d912f51131ed787b91.en.es
TRANSCRIPT

Métodos, Modelos y Técnicas
De alto rendimiento de secuenciación de ADN, conceptos y limitaciones
Martin Kircher y Janet Kelso !
Introducción
En 1977, el firstgenome, thatofthe
5.386 nucleótidos (nt), de un solo strandedbacteriophagewX174, era completamente
secuenciada [1] usingatechnologyinvented sólo unos pocos años antes [2-5] .Sincethen thesequencing ofwholegenomes, así como de los genes individuales Regiones, así como se ha convertido en un foco importante de
la biología moderna y completamente trans-forman el campo de la genética.
ATTHE tiempo Ofthe de secuenciación
wX174, y durante casi una década,
Secuenciación de ADN Wasa apenas automático acoplado y proceso muy tedioso whichinvolved determinar sólo unos pocos nucleótidos Hun-Dred a una time.In los late1980s, semi-automatedsequencerswith mayor throughputbecame vano-poder [6, 7], stillonly capaz de determinea pocas secuencias ATA tiempo .Es posible el avance en 1990swasthedevelopment theearly del conjunto de capilares electro-foresis y apropiados de detección sys-tems [8-12] .Como reciente como 1996, thesedevelopmentsconverged en ThePro-ducción de un solo capillarysequencer comercial (ABI Prism 310). En 1998, Thege Salud MegaBACE 1000 y theABI Prism 3700 DNA Analyzer becamethefirstcommercial96capillarysequencers, un desarrollo que wastermed secuenciación de alto rendimiento.
Overthelastdecade, alternativa
sequencingstrategieshavebecomeavailable [13-18], que nos obliga a com-pletelyredefine''high-throughputsequencing. '' Estos technologiesout-realizar-theolderSanger sequencingtechnologies por un factor of100-1,000in rendimiento diario, y sametimereducethecostofsequencingonemillionnucleotides atthe (1 Mb) to4-0.1% de la asociada con Sangersequencing.Toreflectthesehugechanges, severalcompanies, investigación-res, y las recientes revisiones [19-24] el uso theterm''next-generationsequencing''instead de secuenciación de alto rendimiento, sin embargo, este término en sí mismo puede ser muy pronto outdatedconsideringthespeedofongoingdevelopments.
DOI 10.1002 / bies.200900181
Departamento de Genética Evolutiva del Instituto Max Planck de Antropología Evolutiva, en Leipzig, Alemania
*Autor correspondiente:
Janet Kelsoe-mail: [email protected]
Abreviaturas: A / C / G / T, Desoxiadenosina, desoxicitosina, desoxi-guanosina, desoxitimidina;ATP,Trifosfato de adenosina; dATPunaS, Desoxi-adenosina-5 '- (alfa-tio) trifosfato; CCD, Dispositivo con carga acoplada,
es decir, el dispositivo semiconductor utilizado en las cámaras digitales;
Chip-Sec, Cromatina inmunoprecipitación Sequen-cing; CNV, Número de copias variación; dNTP / PNT, Desoxi-nucleótidos;ddNTPs, Didesoxi-nucleótidos (nucleótidos modificados que faltan un hidroxilo
grupo en
el tercer átomo de carbono Ofthe azúcar);Georgia, ShortforIllumina Analizador Genoma;InDel, Inserción / deleción;kb / Mb / Gb, Kilobase (l03nt) / megabase (106nt) / base giga (l09Nuevo Testamento);MeDIP-Sec, Metil-ación-DependentImmuno-Precipitationsequen- cing; nucleótido (s) nt;PCR,Reacción en cadena de la polimerasa;RNA-Seq, SequencingofmRNAs / transcripciones;SABIOExpresión SerialAnalysisofGene;SNP,Polimorfismo de nucleótido simple; ARNm, El ARN mensajero / transcripciones.
Los recientes avances en la secuenciación del ADN han revolucionado el campo de la genómica, por lo que es posible que incluso los grupos de investigación individuales para generar grandes cantidades de datos ofsequence muy rápidamente y ATA tecnologías de alto throughputsequencing sustancialmente lowercost.These hacer profunda transcriptquantification transcriptoma sequencingand, y la secuenciación del genoma completo de resequencingavailable muchos más investigadores y projects.However, mientras que el tiempo costand se han reducido en gran medida, los perfiles de error y las limitaciones de las plataformas TheNew difieren significativamente de las de los anteriores de secuenciación Technol-gías. La selección de una plataforma de secuenciación apropiado para determinados experimentos TIPOSDE es una consideración importante,
y requiere una comprensión detallada
pie de las tecnologías disponibles; incluyendo fuentes de error, tasa de error, aswell como la velocidad y el coste de la secuenciación. Se revisan los conceptos relevantes andcompare las cuestiones planteadas por los sequencingtechnologies currenthigh-throughputDNA. Analizamos cómo los futuros desarrollos pueden superar estos Limita-ciones y lo que sigue habiendo dificultades.
Palabras clave:.ABI / vida Sólidos tecnologías; Helicos HeliScope; Illumina Genoma Analizador; Roche / 454 GS FLX Titanium; secuenciación capilar Sanger
524www.bioessays-journal.com Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas

A continuación repasamos los cinco secuenciación
las tecnologías actualmente disponibles en themarket (capillarysequencing, pyrose-secuenciación, reversibleterminatorchem-terio, secuenciación por ligación, y la química virtualterminator), discuten las limitaciones intrin-sic ofeach, y proporcionan anoutlook en la nueva technologieson thehorizon.Weexplainhow thevastincreases en throughputare associatedwith tanto nuevos y viejos tipos de problemsin los datos de la secuencia resultante, y howtheselimitthepotentialapplicationsand plantean desafíos para el análisis de datos.
secuenciación capilar Sanger
CurrentSangercapillarysequencingsystems, al igual que el instrumento AppliedBiosystems3xxxseriesortheGEHealthcare MegaBACE ampliamente utilizado,
son
aún basado en la misma schemeapplied en general en 1977 para lawX174 genoma [1,3] .First, millones ofcopies ofthesequence que se determine se purifiedor amplificada, dependiendo de la fuente Ofthe synthesisis cadena sequence.Reverse realizado en estas copias utilizando aknown cebado secuencia ascendente Ofthe secuencia para determinar y amixture ofdeoxy-nucleótidos ( dNTPs, thestandardbuildingblocksofDNA) y didesoxi-nucleótidos (ddNTP,
nucleótidos modificados falta un grupo hidro-Xil en el tercer átomo de carbono de thesugar) .ThedNTP / ddNTPmixturecauses al azar, no reversible termin-ationoftheextensionreaction, la creación de los diferentes ejemplares moles eculesextended a differentlengths.Following desnaturalización y nucleótidos upoffree limpias, cebadores, y theenzyme, theresulting moleculesaresorted por su peso molecular (corre-corres- al pointoftermination) y la etiqueta pegada a la ddNTPs terminat-ción se lee de forma secuencial enel orden creado por la etapa de clasificación. representación Aschematic de disponible en la Fig este processis. 1.
Clasificación por molecularweightwas
originalmente realizada usando gel electro-
foresis, pero se lleva a cabo hoy en día bycapillaryelectrophoresis [7, 25].
Originalmente, radiactivo o opticallabelswere aplicado en cuatro terminatorreactions diferentes (cada uno ordenados y leer outseparately), pero hoy en día cuatro de gripe orophores diferentes, uno por cada nucleótido (A, C, G y T) se utilizan en una sola reacción [6] .Additionally , el advenimiento de más sensi-tivedetectionsystemsandseveralrounds de extensiones de cebadores (equivalenttoalinearamplification) cantidades permitsmaller de ADN de partida a beused para reacciones de secuenciación modernos.
Por desgracia, hay stilllittle forcreation auto-mación Ofthe alta copyinputDNA con cebado conocida sites.Typically esto se hace mediante la clonación,es decir., La introducción de la secuencia diana en la secuencia del vector aknown mediante ligadura restrictionand proceduresand usando abacterialstrain para amplificar el targetsequence en vivo - Por lo tanto exploitingthelow amplificationerrorduetoinherent corrección de pruebas y reparación mech-anisms.However, este proceso es verytediousand issometimeshamperedby dificultades tales como la clonación specificsequencesdue a theirbase compo-sición, la longitud, y se utiliza interactionswiththebacterialhostsystem.Althoughnotyetwidely, integrado micro-fluidicdeviceshavebeen developedwhich tienen como objetivo automatizar el ADN extrac -ción, in vitro amplificación, y sequenc-ción en el mismo chip [26-29].
UsingcurrentSangersequencing
tecnología, itistechnically possibleforup 384 secuencias [29,30] ofbetween 600 y 1.000 longitud ntin [23,31] para ser secuenciados en parallel.However, éstos systemsare 384-capilar rara. El más estándar de 96 capillaryinstrumentsyieldamaximum ofapproximately 6 Mb ofDNA sequenceperday, con costsforconsumablesamounting a alrededor de $ 500 por
1 Mb.
Figura 1.Representación esquemática de la secuenciación de Sanger proc-ess. Entrada de ADN se fragmenta y se clonó en bacterias vectores para
en vivoamplificación. síntesis de la cadena inversa se lleva a cabo en las copias theobtained a partir de una secuencia de cebado conocido y utilizando amixture ofdeoxy-nucleótidos (dNTPs) y didesoxi-nucleótidos (ddNTPs). La mezcla de dNTP / ddNTP provoca la extensión azar
para ser terminado no reversible, creando de manera diferente extendida mol-ecules.Subsequently, después de la desnaturalización, limpiar offree nucleoproteínas mareas, los cebos, y la enzima, las moléculas resultantes se sortedusing electroforesis capilar por theirmolecularweight (corre-corres- al pointoftermination) y la fluorescentlabelattached a los ddNTPs de terminación se lee de forma secuencial.
