estudio de la funcionalidad de los linfocitos t en pacientes infectados con el virus de...
TRANSCRIPT

ESTUDIO DE LA FUNCIONALIDAD DE LOS LINFOCITOS T EN PACIENTES
INFECTADOS CON EL VIRUS DE INMUNODEFICIENCIA HUMANA-1 Y SEROLOGÍA
POSITIVA PARA Toxoplasma gondii Y EVALUACIÓN DE EFECTOS
NEUROTÓXICOS DE AMBOS AGENTES PATÓGENOS EN CÉLULAS DEL SISTEMA
NERVIOSO CENTRAL
Por
Edwin Eliel Escobar Guevara
Tesis de Grado presentada como requisito parcial para optar
al título de Doctor en Ciencias, mención Inmunología.
INSTITUTO VENEZOLANO DE INVESTIGACIONES CIENTÍFICAS
I.V.I.C.
CENTRO DE ESTUDIOS AVANZADOS
ALTOS DE PIPE
SEPTIEMBRE, 2014

La Tesis de Grado de Edwin Eliel Escobar Guevara, titulada “ESTUDIO DE LA
FUNCIONALIDAD DE LOS LINFOCITOS T EN PACIENTES INFECTADOS CON EL
VIRUS DE INMUNODEFICIENCIA HUMANA-1 Y SEROLOGÍA POSITIVA PARA
Toxoplasma gondii Y EVALUACIÓN DE EFECTOS NEUROTÓXICOS DE AMBOS
AGENTES PATÓGENOS EN CÉLULAS DEL SISTEMA NERVIOSO CENTRAL”, ha sido
aprobada por el jurado, quien no se hace responsable de su contenido, pero la ha
encontrado correcta en su calidad y en su forma de presentación, en fe de lo cual
firman,
Dr. Andrés Soyano Dr. Héctor Rangel IVIC IVIC Dr. Dimas Hernández Dra. Nathalie Gago UCV UCV Dra. Mercedes Fernández-Mestre Tutora de la Tesis de Grado IVIC Dr. Miguel Alfonzo Director de la Tesis de Grado UCV
Centro de Estudios Avanzados, IVIC. Altos de Pipe, Septiembre de 2014.

Resumen de la Tesis de Grado presentada como requisito parcial para optar
al título de Doctor en Ciencias, mención Inmunología
ESTUDIO DE LA FUNCIONALIDAD DE LOS LINFOCITOS T EN PACIENTES
INFECTADOS CON EL VIRUS DE INMUNODEFICIENCIA HUMANA-1 Y SEROLOGÍA
POSITIVA PARA Toxoplasma gondii Y EVALUACIÓN DE EFECTOS
NEUROTÓXICOS DE AMBOS AGENTES PATÓGENOS EN CÉLULAS DEL SISTEMA
NERVIOSO CENTRAL
Por
Edwin Eliel Escobar Guevara
CENTRO DE ESTUDIOS AVANZADOS
INSTITUTO VENEZOLANO DE INVESTIGACIONES CIENTÍFICAS
I.V.I.C.
ALTOS DE PIPE, SEPTIEMBRE, 2014
Mercedes Fernández Mestre
Tutora de la Tesis de Grado
Miguel Alfonzo Díaz
Director de la Tesis de Grado
El Virus de Inmunodeficiencia Humana (VIH)-1 y el protozoo Toxoplasma gondii
son agentes causales de dos infecciones que afectan a un número importante de
individuos a escala mundial. Si bien en individuos inmunocompetentes T. gondii
causa generalmente infecciones crónicas latentes (prácticamente asintomáticas), la
inmunodeficiencia que se asocia a la infección por VIH-1 puede permitir la
reactivación de la infección parasitaria latente, especialmente en el sistema
nervioso central, provocando patologías graves, como la encefalitis toxoplásmica.
Igualmente, el VIH-1 posee tropismo por el sistema nervioso central, causando
importantes alteraciones neurológicas en los individuos infectados. Por otra parte,
ambos agentes tienen la capacidad de modificar activamente la respuesta
inmunitaria de sus hospedadores, afectando la funcionalidad de las células del
sistema inmunitario. El objetivo del presente trabajo consistió en evaluar dos
grandes aspectos de la infección por VIH-1 y T. gondii en un mismo hospedador: 1.-
la funcionalidad de linfocitos T de individuos infectados con VIH-1 y serología
positiva para T. gondii, en distintas fases de la infección viral, y 2.- el efecto
neurotóxico de ambos patógenos sobre cultivos primarios de células nerviosas de
rata. El fenotipo y la funcionalidad de los linfocitos T de los pacientes infectados con

VIH-1 y seropositivos para T. gondii no presentaron diferencias significativas con
respecto al de los pacientes infectados con VIH-1 y serología negativa para el
parásito, indicando que las infecciones latentes y las infecciones resueltas por T.
gondii no generarían cambios adicionales en estos parámetros a los inducidos por la
sola infección con VIH-1. Sin embargo, en los pacientes infectados con VIH-1 y
seropositivos para T. gondii se observaron fallas en la producción de IL-2, TNF-α e
IFN-γ en respuesta a antígenos de T. gondii desde las etapas tempranas de la
infección viral, sugiriendo que en estos pacientes la capacidad de control de las
infecciones latentes por T. gondii está comprometida, aumentando el riesgo de que
se presenten reactivaciones, aunque sean limitadas, de dichas infecciones, lo que
podría causar daños acumulativos en los tejidos donde se encuentre el parásito,
como el sistema nervioso central, incluso antes de llegar a la etapa de SIDA. Los
experimentos realizados en cultivos primarios de células nerviosas mostraron que
la interacción del virus y las moléculas virales con estas células, incluso sin infección
productiva, era capaz de inducir respuestas que modificaban el microambiente,
incluyendo la producción de óxido nítrico y citoquinas pro-inflamatorias, con serias
consecuencias para la funcionalidad y sobrevida celular. Asimismo, la presencia del
VIH-1 y de la glicoproteína viral de envoltura, gp41, se asociaron con una inhibición
significativa de la replicación de T. gondii. Por otra parte, en los cultivos realizados
en presencia simultánea de ambos patógenos, se indujo un microambiente más pro-
inflamatorio, con mayor destrucción celular (disminución de los recuentos y
aumento de la apoptosis de las células nerviosas), indicando un mayor efecto
neurotóxico. Estos resultados sugieren que, en pacientes co-infectados, el sistema
nervioso central podría estar más afectado por la presencia simultánea de VIH-1 y
T. gondii, incluso con una replicación parasitaria limitada.

Dedico este trabajo a mi esposa Beatriz Elena y
a mis hijos Daniel Eduardo y Ana Isabel,
quienes han compartido conmigo su gestación
e igualmente comparten la alegría de ver culminada esta etapa.

AGRADECIMIENTOS
Al Dr. Miguel Alfonzo, director de este trabajo, quien combinando la firmeza con
la flexibilidad, lo académico y científico con lo amistoso, familiar y hasta paternal, ha
guiado esta etapa de mi formación científica y profesional.
A la Dra. Mercedes Fernández, tutora, profesora y amiga, que en medio de mil
ocupaciones dedicó tiempo precioso para orientar, revisar, corregir y dar forma a
este trabajo.
A los compañeros del Laboratorio de Inmunofisiología Celular, Escuela “J.M.
Vargas”, UCV, licenciados Alexandra Díaz, Josibel Camacho, Riward Campelo,
Alexandra Rodríguez, Gustavo Rico, Ydelys Fuentes, José A. Carrero, Wolfgang Vivas,
Eduardo Navarro, quienes compartieron distintas facetas de este trabajo.
A las Dras. Yajhaira Roldán (Hospital “José Ignacio Baldó”, Caracas) y Belkisyolé
Alarcón de Noya (Instituto de Medicina Tropical, UCV), por referir a los pacientes
participantes.
Al Laboratorio Clinifar, dirigido por la Dra. Mélida Bermúdez, donde se
realizaron las determinaciones de citometría de flujo, así como parte importante de
los análisis microscópicos.
Al Laboratorio de Biología Molecular del Instituto de Biomedicina de la UCV,
dirigido por el Dr. Félix Tapia, donde recibí la valiosa asistencia de la Dra. Nilka Díaz
y la Lic. Yisis López, para realizar los procedimientos de inmunohistoquímica y
microfotografía de cultivos de células nerviosas.
A los Bioterios del IVIC, Instituto de Biomedicina y Escuela de Medicina “J. M.
Vargas”, dirigidos en su momento por la Lic. Ingrid Kalchbrenner, Dra. Nadia Milani
y Dra. Angela de Martínez, por proporcionar los animales y facilidades necesarias
para la realización de parte importante de los experimentos.
A la Dra. Belkisyolé Alarcón de Noya, del Instituto de Medicina Tropical, UCV,
por proporcionar la cepa RH de Toxoplasma gondii y el antígeno parasitario.
Al Dr. Héctor Rangel, por proporcionar la cepa de referencia de VIH-1, la línea de
células MT4 y las facilidades del Laboratorio de Virología Molecular, CMBC, IVIC,
para realizar la replicación y purificación viral. En este último aspecto fue valiosa la
asistencia del Lic. Domingo J. Garzaro y Miguel Barrios.
Al Instituto Nacional de Higiene “Rafael Rangel”, especialmente a la Lic. Gladys
Amely, por la determinación de la carga viral de los sobrenadantes de cultivo.
A la Dra. Alida Hung, en cuyo laboratorio (Cátedra de Bioquímica, Escuela “J.M.
Vargas”) se realizó la determinación de óxido nítrico.
Al IVIC y FONACIT, por proporcionar becas y otras asistencias económicas en
distintas etapas.
Al personal de seguridad de la Escuela de Medicina “José María Vargas”, UCV,
por permitirme el acceso y permanencia en la Escuela en los diferentes horarios que
se requirieron para la realización de los experimentos.
vi

ÍNDICE GENERAL
Página
Resumen..................................................................................................................................... ... iii
Agradecimientos……………………………………...…………………..……………..…...……… vi
Lista de Tablas……………………………………………..…………..……………………..………. x
Lista de Figuras………………………………………..……...…………..………………………….. xi
Introducción……………………………………………………..……...……………………………... 1
I. Infección por el Virus de Inmunodeficiencia Humana-1 (VIH-1)
A. Epidemiología………………………………………………………………………………… 2
B. Estructura y organización del VIH-1……………………………….……………….. 3
C. Proceso de invasión viral………………………………………………………………… 6
D. Proceso de replicación viral………………………………………………….....……… 9
E. Curso clínico de la infección por VIH-1…………………………………….……… 10
F. Inmunopatogénesis de la infección por VIH-1……………………………..…… 13
G. Infección del sistema nervioso central por VIH-1………..……………….…… 17
II. Infección por Toxoplasma gondii A. Epidemiología………………………………………………………………….………..…… 20
B. Estructura y ciclo de vida de Toxoplasma gondii..……………………...………20
C. Respuesta inmunitaria contra Toxoplasma gondii…………………...……….. 24
D. Inmunopatogénesis de la encefalitis toxoplásmica.………………..………… 27
III. Modelos in vitro para estudio de neurotoxicidad……………...………...…….30
IV. Planteamiento del problema de estudio……………...……….……………….….. 31
Hipótesis.………………………………………………………………….………..………..… 31
Objetivos generales.……………………............................................................….…… 32
Objetivos específicos.……………………………………….…...………………….…….. 32
Materiales y métodos
I. Estudio de la funcionalidad de los linfocitos T en pacientes infectados con
VIH-1 y serología positiva para Toxoplasma gondii
A. Sujetos de estudio………………………………………………………………………….….33
B. Estudio fenotípico de linfocitos T de sangre periférica………………………..34
C. Estudios de funcionalidad de linfocitos T…………………...…..…..……………....35
II. Evaluación de efectos neurotóxicos de VIH-1 y Toxoplasma gondii en células
del sistema nervioso central A. Modelo in vitro de neurotoxicidad……………………………………………………..37
B. Evaluación de efectos neurotóxicos…………………………………………….……..40
vii

C. Análisis estadístico…………………………………………………………………………..41
Resultados
I. Estudio de la funcionalidad de los linfocitos T en pacientes infectados con
VIH-1 y serología positiva para Toxoplasma gondii
A. Sujetos de estudio………………………………………………………...………………… 42
B. Estudio fenotípico de linfocitos T de sangre periférica…….……………..… 44
Subpoblación de linfocitos T CD4+………………………………………….…….. 45
Subpoblación de linfocitos T CD8+………………………………………….……… 46
Relación CD4/CD8………………………………………………………………….…… 47
Carga viral…………………………………………………………………………….…… 48
Expresión de HLA-DR……………………………………………………………...…… 50
Expresión de PD-1…………..………………………………………………………….... 51
Expresión de IL-7Rα…………..……………..…………………………………..……… 52
Expresión de CD95……….……………………………………………………………… 53
Apoptosis ex vivo…………………………………………………………………...……. 55
Expresión de CD45RA y CD45RO.………………….………………………..……… 57
C. Estudios de funcionalidad de linfocitos T
Proliferación de linfocitos T CD4+………………………………………………..……61
Proliferación de linfocitos T CD8+…………………………………………………..…62
Producción de perforina por linfocitos T CD8+………………………….………… 63
Apoptosis y necrosis de linfocitos T CD4+……………………………………..…… 64
Apoptosis y necrosis de linfocitos T CD8+……………………………………..…… 66
Producción de citoquinas ex vivo e in vitro……………………………………...…69
II. Evaluación de efectos neurotóxicos de VIH-1 y Toxoplasma gondii en células
del sistema nervioso central
A. Evaluación de efectos neurotóxicos
Recuento de células nerviosas……………………………………………………..… 76
Viabilidad de células nerviosas…………..……………………………………..…… 78
Necrosis de células nerviosas………………………………………………...……… 79
Apoptosis de células nerviosas………………………………………………….……80
Producción de óxido nítrico…………………………………………………..……… 81
Producción de citoquinas……………………………………………………………… 82
B. Infección por T. gondii
Recuento de taquizoítos libres………………………………………………….…… 87
Porcentaje y recuento de células nerviosas infectadas…………………..…… 89
Número de taquizoítos por célula infectada…………………………………...… 91
viii

C. Subpoblaciones de células nerviosas en cultivos primarios
Porcentaje de astrocitos, neuronas y células de la microglía………..……… 94
Astrocitos, neuronas y células de la microglía infectados por T. gondii….96
D. Análisis microscópico – morfológico…………………………………………..…… 99
Discusión
Fenotipo de linfocitos T de sangre periférica……………………………...……. 101
Funcionalidad de linfocitos T………………………………………..……………..….. 110
Efectos neurotóxicos de VIH-1 y T. gondii…………………………………..……. 118
Conclusiones………………………………………………………………..……………………..……131
Bibliografía………………………………………………………………………...…………………… 132
Hoja Curricular…………………………………………………………..…………………………… 176
ix

LISTA DE TABLAS
Tabla Página
I Descripción de los sujetos de estudio…………………………………… 43
II Porcentaje de células nerviosas con [0-4], [5-15], [16-30]
ó [>30] parásitos en su citoplasma………………………………………. 93
III Porcentaje de astrocitos, neuronas y células de la microglía
infectados por T. gondii……………………………………………………….. 96
x

LISTA DE FIGURAS
Figura Página
1. Número de personas infectadas por VIH-1 en distintas regiones del
mundo…………………………………………………………………………………….…….. 2
2. Estructura del VIH-1…………………………………………………………………………. 3
3. Organización genómica del VIH-1………………………………………………………. 4
4. Relación filogenética entre lentivirus humanos y lentivirus de
primates no humanos…………………………………………………………………….. 6
5. Glicoproteínas de envoltura gp120 y gp41……………………………………..…... 7
6. Proceso de invasión del VIH-1…………………………………………………...………. 8
7. Ciclo de replicación del VIH-1……………………………………………………………. 10
8. Curso clínico de la infección por VIH-1………………………………………………. 11
9. Diferentes respuestas a infecciones virales………………………………………… 16
10. Modelo de neurotoxicidad mediada por VIH-1………………………………..…. 18
11. Vías de señalización celular asociadas a neurotoxicidad por VIH-1…...… 19
12. Ooquistes y esporozoítos de Toxoplasma gondii…………………………………. 21
13. Taquizoítos y bradizoítos de Toxoplasma gondii………..………………………. 22
14. Quistes de Toxoplasma gondii en cerebro de ratón………………………...…… 23
15. Ciclo de vida de Toxoplasma gondii……………………………………...…………….. 24
16. Inmunología de la infección por Toxoplasma gondii……………………………. 25
17. Quistes cerebrales de Toxoplasma gondii…………………………………………… 28
18. Encefalitis toxoplásmica…………………………………………………………………… 29
19. Subpoblaciones de linfocitos T en sangre periférica - Determinación
por citometría de flujo……………………………………………………………………. 44
20. Porcentaje y recuento de linfocitos T CD4+ en sangre periférica…………. 45
21. Porcentaje y recuento de linfocitos T CD8+ en sangre periférica…………. 46
22. Relación CD4/CD8……………………………………………………………………………. 47
23. Carga viral de VIH-1…………………………………………………………………………. 48
24. Fenotipaje de linfocitos T en sangre periférica - Determinación por
citometría de flujo………………………………………………………………………….. 49
25. Expresión de HLA-DR en linfocitos T CD4+ y T CD8+ de sangre
periférica………………………………………………………………………………………. 50
26. Expresión de PD-1 en linfocitos T CD4+ y T CD8+ de sangre
periférica………………………………………………………………………………………. 51
27. Expresión de IL-7Rα en linfocitos T CD4+ y T CD8+ de
sangre periférica……………………………………………………………………………. 52
28. Expresión de CD95 en linfocitos T CD4+ y T CD8+ de sangre
periférica………………………………………………………………………………………. 53
29. Apoptosis de linfocitos T – Análisis por citometría de flujo…………………. 54
xi

Figura Página
30. Apoptosis ex vivo de linfocitos T CD4+ y T CD8+ de sangre
periférica………………………………………………………………………………………. 55
31. Expresión de CD45RA y CD45RO en linfocitos T de sangre
periférica – Análisis por citometría de flujo……………………………………… 56
32. Expresión de CD45RA y CD45RO en linfocitos T CD4+ de sangre
periférica………………………………………………………………………………………. 58
33. Expresión de CD45RA y CD45RO en linfocitos T CD8+ de sangre
periférica………………………………………………………………………………………. 59
34. Relación entre la expresión de CD45RA y CD45RO en linfocitos
T CD4+ y T CD8+ de sangre periférica……………………………………………… 60
35. Proliferación de linfocitos T CD4+……………………………………………………… 61
36. Proliferación de linfocitos T CD8+……………………………………………………… 62
37. Producción de perforina en linfocitos T CD8+……………………………………. 63
38. Apoptosis y necrosis de linfocitos T CD4+…………………………………………. 65
39. Apoptosis y necrosis de linfocitos T CD8+…………………………………………. 67
40. Determinación de citoquinas por citometría de flujo………………………….. 68
41. Determinación de citoquinas en plasma sanguíneo……………………………. 69
42. Determinación de IL-2 en sobrenadantes de cultivo de PBMC…………….. 70
43. Determinación de IL-4 en sobrenadantes de cultivo de PBMC…………….. 71
44. Determinación de IL-6 en sobrenadantes de cultivo de PBMC…………….. 72
45. Determinación de IL-10 en sobrenadantes de cultivo de PBMC…………… 73
46. Determinación de TNF-α en sobrenadantes de cultivo de PBMC…………. 74
47. Determinación de IFN-γ en sobrenadantes de cultivo de PBMC…………... 75
48. Recuento de células nerviosas vivas en cultivos primarios de
células nerviosas de rata en presencia de gp41, VIH-1 y/o T. gondii….. 77
49. Viabilidad en cultivos primarios de células nerviosas de rata
en presencia de gp41, VIH-1 y/o T. gondii ………………………………………. 78
50. Necrosis en cultivos primarios de células nerviosas de rata
en presencia de gp41, VIH-1 y/o T. gondii ………………………………………. 79
51. Apoptosis temprana en cultivos primarios de células nerviosas
de rata en presencia de gp41, VIH-1 y/o T. gondii……………………………. 80
52. Producción de óxido nítrico y correlación “apoptosis/óxido
nítrico” en cultivos primarios de células nerviosas de rata en
presencia de gp41, VIH-1 y/o T. gondii……………………………………………. 81
53. Producción de IL-1α en cultivos primarios de células nerviosas
de rata en presencia de gp41, VIH-1 y/o T. gondii……………………………. 82
54. Producción de IL-6 en cultivos primarios de células nerviosas
de rata en presencia de gp41, VIH-1 y/o T. gondii……………………………. 83
xii

Figura Página
55. Producción de IL-10 en cultivos primarios de células nerviosas
de rata en presencia de gp41, VIH-1 y/o T. gondii……………………………. 84
56. Producción de TNF-α en cultivos primarios de células nerviosas
de rata en presencia de gp41, VIH-1 y/o T. gondii……………………………. 85
57. Producción de IFN-γ en cultivos primarios de células nerviosas
de rata en presencia de gp41, VIH-1 y/o T. gondii……………………………. 86
58. Recuento de taquizoítos libres en sobrenadante de cultivos
primarios de células nerviosas de rata infectados con T. gondii
en presencia de gp41 ó VIH-1…………………………………………………………. 88
59. Porcentaje y recuento de células nerviosas de rata infectadas por
T. gondii en presencia o no de gp41 o VIH-1…………………………………….. 90
60. Número de parásitos en células nerviosas infectadas por T. gondii
en presencia o no de VIH-1 - Micrografías……………………………………….. 92
61. Número de parásitos en células nerviosas infectadas por T. gondii
en presencia o no de VIH-1……………………………………………………………… 93
62. Porcentaje de astrocitos, neuronas y células de la microglía en
cultivos primarios de células nerviosas de rata en presencia de
VIH-1 y/o T. gondii…………………………………………………………………………. 95
63. Porcentaje total de astrocitos, neuronas y células de la microglía
infectados por T. gondii, en presencia o no de VIH-1, en cultivos
primarios de células nerviosas de rata…………………………………………….. 97
64. Correlación entre el porcentaje de cada linaje en la población de
células nerviosas y el porcentaje de infección por T. gondii……………… 98
65. Estudio microscópico de cultivos primarios de células nerviosas
de rata en presencia simultánea de T. gondii y gp41 o VIH-1…………….. 100
xiii

INTRODUCCIÓN El 5 de Junio de 1981 Gottlieb et al.1 reportaron 5 casos inusuales de neumonía
causada por Pneumocystis carinii* en pacientes homosexuales masculinos jóvenes
previamente saludables, que fueron atendidos en distintos hospitales de Los
Ángeles, California. Este tipo de neumonía se presenta casi exclusivamente en
pacientes con inmunodeficiencia severa, como aquellos tratados con drogas
inmunosupresoras, por lo que estos casos se consideraron excepcionales. A tres de
estos pacientes se les evaluó la funcionalidad linfocitaria in vitro, mostrando
profunda disminución de la capacidad proliferativa en respuesta a mitógenos y
antígenos. Los autores sugirieron que la disfunción observada en la respuesta
inmunitaria celular de estos pacientes podría estar relacionada con la exposición a
un factor común, posiblemente asociado al estilo de vida homosexual, que los hacía
susceptibles a sufrir infecciones oportunistas. Éste sería el primero de una serie de
reportes4-7 que darían a conocer un tipo de inmunodeficiencia no descrita
previamente, que posteriormente recibió el nombre de Síndrome de
Inmunodeficiencia Adquirida (SIDA)8, 9, y se convertiría en un problema de salud
pública de alcance mundial8. Aunque la enfermedad fue inicialmente descrita en
pacientes homosexuales masculinos jóvenes1, 4-7, los grupos en riesgo pronto
incluyeron a individuos adictos a drogas inyectables6, receptores de transfusiones11
o productos sanguíneos, como pacientes con hemofilia A12, infantes nacidos de
madres en grupos de riesgo13, parejas sexuales femeninas de hombres
sintomáticos14, 15 y población heterosexual promiscua en África16, 17, 18. Otros
cuadros clínicos que fueron descritos en los pacientes afectados incluían sarcoma
de Kaposi, infecciones fúngicas invasivas, toxoplasmosis, infecciones por
micobacterias y Linfoma No Hodgkin19. Numerosas fueron las teorías que trataron
de explicar las causas del SIDA, hasta que el 20 de Mayo de 1983 Barré-Sinoussi et
al. notificaron el aislamiento de un nuevo retrovirus relacionado con los Virus de
Leucemia de Células T Humanas (HTLV), en un paciente con sintomatología
asociada al SIDA20. Trabajos subsiguientes confirmaron este hallazgo21, 22,
identificando al virus que ahora conocemos como Virus de Inmunodeficiencia
Humana (VIH)-123, como el agente causal de la infección cuya etapa final es el
SIDA24, 25, que para el año 1993 pasaría a ser la principal causa de muerte en
hombres de 25 a 44 años de edad en Estados Unidos26, y para el año 2012 habría
causado 36 millones de muertes en el mundo27.
* Conocido ahora como Pneumocystis jirovecii2, 3.
1

I. Infección por el Virus de Inmunodeficiencia Humana-1
A. Epidemiología
De acuerdo al informe sobre la epidemia mundial de SIDA publicado en el año
2013 por el Programa Conjunto de las Naciones Unidas sobre VIH/SIDA (ONUSIDA)28
se estima que existen 35,3 millones de personas infectadas por el VIH-1 a nivel
mundial, siendo la región de África sub-sahariana la que sobrelleva la mayor carga, con
unos 25 millones de casos, mientras que en América Latina habría 1,5 millones (Fig.
1). Se estima que durante el año 2012, a nivel mundial se contrajeron 2,3 millones de
nuevas infecciones y 1,6 millones de personas murieron por causas asociadas al SIDA.
En Venezuela, por su parte, se estima que 110.000 personas (38.000 mujeres)
están infectadas por el virus, lo que representa una prevalencia del 0.6% para la
población general28, si bien en poblaciones vulnerables, como la de hombres
homosexuales, la prevalencia puede ser superior al 5%29. En el país, 43.032 personas
recibieron tratamiento antirretroviral en el año 2012, lo que representa una cobertura
del 72% de la población en necesidad de ser tratada. Por otra parte, 6.600 nuevas
infecciones y 3.800 muertes asociadas al SIDA se estimaron para el mismo año28.
Figura 1. Número de personas infectadas por VIH-1 en distintas regiones del mundo: Se
presenta el número estimado de personas (adultos y niños) infectados por VIH-1 en distintas
regiones del mundo para el año 2012, colocando dentro de corchetes los rangos estimados basados
en la mejor información disponible. En el óvalo se presentan los datos para Venezuela. (Fuente:
“UNAIDS: Epidemiology 2013” y “ONUSIDA: Informe mundial sobre la epidemia de SIDA 2013”28,
disponibles en http://www.unaids.org).
1,3 millones [1,0 – 1,7 millones]
Asia Central Europa Oriental &
3,9 millones [2,9 – 5,2 millones]
Sur-oriental Asia del Sur &
Centro-occidental Europa
860.000 [800.000 – 930.000]
Oceanía 51.000
[43.000 – 59.000]
África Subsahariana 25,0 millones
[23,5 – 26,6 millones]
Norteamérica 1,3 millones
[980.000 – 1,9 millones]
Caribe 250.000
[220.000 – 280.000]
América Latina 1,5 millones
[1,2 – 1,9 millones]
Venezuela 110.000
[74.000 – 160.000]
Personas Infectadas por VIH-1 Año 2012
UNAIDS: Epidemiology 2013
Asia Oriental 880.000
[650.000 – 1,2 millones]
260.000 [200.000 – 380.000]
África del Norte Medio Oriente &
2

B. Estructura y organización del VIH-1
El VIH-1 pertenece a la familia Retroviridae, subfamilia Lentivirinae, género
Lentivirus30, 31. Su genoma consiste en 2 hebras idénticas de ARN de cadena sencilla y
sentido positivo, de aproximadamente 9.700 bases, que se asocian estrechamente con
las enzimas transcriptasa inversa e integrasa y con la nucleocápside (proteína p7), que
protege al ARN de ser degradado por nucleasas (Fig. 2). El genoma está rodeado por
una cápside cónica y una matriz formadas por las proteínas p24 y p17,
respectivamente. La envoltura viral es la capa externa del virión, y está formada por
una bicapa lipídica derivada de la célula hospedadora, en la que están insertadas
proteínas de dicha célula32, 33, además de las glicoproteínas virales de envoltura,
gp120 y gp41. Las proteínas Vif, Vpr, Nef, p7 y la proteasa viral también forman parte
del virión.
El genoma del VIH-1 está organizado de forma extremadamente eficiente31, 34, con
tres marcos de lectura que permiten el solapamiento de genes en la secuencia
nucleotídica. Para el VIH-1 se han descrito nueve genes, que codifican para proteínas
estructurales, reguladoras y accesorias (Fig. 3)24, 25.
Figura 2. Estructura del VIH-1: el VIH-1 tiene un diámetro de unos 120 nm, un genoma compuesto
por dos hebras idénticas de ARN monocatenario de sentido positivo, asociado a la nucleocápside
(p7) y a las enzimas transcriptasa inversa e integrasa. Posee una cápside cónica (p24), una matriz
(p17) y una envoltura formada a partir de la membrana de la célula hospedadora, en la que están
insertadas las glicoproteínas gp120 y gp41. (Fuente: National Institute of Health, USA; disponible
en: http://commons.wikimedia.org/wiki/File:HIV_Virion-en.png).
3
Vif, Vpr, Nef y p7
Proteasa
Envoltura
Viral
ARN (Genoma Viral)
Transcriptasa
Inversa
Integrasa Matriz
(p17)
Cápside (p24) Nucleocápside
(p7)
gp41
Proteína
Transmembrana
gp120 Proteína de Superficie

Regiones del genoma del VIH-1 que codifican para proteínas estructurales:
Gag (group-specific antigen): esta región codifica para una poliproteína precursora
que es procesada por la proteasa viral, para formar la nucleocápside (p7), la
cápside (p24) y la matriz (p17), que rodean al genoma viral.
Pol (polymerase): este gen codifica para los precursores de las enzimas
transcriptasa inversa, integrasa y proteasa, necesarias para transcribir el ADN
proviral a partir del ARN genómico, integrar la doble cadena de ADN proviral al
genoma celular y procesar la poliproteína Gag, respectivamente.
Env (envelope): codifica para la glicoproteína gp160, que al ser escindida por una
proteasa celular tipo furina, da origen a las glicoproteínas de envoltura, gp120
y gp41.
Regiones del genoma del VIH-1 que codifican para proteínas reguladoras:
Tat (transactivator of transcription): codifica para el trans-activador viral, una
proteína que se une a una región específica (TAR) en las secuencias largas
repetitivas (LTR), aumentando la transcripción de ARN viral, favoreciendo la
síntesis de proteínas reguladoras y accesorias.
Rev (regulator of expression of virion proteins): codifica para una proteína que se une a
secuencias específicas (RRE) de ARN, estabilizando los ARN mensajeros de
proteínas estructurales y facilitando su paso del núcleo al citoplasma celular.
Regiones del genoma del VIH-1 que codifican para proteínas accesorias:
Nef (negative factor): codifica para una proteína que se ha asociado con la
patogénesis de la infección viral. Esta proteína se ancla a la cara interna de la
membrana de la célula infectada, e interacciona con componentes de vías de
señalización que favorecen la activación celular. Disminuye la expresión de
CD4, CD28 y proteínas del complejo principal de histocompatibilidad (MHC)-I
en las células infectadas. Está presente en los viriones y parece potenciar el
Figura 3. Organización genómica del VIH-1: las secuencias genéticas que codifican para
proteínas estructurales se representan en color anaranjado, en color azul para proteínas
reguladoras y en color verde para proteínas auxiliares. Las posiciones verticales representan los
tres marcos de lectura. Las secuencias largas repetitivas (LTR) se representan en color gris.
(Emerman M; Current Biology 1996; 6(9):1096-1103).
Tat. The genomes of oncogenic retroviruses have only three genes: gag, pol and env.
4

proceso de fusión de la envoltura viral con la membrana celular al momento de
la infección.
Vif (viral infectivity factor): codifica para una proteína asociada a infectividad viral,
que se une al ARN y participa en los procesos de ensamblaje y maduración. Por
otra parte, Vif contrarresta la acción de APOBEC3G, un factor de restricción
celular con actividad desaminasa de citidina, que modifica la primera hebra de
ADN proviral al momento de la retrotranscripción inversa, haciéndola blanco
de digestión endonucleolítica, o favoreciendo hipermutagénesis a niveles
incompatibles con la viabilidad viral.
Vpr (viral protein r): codifica para la proteína viral R, que induce el arresto del
ciclo celular en la fase G2, aumentando la producción viral. Vpr promueve la
entrada del ADN viral al núcleo celular. Asimismo, Vpr contrarresta la acción de
SAMHD1, un factor de restricción celular que bloquea la transcripción inversa
en células mieloides y linfocitos T en reposo.
Vpu (viral protein u): codifica para una proteína requerida para la liberación
eficiente y maduración de viriones. Vpu contrarresta la acción de la teterina
(BST-2/CD317), una proteína transmembrana inducida por interferón, que
restringe la liberación de viriones. Vpu también induce la degradación de la
molécula CD4.
El ciclo de replicación del VIH-1 requiere la transcripción inversa del ARN
genómico para sintetizar ADN proviral. Debido a que la transcriptasa inversa viral es
una enzima propensa a cometer errores y carece de actividad correctora, por cada
genoma copiado se introduce por lo menos una mutación, lo que genera una población
viral con alta complejidad genética35. El análisis y comparación de secuencias
nucleotídicas ha permitido identificar tres grupos genéticos del VIH-1: el grupo M (del
inglés main, principal), el grupo O (outlaier, externo) y el grupo N (new, nuevo); siendo
el grupo M el más ampliamente diseminado y responsable de la pandemia10, 35 (Fig. 4).
Evidencias genéticas y epidemiológicas sugieren que los tres grupos genéticos del VIH-
1 son el resultado de introducciones independientes, por salto de especie, de Virus de
Inmunodeficiencia Simia (VIS) a hospedadores humanos, a partir de chimpancés
(VIScpz)36, 37. Asimismo, el VIH-2, un virus relacionado al VIH-1, con diseminación
geográfica más limitada y menor virulencia 38, se habría originado por salto de especie
del SIV a partir de mangabeyes grises39. El grupo M del VIH-1 se ha subdividido en
subtipos (A-K), cada uno de los cuales tiene una distribución geográfica particular. En
Europa, en América del Norte y en buena parte de América Central y del Sur el subtipo
B es el que predomina10, 35. En Venezuela más del 99% de las infecciones son causadas
por el subtipo B40, 41. El subtipo C, presente en forma predominante en las epidemias
de África subsahariana, India y China, es responsable de 55-60% de todas las
infecciones por VIH-1 a nivel mundial10, 35. También se ha descrito la generación de
5

diversos recombinantes virales entre los distintos subtipos del grupo M, lo cual
aumenta la complejidad de la epidemia10, 35.
C. Proceso de invasión viral
El proceso de invasión a la célula blanco por VIH-1 es mediado por las
glicoproteínas de envoltura, gp120 y gp41, en el cual la primera se une a las moléculas
receptoras y correceptoras en la célula blanco y la segunda media la fusión de la
envoltura viral a la membrana celular, para permitir el ingreso del material genético
del virus.
Antes de describir el proceso de invasión viral propiamente, analicemos la
estructura de las glicoproteínas de envoltura42, 43. La gp120 y la gp41 son
sintetizadas a partir de una proteína precursora, la gp160, después de una serie de
pasos de maduración que incluyen la formación de puentes disulfuro, glicosilación
extensiva y escisión por una proteasa celular tipo furina. Estas glicoproteínas
VIH-1 Grupo N
VIH-1 Grupo O
VIH-2 Grupo A
VIH-1 Grupo M
VIH-2 Grupo B
No epidémicos Fuente de aislados
Epidémicos Fuente de aislados
0.05 Sustituciones/sitio
Figura 4. Relación filogenética de lentivirus humanos y de primates no humanos: la pandemia
por VIH-1 está asociada a aislados virales pertenecientes al grupo M (círculo azul), siendo el subtipo
C (representado en color rojo) el que a nivel mundial tiene mayor prevalencia. Las formas
recombinantes virales forman parte del grupo M, pero por razones de claridad no se muestran.
(Simon V et al.; Lancet 2006; 368:489-50410).
Tat. The genomes of oncogenic retroviruses have only three genes: gag, pol and env.
6

forman complejos no covalentes en la envoltura viral, y se agrupan formando
heterotrímeros.
La gp120 está formada por cinco dominios constantes (C1, C2, C3, C4 y C5) y
cinco dominios variables (V1, V2, V3, V4 y V5) (Fig. 5A, C). La gp120, como se ha
mencionado, interacciona con el receptor viral en la célula blanco, la molécula
CD444, 45, así como con los correceptores, las moléculas CXCR4 y CCR546, 47. En base
a la afinidad que tenga por estas últimas, la gp120 define el tropismo viral48, 49, el
cual puede ser X4, R5 o doble, según su afinidad por CXCR4, CCR5 o ambos.
Independientemente de la ruta de transmisión, la mayoría de las nuevas infecciones
involucran variantes virales que utilizan CCR5 como correceptor50, 51. Los virus con
tropismo X4 generalmente aparecen en etapas avanzadas de la infección y se han
asociado con aumento de la patogenicidad y progresión de la enfermedad52.
La glicoproteína gp41, por otra parte, representa la maquinaria molecular que
facilita la fusión de membranas42, 43. Esta glicoproteína posee una región
extracelular o ectodominio, una región transmembrana, que la ancla a la envoltura
viral y un dominio citoplasmático (Fig. 5B, D). El ectodominio tiene una región
hidrofóbica, el péptido de fusión N terminal, una hélice N, un lazo disulfuro, una
hélice C y una región proximal a la membrana.
Figura 5. Glicoproteínas de envoltura gp120 y gp41: A. La gp120 tiene 5 dominios constantes
(C1-C5) y 5 variables (V1-V5). Las posiciones de N-glicosilación se indican con estructuras
ramificadas. Las regiones representadas en color rosado en C1, C2 y C4 se reordenan luego de la
unión a CD4 y forman el minidominio de conexión. B. La gp41 tiene el péptido de fusión (FP) en el
extremo N terminal, la hélice N (HR1), el lazo disulfuro, la hélice C (HR2), la región proximal a la
membrana (MPR), el dominio transmembrana (TM) y el citoplasmático en el extremo C terminal
(representado en color gris claro). C. Diagrama de cintas de la gp120. D. Diagrama de cintas de un
trímero del ectodominio de la gp41 de VIS, formando un haz de 6 hélices. HR1, el lazo disulfuro y
HR2 están representados en color azul claro, amarillo y rojo, respectivamente. (A y B: Tilton &
Doms; Antiviral Res 2010; 85:91-10043; C y D: Caffrey M; Trends Microbiol 2011; 19 (4):191-
19742).
D
A gp120
B gp41
C
Lazo Disulfuro MPR
Ectodominio
7

La infección por VIH-1 es un proceso complejo, que se verifica en varias etapas42, 43
(Fig. 6). La interacción inicial entre el virus y su célula blanco es facilitada por
interacciones no específicas entre dominios con carga positiva de la gp120 y
proteoglicanos negativamente cargados de la membrana celular, o por interacciones
específicas entre gp120 y DC-SIGN53. El receptor primario para VIH-1 es la molécula
CD4, la cual forma parte de la superfamilia de las inmunoglobulinas y se expresa en
monocitos, macrófagos, células dendríticas y una subpoblación de linfocitos T, por lo
que serán éstas las poblaciones celulares susceptibles de ser infectadas por el virus54-
57. La región de la gp120 que interacciona con CD4 es altamente conservada, no
glicosilada y está formada por epítopos discontinuos en la secuencia primaria de la
proteína. La unión a CD4 provoca cambios conformacionales en gp120 que posibilitan
su unión a la molécula correceptora (Fig. 6). Estos cambios incluyen: primero, la
formación del minidominio de conexión, por el acercamiento de dos regiones de hojas
β-plegadas que están separadas en la glicoproteína no unida; segundo, la exposición
de los dominios V1/V2 y V3; y tercero, un cambio en la orientación de gp120 que
dirige al minidominio de conexión y a V3 hacia la membrana celular, lo que permite
que éstos puedan interaccionar con el correceptor.
Los correceptores para VIH-1 son las moléculas CCR5 y CXCR4, receptores de
quimioquinas, miembros de la familia de receptores acoplados a la proteína G, con 7
dominios transmembrana. En la región extracelular de estas proteínas están ubicados
el extremo N terminal y tres lazos, los cuales interactúan con distintas regiones de
gp120. El extremo N terminal interactúa con el minidominio de conexión y la base de
V3, mientras que los lazos extracelulares lo hacen con la parte superior de V3 (Fig. 6).
Haz de 6 hélices
correceptor
Dominios
Variables
Trímero Unión a CD4
Unión al Correceptor
Fusión de
Membranas
Figura 6. Proceso de invasión del VIH-1: En la parte inferior de la figura se representa la
membrana de la célula blanco, en la cual se localizan el receptor viral (la molécula CD4) y los
correceptores (CCR5 o CXCR4), mientras que en la parte superior se representan gp120 y gp41
asociadas a la envoltura viral. Se ilustra la unión secuencial de gp120 al receptor y al correceptor,
así como el proceso de fusión de membranas mediado por gp41. Los dominios de gp120 y gp41
están representados con los mismos colores que en la Figura 5 A y B. (Tilton & Doms; Antiviral Res
2010; 85:91-10043).
8

La unión de gp120 al correceptor provoca cambios conformacionales adicionales en el
trímero que resultan en la exposición del péptido de fusión de la gp41 y su inserción
en la membrana celular, luego de lo cual se lleva a cabo un reordenamiento espacial
energéticamente favorable de las hélices N y C, plegándose éstas una sobre la otra
(Fig. 6). En un trímero funcional, el plegamiento de las hélices N y C forma el haz de
seis hélices, con las tres hélices N ubicadas en la parte central del haz y las 3 hélices C
en la parte exterior (Fig. 5D). Este reordenamiento espacial acerca la región
transmembrana de gp41, que está insertada en la envoltura viral, al péptido de fusión,
que está insertado en la membrana celular (Fig. 6), una yuxtaposición que resulta en
la formación del poro de fusión, permitiendo que la cápside viral ingrese a la célula
blanco.
A. Proceso de replicación viral
Una vez que la cápside viral alcanza el citoplasma celular, la enzima transcriptasa
inversa viral libera la hebra sencilla de ARN genómico de las proteínas unidas a ésta, y
procede a copiar una hebra complementaria de ADN (ADNc), en un proceso conocido
como transcripción inversa. La transcriptasa inversa tiene actividad ribonucleasa, que
degrada al ARN viral utilizado como molde, y actividad polimerasa de ADN
dependiente de ADN, que sintetiza una hebra complementaria de ADN a partir del
ADNc, formando así una molécula de doble cadena. El ADN viral se asocia entonces
con proteínas celulares y virales (Vpr, p17 de matriz, integrasa) para formar el
complejo de pre-integración (PIC)58, que será transportado al núcleo celular. En el
punto central de la infección, la enzima integrasa viral, en conjunto con enzimas
reparadoras de ADN de la propia célula, insertan el genoma viral (provirus) en el ADN
cromosomal de la célula hospedadora, en segmentos genéticos que estén en activa
transcripción59. La integración transforma irreversiblemente a la célula infectada en
una potencial productora de virus. El ADN proviral puede permanecer latente o
transcribirse para generar nuevas partículas virales. En las secuencias repetitivas
largas (LTR) del genoma viral (Fig. 3 y 7) existen regiones reguladoras y promotoras
que son reconocidas por factores de transcripción celulares, como NF-κB, lo cual
explica porque la replicación del VIH-1 se optimiza en linfocitos T activados24. Los
bajos niveles de transcripción inducidos por LTR permiten la producción de la
proteína Tat, la cual potenciará la transcripción de ARN viral, aumentando la
producción de proteínas reguladoras que incluyen a Rev34. La proteína Rev, a su vez,
se unirá a las cadenas nacientes de ARN viral, promoviendo su exportación al
citoplasma celular para la traducción de proteínas estructurales34 y formación de ARN
genómico, que se transportarán a la cercanía de la membrana celular para ensamblar
nuevas partículas virales. La salida del virus de la célula infectada se produce por un
proceso de gemación, no lítico, que aprovecha la maquinaria celular de formación de
9

vesículas60. Finalmente la escisión de las poliproteínas Gag y Pol por la proteasa viral
producirá viriones maduros, capaces de infectar nuevas células.
B. Curso clínico de la infección por VIH-1
Fase aguda - Infección primaria: Aproximadamente 3 a 6 semanas luego de la
exposición e infección primaria con VIH-1, 50-70% de los individuos experimentan un
síndrome agudo, autolimitado, de severidad variable, que persiste por una o varias
semanas y se resuelve espontáneamente61. Este período se asocia con concentraciones
muy altas de carga viral y disminución importante de los recuentos de linfocitos T
CD4+ en sangre periférica (Fig. 8). Al resolverse el cuadro agudo, los recuentos de
células T CD4+ regresan a niveles normales o ligeramente disminuidos. Los recuentos
de linfocitos B y T CD8+ también disminuyen durante la infección aguda, sin embargo
los recuentos de estos últimos aumentan a concentraciones mayores que los niveles
basales unas 3 o 4 semanas luego del inicio del cuadro agudo. Debido a que el aumento
Inhibidores de
Proteasa
NNRTI NRTI
Inhibidores de
Integrasa
Inhibidores
de Entrada
Inhibidores
de Fusión
Fusión
Unión
Maduración
Gemación
VIH-1
Liberación del Genoma Viral
Integración
Transcripción
Citoplasma
ARNm al Núcleo
Traducción Ensamblaje
Transcripción
Inversa
No Infecciosas Partículas Virales
Infecciosas Partículas Virales
Provirus
Genoma Celular
10
Figura 7. Ciclo de replicación del VIH-1: Se ilustran los principales pasos del ciclo de replicación
del VIH-1: la unión al receptor CD4 y al correceptor, la fusión a la membrana celular, ingreso de la
cápside, liberación del ARN genómico y las proteínas virales en el citoplasma, la transcripción
inversa, la formación del complejo de pre-integración (PIC), su traslado al núcleo e integración al
genoma celular, su posterior transcripción y traducción, el ensamblaje y liberación de nuevas
partículas virales y la maduración de las mismas. Se muestran las principales familias de drogas
antirretrovirales (en color verde) y los pasos que ellas bloquean. Asimismo, se presentan los
factores de restricción celular contra VIH-1 (TRIM5α, APOBEC3G, SAMHD1 y teterita, en color rojo)
y sus antagonistas virales correspondientes (en color azul). CCR5, receptor 5 de quimioquinas CC;
LTR, secuencias largas repetitivas; NRTI, nucleósidos inhibidores de transcriptasa inversa; NNRTI,
no nucleósidos inhibidores de transcriptasa inversa. (Barré-Sinoussi F et al.; Nat Rev Microbiol 2013;
11:877–883).
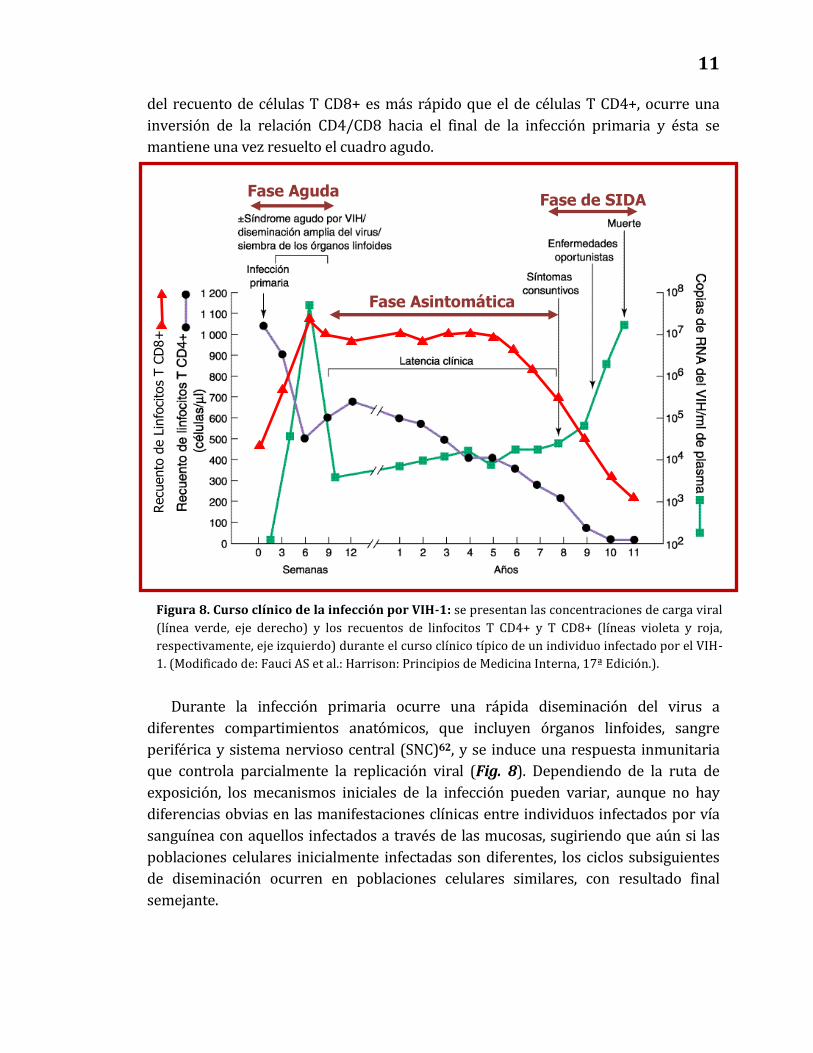
del recuento de células T CD8+ es más rápido que el de células T CD4+, ocurre una
inversión de la relación CD4/CD8 hacia el final de la infección primaria y ésta se
mantiene una vez resuelto el cuadro agudo.
Durante la infección primaria ocurre una rápida diseminación del virus a
diferentes compartimientos anatómicos, que incluyen órganos linfoides, sangre
periférica y sistema nervioso central (SNC)62, y se induce una respuesta inmunitaria
que controla parcialmente la replicación viral (Fig. 8). Dependiendo de la ruta de
exposición, los mecanismos iniciales de la infección pueden variar, aunque no hay
diferencias obvias en las manifestaciones clínicas entre individuos infectados por vía
sanguínea con aquellos infectados a través de las mucosas, sugiriendo que aún si las
poblaciones celulares inicialmente infectadas son diferentes, los ciclos subsiguientes
de diseminación ocurren en poblaciones celulares similares, con resultado final
semejante.
Recu
ento
de L
info
cito
s T C
D8+
Fase de SIDA
Fase Asintomática
Fase Aguda
Figura 8. Curso clínico de la infección por VIH-1: se presentan las concentraciones de carga viral
(línea verde, eje derecho) y los recuentos de linfocitos T CD4+ y T CD8+ (líneas violeta y roja,
respectivamente, eje izquierdo) durante el curso clínico típico de un individuo infectado por el VIH-
1. (Modificado de: Fauci AS et al.: Harrison: Principios de Medicina Interna, 17ª Edición.).
11

Fase asintomática – Infección crónica/persistente: El VIH-1 es un lentivirus, un
grupo de virus caracterizados por causar infecciones crónicas persistentes, con
manifestaciones patológicas que se presentan en los estadios tardíos de la infección. A
pesar de la potente respuesta inmunitaria humoral y celular que se induce durante la
infección primaria, el VIH-1 no es completamente erradicado del individuo infectado y
durante todo el curso de la infección crónica se puede detectar replicación viral, tanto
en sangre periférica como en tejido linfoide63-65. Así, en individuos infectados por VIH-
1, la infección persistente es la norma, y la respuesta inmunitaria no es completamente
protectora. Entre los factores que contribuyen para que el VIH-1 evada la respuesta
inmunitaria están su rápida cinética de replicación66 y la alta tasa de error de su
transcriptasa inversa67, lo cual permite la constante generación y acumulación de
mutaciones, con la resultante variación antigénica.
El tiempo promedio entre la infección primaria y el desarrollo del SIDA es de 10
2 años68, 69. Durante este período los recuentos de linfocitos T CD4+ disminuyen
gradualmente, hasta llegar a un punto en que el riesgo de sufrir enfermedades
oportunistas es alto70, 71.
Fase de SIDA – Infección tardía: Un paciente infectado con VIH-1 se considera en
etapa de SIDA72 cuando su recuento de linfocitos T CD4+ en sangre periférica
disminuye por debajo de 200 células/µL, o cuando su condición clínica se deteriora en
forma grave, evidenciando defectos en su respuesta inmunitaria celular, con
manifestaciones que incluyen:
Infecciones causadas por:
o Hongos: neumonía por P. jirovecii; candidiasis esofágica o pulmonar;
criptococosis, histoplasmosis o coccidioidomicosis extrapulmonar
o Bacterias: tuberculosis pulmonar o extrapulmonar; neumonías bacterianas
recurrentes; septicemia recurrente por Salmonella
o Virus: encefalitis, retinitis, esofagitis, neumonía, pancreatitis por
citomegalovirus; úlceras crónicas, bronquitis, esofagitis por Virus del Herpes
Simple
o Protozoarios: encefalitis toxoplásmica, síndrome gastrointestinal por
Cryptosporidium o Isospora
Cierto tipo de neoplasias: sarcoma de Kaposi, carcinoma cervical invasivo,
linfoma de Burkitt, inmunoblástico o primario del SNC
Encefalopatía asociada al VIH-1
Leucoencefalopatía multifocal progresiva
Síndrome de desgaste asociado al VIH-1
El curso natural de la infección por VIH-1 ha sido modificado con la introducción
de la terapia antirretroviral de gran actividad o TARGA (highly active antiretroviral
therapy – HAART), la cual es una combinación de al menos tres drogas que bloquean
12

diferentes pasos del ciclo de replicación viral (dos inhibidores de la transcriptasa
inversa y un inhibidor de la proteasa, por ejemplo) (Fig. 7), y que permite, en la
mayoría de los pacientes que la reciben, reducir significativamente la carga viral,
aumentar los recuentos de linfocitos T CD4+73 y disminuir tanto la morbilidad como la
mortalidad asociadas a la infección74. La recuperación, al menos parcial, de la
capacidad de respuesta del sistema inmunitario es evidente por la seguridad con que
pueden descontinuarse tratamientos profilácticos primarios o secundarios contra
encefalitis toxoplásmica, neumonía por Pneumocistis jirovecii, o retinitis por
Citomegalovirus75, 76. Sin embargo, y a pesar de los indudables beneficios que le
brindan al paciente, la TARGA no logra erradicar al VIH-1, pudiendo evidenciarse ARN
viral en pacientes con cargas virales “indetectables”77, y observándose
concentraciones de carga viral similares a las exhibidas antes del tratamiento
antirretroviral en pacientes que lo interrumpen después de años de supresión viral
exitosa78. Así, existen reservorios de replicación viral activa que persisten en presencia
de la terapia antirretroviral.
C. Inmunopatología de la infección por VIH-1
La interacción entre el VIH-1 y el sistema inmunitario que conduce a la pérdida de
control de múltiples agentes patógenos y a la aparición de neoplasias oportunistas, se
ha denominado inmunopatogénesis del SIDA79. Si bien unas pocas semanas luego de la
infección primaria la respuesta inmunitaria induce una significativa disminución de la
carga viral, la contención del virus es incompleta25, de manera que la replicación viral
persiste durante todas las etapas de la infección63-65, provocando la pérdida de la
población de linfocitos T CD4+, que ocurre en su mayor parte durante la etapa
clínicamente asintomática25. Son múltiples los mecanismos que se han propuesto para
explicar la disminución de la población de células T CD4+, entre los cuales se pueden
mencionar:
Destrucción de células infectadas por efectos citopáticos del VIH-1: desde los
primeros estudios, que condujeron a la identificación del VIH-1 como el agente
causal del SIDA20-22, se ha reconocido la capacidad que tiene el virus para
destruir a las células T CD4+ infectadas. Estudios posteriores han hecho
evidente que la mayor destrucción de estas células tiene lugar en el tejido
linfoide asociado a la mucosa gastrointestinal80-82.
Disminución de la vida media de células T: durante la infección por VIH-1 la vida
media de los linfocitos T CD4+ (también los T CD8+) está significativamente
disminuida83-85. En el caso de la población T CD8+, un mayor recambio celular
(turnover)84, 85 podría explicar el aumento de esta población en las etapas
iniciales de la infección. Por su parte, entre los factores contribuyen a la
disminución de la vida media celular, están el aumento de la susceptibilidad a
13

sufrir apoptosis y los defectos en la recepción de señales de supervivencia (ver
más adelante).
Disminución de la producción de nuevas células T: la regeneración de linfocitos T
está disminuida durante la infección por VIH-184, 85. Entre los factores que
favorecen esta disminución están la pérdida de la arquitectura y microambiente
de los nódulos linfáticos86, 87, la disminución de la funcionalidad tímica, con
involución avanzada y menor número de timocitos88, 89, y los defectos en la
producción de precursores de médula ósea90, 91.
Aumento de la susceptibilidad a sufrir apoptosis: los linfocitos T de los pacientes
infectados con VIH-1, incluso en la etapa asintomática de la infección, son más
propensos a sufrir apoptosis92, 93, tanto en forma espontánea, como al ser
activados por diversos estímulos94, 95. La proporción de células que sufren
apoptosis es mayor a medida que la infección progresa95. Los linfocitos T de
los pacientes infectados con VIH-1 poseen una expresión incrementada de
CD9595, 96 (un receptor que activa vías de señalización asociadas a
apoptosis)97 y un aumento de la susceptibilidad a sufrir apoptosis mediada
por este receptor96, susceptibilidad que está positivamente correlacionada
con la progresión de la infección95, 96, 98.
Disminución en la captación de señales de supervivencia: la IL-7 es un factor clave
en la homeostasis de los linfocitos T, al favorecer la maduración ontogénica de
timocitos, así como la proliferación y sobrevida de células T en la periferia99, 100.
La IL-7 interacciona con las células a través de la molécula IL-7Rα, la cual forma
un heterodímero con CD132, una cadena común a varios receptores de
citoquinas101. En los pacientes infectados con VIH-1 se ha descrito una
disminución de la expresión de IL-7Rα en los linfocitos T, la cual se ha
relacionado con la pérdida de la población CD4+102.
Aumento de recepción de señales de co-estimulación negativa: múltiples
moléculas de superficie pueden transmitir señales reguladoras tanto
positivas como negativas a las células T. El receptor de membrana PD-1
(Programmed Death–1) se ha relacionado con regulación negativa de la
respuesta inmunitaria, con especial implicación en el mantenimiento de la
tolerancia periférica103, 104. En el contexto de la infección por VIH-1 se ha
descrito un aumento de la expresión de PD-1 en las células T, especialmente
aquellas específicas para el virus105-107, lo cual se correlaciona con menor
sobrevida celular y mayor susceptibilidad a sufrir apoptosis105, además de
disminución de la proliferación celular y de la producción de citoquinas en
respuesta a la estimulación antigénica. Con respecto a este último efecto, es
resaltante que la infección por VIH-1 se asocia con la alteración funcional de los
linfocitos T, aun en etapas asintomáticas79, 108-110. La expresión de PDL-1, uno
14

de los ligandos de PD-1, también está incrementada el contexto de la infección
por VIH-1111.
Activación crónica del sistema inmunitario: Un mecanismo que se ha
relacionado tanto con la alteración numérica como funcional de los linfocitos
T en los pacientes infectados con VIH-1, es la activación crónica del sistema
inmunitario112, 113, la cual es prácticamente una característica patognomónica
de una infección progresiva79, y uno los mayores predictores de sobrevida
corta114. La replicación viral persistente, unida a la alta tasa de mutación,
resulta no solo en una presentación antigénica mantenida, sino también en la
constante variación antigénica de la población viral, lo cual provoca una
continua estimulación del sistema inmunitario, que puede llevar a su
agotamiento115. La masiva destrucción de linfocitos T CD4+ que se verifica en
la mucosa gastrointestinal80-82, también se asocia con daño en la propia
mucosa79, lo que puede permitir el paso de productos microbianos, como el
lipopolisacárido (LPS), a la circulación general, contribuyendo a perpetuar la
activación inmunitaria116. Asimismo, los linfocitos T activados son más
susceptibles de ser infectados por el VIH-1, por lo que se favorecería la
replicación viral79. Las células T activadas, además de tener defectos
funcionales92, son más propensa a sufrir apoptosis94, 95. HLA-DR y CD38 son
dos moléculas que se han utilizado para identificar a los linfocitos T activados
en el contexto de la infección por VIH-1, pues su expresión está incrementada
en estas células95, 117, 118. Boasso y Shearer, reflexionando sobre el papel de la
activación crónica en la patogénesis de la infección por VIH-1, comparan el
desenlace de distintas infecciones virales119. En el caso de las infecciones
exitosamente controladas (Fig. 9A), la replicación viral es inicialmente
contenida por acción de la respuesta inmunitaria innata, con mecanismos que
incluyen la producción moléculas de interferón tipo I (IFN-α e IFN-β), los
cuales favorecen el establecimiento de una respuesta inmunitaria específica,
que reemplaza a la respuesta innata y erradica al virus invasor. Una vez
resuelta la infección, la respuesta adaptativa vuelve a sus niveles basales. En
infecciones por el Virus de Inmunodeficiencia Simia (VIS) en hospedadores
naturalmente resistentes (chimpancés y mangabeyes grises), aunque la
respuesta inmunitaria no es capaz de erradicar al virus, no se observa
activación crónica, ni pérdida de funcionalidad inmunitaria, y la infección no
causa efectos patológicos a pesar de encontrarse valores persistentemente
altos de carga viral (Fig. 9B). El cuadro cambia en el contexto de infecciones
por VIH-1 y SIV en hospedadores susceptibles (hombre y macaco,
respectivamente), en donde, además de observarse valores persistentemente
altos de carga viral, el sistema inmunitario, tanto en su componente innato
como en de la inmunidad adquirida, evidencia un estado de activación
15
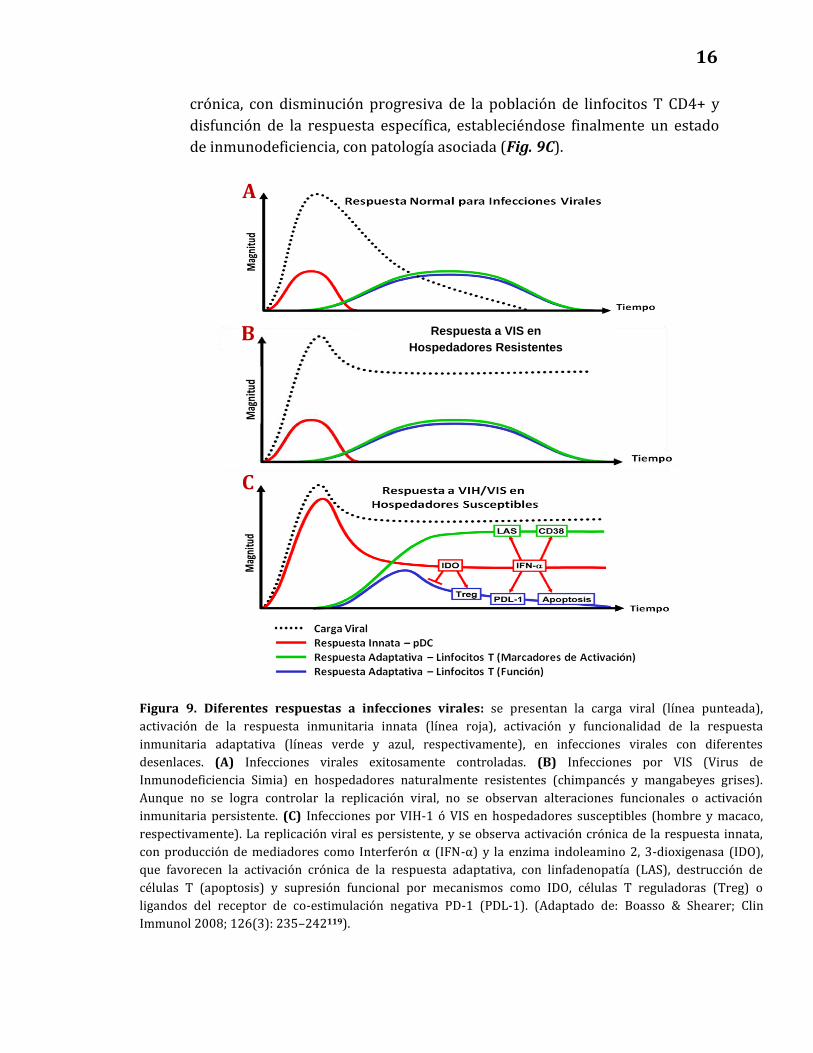
crónica, con disminución progresiva de la población de linfocitos T CD4+ y
disfunción de la respuesta específica, estableciéndose finalmente un estado
de inmunodeficiencia, con patología asociada (Fig. 9C).
A
Respuesta a VIS en
Hospedadores Resistentes B
C
Figura 9. Diferentes respuestas a infecciones virales: se presentan la carga viral (línea punteada),
activación de la respuesta inmunitaria innata (línea roja), activación y funcionalidad de la respuesta
inmunitaria adaptativa (líneas verde y azul, respectivamente), en infecciones virales con diferentes
desenlaces. (A) Infecciones virales exitosamente controladas. (B) Infecciones por VIS (Virus de
Inmunodeficiencia Simia) en hospedadores naturalmente resistentes (chimpancés y mangabeyes grises).
Aunque no se logra controlar la replicación viral, no se observan alteraciones funcionales o activación
inmunitaria persistente. (C) Infecciones por VIH-1 ó VIS en hospedadores susceptibles (hombre y macaco,
respectivamente). La replicación viral es persistente, y se observa activación crónica de la respuesta innata,
con producción de mediadores como Interferón α (IFN-α) y la enzima indoleamino 2, 3-dioxigenasa (IDO),
que favorecen la activación crónica de la respuesta adaptativa, con linfadenopatía (LAS), destrucción de
células T (apoptosis) y supresión funcional por mecanismos como IDO, células T reguladoras (Treg) o
ligandos del receptor de co-estimulación negativa PD-1 (PDL-1). (Adaptado de: Boasso & Shearer; Clin
Immunol 2008; 126(3): 235–242119).
16

