Download - Demencias preseniles

DEMENCIAS PRESENILES

DEFINICIÓN
• Demencia Presenil o temprana o de inicio precoz.
Generalmente con clara relación familiar. Comienza antes
de los 65 años, es de curso más rápido y no supera el 1%
de los casos en la mayoría de las estadísticas.

ENFERMEDAD DE ALZHEIMER FAMILIAR (EA)
• La enfermedad de Alzheimer se caracteriza por la acumulación en el
cerebro de depósitos de pequeños péptidos de diferentes tamaños
llamados beta-amiloide, que son derivados del fraccionamiento
proteolítico de la proteína APP.

• Karlinsky et al. (1992) informaron de una familia de Toronto con la
enfermedad de Alzheimer con herencia autosómica dominante.
• El trastorno se caracteriza por la aparición temprana de los déficits de
memoria.
• El análisis genético identificó una mutación en el gen APP(Proteína
precursora de beta amiloide)

BASES MOLECULARES DE EA
• El gen APP codifica un receptor de superficie celular.
• Contiene 18 exones y probablemente origina una familia de al
menos 8 isoformas transmembranales diferentes, las cuales se
diferencian por la presencia o ausencia de los exones 7, 8, 9 y 15.

• Las isoformas, que se expresan en las neuronas (695, 714, 751 y 770
a.a) que contienen el exón 15 son más amiloidogénicas y liberan
mucho más péptido ßA (42) que las isoformas no neuronales.
Menéndez et al. 2002. Péptido beta amiloide, proteína tau y enfermedad de Alzheimer

• Se ha reportado al menos la existencia de 6 mutaciones de cambio de
sentido, que se manifiesta en un cambio de aminoácido por otro.
Menéndez et al. 2002. Péptido beta amiloide, proteína tau y enfermedad de Alzheimer

En el estado normal, APP se
escinde inicialmente por α-
secretasa para generar sAPP y un
fragmento carboxiterminal C83.
La presencia de sAPP se asocia
con la señalización sináptica
normal, aprendizaje, memoria,
comportamientos emocionales y
la supervivencia neuronal.
http://www.cellsignal.com/common/content/content.jsp?id=pathways-alz
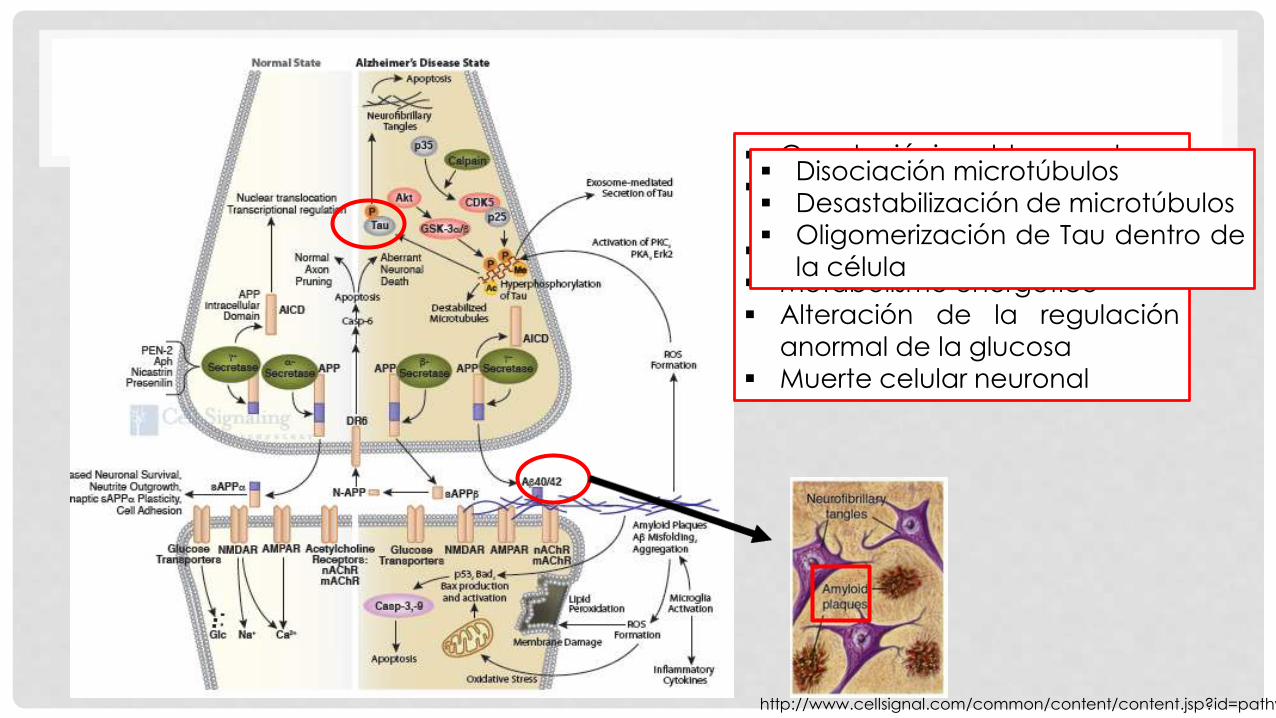
Canales iónicos bloqueados Alteración de la homeostasis
del calcio
Estrés oxidativo mitocondrial
Metabolismo energético
Alteración de la regulación
anormal de la glucosa
Muerte celular neuronal
Disociación microtúbulos
Desastabilización de microtúbulos
Oligomerización de Tau dentro de
la célula
http://www.cellsignal.com/common/content/content.jsp?id=pathways

INVESTIGACIONES RECIENTES
• Resulta que la APP es una oxidasa de hierro, cuyo trabajo es convertir
el hierro a una forma segura para su transporte o almacenamiento.
• Cuando APP no funciona correctamente, como lo hace en la
enfermedad del Alzheimer, los niveles de hierro dentro de las
neuronas llegan a niveles tóxicos.
Everett et al. 2014 Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer's disease peptide β-amyloid (1-42).

