control teórico 4_leucemias
DESCRIPTION
leucemiasTRANSCRIPT

Atlas de Hematología – Leucemias Prof. Carmen Gloria Artigas 2005 LEUCEMIAS AGUDAS
Las Leucemias Agudas (LA) constituyen un grupo heterogéneo de neoplasias que afectan a las células madres hematopoyéticas de la médula ósea (MO). La leucemia surge tras la transformación maligna de un solo progenitor hematopoyético y puede ocurrir en diversas fases de la diferenciación celular, seguida por la replicación celular y la expansión del clon transformado. Las células leucémicas en proliferación se acumulan en la MO suprimiendo la hematopoyesis normal. La consiguiente escasez de progenitores normales da lugar a anemia, infecciones y complicaciones hemorrágicas que caracterizan a la enfermedad. Las células leucémicas proliferan en primer lugar en la MO, circulan por la sangre periférica (SP) y pueden infiltrar otros tejidos tales como: ganglios linfáticos, hígado, bazo, piel, encías, vísceras y sistema nervioso central (SNC). Las leucemias difieren entre sí respecto al origen celular, presentación clínica, evolución, manejo terapéutico e implicancias pronósticas.
Laboratorio: En el 95% de los casos se observa una anemia normocítica, normocrómica y arregenerativa, que obedece a la anulación del tejido hematopoyético medular por la proliferación leucocítica. A veces presenta un matiz megaloblástico, debido al excesivo consumo de ácido fólico por parte de las células leucémicas. La trombocitopenia está presente en el 80-90% de todas las LA, debido a la disminución de precursores medulares. La cifra de leucocitos sufre amplias variaciones; generalmente las formas hiperleucocitarias tienen peor pronóstico. La neutropenia es constante y faltan los elementos de estadio madurativo intermedio (hiatus leucémico), dominan en el hemograma las células blásticas, en porcentaje variable, con aspecto morfológico y comportamiento citoquímico distinto según el tipo de LA. La LA puede transcurrir también de forma aleucémica, donde no se detectan blastos periféricos. El aspecto de las células blásticas periféricas puede variar con respecto al de las células de MO, p.ej: los linfoblastos periféricos suelen ser más pequeños que los medulares, y el carácter monocitoide de la leucemia mielomonocítica es más acusado en la periférica que en la MO.
Diagnóstico: Requiere la demostración de células leucémicas en MO y/o SP. La MO es típicamente hipercelular, con una infiltración monomórfica de blastos leucémicos y una reducción marcada de los elementos normales de la médula. El diagnóstico del tipo de leucemia se basa actualmente en una combinación de características morfológicas, citoquímicas e inmunofenotípicas, así como en la evidencia de translocaciones genéticas específicas.

La blastosis medular suele oscilar entre el 50-100% de la totalidad celular, y su aspecto varía, según el tipo morfológico. Se constata una disminución de las células hematopoyéticas normales. Algunos pacientes con LA y pancitopenia tienen una médula hipocelular, lo que indica que las células leucémicas pueden inhibir directamente la hematopoyesis normal por mecanismos celulares o humorales. Las células hematopoyéticas primitivas normales permanecen en la MO y son capaces de proliferar y restablecer la hematopoyesis después de un tratamiento antileucémico eficaz.
Clasificación: La clasificación de las leucemias está relacionada con el tipo de célula involucrada y con la rapidez del curso clínico (leucemias agudas o crónicas). En 1976 y posteriormente en 1985, el grupo cooperativo franco-americano-británico(FAB), elaboró una clasificación para las leucemias agudas basándose en el análisis citomorfológico de la SP , MO y en el comportamiento citoquímico de los blastos leucémicos. Esta clasificación de las LA ampliamente aceptada, se conoce como: Clasificación FAB: Se distinguen dos tipos clásicos de LA: * Linfoblásticas con tres subtipos linfoides (L1, L2, L3). * Mieloblásticas o no linfoblásticas inicialmente con seis subtipos mieloides (M1, M2, M3, M4, M5, M6), incluyendo posteriormente los sutipos M0 y M7. Mediante la clasificación FAB, en la mayoría de los casos de LA, se logra establecer el diagnóstico, sin embargo, la heterogeneidad de las células mieloides y la distinta funcionalidad de las células linfoides, limita el correcto reconocimiento de los blastos leucémicos; por ello, en la actualidad la clasificación de las LA incluye además, el inmunofenotipo, estudio citogenético y molecular. LEUCEMIAS AGUDAS LINFOBLASTICAS (LAL)
Se distingue por su origen en precursores linfopoyéticos primitivos provenientes de la MO, timo o ganglios linfáticos y por su marcada tendencia a afectar especialmente a los niños. Las manifestaciones clínicas se deben a la falta de producción de células normales en la MO, causada por la infiltración de blastos leucémicos. También puede haber infiltración de blastos en el hígado, bazo, ganglios linfáticos y SNC. La falla medular se traduce en la aparición de la anemia, púrpura, ulceraciones bucales y fiebre. Con frecuencia, la primera manifestación ha sido la instalación de molestias sugerentes de una infección respiratoria alta, que persiste por más tiempo de lo esperado.
Laboratorio: El hemograma, por lo habitual muestra anemia normocítica, normocrómica y no regenerativa, neutropenia, trombocitopenia y la presencia de linfoblastos. La cifra de leucocitos es muy variable, puede ser normal o bien detectarse leucopenia o leucocitosis; en el 30% de los enfermos con LAL se obtienen cifras menores de 5x109/l, en el 20% los recuentos son normales y en el 50% hay leucocitosis con un promedio de

50x109/l, pudiendo superar los 100x109/l . La proporción de blastos en la sangre periférica es asimismo variable, ya que pueden no observarse (LAL aleucémicas) o constituir la totalidad de las células. En las LAL aleucémicas, la fórmula leucocitaria es anormal (a menudo hay granulocitopenia) y pueden observarse eritroblastos circulantes, dato que sugiere la infiltración de la MO por células extrañas. La cifra de plaquetas suele hallarse disminuida en la mayoría de los casos, con frecuencia por debajo de 50x10 9/l. Desde un punto de vista morfológico, se caracteriza por la ausencia de elementos de maduración mieloide, ya sea en las tinciones corrientes o en las citoquímicas, entre estas últimas la más importante es la negatividad de la mieloperoxidasa.
Diagnóstico: Como en toda leucemia aguda, el diagnóstico se establece por la presencia de más de un 30% de blastos en el total de las células nucleadas de la MO. Habitualmente la MO es hipercelular, la infiltración por blastos es masiva y no se observan células maduras de la hematopoyesis normal. Aunque la observación morfológica y el estudio citoquímico convencional permiten diagnosticar y diferenciar la gran mayoría de LAL según la clasificación FAB, es obligatorio efectuar en forma sistemática el estudio inmunofenotípico y citogenético de los blastos.
Inmunofenotipo: Este estudio ha permitido subdividir a las LAL en dos grandes grupos: LAL-B y LAL-T. Se considera que el 80% de las LAL tienen origen B y un 15-25% tienen origen T. Dentro de la estirpe B, se incluyen la LAL pre-pre-B (que incluye a la LAL “nula” y LAL “común”), la LAL pre-B y la LAL-B madura. Dentro de las LAL-T se consideran las pre-T y la T o tímicas (cortical y madura). Por último, hasta un 15% de las LAL del adulto presentan marcadores mieloides.
Citogenética: Se han encontrado anomalías cromosómicas en el 75-90% de las LAL. Entre ellas cabe destacar las numéricas (aneuploidías) y las estructurales (en especial las translocaciones, deleciones e inversiones). Se encuentra hiperdiploidía en un 40% de los casos de LAL y suele ser de buen pronóstico (excepto cuando el número de cromosomas es casi tetraploide, es decir, entre 82 y 92). A su vez, un 5-10% de LAL presentan hipodiploidía, suele asociarse a un pronóstico desfavorable. Las alteraciones estructurales se dan en el 40-50% de LAL y las más frecuentes son las translocaciones, cuya existencia es signo en general de mal pronóstico. Entre ellas destacan la t(1;19), t(9;22) y t(4;11); coincidiendo con origen B. La t(1;19) se da en LAL pre-B, habitualmente tiene morfología L1 (15% de los casos) y es de pronóstico muy desfavorable. La t(9;22) (Ph1+) se presenta en el 5% de los niños y en más del 20% de los adultos, la morfología generalmente es L2 y ocasionalmente L1. Las LAL Ph1+ tienen un pronóstico muy desfavorable. La t(4;11) se da, sobre todo, en LAL infantiles y se asocia a leucocitosis, infiltración del SNC, fenotipo mixto (linfoide y mieloide o monocítico); su pronóstico también es muy desfavorable; presentan morfología L1 o L2 y fenotipo de precursores B
precoz. La t(8;14) se encuentra en las LAL-L3 y fenotipo B maduro. La t(11;14) es específica de las LAL-T.
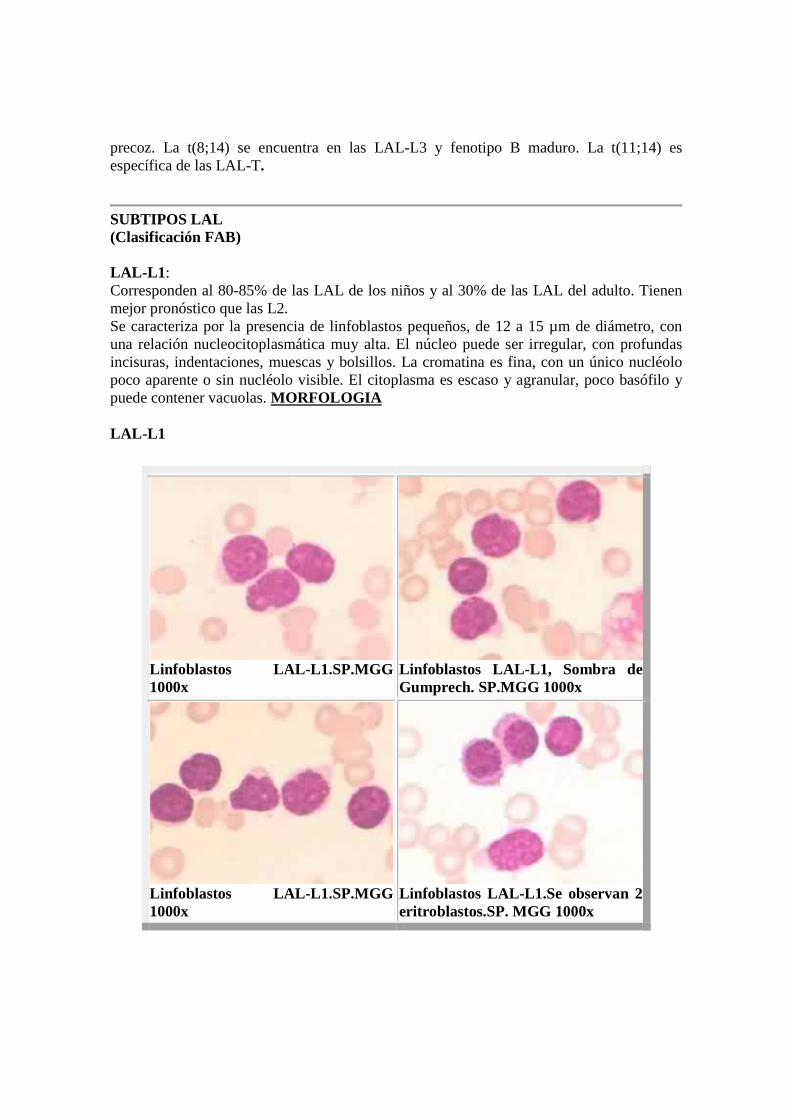