......Métodos, Modelos y Técnicas M. y J. Kircher Kelso
Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc. 525
Méto
dos, M
odelo
s y Té
cnica
s

ThesequencingerrorobservedforSangersequencing duetoerrors ismainly en la etapa de amplificación (a Baja Velocidad cuando se hace en vivo), Naturalvar-iance, y la contaminación en el sampleused, así como el deslizamiento de la polimerasa atlow secuencias complejidad como simplerepeats (número corto tandemrepeats variables) y homopolímeros (stretchesof el mismo nucleótido). Además, lowerintensitiesandmissingterminationvariantstend a conducir a la acumulación de sequencingerrors hacia el final oflong combinación sequences.In separación withreduced por el electrophore-sis, basemiscalls [32] y deletionsincreasewith leer length.However, la tasa de error promedio (promedio de las bases generales de una secuencia) después sequenceend recorte es normalmente muy baja, withan error cada 10.000-100.000 nt [33].
Roche / 454 GS FLX Titaniumsequencer
La plataforma de secuenciación 454 era thefirstofthenewhigh rendimiento
themarket secuenciación platformson (publicado en octubre de 2005). Esta basado
en el enfoque de pirosecuenciación desa-desa- por Pal Nyre'n y Mostafa Ronaghiatthe RoyalInstitute tecnologia, Estocolmo en 1996 [34] .En la tecnología contrasttothe Sanger, pyrosequencingis basado en iterativa complementingsinglestrandsandsimultaneouslyreading fromthenucleotidebeingincorporated (también llamada secuenciación por síntesis, secuenciación durante la extensión) outthesignalemitted .
Electro-
foresis tanto, ya no requiredto generar una ordenada lectura de thenucleotides, Asthe leer outisnowdone simultáneamente con la sequenceextension.
Inthepyrosequencingprocess
(Fig.2), un nucleótido tiempo ata iswashedoverseveralcopiesofthesequenceto bedetermined, causingpolymerases para incorporar el núcleo-marea si es complementario a la cadena tem-placa. La incorporación se detiene Siel tramo longestpossible ofcomp-lementarynucleotideshasbeen
sintetizado por la polimerasa. En el
proceso de incorporación, uno pyrophos-fosfato pernucleotide isreleased andconverted de ATP por un ATP sulfurylase.The ATP impulsa la reacción luz de Luci-ferases presentand la lightsignalismeasured.TopreventthedATP emitida proporcionan para reactionfrom secuenciación que se utiliza directamente en la lightreaction, desoxi-adenosina-5
0- (unatio) -
trifosfato (dATPunaS), que no es asubstrateoftheluciferase, isusedforthebaseincorporation reaction.Standard nucleótidos de desoxirribosa
son
utilizado para todos los otros nucleótidos. Después de casquillo-Turing la intensidad de la luz, los nucleótidos se remainingunincorporated washedawayandthenextnucleotideisprovided.
En 2005, la tecnología de pirosecuenciación
fue paralelizado en una placa picotituladora by454 Ciencias de la Vida (más tarde comprada por Roche Diagnostics) para permitir alta throughputsequencing [16] .El secuenciación platehas unos dos millones de pozos - cada uno ofthem abletoaccommodateexactly
un 28-mtalón m de diámetro cubierto con
Figura 2.El proceso de pirosecuenciación. Uno de los cuatro nucleótidos iswashed secuencialmente sobre copias de la secuencia que se determine, haciendo que las polimerasas de incorporar incorporación nucleotides.The complementaria se detiene si el tramo más largo posible del vano-ablenucleotidehasbeensynthesized.Intheprocessof
incorporación, uno pernucleotide pirofosfato se libera andconverted de ATP por un ATP sulfurylase.The ATP impulsa el lightreaction ofluciferasespresentand un lightsignalproportional (dentro de unos límites) a las incorporaciones numberofnucleotide puede bemeasured.
M. y J. Kircher Kelso Métodos, Modelos y Técnicas
.....
526 Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas

se determinarán copias de una sola hebra de la sequenceto. Las cuentas son incubatedwithapolymeraseandsingle-strandbindingproteinsand, perlas de togetherwithsmaller que llevan las luciferasas de azufre-ylasesand ATP, gravitationallydeposited en los pocillos. nucleotidesare libre luego se lava sobre el flujo de células yla luz emitida durante el Incorpora-ción se captura para todos los pozos en parallelusing una alta resolución de carga-coupleddevice (CCD), características exploitingthelight-transporte de
el plato
usado.
Una Ofthe principales requisitos para
la aplicación de esta serie basada en pyrosequenc-ingapproach iscoveringindividualbeads con múltiples copias del samemolecule. Esto se realiza por primera creatingsequencinglibrariesin que everyindividualmolecule recibe dos differentadaptersequences, uno atthe 50endand uno en el 30final de la molecule.In el caso de la / Roche preparación sequencinglibrary 454 [16], esto se hace bysequentialligation oftwo oligos pre-Synthes-zados. Uno de los adaptadores añadió iscomplementary a oligonucleótidos enel perlas de secuenciación y, por tanto allowsmolecules para ser unido a las perlas byhybridization.Low molécula-a-beadratios y la amplificación de la secuencia de doble cadena hybri-dized en thebeads (keptseparate usando emulsionPCR) hace itpossible para crecer beadswith miles ofcopies OFA singlestartingmolecule.Usingthesecondadapter, perlas cubiertas con moleculescan beseparated de perlas vacías (usando perlas de captura especiales con oligo-nucleótidos complementarios al adaptador sec-ond) y arethen utilizados enla asdescribedabove reacción de secuenciación.
La sustitución promedio (excluyendo
inserción / deleción, InDel tasa de error) es enel intervalo de 10 "3 -10"4[16, 35], que es
higherthantheratesobservedforSangersequencing, butis la sustitución ERRORRATE lowestaverage ofthenew tecnologías de secuenciación discussedhere.As mencionados earlierfor Sangersequencing, in vitro amplificaciones por-formados para la secuenciación preparationcause una tasa de error de fondo superior,
es decir., El error introducido en la samplebeforeitentersthesequencer.Inaddition, inbeadpreparation (es decir., PCR en emulsión) una fracción Ofthe beadsend hasta carryingcopiesofmultiple
differentsequences.These''mixedbeads '' van a participar en un alto incorporaciones numberof por ciclo de flujo, como resultado-ción en la secuenciación readsthatdo notreflectrealmolecules.Mostofthesereads se filtran automáticamente duringthe software de post-procesamiento del filtrado data.The de granos mixtos puede, cómo-ever , causar un agotamiento de sequenceswith real de una alta fracción ofincorporationsper ciclo de flujo.
Alargefractionoftheerrors
observadas para este instrumento son smallInDels, en su mayoría derivados de inaccuratecallingofhomopolymerlength, andsingle deleciones de pares de bases o insertionscaused por thresholdingissues señal-ruido [35]. La mayoría de estos problemas Canbe resuelto por una mayor cobertura. Por mucho(>10 nt) homopolímeros, sin embargo, a menudo Thereis una longitud miscall coherente thatis notresolvable por la cobertura [35-37] .Stronglightsignalsin onewellofthe PicoTiterPlate también puede resultininsertions en secuencias en neighboringwells. Si el pozo vecino está vacía, thiscangenerateso-calledghostwells,es decir., Pozos para el cual un signalisrecorded pesar de que contienen plantilla noSequence; por lo tanto, los Inten-sidades medidos son completamente causedby sangrado a lo largo de la señal del vecino-ing wells.Computationalpost-proceso-ción puede corregir estos artefactos [38] .Como para la secuenciación de Sanger, el rateincreaseswiththepositioninthesequence error. En el caso de la secuencia 454, esto es causado por una reducción en enzymeefficiency o pérdida de enzimas (resultingin una reducción de la intensidad de señal), algunas moléculas ya no ser elong-ado y byan increasingphasingeffect.Phasingisobserved cuando apopulation de moléculas de ADN amplifiedfrom thesamestartingmolecule ( ensemble) se secuencia, y describesthe proceso en el que no todas las moléculas intheensembleareextended en everycycle. Esto hace que las moléculas en theensemble perder sincronía / fase, andresultsin un oftheprecedingcycles de eco que se añaden a la señal como el ruido.
Thecurrent454 / RocheGSFLX
plataforma de titanio hace posible tosequence alrededor de 1,5 millones de tales beadsin un solo experimento y para determinesequences oflength entre 300 and500 nt. La longitud de la lee es deter-minado por el número de ciclos de flujo (thenumberoftimesallfournucleotides
se lavan sobre la placa) como wellasby la composición de base y el orden Ofthe bases en la secuencia a disuadir-minadas. Actualmente, 454 / Roche limita thisnumber a 200 ciclos de flujo, lo que resulta Inan longitud averageread esperado ofabout400 nt.Thisislargely debido tolimitationsimposed por los efficiencyofpolymerases y luciferasas,
cual
cae sobre la carrera de secuenciación, resultingin disminuyó plataformas qualities.Currentlythe base permite about750 secuencia Mb ofDNA que se creará perdaywith costes de unos 20 $ / Mb.
