contribuciÓn al estudio de transportadores en...
TRANSCRIPT

1
CONTRIBUCIÓN AL ESTUDIO DE TRANSPORTADORES EN
TRIPANOSOMÁTIDOS: CLONACIÓN DE POR LO MENOS UN GEN CODIFICANTE
PARA PROTEÍNAS TRANSPORTADORAS DE NAD+
DAVID SANTIAGO MORALES HERRERA
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS
FACULTAD DE CIENCIAS Y EDUCACIÓN
PROYECTO CURRICULAR DE LICENCIATURA EN QUÍMICA
BOGOTÁ, D.C.
2016

2
CONTRIBUCIÓN AL ESTUDIO DE TRANSPORTADORES EN
TRIPANOSOMÁTIDOS: CLONACIÓN DE POR LO MENOS UN GEN CODIFICANTE
PARA PROTEÍNAS TRANSPORTADORAS DE NAD+
DAVID SANTIAGO MORALES HERRERA
CÓDIGO: 20101150035
Trabajo de grado presentado para optar el título de
Licenciado en Química
Directora Externa
CLAUDIA CONSUELO RUBIANO, PhD.
Directora Interna
ADIS AYALA FAJARDO, MSc.
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS
FACULTAD DE CIENCIAS Y EDUCACIÓN
PROYECTO CURRICULAR DE LICENCIATURA EN QUÍMICA
BOGOTÁ, D.C.
2016

3
Nota de aceptación
______________________________________
______________________________________
______________________________________
______________________________________
______________________________________
______________________________________
______________________________________
Firma del Director externo
______________________________________
Firma del Director interno
______________________________________
Firma del jurado

4
AGRADECIMIENTOS
En primer lugar a mi familia quien estuvo siempre a mi lado dándome ánimo para continuar en
esos momentos en los que todo parecía ir de mal en peor, por sus constantes palabras de aliento y
sus brazos tendidos para levantarme en cada tropezón. Verdaderamente agradezco a mi Madre, mi
Tía, mi Hermano Nico y mi Padre sin los cuales este proceso no hubiese sido el mismo y quizás
no hubiese sucedido.
Adicionalmente a mis directoras Adis Ayala Fajardo de la Universidad Distrital Francisco José de
Caldas y Claudia Consuelo Rubiano de la Universidad Nacional de Colombia por su enseñanzas
y arduo trabajo de la mano de sus inigualables consejos y el empeño dispuesto para la elaboración
de esta investigación. Por último a la profesora María Helena Ramírez de la Universidad Nacional
de Colombia quien, sin la obligación de participar en el proyecto colaboró en su exitosa ejecución.
Por otra parte agradezco al Laboratorio de Investigaciones Básicas en Bioquímica (LIBBIQ) por
permitirme desarrollar este proyecto en sus instalaciones y por confiar en mis capacidades desde
el primer instante y permitirme hacer parte del grupo de investigación en mis estudios de maestría
en ciencias Bioquímica. Principalmente agradezco a Carlos Alfonso Nieto, quien fue mi tutor y
hasta la fecha un gran amigo y compañero de trabajo caracterizado por su humildad e inteligencia.
Agradezco inmensamente a mi Alma mater la Universidad Distrital Francisco José de Caldas quien
me educó y edificó como profesional y persona y a la Universidad Nacional de Colombia la cual
abrió sus puertas para experimentar un año de intercambio y ejecución de este proyecto sin recibir
ningún tipo de beneficio.
Por último, pero no menos importante, a mis amigas Tatiana Castillo y Adriana Martínez con las
cuales disfruté este recorrido por la universidad y quienes estuvieron siempre pendientes de mi
bienestar, quienes hoy en día siguen siendo un pilar fundamental en mi formación como persona
y con las que he vivido experiencias inolvidables.
“La vida tiene sus matices claros y oscuros, está en tus manos escoger el que quieras vivir”

5
TABLA DE CONTENIDO
1. INTRODUCCIÓN ............................................................................................................... 13
2. JUSTIFICACIÓN ................................................................................................................ 15
3. ANTECEDENTES .............................................................................................................. 17
4. OBJETIVOS ........................................................................................................................ 20
4.1. General ........................................................................................................................ 20
4.2. Específicos .................................................................................................................. 20
5. MARCO TEÓRICO ............................................................................................................ 21
5.1. Nicotinamida adenina dinucleótido NADH/NAD+ ................................................... 21
5.1.1. Biosíntesis del NAD+ ......................................................................................... 22
5.1.2. Papel no metabólico del NAD+ .......................................................................... 24
5.1.3. Papel metabólico del NAD+ ............................................................................... 26
5.2. Parasitismo.................................................................................................................. 31
5.2.1. Clasificación de los parásitos ............................................................................. 32
5.2.2. Adaptaciones biológicas evolutivas de los parásitos ......................................... 33
5.2.3. Hospedero y vector ............................................................................................. 34
5.2.4. Reproducción ...................................................................................................... 34
5.3. Leishmania braziliensis .............................................................................................. 35
5.4. Ciclo de vida Leishmania braziliensis ........................................................................ 39
5.5. Leishmaniasis cutánea ................................................................................................ 40
5.5.1. Tratamiento leishmanicida ................................................................................ 41
5.6. Proteínas transportadoras de nucleótidos ................................................................... 43
5.6.1. Tipos de transportadores de membrana ............................................................ 44
5.7. Técnicas de biología molecular .................................................................................. 47
5.7.1. Amplificación por PCR ...................................................................................... 47
5.7.2. Clonación en Escherichia coli in vivo ............................................................... 48
5.8. Métodos computacionales .......................................................................................... 52
5.8.1. Análisis de estructura primaria ......................................................................... 52
5.8.2. Análisis de estructura secundaria ...................................................................... 54
5.8.3. Análisis de estructura terciaria .......................................................................... 55
6. METODOLOGÍA ................................................................................................................ 58
6.1. Métodos computacionales. ......................................................................................... 58
6.1.1. Análisis de estructura primaria. ........................................................................ 59
6.1.2. Predicción de estructura secundaria. ................................................................ 60

6
6.1.3. Predicción de la estructura terciaria. ................................................................ 61
6.2. Métodos experimentales. ............................................................................................ 61
6.2.1. Evaluación de la integridad del ADNg de Leishmania braziliensis. ................ 61
6.2.2. Diseño y estandarización de Tm de los cebadores mediante PCR. .................. 62
6.2.3. Estandarización de la amplificación por PCR con Pfu. ................................... 62
6.2.4. Obtención de vectores recombinantes LbTNT´s /pET100 y LbTNT´s
/pBAD202. ........................................................................................................................... 63
6.2.5. Expresión y detección de las proteínas recombinantes 6xHisLbTNT´s y
TrxLbTNT´s por electroforesis SDS-PAGE y Western blot. ............................................. 66
7. RESULTADOS Y DISCUSIÓN ......................................................................................... 68
7.1. Métodos computacionales .......................................................................................... 68
7.1.1. Análisis de estructura primaria ......................................................................... 68
7.I.2. Predicción de estructura secundaria ................................................................. 80
7.I.3. Predicción de la estructura terciaria ................................................................. 86
7.2. Métodos experimentales ............................................................................................. 96
7.2.1. Evaluación de la integridad del ADNg de Leishmania braziliensis ................. 96
7.2.2. Diseño y estandarización de Tm de los cebadores mediante PCR ................... 96
7.2.3. Estandarización de la amplificación por PCR con Pfu .................................... 98
7.2.4. Obtención de vectores recombinantes LbTNT´s/pET100 y LbTNT´s/pBAD202
101
7.2.5. Expresión y detección de las proteínas recombinantes 6xHisLbTNT´s y
TrxLbTNT´s por electroforesis SDS-PAGE y Western blot ............................................ 110
8. CONCLUSIONES ............................................................................................................. 119
9. PERSPECTIVAS............................................................................................................... 121
10. ANEXOS ........................................................................................................................... 122
11. REFERENCIAS ................................................................................................................ 127

7
ÍNDICE DE TABLAS Tabla 1. Reacciones que utilizan como coenzima el NAD(P)+. .............................................................................. 28 Tabla 2. Clasificación taxonómica de Leishmania braziliensis. ............................................................................. 35 Tabla 3. Especies de Cinetoplastidos de mayor importancia a nivel médico y veterinario.................................... 37 Tabla 4. Clasificación taxonómica del género Lutzomyia. ..................................................................................... 40 Tabla 5. Estructuras desarrolladas de cada uno de los medicamentos leishmanicidas disponibles. ..................... 42 Tabla 6. Miembros de la familia de transportadores de solutos relacionados con el transporte de nucleótidos. . 45 Tabla 7. Vectores más utilizados en clonación. .................................................................................................... 48 Tabla 8. Posición de los diferentes genes que componen en vector pET100/D-TOPO. ......................................... 50 Tabla 9. Selección de candidatos a transportadores de NAD+ mediante la herramienta BLAST. .......................... 59 Tabla 10. Características de los cebadores utilizados para amplificar los candidatos a transportadores de NAD+.
.................................................................................................................................................................. 62 Tabla 11. Enzimas de restricción utilizadas para la digestión de los plásmidos recombinantes. ........................... 65 Tabla 12. Especificaciones de las secuencias candidatas como transportadores mitocondriales de NAD+/NADH.68 Tabla 13. Parámetros fisicoquímicos de los candidatos como transportadores mitocondriales de NAD+ evaluadas
con la herramienta ProtParam. ................................................................................................................. 69 Tabla 14. Predicción de motivos y dominios de los candidatos mediante MotifFinder e InterPro. ....................... 71 Tabla 15. Predicción de hélices transmembranales y topología para los candidatos por los servidores TMHMM
2.0, Phobius y TOPCONS. ........................................................................................................................... 74 Tabla 16. Residuos conservados en las secuencias de los candidatos LbTNTA, LbTNTB y LbTNTC y las plantillas
utilizadas. .................................................................................................................................................. 76 Tabla 17. Predicción de sitios de fosforilación y acetilación con las herramientas NetPhos 2.0 y NetAcet 1.0
respectivamente para los 3 candidatos. .................................................................................................... 78 Tabla 18. Predicción de solubilidad de las proteínas recombinantes de los tres candidatos obtenidas con los
vectores pBAD201-d-TOPO, pCOLD, pETSUMO, pET100-d-TOPO y pMAL-c5X. .......................................... 79 Tabla 19. Determinación de la topología del candidato LbTNTA por los servidores TMHMM, Phobius y TOPCONS.
.................................................................................................................................................................. 81 Tabla 20. Determinación de la topología del candidato LbTNTB por los servidores TMHMM, Phobius y TOPCONS.
.................................................................................................................................................................. 83 Tabla 21. Determinación de la topología del candidato LbTNTC por los servidores TMHMM, Phobius y TOPCONS.
.................................................................................................................................................................. 85 Tabla 22. Predicción de estructura terciaria por homología mediante el servidor Swiss-Model. .......................... 87 Tabla 23. Valores C obtenidos para los modelos construidos por Threading para la secuencia completa del
candidato LbTNTA por I-TASSER. ............................................................................................................... 88 Tabla 24. Valores C obtenidos para los modelos construidos por Threading para la secuencia completa e
incompleta del candidato LbTNTB por I-TASSER. ....................................................................................... 91 Tabla 25. Valores C obtenidos para los modelos construidos por Threading para la secuencia completa e
incompleta del candidato LbTNTC por I-TASSER. ....................................................................................... 94 Tabla 26. Valores de absorbancia obtenidos de la cuantificación espectrofotométrica de ADN. .......................... 96 Tabla 27. Análisis de los cebadores diseñados para los candidatos mediante la herramienta OligoAnalyzer 3.1. 97 Tabla 28. Cantidades de inserto necesarias para una ligación con relación inserto:vector de 3:1 en pBAD202/D-
TOPO y pET100/D-TOPO para los tres candidatos. .................................................................................. 102 Tabla 29. Determinación del peso molecular de las proteínas obtenidas a partir de los plásmidos 3 y 5 para
LbTNTB. ................................................................................................................................................... 115 Tabla 30. Determinación del peso molecular de la proteína obtenida a partir del plásmido 1 para LbTNTC. ..... 117

8
ÍNDICE DE FIGURAS
Figura 1. Estructura del NAD(P)+ y el NAD(P)H. _____________________________________________________ 21 Figura 2. Absorción del NAD+ y el NADH en el ultravioleta. ___________________________________________ 22 Figura 3. Rutas de biosíntesis del NAD+.___________________________________________________________ 23 Figura 4. Proceso de mono-ADP-ribosilación y poli-ADP-ribosilación. ___________________________________ 24 Figura 5. Acción catalítica de la ADP-ribosil ciclasa para obtener cADPRP y NAADP. _______________________ 25 Figura 6. Mecanismo de deacetilación regulado por las sirtuínas. ______________________________________ 26 Figura 7. Receptores universales de electrones. ____________________________________________________ 27 Figura 8. Bombeo de protones hacia el espacio intermembranal por la fosforilación oxidativa. _____________ 29 Figura 9. Lanzadera Glicerol-3-Fosfato. ____________________________________________________________ 30 Figura 10. Lanzadera Malato-Aspartato. __________________________________________________________ 31 Figura 11. Reproducción en protozoos. ___________________________________________________________ 35 Figura 12. Estadíos de Leishmania braziliensis. _____________________________________________________ 38 Figura 13. Ciclo de vida de Leishmania braziliensis. __________________________________________________ 39 Figura 14. Proteínas de membrana. ______________________________________________________________ 43 Figura 15. Tipos de transportadores dependientes del gradiente electroquímico. _________________________ 44 Figura 16. Tipos de transportadores. _____________________________________________________________ 45 Figura 17. Modelo de interacción entre los transportadores de nucleótidos identificados en Clamidia
trachomatis.____________________________________________________________________________ 46 Figura 18. Etapas de la amplificación por PCR. ______________________________________________________ 48 Figura 19. Secuencias de los vectores pET100/D-TOPO y pBAD202/D-TOPO. _____________________________ 49 Figura 20. Relaciones óptimas Inserto:Vector para los vectores pET100/D-TOPO y pBAD202/D-TOPO. _______ 51 Figura 21. Mecanismo de ligación en los vectores pET100/D-TOPO y pBAD202/D-TOPO. ___________________ 51 Figura 22. Etapas del modelado de estructuras mediante PHYRE2. _____________________________________ 54 Figura 23. Representación de las etapas definidas por I-TASSER para la predicción de estructura y función de
proteínas. ______________________________________________________________________________ 55 Figura 24. Predicción péptido señal con la herramienta SignalP 4.1. ____________________________________ 70 Figura 25. Predicción de hélices transmembranales para el candidato LbTNTA. __________________________ 74 Figura 26. Alineamiento múltiple de las secuencias de los candidatos y las plantillas mediante PRALINE. _____ 75 Figura 27. Predicción de sitios de fosforilación para las secuencias candidatas con la herramienta NetPhos 2.0. 77 Figura 28. Predicción de la estructura secundaria para el candidato LbTNTA con el servidor PHYRE2. Tomado de L.
Kelley y otros (2015) [58] _________________________________________________________________ 80 Figura 29. Topología predicha para el candidato LbTNTA con el servidor PHYRE2. _________________________ 81 Figura 30. Topología predicha para el candidato LbTNTB con el servidor PHYRE2. ________________________ 83 Figura 31. Topología predicha para el candidato LbTNTC con el servidor PHYRE2. _________________________ 84 Figura 32. Factor B normalizado para la secuencia completa del candidato LbTNTA por I-TASSER. ___________ 87 Figura 33. Modelo de estructura terciaria para el candidato LbTNTA mediante I-TASSER. __________________ 88 Figura 34. Factor B normalizado para la secuencia incompleta del candidato LbTNTA por I-TASSER. __________ 89 Figura 35. Alineamientos estructurales entre el candidato LbTNTA con el transportador mitocondrial de
ADP/ATP mediante el programa UCSF Chimera 1.10.2. _________________________________________ 89 Figura 36. Factor B normalizado para la secuencia completa del candidato LbTNTB por I-TASSER. ___________ 90 Figura 37. Modelo de estructura terciaria para el candidato LbTNTB mediante I-TASSER. __________________ 91 Figura 38. Alineamientos estructurales entre el candidato LbTNTB con el transportador mitocondrial de
ADP/ATP mediante el programa UCSF Chimera 1.10.2. _________________________________________ 92 Figura 39. Factor B normalizado para la secuencia completa del candidato LbTNTC por I-TASSER. ___________ 93 Figura 40. Modelo de estructura terciaria para el candidato LbTNTC mediante I-TASSER. __________________ 94 Figura 41.Alineamientos estructurales entre el candidato LbTNTC con el transportador mitocondrial de ADP/ATP
mediante el programa UCSF Chimera 1.10.2. _________________________________________________ 95 Figura 42. Evaluación de la integridad del ADN extraído de Leishmania braziliensis. _______________________ 96 Figura 43. Estandarización de Tm para los candidatos LbTNTA, LbTNTB y LbTNTC mediante PCR. ____________ 98 Figura 44. Amplificación de los candidatos LbTNTA, LbTNTB y LbTNTC con taq polimerasa. _________________ 98

9
Figura 45. Estandarización Tm y concentración de MgSO4 para el candidato LbTNTA con Pfu polimerasa. _____ 99 Figura 46. Confirmación de la identidad del amplificado del candidato LbTNTA mediante restricción con AluI. 100 Figura 47. Amplificación de los candidatos LbTNTA, LbTNTB y LbTNTC con Pfu polimerasa. ________________ 101 Figura 48. PCR de colonia para rastrear los recombinantes LbTNT´s/pBAD202. __________________________ 102 Figura 49. PCR de colonia para LbTNTB/pBAD202 y LbTNTC/pBAD202 con pools separados. _______________ 103 Figura 50. PCR de plásmido para verificar los recombinantes LbTNT´s/pBAD202. ________________________ 104 Figura 51. Confirmación de la presencia del gen de LbTNTA en pBAD202 mediante restricción del plásmido 1 con
BamHI. _______________________________________________________________________________ 104 Figura 52. Confirmación de la presencia del gen de LbTNTB en pBAD202 mediante restricción del plásmido 7 con
SphI. _________________________________________________________________________________ 105 Figura 53. Confirmación de la presencia del gen de LbTNTC en pBAD202 mediante restricción de los plásmidos 1,
2, 3 y 4 con NcoI y EcoRV. ________________________________________________________________ 106 Figura 54. PCR de colonia para rastrear los recombinantes LbTNT´s/pET100. ___________________________ 107 Figura 55. PCR de plásmido para verificar los recombinantes LbTNT´s/pET100. __________________________ 107 Figura 56. Confirmación de la presencia del gen de LbTNTB en pET100 mediante restricción de los plásmidos 1 –
5 con EcoRV. __________________________________________________________________________ 108 Figura 57. Confirmación de la presencia del gen de LbTNTC en pET100 mediante restricción de los plásmidos 1 –
5, 8, 9 y 10 con NcoI y EcoRV. _____________________________________________________________ 109 Figura 58. Punto de corte de NcoI en el vector recombinante LbTNTC/pET100. __________________________ 110 Figura 59. Expresión de la proteína recombinante TrxLbTNTA en células BL21 DE3. ______________________ 111 Figura 60. Estandarización de la expresión de la proteína recombinante TrxLbTNTA en células BL21 DE3. ____ 112 Figura 61. Fracciones soluble e insoluble de la expresión de la proteína recombinante TrxLbTNTA en células BL21
DE3. _________________________________________________________________________________ 113 Figura 62. Expresión de la proteína recombinante TrxLbTNTC en células BL21 DE3. ______________________ 114 Figura 63. Expresión de la proteína recombinante 6xHisLbTNTB en células BL21 DE3. ____________________ 116 Figura 64. Expresión de la proteína recombinante 6xHisLbTNTC en células BL21 DE3. ____________________ 117 Figura 65. Fracciones soluble e insoluble de la expresión de las proteínas recombinantes 6xHisLbTNTB y
6xHisLbTNTC en células BL21 DE3. _________________________________________________________ 118

10
ÍNDICE DE ANEXOS
Anexo 1. Secuencia de nucleótidos para los candidatos LbTNTA, LbTNTB y LbTNTC. ......................................... 122 Anexo 2. Secuencia de aminoácidos para los candidatos LbTNTA, LbTNTB y LbTNTC. ........................................ 123 Anexo 3. Especificidad de los cebadores propuestos para cada candidato mediante la herramienta BLAST de
NCBI. ....................................................................................................................................................... 123 Anexo 4. Predicción de las hélices transmembranales para el candidato de 318aa. ........................................... 124 Anexo 5. Predicción de las hélices transmembranales para el candidato de 337aa. ........................................... 125 Anexo 6. Predicción de la estructura secundaria para los candidatos LbTNTB y LbTNTC con el servidor PHYRE2.
................................................................................................................................................................ 126

11
RESUMEN
La Leishmaniasis cutánea es producida principalmente por el parásito Leishmania braziliensis, el
cual afecta a los mamíferos al ingresar en su torrente sanguíneo es trasmitido por medio de los
insectos hematófagos del género Lutzomyia. De acuerdo con la organización mundial de la salud
(OMS) se estima que en todo el mundo se presentan entre 0,7 y 1,3 millones de casos nuevos al
año, de los cuales más de dos terceras partes son reportados en Afganistán, Argelia, Brasil,
Colombia, República Islámica del Irán y República Árabe Siria, por lo que se ha convertido en
problema de salud pública en la actualidad y de interés para identificar posibles blancos
terapéuticos.
El estudio del metabolismo energético de Leishmania braziliensis ha demostrado que la enzima
Nicotinamida Mononucleótido Adenililtransferasa (NMNAT) es de vital importancia para el
organismo debido a que cataliza la condensación reversible del ATP con el Mononucleótido de
Nicotinamida (NMN) o con el ácido nicotínico (NaMN) para formar el dinucleótido de
nicotinamida (NAD+) tanto en la ruta de novo como en la de reciclaje. Asimismo, el NAD+ tiene
tanto un papel metabólico en el metabolismo celular en el cual no es consumido sino que participa
en reacciones de oxidoreducción como aceptor de electrones y uno no metabólico en el cual si se
consume, lo que obliga a la célula a sintetizarlo continuamente para mantener niveles óptimos
tanto en el citosol como en otros compartimentos subcelulares como la mitocondria. A pesar de
que las NMNAT’s tienen un papel clave en la síntesis del NAD+ se ha identificado que este
parásito al igual que otros parásitos intracelulares como Tripanosoma cruzi y Plasmodium
falciparum únicamente cuentan con una isoenzima; en comparación se han descrito dos isoenzimas
(GlNMNATa y BLNMNATb) en parásitos extracelulares tales como Giardia duodenalis como en
organismos de vía libre como el Homo sapiens en el que se han reportado con tres isoenzimas
localizadas en el núcleo, el aparato de Golgi y en la mitocondria.
A partir de estos resultados se propuso que la diferencia entre el medio ambiente de organismos
de vía libre con respecto a los parásitos intracelulares ocasiona que los procesos de biosíntesis de
nucleótidos hayan sido reemplazados por los de transporte para obtener nutrientes
macromoleculares como las coenzimas desde las células del hospedero en estos últimos como se
ha demostrado en estudios de algunas especies parasitarias intracelulares pertenecientes al género
Clamidia las cuales han perdido la capacidad de sintetizar el NAD+ por la ruta de novo y como
respuesta han desarrollado proteínas encargadas del transporte tanto de nucleótidos (ATP, GTP,
CTP y UTP) como del dinucleótido de adenina. Por lo tanto, el objetivo de este estudio fue el de
caracterizar, identificar y clonar por lo menos uno de los genes candidatos que codifiquen un
análogo de estas proteínas con función transportadora de NAD+ en el parásito intracelular
Leishmania braziliensis mediante técnicas computacionales para el análisis de la estructura
primaria como la predicción de propiedades fisicoquímicas, motivos y dominios, modificaciones
post-traduccionales, hélices transmembranales, solubilidad de las proteínas recombinantes a partir
de diferentes vectores, presencia de péptido señal, posible localización subcelular y determinación
de los residuos conservados, para la predicción de la estructura secundaria y para la obtención de
modelos predictivos por threading de la estructura terciaria así como mediante el uso de técnicas
experimentales como la amplificación por PCR de cada gen a partir de ADN genómico del
parásito, clonación de estos en los vectores pBAD202/D-TOPO y pET100/D-TOPO,
comprobación mediante PCR y digestión con enzimas de restricción de los vectores

12
recombinantes, expresión, solubilización y finalmente detección por medio de Western blot de las
proteínas recombinantes obtenidas.
A partir de los métodos computacionales se identificaron 19 residuos altamente conservados
presentes en las secuencias de los 3 candidatos LbTNTA, LbTNTB y LbTNTC de los cuales es
importante resaltar la presencia del residuo de lisina K289 y el de ácido glutámico E361
(posiciones en el alineamiento global) relacionados con el transporte de nucleótidos de adenina,
asimismo el estudio de las modificaciones postraduccionales halló que las 3 proteínas tienen alta
probabilidad de presentar una regulación por fosforilación/desfosforilación pero no por
acetilación/desacetilación. Adicionalmente se comprobó en los 3 péptidos la presencia del dominio
característico de la familia de “Transportadores mitocondriales” (PF000153 en Pfam)
caracterizado por la presencia de 6 hélices α transmembranales, las cuales también fueron
reconocidas en las 3 secuencias evaluadas mediante el análisis de estructura primara, secundaria y
adicionalmente en los modelos predictivos de estructura terciaria, a partir de los cuales se
comprobó una homología estructural con el transportador mitocondrial de ADP/ATP de la especie
Bos taurus con un valor C de 0.958 para LbTNTA de 0.847 para LbTNTB y de 0.961 para
LbTNTC, lo cual sugiere una posible homología funcional y por lo tanto la capacidad de
transportar nucleótidos de adenina, como también una psobile función de transporte de NAD+.
Finalmente se determinó una localización mitocondrial o peroxisomal para los 3 candidatos.
Experimentalmente se comprobó la presencia de los genes correspondientes a los 3 candidatos con
un tamaño de 1.089 pb para LbTNTA, 1.014 pb para LbTNTB y 957 pb para LbTNTC en el
genoma de Leishmania braziliensis, con los cuales se obtuvieron 25 colonias para LbTNTA, 9
para LbTNTB y 11 para LbTNTC producto de la ligación en el vector pBAD202/D-TOPO y 1
para LbTNTA, 9 para LbTNTB y 26 para LbTNTC con pET100/D-TOPO. De los clones obtenidos
con pBAD202/D-TOPO se identificó que el plásmido recombinante número 1 para el candidato
LbTNTA expresó la proteína TrxLbTNTA con un peso aproximado de 52 kDa y que esta se
encontraba presente principalmente en la fracción insoluble y que por el contrario no se obtuvieron
clones viables para las otras dos proteínas. Por otra parte la clonación en pET100/D-TOPO
demostró que los vectores recombinantes número 3 y 5 para LbTNTB expresaban una proteína
recombinante con un peso aproximado de 38.51 y 39.25 kDa y que el clon número 1 para LbTNTC
producía una proteína de aproximadamente 36.37 kDa los cuales son pesos moleculares menores
a los esperados para 6xHisLbTNTB (40.8 kDa) y para 6xHisLbTNTC (38.3 kDa). Finalmente se
determinó que ambos péptidos se encuentran únicamente en la fracción proteica insoluble. De
acuerdo con esto se sugiere realizar la secuenciación de los vectores recombinantes número 3 y 5
para LbTNTB/pET100 y número 1 para LbTNTC/pET100 con el objeto de confirmar la secuencia
de los genes de las proteínas recombinantes 6xHisLbTNTB y 6xHisLbTNTC
Mediante este trabajo se obtuvieron las proteínas recombinantes TrxLbTNTA, 6xHisLbTNTB y
6xHisLbTNTC las cuales permitirán llevar a cabo ensayos para la producción de anticuerpos
específicos con los cuales se podrán desarrollar estudios de inmunodetección por Western blot en
los extractos de Leishmania braziliensis como también de localización subcelular in vivo de las 3
proteínas endógenas candidatas a transportadores de NAD+. Igualmente los constructos obtenidos
pueden ser utilizados para evaluar la capacidad transportadora de NAD+ de las proteínas en
modelos procariotas como Escherichia coli, sin embargo se recomienda realizar la subclonación
de estos genes en vectores de organismos eucariotas como levaduras para garantizar su adecuada
expresión.

13
1. INTRODUCCIÓN
Se ha demostrado que el parásito Leishmania braziliensis posee diversas proteínas que interactúan
con las especies NAD+/NADH, ya que son importantes en la producción de energía en la
mitocondria, por lo tanto, su estudio constituye un avance importante para establecer posibles
blancos terapéuticos que permitan combatir a este microorganismo, ya que se estima que en el
mundo se producen entre 0,7 y 1,3 millones de casos nuevos y un número de víctimas mortales
entre 20.000 y 30.000 al año. En el caso particular de Colombia existen aproximadamente 11
millones de personas en riesgo de sufrir la enfermedad, que afecta generalmente solo la piel
ocasionando lesiones que pueden variar desde úlceras y nódulos lisos hasta lesiones
hiperqueratosicas.
Actualmente no hay suficientes estudios relacionados con el transporte NAD+/NADH en el género
Leishmania, a pesar de esto se ha encontrado evidencia de enzimas que interactúan con estas
especies como las isoenzimas de la fumarato reductasa dependiente de NADH presentes en el
glicosoma y en la matriz mitocondrial, las cuales catalizan la conversión de fumarato en succinato
y una Nicotinamida Mononucleótido Adenililtransferasa (NMNAT) en L. braziliensis que sintetiza
el NAD+ a partir de la condensación reversible del ATP con el Mononucleótido de Nicotinamida
(NMN) o de ácido nicotínico (NaMN) en la biosíntesis del NAD+ tanto en la ruta de novo como
en la de reciclaje, adicionalmente se ha estudiado un transportador de Ca+2 relacionado con el
potencial electroquímico del gradiente de protones de la fosforilación oxidativa.
Este trabajo tiene por objetivo la caracterización in-sílico de una serie de proteínas candidatas a
transportadores de NAD+, las cuales fueron seleccionadas por el Laboratorio de Investigaciones
Básicas en Bioquímica (LIBBIQ) de la Universidad Nacional de Colombia a partir de los
transportadores de NAD+ presentes en Arabidopsis thaliana y Saccharomyces cerevisiae y
obtener por lo menos un clon en un vector de expresión de uno de estos candidatos para dar paso
a su futura caracterización experimental.
Investigaciones realizadas demuestran la presencia de enzimas que interactúan con el
NADH/NAD+ tanto en el citosol como en la matriz mitocondrial del parásito. Adicionalmente se
ha identificado una sola isoenzima de la NMNAT en L. braziliensis (al igual que en otros parásitos
intracelulares como Tripanosoma cruzi y Plasmodium falciparum) en comparación con especies
extracelulares o de vida libre como Giardia duodenalis y en Homo sapiens, lo que indica
posiblemente que los mecanismos de biosíntesis de estos dinucleótidos son sustituidos por los de
transporte, ya que poseen el medio (célula del hospedero) para obtenerlos fácilmente. Sin embargo,
no existen reportes que demuestren mecanismos de transporte de estas especies hacia dentro o
fuera del parásito o de la mitocondria, como por ejemplo el sistema de lanzaderas presente en otros
organismos eucariotas superiores o de transportadores transmembranales específicos, por lo tanto
su estudio es importante para identificar posibles blancos terapéuticos en Leishmania braziliensis.
Por otra parte, el estudio de las proteínas implicadas en los procesos de transporte de las especies
NADH/NAD+ en este modelo eucariota contribuye a identificar el modo en el que posiblemente
ingresan a la mitocondria o a la célula estas especies esenciales en el balance redox y sirven como
modelo para otros organismos eucariotas más complejos, para los cuales se desconocen estos
procesos de movilización.

14
Este proyecto contribuyó con el análisis computacional y el modelamiento tridimensional de 3
proteínas seleccionadas como candidatos a transportadores de NAD+ en Leishmania braziliensis
y adicionalmente a la detección y clonación de los genes de cada una de ellas en los vectores
pBAD202/D-TOPO y pET100/D-TOPO. Asimismo este trabajo permitió identificar residuos
altamente conservados para los tres péptidos que pueden tener una relación directa en la interacción
con el NAD+ por lo que se sugiere estudiar su influencia en el posible proceso de transporte. Por
último el desarrollo experimental de esta investigación permitió obtener las proteínas
recombinantes TrxLbTNTA, 6xHisLbTNTB y 6xHisLbTNTC las cuales pueden ser utilizadas
para llevar a cabo procedimientos de identificación de cada una de las proteínas endógenas en el
parásito y así mismo su localización subcelular, por otra parte provee el material de partida para
realizar la subclonación de cada gen en vectores de organismos eucariotas como por ejemplo en
levaduras mediante los cuales evaluar la capacidad transportadora de cada candidato.

15
2. JUSTIFICACIÓN
A finales del siglo XIX Cunningham, Borovsky, Leishman, Donovan, Wright, Lindenberg y
Vianna identificaron el parásito causante de la Leishmaniasis, al que Ronald Ross dio el nombre
genérico de Leishmania. En 1912, en Brasil, Carini identificó Leishmania en las lesiones mucosas
de pacientes con leishmaniasis. En 1914 Yakimoff y Shakor distinguieron los parásitos causantes
de la forma urbana y rural de la leishmaniasis cutánea en Asia Central. A principios de los años
cuarenta, Swaminath, Shortt y Anderson, en la India, y Adler y Ber, en Palestina, demostraron la
transmisión de L. donovani y L. tropica por los miembros de la sub-familia de los flebótomos
(moscas de la arena) [1].
Es importante resaltar que los tripanosomátidos primitivos eran parásitos monogenéticos que
infectaban insectos no hematófagos. Sin embargo, los procesos adaptativos de los insectos
ocasionan que adquieran la habilidad para alimentarse de sangre por succión [2]. A partir de este
cambio, los tripanosomátidos sufrieron cambios morfológicos y funcionales como el desarrollo de
un flagelo y una membrana ondulante para facilitar el movimiento en el torrente sanguíneo de los
vertebrados.
Se considera que la infección en humanos se presentó debido a la deforestación producida por el
hombre para generar terrenos aptos para la agricultura y la cría de ganado en los últimos 200 a 300
años en América Latina, por lo cual los dipteros que se quedaron sin su fuente de alimento, debido
a la disminución de los animales salvajes, colonizaron las áreas circundantes a las viviendas
humanas e inclusive estas. A partir de estos cambios los insectos comenzaron a alimentarse de la
sangre tanto de los animales domésticos como la de los humanos [2]. Asímismo, de acuerdo con
la organización mundial de la salud (OMS) se estima que en todo el mundo se presentan entre 0,7
y 1,3 millones de casos nuevos al año, de los cuales más de dos terceras partes son reportados en
Afganistán, Argelia, Brasil, Colombia, República Islámica del Irán y República Árabe Siria, por
lo que se ha convertido en problema de salud pública en la actualidad y de interés para identificar
posibles blancos terapéuticos [1].
El genoma de Leishmania braziliensis tiene un tamaño de aproximadamente 32’005.207 pb de
ADN linear contenidos en 35 cromosomas organizados en 8.153 genes y 161 pseudogenes, los
cuales traducen un total de 8.333 proteínas. [3]. Al igual que otros cinetoplástidos, se caracteriza
por tener sus genes organizados en unidades de transcripción policistrónica y por la presencia de
pocos intrones, además carece de regiones subtelomericas grandes, por lo tanto, aproximadamente
el 50% de su información genética consiste en una secuencia repetitiva [4]. En este organismo el
ADN se encuentra organizado en dos agrupaciones principales: el genoma nuclear y el genoma
mitocondrial o cinetoplasto, organelo del cual proviene su clasificación.
Diversos estudios han demostrado que las Nicotinamida Mononucleótido Adenililtransferasas
(NMNAT´s) son las encargadas de la síntesis de NADH mediante la condensación reversible del
ATP con el Mononucleótido de Nicotinamida (NMN) o de ácido nicotínico (NaMN). Estas son
enzimas importantes en el metabolismo celular, sin embargo, es evidente que organismos de vía
libre como lo son el Homo sapiens y el parásito extracelular Giardia duodenalis poseen más de
una isoenzima, por el contrario en parásitos intracelulares únicamente se ha identificado una sola,
esto conllevó a proponer que en estos organismos no es necesario tener una enzima en cada

16
compartimento subcelular debido a que nutrientes y macromoléculas, incluido el NAD+/NADH
que necesitan para su supervivencia son tomados del medio intracelular de su hospedero a
diferencia de los organismos de vía libre en los que es necesaria su síntesis. Para realizar este
proceso vital para su supervivencia, los parásitos intracelulares probablemente poseen proteínas
transportadoras de nucleótidos en la membrana celular, lo que las convierte en un importante
objeto de estudio desde su identificación hasta su caracterización bioquímica y biofísica para
identificar posibles blancos terapéuticos en Leishmania braziliensis.
Adicionalmente se ha demostrado la presencia de las especies NADH/NAD+ tanto en el citosol
como en la matriz mitocondrial del parásito. Sin embargo, no existen publicaciones que evidencien
mecanismos de transporte de estas especies en el parásito, pero si se ha demostrado en otras
especies como el transportador CtNTT4 de Clamidia trachomatis que se encarga de realizar el
intercambio antiporte entre NAD+/ADP.
Por otra parte, el estudio de las proteínas implicadas en los procesos de transporte de las especies
NADH/NAD+ en este modelo eucariota contribuye a identificar el modo en el que posiblemente
ingresan a la mitocondria o a la célula estas especies esenciales en el balance redox y sirven como
modelos para otros organismos eucariotas más complejos, en los cuales se desconocen estos
procesos de movilización.

17
3. ANTECEDENTES
Las especies NAD+/NADH son importantes en la producción de energía para la célula siendo
transportadores de electrones entre los metabolitos presentes en los diferentes compartimentos
celulares como el núcleo, el citosol y la mitocondria en su papel como coenzima para muchos
polipéptidos así como su papel como sustrato en reacciones de modificaciones postraduccionales
y de señalización celular en donde se consume la mayor parte del NAD+ presente en la célula. Su
síntesis tanto en la ruta de novo como en la de reciclaje es regulada por la enzima Nicotinamida
Mononucleótido Adenililtransferasa (NMNAT), la cual cataliza la condensación reversible del
ATP con el Mononucleótido de Nicotinamida (NMN) o de ácido nicotínico (NaMN), por lo tanto
las NMNAT´s son enzimas clave en el mantenimiento de concentraciones óptimas de esta
coenzima al interior de la célula como lo han demostrado diferentes estudios en distintos
organismos.
La importancia de estas rutas de biosíntesis se han estudiado en el Homo sapiens y se han
identificado 3 isoenzimas de la NMNAT ubicadas en el núcleo (NMNAT1), en el aparato de Golgi
(NMNAT2) y en la mitocondria (NMNAT3), esta última presenta una alta tolerancia a las
modificaciones que pueda presentar el sustrato, mientras que la NMNAT1, muestra una
preferencia por la síntesis de NAD+ (forma oxidada) las otras dos isoenzimas lo hacen por la del
NADH (forma reducida). Adicionalmente se ha demostrado que cada una de las isoenzimas se
encuentran relacionadas con funciones específicas en cada compartimiento subcelular [5].
En organismos más sencillos como el parásito extracelular Giardia duodenalis se identificaron in-
silico dos isoenzimas, la GlNMNATa y la GlNMNATb, de las cuales se comprobó la actividad
adenililtransferasa para la GlNMNATa tanto con el mononucleótido de nicotinamida (NMN) como
con el ácido nicotínico (NAMN) bajo condiciones óptimas de pH y temperatura de 7.3 y 26°C.
Adicionalmente se identificó una cinética de Michaelis-Menten para el NMN y el ATP y una baja
afinidad por el NAMN [6].
De acuerdo con investigaciones del grupo INS – LIBBIQ de la Universidad Nacional de Colombia
se encontró en el 2015 en Leishmania braziliensis (un parásito intracelular) que la NMNAT, cuenta
con una inserción amino terminal específica para el género Leishmania relacionada con su
actividad enzimática, lo que convierte a esta región en un posible blanco terapéutico, [7]. De la
misma manera se identificó en Tripanosoma cruzi esta enzima por medio de alineamientos y se
generó un modelo de la posible estructura terciaria de la proteína y de un mutante delecional en el
extremo amino terminal mediante herramientas bioinformáticas, adicionalmente obtuvieron la
proteína recombinante 6xHisTcNMNAT con la cual se realizaron ensayos de actividad evaluados
por RP-HPLC que demostraron la actividad adenililtransferasa típica de las NMNAT´s [8]. Otras
investigaciones demostraron la actividad enzimática de la NMNAT en Plasmodium falciparum y
su localización en el citoplasma del parásito, asimismo se comprobó la importancia del residuo
conservado Asp110 para su actividad catalítica mediante mutagénesis [9].
A pesar de la importancia que tienen estas dos vías de síntesis de nucleótidos se ha encontrado que
algunos organismos no poseen o han perdido la capacidad de sintetizarlos por la ruta de novo,
como es el caso de la bacteria intracelular Protoclamidia amoebofilia, que posee 5 proteínas
(NTT1 - NTT5) pertenecientes a la familia de transportadores de nucleótidos (NTT) con

18
especificidad para diferentes nucleótidos entre los que se encuentran el ATP, GTP, CTP, UTP y el
NAD+.
Investigaciones en Protoclamidia amoebofilia demostraron que NTT1 se encuentra relacionado
con el parasitismo energético de la bacteria llevando a cabo el ingreso de ATP por intercambio
con ADP; adicionalmente identificaron que NTT2 posee un 47% de identidad con la translocasa
ATP/ADP (AtNTT1) presente en Clamidia trachomatis que tiene la capacidad de transportar ATP,
GTP y UTP. Asimismo se identificó que NTT4 muestra una identidad del 22% con el transportador
ATP/ADP (AtNTT1) de Arabidopsis thaliana y que tiene mayor preferencia con los nucleósidos
difosfato (como el ADP) que por los trifosfato, a pesar de esto, la presencia de NAD+ y NADH
disminuye significativamente su captación, lo que mostró la alta afinidad de este transportador por
NAD+/NADH (tasa de velocidad de 80 nM/mgh) y un intercambio de las especies
NAD+in/ADPout. Por último este estudio postuló un modelo de las interacciones metabólicas entre
estos 3 transportadores en el que NTT1 y NTT2 proveen el ATP para la obtención del ADP que
es intercambiado por NAD+ del medio extracelular por medio de NTT4 [10]. En esta misma
especie se realizaron estudios de tres de las 5 proteínas denominadas PamNTT2, PamNTT3 y
PamNTT5 confirmándose la afinidad de PamNTT2 con los cuatro nucleótidos constituyentes del
ARN y un transporte unidireccional acoplado con protones para PamTT3 y PamNTT5 con afinidad
hacia el UTP para el primero y con GTP y ATP para el segundo [11].
Trabajos en ingeniería genética demostraron que el transportador NTT4 de P. amoebofilia tiene la
capacidad de transportar NAD+, ya que ha sido utilizado para promover y mantener reacciones
dependientes de este dinucleótido, para lo cual fue necesario mantener sus niveles altos al interior
de la célula. En uno de estos estudios se clonó este transportador y la glicerol deshidrogenasa
(GDH) de Gluconobacter oxydans dependiente de NAD+ en bacterias E. coli para la producción
de dihidroxiacetona (DHA) obteniendose altos niveles de producción de este compuesto y una
relación entre las especies NAD+/NADH constante, lo que evidenció la actividad transportadora
de NTT4 [12].
Investigaciones en C. trachomatis, otra bacteria intracelular miembro del género Clamidia han
establecido que esta ha perdido la capacidad de sintetizar nucleótidos por la ruta de novo, pero para
superar esto posee dos transportadores específicos para nucleótidos que presentan poca homología
con la translocasa de NAD/+ADP (PamNTT4) de P. amoebophila. Sin embargo, el primero es una
translocasa de ATP/ADP (CtNPT1) que también tiene la capacidad de transportar NAD+
(velocidad máxima 5.8 nM/mgh) con mayor afinidad hacia este dinucleótido en comparación con
el ATP. El segundo es un transportador uniporte de GTP, UTP, CTP y ATP (CtNPT2) [13].
Este mecanismo también se ha identificado en la levadura Saccharomyces cerevisiae la cual es
utilizada en el laboratorio como modelo biológico de eucariotas. Este organismo sintetiza el NAD+
en el citosol y para su metabolismo debe atravesar la membrana interna de la mitocondria, para
ello posee un transportador específico denominado “Transportador mitocondrial de NAD+”
(ScNdt1p con código NP_010910.1), el cual se caracterizó funcionalmente, fue localizado en la
mitocondria y asimismo se observó que tenía la capacidad de transportar NAD+ y en menor
medida (d)AMP y (d)GMP, sin embargo, no lo hacía con las especies α-NAD+, NADH, NADP+
ni NADPH. Adicionalmente se encontró una isoforma ScNdt2p con un 70% de homología [14].