DEMENCIA PRESENIL
•Alteración de la memoria
•Apraxia, agnosia, afasia y perturbación en el funcionamiento ejecutivo
•Interferencia con las habilidades sociales, laborales e interpersonales
•Antes de los 65 años
Síndrome que
cursa con

AUMENTO EXPONENCIAL CON LA EDAD

ENFERMEDAD DE ALZHEIMER
• Causa más común de demencia 50-90%
• Alois Alzheimer (1864-1915)
demencia presenil que mostró atrofia cortical difusa, pérdida de
células nerviosas, las placas, y ovillos
• Tipo I es familiar
• Campion et al. (1999) prevalencia: de inicio precoz 41,2 y AD 5,3 por
cada 100.000 personas.
• De inicio precoz: < de 61 años y AD de aparición temprana: 3 casos
en 3 generaciones.
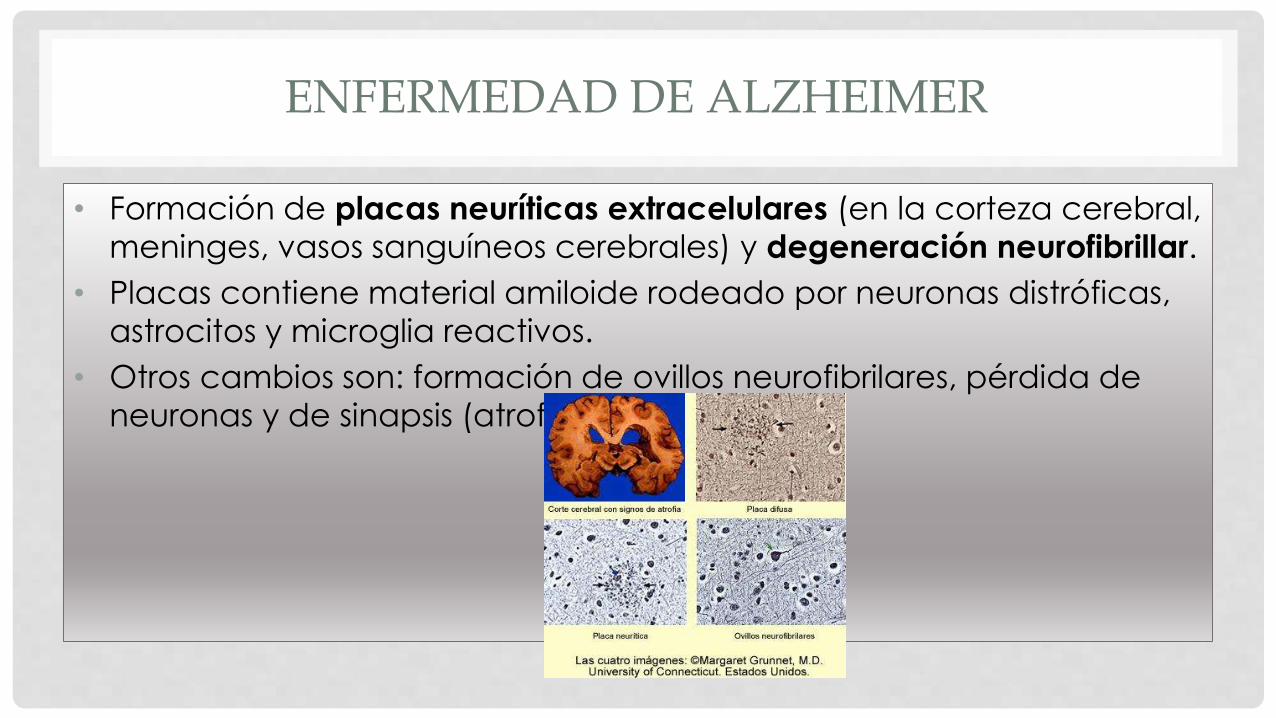
• Formación de placas neuríticas extracelulares (en la corteza cerebral,
