capÍtulo 8: efectos fenotÍpicos de las mutaciones 1 · de mutaciones puntuales, deleciones o...
TRANSCRIPT

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 1
Tema 8. Efectos fenotípicos de las mutaciones
Ganancia y Pérdida de función de un gen. Fenotipos recesivos y pérdida de
función. Fenotipos dominantes por ganancia de función. Haploinsuficiencia
y efecto dominante-negativo. Alteraciones de la impronta genómica.
Ganancia y Pérdida de función de un gen
Cuando un gen está mutado por cualquiera de los mecanismos que hemos visto en el capítulo anterior,
pueden suceder fundamentalmente dos cosas: (a) que esa mutación se silenciosa, no tenga ningún
efecto pernicioso ni provoque ningún fenotipo anómalo; ó (b) que esa mutación provoque la aparición
de un fenotipo aberrante que conduzca al desarrollo de una enfermedad. En este segundo caso, que es
el que nos ocupa en esta asignatura, es importante darse cuenta de que la alteración de un gen puede
provocar un fenotipo anormal mediante dos mecanismos, fundamentalmente: la pérdida de función ó la
ganancia de función. La pérdida de función de un gen equivale a su ausencia total, ya que el gen no
codifica la proteína respectiva. En general, esto da lugar a fenotipos que se heredan de manera
recesiva, ya que es necesario que estén mutados los dos alelos para que se manifieste el fenotipo. Sin
embargo, en aquellos casos en los que hay un efecto de dosis génica, en los que se producen fenotipos
dominantes por un fenómeno llamado haploinsuficiencia, la pérdida de función genera fenotipos de
herencia dominante. En el caso de las mutaciones que provocan una ganancia de función, los
fenotipos resultantes son generalmente de herencia dominante, porque la mutación facilita que el gen
escape a los mecanismos que regulan su expresión, o bien crea una nueva función que tiene un efecto
dominante negativo. Como regla general, podemos decir que cuando las mutaciones puntuales en un
gen producen el mismo fenotipo que las deleciones de todo el gen, lo más probable es que esas
mutaciones provoquen una pérdida de función; en cambio, las mutaciones que operan a través de una
ganancia de función suelen provocar fenotipos distintos a los que genera la deleción o disrupción de ese
gen. A continuación veremos un poco más a fondo cada una de estas dos situaciones.
Fenotipos recesivos y pérdida de función
Un gen puede perder su función de varias maneras:
Mutaciones en el propio gen: cualquier tipo de mutación que interrumpa la capacidad codificante
del gen hará que se pierda su función. Como hemos visto en el capítulo precedente, puede tratarse
de mutaciones puntuales, deleciones o inserciones que interrumpen el marco de lectura, así como
alteraciones de las secuencias de ayuste (splicing). También podemos incluir aquellas mutaciones
que no afectan a la secuencia del transcrito (ARNm) sino a su estabilidad, de forma que el mensajero
se degrada rápidamente y no llega a traducirse en la proteína correspondiente.
Mutaciones que afectan a los elementos que regulan la expresión génica. Un gen puede dejar
de expresarse por mutaciones que afectan al promotor, bien por mutaciones que alteran las
secuencias de unión a factores de transcripción o bien provocando su metilación. Otro mecanismo
que disminuye la expresión génica es el que se conoce como efectos de posición, es decir, una
reordenación cromosómica que mueve un gen de su posición original a otra región del genoma que
está silenciada (una región de heterocromatina, por ejemplo). En ocasiones, esto hace que el gen

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 2
deje de expresarse. Hay varios ejemplos de enfermedades humanas producidas por este mecanismo,
como la Aniridia (por alteraciones del gen PAX6) o la Displasia Campomélica (alteraciones del gen
SOX9), enfermedades en las que muchas veces se observan translocaciones que no interrumpen
directamente los genes respectivos, sino que los separan de su contexto genómico habitual y los
colocan en una región de cromatina silenciada.
Fenotipos dominantes por ganancia de función
Los mecanismos por los que las mutaciones pueden originar una ganancia de función que se manifieste
en un fenotipo dominante son variados, pero podemos señalar tres:
Una mutación que altera la proteína de forma que ésta adquiere una nueva función. El mejor
ejemplo es el alelo "Pittsburgh" de alfa-1 antitripsina, un enzima con actividad anti-elastasa.
Habitualmente, los alelos mutantes de alfa-1 antitripsina producen una pérdida de función, con la
consiguiente disminución de la actividad enzimática y el desarrollo de una enfermedad denominada
enfisema pulmonar. En cambio, el alelo "Pittsburgh" está debido a una mutación que cambia la
metionina en posición 358 a una arginina (M358R). Esto hace que el enzima adquiera una actividad
anti-trombina que antes no tenía, dando lugar a un trastorno de la coagulación en vez de provocar
enfisema pulmonar. Las mutaciones con ganancia de función son muy frecuentes en cáncer, siendo
uno de los principales mecanismos de activación de oncogenes. Por ejemplo, la fusión de los genes
BCR y ABL, en pacientes con leucemia mieloide crónica, es el resultado de una translocación
(9;22) en la que el gen ABL —una tirosina-quinasa— pierde su región reguladora y queda fusionado
con la región 5' del gen BCR, que se expresa en células progenitoras hematopoyéticas. Esto provoca
un nuevo gen de fusión que conduce a una expresión constitutiva y des-regulada de la tirosina-
kinasa ABL en células de la serie mieloide en la médula ósea, lo que lleva el desarrollo de la
leucemia.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 3
Figura 8.1 estructura de los genes BCR y ABL, indicando los exones y las
regiones en las que se agrupan los puntos de rotura que originan la
translocación (9;22) que aparece en individuos con leucemia mieloide
crónica. La translocación da lugar a un cromosoma derivado llamado
Cromosoma Philadelphia (Ph1), que contiene la parte 5' del gen BCR
fusionada con la región 3' del gen ABL (el diagrama inferior de la
figura). Dicho cromosoma produce proteínas de fusión en las que la
actividad tirosina-quinasa de ABL está desregulada, ya que se expresa a
partir del promotor del gen BCR. La imagen está tomada de la página
Web del International Inmunogenetics Information System.