19
Por otra parte estudios realizados en la planta Arabidopsis thaliana identificaron dos isoenzimas
AtNDT1 (NP_566102) y AtNDT2 (NP_564233), las cuales presentaron gran semejanza
estructural con ScNdt1p descrita anteriormente. En esta investigación se encontró que AtNDT1
estaba ubicada en el cloroplasto y AtNDT2 en la mitocondria como también que estas tenían la
capacidad de transportar el NAD+ mediante intercambio siendo el ADP y el AMP los más
eficientes para este proceso [15].
En organismos más complejos evolutivamente como el Homo sapiens se halló un transportador
para las especies CoA, FAD, FMN y AMP y en menor medida de NAD+, PAP (adenosina 3’,5’-
difosfato) y ADP, el cual fue localizado en la membrana del peroxisoma y presenta un transporte
mediante intercambio de CoA, FAD+ y NAD+ hacia el interior del organelo por las especies PAP,
FMN y AMP generadas en su interior [16].
Estos estudios evidencian que las especies extracelulares o de vía libre como Giardia duodenalis
en incluso el Homo sapiens poseen varias isoenzimas de la NMNAT presentes en diversos
compartimientos subcelulares como el núcleo, el citosol o la mitocondria, mientras que, en las
intracelulares se ha identificado únicamente una isoforma, lo que indica posiblemente que los
mecanismos de biosíntesis de estos nucleótidos son relevados (o se encuentran ausentes) por los
de transporte como se ha descrito anteriormente, ya que al encontrarse en un medio (célula del
hospedero) de donde pueden obtenerlos fácilmente desarrollaron transportadores específicos para
cada uno de ellos, específicamente para el NAD+.

20
4. OBJETIVOS
4.1. General
Caracterizar in-silico e identificar al menos uno de los candidatos a proteínas transportadoras de
NAD+ en el parásito Leishmania braziliensis por medio de métodos computacionales y técnicas
de biología molecular a partir de los vectores de expresión pET100/D-TOPO y pBAD202/D-
TOPO.
4.2. Específicos
Caracterizar por métodos computacionales candidatos a proteínas transportadoras de NAD+
mediante análisis de estructura primaria, secundaria y terciaria.
Comprobar la presencia de los genes de los candidatos seleccionados en el genoma del parásito
Leishmania braziliensis.
Realizar un primer acercamiento experimental para obtener un clon de al menos una de las
proteínas candidatas a transportadores de NAD+.
Identificar la expresión de las proteínas recombinantes obtenidas a partir de los vectores
pET100/D-TOPO y pBAD202/D-TOPO.

21
5. MARCO TEÓRICO
5.1. Nicotinamida adenina dinucleótido NADH/NAD+
Es una coenzima que consta de dos nucleótidos unidos a través de sus grupos fosfato presentes en
el carbono 5’ de la ribosa, el primero de ellos contiene la nicotinamida unida al carbono 1 de la
ribosa y el segundo un anillo de adenina en la misma posición. Se puede encontrar en dos formas
en las células, como NAD+ y como NADP+ (formas oxidadas), esta última es producto de la
fosforilación en la posición 2’ de la ribosa de la adenosina del NAD+, siendo capaces cada una de
ellas de aceptar un par electrones en forma del ion hidruro (H-) para dar paso al NADH y al
NADPH (formas reducidas) respectivamente (Figura 1), de esta manera se convierten en
donadores de electrones. El grupo nicotinamida puede estar unido en dos orientaciones al átomo
de carbono 1’ de la ribosa, formándose dos conformaciones anoméricas denominadas α y β. Sin
embargo, la forma β-nicotinamida es la que se encuentra presente en los organismos [17].
Figura 1. Estructura del NAD(P)+ y el NAD(P)H.
Tomado de Pérez, G. 2016 [17].
Estas reacciones de transferencia de electrones son la principal función del NAD+. Sin embargo,
también es utilizado en otros procesos celulares como modificaciones post-traduccionales en
donde es sustrato de enzimas que añaden o eliminan grupos a las proteínas. El NADP+
(nicotinamida adenina dinucleótido fosfato) se utiliza como un agente reductor en reacciones
anabólicas, como la síntesis de lípidos, colesterol y ácidos nucleicos, así como en la elongación de
la cadena de ácidos grasos. Asimismo, es la fuente de equivalentes reductores en la hidroxilación
de compuestos aromáticos, esteroides, alcoholes y otras sustancias.
22
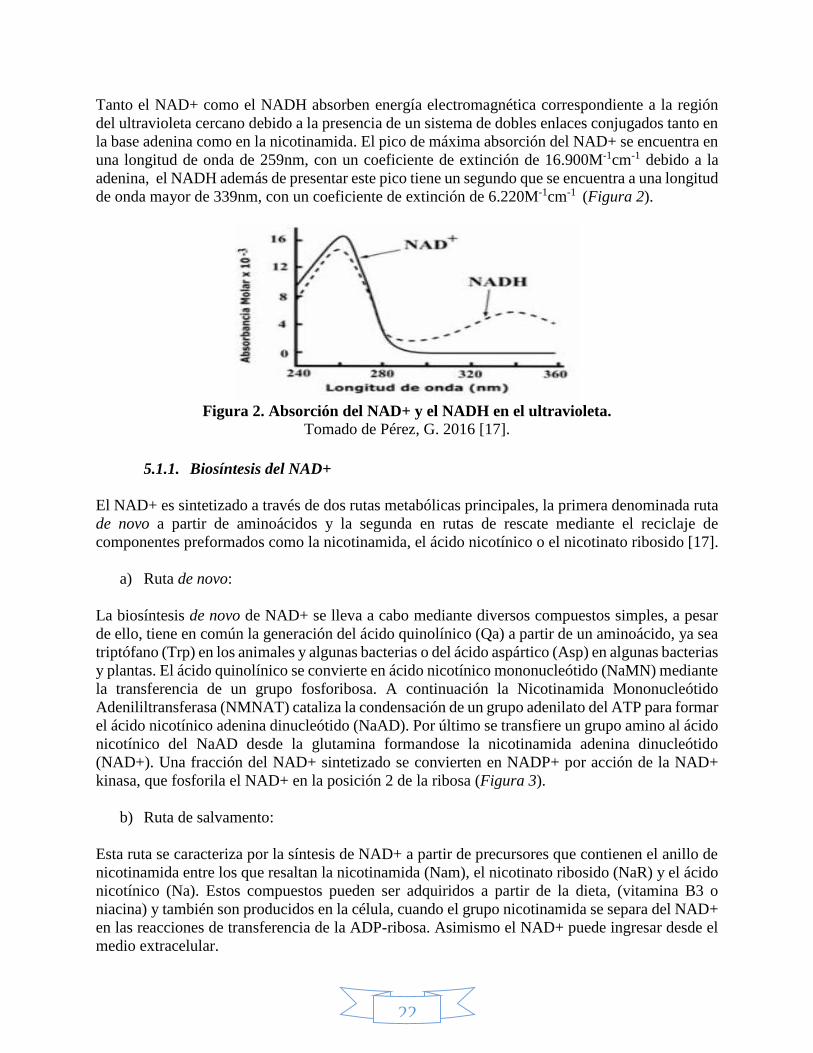
Tanto el NAD+ como el NADH absorben energía electromagnética correspondiente a la región
del ultravioleta cercano debido a la presencia de un sistema de dobles enlaces conjugados tanto en
la base adenina como en la nicotinamida. El pico de máxima absorción del NAD+ se encuentra en
una longitud de onda de 259nm, con un coeficiente de extinción de 16.900M-1cm-1 debido a la
adenina, el NADH además de presentar este pico tiene un segundo que se encuentra a una longitud
de onda mayor de 339nm, con un coeficiente de extinción de 6.220M-1cm-1 (Figura 2).
Figura 2. Absorción del NAD+ y el NADH en el ultravioleta.
Tomado de Pérez, G. 2016 [17].
5.1.1. Biosíntesis del NAD+
El NAD+ es sintetizado a través de dos rutas metabólicas principales, la primera denominada ruta
de novo a partir de aminoácidos y la segunda en rutas de rescate mediante el reciclaje de
componentes preformados como la nicotinamida, el ácido nicotínico o el nicotinato ribosido [17].
a) Ruta de novo:
La biosíntesis de novo de NAD+ se lleva a cabo mediante diversos compuestos simples, a pesar
de ello, tiene en común la generación del ácido quinolínico (Qa) a partir de un aminoácido, ya sea
triptófano (Trp) en los animales y algunas bacterias o del ácido aspártico (Asp) en algunas bacterias
y plantas. El ácido quinolínico se convierte en ácido nicotínico mononucleótido (NaMN) mediante
la transferencia de un grupo fosforibosa. A continuación la Nicotinamida Mononucleótido
Adenililtransferasa (NMNAT) cataliza la condensación de un grupo adenilato del ATP para formar
el ácido nicotínico adenina dinucleótido (NaAD). Por último se transfiere un grupo amino al ácido
nicotínico del NaAD desde la glutamina formandose la nicotinamida adenina dinucleótido
(NAD+). Una fracción del NAD+ sintetizado se convierten en NADP+ por acción de la NAD+
kinasa, que fosforila el NAD+ en la posición 2 de la ribosa (Figura 3).
b) Ruta de salvamento:
Esta ruta se caracteriza por la síntesis de NAD+ a partir de precursores que contienen el anillo de
nicotinamida entre los que resaltan la nicotinamida (Nam), el nicotinato ribosido (NaR) y el ácido
nicotínico (Na). Estos compuestos pueden ser adquiridos a partir de la dieta, (vitamina B3 o
niacina) y también son producidos en la célula, cuando el grupo nicotinamida se separa del NAD+
en las reacciones de transferencia de la ADP-ribosa. Asimismo el NAD+ puede ingresar desde el
medio extracelular.

23
Por otra parte el ácido nicotínico (Na) y el nicotinato ribósido (NaR) se obtienen a partir de la
nicotinamida (Nam) por desamidación y posterior ribosilación respectivamente. Una vez se
obtienen estos precursores son convertidos en el ácido nicotínico mononucleótido (NaMN) por
fosforilación para el caso del NaR y adicional ribosilación para el Na, a partir de este punto
continúan el mismo camino que la ruta de novo. Por último, la nicotinamida es ribosilada para
obtener la nicotinamida mononucleótido (NMN), que posteriormente se une con ATP por la acción
de la NMNAT para finalmente obtener el NAD+ (Figura 3).
Es importante resaltar que a pesar de la existencia de la ruta de novo, la ruta de reciclaje juega un
papel esencial en el metabolismo celular, ya que gran parte del NAD+ producido es consumido en
reacciones no metabólicas como las modificaciones postraduccionales lo que hace necesario su
síntesis en comparación con las reacciones redox en las que no se consume esta coenzima.
Figura 3. Rutas de biosíntesis del NAD+.
DHAP, dihidroxiacetona fosfato; Qa, quinolinato; PRPP, 5′-fosforibosil-1′-pirofosfato; NaMN,
nicotinato mononucleotido; NaAD, nicotinato adenina dinucleotido; (c)ADPR, ADP-ribosa
cíclico; PRO, proteína; Acetyl-H, histona acetilada; Nam, nicotinamida; Na, nicotinato; R1P,
ribosa 1-fosfato; NaR, nicotinato ribosido; QPRT, quinolinato fosforibosiltransferasa; NMNAT,
nicotinato (nicotinamida) mononucleotido adenililtransferasa; NADS, NAD sintasa; NAD ppase,
NAD pirofosfatasa; NADase, NAD glicohidrolasa; PARP, poli(ADP-ribosa)polimerasa;
N(a)PRT, nicotinamida (nicotinato) fosforibosiltransferasa; NIC, nicotinamidasa; NuP, nucleosido
fosforilasa; NaRK, nicotinato ribosido kinasa; NMN, nicotinamida mononucleótido. Tomado de
OXFORD Journals, 2016 [18].

24
Estas dos rutas de biosíntesis presentan una enzima en común, la nicotinato (nicotinamida)
mononucleotido adenililtransferasa o N(a)MNAT con un número de clasificación EC 2.7.7.1
cuando trabaja con la nicotinamida y EC 2.7.7.18 cuando lo hace con el ácido nicotínico [19]. Esta
clasificación enzimática corresponde con:
2. Transferasa
2.7 Transferasa de grupos que contienen fósforo
2.7.7 Nucleotidil transferasas
2.7.7.1 Nicotinamida mononucleótido adenililtransferasa
2.7.7.18 Nicotinato mononucleotido adenililtransferasa
5.1.2. Papel no metabólico del NAD+
El NAD+ cumple diversas funciones en las que no se producen reacciones de óxido reducción,
entre las que se encuentran algunas modificaciones postraduccionales, señalización celular y
deacetilaciones dependientes de NAD+. En este papel no metabólico el NAD+ es consumido y a
partir de algunos de sus subproductos se lleva a cabo la ruta de reciclaje para su regeneración [17].
a) ADP-Ribosilación:
Las reacciones de transferencia de ADP-ribosa son modificaciones postraduccionales
denominadas ADP-ribosilación que son catalizadas por las enzimas ADP-ribosiltransferasas que
transfieren el grupo ADP-ribosa del NAD+ a las proteínas. Esta reacción puede llevarse a cabo
con diversos objetivos, por una parte la adición de un solo grupo ADP-ribosa (mono-ADP-
ribosilación Figura 4) para la señalización celular, o por otra, la transferencia de cadena
ramificadas de poli-ADP-ribosa a las proteínas (poli-ADP-ribosilación Figura 4), reacción que es
catalizada por las polimerasas poli-(ADP-ribosa), las cuales están relacionadas con la regulación
de varios procesos celulares como la reparación del ADN y la apoptosis.
Figura 4. Proceso de mono-ADP-ribosilación y poli-ADP-ribosilación.
Tomado de Hassa, P. et. al. 2006 [20].

25
Este tipo de modificación se lleva a cabo por la transferencia de un grupo ADP-Ribosa desde el
NAD+ hacia receptores como los residuos de arginina, ácido glutámico o aspártico en la proteína
sustrato. Por su parte, la poli-ADP-ribosilación es un proceso que es muy frecuente en organismos
eucariotas pero no en procariotas o levaduras. A su vez, estas reacciones son reversibles por la
acción de las ADP-Ribosil hidrolasas.
Como se puede observar (Figura 4) este es un proceso en el que el NAD+ no participa como un
cofactor para las enzimas sino que tiene un papel como sustrato, lo que ocasiona que se consuma
y que en este caso se descomponga en ADP-ribosa y como subproducto en nicotinamida, uno de
los precursores del NAD+ por la ruta de reciclaje.
b) Movilización de calcio (Ca+2):
Las reservas intracelulares de calcio (Ca+2) son movilizadas a partir de la señalización de tres
moléculas, el inositol 1,4,5-trisfosfato (IP3), la ADP-fosfato-ribosa cíclica (cADPPr) y el ácido
nicotínico adenina dinucleótido fosforilado (NAADP) mediante señales internas producto su unión
y posterior apertura de canales de calcio denominados receptores de rianodina, que se encuentran
en las membranas de los organelos como el retículo endoplasmático [21].
El NADP+ es el sustrato de las ADP-ribosil ciclasas para dar lugar a la ADP-fosfato-ribosa cíclica
(Figura 5) proceso en el cual consumen este dinucleótido, generando por una parte la ciclación del
grupo ADP-ribosa con funciones de mensajero para la movilización de calcio y como subproducto
la nicotinamida que, como se mostró anteriormente es uno de los precursores de NAD+. Asimismo
estas ciclasas tienen la capacidad de intercambiar la nicotinamida por el ácido nicotínido en el
NAD+ para formar otro de las tres moléculas señalizadoras, el NAADP [22].
Figura 5. Acción catalítica de la ADP-ribosil ciclasa para obtener cADPRP y NAADP.
Tomado de Patel, S. Churchill, G; Galione, A. 2001 [21]

26
c) Deacetilación:
Por otra parte, el NAD+ también es consumido por las sirtuinas, un grupo de enzimas deacetilasas
dependientes de NAD+ que actúan mediante la transferencia de un grupo acetilo de los residuos
de lisina de la proteína sustrato al grupo ADP-ribosa del NAD+, ocasionando que este se rompa y
se libere la nicotinamida y la O-acetil-ADP-ribosa (3(2)’OAADPr ). Estas enzimas parecen estar
relacionadas con la regulación de la transcripción a través de histonas deacetilantes y la alteración
de la estructura del nucleosoma.
Figura 6. Mecanismo de deacetilación regulado por las sirtuínas.
Tomado de Feldman, J; Baeza, J; Denu, J. 2013 [23].
Las sirtuinas son enzimas que se encuentran relacionadas con procesos vitales de la célula como
la transcripción, la reparación del ADN y la resistencia al estrés, mediante el uso del NAD+ como
sustrato, consumiéndolo en su acción catalítica y obteniéndose como subproducto un precursor de
la biosíntesis de este dinucleótido por la ruta de salvamento (la nicotinamida), al igual que la ADP-
ribosilación [23].
Las ADN ligasas bacterianas son enzimas dependientes de NAD y se encargan de unir dos
extremos de ADN escindidos a partir de la donación del grupo adenosina monofosfato (AMP) del
NAD+ al fosfato 5' de un extremo de ADN. Este intermediario interactúa con el grupo hidroxilo
3' del otro extremo de ADN, formando un nuevo enlace fosfodiéster.
5.1.3. Papel metabólico del NAD+
El proceso metabólico de la célula acarrea el intercambio de electrones desde diversas especies
químicas entre los diferentes compartimientos celulares, dichos procesos son mediados por
deshidrogenasas presentes en las vías catabólicas que se encargan de catalizar el transporte de
electrones hacia sus receptores universales, los dinucleótidos de nicotinamida NAD+ o NADP+
(Figura 7) y los nucleótidos de flavina FMN o FAD (Figura 7), siendo partícipes en las siguientes
reacciones [24]:

27
Sustrato reducido + NAD+ ⇌ Sustrato oxidado + NADH + H+
Sustrato reducido + NADP+ ⇌ Sustrato oxidado + NADPH + H+
Sustrato reducido + FAD+ ⇌ Sustrato oxidado + FADH2
Sustrato reducido + FMN ⇌ Sustrato oxidado + FMNH2
Figura 7. Receptores universales de electrones.
a) NAD+, b) NADP+, c) FMN y d) FAD.
Tomado de Nelson, D. Co, M. 2008 [24]
Sin embargo, es importante tener en cuenta que la mayoría de las deshidrogenasas son específicas
para el NAD+ o presentan mayor afinidad con esta coenzima en comparación con los otros
receptores. Asimismo su distribución en la célula es diversa ya que algunas se encuentran
únicamente en el citosol, otras únicamente en la mitocondria y algunas de ellas poseen isoenzimas
en ambos compartimentos subcelulares. El complejo NAD-Deshidrogenasa remueve 2 átomos de
hidrógeno de los sustratos, uno de ellos se transfiere como un hidruro (:H-) al NAD+ y el otro es
liberado al medio como H+.
Vías importantes como el ciclo del ácido cítrico (ciclo de Krebs), la β–oxidación (oxidación de
ácidos grasos), la oxidación de aminoácidos y la descarboxilación oxidativa del piruvato
(catalizada por la piruvato deshidrogenasa) se llevan a cabo en la matriz mitocondrial (Tabla 1) y
son procesos en los que interviene el NAD+ como coenzima. Las principales reacciones en las que
el NAD(P)+ actúa como coenzima son las siguientes (Tabla 1):

28
Tabla 1. Reacciones que utilizan como coenzima el NAD(P)+.
COFACTOR REACCIÓN LOCALIZACIÓN
NAD+
α-cetoglutarato + CoA + NAD+ ⇌ Succinil-CoA + CO2 +
NADH + H+ Mitocondria
L-Malato + NAD+ ⇌ Oxalacetato + NADH + H+ Mitocondria y citosol
Piruvato + CoA + NAD+ ⇌ Acetil-CoA + CO2 + NADH +
H+ Mitocondria
Gliceraldeido 3-Fosfato + Pi + NAD+ ⇌ 1,3-
Bisfosfoglicerato + NADH + H+ Citosol
Lactato + NAD+ ⇌ Piruvato + NADH + H+ Citosol
β-Hidroxiacil-CoA + NAD+ ⇌ β-Cetoacil-CoA + NADH +
H+ Mitocondria
NADP+
Glucosa 6-Fosfato + NADP+ ⇌ 6-Fosfogluconato +
NADPH + H+ Citosol
6-Fosfogluconato + NADP+ ⇌ Ribosa-5-Fosfato + CO2 + NADPH + H+
Citosol
NAD+ o
NADP+
L-Glutamato + H2O + NAD(P) + ⇌ α-Cetoglutarato + NH4+
+ NAD(P)H Mitocondria
Isocitrato + NAD(P) + ⇌ α-Cetoglutarato + CO2 +
NAD(P)H + H+ Mitocondria y citosol
Tomado de Tomado de Nelson, D. Co, M. 2008 [24]
El transporte de electrones tiene por objetivo por una parte mantener el balance redox de la célula
y por otra la obtención de energía en la mitocondria mediante la vía metabólica denominada
fosforilación oxidativa, la cual inicia cuando los electrones llegan a la cadena respiratoria e
impulsan la síntesis de ATP. Los complejos enzimáticos encargados de estas tareas se encuentran
ubicados en la membrana interna de este organelo, la cual es impermeable a moléculas como los
H+, pero posee transportadores específicos para algunas moléculas como el piruvato, los ácidos
grasos, los aminoácidos y sus α–ceto derivados por medio de los cuales pueden atravesarla. En los
organismos aeróbicos más del 90% del ATP que se necesita para mantener la vida proviene de las
mitocondrias, el resto se forma en las glicólisis anaeróbica.
La fosforilación oxidativa consiste en una serie de portadores de electrones que actúan
secuencialmente, de los cuales, la mayoría corresponden a los grupos prostéticos de los complejos
proteicos que hacen parte de la cadena respiratoria, los cuales tienen la capacidad de donar/aceptar
1 o 2 electrones. El transporte de electrones se puede clasificar en tres grupos:
1. Transferencia directa de electrones: por ejemplo la reducción de Fe+3 a Fe+2.
2. Transferencia como H (H+ + e-).
3. Transferencia del ion hidruro (:H-), el cual transporta 2 electrones.
Los 2 electrones provenientes del NAD(P)H + H+ finalmente son captados por oxígeno molecular
(proveniente de la respiración celular), el cual se reduce para formar una molécula de agua, de
acuerdo a la siguiente reacción:
NADH + H+ + 1/2O2 NAD+ + H2O

29
Durante el proceso de transporte de 2 electrones por los cuatro complejos de la cadena respiratoria
hasta llegar al oxígeno molecular, son bombeados hacia el espacio intermembranal 10 protones
(H+), como sigue [24]:
1. 4 Protones son bombeados por el complejo I.
2. 4 Protones son bombeados por el complejo III.
3. 2 Protones son bombeados por el complejo IV.
Este proceso (Figura 8) se puede resumir en la siguiente ecuación:
NADH + 11H𝑁+ + 1/2O2 NAD+ + 10H𝑝
+ + H2O
Las letras N y P simbolizan la matriz mitocondrial y el espacio intermembranal respectivamente.
Figura 8. Bombeo de protones hacia el espacio intermembranal por la fosforilación
oxidativa.
De izquierda a derecha se muestran los complejos enzimáticos que conforman el ciclo de la
fosforilación oxidativa, evidenciándose el bombeo de 4 protones por el complejo I, 4 por el
complejo III y 2 por el complejo IV. Tomado de Nelson, D. Co, M. 2008 [24]
De esta manera el espacio intermembranal adquiere una alta concentración de H+, mientras que la
matriz una baja concentración, esto genera una diferencia de potencial en la membrana interna
mitocondrial lo que favorece la síntesis de ATP por el complejo multienzimático denominado ATP
sintasa, a partir de la energía generada por el transporte pasivo de 4 protones del espacio
intermembranal hacia la matriz mitocondrial, de acuerdo a la siguiente ecuación [24]:
ADP + Pi + 4H𝑝+ ATP + H2O + 4H𝑁
+
En estos dos procesos se evidencia que el NADH + H+ produce más energía que el FADH2 debido
a que el primero ingresa a la cadena de transporte de electrones por el complejo I, mientras que el

30
segundo lo hace por el complejo II, por lo tanto, existe en una diferencia de 4 protones que no son
bombeados al espacio intermembranal por el nucleótido de flavina. La cantidad de ATP sintetizado
por la fosforilación oxidativa de ambos receptores de electrones se muestra a continuación:
1. NADH + H+:
𝐻+ =10 𝐻+𝐸𝑠𝑝𝑎𝑐𝑖𝑜 𝑖𝑛𝑡𝑒𝑟𝑚𝑒𝑚𝑏𝑟𝑎𝑛𝑎𝑙
4 𝐻+ 𝐸𝑠𝑝𝑎𝑐𝑖𝑜 𝑖𝑛𝑡𝑟𝑎𝑚𝑖𝑡𝑜𝑐𝑜𝑛𝑑𝑟𝑖𝑎𝑙= 2.5 𝐴𝑇𝑃
2. FADH2:
𝐻+ =6 𝐻+𝐸𝑠𝑝𝑎𝑐𝑖𝑜 𝑖𝑛𝑡𝑒𝑟𝑚𝑒𝑚𝑏𝑟𝑎𝑛𝑎𝑙
4 𝐻+ 𝐸𝑠𝑝𝑎𝑐𝑖𝑜 𝑖𝑛𝑡𝑟𝑎𝑚𝑖𝑡𝑜𝑐𝑜𝑛𝑑𝑟𝑖𝑎𝑙= 1.5 𝐴𝑇𝑃
Como se mostró anteriormente (Tabla 1) algunos procesos que generan NADH + H+ ocurren en
el citoplasma, por lo tanto, este receptor de electrones debe ingresar a la mitocondria para ser
utilizado en la cadena de transporte de electrones para su oxidación aeróbica. A pesar de esto no
se ha evidenciado la presencia de una proteína transportadora de NADH/NAD+ en la membrana
interna mitocondrial, por lo cual los electrones provenientes del NADH citosólico son
transportados al interior de la mitocondria por sistemas de lanzaderas, de los cuales se han
identificado los siguientes:
a) Lanzadera Glicerol-3-Fosfato
En este sistema de lanzadera se aprovecha el NAD+ generado en la hidrogenación de la
dihidroxiacetona fosfato para obtener gricerol-3-fosfato en el citosol, debido a que estos dos
sustratos tienen transportadores específicos en la membrana interna de la mitocondria pueden
acceder a la matriz mitocondrial, en donde se cataliza la reacción inversa utilizando como co-factor
el FAD+ (Figura 9), sin embargo esta lanzadera ocasiona una pérdida de energía al utilizar FAD+
como aceptor de electrones en lugar de NAD+ al interior de la mitocondria como se mostró
anteriormente.
Figura 9. Lanzadera Glicerol-3-Fosfato.
Tomado de Gallardo, M. 2011 [25].

31
b) Lanzadera Malato-Aspartato
En este sistema de lanzadera se aprovecha el NAD+ generado en la hidrogenación del oxalacetato
(proveniente de la desaminación del aspartato) para obtener malato en el citosol, debido a que el
malato y el aspartato tienen transportadores específicos en la membrana interna de la mitocondria
pueden acceder a la matriz mitocondrial, en donde se cataliza la reacción inversa utilizando NAD+
como coenzima aceptora de los electrones provenientes del citosol (Figura 10).
Figura 10. Lanzadera Malato-Aspartato.
Tomado de Gallardo, M. 2011 [25].
Este proceso inicia cuando el oxalacetato es reducido a malato por el NADH + H+ proveniente de
la reacción citosólica que genera 1,3-Bifosfoglicerato a partir de gliceraldehido-3-fosfáto (Figura
10, paso 1). La membrana interna mitocondrial posee el transportador malato-α-cetoglutarato por
el cual ingresa el malato a la matriz (Figura 10, paso 2), en donde es oxidado a oxalacetato por
NAD+ (Figura 10, paso 3), a continuación el oxalacetato reacciona con el glutamato mediante una
reacción de transaminación y produce el ácido aspártico y α-cetoglutarato (Figura 10, paso 4), el
ácido aspártico atraviesa la membrana interna por el transportador glutamato-aspartato (Figura
10, paso 5) para salir al espacio intermembranal en donde reacciona con el α-cetoglutarato
mediante una transaminación que genera glutamato y oxalacetato (Figura 10, paso 6), permitiendo
captar nuevamente electrones provenientes del proceso glicolítico.
5.2. Parasitismo
El parasitismo es una asociación biológica en donde un ser vivo (denominado parásito) se aloja en
otro de diferente especie (denominado hospedero o huésped) del cual obtiene beneficios como su
óptimo desarrollo biológico. Esta asociación causa daño al hospedero y se dice que si el parásito

32
causa el menor daño posible este se encuentra adaptado al ambiente de su hospedero; por el
contrario los parásitos menos adaptados son aquellos que pueden llegar a causar la muerte de su
hospedero [26].
5.2.1. Clasificación de los parásitos
Los parásitos se clasifican en categorías de acuerdo a los siguientes aspectos, teniendo en cuenta
que una misma especie parasitaria puede pertenecer a varias de ellas [27]:
1. De acuerdo a su naturaleza:
a) Zooparásitos: parásitos pertenecientes al reino animal o que invaden alguna de sus
especies.
b) Fitoparásitos: parásitos pertenecientes al reino vegetal o que invaden alguna de sus
especies.
2. De acuerdo a su localización:
a) Edoparásitos: parásitos que se encuentran en el interior del organismo hospedero, ya
sea en cavidades orgánicas (celozoicos o cavitarios), en la vísceras, bien sea en el lumen
(intestinales), o en el seno de los tejidos (histozoicos), en el ambiente intracelular o
extracelular (plasmazoicos), o en el núcleo (cariozoicos).
b) Ectoparásitos: parásitos que se encuentran en la parte externa del organismo hospedero
o en cavidades que comunican directamente con el exterior.
3. De acuerdo a su especificidad: teniendo en cuenta las especies de hospederos adultos que
pueden ser infectadas.
a) Eurixenos (eurys, ancho y xenos, huésped, extranjero): también conocidos como
polixenos ya que pueden infectar un gran número de especies debido a que poseen baja
especificidad.
b) Estenoxenos o estenoicos (stenós, estrecho): parásitos que tienen la capacidad de
infectar muy pocas especies.
c) Monoxenos o monoicos: parásitos que infectan únicamente una especie ya que poseen
una especificidad muy alta.
d) Oligoxeno (oligo, poco, escaso): parásitos que poseen una especificidad intermedia con
relación a los parásitos eurixenos y los estenoxenos.
e) Hiperparásitos: parásitos cuyos hospederos son otros parásitos.
4. De acuerdo a su dependencia al parasitismo: teniendo en cuenta la dependencia metabólica
del parásito hacia su hospedero.
a) Parásitos obligados: parásitos que tienen dependencia metabólica estricta y selectiva
hacia su hospedero, son incapaces de vivir libremente.
b) Parásitos facultativos: parásitos que viven libremente y al mismo tiempo pueden
adaptarse al parasitismo.

33
c) Accidentales u ocasionales: parásitos que presenta un parasitismo de muy corto tiempo
debido a un encuentro incidental con el hospedero.
d) Hemiparásitos: parásitos que solamente adquieren de su hospedero parte de los
nutrientes que necesitan.
5. De acuerdo a la duración de la estancia en el hospedero:
a) Temporales: parásitos que establecen contacto con su hospedero el tiempo necesario
para alimentarse (muy poco tiempo).
b) Estacionarios: parásitos que establecen contacto con su hospedero por un tiempo
prolongado mayor al de los parásitos temporales.
6. De acuerdo a la fase parasitaria implicada: teniendo en cuenta el estadío de su ciclo de vida
en el cual son parásitos:
a) Periódicos: parásitos en los que únicamente en un estadío lleva una vida parasitaria.
b) Permanentes: parásitos en los que en todos los estadíos lleva una vida parasitaria.
5.2.2. Adaptaciones biológicas evolutivas de los parásitos
Los parásitos se encuentran en una estrecha asociación biológica con sus hospederos, lo que
ocasiona que ambos organismos co-evolucionen, de esta manera la transformación de los factores
relacionados con resistencia y susceptibilidad dependen de la actividad del parásito sobre el
hospedero de la misma manera en que la virulencia del parásito progresa. Así mismo es importante
resaltar que si los cambios conllevan un gasto energético que afecte la habilidad de dejar una
descendencia viable es poco probable que sucedan [28].
Dentro de las adaptaciones de los parásitos a su hospedero dependen principalmente de un
conjunto de aspectos, tanto positivos como negativos, propios del parasitismo [27]:
Positivos:
Ausencia o limitada competencia interespecífica por espacio, alimento, entre otros.
Abundancia de alimento y uniformidad de las condiciones físicas, lo que genera un
ahorro de energía para su búsqueda.
Facilidad para la reproducción.
Negativos:
Modificaciones especializadas de la superficie dérmica, cutícula, tegumento activo,
ya presentes como condición de preadaptación.
Desarrollo de órganos especializados para la fijación: Ganchos, ventosas, armadura
bucal, musculatura somática, entre otras.
Pérdida de órganos, ambulatorios, sensoriales, digestivos, entre otros.
Aumento del potencial biótico: gran producción de huevos.

34
Todos los cambios evolutivos que sean ocasionados por el parasitismo conllevan a reducir el
impacto negativo que tenga el parásito hacia su hospedero, ya que si este muere, el parásito por
consiguiente morirá. Sin embargo, se observa que para que los parásitos logren tener una
descendencia viable tienden a convertirse en más virulentos, por consiguiente llegar a causa la
muerte de su hospedero, a pesar de ello se presenta una co-evolución en la que intenta adquirir
más recursos de su hospedero para producir su descendencia mientras que el hospedero desarrolla
mecanismos para reducir el impacto del parásito y eliminarlo [28].
El hospedero por su parte, al poseer un ciclo de vida más extenso, a comparación de los parásitos,
posee un sistema inmune que es capaz de identificar una gran cantidad de materiales extraños para
el organismo, como es el caso de diversos tipos de microorganismos, por lo tanto, el hospedero no
es un ambiente constante para el parásito.
A largo plazo, la reproducción sexual en los hospederos es un factor que contribuye a la
combinación de alelos de resistencia frente a las infecciones parasitarias, por lo tanto permite una
mayor diversidad que la herencia en la reproducción asexual (mecanismo reproductivo en la
mayoría de los parásitos), convirtiéndose de esta manera las infecciones parasitarias en un factor
de la evolución hacia la reproducción sexual y su mantenimiento a lo largo de los años [28].
5.2.3. Hospedero y vector
El hospedero es el organismo que recibe al parásito y existen diferentes tipos de hospederos: el
definitivo es aquel en el que reside el estado adulto o sexual del parásito, el intermediario es aquel
en el que se encuentran las formas larvarias en desarrollo o en el cual el parásito se reproduce de
manera asexual y por último el paraténico o transportador en el cual el parásito se encuentra en
su forma larvaria que no se desarrolla [26].
Adicionalmente se considera vectores a los artrópodos (principalmente) o cualquier otro animal
invertebrado que transmite un parásito al hospedero ya sea de manera mecánica (el parásito no
sufre ningún tipo de modificación al interior del vector ni se reproduce) o biológica, en la cual se
presentan diferentes modalidades [27]:
Ciclomultiplicadores: El parásito evoluciona en sus estados y reproduce o multiplica.
Cicloevolutivos: El parásito evoluciona hasta su estado infectivo pero no se reproduce o
multiplica.
Multiplicador: El parásito únicamente se reproduce o multiplica.
5.2.4. Reproducción
Los parásitos, principalmente los protozoarios se multiplican por reproducción asexual y un
pequeño porcentaje por reproducción sexual (Figura 11) [26]:
1. Reproducción asexual: se puede presentar en tres modalidades:
a) División binaria: consiste en la división longitudinal o trasversal de las formas
vegetativas, dando como resultado dos individuos idénticos al inicial.

35
b) División múltiple: a partir de un progenitor se obtienen varias formas vegetativas.
c) Endodiogenia: formación de dos células hijas al interior de la célula madre.
2. Reproducción sexual: se puede presentar en dos modalidades:
a) Reproducción esporogónica: los trozofoitos sufren una serie de diferenciaciones
morfológicas, transformándose en gametocitos, los cuales al madurar sexualmente se
transforman en los gametos que al unirse forman el zigoto y por último dan origen a
nuevos individuos.
b) Conjugación: consiste en la unión de dos células entre las cuales se forma un puente
citoplasmático por donde intercambian material genético, a continuación se separa y
dan origen a nuevos individuos por división binaria.
Figura 11. Reproducción en protozoos.
Modalidades de 1. Reproducción asexual: a. División binaria, b. División múltiple, c.
Endodiagenia y 2. Reproducción sexual: a. Esporogónica y b. Conjugación. Tomado de D. Botero
y M. Restrepo 2012 [26].
5.3. Leishmania braziliensis
Leishmania braziliensis es un organismo cuya clasificación taxonómica lo ubica entre los
protozoos, en el orden cinetoplastida y en la familia de los Tripaposomátidos (Tabla 2). Estas
categorías tienen características bien definidas que contribuyen a diferenciar este parásito de otros
organismos, de esta manera en los párrafos siguientes se presentará una breve descripción de cada
una de ellas.
Tabla 2. Clasificación taxonómica de Leishmania braziliensis.
Reino Eucariota
Subreino Protozoo
Filo Sarcomastigophora
Subfilo Mastigophora
Clase Zoomastigophorea
Orden Kinetoplastida
Familia Trypanosomatidae
Género Leishmania
Subgénero Leishmania
Especie L. braziliensis
Tomado de Botero, D; Restrepo, M. 2012 [26].

36
En primer lugar, los protozoos son organismos unicelulares que presentan asociaciones biológicas
de mutualismo, comensalismo y parasitismo. Constituyen un grupo de estudio de gran importancia
debido a que generan un gran número de enfermedades en animales y en el hombre. El estadío
adulto que presentan los protozoos se denomina trofozoito, el cual se convierte en quiste para
protegerse rodeándose de una capa de grasas; algunos protozoos adoptan la forma de espora para
resistir cambios en el ambiente que los rodea y para dispersarse justo antes de la reproducción
sexual. Los protozoos poseen las siguientes características [29]:
1. Membrana: todos los protozoos poseen membrana celular la cual varía su grosor
dependiendo de la especie, confiriéndole diversas propiedades entre ellas, la más
importante, el cambio de forma.
2. Citoplasma: en los protozoos se puede distinguir dos regiones del citoplasma, el ectoplasma
periférico y el endoplasma central, los cuales varían su conformación de acuerdo a la
especie. En él se encuentran organelos como mitocondrias, vacuolas digestivas y
contráctiles, órganos de locomoción y se desprenden los flagelos y cilios en los organismos
más complejos.
3. Núcleo: todos los protozoos poseen uno o más núcleos definidos. De acuerdo a su
morfología se pueden distinguir los núcleos vesiculares (membrana nuclear visible y la
cromatina se encuentra en cúmulos) propio de las amebas y los flagelados y los núcleos
compactos (membrana nuclear no visible y la cromatina se encuentra dispersa) propio de
los ciliados.
A este grupo pertenece el subfilo mastigophora o los mastigóforos que reúne a los organismos que
en su fase adulta (trofozoito) poseen uno o más flagelos, por lo cual se les suele denominar
flagelados. Es el subfilo más grande de los protozoos y la mayoría de las especies son de vía libre,
sin embargo algunos de ellos son parásitos de vertebrados e invertebrados. Los aspectos más
importantes de su morfología son [29]:
1. Flagelo: desde el punto de vista funcional pueden ser de dos tipos, los tractelos, situados
en la parte posterior de la célula de tal manera que la célula se desplaza en la dirección del
flagelo y los pulselos, situados en el centro o en el extremo superior de tal modo que
empujan a la célula. La mayoría de las especies presentan flagelos tractelos. Cada flagelo
termina en un granulo basal (cinetosoma) que en algunas especies se encuentra próximo a
una estructura denominada cinetoplasto.
2. Axostilo: corresponde a una estructura similar al citoesqueleto situada en la dirección del
eje longitudinal del parásito.
3. Membrana ondulante: algunas especies poseen una membrana ondulante unida al flagelo
y a la membrana celular.
Por su parte, el orden cinetoplástida o los cinetoplastidos se encuentran incluidos en este subfilo y
son organismos que se diferencian de los demás mastigóforos debido a que presentan una
estructura intracelular única denominada cinetoplasto la cual se encuentra al interior de una
mitocondria redondeada, el cual es una estructura altamente compleja compuesta de círculos de

37
ADN de diferentes tamaños concatenados en cadenas entrelazadas (ADNk) que componen entre
el 10 y el 20% del contenido total de ADN del organismo.
Del ADN del cinetoplasto se pueden identificar dos estructura principalmente: los maxicirculos y
los minicirculos; los maxicirculos son el equivalente al ADN mitocondrial de los eucariontes más
evolucionados, de él se codifican ARN ribosómico (ARNr), ARN transferente (ARNt), y ARN
codificador de proteínas; los minicirculos son la mayor proporción del ADN del organismo y
modifican el ARNm mitocondrial. Esta estructura se ubica debajo del flagelo en su estadío de
trofozoito [30, 31]. [30] [31]
Adicionalmente se evidencia la presencia de un organelo único denominado glicosoma, el cual
posee enzimas glicolíticas, por lo cual se identifica como el organelo en donde se lleva a cabo el
metabolismo de la glucosa. Al orden cinetoplastida pertenecen numerosos géneros, entre los que
tienen mayor importancia a nivel médico y veterinario son el género Leishmania, el Trypanosoma
y el Phytomona (Tabla 3) [28].
Los géneros Leishmania y Trypanosoma se encuentran clasificados en la familia de los
Tripanosomátidos, de la cual todos sus miembros son parásitos, poseen un único flagelo tractelo
que tiene su terminación en el gránulo basal, próximo al cinetoplasto. El flagelo puede formar
parte de una membrana ondulante, estar libre o encontrarse reducido a su trayecto intracelular. Su
ciclo de vida es polimórfico, lo que quiere decir que presenta transformaciones morfológicas a lo
largo de este. Adicionalmente, todos los miembros de la familia sin importar su hospedero final
tienen una o más fases en el intestino de un artrópodo (generalmente un insecto) [29].
Tabla 3. Especies de Cinetoplastidos de mayor importancia a nivel médico y veterinario.
GÉNERO EJEMPLO HOSPEDERO VECTOR ENFERMEDAD
Leishmania
L. donovani Humano, perro y rata Phlebotomus
sandflies
Leishmaniasis
intestinal
L. major Humano, mono,
perro y roedores
Phlebotomus
sandflies
Leishmaniasis
cutánea
L. tropica Humano, mono,
perro y roedores
Phlebotomus
sandflies
Leishmaniasis
cutánea
L. braziliensis Humano, mono,
zarigüeyas, etc. Lutzomyia sandflies
Leishmaniasis
cutánea
Trypanosoma
T. brucei
gambesiense Humano
Tsetse flies (Glossina
spp.)
Tripanosomiasis
africana
(Enfermedad del
sueño)
T. congolense Ganado Tsetse flies (Glossina
spp.)
Tripanosomiasis
africana (Nagana)
T. equiperdum Caballo Sexual Durina
T. cruzi Humano, perro, gato,
rata, etc. Triatominos
Tripanosomiasis
americana
Phytomona P. staheli Palma de coco Lincus lobuliger Hartroot
Tomado de Gunn A. y Pitt, S. 2012 [28].

38
Específicamente, todos los miembros del género Leishmania presentan un hospedero final
vertebrado (frecuentemente un mamífero) y la mayoría tienen vectores invertebrados, ocasionando
diversas enfermedades conocidas como Leishmaniasis. En ambos hospederos se lleva a cabo la
reproducción del parásito, siendo así un parásito heteroxeno y se pueden identificar dos subgéneros
principalmente [28]:
1. Subgénero Leishmania: parásitos que se desarrollan en el intestino medio del vector y a
continuación se movilizan hacia su faringe en donde se transmiten por una picadura.
2. Sección Viannia: parásitos que se desarrollan en el intestino posterior del vector y a
continuación se movilizan hacia su faringe.
Se pueden distinguir dos grupos dentro de este género, en primer lugar los que invaden órganos
conocidos como viscerales y los que afectan la superficie del cuerpo conocidos como cutáneos.
Actualmente se conocen más de 30 especies de parásitos pertenecientes a este género, de los cuales
10 representan gran importancia médica y veterinaria.
Los tripanosomátidos presentan diversos estadíos que se diferencian por la posición del
cinetoplasto en relación al núcleo y la presencia de una membrana ondulante como ocurre en todos
los miembros de la familia. En el caso específico de la especie L. braziliensis se pueden identificar
dos estadíos bien diferenciados morfológicamente (Figura 12) [28]:
1. Amastigote: es un estadío intracelular multiplicativo, tiene forma ovoide o fusiforme y
achatado; mide entre 3 y 4 μm de largo. Se caracterizan porque presentan un núcleo de
gran tamaño, redondo y excéntrico y el cinetoplasto es muy notorio y no posee flagelo.
2. Promastigote: es un estadío extracelular multiplicativo, tiene forma alargada, mide hasta
20 μm. El cinetoplasto se localiza cerca del núcleo en la parte anterior en donde el flagelo
emerge libre sin membrana ondulante.
Figura 12. Estadíos de Leishmania braziliensis.
Tomado de Walsh, M. 2011 [32].
L. braziliensis posee un ciclo de vida heterxeno en el cual precisa de un hospedero intermediario
en donde se desarrolla uno de sus estadíos, los vectores de este parásito son los insectos
(artrópodos) hematófagos pertenecientes al género Lutzomyia, orden Diptera, principalmente las
especies Lutzomyia sandflies, Lutzomyia spinicrassa, Lutzomyia gomezi, Lutzomyia ovallesi,
Lutzomyia longiflocosa, Lutzomiyia youngi, Lutzomyia scorzai, Lutzomyia lichyi y Lutzomyia
columbiana [33]. En su estadío infectivo es capaz de invadir las células que constituyen el sistema

39
retículo-endotelial de la piel y mucosas y los leucocitos, ocasionando la enfermedad conocida
como la Leishmaniasis cutánea [29].
5.4. Ciclo de vida Leishmania braziliensis
La infección es transmitida por los dipteros (hembras), el ciclo comienza cuando un insecto que
anteriormente se alimentó de sangre infectada con el parásito Leishmania braziliensis se alimenta
de un nuevo individuo y regurgita al mismo tiempo (Figura 13, paso 1), de esta manera, el estadío
promastigote (etapa infectiva) presente en los restos infectados penetra las capas de la piel por las
porosidades o anteriores picaduras e ingresa al torrente sanguíneo, a continuación el parásito es
fagocitado por los leucocitos del hospedero (Figura 13, paso 2), una vez en el interior de la célula,
el parásito se transforma a su estadío de amastigote (Figura 13, paso 3) para posteriormente
reproducirse asexualmente mediante fisión binaria (Figura 13, paso 4); cuando se alcanza un
número suficiente de individuos al interior de la célula esta se lisa y los parásitos son liberados al
medio extracelular (Figura 13, paso 5).
De esta manera el parásito nuevamente se encuentra en el torrente sanguíneo en donde puede
infectar nuevas células al interior del hospedero o puede ser ingerido por otro insecto al alimentarse
mediante una picadura (Figura 13, paso 6), así, el parásito ingresa al intestino medio anterior del
insecto en donde se moviliza al intestino medio posterior y se transforma a su estadío promastigote
(Figura 13, paso 7), una vez en este estadío se reproduce asexualmente por fisión binaria
longitudinal (Figura 13, paso 8) y se moviliza hacia el intestino medio anterior (Figura 13, paso
9) para nuevamente dar inicio al ciclo.
Figura 13. Ciclo de vida de Leishmania braziliensis.
Se muestran los estadíos presentes en invertebrados (izquierda) y en mamíferos (derecha) siendo
la picadura y la regurgitación del invertebrado los causantes de la transmisión. Elaborado por el
autor.

