bol_soc_esp_min_26_ocr.pdf
TRANSCRIPT

Boletín de . ISSN 0210-6558
la Sociedad Española de
Mineralogía Una revista europea de Mineralogía, Petrología,
Geoquímica y Yacimientos Minerales

Boletín de la
Sociedad Española de Mineralogía
Publicado por la Sociedad Española de Mineralogía (integrada en la "European Mincralogical Union")
Directora
P. Fenoll Hach-Alí Universidad de Granada [email protected]
Comité Editorial (2003)
E. Cardellach López C. Casquet Martín
E. Galán Huertos J.M. González López
J. M. Herrero Rubio A. López Galindo
Univ. Autónoma Barcelona Univ. Complutense Madrid Univ. Sevilla Univ. Zaragoza Univ. Pais Vasco Univ. Granada
Suscripciones
Esta revista se envía directamente a todos los miembros de la Sociedad Española de Mineralogía que mantienen actualizada su cuota. Sin embargo, los miembros no asociados y
entidades que deseen obtenerla pueden solicitar su recepción mediante el pago de un abono de 7000 ptas por ejemplar, más gastos de envio, en concepto de gastos de edición, dirigiendose a la Secretaría de la Sociedad Española dc Mineralogía, e/ Alenza, 1, 28003 MADRID.
Para inscribirse como socio los candidatos deberán solicitar el Boletín de Inscripción obligandosc a pagar la cuota anual. Ello les dará derecho a asistir a las reuniones de la Sociedad y recibir la documentación que periodicamente se distribuye entre los socios. incluido el Boletín de la Socíedad Española de Mineralogía.
Todo cambio de dirección deberá ser notificado al secretario de la Sociedad para cvitar la devolución de correspondencia.
Para otra infonnación dirigirse a: Sociedad Española de Mineralogía, e/ AJenza, 1,
28003 Madrid, www.chu.es/som

Vol. 26
Boletín de la
Sociedad Española de Mineralogía
Volumen 26, 2003 (Enero-Diciembre)
Publicado por la Sociedad Española de Mineralogía (inlegrada en la "European Mineralogical Un Ion")
Sociedad Española de Mineralogia www.ehu.es/sem
Alenz8, 1 . 28003 Madrid
ISSN 0210-6558

Boletín de la Sociedad Española de Mineralogía, 2003; 26 ( ENE-DIC)
Índice
Aportacionnes de la mineralogía a la evaluación y tratamientos de suelos ysedimentos contaminados por elementos traza / Galán Huertos, Emilio
1-28
Las mineralizaciones de pirita de la cuenca de Cameros en su contexto geológico / Alonso-Azcárate, Jacinto
29-44
Caracterización textural y mineralógica del gossan de Filón Sur (Tharsis,Huelva) / Capitán, Angeles / Nieto, José M. / Sáez, Reinaldo / Almodóvar, R.
45-58
Caracterización mineralógica y técnica de materiales zeolíticos naturales deuso comercial / Flores Macías, Antonio / Bosch Giral, Pedro / Lara Corona, Victor / Ordaz Chaparro, Victor / Cortés Flores, José I.
59-67
Síntesis y caracterización de monocristales de materiales zeolíticos / Mateo González, Ester / Vilaseca Llosada, Montserrat / Paniagua Condado, Andrés / Coronas Ceresuela, Joaquín / Santamaría Ramiro, Jesús
68-78
Inclusiones fluidas en venas auríferas del yacimiento Paua-Pique, suroeste del Cantón Amazónico, Brasil / Pulz, Gênova M. / Barboza, Elzio S. / Pinho,Francisco E. / Ronchi, Luis H. / Jelinek, Andréa R. / Duarte, Lauren da C.
79-92
Melt inclusions in quartz from subvolcanic sills of the Iberian Pyrite Belt:Implications for magma evolution and hydrothermal alteration / Tornos, Fernando / Simonov, Vladimir / Kovyazin, Sergei
93-105
Sulfosales de Ag-Bi-Pb de Bustarviejo (Sierra de Guadarrama, Madrid) / Martín Crespo, Tomás / Vindel, Elena / López García, José A.
107-112
Policromías de la Iglesia del Monasterio de San Jerónimo de Granada: composición, alteración y técnicas de ejecución / Cardell-Fernández, Carolina / Rodríguez-Gordillo, José
113-121
Características metamórficas del Triásico Maláguide en las unidadesintermedias del sector de Diezma (Sierra Arana, Cordillera Bética) / Lázaro, Concepción / Ruiz Cruz, María Dolores / Sanz de Galdeano, Carlos
123-136
Alteración de la cristalinidad del grafito por cizalla simple: comparación entreprocesos experimentales y naturales / Crespo Feo, Elena / Luque del Villar, Francisco Javier / Rodas González, Magdalena
137-153

Efectos de la molienda en la caracterización mineralógica de materialessepiolíticos y esmectíticos / Ruiz de León, David / Cuevas, Jaime / Fernández, Raúl / Sánchez, Laura / Leguey, Santiago
155-165
Caracterización mediante microscopía electrónica de barrido de partículasatmosféricas del área industrial de Huelva (SW de España) / Bernabé, José M./ Carretero, M. Isabel
167-177
New minerals approved in 2002. Nomenclature modifications approved 1998-2002 by the commission on new minerals and mineral names internationalmineralogical association./ Grice, Joel D. / Ferraris, Giovanni
179-186

Un poco de historia
Desde la fundación de la Sociedad Española de Mineralogía. hasta que nos dejó en 1981, fue José Marra Melgar y Escrivfi de Romaní el director de nueSlro Boletín, momenlO en el cual
asumf dicha responsabilidad hasta 2003.
Durante estos años la edición del BoleHn ha pasado por diversas vicisitudes, barajándose
incluso la posibilidad de su desaparición. debido principalmente a dos razones fundamentales: el factor económico y la falta de aportaciones periódicas por parle de nuestra comunidad científica, lo que hada imposible que se mantuviera la regularidad de la publicación. tal y
como se tenía previsto.
Este momento crítico se superó. sin embargo. gracias al patrocinio económico del entonces Institulo Geológico y Minero de Espai'ia, por una parle, y a la positiva respuesta de los
investigadores mediante su participación regular y asidua a las Reuniones y/o Congresos organizados anualmente por la Sociedad Española de Mineralogía, que sirvió de fuente de
abastecimiento de trabajos posteriormente publicados en el Boletín de la SEM, bien como
resúmenes o como trabajos completos. A éstos se unían airas aportaciones que, de forma
esporádica, eran enviadas directamente para su publicación
Con la integración de nuestra Sociedad en el entonces llamado Grupo Europeo de
Mineralogía (actual European Mineralogical Un ion) se homologó el formato de nuestro Boletín y se creó un comité de redacción, asesorado a su vez por revisores externos.
Con el esfuerzo de todos, la calidad de los artículos científicos publicados en el Boletín de
la SEM fue en aumento, alcanzando un cierto prestigio en nuestro campo de investigación. Sin embargo, la imposición en la década de los 90 de criterios basados casi en exclusividad en los
índices de impacto de las revistas a nivel internacional hizo disminuir drásticamente el interés
por publicar en nuestro Boletín.
Con objeto de adecuar nuestra publicación a esta nueva situación, se iniciaron una serie de gestiones para integrar el Boletín de la SEM en el European 10urnal of Mineralogy, y no ha sido hasta 2003 cuando se ha conseguido finalmente dicha integración, lo cual implica la desaparición de nuestro boletín como revista independiente a partir de 2004.
Por ello, con este volumen correspondiente al 2003 damos por terminado un ciclo. en la esperanza y certeza de que en la nueva etapa que se nos avecina nuestra Sociedad se verá bien
reflejada tanto en el ámbito internacional como a nivel nacional.
Finalmente deseo expresar mi agradecimiento a nuestros socios, de quienes espero hayan
sabido disculpar todos mis errores. y mostrar la satisfacción de haber aprendido mucho gracias a la ayuda y colaboración de todos.
Purificación Fellofl Hach·Alí

Sociedad Espafiola de Mineralogía
Junta Directiva de la SEM 2003-2004 (elegida en Asambla General de Socios celebrada en Lograña 16 de septiembre de 2002)
Presidente: Emilio Galán Huertos Universidad de Sevilla [email protected]
Vicepresidente: Guillenno Corretgé CastañoR Universidad de Oviedo [email protected]
Tesorero: Rafael Arana Castillo Universidad de Murcia [email protected]
Secretario: Carlos Sánchez Jiménez Universidad de Castilla -La Mancha [email protected]
Vicesecretaria: Carnen Galindo Francisco Universidad Complutense, Madrid [email protected]
Vocales
Benjamín Calvo Pérez Universidad Politécnica de Madrid [email protected]
Angels Canals Sabaté Universidad de Barcelona [email protected]
Constanza Femandez-Nieto Femandez Universidad de Zaragoza [email protected]
José López Ruiz CSIC, Madrid [email protected]
Fernando Nieto Garcia Universidad de Granada [email protected]
Manuel Prieto Rubio Universidad de Oviedo [email protected]
Magdalena Rodas GonlAlez Universidada Complutense. Madrid [email protected]
Fernando Rull Perez Universidad de Valladolid [email protected]
Gabriel Ruiz de Almodovar Sel Universidad de Huelva [email protected]
Francisco Velasco Roldan Universidad del Pais Vasco. Leioa [email protected]
••••••••••••

Boletfn de la Sociedad Espmiola de Milluafog(a. 26 (2ooJ). ¡·28
Aportaciones de la mineralogía a la evaluación y trata
mientos de suelos y sedimentos contaminados por elementos traza. *
Emilio GALÁN HUERTOS. Deparlamenlo de Crislalografía, Mineralogía y Química Agrícola. Facullad de Química. Universidad de Sevilla
Preámbulo.
La Mineralogía Ambienlal es un campo nuevo en el que anualmenle crece la lileralura cienlífica de forma exponencial. Ya exislen libros, monografías y una gran canlidad de arlículos dispersos en reviSlas de distinlOs li· pos, que tratan sobre aspectos mineralógicos del medio ambienle. Inclusive las revistas American Minernlogist y Mineralogical Ma· gazine editan un "número verde" sobre Mineralogía Ambiental que se publica de forma adicional.
En eSlos momentos en que algunas de 13s ramas tradicionales de la Mineralogra están en franca decadencia. los mineralogistas tenemos una oporlunidad de hacer valer nuestros cono· cimientos en una ciencia tan imporlante y mullidisciplinar como es el Medio Ambiente.
El mineralogista posee la visión y el co· nocimienlO de la evolución de los minerales en la superficie lerrestre. Sus interacciones con la biosfera. la hidrosfera y la atmósfera. Podemos pasar de la escala atómica y molecular, de la superficie de los minerales, en donde se producen la mayor parte de los
·Confereneia Inaugural de la XXII Reunión de la Socied:ld Esp:ll\ol:l de Miner:llogfa celebrnd:l en LogToña (14·17 Septiembre. 2002)
fenómenos que inleresan al medio ambiente, a los grandes ciclos geoqufmicos. Conocemos las técnicas .que nos permilen caracterizar e identificar las fases minerales y sus producloli de alteración.
Podemos simular en el laboratorio expe· rimentos que nos aproximen a los sistemas reales. y modelizar mediante la informática lo que puede ser el problema actual y su futura evolución.
Int ... oducciÓn.
Degradar un suelo es modificar su equili· brio natural con efectos negativos; consecuencia directa de la utilización (agrícola. forestal, ganadera. industrial. etc.) o de procesos nalurales (desenización. aclividad volcánica, lixiviados de mineralizaciones. etc.). Un tipo especial de degradación es la producida por la presencia de sustancias químicas. que en ciertas concentraciones tienen efectos nocivos.
Un cOlllum;IIUllte es un elemento o compuesto químico presente en un suelo en con· centraciones mayores de las habituales. y que en general tienen un efecto adverso sobre algunos organismos cuando está disponible (biodisponible). Por su origen puede ser geogénico o antropogénico. Los primeros pue· den proceder de la roca madre. de la actividad

2 GALÁN HUERTOS. E.
volcánica o del lixiviado de mineralizaciones, mientras los antropogénicos son consecuencia de actividades mineras, industriales. agrícolas o urbanas. La posibilidad de que un elemento o compuesto quede libre y pase al suelo se llama disponibilidad, y si puede ser asimilado por un organismo se denomina biodispoII ibilidad.
Respecto a los contaminantes denominados metales pesados, la labIa periódica incluye unos 70 elementos metálicos. y de ellos 59 pueden ser considerados "metales pesados", que son aquellos con peso atómico mayor que el del hierro (55.85). Pero otros más ligeros pueden también ser metales contaminantes. como V (50.95), Mn (54,44) o Cr (52.01). Además OIrOS que no son metales como As, F y P, también pueden llegar a ser elementos contaminantes, por lo que en general resulta mejor hablar de "elememo traza" y no de "metales pesados", si bien hay que reconocer que la mayorra de los contaminantes inorgánicos son "metales pesados".
Por otra parte el término "elemento traza" lleva siempre una connotación sobre su baja concentración en el medio, y a veces la contaminación puede ser por un metal pesado o no (Fe, Al) que puede ser elemento mayoritario del suelo. Además no hay que olvidar que muchos elementos traza que llegan a ser tóxicos en exceso, son al mismo tiempo micronutrientes y una deficiencia en la dieta puede causar enfermedades (Tabla 1). Por todo ello, debe prevalecer la idea de que un COlltaminame supone la presencia en exceso de /111
elemelllo COII efecto nocivo, sobre la precisión del término empleado (elemento traza, metal pesado, traza inorgánica, etc.)
El suelo actúa como una barrera protectora de otros medios más sensibles (hidrológicos y biológicos), filtrando, descomponiendo, neutralizando o almacenando contaminantes y evitando en gran parte su biodisponibilidad. La cantidad máxima admisible de un contaminante. a partir de la cual el suelo se convierte en contaminado, se llama carga crÍlica y marca el umbral de toxicidad.
Desde el punto de vista del ciclo de los elementos en el suelo. la capacidad de carga de lUZ SI/e/O agrícola para los metales pesados (LCASHM. Load Capadty of Agricultural Soil for Heavy Metals), se puede definir como la cantidad máxima admitida de metales pesados en un suelo agrícola para que se conserve el ciclo de los elementos en el suelo y se limiten los efectos adversos en la biosfera. hidrosfera, atmósfera y litosfera (Chen et al., 2001). Esta capacidad de carga depende de una combinación de factores tales como las características del suelo. los organismos indicadores de la toxicidad, la forma e historia de los contaminantes. y otros parámetros ambientales. El valor para cada elemento no es por tanto fijo y en general se suele dar un rango. Por ejemplo para China (Chen et aL, 2001) dan los siguientes valores:
Cd, 23-37 g/hala Pb: 6750-10.125 g/h:tla eu: 687-2812 g/ha/a As: 450-675 g/ha/a
Tabla L- Micronutrientes y macronutrientes para el óptimo funcionamiento de los organismos vivos (Recopilado de distintas fuentes por Siegel, 2002)
Micronutrientes esenciales (mg Ó )J.gldía) As, Ca, Cr, Cu, Fe, Mn, Mo, Se, V, Zn, F, J, Si
Macronutrientes (100 mg o más por dla) Ca, CI, Mg, P, K, Na, S
Metales pesados no esenciales· Be, Cd, Hg, (NO, Pb, Sb, (Sn), Ti
• Los metales entre paréntesis pueden ser esenciales.

Aportaciones de la mineralogía a la evaluación y tratamientos . . . 3
De todos los elementos traza encontrados en suelos, 17 se consideran como muy tóxicos y a la vez fácilmente disponibles en muchos suelos en concentraciones que sobrepasan los niveles de toxicidad. Estos elementos son: Ag, As. Bi, Cd, Ca, Cu, Hg. Ni, Pb, Pd, Pt, Sb. Se. Sn. Te, TI y Zn. De ellos 10 son fácilmente movilizados por la actividad hu· mana: Ag, As, Cd, CUt Hg, Ni, Pb. Sb, Sn y TI (Novotny. 1995). La EPA (U.S. Environmental Protection Agency) induye en la lista de contaminantes prioritarios a los siguientes trece elementos: antimonio, arsénico. berilio, cadmio, cromo, cobre. mercurio. níquel, plata, plomo. selenio. talio y zinc.
Los elementos traza más abundantes en los suelos son (Bowen, 1 979):
Cr. Ni, Pb y Zn (1·7500 mg/kg) As, Ca, Cu (0.1-250 rr.gJkg) Cd y Hg (0.01·2 mg/kg)
No obstante. se deben considerar rangos más estrictos para las concentraciones normales de los elementos traza, puesto que las anomalías aunque puedan llegar hasta niveles muy altos no son habituales (Tabla 2).
Fondos gcoquímicos de los suelos
La contaminación originada por el creci· miento de la población y su desarrollo socioeconómico es una gran amenaza para el equilibrio ecológico y para el desarrollo sostenible de los recursos naturales. La dimensión y velocidad de los impactos de la actividad humana es tan grande que existe peligro de que en pocos años se produzca un desastre ecológico global. Conscientes de esta situación se están realizando numerosas investigaciones a diferentes niveles sobre el cambio climático, la biodiversidad o la calidad del agua, pero todavía hay relativamente pocos trabajos sobre los problemas de contaminación de los suelos y las consecuencias tóxicas que se pueden derivar para los distintos ecosistemas.
En lo relativo a los elementos traza, y especialmente sobre los metales pesados, se está avanzando bastante en los últimos años en definir los fondos geoquímicos naturales de los distintos suelos y contextos geológicos, como base para documentar cualquier cambio
Tabla 2.- Concentración de elementos traza en suelos en niveles normales y como anomalías geoquímicas (Bowie & Thornton, 1985).
Elemento Niveles normales (mg/kg) Anomalías geoquímicas (mg/kg)
As <5-40 hasta 2500 Cd <1-2 hasta 30 Cu 2-60 hasta 2000 Mo <1-5 10-100 Ni 2-100 hasta 8000 Pb 10-150 10000 ó más
Se <1-2 hasta 500 Zn 25-200 10000 Ó más

4 GALÁN HUERTOS. E.
¡¡clual o futuro, que conduzca a considerar un suelo como contaminado.
La geoquímica puede identificar no sólo la cantidad total de un elemento en el suelo sino también la cantidad biodisponible, lo que es más importante que la cantidad total pero menos fácil de establecer. En general la biodisponibilidad de un elemen:o depende de su forma O especiación química.
Actualmente, la mayoría de los datos que se tienen de un país o de una región están incompletos, porque las muestras no se han tomado con la finalidad de establecer los fondos regionales. sino más bien para prospecciones de distinlos tipos de yacimientos, por eso es necesario seguir una metodología común general para que los datos obtenidos puedan ser comparables en los distintos países y puedan ser integrados.
Los principales problemas a tener en cuenta derivan de:
a) La escala del muestreo que debe ser acorde con el área de investigación.
b) La elección de los puntos de muestreo que deberían estar exentos de contaminación antropogénica.
c) La armonización de la metodología: forma y profundidad de la muestra, protocolo analítico y técnicas a usar, etc.
En general el "folldo geoqllímico /la/Ifral" representa la concentración de un elemento en suelos no contaminados (equivale al concepto de "geochemical background"). La determinación de este fondo es prácticamente imposible, porque incluso en las zonas aparentemente vírgenes de parques nacionales existe una cierta contaminación de origen eólico. Sin embargo, es más práctico hablar de "lIiveles de fOlldos geoqllimicos" (geochemical baselines), que representan las variaciones de las concentraciones de los distintos elementos químicos en los suelos (Sal minen y Gregorauskiené, 2000), tal y como se encuentran en el momento de realizar la toma de muestra. Estos "lIiveles de fOlldos" para un territorio representan una
medida de las variaciones geoquímicas superficiales, y están lógicamente influenciados por la litología del subsuelo.
Los lIiveles de fOlldo regionales, se pueden describir como el rango de concentraciones naturales (fondo geoquímico natural), propios de los niveles inferiores de los suelos. más las concentraciones antropogénicas difusas del nivel superior. Un nivel de fondo regional debe ser determinado separadamente para cada elemento en cada diferente contexto geológico (Salminen & Tarvainen, 1997).
El nivel de fondo para un elemento se suele representar por un sól0 valor correspondiente a la media de los valores encontrados para ese elemento en niveles superiores de los suelos de una región. Esto es válido sólo para poblaciones con distribución normal, pero dado que frecuentemente no se da este tipo de distribución, parece más adecuado utilizar la mediana (Rack, 1988; Sal minen y Tarvainen, 1997). o el margen de variación. La mediana es más representativa que la media al eSt3r menos afectada por los valores extremos.
Los valores de niveles de fondo para grandes áreas (países. por ejemplo), son muy poco representativos. Suele ser más interesante los fondos regionales referidos a áreas geológicas. donde las anomalías geoquímicas no sean subestimadas. infravaloradas o enmascaradas entre un número elevado de datos de un área heterogénea desde el punto de vista geológico.
También es muy importante definir los "niveles gllía ". Se trata de valores fijados por las administraciones para controlar la posible contaminación admisible en un suelo. En general, existe siempre un "lIivel genérico ,le referencia" que corresponde a la concentración a partir de la cual se puede perder prestaciones del suelo. y constituye un nivel de seguridad. Define la máxima concenlraci6n que puede admitir un suelo sin que se produzcan efectos nocivos. Este "nivel de referencia" se corresponde en algunos paises con el nivel de fondo, o sea con [a mediana de la población estudiada. pero puede ser calcula-

Aportaciones de la mineralog!a a la evaluación y tratamientos . . . 5
do para cada elemento mediante formulaciones adecuadas en cada escenario.
Eltisten otros valores críticos: • El nivel de invesligació/l (threshold
value). que delimita la concentración máltima admisible. a partir de la cual probablemente se pueden producir efectos nocivos. pero a un nivel tolerable. Se suele obtener a partir del percentil 95 (o 90). Entre el nivel de referencia y el de investigación se recomienda una investigación para conocer los factores de riesgo.
• El nivel de imervención supone que el suelo presenta un riesgo confirmado y han de tomarse medidas para su recuperación. Entre este nivel y el de investigación. es obligatoria una investigación detallada. que puede conducir a que incluso se califique el suelo como contaminado. o se rebaje la valoración de la peligrosidad.
Todos estos niveles están establecidos a partir de valores de concentraciones totales de los elementos traza en el suelo. cuando es conocido que lo que realmente puede producir un peligro para el ecosistema es la movilidad del metal y su biodisponibilidad. Por lo estos ensayos.junlo con los de especiación. deben realizarse para un diagnóstico definitivo.
Finalmente. insistir en la necesidad de dar en las legislaciones diferente valores guías de acuerdo con las distintas situaciones geológicas del país (región. provincia. etc.). Definir los niveles con un solo valor puede conducir a que ciertas áreas den anomalías geog6nicas naturales superiores a los valores oficiales. provocando alarmas injustificadas.
Origen natural (geogénico) y antropogénico de los elementos Cra:r.a en los suelos.
El origen natural de los elementos en los suelos es muy poco significativo en relación con las aportaciones antropogénicas. y en general se encuentran en formas estables y por tanto poco disponibles. disminuyendo hacia los niveles superiores las concentraciones derivadas de
la roca madre. Por el contrario. los contenidos en metales pesados derivados de acciones antropogénicas son más abundantes en los primeros centlmelros del suelo o sedimento.
Todos los elementos están implicados en ciclos geoqufmicos y biogeoquímicos característicos de la dinámica terrestre. De acuerdo con estos ciclos los metales se pueden concentrar o dispersar en la litosfera y moverse en la hidrosfera, atmósfera y biosfera o en la interfase entre ellas. que son los suelos. En estos ciclos geoqufmicos ha intervenido también el hombre en los últimos 300 anos, modificando sustancialmente con su actividad el ciclo de alguno de los elemenlOS. Por ejemplo. más del 80% del cadmio liberado a la atmósfera proviene de la actividad humana (fusión de metales. combuslión del petróleo) y sólo un 20% deriva de las emisiones volcánicas.
El origen geoginico de todos los elementos en el suelo está. en una primera aproximación. en su liberación y movilización tras la destrucción de los minerales que los contienen. En este proceso el agua juega un papel muy importante, como medio y como reaclivo. para que se produzcan reacciones de disolución. hidrólisis y precipilación. El agua es además esencial para el crecimienlo de los organismos y por ello se favorecerá tambitn la meleorización biológica. Las principales reacciones físico-químicas que lienen lugar durante la meleorización de los minerales por la acción del agua y COJ son [a disolución e hidrólisis. junto con los procesos de oxidación-reducción y de acomplejamienlo cuando está presente la materia orgánica.
En general la a1!eración de los mineraJes pelTogtnicos es inversa a la secuencia de cris· lalización sugerida por las series de reacción de Bowen. La explicación de esta aherabilidad de los minerales eSlá en su eslruclura y en la estabilidad lermodinámica de los enlaces que existen en el mineral. Los silicatos con esIructuras más polimerizadas son más estables, El orden de eSlabilidad de los enlaces en las estructuras es Si-O>AI·O>X-O (X= K'. Na+,

6 GALÁN HUERTOS, E,
Calo, CIC.) Así, el CUilrzo con s610 enlaces SiO es el más estable. mientras aquellos con s610 tetraedros SiO/" aislados son menos estables (Tabla 3).
En la meteorización y liberación de los elementos de los minerales innuyen las siguientes condiciones y parámetros: a) drenaje (sistema cerrado/abierto), b) clima e) topografía, d) tiempo, e) actividad biológica.
En relación con este último aspecto, quizás el menos conocido. es cada vez más valorado el papel que juegan [os microorganismos en la Imnsformaci6n y degradación de los minerales (Figura 1). Las interacciones microbio-mineral se dan a escala micrométrica. Se conoce desde antiguo que los minerales pClrogénclicos son la primera fuente de nutrientes (K, Ca. Mg, Fe. p. Cu, Zn) desde hace más de 3.000 m.a. Asf las bacterias. y posteriormente hongos y líquenes. por exudación de ácidos orgánicos atacan la superficie de las rocas, solubilizan minr:rales y quelalan iones (biodegradación), que son absorbidos por las plantas y así pasan a la cadena Irófica.
Los microorganismos están en el ciclo biogeoqufmico de los principales nutrientes, incluidos los elementos traza. Incluso este papel de los microorganismos se utiliza indus-
trialmente. Son conocidas las actividades bacterianas en la bioxidación de piritas, en la biolixivación industrial de metales o en la bioregeneración. Por otra parte, la biodegradación de la piedra usada en el patrimonio cultural se debe a la acción biológica, especialmente de microorganismos.
Este campo de investigación de la interacción de los microorganismos y los minerales puede considerarse como una nueva línea de investigación en Mineralogfa Ambiental de gran porvenir.
Figura l .- Vermiculitización de una nogopil:t por acción de los microbios asociados:t un pino (Gadd. 2000)
Tabla 3.- Vida media de un cristal de 1 mm de varios minerales en solución acuosa a 2jOC
y pH 5 (Lasaga y Berner, 1998)
Mineral Vida media en aftos Mineral Vida media en aftos
Cuano 34000000 Sanidina 291000
Caolinita 6000000 Enstatita 10100
Moscovita 2600000 Diopsido 6800
Epidota 923000 Forstenta 2300
Microclina 921000 Wollastonita 79
Albita 575000 Calcita 0,43

Aportaciones de la mineralogía a la evaluación y tratamientos . . . 7
En cuanto a los metales pesados. las principa[es fuentes son desde luego los varios tipos de menas (sulfuros y óxidos) y de otros minerales acompañantes en los yacimientos metálicos. Una vez destruidos los minerales durante la meteorización. los metales pueden incorporarse a los suelos y sedimentos y a las aguas superficiales y subterráneas. En el caso de la meteorización de sulfuros se produce además la acidificación de las aguas. Los metales se pueden transportar en solución como complejos. o en forma coloidal. o por el viento (Figura 2). Durante esta movilización pueden ser adsorbidos por otros minerales y coloides (minerales de la arcilla. hidróxidos de hierro) o precipitar formando otros compuestos más estables. De esta forma se incorporan a suelos y sedimentos. ríos. lagos. costas y glaci.:lres. En diferentes etapas los metales pueden estar más o menos disponibles para ser captados por las plantas u otros organismos superiores.
La verdadera comprensión de [os fenómenos de meteorización se tiene cuando se estudia la superficie de los minerales a nivel atómico y molecular. lo que ha comenzado hace sólo unos pocos años. gracias a ciertas técnicas como la microscopía de barrido con efecto túnel (STM). la microscopía de barrido con microsonda (SPM) y el microscopio de fuerza
atómica (AFM). que directamente permiten observar los fenómenos con resolución atómica (Figura 3). Hasta estos momentos se han estudiado la superficie de alteración de Fep). Fe,ol' FeTiO). Ti0l' FeS!. PbS. CuS y CuFeS. Otras técnicas espectroscópicas tales como la espectrometría de absorción de rayos-X (EXAFS y XANES) o el XPS (epectrometría de fotoelectrones de rayos-X) se han usado para a partir de sus espectros poder modelizar los fenómenos de degradación y de adsorción en las superficies de los minerales (pirita. pirrotina. galena. calcopirita). tales como la relajación de átomos en superficie. el papel de los defectos y huecos. [a adsorción de cationes. la migración de elementos en la superficie. etc. (ver Wogelius y Vaughan. 2000; Keilh y Vaughan. 2000; V:.ughan �t al. 2002. p:.r:. referencias y detalles)
Un ejemplo de este tipo de estudio es de la oxidación de la calcopirita en una solución alcalina a pH 9.2. típica de un proceso de flotación. utilizando una combinación de dalas electroquímicos obtenidos con un voltograma cfclico y datos de XPS y XAS (Yin el al .• 1995. 2000; Vaughan el al.. 1995). Se han supuesto una serie de reacciones para explicar las transformaciones observadas en superficie. que conducen a la formación de oxihidróxidos de hierro y CuSz (fase mctaestable). Con una
Imnrpontdón directa
a sutloslsedlmentDs
I ." """""""" ¡o. lo< mJ�'" � L--'-'-"-T'=---' ,.. � Olsperslón aérea
de partículas
Transporte de 105 metales en soIudónes (ácidM), como Qtfones. complejos o coloides
Il'ICOfpOI'adOn a glaciares, Iios, legos, mares, suelos
I """"""'" • ríos. lagos, costas, acuíferos I� I suelos V sedImentos
Proce5O$ de AcIsordón Precipitación (Minerales de la Ardlla, hidro_idos de Fe, Mn)
Figurll. 2.- Movilidad de los metales pesados y evolución en los suelos
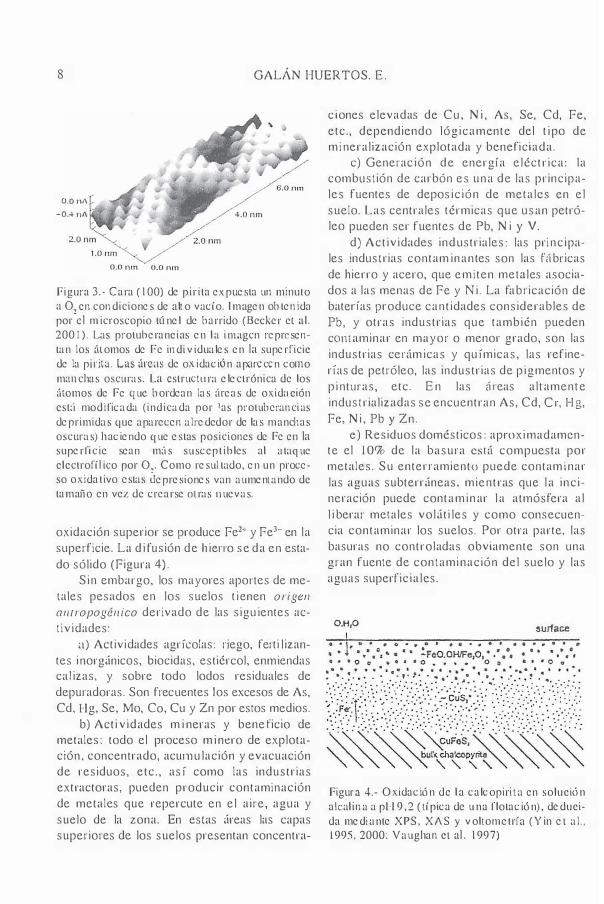
8 GALÁN HUERTOS. E .
0.0"" -OA 111\
Figura 3.- Cara (100) de pirita expuesta un minuto a O!cn condiciones de alto vacío. I m agen obtenida por el microscopio túnel de barrido (Beder el al. 2001). Las protub eranci as en l a inwgcn representan los {\tomos de Fe indi viduales en la superficie de la pi !"i!". Las úrc�ls de oxidación aparecen como m;l1lchas oscuras. La cstruClllra electrónica de los álomos de Fe que bordean las {¡reas de oxidílción cst{¡ modificada (indicada por las protuberanci as deprimidas que aparecen alrededor dc las manchas oscuras) h aciendo que estas posiciones de Fe en la superfic ie sean m{¡s susceptibles al ataque clcctrofílico por O�. Como resultado, en un proceso oxidativo estas depresiones van aumentando de tamaño en vez de crearse OLras lluevas.
oxidación superior se produce Fe2-+ y Fe3-+ en la superficie. La d i fusión de h iclTo se da en estado sólido (Figura 4).
Sin embargo, los mayores apones de metales pesados en los suelos t ienen origen
lIlllropogénico derivado de las sigu ientes act iv idades:
<1) Actividades agrícolas: riego, ferti l izantes inorgánicos, biocidas, estiércol, enmiendas calizas, y sobre todo lodos residuales de depuradoras. Son frecuentes los excesos de As, Cd, Hg, Se, Mo, Ca, eu y Zn por estos medios.
b) Acti vidades m i neras y beneficio de metales: todo el proceso m in�ro de explotación, concentrado, aculllu lación y evacuación de residuos, etc., así como las industrias extractoras, pueden producir contaminación de metales que repercute en el aire, agua y suelo de la zona. En estas áreas las capas superiores de los suelos presentan concentra-
c iones elevadas de Cu, N i , As, Se, Cd, Fe, etc., dependiendo lógicamente del t ipo de m ineralización explotada y beneficiada.
c) Generac ión de energía eléctrica: la combustión de carbón es una de las principales fuentes de deposic ión de metalcs en el suelo. Las centrales térmicas que usan petróleo pueden ser fuentes de Pb, N i Y V.
d) Act iv idades industriales: las pri ncipales industrias contam inantes son las f;.íbricas de hierro y acero, que emiten metales asociados a las menas de Fe y Ni . La fabricación de baterías produce cantidades considerables de Pb, y otras industrias que también pueden cOlllaminar en mayor o menor grado, son las industrias cenímicas y químicas, las refinerías de petróleo, las industrias de p igmentos y p inturas, etc. E n las <Íreas altamente industrializadas se encuentran As, Cd, er, Hg, Fe, N i , Pb Y Zn.
e) Residuos domésticos: aproximadamcnte el 10% de la basura CSl<í compuesta por metales. Su enterramiento puede contaminar las aguas subterráneas, mientras que l a inc ineración puede contaminar l a atmósfera al l i berar metales voláti les y como consecuencia contaminar los suelos. Por otra parte, las basuras no controladas obviamente son una gran fuente de contaminación del suelo y las aguas superfic ia les.
O.H,O surface
Figura 4.· Oxidaci6n d e l a calcopirita en solución alcal ina a pl-l 9.2 (típica de u n a flotaci6n), deducida mediante XPS. XAS y voltometría (Yin el al., t995, 2000: Vaughan el al . 1997)

Aportaciones de la mineralogía a la evaluación y tratamientos . . 9
El drenaje ácido de rocas (ARD) como fuente de contaminación.
Uno de los casos más interesantes de contaminación es el derivado de la lixiviación de mineralizaciones aflorantes de sulfuros, O lo que es más habitual de residuos de diversas clases que se acumulan en una explotación de sulfuros complejos. Este fenómeno es conocido internacionalmente como acid mil/e draillage (AMD) o de forma más general como acid rock draillage (ARD). Las aguas generadas son de carácter ácido y contienen normalmente elevadas concentraciones de metales tóxicos (As, Cd, Cu, Hg, Sb. Se. Pb. Zn). Desgraciadamente este fenómeno de drenaje ácido es bastante abundante porque globalmeOle se producen anualmente miles de Tm de estériles que contienen sulfuros, especialmente pirita. Así, por ejemplo, en la explotación de yacimientos tipo "porphyry copper", con = 1% de Cu, se producen"" 980 kg de estériles por tonelada de todo-uno, ricos en sulfuros diversos.
Los drenajes ácidos de rocas son originados por reacciones qufmicas y procesos de oxidación bateriana. En la composición de la roca es esencial la existencia de sulfuros. La pirita es el más común de los sulfuros implicados en la generación de ARO (Gray, 1996), aunque exiSlen otros sulfuros que también pueden estar implicados. El agua y el oxígeno son también esenciales, proque proporcionan las condiciones de oxidación necesarias para la reactividad de los sulfuros, asf como mecanismos de transporte de los micronutrientes los cuales suministran las bacterias que pueden catalizar las reacciones de oxidación. Las concentraciones de oxígeno taOlo en el agua como en el aire son críticas, porque sin oxígeno, ninguna de las reacciones del ARO tiene lugar y las bacterias mueren debido a la falta de oxígeno (Salomons, 1995).
El drenaje ácido de rocas (ARD) se caracteriza por la presencia tanto de fluidos como de partículas sólidas, en particular óxidos de
hierro hidratados "ocres". Los fluidos tienen bajos pH (2.1-6.5), alto Eh. alta conductividad de 800-6500 �S/cm (100 veces mayores que los de las aguas subterráneas). elevadas concentraciones de partículas finas, e iones (H. Fe, Al, Mn, Pb. Hg, Cd).
Existen varias condiciones necesarias para que las reacciones que generan el ARO tengan lugar, y están principalmente asociadas a la fuente de materiales y a la geoquímica medioambiental. Estas condiciones se pueden resumir de la siguiente forma:
• La fuente de material debe contener bastante sulfuros como para reaccionar y formar fluidos ácidos en un rango que exceda la capacidad de neutralización de cualquier compuesto alcalino (como los carbonatos) contenido en el sistema (Belzile et al .• 1997).
• Los materiales deben ser tales que permitan la incorporación del agua y aire necesa: rios para soportar las reacciones químicas, incluidas aquellas promovidas por la actividad bacteriana. Así los materiales de grano' fino. cuando se compactan. limitan la cantidad de odgeno entrante, mientras que los materiales de grano grueso promueven la difusión de oxígeno (Salomons, 1995). Los materiales de pequeño tamaño también ofrecen una mayor superficie de reacción.
• El clima debe ser tal que exista suficiente lluvia e infiltraciones dentro de los materiales del sistema ARO. Es decir asegurar un buen aporte de agua. oxígeno y nutrientes capaz de acidificar el agua para moverla a través del medio (Belzile et al. 1997).
• Aunque existen varios cientos de sulfuros metálicos, sólo cinco de estos minerales son lo suficientemente abundantes como para ser clasificados "minerales formadores de rocas" (Deer et al., 1992). Estos son pirita, pirrotita. galena. calcopirita y esfalerita. y de ellos dos (pirita y pirrotina) son responsables de la mayoría de los drenajes ácidos del mundo (Jambor y Blowes. 1998).
La reactividad de los sulfuros es com-

10 GALÁN HUERTOS. E.
pleja y variable. En la tabla 4 se ilustra la reaclividad relativa de los sulfuros más comunes obtenidas en distintos laboratorios (Jambar. 1994). Estas diferencias renejan parcialmente diferentes condiciones de oxidación y están claramente relacionadas con el tamaño de grano y las asociaciones minerales. A pesar de estas complej idades, bajo las condiciones dominantes en un sistema ARO. parece que la pirita es generalmente más estable. y la pirrotina es altamente reactiva seguida de galena y esfaJeTÍta. De tal manera que Jambor (1994) indicó que la secuencia de reactividad (de mayor a menor) en balsas de residuos es normalmente:
Pirrotina > galena - esfalerita > piritaarsenopirila > calcopirita
La marcasita es una fase metaestable que también es altamente reactiva. pero debido a que la pirita es el sulfuro más abundante y uno de los más reactivos. a continuación se van a analizar con cierto detalle las úHimas investigaciones realizadas sobre este mineral en relación con la producción de drenajes ácidos.
Pirita La pirita presenta una estructura cristali
na bien conocida aunque la superficie atómica de la estructura ha sido objeto de estudios limitados debido a problemas para obtener una superficie lisa. Sin embargo. estudios recientes por LEED (low-er.ergy electron diffraction) y STM (scanning tunnelling microscopy) sobre una superficie {lOO) limpia, generada por limpieza en vacío, han marcado un gran avance (Russo et al., 1999). Este hecho unido a la aplicación de técnicas de modelización en mecánica cuántica ha llevado a sugerir que en la superficie (lOO) se da un pequeño "relajamiento" y que puede parecerse a una simple terminación de la estructura global a lo largo del plano de exfoliación Fe-S. Aunque la estructura atómica en la superficie parece diferenciarse ligeramente de la estructura interior. esta pérdida de coordinación en la superficie necesariamente produce cambios en el enlace (o estructura electrónica). Esto conduce a que las posiciones ocupadas en la superficie por el Fe sean
Tabla 4.- Reactividad relativa de los sulfuros (Jambor, 1994)
esfalerita> galena> calcopirita> pirita ,,,1. 1 calculada (1991)
2 marcasita> pirrotina> calcopirita> pirita = arsenopirila Residuos de Kwong & Ferguson
3 pirrotina> calcopirita> pirita fina> esfalerita > galena > pirita gruesa
4 pirrolina> arsenopirita > pirita> calcopirita> esfalerita > galena
S pirrotina > pirrotina-pirita > pirrotina-aneno > anenopirita> pirita> calcopirita> esfalerita > galena> calcosina
6 pirrotina > calcosina > tetrahedrita > galena> arsenopirita > esfalerita > pirita > marcasita > calcopirita
7 esfalerita > grupo de la letraedrita > calcopirita> Bi-Sb sulfosales > galena> arsenopirita > pirita
8 pirita> calcopirita> galena> esfalerita
rocas (1990)
Gossan
Laboratorio, pH 2-6
Gossan
Atmósfera
Andrew(1984)
Kakovsky & Kosikov (1975) Flann & Lucaszenwski (1970)
Brock et al. (1984)
Boyle (1994)
Brion (1980)

Aportaciones de la mineralogía a la evaluación y tratamientos . . . 1 1
energéticamente más reactivas que aquellas ocupadas por el Sz y por lo tanto intervendrán en reacciones redox que llevarán a la aparición de nuevas especies en la superficie. Estudios recientes indican que la complejidad de los problemas asociados a la reactividad superficial de la pirita son tales, que se pueden identificar diferentes lugares activos e introducir la posibilidad de distintos mecanismos que pueden tener lugar de forma simuhánea a lo largo de la superficie. Por ejemplo se han identificado posiciones reactivas de S por la rotura de enlaces S-S (Nesbitt et aL, 1998).
Aunque son numerosos los estudios realizados sobre la oxidación de la pirita, aún existen sin resolver aspectos claves de la misma. Esto se debe en primer lugar al gran número de variables que están asociadas a este proceso de oxidación (medio de oxidación, pH, Eh, temperatura, existencia de ciertas bacterias), que pueden afectar significativamente la velocidad e intensidad de la oxidación. Por ejemplo, la presencia de bacterias puede acelerar la reacción en varios órdenes de magnitud. Por otra parte la oxidación de la pirita a sulfato requiere que se transfieran 8 electrones por átomo de S y esto debería darse a través de varias reacciones (aún sin resolver) que implicarán estados intermedios de oxidación para
S. En conjunto Biegler & Swift ( 1979) sugieren que en agua pura, la pirita se oxida mediante una combinación de las siguientes reacciones: FeS1 + 8Hp � FeJo + 2S0� l· + 16H' + 15 eFeS) � Fe)� + 2S + 3e-
Donde el Eh y pH determinan el camino predominante, y asr la ruta del sulfato domina en condiciones ambientes. A pH bajo la pirita puede oxidarse con 0, y con Fe)� (McKibben y Barnes, 1986) siendo las reacciones: FeSz + 7/2 02 + H20 � Felt + 2S0t +2H+ FeS + 14 Fe)' + 8H ° � 15 Fe2+ + 2S0 l· , , . +16H'
Estas reacciones pueden ser cata!izadas por la actividad de microorganismos, por ejemplo el Thiobacillus ferrooxidans acelern la alteración de la pirita por catálisis de la oxidación de Feh a Fel' usando oxígeno libre como aceptar de electrones. La interacción dinámica entre el oxígeno libre, el hierro y la pirita disuelta se esquematiza en la figura 5. donde se indica que la pirita es oxidada a sulfato por la reacción A. Alternativamente la reacción B muestra la superficie de la pirita oxidada por oxígeno libre. En el paso e el Ión ferroso es oxidado y puede precipitar como hidróxido de hierro insoluble (paso E), y al· ternativamente el Fe3+ puede disolver la pirita (paso D).
Figura 5.- Posibles reacciones para la oxidación dc la pirita (Banks el al., 1997)

12 GALÁN HUERTOS. E.
Evaluación de la contaminación por elementos traza
El análisis químico de elementos totales del un suelo es una medida poco representativa de la peligrosidad de los posibles contaminantes. Indica en tocio caso la peligrosidad potencial o futura, pero no la actual, de los elementos determinados, con referencia a ciertos valores acordados previamente, los cuales no deben ser superados (niveles guías). Además de este análisis químico se debe disponer de datos sobre las fracciones asimilables de los elementos, que es una medida directa de la peligrosidad real.
La fracción asimilable por las plantas de un determinado elemento de un suelo depende de su especiaci6t1 qll[mica, o sea de su distribución entre sus formas químicas o especies. La toxicidad de un elemento es muy distinta dependiendo de su presentación, lo que va a regular no sólo su disponibilidad sino también su grado de toxicidad. Según se encuentre el metal retenido en el suelo, así será su disponibilidad relativa por las plantas y por tanto la incorporación a los organismos (Tabla 5).
Pero la disponibilidad de un metal no depende sólo de su especiación química, sino de su especiación milleralógica. No todos los caliones de cambio están igualmente disponi-
bies, depende del mineral del que está formando parte como complejo de cambio. Así no será igual si se encuentra en una esmectita o en una vermiculita. Cuando el metal está precipitado no se comportará igual si lo hace como carbonato, sulfato o fosfato. Tampoco será lo mismo que el metal se encuentre formando parte de un sulfuro (relativamente oxidable y solubilizable) que de un silicato (prácticamente resistente en todos los medios).
Independientemente de su especiación la movilidad de los metales pesados es en general muy baja, quedando acumulados en los primeros centímetros del suelo, siendo lixiviados a los horizontes inferiores en muy pequeñas cantidades. Por eso la presencia de altas concentraciones en el horizonte superior cae drásticamente en profundidad. cuando la contaminanción es antrópica.
Esto sucede porque la disponibilidad de un elemenJo depende también de las características del suelo en donde se encuentra. Los parámetros geoedáficos llegan a ser esenciales para valorar la sensibilidad de los suelos a la agresión de los contaminantes. En concreto:
• pH. La mayoría de los metales tienden a estar más disponibles a pH ácido porque son menos fuertememe adsorbidos, excepto As, Mo, Se y Cr que son mas móviles a pH alcalinos.
• Textura. Los suelos arcillosos retienen
Tabla 5.- Disponibilidad relativa de los metales retenidos en el suelo por las plantas.
Forma del metal en el suelo Iones simples o complejos en solución Cationes de cambio Metales quelatados por compuestos orgánicos
Movilidad-Disponibilidad relativa Fácil Media
Menos disponibles
Metales adsorbidos sobre partículas del suelo Menos disponibles
Compuestos metálicos precipitados sobre Disponibles cuando se disuelve el partlculas del suelo compuesto Metales asociados o incorporados a una Disponibles cuando se descompone matriz biológica Metal asociado o formando parte de la Disponible cuando se meteoriza/destruye estructura de un mineral el mineral

Aportaciones de la mineralogía a la evaluación y tratamientos . . . 13
más metales por adsorción o en el complejo de cambio de los minerales de la arci1Ja. Por el contrario, los arenosos carecen de capacidad de fijación y puede contaminarse el nivel freático.
• Mineralogía de arcillas. Cada mineral de la arcilla tiene unos determinados valores de superficie específica y de descompensación eléctrica. Cuanto mayor es la superficie activade un filosilicato mayores son sus posibilidades de adsorber metales (Tabla 6). Este poder de adsorción será máximo en el punto de carga cero superficial, cuando su competencia con los H' es mínima, lo que se consigue a diferentes pH, según el mineral (Sposito. 1989) (Tabla 7). Sin embargo la importancia de los minerales de la arcilla como adsorbentes es secundaria cuando en un suelo existe abundante materia orgánica y/o oxi-hidroxidos de hierro, componentes más competitivos (Oa. hin. 2000).
• Materia orgánica. Reacciona con los metales formando complejos de cambio o quel:lIos. La adsorción puede ser tan fuerte que queden estabilizados, como el caso del Cu o formen quelatos también muy estables como puede pasar con el Pb y Zn. En muchos casos se forman complejos organo-metálicos lo que facilita la solubilidad del metal, la disponibilidad y dispersión porque puede de· gradarse por los organismos del suelo. Esto conduce a una persistencia de la toxicidad.
• Capacidad de cambio. Es función del contenido de arci1Ja y de la materia orgánica.
Tabla 6.- Area superficial típica de minerales del suelo (Bourg, 1995).
Minerales del sudo Caolinita
lIIita Montmorillonita 6l\idos de manganeso Ooethita Carbonatos/arenas
An:a superficial (ml/g) 7-30
65·100
700-800
30-300
40-80
0,5-5
Tabla 7.- Punto cero de carga (PZC) superficial (Sposito, 1989)
Mineral pH
Cuarzo/sílice 2-3 Caolinita 4,0-4,5 Goethita 7,0-8,0 Hematites 8,0-8,5 Gibbsita 9,0-9,5 Humus 4,0-4,5
El poder de intercambio catiónico depende del tipo de minerales de la arcilla, de la materia orgánica y de la valencia y del radio iónico hidratado del metal. A mayor tamaño y menor valencia de los cationes, menos frecuentemente q1ledan retenidos. Respecto a los minerales de la arcilla, la retención es mínima para los minerales del grupo del caoHn, baja para las illitas, alta para las esmectitas y máxima para las vermiculitas.
• Condiciones redox. El potencial de oxidación-reducción es responsable de que el metal se encuentre en estado oxidado o reducido. Los diagramas Eh-pH (Fig. 6) se utilizan para mostrar la estabilidad de los compuestos metálicos y proporcionan un método fácil para predecir el comportamiento de los metales pesados frente a un cambio de las condiciones ambientales.
• Carbonatos. La presencia de carbonatos garantiza el mantenimiento de los altos pHs, y en estas condiciones tiende .. a precipitar los metales pesados. El Cd y otros metales tienden a quedar adsorbidos por los carbonatos.
• Óxidos e hidróxidos de Fe y Mn. Juegan un importante papel en la retención de metales pesados y en su inmovilización. Se encuentran finamente diseminados en la masa de suelo por lo que son muy activos. Por su baja cristalinidad y pequeño tamaño de partícula, tienen una alta capacidad sorcitiva para

14 GALÁN HUERTOS. E.
u u
" ...
.. ..
.. o.,
o. •• ... fu �0-2 � , •
.. • 0
... -u
-... -..
... -o •
• • . .. '
• • • • • ' ..
• G
Figura 6.- Solubilidad de metales pesados en función del pH '1 del Eh (en ausencia de maleri3. orgánica disuelta o sólida) (a) 10$ principales minerales controlan la solubilidad de los metnles pesados: (b) tendencia de incremento de la solubilid:r.d (FOstner. 1987)
adsorber metales divalenles, especialmente Cu y Pb, Y en menor extensión Zn. Co, er, Mo, Ni Y también As.
• Salinidad. El aumento de la salinidad puede incrementar la movilización de metales y su retención por dos mecanismos. Primeramente los cationes Na y K pueden reemplazar a metales pesados en lugares de intercambio catiÓnico. En una segunda fase los aniones cloruros y sulfatos pueden formar compuestos más estables con metales tales como Pb, Zn. Cu, Cd y Hg. Por otra parte las sales normalmente dan pH alcalino.
Teniendo en cuenta las caracterfsticas de los distintos metales y la influencia de todos estos parámetros geoedáficos, se puede predecir el comportamiento de un elemento en el suelo. Son elementos traza móviles Zn y Cd, que están ligados a la materia orgánica o como cationes de cambio y son sólo relativamente móviles Pb. Ni Y Cr que en su mayor parte están en forma de sulfuros o silicatos. En la práctica, para conocer esta afinidad de los metales por los distintos componentes del suelo y conocer su movilidad. se siguen mttodos de
extracciones secuenciales. de los que puede derivarse una especiaci611 qllímica (Figura 7).
Con esta metodología se mide la concentración total de los elementos y distribución selectiva según diferentes fracciones con distinto comportamiento. La selectividad de estos mttodos depende de la rapidez de las reacciones en la formación o disociación de las especies que se miden. Se necesitan criterios para distinguir entre las fases lábiles y cinética mente inertes, en cada paso sucesivo de la extracción (Tessier et al. 1979, Tessier et al. 1985, Campbell & Tessier 1987). En esencia el procedimiento consiste en obtener varias fracciones utilizando extractantes diferentes, de forma sucesiva. Un esquema simplificado puede ser el siguiente (Tabla 8): la fracción I es la tratada con acetato amónico, la fracción 2 obtenida en un medio reductor con clorhidrato de hidroxilamina y ácido acttico, la fracción 3 obtenida con ácido nftrico y agua oxigenada y la cuarta es la fracción insoluble
F1 corresponde a los metales del complejo de cambio y a los que están formando (o

Aportaciones de la mineralogía a la evaluación y tnltamientos ... 15
adsorbidos en carbonatos. Este paso se puede subdividir en dos, un tratamiento con MgCI1 1M, Ih. para extraer los cationes de cambio. y un segundo tratamiento con CHlCOONa. pH 5 con ácido acético. 5h., para hacer el atuque de los carbonatos. En F2 se concentT3n los metales asociados a los compuestos reduci-
bies (óxidos de Mn y oxi-hidróxidos de Fe amorfos o pobremente cristalinos). F3 es la fase oxidable y en ella estarán los cationes ligados a la materia orgánica y a los sulfuros (parcialmente) y F4 es la fracción residual, donde los metales están en las estructuras cristalinas de los minerales resistentes.
Mo Zn Cd Cu Pb Ni Cr
(. __ ;w. .. _.
0"00'''''''-
� F .. «OIn ..... . ..-.. ... 0 ......
• , .... loI<I •• oc .... . _.�h)
mi " ... ;w. __
• f .... _ , •• _._
Figura 7.- Ejemplo de especiaci6n de elementos tTaz:! en suelos (Kabata-Pendias. 1992)
Tabla 8.- Esquema de extracción secuencial para la evaluación de la distribución de metales en sedimentos (0.5 g de muestra) (Galán et al. 2000)
Fracción
Ácida(FI)
Reductora (F2)
Oxidante (F3)
Residuo (F4)
Procedimiento
NH4Ac 1M (35 mi), pH 5, lh, 20°C, agitación continua (volteador 40 rpm)
NH10H.HCI O,4M en ácido acético al 25% (20 mi), 6h, 96°C, agitación manual cada 30 mino
HNO, 0,2M (3 mi) + H,O, 30% (S mi), pH 2 con HNO" 2h, 85°C, agitación manual cada 30 min.; Madir H102 30% (3ml). pH 2 con HNO" 3h, 8S·C, NH,OAc 3,2 M (Sml) v/v en HNO, diluida en 20 mi con H10l, 30 min .• 200<:, agitación contInua (volteador 40 rpm)
HFIHNO,lHCI I0/3/2,S (20 mi), 2h a sequedad
16 GALÁN HUERTOS. E.
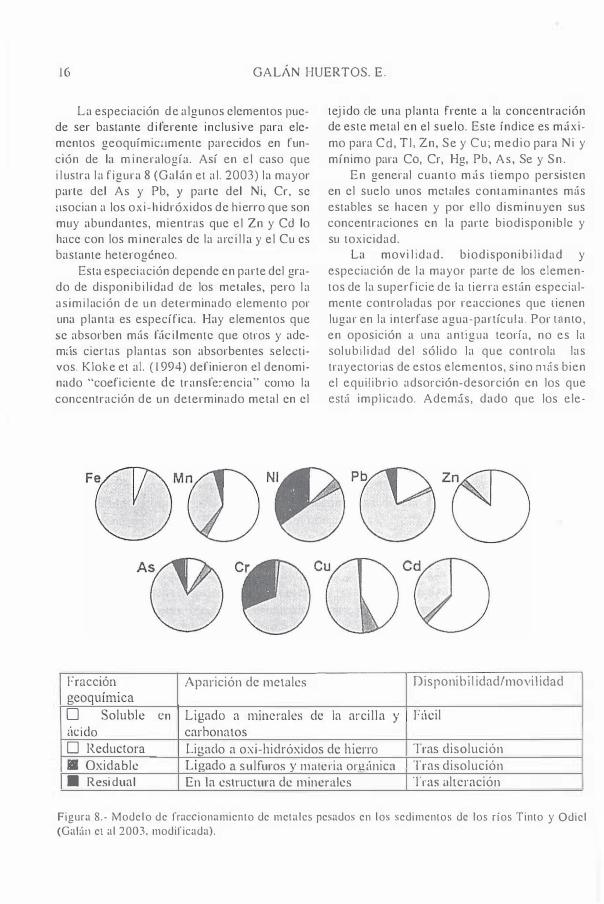
La especiación de algunos elementos puede ser bastante d i ferente inclusive para elementos geoquímic<lmcnte parecidos en runción de la m ineralogía. Así en el caso que i l ustra l a figura 8 (Gal¡ín et al . 2003) la mayor parte del As y Pb, Y parte del Ni, Cr, se :lsocian a los oxi-h idróxidos de hierro que son muy abundantes, mientras que el Zn y Ce! lo hace con los m inerales de la arc i l l a y el Cu es bastante heterogéneo.
Esta especi .. ¡ción depende en parte del grado de disponib i l idad de los metales, pero l a as imi lación de un determinado elemento por una planta es específica. Hay elementos que se absorben más f;ki lmcntc que otros y adem:ís ciertas plantas son absorbentes selectivos. Kloke et al. ( 1 994) definieron el denominado "coeficiente de transfe:-encia" C0l110 la concentración de un determinado metal en el
Fracción Aparición de metales geoquímiea O Soluble en Ligado a minerales tic ido carbonatos
de
tej ido ele una planta frente a la concentración de este metal en el sucIo. Este índice es máximo para Cd, TI , Zn, Se y Cl!; medio para Ni y mínimo para Ca, Cr, Hg, Pb, As, Se y Sn .
En general cuanto más tiempo persisten en el suelo unos mcwles contaminantes más estables se hacen y por el lo dism inuyen sus concentraciones en la parte biodisponiblc y su toxicidad.
La movi l idad. biodisponibi l i dad y cspeciación de l a mayor parle de los elementos de la superficie de la tierra están especialmente controladas por reacciones que tienen lugar en la interfase agua-partícu la . Por I<InIO, en oposición a una ant igua teoría, no es la solubi l idad del sólido la que controla las trayectorias de estos clementos, s ino nHís bien el equilibrio adsorción-desorción en los que eSlií impl icado. Adem,ís, dado que los ele-
Di spon ibi I idad/movi I idad
la arci l la y Fúeil
O Reductora Ligado a oxi-hidróxidos de h ierro Tras disolución a Oxidable Ligado a sulfuros y materia orgánica Tras disolución • Resi dua l En la estructura de minerales Tras altcración
Figura 8.- Modelo de fraccionamiento de metalcs pesados en los sedimentos dc los ríos Tinto y Odicl (Gal:'1Il el al 2003. modificada).