D. Infección del sistema nervioso central por VIH-1
El VIH-1 es un virus neurotrópico120, 121 que ingresa en el sistema nervioso central
(SNC) poco después de iniciada la infección sistémica122, 123. En un número importante
de pacientes, la infección por VIH-1 se asocia con trastornos sensitivos, motores,
cognitivos y conductuales de diferente grado, lo que se ha denominado demencia
asociada al VIH (HAD, HIV-associated dementia)124, correlacionándose el inicio y la
severidad de los síntomas con altas concentraciones de carga viral. La introducción de
la terapia antirretroviral de gran actividad (TARGA) ha permitido disminuir las formas
más graves de HAD. Sin embargo, debido a que no todas las drogas antirretrovirales
ingresan eficientemente al cerebro y, como ya se ha mencionado, aún en presencia de
TARGA la replicación viral persiste (aunque a muy bajos niveles); se ha dado paso a
una forma más sutil de disfunción del SNC, la enfermedad cognitiva motora menor
(MCMD, minor cognitive motor disorder), en la cual la pérdida de la memoria y la
disminución de las habilidades de cálculo y otras funciones corticales superiores es
mucho menos pronunciada124.
Para ingresar al SNC, el VIH-1 debe atravesar la barrera hematoencefálica, una
capa selectivamente permeable, formada por las células endoteliales de la
microvasculatura cerebral, las cuales se conectan unas con otras por medio de uniones
estrechas, regulando el paso de células y sustancias. Se ha propuesto que el VIH-1
invade el cerebro por medio de monocitos infectados que ingresan como parte del
recambio normal de macrófagos perivasculares (hipótesis del caballo de Troya)124-126.
Los macrófagos perivasculares y las células de la microglía son las poblaciones
celulares del sistema nervioso central que expresan tanto el receptor (CD4) como los
correceptores (CXCR4 y CCR5) para el VIH-1124, y son productivamente infectadas por
el virus120, 127, 128. Por su parte, las neuronas, los astrocitos, los oligodendrocitos y las
células endoteliales de la microvasculatura cerebral expresan los receptores de
quimioquina CXCR4 y CCR5, pero no la molécula receptora CD4, y no son
productivamente infectados124. Se ha descrito, sin embargo, una limitada presencia del
virus en astrocitos120, 129, 130, no asociada a replicación viral productiva, pero sí
implicada en la patogénesis de la HAD124, 129 y la propagación del virus a células no
infectadas130. Los linfocitos T CD4+ también ingresan al parénquima cerebral como
parte de los mecanismos de vigilancia inmunitaria131, 132, pero su papel en la
producción local de virus no está claramente establecida124.
Debido a que el VIH-1 no infecta productivamente a las neuronas, otros
mecanismos deben estar implicados en los efectos neurotóxicos asociados al virus. La
infección de macrófagos perivasculares y células de la microglía, así como la presencia
del virus en astrocitos, puede causar alteración del microambiente y la funcionalidad
del SNC por mecanismos directos (mediados por proteínas virales) e indirectos
(mediados por mecanismos inflamatorios). La gp120 viral, por ejemplo, puede
17
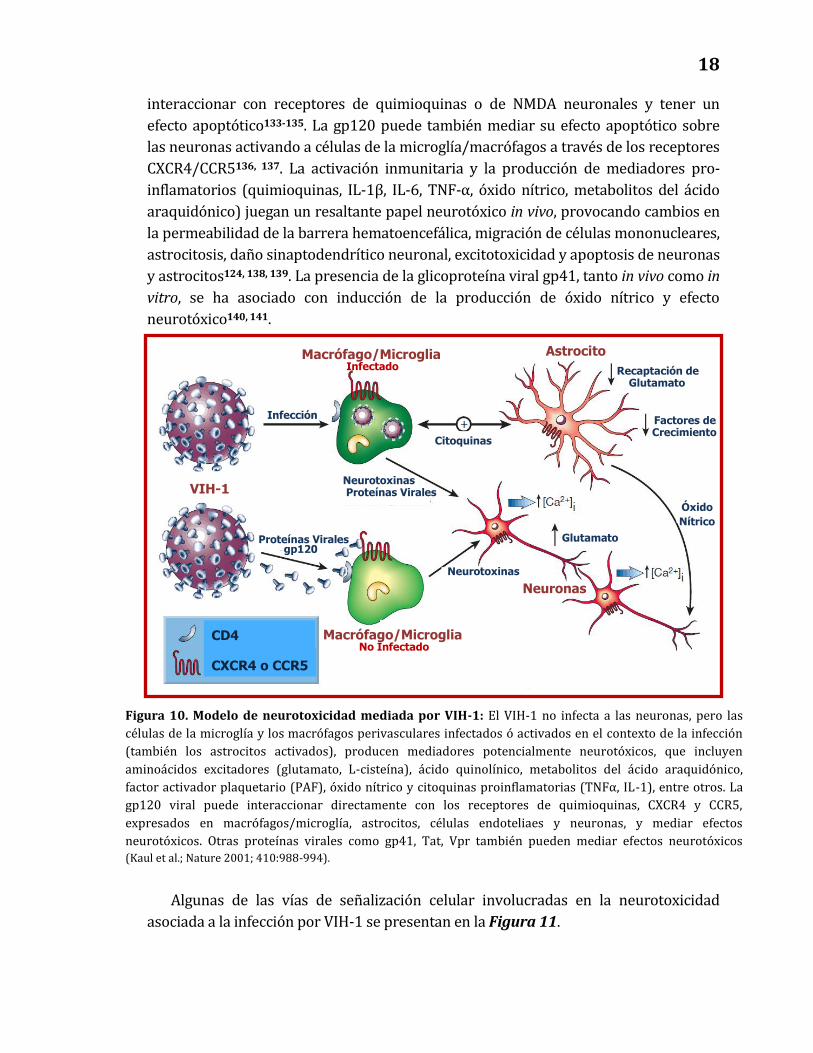
interaccionar con receptores de quimioquinas o de NMDA neuronales y tener un
efecto apoptótico133-135. La gp120 puede también mediar su efecto apoptótico sobre
las neuronas activando a células de la microglía/macrófagos a través de los receptores
CXCR4/CCR5136, 137. La activación inmunitaria y la producción de mediadores pro-
inflamatorios (quimioquinas, IL-1β, IL-6, TNF-α, óxido nítrico, metabolitos del ácido
araquidónico) juegan un resaltante papel neurotóxico in vivo, provocando cambios en
la permeabilidad de la barrera hematoencefálica, migración de células mononucleares,
astrocitosis, daño sinaptodendrítico neuronal, excitotoxicidad y apoptosis de neuronas
y astrocitos124, 138, 139. La presencia de la glicoproteína viral gp41, tanto in vivo como in
vitro, se ha asociado con inducción de la producción de óxido nítrico y efecto
neurotóxico140, 141.
Algunas de las vías de señalización celular involucradas en la neurotoxicidad
asociada a la infección por VIH-1 se presentan en la Figura 11.
VIH-1
Macrófago/Microglia Infectado
Macrófago/Microglia No Infectado
Astrocito
Neuronas
Infección
Citoquinas
Neurotoxinas Proteínas Virales
Neurotoxinas
Glutamato gp120
Proteínas Virales
Óxido
Nítrico
Glutamato Recaptación de
Crecimiento Factores de
CD4
CXCR4 o CCR5
Figura 10. Modelo de neurotoxicidad mediada por VIH-1: El VIH-1 no infecta a las neuronas, pero las
células de la microglía y los macrófagos perivasculares infectados ó activados en el contexto de la infección
(también los astrocitos activados), producen mediadores potencialmente neurotóxicos, que incluyen
aminoácidos excitadores (glutamato, L-cisteína), ácido quinolínico, metabolitos del ácido araquidónico,
factor activador plaquetario (PAF), óxido nítrico y citoquinas proinflamatorias (TNFα, IL-1), entre otros. La
gp120 viral puede interaccionar directamente con los receptores de quimioquinas, CXCR4 y CCR5,
expresados en macrófagos/microglía, astrocitos, células endoteliaes y neuronas, y mediar efectos
neurotóxicos. Otras proteínas virales como gp41, Tat, Vpr también pueden mediar efectos neurotóxicos
(Kaul et al.; Nature 2001; 410:988-994).
18
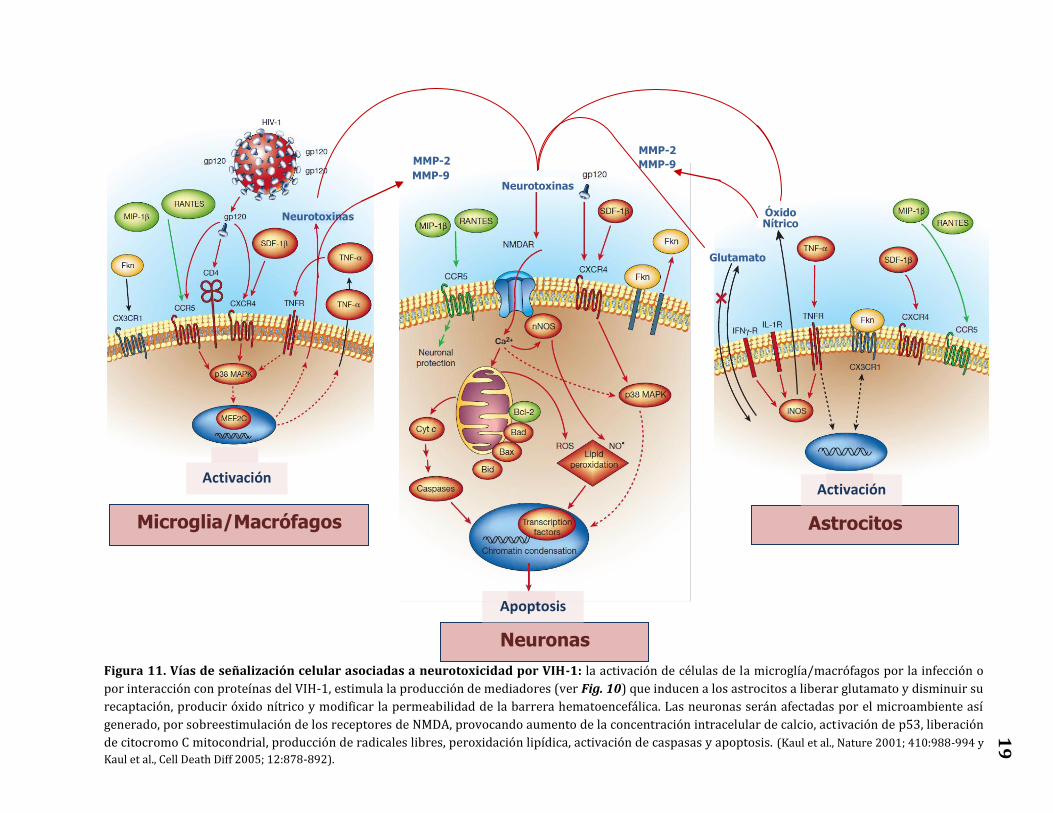
Figura 11. Vías de señalización celular asociadas a neurotoxicidad por VIH-1: la activación de células de la microglía/macrófagos por la infección o
por interacción con proteínas del VIH-1, estimula la producción de mediadores (ver Fig. 10) que inducen a los astrocitos a liberar glutamato y disminuir su
recaptación, producir óxido nítrico y modificar la permeabilidad de la barrera hematoencefálica. Las neuronas serán afectadas por el microambiente así
generado, por sobreestimulación de los receptores de NMDA, provocando aumento de la concentración intracelular de calcio, activación de p53, liberación
de citocromo C mitocondrial, producción de radicales libres, peroxidación lipídica, activación de caspasas y apoptosis. (Kaul et al., Nature 2001; 410:988-994 y
Kaul et al., Cell Death Diff 2005; 12:878-892).
Microglia/Macrófagos
Neuronas
Astrocitos
Apoptosis
Activación Activación
Neurotoxinas
Neurotoxinas
Glutamato
Óxido Nítrico
MMP-9 MMP-9 MMP-2 MMP-2
19

Además de la encefalopatía ocasionada por el VIH-1 y descrita en los párrafos
anteriores, las infecciones oportunistas del SNC son una complicación común en los
pacientes en etapa de SIDA, y en 10 a 20% de los pacientes la enfermedad neurológica
es la primera manifestación de su infección por VIH-1142. Una de estas afecciones es la
encefalitis toxoplásmica, cuyo agente causal es Toxoplasma gondii, que a continuación
estudiaremos en más detalle.
II. Infección por Toxoplasma gondii
Toxoplasma gondii es un protozoario ubicuo, parásito de animales y seres
humanos, descubierto en 1908 por Nicolle y Manceaux143, que pertenece al Phylum
Apicomplexa, clase Sporozoa, subclase Coccidia144.
La infección por T. gondii en seres humanos inmunocompetentes es
generalmente asintomática o de manifestación benigna y autolimitada145. En
contraste, la infección congénita y en individuos inmunocomprometidos puede
dejar serias secuelas y causar la muerte145. En pacientes con SIDA, T. gondii se
comporta como un patógeno oportunista, afectando especialmente al SNC, siendo la
causa más frecuente de lesiones focales intracerebrales en estos pacientes142, 145. La
encefalitis toxoplásmica en pacientes con SIDA es casi siempre el resultado de la
reactivación de una infección latente, como resultado del deterioro progresivo de la
función inmunitaria celular142, 146.
A. Epidemiología
En los seres humanos la seroprevalencia de anticuerpos específicos contra T.
gondii se incrementa con la edad, no presenta diferencias significativas entre hombres
y mujeres, y es menor en regiones frías, en zonas áridas y calientes, y en áreas situadas
a grandes alturas147. Si bien la infección está ampliamente distribuida a nivel mundial,
la prevalencia varía entre distintas poblaciones y localizaciones geográficas. Así, se ha
descrito una seroprevalencia de 10-12% en individuos de 6-49 años de edad en
Estados Unidos148; 14-26% en mujeres embarazadas de Suecia149; 42-48% en
Colombia150 y 65-69% en Brasil151, también en mujeres embarazadas.
La prevalencia de anticuerpos contra T. gondii en la población venezolana ha sido
reportada en 23-65% en individuos de 1-65 años152-154; 41-61% en mujeres
embarazadas155, 156; 37-79% en niños de 0-15 años157 y 38-88% en población
indígena/autóctona158, 159.
B. Estructura y ciclo de vida de T. gondii
Toxoplasma gondii es un parásito intracelular obligado que existe en tres formas:
ooquiste, taquizoíto y bradizoíto145, 147, 160.
Ooquiste: es la forma parasitaria resistente al medio ambiente y
diseminada por los felinos (hospedadores definitivos de T. gondii, en cuyo
intestino ocurre la reproducción sexual del parásito) en sus heces. Los
20
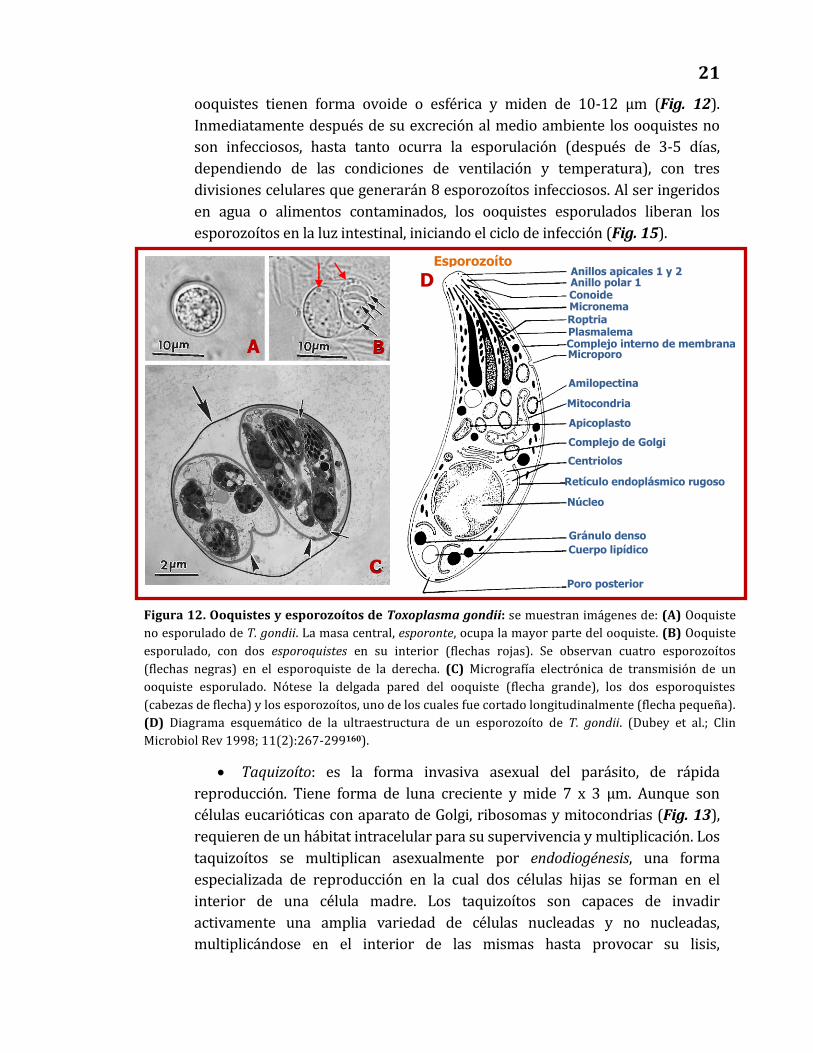
ooquistes tienen forma ovoide o esférica y miden de 10-12 µm (Fig. 12).
Inmediatamente después de su excreción al medio ambiente los ooquistes no
son infecciosos, hasta tanto ocurra la esporulación (después de 3-5 días,
dependiendo de las condiciones de ventilación y temperatura), con tres
divisiones celulares que generarán 8 esporozoítos infecciosos. Al ser ingeridos
en agua o alimentos contaminados, los ooquistes esporulados liberan los
esporozoítos en la luz intestinal, iniciando el ciclo de infección (Fig. 15).
Taquizoíto: es la forma invasiva asexual del parásito, de rápida
reproducción. Tiene forma de luna creciente y mide 7 x 3 µm. Aunque son
células eucarióticas con aparato de Golgi, ribosomas y mitocondrias (Fig. 13),
requieren de un hábitat intracelular para su supervivencia y multiplicación. Los
taquizoítos se multiplican asexualmente por endodiogénesis, una forma
especializada de reproducción en la cual dos células hijas se forman en el
interior de una célula madre. Los taquizoítos son capaces de invadir
activamente una amplia variedad de células nucleadas y no nucleadas,
multiplicándose en el interior de las mismas hasta provocar su lisis,
A B
C
D Anillos apicales 1 y 2
Conoide
Roptria
Microporo
Amilopectina
Apicoplasto
Centriolos
Núcleo
Poro posterior
Gránulo denso
Cuerpo lipídico
Retículo endoplásmico rugoso
Complejo de Golgi
Mitocondria
Complejo interno de membrana
Micronema
Anillo polar 1
Plasmalema
Esporozoíto
Figura 12. Ooquistes y esporozoítos de Toxoplasma gondii: se muestran imágenes de: (A) Ooquiste
no esporulado de T. gondii. La masa central, esporonte, ocupa la mayor parte del ooquiste. (B) Ooquiste
esporulado, con dos esporoquistes en su interior (flechas rojas). Se observan cuatro esporozoítos
(flechas negras) en el esporoquiste de la derecha. (C) Micrografía electrónica de transmisión de un
ooquiste esporulado. Nótese la delgada pared del ooquiste (flecha grande), los dos esporoquistes
(cabezas de flecha) y los esporozoítos, uno de los cuales fue cortado longitudinalmente (flecha pequeña).
(D) Diagrama esquemático de la ultraestructura de un esporozoíto de T. gondii. (Dubey et al.; Clin
Microbiol Rev 1998; 11(2):267-299160).
21
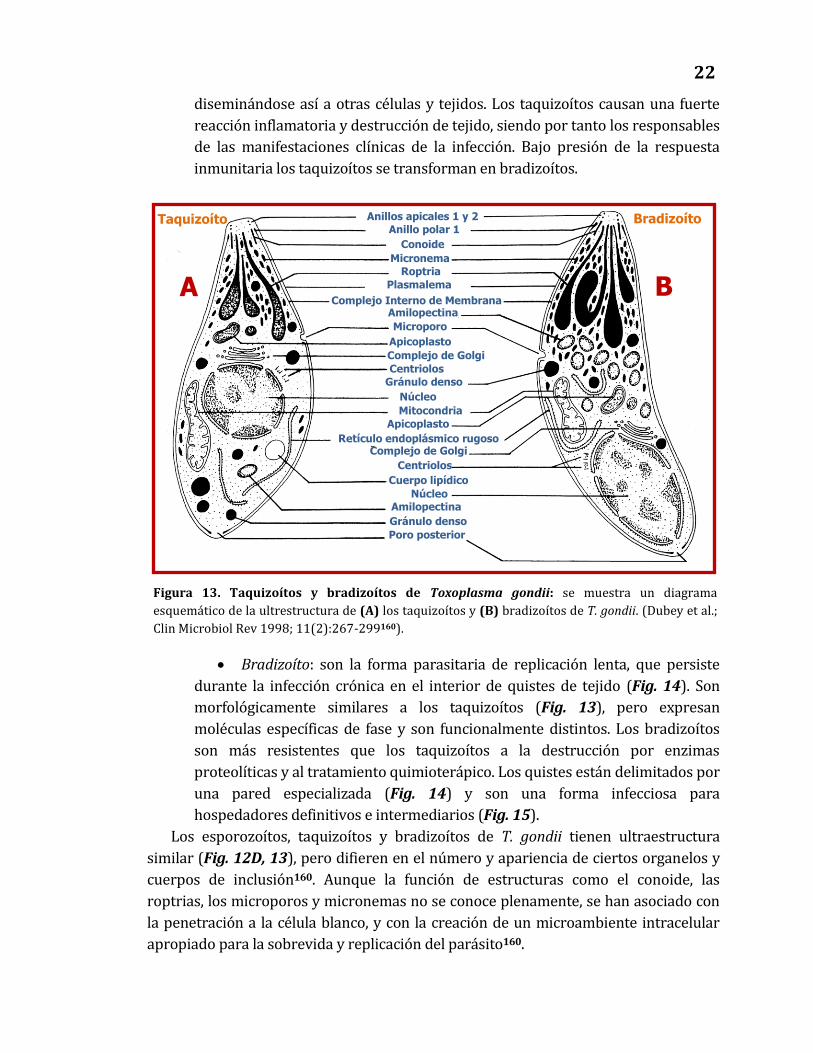
diseminándose así a otras células y tejidos. Los taquizoítos causan una fuerte
reacción inflamatoria y destrucción de tejido, siendo por tanto los responsables
de las manifestaciones clínicas de la infección. Bajo presión de la respuesta
inmunitaria los taquizoítos se transforman en bradizoítos.
Bradizoíto: son la forma parasitaria de replicación lenta, que persiste
durante la infección crónica en el interior de quistes de tejido (Fig. 14). Son
morfológicamente similares a los taquizoítos (Fig. 13), pero expresan
moléculas específicas de fase y son funcionalmente distintos. Los bradizoítos
son más resistentes que los taquizoítos a la destrucción por enzimas
proteolíticas y al tratamiento quimioterápico. Los quistes están delimitados por
una pared especializada (Fig. 14) y son una forma infecciosa para
hospedadores definitivos e intermediarios (Fig. 15).
Los esporozoítos, taquizoítos y bradizoítos de T. gondii tienen ultraestructura
similar (Fig. 12D, 13), pero difieren en el número y apariencia de ciertos organelos y
cuerpos de inclusión160. Aunque la función de estructuras como el conoide, las
roptrias, los microporos y micronemas no se conoce plenamente, se han asociado con
la penetración a la célula blanco, y con la creación de un microambiente intracelular
apropiado para la sobrevida y replicación del parásito160.
Figura 13. Taquizoítos y bradizoítos de Toxoplasma gondii: se muestra un diagrama
esquemático de la ultrestructura de (A) los taquizoítos y (B) bradizoítos de T. gondii. (Dubey et al.;
Clin Microbiol Rev 1998; 11(2):267-299160).
Conoide
Roptria
Microporo
Núcleo
Cuerpo lipídico
Centriolos
Núcleo
Micronema
Anillos apicales 1 y 2
Anillo polar 1
Plasmalema
Apicoplasto
Apicoplasto
Complejo Interno de Membrana
Centriolos
Complejo de Golgi
Gránulo denso
Gránulo denso
Retículo endoplásmico rugoso
Poro posterior
Amilopectina
Amilopectina
Mitocondria
Complejo de Golgi
Taquizoíto
B A
Bradizoíto
22

Las principales formas de transmisión de T. gondii a los seres humanos son los
quistes de tejido (que contienen bradizoítos) y los ooquistes esporulados (que
contienen esporozoítos). La infección se inicia generalmente por vía oral, por la
ingestión de carne cruda o mal cocida (especialmente cerdo y cordero) que
contenga quistes, o de agua o alimentos contaminados con ooquistes. Una vez
ingeridos, los quistes u ooquistes sufren la degradación enzimática de sus paredes
externas, permitiendo la liberación a la luz intestinal de las formas infecciosas
(bradizoítos y esporozoítos, respectivamente) que rápidamente invadirán y se
multiplicarán en las células intestinales, donde se convertirán en taquizoítos. A
partir de ese momento, la propagación de los taquizoítos ocurrirá por lisis de las
células infectadas, seguida de invasión de células vecinas, o vía sanguínea/linfática,
especialmente en el interior de células fagocíticas, para alcanzar tejidos más
distantes, resultando en amplia diseminación del parásito. T. gondii puede infectar
virtualmente cualquier célula o tejido del organismo. La aparición de una respuesta
inmunitaria contra el parásito se asocia con la interrupción de la replicación e
invasión de los taquizoítos, que se convertirán en bradizoítos, induciéndose la
A B C
D E F
Figura 14. Quistes de Toxoplasma gondii en cerebro de ratón: (A) Quiste con 3 bradizoítos, cada
uno con un núcleo terminal (flechas). Nótese la delgada pared del quiste (cabeza de flecha). (B) Tres
quistes con pared bien definida (cabeza de flecha). Uno de los quistes contiene dos bradizoítos, cada
uno con núcleo terminal (flechas). (C) Sección de un quiste intracelular. Nótese la delgada pared del
quiste (flecha) y el núcleo de la célula hospedadora (cabeza de flecha). (D) Quiste de tejido con
numerosos bradizoítos, positivos para tinción de PAS (cabezas de flecha), rodeados por la pared del
quiste, negativa para tinción de PAS (flecha). (E) Imagen en fresco de un quiste liberado de cerebro de
ratón. Nótese la pared del quiste (flecha) rodeando a cientos de bradizoítos. (F) Micrografía electrónica
de transmisión, mostrando un quiste en el cerebro de un ratón, inoculado 8 meses antes con ooquistes
de T. gondii, cepa VEG. Esta sección ultrafina muestra la pared del quiste (flecha negra), con
aproximadamente 110 bradizoítos en su interior (flechas rojas). El quiste se encuentra en el interior de
una célula hospedadora, cuyo núcleo se observa en la parte superior izquierda de la micrografía (flecha
azul). (Dubey et al.; Clin Microbiol Rev 1998; 11(2):267-299160).
23

formación de quistes. Si bien los quistes se pueden formar en cualquier órgano,
éstos son más prevalentes en tejido nervioso y muscular, incluyendo cerebro, ojo,
músculo esquelético y cardíaco160, 161.
Si una mujer se infecta con T. gondii durante el embarazo o poco antes de la
concepción, la placenta puede ser invadida por el parásito durante la fase de rápida
proliferación, resultando en la infección del embrión/feto145, 147. Otras fuentes de
infección en humanos son las transfusiones sanguíneas162, el transplante de
órganos163, 164, y la inoculación accidental en laboratorios165-167.
C. Respuesta inmunitaria contra T. gondii
El protozoario T. gondii causa infecciones asintomáticas en la mayoría de sus
hospedadores inmunocompetentes, debido, en gran parte, a la rápida inducción de
una fuerte respuesta inmunitaria celular168, que resulta en el control de la replicación
Hospedador definitivo Reproducción sexual del parásito
Hospedador intermediario Portadores
Infección en humanos Horizontal y vertical
Bradizoítos Quistes
en tejidos
Bradizoítos Quistes
en tejidos Bradizoítos
Quistes en tejidos
Heces Liberado en
Esporulación En el medio ambiente
Contaminación Suelo, agua, alimentos
Ooquiste No esporulado
Ooquiste Esporulado
Esporozoítos Ooquistes
s
Taquizoítos Placenta
Figura 15. Ciclo de vida de Toxoplasma gondii: Los taquizoítos, bradizoítos y esporozoítos son las tres
formas infecciosas de T. gondii. Los seres humanos y animales se infectan primordialmente por la
ingestión de bradizoítos u ooquistes. Una vez ingeridos, los bradizoítos y esporozoítos se transforman
en taquizoítos, invadiendo los tejidos. La interconversión de taquizoítos a bradizoítos (al pasar una
infección de fase aguda a crónica) y de bradizoítos a taquizoítos (al reactivarse una infección crónica)
tiene importancia tanto biológica como clínica. Los felinos son los hospedadores definitivos, en cuyo
intestino la reproducción sexual del parásito tiene lugar. En humanos la infección puede transmitirse
verticalmente, de una madre con infección activa a su embrión/feto. (Modificado de: The Queen's Printer's
of Ontario; disponible en: http://www.omafra.gov.on.ca/english/livestock/swine/facts/04-055.htm).
24
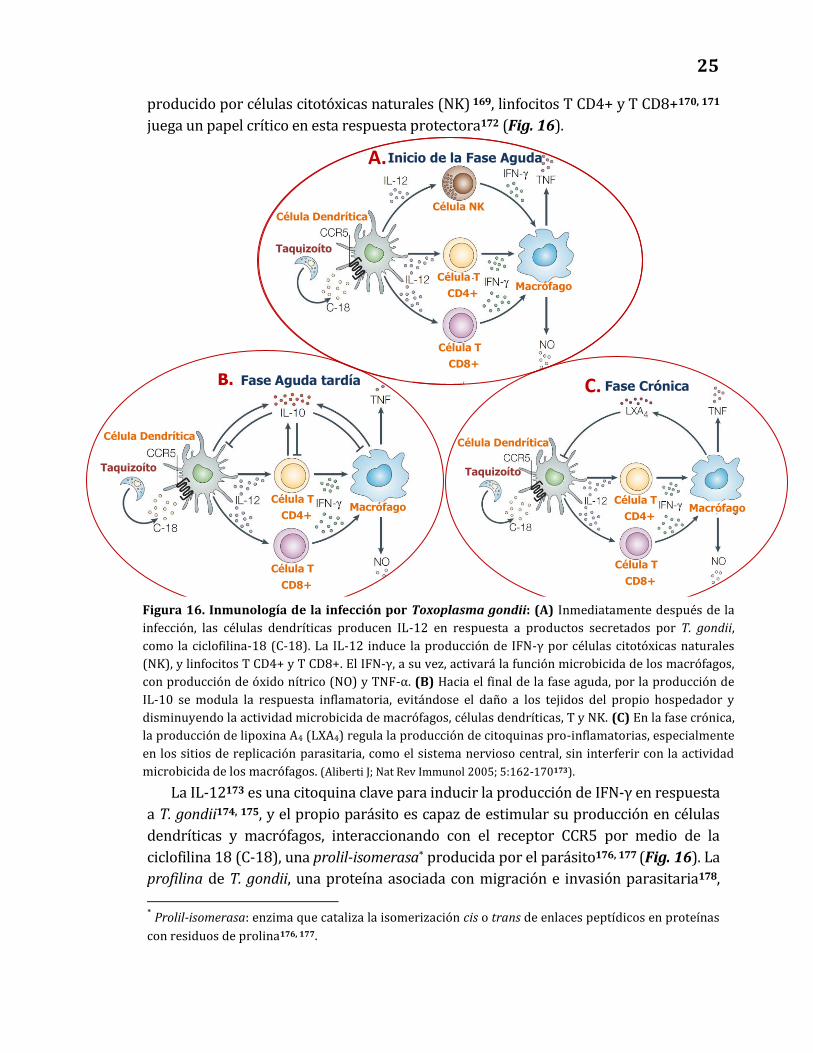
producido por células citotóxicas naturales (NK) 169, linfocitos T CD4+ y T CD8+170, 171
juega un papel crítico en esta respuesta protectora172 (Fig. 16).
La IL-12173 es una citoquina clave para inducir la producción de IFN-γ en respuesta
a T. gondii174, 175, y el propio parásito es capaz de estimular su producción en células
dendríticas y macrófagos, interaccionando con el receptor CCR5 por medio de la
ciclofilina 18 (C-18), una prolil-isomerasa* producida por el parásito176, 177 (Fig. 16). La
profilina de T. gondii, una proteína asociada con migración e invasión parasitaria178,
* Prolil-isomerasa: enzima que cataliza la isomerización cis o trans de enlaces peptídicos en proteínas
con residuos de prolina176, 177.
25
A. Inicio de la Fase Aguda
Taquizoíto
Célula Dendrítica Célula NK
Célula T
CD4+
Célula T
CD8+
Macrófago
B. Fase Aguda tardía
Taquizoíto
Célula Dendrítica
Célula T
CD4+
Célula T
CD8+
Macrófago
Taquizoíto
C. Fase Crónica
Macrófago
Célula Dendrítica
Célula T
CD4+
Célula T
CD8+
A.
C.
Figura 16. Inmunología de la infección por Toxoplasma gondii: (A) Inmediatamente después de la
infección, las células dendríticas producen IL-12 en respuesta a productos secretados por T. gondii,
como la ciclofilina-18 (C-18). La IL-12 induce la producción de IFN-γ por células citotóxicas naturales
(NK), y linfocitos T CD4+ y T CD8+. El IFN-γ, a su vez, activará la función microbicida de los macrófagos,
con producción de óxido nítrico (NO) y TNF-α. (B) Hacia el final de la fase aguda, por la producción de
IL-10 se modula la respuesta inflamatoria, evitándose el daño a los tejidos del propio hospedador y
disminuyendo la actividad microbicida de macrófagos, células dendríticas, T y NK. (C) En la fase crónica,
la producción de lipoxina A4 (LXA4) regula la producción de citoquinas pro-inflamatorias, especialmente
en los sitios de replicación parasitaria, como el sistema nervioso central, sin interferir con la actividad
microbicida de los macrófagos. (Aliberti J; Nat Rev Immunol 2005; 5:162-170173).

puede también inducir la producción de IL-12 por su interacción con los receptores
tipo Toll179, 180 TLR11181 y TLR12182. La IL-12 activa la producción de IFN-γ por
células NK y linfocitos T, lo cual se requiere para inducir y mantener el control de la
infección aguda y crónica169-172. El IFN-γ, a su vez, activa la producción de óxido nítrico
(NO) por parte de macrófagos, el cual tiene un efecto microbicida y microbiostático
sobre el parásito intracelular183-185. El TNF-α también tiene participación resaltante en
la respuesta inmunitaria contra T. gondii186, 187. Sin embargo, a pesar de que se ponen
en acción mecanismos microbicidas potentes, la respuesta no logra una completa
eliminación del parásito168, y este persiste por largos períodos en forma de quistes en
los tejidos.
El control de la respuesta inflamatoria inducida por la infección parasitaria es de
suma importancia, y la IL-10 se ha identicado como un importante mediador de este
control, tanto en la fase aguda de la infección188, 189 como en la crónica 190 (Fig. 16B).
En modelos múridos, la ausencia de IL-10, o el bloqueo experimental de su actividad,
se asocia con exacerbada producción de IL-12, INF-γ, TNF-α, IL-1 e IL-6, es decir,
potenciación de la respuesta inflamatoria, lo que resulta en daño severo/necrosis de
los tejidos del hospedador (mucosa intestinal189, hígado188, 189, cerebro190),
provocando la muerte del animal infectado. La IL-10 también disminuye parcialmente
la actividad anti-parasitaria de la respuesta inmunitaria177, 190, sin embargo su efecto
neto sobre el hospedador se considera beneficioso/protector188-190.
Otro potente mediador anti-inflamatorio que se pone en acción durante la
infección por T. gondii es la lipoxina A4 (LXA4), eicosanoide derivado del ácido
araquidónico191, 192 (Fig. 16C), que inhibe la producción de IL-12 y por ende la de IFN-
γ, así como la expresión de CCR5 y la función migratoria de células dendríticas,
previniendo la histopatología asociada a una respuesta inflamatoria exacerbada. Si
bien las actividades de IL-10 y LXA4 se solapan parcialmente, éstas no son
completamente redundantes. Por ejemplo, en ratones deficientes de lipoxigenasa-5
(una enzima necesaria en la vía de síntesis de LXA4), el tratamiento con IL-10 no logra
rescatar a los animales de la mortalidad asociada a la infección por T. gondii, mientras
los análogos de LXA4 les permiten sobrevivir, evitando el extensivo daño a los
tejidos192. Por otra parte, la LXA4, a diferencia de la IL-10, no disminuye la actividad
microbicida de los macrófagos191. La LXA4, además, pareciera ejercer su papel
protector principalmente a nivel del cerebro, evitando la encefalitis asociada a la
infección parasitaria, especialmente en la fase crónica192. Asimismo, es resaltante que
T. gondii exprese una proteína con actividad de lipoxigenasa-15, lo que le permite
interaccionar con la vía de síntesis de LXA4, favoreciendo su producción193.
Así, por la presencia de proteínas como ciclofilina 18 y profilina, T. gondii puede
favorecer una potente respuesta inflamatoria aguda, evitando que su multiplicación
exagerada provoque la muerte de su hospedador, pero también, por moléculas como
la lipoxigenasa-15 parasitaria, puede modular la respuesta inflamatoria inicial,
26

evitando la inmunopatología que igualmente comprometería la sobrevida de su
hospedador. Además, la infección por T. gondii interfiere con la funcionalidad de
macrófagos194, 195, inhibiendo la producción de IL-12 y TNF-α, y aumentando la
producción de IL-10, inducida por la estimulación de TLR4 con lipopolisacárido (LPS)
bacteriano196. En conclusión, T. gondii es capaz de modular/modificar activamente la
respuesta inmunitaria197-199, lo cual se ha asociado con sobrevida, tanto del parásito
como del hospedador.
D. Inmunopatogénesis de la encefalitis toxoplásmica
Una vez que ingresa al organismo, T. gondii invade células circulantes, como
macrófagos y células dendríticas, y las utiliza como caballo de Troya para ganar acceso
a sitios privilegiados como el cerebro200-202, en el cual puede infectar una variedad de
células, que incluyen neuronas, astrocitos y células de la microglía203-207. En el cerebro,
en forma similar a la respuesta observada en la periferia, la proliferación de
taquizoítos es suprimida principalmente por una respuesta inmunitaria celular
dependiente de IFN-γ170-172, con una contribución secundaria de la respuesta
humoral208-210. Esto conduce al establecimiento de una infección crónica, clínicamente
asintomática*, induciéndose la formación de quistes que pueden persistir de por vida.
El IFN-γ es esencial para prevenir la reactivación y el desarrollo de la encefalitis
toxoplásmica171, 215, y sus principales productores son los linfocitos T que infiltran el
cerebro en respuesta a la infección216-218. Si bien macrófagos y células de la microglía
son también capaces de producir IFN-γ219, la presencia de células T es fundamental
para el establecimiento de una respuesta protectora220. Además del IFN-γ, durante la
infección cerebral por T. gondii se induce la producción, por parte de neuronas,
astrocitos y células de la microglía, de otras citoquinas que promueven (IL-1, IL-6,
TNF-α) o suprimen (IL-10, TGF-β) la respuesta inflamatoria221-223. En seres humanos,
el IFN-γ, el TNF-α y la IL-6, participan en la estimulación de las células de la microglía,
para que éstas inhiban la replicación intracelular de T. gondii224. Asimismo, el IFN-γ y
la IL-1β activan a los astrocitos para que éstos inhiban la replicación del parásito por
producción de óxido nítrico225. El IFN-γ y el TNF-α inducen en astrocitos y células de
glioblastoma humano la expresión de indoleamino 2, 3-dioxigenasa (IDO), una enzima
que tiene una fuerte actividad toxoplasmostática, por degradación del triptófano, un
aminoácido esencial para el parásito226. Aunque las células T son la población linfoide
predominante en el cerebro de animales infectados, también los linfocitos B227, las
células citotóxicas naturales (NK)219, los macrófagos219 y las células dendríticas201, 219
migran al cerebro en respuesta a la infección.
Como ya se ha mencionado, la respuesta inmunitaria es esencial para mantener la
latencia de la infección crónica. En modelos de infección crónica en roedores, los * Esta concepción de infección crónica asintomática está siendo reconsiderada211-214.
27

quistes intactos no se asocian con reacción inflamatoria local, el tejido alrededor del
quiste aparece indenme y el animal infectado no presenta sintomatología aparente160
(Fig. 17A). En estos animales la ruptura de quistes es un evento sumamente raro
(frecuencia aproximada: 0,27%), y se asocia a una rápida respuesta inmunitaria
celular, formando nódulos microgliales/inflamatorios, con infiltrado de macrófagos
que fagocitan y degradan a los parásitos liberados (Fig. 17B y C); mostrando que en
individuos inmunocompetentes la eventual diseminación de parásitos, con su
potencial daño a tejidos, es contenida por la respuesta inmunitaria228.
Por el contrario, cuando en estos modelos de roedores se induce inmunosupresión
por diversos tratamientos, el resultado es una pérdida de control de la infección
crónica, con rompimiento de las paredes de los quistes, reactivación de la replicación
parasitaria y patología asociada, con exacerbación de la respuesta inflamatoria y
necrosis del tejido cerebral (Fig. 18A), que puede causar la muerte del animal
infectado161, 162, 215, 229. En congruencia con estos modelos, los seres humanos que
presentan defectos en su respuesta inmunitaria celular, como el caso de pacientes con
enfermedades neoplásicas, incluyendo enfermedad de Hodgkin y Linfoma No Hodgkin,
así como aquellos que reciben terapia inmunosupresora, están en riesgo de presentar
reactivación de infecciones por T. gondii a nivel cerebral203, 230, 231. Asimismo, la
incidencia de encefalitis toxoplásmica está incrementada en pacientes infectados con
A
B
C 10 µm
Figura 17. Quistes cerebrales de Toxoplasma gondii: (A) Grupo de quistes de T. gondii (flechas rojas)
en el cerebro de un ratón que en vida no mostraba sintomatología. Nótese que las paredes de los quistes
están conservadas, que no hay infiltrado de células inflamatorias y el tejido cerebral está indemne. (B)
Micrografía de luz de un quiste roto (flechas) rodeado de células inflamatorias (x800). (C) Micrografía
electrónica del quiste roto mostrado en B, ilustrando los restos de la pared del quiste (flecha
anaranjada), numerosos parásitos (flechas rojas) y macrófagos (flechas azules) (x3000). (Dubey et al.; Clin
Microbiol Rev 1998; 11(2):267-299160; Fergunson DJ et al.; Parasitol Res 1989; 75:599-603228).
28
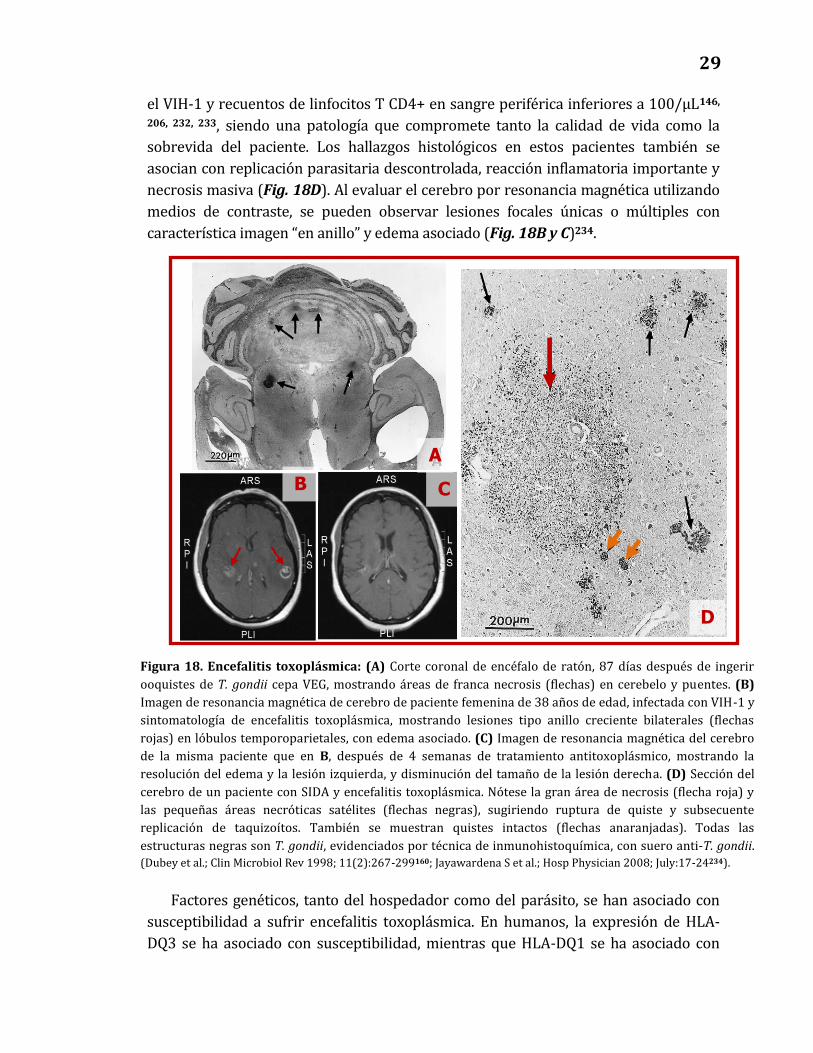
el VIH-1 y recuentos de linfocitos T CD4+ en sangre periférica inferiores a 100/µL146,
206, 232, 233, siendo una patología que compromete tanto la calidad de vida como la
sobrevida del paciente. Los hallazgos histológicos en estos pacientes también se
asocian con replicación parasitaria descontrolada, reacción inflamatoria importante y
necrosis masiva (Fig. 18D). Al evaluar el cerebro por resonancia magnética utilizando
medios de contraste, se pueden observar lesiones focales únicas o múltiples con
característica imagen “en anillo” y edema asociado (Fig. 18B y C)234.
Factores genéticos, tanto del hospedador como del parásito, se han asociado con
susceptibilidad a sufrir encefalitis toxoplásmica. En humanos, la expresión de HLA-
DQ3 se ha asociado con susceptibilidad, mientras que HLA-DQ1 se ha asociado con
A
B C
D
Figura 18. Encefalitis toxoplásmica: (A) Corte coronal de encéfalo de ratón, 87 días después de ingerir
ooquistes de T. gondii cepa VEG, mostrando áreas de franca necrosis (flechas) en cerebelo y puentes. (B)
Imagen de resonancia magnética de cerebro de paciente femenina de 38 años de edad, infectada con VIH-1 y
sintomatología de encefalitis toxoplásmica, mostrando lesiones tipo anillo creciente bilaterales (flechas
rojas) en lóbulos temporoparietales, con edema asociado. (C) Imagen de resonancia magnética del cerebro
de la misma paciente que en B, después de 4 semanas de tratamiento antitoxoplásmico, mostrando la
resolución del edema y la lesión izquierda, y disminución del tamaño de la lesión derecha. (D) Sección del
cerebro de un paciente con SIDA y encefalitis toxoplásmica. Nótese la gran área de necrosis (flecha roja) y
las pequeñas áreas necróticas satélites (flechas negras), sugiriendo ruptura de quiste y subsecuente
replicación de taquizoítos. También se muestran quistes intactos (flechas anaranjadas). Todas las
estructuras negras son T. gondii, evidenciados por técnica de inmunohistoquímica, con suero anti-T. gondii.
(Dubey et al.; Clin Microbiol Rev 1998; 11(2):267-299160; Jayawardena S et al.; Hosp Physician 2008; July:17-24234).
29

resistencia235. En cuanto al parásito, de los tres genotipos descritos236, las cepas
causantes de encefalitis toxoplásmica en pacientes inmunocomprometidos (infectados
o no con VIH-1) generalmente pertenecen al tipo II236-238.
III. Modelos in vitro para el estudio de neurotoxicidad
En las dos primeras secciones se ha referido que VIH-1 y T. gondii tienen la
capacidad de invadir el cerebro, causar infecciones crónicas y afectar la funcionalidad
del mismo. Son diversos los modelos in vitro que se han utilizado para estudiar los
mecanismos que se activan durante la infección con estos agentes y cómo éstos se
asocian con los efectos neurotóxicos observados.
Por ejemplo, para evaluar la inducción de apoptosis por la infección con diferentes
aislados de VIH-1, Ohagen et al. utilizaron cultivos primarios de células de cerebro de
feto humano, los cuales estaban formados por poblaciones mixtas de células nerviosas
que incluían astrocitos (70-90%), neuronas (10-30%), células de la microglía (1-5%) y
fibroblastos (1-5%). En este modelo las células de la microglía eran infectadas
productivamente por los aislados virales, y se pudo determinar que su capacidad de
inducir apoptosis estaba asociada con la molécula gp120 y su capacidad de unirse a los
correceptores CCR5 o CXCR4239. En otra aproximación, Koka et al., evaluaron efectos
neurotóxicos no dependientes de la infección viral propiamente (mecanismos
indirectos) utilizando cultivos primarios de células gliales, derivados de cerebro fetal,
neonatal y adulto humano. Estos cultivos, formados principalmente por astrocitos y
células de la microglía, fueron expuestos a las glicoproteínas virales gp120 y gp41,
pudiéndose comprobar que esta interacción inducía la producción de óxido nítrico, IL-
1 y TNF-α240. Es notable que cultivos primarios de células nerviosas de ratón y de rata
expuestos a gp120136, 137, 241 y a gp41140, 242, también respondan con producción de IL-
1, TNF-α, óxido nítrico y apoptosis, evidenciando que existen vías comunes a la
humana en estas células nerviosas de roedores. De hecho, astrocitos, neuronas y
células de la microglía de ratón y rata expresan los correceptores CXCR4 y CCR5, al
igual que las humanas, y pueden interaccionar con la gp120 del VIH-1 y con las
quimioquinas humanas MIP-1β, SDF-α y SDF-β136, 137. Otros autores han utilizado
líneas celulares humanas243 para la evaluación de los efectos neurotóxicos del VIH-1,
así como cultivos primarios de linajes aislados de células nerviosas humanas y de
roedores244-246, sin embargo, los cultivos mixtos que incluyen los linajes y
proporciones celulares que se encuentran en los tejidos in vivo, tiene la ventaja de que
permiten evaluar la interrelación que se dan entre los distintos linajes en presencia del
virus136, 137. Para evaluar los efectos de la infección por T. gondii en células del sistema
nervioso central, también han sido utilizados cultivos primarios de células de cerebro
humano y de roedores204, 247-250. El parásito es capaz de infectar y multiplicarse en
neuronas, astrocitos y células de la microglía de humanos y roedores. Estos modelos
30

han proporcionado valiosa información sobre la compleja interacción entre los
agentes patógenos y las células del SNC.
IV. Planteamiento del problema de estudio
Hemos descrito dos patógenos, VIH-1 y T. gondii, que infectan un número
importante de individuos a nivel mundial, causando infecciones que se prolongan por
años, que tienen la capacidad de modificar activamente la respuesta inmunitaria de
sus hospedadores, que invaden tempranamente el sistema nervioso central y que
pueden modificar la funcionalidad del mismo, causando cuadros que afectan la calidad
de vida y comprometen la sobrevida de los pacientes. Ahora bien, si estos agentes por
separado pueden mediar estos efectos, ¿cómo interaccionarán en caso de infectar
simultáneamente a un mismo hospedador? Como es bien sabido, la inmunodeficiencia
provocada por la infección con VIH-1 permite la reactivación de infecciones latentes
por T. gondii, especialmente a nivel cerebral, pero, ¿se generarán modificaciones en el
control de la infección parasitaria en etapas previas al SIDA? ¿Afectará la presencia del
parásito a la respuesta inmunitaria contra el virus? ¿Se asociará la presencia
simultánea de ambos agentes con mayores defectos en la respuesta inmunitaria? ¿Será
mayor el efecto neurotóxico en presencia de ambos?
El presente trabajo se diseñó para evaluar dos grandes aspectos de la infección por
VIH-1 y T. gondii en un mismo hospedador: por una parte estudiar, mediante un
modelo in vitro, los efectos neurotóxicos de la presencia simultánea de los dos
patógenos sobre células del sistema nervioso central; y por la otra, evaluar la
respuesta inmunitaria, con especial énfasis en la funcionalidad de linfocitos T, en
pacientes infectados con VIH-1 y serología positiva para T. gondii, en diferentes fases
de la infección viral, en la búsqueda de una mayor comprensión de mecanismos
inmunopatológicos, que permitiesen eventualmente optimizar la intervención
terapéutica, mejorando la calidad de vida y aumentando la sobrevida de los pacientes.
A continuación se presentan las hipótesis y objetivos del trabajo:
Hipótesis
I. En pacientes infectados con VIH-1, seropositivos para T. gondii, la
inmunodeficiencia causada por la progresión de la infección viral afectará la
respuesta específica contra T. gondii.
II. La presencia simultánea de VIH-1 y taquizoítos de T. gondii en cultivos
primarios de células del sistema nervioso central potenciará los efectos
neurotóxicos de ambos patógenos.
31

Objetivos generales
1. Estudiar aspectos de la respuesta inmunitaria de linfocitos T en pacientes
infectados con VIH-1 y serología positiva para T. gondii.
2. Evaluar los efectos neurotóxicos de la presencia simultánea de T. gondii y
VIH-1 sobre células nerviosas de feto de rata.
Objetivos específicos
1. Estudiar el fenotipo de la población de linfocitos T de sangre periférica de
pacientes infectados con VIH-1 y serología positiva para T. gondii.
2. Estudiar la funcionalidad de estos linfocitos T en respuesta a estímulos
específicos (antígenos de VIH-1 y T. gondii) y no específicos (mitógenos).
3. Correlacionar el fenotipo y funcionalidad de los linfocitos T con la carga viral
y el estadio de la infección.
4. En cultivos primarios de células nerviosas de feto de rata, estudiar los
efectos de la presencia simultánea de VIH-1 y T. gondii en la sobrevida,
apoptosis y necrosis de células nerviosas, así como en la producción de
citoquinas y óxido nítrico.
32

MATERIALES Y MÉTODOS
I. ESTUDIO DE LA FUNCIONALIDAD DE LOS LINFOCITOS T EN
PACIENTES INFECTADOS CON VIH-1 Y SEROLOGÍA POSITIVA PARA
Toxoplasma gondii
A. SUJETOS DE ESTUDIO
En el estudio se incluyeron 105 individuos adultos, referidos por el Servicio
de Infectología del Hospital “José Ignacio Baldó” (El Algodonal, Caracas) y el Servicio
de Inmunología del Instituto de Medicina Tropical (Universidad Central de
Venezuela, Caracas), que aceptaron en forma voluntaria participar en el estudio y
dieron su consentimiento por escrito. Este trabajo fue aprobado por los Comités de
Bioética del IVIC y del Hospital “José Ignacio Baldó”.
El despistaje serológico de la infección por el Virus de Inmunodeficiencia
Humana-1 (VIH-1) se realizó por ELISA y Western Blot y el de la infección por
Toxoplasma gondii (T. gondii) por ELISA y Avidez. Los individuos participantes no
habían recibido terapia anti-retroviral para la infección por VIH-1, ni profilaxis
contra toxoplasmosis al momento del estudio, y se dividieron en cuatro grupos
principales:
Grupo Control 1 (C1): individuos aparentemente sanos, con serología
negativa para VIH-1 y T. gondii (n=11).
Grupo Control 2 (C2): individuos aparentemente sanos, con serología
negativa para VIH-1 y positiva para T. gondii (n=13).
Grupo de Pacientes 1 (P1): individuos con serología positiva para VIH-1 y
negativa para T. gondii (n=37). Basado en el recuento de linfocitos T CD4+ en
sangre periférica, este grupo fue sub-dividido de la siguiente manera:
P1A: individuos con recuento de linfocitos T CD4+ > 350/μL (n=12).
P1B: individuos con recuento de linfocitos T CD4+ entre 200 y 350/μL (n=16).
P1C: individuos con recuento de linfocitos T CD4+ < 200/μL (n=9).
Grupo de Pacientes 2 (P2): individuos con serología positiva para VIH-1 y T.
gondii (n=44). Basado en el recuento de linfocitos T CD4+ en sangre
periférica, este grupo fue sub-dividido de la siguiente manera:
P2A: individuos con recuento de linfocitos T CD4+ > 350/μL (n=15).
P2B: individuos con recuento de linfocitos T CD4+ entre 200 y 350/μL (n=17).
P2C: individuos con recuento de linfocitos T CD4+ < 200/μL (n=12).
Se consideraron como criterios de exclusión: antecedentes de consumo de
cocaína o sus derivados, alcoholismo, antecedentes de traumatismos cráneo-
encefálicos con conmoción cerebral, convulsiones u otras enfermedades del SNC
33

(Alzheimer, Parkinson), enfermedades autoinmunes (Lupus Eritematoso Sistémico,
Artritis Reumatoide), cáncer y tratamiento con inmunosupresores.
La atención médica, la historia clínica de los pacientes, así como la clasificación
clínica de la infección por VIH-172, fue realizada en los servicios de Infectología e
Inmunología de los centros de referencia de los pacientes (Hospital “José Ignacio
Baldó” e Instituto de Medicina Tropical). Si bien el haber recibido tratamiento
específico contra VIH o contra T. gondii fueron criterios de exclusión, la participación
en el protocolo no fue causa de omisión o demora del tratamiento que necesitó cada
paciente. La atención médica y el seguimiento o tratamiento que requirió cada
paciente dependió de sus necesidades y de la dinámica propia del servicio de cada
hospital y no de su participación en el estudio. El paciente fue informado de su
situación clínica por su médico tratante.
A cada participante se le tomó una muestra de 15-20 ml de sangre periférica,
utilizando EDTA como anticoagulante. Esta muestra fue utilizada para el estudio de
subpoblaciones linfocitarias, obtención de células mononucleares de sangre periférica
(PBMC) para su cultivo y obtención de plasma para la determinación de carga viral* y
citoquinas.
B. ESTUDIO FENOTÍPICO DE LINFOCITOS T DE SANGRE PERIFÉRICA
Utilizando citometría de flujo (Citómetro FACScalibur y programa Cell Quest,
Becton Dickinson), se evaluaron en la muestra de sangre periférica tomada de los
participantes los siguientes parámetros ex vivo:
Subpoblaciones de linfocitos T: Los porcentajes de linfocitos T totales
(población linfoide CD3+), linfocitos T CD4+ (población linfoide CD3+/CD4+) y
linfocitos T CD8+ (población linfoide CD3+/CD8+) se determinaron por
citometría de flujo, mezclando en forma apropiada 50 μL de sangre periférica
de los participantes, con 1 μL de cada uno de los siguientes anticuerpos
monoclonales: anti-CD3 conjugado con el fluorocromo Proteína Clorofila
Peridina-PerCP (CD3-PerCP, Becton Dickinson), anti-CD4 conjugado a
Aloficocianina-APC (CD4-APC, Becton Dickinson) o anti-CD8 conjugado a APC ó a
Isotiocianato de Fluoresceína-FITC (CD8-APC y CD8-FITC, Becton Dickinson),
incubando a temperatura ambiente y en oscuridad por 15-30 minutos,
realizando lisis de glóbulos rojos, lavado y fijación (paraformaldehido 1%) de
las muestras y finalmente adquisición/análisis en el citómetro de flujo. Para
calcular los valores absolutos (células por microlitro de sangre periférica) de
estas poblaciones, se utilizaron los porcentajes obtenidos en el análisis
* La determinación de la carga viral fue realizada en el Instituto Nacional de Higiene “Rafael Rangel”,
MPPSDS, Caracas, utilizando el método Branched DNA (bDNA), con capacidad de detección entre 50 y
500.000 copias de RNA de VIH-1/mL (1.7 – 5.7 Log de copias de RNA de VIH-1/mL).
34

citométrico, junto al recuento de leucocitos y porcentaje de linfocitos totales
suministrados por el estudio hematológico (Hematología Completa).
Combinando los anticuerpos ya mencionados con otros que mencionaremos a
continuación, fue posible evaluar la expresión de marcadores adicionales:
Expresión de HLA-DR: Para medir la expresión de la molécula HLA-DR se
utilizó un anticuerpo monoclonal anti-HLA-DR conjugado a Ficoeritrina-PE
(HLA-DR-PE, Becton Dickinson).
Expresión de Fas (CD95): Para medir la expresión de Fas se utilizó un
anticuerpo monoclonal anti-CD95 conjugado a FITC (CD95-FITC, Becton
Dickinson).
Expresión de la subunidad alfa del receptor de IL-7 (IL-7Rα/CD127): Para
medir esta expresión se utilizó un anticuerpo monoclonal anti-IL-7Rα
conjugado a PE (CD127-PE, Becton Dickinson).
Expresión de PD-1 (CD279): Para estudiarla se utilizó un anticuerpo
monoclonal anti- PD-1 conjugado a PE (PD-1-PE, Becton Dickinson).
Células vírgenes y de memoria: Mediante la expresión de las moléculas
CD45RA y CD45RO, se hizo una aproximación a la identificación de las
poblaciones de células vírgenes y de memoria251, respectivamente, utilizando
anticuerpos monoclonales conjugados a FITC y PE (CD45RA-FITC y CD45RO-PE,
Becton Dickinson).
Apoptosis ex vivo: la apoptosis y necrosis ex vivo de linfocitos T CD4+ y T
CD8+, se determinó utilizando y siguiendo las instrucciones de un estuche
comercial (Annexin V-FITC Apoptosis Detection Kit; Becton-Dickinson),
fundamentado en la detección de la traslocación de la fosfatidilserina a la cara
externa de la membrana celular en las fases tempranas de la apoptosis252, 253,
mediante el uso de la Anexina V, conjugada a FITC253.
C. ESTUDIOS DE FUNCIONALIDAD DE LINFOCITOS T
Las células mononucleares de sangre periférica (PBMC) de los pacientes y
controles se separaron por gradiente de densidad (Histopaque 1077, Sigma-Aldrich) y
se cultivaron en placas de 96 pozos (Nunclone, Nunc), a razón de 1 x 105 células/pozo,
en un volumen final de 200 μl/pozo, por 72 horas, a 37°C y 5% de CO2, en medio
completo RPMI*(RPMI 1640, Gibco) y 10% de suero fetal bovino (SFB), en triplicado (3
pozos por cada condición), en cuatro condiciones básicas de estimulación:
Control: PBMC cultivadas sin estimulación, sólo con medio de cultivo
* El medio completo RPMI utilizado para los distintos procedimientos de cultivo, se preparó con
RPMI 1640 (Gibco); Tampón HEPES 10 mM (Gibco); Piruvato de Sodio 1 mM (Sigma); solución de
Aminoácidos No Esenciales, 1x (Gibco); Glutamina 2 mM (Gibco); solución de Antibióticos (Penicilina,
100 U/mL; Estreptomicina, 100 mg/mL; Gibco) y suero fetal bovino (SFB) 10%.
35

PHA: PBMC estimuladas con el mitógeno fitohemaglutinina (PHA, Sigma, 5
µg/mL)
SATg: PBMC estimuladas con antígenos solubles de taquizoítos de T. gondii
(SATg, amablemente cedido por la Dra. Belkisyolé Alarcón de Noya, Instituto de
Medicina Tropical, UCV, 1 µg/mL)
Env: PBMC estimuladas con péptidos sintéticos gp41 de la envoltura viral del
VIH-1 (ENV-gp41 recombinante, Chemicon, 1 µg/mL)
Luego de 72 horas de cultivo, se recuperaron y congelaron (-80°C) los
sobrenadantes de los cultivos, para la posterior determinación de citoquinas (ver más
adelante) y las células fueron procesadas para estudiar los diferentes parámetros de
funcionalidad, utilizando citometría de flujo. Para estos experimentos (excepto para la
determinación de Apoptosis/Necrosis), las células fueron fijadas con
paraformaldehido al 1% en PBS y permeabilizadas con solución de saponina al 0.5%
(preparada en PBS, 5% SFB), antes de su incubación con 1 μL de los distintos
anticuerpos monoclonales. Las poblaciones de células T CD4+ y T CD8+ se
identificaron por medio de los anticuerpos monoclonales anti-CD3-PerCP (Beckton
Dickinson), anti-CD4-APC (Beckton Dickinson) y anti-CD8-APC (Beckton Dickinson),
combinados en tubos correspondientes con anticuerpos relevantes a la función a
medir, de la siguiente manera:
Proliferación: se evaluó en las poblaciones de linfocitos T CD4+ y T CD8+ totales.
Para evaluar la proliferación se utilizó un nucleósido sintético análogo de la
timidina, la bromodeoxiuridina (BrdU, Sigma), la cual se incorpora en el ADN de las
células en replicación (fase S del ciclo celular). La BrdU se agregó a las células en
las últimas 4-6 horas de cultivo, a una concentración final de 10 µM. Las células en
proliferación se identificaron utilizando un anticuerpo monoclonal anti-BrdU
conjugado a FITC (BrdU-FITC), con un reactivo comercial que incluía la enzima
ADNasa254 (Becton-Dickinson).
Producción de perforina por linfocitos T CD8+: la detección de perforina255
intracelular en la población de linfocitos T CD8+ totales, como una forma indirecta
de estudiar la capacidad citotóxica de estas células, se realizó tratando con
Brefeldina A, un bloqueador de la exocitosis de proteínas256 (10 µg/mL,
concentración final) en las últimas 4-6 horas de cultivo, y marcando con un
anticuerpo anti-perforina conjugado con PE (Perforina-PE, Becton-Dickinson).
Apoptosis y necrosis de PBMC cultivadas: la apoptosis y necrosis de las PBMC se
determinaron al final del período de cultivo, utilizando el estuche comercial
Annexin V-FITC Apoptosis Detection Kit (Becton-Dickinson), con el fundamento
descrito anteriormente. La necrosis se detectó por medio del yoduro de propidio,
que se une a los ácidos nucleicos (ADN, ARN) de las células cuya integridad de
membrana se ha perdido (células necróticas).
36

Producción de citoquinas ex vivo e in vitro: la cuantificación de IL-2, IL-4, IL-6,
IL-10, TNF-α e IFN-γ en el plasma (producción ex vivo) y los sobrenadantes de
PBMC cultivadas por 72 horas en las distintas condiciones de estimulación
(producción in vitro) se hizo por citometría de flujo, utilizando y siguiendo el
protocolo de un estuche comercial (Human Th1/Th2 Cytokine Kit, Cytometric
Bead Array, Becton-Dickinson). Brevemente, este estuche utiliza múltiples
poblaciones de perlas de captura con intensidad de fluorescencia discreta, que
pueden discriminarse en el canal de fluorescencia FL3 del citómetro. Cada
población de perlas está conjugada a anticuerpos específicos a una de las
diferentes citoquinas a evaluar, de manera que al incubarse con las muestras
“capturarán”, es decir, se unirán a las citoquinas presentes en la muestra. Para
evidenciar la interacción “perla de captura-citoquina”, se utilizan anticuerpos de
detección, específicos a cada citoquina y conjugados a PE. De esta forma la
intensidad de fluorescencia para PE (canal FL2 del citómetro) será proporcional
a la cantidad de cada citoquina presente en la muestra y unida a la perla de
captura. Finalmente, mediante curvas de calibración realizadas con patrones de
concentración conocida, se pueden calcular las concentraciones de las muestras
experimentales.
II. EVALUACIÓN DE EFECTOS NEUROTÓXICOS DE VIH-1 Y
Toxoplasma gondii EN CÉLULAS DEL SISTEMA NERVIOSO CENTRAL
MODELO IN VITRO DE NEUROTOXICIDAD
Para la evaluación in vitro de efectos neurotóxicos se utilizaron células nerviosas
de feto de rata Sprague Dawley cultivadas sin estimulación/infección (condición
Control), o en presencia de viriones de VIH-1, glicoproteína viral gp41, taquizoítos
de la cepa RH de T. gondii, o una combinación de éstos. A continuación se describe
como se obtuvieron los distintos componentes de este modelo in vitro de
neurotoxicidad.
Cultivos primarios de células nerviosas de feto de rata: las células
nerviosas se obtuvieron de cerebros de feto de ratas Sprague-Dawley, de 17-19
días de gestación. Tras anestesiar a la rata gestante (Ketamina 40-80 mg/kg;
Xilacina 5-10 mg/kg), se procedió a la extracción por cesárea de los fetos,
sacrificando seguidamente a la madre por ruptura cervical, mientras se
encontraba anestesiada. Los hemisferios cerebrales fetales se disecaron en
condiciones estériles, separándolos de cerebelo, tronco cerebral y meninges. Se
disociaron mecánicamente utilizando pipetas de Pasteur y se cultivaron en
37

placas de Petri previamente tratadas con poli-lisina (0.1mg/mL)*, en RPMI con
SFB al 10%, a 37°C y 5% de CO2, a razón de 15-20 x 106 células/placa. Después
de 7 a 15 días de cultivo se rompió la adherencia de las células nerviosas a la
superficie de la placa por tratamiento con tripsina (Tripsina-EDTA 0.5%,
Gibco/Invitrogen), para recuperarlas y sembrarlas en placas de cultivo de 12 ó
24 pozos, tratadas previamente con poli-lisina (0.1mg/mL), a una
concentración de 2 - 3 x 105 células/pozo. Se cultivaron las células por 7 a 15
días, para permitir la formación de una monocapa confluyente, y se realizaron
los distintos experimentos.
Péptidos sintéticos de VIH-1: se utilizó la proteína recombinante gp41 (Env,
Chemicon International). La gp41 se utilizó en los experimentos a una
concentración de 10 nM, en base a resultados obtenidos en experimentos
preliminares, donde se evaluaron varias concentraciones de gp41.
VIH-1: se utilizaron como fuente de viriones (partículas virales completas) de
VIH-1 los sobrenadantes de cultivos de células MT4 (línea celular de linfocitos
T CD4+ humanos, procedentes de un paciente con Leucemia de Células T del
Adulto, amablemente cedido por el Dr. Héctor Rangel, CMBC, IVIC) infectadas
in vitro con la cepa de VIH-1IIIB (amablemente cedido por el Dr. Héctor Rangel,
CMBC, IVIC). Brevemente, se incubaron 1 x 106 células MT4 con VIH-1IIIB, en un
volumen de 1 mL de medio de cultivo RPMI, SFB al 10%, por 3 horas, a 37°C y
5% de CO2, para permitir el establecimiento de la infección. Luego de estas 3
horas de incubación, se completó con medio de cultivo hasta 30-35 mL y se
cultivó. Una o dos veces por semana se recuperaron y congelaron (-80°C) los
sobrenadantes (donde se encontraban las partículas virales) y se agregaron
medio y células frescas a los cultivos. Finalmente, se mezclaron todos los
sobrenadantes obtenidos durante el proceso de infección in vitro. Para separar
el virus generado, la mezcla de sobrenadantes se sometió a ultracentrifugación
(26.000 G, 2 horas), recuperando el sedimento con el virus concentrado, el cual
se resuspendió en medio fresco (RPMI, 10% SBF), se separó en alícuotas de 1,5
mL y se congeló a -80°C. Una de estas alícuotas se envió al Instituto Nacional de
Higiene “Rafael Rangel”, MPPSDS, Caracas, para determinar la carga viral. Este
virus fue utilizado para los experimentos con células nerviosas a una
concentración de 1,9 x 109 copias/mL, en base a resultados obtenidos en
* Tratamiento de placas de cultivo con poli-L-lisina: para favorecer la adhesión celular a la superficie de la
placa de cultivo, éstas se cubren, en condiciones estériles, con una solución de poli-L-lisina (Sigma, 0.1mg/mL)
por 5 minutos. Luego de este tiempo, la solución se retira y se dejan secar espontáneamente las placas,
manteniendo las condiciones de esterilidad. Las placas así tratadas, se utilizan para realizar los cultivos
celulares.
38

experimentos preliminares, donde se evaluaron varias concentraciones virales
de trabajo.
Taquizoítos de T. gondii: se obtuvieron a partir del líquido ascítico de ratones
BALB/c infectados previamente por inoculación intraperitoneal de taquizoítos
de T. gondii de la cepa RH (amablemente cedidos por la Dra. Belkisyolé Alarcón
de Noya, Instituto de Medicina Tropical, UCV). Los animales se sacrificaron por
ruptura cervical 3 días después de la infección y los parásitos se recuperaron
en condiciones estériles, de la cavidad abdominal, utilizando una jeringa de
insulina. Los parásitos obtenidos se lavaron dos veces con PBS, para descartar
fluido ascítico y restos/detritus celulares. Después de centrifugar y descartar el
sobrenadante, se resuspendieron los taquizoítos en medio de cultivo (RPMI,
10% SFB) y se procedió a hacer el recuento de parásitos viables, utilizando azul
de tripano. Estos parásitos se utilizaron para la infección in vitro de células
nerviosas.
Cultivo de células nerviosas de rata en presencia de VIH, gp41 y/o T.
gondii: las células nerviosas de feto de rata se cultivaron en un volumen final
de 1 mL de medio de cultivo RPMI, 10% de SFB, a 37°C y 5% de CO2, por 3, 5 y
7 días. Las condiciones de estimulación/infección fueron las siguientes:
1. Condición Control: células nerviosas cultivadas en ausencia de VIH-1,
gp41 y T. gondii.
2. Condición gp41: células nerviosas cultivadas en presencia de gp41
10nM.
3. Condición VIH: células nerviosas cultivadas en presencia de 1,9 x 109
copias/mL de VIH-1IIIB.
4. Condición T. gondii: células nerviosas cultivadas en presencia de
taquizoítos de la cepa RH de T. gondii. La infección in vitro se hizo
utilizando una relación parásito/célula blanco: 1/250*, incubando por
cuatro horas en un volumen de 300 μL de medio RPMI y SFB al 10%, a
37°C y 5% de CO2. Al finalizar las cuatro horas de incubación, se
realizaron dos lavados con PBS, para descartar los parásitos no unidos,
y se completó el volumen final a 1 mL con medio de cultivo.
5. Condición gp41/T. gondii: células nerviosas cultivadas en presencia
simultánea de gp41 (10nM) y de T. gondii (1/250).
6. Condición VIH/T. gondii: células nerviosas cultivadas en presencia
simultánea de VIH-1IIIB (1,9 x 109 copias/mL) y de T. gondii (1/250).
* Esta relación se escogió en base a los resultados obtenidos en experimentos preliminares, para lograr una
cinética de infección parasitaria lo suficientemente “lenta”, que permitiera evaluar el efecto de la presencia
simultánea de VIH-1 ó gp41, en cultivos que debían prolongarse hasta por 7 días.
39