Illumina Genome Analyzer II / IIx
Thereversibleterminatortechnologyused por el Illumina Genome Analyzer (GA) employsasequencing-por-sin-tesis conceptthatis similarto thatused en Sangersequencing,es decir.theincorporation reacción se detuvo aftereach base, la etiqueta de la base incorp corporado-se lee con tintes fluorescentes, y la reacción de secuenciación es entonces con-continuó con la incorporación ofthenext de base [13, 39] (Fig. 3).
Like454 / Roche, theIllumina
protocolrequires secuenciación thatthesequences que se determinen son acondicionado verted en una biblioteca de secuenciación especial, lo que les permite ser amplificado andimmobilized para la secuenciación [13, 40]. Forthis propósito dos adaptadores diferentes areadded al 50y 30extremos de todos MOL-ecules utilizando la ligadura de los llamados forkedadapters.1La biblioteca es entonces amplifiedusing secuencias de cebador más largo,
cual
extendandfurtherdiversifytheadapters para crear la finalsequenceneeded en pasos posteriores.
Thisdouble-strandedlibraryis
meltedusingsodium hydroxidetoobtainsingle-strandedDNAs, whichare luego se bombea a un muy bajo concen-tración a través de los canales de una célula de flujo. Esta celda de flujo tiene en su superficie twopopulationsofimmobilizedoligonu-cleotidescomplementarytothetwodifferentsingle hebra adapterendsof la secuenciación library.These oligo-nucleotideshybridizetothesingle-
1 Los híbridos de oligonucleo- parcialmente complementaria
mareas creando un extremo de doble cadena con un Toverhang, con un de cadena sencilla y un differentsequence en el otro extremo.
......Métodos, Modelos y Técnicas M. y J. Kircher Kelso
Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc. 527
Méto
dos, M
odelo
s y Té
cnica
s

moléculas de la biblioteca varados.por inversa
síntesis de la cadena a partir de la parte hybri-dized (de doble cadena), el newstrandbeingcreatediscovalentlybound a la celda de flujo. Si este nuevo strandbends más y se une a otro oli-gonucleotide complementaria a la secuencia adaptador sec-ond en el extremo libre Ofthe hebra, que puede ser utilizado para sintetizar una segunda strand.Thisprocessofbending inverso unido covalentemente y síntesis reversestrand, llamado puente amplifi-cación, se repite severaltimes andcreates grupos de varios 1.000 copiesofthe originalsequence en muy closeproximity de la otra en la celda de flujo [13, 40].
Estos grupos distribuidos al azar
contienen moleculesthatrepresenttheforwardaswellasreversestrandsof las secuencias originales. Antes de disuadir a-la minería de la secuencia, uno de los strandshastoberemovedtopreventitfrom obstaculizando los reactionstericallyorbycomplementarybasepairing.Strands de extensión son modificaciones de forma selectiva cleavedatbase ofoligonucleoti-des en el strandremoval cell.Following flujo, cada grupo en los cellconsists flujo deHabitación trenzado, copias identicallyoriented Ofthe misma secuencia; que puede ser secuenciado por hibridación
el cebador de secuenciación en los adaptersequences y comenzar la química reversibleterminator.
'' '', Solexasequencing asitwas
introducedinearly2007, initiallyallowed para la administración simultánea sequenc-ción de varios millones de Sequen-ces muy cortos (nt atmost26) inasingleexperiment.In los últimos años se havebeen severaltechnical, química, andsoftware updates.The producto, whichisnow llama theIlluminaGenomeAnalyzer, ha aumentado clusterdensities celda de flujo (más de 200 millones clus-tersperrun), un widerrangeoftheflow cellisimaged, andsequencereads de hasta 100 nt pueden ser generated.A technicalupdate también permitió thesequencingofthereversestrand ofeach molecule.Thisisachieved bychemicalmelting y lavado awaythe secuencia sintetizada,
la repetición de una
síntesis de la cadena bridgeamplificationcyclesforreverse pocos, y luego selec-tivelyremovingthestartingstrand (de nuevo usando modificaciones de bases de
el
celloligonucleotide poblaciones de flujo), beforeannealing anothersequencingprimer para el segundo read.Using this''paired-endsequencing''approach, approximatelytwicetheamountofdata puede haber generated.The Illumina
libraryandflowcellpreparationincludes variosin vitro amplificationsteps, lo que causa un alto grado backgrounderror y contribuir a la tasa ofabout10 averageerror"2-10"3[41,42] .Further, theflow fracción cellpreparationcreatesa ofordinary-lookingclustersthatare inició a partir de secuencias de calidad mostlylow uno signalsand individualsequence.Theseresultsin mixta morethan de estos clusters.Similarto the454 ghostwells, theIllumina identifychemistrycrystals imagen analysismay, polvo, andlintparticles como racimos y llaman sequencesfrom these.In tales casos, las resultingsequences típicamente parecen ser de complejidad lowsequence.
Como es el caso para las otras plataformas,
La tasa de error aumenta con increasingposition en los sequence.This determinada se debe principalmente a la eliminación gradual, proceso de secuenciación whichincreasesthebackgroundnoiseassequencingprogresses.Whiletheensemble de piro-sequencingcreatesuni-directionalphasing, crea reversible terminador sequenc-ción bi-directionalphasing [41,43] como algunos incorporan también nucleotidesmay fallar a darse por terminada correctamente -permitiendo la extensión de la secuencia
por otro nucleótido en el mismo ciclo.
Figura 3.Terminator química reversible aplicada por los cebadores Illumina GA.Sequencing se recuecen a los adaptadores de la sequencesto determinar. Las polimerasas se utilizan para ampliar las sequencingprimers por incorporación offluorescently etiquetados y terminatednucleotides.The incorporación se detiene inmediatamente después de The firstnucleotide debido a la terminators.The polymerasesand libre
nucleótidos son lavados y la etiqueta de las bases incorporatedfor cada secuencia se lee con cuatro imágenes tomadas a través differentfilters (filtro de nucleótido T se indica en la figura)
y el uso de dos
láseres diferentes (rojo: A, C y verde: G, T) para iluminar fluorophores.Subsequently, los fluoróforos y terminadores se eliminan yla secuenciación continuó con la incorporación de la base siguiente.
M. y J. Kircher Kelso Métodos, Modelos y Técnicas
.....
528 Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas

Con increasingcyclenumbers, theintensities extraen de la clustersdecline [41, 43, 44]. Esto es debido a fewermolecules que participan en el extensionreaction como resultofnon-reversibletermination, o debido a la atenuación effectsof los fluoróforos de secuenciación. En earlyversions Ofthe química, uno ofthefluorophorescould becomestucktothe grupos que crean otra fuente ofincreased ruido de fondo [41].
la
simultaneousidentificationoffourdifferentnucleotides es también un cuestión.La GA utiliza fourfluorescentdyes todistinguish los fournucleotides A, C,
G, y T.Of estos, dos pares (A / C y G / T) excitado usando el mismo láser, son
similarin theiremission espectros andshow única separación limitado el uso de filtros Opti-cal. Por lo tanto, las mayores errores substi-tución son observadas entre A / Cy G / T [41, 42].
A pesar de que el Illumina GA lee
mostrar ahigheraverageerrorrate, un errorrange wideraverage, y areconsiderablyshorterthan 454 / Rochereads, el GA instrumentdeterminesmore de 5.000 Mb / día con un priceofabout0.50 $ / Mb.This es más thansix veces mayor rendimiento diario
y
foraconsiderablylowerpricepermegabase.
Applied Biosystems SóLIdAS
TheprototypeofwhatwasfurtherdevelopedandlatersoldbyLifeTechnologies / Applied Biosystems (ABI) como la plataforma de secuenciación sólida, wasdeveloped por Harvard MedicalSchoolandtheHowardHughesMedicalInstitute y publicados en 2005 [17] .Con itscommercialreleasein finales
2007, sólido fue sólo el tercer newhigh-throughputsystem mercado enteringahighly competitiva con todo threevendorsselling theirinstrumentsforaroundhalfamilliondollars.TheChurch laboratorio de la Universidad de Harvard MedicalSchoolcontinuedthedevelopmentofthesystem andnow offersacheaper (<$ 200.000) SourceVersion abierta Ofthe sistema (llamado Polonator) en colab-oración con el sistema de Dover. En el thirdquarter de 2008, un com-biotecnología empresa de Mountain View, California, namedCompleteGenomicsstartedoffering una tecnología genoma humano sequencingservice.Their se basa también
ontheChurchlabsequencing por ligadura de concepto, pero itwitha combina nueva estrategia libraryconstruction ofsequencing y la secuencia immobiliz-ación utilizando círculo amplificación de laminación [45]. Aquí, nos centramos en el sistema commercialSOLiD ya que esta es la aplicación más extendida de este concepto.