Figura 8.2 Ensayo de FISH: en las células que llevan la translocación hay
una fusión de la señal verde (del gen BCR) y la señal roja (del gen ABL),
dando como resultado una señal amarilla que indica la presencia de la
translocación. En ambos núcleos (con fluorescencia azul debida a la
tinción con DAPI), aparece una señal verde (correspondiente al
cromosoma 22 normal), una señal roja (correspondiente al cromosoma
9 normal) y una señal amarilla que corresponde al cromosoma
Philadelphia con la translocación.
Ganancia de función también por la sobre-expresión de un gen, como resultado de duplicaciones
o amplificaciones génicas: un segmento genómico que se copia varias veces, de forma que los genes
contenidos en ese segmento estarán en un número de copias muy superior al normal. Este también
es un mecanismo frecuente en distintos tipos de cáncer. Por ejemplo, el oncogén C-MYC está
amplificado en varios tumores (sobre todo en neuroblastomas), C-ERBB2 en tumores de mama, etc.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 4
Aunque estas amplificaciones pueden verse en un cariotipo convencional como regiones de tinción
homogénea (Homogenous Staining Regions, HSR), o también como "Dobles Diminutos" (cromosomas
minúsculos), la técnica de FISH las detecta con mucha mayor sensibilidad y permite además contar
el número de veces que el gen se ha amplificado.
Figura 8.3 Ensayo de FISH para la detección del número de copias del gen
ERBB2 en cortes de parafina de tumores de mama. Como se ve en esta
imagen, algunos tumores tienen una amplificación del gen (muchas
señales verdes por cada núcleo). Esto tiene importancia clínica, porque los
tumores de mama con amplificación de ERBB2 responden a un fármaco
llamado Herceptin, mientras que los tumores de mama sin amplificación de
ERBB2 no responden a este fármaco.
Haploinsuficiencia y efecto dominante-negativo
Aunque la pérdida de función de un gen suele desencadenar fenotipos recesivos, a veces se obtienen
fenotipos con herencia dominante. Esto sucede en aquellos casos en los que la disminución de la dosis
génica al 50% de lo que es habitual ya es capaz de provocar el fenotipo patológico. Como es sabido,
los genes autosómicos y el cromosoma X en mujeres están en doble dosis génica (hay dos alelos para
cada gen), lo que hace que en circunstancias normales se produzcan unos niveles determinados de
producto proteico. Habitualmente, la pérdida de uno de los alelos no tiene ningún efecto patológico:
aunque sólo se produce la mitad de proteína de lo que sería normal, la cantidad producida por el alelo
normal es suficiente para realizar la función respectiva. Sin embargo, en algunos casos la simple
disminución de la dosis génica a la mitad, por la inactivación de uno de los alelos mediante cualquier

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 5
mecanismo mutacional de los que se han estudiado, provoca el desarrollo del fenotipo patológico.
Cuando esto sucede, se habla de enfermedades causadas por haploinsuficiencia o insuficiencia
haploide, que lógicamente se heredan con un patrón dominante.
Figura 8.4 Haploinsuficiencia. En condiciones normales, una mutación con
pérdida de función sólo produce efectos fenotípicos cuando ambos alelos
están mutados (genotipo aa, umbral inferior). En ocasiones, cuando el
umbral necesario para la correcta funcionalidad de la proteína es más alto
(umbral superior en la gráfica de la derecha), la mutación de un solo alelo
(Aa) es suficiente para provocar la enfermedad.
Aunque hay bastantes enfermedades humanas causadas por haploinsuficiencia, un buen ejemplo es el
Síndrome de Turner (45,X0), en el que basta la pérdida de una copia de los genes implicados para
que se desarrollen las características fenotípicas propias de esta enfermedad. En concreto, se ha visto
que uno los genes responsables de esta enfermedad (SHOX) se localiza en la región pseudoautosómica
del cromosoma X y habitualmente escapa a la inactivación del cromosoma X, por lo que en condiciones
normales está presente en doble dosis génica. Cuando se pierde una copia de este gen (por la ausencia
de uno de los cromosomas X), la única copia que queda no es suficiente para llevar a cabo
correctamente su función y esto parece ser la causa de la talla corta de las mujeres con Síndrome de
Turner.
Otro modo por el que mutaciones con pérdida de función pueden dar lugar a fenotipos de herencia
dominante es el efecto dominante-negativo. Consiste en que ciertos tipos de genes codifican
moléculas que funcionan como dímeros o forman parte de complejos multiméricos, de modo que basta
la mutación en uno de los alelos del gen para que las moléculas proteicas mutantes desestabilicen
totalmente los complejos de los que forman parte. Hay dos ejemplos típicos que ilustran muy bien
este fenómeno:

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 6
A. Enfermedades que afectan a los genes que codifican alguna de las cadenas de colágeno. La
molécula de pro-colágeno es un trímero formado por distintos tipos de cadenas, cuya combinación da
lugar a los diferentes tipos de colágeno que existen. De modo general, una mutación en el gen de una
de las cadenas provocará la desestabilización de todo el complejo trimérico, por lo que basta la mutación
en un solo alelo para que se desarrolle la enfermedad. En estos casos, puede darse la circunstancia de
que una mutación puede tener distinta gravedad en función del número de moléculas mutantes y
nativas que se generan.