SUBTIPOS LAL (Clasificación FAB) LAL-L1 : Corresponden al 80-85% de las LAL de los niños y al 30% de las LAL del adulto. Tienen mejor pronóstico que las L2. Se caracteriza por la presencia de linfoblastos pequeños, de 12 a 15 µm de diámetro, con una relación nucleocitoplasmática muy alta. El núcleo puede ser irregular, con profundas incisuras, indentaciones, muescas y bolsillos. La cromatina es fina, con un único nucléolo poco aparente o sin nucléolo visible. El citoplasma es escaso y agranular, poco basófilo y puede contener vacuolas. MORFOLOGIA LAL-L1
Linfoblastos LAL- L1.SP.MGG 1000x
Linfoblastos LAL- L1, Sombra de Gumprech. SP.MGG 1000x
Linfoblastos LAL- L1.SP.MGG 1000x
Linfoblastos LAL- L1.Se observan 2 eritroblastos.SP. MGG 1000x
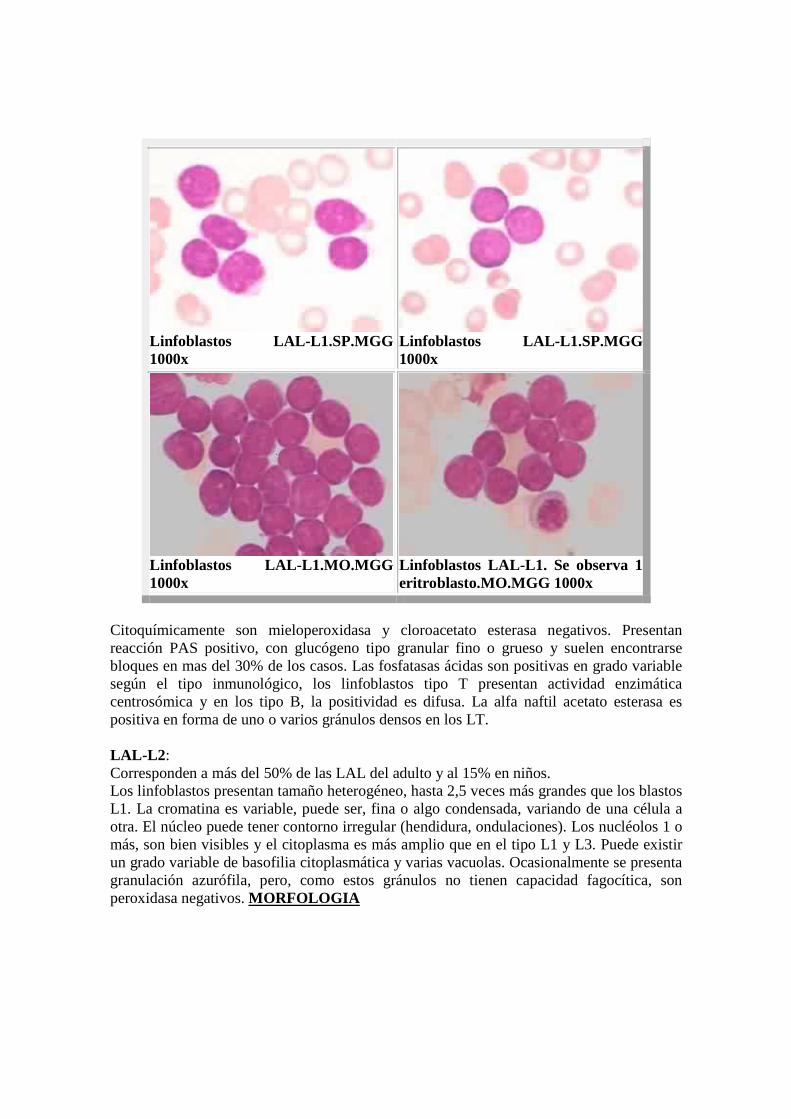
Linfoblastos LAL- L1.SP.MGG 1000x
Linfoblastos LAL- L1.SP.MGG 1000x
Linfoblastos LAL- L1.MO.MGG 1000x
Linfoblastos LAL- L1. Se observa 1 eritroblasto.MO.MGG 1000x
Citoquímicamente son mieloperoxidasa y cloroacetato esterasa negativos. Presentan reacción PAS positivo, con glucógeno tipo granular fino o grueso y suelen encontrarse bloques en mas del 30% de los casos. Las fosfatasas ácidas son positivas en grado variable según el tipo inmunológico, los linfoblastos tipo T presentan actividad enzimática centrosómica y en los tipo B, la positividad es difusa. La alfa naftil acetato esterasa es positiva en forma de uno o varios gránulos densos en los LT. LAL-L2 : Corresponden a más del 50% de las LAL del adulto y al 15% en niños. Los linfoblastos presentan tamaño heterogéneo, hasta 2,5 veces más grandes que los blastos L1. La cromatina es variable, puede ser, fina o algo condensada, variando de una célula a otra. El núcleo puede tener contorno irregular (hendidura, ondulaciones). Los nucléolos 1 o más, son bien visibles y el citoplasma es más amplio que en el tipo L1 y L3. Puede existir un grado variable de basofilia citoplasmática y varias vacuolas. Ocasionalmente se presenta granulación azurófila, pero, como estos gránulos no tienen capacidad fagocítica, son peroxidasa negativos. MORFOLOGIA
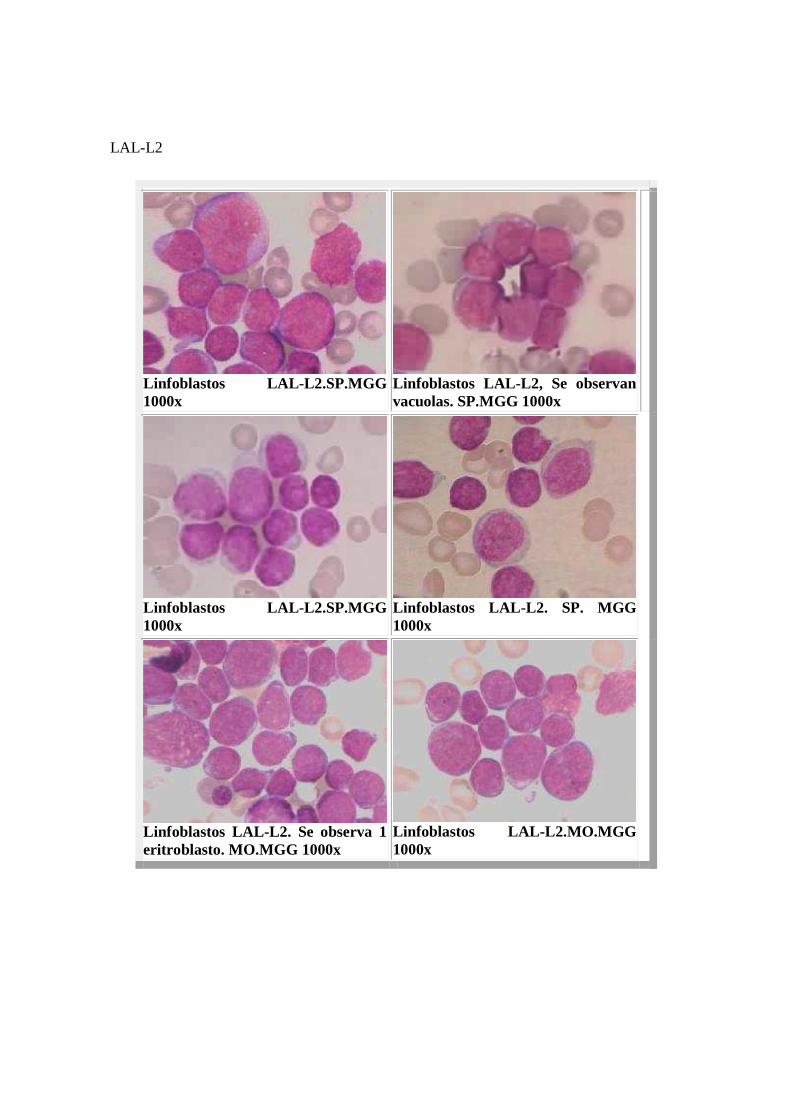
LAL-L2
Linfoblastos LAL- L2.SP.MGG 1000x
Linfoblastos LAL- L2, Se observan vacuolas. SP.MGG 1000x
Linfoblastos LAL- L2.SP.MGG 1000x
Linfoblastos LAL-L2. SP. MGG 1000x
Linfoblastos LAL- L2. Se observa 1 eritroblasto. MO.MGG 1000x
Linfoblastos LAL- L2.MO.MGG 1000x
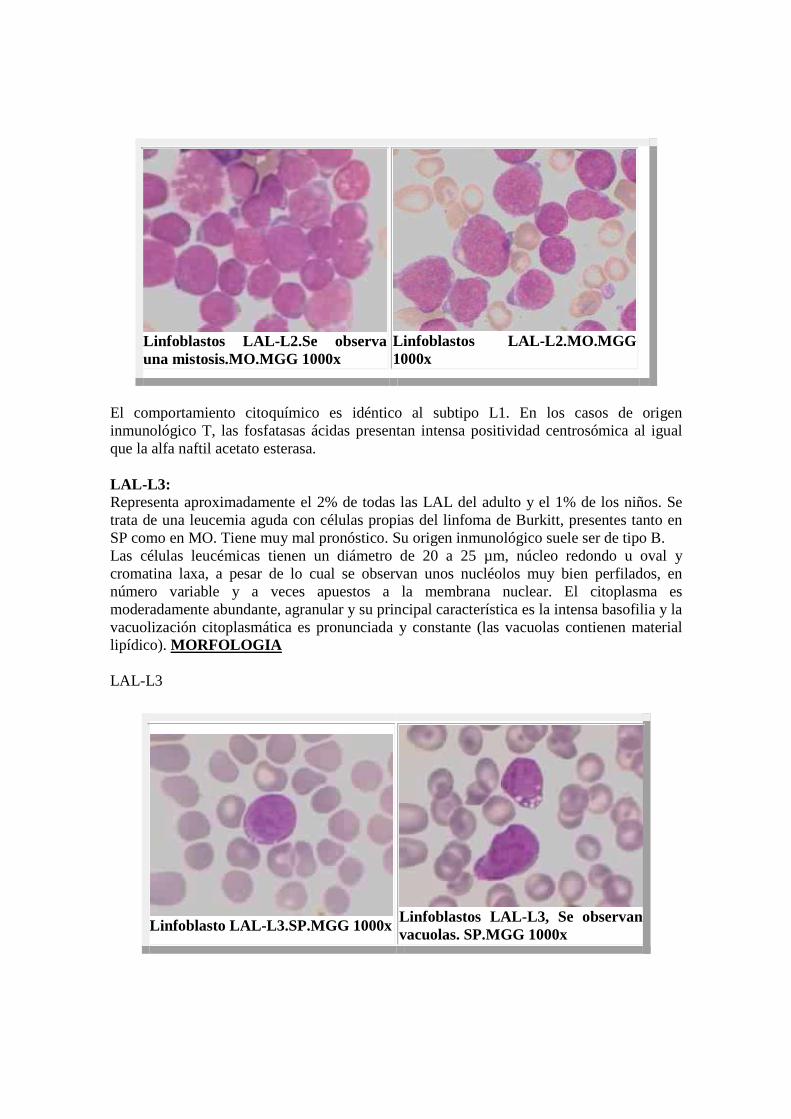
Linfoblastos LAL- L2.Se observa una mistosis.MO.MGG 1000x
Linfoblastos LAL- L2.MO.MGG 1000x
El comportamiento citoquímico es idéntico al subtipo L1. En los casos de origen inmunológico T, las fosfatasas ácidas presentan intensa positividad centrosómica al igual que la alfa naftil acetato esterasa. LAL-L3: Representa aproximadamente el 2% de todas las LAL del adulto y el 1% de los niños. Se trata de una leucemia aguda con células propias del linfoma de Burkitt, presentes tanto en SP como en MO. Tiene muy mal pronóstico. Su origen inmunológico suele ser de tipo B. Las células leucémicas tienen un diámetro de 20 a 25 µm, núcleo redondo u oval y cromatina laxa, a pesar de lo cual se observan unos nucléolos muy bien perfilados, en número variable y a veces apuestos a la membrana nuclear. El citoplasma es moderadamente abundante, agranular y su principal característica es la intensa basofilia y la vacuolización citoplasmática es pronunciada y constante (las vacuolas contienen material lipídico). MORFOLOGIA LAL-L3
Linfoblasto LAL-L3.SP.MGG 1000x
Linfoblastos LAL- L3, Se observan vacuolas. SP.MGG 1000x
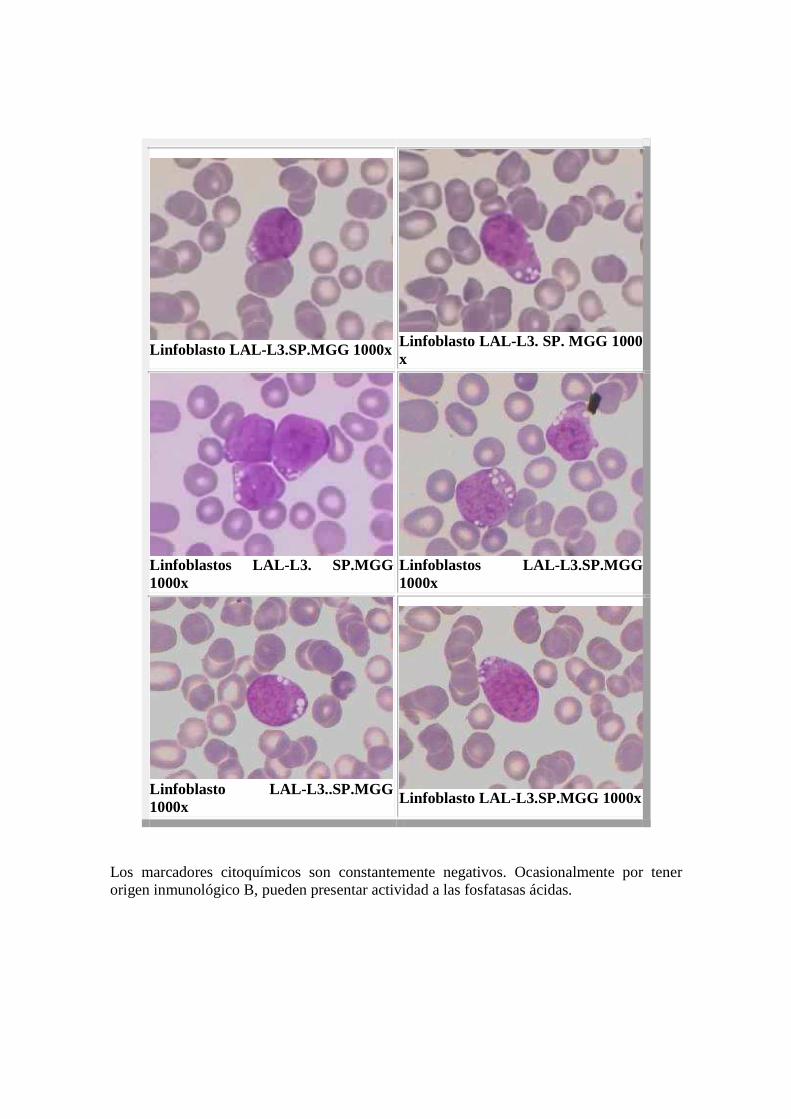
Linfoblasto LAL-L3.SP.MGG 1000x Linfoblasto LAL-L3. SP. MGG 1000
x
Linfoblastos LAL-L3. SP.MGG 1000x
Linfoblastos LAL- L3.SP.MGG 1000x
Linfoblasto LAL- L3..SP.MGG 1000x Linfoblasto LAL-L3.SP.MGG 1000x
Los marcadores citoquímicos son constantemente negativos. Ocasionalmente por tener origen inmunológico B, pueden presentar actividad a las fosfatasas ácidas.

LEUCEMIAS AGUDAS MIELOBLASTICAS (LAM)
Las leucemias agudas mieloblásticas o no linfoblásticas constituyen un grupo de proliferaciones neoplásicas de las células mielopoyéticas primitivas, con características clínicas similares, pero con caracteres morfológicos, citoquímicos y citogenéticos diferentes. El cuadro clínico está en relación con la infiltración de la MO y de tejidos extramedulares por las células leucémicas, así como con el número de dichas células en la sangre y liberación de sustancias intracelulares. Los enfermos presentan un cuadro hemorrágico o infeccioso de instauración rápida con afectación del estado general. La infiltración de la MO produce un desplazamiento de las células hematopoyéticas normales, lo que conlleva un déficit en la producción de eritrocitos, leucocitos y plaquetas, que se manifiesta clínicamente en tres grandes síndromes: anémico, febril y hemorrágico. Corresponden al 80% de los casos de LA del adulto y al 20-25% de las LA de los niños.
Laboratorio: Los hallazgos característicos son anemia, neutropenia y trombocitopenia. Habitualmente presentan anemia normocítica, normocrómica y discretamente hiporregenerativa. En el 80-90% de los pacientes la hemoglobina es <11 g/dl, en más de la mitad <9 g/dl y <7 g/dl en el 15-20%. La mayoría de los pacientes presenta leucocitosis >20x109/l, y en el 20-25% de los casos se observan cifras >100x109/l, esta hiperleucocitosis en base a blastos en SP, es considerada un factor desfavorable por producir aumento de la viscosidad, lo que dificulta la circulación sanguínea (leucostasis), por formación de agregados de células blásticas poco deformables y/o invasión de la pared vascular; los órganos más afectados son el SNC y los pulmones, originando una insuficiencia respiratoria aguda y/o un cuadro neurológico. Algunos pacientes tienen cifras de leucocitos normales o inferiores a 5x109/l. Junto a la neutropenia, el 80-85% de los pacientes presentan blastos. Pueden observarse alteraciones displásicas de los neutrófilos, de las plaquetas, así como macroovalocitosis en los hematíes. La cifra de plaquetas generalmente es <50x109/l y frecuentemente <20x109/l. En algunos casos puede presentarse sin anemia o trombocitopenia. Hay elevación de LDH, ácido úrico, lisozima, hipopotasemia, hipomagnesemia, hipocalcemia (hiperfosforemia) o hipercalcemia. En caso de hiperleucocitosis, se detectan falsas hipoglucemias, hipoxia e hiperpotasemia
Diagnóstico: Generalmente puede establecerse el diagnóstico por el examen citomorfológico de SP y MO, junto con algunas reacciones citoquímicas y una historia clínica detallada; sin embargo, muchas veces para 9/lidentificación precisa del subtipo mieloide, se hace necesario el estudio inmunofenotípico, citogenético y molecular. El diagnóstico se plantea inicialmente, al encontrar un 30% o más de blastos mieloides en la MO.

La MO es hipercelular, con ausencia o disminución del número de megacariocitos, las células blásticas(>30%) desplazan a la hematopoyesis normal. Sólo un 5-10% de los pacientes con LAM muestran un aspirado hipocelular. Inmunofenotipo: La determinación del inmunofenotipo ha sido de gran ayuda para identificar casos de morfología indiferenciada, muy atípica y bifenotípica. Los anticuerpos monoclonales (AcMo) más utilizados para la diferenciación mieloide son: CD13 y CD33 para la línea granulocítica; CD14 y CD33 para la línea monocítica; CD34 como Ag precoz para los blastos más inmaduros. Para la diferenciación eritroblástica se usa antiglicoforina A y para la diferenciación megacariocítica CD41(antiglicoproteína IIb/IIIa) y CD42 (glicoproteína Ib). Citogenética: Este estudio ha adquirido gran importancia por existir cierta concordancia entre cambios cromosómicos y la localización de oncogenes celulares, que estarían comprometidos en la patogénesis de la enfermedad. En el 70-85% de los pacientes es posible detectar alteraciones cromosómicas, siendo las más frecuentes la trisomía 8 y anomalía del cromosoma 7. Existe una correlación de la morfología FAB con las alteraciones citogenéticas: M1 con t(9;22), M2 con t(8;21), M3 con t(15;17), M4 eosinófila con inversión del cr.16, M4 ó M5 con t(1;11) y t(9;11), M5a con t(9;11). Las alteraciones cromosómicas son un factor pronóstico para los pacientes, pudiendo ser factores favorables o desfavorables. SUBTIPOS MIELOIDES (Clasificación FAB) LAM-M0 o Mieloblástica indiferenciada: Puede representar un 2-3% de todas las LAM. Se define como una leucemia aguda con un grado de diferenciación mínima, morfología L2 o indiferenciada, negativa para la tinción citoquímica de mieloperoxidasa y esterasa. Además presentan negatividad a todas las reacciones citoquímicas. El compromiso mieloide se basa en la expresión de uno o más marcadores mieloides (CD13, CD33 o MPO), mientras que los marcadores específicos de la línea linfoide son negativos. Los blastos son preferentemente de tamaño mediano y poseen un núcleo redondo con cromatina fina y citoplasma basófilo agranular; una proporción de blastos muestra nucléolos prominentes. Para el diagnóstico de la LAM-M0 es conveniente realizar además estudio citogenético o molecular de la población leucémica. En general este tipo de leucemia presenta un pronóstico desfavorable, uno de los factores de mal pronóstico es el hallazgo frecuente de alteraciones cromosómicas.