Theprinciplebehind sequencing-
por la ligadura isvery differentFrom theapproachesdiscussedthusfar.Thesequence reacción de extensión no es coche-Ried polimerasas outby butrather byligases [17] (véase la Figura 4 para un schematicrepresentation Ofthe plat-forma sólida 2/3) .Inthesequencing por ligationprocess, es un cebador de secuenciación hybri-dized de copias de una sola hebra de
el
moléculas de la biblioteca para ser sequenced.Amixture sondas OF8-mer que llevan fourdistinctfluorescentlabels compiten forligation al cebador de secuenciación.
la
codificación fluoróforo, que es BasedOn los dos 30-mostnucleotides oftheprobe, se ofrecen las bases read.Three incluidoel tinte se escinden del 5
0 final de
la sonda, dejando una libre 50 fosfato de
la extendida (por cinco nucleótidos) pri-mer, que a su vez está disponible para furtherligation.After múltiples ligaduras (typi-camente hasta 10 ciclos), el synthesizedstrandsaremelted y theligationproductis arrastrado ante un newsequencingprimer (shiftedbyonenucleotide) se annealed.Starting fromthe nueva secuenciación el cebador ligationreaction se repite. El mismo proceso isfollowed para otros tres cebadores, facili-litar la lectura de la dinucleotideencoding para cada posición de inicio en thesequence.Usingspecificfluorescentlabelencoding, el colorante salidas leídas (es decir.colors) se puede convertir en una secuencia [46]. Esta conversión de tosequence espacio de color requiere un firstbase conocido, que es la última base de la libraryadaptersequence.Given utilizado un referencesequence, esto allowsdetection sistema de codificación ofmachineerrorsand theapplication ofan errorcorrection toreduce la theabsence promedio errorrate.In de una secuencia de referencia, la forma de la historia, color la conversión falla con un errorin el tinte leer outand causesthesequence aguas abajo Ofthe errortobe incorrecta.
Para la paralelización, la secuenciación
copias processusesbeadscoveredwithmultiple Ofthe secuencia para bedetermined. Estas perlas se crean en
de una manera similar a la descrita oído-lier para la plataforma 454 / Roche. En contraste con-a la tecnología 454 / Roche,
el
sólido sistema hace un Nouse forfixation PicoTiterPlate Ofthe granos en proceso de thesequencing; en lugar de la 3
0 extremos
de las secuencias en las perlas se modi-ficado de manera thatallows obligado a becovalently en un glassslide.As para el sistema de Illumina GA, este cre-ates una dispersión aleatoria de las cuentas enla cámara de secuenciación y permite densidades de carga forhigher. Sin embargo dispersión, Ran-dom complica la identi-ficación de las posiciones de cuentas de las imágenes, y da lugar a la posibilidad de que los cristales químico-cal, polvo y partículas de pelusa canbe identificado erróneamente asclusters.Further, la dispersión de los resultados de los granos en una distancias widerangeofinter-grano , whichthen tienen diferente susceptibilidad a beinfluenced por señales de neighboringbeads.
Tipos y causas de los errores de secuencia
son diversos: primero, la in vitro pasos amplifica-ción causan un mayor backgrounderror rate.Secondly, perlas que llevan amixture de secuencias y los granos en closeproximity a falsereads uno anothercreate y baja bases.Further calidad, disminución de la señal, una pequeña phasingeffect regular, andincompletedyeremovalresult en el aumento de error como el ligationcyclesprogress [47] .Phasing, asdescribed anteriormente, es una plataforma de minorissue onthis como secuencias no extendedin el último ciclo son no reversible ter-minatedusingphosphatases.Sincehybridization proceso estocástico isa, esto provoca una reducción considerable enel número ofmolecules participatingin subsequentligation reacciones, andthereforesubstantialsignaldecline.On Por otro lado, dado el efficiencyof fosfatasas la phasingeffectcanbeconsideredverylow.However restante, ofthedyes de escisión incompletas pueden permitir la escisión en la reacción nextligation, que luego allowsfor la extensión en el nextbutonecycle.This causa una additionalnoise differentphasingeffectand de colorantes de theprevious de ciclo en el proceso de identi-ficación colorante.
El sólido sistema permite en la actualidad
ofmore secuenciación de 300 millionbeads en paralelo, con un typicalreadlength de entre 25 y 75 nt.
En el
momento de escribir, el sólido sistema ABI istherefore comparable a la Illumina GA
......Métodos, Modelos y Técnicas M. y J. Kircher Kelso
Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc. 529
Méto
dos, M
odelo
s y Té
cnica
s

sistema en términos de rendimiento y pricepermillionnucleotides (#5,000 Mb / día,#0,50 $ / Mb) .Average errorratesare, sin embargo, que dependen del genoma de referencia OFA-vano capacidad para errorcorrection (10
"3 -10"4vs. 10"2-10"3). En
theabsenceofa referencegenome, montaje y consensuscalling El procedimiento puede realizarse sobre la base de tinte leer outs (los llamados colorspacesequences) toreduce los errores antes de la conversión enel secuencia de nucleótidos. Si hay referencegenome está disponible para la corrección de errores, y no hay consenso callingis montaje y realiza, a continuación, el error medio rateis superior a la de la Illumina GA.
Helicos HeliScope
Helicos es la firstcompany a sellasequencer capaz de secuenciar individualmolecules en lugar ofmolecule ENSEM-bles creados por una amplificación proc-ess. la secuenciación de una sola molécula tiene bybiases theadvantagethatitisnotaffected o errores introducidos en un paso librarypreparation o amplificación,
y
puede facilitar ofminimalamounts de secuenciación de entrada DNA.Using methodsable para detectar los nucleótidos no estándar, sino que también podría tener en cuenta las modificaciones del ADN identificationof, perdió comúnmente enel in vitro proceso de amplificación.
El HeliScope, como el sequenceris Helicos llamada, se vendió por primera vez en marzo de 2008, y para el final Ofthe firstquarter of2009 única fourmachineshavebeeninstalled en todo el mundo. Esto podría ser sur la extracción con palanca dadas las ventajas de
soltero
moleculesequencing, butprobablyreflects tanto las limitaciones específicas ofthisplatform, theprice (dólares aboutonemillion), y un relativamente smallmarket que ya ha invertido exten-vamente en las nuevas tecnologías de secuenciación.
Thetechnologyapplied (figura 5)
que podría denominarse virtualterminatorchemistry asíncrono [15] .InputDNAisfragmentedandmeltedbefore
una
Figura 4.secuenciación de Solid Applied Biosystems por imprimación ligation.Asequencing se hibrida con las copias de una sola hebra de Sequen-ces que se determinen. Octamer sondas se hibridan, se ligan a thesequencing cebador, y un tinte fluorescente en el 5
0 final del ligado
8-mer sondas, la codificación de los dos 30nucleótidos -la mayoría de la sonda, es
leer dephosphorylated.Threenucleotides primersare extendidos-out.Non de la sonda que incluye el colorante se escinden, creando un
libre 50fosfato para más ligaduras múltiples ligations.After, hebras thesynthesized se funde y el producto de ligación es washedaway antes de una nueva, nucleótidos desplazada por un cebador de secuenciación isannealed.Starting de la nueva ligationreaction secuenciación primerthe se repeated.The mismo se hace forthree otherprimers , lo que permite la lectura de la etiqueta dinucleótido
para cada posición en
la secuencia.
M. y J. Kircher Kelso Métodos, Modelos y Técnicas
.....
530 Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas

poli-A-tailissynthesized molécula ontoeachsingle hebra usando una polimerasa poli-ciclasa. En la última etapa ofpolyadenylation, se añade un labeledadenine con fluorescencia. La biblioteca es washedover un flujo cellwhere poli-A tailsbind a poli-T oligonucleotides.Thebound coordenadas en la celda de flujo aredetermined utilizando una fluorescencia de basedread fuera del flujo thesecoordinates cell.Having detectadas, la fluorescentlabel de la 30adenina se elimina andthesequencingreactionstarted.Polymerasesare lavó a través de theflow célula con un tipo de nucleótido fluorescentlylabeled (A, C, G, o T) en atime y las polimerasas se extienden thereverse hebra de las secuencias startingfrom la incorporación de poli-T oligonucleotides.Thenucleotide Ofthe poli-merases se ralentiza por el etiquetado fluor-escent y permite como máximo
una constitución antes de polímero-asa se elimina por lavado junto con los nucleótidos Thenon-Incorporated (termedvirtualtermination [48,49]). El flowcellisthen fotografiado de nuevo, los tintes Fluor-escent se eliminan, y la reac-ción continuó con otra nucleotide.By este molécula proceso notevery isextended en cada ciclo, que es whyit es una secuenciación proc-ess asíncrono que resulta en secuencias ofdifferentlength (como es el caso para el 454 / Rocheplatform).
Ya que soltero moleculesare
secuenciado, siendo débil measuredare las señales, y no hay posibilidad thatmisincorporationerrorscan Becor-rected por un conjunto effect.Due enel hecho de que las moléculas se unen enel celda de flujo sólo por hibridación, thereis la posibilidad de que las moléculas plantilla canbe pierden en el lavado steps.In adición,
moleculesmay beirreversibly termi-nado por la incorporación de nucleótidos incorrectlysynthesized.
En general, se lee
tienen entre 24 y 70 nt de longitud (nt average32) [50] y por lo tanto más corto que para theother plataformas. Debido a la mayor num-ber de secuencias determinadas en paralelo, el rendimiento total por día (4150 Mb / daywith un coste de #0,33 $ / Mb [50]) está en thesame rango que forthe GA y SOLiDsystems.The tasa de error promedio, whichisin la gama OFA pequeño tanto por ciento, isslightly superior a la de todos los demás instru-mentos y sesgada hacia Indeles sustituciones mas bien que.
