípicas de tipo
papuloso
Distribución Escasas, simétricas y localizadas en
extremidades (+ distal)
NO mucosas (o solo oral)
Más numerosas y localizadas en extremidades,
cara y tronco
SI mucosas (moderado)
Extensión Menos del 10% de superficie corporal
Comp. Gral. NO SI (leve a importante)
etiología VHS I-II, Mycoplasma Pneumoniae, otras
Evolución Autorresolutiva – Recurrente (+)
2 - 4 semanas 4 - 6
semanas
Tratamiento Aciclovir en el recurrente
800 mg/día por tiempo prolongado
Corticoides precoz 0.5 mg/kg 5-7 días
Asociar Aciclovir o azitromicina

EXAMENES COMPLEMENTARIOS
� LABORATORIO: siempre útil serologia para herpes. M pneumoneae: confirmación con Ig M + o seroconversion con Ig G.
� ANATOMIA PATOLOGICA:
-temprano: infiltrado linfocitos en unión dermoepidérmica, linfocitos unidos a queratinocitos necróticos, espongiosis, degeneración vacuolar de cel. basales y hendiduras en unión dermoep. Y surepidermica. Dermis con infiltrado denso mononuclear. IF – o inespecifica
-Avanzado: ampollas subepidermica. Es Rara la necrosis de todo el espesor epidérmico
Diferencia AP con NET y SSj: El infiltrado inflamatorio en estas es moderado a nulo y la necrosis epidérmica es mucho mas acentuada.

Infiltrado dermico superficial de linfocitos que oscurece la delimitacion del estrato basal de la epidermis

Detalle del estrato basal de la epidermis donde se observa exocitosis de linfocitos, daño valuolar y disposición en empalizada de queratinocitos

DIAGNOSTICOS DIFERENCIALES
� Urticaria crónica
� Erupción medicamentosa
� Dermatitis herpetiforme
� Penfigoide ampollar
� Penfigo paraneoplasico
� Sífilis secundaria
� Exantemas virales
� Micosis fungoide

DIAGNOSTICOS DIREFENCIALES
mucoso clinica A.P laborat. evolucion
URTICARIA NO Pápula dérmica edematosa pruriginosa
Edema + inf. inflamatorio
------ Mas agudo que EM (1 a 24 hs.)
Erupción medicamentosa maculopapular
Rara (labios)
Lesiones polimorfas generalizadas tipo diana macula papulao placa
inespecifica Subaguda o cronica
PENFIGOIDE RARO Placas eritemat. circinadas
Ampolla subepidermic
Ac presentes cronica
PENFIGO PARANEOPLASICO
SI, precoz y grave
Lesion tipo EM mas papulas liquenoides. Nikolsky +
AcantolisisIFD +
Ac presentes cronica
SSJ constante Pequeñas ampollas generalizadasDianas atípicasSint. generales
Dermatitis de la interfaseNecrosis epidérmica
aguda

Tratamiento
� enjuagues bucales con anestésicos, dieta blanda, evitar comidas picantes y aumentar la ingesta de líquidos.
� También pueden administrarse corticoides tópicos y antibioterapia para prevenir infecciones
� En pacientes adultos con historia de infección por VHS, una terapia antivírica puede producir beneficios, sobre todo previniendo recurrencias cuando se aplica de forma profiláctica.
� Las dosis varían algo según distintos autores, 200-800 mg de aciclovir/día para ir disminuyendo progresivamente hasta encontrar la dosis menor capaz de prevenir la recidiva durante periodos prolongados de cinco o seis meses
� El tratamiento del episodio agudo con aciclovir sólo está indicado cuando se instaura muy precozmente.

� Existe controversia respecto al uso de corticoides sistémicos para el tratamiento del EM, sobre todo en cuadros clínicos moderados y los EM recurrentes debidos a herpes simple.
� Las dosis que proponen los distintos autores también varían. 30-50 mg al día de prednisona o metilprednisolona durante varios días disminuyendo la dosis paulatinamente.
� Otro esquema 0,5-1 mg/Kg/día durante una semana y luego disminuyen progresivamente.