Aportaciones de l a m ineralogía a l a evaluación y tratamientos. 1 7
mentas o iones adsorbidos sobre u n sólido pueden oxidarse o reducirse, las reacciones de superficie pueden también cambiar su solubil idad y toxicidad. Entre las partículas naturales, los oxi-hiclróxidos de hierro y manganeso se han reconocido como los que juegan un papel m<ls importante en controlar los nutrientes y elementos traza en los suelos, aguas superficiales y subterdneas y sedimentos. Este papel se debe esencialmente a su alto desorden estructural, abundancia y habi l idad para catalizar reacciones redox (Wherli et a l . , 1 989).
A modo de resumen y como conclusión, la evaluación de l a contaminación por elementos traza cn sucios y sedimentos se debe hacer en varias fases. En una primera fase se debe comparar el contenido total de cada elemento con el fondo geoquímico y con n iveles guías, lo que podd indicar si existe una potencial contaminación. En caso positivo se debe pasar a una segunda fase en la que se
debe investigar la fracción as imi lable. Para el lo, se puede recurrir a extracciones selectivas y a l a especiación química. Las extracciones mtts comunes son: a) con agua, b) con agua <lcida y c) con EDTA (fracción biodisponible para plantas). La especiación química nos indicar<l l a distribución según distintas matrices, que van desde las muy I{¡biles (f<Íci l mente l i beran los metales asociados) a las muy estables. con lo que obtendremos un panorama de cómo los distintos componentes del suelo o sedimento i n fl uyen en la retención de los metales y en su probable estabilidad. F inalmente una determinación de los principales panímetros y una combinación de protocolos químicos y m ineralógicos. podd asegurar razonablemente el riesgo de contaminación a corto y medio plazo en un suelo/ sedimenw. Un esquema para un protocolo de espec iación químico-minernlógica se describe en la figura 9.
M UEfstra •
Granulometría �
Separación fe fracciones < 2mm
� < 2�m .---------1f-------·
� Separa �ón con Estudio DRX, IR brom9 ormo* AQ elem tr tata les
� . , SEM, AQ elem tr ¡ !
� Ligera Extracción ox. Fe �
DRX, IR, SEM
l Pesada --o
Probeta � M icroscopia
Espe laclon
DRX
SEM, Mapas elem tr �
AnáliSis EMPA, (�aman) de M . P . •
*Alternativamente a la separación de MP con bromoformo se puede hace una separación magnética
Figura 9 ." Esquema d e trabajo para l a espcciacióll químico " mineralógica

18 GALÁN HUERTOS. E.
Contaminación de sedimentos recientes.
Los sedimentos recientes comprenden una mezcla de minerales detríticos y meteorizados amorfos o pobremente cristalinos. material biogénico. materia orgánica y agua intersticial. Esta mezcla es inestable y progresivamente va alcanzando el equilibrio a través de una serie de reacciones diagenéticas.
El origen de estos materiales está básicamente relacionado con las áreas continentales, su naturaleza, el tipo de meteorización y los procesos hidrológicos. Las partículas son transportadas a Jos lagos. costas, estuarios. etc. por los ríos y olras aguas de escorrentía, y por el viento. La composición química es un reflejo de esta composición mineralógica y también de los materiales contaminados que drenan los ríos, o las aguas de lixiviación que a ellos llegan y los efluentes industriales y mineros. Los contaminantes más comunes son: elementos traza, nutrientes, productos derivados del petróleo, residuos de distintos orígenes (industrial, urbano), peslicidas, lluvia ácida y radionucJeidos.
Muchos de los metales que se encuentran en sedimentos, rfos, lagos y costas proceden de la minerfa y de la industria extractiva. Los sedimentos que han recibido estos aportes se convierten en potencialmente contaminados de una forma más estable que las láminas de agua. El que un elemento traza tóxico se estabilice en el sedimento o se movilice fácilmente y
pase al agua, depende de su especiación. El sedimento no puede considerarse como contaminado si no contamina a las aguas que están íntimamente en contacto, y esto no es fácil de evaluar. Los efectos de los elementos traza en la biola (Tabla 9) se manifiestan generalmente por una reducción de la diversidad, productividad y densidad de organismos (fitoplacton, plantas, invertebrados y peces).
La mayor parte de los elementos traza que se incorporan a lagos, estuarios y plataforma continental son rápidamente captados por el sedimento (carbonatos, oxi-hidróxidos de Fe y Mn, arcillas, rr.ateria orgánica, fosfatos). Al igual que para un suelo, el análisis químico total es una expresión pobre de lo que puede estar biodisponible. De nuevo una extracción secuencial puede predecir mejor el potencial tóxico del sedimento.
Los elementos biodisponibles están esencialmenle relacionados con su solubil idad en agua, que depende del pH y Eh. Por ejemplo, la biodisponibilidad del Cd es baja cuando las condiciones son anaerobicas, probablemente porque se produce CdS insoluble en ambiente reductor; el Se puede aparecer como seleniuro Sel., Seo, selinito Se/' con Se4+, y seleniato SeO/' con Se6', este último es mucho más biodisponible que las formas más reducidas.
Otro caso interesante es el del arsénico. La movilidad y biodisponibilidad de este elemento en el sistema sedimento-agua, y por tanto su toxicidad, depende de su eSlado de
Tabla 9.- Organismos más frecuentemente afectados por la toxicidad de algunos elementos traza (Page, 1992).
Especies adversamente afectadas Elemento Humanos Animales Organismos acuáticos Pájaros Plantas
e. x x x x X As, Pb, Hg, Cr, Se X X X X Cu, Ni, Zo X X Mo. F,Co X B X

Aportaciones de la mineralogfa a la evaluación y tratamientos. 19
oxidación y especiación química. En los sedimentos (y suelos) se encuentra Asl. (arsenito) y As)· (arseniato), siendo la forma más reducida mucho más tóxica (Ferguson y Davis, 1972), porque es más soluble y móvil. Pero los óxidos de Fe y Mn promueven la oxidación de Asl• a Ass., y además minerales de Mn captan fácilmente ambos tipos de As, por [o que este elemento en presencia de estos óxidos, reducen la concentración de As (Deschamp et al., 2003).
A[ igual que ocurría en los suelos, el pH tiene una gran imponancia en la biodisponibi lidad de metales. En general aumenta cuando disminuye el pH, pero el efecto sobre oxi-aniones es muy variable. Así As, Mo, Se y algunas formas de Cr pueden estar más disponibles cuando aumenta el pH. La existencia de protones o de hidroxilos pueden competir con los elementos traza en procesos de adsorción y de acomplajamiento, y pueden alterar la distribución de elementos traza entre el agua y el sedimento. La biodisponibilidad de Cd, Cu y Ni, por ejemplo disminuye con el aumento del pH.
Otros dos factores de gran importancia en el control de la movilidad de elementos traza en un sedimento son los minerales de la arcilla y la materia orgánica. Los minerales de la arcilla pueden actuar como adsorbentes e inlercambiadores de iones con el medio. controlando la movilidad de los metales, y la materia orgánica puede formar complejos organo-metálicos, que sólo si son destruidos por oxidación liberan el metal. Estos componentes del sedimento controlan esencialmente Cd, Zn, Cu, Ca y Ni.
Un caso estudiado por nosotros ha sido la caracterización y delimitación de la contaminación en los sedimienlOS recientes de la ría de Huelva (Fernández Caliani et al., 1997).
El estuario de Huelva es uno de los sistemas acuáticos más contaminados de Europa, debido a los efectos de drenaje ácido de las minas de la Faja Pirílica Ibérica y a los efluentes de procesos industriales del Polo
Químico de Huelva (Figura 10). Los sedimentos se caracterizan por una distribución granulométrica poli modal, predominando los limos y arcillas en el estuario y aumentando progresivamente e[ tamaño de grano hacia la plataforma adyacente donde los sedimentos son arenosos.
La composición mineralógica global consiste en cuarzo, feldespatos, carbonatos, filosilicatos y ocasionalmenle yeso y oxihidróxidos de hierro de baja cristalinidad. As! mismo, existe una amplia variedad de minerales accesorios, entre los que destacan ilmenita, hematites, magnetita, pirita, rutilo y zircón. Los filosilicatos son los componentes esenciales de los sedimentos estuarinos y la iIIita es el mineral más abundante (70-80%) con caolinita entre 15 y 36% Y clorita como accesorio (<5%). En general, la relación de abundancia entre ilita y caolínita aumenta gradualmente hacia el mar exterior. A diferencia de otros sistemas costeros del suroeste de España, como la desembocadura del Guadalquivir o la Bahía de Cádiz, conviene señalar la ausencia de esmectilas en los sedimentos del estuario de Huelva, debido a la disolución que experimentan al entrar en contacto con las aguas ácidas de los ríos Tinto y Odiel (Galán et al., 1 999).
La distribución de metales pesados en el estuario lambién está condicionada por la granulometr!a de los sedimentos. Las mayores concentraciones de elementos potencialmente tóxicos se detectan en la confluencia de los ríos Tinto y Odiel, donde los sedimentos arcillosos contienen hasta 1830 ppm de Cu, 926 ppm de Pb. 2300 ppm de Zn y 850 ppm de As (Figura 1 1). Estas concentraciones se corresponden con índices de geoacumulación carac· terísticos de sedimentos extremadamente conlaminados.
Los mela les pesados disminuyen consi· ·derablemente hacia el medio marino, hasta llegar a valores normales en los sedimentos de la plataforma. Los metals pesados muesIran una gran afinidad por los sedimentos más

20
Piedras River
,o
'----20
,--- LEGEND ----D Ncogcne-Quaternary scdimcnts
EJ Sall marshes
• Industrial arcas
Industrial proccssing waS1Cs
.... \0- Isohaths (in melrcs) O Sampling statiol1s
GALÁN HUERTOS. E .
� Moguer
Padre Santo Channel
S ,-_'_' _' _-<:." o o 06
ATLANTlC OCEAN
® Mazag6n
' 1 1 , 1 2
N
5 Km
Figura 10.- Mapa del estuario de ¡-llIclva y platafoma adyacente (Fcrn(lIldcz Caliani el al.. [997).
N 5 Km
�.
Umbría
[illG8Y�
Figura 11.- Distribución cspaciai de las concentraciones de mcwlcs traza asociados a sul furos (Fcrnándcz Caliani e' al.. 1997).

Aporlaciones de la mineralogía a la evaluaci6n y tratamienlos . . . 21
finos del eSluario (Figura 12). Sin embargo debido a la reducida superficie especffica y la escasa capacidad de cambio iónico de los minerales arcillosos presenles (illila y caolinila). parece razonable asumir que la mayor proporci6n de metales se encuenlra adsorbidos específicamenle sobre los oxi-hidr6xidos de hierro. como se ha demostrado mediante exIracciones secuenciales (Galán el al.. 2003).
ESle es un caso típico de dislribución de minerales y metales pesados en un eSlUario y en la plataforma conlinenlal adyacente donde se refleja la interacción de complicados procesos hidrodinámicos e hidroquímicos en un medio litoral severamente eSlresado por recurrenles impactos ambienlales de origen anlr6pico.
Los minerales en el control y tratamiento de sucios contaminados.
Los minerales pueden desempeñar diSlintos papeles en el conlJol de contaminantes cuando: 1 ) pueden controlar los conlaminanles mediante ¡nleracciones de superficie ya sea por una atracci6n superficial reversible (adsorci6n) o por un proceso de precipilación helerogénea (por ejemplo. inducido por la superficie mineral) de mayor duraci6n; 2) pueden producir una precipitación homogénea (por ejemplo. di reciamente desde una soluci6n) para formar una fase eSlable. caplUrando conlaminantes denlro de la eSlructura mineral neoformada; o 3) pueden relener o liberar a los conlaminanles desde su estruclUra por inlercambio i6nico con una soluci6n.
Todas estas reacciones están controladas mayorilariamente por la superficie mineral. que es la ¡nlerfase con el aire. agua. moléculas orgánicas/inorgánicas y (micro-) organismos. Pero lo más imporlante es que las propiedades de la superficie mineral son subslancialmente diferentes de aquellas de la masa mineral global. por lo que debe considerarse la superficie como una entidad aparle.
La superficie mineral represenla una inlerrupción de la configuraci6n tridimensional de la red cristalina por 10 que se producen cambios en la química de coordinación alrededor de los iones expuestos en la superficie. Cuando la superficie ionizada entra en contacto con el agua la consecuencia directa es un cambio de la carga electrostática superficial. Este cambio. unido a la heleregenoidad topográfica. produce superficies reactivas. La reactividad está además innuenciada por el pH de la fase acuosa. la lemperatura. composici6n de la soluci6n. tamaño de partícula y cristalinidad (Sung & Margan, 1996; Slipp el al.. 1999).
Ciertos grupos de minerales son dignos de mención debido a su importante significado medioambiental y a su relevancia en el inlercambio de los contaminantes en ambienles contaminados. Esta imporlancia se debe a una de las siguientes propiedades: 1) alla solubilidad del mineral que contiene al ele-
Frldsptm """"";;'''.,1
-05
F.ador 1 Figura 12.- Representaeión de factores principales obtenidos de datos analltieos de muestras del estuario de Huelva y plataforma continental (Fern:'indez Caliani et al.. 1997). GR. MS. CS. VCS: grava y arena gruesa: FS y VFS: arena fina.

22 GALÁN HUERTOS. E.
mento contaminante (por ejemplo. carbonatos). 10 que conducirá a la liberación del contaminante; 2) baja solubilidad del mineral que contiene el contaminante huésped. lo que reducirá la disponibilidad del contaminante (por ejemplo. fosfatos); 3) área superficial grande, propiedad de gran importancia puesto que la liberación está bastante controlada por la superficie (los minerales claves aquí son los axihidr6xidos de hierro. que a menudo son fases pobremenle cristalizadas. formadas por muy pequei'ios cristalitos y que como consecuencia poseen área superficial muy alta; 4) estructuras cristalinas laminares. propiedad que junio al área superficial caracteriza a los minerales de la arcilla (esta estructura laminar hace que los minerales de la arcilla tengan una capacidad única para intercalar contaminantes entre láminas); 5) estructura� abienas, una propiedad característica de las zeolitas, las cuales pueden actuar como tamices moleculares y atrapar contaminantes (Valsani-Jones, 2000).
Los minerales o grupos que presentan alguno o varios de los requisitos necesarios para neutralizar/disminuir/controlar la contaminación son los siguientes: minerales de la arcilla, oxi-hidróxidos de hierro, manganeso y aluminio, zeolitas, fosfatos, carbonatos, glauconita, jarosita e hidrotalcitas. Además las rocas volcánicas pueden también ser bastante eficaces. En la figura 13 se puede observar el efecto de la adsorción de Ca por montmorillonila y óxidos de Fe y Mn. Estos últimos son mucho más eficaces porque el área superficial es bastante mayor que el de la montmorillonita.
De forma industrial un material usado en la gestión de residuos debe presentar la mayor parte de las siguientes propiedades: a) capacidad de neutralización, b) retención de agua, c) resistencia a la difusión, d) plasticidad y resistencia mecánica, e) capacidad de adsorción, f) capacidad de intercambio catiónico. y g) estabilidad (resistencia a la alteración). Varias de estas características son interactivas.
Cuando los minerales se usan para preparar barreras, deben colaborar a que éstas cumplan los requisitos fundamentales: encerrar y retener contaminantes; retener, recoger, controlar y eliminar las aguas superficiales y lixiviados; garantizar la estabilidad del depósito, y controlar el emplazamiento de los residuos y la evolución con el tiempo. Como se sabe una barrera técnica es una medida preventiva para proteger a bio y geoesferas de las sustancias peligrosas que emanen del depósito (Hermanns Stengele & PI�tze. 2000). De nuevo los minerales más idóneos para el control de la contaminación son los minerales de la arcilla y especialmenle las bentonitas.
Neutralización de la contaminación
Una de las cosas más interesantes de la utilidad de los minerales en el control de la contaminación es el que tiene lugar cuando de forma natural o provocada. se mitiga o neutraliza la producción de drenajes ácidos de rocas y estériles de minas.
En los residuos minerales. especialmente los derivados de la explotación de sulfuros,
Óxidos de f� y Mn
"
ÁcidCI h"mito
Mu .. lmorillonila
,. , , . , Co,," $OluciólI en eouilibrio (1'lI km))
Figura 13.- Curvas de adsorción de Ca por óxidos de Fe y Mn. ácido húmico y montmoril1onita (McLaren el al .. 1986).

Aporraciones de la mineralogía a la evaluación y tratamientos . . . 23
se producen lixiviados ácidos por oxidación de pirita y otras sulfuros, de acuerdo con la reacción: FeS} + 15/4 02 + 7n HlO _ Fe(OH)J + 2S0/ + 4W ( 1 ) que puede se atenuada o neutralizada por la existencia en la masa de estériles de
-otros
minerales. Se tendrá entonces un cierto potencial de ácidez. (PA) que podrá ser neutralizado según el potellcial de IIel/tralización (PN) que tenga el residuo. Si PA>PN se producirá el drenaje ácido.
El potencial de neutralización de una roca o estériles se expresa en Kg de CaCOJ por Tm de material. La relación PN/PA debe ser 2 ó 3 para estar seguro de prevenir la acidez (Lawrence & Wang 1996; Jambor & Blowes, ¡ 998).
Teniendo en cuenta la reacción (1) y que: CaCO) + 2 H· _ Cal. + COl + H20 Como por cada átomo de S se producen 2
W, 1 mol de S es equivalente a 1 mol de CaCO]'
PA",(XlIOO) x 1000 kg x (peso molecular CaCO/peso atómico S)
PA", %S x 31 .25 kg CaCO/fm Los minerales que existen (o pueden ser
añadidos) en los residuos controlan potencialmente la acidez de acuerdo con su velocidad de alteración. Sverdrup ( 1990) agrupó los minerales en seis categorías para los que Know ( 1993) dio valores numéricos (Tabla 10). Estos datos. con cierta prudencia, pueden ser usados para recalcular el PN de residuos mineros en los que se están generando aguas ácidas.
La interpretación de los valores de PN se facilita con los datos de meterorización de minerales en suelos y rocas, a partir del Programa PROFILE (Sverdrop y Warfinge, 1993, 1995; Hodson et al. 1997), que establece además un orden relativo para la modelización en la velocidad de meteorización de los minerales.
Algunos de estos minerales pueden añadirse a suelos como enmiendas, pero hay que tener en cuenta que su descomposición pueden generar ciertos metales en exceso (K, Al. Fe).
Tabla 10.- Agrupación de minerales según la capacidad de neutralización (Knowg. 1993; Sverdrup, 1990).
1.- Disolución
2.- Meteorización rápida
3.- Meteorización intennedia
4.- Meteorización lenta
5.- Meteorización muy lenta
6.- Inerte
Minerales Típicos
Calcita, aragonito, dolomita, magnesita, brucita
Anortita, nefelina, olivino, granate, jadeita, leucita, espodumena, diopsido, wollastonita
Epidota, zoisita, enstatia, hiperstena, augita, hedenbergita, homblenda, glaucofana, tremol ita, actinolita, antofillita, serpentina, crisotilo, talco,
clorita, biotita
Albita, oligoclasa, labradorita, vermiculita, montmorillonita, gibsita, caolinita
Feldespato K, moscovita
Cuarzo, rutilo, circón
Reactividad Relativa
1.0
0,6
0,4
0,02
0,01
0,004
Reaetividad Relativa sobre la base de suelos monomincrálicos con el tOO% de mineral individual.

24 GALÁN HUERTOS. E.
Es"¡ es uno de las investigaciones aplicadas más atractivas y con ruturo para un mineralogista ambiental.
Conclusiones.
La aportación de los mineralogistas a la geoquímica ambiental, especialmente en los temas relativos a la contaminación de los suelos y sedimentos en lagos. ríos y plataforma continental. está colaborando decisivamente a la evaluación de los riesgos reales a corto y medio plazo de la presencia de concentraciones anómalas de ciertos elementos traza y al control y tratamiento.
Las posibilidades de estudio en Mineralogía Ambiental cubren aspectos que van desde la invesligación de los fenómenos a escala atómica en la superficie de los cristales (procesos de disolución. adsorción. interacciones microbianas, etc.), a la determinación de "fondos geoquímicos", de "niveles de referencia" y de "niveles guías" para evaluar el origen y potencial contaminación por elementos traza en primera aproximación, y la investigación detallada de los riesgos a corto y medio plazo mediante la especiación químico-mineralógica.
En un futuro grandes superficies mineras abandonadas deberán ser objeto de investigación y control para evitar desastres ecológicos, y para, si es posible, recuperar el suelo para un uso agrícola, ganadero o forestal, y el papel del mineralogista en esta tarea puede ser fundamental.
Por OlTa parte, en el ámbito docente, estas perspectivas deben impartirse a nivel de "master", lo que proporcionará al licenciado una nueva especialización profesional.
Agradecimientos.
Muchas de las ideas aquí expuestas han surgido de la discusión con los integrantes del
Grupo de Investigación RNM 135 "Mineralogía Aplicada", a quienes deseo dedicar y agradecer esta conferencia. Así mismo, agradecer a los organizadores de la XXII Reunión de la Sociedad Española de Mineralogía la oportunidad que me han dado de hacer públicas estas reflexiones, invitándome a dar la Conferencia Inaugural del Congreso celebrado en Logroño.
Bibliografía.
Andrew R.L ( 1984) Supergene alteration and gossan textures of base-metal ores in sourthern Afdca. Mil/erais Sci. ElIg., 12,193-215.
Banks O. Younger P.L., Arnesen R-T., Iversen E.R., Banks S.B ( 1997) Mine-water chemistry: the good, (he bad and the ugly. Envirol/. Geof. 32: 157-173.
Becker P.C., Rasso K.M., Hochella M.F. (2001 ) The proximity effect on semiconducting mineral surfaces: A new aspect of mineral surface reactivity and surface complexation therory? Geochim. Cos/llocl¡jm. Acta, 65: 2641-2649.
Belzile N., Maki S .• Chen Y·W, Goldsack O. ( 1997) Inhibition of pyrite oxidation by surface trealmen!. Sci. Tot. Environ., 196: 177-186.
Biegler T., Swift O.A. (1979) Anoxic behaviour of pyrite oxidalion in acid solutions. Electrochim. Acta, 24: 415-420.
Bourg A.C.M. ( 1995) Speciation of heavy metal s in soils and groundwater and implications for their natural and provoked mobility. In .. Heavy Metals". W. Salomons, U. Forstner & P. Mader (Eds.). Springer-Verlag. Berlin. Pp: 19-3 1 .
Bowen H.J.M. ( 1979) Enviromental chemistry ofthe elements. Academic Press. London: 333 pp.
Bowie S.H.U., Thornton (Eds). ( 1985) Environmental Geochemistry and Health. Kluwer Academic Pub!.. Hingham, MA.
Boyle O.R. ( 1994) Oxidation of massive

Aportaciones de la mineralogía a la evaluación y trlllamienlos . . . 25
sulfide deposils in the BarthurSI Mining Camp, New Brunswick. Pp. 535-550 in: Environmental Geochemistry of Sulfide Qx.idalion (C.N. Alpers and D. W. Blowes, editors), American Chemical Society, Symp. Series, SSO.
Brion D. (1980) Etude par spectroscopy de photoeleclrons de la degradation superficialle de FeSl, CuFeSl, ZnS and PbS a lairs dans. Appf. SlIr! Sci., S: 133-152.
Brock T.P., Smilh q.W., Madigan M.T. (1984) Biology of Micro-organism. Prenlice-Hall, Englewood Cliffs, New Jersey.
Campbell P.G.C., Tessier A. ( 1987) Current Slatus of metal speciation Sludies. In: "Metal Specialion. Separation and Recovery". JW PaUerson & R. Passino (Eds.). Lewis Publ. Chrlsea, Michigan. Pp: 201-224.
Chen H.M .• Zheng C.R., Tu C .• Zhou D.M. (2001) Sludies on loading capacity of agricultural soils for heavy metals and iIS applicalions in China. App. Geocllem. 16: 1397-1403.
Deer W.A . • Howie R.A., Zussman J. (1992) An introduclion lO rock-forming minerals. Longman Scientific & Technical. Harlow. Essex., UK.
Deschamps, E.; Ciminelli, S.T.; Weidler, P.G. and Ramos, A. Y. (2003). Arsenic sorption onto soils enriched in Mn and Fe minerals. Cfays Clay Mil/u., SI, 197-204.
Ferguson. J.G. and Davis. J. ( 1972). A review of arsenic cycJe in nalural waters. Water ResOllrces. 6. 1259-1274.
Fernánde:z. Caliani J.C . • Ruiz Muñoz F., Galán E. (1997) Clay mineral and heavy metal distribution in the coger estuary of Huelva and adyacent Atlantic shelf. SW. Spain. Sci. Toral El/v. 198: 1 8 1-200.
Flann R.C., Lukaszewski G.M. ( 1970) The oxidation of pyrrhotite in ores and concentrates. Presentation al Austral. lnsl. Mining Metal!., Regional Mtg., Tennant Creek, Australia. Pp. 59-102 in: Environmental Geochemislry of Sulfide
Minewastes O.L. Jambor and D.W. Blowes, editors). Short Course. 22. Mineralogical Association of Canada.
F�sler U. ( 1987) Changes in metal mobilities in aqualic and terrestial cycles. In " Hcavy Melals". W. Salomons, U. F�rslner & P. Mader (Eds.). Springer-Verlag. Berlin. Pp: 3-26.
Gaad G. (2000) Heterotrophic solubilization of metal-bearing minerals in fungi. Pp 57-75 en: Environmenlal Mineralogy: Microbial Inleations, Antrophogenic Influences, Contaminated Land and Wasle Management (1.0. Couer-Howells, L.S. Campbell, E. Valsami-Jones and M. Batchelder, editors). Mineralogical Society Series, 9. Mineralogical Society, Landon.
Galán E. (2000) The role of clay minerals in removing and immobilising heavy metals from contaminated soils. In Proceedings ofthe 1" Latin American Clay Conference. C. Gomes (Ed.) Funchal 2000, vol. 1, pp: 351-361.
Galán E., Carretero M.I.. Fernández Caliani J.C. (1999) Effects of acid mine drainage on clay minerals su pended in Ihe Tinto River (Rio Tinto, Spain). An experimental approach. C/ay Miner., 34: 99-108.
Galán E., Gómez Ariza J.L., González l., Fernández Caliani J.C., Morales E., Gidldez J. (2000) Utilidad de las técnicas de extracción secuencial en la mejora y caracterización mineral6gica por DRX de suelos y sedimentos con altos contenidos de óxidos de hierro. En: "Integración de Ciencia y Tecnologfa de Arcillas en el Contexto Tecnológico-Social del Nuevo Milenio» Pascual J .• Zapatero J. Ramfre:z. del Valle AJ .• Moya Garcfa M.V. (Eds). Sociedad Española de Arcillas y Diputación Provincial de Málaga. Pp: 337-347.
Galán E., Gómez Ariza J.L., González l., Fernández Caliani J.c.. Morales E., Giráldez 1. (2003) Heavy metal partilioning in river sediments severely polluted by acid mine dreainage in the

26 GALÁN HUERTOS. E.
Iberian Pyrite bell. Appf. Geoc/¡em., 18: 409·421 .
Gray N.F. ( 1996) Field assessmenl of acid mine drainage contamination in surface and ground water. El/virOI/. Geof. , 27 : 358-361.
Hermanns Stengele R., P!tUze M. (2000) Suitability of minerals for conlrolled landfill and conlainment. Pp: 291-331 en: Environmental Mineralogy (Vaughan D.J . • Wogelius R.A. editors). European Mineralogical Union. Budapest. Hugary.
Hodson M.E., Langan S,J" Wilsoln MJ. ( 1997) A critica] evaluation of ¡he use of ,he profiJe model in calculating mineral wealhering mies. Water, Air, SaO. Polllu., 98: 79-104
Jambor J.L. ( 1994) Mineralogy of sulpohiderieh lailings and their oxidation prodUCIS. Pp: 59·102 in: Environmental Geochemistry of Sulfide MinewaSles (J.L. Jambor and O.W. Blowes, OOitors). Short Course, 22. Mineralogical Associ:nion of Canada.
Jambor J.L., Blowes oF.W. ( 1998) Theory and applieations of mineralogy in environmenlal sludies of sulfide-bearing mine wastes. Pp. 367-402 in: Modern Approaehes 10 Ore and Environmenlal Mineralogy (LJ. Cabri and o.J. Vaughan, editors). Shorl Course, 27. Minernlogical Association of Canada.
Kabata-Pendias A., Pendias H. (1992) Trace elements in soils and plants. 2001 edition. CRC Press. Ine. Boca Raton, Florida. Pp: 365.
Kakovsky I.A., Kosikov Y.M. ( 1975) Sludy of kinelies of oxidation of sorne sulfide minerals. Obogasheh. Rud., 20: 18-21-
Keith C.N., Vaughan, oJ. (2000) Meehanisms and rales of sulphide oxidation in relalion 10 Ihe problems of aeid roek (mine) drainage. Pp 1 17-140 en: Environmental Mineralogy: Microbial Inteations, Antrophogenie Influences, Conlaminated Land and Waste Managemenl (J.o. ColterHowells, L.S. Campbell, E. Valsami-Jones and M. Batchelder, editors). Minernlogical
Soeiety Series, 9. Mineralogieal Society, London.
Kloke A., Sauerbeek o.R., Vetler H. ( 1994) In "Changing metals eycles and human health". J. Nriagu (Ed.) Springer-Verlag, Berl{n.
Kwong Y.TJ., Ferguson K.O. ( 1990) Water chemistry and mineralogu at Moun! Washinglon: implications to acid generation and metal leaching. Pp. 217-230 in: Aeid Mine orainage: oesigning for Closure (J.W. Gadsby, J.A. Maliek and S.J. oal, editors). Bilech Publishers, Vancouver, British Columbia.
Kwong YTJ. (1993) Prediction and prevention of acid mine drainage from a geologieal and mineralogieal perspeelive. Projeet 1.32.1, MENO, Natural Resources Canada, OUawa.
Lasaga A.C, Berner, R.A. ( 1998) Fundamental aspeels of quantitative models for geochemical cycles. C/¡em. Geol. , 145:
161-175. Lawrenee R.W., Wang Y. (1996) oetermination
of neutralization potential for acid rod drainage predietion. Draft Report, MENO projeel 1 . 16.3. OUawa (Ont.): MENO, Natural Resourees Canada.
McKibben M.A., Barnes H.L. ( 1986) · Oxidalion of pyrite i n low lemperature aeidic consideralions. Geocllim. Cosmochim. Acra, 51 : 3 1 93-3199.
MeLaren R.O., Lawson D.M., Swifi R.S. (/986) Sorption and desorption of eobal! by soils and soil components. J. SoU ser 37: 41 3-426.
Nesbi!t H.W., Bancroft G.M., Pratl A.R., Seaini M.J, (1998) Sulfur and iron surface slates on fraelured pyrile surfaces. Amt"r. Minual., 83: 1067- 1076.
Novolny V. ( 1 995) Oiffuse sources of pollution by loxic melals and impael on receiving waters. I n " Heavy Metals". W. Salomons. U. FOrstner & P. Mader (Eds.). Springer-Verlag. Berlin. Pp: 33-52.
Page, H.L. ( 1992) In: Pierzynski. O.M.; Sims.

Aponaeiones de la mineralogía a la evaluación y tratamientos . . . 27
J.T. and Vanee, G. ( 1 994). Soils and Environmenlal Quality . Lewis Pub. London.
Rock N.M.S. (1988) Numerical Geology. Leeture Notes in Earth Science, 18.
Springer. Berlin. 427 pp. Russo K.M., Becker U., Hochella M.J. Jr.
( 1999) The interaclion of pyrile { lOO) surface wilh 02 and H10: fundamental oxidalions mechanisms. Amer. Mil/eral. , 82: 1549- 1561.
Sal minen R., Gregorauskiené V. (2000) Considerations regarding the definilion of a geochemical baseline of elemenls in Ihe surficia! materials in areas differeing in basic geology. App. Geochem. 15: 647-653.
Sal minen R., Tarvainen T. ( 1997) The problem of defining geochemical baselines. A ease study of selected etements and geological materials in Fintand. J. Geoc/¡em. Explor. 60, 9 1 ·98.
Salomons W. ( 1995) Environmenlal impaels of melals derived from mining aetivities : processes, prediclions, prevenlion. J. Geochem. Expl., 52: 5-23.
Sarveswara Rao, R.K., Das R.P., Ray H.S. (1991) Study of leaehing of mullimetal sulphides Ihrough an interdisciplinary approaeh. Mil/eral. Process. Ex/r. Metal/. Re .... , 7: 209-233.
Siegel, F.R. 2002. Environmental Geoehemislry of Potenti lly Toxic Metals. Springer, 2 18 pp.
Sposito G. (1989) The chemislrY of soils. Oxford Univ. Press: 234 pp.
Stipp S.L.S., Brady P.V., Ragnarsdottir K.V., Charlet L. (Edilors). ( 1999) Oeochemistry in aqueous systems. A special issuc in honour of Werner Stumm. GeocJ¡¡m. Cosllloc/¡im. Acta, 63: 2891-3497.
Sung, W and Morgan, 1.1. (1981). Oxidative remova1 of Mn (11) from solution catalysed by the lepidocrucite surface. Geoe/lim. COSllloe/Jim. Acta. 45, 2377-2383.
Sverdrop H., Warfinge P. ( 1993) Caleulating
field weathering rales using a mechanistic geochemieal model PROFILE. App. Geochem., 8: 273-283.
Sverdrop H., Warfinge P. ( 1995) Estimating field wealhering rates using laboratory kineties. Pp. 485-541 en: Chemieal Weathering Rates of Silicale Minerals (A.F. White and S.L. Brantley, editors). Review in Mineralogy, 31. Mineralogical Society of America, Washington D.C.
Sverdrup H.U. ( 1990) The kinetics of base cal ion relense due to ehemiea! weathering. Lund UniversilY Press, Lund, Sweden.
Tessier A., Campbell POC, Bisson M. ( 1979) Sequential extraction procedure for the speciation of parliculate trace metals. Al/al. Cllem. 51: 844-851.
Tessier A., Campbell POC, Bisson M. (1985) Trace elements in oxic lake sediments: possible adsorption of oxy-hydroxides. Geocllim. Cosmoc/¡im. Acta. 49: 183- 194.
Valsami-Jones E. (2000) Section 3: Minerals in contaminated environments. Pp. 201-205 in: Environmental Mineralogy: Microbial Interactions, Anlropogenie Influences, Contaminated Land and Waste Management (J.D. Cotler-Howells, L.S. Campbell, E. Valsami-Jones and M. Batehelder, editors). Mineralogical Society Series, 9. Mineralogical Sociey, London.
Vaughan DJ., England K.E.R., Kelsall G.H., Yin Q. (1995) Eleetrochimical oxidation of chalcopyrile (CuFeS) and the related metal-enriched derivatives Cu�Fe,SI' CugFe9S16 and Cu9Fe.SI6• Amer. Mi/leral., SO: 725-773.
Vaughan DJ., Becker U., Wright K. ( 1997) Sulfide mineral surfnces: Iheory and experiment. /IIf. J. Millelal Proc. 51: 1-14.
Vaughan DJ., PaUrick R.A.D., Wogelius KA. (2002) Minerals. metal s and moleeules: ore and environmentnl mineralogy in the new mil1ennium. Mi/leral. Magazine, 66: 653·676.
Wehrli B., Sulzberger B., Stumm W. (1989) Redox processes catalyzed by hidrous

28 GALÁN HUERTOS. E.
oxide surface. Chem. Geol., 78: 167·179. WogeliusR. A .• VaughanD.J. (2000) Analytical,
experimental and computationnl methods in environmental mineralogy. Pp: 7·88 En: Environmental Mineralogy (Vaughan D.J .• Wogelius R.A. editors). European Mineralogical Union. Budapest. Hugary.
Yin Q., Kelsall G.H .• England K .• Vaughan •
50lulion5. J. Eleclrochem. Soc . • 147: 2945· 295 1 .
Vin Q., Kelsall a.H., Vaughan D.J . • England K.E.R. ( 1 995) Atmospheric and electrochemical oxidation of the surface of chalcopyrite (CuFeSJ). Geoc/¡im. Cosmoclrim. Acta. 59: 1 091- 1 100.
OJ. (2000) Surface Qxidation of Recibido: Diciembre 2003 chalcopyrile (CuFeSJ) in alkaline Aceptado: Diciembre 2003

Boletín de la Sociedad Espmiola de Mil/eralogía, 26 (2003), 29.44 29
Las mineralizaciones de pirita de la cuenca de Cameros en su contexto geológico
Jacinto ALONSQ-AZCÁRATE Facultad de Ciencias del Medio Ambiente, Fábrica de Armas. Universidad de Castilla - La
Mancha. -45071-Toledo, España. (e-mail: [email protected])
Abslract: The low-grade metasediments of (he Cameros basin, NE Spain. host a numbcr of deposits of speetacular quality pyrite mineralization. The deposits wer� formed at, or close 10, the peak of metamorphism and are always related to sandstone units in the mainly lutitic sequence. Iron remained immobile and conservalive, pyrite-iron being derived by sulphidation of chlorite in (he host metapelites. Reduced sulphur however, was supplied from twoexlernal sources: Ihermochemical reduction ofsulphate and release of sulphur during metamorphism of sedimentary sulphides. These sources provided isolopieally heavy and lighl sulphur. respectively, with variation in pyrile isotopic compasilion bctween different deposits resulting from differences in their relative importance at each site. During melamorphism the sandstone units acted as aquifers, carrying the sulphidic pare-watcrs 10 locations where permeability provided by syndcpositional fractures allowcd ilS imeraction with the mClapelites. Morphological varialion on pyrite cryslals is related 10 changes in sedimentary sulphur availability and melamorphic fluids chemistry. High supersaluration morphologies are rclated 10 fluvio-deltaic sedimentary facies with high concenlrations of sedimentary pyrite and sulphales. Moreover, reactions involving sulphate during melamorphism may have modified fluid chemistry. whieh would also aet to produce higher degrees of pyrite saturation i n fluids. Low supersaluration morphologies appean i n meandriform sedimentary facies where the availability of sulphur is low.
Key words: pyrite, hydrothermal melamorphism. sulphur isotopes, fluid flow, crystal morphology, Cameros Basin: Spain.
Resumen: La cuenca de Cameros presenta una serie de mineralizaciones de pirita conocidas mundialmente debido a la calidad de sus cristales. Las mineralizaciones se formaron durante el pico de metamorfismo que afecto a los materiales del sector oriental de la cuenca y están localizadas en niveles lutllicos siempre en relación con potentes niveles de arenisca. Las lutitas que forman la matriz constituyen la fuente del hierro, el cual se libera medi:lnle un proceso de sulfurización de las cloritas del propio sedimento. El azufre reducido tiene un origen externo y deriva de dos fuentes diferentes: la reducción termoquímica de sulfatos sedimentarios y la rotura térmica de la pirita de los sedimentos de la cuenca. Estas dos fuentes suministraron azufre isotópicamente pesado y ligero respectivamente, estando las composiciones isotópicas de cada depósito determinadas par la importancia relativa de cada fuente. Durante el metamorfismo, los niveles de arenisca actuaron como un acuífero de alta permeabilidad, transportando nuidos ricos en azufre hasta que alcanzaron los niveles lutCtieos. Las lutitas presentaban una gran permeabilidad generada por la presencia de fracturas, de esta forma, los nuidos ricos en azufre interaccionaron con las lutitas generando las mineralizaciones, Las variaciones morfológicas que se observan en los cristales de pirita de las diferentes mineralizaciones están relacionadas con cambios en la disponibilidad de azufre y la química de los fluidos metamórficos. Las morfologCas de alta sobresaturación aparecen en materiales fluvio-deltaicos con altas concentraciones de pirita sedimentaria y sulfalOS. Además, las reacciones metamórficas en las que participaron los sulfatos: debieron tener un profundo

lO ALONSO.AZCÁRATE. J
efecto en la qufmica de los nuidos metamórficos, haciendo aumentar el grado de saturación en pirita. Las morfologras de baja sobresaturación nparcccn en f3cic$ sedimentarias de tipo meandriforme donde la disponibilidad de azufre es menor.
.
Palabras clave: pirita, metamorfismo hidrotcrmal, isótopos de azufre. nujo de nuidos, morfologra cristalina, cuenca de Cameros; Espili"ia.
Introducción
Un aspecto de gran interés en relación con la cuenca de Cameros. son las mineralizaciones de pirita que aparecen en el sector oriental de [a cuenca. Estas mineralizaciones son mundialmente conocidas por la calidad de sus ejemplares de pirita. los cuaJes pueden encontrarse en los mejores museos de mineralogía del mundo. A pesar de la notoriedad de estos yacimientos hasta hace poco tiempo no existía ningún estudio sistemático sobre su formación que los relacionase con la gtnesis y evolución de la Cuenca de Cameros.
,. I
CIJO."
,. r I
Características geológicas de la cuenca de Cameros
La cuenca de Cameros está situada en el extremo noroccidental de la Cordillera Ibérica (Fig. 1) , encontrándose al NW de tsta el macizo paleozoico de la sierra de la Demanda. Está limitada por las cuencas terciarias del Ebro al N y del Duero y Almazán al Sur. La cuenca puede dividirse en dos subcuo!ncas de morfología romboidal: Cameros W y Cameros E, siendo en esta última donde se localizan las mineralizaciones de pirita (Fig. 2).
.. I
r I
N
A
,.
_ 410
- 'O'
D CUf'.NCAS Tf.RCIARJAS O UNlf)M)f';.'; Mf.'IOZOI� UHllllU>tS lIudNlCAS
Figura 1.- Locali1.3ción de la cuenca de Cameros.

Las mineralizaciones de pirita de la cuenca de Cameros . . . 31
La cuenca de Cameros presenta una serie de peculiaridades que la distinguen claramente del resto de cuencas mesozoicas de la Cadena. Presenta una alta velocidad de subsidencia y lasa de sedimentación. acumulándose 5000 m de espesor vertical de sedimentos. que representan hasta 9000 m de registro estratigráfico en el sentido de desplazamiento de los depocentros de las sucesivas secuencias de depósito. comprendidas entre el Titónico y el Albiense inferior (Mas et al.. 1993).
Por otro lado. la cuenca de Cameros es la única de la cadena ibérica en la que parte de sus materiales están afectados por metamorfismo de grado bajo. el cual ha sido caracterizado como de tipo hidrotermal (Casque! et al .• 1992; Barrenechea et al .• 1995: Alonso-Azcárate et al.. 1995). A partir de dataciones en 1Iitas autigénicas. se obtienen edades que oscilan entre los 108 a 86 Ma para el metamorfismo, siendo estas edades clara-
DURCOS •
mente posteriores al relleno de la cuenca. Las temperaturas máximas alcanz.adas por estos materiales fueron de aproximadamente 360°C (Alonso-Azcárale. 1997) y las presiones de aproximadamente 1 Kb (Casquet et al .• 1992).
Desde el punto de vista estratigráfico. la megasecuencia de relleno de la cuenca de Cameros ha sido subdividida en seis secuencias deposicionales (SO 1 a SD6: Mas et al.. 1993). constituidas fundamentalmente por sedimentos continentales en facies nuviales y lacustres con esporádicas incursiones marinas.
Estructuralmente. la cuenca de Cameros es interpretada como una cuenca sinclinal. formada sobre una rampa de buzamiento sur que conecta dos rellanos de una falla extensiva, situada a varios kilómetros de profundidad dentro del zócalo (Mas el al.. 1993). Todas estas características se ajustarían al modelo de una cuenca de rampa extensional (Guimerá et al., 1995). Durante la compresión terciaria
• LOGRON'O
• "medo
N
t
,
� ......,,, R ;� , ---. Cn:tXico Suporior , . o " " ,. " ,. "'"
rm Hcn:iaico
Tri.»ico �. Jur.ó$ico - _ .. '1 --
D Fm. Utnlbs � l · • !! T"",i. .. io
Figuril 2.- M:lp:l geológico dct:lll:ldo de la cucnc:l de C:lmcros con l:l loc:lliz:¡ción dc los yacimientos de piril:l. A: Munilla; 8: Valdcnegrillos; C: Navajún: D: Valoria; E: Valdeperillos; F: Ambasaguas-A. Canadillas.

32 ALONSO-AZCÁRATE. J
(pale6geno-Mioceno inferior), se produjo la inversi6n de la cuenca, mediante un cabalgamiento neoformado en su margen norte sobre la Cuenca del Ebro, con un desplazamiento de hasta 30 Km y un sistema de cabalgamientos en su borde sur sobre las cuencas del Duero y Almazán (Mas el al.. 1993).
Los materiales que contienen las mineralizaciones fueron depositados en dos ambientes sedimentarios diferentes. Los sedimentos de las mineralizaciones de Ambasaguas y Arroyo Canadillas (Fig.2) fueron depositados en llanuras deltaicas con frecuentes episodios lacustres, los cuales se caracterizan por la presencia de contenidos relativamente altos de materia orgánica y minerales de hierro reactivos. En estos sedimentos lacustres la presencia de pirita sedimentaria es rrecuenle así como la de sulratos sedimentarios. Por otro lado, las mineralizaciones de Navajlln. Valdeperillos. Valdenegrillos, Valoria y Munilla (Fig. 2) encajan en sedimentos depositados en sistemas fluviales de tipo meandrirorme. caracterizados por prcsenlar contenidos bajos en pirita sedimentaria y sulratos.
Materiales y métodos
Se estudiaron un número representativo de cristales de pirita de cada yacimiento (Fig. 2). asf como sus materiales encajantes. También se analizaron sedimentos lulfticos y margosos del borde del sector occidental de la cuenca y sedimentos cercanos a los yacimientos de pirita del sector oriental de la cuenca. La composición mineralógica global de las muestras lutíticas es: cuarzo + feldespatos + filosilicatos ± calcita ± dolomita. Los filosilicatos presentes en la rracción fina son: ilila ± clorita ± interestratiricados (principalmente ilitaJclorita e ilita lesmectita).
Los sulfuros sedimentarios se extrajeron químicamente mediante el método propuesto por Candfield et al. ( 1986), modificado por Newton et al. ( 1995). Con este método, en una
primera etapa se extraen los monosulfuros de Fe (pirrotita) mediante una reacción con HCI denominada A VS (acid volotile sufpllide). A continuación, utilizando dicloruro de cromo (CrCl1), se realiza la extracción de la pirita.
Lns determinaciones de las relaciones isotópicas de S (�SplS) se realizaron sobre SOl gas. preparado por oxidación del sulruro con óxido cuproso (Robinson y Kusakabe. 1975). El S0l rue analizado en un espectrómetro de masas VG Isogas SIRA 10. Las muestras y los estándars internos (British Geological Survey Chalcopyrite Cp-I) se analiznron con un gas de rererencia interno. Los valores de &]..IS ;e calcularon usando procedimientos de corrección estándar (Craig. 1957; Coleman. 1980) y se expresan en %o relativos al estándar de troilita (FeS) del Cañón del Diablo (CDT). Los análisis de SuS para la pirita mediante microsonda láser se realizaron usando la técnica descrita por Kelly y Fallick ( 1990). Los análisis de &]..15 de las inclusiones de anhidrita mediante microsonda láser se realizaron con un láser de COl como se describe en Alonso-Azcárale et ni. ( 1999b).
Las muestras lutíticas rueron analizadas mediante técnicas de dirracción de rayos X. Se utilizó un difractómetro Philips PW 1 730/ 90. usando radiación Cu-K". monocromador de grafito. rendijas de 1°_0.2 mm_1°. intensidad de 1 x lOl a 1 x 104 c.p.s. con una constante de tiempo de l . 40Kv, 30 mA y una velocidad de barrido de 2°6.29min.
La concentración en elementos menores y trazas en lutitas y piritas rue determinada mediante espectroscopfa de plasma inducido (lCP-AES) disolviendo las muestras a presión en una disolución de HFIHC10iHNO).
Resultados
Las mineralizaciones de pirita aparecen exclusivamente en el sector oriental de la cuenca (Fig. 2). dentro de lutitas afectadas por metamorfismo hidrotermal de bajo grado.

Las mineralizaciones de pirita de la cuenca de Cameros . . . 33
Aunque todas las secuencias deposicionales presentan pequeñas mineralizaciones de pirita, los depósitos mayores están localizados en las secuencias deposicionale¡; 4 y 5.
Las mineralizaciones se localizan en niveles lutfticos los cuales presentan siempre a techo potentes niveles de arenisca. Los niveles de arenisca raramente contienen cristales de pirita. Ocasionalmente es posible encontrar pequeñas mineralizaciones asociadas a margas o calizas pero de nuevo siempre asociadas con niveles de arenisca.
Los niveles lutiticos mineralizados presentan una serie de fracturas hidroplásticas, probablemente de origen edárico, que se forman muy tempranamente durante la compactación. Las superficies de fractura son alabeadas de aspecto satinado y son muy frecuentes en toda la cuenca (Guiraud. 1983). Estas fracturas van a controlar el estilo de la mineralización cambiando la densidad de ésta e incluso el hábito de los cristales entre fracturas contiguas.
Otra característica importante común a todos los yacimientos. son las fuertes decoloraciones que presentan las lulilas mineralizadas que tienen colores blanquecinos con respecto a [as tonalidades verdosas que presentan generalmente los sedimentos lutilicos en la cuenca. Estas decoloraciones van a estar relacionadas con la ausencia de clorita en estos sedimentos.
Los cristales de pirita aparecen generalmente rodeados de una envuella isópaca formada fundamentalmente por cuarzo y cookeita y en menor proporción por calcita y caolinita. En los yacimientos cuya roca encajante aparece deformada, estas envueltas aparecen formando sombras de presión alrededor de los cristales de pirita.
A pesar de las superficies limpias de los cristales. éstos presentan una gran cantidad de inclusiones en su interior. Las mineralizaciones en las que sus sedimentos alcanzaron condiciones de epizona presentan como inclusiones cristales de cJoritoide y en
general en todos los depósitos vamos a encontrar inclusiones de diversos minerales (iIlita. clorita, cuarzo, calcopirita. blenda) entre los que cabe destacar la presencia de anhidrita debido a su importancia para la rcalización de cálculos geotermométricos.
También se realizó un estudio de los sulfuros diseminados en los sedimentos. los cuales van a jugar un papel imponante en la génesis de las mineralizaciones. Se estudiaron muestras tanto de la zona oriental como de la occidental de la cuenca. Los sedimentos presentan un contenido medio de sulfuros bajo. aunque es bastante variable (0.02- 2.3 % $). En algunas muestras junto a [a pirita encontramos también pequeñas cantidades de pirrotina siempre en menor proporción que ésta. La pirita aparece en [os sedimentos en forma de frambóides. cristales euhedra[es o sustituyendo la concha de fósiles (AlonsoAzcárate et al.. 1999c).
En la Tabla ¡ se presenta el contenido medio en Fe para la matriz lutítica de las diferentes mineralizaciones, también aparece una muestra lulftica no mineralizada representativa de las lutilas que aparecen en la cuenca, asr como el contenido medio en rocas
Tabla 1.- Contenido en Fe de las lutilas de las diferentes mineralizaciono&. un sedimento no mineralizado y una ¡utita estándar (Carmichael. 1989).
Yacimiento Valdeperillos Navajúo VaJdenegrillos Valoria Munilla Ambasaguas A. Canadillas Lutita no mineralizada Lutita estandar
0.66 0.87 0.65 0.74 2.37 0.76 0.68 3.84 2.80

34 ALONSO·AZCÁRATE. J
pelíticas. En general los contenidos en Fe de la matriz son menores que los valores medios en rocas pelfticas y que en la ¡utila no mineralizada.
Otro aspecto importante de estos yacimientos es la variación en la morfologí:1 de los cristales de pirita entre los diferentes yacimientos de la cuenca. Por un lado. tenemos los yacimientos de Ambasaguas y Arroyo Canildillas (grupo 1). en los cuales los cristales presentan morfologías de piritoedro y cubopiriloedro. con las caras fuertemente estriadas. así como cristales con morfologí:lS de crecimiento cuarteado y agregados de cristales de pirita de grano fino. Por Olro ludo. tenemos los yacimientos de N,lVujún, Valdeperillos, Valdenegrillos. Valoría y Munilla (grupo 2) en los cUlIles encontrllmos cristllles cúbicos de caras lislls y más TlIramente cristales con morfologfas aplanadas y alargadas y cubooctaédricos.
DEPÓSITOS
Dalas de isótopos de azufre En la Tabla 2 se presentan los resultados
de los análisis isotópicos en las piritas y pirrotinns de los sedimentos. Existe una gran diferencia entre los valores de la zona occidental de la cuenca. con valores muy negativos, mientras que los de la zona oriental son positivos con un rango de variación amplio. En las muestTlls con pirita y pirrotina juntas. la pirrotina presenta composiciones isotópicas siempre más pesadas que la pirita acompañante.
En la Figura 3 aparecen los valores de B14S para los cristales de pirita de los yacimientos. Se puede observar que existe una gran dispersión de valores entre los diferentes yacimientos pero que dentro de cada depósito el rango de variación es muy pequeño.
También se realizaron análisis seriados en varios cubos de pirita para intentar observar las posibles zonaciones isotópicas de és-
A. C�nlluillllll - - - _ . _ _ . _ - - - - - - - _ . . . . - - - - - _ . _ - - � - � -� - - _ . _ - _ .
- - - - - - - - - - - . - - - - - - - - - - -� - -ar - - - · - - - · - - - - - - - ·
- - - - - - - - - - - - - - - II - � - - - - - - - - - - - - - - _ · _ - - - - - _ ·
VllldcnC'gril1o} - - - - - - · - - - · ·EEII - - II - - - - - - - - - - - - - - · - - - - - - - - - - - ·
VlIlorill - - - - - - - - _ .�- - - - - - _ . _ - - - _ . . _ _ . _ - - - _ . . _ - _ . _ . _ .
Munilb. - - - - - - - - - - - - II � · · - · - - - - _ · _ - - - - - - - - - - - - - - - - _ ·
Valdc¡lerillos _ _ _ _ . _ _ _ _ _ - - - - - - - - - - - - - - a BI-- - - - - - - - . - - - - - - - .
Y.n�:u - - - - - - - - - - - - - - - - - - - - - - - - - _ . _ - - - - - - - - _ . _ -� - - �.
. ., ·10 ., o , 10 .,
/) " S
Figura 3.- Composición isotópica de las piritas de los diferentes depósitos.