40
L. braziliensis en su ciclo de vida tiene como vectores los miembros del orden Diptera (dípteros),
el cual es uno de los más grandes entre los insectos, ya que contiene más de 120.000 especies
destacándose su variedad morfológica e importancia en los ecosistemas, las cuales son fitófagas,
detritívoras de materia orgánica de origen vegetal y animal, polinizadoras, predadoras, parasitoides
o hematófagas.
Constituyen uno de los grupos predominantes dentro de los insectos del estiércol y la carroña, ya
que explotan rápidamente estos recursos, encontrándose especies coprófagas, necrófagas,
predadoras y también omnívoras. Los excrementos y cadáveres son utilizados por las especies
coprófagas y necrófagas (respectivamente) como fuente de proteínas para la maduración de sus
huevos y/o como alimento para el desarrollo de las larvas [34].
Se ha identificado que las especies del género Lutzomyia perteneciente a los dípteros (Clasificación
taxonómica Tabla 4), son los vectores de L. braziliensis más frecuentes dentro de este orden. Los
miembros de este género son insectos pequeños con muy poca capacidad de vuelo y con el cuerpo
y alas cubiertas de pelos y un período de vida de 40 a 50 días, todos hematófagos. La mayoría de
las hembras ingieren sangre de 1 a 4 días luego de emerger de la pupa. Sus huevos son oscuros,
elípticos y su superficie surcos y otras protuberancias que forman patrones típicos de la especie o
del complejo de especies. La cantidad de huevos varía de 40 a 70 según la especie, tamaño y
calidad del alimento [35].
Tabla 4. Clasificación taxonómica del género Lutzomyia.
Reino Animalia
Filo Artrophoda
Clase Insecta
Orden Diptera
Familia Psychodidae
Subfamilía Phlebotominae
Género Lutzomyia
Tomado de Sierra, D; Vélez, I; Uribe, S. 2000 [35]
5.5. Leishmaniasis cutánea
Los humanos pueden ser portadores de algunas especies de Leishmania sin presentar síntomas
durante largos períodos de tiempo y sin enfermarse, variando el período de incubación de 1 a 2
semanas hasta de varios meses cuando es causada por las especies del Nuevo Mundo y de hasta 3
años en el caso de las especies del Viejo Mundo. Es importante resaltar que la forma de la
enfermedad y los signos clínicos varían con las especies de Leishmania, llegando inclusive a
presentarse infecciones asintomáticas [36].
La Leishmaniasis cutánea afecta generalmente solo a la piel y el número de lesiones puede variar
según la especie involucrada así como la gravedad de estas que van desde úlceras, nódulos lisos
hasta lesiones hiperqueratosicas. El parásito suele invadir una región delimitada de la piel, sin

41
embargo en ocasiones, se puede propagar a diferentes partes, incluyendo las mucosas.
Frecuentemente es una afección indolora y la infección no afecta los tejidos subcutáneos, aunque
las cicatrices tienen un tiempo de curación desde varios meses a un año. Se reconocen dos
variedades [37]:
1. La variedad del Viejo Mundo está causada por las especies L. trópica, L. major, L. infantum
y L. aethiopica y es endémica en la zona mediterránea, Asia occidental, las regiones
occidentales del subcontinente indio y África oriental y occidental.
2. La variedad del Nuevo Mundo está causada por L. amazonensis, L. mexicana, L. peruviana,
L. guyanensis, L. panamensis y L. braziliensis y es endémica en América Central y del Sur.
A medida que se desarrolla la inmunidad sobreviene la curación por fibrosis, la cual deja una
cicatriz prominente, teniendo en cuenta que la variedad del Nuevo Mundo tiende a ser más grave
y tarda más en curar.
5.5.1. Tratamiento leishmanicida
El diagnóstico y tratamiento oportuno en la fase aguda de la infección permite curar o mejorar la
calidad de vida de los pacientes de la Leishmanisis cutánea. A continuación se muestran los
medicamentos disponibles con los que se lleva a cabo el tratamiento (Tabla 5) [38]:
1. Sales de Antimonio pentavalente (Sb+5): en este grupo de medicamentos se encuentra por
un lado el antimoniato de meglumina (glucantime®) cuyo principio activo son los
complejos de antimoniato de N-metil glucamina y por el otro el estibogluconato de sodio
(pentostam®). Ambas sales tienen efecto cuando el ion Sb+5 es reducido a la forma Sb+3
que es tóxica para los amastigotes ya que se une inespecíficamente a los grupos sulfhidrilos
de las proteínas del parásito.
2. Anfotericina B: es un macrólido heptaénico, rígido y cilíndrico. Tienen su efecto debido a
que presentan gran afinidad por el ergosterol de la membrana celular del parásito
interfiriendo en su dinámica de intercambio de agua e iones pequeños llegando a ocasionar
daño oxidativo a la célula.
3. Miltefosina: es un análogo de la alquilfosfocolina que actúa alterando el metabolismo de
éster-lípidos, el envío de señales celulares y la síntesis del mecanismo de anclaje
glucosilfosfatidilinositol.
4. Paramomicina o aminosidina: es un aminoglucósido que consta de tres amino azúcares
unidos por enlaces glucosídicos a un núcleo de 2-desoxiestreptamina. Su acción consiste
en la unión con la subunidad ribosomal 30S bloqueando la síntesis de las proteínas y
disminuyendo la fidelidad de la traducción.
5. Pentamidina: este compuesto es una diamina aromática, su acción consiste en acumularse
al interior de la mitocondria y alterar el potencial de su membrana al mismo tiempo que
ocasiona la desintegración del cinetoplasto.
6. Ketoconazol: es un imidazol antifúngico. Su mecanismo de acción consiste en inhibir la
enzima 14-α-demetilasa por lo cual altera la biosíntesis de esteroles de membrana llevando
a la acumulación de 14-α-metilesteroles.
7. Fluconazol: es un bistriazol fluorado que tiene el mecanismo de acción del ketoconazol.
8. Lidocaína: es una aminoetilamida de la cual no se conoce su mecanismo de acción.
9. Alopurinol: es un análogo de la hipoxantina, su mecanismo de acción está relacionado con
el ADN del parásito.

42
Tabla 5. Estructuras desarrolladas de cada uno de los medicamentos leishmanicidas disponibles.
NOMBRE ESTRUCTURA
Estibogluconato
de sodio
Anfotericina B
Miltefosina
Paramomicina
o aminosidina
Petamidina
Ketoconazol
Fluconazol
Lidocaína
Alopurinol
Tomado de Vasquez, L. 2009 [38].

43
Los diferentes medicamentos que son utilizados para el tratamiento de la Leishmaniasis cutánea
presentan algunos efectos adversos como se indica a continuación:
1. Sales de Antimonio pentavalente (Sb+5): vomito, nauseas, mialgias, artralgias, y cefalea.
Se presentan efectos tóxicos sobre riñón, hígado, corazón y páncreas. Su punto máximo de
presentación ocurre entre los 7 y 14 días.
2. Anfotericina B: Fiebre, anorexia, nauseas, vómitos, astenia, adinamia, flebitis,
hipocalemia, reacciones anafilácticas y alteraciones cardiacas.
3. Miltefosina: efectos adversos gastrointestinales leves como nauseas, vómito y diarrea,
elevación de transaminasas o creatinina.
4. Pentamidina: efectos leves como dolor y edemas en el sitio de aplicación, abscesos, mareo,
fiebre, cefalea, adinamia, náuseas y dolor articular, adicionalmente efectos
cardiovasculares similares a los producidos por las sales antimoniales pentavalentes.
5.6. Proteínas transportadoras de nucleótidos
Las proteínas, al igual que los lípidos, constituyen un porcentaje importante de la membrana
celular, asociadas a esta por medio de interacciones no covalentes. Dependiendo de su ubicación
se distinguen [39]:
a) Proteínas integrales o intrínsecas: aquellas en las que una porción de su cadena se encuentra
insertada en la bicapa (Figura 14), se evidencia que los segmentos insertados o dominios
transmembranales presentan mayor cantidad de aminoácidos hidrofóbicos que el resto de
la secuencia y principalmente se encuentran configurados en forma de hélice alfa, sin
embargo existen algunas que forman canales constituidos por múltiples láminas beta
dispuestas en forma de barril.
b) Proteínas periféricas o extrínsecas: aquellas que se encuentran ubicadas sobre cualquiera
de las dos superficies de la membrana (Figura 14), poseen una mayor cantidad de
aminoácidos hidrofílicos que los dominios transmembranales de las proteínas integrales.
Adicionalmente tienen la capacidad de desplazarse lateralmente o rotar sobre su eje.
Figura 14. Proteínas de membrana.
Proteínas integrales (izquierda) y proteínas periféricas (derecha). Tomado de Blanco, A. [39]

44
Las proteínas de membrana permiten que las células funcionen como miembros de un tejido, entre
ellas se encuentran [40]:
a) Proteínas de adhesión: se encuentran a lo largo de la superficie de la célula y permiten el
contacto entre ellas.
b) Proteínas de transporte: regulan la entrada y salida de nutrientes.
c) Receptores: permiten que las células respondan a hormonas u otras moléculas
señalizadoras.
d) Proteínas citoesqueléticas: corresponden a proteínas periféricas que se adhieren a la
membrana y regulan su forma y estabilizan su estructura.
5.6.1. Tipos de transportadores de membrana
El transporte por parte de las proteínas de membrana puede ser pasivo, siempre que sea a favor de
un gradiente de concentración electroquímico, o activo, cuando se da en contra de uno, lo que hace
necesario un gasto energético proveniente de diversas fuentes. Los principales tipos de
transportadores son los siguientes (Figura 16) [41]:
Poros y canales: no requieren un cambio conformacional de la proteína para que se efectúe
el transporte por difusión facilitada, estas crean un conducto hidrofílico que atraviesa la
membrana, el cual puede ser atravesado por solutos polares o iónicos.
Transportadores dependientes del gradiente electroquímico: llevan a cabo procesos de
transporte empleando como fuente de energía el potencial electroquímico de al menos uno
de los solutos transportados y mediante un cambio conformacional de la proteína, tras la
formación de un complejo reversible transportador-soluto. Puede ocurrir de dos maneras
desde un punto de vista energético:
a) Uniporte: se transporta un único soluto a favor de su potencial electroquímico (difusión
facilitada).
b) Cotransporte: transporte acoplado de solutos o transporte activo secundario. Al menos
uno de los solutos se moviliza a favor de su potencial electroquímico, lo que suministra
la energía necesaria para acarrear al segundo soluto en contra de su potencial
electroquímico. Si se transportan ambos solutos en el mismo sentido se trata de
Sinporte; si se transportan en sentidos opuestos, Antiporte (Figura 15).
Figura 15. Tipos de transportadores dependientes del gradiente electroquímico.
Tomado de Sancho López, P. 2009 [42].

45
Transportadores activos (bombas): usan una fuente de energía externa independiente del
potencial electroquímico para transportar solutos en contra este. Se forma un complejo
bomba-soluto, análogo a los complejos proteína –ligando, por lo que este tipo de transporte
presenta saturación. Se puede encontrar bombas que utilizan la energía de la hidrólisis de
un enlace pirofosfato (del ATP, de otro nucleosido trifosfato o del pirofosfato inorgánico),
las que utilizan la suministrada por una reacción redox y las que utilizan la suministrada
directamente por la luz.
Figura 16. Tipos de transportadores.
Tomado de BioROM 2011 [41].
El transporte de nucleótidos se realiza mediante las proteínas que se encuentran en la familia de
transportadores de solutos, se conocen alrededor de 300 miembros organizados en 52 familias. Son
proteínas integrales que poseen dominios transmembranales principalmente en estructuras tipo
hélice α con residuos hidrofóbicos en su estructura. La mayoría de los miembros de esta familia se
encuentran localizados en la membrana celular, algunos en la membrana de la mitocondria (familia
25, Tabla 6) y algunos en la membrana de otros organelos.
Tabla 6. Miembros de la familia de transportadores de solutos relacionados con el
transporte de nucleótidos.
FAMILIA MIEMBROS
CÓDIGO NOMBRE
25. Transportadores
mitocondriales
SLC25A3 Transportador de fosfato
SLC25A4 Translocasa de ADP/ATP 1
SLC25A5 Translocador de adenina nucleótido
SLC25A6 Translocasa de ADP/ATP 3
SLC25A31 Translocasa de ADP/ATP 4
29. Transportadores de
nucleósidos
SLC29A1 Transportador equilibrado de nucleósidos 1
SLC29A2 Transportador equilibrado de nucleósidos 2

46
Los transportadores de nucleótidos se han definido en diversos organismos, principalmente las
translocasas de ADP/ATP que se encuentran relacionadas con la síntesis del ATP. En Clamidia
amoebophila se han identificado 5 isoenzimas encargadas de este proceso, ya que esta bacteria ha
perdido la capacidad de sintetizar estos compuestos de novo, adicionalmente se ha propuesto un
modelo en el que se muestran las relaciones que existen entre estos 5 transportadores (Figura 17).
Es importante resaltar que no solamente se han identificado transportadores de mononucleótidos,
sino que también un transportador del dinucleótido de adenina nicotinamida (NAD+) en esta
misma especie que se observó lleva a cabo un transporte de tipo antiporte entre el NAD+/ADP. En
Clamidia trachomatis se identificó que su translocasa ATP/ADP presenta una mayor afinidad por
el NAD+ que por el ATP, proponiéndose como una posible proteína transportadora de este
dinucleótido [13].
Figura 17. Modelo de interacción entre los transportadores de nucleótidos identificados en
Clamidia trachomatis.
Convención numérica para los transportadores: 1. CtNTT1, 2. CtNTT2, 3. CtNTT3, 4. CtNTT4.
Tomado de Haferkamp, I. et. al. 2006 [11].
La familia de transportadores de nucleótidos puede ser dividida en tres clases de acuerdo al modo
en el que realizan el transporte en:
a. Clase I: Catalizan el transporte de nucleótidos de modo “counter exchange”. En esta
categoría se encuentran los homólogos de NTT1 de P. amoebophila.
b. Clase II: Catalizan la importación de nucleótidos unidireccional impulsada por un gradiente
de protones.
c. Clase III: Catalizan el transporte modo “counter exchange” de NAD+/ATP.

47
Se ha identificado que algunos residuos en las regiones transmembranales son importantes para el
transporte de los nucleótidos y que, adicionalmente son muy conservados entre diversas secuencias
de proteínas con estas funciones. En la translocasa de ADP/ATP presente en Arabidopsis thaliana
los residuos cargados K155, E245, E385 y K527 se encuentran relacionados con el transporte de
nucleótidos de adenina, cuando estos son delecionados o intercambiados por residuos sin carga, el
transporte de este nucleótido disminuye considerablemente e inclusive el proceso se ve
interrumpido, teniendo en cuenta que la orientación y expresión de los mutantes con relación a las
proteínas nativas no se ven afectadas [43].
Investigaciones realizadas han demostrado que el residuo de lisina K446 de PamNTT1 (K527 en
NTT1de A. thaliana) tiene una gran importancia en el proceso de transporte de nucleósidos
trifosfato pero no en el de nucleósidos difosfato, también se encontró que PamNTT4 no posee el
residuo en esta posición y como se demostró experimentalmente no tiene la capacidad de
transportar ATP pero si ADP. Por otra parte el estudio halló que los demás transportadores
evaluados (PamNTT1, PamNTT2, PamNTT3 y PamNTT5) en P. amoebophila si poseen este
residuo de lisina y por consiguiente tienen alta afinidad por los nucleósidfos trifosfato (Figura 17).
Estos resultados evidencian que la presencia de ciertos residuos en determinadas posiciones provee
mayor información para la función del NTT y de la especificidad de sustrato en comparación con
la que brinda la homología global de la secuencia primaria entre los candidatos [11].
5.7. Técnicas de biología molecular
Entre las técnicas de biología molecular que se utilizarán se encuentra la extracción de ADN
genómico de los parásitos mediante tratamiento con bromuro de hexadecil-trimetil-amino,
amplificación por PCR, electroforesis horizontal en gel de agarosa, ligación entre inserto y el
vector pGEM-T, transformación y clonación en bacterias de Escherichia coli e identificación de
colonias recombinantes por PCR de colonia. A continuación se muestran algunas de estas técnicas:
5.7.1. Amplificación por PCR
La técnica PCR o de reacción en cadena de la polimerasa es una tecnología utilizada para
multiplicar (sintetizar) in vitro fragmentos específicos de ADN con la finalidad de detectar una
secuencia o gen de interés en el genoma del individuo estudiado. Se basa en la amplificación de
fragmentos de ADN a partir de secuencias de nucleótidos denominadas “cebador” (primer), que
son capaces de reconocer una secuencia blanco para la cual es complementaria, obteniéndose así
un aumento exponencial en el número de copias del gen que se sometió a este proceso [44].
En forma general, los principales pasos del PCR son los siguientes: se extrae el ADN del material
a analizar, se separa la molécula del ADN en dos hebras (desnaturalización), se induce el
alineamiento o reconocimiento del cebador con las secuencias blanco complementarias o molde
del ADN, se adicionan los dNTP (dATP, dCTP, dGTP, dTTP) y por medio de la enzima Taq
polimerasa, la cual reconoce el extremo 3´ del cebador y lleva a cabo su extensión o alargamiento
en sentido 5´ 3´ copiando la información de la cadena molde. Este proceso se realiza en un
termociclador, el cual se encarga de realizar los cambios de temperatura necesarios para que se
desarrollen las etapas o ciclos anteriormente mencionados (denaturación, unión del cebador y
amplificación). Los ciclos se repiten la cantidad de veces que sea necesario, hasta obtener en forma
exponencial un gran número de copias de ADN (Figura 18) [45].

48
Figura 18. Etapas de la amplificación por PCR.
1). Denaturación a una temperatura de 98°C. 2). Alineación de primers de acuerdo a su Tm. 3).
Extensión del primer por la Taq polimerasa, obteniéndose así una 4). Amplificación exponencial
por la repetición de cada ciclo térmico. Esquina superior izquierda termociclador. Tomado de
New Englands Biolabs inc. 2015 [46]
5.7.2. Clonación en Escherichia coli in vivo
La clonación consiste en insertar ADN amplificado mediante PCR, un gen, un fragmento de un
gen, entre otros, denominado inserto en un plásmido apropiado, denominado vector previamente
linearizado (digestión o restricción), mediante un proceso denominado ligación, Para que esta
construcción (ADN recombinante) se mantenga y amplifique in vivo es necesario que sea
introducida en un hospedador apropiado (transformación), este proceso puede ser llevado a cabo
por métodos físicos y químicos entre los que se encuentran el choque térmico o la electrofusión,
con los que se altera la permeabilidad de la membrana celular mediante cambios bruscos de
temperatura o del potencial eléctrico del espacio extracelular respectivamente [47]. Entre los
vectores más utilizados (Tabla 7) para sobrexpresión de proteínas se encuentran pET100/D-TOPO,
pBAD/D-TOPO, pMAL-c5x, pCOLD y pET-SUMO.
Tabla 7. Vectores más utilizados en clonación.
VECTOR pET100/D-
TOPO
pBAD/D-
TOPO pMAL-c5x pCOLD pET-SUMO
TAMAÑO (pb) 5764 4400 5752 4407 5643
PROMOTOR T7 araBAD LacIq cspA - LacI T7
OPERADOR LacO Arabinosa O1
y O2 LacI Lac LacO
INDUCTOR IPTG Arabinosa IPTG IPTG IPTG
GEN DE
RESISTENCIA Ampicilina Kanamicina Ampicilina Ampicilina Kanamicina
TAG
N-TERMINAL 6xHis
His-
Tiorredoxina MBP 6xHis
Epitope HisG
Prot. SUMO
TAG
C-TERMINAL -
Epitope V5
6xHis - - -

49
Entre los sistemas de expresión se pueden encontrar bacterias, levaduras, hongos, células de
insectos o de mamíferos, siendo Escherichia coli el sistema más utilizado, ya que es muy
económico, eficiente y rápido en comparación con los demás, adicionalmente posee un medio de
cultivo muy sencillo y una alta cinética de crecimiento hasta fase estacionaria hasta llegar a la fase
logarítmica, en donde el número de células se duplica cada 20 minutos aproximadamente.
Para garantizar una adecuada transformación las cepas de Escherichia coli, estas deben hacerse
“competentes” previamente para favorecer la entrada del ADN recombinante, esto se logra
alterando la permeabilidad de la membrana celular como por ejemplo realizando lavados a baja
temperatura con sales como el cloruro de calcio.
Las cepas de E. coli más utilizadas son las Top 10 las cuales se utilizan para el mantenimiento y
propagación de plásmidos recombinantes debido a que no posee la maquinaria necesaria para la
expresión de proteínas (ARN polimerasa) a diferencia de la cepa BL21 (DE3) que contiene al fago
lambda DE3 que a su vez posee el constructo lac el cual está conformado por el gen lacI que
codifica el represor Lac, el gen de la ARN polimerasa T7 controlada por el promotor lacUV5 y
una pequeña porción del gen lacZ, la regulación de la transcripción de la polimerasa ocurre por la
unión del represor Lac con el operador lac que se encuentra corriente abajo del promotor T7 [48].
La cepa BL21 (DE3) adicionalmente posee la mutación rne131 que codifica una RNasa E truncada
sin su extremo C-terminal (477aa) que está relacionado con la degradación del ARNm, mientras
que el extremo N-terminal codificado (584aa) se encarga de la maduración del ARNr y contribuye
con el crecimiento celular, lo cual contribuye a que el ARNm codificado en esta sepa posea mayor
estabilidad en comparación con otras [48].
A continuación se lleva a cabo la selección de aquellas células recombinantes con base a la
resistencia a algún(os) antibiótico(s) debida al vector introducido, de esta manera aquellas células
en las que el proceso de transformación del plásmido haya sido satisfactorio sobrevivirán en un
medio de cultivo con el antibiótico correspondiente. Posteriormente se debe realizar un rastreo de
las colonias mediante PCR para identificar la presencia del inserto y finalmente extraer el ADN
plasmídico y obtener la proteína recombinante correspondiente en grandes cantidades.
Figura 19. Secuencias de los vectores pET100/D-TOPO y pBAD202/D-TOPO.
Tomado de Thermofisher 2015 [48]

50
El plásmido pET100/D-TOPO (Figura 19) contiene el gen regulador de la expresión T7 con el
cual se obtiene una eficiencia mayor al 90% al mismo tiempo la clonación en este vector adiciona
una secuencia de aminoácidos o proteína fusión a la proteína de interés en su extremo N-terminal
con un peso de 3 kDa correspondiente a un tag de 6 histidinas [48]. Para clonar en este vector es
necesario incluir en el extremo 5’ del primer directo la secuencia CACC que le otorga a la
clonación la direccionalidad correcta, teniendo en cuenta la posición de cada uno de los genes del
vector pET100/D-TOPO (Tabla 8). Por último se debe tener en cuenta que este vector se encuentra
linearizado por lo que no es necesario realizar un tratamiento con enzimas de restricción.
En el caso del vector pBAD202/D-TOPO (Figura 19) la expresión está regulada por el gen
araBAD en presencia o ausencia de arabinosa (inductor). Este vector adiciona una secuencia de
aminoácidos o proteína fusión a la proteína de interés en su extremo N-terminal con un peso de 13
kDa correspondiente a un tag de His-Tiorredoxina, una secuencia de tiorredoxina en la que se han
realizado las modificaciones de los residuos de glutamato E32H y glutamina Q64H, las cuales
interactúan con la histidina H8 nativa y presentan una gran afinidad por iones divalentes lo que
facilita la purificación de la proteína fusión. Asimismo adiciona en el extremo C-terminal un tag
de 6 histidinas y el epitope V5 (Gly-Lys-Pro-Ile-Pro-Asn-ProLeu-Leu-Gly-Leu-Asp-Ser-Thr) con
un peso de 3 kDa. Al igual que en pET100, en este vector se debe adicionar la secuencia CACC
en el extremo 5’ del primer directo para asegurar una direccionalidad adecuada [49]. Al igual que
pET100 este vector se encuentra linearizado por lo que no es necesario realizar un tratamiento con
enzimas de restricción.
Tabla 8. Posición de los diferentes genes que componen en vector pET100/D-TOPO.
GEN POSICIÓN
Promotor T7 209 – 225
Operador lac (lacO) 228 – 252
Sitio de unión al ribosoma (RBS) 282 – 288
Codón de inicio ATG 297 – 299
Región de polihistidina (6xHis) 309 – 326
Epitope Xperss 366 – 389
Sitio de reconocimiento de la EK 375 – 389
Sitio de clonación TOPO
(Direccionalidad) 396 – 409
Región de terminación de la
transcripción T7 427 – 555
Promotor bla 856 – 954
Resistencia a la ampicilina (bla) 955 – 1815
Origen de replicación pBR322 2022 – 2757
LacI ORF 4507 – 5595
Tomado de Thermofisher 2015 [48].
La relación molar inserto:vector óptima en ambos vectores varía entre 0.5:1 y 4:1 (Figura 20). Para
calcular la cantidad apropiada de inserto que se debe usar en la ligación se utiliza la siguiente
ecuación:

51
(𝑛𝑔 𝑉𝑒𝑐𝑡𝑜𝑟)𝑥(𝐾𝑏 𝐼𝑛𝑠𝑒𝑟𝑡𝑜)
𝐾𝑏 𝑉𝑒𝑐𝑡𝑜𝑟𝑥 𝑅 = 𝑛𝑔 𝐼𝑛𝑠𝑒𝑟𝑡𝑜
Figura 20. Relaciones óptimas Inserto:Vector para los vectores pET100/D-TOPO y
pBAD202/D-TOPO.
Tomado de Thermofisher 2015 [49].
La ligación en estos vectores es llevada a cabo mediante la enzima Topoisomerasa I del virus
Vaccinia, la cual reconoce la secuencia 5′-CCCTT y rompe el enlace fosfodiéster de una de las
hebras de ADN después de esta secuencia, posteriormente la energía de este enlace es utilizada
para formar uno nuevo entre el grupo fosfato 3’ de la cadena escindida y el residuo de tirosina
Y274 de la Topoisomerasa I (Figura 21). Los vectores pET100/D-TOPO y pBAD202/D-TOPO
poseen esta secuencia tanto en la hebra 5’ – 3’ como en la complementaria, para la hebra con
sentido en las posiciones 396 – 400 y 718 – 722 y en la antisentido entre 405 – 409 y 727 – 731
para cada vector respectivamente por consiguiente se encuentran linearizados [48] [49].
Figura 21. Mecanismo de ligación en los vectores pET100/D-TOPO y pBAD202/D-TOPO.
Inicialmente la Topoisomerasa I reconoce la secuencia 5’-CCCTT en las hebras de ADN y escinde
el enlace fosfodiéster en una de ellas formando uno nuevo entre el grupo fosfato 3’ de la cadena
escindida y el residuo Y274 de la enzima, a continuación el producto amplificado por PCR ataca
este enlace y se une al vector.
(1)

52
Entre los dos puntos de corte se encuentra la secuencia 5′-CACC y su complementaria 3’-GTGG,
por lo tanto se asegura la correcta direccionalidad de los productos de ligación adicionando la
secuencia 5’-CACC al extremo 5’ de los cebadores directos, de esta manera el grupo hidroxilo 5’
del amplificado puede atacar el enlace entre la cadena de ADN y el residuo Y274 de la enzima
revirtiendo la reacción y liberando la Topoisomerasa, a continuación los nucleótidos adicionados
al cebador se unen por complementariedad a la hebra antisentido del vector. El extremo 3’ del
amplificado se une al vector de igual manera, teniendo en cuenta que la unión se lleva a cabo entre
el extremo 5’ de la cadena antisentido del amplificado y el 3’ del vector. Estas reacciones
circularizan el vector nuevamente.
5.8. Métodos computacionales
Los métodos computacionales más utilizados para el análisis de macromoléculas en específico de
proteínas se realizan inicialmente a partir del análisis de la estructura primaria como la predicción
de propiedades fisicoquímicas, péptido señal, localización subcelular, motivos y dominios
conservados, modificaciones postraduccionales y solubilidad. Asimismo se realiza una predicción
de la estructura secundaria y la topología y por último de la estructura tridimensional y posibles
interacciones con otras moléculas. A continuación se describen algunas de las herramientas,
plataformas y servidores utilizados para estos análisis:
5.8.1. Análisis de estructura primaria
El análisis de la estructura primaria comprende la identificación de motivos y dominios
conservados. Los primeros corresponden con un segmento conservado de la secuencia de
aminoácidos de la proteína que generalmente se encuentran asociados a una función concreta, y
los segundos se relacionan con la región que posee interés biológico, funcional o estructural. Este
análisis se puede llevar a cabo mediante dos servidores, el primero de ellos es MotifFinder, el cual
evalúa la secuencia de aminoácidos y el score de coincidencia con los motivos de la base de datos
de Pfam mediante el valor-E (mientras menor es el valor, mayor coincidencia), de igual manera lo
hace con la información contenida en la Base de datos de Dominios Conservados (CDD). El
segundo servidor es InterPro, el cual clasifica las secuencias proteicas en familias y predice la
presencia de dominios y sitios importantes en ella, a partir de la información contenida en varias
bases de datos que hacen parte del consorcio InterPro como PROSITE, Pfam, PRINTS,
SUPERFAMILY, entre otras.
Para proteínas integrales de membrana se realiza la predicción de las hélices transmembranales
que posee en su estructura, para esto se utilizan servidores como TMHMM 2.0, Phobius, Philius y
SPOCTOPUS. El servidor TMHMM 2.0 se basa en un Modelo Oculto de Márkov (HMM) el cual
reconoce patrones a partir de los cuales predice un modelo probabilístico, en este caso la topología
a partir de la gramática de la secuencia de aminoácidos en donde los bucles citoplasmáticos y no
citoplasmáticos se encuentran intercalados, adicionalmente posee modelos especializados en
diferentes regiones de las proteínas de membrana como lo son los extremos de las hélices α, su
región intermedia y su longitud, las regiones próximas a la membrana y los dominios tipo bucle,
asimismo incluye el perfil de hidrofobicidad (dominio transmembranal) y la polarización debida a
residuos cargados positivamente (bucles citoplasmáticos) organizados en clases estructurales [50].

53
El servidor Phobius utiliza un algoritmo que combina el modelo de predicción de péptido señal de
la herramienta SignalIP con el de predicción de topología transmembranal de TMHMM lo que
mejora la precisión global en la detección y diferenciación de proteínas que presentan péptido
señal en su extremo N-terminal y aquellas con segmentos transmembranales. Asimismo Philius
combina estos dos modelos y adicionalmente hace uso de las Redes Bayesianas Dinámicas (DBN),
las cuales modelan un fenómeno mediante un conjunto de variables y sus relaciones de
dependencia, por lo tanto estiman la probabilidad de una variable desconocida analizando
conjuntamente otras dimensiones como por ejemplo la posición en la secuencia de la proteína, por
consiguiente aborda el problema de diferenciar entre cuatro tipos de proteínas: globulares (G),
globulares con un péptido señal (SP + G), transmembranales (TM) y transmembranales con un
péptido señal (SP + TM) y de predecir la topología de cada segmento por separado (transmembrana
y bucles en la cara interna o externa de la membrana) [51]. De la misma manera, PolyPhobius se
basa en las predicciones de Phobius, sin embargo adiciona la información estructural que brinda
el alineamiento con secuencias homologas [52].
El servidor SPOCTOPUS adiciona el modelo de predicción de péptido señal a la predicción del
servidor OCTOPUS [53] que analiza la topología mediante una combinación entre los puntajes de
las preferencias de cada residuo (ubicación en membrana, en su cara interna o la externa) obtenidos
mediante una Red Neural Artificial (ANN) con el algoritmo de predicción global del HMM e
incluye en la gramática topológica regiones que se encuentran inmersas en la membrana así como
horquillas helicoidales que no la atraviesan completamente [54].
Debido a que para la caracterización bioquímica y biofísica de las proteínas es necesario obtenerlas
de manera solubles (lo que favorece su adecuado plegamiento) se realiza la predicción de la
solubilidad de diferentes proteínas fusión para una misma secuencia para así determinar el mejor
sistema de expresión con el cual trabajar de acuerdo a la naturaleza de la proteínas a estudiar. Para
este análisis se utilizan tres servidores principalmente: PROSO II, SOLpro y ESPRESSO.
Para realizar la predicción PROSO II se basa en la información contenida en la base de datos
TargetDB y Protein Data Bank (PDB) realizando la predicción mediante un algoritmo con dos
niveles: El primero basado en una “Máquina de Vectores de Soporte” (SVM – Conjunto de
proteínas solubles e insolubles) y el segundo en un “Clasificador Bayesiano Ingenuo” (se asume
que la presencia o ausencia de una característica particular no está relacionada con la presencia o
ausencia de cualquier otra característica) identificándose que los aminoácidos R, D, E, G, S, C, M
y L y los dipéptidos ER, EG, KG, QA, HM contribuyen en la solubilidad global del péptido, por
lo tanto se evidencia la relación directa entre la estructura primaria y la solubilidad [55].
De la misma manera SOLpro realiza la predicción teniendo en cuenta estos mismos parámetros y
adiciona en su algoritmo algunas características derivadas de la estructura primaria de la proteína
como su longitud, peso molecular, índice de hidropaticidad, índice alifático, fracción de
aminoácidos que presentan torsión, carga absoluta por residuo y la fracción de residuos expuestos.
A pesar de tener una estructura similar a la de PROSO II, SOLpro disminuye la redundancia de
las secuencias presentes en su base de datos [56].
Finalmente el servidor ESPRESSO utiliza dos tipos de métodos de predicción: el primero basado
en la estructura predicha a partir de la secuencia y el segundo en patrones de secuencia, como

54
respuesta a la limitación de los servidores que utilizan como modelo únicamente a E. coli y a que
la mayoría no consideran las variables que influyen en la expresión de la proteína y su solubilidad,
por lo tanto brinda la posibilidad de predecir estas propiedades en diferentes sistemas [57].
5.8.2. Análisis de estructura secundaria
El análisis de la estructura secundaria de una proteína se puede llevar a cabo mediante diversos
servidores como PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/), Predict Protein
(http://ppopen.informatik.tu-muenchen.de/), JPred3 (http://www.compbio.dundee.ac.uk/www-
jpred/), PBIL-NPS@ (http://pbil.ibcp.fr/htm/index.php?page=pbil_index.html) y Phyre2
(http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index). De estos Phyre2 es uno de los más
utilizados debido a su fácil manejo e interpretación de los resultados y así mismo permite el
modelado de la estructura secundaria por medio de cuatro etapas consecutivas (Figura 22) [58]:
Figura 22. Etapas del modelado de estructuras mediante PHYRE2.
1. Recopilación de secuencias homologas. 2. Escaneo en la biblioteca de pliegues. 3.
Modelamiento de bucles 4. Colocación de la cadena lateral. Tomado de Kelley, L. 2015 [58].
1. Recopilación de secuencias homologas: en esta etapa se construye un perfil evolutivo de la
secuencia para identificar los residuos que se mantienen conservados en diferentes proteínas
realizando discriminaciones de acuerdo a los posibles perfiles de los 20 aminoácidos en cada
una de las posiciones de las diferentes secuencias (HHblits). Mediante este perfil se realiza la
búsqueda de homólogos con una identidad mayor al 20%. De la misma manera se predice la
estructura secundaria mediante PSIPRED el cual utiliza redes neuronales que analizan perfiles
de secuencia de proteínas para identificar la presencia de hélices α, láminas β y bucles con
una precisión del 75-80%. El perfil creado y la estructura secundaria predicha son convertidos
finalmente en un modelo oculto de Márkov (HMM).
2. Escaneo en la biblioteca de pliegues: el HMM creado en la primera etapa es escaneado por
medio de la comparación (HMM-HMM) del modelo de la proteína analizada contra una base

55
de datos de HMM’s de estructura conocida (biblioteca pliegues) que contiene un conjunto
representativo de proteínas cuya estructura ha sido determinada por métodos experimentales
y cuyos perfiles han sido calculados como en la primera etapa. El algoritmo de homología
utilizado en esta etapa es HHsearch y genera una lista de alineamientos con sus respectivas
plantillas, clasificadas de acuerdo a sus probabilidades.
3. Modelamiento de bucles: las mutaciones se analizan de acuerdo a su naturaleza, para el caso
de las inserciones o deleciones se utiliza una librería construida a partir de fragmentos de 2 –
15 aminoácidos de proteínas con estructura conocida producto de su fragmentación y posterior
agrupación estructural, asimismo se considera la distancia entre los puntos finales para cada
mutación.
4. Colocación de la cadena lateral: Esta etapa se lleva a cabo utilizando el protocolo de R3, que
involucra una técnica basada en gráficos y una biblioteca de rotámeros de cadena lateral para
colocar cada una en su más probable rotámero, evitando choques estéricos. Esta técnica tiene
aproximadamente un 80% de precisión, siempre y cuando el esqueleto productos de las etapas
anteriores sea adecuado.
5.8.3. Análisis de estructura terciaria
Una de las plataformas más utilizadas para realizar la predicción de la estructura terciaria de una
proteína es I – TASSER, la cual está integrada por un predictor automatizado de estructura de
proteínas basado en el modelado por homología haciendo uso de Threading de alineamientos
múltiples. Los resultados obtenidos a partir de esta plataforma para una secuencia en específico
contienen una predicción de su estructura secundaria y terciaria, su función mediante homología
estructural con modelos 3D de otras proteínas conocidas, anotaciones de sitios de unión a ligandos,
clasificación de acuerdo a la comisión enzimática y ontología de genes. Este procedimiento tiene
cuatro etapas bien definidas (Figura 23): threading, ensamblaje estructural, selección del modelo
y refinamiento y por último anotaciones funcionales basadas en estructura
Figura 23. Representación de las etapas definidas por I-TASSER para la predicción de
estructura y función de proteínas.
Tomado de Roy, A. Kucukural, A. Zhang, Y. 2010 [59].

56
1. Threading: la palabra threading hace referencia al procedimiento bioinformático en el que se
realiza una búsqueda de una plantilla en una base de datos de proteínas con estructura 3D
elucidada experimentalmente, que posea una estructura o motivo estructural similar al de la
secuencia analizada. En primer lugar se realiza un alineamiento múltiple mediante PSI-
BLAST para identificar parientes evolutivos, a partir de los cuales se construye un perfil de la
secuencia y se predice la estructura secundaria mediante PSIPRED.
A partir de los perfiles y la predicción de estructura secundaria se realiza un análisis a partir
de la librería PDB usando LOMETS, un servidor que reúne 7 programas de threading
(FUGUE, HHSEARCH, MUSTER, PROSPECT, PPA, SP3 y SPARKS), que clasifican las
posibles plantillas mediante un conjunto de puntajes basados en secuencia y estructura, de los
cuales se seleccionan los de mayor puntaje en cada programa. La calidad de los alineamientos
de cada plantilla se analiza con la significación estadística de la mejor alineación de
Threading, es decir, el puntaje Z (puntuación de energía en unidades de desviación estándar
respecto a la media estadística de todos los alineamientos).
2. Ensamblaje estructural: los fragmentos de los alineamientos de Threading son escindidos de
las estructuras de plantilla y son utilizados para ensamblar conformaciones estructurales entre
las secciones correctamente alineadas con las regiones no alineadas (bucles o colas). Para
mejorar la eficiencia de esta búsqueda I-TASSER adopta un modelo reducido para representar
la cadena de la proteína con cada residuo descrito por su átomo de Cα y el centro de masa de
su cadena lateral. En este punto se observa que los modelos de las regiones correctamente
alineadas tienen alta precisión y se mantienen rígidos durante las simulaciones para conservar
la alta resolución de la plantilla, mientras que las que no se alinearon presentan baja precisión,
sin embargo se escoge la conformación con menor entropía posible. El ensamblaje de estos
fragmentos se realiza mediante una técnica de simulación de Monte Carlo, que implementa
varias simulaciones de réplicas en paralelo a diferentes temperaturas.
Las conformaciones generadas en las réplicas de baja temperatura durante la refinación por
simulación son agrupadas por SPICKER con el fin de identificar los estados con menor
energía libre. En este punto se realizan agrupaciones o clusters promediando las coordenadas
3D de cada ensamblaje.
3. Selección del modelo y refinamiento: en esta etapa se realiza una nueva simulación de los
ensamblajes utilizando los clústeres seleccionados en la etapa anterior, con lo que se
seleccionan las estructuras en PDB con mayor cercanía estructural a ellos mediante TM-aling.
Esta segunda simulación se realiza con el objetivo de eliminar los choques estéricos y para
refinar la topología global de los clusters. A continuación se agrupan nuevamente los
ensamblajes y se escogen aquellos con menor energía para analizarlos con la herramienta
REMO que genera los modelos estructurales finales mediante la construcción de modelos de
todos los Cα a través de la optimización de las redes de enlaces de hidrógeno.
4. Anotaciones funcionales basadas en estructura: la función de la secuencia analizada es inferida
mediante la coincidencia estructural de los modelos 3D predichos con las proteínas de
estructura y la función conocida en PDB. Para esto se construyeron tres librerías de
estructura/función de proteínas con entradas no redundantes, una de 5,798 con números de la

57
Comisión Enzimática (EC), una de 26,045 con términos de Ontología de Genes (GO) y otra
de 19,658 con sitios de unión a ligandos.
Los análogos estructurales en la librería de GO son seleccionados por su topología global
mediante TM-aling, mientras que los análogos de EC y sitios de unión a ligando se basan tanto
en la similitud global y local.