En las condiciones de incubación simultánea “gp41/T. gondii” y “VIH/T. gondii”,
se realizaba primero la infección parasitaria por cuatro horas, de acuerdo al
protocolo descrito, y luego de los dos lavados con PBS, se agregaba, según
correspondiese, la gp41 o el VIH-1IIIB, iniciándose el co-cultivo.
Al finalizar el tiempo de cultivo, se recuperaron y conservaron los
sobrenadantes (-80°C) para la posterior medición de nitritos (óxido nítrico) y
citoquinas, y se trataron las células con tripsina (0.5%) por 10-15 minutos,
para romper la adherencia y recuperarlas para la evaluación de mecanismos
neurotóxicos.
Cultivos sobre laminillas para estudio microscópico: Para algunos de los
experimentos las células nerviosas se cultivaron sobre laminillas
cubreobjeto, tratadas previamente con poli-lisina y colocadas en los pozos de
las placas de cultivo, siguiendo el mismo protocolo general ya descrito, de
forma que se pudiera realizar un análisis microscópico de las monocapas
celulares en las distintas condiciones. En estos experimentos sobre
laminillas, al finalizar los tiempos de cultivo las células eran fijadas con una
solución de paraformaldehido al 4% y posteriormente lavadas con PBS, y
conservadas para su procesamiento por diversas coloraciones/técnicas
(Wright-Giemsa o inmunocitoquímica).
B. EVALUACIÓN DE EFECTOS NEUROTÓXICOS
Recuento y viabilidad de células nerviosas y taquizoítos libres: luego de
recuperar las células nerviosas de cada pozo en las distintas condiciones de
cultivo, se hizo un recuento (cámara de Neubauer) con azul de tripano, para
determinar tanto el número de células como la viabilidad de las mismas. En
las condiciones que correspondía, simultáneamente al recuento de células
nerviosas, se hizo también el recuento de taquizoítos de T. gondii libres
(parásitos extracelulares) presentes en los sobrenadantes recuperados, dato
que fue utilizado como una medida indirecta de la progresión de la infección
parasitaria in vitro.
Apoptosis y necrosis de células nerviosas: utilizando el estuche comercial
Annexin V-FITC Apoptosis Detection Kit (Becton-Dickinson) se determinaron
la apoptosis y necrosis de las células nerviosas.
Producción de óxido nítrico: la determinación de óxido nítrico en los
sobrenadantes de cultivo se hizo en forma indirecta, midiendo la
concentración de nitritos (NO2-) en las muestras, utilizando el método y
reactivo de Griess257. Brevemente, 100 μL de las muestras a analizar se
incubaron con 300 μL de reactivo de Griess (Sulfanilamina 1%; Naftil-etilen-
diamino-dihidroclorhídrico 0,1%, Ácido Fosfórico 2,5%) por 10 minutos, a
temperatura ambiente. La reacción colorimétrica se midió a 570 nm. La
40

concentración se calculó utilizando una curva de calibración, realizada con
patrones de nitrito de sodio (NaNO2).
Producción de citoquinas en cultivos de células nerviosas: la
determinación de IL-1α, IL-6, IL-10, TNF-α e IFN-γ en los sobrenadantes de
cultivo de células nerviosas de feto de rata se realizó por citometría de flujo,
utilizando y siguiendo el protocolo de un estuche comercial (Cytometric Bead
Array, Rat Flex Sets, Becton Dickinson), con un fundamento similar al descrito
anteriormente para la determinación de citoquinas humanas en plasma y
sobrenadantes de cultivo.
Análisis microscópico de los cultivos de células nerviosas: los cultivos
realizados sobre laminillas fueron sometidos a procesos de coloración con la
técnica Wright/Giemsa o por procedimientos de inmunocitoquímica,
utilizando anticuerpos anti-Proteína Asociada a los Microtúbulos-2 (MAP2,
Sigma) para la identificación de neuronas, anti-Proteína Acida Fibrilar de la
Glía (GFAP, Becton Dickinson) para la identificación de astrocitos, y anti
CD11b/c (Becton Dickinson) para la identificación de células de la microglía.
El análisis microscópico de las laminillas así procesadas permitió determinar
el porcentaje de cada linaje celular (astrocitos, neuronas y microglía) en la
población de los cultivos primarios, el porcentaje de células infectadas, así
como el número de parásitos presentes en el citoplasma de dichas células.
C. ANÁLISIS ESTADÍSTICO
El análisis estadístico de los datos se hizo utilizando el programa SigmaStat
para Windows (Jandel Corporation). La comparación de los grupos de estudio se
hizo por medio de la prueba de ANOVA, para aquellos datos que se distribuyeron en
forma normal, y aplicando la prueba de Kruskal-Wallis, para aquellos casos que no
superaron la prueba de normalidad, en combinación con las pruebas de
comparaciones múltiples de Bonferroni y Dunn, respectivamente. Las correlaciones
se analizaron por la prueba de Pearson, para datos con distribución normal y la
prueba de Spearman, para datos no paramétricos. Las pruebas se consideraron
estadísticamente significativas para valores de p menores a 0,05.
41

RESULTADOS
I. ESTUDIO DE LA FUNCIONALIDAD DE LOS LINFOCITOS T EN
PACIENTES INFECTADOS CON VIH-1 Y SEROLOGÍA POSITIVA
PARA Toxoplasma gondii
A. SUJETOS DE ESTUDIO
En la Tabla I se presentan las características generales de los grupos de estudio.
Los pacientes, tanto P1 (VIH+/T. gondii-) como P2 (VIH+/T. gondii+), se clasificaron
en base a su recuento de linfocitos T CD4+ en sangre periférica en los subgrupos A
(CD4>350/μL), B (CD4:200-350/μL) y C (CD4<200/μL) y basados en esta
clasificación se presentan los distintos resultados y análisis. En vista de que la
progresión de la infección por VIH-1 se asocia con una disminución gradual en el
recuento de linfocitos T CD4+ de sangre periférica (Fig. 8), la agrupación de los
pacientes basada en este parámetro nos permite analizar en una aproximación
secuencial a su progresión, de etapas más tempranas (sub-grupo A) a etapas más
tardías (sub-grupo C) de la infección viral, o, dicho en otras palabras, desde los
pacientes más inmunocompetentes (mayores recuentos de linfocitos T CD4+) a los
más inmunocomprometidos (menores recuentos de linfocitos T CD4+). Es preciso
enfatizar que este es un enfoque poblacional de los datos de los grupos de estudio,
ya que desde el punto de vista individual, un determinado valor de recuento de
linfocitos T CD4+ de sangre periférica no significa exactamente lo mismo para cada
paciente.
Edad: no se observaron diferencias estadísticamente significativas de edad
entre los distintos grupos de estudio.
Sexo: al discriminar por sexo los distintos parámetros evaluados en este
trabajo, no se observaron diferencias significativas entre los individuos de
sexo masculino y los de sexo femenino.
42

43
Tabla I- Descripción de los sujetos de estudio
Grupos n Edad
(años)
Sexo
(M/F)
%
Leuc %Linf Linf/uL %CD3 CD3/uL %CD4 CD4/uL %CD8 CD8/uL
Carga
Viral
(Log)
C1 11 31 ± 8 36/64 6.782
± 1.518 39 ± 5
2.616
± 564 63 ± 9
1.644
± 367 36 ± 4
938
± 244 23 ± 8
592
± 198 N/A
C2 13 33 ± 12 64/36 7.357
± 2.312 32 ± 9
2.202
± 473 63 ± 10
1.398
± 411 33 ± 9
733
± 238 23 ± 8
517
± 269 N/A
P1A 12 31 ± 8 58/42 6.300
±1.226 39 ± 9
2.421
± 562 70 ± 9
1.685
± 423 24 ± 6
563
± 177 41 ± 9
994
± 340
3.85
± 0.85
P1B 16 35 ± 10 75/25 5.725 ± 1.184
34 ± 12 1.918
± 668 68 ± 12
1.354
± 705 15 ± 5
261
± 46 48 ± 14
991
± 635
4.45
± 0.59
P1C 9 35 ± 10 89/11 4.533 ± 2.336
38 ± 13 1.597
± 577 63 ± 13
1.010
± 417 9 ± 9
116
± 68 49 ± 16
828
± 411
5.05
± 0.66
P2A 15 32 ± 10 80/20 6.120 ± 1.755
45 ± 14 2.698
± 1.113 75 ± 10
2.062
± 1.026 23 ± 9
581
± 277 46 ± 13
1.324
± 971
3.87
± 0.56
P2B 17 36 ± 8 76/24 6.365
± 1.540 35 ± 11
2.148
± 522 67 ± 12
1.456
± 446 14 ± 4
280
± 51 48 ± 12
1.059
± 418
4.43
± 0.75
P2C 12 40 ± 11 92/8 3.983 ± 1.079
36 ± 11 1.405
± 458 72 ± 11
1.018
± 422 7 ± 4
86
± 51 58 ± 11
837
± 397
4.65
± 0.52
C1 (VIH-/T. gondii-)
C2 (VIH-/T. gondii+)
P1A (VIH+/T. gondii-; CD4>350/L)
P1B (VIH+/T. gondii-; CD4:200-350/L)
P1C (VIH+/T. gondii-; CD4<200/L)
P2A (VIH+/T. gondii+; CD4>350/L)
P2B (VIH+/T. gondii+; CD4:200-350/L)
P2C (VIH+/T. gondii+; CD4<200/L)
Los datos están expresados en promedio ± desviación
estándar
n: número de pacientes por grupo
Sexo: está expresado en porcentaje de individuos
Masculinos/porcentaje de individuos Femeninos
N/A: no aplicable
Carga Viral: está expresada en logaritmo de copias de
RNA de VIH-1/mL de plasma
Leuc: recuento de leucocitos en sangre periférica (/L)
%Linf, %CD3, %CD4, %CD8: porcentaje de linfocitos
totales, linfocitos T totales, linfocitos T CD4+ y linfocitos T
CD8+, respectivamente, en la población de leucocitos de
sangre periférica
Linf/μL, CD3/μL, CD4/μL, CD8/μL: recuento de linfocitos
totales, linfocitos T totales, linfocitos T CD4+ y linfocitos T
CD8+, respectivamente, en sangre periférica (/L)
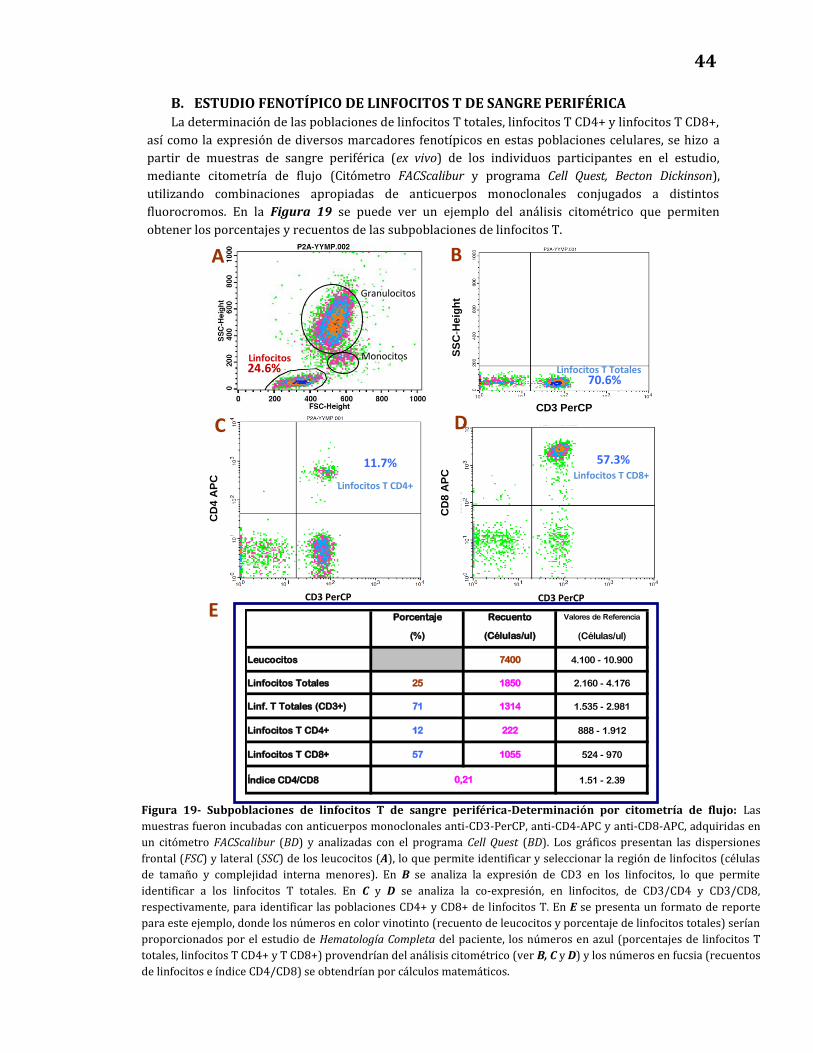
B. ESTUDIO FENOTÍPICO DE LINFOCITOS T DE SANGRE PERIFÉRICA La determinación de las poblaciones de linfocitos T totales, linfocitos T CD4+ y linfocitos T CD8+,
así como la expresión de diversos marcadores fenotípicos en estas poblaciones celulares, se hizo a
partir de muestras de sangre periférica (ex vivo) de los individuos participantes en el estudio,
mediante citometría de flujo (Citómetro FACScalibur y programa Cell Quest, Becton Dickinson),
utilizando combinaciones apropiadas de anticuerpos monoclonales conjugados a distintos
fluorocromos. En la Figura 19 se puede ver un ejemplo del análisis citométrico que permiten
obtener los porcentajes y recuentos de las subpoblaciones de linfocitos T.
Figura 19- Subpoblaciones de linfocitos T de sangre periférica-Determinación por citometría de flujo: Las
muestras fueron incubadas con anticuerpos monoclonales anti-CD3-PerCP, anti-CD4-APC y anti-CD8-APC, adquiridas en
un citómetro FACScalibur (BD) y analizadas con el programa Cell Quest (BD). Los gráficos presentan las dispersiones
frontal (FSC) y lateral (SSC) de los leucocitos (A), lo que permite identificar y seleccionar la región de linfocitos (células
de tamaño y complejidad interna menores). En B se analiza la expresión de CD3 en los linfocitos, lo que permite
identificar a los linfocitos T totales. En C y D se analiza la co-expresión, en linfocitos, de CD3/CD4 y CD3/CD8,
respectivamente, para identificar las poblaciones CD4+ y CD8+ de linfocitos T. En E se presenta un formato de reporte
para este ejemplo, donde los números en color vinotinto (recuento de leucocitos y porcentaje de linfocitos totales) serían
proporcionados por el estudio de Hematología Completa del paciente, los números en azul (porcentajes de linfocitos T
totales, linfocitos T CD4+ y T CD8+) provendrían del análisis citométrico (ver B, C y D) y los números en fucsia (recuentos
de linfocitos e índice CD4/CD8) se obtendrían por cálculos matemáticos.
57.3% Linfocitos T CD8+
D C
D8 A
PC
CD3 PerCP
70.6%
Linfocitos T Totales
B
CD3 PerCP
SS
C-H
eig
ht
C
11.7%
Linfocitos T CD4+
CD
4 A
PC
CD3 PerCP
Granulocitos
Monocitos Linfocitos 24.6%
A
Porcentaje Recuento Valores de Referencia
(%) (Células/ul) (Células/ul)
Leucocitos 7400 4.100 - 10.900
Linfocitos Totales 25 1850 2.160 - 4.176
Linf. T Totales (CD3+) 71 1314 1.535 - 2.981
Linfocitos T CD4+ 12 222 888 - 1.912
Linfocitos T CD8+ 57 1055 524 - 970
Índice CD4/CD8 1.51 - 2.390,21
E
44

Subpoblación de linfocitos T CD4+: Los grupos de pacientes (P1 y P2)
presentaron valores de recuento y porcentaje de linfocitos T CD4+ en sangre
periférica significativamente menores a los de los grupos Control (C1 y C2) (Fig. 20A
y 20B) desde las etapas más tempranas de la infección viral. Los valores de
porcentaje de linfocitos T CD4+ disminuyeron progresivamente a medida que los
recuentos eran menores. Al comparar los valores de porcentaje y recuento de
linfocitos T CD4+ de sangre periférica entre los subgrupos homólogos de controles y
pacientes (C1 vs. C2, P1A vs. P2A, P1B vs. P2B y P1C vs. P2C), no se observaron
diferencias estadísticamente significativas.
Recu
en
to (
cé
lula
s/
l)
0
200
400
600
800
1000
1200
1400
1600* p<0.05
C1 C2 P1A P1B P1C P2A P2B P2C
**
**
**
*
**
*
*
*
B
Linfocitos T CD4+
Po
rce
nta
je (
%)
0
10
20
30
40
50
C1 C2 P1A P1B P1C P2A P2B P2C
**
*
**
*
**
*
**
*
A Figura 20- Porcentaje
y recuento de
linfocitos T CD4+ en
sangre periférica: se
presentan los resultados
(las líneas negras
horizontales representan
los promedio) del
porcentaje (A) y recuento
(B) de las poblaciones de
linfocitos T CD4+ en sangre
periférica, en los distintos
grupos de estudio.
Controles: C1 (VIH-/T.
gondii-; n=11) y C2 (VIH-
/T. gondii+; n=13);
Pacientes: P1A (VIH+/T.
gondii-; CD4>350/L;
n=12), P1B (VIH+/T.
gondii-; CD4:200-350/L;
n=16), P1C (VIH+/T.
gondii-; CD4<200/L; n=9),
P2A (VIH+/T. gondii+;
CD4>350/L; n=15), P2B
(VIH+/T. gondii+; CD4:200-
350/L; n=17), P2C
(VIH+/T. gondii+;
CD4<200/L; n=12). Los
grupos se compararon
utilizando las pruebas de
ANOVA o Kruskal-Wallis,
con pruebas de
comparación múltiple de
Bonferroni o Dunn,
respectivamente, según
fuese apropiado. Se
consideraron significativos
valores de p<0.05.
45
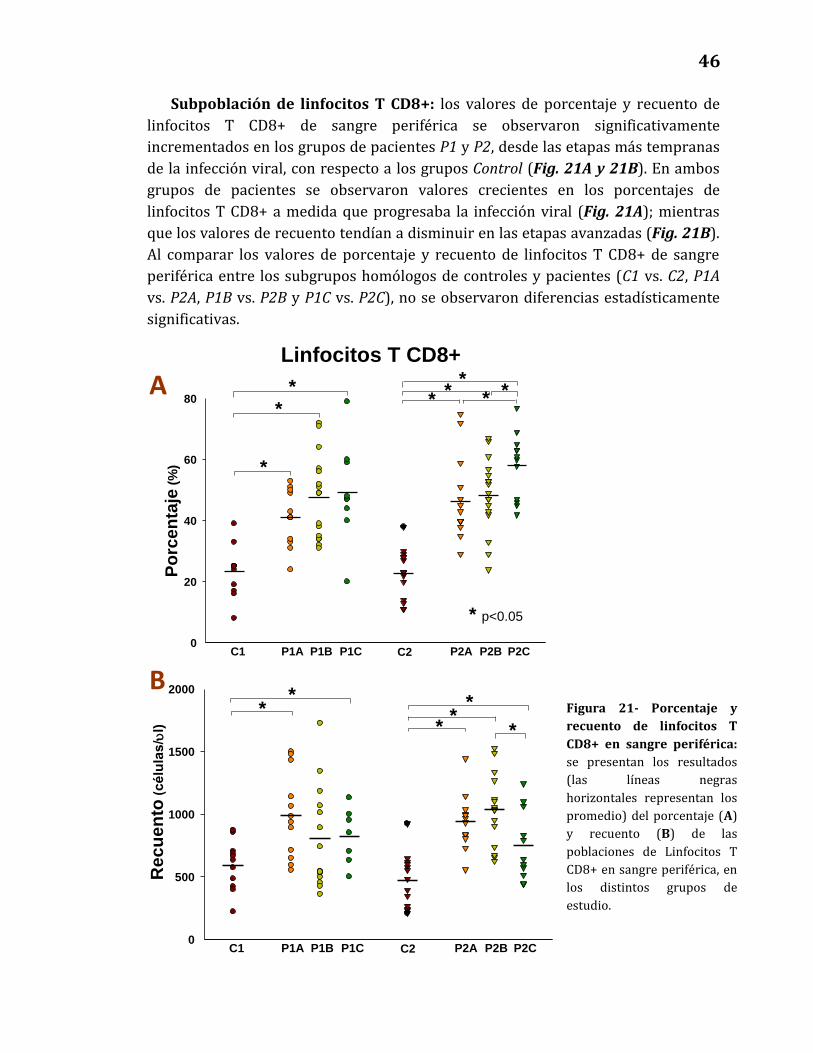
Subpoblación de linfocitos T CD8+: los valores de porcentaje y recuento de
linfocitos T CD8+ de sangre periférica se observaron significativamente
incrementados en los grupos de pacientes P1 y P2, desde las etapas más tempranas
de la infección viral, con respecto a los grupos Control (Fig. 21A y 21B). En ambos
grupos de pacientes se observaron valores crecientes en los porcentajes de
linfocitos T CD8+ a medida que progresaba la infección viral (Fig. 21A); mientras
que los valores de recuento tendían a disminuir en las etapas avanzadas (Fig. 21B).
Al comparar los valores de porcentaje y recuento de linfocitos T CD8+ de sangre
periférica entre los subgrupos homólogos de controles y pacientes (C1 vs. C2, P1A
vs. P2A, P1B vs. P2B y P1C vs. P2C), no se observaron diferencias estadísticamente
significativas.
Re
cu
en
to (
cé
lula
s/
l)
0
500
1000
1500
2000
C1 C2 P1A P1B P1C P2A P2B P2C
***
*
*
*B
Linfocitos T CD8+
Po
rcen
taje
(%
)
0
20
40
60
80
C1 C2 P1A P1B P1C P2A P2B P2C
*
* p<0.05
** *
** * *A
Figura 21- Porcentaje y
recuento de linfocitos T
CD8+ en sangre periférica:
se presentan los resultados
(las líneas negras
horizontales representan los
promedio) del porcentaje (A)
y recuento (B) de las
poblaciones de Linfocitos T
CD8+ en sangre periférica, en
los distintos grupos de
estudio.
46

Relación CD4/CD8*: Así, de acuerdo a los resultados descritos hasta ahora, en la
población de linfocitos T CD4+, la disminución en los valores absolutos se correlacionaba
estrechamente con disminución en el porcentaje de la misma población celular (Fig. 20).
Por el contrario, a medida que disminuía el recuento de linfocitos T CD4+, aumentaba
gradualmente el porcentaje de linfocitos T CD8+ de sangre periférica (correlación negativa
significativa), alcanzando su mayor valor en los grupos de pacientes más
inmunocomprometidos (Fig. 21A). Por su parte, el recuento de linfocitos T CD8+ seguía una
dinámica distinta al porcentaje de la misma población, pues, a pesar de que los pacientes
presentaban valores mayores que los grupos Control desde etapas iniciales de la infección
viral, dichos valores no aumentaban progresivamente al progresar la infección, sino que en
la etapa avanzada tendían a ser menores (Fig. 21B), indicando una disminución de ambas
subpoblaciones de linfocitos T (CD4+ y CD8+) en las etapas avanzadas de la infección viral,
si bien la población CD4+ presentaba la disminución proporcionalmente mayor. De hecho,
mientras que en los grupos de individuos no infectados por VIH-1 la relación CD4/CD8 fue
>1, en los pacientes infectados esta relación se invirtió, es decir, fue <1, y disminuyó en
forma significativa a medida que progresaba la infección viral (Fig. 22).
* Relación CD4/CD8: relación matemática de dividir el número absoluto de linfocitos T CD4+ de
sangre periférica (recuento), entre el número absoluto de linfocitos T CD8+.
Relación CD4/CD8
Rela
ció
n C
D4/C
D8
0,0
0,5
1,0
1,5
2,0
2,5
* p<0.05
C1 C2 P1A P1B P1C P2A P2B P2C
** *
*
**
*
**
*
**
Figura 22- Relación CD4/CD8: se presentan los resultados (las líneas negras horizontales representan
los promedio) de la relación entre las poblaciones de Linfocitos T CD4+ y T CD8+ de sangre periférica, en los
distintos grupos de estudio.
47
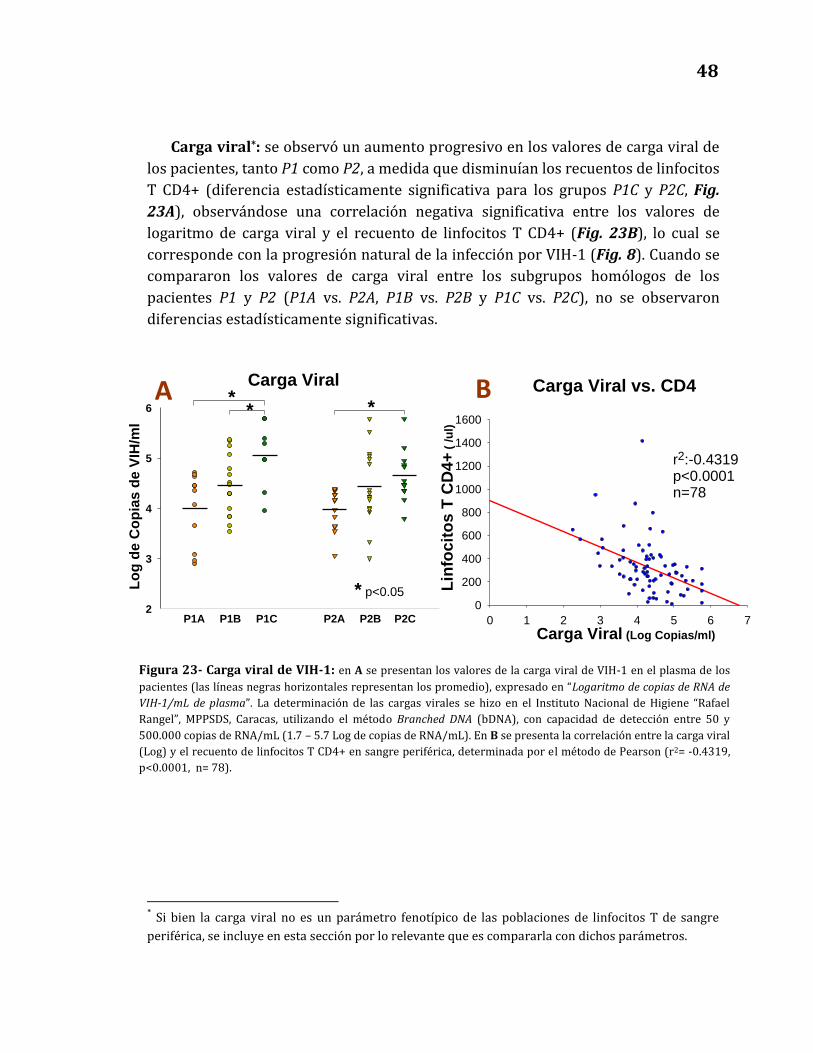
Carga viral*: se observó un aumento progresivo en los valores de carga viral de
los pacientes, tanto P1 como P2, a medida que disminuían los recuentos de linfocitos
T CD4+ (diferencia estadísticamente significativa para los grupos P1C y P2C, Fig.
23A), observándose una correlación negativa significativa entre los valores de
logaritmo de carga viral y el recuento de linfocitos T CD4+ (Fig. 23B), lo cual se
corresponde con la progresión natural de la infección por VIH-1 (Fig. 8). Cuando se
compararon los valores de carga viral entre los subgrupos homólogos de los
pacientes P1 y P2 (P1A vs. P2A, P1B vs. P2B y P1C vs. P2C), no se observaron
diferencias estadísticamente significativas.
* Si bien la carga viral no es un parámetro fenotípico de las poblaciones de linfocitos T de sangre
periférica, se incluye en esta sección por lo relevante que es compararla con dichos parámetros.
Figura 23- Carga viral de VIH-1: en A se presentan los valores de la carga viral de VIH-1 en el plasma de los
pacientes (las líneas negras horizontales representan los promedio), expresado en “Logaritmo de copias de RNA de
VIH-1/mL de plasma”. La determinación de las cargas virales se hizo en el Instituto Nacional de Higiene “Rafael
Rangel”, MPPSDS, Caracas, utilizando el método Branched DNA (bDNA), con capacidad de detección entre 50 y
500.000 copias de RNA/mL (1.7 – 5.7 Log de copias de RNA/mL). En B se presenta la correlación entre la carga viral
(Log) y el recuento de linfocitos T CD4+ en sangre periférica, determinada por el método de Pearson (r2= -0.4319,
p<0.0001, n= 78).
Carga Viral
Lo
g d
e C
op
ias d
e V
IH/m
l
2
3
4
5
6
* p<0.05
P1A P1B P1C P2A P2B P2C
** *
A Carga Viral vs. CD4
Carga Viral (Log Copias/ml)
0 1 2 3 4 5 6 7
Lin
foc
ito
s T
CD
4+
( /
ul)
0
200
400
600
800
1000
1200
1400
1600
r2:-0.4319 p<0.0001n=78
B
48
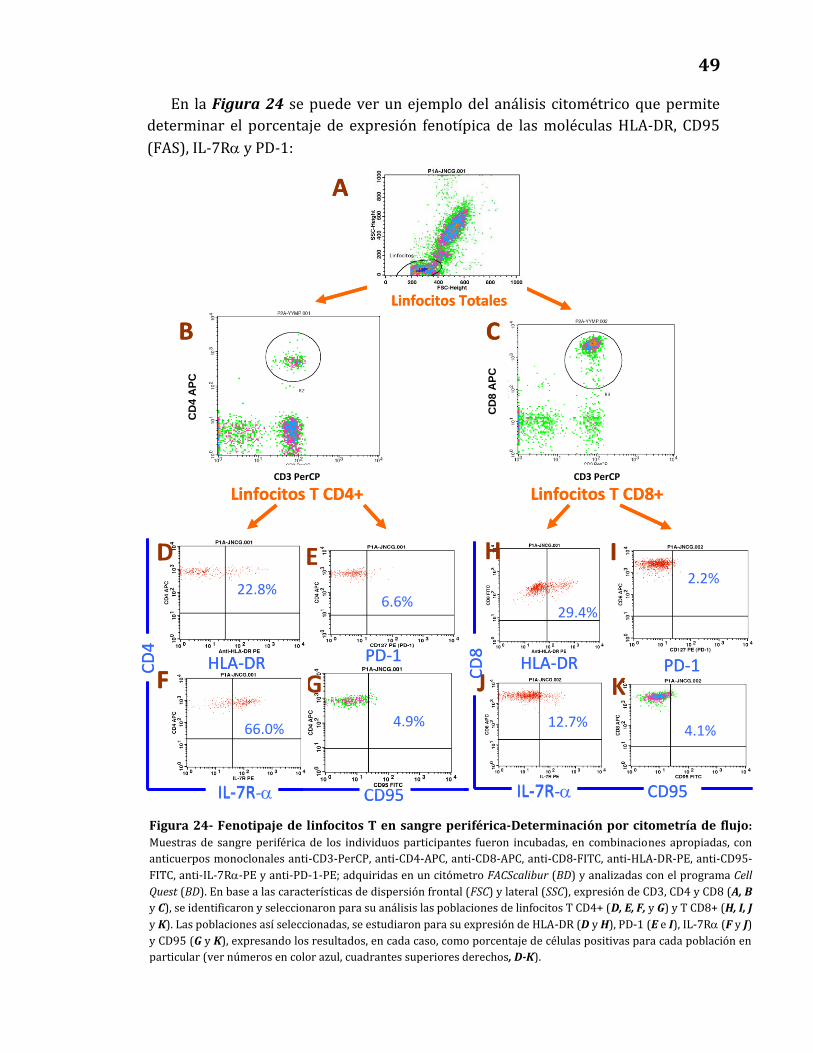
En la Figura 24 se puede ver un ejemplo del análisis citométrico que permite
determinar el porcentaje de expresión fenotípica de las moléculas HLA-DR, CD95
(FAS), IL-7R y PD-1:
Figura 24- Fenotipaje de linfocitos T en sangre periférica-Determinación por citometría de flujo:
Muestras de sangre periférica de los individuos participantes fueron incubadas, en combinaciones apropiadas, con
anticuerpos monoclonales anti-CD3-PerCP, anti-CD4-APC, anti-CD8-APC, anti-CD8-FITC, anti-HLA-DR-PE, anti-CD95-
FITC, anti-IL-7R-PE y anti-PD-1-PE; adquiridas en un citómetro FACScalibur (BD) y analizadas con el programa Cell
Quest (BD). En base a las características de dispersión frontal (FSC) y lateral (SSC), expresión de CD3, CD4 y CD8 (A, B
y C), se identificaron y seleccionaron para su análisis las poblaciones de linfocitos T CD4+ (D, E, F, y G) y T CD8+ (H, I, J
y K). Las poblaciones así seleccionadas, se estudiaron para su expresión de HLA-DR (D y H), PD-1 (E e I), IL-7R (F y J)
y CD95 (G y K), expresando los resultados, en cada caso, como porcentaje de células positivas para cada población en
particular (ver números en color azul, cuadrantes superiores derechos, D-K).
12.7%
J
29.4%
H
HLA-DR
CD95
4.1%
K
IL-7R-
CD
8
PD-1
I 2.2%
4.9%
G
CD95
22.8%
D
HLA-DR
IL-7R-
66.0%
F
6.6%
E
PD-1
CD
4
Linfocitos Totales
A
B
CD3 PerCP
CD
4 A
PC
Linfocitos T CD4+
C
CD3 PerCP
CD
8 A
PC
Linfocitos T CD8+
12.7%
J
29.4%
H
HLA-DR
CD95
4.1%
K
IL-7R
CD
8
PD-1
I 2.2%
4.9%
G
CD95
22.8%
D
HLA-DR
IL-7R
66.0%
F
6.6%
E
PD-1
CD
4
Linfocitos Totales
A
B
CD3 PerCP
CD
4 A
PC
Linfocitos T CD4+
C
CD3 PerCP
CD
8 A
PC
Linfocitos T CD8+
49
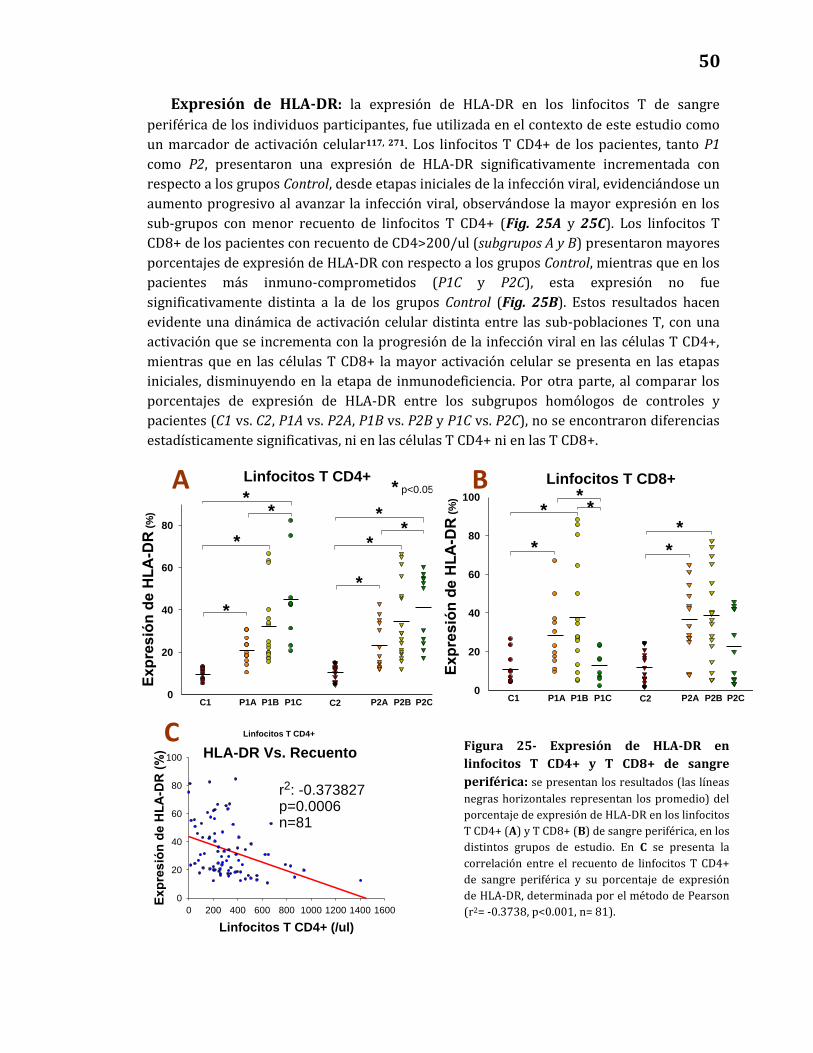
Expresión de HLA-DR: la expresión de HLA-DR en los linfocitos T de sangre
periférica de los individuos participantes, fue utilizada en el contexto de este estudio como
un marcador de activación celular117, 271. Los linfocitos T CD4+ de los pacientes, tanto P1
como P2, presentaron una expresión de HLA-DR significativamente incrementada con
respecto a los grupos Control, desde etapas iniciales de la infección viral, evidenciándose un
aumento progresivo al avanzar la infección viral, observándose la mayor expresión en los
sub-grupos con menor recuento de linfocitos T CD4+ (Fig. 25A y 25C). Los linfocitos T
CD8+ de los pacientes con recuento de CD4>200/ul (subgrupos A y B) presentaron mayores
porcentajes de expresión de HLA-DR con respecto a los grupos Control, mientras que en los
pacientes más inmuno-comprometidos (P1C y P2C), esta expresión no fue
significativamente distinta a la de los grupos Control (Fig. 25B). Estos resultados hacen
evidente una dinámica de activación celular distinta entre las sub-poblaciones T, con una
activación que se incrementa con la progresión de la infección viral en las células T CD4+,
mientras que en las células T CD8+ la mayor activación celular se presenta en las etapas
iniciales, disminuyendo en la etapa de inmunodeficiencia. Por otra parte, al comparar los
porcentajes de expresión de HLA-DR entre los subgrupos homólogos de controles y
pacientes (C1 vs. C2, P1A vs. P2A, P1B vs. P2B y P1C vs. P2C), no se encontraron diferencias
estadísticamente significativas, ni en las células T CD4+ ni en las T CD8+.
Figura 25- Expresión de HLA-DR en
linfocitos T CD4+ y T CD8+ de sangre
periférica: se presentan los resultados (las líneas
negras horizontales representan los promedio) del
porcentaje de expresión de HLA-DR en los linfocitos
T CD4+ (A) y T CD8+ (B) de sangre periférica, en los
distintos grupos de estudio. En C se presenta la
correlación entre el recuento de linfocitos T CD4+
de sangre periférica y su porcentaje de expresión
de HLA-DR, determinada por el método de Pearson
(r2= -0.3738, p<0.001, n= 81).
Linfocitos T CD4+
HLA-DR Vs. Recuento
Linfocitos T CD4+ (/ul)
0 200 400 600 800 1000 1200 1400 1600
Ex
pre
sió
n d
e H
LA
-DR
(%
)
0
20
40
60
80
100
r2: -0.373827p=0.0006n=81
C
Linfocitos T CD4+
Ex
pre
sió
n d
e H
LA
-DR
(%
)
0
20
40
60
80
* p<0.05
C1 C2 P1A P1B P1C P2A P2B P2C
*
**
* **
*
*
A Linfocitos T CD8+
Ex
pre
sió
n d
e H
LA
-DR
(%
)
0
20
40
60
80
100
C1 C2 P1A P1B P1C P2A P2B P2C
*
**
*
**
B
50
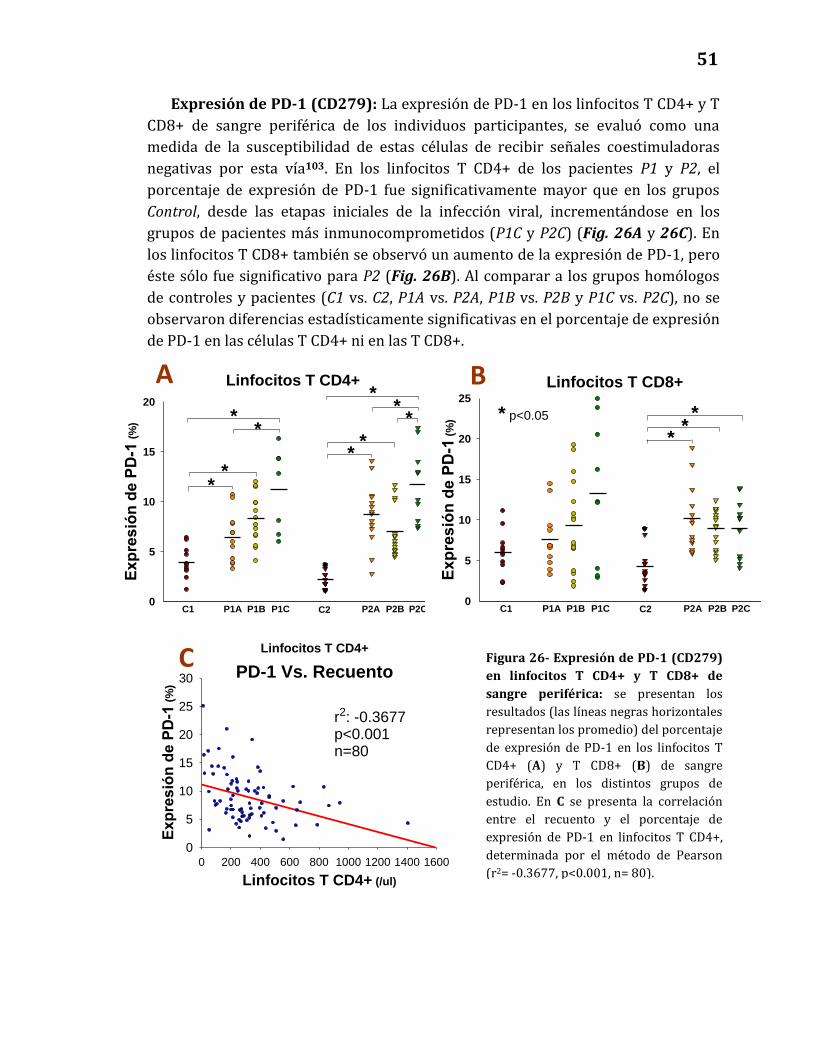
Expresión de PD-1 (CD279): La expresión de PD-1 en los linfocitos T CD4+ y T
CD8+ de sangre periférica de los individuos participantes, se evaluó como una
medida de la susceptibilidad de estas células de recibir señales coestimuladoras
negativas por esta vía103. En los linfocitos T CD4+ de los pacientes P1 y P2, el
porcentaje de expresión de PD-1 fue significativamente mayor que en los grupos
Control, desde las etapas iniciales de la infección viral, incrementándose en los
grupos de pacientes más inmunocomprometidos (P1C y P2C) (Fig. 26A y 26C). En
los linfocitos T CD8+ también se observó un aumento de la expresión de PD-1, pero
éste sólo fue significativo para P2 (Fig. 26B). Al comparar a los grupos homólogos
de controles y pacientes (C1 vs. C2, P1A vs. P2A, P1B vs. P2B y P1C vs. P2C), no se
observaron diferencias estadísticamente significativas en el porcentaje de expresión
de PD-1 en las células T CD4+ ni en las T CD8+.
Figura 26- Expresión de PD-1 (CD279)
en linfocitos T CD4+ y T CD8+ de
sangre periférica: se presentan los
resultados (las líneas negras horizontales
representan los promedio) del porcentaje
de expresión de PD-1 en los linfocitos T
CD4+ (A) y T CD8+ (B) de sangre
periférica, en los distintos grupos de
estudio. En C se presenta la correlación
entre el recuento y el porcentaje de
expresión de PD-1 en linfocitos T CD4+,
determinada por el método de Pearson
(r2= -0.3677, p<0.001, n= 80).
Linfocitos T CD4+
PD-1 Vs. Recuento
Linfocitos T CD4+ (/ul)
0 200 400 600 800 1000 1200 1400 1600
Ex
pre
sió
n d
e P
D-1
(%
)
0
5
10
15
20
25
30
r2: -0.3677p<0.001n=80
C
Linfocitos T CD4+
Ex
pre
sió
n d
e P
D-1
(%
)
0
5
10
15
20
C1 C2 P1A P1B P1C P2A P2B P2C
*
**
**
**
**
A Linfocitos T CD8+
Ex
pre
sió
n d
e P
D-1
(%
)
0
5
10
15
20
25
C1 C2 P1A P1B P1C P2A P2B P2C
*
* p<0.05**
B
51
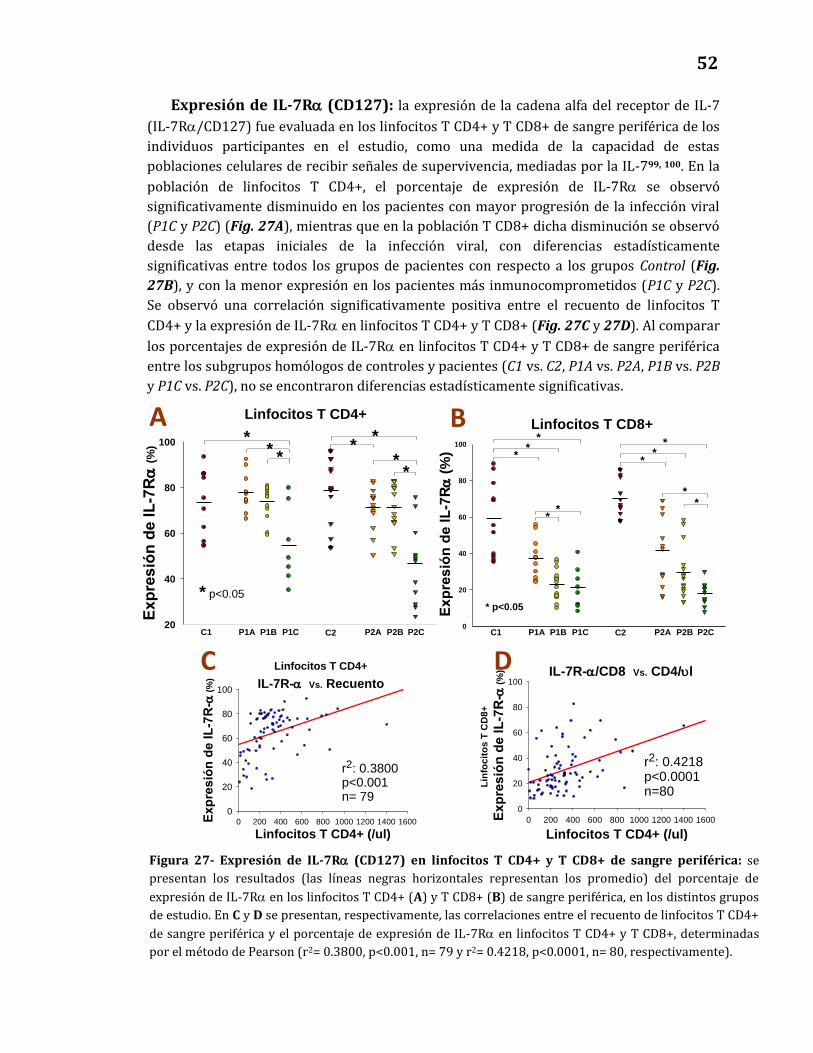
Expresión de IL-7R (CD127): la expresión de la cadena alfa del receptor de IL-7
(IL-7R/CD127) fue evaluada en los linfocitos T CD4+ y T CD8+ de sangre periférica de los
individuos participantes en el estudio, como una medida de la capacidad de estas
poblaciones celulares de recibir señales de supervivencia, mediadas por la IL-799, 100. En la
población de linfocitos T CD4+, el porcentaje de expresión de IL-7R se observó
significativamente disminuido en los pacientes con mayor progresión de la infección viral
(P1C y P2C) (Fig. 27A), mientras que en la población T CD8+ dicha disminución se observó
desde las etapas iniciales de la infección viral, con diferencias estadísticamente
significativas entre todos los grupos de pacientes con respecto a los grupos Control (Fig.
27B), y con la menor expresión en los pacientes más inmunocomprometidos (P1C y P2C).
Se observó una correlación significativamente positiva entre el recuento de linfocitos T
CD4+ y la expresión de IL-7R en linfocitos T CD4+ y T CD8+ (Fig. 27C y 27D). Al comparar
los porcentajes de expresión de IL-7R en linfocitos T CD4+ y T CD8+ de sangre periférica
entre los subgrupos homólogos de controles y pacientes (C1 vs. C2, P1A vs. P2A, P1B vs. P2B
y P1C vs. P2C), no se encontraron diferencias estadísticamente significativas.
Figura 27- Expresión de IL-7R (CD127) en linfocitos T CD4+ y T CD8+ de sangre periférica: se
presentan los resultados (las líneas negras horizontales representan los promedio) del porcentaje de
expresión de IL-7R en los linfocitos T CD4+ (A) y T CD8+ (B) de sangre periférica, en los distintos grupos
de estudio. En C y D se presentan, respectivamente, las correlaciones entre el recuento de linfocitos T CD4+
de sangre periférica y el porcentaje de expresión de IL-7R en linfocitos T CD4+ y T CD8+, determinadas
por el método de Pearson (r2= 0.3800, p<0.001, n= 79 y r2= 0.4218, p<0.0001, n= 80, respectivamente).
Linfocitos T CD4+
IL-7R- Vs. Recuento
Linfocitos T CD4+ (/ul)0 200 400 600 800 1000 1200 1400 1600Exp
resió
n d
e IL
-7R
- (
%)
0
20
40
60
80
100
r2: 0.3800p<0.001n= 79
C IL-7R-/CD8 Vs. CD4/l
Linfocitos T CD4+ (/ul)
0 200 400 600 800 1000 1200 1400 1600
Lin
focit
os T
CD
8+
Ex
pre
sió
n d
e I
L-7
R-
(%
)
0
20
40
60
80
100
r2: 0.4218 p<0.0001n=80
D
Linfocitos T CD4+
Ex
pre
sió
n d
e IL
-7R
(%
)
20
40
60
80
100
* p<0.05
C1 C2 P1A P1B P1C P2A P2B P2C
*
**
* ** *
A Linfocitos T CD8+
Ex
pre
sió
n d
e I
L-7
R
(%
)
0
20
40
60
80
100
C1 C2 P1A P1B P1C P2A P2B P2C
**
*
**
**
* p<0.05
* *
*
B
52

Expresión de CD95 (FAS): la expresión de CD95, como un marcador de la
susceptibilidad a sufrir apoptosis, mediada por este receptor96, 97, fue evaluada en los
linfocitos T CD4+ y T CD8+ de sangre periférica de los individuos participantes. En la
población de células T CD4+ de los pacientes se observó un discreto aumento de la
expresión de CD95 en las etapas iniciales de la infección viral (diferencia significativa para
P2A y P2B) al compararla con los grupos Control. Fue resaltante, sin embargo, que en los
pacientes más inmunocomprometidos (P1C y P2C) los porcentajes de expresión de CD95
alcanzaron su máximo valor, con diferencias significativas al resto de los grupos, siendo, en
promedio, 3 veces mayores a los de los grupos Control (Fig. 28A). Al comparar los valores
de recuento y expresión de CD95 en la población CD4+, se evidenció una significativa
correlación negativa (Fig. 28C). Por su parte, la expresión de CD95 en los linfocitos T CD8+
de sangre periférica de los pacientes (P1 y P2), presentó valores discretamente mayores a
los grupos Control, con diferencias significativas para P2A y P2B (Fig. 28B). Al comparar los
porcentajes de expresión de CD95 en linfocitos T CD4+ y T CD8+ de sangre periférica entre
los subgrupos homólogos de controles y pacientes (C1 vs. C2, P1A vs. P2A, P1B vs. P2B y P1C
vs. P2C), no se encontraron diferencias estadísticamente significativas.
Figura 28- Expresión de CD95 (FAS) en
linfocitos T CD4+ y T CD8+ de sangre
periférica: se presentan los resultados (las
líneas negras horizontales representan los
promedio) del porcentaje de expresión de
CD95 en los linfocitos T CD4+ (A) y T CD8+
(B) de sangre periférica, en los distintos
grupos de estudio. En C se presenta la
correlación entre el recuento de linfocitos T CD4+
de sangre periférica y su porcentaje de expresión
de CD95, determinada por el método de Pearson
(r2= -0.3580, p=0.00103, n= 81).
Linfocitos T CD4+
CD95 Vs. Recuento
Linfocitos T CD4+ (/ul)
0 200 400 600 800 1000 1200 1400 1600
Exp
resió
n d
e C
D95
(%
)
0
10
20
30
40
50
r2: -0.35796
p=0.00103n=81
C
Linfocitos T CD4+
Ex
pre
sió
n d
e C
D9
5 (
%)
0
5
10
15
20
25
30
* p<0.05
C1 C2 P1A P1B P1C P2A P2B P2C
*
*
**
*
**
*
A Linfocitos T CD8+E
xp
res
ión
de
CD
95
(%
)
0
2
4
6
8
10
C1 C2 P1A P1B P1C P2A P2B P2C
**
B
53
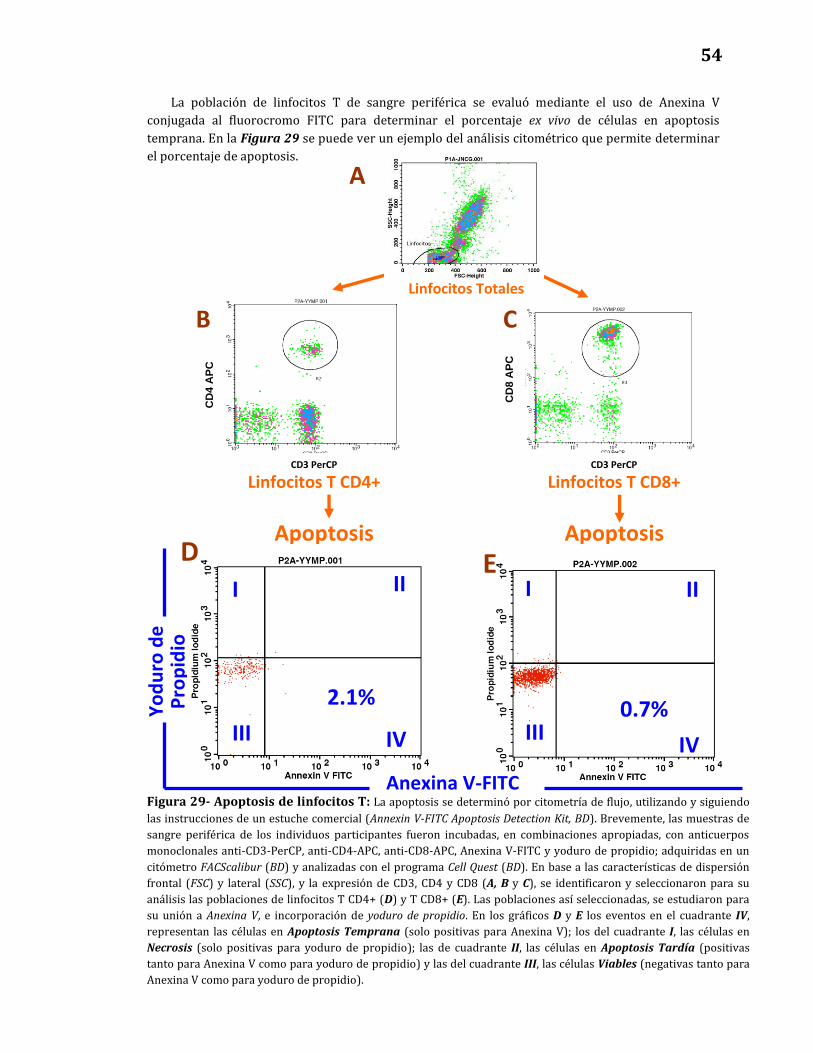
La población de linfocitos T de sangre periférica se evaluó mediante el uso de Anexina V
conjugada al fluorocromo FITC para determinar el porcentaje ex vivo de células en apoptosis
temprana. En la Figura 29 se puede ver un ejemplo del análisis citométrico que permite determinar
el porcentaje de apoptosis.
Figura 29- Apoptosis de linfocitos T: La apoptosis se determinó por citometría de flujo, utilizando y siguiendo
las instrucciones de un estuche comercial (Annexin V-FITC Apoptosis Detection Kit, BD). Brevemente, las muestras de
sangre periférica de los individuos participantes fueron incubadas, en combinaciones apropiadas, con anticuerpos
monoclonales anti-CD3-PerCP, anti-CD4-APC, anti-CD8-APC, Anexina V-FITC y yoduro de propidio; adquiridas en un
citómetro FACScalibur (BD) y analizadas con el programa Cell Quest (BD). En base a las características de dispersión
frontal (FSC) y lateral (SSC), y la expresión de CD3, CD4 y CD8 (A, B y C), se identificaron y seleccionaron para su
análisis las poblaciones de linfocitos T CD4+ (D) y T CD8+ (E). Las poblaciones así seleccionadas, se estudiaron para
su unión a Anexina V, e incorporación de yoduro de propidio. En los gráficos D y E los eventos en el cuadrante IV,
representan las células en Apoptosis Temprana (solo positivas para Anexina V); los del cuadrante I, las células en
Necrosis (solo positivas para yoduro de propidio); las de cuadrante II, las células en Apoptosis Tardía (positivas
tanto para Anexina V como para yoduro de propidio) y las del cuadrante III, las células Viables (negativas tanto para
Anexina V como para yoduro de propidio).
Apoptosis Apoptosis
2.1%
I
III
II
IV
D
0.7%
II
IV
I
III
E
Yo
du
ro d
e
P
rop
idio
Anexina V-FITC
Linfocitos Totales
A
B
CD3 PerCP
CD
4 A
PC
Linfocitos T CD4+
C
CD3 PerCP
CD
8 A
PC
Linfocitos T CD8+
54
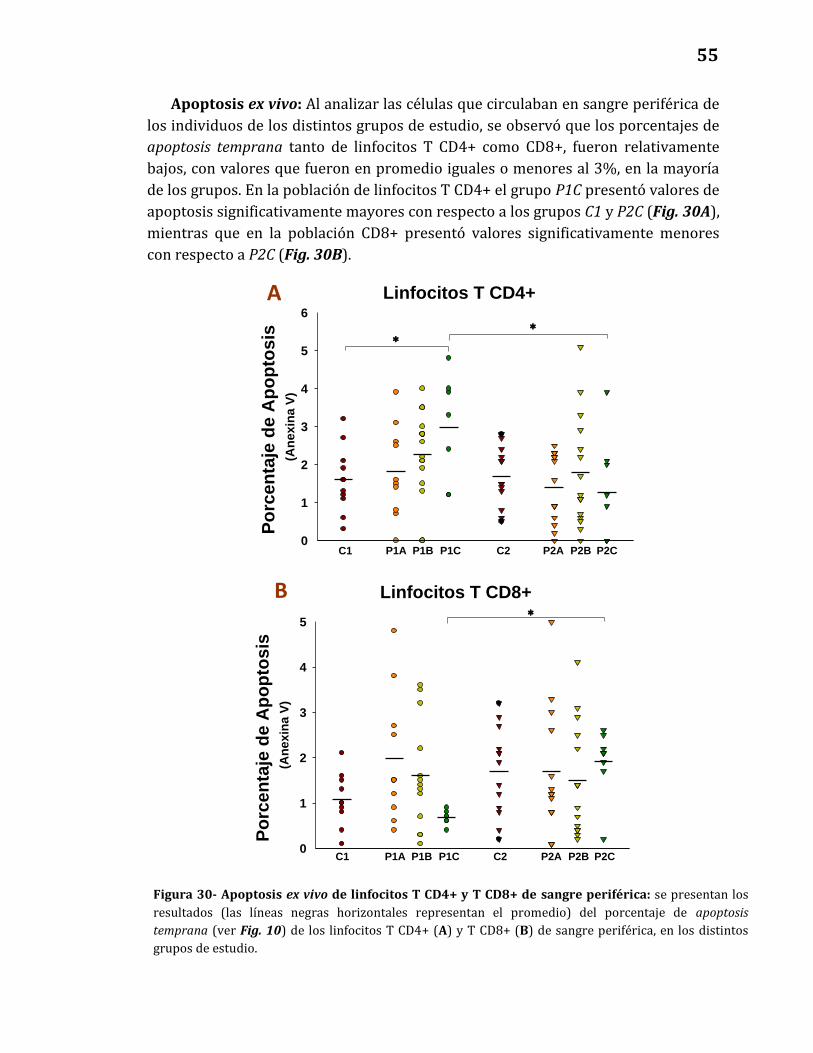
Apoptosis ex vivo: Al analizar las células que circulaban en sangre periférica de
los individuos de los distintos grupos de estudio, se observó que los porcentajes de
apoptosis temprana tanto de linfocitos T CD4+ como CD8+, fueron relativamente
bajos, con valores que fueron en promedio iguales o menores al 3%, en la mayoría
de los grupos. En la población de linfocitos T CD4+ el grupo P1C presentó valores de
apoptosis significativamente mayores con respecto a los grupos C1 y P2C (Fig. 30A),
mientras que en la población CD8+ presentó valores significativamente menores
con respecto a P2C (Fig. 30B).
Figura 30- Apoptosis ex vivo de linfocitos T CD4+ y T CD8+ de sangre periférica: se presentan los
resultados (las líneas negras horizontales representan el promedio) del porcentaje de apoptosis
temprana (ver Fig. 10) de los linfocitos T CD4+ (A) y T CD8+ (B) de sangre periférica, en los distintos
grupos de estudio.
Linfocitos T CD8+
Po
rcen
taje
de A
po
pto
sis
(A
nexin
a V
)
0
1
2
3
4
5
C1 C2 P1A P1B P1C P2A P2B P2C
B
Linfocitos T CD4+
Po
rcen
taje
de A
po
pto
sis
(A
ne
xin
a V
)
0
1
2
3
4
5
6
C1 C2 P1A P1B P1C P2A P2B P2C
A
55
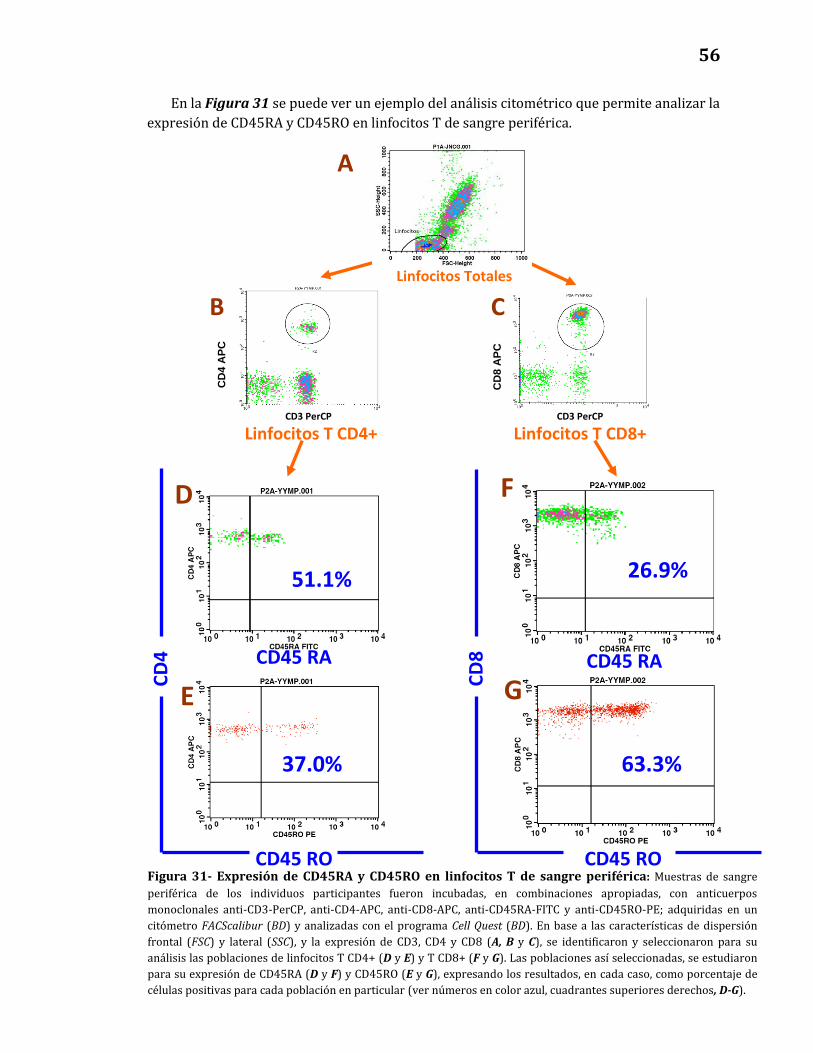
En la Figura 31 se puede ver un ejemplo del análisis citométrico que permite analizar la
expresión de CD45RA y CD45RO en linfocitos T de sangre periférica.
Figura 31- Expresión de CD45RA y CD45RO en linfocitos T de sangre periférica: Muestras de sangre
periférica de los individuos participantes fueron incubadas, en combinaciones apropiadas, con anticuerpos
monoclonales anti-CD3-PerCP, anti-CD4-APC, anti-CD8-APC, anti-CD45RA-FITC y anti-CD45RO-PE; adquiridas en un
citómetro FACScalibur (BD) y analizadas con el programa Cell Quest (BD). En base a las características de dispersión
frontal (FSC) y lateral (SSC), y la expresión de CD3, CD4 y CD8 (A, B y C), se identificaron y seleccionaron para su
análisis las poblaciones de linfocitos T CD4+ (D y E) y T CD8+ (F y G). Las poblaciones así seleccionadas, se estudiaron
para su expresión de CD45RA (D y F) y CD45RO (E y G), expresando los resultados, en cada caso, como porcentaje de
células positivas para cada población en particular (ver números en color azul, cuadrantes superiores derechos, D-G).
CD
4
51.1%
D
CD45 RA
CD
8
26.9%
F
CD45 RA
63.3%
G
CD45 RO
37.0%
E
CD45 RO
Linfocitos Totales
A
B
CD3 PerCP
CD
4 A
PC
Linfocitos T CD4+
C
CD3 PerCP
CD
8 A
PC
Linfocitos T CD8+
56