Aplicaciones y generalconsiderations
Allcurrenthigh-throughputtechnol-gías tienen un error medio tasa que
Figura 5.la química virtualterminator asíncrono realizado byThe ADN HeliScope.Input se fragmenta, se derritió, y RELAClONADAS-polyadeny. Un adenina marcado con fluorescencia se añade en el último paso. Thissingle de hebra de ADN se lava a través de una celda de flujo
con poli-T oligo-
nucleótidos que permiten la hibridación. Las coordenadas consolidados sobre la
cellare flujo determina utilizando el marcado con fluorescencia adenines.Having las coordenadas identificadas, el fluorescentlabelofthe 30adeninas se removed.Polymerases se lavan a través con una
escriba nucleótidos de la etiqueta fluorescente (A, C, G y T) a la vez, y las polimerasas se extienden la cadena inversa de las secuencias de
a partir de los oligonucleótidos de poli-T. El nucleótido Incorpora-ción de las polimerasas se ralentiza por la labelingand fluorescente permite a lo sumo una incorporación antes de la polimerasa iswashed away.The Cellis flujo entonces fotografiado, la fluorescentdyesremoved, y la reacción continuó con otro nucleótido.
......Métodos, Modelos y Técnicas M. y J. Kircher Kelso
Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc. 531
Méto
dos, M
odelo
s y Té
cnica
s

es considerablemente más alto que el / 10,000to1 / 100,000observedforhigh calidad typical1
Sanger secuencias.
Además, theGSFLX titanio, GA, y plataformas HeliScope SOLiD eachhave Limita-ciones biasesand muy específicas, por lo que es necesario escoger aplatform apropiado para una aplicación específica proyec-ector (para un resumen verTabla 1). Una combinación de tecnologías [51-54] y protocolos experimentales [55-57] También puede ser apropiado, y evencomplementary, para proyectos específicos.
De alto qualitySangersequencing
isnow commonlyused a generatelow-cobertura de la secuenciación de individualpositions y regiones (p.ej., Diagnosticgenotyping) genomes.AstheSangersequencelengthislongerthanmostabundantshortrepeat clases enteras y orthesequencingofvirus--fago de tamaño, que permite a los unambigu-ousassemblyofmostgenomicregions - algo thatis generallynotpossibleusing theshorterreadplatforms.However, la tecnología y isexpensive demasiado lento para las muestras sequencinglarge, extendidos genómicos re-regiones, o las muchas moléculas requiredforquantitativeapplications [p.ej., La expresión génica cuantificación; cro-Matin inmunoprecipitación sequenc-ing (chip-ss.), Andmethylation dependiente de inmunoprecipitación Sequen-cing (MeDIP-Sec)] termsofsequencenumberandhastheadvantage ForquantitativeapplicationstheHeliScopeprovidesthehighestthroughputin de no requerir un protocolo de preparación multisteplibrary. En el AT-herhand, el HeliScope ofrece thelowest resolución en la cartografía accuracyfor genomas complejos debido a su corta readlength y errorprofile.TheGA orSOLiD resultados cuantitativos para platformsmaythusprovideequivalent dantes-cationes, mientras que proporciona menos pero lon-gerreadsandrequiringamoreelaborate preparación biblioteca.
A pesar de que aún no ha sido totalmente analógica
lyzed, es posible (e incluso probable) thatlibrary protocolos de preparación podrían biasthe representación secuencia en una muestra [42, 58, 59], por lo que la sustitución ofthisstep un importantgoal.Further, de múltiples etapas biblioteca preparación protocolsrequirehigheramountsofinputmaterial, limitando theirgeneralappli-cation.However, protocolsforlibraryconstructionfromlimitedsample
cantidades están disponibles orbeing desa-desa- para cada una de las plataformas, y Pub-publica- demuestran que, si bien vendorprotocols indican la necesidad forhighersamplequantities (rango microgramos), muchos usuarios están procediendo cantidades de ADN de entrada successfullywith bajas (nanogramto rango de picogramos), como, por ejemplo, , las muestras de ADN fromancient [60-62].
Al igual que la secuenciación de Sanger, el GS FLX
El titanio proporciona una longitud de leer lapso de Ning muchas de las secuencias repetidas cortas
- Un assemblyofgenomes importantfeatureforaccuratesequencemappingand [63] .A pesar de los errores indel, esta tecnología tiene índices muy bajos ofmisidentifying bases individuales, makingit perfectamente adecuado para los identificationofsinglenucleotidepolymorphisms (SNP). También dirigido a los identificationofSNPs, atleastfor muestras con anexistingreferencegenome, instrumento istheSOLiD con su dinucleotideencodingscheme [46] .Considerablyhighercoverage se necesita para llamar performSNP con una precisión similar usingtheIlluminaGA [64] .NeithertheIllumina GA ni los sistemas sequenc-ing SOLiD ABI son propensos a generar highrates de indeles pequeñas, haciéndolos wellsuited para el estudio de la variación InDel.
Como se mencionó anteriormente, el inconveniente
ofshortreads (por debajo de about75 nt) obtenidos a partir de Helicos, sólido, orGAinstrumentsisin genomeassemblyand aplicaciones de mapas, donde theplacementofrepeated orvery similarsequences no se pueden resolver UNAMBIG-nuamente. La colocación correcta se furthercomplicated por altas tasas de error intro-ducing una distancia requirementfora minimumsequence ofan unambiguousplacement. -Extremo emparejado o de yerba mate de par ayuda a favor de los protocolos para superar algunos de theselimitations de corta lee [65] por la información Provid-ción acerca de la orientación relativa locationand ofapairofreads.Currently un protocolo de extremo emparejado se aplica onlycommonly en el GA, whilemate-pairprotocolsareavailableforSOLiD, GS FLX Titanium, y GA.Inpaired de fin de secuenciar el real
extremos
moléculas de ADN de más bien cortas (<1 kb) aredetermined, whilemate-pairsequencing requiere la preparación deOferta bibliotecas. En estos protocolos, theends de moléculas más largas, tamaño seleccionado (p.ej., 8, 12, o 20 kb) están conectados withan secuencia adaptador interno en un circu-larizationreaction.Thecircular
molécula se procesa a continuación, utilizando enzimas restric-ción orfragmentation beforeouter adaptadores biblioteca se añaden dos extremos del aroundthe molécula combinada.
la
adaptador interno se puede entonces utilizar como moléculas additionalsequencingreactiononthesameimmobilized una segunda sitio de cebado FORAN. Por lo tanto, compañero-pairsequencingprovidesdistanceinfor-mación usefulforassembly, butdoesnot permitir la fusión de los dos se superpone a finales lee, ya que por themolecules de diseño no se solaparán en sequenc-ing.However, la fusión de los dos se superponen-ping avance y retroceso endreads pareadas de corto inserto bibliotecas allowsthe reconstrucción OFA completa con-secutivemoleculesequence, longerthan la longitud individualread, ycon las tasas de error promedio de reducción en theoverlapping secuencia de parte [60, 66].
Debido a las grandes cantidades de secuencial
ces creadas, hay interés en regiones seleccionadas sequenc-ing (p.ej.agenomiclocus, desde sequencecaptureexper-iments [67-69]) en múltiples individuos / samplesinsteadofsequencingonesamplein excessivedepth.Alltech-gías, por tanto, proporcionan una placa separationoftheirsequencing en definedregions o channels.However, como máximo, 16 tales regiones / canales están disponibles (GS FLX Titanium y placas HeliScope), que, no tengan el sufficientforsomeapplications. El uso de diferentes bibliotecas con-structionprotocols, además someplatformsallow de muestra de código de barras específico (a veces llamado '' index '') secuencias enel molecules.These biblioteca moleculescan entonces besequenced en thesameregion / canal, andlaterseparated (computacionalmente) theirbar basa en la secuencia de código [70-73 ].
esto facilita
altamente parallelsequencing LargeNumber OFA de las muestras más allá de que los possibleusing (protocolos mostlynon proveedores) physicallane / channelsepar-ation.Currently tales protocolos disponibles forthe GS FLX Titanium, GA, y SOLiDinstrument.
Aunque los precios de secuenciación por giga-
de base han disminuido considerablemente en los últimos años a, haciendo Proyecto Genoma projectslikethe1000Human Variación, 1001Arabidopsis thaliana Proyecto genomas, theMammalianGenomeProject, ortheInternationalCancerGenomeConsortium posible, de alta throughputsequencing adquisición stillhashigh, el funcionamiento y los costes de mantenimiento,
cual
M. y J. Kircher Kelso Métodos, Modelos y Técnicas
.....
532 Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas
no se incluyen en la Tabla 1. Además, eachof estas plataformas requiere una managementandanalysis datos substantialinvestmentin, tiempo y personal [74-77] grupos de investigación .Smaller todavía puede
encontrar
prohibitivos los costos de la infrastructureneeded para almacenar, manipular y ana-lizarla severaltens de gigabytes de datos y terabytes de puresequence
varios
miles de archivos intermedios generados instrumentos bythese cada semana. Incluso para
mayores, centros de genoma experimentados thisaspect sigue siendo una cada vez mayores Chal-lengefortheongoinguseoftheseplatforms.
próximas promociones
Motivado bythegoalofa $ 1.000 genoma Fijado por el NIH / NHGRIto enablepersonalized la medicina, los throughputofallsystems descrito es constante
y el aumento de los números dados herearerapidlyoutdated.However, inaddition a las mejoras de currenttechnologies, includingtheJanuary2010 announcementoftheIlluminaHiSeq sistema de 2000, que determinessequences de clusters en el fondo y topof la celda de flujo y procesa dos celdas de flujo en paralelo, una nueva generación ofsequencers ya está en el horizonte.
