cambios del hepatocito en el estado de resistencia a la insulina
TRANSCRIPT

CAMBIOS DEL HEPATOCITO EN EL ESTADO DE RESISTENCIA A LA INSULINA
Dra. Ma. Ludivina Robles OsorioDepartamento de Investigación Biomédica Facultad de Medicina Universidad Autónoma de Querétaro

AGENDA
Prevalencia de las alteraciones hepáticas
Introducción
Diferentes mecanismos fisiopatológicos de daño al hepatocito
Conclusiones

PREVALENCIA
La prevalencia reportada es de 30% en adultos y 13% en niños
En los pacientes con DM2 la EHNA parece ser una consecuencia de la obesidad
En obesos la prevalencia de la EHNA es de 95% Factores que contribuyen son:
Sedentarismo Dieta alta en grasa y miel de maíz alta en fructosa
(dieta de cafetería) Resistencia a insulina
World J Gastroenterol 2010; 16: 2223-2226

DEFINICIÓN
Acúmulo de grasa en los hepatocitos
La grasa constituye más de 5% del peso del hígado
Efectos deletereos en organelos del hepatocito
Características típicas de obesidad central y resistencia a la insulina
World J Gastroenterol 2010; 16: 2223-2226

INTRODUCCIÓN
La resistencia a la insulina tiene efectos en los hepatocitos y su metabolismo que resultan en:
Concentraciones elevadas de ácidos grasos libres que son sustratos para la formación de triglicéridos por el hígado.
Los ácidos grasos libres (AGL) pueden ser citotóxicos.
Los hepatocitos sufren apoptosis.
La apoptosis es una característica clave en la esteatosishepática no alcohólica.

Introducción
Existe además inflamación y fibrosis progresiva
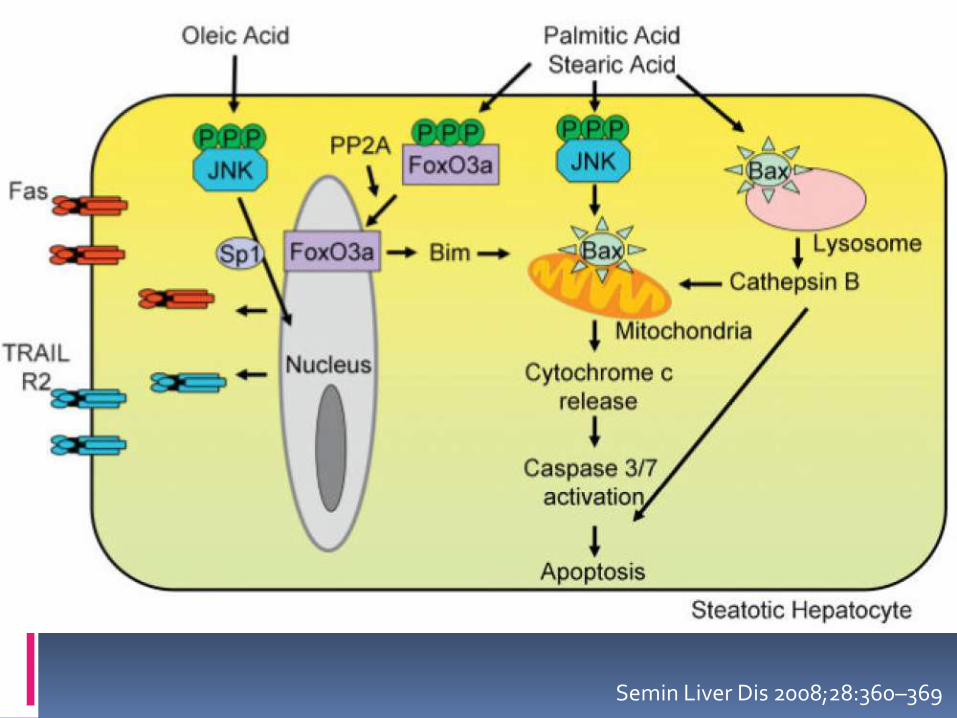
Los ácidos grasos libres están implicados en la maquinaria apoptótica al activar Bax una proteína apoptótica mediante la activación de c-jun N-la cinasaterminal
Los AGL también activan la vía lisosómica de la muerte celular y regulan la expresión de genes relacionados con muerte celular
El retículo endoplásmico liso desarrolla estrés oxidativotambién como parte de la fisiopatología del daño hepático

ESTEATOSIS HEPATICA NO ALCOHÓLICADEFINICIÓN Y CONSECUENCIAS
LIPOTOXICIDAD:
-Definida como la toxicidad celular por la presencia de acúmulo anormal de grasa
-En el hígado el acúmulo de grasa se define comoesteatosis hepática
-El tejido adiposo sirve como reservorio del exceso en la ingesta calórica. Los cambios son característicos del adipocito con resistencia a la insulina