Las mineralizaciones de pirita de la cuenca de Cameros . . . 35
tos (Fig. 4), no observándose ninguna tendencia general definida.
En el interior de algunos cristales de pirita se realizaron análisis isotópicos medianle microsonda láser de inclusiones de anhidrita, asf como de la pirita alrededor de estas inclusiones (Tabla 3).
En la Tabla 4 encontramos las composiciones isotópicas y el contenido en arsénico en relación con las morfologías de los diferentes tipos de cristales en los dos grupos de mineralizaciones.
Por ultimo, se analizaron los sulfatos de la matriz de las mineral izaciones y de los sedimentos, obteniéndose resultados similares con un valor medio de + 12.0 %0.
Al
el
·u · u
-u ..
"
u >
•• u'5.1
,.,11
Tabla 2.- Composición isotópica de las piritas y pirrotinas de los sedimentos.
GRA-S- 13.0 47.4 GRA-6- 9.2
ZONA ORA- l I - 16.9 21.4 ORIENTAL ORA-19- 16.8 26.9
ORA-21- 19.7 26.8 GRA-27- 23.9 39.8 ORA-38- 2\.9 AMB-4- 2 .• 18.1 AMB-II- 12.4
ZONA AZUD-2- -17.9 OCCIDENTAL AZUD-J- -1 8.6 39.1
GOLMAYO -17.6
BI
·u
DI
..
.,. ,
,. .O,l,.7 o.,
Figura 4.- Valores de d:H$ a lo largo de dos secciones perpendiculares en cristales de pirita. ( o centro del cubo. A: Navajún. B: Valoria. C: Valdeperillos. D: Ambasaguas)

36 ALONSO-AZCÁRATE. J
Tabla 3.- D:lIas isotópicos de los pares pirita-anhidrita utilizados para la realización de cálculos geotermométricos.
Muestn Py 6JlS Anh 6JlS .1 611S T'C Navajún- I_ -0.7 16.1 16.8 '" NavljÚJI·2_ -8.S 7.' 16.1 l7I Navajún-). -7.8 '.2 16.0 J74 ValdeperiJlos-l. -2.1 14.4 16.5 364
Discusión
Origell del azufre Si re:llizamos un simple balance de masas
del contenido de S de los depósitos y lo comparamos con el contenido de S en los sedimentos, vemos que la matriz de las mineralizaciones solo podría suministrar una muy pequeña parle del S necesario para formar 10$ depósitos. De esta forma el azufre tuvo que ser movilizado e introducido en las lutilas que forman la matriz de las mineralizaciones durante el proceso de formación de los depósitos.
El amplio r�lngo de composiciones isotópicas que muesman las diferentes mineralizaciones (Fig. 3) parece indicar que deben existir diferentes fuentes de azufre con distinta signatura isotópica. Las condiciones metamórficas alcanzadas por los sedimentos encajantes de estas mineralizaciones fueron muy similares (Alonso-Azcárate et al., 1999a), así las diferencias en temperatura y qufmica de los nuidos sólo podrfan producir pequenas variaciones en la composición isotópica entre
mineralizaciones con una únicn fuente de azufre (Ohomoto y Rye, 1979). El rango de composiciones isotópicas encontrado parece indicar In mezcla de varins fuentes de nzufre con diferente composición isotópica. Debido a que los yacimientos de pirita no están asociados con ningún accidente tectónico importante que pudiera introducir azufre del exterior del sistema. las fuentes de nzufre deben de encontrarse en los sedimentos de la cuenca. Las dos fuentes más probables son los sulfatos y sulfuros presentes en los sedimentos normales de la cuenca.
La primera fuente seria la reducción termoquímica de sulfatos (RTS). Los sulfatos que presentan las diferentes formaciones de la cuenca (GÓmez-Fernández. 1992) tienen su origen primario en los sulfatos Triásicos. los cuales durante la sedimentación de los materiales de la cuenca de Cameros ya anOTaban, proporcionando materiales a los sistemas de depósito (Mas el al.. 1993).
El proceso de lermoreducción en ambientes metamórficos de este tipo es un proceso muy rápido, en el que independientemente del fraccionamiento cin6tico del proceso. que puede ser variable. al reducirse todo el sulfato de un reservorio determinado la composición isot6picn del sulfato inicial y del sulfuro final va a ser id6ntica (Krouse. 1977; Machel et al., 1995). Asf. teniendo en cuenla el valor medio de los sulfatos Triásicos de la zona (+13.4±1 .6%o, Utrilla et al.. 1992) y los valores obtenidos en este
Tabla 4.- Contenido en As. composición isotópica y grupo de depósito para una serie de cristales seleccionados de cada tipo morfológico.
Habito C. cuarteados Piritoedro Cubo-piritoedro Cubo-octaedro Cubo Cristales aplanados Cristales alargados
As (ppm) 125.3 124.6 128.9
1 16.1 124.9. 138.2, 1 3 1 .0, 132,2
1 32,5, 1 14.4 127.0, 127.4
ó"s 7.7,8.6, 9.6, 7.8, 8.1 1 . 1 , 6.5, 7.4, 7.1, 0.8 0.2, 5.8, 6 . 1 , 6.2, 5.6, 5.9, 6.
-0.5, -LO, -0.7, -0.9
-4.4, -4.3, -4.9, -5.9, -7.3
3.1, 1 .9, -7.0, -10.5 -7.6, 3.0, -5.8, -6.3, -2.6
Depósito
Grupo 11
Grupo 1

Las mineralizaciones de pirita de la cuenca de Cameros . . . 37
estudio. este proceso de termoreducción generaría un S isotópicamente pesado con una composición media de 12- I 3 %c. La T mínima a la que se produce este proceso es de 100-140 oC (Heydari y Moore. 1988; Worden et al.. 1995). aunque a tasas geológicamente significativas a temperaturas superiores a 2500 C (Kiyosu. 1980). las cuales fueron alcanzadas por este sector de la cuenca donde se localizan las mineralizaciones de pirita (Alonso-Azcárate et al.. 1999a).
La fuente de S isotópicamente ligero debe encontrarse en las piritas sedimentarias que aparecen en los sedimentos normales de la cuenca. La rotura térmica de la pirita sedimentaria para formar pirrotina en presencia de carbono orgánico. produce un fluido rico en H¡S mediante reacciones del tipO (Ferry. 1 98 1 ; Oliver et al.. 1992):
La generación de fluidos ricos en azufre es más probable que se produzca mediante reacciones similares a ( 1 ) que mediante la simple disolución de los sulfuros sedimentarios ya que la solubilidad de los sulfuros en fluidos metamórfico-hidrotermales es baja (Barnes. 1979; Holland y Malinin. 1979). Muchas de las lutitas analizadas en la cuenca presentan pirita y pirrotina (Tabla 2) lo cual puede !Ornarse como evidencia de que la reacción ( 1 ) ha tenido lugar. ya que la pirrotina aparece muy raramente como sulfuro diagenético.
Durante el proceso de rotura térmica de la pirita existe un efecto isotópico cinético que produce un enriquecimiento en }.lS en la pirrotina resultante y por lo tanto. para que se conserve el balance isotópico. se genera azufre reducido isotópicamente ligero (Kajiwara et al.. 1981; Yamamoto. 1984). El mayor contenido en �S de la pirrotina con respecto a la pirita en las muestras de la Tabla 2 corrobora esta hipótesis. La mayor proporción de )�S en las piritas sedimentarias del sector oriental de la cuenca con respecto al sector occiendental (Tabla 2), es
en parle el resultado de la progresiva volatilización del lIS durante el metamorfismo de bajo grado y la subsiguiente retrogradación de la pirrotina resultante (enriquecida en �S) a pirita (Alonso-Azcárate el al.. 1999c).
La composición isotópica en cada mineralización es bastante homogénea pero entre los diferentes depósitos el rango de variación es muy grande (Fig. 3). Esto implica la existencia de fuentes locales de azufre para cada mineralización. La composición isotópica concreta en cada mineralización estará determinada por la proporción de azufre procedente de cada fuente. La existencia de una variación sin ninguna lendencia definida en las composiciones isotópicas dentro de un cristal de pirita (Fig. 4) también parece indicar la presencia de fuentes locales de azufre y su variación durante el proceso de mineralización.
La proporción de azufre en cada depósito derivada de la reducción termoquímica de sulfatos y de la rotura térmica de la pirita sedimentaria puede ser estimada si conocemos la composición isotópica de azufre de cada fuente. El azufre derivado de la RTS tiene un valor medio de 1 2 %O. como se indicó anteriormente. La composición isotópica del azufre procedente de la rotura térmica de la pirita es más difícil de estimar y podría ser diferente para cada mineralización debido a variaciones locales en la composición isotópica de la pirita sedimentaria original. Se ha estimado como valor la composición isotópica más ligera de las piritas de todos los yacimientos (-10.3 %e). ya que en este depósito la composición isotópica del azufre procedente de las piritas sedimentarias debió de ser este valor o algo más ligero. En la Tabla 5 se presentan las proporciones de azufre derivadas de cada fuente para las diferentes mineralizaciones estudiadas en función de la composición isotópica media de cada depósito. Hay que señalar que los depósitos que presentan elevadas proporciones de azufre derivado de RTS se encuentran en secuencias deposicionales en las cuates los sulfatos sedimentarios son abundantes.

38 ALONSO-AZCÁRATE. J
Tabla 5.- Composición isotópica de azufre media para cada mineralización y la proporción de azufre derivada de la reducción lermoqufmica de sulfatos (RTS) y de la rotura t�rmica de la pirita (Py -, Po).
Yacimiento A.C&nadillas Ambasaguas Navajún Vlldenegrillos Valoria Munilla Valde�riJlos
6US medio 7.5 l.'
-2.4 -5.1 ·6.8 -5.\ -2.7
Origen de/ hierro
% TRS % I')'-Po 85 15 67 J3 54 46 45 " 3' 61 45 " 53 47
Es posible realizar un simple balance de masas para comprobar si el Fe disponible en las cloritas de los sedimentos sin alterar es suficiente par3 formar la cantidad de pirita que encontramos en las mineralizaciones. La densidad media de mineralización en Nnvajún es de lOO Kglml (MINAS VICTORIA SL.), presentando este depósito la mayor concentración de pirita de todos los estudiados. Por lo tanto. este valor puede ser considerado como la densidad de mineralización media máxima. La densidad media de la piritól y de una lutita estándar es de 5.01 y 2.67 gr/cm) respectivamente (Carmichael. 1989). Así. si consideramos la cantidad de Fe presente en el sedimento no mineralizado, tendremos 0.038 Kg de Fe en 1 Kg de sedimento. Por lo tanto en 1 ml de sedimento (2670 Kg) tendremos 102.5 Kg de Fe. Esta cantidad de Fe si se piritiza totalmente formada 220.4 Kg de pirita en un ml de sedimento. Esta cantidad de pirita que se puede formar sobrepasa en 120 Kg el contenido medio de pirita en el depósito con la mayor concentración en este mineral. Por lo tanto. la movilización del Fe contenido en el sedimento es suficiente para generar el volumen de mineralización que encontramos en los diferentes depósitos.
Como indicábamos anteriormente las lutitas mineralizadas presentan unas fuertes
decoloraciones, no presentando clorita y su contenido en Fe es muy inferior al de las lutitas normales de la cuenca (Tabla 1). Esto es debido a que durante el proceso de mineralización los fluidos metamórficos desestabilizaron las cloritas. que son las fases que contiene más Fe en los sedimentos, estando así disponible para la formación de los cristales de pirita.
De esta forma, mediante la acción de los fluidos hidrotermales se produce un proceso de sulfurización de las cloritas de los sedimentos y se genera pirita. La sulfurización de silicatos ferromagnesianos ha sido demostrada tanto experimentalmente (Tso et al.. 1979) como en medios naturales ( MaUio y Gheith. 1972). La sulfurización metamórfica de las cloritas ha sido descrita por Phillips y Graves ( 1984) mediante las siguientes reacciones:
3 Fe�M glS i ,0 I O( O H ) .+ 1 2 H lS + 30 2-6FeS1 +2Fe)Mg)Si .Olo(OH),+4Si01+ 16Hp (2)
Fe�SiP!o(OH).+12H1S+30J - 6FeS1 + 45i01 + 16HlO (3)
En estas reacciones metamórficas se genera. además de pirita. cuarzo (3) o clorita magnésica y cuarzo (2). En los depósitos estudiados los cristales de pirita están rodeados por unas envueltas formadas fundamentalmente por cuarzo y cookeita. Esta cookeita es una clorita diOClólédrica lítico-alumínica sin hierra y con un contenido en magnesio muy bajo (Alonso-Azcárate. 1997). Estas envueltas se interpretan como los subproductos de las reacciones de sulfurización de las cloritas de los sedimentos mediante las reacciones (2) (envueltas de cookeita + cuarzo) y (3) (envueltas de cuarzo).
LocalizaciólI de las ",i/!uafit,aciolles de pirita
El punto concreto donde se forman las mineralizaciones de pirita debe presentar alta permeabilidad y unas características quími-

Las mineralizaciones de pirita de la cuenca de Cameros . . . 39
cas que favorezcan las reacciones entre el fluido y la roca encajante (Hobb, 1987). En este caso los sedimentos que albergan las mineralizaciones presentan una alta permeabilidad generada mecánicamente debido a la presencia de las fracturas hidroplásticas generadas muy tempranamente. Estas fracturas son la vía de entrada de los fluidos a los lutitas, las cuales sin ellas presentarfan bajos valores de permeabilidad. De hecho, estas fracturas controlan la geometría de las mineralizaciones, cambiando la densidad e incluso el hábito y tamaño de los cristales entre fracturas contiguas. Las lutitas que contienen las mineralizaciones. en origen, debieron tener altos contenidos en clorita capaz de suministrar el hierro necesario para fijar el sulfuro de los fluidos metamórficos como pirita.
Variaciones ell fa morfología de fas cristales de pirila
Las morfologías que presentan los cristales de pirita van a eSlar relacionadas con el tipo de mecanismo de crecimiento que las ha generado. La temperatura y la sobresaturación son los factores principales que determinan el tipo de crecimiento (Murowchick y Sarnes, 1987). En el caso de la pirita. las variaciones de temperatura necesarias para que se produzcan los cambios morfológicos observados son muy grandes (Murowchick y Sarnes, 1987). Estas variaciones no se dieron entre estos yacimientos, ya que las condiciol'es metamórficas alcanzadas por todos los depósitos fueron similares (Alonso-Azcárate et al.. 1999a). Es más. los yacimientos de Ambasaguas y A. Canadillas, Jos cuales presentan las morfologías de más alta temperatura, alcanzaron condicio· nes de anquizona, las más bajas dentro de todos los yacimientos estudiados (AlonsoAzcárate et al.. 1999a). Asr, las variaciones en la sobresaturación de los fluidos van a ser la causa de estos cambios morfológicos.
Otro factor que puede afectar la morfología cristalina es la presencia de impurezas en el medio de crecimiento. ya que afectan a la
rugosidad de la superficie del cristal. En el caso de la pirita. la presencia de As parece promover la presencia de caras al J l l } (Hayashida y Muta. 1952; Sunagawa y Takahashi, 1955), aunque estos autores señalan que la influencia de otros factores puede limitar este efecto. En nuestro caso, sólo encontramos cristales de pirita con caras { l l l J en el yacimiemo de Navajún. presentando estos cristales los contenidos en As más bajos de todas las diferentes morfologías (Tabla 4). Por lo tanto. en este caso la presencia de As no parece estar implicada en el desarrollo de caras { 1 1 1 J .
En la Figura 5 se muestran los diferentes tipos de morfologías de cristales de pirita en función de la sobresaturaciÓn. Cuando las sobresaturaciones son muy altas. la interfase de crecimiento es rugosa y las unidades de cristalización que llegan son incorporadas rápidamente al cristal, generándose los cristales con morfología de crecimiento cuarteado (Prieto, 1994). Cuando la sobresaturaci6n es algo menor, tenemos un mecanismo de crecimiento capa a capa por nuc!eación bidimensional (Kossel. 1930) donde se generan los cristales con morfologf3s de piritoedro. En condiciones de sobresaturación algo mas bajas el crecimiento se produce por la aparición de dislocaciones helicoidales en las caras (Burton et al.. 1951). De este modo, los cristales con formas alargadas se producen por la sola presencia de un par de dislocaciones helicoidales activas en sus caras más pequeñas (Sunagawa. 1987).
Si consideramos el tipo de facies en el que encajan estas mineralizaciones se puede eltplicar las variaciones en la sobresaturaciÓn de las soluciones que generan los depósitos. Los yacimientos de Ambasaguas y A.Canadillas se encuentran en medios de depósito de llanura deltaica, los cuales van a ser ricos en materia orgánica. presentando abundante pirita sedimentaria y sulfatos. Asf. vamos a disponer de un importante reservaría de S para formar las mineralizaciones. Además, la presencia de abundantes sulfatos ten-

40 ALONSO-AZCÁRATE. J
Crecimiento controlado por la superficie
Crecimiento controlado por difusión ,
BAJA MEDIA
SOBRESATURACIÓN ALTA
Figura 5.- Rel3ción entre el h.1bilo de las piritas, el grado de sobrcsaluración y el mcc:¡nismo de crecimiento cristalino.
drá un profundo efecto en la química de los fluidos durante el metamorfismo al producir. se reacciones de termoreducción de sulfatos del tipo (Alonso-Azcarate el al .• 2001):
SO l· + W + 4H � 4H O + HS- (4) , " SO/" + 2W + 4Hz � 4H10 + H1S (5)
que influenci.uán tanto el potencial redox (fHz) y el pH como la generación de S reducido. De esta forma, los fluidos generados en litologías ricas en sulfatos tendrán una menor mI y una baja nH+ cuyo efecto en las reacciones de precipitación de pirita será el incremento del grado de saturaci6n en pirita:
Fe!· + 2HS· � FeS1 + Hl (6). Fe:' + 2H1S � FeS: + 2H* + Hz (1).
Sin embargo. el resto de los yacimientos se sitúan en materiales depositados en sistemas meandriformes en los que la formación de piritas sedimentarias seria menor y no presentan concentraciones de sulfatos importantes.
Esta hipótesis puede ser corroborada mediante las composiciones isotópicas de las piritas en ambos grupos de depósitos. Las reacciones (4) y (5) suministran azufre reducido
a los fluidos metamórficos mediante la reducción termoquímica de sulfatos sedimentarios. siendo este azufre isotópicamente pesado. como se indicó anteriormente. De esta forma. si las reacciones anteriores han controlado la química de los fluidos metamórficos. los cristales de piritu de los yacimientos de Ambasaguus y Arroyo Canadillas (grupal ) deberían estar enriquecidos en }.IS respecto a los otros. En la tabla 4 se puede comprobar como los yacimientos del grupo 1 presentan composiciones isotópicas significativamente más pesudas que las del grupo 2. Así. podemos concluir que la mayor disponibilidad de azufre asl como los cambios en las condiciones redox y pH de los fluidos como resultado de las reacciones (4) y (5) fueron los factores dominantes en el control de la sobresaturaci6n en pirita de los nuidos metamórficos.
Cálculos geotermomélricos Mediante los análisis puntuales con la
microsonda láser de las inclusiones de anhidrita y la pirita que la rodeaba (Tabla 3) se realizaron dlculos geolermomélricos con el par sulfato-sulfuro. usando la ecuación de fraccionamiento isotópico en el equilibrio de Ohornoto y Lasaga ( 1982). obteniéndose unas

Las mineralizaciones de pirita de la cuenca de Cameros . . . 41
temperaturas de equilibrio de 367±6DC. Esto sugiere que se alcanzó el equilibrio isotópico entre las inclusiones de anhidrita y la pirita que las contiene y que no fue afectado por reacciones de reequilibrio (Alonso-Azcárate et al., 1999b). Estas T coinciden con las máximas T estimadas para el metamorfismo de la cuenca durante las cuales se produjo la formación del cloritoide en los sedimentos. De esta forma, la presencia de inclusiones de cloritoide en las piritas así como las T de formación calculadas, indican que la formación de eSltlS mineralizaciones debió de ser coetánea con el pico de metamorfismo.
Conclusiones
Estos yacimientos pueden ser clasificados como yacimientos melamorfogtnicos (Pohl, 1992) formados como resultado de un proceso de movilización. Aunque en este caso
--�------�-:-:-
,, - . . ' : : ' : : : : : -' : - : : - ' : . , , ' . " " " , .
:-!-: -=-=: ::�-:::
no se movilizan los metales, como generalmente ocurre en este tipo de yacimientos. sino que lo que se moviliza es el S.
Así, durante el metamorfismo hidrotermal que afecto a estos materiales del sector oriental de la cuenca, se genero un fluido rico en S procedente de la rotura térmica de piritas sedimentarias y la reducci6n termoquímica de sulfatos Triásicos reciclados (Figrua 6). Estos fluidos circulaban a través de las areniscas permeables, confinadas entre materiales de baja permeabilidad como son las lutitas, hasta encontrar un punto con alta porosidad y permeabilidad, que en este caso serían 100S lutitas que constituyen la matriz. en las cuales su permeabilidad ha sido aumentada mecánicamente por la presencia de las fracturas hidroplásticas, De esta forma, estas fracturas son la vía de entrada de los fluidos a estos niveles, los cuales presentaban minerales de Fe reactivos como la clorita, que mediante reacciones de sulfurizaci6n formarían los cristales de pirita,
I I l I 0 " ,- ::; . . . . . . . ' .:.: ,�.!;
• ' o o , • • • o . , ' • • •
'7 27 ¿ 'Z
: F[u[dj) �o.;S : , : :
:� _ F, _ E.!� ......
+ T, t
Fe ClorIUl I .......
e PI,III udlmena,1a eu_rtee O PlrIIa m.lI-m'rfIca� Figura 6.- Diagrama csquem:itico del proceso de formación de las mineralizaciones de pirita,

42 ALONSO-AZCÁRATE. J
Las importantes variaciones en las composiciones isotópicas entre depósitos muy cercanos, indican la presencia de fuentes locales de azufre y su variación durante el proceso de mineralización. La presencia de inclusiones de cloriloide en las piritas así como las temperatura de formación calculadas indican que la formación de estas mineralizaciones debió de ser coetánea con el pico de metamorfismo.
Las variaciones morfológicas en los cristales de pirita entre las diferentes mineralizaciones están comroladas por el grado de sobresaturación en azufre de Jos fluidos metamórficos. Éste a su vez está controlado por el tipo de facies sedimentarias en el cual encajan los diferentes depósitos. Por un lado, las morfologías de alta sobresaluración aparecen en las mineralizaciones cuyos sedimentos presentaban una mayor disponibilidad de azufre sedimentario en forma de pirita y sulfatos (facies de llanura deltaica lacustre). Además, las reacciones mf',tamórficas en las cuales participan sulfatos producen fluidos más oxidados , con mayor pH y con mas altos contenidos en azufre reducido, lo que genera una mayor saturación en pirita. Por otro lado, las morfologías de baja sobresaturación aparecen en depósitos que se encuentran en facies sedimentarias de tipo meanclriforme donde la disponibilidad de azufre sedimentario es más baja.
Agradecimientos
Este trabajo ha sido financiado por los proyectos PR 179/91-3469 de [a Universidad Complutense de Madrid, PB94-0054 de la DGICYT, PB97-0298 de la D.G.E.S. y BTE2001-026. Se agradece la inestimable colaboración de todos mis compañeros de la Universidad Complutense y la Universidad de Leeds en la realización de este trabajo.
Bibliografía
Alonso-Azcárate, J. ( 1997): Evolucion de los filosilicatos y genesis de [os yacimientos
de pirita en la Cuenca de Cameros: su relacion con las facies sedimentarias y el metamorfismo. Cretacico Inferior. La Rioja-Soria. Tesis Doctoral, Universidad Complutense, Madrid.
A!onso-Azcárate, J., Barrenechea, J.F., Rodas, M., Mas, R. ( 1995): Comparative study of the transition between very low grade and low grade metamorphism in siliciclastic and carbonate sediments, Early Crelaceous, Cameros Basin (North Spain). C/ay Mil/erais, 30, 409-422.
Alonso-Azcárale J., Rodas M., Bottrell S.H., Raiswell R., Velasco F., Mas J.R.
( 1999a): Pathways and dislances offluid flow during !ow-grade metamorphism: evidence from pyrile deposits of the Cameros Basin, Spain JOllmal o[ Melamorphic Ge% gy 17. 339-348.
A!onso-Azcárate J., Boyce AJ., Bottrell S.H., MacAulay C., Rodas M., Fallick A.E., Mas, J.R. ( l 999b): Devc!opment and use of ill sitll laser sulfur isotope analyses for pyrite anhydrite geothermometry: an examp!e from Ihe pyrite deposits of the Cameros Basin, NE Spain. Geochimica el Cosmoclzimica Acta 63, 509-5 13.
Alonso-Azcárate, J., Rodas, M., Boltrell, S.H., Mas, J.R., Raiswell. R. ( 1999c): Estudio textural e isotópico de [os sulfuros diseminados en los sedimentos de la cuenca de Cameros (La Rioja, España). Re\!. Soco Geol. Espmia, 12, 241-249.
Alonso-Azcárate. J .• Rodas. M., FernándezDíaz. L., Boure!l. S .H .• Mas, J.R. y LópezAndrés. (2001): Causes of variation in crystal morphology in metamorphogenic pyrite deposits of the Cameros Basin (N Spain). Geofogical Joumaf, 36. 159-170.
Barnes, H. L. ( 1 979): So!ubilities of ore minerals. In: (H. L. Bornes ed.) Geocllelllistry o[ IIydrotllermol ore deposits. Wifey, New York, N. Y, 2nd ed., 404-460.
Barrenechea. J.F., Rodas. M., Mas, J.R. ( 1995): Clay mineral variation associated

Las mineralizaciones de pirita de la cuenca de Cameros . . . 43
to diagenesis and low grade metamorphism of early Creataceous sediments in the Cameros basin, Spain. Clay Minera/s, 30, 89-J03.
Burton, W. K., Cabrera, N., Frank, F.C. (1951): The growth of cryslals and equilibrium slruclures of their surfaces. Pllilosofieal Trallsactiolls 243, 299-358.
Candfield, O.E., Raiswell, R., Westrich, J.T .• Reaves. C.M .• Bemer, R.A. (1986): The use of chromium reduction in Ihe analysis of reduced inorganic sulfur in sediments and shales. C/¡emicaf Geology. 54, 149-155.
Carmichael. R. S. ( 1 989): Practical Handbook of Physical Properties of Rocks and Minera1s. CSC Press, 741 p.
Casquel. C., Galindo, C., González Casado, J. M., Alonso, A., Mas, R . • Rodas, M . • García, E., Barrenechea, J. F. ( 1 992): El metamorfismo en la Cuenca de los Cameros. Geocronología e implicaciones tectónicas. Geogaceta. 11. 22-25.
Coleman. M.L. ( 1980): Corrections for mass spectrometry analysis of sulphur dioxide. 1solope Geology Ullit, Stable Isotope Repon, 45 pp.
Craig, H. (1957): Isolopic slandards for carOOn and oxygen isolope correclion faclors for rnass-spectrometric analysis of carbon dioxide. Geochim. Cosmoe/¡jm. Acta. 12, 133-149.
Ferry. J.M. ( 1981): Petrology of graphitic sulfide-rich schists from soulh-central
Maine: an example of desulphidation during prograde regional melamorphisrn.
Amerieall Milleralogist. 65, 720-732. Górnez Fernández, J.C. (1992): Análisis de la
Cuenca sedimentaria de los Cameros durante sus etapas iniciales de relleno en relación con su evolución paleogeográfica.Tesis Doctoral. Univ. Compl/lte/lSe de Madrid, 343 p. Inédita.
Guimerá. J., Alonso. A.. Mas. J. R. ( 1 995): Inversion of an extensional-ramp basin by a newly formed thrust: the Cameros basin (N. Spain). (Buc/¡anall, J.G. &
Blle/IO/lall P. G. ed.) Basiu 1l/versioll, Geological Society Special Pl/bUcalioll. 88. 433-453.
Guiraud. M. ( 1983): Evolution tectonosédimenlaire du basin Wealdien (Crétacé inféTieur) en reIais de décrochements de Logroño-Soria (NW Espagne). Tesis doctota!. MOlllpellier, Ullil'ersité des Seiellces el Teclllliqlles de ulIIglledoc. 183 pp.
Hayashida, S. y Muta. K. (1952): Relation of trace-elernent content and crystal form in pyrite. JOllfllal 01 tire Mil/illg JIIslilllte 01 KYllsfw, 20: 233-238.
Heydari. E. y Moore. e.H. ( 1989): Burial diagenesis and thermochemical sulfate reduction, Smackover Formation, southeastern Mississippi Salt Basin. Geology, 12, 1080-1084.
Hobbs, B. E. ( 1 987): Principies involved in mobilization and remobilization. Ore Geology Rel'iews. 3, 37-45.
Holland, H. O. y Malinin, S. O. ( 1979): The solubility and occurrences of non-ores minerals. In: (H. L. Bames eJ.) Geocliemistry 01 /¡ydrol/¡ermal ore deposits. Wiley. New York. N. Y . . 461-508.
Kajiwara, Y., Sasaki, A. & Matsubaya, O .• 1981. Kinelic sulfur isotope effects in the Ihermal decomposilion of pyrile. Geochemical JOllmal.15. 193-197.
Kelley, S.P. y Fallick. A.E. ( 1990): High precision. spatially resolved analysis of l�S in sulfides using a laser extraction technique. Geocllim. CosllloclJim. Acta 54. 883-888.
Kossel. W. ( 1930): Uber Krislallwachslrum. Naturwiss. , 18,901-910.
Krouse. H.R. ( 1 977): Sulfur isotope studies and Iheir role in petroleum exploration. JOllmal 01 Geoc/¡emieal Exploratioll. 7, 1 89-2 1 1 .
Kiyosu. Y. ( 1980): Chemical reduction and sulfur isolope effects of sulfate by organic malter under hydrothermal conditions. CIJemica{ Geology, 30, 47-56.
Mache!, H. G., Krouse. H. R . • Sassen. R.

44 ALONSO-AZCÁRATE. J
(l995): Producls and distinguishing crileria of bacterial and thermochemical sulfate reduction. Applied Geochemistry, 10. 373-389.
Mallio. W. J. Y Gheilh, M. A. ( 1972): Tclttural and chemical evidence bearing on sulfidesilicale reactions in melasedimenls. Millerafium Deposita, 7. 13-17.
Mas. J.R., Alonso. A., Guimerá. J. (1993): Evolución tectonosedimcntaria de una cuenca e",tcnsional ¡ntraplaca: la cuenca finijurásica-eocretácica de Los Cameros (La Rioja-Soria). Revista de la Sociedad Geológica de Espmia, 6 (3-4), 129-144.
Mas, J.R., Guimerá, J., Alonso, A. ( 1997): Evolution of a mesozoic intraplate extcnsional basin: Ihe Cameros Basín (Norlh Spain)>>, Amwaf Meeting oi JGep Project No. 369 - Compararive EVO{lIIioll 01 PeriTerllyan Rift Basills, 36-39.
Murowehiek JB, Barnes HL. 1987. Effeets of temperature and degree of supersnturation on pyrite morphology. American Millera{ogisr72: 1241-1250.
Newlon, R.J., Bonrell, S.H., Oenn. S.P. Hatfield. D. , Raiswell, R. (1995): An investigntion of the use of ehromous ehloride digestion in the isotopie analysis of pyrite in roeks and sediments. Chem. GeoJ., 125, 3 17-320.
Ohmoto, H. y Rye, R. O. ( 1979): ISOlopes of sulfur and earbon. In Geoc/¡emisrry 01 hydrotllermal ore deposils. (H. L Bames ed.) 10/111 Wiley & Sonso 509-567.
Oliver, N.H.S., Hoering, T.e., Johnson, T.W., Rumble, D., Shanks, w.e. (1992):
Sulfur isotopie disequilibrium and nuid-rock interaetion during metamorphism of sulfidic black shales from the WatervilleAugusta area, Maine, USA. Geoc¡'¡mica el Cosmochimica Acta, 56, 4257-4265.
Ohmoto, H. y Lasaga, A.C. (1982): Kinetics of reactions between llqueous sulfates and sulfides in hydrothermlll sySlems. Geoe/rim. COSII/ochim. Acta. 46, 1727-1745.
Phillips, G. N. Y Groves, O. 1. (1984): Fluid access and nuid-wall rock interaction in ¡he genesis of ¡he Arehllean gold-qunrtz vein deposit at Hunt Mine. Kambalda, Western Australia. Gold'82: Tire geology, geochemistry alld gel/esis 01 gold deposits (R.P. Foster ed.), 390-416.
Pohl, W. ( 1992): Defining Metamorphogenic Mineral Deposits-an introduction. Mineralogy Glld Petr% gy, 45, 145-152.
Prieto, M. ( 1994): Fundamentos de onlogenia mineral. En: Yacimielllos Minera/es. (ed. Lunar, R.; Oyllrzun, R.), 3-3 1 .
Robinson, B.W. y Kusakabe. M. ( 1 975): Quantitative preparalion of sulfurdioxide. for HS/nS analyses. from sulfides by combustion with euprous oxide. Ana/yliea/ Clremislry, 47. 1 179- 1 1 8 1 .
Sunagawa. l . ( 1987): Morphology ofMinerals. In: Morp//Ology o[ Crystals, Sunagawa I (ed). Terrapub. pp 509-587.
Sunagawa. 1. y Takahashi. K. ( 1955): Preliminary reporl on the relation between the 0(1 1 1 ) face of pyrite crystals and its minor conlents of arsenic. Geological Society ollapal/ BI/lletill 6, 1-10.
Tso. J. L., Gilbert. M. C., eraig, J. R. ( 1979): Sulfidation of synthetic biotites. American Minera/ogist, 64, 304-316.
Ulrilla, R., Pierre, c., Orli, F., Pueyo, J. J. ( 1992): Oxigen and sulphur ¡solope compositions as indicators of the origin ofMesozoic and Cenozoic evaporites from Spain. Clremical Geology (isotope geosciellce sectioll). 102. 229-244.
Worden, R.H .. Smalley, P.C., Oxtoby, N.H. ( 1995): Gas souring by Ihermochemical sulfate reduction al 140°C. Am. Ass. Petrol. Geol. Bull., 79. 854-863.
Yamamoto, M., 1984. Sulfur iSOIOpe effects in (he Ihermal breakdown of pyrite. Eartlr alld Planelary Sciellce Letters, 69, 335-340.
Recibido: Enero 2003 Aceptado: Marzo 2003

BoletElI de la Sociedad Espa,io/a de Minua/ogfa, 26 (2003), 45-58 45
Caracterización textural y mineralógica del gossan de Fi
lón Sur (Tharsis, Huelva)
Ángeles CAPITÁN, José M. NIETO, Rein::.ldo SÁEZ y G::.briel R. ALMODÓVAR Dep::.rlamenlo de Geologf::., Facull::.d de Ciencias Experimentales, Universidad de Huelva, 21071 Huelva
Ahslruct: The Filón Sur dcposil is loc�tcd in the Thnrsis mining districl. in Ihc centra! pan of the Iberian Pyrile Beh, about 50 Km to thc north of the city 01' Huelva, A genelic hypothesis for the gossan evolution has becn estabished from Iheir textural and mineralogieal charaeterization. Two main stages have been rccognised in Ihe gossan formation process. Thc I1rst one is associatcd 10 the oxidation of thc primary sulfhidc mineralization above the water table, giving rise to the formalion of goclhile, bcudantite·corkile, and aceesory iodargirite. chlorargirite, silver, gold. barite and eerussite. The second stage is related 10 the evolution of the gossan aOer Ihe complete oxidation of Ihe sulphide mineralization took place. In Ihis stage gocthite and hematite are formed from destabilization ofbcudantile and dehydration of gocthite.
Key words: gossan mineralogy, gold, Filón Sur, Tharsis, lberian Pyrite Bell
Resumen: El yacimiento de Filón Sur se localiza en el distrito minero de Tharsis. en la parte central de la Faja Pirltica lbtrica. a unos 50 Km al norte de la ciudad de Huelva. Se ha establecido una hipótesis ge.nética para la evolución del gossan a partir de su caracterizaeión textural y mineralógica. Se han reconocido dos etapas en el proceso de formación del gossan. La primera de el1as asociada a la oxidación de la mincralización primaria de sulfuros por encima del nivel freático. dando lugar a la formación de goelhila, beudantita·corkita. iodargirila. clorargirita. plata, oro, barita y cerusita. La segunda etapa está relaciollada con la evolución del gossan, tras la oxidación completa de la mineralización primaria de sulfuros. En esta etapa se formaron goethita y hematites por desestabilización de la beudantita y por deshidr:llac:ión de la gocthita.
Palabras clave: mineralogfa de gossan, oro. Filón Sur. Tharsis, Faja Pirftica Ibérica
Introducción
La Faja Pirítica Ibérica se caracteriza por la existencia de un elevado número de yacimientos de sulfuros masivos poli metálicos (Almodóvar y Sáez, 1992; Leistel et al., 1998; Sáez et al., 1999). La meteorización de estas mineralizaciones ha dado lugar a potentes monteras de hierro enriquecidas en ciertos elementos, entre ellos metales preciosos como Au y Ag. Hoy en día estas monteraS, conocidas con el nombre de gossans, están desmanteladas casi en su totalidad debido a las ¡nten-
sas labores mineras que desde épOcas prehistóricas se han desarrollado en toda la región (Pinedo Vara, 1963).
En la Faja Pirítica Ibérica los datos bibliográficos existentes sobre gossans están vinculados básicamente con la minerí::. y metalurgia de estos depósitos. En COnsecuencia, los estudios mineralógicos y geoqurmicos realizados hast::. el momento se han centrado fundamentalmente en la recuperación de metales (Roca el al., 1999), y en la estimación de reservas de Au y Ag (Garda Palomero et al., 1986), sobre todo a partir de los años ochenta

46 CAPITÁN, A. et a l .
como consecuencia de la caída del precio del Cu (Arribas, 1998) . Las características m i neralógicas, texturales y l i tológicas en este tipo de formaciones se conocen con menos detalle. Por tanto, el objetivo de este trabajo es la caracterización m i neralógica y petrogrMica de los dist i ntos tipos l i tológicos de gossan d iferenciados en Filón Sur (Tharsis), así como el estudio de la distribución de las fases mi nerales a lo largo de un perfil COIll� piCIO. con l a I'ina l idad de establecer las relaciones existentes entre m ineralogía, l i tología y procesos de evolución en el gossan.
Contexto geológico regional
La FPI ocupa una franja de aproximadamente 230 Km de longitud y 40 Km ele anchura media, desde Sev i l l a hasta la costa at[(¡nli - . ca portuguesa. Es!¡í constituida por materiales sedimentarios y volci.ln icos de edad Paleozoico superior (Figura 1 ) Y su estratigrafía está bien establecida a pesar de
o 20
N
A
40 Km.
ID GRUPOCULI.1
FPI O COMPLEJO VOLCANO-SEOIMENTARlO
GRUPOPQ
su complejidad (Schennerhorn, 1 97 1 ). S in considerar los terrenos de l a Zona de OssaMorena y del Grupo Pulo do Lobo, ni los sedimentarios recientes del Va l le del Guadalqu iv ir, son tres las un idades principales d i ferenciadas en la Faja Pirítica Ibérica: Grupo Pizarroso-Cuarcít ico, Complejo Vol canoSedimentario y Grupo Cu lm.
El Grupo Pizarroso-Cuarcítico es de edad Devónico medio-superior. Está constituido por secuencias de pizarras y cuarcitas con potencias de hasta 3000 m (Strauss, 1 970), predom inando las cuarcitas sobre las pi zarras a techo de dichas secuencias (Moreno y S¡íez, 1 990). Estos maleriales son los más ant iguos que se han identi ficado en la FPI.
La edad del Complejo VolcanoSed i mentario es Devónico superior Carbonífero inferior. Se caracteriza por ser una unidad muy heterogénea cuya potencia varía entre 50 y 750 J1l (Schermerhorn, 197 1 ) , habiéndose descrito importantes acuñamientos laterales. Está compuesto por varios episodios magmálicos félsicos y máficos i n tercala-
D COBERTERA POSTHERCINICA
l.J:2I ROCAS PLUTÓNICAS
�:OOi GRUPO PULO 00 LOBO
Madrid .
* • . SULFUROS MASIVOS
Figura 1.- Mapa geológico regional de l a Faja Pirítica Ibérica en el que están localizados los principales yacimientos de sulfuros masivos. Modificado de Sáez et al . ( 1 996).

Caracterización textural y mineralógica del gossan de Filón Sur . . . 47
dos en una secuencia sedimentaria constituida por rocas detríticas (en sentido estricto) y vulcanodetrfticas, así como depósitos de origen químico. Entre estos últimos, se incluyen niveles si1fceos de chert y jaspes manganesíferos y los sulfuros masivos. que muestran relaciones estratigráficas diversas con las rocas encajantes (Sáez et al., 1996).
El Grupo Culm, de edad Carbonífero medio-superior. se compone de pizarras, areniscas y conglomerados que muestran caracterrsticas de facies nysh y representan el relleno de una cuenca subsidente por sedimentación turbidrtica, cuyas áreas fuente habrían sido tanto la propia FPI como las unidades del norte. incluida la zona de Ossa-Morena (Moreno, 1993).
Distrito minero de Tharsis
El distrito minero de Tharsis lo componen 16 mineralizaciones conocidas de sulfuros masivos asociadas al volcanismo ácido inicial del Complejo Volcano-Sedimentario. Estas mineralizaciones se localizan en el Anticlinal de Tharsis. que constituye el cierre oriental del Anticlinorio de Puebla de Guzmán (Strauss y Madel, 1974).
En todo el distrito las mineralizaciones de sulfuros importantes económicamente son cuerpos de morfología estraliforme encajados en pizarras negras. Filón Norte es el mayor de los yacimientos de este distrito y además uno de los depósitos de sulfuros masivos más grandes del mundo. Estructuralmente, consiste en un sistema imbricado de cabalgamientos de bajo ángulo que buzan al norte, en el que tres zonas de falla muy potentes separan las tres unidades principales diferenciadas por sus rasgos lilológicos.e hidrotermales (Tornos et aL. 1998). La edad de la mineralización primaria ha sido determi nada por métodos isotópicos (Mathur et al .• 1999) y mediante palinomorfos (González et al., 2002). En ambos casos. los resultados indican una edad próxima al límite Devónico-Carbonífero.
El yacimiento de Filón Sur, constituido por pequeñas masas lenticulares de sulfuros masivos con longitudes entre 100 y 150 m y potencias de 20 a 30 m, está encajado en pizarras negras (Figura 2). Existen también mineralizaciones de tipo stockwork y diseminaciones de sulfuros en pizarras, oscilando el grado de mineralización desde muy intenso a débil. Este conjunto de materiales, sería equivalente a la Unidad Inferior diferenciada en Filón Norte por Tornos et al. ( 1 998).
Los procesos de oxidación ;11 silu de los yacimientos de sulfuros masivos originaron los gossans masivos existentes en esta zona. En ellos se centró la actividad minera desde épocas prehistóricas e históricas para la obtención de Cu. Au y Ag. Los gossans de la FPI. principalmente en Rro Tinto y Tharsis, han sido una importante fuente de Au para Europa a partir de 1970. En estos yacimientos secundarios el factor medio de enriquecimiento en Au es aproximadamente 6, con leyes medias que oscilan entre 1 y 3 gil.
Metodología
Se ha seleccionado un perfil completo de gossan itl SiIU, localizado sobre sulfuros masivos, en el que se han recogido un número representati va de muestras en cada uno de los niveles diferenciados en dicho perfil.
El estudio mineralógico y textural se ha realizado combinando los resultados obtenidos mediante microscopfa óptica de luz reflejada, microscopfa electrónica de barrido, difracción de Rayos X y microsonda electrónica. Las muestras se han analizado mediante difracción de Rayos X para la identificación de las fases mayoritarias, utilizando para ello un difractómetro Siemens modelo 0-501 , con radiación CuKa filtrada por Ni y monocromador de grafito. Las condiciones de análisis han sido 40 Kv Y 20 mA. con un barrido 29 entre 2° y 70°. Para [a identificación de las fases, se han empleado ficheros

48 CAPITÁN , A. el a l .
JCPDF (Jo inl Commillee For Po\Vder Diffraclion F i le) . Mediante m icroscopía elec� trónica de barrido (MEB) se ha realizado un estudio microtextural de deta l le y ha sido posible la caracterización qu ímica preliminar de la m ineralogía mayoritaria y m inoritaria, obteniéndose anúl is is cua l i tativos y semicuantita:ivos por espectrometría de dispersión de energía (EDS) en condiciones de 20 Kv de voltaje de aceleración. Los hidróxisu lfatos de Fe se han caracterizado cual i tativamente mediante MEB-EDS, siendo necesario introducir correcciones en los anál isis obtenidos a panir de los resultados de m icrosonda electrónica. El estudio químico cunntitativo de las fases minerales se ha realizado con una microsonda CAMECA SX50 equipada con cuatro espectrómetros de dispersión de longitud de onda. Se han seguido dos rutinas de análisis, una para óxi-hidróxidos de Fe y otra para hidróxi-sulfalos de Fe. En ambas rutinas se ha trabajado con un voltaje de aceleración de 20 Kv y una corriente de sonda de 30 nA.
Descripción del pe/Jil del gossw/ y dife
renciación de facies
La secuencia de l i tofacies de l a zona de alteración del yaci m iento de F i lón Sur se h a establecido tras e l estudio y caracterización de un perfil completo del gossan masivo, en el que no existen materiales suprayacentes al n ivel superior del mismo y el muro se encuentra en contacto directo con los su l furos masivos precursores (Figura 3 ) . Se han d i ferenciado, de techo a muro, tres l i tofacies a las que se ha denominado: Gossan Hematítico, Gossan Goethítico y Gossan Jarosítico. La terminología ha sido establecida en función del mineral primario que predomine entre las fases mayoritarias de cada I i tofacies. La hematites, la goethita y los m i nerales del grupo de [a jarosita son las fases m¡'is abundantes respectivamente en cada una de las l i tofacies, las cuales se disponen siempre en el mismo orden secuencial con importantes variaciones laterales de espesor. E l gossan goethíl ico presenta las mayores potencias de todo el perfi l . Los contactos no son netos y ex isten cambios
o m 20
Figura 2.- Mapa geológico del entorno de la corta de Filón Sur. a) Cuarcitas del PQ. b) Pizarras del PQ. e) Roeas volcánicas. d) Pizarras negras. e) Sulfuros masivos. f) Pizarras gossanizadas. g) Gossan masivo.
h) Niveles de sulfatos. i ) Gossan transportado. j) Pizarras meleorizadas. Modi ficado del mapa realizado por la Compañía de Azufre y Cobre de Tharsis para el proyecto Filón Sur (anterior a la explotación) . La Iínc.t discontinúa representa la situación actual de la corta.

Caracterización textural y mineralógica del gossan de Filón Sur. . . 49
graduales en la abundancia relativa de los minerales mayoritarios que permiten la diferenciación de las litofacies.
En el Gossan Hematítico son la goethita y la hematites los minerales más abundantes. Además se han identificado cuarzo y beudantita, esta última formando agregados mili métricos de cristales de color amarillo intenso. Dentro del gossan hematítico se diferencian a su vez dos variedades textura les, facies brechoides y facies coloformes, que también se distinguen en el gossan goethítico. Las variedades coloformes son las características propiamente de este tipo de yacimientos de alteración, en las que los óxi-hidróxidos de hierro presentan textura coloforme y tienen un aspecto tornasol irisado. Las variedades brechoides corresponden a facies con texturas heredadas. Los fragmentos de la brecha son de cuarzo, que es el mineral más abundante en estas facies, con morfologra desde angular a subredondeada y tamaño entre centimétrico y micrométrico. Dichos fragmentos están cementados por óxi-hidróxidos de hierro también con textura coloforme, en el gossan hematítico mayoritariamente por he-
matiles y en el gossan goethítico, por goethita. La brecha es anterior al proceso de formación del gossan, de modo que en la parte superior de la misma los procesos de oxidación están más evolucionados que en su parte inferior, mostrando la matriz la misma variación mineralógica que las litofacies en las que está intercalada, de hematflica a goethítica.
La facies brechoide correspondiente al gossan goethftico se restringe sólo a la zona superior del mismo, donde se intercala con la facies coloforme. Esta última se compone por una matriz limonftica que está cementada por costras goethfticas con textura coloforme. Las costras goethíticas coloformes son muy abundantes y caracterrsticas de la parte superior de esta lilOfacies, pero desaparecen de forma progresiva hacia la parte inferior de la misma. donde la matriz presenta un aspecto menos masivo. La matriz terrosa de la parle superior presenta un tamaño de grano más fino « I 1-1-) y homogéneo que la de la parte inferior. También hacia la parte más alta, por debajo del gossan hematftico, existen niveles de goethita iridiscente que marcan el tránsito entre el gossan hematftico y el goethftico, en los que
� Facies Brechoide
� Facies Hematltica Colo/arme
E:�j Facies Goethltlca CoIoforme
t-_-:J Facies GoalhrUca Masiva
� AIoocIIIos de sulfatos
� Facies Jarosillca
.. Sunuro masivo
Figura 3.- Columna sintética del perfil del gossan de Filón Sur donde se muestra la disposición de las lilofacies diferenciadas.

50 CAPITÁN. A. el al.
es frecuente la cristalización en los huecos de este mineral formando estalactitas de tamaño cenlimétrico. Por debajo de dichos niveles es además característica la presencia de filoncillos de sulfatos (principalmente beudantita) de espesor entre 0.5-1 cm.
La denominación "Gossan Jarosftico" hace referencia al predominio de los sulfatos del grupo de la jarosita. siendo la beudantit3 el mineral más abundante. A escala macroscópica. se observan agregados de beudantila de color amarillo dentro de una matriz terrosa de tamaño de grano grueso, tonalidades abigarradas y gran densidad. La parte superior presenta una estructura masiva debido a la cementación mientras que la parte inferior presenta una aspecto terroso.
Mineralogía
Las fases minerales mayoritarias identificadas en el gossan de Filón Sur son: óxihidróxidos de Fe, cuarzo y sulfatos de la familia de la jarosita. Dentro de estos últimos, el ttrmino identificado en todo el perfil es la beudantita (Figuras 4 y 5), aunque en las facies hematítica coloforme y hematítica brechoide (en el gossan hematítico) tambitn se ha reconocido corkita (Figura 4), según los criterios de clasificación de Scon (1987) y Dutrizac & Jambor (2000). La composición de la beudantita en los distintos tipos de gossan es, en general, homogénea.
Los análisis de goethita y hematites, obtenidos tanto por EPMA como por MEB-EOS, muestran contenidos en FeUl inferiores a los que corresponden estequiomttricamente a estos minerales. Este dUicit de FeUlen la goethita se debe a imperfecciones estructurales, mientras que en la hematites se debería a la sustitución parcial de Ol- por grupos OH", lo que provoca vacancias en posiciones catiónicas. A pesar de que mediante DRX se ha identificado hematites, los datos composicionales obtenidos por MEB-EDS y EPMA, han permitido
diferenciar dos variedades de este mineral, protohematites e hidrohematites. Hidrohematites y protohematites son dos variedades de hematites que se diferencian composicionalmente entre sí por el grado de hidratación respecto a la hematites como fase pura (Wolska & Schwertmann, 1989). En realidad, por el grado de deshidratación de la goethita original, ya que ambas son fases transicionales en el proceso de deshidroxilación y I o deshidratación de
·Ia goethita a baja temperatura. Protohematites es la fase que se forma en los primeros estadios de este proceso, siendo su comportamiento metaestable respecto a hidrohematites. Este último se forma a medida que el proceso continua y aumenta la proporción de 01. respecto a la de OH·. Según Wolska & Schwertmann (1989) ambas fases pueden diferenciarse en función de la proporción de ()l' sustituidos por grupos OH·, ya que tienen espectros de absorción de infrarrojos característicos. Determinando así el grado de sustitución en cada caso, establecen la fórmula estructural FeJ_� �(OH).OJ .• y determinan el valor de x para cada fase.
Al tratarse de un proceso continuo de deshidratación es prácticamente imposible establecer diferenciaciones texturales claras entre
so,
� o>
Beudantita
Klntorelta Segnltita
PO,
Figura 4.· Diagrama de Sean ( 1987) para la clasificación de los minerales del grupo alunita-jarosita.