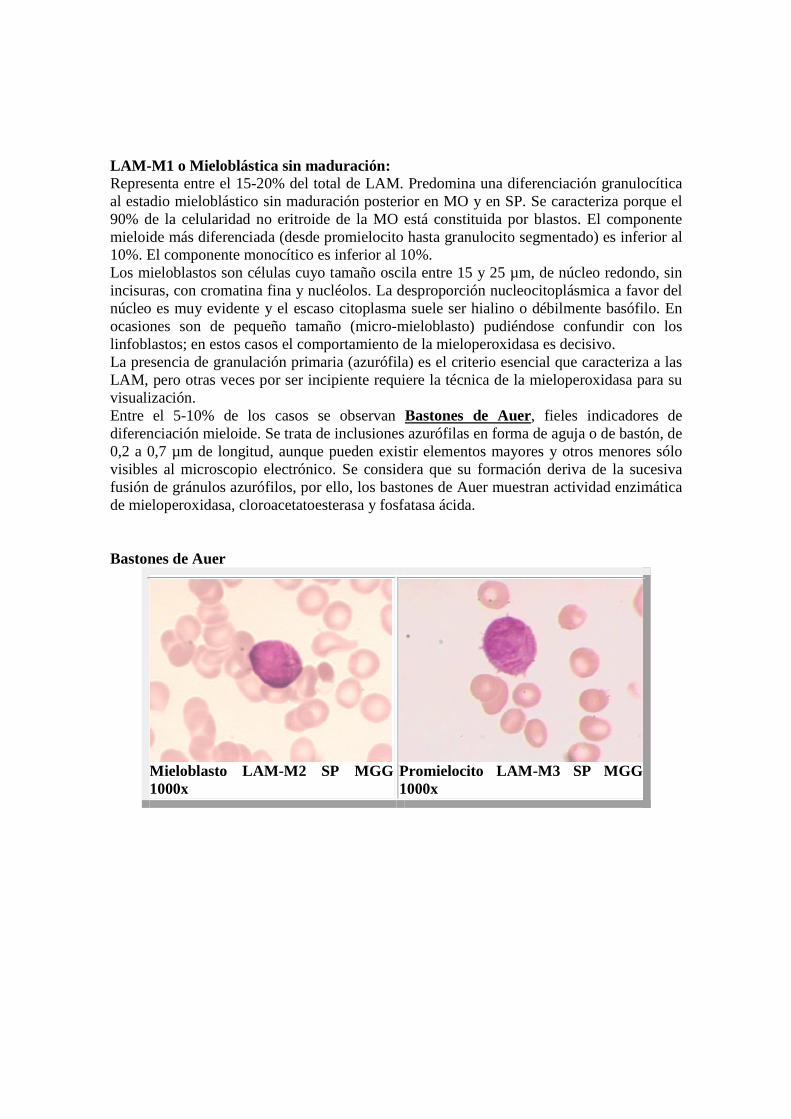
LAM-M1 o Mieloblástica sin maduración: Representa entre el 15-20% del total de LAM. Predomina una diferenciación granulocítica al estadio mieloblástico sin maduración posterior en MO y en SP. Se caracteriza porque el 90% de la celularidad no eritroide de la MO está constituida por blastos. El componente mieloide más diferenciada (desde promielocito hasta granulocito segmentado) es inferior al 10%. El componente monocítico es inferior al 10%. Los mieloblastos son células cuyo tamaño oscila entre 15 y 25 µm, de núcleo redondo, sin incisuras, con cromatina fina y nucléolos. La desproporción nucleocitoplásmica a favor del núcleo es muy evidente y el escaso citoplasma suele ser hialino o débilmente basófilo. En ocasiones son de pequeño tamaño (micro-mieloblasto) pudiéndose confundir con los linfoblastos; en estos casos el comportamiento de la mieloperoxidasa es decisivo. La presencia de granulación primaria (azurófila) es el criterio esencial que caracteriza a las LAM, pero otras veces por ser incipiente requiere la técnica de la mieloperoxidasa para su visualización. Entre el 5-10% de los casos se observan Bastones de Auer, fieles indicadores de diferenciación mieloide. Se trata de inclusiones azurófilas en forma de aguja o de bastón, de 0,2 a 0,7 µm de longitud, aunque pueden existir elementos mayores y otros menores sólo visibles al microscopio electrónico. Se considera que su formación deriva de la sucesiva fusión de gránulos azurófilos, por ello, los bastones de Auer muestran actividad enzimática de mieloperoxidasa, cloroacetatoesterasa y fosfatasa ácida. Bastones de Auer
Mieloblasto LAM- M2 SP MGG 1000x
Promielocito LAM- M3 SP MGG 1000x
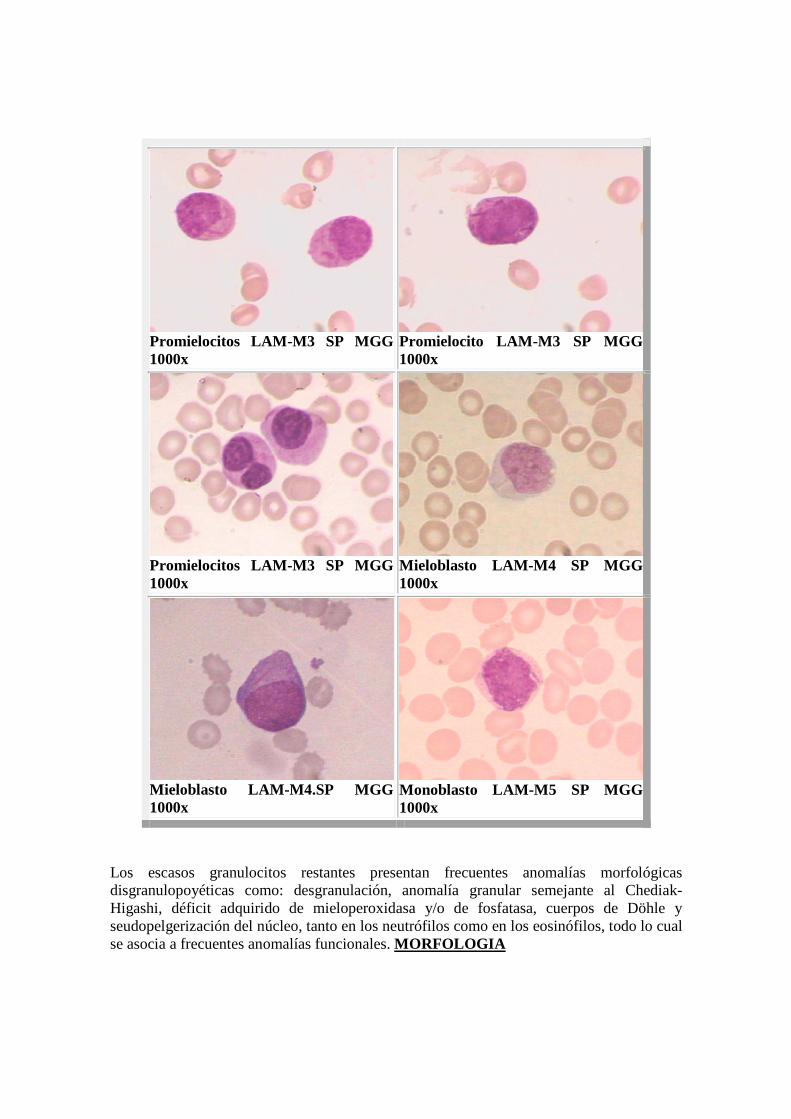
Promielocitos LAM-M3 SP MGG 1000x
Promielocito LAM- M3 SP MGG 1000x
Promielocitos LAM-M3 SP MGG 1000x
Mieloblasto LAM- M4 SP MGG 1000x
Mieloblasto LAM- M4.SP MGG 1000x
Monoblasto LAM-M5 SP MGG 1000x
Los escasos granulocitos restantes presentan frecuentes anomalías morfológicas disgranulopoyéticas como: desgranulación, anomalía granular semejante al Chediak-Higashi, déficit adquirido de mieloperoxidasa y/o de fosfatasa, cuerpos de Döhle y seudopelgerización del núcleo, tanto en los neutrófilos como en los eosinófilos, todo lo cual se asocia a frecuentes anomalías funcionales. MORFOLOGIA
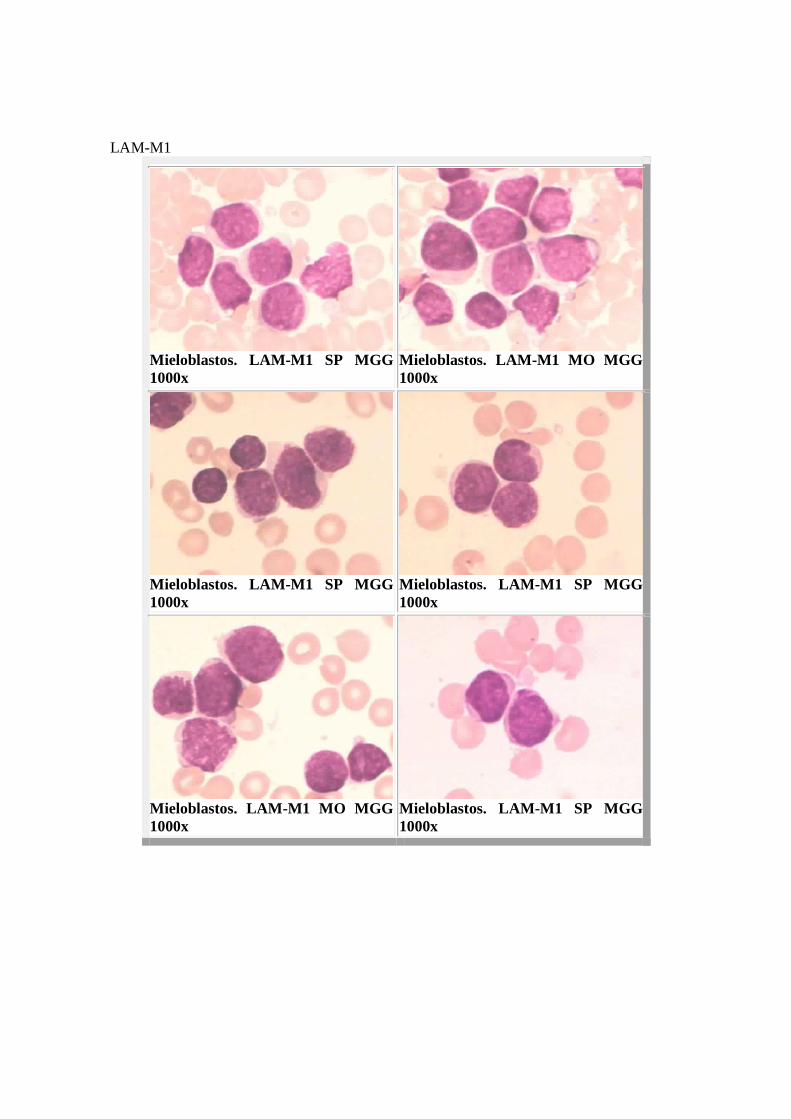
LAM-M1
Mieloblastos. LAM-M1 SP MGG 1000x
Mieloblastos. LAM-M1 MO MGG 1000x
Mieloblastos. LAM-M1 SP MGG 1000x
Mieloblastos. LAM-M1 SP MGG 1000x
Mieloblastos. LAM-M1 MO MGG 1000x
Mieloblastos. LAM-M1 SP MGG 1000x

Mieloblastos. LAM-M1 MO MGG 1000x
Mieloblastos. LAM-M1 MO MGG 1000x
En las reacciones citoquímicas para la clasificación mieloide, se exige que más del 3% de los blastos sean positivos a la mieloperoxidasa o al negro sudán B y a la cloroacetato esterasa, puesto que un porcentaje inferior podría corresponder a algún mieloblasto de la hematopoyesis normal residual de una LAL. La reacción de PAS es positiva débil y difusa. También presentan positividad difusa a las fosfatasas ácidas y son tartratosensible.
Constituyen el 30% de todas las LAM. Se caracteriza por la maduración hasta el estadio promielocítico o más allá. Los mieloblastos y promielocitos superan el 40-50% de la celularidad no eritroide, el componente granulocítico semimaduro es superior al 10% y con frecuencia corresponden a la serie eosinófila. Los mieloblastos presentan las mismas características morfológicas referidas para el subtipo M1, donde destaca la presencia de abundante granulación azurófila, tanto en mieloblastos como en promielocitos. Habitualmente hay de bastones de Auer. Los mielocitos, juveniles y granulocitos maduros muestran a menudo intensos rasgos disgranulopoyéticos, tales como desgranulación, cuerpos de Döhle y el aspecto seudo-Pelger del núcleo. MORFOLOGIA
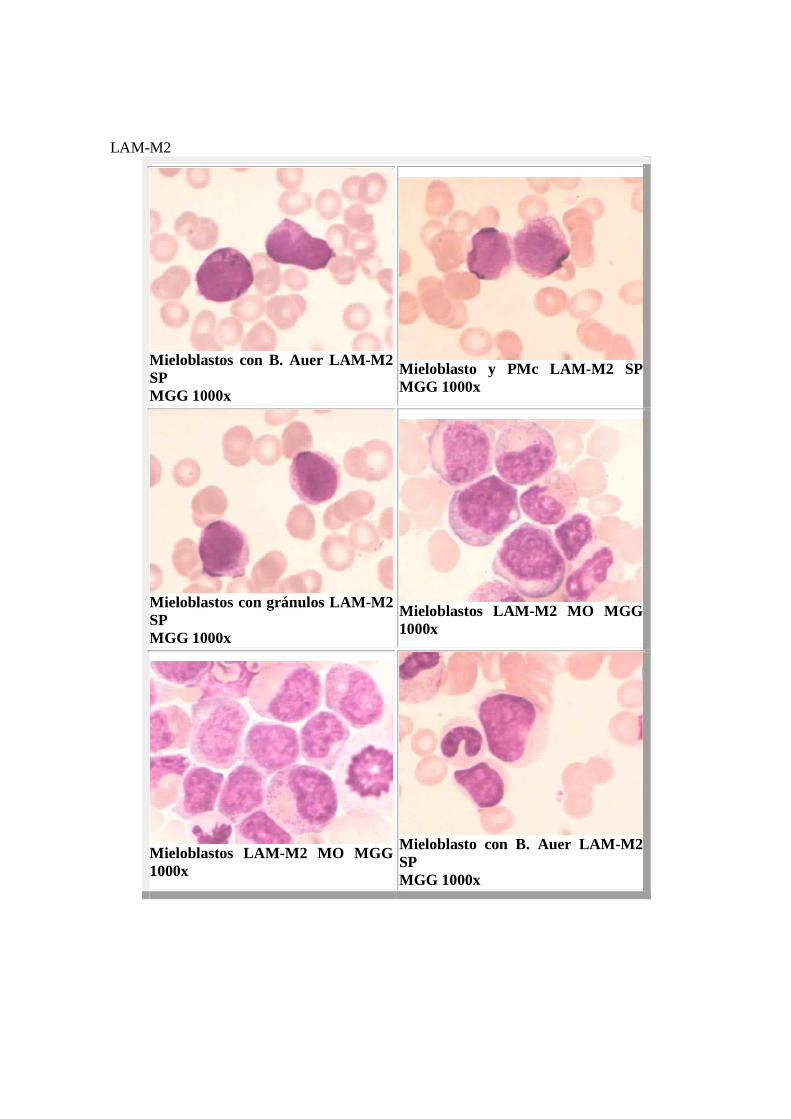
LAM-M2
Mieloblastos con B. Auer LAM-M2 SP MGG 1000x
Mieloblasto y PMc LAM- M2 SP MGG 1000x
Mieloblastos con gránulos LAM-M2 SP MGG 1000x
Mieloblastos LAM-M2 MO MGG 1000x
Mieloblastos LAM-M2 MO MGG 1000x
Mieloblasto con B. Auer LAM-M2 SP MGG 1000x
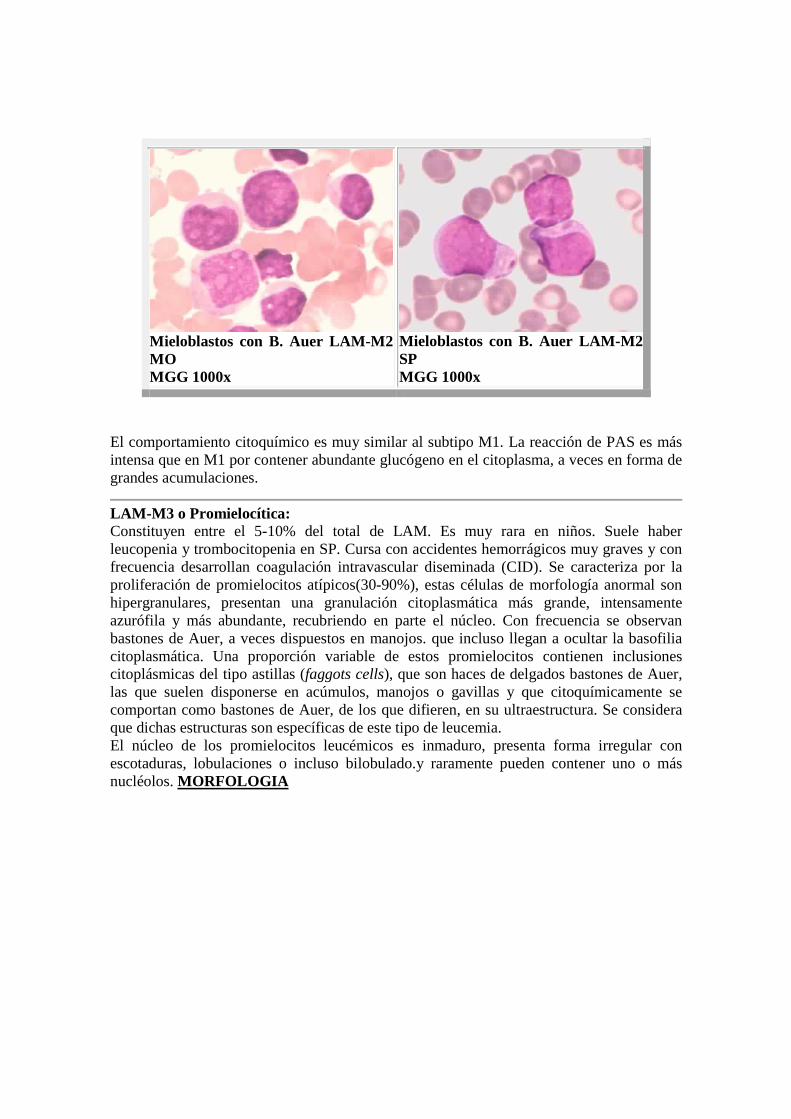
Mieloblastos con B. Auer LAM-M2 MO MGG 1000x
Mieloblastos con B. Auer LAM-M2 SP MGG 1000x
El comportamiento citoquímico es muy similar al subtipo M1. La reacción de PAS es más intensa que en M1 por contener abundante glucógeno en el citoplasma, a veces en forma de grandes acumulaciones.