Expresión de CD45RA y CD45RO en linfocitos T: La expresión de CD45RA y
CD45RO en linfocitos T se utilizó como una medida relativa de la población de
células vírgenes y de memoria, respectivamente251. Al evaluar los porcentajes de
expresión de CD45RA en la población de linfocitos T CD4+ de sangre periférica, se
observaron valores significativamente menores en los grupos con mayor progresión
de la infección viral (P1C y P2C) con respecto a los grupos con menor progresión
(P1A y P2A, respectivamente, Fig. 32A). Por su parte, en todos los grupos de
pacientes se observaron mayores valores en los porcentajes de expresión de
CD45RO en la población de linfocitos T CD4+ que en los grupos Control (diferencias
estadísticamente significativas para los grupos P2B y P2C, Fig. 32B). La dinámica de
la expresión de las moléculas CD45RA y CD45RO en la población CD4+ (Fig. 32A y
32B), sugiere que la disminución de células vírgenes en esta población es mayor
que la de células de memoria, al progresar la infección viral, con disminución de la
relación CD45RA/CD45RO (Fig. 34A). Cuando se compararon los valores de
recuento de linfocitos T CD4+/CD45RA+ y T CD4+/CD45RO+ de sangre periférica,
se pudo constatar que todos los grupos de pacientes infectados con VIH-1
presentaron valores menores a los de sus grupos Control respectivos (Fig. 32C y D),
lo cual se corresponde con la disminución de la población de células T CD4+ totales
(Fig. 20B). Al evaluar la población de linfocitos T CD8+, los porcentajes de
expresión de la molécula CD45RA fueron menores en los pacientes VIH-1+
(diferencias significativas para P1A, P1B, P1C y P2C, Fig. 33A) y los de CD45RO
mayores (diferencias significativas para todos los grupos de pacientes, Fig. 33B)
con respecto a sus grupos Control, con valores de relación CD45RA/CD45RO
menores en todos los grupos de pacientes, evidenciando una mayor proporción de
células de memoria en éstos, con respecto a sus grupos Control (Fig. 34B). Los
recuentos de linfocitos T CD8+ que expresaban CD45RA presentaron un discreto
aumento en los pacientes infectados con VIH-1, con diferencia significativa con
respecto al Control solo en los grupos P2A y P2B (Fig. 33C). Por su parte los
recuentos de linfocitos T CD8+ que expresaban la molécula CD45RO, fueron
consistente y significativamente mayores en todos los grupos de pacientes
infectados con VIH-1 al compararlos con los individuos no infectados (Fig. 33D),
evidenciando que el principal grupo celular que contribuye con el aumento del
recuento de células T CD8+ asociado a la infección por VIH-1 posee marcadores
fenotípicos de células de memoria. Por último, al comparar los porcentajes y
recuentos de linfocitos T CD4+ y T CD8+ de sangre periférica que expresaban las
moléculas CD45RA y CD45RO, no se observaron diferencias significativas al
comparar a los grupos de individuos con serología positiva para T. gondii y sus
grupos homólogos con serología negativa para el parásito (C1 vs. C2, P1A vs. P2A,
P1B vs. P2B y P1C vs. P2C).
57
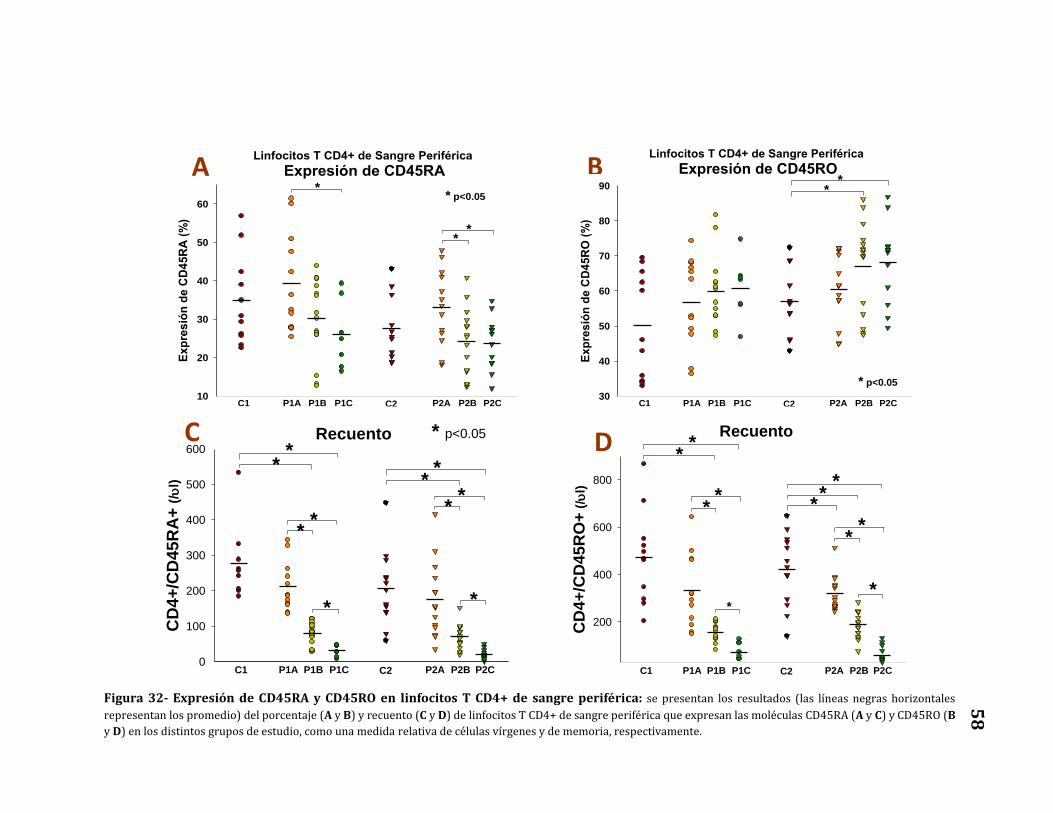
Linfocitos T CD4+ de Sangre Periférica
Expresión de CD45RAE
xp
res
ión
de
CD
45
RA
(%
)
10
20
30
40
50
60
(VIH+/T.gondii-) (VIH+/T.gondii+)C1 C2 P1A P1B P1C P2A P2B P2C
(Control)(Control)
*
**
* p<0.05
A Linfocitos T CD4+ de Sangre Periférica
Expresión de CD45RO
Ex
pre
sió
n d
e C
D4
5R
O (
%)
30
40
50
60
70
80
90
(VIH+/T.gondii-) (VIH+/T.gondii+)C1 C2 P1A P1B P1C P2A P2B P2C
(Control)(Control)
**
* p<0.05
B
Recuento
CD
4+
/CD
45
RA
+ (
/l)
0
100
200
300
400
500
600
C1 C2 P1A P1B P1C P2A P2B P2C
**
* p<0.05
*
**
*
**
**
C Recuento
CD
4+
/CD
45
RO
+ (
/l)
200
400
600
800
C1 C2 P1A P1B P1C P2A P2B P2C
**
**
*
**
**
*
*
D
Figura 32- Expresión de CD45RA y CD45RO en linfocitos T CD4+ de sangre periférica: se presentan los resultados (las líneas negras horizontales
representan los promedio) del porcentaje (A y B) y recuento (C y D) de linfocitos T CD4+ de sangre periférica que expresan las moléculas CD45RA (A y C) y CD45RO (B
y D) en los distintos grupos de estudio, como una medida relativa de células vírgenes y de memoria, respectivamente.
58
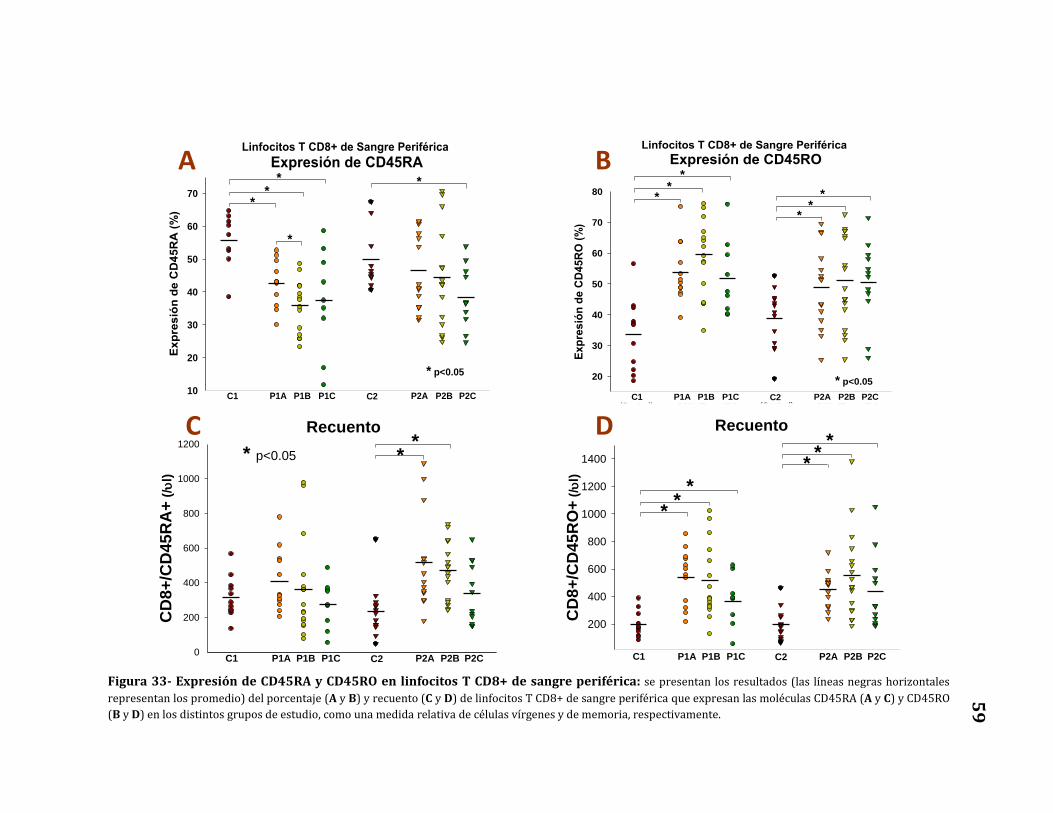
Linfocitos T CD8+ de Sangre Periférica
Expresión de CD45RAE
xp
res
ión
de
CD
45
RA
(%
)
10
20
30
40
50
60
70
(VIH+/T.gondii-) (VIH+/T.gondii+)C1 C2 P1A P1B P1C P2A P2B P2C
(Control)(Control)
*
* p<0.05
**
*
*
A Linfocitos T CD8+ de Sangre Periférica
Expresión de CD45RO
Ex
pre
sió
n d
e C
D45
RO
(%
)
20
30
40
50
60
70
80
(VIH+/T.gondii-) (VIH+/T.gondii+)C1 C2 P1A P1B P1C P2A P2B P2C
(Control)(Control)
* p<0.05
**
*
**
*
B
Recuento
CD
8+
/CD
45
RO
+ (
/l)
200
400
600
800
1000
1200
1400
C1 C2 P1A P1B P1C P2A P2B P2C
**
*
**
*
D Recuento
CD
8+
/CD
45
RA
+ (
/l)
0
200
400
600
800
1000
1200
C1 C2 P1A P1B P1C P2A P2B P2C
* p<0.05*
*
C
Figura 33- Expresión de CD45RA y CD45RO en linfocitos T CD8+ de sangre periférica: se presentan los resultados (las líneas negras horizontales
representan los promedio) del porcentaje (A y B) y recuento (C y D) de linfocitos T CD8+ de sangre periférica que expresan las moléculas CD45RA (A y C) y CD45RO
(B y D) en los distintos grupos de estudio, como una medida relativa de células vírgenes y de memoria, respectivamente.
59
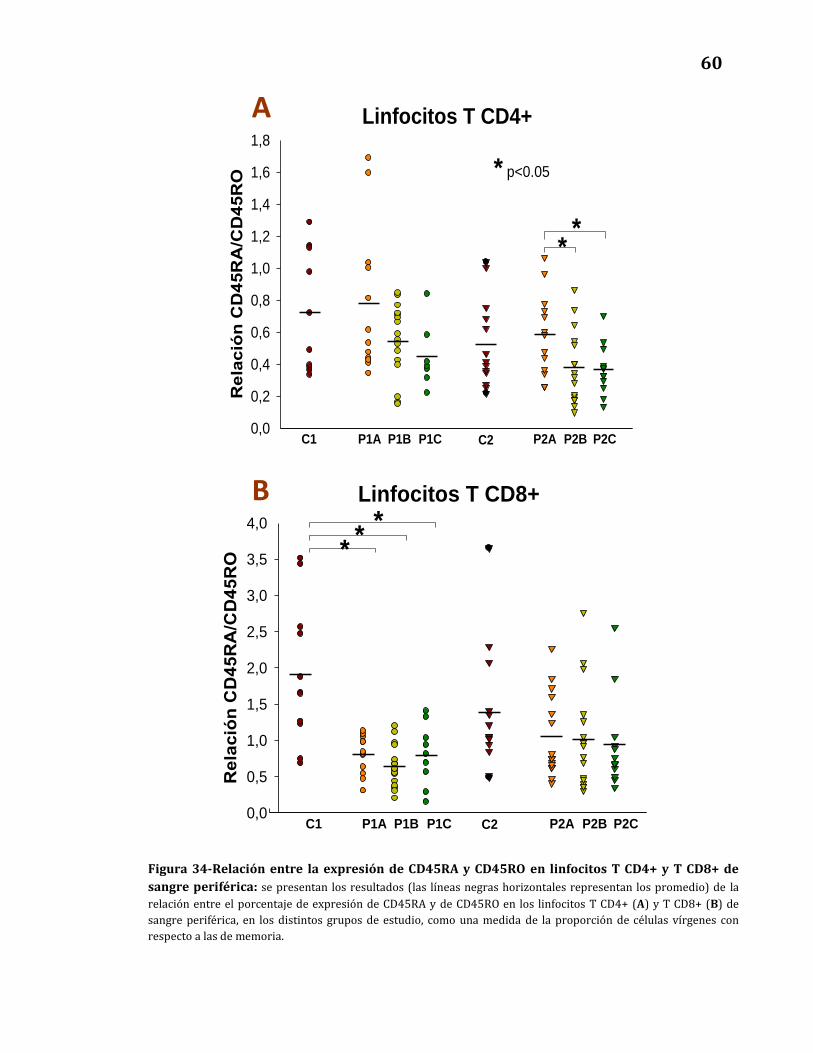
Figura 34-Relación entre la expresión de CD45RA y CD45RO en linfocitos T CD4+ y T CD8+ de
sangre periférica: se presentan los resultados (las líneas negras horizontales representan los promedio) de la
relación entre el porcentaje de expresión de CD45RA y de CD45RO en los linfocitos T CD4+ (A) y T CD8+ (B) de
sangre periférica, en los distintos grupos de estudio, como una medida de la proporción de células vírgenes con
respecto a las de memoria.
Linfocitos T CD4+
Rela
ció
n C
D45R
A/C
D45R
O
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
C1 C2 P1A P1B P1C P2A P2B P2C
**
* p<0.05
A
Linfocitos T CD8+
Re
lac
ión
CD
45
RA
/CD
45
RO
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
C1 C2 P1A P1B P1C P2A P2B P2C
**
*B
60
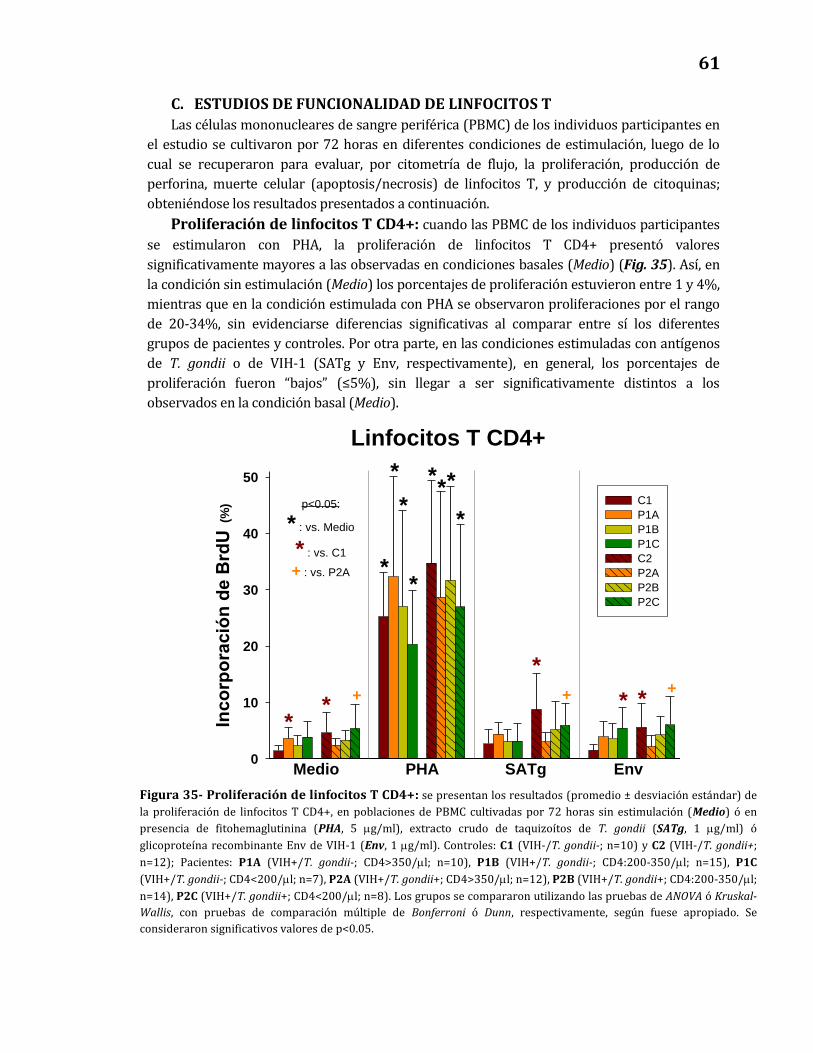
C. ESTUDIOS DE FUNCIONALIDAD DE LINFOCITOS T
Las células mononucleares de sangre periférica (PBMC) de los individuos participantes en
el estudio se cultivaron por 72 horas en diferentes condiciones de estimulación, luego de lo
cual se recuperaron para evaluar, por citometría de flujo, la proliferación, producción de
perforina, muerte celular (apoptosis/necrosis) de linfocitos T, y producción de citoquinas;
obteniéndose los resultados presentados a continuación.
Proliferación de linfocitos T CD4+: cuando las PBMC de los individuos participantes
se estimularon con PHA, la proliferación de linfocitos T CD4+ presentó valores
significativamente mayores a las observadas en condiciones basales (Medio) (Fig. 35). Así, en
la condición sin estimulación (Medio) los porcentajes de proliferación estuvieron entre 1 y 4%,
mientras que en la condición estimulada con PHA se observaron proliferaciones por el rango
de 20-34%, sin evidenciarse diferencias significativas al comparar entre sí los diferentes
grupos de pacientes y controles. Por otra parte, en las condiciones estimuladas con antígenos
de T. gondii o de VIH-1 (SATg y Env, respectivamente), en general, los porcentajes de
proliferación fueron “bajos” (≤5%), sin llegar a ser significativamente distintos a los
observados en la condición basal (Medio).
Linfocitos T CD4+
Inc
orp
ora
ció
n d
e B
rdU
(%
)
0
10
20
30
40
50
C1
P1A
P1B
P1C
C2
P2A
P2B
P2C
Medio PHA SATg Env
*
+
p<0.05:
* : vs. Medio
* : vs. C1
+ : vs. P2A
*
**
* *
*
**
*
*
+ *+
*
Figura 35- Proliferación de linfocitos T CD4+: se presentan los resultados (promedio ± desviación estándar) de
la proliferación de linfocitos T CD4+, en poblaciones de PBMC cultivadas por 72 horas sin estimulación (Medio) ó en
presencia de fitohemaglutinina (PHA, 5 g/ml), extracto crudo de taquizoítos de T. gondii (SATg, 1 g/ml) ó
glicoproteína recombinante Env de VIH-1 (Env, 1 g/ml). Controles: C1 (VIH-/T. gondii-; n=10) y C2 (VIH-/T. gondii+;
n=12); Pacientes: P1A (VIH+/T. gondii-; CD4>350/l; n=10), P1B (VIH+/T. gondii-; CD4:200-350/l; n=15), P1C
(VIH+/T. gondii-; CD4<200/l; n=7), P2A (VIH+/T. gondii+; CD4>350/l; n=12), P2B (VIH+/T. gondii+; CD4:200-350/l;
n=14), P2C (VIH+/T. gondii+; CD4<200/l; n=8). Los grupos se compararon utilizando las pruebas de ANOVA ó Kruskal-
Wallis, con pruebas de comparación múltiple de Bonferroni ó Dunn, respectivamente, según fuese apropiado. Se
consideraron significativos valores de p<0.05.
61
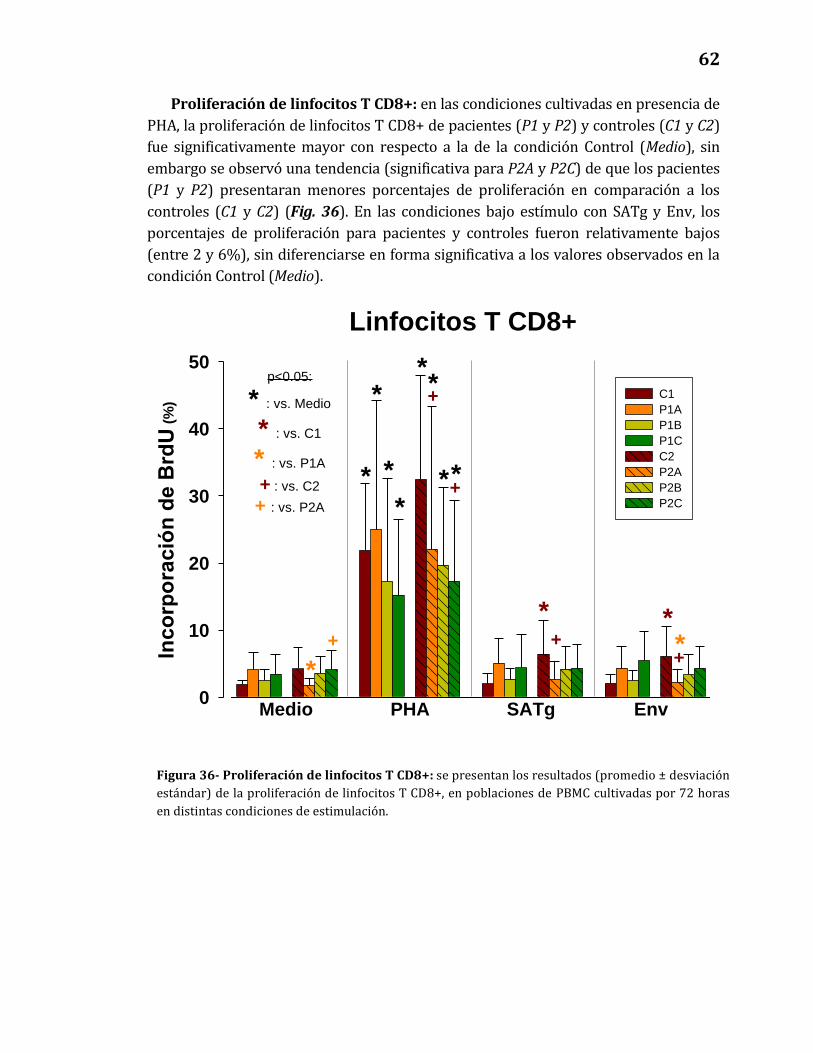
Proliferación de linfocitos T CD8+: en las condiciones cultivadas en presencia de
PHA, la proliferación de linfocitos T CD8+ de pacientes (P1 y P2) y controles (C1 y C2)
fue significativamente mayor con respecto a la de la condición Control (Medio), sin
embargo se observó una tendencia (significativa para P2A y P2C) de que los pacientes
(P1 y P2) presentaran menores porcentajes de proliferación en comparación a los
controles (C1 y C2) (Fig. 36). En las condiciones bajo estímulo con SATg y Env, los
porcentajes de proliferación para pacientes y controles fueron relativamente bajos
(entre 2 y 6%), sin diferenciarse en forma significativa a los valores observados en la
condición Control (Medio).
Figura 36- Proliferación de linfocitos T CD8+: se presentan los resultados (promedio ± desviación
estándar) de la proliferación de linfocitos T CD8+, en poblaciones de PBMC cultivadas por 72 horas
en distintas condiciones de estimulación.
Linfocitos T CD8+
Inco
rpo
ració
n d
e B
rdU
(%
)
0
10
20
30
40
50
C1
P1A
P1B
P1C
C2
P2A
P2B
P2C
Medio PHA SATg Env
+
*
* *
+
p<0.05:
* : vs. Medio
* : vs. C1
* : vs. P1A
+ : vs. C2
+ : vs. P2A
**
*
**
+
*+
*+
*
*
62
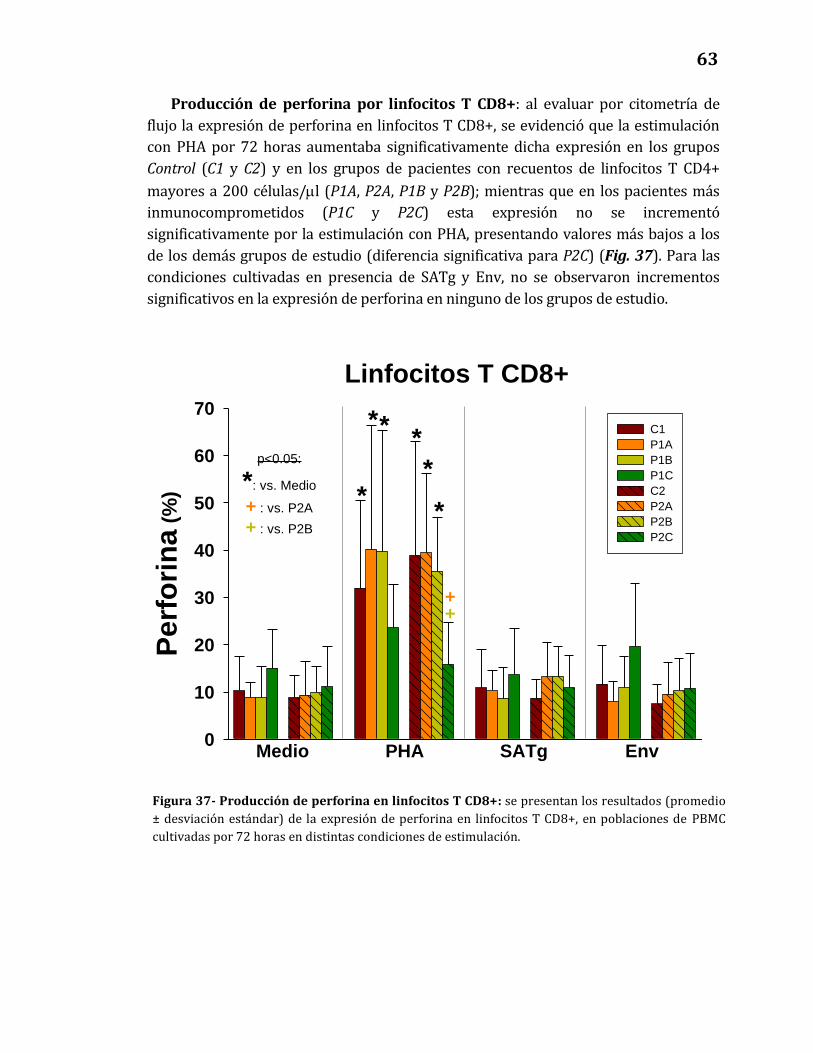
Producción de perforina por linfocitos T CD8+: al evaluar por citometría de
flujo la expresión de perforina en linfocitos T CD8+, se evidenció que la estimulación
con PHA por 72 horas aumentaba significativamente dicha expresión en los grupos
Control (C1 y C2) y en los grupos de pacientes con recuentos de linfocitos T CD4+
mayores a 200 células/l (P1A, P2A, P1B y P2B); mientras que en los pacientes más
inmunocomprometidos (P1C y P2C) esta expresión no se incrementó
significativamente por la estimulación con PHA, presentando valores más bajos a los
de los demás grupos de estudio (diferencia significativa para P2C) (Fig. 37). Para las
condiciones cultivadas en presencia de SATg y Env, no se observaron incrementos
significativos en la expresión de perforina en ninguno de los grupos de estudio.
Linfocitos T CD8+
Pe
rfo
rin
a (
%)
0
10
20
30
40
50
60
70C1
P1A
P1B
P1C
C2
P2A
P2B
P2C
Medio PHA SATg Env
p<0.05:
*: vs. Medio
+ : vs. P2A
+ : vs. P2B
*
++
*
*
*
*
*
Figura 37- Producción de perforina en linfocitos T CD8+: se presentan los resultados (promedio
± desviación estándar) de la expresión de perforina en linfocitos T CD8+, en poblaciones de PBMC
cultivadas por 72 horas en distintas condiciones de estimulación.
63

Apoptosis y necrosis de Linfocitos T CD4+: los resultados se presentan, de
acuerdo al análisis citométrico presentado en la Figura 29, como apoptosis temprana
(eventos solamente positivos para Anexina V), necrosis (eventos solamente positivos
para yoduro de propidio), apoptosis tardía (eventos positivos tanto para Anexina V
como para yoduro de propidio) y mortalidad total (referida en esta sección como la
sumatoria de apoptosis temprana + apoptosis tardía + necrosis).
Apoptosis Temprana: La estimulación con PHA se asoció con aumentos
significativos de la apoptosis temprana de los linfocitos T CD4+ de pacientes (P1 y
P2) y controles (C1 y C2) al comparar con la condición sin estimulación (Medio)
(Fig. 38A). En las demás condiciones de cultivo, incluyendo la condición sin
estimulación (Medio), los pacientes con menores recuentos de linfocitos T CD4+
presentaron mayores valores de apoptosis temprana que el resto de los grupos de
estudio (diferencia significativa para P2C en las condiciones Medio y Env).
Necrosis: La estimulación con PHA se asoció con la tendencia presentar mayores
porcentajes de necrosis en linfocitos T CD4+ de pacientes y controles (diferencia
significativa para C1, C2, P2A y P2B) al comparar con la condición sin estimulación
(Medio) (Fig. 38B). En las condiciones Medio, SATg y Env se observó la tendencia a
que los pacientes con menores recuentos de linfocitos T CD4+ presentaran
mayores porcentajes de necrosis de linfocitos T CD4+ (diferencia significativa para
P2C en la condición Medio).
Apoptosis Tardía: La apoptosis tardía de linfocitos T CD4+ de pacientes y
controles fue significativamente mayor en los cultivos estimulados con PHA, al
compararla con la condición sin estimulación (Medio) (Fig. 38C). Así mismo, en las
condiciones Medio, SATg y Env, los pacientes con menores recuentos de linfocitos T
CD4+ presentaron porcentajes de apoptosis tardía de linfocitos T CD4+
significativamente mayores al resto de los grupos de estudio.
Mortalidad Total: la mortalidad total de linfocitos T CD4+ (sumatoria de apoptosis
temprana + apoptosis tardía + necrosis) presentó el mismo esquema de los eventos
anteriormente descritos, es decir, valores significativamente mayores para
pacientes y controles al ser estimulados con PHA y valores significativamente
mayores para los pacientes con menores recuentos de linfocitos T CD4+ en las
condiciones Medio, SATg y Env (Fig. 38D).
64
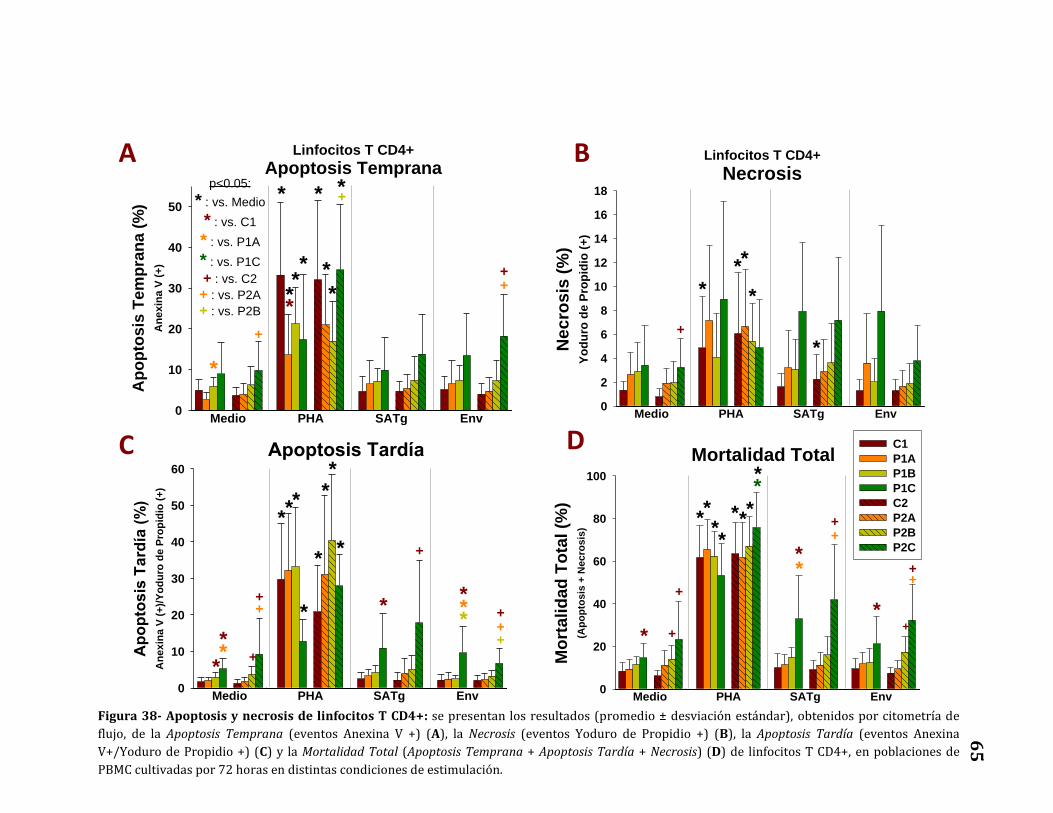
Linfocitos T CD4+
Necrosis
Ne
cro
sis
(%
)Y
od
uro
de P
rop
idio
(+
)
0
2
4
6
8
10
12
14
16
18
Medio PHA SATg Env
*
+
**
*
*
B
Mortalidad Total
Mo
rtalid
ad
To
tal (%
)(A
po
pto
sis
+ N
ec
ros
is)
0
20
40
60
80
100
C1
P1A
P1B
P1C
C2
P2A
P2B
P2C
Medio PHA SATg Env
*
+
*
*
*
**
**
*
+
+
**
**
+
++
+
D
Linfocitos T CD4+
Apoptosis Temprana
Ap
op
tosis
Tem
pra
na (
%)
An
ex
ina
V (
+)
0
10
20
30
40
50
Medio PHA SATg Env
*
+
*
**
*
*
+
++
p<0.05:
* : vs. Medio
* : vs. C1
* : vs. P1A
* : vs. P1C
+ : vs. C2
+ : vs. P2A
+ : vs. P2B
*
**
*
A
Apoptosis Tardía
Ap
op
tos
is T
ard
ía (
%)
An
ex
ina
V (
+)/
Yo
du
ro d
e P
rop
idio
(+
)
0
10
20
30
40
50
60
Medio PHA SATg Env
*
+
*
*
*
*
+
*
+
*
*
*
*
*
*
+
***
+++
C
Figura 38- Apoptosis y necrosis de linfocitos T CD4+: se presentan los resultados (promedio ± desviación estándar), obtenidos por citometría de
flujo, de la Apoptosis Temprana (eventos Anexina V +) (A), la Necrosis (eventos Yoduro de Propidio +) (B), la Apoptosis Tardía (eventos Anexina
V+/Yoduro de Propidio +) (C) y la Mortalidad Total (Apoptosis Temprana + Apoptosis Tardía + Necrosis) (D) de linfocitos T CD4+, en poblaciones de
PBMC cultivadas por 72 horas en distintas condiciones de estimulación.
65

Apoptosis y necrosis de linfocitos T CD8+: los resultados para linfocitos T CD8+
también se presentan como apoptosis temprana (eventos solamente positivos para
Anexina V), necrosis (eventos solamente positivos para yoduro de propidio), apoptosis
tardía (eventos positivos tanto para Anexina V como para yoduro de propidio) y
mortalidad total (referida en esta sección como la sumatoria de apoptosis temprana +
apoptosis tardía + necrosis).
Apoptosis Temprana: La estimulación con PHA se asoció con aumentos
significativos de la apoptosis temprana de los linfocitos T CD8+ de pacientes (P1 y
P2) y controles (C1 y C2) al comparar con la condición sin estimulación (Medio)
(Fig. 39A). En las demás condiciones de cultivo (Medio, SATg y Env) no se
observaron diferencias significativas para los valores de apoptosis temprana de
linfocitos T CD8+ entre los distintos grupos de estudio.
Necrosis: Los linfocitos T CD8+ de pacientes y controles presentaron bajos
porcentajes de necrosis (promedios <8%) en todas las condiciones de estimulación
estudiadas, sin presentar, en general, diferencias significativas entre los diferentes
grupos de estudio y tampoco al comparar con la condición sin estimulación
(Medio) (Fig. 39B).
Apoptosis Tardía : La apoptosis tardía de linfocitos T CD8+ de pacientes y
controles fue significativamente mayor en los cultivos estimulados con PHA, al
comparar con la condición sin estimulación (Medio) (Fig. 39C). En las condiciones
Medio, SATg y Env, en general, no se observaron diferencias significativas en los
valores de apoptosis tardía de linfocitos T CD8+ entre los distintos grupos de
estudio.
Mortalidad Total: la mortalidad total de linfocitos T CD8+ (sumatoria de apoptosis
temprana + apoptosis tardía + necrosis) presentó, así mismo, valores
significativamente mayores en pacientes y controles al ser estimulados con PHA
(Fig. 39D). Por otra parte, en las condiciones Medio, SATg y Env, en general, no se
observaron diferencias significativas entre los distintos grupos de estudio.
66
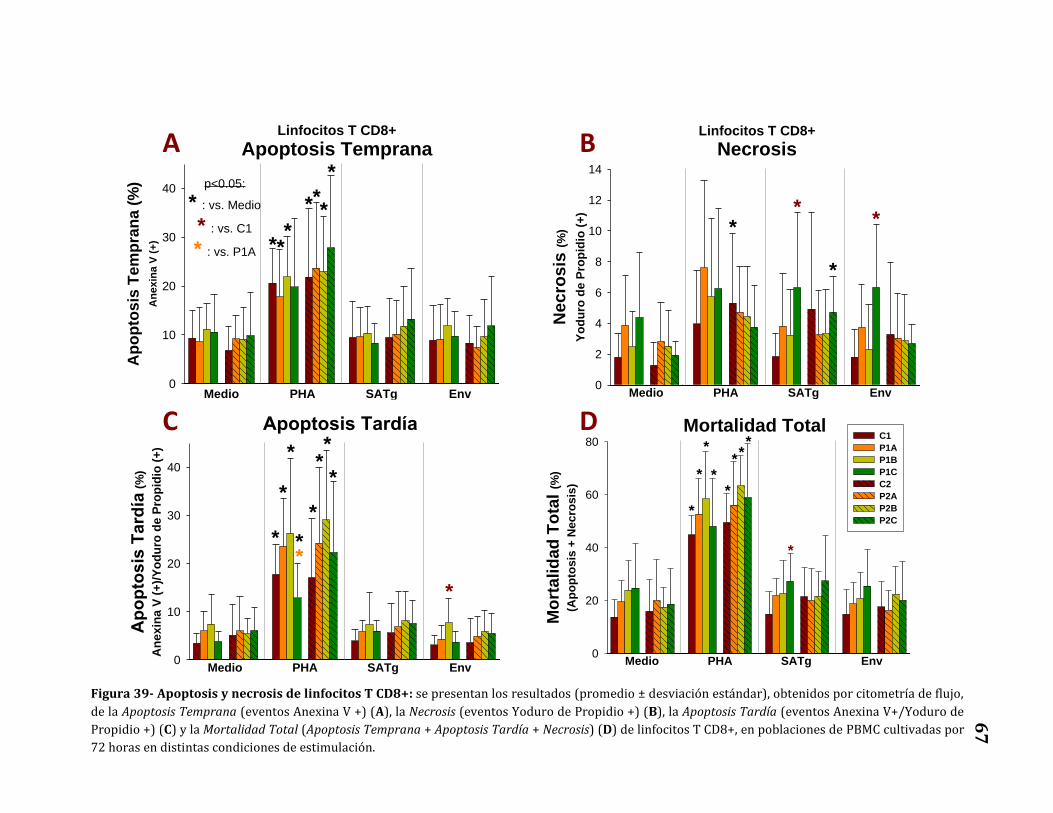
Linfocitos T CD8+
Apoptosis TempranaA
po
pto
sis
Tem
pra
na (
%)
An
exin
a V
(+
)
0
10
20
30
40
Medio PHA SATg Env
*
**
p<0.05:
* : vs. Medio
* : vs. C1
* : vs. P1A
*
*
*
*
A Linfocitos T CD8+
Necrosis
Ne
cro
sis
(%
)
Yo
du
ro d
e P
rop
idio
(+
)
0
2
4
6
8
10
12
14
Medio PHA SATg Env
*
*
**
B
Apoptosis Tardía
Ap
op
tos
is T
ard
ía (
%)
An
exin
a V
(+
)/Y
od
uro
de P
rop
idio
(+
)
0
10
20
30
40
Medio PHA SATg Env
*
**
*
*
*
***
*
C Mortalidad Total
Mo
rta
lid
ad
To
tal
(%)
(Ap
op
tos
is +
Ne
cro
sis
)
0
20
40
60
80C1
P1A
P1B
P1C
C2
P2A
P2B
P2C
Medio PHA SATg Env
*
**
*
*
**
**
D
Figura 39- Apoptosis y necrosis de linfocitos T CD8+: se presentan los resultados (promedio ± desviación estándar), obtenidos por citometría de flujo,
de la Apoptosis Temprana (eventos Anexina V +) (A), la Necrosis (eventos Yoduro de Propidio +) (B), la Apoptosis Tardía (eventos Anexina V+/Yoduro de
Propidio +) (C) y la Mortalidad Total (Apoptosis Temprana + Apoptosis Tardía + Necrosis) (D) de linfocitos T CD8+, en poblaciones de PBMC cultivadas por
72 horas en distintas condiciones de estimulación.
67

Producción de citoquinas ex vivo e in vitro: La determinación de citoquinas se
hizo por citometría de flujo, utilizando y siguiendo las instrucciones de un estuche
comercial (Fig. 40).
Perlas
FSC
A
SSC
MFI
(FL
2)
Concentración (pg/ml)
Curva de Calibración - IFN-
E
FL3
FL2
B Patrón 1 (0 pg/ml) C D Patrón 4 (80 pg/ml) Patrón 9 (2.500 pg/ml)
IL-2
IL-6
IFN-
IL-4
IL-6
IL-10
TNF-
IFN-
IL-6
IL-10
TNF-
IL-2
IL-4
IL-10
IL-2
IL-4
IFN-
TNF-
Figura 40-Determinación de citoquinas por citometría de flujo: La determinación de citoquinas
en plasma y sobrenadante de cultivos de células mononucleares de sangre periférica de los individuos
participantes, se hizo por citometría de flujo, utilizando y siguiendo las instrucciones de un estuche
comercial (Human Th1-Th2 Cytokine Kit-CBA-Becton Dickinson). Brevemente, el estuche comercial provee 6
poblaciones de perlas (A1, A2, A3, A4, A5 y A6), con fluorescencias discretas que pueden ser resueltas en el
canal de fluorescencia FL3 del citómetro. Cada población de perlas tienen unida a su superficie anticuerpos
de captura específicos a una de las 6 citoquinas a evaluar (IL-2, IL-4, IL-6, IL-10, TNF- e IFN-,
respectivamente). El estuche comercial también provee anticuerpos detectores, específicos para cada una
de las seis citoquinas, los cuales están acoplados al fluorocromo ficoeritrina (PE), por lo que pueden ser
resueltos en el detector de fluorescencia FL2 del citómetro. El principio del ensayo es que al incubar
simultáneamente las muestras a evaluar con las perlas de captura y los anticuerpos detectores, las
citoquinas presentes en las muestras se unen a su población de perlas específicas por medio de los
anticuerpos de captura y dicha unión se evidencia por los anticuerpos detectores, que también se unen al
complejo. En la figura se presenta (A) un gráfico de puntos con las señales de dispersión frontal (FSC) y
lateral (SSC) de las perlas de captura, que permite seleccionar la región o eventos a analizar en los
siguientes gráficos (región de Perlas en color rojo). La región de Perlas así seleccionada es entonces
analizada en el detector de fluorescencia FL3 (B, C y D; eje vertical de los gráficos), lo que permite separar
seis poblaciones discretas de perlas, correspondientes a cada citoquina a evaluar (representadas como
eventos de color verde claro, fucsia, azul claro, anaranjado, azul oscuro y verde oscuro). La intensidad de
fluorescencia promedio (MFI) de cada población en FL2 (B, C y D; eje horizontal de los gráficos), será
proporcional a la cantidad de citoquina presente en la muestra (en B, C y D se representan,
respectivamente, los resultados de tres Patrones de Calibración, con concentraciones conocidas de 0, 80 y
2.500 pg/ml para cada citoquina). La utilización de Patrones de Calibración permite calcular la
concentración presente en las muestras (E, curva de calibración típica para IFN-).
68
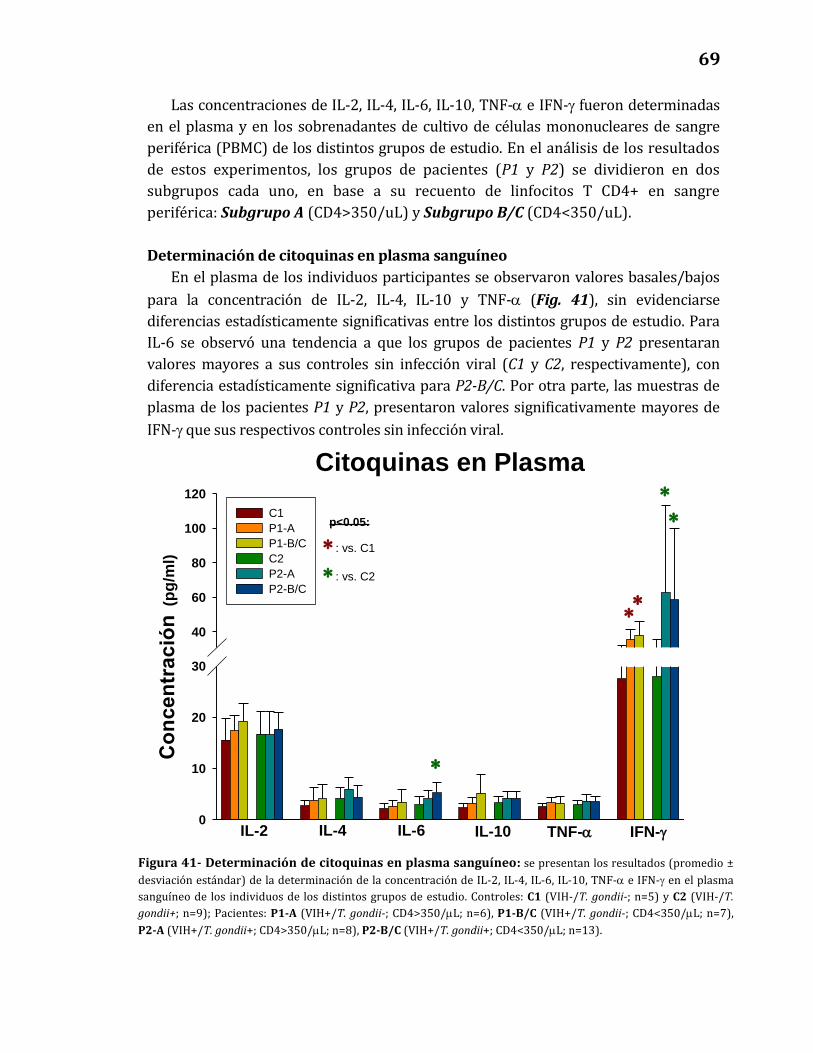
Las concentraciones de IL-2, IL-4, IL-6, IL-10, TNF- e IFN- fueron determinadas
en el plasma y en los sobrenadantes de cultivo de células mononucleares de sangre
periférica (PBMC) de los distintos grupos de estudio. En el análisis de los resultados
de estos experimentos, los grupos de pacientes (P1 y P2) se dividieron en dos
subgrupos cada uno, en base a su recuento de linfocitos T CD4+ en sangre
periférica: Subgrupo A (CD4>350/uL) y Subgrupo B/C (CD4<350/uL).
Determinación de citoquinas en plasma sanguíneo
En el plasma de los individuos participantes se observaron valores basales/bajos
para la concentración de IL-2, IL-4, IL-10 y TNF- (Fig. 41), sin evidenciarse
diferencias estadísticamente significativas entre los distintos grupos de estudio. Para
IL-6 se observó una tendencia a que los grupos de pacientes P1 y P2 presentaran
valores mayores a sus controles sin infección viral (C1 y C2, respectivamente), con
diferencia estadísticamente significativa para P2-B/C. Por otra parte, las muestras de
plasma de los pacientes P1 y P2, presentaron valores significativamente mayores de
IFN- que sus respectivos controles sin infección viral.
Figura 41- Determinación de citoquinas en plasma sanguíneo: se presentan los resultados (promedio ±
desviación estándar) de la determinación de la concentración de IL-2, IL-4, IL-6, IL-10, TNF- e IFN- en el plasma
sanguíneo de los individuos de los distintos grupos de estudio. Controles: C1 (VIH-/T. gondii-; n=5) y C2 (VIH-/T.
gondii+; n=9); Pacientes: P1-A (VIH+/T. gondii-; CD4>350/L; n=6), P1-B/C (VIH+/T. gondii-; CD4<350/L; n=7),
P2-A (VIH+/T. gondii+; CD4>350/L; n=8), P2-B/C (VIH+/T. gondii+; CD4<350/L; n=13).
Citoquinas en Plasma
Co
nc
en
tra
ció
n (p
g/m
l)
0
10
20
30
40
60
80
100
120
C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
IL-4 IL-6 IL-10 TNF- IFN-IL-2
p<0.05:
: vs. C1
: vs. C2
69
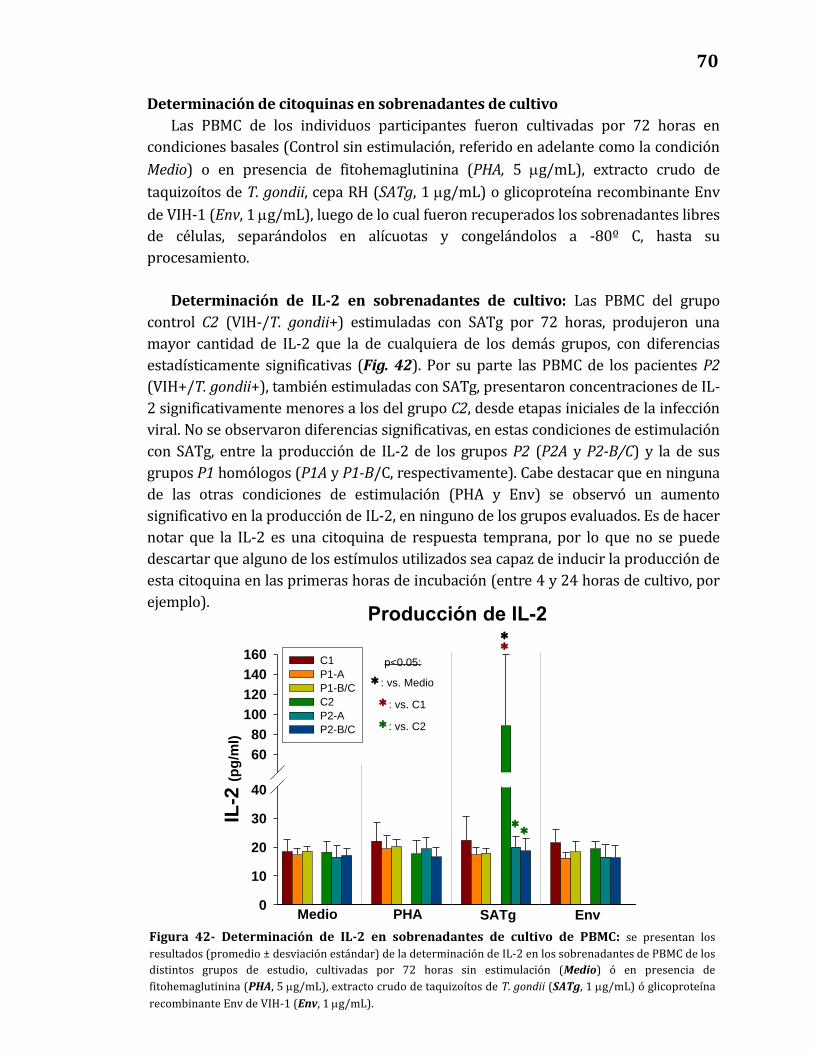
Determinación de citoquinas en sobrenadantes de cultivo
Las PBMC de los individuos participantes fueron cultivadas por 72 horas en
condiciones basales (Control sin estimulación, referido en adelante como la condición
Medio) o en presencia de fitohemaglutinina (PHA, 5 g/mL), extracto crudo de
taquizoítos de T. gondii, cepa RH (SATg, 1 g/mL) o glicoproteína recombinante Env
de VIH-1 (Env, 1 g/mL), luego de lo cual fueron recuperados los sobrenadantes libres
de células, separándolos en alícuotas y congelándolos a -80º C, hasta su
procesamiento.
Determinación de IL-2 en sobrenadantes de cultivo: Las PBMC del grupo
control C2 (VIH-/T. gondii+) estimuladas con SATg por 72 horas, produjeron una
mayor cantidad de IL-2 que la de cualquiera de los demás grupos, con diferencias
estadísticamente significativas (Fig. 42). Por su parte las PBMC de los pacientes P2
(VIH+/T. gondii+), también estimuladas con SATg, presentaron concentraciones de IL-
2 significativamente menores a los del grupo C2, desde etapas iniciales de la infección
viral. No se observaron diferencias significativas, en estas condiciones de estimulación
con SATg, entre la producción de IL-2 de los grupos P2 (P2A y P2-B/C) y la de sus
grupos P1 homólogos (P1A y P1-B/C, respectivamente). Cabe destacar que en ninguna
de las otras condiciones de estimulación (PHA y Env) se observó un aumento
significativo en la producción de IL-2, en ninguno de los grupos evaluados. Es de hacer
notar que la IL-2 es una citoquina de respuesta temprana, por lo que no se puede
descartar que alguno de los estímulos utilizados sea capaz de inducir la producción de
esta citoquina en las primeras horas de incubación (entre 4 y 24 horas de cultivo, por
ejemplo).
Producción de IL-2
IL-2
(p
g/m
l)
0
10
20
30
40
60
80
100
120
140
160 C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
PHA SATg EnvMedio
p<0.05:
: vs. Medio
: vs. C1
: vs. C2
Figura 42- Determinación de IL-2 en sobrenadantes de cultivo de PBMC: se presentan los
resultados (promedio ± desviación estándar) de la determinación de IL-2 en los sobrenadantes de PBMC de los
distintos grupos de estudio, cultivadas por 72 horas sin estimulación (Medio) ó en presencia de
fitohemaglutinina (PHA, 5 g/mL), extracto crudo de taquizoítos de T. gondii (SATg, 1 g/mL) ó glicoproteína
recombinante Env de VIH-1 (Env, 1 g/mL).
70
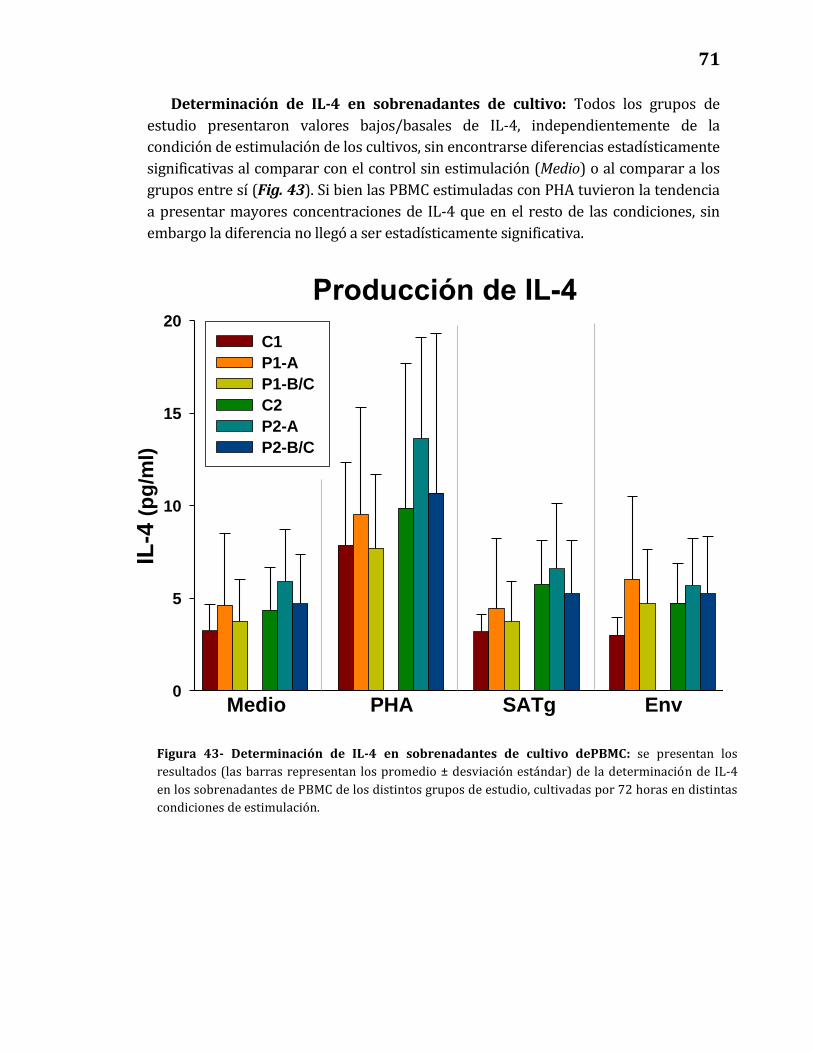
Determinación de IL-4 en sobrenadantes de cultivo: Todos los grupos de
estudio presentaron valores bajos/basales de IL-4, independientemente de la
condición de estimulación de los cultivos, sin encontrarse diferencias estadísticamente
significativas al comparar con el control sin estimulación (Medio) o al comparar a los
grupos entre sí (Fig. 43). Si bien las PBMC estimuladas con PHA tuvieron la tendencia
a presentar mayores concentraciones de IL-4 que en el resto de las condiciones, sin
embargo la diferencia no llegó a ser estadísticamente significativa.
Producción de IL-4
IL-4
(p
g/m
l)
0
5
10
15
20
C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
PHA SATg EnvMedio
Figura 43- Determinación de IL-4 en sobrenadantes de cultivo dePBMC: se presentan los
resultados (las barras representan los promedio ± desviación estándar) de la determinación de IL-4
en los sobrenadantes de PBMC de los distintos grupos de estudio, cultivadas por 72 horas en distintas
condiciones de estimulación.
71
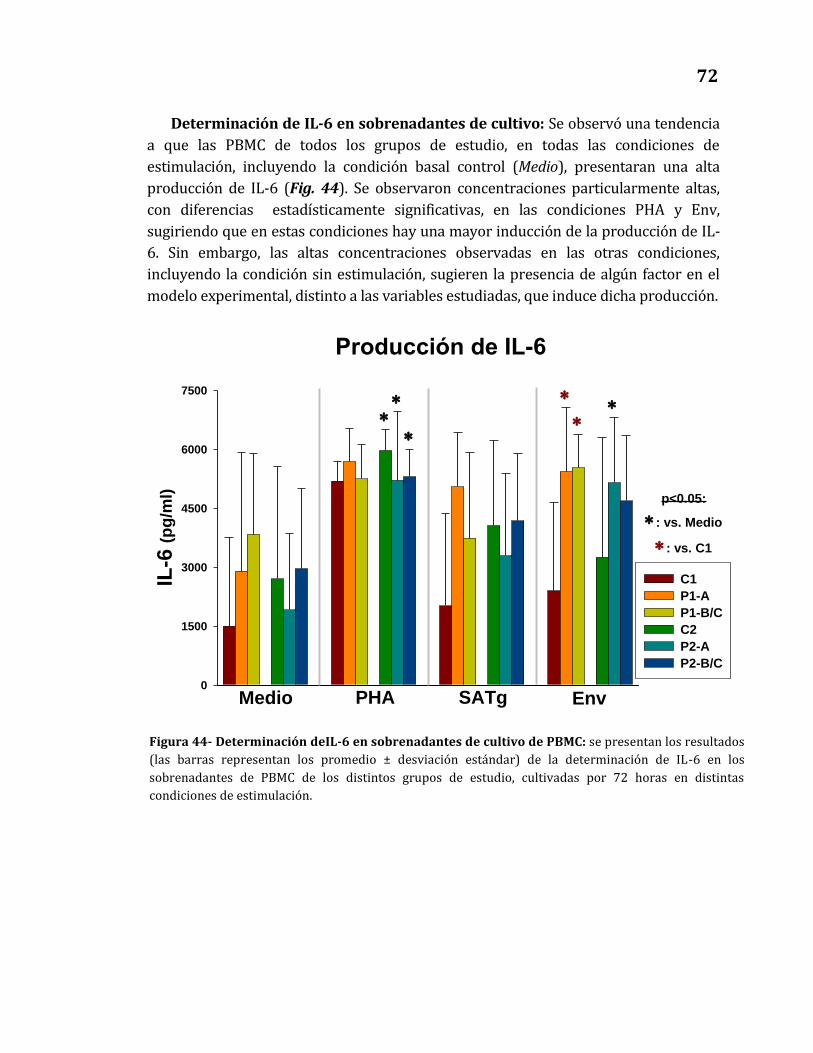
Determinación de IL-6 en sobrenadantes de cultivo: Se observó una tendencia
a que las PBMC de todos los grupos de estudio, en todas las condiciones de
estimulación, incluyendo la condición basal control (Medio), presentaran una alta
producción de IL-6 (Fig. 44). Se observaron concentraciones particularmente altas,
con diferencias estadísticamente significativas, en las condiciones PHA y Env,
sugiriendo que en estas condiciones hay una mayor inducción de la producción de IL-
6. Sin embargo, las altas concentraciones observadas en las otras condiciones,
incluyendo la condición sin estimulación, sugieren la presencia de algún factor en el
modelo experimental, distinto a las variables estudiadas, que induce dicha producción.
Producción de IL-6
IL-6
(p
g/m
l)
0
1500
3000
4500
6000
7500
C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
PHA SATg EnvMedio
p<0.05:
: vs. Medio
: vs. C1
Figura 44- Determinación deIL-6 en sobrenadantes de cultivo de PBMC: se presentan los resultados
(las barras representan los promedio ± desviación estándar) de la determinación de IL-6 en los
sobrenadantes de PBMC de los distintos grupos de estudio, cultivadas por 72 horas en distintas
condiciones de estimulación.
72
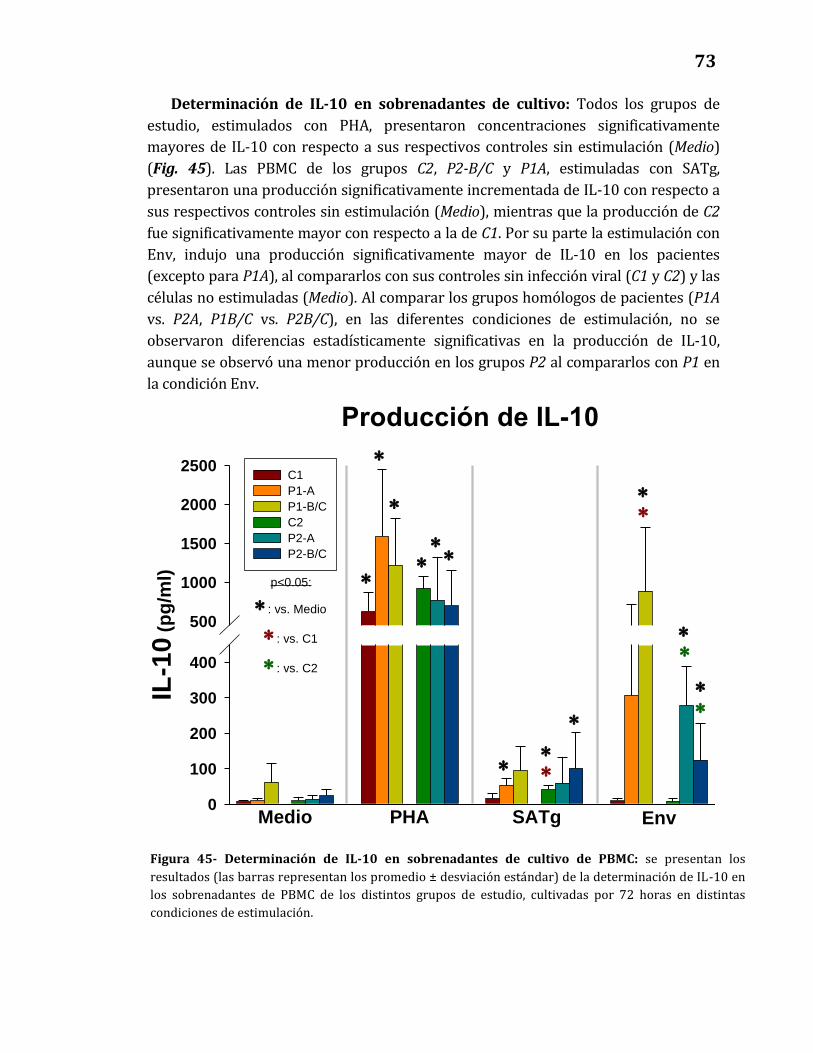
Determinación de IL-10 en sobrenadantes de cultivo: Todos los grupos de
estudio, estimulados con PHA, presentaron concentraciones significativamente
mayores de IL-10 con respecto a sus respectivos controles sin estimulación (Medio)
(Fig. 45). Las PBMC de los grupos C2, P2-B/C y P1A, estimuladas con SATg,
presentaron una producción significativamente incrementada de IL-10 con respecto a
sus respectivos controles sin estimulación (Medio), mientras que la producción de C2
fue significativamente mayor con respecto a la de C1. Por su parte la estimulación con
Env, indujo una producción significativamente mayor de IL-10 en los pacientes
(excepto para P1A), al compararlos con sus controles sin infección viral (C1 y C2) y las
células no estimuladas (Medio). Al comparar los grupos homólogos de pacientes (P1A
vs. P2A, P1B/C vs. P2B/C), en las diferentes condiciones de estimulación, no se
observaron diferencias estadísticamente significativas en la producción de IL-10,
aunque se observó una menor producción en los grupos P2 al compararlos con P1 en
la condición Env.
Producción de IL-10
IL-1
0 (
pg
/ml)
0
100
200
300
400
500
1000
1500
2000
2500C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
PHA SATg EnvMedio
p<0.05:
: vs. Medio
: vs. C1
: vs. C2
Figura 45- Determinación de IL-10 en sobrenadantes de cultivo de PBMC: se presentan los
resultados (las barras representan los promedio ± desviación estándar) de la determinación de IL-10 en
los sobrenadantes de PBMC de los distintos grupos de estudio, cultivadas por 72 horas en distintas
condiciones de estimulación.
73
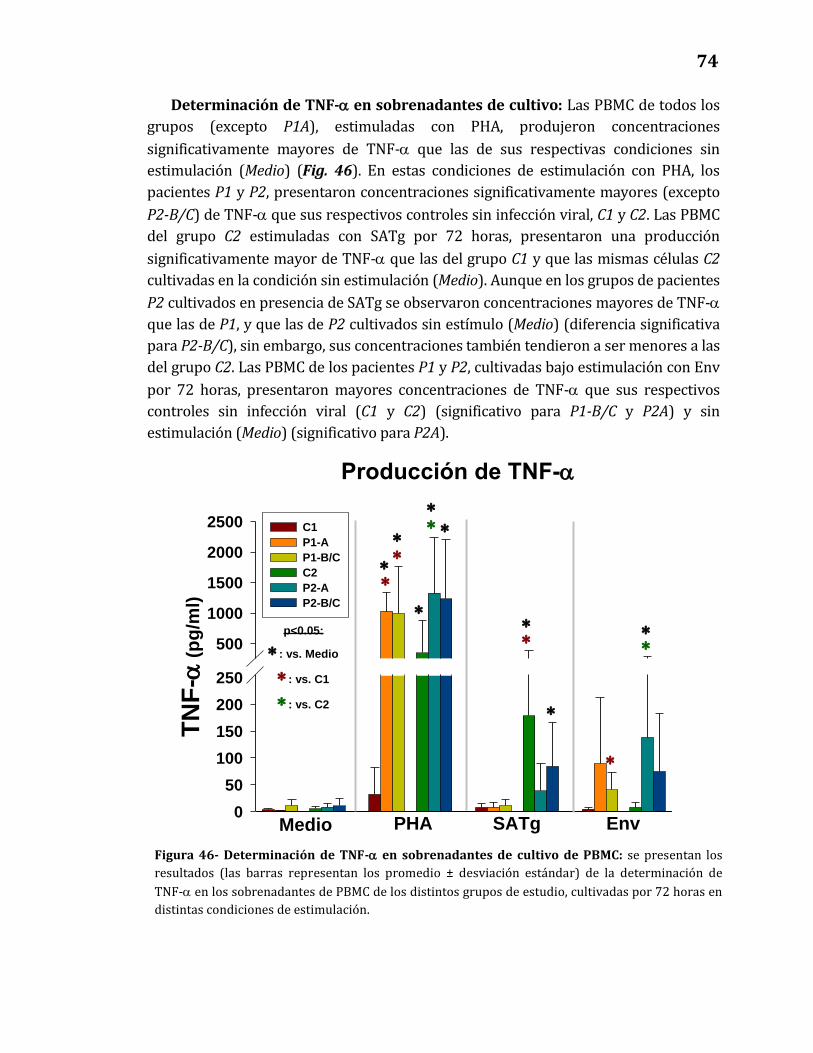
Determinación de TNF- en sobrenadantes de cultivo: Las PBMC de todos los
grupos (excepto P1A), estimuladas con PHA, produjeron concentraciones
significativamente mayores de TNF- que las de sus respectivas condiciones sin
estimulación (Medio) (Fig. 46). En estas condiciones de estimulación con PHA, los
pacientes P1 y P2, presentaron concentraciones significativamente mayores (excepto
P2-B/C) de TNF- que sus respectivos controles sin infección viral, C1 y C2. Las PBMC
del grupo C2 estimuladas con SATg por 72 horas, presentaron una producción
significativamente mayor de TNF- que las del grupo C1 y que las mismas células C2
cultivadas en la condición sin estimulación (Medio). Aunque en los grupos de pacientes
P2 cultivados en presencia de SATg se observaron concentraciones mayores de TNF-
que las de P1, y que las de P2 cultivados sin estímulo (Medio) (diferencia significativa
para P2-B/C), sin embargo, sus concentraciones también tendieron a ser menores a las
del grupo C2. Las PBMC de los pacientes P1 y P2, cultivadas bajo estimulación con Env
por 72 horas, presentaron mayores concentraciones de TNF- que sus respectivos
controles sin infección viral (C1 y C2) (significativo para P1-B/C y P2A) y sin
estimulación (Medio) (significativo para P2A).
Producción de TNF-
TN
F-
(p
g/m
l)
0
50
100
150
200
250
500
1000
1500
2000
2500 C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
PHA SATg EnvMedio
p<0.05:
: vs. Medio
: vs. C1
: vs. C2
Figura 46- Determinación de TNF- en sobrenadantes de cultivo de PBMC: se presentan los
resultados (las barras representan los promedio ± desviación estándar) de la determinación de
TNF- en los sobrenadantes de PBMC de los distintos grupos de estudio, cultivadas por 72 horas en
distintas condiciones de estimulación.
74
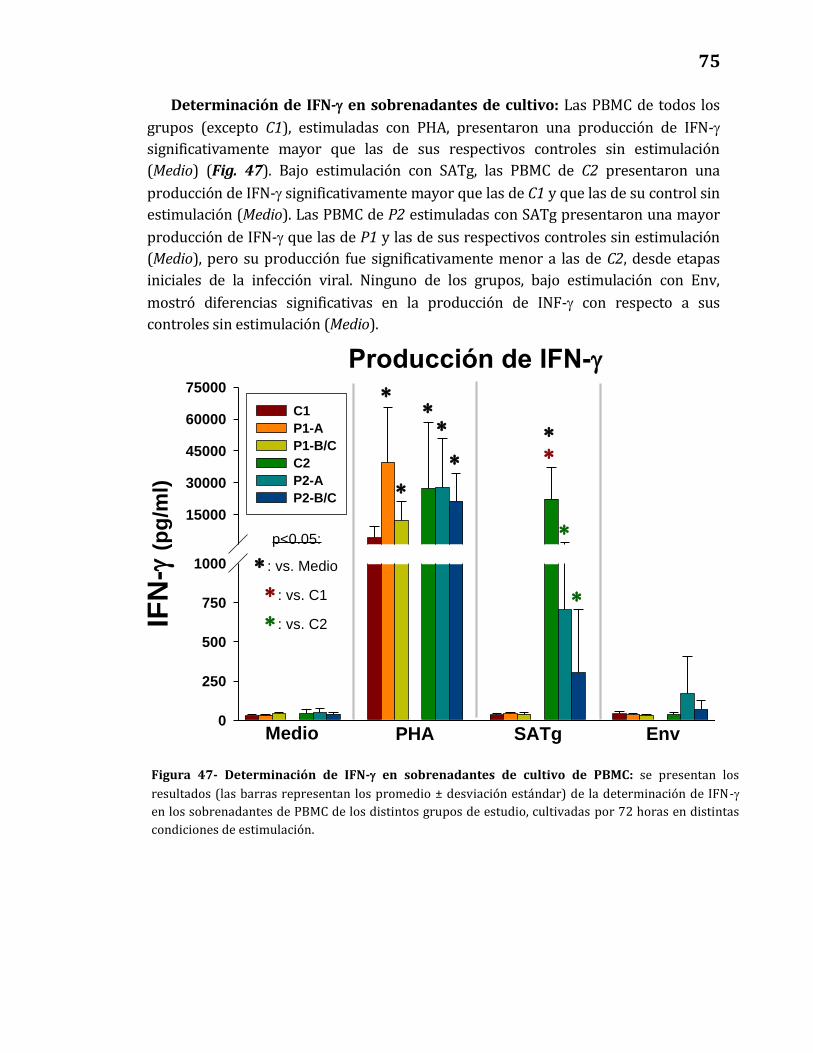
Determinación de IFN- en sobrenadantes de cultivo: Las PBMC de todos los
grupos (excepto C1), estimuladas con PHA, presentaron una producción de IFN-
significativamente mayor que las de sus respectivos controles sin estimulación
(Medio) (Fig. 47). Bajo estimulación con SATg, las PBMC de C2 presentaron una
producción de IFN- significativamente mayor que las de C1 y que las de su control sin
estimulación (Medio). Las PBMC de P2 estimuladas con SATg presentaron una mayor
producción de IFN- que las de P1 y las de sus respectivos controles sin estimulación
(Medio), pero su producción fue significativamente menor a las de C2, desde etapas
iniciales de la infección viral. Ninguno de los grupos, bajo estimulación con Env,
mostró diferencias significativas en la producción de INF- con respecto a sus
controles sin estimulación (Medio).
Producción de IFN-
IFN
- (
pg
/ml)
0
250
500
750
1000
15000
30000
45000
60000
75000
C1
P1-A
P1-B/C
C2
P2-A
P2-B/C
PHA SATg EnvMedio
p<0.05:
: vs. Medio
: vs. C1
: vs. C2
Figura 47- Determinación de IFN- en sobrenadantes de cultivo de PBMC: se presentan los
resultados (las barras representan los promedio ± desviación estándar) de la determinación de IFN-
en los sobrenadantes de PBMC de los distintos grupos de estudio, cultivadas por 72 horas en distintas
condiciones de estimulación.
75