Caracterización textural y mineralógica del gossan de Filón Sur . . . 5 1
ambas variedades, refiriéndonos a estas fases como hematites, tal como se ha caracterizado mediante difracción de Rayos X . Sin embargo, sí puede apreciarse una mayor abundancia de la variedad hidrohematites en la parte superior del perfil respecto a la parte baja del
mismo, donde predomina la protohematites. Este hecho seria coherente con estadios más avanzados de oxidación de la parle alta respecto a la parte baja del perfil de oxidación estudiado.
Los análisis realizados (Tabla 1 ) indican,
1.0 ,---------....,.---..., 0.8
0.4
0.2
Grupo Jarosita
Grupo Lusungita
o � __ �--__ ���--____ �� O 0.2 004 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0
(X04)3. (mol)
Figura 5.- Diagrama SO.-PO.-AsO. para la clasificación de los miembros ricos en Pb del supcrgrupo jarosila. Modificado de Dutrizac & Jambor. (2000).
Tabla 1.- Análisis de elementos mayores y trazas en hematites y goethita obtenidos mediante EPMA. (G.H. = Gossan Hematítico; G.G. = Gossan Goethílico¡ GJ. = Gossan Jarosítico).
�" 0'.10 03.3$ U.M 89.12 0.10 0.11 O.� O." 0.0< O ... ", o." o.ro o.ro
05.41 93.75 ... " �.U � ... 93.15 83.4g Ekme-alo. AcUorbldos
...., O.ro O.ro O.ro o.,. O.ro 0.02 O.ro O.ro O.ro o.,. O.ro 0.0' ." O.ro o.,. 0.0> O.,. 0.01 O.ro
"'" 0.0< O.ro O.ro 0.01 O.ro o,ro 0.03 o.,. o,ro 0.01 o,ro 0.0' 0,01 o,ro 0.01 O,M 0,0< O,,, o,ro
"" 0.0> O.ro O.Of 0,0< 0,0< 0,0< O.OS O,,. o,ro O.ro O.ro 0.0\ ." 0,0< o,ro 0.10 0.0> MI O.ro "'" 1,," 1,74 O ... MI ',," 2.32 O,U 0.e5 O.� o ... O,H 3.40 ',," O,N 0.32 1,1' ,,, 1," 1.40 "'" o,ro .ro o.,. o,ro o,ro o,ro o,ro o.,. o,ro o,ro 0,0< o ... O.ro o,ro o.,. o,ro o,ro o,ro o,ro
""" 0,0> 0.01 o,ro 0.01 0.01 o,ro o ... 0.11 0,0> ,0< 0.02 O ... o,ro 0.07 0.0< 0.02 0,0< o,ro 0.07 ...." o.n .� 0.47 O.U 1.57 1,0< 0.18 0.24 0.11 o.Ie 0.11 o,n o,n o,ro o,n o.n 1.16 o ... 0,0> .", ." O."' 0.01 O,", 0.02 0.01 0,0> 0.0< 0.03 ." 0.01 0.01 0.03 O.ro 0.02 O." 0.02 "" 0,0>
COIDJMnIclón !l"rmlc. de la ll.otIhlta �� G.H. G.H G.H. GJ-I. G,H. G,H. G.G. G.G: G.G. G.G. G.G. G,G. G.G. G.J. G,J . . ,. " . . , . . ,.
'0,0, aMI 83.8$ 81.33 81.51 al,\2 aU2 fUO fUI 8:UI 77.$11 MI.12 al.12 n.� ... " a7,47 80,05 n.5i 83.5i MM ."" O,M 0.0' 0.10 0.47 O." 0.�8 O,q O.� O'" 1,15 0.0< O." 0.71 0.\$ 0,111 0.0> 1.12 1,," O." no. o,ro O." o.ro 0,0\ 0.01 0.02 o,ro o,ro 0.02 0.0\ .ro 0.01 o.ro 0.02 O.le 0,00 O." 0,00 o,ro
Total ",O< ",," '1.43 81.119 81,73 �,. oo,," oo,," 82.$7 l"iI.14 1I$,\a e1.73 77,96 88.22 811.24 80.97 15.33 114,119 87.011 Elanfnlos AdsorbldOl
""" o,ro "'O O ... 0.00 O.ro 0.00 O.ro 0,00 O.ro 0.01 0.01 o,ro o,ro o,ro O.ro 1," o,ro O.ro O,� '"0 o,ro 0.0> O.ro 0.0> O,� O.M 0.0< 0,00 O.M 0.01 0.01 0,00 0,12 0,00 O.ro 0,0< 0.25 0.14 O." "" 0.01 O,M O,M 0,17 0.12 0,\5 0,\5 O,tl 0,14 O.U 0.07 0.12 0,10 0.31 0.2. O,e7 O.� O,� O,M "'" U. .." ." O." O,� 0 .• 3 1,75 1,M 1,M 2,81 ,,� O,� 3,10 ,,� 1," �.�S D.53 O." '" """ o,ro o,ro O.ro o,ro 0,00 0,00 O.ro 0.01 0.00 O'" O.ro 0.00 o,ro o,ro o,ro o,ro 0,00 ... O.ro """ 0.01 O'" 0.01 o,ro 0,00 0.01 0,01 o,ro 0.01 O'" o,ro 0.00 0,01 0.01 0,0< O.� 0.02 O." O." .<0,0, 1,n " . ". O,� 0.�2 O,� O.�, O,e7 O,M '.� 0,82 0.'2 1.M 0.0< 0,00 0.4' O,� ". Q.e7 "'" O'" ,0> 0,0< 0.03 0,03 0,01 0.03 0,011 0.03 0.0> 0.03 0,03 0.03 e.02 0.0< 0.01 0.01 0.0> 0.01

52 CAPITÁN, A. el al.
además. la presencia de elementos trazas 5ig· nificativos. tanto en la goelhila como en la hematites. Entre ellos, los más importantes son As. Sb y Pb. Teniendo en cuenta la dificultad de entrada de estos elementos en la estructura de los óxi-hidróxidos citados. se considera que su presencia estaría asociada a procesos de adsorción, siendo estos procesos más eficaces en la goelhila que en la hematites.
Aunque en todo el gossan los minerales mayoritarios son esencialmente los mismos. cada facies difiere de las demás en la abundancia relativa de las fases mayoritarias y en la presencia de minerales accesorios, como puede verse de forma sistemática en la Tabla 2. Asr. en la facies colofarme del gossan hematÍlico el cuarzo « 1%) es minoritario, y las fases mayoritarias son la beudantita-corkita (10%), la goethita (10%) Y la hematites (80%). En la facies brechoide el cuarzo (75%) es
siempre el mineral más abundante. En el gossan hematítico, estn facies es más rica en hematites (15%) que en goethita (5%) y se han identificado beudantita y corkita (!!> 5%). Sin embargo, en el gossan goethítico es más abundante la goethita (15%) que la hematites (5%) y sólo se ha identificado beudantita (!!> 5%). En la facies coloforme del gossan goethítico, el óxi-hidróxido más abundante es la goethita (50%), mientras que la hematites (!!> 5%) Y la beudantita (!!> 5%) aparecen de forma mayoritaria sólo en la parle superior del mismo. El cuarzo (40%) está presente en toda esta parte del perfi l de alteración. La mineralogía mayoritaria del gossan jarosítico se compone también de cuarzo (20%), beudantita (40%), goethita (15%) y hematites (25%), mostrando la beudantita las abundancias más altas de todo el perfi l.
En función del origen se han distinguido
Tnbla 2.- Mineralogía de las litofacies diferenciadas en el gossan de Filón Sur.
Naolormación MII)'OriI&rbl
, I(
ResidualeS Mlil8tales�
NeolormaciOn , .. -
Residuales Mlneralt$ �
Aesidllll!es
NllformaciOn
Resldualos

Caracterización textural y mineralógica del gossan de Filón SUL . . 53
dos tipos de minerales minoritarios: los residuales y los de neoformación. La distribución de unos y otros en las diferentes lilOfacies es heterogénea. Considerando en primer lugar los de origen residual, el rutilo y el circón están presentes a 10 largo de todo el perfil. La casiterita se ha identificado en todo el gossan excepto en el nivel goethítico. La xenotima está presente en las facies brechoides y la monacita en la goethítica colo forme. Como minerales minoritarios de neoformaci6n más importantes, se han identificado haluros de plata (AgCI, AgI), óxi-hidróxidos de baja cristalinidad (Fe-Ti, Fe-Sn, Fe-Sb), cerusita, barita, caolinita, oro y plata. La iodargirita está presente en todo el perfil. mienlras que la clorargirita y la plata sólo se han identificado en el gossan goethítico. El oro, como granos visibles mediante MES, sólo se ha reconocido en la facies goethítica coloforme asociado normalmente a cristales de iodargirita.
Estudio microtextural
Los patrones textura les de las fases minerales formadas durante la oxidación de los sulfuros, son muy homogéneos en todo el gossan de Filón Sur. Predominan las texturas de transformación y reemplazamiento entre los minerales presentes, aunque cada facies presente además rasgos textura les propios.
Facies Breclloide. Los fragmentos de la brecha están cementados por una matriz de cristales de óxi-hidróxidos de Fe inferiores a I IJ. con textura masiva, o por goethita (facies goethítica brechoide) o hematites (facies hematítica brechoide) con textura coloforme. Esta textura sólo se desarrolla en el contorno de los fragmentos de cuarzo y presenta a veces evidencias de reemplazamiento de goethita por hematites, conservando esta última la textura coloforme original de la goethita. Las texturas de reemplazamiento son muy frecuentes también dentro de la matriz, siendo uno de los principales rasgos texturales la transfor-
mación de beudantita a goethita y I o hematites y la de goethita a hematites.
Facies Hematítica Colo/arme. En la que predomina la textura coloforme, siendo característica la presencia de un microbandeado como resultado de la transformación progresiva de goethita a hematites. En los huecos, cristaliza beudantita subidiomorfa, con evidencias de reemplazamiento parcial por goethita y hematites (Figuras 6a y 6b). De forma general, la secuencia de transformación que muestran estas texturas es: beudantita � goethita � hematites. A pesar de que la secuencia de transformación anterior es la más común, existen evidencias de reemplazamiento de beudantila por hematites (Figura 6c). En las dos facies diferenciadas en el gossan hematftico, la matriz de textura masiva está constituida por hematites y goethita. En ella, los cristales subidiomorfos de beudantita se distribuyen mostrando texturas propias de procesos de desestabilización y transformación a 6xi-hidróxidos de hierro.
Facies Goetflít;ca Coloforme. La textura colofarme característica de la parte superior de este nivel, presenta un microbandeado en el que la goethila ocupa siempre la parle más interna (Figura 6d). Hacia el borde de esta textura alternan goethita y hematites. con predominio de la última en las bandas más externas y en las zonas más porosas. En algunos casos. la última banda la compone beudantita y en ella se puede observar el reemplazamiento parcial de este mineral por goethita (Figura 7a). Las zonas masivas de la matriz están formadas por agregados de goethita acicular de tamaño de grano superior a I ).L, a diferencia del resto de las l itofacies en las que la goethila presenta un tamaño inferior. Al igual que en el gossan hemalÍ1ico, el microbandeado es el resultado de los procesos de transformación de beudantita a goethita y de ésta a hematites. Los agregados aciculares de óxihidróxidos de Fe que componen la matriz, también se disponen en agregados nodulares que se asemejan a 105 típicos framboides de

54 CAPlTÁN, A. el a l .
pir i ta . Aunque estos nódulos son mayoritariamente goethíticos. en determinadas muestras presentan núcleos de beudantita y bordes hematíticos.
Facies Jarosítica. Las relaciones texturales entre l as fases mayoritarias son muy diferentes desde la parte superior de esta l i toracies hacia el contacto con los sulfuros masivos. En la zona de transición entre el gossan gocthítico y el jarosílico, las texturas, aunque más simples, son simi lares a [as descrilas anteriormente. La matriz de óxi-hidróxiclos
de Fe, de textura masiva, cementa granos de cuarzo y cristales relictos de ruti lo, circón y casiterita. La ausencia generalizada de texturas colo formes caracteriza al gossan jarosítico masivo. La beudantita es muy abundante y se concentra principalmente como agregados cristalinos en los huecos entre los granos de cuarzo mostrando evidencias de reemplazamiento por goethita (Figura 7b), la cllal est<Í a su vez reemplazada por hematites (Figura 7c). En algunos casos, la hematites se forma por desestabilización de beudallt i ta (Figura 7d) .
Figura 6. a) Gossan hematít ico coloformc: microbandcado coloformc desarrollado por transformación de goethi ta (goe) a hematites, con cristalización dc bClIdantita (be) en los huecos. b) Gossan hcmatít ico coloforme: cristal idiomort"o de beudantita parcialmente reemplazado por goeth ita y hemati tes . Transformación gradual de l a gocthita a hematites. e) Gossan hcm:Hítica co lo forme : nive les de bcudanti ta parcialmente reemplazados por hematites. d) Gossan goethítico colot"orme: microbandeado coloforme por transfotmación de goe th i ta (gac) n hematites (hm) Cll l a p artc más cxtcrna de esta tcxtura. La última banda está constituida por bClIdantita (be) parcialmente transformada a goclh i ta.

Caracterización textural y mineralógica del gossan de Fi lón Sur . 55
Discusión y conclusiones
El estudio mineralógico y microtextural del gossan de Fi lón Sur ha permitido reconocer cinco etapas de cristalización que se incluyen en dos fases principales de alteración de los sulfuros masivos (Tabla 3). La primera fase comprende los procesos de oxidación de sulfuros sellSI/ slricro, consistentes en la transformación de su l furos a óxi-h idróxidos e hidróxi-sulfalos (Etapas I a 3) , mientras que la segunda fase (Etapas 4 y 5 ) está relacionada con la evolución posterior del gossan tras su formación, y consiste en la transformación de
las asociaciones originadas en la primera fase, La primera fase comienza con la oxida
ción de los su l furos, evolucionando el pH progresivamente hasta valores de acidez moderados. In ic ia lmente (Etapa ¡ ) , el pI-! moderadamente ácido (>3) y la baja concentración de SO/o en las soluciones, favoreció que sólo se rorlllíua goelh i la por oxidación de la pir i ta , En un estadio más avanzado del proceso, cuando la proporción de su l furos oxidados es superior, d isminuye el pl-l de las soluciones y aumenta progres ivamente l a concentración de SO}, en las mismas, lo que favorece la estabil i z¡¡ción de la beudantita (Brown, 197 1 ) . Du-
Figura 7, a) Gossan gocthítico colofarme: detalle de la rolo 6-d donde se observa la transformación de bcudantita a gocthita, b) Gossan jarosítico: textura de reemplazamiento entre bcudantita y gocthita. e) Gossan jarosísl ica: textura de reemplazamiento entre goclhita y hematites. d) Goss::tJl jarosísl ica: cristales idiomorfos de bcudantita en la matriz parcialmente reemplazados por hematites.

56 CAPITÁN, A. el al.
Tabla 3.- Paragénesis minerales correspondientes a las distintas elapas de alteración diferenciadas en el gossan de Filón Sur.
Primera Fase l · Segunda Fase Etapa 1 I Etapa 2
GOETHITA 1_ BEUOANTITA , IOOARG1RITA
BARITA: " GOETHITA 2 HEMATITES
rante un tiempo. se forman simuJláneamenle goethita y beudantita (Etapa 2). aunque en condiciones de pH inferiores o próximas a tres, la goethita deja de ser estable, favoreciendo la cristalización de beudantita (Elapa 3). En esta misma elapa, aunque no necesariamente de forma simultánea, se formaron otros minerales como iodargirita, clorargirita y barita. Esta primera fase finaliza cuando se completan los procesos de oxidación de sulfuros por encima del nivel freático.
A partir de ese momento comienza lo que hemos denominado segunda fase, en la que las soluciones percal antes evolucionan progresivamente a más oxidantes y menos ácidas. Al no quedar sulfuros que oxidar, se interrumpe el proceso de generación de acidez y el consumo de oxígeno, y al llegar de nuevo a condiciones de pH superiores a tres (Etapa 4), la beudantita deja de ser estable, y comienza a transformarse en goelhita. A su vez, la goethita se comporta de forma metaestable, iniciando su deshidratación a baja temperatura y la transformación parcial a hematites. Esta transformación pudo ser simultánea, durante un tiempo, con la desestabi lización de beudantita a goethita, o bien ocurrir posteriormente, ya que no existen evidencias texturales claras en este sentido. En un último estadio de esta segunda fase (Etapa 5), la desestabilización generalizada de beudantita y de goethila da como producto final hematites. Las texturas de transformación gradual y de reemplazamiento de goethita y beudanlita a
I Etapa 3 I Etapa 4
- - - -
Etapa 5
hematites, son las que predominan en todo el gossan de Filón Sur.
La segunda fase en el origen y evolución del gossan de Filón Sur continúa en la actualidad, puesto que existen en desequilibrio beudantita, goethita y hematites. La desestabilización de la beudantita está controlada por el aumento progresivo del pH, ya que este mineral se forma exclusivamente en condiciones oxidantes y ácidas (Brown, 1971). El campo de estabilidad de este mineral aparece para condiciones de pH inferiores a tres, mientras que el de los óxi-hidróxidos de Fe lo hace por encima de este valor. Sin embargo, dado que la desestabilización de beudantita a pH superior a tres es relativamente lenta, la existencia de beudantita junto con goethita y I o hematites es habitual.
Por otro lado, en condiciones naturales se produce un "envejecimiento" de la goethita y su transformación progresiva a hematites por procesos muy lentos de deshidratación (Alpers y Brimhall, 1989). Por esta razón, la goelhila es muy frecuente en sedimentos recientes pero no en rocas sedimentarias más antiguas, en las que predomina la hematites. La formación de la hematites en el gossan de Filón Sur se produce siempre por deshidratación de la goethita o por la desestabilización de la beudantita. ya que no se ha identificado hematites como fase pura, sino dos variedades parcialmente hidratadas: protohematites e hidrohematites. Por otra parte. el mayor contenido en elemen-

Caracterización texlUraJ y mineralógica del gossan de Filón Sur . . . 57
tos adsorbidos que presenta la goethita frente a la hematites. se relaciona con el proceso de formación de cada mineral. La goethita se forma a partir de soluciones ricas en metales durante los procesos de alteración de sulfuros. adsorbiendo parcialmente dichos elementos en solución. En cambio. la hematites se forma por deshidratación de la goethita. siendo movilizados en este proceso parte de los elementos adsorbidos inicialmente por la goethita.
La transformación de goethita a hematites está también condicionada por otros factores. como la temperatura y el tamaño de los cristales. De hecho. existen diferencias entre los niveles del perfil de gossan en Filón Sur, que se relacionan con el tamaño original de los cristales de goethita. Así, en el gossan goethftico, con un tamaño medio de los cristales de goethita superior a 1 J-l, es donde menos a progresado la transformación de goethita a hematites, siendo muy generalizada en el resto de niveles definidos, que muestran un tamaño medio de cristales inferior a l J-l. Según Langmuir (1971). la transformación de goethita a hematites se favorece con el aumento de temperatura, aunque el límite térmico de estabilidad entre ambas fases es variable y función directa del tamaño de los cristales. por lo que a igualdad de temperatura se favorece la transformación a hematites en aquellos niveles en los que la goethita presenta un tamaño de grano inferior.
Agradecimientos
Este trabajo ha sido financiado por el ministerio de Ciencia y Tecnología a través del proyecto REN2000-1003-C03-03 y por la Junta de Andalucía (Grupo de Investigación RNM 198).
Bibliografia
Almod6var, G.R. Y Sáez, R. (1992): Los yacimientos de sulfuros masivos de la Faja
Pirítica Sur-Ibérica. In: "Recursos Minerales de España", J. Guinea y J. Martínez Frías, eds. CSIC. Madrid, 1309-1324.
Alpers. C.N. y Brimhall, G.H. (1989): Paleohydrologic evolution and geochemical dinamics of cumulative super gene metal enrichment al La Escondida. Atacama Desert. nothern Chile. EcolI. Geology 84, 229-255.
Arribas. A. ( 1 998): Los Yacimientos de Oro asociados con las monteras limonÍlicas de la FPI. Bolelfn Geológico y Mil/ero 109.
429·434. Brown, B . (1971): Jarosite - Goethite
stabilities at 25 oC, 1 atm. Mineral.
Deposita 6, 245-252. Dutrizac, lE. y Jambor. J.L. (2000): Jarosites
and their application in hydrometallurgy. In: "Sulfate Minerals: crystallography, geochemistry & environmental significance", C.N. Alpers, J.L. Jambor y O.K. Nordstrom, eds. Reviews in Mineralogy & Geochemistry, vol. 40, 405-452.
García Palomero, F., Bedia Fernández, J.L.. García Magariño. M. y Sides. EJ. (1986): Nuevas investigaciones y trabajos de evaluación de reservas de gossan en Minas de Riotinto. Bol. Geol. Mili. T. 97. 622-642.
Gonzáles. F., Moreno, C., Sáez, R. Y Clayton, G. (2002): Ore genesis age of the Tharsis Mining District (Ibrian Pyrite Bell): a palynological approach. Geol. Soco
l..o/UJOII 159, 229-232. Langmuir. D. (1971) : Panicle size effect on
the reaction: goethite = hematite + water. Am. lour. Sc;. 271. 147-156.
Leistel. J.M .• Marcoux. E . • Thiéblemont. D . • Quesada. C . • Sánchez, A., Almodóvar. G.R., Pascual, E. y Sáez. R. ( 1 998) : The volcanic-hosted massive sulphide deposits of ¡he Iberian Pyrite Beh. Review and preface to the Thematic Issue. Mineral. Deposita 33, 2-30.
Mathur. R., Ruiz, J. y Tornos, F. ( 1999): Age and sources of the ore al Tharsis and Río

58 CAPITÁN, A. el al.
Tinto. Iberian Pyrite Bell, from Re-Os ¡solOpes. Mineral. Deposita 34, 790-793.
Moreno, C. (1993): Postvolcanic Paleozoic of {he Iberian Pyritc Belt: nn example of basin morphologic control on serliment distribution in a turbiditic basin. Jour. Sed. Petrol. 63, 1 1 18-1 128.
Moreno. C. y Sáez, R. (1990): Sedimentación marina somera en el Devónico del Anticlinorio de Puebla de Guzmán. Faja Pirítica Ibérica. Geogaceta 8. 62-64.
Pineda Vara, I. (1963): Piritas de Huelva: Su historia, minería y aprovechamiento. Ed. Summa, Madrid, 1003 pp.
Roca. A., Viñals, J . , Arranz, M. y Calero, J. (1999): Characterization and alkatine decomposition/cyanidation of beudantitejarosite rnaterials from Río Tinto gossan ores. Call. Meral{. Q//art. 38, 93-103.
Sáez. R . • Almodóvar, O.R. y Pascual, E. (1996): Oeological constraints on massive sulphide genesis in the Iberian Pyrite Belt. Ore Ceo/. Rev. 11, 429-45 1 .
Sáez, R . • Pascual, E., Toscano, M. y Almodóvar, O.R. (1999): The Iberian type volcano-sedimentary massive sulphide deposits. Mi/leral. Deposifa 34, 549-570.
Schermerhorn. L.J.C. (1971); An outline stratigraphy of ¡he Iberian Pyrite Belt. Bol. Ceo!. Mili. 82, 239-268.
Scon. KM. (1987): Salid solution in. and
classification of, gossan-derived members of the alunite-jarosite family, northwest Queensland, Australia Am. Milleral. 72, 178-187.
Strauss. O.K ( 1970): Sobre la geología de la provincia piritífera del Suroeste de la Península Ibérica y sus yacimientos, en especial sobre la mina de Lousal (Portugal). Mem. lim. Geot. Mili. Espalia 77, 266 pp.
Strauss. O.K. y Madel, J. (1974): Geology of massive sulphide deposits in the Spanish - Portuguese Pyrite Belt. Ceo/. RlllldscJ¡. 63. 191-2 1 1 .
Strauss. G.K. y Beck, J.S . ( 1990): Oold mineralizations in the SW Iberian Pyrite Bel!. Mineral. Deposita 25, 237-245.
Tornos. F . • González Clavijo, E. y Spiro, B. ( 1998): The Filon Norte orebody (Tharsis Iberian Pyrite Belt): a proximal lowtemperalure shale-hosted massive sulphide in a thin-skinned tectonic bell. Mineral. Deposita 33, 150-169.
Wolska. E. y Schwerlmann, U. (1989):
Nonstoichiometric structures during dehydroxylation of goethite. Z. Kristallogr. 189, 223-237.
Recibido: Abril 2003
Aceptado: Agosto 2003

Bolea" de la Sociedad Espmiola de Mineralogía, 26 (2003), 59-67 59
Caracterización mineralógica y técnica de materiales zeoIíticos naturales de uso comercial.
Antonio FLORES MACiAS', Pedro BOSCH GIRALl , Victor LARA CORONAl, Víctor ORDAZ CHAPARRO�, José 1. CORTÉS FLORES�, Ferna'ndo DE LEÓN GONZÁLEZ', 'Laboratorio de Fisiología y Tecnología de Cultivos, Universidad Autónoma MetropolitanaXochimilco, Calzada del Hueso 1 100, Col. Villa Quietud, 04960 México D.F., México. lInslituto de Investigaciones en Materiales, Ciudad Universitaria, Circuito Exterior, México D. F., México lDeparlamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Michoacán y La Purfsima, México D.F., México. 41nsl11uto de Recursos Nalurales. Colegio de Poslgraduados. Km. 35.5 Carr. México-Texcoco. Montecillo, Edo de México
Abstraet: One of Ihe mosl common limilalions of Ihe research work on zeolites as ionie exchangers is ¡hat the used natural zeolite (eatures are unknown. This work compares Ihe cryslallographic and morphological properties as well as Ihe semiquantitative chemical eomposilion of IWO materials. The used techniques were X-ray diffraction (XRD), scanning electron microscopy (SEM), and energy dispersive X-ray Speclroscopy (EDXS). The cation exchange capacity (CEC) values obtained through the saluralion techniques wilh ammonium acctate (SAM). sodium acetate (SAS) and modified ammonium acetate (SAMM) are also shown. Thc XRD study of both zeolites showed the presenee of clinoplilolite (Zep) in a percentage of 90% and 45%. The CEC values oblained Ihrough SAM. SAS and SAMM are substanlially different, the lower values were obtained using Ihe SAM and SAS techniques. Unexpectedly, Ihe highcr clinoptilolite-zeolite content showed lower CEC values, while the less one registered the highest values. CEC results suggest that the use of one or the other technique might underestimate or overestimate the ionk exchange interactions when the zeolite was meant to function as a cation exchanger. According to the XRD and CEC data. the clinoptilolite canten! of a zeolite material is not always related lo a high CEC levc!.
Key words: Clinoptilolile. Mordenite, Nalural zeolites, XRD. SEM, Ion exchanger. Cation exchange capacilY (CEe).
Resumen: Es frecuente que en investigaciones que experiment:m con zeolilas como intereambiadores iónicos no se conozcan sus características. Este trllbajo compara los resultados mineralógicos. morfológicos y de composición química de dos materillles zeolfticos car3cterizados mediante difracción de rayos X (DRX), microscopia electrónica de barrido (MEB) y encrgla dispersiva de rayos X (EDS). Se discuten los valores de capacidlld de intercambio catiónico (CIC) obtenidos mediante las técnicas de saturación con acetato de amonio (SAM). lIeetato de sodio (SAS) y acetato de amonio modificado (SAMM). El estudio mediante DRX de ambos materiales zeoJ[ticos mostró la presencia de clinoptilolita (Zcp) en grados del 90% y del 45%. Los valores de CIC obtenidos son significativamente diferentes, encontrándose los valores más bajos con las técnicas de SAM y SAS. Contrariamente a lo esperado. la muestra con mayor contenido en elinoptilolit3 presentó los valores menores de CIC, mientras que la de menor contenido mostró los valores más 3ltos. Los resultados de ClC sugieren que la utilización de una u otra técnica

60 FLORES MACIAS. A. el al.
podrfa subestimar o sobreestimar [as interacciones de intercambio iónico cuando el material zeolftico fuera destinado a una aplicación como intcrcambiador catiónico. Los datos de DRX y elc indican que un elevado contenido mineralógico de dinoptilolit3 no siempre está asociado con una alta CIC.
Palabras clave: Clinoptilolit3, mordcnita, DRX. MEB, zcoHtas naturales. ¡ntercambiador iónico. capacidad de intercambio catiónico (CIC).
Introducción.
Las zeolitas son un grupo de aluminosilicatos hidratados con estructura cristalina tridimensional formada por tetraedros de SiO/ .. en tos que la sustitución de Si'· por AP· crea un valor de carga negativo que le confiere al mineral una aJla capacidad de intercambio catiónico. En las zeolitas naturales. el intercambio catiónico ocurre en la superficie externa e interna del cristal, lo que las coloca como uno de los mejores intercambiadores ya que sus capacidades son del orden de 2 a 3 meq/g (Mumpton, 1983 y 2000).
A partir de 1950 varias investigaciones relacionadas con las zeolitas han aparecido en la literatura técnica. Se publican datos sobre su aparición en rocas sedimentarias deorigen volcánico, sus valiosas propiedades físicas, qufmicas y su potencial presente y futuro en aplicaciones tecnológicas en diversas áreas industriales (Galindo etal .• 2000; Colela el al., 2001) o agrícolas (Buondonno el al .• 2000). Sin embargo, uno de los defectos más comunes en la información recogida es que no se conocen las caracterfsticas de las zeolitas utilizadas. En otras ocasiones, los resultados son de limitado valor debido a que las muestras se prepararon en forma inadecuada, se usaron mueslras diferentes considerándolas como idénticas, o bien, no hubo un conocimiento del comportamiento químico de las zeolitas, todo lo cual ha contribuido a dificultar la interpretación y reproducción de los resultados experimentales (Sheppard,1983; Seff, 1996).
Métodos como la DRX (Hoof y Roelofsen, 1991), MEB Y el estudio de propiedades especificas, como la capacidad de intercambio catiónico son los más util izados para caracterizar una zeolita (Pe-Piper, 2000). Para la
determinación de esta última propiedad en zeolitas, se suele utilizar el método de saturación con acetato de amonio (SAM) como si se tratara de arcillas (Kitsopoulos, 1999), aunque también existen el de saturación con acetato de sodio (SAS) y el modificado de acetato de amonio (SAMM).
En el presente trabajo, se muestran los resultados obtenidos al estudiar muestras de material zeoHtico que, por sus propiedades, están siendo utilizados como intercambiadores iónicos en el tratamiento de aguas residuales y en aplicaciones agropecuarias. Los objetivos del estudio incluyeron: 1 ) Determinar la composición mineralógica de la fase zeolítica de los dos materiales estudiados, 2) comparar la composición de la fase zeolítica con los valores de CIC determinados mediante tres técnicas de laboratorio y 3) Conocer, en ambas muestras, algunas propiedades químicas y físicas de interés en investigación aplicada.
Materiales y Métodos.
Una de las muestras de material zeolítico investigadas procede de Cuba (Zcu) y la otra de México (Zme). La primera, fue proporcionada por la empresa Productos Siderúrgicos Coper S.A., la segunda provino de un yacimiento explotado por Mina Monte Carla S.A. de C.V. ubicado en, Sinaloa (México).
Zme es el producto de actividad volcánica de un ambiente geológico esencialmente integrado por rocas volcánicas sin diferenciar y deformadas del Cenozoico, que se distinguen por estar abiertas y deformes. Principalmente, se aprecian andesitas con zeolitas. tobas y brechas; localmente riolitas. ignimbritas y areniscas del Terciario inferior continental (CRM . • 1991).

Caracterización de materiales zeolÍlicos comerciales 61
Zcu procede de depósitos de zeolitas vinculados a las secuencias de las cuencas de retroarco de edad Cretácico y PaleocenoEoceno y a las secuencias superiores de la zona axial del arco Cretácico. Se forman clinoptilolita, mordenita y, en menor medida, montmorilJonita por la transformación del vidrio volcánico contenido en las tobas, de composición intermedia a ácida, La clinoptilolita sustituye pseudomórficamente los fragmentos de vidrio volcánico, mientras que la mordenita se forma posteriormente a ésta (Orozco y Rizo, 1998).
Para su caracterización, se realizaron por triplicado los diferentes análisis, siendo elegidas al azar las muestras obtenidas de los lotes proporcionados por los proveedores. El estudio cristalográfico fue realizado por DRX mediante un difractómetro Siemens D 500 acoplado a un tubo de rayos X con ánodo de cobre, y a un monocromador de haz difractado, utilizando para las identificaciones las fichas del Joint Committee of Powder Diffraction Standards (1CPDS). La velocidad angular para obtener el difractograma fue de 0.020 gradosl segundo.
Se determinó la morfología utilizando un microscopio electrónico de barrido Philips XL30 y la composición química mediante analizador EDAX con detector de zafiro. Los grá· nulos utilizados en el estudio fueron momados en un portamuestras y sometido a un baño de evaporación de oro en frfo (spuuering), en cámara de vació marca Dentan Vacum Desk 2, con la finalidad de mejorar su conductividad eléctrica. En cada muestra estudiada se realizaron las determinaciones en un área de barrido de un milímetro cuadrado, eligiendo al azar tres puntos de observación. Para la determinación química semicuantitativa se utilizó la técnica de energfa dispersiva de rayos X (EDS). Las muestras para las determinaciones elementales fueron igualmente elegidas al azar del lote de zeolita, molida en mortero hasta tamaño de malla 120 y compactada en prensa manual hasta obtener una pastilla de cinco
milímetros de diámetro por tres de espesor. A cada material zeolítico se le determinó
por triplicado su capacidad de intercambio catiónico (CIC) mediante el método de saturación con :acetato de amonio (SAM 24 h) (Chapman, 1965), con :acetato de sodio (S AS 24 h) (Chapman, 1965) y con acetato de :amonio modific:ado (SAMM 240 ' h) (Kitsopoulos, 1999). También. utilizando técnicas convencion:ales de laboratorio (SEMARNAP, 2001), se estudiaron el pH, la conductividad eléctrica, los cationes intercambiables, la densidad aparente, la densidad real, la capacidad de campo, el punto de marchitamiento permanente, el punto de salUración y la granulometría.
Resultados y discusión.
La composición mineralógica de ambas muestras presenta un alto contenido de fase zeolítica. Ambos difractogramas (Fig. 1 Y 2) coinciden con la ficha de identificación de la clinoptilolita 25-1349 (JCPDS). Además de la clinoptilolita, la muestra Zcu presenta picos que corresponden a la mordenitQ (ficha 29-1257). Estimaciones cuantitativas a partir de la intensidad de los picos de difracción (Hoof y Roelorsen, 1991; Sands, 1993; Borchardt, 1995) indican que Zcu está formada por alrededor de un 45% de clinoptilolita (Zcp) y de un 55% de mordenita (Zmo).
La composición mineralógica de Zme muestra una fase zeolítica constituida de clinoptilolita en cantidad cercana al 90%, estando constituido el 10% restante por cuarzo y otros compuestos amorfos.
El estudio morfológico mediante MEB de la Zcu (Fig. 3). no mostró las formaciones típicas de placas de la clinoptilolita y tampoco las formaciones filamentosas características de mordenita (Mumpton y Ormsby, 1976). Sin embargo, pudieron apreciarse morfologías similares a lajas, aparentemente cubiertas por partículas más pequeñas de algún otro mate-

62 FLORES MACiAS. A. el al.
rial. seguramente mordenita. La Zme (Fig. 4) muestra formaciones de placa cuyo tamaño varfa entre 5 y 8 micrones con un espesor de 1 a 3 micrones. en las que se aprecia la característica forma de "ataúd" de la cJinoptilolila (Mumpton y Ormsby, 1976).
El espectro EOS de composición qufmica semicuaOlitativa de ambas zeolilas (Tabla 1, Fig. 5), muestra la presencia de los cationes típicos de éstos minerales, sin encontrarse entre estos formas nitrogenadas, lo que coincide
con lo indicado por otros autores (Lewis, 1981; Pirela el al. , 1983; Pe-Piper, 2000).
La presencia de una alta relación molar Al/Si en la composición química en ambas muestras zeolílicas. permite esperar una elevada estabilidad térmica y resistencia a los agentes ácidos. característica deseable en apl icaciones prácticas.
Como es característico de las zeolitas calcico-sódicas. Zcu (Tabla 11) contiene predominanlemente calcio y sodio en las posi-
Figura l. Difraclograma dc la zcolita natural Zcu
'"
I r J i ,�
•
,�
• "
,�
" " " " " " "
Figura 2. Dirractograma dc la zcolila natural Zme
..
----,--- .-.--_.-,.. 43 �2tet.

Caracterización de materiales zeolíticos comerciales 63
Figura 3 . Micrografía de ZClt en la que se aprecian algunas formas parecidas a lajas recubiertas por partículas mús pequeñas.
ciones intercambiables, lo cua l coincide con los valores de los cationes más abundantes reportados para la especies mordenita y c l i nopt i lo l i ta , (Gottard i , 1 978) . El pl-l de alcal in idad med ia , de este material, es i n rlui· do por l a presencia abundante de los cationes mencionados, en forma s imi lar al reponado por otros autores (Ferguson el al. , 1 986).
En comparación con la ZClI, la Zme mues· tra cantidades considerablemente menores de cationes, lo que al parecer, est;í más asociado con una baja capacidad de intercambio, que a l a no presencia de estos en el m ineral. E l anál is is granulométrico i ndica que ambas muestras están constituidas a l menos en un
Tabla 1 . Composición elemental ('70 peso)
Elemento Zme Zcu
MgO 1 . 8 0.8 AI,O, 8 .7 7.7 SiO, 35.9 39.8 K,O 0.6 1 . 5 CaO 3.8 3.4 TiO 0.2 0.21
Fe,O, 1 . 5 1 . 9 Na,O 1 .2
Pigura 4. Micrograría de Zme en la que se apre· cian placas desarroll:Jdas con la morfología típica de "ataúd" de la especie c l inoptilolita.
90% por gránulos comprendidos entre 0.99 y 2.0 111111, por lo que el t ipo de grano se asemeja al de una arena gruesa (Brad)' )' \Vei l , 1999). Sin embnrgo. la Zcu muestrn una mayor proporción de granos pequeños que deben favorecer valores mayores en su ele (Nus y Brauen, 1 99 1 ) .
Se encontraron d i ferencias s ign i l'icntivas para los valores de CIC (Tabla 1fI), determinados mediante las técnicas SAM, SAS y SAMM.
La Zcu muestra diferencias altamente sign i ficativas entre los valores de Cle obtenidos por las tres técnicas. La d i ferenci a significativamente menor d e CIC entre l a técnica de saturación con acetato de amonio y l a que uti l iza acetato de sodio, podría expl icarse al suponer que el potasio existente en la zeolita no fue reemplazado por e l sodio de la solución de saturación, ya que este ú l t imo ocupa una posición más baja en l a secuencia de selectividad catiónica de la c l i nopti lol i ta (Huang )' Petrovic, 1 994); también, podría no haber sido suficiente la cantidad de acetato de amonio uti l izada en SAS para reemplazar el sodio procedente de la solución de saturación NaOAc y el contenido en l a zeolita (0.5434 meq/g). El valor de CIC obtenido mediante SAM es significativamente menor al obtenido mediante SAMM como resultado del menor tiempo

64 FLORES MACiAS. A. el al.
zc. ZM •
•• ••
• • ••• , .• . ... .... . ... . ... . ... .... .... .... .... �K C"I� • • ••
1 . " • . •• ' , " � ... S . " .... • ... . . .. . . ..
Figura 5. Espectro EDS de composición química semicuanlitativa de Zcu y de Zme.
Tabla 11. Propiedades de la zeolita cubana Zeu y de la mexicana Zme determinadas en laboratorio (Norma Oficial Mexicana NOM-021-RECNAT· 2001). Se presentan dalOs de una zeolita sódica.
Propiedades
pH en agua (1 :2) Conductividad eléctrica (dS m" )
Na' Cationes Ca·· Intercambiables
MgH (meq/g) K'
Densidad aparente (9 cm-l) Densidad real (g cm.) Capacidad de campo 0.3 bar Punto de marchitamiento permanente 15.0 bar Punto de saluración
>2.00 mm Temallo
1.00 a 1.99 mm de partlcula 0.425 a 0.999 mm (%) < 0.425 mm
·Zeolila dinoptilolita sódica (FerglolSOtl el el, '986)
Zcu
8.02
0.940
.5434
.668
.232
.135
0.96
2.24
19.5
17.8
42
6.9
63.2
27.5
2.4
Zme
7.70
0.480
0.7642
.0665
.0390
.0046
0.96
2.20
19.34
12.75
39.34
2.4
41.3
53.7
2.7
Zcp·
6.2
0.2
126
.020
.04
.46
2.10
64.6
13.9
1.5
Tabla III. Promedios y desviaciones estándar de los valores de CIC (meq/g) analizados mediante tres técnicas.
Técnica Tiempo de Zeu Zme saturación
SAM 24 h 1 .4206 . 0.72 0.1443 . 0.05 SAS 24 h 1.0595 . 0.68 0.1535 . 0.07 SAMM 240 h 1.7600 ± 2.00 0.8666 . 5.68

Caracterización de materiales zeolíticos comerciales 65
utilizado en la saturación con acetato de amonio. 24 h Y 240 h respectivamente. El tiempo es una variable importante para que ocurra el reemplazamiento de los cationes adsorbidos a la zeolita (Kitsopoulos, 1999).
El valor de ele tan bajo (SAM y SAS) y bajo (SAMM) de Zme. coincide con los valores registrados por otros investigadores' para materiales que contienen clinoptilolita, pero que no indican el contenido porcentual de ésta (Broschat, 2001). La diferencia altamente significativa entre los valores de CIC obtenidos por SAM y SAS y los encontrados con SAMM se debe al tiempo de saturación, considerablemente mayor para este último método. Sobresale el hecho de que Zme presente una alta proporción de clinoptilolita pero, al contrario de lo esperado, el valor de elc es muy bajo. Este material, cuyo análisis difractométrico muestra un contenido de clinoptilolita del 90%, presenta valores de CIC por debajo del rango de entre 1 .60 y 3.00 meq/g que caracteriza a la especie clinoptilolita (Lewis et al., 1983. Ferguson et al., 1986); ello. podrfa estar relacionado con el bloqueo de anillos y canales internos de la zeolita. Las zeolitas pueden sufrir este fenómeno, perdiendo su utilidad como intercambiadores cuando iones o moléculas congestionan el espacio interno e impiden la difusión y el intercambio a través de éste (Seff, 1996). Los datos de densidad aparente y densidad real, que coinciden con lo reportado por otros autores (Tabla 1), indican que ambas zeolitas presentan un espacio poroso similar (56%), lo que sugiere que la superficie interna de la Zme no es accesible y por lo tanto no ocurren la difusión y el intercambio antes mencionados.
Valores mayores de CIC se encontraron en Zcu (contenido en clinoptilolita 45%), que coinciden con los reportados por diversos investigadores (Ferguson el al., 1 986; Allen et aL, 1996). El bajo contenido de clinoptilolita de este material no disminuyó el valor de ele debido a que estaba combinada con mordenita,
zeolita que en estado de alta pureza también presenta valores de CIC tan elevados como ±2.30 meq/g (Barrer, 1976), no es sorprendente enlOnces que aún sin ser una clinoptilolita pura muestre un elevado valor de cle.
Finalmente, los datos de capacidad de campo y punto de marchitamiento permanente (Tabla 11) muestran que la Zme presenta un mayor porcentaje de agua disponible que la Zcu, característica deseable en aplicaciones agrícolas.
Conclusiones.
La determinación mineralógica realizada mediante DRX resultó ser el principal método para reconocer la presencia de zeolita en materiales que contenían OIrOS minerales.
La muestra zeolftica que tuvo mayor contenido en clinoptilolita y la morfología típica de esta especie mineral, presentó un valor relativamente bajo de Cle, probablemente como resultado de un bloqueo de los anillos y de los canales internos del material. Materia� les que presentan este fenómeno, no cumplen con las características de un intercambiador iónico. Por 10 tanto, no debe asumirse que la riqueza en clinoptilolita de un material zeolítico es un indicador correlacionado con una alta CIC. Lo anterior indica que para la utilización de zeolitas como intercambiadores iónicos debe de realizarse. además de la correcta caracterización mineralógica, la determinación de su CIC.
Las tres técnicas usadas para determinar la elC dan resultados significativamente di� ferentes, lo que indica que las técnicas utilizadas para determinar esta propiedad en la arcillas (SAM y SAS) y que han sido utilizadas comúnmente en zeolitas, no son igualmente útiles, ya que estás últimas requieren un periodo mayor de saturación.
La presencia abundante de sodio y de calcio como cationes intercambiables en una de

66 FLORES MACÍAS. A. el al.
los materiales zeolílicos (Zcu) podrfa ser un factor limitante para utilizarla en aplicaciones en las que la liberaci6n de estos elementos pudieran ocasionar problemas al sistema estudiado.
Bibliografía.
Allen, E., Hossner, L., Ming. D., Henninger. D. (1996): Release Tates of phosphorus. ammonium, and pOlassium In clinoptilolite-phospate rack syslems. Soil Sci. Soco Am. 1. 60. 1467-1472.
Barrer. R. ( 1976): Cation-exchange equilibria in zeolites and feldspalhoids. In: Natural Zeoliles. Ocurrence. Properlies. Use. Sand L.B. and F.A. Mumplon eds. Pergamon Press 1978. New York., 385·397.
Borchardt. W. (1995): Crylallography. Ed. Springer-Verlag. N. York.
Brady, N.C. y Weil, R. (1999): The natufe and properlies of 50i15. Prenlice-Hall lnc. New Jersey.
Broschal, T. (2001) : Sub51rate nutrienl relenlion and growth of container-grow plants in clinoptilolitic zeolite-amended substrates. HorTeclmology 1 1 ,75-78.
Buondonno, A . • Coppola. E:. Cninniello, A .• Di Sarno, 1.. Langella. A., Mnrino, R. (2000): Short-term effecls of Italian zeolitic tuffs on the availability of beneficial and toxic elemenlS. In: Natural Zeolites for the Third Millenium. C. Colella and F. A. Mumpton eds., 2000 De Frede Editore, Nápoles, Italia, 459-470.
Chapman. H. ( 1 965): Cation exchange capacity. In Methods of soU analysis (number 9 in Series Agronomy), Parl 1 , A. Black, (Editor). American Sociely of Agronomy. Madison, Wisconsin, 891·901.
CRM., 1991. Monograffa geológica minern del estado de Sinaloa. Consejo de Recursos Minerales. México
Ferguson, G.A., Pepper, I.L., Kneebone. W.R. (1986): Growth of creeping benlgrass on
a new media for turfgrass growth: clinoptilolite zeolite-amended sand. Agroll. J. 78,969-980.
Galindo, C., Ming, D. W., Carr, M. J., Morgan, A., Pickering, K. D. (2000): Use of Caexchanged clinoptilolile for ammonium removal from NASA 's advanced life· support wastewater Sy5tem. In: Natural Zeolites for the Third Millenium. C. Colella and F. A. Mumpton eds., 2000 De Frede Editore, Nápoles, Italia, 363-371.
Gotardi, G. (1978): Mineralogy and cristal chemistry ofzeolites. In: Natural Zeolites. Ocurrence, Properties, Use. Sand L.B. and F.A. Mumplon eds. Pergamon Press 1978. New York. , 31-44.
Hoff. J.H., Roelofsen, J.W. (1991): Techniques ofzeolile characterization. Jn: Introduclion 10 zeolile science and practice. H. Bekkum, E.M. Flanigen, J.C. Jansen eds. Elsevier Science Publishing Company INC. 1991. New York, 241-283.
Huang, Z., Petrovic, A. ( 1994): Clinoplilolile zeolile influence on nilrate leaching and nitrogen use efficiency in simulaled sand bases golf green. J. Ellviromellfal. Q/lal. 23.1190-1194.
Kitsopoulos, K. ( 1 999): Cal ion exchange capacily (CEC) of zeolilic volcaniclastic material s: applicability of the ammonium ncelale saturalion (AMAS) method. Clays alld elay Minera/s. 46, 688-696.
Lewis, M. (1981): Clinoptilolite as a N, K and Zn source of plants: M.S. thesis, Colorado State University, FOTt Collins, Colorado, 1 1 8p
Lewis. M., Moore, F., Goldsberry, K. (1983): Ammonium-exchanged clinoptilolile and granulated clinoplilolite with urea as nitrogen ferti1izers. ln: Zeo-Agricullure: Use of Natural Zeoliles in Agriculture and Aquaculture. G. Pond and A. Mumplon eds. Weslview Pres5 1984. Colorado. 105- 1 1 1 .
Mumplon, F.A. (1983): Natural zeolites. In: Zeo-Agriculture: Use of Natural Zeolites in Agriculture and Aquaculture. G. Pond

Caracterización de materiales zeolíticos comerciales 67
and A. Mumpton eds. Weslview Press 1984. Colorado, 33-43.
Mumpton, F.A. (2000): Natural zeoliles: where have we been, where are we going. In: Natural Zeoliles for the Third Millenium. C. Colella and F. A. Mumpton eds., 2000 De Frede Editore, Nápoles, lIalia, 19-34.
Mumpton, F.A., Ormsby, W. (1976): Morphology of zeolites in sedimentary rocks by scanning electrón microscopy. Clays alld e/ay millera/s. Vol. 24, 1-23.
Nus, J.L. y Brauen, S.E. (1991): Clinoptilolitic zeolite an amendment for establishment of creeping benlgrass on sandy media. HortSciellce, 26(2), 1 17 - 1 19.
Orozco, G .. Rizo, R. (1998): Depósitos de zeolitas naturales de Cuba. Acta Geol6gica Hispdl/;ca, 1998,33( 1-4):335-349
Pe-Piper, G. (2000): Mode of occurrence, chemical vanallon and genesis of mordenite and associated zeolites from the Morden area, Nova SC01ia. Canada. Tlle Cal/(ldiall Milleralogist 38, 1215-1232.
Pirela, H., Westfall. D., Barbarie", K.A. ( 1983): Use of elinoptilolite in combination with nitrogen fertilizalion to increase plant growth. In: Zeo-
Agriculture: Use of Natural Zeolites in Agriculture and Aquaculture. G. Pond and A. Mumplon eds. Westview Press 1984. Colorado,113-124.
Sands, D. ( 1 993): Introduction 10 cryslallography. Dover Publications Inc. N. York.
SEMARNAP, 2001. Norma Oficial Mexicana NOM-021-RECNAT- 2001 . Ley Federal sobre Metrología y Normalización. Secretaría del Medio Ambiente y Recursos Naturales. Diario Oficial de la Federación, 17-X-2000.
Seff, K. (1996): What can be in the channels and cavilies of zeolites? In: Recen Advances and New Horizons in Zeolile Science and Technology. H. Chon. S.1. Woo and S.E. Park eds. Elsevier Science B.V. 1996. The Netherlands. 267-293.
Sheppard. R. ( 1983): Characteriza�ion of zeolitic material in agricultural research. In: Zeo-Agriculture: Use 'of Natural Zeoliles in Agriculture and Aquaculture. G. Pond and A. Mumpton Eds. Weslview Press 1984. Colorado 79-87.
Recibido: Marzo 2003 Aceptado: Octubre 2003

Bol�tíll de la Soci�dad Espallola de Millualogía, 26 (2003), 69-78 69
Síntesis y caracterización de mono cristales de materiales zeolíticos
Ester MATEO GONZÁLEZ(I,2), Montserral VILASECA LLOSADA(2), Andrés PANIAGUA CONDADO(I), Joaqufn CORONAS CERESUELA(2), Jesús SANTAMARfA RAMIRO(2). (l)Dpto. Ciencias de la Tierra. Área de Cristalografía y Mineralogra (2)Dpto. Ingenierra Qurmica y Tecnologías del Medio Ambiente. Facultad de Ciencias. Universidad de Zaragoza. cl Pedro Cerbuna, 12.
Abstraet: Although natural zeolites can occur as Jarge single cryslal, the synthesis of large zeolite single cryslals requires a special care in the nucleation and crystal growth steps.
In lhis work silicalite, analcime and cancrinite single crystals with a size up to 3, 2 and 0,008 mm respeclively have been prepared by hydrothermal synthesis and eharacterized using several microscopic and XRD techniques.
Key words: zeolites. large crystal. hydrothcrmal synthesis. nuclcalion, crystal growth.
Resumen: Aunque en la no.turateza pueden encontrarse cristales de zeolila de grandes dimensiones, la srntesis de monocristales de zeolllas de unos pocos milfmetros requiere un control preciso de los procesos de nucleación y crecimiento cristalino. En eSle trabo.jo se han preparado por sfnlesis hldrotermal y caracterizado usando diferentes técnicas microscópicas y difracción de rayos X, monocristales de siliealita, analeima y cancrinita de ho.sta 3, 2 Y 0,008 mm. respectivamente.
Palabras dave: zeolilas, monocristales de zeolilas. sfntesis hidrotermal. nuclellción, crecimiento cristalino.
Introducción
El término "zeolita" se ha utilizado para designar a una familia de minerales naturales que presentan como propiedades particulares el intercambio de iones y la desorción reversible de agua.
Hoy en dla, dicho término engloba a un gran número de minerales y fases sintéticas que presentan caracterrsticas estructurales comunes. Existen unas 135 estructuras zeolíticas o zeotipos distintos. tal y como se describen en el Atlas de las Zeolitas (Meier y Olson, 1m; www.iza-structure.orgldatabases). De una forma precisa se puede definir a las zeolitas como aluminosilicatos microporosos
e hidr:lIados basados en una red cristalina formada por la combinación tridimensional de tetraedros TO� (T= Si, Al) unidos por sus vértices. formando puentes de oxrgeno no lineales. A diferencia de otros tectosilicatos, la estructura de las zeolitas presenta canales y cavidades de dimensiones, por 10 general, subnanomélricas, en los cuales se encuentran los denominados cationes de compensación, moléculas de agua u otros absorbatos y sales. Este tipo de estructura permite la transferencia de materia entre el espacio intercristalino y el medio que lo rodea. La tabla I resume las propiedades generales de las zeolitas.
Impulsado por las clásicas (adsorción. intercambio iónico y catálisis) y por las nuevas

70 MATEO GONZÁLEZ. E. el al.
(membranas zeolititas, sensores y microsistemas) aplicaciones de las zeolilas (Auerbach el aL, 2003), en los últimos años se ha realizado un gran avance en la síntesis de nuevas zeolitas (sin contraparte en la naturaleza) y de los materiales relacionados (SAPO}. AIPO). titanosilicatos. materiales mesoporosos de estruclUra regular. elc.) (Da vis. 2002; Cundy y Cox. 2003). En la
figura 1 se representa un esquema del meca· nismo de la síntesis de zeolilas: la estructura del gel se debe a una reacción de polimerización. en la cual se forman moléculas grandes a partir de otras nuevas tales como aluminatos y silicatos. Los geles utilizados se preparan a panir de aluminato de sodio, silicato de sodio e hidróxido de sodio. especies todas solubles en agua.
Tabla l o Propiedades generales de las zeolitas.
f)ensidad Durc7.B.
Diámetro de poro . "Diámetro de caVidades-
1,9 a 2,3 g/cc 4 a 5 en la escala de Mohs
2 3 12 A . .
6 a li A"" ·-·--Sú·Pcrrrcic'iniériiii - - - ¡ . . ., Varios dcñtos de rib¡; Capacidad de intercambiocatiónToo·- í---- -Óa 650 meq/fOOg -- ---
-. --.. ----------.---.. ---.. ---.-.. -- .- - J.-.----... Capacidad de adsorción I < 0,35 cm Ig - Estabilidad téñii"¡"Ci .-- ¡-Desde 6o"C haita mIs de IOOOOC--��=�--=="'"""....",====-=,=--====---
---
---Silicatos y aluminatos solubles
I
I Gel
- _ .. _-_._- _ .. _---I
Despolimerización de cadenas de alumlnoslllcatos
I Unidades de construcción
primarias
I Unidades de construcción
secundarias
I 1 __ __ P
_
o�ledros __ _ _
I Zeollta
Figura l . Srntesis de una 2colit3; mecanismo de cristalización (Garera. 2000).