WhatstartedwiththeHelicos
sistema - la secuencia de un solo mol-ecules sin amplificación previa preparationor biblioteca - probablemente se convierta en apopular paradigma. En concreto, tres OT-sus sistemas han captado la atención de medios andscientific WELLIN avance oftheir
real Disponibilidad: Pacific
nanoporos Bioscience'sSingleMoleculeRealTime (SMRT) sequencingtechnology [18], BASE-Technol gía de Oxford nanoporos [14] y, recientemente, la propuesta de IBM-ofsilicon basa [78].
PacificBiosciences'SMRT technologyperformsthe reacción de secuenciación onsilicon dióxido de chipswith un 100 nmmetalfilm containingthousandsoftens-de-nanometerdiameterholes, los llamados guías de onda en modo cero (ZMWs) [79]. Cada ZMW se utiliza como una cámara de nano-visual-ización, proporcionando un detectionvolume de 20 zeptoliters (10
"21 l). en este
volumen, una sola molécula puede ser illu-minated excluyendo otros labelednucleotides en el fondo - savingtime y química de secuenciación por omitir-ting etapas de lavado. Un ADN único polímero-asa está fijada a la parte inferior de la surfacewithinthedetectionvolume, andnucleotides, con
differentdyes
unido a la cadena de fosfato, areusedinconcentrationsallowingnormal procesividad de la enzima. Como las pol-ymeraseincorporatescomplementarynucleotides, thenucleotideisheldwithin Fortens volumen de detección
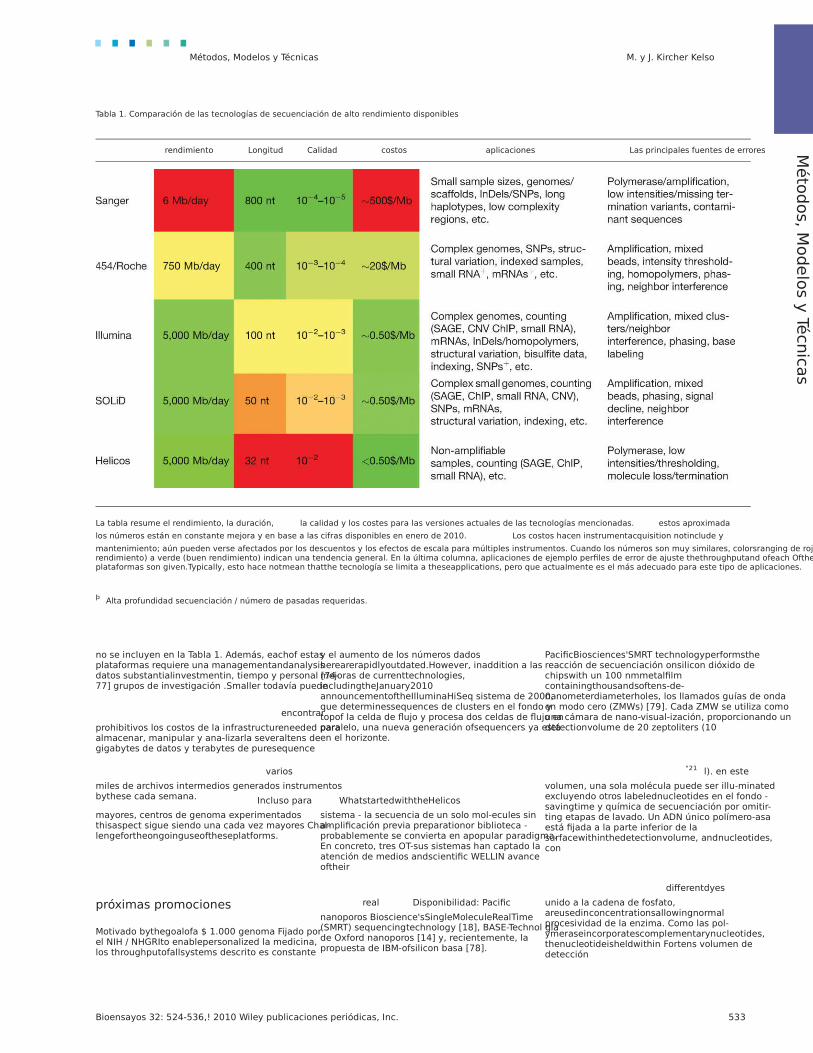
Tabla 1. Comparación de las tecnologías de secuenciación de alto rendimiento disponibles
rendimiento Longitud Calidad costos aplicaciones Las principales fuentes de errores
La tabla resume el rendimiento, la duración, la calidad y los costes para las versiones actuales de las tecnologías mencionadas. estos aproximada
los números están en constante mejora y en base a las cifras disponibles en enero de 2010. Los costos hacen instrumentacquisition notinclude y
mantenimiento; aún pueden verse afectados por los descuentos y los efectos de escala para múltiples instrumentos. Cuando los números son muy similares, colorsranging de rojo (bajo rendimiento) a verde (buen rendimiento) indican una tendencia general. En la última columna, aplicaciones de ejemplo perfiles de error de ajuste thethroughputand ofeach Ofthe plataformas son given.Typically, esto hace notmean thatthe tecnología se limita a theseapplications, pero que actualmente es el más adecuado para este tipo de aplicaciones.
þ Alta profundidad secuenciación / número de pasadas requeridas.
......Métodos, Modelos y Técnicas M. y J. Kircher Kelso
Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc. 533
Méto
dos, M
odelo
s y Té
cnica
s

ofmilliseconds, ordersofmagnitudelongerthan forunspecificdiffusionevents. De esta manera el tinte fluorescente oftheincorporatednucleotidecanbeidentified durante la síntesis normal de reversestrand velocidad [79] .En pilotexper-iments, Pacific Biosciences tiene shownthatitstechnology allowsfordirectsequencing OFA pocos miles basesbeforethepolymeraseisdenatureddue a la lectura láser outofthe tecnología dyes.The SMRT está destinado forrelease en 2010.Even aunque se necesita furtherdevelopment para crear un morerobustsystem, la omisión oflibrarypreparation y amplificación así como seguir las secuencias largas generadas willundoubtedlyprovideanadvantageoverthecurrentsystemsformanyapplications.
BASE tecnología de Oxford nanoporos
es poco probable que sea lanzado pronto como modificaciones astheSMRTtechnology.BASEoffersthepotentialtoidentifyindividualnucleotide (p.ej.
5-metil-
citosinavs.cytosine) proceso duringthesequencing [14]. La idea behindthis tecnología es la ofindividualnucleotides de identificación utilizando un changein el potencial de membrana, ya que de paso a través de una modificación una-hemolisina de poro mem-brana con un sensor de ciclodextrina [14, 80]. Sin embargo, la aplicación de este Technol-logía para la secuenciación, el poro tiene que befused a una exonucleasa, que degradessingle-strandedDNAsequencesandreleases individualnucleotides en thepore. Además, la tecnología needsto ser paralelizado en formato de matriz, beforeits liberación como un alto rendimiento sequenc-ción platform.While la sensibilidad forindividualnucleotidemodificationsseemsto ser un majoradvantage, thedestructivefashionoftheoutlinedsequencing proceso podría ser forApplications considereda obstáculo con muestras de pre-preciosas, y que no permite una segunda ciclo de lectura para la reducción de error.
A principios de octubre de 2009, IBM emitió una
Comunicado de prensa [78] describe un método toslow la velocidad ofan individualDNA cadena que pasa a través de un nano-pore.For este fin developeda de múltiples capas de metal / dieléctrico nanoporedevice que utiliza la interacción de cargas theDNA columna vertebral con un campo eléctrico RELAClONADAS-modu para atrapar y slowlyreleases una tecnología individualDNA molecule.The descrito podría Theor-éticamente combinarse con, por ejemplo,
las tecnologías desarrolladas Nanopore atHarvard Universidad [81] o la base previouslydescribed technologywhereitmay superar la destructiva approachfollowed hasta ahora.
Conclusión
Currenthigh-throughputsequencingtechnologies proporcionan una gran variedad ofsequencingapplicationstomanyresearchersandprojects.Giventheimmense la diversidad, hemos notdis-terco estas aplicaciones en profundidad aquí, otras opiniones con unas fuertes aplicaciones onspecific focales y analysisare datos disponibles [24, 82-88]. Los discussedtechnologies hacen posible que evensingle grupos de investigación para generar largeamounts de muy rapidlyand atsubstantially lowercosts thantraditionalSangersequencing.Whilecosts se han reducido a menos than4-0.1% y el tiempo ha sido un factor de shortenedby 100-1.000 sobre la base de datos de secuencias, dailythroughput theerrorprofilesandlimitations observados para el plat nuevas formas de differsignificantly Sangersequencing y entre approaches.Further, cada uno de estos nuevos sequencingplatforms requiere additionalinvestments sustanciales - factores thathave oftennot hacer hincapié suficiente en researchpublications que describe una específica apli-cation.Somevendorshaverecentlystarted para ofrecer versiones de presupuesto de theirinstruments (p.ej. Illumina GA IIe o 454 / Roche GS Junior) con sequencingcapacity.However menor, precio whiletheinstru-ment es más bajo, la financiera invertirá-ment permanece high.Costs por base aregenerally superior a la de la standardinstrument, y verysimilaroverallinfrastructureisstillrequired.Oftenthe elección de una adecuada sequencingplatform es projectspecific y algunos timescombinationscanbeadvan--tageous.Thismayopen themarketfurtherto
companiesproviding
secuenciación-en-demandservices, butwill no reemplaza la necesidad de laboratoriesto invertir un tiempo considerable y exper-Tise tanto en la producción de análisis librariesand Ofthe vastquantities ofdata que se generará.