II. EVALUACIÓN DE EFECTOS NEUROTÓXICOS DE VIH-1 Y
Toxoplasma gondii EN CÉLULAS DEL SISTEMA NERVIOSO
CENTRAL Para evaluar los efectos neurotóxicos producidos por la presencia simultánea de
VIH y T. gondii en células del sistema nervioso central (CNS) se utilizaron cultivos
primarios de células nerviosas de feto de rata Sprague-Dawley. Estos cultivos
consistían en poblaciones heterogéneas de células nerviosas que crecían en forma
de monocapa confluente. Las células se cultivaban por 3, 5 y 7 días, en seis
condiciones distintas: la primera era la condición Control (referida también como
Medio), en la cual los cultivos primarios no se sometían a estimulación/infección
por los agentes patógenos, sino que se cultivaban en condiciones basales, en medio
RPMI-10% SFB, a una temperatura de 37º C y una atmósfera con 5% de CO2. La
segunda condición se realizaba en presencia de la glicoproteína de la envoltura
viral, gp41, a una concentración 10nM. La tercera condición era en presencia de
VIH-1IIIB, con 1.9 x 109 partículas virales/ml. En la cuarta condición las células eran
infectadas con taquizoítos de la cepa RH de T. gondii, a una relación parásito/célula
nerviosa de 1/250. La quinta condición se realizaba en presencia simultánea de
gp41 (10 nM) y T. gondii (1/250) y la sexta condición en presencia simultánea de
VIH-1IIIB (1.9 x 109 partículas virales/ml) y T. gondii (1/250). Se evaluó entonces,
cómo se modificaba el microambiente de los cultivos en las distintas condiciones,
con especial énfasis en la producción de mediadores pro-inflamatorios (óxido
nítrico y diversas citoquinas), y cómo estos cambios en el microambiente afectaban
la sobrevida celular; parámetro que se evaluaba a través de la determinación del
recuento, viabilidad, apoptosis y necrosis de las células nerviosas. También se
evaluó cómo se modificaba la progresión de la infección de las células nerviosas por
T. gondii cuando dicha infección se realizaba en presencia simultánea de gp41 ó de
VIH-1. Esto se hizo mediante el recuento de los taquizoítos libres en los
sobrenadantes de los cultivos y la determinación, por análisis microscópico, del
porcentaje de células infectadas y del número de taquizoítos presentes en dichas
células infectadas. Los resultados obtenidos en estos experimentos se presentan a
continuación.
A. EVALUACIÓN DE EFECTOS NEUROTÓXICOS
Recuento de células nerviosas: Las células nerviosas de feto de rata cultivadas
en presencia únicamente de gp41, VIH-1 o T. gondii, presentaron disminución en el
recuento de células vivas, con diferencias estadísticamente significativas con
respecto a la condición Control, desde el día 5 de cultivo (Fig. 48A), indicando que la
presencia de estos agentes induce un efecto citotóxico. Sin embargo, cabe destacar,
que en las condiciones donde las células se cultivaron en presencia simultánea de
76
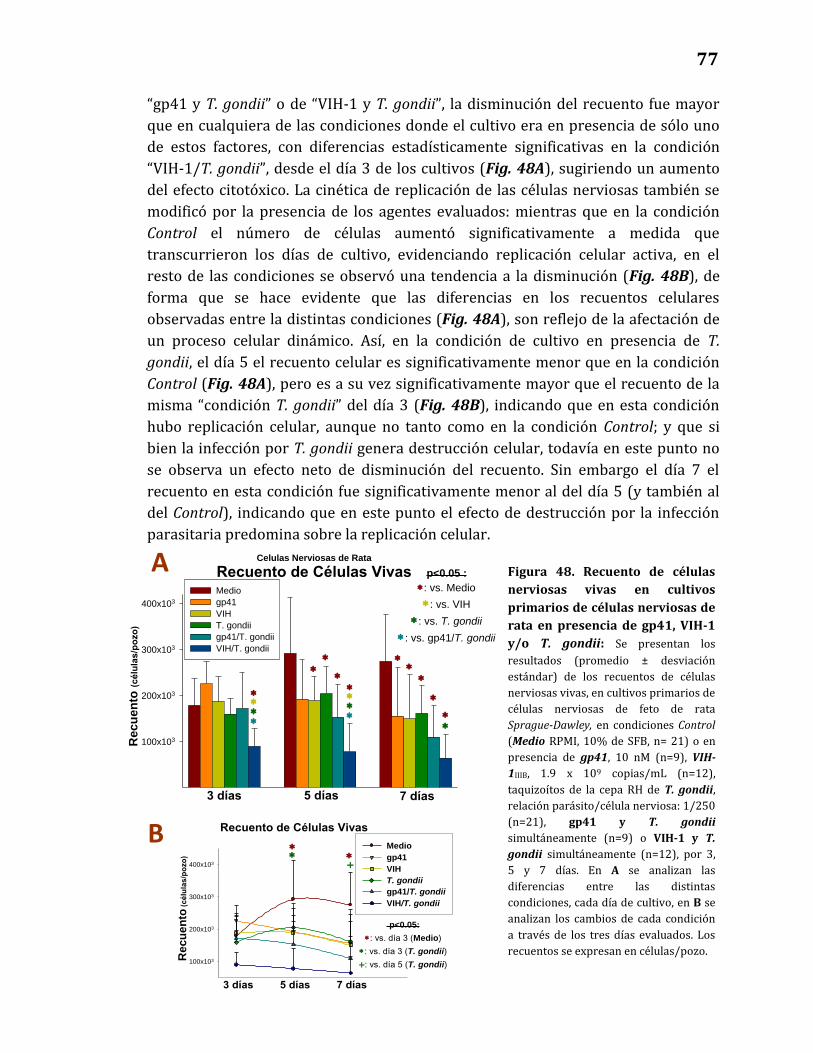
“gp41 y T. gondii” o de “VIH-1 y T. gondii”, la disminución del recuento fue mayor
que en cualquiera de las condiciones donde el cultivo era en presencia de sólo uno
de estos factores, con diferencias estadísticamente significativas en la condición
“VIH-1/T. gondii”, desde el día 3 de los cultivos (Fig. 48A), sugiriendo un aumento
del efecto citotóxico. La cinética de replicación de las células nerviosas también se
modificó por la presencia de los agentes evaluados: mientras que en la condición
Control el número de células aumentó significativamente a medida que
transcurrieron los días de cultivo, evidenciando replicación celular activa, en el
resto de las condiciones se observó una tendencia a la disminución (Fig. 48B), de
forma que se hace evidente que las diferencias en los recuentos celulares
observadas entre la distintas condiciones (Fig. 48A), son reflejo de la afectación de
un proceso celular dinámico. Así, en la condición de cultivo en presencia de T.
gondii, el día 5 el recuento celular es significativamente menor que en la condición
Control (Fig. 48A), pero es a su vez significativamente mayor que el recuento de la
misma “condición T. gondii” del día 3 (Fig. 48B), indicando que en esta condición
hubo replicación celular, aunque no tanto como en la condición Control; y que si
bien la infección por T. gondii genera destrucción celular, todavía en este punto no
se observa un efecto neto de disminución del recuento. Sin embargo el día 7 el
recuento en esta condición fue significativamente menor al del día 5 (y también al
del Control), indicando que en este punto el efecto de destrucción por la infección
parasitaria predomina sobre la replicación celular.
Figura 48. Recuento de células
nerviosas vivas en cultivos
primarios de células nerviosas de
rata en presencia de gp41, VIH-1
y/o T. gondii: Se presentan los
resultados (promedio ± desviación
estándar) de los recuentos de células
nerviosas vivas, en cultivos primarios de
células nerviosas de feto de rata
Sprague-Dawley, en condiciones Control
(Medio RPMI, 10% de SFB, n= 21) o en
presencia de gp41, 10 nM (n=9), VIH-
1IIIB, 1.9 x 109 copias/mL (n=12),
taquizoítos de la cepa RH de T. gondii,
relación parásito/célula nerviosa: 1/250
(n=21), gp41 y T. gondii
simultáneamente (n=9) o VIH-1 y T.
gondii simultáneamente (n=12), por 3,
5 y 7 días. En A se analizan las
diferencias entre las distintas
condiciones, cada día de cultivo, en B se
analizan los cambios de cada condición
a través de los tres días evaluados. Los
recuentos se expresan en células/pozo.
Recuento de Células Vivas
Re
cu
en
to (
cé
lula
s/p
ozo
)
100x103
200x103
300x103
400x103
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (Medio)
: vs. día 3 (T. gondii)
: vs. día 5 (T. gondii)
B
Celulas Nerviosas de Rata
Recuento de Células Vivas
Recu
en
to (
célu
las
/po
zo
)
100x103
200x103
300x103
400x103
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05 :
: vs. Medio
: vs. VIH
: vs. T. gondii
: vs. gp41/T. gondii
A
77
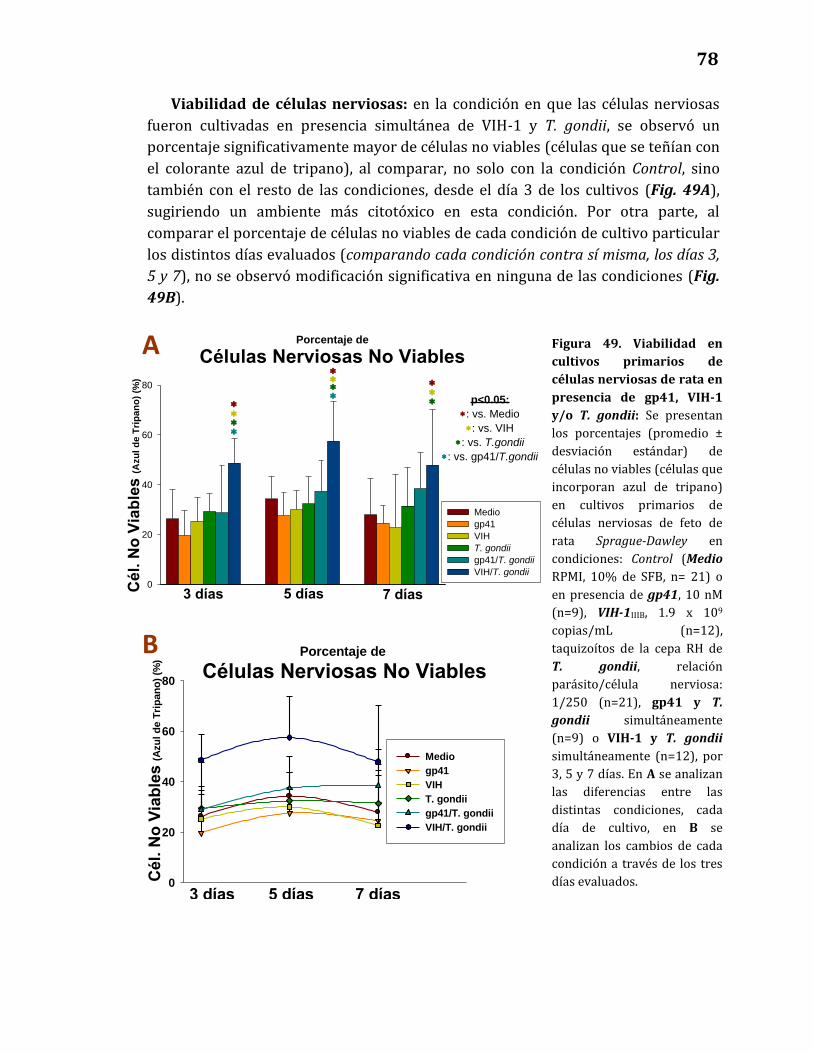
Viabilidad de células nerviosas: en la condición en que las células nerviosas
fueron cultivadas en presencia simultánea de VIH-1 y T. gondii, se observó un
porcentaje significativamente mayor de células no viables (células que se teñían con
el colorante azul de tripano), al comparar, no solo con la condición Control, sino
también con el resto de las condiciones, desde el día 3 de los cultivos (Fig. 49A),
sugiriendo un ambiente más citotóxico en esta condición. Por otra parte, al
comparar el porcentaje de células no viables de cada condición de cultivo particular
los distintos días evaluados (comparando cada condición contra sí misma, los días 3,
5 y 7), no se observó modificación significativa en ninguna de las condiciones (Fig.
49B).
Figura 49. Viabilidad en
cultivos primarios de
células nerviosas de rata en
presencia de gp41, VIH-1
y/o T. gondii: Se presentan
los porcentajes (promedio ±
desviación estándar) de
células no viables (células que
incorporan azul de tripano)
en cultivos primarios de
células nerviosas de feto de
rata Sprague-Dawley en
condiciones: Control (Medio
RPMI, 10% de SFB, n= 21) o
en presencia de gp41, 10 nM
(n=9), VIH-1IIIB, 1.9 x 109
copias/mL (n=12),
taquizoítos de la cepa RH de
T. gondii, relación
parásito/célula nerviosa:
1/250 (n=21), gp41 y T.
gondii simultáneamente
(n=9) o VIH-1 y T. gondii
simultáneamente (n=12), por
3, 5 y 7 días. En A se analizan
las diferencias entre las
distintas condiciones, cada
día de cultivo, en B se
analizan los cambios de cada
condición a través de los tres
días evaluados.
Porcentaje de
Células Nerviosas No Viables
Cél.
No
Via
ble
s (
Azu
l d
e T
rip
an
o)
(%)
0
20
40
60
80
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
Cél. N
o V
iab
les (
Azu
l d
e T
rip
an
o)
(%)
B
Porcentaje de
Células Nerviosas No Viables
Cé
l. N
o V
iab
les
(A
zu
l d
e T
rip
an
o)
(%)
0
20
40
60
80
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. Medio
: vs. VIH
: vs. T.gondii
: vs. gp41/T.gondii
A
78
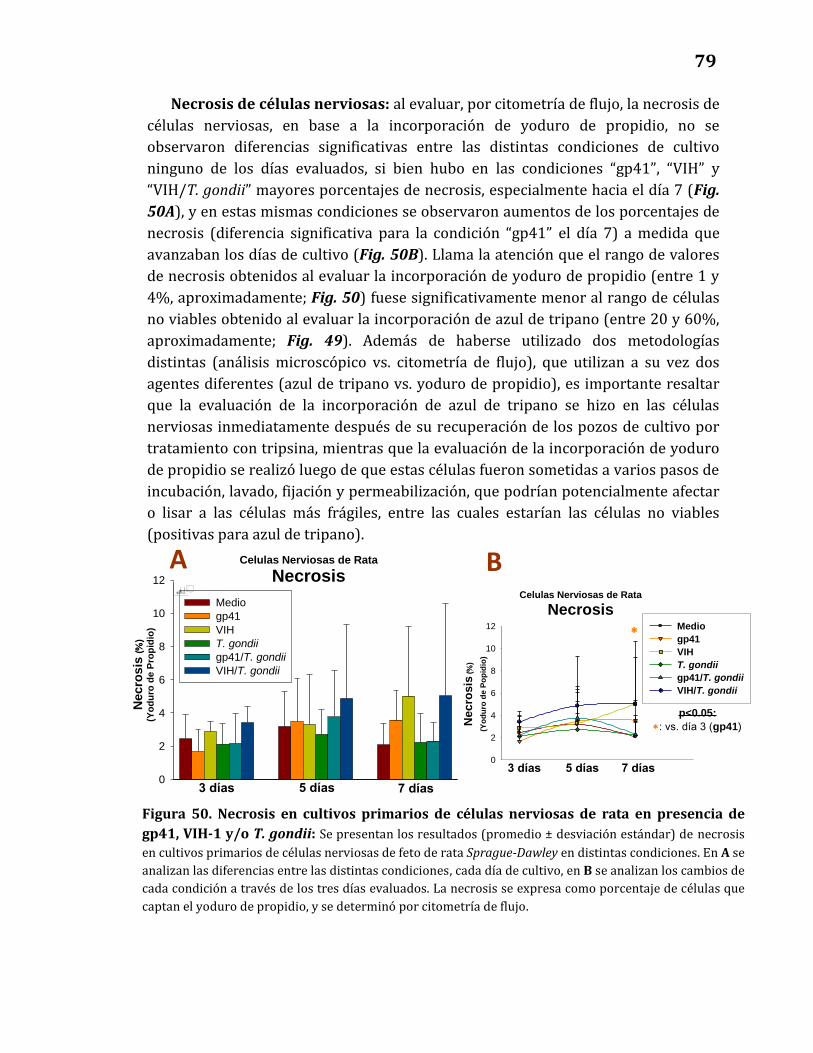
Necrosis de células nerviosas: al evaluar, por citometría de flujo, la necrosis de
células nerviosas, en base a la incorporación de yoduro de propidio, no se
observaron diferencias significativas entre las distintas condiciones de cultivo
ninguno de los días evaluados, si bien hubo en las condiciones “gp41”, “VIH” y
“VIH/T. gondii” mayores porcentajes de necrosis, especialmente hacia el día 7 (Fig.
50A), y en estas mismas condiciones se observaron aumentos de los porcentajes de
necrosis (diferencia significativa para la condición “gp41” el día 7) a medida que
avanzaban los días de cultivo (Fig. 50B). Llama la atención que el rango de valores
de necrosis obtenidos al evaluar la incorporación de yoduro de propidio (entre 1 y
4%, aproximadamente; Fig. 50) fuese significativamente menor al rango de células
no viables obtenido al evaluar la incorporación de azul de tripano (entre 20 y 60%,
aproximadamente; Fig. 49). Además de haberse utilizado dos metodologías
distintas (análisis microscópico vs. citometría de flujo), que utilizan a su vez dos
agentes diferentes (azul de tripano vs. yoduro de propidio), es importante resaltar
que la evaluación de la incorporación de azul de tripano se hizo en las células
nerviosas inmediatamente después de su recuperación de los pozos de cultivo por
tratamiento con tripsina, mientras que la evaluación de la incorporación de yoduro
de propidio se realizó luego de que estas células fueron sometidas a varios pasos de
incubación, lavado, fijación y permeabilización, que podrían potencialmente afectar
o lisar a las células más frágiles, entre las cuales estarían las células no viables
(positivas para azul de tripano).
Celulas Nerviosas de Rata
Necrosis
Necro
sis
(
)
(Yo
du
ro d
e P
rop
idio
)
0
2
4
6
8
10
12
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
ANecrosis de Células Nerviosas
(Cultivos Primarios de Celulas Nerviosas de Rata)
Ne
cro
sis
(
)
(Yo
du
ro d
e P
op
idio
)
0
2
4
6
8
10
12
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (gp41)
Celulas Nerviosas de Rata
Necrosis
Ne
cro
sis
(
)
(Yo
du
ro d
e P
op
idio
)
0
2
4
6
8
10
12 Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (gp41)
BNecrosis de Células Nerviosas
(Cultivos Primarios de Celulas Nerviosas de Rata)
Ne
cro
sis
(
)
(Yo
du
ro d
e P
op
idio
)
0
2
4
6
8
10
12
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (gp41)
Figura 50. Necrosis en cultivos primarios de células nerviosas de rata en presencia de
gp41, VIH-1 y/o T. gondii: Se presentan los resultados (promedio ± desviación estándar) de necrosis
en cultivos primarios de células nerviosas de feto de rata Sprague-Dawley en distintas condiciones. En A se
analizan las diferencias entre las distintas condiciones, cada día de cultivo, en B se analizan los cambios de
cada condición a través de los tres días evaluados. La necrosis se expresa como porcentaje de células que
captan el yoduro de propidio, y se determinó por citometría de flujo.
79
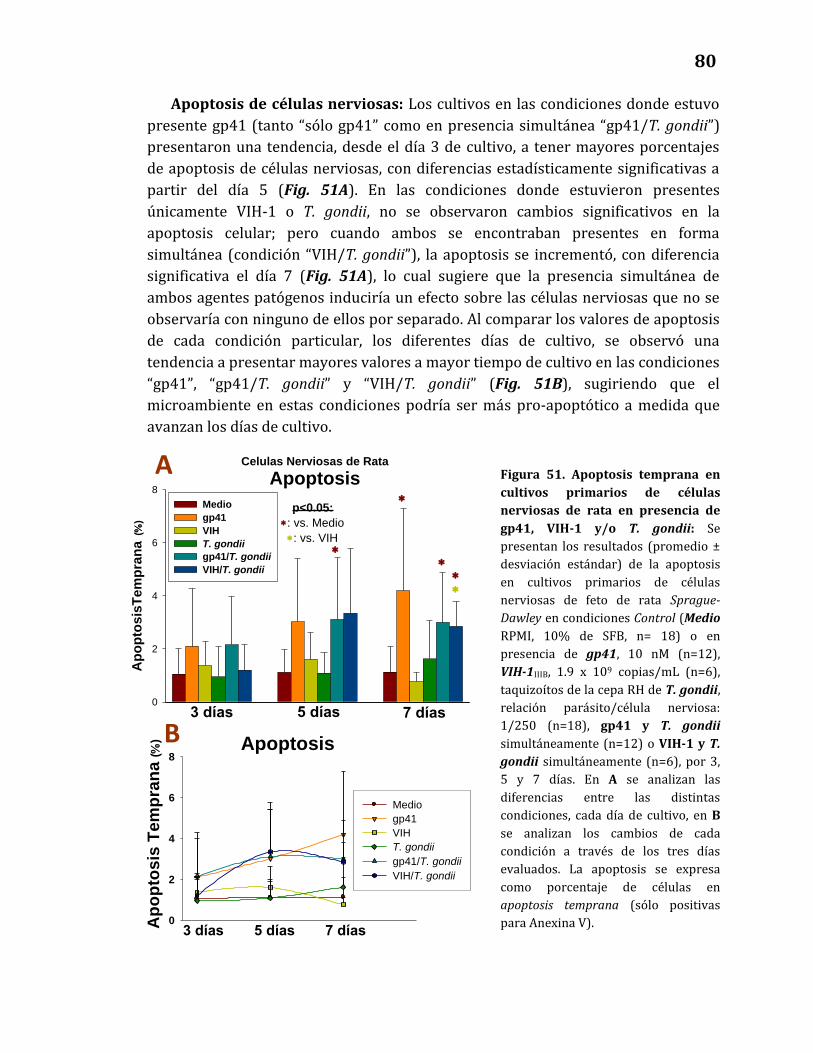
Apoptosis de células nerviosas: Los cultivos en las condiciones donde estuvo
presente gp41 (tanto “sólo gp41” como en presencia simultánea “gp41/T. gondii”)
presentaron una tendencia, desde el día 3 de cultivo, a tener mayores porcentajes
de apoptosis de células nerviosas, con diferencias estadísticamente significativas a
partir del día 5 (Fig. 51A). En las condiciones donde estuvieron presentes
únicamente VIH-1 o T. gondii, no se observaron cambios significativos en la
apoptosis celular; pero cuando ambos se encontraban presentes en forma
simultánea (condición “VIH/T. gondii”), la apoptosis se incrementó, con diferencia
significativa el día 7 (Fig. 51A), lo cual sugiere que la presencia simultánea de
ambos agentes patógenos induciría un efecto sobre las células nerviosas que no se
observaría con ninguno de ellos por separado. Al comparar los valores de apoptosis
de cada condición particular, los diferentes días de cultivo, se observó una
tendencia a presentar mayores valores a mayor tiempo de cultivo en las condiciones
“gp41”, “gp41/T. gondii” y “VIH/T. gondii” (Fig. 51B), sugiriendo que el
microambiente en estas condiciones podría ser más pro-apoptótico a medida que
avanzan los días de cultivo.
Figura 51. Apoptosis temprana en
cultivos primarios de células
nerviosas de rata en presencia de
gp41, VIH-1 y/o T. gondii: Se
presentan los resultados (promedio ±
desviación estándar) de la apoptosis
en cultivos primarios de células
nerviosas de feto de rata Sprague-
Dawley en condiciones Control (Medio
RPMI, 10% de SFB, n= 18) o en
presencia de gp41, 10 nM (n=12),
VIH-1IIIB, 1.9 x 109 copias/mL (n=6),
taquizoítos de la cepa RH de T. gondii,
relación parásito/célula nerviosa:
1/250 (n=18), gp41 y T. gondii
simultáneamente (n=12) o VIH-1 y T.
gondii simultáneamente (n=6), por 3,
5 y 7 días. En A se analizan las
diferencias entre las distintas
condiciones, cada día de cultivo, en B
se analizan los cambios de cada
condición a través de los tres días
evaluados. La apoptosis se expresa
como porcentaje de células en
apoptosis temprana (sólo positivas
para Anexina V).
Celulas Nerviosas de Rata
Apoptosis
Ap
op
tosis
Tem
pra
na
(
)
0
2
4
6
8
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. Medio
: vs. VIH
A
Apoptosis
Ap
op
tos
is T
em
pra
na
(
)
0
2
4
6
8
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
B
80
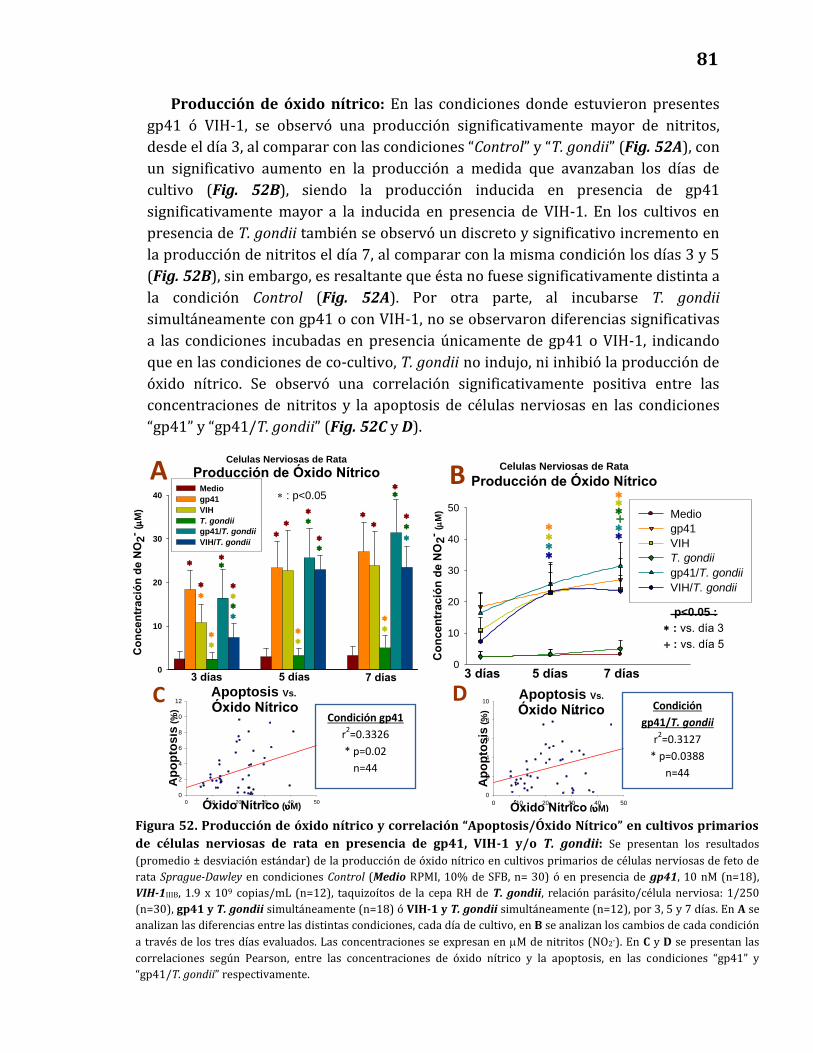
Producción de óxido nítrico: En las condiciones donde estuvieron presentes
gp41 ó VIH-1, se observó una producción significativamente mayor de nitritos,
desde el día 3, al comparar con las condiciones “Control” y “T. gondii” (Fig. 52A), con
un significativo aumento en la producción a medida que avanzaban los días de
cultivo (Fig. 52B), siendo la producción inducida en presencia de gp41
significativamente mayor a la inducida en presencia de VIH-1. En los cultivos en
presencia de T. gondii también se observó un discreto y significativo incremento en
la producción de nitritos el día 7, al comparar con la misma condición los días 3 y 5
(Fig. 52B), sin embargo, es resaltante que ésta no fuese significativamente distinta a
la condición Control (Fig. 52A). Por otra parte, al incubarse T. gondii
simultáneamente con gp41 o con VIH-1, no se observaron diferencias significativas
a las condiciones incubadas en presencia únicamente de gp41 o VIH-1, indicando
que en las condiciones de co-cultivo, T. gondii no indujo, ni inhibió la producción de
óxido nítrico. Se observó una correlación significativamente positiva entre las
concentraciones de nitritos y la apoptosis de células nerviosas en las condiciones
“gp41” y “gp41/T. gondii” (Fig. 52C y D).
Figura 52. Producción de óxido nítrico y correlación “Apoptosis/Óxido Nítrico” en cultivos primarios
de células nerviosas de rata en presencia de gp41, VIH-1 y/o T. gondii: Se presentan los resultados
(promedio ± desviación estándar) de la producción de óxido nítrico en cultivos primarios de células nerviosas de feto de
rata Sprague-Dawley en condiciones Control (Medio RPMI, 10% de SFB, n= 30) ó en presencia de gp41, 10 nM (n=18),
VIH-1IIIB, 1.9 x 109 copias/mL (n=12), taquizoítos de la cepa RH de T. gondii, relación parásito/célula nerviosa: 1/250
(n=30), gp41 y T. gondii simultáneamente (n=18) ó VIH-1 y T. gondii simultáneamente (n=12), por 3, 5 y 7 días. En A se
analizan las diferencias entre las distintas condiciones, cada día de cultivo, en B se analizan los cambios de cada condición
a través de los tres días evaluados. Las concentraciones se expresan en M de nitritos (NO2-). En C y D se presentan las
correlaciones según Pearson, entre las concentraciones de óxido nítrico y la apoptosis, en las condiciones “gp41” y
“gp41/T. gondii” respectivamente.
Celulas Nerviosas de Rata
Producción de Óxido Nítrico
Co
ncen
tració
n d
e N
O2-
(M
)
0
10
20
30
40
50Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05 :
: vs. día 3
: vs. día 5
B
Apoptosis Vs.
Óxido Nítrico
Óxido Nítrico (M)0 10 20 30 40 50
Ap
op
tosis
(%
)
0
2
4
6
8
10
12C Condición gp41
r2=0.3326
* p=0.02
n=44
Apoptosis Vs.
Óxido Nítrico
Óxido Nítrico (M)0 10 20 30 40 50
Ap
op
tosis
(%
)
0
2
4
6
8
10D Condición
gp41/T. gondii
r2=0.3127
* p=0.0388
n=44
Celulas Nerviosas de Rata
Producción de Óxido Nítrico
Co
nc
en
tra
ció
n d
e N
O2-
(M
)
0
10
20
30
40Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
: p<0.05
A
81

Producción de citoquinas en cultivos primarios de células nerviosas
Producción de IL-1: Las presencias exclusivas de gp41 o de VIH-1 se
asociaron con un aumento significativo en la producción de IL-1, con respecto a la
condición Control (Fig. 53A), mientras que las condiciones cultivadas en presencia
exclusiva de T. gondii no presentaron una producción significativa. Sin embargo,
cuando las células eran cultivadas en presencia simultánea de “VIH-1 y T. gondii”, la
producción de IL-1 era aproximadamente 2 ó 3 veces mayor a la de la condición
“solo con VIH-1”. Por otra parte, en las condiciones donde estuvo presente VIH-1 la
producción de IL-1 se incrementó significativamente a medida que avanzaban los
días de cultivo (Fig. 53B).
Celulas Nerviosas de Rata
Producción de IL-1
IL-1
(p
g/m
l)
0
50
100
150
200
300
400
500
600
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
*: vs. Medio
*: vs. gp41
*: vs. VIH
*: vs. T.gondii
*: vs. gp41/T.gondii
A
IL-1
(p
g/m
l)
60
90
120
150
200
300
400
500
600 Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3
+ : vs. día 5
Producción de IL-1B
Figura 53. Producción de IL-1
en cultivos primarios de células
nerviosas de rata en presencia
de gp41, VIH-1 y/o T. gondii: Se
presentan los resultados (promedio ±
desviación estándar) de la producción
de IL-1 en cultivos primarios de
células nerviosas de feto de rata
Sprague-Dawley en condiciones
Control (Medio RPMI, 10% de SFB, n=
12) ó en presencia de gp41, 10 nM
(n=6), VIH-1IIIB, 1.9 x 109 copias/mL
(n=6), taquizoítos de la cepa RH de T.
gondii, relación parásito/célula
nerviosa: 1/250 (n=12), gp41 y T.
gondii simultáneamente (n=6) o
VIH-1 y T. gondii simultáneamente
(n=6), por 3, 5 y 7 días. Las
diferencias entre las condiciones se
analizaron utilizando ANOVA o
Kruskal-Wallis, según fuese
apropiado, con pruebas de
comparación múltiple de Bonferroni o
Dunn, respectivamente,
considerándose significativos valores
de p<0.05. En A se analizan las
diferencias entre las distintas
condiciones, cada día de cultivo, en B
se analizan los cambios de cada
condición a través de los tres días
evaluados. Las concentraciones se
expresan en pg/mL.
82

Producción de IL-6: La producción de IL-6 aumentó significativamente en las
condiciones con presencia de gp41 o de VIH-1, con respecto a la condición Control
(Fig. 54A), manteniéndose “alta” durante los tres días evaluados, con una
producción máxima el día 5 para la condición “VIH-1/T. gondii” (Fig. 54B), siendo la
producción en presencia de gp41 mayor que en presencia de VIH-1. Por su parte, las
condiciones cultivadas en presencia exclusiva de T. gondii no presentaron aumento
significativo en esta producción, y cuando las células nerviosas se cultivaron en
presencia simultánea de “gp41/T. gondii” o de “VIH-1/T. gondii” no se observó
modificación a la producción observada en presencia exclusiva de gp41 o de VIH-1
(excepto el día 3 para “VIH-1/T. gondii”), respectivamente, indicando que, en
general, la presencia de T. gondii no indujo ni inhibió la producción de IL-6.
Figura 54. Producción de IL-6 en cultivos primarios de células nerviosas de rata en
presencia de gp41, VIH-1 y/o T. gondii: Se presentan los resultados (promedio ± desviación
estándar) de la producción de IL-6 en cultivos primarios de células nerviosas de feto de rata
Sprague-Dawley en distintas condiciones. En A se analizan las diferencias entre las distintas
condiciones, cada día de cultivo, en B se analizan los cambios de cada condición a través de los tres
días evaluados. Las concentraciones se expresan en pg/mL.
Celulas Nerviosas de Rata
Producción de IL-6
IL-6
(p
g/m
l)
0
2000
4000
6000
8000
10000
12000
14000
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (VIH/T. gondii)
+: vs. día 5 (VIH/T. gondii)
B
Celulas Nerviosas de Rata
Producción de IL-6
IL-6
(p
g/m
l)
0
2000
4000
6000
8000
10000
12000
14000Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. Medio
: vs. gp41
: vs. VIH
: vs. T. gondii
: vs. gp41/T. gondii
A
83
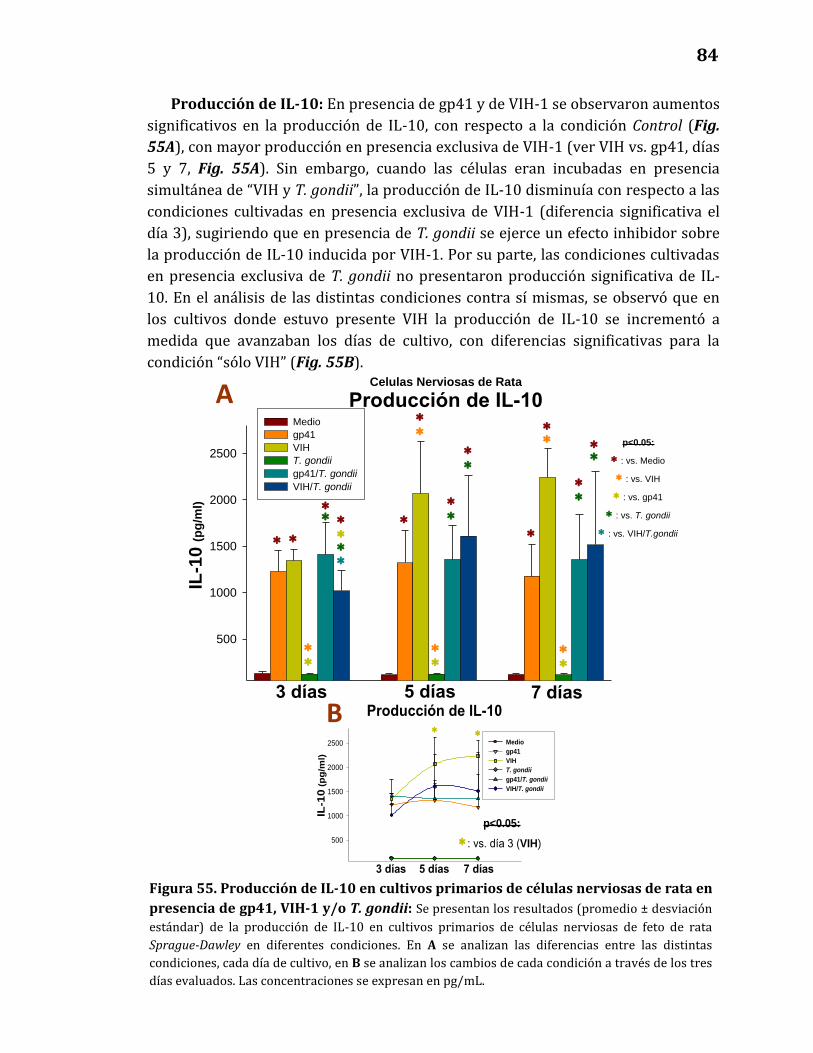
Producción de IL-10: En presencia de gp41 y de VIH-1 se observaron aumentos
significativos en la producción de IL-10, con respecto a la condición Control (Fig.
55A), con mayor producción en presencia exclusiva de VIH-1 (ver VIH vs. gp41, días
5 y 7, Fig. 55A). Sin embargo, cuando las células eran incubadas en presencia
simultánea de “VIH y T. gondii”, la producción de IL-10 disminuía con respecto a las
condiciones cultivadas en presencia exclusiva de VIH-1 (diferencia significativa el
día 3), sugiriendo que en presencia de T. gondii se ejerce un efecto inhibidor sobre
la producción de IL-10 inducida por VIH-1. Por su parte, las condiciones cultivadas
en presencia exclusiva de T. gondii no presentaron producción significativa de IL-
10. En el análisis de las distintas condiciones contra sí mismas, se observó que en
los cultivos donde estuvo presente VIH la producción de IL-10 se incrementó a
medida que avanzaban los días de cultivo, con diferencias significativas para la
condición “sólo VIH” (Fig. 55B).
Figura 55. Producción de IL-10 en cultivos primarios de células nerviosas de rata en
presencia de gp41, VIH-1 y/o T. gondii: Se presentan los resultados (promedio ± desviación
estándar) de la producción de IL-10 en cultivos primarios de células nerviosas de feto de rata
Sprague-Dawley en diferentes condiciones. En A se analizan las diferencias entre las distintas
condiciones, cada día de cultivo, en B se analizan los cambios de cada condición a través de los tres
días evaluados. Las concentraciones se expresan en pg/mL.
Producción de IL-10
500
1000
1500
2000
2500 Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (VIH)
IL-1
0 (
pg
/ml)
B
Celulas Nerviosas de Rata
Producción de IL-10
IL-1
0 (
pg
/ml)
500
1000
1500
2000
2500
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. Medio
: vs. VIH
: vs. gp41
: vs. T. gondii
: vs. VIH/T.gondii
A
84
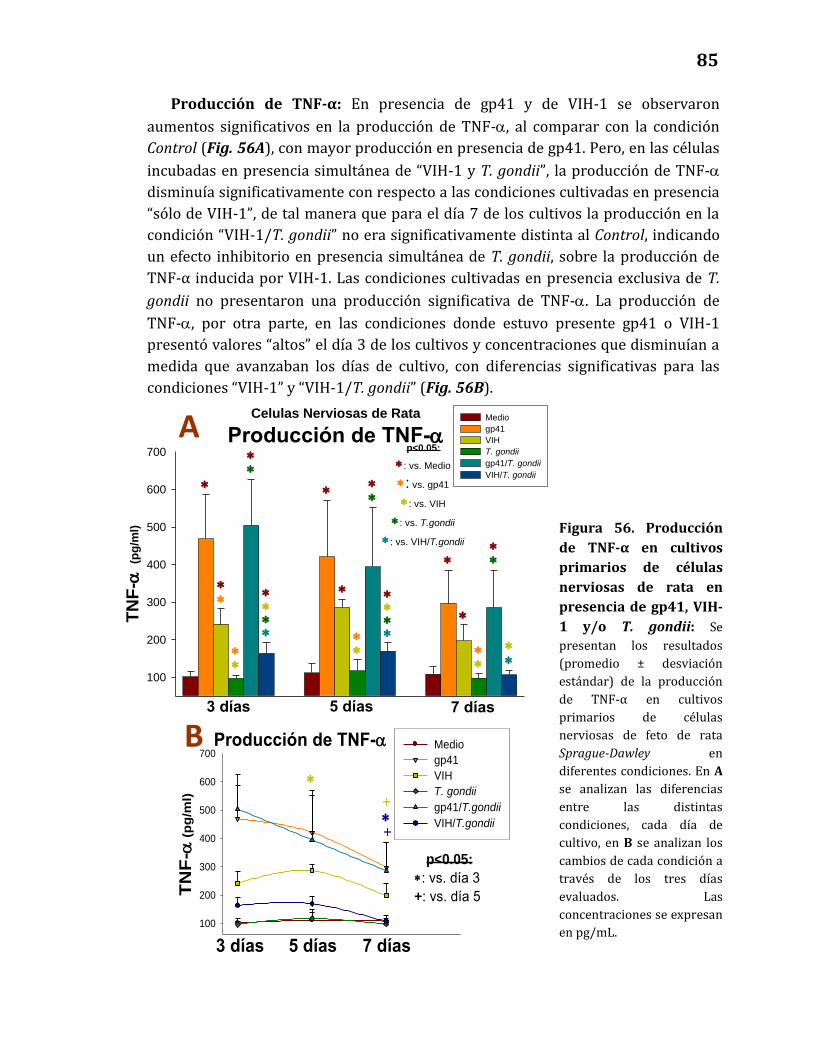
Producción de TNF-α: En presencia de gp41 y de VIH-1 se observaron
aumentos significativos en la producción de TNF-, al comparar con la condición
Control (Fig. 56A), con mayor producción en presencia de gp41. Pero, en las células
incubadas en presencia simultánea de “VIH-1 y T. gondii”, la producción de TNF-
disminuía significativamente con respecto a las condiciones cultivadas en presencia
“sólo de VIH-1”, de tal manera que para el día 7 de los cultivos la producción en la
condición “VIH-1/T. gondii” no era significativamente distinta al Control, indicando
un efecto inhibitorio en presencia simultánea de T. gondii, sobre la producción de
TNF-α inducida por VIH-1. Las condiciones cultivadas en presencia exclusiva de T.
gondii no presentaron una producción significativa de TNF-. La producción de
TNF-, por otra parte, en las condiciones donde estuvo presente gp41 o VIH-1
presentó valores “altos” el día 3 de los cultivos y concentraciones que disminuían a
medida que avanzaban los días de cultivo, con diferencias significativas para las
condiciones “VIH-1” y “VIH-1/T. gondii” (Fig. 56B).
Figura 56. Producción
de TNF-α en cultivos
primarios de células
nerviosas de rata en
presencia de gp41, VIH-
1 y/o T. gondii: Se
presentan los resultados
(promedio ± desviación
estándar) de la producción
de TNF-α en cultivos
primarios de células
nerviosas de feto de rata
Sprague-Dawley en
diferentes condiciones. En A
se analizan las diferencias
entre las distintas
condiciones, cada día de
cultivo, en B se analizan los
cambios de cada condición a
través de los tres días
evaluados. Las
concentraciones se expresan
en pg/mL.
Producción de TNF-
TN
F-
(p
g/m
l)
100
200
300
400
500
600
700Medio
gp41
VIH
T. gondii
gp41/T.gondii
VIH/T.gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3
+: vs. día 5
B
Celulas Nerviosas de Rata
Producción de TNF-
TN
F- (
pg
/ml)
100
200
300
400
500
600
700
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. Medio
: vs. gp41
: vs. VIH
: vs. T.gondii
: vs. VIH/T.gondii
A
85
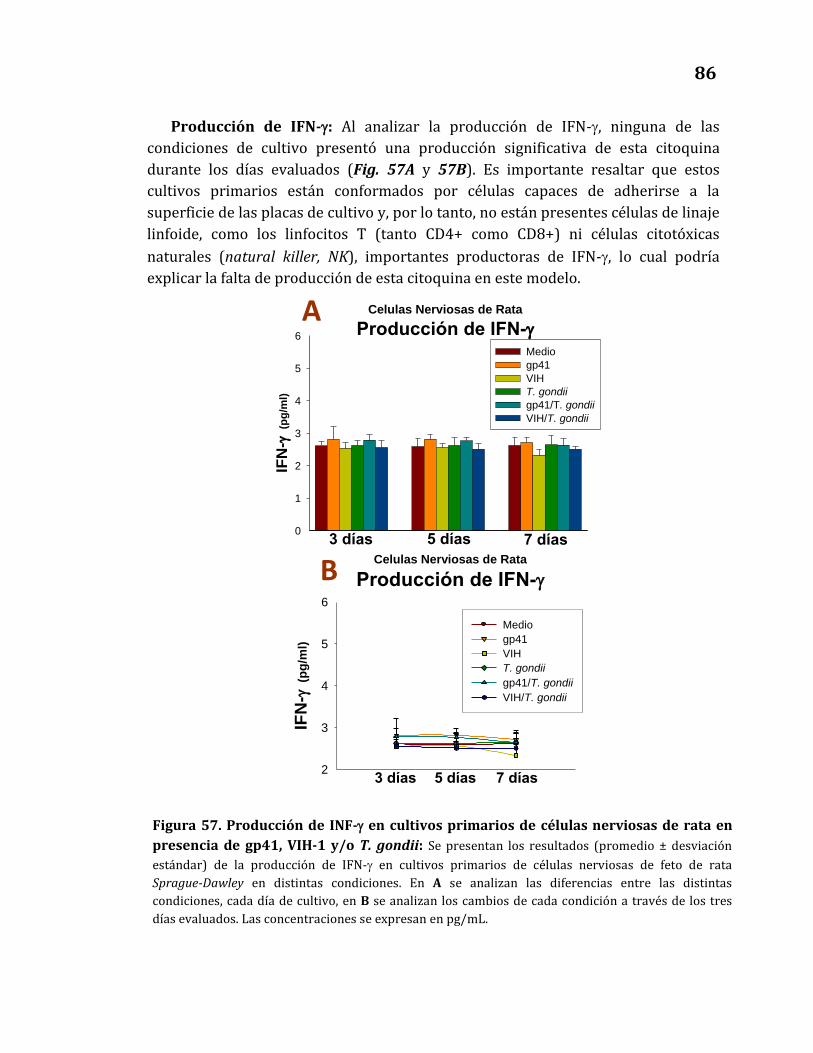
Producción de IFN-: Al analizar la producción de IFN-, ninguna de las
condiciones de cultivo presentó una producción significativa de esta citoquina
durante los días evaluados (Fig. 57A y 57B). Es importante resaltar que estos
cultivos primarios están conformados por células capaces de adherirse a la
superficie de las placas de cultivo y, por lo tanto, no están presentes células de linaje
linfoide, como los linfocitos T (tanto CD4+ como CD8+) ni células citotóxicas
naturales (natural killer, NK), importantes productoras de IFN-, lo cual podría
explicar la falta de producción de esta citoquina en este modelo.
Figura 57. Producción de INF- en cultivos primarios de células nerviosas de rata en
presencia de gp41, VIH-1 y/o T. gondii: Se presentan los resultados (promedio ± desviación
estándar) de la producción de IFN- en cultivos primarios de células nerviosas de feto de rata
Sprague-Dawley en distintas condiciones. En A se analizan las diferencias entre las distintas
condiciones, cada día de cultivo, en B se analizan los cambios de cada condición a través de los tres
días evaluados. Las concentraciones se expresan en pg/mL.
Celulas Nerviosas de Rata
Producción de IFN-
IFN
-
(pg
/ml)
2
3
4
5
6
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
B
Celulas Nerviosas de Rata
Producción de IFN-
IFN
- (
pg
/ml)
0
1
2
3
4
5
6
Medio
gp41
VIH
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
A
86

B. INFECCIÓN DE CÉLULAS NERVIOSAS POR T. gondii
Recuento de taquizoítos libres: los taquizoítos de T. gondii se replican
activamente en el citoplasma de las células nerviosas infectadas, provocando la lisis
de éstas, liberando taquizoítos recién replicados, los cuales infectan nuevas células.
Se generan, entonces, ciclos consecutivos de infección/lisis celular/liberación de
taquizoítos/infección de nuevas células, lo cual resulta en el aumento del número de
parásitos y la destrucción progresiva de la monocapa celular. El recuento de los
taquizoítos libres en los sobrenadantes de cultivo se consideró, pues, como un
indicador de la progresión de la infección parasitaria. En congruencia con esto, las
condiciones donde las células nerviosas fueron infectadas con T. gondii (“sólo T.
gondii”), presentaron recuentos de taquizoítos libres significativamente mayores a
medida que avanzaban los días de cultivo (Fig. 58B). También en las condiciones
donde las células fueron cultivadas en presencia simultánea de “gp41 y T. gondii”, se
observaron aumentos significativos en los recuentos de taquizoítos libres al
aumentar los días de cultivo (Fig. 58B), pero es resaltante que cada día evaluado
estos recuentos fueron significativamente menores a los observados en presencia
exclusiva de T. gondii (Fig. 58A), sugiriendo que la presencia de gp41 en los cultivos
de alguna forma interfería con el proceso de replicación parasitaria. Por otra parte,
al cultivar las células nerviosas en presencia simultánea de “VIH-1 y T. gondii”,
aunque se observó el día 3 de los cultivos una replicación parasitaria similar a las
condiciones donde estaba presente “sólo T. gondii” (Fig. 58A), los días 5 y 7 se
observó prácticamente una inhibición total de la subsecuente replicación
parasitaria en la condición “VIH-1/T. gondii”, manteniéndose los recuentos de
taquizoítos libres sin cambios significativos al compararlos con la misma condición
el día 3 de cultivo (Fig. 58B), evidenciando que en presencia de VIH-1 la
interferencia con la replicación de T. gondii fue aún mayor que la observada en
presencia de gp41, aunque con una cinética distinta, con una inhibición más bien
“tardía” (Fig. 58A).
87
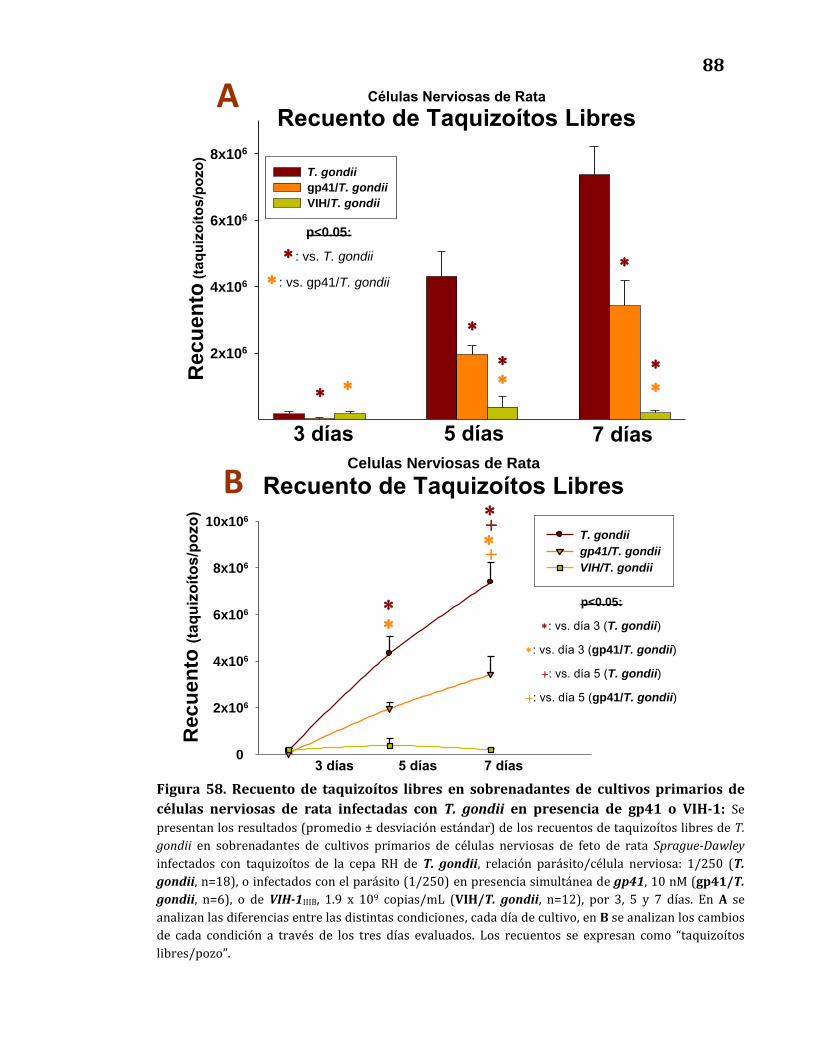
Figura 58. Recuento de taquizoítos libres en sobrenadantes de cultivos primarios de
células nerviosas de rata infectadas con T. gondii en presencia de gp41 o VIH-1: Se
presentan los resultados (promedio ± desviación estándar) de los recuentos de taquizoítos libres de T.
gondii en sobrenadantes de cultivos primarios de células nerviosas de feto de rata Sprague-Dawley
infectados con taquizoítos de la cepa RH de T. gondii, relación parásito/célula nerviosa: 1/250 (T.
gondii, n=18), o infectados con el parásito (1/250) en presencia simultánea de gp41, 10 nM (gp41/T.
gondii, n=6), o de VIH-1IIIB, 1.9 x 109 copias/mL (VIH/T. gondii, n=12), por 3, 5 y 7 días. En A se
analizan las diferencias entre las distintas condiciones, cada día de cultivo, en B se analizan los cambios
de cada condición a través de los tres días evaluados. Los recuentos se expresan como “taquizoítos
libres/pozo”.
Células Nerviosas de Rata
Recuento de Taquizoítos Libres
Re
cu
en
to (
taq
uiz
oít
os
/po
zo
)
2x106
4x106
6x106
8x106
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. T. gondii
: vs. gp41/T. gondii
A
Celulas Nerviosas de Rata
Recuento de Taquizoítos Libres
Re
cu
en
to (
taq
uiz
oít
os
/po
zo
)
0
2x106
4x106
6x106
8x106
10x106
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
p<0.05:
: vs. día 3 (T. gondii)
: vs. día 3 (gp41/T. gondii)
: vs. día 5 (T. gondii)
: vs. día 5 (gp41/T. gondii)
B
88

Porcentaje y recuento de células nerviosas infectadas por T. gondii: algunos
de los experimentos se realizaron sobre laminillas cubreobjeto, tratadas
previamente con poli-lisina y colocadas en los pozos de las placas de cultivo,
siguiendo el mismo protocolo general ya descrito, de forma que se pudiera realizar
un análisis microscópico de las monocapas celulares en las distintas condiciones. Al
finalizar los tiempos de cultivo, las células eran fijadas con una solución de
paraformaldehido al 4%, posteriormente lavadas con PBS, y conservadas para su
coloración (Wright-Giemsa) o procesamiento por técnicas de inmunocitoquímica.
Por el análisis microscópico de estas laminillas se determinó el porcentaje de
células nerviosas infectadas por T. gondii (Fig. 59A y 59B), y con los promedios de
los recuentos de células nerviosas totales de cada condición (Fig. 48) se calculó el
recuento de células infectadas (“infección absoluta”, Fig. 59C y 59D). En estos
análisis se pudo evidenciar que en la condición “VIH-1/T. gondii”, tanto el
porcentaje como el recuento de células infectadas eran significativamente menores
al de las otras condiciones (Fig. 59A y 59C), lo cual es congruente con los resultados
de recuentos de taquizoítos libres (Fig. 58), sugiriendo una inhibición “tardía” de la
infección parasitaria en presencia de VIH-1 (la condición “VIH-1/T. gondii” no fue
significativamente distinta a la condición “T. gondii” el día 3 de cultivo, pero sí los
días 5 y 7; Fig. 58A, 59A y 59C). Por otra parte, no se observaron diferencias
significativas en el porcentaje y recuento de células infectadas entre las condiciones
“T. gondii” y “gp41/T. gondii”, si bien el recuento de células infectadas tendió a ser
menor en la condición “gp41/T. gondii” los días 5 y 7 de cultivo (Fig. 59C).
Finalmente, al analizar cada condición contra sí misma, los distintos días evaluados,
se evidenció que tanto el porcentaje como el valor absoluto de las células infectadas
por T. gondii se incrementaban significativamente al aumentar los días de cultivo en
las tres condiciones donde estaba presente el parásito (Fig. 59B y 59D).
89
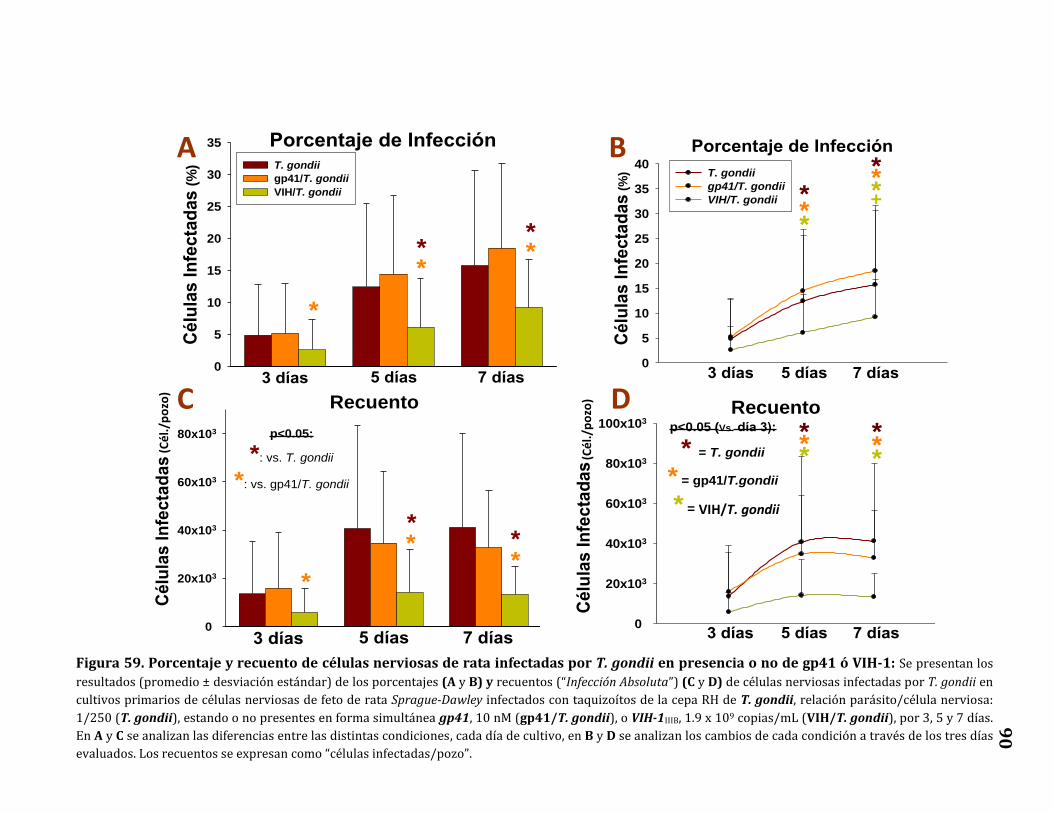
0
5
10
15
20
25
30
35
40T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
Porcentaje de Infección
Cé
lula
s I
nfe
cta
da
s (
%) *
***
**
+
B
0
20x103
40x103
60x103
80x103
100x103
3 días 5 días 7 días
Recuento
Cé
lula
s In
fec
tad
as
(/
l) p<0.05 (Vs. día 3):
* = T. gondii
* = gp41/T.gondii
* = vih/T.gondii
**
* ** *
D
Porcentaje de Infección
Cé
lula
s I
nfe
cta
da
s (
%)
0
5
10
15
20
25
30
35
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
*
** *
*
A
Recuento
Cé
lula
s In
fec
tad
as
(/
l)
0
20x103
40x103
60x103
80x103
3 días 5 días 7 días
*
**
**
p<0.05:
*: vs. T. gondii
*: vs. gp41/T. gondii
C
VIH/T. gondii
(Cé
l./p
ozo
)
(Cé
l./p
ozo
)
Figura 59. Porcentaje y recuento de células nerviosas de rata infectadas por T. gondii en presencia o no de gp41 ó VIH-1: Se presentan los
resultados (promedio ± desviación estándar) de los porcentajes (A y B) y recuentos (“Infección Absoluta”) (C y D) de células nerviosas infectadas por T. gondii en
cultivos primarios de células nerviosas de feto de rata Sprague-Dawley infectados con taquizoítos de la cepa RH de T. gondii, relación parásito/célula nerviosa:
1/250 (T. gondii), estando o no presentes en forma simultánea gp41, 10 nM (gp41/T. gondii), o VIH-1IIIB, 1.9 x 109 copias/mL (VIH/T. gondii), por 3, 5 y 7 días.
En A y C se analizan las diferencias entre las distintas condiciones, cada día de cultivo, en B y D se analizan los cambios de cada condición a través de los tres días
evaluados. Los recuentos se expresan como “células infectadas/pozo”.
90

Infección de células nerviosas por T. gondii – Número de taquizoítos por
célula infectada: T. gondii infecta a las células nerviosas y se replica en su
citoplasma, por lo que es posible observar un número creciente de parásitos en el
interior de las células a medida que la infección progresa. Como, en un momento
dado, no todas las células infectadas se encuentran en una misma etapa de
infección, será posible observar números variables de parásitos en las distintas
células (Fig. 60), y la proporción de éstas nos pueden orientar en cuanto a la
progresión de la infección. Por esta razón, en las distintas condiciones de cultivo, las
células infectadas fueron analizadas para determinar el grado de infección, esto es,
si el número de parásitos dentro de la célula estaba entre 0 y 4, entre 5 y 15, entre
16 y 30 o si era mayor de 30. En la Figura 61 y en la Tabla II se presentan los
resultados de estos análisis. En la condiciones donde las células nerviosas eran
incubadas sólo con T. gondii, era posible observar el día 3 de los cultivos un
predominio significativo de las células con 5-15 parásitos en su citoplasma; para el
día 5 predominaban significativamente células con 5-30 parásitos, mientras que el
día 7 el predominio era para el grupo con 16-30 parásitos por célula infectada. Esto
sugiere un desplazamiento progresivo del grado de infección, con número creciente
de parásitos intracelulares al ir avanzando los días de cultivo. Sin embargo esta
cinética de infección parasitaria se modificó cuando en los cultivos se incorporaba
simultáneamente al VIH-1 (condición “VIH-1/T. gondii). Los días 3 y 5 de los
cultivos predominaron significativamente las células con 5 a 30 parásitos en su
citoplasma, pero para el día 7 el grupo celular que predominó en forma significativa
fue el que tenía de 5 a 15 parásitos, lo que indica un desplazamiento significativo
hacia una menor replicación parasitaria el día 7, sugiriendo un efecto tardío de
inhibición de la replicación, en congruencia con los resultados presentados en las
páginas anteriores (Fig. 58 y 59). Así, mientras que para el día 3, en la condición “T.
gondii” cerca del 70% de las células infectadas tenían 15 parásitos intracelulares o
menos, en la condición “VIH-1/T. gondii” más del 50% de las células infectadas
tenían 16 parásitos o más. A partir del día 5, sin embargo, la situación se invierte,
con 16 parásitos o más para 60% de las células infectadas en la condición “T. gondii”
y 15 parásitos o menos para más del 50% de las células en la condición “VIH-1/T.
gondii”. Por su parte, la condición “gp41/T. gondii” mostró una cinética de
replicación parasitaria que no fue significativamente distinta a la de la condición “T.
gondii”, excepto el día 3 de cultivo (Fig. 61, Tabla II).
91
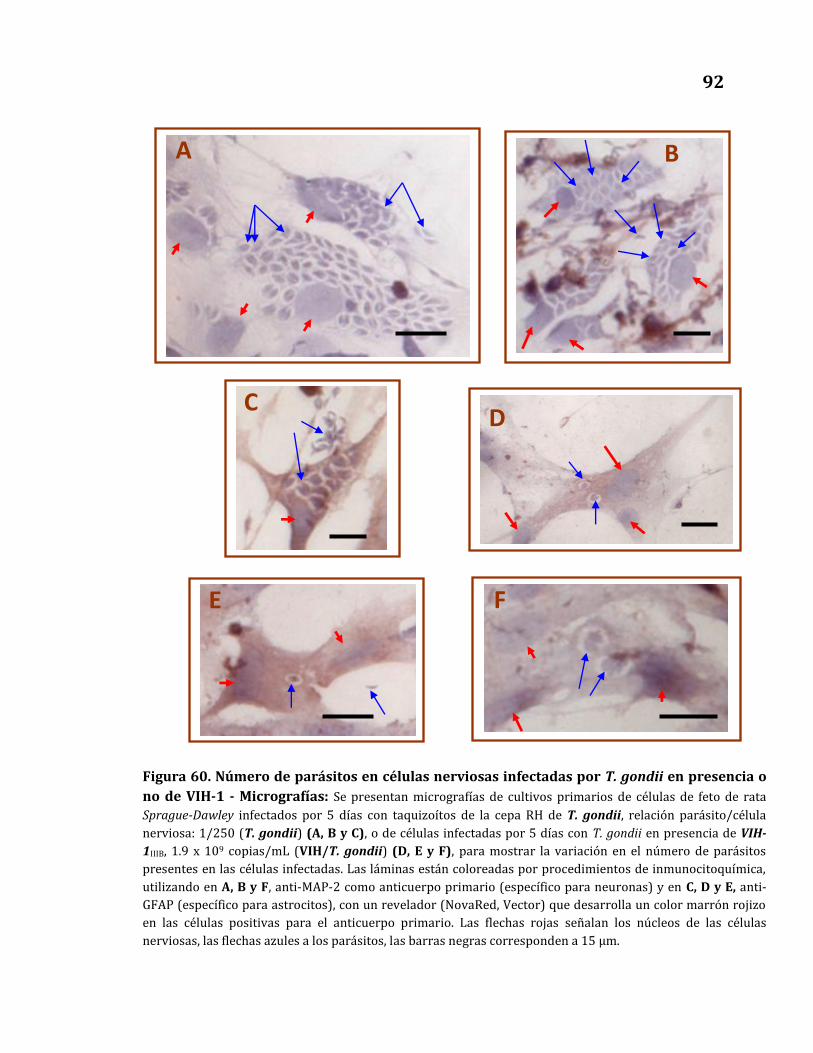
Figura 60. Número de parásitos en células nerviosas infectadas por T. gondii en presencia o
no de VIH-1 - Micrografías: Se presentan micrografías de cultivos primarios de células de feto de rata
Sprague-Dawley infectados por 5 días con taquizoítos de la cepa RH de T. gondii, relación parásito/célula
nerviosa: 1/250 (T. gondii) (A, B y C), o de células infectadas por 5 días con T. gondii en presencia de VIH-
1IIIB, 1.9 x 109 copias/mL (VIH/T. gondii) (D, E y F), para mostrar la variación en el número de parásitos
presentes en las células infectadas. Las láminas están coloreadas por procedimientos de inmunocitoquímica,
utilizando en A, B y F, anti-MAP-2 como anticuerpo primario (específico para neuronas) y en C, D y E, anti-
GFAP (específico para astrocitos), con un revelador (NovaRed, Vector) que desarrolla un color marrón rojizo
en las células positivas para el anticuerpo primario. Las flechas rojas señalan los núcleos de las células
nerviosas, las flechas azules a los parásitos, las barras negras corresponden a 15 μm.
C
E
F
D
A
B
92
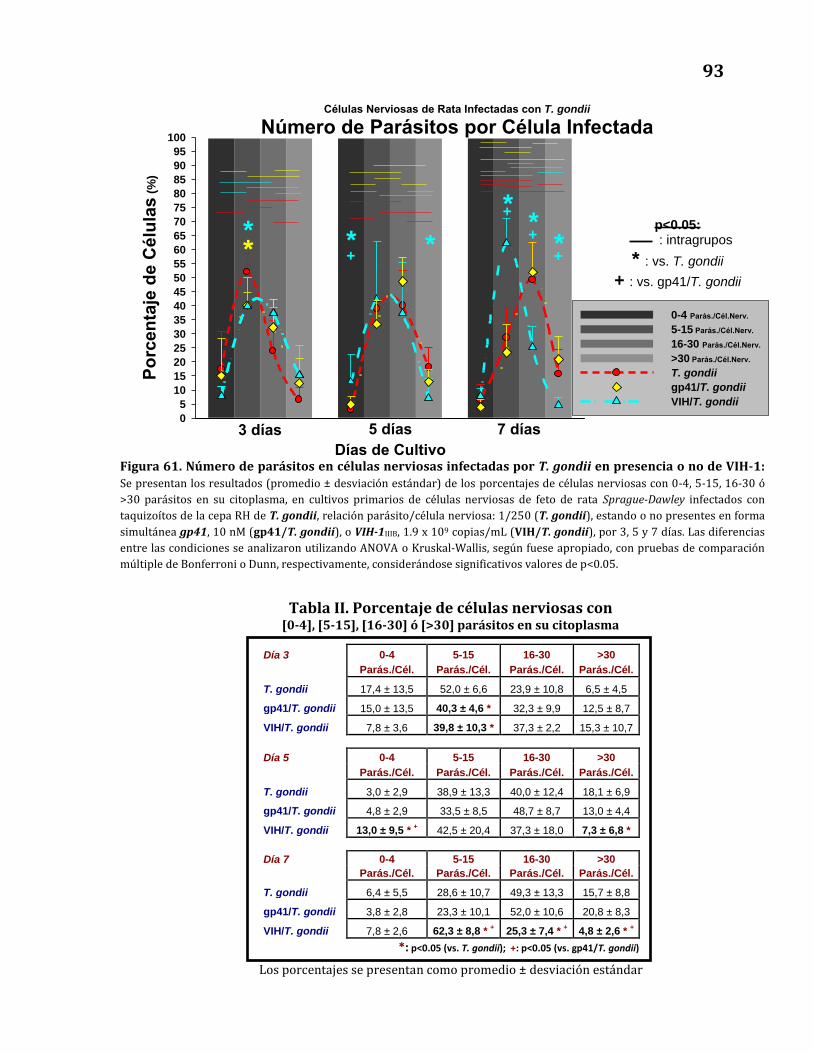
Figura 61. Número de parásitos en células nerviosas infectadas por T. gondii en presencia o no de VIH-1:
Se presentan los resultados (promedio ± desviación estándar) de los porcentajes de células nerviosas con 0-4, 5-15, 16-30 ó
>30 parásitos en su citoplasma, en cultivos primarios de células nerviosas de feto de rata Sprague-Dawley infectados con
taquizoítos de la cepa RH de T. gondii, relación parásito/célula nerviosa: 1/250 (T. gondii), estando o no presentes en forma
simultánea gp41, 10 nM (gp41/T. gondii), o VIH-1IIIB, 1.9 x 109 copias/mL (VIH/T. gondii), por 3, 5 y 7 días. Las diferencias
entre las condiciones se analizaron utilizando ANOVA o Kruskal-Wallis, según fuese apropiado, con pruebas de comparación
múltiple de Bonferroni o Dunn, respectivamente, considerándose significativos valores de p<0.05.
Células Nerviosas de Rata Infectadas con T. gondii
Número de Parásitos por Célula Infectada
Días de Cultivo
Po
rcen
taje
de C
élu
las
(%
)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
0-4 Parás./Cél.Nerv.
5-15 Parás./Cél.Nerv.
16-30 Parás./Cél.Nerv.
>30 Parás./Cél.Nerv.
T. gondii
gp41/T. gondii
VIH/T. gondii
3 días 5 días 7 días
**
p<0.05: : intragrupos
* : vs. T. gondii
+ : vs. gp41/T. gondii
+* *
*+*+
*+
Los porcentajes se presentan como promedio ± desviación estándar
Día 3 0-4 5-15 16-30 >30
Parás./Cél. Parás./Cél. Parás./Cél. Parás./Cél.
T. gondii 17,4 ± 13,5 52,0 ± 6,6 23,9 ± 10,8 6,5 ± 4,5
gp41/T. gondii 15,0 ± 13,5 40,3 ± 4,6 * 32,3 ± 9,9 12,5 ± 8,7
VIH/T. gondii 7,8 ± 3,6 39,8 ± 10,3 * 37,3 ± 2,2 15,3 ± 10,7
Día 5 0-4 5-15 16-30 >30
Parás./Cél. Parás./Cél. Parás./Cél. Parás./Cél.
T. gondii 3,0 ± 2,9 38,9 ± 13,3 40,0 ± 12,4 18,1 ± 6,9
gp41/T. gondii 4,8 ± 2,9 33,5 ± 8,5 48,7 ± 8,7 13,0 ± 4,4
VIH/T. gondii 13,0 ± 9,5 * + 42,5 ± 20,4 37,3 ± 18,0 7,3 ± 6,8 *
Día 7 0-4 5-15 16-30 >30
Parás./Cél. Parás./Cél. Parás./Cél. Parás./Cél.
T. gondii 6,4 ± 5,5 28,6 ± 10,7 49,3 ± 13,3 15,7 ± 8,8
gp41/T. gondii 3,8 ± 2,8 23,3 ± 10,1 52,0 ± 10,6 20,8 ± 8,3
VIH/T. gondii 7,8 ± 2,6 62,3 ± 8,8 * + 25,3 ± 7,4 *
+ 4,8 ± 2,6 *
+
*: p<0.05 (vs. T. gondii); +: p<0.05 (vs. gp41/T. gondii)
Tabla II. Porcentaje de células nerviosas con [0-4], [5-15], [16-30] ó [>30] parásitos en su citoplasma
93