Síntesis y caracterización de monocristales de materiales zeolíticos 7 1
E n condiciones alcalinas s e obtienen cristales de pequei\o tamaño, varios micrómetros, siendo difícil sintetizar cristales de mayores dimensiones (mil im6tricos). Sin embargo, los cristales zeollticos de grandes dimensiones son muy útiles ya que permiten determinar la estructura interna de las distintas zeolitas. su anisotropía el6ctrica, estudiar los mecanismos de crecimiento. adsorción y difusión. as! como propiedades ópticas y magn6ticas, ele.
Han sido varios los autores que han estudiado las condiciones más favorables para conseguir cristales de gran tamai\o. La primera esttlltegia fue propuesta por Charnell en 1971. quien sintetizó cristales de zeolita A (LTA) Y zeolita X (FAU) con dimensiones de 65 y 140 11m respectivamente. bajo condiciones hidrolermales. Este autor añadió trietanolamina (TEA) al gel. que tiene un efecto estabilizan te y amortiguador en el crecimiento del cristal, ya que inhibe la nucleación. En 1985, Bibbí Y Dale realizan sfntesis de sodalitll usando disol ventes orgánicos. abriendo as! una nueva ruta para la síntesis de zeolitas. especialmente para la formación de cristales grandes. Kuperman et al. (1993) sintetiza silicatos. aluminosil iC:lIos y aluminofosfatos en medios no acuosos con dimensiones entre 0,4 y 5 mm. En 1993. Mostowicz et al. desarrollan nuevas síntesis de zeolitas en presencia de HF. favoreciendo la mineralización ya que tiene un poder mineralizante. comparable al de OH-, a la vez que aumenta la velocidad de crecimiento cristalino. Sun et al. ( 1995) sintetizaron cristales de mordenitll de O, 185 mm a I 500C durante 15 dras. utilizando dos tipos de fuentes de sílice (Aerosil y silicato de sodio) y cloruro de sodio. Shimizu y Hamada (2001) obtuvieron crist:lles de silicalita de 4 mm, de analcima con tamaños alrededor de 3 mm. cancrinita, zeolita JWB y sodalita con tamai\os de 0.6 mm. en medio acuoso controlando la solubilidad en sistemas hidrotermales; utilizando tubos de cuarzo como ruente de Si y ácido fluorhídrico; 6ste favorece la disolución lenta de la fuente de Si que per-
mile comrolar la cinética de cristalización. Un método similar fue propuesto por Gao et al. (2001) obteniendo cristales MFI de hasta 2 mm y usando como fuente de Si una lámina de silicio monocristalino y también ácido fluorhídrico.
El objetivo de este trabajo es sintetizar monocristales de varias zeolitas (silicalit:l. analcima y cancrinita) que puedan tener aplicaciones en campos diversos, como son la caracterización de zeolitas, la formación de membranas o el de los sensores ópticos.
La estructura de la silicalita (Nan(HP)16[AlnSi96.nOI91J), de simetría rómbica. puede describirse como un sistema tridimensional con dos tipos de canales: unos lineales con sección elíptica de 0.5 I x 0,55 nm en la dirección y. y otros en zigzag. con sección casi circular de 0.53 x 0.56 nm en el plano x-z. Por tanto. presentan anisotropía bidimensional en su estructura.
La analcima tiene una simetría cúbica (Na [AISiP.1"HP) y su estructura está representada por canales irregulares de 4,2 x 1,6 nm en el plano ( 1 10). La cancrinita es hexagonal (Nu,Ca1AI6Si,Ou(CO) 2(HJO)1 y presenta una estructura caracterizada por contener canales de 5,9 x 5.9 nm.
Experimental
Sílllesis de fas cristafes de zeofitas Se utilizan autoclaves de acero inoxida
ble (SS 3 J 6L) con un volumen de 45 cm)
(Figura 2). Principalmente consta de un recipiente cilíndrico que contendrá las partes de tenón, dos discos metálicos (uno de ruptura y airo anticorrosión) y dos piezas que encierran un muelle que permite soportar al sistema una presión mhima de 1200 kPa.
En este trabajo se han realizado síntesis hidrotermales de silicalila, analcima y cancrinita basándose en la metodología de Shimizu y Hamada (20'01). Como fuente de Si se han utilizado tubos de cuarzo de 26 mm

72 MATEO GONZÁLEZ, E. et a l .
---.-JI Muelle -1
I L Pieza de sellado
Tap¡l roscada -1 1-
1---:: I 7r-
Asiento
Tapa de tenón
de longitud y 1 0 Y 8 mm de diámetro externo e interno, respect ivamente ( 1 ,59 g), y como fuente de Al, láminas de alúmina (Rubal i t 708S, 96 % A1203) de dimensiones 25 x 4 x 0,5 mm (0,23 g). Respecto a las condiciones de las síntesis se ha trabajado a temperaturas de 200 y 220"C (obtenidas al introducir los autoclaves en el interior de una estura de convección forzada) y tiempos entre 7 y 46 días. La tabla 2 resume las condiciones de síntesis ut i l izadas para cada una de las zeolilas estudiadas en este trabajo.
-Cuerpo cilindrico
o Disc bas e
,.
1
l iJ
1
Cuerpo de tcnón
Caracterización de los cristales zeolíticos Di fracción de Rayos X
Figura 2. Carcasas metálicas, recubrimientos internos de tcnón y piezas constituyentes de los autoclaves empIcados en la síntesis de·zeolilas.
Los espectros de dirracción que se muestran en este trabajo se obtuvieron en un di fractómctro Phi l ips PW I 7 1 0, con monocromador de grafito, rendija automática y radiación CuKa 1 ,54 1 8 Á. El registro de los diagramas se ha realizado mediante el programa inform(¡tico POLVO (Martín Ramos, 1990). La difracción de Rayos X se ut i l izó para anali-
Tabla 2. Composiciones molares de los geles de síntesis, condiciones de síntesis y tamaños obtenidos en los distintos experimentos. (*) Gel recogido después una primera síntesis, tras ser filtrado.
r CristaJ i;,r.ación Zcolita Fuente I Gel Temperatura Tiempo Tamano (mm.)
("el (dias)
I ·nIAOH/HF/H:O 200 I 34 3,Ox l ,9 x l ,'1
I 24/38/1000 220 34 0,3 - t,O
Silicalita Si: Tubo de euar/.o I TPAOH/HI:IH2O 200 46 2,9:-.:2,Ox 1,5 10/1611000
Gel slnlcsis <Interior') 200 1 0 3,Ox2,Ox 1 ,5
NaOH/H¡O 200 7 1 ,2 - 2,0 Si: Tubo de euarLO 24/tOOO Analeima Al: Lá mina de alumina Gel síntesis anterior\'¡ 200 t O 1 ,2 - 2,0
Canerinita ¡ Si : Tubo d e cuarLO NaOHlH,O 200 7 25x28x75 plll. Al; 1.ámin¡¡ de ¡¡Iumina 75/1 000 ---

Síntesis y caracterización de monocristales de materiales zeolíticos 73
zar el producto obtenido en la síntesis de los cristales, comparándolo con los patrones.
Microscopía -Microscopfa óptica Las muestras se han estudiado mediante
estereomicroscopfa en un esteromicroscopio Leica MZ7j, y microscopía óptica de campo claro. campo oscuro y polarización por reflexión en un microscopio Olympus BX41.
-Microscopía electrónica de barrido (SEM)
Se utilizó un microscopio electrónico de barrido lEOL lSM-6400. Las muestras son previamente recubiertas por una delgada capa de grafito para mejorar el contraste, utilizando para ello una unidad de re'cubrimiento por evaporación en vacfo marca Balzers modelo MEO 010. Se obtuvieron imágenes de electrones secundarios, para estudiar aspectos morfológicos de los cristales y sus modos de agregación.
-Microscopía de fuerza atómica (AFM) Se obtuvieron imágenes de contraste de
fase en un equipo AFM modelo Nanoscope lIlA de Digital Instruments, en modo de contacto intermitente. para su comparación con imágenes equivalentes sobre nanorases de las mismas zeolilas.
Espectrometría de emisión alómica acoplada a plasma (lCP-AES)
Se ha utilizado un espectrógrafo de dispersión cruzada Echelle y un detector CID 38. de 16 mm2• Hemos utilizado esta técnica para analizar la composición de los geles tanto antes como después de la síntesis y así poder deducir el grado de disolución de las fuentes de Si y de Al.
Resullados
En general, la síntesis hidrotermal de zeolitas consta de varios pasos que convierten una solución sobresaturada en un compuesto cristalino microporoso. La composición de los geles precursores para la sfntesis de zeolilas
viene definida por relaciones molares. las cuales tienen una influencia fundamental sobre las características de la cristalización (nucleación y crecimiento cristalino).
Las emategias que se han seguido para producir monocristales zeolfticos conslan de los siguientes elementos: i) sustitución de las fuentes tfpicas solubles de Si (silicatos. sales de sílice. etc.) y de Al (alúmina en polvo, aluminato de sodio. etc.) por otros mucho menos solubles, como tubos de cuarzo para el Si y placas de a-alúmina para el Al (Shimizu y Hamada, 2001). Al controlar la disoluciÓn de las fuentes precursores se controla la sobresaturación y, por tanto, la velocidad de nucleación; ii) utilización de un medio fluoruro en el que se consigue una mineralización semejante a la lograda con los iones OH - a la vez que aumenta la velocidad de crecimiento y se logran cristales mayores (Moslowicz et al.. 1993); iii) subiendo la temperatura porencima de la habitual ( 1 70-1800C) para aumentar también la cinética del crecimiento cristalino.
Si licalita Después de 34 días de cristalización a
2000C (TPAOH/HFlHp=24/3811000) se obtienen cristales tanto en la superficie del tubo de cuarzo como en las paredes y el fondo del autoclave. Todos ellos presentan la típica morfología en forma de ataúd de la silicalita y se identifican como silicalita (www.izastructure.org/databases) por Difracción de Rayos X de polvo (Figura 3). Los tamaños de los cristales obtenidos son variables, con valores máximos de 3.0 x 1.9 x 1,4 mm. Algunos de estos cristales se encuentran maclados y zonados y, en ocasiones. una de las caras presenta escalones de crecimiento, debido probablemente a efectos de anisotropía en el flujo de nutrientes.
Al aumentar la temperatura hasta 2200C, manteniendo las condiciones anteriores, los cristales presenlan aristas menos marcadas y tamaños menores que oscilan entre 0,3 y 1,0

74 MATEO GONZÁLEZ, E. et a l .
mm. Es decir, un aumento de 20°C en la temperatura no ofrece u n mejor resultado, como se hubiera esperado, lo que podría deberse a los cambios producidos en l a composición del medio de reacción .
Si se cambia l a relación molar del gel (TPAOI-IIHF/H,O= I 0/ 1 6/ 1 000) Y se aumenta el número de días hasta 46, manteniendo l a temperatura a 20Qoe, se obtienen cristales de s i l ical i ta idiomorfos con tamaños parecidos a los anteriores.
Al realizar una segunda síntesis con el gel y las condiciones descritas anteriormente, y
añadiendo nuevos tubos de cuarzo y algunos de los cristales de las síntesis previas, se ob-
t:l « t:l .... Ul Z w .... Z ....
o 10 20 30
serva que el creci m iento no contintla sino que estos cristales sirven de núcleo para nuevos cristales e incluso, en algún caso, se observan procesos de disolución. Por el contrario, si realizamos esta segunda síntesis con el gel resultante tras la primera síntesis, el cristal sigue crec iendo; se observan nuevos escalones de crecimiento (Figura 4.A y B). Aunque en estas condiciones, también se observan nuevos cristales esferoidales de 20 ).1111 que se desarrol lan sobre el cristal previo, son más escasos que en las síntesis con geles frescos.
Al analizar mediante lep el gel fresco y
las disoluciones tras l a primera y l a segunda síntesis, se ve que el contenido en Si va au-
40 20
50 60 70 80
Figura 3. Difracción de rayos X de los cristales de silicalita obtenidos.
Figura 4.A) Imagen SEM donde se observ.m escalones de lluevo crecimiento. B) Imagen SEM, detalle de la figura A.

Síntesis y caracterización de monocristales de materiales zeolíticos 75
mentando con las síntesis y que la disolución va adquiriendo un carácter menos ácido, aunque s in l l egar a ser básico (Tabla 3) . Esto está de acuerdo en l a "observación general de que el pl-I para l a síntesis de s i l ical ita puede ser mucho más bajos en medios F- , ya que estos iones actúan también como agentes mineralizantes (Aiel lo et a l . , 1 999).
Microscopía de fl/erza mómictl Los cristales de s i l ica l i ta se han ut i l izado
para comparar tamaños de poro de partículas de 150 nm y monocristales. Con el propósito de estudiar las et.apas iniciales del mecanismo de formación de capas zeo l i t i las, se prepararó zeolita tipo MFI sobre soportes de a-al úmina mediante síntesi� hidrotennal in-situ. En este caso, con un gel de composición molar: TPAOHrrEOS/H,o= 0,32/ 1 1 1 65 (Wang y Yan, 200 1 ) , a 1 65°C durante 2 horas. Las m uestras se lavaron con agua desionizada después de la síntesis y se calcinaron a 480uC durante 8 horas para eliminar el agente estrucluranle, con rampas de calentamiento y enfriamiento de 0,5 y 1 °C/mi n . , respectivamente.
Tras estudios microscópicos, se observa una capa de cristales de s i l ica l i ta formada sobre un sustrato de a lúmina , con orientación en la dirección [0 1 0] . Observando la superficie de estas capas mediante un AFM se detectan partículas esféricas de unos I SO n m sobre los cristales de s i l ical i ta (Figura 5) . U l i l i zando un monocristal de s i l ical i ta como patrón se
Tabla 3. Determinación de Si (ICP) y de pH de los geles precursores de s i l ical ita.
r Sil icalita -
I Concentración Si (mglL) r pH fG"'cI fresco 1 I 1,5
Gcl I1 sintcsis I 659,33 f s.s Gcll" Sinlcsis I 692,09 I 5,5
compararon las estructuras porosas de las partículas con las de un cristal grande confirmando que corresponde al mismo material . Las distancias entre centros de poros' diagonales (promedio ele 1 0 medidas) son de' 0,7 1 ± 0,03 n!TI en el cristal grande y 0,54 ± 0,06 nm en las partícu las de 150 nm, siendo 0,68 nm para la estructura MFI . De esta forma, al comprobar que estas partículas son núcleos zeolílicos, deducimos que en la formación de capas soportadas es importante l a nucleación sobre e l sustrato y los cristales formados, no sólo en el seno de la disolución.
Ana/ci/l/a y ctlncril/itn Cuando se ut i l iza un tubo de cuarzo como
fuente de S i y una lúmina de a lúmina como fuente de Al, se obtienen distintas zeolitas (analcima o cnllcrinita) en función de la relación molar del gel .
S i se ut i l iza l a relación molar NaOH/ H,o=24/ 1 000 Y se rea l iza la síntesis a 200·C durante 7 días, se obtienen cristales de analcima con simetría 2/m3y morfologías de trapezoedro (hkk). Estos cristales, pulverizados, han sido anal izados por difracción de Rayos X coinci-
Figura 5. Imagen AFM. fase. rango Z 100 cleg.

76 MATEO GONZÁLEZ, E. el a l .
el " el � VI Z w .... Z �
o 10 20 30 40 20
50 60 70 80
Figura 6. Difracción de Rayos X de los cristales de 'lIlalcima obtenidos.
diendo sus picos con los del patrón de analcima (Figura 6). En estas síntesis, el tubo de cuarzo se d isuelve completamente, debido a que las condiciones son muy b;:ísicas (pl -l= 12,7), mien· tras que la lámina de alúmina sirve de soporte para los nuevos cristales, cuyos tamaños oscilan entre 1 ,2 y 2.0 �lm.
Si se real iza una segunda síntesis con el gel sobrante de la primera, se forma sobre los monocristales anteriores tina segunda generación de crislales de unos 40 �lIn (Figura 7) . Al analizar los geles de síntesis. antes y después de las sín tesis de analc ima, se observa que el contenido de Al va dismi nuyendo con las síntesis, mientras que cl contcnido en Si va au-
Figura 7. Imagen SEM de un cristal de analcima con ulla segunda generación tle cristales.
mentando. El medio cada vez se hace menos básico (Tabla 4).
En cambio, al ut i l izar la relación mo lar NaOHIH,O=75/l 000, se obtienen agregados de cristales fibrosos con base hexagonal. Su <In.í1 isis por d ifracción de Rayos X, permite concluir que se trata de cancrinita (Figura 8) . Los tamaños de estos cristales son, aproximad'lmente, de 2S x 28 x 7S ¡..tm.
Conclusiones
El ajuste de las condiciones de síntcsis permite controlar las condiciones de nucleación y crecimiento cristal i no, de modo que se puedan producir nuevos cristales de zcolitas de d imensiones m i l imétricas. De este modo, monocristales id iomorfos de s i l ica l i ta con tamaños de 3,0 x 1 ,9 x 1 ,4 mm han sido si ntetizados, tras una segunda síntesis con el gel sobrante de l a síntesis anterior, se observa como predomina el mecanismo de crecimiento frente al de Illlcleación. También se han si ntetizado cristales de analcima de 2 mm y de cancrinita de 75 )lm controlando l a cinética de crecimiento.

Sfntesis y caracterización de monocristales de materiales zeolíticos 17
Tabla 4. Determinación de Al y Si (lCP) y de pH de los geles precursores de analcima.
¡-Anaicirñl' -r Contenncl6n Aí(ñ1g1L) � Coneenuwci6n Si (iñ¡/L) ! . pH ..
I Gel (rCiCo . ' 1 u - - � . . .. , •• -- � ;' 12.70 ' , � I
fdciT"iIñ,esiJ j 6lji I
11914'· 'r 1 1 .49 -
r GcÍi" iiñi"csis I 46}6- 15'912 T I I.30 -
- -_.�-�---.. -
o 10 20 30 40 20
so 60 70 80
Figura 8. Dirracción de Rayos X de los cristales de cancrinita obtenidos.
Bibliografia
AieJlo. R .• Crea, F., Nigro, E .• Testa. F . • Mostowicz. R . • Fonseca. A. y Nagy, LB. (1999): Synthesis or large single crystals or (he large-poro aluminophosphate molecular sieve VP¡-5. Micropor. Mesopor. Mat . • 28. 241-259.
Auerbach. S.M., Carrado. K.A. y Duna. P.K. (2003): Handbook or zeolite science and lechnology. Marcel Dekker ed .• New York. 1 184 p.
Bibby. D.M. Y Dale. M.P. (1985): Synthesis or silica-sodalite rrom non�aqueous syslem. Nature, 317, 157-158.
Charnell. J.F. (1971): Gel growlh or large cryslal or sodium A and sodium X zeoliles. J. Crysl. Growth. 8. 291 -294.
Cundy. C.S. y Cox. P.A. (2003): The hydrolhermal synthesis orzeoliles: hislory
and developmenl from Ihe earliest days 10 Ihe present time. Chem. Rev .• 103. 663-70l .
Davis. M.E. (2002): Ordered porous materials ror emerging applications. Nature. 417. 813-82l .
Garda. J. (2000): Sfntesis y caracterización de zeolilas y materiales compuestos zeolila!carbón. Aplicaciones para la eliminación de S02. Tesis Doctoral. Universidad de Alicante (Espai\a).
Gao. F .• Zhu. G .• Li. X . • Li. B., Terasaki. O. y Qiu. S. (2001): Synthesis or a high-quality host material: zeolile MFI giant single crystal from monocrystalline siticon si ice. J. Phys. Chem. B., 105. 12704-12708.
Kuperman. A . • Nadami. S., Oliver, S .• Ozin, G., y Garcés. J.M. (1993): Non-aqueous synthesis or giant cryslals or zeolites and molecular sieves. Nature, 365, 239-242.

78 MATEO GONZÁLEZ, E. el al.
Martín Ramos. D. (1990): Un programa e interfase inteligente para control del difract6metro PHILPIS PW 1710 que incluye análisis de perfil de linca y determinación de fases de mezclas policrislalinas. Dep. Leg. M-11719.
Meier, W.M. y 0lson. D. H. (1992): AlIas of zeolite struclure types. ButtcrworthHeineman ed., USA, 200 p.
Moslowicz. R., Crea, F. y Nagy, J.B. (1993): Crystallization of silicalite-l in Ihe presence of fluoride ions. Zeolilcs, 13, 678-684.
Shimizu, S. y Hamada, H. (2001): Synthesis
of gillnt zeolite cryslals by a bulk material dissolution technique. Micropor. Mesopor. Mal.. 48. 39-46.
Sun. Y .. Song, T .. Qiu, S .. Shen. W.P. y Yue. DJ. (1995): Synthesis of mordenitc single cryslals using tWQ silica sources. Zeolites, 15, 745-753.
Wang, Z. y Yan, Y. (2001): Oriented zeolile MFI monolayer films on melal substrates by in Silu crystallizalion. Micropor. Mesopor. Mat., 48, 229-238.
Recibido: Noviembre 2003 Aceptado: Diciembre 2003

Boletíll de la Sociedad Espmiola de Milleralogía, 26 (2003), 79-92 79
Inclusiones fluidas en venas auríferas del yacimiento Pau
a-Pique, suroeste del Cantón Amazónico, Brasil
Genova M. PULZ'; Elzio S. BARBOZN; Francisco E. PINHOl; Luis H. RONCHI4; Andréa R. JELlNEKI. Lauren da C. DUARTEl 1 . Centro de Estudos em Petrologia e Geoquímica, Instituto de Geociencias. Universidade Federal do Rio Grande do Sul. Porto Alegre. Rio Grande do Sul. Brasil. Caixa Postal 1500!. Cep: 91501-970 E-mail: [email protected] 2. Programa de P6s-Graduar;ao em Geociéncias. Instituto de Geociencias, Universidade Federal do Rio Grande do Sul, Porto Alegre, Rio Grande do Sul. Brasil. Caixa Postal 15001. Cep: 91501-970 E-mail: [email protected]. [email protected] 3. Curso de Geologia. Institulo de Ciencias Exatas e da Terra. Universidade Federal do Mato Grosso. Cuiabá. Estado do Mato Grosso. Brasil. E-mail: [email protected] 4. Programa de P6s-Graduar;3o em Geociencias. Universidade do Vale do Rio dos Sinos. Sao Leopoldo. RS, Caixa Postal 275, Cep 93022-000 [email protected]
Abslracl: Gold ore from Pau-a-Pique deposit is associaled to quartz veins hosted in Prolerozoic melaconglomerale (Aguapcf Group) in soulhwcslern of Amazonic eralon (Bralil). Quartz veins fill fractures (R. R', D and T), which are related to regional ruptil-ductil deformalion. $hear zones crosscul the metassediments of Aguapeí Group. Four mineralization slages \Vere identified: earl)' (p)'rrholile. chalcop)'rite and galena), inlermediate (pyrile 1. native gold (1) and native sil ver (1» , late (pyrile 11, native gold and nalive silver) and weathered (hematite, magnetile, martile). Petrographic and nuid inclusion sludies were carrying out to aim characteriled Ihe ore solutions trappcd in quartz veins. Threc populations of Ouid inclusions \Vere recognised: Typc l comprises triphasic aquo-carbonic inclusions. The salinit)' of majorit)' of inclusions this population is situated betwcen 5 ano 6 % weight eq. NaC!, and homogenizalion temperature varies betlVeen J90 and 230 oc. T)'pe 2 consisls in biphasic aqueous inc1usions. wilh salinit)' values concenlrated mainl)' in Ihe interval between 160 and 180 OC. T)'pe 3 inclusions are represcnted b)' monophasic inclusions of di minute sile « I mm). The association of native gold grains and l)'pe 2 inc1usions suggcsI Ihal Ihe gold deposition is related to pcrcolation of aqueous solutions in the late stages of hydrothermul alteration. In aooition. microthermometric data indicate Ihat golo deposition in the ore struelures is related to boiling as well as decreasing of temperature and pressure of hydrothermal s)'stem. The results show Ihat Pau-a-Pique deposil is an e;ll;ample of epigenetic gold mineralization hosleo in Ihe Amazonic Craton.
Key words: Fluid inelusion, gold, mesothermal. metaconglomerate, Brazil.
Resumen: La mineralización del oro en el yacimiento Pau-a-Pique esta asociada con venas de cuarzo en milonitas de metaconglomerados protero7.0icos en el suroeste del Cratón Amazónico (Brasil). La orientación de las venas mineralizadas es controlada por fracturas (R, R', D Y T), relacionadas con el cizallamiento regional frágil-dúctil que corla los metasedimentos del Grupo Aguapef. La orden de sucesión de los minerales metálicos permite el reconocimienlO de diferentes etapas de mineralización: temprana (piTTotina, calcopirita y galena). intermediaria (pi rila 1. oro nativo (iJ y plata nativa (¿n, tardfa (pirita 1 1 , oro nativo )' plata nativa) y intempcrica (hemalita. magnetita )' martita). La pelrografía )' la

80 PULZ. G. M. el al.
microtermometrfa de las inclusiones nuidas atrapadas en las venas mineralizadas fueron las técnicas empleadas para caracterizar los fluidos que acampanaron la precipitación del oro en el yacimiento eSlUdiado. Tres familias de inclusiones fluidas fueron identificadas. Tipo 1 : inclusiones trifásicas acuoso-carbónicas; la salinidad de la mayoria de las inclusiones de esta familia es de 5 y 6 % peso eq. de NaCI y la temperatura de homogenizaci6n varia entre 190 y 230 oc. Tipo 2: inclusiones bifásicas acuosas, con salinidad entre 4 y 8 % peso eq. de NaCl y temperatura de homoge.nización entre 160 y 180 OC. La asociación del oro nativo con inclusiones del tipo 2 en fracturas sugiere que la deposición del oro puede ser asociada a la infiltración de soluciones acuosas en etapas tardras. Tipo 3: inclusiones monofásicas secundarias de tamano inferior a 1 mm. Los resultados microtermométricos sugieren que la deposición de oro nativo en las venas de cuarzo está relacionada con la ebullición y disminución de la temperatura y presión del sistema hidrotermaJ. En conjunto, los dados sugieren que el yacimiento Pau-a· Pique constituye un ejemplo de mineralización del oro cpigcnélico en el Cralón Amazónico.
Palabras dave: Inclusiones fluidas, oro, metaconglomerado, Cratón Amazónico, Brasil.
Introducción
El Cratón Amazónico se distingue en el territorio brasilero por su potencial metalogenético representado por importantes mineralizaciones económicas de oro, hierro y cobre conforme registrada en la revisión de Schobbenhaus y Coelho (1988). Según el Ministerio de Minas y Energía de Brasil, en el período de 1990 a 1995 fueron extraídas aproximadamente 33 millones de toneladas de minério. con tenor de 0.41 ppm de oro en esta región (Anuário Mineral, 1997).
En el limite suroeste del Cratón Amazónico se localiza la Provincia Aurífera Alto Guaporé. la cual aloja innumerables prospectos de oro en metaconglomerados del Grupo Aguapeí, tales como el yacimiento Pau-aPique. entre otros (Fig. 1) .
En la década de los 80. varios prospectos de oro alojadas en metaconglomerados del Grupo Aguapeí localizados en el limite SW del Cratón Amazónico. tales como Mina Sao Vicente. Ruínas Sao Francisco. Rio Sararé, lncra, Córrego da On�a, Lavrinha, Bananal-Rio Ale· gre y Pau-a-Pique (Fig. 1). A pesar de la importancia económica de estos prospectos hay pocos estudios detallados sobre cada yacimiento. en especial. sobre inclusiones fluidas.
Este estudio presenta los resultados de los ensayos microtermométricos de las inclusiones fluidas atrapadas en las venas audfe-
ras del yacimiento Pau-a-Pique, con el objetivo de contribuir en la caracterizaciÓn del fluido hidrotermal relacionado con la mineralización de oro.
Contexto geológico
El yacimiento Pau-a-Pique se encuentra en la zona centro-oeste de lo Brasil a unos 440 km de Cuiabá (capital del Estado de Mato Grosso). Este yacimiento está alojado en el contexto del Grupo Aguapeí en la región suroeste del erutÓn Amazónico. La región engloba terrenos proterozoicos de 1.33 a 0.99 Oa. conforme n estimaciones U-Pb (SHRIMP) en circón y Pb-Pb en roca total (Santos. 1999; Tassinari y Macambira, 1999).
La porción suroeste del Cratón Amazónico fue afectada por la tectÓnica distensiva del Proterozoico, resultando en ambientes de rilr continental con la formación de aulacógeno (Carneiro et al., 1992). En el interior del aulacógeno fueron depositados sedimentos terrígenos y marinos (Litherland et al., 1985, 1989) de los grupos Aguapeí (en Brasil) y Sunsb (en Bolivia). Datos K·Ar de filosilicatos del Grupo Aguapeí indican que la inversión del proceso de abertura del sistema de rift ocurrió entre 1000 y 900 Ma durante la Oro génesis Sunsás (Geraldes, 2000. Litherland et al., 1989).

60'
10
60' __ 16'
480m
o
Inclusiones fluidas en venas auríferas , cr{¡ton a mazónico.
'-
�aaal S I /I:( uc O
Región del yacimiento Pau-a-Pique
1 000 km
I,.j'f.; I Halo h idrotennal y venas de cuarzo
� Fallas inversas L.é-J D Grupo Aguapcí
ITIIIIll Rocas vulcano-sedimcntares
l' ,>S:IGranito-gncises Santa Helena
3 km -----------+. Y'
8 1
Figura 1 - Mapa geológico del suroeste del Cratón Amazónico destacando l a localización de los yacimientos de oro en el contexto regional del Grupo Aguapeí. A) Mina S50 V icente . B) Ruínas S50 Francisco. C) Río Sararé. O) Inera. E) Córrcgo da 011l;a . F) Lavrinha. G) Bananal-Rio Alegre. 1-1) PalHlPique.

82 PULZ, G. M . et a l .
La base del Grupo Aguapcí eSl;:í formada por mctaconglomerados ol igomícticos con i n terca lac iones de metarenitas orlocuarcít icas de la Formación Fortuna (Saes y Fragoso Cesar, 1 994). Esta unidad está sobrepuesta por l a Formación Vale da Promissao, la cua l est:í const i tu ida por metal i moni tas, pizarras, fi l i las y mctarenitas finas. Estas rocas hac ia para la Formación Morro Crista l i no por i n lcrcl ig i taciones de mcta l imonitas , metarenitas y metacanglornerados o l igomícticos.
Las rocas del Grupo Aguapeí cabalgaron sobre los litolip05 del basamento durante la Orogénesis SUllsás. El basamento del Grupo Aguapeí esti.Í representado por los granitosgneises Santa Helena en el tírea estudiada. Determinaciones radiométricas Rb-Sr en estas ro-
cas proporcionaron valores entre 1 308 ± 1 3 M a y 1 3 1 8 ± 24 M a (Menezes et a l . , 1993).
Se observa en el frente de exploración del yacimiento Pau-a-Pique, que l a Formación Fortuna está cruzada por fajas m i l oníticas, Ins cuales están relacionadas con la deformación progres iva i mpuesta por l a Zona de Cizal lam ienlO Corredor (Fernandes el a ! . , 1 998). Esta estructura tectónica coloca los met<lsedimcnlos de la Formación Fortuna en contacto con los granito-gneises Santa Helena. Las m i lon i tas de la Formación Fortuna también est¡ín en contacto con una roca tonalítica, transformada en m i lonita con anfíbol-bioti taclorita (Fig. 2) .
Fajas irregulares, de espesor centi l11etrico, de s i l icificación y moscotizaci6n han sido observadas adyacentes a algunas venas de
50 In
I:s:! VenAS de cu:m;o
D MCIn.conglomc fado (Frn. Fonul1:l)
O MilonitA con nnfibo\·biotitn.·c!orila
� Curvas de nivcJ
o Lugo
FigUf<l 2 - Esbozo geológico del yacimiento Pau-a-Pique, destacando la locnlización de las venas estudiadas para el estudio dc inclusioncs nuidas.

Inclusiones fluidas en venas auríferas, cráton amazónico . . . 83
cuarzo. Por otra parte, al rededor de las estructuras mineralizadas predominan diseminaciones de sulfuros, óxidos de hierro, turmalina, biotita. clorita y epidOla. Los sulfuros también aparecen diseminados junto al contacto del metaconglomerado mi[onitizado y [a milonita con anfíbol-biotitaclorita. Determinaciones K-Ar en mica blanca y Pb-Pb en galena, de algunos indicios de oro en el Grupo Aguapd, sugieren que el evento mineralizante en la región estudiada ocurrió entre 964 ± 42 a 9 1 8 ± 10 Ma (Geraldes et al., 1997) durante [a Orogénesis Sunsás.
Procedimiento analítico
El estudio microtermométrico se ha efectuado en inclusiones fluidas atrapadas en cuarzo de venas con una platina Chaixmeca� acopiada al microscopio petrográfico Zeiss� utilizando objetivos con aumentos de 25 e 50 veces y oculares de 10 veces. Las muestras fueron enfriadas a - 1 90 oC con nitrógeno [íquido (N1) y calentadas hasta +400 oC controladas con un sistema digital EurotermOl• El aparato fue calibrado con una inclusión natural acuoso-carbónica trifásica (-56,6 OC), agua desmineralizada (O oC) y productos sintéticos MerckOl• La reproducibilidad de las medidas se ha calculado en +0.2 ·C. La salinidad de las inclusiones acuosas se ha calculado por la temperatura de fusión del hielo. conforme el procedimiento sugerido por Bodnar y Vitik ( 1994). La densidad de las inclusiones acuosas se ha determinada según Shepherd et al. ( 1985). La salinidad y la densidad de las inclusiones acuoso-carbónicas fueron estimadas por el método de Brown y Hagemann ( 1994).
Las texturas de los minerales metálicos fueron estudiadas con el microscopio petrográfico ZeissOl de luz transmitida. Las observaciones petrográficas fueron complementadas por imágenes de microscopio electrónico de barrido y análisis de espectrometría de dispersión de energía con el equipamiento
Jeol Scan Microscope 5800� del Centro de Microscopía Electrónica, Universidad Federal de lo Rio Grande do Sul (UFRGS). Los análisis semicuantitativos de EDS se han obtenidas con unas condiciones instrumentales de 25 keV. I nA de excitación. 1 0 ¡lm de ancho del canal y ángulo de 35°.
Las composiciones de los sulfuros se han obtenido con una Microsonda CamecaOl Sx50. Las condiciones experimentales han sido las siguientes: haz de electrones de 5 ]..lm, tensión de 3celeración de 20 keV. corriente de 25 nA y tiempo de contaje de 20 a 30 segundos. El equipamiento fue calibrado con CuFeS? natural; ZnS sintética; As metálico; Te sintético en un compuesto de Au. Ag y Te; Ag sintética en un compuesto de Ag, Bi Y Se. Los análisis del microsonda electrónica y microtermométricos fueron realizados en el Centro de Estudios en Petrología y Geoquímica (Instituto de Geociencias. UFRGS).
Características de la mineralización
En el yacimiento Pau-a-Pique aparecen principalmente venas rellenando fracturas R y D (Fig. 3), según la cl3sificación de Tchalenko ( 1968). Además, algunas venas rellenando fracturas R ' Y T han sido observadas en la área del yacimiento estudiado. Adyacente a las ven3S, se puede observar. en general, una estrecha banda micácea paralela a la foliación milonítica. Todos los tipos estructurales de venas se encuentran boudinados 3 lo largo de su dirección y el espesor de las venas v3TÍ3 desde milímetros hasta 2 metros.
Las venas D se posicionan subparalelas (O a 5°) a los planos de cizalla según N15-50W/55-83 SW en los metaconglomerados, y según N lOW/60-66 SW o N42W/80 NE en las milonitas con anfíbol-biolita-cloTÍta. Estas venas están rellenas de cuarzo con texturas masiva. ribbOI1 o sacaroideo, conforme la nomenclatura de Dowling y Morrison ( 1989).
Las venas R forman un ángulo que varía

84 PULZ. G. M. el al.
de 5 a 2S· con la foliación milonítica, según NI5·50W/ 66·86 SW. Estas venas están rellenas por drusas de cuarzo incoloro. que confieren a la mineralización una textura en peine. A veces, diferentes tipos Icxlurales aparecen a lo largo de la extensión de una misma vena resultando en texturas compuestas (tipo ribbon-sacaroideo y masivasacaroideo), probablemente, debido a repelición de los episodios de apertura y relleno de las fracturas R.
Venas R' seccionan los planos e con un ángulo de 6S a 90" con los planos C. Estas venas están orientadas según N20WI 60-75 SW en el anrrbol-biotita-clorila milonita. El cuarzo que rellena estas estructuras presenta textura masiva.
Las venas T son estructuras dilatacionales que rellenan fracturas de extensión. Estas venas forman un ángulo de 40 a 60" con la foliación milonítica. El cuarzo sacaroidal rellena parcialmente estas estructuras formando venas discontinuas, según N40W/ 80-86 SW.
Los minerales del yacimiento están diseminados en las venas y en el halo de alteración. La composición y relación de texturas de los minerales metálicos (Tab. 1 , Fig. 4). en conjunto con los resultados microlermométricos de las inclusiones fluidas. posibilitaron el reconocimiento de cuatro etapas de
@
mineralización en el yacimiento Pau-a-Pique (Tab. 2), los cuales son:
Etapa temprana: pirrotina, calcopirita y galena.
Etapa intermediaria: pirita-I, oro nativo (1) y plata nativa (1).
Etapa tardía: pirita-JI, oro nativo y plata nativa.
Etapa intemperica: hematita. magnetita y martita.
La pirita es el sulfuro predominante en la mineralización (Barbaza, 2001), en cual se presenta en dos variedades texturales. La pirita-I es sintectónica, como indica su textura cataclástica, mientras la pirita-U se presenta en cubos perfectos, exentos de textUrllS de deformación, indicando que su crecimiento ocurrió al final del cizallamiento. El oro nativo (Fig. 4) se encuentra asociado a la pirita-!. La plata nativa ocurre incluida o rellenando fracturas en la pirita-lI.
La calcopirita y la pirrotina monoclfnica aparecen incluidas en la pirita-J. La galena, que se presente en pequeñas cantidades en el yacimiento. sobre la forma de inclusiones en la pirita-J!. Los análisis de Au. Zn y As en los diferentes sul furos analizados se sitúan próximos al límite de detección por microsonda.
La hematites es el óxido más abundante en el yacimiento. Este mineral sustituye los
NE sw
Figura 3 - A) Modelo teórico de la orientación de las fracturas en "lonas de cizallamiento rúptil-dúctil (Harris (988). B) Croquis del perfil N80E en la región del yacimiento pau-a-Pique destacando las venas de cuarzo en rracturas R. O Y T.
Inclusiones fluidas en venas auríferas, cráton amazónico . . . 85
Tabla 1- Composición representativa de los sulfuros del yacimiento Pau-a-Pique obtenidos por microsonda electrónica. A) pirrotinn (Po). B) calcopirita (Cpy). C) pirita-I (Py). (nd: no detectado).
A e Grano P04 Po5 Po6 Po7 Po8 P09 PolO Grano Pl:I Pl:2 Pl:3
Fe 60,S7 S9,7660,71 60,41 60,3660,7261, 17 F. 46,S6 47,44 47,01 Cu 0,0\ 0,03 0,03 0,01 0,02 0,21 0,2 Cu 0,03 od od S 39,5339,43 39,5 40,0139,5739,1539,01 S 54,05 53,9 54,45
Au 0, 1 1 od od od 0,07 od od Au od od Nd As 0,05 0,03 od od od 0,1 0,12 As 0,1 od 0,07 Zo od 0,01 od od od 0,05 0,1 Zo 0,01 0,03 Nd
Proporción atómica Proporción atómica Fe 46,77 46,S I 46,8646,4346,6746,9847 ,2S F. 33,06 33,55 33,13 Cu 0,01 0,03 0,02 0,01 0,02 O,IS 0,14 Cu 0,02 0,03 0,01 S S3,I6 S3,44 S3,I I 53,56 S3,29 52,77 S2,48 S 66,85 66,4 66,83
Au 0,03 od od od 0,02 od od Au od od od As 0,03 0,02 od od od 0,06 0,07 As 0,06 od 0,04 Zo od 0,01 od od od 0,03 0,07 Zo 0,01 0,02 od
B Grano CEl:l l CEl:12Ce:z:13 Cel:14Cel:ISCEl:16Ce:z:17Ce:z:18Ce:z:19Ce:z:20Ce:z:21 Ce:z:22
Fe 30,04 30,45 30,45 30,34 30,93 30,34 30,93 30,22 30,S I 30,43 30,72 30,45 Cu 34,43 34,48 34,24 34,79 34,OS 34,79 34,05 34,31 34,45 34,68 34,31 34,57 S 35,69 36,09 34,81 35,31 35,45 35,31 35,45 34,75 35,33 35,37 35,47 35,2
Au od 0, 1 1 0,09 od od od od od 0,14 0,04 od 0,05 As od 0,04 od 0,01 0,03 0,01 0,03 0,01 0,02 od od od Zo od 0,03 od od od od od od 0,01 0,01 od 0,02
Proporción atómica F. 24,53 24,61 25,12 24,78 25,22 24,78 25,22 24,99 24,93 24,83 25,04 24,92
Cu 24,71 24,49 24,83 24,97 24,41 24,97 24,41 24,94 24,74 24,87 24,58 24,67 S 50,76 50,81 50,02 50,24 50,35 50,24 50,35 50,06 50,27 50,27 50,37 50,17
Au od 0,03 0,02 od od od od od 0,03 0,01 od 0,01 As od 0,03 od 0,01 0,02 0,01 0,02 0,01 0,02 od od od Zo "d 0,03 nd nd nd nd nd nd 0,01 0.01 Nd 0,02

86 PULZ, G. M . el a l .
su l furos como resultado de l a alteración supergénica y se encuentra también como una fina película cubriendo los cristales de cuarzo (Fig. 4), a veces formando concreciones esferoidales. La magnetita aparece intercrecida con la hematita formando martita. que también sustituye los su l furos.
Características y tipos de inclusiones tlllidas
Solamente las inc lusiones nuidas que ocurren en las venas R , D Y T de la corta principal del yacimiento Pau-a-Piquc fueron investigadas en este estudio. Una característica co-
l11ún a lodas las venas es l a concentración de las inclusiones fl uidas en agrupaciones planeares, lo que permite considerarlas secundarias, conforme a la c l as i ficación de Roedder ( 1 979). También, las evidencias de crepitación y estrangu lamiento son comunes en bs inclusiones de todas las venas estudiadas (I?arboza el a l . , 2000). El número de fases de las inclus iones fluidas en temperatura ambiente y su distribución en cuarzo posibilitaron l a ident iricación de tres variedades pctrogr{¡ficas de inclusiones (Fig. 5). Una síntesis de los resultados de los experimentos son mostradas en la tabla 3 .
Figura 4 - Imágenes electrónica de barrido (BSE) de los minerales Illct{lIicos e n e l yacimiento Pau-aPique. A) Oro nativo en ffrlcturas en la pirita-! . B) Agregados ovoides de hcmatita sobre drusas de cuarzo en peine. C) Magnetita cuhédrica. O) Microcristalcs de galena incluida en la pirita-!.
Inclusiones fluidas en venas aurlferas, cráton amazónico . . . 87
Tabla 2- Características de las etapas de mineralización del yacimiento Pau-a-Pique (Po: pirrotina; Cpy: calcopirita; Pyl: pirita-l; Py!I: pirita-tI; Au: oro nativo; Ga: galena; Hem: hematites; Mar: martita).
Etapa IDclusioDes MiDerales
Fluidas Metálicos VeDas
Acuoso-carbónicas Po R-R' Temprana ,
y acuosas Cpy , Compuestas _ . ..
Pyl R-R'-D
Intennediaria Acuoso-carbónicas Au I
Ribbon y acuosas Ag Sacaroideo
Ga I Compuestas - __ o ._-. . - - _. -�._- - . ----
Au D-T Tardla Acuosas Ag Masiva
Pyll Sacaroideo __ l�.!'!�uestas
, Intempérica Monofásicas Hem
I En peine I Mar I
Tabla 3 - Resumen de los resultados microtermométricos obtenidos en las inclusiones acuoso-carbónicas (Tipo 1) y acuosas (Tipo 2) de las venas de cuarzo del yacimiento Pau-aPique. Th: temperatura de homogeneización total; TMH10: temperatura de fusión del hielo; TMCO,: temperatura de fusión del COl; ThC01: temperatura de homogeneización del COl; Tdclat: temperatura de disolución del c1atrato.
Th (OC) TMH,O (oC) TMCO, (oC) ThCO, (oC) Tdclat (oC)
TIPO ] 153 - 343
-6,6 - -0,6 -58 - -56,6
1 5 - 3 1 6,4 - 8,4
IiruLl: inclusiones trifásicas compuestas por una fase carbónica vapor (V) envuelta por dos Ifquidos inmiscibles, denominados L I (acuoso) y L2 (carbónico), respectivamente (Fig. 5). Presentan formas variables desde cristal negativo a irregulares, con dimensiones entre 4 e 20 J.lm. La fase carbónica (L2+ V) ocupa de 15 a 55 % del volumen total de la
TIPO ] 98,4 - 396
-19,0 - -0,3
inclusión. Este tipo de inclusión se observa a lo largo de hileras. a veces asociada con inclusiones de tipo 2 y 3. Las inClusiones acuoso-carbónicas del tipo 1 son aquí consideradas precoces, pues fueron encontradas en las venas R, que son las primeras en formarse durante la evolución del cizalla miento (Ramsay y Huber. 1983).

88 PULZ, G. M . et a l .
Durante los ensayos microlermométricos, la mayoría de las inclus iones acuoso-carbónicas del tipo 1 mostraron la fusión del CO") entre -57,0 y -56,8 oC, indicando que la fas�
carbónica no es pura (Fig. 6). No obstante, la fusión del CO2 sólido de algunas inclusiones del tipo 1 ocurrió en -56,6 oC, demostrando la existencia de fluidos acuoso-carbónicos puros en las mismas venas. La moda de la disolución del clatrato en las inclusiones acuoso-carbónicas fue observada entre 7,2 y 7,4 oC, lo que implica salinidad de 5 a 6 % peso equivalente
f"':Tipo 3 -15 um
NaCI (Tab. 3) . La homogeneización de l ca, (tanto en l a fase líquida, como vapor) ocurriÓ
en proporciones equivalentes, predominantemente entre 25 y 29 oC. La homogeneización total de las inclusiones acuoso-carbónicas ( fase líquida o vapor) eSli:í comprendida, principalmente, entre 1 90 y 230 "C (Fig. 6).
Tipo 2 : inc lusiones bifásicas acuosas (líquido + vapor), con d imensiones de 4 a 35 �lln, presentándose ora como cristales negativos, ora irregulares O alongadas (Fig. 5). El
. (
• •
Figura 5 - Inclusiones fluidas en el cuarzo de las venas del yacimiento Pau-a-Pique. A) Representación esquemática de la clasificación de las inclusiones adoptada en este estudio, considerando el modo de ocurrencia de las inclusiones cn relación con la orientación de las fracturas. B) FOlomicrografin de una inclusión trirúsic<1 acuoso-carbónica. (L 1 : H,O líquido, L2: CO� líquido y V: CO� vapor). C) Fotomicrografia de inclusiones bifásicas acuosas. D) f-olomicrografia de incltisioncs monofásicas vapor.
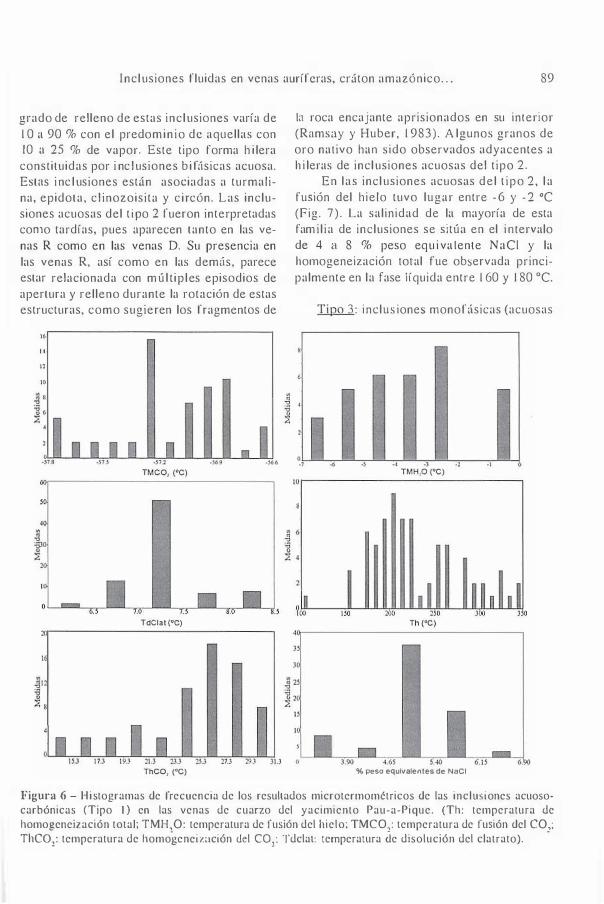
Inclusiones fluidas en venas auríferas, cr<Íton amazónico . . . 89
grado de relleno de estas inclusiones varía de 1 0 a 90 % con el predomin io de aquellas con 10 a 25 % de vapor. Este tipo forma h i lera constituidas por i nc l usiones b ifí.Ísicas acuosa. ESlrls i nc l usiones est<Ín asociadas a turmalina, epidota, c l inozoisita y c i rcón. Las inclusiones acuosas del t i po 2 fueron interpretadas corno tardías, pues aparecen tanto en las venas R como en las venas D. Su presencia en las venas R, así como en las demí.Ís, parece estar relacionada con mú l t ip les episodios de apertura y relleno durante la rotación de estas estructuras, como sugieren los fragmentos de
"r-------------------, " " "
TdClat (OC)
la roca encajante aprisionados en su interior (Ramsay y Huber, 1 983) . A lgunos granos de oro nativo han sido observados adyacentes a h i leras de inclusiones acuosas del t ipo 2.
En las inclusiones acuosas del t ipo 2, l a fusión del hielo tuvo lugar entre -6 y -2 oC (Fig. 7) . La salinidad de la mayoría de esta fami l ia de inclusiones se sitúa en el i ntervalo de 4 a 8 % peso equ iva lente NaCI y la homogeneización total fue observada principalmente en la ¡ase líquida entre 1 60 y 1 80 oC.
•
•
• ,
• .,
,
•
•
•
,
V n
Tipo 3 : inclus iones monof<Ísicas (acuosas
�
• ., • o .. .,
.1 IIn .1 '" 250 lOO llO
% peso equivalentes de NaCI
Figura 6 - Histogramas de rrecuencia de los resultados microtermométricos de las inclusiones acuosocarbónicas (Tipo 1 ) en las venas de cuarzo del yacimiento Pau-a-Pique. (Th: temperatura de homogeneización total; TMH,O: temperatura de rusión del hielo; TMCO, : temperatura de fusión del CO,; ThC02: temperatura de homogeneiz�lción del ca!: Tdclal: temperatura de disolución del clatrato).
-

90 PULZ, G. M. et a l .
y vapor) o a ú n por l a asociación de monofásicas y bifásicas acuosas a lo largo de una misma hilera. Las inclusiones de esta fami l ia presentan d iámetro i n ferior a I �lJl1 Y a veces se muestran vacías, lo que d i ficultó la observación de los cambios de fase durante los ensayos microtermométricos. Por eso, esta fam i l ia de inclusiones no será considerada en la discusión siguiente. Las inclusiones del tipo 3 son mas jóvenes que aquellas de los tipos 1 y 2, pues ocurren en h i leras que cortan los límites de los granos. No se ha observado ninguna evidencia
120)�------------------,
20
120
'00
., • '¿60
;¡ "
20
·20 ·18 -16 ·14
"" '"
n ·12 -ID ·8 .,.,
TMII:OI"C)
2().1 254 )0-1 ThI'Cl
.,
3.11 "Al 9 2 1 11.11 1).79 16.Q.l 17.92 19-62 2UO 2260
Y. pC:IO tquivalclltc. de ",CI
Figunl 7 - HislOgrarnas de frecuencia de los resultados microterl11ométricos de las inclusiones ;:ICUOsas (Tipo 2) en las venas de cuarzo del yacimiento Puu-a-Pique. Th: temperatura de homogeneización . total ; TMI-I�O: temperatura de fusión del hie lo.
textural de esta fam i l i a de inclusiones Ouidas con los granos d e oro nativo.
Consideraciones finales
Las venas de cuarzo del yacimiento Palla-Pique contiene granos de oro nativo asociado con p i r i ta e i n c l u s iones aCUOS3S de baja s a l i n idad y moderada temperatura ( l sO y 2 1 0 OC). Basado en estudios experimentales, Seward ( 1 97 3 ) concluyó q ue soluciones d e baja sal i n i dad c o n tem peratura entre 1 5 0 y 300 oC favorecen el transporte de oro por tilia-complejos.
Las característ icas petrográficas y micro termométricas de las i n c l usiones fl u idas en las venas estudiadas indi can que en la evolución del f lu ido h i drotermal en el yacimiento Pau-a-Pique tu vicran l u gar fenómcnos de ebu l l ic ión (boiling). Las principales evidencias de ebul l ic ión d e lo estudio d e las inclusiones acuoso-carbónicas (t ipo 1 ) son las siguientes: 1) l a variación del grado de relleno ( 1 0 a 90 %) a lo largo d e una m isma agrupación de inclus i ones (Cr.:twford, 1 98 1 ; Roedder, 1 984) ; 2) el m a n t e n i m iento de la temperatura de homogeneización IOtal dc las inclusiones en un m is m o i n tervalo de temperatura, lanto si homogeneizan en l a fase l íq u i d a , como si la hacen a fase vapor; 3 ) la aso� ciación de las inclus i ones de los t i pos I y 2 en un m i s m o grano de cuarzo. Los fenómenos de ebul l ición pueden haber contribuido para l a inmi scibi l idad d e los fl uidos .:tcuoso-carbónicos, presum iblemente homogéneos en origen, durante el descenso d e presión provocado por el ascenso crustal de los mctasedi mentos y de las rocas m i loníticas a lo largo de planos de cabalgamiento regionales.
Por otra parte, también se han observado evidencias de necking d01V1I en las venas del yacim iento Pau-a-Pique. En eSle caso, a l a ampl ia variación d e l gracia d e relleno s e asocian i m portantes variaciones en l a homogeneización lotal , combinadas c o n l a

Inclusiones fluidas en venas auríferas, cráton amazónico . . . 9 1
salinidad constanle de algunas inclusiones acuosas, que claramente indican procesos de neckillg d01Y1I durante la percolación de fluidos en el yacimiento estudiado.
Teniendo en consideración los resultados obtenidos en este estudio se concluye que el yacimiento Pau-a-Pique constituye un ejemplo en el suroeste del Cratón Amazónico de venas epi genéticas de cuarzo y oro nativo relacionados con la deformación proterozoica. Los datos microtermométricos de las inclusiones fluidas de las venas estudiadas sugieren que el transpone y deposición del oro envolvió la percal ación de fluidos acuosos de baja a moderada temperatura y baja salinidad.
Agradecimientos
Los autores agradecen a la Oeol. Msc. Isabel Medina Yarza, OeoL Msc. Marlene Blanco y Msc. Noelia Del Valle Franco Rondom por la ayuda en la traducción del manuscrito a la lengua española. Esta investigación ha sido financiada por los proyectos PADCT 03-GTM-OI/97-02/02-7 (FINEP 88.98.0344-00) y PRONEXIIG/UFRGS.
Bibliografía
Anuário Mineral Brasileiro (1987): ll.lUtil www.dnpm.gov.br (en 08/09/1999).
Barboza, E.S. (200 1): Geoquímica e microtermometria dos fluidos mineralizantes do depósito Pau-a-Pique, sudoeste do Estado de Mato Grosso. M. S. Thesis, Univ. Federal do Río Grande do Su!, Porto Alegre, Brasil.
Barboza, E. S., Pulz, G. M . • Ronchi, L. H., Pinho, E. F., Quadros, A. P. (2000): Estudo preliminar das inclusCles fluidas nos veios de qua riZO do depósito aurífero Pau-a-Pique, sudoeste do estado de Mato Orosso. 5 Congo de Mineralogia y
Metalogenia. Univ. de La Plata Publicación 6, 40-46.
Bodnar, RJ. y Vitik. M.O. ( 1994): Interpretation of microthermometric for Hp-NaCI fluid inclusions. in: "Fluid Inclusions in minerals: Methods and applications", De Vivo. B. y Frezzotti. M.L., eds. Virginia Tech, Blacksburg, l l7-l 30.
Brown, P.E. y Hagemann, S.G. (1994): MacFlinCor: a computer program for fluid inclusion data reduction and manipulation. in: "Fluid inclusions in minerals: methods and applications", De Vivo, B. y Frezzoui, M.L.. eds. Virginia Tech. Blacksburg, 231-250.
Carneiro, M.A., Ulbrich, H.H.GJ., Kawashita, K. (1992): Proterozoic crustal evolution at the southern margin of the Amazonian cralon in the state of Mato Orosso. Brazil: Evidence from Rb/Sr and K/Ar dala. Precamb. Res. 59, 263-282.
Crawford, M.L. (1981): Fluid inclusions in metamorphic rocks low and medium grade. Mineral. Assoc. Canada Short Course 6, 157- 181 .
Dowling, K. y Morrinson, G. ( 1989): Application of quartz teltlureS to the classification of gold deposits using North Queensland eltamples. Eco". Geol. MOllograp/¡ 6. 342-355.
Fernandes,C. J., Pulz, G. M., Pinho, F. E .• Quadros, A. P., Oliveira, A. S., Santos. A. F. ( 1998): Zona de Cisalhamento Corredor e sua relalJao com o depósito aurífero Pau-a-Pique, sudoeste do Estado de Mato Orosso. 40 Congo Bras. OeoL, Soco Bras. Oeo!., 165.
Geraldes, M. C. (2000): Oeoqufmica e Geocronologia do Plutonismo Granítico Mesopro!erozóico do SW do Estado de Mato Grosso (SW do Cráton Amazónico). PhD Thesis. Univ. de Sao Paulo, Sao Paulo, Brasil.
Oeraldes, M. C., Figueiredo, B. R . • Tassinari, C. C. G .. Ebert. H. D. ( 1997): Middle Proterozoic Vein-Hosted Gold Deposits

92 PULZ. G. M. el al.
in (he Pantes e Lacerda Region, Southwestern Amazonian eralan. Brazil. Intern. Geo/. Review 39, 438·448.
Harris, L. (l98B) : A muctural approach lo ¡inearnen! analysis. in: "Struclural Geology Workshop - Gold Mineralizatian in shear loanes", Baxter, J.L.. ed. Hermilage Holdings Ply. LId. Tapie 10. 2 1·23.
Lilherland. M., Klink. B.A., OTonnar, E.A., Pitfield, P.E.J. (1985): Andean trending mobile belt in Brazilian Shield. Nalure 314, 345-348.
Litherland, M., Annels. R.N., Darbyshire. D.P.E, Flethcher, C.l.N., Hawkins. M.P .• Klink. B .A . • Mitchel, W.I., O'Connor, E.A., Pitfield. P.EJ., Power, G., Webb, S.C. (1989): The Proterozoic of eastern Bolivia and ils relationship to ¡he Andean mobile beh. PrecQmb. Res. 43. 157-174.
Menez�. R.G.; Silva. P.C.S.; Silva. L.C.; Takzhashi. A. T.; LopesJr .• I.; Bezerra.J.R.L. (1993): Pontes e Lacerda- Folha SD-21-YC-J1. Programa de Levantamento Geológico Básico do Brasil. Departamento Nacional da Produ�ao Mineral- Companhia de Pesquisas de Recursos Minerais. Brasília. 131.
Ramsay. J. G. Y Huber. M. 1. ( 1983): The Techniques of Modern Structural Geology: Slrain Analysis. Acad. Press Inc .• London. v. 1 . 308.
Roedder. E. (1979): Fluid inclusions as samples of ore fluids. in: "Geochemistry of Hydrothermal Ore Deposits". Barnes. H. L.. ed . • Second Edition. John Wiley y Sons. 684-737.
Roedder. E. (1984): Fluid inclusions. Rev. Mineral. 12, 664.
Saes. G. S. y Fragoso Cesar. A. R. S. (1994): The Aguapef Basin (Southwest Amazonia): A Greenville age aulacogen of the Sunsas Orogen. 38 Congo Bras. Geol.. 1 . 207-209.
Santos. J. O. S. ( 1999): New understanding o( the Amazon Craton Gold Provinces. in: "New Developments in Economic Geology". Univ. Western Australia. Perth. Un pub. Report. 10.
Schobbenhaus. C. y Coelho. C.E.S. (1988). Principais depósitos minerais do Brasil: Metais básicos nao ferrosos. ouro e alumfnio. Departamento Nacional da Produ�¡¡o Mineral. Rio de Janeiro. v.3. 670.
Seward. T.M. ( 1973): This complex is of gold in hydrothermal ore solulions. Geochim. Cosmochim. ACla 37, 379-399.
Shepherd, T. J., Rankin, A. R, Alderlon, D. H. M. ( 1985): A Practical Guide lo Fluid
• Inclusion Sludies. Blackie and Son .• Glasgow . 239.
Tassinari, C. C. G. y Macambira. M. J. B . (1999): Geochronological provinces ofthe Amazonian Cralon. Episodes22. 174-182.
Tchalenko. 1.S. (1968): The evolulion of kink bands and the development of compressive textures in sheared days. Tectollopllysics 6. 159-174.
Recibido:Diciembre 2002 Aceptado: Noviembre 2003