meninges, vasos sanguíneos cerebrales) y degeneración neurofibrillar.
• Placas contiene material amiloide rodeado por neuronas distróficas,
astrocitos y microglia reactivos.
• Otros cambios son: formación de ovillos neurofibrilares, pérdida de
neuronas y de sinapsis (atrofia cerebral).
ENFERMEDAD DE ALZHEIMER
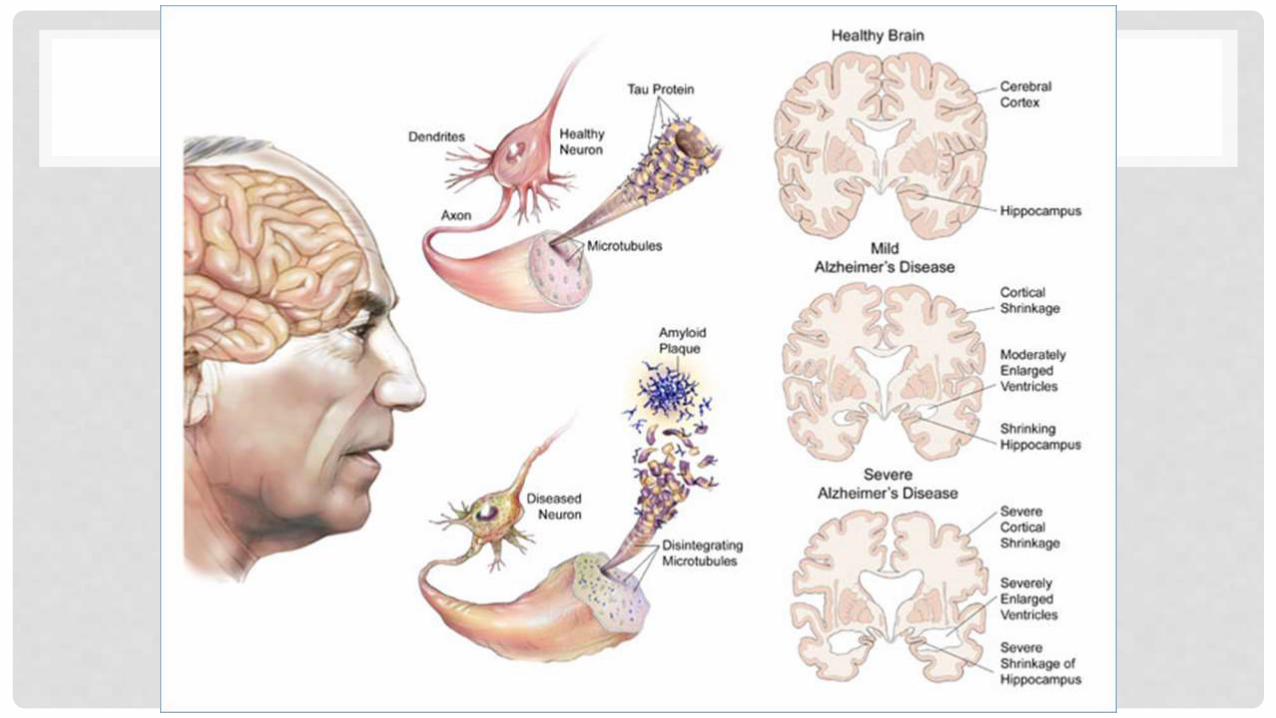
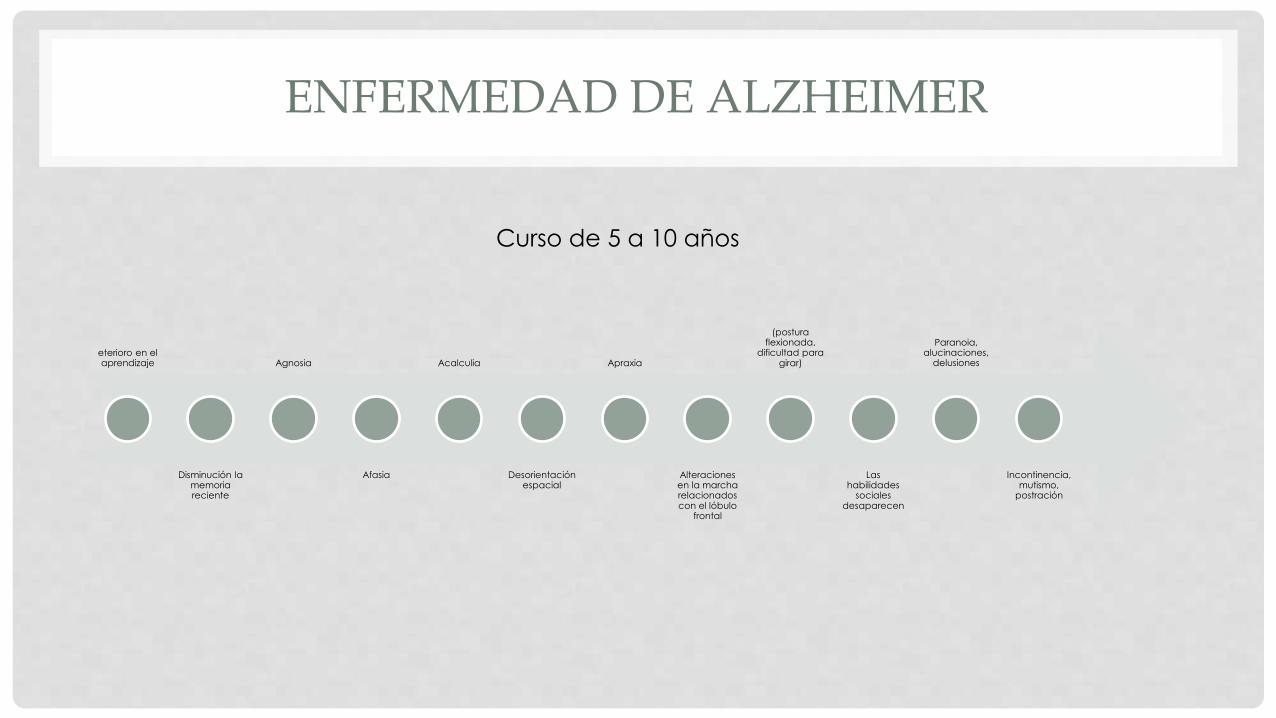
ENFERMEDAD DE ALZHEIMER
eterioro en el aprendizaje
Disminución la memoria reciente
Agnosia
Afasia
Acalculia
Desorientación espacial
Apraxia
Alteraciones en la marcha relacionados con el lóbulo
frontal
(postura flexionada,
dificultad para girar)
Las habilidades
sociales desaparecen
Paranoia, alucinaciones,
delusiones
Incontinencia, mutismo,
postración
Curso de 5 a 10 años