C. SUBPOBLACIONES DE CÉLULAS NERVIOSAS EN CULTIVOS PRIMARIOS
Porcentaje de astrocitos, neuronas y células de la microglía en cultivos
primarios: para estudiar qué poblaciones (linajes) de células nerviosas estaban
presentes en los cultivos primarios, algunos de los experimentos fueron realizados
sobre laminillas cubreobjeto y se procesaron por técnicas de inmunocitoquímica,
utilizando anticuerpos primarios anti-MAP-2, anti-GFAP y anti-CD11b/c, para la
identificación de neuronas, astrocitos y células de la microglía, respectivamente. El
análisis de las laminillas así procesadas, permitió determinar el porcentaje de cada
población celular evaluada, así como el porcentaje de infección por linaje. Por
razones técnicas no fue posible procesar por inmunocitoquímica a las células
cultivadas en presencia de “gp41” y de “gp41/T. gondii” (éstas sólo pudieron ser
evaluadas por tinción Wright/Giemsa). Este análisis permitió conocer que en las
condiciones “Control”, “VIH-1”, “T. gondii” y “VIH-1/T. gondii”, el porcentaje de
astrocitos oscilaba entre el 66 y 80%, el de neuronas entre 10 y 35%, y el de células
de la microglía entre el 1 y 10%, manteniéndose estas proporciones básicamente
estables durante los días evaluados, en las cuatro distintas condiciones de cultivo
(Fig. 62). Estos resultados muestran claramente que los cultivos primarios estaban
compuestos por una población mixta de células, con una proporción similar a la
descrita por otros autores136, 137, 258, representando subpoblaciones (linajes) y
proporciones presentes en el sistema nervioso central intacto137, 258, permitiendo,
durante la realización de los experimentos, la interacción entre los distintos linajes
celulares, y entre éstos y los agentes patógenos evaluados, permitiendo una visión
más integral, un mayor acercamiento al fenómeno in vivo.
94
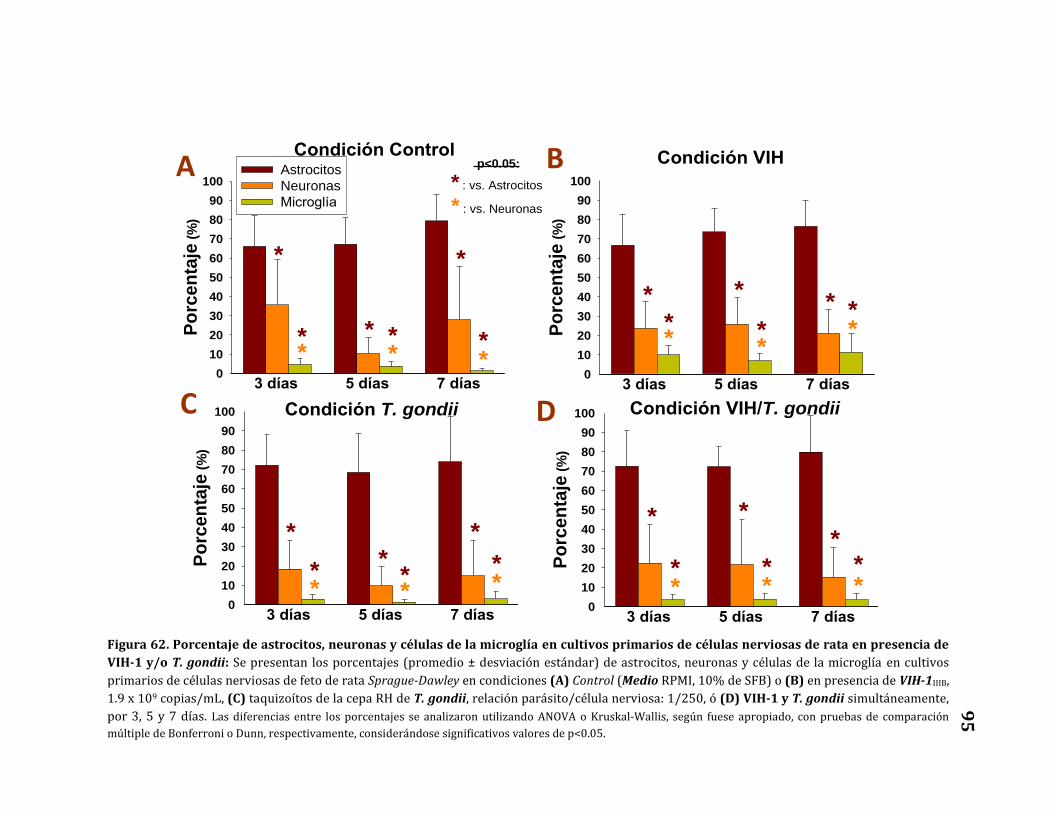
Condición T. gondii
Po
rcen
taje
(%
)
0
10
20
30
40
50
60
70
80
90
100
3 días 5 días 7 días
*
*
*
*
*
** *
*
C Condición VIH/T. gondii
Po
rce
nta
je (
%)
0
10
20
30
40
50
60
70
80
90
100
3 días 5 días 7 días
**
*
*
*
*
** *
D
Condición Control
Po
rce
nta
je (
%)
0
10
20
30
40
50
60
70
80
90
100Astrocitos
Neuronas
Microglía
3 días 5 días 7 días
p<0.05:
* : vs. Astrocitos
* : vs. Neuronas
*
*
****
*
* *
A Condición VIH
Po
rcen
taje
(%
)
0
10
20
30
40
50
60
70
80
90
100
3 días 5 días 7 días
**
*
*
*
*
***
B
Figura 62. Porcentaje de astrocitos, neuronas y células de la microglía en cultivos primarios de células nerviosas de rata en presencia de
VIH-1 y/o T. gondii: Se presentan los porcentajes (promedio ± desviación estándar) de astrocitos, neuronas y células de la microglía en cultivos
primarios de células nerviosas de feto de rata Sprague-Dawley en condiciones (A) Control (Medio RPMI, 10% de SFB) o (B) en presencia de VIH-1IIIB,
1.9 x 109 copias/mL, (C) taquizoítos de la cepa RH de T. gondii, relación parásito/célula nerviosa: 1/250, ó (D) VIH-1 y T. gondii simultáneamente,
por 3, 5 y 7 días. Las diferencias entre los porcentajes se analizaron utilizando ANOVA o Kruskal-Wallis, según fuese apropiado, con pruebas de comparación
múltiple de Bonferroni o Dunn, respectivamente, considerándose significativos valores de p<0.05.
95

Porcentaje de astrocitos, neuronas y células de la microglía infectados por
T. gondii en cultivos primarios: las láminas procesadas por inmunocitoquímica,
también permitieron, como ya se mencionó, determinar el porcentaje de infección
parasitaria por linaje de células nerviosas en las condiciones cultivadas en
presencia exclusiva de T. gondii, y en las condiciones cultivadas en presencia
simultánea de “T. gondii y VIH-1” (Fig. 63). Fue así posible evidenciar que para el
día 3 de los cultivos, los astrocitos infectados por T. gondii representaban 0.6 1.5%
de la población total de células nerviosas, 6.8 8.6 % el día 5 y 12.3 11.0% el día
7. Las neuronas, por su parte, representaban el 1.3 2.6% el día 3; 2.5 4.1 el día 5
y 2.5 5.0 el día 7. Para estos mismos días los porcentajes de células de la microglía
infectadas era de 0.6 1.3; 0.4 1.0 y 0.5 1.3, respectivamente (Tabla III). Los
porcentajes de astrocitos, neuronas y células de la microglía infectados por T. gondii
no fueron significativamente distintos, ninguno de los tres días evaluados, al
comparar las condiciones “T. gondii” vs. “VIH-1/T. gondii” (Fig. 63C, 63D y 63E).
Solo en el linaje de astrocitos se observó un aumento significativo en el porcentaje
de células infectadas a medida que transcurría los días de cultivo (Fig. 63A y 63B,
inserto). Cuando se analizaron los porcentajes de infección de cada linaje celular
con respecto al porcentaje que dicho linaje representaba en la población de células
nerviosas, se evidenció, en general, una significativa correlación positiva entre
ambas variables (Fig. 64), sugiriendo que T. gondii infectaría a más células del linaje
que fuese proporcionalmente más abundante, que en el caso de estos cultivo fue el
de astrocitos.
Los porcentajes (con respecto a la población total de células nerviosas)
se presentan como promedio ± desviación estándar
*: p<0.05 (vs. Día 3)
Tabla III. Porcentaje de astrocitos, neuronas y células de la microglía infectados por T. gondii
Astrocitos Neuronas Microglía
Día 3 0,6 ± 1,5 1,3 ± 2,6 0,6 ± 1,3
Día 5 6,8 ± 8,6 * 2,5 ± 4,1 0,4 ± 1,0
Día 7 12,3 ± 11,0 * 2,5 ± 5,0 0,5 ± 1,3
96
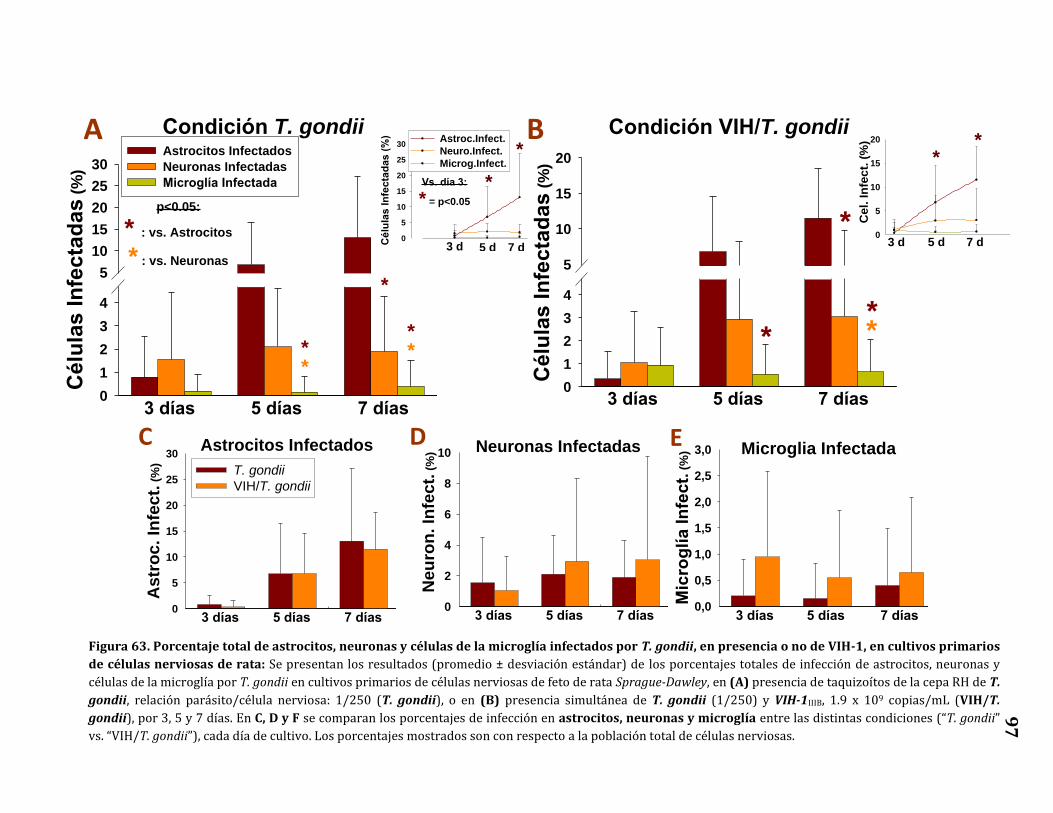
Figura 63. Porcentaje total de astrocitos, neuronas y células de la microglía infectados por T. gondii, en presencia o no de VIH-1, en cultivos primarios
de células nerviosas de rata: Se presentan los resultados (promedio ± desviación estándar) de los porcentajes totales de infección de astrocitos, neuronas y
células de la microglía por T. gondii en cultivos primarios de células nerviosas de feto de rata Sprague-Dawley, en (A) presencia de taquizoítos de la cepa RH de T.
gondii, relación parásito/célula nerviosa: 1/250 (T. gondii), o en (B) presencia simultánea de T. gondii (1/250) y VIH-1IIIB, 1.9 x 109 copias/mL (VIH/T.
gondii), por 3, 5 y 7 días. En C, D y F se comparan los porcentajes de infección en astrocitos, neuronas y microglía entre las distintas condiciones (“T. gondii”
vs. “VIH/T. gondii”), cada día de cultivo. Los porcentajes mostrados son con respecto a la población total de células nerviosas.
Condición T. gondii
Célu
las I
nfe
cta
da
s (
%)
0
1
2
3
4
5
10
15
20
25
30Astrocitos Infectados
Neuronas Infectadas
Microglía Infectada
3 días 5 días 7 días
p<0.05:
* : vs. Astrocitos
* : vs. Neuronas
**
**
*
A
0
5
10
15
20
25
30Astroc.Infect.
Neuro.Infect.
Microg.Infect.
3 d 5 d 7 d
Cé
lula
s I
nfe
cta
da
s (
%)
Vs. día 3:
* = p<0.05
*
*Condición VIH/T. gondii
Cé
lula
s I
nfe
cta
da
s (
%)
0
1
2
3
4
5
10
15
20
3 días 5 días 7 días
**
*
*
B
0
5
10
15
20
3 d 5 d 7 d
Cel. In
fec
t. (
%)
**
Microglia Infectada
Mic
rog
lía
In
fec
t. (
%)
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3 días 5 días 7 días
E Neuronas InfectadasN
eu
ron
. In
fect.
(%
)
0
2
4
6
8
10
3 días 5 días 7 días
D Astrocitos Infectados
As
tro
c.
Infe
ct.
(%
)
0
5
10
15
20
25
30
T. gondii
VIH/T. gondii
3 días 5 días 7 días
C
97
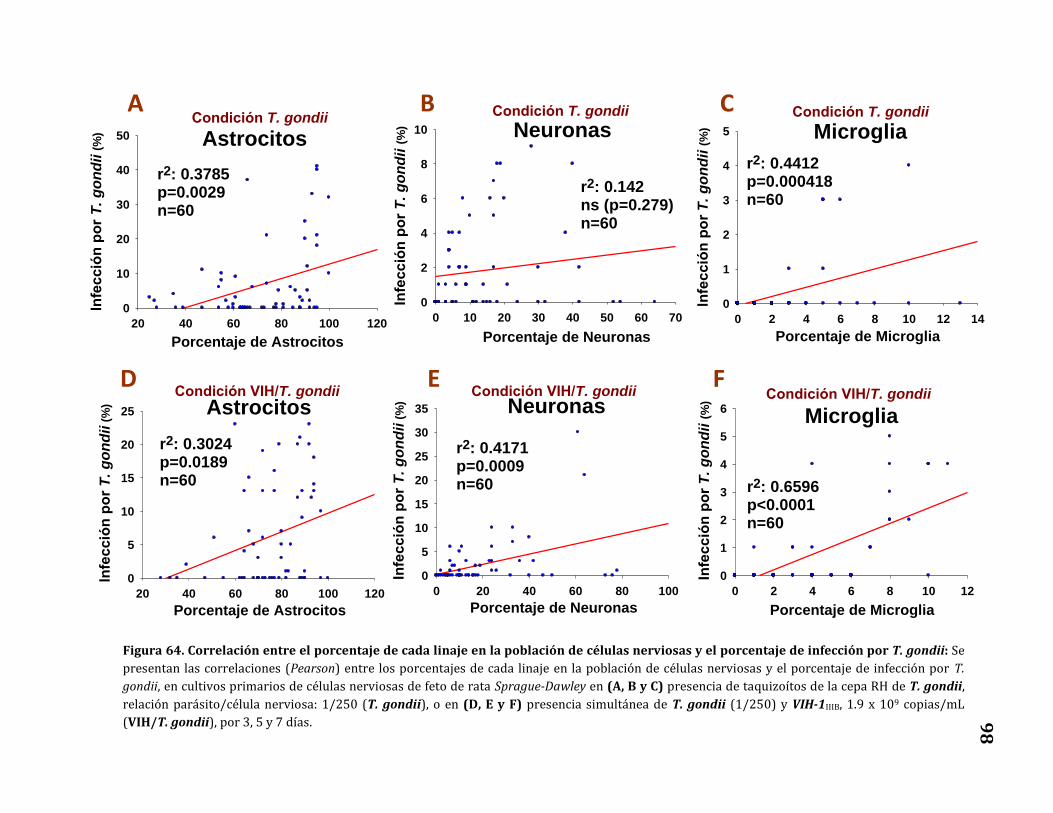
Figura 64. Correlación entre el porcentaje de cada linaje en la población de células nerviosas y el porcentaje de infección por T. gondii: Se
presentan las correlaciones (Pearson) entre los porcentajes de cada linaje en la población de células nerviosas y el porcentaje de infección por T.
gondii, en cultivos primarios de células nerviosas de feto de rata Sprague-Dawley en (A, B y C) presencia de taquizoítos de la cepa RH de T. gondii,
relación parásito/célula nerviosa: 1/250 (T. gondii), o en (D, E y F) presencia simultánea de T. gondii (1/250) y VIH-1IIIB, 1.9 x 109 copias/mL
(VIH/T. gondii), por 3, 5 y 7 días.
Neuronas
Porcentaje de Neuronas
0 20 40 60 80 100
Infe
cció
n p
or
T.
go
nd
ii (
%)
0
5
10
15
20
25
30
35
r2: 0.4171p=0.0009n=60
Condición VIH/T. gondiiE
Astrocitos
Porcentaje de Astrocitos
20 40 60 80 100 120
Infe
cció
n p
or
T.
go
nd
ii (
%)
0
5
10
15
20
25
r2: 0.3024p=0.0189n=60
Condición VIH/T. gondiiD
Neuronas
Porcentaje de Neuronas
0 10 20 30 40 50 60 70
Infe
cc
ión
po
r T
. g
on
dii
(%
)
0
2
4
6
8
10
r2: 0.142ns (p=0.279)n=60
Condición T. gondiiB
Astrocitos
Porcentaje de Astrocitos
20 40 60 80 100 120
Infe
cció
n p
or
T.
go
nd
ii (
%)
0
10
20
30
40
50
r2: 0.3785p=0.0029n=60
Condición T. gondiiA
Microglia
Porcentaje de Microglia
0 2 4 6 8 10 12 14
Infe
cc
ión
po
r T
. g
on
dii (
%)
0
1
2
3
4
5
r2: 0.4412p=0.000418n=60
Condición T. gondiiC
Microglia
Porcentaje de Microglia
0 2 4 6 8 10 12
Infe
cc
ión
po
r T
. g
on
dii (
%)
0
1
2
3
4
5
6
r2: 0.6596p<0.0001n=60
Condición VIH/T. gondiiF
98

D. ANÁLISIS MICROSCÓPICO - MORFOLÓGICO
Las células nerviosas de los cultivos primarios crecen formando una monocapa
celular con alta confluencia, con un contacto célula-célula tan estrecho, que
prácticamente no se pueden distinguir los límites entre células vecinas (Fig. 65A,
condición Control). El examen microscópico demostró que los patógenos afectan a
las células en forma distinta. T. gondii forma focos infecciosos, con presencia de
taquizoítos y destrucción/lisis de las células donde éstos se han replicado, sin
causar alteraciones morfológicas en regiones de la monocapa no infectadas (Fig.
65D). Así, en la condición con presencia exclusiva del parásito, se observan zonas de
destrucción en la monocapa (“huecos”), que afectan un área mayor a medida que
aumentan los días de cultivo. Este carácter “focal” sugiere la importancia de la
confluencia celular de las monocapas en cultivos primarios para favorecer la
infección, pues al romperse una célula infectada y liberar los taquizoítos que se han
replicado en su interior, éstos podrán acceder más fácilmente a las células vecinas e
iniciar nuevos ciclos de infección. La liberación cíclica de taquizoítos se utilizó en
este trabajo para evaluar la progresión de la infección parasitaria (recuento de
taquizoítos libres, Fig. 58), asumiendo que a medida que progresa la infección, más
células serán infectadas, liberando al medio un número mayor de taquizoítos. Por su
parte, la presencia de VIH-1 en los cultivos se asoció con afectación de toda la
monocapa simultáneamente (Fig. 65C), provocando vacuolización y retraimiento de
los citoplasmas celulares, resultando en un menor contacto célula-célula, lo cual
podría explicar en parte el efecto inhibidor sobre la infección parasitaria asociado
con la presencia del virus en estos experimentos, pues al estar presente, disminuiría
la confluencia de la monocapa celular, creando un problema espacial, pues los
taquizoítos liberados al romperse una célula infectada, tendrían mayor dificultad
para alcanzar a una nueva célula blanco, y así iniciar otro ciclo de infección. De
hecho, cuando están presentes en forma simultánea VIH-1 y T. gondii, se observa
una morfología similar a la observada en presencia exclusiva de VIH-1, con
afectación de toda la monocapa y retraimiento de los citoplasmas celulares (Fig.
65F). La presencia de gp41, por su parte, también se asocia a modificación de la
monocapa, con retraimiento de los citoplasmas celulares, aunque el efecto es más
discreto al observado en presencia de VIH-1, y si bien se observa inhibición de la
replicación parasitaria en presencia de gp41 (Fig. 58), ésta es significativamente
menor a la observada en presencia de VIH-1 (Fig. 58, 59).
99
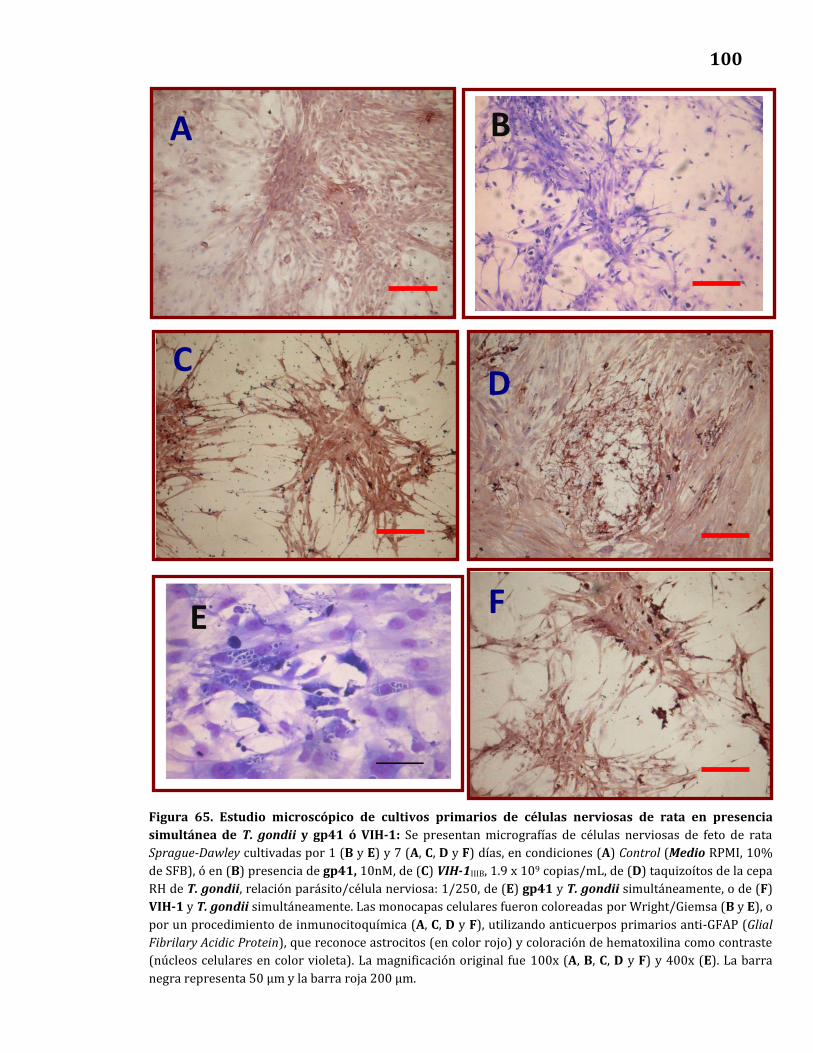
Figura 65. Estudio microscópico de cultivos primarios de células nerviosas de rata en presencia
simultánea de T. gondii y gp41 ó VIH-1: Se presentan micrografías de células nerviosas de feto de rata
Sprague-Dawley cultivadas por 1 (B y E) y 7 (A, C, D y F) días, en condiciones (A) Control (Medio RPMI, 10%
de SFB), ó en (B) presencia de gp41, 10nM, de (C) VIH-1IIIB, 1.9 x 109 copias/mL, de (D) taquizoítos de la cepa
RH de T. gondii, relación parásito/célula nerviosa: 1/250, de (E) gp41 y T. gondii simultáneamente, o de (F)
VIH-1 y T. gondii simultáneamente. Las monocapas celulares fueron coloreadas por Wright/Giemsa (B y E), o
por un procedimiento de inmunocitoquímica (A, C, D y F), utilizando anticuerpos primarios anti-GFAP (Glial
Fibrilary Acidic Protein), que reconoce astrocitos (en color rojo) y coloración de hematoxilina como contraste
(núcleos celulares en color violeta). La magnificación original fue 100x (A, B, C, D y F) y 400x (E). La barra
negra representa 50 µm y la barra roja 200 µm.
C
E
A
D
F
B
100

DISCUSIÓN
Con el presente trabajo se ha buscado esclarecer los mecanismos
inmunológicos que se desencadenan cuando un mismo hospedador humano es
infectado por VIH-1 y T. gondii, utilizando una doble aproximación al problema de
estudio; por una parte evaluando la respuesta de linfocitos T en pacientes y grupos
Control relevantes, y por otra, estudiando en un modelo in vitro los efectos tóxicos
de estos agentes patógenos sobre células del sistema nervioso central.
Fenotipo de linfocitos T de sangre periférica
Los linfocitos T expresan en forma dinámica distintas moléculas en su
membrana celular, que reflejan características tan diversas como el estado de
activación celular112, 117; o la capacidad para dar y recibir señales de
estimulación259, supresión260, supervivencia102 o muerte celular95. La evaluación
fenotípica de estas moléculas permite determinar el estado funcional de estas
células en un momento dado. Mediante esta aproximación se estudió la expresión ex
vivo de diversas moléculas en los linfocitos T de sangre periférica de los diferentes
grupos de pacientes y controles, con especial atención en los pacientes infectados
con VIH-1 y serología positiva para T. gondii. Para realizar este análisis, los
pacientes fueron agrupados en base a su recuento de linfocitos T CD4+ en sangre
periférica, de acuerdo a lo descrito en la sección de Materiales y métodos. Esto, como
fue mencionado, permitió que el análisis tuviese una aproximación secuencial de la
progresión de la infección por VIH-1.
Al evaluar la dinámica de las poblaciones de linfocitos T en los pacientes
infectados por el virus, se hizo evidente que la pérdida progresiva en el número de
linfocitos T CD4+, se correlacionaba con otros cambios numéricos y fenotípicos en
las células T. Así, mientras menores eran el recuento y porcentaje de linfocitos T
CD4+ (Fig. 20), mayor era el porcentaje de linfocitos T CD8+ (Fig. 21A). El recuento
de células T CD8+, por su parte, era mayor en pacientes que en controles, pero
tendía a disminuir en etapas tardías de la infección viral (Fig. 21B). Desde los casos
iniciales descritos en el año 1981, cuando se hizo evidente la presencia de un tipo de
inmunodeficiencia adquirida no descrita previamente, la afectación numérica y
funcional de la población T fue reconocida como parte fundamental del síndrome1, 4-
7. De hecho, la disminución que presentaban los pacientes en su población de
linfocitos T CD4+ de sangre periférica y los defectos funcionales asociados a ésta,
orientó en la búsqueda de un agente causal infeccioso que afectara en forma
específica a esta población, dando como resultado el descubrimiento del virus que
hoy conocemos como VIH-120-23. La inversión de la relación CD4/CD8 observada en
los pacientes, refleja la mayor disminución en el recuento de la población T CD4+
durante todo el curso de la infección (Fig. 22)261. En infecciones causadas por virus
distintos al VIH-1, como Citomegalovirus262 y Virus de Hepatitis B263, por ejemplo,
101

la respuesta antiviral incluye un aumento en el valor absoluto de la población T
CD8+, por lo que puede observarse una inversión de la relación CD4/CD8, sin
asociarse ésta a una disminución absoluta de la población T CD4+262, 263. En la
respuesta anti VIH-1, por su parte, como se hace evidente en la evaluación de los
pacientes de este estudio, también hay un aumento significativo en el número de
linfocitos T CD8+, pero simultáneamente se está produciendo una disminución
progresiva, no solo de los porcentajes sino también de los recuentos de la población
T CD4+ (Fig. 20), lo cual es una característica distintiva de esta infección261,
resultando en la mencionada inversión de la relación CD4/CD8, incluso en la etapa
más avanzada de la infección viral, a pesar de que en dicha etapa también
disminuyen los recuentos de células T CD8+. Debido a que el VIH-1 infecta a los
linfocitos T CD4+57, inicialmente se asumía que la disminución de estas células era
resultado directo de la infección viral21. Sin embargo, estudios subsiguientes
demostraron que en los pacientes la mayoría de las células destruidas son células
no infectadas264, indicando que existen mecanismos indirectos en el contexto de la
infección que provocan la destrucción de esta población celular. Cabe destacar que
la disminución en el recuento de linfocitos T CD4+ se correlaciona negativamente
con los valores de carga viral, tal como fue observado en este trabajo (Fig. 23),
enfatizando por una parte el efecto deletéreo que tiene la presencia del virus en la
sobrevida del linfocito T CD4+21 y por otra, la importancia de la población de células
T en el control de la replicación viral265-267. En este punto cabe destacar que, en los
pacientes infectados con VIH-1 evaluados en este estudio, no se presentaron
diferencias significativas entre aquéllos con serología positiva para T. gondii (P2), y
los que presentaban serología negativa (P1), con respecto a sus recuentos y
porcentajes de linfocitos T CD4+ y T CD8+, y sus valores de carga viral. Este hallazgo
sugiriere que las infecciones por T. gondii resueltas o en etapas asintomáticas, no
afectan el curso de la infección por VIH-1, con respecto al número de células T en
sangre periférica, ni la carga viral.
De acuerdo a lo descrito, es evidente que la infección viral está asociada a la
destrucción de células T CD4+261; que esta destrucción está relacionada con la
presencia del propio virus21, observándose en los resultados de este trabajo, que
mientras mayor era la carga viral, menor era recuento de células T CD4+; que
también hay cambios asociados en la población T CD8+, con aumento numérico que
tiende a disminuir en la etapa avanzada de la infección viral; y que la mayoría de las
células que son destruidas no están productivamente infectadas264. Interesa
entonces conocer cuál es la causa de la destrucción celular y por qué la respuesta
inmunitaria, que incluye aumento de la población de células T CD8+, no logra
contener/erradicar al virus. Los resultados obtenidos al evaluar los diferentes
marcadores fenotípicos proporcionan importante información al respecto.
102

La evaluación de la expresión de la molécula HLA-DR permitió conocer datos
importantes con respecto al estado de activación de las células T durante la
infección viral. Mientras en la población T CD4+ el porcentaje de células que
expresaban HLA-DR aumentaba a medida que progresaba la infección viral (Fig.
25A), en la población T CD8+ el porcentaje de células HLA-DR+ aumentaba con
respecto al Control sólo en las etapas iniciales, disminuyendo hasta valores basales
(similares al Control) en la etapa avanzada (Fig. 25B). Es preciso enfatizar la
dependencia que la población T CD8+ tiene de la población T CD4+ para cumplir sus
funciones, lo cual incluye la activación celular265, 266. En la etapa de
inmunodeficiencia, la disminución de las células T CD4+ podría limitar su capacidad
de estimulación a la población T CD8+265, resultando en la no activación de la
misma. Esta falta de activación de la población T CD8+ también podría estar
involucrada con el propio estado de inmunodeficiencia, al limitar su capacidad
antiviral267, 268. Pantaleo ha descrito que la población T CD8+/HLA-DR+, presenta
actividad anti-VIH-1 in vitro269, de allí la importancia funcional que su disminución
pudiera significar. Es necesario hacer énfasis, sin embargo, que la expresión de HLA-
DR no es el único marcador de activación celular disponible. Otra molécula cuya
expresión también ha sido utilizada para identificar células activadas es CD38270.
Kestens et al. describieron los cambios que se presentan en la expresión de HLA-DR
y CD38 en linfocitos T CD8+ en diferentes etapas de la infección por VIH-1271. Estos
autores muestran que en etapas iniciales de la infección viral, la expresión de ambas
moléculas está elevada en esta población celular, pero en la etapa de SIDA la
población HLA-DR+/CD38- disminuye, mientras que la HLA-DR-/CD38+ se eleva
significativamente, evidenciando un desacoplamiento en la expresión de estas
moléculas de activación, lo cual es altamente sugestivo de que exista también
desacoplamiento en el estado de activación mismo269, 271, lo que podría en parte
explicar la defectuosa funcionalidad y el establecimiento de la inmunodeficiencia.
Nuevamente, destaca que los pacientes con serología positiva para T. gondii (P2)
presentaran porcentajes de expresión de la molécula HLA-DR en sus linfocitos T
CD4+ y T CD8+ similares a los de los pacientes con serología negativa (P1),
mostrando que el reto antigénico con el protozoario, en estos pacientes P2, no
indujo cambios significativos en el estado de activación de su población de células T
periféricas, con respecto a la expresión de esta molécula. Ahora bien, T. gondii es
capaz de modificar la expresión de HLA-DR en las células que infecta. Así, se conoce
que ésta aumenta en células dendríticas272, macrófagos y linfocitos B infectados273.
Por otra parte, se ha demostrado que la infección por T. gondii se asocia con
disminución de la expresión de HLA-DR inducida por IFN- e IL-6, restringiendo la
presentación antigénica274, 275. En todo caso, estos cambios fenotípicos se observan
en células infectadas por el parásito272-275. En sangre periférica, durante la etapa
asintomática de la infección parasitaria, se ha evidenciado que la proporción de
103

células infectadas es mínima276. En los pacientes VIH-1+/T. gondii+ (P2) evaluados
en este trabajo, asintomáticos para la infección por el protozoario, no se esperaría
que una proporción significativa de la población T de sangre periférica esté
infectada por el parásito, lo que explicaría que su expresión de HLA-DR no sea
distinta a la de los pacientes con serología negativa (P1).
Se conoce que para que el linfocito T CD8+ se pueda activar y cumplir con su
función antiviral, son importantísimas las señales que éste reciba, no solo de
reconocimiento antigénico, a través de su receptor de célula T (TCR)277, sino
también a través de receptores de señales co-estimuladoras secundarias, que bien
pueden propiciar una respuesta específica activa259, o favorecer una respuesta de
tolerancia o anergia260, 278. Así mismo, para que el linfocito T CD4+ pueda mediar la
activación del linfocito T CD8+ y de otras células efectoras de la respuesta antiviral,
debe también recibir señales específicas y secundarias apropiadas277. En este
trabajo, para evaluar la presencia de receptores de señales co-estimuladoras
negativas en las células T, se midió la expresión de PD-1105-107, 260. En la población T
CD4+ se pudo observar un aumento significativo en el porcentaje de células que
expresaban esta molécula desde etapas iniciales de la infección viral, porcentaje que
alcanzaba su máximo valor en la etapa más avanzada (Fig. 26A). Esta expresión
comprometería adicionalmente la funcionalidad de la población T CD4+, que no solo
estaría numéricamente disminuida, sino que tendría mayor propensión a recibir
señales co-estimuladoras negativas, disminuyendo su capacidad de mediar
funciones colaboradoras con otras poblaciones celulares, incluyendo la T CD8+,
para propiciar el establecimiento de una respuesta antiviral efectiva103, 260. Por su
parte el porcentaje de expresión de PD-1 en la población T CD8+ de los pacientes
también estuvo incrementado (Fig. 26B), sugiriendo que la capacidad de respuesta
de esta población estaría limitada. La relevancia de la vía de PD-1 y sus ligandos en
el contexto de la infección por VIH-1 ha sido ampliamente demostrada, como lo
ilustra el trabajo de Day et al.107. Estos autores describen, por una parte, que la
expresión de PD-1 está significativamente aumentada en las poblaciones totales de
células T CD4+ y T CD8+ de pacientes infectados por VIH-1, que el aumento es
significativamente mayor en las células con especificidad anti-VIH-1 y que se
correlaciona con defectos de proliferación y producción de citoquinas ante retos
antigénicos. Más aún, demostraron que el bloqueo in vitro de la vía de señalización
entre PD-1 y uno de sus ligandos (PDL-1), permite la restauración de la capacidad
de proliferación y producción de citoquinas tanto de células T CD4+ como de células
T CD8+, evidenciando no solo la existencia de clones con especificidad anti-VIH-1 en
los pacientes evaluados, sino también que manipulando las vías de señalización es
posible restaurar la capacidad de respuesta de estos clones. Estos hallazgos son de
importancia trascendental, no solo en el entendimiento de los defectos funcionales
de la respuesta específica durante la infección por VIH-1, sino también porque
104

abren la posibilidad de intervención terapéutica para restaurar la respuesta
antiviral efectiva en los pacientes infectados. Aunque son pocos los trabajos que
miden la expresión de PD-1 durante la infección por T. gondii, en modelos animales
se ha descrito un incremento en el porcentaje de expresión de esta molécula en los
linfocitos T CD8+276, 279, que se asocia con pérdida de la funcionalidad de esta
población276, 280, siendo posible recuperar la capacidad de respuesta específica al
bloquear la vía de señalización PD-1/PDL-1, logrando incluso evitar la mortalidad
asociada a la reactivación de la infección276. Estos resultados276, 279, 280, aunque
reflejan una dinámica de expresión de PD-1 en la población de células T durante la
infección por T. gondii muy similar a la descrita para pacientes infectados con VIH-
1105, 107, aún esperan por ser verificados en seres humanos. Mientras tanto, cabe
resaltar que el modelo múrido utilizado en los trabajos citados, fue el de ratones
C57BL/6, infectados con una cepa tipo II de T. gondii, la ME49. En este modelo, los
ratones tenían lesiones cerebrales asociadas a encefalitis toxoplásmica cuatro
semanas después de ser infectados279, y la muerte ocurría entre los 49 y los 125
días post-infección276, 280. Es claro que este modelo difiere del curso de la infección
por T. gondii en seres humanos, donde, en general, los hospedadores
inmunocompetentes logran controlar la infección aguda, y la reactivación sólo se
presenta en asociación a estados de inmunodeficiencia233; diferencia que limitarían
la extrapolación a la infección en seres humanos de los resultados obtenidos en
estos modelos múridos. En los individuos evaluados en este trabajo no se observó
diferencia significativa en el porcentaje de expresión de PD-1 en los linfocitos T
CD4+ y T CD8+ entre los grupos con serología positiva para T. gondii (C2, P2A, P2B y
P2C) y los grupos con serología negativa (C1, P1A, P1B y P1C), si bien sólo en los
pacientes P2 se observó consistentemente diferencia significativa con respecto al
Control en la expresión de esta molécula en la población T CD8+ (Fig. 26B). Estos
resultados sugieren que en seres humanos las infecciones por T. gondii latentes o
resueltas, no modificarían la expresión de PD-1 en los linfocitos T de sangre
periférica.
En otro orden de ideas, el equilibrio homeostático de la población de
linfocitos T requiere un balance entre la proliferación, la sobrevida y la muerte
celular de esta población281. Como componente central en el mantenimiento de este
equilibrio están las señales recibidas a través de la cadena α del receptor de la IL-7
(IL-7Rα/CD127), que median sobrevida y proliferación homeostática de estas
células101, 282. Para evaluar la capacidad de los linfocitos T de sangre periférica de
los individuos participantes en este estudio de recibir las señales de supervivencia
proporcionadas por esta vía, se midió la expresión de IL-7Rα en estas células. En los
pacientes en la etapa más avanzada de la infección por VIH-1, se presentó una
disminución significativa en el porcentaje de células que expresaban esta molécula
en la población de linfocitos T CD4+ al comparar con el resto de los grupos (Fig.
105

27A), mientras que este porcentaje en la población T CD8+ se observó
significativamente disminuido desde las etapas iniciales (Fig. 27B). El porcentaje de
expresión de IL-7Rα en las poblaciones celulares T CD4+ y T CD8+ se correlacionó
en forma significativa con el recuento de linfocitos T CD4+ de sangre periférica (Fig.
27C y 27D), indicando su relación con la progresión de la infección viral, tal como ya
ha sido descrito283, 284. Es evidente, pues, que en los pacientes infectados por VIH-1
evaluados en este estudio, tanto la población T CD4+ como la T CD8+ tienen
disminuida su capacidad de recibir las señales de supervivencia mediadas por la IL-
7282, pero esta disminución tendría un inicio más temprano y una mayor magnitud
en la población T CD8+283 (Fig. 27B). La disminución de la expresión de IL-7Rα en
la población de linfocitos T se ha asociado con baja expresión de la proteína
antiapoptótica Bcl-2285 y con aumento de la apoptosis283, por lo que se ha sugerido
que sea uno de los mecanismos responsables en la disminución de la población T
CD4+ durante la infección por VIH-1286. Llama la atención, sin embargo, que en los
pacientes evaluados en este trabajo la pérdida en la expresión de esta molécula se
haya presentado en etapas más tempranas de la infección viral y con una diferencia
proporcional mayor con respecto al Control, en la población T CD8+ (Fig. 27B)
comparada con la T CD4+ (Fig. 27A), a pesar de que la pérdida de células sea mayor
en esta última población, lo cual evidencia que la sola disminución en la expresión
de IL-7Rα no es suficiente para explicar la destrucción de las poblaciones T en la
progresión de la infección por VIH-1. Es también de particular importancia el hecho
de que los linfocitos T CD4+ y T CD8+ de memoria central y de memoria efectora287,
sean especialmente susceptibles a la disminución de la expresión de IL-7Rα283,
estando la población T de memoria central especialmente afectada283, 284, 288, con la
potencial consecuencia que esto pueda tener para el mantenimiento de la población
específica de memoria a largo plazo286, 287. Por otra parte, en el contexto de la
infección por T. gondii, la IL-7 tiene un papel muy importante en la estimulación de
una respuesta inmunitaria protectora289, 290. La administración de IL-7 in vivo
permite la sobrevida de ratones inoculados con dosis letales del parásito,
estimulando la respuesta de células NK y de linfocitos T CD8+ citotóxicos, y
aumentando la producción de IFN-γ e IL-2289. Así mismo, la eliminación in vivo de
esta citoquina aumenta la susceptibilidad a la infección289. Es claro, pues, que en el
establecimiento y mantenimiento de una respuesta anti-T. gondii efectiva son
fundamentales las señales recibidas a través de IL-7Rα; de allí la importancia
funcional que podría tener la disminución de su expresión observada en los
pacientes de este estudio, y la posibilidad, que se incrementaría con la progresión
de la infección viral, de perder la respuesta anti-parasitaria específica. En este
estudio no se observaron diferencias significativas en la expresión de IL-7Rα en los
linfocitos T CD4+ y T CD8+ entre los pacientes infectados con VIH-1 y serología
positiva para T. gondii (P2) y aquellos con serología negativa (P1) (Fig. 27A y 27B),
106

indicando que el reto antigénico con el parásito, en infecciones resueltas o latentes,
no induciría cambios adicionales en la expresión de IL-7Rα en linfocitos T de sangre
periférica a los inducidos por la infección por VIH-1, si bien, como se expresó antes,
la respuesta antiparasitaria podría estar comprometida, aumentando la
susceptibilidad a la infección por T. gondii.
Otro de los aspectos clave en el mantenimiento del equilibrio homeostático
de la población de linfocitos T, como se mencionó en el párrafo anterior, es la
muerte celular, siendo la apoptosis un mecanismo crucial en este proceso291. La
infección por VIH-1 se ha asociado con aumento de la apoptosis de los linfocitos T92-
95, con especial contribución de la molécula CD9596-98 en la inducción de la misma.
Durante la infección por VIH-1 se observa un aumento en el porcentaje de linfocitos
T que expresan CD9595, 98, y en estas células la susceptibilidad de sufrir apoptosis
por estimulación de este receptor está aumentada96, 98. En los pacientes infectados
con VIH-1 evaluados en este trabajo, el porcentaje de linfocitos T CD4+ que
expresaban CD95 estuvo significativamente aumentado en la etapa más avanzada
de la infección viral (Fig. 28A), observándose una correlación significativamente
negativa entre esta expresión y el recuento de linfocitos T CD4+ (Fig. 28C),
indicando su relación con la progresión95. La población T CD8+ que expresaba CD95
presentó un pequeño aumento en todos los grupos de pacientes infectados con VIH-
1 al compararlos con su respectivo grupo Control, pero la diferencia sólo fue
significativa para los pacientes infectados con VIH-1 seropositivos para T. gondii
que se encontraban en las primeras etapas de la infección viral (P2A y P2B, Fig.
28B). Cuando se comparó la expresión de CD95 en los linfocitos T CD4+ y T CD8+ de
sangre periférica de los individuos con serología positiva para T. gondii (C2, P2A,
P2B y P2C) con la de aquellos con serología negativa (C1, P1A, P1B y P1C), no se
observaron diferencias significativas, sugiriendo que la infección por T. gondii
resuelta o latente no modifica dicha expresión.
En en este estudio también se evaluó el porcentaje de apoptosis temprana ex
vivo de linfocitos T, es decir, el porcentaje de células T de sangre periférica positivas
para Anexina V, evaluadas inmediatamente después de la toma de muestra. En estos
ensayos, tanto los pacientes infectados con VIH-1 como los individuos de grupos
Control presentaron valores bajos, en general inferiores al 3% de las células de cada
población. Sólo se observaron diferencias significativas en el grupo de pacientes
infectados con VIH-1 y seronegativos para T. gondii en la etapa más avanzada de la
infección viral (P1C), cuyas células T CD4+ presentaron valores significativamente
mayores de apoptosis temprana a los de su grupo Control (C1) y a los de su grupo
P2 homólogo (P2C), mientras que en las células T CD8+, presentaron valores
significativamente menores a los del grupo P2C (Fig. 30). Sin embargo, aún en estos
grupos que presentaron diferencias significativas, los porcentajes de apoptosis
espontánea ex vivo fueron bajos, sugiriendo que no juegan un papel relevante en la
107

destrucción celular en el contexto de la infección viral. Gougeon et al. también
encontraron valores de apoptosis ex vivo sumamente bajos en linfocitos T CD4+ y T
CD8+ de sangre periférica, con promedios menores al 2% en la población evaluada,
sin diferencias significativas entre los pacientes infectados con VIH-1 y los
individuos no infectados95. Es preciso diferenciar esta apoptosis espontánea ex vivo,
de la apoptosis espontánea después de 24 horas de cultivo, por ejemplo, y también
de la apoptosis inducida por estimulación de diversos receptores celulares, como
CD3 o CD95, donde sí se han descrito valores significativamente incrementados en
los pacientes infectados con VIH-194-96, 98. Por su parte, la infección por T. gondii se
ha asociado con inhibición de la apoptosis mediada por distintos mecanismos292,
incluyendo la inducida por CD95292, 293. Los mecanismos hasta ahora descritos,
responsables de esta inhibición, incluyen la vía de amplificación mitocondrial con
bloqueo de la liberación294 y actividad295 del citocromo C, alteración del balance
entre proteínas pro-apoptóticas y anti-apoptóticas de la familia Bcl-2294, y el
bloqueo de la vía independiente a la mitocondria, con degradación de la caspasa
8293. Como otros de los efectos provocados por T. gondii y descritos en los párrafos
anteriores, esta inhibición de la apoptosis celular se observa en células activamente
infectadas por el parásito292, de allí, que en condiciones asintomáticas, donde no se
espera que haya un número significativo de células infectadas por el parásito en
sangre periférica276, no se observen tampoco diferencias en la apoptosis ex vivo de
linfocitos T, entre los individuos con serología positiva para el parásito (C2, P2A,
P2B) y aquellos con serología negativa (C1, P1A, P1B), con la limitada excepción de
los grupos P1C y P2C, ya mencionada.
Pasando a otro punto, entre los antígenos de membrana que se expresan
diferencialmente en subpoblaciones de células T, están las moléculas CD45RA y
CD45RO, isoformas de la familia del antígeno común leucocitario (CD45) que se han
utilizado ampliamente para identificar las células T vírgenes y de memoria,
respectivamente251. Se ha demostrado que las células T que expresan CD45RA, al
ser estimuladas in vitro con mitógenos como el PHA, pierden gradualmente la
expresión de CD45RA, mientras comienzan, en forma recíproca, a expresar
CD45RO296. La evaluación en las células T CD45RO+ de la expresión de moléculas
adicionales como CCR7 y CD62L, permite identificar poblaciones especializadas
dentro de la población de célula T de memoria297. Así las células T
CD45RO+/CCR7+/CD62L+, llamadas de memoria central, identifican una población
con migración preferencial a los ganglios linfáticos, y que en respuesta a la
estimulación antigénica entran en un proceso de proliferación activa, produciendo
inicialmente IL-2, y posteriormente IFN-γ o IL-4297. Por su parte las células T
CD45RO+/CCR7- son una población con migración preferencial a los tejidos
periféricos inflamados, función efectora inmediata y rápida producción de IFN-γ o
IL-4, de allí que se les denomine de memoria efectora297. En los pacientes infectados
108

con VIH-1 la mayoría de las células T CD8+ en circulación, con especificidad anti-
VIH-1, tienen fenotipo de memoria efectora107, 298. Al evaluar la expresión de las
moléculas CD45RA y CD45RO en la población de linfocitos T CD4+ de sangre
periférica en los individuos participantes en este trabajo, se pudo observar una
disminución en el número tanto de células vírgenes como de células de memoria, en
los pacientes infectados con VIH-1 al comparar con sus grupos Control respectivos
(Fig. 32C y D), siendo la disminución de células vírgenes proporcionalmente mayor
con respecto a las células de memoria, a medida que avanzaba la infección viral
(Fig. 34A). Esta disminución en la relación CD45RA/CD45RO en la población T
CD4+ podría explicarse por cambios en el fenotipo de la población virgen, que al
recibir señales de estimulación, en el contexto de una infección viral asociada a
inflamación crónica, dejaría de expresar CD45RA y comenzaría a expresar
CD45RO296, incrementándose así la población de memoria. También es necesario
tomar en cuenta que al aumentar la edad del individuo, disminuye la capacidad del
timo para regenerar la población de células vírgenes de sangre periférica88, 89, lo
cual también podría contribuir con la disminución preferencial de esta población.
Más aún, la infección por VIH-1 se asocia con cambios morfológicos anormales del
timo, que incluyen disminución de timocitos e involución avanzada88, 89,
comprometiendo adicional y seriamente la capacidad funcional de este órgano,
incluyendo la generación de células T vírgenes. Por su parte, la evaluación de la
expresión de CD45RA y CD45RO en la población T CD8+, demostró un significativo
incremento en la población de células de memoria en todos los grupos de pacientes
infectados con VIH-1, al compararlos con sus respectivos grupos Control (Fig. 33D y
34B), con recuentos de población T CD8+ virgen similares o discretamente mayores
que en los individuos no infectados por el virus (Fig. 33B). Esta dinámica en la
población T CD8+ durante la infección por VIH-1, se relaciona, por una parte con un
aumento en el número de linfocitos T CD8+ totales (Fig. 21B), tal como ocurre en
otras infecciones virales262, 263, y por otra, con el predominio de células de memoria,
especialmente de memoria efectora, que se ha asociado con esta infección284, 298. Es
resaltante que se haya descrito que esta población T CD8+ de memoria efectora
posea una expresión de IL-7Rα significativa disminuida283, lo cual es congruente
con los resultados obtenidos en este trabajo (Fig. 27B). Paiardini et al.299
observaron en individuos infectados con VIH-1 que las células T CD8+ con pérdida
en la expresión de IL-7Rα identifican a una población con marcadores fenotípicos
(CCR7-/CD62L-) y funcionales (producción preferencial de IFN-γ y no de IL-2,
menor capacidad proliferativa, mayor susceptibilidad a apoptosis) de células de
memoria efectora. También observaron que el aumento de esta población en los
pacientes infectados con VIH-1, se correlacionaba con la expresión de marcadores
de activación en la población de células T, apoptosis, carga viral y destrucción de
linfocitos T CD4+283, 299, en congruencia con los resultados obtenidos en este
109

trabajo. Finalmente, al comparar la expresión de las moléculas CD45RA y CD45RO
en las poblaciones de linfocitos T CD4+ y T CD8+ de sangre periférica de los grupos
con serología positiva para T. gondii con la de sus grupos homólogos y serología
negativa, no se observaron diferencias significativas, sugiriendo que la infección por
T. gondii, crónica o resuelta, no modificaría la proporción ni el recuento de células T
vírgenes y T de memoria en sangre periférica.
Funcionalidad de linfocitos T
Las PBMC de los individuos participantes en el estudio fueron cultivadas por
72 horas bajo estimulación con la proteína recombinante Env del VIH-1, ó con una
preparación de antígenos solubles de T. gondii (SATg), para evaluar respuesta
específica, utilizando células no estimuladas y células cultivadas en presencia de
PHA, como controles negativos y positivos, respectivamente. Las variables
estudiadas fueron proliferación, expresión de perforina, apoptosis, necrosis y
producción de citoquinas.
Proliferación de linfocitos T
Cuando las células de los individuos participantes en este estudio fueron
cultivadas en presencia de PHA, se demostró, tanto en los linfocitos T CD4+ como en
los T CD8+, una proliferación significativamente mayor a la de las células cultivadas
sin estimulación (condición Medio), lo cual se observó en todos los grupos de
estudio, si bien los pacientes infectados con VIH-1, especialmente aquellos con
mayor progresión de la infección viral, presentararon valores de proliferación
menores a los de su grupo Control (Fig. 35 y 36). Sin embargo, al evaluar la
proliferación en respuesta a la estimulación específica, tanto con el antígeno viral
como con el parasitario, no fue posible detectar respuestas que fuesen distintas a las
de la condición sin estimulación (Medio) en ninguno de los grupos de estudio. La
estimulación con SATg de las PBMC de los individuos del grupo Control C2 (VIH-/T.
gondii+) indujo una proliferación significativamente mayor, tanto en las células T
CD4+ como en las T CD8+, a los del grupo C1 (VIH-/T. gondii-), lo cual pudiera
sugerir una respuesta específica al parásito en los individuos previamente
expuestos al mismo; sin embargo, una respuesta similar se observó en la condición
de cultivo sin estimulación (Medio) y en los cultivos bajo estimulación con Env, lo
cual sugiere que no fue una respuesta específica al antígeno parasitario lo que
indujo el aumento de la proliferación observado en este grupo.
Evidentemente, el protocolo utilizado en este trabajo para evaluar la
proliferación de linfocitos T CD4+ y T CD8+, en células cultivadas por 72 horas,
fundamentado en la incorporación de bromodeoxiuridina, pudo detectar respuestas
inducidas por estimulación policlonal, pero no las inducidas por estimulación de
clones específicos. Así, cuando las PBMC fueron cultivadas sin estimulación,
110

estimuladas con antígenos virales o parasitarios, los porcentajes de proliferación
oscilaron entre 1 y 8%, mientras que en las estimuladas con PHA, el rango se ubicó
entre 15 y 45%. Para entender la razón por la que no fue posible detectar la
proliferación en condiciones de estimulación antigénica, se comparó el protocolo
utilizado en este trabajo, con otros descritos en la literatura. Otros autores,
utilizando la incorporación de timidina tritiada como marcador de proliferación, en
este caso no de poblaciones linfoides T particulares, sino en el grupo total de PBMC,
utilizando mayores tiempos de cultivo (5300 y 7 días301) y mayor concentración de
SATg (5300 y 50 μg/ml301), han logrado discriminar entre la respuesta de individuos
asintomáticos con serología positiva y serología negativa para T. gondii. En sus
ensayos la respuesta proliferativa frente a SATg fue clara y significativamente
mayor en los individuos con serología positiva. Utilizando estos protocolos,
pudieron determinar que los pacientes infectados con VIH-1 y serología positiva
para T. gondii, presentaban una proliferación significativamente menor con
respecto a los individuos sin infección por el virus y serología positiva para el
parásito300, 302. Incluso evidenciaron que la pérdida de la respuesta era mayor en los
pacientes VIH+/T. gondii+ con recuentos de linfocitos T CD4+ menores a 200/μL,
especialmente en aquellos con episodios presentes o pasados de encefalitis
toxoplásmica302, mientras que la capacidad de respuesta anti-T. gondii era
parcialmente recuperada por los pacientes que recibían terapia antirretroviral de
alta eficiencia300, 302. En cuanto a la proliferación por estimulación con PHA, estos
autores describieron valores de proliferación en los individuos infectados con VIH-1
similares a los de los individuos no infectados300, 302, pero con una disminución
significativa en pacientes con recuentos de linfocitos T CD4+ en sangre periférica
<200/μl y episodios de encefalitis toxoplásmica302, en una dinámica similar a la
observada en este trabajo (Fig. 35 y 36). Por otra parte, utilizando ester de
succinimidil 5, 6-carboxifluorescein diacetato (CFSE) como marcador de
proliferación, en cultivos de 6 días, Migueles et al. describieron una respuesta de
proliferación en respuesta al VIH-1 entre el 1 y el 12% en linfocitos T CD8+ de
individuos infectados con el virus110. Estos autores observaron que un grupo
especial de pacientes infectados con VIH-1, que lograban controlar la replicación
viral hasta concentraciones indetectables (menos de 50 copias de RNA viral por ml
de plasma) por largos períodos de tiempo (algunos, más de 20 años) en ausencia de
terapia anti-retroviral, los así denominados No Progresores a Largo Plazo (LTNP),
tenían respuestas de proliferación ante estimulación antigénica viral
significativamente mayores al resto de los pacientes (los llamados Progresores, que
conforman la mayoría de la población infectada por el virus), con rangos que
oscilaban entre el 30 y el 70% de sus linfocitos T CD8+110. Así, pudieron establecer
que la capacidad de proliferación en respuesta al VIH-1 está estrechamente
asociada al control de la replicación viral y a la sobrevida libre de enfermedad en
111

ausencia de tratamiento específico110, resaltando la importancia de esta función
para el control de la infección, capacidad que está disminuida en la mayoría de los
pacientes infectados. Los resultados mencionados anteriormente, con respecto a la
mayor disminución de la respuesta de proliferación específica en individuos con
historia de encefalitis toxoplásmica302, también enfatizan la importancia de esta
capacidad funcional para el control de la infección por T. gondii. Así, vemos que con
protocolos que utilizan un mayor tiempo de cultivo (5, 6 y 7 días), otros marcadores
(timidina tritiada, CFSE) u otras preparaciones antigénicas, es posible evaluar in
vitro las respuestas de proliferación específicas anti-T. gondii y anti-VIH-1, las
cuales se correlacionan con el control de estas infecciones y se pierden
significativamente en los pacientes infectados con VIH-1. Como se mencionó
anteriormente, el aumento de la expresión del receptor de señales de co-
estimulación negativa, PD-1, en las poblaciones de células T, ha demostrado tener
una relación negativa con la capacidad de respuesta específica de estas células 107,
276, 280, pudiendo por lo tanto ser éste uno de los factores que está contribuyendo
con la pérdida de la capacidad de proliferación observada en el contexto de la
infección por VIH-1.
Producción de perforina por linfocitos T CD8+
La perforina tiene una participación crítica en la actividad citotóxica de los
linfocitos T, al mediar la entrada de granzimas a la célula blanco, de manera que
éstas inicien cascadas de degradación de ADN, provocando la muerte celular303. La
actividad citotóxica de los linfocitos T ha demostrado ser importante en el control
de las infecciones por VIH-1304 y T. gondii305. Para evaluar la capacidad citotóxica en
la población T CD8+ en este estudio, se midió la expresión de perforina bajo
distintas condiciones de estimulación. Cuando las PBMC de los distintos grupos de
estudio fueron estimuladas con PHA por 72 horas, la expresión de perforina en los
linfocitos T CD8+ se incrementó significativamente, al comparar con las células
cultivadas sin estimulación (Medio), en los individuos no infectados por VIH-1 (C1 y
C2) y en los pacientes infectados con VIH-1 con recuentos mayores a 200 linfocitos
T CD4+ por μL de sangre periférica (P1A, P1B, P2A y P2B) (Fig. 37). Por el contrario,
en los individuos infectados con VIH-1 y recuentos de linfocitos T CD4+ menores a
200/μL (P1C y P2C), la expresión de perforina bajo estimulación con PHA no fue
significativamente distinta a la de la condición sin estimulación, lo cual sugiere que
los pacientes de estos grupos no son capaces de aumentar la capacidad citotóxica de
sus linfocitos T CD8+ por encima de los valores basales, incluso bajo estimulación
policlonal, lo cual los colocaría en desventaja para controlar infecciones por
patógenos intracelulares como VIH-1304, 306 y T. gondii305, donde la actividad
citotóxica es parte importante de la respuesta inmunitaria efectiva. Por otra parte,
al estimular las PBMC con Env o SATg no se evidenció aumento de la expresión de
112

perforina en ninguno de los grupos de individuos participantes, lo cual sugiere que,
al igual que en la evaluación de la proliferación celular por estimulación antigénica,
el protocolo utilizado en este trabajo no tuvo la sensibilidad necesaria para detectar
producción de perforina bajo estimulación específica. Migueles et al., con el
protocolo utilizado en el trabajo citado en la sección anterior110, pudieron
determinar que la expresión de perforina estaba estrechamente asociada con la
capacidad de proliferación de los linfocitos T CD8+, y que ésta era
significativamente mayor en el grupo de pacientes LTNP. Esto sugiere que, en este
trabajo, la falta de sensibilidad para detectar proliferación en las condiciones de
estimulación específica, haya también impedido la detección de producción de
perforina asociada a ésta110. El aumento en la expresión de PD-1, con la posibilidad
de disminuir la capacidad proliferativa de los linfocitos T, también podría estar
asociado a la no detección de perforina. De hecho, Day et al. encontraron baja
expresión de perforina en las células T CD8+ específicas para VIH-1 que tenían
aumentada la expresión de PD-1107, mientras que Bhadra et al. encontraron una
asociación entre el aumento de la expresión de PD-1 en los linfocitos T CD8+ y la
pérdida de la capacidad polifuncional de éstos (incluyendo capacidad citotóxica) en
el contexto de la infección por T. gondii280. El defecto en la producción de perforina
es por tanto relevante en la capacidad de control de las infecciones por VIH-1 y T.
gondii evaluadas en este estudio.
Apoptosis y necrosis de linfocitos T
Una característica distintiva de la infección por VIH-1 es la pérdida gradual
de linfocitos T CD4+ a medida que la infección progresa, lo cual está estrechamente
asociado con el establecimiento del estado de inmunodeficiencia (SIDA) en los
pacientes307. Siendo la pérdida de linfocitos T un aspecto crítico en la patogénesis
de la infección por VIH-1, en este trabajo se evaluó la muerte celular en distintas
condiciones de cultivo, discriminando dicha muerte como apoptosis temprana
(células con expresión positiva de Anexina V), necrosis (células positivas para la
incorporación de yoduro de propidio), apoptosis tardía (células positivas tanto para
Anexina V como para yoduro de propidio) y mortalidad total (referida como la
sumatoria de apoptosis temprana, apoptosis tardía y necrosis) (Fig. 29). Cuando las
PBMC fueron cultivadas por 72 horas en presencia de PHA, se observó un
incremento significativo en el porcentaje de linfocitos T CD4+ y T CD8+ en
apoptosis temprana, en apoptosis tardía y en mortalidad total, en todos los grupos
de estudio, al compararlas con las células cultivadas sin estimulación (Medio) (Fig.
38 y 39). Dado que esta condición de cultivo con PHA estuvo asociada con la mayor
proliferación y producción de perforina, y tomando en cuenta la naturaleza
policlonal de este estímulo, puede inferirse que el aumento de apoptosis observada
en esta condición es muerte celular inducida por activación (AICD)308. Se observó
113

también, en esta condición de cultivo con PHA, la tendencia a que los porcentajes de
muerte celular observados, fueran mayores para la población de células T CD4+
(3020, 2716 y 6318, para apoptosis temprana, apoptosis tardía y muerte total,
respectivamente, para los grupos Control) que para la población T CD8+ (2412,
1914 y 4813), sugiriendo mayor susceptibilidad de las células T CD4+ a la AICD,
lo cual sería relevante en el contexto de la infección por VIH-1, por la mayor pérdida
de esta población, asociada a un ambiente de activación crónica94 (Fig. 20, 21, 22 y
25). En condiciones de estimulación por antígenos virales (Env) y parasitarios
(SATg) las células T CD8+, en general, no mostraron diferencias en los porcentajes
de muerte celular con respecto a la condición sin estimulación, ni entre los distintos
grupos de estudio; mientras que los linfocitos T CD4+ de los grupos con mayor
progresión de la infección viral (P1C, P2C) presentaron mayores porcentajes de
apoptosis temprana y necrosis y valores significativamente mayores de apoptosis
tardía y muerte total que sus grupos Control respectivos. Es resaltante que la
población T CD4+ de los pacientes más inmunocomprometidos (P1C, P2C), presentó
en los análisis fenotípicos ex vivo un aumento significativo en la expresión de CD95
(receptor de señales de apoptosis, Fig. 28), HLA-DR (marcador de activación, Fig.
25) y PD-1 (receptor de señales de co-estimulación negativa, Fig. 26), y una
significativa disminución en la expresión de IL-7Rα (receptor de señales de
supervivencia, Fig. 27), enfatizando lo relevante que serían estos marcadores
fenotípicos en definir la susceptibilidad de los linfocitos T CD4+ a sufrir muerte
celular, tanto en forma espontánea (incluso en la condición sin estimulación se
observó aumento de muerte celular) como en condiciones de estimulación
específica en cultivo y cómo esto pudiera afectar la capacidad de respuesta ante
retos antigénicos en los pacientes en esta etapa de la infección viral. Finalmente, los
individuos con serología positiva para T. gondii (C2, P2A, P2B y P2C) no presentaron
valores de muerte celular en su población de linfocitos T significativamente
distintos a los de los individuos con serología negativa (C1, P1A, P1B y P1C), en
ninguna de las condiciones de estimulación evaluadas en los cultivos, sugiriendo
que la infección por T. gondii resuelta o asintomática no influye en este aspecto de la
funcionalidad de las células T.
Producción de citoquinas ex vivo e in vitro
Las citoquinas son proteínas capaces de activar y modular la respuesta
inmunitaria; y tanto el control como la patogénesis de las infecciones por VIH-1 y T.
gondii han sido vinculados con la producción de algunas de estas proteínas309, 310.
Así, la evaluación de la producción de citoquinas por los individuos participantes en
este estudio, tanto ex vivo como in vitro, ha proporcionado importante información
sobre la capacidad de respuesta específica y el microambiente generado en el
contexto de estas infecciones.
114