Figura 8.5 El video ilustra la diferencia entre mutaciones con pérdida de
función y mutaciones con ganancia de función en un mismo gen, utilizando
el ejemplo del procolágeno tipo I. Cada molécula de colágeno tipo I es un
trímero formado por dos cadenas alfa-1 (codificadas por el gen COL1A1) y
por una cadena alfa-2 (codificada por el gen COL1A2). En circunstancias
normales, se mantiene un equilibrio tal que el locus COL1A1 produce el
doble de cadenas que el locus COL1A2, con lo que se forma el procolágeno
I sin problemas.
Una mutación con ganancia de función en uno de los alelos del gen COL1A1 provocará que la mitad de
las cadenas alfa-1 sean anormales, de manera que la mayoría de los trímeros estarán alterados y la
cantidad de procolágeno I normal será muy baja (de hecho, muy inferior al 50%). Esto causa una
enfermedad llamada Osteogénesis Imperfecta tipo I. En cambio, una mutación que anule uno de los
alelos del gen COL1A1 (pérdida de función) creará una situación en la que el número total de cadenas
alfa-1 será la mitad del normal. El resultado final es la formación de trímeros de procolágeno I normales,
pero en número equivalente al 50% de las personas sanas. Esto provoca una osteogénesis imperfecta
mucho más leve que en el caso anterior.
B. Receptores de las vías de transducción de señales: constituyen otro gran grupo de proteínas
que suelen dar lugar a fenotipos de herencia dominante, habitualmente por un efecto dominante
negativo de las moléculas mutadas. A continuación se señalan dos de los ejemplos más ilustrativos:
Los genes que codifican receptores para el factor de crecimiento fibroblástico (FGF). Tras la
unión del ligando extracelular al receptor, unión mediada por heparan-sulfato de la matriz
extracelular, los receptores forman homo- o heterodímeros, se transfosforilan en el dominio quinasa
intracelular y a continuación fosforilan proteínas intracelulares implicadas en diversas vías de
transducción de señales. Las mutaciones que afectan a los genes de estos receptores (FGFR1,
FGFR2 y FGFR3) causan varios síndromes de craniosinostosis y estatura baja cuyas bases
moleculares están comenzando a comprenderse. Por ejemplo, la Acondroplasia (la causa más
frecuente de estatura baja) es una enfermedad de herencia autosómica dominante debida a
mutaciones en el gen del receptor del Factor de Crecimiento Fibroblástico-3 (FGF3). Este receptor
forma un homodímero que se activa en respuesta a su ligando natural. En cambio, en los pacientes
con acondroplasia se encuentran mutaciones en uno de los alelos del gen, de forma que la molécula
mutada conduce a la dimerización del receptor incluso en ausencia de ligando. Como resultado, se
obtiene una activación constitutiva de la cascada de señales iniciada por el receptor, que en último
término provoca el cierre prematuro del cartílago de crecimiento de los huesos largos y la baja talla
característica de esta enfermedad. La mutación más frecuente en casos de acondroplasia es la
mutación G380R del gen FGFR3, que afecta al dominio transmembrana del receptor.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 7
Curiosamente, mutaciones en otras regiones de este mismo gen causan fenotipos similares pero no
idénticos, como por ejemplo la displasia tanatofórica (micromelia e hipoplasia de costillas con
fallo respiratorio). Estas enfermedades son de herencia dominante por el efecto dominante
negativo de estas mutaciones, que hace que con una sola molécula mutada ya que se produzca la
activación constitutiva del receptor en ausencia de ligando. Respecto a mutaciones en otros genes
que codifican receptores de FGF (FGFR1 y FGFR2), también causan enfermedades con alteraciones
de huesos y cartílagos, como el Síndrome de Crouzon (craniosinostosis, bien aislada o bien
asociada con otros defectos esqueléticos), el Síndrome de Pfeiffer (pulgares y primer dedo del pie
anchos con anquilosis interfalángica), el Síndrome de Jackson-Weiss (lo anterior más
coalescencia tarso-metatarsiana) y el Síndrome de Apert (sindactilia severa).
Mutaciones que afectan al gen RET. Este gen codifica un receptor tirosina-quinasa, cuyo ligando
es el GDNF (Glial cell line-Derived Neurotrophic Factor). Las mutaciones de RET son muy
interesantes, ya que muestran cómo un mismo gen puede sufrir mutaciones que provocan
pérdida o ganancia de función, dando lugar a fenotipos diferentes. En el caso concreto de RET,
algunas mutaciones con cambio de sentido afectan a aminoácidos del dominio extracelular o del
dominio quinasa, y provocan la activación del receptor de manera independiente de ligando. Estas
mutaciones carcinoma medular de tiroides familiar u otros cuadros de cáncer familiar
denominados MEN2 (Multiple Endocrine Neoplasia), que son de herencia dominante porque un solo
alelo mutado desencadena la enfermedad por un efecto dominante negativo. Por el contrario,
algunas mutaciones de RET que llevan a la pérdida de función (mutaciones sin sentido, cambios del
marco de lectura, deleciones, etc) provocan la ausencia de las moléculas del receptor codificadas
por el alelo mutado, con el resultado de que la intensidad de la señalización de esta vía se ve
disminuida. Esto provoca un déficit en la migración de precursores neurales al tubo digestivo
durante el desarrollo embrionario, lo que da lugar a la Enfermedad de Hirschsprung (megacolon
agangliónico: ausencia congénita de los plexos submucosos y mientérico del colon distal). Esta
enfermedad se hereda con un patrón dominante, aunque también se han identificado mutaciones en
otros genes que causan la misma enfermedad con herencia recesiva, como el gen EDNRB del
receptor para endotelina B (receptor de proteinas G) ó también el gen de su ligando EDN3
(endotelina 3).