58
6. METODOLOGÍA
6.1. Métodos computacionales.
El estudio del metabolismo energético de diversos parásitos ha demostrado que las NMNAT son
enzimas clave en su ciclo de vida puesto que catalizan la síntesis del NAD+, uno de los principales
receptores de electrones en la célula. Sin embargo, las especies extracelulares como Giardia
dueodenalis, al igual que en el Homo sapiens, poseen varias isoformas de esta enzima presentes
en diversos compartimientos subcelulares como el núcleo, el citosol o la mitocondria, mientras
que, en las intracelulares como Leishmania braziliensis, Tripanosoma cruzi y Plasmodium
falciparum se ha identificado únicamente una isoforma, lo que puede deberse a que estos
organismos adquieren el NAD+ del medio intracelular de su hospedero mediante transportadores
específicos para este dinucleótido.
Este mecanismo se ha identificado en la levadura Saccharomyces cerevisiae la cual es utilizada en
el laboratorio como modelo biológico de eucariotas, ya que este organismo sintetiza el NAD+
fuera de la mitocondria y para su metabolismo debe atravesar la membrana interna de este
organelo, para esto posee un transportador específico denominado “Transportador mitocondrial de
NAD+” (ScNdt1p con código NP_010910.1), el cual fue caracterizado funcionalmente
identificándose que tiene la capacidad de transportar NAD+ y en menor medida (d)AMP y
(d)GMP, sin embargo no lo hace con las especies α-NAD+, NADH, NADP+ ni NADPH;
adicionalmente se identificó que este se encuentra en la mitocondria y que posee una isoforma
ScNdt2p con un 70% de homología [14].
Por otra parte, en la planta Arabidopsis thaliana se identificaron dos isoformas AtNDT1
(NP_566102) y AtNDT2 (NP_564233), las cuales presentan gran semejanza estructural con
ScNdt1p descrita anteriormente. Se encontró que AtNDT1 está ubicada en el cloroplasto y
AtNDT2 en la mitocondria y tienen la capacidad de transportar el NAD+ mediante intercambio
siendo el ADP y el AMP los más eficientes para este proceso [15].
En el Homo sapiens se identificó un transportador de CoA, FAD, FMN y AMP y en menor medida
de NAD+, PAP (adenosina 3’,5’-difosfato) y ADP, el cual fue localizado anteriormente en la
membrana del peroxisoma y presenta un transporte mediante intercambio de CoA, FAD y NAD+
hacia el interior del organelo por las especies PAP, FMN y AMP generadas en su interior [16].
De acuerdo con esta información se seleccionaron las secuencias de los transportadores presentes
en las bacterias Protoclamidia amoebofilia y Clamidia tracomitis, en la planta Arabidopsis
thaliana, en el hongo Saccharomyces cerevisiae y en el eucariota Homo sapiens con los cuales se
llevó a cabo un alineamiento múltiple mediante el servidor PRALINE
(http://www.ibi.vu.nl/programs/pralinewww/) a partir del cual se obtuvo un consenso que fue
utilizado para identificar homólogos en el genoma de Leishmania braziliensis MHOM/BR/75/
mediante la herramienta BLAST.
A partir de este consenso no se identificó ningún homólogo, por lo tanto se realizaron
combinaciones entre cada una de las secuencias de los transportadores identificándose que el
consenso obtenido a partir de las secuencias de Arabidopsis thaliana (NP_564233) y de
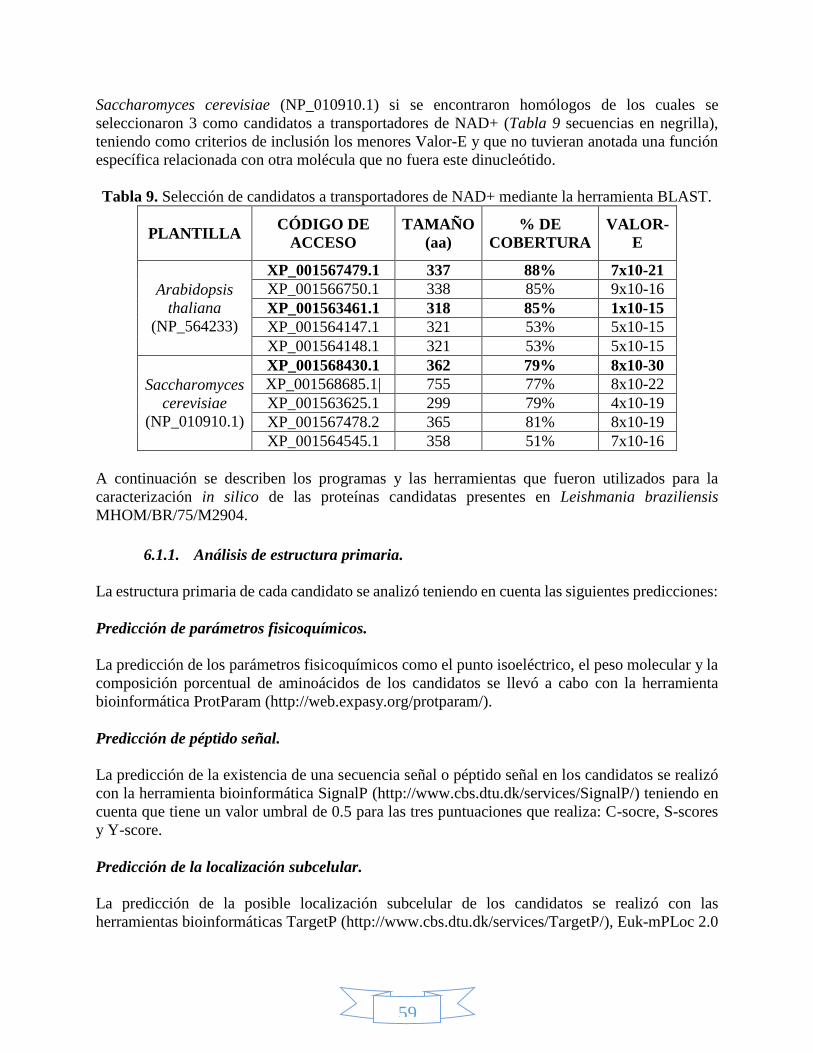
59
Saccharomyces cerevisiae (NP_010910.1) si se encontraron homólogos de los cuales se
seleccionaron 3 como candidatos a transportadores de NAD+ (Tabla 9 secuencias en negrilla),
teniendo como criterios de inclusión los menores Valor-E y que no tuvieran anotada una función
específica relacionada con otra molécula que no fuera este dinucleótido.
Tabla 9. Selección de candidatos a transportadores de NAD+ mediante la herramienta BLAST.
PLANTILLA CÓDIGO DE
ACCESO
TAMAÑO
(aa)
% DE
COBERTURA
VALOR-
E
Arabidopsis
thaliana
(NP_564233)
XP_001567479.1 337 88% 7x10-21
XP_001566750.1 338 85% 9x10-16
XP_001563461.1 318 85% 1x10-15
XP_001564147.1 321 53% 5x10-15
XP_001564148.1 321 53% 5x10-15
Saccharomyces
cerevisiae
(NP_010910.1)
XP_001568430.1 362 79% 8x10-30
XP_001568685.1| 755 77% 8x10-22
XP_001563625.1 299 79% 4x10-19
XP_001567478.2 365 81% 8x10-19
XP_001564545.1 358 51% 7x10-16
A continuación se describen los programas y las herramientas que fueron utilizados para la
caracterización in silico de las proteínas candidatas presentes en Leishmania braziliensis
MHOM/BR/75/M2904.
6.1.1. Análisis de estructura primaria.
La estructura primaria de cada candidato se analizó teniendo en cuenta las siguientes predicciones:
Predicción de parámetros fisicoquímicos.
La predicción de los parámetros fisicoquímicos como el punto isoeléctrico, el peso molecular y la
composición porcentual de aminoácidos de los candidatos se llevó a cabo con la herramienta
bioinformática ProtParam (http://web.expasy.org/protparam/).
Predicción de péptido señal.
La predicción de la existencia de una secuencia señal o péptido señal en los candidatos se realizó
con la herramienta bioinformática SignalP (http://www.cbs.dtu.dk/services/SignalP/) teniendo en
cuenta que tiene un valor umbral de 0.5 para las tres puntuaciones que realiza: C-socre, S-scores
y Y-score.
Predicción de la localización subcelular.
La predicción de la posible localización subcelular de los candidatos se realizó con las
herramientas bioinformáticas TargetP (http://www.cbs.dtu.dk/services/TargetP/), Euk-mPLoc 2.0

60
(http://www.csbio.sjtu.edu.cn/cgi-bin/EukmPLoc2.cgi) y iLoc-Animal (http://www.jci-
bioinfo.cn/iLoc-Animal).
Predicción de motivos y dominios.
La predicción de motivos en los candidatos se realizó con la herramienta bioinformática MOTIF
Search (http://www.genome.jp/tools/motif/) y la de los dominios con InterPro
(http://www.ebi.ac.uk/interpro/).
Predicción de hélices transmembranales y topología.
La predicción de la presencia de hélices transmembranales en los candidatos se realizó con las
herramientas TMHMM (http://www.cbs.dtu.dk/services/TMHMM-2.0/), Phobius y PolyPhobius
(http://phobius.sbc.su.se/) y la topología por medio de las herramientas compiladas en Topcons
(http://topcons.cbr.su.se/) como OCTOPUS y SPOCTOPUS (http://octopus.cbr.su.se/) y Philius
(http://noble.gs.washington.edu/proj/philius).
Predicción de residuos conservados.
La predicción de residuos altamente conservados se llevó a cabo mediante un alineamiento
múltiple entre las secuencias de los candidatos LbTNTA, LbTNTB y LbTNTC con las de las
plantillas utilizadas para realizar su búsqueda bioinformática (NP_564233 y NP_010910.1) con el
servidor PRALINE (http://www.ibi.vu.nl/programs/pralinewww/).
Predicción de las modificaciones post-traduccionales.
La predicción de las posibles modificaciones post-traduccionales de los candidatos se realizó con
las herramientas bioinformáticas NetPhos 2.0 http://www.cbs.dtu.dk/services/NetPhos/) para
fosforilaciones en serinas, treoninas y tirosinas y NetAcet 1.1
(http://www.cbs.dtu.dk/services/NetAcet/) para acetilaciones.
Predicción de la solubilidad de las proteínas recombinantes.
La predicción de la solubilidad de las proteínas recombinantes obtenidas con el software snapgene
2.8 en los vectores pBAD202-d-TOPO, pCOLD-I, pET SUMO, pET100-d-TOPO y pMAL-c5X
se realizó mediante las herramientas PROSO II (http://mips.helmholtz-
muenchen.de/prosoII/prosoII.seam), SOLpro (http://scratch.proteomics.ics.uci.edu/) y
ESPRESSO (http://mbs.cbrc.jp/ESPRESSO/TopPage.html).
6.1.2. Predicción de estructura secundaria.
La predicción de la estructura secundaria y de la topología de las proteínas seleccionadas como
candidatos a transportadores de NAD+ se llevó a cabo mediante el servidor PHYRE2
(http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index).

61
6.1.3. Predicción de la estructura terciaria.
A partir de las secuencias que presentaron la misma arquitectura de dominio de los candidatos y
que tenían una estructura 3D determinada experimentalmente en PDB (http://www.wwpdb.org/)
se intentó realizar la predicción de la estructura secundaria mediante homología con la herramienta
Swiss-Model (http://swissmodel.expasy.org/), sin embargo, no se encontraron plantillas con una
homología superior al 30% por lo tanto se construyó un modelo predictivo mediante Threading
haciendo uso del servidor I – TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) y el
software UCSF Chimera. Para refinar estos modelos se utilizó el servidor 3Drefine
(http://sysbio.rnet.missouri.edu/3Drefine/).
6.2. Métodos experimentales.
A continuación se presentan los procedimientos de biología molecular que se llevaron a cabo para
la obtención in vitro de las proteínas candidatas presentes en el parásito Leishmania Braziliensis:
6.2.1. Evaluación de la integridad del ADNg de Leishmania braziliensis.
Se trabajó con ADNg extraído en enero de 2016 mediante el kit Wizard® Genomic DNA
Purificatión Kit (Promega) [60] a partir de promastigotes de Leishmania braziliensis (M2904
MHOM/BR/75M2904) recolectados por centrifugación a 8.000 r.p.m por 10 min a 4°C
previamente cultivados en medio LIT (liver infusion-tryptose) suplementado con suero fetal
bovino (SFB) al 10% (v/v) a 26ºC en frascos de 25 cm2 (T25).
La integridad del ADNg se verificó mediante electroforesis horizontal en gel de agarosa al
1,0%(p/v) en TBE 0.5X con un voltaje de 100V durante 40 minutos, adicionalmente se determinó
su concentración mediante espectrofotometría, haciendo uso de las siguientes ecuaciones:
[ADNg] = (Abs260nm – Abs320nm) x 50µg/mL x Fd
En donde:
[ADNg] = Concentración ADN genómico.
Abs260nm = Absorbancia de la muestra a 260nm.
Abs320nm = Absorbancia de la muestra a 320nm.
Fd = Factor de dilución de la muestra.
Asimismo se determinó la pureza de la muestra con las siguientes ecuaciones:
𝑅𝑎𝑡𝑖𝑜 (𝑅) = 𝐴𝑏𝑠260𝑛𝑚 − 𝐴𝑏𝑠320𝑛𝑚
𝐴𝑏𝑠280𝑛𝑚 − 𝐴𝑏𝑠320𝑛𝑚
% 𝑃𝑢𝑟𝑒𝑧𝑎 =𝑅𝑎𝑡𝑖𝑜
1.8𝑥 100
(2)
(3)
(4)

62
6.2.2. Diseño y estandarización de Tm de los cebadores mediante PCR.
El diseño de cebadores se realizó mediante la herramienta bioinformática Primer 3 Plus
(http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi), la temperatura de anillaje
(Tm) se determinó con la herramienta OligoAnalyzer 3.1 (https://www.idtdna.com/calc/analyzer)
y se comprobó la especificidad de los cebadores mediante un BLAST en NCBI
(http://blast.ncbi.nlm.nih.gov/Blast.cgi) contra todo el genoma de Leishmania braziliensis
MHOM/BR/75/M2904, teniendo como base la secuencia del gen que codifica cada una de las
proteínas candidatas.
Tabla 10. Características de los cebadores utilizados para amplificar los candidatos a
transportadores de NAD+.
CANDIDATO PRIMER SECUENCIA
5´ - 3´
TEMPERATURA
DE ANILLAJE
(°C)
TAMAÑO
ESPERADO
(pb)
LbTNTA LbTNTAd CACCATGACGCAGTCTTTCTCATCA
50 1.089 LbTNTAr CTACTGGAACTGAGGCTGTCCCACA
LbTNTB LbTNTBd CACCATGAGAGAGAAGAAGTCAGCA
53 1.014 LbTNTBr TCATGAGATGTACTTTGCCACCTTT
LbTNTC LbTNTCd CACCATGGCATCTACGTCGCCCGCA
57 957 LbTNTCr CTACTCCATCGTCGATTCATCGGAC
La temperatura óptima de anillaje para la amplificación de cada candidato se evaluó para las
temperaturas 50, 53, 55 y 57°C para los tres pares de cebadores (adicionalmente una Tm de 60°C
para los que amplifican LbTNTC) mediante una amplificación por PCR con una mezcla de
reacción que contenía buffer de PCR 1X (KCl 50 mM, Tris-HCl 10 mM pH 8.3 y Tritón 0,1 %),
cebadores directo y reverso 0.2 µM (Tabla 10), cloruro de magnesio 2.0 mM, dNTPs (dATP,
dCTP, dGTP, dTTP) 0.2 mM, 1U de Taq polimerasa y 1 µL de plantilla de ADNg 50 ng/µL de
Leishmania Braziliensis en un volumen final de 15 µL. Las condiciones del ciclo térmico se
establecieron en un primer ciclo de desnaturalización inicial a 94°C por 10 minutos, seguido por
30 ciclos de amplificación con una programación de desnaturalización a 94°C por 30 segundos,
anillaje de cebadores a 50°C por 1 minuto y extensión a 72°C por 1 minuto, finalmente se realizó
un último ciclo de extensión final a 72°C por 10 minutos. Como control positivo se utilizó el gen
de la NMNAT de L. braziliensis con una Tm de 56°C y como control negativo agua DEPC
(Dietilpirocarbonato la cual inactiva las RNasas).
La verificación de cada PCR se realizó mediante electroforesis horizontal en gel de agarosa al
1.5% (p/v) en TBE 0,5X con un voltaje de 100 V durante 40 minutos.
6.2.3. Estandarización de la amplificación por PCR con Pfu.
La estandarización de la amplificación por PCR con Pfu polimerasa se llevó a cabo con el
candidato LbTNTA, para la cual se tuvieron en cuenta la temperatura de anillaje de los cebadores
y la concentración de sulfato de magnesio (MgSO4) debido a que estos son los principales factores
que afectan la especificidad de los cebadores y de la enzima. Para los ensayos de temperatura de
anillaje se utilizaron como controles positivos el gen de la LbNMNAT amplificado desde ADNg

63
de L. braziliensis y a partir del vector recombinante LbNMNAT/pET100 construido en un trabajo
previo [7]. El gen de la LbNMNAT tiene un tamaño de 927 pb y se amplificó con un Tm de 56°C.
En primer lugar se evaluaron las temperaturas de anillaje de los cebadores 50, 53, 55 y 57
(adicionalmente 60°C para LbTNTC) con una mezcla de reacción que contenía buffer de PCR 1X
(KCl 50 mM, Tris-HCl 10 mM pH 8.8), cebadores directo y reverso 0.2 µM, sulfato de magnesio
10.0 mM, dNTPs (dATP, dCTP, dGTP, dTTP) 0.2 mM, 1U de Pfu polimerasa y 1 µL de plantilla
de ADNg 50 ng/µL de Leishmania Braziliensis. Las condiciones del ciclo térmico son las mismas
que se establecieron en el numeral 6.2.2 con Taq polimerasa teniendo en cuenta que se llevó a cabo
un gradiente de temperatura para el paso de anillaje de cebadores como se mencionó anteriormente.
A continuación se evaluó la concentración de MgSO4 con valores de 10.0, 5.0, 2.5, 2.0 y 1.5 mM
a una temperatura de anillaje de 57°C y manteniendo constantes las demás condiciones
anteriormente mencionadas.
Para identificar la identidad del amplificado de LbTNTA se realizó una digestión de 2 µL del
producto de PCR con 0.5 µL de la enzima de restricción AluI, 1 µL de buffer Tango 1X (33 mM
Tris-acetato (pH 7.9), 10 mM acetato de magnesio, 66 mM acetato de potasio, 0.1 mg/mL BSA) y
6 µL de agua DEPC y se incubó durante 4 horas a 37°C, el perfil de digestión se chequeó por
electroforesis horizontal en gel de agarosa al 1.5% (p/v) en TBE 0,5X con un voltaje de 100 V
durante 40 minutos. Una vez identificado el perfil del producto de la amplificación de LbTNTA
se amplificaron los candidatos LbTNTB y LbTNTC por PCR con Pfu con una mezcla de reacción
que contenía buffer de PCR 1X (KCl 50 mM, Tris-HCl 10 mM pH 8.8), cebadores directo y reverso
0.2 µM, sulfato de magnesio 1.5 mM, dNTPs (dATP, dCTP, dGTP, dTTP) 0.2 mM, 1U de Pfu
polimerasa y 1 µL de plantilla de ADNg 50 ng/µL de Leishmania Braziliensis con una temperatura
de anillaje de 57°C para ambos candidatos.
El chequeo de cada PCR se realizó mediante electroforesis horizontal en gel de agarosa al 1.5%
(p/v) en TBE 0,5X con un voltaje de 100 V durante 40 minutos.
6.2.4. Obtención de vectores recombinantes LbTNT´s /pET100 y LbTNT´s /pBAD202.
La clonación de los 3 candidatos se llevó a cabo en los vectores pET100/D-TOPO y pBAD202/D-
TOPO realizando las etapas que se describen a continuación:
Ligación inserto-vector.
La cantidad de inserto necesaria para la ligación se determinó mediante la ecuación 1, a partir de
la concentración de cada producto de PCR la cual se calculó por densitometría en comparación
con el marcador de peso molecular manteniendo una relación 3:1 inserto:vector.
La mezcla de reacción contenía 1 µL de solución salina 1X (1,2 M de NaCl y 0.06 M de MgCl2),
1 µL del vector pET100/D-TOPO (en buffer 50 mM Tris-HCl (pH 7,4), 2 mM de DTT, 1 mM de
EDTA, 0.1% de Triton X-100, 100 μg/ml de BSA, 30 μM de azul de bromofenol y 50% de glicerol)
o pBAD202/D-TOPO (en buffer 50 mM Tris-HCl pH 7,4, 1 mM de DTT, 1 mM de EDTA, 0.1%
de Triton X-100, 100 μg/ml de BSA, 30 μM de azul de bromofenol y 50% de glicerol), el volumen
anteriormente calculado con la ecuación 1 de producto de PCR sin purificar y se completó a un
volumen de 6 µL con agua estéril, a continuación se incubó a 37°c durante 15 minutos.

64
Transformación en Escherichia coli.
Se llevó a cabo en células de Escherichia coli Top10 (cepa de mantenimiento) químicamente
competentes, las cuales se obtuvieron cultivando a 37°C durante toda la noche 1 µL de inóculo de
células con medio LB autoclavado (bactotriptona 1% p/v, cloruro de sodio 1% p/v y extracto de
levadura 0.5% p/v a pH 7.0), posteriormente se realizó una dilución 1:50 en el mismo medio y se
incubó nuevamente a 37°C hasta alcanzar una densidad óptica entre 0.3 – 0.4 a 600 nm. Una vez
alcanzada esta densidad óptica se centrifugó la muestra a 5.000 r.p.m por 10 minutos a 4°C, se
separó el medio del pellet de células y se le adicionó CaCl2 100 mM frío estéril, luego se incubó
en baño de hielo por 10 minutos. Posteriormente se agitó suavemente hasta disolución y se repitió
la adición de CaCl2 una vez más hasta obtener el pellet que se resuspendió en 1.750 µL de CaCl2
100 mM y 750 µL de glicerol estéril al 30% (v/v).
A estas células Top 10 químicamente competentes se adicionó 3µL del producto de la reacción de
ligación y se llevó a cabo la transformación mediante choque térmico iniciando un ciclo en baño
de hielo por 30 minutos, a continuación uno a 42°C por 42 segundos y finalmente en baño de hielo
por 5 minutos. Posteriormente se incubaron las células en LB frío (medio de recuperación) a 37°C
por 1 hora. Una vez las células se recuperaron se platearon 300 µl del producto en cajas de Petri
con medio de cultivo sólido (LB-agar) previamente autoclavado al cual se adicionó ampicilina a
una concentración final de 100 µg/mL o kanamicina a una concentración final de 50 µg/mL para
los recombinantes de pET100 y pBAD202 respectivamente y se incubaron a 37°C toda la noche.
Rastreo de colonias recombinantes.
La evaluación de los recombinantes construidos a partir del vector pBAD202/D-TOPO se llevó a
cabo agrupando las 25 colonias para LbTNTA de la siguiente manera: en los pools A1 (2 – 5), A2
(6 – 10), A3 (11 – 15), A4 (16 – 20) y A5 (21 – 25) dejando 1 sin agrupar (C1 para la colonia 1).
Para LbTNTB se agruparon las 9 colonias en los pools B1 (1 y 2), B2 (3 y 4), B3 (5 y 6), B4 (7 y
8) y 1 sin agrupar (B5 para la colonia 9) y para LbTNTC las 11 fueron mezcladas en tres pools;
C1 (2 – 5), C2 (6 – 9) y C3 (10 y 11) dejando 1 sin agrupar (1 para la colonia 1). Solamente para
las colonias recombinantes que dieron resultados positivos mediante PCR para la detección de los
genes de LbTNTB y LbTNTC se abrieron los pools B4 y C1 respectivamente con el fin de
establecer cuales eran las colonias positivas. Para el caso de la clonación en el vector pET100/D-
TOPO se analizó la única colonia obtenida mediante transformación para el candidato LbTNTA,
5 para LbTNTB y 10 para LbTNTC las cuales fueron seleccionadas al azar en ambos casos. El
rastreo se llevó a cabo mediante PCR de colonia, en la cual, cada colonia se resuspendió en 10 µL
de agua DEPC y se lisó a 95°C en baño María. A partir de este lisado se realizó una PCR bajo las
mismas condiciones estandarizadas en el numeral 6.2.2 con Tm de 50, 53 y 57°C para LbTNTA,
LbTNTB y LbTNTC respectivamente y Taq polimerasa. Una vez obtenidos los productos de
amplificación se chequearon por electroforesis horizontal en gel de agarosa al 1.5% (p/v) en TBE
0,5X con un voltaje de 100 V durante 40 minutos.

65
Evaluación de plásmidos recombinantes.
La colonia número 1 para LbTNTA, la 7 para LbTNTB y las 1, 2, 3 y 4 para LbTNTC obtenidas
a partir del vector pBAD202/D-TOPO fueron inoculadas en 10 mL de medio LB líquido con
kanamicina a una concentración final de 50 µg/mL y las colonias 1 – 5 para LbTNTB y 1 – 5, 8,
9 y 10 para LbTNTB a partir del vector pET100/D-TOPO en 10 mL de medio LB líquido con
ampicilina a una concentración final de 100 µg/mL y se incubaron a 37°C toda la noche. A
continuación se centrifugó el inóculo a 5.000 r.p.m por 10 minutos y se descartó el sobrenadante,
el pellet de células obtenido se resuspendió en 300 µL de solución isotónica (Glucosa 50 mM, Tris
HCl 25 mM pH 8,0, EDTA 2,5 mM pH 8,0), la cual se incubó por 5 minutos a 4°C. Una vez
transcurrido este tiempo se adicionaron 300 µL de la solución de lisis (NaOH 0,2 N, SDS 1% p/v),
se mezcló por inversión y se incubó a 4°C por 5 minutos, posteriormente se adicionaron 300 µL
de solución de neutralización (acetato de potasio 3M, pH 4,8), se centrifugó a 11.000 r.p.m por 10
minutos y se recuperó el sobrenadante y se transfirió a un tubo nuevo, al cual se le adicionó 1 µL
de RNasa (10 ng/mL) y se incubó a 37°C por 30 minutos. A este producto se adicionó un volumen
(800 µL) de la mezcla fenol-cloroformo-alcohol Isoamílico (25:24:1), se homogenizó por
inversión 10 veces y se incubó nuevamente a 4°C por 10 minutos con agitación constante,
posteriormente se centrifugó a 11.000 r.p.m por 5 minutos, se recuperó la fase acuosa y se
agregaron 2.5 volúmenes de etanol absoluto frío (1 mL) 1/10 volúmenes de acetato de sodio 3 M
pH 5,2 frio (40 µL); la mezcla resultante se precipitó a -80°C. A continuación se centrifugó por 10
minutos a 12.000 r.p.m, se descartó el sobrenadante y se realizaron 2 lavados al precipitado
obtenido con 1 mL de etanol al 70% frio, finalmente se dejó evaporar el etanol y se resuspendió el
pellet en 50 µL de agua DEPC.
Los plásmidos recombinantes obtenidos se evaluaron mediante PCR bajo las mismas condiciones
estandarizadas en el numeral 6.2.2 con Tm de 50, 53 y 57°C para LbTNTA, LbTNTB y LbTNTC
respectivamente y Taq polimerasa utilizando como plantilla el plásmido extraído. Una vez
obtenidos los productos de amplificación se chequearon por electroforesis horizontal en gel de
agarosa al 1.5% (p/v) en TBE 0,5X con un voltaje de 100 V durante 40 minutos.
Para confirmar la presencia de los candidatos en cada vector se llevó a cabo una digestión de los
plásmidos recombinantes con las enzimas de restricción EcoRV, NcoI, BamHI, SphI y una doble
digestión con NcoI y EcoRV (Tabla 11), en una mezcla de reacción con 3,0 μL de plásmido, 0,5μL
de cada enzima, 1 μL de cada uno de los buffer correspondientes y se completó a un volumen final
de 10 μL con agua DEPC, esta mezcla se incubó por 6 horas a 37°C para todas las enzimas.
Tabla 11. Enzimas de restricción utilizadas para la digestión de los plásmidos recombinantes.
PLÁSMIDO
RECOMBINANTE
ENZIMAS DE
RESTRICCIÓN BUFFER COMPOSICIÓN BUFFER
SITIO DE
RECONOCIMIENTO
LbTNTA/pET100 EcoRV R 1X
10 mM Tris-HCl (pH 8.5), 10
mM MgCl2, 100 mM KCl,
0.1 mg/mL BSA LbTNTB/pET100
LbTNTC/pET100 NcoI – EcoRV Tango 1X
33 mM Tris-acetato (pH 7.9),
10 mM acetato de magnesio,
66 mM acetato de potasio,
0.1 mg/mL BSA.
EcoRV
NcoI
66
PLÁSMIDO
RECOMBINANTE
ENZIMAS DE
RESTRICCIÓN BUFFER COMPOSICIÓN BUFFER
SITIO DE
RECONOCIMIENTO
LbTNTA/pBAD202 BamHI BamHI
1X
10 mM Tris-HCl (pH 8.0), 5
mM MgCl2, 100 mM KCl,
0.02% Triton X-100, 0.1
mg/mL BSA.
LbTNTB/pBAD202 SphI B 1X 10 mM Tris-HCl (pH 7.5), 10
mM MgCl2, 0.1 mg/mL BSA.
LbTNTC/pBAD202 NcoI – EcoRV Tango 1X
33 mM Tris-acetato (pH 7.9),
10 mM acetato de magnesio,
66 mM acetato de potasio,
0.1 mg/mL BSA.
EcoRV
NcoI
Una vez obtenidos los productos de restricción se chequearon los perfiles de digestión por
electroforesis horizontal en gel de agarosa al 1.0% (p/v) en TBE 0,5X con un voltaje de 100 V
durante 40 minutos. Finalmente, aquellos plásmidos que presentaron el perfil de digestión
esperado fueron crio-conservados en medio LB con glicerol estéril al 40% a -80°C.
6.2.5. Expresión y detección de las proteínas recombinantes 6xHisLbTNT´s y
TrxLbTNT´s por electroforesis SDS-PAGE y Western blot.
La transformación se llevó a cabo por choque térmico en células BL21 (DE3) (cepa de expresión)
químicamente competentes con los vectores recombinantes como se especificó en el numeral 6.2.4.
A partir de las bacterias transformadas se realizaron inóculos líquidos usando medio LB con
ampicilina a una concentración final de 100 µg/mL o kanamicina a una concentración final de 50
µg/mL los cuales fueron incubados toda la noche a 37°C con agitación constante, a continuación
se realizaron diluciones 1:50 y se continuaron incubando a la misma temperatura hasta alcanzar
una densidad óptica de 0,6 a 600 nm, momento en el cual se indujo la expresión de la proteína
recombinante con isopropil β-D-1-tiogalactopiranósido (IPTG) a una concentración final de 1 mM
para los clones de pET100 y con una concentración final de arabinosa de 0.02% (p/v) para
pBAD202, a continuación se incubó a 37°C con agitación constante durante toda la noche,
tomando muestras al tiempo cero y a las 24 horas.
La inducción de la proteína recombinante 6xHisLbTNTA fue estandarizada teniendo en cuenta las
variables de temperatura (20 y 37°C), tiempo de inducción (0, 2, 4, 6 y 24 horas) y concentración
de arabinosa (0.002, 0.02 y 0.2% p/v).
Una vez finalizada la inducción se centrifugó el cultivo a 6000 rpm por 10 minutos para obtener
las fracciones soluble e insoluble a partir de la lisis de los pellets resultantes que se suspendieron
en 250 µL de buffer de lisis 1 (NaH2PO4 50 mM, NaCl 300 mM, Imidazol 10 mM a pH 8.0) para
los recombinantes LbTNT´s/pBAD202 y buffer de lisis 2 (NaH2PO4 50 mM, NaCl 400 mM, KCl
100 mM, Imidazol 100 mM, glicerol al 10% y Triton X-10 al 0.5% a pH 7.78), a continuación se
adicionó lisozima (concentración final 1 mg/mL) e inhibidores de proteasas con una dilución 1:400
(Sigma P8340: 1 mM AEBSF, 14 μM E64, 15 μM Pepstatin A, 40 μM Bestatin, 20 μM Leupeptin,
0,8 μM Aprotinin), la mezcla resultante se incubó a 4°C por 40 minutos con agitación constante y
se sonicaron durante 30 minutos con pulsos de 30 segundos, descanso de 15 y con una amplitud

67
del 50%. A continuación se centrifugó la mezcla a 12.000 r.p.m por 20 minutos y se separó el
sobrenadante (fracción soluble) del pellet (fracción insoluble). Para obtener las proteínas totales
se obtuvieron las bacterias por centrifugación de una alícuota del cultivo a 6000 r.p.m durante 10
minutos a 4°C y se descartó el sobrenadante.
A cada una de las muestras de la fracción insoluble y proteínas totales se adicionó 50 – 250 µl
buffer de carga 1X (Tris 0,083 M, SDS 1,74% (p/v), Glicerol 5% (v/v), β-mercaptoetanol 5,5%
(v/v) y azul de bromofenol 0,002% p/v) de acuerdo al tamaño a cada pellet y 10 µL de buffer de
carga 6X a 50 µL de sobrenadante y se llevó a baño María durante 15 minutos.
Las muestras fueron separadas y analizadas mediante electroforesis SDS-PAGE en la cual se cargó
15 μL de las fracciones soluble e insoluble y proteínas totales de cada muestra en un gel de
acrilamida discontinuo con un gel concentrador del 3,9% (Tris 0,125 M pH 6,8 SDS 0,1% p/v) y
un gel separador del 12% (Tris 0,375M pH 8,8 SDS 0,1% p/v). Se aplicó una diferencia de
potencial de 108 V en buffer de corrida Tris-Glicina (Tris 25 mM, Glicina 192 mM, SDS 0,1%
p/v) durante 2 horas y se reveló el gel obtenido mediante tinción en solución de azul de Coomassie
R250 (0,05% (p/v), isopropanol 25% (v/v), ácido acético 11,2% (v/v)) durante 2 horas y se
decoloró durante toda la noche en solución decoloradora ().
Para la inmunodetección (Western blot) las proteínas separadas por SDS-PAGE se transfirieron a
membranas de nitrocelulosa (previamente equilibrada en buffer de transferencia), usando el
método de electrotransferencia húmeda con buffer de transferencia (Tris base 25 mM, Glicina 192
mM y Metanol 10% V/V, pH 8,3) a 200mA por 2 horas.
Para detectar la proteína recombinante se empleó el sistema Biotina-estreptavidina conjugada a
fosfatasa alcalina para lo cual se bloqueó la membrana por una hora en buffer TBST-L (leche 5%,
Tris-HCl 20 mM pH 7,5, NaCl 150mM y Tween 20 0,1% V/V) y se incubó durante una hora con
anticuerpo primario monoclonal de ratón Anti6xHis (diluido 1:3000 en TBST) para las proteínas
recombinantes obtenidas con pET100/D-TOPO o anticuerpo primario monoclonal de ratón
AntiTioredoxina (diluido 1:3000 en TBST) para las proteínas recombinantes obtenidas con
pBAD/D-TOPO. A continuación se realizaron tres lavados cada uno de 10 minutos con TBST-L
y se incubó durante una hora con el anticuerpo secundario biotinilado anti-IgG de ratón (diluido
1:5000 en TBST), posteriormente se realizaron tres lavados cada uno de 10 minutos con TBST-L
y se incubó la membrana durante 30 minutos con estreptavidina fosfatasa alcalina en TBST
(diluido 1:3000) y se realizaron 3 lavados cada uno de 10 minutos con TBST.
Para revelar la membrana se utilizaron 16.5 µL de los sustratos cromogénicos NBCIP (5-Bromo-
4-Chloro-3’-Indolyphosphate p-Toluidine Salt, S381C Promega) y NBT (Nitro-Blue Tetrazolium
Choride, S380C, Promega) en buffer de reacción (Tris-HCl 100 mM pH 9,0, NaCl 150 mM, MgCl2
1 mM). La detección inmunológica se detuvo con agua destilada cuando se evidenciaron productos
coloreados sobre la membrana.

68
7. RESULTADOS Y DISCUSIÓN
Los análisis realizados a los candidatos a transportadores de NAD+ consistieron en estudios
bioinformáticos para la caracterización de las proteínas y de la clonación de cada una de estas.
7.1. Métodos computacionales
Como se mencionó en la metodología se escogieron 3 secuencias de proteínas como candidatos a
“Transportadores de NAD+ Transmembranales de Leishmania braziliensis” (LbTNT´s) los cuales
corresponden con genes de 1.089, 1.014 y 957 pb (Anexo 1) que codifican proteínas de 362, 337 y
318 aminoácidos (Anexo 2) fueron denominados LbTNTA, LbTNTB y LbTNTC respectivamente
(Tabla 12).
Tabla 12. Especificaciones de las secuencias candidatas como transportadores mitocondriales de
NAD+/NADH.
PARÁMETRO LbTNTA LbTNTB LbTNTC
Nombre
Putative
Mitochondrial Carrier
Protein
Mitochondrial Carrier
Protein-like Protein
Putative
Mitochondrial Carrier
Protein
Ubicación
cromosomal 35:1270427...1271515 32:447043...448056 15:38578...39534
Gen ID/
Prot ID/
UniProtKB
XM_001568380.1/
XP_001568430.1/
A4HN00
XM_001567429.2/
XP_001567479.1/
A4HK94
XM_001563411.1/
XP_001563461.1/
A4H7Y2
Tamaño del gen
(pb) 1.089 1.014 957
Tamaño de la
proteína (aa) 362 337 318
Peso molecular
proteína (Da) 39.827,90 37.851,82 35.276,84
Con la secuencia de aminoácidos de los tres candidatos (Anexo 2) se realizaron los estudios
bioinformáticos que se muestran a continuación:
7.1.1. Análisis de estructura primaria
Mediante la herramienta ProtParam se determinó el peso molecular, el punto isoeléctrico teórico
y la composición porcentual de aminoácidos para cada una de las proteínas (Tabla 13)
evidenciándose que el transportador LbTNTA presenta el mayor PI (10,07), seguido por el
LbTNTB (9,93) y por último el LbTNTC (8,94), estos los tres valores se encuentran por encima
del pH que poseen las caras interna y externa de la membrana interna de la mitocondria como se
ha reportado por Saenz Peña, C. en donde se determinó un pH de 8.5 y 5.5 respectivamente debido
al gradiente de protones (H+) generado por la fosforilación oxidativa [61].

69
Por lo tanto al encontrarse el pH del medio por debajo del punto isoeléctrico estimado para los tres
candidatos tendrían una carga eléctrica positiva lo que no permitiría una interacción entre las tres
proteínas y el NAD+ ya que este dinucleótido posee la misma carga, sin embargo, se puede sugerir
que como posibles transportadores transmembranales la mayor parte de su estructura se encuentra
integrada en la membrana y por lo tanto un pH local de esta estructura permitiría la interacción
con el dinucleótido.
En cuanto a la composición porcentual de aminoácidos, se determinó que el candidato LbTNTC
tiene mayor proporción de valina, LbTNTB de leucina y LbTNTA de glicina, los cuales le dan a
la proteína un carácter apolar, como se determinó para proteínas transmembranales ya que en el
interior de la membrana se encuentra la parte hidrofóbica de los lípidos que interactúan con este
tipo de aminoácidos y contribuyen a su sujeción. Adicionalmente se identificó que la alanina y la leucina (Tabla 13 aminoácidos en rojo) se
encuentran entre los residuos de mayor proporción en las tres proteínas, lo que concuerda con la
composición típica de las conformaciones tipo hélice alfa presentes en gran medida en proteínas
transmembranales [62], a su vez, las hélices pueden formar estructuras de canal con residuos
hidrofílicos en la cara interna del canal como la treonina, la cisteína y la arginina que se encuentran
en una alta proporción en los tres candidatos (Tabla 13 aminoácidos en azul) junto con los
hidrofóbicos como la valina y la glicina (Tabla 13 aminoácidos en verde) en la cara externa que
interactúan con los componentes apolares del interior de la bicapa. Esta tendencia en la
composición es un indicio clave que relaciona a los tres candidatos con proteínas integrales de
membrana [63].
Tabla 13. Parámetros fisicoquímicos de los candidatos como transportadores mitocondriales de
NAD+ evaluadas con la herramienta ProtParam.
En rojo los residuos más comunes en conformaciones tipo hélice α, en azul los hidrofílicos y en
verde los hidrofóbicos.
ANÁLISIS LbTNTA LbTNTB LbTNTC
Tamaño (aa) 362 337 318
pI 10,07 9,93 8,94
MW 39.827,90 37.851,82 35.276,84
Composición
porcentual de
aminoácidos
Asp 5 1.40% Met 6 1.8% Trp 3 0.9%
Cys 6 1.70% Asn 6 1.8% Cys 5 1.6%
Trp 7 1.90% Trp 7 2.1% Gln 5 1.6%
Tyr 7 1.90% Cys 8 2.4% Asn 10 3.1%
Lys 10 2.80% Pro 9 2.7% Lys 10 3.1%
Asn 12 3.30% His 9 2.7% Asp 11 3.5%
His 13 3.60% Asp 10 3.0% His 11 3.5%
Pro 14 3.90% Glu 11 3.3% Tyr 11 3.5%
Ile 15 4.10% Ile 11 3.3% Met 12 3.8%
Phe 16 4.40% Lys 11 3.3% Pro 13 4.1%
Gln 16 4.40% Tyr 12 3.6% Glu 16 5.0%
Glu 16 4.40% Phe 15 4.5% Ile 16 5.0%
Met 17 4.70% Gln 18 5.3% Phe 17 5.3%
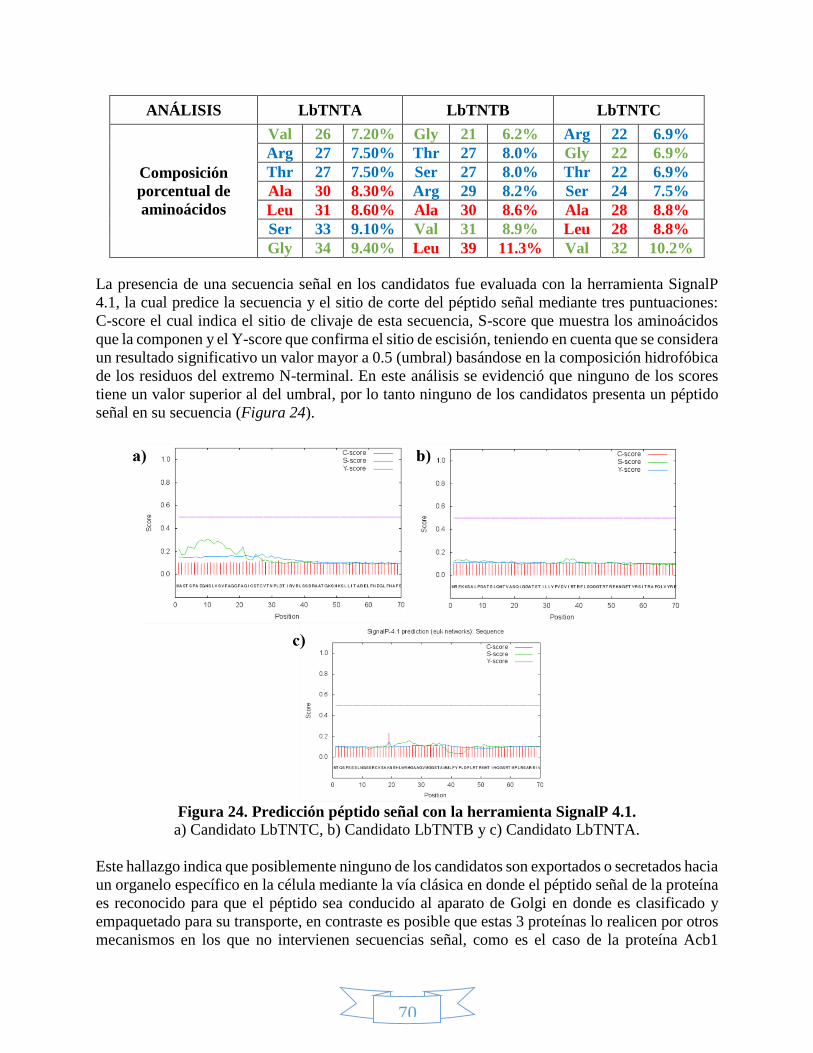
70
ANÁLISIS LbTNTA LbTNTB LbTNTC
Composición
porcentual de
aminoácidos
Val 26 7.20% Gly 21 6.2% Arg 22 6.9%
Arg 27 7.50% Thr 27 8.0% Gly 22 6.9%
Thr 27 7.50% Ser 27 8.0% Thr 22 6.9%
Ala 30 8.30% Arg 29 8.2% Ser 24 7.5%
Leu 31 8.60% Ala 30 8.6% Ala 28 8.8%
Ser 33 9.10% Val 31 8.9% Leu 28 8.8%
Gly 34 9.40% Leu 39 11.3% Val 32 10.2%
La presencia de una secuencia señal en los candidatos fue evaluada con la herramienta SignalP
4.1, la cual predice la secuencia y el sitio de corte del péptido señal mediante tres puntuaciones:
C-score el cual indica el sitio de clivaje de esta secuencia, S-score que muestra los aminoácidos
que la componen y el Y-score que confirma el sitio de escisión, teniendo en cuenta que se considera
un resultado significativo un valor mayor a 0.5 (umbral) basándose en la composición hidrofóbica
de los residuos del extremo N-terminal. En este análisis se evidenció que ninguno de los scores
tiene un valor superior al del umbral, por lo tanto ninguno de los candidatos presenta un péptido
señal en su secuencia (Figura 24).
Figura 24. Predicción péptido señal con la herramienta SignalP 4.1. a) Candidato LbTNTC, b) Candidato LbTNTB y c) Candidato LbTNTA.
Este hallazgo indica que posiblemente ninguno de los candidatos son exportados o secretados hacia
un organelo específico en la célula mediante la vía clásica en donde el péptido señal de la proteína
es reconocido para que el péptido sea conducido al aparato de Golgi en donde es clasificado y
empaquetado para su transporte, en contraste es posible que estas 3 proteínas lo realicen por otros
mecanismos en los que no intervienen secuencias señal, como es el caso de la proteína Acb1
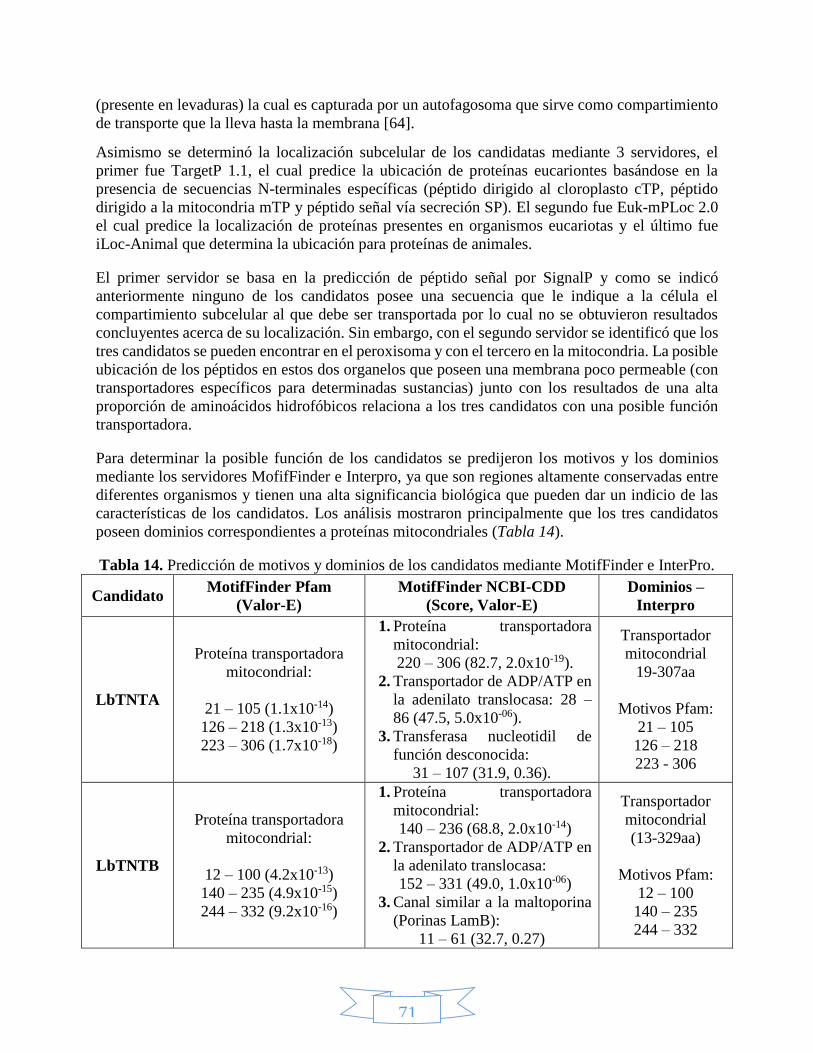
71
(presente en levaduras) la cual es capturada por un autofagosoma que sirve como compartimiento
de transporte que la lleva hasta la membrana [64].
Asimismo se determinó la localización subcelular de los candidatas mediante 3 servidores, el
primer fue TargetP 1.1, el cual predice la ubicación de proteínas eucariontes basándose en la
presencia de secuencias N-terminales específicas (péptido dirigido al cloroplasto cTP, péptido
dirigido a la mitocondria mTP y péptido señal vía secreción SP). El segundo fue Euk-mPLoc 2.0
el cual predice la localización de proteínas presentes en organismos eucariotas y el último fue
iLoc-Animal que determina la ubicación para proteínas de animales.
El primer servidor se basa en la predicción de péptido señal por SignalP y como se indicó
anteriormente ninguno de los candidatos posee una secuencia que le indique a la célula el
compartimiento subcelular al que debe ser transportada por lo cual no se obtuvieron resultados
concluyentes acerca de su localización. Sin embargo, con el segundo servidor se identificó que los
tres candidatos se pueden encontrar en el peroxisoma y con el tercero en la mitocondria. La posible
ubicación de los péptidos en estos dos organelos que poseen una membrana poco permeable (con
transportadores específicos para determinadas sustancias) junto con los resultados de una alta
proporción de aminoácidos hidrofóbicos relaciona a los tres candidatos con una posible función
transportadora.
Para determinar la posible función de los candidatos se predijeron los motivos y los dominios
mediante los servidores MofifFinder e Interpro, ya que son regiones altamente conservadas entre
diferentes organismos y tienen una alta significancia biológica que pueden dar un indicio de las
características de los candidatos. Los análisis mostraron principalmente que los tres candidatos
poseen dominios correspondientes a proteínas mitocondriales (Tabla 14).
Tabla 14. Predicción de motivos y dominios de los candidatos mediante MotifFinder e InterPro.
Candidato MotifFinder Pfam
(Valor-E)
MotifFinder NCBI-CDD
(Score, Valor-E)
Dominios –
Interpro
LbTNTA
Proteína transportadora
mitocondrial:
21 – 105 (1.1x10-14)
126 – 218 (1.3x10-13)
223 – 306 (1.7x10-18)
1. Proteína transportadora
mitocondrial:
220 – 306 (82.7, 2.0x10-19).
2. Transportador de ADP/ATP en
la adenilato translocasa: 28 –
86 (47.5, 5.0x10-06).
3. Transferasa nucleotidil de
función desconocida:
31 – 107 (31.9, 0.36).
Transportador
mitocondrial
19-307aa
Motivos Pfam:
21 – 105
126 – 218
223 - 306
LbTNTB
Proteína transportadora
mitocondrial:
12 – 100 (4.2x10-13)
140 – 235 (4.9x10-15)
244 – 332 (9.2x10-16)
1. Proteína transportadora
mitocondrial:
140 – 236 (68.8, 2.0x10-14)
2. Transportador de ADP/ATP en
la adenilato translocasa:
152 – 331 (49.0, 1.0x10-06)
3. Canal similar a la maltoporina
(Porinas LamB):
11 – 61 (32.7, 0.27)
Transportador
mitocondrial
(13-329aa)
Motivos Pfam:
12 – 100
140 – 235
244 – 332