ENFERMEDAD DE ALZHEIMER

ENFERMEDAD DE ALZHEIMER


Reduccion de la acetiltransferasa de la colina
Disminucion de la acetil colina
Disminucion de neuronas mono aminergicas
Desbalance de Glutamato
ENFERMEDAD DE ALZHEIMER
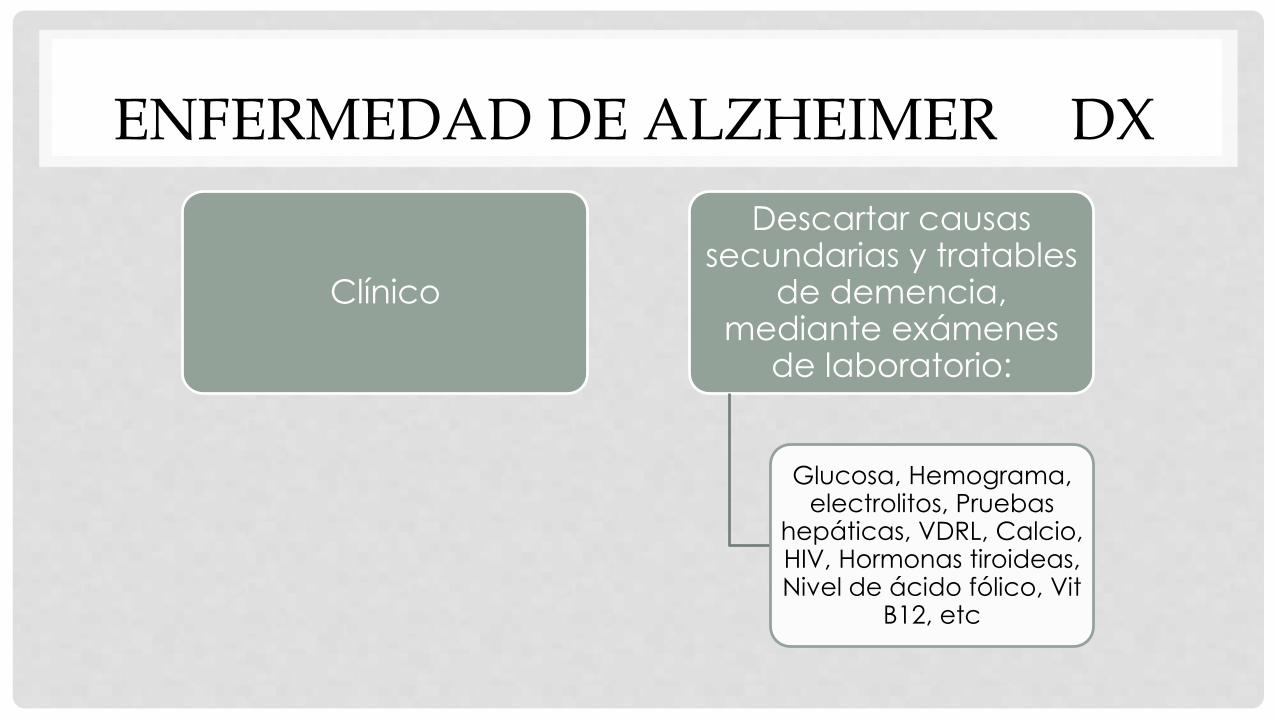
Clínico
Descartar causas secundarias y tratables
de demencia, mediante exámenes
de laboratorio:
Glucosa, Hemograma, electrolitos, Pruebas
hepáticas, VDRL, Calcio, HIV, Hormonas tiroideas, Nivel de ácido fólico, Vit
B12, etc
ENFERMEDAD DE ALZHEIMER DX

• TAC: Atrofia cortical frontal, temporal y
parietal. hidrocefalia
• MRI
• PET: disminucion del metabolismo en
lobulos temporal y parietal
• SPECT: disminucion del flujo sanguineo en
lobulos temporal y parietal
ENFERMEDAD DE ALZHEIMER

TRATAMIENTO
El objetivo es mejorar las funciones cognitivas,
mediante el uso de:
Anticolinesterásicos (Rivastigmina,
Donepezilo) que incrementan el nivel
de acetilcolina
Antioxidantes (Alfa tocoferol: Vit E) que disminuye la lesión neuronal mediado
por oxidantes