Figura 8.6 Este video muestra la familia de receptores tirosina-quinasa,
que ilustra los efectos de distintas mutaciones activadoras (con
ganancia de función) e inactivadoras (con pérdida de función) en un
mismo gen, y el efecto dominante negativo de las mutaciones
activadoras. Varios receptores de esta familia, como FGFR3 ó RET, dan
lugar a fenotipos dominantes.
Alteraciones de la impronta genómica
El imprinting o impronta genómica es la marca epigenética que define una región genómica como
materna ó paterna en el cigoto. La existencia de este fenómeno se descubrió al crear embriones de
ratón por transferencia nuclear: cambiando un pronúcleo masculino por un segundo pronúcleo femenino
se obtiene un ginogenonte; reemplazando el pronúcleo femenino por un segundo pronúcleo masculino se
obtiene un androgenonte. En teoría, estos embriones (excepto los androgenontes YY) deberían ser

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 8
viables, pero en cambio se observó que estas modificaciones son letales en el periodo embrionario, y
que esa letalidad tiene dos formas diferentes: los ginogenontes muestran un embrión normal con falta
de desarrollo de tejidos extraembrionarios, mientras que los androgenontes muestran más
alteraciones en el embrión que en tejidos extraembrionarios. En humanos, las concepciones
uniparentales androgenéticas, que se originan raramente por la pérdida de los cromosomas maternos
poco después de la fertilización, se manifiestan en una estructura que se conoce como mola
hidatidiforme completa. Por el contrario, los oocitos que se dividen por partenogénesis sólo contienen
material materno, y dan lugar a quistes dermoides. Estos hallazgos se explican actualmente por la
existencia en el genoma de genes "improntados" (sometidos al fenómeno de la impronta), en los que
de algún modo se reconoce el origen parental de cada alelo y se produce el silenciamiento selectivo de
uno de ellos: en unos casos siempre se silencia el materno, en otros genes siempre se silencia el alelo
paterno. Por tanto, cuando el núcleo del cigoto sólo contiene material de uno de los progenitores, pero
no una mezcla de ambos, los embriones resultantes son inviables. Esto sugiere la existencia de una
asimetría de los genomas materno y paterno, en cuanto a que ambos genomas tienen silenciados
distintos grupos de genes. Esta asimetría es necesaria para el correcto desarrollo embrionario.
Dado que ambos alelos de los genes sometidos a imprinting son idénticos y capaces de interaccionar con
los mismos factores de transcripción, el mecanismo que silencia uno de ellos de manera estable y
heredable debe ser un mecanismo epigenético, que haga posible que el imprinting pueda borrarse
durante la gametogénesis y re-establecerse tras la fecundación. La metilación, como mecanismo
silenciador de la expresión génica, cumple la mayor parte de estos requisitos, y hay bastantes líneas de
evidencia que apoyan el papel de la metilación en el fenómeno de imprinting: 1) todos los genes
improntados descritos hasta el momento -excepto uno- muestran regiones con metilación diferencial
(abreviadas DMR en inglés, regiones que están metiladas en uno de los alelos pero no en el otro), cuyos
patrones de metilación se establecen durante la gametogénesis y se mantienen gracias a la metilación
específica de ADN hemi-metilado; 2) los embriones de ratón dnmt1-/- (que carece de ADN-
metiltransferasa-1) muestran expresión bi-alélica de genes improntados, es decir, en ausencia de la
metilasa de mantenimiento se re-expresa el alelo que estaba silenciado; 3) se ha descrito el caso de una
mujer con mutaciones en el gen DNMT3L (que codifica una metilasa) que tiene infertilidad porque los
embriones fallecen al implantarse en el útero; y 4) no se ha encontrado un fenómeno similar al
imprinting en especies incapaces de llevar a cabo metilación.
Igualmente, se ha comprobado que los genes sometidos a imprinting también se replican de manera
asincrónica: por FISH se suelen ver dos señales de hibridación correspondientes a las dos cromátides
que se forman durante la fase S del ciclo celular, pero en genes improntados se ve un cromosoma con
dos señales y el otro cromosoma homólogo con una sola señal. Esto sugiere que el alelo específicamente
silenciado se replica más tarde.