Ex vivo: La determinación de citoquinas en el plasma sanguíneo mostró
concentraciones bajas en todos los grupos de estudio, con valores de menos de 20
pg/mL para IL-2 y de menos de 8 pg/mL para IL-4, IL-6, IL-10 y TNF-α, con una
discreta elevación de IL-6 en el grupo de pacientes P2B/C (Fig. 41). Por su parte,
todos los grupos de pacientes infectados con VIH-1 presentaron concentraciones de
IFN-γ en plasma significativamente mayores a los de los individuos sin infección por
el virus, indicando por una parte la capacidad de los pacientes de producir esta
citoquina, que se ha asociado con actividad anti-VIH-1309, y por otra parte
sugiriendo un microambiente particular en los pacientes infectados, desde etapas
tempranas hasta etapas avanzadas de la infección viral. Si bien se observó una
tendencia en los pacientes infectados con VIH-1 y serología positiva para T. gondii
(P2) a presentar concentraciones mayores de IFN-γ en plasma que los pacientes con
serología negativa para el parásito (P1), la diferencia no fue estadísticamente
significativa, por lo que se sugiere que infecciones por el parásito latentes o
resueltas, no modificarían los valores de IFN-γ generados en el contexto de la
infección por VIH-1, evidenciados en el plasma de los pacientes. La producción de
IFN-γ, por su asociación con una respuesta antiviral, citotóxica, ha sido uno de los
mediadores que ha recibido especial atención en el contexto de la infección por VIH-
1. Ya desde el año 1984, Murray et al. habían descrito defectos en la producción de
esta citoquina asociados con la infección viral, en experimentos de estimulación
antigénica específica in vitro311. En base a antecedentes como este, se hace
especialmente significativo el hallazgo de este trabajo, de que los pacientes
infectados por el virus presenten concentraciones significativamente mayores de
IFN-γ plasmático que los individuos no infectados. Como se mencionó
anteriormente, esto demuestra que esta capacidad de producción no se ha perdido
en los pacientes, ni siquiera en aquellos en etapas más avanzadas de la infección. A
pesar del aumento en la producción de IFN-γ, es claro que éste no es suficiente para
detener la replicación del virus, como lo demuestran los valores de carga viral de
estos mismos pacientes (Fig. 23), si bien puede sugerir una respuesta inmunitaria
que trata de contenerla. El hecho de que sea en plasma donde se observó el
aumento de concentración de esta citoquina, sugiere un aspecto sistémico de la
respuesta inmunitaria, probablemente asociado al estado de activación crónico
generalizado, característico de la infección, sin reflejar necesariamente la respuesta
específica en el microambiente donde se esté replicando activamente el virus,
recordando, de hecho, que son las células con especifidad anti-VIH-1 las que tienen
mayor expresión de la molécula co-estimuladora negativa PD-1 y capacidad
limitada de producir IFN-γ en respuesta a la estimulación antigénica105-107, 312, lo
cual puede ser un factor determinante en la incapacidad de detener la replicación
viral110. En todo caso, la elevación de IFN-γ en el plasma de los pacientes, reitera
115

que la presencia de este solo mediador de la respuesta inmunitaria no basta para
contener la progresión de la infección.
PHA: Al evaluar la producción de citoquinas por PBMC en respuesta a la
estimulación con PHA por 72 horas, se observaron valores de concentración de IL-
10, TNF-α e IFN-γ significativamente mayores a los de las células cultivadas sin
estimulación, en todos los grupos de estudio (Fig. 45, 46 y 47), mostrando la
capacidad de las PBMC de los pacientes infectados con VIH-1 de producir estas
citoquinas bajo estimulación policlonal, incluso en etapas avanzadas de la infección
viral, a pesar de las alteraciones en la respuesta inmunitaria que se observan en
estas etapas. En el caso de TNF-α, los pacientes infectados con VIH-1 (P1 y P2),
presentaron concentraciones significativamente mayores a los individuos no
infectados (C1 y C2), sugiriendo una modificación en el estado de respuesta de esta
población celular en el contexto de la infección, quizá motivado al hecho de venir
“pre-estimuladas ex vivo” por la infección viral misma, generando in vitro respuestas
de producción mayores a las de los grupos Control. No se observaron diferencias
significativas en la producción de estas citoquinas, bajo estimulación con PHA, entre
los grupos P1 y P2, sugiriendo que la infección por T. gondii pasada o asintomática,
no modifica esta producción.
Env: Las PBMC de los pacientes infectados con VIH-1 (P1 y P2) cultivadas en
presencia del antígeno viral Env por 72 horas, presentaron concentraciones de IL-
10 y TNF-α significativamente mayores a las de las células de los individuos no
infectados (C1 y C2), y a las de las células sin estimulación (Medio), si bien no fueron
capaces de producir cantidades significativas de IFN-γ (Fig. 45, 46 y 47). El
aumento en la producción de IL-10, con su capacidad inmunomoduladora, unido a
la no producción de IFN-γ, sugiere un perfil de inhibición de la respuesta
inmunitaria anti-VIH-1, consistente con la falla en el control de la replicación viral
(Fig. 23). Ya se ha mencionado que en el contexto de la infección por VIH-1 existe
un defecto en la producción de IFN-γ bajo estimulación in vitro con antígenos del
virus311, 312, defecto que puede revertirse por el bloqueo de la vía de señalización
PD-1/PDL-1106, 107, 312, haciendo patente cómo en el contexto de la infección la
propia respuesta anti-viral está comprometida, pero, cómo, a su vez, existen
posibles vías para revertir este compromiso106, 312, 313. En cuanto a la producción de
IL-10, la evidencia experimental nos muestra un cuadro complejo de la respuesta
inmunitaria y de su control, pues si bien el bloqueo in vitro de la interacción de la IL-
10 con su receptor (IL-10Rα) aumenta la producción de IL-2 e IFN-γ por linfocitos T
CD4+ con especificidad anti-VIH-1, así como la proliferación de éstos y de linfocitos
T CD8+ en respuesta a la estimulación con antígenos virales, evidenciando que la
presencia de IL-10 está inhibiendo la respuesta anti-VIH-1 en los pacientes
infectados314; por otra parte también hay evidencias de que el aumento en la
concentración de IL-10 en pacientes VIH-1+ se correlaciona con el control de la
116

replicación viral315. Adicionalmente, debido al papel crucial que tiene la activación
inmunitaria crónica en la progresión de la infección por VIH-1113, es posible que, a
pesar de tener un efecto inhibitorio sobre la respuesta antiviral específica, la IL-10
tenga un acción beneficiosa neta, al limitar la activación celular y disminuir la
pérdida de linfocitos T CD4+315. Finalmente, la infección por T. gondii crónica
asintomática o la infección resuelta, no modifican la producción de IL-10, TNF-α o
IFN-γ por PBMC en respuesta a la estimulación con el antígeno Env del VIH-1 por 72
horas en pacientes VIH-1+, según lo evidencia la ausencia de diferencias
significativas en estos parámetros entre los grupos P1 y P2.
SATg: La estimulación por 72 horas con el extracto antigénico soluble de T.
gondii, SATg, de PBMC de individuos no infectados con VIH-1, pero seropositivos
para T. gondii (C2), resultó en una significativa y consistente producción de IL-2, IL-
10, TNF-α e IFN-γ, al comparar con el grupo Control seronegativo para el parásito
(C1) y con las células cultivadas sin estimulación (Medio) (Fig. 42, 45, 46 y 47).
Estos hallazgos muestran que las condiciones de cultivo de las PBMC fueron
adecuadas para discriminar entre la respuesta anti-T. gondii de producción de estas
citoquinas por individuos con serología positiva para el parásito, y la respuesta de
individuos con serología negativa (C1 vs. C2). La infección por VIH-1, por su parte,
indujo significativos cambios en estos parámetros de funcionalidad en los
individuos con serología positiva para T. gondii, como lo demuestran los resultados
de los pacientes del grupo P2 (VIH-1+/T. gondii+). En la producción de IL-2, por
ejemplo, el grupo P2, no presentó concentraciones que difirieran significativamente
de los del grupo P1, ni de las células sin estimulación (Medio), siendo a su vez
significativamente menores a los del grupo C2 (Fig. 42). En cuanto a la producción
de TNF-α e IFN-γ, si bien el grupo P2 presentó concentraciones mayores a las del
grupo P1, estas concentraciones fueron consistentemente menores a las del grupo
C2, con diferencias significativas para IFN-γ (Fig. 46 y 47), evidenciando defectos en
la producción de estas citoquinas en los pacientes infectados con VIH-1, desde las
etapas tempranas de la infección viral. Estos defectos en la producción de IL-2, TNF-
α e IFN-γ en pacientes infectados por VIH-1, presentes desde etapas iniciales de la
infección viral, comprometerían la capacidad de control de infecciones por T. gondii 172, 186, 187, 316, 317, aumentando el riesgo de que en los pacientes con infecciones
latentes se reactivara la replicación parasitaria, aunque fuese de forma limitada, con
la posibilidad de ir provocando daños acumulativos en órganos como el cerebro318,
319, por el cual el parásito tiene especial tropismo, incluso antes de llegar a etapas
francamente sintomáticas, como la encefalitis toxoplásmica. De hecho, y en
congruencia con este razonamiento, la evaluación neurofisiológica de estos
pacientes evidenció que los individuos del grupo P2 (VIH-1+/T. gondii+) presentaron
una deficiencia significativamente mayor en la memoria a corto plazo, más
trastornos en el procesamiento cognitivo de estímulos visuales (P300 visual) y
117

mayor alteración de las funciones ejecutivas del lóbulo frontal (Prueba de
Clasificación de Tarjetas de Wisconsin) que los pacientes del grupo P1 (VIH-1+/T.
gondii-), sugiriendo que en estos pacientes con serología positiva para T. gondii la
neurotoxicidad estaría aumentada320, 321. Así, la evaluación de producción de
citoquinas in vitro mostró que la infección por VIH-1 se asocia con defectos en la
respuesta anti-T. gondii desde las primeras etapas de la infección viral; mientras
que la evaluación neurofisiológica mostró una mayor alteración funcional en el
grupo de pacientes que podría incluir a los individuos con infecciones latentes por
el parásito (P2), lo cual refuerza la relevancia que puede tener la pérdida gradual
del control de tales infecciones latentes, con posibilidad de reactivaciones
subclínicas/limitadas de la replicación parasitaria en el cerebro de estos pacientes,
causando daños acumulativos, que irían gradualmente comprometiendo la
funcionalidad del sistema nervioso central, adicionalmente a la afectación que
pueda generar el propio VIH-1120, 121, 124, complicando el cuadro en pacientes co-
infectados.
Efectos neurotóxicos de VIH-1 y T. gondii
La evaluación de los efectos neurotóxicos de VIH-1 y T. gondii se realizó
utilizando cultivos primarios de células de cerebro de feto de rata Sprague-Dawley,
los cuales estaban formados por poblaciones heterogéneas de células nerviosas
(astrocitos, neuronas, células de la microglía) que fueron cultivadas en presencia
del parásito, del virus (o gp41), o de ambos patógenos en forma simultánea. Estos
cultivos permitieron evaluar la interacción de los agentes infecciosos con los
distintos linajes celulares, y la interacción de los distintos linajes entre sí en el
contexto de la respuesta ante los agentes infecciosos. Determinando el recuento, la
viabilidad, la apoptosis y necrosis de las células nerviosas, se evaluó el efecto
neurotóxico en las distintas condiciones. Midiendo en los sobrenadantes de cultivo
los mediadores solubles producidos, incluyendo óxido nítrico, IL-1α, IL-6, IL-10,
TNF-α e IFN-γ, se estudiaron las posibles causas de los efectos neurotóxicos
observados. Además, al evaluar el porcentaje y linaje de células nerviosas infectadas
por el parásito, así como el número de taquizoítos libres, se pudo seguir el curso de
la infección parasitaria y elucidar cómo ésta era afectada por la presencia
simultánea de VIH-1 o gp41. Así, este sistema nos permitió evaluar múltiples
variables en compleja interacción. Es oportuno enfatizar que la inducción de la
producción de los distintos mediadores evaluados en este trabajo se presenta en
ausencia de infección viral, ya que el VIH-1 no infecta a las células nerviosas de rata,
y en el caso de los cultivos en presencia de gp41, la proteína recombinante estaba
en una solución libre de partículas virales. Así, los efectos inducidos serían
resultado de la interacción molecular con las células de las monocapas, pero no de
infección productiva de las mismas322, enfatizando la capacidad del virus, o de su
118

glicoproteína de envoltura, la gp41, de generar respuestas en células nerviosas no
infectadas por el mismo, bien directamente (interacción “virus-célula” o “gp41-
célula”) o a través de los mediadores cuya producción se induce por dicha
interacción322, hecho que tiene gran relevancia en la neuropatología de pacientes
infectados con VIH-1, donde la infección de un número limitado de macrófagos
cerebrales y células de la microglía, ejerce un efecto deletéreo sobre neuronas y
otras células no infectadas258.
Al estudiar los efectos causados por la presencia exclusiva de T. gondii sobre
las células nerviosas, se hizo evidente que el parásito es capaz de infectar a estas
células y replicarse activamente en las mismas. Los tres linajes de células nerviosas
evaluados, astrocitos, neuronas y células de la microglía, fueron susceptibles a la
infección parasitaria (Fig. 63), lo cual es congruente con los hallazgos descritos en
humanos203, 204 y roedores250, tanto in vivo como in vitro: T. gondii es capaz de
infectar múltiples linajes celulares del sistema nervioso central. La significativa
correlación positiva observada en este trabajo entre los porcentajes de cada linaje
en la población de células nerviosas y su respectivo porcentaje de infección (Fig.
64), sugiere que T. gondii infectaría a las células en los cultivos de acuerdo a su
disponibilidad como célula blanco. Precisamente, siendo los astrocitos las células
representadas en mayor proporción en estos cultivos primarios (Fig. 62), fueron las
células que presentaron un mayor porcentaje de infección (Fig. 63).
La presencia exclusiva de T. gondii se asoció con inhibición del aumento de
los recuentos celulares observados en condiciones basales, a medida que
transcurrían los días de cultivo (Fig. 48B), sugiriendo inhibición de la proliferación
y/o destrucción celular que contrarrestaba dicha proliferación. El análisis
microscópico mostró que la infección se localiza en focos de la monocapa, con
replicación parasitaria activa en el citoplasma de las células nerviosas y lisis de
dichas células al aumentar el número de parásitos intracelulares (Fig. 65D). La
disposición focal de la infección parasitaria en las monocapas celulares, recuerda la
presentación típica de la encefalitis toxoplásmica, la cual se caracteriza por la
presencia de lesiones de localización focal que van invadiendo progresivamente el
tejido cerebral, como lo evidencian los estudios anatomopatológicos tipo biopsias y
necropsias (Fig. 18A y D), y los estudios imagenológicos, tipo tomografía
computarizada y resonancia magnética (Fig. 18 B y C), tanto en humanos como en
roedores160, 234. Así, en presencia del parásito, la destrucción activa de células
infectadas contribuyó con la detención en el aumento de los recuentos de las
mismas. De hecho, cuando las células se cultivaron en presencia únicamente de T.
gondii, no se detectó una producción significativa de óxido nítrico ni de ninguna de
las citoquinas evaluadas, lo cual sugiere que la destrucción celular en presencia del
parásito no estaba dada por la acción de mediadores solubles, sino primordialmente
por la lisis de células infectadas.
119

Llama la atención que las células nerviosas cultivadas en presencia de T.
gondii no hayan respondido con la producción de mediadores solubles ante una
infección parasitaria en activa progresión, con significativa destrucción celular. En
modelos múridos de encefalitis toxoplásmica, se ha demostrado que los linfocitos T
CD4+ y T CD8+ infiltran el tejido cerebral luego de la infección216-218, y que la
presencia de estas células es imprescindible para generar una respuesta
inmunitaria efectiva171, 220, en la cual la producción de IFN-γ es crucial172. En el
contexto de la infección cerebral, otras células que participan en la producción de
IFN-γ junto a los linfocitos T, son las células citotóxicas naturales (NK), los
macrófagos y las células de la microglía219, 220, 323, 324. Sin embargo, en modelos
múridos se ha demostrado que en ausencia de células T, la producción de IFN-γ por
estos otros linajes celulares es insuficiente para detener la infección220. Los cultivos
primarios utilizados en este trabajo están formados por células con capacidad de
adherirse a la superficie de las placas de cultivos, por lo que las poblaciones
linfoides T y NK estarían ausentes, lo cual podría explicar que en las condiciones de
estos experimentos no se hayan generado los mediadores estudiados. Se ha
demostrado, tanto en la respuesta periférica, como en el contexto de infección en el
sistema nervioso central, que un paso importante en el inicio de la respuesta
inmunitaria anti-T. gondii es la producción de IL-12 por las células dendríticas177,
325, la cual estimularía la producción de IFN-γ por linfocitos T y células NK (Fig. 16).
Sin embargo, aun si las células dendríticas estuviesen presentes en los cultivos
primarios, y en presencia de T. gondii produjesen IL-12, seguirían faltando en el
sistema los linfocitos T y las células NK, para recibir la señal mediada por la IL-12
que indujese la producción de IFN-γ. La importancia de la población de linfocitos T
para el control del parásito en el sistema nervioso central también es sugerida en la
infección humana, pues en pacientes que desarrollan deficiencias en esta población
celular debido a enfermedades neoplásicas, como el linfoma de Hodgkin, se puede
presentar reactivación de infecciones latentes por T. gondii203, 230. Asimismo, en
pacientes infectados con VIH-1 existe una estrecha relación entre la disminución del
recuento de linfocitos T de sangre periférica y el desarrollo de la encefalitis
toxoplásmica146, la cual generalmente se presenta en pacientes con recuentos de
linfocitos T CD4+ menores a 100/μL, y reducciones correspondientes en la
población T CD8+232, 233. Además del descenso numérico en la población de células
T en el contexto de la infección por VIH-1, es igualmente relevante para el control in
vivo de la infección cerebral por T. gondii la pérdida de la capacidad funcional de
estas células, que incluyen defectos en la producción de IFN-γ en respuesta al
parásito311.
Con respecto a los efectos ejercidos sobre los cultivos primarios por la
presencia de VIH-1 o de gp41, se evidenció que estos agentes indujeron también
una inhibición en el aumento de los recuentos de las células nerviosas observado en
120

condiciones basales. En presencia únicamente de gp41 se observó un significativo
aumento en la apoptosis de las células nerviosas, lo cual sugiere que este proceso
estaría contribuyendo con la ausencia de aumento en los recuentos. Sin embargo,
dado que el porcentaje de células en apoptosis era relativamente bajo (3-4%),
mecanismos adicionales deben estar participando para explicar los efectos
observados. El análisis microscópico reveló que en presencia de VIH-1 o de gp41, la
morfología de las monocapas se modificaba notablemente, con retraimiento de los
citoplasmas y disminución de la confluencia entre las células, presentándose esta
modificación en toda la población celular, sugiriendo un efecto neurotóxico
generalizado en estas condiciones de cultivo. Los mediadores generados en
presencia de estos agentes patógenos, pueden orientar en cuanto a los posibles
mecanismos que están involucrados en los efectos observados.
Los cultivos realizados en presencia de gp41 o de VIH-1, se asociaron con un
aumento significativo en la producción de óxido nítrico. En el sistema nervioso
central el óxido nítrico cumple importantes funciones fisiológicas y de protección
neuronal, pero el aumento exacerbado en su producción se asocia a diversos
mecanismos neurotóxicos326, 327. Por ejemplo, en condiciones de estrés oxidativo el
óxido nítrico puede reaccionar con el anión superóxido (O2-) y formar peroxinitrito
(ONOO-), un anión con fuerte poder oxidativo que puede mediar tanto apoptosis
como necrosis neuronal326, 328. El estrés oxidativo asociado con aumentos en la
concentración de óxido nítrico327 también puede alterar el metabolismo lipídico,
aumentando la concentración de ceramida, un producto del metabolismo de la
esfingomielina que funciona como segundo mensajero y se ha asociado con
activación de vías neurotóxicas en la infección por VIH-1329, 330. El óxido nítrico
también puede mediar S-nitrosilación, esto es, la transferencia de NO a residuos
críticos de cisteína, mediando la activación de la metaloproteasa de matriz (MMP)-
9, y quizá también de otras MMP, que a su vez mediarán la muerte neuronal por
alteración de mecanismos de unión de la neurona a la matriz extracelular o a otras
neuronas331. En pacientes infectados con VIH-1 se ha observado un aumento
significativo de la actividad de MMP-9 en líquido cefalorraquídeo, que se ha
asociado con facilitación de la entrada del virus en el sistema nervioso central por
migración transendotelial de células infectadas a través de la barrera
hematoencefálica, cuyos componentes de matriz extracelular habrían sido
degradados por la MMP-9332. Adicionalmente, el óxido nítrico puede afectar la
función mitocondrial de la neurona mediando efectos excitotóxicos asociados al
glutamato333. Así, son múltiples los mecanismos neurotóxicos que pueden ser
mediados por el óxido nítrico, de allí la importancia que pueda tener para la
sobrevida y funcionalidad de las células nerviosas el aumento de su concentración.
En pacientes con demencia severa asociada al VIH-1 se ha observado en necropsias
de tejido cerebral un significativo aumento en la expresión de la enzima óxido
121

nítrico sintasa inducible (iNOS), responsable de la síntesis del óxido nítrico, y esta
expresión se ha asociado con incrementos en la concentración de la gp41 viral en
dichos tejidos140. En congruencia con esto, los cultivos realizados en presencia de
gp41 presentaron una significativa correlación positiva entre la concentración de
óxido nítrico y la apoptosis de células nerviosas (Fig. 52C y D), sugiriendo la
asociación de éste con el proceso de muerte celular326. Sin embargo, es necesario
enfatizar que el aumento en la concentración de óxido nítrico, por sí sola, no fue
suficiente en las condiciones de estos ensayos para generar apoptosis, como lo
demuestran los hallazgos en la condición de cultivo en presencia exclusiva de VIH-1,
donde dicho aumento no se asoció con mayores porcentajes de apoptosis (Fig. 51A
y 52A). De manera que si bien el óxido nítrico puede mediar procesos de muerte
celular, no es el único factor involucrado en estos procesos.
En los experimentos donde las células nerviosas fueron cultivadas en
presencia de VIH-1 o de gp41 también se observó una producción significativa
mayor de IL-1α, IL-6, IL-10 y TNF-α que en la condición Control. Se ha descrito que
aumentos en las concentraciones de TNF-α e IL-1 pueden mediar la activación de
astrocitos258, induciendo que éstos liberen una mayor cantidad de glutamato al
medio y que a su vez presenten una menor recaptación de este aminoácido
excitador258, 334, favoreciendo así un ambiente excitotóxico, es decir, de daño
neuronal por exceso de estimulación335. La activación de los astrocitos inducida por
el TNF-α y la IL-1 estimula también la expresión de iNOS, con la subsecuente
producción de óxido nítrico258, promoviendo los efectos neurotóxicos ya descritos.
El TNF-α y la IL-1 pueden además mediar la liberación de L-cisteína por parte de los
macrófagos, que al estimular los receptores NMDAR puede provocar apoptosis
neuronal336. Asimismo, el TNF-α se ha asociado con la inducción de apoptosis
neuronal por modificación de la relación entre proteínas pro-apoptóticas y anti-
apoptóticas337, y por activación de una vía que incluye la formación de especies
reactivas del oxígeno (ROS)338. Llama la atención, a propósito de estos hallazgos,
que en las condiciones cultivadas en presencia de gp41, que presentaron
concentraciones significativamente mayores de TNF-α (Fig. 56), fue donde se
observaron también porcentajes significativamente mayores de apoptosis (Fig. 51).
Con respecto a la importancia de estos resultados en el contexto de la
neuropatogénesis de la infección viral, es oportuno señalar que en el cerebro de
pacientes con demencia asociada al VIH-1 (HAD), en estudios post mortem, se ha
observado un aumento de la apoptosis en neuronas, astrocitos y células
endoteliales339, 340, así como también una elevación en la expresión del ARN
mensajero específico para TNF-α en astrocitos y células de la microglía,
correlacionándose esta elevación con los síntomas clínicos de la HAD341.
En presencia de VIH-1 y gp41, los cultivos además mostraron aumentos en la
concentración de IL-6, lo cual se ha asociado a respuesta de las células nerviosas a
122

estímulos tóxicos342. Neuronas, astrocitos y células de la microglía son capaces de
producir IL-6 al ser estimuladas por IL-1 y TNF-α, y también en el contexto de
infecciones virales. La IL-6 cumple importantes funciones fisiológicas en el sistema
nervioso central, donde se ha asociado con diversos mecanismos neurotrópicos,
mediando además el enlace entre la respuesta innata y la respuesta adaptativa342. El
posible papel neuropatogénico de la IL-6 también ha sido sugerido por su
asociación a respuestas proinflamatorias y la modificación en su producción que se
ha observado en algunas enfermedades degenerativas del sistema nervioso
central342. En pacientes infectados con VIH-1, especialmente aquellos en etapa de
SIDA o complejo relacionado al SIDA (ARC), se ha observado un aumento de la
concentración de IL-6 en líquido cefalorraquídeo343. El aumento en las
concentraciones de IL-6 también se ha asociado con defectos en la memoria344, 345,
lo cual puede ser relevante en los pacientes infectados con VIH-1. De hecho, en los
pacientes evaluados en este trabajo una de las funciones que se vio afectada fue la
de memoria a corto plazo320.
La IL-10 fue otro de los mediadores cuya concentración aumentó en
presencia de VIH-1 y gp41, siendo el aumento en presencia de VIH-1
significativamente mayor que en presencia de gp41. Esta citoquina se ha asociado
con el control de la respuesta inflamatoria346, 347, en la cual, si bien la limitación de
la invasión y replicación de agentes patógenos es sumamente importante,
igualmente lo es evitar que los mecanismos utilizados para controlar y/o eliminar al
agente invasor causen daño de los propios tejidos del hospedador346, 347. Los
hallazgos señalan que en cultivos de células nerviosas la IL-10 puede regular la
producción de los mediadores proinflamatorios, incluidos la IL-1, IL-6, TNF-α y
óxido nítrico348, 349. En el contexto de los experimentos de este trabajo la
producción de IL-10 en presencia de VIH-1 y gp41 puede por tanto interpretarse
como una respuesta de equilibrio entre mediadores pro-inflamatorios y anti-
inflamatorios, reforzando la importancia del control de la respuesta inmunitaria.
Pasemos ahora a analizar cómo el VIH-1 y la gp41, en ausencia de infección
viral productiva, pueden inducir en los cultivos de células nerviosas la producción
de mediadores solubles y provocar los efectos neurotóxicos descritos en las páginas
previas. Revisemos cómo la interacción molecular entre el virus (o sus
componentes) y las células nerviosas pueden activar vías de señalización o generar
las modificaciones necesarias para explicar estos hallazgos.
Examinemos primero a la gp41, una glicoproteína de la envoltura del VIH-
142. Durante el proceso de entrada del virus a su célula blanco, la gp41 sufre
modificaciones conformacionales que permiten que la región de la glicoproteína
denominada péptido de fusión N terminal se inserte en la membrana celular y ésta se
fusione a la membrana viral, dando paso a la formación de un poro por el cual
ingresa el material genético del virus al interior de la célula blanco242, 350 (Fig. 6). La
123

fusión de membranas es un proceso secuencial que incluye los pasos intermedios de
hemifusión (unión de las capas externas de las membranas sin intercambio de
contenido) y formación de pequeños poros351. Actualmente se reconoce la
importancia de los procesos de fusión y hemifusión de membranas mediados por la
gp41, no solo para el ingreso del virus a su célula blanco, sino también en la
afectación de células no productivamente infectadas351, 352. Así, se ha descrito que la
interacción de linfocitos T CD4+ no infectados (células blanco) con la gp41 presente
en la membrana de células transfectadas (células efectoras), provoca la apoptosis de
las células blanco por hemifusión, en un proceso que incluye la activación de
caspasa 3, despolarización mitocondrial y formación de especies reactivas del
oxígeno353.
Los hallazgos en los experimentos de cultivos primarios sugieren que la gp41
pueda estar ejerciendo en las células nerviosas un efecto similar al descrito en
células T353. De hecho la gp41 es capaz de inducir apoptosis, asociada a la
producción de IL-1, TNF-α y óxido nítrico en células nerviosas tanto de roedores
como humanas240, 242, y la región de la molécula principalmente responsable de este
efecto, denominado dominio neurotóxico, se ubica hacia el extremo amino
terminal240, 242, cerca del péptido de fusión N terminal (Fig. 5B), la región de la
molécula que interacciona con la membrana de la célula blanco (Fig. 6), lo cual
colocaría a este dominio neurotóxico en una posición ideal para generar un efecto
en dicha célula242. Asimismo una región del dominio citoplasmático de la gp41 (Fig.
5B) denominada Péptido Lítico Lentiviral-1 (LLP-1) 354 se ha asociado con procesos
neurotóxicos. El LLP-1 puede causar la muerte neuronal por disminución del
glutatión intracelular (una molécula con importante función antioxidante),
provocando daño mitocondrial354. Otro efecto neurotóxico mediado por el LLP-1 es
la alteración del transporte de aminoácidos excitadores por parte de los astrocitos;
en esta población celular el LLP-1 reduce la recaptación de aspartato e induce la
liberación de glutamato, favoreciendo un ambiente excitotóxico355. Por otra parte se
ha descrito que en condiciones fisiológicas, la gp41 forma espontáneamente
agregados estables de alto peso molecular, compuestos por números variables
(usualmente entre 7 y 70) de trímeros de gp41356. Se ha propuesto que la
acumulación de estos agregados pueda provocar daño neurológico como el
observado en otras enfermedades del sistema nervioso central asociadas a
depósitos de proteínas (Enfermedad de Creutzfeldt-Jakob y Alzheimer)356. En
congruencia con este razonamiento, en algunos casos de HAD se ha reportado una
encefalopatía espongiforme357, 358, indistinguible de la asociada a la enfermedad de
Creutzfeldt-Jakob, esta última causada por la acumulación de proteínas priónicas en
forma de placas356. En pacientes infectados con el VIH-1 el grado de acumulación de
gp41 en el tejido cerebral se ha relacionado con el grado o severidad de la demencia
asociada a la infección141, 356, 359.
124

La gp120 es otra molécula estructural del VIH-1 que ha sido ampliamente
estudiada por su capacidad de generar efectos neurotóxicos136, 137, 241, 336, y podría
estar contribuyendo con la producción de los efectos observados en los cultivos
primarios. En el cerebro humano y de roedores, la gp120 puede interaccionar con
las células de la microglía y los macrófagos perivasculares a través de la molécula
CD4 y los receptores de quimioquina CXCR4 y CCR5 que estas expresan. Estas
moléculas son reconocidas por regiones específicas de la gp120 viral42, 137, 258. La
glicoproteína también puede interaccionar con astrocitos y neuronas, que expresan
CXCR4 y CCR5.
La interacción de la gp120 (unida o no al virus) con las células de la microglía
activa la vía de señalización de la p38 MAPK (Proteína Quinasa Activada por
Mitógeno p38), que por intermedio del factor de transcripción MEF2C (Factor
Potenciador Específico del Miocito-2C), induce la producción y liberación de
múltiples mediadores neurotóxicos, entre los que están los aminoácidos excitadores
(glutamato, L-cisteína), el ácido araquidónico, el ácido quinolínico, el PAF (Factor de
Activación Plaquetaria), el TRAIL (Ligando Inductor de Apoptosis Relacionado a
TNF), la MMP-2 (Metaloproteasa de Matriz-2), la MMP-9 y las citoquinas pro-
inflamatorias (IL-1, TNF-α)124, 258. Estos mediadores ponen en acción diversas vías
de retroalimentación y afectan a otras poblaciones de células nerviosas como
neuronas y astrocitos. Así, por acción de las citoquinas proinflamatorias y el ácido
araquidónico generados por la microglía, se induce en los astrocitos la expresión de
iNOS, con la consecuente producción de óxido nítrico, molécula que como se ha
descrito en párrafos previos puede mediar daño a las células nerviosas. Asimismo
las citoquinas proinflamatorias y el ácido araquidónico provocan que los astrocitos
aumenten la liberación de glutamato y disminuyan la recaptación del mismo,
favoreciendo un ambiente excitotóxico. La excesiva estimulación del receptor
NMDAR en la población neuronal por la acción del glutamato y otras moléculas
como la L-cisteína y el ácido quinolínico, provoca la entrada masiva de calcio a
través del canal iónico asociado a este receptor360. El aumento en la concentración
de calcio intracelular provoca la activación de p38 MAPK y p53, la sobrecarga
mitocondrial de calcio, con pérdida del potencial de membrana mitocondrial, la
disminución en la síntesis de ATP, la liberación del citocromo C y el AIF (Factor
Inductor de Apoptosis), la activación de la caspasa 3, la produción de especies
reactivas del oxígeno (ROS) y de óxido nítrico, la peroxidación lipídica, el daño al
citoesqueleto y la apoptosis258, 360. En este trabajo, el cultivo de células nerviosas en
presencia del VIH-1, en cuya envoltura se expresa la gp120, se asoció con un
ambiente proinflamatorio (producción aumentada de IL-1, IL-6, TNF- y óxido
nítrico) así como de efectos neurotóxicos (disminución en los recuentos,
alteraciones morfológicas), lo cual es congruente con que los mecanismos descritos
en la literatura estén activos en este modelo, representando éstos posibles rutas de
125

inducción en la producción de los distintos mediadores y efectos tóxicos
observados.
Otro mediador que podría estar generando daño a las células nerviosas de
los cultivos primarios es la α quimioquina SDF-1 (Factor Derivado de la Célula
Estromal-1), el ligando natural de CXCR4, que en el sistema nervioso central puede
ser producido por astrocitos, neuronas y macrófagos258. En pacientes infectados por
VIH-1 y enfermedad cerebral avanzada se ha descrito un significativo aumento de
SDF-1 en neuronas361. En cultivos primarios de células nerviosas de roedores, el
SDF-1 puede mediar apoptosis de neuronas uniéndose a su receptor CXCR4136, 137.
Sin embargo, es necesario mencionar que en algunos reportes se ha asociado la
presencia de SDF-1 a función neurotrópica362. Por otra parte, el SDF-1 puede sufrir
proteólisis por la MMP-2 (cuya concentración también se encuentra aumentada en
la infección por VIH-1363), generando un péptido que ya no es capaz de unirse a
CXCR4, pero que es mucho más neurotóxico que la quimioquina original364.
Aparte de la vía indirecta de generación de neurotoxinas a través de la
activación de células de la microglía y macrófagos, se ha descrito que la gp120 es
capaz de interaccionar directamente con las neuronas en ausencia de células
gliales360, uniéndose a los receptores de quimioquinas expresados en estas células
en forma independiente a CD4134, provocando la muerte neuronal133.
Adicionalmente, se ha reportado que concentraciones nanomolares de gp120
pueden interaccionar con el sitio de unión de la glicina en el receptor NMDAR365,
sugiriendo otro mecanismo por el cual la glicoproteína viral puede afectar
directamente a la neurona. Más aún, en neuronas del hipocampo de rata la gp120
puede ejercer un efecto excitotóxico mediado por el receptor NMDAR366. En
astrocitos humanos y de rata también se ha descrito un efecto directo de la gp120,
inhibiendo la recaptación de glutamato por modificación de los mecanismos de
intercambio Na+/H+367. Sin embargo, estos efectos directos han sido observados en
experimentos in vitro con linajes celulares aislados. Los resultados obtenidos en
cultivos con poblaciones mixtas de células nerviosas sugieren que la ruta indirecta
de daño neuronal, a través de mediadores producidos por la microglía infectada y/o
activada, es la que predominaría136, 137, 258, 360.
Además de las glicoproteínas de envoltura gp120 y gp41, se ha demostrado
que otras moléculas del VIH-1 pueden mediar efectos neurotóxicos368, por lo que
podrían estar contribuyendo con los resultados observados en los experimentos de
este trabajo. Por ejemplo, Tat (Transactivador de la Expresión Génica), una proteína
reguladora de la replicación viral, puede interaccionar directamente con la
membrana de la neurona y causar despolarización369, aumentando la concentración
citoplasmática de calcio, provocando sobrecarga mitocondrial, producción de ROS,
activación de caspasas y eventualmente apoptosis370, 371. Tat también puede inducir
la producción de citoquinas pro-inflamatorias y MMP en células gliales372-374. La Vpr
126

(Proteína Viral R), una proteína viral que regula el transporte del material genético
viral hacia el núcleo de la célula infectada375, es también capaz de causar la muerte
neuronal por la formación de canales permeables a cationes en la membrana de
éstas376. La región básica de la proteína Rev (Regulador de la Expresión de Proteínas
del Virión, que participa en la exportación del ARN mensajero viral del núcleo al
citoplasma de la célula infectada377), puede interaccionar con fosfolípidos de
membrana e inducir conformación tipo hélice . Esta modificación conformacional
inducida por Rev puede trastornar la integridad de la membrana e inducir
neurotoxicidad368, 378. La proteína reguladora viral Nef (Factor de Regulación
Negativa) también puede mediar efectos tóxicos en neuronas y células gliales379, así
como la producción de MMP-9 y daño a la barrera hematoencefálica380. Así,
múltiples proteínas virales, tanto estructurales como no estructurales, pueden
mediar inflamación y efectos neurotóxicos368, incluso en ausencia de infección viral
productiva, todo lo cual ilustra las posibles vías que en los experimentos de este
trabajo estén generando la producción de los mediadores evaluados, así como los
efectos tóxicos observados. Más aún, estos hallazgos permiten explicar cómo la
infección de un número limitado de células nerviosas en los pacientes, al favorecer
la formación de factores que pueden afectar a otras células no infectadas, puede
generar las marcadas alteraciones en la funcionalidad del sistema nervioso central
asociadas a la infección viral.
Haciendo un recuento, es evidente que la presencia exclusiva de T. gondii, del
VIH-1 ó de la gp41 en cultivos primarios de células nerviosas crea condiciones
desfavorables para éstas, con disminución de los recuentos celulares al comparar
con la condición Control. De acuerdo a los hallazgos, en las condiciones de estos
experimentos la disminución de células nerviosas en presencia del parásito estaría
mediada principalmente por la lisis de células infectadas, mientras que el virus y su
glicoproteína de envoltura, mediarían sus efectos citotóxicos a través de la
generación de factores solubles, incluyendo citoquinas proinflamatorias (IL-1α, IL-
6, TNF-α) y óxido nítrico, pero posiblemente también, según los reportes de la
literatura, aminoácidos excitadores (glutamato, L-cisteína, aspartato), ácido
araquidónico, ácido quinolínico, ROS, PAF, TRAIL, MMP-2, MMP-9, SDF-1, además
de daño directo a la membrana celular. Quedaría entonces, por analizar qué
modificaciones se generan en los cultivos primarios cuando están presentes en
forma simultánea el VIH-1 (o la gp41) y T. gondii.
De acuerdo a los resultados, es claro que la presencia del virus inhibe la
replicación del parásito, como lo indican la disminución significativa en el número
de taquizoítos libres en los sobrenadantes de células nerviosas infectadas con T.
gondii en presencia simultánea de VIH-1 o gp41 (Fig. 58)381 y la disminución
significativa del porcentaje y valor absoluto de células infectadas (Fig. 59), así como
del número de parásitos en el citoplasma de dichas células, en presencia del VIH-1
127

(Fig. 61, Tabla II)382. Llama la atención que no se hayan observado diferencias
significativas para el porcentaje y para el valor absoluto de infección, entre los
cultivos en presencia únicamente de T. gondii y aquellos en presencia simultánea de
“gp41/T. gondii”, si bien los valores absolutos de infección tendieron a ser menores
en esta última condición (Fig. 59). Esto sugiere que, en las condiciones de estos
ensayos, la presencia de VIH-1 ejercería un mayor efecto inhibitorio sobre la
infección parasitaria, que el ejercido por la glicoproteína viral gp41. Varios
mecanismos pueden ser responsables de la inhibición observada en la infección
parasitaria. El óxido nítrico, que se genera en estos cultivos, tanto en presencia del
VIH-1 como de gp41140, 322, 383 (Fig. 52), por su capacidad toxoplasmicida187, 384, es
uno de los factores que puede estar disminuyendo la replicación parasitaria184.
También las citoquinas proinflamatorias IL-1α, IL-6 y TNF-α producidas en los
cultivos322 (Fig. 53, 54, 56), pueden ejercer un efecto inhibitorio sobre la
replicación de T. gondii385, 386. Las modificaciones morfológicas de las células
nerviosas observadas en presencia del VIH-1 y la gp41 (Fig. 65), con retraimiento
de los citoplasmas celulares y menor contacto célula-célula, pueden también
dificultar la invasión de nuevas células por el parásito. Asímismo, la destrucción
celular en presencia del virus, disminuiría el número de células blanco disponibles
para el parásito, lo cual sería otro factor en contra de su replicación. Esta inhibición
in vitro de la replicación parasitaria en presencia del VIH-1 es opuesta a la
reactivación de las infecciones latentes que se observan en los pacientes en etapa de
SIDA, indicando diferencias en las condiciones in vivo, entre las que pudieran estar
la mayor disponibilidad de células blanco para el parásito, y el control de la
replicación ejercido por los linfocitos T en etapas tempranas, que se pierde en la
etapa de inmunodeficiencia, al disminuir esta población linfocitaria.
A pesar de que la replicación parasitaria haya estado inhibida, es resaltante
que la presencia simultánea de T. gondii y VIH-1 se asociara con mayor destrucción
celular, como lo indican el recuento significativamente menor y el mayor porcentaje
de células no viables y apoptóticas (Fig. 48, 49 y 51)387, indicando un mayor efecto
neurotóxico, sugiriendo que la destrucción directa de las células por el parásito no
es el mecanismo responsable del efecto observado. La producción de óxido nítrico
en los cultivos puede ejercer no solo un efecto inhibidor sobre la replicación
parasitaria, como se mencionó anteriormente, sino que también puede ser tóxico
para las células nerviosas, provocando su muerte258, 326, hecho que está
correlacionado también con el aumento de la apoptosis celular en estas
condiciones383 (Fig. 51, 52). El aumento en la producción de IL-1α, IL-6 y TNFα, así
como la disminución de la citoquina inmunomoduladora IL-10322, 385, 388, al
favorecer un ambiente pro-inflamatorio, puede también ejercer un efecto deletéreo
sobre la célula nerviosa190, 258. Estos resultados evidencian una mayor afectación de
las células nerviosas en presencia simultánea de VIH-1 y T. gondii, incluso con una
128

replicación parasitaria disminuida, lo cual podría ser relevante para pacientes
infectados con ambos agentes en forma simultánea, donde el compromiso de su SNC
podría ser mayor. En este punto, es oportuno recordar que en los pacientes
infectados con VIH-1 y serología positiva para T. gondii evaluados en este trabajo, se
evideció mayor afectación en la funcionalidad de su sistema nervioso central320, 321,
sugiriendo que el aumento de la neurotoxicidad evidenciada en los experimentos in
vitro en presencia de ambos agentes infecciosos385, 387, pudiera también estar
presente in vivo en los pacientes co-infectados320, 321.
Cabe destacar que en presencia simultánea de VIH-1 y T. gondii, mientras se
mantenía aumentada la producción de IL-6 y óxido nítrico con respecto a la
condición Control, la producción de IL-1α fue significativamente mayor y la de IL-
10, fue significativamente menor a la de la condición en presencia únicamente de
VIH-1, sugiriendo, como ya se ha mencionado, un aumento del carácter
proinflamatorio del microambiente. Consideremos algunas posibles vías para que la
presencia de T. gondii, incluso con una replicación restringida, pueda aumentar el
efecto pro-inflamatorio ejercido por VIH-1. Varias moléculas del parásito son
capaces de interaccionar con receptores en las células de su hospedador y modular
la respuesta de éstas389. La profilina del parásito es capaz de interaccionar con los
receptores tipo Toll (TLR) 11 y TLR12 e inducir la producción de IL-12181, 182. En el
cerebro de roedores TLR 11 es expresado en neuronas y células de la microglía390.
Otra molécula del parásito, la Ciclofilina-18, es capaz de interaccionar con el
receptor CCR5 de humanos y roedores, induciendo también la producción de IL-12
por células dendríticas176. Como ya se ha mencionado, astrocitos, neuronas y células
de la microglía expresan CCR5137. En la respuesta in vivo ante T. gondii, la
producción de IL-12 por parte de las células dendríticas, se ha descrito como uno de
los pasos iniciales fundamentales para el control de la infección, al inducir la
producción de IFN-γ por parte de linfocitos T y células NK177 (Fig. 16). Si bien estas
poblaciones linfoides estarían ausentes en los cultivos primarios utilizados en este
trabajo, las células de la microglía, en determinadas circunstancias, pueden producir
IL-12391, 392, y podrían favorecer el ambiente proinflamatorio en respuesta a la
Ciclofilina-18 y/o a la Profilina, sin descartarse la posibilidad de interacción de estas
moléculas parasitarias con neuronas y astrocitos, añadiendo otras posibles vías de
modificación del microambiente por las proteínas del parásito. Además, se ha
descrito que CCR5 puede mediar la transmisión de señales neuroprotectoras137, por
lo que su bloqueo por la Ciclofilina-18 podría evitar la recepción de tales señales,
resultando en aumento de la neurotoxicidad en estas condiciones de cultivo en
presencia de simultánea de VIH-1 y T. gondii.
La producción de TNF-α también estuvo significativamente disminuida en
presencia simultánea de VIH-1 y T. gondii, al comparar con la condición “sólo VIH-
1”, mostrando una compleja y dinámica modificación del microambiente en
129

presencia de ambos patógenos. Se ha descrito que T. gondii puede interferir con la
fosforilación y acetilación de la histona H3, para prevenir la remodelación de
cromatina en la región promotora del gen de TNF-α, inhibiendo la transcripción y
producción de esta citoquina en las células infectadas389, 393, lo cual explicaría la
menor producción en presencia de ambos patógenos.
Finalmente, los experimentos realizados en cultivos primarios de células
nerviosas muestran que la sola interacción del virus y las moléculas virales con las
células nerviosas o sus receptores, incluso sin infección productiva, es capaz de
inducir en estas poblaciones celulares respuestas que modifican el microambiente,
con serias consecuencias para su funcionalidad y sobrevida. Asimismo, la
modificación del microambiente en presencia simultánea de ambos patógenos y la
mayor destrucción celular asociada a éste, evidencia que un ambiente más pro-
inflamatorio es también un ambiente más neurotóxico. Estos resultados enfatizan la
importancia que para los pacientes pueda tener la presencia simultánea de ambos
agentes patógenos en el contexto de una co-infección, y cómo el sistema nervioso
central podría estar más afectado en esta situación, incluso con una replicación
parasitaria limitada.
130

CONCLUSIONES Con base en los resultados en los experimentos de fenotipaje de linfocitos T de
sangre periférica, la carga viral, la proliferación y producción de perforina por
linfocitos T y la producción de citoquinas ex vivo e in vitro en los pacientes
evaluados, se concluye que:
1. La infección resuelta y la infección crónica asintomática por T. gondii no
modifican el fenotipo y la funcionalidad de linfocitos T inducidos por la
infección por el VIH-1, ni la respuesta específica contra el virus en pacientes
infectados con VIH-1.
Con base en los resultados en los experimentos in vitro de producción de IL-2,
TNF-α e IFN-γ por PBMC en presencia de antígenos solubles de T. gondii, en
pacientes infectados con VIH-1 y serología positiva para T. gondii, se concluye que:
2. La infección por VIH-1 afecta la respuesta inmunitaria anti-T. gondii desde
las etapas iniciales de la infección viral, incrementando el riesgo en los
pacientes co-infectados de presentar reactivaciones limitadas de infecciones
parasitarias latentes, incluso en las etapas de la infección viral consideradas
“asintomáticas”, lo que podría causar daños acumulativos en los tejidos
donde se encuentre el parásito, siendo de especial interés el sistema
nervioso central*.
En base a los resultados en los experimentos de cultivos primarios de células
nerviosas de feto de rata Sprague-Dawley en presencia simultánea de VIH-1 y T.
gondii, se concluye que:
3. La presencia simultánea de VIH-1 o de su glicoproteína de envoltura gp41 en
cultivos primarios de células nerviosas de feto de rata inhibe
significativamente la replicación de T. gondii en dichos cultivos.
4. La presencia simultánea de VIH-1 y T. gondii en cultivos primarios de células
nerviosas favorece un ambiente más pro-inflamatorio y mayor destrucción
celular (disminución de los recuentos y aumento de la apoptosis de células
nerviosas), incluso con replicación parasitaria restringida, sugiriendo que la
presencia de ambos patógenos aumenta el efecto neurotóxico, lo cual puede
ser de gran relevancia para la funcionalidad del sistema nervioso central en
pacientes co-infectados.
* Esta conclusión está adicionalmente sustentada por los hallazgos en la evaluación neurofisiológica
de estos mismos pacientes320, 321.
131

BIBLIOGRAFÍA 1. Centers for Disease Control (CDC): Pneumocystis pneumonia - Los Angeles.
MMWR Morb Mortal Wkly Rep 1981; 30(21):250-252. (Disponible en:
http://stacks.cdc.gov/view/cdc/1261/cdc_1261_DS1.pdf)
2. Stringer JR, Beard CB, Miller RF, Wakefield AE: A new name for Pneumocystis
from humans and new perspectives on the host-pathogen relationship. Emerg
Infect Dis 2002; 8(9): 891–896.
3. Stringer JR, Beard CB, Miller RF: Spelling Pneumocystis jirovecii. Emerg Infect
Dis 2009; 15(3): 506.
4. Centers for Disease Control (CDC): Kaposi's sarcoma and Pneumocystis
pneumonia among homosexual men-New York City and California. MMWR
Morb Mortal Wkly Rep 1981; 30(25):305-308. (Disponible en:
http://stacks.cdc.gov/view/cdc/1265/cdc_1265_DS1.pdf)
5. Gottlieb MS, Schroff R, Schanker HM, Weisman JD, Fan PT, Wolf RA, Saxon A:
Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy
homosexual men — Evidence of a new acquired cellular immunodeficiency. N
Engl J Med 1981; 305:1425-1431.
6. Masur H, Michelis MA, Greene JB, Onorato I, Vande Stouwe RA, Holzman RS,
Wormser G, Brettman L, Lange M, Murray HW, Cunningham-Rundles S: An
outbreak of community-acquired Pneumocystis carinii pneumonia — Initial
manifestation of cellular immune dysfunction. N Engl J Med. 1981; 305:1431–
1438.
7. Siegal FP, Lopez C, Hammer GS, Brown AE, Kornfeld SJ, Gold J, Hassett J,
Hirschman SZ, Cunningham-Rundles C, Adelsberg BR, Parham DM, Siegal M,
Cunningham-Rundles S, Armstrong D: Severe acquired immunodeficiency in
male homosexuals, manifested by chronic perianal ulcerative Herpes Simplex
lesions. N Engl J Med 1981; 305:1439-1444.
8. Marx JL: New disease baffles medical community. Science 1982;
217(4560):618-621.
132

9. Centers for Disease Control (CDC): Update on acquired immune deficiency
syndrome (AIDS)-United States. MMWR Morb Mortal Wkly Rep 1982;
31(37):507-508, 513-514.
10. Simon V, Ho DD, Abdool Karim Q: HIV/AIDS epidemiology, pathogenesis,
prevention and treatment. Lancet 2006; 368:489-504.
11. Centers for Disease Control (CDC): Possible transfusion-associated acquired
immune deficiency syndrome (AIDS) - California. Morb Mortal Wkly Rep 1982;
31(48):652-654.
12. Centers for Disease Control (CDC): Pneumocystis carinii pneumonia among
persons with hemophilia A. MMWR Morb Mortal Wkly Rep 1982; 31(27):365-
367.
13. Centers for Disease Control (CDC): Unexplained immunodeficiency and
opportunistic infections in infants-New York, New Jersey, California. MMWR
Morb Mortal Wkly Rep 1982; 31(49):665-667.
14. Masur H, Michelis MA, Wormser GP, Lewin S, Gold J, Tapper ML, Giron J, Lerner
CW, Armstrong D, Setia U, Sender JA, Siebken RS, Nicholas P, Arlen Z, Maayan S,
Ernst JA, Siegal FP, Cunningham-Rundles S: Opportunistic infection in
previously healthy women. Initial manifestations of a community-acquired
cellular immunodeficiency. Ann Intern Med 1982; 97(4):533-539.
15. Centers for Disease Control (CDC): Immunodeficiency among female sexual
partners of males with acquired immune deficiency syndrome (AIDS) - New
York. MMWR Morb Mortal Wkly Rep 1983; 31(52):697-698.
16. Clumeck N, Mascart-Lemone F, de Maubeuge J, Brenez D, Marcelis L: Acquired
immune deficiency syndrome in Black Africans. Lancet 1983; 1(8325):642.
17. Piot P, Quinn TC, Taelman H, Feinsod FM, Minlangu KB, Wobin O, Mbendi N,
Mazebo P, Ndangi K, Stevens W, et al.: Acquired immunodeficiency syndrome in
a heterosexual population in Zaire. Lancet 1984; 2(8394):65-69.
18. Van de Perre P, Rouvroy D, Lepage P, Bogaerts J, Kestelyn P, Kayihigi J, Hekker
AC, Butzler JP, Clumeck N: Acquired immunodeficiency syndrome in Rwanda.
Lancet 1984; 2(8394):62-65.
133

19. Sepkowitz KA: AIDS-the first 20 years. N Engl J Med 2001; 344(23):1764-1772.
20. Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J,
Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozenbaum W,
Montagnier L: Isolation of a T-lymphotropic retrovirus from a patient at risk
for acquired immune deficiency syndrome (AIDS). Science 1983;
220(4599):868-871.
21. Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker
TJ, Redfield R, Oleske J, Safai B, White G, Foster P, Markham PD: Frequent
detection and isolation of cytopathic retroviruses (HTLV-III) from patients with
AIDS and at risk for AIDS. Science 1984; 224(4648):500-503.
22. Levy JA, Hoffman AD, Kramer SM, Landis JA, Shimabukuro JM, Oshiro LS:
Isolation of lymphocytopathic retroviruses from San Francisco patients with
AIDS. Science 1984; 225(4664):840-842.
23. Coffin J, Haase A, Levy JA, Montagnier L, Oroszlan S, Teich N, Temin H,
Toyoshima K, Varmus H, Vogt P: Human immunodeficiency viruses. Science
1986; 232(4751): 697.
24. Flint SJ, Enquist LW, Racaniello VR, Skalka AM: Human Immunodeficiency Virus
pathogenesis. En: Principles of virology, 2nd ed., Washington DC: ASM Press,
2004: 622-653.
25. Letvin NL: Immunology of HIV infection. En: Paul WE (Eds.): Fundamental
Immunology, 6th ed., Philadelphia: Wolters Kluwer/Lippincott Williams &
Wilkins, 2008: 1204-1232.
26. Centers for Disease Control and Prevention (CDC): Update: mortality
attributable to HIV infection among persons aged 25-44 years-United States,
1994. MMWR Morb Mortal Wkly Rep 1996; 45(6):121-125.
27. UNAIDS: Global report 2013 - Fact sheet. Consultada el 2 de Abril de 2014.
(Disponible en:
http://www.unaids.org/en/resources/campaigns/globalreport2013/factsheet
/)
134

28. ONUSIDA: Informe sobre la epidemia mundial de SIDA 2013. (Disponible en:
http://www.unaids.org/en/media/unaids/contentassets/documents/epidemi
ology/2013/gr2013/UNAIDS_Global_Report_2013_es.pdf)
29. MPPPS/Programa Nacional SIDA/ITS, MPPPRE: Informe nacional de avances
en la implementación de la declaración de compromisos sobre VIH/SIDA
(2001) y la declaración política sobre VIH/SIDA (2006 y 2011). República
Bolivariana de Venezuela; Marzo 2012. (Disponible en:
http://www.unaids.org/en/dataanalysis/knowyourresponse/countryprogress
reports/2012countries/ce_VE_Narrative_Report%5B1%5D.pdf).
30. Chiu IM, Yaniv A, Dahlberg JE, Gazit A, Skuntz SF, Tronick SR, Aaronson SA:
Nucleotide sequence evidence for relationship of AIDS retrovirus to
lentiviruses. Nature. 1985; 317(6035):366-368.
31. Shehu-Xhilaga M, Oelrichs R: Basic HIV virology. En: HIV Management in Australasia: a guide for clinical care. Darlinghurst, NSW: Australasian Society for HIV Medicine, 2009: 9-18. (Disponible en: http://www.ashm.org.au/images/Publications/Monographs/HIV_Management_Australasia/HIV-Management-Australia-2009.pdf).
32. Meerloo T, Sheikh MA, Bloem AC, de Ronde A, Schutten M, van Els CA, Roholl PJ,
Joling P, Goudsmit J, Schuurman HJ: Host cell membrane proteins on human
immunodeficiency virus type 1 after in vitro infection of H9 cells and blood
mononuclear cells. An immuno-electron microscopic study. J Gen Virol 1993;
74 (Pt 1):129-135.
33. Cantin R, Méthot S, Tremblay MJ: Plunder and stowaways: incorporation of
cellular proteins by enveloped viruses. J Virol 2005; 79(11):6577-6587.
34. Cullen BR: Human immunodeficiency virus as a prototypic complex retrovirus.
J Virol. 1991; 65(3):1053-1056.
35. Kanki PJ, Essex ME: Virology. En: Mayer KH, Pizer HF (Eds): The AIDS
pandemic: impact on science and society. London: Elsevier Academic Press,
2005: 13-35.
36. Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB,
Arthur LO, Peeters M, Shaw GM, Sharp PM, Hahn BH: Origin of HIV-1 in the
chimpanzee Pan troglodytes troglodytes. Nature 1999; 397(6718):436-441.
135

37. Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-
Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte
E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH: Chimpanzee
reservoirs of pandemic and nonpandemic HIV-1. Science 2006;
313(5786):523-526.
38. Reeves JD, Doms RW: Human immunodeficiency virus type 2. J Gen Virol 2002;
83(Pt 6):1253-1265.
39. Hirsch VM, Olmsted RA, Murphey-Corb M, Purcell RH, Johnson PR: An African
primate lentivirus (SIVsm) closely related to HIV-2. Nature 1989;
339(6223):389-392.
40. Rangel HR, Garzaro DJ, Pujol FH: Diversidad genética del Virus de
Inmunodeficiencia Humana en las Américas, con énfasis en la epidemia
venezolana. Interciencia 2009; 34(3):163-169.
41. Rangel HR, Garzaro D, Gutiérrez CR, Vásquez L, Guillen G, Torres JR, Pujol FH:
HIV diversity in Venezuela: predominance of HIV type 1 subtype B and genomic
characterization of non-B variants. AIDS Res Hum Retroviruses 2009;
25(3):347-350.
42. Caffrey M: HIV envelope: challenges and opportunities for development of
entry inhibitors. Trends Microbiol 2011; 19(4): 191–197.
43. Tilton JC, Doms RW: Entry inhibitors in the treatment of HIV-1 infection.
Antiviral Res 2010; 85(1):91-100.
44. Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA:
The CD4 (T4) antigen is an essential component of the receptor for the AIDS
retrovirus. Nature 1984; 312:763–767.
45. Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T,
Gluckman JC, Montagnier L: T-lymphocyte T4 molecule behaves as the receptor
for human retrovirus LAV. Nature 1984; 312:767–768.
46. Feng, Y, Broder CC, Kennedy PE, Berger EA: HIV-1 entry cofactor: functional
cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science
1996; 272:872–877.
136

47. Weiss RA: Thirty years on: HIV receptor gymnastics and the prevention of
infection. BMC Biol 2013; 11: 57.
48. Shioda T, Levy JA, Cheng-Mayer C: Small amino acid changes in the V3
hypervariable region of gp120 can affect the T-cell-line and macrophage
tropism of human immunodeficiency virus type 1. Proc Natl Acad Sci USA.
1992; 89(20):9434-9438.
49. Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, Kabat D, Chesebro B,
Goldsmith MA: Selective employment of chemokine receptors as human
immunodeficiency virus type 1 coreceptors determined by individual amino
acids within the envelope V3 loop. J Virol. 1997; 71(9):7136-7139.
50. Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD: Genotypic and
phenotypic characterization of HIV-1 patients with primary infection. Science
1993; 261(5125):1179-1181.
51. van't Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-van Lent N, H J
Scherpbier HJ, Veenstra J, Boer K, Coutinho RA, Miedema F, Schuitemaker H:
Macrophage-tropic variants initiate human immunodeficiency virus type 1
infection after sexual, parenteral, and vertical transmission. J Clin Invest 1994;
94(5):2060–2067.
52. Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR: Change in coreceptor
use correlates with disease progression in HIV-1-infected individuals. J Exp
Med 1997; 185(4):621-628.
53. Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel
J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, Figdor CG, van
Kooyk Y: DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances
trans-infection of T cells. Cell 2000; 100(5):587-597.
54. Nicholson JK, Cross GD, Callaway CS, McDougal JS: In vitro infection of human
monocytes with human T lymphotropic virus type III/lymphadenopathy-
associated virus (HTLV-III/LAV). J Immunol 1986; 137(1):323-329.
55. Tschachler E, Groh V, Popovic M, Mann DL, Konrad K, Safai B, Eron L, diMarzo
Veronese F, Wolff K, Stingl G: Epidermal Langerhans cells--a target for HTLV-
III/LAV infection. J Invest Dermatol 1987; 88(2):233-237.
137

56. Patterson S, Knight SC: Susceptibility of human peripheral blood dendritic cells
to infection by human immunodeficiency virus. J Gen Virol 1987; 68(Pt
4):1177-1181.
57. Klatzmann D, Barré-Sinoussi F, Nugeyre MT, Danquet C, Vilmer E, Griscelli C,
Brun-Veziret F, Rouzioux C, Gluckman JC, Chermann JC, et al.: Selective tropism
of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes.
Science 1984; 225(4657):59-63.
58. Piller SC, Caly L, Jans DA: Nuclear import of the pre-integration complex (PIC):
the Achilles heel of HIV? Curr Drug Targets 2003; 4(5):409-429.
59. Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F: HIV-1 integration
in the human genome favors active genes and local hotspots. Cell 2002;
110(4):521-529.
60. Martin-Serrano J, Zang T, Bieniasz PD: Role of ESCRT-I in retroviral budding. J
Virol 2003; 77(8):4794-4804.
61. Dybul M, Connors M, Fauci AS: Immunology of HIV infection. En: Paul WE
(Eds): Fundamental immunology, 5th ed., Philadelphia: Lippincott Williams &
Wilkins, 2003: 1285-1318.
62. Fauci AS, Desrosiers RC: Pathogenesis of HIV and SIV. En: Coffin JM, Hughes
SH, Varmus HE (Eds): Retroviruses. Cold Spring Harbor (NY): Cold Spring
Harbor Laboratory Press; 1997. (disponible en:
http://www.ncbi.nlm.nih.gov/books/NBK19374/)
63. Michael NL, Vahey M, Burke DS, Redfield RR: Viral DNA and mRNA expression
correlate with the stage of Human Immunodeficiency Virus (HIV) type 1
infection in humans: evidence for viral replication in all stages of HIV disease. J
Virol 1992; 66(1):310-316.
64. Piatak M Jr, Saag MS, Yang LC, Clark SJ, Kappes JC, Luk KC, Hahn BH, Shaw GM,
Lifson JD: High levels of HIV-1 in plasma during all stages of infection
determined by competitive PCR. Science 1993; 259(5102):1749-1754.
65. Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein
JM, Kotler DP, Fauci AS: HIV infection is active and progressive in lymphoid
138

tissue during the clinically latent stage of disease. Nature 1993;
362(6418):355-358.
66. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M: Rapid
turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature
1995; 373(6510):123-126.
67. Roberts JD, Bebenek K, Kunkel TA: The accuracy of reverse transcriptase from
HIV-1. Science 1988; 242(4882):1171-1173.
68. Moss AR, Bacchetti P: Natural history of HIV infection. AIDS 1989; 3(2):55-61.
69. Lemp GF, Payne SF, Rutherford GW, Hessol NA, Winkelstein W Jr, Wiley JA,
Moss AR, Chaisson RE, Chen RT, Feigal DW Jr, et al.: Projections of AIDS
morbidity and mortality in San Francisco. JAMA 1990; 263(11):1497-1501.
70. Masur H, Ognibene FP, Yarchoan R, Shelhamer JH, Baird BF, Travis W,
Suffredini AF, Deyton L, Kovacs JA, Falloon J, et al.: CD4 counts as predictors of
opportunistic pneumonias in human immunodeficiency virus (HIV) infection.
Ann Intern Med 1989; 111(3):223-231.
71. Crowe SM, Carlin JB, Stewart KI, Lucas CR, Hoy JF: Predictive value of CD4
lymphocyte numbers for the development of opportunistic infections and
malignancies in HIV-infected persons. J Acquir Immune Defic Syndr 1991;
4(8):770-776.
72. Centers for Disease Control (CDC): 1993 revised classification system for HIV
infection and expanded surveillance case definition for AIDS among
adolescents and adults. MMWR Recomm Rep 1992; 41(RR-17):1-19.
(Disponible en:
http://www.cdc.gov/mmwr/preview/mmwrhtml/00018871.htm)
73. Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron
JJ Jr, Feinberg JE, Balfour HH Jr, Deyton LR, Chodakewitz JA, Fischl MA: A
controlled trial of two nucleoside analogues plus indinavir in persons with
human immunodeficiency virus infection and CD4 cell counts of 200 per cubic
millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Eng J Med
1997; 337(11):725-733.
139

74. Palella FJ Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA,
Aschman DJ, Holmberg SD: Declining morbidity and mortality among patients
with advanced human immunodeficiency virus infection. HIV Outpatient Study
Investigators. N Engl J Med 1998; 338(13):853-860.
75. Mussini C, Pezzotti P, Govoni A, Borghi V, Antinori A, d'Arminio Monforte A, De
Luca A, Mongiardo N, Cerri MC, Chiodo F, Concia E, Bonazzi L, Moroni M, Ortona
L, Esposito R, Cossarizza A, De Rienzo B: Discontinuation of primary
prophylaxis for Pneumocystis carinii pneumonia and toxoplasmic encephalitis
in human immunodeficiency virus type I-infected patients: the changes in
opportunistic prophylaxis study. J Infect Dis 2000; 181(5):1635-1642.
76. Whitcup SM, Fortin E, Lindblad AS, Griffiths P, Metcalf JA, Robinson MR,
Manischewitz J, Baird B, Perry C, Kidd IM, Vrabec T, Davey RT Jr, Falloon J,
Walker RE, Kovacs JA, Lane HC, Nussenblatt RB, Smith J, Masur H, Polis MA:
Discontinuation of anticytomegalovirus therapy in patients with HIV infection
and cytomegalovirus retinitis. JAMA 1999; 282(17):1633-1637.
77. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak
MA, Fauci AS: Presence of an inducible HIV-1 latent reservoir during highly
active antiretroviral therapy. Proc Natl Acad Sci USA 1997; 94(24):13193-
13197.
78. Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V,
Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE,
Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC: HIV-1 and
T cell dynamics after interruption of highly active antiretroviral therapy
(HAART) in patients with a history of sustained viral suppression. Proc Natl
Acad Sci USA 1999; 96(26):15109-15114.
79. Douek DC, Roederer M, Koup RA: Emerging concepts in the
immunopathogenesis of AIDS. Annu Rev Med 2009; 60:471-484.
80. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen
PL, Khoruts A, Larson M, Haase AT, Douek DC: CD4+ T cell depletion during all
stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp
Med 2004; 200(6):749-759.
140

81. Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, Dandekar S:
Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human
immunodeficiency virus type 1 infection and substantial delay in restoration
following highly active antiretroviral therapy. J Virol 2003; 77(21):11708-
11717.
82. Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden
D, Racz P, Markowitz M: Primary HIV-1 infection is associated with preferential
depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal
tract. J Exp Med 2004; 200(6):761-770.
83. Ho DD: Perspectives series: host/pathogen interactions. Dynamics of HIV-1
replication in vivo. J Clin Invest 1997; 99(11):2565-2567.
84. Wolthers KC, Bea G, Wisman A, Otto SA, de Roda Husman AM, Schaft N, de Wolf
F, Goudsmit J, Coutinho RA, van der Zee AG, Meyaard L, Miedema F: T cell
telomere length in HIV-1 infection: no evidence for increased CD4+ T cell
turnover. Science 1996; 274(5292):1543-1547.
85. Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E,
Schmidt D, Hoh R, Neese R, Macallan D, Deeks S, McCune JM: Directly measured
kinetics of circulating T lymphocytes in normal and HIV-1-infected humans.
Nat Med 1999; 5(1):83-89.
86. Pantaleo G, Graziosi C, Demarest JF, Cohen OJ, Vaccarezza M, Gantt K, Muro-
Cacho C, Fauci AS: Role of lymphoid organs in the pathogenesis of human
immunodeficiency virus (HIV) infection. Immunol Rev 1994; 140:105-130.
87. Schacker TW, Brenchley JM, Beilman GJ, Reilly C, Pambuccian SE, Taylor J,
Skarda D, Larson M, Douek DC, Haase AT: Lymphatic tissue fibrosis is
associated with reduced numbers of naive CD4+ T cells in human
immunodeficiency virus type 1 infection. Clin Vaccine Immunol 2006;
13(5):556-560.
88. Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP: The role of the
thymus in immune reconstitution in aging, bone marrow transplantation, and
HIV-1 infection. Annu Rev Immunol. 2000; 18:529-560.
141

89. Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA,
Haase AT, Feinberg MB, Sullivan JL, Jamieson BD, Zack JA, Picker ZJ, Koup RA:
Changes in thymic function with age and during the treatment of HIV infection.
Nature 1998; 396(6712):690-695.
90. Moses A, Nelson J, Bagby GC Jr: The influence of human immunodeficiency
virus-1 on hematopoiesis. Blood 1998; 91(5):1479-1495.
91. Tripathi AK, Misra R, Kalra P, Gupta N, Ahmad R: Bone marrow abnormalities
in HIV disease. J Assoc Physicians India 2005; 53:705-710.
92. Ameisen JC, Capron A: Cell dysfunction and depletion in AIDS: the programmed
cell death hypothesis. Immunol Today 1991; 12(4):102-105.
93. Ameisen JC, Estaquier J, Idziorek T, DeBels F: The relevance of apoptosis to
AIDS pathogenesis. Trends Cell Biol 1995; 5: 27-32.
94. Groux H, Torpier G, Monte D, Mounton Y, Capron A, Ameisen JC: Activation-
induced death by apoptosis in CD4+ T cells from HIV-infected individuals. J Exp
Med 1992; 175: 331-340.
95. Gougeon ML, Lecouer H, Dulioust A, Enouf MG, Crouvoisier M, Goujard C,
Debord T, Montagnier L: Programmed cell death in peripheral lymphocytes
from HIV-infected persons. Increased susceptibility to apoptosis of CD4 and
CD8 T cells correlates with lymphocyte activation and with disease
progression. J Immunol 1996; 156(9):3509-3520.
96. Katsikis PD, Wunderlich ES, Smith CA, Herzenberg LA, Herzenberg LA: Fas
antigen stimulation induces marked apoptosis of T lymphocytes in human
immunodeficiency virus-infected individuals. J Exp Med 1995; 181:2029-2036.
97. Krammer PH: CD95's deadly mission in the immune system. Nature 2000;
407(6805):789-795.
98. Bohler T, Baumler C, Herr I, Groll A, Kurz M, Debatin KM: Activation of the
CD95 system increases with disease progression in human immunodeficiency
virus type 1-infected children and adolescents. Pediatr Infect Dis J 1997;
16:754-759.
142

99. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD: IL-7 is
critical for homeostatic proliferation and survival of naive T cells. Proc Natl
Acad Sci USA 2001; 98(15):8732-8737.
100. Beq S, Delfraissy JF, Theze J: Interleukin-7 (IL-7): immune function,
involvement in the pathogenesis of HIV infection and therapeutic potential. Eur
Cytokine Netw 2004; 15(4):279-289.
101. Mazzucchelli R, Durum SK: Interleukin-7 receptor expression: intelligent
design. Nat Rev Immunol 2007; 7: 144-154.
102. Rethi B, Fluur C, Atlas A, Krzyzowska M, Mowafi F, Grützmeier S, De Milito A,
Bellocco R, Falk KI, Rajnavölgyi E, Chiodi F: Loss of IL-7Ralpha is associated
with CD4 T-cell depletion, high interleukin-7 levels and CD28 down-regulation
in HIV infected patients. AIDS 2005; 19(18):2077-2086.
103. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ,
Malenkovich N, Okasaki T, Byrne MC, Hoton HF, Fouser L, Carter L, Ling V,
Bowman MR, Carreno BM, Collins M, Wodd CR, Honjo T: Engagement of the PD-
1 immunoinhibitory receptor by a novel B7 family member leads to negative
regulation of lymphocyte activation. J Exp Med 2000; 192(7):1027-1034.
104. Greenwald RJ, Freeman GJ, Sharpe AH: The B7 family revisited. Annu Rev
Immunol 2005; 23:515–548.
105. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio
ML, Schacker T, Roederer M, Douek DC, Koup RA: PD-1 is a regulator of virus-
specific CD8+ T cell survival in HIV infection. J Exp Med 2006; 203:2281-2292.
106. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel
MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, Sekaly RP:
Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to
reversible immune dysfunction. Nat Med 2006; 12(10):1198-1202.
107. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW,
Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q,
Altfeld M, Wherry EJ, Coovadia HM, Goulder PJR, Klenerman P, Ahmed R,
Freeman GJ, Walker BD: PD-1 expression on HIV-specific T cells is associated
with T-cell exhaustion and disease progression. Nature 2006; 443:350-354.
143

108. Clerici M, Stocks NI, Zajac RA, Boswell RN, Lucey DR, Via CS, Shearer GM:
Detection of three distinct patterns of T helper cell dysfunction in
asymptomatic, human immunodeficiency virus-seropositive patients.
Independence of CD4+ cell numbers and clinical staging. J Clin Invest 1989;
84(6):1892-1899.
109. Clerici M, Hakim FT, Venzon DJ, Blatt S, Hendrix CW, Wynn TA, Shearer GM:
Changes in interleukin-2 and interleukin-4 production in asymptomatic, human
immunodeficiency virus-seropositive individuals. J Clin Invest 1993;
91(3):759-765.
110. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW,
Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S,
Connors M: HIV-specific CD8+ T cell proliferation is coupled to perforin
expression and is maintained in nonprogressors. Nat Immunol 2002;
3(11):1061-1068.
111. Trabattoni D, Saresella M, Biasin M, Boasso A, Piacentini L, Ferrante P, Dong H,
Maserati R, Shearer GM, Chen L, Clerici M: B7-H1 is upregulated in HIV
infection and is a novel surrogate marker of disease progression. Blood 2003;
101: 2514-2520.
112. Bentwich Z, Kalinkovich A, Weisman Z, Grossman Z: Immune activation in the
context of HIV infection. Clin Exp Immunol 1998; 111: 1-2.
113. Sodora DL, Silvestri G: Immune activation and AIDS pathogenesis. AIDS 2008;
22(4):439-446.
114. Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, Shih R,
Lewis J, Wiley DJ, Phair JP, Wolinsky SM, Detels R: Shorter survival in advanced
human immunodeficiency virus type 1 infection is more closely associated with
T lymphocyte activation than with plasma virus burden or virus chemokine
coreceptor usage. J Infect Dis 1999; 179:859–870.
115. Munier ML, Kelleher AD: Acutely dysregulated, chronically disabled by the
enemy within: T-cell responses to HIV-1 infection. Immunol Cell Biol 2007;
85:6–15.
144

116. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z,
Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson
L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek
DC: Microbial translocation is a cause of systemic immune activation in chronic
HIV infection. Nature Med 2006; 12(12):1365-1371.
117. Kamoun M, Zerva L, Sloan S, Zmijewski C, Monos D, Trinchieri G: Induction of
HLA class II molecules on human T cells: relationship to immunoregulation and
the pathogenesis of AIDS. DNA Cell Biol 1992; 11(3):265-268.
118. Liu Z, Cumberland WG, Hultin LE, Prince HE, Detels R, Giorgi JV: Elevated CD38
antigen expression on CD8+ T cells is a stronger marker for the risk of chronic
HIV disease progression to AIDS and death in the Multicenter AIDS Cohort
Study than CD4+ cell count, soluble immune activation markers, or
combinations of HLA-DR and CD38 expression. J Acquir Immune Defic Syndr
Hum Retrovirol 1997; 16:83–92.
119. Boasso A, Shearer GM: Chronic innate immune activation as a cause of HIV-1
immunopathogenesis. Clin Immunol 2008; 126(3): 235-242.
120. Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P: The brain in
AIDS: central nervous system HIV-1 infection and AIDS dementia complex.
Science 1988; 239(4840):586-592.
121. Gabuzda DH, Hirsch MS: Neurologic manifestations of infection with human
immunodeficiency virus. Clinical features and pathogenesis. Ann Intern Med
1987; 107(3):383-391.
122. Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA,
Mills RG, Wachsman W, Wiley CA: Early viral brain invasion in iatrogenic
human immunodeficiency virus infection. Neurology 1992; 42(9):1736-1739.
123. An SF, Groves M, Gray F, Scaravilli F: Early entry and widespread cellular
involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic
individuals. J Neuropathol Exp Neurol 1999; 58(11):1156-62.
124. González-Scarano F, Martín-García J: The neuropathogenesis of AIDS. Nat Rev
Immunol 2005; 5: 69-81.
145

125. Peluso R, Haase A, Stowring L, Edwards M, Ventura P: A Trojan Horse
mechanism for the spread of visna virus in monocytes. Virology 1985;
147(1):231-236.
126. Haase AT: Pathogenesis of lentivirus infections. Nature 1986; 322(6075):130-
136.
127. Takahashi K, Wesselingh SL, Griffin DE, McArthur JC, Johnson RT, Glass JD:
Localization of HIV-1 in human brain using polymerase chain reaction/in situ
hybridization and immunocytochemistry. Ann Neurol 1996; 39(6):705-711.
128. Cosenza MA, Zhao ML, Si Q, Lee SC: Human brain parenchymal microglia
express CD14 and CD45 and are productively infected by HIV-1 in HIV-1
encephalitis. Brain Pathol 2002; 12(4):442-455.
129. Eugenin EA, Clements JE, Zink MC, Berman JW: Human immunodeficiency virus
infection of human astrocytes disrupts blood-brain barrier integrity by a gap
junction-dependent mechanism. J Neurosci 2011; 31(26):9456-9465.
130. Gray LR, Turville SG, Hitchen TL, Cheng WJ, Ellett AM, Salimi H, Roche MJ,
Wesselingh SL, Gorry PR, Churchill MJ: HIV-1 entry and trans-infection of
astrocytes involves CD81 vesicles. PLoS One 2014; 9(2):e90620.
131. Hickey WF, Hsu BL, Kimura H: T-lymphocyte entry into the central nervous
system. J Neurosci Res 1991; 28(2):254-260.
132. Hickey WF: Leukocyte traffic in the central nervous system: the participants
and their roles. Semin Immunol 1999; 11(2):125-137.
133. Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ: Chemokines
regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl
Acad Sci USA 1998; 95(24):14500-14505.
134. Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL,
Taub D, Horuk R: CD4-independent association between HIV-1 gp120 and
CXCR4: functional chemokine receptors are expressed in human neurons. Curr
Biol 1997; 7(2):112-121.
146

135. O'Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson DL:
Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the
NMDA receptor subtypes. J Neurosci 2006; 26(3):981-990.
136. Kaul M, Lipton SA: Chemokines and activated macrophages in HIV gp120-
induced neuronal apoptosis. Proc Natl Acad Sci USA 1999; 96(14):8212-8216.
137. Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA: HIV-1 coreceptors CCR5 and
CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also
contribute to protection. Cell Death Differ 2007; 14(2):296-305.
138. Ellis R, Langford D, Masliah E: HIV and antiretroviral therapy in the brain:
neuronal injury and repair. Nat Rev Neurosci 2007; 8(1):33-44.
139. Rausch DM, Davis MR: HIV in the CNS: pathogenic relationships to systemic HIV
disease and other CNS diseases. J Neurovirol 2001; 7(2):85-96.
140. Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI,
Dawson TM, Dawson VL: Immunologic NO synthase: elevation in severe AIDS
dementia and induction by HIV-1 gp41. Science 1996; 274(5294):1917-1921.
141. Adamson DC, McArthur JC, Dawson TM, Dawson VL: Rate and severity of HIV-
associated dementia (HAD): correlations with gp41 and iNOS. Mol Med 1999;
5(2):98-109.
142. Mamidi A, DeSimone JA, Pomerantz RJ: Central nervous system infections in
individuals with HIV-1 infection. J Neurovirol 2002; 8(3):158-167.
143. Ajioka JW, Morrissette NS: A century of Toxoplasma research. Int J Parasitol
2009; 39(8):859-860.
144. Levine ND, Corliss JO, Cox FE, Deroux G, Grain J, Honigberg BM, Leedale GF,
Loeblich AR 3rd, Lom J, Lynn D, Merinfeld EG, Page FC, Poljansky G, Sprague V,
Vavra J, Wallace FG: A newly revised classification of the protozoa. J Protozool
1980; 27(1):37-58.
145. Wong SY, Remington JS: Biology of Toxoplasma gondii. AIDS 1993; 7(3):299-
316.
147

146. Luft BJ, Remington JS: AIDS commentary. Toxoplasmic encephalitis. J Infect Dis
1988; 157:1-6.
147. Montoya JG, Liesenfeld O: Toxoplasmosis. Lancet 2004; 363: 1965-1976.
148. Jones JL, Kruszon-Moran D, Sanders-Lewis K, Wilson M: Toxoplasma gondii
infection in the United States, 1999-2004, decline from the prior decade. Am J
Trop Med Hyg 2007; 77(3):405-410.
149. Petersson K, Stray-Pedersen B, Malm G, Forsgren M, Evengård B:
Seroprevalence of Toxoplasma gondii among pregnant women in Sweden. Acta
Obstet Gynecol Scand 2000; 79(10):824-829.
150. Rosso F, Les JT, Agudelo A, Villalobos C, Chaves JA, Tunubala GA, Messa A,
Remington JS, Montoya JG: Prevalence of infection with Toxoplasma gondii
among pregnant women in Cali, Colombia, South America. Am J Trop Med Hyg
2008; 78(3):504-508.
151. Lago EG, Conrado GS, Piccoli CS, Carvalho RL, Bender AL: Toxoplasma gondii
antibody profile in HIV-infected pregnant women and the risk of congenital
toxoplasmosis. Eur J Clin Microbiol Infect Dis 2009; 28(4):345-351.
152. Chacín-Bonilla L, Sánchez-Chávez Y, Estévez J, Larreal Y, Molero E: Prevalence of
human toxoplasmosis in San Carlos Island, Venezuela. Interciencia 2003; 28(8):
457-462.
153. Bonfante Garrido R, Álvarez N, Anzola N, Crespo L, Bonfante de Peñaloza S:
Toxoplasmosis en pacientes de 14 estados de Venezuela. Bol Ofic Sanit Panam
1984; 96:502-510.
154. Serrano H: Estudios sobre la incidencia de anticuerpos séricos para Toxoplasma
en las poblaciones de Maracaibo y un pueblo rural del Estado Zulia y comparación
de tres métodos serológicos distintos. Kasmera 1974; 5: 75-101.
155. Figallo L, Maekelt GA: Anticuerpos de toxoplasmosis en parturientas y recién
nacidos de la maternidad “Concepción Palacios” de Caracas, Venezuela. Arch
Venez Med Trop Parasitol Med 1962; 4: 289-299.
148

156. Maekelt GA, Gómez Z: Primeras experiencias con la prueba Sabin-Feldman para el
diagnóstico de toxoplasmosis. Arch Venez Med Trop Parasitol Med 1962; 4: 265-
275.
157. Vargas de Caminos N: Títulos de anticuerpos para Toxoplasma en una población
pediátrica de Maracaibo, Venezuela. Kasmera 1982; 10: 72-81.
158. De La Rosa M, Bolívar J, Pérez HA: Toxoplasma gondii infection in Amerindians of
Venezuela Amazon. Medicina 1999; 59: 759-762.
159. Chacín-Bonilla L, Sánchez-Chávez Y, Monsalve F, Estévez J: Seroepidemiology of
toxoplasmosis in Amerindians from western Venezuela. Am J Trop Med Hyg
2001; 65: 131-135.
160. Dubey JP, Lindsay DS, Speer CA: Structures of Toxoplasma gondii tachyzoites,
bradyzoites, and sporozoites and biology and development of tissue cysts. Clin
Microbiol Rev 1998; 11(2):267-299.
161. Dubey JP: Long-term persistence of Toxoplasma gondii in tissues of pigs
inoculated with T gondii oocysts and effect of freezing on viability of tissue
cysts in pork. Am J Vet Res 1988; 49(6):910-913.
162. Siegel SE, Lunde MN, Gelderman AH, Halterman RH, Brown JA, Levine AS, Graw
RG Jr: Transmission of toxoplasmosis by leukocyte transfusion. Blood 1971;
37(4):388-394.
163. Ryning FW, McLeod R, Maddox JC, Hunt S, Remington JS: Probable transmission
of Toxoplasma gondii by organ transplantation. Ann Intern Med 1979;
90(1):47-49.
164. Luft BJ, Naot Y, Araujo FG, Stinson EB, Remington JS: Primary and reactivated
toxoplasma infection in patients with cardiac transplants. Clinical spectrum and
problems in diagnosis in a defined population. Ann Intern Med 1983; 99(1):27-
31.
165. Rawal BD: Laboratory infection with Toxoplasma. J Clin Pathol 1959; 12(1):59-
61.
149

166. Herwaldt BL, Juranek DD: Laboratory-acquired malaria, leishmaniasis,
trypanosomiasis, and toxoplasmosis. Am J Trop Med Hyg 1993; 48(3):313-323.
167. Parker SL, Holliman RE: Toxoplasmosis and laboratory workers: a case-control
assessment of risk. Med Lab Sci 1992; 49(2):103-106.
168. Yap GS, Sher A: Cell-mediated immunity to Toxoplasma gondii: initiation,
regulation and effector function. Immunobiology 1999; 201(2):240-247.
169. Sher A, Oswald IP, Hieny S, Gazzinelli RT: Toxoplasma gondii induces a T-
independent IFN-gamma response in natural killer cells that requires both
adherent accessory cells and tumor necrosis factor-alpha. J Immunol 1993;
150(9):3982-3989.
170. Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A: Synergistic role of CD4+
and CD8+ T lymphocytes in IFN-gamma production and protective immunity
induced by an attenuated Toxoplasma gondii vaccine. J Immunol 1991;
146(1):286-292.
171. Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A: Simultaneous depletion of CD4+
and CD8+ T lymphocytes is required to reactivate chronic infection with
Toxoplasma gondii. J Immunol 1992; 149(1):175-180.
172. Suzuki Y, Orellana MA, Schreiber RD, Remington JS: Interferon-gamma: the
major mediator of resistance against Toxoplasma gondii. Science 1988;
240(4851):516-518.
173. Biron CA, Gazzinelli RT: Effects of IL-12 on immune responses to microbial
infections: a key mediator in regulating disease outcome. Curr Opin Immunol
1995; 7(4):485-496.
174. Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri
G, Sher A: Parasite-induced IL-12 stimulates early IFN-gamma synthesis and
resistance during acute infection with Toxoplasma gondii. J Immunol 1994;
153(6):2533-43.
175. Wilson DC, Matthews S, Yap GS: IL-12 signaling drives CD8+ T cell IFN-gamma
production and differentiation of KLRG1+ effector subpopulations during
Toxoplasma gondii Infection. J Immunol 2008; 180(9):5935-5945.
150

176. Aliberti J, Valenzuela JG, Carruthers VB, Hieny S, Andersen J, Charest H, Reis e
Sousa C, Fairlamb A, Ribeiro JM, Sher A: Molecular mimicry of a CCR5 binding-
domain in the microbial activation of dendritic cells. Nat Immunol 2003;
4(5):485-490.
177. Aliberti, J: Host persistence: exploitation of anti-inflammatory pathways by
Toxoplasma gondii. Nat Rev Immunol 2005; 5:162-170.
178. Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, Soldati-Favre
D: Toxoplasma profilin is essential for host cell invasion and TLR11-dependent
induction of an interleukin-12 response. Cell Host Microbe 2008; 3(2):77–87.
179. Yarovinsky F: Toll-like receptors and their role in host resistance to
Toxoplasma gondii. Immunol Lett 2008; 119(1-2):17-21.
180. Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov
R, Sher A: Cutting edge: MyD88 is required for resistance to Toxoplasma gondii
infection and regulates parasite-induced IL-12 production by dendritic cells. J
Immunol 2002; 168(12):5997-6001.
181. Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS,
Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A: TLR11 activation of
dendritic cells by a protozoan profilin-like protein. Science 2005;
308(5728):1626-1629.
182. Raetz M, Kibardin A, Sturge CR, Pifer R, Li H, Burstein E, Ozato K, Larin S,
Yarovinsky F: Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12
response to Toxoplasma gondii profilin. J Immunol 2013 Nov 1; 191(9):4818-
4827.
183. James SL: Role of nitric oxide in parasitic infections. Microbiol Rev 1995;
59(4):533-547.
184. Bohne W, Heesemann J, Gross U: Reduced replication of Toxoplasma gondii is
necessary for induction of bradyzoite-specific antigens: a possible role for
nitric oxide in triggering stage conversion. Infect Immun 1994; 62(5):1761-
1767.
151

185. Scharton-Kersten TM, Yap G, Magram J, Sher A: Inducible nitric oxide is
essential for host control of persistent but not acute infection with the
intracellular pathogen Toxoplasma gondii. J Exp Med 1997; 185(7):1261-1273.
186. Langermans JA, Van der Hulst ME, Nibbering PH, Hiemstra PS, Fransen L, Van
Furth R: IFN-gamma-induced L-arginine-dependent toxoplasmastatic activity
in murine peritoneal macrophages is mediated by endogenous tumor necrosis
factor-alpha. J Immunol 1992; 148(2):568-574.
187. Gazzinelli RT, Eltoum I, Wynn TA, Sher A: Acute cerebral toxoplasmosis is
induced by in vivo neutralization of TNF-alpha and correlates with the down-
regulated expression of inducible nitric oxide synthase and other markers of
macrophage activation. J Immunol 1993; 151(7):3672-3681.
188. Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kühn R,
Müller W, Trinchieri G, Sher A: In the absence of endogenous IL-10, mice
acutely infected with Toxoplasma gondii succumb to a lethal immune response
dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-
gamma and TNF-alpha. J Immunol 1996; 157(2):798-805.
189. Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, Fort M, Kang H, Gufwoli
E: IL-10 is required for prevention of necrosis in the small intestine and
mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice
following peroral infection with Toxoplasma gondii. J Immunol 2000;
164(10):5375-5382.
190. Wilson EH, Wille-Reece U, Dzierszinski F, Hunter CA: A critical role for IL-10 in
limiting inflammation during toxoplasmic encephalitis. J Neuroimmunol 2005;
165(1):63-74.
191. Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A: Lipoxin-mediated
inhibition of IL-12 production by DCs: a mechanism for regulation of microbial
immunity. Nat Immunol 2002; 3(1):76-82.
192. Aliberti J, Serhan C, Sher A: Parasite-induced lipoxin A4 is an endogenous
regulator of IL-12 production and immunopathology in Toxoplasma gondii
infection. J Exp Med 2002; 196(9):1253-1262.
152

193. Bannenberg GL, Aliberti J, Hong S, Sher A, Serhan C: Exogenous pathogen and
plant 15-lipoxygenase initiate endogenous lipoxin A4 biosynthesis. J Exp Med
2004; 199(4):515-523.
194. Denkers EY, Kim L, Butcher BA: In the belly of the beast: subversion of
macrophage proinflammatory signalling cascades during Toxoplasma gondii
infection. Cell Microbiol 2003; 5(2):75-83.
195. Shapira S, Harb OS, Caamano J, Hunter CA: The NF-kappaB signaling pathway:
immune evasion and immunoregulation during toxoplasmosis. Int J Parasitol
2004; 34(3):393-400.
196. Lee CW, Bennouna S, Denkers EY: Screening for Toxoplasma gondii-regulated
transcriptional responses in lipopolysaccharide-activated macrophages. Infect
Immun 2006; 74(3):1916-1923.
197. Lang C, Gross U, Lüder CG: Subversion of innate and adaptive immune
responses by Toxoplasma gondii. Parasitol Res 2007; 100(2):191-203.
198. Buzoni-Gatel D, Werts C: Toxoplasma gondii and subversion of the immune
system. Trends Parasitol 2006; 22(10):448-452.
199. Sacks D, Sher A: Evasion of innate immunity by parasitic protozoa. Nat
Immunol 2002; 3(11):1041-1047.
200. Da Gama LM, Ribeiro-Gomes FL, Guimarães U Jr, Arnholdt AC: Reduction in
adhesiveness to extracellular matrix components, modulation of adhesion
molecules and in vivo migration of murine macrophages infected with
Toxoplasma gondii. Microbes Infect 2004; 6(14):1287-1296.
201. Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gâtel D, Tardieux I: CD11c- and
CD11b-expressing mouse leukocytes transport single Toxoplasma gondii
tachyzoites to the brain. Blood 2006; 107(1):309-316.
202. Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A: Induction of
dendritic cell migration upon Toxoplasma gondii infection potentiates parasite
dissemination. Cell Microbiol 2006; 8(10):1611-1623.
153

203. Ghatak NR, Sawyer DR: A morphologic study of opportunistic cerebral
toxoplasmosis. Acta Neuropathol 1978; 42(3):217-221.
204. Halonen SK, Lyman WD, Chiu FC: Growth and development of Toxoplasma
gondii in human neurons and astrocytes. J Neuropathol Exp Neurol 1996;
55(11):1150-1156.
205. Powell HC, Gibbs CJ Jr, Lorenzo AM, Lampert PW, Gajdusek DC: Toxoplasmosis
of the central nervous system in the adult. Electron microscopic observations.
Acta Neuropathol 1978; 41(3):211-216.
206. Bertoli F, Espino M, Arosemena JR 5th, Fishback JL, Frenkel JK: A spectrum in
the pathology of toxoplasmosis in patients with acquired immunodeficiency
syndrome. Arch Pathol Lab Med 1995; 119(3):214-224.
207. Ghatak NR, Zimmerman HM: Fine structure of Toxoplasma in the human brain.
Arch Pathol 1973; 95(4):276-283.
208. Kang H, Remington JS, Suzuki Y: Decreased resistance of B cell-deficient mice to
infection with Toxoplasma gondii despite unimpaired expression of IFN-
gamma, TNF-alpha, and inducible nitric oxide synthase. J Immunol 2000;
164(5):2629-2634.
209. Frenkel JK, Taylor DW: Toxoplasmosis in immunoglobulin M-suppressed mice.
Infect Immun 1982; 38(1):360-367.
210. Johnson LL, Sayles PC: Deficient humoral responses underlie susceptibility to
Toxoplasma gondii in CD4-deficient mice. Infect Immun 2002; 70(1):185-191.
211. Carruthers VB, Suzuki Y: Effects of Toxoplasma gondii infection on the brain.
Schizophr Bull 2007; 33(3):745-751.
212. Dickerson F, Boronow J, Stallings C, Origoni A, Yolken R: Toxoplasma gondii in
individuals with schizophrenia: association with clinical and demographic
factors and with mortality. Schizophr Bull 2007; 33(3):737-740.
213. Schwarcz R, Hunter CA: Toxoplasma gondii and schizophrenia: linkage through
astrocyte-derived kynurenic acid? Schizophr Bull 2007; 33(3):652-653.
154

214. Flegr J: Effects of toxoplasma on human behavior. Schizophr Bull 2007;
33(3):757-760.
215. Suzuki Y, Conley FK, Remington JS: Importance of endogenous IFN-gamma for
prevention of toxoplasmic encephalitis in mice. J Immunol 1989; 143(6):2045-
2050.
216. Suzuki Y, Rani S, Liesenfeld O, Kojima T, Lim S, Nguyen TA, Dalrymple SA,
Murray R, Remington JS: Impaired resistance to the development of
toxoplasmic encephalitis in interleukin-6-deficient mice. Infect Immun 1997;
65(6):2339-2345.
217. Wang X, Kang H, Kikuchi T, Suzuki Y: Gamma interferon production, but not
perforin-mediated cytolytic activity, of T cells is required for prevention of
toxoplasmic encephalitis in BALB/c mice genetically resistant to the disease.
Infect Immun 2004; 72(8):4432-4438.
218. Denkers EY, Yap G, Scharton-Kersten T, Charest H, Butcher BA, Caspar P, Heiny
S, Sher A: Perforin-mediated cytolysis plays a limited role in host resistance to
Toxoplasma gondii. J Immunol 1997; 159(4):1903-1908.
219. Suzuki Y, Claflin J, Wang X, Lengi A, Kikuchi T: Microglia and macrophages as
innate producers of interferon-gamma in the brain following infection with
Toxoplasma gondii. Int J Parasitol 2005; 35(1):83-90.
220. Kang H, Suzuki Y: Requirement of non-T cells that produce gamma interferon
for prevention of reactivation of Toxoplasma gondii infection in the brain. Infect
Immun 2001; 69(5):2920-2927.
221. Schlüter D, Kaefer N, Hof H, Wiestler OD, Deckert-Schlüter M: Expression
pattern and cellular origin of cytokines in the normal and Toxoplasma gondii-
infected murine brain. Am J Pathol 1997; 150(3):1021-1035.
222. Fischer HG, Nitzgen B, Reichmann G, Hadding U: Cytokine responses induced
by Toxoplasma gondii in astrocytes and microglial cells. Eur J Immunol 1997;
27(6):1539-1548.
223. Schlüter D, Deckert M, Hof H, Frei K: Toxoplasma gondii infection of neurons
induces neuronal cytokine and chemokine production, but gamma interferon-
155

and tumor necrosis factor-stimulated neurons fail to inhibit the invasion and
growth of T. gondii. Infect Immun 2001; 69(12):7889-7893.
224. Chao CC, Gekker G, Hu S, Peterson PK: Human microglial cell defense against
Toxoplasma gondii. The role of cytokines. J Immunol 1994; 152(3):1246-52.
225. Peterson PK, Gekker G, Hu S, Chao CC: Human astrocytes inhibit intracellular
multiplication of Toxoplasma gondii by a nitric oxide-mediated mechanism. J
Infect Dis.1995; 171(2):516-518.
226. Däubener W, Remscheid C, Nockemann S, Pilz K, Seghrouchni S, Mackenzie C,
Hadding U: Anti-parasitic effector mechanisms in human brain tumor cells: role
of interferon-gamma and tumor necrosis factor-alpha. Eur J Immunol 1996;
26(2):487-492.
227. Schlüter D, Bertsch D, Frei K, Hübers SB, Wiestler OD, Hof H, Fontana A,
Deckert-Schlüter M: Interferon-gamma antagonizes transforming growth
factor-beta2-mediated immunosuppression in murine Toxoplasma
encephalitis. J Neuroimmunol 1998; 81(1-2):38-48.
228. Ferguson DJ, Hutchison WM, Pettersen E: Tissue cyst rupture in mice
chronically infected with Toxoplasma gondii. An immunocytochemical and
ultrastructural study. Parasitol Res 1989; 75(8):599-603.
229. Gazzinelli RT, Denkers EY, Sher A: Host resistance to Toxoplasma gondii: model
for studying the selective induction of cell-mediated immunity by intracellular
parasites. Infect Agents Dis 1993; 2(3):139-149.
230. Israelski DM, Remington JS: Toxoplasmosis in the non-AIDS
immunocompromised host. Curr Clin Top Infect Dis 1993; 13:322-356.
231. Derouin F, Devergie A, Auber P, Gluckman E, Beauvais B, Garin YJ, Lariviere M:
Toxoplasmosis in bone marrow-transplant recipients: report of seven cases
and review. Clin Infect Dis 1992; 15(2):267-270.
232. Porter SB, Sande M: Toxoplasmosis of the central nervous system in Acquired
Immunodeficiency Syndrome. N Engl J Med 1992; 327:1643-1648.
156

233. Hunter CA, Remington JS: Inmunopathogenesis of toxoplasmic encephalitis. J
Infect Dis 1994; 170: 1057-1067.
234. Jayawardena S, Singh S, Burzyantseva O, Clarke H: Cerebral toxoplasmosis in
adult patients with HIV infection. Hosp Physician 2008; July: 17-24. (Disponible
en: http://www.turner-
white.com/memberfile.php?PubCode=hp_jul08_toxoplasmosis.pdf).
235. Suzuki Y, Wong SY, Grumet FC, Fessel J, Montoya JG, Zolopa AR, Portmore A,
Schumacher-Perdreau F, Schrappe M, Köppen S, Ruf B, Brown BW, Remington
JS: Evidence for genetic regulation of susceptibility to toxoplasmic encephalitis
in AIDS patients. J Infect Dis 1996; 173(1):265-268.
236. Howe DK, Sibley LD: Toxoplasma gondii comprises three clonal lineages:
correlation of parasite genotype with human disease. J Infect Dis 1995;
172(6):1561-1566.
237. Honoré S, Couvelard A, Garin YJ, Bedel C, Hénin D, Dardé ML, Derouin F:
Genotyping of Toxoplasma gondii strains from immunocompromised patients.
[Artículo en Francés] Pathol Biol (Paris) 2000; 48(6):541-547.
238. Howe DK, Honoré S, Derouin F, Sibley LD: Determination of genotypes of
Toxoplasma gondii strains isolated from patients with toxoplasmosis. J Clin
Microbiol 1997; 35(6):1411-1414.
239. Ohagen A, Ghosh S, He J, Huang K, Chen Y, Yuan M, Osathanondh R, Gartner S,
Shi B, Shaw G, Gabuzda D: Apoptosis induced by infection of primary brain
cultures with diverse human immunodeficiency virus type 1 isolates: evidence
for a role of the envelope. J Virol 1999; 73(2):897-906.
240. Koka P, He K, Zack JA, Kitchen S, Peacock W, Fried I, Tran T, Yashar SS, Merrill
JE: Human immunodeficiency virus 1 envelope proteins induce interleukin 1,
tumor necrosis factor alpha, and nitric oxide in glial cultures derived from fetal,
neonatal, and adult human brain. J Exp Med 1995; 182(4):941-951.
241. Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, Friedlander RM,
Yuan J, Masliah E, Lipton SA: Caspase cascades in human immunodeficiency
virus-associated neurodegeneration. J Neurosci 2002; 22(10):4015-4024.
157

242. Adamson DC, Kopnisky KL, Dawson TM, Dawson VL: Mechanisms and
structural determinants of HIV-1 coat protein, gp41-induced neurotoxicity. J
Neurosci 1999; 19(1):64-71.
243. Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL,
Taub D, Horuk R: CD4-independent association between HIV-1 gp120 and
CXCR4: functional chemokine receptors are expressed in human neurons. Curr
Biol 1997; 7(2):112-121.
244. Lannuzel A, Lledo PM, Lamghitnia HO, Vincent JD, Tardieu M: HIV-1 envelope
proteins gp120 and gp160 potentiate NMDA-induced [Ca2+]i increase, alter
[Ca2+]i homeostasis and induce neurotoxicity in human embryonic neurons.
Eur J Neurosci 1995; 7(11):2285-2293.
245. Müller WE, Schröder HC, Ushijima H, Dapper J, Bormann J: gp120 of HIV-1
induces apoptosis in rat cortical cell cultures: prevention by memantine. Eur J
Pharmacol 1992; 226(3):209-214.
246. Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR,
Pert CB: Neuronal cell killing by the envelope protein of HIV and its prevention
by vasoactive intestinal peptide. Nature 1988; 335(6191):639-642.
247. Brenier-Pinchart MP, Blanc-Gonnet E, Marche PN, Berger F, Durand F,
Ambroise-Thomas P, Pelloux H: Infection of human astrocytes and
glioblastoma cells with Toxoplasma gondii: monocyte chemotactic protein-1
secretion and chemokine expression in vitro. Acta Neuropathol 2004;
107(3):245-249.
248. Peterson PK, Gekker G, Hu S, Chao CC: Intracellular survival and multiplication
of Toxoplasma gondii in astrocytes. J Infect Dis 1993; 168(6):1472-1478.
249. Schlüter D, Deckert M, Hof H, Frei K: Toxoplasma gondii infection of neurons
induces neuronal cytokine and chemokine production, but gamma interferon-
and tumor necrosis factor-stimulated neurons fail to inhibit the invasion and
growth of T. gondii. Infect Immun 2001; 69(12):7889-7893.
250. Lüder CG, Giraldo-Velásquez M, Sendtner M, Gross U: Toxoplasma gondii in
primary rat CNS cells: differential contribution of neurons, astrocytes, and
158

microglial cells for the intracerebral development and stage differentiation.
Exp Parasitol 1999; 93(1):23-32.
251. Clement LT: Isoforms of the CD45 common leukocyte antigen family: markers
for human T-cell differentiation. J Clin Immunol 1992; 12(1):1-10.
252. Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace
DM, Green DR: Early redistribution of plasma membrane phosphatidylserine is
a general feature of apoptosis regardless of the initiating stimulus: inhibition
by overexpression of Bcl-2 and Abl. J Exp Med. 1995; 182(5):1545-1556.
253. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C: A novel assay for
apoptosis. Flow cytometric detection of phosphatidylserine expression on early
apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;
184(1):39-51.
254. Gonchoroff NJ, Katzmann JA, Currie RM, Evans EL, Houck DW, Kline BC, Greipp
PR, Loken MR: S-phase detection with an antibody to bromodeoxyuridine. Role
of DNase pretreatment. J Immunol Methods 1986; 93(1):97-101.
255. Young JD, Liu CC, Leong LG, Cohn ZA: The pore-forming protein (perforin) of
cytolytic T lymphocytes is immunologically related to the components of
membrane attack complex of complement through cysteine-rich domains. J Exp
Med 1986; 164(6):2077-2082.
256. Misumi Y, Misumi Y, Koichiro Miki K, Takatsuki A, Tamura G, Ikehara Y: Novel
blockade by Brefeldin A of intracellular transport of secretory proteins in
cultured rat hepatocytes. J Biol Chem 1986; 261(24): 11398-11403.
257. Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR:
Analysis of nitrate, nitrite, and [15N]nitrate in biological samples. Anal Biochem
1982; 126(1):131-138.
258. Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA: HIV-1 infection and
AIDS: consequences for the central nervous system. Cell Death Differ 2005;
12(Suppl 1):878-892.
259. Keir ME, Sharpe AH: The B7/CD28 costimulatory family in autoimmunity.
Immunol Rev 2005; 204:128-143.
159

260. Zha YY, Blank C, Gajewski TF: Negative regulation of T-cell function by PD-1.
Critical Rev Immunol 2004; 24(4):229-237.
261. Fahey JL, Prince H, Weaver M, Groopman J, Visscher B, Schwartz K, Detels R:
Quantitative changes in T helper or T suppressor/cytotoxic lymphocyte subsets
that distinguish acquired immune deficiency syndrome from other immune
subset disorders. Am J Med 1984; 76(1):95-100.
262. Carney WP, Rubin RH, Hoffman RA, Hansen WP, Healey K, Hirsch MS: Analysis
of T lymphocyte subsets in cytomegalovirus mononucleosis. J Immunol 1981;
126(6): 2114-2116.
263. Thomas HC: T cell subsets in patients with acute and chronic HBV infection,
primary biliary cirrhosis and alcohol induced liver disease. Int J
Immunopharmacol 1981, 3(3): 301–305.
264. Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, Baba
TW, Ruprecht RM, Kupfer A: Apoptosis occurs predominantly in bystander cells
and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat
Med 1995; 1(2):129-134.
265. Matloubian M, Concepcion RJ, Ahmed R: CD4+ T cells are required to sustain
CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 1994;
68(12): 8056-8063.
266. Phares TW, Stohlman SA, Hwang M, Min B, Hinton DR, Bergmann CC: CD4 T
cells promote CD8 T cell immunity at the priming and effector site during viral
encephalitis. J Virol 2012; 86(5): 2416-2427.
267. Walker CM, Moody DJ, Stites DP, Levy JA: CD8+ lymphocytes can control HIV
infection in vitro by suppressing virus replication. Science 1986;
234(4783):1563-1566.
268. Tsubota H, Lord CI, Watkins DI, Morimoto C, Letvin NL: A cytotoxic T
lymphocyte inhibits acquired immunodeficiency syndrome virus replication in
peripheral blood lymphocytes. J Exp Med. 1989; 169(4): 1421–1434.
269. Pantaleo G: Mechanisms of CD8+ cell dysfunction in HIV infection. En: Fauci AS,
Schnittman SM, Poli G, Koenig S, Pantaleo G: NIH conference.
160

Immunopathogenic mechanisms in human immunodeficiency virus (HIV)
infection. Ann Intern Med 1991; 114(8):678-693
270. Funaro A, Spagnoli GC, Ausiello CM, Alessio M, Roggero S, Delia D, Zaccolo M,
Malavasi F: Involvement of the multilineage CD38 molecule in a unique
pathway of cell activation and proliferation. J Immunol 1990; 145(8): 2390-
2396.
271. Kestens L, Vanham G, Gigase P, Young G, Hannet I, Vanlangendonck F, Hulstaert
F, Bach BA: Expression of activation antigens, HLA-DR and CD38, on CD8
lymphocytes during HIV-1 infection. AIDS 1992; 6(8):793-797.
272. Subauste CS, Wessendarp M: Human dendritic cells discriminate between
viable and killed Toxoplasma gondii tachyzoites: dendritic cell activation after
infection with viable parasites results in CD28 and CD40 ligand signaling that
controls IL-12-dependent and -independent T cell production of IFN-gamma. J
Immunol 2000; 165(3):1498-1505.
273. Subauste CS, de Waal Malefyt R, Fuh F: Role of CD80 (B7.1) and CD86 (B7.2) in
the immune response to an intracellular pathogen. J Immunol 1998;
160(4):1831-1840.
274. Yang TH, Aosai F, Norose K, Ueda M, Yano A: Differential regulation of HLA-DR
expression and antigen presentation in Toxoplasma gondii-infected melanoma
cells by interleukin 6 and interferon gamma. Microbiol Immunol 1996;
40(6):443-449.
275. Lüder CG, Lang C, Giraldo-Velasquez M, Algner M, Gerdes J, Gross U:
Toxoplasma gondii inhibits MHC class II expression in neural antigen-
presenting cells by down-regulating the class II transactivator CIITA. J
Neuroimmunol 2003; 134(1-2):12-24.
276. Bhadra R, Gigley JP, Weiss LM, Khan IA: Control of Toxoplasma reactivation by
rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc
Natl Acad Sci USA 2011; 108(22):9196-9201.
277. Weiss A, Samelson LE: T-lymphocyte activation. En: Paul WE (Eds.);
Fundamental Immunology, 5th ed., Filadelfia: Lippincott Williams & Wilkins,
2003; 321-363.
161

278. Khoury SJ, Sayegh MH: The roles of the new negative T cells costimulatory
pathways in regulating autoimmunity. Immunity 2004; 20:529-538.
279. Wilson EH, Harris TH, Mrass P, John B, Tait ED, Wu GF, Pepper M, Wherry EJ,
Dzierzinski F, Roos D, Haydon PG, Laufer TM, Weninger W, Hunter CA:
Behavior of parasite-specific effector CD8+ T cells in the brain and visualization
of a kinesis-associated system of reticular fibers. Immunity 2009; 30(2):300-
311.
280. Bhadra R, Gigley JP, Khan IA: PD-1-mediated attrition of polyfunctional
memory CD8+ T cells in chronic toxoplasma infection. J Infect Dis 2012;
206(1):125-134.
281. Surh CD, Sprent J: Regulation of mature T cell homeostasis. Semin Immunol
2005; 17(3):183-191.
282. Fry TJ, Mackall CL: The many faces of IL-7: from lymphopoiesis to peripheral T
cell maintenance. J Immunol 2005; 174(11):6571-6576.
283. Koesters SA, Alimonti JB, Wachihi C, Matu L, Anzala O, Kimani J, Embree JE,
Plummer FA, Fowke KR: IL-7Ralpha expression on CD4+ T lymphocytes
decreases with HIV disease progression and inversely correlates with immune
activation. Eur J Immunol 2006; 36(2):336-44.
284. Dunham RM, Cervasi B, Brenchley JM, Albrecht H, Weintrob A, Sumpter B,
Engram J, Gordon S, Klatt NR, Frank I, Sodora DL, Douek DC, Paiardini M,
Silvestri G: CD127 and CD25 expression defines CD4+ T cell subsets that are
differentially depleted during HIV infection. J Immunol 2008; 180(8):5582-
5592.
285. Jiang Q, Li WQ, Hofmeister RR, Young HA, Hodge DR, Keller JR, Khaled AR,
Durum SK: Distinct regions of the Interleukin-7 Receptor regulate different
Bcl2 family members. Mol Cell Biol 2004; 24(14): 6501–6513.
286. Kiazyk SA, Fowke KR: Loss of CD127 expression links immune activation and
CD4 (+) T cell loss in HIV infection. Trends Microbiol 2008; 16(12):567-573.
287. Lanzavecchia A, Sallusto F: Understanding the generation and function of
memory T cell subsets. Curr Opin Immunol 2005; 17(3):326-332.
162

288. Ladell K, Hellerstein MK, Cesar D, Busch R, Boban D, McCune JM: Central
memory CD8+ T cells appear to have a shorter lifespan and reduced abundance
as a function of HIV disease progression. J Immunol 2008; 180(12):7907-7918.
289. Kasper LH, Matsuura T, Khan IA: IL-7 stimulates protective immunity in mice
against the intracellular pathogen, Toxoplasma gondii. J Immunol 1995;
155(10):4798-4804.
290. Bhadra R, Guan H, Khan IA: Absence of both IL-7 and IL-15 severely impairs the
development of CD8 T cell response against Toxoplasma gondii. PLoS One
2010; 5(5):e10842.
291. Opferman JT, Korsmeyer SJ: Apoptosis in the development and maintenance of
the immune system. Nat Immunol 2003; 4(5):410-415.
292. Nash PB, Purner MB, Leon RP, Clarke P, Duke RC, Curiel TJ: Toxoplasma gondii-
infected cells are resistant to multiple inducers of apoptosis. J Immunol 1998;
160(4):1824-1830.
293. Vutova P, Wirth M, Hippe D, Gross U, Schulze-Osthoff K, Schmitz I, Lüder CG:
Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant
processing and degradation of caspase 8. Cell Microbiol 2007; 9(6):1556-1570.
294. Goebel S, Gross U, Lüder CG: Inhibition of host cell apoptosis by Toxoplasma
gondii is accompanied by reduced activation of the caspase cascade and
alterations of poly(ADP-ribose) polymerase expression. J Cell Sci 2001; 114(Pt
19):3495-3505.
295. Keller P, Schaumburg F, Fischer SF, Häcker G, Gross U, Lüder CG: Direct
inhibition of cytochrome c-induced caspase activation in vitro by Toxoplasma
gondii reveals novel mechanisms of interference with host cell apoptosis. FEMS
Microbiol Lett 2006; 258(2):312-319.
296. Akbar AN, Terry L, Timms A, Beverley PCL, Janossy G: Loss of CD45R and gain
of UCHL1 reactivity is a feature of primed T cells. J. Immunol 1988; 140: 2171-
2178.
163

297. Sallusto F, Geginat J, Lanzavecchia A: Central memory and effector memory T
cell subsets: function, generation, and maintenance. Annu Rev Immunol 2004;
22:745-763.
298. Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, Appay V,
Rizzardi GP, Fleury S, Lipp M, Förster R, Rowland-Jones S, Sékaly RP, McMichael
AJ, Pantaleo G: Skewed maturation of memory HIV-specific CD8 T lymphocytes.
Nature 2001; 410(6824):106-111.
299. Paiardini M, Cervasi B, Albrecht H, Muthukumar A, Dunham R, Gordon S,
Radziewicz H, Piedimonte G, Magnani M, Montroni M, Kaech SM, Weintrob A,
Altman JD, Sodora D, Feinberg MB, Silvestri G: Loss of CD127 expression
defines an expansion of effector CD8+ T cells in HIV-infected individuals. J
Immunol 2005; 174: 2900–2909.
300. Alfonzo M, Blanc D, Troadec C, Huerre M, Eliaszewicz M, Gónzalez G, Koyanagi
Y, Scott-Algara D: Temporary restoration of immune response against
Toxoplasma gondii in HIV-infected individuals after HAART, as studied in the
hu-PBMC-SCID mouse model. Clin Exp Immunol 2002; 129(3): 411–419.
301. Lejeune M, Miró JM, Haumont M, Martínez E, Martínez C, Claramente X, Morillas
S, García F, Plana M, Valls ME, Fumarola T, Jacquet A, Gatell JM, Gallart T: T-cell
response to Toxoplasma gondii (Tg) antigens discriminates healthy individuals
with and without chronic infection. Its interest to evaluate the reconstitution of
Tg-specific CD4+ T cells in Tg-coinfected AIDS patients receiving antiretroviral
therapy. Inmunología 2001; 20(1):1-11. Disponible en:
http://revista.inmunologia.org/Upload/Articles/5/2/521.pdf.
302. Lejeune M, Miró JM, De Lazzari E, García F, Claramonte X, Martínez E, Ribera E,
Arrizabalaga J, Arribas JR, Domingo P, Ferrer E, Plana M, Valls ME, Podzamczer
D, Pumarola T, Jacquet A, Mallolas J, Gatell JM, Gallart T, Spanish Toxoplasma
gondii Study Group: Restoration of T cell responses to Toxoplasma gondii after
successful combined antiretroviral therapy in patients with AIDS with previous
toxoplasmic encephalitis. Clin Infect Dis 2011; 52(5):662-670.
303. Russell JH, Ley TJ: Lymphocyte-mediated cytotoxicity. Annu Rev Immunol
2002; 20:323-370.
164

304. Lubaki NM, Shepherd ME, Brookmeyer RS, Hon H, Quinn TC, Kashamuka M,
Johnson M, Gottle R, Devers J, Lederman HM, Bollinger RC: HIV-1-specific
cytolytic T-lymphocyte activity correlates with lower viral load, higher CD4
count, and CD8+CD38-DR- phenotype: comparison of statistical methods for
measurement. J Acquir Immune Defic Syndr. 1999; 22(1):19-30.
305. Suzuki Y, Wang X, Jortner BS, Payne L, Ni Y, Michie SA, Xu B, Kudo T, Perkins S:
Removal of Toxoplasma gondii cysts from the brain by perforin-mediated
activity of CD8+ T cells. Am J Pathol. 2010; 176(4):1607-1613.
306. Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM,
Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman
MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR: Perforin expression
directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control.
PLoS Pathog 2010 May 27; 6(5):e1000917.
307. Alimonti JB, Ball TB, Fowke KR: Mechanisms of CD4+ T lymphocyte cell death
in human immunodeficiency virus infection and AIDS. J Gen Virol 2003;
84(7):1649-1661.
308. Green DR, Droin N, Pinkoski M: Activation-induced cell death in T cells.
Immunol Rev 2003; 193:70-81.
309. Clerici M: Beyond IL-17: new cytokines in the pathogenesis of HIV infection.
Curr Opin HIV AIDS 2010; 5(2):184-188.
310. Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley LD: Acute
toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol
2001; 167(8):4574-4584.
311. Murray HW, Rubin BY, Masur H, Roberts RB: Impaired production of
lymphokines and immune (gamma) interferon in the acquired
immunodeficiency syndrome. N Engl J Med 1984; 310(14):883-889.
312. Zhang JY, Zhang Z, Wang X, Fu JL, Yao J, Jiao Y, Chen L, Zhang H, Wei J, Jin L, Shi
M, Gao GF, Wu H, Wang FS: PD-1 up-regulation is correlated with HIV-specific
memory CD8+ T-cell exhaustion in typical progressors but not in long-term
nonprogressors. Blood 2007; 109(11):4671-4678.
165

313. Riley JL, June CH: The road to recovery: translating PD-1 biology into clinical
benefit. Trends Immunol 2007; 28(2):48-50.
314. Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, Porichis F, Le
Gall S, Waring MT, Moss K, Jessen H, Pereyra F, Kavanagh DG, Walker BD,
Kaufmann DE: IL-10 is up-regulated in multiple cell types during viremic HIV
infection and reversibly inhibits virus-specific T cells. Blood. 2009; 114(2):346-
356.
315. Andrade RM, Lima PG, Filho RG, Hygino J, Milczanowski SF, Andrade AF, Lauria
C, Brindeiro R, Tanuri A, Bento CA: Interleukin-10-secreting CD4 cells from
aged patients with AIDS decrease in-vitro HIV replication and tumour necrosis
factor alpha production. AIDS. 2007; 21(13):1763-1770.
316. Dupont CD, Christian DA, Hunter CA: Immune response and immunopathology
during toxoplasmosis. Semin Immunopathol 2012; 34(6):793-813.
317. Schlüter D, Kwok LY, Lütjen S, Soltek S, Hoffmann S, Körner H, Deckert M: Both
lymphotoxin-alpha and TNF are crucial for control of Toxoplasma gondii in the
central nervous system. J Immunol 2003; 170(12):6172-6182.
318. Escobar Guevara EE, Alfonzo Díaz MA, Fernández-Mestre M, Camacho
Velásquez JC, Roldán Dávila YB, Alarcón de Noya B, De Quesada ME:
HIV/Toxoplasma gondii co-infected patients produce lower levels of IFN-γ in
response to T. gondii antigens, even in the asymptomatic stage of viral
infection. 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention,
2009; Abstract no. WELBA104. (Disponible en:
http://www.iasociety.org/Default.aspx?pageId=11&abstractId=200722748).
319. Escobar-Guevara EE, Alfonzo-Díaz MA, Camacho-Velásquez JC, Roldán-Dávila
YB, Alarcón de Noya B: HIV/Toxoplasma gondii coinfected patients modify their
IL-2 and IL-10 production in response to T. gondii and HIV antigens, even in the
early stage of viral infection. 9th Latin American Congress of Immunology,
2009; Abstract No. 354.
320. De Quesada ME, Marín H, Fuentes Alvarado YJ, Escobar Guevara EE, Roldán
Dávila YB, Alfonzo Díaz MA: Disorders on attention, short-term memory and
executive functions in HIV/Toxoplasma gondii co-infected patients, in the
asymptomatic stage of viral infection. 6th IAS Conference on HIV Pathogenesis,
166

Treatment and Prevention, 2011; Abstract no. CDB224. (Disponible en:
http://www.iasociety.org/Default.aspx?pageId=11&abstractId=200742783).
321. De Quesada ME, Fuentes Alvarado YJ, Marín H, Escobar Guevara EE, Roldán
Dávila YB, Alfonzo Díaz MA: Visual and auditory event related potentials in
HIV/Toxoplasma gondii co-infected patients, in the asymptomatic stage of viral
infection. 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention,
2011; Abstract no. CDB225. (Disponible en:
http://www.iasociety.org/Default.aspx?pageId=11&abstractId=200742799).
322. Escobar-Guevara E, Alfonzo-Díaz M: HIV induces a pro-
inflammatory/neurotoxic response in primary cultures of nervous cells, even
without infection. Symposium “30 Years of HIV Science: Imagine the Future”,
Institute Pasteur, 2013; Abstract No. 70/14PS. (Disponible en:
http://www.30yearshiv.org/Images/Public/30Y-HIV2013-Abstract-book.pdf,
Pág. 93).
323. Schlüter D, Hein A, Dörries R, Deckert-Schlüter M: Different subsets of T cells in
conjunction with natural killer cells, macrophages, and activated microglia
participate in the intracerebral immune response to Toxoplasma gondii in
athymic nude and immunocompetent rats. Am J Pathol 1995; 146(4):999-1007.
324. Suzuki Y: Immunopathogenesis of cerebral toxoplasmosis. J Infect Dis 2002;
186(Suppl 2): S234-S240.
325. Fischer HG, Bonifas U, Reichmann G: Phenotype and functions of brain
dendritic cells emerging during chronic infection of mice with Toxoplasma
gondii. J Immunol 2000; 164(9):4826-34.
326. Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Giuffrida AM:
Nitric oxide in the central nervous system: neuroprotection versus
neurotoxicity. Nat Rev Neurosci 2007; 8(10): 766-775.
327. Wei T, Chen C, Hou J, Xin W, Mori A: Nitric oxide induces oxidative stress and
apoptosis in neuronal cells. Biochim Biophys Acta 2000; 1498(1):72-79.
328. Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA: Apoptosis and
necrosis: two distinct events induced, respectively, by mild and intense insults
167

with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures.
Proc Natl Acad Sci USA 1995; 92(16): 7162–7166.
329. Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J,
Nath A, Mattson MP: Perturbation of sphingolipid metabolism and ceramide
production in HIV-dementia. Ann Neurol. 2004; 55(2):257-267.
330. Mattson MP, Haughey NJ, Nath A: Cell death in HIV dementia. Cell Death Differ
2005; 12(Suppl 1):893-904.
331. Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton
SA: S-nitrosylation of matrix metalloproteinases: signaling pathway to
neuronal cell death. Science 2002; 297(5584):1186-1190.
332. Liuzzi GM, Mastroianni CM, Santacroce MP, Fanelli M, D'Agostino C, Vullo V,
Riccio P: Increased activity of matrix metalloproteinases in the cerebrospinal
fluid of patients with HIV-associated neurological diseases. J Neurovirol 2000;
6(2):156-163.
333. Almeida A, Bolaños JP, Medina JM: Nitric oxide mediates glutamate-induced
mitochondrial depolarization in rat cortical neurons. Brain Res 1999;
816(2):580-586.
334. Jiang ZG, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, Zheng J,
Gendelman HE, Markey SP: Glutamate is a mediator of neurotoxicity in
secretions of activated HIV-1-infected macrophages. J Neuroimmunol 2001;
117: 97–107.
335. Erdmann NB, Whitney NP, Zheng J: Potentiation of excitotoxicity in HIV-1
Associated Dementia and the significance of glutaminase. Clin Neurosci Res
2006; 6(5):315-328.
336. Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, Lipton SA:
Cytokine-stimulated, but not HIV-infected, human monocytederived
macrophages produce neurotoxic levels of L-cysteine. J Immunol 2000; 164:
4265–4270.
168

337. Pulliam L, Zhou M, Stubblebine M, Bitler CM: Differential modulation of cell
death proteins in human brain cells by tumor necrosis factor alpha and platelet
activating factor. J Neurosci Res 1998; 54(4):530-538.
338. Talley AK, Dewhurst S, Perry SW, Dollard SC, Gummuluru S, Fine SM, New D,
Epstein LG, Gendelman HE, Gelbard HA: Tumor necrosis factor alpha-induced
apoptosis in human neuronal cells: protection by the antioxidant N-
acetylcysteine and the genes Bcl-2 and crma. Mol Cell Biol 1995; 15: 2359–
2366.
339. Shi B, De Girolami U, He J, Wang S, Lorenzo A, Busciglio J, Gabuzda D: Apoptosis
induced by HIV-1 infection of the central nervous system. J Clin Invest. 1996;
98(9): 1979–1990.
340. Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F:
Neuronal apoptosis in HIV infection in adults. Neuropathol Appl Neurobiol
1995 Jun; 21(3):218-227.
341. Wesselingh SL, Takahashi K, Glass JD, McArthur JC, Griffin JW, Griffin DE:
Cellular localization of tumor necrosis factor mRNA in neurological tissue from
HIV-infected patients by combined reverse transcriptase/polymerase chain
reaction in situ hybridization and immunohistochemistry. J Neuroimmunol
1997; 74: 1–8.
342. Erta M, Quintana A, Hidalgo J: Interleukin-6, a major cytokine in the central
nervous system. Int J Biol Sci 2012; 8(9):1254-1266.
343. Laurenzi MA, Sidén A, Persson MA, Norkrans G, Hagberg L, Chiodi F:
Cerebrospinal fluid interleukin-6 activity in HIV infection and inflammatory
and noninflammatory diseases of the nervous system. Clin Immunol
Immunopathol 1990; 57(2):233-241.
344. Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW:
Interleukin-6 facilitates lipopolysaccharide-induced disruption in working
memory and expression of other proinflammatory cytokines in hippocampal
neuronal cell layers. J Neurosci 2006; 26(42):10709-10716.
169

345. Elderkin-Thompson V, Irwin MR, Hellemann G, Kumar A: Interleukin-6 and
memory functions of encoding and recall in healthy and depressed elderly
adults. Am J Geriatr Psychiatry 2012; 20(9):753-763.
346. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A: Interleukin-10 and the
interleukin-10 receptor. Annu Rev Immunol 2001; 19:683-765.
347. Couper KN, Blount DG, Riley EM: IL-10: the master regulator of immunity to
infection. J Immunol 2008; 180(9):5771-5777.
348. Ledeboer A, Brevé JJ, Poole S, Tilders FJ, Van Dam AM: Interleukin-10,
interleukin-4, and transforming growth factor-beta differentially regulate
lipopolysaccharide-induced production of pro-inflammatory cytokines and
nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 2000;
30(2):134-142.
349. Sheng WS, Hu S, Kravitz FH, Peterson PK, Chao CC: Tumor necrosis factor alpha
upregulates human microglial cell production of interleukin-10 in vitro. Clin
Diagn Lab Immunol 1995; 2(5):604-608.
350. Blumenthal R, Clague MJ, Durell SR, Epand RM: Membrane fusion. Chem Rev
2003; 103(1):53-69.
351. Garg H, Blumenthal R: Role of HIV gp41 mediated fusion/hemifusion in
bystander apoptosis. Cell Mol Life Sci 2008; 65(20):3134-3144.
352. Perfettini JL, Castedo M, Roumier T, Andreau K, Nardacci R, Piacentini M,
Kroemer G: Mechanisms of apoptosis induction by the HIV-1 envelope. Cell
Death Differ 2005; 12(Suppl 1):916-923.
353. Garg H, Blumenthal R: HIV gp41-induced apoptosis is mediated by caspase-3-
dependent mitochondrial depolarization, which is inhibited by HIV protease
inhibitor nelfinavir. J Leukoc Biol 2006; 79(2):351-362.
354. Sung JH, Shin SA, Park HK, Montelaro RC, Chong YH: Protective effect of
glutathione in HIV-1 lytic peptide 1-induced cell death in human neuronal cells.
J Neurovirol 2001; 7(5):454-465.
170

355. Kort JJ: Impairment of excitatory amino acid transport in astroglial cells
infected with the human immunodeficiency virus type 1. AIDS Res Hum
Retroviruses. 1998;14(15):1329-1339.
356. Caffrey M, Braddock DT, Louis JM, Abu-Asab MA, Kingma D, Liotta L, Tsokos M,
Tresser N, Pannell LK, Watts N, Steven AC, Simon MN, Stahl SJ, Wingfield PT,
Clore GM: Biophysical characterization of gp41 aggregates suggests a model for
the molecular mechanism of HIV-associated neurological damage and
dementia. J Biol Chem 2000; 275(26):19877-19882.
357. Schwenk J, Cruz-Sanchez F, Gosztonyi G, Cervos-Navarro J: Spongiform
encephalopathy in a patient with acquired immune deficiency syndrome
(AIDS). Acta Neuropathol 1987; 74(4):389-392.
358. Artigas J, Niedobitek F, Grosse G, Heise W, Gosztonyi G: Spongiform
encephalopathy in AIDS dementia complex: report of five cases. J Acquir
Immune Defic Syndr 1989; 2(4):374-381.
359. Rostasy K, Monti L, Yiannoutsos C, Kneissl M, Bell J, Kemper TL, Hedreen JC,
Navia BA: Human immunodeficiency virus infection, inducible nitric oxide
synthase expression, and microglial activation: pathogenetic relationship to the
acquired immunodeficiency syndrome dementia complex. Ann Neurol 1999;
46:207–216.
360. Kaul M, Lipton SA: The NMDA Receptor - Its Role in neuronal apoptosis and
HIV-Associated Dementia. NeuroAids 2000; 3(6). (Disponible en:
http://aidscience.org/neuroaids/zones/articles/2000/11/NMDA/).
361. Rostasy K, Egles C, Chauhan A, Kneissl M, Bahrani P, Yiannoutsos C, Hunter DD,
Nath A, Hedreen JC, Navia BA.: SDF-1alpha is expressed in astrocytes and neurons
in the AIDS dementia complex: an in vivo and in vitro study. J Neuropathol Exp
Neurol 2003; 62(6):617-626.
362. Langford D, Sanders VJ, Mallory M, Kaul M, Masliah E: Expression of stromal cell-
derived factor 1alpha protein in HIV encephalitis. J Neuroimmunol. 2002; 127(1-
2):115-126.
171

363. Johnston JB, Jiang Y, van Marle G, Mayne MB, Ni W, Holden J, McArthur JC,
Power C: Lentivirus infection in the brain induces matrix metalloproteinase
expression: role of envelope diversity. J Virol 2000; 74(16):7211-7220.
364. Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM, Power C: HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci 2003; 6(10):1064-1071.
365. Fontana G, Valenti L, Raiteri M: Gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J Neurosci Res 1997; 49(6):732-738.
366. Lo TM, Fallert CJ, Piser TM, Thayer SA: HIV-1 envelope protein evokes intracellular calcium oscillations in rat hippocampal neurons. Brain Res. 1992; 594(2):189-196.
367. Patton HK, Zhou ZH, Bubien JK, Benveniste EN, Benos DJ: gp120-induced alterations of human astrocyte function: Na(+)/H(+) exchange, K(+) conductance, and glutamate flux. Am J Physiol Cell Physiol. 2000; 279(3):C700-C708.
368. Nath A: Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002; 186(Suppl 2):S193-S198.
369. Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A: Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995; 37(3):373-380.
370. Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G: HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001; 288(2):301-308.
371. Kruman II, Nath A, Maragos WF, Chan SL, Jones M, Rangnekar VM, Jakel RJ, Mattson MP: Evidence that Par-4 participates in the pathogenesis of HIV encephalitis. Am J Pathol. 1999; 155(1):39-46.
372. Chen P, Mayne M, Power C, Nath A: The Tat protein of HIV-1 induces tumor necrosis factor-alpha production. Implications for HIV-1-associated neurological diseases. J Biol Chem 1997; 272(36):22385-22388.
172

373. Kutsch O, Oh J, Nath A, Benveniste EN: Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 tat in astrocytes. J Virol 2000; 74(19):9214-9221.
374. Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C: HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol 2001; 49(2):230-241.
375. Bukrinsky M, Adzhubei A: Viral protein R of HIV-1. Rev Med Virol 1999; 9(1):39-49.
376. Piller SC, Jans P, Gage PW, Jans DA: Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc Natl Acad Sci USA 1998; 95(8):4595-4600.
377. Strebel K: Virus-host interactions: role of HIV proteins Vif, Tat, and Rev. AIDS 2003; 17(Suppl 4):S25-S34.
378. Mabrouk K, Van Rietschoten J, Vives E, Darbon H, Rochat H, Sabatier JM: Lethal neurotoxicity in mice of the basic domains of HIV and SIV Rev proteins. Study of these regions by circular dichroism. FEBS Lett 1991; 289(1):13-17.
379. Trillo-Pazos G, McFarlane-Abdulla E, Campbell IC, Pilkington GJ, Everall IP: Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res 2000; 864(2):315-326.
380. Sporer B, Koedel U, Paul R, Kohleisen B, Erfle V, Fontana A, Pfister HW: Human immunodeficiency virus type-1 Nef protein induces blood-brain barrier disruption in the rat: role of matrix metalloproteinase-9. J Neuroimmunol 2000; 102(2):125-130.
381. Escobar-Guevara E, Alfonzo-Díaz M: VIH-1 inhibe la replicación de taquizoítos de Toxoplasma gondii en cultivos primarios de células nerviosas. Memorias de LXII Convención Anual de AsoVAC, UNIMET, Caracas 2012 (Disponible en: https://docs.google.com/file/d/0B5kEi2NiEC1sY0t0Y1g4TXc0TTA/edit?pli=1; pág. 258).
382. Escobar E, Alfonzo M: VIH-1 inhibe el porcentaje de infección por Toxoplasma gondii en cultivos primarios de células nerviosas. Acta Cient Venez 2013; 63(Sup.1):72-73. (disponible en: http://asovaccarabobo.org.ve/pdf/librolxiii.pdf)
173

383. Carrero J, Escobar E, Fuentes Y, Alfonzo M: Producción de óxido nítrico e inducción de apoptosis en células nerviosas de feto de rata Sprague-Dawley incubadas con gp41 de VIH y Toxoplasma gondii. Acta Cient Venez 2011; 62(Sup. 1):64.
384. Khan IA, Schwartzman JD, Matsuura T, Kasper LH: A dichotomous role for nitric
oxide during acute Toxoplasma gondii infection in mice. Proc Natl Acad Sci USA
1997; 94(25): 13955–13960.
385. Escobar EE, Alfonzo MA: A more pro-inflammatory environment is generated
in nervous cells cultures in the simultaneous presence of HIV-1 and
Toxoplasma gondii, even with a lower parasite replication. Front Immunol
2013; Conference Abstract: 15th International Congress of Immunology.
(http://www.frontiersin.org/10.3389/conf.fimmu.2013.02.01122/event_abstr
act)
386. Benedetto N, Folgore A, Romano Carratelli C, Galdiero F: Effects of cytokines
and prolactin on the replication of Toxoplasma gondii in murine microglia. Eur
Cytokine Netw 2001; 12(2):348-358.
387. Escobar Guevara EE, Fuentes Alvarado YJ, Carrero Salas JA, Roldán Dávila YB,
Alarcón de Noya B, De Quesada ME, Alfonzo Díaz MA: Destruction of nervous
cells is potentiated in the simultaneous presence of gp41 and Toxoplasma
gondii. 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention
2011; Abstract no. WEPDA0105. (Disponible en:
http://www.iasociety.org/Default.aspx?pageId=11&abstractId=200742379).
388. Escobar-Guevara E, Alfonzo-Díaz M: Perfil de citoquinas de células nerviosas
cultivadas en presencia simultánea de VIH-1 y Toxoplasma gondii sugiere
ambiente más proinflamatorio y menos control de patógenos. Memorias de
LXII Convención Anual de AsoVAC. UNIMET, Caracas 2012 (Disponible en:
https://docs.google.com/file/d/0B5kEi2NiEC1sY0t0Y1g4TXc0TTA/edit?pli=1;
pág. 257).
389. Pollard AM, Knoll LJ, Mordue DG: The role of specific Toxoplasma gondii
molecules in manipulation of innate immunity. Trends Parasitol 2009;
25(11):491-494.
390. Mishra BB, Gundra UM, Teale JM: Expression and distribution of Toll-like
receptors 11–13 in the brain during murine neurocysticercosis. J
174

Neuroinflammation 2008; 5:53. (Disponible en:
http://www.jneuroinflammation.com/content/pdf/1742-2094-5-53.pdf)
391. Aloisi F, Penna G, Cerase J, Menéndez Iglesias B, Adorini L: IL-12 production by
central nervous system microglia is inhibited by astrocytes. J Immunol 1997;
159(4):1604-1612.
392. Taoufik Y, de Goër de Herve MG, Giron-Michel J, Durali D, Cazes E, Tardieu M,
Azzarone B, Delfraissy JF: Human microglial cells express a functional IL-12
receptor and produce IL-12 following IL-12 stimulation. Eur J Immunol 2001;
31(11):3228-3239.
393. Leng J, Butcher BA, Egan CE, Abi Abdallah DS, Denkers EY: Toxoplasma gondii
prevents chromatin remodeling initiated by TLR-triggered macrophage
activation. J Immunol 2009; 182(1): 489–497.
175

Curriculum Vitae
Nombre: Edwin Eliel Escobar Guevara
Lugar y fecha de nacimiento: Carúpano, Estado Sucre, Venezuela; 24 de Enero de
1966.
Nacionalidad: Venezolana.
Estudios realizados:
Especialista en Inmunología de Laboratorio (1997-2004): Universidad Central de
Venezuela, Caracas, Venezuela.
Licenciado en Bioanálisis (1984-1990: Universidad Central de Venezuela, Caracas,
Venezuela.
Cargos desempeñados:
Estudiante Graduado, Doctorado en Inmunología, (2006-2014), Centro de Medicina
Experimental, Instituto Venezolano de Investigaciones Científicas (IVIC).
Tutora: Dra. Mercedes Fernández Mestre.
Bioanalista, citometría de flujo (2000-2006), Laboratorio BioCell 220 C.A. Caracas,
Venezuela. Supervisor: Dr. Pedro Sánchez Llamozas.
Bioanalista (1998-2001), Policlínica CABISOGUARNAC, Caracas, Venezuela.
Supervisor: Coronel Gregorio Arocha.
Bioanalista (1990-1997), Dispensario Adventista, Caracas, Venezuela. Supervisor:
Lic. José Emmons.
Campos en que ha trabajado y/o publicado: Inmunología, hemato-oncología,
citometría de flujo, laboratorio clínico.
Honores y distinciones:
Beca IVIC (2011-2012), Instituto Venezolano de Investigaciones Científicas, Altos de
Pipe, Estado Miranda, Venezuela.
Beca Misión Ciencia (2006-2010), FONACIT, Ministerio del Poder Popular Para
Ciencia y Tecnología, Caracas, Venezuela.
Beca MSAS (1997-1998), Ministerio de Sanidad y Asistencia Social, Caracas,
Venezuela.