Boletíll de la Sociedad Espaiiola de Mineralogia, 26 (2003), 93-/05 93
MeU incIusions in quartz from subvoIcanic sills of the Iberian Pyrite BeU: Implications for magma evolution and hydrothermal alteration
Fernando TORNOS l. Ylad imir SIMONoyl and Sergei KOYYAZINl I InslilulO Geológico y Minero de España. e/Azafranal 48, 37002 Salamanca (Spain) [email protected] 1 United Instilute of Geology, Geophysies and Mineralog)'. Russian Aeademy of Scienee. Academic Koplug ave., 3. 630090 Novosibirsk (Russia) [email protected]
Abstracl: Melt inclusions in qurutz phcnocrysts ofmassive felsic sills of lhe Iberian Pyrite Belt homogenize to liquid at tempcraturcs betwccn 10250 and > 1 1 50GC. Thcy show a chcmkal compasitlon consistent with that of their peraluminous and ealc-alkaline host rocks . On the basis of the melt inc1usion compasition two types of quartz phenocrysts can be distinguishcd. The first g10up is dominant and has inclusions wilh compositions consistent with (hat of apogranites. They are in!erpreted as reflecting \he me1ting of an arkosic protolith or, more likely, are inherited f10m a deep leueogranite that could be the source of the tin anomalies wilhin \he lberian Pyrite Bell. The seeond group has melt inc1usions with a composition consisten! with that ofthe host rocks, wilh ils trapping synchronously wilh \he prceipitation of feldspars and mafic minerals.
The abnormally high temperature of mell homogenization is interpreted as a primary feature, renecting the existence of deep superhealcd and dry magmas. Panial melting of a water-poor granulilic crust by underplating mafic magmas produeed watcr·deficient intcrmediale-acid roeks that rose up 10 (sub)volcanic levels in the crust.
Comparison of the composilion of the mell inc1usions with differcnt chemical alteration vectors proposed for hydrothermally altered volcanic rocks suggest ¡hat these equations do nOI unequivocally represent the degree of altcration.
Key words: Melt inclusions, dacite, geochemistry, lherian Pyrite Belt
Resumen: Las inclusiones vítreas en los fenocristales de cuarzo de un sill félsico en el Complejo Yolcano Sedimemario de la Faja Pirltica Ibérica homogeneizan a Hquido a temperaturas entre 10250 y >1 150°C. Muestran una composición qufmica consistente con el carácter caJcoalcalino y peralumfnico de las rocas encajantes. Hay dos tipos dc fenocristalcs dc cuarzo. Los más abundantcs tienen inclusiones vltrcas con una composición tfpica de apogranitos que puede ser derivadas de la fusión de un prololilo arcósico o, más probablemente, ser heredados dc un leucogranito profundo. El scgundo grupo de fenocristales tiene inclusiones con composicioncs similares a las de la roca de caja e indican que la cristalización del cuarzo fue sincrónica con la precipitación de feldespatos y minerales miificos.
La elevada temperatura de homogeneización/fusión, interpretada como primaria, es indicativa de la existencia de magmas supercalentados y anhidros en profundidad. La fusión parcial de una coneza granuHtica deficiente en agua producida por la intrusión de magmas miificos profundos generó magmas de composición intermedia a ácida que ascendieron hasta niveles (sub)volciinicos de la corteza.
La comparación de la composición de las inclusiones vltreas con diferentes vectores qulmicos de alteración hidrotermal de rocas volcánicas sugiere que estas ecuaciones pueden no representar distintos grados de alteración hidrotermal, sino caracteres geoquímicos primarios.
Palabras clave: Inclusiones vftreas, dacita, geoqulmica, Faja Pirítica Ibérica

94 TORNOS, F. el al.
Introduction
The Iberian Pyrite Beh (lPB) hasts abundan! massive sulphide orebodies intimately associated with bimodal (basaltrhyolite) ¡gncoos racks and interbedded siliciclastic sediments. As in equivalen! ore disldels, sorne of the orebodies have a direct spatial and perhaps genelic relationship with felsic subvolcanic and extrusive magma tic rocks (e.g .• Rio Tinto. Aljuslrel, Neves Corvo, Aguas Teñidas; Boulter, 1993; Sáez el 011., 1996; Carvalho el aL, 1997; Leistel el al.. 1998). These hasl racks have endured a polyphase pervasive hydrothermal alleration and subsequent Variscan metamorphism Iha! have dramatically modified ils original geochemislry, leading 10 pronounced changes in mobile major and Irace elemenls (e.g . • Sánchez Espai'ia el al.. 2000).
In Ihis contexl, melt inelusions are unvaluable Iracers of Ihe geochemislry of Ihe magmas since Ihey usually relain Iheir originnl meh composilion when Ihe enelosing mineral cryslallized. Somelimes Ihey are Ihe unique source of informalion aboul Ihe condilions of emplacemenl of Ihe magmatic rocks, Ihe processes ¡nvolved in Ihe magmalic evolulion, and the exlenl and Iype of hydrothermnl alteralion (Audelal el al., 1998). However, melt inclusion sludies are scarce in areas where volcanic-hosled massive sulphide deposits are found. There are only some references on sorne russian deposits (Babansky el al., 1995; TilOv etal., 1996; Simonovet al..1999) as well in Ihe present day syslem af Ihe Lau basin (Yang and SCOlI. 1996; Kamenetsky el al., 1999). In Ihis work we present Ihe firsl microlhermomelric and geochemical results of melt inclusions in quartz phenocrysls from subvolcanic massive quartz porphyritic felsic sills in Ihe lberian Pyrile Bell.
Geologic seUlng
The Iberian Pyrite Beh is characterized
by a fairly simple slratigraphy, wilh rocks ranging in age from Ihe Upper Devonian 10 the Lower Cnrboniferous. The lawermost sequence is formed by silicielaslic sedimenls (shnles and sandslones) deposiled in nn epicontinenlnl platform (Upper Devonian). Above il Ihere is a rnlher Ihin « 1000 m) sequence of volcanic, volcanielnstic rocks and siliciclastic sedimenlS (mainly shales) wilh sorne jnsper levels (Volcano Sedimenlary Complex - VSC). The sequence ¡ncludes abundant dark shales and volcanielastic rocks, typically Ihick. pumice-bearing submarine mass flows. Inlerbedded massive ¡gneous rocks inelude subvolcanic sills and cryplodames of felsic (dacite-rhyolile) composilion as well as si lis and • locally pil lowed - lavn flows of mafic (basaltic) compasilion. These mafic rocks are continental Iholeiitic and wilhin-plate continenlal basalts while the felsic rocks are low AIJOJ high Yb calc-alkaline anes (Munhá, 1983; Mitjavila el al.. 1997; Thieblemont el al., 1998). The andesiles are very scarce. This Complex Is chnracterized by abundant changes of facies and ranges in age between the Upper Fammenian and Ihe Lale Visean.
All lhese rocks are pervasively altered by syn- lO post-magmalic hydrolhermal fluids. There was an early submarine hydrolherrnal alteration (hydrolhermal melamorphism of Munhá el al., 1986). In close spatinl associalion wilh the massive sulphides, there is a superimposed alteration, Ihat produced hydrothermal hnlos with an exlernal zone of sericillzation and an internnl chlorilization relaled wilh Ihe massive sulphide feeder zone.
The deformation in Ihe Iberian Pyrile 8eh is of Ihe thin skinned Ihrust and fold Iype, wilh norlhward dipping majar Ihrusls and overturned folds. The melamorphic grade is of low 10 very low grnde (Munhá, 1990; Silvn et aL, 1990; Quesndn, 1998).
Massive bodies of felsic rocks ranging in composilion belween daciles and rhyolites are very common in the VSC. Perhaps, one of lhe

Melt inclusions Iberian Pyrite Belt 95
more studied ones is that located in Ihe Rio Tinto-Odiel area (e.g., Boulte.r, 1993; Valenzuela et aL, 2000. Here, the felsic rocks occur in a E-W trending body of more than 30 km long and between 500 m and I km in thickness located on the northern limb of a majar antieline: the sequence ineludes yolcaniclastic pumice-rich rocks intruded by sills. It is close to the contact between two major tectonic unils, the hosting Valyerde Unít that is thrusled by Ihe Berrocal Unit, consisting of lurbiditic sediments of Ihe Culm Group (Figure 1 ; Quesada, 1998).
The studied samples are from one of Ihe areas where Ihere are massiye rhyodaciles wilh conspicuous flow banding and columnar jointing; they haye inlrusiye, locally peperilic. contacts with both the oyer and underlying grey shales. The sample were colJected about 50 m below thal upper intrusiye contacto These rhyodacites haye unoriented microporphyritic ameboidal quarlz (0.8-1 mm) and some fresh plagioelase (replacement by hydrothermal
l' '1
al bite?) 1 - 1 .2 mm-sized phenocrysts in a recrystallized and strongly hydrothermally altered matrix, made up of microcryslalJine quartz, feldspars (sericitized) and minor mafic minerals replaced by chlorile and sericite; no biotite phenocrysls. eyen replaced. haye been observed. The scarce presence of K-feldspar phenocrysts seem lO be a Iypical fealure of felsic rocks in the IPB (Thieblemont et aL, 1998). Whole-rock analyses of samples belonging lo Ihis unit are shown in the Table l . Aboul 15 km lo the east, Ihe sequence hosts Ihe massiye sulphides and related slockwork of the Rio Tinto mine.
Melt inclusions
The silicale melt inclusions are found ;so[ated in regular interyals within ¡he individual magmatic quartz phenocrysts, with sizes rangiog between 10 and 50 microns (Figure 2). They usually have rounded shapes, bUI
----
flySCn Group
?!.rrple mies felsIc massIve & voIconlclos1lc ocks Molle $/lis
PQGroup
""""',
Figure 1. Geological map of the Rio Tinto-Odiel area showing Ihe location of the porphyrytic rocks, studied samples and of the majar ore deposits. Modified from lOME ( 1978).

96 TORNOS, F. et a l .
Table 1 . Chemieal eomposition of ( 1 994) and Titov et a l . ( 1 996). A key aspeet rhyo l i tes
Sample
Sia, TiO, AI,O) Fe,O) MnO MgO CaO Na,O K,O P,O, LO!
78 73 .99 0.20
1 2.29 3 .06 0.02 2.82 0 . 3 1 0.99 2.66 0 . 06 3 . 1 2
79 73.96 0. 1 8
1 3 . 1 6 2.39 0.03 0.77 1 .79 3 .89 2.48 0.06 0.90
Analysis JGME. LOI: Loss on ignition. Total Fe as Fe103
sometimes hexagonal edges can be observed. The inclusions contain a dominant se! of fine brownish amorphous phases (metastable s i l icate glass), wit il Dne 01" more shrinkage bubbles. The presenee o f erystals in the edge of Ihe inclusions nor other cvidences 01' recrystall ization or interaction with the h051 mineral could be observed. Local l y , thefe are sorne minute ( 1 -2 microns) daughter opaque phases that probably eorrespond 10 sulphides. rhe characteris t i c s o f lhe incl usions are indicati ve 01' q u i c k quenching and correspond lO low or non evolved incl usions in Ihe sense of Cloehiatti ( 1 975), typieal of rocks emplaeed in s h a l l ow condit ions (Lin Qui X i a and Cloehiatt i , 1 985; Mangas et a l . , 1993). The distribution and charactcristics of lhe inclusions suggesl ¡hat they are primary _ in origin (Roedder, 1 984; Lowenstern, 1 995).
The melt inclusions were studied in double polished t h i c k sections \Vith a h i gh tcmperature heati n g stage w i l h an i nert med i u m (Sobolev and S l lltsky, 1984) fol lowing lhe procedures described III Simonov ( 1 993), Sobolev <lnd DanYllshevsky
of ¡hese runs i s lhe dete r m i nation of rhe homogen isation tCl11perature. Long heating t i mes produce a conta m i natíon o f lhe melt inclusion by lhe hOSl quartz. Inversely, q u i ck heati ng \Vi i I ¡cad lo abnofm a l l y h igh homogcnisation tcmperatures. The experience indicates ¡hat me!! i n c l u sions in quartz can be heated slowcr than [hose in pyroxene and ol ivine, but lhe heating time must be smal ler ¡han about 1 .5 hours in arder lO avoid contam ination. The absence 01' a n inverse correlation between s i l i c a and the other elements (see below) exclude a significative interchange w i th the host rack.
During heating, at about 500°C, lhe s a l i d I'raclion of inclus ions became d a r k . Al aboul 900°C a transparent l i q u i d started 10 be v i s i-
•
-10 P 111
Figure 2. Aspect 01' a Illclt inclusions in a quartz phcnocrysl from the felsic s i l ls .

Melt inclusions Iberian Pyri¡e Belt 97
ble and near 950°C some vapour bubbles could be observed wilhin (he molten fraction. The complete homogenizalion with dissolution of the bubble into the liquid occurred in a range between 1 025-1075°C. However, sorne inclusions did not homogenize a¡ temperatures near 1 150°C. ¡he highesl measurable temperature (Table 2). Al this temperalure, these inclusions consisted of a transparent mel¡
with small and dispersed vapour bubbles. The measured homogenization temperatures could
correspond lO Ihe approxirnate liquidus temperature al ¡he time of trapping, represenling ¡he mínimum temperature of forrnation of these phenocrysls (Roedder, 1984).
Geochemistry
Afler healing, Ihe melt inclusions \Vere cooled fasl in order to produce a homogenous
glass mling all the cavily. The samples \Vere cut and Ihe previously measured melt inclusions were analysed by electron microprobe following conventional procedures. The measured contents of Na, Mg, and K can be low due 10 their 10ss below the beam. In order 10 minimize this error. the alkalies \Vere analyzed firsl with short integralion limes. The compasilion of the melt inclusions is shown in the Table 2.
The melt inclusions are usually interpreted as represenlative of the composilion of the magma at the time of trapping along the quartz liquidus. However, that only holds true for the largest inclusions. According 10 experimental \Vork with artifi· cial minerals, only the inclusions larger ¡han about 10-20 microns have ¡he composition equivalent to that of the corresponding mag
ma. Thus, only the inclusions fiuing that criteria were analyzed. Notwilhstanding, they do nOI represenl the composilion of the hosl
Table 2. Chemical composilion and homogenization temperalUres of melt inclusions.
Sample 1 - 1 1-2 1-3 2-5 2-6 2-7 3-8 3-9 4-10 4-11 4-12 4-14 4- I S S-16 S-17 5-18
r"" 11 11 11 1 1 1 1 1 1 1
Si01 70.10 76.68 72.38 68.27 67.82 67.9266.10 68.74 70.42 71.5 73.16 62.32 69.23 72.24 73.47 73.53
TiO¡ 0.52 0.42 0.49 0.01 0.00 0.00 0.01 0.01 0.08 0.1 0.03 0.02 0.00 0.02 0.03 0.01
A110) 13.91 10.09 12.85 19.04 19.45 19.20 19.7\ 11.92 \7.56 \6.8 15.83 22.09 18.80 19.39 18.29 \8.40
F,O 3.92 3.23 3.77 0.54 0.33 0.38 0.69 0.40 0.62 0.62 0.51 0.73 0.61 1 . 1 1 1 .02 1 .0\
MnO 0.08 0.04 0.08 0.03 0.05 0.05 0.04 0.04 0.02 O 0.02 0.03 0.02 0.05 0.03 0.03
M80 1.33 0.93 1.I7 0.06 0.06 0.06 0.01 0.06 0.09 0.1 0.07 0.45 0.33 0.06 0.05 0.04
e.o 0.38 0.23 0.36 0.94 1 .00 0.97 1 . 15 1.02 0.93 0.79 0.67 0.18 0.19 1 . 1 7 1 .00 1 .04
Na�O 2.17 2.34 1.6\ 3.29 3.23 3.37 3.52 3.56 2.32 1.73 2.13 221 2.17 2.16 2.15 1.37
K¡O 5.47 4.94 5.38 5.84 5.80 5.88 6.09 5.98 4.74 4.7 4.80 6.62 7.27 3.63 3.72 3.60
TOla! 97.89 98.9098.08 98.0097.73 97.82 97.4 91.72 96.77 96.3 97.21 94.65 98.62 99.83 99.75 99.03
AI-! 72.70 69.53 76.91 58.23 58.12 57.76 56.8956.88 59.79 63.5 63.52 74.7\ 76.34 52.56 54.48 60.2
AI-2 43 38.5 43.8 6.97 4.93 5.35 8.36 5.32 10.2 9.08 8.59 12.8 9.83 18.8 11.1 19.3
T.�L) 'C 1025 1025 1025 1050 1050 1050 1075 1075 1060 1060 1060 1060 1060 n.h. n.h. n.h.
Analysis by X-Ray microanalyzcr Cameh3x-micro 31 1he Unitcd Instilule of Gcology, Gcophysics 3nd Mincralogy Russi3n Academy of Scicnec, Novosibirsk, Russi3. AI·I : Altcralion Index or Ishikawa el al. ( 1976); Al-U: Allcration Index of Sánchez Espai'lael al. (2000). n.h.: nOI homogenizcd al 1 150°C.

98 TORNOS, F. el al.
rack because the bulk compasilion orlhe magma gradually changes due 10 ffaclional crystallization ar mixing processes and the inc\usions only represenl instantaneous ("frozen") compositions (Roedder, 1984).
Postmagmatic processes can modify the original compasilion of the mel! inclusions ir Ihe has! mineral is altered ar Ihe inclusions are Iinked with the surface of the crystuts via microcracks. Careful examination of ¡he sections suggesl Iha! Ihe studied quaTlZ phenocrysts hu ve behaved as closed systems, al leasl ror ¡he majar oxides. There is no evidence of microcracks in the quartz neaT Ihe ¡nclusioos; also, ¡he microprobe analyses are internally consisten! and reproducible.
The measured inclusions are characlerized by very variable SiOl contenls (62.3-76.7 wt.%), ranging in composition from daciles 10 rhyolites. They define a trend eorresponding 10 Ihat of subalkaline rocks of eale-alkaline eomposition (Figure 3c), a fealure Iha! confirms that the ealc-alkaline eharacter of Ihese rocks is magmalic and nol due lO superimposed hydrolhermal modificalions. They have high NalO+Kp (4.97-9.61%) bul low CaO (0.18-1.17%) eonlenlS. All lhe mell inclusions have normative corundum. The only observed Irend is tha! there seems lo be a drop in ¡he alkalies and Alp) with the silica conlenl (Figure 3a and b). These inclusions have compositions equivalenl lo Ihose of apogranitic syslems, with ab-an-or normalive ralios close 10 the cotectie line at 1 kb (Winkler, 1978).
The chemical composilion shows Ihat Ihe melt inclusions can be grouped in IwO distinet types. The Type I group mosl ofthe inclusions (n=13). They are characlerized by very low MgO « 0.5%, usually less (han 0.07%), FeO « l.1%), and TiOl « 0.08%), bul high Alp, (>16%) contenlS (Table 2). The second group ineludes Ihree inelusions in a quartz phenocryst (S-l) and are eharaclerized by high MgO (>0.9%), FeO (>3%) and TiOl (>0.4%), bul low Alp, contents « 14%); they have normative enslalile and ferrosilile. The high silica content of the melt inclusions suggesl
thal Ihe high SiO} eontenl of the felsic rocks in Ihe Iberian Pyrile SeU is a primary and nOI an hydrothermal alteration feature.
Fluid inc!usions
The quartz phenocrysts also host abundant fluid inclusions wilh a dominant liquid phase and a subordinale vapor bubble. They are small, rangjng in size between 3 and 1 5 microns and are grouped io cluSlers aod along healed cracks, iodieating a secondary origino They are usually irregular but sometimes idiomorphie oear the edge of the crystals.
Microthermomelric studies were carried on a heating-freezing equipment of original desigo (Simonov, 1993). The ioclusions yield firsl melting lemperatures belween -22.5 and _22.0oC, indicatiog Ihal Ihe fluid was dominaled by ¡he Hp-NaCI-(KCl) system. The lasl melling temperalure of ice ranges between -3.8 and _2.0°C, suggesting salinities of 3-6 wl.% NaCI eq. Final homogenization occurred al temperatures between 1440 and 171°C. No traces of COl were found.
These results are very similar lo Ihose found by Sánchez España et a.1. (2000) in (he lale, synorogeoic fluids eireulating io (he IPS during Variscan limes, beiog aqueous fluids with salioities clase lO 2-6 wt%NaCI eq. and homogeniza.tion temperalures of 130-1 80°C. However, they are different from the dominan! fluids related with the massive sulphides, with homogenizalion temperatures io the 130-380°C raoge and salinities up 10 23 WI.% NaCI eq. This suggests tha! hydrothermal fluids leading 10 Ihe formation of the massive sulphides did nol eireulate wilhin these ¡gneous rocks, since probably were focused along the majar feeder zanes.
Discussion
The sludied quartz phenacrysts are aplically similar and no ehronalogical nor

Melt inclusions Iberian Pyrite Belt 99
geochemical evolulion could be eSlablished between them. However. lhe chemical dala of Ihe melt inclusions suggesl ¡hat Ihere are two independent types of quartz phenocrysts. Ihat reflee! ¡he growth in different settings.
The Type 1 melt inclusions are significantly depleted in MgO. Fea,. CaO. and Tia! and enriched in AlP3 when compared wilh average dala of daciles and
f :�::I f'" : . . .': ... . ' .. . $ .t.HOII rod<s I!I :.'
o .:' y,!'I���,��!f>B '.' .10 • • • ' . .. .: . . :.· �.t ,...
+ •••••• 'fl. � . '
A
o J • ' .. " • '" .. , .. .......... :-.. .. :E , , " ""1 . ,
• • • ",,,.".:! + ,.+ . + +.: . ....... ,.
• ':----::----:"-'---::--_---l IKI " SIO, ,g ,. IKI
• .. .. .. $10,
" ..
" ,----------------------, • •
• •
• • • •
. ".
E
•
1·;:11 . " • •
".lc-_-_--'--_-------1 .. .. • SIO,
" ..
rhyolites of Ehlers & Blal! ( 1982) and Middlemost (1985) as well the regional data of the lberian Pyrite Bell (ITGE. 1997). The inclusions were trapped during the crystallization of the feldspar and quartz phenocrysts, as suggested by the drop of alkalies and AIlO) wilh an increase in silica. This chemical trend is very similar 10 that of apogranites and could reflect the mehing of a
.,-----------��F-�_n • Type 1 B .T'fPtI 1I .t. HO$1 roeks • Volunic rocks 1P8 • "
+ .. . . �I .. .. , . . •
1,,' 'l' " , • . . -.. � .. . .
I .., ... ....... ot.'" ••• •. • ( " \ . ' . . . .. .. .. .. . ,'it' � ' l · " .. ... ,. . . ' . . .. . "'-( '" • ' ''' .... .fIf,� ...
":--�7----':---:'c' -"O-!..�. IKI " sr&. '$ 110
'� rc�=o---------� I I.Type l l O I I.TYIle 11I 1 • •
n.
• • •
• • • • •
• • • '�+------e------__________ �
.. .. "
"r==-------, 1.r;11 • • F . T 11 •
. , • .
,. . .. .
.�--�----�------� 110 H '0 " .. 510,
Figure 3. Binary geochemieal diagrams of the mel! inelusions. For eomparison, data from regional volcanie roeks of the Iberian Pyrite 6eh (ITGE. 1997) have been included, The dala are from Table 1 and 2. The alkaline-subalkaline lields or Figure 3c are rrom Irvine and Baragar ( 1 971). The Alteration lndex 1 is rrom lshikawa (1977) and the Alteration lndex 2 is rrom S:l.nehez España el al. (2000).

100 TORNOS, F. el al.
prololith with an arkosic compaslllon. However. such rocks are nO;t known in the basement oflhe Iberian Pyrite Belt. The quartz phenocrysls could al so be derived from deep unexposed apogranites. The hypolhetical presence of deep granites has beco proposed by several authors (Relvas, 2000; Sánchez España el al., 2000; Solomon el al.. 2002) as the source of magmatic fluids and of the high So contents of many of the massive sulphide deposits in Ihe Iberian Pyrite Belt including Neves Corvo, ane of the world-class tin deposits. Geochemically equivalent inclusions have beco found by Dietrich el al. (2000) in Sn-bearing porphyries in Bolivia
The Type 11 inclusions ha ve brandly equivalen! compositions 10 Ihese average composilions and reflecl Ihe growlh or sorne quarlz phenocrysls in equiJibrium wilh the host rocks (Table 2; Figure 3). The drop in K10, AI10). Fe and Mg synchronously with the iocrease in silica is here consistent with the precipitalion orfeldspars and mafic phases. very likely biotite. A similar behaviour has been observed elsewhere (Zhaolin, 1994).
The melt inclusions in volcanic quarlz phenocrysts usually homogenize in a range belween 800 and 1200°C, bul values higher Ihal 1000°C are uncommon (Zhaolin, 1994); in fact, lhese values are commonly interpreted as unrealislically high (Audetat et al., 2000) and related 10 post-Irapping modifications with waler loss and/or filling of Ihe shrinkage bubble at ambient pressure. The lemperalure of homogenization of the studied inclusions (>JOOooq is significanlly higher Ihan the sol idus of the granodiorile and granile, Ihal is slrongly dependa ni on the pressure and composilion bul almosl independent of Ihe X(H10). The solidus of granitic rocks is eslimaled lO be around 750-800°C at I kb, 705°C al 2 kb and 630°C al 8 kb (e.g., Whitney, 1989 and references therein). The crystallizalion of the quartz phenocrysls is strongly dependant on Ihe XHzO; for example, al pressures or 2 and 8 kb bul at low waler
contents « 1 wl% HzO) it can crystallize at lemperalures as high as 980- 1 120°C, respectively (Whitney, 1989). It always posldales plagioclase but at high pressures it can also predate ¡he formalion of K feldspar. Thus, if Ihe melt inclusions represenl true quartz liquidus lemperalures, then Ihe syslem was a strongly water-depleted one. Equivalent high lemperatures ha ve been locally quoted in sorne felsic melis (e.g., Zhaolin, 1994; Babansky el al., 1995; Titov et al., 1996; Simonov et al.,1999) including ¡he felsic rocks hosting the Kidd Creek gianl massive sulphide deposit (Barrie, 1995).
As quoted aboye. ¡he water-undersalurated character of Ihe melt inclusions can be a postIrapping fealure. There are evidences Ihal high temperature melt and fluid inclusions can loose small quanlilies of waler due to an outwards diffusion through {he hosl rocks (e.g, Mavrovenges and Bodnar, 1 994) and later yielding high homogenization temperalures due 10 the relaled raise in the liquidus curve. However, there is a consislency between Ihe homogenizalion lemperalures and Ihe chemical composilion or the melt inclusions, wilh Ihe lower homogenizalion lemperatures in Ihe Type 11 inclusions (Table 2). These data slrongly suggest Ihal these lernperatures reflect a primary feature and are nOI due 10 posl-trapping effecls.
The melt inclusions can provide impOrlaOl informal ion aboul the volalile content or the magmas (Lowenstern, 1995; Kamenilsky el al., 1999). The reviews of Clernens ( 1984) and Lowenstern ( 1 995) show thal most rhyolites ha ve waler conlents between 0.5 and 7 wt% while Thomas ( 1994) suggests that ¡he water content in granites is between 2.5 and 9%Hp.
Usually, the difference lO 100 wl % oflhe melt inelusion analysis is assumed as indicalive of ¡he volatile conlent of Ihe inelusion. Total sums of the studied melt inclusions are belween 94.65 and 99.83%, wilh an average value of 97.83%. However, Ihese

Melt inclusions Iberian Pyrite Belt 101
values are probably very approximate since. as quoted aboye. there can be significant analytical errors or a wening effect during trapping plus an unknown post-lrapping loss of water due to diffusion during heating (Audetat et aL. 2000).
Simple numerical estimations suggesl thal if most of the mafic hydrous minerals formed earlier than the quartz. then Ihe amount of magmatic fluids that can be exsolved from the porphyries is volumelrically significant and more than enough than thal needed for forming the supergiant massive sulphide orebodies. Recently, Relvas (2000) and Sánchez España et al. (2000) have shown Ihal there are isotopic and fluid inclusion data thal can be consistent wilh the presence of a magmalic input in the hydrothermal fluids wilhin Ihe Iberian Pyrite Bell.
The studied melt inclusions show no evidence of fluid unmixing and Ihere are no geologic indications of exsolulion of magmatic f1uids from Ihese sills. The absence of aqueous fluids is consistent with the melIS being trapped as a homogeneous magma before significanl water exsolution (Lowenstern, 1995). Also, Ihere is no geologic evidence of an early high temperalure, potassic. hydrolhermal alteralion as is found in porphyry-like or granite-related systems (e.g, Beane and Titley. 198 1 ; Jackson et aL, 1989; Pollard and Taylor, 1986) nor magmatic breccia pipes tha! could beexpected in shallow magmatic hydrothermal sySlems (Sillitoe, 1985). The secondary fluid inclusions are low salinity COl-poor walers, probably of connale or sea water origin, with variable inleraction with the hosl rocks and very different 10 the C01-rich or very saline brines of magmatic origino The absence of K feldspar phenocrysls in the hosl rocks, tha! is abunda nI in the matrix, suggesl that the quartz and plagioclase phenocrysts formed al rnlher high temperatures (>IOOO°C for les S than 2 wt% H¡O) and pressures (>2 kb) at waterundersaturated conditions (e.g., Naney, 1983).
In fact, the late precipitation of biotite after quarlz is also typical of low-water systems « 1.5% al 2 kb; Naney, op.CI.).
Emplacement at undersaturated conditions imply Ihal Ihe magma was generated under relatively dry granulitic conditions, since usually they lend 10 approach water saturation during the late cryslallization (e.g., Clarke. 1992). These resulls are consistent with the formation of dry (< I wt% HP) intermediate felsic melis from a quartz-feldspathic source in a high-Iemperature low-pressure regime. These superheated and dry magmas are able 10 rise to very shallow crustal levels (Clemens. 1984; Clarke, 1992; Lenlz, 1999). The ultimate source could be the upper crust, wilh anatexis or granulitic lower crust generated by conductive heating due lo underplated deep mafic magmas (Mitjavila et al.. 1997; Thieblemont et al., 1998) generated during Ihe Iranspressional deformalion and concomitant deep crustal faulting wilhin Ihe Iberian Pyrite Belt during Variscan times.
rile qlllllltifica/iol! o[ lIle alterario" processes
The degree of hydrothermal alteration of vo!canic rocks is usually measured using Ihe Alteration Index (Al) of Ishikawa ( 1976); recently. Sánchez España et al. (2000) have proposed a different equation for the felsic igneous rocks of Ihe Iberian Pyrite Bel!. Ifthe melt inclusions represent pristine portions of unaltered magma, then their alteralion index should be near O. Recently, Large el al. (2001 ) have shown thal unaltered vo!canic rocks have Al values lower Ihan 60 while their hydrothermally altered counterparts have A l indexes between 50 und 100.
The igneous rocks of the IPB ha ve elevated Ishikawa ( 1976) A l values. ranging belween 4 and 99 with an average of 50.3. The melt inclusions hu ve also high alteration indexes, belween 52.6 and 76.9. despile Ihe absence of evidence of severe hydrothermal alteralion. The recent sludy by Sánchez Espa-

102 TORNOS, F. el al.
i'la el al. (2000) suggests a differenl Alteratian Index for the Iberian Pyrite Bell. based on (he geochemistry of altered and unaltered felsic rocks (AImajor = (FeZO) '010' + MgO + MnO)! (Fc10" OI., + MgO + MnO + Nap + K10); this index is broadly equivalent 10 that recenlly suggested by Large el al. (2001). The index when applied to the melt inclusions shows an irregular behaviour. The sludied melt inclusions have low Al values, belween S and 43. bUI with a marked contrast betwecn Ihe Type 1 (5-19) and 11 (38-44) inclusions. indicating thal lhere is a significant ¡nfluence of the original FeO and Mg O contenl of Ihe magmas in the Altcration Index. These rcsults $ugges! that the Alteration Index can give systcmatic crrors in rocks charaeterized by high K,0 but low MgO values in which there seems not to be a drama!ie KP mobility.
Conclusions
The melt inclusions in quarlz phenocrysts of felsic subvoleanic si lis indiente (he existence of. al least. !wo !ypes of quartz phenocrysls that grew a! different stages of magma evolution. The first group (Type 1) have compositions close to that of apogranites and hypothetically could be derived (rom deeper apogranites. The seeond group o( quartz phenocrysts grew in equilibrium with the hos! roek and synehronously with the crystnllization of biotite and plagioclase.
The composition and homogenization temperatures of the melt inclusions are consistent with the shallow. waterundersatured emplaeement of the felsic magmas in the area. which were probably derived from the partial melting of a waterdendent granulitie erust by heat transfer from underplated manc magmas.
These shallow systems were probably unable lo exsolve magma tic fluids ando thus. seem not to have any chemical affinity with the nearby massive sulphides. However, they
were probably able to generate large convective hydrothermal systems dominaled by external connate or marine waters that leached metal s from the host roeks. A magmatic contribution. i f any. should be derived (rom large. deeper and more waterrich felsic rocks perhaps parental to these sills. The superheated emplacement of the rhyolites was probably related to the abnormal high heal flow in Ihe IPB and related to the transpressional deformation and concomitan! igneous activity. Perhaps, the size of the massive sulphides in the ¡PB and their cluster around spedfic zones can be related with Ihe generalion of large focussed Iherma! anomalies' related wilh deep intrusions.
The composition of the melt ¡nelusions sugges! Ihal the Alteration Index commonly used in the lilerature for estimaling the degree of hydrotherma! alteration of voleanie rocks cannol be applied lo the fe!sie ¡gneous rocks of ¡he Iberian Pyrile Beh.
Aeknowledgements
The analylical work has becn earried oul in the Uniled InstituleofGeology, Geophysies and Mineralogy Russian Academy ofSdence. Novosibirsk, Russia. This work has been founded by Ihe INTAS EC project 1699 and the Science and Technology Spanish Department Project number BTE2000-0161-C02 and is a contribution 10 the GEODE project (European Science Foundation). We acknowledge Drs. César Casquet. Emilio Pascual and Dimitry Kamenetsky their . comments about the problematic aspeets of Ihese rocks and David Lentz for the eareful review of the English and the texto
References
Audetat. A., Gunlher. D. y Heinrich. C.A. ( 1 998): Formation of a magmatic-

Melt inelusions Iberian Pyrite Belt 103
hydrothermal ore deposit: Insights with LA-ICP-MS analysis of fluid inelusions. Seienee. 279. 2091-2094
Babansky. A . . D... Ashikehmina. N.A . • Kovalenko. V.I.. Lyatifova. E .. N. y Kononkova. N .. N .. (1995): Initíal magma of Verkchnechegemsky caldera komplex (Norlh Caucasus) on the dala ofinelusions in minerals study. Dok/. RAS. 344-22. 226-228 (en ruso).
Barrie. C.T. ( 1 995): Zircon Ihermometry or high temperature rhyolites near volcanicassociated massive sulfide deposits. Abitibi Province. Canada. Ge% gy, 23-2. 1 69-172
Beane. R,E, y Titley. S.R. (1981): Porphyry Copper deposits. part 11,- Hydrolhermal alteration and mineralizalion. En: Eeollomie Oe% gy, 75 aJmivers(¡ry vo/ume. B.E. Skinner. (ed.). 235-269
Boulter, C,A. (1993): Compurison of Rio Tinto. Spain and Guaymas Basin, Gulf of California: nn explanation of a supergianl massive sulfide deposil in an ancient sillsediment complex .. Oe% gy, 21. 801-804
Carvalho. D., Barriga, F. y Munhá. J. (1997): Bimodal Silicielastic systems: The case of the Iberian Pyrite Belt. En: Vo/eanie associated massi"e sl/lfide deposils: Processes alld examp/es ill modem (¡lid allcielll seltings. C.T. Barrie. M.O. Hannington (eds.), Reviews Economic Ge% gy. 8, 375-408
Clarke. D.B. ( 1992): Granitoid Rocks. Chapman & Hall. Landon. 283 pp.
Clemens, J.D. ( 1984): Water contents ofsilicic to inlermediale magmas. Lit/lOs. 17. 273-287
Clocchiatti. R. ( 1975): Les inclusions vitreuses du quartz: donnees opliques, thermometriques et chimiques. Mem. Soco Gcot. Franee. 122, 1-96
Dietrich, A .• Lehmann, B. y Wallianos, A. (2000): Bulk rock and melt inelusion geochemistry of bolivian tin porphyry systems. Ecollomie Ge% gy, 95-2.: 3 13·326
Ehlers, A.G. Y Blat!. H. ( 1 982): Petrology; igneous. sedimenlary and metamorphic. Freeman & Co, San Francisco. 732 pp.
Irvine, T.N. y Baragar. W.R.A. (1971): A guide to the chemicnl classification of Ihe common volcanic rocks. Calladiall ¡al/mal Earlfl Sciellees, 8 , 523-548
Ishikawa, Y., Sawaguchi, T., Iwaya, S. y Horiuchi, M. ( 1976): Delineation of prospecting targets for kuroko deposits based on modes of volcanism of underlying dacite and alteration halos. Millillg Ge% gy, 26, 105- 1 17
ITGE (1997): Proyecto Inlegrado Faja Piritica. Informe Interno. 7 volúmenes. Centro de Documenl<lci6n IGME.
hckson, NJ., Willis-Richards, J . • Manning. D.A. y Sams, M.S. ( 1989): Evolution of the Cornubian ore field, SW England: Part 1. Mineral deposils and ore forming processes. Eeollomic Ce% gy, 84-5, 1 101. 1 133
Kamenetsky. V .S., Wolfe, R.e.. Eggins, S.M .• Mernagh. T.P. y Baslrakov, E. (1999): Volatile exsolution at the Dinkidi Cu-Au porphyry deposito Philippines: a melt inelusion record of initial ore-forming processes. Ge% gy, 27-8, 691-694
Large. R.R., Gemmell, J.B. y Paulick. H. (2001): The alteration box plol: A simple approach to understanding Ihe relationship between alteralion mineralogy and lilhogeochemistry associated with volcanic-hosled massive sulphides. EeOIlO/llic Geology. 96. 957-971
Leistel, J.M., Marcoux. E . • Thieblemont. D . • Quesada, C., Sánchez, A., Almodovar. G.R., Pascual. E. y Sáez. R ( 1998): The volcanic-hosted massive sulphide deposits of the Iberian Pyrite Beh. Review and preface lo the spedal issue. Millerafium Deposila. 33, 1-2. 2-30
Lenlz. D.R. ( 1 999): Pelrology, geochemislry and oxygen isolope inlerpretation of felsic volcanic and relaled rocks hosling the Brunswick 6 and 12 massive sulfide

104 TORNOS, F. el al.
deposits (Brunswick Belt), Bathurst Mining Camp, New Brunswick. Canada. Ecollomic Geofogy, 94.1, 57-86.
Un Qi Xia y Clocchiatti, R. ( 1985): A new classification of silicale mel! inclusions. Kexlle TOl/bao. 30, 488-492
Lowcnslern, 1.B. ( 1995): Applications of silicate-melt inc\usions lO ¡he study of magmatic volatiles. En: Maglllas, fluid! alld ore deposits. J.F.H. Thompson (ed.) Mineralogical Associatioll Cal/ada SllOrl Course. 23, 71-99
Mangas, J., Pérez Torrado, F.J., Massare, D. y Clocchiaui, R. ( 1993): Phonolitic origin of Roque Nublo ignimbrites of Gran Canaria (Canary Islands, Spain) (rom ctinopyroxene me!' inclusion studies. EI/ropean Joumal Mil/era/ogy, S, 97-106
Mavrovenges.JA y BocInar, RJ. (1994): Hydrogen movement into and out of fluid inclusions in quartz: experimental evidencc and geological implications. Geochilllica. COSlllochimica Acta, 58. 141·148.
Middlemost, E.A.K. (1985): Magmas and magmatic rocks. Longman, London, 266 pp
Mitjavila, J., Mard, J. y Soriano, C. ( 1997): Magmatic cvolution and tcctonic setting of the Iberian Pyrile Selt volcanism. Joumal Petrology, 38·6, 727-755
Munhá, J. (1983): Hercynian magmatism in the Ibcrian Pyrilc Sclt. Mem.Servicio Geo{ogico Portugal, 29, 39·81
Munhá, J. ( 1990): Metamorphic evolution of the South Porluguese/Pulo do Lobo Zane. En: Pre-Mesozoic evo/utioll oi Iberia. R.O. Dallmeyer y E. Martinez.Garcia (eds.) Springer Verlag, 363·368
Munhá, J .• Barriga, F.J.A.S. y Kerrich, R. ( l 986): High 110 ore forming fluids in volcanic hosted base metal massive sulfide deposits: Geologic, Ilorr.o, and D/H evidence from the Iberian Pyrite Belt, Crandon, Wisconsin and Blue Hill, Maine. ECOllomic Geology, 81·3, 530-552
Naney, M.T. (1983): Phase equlibria of rock forming ferromagnesian silicates in
granltlc systems. Americall JOl/rllal Sciellce, 283, 993-1033
Pollard, P.1., Taylor, R.G. ( 1986): Progressive evolution of alteration and lin mineralization: controls by interstitial permeability and fracture related tapping ofmagmatic fluid reservoirs in lin granites. ECOllomic Geofogy, 81-7, 1795-1800
Quesada, C. (1998): A reappraisal of the struclUre of the spanish segmenl of the Spanish segment ofthe Iberian Pyrite Belt. Milleraliul/I Deposita, 33, 1-2, 3 1 ·44
Relvas, J. (2000): Geology and metallogeny at the Neves Corvo deposit, Portugal. Tesis Doctoral, Univ. Lisboa, 3 19 pp
Roedder, E . ( 1984): Fluid inclusions. Reviews Mil/eralogy, 12. 644 pp.
Sáez, R., Almod6var, G.R. y Pascual E ( 1996): Geological constraints on massive sulphide genesis in the Iberian Pyrite Belt. Ore Geology Reviews, 11, 429·45 1
Sánchez España, J., Velasco, F. y Yusla, r. (2000): Hydrothermal alteration of fetsic volcanic rocks associated with massive sulphide deposition in the Northern lberian Pyrite Beh (SW Spain). Applied Geocl!emistry, 15, 1265- 1290
Sillitoe, R.H. ( 1985); Ore related breccias in volcanoplutonic ares. ECOllomic Geology, 80, 1467· 1514
Silva, J.B., Oliveira, J.T. y Ribeiro. A. ( 1990): Structural outline of the Soulh Portuguese Zone. En: PreMesozoic Geology oflberia, R.O. Dallmeyer y E. Martinez Garda (eds.) Springer Verlag, 348-362
Simonov. V.A. ( 1993): Petrogenesis of ophiolites (thermobarogeochemical investigations). UIGGM SB RAS Publishing House. Novosibirsk, 247 p. (en' ruso).
Simonov, V.A., Zaikov V.V. y Kovyazin S.V. ( 1999): Paleogeodynamic conditions of Kyzyl-Tashtyg deposit hydrothermal systems (Earsl Tuva). Metallogeny of ancient and modern oceans - 99. Orebearing hydrothermal systems. Miass: IMIN UrB RAS. 1 6-23 (en ruso).

Me!! inclusions Iberian Pyrite Belt 105
Sobolev, A.V., Danyushevsky, L.V. (1994): Petrology and Geochemistry of Boninites from rhe North Terminarion of ¡he Tonga Trench: Constraints on the Generation Conditions of Primary High�Ca Boninile Magmas. JO/lr. Petrol. 35, 1 183- 12 1 1 .
Sobolev, A.V. y Slutsky A.B. ( l984): Composilion and crystallization condirions of ¡nirial melt of siberian meimechires in connelion wilh general problem of ultramafic magma. Ge% gy Ceop"ysics, 12: 97- 1 10 (en ruso).
Solomon, M., Tornos. F. y Gaspar. O.F. (2002): Explanarion for many of ¡he unusual features of Ihe massive sulfide deposits of the Iberian pyrite belt. Ceology, 30-1. 87·90
Titov. A.V., Vladimirov, A.G .• Kruk, N.N. y Palessky S.V. ( 1996): Petrogenesis and age of volcanic rocks of Kyzyldarabat strulure (South-Earst Pamir). Ce% gy Ceop/¡ysics, 37·5. 62-72 (en ruso).
Thieblemont, D., Pascual, E. y Slein, G. ( 1998): Magmatism in the lberian Pyrite Belt: petrological constraints on a metallogenic model. Minera/illlll Deposita, 33, 1·2, 98� 1 1 0
Thomas, R . ( 1994): Estimatíon of Ihe viscosilY and the water conlent of silicale melts from melt inclusion data. Ellropeall JOllmaf Mil/era/ogy, 6, 5 1 1·535
Valenzuela. A., Donaire, T. y Pascual, E. (2001) : The Odiel River section: an example of complexities in volcanic evolution of ¡he Volcano Sedimentary Complex in {he Iberian Pyrile Belt, Spain. En: "GEODE Workshop: Massive sulphide deposilS in Ihe Iberian Pyrite Belt: New advances and comparison with equivalent syslems", FTornos, E.Pascual, R.Saez y R. Hidalgo (eds.), 61-62.
Whitney, J.A. (1989): Origin and evolution of silicic magmas. En: Ore depositiol/ associalel! witli lI/a8l11as, J.A. Whitney y AJ. Naldrell (eds.), Rel'iews Ecollomic Ce% gy, 4 , 183-202
Winkler. H.G.F. (1978): Petrogénesis de rocas metamórficas. Ed.Blume, Madrid, 346 p.
Yang, K. y Scon, S.D. ( 1 996): Possible contribution of a metal-rich magmalic fluid 10 a sea-noor hydrothermal system. Natllre, 383, 420-423
Zhaolin. L. ( 1994): The silicale melt inclusions in igneous rocks. En: Flllid Incfll$iollS ;" 1/I;lIerafs: Metlwc/s (J1U1 applicaliol/s, B. Vivo, M.L. Frezoni (eds.), Sllort COllrse Jllfematiollal Mil/eralogical Assoc. , 73-94
Recibido: Enero 2003 Aceptado: Julio 2003

Bolelill de la Sociedad Espa/1ola de Mi/leralogía. 26 (2003). 107-112 107
Sulfosales de Ag-Bi-Pb de Bustarviejo (Sierra de Guadarrama, Madrid)
Tomás MARTfN CRESPO·, Elena VINDELl & José A. LÓPEZ GARCfAl
• Dpto. Matemáticas y Física aplicadas y Ciencias de la Naturaleza. ESCET, Univ. Rey Juan Carlos, CI Tulipán sIn, 28933 M6stoles. (Madrid) 1 Dpto. Cristalografía y Mineralogfa. F. C.C. Geol6gicas. Univ. Complutense, Ciudad Universitaria sIn, 28040 Madrid
Abstracl: Ag-Bi-Pb sulphosahs from Ihe Sierra de Guadarrama (Bustarviejo. Madrid) have been firsl characterised in this work. The most abundant Ag-Bi-Pb sulphosalts have been c1assified as "schirmer1te" in addition to the Ag-Bi sulphosal¡s (matildite) previously reporled. A conlinuous composition:r.l trend in Ihe Pb-Ag-Bi minerals has been defined: pure galena, Ag-Bi·bearing galena, AgBi-Pb sulphosalts, Ag-Bi sulphosalts and native bismuth. This trend suggesls a mineralised nuids evolution characterised by impoverish in Pb and enriehmenl in Ag and Bi.
Key words: Sulphosalt, Sierra de Gu:r.d:r.rr:r.ma, matildite, schirmerite, mincralisation
Resumen: En el presente trabajo se han idcntificado por primera vez en la Sierra de Guadarrama (Bustarviejo, Madrid) sulfosales de Ag-Bi·Pb. que se pueden clasific:r.r como "schirmerita". Estas sulfosales son mb frecuentes que otras sulfosales de Ag-Bi pobres en Pb (matilditas) definidas hasta el momento. Se h:r. delermin:r.do una v:r.riaciÓn composicional eontrnua en los minerales de Pb-Ag-Bi: galena practicamenle pura, galena con cantidades apreciables de Ag y Bi, sulfosales dc Ag-Bi-Pb. 5ulfosales de Ag-Bi y bismuto nativo. Este hecho sugiere un empobrecimiento en Pb a la vez que un enriquecimiento en Ag y ai de los fluidos mineralizadores.
Palabras dave: Sulfosal, Sierra de Guadarrama. matildita. schirmerita. mineralización
Introducción
Las sulfosales de Ag-Bi de la Sierra de Guadarrama (Sistema Central) fueron citadas por primera vez por Cánepa ( 1968) en la mina Mónica, Buslarviejo (Madrid). En este primer trabajo. fueron definidas como matilditas (AgBiS1), y su presencia en Bustarviejo fue reiterada por P. Ramdohr en su manual sobre identificación de minerales opacos al microscopio y sus intercrecimientos (Ramdohr, 1969). La clasificación de estas sulfosales como matilditas (tambitn denominadas schapbachitas) se confirmó en trabajos poste-
riores (Samper ( 1977). Vindel ( 1982) y MarHnez Frras el al. (1984)]. Por otro lado. Mayor et al. ( 1 986) tambitn reconoció la presencia de matilditas en las mineralizaciones argentíferas de la zona de La AcebedaRobregordo (Madrid). al Este de Bustarviejo. En estos trabajos, la clasificación de las sulfosales de Ag-Si como matilditas atendió a criterios texturale$ (Cánepa, 1968; Ramdohr. 1969; Samper. 1977) o, posteriormente. a la interpretación de los análisis de microsonda electrónica (Vindel. 1982; Martínez Frfas el al.. 1984; Mayor el al.. 1986).
Actualmente, el análisis de fases minera-

108 MARTÍN CRESPO, T. el al.
les utilizando microsonda electrónica con ]f. mites de detección más bajos, menor tamaño de haz de electrones y la posibilidad de obtener imágenes con electrones retrodispersados. permite una caracterización más exacta de las mismas. Un estudio de detalle sobre la mineralización de Buslarviejo sugiere la posibilidad de que las fases clasificadas anteriormente como matildilas correspondan en su mayoría a otras sulfosales de Ag-Bi. En el presente trabajo se han analizado 14 nuevas muestras de la mineralización de As-(Ag) de Buslarviejo, se han identificado nuevas sulfosales de Ag-Bi-(Pb) además de matildita y se han comparado con los datos publicados de la mineralización As-(Ag) de La AcebedaRobregordo.
Sulfosales de Ag.Bi-Pb
M in era I i¡aciones La mineralización de As·(Ag) de
Bustarviejo rellena pequeños filoncillos « IOcm) y cavidades que siguen una dirección predominante N30-35°E y que encajan en ortogneises bandeados de edad Preordovícica (Martín Crespo et al., 2003). Tanto la roca de caja como las venas mineralizadas presenlan una brechificación característica y se reconoce una importante sericitización y cloritización como resultado de la interacción fluido-roca. La mineralización está caracterizada por la circulación y precipitación de tres estadios hidrotermales: estadio 1 (As-Fe), constituido por arsenopirita, pirita J, moscovita y cuarzo J; estadio 11 (Zn-Cu-Sn), en el que se han formado esfalerita, calcopirita. pirrotina, estannita, y pirita 11, y estadio III (Pb-Ag-Bi), represenlado por la presencia de galena, sulfosales de Ag-Bi. bismuto nativo y cuarzo 11.
La zona de La Acebeda-Robregordo está caracterizada por la presencia de varios filones mineralizados de escasa potencia (inferior a 1 m), direcciones variables (N30-35°E, N80"E y N 1 30"E) Y buzamientos subverticales,
que encajan de manera discordante en gneisses glandulares. y metasedimentos (Mayor et al.. 1986). Son frecuentes los fenómenos de alteración hidrotermal tales como silicificación, caolinización y turmalinizaciÓn. La asociación mineral es semejante a la de la mineralización de Bustarviejo, en donde se han identificado los mismos estadios hidrotermales: estadio (As-Fe), con arsenopirita. pirita, pirrotina y cuarzo 1; estadio 11 (Cu-Zn), con la presencia de esfalerita, calcopirita y freibergita. y estadio 111 (Pb-Ag-8i), caracterizado por galena, pirargirita, matildita, bismuto nativo y cuarzo 11.
En ambas mineralizaciones se reconoce un último estadio de alteración supergénica. que provoca la formación de marcasita. covellina. escorodila y goethita.
Estas mineralizaciones se caracterizan por el desarrollo de numerosas texturas e intercrecimientos entre sulfuros. Destacan en el estadio li la presencia de gotas y laminillas de calcopirita, estannita y pirrotina incluidas en esfalerita. así como gotas y estrellas de esfalerita y estannita en calcopirita. El estadio 111 está caracterizado por la presencia de sulfosales de Ag-Si siempre asociadas a galena, en forma de granos subredondeados o de laminillas dispuestas según los planos ( 1 1 1) de la galena. Esta textura en enrejado se ha interpretado tradicionalmente como producto de procesos de exsolución (Ramdohr. 1969).
Caracteriz.ación de las mlfosales Para la realización de los análisis del pre
sente trabajo se ha utilizado la microsonda electrónica de la casa JEOL del Centro de Microscopia Luis Bru (Universidad Complutense de Madrid). Se empleó un voltaje de 20 kV. un haz de electrones de 1 mm, y galena, anglesita. niquelina, AsGa, HR-160(CoNi·Cr). calcopirita. Zn. Mo. Ag, Cd, Sb, Bi2Te}, cinabrio y hutchinsonita como patrones.
Los datos de microsonda de las sulfosales de Ag·Si llevados a cabo están recogidos en la Tabla 1 . A partir de los análisis microsc6-

Sulfosales de Ag-Bi-Pb de Bustarviejo (Sierra de Guadarrama, Madrid) 109
picos y microanalíticos se han reconocido sulfosales de Ag-Bi-Pb. sulfosales de Ag-Bi. galena con cantidades apreciables de Bi y Ag. Y bismuto nativo.
Suljosales de Ag-Bi-Pb Las sulfosales de Ag-Bi-Pb más frecuen
tes en la mineralización de Bustarviejo corresponden a composiciones ricas en Pb (23,23-31.50 %peso) y. de acuerdo con Cook (1998), se proyectan en el campo de schirmerita en el diagrama triangular Ag-BiPb (Fig. l) . El contenido en Ag de estas sulfosales oscila entre 8.10 y 12.05 %peso. Se trata de cristales o agregados de cristales con hábito tabular (Fig. 2a. 2b), siempre asociados a galena y que se diferencian de ésta por su fuerte anisotropfa. Los datos de microsonda muestran una composición más o menos homogénea (Tabla 1) que, dentro del campo de schirmerita. se proyectan muy cercanas a los términos composicionales de tipo eskimoita, vikingita y/o gusta 'lita (Fig.1).
Si
.,
.,
.,
.,
..
SI/ljosafes de Ag-Bi A diferencia de lo que se ha publicado
hasta la fecha. en la mineralización de Buslarviejo las su1fosales sin Pb (matildita) son accesorias en comparación con aquellas ricas en Pb. En las 14 muestras estudiadas en eSle trabajo se han encontrado unicamente 4 granos que correspondan a verdaderas matilditas. Se trata de intercrecimientos de pequeñas laminillas de matildita con galena (Fig. 2c,d), que se disponen según las direcciones ( 1 1 1) de la misma, y que destacan por su elevada anisolropía. Su composición (Tabla 1 ; Fig. 1) es relativameante homogénea, con una proporción de hasta 3,29 %peso de Pb en las escasa matilditas encontradas en Bustarviejo. En los análisis que se realizaron en la mineralización de la AcebedaRobregordo (Mayor et al., 1986) no figura el contenido en Pb. El contenido en Ag (>20 %peso) de las fases minerales que se pueden considerar verdaderas matilditas en Bustarviejo (Tabla 1, análisis 12 y 13), es
......... ,kja • SUlh....!"�.IIi."', 0 " .. 1, ..... ..... _ ........ ..--... o �"", .... ,
•
Figura 1.- Análisis de microsonda electrónica de las sulfosalcs de Ag-BHPb) de Bustarviejo y La Acebeda-Robrcgordo proyectados en el diagrama ternario Ag-Bi-Pb. Términos teóricos segun Foord y Shawe ( 1989).