Existen varios modelos para explicar cómo la metilación diferencial da lugar a las diferencias de
expresión entre alelos paterno y materno, y cómo se regulan genes vecinos que tienen patrones de
imprinting opuestos. Parece claro que en algunos casos es muy importante la proteína CTCF (CTC-
binding factor) una proteína que se une a la las regiones de metilación diferencial (DMR) únicamente
cuando éstas no están metiladas. Por ejemplo, se ha visto que la regulación de los genes Igf2 y H19 en
ratón (que tienen patrones de imprinting opuestos) se lleva a cabo por este mecanismo, ya que CTCF
actúa como aislador (insulator) del promotor de Igf2 de unos potenciadores lejanos.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 9
Figura 8.7 Tres modelos de regulación de los genes que están sometidos a
imprinting. En la figura superior vemos el caso más sencillo en que una
Región de Metilación Diferencial (DMR), representada como un rectángulo
naranja, regula la expresión de un único gen: si la DMR está metilada
(círculo azul), el gen está silenciado y no se expresa. En el medio se
representa otro modo de co-regulación de dos genes: uno de ellos se
expresa en un alelo pero está silenciado en el otro alelo (línea terminada
en punto). El otro gen del cluster tiene el patrón de expresión recíproco,
debido a la metilación de una DMR en uno de los genes, cuya expresión
interfiere con la expresión del otro ya que sus regiones codificantes se
solapan: cuando el gen de la izquierda se transcribe, esto interfiere con la
transcripción del gen de la derecha, que queda por tanto silenciado. El
tercer modo de co-regulación de genes con impronta recíproca se debe a la
unión de "aisladores" ó elementos "frontera" a una DMR. Por ejemplo, en
la figura inferior se representa uno de estos elementos (óvalo azul), que
únicamente puede unirse a una DMR cuando no está metilada (alelo 1), de

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 10
modo que el aislador bloquea la acción de un potenciador lejano
(rectángulo azul) sobre el gen de la izquierda; por tanto este gen está
silenciado, mientras que el gen de la derecha se expresa. En cambio, la
metilación de la DMR (alelo 2) silencia la expresión del gen de la derecha
pero además impide la unión del aislador; como consecuencia, el
potenciado puede ahora actuar sobre el promotr del gen de la izquierda y
hacer que éste se exprese.
La importancia de la proteína CTCF se ha puesto de manifiesto gracias a un hallazgo sorprendente, al
conseguir ratones partenogenéticos viables cuando se deleciona de su genoma el gen H19 y la
región de metilación diferencial a la que se une CTCF. Inicialmente, un ratón partenogenético lleva
dos copias del genoma materno, por lo que tendrá expresión bialélica del gen H19 y silenciamiento de
ambos alelos de Igf2, de ahí que sean inviables. La deleción de una de las copias del H19 reestablece la
expresión monoalélica de este gen, pero todavía hay silenciamiento total de Igf2e inviabilidad
embrionaria. En cambio, si además se deleciona la región de metilación diferencial a la que se une CTCF,
ahora los potenciadores pueden promover la expresión de Igf2 en este cromosoma, con lo que se
reestablece la expresión monoalélica de Igf2. El resultado final es la expresión monoalélica de H19 de
uno de los cromosomas y la expresión monoalélica de Igf2 del cromosoma homólogo, que es la situación
normal. Esto ilustra la importancia que tiene la impronta genómica en el desarrollo embrionario, y
sugiere que la ineficacia de los procesos de clonación de mamíferos probablemente se deba a que
los núcleos somáticos utilizados para la transferencia nuclear no tienen los patrones de imprinting
necesarios para la viabilidad embrionaria.
Figura 8.8 El video muestra la importancia del fenómeno del imprinting en
el desarrollo embrionario. Los genes Igf2 y H19 forman un cluster de
impronta recíproca en ratón, de forma que Igf2 se expresa sólo en el
cromosoma paterno y H19 se expresa sólo en el cromosoma materno. La
regulación de este cluster se lleva a cabo por un factor aislador llamado
CTCF, que se une a una región de metilación diferencial (el último modelo
explicado en la figura anterior). Como muestra el video, ratones obtenidos
por partenogénesis, que llevan dos copias del genoma materno, son
inviables porque Igf2 está silenciado en ambos cromosomas. En cambio, la
deleción de la región de metilación diferencial en uno de los cromosomas
hace que se recupere la expresión monoalélica de ambos genes y que los
embriones de estos ratones sean viables.
En cada ciclo reproductivo, los patrones de imprinting han de establecerse de nuevo, puesto que lo que
en una meiosis fue material de origen paterno (transmisión de un varón a su hija, por ejemplo) en la
siguiente meiosis pasará a ser marcado como materno (cuando esa hija tenga descendencia). Para
poder cambiar el imprinting es necesario borrar toda la información previa sobre cuál de los alelos debe
silenciarse. Como el genoma está muy desmetilado en los gametos, el cambio en los patrones de
imprinting debe de tener lugar durante la gametogénesis. Con los datos actuales, la hipótesis más
aceptada para explicar el borrado y re-establecimiento de los patrones de metilación durante la

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 11
gametogénesis es la existencia de proteínas capaces de unirse a las DMR no-metiladas (y así protegerlas
de la metilación), junto con otros factores que se unen a las DMR metiladas y ayudan a mantener el
estado silenciado. Se piensa que sólo una de las líneas germinales (masculina ó femenina) es
capaz de producir los factores que protegen de la metilación a las regiones no-metiladas. Así,
estas regiones se mantendrán des-metiladas en la línea germinal que produzca estos factores. Por el
contrario, estas regiones se metilarán y silenciarán en la línea germinal que no los produce. Por lo que
respecta a la identidad de dichos factores, los candidatos ideales son las proteínas que se unen
específicamente a regiones metiladas (MeCP2, HDAC, etc) ó a regiones no metiladas (CTCF, factores de
transcripción que se unen a CpGs no metilados, etc).