ENFERMEDAD DE PICK
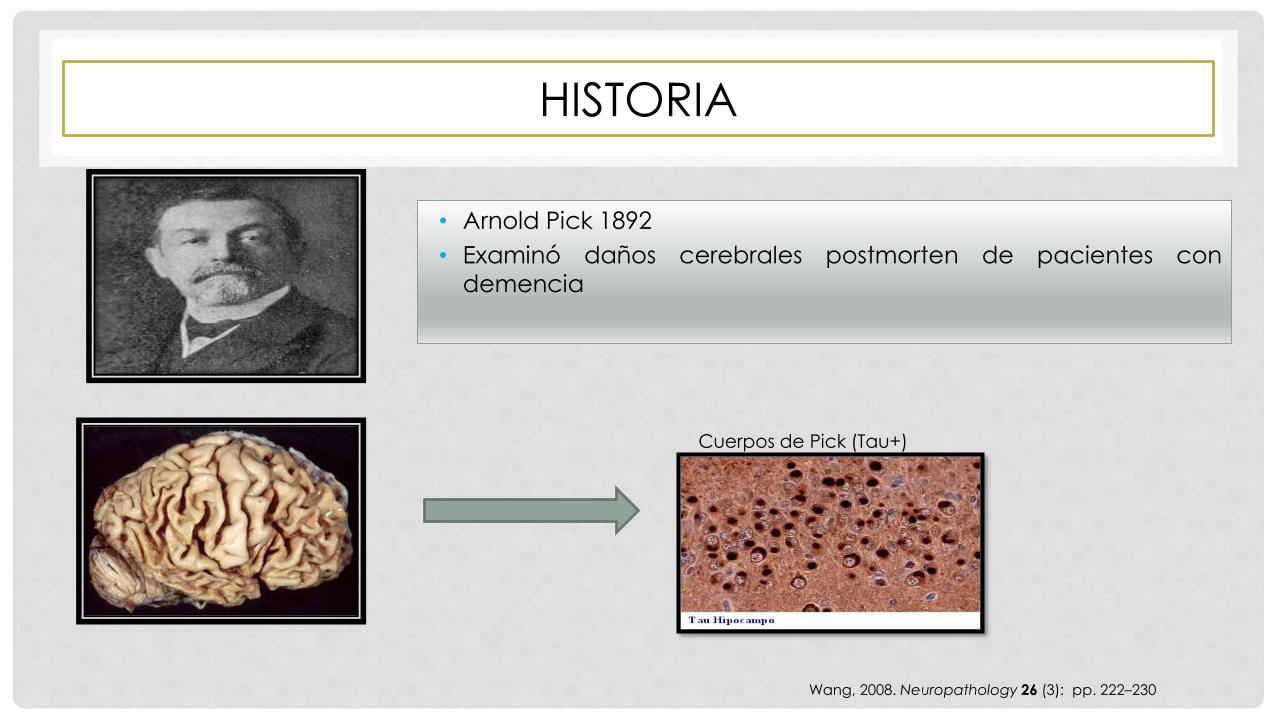
HISTORIA
• Arnold Pick 1892
• Examinó daños cerebrales postmorten de pacientes con
demencia
Wang, 2008. Neuropathology 26 (3): pp. 222–230
Cuerpos de Pick (Tau+)

GENÉTICA DE LA ENFERMEDAD DE PICK
Locus Gene
14q24.2 PSEN1
17q21.31 MAPT
Mecanismo molecular desconocido, algunos genes implicados
MIM #172700

PSEN1
GENÉTICA DE LA ENFERMEDAD DE PICK
60 kb
MIM #172700

Estructura proteica de la presenilina 1
Parte del complejo presenilina
Sitio catalítico de gamma
secretasa o cofactor?
Hyslop P, 2012; De Strooper B 2007
SNP G/T en exon 6 de PSEN1gly183-val (G183V)

COMPLEJO GAMMA SECRETASA
Generación de β amiloide a partir del precursor proteico amiloide (APP)
β amiloide
Agregados
amiloides
Formación de cuerpos de
pick?
Hyslop P, 2012
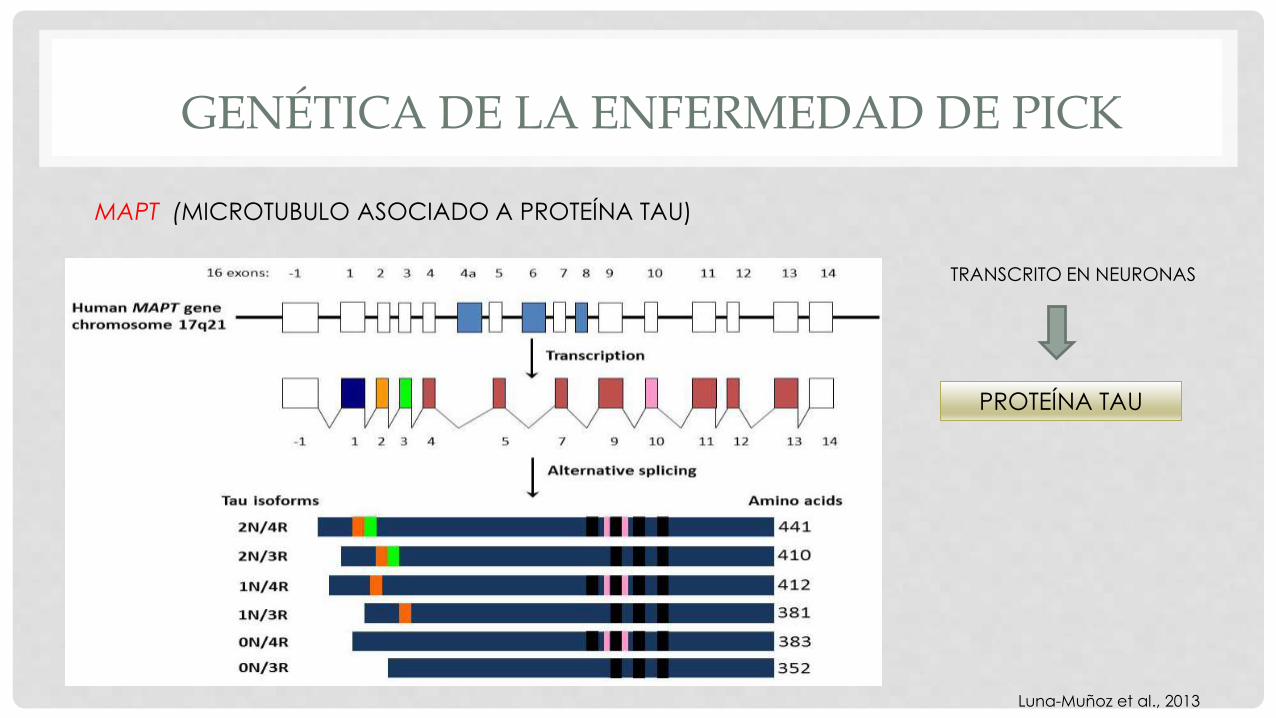
MAPT (MICROTUBULO ASOCIADO A PROTEÍNA TAU)
TRANSCRITO EN NEURONAS
PROTEÍNA TAU
Luna-Muñoz et al., 2013
GENÉTICA DE LA ENFERMEDAD DE PICK