Las nuevas tecnologías en el horizonte,
SMRT por Pacific Biosciences, BASE byOxford nanoporos, y othertechnol-gías como thatsuggested por IBM,
demostrar el futuro importante directionsin el campo de la secuenciación de ADN: el Abil-dad de usar moléculas individuales withoutany preparación biblioteca o la amplificación, la identificación de nucleotidemodifications específicos, y la capacidad de gener comió-secuencia más larga lee. Estos debuta de desa- facilitarán futuras investigaciones inmany campos, hacer más fácil el análisis de datos, y los costos de secuenciación furtherreduce, hopefullyachievingtheaim OFA $ 1.000 genoma humano sugiere byNIH / NHGRI que se requiere para la medicina-zado personal.
Expresiones de gratitud
Agradecemos a los miembros Ofthe Salida-mentofEvolutionaryGenetics, los miembros de la andparticularly sequencingGroup, forproviding datafrommultipleplatforms de secuenciación, aswellasinteresting usefulinsights.We discussionsand también están en deuda con A.Wilkinsandthethreeanonymousreviewersforcriticalreadingofthemanuscriptand thoughtfulcomments.This trabajo fue apoyado por la Sociedad Max Planck.
referencias
1.Sanger F,GM aire,BarrellBG,et.1977.Nucleotide secuencia de ADN ofbacteriophage phiX174. Naturaleza 265: 687-95.
2.Gilbert W,Maxam A.nucleotidesequence 1973.The del operador lac. Proc Natl AcadSciUSA 70: 3581-4.
3.SangerF,Nicklen S,Coulson AR.1977.DNAsequencingwithchain-terminatinginhibitors.ProcNatlAcadSciUSA 74:
5463-7.
4.Sanger F, Coulson AR. 1975. Una rápida methodfor determinar las secuencias en el ADN por primedsynthesis con ADN polimerasa. J Mol Biol 94:
441-8.
5.Wu R, Kaiser AD. 1968. Estructura y basesequence en los extremos cohesivos de ADN lambda-fago ofbacterio. J MOLBIOL35: 523-37.
6.Smith LM,Sanders JZ,KaiserRJ,et.
detección 1986.Fluorescence en análisis de la secuencia automatedDNA. Naturaleza 321: 674-9.
7.Swerdlow H,Gesteland R.1990.Capillarygelelectrophoresis para la secuenciación rápida resolutionDNA, alta.NucleicAcidsRes18:
1415-9.
8.Zagursky RJ,McCormick RM.1990.DNAsequencing separaciones en geles capilares en amodified instrumento de secuenciación de ADN comercial
ambiente. Biotechniques 9: 74-9.
9.Huang XC, Quesada MA, Mathies RA. 1992.DNA secuenciación capilar usando matriz electro-foresis. AnalChem 64: 2149-54.
10.Kambara H,TakahashiS.analizador de capillaryarrayDNA 1993.Multiple-sheathflow.
Naturaleza 361: 565-6.
M. y J. Kircher Kelso Métodos, Modelos y Técnicas
.....
534 Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas

11.Ueno K,Yeung ES.fragmentos 1994.Simultaneousmonitoring ofDNA separados por
electroforesis en una matriz de multiplexado de 100capillaries. AnalChem 66: 1424-1431.
12.kimś,YooHJ,HahnJH.1996.Postelectrophoresiscapillary
exploración
secuenciación método forDNA.AnalChem68: 936-9.
13.Bentley DR, balasubramanian S, SwerdlowHP,et.2008.Accuratewholehuman
la secuenciación del genoma usando la química termin ator reversible. Naturaleza 456: 53-9.
14.Clarke J, Wu HC, Jayasinghe L, et al. identificación de la base 2009.Continuous para single-mol
eculenanoporeDNAsequencing.NatNanotechnol4: 265-70.
15.Harris TD,Buzby PR,Babcock H,et.
La secuenciación del ADN de una sola molécula de 2008. de aviralgenome. ciencia 320: 106-9.
dieciséis.Margulies M, Egholm M, WE Altman, et al.
2005. La secuenciación del genoma en microfabricatedhigh densidad picolitros reactores.Naturaleza 437: 376-80.
17.Shendure J, Porreca GJ, REPPAS NB, et al.
2005. Accurate multiplex polony sequencingof un genoma bacteriano evolucionado. ciencia 309: 1728-1732.
18.Korlach J, marcas PJ, Cicero RL, et al. pasivación de aluminio para 2008.Selective dirigida
inmovilización de ADN polimerasa MOL-ecules individuales en cero en modo de guía de ondas nanostruc-turas. Proc NatlAcad SciUSA 105: 1176-1181.
19.Ansorge WJ.2009.Next generación, técnicas de Secuenciación del ADN.NatBiotechnol25:
195-203.
20.MardisER.2008.Next generación, métodos Secuenciación del ADN.Annu Rev Genómica
Hum Genet 9: 387-402.
21.Schuster CAROLINA DEL SUR.2008.Next generación
secuenciación transforma la biología de hoy.NatMethods 5: 16-8.
22.Shendure J,JIH.2008.Next generación, la secuenciación del ADN.NatBiotechnol26: 1135-
45.
23.Shendure JA,Porreca GJ, Iglesia GM.
2008.Overview ofDNA secuenciación Strat-tegias.Curr protoc MOLBIOLCapítulo 7: Unidad
7.1.
24.Metzker ML. 2010. Las tecnologías de secuenciación
- la próxima generación. Nat Rev Genet 11: 31-
46.
25.George KS, Zhao X, Gallahan D, et al. 1997.Capillaryelectrophoresismethodologyfor
identificación de genes relacionados con el cáncer patrones expre-sión de reacción en cadena de displaypolymerase diferencial fluorescente.J Chromatogr BBiomed SciAppl695: 93-102.
26.BlazejRG,P Kumaresan,MathiesRA.
2006.Microfabricated bioprocessor para inte-rallado nanolitros escala de ADN Sanger sequenc-ción. Proc NatlAcad SciUSA 103: 7240-5.
27.Mariella R Jr. 2008. Preparación de la muestra: theweak eslabón de bio-detección basada en la microfluídica.
Microdispositivos Biomed 10: 777-84.
28.Roper MG,Easley CJ,Legendre LA,et al.
2007. sistema de control de temperatura por infrarrojos fora chainreaction completamente noncontactpolymerase en chips de microfluidos.AnalChem79: 1294-1300.
29.Emrich CA, Tian H, Medintz IL, et al. 2002.Microfabricated 384 carriles conjunto de capilares elec-
Bioanalyzer trophoresis para el análisis de ultra-alta-a través putgenetic.AnalChem 74: 5076-
83.
30.Shibata K,Itoh M,Aizawa K,et.2000.RIKEN Análisis de la secuencia integrada (RISA)
sistema - 384 formato de la tubería con la secuenciación
384 secuenciador multicapilar.Genome Res10: 1757-1771.
31.HertDG,Fredlake CP,Barron AE.2008.Advantages y limitaciones ofnext-generación
tecnologías de secuenciación ación: a comparisonofelectrophoresis y no electrophoresismethods. electroforesis 29: 4618-26.
32.Ewing B,Hillier L,WendlMC,et.1998.Base Insultos de trazas secuenciador automático
usingphred.I.Accuracyassessment.Genome Res 8: 175-85.
33.Ewing B,P. Verde1998.Base-llamando sequencertraces ofautomated utilizando Phred.
II.Errorprobabilities.GenomeRes8: 186-94.
34.RonaghiM,Karamohamed S,Pettersson
segundo,etTiempo de liberación .1996.Real ofpyrophosphate detección sequencingusing ADN.AnalBiochem 242: 84-9.
35.Quinlan AR,StewartDA,Stromberg MP,et.2008.Pyrobayes: Una base mejorada
descubrimiento callerforSNP en pyrosequences.Métodos Nat 5: 179-81.
36.T mimbre, E Schlagenhauf, Un Graner, et al.
2006. 454 secuenciación puso a prueba utilizando thecomplex genoma de la cebada. BMC Genomics 7:
275.
37.RE verde, Malaspina AS, Krause J, et al.
2008. Una completa Neandertalmitochondrialgenomesequencedeterminedbyhigh-throughputsequencing.Celda134: 416-
26.
38.RE verde,Krause J,Ptak SE,et.2006.Analysisofonemillionbasepairsof
NeanderthalDNA. Naturaleza 444: 330-6.
39.TurcattiG,Un romieu,Fedurco M,et.
2008. Una nueva clase fluorescentnucleotides ofcleavable: síntesis y optimización de síntesis terminadores asreversible sequencingby forDNA. Nucleic Acids Res 36: E25.
40.Fedurco M,Un romieu,Williams S,et.
2006.BTA, un novelreagent para el ADN adjuntar-menton vidrio y colonias de ADN amplificados efficientgeneration fase ofsolid. NucleicAcids Res 34: E22.
41.KircherM,StenzelU,KelsoJ.aprendizaje automático 2009.ImprovedbasecallingfortheIlluminaGenome Analyzerusing
estrategias. Biol genoma10: R83.
42.Dohm JC, Lottaz C, Borodina T, et al. 2008.Substantialbiases de datos ultra-shortread
establece a partir de la secuenciación de ADN de alto rendimiento.Nucleic Acids Res 36: E105.