LAM-M3 o Promielocítica: Constituyen entre el 5-10% del total de LAM. Es muy rara en niños. Suele haber leucopenia y trombocitopenia en SP. Cursa con accidentes hemorrágicos muy graves y con frecuencia desarrollan coagulación intravascular diseminada (CID). Se caracteriza por la proliferación de promielocitos atípicos(30-90%), estas células de morfología anormal son hipergranulares, presentan una granulación citoplasmática más grande, intensamente azurófila y más abundante, recubriendo en parte el núcleo. Con frecuencia se observan bastones de Auer, a veces dispuestos en manojos. que incluso llegan a ocultar la basofilia citoplasmática. Una proporción variable de estos promielocitos contienen inclusiones citoplásmicas del tipo astillas (faggots cells), que son haces de delgados bastones de Auer, las que suelen disponerse en acúmulos, manojos o gavillas y que citoquímicamente se comportan como bastones de Auer, de los que difieren, en su ultraestructura. Se considera que dichas estructuras son específicas de este tipo de leucemia. El núcleo de los promielocitos leucémicos es inmaduro, presenta forma irregular con escotaduras, lobulaciones o incluso bilobulado.y raramente pueden contener uno o más nucléolos. MORFOLOGIA
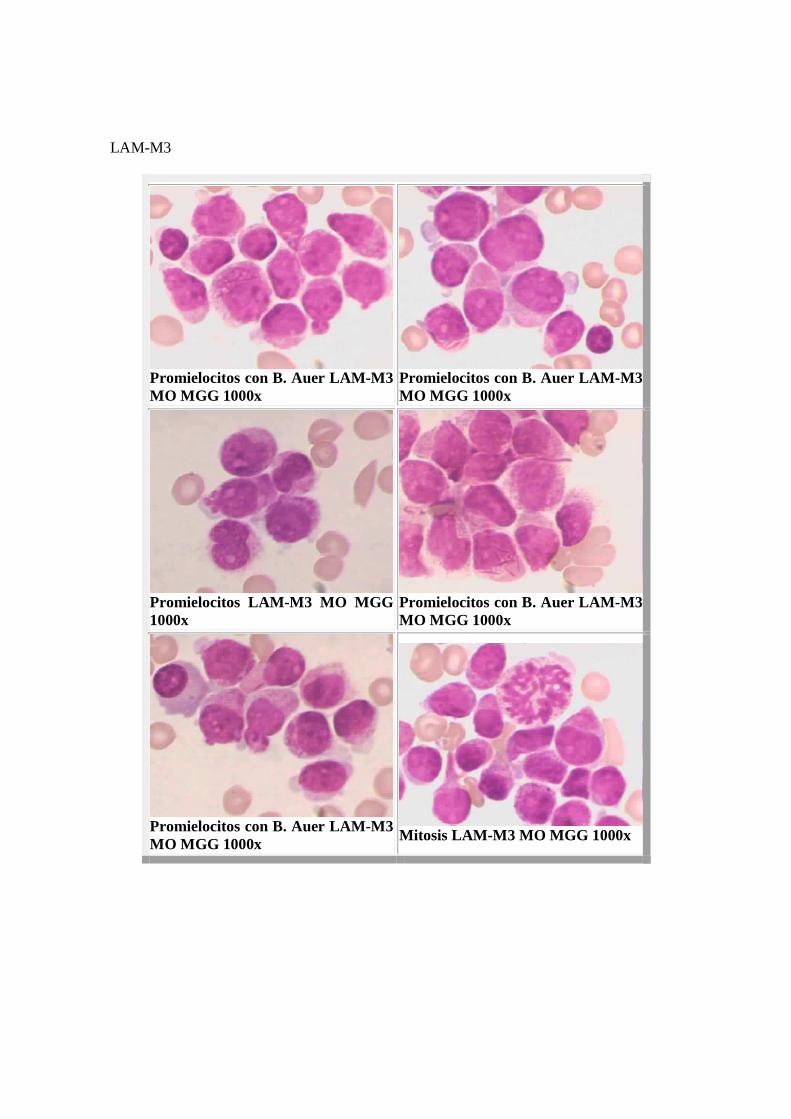
LAM-M3
Promielocitos con B. Auer LAM-M3 MO MGG 1000x
Promielocitos con B. Auer LAM-M3 MO MGG 1000x
Promielocitos LAM-M3 MO MGG 1000x
Promielocitos con B. Auer LAM-M3 MO MGG 1000x
Promielocitos con B. Auer LAM-M3 MO MGG 1000x
Mitosis LAM-M3 MO MGG 1000x
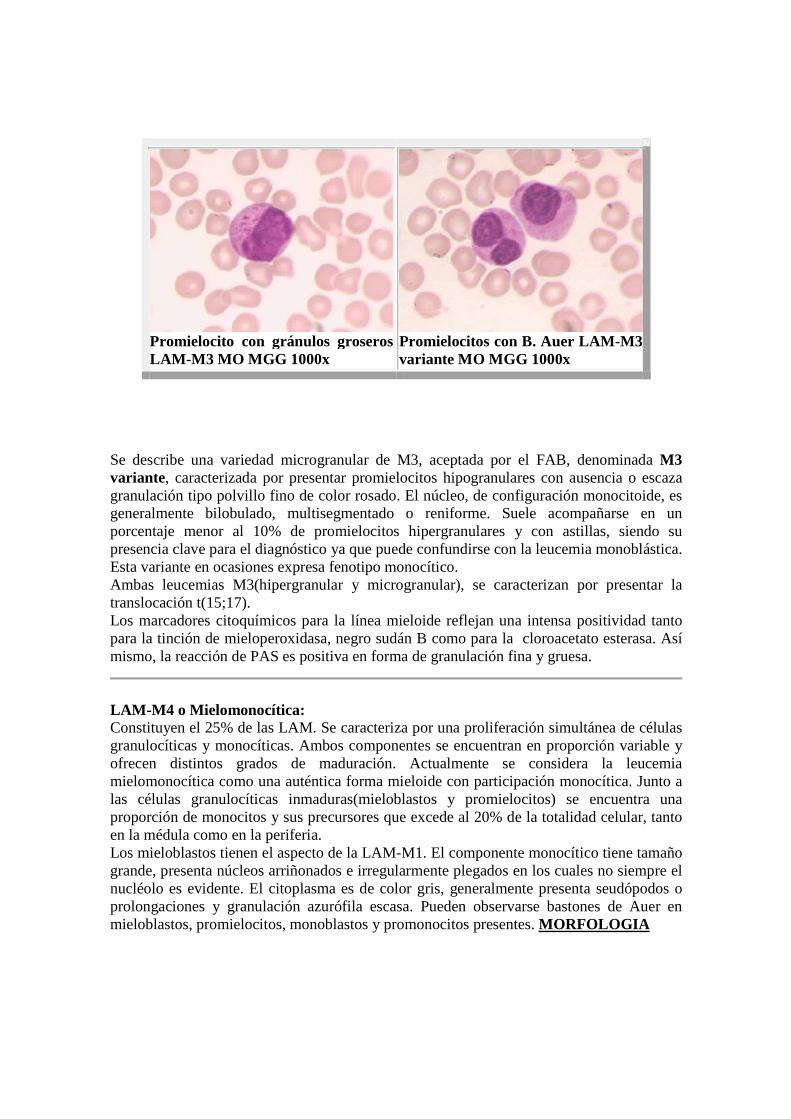
Promielocito con gránulos groseros LAM-M3 MO MGG 1000x
Promielocitos con B. Auer LAM-M3 variante MO MGG 1000x
Se describe una variedad microgranular de M3, aceptada por el FAB, denominada M3 variante, caracterizada por presentar promielocitos hipogranulares con ausencia o escaza granulación tipo polvillo fino de color rosado. El núcleo, de configuración monocitoide, es generalmente bilobulado, multisegmentado o reniforme. Suele acompañarse en un porcentaje menor al 10% de promielocitos hipergranulares y con astillas, siendo su presencia clave para el diagnóstico ya que puede confundirse con la leucemia monoblástica. Esta variante en ocasiones expresa fenotipo monocítico. Ambas leucemias M3(hipergranular y microgranular), se caracterizan por presentar la translocación t(15;17). Los marcadores citoquímicos para la línea mieloide reflejan una intensa positividad tanto para la tinción de mieloperoxidasa, negro sudán B como para la cloroacetato esterasa. Así mismo, la reacción de PAS es positiva en forma de granulación fina y gruesa.
LAM-M4 o Mielomonocítica: Constituyen el 25% de las LAM. Se caracteriza por una proliferación simultánea de células granulocíticas y monocíticas. Ambos componentes se encuentran en proporción variable y ofrecen distintos grados de maduración. Actualmente se considera la leucemia mielomonocítica como una auténtica forma mieloide con participación monocítica. Junto a las células granulocíticas inmaduras(mieloblastos y promielocitos) se encuentra una proporción de monocitos y sus precursores que excede al 20% de la totalidad celular, tanto en la médula como en la periferia. Los mieloblastos tienen el aspecto de la LAM-M1. El componente monocítico tiene tamaño grande, presenta núcleos arriñonados e irregularmente plegados en los cuales no siempre el nucléolo es evidente. El citoplasma es de color gris, generalmente presenta seudópodos o prolongaciones y granulación azurófila escasa. Pueden observarse bastones de Auer en mieloblastos, promielocitos, monoblastos y promonocitos presentes. MORFOLOGIA
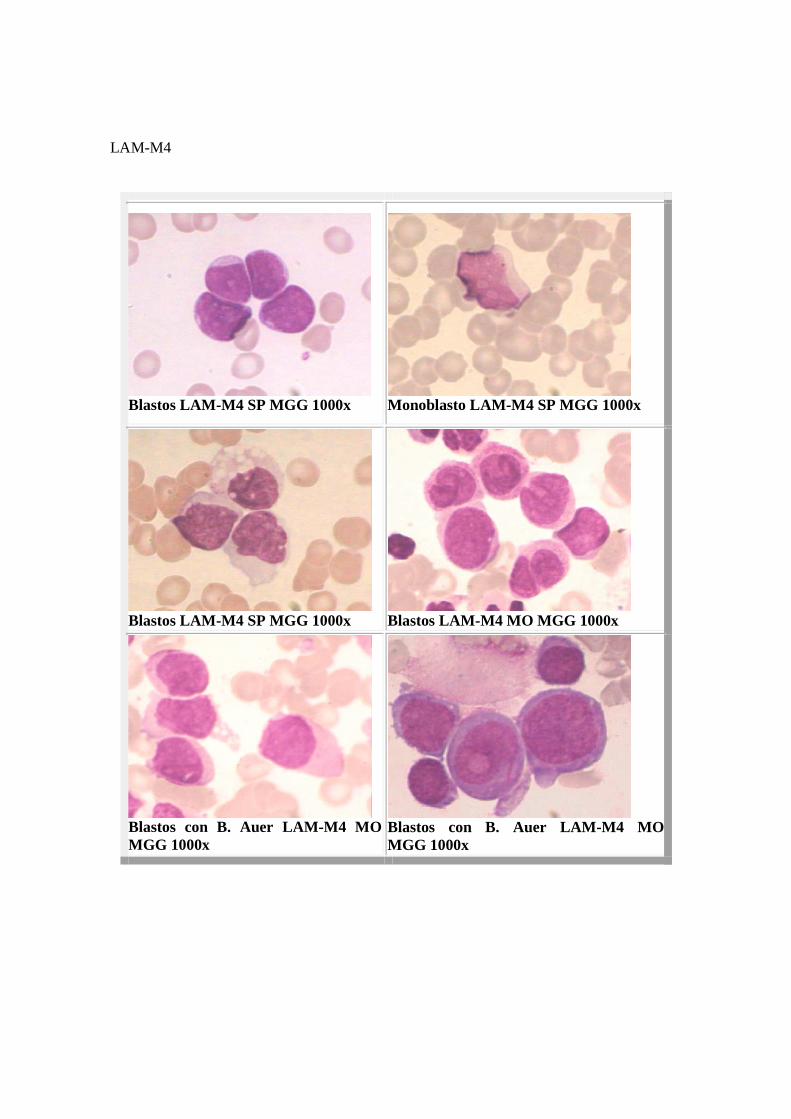
LAM-M4
Blastos LAM-M4 SP MGG 1000x
Monoblasto LAM-M4 SP MGG 1000x
Blastos LAM-M4 SP MGG 1000x
Blastos LAM-M4 MO MGG 1000x
Blastos con B. Auer LAM-M4 MO MGG 1000x
Blastos con B. Auer LAM-M4 MO MGG 1000x
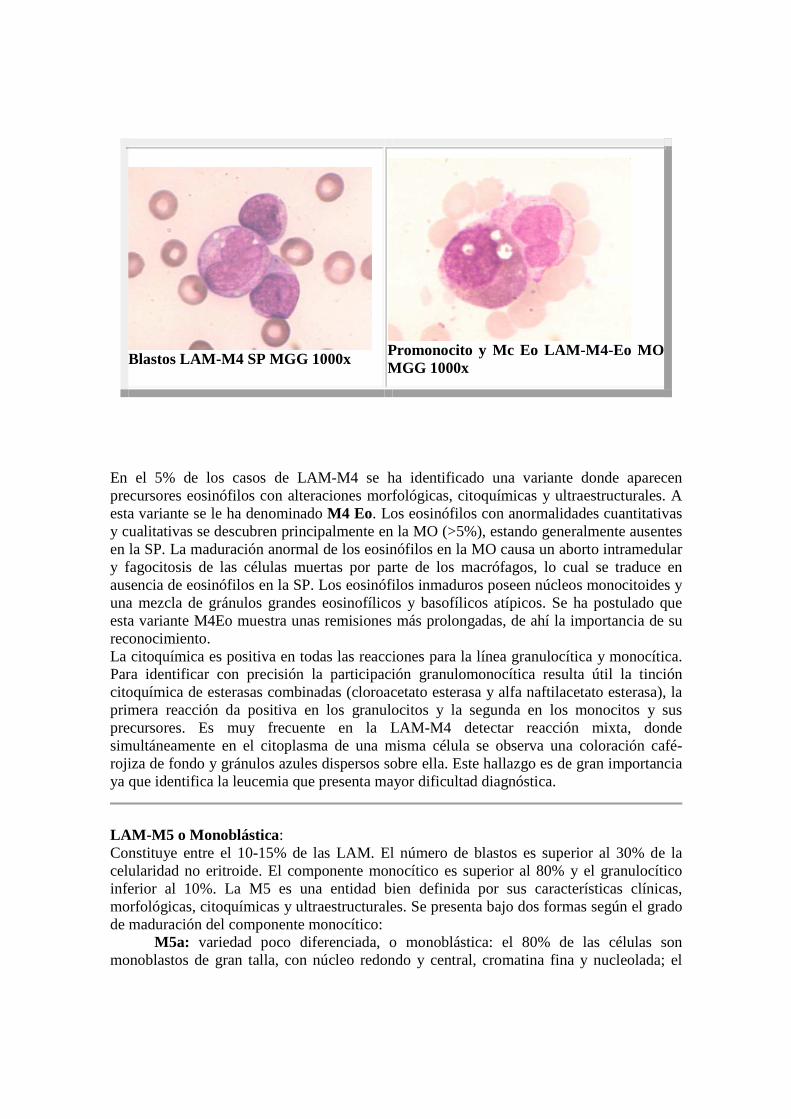
Blastos LAM-M4 SP MGG 1000x
Promonocito y Mc Eo LAM-M4-Eo MOMGG 1000x
En el 5% de los casos de LAM-M4 se ha identificado una variante donde aparecen precursores eosinófilos con alteraciones morfológicas, citoquímicas y ultraestructurales. A esta variante se le ha denominado M4 Eo. Los eosinófilos con anormalidades cuantitativas y cualitativas se descubren principalmente en la MO (>5%), estando generalmente ausentes en la SP. La maduración anormal de los eosinófilos en la MO causa un aborto intramedular y fagocitosis de las células muertas por parte de los macrófagos, lo cual se traduce en ausencia de eosinófilos en la SP. Los eosinófilos inmaduros poseen núcleos monocitoides y una mezcla de gránulos grandes eosinofílicos y basofílicos atípicos. Se ha postulado que esta variante M4Eo muestra unas remisiones más prolongadas, de ahí la importancia de su reconocimiento. La citoquímica es positiva en todas las reacciones para la línea granulocítica y monocítica. Para identificar con precisión la participación granulomonocítica resulta útil la tinción citoquímica de esterasas combinadas (cloroacetato esterasa y alfa naftilacetato esterasa), la primera reacción da positiva en los granulocitos y la segunda en los monocitos y sus precursores. Es muy frecuente en la LAM-M4 detectar reacción mixta, donde simultáneamente en el citoplasma de una misma célula se observa una coloración café-rojiza de fondo y gránulos azules dispersos sobre ella. Este hallazgo es de gran importancia ya que identifica la leucemia que presenta mayor dificultad diagnóstica.