Tratamiento
� EMm
� No se indica tratamiento por ser autorresolutivo
� En la variante recurrente, para prevenir los rebrotes de herpes simple se usa como dosis de ataque: aciclovir 200 mg 5 Veces /Día x 5 días
� De manera profiláctica: aciclovir 400 mg 1 o 2 veces por día por un lapso de 6 meses a 2 años

Tratamiento
� EMM
� Prednisona o metilprednisolona VO 0,5mg/kg/día, durante 5 – 7 días
� enjuagues bucales con anestésicos, dieta blanda, evitar comidas picantes y aumentar la ingesta de líquidos.

� Los casos sintomáticos de infecciones por M. pneumoniae deben tratarse con antibióticos (macrólidos o quinolonas)
� No hay evidencia que indique si esto mejora la evolución del EM asociado.
� Por lo tanto cuando la serología sugiera infección asintomática, el tratamiento no es obligatorio.


SSJ (1920) NET (Lyell 1956)
tipo Escarapela atípica
Máculas eritemato-purpúricas-ampollares
Máculas eritemato-purpúricas y grandes napas de piel denudada, con o sin
escarapelas atípicas
Distribución Amplia distribución en tronco
Gran compromiso mucoso +++ (+ ocular)
Más generalizado
(cara, CC y extremidades suelen estar conservadas)
Compromiso mucoso ++
Extensión < 10% > 30%
Compr. Gral. Si. Importante Si. serio
Órganos internos Renal – Respiratorio – Hepático – Cardíaco – Gástrico - Articular
HP Necrosis masiva de queratinositos. (evaluar infiltrado inflamatorio)
Etiología Fármacos – otras
Pronóstico Severo (fatal 5 – 10%) Serio ( fatal 30%)
Asociación Colagenopatías (5%), tumores (2 -10%) y HIV (5 – 8%) (no cambia el pronóstico)
tratamiento Cuidados generales
Corticoides a altas dosis (1 mg/kg) hasta la resolución
Cuidados generales, tratarlo como un gran quemado
Inmuoglobulinas – plasmaferesis – anti TNF α


� Mujeres : Varones 1,5 : 1
� Incidencia:
� SSJ: 1-6/1,000,000 población/año
� NET: 0,4-1,2 /1,000,000 población/año
� Más frecuente en pacientes HIV+, trasplantados, colagenopatías
� Asociado a HLA B12
� Casi todos desencadenados por fármacos
� El fármaco sospechoso introducido 7 a 21 días previos� No es dosis dependiente


Patogenia
� Enzimas metabolizantes oxidativas (Citocromo P450 y otras) generan metabolitos de las drogas (fase I), los que son desintoxicados por enzimas como la epoxido-hidrolasa o glutation S transferasa (fase II).
� La reacción adversa por fármacos se desencadenaría por falla en estos pasos y el defecto seria constitucional y hereditario.


� NET
� La apoptosis de queratinocitos es mediada por la vía Fas-FasL.
� Cuando se estimulan los receptores de la superficie de los queratinocitos, por sobreexpresión de FasL, se inicia una cascada que transforma
proenzimas (procaspasa) en enzimas activas (caspasa), liberando la nucleasa de su inhibidor, con la consiguiente fragmentación del ADN nuclear.
� NET hay niveles aumentados (3-4 veces) de FasL