FUNCIONES DE TAU
• Estabilidad de microtúbulos
• Transporte de nutrientes
Luna-Muñoz et al., 2013

MUTACIONES DE MAPT EN ENFERMEDAD DE PICK
• PROTEÍNA ANÓMALA
• Mutaciones reducen o aumentan la eficacia de tau conllevando a
enfermedad.
• Más de 50 mutaciones diferentes en el gen de tau se han asociado con
demencias frontotemporal hereditaria (FTD).
Mutación: G272V tau
Bronner IF et al., 2005
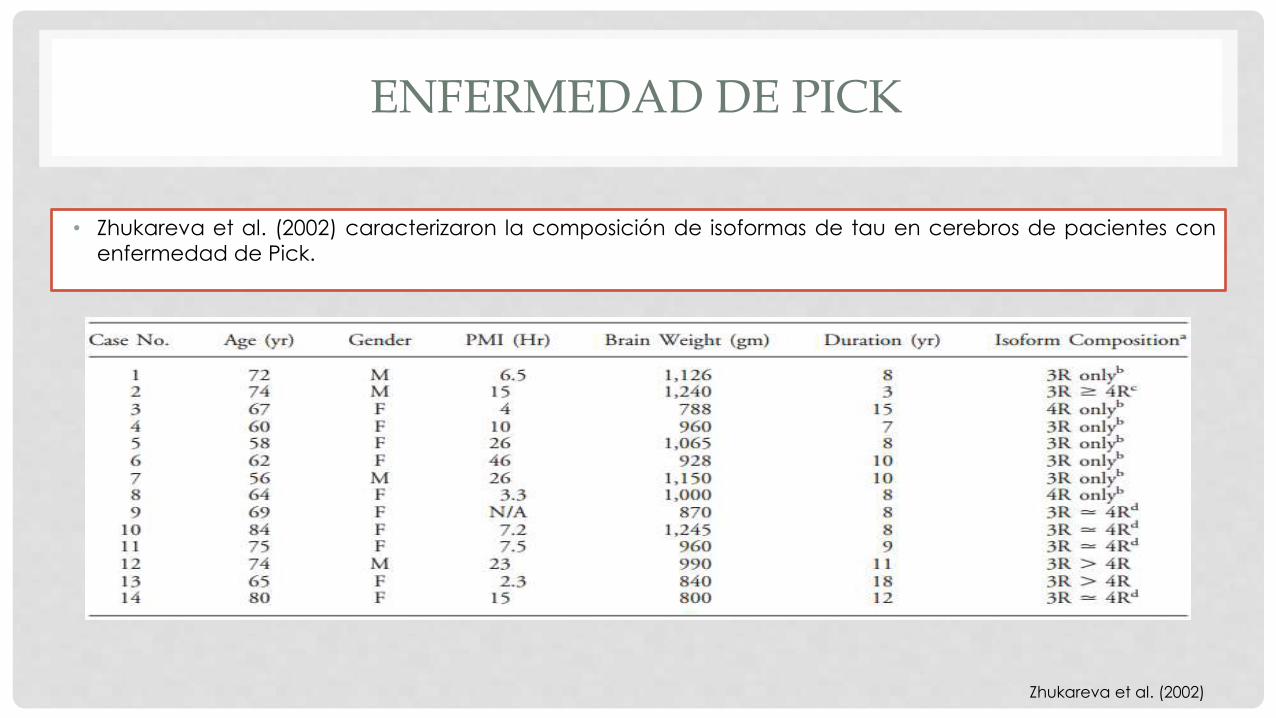
ENFERMEDAD DE PICK
• Zhukareva et al. (2002) caracterizaron la composición de isoformas de tau en cerebros de pacientes con
enfermedad de Pick.
Zhukareva et al. (2002)

DEMENCIA PRESENIL
•Alteración de la memoria
•Apraxia, agnosia, afasia y perturbación en el funcionamiento ejecutivo
•Interferencia con las habilidades sociales, laborales e interpersonales
•Antes de los 65 años
Síndrome que
cursa con

ENFERMEDAD DE PICK OMIM 172700
• Sanders 1939
AD
Casos aislados
• La menos frecuente
• Pertenece demencia frontotemporal
• Aparece entre al 4ta y 5ta década de la vida
• Edad de aparición y la muerta varia en entre 2 a 16 años.
• Cambios precoces y lentamente progresivos del carácter y alteraciones del comportamiento.
• Atrofia lobar del cerebro e inclusiones citoplasmáticas
• Pertenece a las demencias fronto temporales
• Deterioro de la inteligencia, de la memoria y del lenguaje, acompañado de apatía, de euforia y signos extrapiramidales.