La impronta es, por tanto, un mecanismo fisiológico normal. Sin embargo, algunas alteraciones
genéticas que afectan a regiones sometidas a imprinting pueden dar lugar a enfermedades. Este es el
caso de la llamada disomía uniparental, situación en la que ambas copias de de un cromosoma o de
una región cromosómica proceden del mismo progenitor. Si esa región contiene genes “improntados”, el
resultado puede ser la presencia de dos copias “silenciadas” de un gen, con lo que el efecto viene a ser
similar a una deleción homocigótica de ese locus. Hoy en día conocemos una larga lista de enfermedades
y tumores humanos debidos a disomía uniparental de loci sometidos a impronta. Además, se han
detectado regiones improntadas en los cromosomas 11, 15, 7 y 14, y actualmente se conocen unos 30
genes improntados en ratón y humanos.
Los loci sometidos a impronta tienden a aparecer agrupados en determinadas regiones cromosómicas,
de manera que —dentro de cada uno de estos grupos o clusters— unos genes muestran expresión
materna y otros genes muestran expresión paterna. Esto se ilustra con el ejemplo de dos enfermedades
llamadas Síndrome de Prader-Willi y Síndrome de Angelman, originados por alteraciones en el
grupo de genes improntados que se encuentran en 15q11-q13. Todos los genes improntados en esta
región muestran expresión específica del alelo paterno excepto el gen UBE3A, que muestra
expresión materna en cerebro. Parece que la expresión de UBE3A depende de la expresión de un
transcrito antisentido que comienza 6,5 kb en dirección 3’ del codón de parada de UBE3A y que
alcanza hasta la mitad 3’ de UBE3A, de manera que en la mayoría de los tejidos somáticos este
transcrito no se expresa y por tanto la expresión de UBE3A en estos tejidos es bialélica. Por el contrario,
el transcrito antisentido se expresa en células de Purkinje, neuronas del hipocampo y células mitrales
olfatorias, silenciando el alelo paterno y originando el imprinting de UBE3A en estos tejidos.
Por estudios en pacientes con microdeleciones, sabemos que el centro de imprinting de esta región es
bipartito y cubre en total unas 100 kb. Dentro de esta región, el centro de imprinting para Angelman
cubre 1,15 kb, mientras que el centro de imprinting Prader-Willi es más distal y cubre unas 4,3 kb
(incluyendo el promotor y el exón 1 del gen SNRPN). Se postula que durante la gametogénesis estos
centros fijan los patrones de expresión de toda esta región, de forma que en la oogénesis se activa el
centro de imprinting AS —que, a su vez, bloquea la función del centro de imprinting PW. Como
resultado, los gametos femeninos sólo expresan el gen UBE3A. En cambio, en la espermatogénesis el
centro AS estaría bloqueado, de manera que el centro de imprinting PW es ahora activo y promueve la
expresión de todos los genes de expresión paterna, incluído el gen antisentido que bloquea la expresión
de UBE3A. De este modo se crean los epigenotipos paterno y materno que se mantendrán durante el
desarrollo embrionario y en células somáticas adultas.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 12
Figura 8.9 se muestra un esquema de la organización de este cluster, con
los genes de expresión paterna en rojo y los genes de expresión materna
(UBE3A) en amarillo. El centro de imprinting, localizado en la región 5' del
gen SNRPN, se representa como dos óvalos verdes. En el cromosoma
paterno, el centro de imprinting de Prader-Willi favorece la expresión de
los genes en rojo, incluído el gen "Antisense", cuya expresión impide la
transcripción de UBE3A. En el cromosoma materno, el centro de imprinting
de Angelman inhibe la función del centro de imprinting de Prader-Willi,
silenciando los genes en rojo y permitiendo la expresión de UBE3A.
El Síndrome de Prader-Willi, cuya incidencia estimada es de 1/25.000, se caracteriza por disminución
de la actividad fetal, obesidad, hipotonía muscular, retraso mental, hipogonadismo hipogonadotrófico,
manos y pies pequeños, hipopigmentación, baja estatura y rasgos dismórficos. Se debe a la falta de
expresión de los genes específicos del cromosoma paterno dentro de esta región. Por el contrario, el
Síndrome de Angelman tiene una incidencia de 1/15.000 nacimientos y está caracterizado por un
fenotipo distinto al de Prader-Willi: retraso mental severo, temblor, ataxia, marcha anormal, tendencia a
la risa, transtornos del sueño y convulsiones. Está provocado por la falta de expresión del gen UBE3A,
que se expresa específicamente en el cromosoma materno. Las alteraciones de la impronta de todos
estos genes pueden tener lugar por varios mecanismos:
1) Deleciones grandes de la región: producirán Síndrome de Prader-Willi (SPW) si la deleción afecta al
cromosoma paterno, ó Síndrome de Angelman (AS) si la deleción afecta al cromosoma materno.
Constituyen la principal causa de estas enfermedades (70% de todos los casos).
2) Disomía uniparental (UPD), debida a no-disyunción durante una de las meiosis parentales. En
efecto, la no-disyunción puede originar gametos disómicos o nulisómicos, que originan cigotos
trisómicos o monosómicos. En estas circunstancias, la disomía uniparental puede ser resultado tanto
de la recuperación de un cigoto trisómico por pérdida de uno de los cromosomas, como de la
duplicación completa de un cromosoma para recuperar un cigoto monosómico. En este último caso
vemos una isodisomía (ambos cromosomas provienen de la duplicación de un mismo cromosoma

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 13
parental), mientras que un 70% de los casos de reducción de cigotos trisómicos dan lugar a
heterodisomía (presencia de los dos cromosomas del mismo progenitor). La presencia de dos
cromosomas 15 paternos da lugar a un 2% de los casos de Síndrome de Angelman, mientras que el
25% de los casos de Síndrome de Prader-Willi se deben a la presencia de dos cromosomas 15
maternos.