Una característica importante es la apoptosis del adipocito
Los cambios incluyen incremento de la lipólisis, con liberación de AGL o tambiénllamados ácidos grasos no esterificados
También la liberación de adipocinas y citocinas inflamatorias
ESTEATOSIS HEPATICA NO ALCOHÓLICA DEFINICIÓN Y CONSECUENCIAS

Mecanismo de daño del hepatocito
AGL CC
C
C
C GRASA
ESTRES
APOPTOSIS
AGL
C
Resistencia a Insulina
Resistencia a Insulina

La resistencia a la insulina junto con los cambios antes descritos promueven el desarrollo de esteatosis del hepatocito (Esteatosis hepática no alcohólica)
La EHNA es un depósito progresivo de triglicéridos queexcede la capacidad de hígado de exportar VLDL y la beta oxidación de los AGL
Una proporción de los pacientes con EHNA desarrollaninflamación hepática conocida como esteatohepatitis no alcohólica la cual puede progresar a cirrosis.
ESTEATOSIS HEPATICA NO ALCOHÓLICA DEFINICIÓN Y CONSECUENCIAS

MECANISMOS DE DAÑO REPORTADOS
Mecanismos posibles de daño al hígado son el incremento en la oxidación de ácidos grasos y estrés oxidativo
Alteración en la composición de los ácidos grasos y fosfolípidos de la membrana
Alteración en el contenido de colesterol
Alteraciones en la vía de señalización de las ceramidas
Toxicidad directa de los ácidos grasos libres
Semin Liver Dis 2008;28:360–369

ACIDOS GRASOS LIBRES
Tres fuentes:
Del tejido adiposo y de la dieta y lipogénesis
El tejido adiposo contribuye en un 82 a 62% de las grasas en ayuno y posprandial
A su vez proveen el 59 y 62% de los AGL en el hígado y lipoproteinas ricas en triglicéridos.
J Clin Invest 2005;115(5):1343–1351.

ACIDOS GRASOS LIBRES
Los triglicéridos hepáticos son derivados en su mayoría de la lipólisis de adipocitos vienen de los carbohidratos de la dieta de la lipogenesis de novo (aminoácidos)
Los triglicéridos provenientes de los AGL circulantes se generan en su mayoría de la lipólisis de adipocitos(característico de la resistencia a la insulina y Sxmetabólico)
J Clin Invest 2005;115(5):1343–1351.

LÍPIDOS INTRAHEPÁTICOS
Puri y colaboradores en 2007 midieron la concentración de lípidos y trigliceridos en 9 sujetos con y 9 sin EHNA
Los lípidos totales y triglicéridos estaban incrementados , comparados con controles de la misma edad, sexo e IMC
En la composición de los triglicéridos y diglicéridoscirculantes se encontraron elevados: el ácido palmítico(saturado) y el oléico (poliinsaturado)
Hepatology 2007;46:1081–1090Clin Nutr 2002;21:219–223.J Hepatol 2008;48: 300–307.

LÍPIDOS INTRAHEPÁTICOS
El ácido palmítico y oléico se encuentran elevadosen suero de los pacientes con EHNA
Los AGL no están elevados en el hígadoprobablemente porque se esterifican para formartriglicéridos
Hepatology 2007;46:1081–1090Clin Nutr 2002;21:219–223.J Hepatol 2008;48: 300–307.

LIPOAPOPTOSIS
La apoptosis es una característica morfológica y patogénica del EHNA
Es secundaria al exceso de depósito de lípidos
Se puede desencadenar por los ligandos de la muertecomo Fas y TNFα
Se inicia por el ligando relacionado con apoptosis (TRAIL) o por la vía intrínseca, estrésintracelular, lisosomas, RE y mitocondria
Gastroenterology 2003;125:437–443. Dig Liver Dis 2001;33:768–777.Am J Gastroenterol 2004;99:1708–1717.

LIPOAPOPTOSIS
Al activarse la cascada de apoptosis lasmitocondrias se permeabilizan, activan lascaspasas 3 y 7. Se desencadena la activación y liberación de las proteínasproapoptóticas: Bcl-2, Bid,Noxa, Puma, Bim, Bmf, Bik, Hrk y el dominio BH-3
El grado de apoptosis coincide con el grado de inflamación y fibrosis
Gastroenterology 2003;125:437–443. Dig Liver Dis 2001;33:768–777.Am J Gastroenterol 2004;99:1708–1717.

ACTIVACIÓN DE LOS LISOSOMAS
Los ácidos grasos libres pueden activar tambiénla permeación de los lisosomas para conducir a apoptosis
Debido a que los lisosomas tienen enzimashidrolíticas y éstos son activados por las altasconcentraciones de ácido oleico y palmítico, lascuales son tóxicas y desencadenan la permeabilización de los lisosomas por la activación de Bax y liberación secundariamentede la catepsina B
Am J Physiol Gastrointest Liver Physiol 2006;290:G1339–G1346. Hepatology 2004;40:185–194.Oncogene 2004;23:2881–2890.