Tabla l . Análisis representativos obtenidos con microsonda electrónica (% peso) y fórmulas minerales calculadas de sulfosales de Ag-Bi-Pb de Bustarviejo y La Acebeda-Robregordo, y galena de Bustarviejo. o
Sulrosales Ag-Bi-{Pb) Galena
Aru'ilisis Bust. Bust. Bust. Busl. Bust. Bus!. Bust. S"". Bust. Busl. Busl. Bus!. BuS!. Aceb. Accb. Accb Aceb Bust. Bus!. Bust.
(1) (2) (3) (4) (') (6) (7) (') (9) (10) ( 1 1) (12) (13) (14)' (IS)' (16)* (17)' (IS) (19) (20)
F. 0.00 0,05 O,OS 0.01 0,03 0,01 0,01 0,01 0,03 0,02 0.00 0,01 0,01 n.a. n.a. n.a. n.a. 0.02 0,05 0,01
Ag '.54 '.90 S,S2 9.76 9.60 S.IO 1 1 .73 10,OS S.5S 10.55 12,05 2 1 .41 22,54 2S,18 26.68 24,90 36.47 1,01 1 . 1 1 0.00
Pb 31,50 26,41 29,15 25,61 27,52 25.84 31,41 25,72 29,90 29,66 23,23 3,29 1,14 n.a. n.a. n.a. n.a. 81,88 SI,31 SS,57
Si 44,39 46.73 45,62 4S,I3 4S,81 49,25 39.35 47.39 45,26 43,12 46,26 55,53 56,17 55,13 56,10 S7,3S 42,98 3,10 J." 0,20 :;: »
S 16,03 16.29 16,06 16,54 16,36 16.66 IS,SO 16,29 16,06 16,26 16,54 17,09 I7,OS 16,6S 17,22 17,75 20,SS 13,53 13,57 13,35 '" So 0.00 0.03 0,04 0,03 0,02 0.01 om 0,00 0,02 0.03 0.00 0,02 0.00 0.01 0,00 0,00
.... n.a. n.a. n.a. n.a. -Z
T. 0.15 0,10 0,16 0.11 0.15 0,19 0,16 0,11 0,14 0,17 0,14 0,12 0,16 n.a. n.a. n.a. u 0,15 0,14 0, 1 1 () '" rn
Total 100,61 9S,51 99,90 100,20 99,48 100,06 98,46 99,60 99,96 99,79 9S,23 97,47 97,10 99,99 lOO lOO 100 199,70 99,87 99,24 '" 'O O
.... Formula: (6) Ar.o.l6Pb, .. ,Biu,(S,. .... Teom) ( 12) ASo."Pbo.cr,Bio..,S¡ (IS) Pbo."ASo.o!Bio.o,.s 2 (1) AGo."Pb,."B¡uo(S�."Tc,w,) (7) Ag,_t!Pb, ... Bil.ltCS.l.91 Teo.Ol) (13) AknPbo.mBi,.II'S¡ (19) Pbo.9)A8o.olBioJl1S "-
(2) Feu,A80.91Pb,J08i:.6J(S.l.99 Tc,w,) (8) Ag, . .J>b,...aiu�S,.99 Te..u,,) (14) Ag,.ooBi,.o,S¡ (20) PbS (3) Ag.,.9IPb, .... Bi¡...o(S.I..SCU,TCo,o,) (9) Fea.o,Al:o.nPb,.nBiutCS.l.99 TCo.l1') (IS) A&,9¡Bi,.oaS¡ n.a.: no analizado
(4) Ag,JI1Pb, .• ,Biw(S,.",TCo,o,) (lO) Ag,." Pb',69Bi¡,o(S'.9I TCo.ozl (16) ASo,uBia,,,.51 • Según Mayor el al. (1986)
(S) Fe'I.IIIAg,�Pb,,,.Bi!,)l(S,.,,, Tc,w,) ( 1 1) Ag'.l'IPb,�i2.!1(S"99 Tc,w,) (17) Ag'.olBiu"S¡

Sll l fosales de Ag-Bi-Pb de Bustarviejo (Sierra de Glladarrama, Madrid) 1 1 ¡
sensiblemente mayor que el de las otras sulfosales anal izadas « 1 2 %peso), mas ricas en Pb (Tabla 1, anál is is 1 al 1 1 ) . Las mat i ld i tus
de l a m i neral i zación de La AcebedaRobregordo muestran unos contenidos en Ag (>25 %peso) aün mayores (Tabla ¡ , "n¡¡l is is 14 a l 17) .
lV/¡nera/es asociados A destacar es la presencia de galena con
unos contenidos anormalmente altos en B i y Ag, Y otras casi l i bres de estos elementos (Fig. 1). Las primeras (Tabla 1 , anál is is 18 y 1 9) muestran unas propiedades ópticas l igeramente d i ferentes a las de las galenas puras, tales
Figura 2.- (n) Cristales tabulares de sul fosalcs de Ag-Bi-Pb (gris claro) incluidos en galena (luz rellejada, N.C.), (b) Misma imagen que Ca) con microscopía electrónica en imagen de electroncs rctrodispcrsados (gris oscuro: sulrosalcs dc Ag·Bi.Pb). (e) Laminillas de matildita (gris claro) en galena (luz renejada, N.C.), y (el) Misma zona que (e), obtenida con microscopía electrónica en imagen de electrones retrodispersados (gris oscuro: matildita).

1 12 MARTíN CRESPO, T. el al.
como un color mas azulado, ausencia de figuras de exfoliación e inclusiones de bismuto nativo. Las sulfosales de Ag-Bi-(Pb) se encuentran intcrcrecidas con este tipo de galena.
Discusión y conclusiones
Las sulfosales de Ag-Bi predominantes en la mineralización de Buslarviejo son las (ases ricas en Pb (schirmcrita), mientras que las pobres en Pb (mulildita) se encuentran únicamente como fases minerales accesorias. La última etapa mincralizadora (estadio 111: Pb-Ag-Bi) muestra una variación composicional conlínua desde galenas practicamente puras, galenas con cantidudes apreciables de Ag y Bi, sulfosales de Ag-BiPb. hasta sulfosales de Ag-Si y bismuto nativo (Fig. l ) . Esto sugiere un empobrecimiento en Pb de los fluidos mineralizadores así como un enriquecimiento en Ag y Bi de las fases finales del proceso mineralizador, coherente con el desarrollo de procesos de ex solución de unas fases en otras, como el de las sulfosales de Ag·Bi-Pb en galena. La ausencia de análisis de Pb en las sulfosales de la mineralización de La Acebeda·Robregordo, debido a las limitaciones analíticas del momento en que se realizó el trabajo, probablemente no permitió caracterizar adecuadamente las sulfosales presentes. Este hecho, unido a las similitudes mineralógicas con la mineralización de Bustarviejo, permite suponer la presencia de una mayor variedad de sulfosales de Ag-BiPb en La Acebeda-Robregordo y. por tanto. en otras mineralizaciones del mismo tipo de la Sierra de Guadarrama. tales como Tamajón y Prádena del Rincón (Martínez Frías. 1986).
Bibliografia
Cánepa. C. (1967): Contribución a la metalogenia de la Sierra de Guadarrama (Hojas 484 y 509. Prov. Madrid). Ph D
Thesis. Univ. Complutense. 109 p. Cook, NJ. ( 1998): Bismsuth sulphosalts from
hydrothermal vein deposits of Neogene age. N.W. Romania. Mmit.Osterr. Miner. Ges. 143, 19-39.
Foord. E.E. y Shawe, D.R. ( 1989): The Pb-BiAg-Cu-(Hg) chemistry of galena and some associated sulphosalts: a review and some new data from Colorado. California and Pennsylvania. Call. Mil/eral. 27. 363-382.
Marlínez Frfas, J .• Vindel. E., Lunar, R. ( 1984): Estudio textural y metalogénico de la mineralización de Bustarviejo (Sierra de Guadarrama). Rev. Mar. Proc. Geo!. 2. 177-192.
Martínez Frias. J. ( 1986): Mineralogía y metalogenia de las mineralizaciones de Ag del Sector Oriental del Sistema Cenlral. Ph D Thesis. Univ. Complutense. 379 p.
Mayor. N . • VindeJ, E., Lunar, R. ( 1986): Metalogenia de las mineralizaciones argentiferas del Sistema Central: Zona de La Acebeda-Robregordo. Bol. Geof. Min . 96. 473-485.
Ramdohr, P. ( 1969): The ore minerals and their intergrowlhs. Pergamon Press. 1 174 p.
Samper. J. ( 1977): Estudio metalogénico y evolución de la minería en la mina Mónica de Bustarviejo. Teclliterrae 19. 14-22.
Yindel. E. ( 1982): Estudio mineralógico y metalogénico de las mineralizaciones de la Sierra de Guadarrama (Sistema Cenlral Español). Parte 1. Bol. Geol. Min. 93. 33-58.
Recibido: Junio 2003 Aceptado: Noviembre 2003

Boletfn de la Sociedad espm-'ola de Mi/lera/ogia, 26 (2003), / J 3-/2/ 1 13
Policromías de la Iglesia del Monasterio de San Jerónimo de Granada: composición, alteración y técnicas de ejecución.
Polychromy in the Church oC San Jerónimo Monastery in Grana·
da: nature, alteration and painting techniques
Carolina CARDELL-FERNÁNDEZ' y José RODRfaUEZ-GORDILLOz l.-Opto. Geología. Área Edafologra y Química Agrícola, Facultad de Ciencias Experimenta
les. Paraje Las Lagunillas sIn. Universidad Jaén, 23071 Jaén. 2.-0pto. Mineralogía y Petrología, Facultad de Ciencias, Av. Fuentenueva sIn. Universidad Granada, 18004 Granada.
Abstrad: The inleriorof the Chureh ofSan Jerónimo monastery in Granada was complelely eovcred with mural paintings in the 18'� eemury, as well as polychrome seulptures in the upper altar-piecc. Massive salt crystaJlization, due to intense infiltration by groundwatcr and precipitation, has lead to sometimes irreversible alteration of the murals and their supponing materials (calearenites, rendering ... ). This study examines the state of alteration of the murals and polychromy in sculpture, as well as composition and painting techniqucs, with an ultimale goal of facilitating eonservalion.
Murals were painted following various /releo UCO techniques, suggesling that yarious pcriads or artists are rcpresenled. The sometime use of yeUo\V chrOl/lt (a pigment available commercially beginning only in 1797) supports this theory. The sculptures were polychromed vía the Itlllplt technique. In neilher murals nor sculplUrcs werc lhe calcarenites protected; rather. gypsum-prcparation and clay primer layers (minerals that are Yery absorbent of humidity) were directly applied, thus fayouring the weathering of the paintings. The reaetion between sulphates (from gypsum) and the mineral pigment cations - e.g., clays. lead carbonates, and iron oxides - led to the formation of Tul1011 So/u and lead chlorides.
Key words: polychromy, seulptures, mural paintings, optical microscope. SEM-EDX. alteration. salts. painting teehniques.
Resumen: El interior de la Iglesia de San Jerónimo se cubrió totalmente en el siglo XVIII con pinturas murales, incluidas las esculturas de la zona superior del retablo mayor. Debido a la intensa infiltración de agua del terreno y de precipitación. cristalizan sales de forma masiva provocando la alteración (a veces irreversible) de dichas pinturas y sus materiales soporte (ca1earenita, revocos y enlucidos). Este trabajo aborda el estudio de Ills pinturas mUTlllcs y la policromfa de esculturas, su composición. técnicas de ejecución y estado de alteración, en aras a su conservación.
La pintura mural fue ejecutada al/reseo seco según distintas técnicas, sugiriendo que fue ejecutada en direrentes épocas o por diversos artistas. El uso de amarillo de cromo (pigmento comercializado a partir de 1797) apoya esta teorra. Las esculluras fueron policromadas al ll.'mple. En ambos casos la calearenita no se protegió con mordiente. llplicando directamente preparaciones de yeso e imprimaciones de arcilla (soportes porosos y absorbentes de humedad). potenciando asr la alteración de IllS pinturas. La reacción entre sulfatos (del yeso) y los cationes procedentes de los minerales de los pigmentos. e.g. arcillas, carbonato de plomo y oxi-hidróxidos de hierro. ha originado la formación de Soles de TUI/o/I y cloruros de plomo.
Palabras clave: Policromfa. esculturas. pintura mural, microscopio óptico, SEM-EOX. alteración. sales, técnicas pictóricas.

1 14 CARDELL FERNÁNDEZ, C. el a l .
Introducción
La construcción d e l a Iglesia del Monasterio de San Jeró n i m o se i nic ió e n e l primer tercio del s i g l o XVI e n un e s t i l o gótico isabelino senc i l lo. L a intervención del arquitecto Diego de S i loé convirtió a l templo en una de las más notables obras del renacimiento español. La i g l es i a consta de una única nave con cabecera semioctogonal (ábside) y cuatro c a p i l l as en ambos brazos del crucero, estando el retablo mayor rlanq ucaclo a cada lado por u n a h a b i ta c i ó n (Col i na M u nguía, 1 986) (Fig. I ) . El relablo fue comenzado en el s i glo XVI ( 1 570) por Juan d e Aragón, y const i tuye una d e las obras maestras d e l a imagi nería española, siendo punto de arranque de las esclIelas d e escultura de Sevi l l a y Granada (Camón, 1 9 8 1 ) . El fondo del presbiterio y bóvedas central y laterales están cubiertos por hornacinas ornamentadas con figuras de santos, héroes, bustos, querubines, monstruos, etc. En el s i g l o X V I I I todas las naves se cubrieron con p i n t ur a s murales al Fesco realizadas por Juan de Med i n a ( 1 723-1 735), i n c l uidas las esculturas de S i loé (Colina M u n g u ía, 1 986).
E l exterior d e l a Iglesia se d i vide en dos cuerpos coronados por un c i m borrio. El primer cuerpo (de unos 15 m d e a l t ura) está construido pr incipa l mente con c a l i za de S ierra E l vira y travert i n o de A l facar (Gallego y B urín, 1 982), estando el segundo cuerpo y el c i m borrio construidos por calcarenita d e las canteras de Santa Pudia (Rodríguez Navarro el a l . , 1 99 1 ). La ig les ia CSIi.í cubierta en su i nterior por revocos d e d i s t i n ta naturaleza (yeso, yeso y cal, y cal y arena). ESlUdios sobre el estado d e conservación de la Iglesia d e San Jerónimo revelan l a existenc i a mas i va de sales en el i nterior del edificio. afectando tanto a m a t e r i a l es decorativos (revocos, enlucidos, policromías y p i nturas murales) como a constructivos (Cardell-Fern;índez y Rodríguez Gord i l lo, 1 992, 2003). Las sales se local i z a n principal mente en la zona baja del
;íbside y sus dos habitaciones laterales (consecuencia del ascenso capi lar d e humedad del terreno), y e n l a base de a l g unas esculturas de l a zona superior del ábside, a linos 15 m de altura (debido a i n fi l tración de agua de l l u via). El resultado en ambos casos es l a arenización y/o desmoronamiento (sacarificación) de s i l l ares de calcarenita y el desprend i miento d e enlucidos y capas pictóricas.
La pr:ktica ele policromar esculturas de piedra estuvo m uy d i fu n d i d a entre los pueblos pr imit i vos, las c i v i l i zaciones no europeas, el mundo ant i guo preclásico y c l <ísico y la Edad Med i a rom á n i c a y pregótica. Esta práctica cayó en desuso a parti r del s . XVI (Mallese, 1 973). Existen documenlos que men-
D Fig. l . Plallla del Monasterio e Iglesia de San Jerónimo de Granada. Zonas de muestreo: ábside ( 1 6) : zoníl superior del retablo con orienlaeión E (esculturas A , B Y C) y orientación W (esculturas D, E Y F); parte i n ferior del retablo: zona Este y Oeste del altar mayor y habitaciones E y W adyacentes al mismo.

Policromías de la Iglesia de San Jerónimo de Granada 1 15
cionan la importancia concedida a la policromfa de las esculturas durante ciertas épocas de la historia del arte. De hecho, el trabajo se encargaba a pintores famosos, a veces mejor pagados que el propio escultor (Libro de Metiers escrito en 1268, en Aubert. 1964). A pesar de ello, los recetarios y tratados técnicos medievales (y de otros períodos) dicen poco o nada sobre los materiales y los métodos para policromar esculturas pétreas. En general. coinciden en que las esculturas siempre debían policromarse, y la policromía tenía que aplicarse sobre una capa de imprimación (Libro de Mefiers op.cit.; Tratado de Erq,cfio del s. XIII, en Merrifield, 1967; El libro del arte del s. XVI en Cennini, ed. 1988). La.información sobre los materiales y las técnicas de ejecución empleadas en policromar esculturas es escasa. particularmente la referente a la escultura monumental y a los espacios exteriores de monumentos arquitectónicos. Han sido las restauraciones de finales del siglo XX y los estudios analíticos de policromías (Rossi Manaresi, 1987) similares a éste que aquí se presenta, los que arrojan luz sobre dicho tema.
En este trabajo se aborda el estudio de la pintura mural y de las policromía de las esculluras de la Iglesia de San Jerónimo en Granada: su naturaleza, estado de conservación y técnicas de aplicación. Dichas policromías constituyen una muestra de la calidad anfstica del barroco español, lo que justifica la necesidad de su estudio en aras a su preservación.
Muestreo y técnicas analíticas
En el muestreo se ha considerado la gama de colores empleada en las policromías y sus signos de alteración: variaciones cromáticas, fisuras, hinchamientos, desprendimientos, presencia de sales ... Se han tomado 23 muestras de policromía: ocho de las esculturas de la zona superior del ábside (PE3, PE4, PE6. PE7, PEI3. PE14, PE15 Y PE17) Y el resto de di-
versas pinturas murales: siete muestras de las paredes de las hornacinas donde se ubican dichas esculturas (PMI. PM2, PM5, PM8 a PMII) y ocho de las pinturas murales de las habitaciones laterales del ábside (PM-22, PM-23, PM-24, PM-27, PM-30, PM-32, PM-33 Y PM-34) (Fig. l .). La ubicación de las muestras, su color en superficie, el substrato sobre el que se aplicaron y su grado de alteración se recoge en la Tabla l . Las seis esculturas muestreadas en el retablo superior (ábside) se han nombrado con una letra: A, B, Y e para las esculturas situadas en la zona este del retablo, y D, E Y F para aquellas ubicadas en la zona oeste (Fig. 1 . n.16 = retablo mayor).
El examen preliminar de las muestras pictóricas se ha realizado mediante lupa binocular (Olympus SZHlO) para determinar globalmente su estructura interna, técnica de ejecución y estado de conservación. A continuación se han elaborado láminas delgado pulidas que permiten el estudio de las diversas estratigrafías pictóricas. La composición y microestructura de las estratigrafías pictóricas se ha analizado mediante microscopía óptica de luz polarizada (microscopio Carl Zeiss modelo Jenapol U con luz transmitida y reflejada) y con microscopía electrónica de bnrrido (SEM) (microscopio Zeiss DSM 950 con microanalizador Link QK 2000). La descripción de las capas pictóricas comienza siempre desde la zona más interna hasta la superficie. Así, la última capa pictórica descrita corresponde al color en superficie de la muestra.
Resultados
Pilltura lIlural y policromía de escultllras el! fa zona superior del ábside
En las esculturas policromadas se han identificado una capa de preparación gris, varias capas de imprimación coloreadas, y las capas pictóricas de color gris, amarillo, rojo, azul claro y azul oscuro:
Capa de preparación: elaborada con yeso

1 16 CARDELL FERNÁNDEZ, C. et al.
y algo de celestina (espesor = 100·300 ¡.tm) mezclados con abundante cola. Los cristales de yeso presentan hábito en «punta de necha» con tamaños comprendidos eOlre 1 0·65 ¡.too. La disolución del yeso ha favorecido un aumento de la porosidad, observándose yeso de recristalizaci6n. Los poros y microfisuras son frecuentes y originan despegues en la propia capa y respeclo a las superiores.
La/las capas de imprimación son de tonos tostados gradando de rojizos a amarillos (espesor = 120-230 ¡.tm). Se hao elaborado con arcillas ricas en hierro. oxi-hidróxidos de hierro, carbonato cálcico, yeso, cuarzo y carbonato de plomo (blallco de plomo). También se ha detectado sulfato de hierro (producto de alteración). El tamaño de los pigmentos es grande, alcanzando hasta 230 J.lm (PEI7). El
Tabla 1 : Muestras pictóricas estudiadas en [a Iglesia de San Jerónimo de Granada.
MUESTRA U81CACIÓN COLOR SUBSTRATO CONSERVACIÓN
HA8ITACIÓN E
PM-22 Acc;:eso habital;:ión Gris mct&Iico Cta + P de yeso Y caCo, S8: SO: de Fe. QIZOIcta5., desp1'aIdimicntO'S
PM-23 "km Verde Revoco cal S8: CI' y COJ- de Pb
PM-24 Id= Rojo Muy alterada
Da + P de yeso Idem + escamas
HABITACiÓN W
PM-27 Pared NW Gris Revoco cal y arena EfloresccncillS y SB PM-30 P""" E Gris Idem 'dom PM-)2 P"'" W Qri, Id= Id= PM-)) Id= on. ld= Id= PM-)4 Idem Gris Idem ,-
ABSIDE SUPERIOR W
PM·I Hornacina Violeta P de yeso M,y alterada: cazoletas, desprendimiollOS
PM·2 Hornacina escultura F Do�do Id= 'd<m PM-S Hornacina escultura F Gris a. 58: so; de Fe PM-8 Hornacina eseultura O "'� P deycso Idem PM-9 Hornacina escultura O "'� Idcm Id= PM·IO Base escultura O "'� Id= Muy a1tcJada: 59: SO; de Fe;
50/ yCo,' de Na PM-11 8ase escultura O Gris a. Idem PE-, Vestido escultura F Azul oscuro '<km Alteración moderada PE-< Pie escultura F Camación 'dom ldem PE� Pie escultura E Carnación Id= Cta pulverizada PE·7 Faldón escultura E Azul claro Idem Alteración moderada
ABSIOE SUPERIOR E
PE-13 Manto escultura C Gris Cta + P de yeso Cawletas, 58. PE-14 Faldón escultura B Gris-uulado 'don SB PE·IS Faldón escultura B Amarillo Id"" Cawletas, 58. PE-17 Pie escultura A Carnación Idcm Cta pulvenuda
P.M = Pintura Mural; P.E = Policromía escullur¡¡; Ct¡¡ = calcarcni\¡¡; P = C¡¡P¡¡ de prep¡¡radón; S B :> subcfloresccncias; • cazoletas = grietas en la capa de pintura en form� de pc:quci'l�s cazoletas cóncavas enn bordes levantados (Diaz M¡¡f\ns. 1978).

Policromías de la Iglesia de San Jerónimo de Granada 1 17
pigmento elaborado a panir de arcillas ricas en hierro se denomina Tierra, y presenta tonalida· des que van del amarillo al rojo. En las escullu· ras se han empleado principalmente la Tierra de Siella /latural y la Tierra de Siena tostada, ade· más del ocre rojo y ocre amarillo (ambos oxihidróxidos de hierro). Son frecuentes los poros de lO-lOO �m, las fisuras y los despegues entre estas capas y las capas adyacentes.
El color &r.i.l se ha obtenido aplicando sobre la imprimación rojiza una capa de yeso con abundantes impurezas de cuarzo y oxi· hidróxidos de hierro (PE 13).
Para el color amarillo se aplicaron dos capas de yeso (200 �m) sobre la imprima. ciÓn. En la capa superior el yeso se mezcló con carbonato cálcico. carbonato de plomo, y oxi-hidróxidos de hierro, proporcionando éso los últimos el color amarillo (PEI5). Se de· tectan despegues y poros inducidos por la disolución del yeso.
Los dos tonos de m.i.D. corresponden a carnaciones de los pies de dos esculturas. En PE6 la carnación es de tonalidad clara; se obtuvo al aplicar una capa rojiza sobre una capa blanca de yeso. En PEI7 la carnación es oscura; se aplicaron dos capas rojizas sobre la imprimación también roja (Fig. 2. A). Ambas carnaciones están elaboradas principalmente con blanco de plomo al que se ha añadido óxido de plomo (rojo llJi/lio) en PE6 y en PE17 rojo de óxido de hierro. además de pigmentos azules y negros, para imitar el color de la carne (éstos dos últimos no analizados por su naturaleza orgánica).
El .uu.l. se elaboró con awl de esmalte (vidrio silicatado de caballo). El tono claro se consiguió aplicando una 'capa de 45 �m de espesor de este pigmento sobre la preparación de yeso, y el azul oscuro aplicando una capa azul de esmalte de 90 �m sobre la imprima· ción rojiza.
De las pinturas murales que cubren las hornacinas y otros elementos decorati vos donde se ubican las esculturas se han estudiado el color gris, ocre, violeta y dorado:
El &W. se elaboró con yeso (cristales con hábito en «punta de flecha� y tamaños de 120x30 J.1m). correspondiendo a la capa de preparación (espesor = 140 �m) que se aplicó directamente sobre la calcarenita (PM5 y PMII). Además se han identificado oXlhidróxidos de hierro, y en las zonos más alleradas sulfato de hierro como producto de reacción entre el yeso y los oxi-hidróxidos de hierro. La disolución del yeso ha favorecido el desarrollo de microfisuras y despegues.
El color � se obtuvo de dos formas: i) aplicando sobre la calcarenita una capa (200 Ilm� compuesta principalmente por yeso y oxi-hidróxidos de hierro; además se ha detectado cal (grumos agrietados) y granos de dolomita, cuarzo, micas y oxi-hidróxidos de hierro, responsables estos últimos de la coloración amarilla. En la muestra PM8 (muy alterada) se han detectado diversas sales, como sulfato de sodio con morfologías aciculares, halila, carbonato sódico. mezclas de estas sales y cristales de sulfato de hierro; ii) aplicando sobre la calcarenita una capa de yeso y sobre éSla otra capa de yeso con abundantes oxi-hidróxidos de hierro y algo de calcita y micas. El espesor total de las dos capas es de 800 J.1m. presentando el yeso hábito en �punta de flecha,. y tamanos de hasta 45 �m. El aglutinante es abundante y los escasos poros aparecen coneclados formando microfisuras. Se han detectado sulfatos de hierro y sulfatos de sodio.
Violeta: se observaron las siguientes capas en la estratigraffa pictórica: preparación de yeso, imprimación rojiza de Tierra de Siena loslada. capa de yeso, capa de Tierra de Siena tostada y por último una lámina de estai'o (65 �m) muy alterada formando cazoletas de tono oscuro por la oxidación del estaño (Fig. 2. B). Las capas de yeso presentan abundantes poros originados por la disolución del yeso, dan· do lugar a desprendimientos entre capas.
Para el d.aW2. se aplicó sobre la calcarenita una capa de yeso, un bol rojo (arcillas ricas en hierro), tres capas de espesor

1 18 CARDELL FERNÁNDEZ. C. el al.
comprendido entre 50 y 200 Ilrn elaboradas con una mezcla de yeso y carbonilto cálcico, y por último una lámina de 5 Ilrn de oro. En gran parte el oro se ha perdido y el que permanece está muy oscurecido por la suciedad.
Pilllura$ mllrales de la lOlla inferior del dbside
En la habitación W se ha estudiado el color gris. Se obtuvo aplicando una capa (235 J.1m de espesor) compuesta principalmente por yeso más calcita. cuarzo, y escasos pigmenlos rojos de óxidos de hierro y azul de "bramar (azul de lapislázuli) sobre un revoco de cal y arena. En la habitacjón E se han muestreado los colores gris oscuro, verde y rojo oscuro:
El &ri1 se obtuvo aplicando sobre la calcarenila una capa de carbonato cálcico con algo de yeso y arcillas. y encima una capa rojiza de arcillas ricas en hierro y carbonato cálcico. Por último aparecen hasta tres láminas de estaño. desprendidas del sustrato y formando cazoletas. En la capa de arcillas se han detectado sulfatos de hierro.
El estudio del color � revela una capa muy alterada. donde se distinguen grumos amarillentos y verdosos. aplicada sobre un revoco de cal (Fig. 2. C). El SEM ha revelado la existencia en esta capa pictórica de carbonato de plomo. grumos verdosos compuestos por Cr. Pb y CI. otros nmnrillentos de Cr. Pb y Ca y cristales aciculares de cloruro de plomo (Fig. 2. O). El avanzndo estado de alteración de la capa pictórica dificulta establecer qué pigmento/s fueron los originalmente empleados. Se ha concluido que el pigmento utilizado fue el amarillo de cromo. un cromato de plomo que se vuelve verdoso con el tiempo. La masa de cristalitos compuesta por CI. Cr y Pb es producto de la reacción entre el cloro procedente de la halita (muy abundante en la zona baja de la iglesia) y amarillo de cromo.
El color [QjQ oscuro se consiguió aplicando dos capas de blallco de plomo con pigmentos rojos de óxido de hierro. sobre dos capas de yeso con impurezas de arcillas. Se
han detectado sales de cloruro de plomo como producto de alteración.
Discusión y conclusiones
ESC/l/wras policromadas La ttcnica de ejecución de la policromía
empleada en las esculturas de la Iglesia de San Jerónimo es el temple. A la ca\carenila se aplicó una preparación de yeso. encima varias capas de imprimación coloreadas y finalmente las capas pictóricas al óleo. El artista (o artistas) empleó una paleta de colores muy reducida: blanco de yeso para la preparación y blallca de plomo para las carnaciones; Tierra de Siel/a 1I0t/lral y Tierra de Siena tostada para las imprimaciones; az.¡¡1 de esmalte, ocre amarillo. mil/io. oro y estmio para las capas pictóricas. Dado que se trata de un temple no hay capas de barniz. La policromía se ejecutó según las reglas sugeridas por Cennini (Cennini, 1988). Respecto a las características de los pigmentos y la ejecución de la policromía, hay que mencionnr varios hechos:
i) Las Tierras precisan la máxima pureza para pmenciar la intensidad de su tono y resistencia a la alteración. Este trabajo ha puesto de manifiesto la escasa minuciosidad en la elaboración de las Tierras empleadas en estas policromías. como se desprende por la presencia de minerales como el carbonato cálcico. cuarzo y yeso, además de las arcillas (componentes principales de las Tierras) y el gran tamaño de los pigmentos (hasta 230 Ilm en PEI?).
ii) Las distintas tonalidades de azul se consiguieron jugando con el color de las capas inferiores y el grosor de la capa pictórica elaborada con awl de esmalte (pigmento artificial fabricado en el s. XVII y en desuso a mediados del s. XIX). Así, el azul oscuro se obtuvo aplicando una capa azul de unos 80-100 Ilm de espesor sobre una imprimación roja. de extendido uso en los s. XVII Y XVIII (Draz-Martos. 1978). Para el azul claro se

Pol icromías de la Iglesia de San Jerónimo de Granada 1 1 9
Fig. 2 . A , B y C. Microfotografías realizadas con microscopio óptico. Descripción de las estratigrafí<ls pictóricas de abajo hacia arriba. Barra ;; 0.5 mm. A) Preparación de yeso, imprimación rojiza, capa pictórica de color carne (luz transmitida y polarizadores 11). B) Preparación de yeso. imprimación rojiza, preparación de yeso, imprimación rojiza y lámina de estaño (Sil) (luz renejada y polarizadores #). C) Revoco de cal y capa pictórica amarillenta (luz renejada y polarizadores U). D) Microfotografía de SEM de la capa pictórica de la muestra C). Cristales aciclllares de cloruro de plomo prodllclO de reacción eIllre el pigmento amarillo de cromo y sales de halita (NaCI). p ;; capa de preparación: i ;; capa de imprimación, c.p. ;; capa pictórica; r ;; revoco.
apl icó una delgada capa azul (40 11m) sobre una preparación blanca.
i i i ) Las carnaciones mates se elaboraron con blanco de plomo. Los d iversos tonos de carne se obtuvieron añadiendo pigmentos de d iferente naturaleza y apl icando capas de imprimación de d i ferentes tonalidades.
iv) El método de dorar fue el dorado eOIl aceite también l l amado dorado (l/ lIIixtiólI. El procedimiento habitual consiste en encolar la superficie para conseguir una capa l isa y fina, esperar hasta obtener el grado de sequedad y adherencia correcto y aplicar el pan de oro (Mayer, 1 988). E n el presente caso, la lámina
de oro se aplicó sobre varias capas de yeso bien pulidas. En grandes superficies se empicaron láminas de estaño (falso oro) para im itar el oro, ya que era más económico (Matteini y Moles, 1 989). El estaño es muy sensible a los procesos de oxidación, por lo que debe protegerse con lacas o barnices. La ausencia de tal protección en estas pol icromías ha provocado su desprendim iento y oscureci-111 iento genera I izado.
La presencia de preparaciones de yeso (en contacto d i recto con la caJcarenita) e imprimaciones elaboradas con arcil las, ha potenciado el estado de degradación de las

120 CARDELL FERNÁNDEZ. C. el al.
policromías de las esculturas. Los fondos de yeso son muy porosos y absorbentes de la humedad (por lo que deben eSI:lr totalmente secos antes de trabajar con ellos) y las arcillas retienen el agua largo tiempo y poseen lendencia a meleorizarse (Doerncr, 1921). Mediante microscopía óptica y SEM se ha observado en todas las estratigrafías pictóricas fenómenos de disolución del yeso y de alteración en las arcillas. Los sulfatos libres en el medio (procedentes del yeso) se han combinado con los cationes procedentes de los minerales que constituyen los pigmentos. más los aportados por el agua de infiltración. Así, es frecuente encontrar sales del grupo de los sulfatos. Estas sajes se denominan Sales de TI/Uol! y constituyen una extensa serie de sulfatos monoclfnicos isomorfos de composición general A'2Bl'(SO)1 ' 6H10 donde A*= K .. NH;, Rb·, Cs', y 81+ = cunlquier catiÓn divalente con radio aproximado a 0.7Á. Los sulfatos de magnesio con 6 y 7 mol�culas de agua (hexahidrita y epsomita respectivamente) son los más abundantes en 13s esculturas. El Mg puede proceder de 135 arcillas de la imprimación, de [a calcaren ita (contenido en MgO entre 0.38 y 5.64 %, Cardel[-Fernández, 2(03) y/o del agua de infiltr3ción. En 13s capas pictóricas elaboradas con oxi-hidróxidos de hierro se han detectado sistemálicamente sulfatos de Fe, evidencia del elevado grado de alleración de las policromías.
Pimura mural La pintura mural es un elemento inte
grante de la arquitectura y no puede ser concebida ni comprendida aislándola de la construcción de que forma parte (Doerner, 1921; Mayer, 1988; 80ntc�, 1989). Este arte era resuelto en la �poca de esplendor de la pintura italiana por dos m�todos diferentes: i) el buon fresco (el verdadero fresco) en el que los pigmentos se aplican sobre una capa de cal húmeda. Una vez seco se obtiene una superficie vítrea y el color se conserva cnsi en forma permanente debido a la estabilidad de
los pigmentos; ji el secco fresco (fresco seco) en el que la pintura se ejecuta en una superficie de argamasa seca. Los colores se mezclan con agun y una solución de caseínn, cola o yema de huevo. El término secco se utiliz3 para describir cualquier método de pintura mural en seco en oposición alfresco O pintura sobre argamasa húmeda.
Las fuentes bibliográficas consultadas apuntan que las pinturas que recubren todas las paredes de la Iglesia de San Jerónimo son pinturas al fresco realizadas por Juan de Medina entre 1723 y 1735 (Gómez MorenoGonzález, 1892; Gallego Burín, 1982; Colina Munguía, 1986). En este trabajo se demuestra que el método de pintura mural empleado en la zona baja de la iglesia, y que puede hacerse extensivo al resto del Templo, ha sido elfresca seco, distingui�ndose varias técnicas. Así. en la habitación E la pintura se ha realizado al temple sobre la pared seca. Los pigmentos son muy escasos y aparecen englobados en la capa de yeso. En la habitación W la pintura se ha ejecutado: i) al temple propiamente dicho. aplicando sobre la preparación de yeso las capas pictóricas al óleo; ii) superponiendo las capas pictóricas sobre varias capas de carbonato cálcico. y iii) pintando directamente sobre el revestimiento de cal. Esta variedad de técnicas al fresco seco sugiere que las pinturas fueron ejecutadas O en épocas distintas o por diferentes artistas. Por otra parte. la identificación del pigmento amariflo de cromo ha permitido establecer que. al menos alH donde se localiza este pigmento. la ejecución de la pintura mural es posterior a las fechas arriba mencionadas. ya que el amarillo de crOIllO es de origen artificial y se comenzó a comercializar a partir de 1797 según Mayer (Mayer, 1988) y a partir de 1818 según Matteini y Moles (Matteini y Moles. 1989).
El principal agente de alteración de estas pinturas murales es el. agua del terreno que asciende por capilari�ad. En la habitación W. la dolomitn presente en el árido del revestimiento de cal (sob,� el que se aplicó la pintu-

Policromías de la Iglesia de San Jerónimo de Granada 121
ra) ha reaccionado con el yeso originando subeflorescencias de sulfato de magnesio. Las presiones de cristalización yfo hidratación que ejercen estas sales sobre las capas de pintura provocan el desprendimiento de la policromía. En la habitación E, el agua rica en sales cloruradas ha reaccionado con los pigmentos, principalmente los elaborados a base de pIorno, formándose sales de cloruro de plomo. Por tanto, en la lOna inferior de la iglesia la composición de las pinturas murales y la naturaleza de los materiales del substrato influyen directamente en el tipo de las sales originadas.
Agradecimientos
Este trabajo ha sido subvencionado por el Grupo de Investigación de la Junta de Andalucía RNM 176. Los datos de microscopía electrónica de barrido se obtuvieron en el Centro de Instrumentación Científica (ele) de la Universidad de Granada.
Bibliografía
Aubert. M. (1964): Trionfo del gotico. Milano. 7 1 -74.
Bontcé, J. (1989): Técnicas y secretos de la pintura. LEDA ed .• Barcelona. 176.
Camón Aznar, J. (1981): «Summa Artis», Historia General del Arte. Vol. XVIII. Espasa-Ca/pe, S.A. etl . • Madrid, 425.
Cardell Fernández, C. (2003). Cristalización de sales en calcaren itas: aplicación al Monasterio de San Jerónimo. Gramada. Depto. Mineralogía y Petrología. Universidad de Granada. 219.
Cardell Fernández, C. y Rodríguez Gordillo. J. (1992): Formación de sales en materiales pétreos (Monasterio de San Jerónimo. Granada). Origen y procesos de deterioro asociados. 111 COI/gr. Ceo/. de Espal1a y VIII COllgr. La¡illoamericano de Geol. Salamanca (España) 3, 46-48.
Cardell Fernández. C. y Rodríguez Gordillo. J. (2003). Estudio de policromías de la Iglesia del Monasterio de San Jerónimo de Gmnada. Estado de conservación. XXIII Reullión de la S.E.M. Puerto de Sama María. Cádiz, 1 , 1 1 1- 1 12.
Cennini, C. (1988): El libro del arte. Akal ed . • Madrid. 264
Colina Munguía, S. (1986): El Monasterio de San Jerónimo de Granada. El'eresl, S.A. ell., Madrid. 61
Diaz Marlos. A. (1978). Restauración y conservación del arte pictórico. Af/e Res/al/ro, S.A. Madrid. 2 13pp.
Doerner, M. (1921): Los materiales de pintura y su empleo en el arte. Reverté. S.A. ed., Barcelona, 460
Gallego y Burín. A. ( 1982): Granada. guía artística e histórica de la ciudad. Don Quijote. ed .• Madrid, 433
Gómez-Moreno González, M. ( 1 892). «Guía de Granada». Imp. IlIdlllecio Velllllra. Granada. 530.
Maltese. C. ( 1973): Las técnicas artísticas. Manuales Arle Cátedra ed., Madrid, 479
Mallein;, M. y Moles, A. (1989): La chimica nel restauro. 1 materiali dell' arte piltorica. Nardilli ed., 379
Mayer. R. (1988): Materiales y técnicas del arte. Hermallll Blullle ed., Madrid. 687
Merrifield. M.P. ( 1967): Original treatises on the arts of painting. New York, 1 82-257.
Rodrfguez Navarro. C .• Sebastián, E., Zezza, U., De la Torre M. J., Cardell-Fernández, C. (1991): Caracterización mineralógica y petrofisica de los materiales biocalcareníticos utilizados en la construcción de monumentos históricos de Granada. Bol. Soco Esp. Mili. 14-1. 25-26
Rossi Manaresi, R. ( 1987): Considerazioni tecniche sulla scultura monumental e policromata. romanica e gOlica. Bol/etillo d'arte. Supplemento n"41 . Vol. n, 173-1861
Recibido: Julio 2003 Aceptado: Diciembre 2003

Boletí" de la Sociedad EspOIiola de Mi/leralogía, 26 (2003), /23-/36 123
Características metamórficas del Triásico Maláguide en
las unidades intermedias del sector de Diezma (Sierra
Arana, Cordillera Bética)
i1IConcepción LÁZARO, tlIMaría Dolores RUIZ CRUZ y (}ICarlos SANZ DE GALDEANO i1lDepartamento de Mineralogía y Petrología, Facultad de Ciencias, Campus de Fuentenueva. Universidad de Granada. 18002 Granada mOepartamento de Química Inorgánica. Cristalografía y Mineralogfa. Facultad de Ciencias. Campus de Teatinos. Universidad de M:ílaga. 29071 Málaga OJlnstituto Andaluz de Ciencias de la Tierra. CSIC. Universidad de Granada, 18002 Granada
Abstraet: The met:lmorphic assemblages characteristic of the triassic rocks from mal:iguidealpujárride intermedi:lte units of the central sector of Ihe Betic Cordillera h:lve been studied by X-ray dirrraction, optieal microscopy and eleetron microprobe. The most ch:lnleteristie mineral assemblages are: illite + diekite: illite + dickile + pyrophyllite; and illite + dickite + sudoile. These assemblages imply a series of successive low-grade metamorphic reactions that inelude: a) Ihe kaolinite-to-dickite transformation; b) Ihe ¡Hitization of dickite; c) the dickite-to-pyrophyllile transrarmation; and d) the dickite -i sudoile reactian. Both Ihe presence of sudoite and pyrophyl1ite and Ihe erystallochemical characteristies or the several phyllosilieales indicale ror Ihese unils an alpine metamorphism wilh grade higher than thal determined in the maláguide unils. Minimum lemperatures in the order or 300 oC and intermediate pressures were estimateu.
Key words: Alpujárride Complex, Maláguitle Complex, Intermediate unilS, dickite. illite, pyrophyllite, sudoite. X -ray diffraction, electron microprobe.
Resumen: Las asociaciones metamórficas presentes en rocas triásieils de unidades intermedias mal:iguide-alpujárride del sector central de la Cordiller:l Bética se han estudiado mediante difracción de rayos X, microscopia óptica y microsonda electrónica. LilS asociaciones más caracterfsticas son: ilita + dickita, ilita + dickita + pirofilita e ilita + dickila + sudoita. Estas asociaciones ponen de manifiesto una serie de reilcciones metamórficas sucesivas de bajo grildo, que incluyen: a) la transición caolinila -i dickita: b) la ititización de la dickila: c) la transformación dickita -i pirofilit:l: y d) la transformación dickita 1I sudoita. La presencia de fases tales como pirofilita y sudoita junto con las caracterfsticas crislaloqurmicas de los filosilicatos indican que estas unidades fueron ilfectadas por un metamorfismo alpino de mayor grado que el determinado en unidades claramente mal:1guides. Se han estimado temperaturas del orden de 300 "C y presiones intermedias.
Palabras clave: Complejo Maldguide, Complejo Alpuj5.rride. unidades intermedias. dickita, ilila, pirofilila, sudoita, difracción de r:lyos X. microsonda electrónica.
Introducción
Los complejos Maláguide y Alpujárride, pertenecientes a las zonas inlernas de la Cordillera Bética. suelen diferenciarse sobre la
base de sus características Iitoestratigráficas, grado metamórfico y posición tectónica.
El complejo Alpujárride está formado por una sucesión sedimentaria que abarca del Paleozoico inferior al Triásico, Las secuen-

124 LÁZARO. C. el :11.
das triásicas están generalmente constituidas. de abajo hacia arriba. por esquistos. Filitas y carbonatos. En este complejoeJ metamorfismo alpino incluye un episodio de alla presiónl bnja temperatura, seguido por un episodio de bajn presión/alta temperatura (Azañón. 1994).
El complejo Maláguide comprende unidades scdimenl:lrias que incluyen materiales del Paleozoico al Mioceno. Sus secuencias triásicas están formadas por conglomerados. areniscas y ¡utilas. Las intercalaciones carbonatadas son también comunes a nivel del Triásico medio y superior. En este complejo se ha identificado un episodio metnmórrico herciniano. en gran parte enmascarado, en los materiales paleozoicos inferiores, por el metamorfismo alpino (Ruiz Cruz. 1997). El metamorfismo alpino. que afectó a los materiales triásicos. muestra una evolución que abarca de la diagénesis temprana (en los materiales triásicos superiores) a la anquizona (en los materiales triásicos inferiores).
La diferenciación entre ambos complejos suele ser fácil a nivel de los materiales triásicos y del Palezoico superior pero resulta muy complicada en materiales del Paleozoico inferior. donde los esquistos de ambos complejos son muy similares. Además. incluso a nivel del Triásico. se han descrito unidades intermedias entre ambos complejos. en diferentes zonas de la Cordillera (Sanz de Galdeano el al., 2001). Estas unidades intermedias se caracterizan por: a) estar situadas tectónica mente entre ambos complejos; b) presentar un grado metamórfico más bajo que las unidades alpujárrides situadas por debajo y más alto que las unidades maláguides situadas encima; y c) tener rasgos litológicos intermedios entre las series alpujárrides y maláguides.
Dado que la evolución metamórfica de ambos complejos muestra cllracterfsticas diferentes, estas áreas representan enclaves geológicos privilegiados para investigar la evolución de las condiciones metamórficas entre ambos complejos, especialmente II nivel de las series triásicas. Este estudio requiere la
identificación de las asociaciones minerales de bajo grado y de las caracterfsticas cristaloqufmicas de los filosil icatos en maleriales triásicos de las unidades intermedias y su companción con las asociaciones mineralógicas descritas en series IfpicllS maláguides y alpujárrides.
Este estudio se ha iniciado en Sierra Arana, al NEde Granllda, donde Sanz de Galdeano et af. (1995 a, b, c) han señalado III presencia de estas unidades intermedias.
Características del área estudiada, muestras y metodología
En el sector de Diezma, en Sierra Arana (Fig. I A). las unidades intermedias maláguidealpujárride aparecen como una serie de escamas tectónicas superpuestas. Las superiores. situadas más al norte, son similares a las del complejo Maláguide. en el que sedimentos triásicos rojos recubren discordante mente un basamento Paleozoico. A medida que las escamas se sitúan en posiciones inferiores, las lutitas rojas y las areniscas típicas del complejo Maláguide evolucionan hacill fililllS púrpura y azules. a la vez que las caracterfsticas texturales indican un mayor grado metamórfico. En esta zona se han muestreado diez escamas (Fig. 1 B). De éstas. sólo las dos más llltas tectónicamenle mueSlran características litol6gicas tfpicamente maláguides y son el objetivo de este estudio.
Las rocas IriásicllS maláguides de las escamas superiores incluyen varios tipos de llTeniscas rojas. conglomerados y lutitlls y pueden adscribirse a los miembros triásicos inferiores definidos por M:tkel (1985): el miembro conglomerático y el miembro de areniscas. Se han estudiado dos conglomerados, cuatro niveles de areniscas y tres de lulilas.
Las muestras se estudiaron por microscopIa ópticll y d.ifracción de rayos X, con el objetivo de determinar la composición mineralógica y las relaciones textura les. So-

Metamorfismo del Triásico maláguide en u n idades i ntermedias 125
[] Ncógcno-Cuaternario
D Brechas ncógcnas
D Unidades de la Do/sal
O Complejo Malaguidc
� Unid;ldc� de ll;lllsicjón entre el L..:!.::J M;;If;igulcle y el Alpuj;\¡llde G Unidad de la Plata
EJ Unidml de los Bla nquizares
� Unidad de la Mora-Padules
A
o 2km
� Cabalgamientos
� Falla normal
- - - - - -- Contacto disco rdante
AA'; Corte de muestreo
Figura 1 .- A: Esquema geológico de la zona de Diezma (Sierra Arana).
B Figura 1 . - B : Columna esquemálica de las escamas lcctóni¡;<!s !lHlcstrcadas en el Sector de Diezma (Corte
A-A').

126 LÁZARO. C. el al.
bre la base de este estudio. la composición química de las diferentes fases minerales se determinó. cuando el tamaño de grono lo permitió. por microsonda electrónica.
Por difracción de rayos X (DRX) se estudiaron lanlO las muestras totales como las fracciones de tamaño 2-20 J..lm y <2 J..lffi. Los diagramas se obtuvieron en la Universidad de Málaga, con un difraclómetro Siemens 0-5000 con radiación CuKa y monocromador de grafito. operando a 40 mA y 40 kV. El tamaño de paso fue de 0.010 y el tiempo de conlaje por p<lSQ de I s. Las diferentes fracciones de tamaño se obtuvieron por sedimentación. Se utilizaron muestras desorientadas para la determinación semicuantitativa de la composición mineralógica y para determinar el pnrámetro b de los filosilicntos y [os politipos presentes. Ln determinación del politipo de las micas blancas se basó en la presencia de dos reflexiones características: la reflexión 1 14 (a 3.50 A}.!..exclusiva del politipo 2M,. Y la reflexión 1 1 2 (a 3.66 A). caracterfstica del politipo 1M. La identificación de la dickita se basó igualmente en la presencia y posición de sus reflexiones caracterfsticas: 1 1 I (a 3.95 A); 002 (a 3.79 A). y especialmente la reflexión 132-204 (a 3.32 A) (Bailey. 1980). Para la identificación y cuantificación de los filosilicatos se util izaron agregados orientados secados al aire. tras la solvatación con etiltn-glicol y tras el calentamiento a 550 OC. La cristalinidad de la ilita (IC) se midió en ambas fracciones. utilizando el método de KUbler ( 1968). tanto en los diagramas obtenidos a partir de las muestras secadas al aire como en los obtenidos tras el tratamiento con etiltn-glicol. siguiendo las recomendnciones de Kisch ( 1991). Nuestros valores (y) se transformaron en valores standard (C.S.I., Warr y Rice. 1994) (x) mediante la ecuación y = 1 .23x-0-07. La cristalinidad de In clorita (CC) y de la dickita se determinaron a partir de la reflexión a 7 A. en las mismas condiciones que en el caso de la ilita. La identificación de la sudoita se basó en la posición de la re-
flexión 060 Y en la relación de intensidades basales. especialmente en la intensidad de la reflexión 003 (Eggleston y Sailey. 1967; Bailey y Lister. 1989).
El estudio mediante microsonda electró· nica (EPMA) se realizó en la Universidad de Granada. con un equipo Carneca SX-50. El vohaje utilizado fue 20 kV Y la intensidad de la corriente de 20 nA. El aparato había sido calibrado con los siguientes patrones: wollastonita (para Si y Ca). AIPJ (para Al). ortoclasa (para K), albita (para Na), FeZOJ sintético (para Fe). periclasa (para Mg) y MnTiO) sintttico (para Mn y Ti).
Resultados
Observaciolles petrofógicas )1 resllltados de microsollda
Tanto los conglomerados como las arenicas se estudiaron al microscopio óptico. Los conglomerados. con una pobre selección. aparecen constituidos por granos detríticos de cuarzo monocristalino y policristalino y una pequena proporción de mica blanca. El cemento está constituido por cuarzo microcristalino, dickita y óxidos de hierro. La dickila. cuya identificación se basó en los datos de DRX. aparece como grandes paque· tes de láminas paralelas (del orden de 20 mm). que ocasionalmente reemplazan a los granos de cuarzo (Fig. 2A). Los paquetes de dickita aparecen recubiertos por una delgada película de óxidos de hierro. que permite su fácil identificación y la diferenciación del cemento de cuarzo microcristalino. que presenta una birrefringencia similar. Las relaciones textura les, especialmente el reemplazamiento de los recrecimientos de cuarzo. indican que este mineral creció durante una elapa posterior a la diagénesis de enterramiento.
Las areniscas estudiadas se pueden clasificar, de acuerdo con Fuchtbauer (l983). en dos grupos principales: areniscas arcillosas. con >15% de matriz o cemento arcillosos, y

Metamorfismo del Triásico mal{¡guide en unidades intermedias 1 27
Figura 2.- A : Microrotografía obtenida con nicoles cruz;¡dos, mostrando el cemento de dickita (Ok) y e l inicio de la transf'orm,¡ción de este mineral e n ¡ l i ta (11) e n u n conglomerado. 13: Microfotografía obtenida con nicolcs cruzados, mostrando el desarrollo de sudoit;:¡ (Sud). su transformación avanzada en i l i ta (11) y l a presencia de escasos restos de dickita (Ok). en una arenisca carbonática,
Tabla 1 .- An¡í l is is químicos representativos de i l i t a y sudoita
Óxidos Ms Ms Ms Ms Sud Sud (% peso)
SiO, 4 8. 1 1 5 50.87 1 49.941 5 1 .9 1 1 33.542 36.828
AI,O, 34.67 1 33.654 33 .642 34.709 34. 1 53 33.952
TiO, 0.030 0.065 0.042 0. 1 8 1 0.0 1 3 0.000
FeO 1 .286 1 .573 0.923 1 . 5 1 7 6.873 6.3 0 1
MnO 0.000 0.0 1 0 0.001 0 .071 0.004 0.003
MgO 0.398 0.526 0.754 0.5 1 5 1 0.895 9.905 CaO 0.228 0.2 1 4 0.070 0 . 1 44 0.084 0.066 BaO 0. 1 02 0. 1 1 4 0.000 0.000 0.021 0.000 Na,O 0.723 0.593 0.000 0.738 0.655 0.008 K,O 8.227 8.038 1 0.0 1 5 7 .891 0.032 0.055 P,O, 0.0 1 9 0.056 0.03 1 0.0 1 4 0.00 1 0.030
Total 93.800 95. 7 1 7 95.594 95.230 85 .649 87. 1 33
Fórmulas estructurales Si 3. 1 9 3 .30 3.25 3.27 3 . 1 4 3.35 AI1V
0.8 1 0.70 0.75 0.73 0.86 0.65 A lv 1 1 .90 1 .88 1 .70 1 . 85 2.9 1 2.99 Fe 0.07 0.09 0 . 1 5 0 . 1 2 0.54 0.50 Mg 0.04 0.05 1 .52 0. 1 1 0.03 1 .3 4 C a 0.02 0.0 1 0.0 1 0.02 0.03 0.00 Na 0.09 0.07 0.09 0.08 0.00 0.00 K 0.69 0.66 0.93 0.7 1 0.00 0.0 1 O 1 1 I 1 I I 1 1 1 4 1 4

128 LÁZARO. C. el 0.1.
areniscas calcáreas, con >20% de cemento carbonatado o de fragmentos de rocas carbonatadas. Los minerales detrfticos dominantes en las areniscas arcillosas son cuarzo mono y policristalino y mica blanca. La matriz detrítica parece ser escasa pero el contcnido en cemento silíceo y arcilloso es a me· nudo >25%. Estas areniscas contienen escasos granos de feldespatos. que sugieren. de acuerdo con las observaciones en otras áreas de la Cordillera (Ruiz Cruz. 1996) un importante proceso de disolución durante la diagénesis temprana, que proporcionaría el Si y Al necesarios para el desarrollo de caolinita. La presencia de conlactos suturados y de recrecimientos alrededor de los granos de cuarzo detrÍlico indican. por otra parte. que la solución por presión fue otro proceso generalizado durante la diagéneis de enterramiento. El cemento actual está constituido esencialmente por ilita y pirofilita. Aunque algunas de estas areniscas contienen dickita. este mineral es diffcilmente distinguible por microscopfa óptica. ya que aparece casi tolalmente transformado en mica y pirofilita.
En las areniscas calcfireas, de tamaño de grano fino a medio, los fragmentos de rocas carbonatadas son constituyentes detríticos esenciales junto con cuarzo y mica blanca. Estas areniscas presentan además un cemento calcáreo bien desarrollado. El cemento arcilloso aparece formado por sudoita. ilita y pirofilita. Con nicoles paralelos. la sudoita aparece como agregados laminares de hasta 50 J.Lm de longitud. Con nicoles cruzados la
sudoita se caracteriza por el color de interferencia blanco. La sudoita muestra un grado muy variable de transformación en iJjla y. probablemente. en pirofilila (Fig. 2B).
En la Tabla 1 se recogen algunos análisis representativos de ilita y sudoita. Las fórmulas de la mica. calculadas para 1 1 oxígenos. muestran altos contenidos en Si (3.20-3.30 apfll). contenidos en Fe+Mg del orden de 0.15 apfll y un ligero déficit de carga inlerlaminar. que indican su naturaleza ilítica. El contenido en Na es del orden de 0.1 apfll. Los análisis de sudoita muestran valores muy homogéneos de los contenidos en AI.Fe y Mg Y contenidos más variables en Si. Las fórmulas estructurales se han calculado para 14 oxígenos y suponiendo todo el Fe como ferroso. aunque datos previos (p. ej.. Fransolet y Bourguignon. 1978; BilIault el al., 2(02) indican que el contenido en Fe férrico puede representar hasta el 60% en sudoilas ricas en Fe. Las fórmulas obtenidas son consistentes con una estructura di.trioctaédrica y coinciden con el rango composicional de las sudoitas ricas en Fe (p. ej .• Daniels y Allaner. 1990; Billault el al., 2002).
Eswdio por difracciól/ de rayos X En la Tabla 2 se recoge la composición
mineralógica de varias muestras representativas de las diferenles litologías triásicas, deducidas de los datos de DRX. Algunos diagramas representativos se muestran en las figuras 3-6.
Los diagramas de DRX de las dos fracciones de tamaño de los conglomerados reve·
Tabla 2.- LilOlogla y mineralogía de rocas triásicas representativas.
MinmIoI!! de Ip fOS! 10111 AK!CQci6n "'narn6ñIS' le ce º" FA e Ejl
Coallomc1a4os " " !til> . dickila 0.44 AmliIc:as millosM " " llill . piro/ilil. + dódill 0.1141.'1 ArcniKUu�u " " " Hill + pirofilila • sudoiui O.)I·O.�9 O.2�-O.Jl
Hitl � pirofim • • dickill
L.lIli!!1 ., , • ., llitl:' RiI:l!{iJ"l! ¡ I!II!��I Il:�� !l,n Qlz: Cuano: Ed: Feld.,pI'os; C: carbon.lOO: Fil: Fiknillcl1oo; IC: Cri.I.linidld d. la lIil.: ce: C,illllittldad d< la crorill (wdoiuo): oc: CrÍSIIllnidad do l. dickill
oc
", 0.21-4.].(

Metamorfismo del Triásico maláguide en unidades intermedias 129
lan que el filosilicato dominante es la dickita aunque el contenido relativo en ilita aumenta ligeramente en la fracción <21J.m (Fig. 3). La anchura (a mitad de la altura) de la renexión a 7 A de la dickita, del orden de 0.22 (6D28), indica una alta cristalinidad. El valor del índi· ce de cristalinidad de la ilita en estas muesIras es del orden de 0.50.
Los diagramas de DRX de las dos fracciones de tamaño de las areniscas arcillosas revelan la presencia bien de ilita sola, bien de la asociación ilita + pirofilita + dickita (Fig. 4). La presencia de una renexión bien definida a 4.72 A. pone de manifiesto, además, la presencia de sudoita. El contenido en dickita es bajo en estas muestras. La anchura de la reflexión 002 es mayor que en los conglomerados (del orden de 0.30) lo que reneja, posiblemente. la presencia una pequeña proporción de sudoita. Los valores del [ndice de cristalinidad de la ilita varían entre 0.38 y 0.5 1 .
Los diagramas de DRX de las fracciones finas de areniscas calcáreas son muy similares a los de las areniscas arcillosas (Fig. 5), aunque la presencia de una intensa reflexión a
nr,j<mr.ó>
4.72 Á revela que la fase a 7 Á no es dickita sino sudoita. Sin embargo, el estudio óptico ha permitido poner de manifiesto la presencia de una pequeña proporción de dickita en estas muestras. La cristalinidad de la sudoita se sitúa entre 0.25 y 0.33, mientras el índice de Kubler varía entre 0.30 y 0.49. Los diagramas de DRX indican de nuevo que el politipo dominante de la ilita es el 2M,.
Los diagramas de DRX de las diferentes fracciones de tamaño de las lotitas (Fig. 6) indican la presencia de cuarzo, feldespato (plagioclasa y microclina) y un bajo contenido en carbonatos (calcita y siderita). La ilila es el filosilicato dominante en estas muestras, acompañada ocasionalmente por una pequeña proporción de sudo ita. El [ndice de cristalinidad de la ilila es del orden de 0.40. La cristalinidad de la sudoila ofrece valores similares.
El polilipo de ilila dominante en todos los litotipos es el 2M l ' Por otra parte. el valor del parámetro b de la ¡lita oscila dentro de un estrecho margen (8.98-9.00 A) en todos los litotipos estudiados.
""- <l¡m ...
A '"
I\.. llI\ A .... '--..
'lOO
J , ..
2-3)¡m
o '--, 10 JO "
Figura 3.- Diagramas de difracción de rayos X obtenidos a panir de ;¡gregados orientados naturales de las fracciones <21!m y 2-20 I!m, de un conglomerado, mostrando la asocinci6n mineral característica. 11: Ilita: Dk: Dickita.