72
Candidato MotifFinder Pfam
(Valor-E)
MotifFinder NCBI-CDD
(Score, Valor-E)
Dominios –
Interpro
LbTNTC
1. Proteína transportadora
mitocondrial:
12 – 99 (5.7x10-16)
108 – 188 (5.3x10-13)
205 – 285 (2.5x10-10)
2. Región transmembranal
sustrato-específica de
captación de cobalto:
73 – 166 (0.03)
1. Proteína transportadora
mitocondrial:
10 – 102 (81.1, 4.0x10-19)
2. Transportador de ADP/ATP en
la adenilato translocasa:
126 – 270 (50.2, 6.0x10-07)
3. Proteína transportadora
mitocondrial provisional:
105 – 260 (34.5, 0.052)
Transportador
mitocondrial
(11-284aa)
Motivos Pfam:
12 – 99
108 – 188
205 – 285
El análisis de MotifFinder mediante la evaluación de las secuencias primarias por Pfam predijo,
para los tres candidatos, la presencia de motivos relacionados con el transporte de solutos
principalmente a través de la membrana interna de la mitocondria y en otros organelos como los
peroxisomas (Tabla 14). Estos motivos que corresponden a la superfamilia de “Proteínas
Transportadoras Mitocondriales” con el código PF00153 en Pfam, presentaron valores-E
significativos (menores a 1.0x10-10) y fueron corroborados al analizar 3 secuencias de las proteínas
con NCBI – CDD, donde se predijo un dominio para los tres candidatos relacionado con esta
función y uno relacionado con un transportador de ADP/ATP en la adenilato translocasa de igual
manera para las tres secuencias (Tabla 14).
El análisis de dominios por medio del servidor InterPro predijo para los tres candidatos la presencia
del “Dominio estructural de Transportador Mitocondrial” (IPR023395 en InterPro), el cual está
compuesto por 6 hélices alfa transmembranales compactas. Adicionalmente se evidencian motivos
repetitivos que concuerdan con los predichos con MotifFinder relacionados con el transporte de
solutos principalmente a través de la membrana interna de la mitocondria y en otros organelos
como los peroxisomas con el código PF00153 en Pfam (Tabla 14).
Los resultados encontrados con respecto a la predicción de la localización y de la composición de
aminoácidos se relacionan con los motivos y dominios encontrados, los cuales ubican a los
candidatos en la familia de transportadores mitocondriales (PF00153 en Pfam) que se caracteriza
por presentar 3 secuencias repetidas en Tándem de aproximadamente 100aa cada una, las cuales
fueron halladas en las 3 secuencias con MotifFinder Pfam (Tabla 14). A su vez estas repeticiones
forman una estructura con 6 hélices alfa transmembranales y se encuentran únicamente en la
membrana de la mitocondria. Adicionalmente se ha reportado que hacen parte de esta familia el
translocador de ADP/ATP y los transportadores de fosfato, glutamato/aspartato, monocarboxilato,
carnitina, glutamato, ornitina, entre otros [65].
Las predicciones demuestran que los 3 candidatos corresponden con transportadores
mitocondriales transmembranales, sin embargo no se encontraron motivos o dominios
relacionados con el transporte de NAD+ en comparación con las secuencias repetitivas
identificadas para la familia de transportadores mitocondriales en las cuales es posible reconocer
tripletes de residuos alineados, que pueden ser simétricos (aminoácidos idénticos) o asimétricos
(aminoácidos diferentes). En donde aquellos que son muy simétricos y conservados están
relacionados con la estructura y/o mecanismo de transporte, mientras que los asimétricos con la
especificidad y/o unión del sustrato [66].

73
En estudios realizados por Montalvo, A. y Palmieri, F en 2004 se han identificado residuos de
prolina y glicina altamente conservados al interior de las secuencias de las proteínas
transportadoras mitocondriales, tanto en las alfa-hélices transmembrana pares como las impares
(tripletes 28 y 83 de prolina y 19 y 28 de glicina). Además, se ha observado que los residuos de
prolina de las hélices impares y los residuos de prolina de las hélices pares están situados por
encima de la matriz y por debajo del citosol, por lo tanto estos autores han propuesto que los
residuos de glicina y de prolina de las hélices pares y los residuos de las hélices impares actuarían
como bisagras para abrir o cerrar el transportador [66].
Debido a que se determinó que los candidatos pertenecen a la familia de transportadores
mitocondriales, los cuales forman una estructura con 6 hélices alfa transmembranales se predijo
su presencia y ubicación mediante los servidores TMHMM 2.0, Phobius y TOPCONS, el cual
reúne la información brindada por Philius, PolyPhobius y SPOCTOPUS (Tabla 15).
Para el candidato LbTNTA se predijo un mínimo de 3 hélices transmembranales y un máximo de
6 (Figura 25), de las cuales fueron coincidentes en los cinco servidores las regiones de los
aminoácidos 25 – 42 (Tabla 15 resaltadas en rojo), 81 – 96 (Tabla 15 resaltadas en azul) y 127 –
147 (Tabla 15 resaltadas en verde) y en 4 de los cinco servidores la región 190 – 208 (Tabla 15
resaltadas en amarillo).
En el caso del candidato LbTNTB se predijo mediante TMHMM que se trata de una proteína
periférica y que se encuentra en la cara externa de la membrana, a pesar de este resultado también
se obtuvo evidencia (aunque con una menor probabilidad) de la existencia de hélices
transmembranales en las posiciones 76 – 99 y 146 – 167 con este mismo servidor (Anexo 4), por
consiguiente se predijo un mínimo de 2 hélices y un máximo de 6, de las cuales fueron coincidentes
en los cinco servidores las regiones de los aminoácidos 75 – 100 (en rojo Tabla 15) y 144 – 168
(en azul Tabla 15), en 3 de los cinco servidores la región 243 – 271 (en amarillo Tabla 15) y en 2
de ellos la de 13 – 37 (en verde Tabla 15) y 206 – 228 (en morado Tabla 15).
En los reportes obtenidos es importante resaltar que mediante TMHMM se predijo que el candidato
LbTNTC es una proteína periférica y que está ubicada en la cara externa de la membrana, a pesar
de esto, en el gráfico proporcionado por el servidor se evidencia que si se presentan hélices
transmembranales en las posiciones 10 – 35, 107 – 132 y 197 – 225 por encima del valor umbral
(Anexo 5), de esta manera se predijo un mínimo de 3 hélices y un máximo de 6, de las cuales
fueron coincidentes en todos los servidores las regiones de los aminoácidos 10 – 35 (en rojo Tabla
15) y 107 – 132 (en azul Tabla 15), en 4 de ellos la región de 197 – 228 (en verde Tabla 15) y en
3 de ellos la de 68 – 91 (en amarillo Tabla 15).
A partir de este estudio se encontró, con una alta probabilidad, que las proteínas candidatas de
LbTNTA y LbTNTC presentan en su estructura un mínimo de 4 hélices transmembranales y la de
LbTNTB un mínimo de 2. Para los tres candidatos se predijeron un máximo de 6 hélices
transmembranales, lo que está en concordancia con una de las características de la familia de
transportadores mitocondriales a la cual, se predijo, que pertenecían mediante el análisis de
dominios y motivos.

74
Figura 25. Predicción de hélices transmembranales para el candidato LbTNTA.
Cada gráfico corresponde con los servidores a) TMHMM 2.0, b) Phobius y c) TOPCONS.
Tabla 15. Predicción de hélices transmembranales y topología para los candidatos por los
servidores TMHMM 2.0, Phobius y TOPCONS.
SERVIDOR LbTNTA LbTNTB LbTNTC
TMHMM
TM1: 22 – 44
TM2: 81 – 103
TM3: 125 – 147
TM4: 186 – 208
TM1: 76 – 99
TM2: 146 – 167
TM1: 10 – 35
TM2: 107 – 132
TM3: 197 – 225
Phobius
TM1: 23 – 42
TM2: 77 – 96
TM3: 127 – 147
TM1: 75 – 100
TM2: 144 – 167
TM3: 243 – 269
TM1: 12 – 32
TM2: 111 – 130
TM3: 197 – 220
Philius
TM1: 23 – 45
TM2: 77 – 99
TM3: 127 – 148
TM4: 190 – 209
TM1: 76 – 100
TM2: 147 – 168
TM1: 12 – 33
TM2: 68 – 89
TM3: 110 – 131
TM4: 197 – 221

75
SERVIDOR LbTNTA LbTNTB LbTNTC
PolyPhobius
TM1: 24 – 45
TM2: 79 – 113
TM3: 127 – 148
TM4: 187 – 209
TM5: 225 – 244
TM1: 13 – 35
TM2: 77 – 100
TM3: 146 – 167
TM4: 207 – 228
TM5: 245 – 265
TM1: 12 – 33
TM2: 72 – 91
TM3: 109 – 130
SPOCTOPUS
TM1: 25 – 46
TM2: 78 – 99
TM3: 126 – 147
TM4: 187 – 208
TM5: 228 – 249
TM6: 286 – 307
TM1: 16 – 37
TM2: 77 – 98
TM3: 147 – 168
TM4: 206 – 227
TM5: 250 – 271
TM6: 308 – 329
TM1: 14 – 35
TM2: 70 – 91
TM3: 107 – 128
TM4: 159 – 180
TM5: 207 – 228
TM6: 268 – 289
De igual manera se realizó un alineamiento múltiple mediante la herramienta PRALINE para
determinar los residuos conservados en las secuencias de cada candidato en comparación con las
secuencias de las proteínas utilizadas como plantillas con el fin de identificar los sitios de posible
interacción con el NAD+, en donde se identificaron 19 residuos altamente conservados (Figura
26 y Tabla 16).
Figura 26. Alineamiento múltiple de las secuencias de los candidatos y las plantillas
mediante PRALINE.
Los 19 residuos altamente conservados que fueron identificados en todas las secuencias se
encontraban distribuidos en 6 glicinas, 3 prolinas, 3 tirosinas, 2 argininas, 1 ácido aspártico, 1
alanina, 1 treonina, 1 lisina y 1 ácido glutámico (Tabla 16).

76
Tabla 16. Residuos conservados en las secuencias de los candidatos LbTNTA, LbTNTB y
LbTNTC y las plantillas utilizadas.
Posición residuo
alineamiento
Posición en
LbTNTA
Posición en
LbTNTB
Posición en
LbTNTC
UBICACIÓN EN
HÉLICE TM
G49 G34 G24 G22 1 – Extremo externo
P58 P43 P33 P31 1 – Extremo interno
D60 D45 D35 D33 1 – Extremo interno
R65 R50 R40 R38 1 – Extremo interno
G105 G81 G80 G72 2 – Extremo interno
Y120 Y96 Y95 G87 2 – Extremo externo
A178 A133 A153 A116 3 – Extremo externo
P188 P143 P163 P126 3 – Extremo interno
T194 T149 T169 T132 3 – Extremo interno
R195 R150 R170 T133 3 – Extremo interno
G228 G183 G202 G154 Bucle interno
G235 G190 G209 G161 4 – Extremo interno
Y254 Y208 Y227 Y180 4 – Extremo externo
K289 K235 K256 K212 5 – Extremo externo
P298 P244 P265 P221 5 – Extremo interno
G334 G278 G300 G256 6 – Extremo interno
Y339 Y283 Y305 Y261 6 – Extremo interno
G341 G285 G307 G263 6 – Extremo interno
E361 E305 E327 E283 6 – Extremo externo
De estos residuos es importante resaltar la presencia del residuo de lisina K289 conservado y el de
ácido glutámico E361 debido a que se ha demostrado que la translocasa de ADP/ATP presente en
Arabidopsis thaliana posee los residuos cargados K155, E245, E385 y K527 relacionados con el
transporte de nucleótidos de adenina [43]. Adicionalmente otros residuos cargados como el ácido
aspártico D60 y las argininas R65 y R195 podrían tener relevancia en la interacción de los tres
candidatos con el NAD+.
De acuerdo con la predicción de hélices transmembranales y al alineamiento múltiple se identificó
en los 3 péptidos de este estudio que los residuos conservados mencionados anteriormente se
encuentran distribuidos en las regiones correspondientes a hélices transmembrana (Tabla 16).
Asimismo se halló que los residuos K289 y E361 se encuentran ubicados en el extremo externo de
las hélices en los tres candidatos lo que probablemente está relacionado con la atracción del
dinucleótido hacia el interior de la estructura y por otra parte es posible que los residuos D60, R65
y R195 en el extremo interno estén relacionados con el transporte hacia la matriz mitocondrial o
el citosol. Adicionalmente, esta ubicación podría permitir la interacción entre los residuos y regular
el ingreso de sustancias por el canal como ocurre en los miembros de la familia de transportadores
mitocondriales, en donde tripletes de residuos con carga alineados desde diferentes hélices
transmembranales forman un puente salino que regula la actividad del transportador [66].
De igual manera en LbTNTA, LbTNTB y LbTNTC se identificaron los residuos conservados de
glicina G49, G105, G228, G235, G334 y G341 y de prolina P58, P188 Y P298 para los cuales se
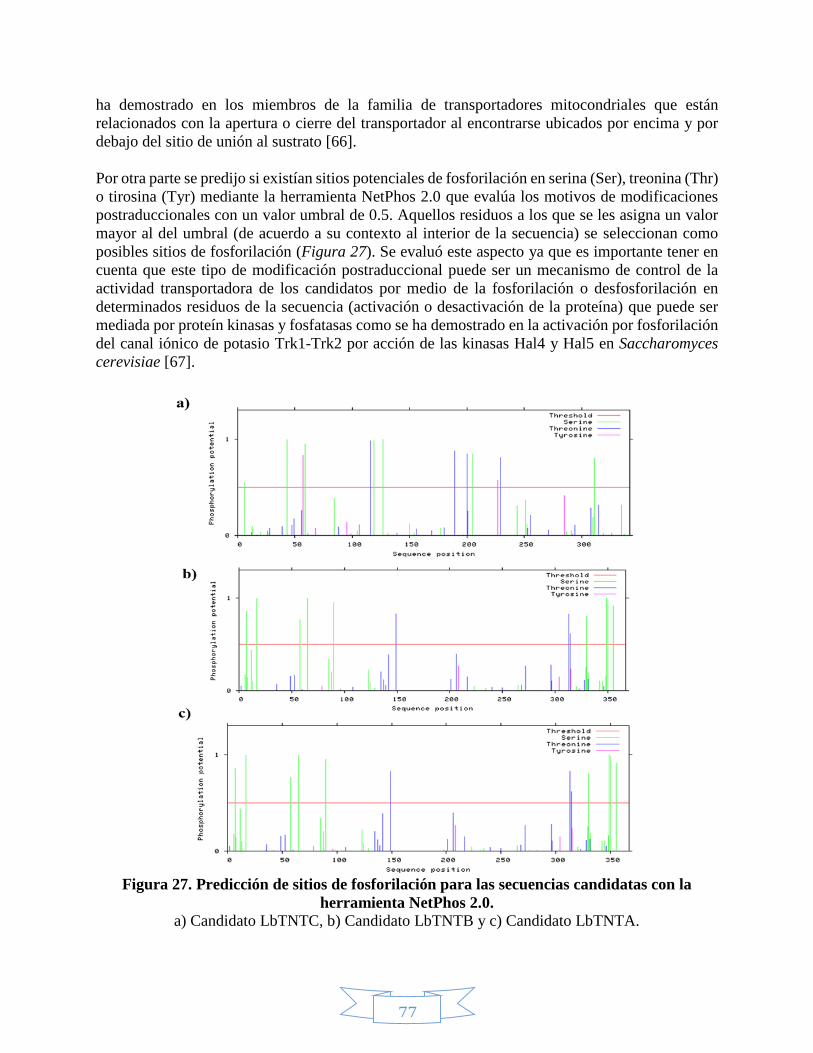
77
ha demostrado en los miembros de la familia de transportadores mitocondriales que están
relacionados con la apertura o cierre del transportador al encontrarse ubicados por encima y por
debajo del sitio de unión al sustrato [66].
Por otra parte se predijo si existían sitios potenciales de fosforilación en serina (Ser), treonina (Thr)
o tirosina (Tyr) mediante la herramienta NetPhos 2.0 que evalúa los motivos de modificaciones
postraduccionales con un valor umbral de 0.5. Aquellos residuos a los que se les asigna un valor
mayor al del umbral (de acuerdo a su contexto al interior de la secuencia) se seleccionan como
posibles sitios de fosforilación (Figura 27). Se evaluó este aspecto ya que es importante tener en
cuenta que este tipo de modificación postraduccional puede ser un mecanismo de control de la
actividad transportadora de los candidatos por medio de la fosforilación o desfosforilación en
determinados residuos de la secuencia (activación o desactivación de la proteína) que puede ser
mediada por proteín kinasas y fosfatasas como se ha demostrado en la activación por fosforilación
del canal iónico de potasio Trk1-Trk2 por acción de las kinasas Hal4 y Hal5 en Saccharomyces
cerevisiae [67].
Figura 27. Predicción de sitios de fosforilación para las secuencias candidatas con la
herramienta NetPhos 2.0. a) Candidato LbTNTC, b) Candidato LbTNTB y c) Candidato LbTNTA.

78
A partir de este análisis se determinó que los tres candidatos presentan posibles sitios de
fosforilación en serinas y treoninas, teniendo en cuenta que para LbTNTA no se predijeron
fosforilaciones en tirosinas en contraste con LbTNTB y LbTNTC, de la misma manera, no se
predijeron parar LbTNTA acetilaciones mientras que para los otros dos candidatos si (Tabla 17).
Tabla 17. Predicción de sitios de fosforilación y acetilación con las herramientas NetPhos 2.0 y
NetAcet 1.0 respectivamente para los 3 candidatos.
ANÁLISIS LbTNTA LbTNTB LbTNTC
Fosforilación
(score)
Ser = 7 (0.857), 17
(0.995), 58 (0.767), 65
(0.992), 90 (0.95), 330
(0.802), 349 (0.998),
350 (0.981) y 355
(0.911)
Ser = 6 (0.551), 43
(0.994), 59 (0.947)),
119 (0.983), 127
(0.997), 205 (0.842) y
311 (0.802)
Ser = 5 (0.851), 14
(0.524), 40 (0.995), 41
(0.995), 49 (0.962),
141 (0.82), 312
(0.985) y 315 (0.984)
Thr = 149 (0.827),
313 (0.827) y 314
(0.616)
Thr = 116 (0.982),
189 (0.872), 200
(0.847) y 229 (0.809)
Thr = 34 (0.97), 46
(0.932) y 56 (0.524)
- Tyr = 57 (0.83) y 227
(0.57) Tyr = 241 (0.978)
Acetilaciones
(score) -
Ala, Gly, Ser o Thr de
las posiciones 1-3 Serina 3 (0.511)
Se predijeron 12 posibles sitios de fosforilación para el candidato LbTNTA distribuidos en 9
serinas y 3 treoninas, para LbTNTB un total de 13 sitios conformados por 7 Serinas, 4 treoninas y
2 tirosinas y para LbTNTC un total de 12 ubicados en 8 serinas, 3 treoninas y 1 tirosina. Esto
indica sitios potenciales de fosforilación para cada candidato por lo cual es muy probable que
presenten una regulación de su actividad por este mecanismo, lo que permite sugerir la hipótesis
de la existencia de una kinasa y una fosfatasa que fosforilen y desfosforilen estas proteínas
convirtiéndolos en un posible objeto de estudio.
De igual manera se predijo para los 3 candidatos los sitios potenciales de acetilación por medio de
la herramienta NetAcet 1.0. la cual se basa en los resultados obtenidos para la N-Acetiltransferasa
A (NatA) de levadura y otros eucariontes que considera como positivos a aquellos residuos con
valores (de acuerdo a su contexto al interior de la secuencia) superiores a un umbral de 0.5 y son
seleccionados como posibles sitios de acetilación, puesto que esta modificación postraduccional
genera cambios estructurales y funcionales de las proteínas, parecidos a los producidos por la
fosforilación, por consiguiente la acetilación y desacetilación son mecanismos de regulación de la
actividad de las proteínas.
Para el candidato de LbTNTA no se predijo ninguna acetilación, mientras que para LbTNTB se
encontraron acetilaciones en alanina, glicina, serina o treonina en las posiciones 1-3 y por último
en LbTNTC se halló un posible sitio de acetilación en serina (Tabla 17). A diferencia de la
fosforilación, la predicción de esta modificación postraduccional demuestra que dos de los tres
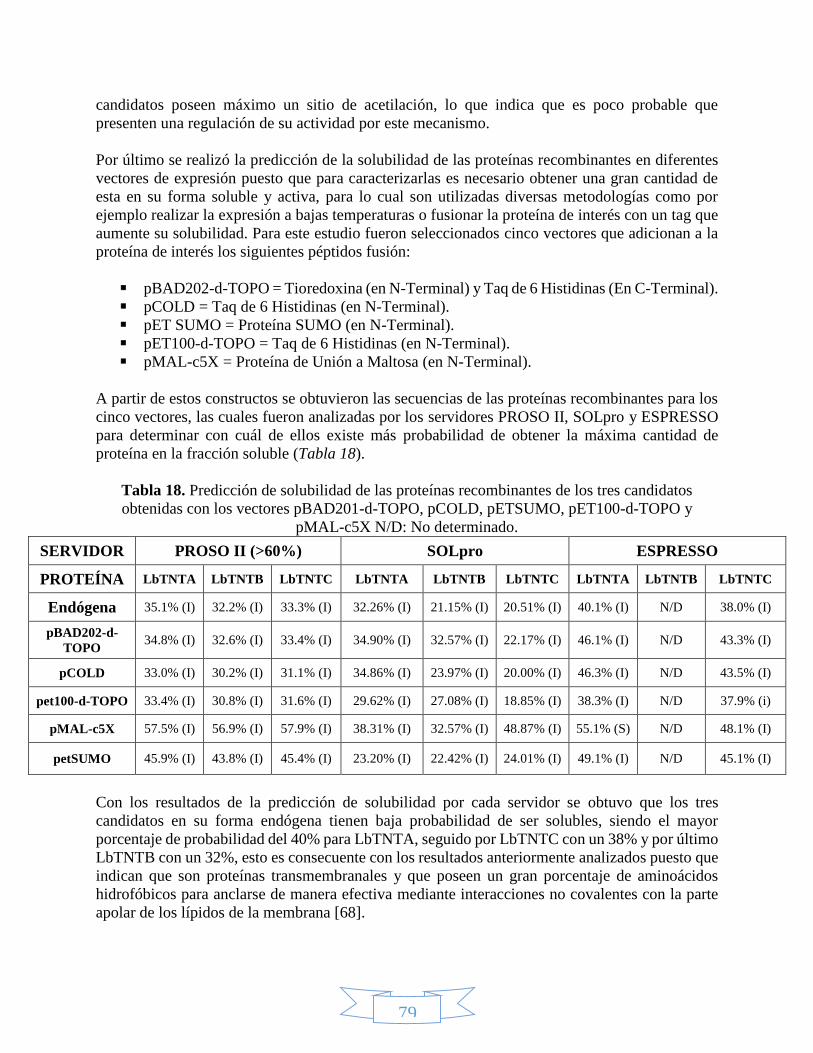
79
candidatos poseen máximo un sitio de acetilación, lo que indica que es poco probable que
presenten una regulación de su actividad por este mecanismo.
Por último se realizó la predicción de la solubilidad de las proteínas recombinantes en diferentes
vectores de expresión puesto que para caracterizarlas es necesario obtener una gran cantidad de
esta en su forma soluble y activa, para lo cual son utilizadas diversas metodologías como por
ejemplo realizar la expresión a bajas temperaturas o fusionar la proteína de interés con un tag que
aumente su solubilidad. Para este estudio fueron seleccionados cinco vectores que adicionan a la
proteína de interés los siguientes péptidos fusión:
pBAD202-d-TOPO = Tioredoxina (en N-Terminal) y Taq de 6 Histidinas (En C-Terminal).
pCOLD = Taq de 6 Histidinas (en N-Terminal).
pET SUMO = Proteína SUMO (en N-Terminal).
pET100-d-TOPO = Taq de 6 Histidinas (en N-Terminal).
pMAL-c5X = Proteína de Unión a Maltosa (en N-Terminal).
A partir de estos constructos se obtuvieron las secuencias de las proteínas recombinantes para los
cinco vectores, las cuales fueron analizadas por los servidores PROSO II, SOLpro y ESPRESSO
para determinar con cuál de ellos existe más probabilidad de obtener la máxima cantidad de
proteína en la fracción soluble (Tabla 18).
Tabla 18. Predicción de solubilidad de las proteínas recombinantes de los tres candidatos
obtenidas con los vectores pBAD201-d-TOPO, pCOLD, pETSUMO, pET100-d-TOPO y
pMAL-c5X N/D: No determinado.
SERVIDOR PROSO II (>60%) SOLpro ESPRESSO
PROTEÍNA LbTNTA LbTNTB LbTNTC LbTNTA LbTNTB LbTNTC LbTNTA LbTNTB LbTNTC
Endógena 35.1% (I) 32.2% (I) 33.3% (I) 32.26% (I) 21.15% (I) 20.51% (I) 40.1% (I) N/D 38.0% (I)
pBAD202-d-
TOPO 34.8% (I) 32.6% (I) 33.4% (I) 34.90% (I) 32.57% (I) 22.17% (I) 46.1% (I) N/D 43.3% (I)
pCOLD 33.0% (I) 30.2% (I) 31.1% (I) 34.86% (I) 23.97% (I) 20.00% (I) 46.3% (I) N/D 43.5% (I)
pet100-d-TOPO 33.4% (I) 30.8% (I) 31.6% (I) 29.62% (I) 27.08% (I) 18.85% (I) 38.3% (I) N/D 37.9% (i)
pMAL-c5X 57.5% (I) 56.9% (I) 57.9% (I) 38.31% (I) 32.57% (I) 48.87% (I) 55.1% (S) N/D 48.1% (I)
petSUMO 45.9% (I) 43.8% (I) 45.4% (I) 23.20% (I) 22.42% (I) 24.01% (I) 49.1% (I) N/D 45.1% (I)
Con los resultados de la predicción de solubilidad por cada servidor se obtuvo que los tres
candidatos en su forma endógena tienen baja probabilidad de ser solubles, siendo el mayor
porcentaje de probabilidad del 40% para LbTNTA, seguido por LbTNTC con un 38% y por último
LbTNTB con un 32%, esto es consecuente con los resultados anteriormente analizados puesto que
indican que son proteínas transmembranales y que poseen un gran porcentaje de aminoácidos
hidrofóbicos para anclarse de manera efectiva mediante interacciones no covalentes con la parte
apolar de los lípidos de la membrana [68].

80
De igual manera se identificó, para los tres candidatos, que las proteínas recombinantes obtenidas
con el vector pMAL-c5X tendrían la mayor solubilidad en comparación con los demás vectores,
debido a que este adiciona como péptido fusión la proteína de unión a maltosa, la cual se expresa
en el citoplasma y presenta un alto porcentaje de aminoácidos polares confiriéndole a los
candidatos mayor solubilidad (Tabla 18), sin embargo es posible que esta misma cantidad de
aminoácidos pueda ocasionar algún efecto de interferencia en la actividad de las proteínas.
7.I.2. Predicción de estructura secundaria
La determinación experimental de la estructura secundaria y terciaria de proteínas de membrana
resulta compleja, desde la obtención de las muestras hasta su análisis lo que da como resultado
modelos de baja resolución tanto por cristalografía de rayos X como por resonancia magnética
nuclear (NMR), sin embargo en las últimas décadas, estas dos técnicas se han desarrollado y han
surgido nuevas metodologías y consorcios dedicados exclusivamente al estudio de estos péptidos.
Con esta información experimental se construyeron bases de datos a partir de las cuales se generan
modelos por homología, asimismo se han construido escalas de hidrofobicidad para la predicción
de segmentos transmembranales e identificación de segmentos con residuos cargados
positivamente para la determinación de la topología. Sin embargo son necesarios nuevos
predictores que mejoren la resolución de los resultados como la discriminación de fragmentos
transmembranales cortos, hélices en la superficie de la membrana [69]. La estructura secundaria
de los tres candidatos a proteínas transportadoras de NAD+ se predijo mediante el servidor
PHYRE2 como se muestra a continuación:
Candidato LbTNTA
Se predijo la estructura secundaria del candidato LbTNTA mediante el servidor PHYRE2, a partir
del cual se identificó que un 71% de la secuencia tiene una estructura de hélice alfa, 0% en
conformación hoja plegada beta y un 25% no corresponde con ninguno de estos dos plegamientos;
asimismo se encontró que el 48% de la secuencia corresponde a hélices alfa transmembranales
(Figura 28).
Figura 28. Predicción de la estructura secundaria para el candidato LbTNTA con el
servidor PHYRE2. Tomado de L. Kelley y otros (2015) [58]

81
De acuerdo con esto, se predijeron un total de 7 hélices alfa transmembranales distribuidas en las
regiones 27 – 52, 76 – 99, 127 – 153, 185 – 207, 228 – 253, 280 – 307 y 332 – 349 (Figura 29)
que a su vez corresponden con las predichas por los servidores utilizados para identificar hélices
transmembranales anteriormente mencionadas, lo que permitió comprobar la presencia de un
mínimo de 4 hélices transmembranales en las regiones 25 – 52, 81 – 96, 127 – 153 y 185 – 208.
Asimismo (al igual que los servidores PolyPhobius y SPOCTOPUS, Tabla 15) se predijeron 2
hélices transmembranales adicionales en las regiones 228 – 253 y 280 – 307 y una tercera
(únicamente predicha por PHYRE2) en la región 332 – 349.
Figura 29. Topología predicha para el candidato LbTNTA con el servidor PHYRE2.
Por otra parte, la predicción de la topología del candidato mostró al extremo N-Terminal en el
espacio extracelular (o intermembranal) al igual que las regiones 100 – 126, 208 – 227 y 308 - 331
y al extremo C-Terminal en el citoplasma (o matriz mitocondrial) al igual que las regiones 53 –
75, 154 – 184 y 254 – 279 (Figura 29).
Los resultados obtenidos a partir de los diferentes servidores demuestran que el extremo N-
Terminal en todos los análisis se encuentra ubicado en los aminoácidos 1 – 23 aproximadamente
en la cara externa de la membrana (espacio intermembranal o extracelular), por el contrario, no se
evidenció un consenso de la ubicación del extremo C-Terminal (Tabla 19).
Tabla 19. Determinación de la topología del candidato LbTNTA por los servidores TMHMM,
Phobius y TOPCONS.
SERVIDOR
TMHMM Phobius Philius PolyPhobius
Afuera: 1 – 21
TM1: 22 – 44
Adentro: 45 – 80
TM2: 81 – 103
Afuera: 104 – 124
TM3: 125 – 147
Adentro: 148 – 185
TM4: 186 – 208
Afuera: 209 – 362
Afuera: 1 – 22
TM1: 23 – 42
Adentro: 43 – 76
TM2: 77 – 96
Afuera: 97 – 126
TM3: 127 – 147
Adentro: 148 – 362
Afuera: 1 – 22
TM1: 23 – 45
Adentro: 46 – 76
TM2: 77 – 99
Afuera: 100 – 126
TM3: 127 – 148
Adentro: 149 – 189
TM4: 190 – 209
Afuera: 210 – 362
Afuera: 1 – 23
TM1: 24 – 45
Adentro: 46 – 78
TM2: 79 – 113
Afuera: 114 – 126
TM3: 127 – 148
Adentro: 149 – 186
TM4: 187 – 209
Afuera: 210 – 224
TM5: 225 – 244
Adentro: 245 – 362

82
Para los segmentos entre hélices de los primeros 200aa se obtuvo la misma predicción en todos los
servidores como se muestra a continuación:
I. Segmento 45 – 76 aproximadamente: Adentro (Matriz mitocondrial o citosol).
II. Segmento 104 – 124 aproximadamente: Afuera (Espacio intermembranal o extracelular).
III. Segmento 149 – 185 aproximadamente: Adentro (Matriz mitocondrial o citosol).
Para los segmentos del residuo 200 en adelante existe una concordancia entre la predicción
mediante PHYRUS2 y PolyPhobius como sigue:
I. Segmento 210 – 224 aproximadamente: Afuera (Espacio intermembranal o extracelular).
II. Segmento 245 – 279 aproximadamente: Adentro (Matriz mitocondrial o citosol).
III. Segmento 308 – 331 aproximadamente: Afuera (Espacio intermembranal o extraceluar).
Únicamente con PHYRE2.
El modelo construido mediante PHYRE2 es un consenso de las predicciones realizadas por los
servidores utilizados en la predicción de hélices transmembranales, por lo tanto los resultados
hallados permiten inferir, con una alta probabilidad, que el candidato LbTNTA presenta 6 hélices
alfa transmembranales y que posee su extremo N-Terminal en la parte externa de la membrana
(espacio intermembranal), a partir del cual se encuentran alternados, en ambos espacios alrededor
de la membrana, los segmentos entre hélices o bucles. Las predicciones obtenidas a partir de este
servidor están de acuerdo con la clasificación del candidato en la familia de los transportadores
mitocondriales, cuyos miembros presentan 6 hélices transmembrana.
Candidato LbTNTB
La estructura secundaria del candidato LbTNTB se predijo de igual manera que la de LbTNTA
mediante el servidor PHYRE2, con el cual se identificó que un 67% de la secuencia tiene una
estructura de hélice alfa, 0% en conformación hoja plegada beta y un 27% no corresponde con
ninguno de estos dos plegamientos relacionados posiblemente con los bucles entre hélices;
asimismo se identificó que el 46% de la secuencia corresponde con hélices alfa transmembranales
(Anexo 6a).
De igual manera se predijeron un total de 6 hélices alfa transmembranales distribuidas en las
regiones 17 – 43, 75 – 98, 148 – 173, 204 – 226, 249 – 274 y 302 – 329 (Figura 30) que a su vez
corresponden con las predichas por los servidores utilizados para identificar hélices
transmembranales anteriormente mencionado, lo que permitió comprobar la presencia de un
mínimo de 3 hélices transmembranales en las regiones 75 – 100, 144 – 173 y 243 – 274. Asimismo
se predijeron 2 hélices transmembranales adicionales (al igual que los servidores PolyPhobius y
SPOCTOPUS, Tabla 15) en las regiones 13 – 43 y 204 – 228 y una tercera (predicha por PHYRE2
y SPOCTOPUS) en la región 302 – 329.

83
Figura 30. Topología predicha para el candidato LbTNTB con el servidor PHYRE2.
Por otra parte la predicción de la topología del candidato LbTNTB mostró al extremo N-Terminal
en el espacio extracelular (o intermembranal) al igual que al extremo C-Terminal como también a
las regiones 99 – 147 y 227 – 248, asimismo se encontró que presentaron una orientación en el
citoplasma (o matriz mitocondrial) las regiones 44 – 74, 174 – 203 y 275 – 301 (Figura 30).
Los resultados obtenidos a partir de los diferentes servidores demostraron que el extremo N-
Terminal en todos los análisis se encontraba en la cara externa de la membrana (espacio
intermembranal o extracelular) a excepción del servidor Philius. Por el contrario, no se evidenció
un consenso de la ubicación del extremo C-Terminal como consecuencia del número de hélices
transmembranales predichas, sin embargo, 4 de los 6 servidores (incluido PHYRE2) lo ubican en
la cara externa (Tabla 20).
Tabla 20. Determinación de la topología del candidato LbTNTB por los servidores TMHMM,
Phobius y TOPCONS.
SERVIDOR
TMHMM Phobius Philius PolyPhobius
Afuera: 1 – 75
TM1: 76 – 99
Afuera: 100 – 145
TM2: 146 – 167
Afuera: 168 – 337
Afuera: 1 – 74
TM1: 75 – 100
Adentro: 101 – 143
TM2: 144 – 167
Afuera: 168 – 242
TM3: 243 – 269
Afuera: 270 – 337
Adentro: 1 – 75
TM1: 76 – 100
Afuera: 101 – 146
TM2: 147 – 168
Adentro: 169 – 337
Afuera: 1 – 12
TM1: 13 – 35
Adentro: 36 – 76
TM2: 77 – 100
Afuera: 101 – 145
TM3: 146 – 167
Adentro: 168 – 206
TM4: 207 – 228
Afuera: 229 – 246
TM5: 245 – 265
Adentro: 266 – 337
De igual manera se identificó que algunos segmentos entre hélices presentan la misma predicción
en la mayoría de los servidores lo que traduce en una alta probabilidad de existencia en la estructura
como se muestra a continuación:
I. Segmento 36 – 76 aproximadamente: Adentro (Matriz mitocondrial o citosol).
II. Segmento 98 – 148 aproximadamente: Afuera (Espacio intermembranal o extracelular).

84
Adicionalmente hubo una concordancia aunque con menor frecuencia (por PolyPhobius y
PHYRE2) para la ubicación de los siguientes segmentos:
I. Segmento 168 – 204 aproximadamente: Adentro (Matriz mitocondrial o citosol).
II. Segmento 226 – 249 aproximadamente: Afuera (Espacio intermembranal o extracelular).
III. Segmento 274 – 302 aproximadamente: Adentro (Matriz mitocondrial o citosol).
Únicamente con PHYRE2.
El modelo construido mediante PHYRE2 es un consenso de las predicciones realizadas por los
servidores utilizados en la predicción de hélices transmembranales, por lo tanto los resultados
hallados permiten inferir, con una alta probabilidad, que el candidato LbTNTB presenta de 5 a 6
hélices alfa transmembranales y que posee su extremo N-Terminal en la parte externa de la
membrana (espacio intermembranal o extracelular), a partir del cual se encuentran alternados, en
ambos espacios alrededor de la membrana, los segmentos entre hélices o bucles. Las predicciones
obtenidas a partir de este servidor están de acuerdo con la clasificación del candidato en la familia
de los transportadores mitocondriales, cuyos miembros presentan 6 hélices transmembrana.
Candidato LbTNTC
La estructura secundaria de LbTNTC se predijo mediante el servidor PHYRE2 al igual que para
los otros dos candidatos, con el cual se identificó que un 76% de la secuencia tiene una estructura
de hélice alfa, 0% en conformación hoja plegada beta y un 17% no corresponde con ninguno de
estos dos plegamientos relacionados posiblemente con los bucles entre hélices; asimismo se
identificó que el 48% de la secuencia corresponde con hélices alfa transmembranales (Anexo 6b).
De igual manera se predijeron un total de 6 hélices alfa transmembranales distribuidas en las
regiones 16 – 41, 67 – 90, 110 – 134, 156 – 179, 205 – 230 y 258 – 284 (Figura 31) que a su vez
corresponden con las predichas por los servidores utilizados para identificar hélices
transmembranales como se mostró anteriormente, lo que permitió comprobar por consenso la
presencia de un mínimo de 3 hélices transmembranales en las regiones 10 – 41, 107 – 134 y 197
– 230 al compararse estos resultados con los obtenidos en el análisis de estructura primaria en el
numeral 6.1.1. Asimismo se predijo 1 hélice transmembranal adicional (al igual que con los
servidores Philius, PolyPhobius y SPOCTOPUS, Tabla 15) en la región 67 – 91 y dos más
(predicha por PHYRE2 y SPOCTOPUS) en las regiones 148 – 180 y 258 – 289.
Figura 31. Topología predicha para el candidato LbTNTC con el servidor PHYRE2.