LAM-M5 o Monoblástica : Constituye entre el 10-15% de las LAM. El número de blastos es superior al 30% de la celularidad no eritroide. El componente monocítico es superior al 80% y el granulocítico inferior al 10%. La M5 es una entidad bien definida por sus características clínicas, morfológicas, citoquímicas y ultraestructurales. Se presenta bajo dos formas según el grado de maduración del componente monocítico: � M5a: variedad poco diferenciada, o monoblástica: el 80% de las células son monoblastos de gran talla, con núcleo redondo y central, cromatina fina y nucleolada; el
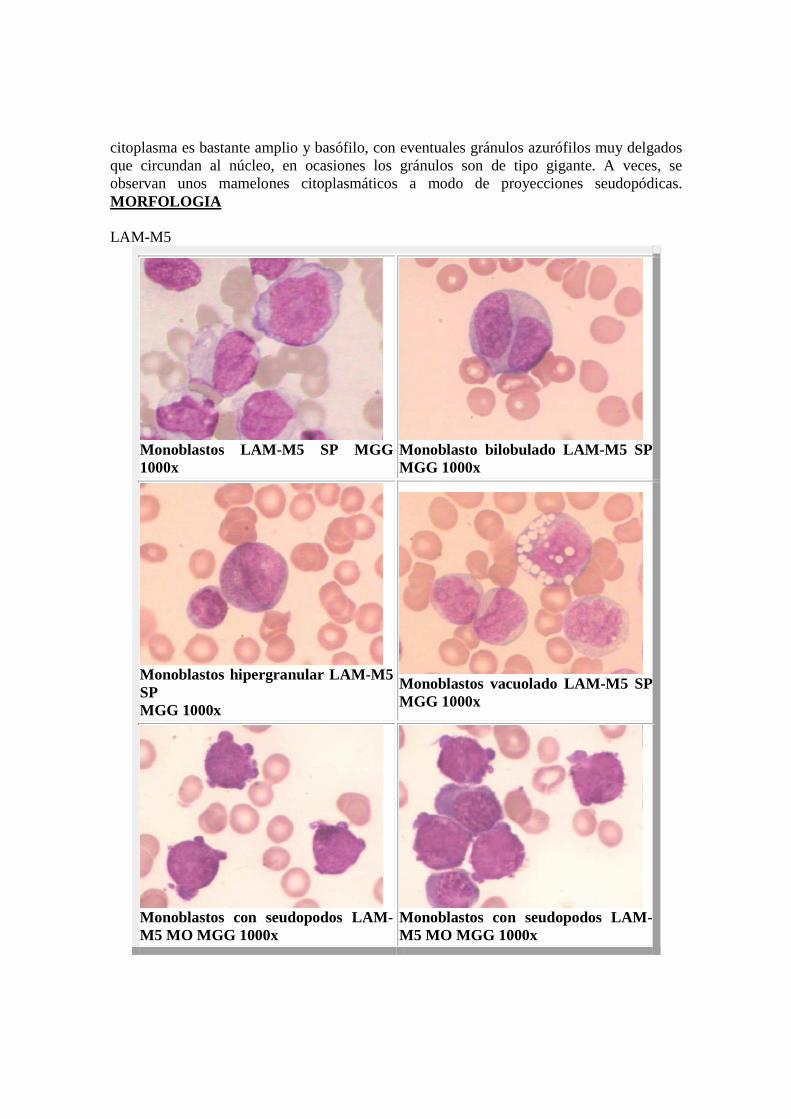
citoplasma es bastante amplio y basófilo, con eventuales gránulos azurófilos muy delgados que circundan al núcleo, en ocasiones los gránulos son de tipo gigante. A veces, se observan unos mamelones citoplasmáticos a modo de proyecciones seudopódicas. MORFOLOGIA LAM-M5
Monoblastos LAM-M5 SP MGG 1000x
Monoblasto bilobulado LAM-M5 SP MGG 1000x
Monoblastos hipergranular LAM- M5 SP MGG 1000x
Monoblastos vacuolado LAM-M5 SP MGG 1000x
Monoblastos con seudopodos LAM-M5 MO MGG 1000x
Monoblastos con seudopodos LAM-M5 MO MGG 1000x
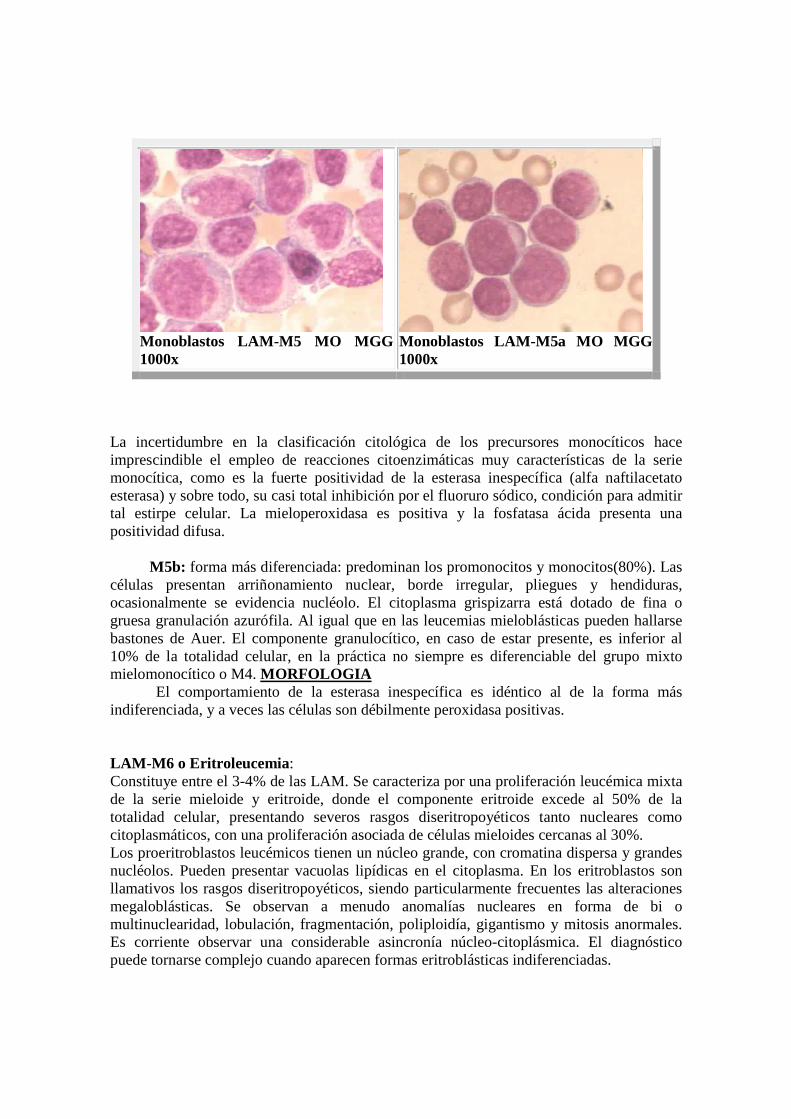
Monoblastos LAM-M5 MO MGG 1000x
Monoblastos LAM-M5a MO MGG 1000x
La incertidumbre en la clasificación citológica de los precursores monocíticos hace imprescindible el empleo de reacciones citoenzimáticas muy características de la serie monocítica, como es la fuerte positividad de la esterasa inespecífica (alfa naftilacetato esterasa) y sobre todo, su casi total inhibición por el fluoruro sódico, condición para admitir tal estirpe celular. La mieloperoxidasa es positiva y la fosfatasa ácida presenta una positividad difusa. � M5b: forma más diferenciada: predominan los promonocitos y monocitos(80%). Las células presentan arriñonamiento nuclear, borde irregular, pliegues y hendiduras, ocasionalmente se evidencia nucléolo. El citoplasma grispizarra está dotado de fina o gruesa granulación azurófila. Al igual que en las leucemias mieloblásticas pueden hallarse bastones de Auer. El componente granulocítico, en caso de estar presente, es inferior al 10% de la totalidad celular, en la práctica no siempre es diferenciable del grupo mixto mielomonocítico o M4. MORFOLOGIA El comportamiento de la esterasa inespecífica es idéntico al de la forma más indiferenciada, y a veces las células son débilmente peroxidasa positivas. LAM-M6 o Eritroleucemia : Constituye entre el 3-4% de las LAM. Se caracteriza por una proliferación leucémica mixta de la serie mieloide y eritroide, donde el componente eritroide excede al 50% de la totalidad celular, presentando severos rasgos diseritropoyéticos tanto nucleares como citoplasmáticos, con una proliferación asociada de células mieloides cercanas al 30%. Los proeritroblastos leucémicos tienen un núcleo grande, con cromatina dispersa y grandes nucléolos. Pueden presentar vacuolas lipídicas en el citoplasma. En los eritroblastos son llamativos los rasgos diseritropoyéticos, siendo particularmente frecuentes las alteraciones megaloblásticas. Se observan a menudo anomalías nucleares en forma de bi o multinuclearidad, lobulación, fragmentación, poliploidía, gigantismo y mitosis anormales. Es corriente observar una considerable asincronía núcleo-citoplásmica. El diagnóstico puede tornarse complejo cuando aparecen formas eritroblásticas indiferenciadas.