3) Mutaciones puntuales en los genes responsables. En el Síndrome de Angelman se detectan
mutaciones en el gen UBE3A (que codifica la E6 ubiquitin-ligasa) hasta en un 20% de los enfermos.
En el caso del Síndrome de Prader-Willi, en cambio, la búsqueda de un gen candidato no ha dado
resultados, aunque el principal gen implicado es SNRPN. En el caso concreto de este gen (SNRPN),
se ha visto que existe otra secuencia codificante en dirección 5’ (SNURF por "SNRPN Upstream
Reading Frame") de manera que, de hecho, ambos forman un único gen bicistrónico: un solo
transcrito del que se traducen dos proteínas. Con los datos actuales, hay que considerar el
Síndrome de Prader-Willi como un síndrome de microdeleción causado por la pérdida de varios
genes contiguos.
4) Mutaciones o microdeleciones que afectan a los centros de imprinting, de manera que no se
podrán re-establecer los patrones durante la gametogénesis en el caso de que haya
transmisión de un sexo al opuesto (madre-hijo ó padre-hija). El resultado funcional en el
cigoto es idéntico a lo que sucede en la disomía uniparental: ambos alelos llevan el mismo patrón de
imprinting. Este tipo de alteraciones son responsables de un 5% de los casos de Síndrome de
Prader-Willi o de Síndrome de Angelman. El ejemplo típico es el de la abuela paterna portadora de
una mutación en el centro de imprinting del cromosoma 15 que lleva marcado como materno. Al
transmitir este cromosoma a un hijo varón, el cromosoma mutado pasará a su hijo marcado como
materno, por lo que no es necesario reestablecer el imprinting y el hijo será normal (llevará un
cromosoma marcado como paterno y otro como materno). En la siguiente generación, en cambio,
este individuo varón deberá pasar su cromosoma 15 marcado como paterno, por lo que la marca
que lo identificaba como materno deberá ser borrada durante la espermatogénesis y sustituida por
la marca que lo identifique como paterno. En este individuo dicho cambio será imposible, debido a la
mutación en el centro de imprinting de su cromosoma 15 materno, por lo que su descendencia
llevará dos cromosomas con epigenotipo materno y desarrollará Síndrome de Prader-Willi. Lo mismo
puede suceder para el Síndrome de Angelman, cuando la mutación del centro de imprinting afecta al
cromosoma paterno.
El diagnóstico genético en estas dos enfermedades va dirigido a la detección de deleciones, de disomía
uniparental o de alteraciones en el centro de imprinting, y se realiza por los siguentes procedimientos:
Cariotipo convencional. Es insuficiente para detectar la microdeleción típica, pero es útil en el
probando y en los padres para detectar posibles translocaciones, ya que un pequeño porcentaje de
las deleciones son fruto de translocaciones no equilibradas (casi siempre de novo). También
detectará translocaciones equilibradas de novo o heredadas, aunque estos casos son excepcionales.
FISH: detecta todas las deleciones, usando sondas frente al gen SNRPN en combinación con una
sonda centromérica del cromosoma 15. La mayoría de las deleciones son intersticiales, siendo la más
frecuente una pequeña deleción de unas 4 Mb de tamaño.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 14
Figura 8.10 Imagen de FISH para la detección de la microdeleción que
causa el Síndrome de Prader-Willi.
Detección de Disomía Uniparental: se usan microsatélites del cromosoma 15 para determinar el
origen parental de cada cromosoma. Lógicamente, es necesario descartar primero la presencia de
deleción, y además este procedimiento viene limitado por la necesidad de obtener muestras del
probando y de ambos progenitores, así como por la posible falsa paternidad.
Figura 8.11 Análisis de microsatélites del cromosoma 15 para detectar
disomía uniparental en el diagnóstico del Síndrome de Prader-Willi.