ACTIVACIÓN DE LOS LISOSOMAS
En pacientes con EHNA la permeabilización de los lisosomas y liberación de catepsina B correlacionancon el grado de actividad inflamatoria
Ratones deficientes de catepsina B estuvieronprotegidos contra el desarrollo de hígado graso, a pesar del desarrollo de obesidad
La permeación lisosomal se asoció con la activacióndel NF-κB, esta activación genera TNF-α
La sobreexpresión de Bcl-xL bloqueo la permeaciónlisosomal
Am J Physiol Gastrointest Liver Physiol 2006;290:G1339–G1346. Hepatology 2004;40:185–194.Oncogene 2004;23:2881–2890.

ESTRES DEL RETÍCULO ENDOPLÁSMICO
Tiene sensores de estrés en la membrana, si hay estréscrónico se descencadena apoptosis: proteína requiriente de inositol-1 (IRE-1), factor activante de la transcripción-6 (ATF-6) y la protein cinasa RNA-like del RE (PERK)
La activación de IRE-1 conduce a activación de JNK y ruptura endonucleolítica de la proteína fijadora de la caja X (uXBP1)
En trabajos hechos con modelos de ratones obesos porgenética y dieta demostraron que el estrés del RE condujo a la activación de JNK y otros marcadores de estrés del RE
Nat Rev Mol Cell Biol 2007;8:519–529. J Hepatol 2006;45:321–333. Science 2004;306(5695):457–461. Aliment Pharmacol Ther 2006;23(1):91–98Endocrinology 2006;147(2):943–951. Am J Physiol EndocrinolMetab 2006;291(2):E275–E281..

ESTRES DEL RETÍCULO ENDOPLÁSMICO La activación del JNK llevo a la fosforilación de serina del IRS-1
y resistencia a la insulina.
Un modelo de ratones con dieta rica en Ac. grasos saturados, activó la vía del estrés del RE e incremento de sXBP-1, BiP/GRP 78 y CHOP, mas porcentaje de apoptosis, comparados con una dieta con ácidos grasos poliinsaturados
En otro estudio se encontró que Ac. grasos saturados (palmítico y estearico) activaron la vía de estrés del RE y condujeron a apoptosis in vitro
En humanos con esteatosis y esteatohepatitis se observa una activación importante del estrés del RE.
Nat Rev Mol Cell Biol 2007;8:519–529. J Hepatol 2006;45:321–333. Science 2004;306(5695):457–461. Aliment Pharmacol Ther 2006;23(1):91–98Endocrinology 2006;147(2):943–951. Am J Physiol EndocrinolMetab 2006;291(2):E275–E281..

RECEPTORES DE LA MUERTE
Los receptores del TNF-α (TNFR 1 y 2) se encuentran en los hepatocitos y activan apoptosis a través de NFκβ activa genes de inflamación e inhibe los de sobrevida celular

TNFα Y MUERTE CELULAR
TNF-α es una citocina proinflamatoria puede inducir muertecelular
La obesidad es un estado inflamatorio crónico con infiltraciónde macrófagos en tejido adiposo y liberación de mediadoresinflamatorios incluyendo TNF-α
La obesidad por alimentos conduce a elevación de TNF-α y resistencia a la insulina
J Clin Invest 2003;112(12):1796–1808.Am J Med Sci 2001;322(2): 75–78. Hepatology 2003;37(2):343–350.

TNFα Y MUERTE CELULAR El TNF-α promueve el desarrollo de esteatosis hepática dado que
aumenta la expresión de genes lipogénicos
Esto corroborado por el descenso de la esteatosis con la inhibiciónfarmacológica (inflixumab)
La expresión del ligando de FAS esta incrementada en pacientes con esteatosis hepática.
J Clin Invest 2003;112(12):1796–1808.Am J Med Sci 2001;322(2): 75–78. Hepatology 2003;37(2):343–350.

BIM Activación JNK Bax Incremento Oxidación
TLR 4 ¿?
Ácidos grasos libres
Permeabilizaciónmitocondrial
Apoptosis de Hepatocitos
Permeabilizaciónlisosomal
Estrés Oxidativo
Estrés RE
Expresión de Genes de Inflamación
Apoptosis deHepatocitos
Fibrosis
CirrosisSemin Liver Dis 2008;28:360–369

CONCLUSIONES
Los cambios en el hepatocito secundarios a las alteraciones metabólicas que genera la obesidad y resistencia a la insulina están relacionados con:
Liberación de ácidos grasos libres
Los ácidos grasos libres general estrés en el RE, así como inducción de proteínas proapoptóticas de la mitocondria y lisosomas
El TNF-α elevado por el proceso inflamatorio genera expresión de proteínas de muerte celular

CONCLUSIONES
El estrés celular que se genera por el proceso inflamatorio lleva a apoptosis del hepatocito, posteriormente fibrosis y eventualmente cirrosis.

GRACIAS