ENFERMEDAD DE PICK
• Atrofia circunscrita del cerebro, lóbulo frontal,
temporal o parietal, con dilatación de los
ventrículos y afectación grave de la corteza
• Generalmente se afecta mas el hemisferio
izquierdo
• Microscópicamente: inclusiones Tau
hiperfosforilada argirófilas (cuerpos de Pick)
Células de pick (neuronas alargadas). En el
hipocampo hay pérdida de astroglia.
• Sin la aparición de placas neuríticas ni
degeneración neurofibrilar.

ENFERMEDAD DE PICK
• Nakamura et al.(1994) utilizó la tinción de anticuerpos para demostrar
la presencia de clatrina cadenas A y B, así como neurofilamentos,
kinesina y sinaptofisina, en los cuerpos de pick
• Esto sugirió deterioro del transporte axonal.
• Se describen 2 alteraciones gliales: Ovillos fibrilares gliales y gránulos
de proteína del complemento C4d en hipocampo

ENFERMEDAD DE PICK
• Kertesz en 2003 señalo:
• Que la demencia fronto temporal es la “enfermedad de Pick clínica”
y la enfermedad de Pick se limita a la comprobación de los cuerpos
de pick.

ENFERMEDAD DE PICK, MANIFESTACIONES PSIQUIÁTRICAS
Falta de motivación
Embotamiento emocional
Risa inapropiada
Apatía
Irritabilidad
Desinhibición
Comportamiento estereotipado
ecolalia
Hiperfagia
hiperoralia

DEMENCIA VASCULAR
15% de todas las demencias
Factores de riesgo: hipertensión, cardiopatías, diabetes y enfermedad cerebral vascular
Inicio abrupto y deterioro progresivo
El deterioro depende del área afectada
Hemorragia, trombosis o isquemia

DEMENCIA VASCULAR
Exageración de los reflejos tendinosos
profundos, respuesta de extensión plantar,
parálisis pseudobulbar
(hablar, masticar y tragar),
anormalidades en la marcha,
adormecimiento de una extremidad
Datos de enfermedad
cerebrovascular: Infartos múltiples que implican la corteza y
materia blanca

DEMENCIA VASCULAR
• Deterioro cognitivo vascular leve - vasos parcialmente obstruidos - lentos
• Demencia multi-infarto
• La demencia vascular debido a un área dañada - "accidente cerebrovascular”
• La demencia vascular debido a infartos lacunares (daño profundo en el cerebro)
• La demencia vascular debido a infartos hemorrágicos - empeoramiento durante horas-días
• Enfermedad de Binswanger - la demencia vascular subcortical y vasos pequeños
• Demencia mixta (lesión vascular más la enfermedad de Alzheimer)
• Arteriopatía cerebral autosómica dominante, con infartos subcorticales y leucoencefalopatia (CADASIL)

CADASIL
Arteriopatía cerebral con infartos subcorticales y leucoencefalopatía
Mutación heterocigota en el gen NOTCH3 cromosoma 19p13
AD
Aparición en la tercera década
43% infartos cerebrales tempranos
Penetrancia completa
Trastorno progresivo de los pequeños vasos arteriales del cerebro
Migraña, accidentes cerebrovasculares y lesiones de la sustancia blanca, con deterioro cognitivo.

CADASIL
Pérdida aguda de la
visión (infarto nervio
óptico)
Infartos subcorticales
recurrentes
Neuropatia óptica
isquémica anterior
Parálisis seudobulbar
Electrorretinograma
anormal
Demencia subcortical
progresiva 6%
Respuestas evocadas
visuales anormal
Migraña 40%
Incontinencia urinaria Convulsiones
Insf venosa periferica Lesiones de
leucoencefalopatia
subcortical

CADASIL HALLAZGOS Y RM
• Entre los 20 y 30 años – lesiones hiperintensas en T2 en frontales y lóbulos temporales de manera anterior
• 30-40 años – áreas hiperintensas periventriculares, cápsula externa, ganglios basales, talamo y tallo cerebral
• > 40 años – infartos lacunares, lesiones hemorrágicas en el interior de la capsula
• > 50 años – infartos lacunares, sangrados microscópicos. Se encuentra vasculopatía de arterias pequeñas que penetran en la sustancia blanca, las de mediano calibre muestran estrechamiento, obliteración
• Las arterias afectadas muestran material granular denso cerca de las membranas celulares de musculo liso y pérdida de las mismas células

CADASIL
Los rastreos con imagen
por emisión de positrones
tomografía por emisión de
fotón único
Mostraran la reducción del flujo sanguíneo
cerebral

DEMENCIA VASCULAR (TRATAMIENTO)
Control de la Hipertensión,
diabetes, hipercolesterolemia.
Dieta, ejercicio, control de peso, evitar el uso de
drogas
Terapia anti-plaquetaria (como
la aspirina, Ticlid, Plavix)
Agentes hemorreológicos
(Trental)
Tratar la depresión
Dejar de fumar