85
Por otra parte, se predijo la topología del candidato, con el extremo N-Terminal en el espacio
extracelular (o intermembranal) al igual que el extremo C-Terminal y las regiones 91 – 109 y 180
– 204, asimismo una ubicación en el citoplasma (o matriz mitocondrial) para las regiones 42 – 66,
135 – 155 y 231 – 257 (Figura 31).
Los resultados obtenidos a partir de los diferentes servidores (Tabla 21) demuestran que el extremo
N-Terminal en todos los análisis se encuentra en la cara externa de la membrana (o espacio
intermembranal). Por el contrario, no se evidenció un consenso de la ubicación del extremo C-
Terminal como consecuencia del número de hélices transmembranales predichas, sin embargo, 3
de los 6 servidores (incluido PHYRE2) lo ubican en la cara externa. De igual manera se identificó
que algunos segmentos entre hélices presentan la misma predicción en la mayoría de los servidores
lo que traduce en una alta probabilidad de existencia en la estructura como se muestra a
continuación:
I. Segmento 33 – 71 aproximadamente: Adentro (Matriz mitocondrial o citosol).
II. Segmento 90 – 110 aproximadamente: Afuera (Espacio intermembranal o extracelular).
Adicionalmente hubo una concordancia aunque con menor frecuencia para la ubicación de los
siguientes segmentos:
I. Segmento 129 – 158 aproximadamente: Adentro (Espacio intermembranal o extracelular).
II. Segmento 169 – 206 aproximadamente: Afuera (Matriz mitocondrial o citosol).
III. Segmento 229 – 267 aproximadamente: Adentro (Espacio intermembranal o extracelular).
Tabla 21. Determinación de la topología del candidato LbTNTC por los servidores TMHMM,
Phobius y TOPCONS.
SERVIDOR
TMHMM Phobius Philius PolyPhobius
Afuera: 1 – 9
TM1: 10 – 35
Adentro: 36 – 106
TM2: 107 – 132
Afuera: 133 – 196
TM3: 197 – 225
Adentro: 226 – 318
Afuera: 1 – 11
TM1: 12 – 32
Adentro: 33 – 110
TM2: 111 – 130
Afuera: 131 – 196
TM3: 197 – 220
Adentro: 221 – 318
Afuera: 1 – 11
TM1: 12 – 33
Adentro: 34 – 67
TM2: 68 – 89
Afuera: 90 – 109
TM3: 110 – 131
Adentro: 132 – 196
TM4: 197 – 221
Afuera: 222 – 318
Afuera: 1 – 11
TM1: 12 – 33
Adentro: 34 – 71
TM2: 72 – 91
Afuera: 92 – 108
TM3: 109 – 130
Adentro: 131 – 318
El modelo construido mediante PHYRE2 es un consenso de las predicciones realizadas por los
servidores utilizados en la predicción de hélices transmembranales, por lo tanto los resultados
hallados permiten inferir, con una alta probabilidad, que el candidato LbTNTC presenta de 5 a 6
hélices alfa transmembranales y que posee su extremo N-Terminal en la parte externa de la
membrana (espacio intermembranal o extracelular), a partir del cual se encuentran alternados, en
ambos espacios alrededor de la membrana, los segmentos entre hélices o bucles. Las predicciones

86
obtenidas a partir de este servidor están de acuerdo con la clasificación del candidato en la familia
de los transportadores mitocondriales, cuyos miembros presentan 6 hélices transmembrana.
7.I.3. Predicción de la estructura terciaria
El modelado por homología es un método que tiene en cuenta la similitud que existe entre la
secuencia por modelar y las que son utilizadas como plantillas (cuentan con una estructura 3D
elucidada experimentalmente) por medio de alineamientos, puesto que algunos de los segmentos
transmembranales generalmente son muy conservados entre diferentes especies. Estos resultados
son valorados mediante matrices que se componen de análisis de la estructura secundaria, la
mutabilidad, el emparejamiento de los residuos y su ambiente, estados de mínima energía, entre
otros. Asimismo se incluyen restricciones experimentales (obtenidas por NMR, cristalografía de
rayos X u otras técnicas), lo que contribuye a obtener modelos de mayor calidad, sin embargo,
estos datos son difíciles de obtener.
Sin embargo, la dificultad de realizar un modelado por este método radica en encontrar una
plantilla (o plantillas) que presente mínimo un 30% de identidad con la secuencia a modelar, para
realizar los alineamientos y así obtener un modelo que tenga una calidad aceptable.
Adicionalmente se ha observado el problema de que las regiones no transmembranales o “bucles”
son poco conservados, por lo tanto su modelado es más complejo [69]. A pesar de esto es uno de
los métodos más utilizado y que presenta buenos resultados en comparación con la evidencia
experimental.
EL modelado por homología se llevó a cabo mediante el servidor Swiss-Model, que utiliza el valor
QMEAN4 para evaluar los modelos generados mediante una puntuación de fiabilidad para todo el
modelo que se puede utilizar con el fin de comparar y clasificar modelos alternativos de la misma
secuencia diana [70]. Es una combinación de 4 descriptores estructurales que utilizan potenciales
estadísticos:
1. La geometría local, la cual es analizada por el ángulo de torsión formado por 3 aminoácidos
consecutivos.
2. Dos potenciales de interacción dependientes de la distancia se utilizan para evaluar las
interacciones de largo alcance: El primero basado únicamente en los átomos de carbono β
y el segundo en todos los átomos de la macromolécula.
3. Un potencial de solvatación investiga el estado de enterramiento de los residuos al interior
de la estructura 3D.
La estimación de calidad del modelo mediante el valor QMEAN4 está relacionada con el valor Z
el cual representa una medida de la calidad absoluta de un modelo y proporciona una estimación
del grado de natividad de las características estructurales observadas en un modelo y la
probabilidad de que este sea de calidad comparable a las estructuras experimentales. De esta
manera, mientras más negativo sea el valor de la puntuación Z QMEAN, menor calidad tendrá el
modelo generado.
En comparación I-TASSER utiliza el valor del RMSD (root-mean-square deviation) para analizar
la calidad del modelado mediante la evaluación de la distancia entre el modelo predicho y las

87
estructuras nativas utilizadas como plantillas, obteniéndose que a mayor valor, la calidad de la
predicción es menor. A su vez, se utiliza el valor C para estimar la calidad de los modelos predichos
por esta plataforma mediante la significancia (valor Z) de los alineamientos hechos con el
programa LOMETS y la convergencia (densidad de clusters o conformaciones estructurales
similares) de las simulaciones de I-TASSER. Los puntajes del valor C se encuentran entre -5 y 2,
donde un valor alto indica mayor calidad en el modelo. Adicionalmente esta plataforma genera un
factor B del modelo predicho, el cual indica la movilidad de los residuos en el péptido y se obtiene
de las proteínas utilizadas como plantillas en el Threading y los perfiles de las secuencias en bases
de datos con un valor umbral de 0, en donde los valores por debajo representan una estructura de
hélice y por encima de cola o hebra para los residuos. La predicción de la estructura terciara de los
tres candidatos se realizó como se muestra a continuación:
Candidato LbTNTA
Para realizar el modelado por homología del candidato TNTA se buscó en el servidor InterPro las
secuencias que tuvieran la misma arquitectura del “Dominio estructural de Transportador
Mitocondrial” (IPR023395 en InterPro), obteniéndose un total de 35.690 proteínas, de las cuales
2 secuencias tienen anotadas una estructura en la base de datos de PDBe (1okc y 2c3e), con las
que se realizó el modelamiento por homología mediante el servidor Swiss-Model, el cual no
produjo un modelo óptimo, obteniendo un porcentaje de identidad del 25,86% para ambas
predicciones y un valor QMEAN4 de -8,24 para el modelo generado con la secuencia 1okc y de -
9,38 para el generado con 2c3e (Tabla 22).
Tabla 22. Predicción de estructura terciaria por homología mediante el servidor Swiss-Model.
Descripción Código PDBe %
Identidad Rango Cobertura QMEAN4
Mitochondrial ADP/ATP
carrier in complex with
carboxyatractyloside
1okc 25,86% 28 – 211 48% -8,04
The bovine mitochondrial
ADP-ATP carrier 2c3e 25,86% 28 – 211 48% -9,38
Debido a que se obtuvieron valores Z QMEAN muy negativos para los dos modelos generados
por homología se decidió rechazarlos y utilizar la plataforma I-TASSER [59], mediante el cual se
obtuvo la gráfica del factor B para el candidato LbTNTA, en donde se especificaron los residuos
que se encuentran conformando un total de 6 hélices α unidas entre sí por varios bucles o colas,
los cuales concuerdan con los predichos con PHYRE2 en las regiones 27 – 52, 76 – 99, 127 – 153,
185 – 207, 228 – 253, 280 – 307 y 332 – 349 (Figura 29 y Figura 32).
Figura 32. Factor B normalizado para la secuencia completa del candidato LbTNTA por I-
TASSER.

88
Los 5 modelos obtenidos mediante I-TASSER presentaron valores C negativo (Tabla 23), lo que
indica una baja calidad de los modelos y por consiguiente baja similitud entre las estructuras de
las plantillas y los modelos generados. Igualmente se obtuvo un RMSD con un rango grande (de
4.2 a 13.4 Å) para el modelo con mayor valor C por lo cual se decidió descartar este resultado ya
que no genera confiabilidad el modelado.
Tabla 23. Valores C obtenidos para los modelos construidos por Threading para la secuencia
completa del candidato LbTNTA por I-TASSER.
MODELO 1 2 3 4 5
VALOR C -0.99 -1.46 -1.60 -1.97 -2.72
RMSD 8.8 ± 4.6 Å - - - -
De acuerdo con la magnitud de los valores C y de RMSD se determinó que ninguno de los modelos
predichos por I-TASSER para la secuencia completa del candidato LbTNTA eran lo
suficientemente buenos para generar la estructura tridimensional, por lo tanto se utilizó la
arquitectura del dominio IPR023395 en InterPro predicha (numeral 7.1.1), ubicada entre los
aminoácidos 19 – 307 para generar un modelo de estructura terciaria de la secuencia sin los
extremos N y C terminal que no están contenidos en dicho dominio (1-18 para el N terminal y 308
– 362 para el C terminal), obteniéndose un péptido de 289 aminoácidos, el cual nuevamente se
evaluó con el servidor I-TASSER dando como resultado un único modelo con un valor C de 1.50
y un RMSD de 3.1 ± 2.2 Å, el cual tiene una mayor calidad al ser comparados con los generados
a partir de la secuencia completa y en donde se evidenció la presencia de 6 segmentos con
conformación tipo hélice α que por su longitud, ubicación y de acuerdo con los análisis de
estructura primaria y secundaria anteriormente mostrados (numerales 7.1.1 y 7.I.2) corresponden,
con una alta probabilidad, con hélices transmembranales (Figura 33 hélices azul oscura, azul clara,
verde oscura, verde clara, amarilla y roja para las hélices 1, 2, 3, 4, 5 y 6 respectivamente).
Figura 33. Modelo de estructura terciaria para el candidato LbTNTA mediante I-TASSER.
a) Vista superior. b) vista frontal y c) vista inferior.
De la misma manera se obtuvo el gráfico del factor B para el modelo predictivo (Figura 34) en
donde se identificaron 6 zonas que se encuentran en conformación tipo hélice α unidas entre ellas
por bucles o colas los cuales concuerdan con los resultados predichos con PHYRE2 en las regiones
27 – 52, 76 – 99, 127 – 153, 185 – 207, 228 – 253 y la región 280 – 307 con en el extremo C
terminal tipo hebra.

89
Figura 34. Factor B normalizado para la secuencia incompleta del candidato LbTNTA por
I-TASSER.
La mayor similitud del modelo generado con las estructuras en 3D elucidadas experimentalmente
de la base de datos PDB la realizó el servidor I-TASSER mediante el valor TM el cual mide la
similitud de pliegues global entre las estructuras y es menos sensible a las variaciones estructurales
locales, con valores umbral entre 0 y 1, siendo 1 el valor para una similitud del 100%. Mediante
este servidor se identificó que el modelo generado tiene un TM del 0.958 con la estructura de la
proteína “Mitochondrial ADP/ATP carrier in complex with carboxyatractyloside” 1okc en PDB
de la especie Bos taurus, como se demuestró en la superposición de ambas estructuras terciarias
con un RMSD de 0.584 angstrom entre 194 parejas de átomos (Figura 35).
Figura 35. Alineamientos estructurales entre el candidato LbTNTA con el transportador
mitocondrial de ADP/ATP mediante el programa UCSF Chimera 1.10.2.
a) Vista frontal. b) vista superior y c) vista frontal invertida. Candidato LbTNTA en azul y el
Transportador mitocondrial de ADP/ATP (1okc en PDB en rojo).
La similitud entre las estructuras tridimensionales de estas dos proteínas indican una posible
homología funcional, por lo tanto se sugiere que es posible que el candidato LbTNTA posea la
capacidad de transportar las especies ADP/ATP. Es importante contrastar estos resultados con los
obtenidos en estudios en organismos del género Clamidia como C. tracomatis en donde se
comprobó experimentalmente que la translocasa de ATP/ADP (CtNPT1) posee la capacidad de
transportar además de ADP/ATP el dinucleótido NAD+, inclusive con mayor afinidad con relación
a los mononucleótidos [13]. Asimismo los resultados permiten proponer que esta proteínas se

90
encuentra relacionada con una posible función como transportador de NAD+ en Leishmania
braziliensis.
Por último se analizó la posible función de acuerdo a los número de la comisión enzimática
predichos por I-TASSER producto del alineamiento del modelo generado con las estructuras de
proteínas elucidadas experimentalmente y de función conocida, de esta manera se tuvieron en
cuenta los alineamientos con mayor valor C, siendo el de mayor magnitud (0.238) con la Xantina
Deshidrogenasa de Rhodobacter capsulatus, seguido por el de la ATPasa de intercambio H+/K+
(0.233) de Sus scrofa.
La Xantina Deshidrogenasa tiene un número de la comisión enzimática (E.C) 1.17.1.4 y la enzima
ATPasa de intercambio H+/K+ tiene un E.C = 3.6.3.10, los cuales corresponden con la siguiente
clasificación:
1 = Oxidoreductasas.
1.17 = Actúan sobre grupos CH o CH2.
1.17.1 = Con NAD+ o NADP+ como
aceptores.
1.17.1.4 = Xantina Deshidrogenasa.
3 = Hidrolasas.
3.6 = Actúan sobre anhídridos de ácidos
carboxílicos.
3.6.3 = Catalizan el movimiento transmembrana
de sustancias.
3.6.3.10 = ATPasa de intercambio H+/K+.
Es importante resaltar que la enzima con la cual se obtuvo el mayor puntaje de alineamiento para
predecir la posible función de LbTNTA usa como cofactor las especies NAD+ o NADP+, lo que
es un indicio de que el candidato interactúa con este dinucleótido específicamente. Asimismo el
alineamiento con la ATPasa de intercambio H+/K+ permitió corroborar la posible interacción del
candidato con nucleótidos de adenina.
Candidato LbTNTB
Para este candidato se decidió utilizar la plataforma I-TASSER directamente para la realizar la
predicción de la estructura terciaria, a partir de la cual se obtuvo la gráfica del factor B (Figura
36), en donde se especifican los residuos que se encuentran conformando un total de 6 hélices α
unidas entre sí por varios bucles o colas, los cuales concuerdan aproximadamente con los predichos
con PHYRE2 en las regiones 17 – 43, 75 – 98, 148 – 173, 204 – 226, 249 – 274 y 302 – 329
(Figura 30).
Figura 36. Factor B normalizado para la secuencia completa del candidato LbTNTB por I-
TASSER.

91
Los modelos 2, 3, 4 y 5 obtenidos mediante I-TASSER para la secuencia completa del candidato
LbTNTB presentaron valores C negativos, lo que indica una baja calidad de los modelos y por
consiguiente baja similitud entre las estructuras de las plantillas y los modelos generados, no
obstante, el mejor modelo construido (número 1) obtuvo una puntuación C de 0.02 y un RMSD
con un rango entre 2.5 y 10.3 Å, a pesar de esto se modeló la estructura terciaria a partir de la
secuencia de 317 aminoácidos relacionada con la arquitectura del dominio IPR023395 en InterPro
predicha para este candidato (numeral 7.1.1), ubicada entre los aminoácidos 13 – 329 para obtener
un mejor modelado, sin embargo ninguno obtuvo mejor puntuación que el obtenido a partir de la
secuencia completa, por lo tanto se seleccionó este como el modelo óptimo (Tabla 24).
Tabla 24. Valores C obtenidos para los modelos construidos por Threading para la secuencia
completa e incompleta del candidato LbTNTB por I-TASSER.
SECUENCIA MODELO VALOR C RMSD
Completa
(337aa)
1 0.02 6.4 ± 3.9 Å
2 -0.79 -
3 -0.58 -
4 -1.61 -
5 -1.55 -
Incompleta
(317aa)
1 0.00 6.3 ± 3.9 Å
2 -0.35 -
3 -0.65 -
4 -1.78 -
5 -4.16 -
En el modelo predictivo número 1 para la secuencia completa se evidenció la presencia de 6
segmentos con conformación tipo hélice α que por su longitud, ubicación y de acuerdo con los
análisis de estructura primaria y secundaria anteriormente mostrados (numerales 7.1.1 y 7.I.2)
corresponden, con una alta probabilidad, con hélices transmembranales (Figura 37b hélices azul
oscura, azul clara, verde oscura, verde clara, amarilla y roja para las hélices 1, 2, 3, 4, 5 y 6
respectivamente), adicionalmente se predijeron 3 hélices de menor tamaño ubicadas hacia el lado
opuesto de los extremos C y N-terminal (naranja, verde claro y azul claro Figura 37c) y que se
encuentran intercaladas entre las hélices 1 – 2, en el caso de la azul clara, entre la 3 – 4 para la
verde clara y entre la 5 – 6 para la naranja. Adicionalmente se identificó una estructura en forma
de cilindro o tubo característica para proteínas transportadoras (Figura 37a y Figura 37c).
Figura 37. Modelo de estructura terciaria para el candidato LbTNTB mediante I-TASSER.
a) Vista superior. b) vista frontal y c) vista inferior.

92
La mayor similitud del modelo generado con las estructuras en 3D elucidadas experimentalmente
de la base de datos PDB fue con la estructura de la proteína “The bovine mitochondrial ADP/ATP
carrier” de la especie Bos taurus con un Tm del 0.847 y código de acceso 2c3e en PDB, como se
demuestra en la superposición de ambas estructuras terciarias con un RMSD de 0.798 angstrom
entre 221 parejas de átomos (Figura 38).
Figura 38. Alineamientos estructurales entre el candidato LbTNTB con el transportador
mitocondrial de ADP/ATP mediante el programa UCSF Chimera 1.10.2.
a) Vista frontal. b) vista superior y c) vista posterior. Candidato LbTNTB en azul y el
Transportador mitocondrial de ADP/ATP (2c3e en PDB en rojo).
La similitud entre las estructuras tridimensionales de estas dos proteínas indican una posible
homología funcional, por lo tanto se sugiere que es posible que el candidato LbTNTB posea la
capacidad de transportar las especies ADP/ATP al igual que el candidato LbTNTA. Es importante
contrastar estos resultados con los obtenidos en estudios en organismos del género Clamidia como
C. tracomatis en donde se comprobó experimentalmente que la translocasa de ATP/ADP
(CtNPT1) posee la capacidad de transportar además de ADP/ATP el dinucleótido NAD+, inclusive
con mayor afinidad con relación a los mononucleótidos [13]. Asimismo los resultados permiten
proponer que esta proteínas se encuentra relacionada con una posible función como transportador
de NAD+ en Leishmania braziliensis.
Por último se analizó la posible función de acuerdo a los número de la comisión enzimática
predichos por I-TASSER producto del alineamiento del modelo generado con las estructuras de
proteínas elucidadas experimentalmente y de función conocida, de esta manera se tuvieron en
cuenta los alineamientos con mayor valor C, siendo el de mayor magnitud (0.180) con la Sintasa
(+)-delta-cadineno de Gossypium arboreum, seguido por el de la ATPasa de intercambio Na+/K+
(0.177) de Sus scrofa.
La Sintasa (+)-delta-cadineno tiene un número de la comisión enzimática (E.C) 4.2.3.13 y por su
parte la ATPasa de intercambio Na+/K+ tiene un E.C 3.6.3.9, los cuales corresponden con la
siguiente clasificación:

93
4 = Liasas (rompen enlaces C-C, C-O, C-N).
4.2 = Liasas Carbono-Oxigeno.
4.2.3 = Actúan sobre grupos fosfato.
4.2.3.13 = Sintasa (+)-delta-cadineno.
3 = Hidrolasas.
3.6 = Actúan sobre anhídridos de ácidos
carboxílicos.
3.6.3 = Catalizan el movimiento transmembrana
de sustancias.
3.6.3.10 = ATPasa de intercambio Na+/K+.
Es importante resaltar que la enzima con la cual se obtuvo el mayor puntaje de alineamiento para
predecir la posible función de LbTNTB no presenta ninguna relación con las especies NAD+ o
NADP+ ni la función transportadora, sin embargo la similitud con la ATPasa de intercambio
Na+/K+ muestra que posiblemente el candidato presenta interacción con nucleótidos de adenina y
que adicionalmente tiene una posible función transportadora.
Candidato LbTNTC
Para este candidato se decidió utilizar la plataforma I-TASSER directamente a partir de la cual se
obtuvo la gráfica del factor B (Figura 39), en donde se especifican los residuos que se encuentran
conformando un total de 7 hélices α unidas entre sí por varios bucles o colas, de las cuales 6
concuerdan aproximadamente con los predichos con PHYRE2 en las regiones 16 – 41, 67 – 90,
110 – 134, 156 – 179, 205 – 230 y 258 – 284 y una adicional de 300 – 316 (Figura 31).
Figura 39. Factor B normalizado para la secuencia completa del candidato LbTNTC por I-
TASSER.
Los 5 modelos obtenidos mediante I-TASSER presentaron valores C negativos, lo que indica una
baja calidad de los modelos y por consiguiente baja similitud entre las estructuras de las plantillas
y los modelos generados. Igualmente se obtuvo un RMSD con un rango grande (de 2.9 a 11.3 Å)
para el modelo con mayor valor C lo que no genera confiabilidad en este modelado (Tabla 25). De
acuerdo con la magnitud de los valores C y de RMSD se determinó que ninguno de los modelos
predichos por I-TASSER para la secuencia completa del candidato LbTNTC eran lo
suficientemente buenos para generar la estructura tridimensional, por lo tanto se utilizó la
arquitectura del dominio IPR023395 en InterPro predicha en el numeral 7.1.1, ubicada entre los
aminoácidos 11 – 284 para generar un modelo de estructura terciaria de la secuencia sin los
extremos N y C terminal que no están contenidos en dicho dominio (1 – 10 para el N terminal y
285 – 318 para el C terminal), con lo cual se obtuvo un péptido de 273 aminoácidos, que fue
evaluado con el servidor I-TASSER dando como resultado 5 nuevos modelos, de los cuales el
mejor presentó un valor C de 1.18 y un RMSD entre 1.1 – 6.1 Å, por lo cual fue seleccionado
como el modelo óptimo de estructura terciaria (Tabla 25).

94
Tabla 25. Valores C obtenidos para los modelos construidos por Threading para la
secuencia completa e incompleta del candidato LbTNTC por I-TASSER.
SECUENCIA MODELO VALOR C RMSD
Completa
(318aa)
1 -0.37 7.1 ± 4.2 Å
2 -0.39 -
3 -1.45 -
4 -1.38 -
5 -2.61 -
Incompleta
(273aa)
1 1.18 3.6 ± 2.5 Å
2 -2.74 -
3 -4.94 -
4 -0.80 -
5 0.13 -
En el modelo predictivo se evidenció la presencia de 6 segmentos con conformación tipo hélice α
que por su longitud, ubicación y de acuerdo con los análisis de estructura primaria y secundaria
anteriormente mostrados en los numerales 7.1.1 y 7.I.2 corresponden, con una alta probabilidad,
con hélices transmembranales (Figura 40b hélices azul oscura, azul clara, verde clara, verde
oscura, amarilla y roja para las hélices 1, 2, 3, 4, 5 y 6 respectivamente), adicionalmente se
predijeron 3 hélices de menor tamaño ubicadas hacia el lado opuesto de los extremos C y N-
terminal (Figura 40c hélices naranja, verde oscura y azul claro) y que se encuentran intercaladas
entre las hélices 1 – 2, en el caso de la azul clara, entre la 3 – 4 para la verde oscura y entre la 5 –
6 para la naranja. Adicionalmente se identificó una estructura en forma de cilindro o tubo
característica para proteínas transportadoras (Figura 40a y Figura 40c).
Figura 40. Modelo de estructura terciaria para el candidato LbTNTC mediante I-TASSER.
a) Vista superior. b) vista frontal y c) vista inferior.
La mayor similitud del modelo generado con las estructuras en 3D elucidadas experimentalmente
de la base de datos PDB fue con la estructura de la proteína “The bovine mitochondrial ADP/ATP
carrier” de la especie Bos taurus con un Tm del 0.961 con código de acceso 2c3e en PDB, como
se demostró en la superposición de ambas estructuras terciarias con un RMSD de 0.661 angstrom
entre 228 parejas de átomos (Figura 41).

95
Figura 41.Alineamientos estructurales entre el candidato LbTNTC con el transportador
mitocondrial de ADP/ATP mediante el programa UCSF Chimera 1.10.2.
a) Vista frontal. b) vista superior y c) vista posterior. Candidato LbTNTC en azul y el
Transportador mitocondrial de ADP/ATP (2c3e en PDB en rojo).
La similitud entre las estructuras tridimensionales de estas dos proteínas indican una posible
homología funcional, por lo tanto se sugiere que es posible que el candidato LbTNTC posea la
capacidad de transportar las especies ADP/ATP al igual que los candidatos LbTNTA y LbTNTB.
Es importante contrastar estos resultados con los obtenidos en estudios en organismos del género
Clamidia como C. tracomatis en donde se comprobó experimentalmente que la translocasa de
ATP/ADP (CtNPT1) posee la capacidad de transportar además de ADP/ATP el dinucleótido
NAD+, inclusive con mayor afinidad con relación a los mononucleótidos [13]. Asimismo los
resultados permiten proponer que esta proteínas se encuentra relacionada con una posible función
como transportador de NAD+ en Leishmania braziliensis.
Por último se analizó la posible función de acuerdo a los número de la comisión enzimática
predichos por I-TASSER producto del alineamiento del modelo generado con las estructuras de
proteínas elucidadas experimentalmente y de función conocida, de esta manera se tuvieron en
cuenta los alineamientos con mayor valor C, siendo el de mayor magnitud (0.225) con el
Citocromo P450 EryK de Saccharopolyspora erythraea, seguido por el de CYP130 (0.221) de
Mycobacterium tuberculosis.
La clasificación de estas dos enzimas de acuerdo al número de la comisión enzimática (E.C) no se
encuentra especificado para cada una en particular, sin embargo ambas estructuras se encuentran
incluidas en la clasificación 1.14 que corresponde con la siguiente clasificación:
1 = Oxidoreductasas.
1.14 = Actúan sobre pares donadores con la reducción del oxígeno molecular.
A pesar de que no se especifica directamente el cofactor que utiliza cada una de estas enzimas, en
este grupo de clasificación se encuentran varias subclases de enzimas que utilizan como donador
al NADH o el NADPH, lo que demuestra que posiblemente el candidato presenta interacción con
estos dinucleótidos.

96
7.2. Métodos experimentales
A continuación se detalla el análisis de los resultados obtenidos a partir de los procedimientos
experimentales efectuados para la detección de las proteínas recombinantes de los candidatos
LbTNTA, LbTNTB y LbTNTC.
7.2.1. Evaluación de la integridad del ADNg de Leishmania braziliensis
Se determinó que el ADN genómico extraído del parásito Leishmania braziliensis el 06 de enero
de 2016 no se encontraba degradado ya que se observó una única banda cerca al punto de siembra
y ningún barrido o fragmentos de menor tamaño producto de una posible degradación (Figura 42
pozo 1).
Figura 42. Evaluación de la integridad del ADN extraído de Leishmania braziliensis.
MW, Marcador de peso molecular Lambda-HindIII; 1, ADNg de L. braziliensis sin diluir.
Una vez se identificó que el ADN genómico de partida se encontraba en óptimas condiciones, se
llevó a cabo su cuantificación espectrofotométrica la cual mostró un valor de 330 µg/mL y una
pureza del 91.67% (Tabla 26).
Tabla 26. Valores de absorbancia obtenidos de la cuantificación espectrofotométrica de ADN.
LONGITUD DE
ONDA (nm) ABSORBANCIA
CONCENTRACIÓN
MUESTRA
PUREZA
MUESTRA
260 0.113
330 µg/mL 91.67% 280 0.087
320 0.047
7.2.2. Diseño y estandarización de Tm de los cebadores mediante PCR
Debido a que la clonación de los genes de los candidatos se realizó en los vectores pET100/D-
TOPO y pBAD202/D-TOPO se adicionó la secuencia 5´ CACC 3´ al extremo 5´ de los tres
cebadores directos diseñados, los cuales se analizaron con la herramienta OligoAnalyzer 3.1
evidenciándose temperaturas de anillaje cercanas para cada par de cebadores (56.6 – 62.0°C),
excepto para los correspondientes a LbTNTC que presentaron una diferencia de temperaturas de
8.5°C entre ellos (Tabla 27).

97
Tabla 27. Análisis de los cebadores diseñados para los candidatos mediante la herramienta
OligoAnalyzer 3.1.
CANDIDATO PRIMER SECUENCIA
5´- 3´
TEMPERATURA
DE ANILLAJE
(°C)
%
CG
LbTNTA LbTNTAd CACCATGACGCAGTCTTTCTCATCA 59.0 48%
LbTNTAr CTACTGGAACTGAGGCTGTCCCACA 62.0 56%
LbTNTB LbTNTBd CACCATGAGAGAGAAGAAGTCAGCA 58.2 48%
LbTNTBr TCATGAGATGTACTTTGCCACCTTT 56.6 40%
LbTNTC LbTNTCd CACCATGGCATCTACGTCGCCCGCA 67.0 64%
LbTNTCr CTACTCCATCGTCGATTCATCGGAC 58.5 52%
Adicionalmente se analizó la especificidad de cada primer mediante la herramienta BLAST contra
todo el genoma de Leishmania braziliensis MHOM/BR/75/M2904, para identificar si los
oligonucleótidos se unían a más de un sitio en la secuencia del ADN del parásito, de esta manera
se identificó que todos los cebadores tenían un sitio de unión único y eran complementarios en un
100% (exceptuando las bases adicionales 5´ CACC 3´ de los cebadores directo) y se localizaban
al inicio y al final de cada uno de los genes de cada transportador. Asimismo se identificaron otras
uniones inespecíficas debidas a apareamientos entre pequeños segmentos al interior de la
secuencia de los cebadores, sin embargo no representan alineamientos significativos (Anexo 3).
La estandarización de la temperatura óptima de anillaje para cada par de cebadores, evidenció que
para el candidato LbTNTA se obtuvieron amplificados únicos a las temperaturas evaluadas de 50,
53, 55 y 57°C (Figura 43a pozos 1 – 4), a pesar de esto se escogió la de 50°C debido a que fue en
la que se observó una mayor concentración del amplificado (Figura 43a pozo 1). Para LbTNTB
se identificó que con las cuatro temperaturas se obtenía un buen amplificado, sin embargo se
obtuvieron amplificados de tamaño inferior al emplear 50, 55 y 57°C (Figura 43b pozos 1, 3 y 4)
a diferencia de la de 50°C en la cual se observó una mayor intensidad del amplificado (Figura 43b
pozo 2). Finalmente la amplificación de LbTNTC con las temperaturas evaluadas no dio como
resultado un amplificado único (Figura 43c), sin embargo, se escogió un Tm de 57°C debido a que
en estas condiciones se obtuvo un perfil de amplificación más limpio en comparación con las
demás (Figura 43c pozo 4). La estandarización del Tm con Taq polimerasa se llevó a cabo teniendo
en cuenta que los ensayos posteriores para identificar los vectores recombinantes requerían la
amplificación con esta enzima.

98
Figura 43. Estandarización de Tm para los candidatos LbTNTA, LbTNTB y LbTNTC
mediante PCR. a) Estandarización de Tm para LbTNTA, b) Estandarización de Tm para LbTNTB y c) Estandarización de
Tm para LbTNTC. MW, Marcador de peso molecular de 1 kb; 2MW, Marcador de peso molecular de 100
pb; C-, control negativo; C+1, Control positivo LbNMNAT a partir de ADNg; C+2, Control positivo
LbNMNAT a partir de LbNMNAT/pET100; 1, 2, 3, 4 y 5, Amplificados a Tm de 50, 53, 55, 57 y 60°C
respectivamente. Se sembraron 2 μL de marcador de peso molecular y 10 μL de cada una de las muestras.
Agarosa al 1.5%, en buffer TBE 0.5X.
Para identificar la diferencia en el tamaño de cada uno de los productos de la amplificación
obtenidos para cada par de cebadores se realizó una electroforesis en gel de agarosa con la cual se
identificaron los tamaños para cada banda que concuerdan con el esperado para los 3 candidatos
con pesos de 1.089, 1.014 y 957 pb para LbTNTa, LbTNTB y LbTNTC respectivamente (Figura
44 pozos 1, 2 y 3 y Tabla 10).
Figura 44. Amplificación de los candidatos LbTNTA, LbTNTB y LbTNTC con taq
polimerasa. MW, Marcador de peso molecular de 1 kb; C-, control negativo; 1, Amplificado LbTNTA (1.089 pb); 2,
Amplificado LbTNTB (1.014 pb), 3, Amplificado LbTNTC (957 pb). Se sembraron 2 μL de marcador de
peso molecular y 10 μL de cada una de las muestras. Agarosa al 1.5%, en buffer TBE 0.5X.
7.2.3. Estandarización de la amplificación por PCR con Pfu
El producto de amplificación que se utilizó para obtener los vectores recombinantes se obtuvo
mediante PCR con Pfu polimerasa, debido a que esta enzima posee la capacidad correctora de
errores 3´ a 5’ durante la amplificación y a que no agrega nucleótidos en ninguno de los extremos
de la secuencia en comparación con la Taq polimerasa, condición necesaria para llevar a cabo la

99
posterior reacción de ligación. Para obtener productos únicos de amplificación se estandarizaron
los factores que se consideraron tienen mayor efecto en la calidad de la amplificación como lo son
la temperatura de anillaje y la concentración de MgSO4, esta estandarización se realizó utilizando
como modelo el candidato LbTNTA.
La determinación de la temperatura óptima de anillaje con pfu polimerasa evidenció una gran
cantidad de productos inespecíficos para todas las temperaturas evaluadas (Figura 45a), sin
embargo se escogió una Tm de 57°C debido a que se observó un amplificado del tamaño esperado
con mayor intensidad y de menor intensidad para las bandas inespecíficas en comparación con las
demás temperaturas probadas (Figura 45a pozos 1 – 5).
En cuanto a la concentración de MgSO4 se observó que la cantidad de productos inespecíficos
disminuyó a medida que lo hizo la concentración la sal, adicionalmente se identificó que se
obtuvieron como resultado tres amplificados de diferente tamaño en la región entre 1.000 – 1.500
pb, el más grande para las concentraciones de 10.0 y 5.0 mM (Figura 45b pozos 4 y 5), el siguiente
para 2.5 y 2.0 (Figura 45b pozos 2 y 3) mM y el menor para 1.5 mM (Figura 45b pozo 1). Para
comprobar cuál de los tres amplificados obtenidos era el que se buscaba se compararon los
resultados obtenidos a partir de dichas concentraciones de MgSO4 con el amplificado
estandarizado previamente con taq polimerasa debido a que con esta enzima, a pesar de que se
varió el Tm, siempre se obtuvo un solo amplificado en la región del tamaño esperado el cual se
consideró como el gen del candidato LbTNTA (Figura 45a). De esta manera se identificó que el
fragmento más pequeño obtenido con 1.5 mM de MgSO4 presentó el mismo tamaño que el
obtenido con taq polimerasa, por lo tanto se escogió esta como la concentración óptima para la
amplificación con pfu polimerasa (Figura 45c pozos 1 y 2).
Figura 45. Estandarización Tm y concentración de MgSO4 para el candidato LbTNTA con
Pfu polimerasa. a) Estandarización de Tm para el candidato LbTNTA: MW, Marcador de peso molecular de 1 kb; C–,
Control negativo; 1, 2, 3, 4, 5, Amplificados a Tm de 50, 53, 55, 57 y 60°C respectivamente. b)
Estandarización de la concentración de MgSO4 para el candidato LbTNTA: MW, Marcador de peso
molecular de 1 kb; C–, Control negativo; 1, 2, 3, 4 y 5, Amplificados a concentraciones de MgSO4 de 1.5,
2.0, 2.5, 5.0 y 10.0 mM respectivamente. c) Evaluación de los amplificados obtenidos para el candidato
LbTNTA: MW, Marcador de peso molecular de 1 kb; C–, Control negativo; 1, Amplificado LbTNTA con
Taq polimerasa a 50°C y 2.0 mM de MgCl2; 2, Amplificado LbTNTA con MgSO4 1.5 mM; 3, Amplificado
LbTNTA con MgSO4 2.5 mM; 4, Amplificado LbTNTA con MgSO4 10.0 mM. Se sembraron 2 μL de
marcador de peso molecular y 10 μL de cada una de las muestras. Agarosa al 1.5%, en buffer TBE 0.5X.

100
Finalmente se evaluó la identidad del amplificado obtenido con Pfu polimerasa para el candidato
LbTNTA mediante la restricción del producto de PCR con la enzima AluI, la cual reconoce 2
puntos de corte en la secuencia de 1.089 pb del gen de LbTNTA (entre los nucleótidos 456 – 457
y 529 – 530), generando como producto tres fragmentos de 73, 456 y 560 pb respectivamente
(Figura 46b), de los cuales fueron reconocidos los de 456 y 560 pb mediante electroforesis
horizontal en gel de agarosa pero no el de menor tamaño ya que posiblemente salió del gel debido
al tiempo de corrida (40 minutos), sin embargo, la presencia y longitud de estos dos fragmentos
correspondieron con el perfil esperado de la digestión predicho con NEBcutter V2.0
(http://nc2.neb.com/NEBcutter2/index.php), por lo tanto se consideró que el producto amplificado
a 57°C y 1.5 mM de MgSO4 correspondía con el gen del candidato LbTNTA.
Figura 46. Confirmación de la identidad del amplificado del candidato LbTNTA mediante
restricción con AluI. a) Perfil de restricción obtenido experimentalmente para el producto de PCR de LbTNTA con AluI: MW,
Marcador de peso molecular de 100 pb; 1, Producto de PCR para LbTNTA digerido con AluI; 2, Inserto
sin digerir. b) Perfil de restricción predicho mediante NEBcutter V2.0 para la digestión del gen LbTNTA
con AluI: MW, Marcador de peso molecular de 100 pb; 1, Gen LbTNTA digerido con AluI. Se sembraron
2 μL de marcador de peso molecular y 10 μL de cada una de las muestras. Agarosa al 2.0%, en buffer TBE
0.5X durante 40 minutos.
A partir de estas condiciones (Tm = 57°C y 1.5 mM de MgSO4) estandarizadas para la
amplificación LbTNTA, se amplificaron los genes de LbTNTB y LbTNTC y se obtuvieron
productos únicos con los cuales llevó a cabo la reacción de ligación (Figura 47 pozos 1, 2 y 3). Es
importante resaltar que se utilizó como modelo a LbTNTA para la estandarización de PCR con
Pfu polimerasa debido a que los tres candidatos poseen una gran similitud entre sus secuencias y
tamaños, por consiguiente se esperaba que las condiciones para su amplificación fueran similares.

101
Figura 47. Amplificación de los candidatos LbTNTA, LbTNTB y LbTNTC con Pfu
polimerasa.
MW, Marcador de peso molecular de 100 pb; C–, Control negativo; 1, Amplificado LbTNTA
(1.089 pb); 2, Amplificado LbTNTB (1.014 pb) 3, Amplificado LbTNTC (957 pb). Se sembraron
2 μL de marcador de peso molecular y 2 μL de cada una de las muestras. Agarosa al 1.5%, en
buffer TBE 0.5X.
7.2.4. Obtención de vectores recombinantes LbTNT´s/pET100 y LbTNT´s/pBAD202
El proceso de clonación de los genes de los tres candidatos se realizó en los vectores pBAD202/D-
TOPO y pET100/D-TOPO. Para llevar a cabo la ligación de cada inserto con los vectores se
calculó la concentración de los productos de PCR mediante densitometría comparando cada una
de las bandas de los amplificados con las del marcador de peso molecular encontrándose similitud
de la intensidad de la banda correspondiente a LbTNTA con la de 1.000 pb y las de LbTNTB y
LbTNTC con la de 3.000 pb (Figura 47). Las bandas de 1.000 y 3.000 pb corresponden con el 12
y el 14% de la cantidad de marcador sembrado respectivamente, teniendo en cuenta que se
sembraron 2 µL del marcador (50 ng/µL) como también de los 3 productos de PCR se determinó
su concentración como sigue:
[ ] 𝐿𝑏𝑇𝑁𝑇𝐴 = 50𝑛𝑔
µ𝐿𝑥 2µ𝐿 = 100𝑛𝑔 𝑥 12% = 12𝑛𝑔/2µ𝐿 = 6.0
𝑛𝑔
µ𝐿
[ ] 𝐿𝑏𝑇𝑁𝑇𝐵 𝑦 𝐿𝑏𝑇𝑁𝑇𝐶 = 50𝑛𝑔
µ𝐿𝑥 2µ𝐿 = 100𝑛𝑔 𝑥 14% = 14𝑛𝑔/2µ𝐿 = 7.0
𝑛𝑔
µ𝐿
Los ng de inserto necesarios para llevar a cabo una reacción de ligación 3:1 inserto:vector se
calcularon con la ecuación 1, teniendo en cuenta que se ligaron 20 ng del vector pBAD202/D-
TOPO que tiene un tamaño de 4.400 pb y la misma cantidad del vector pET100/D-TOPO con un
tamaño de 5.764 pb y que los genes de los candidatos LbTNTA, LbTNTB y LbTNTC tienen
tamaños de 1.089, 1.014 y 957 pb respectivamente. De acuerdo con esto se calculó el volumen
necesario para llevar a cabo la ligación en estos dos vectores (Tabla 28).

102
Tabla 28. Cantidades de inserto necesarias para una ligación con relación inserto:vector de 3:1
en pBAD202/D-TOPO y pET100/D-TOPO para los tres candidatos.
INSERTO CONCENTRACIÓN
(ng/µL)
pBAD202 pET100
ng Inserto Volumen (µL) ng Inserto Volumen (µL)
LbTNTA 6.0 14.85 2.5 11.33 1.9
LbTNTB 7.0 13.83 2.0 10.55 1.5
LbTNTB 7.0 13.05 1.9 9.96 1.4
Cada uno de los productos de ligación se transformó en células Top10 de E. coli químicamente
competentes y se platearon 300 µL en cajas de Petri con medio de cultivo sólido (LB-agar) con
ampicilina a una concentración final de 100 µg/mL o kanamicina a una concentración final de 50
µg/mL para los recombinantes de pET100 y pBAD202 respectivamente.
Clonación de los candidatos en pBAD202/D-TOPO
Se platearon dos volúmenes (600 µL) de cada producto de transformación obteniéndose 41.7
UFC/mL para LbTNTA (25 colonias), 15 UFC/mL para LbTNTB (9 colonias) y 18.33 UFC/mL
para LbTNTC (11 colonias). Estas colonias se rastrearon mediante PCR con Taq polimerasa bajo
las condiciones estandarizadas en el numeral 7.2.2 para determinar en cual se encontraban los
vectores recombinantes. De acuerdo con esto se identificó un amplificado del tamaño esperado
(1.089 pb) en la colonia 1 de LbTNTA con un producto inespecífico de aproximadamente 400 pb
(Figura 48a pozo 1) mientras que en los pozos 2 – 6 no se observó un amplificado cercano a los
1.000 pb pero si una inespecificidad entre 500 y 600 pb en los pools A2 y A3 (Figura 48a pozos
3 y 4). De igual manera se evidenció que el pool de colonias B4 (colonias 7 y 8) para LbTNTB
presentaba un amplificado tenue del mismo tamaño que el control positivo (1.014 pb) junto con
una banda inespecífica entre 300 – 400 pb (Figura 48b pozo 4). Por último se observó que la
colonia 1 y el pool C1 (colonias 2 – 5) para LbTNTC presentaron amplificados con el tamaño
esperado (957 pb) así como bandas inespecíficas de mayor y menor tamaño al igual que el control
positivo (Figura 48c pozos C+, 1 y 2). A partir de estos resultados se decidió continuar analizando
las colonias que presentaron amplificado del tamaño esperado para cada candidato.
Figura 48. PCR de colonia para rastrear los recombinantes LbTNT´s/pBAD202.
a) PCR de colonia LbTNTA: MW, Marcador de peso molecular de 100 pb; C–, Control negativo; C+,
Control positivo LbTNTA a partir de ADNg; 1, Colonia 1 LbTNTA; 2, pool de colonias A1 (2-5) LbTNTA;
3, Pool de colonias A2 (6-10) LbTNTA; 4, Pool de colonias A3 (11-15) LbTNTA; 5, Pool de colonias A4
(16-20) LbTNTA; 6, Pool de colonias A5 (21-25) LbTNTA. b) PCR de colonia LbTNTB: MW, Marcador

103
de peso molecular de 100 pb; C–, Control negativo; C+, Control positivo LbTNTB a partir de ADNg; 1,
Pool de colonias B1 (1 y 2) LbTNTB; 2, Pool de colonias B2 (3 y 4) LbTNTB; 3, Pool de colonias B3 (5 y
6) LbTNTB; 4, pool de colonias B4 (7 y 8) LbTNTB; 5, Colonia 9 LbTNTB. c) PCR de colonia LbTNTC:
MW, Marcador de peso molecular de 100 pb; C–, Control negativo; C+, Control positivo LbTNTC a partir
de ADNg; 1, Colonia 1 LbTNTC; 2, Pool de colonias C1 (2 – 5) LbTNTC; 3, Pool de colonias C2 (6 – 9)
LbTNTC; 4, Pool de colonias C3 (10 y 11) LbTNTC. Se sembraron 2 μL de marcador de peso molecular y
10 μL de cada una de las muestras. Agarosa al 1.5%, en buffer TBE 0.5X.
Debido a que algunos de los amplificados obtenidos correspondían a agrupaciones de diferentes
colonias (pool B4 para LbTNTB y pool C1 para LbTNTC) se procedió a separarlas para identificar
en cuál de las colonias se encontraba el vector recombinante mediante PCR, con lo cual se
identificó que la colonia 7 de LbTNTB y las colonias 2, 3 y 4 de LbTNTC presentaron un
amplificado con el tamaño esperado, sin embargo con una intensidad muy baja y con bandas
inespecíficas de menor tamaño alrededor de 800 pb y superiores a 1000 pb (Figura 49a pozo 1 y
Figura 49b pozos 1, 2 y 3).
Figura 49. PCR de colonia para LbTNTB/pBAD202 y LbTNTC/pBAD202 con pools
separados. a) PCR de colonia LbTNTB: MW, Marcador de peso molecular de 100 pb; C–, Control negativo; C+,
Control positivo LbTNTB a partir de ADNg; 1, 2, Colonias 7 y 8 LbTNTB respectivamente. b) PCR de
colonia LbTNTC: MW, Marcador de peso molecular de 100 pb; C–, Control negativo; C+, Control positivo
LbTNTC desde ADNg; 1, 2, 3 y 4 Colonias 2, 3, 4 y 5 LbTNTC respectivamente. Se sembraron 2 μL de
marcador de peso molecular y 10 μL de cada una de las muestras. Agarosa al 1.5%, en buffer TBE 0.5X.
De acuerdo con estos resultados se eligió la colonia 1 de LbTNTA, la 7 de LbTNTB y las colonias
1, 2, 3 y 4 de LbTNTC para extraer el ADN plasmídico por lisis alcalina, los cuales fueron
evaluados por PCR con Taq polimerasa bajo las condiciones estandarizadas en el numeral 7.2.2
para verificar la presencia del inserto. De esta manera se identificó que el plásmido 1 de LbTNTA
(Figura 50a pozo 1) y el 2 de LbTNTC (Figura 50c pozo 2) presentan un amplificado
correspondiente al tamaño esperado para ambos candidatos (1.089 y 957 pb respectivamente),
mientras que para LbTNTB ninguno de los plásmidos evaluados presentó amplificación por lo que
fueron descartados. Se debe resaltar que el amplificado obtenido para LbTNTC presentó una
intensidad muy leve, sin embargo se continuó con su estudio.