43.Y Erlich,Mitra PP,delaBastide M,et.
2008.Alta-cíclica: la secuenciación Aself-optimizingbasecallerfornext generación.NatMethods 5: 679-82.
44.RougemontJ,Un Amzallag,IseliC,et.
2008.Probabilistic base de datos llamando ofSolexasequencing. BMC Bioinf. 9: 431.
45.R Drmanac,Sparks AB,Callow MJ,et.
2010.Humangenomesequencingusingunchained readson base de nanoarrays auto-assemblingDNA. ciencia 327: 78-81.
46.Applied Biosystems. UN Teórico
Comprensión de2 Base colorcodes andits
Aplicación a Anotación, error
Detección, andErrorCorrection.WhitePaper SóLIdASTMSistema; 2008.
47.Dimalanta ET,Zhang L,Hendrickson CL,et.2009.Increased Leer longitud en el
SólidoTMSequencingPlatform.PosterSOLiDTMSistema.
48.ZhuZ,WaggonerAS.1997.Molecularmechanism el control de la incorporación de
fluorescentnucleotides en el ADN por PCR.citometría 28: 206-11.
49.Bowers J,MitchellJ,cerveza E,et.2009.Virtualterminatornucleotidesfornext-
la secuenciación del ADN generación.NatMethods6: 593-5.
50.Pushkarev D,NeffNF,SR terremoto.2009.Single secuenciación de moléculas individuales ofan
Genoma humano. Nat Biotechnol 27: 847-52.
51.Reinhardt JA,Baltrus DA,Nishimura MT,et al. conjunto de 2009. De novo usando bajo tura
tura datos de la secuencia corta leídos del ricepathogen Pseudomonas syringae pv. oryzae.Genome Res 19: 294-305.
52.DiguistiniS,Liao NY,Platt D,et.2009.De Novo secuencia del genoma de montaje OFA
hongo filamentoso utilizando Sanger, 454 datos de secuencia andIllumina.Biol genoma10: R94.
53.Miller JR, Delcher AL, Koren S, et al. 2008.Aggressiveassemblyofpyrosequencing
lee con compañeros.bioinformática 24: 2818-
24.
54.Chen W, R Ullmann, Langnick C, et al. análisis 2009.Breakpoint del cromosoma equilibrada
reordenamientos de secuenciación de próxima generación de extremo emparejado.Eur J Hum GenetDol: 10.1038 / ejhg .2009.21118 [Epub ahead of print].
55.Zimin AV, Delcher AL, Florea L, et al. 2009.A ensamblaje del genoma completo Ofthe doméstica
vaca, Bos taurus. Biol genoma10: R42.
56.Zhou X,Su Z,Sammons RD,et.2009.Novelsoftware Paquete forcross-platformtranscriptomeanalysis (CPTRA).BMC
Bioinf. 11: S16.
57.Kim JI, Ju YS, parque H, et al. 2009. Un highlyannotatedwhole-genomesequenceofa
Corea del individuo. Naturaleza 460: 1011-5.
58.Linsen SE, de Wit E, Janssens G, et al. 2009.Limitations y posibilidades ofsmallRNA
digitalgeneexpressionprofiling.NatMethods 6: 474-6.
59.QuailMA,Swerdlow H,Turner DJ.2009.Improved protocolos para la Illumina Genoma
Analyzersequencing sistema.CurrProtocHum Genet Capítulo 18: Unidad de 18.2.
60.Briggs AW, Stenzel U, Meyer M, et al. 2009.Removal de citosinas deaminados y detección
ción deen vivo metilación en ancientDNA.Nucleic Acids Res 38(6): e87 [Epub aheadof de impresión].
61.T Maricic,Pääbo S.2009.Optimization of454 preparación biblioteca de secuenciación de pequeños
cantidades de ADN permite secuencia de determi-nación ofboth cadenas de ADN.Biotechniques46: 51-2, 54-7.
62.N Rohland, Horfreiter M.2007. Comparisonand extracción de optimización ofancientDNA.
Biotechniques 42: 343-52.
63.Wheeler DA, Srinivasan M, Egholm M, et al.
2008. El genoma completo de un individualby secuenciación del ADN masivamente paralelo. Naturaleza452: 872-6.
64.O Harismendy, PC ng, Strausberg RL, et al.
2009. Evaluación de los estudios sequenc-ingplatformsforpopulationtargetedsequencing de próxima generación. Biol genoma10: R32.
sesenta y cinco.Chaisson MJ, Brinza D, Pevzner PA. ensamblaje fragmento de novo con 2009.De corta mate-
emparejado lee: ¿Es importante la longitud de leer?Genome Res 19: 336-46.
66.Krause J, Briggs AW, Kircher M, et al. 2009.A ADNmt genoma completo de un mo- temprana
ern humana de Kostenki, Rusia.Curr Biol20: 231-6.
67.Un Gnirke, Un Melnikov, Maguire J, et al. 2009.Solution selección de híbridos con oli- ultra-larga
gonucleotides para masivamente paralelo dirigida
secuenciación. Nat Biotechnol27: 182-9.
......Métodos, Modelos y Técnicas M. y J. Kircher Kelso
Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc. 535
Méto
dos, M
odelo
s y Té
cnica
s

68.Hodges E,Rooks M,Xuan Z,etla selección de los intervalos de .2009.Hybrid genómicas discretas
en microarrays de diseño personalizado Formas-sivelyparallelsequencing.NatProtoc4: 960-74.
69.Briggs AW, bueno JM, RE verde, et al. 2009.Targetedretrievalandanalysisoffive
NeandertalmtDNA genomas.ciencia 325: 318-21.
70.MeyerM,StenzelU,HofreiterM.secuenciación 2008.Paralleltagged en la plataforma 454
formar. Nat protoc 3: 267-78.
71.Meyer M,StenzelU,Myles S,et.2007.Targetedhigh-Throughputsequencingof
etiquetados muestras de ácidos nucleicos.AcidsRes nucleicos 35: E97.
72.Y Erlich,Chang K,A Gordon,et.2009.DNA Sudoku - el aprovechamiento de alto rendimiento
secuenciación de muestras multiplexadas Analy-sis. Genome Res 19: 1243-1253.
73.Meyer M, Kircher M. 2010. Illumina sequenc-ing preparación para la biblioteca altamente multiplexados
orientar la captura y secuenciación.Fría SpringHarb protoc DOI: 10.1101 / pdb.prot5448.
74.pop M,Salzberg SL.2008.Bioinformaticschallenges ofnew tecnología de secuenciación.
Trends Genet 24: 142-9.
75.Richter BG, Sexton DP. 2009. La gestión de andanalyzingnext-generationsequencedata.
PLoS Comput Biol5: E1000369.
76.codornices MA, me Kozarewa, Smith F, et al. mejoras de 2008. Una gran centro al genoma
sistema de secuenciación de Illumina. Métodos Nat 5: 1005-1010.
77.BatleyJ,EdwardsD.2009.Genomesequence datos: gestión, almacenamiento, andvisualization. Biotechniques 46: 333-4, 336.
la investigación tiene como objetivo 78.IBM Research.2009.IBM tobuild secuenciador de ADN para ayudar a nanoescala unidad
costos bajos de análisis genético personalizado. InLoughran M, ed .; Notas de prensa, Vol. 2009.new York: IBM.
79.Eid J, Un Fehr, Gray J, et al. 2009. Real-timeDNA secuenciación de una sola moles polimerasa
ecules. ciencia 323: 133-8.
80.Y Astier, O Braha, Bayley H. 2006. TowardsinglemoleculeDNAsequencing: directa
ofribonucleoside identificación y deoxyri-bonucleoside 50-monophosphates por usingan ingeniería de proteínas nanoporos EQUIPPEDWITH un molecularadapter.Soc J Am Chem128: 1705-1710.
81.Albertorio F, Hughes ME, Golovchenko JA,et.2009.Base DependentDNA-carbono
interacciones de nanotubos: la activación enthalpiesand montaje disassemblycontrol.Nano-tecnología 20: 395.101.
82.Medvedev P,Stanciu M,Brudno M.2009.Computationalmethodsfordiscovering
structuralvariationwithnext-generationsequencing. Métodos Nat 6: S13-20.
83.PepkeS,Wold B,MortazaviA.2009.ComputationforChIP-seqandRNA-seqstudies. Métodos Nat 6: S22-32.
84.FlicekP,BirneyE.2009.Sensefromsequence lee: métodos de alineación y
Asamblea. Métodos Nat 6: S6-12.
85.Parque PJ.2009.ChIP-ss: ventajas andchallengesofa tecnología de maduración.Nat
Rev Genet 10: 669-80.
86.WallPK,Leebens-J Mack,ChanderbaliAS, et al. 2009. Comparación de la próxima generación
sequencingtechnologiesfortranscriptomecharacterization. BMC Genomics 10: 347.
87.Holt RA, SJ Jones. 2008. La nueva secuenciación celda de flujo paradigmof. Genome Res 18: 839-
46.
88.DalcaAV,BrudnoM.2010.Genomevariationdiscoverywithhigh rendimiento
los datos de secuenciación. Breve Bioinf. 11: 3-14.
M. y J. Kircher Kelso Métodos, Modelos y Técnicas
.....
536 Bioensayos 32: 524-536,! 2010 Wiley publicaciones periódicas, Inc.
Méto
dos,
Modelo
s y T
écn
icas