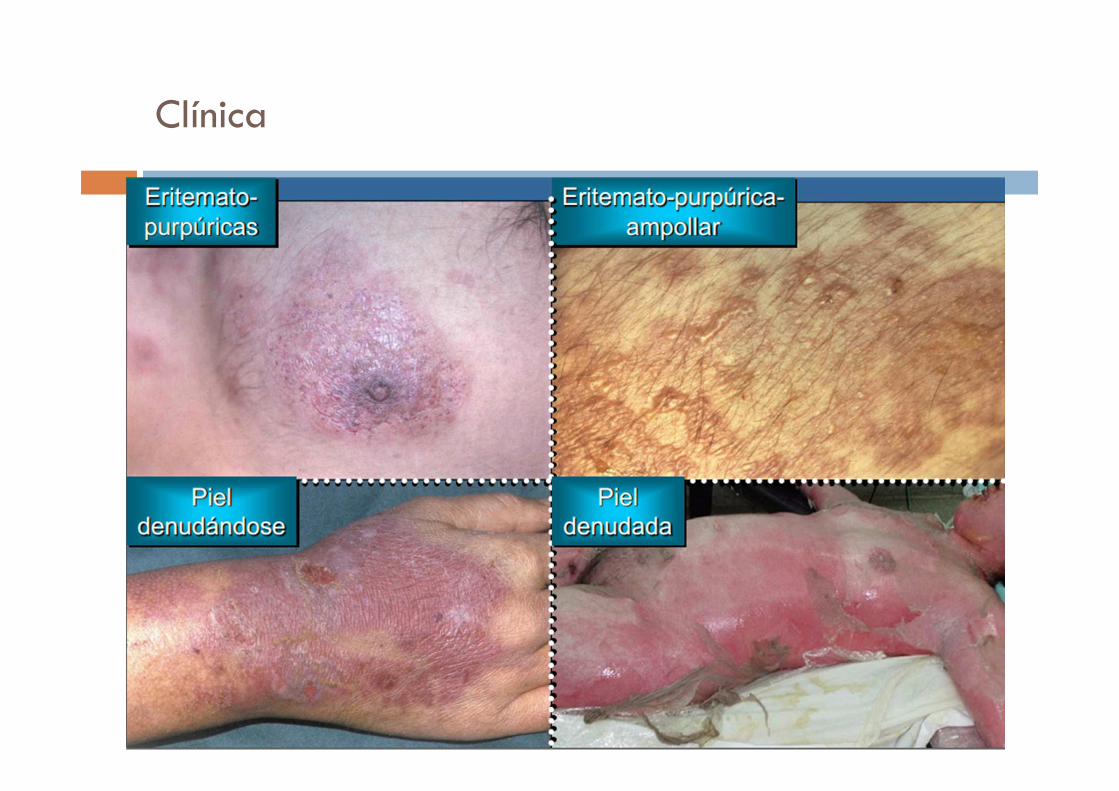
Clínica

� Inicio agudo : fiebre, odinofagia, ardor ocular 1 a 3 días previos, rápidamente progresivo.
� Lesiones cutáneas:
� Localizadas inicialmente en cara y tronco superior
� Progresa en dirección céfalo caudal
� Formas de presentación :
1. Lesiones eritemato purpuricas
2. Ampollas flácidas que se destechan dejando la dermis descubierta
3. Eritema fugaz difuso que se transforma en grandes colgajos


Piel remanente con aspecto de ropa mojadaEpitelio respiratorio: erosiones en tráquea y bronquios en 25%

� Evolución:
� Extensión entre 2 a 14 días
� Reepitelización no menor a 1 mes:
� En napas, deja un patrón discrómico en damero
� Hipo pigmentación residual
� La afectación mucosa suele ser mas severa en la SSJ
� Pronostico:
� Mortalidad entre 30 y 50% por sepsis (S. aureus y Pseudomona
aeruginosa)

Tratamiento
� Suspender el fármaco sospechado
� Internación en UTI
� Venoclísis (medio interno). Incluir coloides
� Laboratorio (control neutropenia)
� Alimentación especial
� Medidas antisépticas (ATB sistémicos si es necesario)
� Desbridamiento quirúrgico. Cubrir con ropas no adhesivas. (membranas de siliconas semipermeables-flexibles)
� Biopsia si es necesario (?)
� Analgésicos. Anticoagulantes. Sedantes.