104
Figura 50. PCR de plásmido para verificar los recombinantes LbTNT´s/pBAD202.
a) PCR de plásmido LbTNTA: MW, Marcador de peso molecular de 100 pb; C–, Control negativo; C+,
Control positivo LbTNTA a partir de ADNg; 1, Plásmido colonia 1 LbTNTA. b) PCR de plásmido
LbTNTB: MW, Marcador de peso molecular de 1 kb; C–, Control negativo; C+, Control positivo LbTNTB
a partir de ADNg; 1, Plásmido colonia 7 LbTNTB. c) PCR de plásmido LbTNTC: MW, Marcador de peso
molecular de 100 pb; C–, Control negativo; C+, Control positivo LbTNTC a partir de ADNg; 1¸ 2, 3 y 4,
Plásmidos colonias 1, 2, 3 y 4 LbTNTC respectivamente. Se sembraron 2 μL de marcador de peso molecular
y 10 μL de cada una de las muestras. Agarosa al 1.5%, en buffer TBE 0.5X.
Paralelamente se realizó la digestión de los plásmidos correspondientes a los tres transportadores
para comprobar la presencia de los insertos en los recombinantes. El plásmido 1 de LbTNTA fue
digerido con la enzima de restricción BamHI la cual reconoce 2 puntos de corte en la secuencia
del vector pBAD202 circularizado (entre los nucleótidos 239 – 240 y 682 – 683), generando como
producto dos fragmentos para el vector vacío de 443 y 4.005 pb y para el vector con el inserto de
443 y 5.094 pb (Figura 51b) los cuales fueron reconocidos mediante electroforesis horizontal en
gel de agarosa tanto para el vector vacío como para el vector recombinante 1 (Figura 51a pozos 1
y 2), con lo que se confirmó la presencia del inserto en el vector.
Figura 51. Confirmación de la presencia del gen de LbTNTA en pBAD202 mediante
restricción del plásmido 1 con BamHI. a) Perfil de restricción obtenido experimentalmente para el plásmido 1 de LbTNTA con BamHI: MW,
Marcador de peso molecular de 1 kb; 1, Vector pBAD202/D-TOPO vacío digerido con BamHI; 1, Plásmido
1 de LbTNTA digerido con BamHI. b) Perfil de restricción predicho mediante NEBcutter V2.0 para la
digestión del plásmido LbTNTA/pBAD202 con BamHI: MW, Marcador de peso molecular de 1 kb; 1,
Predicción de la digestión del vector pBAD202/D-TOPO vacío digerido con BamHI; 2, Predicción de la
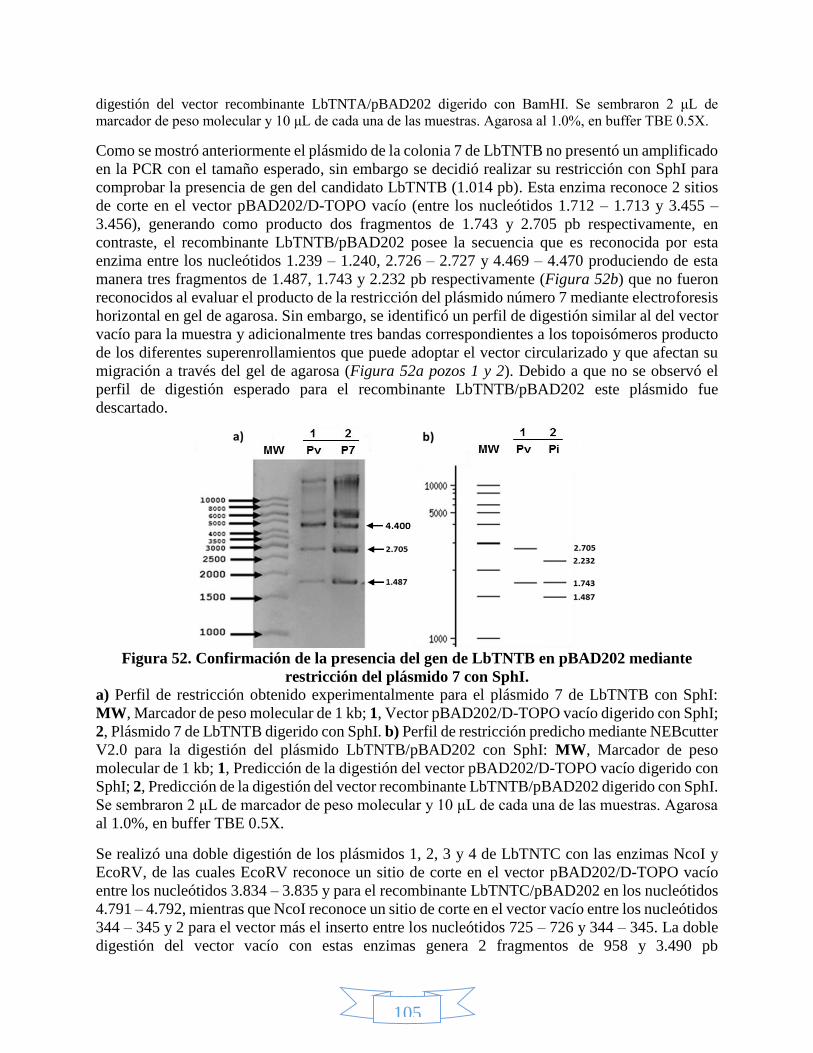
105
digestión del vector recombinante LbTNTA/pBAD202 digerido con BamHI. Se sembraron 2 μL de
marcador de peso molecular y 10 μL de cada una de las muestras. Agarosa al 1.0%, en buffer TBE 0.5X.
Como se mostró anteriormente el plásmido de la colonia 7 de LbTNTB no presentó un amplificado
en la PCR con el tamaño esperado, sin embargo se decidió realizar su restricción con SphI para
comprobar la presencia de gen del candidato LbTNTB (1.014 pb). Esta enzima reconoce 2 sitios
de corte en el vector pBAD202/D-TOPO vacío (entre los nucleótidos 1.712 – 1.713 y 3.455 –
3.456), generando como producto dos fragmentos de 1.743 y 2.705 pb respectivamente, en
contraste, el recombinante LbTNTB/pBAD202 posee la secuencia que es reconocida por esta
enzima entre los nucleótidos 1.239 – 1.240, 2.726 – 2.727 y 4.469 – 4.470 produciendo de esta
manera tres fragmentos de 1.487, 1.743 y 2.232 pb respectivamente (Figura 52b) que no fueron
reconocidos al evaluar el producto de la restricción del plásmido número 7 mediante electroforesis
horizontal en gel de agarosa. Sin embargo, se identificó un perfil de digestión similar al del vector
vacío para la muestra y adicionalmente tres bandas correspondientes a los topoisómeros producto
de los diferentes superenrollamientos que puede adoptar el vector circularizado y que afectan su
migración a través del gel de agarosa (Figura 52a pozos 1 y 2). Debido a que no se observó el
perfil de digestión esperado para el recombinante LbTNTB/pBAD202 este plásmido fue
descartado.
Figura 52. Confirmación de la presencia del gen de LbTNTB en pBAD202 mediante
restricción del plásmido 7 con SphI.
a) Perfil de restricción obtenido experimentalmente para el plásmido 7 de LbTNTB con SphI:
MW, Marcador de peso molecular de 1 kb; 1, Vector pBAD202/D-TOPO vacío digerido con SphI;
2, Plásmido 7 de LbTNTB digerido con SphI. b) Perfil de restricción predicho mediante NEBcutter
V2.0 para la digestión del plásmido LbTNTB/pBAD202 con SphI: MW, Marcador de peso
molecular de 1 kb; 1, Predicción de la digestión del vector pBAD202/D-TOPO vacío digerido con
SphI; 2, Predicción de la digestión del vector recombinante LbTNTB/pBAD202 digerido con SphI.
Se sembraron 2 μL de marcador de peso molecular y 10 μL de cada una de las muestras. Agarosa
al 1.0%, en buffer TBE 0.5X.
Se realizó una doble digestión de los plásmidos 1, 2, 3 y 4 de LbTNTC con las enzimas NcoI y
EcoRV, de las cuales EcoRV reconoce un sitio de corte en el vector pBAD202/D-TOPO vacío
entre los nucleótidos 3.834 – 3.835 y para el recombinante LbTNTC/pBAD202 en los nucleótidos
4.791 – 4.792, mientras que NcoI reconoce un sitio de corte en el vector vacío entre los nucleótidos
344 – 345 y 2 para el vector más el inserto entre los nucleótidos 725 – 726 y 344 – 345. La doble
digestión del vector vacío con estas enzimas genera 2 fragmentos de 958 y 3.490 pb
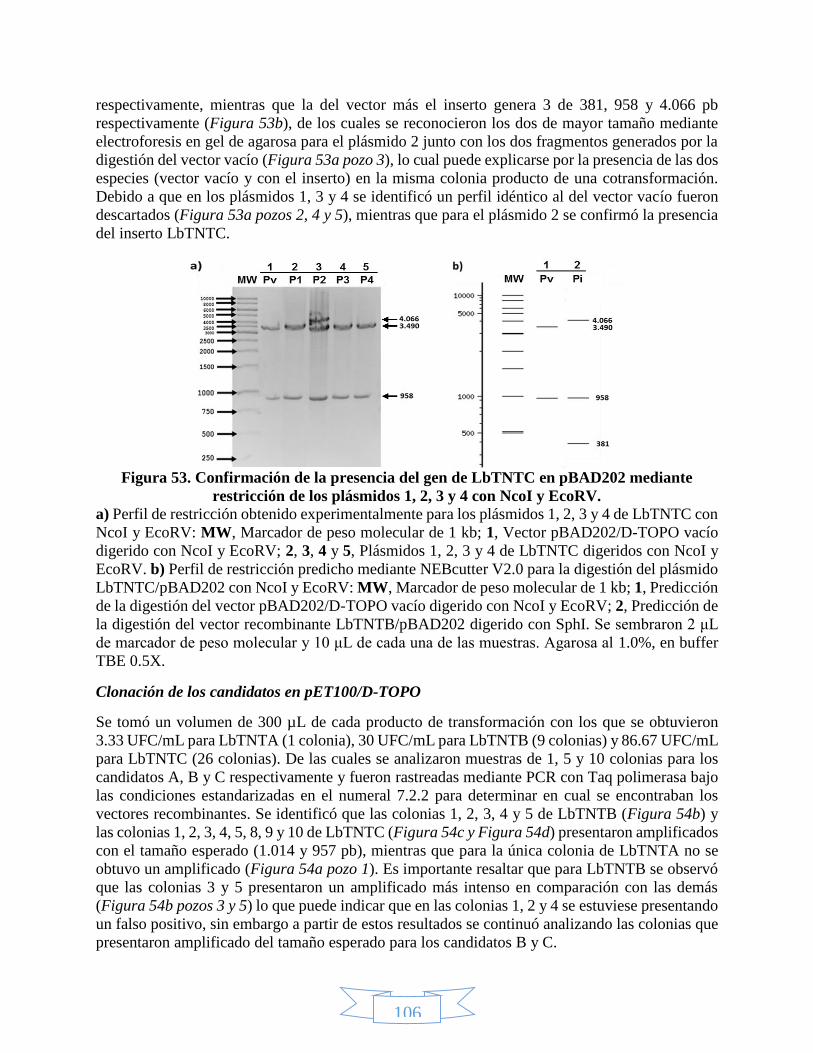
106
respectivamente, mientras que la del vector más el inserto genera 3 de 381, 958 y 4.066 pb
respectivamente (Figura 53b), de los cuales se reconocieron los dos de mayor tamaño mediante
electroforesis en gel de agarosa para el plásmido 2 junto con los dos fragmentos generados por la
digestión del vector vacío (Figura 53a pozo 3), lo cual puede explicarse por la presencia de las dos
especies (vector vacío y con el inserto) en la misma colonia producto de una cotransformación.
Debido a que en los plásmidos 1, 3 y 4 se identificó un perfil idéntico al del vector vacío fueron
descartados (Figura 53a pozos 2, 4 y 5), mientras que para el plásmido 2 se confirmó la presencia
del inserto LbTNTC.
Figura 53. Confirmación de la presencia del gen de LbTNTC en pBAD202 mediante
restricción de los plásmidos 1, 2, 3 y 4 con NcoI y EcoRV.
a) Perfil de restricción obtenido experimentalmente para los plásmidos 1, 2, 3 y 4 de LbTNTC con
NcoI y EcoRV: MW, Marcador de peso molecular de 1 kb; 1, Vector pBAD202/D-TOPO vacío
digerido con NcoI y EcoRV; 2, 3, 4 y 5, Plásmidos 1, 2, 3 y 4 de LbTNTC digeridos con NcoI y
EcoRV. b) Perfil de restricción predicho mediante NEBcutter V2.0 para la digestión del plásmido
LbTNTC/pBAD202 con NcoI y EcoRV: MW, Marcador de peso molecular de 1 kb; 1, Predicción
de la digestión del vector pBAD202/D-TOPO vacío digerido con NcoI y EcoRV; 2, Predicción de
la digestión del vector recombinante LbTNTB/pBAD202 digerido con SphI. Se sembraron 2 μL
de marcador de peso molecular y 10 μL de cada una de las muestras. Agarosa al 1.0%, en buffer
TBE 0.5X.
Clonación de los candidatos en pET100/D-TOPO
Se tomó un volumen de 300 µL de cada producto de transformación con los que se obtuvieron
3.33 UFC/mL para LbTNTA (1 colonia), 30 UFC/mL para LbTNTB (9 colonias) y 86.67 UFC/mL
para LbTNTC (26 colonias). De las cuales se analizaron muestras de 1, 5 y 10 colonias para los
candidatos A, B y C respectivamente y fueron rastreadas mediante PCR con Taq polimerasa bajo
las condiciones estandarizadas en el numeral 7.2.2 para determinar en cual se encontraban los
vectores recombinantes. Se identificó que las colonias 1, 2, 3, 4 y 5 de LbTNTB (Figura 54b) y
las colonias 1, 2, 3, 4, 5, 8, 9 y 10 de LbTNTC (Figura 54c y Figura 54d) presentaron amplificados
con el tamaño esperado (1.014 y 957 pb), mientras que para la única colonia de LbTNTA no se
obtuvo un amplificado (Figura 54a pozo 1). Es importante resaltar que para LbTNTB se observó
que las colonias 3 y 5 presentaron un amplificado más intenso en comparación con las demás
(Figura 54b pozos 3 y 5) lo que puede indicar que en las colonias 1, 2 y 4 se estuviese presentando
un falso positivo, sin embargo a partir de estos resultados se continuó analizando las colonias que
presentaron amplificado del tamaño esperado para los candidatos B y C.
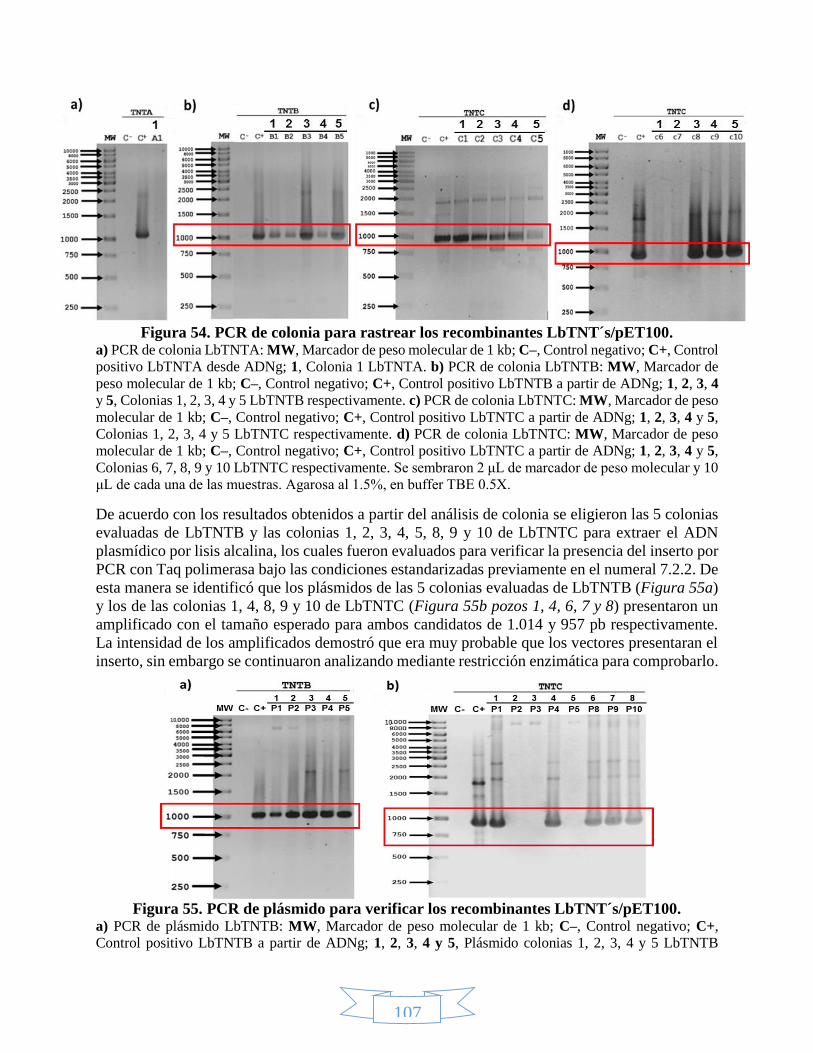
107
Figura 54. PCR de colonia para rastrear los recombinantes LbTNT´s/pET100.
a) PCR de colonia LbTNTA: MW, Marcador de peso molecular de 1 kb; C–, Control negativo; C+, Control
positivo LbTNTA desde ADNg; 1, Colonia 1 LbTNTA. b) PCR de colonia LbTNTB: MW, Marcador de
peso molecular de 1 kb; C–, Control negativo; C+, Control positivo LbTNTB a partir de ADNg; 1, 2, 3, 4
y 5, Colonias 1, 2, 3, 4 y 5 LbTNTB respectivamente. c) PCR de colonia LbTNTC: MW, Marcador de peso
molecular de 1 kb; C–, Control negativo; C+, Control positivo LbTNTC a partir de ADNg; 1, 2, 3, 4 y 5,
Colonias 1, 2, 3, 4 y 5 LbTNTC respectivamente. d) PCR de colonia LbTNTC: MW, Marcador de peso
molecular de 1 kb; C–, Control negativo; C+, Control positivo LbTNTC a partir de ADNg; 1, 2, 3, 4 y 5,
Colonias 6, 7, 8, 9 y 10 LbTNTC respectivamente. Se sembraron 2 μL de marcador de peso molecular y 10
μL de cada una de las muestras. Agarosa al 1.5%, en buffer TBE 0.5X.
De acuerdo con los resultados obtenidos a partir del análisis de colonia se eligieron las 5 colonias
evaluadas de LbTNTB y las colonias 1, 2, 3, 4, 5, 8, 9 y 10 de LbTNTC para extraer el ADN
plasmídico por lisis alcalina, los cuales fueron evaluados para verificar la presencia del inserto por
PCR con Taq polimerasa bajo las condiciones estandarizadas previamente en el numeral 7.2.2. De
esta manera se identificó que los plásmidos de las 5 colonias evaluadas de LbTNTB (Figura 55a)
y los de las colonias 1, 4, 8, 9 y 10 de LbTNTC (Figura 55b pozos 1, 4, 6, 7 y 8) presentaron un
amplificado con el tamaño esperado para ambos candidatos de 1.014 y 957 pb respectivamente.
La intensidad de los amplificados demostró que era muy probable que los vectores presentaran el
inserto, sin embargo se continuaron analizando mediante restricción enzimática para comprobarlo.
Figura 55. PCR de plásmido para verificar los recombinantes LbTNT´s/pET100.
a) PCR de plásmido LbTNTB: MW, Marcador de peso molecular de 1 kb; C–, Control negativo; C+,
Control positivo LbTNTB a partir de ADNg; 1, 2, 3, 4 y 5, Plásmido colonias 1, 2, 3, 4 y 5 LbTNTB

108
respectivamente. b) PCR de plásmido LbTNTC: MW, Marcador de peso molecular de 1 kb; C–, Control
negativo; C+, Control positivo LbTNTC a partir de ADNg; 1, 2, 3, 4, 5, 6, 7 y 8, Plásmido colonias 1, 2, 3,
4, 5, 8, 9 y 10 LbTNTC respectivamente. Se sembraron 2 μL de marcador de peso molecular y 10 μL de
cada una de las muestras. Agarosa al 1.5, en buffer TBE 0.5X.
Para comprobar la presencia del inserto en los plásmidos 1 – 5 seleccionados anteriormente para
el candidato LbTNTB se realizó paralelamente una digestión con la enzima de restricción EcoRV
la cual reconoce 2 puntos de corte en la secuencia del vector pET100/D-TOPO vacío entre los
nucleótidos 555 – 556 y 4.785 – 4.786 generando como producto dos fragmentos de 4.230 y 1.534
pb y para el recombinante LbTNTB/pET100 entre los nucleótidos 1.569 – 1.570 y 5.799 – 5.800
produciendo 2 bandas de 1.534 y 2.548 pb respectivamente (Figura 56b) que fueron reconocidas
en el perfil de digestión de los plásmidos 3, 4 y 5 (Figura 56a pozos 4, 5 y 6), con lo que se
confirmó la presencia del inserto en estos recombinantes. Para los plásmidos 1 y 2 se identificó un
patrón de digestión similar al del vector vacío por lo tanto fueron descartados (Figura 56a pozos
2 y 3).
Figura 56. Confirmación de la presencia del gen de LbTNTB en pET100 mediante
restricción de los plásmidos 1 – 5 con EcoRV. a) Perfil de restricción obtenido experimentalmente para los plásmidos 1 – 5 de LbTNTB con EcoRV: MW,
Marcador de peso molecular de 1 kb; 1, Vector pET100/D-TOPO vacío digerido con EcoRV; 2, 3, 4, 5 y
6, Plásmidos colonias 1, 2, 3, 4 y 5 de LbTNTB digeridos con EcoRV respectivamente. b) Perfil de
restricción predicho mediante NEBcutter V2.0 para la digestión del plásmido LbTNTB/pET100 con
EcoRV: MW, Marcador de peso molecular de 1 kb; 1, Predicción de la digestión del vector pET100/D-
TOPO vacío digerido con EcoRV, 2, Predicción de la digestión del vector recombinante
LbTNTB/pBAD202 digerido con EcoRV. Se sembraron 2 μL de marcador de peso molecular y 10 μL de
cada una de las muestras. Agarosa al 1.0%, en buffer TBE 0.5X.
De igual manera se realizó una doble digestión de los plásmidos 1 – 5 y 8 – 10 de LbTNTC con
las enzimas NcoI y EcoRV, de las cuales EcoRV reconoce dos sitios de corte en el vector
pET100/D-TOPO vacío entre los nucleótidos 555 – 556 y 4.785 – 4.786 y para el recombinante
LbTNTC/pET100 en los nucleótidos 1.512 – 1.513 y 5.742 – 5.743, mientras que NcoI no reconoce
ninguna secuencia de corte en el vector vacío, pero debido a la presencia del inserto se genera una
entre los nucleótidos 403 – 404. La doble digestión del vector vacío genera 2 fragmentos de 1.534
y 4.230 pb respectivamente, mientras que la del vector con el inserto genera 3 de 1.109, 1.382 y
4.230 pb (Figura 57c pozo 2), en comparación, si la digestión se lleva a cabo solamente con EcoRV
el vector con el inserto genera 2 fragmentos de 2.491 y 4.230 pb y sí solamente se lleva a cabo con
NcoI el vector recombinante se linealiza (Figura 57c pozo 3).

109
Se halló que el plásmido 1 fue el único en el que se identificó el perfil de la doble digestión con
NcoI y EcoRV lo que confirma que posee el inserto (Figura 57a pozo 2) al igual que los plásmidos
4, 8, 9 y 10 debido a que estos últimos 4 presentan el perfil de digestión con EcoRV (Figura 57a
pozo 5 y Figura 57b pozos 2, 3 y 4). Sin embargo, la ausencia del fragmento producido por el corte
con NcoI es un indicio de que posiblemente el inserto presenta una mutación en la secuencia 5´
CCATGG 3´ la cual es reconocida por esta enzima o que durante el proceso de ligación ocurrió un
error ya que este mismo segmento coincide con el punto de unión del inserto con el vector (Figura
58), estas dos hipótesis traen como consecuencia que la enzima no pueda realizar el corte, sin
embargo los plásmidos 4, 8, 9 y 10 no se descartaron para comprobar su viabilidad mediante la
expresión de la proteína. A su vez es importante resaltar que esta ubicación adicionalmente
coincide con el codón de inicio para la transcripción de la proteína nativa. Asimismo se identificó
en los plásmidos 2, 3 y 5 un perfil idéntico al del vector vacío por lo cual fueron descartados
(Figura 57a pozos 3, 4 y 6).
Figura 57. Confirmación de la presencia del gen de LbTNTC en pET100 mediante
restricción de los plásmidos 1 – 5, 8, 9 y 10 con NcoI y EcoRV. a) Perfil de restricción obtenido experimentalmente para los plásmidos 1 – 5 y 8 – 10 de LbTNTC con NcoI
y EcoRV: MW, Marcador de peso molecular de 1 kb; 1, Vector pET100/D-TOPO vacío digerido con NcoI
y EcoRV 2, 3, 4, 5, y 6, Plásmidos 1 – 5 de LbTNTC digeridos con NcoI y EcoRV. b) Perfil de restricción
obtenido experimentalmente para los plásmidos 8, 9 y 10 de LbTNTC con NcoI y EcoRV: MW, Marcador
de peso molecular de 1 kb; 1, Vector pET100/D-TOPO vacío digerido con NcoI y EcoRV 2, 3 y 4,
Plásmidos 8, 9 y 10 de LbTNTC digeridos con NcoI y EcoRV. c) Perfil de restricción predicho mediante
NEBcutter V2.0 para la digestión del plásmido LbTNTC/pET100 con NcoI y EcoRV; 1, Predicción de la
digestión del vector pET100/D-TOPO vacío digerido con NcoI y EcoRV. 2, Doble digestión predicha para
el plásmido LbTNTC/pET100 con NcoI y EcoRV; 3, Digestión predicha para el plásmido
LbTNTC/pET100 con EcoRV. Se sembraron 2 μL de marcador de peso molecular y 10 μL de cada una de
las muestras. Agarosa al 1.0%, en buffer TBE 0.5X.

110
Figura 58. Punto de corte de NcoI en el vector recombinante LbTNTC/pET100.
El punto de corte de NcoI (en azul) coincide con la secuencia 5’-CACC adicionada para asegurar
la direccionalidad del inserto después del punto de corte de la Topoisomerasa I (recuadro verde) y
con el codón de inicio de la proteína LbTNTC (corchete rojo).
7.2.5. Expresión y detección de las proteínas recombinantes 6xHisLbTNT´s y
TrxLbTNT´s por electroforesis SDS-PAGE y Western blot
Como se demostró anteriormente el plásmido 1 de LbTNTA/pBAD202, el plásmido 2 de
LbTNTC/pBAD202, los plásmidos 3, 4 y 5 de LbTNTB/pET100 y los plásmidos 1, 8, 9 y 10 de
LbTNTC/pET100 presentaron el inserto en su secuencia, por lo tanto se transformó 3 µL de cada
uno en células BL21 DE3 de E. coli químicamente competentes y se platearon 300 µL en cajas de
Petri con medio de cultivo sólido (LB-agar) con ampicilina a una concentración final de 100 µg/mL
o kanamicina a una concentración final de 50 µg/mL para los recombinantes de pET100 y
pBAD202 respectivamente.
Detección de las proteínas recombinantes TrxLbTNT´s
Las proteínas recombinantes producidas a partir del vector pBAD202/D-TOPO tienen como
péptido fusión en el extremo N-terminal la secuencia His-Tiorredoxina (Trx), la cual consiste en
una secuencia mutada de tiorredoxina en la que se han realizado las modificaciones de los residuos
de glutamato E32H y glutamina Q64H, de esta manera el peso en comparación con las proteínas
nativas aumenta en 13 kDa con este sistema de expresión. De esta manera el peso esperado de la
proteína TrxLbTNTA es de 52.8 kDa y el de TrxLbTNTC de 58.3 kDa
Para evaluar la expresión de la proteína recombinante TrxLbTNTA se recolectaron alícuotas a 0
horas y después de incubación durante toda la noche (18 horas aproximadamente), las cuales se
utilizaron para obtener las proteínas totales de la inducción. Las proteínas totales fueron separadas
mediante electroforesis SDS-PAGE en un gel discontinuo de poliacrilamida al 12% en el cual se
evidenció una banda tenue del tamaño esperado (52.8 kDa) en la alícuota correspondiente a la
inducción durante toda la noche de las bacterias transformadas con el vector recombinante
LbTNTA/pBAD202 (Figura 59a pozo 4). Para verificar si esta banda correspondía con la proteína
recombinante se realizó otra electroforesis SDS-PAGE bajo las mismas condiciones la cual se
transfirió a una membrana de nitrocelulosa por electrotransferencia húmeda para llevar a cabo un
Western Blot revelado por fosfatasa alcalina (Anticuerpo primario monoclonal de ratón α-Trx
(diluido 1:3000 en TBST) y como anticuerpo secundario α-IgG de ratón biotinilado (diluido
1:5000 en TBST), este ensayo permitió reconocer una banda con el peso esperado y varias bandas
de menor tamaño identificadas como productos de degradación de la proteína recombinante con
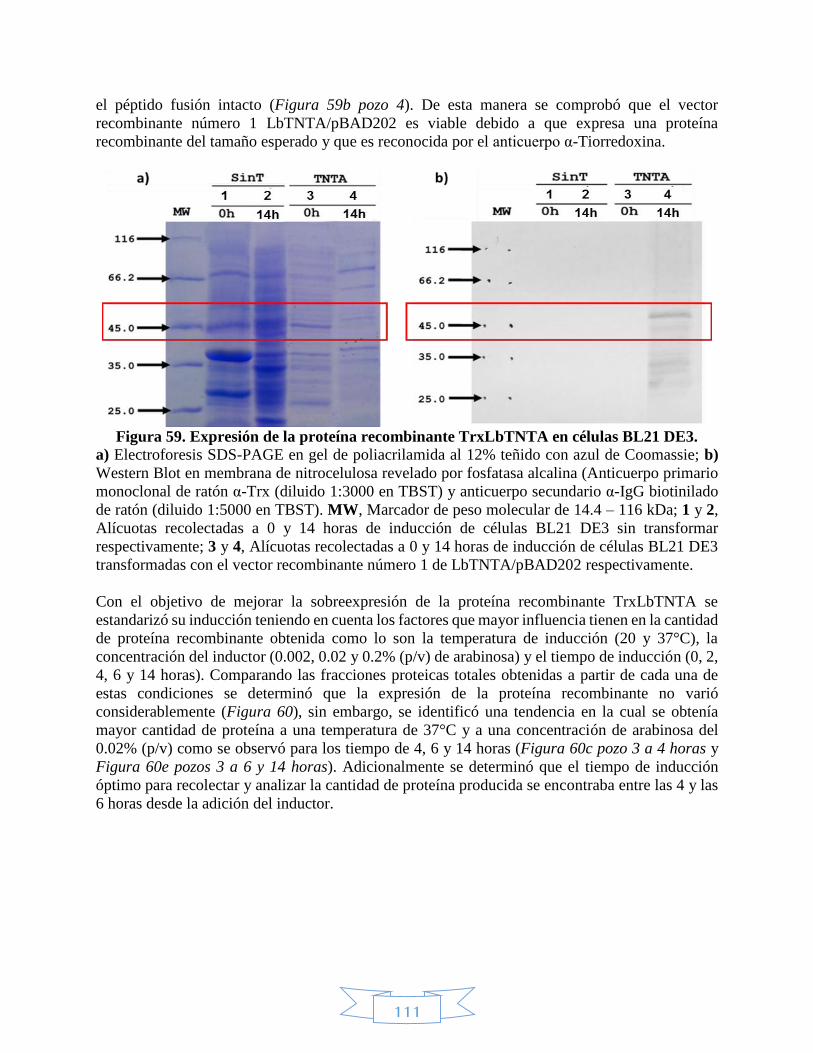
111
el péptido fusión intacto (Figura 59b pozo 4). De esta manera se comprobó que el vector
recombinante número 1 LbTNTA/pBAD202 es viable debido a que expresa una proteína
recombinante del tamaño esperado y que es reconocida por el anticuerpo α-Tiorredoxina.
Figura 59. Expresión de la proteína recombinante TrxLbTNTA en células BL21 DE3.
a) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie; b)
Western Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina (Anticuerpo primario
monoclonal de ratón α-Trx (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG biotinilado
de ratón (diluido 1:5000 en TBST). MW, Marcador de peso molecular de 14.4 – 116 kDa; 1 y 2,
Alícuotas recolectadas a 0 y 14 horas de inducción de células BL21 DE3 sin transformar
respectivamente; 3 y 4, Alícuotas recolectadas a 0 y 14 horas de inducción de células BL21 DE3
transformadas con el vector recombinante número 1 de LbTNTA/pBAD202 respectivamente.
Con el objetivo de mejorar la sobreexpresión de la proteína recombinante TrxLbTNTA se
estandarizó su inducción teniendo en cuenta los factores que mayor influencia tienen en la cantidad
de proteína recombinante obtenida como lo son la temperatura de inducción (20 y 37°C), la
concentración del inductor (0.002, 0.02 y 0.2% (p/v) de arabinosa) y el tiempo de inducción (0, 2,
4, 6 y 14 horas). Comparando las fracciones proteicas totales obtenidas a partir de cada una de
estas condiciones se determinó que la expresión de la proteína recombinante no varió
considerablemente (Figura 60), sin embargo, se identificó una tendencia en la cual se obtenía
mayor cantidad de proteína a una temperatura de 37°C y a una concentración de arabinosa del
0.02% (p/v) como se observó para los tiempo de 4, 6 y 14 horas (Figura 60c pozo 3 a 4 horas y
Figura 60e pozos 3 a 6 y 14 horas). Adicionalmente se determinó que el tiempo de inducción
óptimo para recolectar y analizar la cantidad de proteína producida se encontraba entre las 4 y las
6 horas desde la adición del inductor.
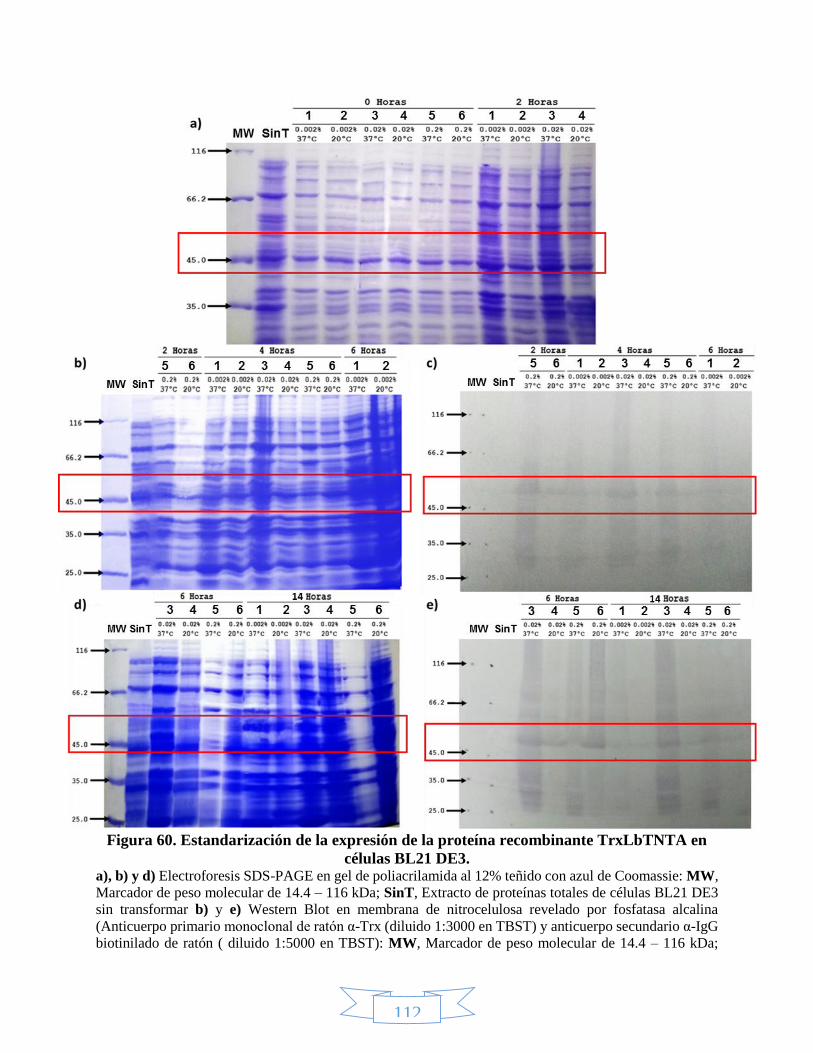
112
Figura 60. Estandarización de la expresión de la proteína recombinante TrxLbTNTA en
células BL21 DE3. a), b) y d) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie: MW,
Marcador de peso molecular de 14.4 – 116 kDa; SinT, Extracto de proteínas totales de células BL21 DE3
sin transformar b) y e) Western Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina
(Anticuerpo primario monoclonal de ratón α-Trx (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG
biotinilado de ratón ( diluido 1:5000 en TBST): MW, Marcador de peso molecular de 14.4 – 116 kDa;
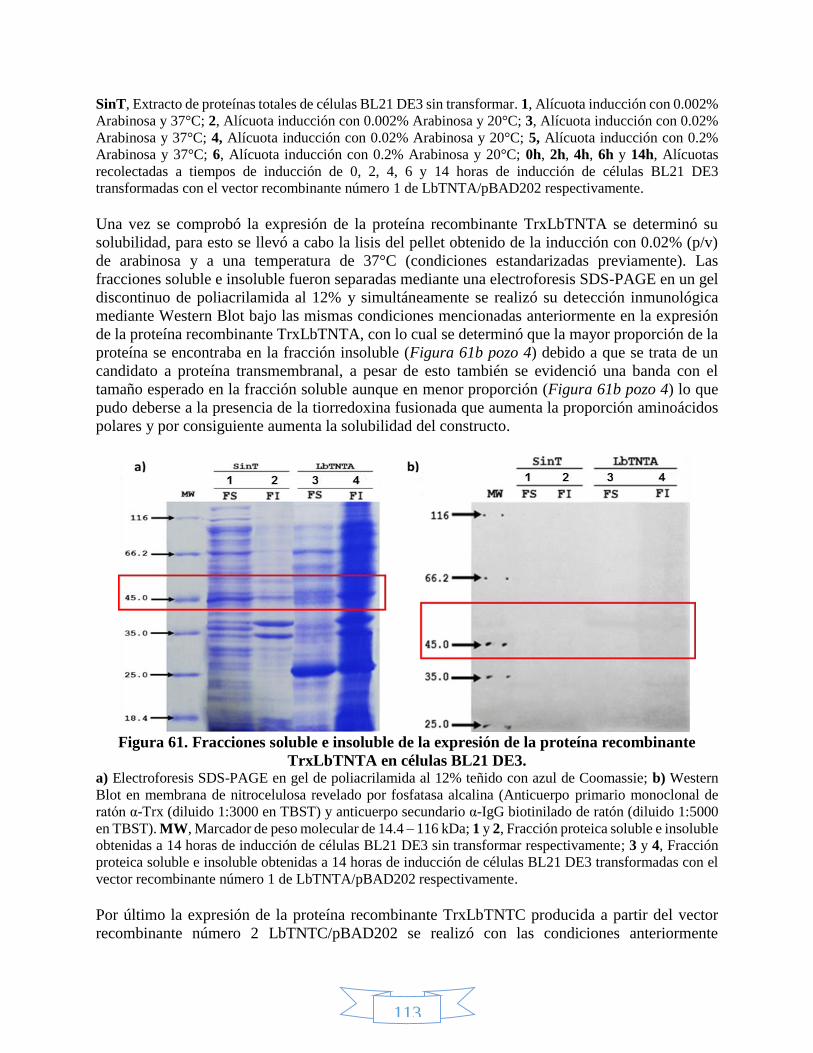
113
SinT, Extracto de proteínas totales de células BL21 DE3 sin transformar. 1, Alícuota inducción con 0.002%
Arabinosa y 37°C; 2, Alícuota inducción con 0.002% Arabinosa y 20°C; 3, Alícuota inducción con 0.02%
Arabinosa y 37°C; 4, Alícuota inducción con 0.02% Arabinosa y 20°C; 5, Alícuota inducción con 0.2%
Arabinosa y 37°C; 6, Alícuota inducción con 0.2% Arabinosa y 20°C; 0h, 2h, 4h, 6h y 14h, Alícuotas
recolectadas a tiempos de inducción de 0, 2, 4, 6 y 14 horas de inducción de células BL21 DE3
transformadas con el vector recombinante número 1 de LbTNTA/pBAD202 respectivamente.
Una vez se comprobó la expresión de la proteína recombinante TrxLbTNTA se determinó su
solubilidad, para esto se llevó a cabo la lisis del pellet obtenido de la inducción con 0.02% (p/v)
de arabinosa y a una temperatura de 37°C (condiciones estandarizadas previamente). Las
fracciones soluble e insoluble fueron separadas mediante una electroforesis SDS-PAGE en un gel
discontinuo de poliacrilamida al 12% y simultáneamente se realizó su detección inmunológica
mediante Western Blot bajo las mismas condiciones mencionadas anteriormente en la expresión
de la proteína recombinante TrxLbTNTA, con lo cual se determinó que la mayor proporción de la
proteína se encontraba en la fracción insoluble (Figura 61b pozo 4) debido a que se trata de un
candidato a proteína transmembranal, a pesar de esto también se evidenció una banda con el
tamaño esperado en la fracción soluble aunque en menor proporción (Figura 61b pozo 4) lo que
pudo deberse a la presencia de la tiorredoxina fusionada que aumenta la proporción aminoácidos
polares y por consiguiente aumenta la solubilidad del constructo.
Figura 61. Fracciones soluble e insoluble de la expresión de la proteína recombinante
TrxLbTNTA en células BL21 DE3. a) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie; b) Western
Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina (Anticuerpo primario monoclonal de
ratón α-Trx (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG biotinilado de ratón (diluido 1:5000
en TBST). MW, Marcador de peso molecular de 14.4 – 116 kDa; 1 y 2, Fracción proteica soluble e insoluble
obtenidas a 14 horas de inducción de células BL21 DE3 sin transformar respectivamente; 3 y 4, Fracción
proteica soluble e insoluble obtenidas a 14 horas de inducción de células BL21 DE3 transformadas con el
vector recombinante número 1 de LbTNTA/pBAD202 respectivamente.
Por último la expresión de la proteína recombinante TrxLbTNTC producida a partir del vector
recombinante número 2 LbTNTC/pBAD202 se realizó con las condiciones anteriormente

114
estandarizadas para TrxLbTNTA con una concentración de arabinosa del 0.02% (p/v) y una
temperatura de incubación de 37°C. Para el análisis de proteínas totales se recogieron alícuotas a
0, 2, 4, 6 y 14 horas. Para detectar la proteína fusión se realizó una electroforesis SDS-PAGE en
un gel discontinuo de poliacrilamida al 12% como se describió para la expresión de la proteína
recombinante TrxLbTNTA. En este gel no se detectó ninguna banda de sobreexpresión con el peso
esperado para la recombinante TrxLbTNTC (58.3 kDa) en ninguno de los tiempos evaluados
(Figura 62a), a pesar de esto, se procedió a realizar un Western Blot debido a que la resolución
del gel de poliacrilamida no era muy buena puesto que se observaron varias proteínas cercanas al
peso molecular esperado, sin embargo en este no se detectó la banda esperada mediante este
procedimiento (Figura 62b), por lo que se concluyó que este plásmido no era viable para producir
la proteína recombinante.
Es importante resaltar que a pesar de que este vector recombinante había sido verificado mediante
PCR y digestión con enzimas de restricción, fue descartado al no ser viable y no producir la
proteína recombinante TrxLbTNTC, esto pudo deberse a mutaciones en la secuencia del codón de
inicio que cambiaron el marco de lectura de la proteína recombinante, ya que no se identificó
ninguna banda mediante Western Blot lo que indica que el péptido fusión no se tradujo. Sin
embargo es posible que no se llevara a cabo la traducción de la proteína fusión a partir del codón
de inicio de su marco de lectura y por lo tanto no fue reconocido por el anticuerpo α-Tiorredoxina
sino que, a partir del marco de lectura de la proteína endógena, de acuerdo con esto se propone la
producción de anticuerpos para reconocer la porción de la proteína endógena y no la del péptido
fusión para llevar a cabo ensayos de inmunodetección.
Figura 62. Expresión de la proteína recombinante TrxLbTNTC en células BL21 DE3.
a) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie; b)
Western Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina (Anticuerpo primario
monoclonal de ratón α-Trx (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG biotinilado
de ratón (diluido 1:5000 en TBST). MW, Marcador de peso molecular de 14.4 – 116 kDa; 1 y 2,
Alícuotas recolectadas a 0 y 14 horas de inducción de células BL21 DE3 sin transformar; 3, 4, 5,
6 y 7 Alícuotas recolectadas a 0, 2, 4, 6 y 14 horas de inducción de células BL21 DE3
transformadas con el vector recombinante número 2 de LbTNTC/pBAD202; C+, Control positivo
TrxLbTNTA (52.8 kDa).