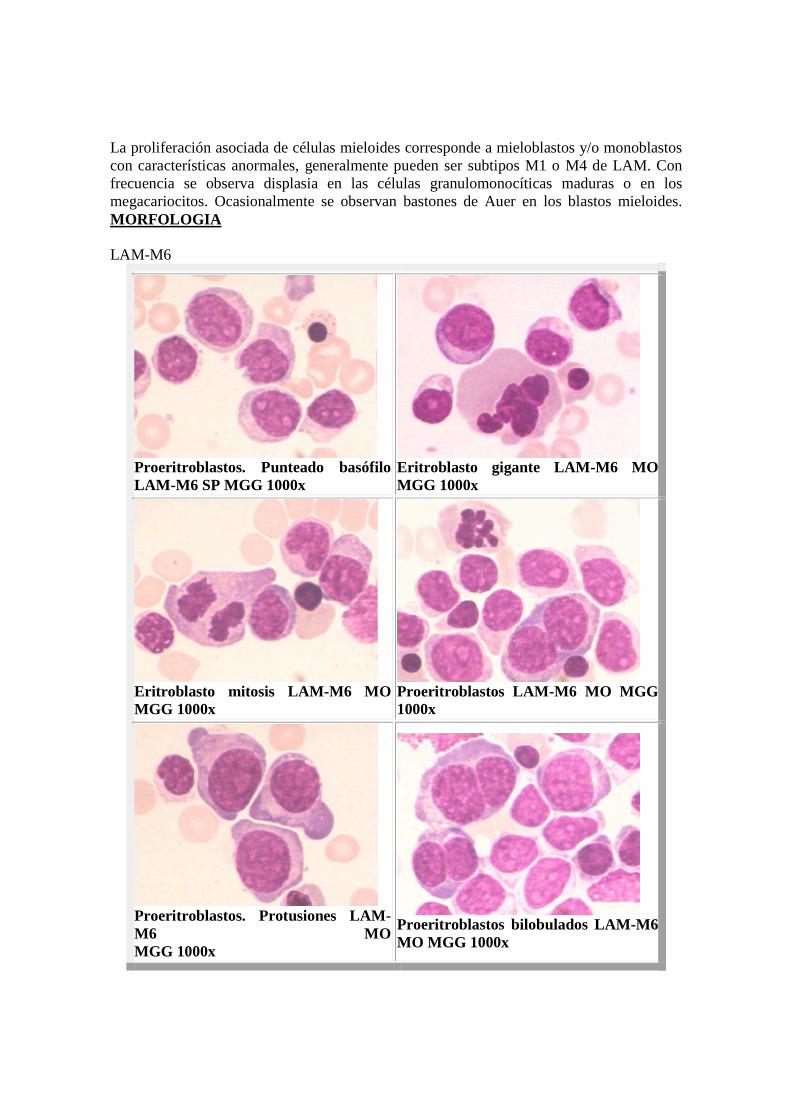
La proliferación asociada de células mieloides corresponde a mieloblastos y/o monoblastos con características anormales, generalmente pueden ser subtipos M1 o M4 de LAM. Con frecuencia se observa displasia en las células granulomonocíticas maduras o en los megacariocitos. Ocasionalmente se observan bastones de Auer en los blastos mieloides. MORFOLOGIA LAM-M6
Proeritroblastos. Punteado basófilo LAM-M6 SP MGG 1000x
Eritroblasto gigante LAM- M6 MOMGG 1000x
Eritroblasto mitosis LAM- M6 MO MGG 1000x
Proeritroblastos LAM- M6 MO MGG 1000x
Proeritroblastos. Protusiones LAM-M6 MO MGG 1000x
Proeritroblastos bilobulados LAM-M6 MO MGG 1000x
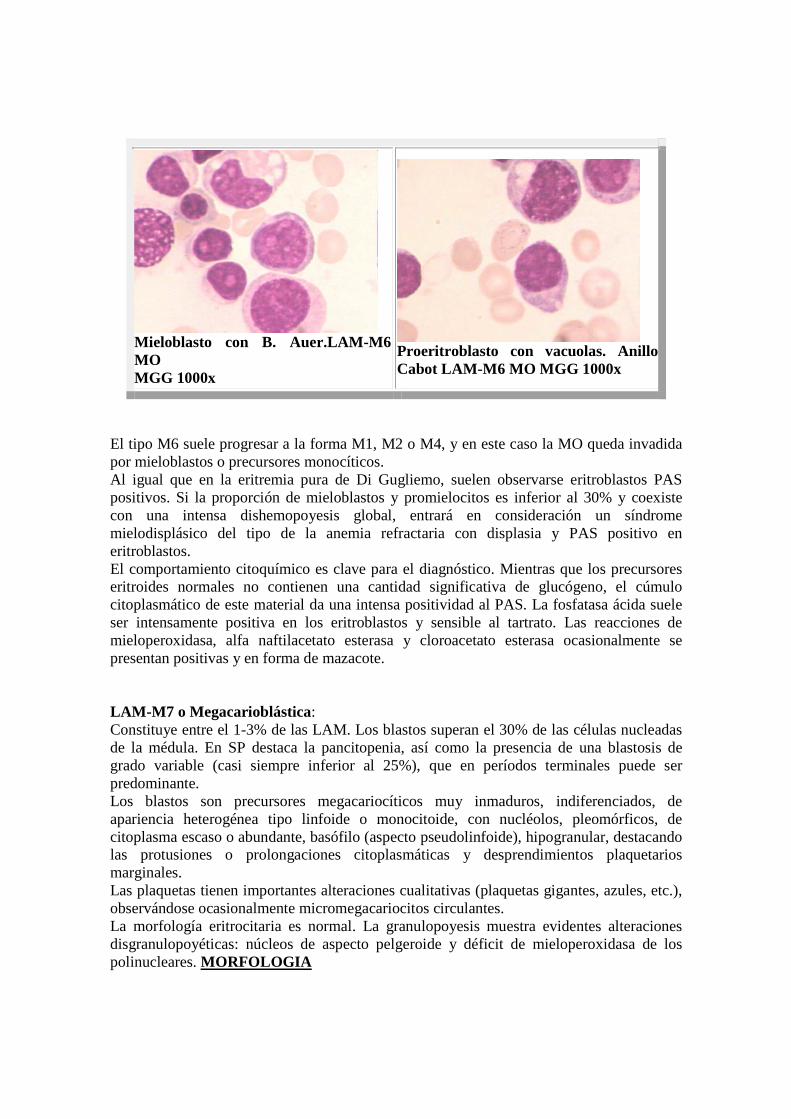
Mieloblasto con B. Auer.LAM-M6 MO MGG 1000x
Proeritroblasto con vacuolas. Anillo Cabot LAM-M6 MO MGG 1000x
El tipo M6 suele progresar a la forma M1, M2 o M4, y en este caso la MO queda invadida por mieloblastos o precursores monocíticos. Al igual que en la eritremia pura de Di Gugliemo, suelen observarse eritroblastos PAS positivos. Si la proporción de mieloblastos y promielocitos es inferior al 30% y coexiste con una intensa dishemopoyesis global, entrará en consideración un síndrome mielodisplásico del tipo de la anemia refractaria con displasia y PAS positivo en eritroblastos. El comportamiento citoquímico es clave para el diagnóstico. Mientras que los precursores eritroides normales no contienen una cantidad significativa de glucógeno, el cúmulo citoplasmático de este material da una intensa positividad al PAS. La fosfatasa ácida suele ser intensamente positiva en los eritroblastos y sensible al tartrato. Las reacciones de mieloperoxidasa, alfa naftilacetato esterasa y cloroacetato esterasa ocasionalmente se presentan positivas y en forma de mazacote. LAM-M7 o Megacarioblástica: Constituye entre el 1-3% de las LAM. Los blastos superan el 30% de las células nucleadas de la médula. En SP destaca la pancitopenia, así como la presencia de una blastosis de grado variable (casi siempre inferior al 25%), que en períodos terminales puede ser predominante. Los blastos son precursores megacariocíticos muy inmaduros, indiferenciados, de apariencia heterogénea tipo linfoide o monocitoide, con nucléolos, pleomórficos, de citoplasma escaso o abundante, basófilo (aspecto pseudolinfoide), hipogranular, destacando las protusiones o prolongaciones citoplasmáticas y desprendimientos plaquetarios marginales. Las plaquetas tienen importantes alteraciones cualitativas (plaquetas gigantes, azules, etc.), observándose ocasionalmente micromegacariocitos circulantes. La morfología eritrocitaria es normal. La granulopoyesis muestra evidentes alteraciones disgranulopoyéticas: núcleos de aspecto pelgeroide y déficit de mieloperoxidasa de los polinucleares. MORFOLOGIA
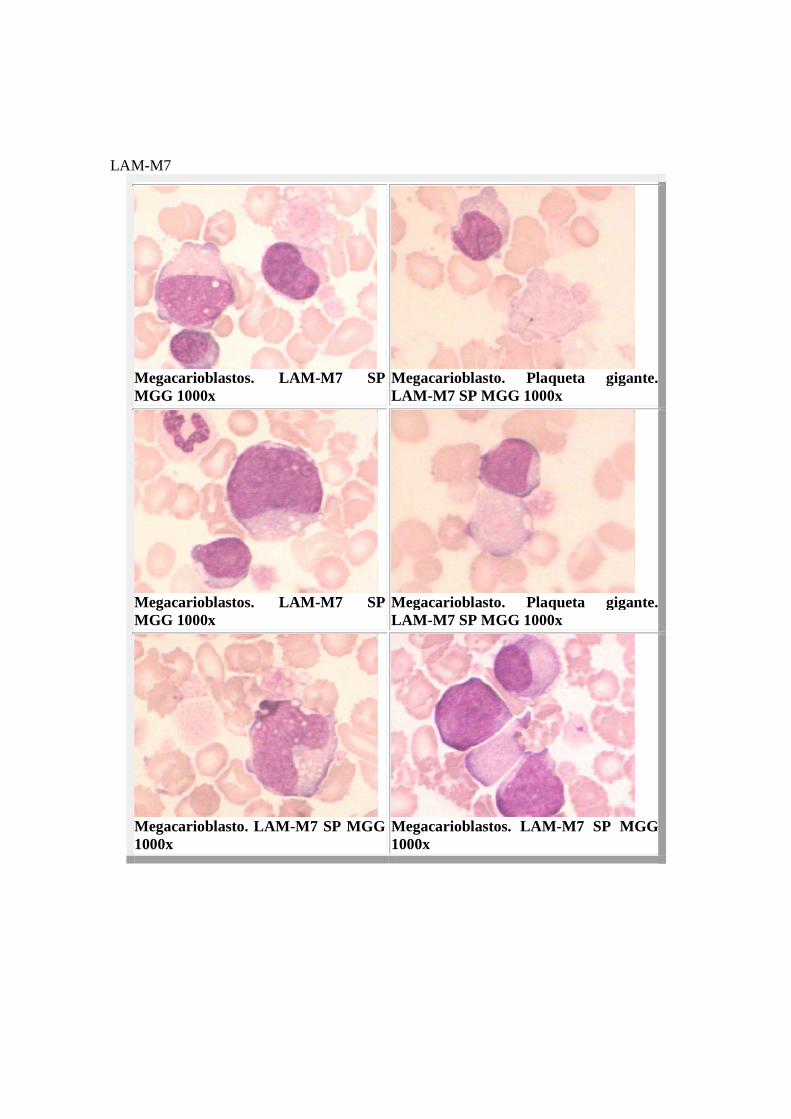
LAM-M7
Megacarioblastos. LAM-M7 SP MGG 1000x
Megacarioblasto. Plaqueta gigante. LAM-M7 SP MGG 1000x
Megacarioblastos. LAM-M7 SP MGG 1000x
Megacarioblasto. Plaqueta gigante. LAM-M7 SP MGG 1000x
Megacarioblasto. LAM-M7 SP MGG 1000x
Megacarioblastos. LAM-M7 SP MGG 1000x
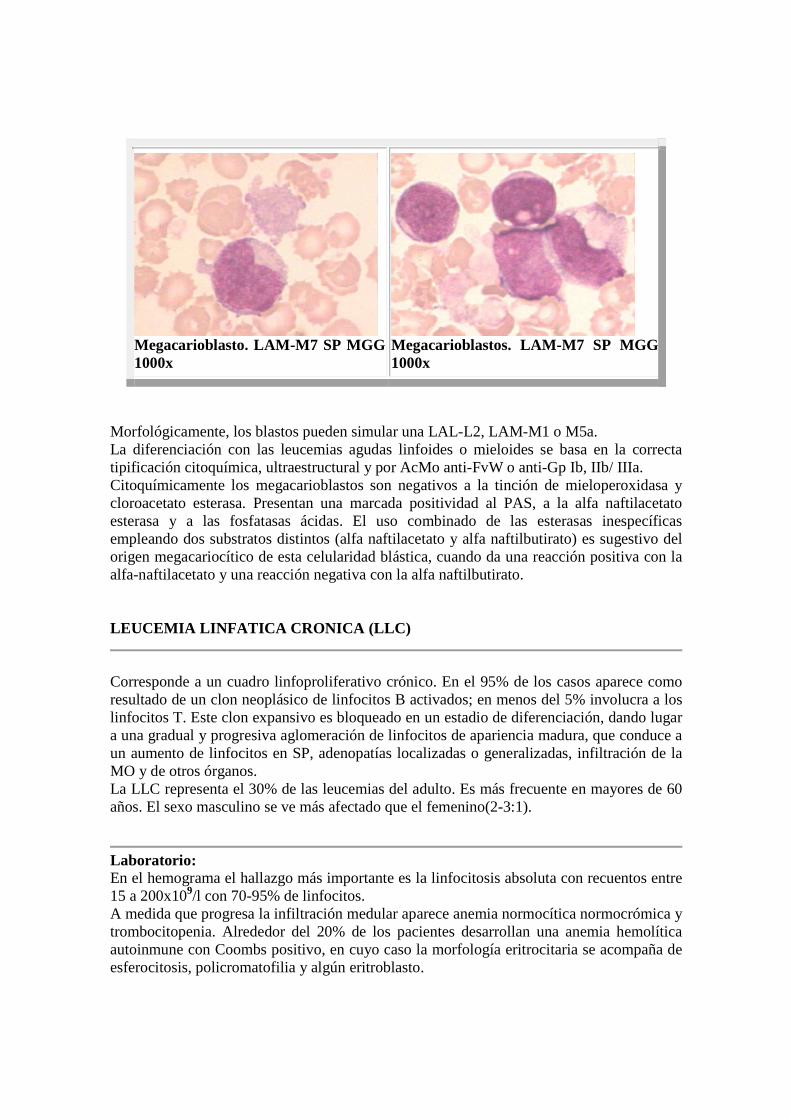
Megacarioblasto. LAM-M7 SP MGG 1000x
Megacarioblastos. LAM-M7 SP MGG 1000x
Morfológicamente, los blastos pueden simular una LAL-L2, LAM-M1 o M5a. La diferenciación con las leucemias agudas linfoides o mieloides se basa en la correcta tipificación citoquímica, ultraestructural y por AcMo anti-FvW o anti-Gp Ib, IIb/ IIIa. Citoquímicamente los megacarioblastos son negativos a la tinción de mieloperoxidasa y cloroacetato esterasa. Presentan una marcada positividad al PAS, a la alfa naftilacetato esterasa y a las fosfatasas ácidas. El uso combinado de las esterasas inespecíficas empleando dos substratos distintos (alfa naftilacetato y alfa naftilbutirato) es sugestivo del origen megacariocítico de esta celularidad blástica, cuando da una reacción positiva con la alfa-naftilacetato y una reacción negativa con la alfa naftilbutirato. LEUCEMIA LINFATICA CRONICA (LLC)
Corresponde a un cuadro linfoproliferativo crónico. En el 95% de los casos aparece como resultado de un clon neoplásico de linfocitos B activados; en menos del 5% involucra a los linfocitos T. Este clon expansivo es bloqueado en un estadio de diferenciación, dando lugar a una gradual y progresiva aglomeración de linfocitos de apariencia madura, que conduce a un aumento de linfocitos en SP, adenopatías localizadas o generalizadas, infiltración de la MO y de otros órganos. La LLC representa el 30% de las leucemias del adulto. Es más frecuente en mayores de 60 años. El sexo masculino se ve más afectado que el femenino(2-3:1).
Laboratorio: En el hemograma el hallazgo más importante es la linfocitosis absoluta con recuentos entre 15 a 200x109/l con 70-95% de linfocitos. A medida que progresa la infiltración medular aparece anemia normocítica normocrómica y trombocitopenia. Alrededor del 20% de los pacientes desarrollan una anemia hemolítica autoinmune con Coombs positivo, en cuyo caso la morfología eritrocitaria se acompaña de esferocitosis, policromatofilia y algún eritroblasto.
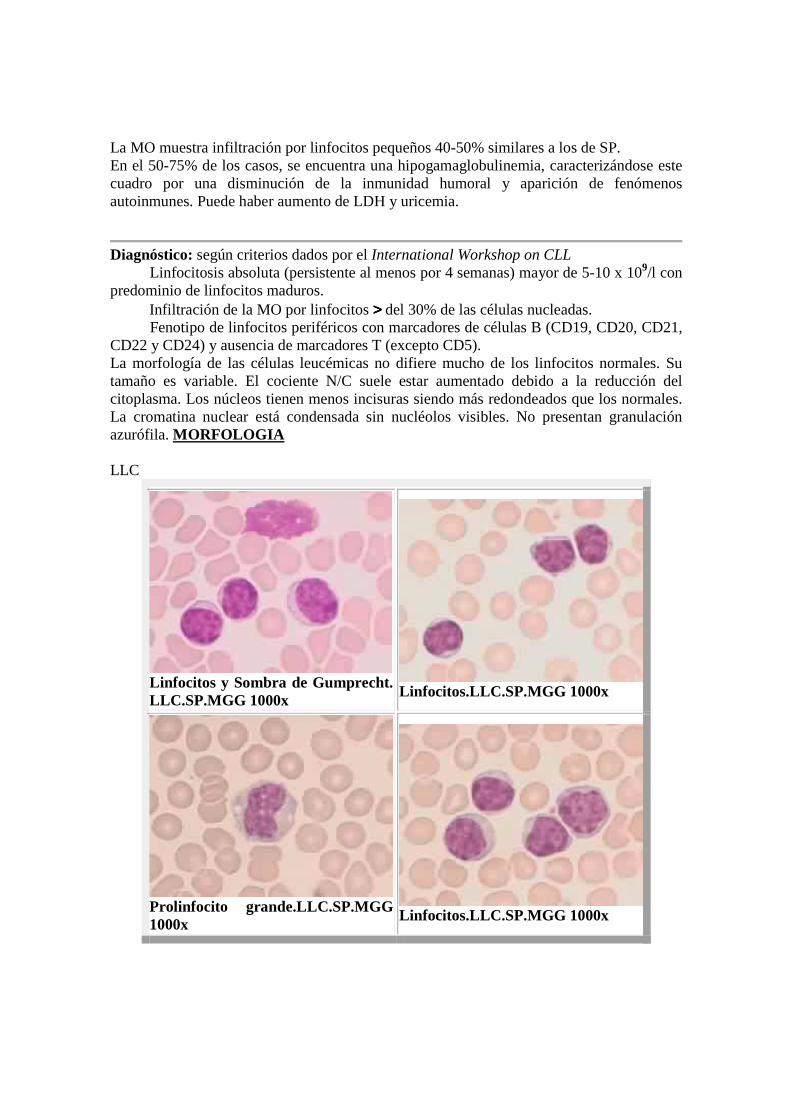
La MO muestra infiltración por linfocitos pequeños 40-50% similares a los de SP. En el 50-75% de los casos, se encuentra una hipogamaglobulinemia, caracterizándose este cuadro por una disminución de la inmunidad humoral y aparición de fenómenos autoinmunes. Puede haber aumento de LDH y uricemia.