Análisis de metilación: puede hacerse bien por Southern blot tras digestión del ADN genómico con
enzimas de restricción sensibles a metilación, o por PCR específica de metilación (MS-PCR). Si el
exón 1 de SNRPN y el centro de imprinting adyacente están metilados en los dinucleótidos CpG, esto
indica un patrón materno; si no están metilados, podemos excluir el silenciamiento de SNRPN y los
demás genes de expresión paterna. Es un análisis que no requiere muestras de los progenitores, y
permite confirmar la existencia de Síndrome de Prader-Willi cuando se observa sólo el patrón de
metilación materno. Tiene la ventaja de que si el análisis es normal (es decir, si se observan dos
Heterodisomía materna (PWS) Isodisomía materna (PWS)
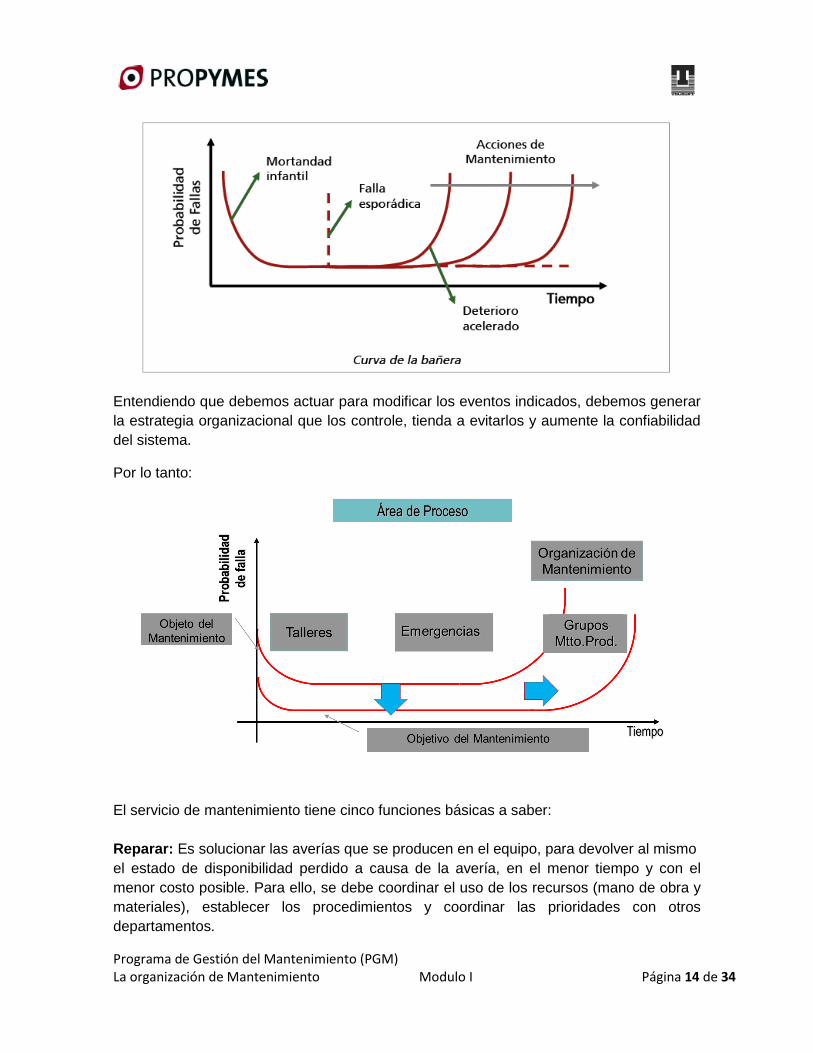
CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 15
patrones distintos de metilación, correspondientes a los cromosomas paterno y materno), esto
automáticamente excluye la presencia de deleción o de disomía uniparental. Cuando esta prueba es
anormal (patrón únicamente materno) y no se detecta deleción ni disomía uniparental, esto sugiere
la existencia de alteraciones en el centro de imprinting.
En el caso concreto del Síndrome de Angelman también es necesario buscar mutaciones en
UBE3A: aunque esto sólo supone un 20% de los pacientes, su detección es de gran importancia por
el aumento dramático en el riesgo de recurrencia de la enfermedad en la misma familia. Se estima
que hasta un tercio de los pacientes que tienen estudios de metilación normales tienen mutaciones
en UBE3A, por lo que la estrategia diagnóstica recomendada es comenzar con estudios de metilación
y después buscar mutaciones de UBE3A en aquellos sujetos con resultados de metilación normales.
Teniendo en cuenta lo anterior, se ha diseñado un árbol de decisión para optimizar la estrategia
diagnóstica en el caso de los síndrome de Prader-Willi y Angelman, de manera que se diagnostique el
mayor número de casos con el menor número de pruebas. En general, cuando los estudios de
metilación son anormales (patrón únicamente materno en el caso del Prader-Willi o patrón
únicamente paterno en el caso del Angelman), podemos continuar investigando la causa de la
enfermedad haciendo FISH para detectar la deleción característica, ya que ésta es la causa
más habitual de estas enfermedades. Si el FISH es normal, se pueden realizar estudios de
microsatélites para confirmar la presencia de disomía uniparental.
Figura 8.12 La estrategia diagnóstica más aceptada para el estudio del
Síndrome de Prader-Willi y del Sindrome de Angelman comienza con los
estudios de metilación de la región correspondiente al centro de
imprinting. Si se detecta un patrón de metilación paterno y otro materno,
esto automáticamente descarta la deleción y la disomía uniparental. En el
caso del Prader-Willi, esto supone que no hay que realizar más estudios,
mientras que en el caso del Angelman habría que buscar mutaciones en el
gen UBE3A, que causan la enfermedad en un 20% de los casos.

CAPÍTULO 8: EFECTOS FENOTÍPICOS DE LAS MUTACIONES 16
En el Síndrome de Prader-Willi el riesgo de recurrencia (un segundo individuo enfermo en la misma
familia) es prácticamente cero en los casos de deleción y de disomía uniparental que no están asociados
a reordenaciones cromosómicas. De todas formas, a efectos de consejo genético se considera un riesgo
de recurrencia del 1%, ya que en una muestra amplia de pacientes se detectó un riesgo de
recurrencia en hermanos de probandos del 1,6% (aunque esto incluía todas las posibles causas). El
mayor riesgo de recurrencia se produce en los casos familiares que están debidos a alteraciones en el
centro de imprinting. En estos casos, el riesgo de recurrencia es del 50% ya que volverá a suceder
siempre que el padre transmita el cromosoma 15 que lleva la mutación. En el Síndrome de Angelman, el
riesgo de recurrencia también se considera habitualmente del 1%, excepto en los casos de mutaciones
del centro de imprinting y de mutaciones en UBE3A, en las que el riesgo de heredar el cromosoma
materno mutado es del 50% (si el cromosoma mutado se hereda del padre no hay enfermedad porque
en ese cromosoma el gen UBE3A no se expresaría en cualquier caso).