SÍNDROME DE CADASIL
• Arteriopatía cerebral autosómica dominante con infartos
subcorticales y leucoencefalopatía (CADASIL)
• Es el trastorno más común de accidente cerebrovascular
hereditario, y se cree que está causada por mutaciones en el gen
Notch 3.
http://ghr.nlm.nih.gov/condition/cerebral-autosomal-dominant-arteriopathy-with-subcortical-infarcts-and-leukoencephalopathy

EPIDEMIOLOGÍA
• Se han publicado casos en todos los grupos
étnicos, aunque los europeos caucásicos
parecen ser los más frecuentes.
• En Finlandia, se ha establecido una
prevalencia de 4 por 100.000 habitantes.
• Hasta el año 2002 existían 100 familias
genéticamente confirmadas en Francia,
Alemania y Reino Unido, estimándose la
existencia de al menos 500 familias en todo el
mundo.
Sandoval et al. 2002. Novedades en CADASIL

GEN NOTCH 3
• 33 exones y codifica una proteína
receptor de 2321 aminoácidos con un
único dominio transmembrana
• 34 repeticiones de Factor de crecimiento
epidérmico
• 6 repeticiones ankirina
Santos et al. 2006. Vía de señalización Notch y nuevas estrategias para el tratamiento del cáncer
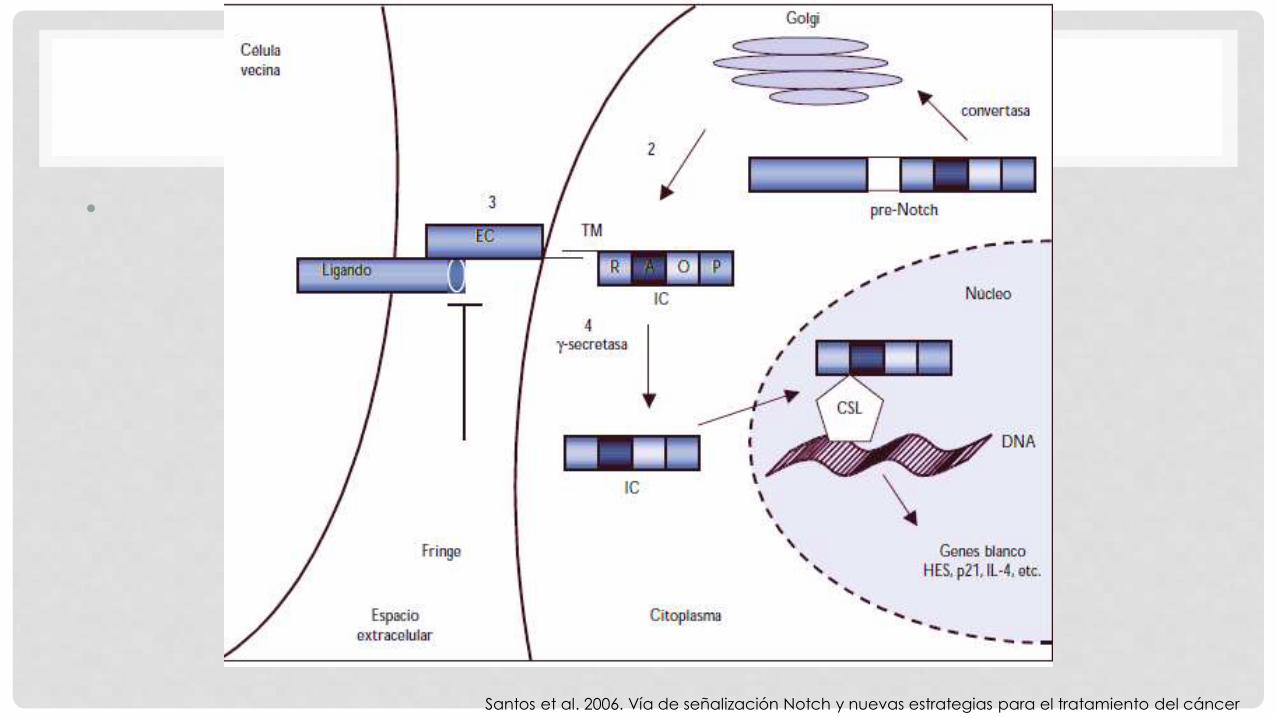
•
Santos et al. 2006. Vía de señalización Notch y nuevas estrategias para el tratamiento del cáncer

• Se han encontrado más de 150 mutaciones
• Más de 95% de los casos de CADASIL se produce por una
mutación puntual en el dominio extracelular del Notch3 , con un
importante acúmulo de mutaciones en el extremo 5’.
• En el 70% de los casos la mutación puntal se encuentra en los
codones 3 y 4, los cuales codifican las 5 primeras repeticiones FCE
Sandoval et al. 2002. Novedades en CADASIL

En vez de 6 cisteínas en la repetición EGF, segenera un número impar de éstas (5 ó 7).
aminoácido por cisteína
Cambio conformacional en el segmento extracelular
Clivaje anómalo.
Acumulación extracelular de una proteína de 210 kDa.
Sandoval et al. 2002. Novedades en CADASIL, Santos et al. 2006. Vía de señalización Notch y nuevas estrategias para el tratamiento del cáncer

• El 5% restante de los casos son producidos por 4 tipos de pequeñas
deleciones que resultan en la pérdida de 1 ó 3 residuos de cisteína,
generando también un número impar.
Sandoval et al. 2002. Novedades en CADASIL, Santos et al. 2006. Vía de señalización Notch y nuevas estrategias para el tratamiento del cáncer