Diagnóstico: según criterios dados por el International Workshop on CLL � Linfocitosis absoluta (persistente al menos por 4 semanas) mayor de 5-10 x 109/l con predominio de linfocitos maduros. � Infiltración de la MO por linfocitos >>>> del 30% de las células nucleadas. � Fenotipo de linfocitos periféricos con marcadores de células B (CD19, CD20, CD21, CD22 y CD24) y ausencia de marcadores T (excepto CD5). La morfología de las células leucémicas no difiere mucho de los linfocitos normales. Su tamaño es variable. El cociente N/C suele estar aumentado debido a la reducción del citoplasma. Los núcleos tienen menos incisuras siendo más redondeados que los normales. La cromatina nuclear está condensada sin nucléolos visibles. No presentan granulación azurófila. MORFOLOGIA LLC
Linfocitos y Sombra de Gumprecht. LLC.SP.MGG 1000x
Linfocitos.LLC.SP.MGG 1000x
Prolinfocito grande.LLC.SP.MGG 1000x
Linfocitos.LLC.SP.MGG 1000x
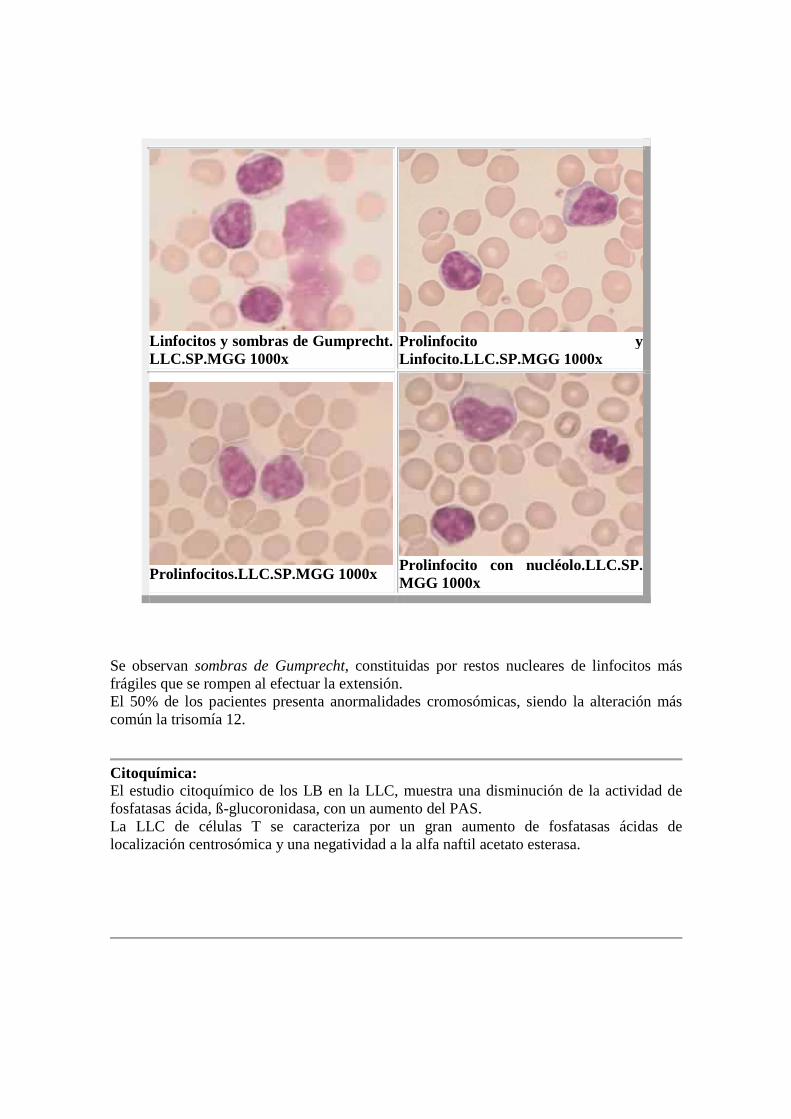
Linfocitos y sombras de Gumprecht. LLC.SP.MGG 1000x
Prolinfocito y Linfocito.LLC.SP.MGG 1000x
Prolinfocitos.LLC.SP.MGG 1000x
Prolinfocito con nucléolo.LLC.SP.MGG 1000x
Se observan sombras de Gumprecht, constituidas por restos nucleares de linfocitos más frágiles que se rompen al efectuar la extensión. El 50% de los pacientes presenta anormalidades cromosómicas, siendo la alteración más común la trisomía 12.
Citoquímica: El estudio citoquímico de los LB en la LLC, muestra una disminución de la actividad de fosfatasas ácida, ß-glucoronidasa, con un aumento del PAS. La LLC de células T se caracteriza por un gran aumento de fosfatasas ácidas de localización centrosómica y una negatividad a la alfa naftil acetato esterasa.
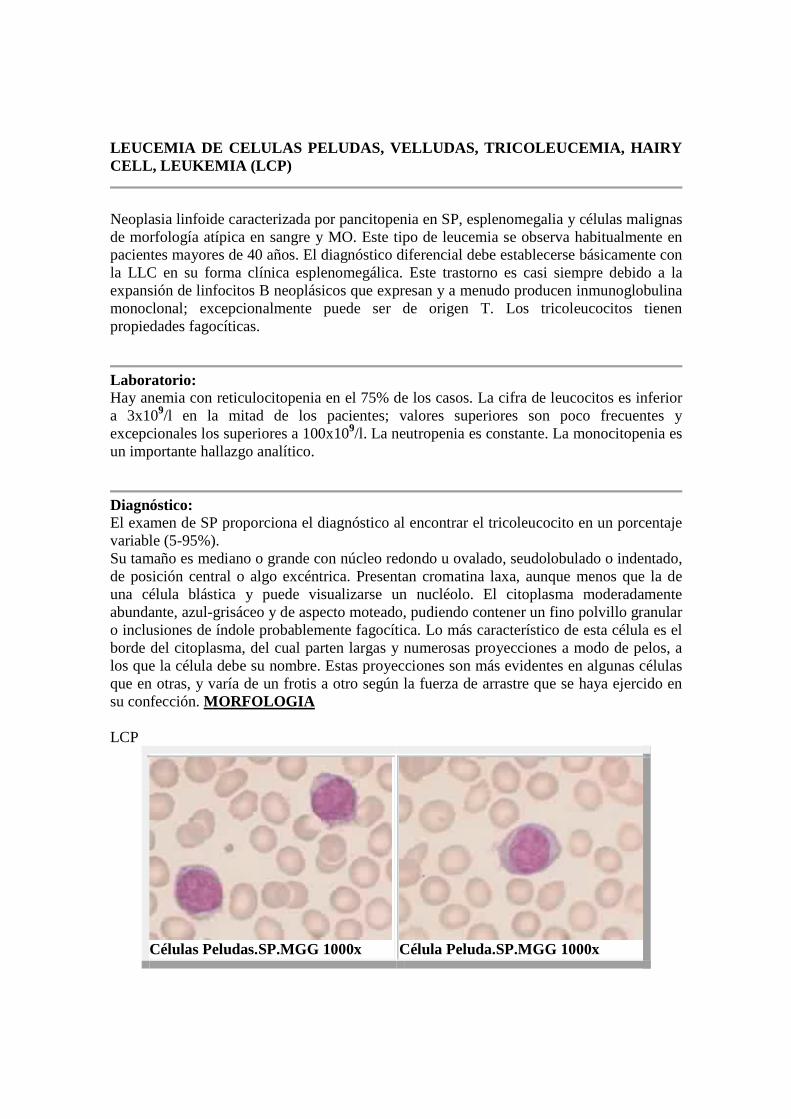
LEUCEMIA DE CELULAS PELUDAS, VELLUDAS, TRICOLEUCEMI A, HAIRY CELL, LEUKEMIA (LCP)
Neoplasia linfoide caracterizada por pancitopenia en SP, esplenomegalia y células malignas de morfología atípica en sangre y MO. Este tipo de leucemia se observa habitualmente en pacientes mayores de 40 años. El diagnóstico diferencial debe establecerse básicamente con la LLC en su forma clínica esplenomegálica. Este trastorno es casi siempre debido a la expansión de linfocitos B neoplásicos que expresan y a menudo producen inmunoglobulina monoclonal; excepcionalmente puede ser de origen T. Los tricoleucocitos tienen propiedades fagocíticas.
Laboratorio: Hay anemia con reticulocitopenia en el 75% de los casos. La cifra de leucocitos es inferior a 3x109/l en la mitad de los pacientes; valores superiores son poco frecuentes y excepcionales los superiores a 100x109/l. La neutropenia es constante. La monocitopenia es un importante hallazgo analítico.
Diagnóstico: El examen de SP proporciona el diagnóstico al encontrar el tricoleucocito en un porcentaje variable (5-95%). Su tamaño es mediano o grande con núcleo redondo u ovalado, seudolobulado o indentado, de posición central o algo excéntrica. Presentan cromatina laxa, aunque menos que la de una célula blástica y puede visualizarse un nucléolo. El citoplasma moderadamente abundante, azul-grisáceo y de aspecto moteado, pudiendo contener un fino polvillo granular o inclusiones de índole probablemente fagocítica. Lo más característico de esta célula es el borde del citoplasma, del cual parten largas y numerosas proyecciones a modo de pelos, a los que la célula debe su nombre. Estas proyecciones son más evidentes en algunas células que en otras, y varía de un frotis a otro según la fuerza de arrastre que se haya ejercido en su confección. MORFOLOGIA LCP
Células Peludas.SP.MGG 1000x Célula Peluda.SP.MGG 1000x
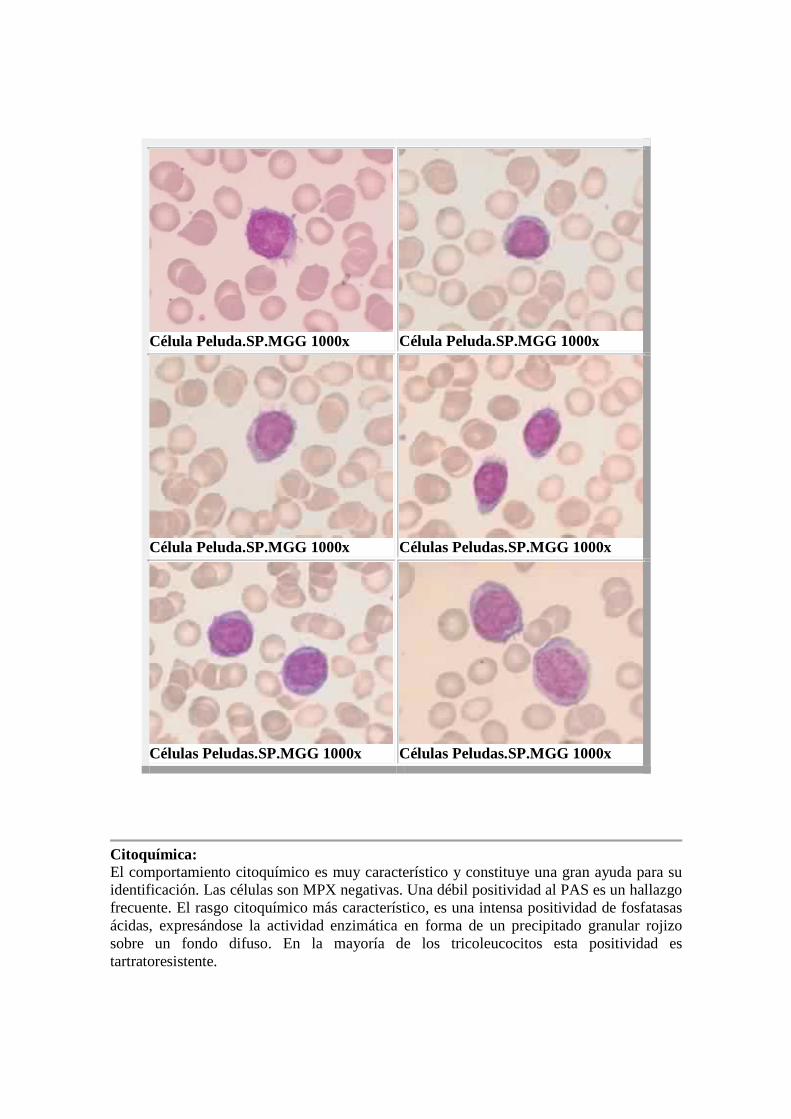
Célula Peluda.SP.MGG 1000x Célula Peluda.SP.MGG 1000x
Célula Peluda.SP.MGG 1000x Células Peludas.SP.MGG 1000x
Células Peludas.SP.MGG 1000x Células Peludas.SP.MGG 1000x
Citoquímica: El comportamiento citoquímico es muy característico y constituye una gran ayuda para su identificación. Las células son MPX negativas. Una débil positividad al PAS es un hallazgo frecuente. El rasgo citoquímico más característico, es una intensa positividad de fosfatasas ácidas, expresándose la actividad enzimática en forma de un precipitado granular rojizo sobre un fondo difuso. En la mayoría de los tricoleucocitos esta positividad es tartratoresistente.
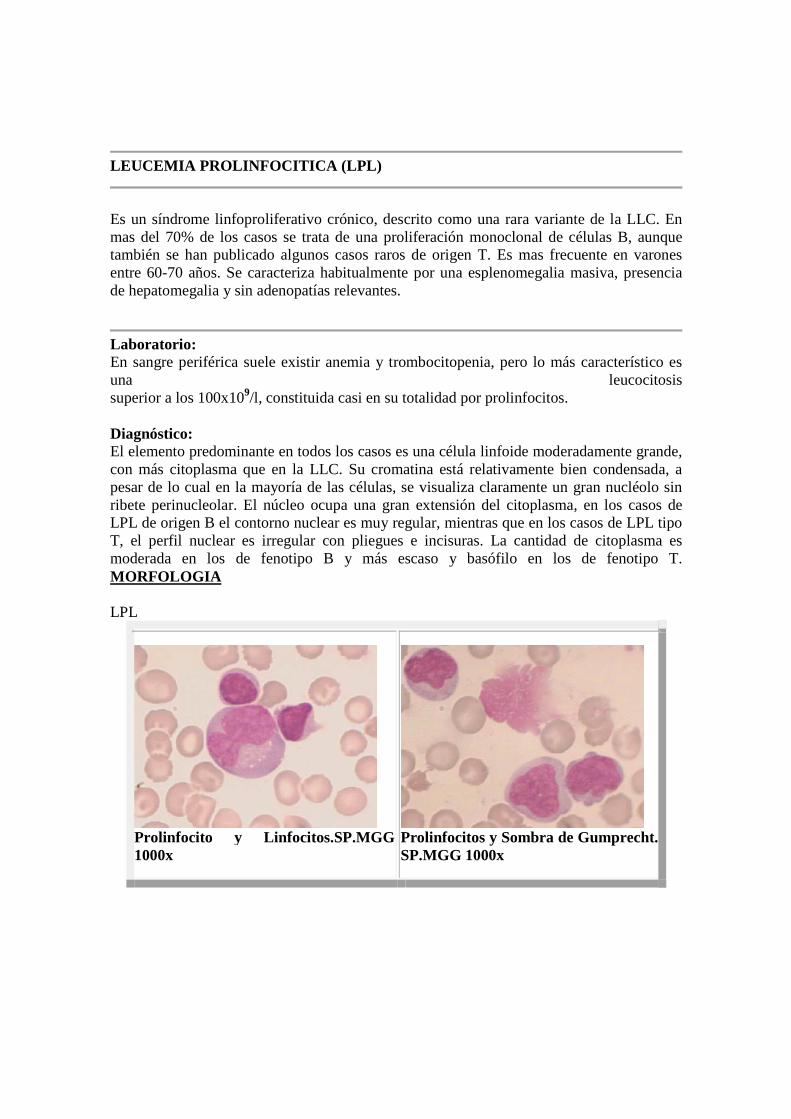
LEUCEMIA PROLINFOCITICA (LPL)
Es un síndrome linfoproliferativo crónico, descrito como una rara variante de la LLC. En mas del 70% de los casos se trata de una proliferación monoclonal de células B, aunque también se han publicado algunos casos raros de origen T. Es mas frecuente en varones entre 60-70 años. Se caracteriza habitualmente por una esplenomegalia masiva, presencia de hepatomegalia y sin adenopatías relevantes.