Corticoides

NET Tratamiento
� Inmunoglobulinas Humanas IV
� Contienen Ac. Anti FAS
� Inhiben la apoptosis mediada por FAS
� Administración precoz. 1gr/kg/día – 3 a 5 días consecutivos
� Asociar N-acetilcistina 300 mg/kg/día IV continua para rescatar los niveles de glutation y poder desintoxicar los metabolitos intermedios.

NET Tratamiento
� Ig – IV
Efectos Adversos� factor de riesgo trombótico (dosis dependiente por aumento de
viscosidad sanguínea y la agregación plaquetaria)
� Falla renal (aguda, por injuria osmótica de túbulos proximales, dando necrosis tubular aguda y nefrosis osmótica)
� Meningitis aséptica
� Trastornos cardiovasculares, hematológicos, dermatológicos y otros.

NET Tratamiento
� Plasmaferesis
� Remueve el fármaco o a los anticuerpos
� Se indicaría cuando las lesiones progresan rápidamente durante el primer día de internación
� El número de tratamientos diarios depende de la progresión: de 1 a 4.

NET Tratamiento
� Anti TNF α
� Infliximab� Etanercept� Pentoxifilina� NO talidomida
� Inmunosupresores (controvertido)
� Ciclofosfamida� Mofetil micofenolato� ciclosporina

Pronóstico - SCORTEN
SCORTEN Tasa de Mortalidad %
0-1 3,2
2 12,1
3 35,8
4 58,3
>5 90

Semimucosa labial con costras serohemáticas

Lesiones eritemato-violaceas, confluentes. Colgajos

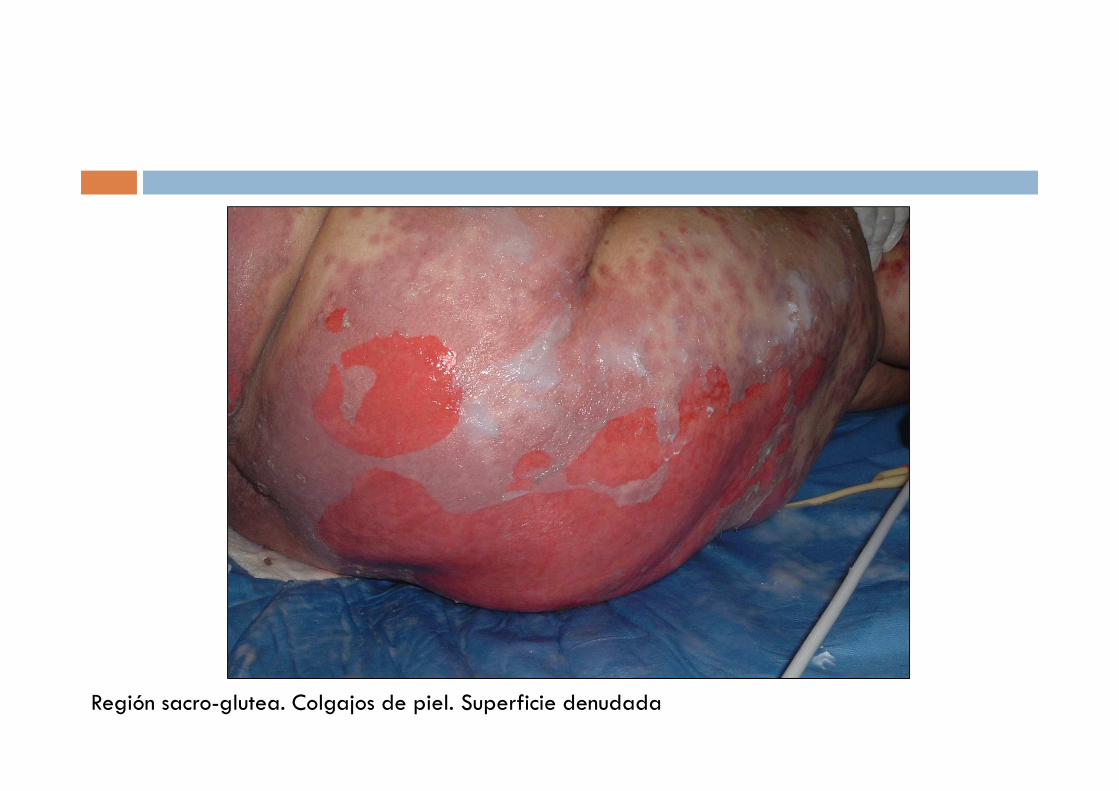
Región sacro-glutea. Colgajos de piel. Superficie denudada