115
Detección de las proteínas recombinantes 6xHisLbTNT´s
Las proteínas recombinantes producidas a partir del vector pET100/D-TOPO tienen como péptido
fusión en el extremo N-terminal una secuencia de 6 histidinas (6xHis) que añaden un peso de 3
kDa a la proteína nativa. De esta manea el peso esperado de la proteína TrxLbTNTB es de 40.8
kDa y el de TrxLbTNTC de 38.3 kDa.
Para evaluar la expresión de las proteínas recombinantes 6xHisLbTNTB y 6xHisLbTNTC
producidas a partir de los vectores recombinantes 3, 4 y 5 para LbTNTB/pET100 y de los vectores
recombinantes 1, 8, 9 y 10 para LbTNTC/pET100 se recolectaron alícuotas a 0 horas y después de
incubación durante toda la noche. Las proteínas totales fueron separadas mediante electroforesis
SDS-PAGE en un gel discontinuo de poliacrilamida al 12%. En el gel obtenido para los vectores
recombinantes LbTNTB/pET100 no se evidenció una banda de sobreexpresión del tamaño
esperado (40.8 kDa) en ninguno de los pozos (Figura 63a). Sin embargo se realizó otra
electroforesis SDS-PAGE bajo las mismas condiciones y se transfirió a una membrana de
nitrocelulosa por electrotransferencia húmeda con la cual se llevó a cabo un Western Blot revelado
por fosfatasa alcalina (Anticuerpo primario monoclonal de ratón α-6xHis (diluido 1:3000 en
TBST) y anticuerpo secundario α-IgG biotinilado de ratón (diluido 1:5000 en TBST) para verificar
la presencia de la proteína recombinante en el cual se reconoció una banda en los carriles
correspondientes a 14 horas de los recombinantes 3 y 5 (Figura 63b pozos 14h para clon 3 y clon
5), sin embargo, estas bandas presentaron un peso de 38.51 kDa para el clon 3 y de 39.25 para el
clon 5 los cuales son menores al esperado (40.8 kDa) como se determinó mediante la ecuación de
la recta con un r2 de 0.9872 para cada una de las bandas analizadas (Tabla 29) es importante resaltar
que el peso calculado para el control positivo 6xHisLbNMNAT fue de 44.0 kDa lo que no
concuerda con el peso esperado (37 kDa).
Tabla 29. Determinación del peso molecular de las proteínas obtenidas a partir de los plásmidos
3 y 5 para LbTNTB.
DISTANCIA (cm) PESO MOLECULAR (kDa) Ln MW
MARCADOR
DE PESO
MOLECULAR
0.6 116 4.75359
1.5 66.2 4.19267
2.65 45 3.80665
3.45 35 3.55535
4.35 25 3.21887
5.25 18.4 2.91235
CLON 3 3.2 38.51 3.65116
CLON 5 3.15 39.25 3.67017
CONTROL
POSITIVO 2.85 44.0 3.78423

116
Figura 63. Expresión de la proteína recombinante 6xHisLbTNTB en células BL21 DE3.
a) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie; b) Western
Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina (Anticuerpo primario monoclonal de
ratón α-6xHis (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG biotinilado de ratón (diluido 1:5000
en TBST). MW, Marcador de peso molecular de 14.4 – 116 kDa; SinT, Extracto de proteínas totales de
células BL21 DE3 sin transformar; LbTNTB, Extracto de proteínas totales de células transformadas con el
vector recombinante LbTNTB/pET100; Clon 3, Clon 4 y Clon 5, Vectores recombinantes 3, 4 y 5
LbTNTB/pET100; 0h y 14h, Alícuotas recolectadas a tiempos de inducción de 0 y 14 horas; C+, Control
positivo LbNMNAT (37.0 kDa).
De igual manera para evaluar la expresión de los vectores recombinantes 1, 8, 9 y 10 de
LbTNTC/pET100 se recogieron alícuotas a 6 y 14 horas, las cuales se separaron posteriormente
mediante electroforesis SDS-PAGE en un gel discontinuo de poliacrilamida al 12% como se
describió anteriormente y simultáneamente se realizó su detección inmunológica mediante
Western Blot en el cual se reconoció una banda del mismo tamaño en los carriles correspondientes
a 0 y 14 horas del recombinante 1 (Figura 64b) en comparación con el SDS-PAGE (Figura 64a),
sin embargo, estas bandas presentaron un peso de 36.37 kDa el cual es menor al esperado (38.3
kDa) como se determinó mediante la ecuación de la recta con un r2 de 0.9893 para cada una de las
bandas analizadas (Tabla 30) es importante resaltar que el peso calculado para el control positivo
6xHisLbNMNAT fue de 40.99 kDa lo que no concuerda con el peso esperado (37 kDa). La
detección de proteínas a tiempo cero pudo deberse a la autoinducción del vector recombinante
como respuesta a una alta densidad poblacional del cultivo anterior a la adición del inductor.
El aumento de la migración para las proteínas recombinantes analizadas para los candidatos
LbTNTB y LbTNTC en comparación a la esperada y de la disminución para el control positivo
correspondiente a la NMNAT de Leishmania braziliensis en relación a la esperada pudo deberse
a la interacción y formación de micelas entre la cadena carbonada del SDS y las regiones
transmembranales de los candidatos como se ha identificado en el monómero de la proteína de
membrana fosfolamban para la cual se diseñaron diversos mutantes que alteraban su
hidrofobicidad determinándose que a mayor contenido de residuos hidrofóbicos la migración
aumentaba con respecto a la de la proteína nativa [71].

117
Tabla 30. Determinación del peso molecular de la proteína obtenida a partir del plásmido 1 para
LbTNTC.
DISTANCIA (cm) PESO MOLECULAR (kDa) Ln MW
MARCADOR
DE PESO
MOLECULAR
0.6 116 4.75359
1.5 66.2 4.19267
2.6 45 3.80665
3.35 35 3.55535
4.2 25 3.21887
5.05 18.4 2.91235
DISTANCIA (cm) PESO MOLECULAR (kDa) Ln MW
CLON 1 3.25 36.37 3.59370
CONTROL
POSITIVO 2.95 40.99 3.71334
Por otra parte no se evidenció ningún reconocimiento para los vectores recombinantes 8, 9 y 10 de
LbTNTC/pET100, esto pudo deberse a que, como se mencionó anteriormente, la secuencia
5´CCATGG 3´ (nucleótidos 403 – 408 en LbTNTC/pET100) reconocida por la enzima NcoI, que
a su vez coincide con el codón de inicio para la transcripción del candidato LbTNTC y es esencial
en el proceso de ligación presenta una mutación o que durante este proceso ocurrió un error en este
mismo punto que ocasionó que la traducción de la proteína recombinante se viera interrumpida y
por consiguiente no se obtuviera un péptido del tamaño esperado para estos plásmidos.
Figura 64. Expresión de la proteína recombinante 6xHisLbTNTC en células BL21 DE3.
a) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie; b) Western
Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina (Anticuerpo primario monoclonal de
ratón α-6xHis (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG biotinilado de ratón (diluido 1:5000
en TBST). MW, Marcador de peso molecular de 14.4 – 116 kDa; SinT, Extracto de proteínas totales de
células BL21 DE3 sin transformar; LbTNTC, Extracto de proteínas totales de células transformadas con
el vector recombinante LbTNTC/pET100; Clon 1, Clon 8, Clon 9 y Clon 10, Vectores recombinantes 1,
8, 9 y 10 LbTNTC/pET100 respectivamente; 0h y 14h, Alícuotas recolectadas a tiempos de inducción de
0 y 14 horas respectivamente; C+, Control positivo LbNMNAT (37.0 kDa).
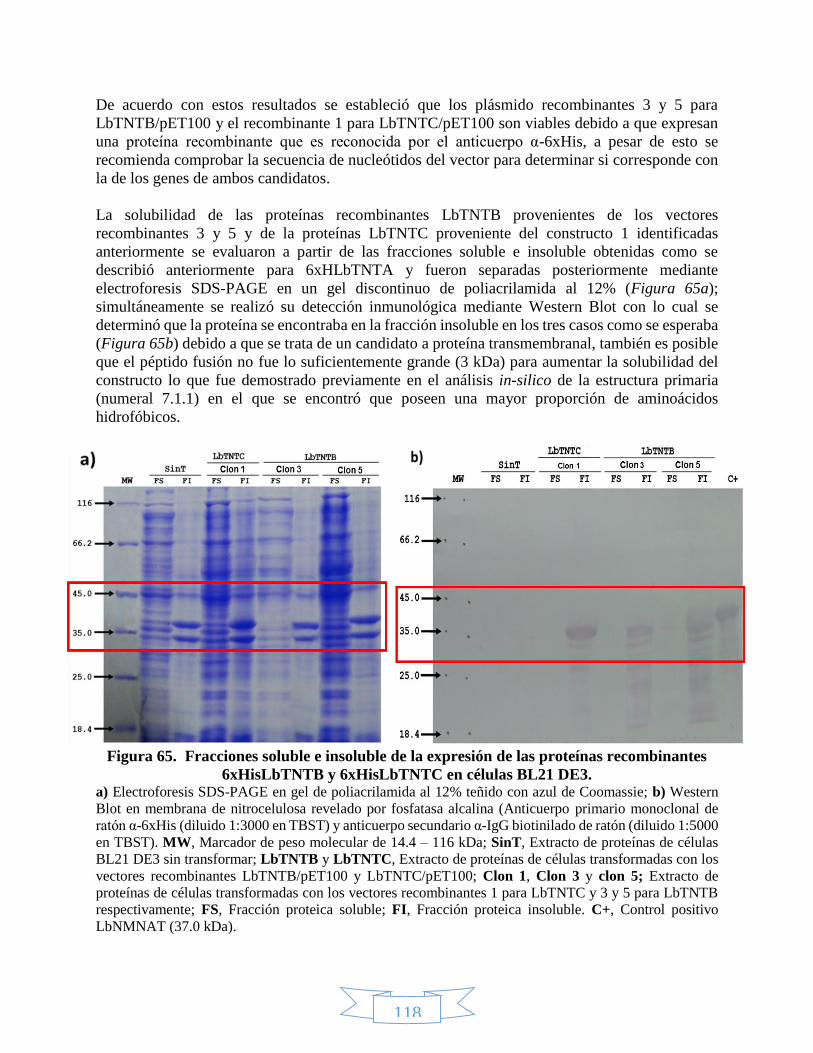
118
De acuerdo con estos resultados se estableció que los plásmido recombinantes 3 y 5 para
LbTNTB/pET100 y el recombinante 1 para LbTNTC/pET100 son viables debido a que expresan
una proteína recombinante que es reconocida por el anticuerpo α-6xHis, a pesar de esto se
recomienda comprobar la secuencia de nucleótidos del vector para determinar si corresponde con
la de los genes de ambos candidatos.
La solubilidad de las proteínas recombinantes LbTNTB provenientes de los vectores
recombinantes 3 y 5 y de la proteínas LbTNTC proveniente del constructo 1 identificadas
anteriormente se evaluaron a partir de las fracciones soluble e insoluble obtenidas como se
describió anteriormente para 6xHLbTNTA y fueron separadas posteriormente mediante
electroforesis SDS-PAGE en un gel discontinuo de poliacrilamida al 12% (Figura 65a);
simultáneamente se realizó su detección inmunológica mediante Western Blot con lo cual se
determinó que la proteína se encontraba en la fracción insoluble en los tres casos como se esperaba
(Figura 65b) debido a que se trata de un candidato a proteína transmembranal, también es posible
que el péptido fusión no fue lo suficientemente grande (3 kDa) para aumentar la solubilidad del
constructo lo que fue demostrado previamente en el análisis in-silico de la estructura primaria
(numeral 7.1.1) en el que se encontró que poseen una mayor proporción de aminoácidos
hidrofóbicos.
Figura 65. Fracciones soluble e insoluble de la expresión de las proteínas recombinantes
6xHisLbTNTB y 6xHisLbTNTC en células BL21 DE3. a) Electroforesis SDS-PAGE en gel de poliacrilamida al 12% teñido con azul de Coomassie; b) Western
Blot en membrana de nitrocelulosa revelado por fosfatasa alcalina (Anticuerpo primario monoclonal de
ratón α-6xHis (diluido 1:3000 en TBST) y anticuerpo secundario α-IgG biotinilado de ratón (diluido 1:5000
en TBST). MW, Marcador de peso molecular de 14.4 – 116 kDa; SinT, Extracto de proteínas de células
BL21 DE3 sin transformar; LbTNTB y LbTNTC, Extracto de proteínas de células transformadas con los
vectores recombinantes LbTNTB/pET100 y LbTNTC/pET100; Clon 1, Clon 3 y clon 5; Extracto de
proteínas de células transformadas con los vectores recombinantes 1 para LbTNTC y 3 y 5 para LbTNTB
respectivamente; FS, Fracción proteica soluble; FI, Fracción proteica insoluble. C+, Control positivo
LbNMNAT (37.0 kDa).

119
8. CONCLUSIONES
Se identificaron 19 residuos altamente conservados presentes en las secuencias de los 3
candidatos LbTNTA, LbTNTB y LbTNTC de los cuales es importante resaltar la presencia
del residuo de lisina K289 y el de ácido glutámico E361 (posiciones en el alineamiento global)
relacionados con el transporte de nucleótidos de adenina. Asimismo otros residuos cargados
como el ácido aspártico D60 y la arginina en las posiciones R65 y R195 las cuales podrían
tener relevancia en la interacción con el NAD+ con los tres candidatos.
Los análisis de modificaciones postraduccionales mostraron que los 3 candidatos tienen alta
probabilidad de presentar una regulación por fosforilación/desfosforilación, ya que se
identificaron 9 sitios potenciales en LbTNTA, 13 en LbTNTB y 12 en LbTNTC.
Se determinó una baja probabilidad de que los candidatos LbTNTB y LbTNTC presenten
regulación por acetilación/desacetilación ya que se encontró un único sitio, mientras que para
LbTNTA no se hallaron.
El estudio bioinformático en los tres candidatos al utilizar la herramienta InterPro demostró
la presencia de un dominio funcional de la familia de “Transportadores mitocondriales”
PF000153 en Pfam, también los motivos correspondientes a 3 repeticiones en Tándem de
aproximadamente 100 aminoácidos característicos del dominio de esta familia.
Se hallaron en los 3 candidatos 6 hélices α transmembranales mediante el análisis de estructura
primara y secundaria, estas también fueron identificadas en los modelos predictivos de
estructura terciaria generados mediante I-TASSER lo cual es característico de los miembros
de la familia de “Transportadores mitocondriales”.
Los 3 candidatos LbTNTA, LbTNTB y LbTNTC mediante las herramientas Euk-mPLoc 2.0
y iLoc-Animal predijeron su localización subcelular en la mitocondria o el peroxisoma.
Se identificó mediante I-TASSER que los 3 modelos generados para cada uno de los
candidatos presentaban una homología estructural con el transportador mitocondrial de
ADP/ATP de la especie Bos taurus con un valor C de 0.958 para LbTNTA de 0.847 para
LbTNTB y de 0.961 para LbTNTC, lo que indica una posible homología funcional.
Este estudio sugiere que las 3 proteínas candidatas poseen la capacidad de transportar
nucleótidos de adenina, como también una posible función de transporte de NAD+.
Se estandarizó la amplificación de los genes correspondientes a los 3 candidatos obteniendo
bandas únicas de 1.089, 1.014 y 957 pb para LbTNTA, LbTNTB y LbTNTC respectivamente
lo cual permitió comprobar la presencia de estas 3 secuencias en el genoma de Leishmania
braziliensis.

120
Se comprobó mediante Western blot la expresión de una proteína de ~52 kDa para
TrxLbTNTA en el plásmido recombinante número 1 obtenido a partir de la ligación del
producto de PCR de este gen con el vector pBAD202/D-TOPO.
Se determinó que la proteína recombinante TrxLbTNTA se encuentra principalmente en la
fracción insoluble lo cual está en concordancia con los resultados de predicción de solubilidad
(46.1%) del servidor ESPRESSO y también se detectó en menor proporción en la fracción
soluble.
Se demostró mediante Western blot la expresión de proteínas con ~38.51 y ~39.25 kDa para
6xHisLbTNTB y 6xHisLbTNTC al clonar los productos de PCR de estos genes 1.014 y 957
pb en el vector pET100/D-TOPO a partir de los clones número 1 para LbTNTC y 3 y 5 para
LbTNTB.
Se observó que las proteínas recombinantes obtenidas a partir de los clones 1 para LbTNTC
y 3 y 5 para LbTNTB se encuentran únicamente en la fracción insoluble.

121
9. PERSPECTIVAS
Se sugiere realizar la secuenciación del segmento correspondiente a los nucleótidos que codifican
las proteínas fusión con los cebadores universales T7 promoter 5’-TAATACGACTCACTAT
AGGG-3’ (directo) y T7 terminator 5’-GCTAGTTATTGCTCAGCGG-3’ (reverso) de los
vectores recombinantes seleccionados para los candidatos LbTNTA/pBAD202, LbTNTB/pET100
y LbTNTC/pET100 con el objeto de confirmar la secuencia de los genes de las proteínas
recombinantes TrxLbTNTA, 6xHisLbTNTB y 6xHisLbTNTC.
Para identificar el papel que juegan los aminoácidos cargados correspondientes a la lisina K289,
el de ácido glutámico E361, el ácido aspártico D60 y la arginina en las posiciones R65 y R195
(posiciones en el alineamiento global) que fueron identificados como residuos altamente
conservados en los tres transportadores se propone la construcción de mutantes delecionales para
cada uno de estos aminoácidos o mutantes en los que estos mismos residuos con carga sean
intercambiados por neutros como la glutamina (Gln).
La obtención de las proteínas recombinantes TrxLbTNTA, 6xHisLbTNTB y 6xHisLbTNTC
permitirán llevar a cabo ensayos para la producción de anticuerpos específicos para estas, con los
cuales se podrán desarrollar estudios de inmunodetección por Western blot en los extractos de
Leishmania braziliensis como también de localización subcelular in vivo de las 3 proteínas
endógenas candidatas a transportadoras de NAD+.
Por otra parte los constructos obtenidos para cada uno de los candidatos pueden ser utilizados para
evaluar la capacidad transportadora de NAD+ de las proteínas en modelos procariotas como
Escherichia coli, sin embargo es recomendable realizar la subclonación de estos genes en vectores
destinados para la expresión en organismos eucariotas como levaduras, en los cuales los
mecanismos de transporte hacia regiones específicas de la célula y de modificaciones
postraduccionales están presentes, con lo cual asegurar la adecuada actividad de las proteínas para
posteriormente evaluarla.

122
10. ANEXOS
Anexo 1. Secuencia de nucleótidos para los candidatos LbTNTA, LbTNTB y LbTNTC.

123
Anexo 2. Secuencia de aminoácidos para los candidatos LbTNTA, LbTNTB y LbTNTC.
CANDIDATO PRIMERS CÓDIGO NCBI
ALINEAMIENTO
% DE
COBERTURA
VALOR-
E PUNTAJE
TNTA
TNTAd
NC_009326 100% 4x10-7 50.1
XM_001568380.1 100% 1x10-4 42.1
XM_001562388.2 60% 1.37 30.2
TNTAr
NC_009326 100% 4x10-7 50.1
XM_001568380.1 100% 4x10-7 50.1
NC_009310 80% 1.5 28.2
TNTB
TNTBd
NC_009324 100% 6x10-6 46.1
XM_001567429.2 100% 1x104 42.1
NC_009319 100% 0.37 30.2
TNTBr
XM_001567429.2 100% 4x10-7 50.1
NC_009324 100% 4x10-7 50.1
NC_009327 100% 0.37 30.2
TNTC
TNTCd
NC_009307 100% 2x10-6 48.1
XM_001563411.1 100% 1x10-4 42.1
NC_009314 100% 0.093 32.2
TNTCr
NC_009307 100% 4x10-7 50.1
XM_001563411.1 100% 4x10-7 50.1
NC_009327 100% 1.5 28.2
Anexo 3. Especificidad de los cebadores propuestos para cada candidato mediante la herramienta
BLAST de NCBI.
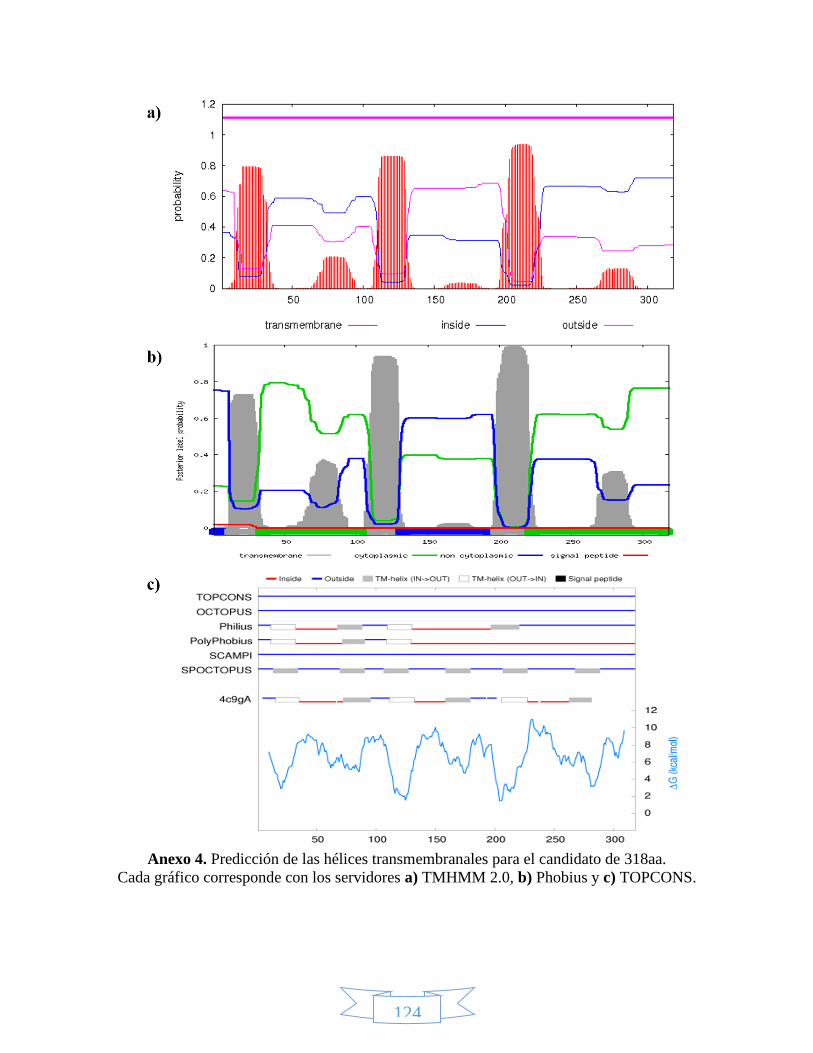
124
Anexo 4. Predicción de las hélices transmembranales para el candidato de 318aa.
Cada gráfico corresponde con los servidores a) TMHMM 2.0, b) Phobius y c) TOPCONS.

125
Anexo 5. Predicción de las hélices transmembranales para el candidato de 337aa.
Cada gráfico corresponde con los servidores a) TMHMM 2.0, b) Phobius y c) TOPCONS.

126
Anexo 6. Predicción de la estructura secundaria para los candidatos LbTNTB y LbTNTC
con el servidor PHYRE2.
a) Predicción de LbTNTB y b) Predicción de LbTNTC

127
11. REFERENCIAS
[1] Organización Mundial de la Salud, «Organización Mundial de la Salud,» 21 Septiembre
2012. [En línea]. Available:
http://apps.who.int/iris/bitstream/10665/82766/1/WHO_TRS_949_spa.pdf. [Último
acceso: 5 Septiembre 2015].
[2] J. Rodrigues Coura y J. Borges-Pereira, «Chagas disease: 100 years after its discovery. A
systemic review,» Elsevier, vol. 115, pp. 5-13, 2010.
[3] Europe PMC Funders Group, «Comparative genomic analysis of three Leishmania species
that cause diverse human disease,» Nat Genet, vol. 39, nº 7, pp. 839-847, 2007.
[4] Cold Spring Harbor Laboratory, «Chromosome and gene copy number variation allow
major structural change between species and strains of Leishmania,» Genome Research,
vol. 21, pp. 2129-2142, 2011.
[5] F. Berger, C. Lau, M. Dahlmann y M. Ziegler, «Subcellular Compartmentation and
Differential Catalytic Properties of the Three Human Nicotinamide Mononucleotide
Adenylyltransferase Isoforms,» The Journal of Biologic Chemistry, 2005.
[6] N. Forero-Baena, D. Sánchez-Lancheros, J. Constanza Buitrago, V. Bustos y M. H.
Ramírez-Hernández, «Identificationofanicotinamide/nicotinatemononucleotide
adenylyltransferaseinGiardialamblia(GlNMNAT),» Biochimie Open, vol. 1, pp. 61-69,
2015.
[7] L. Contreras, R. Neme y M. Ramírez, «Identification and functional evaluation of
Leishmania braziliensis Nicotinamide Mononucleotide Adenylyltransferase,» Protein
Expression and Purification, vol. 115, pp. 26-33, 2015.
[8] C. H. Niño Riveros, Identificación y caracterización de la Nicotinamida Mononucleótido
Adenilil Transferasa (NMNAT) en Trypanosoma cruzi: Enzima clave en el metabolismo del
NAD+, Bogotá, 2014.
[9] J. O´Hara, L. Kerwin, S. Cobbold, J. Tai, T. Bedel, P. Reider y M. Llinás, «Targeting NAD+
Metabolism in the Human Malaria Parasite Plasmodium falciparum,» PLoS ONE, vol. 9, nº
4, 2014.
[10] I. Haferkamp, S. Schmitz-Esser, N. Linka, C. Urbane, A. Collingro, M. Wagner, M. Horn
y H. E. Neuhaus, «A candidate NAD+ transporter in an intracellular bacterial symbiont
related to Chlamydiae,» Nature, vol. 432, nº 7017, pp. 622-625, 2004.
[11] I. Haferkamp, S. Schmitz-Esser, M. Wagner, N. Neigel, M. Horn y E. Neuhaus, «Tapping
the nucleotide pool of the host: novel nucleotide carrier proteins of Protochlamydia
amoebophila,» Molecular Microbiology, vol. 60, nº 6, pp. 1534-1545, 2006.
[12] Y. Zhou, W. Yang, L. Wang, Z. Zhu, S. Zhang y Z. Zhao, «Engineering NAD+ availability
for Escherichia coli whole-cell biocatalysis: a case study for dihydroxyacetone production,»
Microbial Cell Factories, vol. 12, nº 103, 2013.
[13] D. Fisher, R. Fernández y A. Maurelli, «Chlamydia trachomatis Transports NAD via the
Npt1 ATP/ADP trnaslocase,» Journal of Bacteriology, vol. 195, nº 15, pp. 3381-3386,
2013.

128
[14] S. Todisco, G. Agrimi, A. Castegna y F. Palmieri, «Identification of the Mitochondrial
NAD+ Transporter in Saccharomyces cerevisiae,» THE JOURNAL OF BIOLOGICAL
CHEMISTRY, vol. 281, nº 3, p. 1524–1531, 2006.
[15] F. Palmieri, B. Rieder, A. Ventrella, E. Blanco, P. Thi Do, A. Nunes-Nesi, U. Trauth, G. T.
J. Fiermonte, G. Afrimi, S. Kirchberger, E. Paradies, A. Fernie y E. Neuhaus, «Molecular
Identification and Functional Characterization of Arabidopsis thaliana Mitochondrial and
Chloroplastic NAD+ Carrier Proteins,» THE JOURNAL OF BIOLOGICAL CHEMISTRY,
vol. 284, nº 45, p. 31249–31259, 2009.
[16] G. Agrimi, A. Russo, P. Scarcia y F. Palmieri, «The human gene SLC25A17 encodes a
peroxisomal transporter of coenzyme A, FAD and NAD+,» Biochem. J., vol. 443, p. 241–
247, 2012.
[17] G. Pérez, «Coenzima.com,» 2016. [En línea]. Available:
http://www.coenzima.com/coenzimas_nad_y_nadh. [Último acceso: 10 Mayo 2016].
[18] OXFORD University, «Oxfordjournals,» 2016. [En línea]. Available:
http://aob.oxfordjournals.org/content/103/6/819/F1.expansion. [Último acceso: 2 Mayo
2016].
[19] School of Biological and Chemical Sciences, Queen Mary, University of London,
«INTERNATIONAL UNION OF BIOCHEMISTRY AND MOLECULAR BIOLOGY,»
17 Marzo 2016. [En línea]. Available: http://www.chem.qmul.ac.uk/iubmb/enzyme/.
[Último acceso: 15 Mayo 2016].
[20] P. Hassa, S. Haenni, M. Elser y M. Hottiger, «Nuclear ADP-Ribosylation Reactions in
Mammalian Cells: Where Are We Today and Where Are We Going?,» Microbiology and
Molecular Biology, vol. 70, nº 3, pp. 789-829, 2006.
[21] S. Patel, G. Churchill y A. Galione, «Coordination of Ca2+ signalling by NAADP,»
TRENDS in Biochemical Sciences, vol. 26, nº 8, p. 482–489, 2001.
[22] H. Lee, «Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP.,» Physiol
Rev., vol. 77, nº 4, pp. 1133-1164, 1997.
[23] J. Feldman, J. Baeza y J. Denu, «Activation of the Protein Deacetylase SIRT6 by Long-
chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins,» The Journal of
Biological Chemistry, vol. 288, nº 43, p. 31350 –31356, 2013.
[24] D. Nelson y M. Cox, Lehninger: Principles of biochemistry, New York: W H. Freeman and
Company, 2008.
[25] M. Gallardo Gómez, «Blogspot.com,» 25 Noviembre 2011. [En línea]. Available:
http://namibiobigbang.blogspot.com.co/. [Último acceso: 26 Agosto 2015].
[26] D. Botero y M. Restrepo, Parasitosis Humanas, Medellín: Corporación para Investigaciones
Biológicas (CIB), 2012.
[27] M. Cordero del Campillo y F. A. Rojo Vázquez, Parasitología General, España: McGRAW-
HILL-INTERAMERICANA De España, S.A.U., 2007.
[28] A. Gunn y S. Pitt, Parasitology: An Integrated approach, Nueva Deli: Offices, 2012.
[29] C. Perez-Iñigo, Parasitología, España: Hermann Blume Ediciones, 1976.
[30] H. M. Carrasco, «Estudio de la Superóxido Dismutasa en Trypanosoma cruzi: Purificación,
caracterización bioquímica y aplicaciones diagnósticas,» Editorial de la Universidad de
Granada, Granada, 2007.

129
[31] A. I. Zurita Novaro, Búsqueda de proteínas antigénicas de Leishmania braziliensis.
Aislamiento, caracterización génica y utilidad serodiagnóstica de la proteína de choque
térmico de 70 kDa (Hsp70).
[32] M. Walsh, «Infections Landscapes,» 28 Adril 2011. [En línea]. Available:
http://www.infectionlandscapes.org/2011/04/trypanosomiasis-part-2-chagas-disease.html.
[Último acceso: 21 Junio 2015].
[33] Asociación Colombiana de Infectología, Guía 2I Guía de atención a la Leihsmaniasis,
Bogotá, 2013.
[34] M. Remedios, M. Martínez y P. Gonzáles, «Estudio preliminar de los dípteros asociados a
cebos de estiercol y carroña en un bosque serrano de sierra de minas, Uruguay,» Acta
Zoológica Mexicana, vol. 28, nº 2, pp. 378-390, 2012.
[35] D. Sierra, I. Vélez y S. Uribe, «Identificación de Lutzomyia spp. (Diptera: Psychodidae)
grupo verrucarum por medio de microscopía electrónica de sus huevos,» Rev. biol. trop,
vol. 48, nº 2-3, 2000.
[36] Iowa State University of Science and Technology, «The Center for Food Security & Public
Health,» Octubre 2009. [En línea]. Available:
http://www.cfsph.iastate.edu/Factsheets/es/leishmaniasis.pdf. [Último acceso: 13 Agosto
2015].
[37] World Health Organization, «Essential Medicines and Health Products Information Portal,»
2015. [En línea]. Available: http://apps.who.int/medicinedocs/en/d/Jh2924s/2.4.html.
[Último acceso: 26 Agosto 2015].
[38] L. Vásquez de Ricciardi, «Terapéutica antileishmania: revisando el pasado, el presente y el
futuro,» Gac Méd, vol. 117, nº 2, pp. 93-111, 2009.
[39] A. Blanco, Química Biológica.
[40] N. Chandar y S. Viselli, Cell and Molecular Biology, China: Lippincott Williams &
Wilkins, 2010.
[41] BioROM, «BioROM,» 2011. [En línea]. Available:
http://www.biorom.uma.es/contenido/JCorzo/temascompletos/transporte/Clastra.htm.
[Último acceso: 15 Julio 2015].
[42] P. Sancho López, «Campus Universitario de la Universidad de Alcalá,» 2009. [En línea].
Available: http://www3.uah.es/sancho/farmacia/temas%2009-10/Tema%2013b%20-
%20membranas%20transporte%20%20farmacia.pdf. [Último acceso: 13 Julio 2015].
[43] O. Trentmann, C. Decker, H. Winkler y E. Neuhaus, «Charged amino-acid residues in
transmembrane domains of the plastidic ATP/ADP transporter from Arabidopsis are
important for transport efficiency, substrate specificity, and counter exchange properties,»
Eur. J. Biochem., vol. 267, pp. 4098-4105, 2000.
[44] F. Nuez y J. M. Carrillo, Los marcadores genéticos en la mejora vegetal., Valencia: Editorial
de la Universidad Politécnica de Valencia, 2000.
[45] E. Valadez y K. Günter, Huellas de ADN en genomas de plantas (teoría y protocolos de
laboratorio), México: Mundi Prensa, 2000.
[46] New England Biolabs inc., «New England Biolabs inc.,» 2015. [En línea]. Available:
https://www.neb.com/applications/dna-amplification-and-pcr. [Último acceso: 5 Julio
2015].

130
[47] G. Dorado Pérez, «Universdad de Cordoba - Aula Virtual,» 2015. [En línea]. Available:
http://www.uco.es/dptos/bioquimica-biol-
mol/pdfs/45%20CLONACI%C3%93N%20DNA%20AMPLIFICADO%20POR%20PCR.
pdf. [Último acceso: 12 Julio 2015].
[48] Thermofisher, «Invitrogen,» 7 Junio 2010. [En línea]. Available:
https://tools.thermofisher.com/content/sfs/manuals/pettopo_man.pdf. [Último acceso: 22
Agosto 2015].
[49] Thermofisher, «Invitrogen,» 7 Junio 2010. [En línea]. Available:
https://tools.thermofisher.com/content/sfs/manuals/pbad_dtopo_man.pdf. [Último acceso:
28 Febrero 2016].
[50] A. Krogh, B. Larsson, G. Von Heijne y E. Sonnhammer, «Predicting Transmembrane
Protein Topology with a Hidden Markov Model: Application to Complete Genomes,»
Journal of Molecular Biology, vol. 305, pp. 567-580, 2001.
[51] S. Reynolds, L. Käl, M. Riffle, J. Bilmes y W. Stafford Noble, «Transmembrane Topology
and Signal Peptide Prediction Using Dynamic Bayesian Networks,» PLOS: Computational
Biology, vol. 4, nº 11, 2008.
[52] L. Käll, A. Krogh y E. Sonnhammer, «An HMM posterior decoder for sequence feature
prediction that includes homology information,» Bioinformatics, vol. 21, p. i251–i257,
2005.
[53] H. Viklund, A. Bernsel, M. Skwark y A. Elofsson, «SPOCTOPUS: a combined predictor
of signal peptides and membrane protein topology,» Bioinformatics, vol. 24, nº 24, p. 2928–
2929, 2008.
[54] H. Viklund y A. Elofsson, «OCTOPUS: improving topology prediction by two-track ANN-
based preference scores and an extended topological grammar,» Bioinformatics, vol. 24, nº
15, p. 1662–1668, 2008.
[55] P. Smialowski, G. Doose, P. Torkler, S. Kaufmann y D. Frishman, «PROSO II – a new
method for protein solubility prediction,» The FEBS Journal, vol. 279, p. 2192–2200, 2012.
[56] C. Magnan, A. Randall y P. Baldi, «SOLpro: accurate sequence-based prediction of protein
solubility,» Bioinformatics, vol. 25, nº 17, p. 2200–2207, 2009.
[57] S. Hirose y T. Noguchi, «ESPRESSO: A system for estimating protein expression and
solubility in protein expression systems,» Proteomics journal, vol. 13, p. 1444–1456, 2013.
[58] L. Kelley, S. Mezulis, C. Yates, M. Was y M. Sternberg, «The Phyre2 web portal for protein
modeling, prediction and analysis,» Nature Protocols, vol. 10, pp. 845-858, 2015.
[59] A. Roy, A. Kucukural y Y. Zhang, «I-TASSER: a unified platform for automated protein
structure and function prediction,» Nat Protoc., vol. 5, nº 4, p. 725–738, 2010.
[60] Promega, «Promega,» Diciembre 2014. [En línea]. Available:
https://worldwide.promega.com/~/media/files/resources/protocols/technical%20manuals/0
/wizard%20genomic%20dna%20purification%20kit%20protocol.pdf. [Último acceso: 15
Enero 2016].
[61] S. Peña, «Hipertextos del Área de Biología,» Universidad Nacional del Nordeste de
Argentina, 1998-2007. [En línea]. Available:
http://www.biologia.edu.ar/metabolismo/met6.htm. [Último acceso: 5 junio 2016].

131
[62] J. M. Gonzáles Mañas, «Curso de biomoléculas,» [En línea]. Available:
http://www.ehu.eus/biomoleculas/index.htm. [Último acceso: 15 Febrero 2016].
[63] E. Vázques-Contreras, «Bioquímica y Biología Molecular en Línea,» Universidad Nacional
Autónoma de México, 02 Octubre 2003. [En línea]. Available:
http://laguna.fmedic.unam.mx/~evazquez/0403/estructura%20secundaria1.html. [Último
acceso: 12 Octubre 2015].
[64] J. Duran, C. Anjard, C. Stefan, W. Loomis y V. Malhotra, «Unconventional secretion of
Acb1 is mediated by autophagosome,» Journal Cell Biology, 2010.
[65] J. E. Walker, «The mitochondrial transporter family,» Medical Research Council
Laboratory of Molecular Biology, vol. 2, pp. 519-526, 1992.
[66] A. Montalvo y F. Palmieri, «Monografías de la real academia nacional de farmacia,» 28
Noviembre 2004. [En línea]. Available:
http://www.analesranf.com/index.php/mono/article/view/1343/1390. [Último acceso: 2016
Junio 12].
[67] C. Primo Planta, «Repositorio Institucional de la Universitad Politécnica de Valéncia,»
2012. [En línea]. Available:
https://riunet.upv.es/bitstream/handle/10251/15675/tfm_ceciliaprimo.pdf?sequence=1.
[Último acceso: 2016 Junio 5 2016].
[68] R. Cabezas, «Inserrción de proteínas en mebranas: Algunas características del proceso,»
Acta Biológica Colombiana, vol. 9, nº 1, 2004.
[69] K. L. Julia, M. Ulmschneider y J. Gray, «Computational modeling of membrane proteins,»
WILEY PERIODICALS, INC., vol. 83, pp. 1-24, 2014.
[70] SIB, «swissmodel,» [En línea]. Available:
http://swissmodel.expasy.org/workspace/index.php?func=special_help. [Último acceso: 18
12 2015].
[71] A. Rath, M. Glibowickaa, V. Nadeau, G. Chen y C. Deber, «Detergent binding explains
anomalous SDS-PAGE migration of membrane proteins,» Proceedings of the National
Academy of Sciences (PNAS), vol. 106, nº 6, p. 1760 –1765, 2009.
[72] L. A. Bouvier, G. E. Canepa, A. C. Pereira, C. Carrillo y M. R. Miranda, «Trypanosoma
cruzi: Transporte de metabolitos esenciales obtenidos del hospedador,» Medicina, vol. 68,
nº 5, pp. 398-404, 2008.
[73] M. Escalante, M. Montilla, S. Nicholls y P. y. P. C. Del Portillo, «Extracción de ADN de
Trypanosoma cruzi mediante tratamiento con bromuro de hexadecil-trimetil-amonio,»
Biomédica, vol. 17, pp. 120-125, 1997.
[74] Secretaría de Salud de los Estados Unidos Mexicanos, Manual de Diagnóstico y
Tratamiento de la Enfermedad de Chagas, Ciudad de México, 2014.
[75] Human Info NGO, «Essential Medicines and Health Products Information Portal,» 16 Junio
2015. [En línea]. Available: http://apps.who.int/medicinedocs/en/d/Jh2924s/2.11.1.html.
[Último acceso: 28 Junio 2015].
[76] L. F. Jiménez y H. Merchat, Biología celular y molecular, México: Pearson Educación,
2003.

132
[77] Organización Mundial de la Salud, «Oficina Regional para las Américas de la Organización
Mundial de la Salud,» Febrero 2015. [En línea]. Available:
http://www.who.int/mediacentre/factsheets/fs375/es/. [Último acceso: 29 Agosto 2015].
[78] Equipo de Enfermedades Transmitidas por Vectores, «Instituto Nacional de Salud -
Colombia,» 11 Junio 2014. [En línea]. Available: http://www.ins.gov.co/lineas-de-
accion/Subdireccion-
Vigilancia/sivigila/Protocolos%20SIVIGILA/PRO%20Leishmaniasis.pdf. [Último acceso:
29 Agosto 2015].
[79] R. Kizhakkekara y A. B. Santhamma, «Characterization of the respiratory chain of
Leishmania donovani promastigotes,» Molecular and Biochemical Parasitology , vol. 75,
pp. 43-53, 1995.
[80] T. Costa L., F. Ribeiro-Dias, M. Oliveira, J. Bezerra y M. Vinaud, «Energetic metabolism
of axenic promastigotes of Leishmania (Viannia) braziliensis,» Experimental Parasitology,
vol. 128, pp. 438-443, 2011.
[81] G. Benaim, R. Bermudez y J. A. Urbina, «Ca2+ transport in isolated mitochondrial vesicles
from Leishmania braziliensis promastigotes,» Molecular and Biochemical Parasitology,
vol. 39, nº 1, pp. 61-68, 1990.