Laboratorio: En sangre periférica suele existir anemia y trombocitopenia, pero lo más característico es una leucocitosis superior a los 100x109/l, constituida casi en su totalidad por prolinfocitos. Diagnóstico: El elemento predominante en todos los casos es una célula linfoide moderadamente grande, con más citoplasma que en la LLC. Su cromatina está relativamente bien condensada, a pesar de lo cual en la mayoría de las células, se visualiza claramente un gran nucléolo sin ribete perinucleolar. El núcleo ocupa una gran extensión del citoplasma, en los casos de LPL de origen B el contorno nuclear es muy regular, mientras que en los casos de LPL tipo T, el perfil nuclear es irregular con pliegues e incisuras. La cantidad de citoplasma es moderada en los de fenotipo B y más escaso y basófilo en los de fenotipo T. MORFOLOGIA LPL
Prolinfocito y Linfocitos.SP.MGG 1000x
Prolinfocitos y Sombra de Gumprecht.SP.MGG 1000x
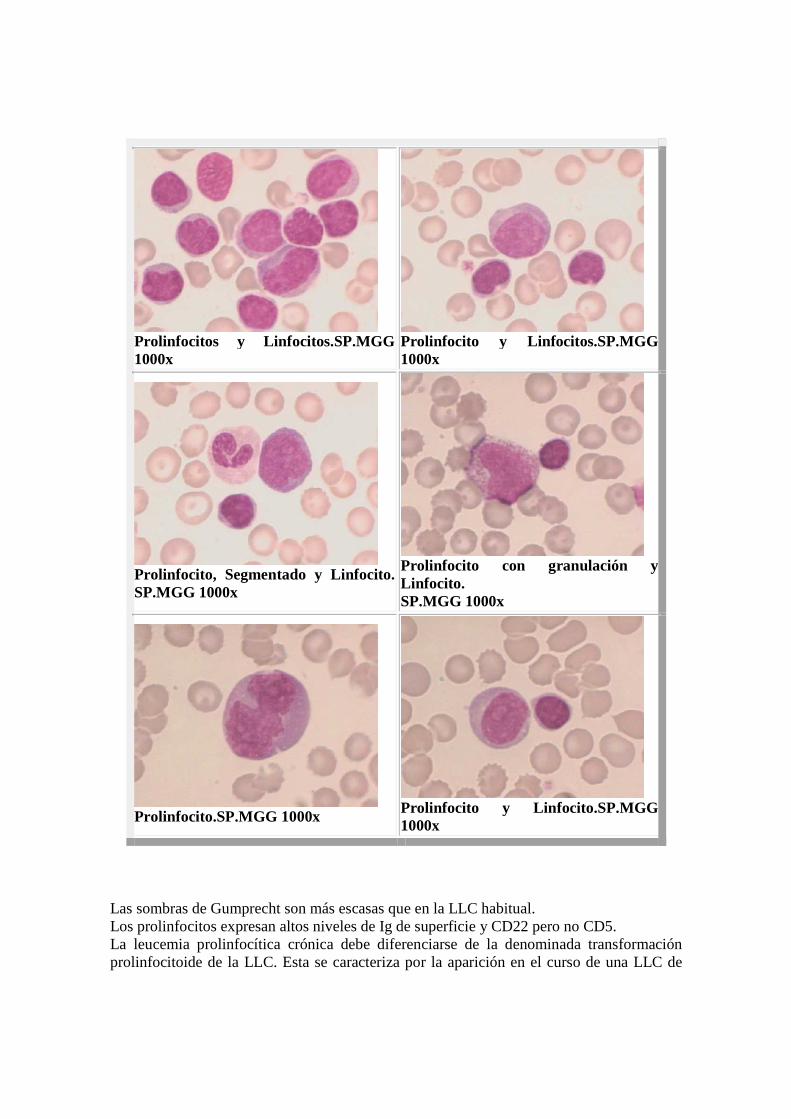
Prolinfocit os y Linfocitos.SP.MGG 1000x
Prolinfocito y Linfocitos.SP.MGG 1000x
Prolinfocito, Segmentado y Linfocito. SP.MGG 1000x
Prolinfocito con granulación y Linfocito. SP.MGG 1000x
Prolinfocito.SP.MGG 1000x
Prolinfocito y Linfocito.SP.MGG 1000x
Las sombras de Gumprecht son más escasas que en la LLC habitual. Los prolinfocitos expresan altos niveles de Ig de superficie y CD22 pero no CD5. La leucemia prolinfocítica crónica debe diferenciarse de la denominada transformación prolinfocitoide de la LLC. Esta se caracteriza por la aparición en el curso de una LLC de

unas células semejantes a los prolinfocitos, asociada a una falta de respuesta al tratamiento de la LLC. El dato morfológico más característico es la presencia en la SP de dos poblaciones celulares bien diferenciadas: los linfocitos de pequeño tamaño, propios de la LLC, y los prolinfocitos.
Citoquímica: Citoquímicamente muestran, como toda celularidad B, disminución de fosfatasas ácidas. Las formas T reaccionan positivamente a ésta enzima con localización centrosómica y dan intensa positividad en forma de precipitado único a la alfa naftil acetato esterasa. LEUCEMIA MIELOIDE CRONICA (LMC)
La LMC es un síndrome mieloproliferativo clonal que afecta a una célula madre pluripotencial hematopoyética muy indiferenciada, común a la serie mieloide (granulo-monocítica, eritrocítica y megacariocítica) y a la linfoide. La clona celular mutada en la MO es proliferativa, con expansión progresiva del pool de precursores granulocíticos, con aumento de su liberación a la circulación y con una masa granulocítica 5-10 veces mayor que lo normal. La LMC constituye el 10-20% de todas las leucemias. Puede aparecer a cualquier edad, siendo más frecuente entre los 30-60 años y muy rara en la infancia. En el 95% de los casos existe una alteración cromosómica, denominado cromosoma Philadelphia (Ph1). Se trata de una translocación recíproca del cromosoma 22 y cromosoma 9, t(9;22), donde se genera el gen quimérico bcr/abl. Dicho oncogén da origen a un RNA mensajero que codifica la síntesis de una proteína con actividad tirosinaquinasa implicada en el control del crecimiento celular. Esta alteración es adquirida y es posible detectarla en los granulocitos, eritroblastos, megacariocitos y linfocitos B; lo que demuestra su origen en la stem cell pluripotencial común a todas las células hematopoyéticas. Aunque el cromosoma Ph1 es considerado el marcador diagnóstico de la LMC no es exclusivo de ella, ya que se ha detectado su presencia en un 20% de los adultos y en un 5% de los niños portadores de LAL, así como también en el 2% de los pacientes con LAM. El cuadro clínico tiene varias etapas de presentación: Fase crónica: de aparición insidiosa, el 20-25% de los pacientes es asintomático en el momento del diagnóstico, el cual se sospecha por el hallazgo de esplenomegalia o alteraciones del hemograma en un examen de rutina. En general, las anormalidades de laboratorio aparecen antes que los síntomas y éstos suelen presentarse sólo después de que los recuentos leucocitarios muestren cifras muy elevadas (30-100x109/l). Esta fase se caracteriza por una superproducción de leucocitos. La blastosis es mínima o inexistente. No se observa hiatus. Entre los promielocitos y los polimorfonucleares aparecen todos los estadios madurativos intermedios. La cifra leucocitaria supera los 50-100x109/l, e incluso llegan a 1000x109/l. La basofilia puede ser importante, existiendo una cierta relación entre el ascenso de los basófilos y la crisis blástica terminal.
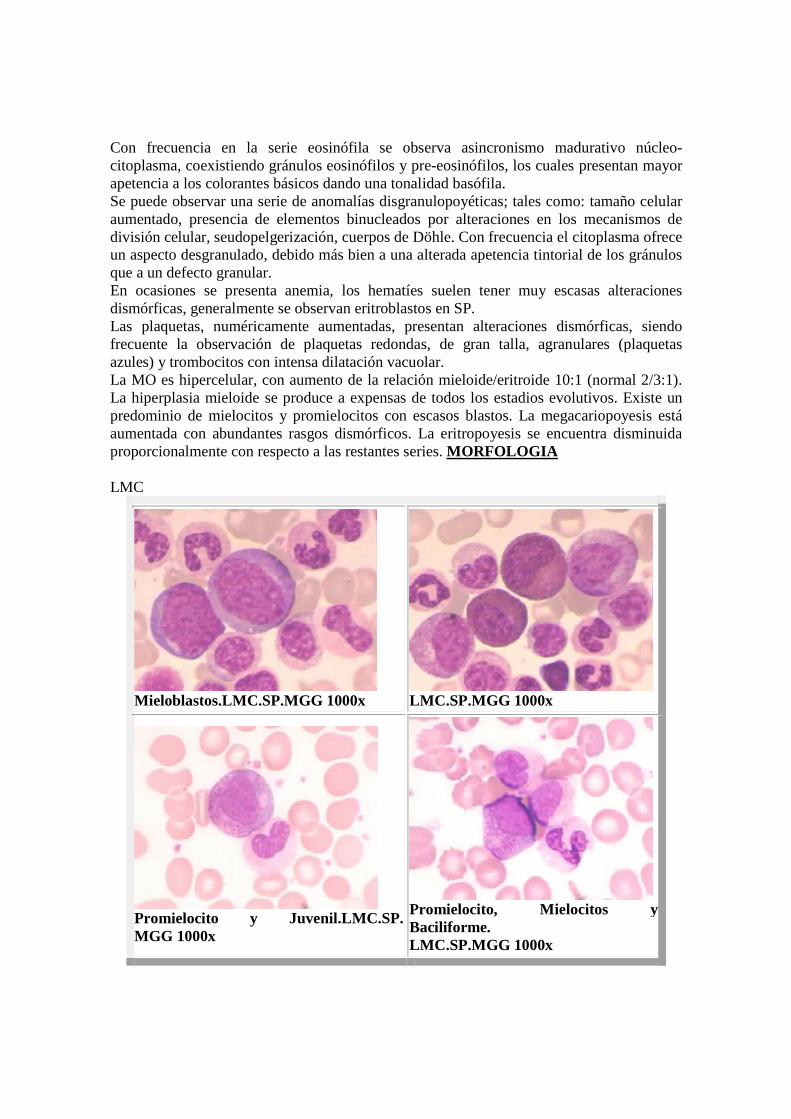
Con frecuencia en la serie eosinófila se observa asincronismo madurativo núcleo-citoplasma, coexistiendo gránulos eosinófilos y pre-eosinófilos, los cuales presentan mayor apetencia a los colorantes básicos dando una tonalidad basófila. Se puede observar una serie de anomalías disgranulopoyéticas; tales como: tamaño celular aumentado, presencia de elementos binucleados por alteraciones en los mecanismos de división celular, seudopelgerización, cuerpos de Döhle. Con frecuencia el citoplasma ofrece un aspecto desgranulado, debido más bien a una alterada apetencia tintorial de los gránulos que a un defecto granular. En ocasiones se presenta anemia, los hematíes suelen tener muy escasas alteraciones dismórficas, generalmente se observan eritroblastos en SP. Las plaquetas, numéricamente aumentadas, presentan alteraciones dismórficas, siendo frecuente la observación de plaquetas redondas, de gran talla, agranulares (plaquetas azules) y trombocitos con intensa dilatación vacuolar. La MO es hipercelular, con aumento de la relación mieloide/eritroide 10:1 (normal 2/3:1). La hiperplasia mieloide se produce a expensas de todos los estadios evolutivos. Existe un predominio de mielocitos y promielocitos con escasos blastos. La megacariopoyesis está aumentada con abundantes rasgos dismórficos. La eritropoyesis se encuentra disminuida proporcionalmente con respecto a las restantes series. MORFOLOGIA LMC
Mieloblastos.LMC.SP.MGG 1000x
LMC.SP.MGG 1000x
Promielocito y Juvenil.LMC.SP. MGG 1000x
Promielocito, Mielocitos y Baciliforme. LMC.SP.MGG 1000x
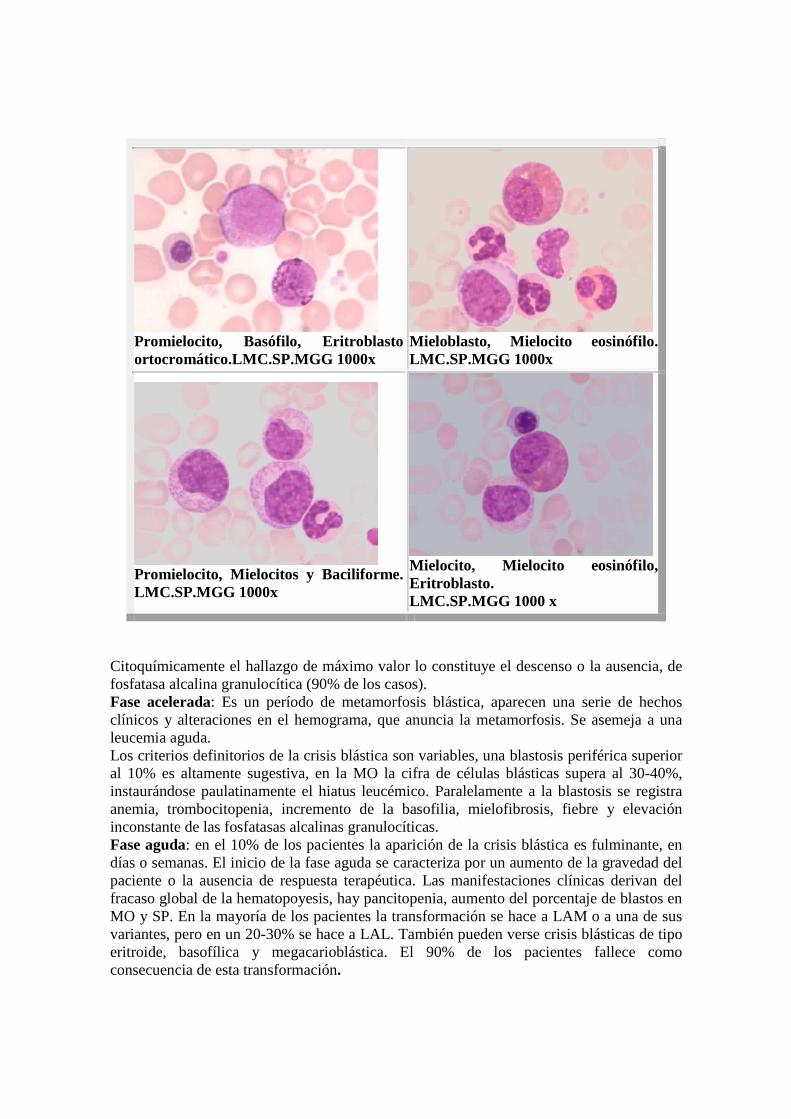
Promielocito, Basófilo, Eritroblasto ortocromático.LMC.SP.MGG 1000x
Mieloblasto, Mielocito eosinófilo.LMC.SP.MGG 1000x
Promielocito, Mielocitos y Baciliforme. LMC.SP.MGG 1000x
Mielocito, Mielocito eosinófilo, Eritroblasto. LMC.SP.MGG 1000 x
Citoquímicamente el hallazgo de máximo valor lo constituye el descenso o la ausencia, de fosfatasa alcalina granulocítica (90% de los casos). Fase acelerada: Es un período de metamorfosis blástica, aparecen una serie de hechos clínicos y alteraciones en el hemograma, que anuncia la metamorfosis. Se asemeja a una leucemia aguda. Los criterios definitorios de la crisis blástica son variables, una blastosis periférica superior al 10% es altamente sugestiva, en la MO la cifra de células blásticas supera al 30-40%, instaurándose paulatinamente el hiatus leucémico. Paralelamente a la blastosis se registra anemia, trombocitopenia, incremento de la basofilia, mielofibrosis, fiebre y elevación inconstante de las fosfatasas alcalinas granulocíticas. Fase aguda: en el 10% de los pacientes la aparición de la crisis blástica es fulminante, en días o semanas. El inicio de la fase aguda se caracteriza por un aumento de la gravedad del paciente o la ausencia de respuesta terapéutica. Las manifestaciones clínicas derivan del fracaso global de la hematopoyesis, hay pancitopenia, aumento del porcentaje de blastos en MO y SP. En la mayoría de los pacientes la transformación se hace a LAM o a una de sus variantes, pero en un 20-30% se hace a LAL. También pueden verse crisis blásticas de tipo eritroide, basofílica y megacarioblástica. El 90% de los pacientes fallece como consecuencia de esta transformación.

Diagnóstico: Para el diagnóstico se considera el cuadro clínico, hemograma, mielograma, biopsia medular, fosfatasa alcalina leucocitaria, detección del cromosoma Ph1 y gen bcr/abl. Se debe diferenciar de otros trastornos mieloproliferativos, ya que cualquiera de ellos puede estar acompañado de leucocitosis, pero usualmente no llegan al grado de la LMC. Además de estos, puede haber una respuesta excesiva de la MO ante procesos infecciosos, inflamatorios o neoplásicos y llegar a producir un cuadro en SP similar a LMC, cuadro conocido como reacción leucemoide, pero en la mayoría de los casos no aparecen mieloblastos, no existe eosinofilia ni basofilia acompañante y el recuento de plaquetas es normal, aunque pudiera estar elevado como reactante de fase aguda frente a otra enfermedad subyacente. En algunos pacientes menores de 5 años de edad se observa una forma rara de LMC juvenil . Esta se distingue de la LMC del adulto por la ausencia del cromosoma Ph1, leucocitosis moderada, hemoglobina fetal elevada, trombocitopenia, monocitosis y ausencia de basofilia. Los pacientes con LMC juvenil rara vez sufren transformaciones blásticas y generalmente mueren por una infección o por insuficiencia de un órgano causada por un infiltrado de monocitos y macrófagos.