� Se indica Gamma globulina hiperinmune 1gr/kg/día por 4 días (EV)
� Curaciones con Sol. fisiológica, sulfadiacina de plata y lidocaína en crema y cobertura con film plástico.
� Día 15

� Día 30

DRESS

� Sme. DRESS:
� Drug reaction/rush with eosinophilia and systemic symptoms.
� Es una rara reaccion inducida por drogas.
� Se caracteriza por fiebre, rash, linfadenopatía, elevación de enzimas hepáticas y leucocitosis con eosinofilia.
� Incidencia: 1:1.000 a 1:10.000
� Otros nombres: � sme. de hipersensibilidad a la fenitoina� Sme. de hipersensibilidad inducido por drogas� Pseudolinfoma inducido por drogas

� Las drogas mas frecuentemente asociadas son los anticonvulsivantes aromáticos y sulfonamidas
� 10 – 20% de los casos la posible droga responsable no se determina

� En cuanto a la patogenia, se cree que un defecto enzimático de causa genética provocaría la acumulación de metabolitos, que actuarían como haptenos para desencadenar una respuesta inmune específica mediada por linfocitos T contra el medicamento.
� También se postula que la reactivación del herpes virus 6 podría favorecer la reacción

Clínica
� Las manifestaciones aparecen típicamente entre la 2º ya 8º semana y generalmente la fiebre precede al rush
� Rush cutáneo:
� morbiliforme más frecuentemente
� Difuso y hasta eritrodermia
� Prurito
� Resuelve con una dermatitis exfoliativa
� 1/3 de los casos se puede asociar con edema facial
� Mucositis (15 – 30%)



Órgano/sistema Alteración Frecuencia %
Piel y mucosa Morbiliforme
Eritrodermia
Pustular
Mucositis
prurito
82.4
7.4 – 18
6 – 7.4
17.6 – 30
35
hepático Alanina aminotransferasa (>100 IU/L)
Aspartato aminotransferasa (>100 IU/L)
Hiperbilirrubinemia
Falla hepatica
100
41.2
64.7
6
Hematológico Leucocitosis
Eosinofilia
Linfocitos atípicos
trombocitopenia
100
30 – 88
12
6
renal Uremia elevada
Creatinina elevada
Proteinuria
Hematuria
Falla renal
64.7
58.8
23.5
17.6
6
otros Fiebre
Linfadenopatía ≥2 sitios
Linfadenopatía generalizada
Edema facial
Taquicardia
Hipotensión
100
35.3
64.7
33 – 59
58.8
17.6
Mortalidad 5 – 10% (1º causa falla hepática)
Compromiso de órganos internos: hepatitis, insuficiencia renal, neumonitis, miocarditis, tiroiditis, DBT tipo 1, encefalitis, serositis, SIHAD, colitis, hemorragia intestinal

� El diagnóstico se realiza por las manifestaciones clínicas, el antecedente de ingesta de dichas drogas, los exámenes complementarios de laboratorio y el estudio histopatológico.
1. Erupción maculopapular que desarrolla despues de 3 semanas de inicio de droga
2. Síntomas clínicos prolongados por más de 2 semanas de suspendida la droga
3. Fiebre > 38 C
4. Adenopatías
5. Alteraciones hepáticas (ALT >100 u/L) (alt. Renal puede sustituir este criterio)
6. Alteraciones leucocitarias:
� Leucocitosis > 11000
� Linfocitos atípicos > 5%
� Eosinofilia > 1500



Muchas gracias