130 LÁZARO. C. el al.
Arenisca arcillosa <000
JOOO <2,..n 003. OOI,OOIN
� 002". ... ""- "" "". .i ,..
'" 2-20 ¡.m
o s " IS " 2S JO
2e(pIos)
Figura 4.- Diagramas de difracción de rayos X obtenidos a partir de agregados orientados naturales de las fracciones <2¡.tm y 2-20 ¡.1m. de unta arenisca arcillosa, mostrando la asoci¡¡ción mineral caractcrfslica. 11: lIita; Ok: Dickita; PrI: Pirofilila.
1100 Arenisca calcárea
'" 1<00
1200
1 '" .>1. <2"", 100
.5 .. 001.
<00 00;.. 200 2-20 ¡.m
o s " IS '" JO
20(�)
Figura 5.- Diagrnmas de difracción de rayos X obtenidos a partir de agregados orientados naturales de las fracciones <2¡.tm y 2-20 ¡.tm, de una arenisca caldrea, que muestra la asociación mineral caracterfSlica. 11: Ilila; PrI: Pirofilita; Sud: Sudoitll.

Metamorfismo del Triásico maláguide en unidades intermedias 131
lID""
(XII,
� \\ """" ' ... ..-.
100
I\... o
s 10
<1¡m
001,
Qz
\. • Á 2-:!)f'l1
IS 2O(¡ja:b¡)
' V
Qz
..v'< � 2S II
Figura 6.- Diagramas de difracción de rayos X obtenidos a panir de agregados orientados naturales de las fracciones <2J.lm y 2-20 J.lm, de una lutita, mostrando la asociación mineral caracterfstica. Una renexión poco intensa a 7 A puede corresponder a dickita (Ok) o sudoitól (Sud). 11: lIitól.
Discusión y conclusiones
La asociación metamórfica de bajo grado y las características cristaloqufmicas de los filosilicatos. determinados en las rocas triásicas con litologías maláguides de las unidades intermedias. permiten su comparación con las asociaciones minerales típicas de las rOCaS triásicas maláguides en otras zonas de la cordillera y la estimación aproximada de las condiciones metamórficas de los materiales triásicos maláguides en la zona de Sierra Arana.
Caracter(sticas diferel/ciales lle los materiales triásicos malágllides del sector de Diezma
Las rocas triásicas estudiadas presentan litologías similares a las que caracterizan al Triásico maláguide en otras zonas de la cordi llera, lo que indica que tanto eráreól fuente como el medio de depósito y los procesos
diagenéticos tempranos fueron también similares.
En las secuencias triásicas maláguides de la Cordillera Bética, la asociación mineralógica característica es ¡¡ita + clorita (trioctaédrica) + dickita. La dickita es un mineral ampliamente desarrollado en los materiales triásicos inferiores, concretamente en los miembros conglomeráticos y de areniscas (Ruiz Cruz, 1996; Ruiz Cruz y Andreo, 1996). en tanto que en los miembros lutíticos superiores, el politipo presente es caolinita. Además, en las rOCas triásicas inferiores. se observa con frecuencia la transformación dickita � ilita y dickita � clorita trioctaédrica. Así pues, las rocas triásicas de las unidades intermedias tienen en común con otras secuencias maláguides la asociación ilita + dickita.
No obstante. este estudio ha puesto de manifiesto varias diferencias significativas entre los materiales triásicos de las escamas superiores de las unidades intermedias en la

132 LÁZARO. C. el al.
zona de Sierra Arana y las series triásicas mnláguides t[picas: l ) Las cristalinidades de la ilitas y. especialmente de la clorita, son mayores (menores valores numéricos) que en otras zonas de la cordillera (Ruiz Cruz, 1996) y similares a las determinadas en unidades intermedias de Sierra Espuña por Abad et al. (2003). 2) En los materiales triásicos maláguides de la zona de Diezma las cloritas trioctaédricas tienen escaso desarrollo. 3) En las unidades intermedias se da. en cambio. un mayor desarrollo de sudoita (al igual que en Sierra Espuña) que, no obstante es un componente minoritario. 4) En las unidades intermedias de Diezma es común la presencia de pirofilita, rase no observada en secuencias maláguides tfpicas ni en Sierra Espui'ia.
Todos estos hechos revelan que las reacciones metamórficas fueron más avanzadas en las unidades intermedias y que el grado metamórfico alcanzado por estas secuencias es superior al detectado en los materiales triásicos maláguides.
Génesis mil/eral en las ullidades imermedias del sector de Diezma
El amplio desarrollo de dickita en areniscas y conglomerados de los miemb¡'os inferiores del Triásico maláguide parece estar relacionado, a lo largo de toda la cordillera, con una secuencia de procesos diagenéticos sucesivos (Ruiz Cruz, 1996): 1) La disolución intensa de los feldespatos y. en menor medida, de las micas detríticas, durante la diagénesis temprana y el desarrollo de una gran cantidad de caolinita, que llenaría la abundante porosidad y reemplazarla a las láminas de mica. Este proceso ha sido frecuentemente reconocido en series triásicas con características litológicas similares (Lonoy el al., 1986; Bjorlikke, 1983, 1986). 2) Durante los procesos diagenéticos relacionados con el enterramiento de estas secuencias ha tenido lugar el desarrollo de cemento de cuarzo microcristalino y el recrecimiento de los granos detríticos de cuarzo. 3) Por último. la transformación caolinita
--t dickita tiene lugar más tarde, en relación con el metamorfismo alpino. Esta secuencia de transformaciones tuvo también lugar en las escamas superiores de las unidades intermedias y pone de manifiesto un medio sedimentario y Un3 historia diagenética común con el resto del maláguide.
La asociación mineral ilita + pirofilita + dickita, identificada en areniscas de las unidades intermedias sugiere que han tenido lugar dos reacciones sucesivas: dickita --t ilita y dickita � pirofilita. La primera es común a lo largo de la cordillera y es una reacción frecuentemente descrita en secuencias diagenéticas en las que e:\iste disponibilidad de potasio (Ehrenberg y Nadeau, 1989). El desarrollo de esta reacción depende, pues. fundamentalmente de la disponibilidad de K. La segunda reacción. claramente metamórfica, aparece en cambio limitada a las unidades intermedias.
La transformación dickita --t clorita trioctaédrica ha sido también descrita en las unidades maláguides a lo largo de la cordillera (Ruiz Cruz, 1996; Ruiz Cruz y Andreo, 1996). En el sector de Diezma no se ha detectado ni a partir del estudio microscópico ni a partir de los datos de microsonda la presencia de clorita trioctaédrica. Se ha observado, en cambio. la transformación de dickita en sudoita. Sobre la base de los datos disponibles en este momento, la formación de sudoita podría ser el resultado de dos procesos diferentes: 1 ) la transformación directa dickita --t sudoita. que podría considerarse equivalente a la reacción dickita --t clorita trioctaédrica, en un medio especialmente rico en Al; y 2) La transformación dickita --t clorita tri octaédrica --t sudoita. El estudio previsto mediante microscopía electrónica de transmisión, permitirá, probablemente, realizar una interpretación más exacta de la génesis de sudoita.
EstimaciólI In, cOlldiciolles metamórficas en el sector de Diezma
La dickita aparece especialmente bien desarrollada en dos medios geológicos: Secuen-

Metamorfismo del Triásico maláguide en unidades intermedias 133
cías sedimentarias sometidas a una diagénesis profunda y alteraciones hidrotermales. En el primer caso, la transición caolinita --+ dickita se ha utilizado como un importante indicador diagenético (p. ej. , ShulOv el al., 1970; Ehrenberg e1 al., 1993; Lanson el al., 1 995; Ruiz Cruz y Moreno Real, 1993; Beaufort el al., 1998). Este mineral se desarrolla especialmente en rocas detríticas ricas en Al, asociado frecuentemente con cuarzo, carbonatos (dolomita o ankerita), feldespatos, ¡lita y clorita trioctaédrica. En el caso de las alteraciones hidrotermales, la dickita aparece generalmente asociada con sulfuros y otros polimorfos del caolín (p. ej. , Brindley y POrler, 1978; Hanson el al., 198; Keller, 1988; Marumo, 1989).
De forma similar, la sudoita aparece también en terrenos diagenéticos o metamórficos de bajo grado (p. ej., Schultz. 1963; Fransolet y Bourguignon, 1970; Daniels y Altaner, 1990; Livi el al., 2002) asr como en alteraciones hidrotermales. asociada a menas metálicas (p. ej., Bailey y Tyler, 1960; Hayashi y Qinuma, 1964; Sudo y Sato, 1966; Alyesheva el al., 1977; Percival y Kodama, 1989). La sudoita es también un mineral típico de rocas ricas en Al, donde coexiste con cuarzo, feldespato, Hita y clorita tri octaédrica. La asociación sudoita + Mg-carfolita + aragonito caracteriza, en cambio, el metamorfismo de alla presión/baja temperatura descrito en las cadenas alpinas perimediterráneas (p. ej. , Theye el al., 1992; Azañón, 1994).
A pesar de que tanto la dickita como la sudoita aparecen en condiciones geológicas similares, la asociación dickita + sudoita no había sido previamente descrita. Este hecho sugiere que determinados factores físicos o geoquímicos deben controlar la formación bien de dickita bien de sudoita.
La transición caolinita --+ dickita se produce a partir de 120 oC durante la diagénesis profunda (Ehrenberg el al., 1993). La ausencia de caolinita en estas secuencias señala, pues, un Hmite mínimo para la temperatura, alrededor de 120 oC, temperatura que puede considerarse como mínima también en las se-
cuencias maláguides típicas. Además, el amplio desarrollo de ilita a partir de dickita sugiere, de acuerdo con los datos de Ehrenberg y Nadeau ( 1989) temperaturas claramente superiores a 140 oC, temperatura que parece indicar el inicio del proceso de ¡Iitización en series con disponibilidad de K. Por otra parte, y aunque la pirofilita puede formarse a partir de 225 oC en presencia de soluciones ricas en metano (Juster el al., 1987), la temperatura normalmente aceptada para el comienzo del desarrollo de pirofilita a partir de minerales del grupo del caolín es de 300 oC (Frey, 1987). Aunque no conocemos con certeza la reacción que ha producido sudoita, diversas asociaciones mineralógicas con sudoita, por ejemplo las asociaciones sudo ita + dolomita + cuarzo + granate (Livi el at., 2002) y sudoita + pirofilita + caolinita + clorita + cloritoide (Theye el al., 1992), proporcionan temperaturas de entre 300 y 360 oC. Aunque no se ha detectado cloritoide ni granate en las muestras del Triásico maláguide, el cloritoide aparece bien desarrollado en filitas alpujárrides de escamas inmediatamente inferiores y apunta, a temperaturas del orden de 300 oC para el metamorfismo alpino que afectó a las escamas superiores de las unidades intermedias en la zona de Sierra Arana. Por otra parte, los valores del parámetro b de la ilita son consistentes con un metamorfismo de presiones intermedias, de acuerdo con Guidotti & Sassi ( 1986).
Las temperaturas eslimadas son claramente superiores a las determinadas en materiales triásicos maláguides en otras áreas de la cordillera, que eran del orden de 150-170 oC (Ruiz Cruz y Andreo, 1996) y sugieren la existencia, en las unidades intermedias, de un metamorfismo de más allo gradiente, siempre en condiciones de presiones relativamente bajas.
Agradecimientos
Los autores agradecen a F. Nieto, I. Abad y un revisor desconocido la cuidadosa revisión efectuada así como las sugerencias y co-

134 LÁZARO. C. el al.
rrecciones, que han mejorado notablemente el manuscrito. Este trabajo se ha realizado con la financiación del Ministerio de Educación y Cultura (proyecto BTE-20aO 1 150) y de la Junla de Andálucía (Grupo de Investigación RNM-199).
Bibliografía
Abad, l.; Nielo. F.; Peacor. O.R.; Velilla, N. (2003) Prograde and retrograde diagenetic and metamorphic evolulion in metapelitic rocks of Sierra Espuña (Spain). C/ay Miner . • 38. 1-23.
Alyesheva, E.I., Rusinova, O.V . • Chekvaidze. V.B. (1977) On sudoites from (he polymetal deposils ofRydnyy Altai. Dok. Acod. NallK. SSSR. 236, 722-724.
Azañón. J.M. (1994) Metamorfismo de alta presión/baja temperall/ra. baja presión! alta temperatura y leetó/lica del Complejo Afpl/járride (cordilleras Bético-Rifelias). Tesis. Universidad de Granada. 331 p.
Bailey, S.W. ( 1980) Structures of Layer Silicates. Pp. 1-124 en (G.W. Brindley y G. Brown eds.): Crystal Structures oJClay Millerals al/d Their X-ray Identiflcatiol/. Mineralogical Society, London.
Bailey, S.W. y Tyler, S.A. (1960) Clay mineral associated with the Lake Superior iron ores. Ecoll. Geol., 55. 150-175.
Bailey, S.W. y Lister. J.S. (1989) Structures, compositions and X-ray identification of dioctahedral chlorites. Clays Clay Miner., 37, 193.202.
Beaufort, D.; Cassagnabere, A.; Petit. S.; Lanson. B.; Berger. G.; Lacharpagne, J.C.; Johansen, H. ( 1998) Kaolinite-to-dickite reaction in sandstone reservoirs. Clay Miner.. 33. 297-316.
Billault, V.; Beaufort, D.; Patrier, P.; Petit, S. (2002) Crystal chemimy of Fe-sudoites from uranium deposits in the Athabasca basin (Saskatchewan. Canada). C/ays Clay Mil/er .. 50. 70-8 1 .
Bjorlikke, ·K. (1983) Diagenetic reactions in sandstones. Pp. 169-213 en: Sedilllellt Diagel/esis (A. Parker y B. Sellwoods eds.). Reidel Pub!. Co., Dordrecht.
Bjorlikke, K. ( 1989) Sedimenrology alld Petrolel/m Ge% gy. Springer-Verlag. Berlin. 363 p.
Brindley, G.W. y Porter, A.R.O. ( 1978) Occurrence o dickite in Jamaica. Ordered and disordered varieties. Am. Mil/eral., 63. 554-562.
Daniels, E.J. y Altaner. S.P. ( 1990) Clay mineral authigenesis in coal and shale from the Anthracite region. Pennsylvania. Am. Mil/eral., 75, 825-839.
Eggleston. R.A. y Bailey, S.W. ( 1967) Structural aspects of dioctahedral chlorite. Am. Mil/era/., 52, 673-689.
Ehrenberg, S.N. y Nadeau, P.H. ( 1989) Formation of diagenetic i Hite in sandstones of the Gran formation. Ha!tenbanken area, mid-Norwegian continental shelf. e/ay Miner., 24, 233-253.
Ehrenberg, S.N.; Aagaard, P.; Wilson. M.; Fraser. A.R.; Outhie, O.M.L. ( 1993) Deplh-dependenl transformation of kaolinite to dickite in sandstones of the Norwegian continental shelf. Clay Miner., 28, 325-352.
Fransolet, A.M. y Bourguignon, P. (1978) Dil trioctahedral chlorite in quartz veins from the Ardennes, Belgium. Call. Mi/lera/., 16. 365·373.
Frey, M. (1987) Very low-grade metamorphism of clastic sedimenlary rocks. Pp. 9-58 In: Low temperatllre Metamorphism (M. Frey ed.) Blackie, Glasgow, U.K.
Fuchtbauer, H. ( 1 983) Facies controls on sandstone diagenesis. Pp. 268-288 en: Sedimellt Diagellesis (A. Parker y B.W. Selwood. eds.) Reidel Pub!. Co., Dordrecht.
Guidolli, C. V. Y Sassi, p.P. ( 1986) Classification and cOITelation of melamorphic facies series

Metamorfismo del Triásico maláguide en unid3des intermedias 135
by means of muscovite b. d3ta from low grade metapeliles. N. Ja"'. Mili. Ab., 153. 363-380.
Hanson, R.F.; Zamora, R.; Keller, W.O. (1981) Nacrite, dickite and kaolinite in one deposit in Nayarit, Mexico. Clays Cfay MÍller . • 29, 451-453.
Hayashi, H. y Oinuma, K. ( 1965) Aluminian chlorite from Kamikita mine, Japan. C/ay Sci., 2, 22-30.
Juster. T.C.; Brown, P.E. & Bailey. S.W. (1987) NH4-bearing iIIite in very lowgrade metamorphic rocks associ3ted with coal, northeastern Pennsylvania. Am. Mineral., 72, 555-565.
Keller, W.O. (1988) Authigenic kaolinite and dickite associated with metal sulfides, probable indieators of a regional thermal evento Cfays Clay Miner., 36. 152-158.
Kiseh. H,J. (1991) lIlite erystallinity; recommendations on sample preparation, X-ray diffraetion seuings. and interlaboratoy samples. J. Melam. Geof., 9. 665-670.
Kubler. B. (1968) Evaluation quantitiltive du métamorphisme par la cistallinité de l'iIlite. Etat des progres réalisés ces derni�res années. BIlIf. Ce/llre Recl!. Pall, S.N.P.A .. 2. 385-397.
Lanson, B.; Beaufort, D.; Beger. G.; Petit, S.; Lacharpacne. J.C. ( 1995) Evolution de la strueture cristallographique des minéraux argileus dans le réservoir gréseux Rotliegende des Pays Bas. Bull. El! Aquitaille, 19, 243-265.
Livi. K.J.T.; Ferry, J.M.; Veblen, D.R.; Frey, M.; Connolly. J.A.D. (2002) Reaclions and physical conditions during melamorphism ofLiassic aluminous black shales and marls in central Swilzerl3nd. Eur. J. Mil/eral., 14, 647-672.
Lonoy, A.; Akselen, J.; Ronning, K. ( 1986) Diagenesis of a deeply buried sandstone reservoir: Hild Field, Northern Nonh Sea. C/ay Miner., 21, 497-51 1 .
Ml1kel, G.H. (1985) The geology of the
Maláguide Complex and i¡s bearing on the geodynamic evolulion of Ihe BelicRif orogen (soulhern Spain and norlhern Morocco). CUA papers Ceof., 22. 263 p.
Marumo. K. (1989) Genesis of kaolin minerals and pyrophyllile in Kuroko deposits of Japan: implications for the origins of Ihe hydrolhermal fluids from mineralogical and Slable isolope dala. Geochim. Cosllloc/¡illl. Acta. 53. 2915-2924.
Percival, J.B. y Kodama, H. ( 1989) Sudoite from Cigar Lake, Saskatchewan. COII. Mineral., 27, 663-64 1 .
Ruiz Cruz, M.O. ( 1 996) Criterios mineralógicos empleados en el análisis del Permotrlas Maláguide. Cllad. Geof. Ibérica, 20, 37-59.
Ruiz Cruz, M.O. ( 1997) Very low-grade chlorile wilh anomalous ehemislry and oplical properlies from Ihe Maláguide Complex, Belic Cordilleras, Spain. Call. Milleral., 35, 923-935.
Ruiz Cruz, M.O. y Moreno Real. L. (1993) Diagenetic kaolinile/dickile (Belic Cordilleras. Spain) Clays C/ay Mjller . • 41.
570-579. Ruiz Cruz, M.O. y Andreo. B. ( 1996) Genesis
and Iransformalion of diekile in PermoTriassic sediments (Betic Cordilleras. Spain). Clay Mjller., 31 , 133-152.
Sanz de Galdeano, C.; Delgado. F.; LópezGarrido, A.C. ( 1995 a) Unidades alpujárrides y maláguides al NE de Granada (Cordillera Bética). Geogacela, 18. 27-29.
Snnz de Galdeano. C.; Delgado, F.; LópezGarrido. A.C. (1995 b) ESlructura del Alpujárride y del Maláguide al NW de Sierra Nevada (Cordillera Bélica). Rev. Soco G�ol. Espatia. 8 (3), 239-250.
Sanz de Galdeano, C.; Delgado. F.; L6pezgarrido. A.C.; Martín-Algarra. A. (1995 e) Appartenance alpujárride propos6 de l'unilé de la Mora nu NE de Grenade (Cordil[�re Bétique, Espagne). C,R. Acad. Sei. Paris, 321 (1101), 893-900.

136 LÁZARO. C. el al.
Sanz de Galdeano. C.; Andreo. B.; GarcíaTorlosa. F.J.; LÓpez-Garrido. A.C. (2001) The Triassic palaeogeographic transition between Ihe Alpujárride and Maláguide comple:r;,es. Belic-Ri( Internal Zane. Pa/aeo. 167, 157-173.
Schultz. L.O. ( 1963) Clay minerals in Triassic rocks of ¡he Colorado plateau. US Geof. Sur\!. Blfll., 1147·C.
Shulov. V.O.; Aleksandrova, A.V.; Losievkaya, S.A. ( 1 970) Genelic inlerprelation of Ihe polytypism of Ihe kaolinite group in sedimentary rocks. Sedimenrology, 15. 69-82.
Sudo, V.O. y Sato. M. ( 1966) Dioctahedral
chlorite. p,.ac. 1m. Cfay COIl! Jeru:rafem. 1 , 33-39.
Theye, T .. Seidel, E. & Vidal, O. ( 1 992) Carpholite, sudoite and chloritoid in lowgrade high-pressure metapelites from Crete and Ihe Peloponnese, Greece. Eur. J. Mineral .. 4, 487-507.
War, L.C. y Rice, H.N. ( 1994) Interlaboratory standarizalion and calibration of c1ay minerals cryslallinily and cryslallile size dala. J. Mera/JI. Ceo/., 12, 141-152.
Recibido: Septiembre 2003 Aceptado: Diciembre 2003

Boletíl/ de la Sociedad Espmiola de Mil/eralogía. 26 (2003), 137-/53 137
Alteración de la cristalinidad del grafito por cizalla simple:
comparación entre procesos experimentales y naturales.
Elena CRESPO FEO. Francisco Javier LUQUE DEL VILLAR y Magdalena RODAS GONZÁLEZ Opto. Cristalografía y Mineralogía. Facultad de Geología. Universidad Complutense de Madrid. CI Ciudad Universitaria sIn. 28040 Madrid. Spain.
Abstracl: Ch:mges in Ihe cryslallinily of graphile due 10 grinding have been sludied by X-ray diffraclion, Raman speelroseopy, high resolution transmission electro" microscopy. and differential Ihermal analyses and Ihermogravimetry. The data presenled in Ihis paper suggest Ihat Ihe alteration of graphile intensifies with inereases grinding lime and depends on the kind of Ihe applied stress. To compare these changes lo those involved in nalural occurrences, a graphite mineralization in which Ihe cryslallinity has been altered by tectonie shear stress has been studied. 80lh artificial and natural shear stress, provoke a decreasc of graphile cryslallinity. The results are discussed in relallon 10 Ihe implications for using graphite as geolhermomeler.
Key words: graphile. eryslallinity, grinding. shear stress. geolhermometry.
Resumen: La variación de la cristalinidad del grafito debido a la inOuencia que la molienda produce sobre la estructura del mineral se ha estudiado utilizando difracción de rayos-X. especlroscopfa Raman. HRTEM y análisis térmico diferencial. Se han observado los efectos de alteración estructural que lienen lugar con el aumento del tiempo de molienda y que dependen del tipo de esfuerzo aplicado. De modo paralelo. se han relacionado los resultados obtenidos con las muestras pertenecienles a una mineralización de grafito en la que se han producido esfuerLCs tectónicos que han modificado su cristalinidad. Comparando ambos procesos se llega a la conclusión de que los esfuerzos de cizalla, ya sean generados de un modo artificial o natural, producen una disminución de los valores de cristalinidad del grafito con las implicaciones que esto puede tener en la utilización de este mineral en geolermomelrfa.
Palabras clave: grafito, crislalinidad. molienda, cizalla, geolermomelrfa
Introducción
La Ifansformación de la materia orgánica presente en los sedimenlOS por efecto del metamorfismo regional o de contacto es el principal mecanismo de generación de grafito. A esta evolución se la conoce como grafitización y supone una serie de transformaciones físicoquímicas que se reflejan en los parámetros estructurales y en l a composición química del mineral. Estos cambios se pueden resumir en: 1) pérdida de compuestos volátiles (H. O Y N)
y enriquecimiento relativo en carbono. 2) formación de anillos hexagonales de átomos de carbono. 3) unión de estos anillos para formar una estructura laminar con simetría bidimensional (crecimiento en el plano a-b), 4) apilamiento de las láminas en una secuencia regular, a lo largo del eje c, que origina la simetrfa tridimensional. De esta forma, el grado de ordenamiento de la estructura del grafito se usa habitualmente como indicador de las condiciones de metamorfismo que lo han producido. La "cristalinidad" o "grado de or-

138 CRESPO FEO. E. el al.
denamiento" del grafito es definido como el grndo de perfección cristalina a 10 largo de los ejes cristalográficos y se puede determi· nar mediante la utilización de diferentes ¡¿cnieas analfticas como la difracción de rayosX. la espectroscopia Raman y la microscopía electrónica de alta resolución. Cada una de ellas nporta información acerca de ti'! estructura cristalina del grafito a diferente escala. Mientras que la primera proporciona una caracterización global de la muestra, las otras dos reflejan variaciones en partfculas individuales y son adecuadas para determinar heterogeneidades estructurales, bien en la dirección de apilamiento. bien a lo largo del plano ¡¡-b (Buseck y Bo-Jun. 1985; Wopenka y Pasteris, 1993).
En el proceso de grafitización intervienen distintos factores como la presión, tipo de precursor orgánico, porosidad y permeabilidad de la roca de caja, y/o duración del metamorfismo, pero sin duda el más importante de todos ellos es la temperatura (Landis, 197 1 ; Kwiecinska, 1980; Wada et al., 1994; entre otros). Shengelia et al. ( 1979) correlacionaron experimentalmente la variación en el valor del parámetro e (e '" 2dool) del grafito con la temperatura, observando que estaba controlado por �sta y que la presión no afectaba, al menos sustancialmente, a los valores obtenidos. El valor de e es un marcador de las condiciones térmicas de formación del grafito y, por tanto, una herramienta adecuada para determinar la temperatura máxima alcanzada por las rocas que lo contienen, posibilitando el uso del grafito como indicador geotermom�trico. Además, el gmdo de ordenamiento estructural del grafito es independiente del retrometamorfismo lo que, junto a la sencillez de los análisis mediante los que se obtienen los parámetros necesarios para la estimación de la temperatura de formación, hace que se le pueda considerar como un geotermómetro monomineral de gran utilidad. En algunos casos, como en los depósitos epigen�ticos en los que solamente aparece
grafito, puede constituir el único indicador geotermométrico disponible. Las estimaciones de temperatura basadas en el grafito pueden ser una alternativa a los geotermómetros de intercambio catiónico o de inclusiones fluidas o, al menos, un método de comprobación fiable (Shengelia et al., 1978; Luque et al., 1993; Malisa, 1998).
Para el estudio específico del grafito se debe separar el mineral de la roca que lo contiene y, para ello, es necesaria la utilización de métodos ffsicos y/o qufmicos que pueden modificar las caracterfsticas cristaloqufmicas del mineral, pudiendo introducir un error en el cálculo de la temperatura de formación. Las posibles modificaciones estructurales que se pueden generar durante la preparación de la muestra y su influencia en las determinaciones geotermométricas, no han sido estudiadas de forma sistemática hasta ahora. En algunas publicaciones se hace notar que un tratamiento inadecuado en la preparación de las muestras puede provocar cambios en la cristalinidad del grafito (Landis, 197 1 ; Pasteris, 1989; Cebulak et al., 1999) e incluso en algunos trabajos se han analizado Jos efectos que provoca la molienda mecánica a nivel de estructura y microtextura sobre diferentes materiales carbonosos, entre ellos grafito cristalino (Nakamizo et al., 1978; Salver-Disma et al., 1999 a, b; Wakayama et al., 1999; Ong y Yang, 2000). Con estos estudios se trata de monitorizar cómo se produce el proceso de destrucción cristalina, principalmente enfocado a la utilización del grafito estructuralmente modificado en la industria tecnológica. En todos estos trabajos los tiempos de molienda exceden en mucho los normalmente requeridos para análisis en geologfa, y únicamente el trabajo de Nakamizo et al. (1978) se realizó sobre muestras de grafito natural.
La alteración de la estructura de minerales laminares, principalmente filosilicatos, tanto por procesos experimentales como naturales ha demostrado variaciones en parámetros estructurales (espaciado basal, "cristalinidad",

Alteración cristalinidad del grafito por procesos cizalla 139
etc.) y térmicos (Pérez-Rodriguez et aL, 1988; Fernández-Cal iani y Galán, 1992; SánchezSoto et aL, 2000; Stepkowska et aL. 200 1). Sin embargo. no existen estudios en los que se comparen ambos procesos.
El presenle trabajo pretende cubrir la laguna experimental existente mediante la modelización de las posibles alteraciones que pueden sufrir las características estructurales y térmicas del grafito debido a los efectos de cizalla provocados por el proceso de molienda, bajo tiempos de duración comunes en la preparación de muestras geológicas, estableciendo los métodos más adecuados en función de las características mineralógicas y texturales de las muestras. El objetivo de este trabajo es doble. Por una parle, se pretende establecer la influencia que la metodología de preparación de las muestras ejerce sobre determinadas características estruclUrales y térmicas del grafila y que son uti lizadas habitualmente para estimar su temperatura de formación. En segundo lugar, se trata de comparar las variaciones observadas a nivel experimental con procesos geológicos análogos (grafito localizado en zonas de cizalla).
Materiales y técnicas
El estudio de la variación de los parámetros estructurales y térmicos del grafi. to se ha realizado sobre una muestra de grafito de Sr¡ Lanka, al que se considera como estándar de alta cristalinidad y cuyos parámetros estructurales son los más cercanos a los del grafito ideal (ficha JCPD 23-64). Las técnicas utilizadas han sido: difracción de rayos-X (XRD), espectroscopia Raman, microscopía electrónica de alta resolución (HRTEM) y análisis térmico diferencial y termogravimetrfa (DT A-TG). Se prepararon 5 muestras para cada tiempo de molienda (l ,
3. 5. 10, 30, 60 Y 120 minutos) en dos tipos de molinos mecánicamente diferentes, un mortero de ágata (Reitsch RMO) y un molino de
aspas especialmente utilizado para materiales de estructura laminar (Culalti MFC CZI3). El tipo de esfuerzo mecánico en el que se basa cada uno de ellos es distinto: el primero genera un esfuerzo normal a la superficie de la partícula (interacción de choque); en el segundo el esfuerzo es tangencial (interacción de corte). Esta es la razón por la que el molino de aspas es el más indicado para minerales laminares, ya que la disgregación de los mismos se produce a favor de las superficies que presentan una mayor debilidad (planos basales).
Para la extrapolación de los resultados experimentales a los medios naturales se ha seleccionado una mineralización de grafito situada en Horcajuelo de la Sierra (Sistema Central, Madrid) donde en una serie de esquistos con sillimanita y biolita aparece intercalado un nivel de cuarcitas grafitosas con un contenido aproximado entre un 10 y un 20% en grafito. La mineralización se desarrolla en el contacto de este nivel de cuarcitas grafitosas con un dique de cuarzo de origen hidrotermal y está intensamente fracturada y brechificada, englobando abundantes fragmentos de cuarcita grafitosa en su interior. El grafito ocupa los planos de fractura generando concentraciones mil imétricas. De esta mineralización se han estudiado, mediante XRD, espectroscopia Raman y DT A-TG, lan(O el grafito que aparece disperso en las cuarcitas como el que se encuentra en las fracturas. A partir de estudios geológicos, mineralógicos e isotópicos previos se ha establecido que la concentración del grafila se ha debido a un proceso de removilización en estado sólido del grafito contenido en las cuarcitas a favor de las zonas de fractura (Crespo el aL. 2002).
Los análisis de XRD se realizaron en un difractómetro Siemens Kristalloflex 810, equipado con monocromador de grafito y utilizando radiación Cu-Ka a 30 kV y 40 mA. El intervalo de exploración fue de 5 a 70 °26, a una velocidad de 0.03 "26/s y una constante

140 CRESPO FEO. E. el al.
de tiempo de I s. Se utilizó silicio como estándar interno, rodándose consecutivamente cada grupo de cinco muestras, correspondientes a un mismo tiempo de molienda. para minimizar y cuantificar los errores.
La espectroscopfa Raman se llevó a cabo en un equipo Dilor XY, constituido por un monocromador doble, un especlrómetro y un sistema de detección multicanal. usando un láser de Argón ionizado (A "" 514,53 nm) de pulso variable. Las condiciones de trabajo fueron en micro Raman (objetivo x 100) con una polencia láser de 100 mW al inicio del haz. y de 3 a 5 mW sobre la muestra. El intervalo de exploración fue de 900 a 1700 cm,l para el espectro Raman de primer orden. y de 2400 a 3200 cm'· para el de segundo orden. De cada una de las muestras se llevaron a cabo cinco análisis que permitieron calcular. al igual que en los análisis de difracción, los valores de la media y desviación estándar del conjunto de los datos.
Los estudios mediante HRTEM fueron efectuados con un microscopio electrónico de transmisión 400Kv JEOL, modelo JEM-4000 EX, del Centro de Microscopía Electrónica "Luis Sru" de la Universidad Complutense de Madrid.
Los DTA y TG para todas las muestras se han efectuado bajo atmósfera de aire dinámico a 50 ml/min. en el rango de temperaturas de 20 a 1000 oC con una velocidad de calentamiento de 10 °C/min .• en un aparato Seiko TGrrDA 320V, utilizando alúmina como material de referencia.
Resultados
En la tabla 1 se muestran los valores obtenidos para los parámetros estructurales medidos mediante XRD y las relaciones determinadas entre ellos. La variación del espaciado dOln y del parámetro e muestra un comportamiento similar para los dos conjuntos de muestras estudiadas, aunque parece que se ve afec-
tOldo más por el tipo de molienda que por la duración de la misma. Los valores son muy semejantes variando dI)(): entre 3.355-3.358 A para las muestras preparadas con el mortero de ágata, y entre 3.353-3.357 A en las muestras procedentes del molino de aspas. La evolución del resto de los parámetros sí que muestra una relación clara entre una molienda más prolongada y la generación de alteraciones en la cristalinidad del grafito. Las muestras preparadas con el mortero de ágata sufren un aumento significativo de la anchura a media ahura del pico (002). expresando un descenso en In cristalinid¡¡d de las mismas, lo que es muy evidente a partir de los 30 minutos de molienda. Las muestras provenientes del molino de aspas presentan la tendencia contraria: mientras que para los tiempos cortos de molienda los valores son mayores que los equivalentes en el mortero de ágata. con el incremento del tiempo de molienda los valores disminuyen no sólo frente al molino de ágata sino. también. respeclo a los tiempos cortos del mismo molino. Los valores correspondientes a la relación looP� pertenecientes al mortero de ágata muestran una tendencia más o menos estable. que sólo a partir de un tiempo de molienda superior a 60 minutos muestra un cambio más acentuado. Para el molino de aspas también se observa un aumento en los valores de esta relación. pasando de un valor de 18 a 2 1 . Lo más significativo es la diferencia entre ambos molinos. ya que las muestras del molino de aspas tienen valores en torno a 2-3 unidades mayores que los correspondientes al mortero de ágata (figura 1). El cálculo del tamai'io de cristal ita a lo largo del eje c (L.) se realiza a partir de la medida de la anchura a media ahura de la reflexión (002) (Wada et al.. 1994). por lo que la tendencia de ambos parámetros es semejante. La evolución de L,. con el aumento de los tiempos de molienda vuelve a reflejar la diferencia que ambos tipos de molinos producen sobre la estructura del mineral. Con el mortero de ágata el tamal'io de cristalito se

Alteración cristaJinidad del grafito por procesos cizalla 141
Tabla 1: Parámetros obtenidos mediante difracción de rayos X (XRD). l/O/:!!: espaciado de la reflexión 002; FWHMO/n: anchura a media altura de la reflexión 002; loo/loo.: relación de intensidades entre la reflexión 002 y la reflexión 004; L.: tamaño de cristal ita a lo largo del eje c (cálculo realizado utilizando la modificación de la fórmula de Scherrer propuesta por Wada et al.. 1994). El dígito entre paréntesis representa la desviación estándar.
MOLIENDA dooJ(Á) FWHMoo1(t.29) /OOJ/OfU L" A) (miD)
1 3,357 (0,002) 0):26 (0,026) 13,86 (2,39) 463,78 (49,81)
� 3 3,358 (0.002) 0):22 (0.01l) 13,12 (1):4) 467,98 (23,91) • :t 5 3,356 (0,002) 0,216 (0,014) 14,34 (IJH) 482,67 (28,60) • �
1 0 3,356 (0,001) 0,222 (0.009) 13,37 (0.53) 469,87 (18.58) e • 30 3,357 (0.001) 0,233 (0.007) 13,25 (1,48) 445,86 (14.04) � o ;¡; 60 3,357 (0,012) 0,263 (0,01l) 15,10 (0,63) 394,91 (19,22)
120 3,355 (0,003) 0,280 (0.006) 18,80 (2,74) 372,84 ( 1 1 ,57)
1 3,3S6 (0,002) 0,244 (0,015) 18,06 (2):2) 428,14 (ll,77)
a 3 3,355 (0,004) 0):47 (0,008) 19,45 ( 1 ,64) 423,46 ( 12):2)
.. 5 3,357 (0,004) 0):65 (0.0tJ9) 21,07 (2,44) 391,52 (14,79) • � 1 0 3,3�3 (0.002) 0,23� (0.006) 20.87 (3):9) 448):0 ( 1 0,64) o
.!! 30 3,356 (0,004) 0,253 (0,017) 17.,86 (3,29) 4 1 1 ,91 (27.64) .. ;¡; 6O 3,355 (0,003) 0,237 (0,014) 21,01 (1,12) 441,16 (25.10)
120 J,35� (0,002) 0,238 ,0,010) '0,21 ,1,50) "41,19 (20,64)
mantiene estable hasta los 10 minutos de mo· lienda en que comienza una disminución muy marcada de estos valores. de hasta 1 10 A. El molino de aspas, sin embargo, muestra una variación poco significativa de apenas 50 A (figura 1). Kwiecinska (1980) estimó que la relación dW! vs. Lr era útil para determinar el grado de evolución de la materia carbonosa. La figura 2 representa esta comparación. ob· servándose que los valores pertenecientes al molino de aspas se concentran en un área restringida reflejando cristalinidades muy se· mejantes. Los valores del mortero de ágata también aparecen agrupados, pero destaca que los puntos correspondientes a las moliendas de 60 y 120 minutos están muy desplazados hacia zonas de menor cristalinidad.
Los parámetros y relaciones obtenidas de los especlros Raman de primer orden apare·
cen en la tabla 2. Los valores de la frecuencia del pico de orden (6uo) para el mortero de ágala no muestran una variación sistemática con la duración de la molienda. mientras que para el molino de aspas se observa una ligera tendencia a la disminución de los valores a partir de los 3 minutos de molienda. Hay que hacer notar los altos valores de desviación estándar que definen las muestras. Éstos re· nejan un bajo nivel de representatividad de los análisis. de modo que se necesila realizar varias repeticiones de las medidas para con· seguir unos valores fiables. La frecuencia de la banda de desorden (Duo) no es un parámelro muy representativo por si sólo. Es mucho más determinante la altura y anchura de la misma, por lo que la relación de inlensidades o áreas entre los picos de desorden y orden aportan más información acerca de la

142 CRESPO FEO. E. el al.
..• -- 1.11_ (Monero Ágata) .�") L. (Mortero de Ágata) ....•... looP .. (Molino Aspas) '*' L. (Molino Aspas)
��--��--�---r----��-----, � 500
"
"
300
,O+---__ --__ --____ �--�--________ __" 2� 3 • ,o � 60 , ..
Tiempo de moliendo. (min)
Figura 1: Variación de los valores de IwJl .. y de L. con cl liempo de molienda. 1",,/1 ... : relación de intcnsidtldes entre la renexión 002 y la 004; L.: tamaño de cr151 .. li10 a lo largo del eje c. Las barras verticales representan la desviación está.ndar.
3.'"
'''' 60' r ilO'
,.'"
•
•
.. Mortero de ÁgafO g Moli"" de Aspu
Figura 2: Comparación de los valores de L, VS. d",,¡ para los distintos tiempos de moliendtl. L.: tamaño de crislalilo ti lo largo del eje e; dOO1: cspaci:ldo de la rcnexión 002. Los valores para tiempos de molienda de 60 y 120 minutos en molino de aspas son casi iguales y por ello están representados por un único punlO.

Alteración cristalinidad del grafito por procesos cizalla 143
cristalinidad del grafito (Wopenka y Pasteris, 1993; Yui el al., 1996). Realmente, la evolución de la posición de la banda O respecto a los cambios en la cristalinidad del grafito no está muy clara, pero en los espectros se observa que estas bandas aparecen cada vez más definidas y con mayor intensidad. Pese a lo que se podría esperar viendo el comportamiento de la posición de la banda O, su anchura experimenta un ligero aumento para ambos tipos de molinos. Los valores varían de 16 a 23 cm·1 en el mortero de ágata, y de 16 a 20 cm·1 en el molino de aspas (tabla 2), y, aunque la variación es muy pequeña, confirma los efectos de la molienda prolongada sobre la cristalinidad del grofito. El valor de FWHM", como ocurría con los valores de FWHMoo1 en XRD, tiende a disminuir con el aumento de la cristalinidad (Pasleris y Wopenka. 1991; Wopenka y Pasteris, 1993; Yui el al.. 1996). Para las relaciones de intensidades (lff/O) y de áreas (Ao/(Ao+Ao)x 100)
entre la banda de orden y la de desorden. una disminución en su valor indicada un mayor grado de cristalinidad del grafito, y, además, las variaciones en la segunda se correlacionan con cambios en el tamaño de cristalito en el plano a-b (L) (Wopenka y Pasteris. 1993).
En el especlro Raman de segundo orden no se observa ninguna variación significativa entre muestras de diferentes tiempos de molienda, ni con molinos distintos. En lodos ellos aparece bien reflejada la banda formada por las frecuencias 2695 y 2735 cm·1 (pico S). El máximo de la banda S se sitúa entre 2720 y 2718 cm·1 y no experimenta variaciones de anchura ni intensidad importantes.
Las imágenes HRTEM de las tres muestras que se seleccionaron para su estudio mediante esta t�cnica ap,necen en la figura 3. Los resultados confirman las variaciones estructurales observadas con la XRD y la espectroscopia Raman. Aunque los valores del espaciado (dOO1 == 3.35 - 3,36 A) y del
Tabla 2: Valores de los padmelros medidos mediante espectroscopia Raman. óu O: frecuencia de la banda de orden, 6.u D: frecuencia de la banda de desorden; FWHMo: anchura a media altura de la banda de orden; 10"/0= relación de inlensidades entre la banda de desorden y [a de orden; AJ(Ao+Aolx 100: relación de áreas entre la banda de desorden y la de orden. El dígito entre pOlr�ntesis representa la desviación estándar.
MOLlE�DA (lnla)
by o (�m" ) by D (�m" ) folla
1 1577,1 (J,2) IlS],J (1,4) 16,5-1 (J,U) 0,14 (o.oJ) 0,13(1,J5)
• f-_"'_--1I-':::'�"C"CIÓ"�')'-II-"C'':''C''"I�'J")-j--='''''�''"I�''�"")-t-:,"�",,17··�"")-t_C'J�·OI::',,.,=.),-� ro 5 1516.1 (2,J) lJSI,J (O) 19,J7 (2,OS) 0,16 (0,01) 4,ZII U,U) •
10 15n,s{l,O) IJ44,6(1,2) 20,01 (1,57) 0,16 (0,03) 4,64(2,71) .1 JO 1577,2 fU) IJSt,O (1,J) 19,0] tl,J') 0,17 (0.05) a,&6 (6,01) 60 1570,\ (5.3) Il�7,4 (l.5) 1 9.01 (1,J1) 0,12 (11.01) 1,57(2.11)
!la 1572,8 (4,J) 1l49,3 ($,J) U,14 (4,0�) 0,10 (0.05) 10,17 O,9J)
J 1510,1 (0,5) IJS5,9(0) 16.53(4,41) 0,11 (0.01) 1,61 (],4S)
¡ f-__ �l __ -"I-":::'=�C·'CIO·'")'-l-'é"="C·C·"·C)--1--='''··éM"(é'·�'·")-t-""�"".IO,,�·'=)-t--"'�J7'(�·="")'-� � 5 1516,2(1") 1346,6 (1,!) IM.JO(Z,Ol) 0,12 (0) 1,01(0)
10 157J,9 U,9) 1)48,9 (O) 10,41 U.aS) 0,14 (O) 4,00 (O) JO 1577,1 (I,J) 1148.S (l,6) 17,59 (I,14) 0,13 (0.06) JM (1,03)
60 1574,6 (1,6) 1348.'(1,.1) 1 1.31 (Z,94) 0,16 (0,1)-1) 5.60(3,7))
UO 1$7I,l t7.31 1J46,4 (J.I) 19,73(4,04) 0,15 (0,01) 5,65(4.34)

144 CRESPO FEO. E. el al.
tamaño de cristalito a lo largo del eje e (L.) indican una alta cristalinidad (Iodos [os ..... 10-res medidos son mayores de 300 A. en las tres muestras estudiadas), las alteraciones que se producen en la superficie de las parlkulas evidencian ciertas transformaciones. En la figura 3a vemos el aspecto que presentan las parlrculas de grafito tras una molienda de 30 minutos en el molino de ágata. El p3ralelismo entre las láminas de átomos de carbono nún se conserva pero la superficie ha dejado de ser un plano horizont:ll. Los efectos de la molienda sobre las partículas de grafito provocan una curvatura que comienza a dtar un aspecto de estructura lurbostrática aunque s610 en superficie (punto 1 ; figura 3a). En las imágenes de In figura 3b y c. correspondientes n la muestrn tratadn durante 120 minutos con el molino de ágata. se observan características similares a la muestra anterior: un paralelismo entre capas con una gran continuidad a lo largo del eje de apilamiento (eje c) que indicarran una alta cristalinidad. pero. también, una modificación del borde ex:terno de la misma que va destruyendo esa disposición horizontaL La figura 3b revela cómo se produce la deslaminnción del grafila en el borde de la partícula. Comienza con la generación de una ondulación que se trnsmite hacia el interior englobando un espesor de 3 nm aproximndamente (punto 2; figura 3b). En el límite con la superficie continua. esa ondulación se traduce en un espacio que continúa abriéndose hasta que se produce la separación de un fragmento (punto 3; figura 3b). Estas láminas independientes oponen menos resistencia al esfuerzo provocado por la molienda que las particulas de mayor tamaño, por lo que su disposición laminar se destruye. formando una madeja de pequeños dominios con estructura ordenada (punto 4; figura 3c). La muestra preparadn en el molino de aspas correspondiente a dos horas de molienda (figura 3d) presenta una estructura más cristalina que su homóloga del mortero de ágata. La disgregación de las partfculas (punto 5; figura 3d) se produce a
favor del plano a-b conservando toda la longitud de la laminación en esa dirección (equivalente al tamaño de cristalito en el plano, L ). Debido a las tensiones provocndns, estos fr
"agmentos se pueden doblar formando codos
como el señalado en el punto 6 (figura 3d). Las zonas más superficiales pueden alterarse algo más. pero en ningún momento llegan a originar morfologías de aspecto turbostrático como las apreciadas en las figura 3a y c.
Respecto a los análisis térmicos, los registros de DTA y TG de las muestras de ambos molinos presentan una bandn exotérmicn muy marcada, acompañada por una fuerte pérdida en peso. que comienza en torno a los 600 oc. Este efecto exotérmico está ligado a la reacción de oxidación (combustión) del grafito y su mhimo se encuentra entre 750 y 820 oC, según la muestra. En los resultados obtenidos por DTA (Fig. 4a), se observó que los valores de temperatura de inicio y fin de In combustión y la temperatura del mhimo exotérmico se mantenían constantes hasta los 10 minutos de molienda. Para intentar monitorizar de manera más precisa esta evolución, se estudió una nueva muestra con 20 minutos de molienda. cuyo comportamiento térmico resultó semejanle a las anteriores. A partir de los 30 minutos. todos los valores de las diferentes temperaturas se reducen sensiblemente.
En el caso del molino de nspns, el descenso en la lempernturn es más evidente a partir de los 60 minutos de molienda. excepto en el caso de la temperatura de inicio de combustión que ya se ve afectada a [os 30 minutos. Pnfa comprobar si estos resultados estaban relacionndos con el tamnño de partfcula obtenido tras cada periodo de molienda. se realizó una serie de análisis que quedan renejados en la figura 4b. La vnriaeión es más evidente en la temperatura del máximo exotérmico. donde se observa un salto claro desde el tamaño <53 llm al de <20 llm. El crecimiento que se produce en el valor de temperatura para tamaños <5 �m puede deberse a que con tamaños

A l teración cristal inidad del grafito por procesos cizal la 145
tan pequeños, las fuerzas electroestáticas provocan que se aglomeren las partículas, y estos conjuntos actúan como partículas i nd i viduales de mayor tamaño que d i ficu ltan la combustión. A partir de l a i nformación aportada por el TG se obtienen datos del contenido en carbono del grafito. Todas las muestras contienen en torno a l 95-98% de e , sin que la molienda produzca , como era de esperar, variaciones en este valor.
En lo referente al estudio estructural y
térmico del grafila procedente de l a m i neralización d e Horcajo. e n la tabla 3 aparecen los valores de los panímetros medidos. En los resultados de XRD son evidentes las d i ferencias entre l as muestras en l as q ue el grafito esuí diseminado en las cuarcitas y las muestras en las que el grafito se concentra en las zonas de fractura (la m i neralización propiamente dicha) . Los valores de dOO2 (y, por tanto, del parámetro e) son menores e n el primer conjunto de muestras i n d icando una
, , -
20 mIl , ,
Figura 3: Imágenes HRTEM . • 1. Conjunto de panículas con aspecto turbostrático de l a muestra de 30 minutos de molienda con mortero de ágata. b. Puralelismo en las láminas de la muestra de 1 20 minutos de molienda en mortero de ágata. c. Curvatura de láminas por efectos de l a molienda en la muestra anterior. d. Aspecto general de una partícula de grafito tras 1 20 minulOs de molienda en molino de aspas. (Para l a explicación de los puntos señalados, ver texto).

146
�
� � � � � � � � e.
�
1000
900
800
700
600
500
- --
---
a
CRESPO FEO. E. el al.
... Mortero de Ágata
final combustión --0-- Molino de Aspas 1=--=f:::::::::�1-"'_A...... _ _ 1I
�...::::: l:'::==':l�.D.--A-"'---11 ' - ' . --11____.. máximo exotérmico �&...--:
... ---= �""::'-Á 11 ·-- 1 �.�
inicio combustión �"--. ... � ...
3 5 10 20 30 60 120
Tiempo de molienda (min)
----- - - ---
ini?O corrburt:n ..........
b
Figura 4: a. Temperaturas de inicio, linal y máximo Cl(otérmico en la curva de ATO. b. Temperaturas de inicio y del máximo exotérmico en funciÓn del tamailo de partfcula para la muestra de 10 minutos de molienda en el mortero de ágata.
Tabla 3: Valores obtenidos para las muestras de la mineralización de Horcajuelo de la Sierra mediante XRD, especlrosCOpra Raman y DT A. T .... : temperatura del máximo exotérmico. El significado de las abreviaturas para los diferentes parámetros estudiados se da en las tablas 1 y 2_
CrOnlO en Cuardlll Grllnlo en Mineralizad n
MUESTRA!
PAR METROS HORC-I HORC-6 HORC-5 HORC-1 HORC-JO "ORe-1J
d.,IA) 3,354 3.355 3,356 3,360 3,358 3.356
� FWHM., (.6."l1t) 0,225 0,198 0,236 0,268 0,210 0,242 e
I .. ,flto• 2 5 1 8 22,36 23 1 0 2080 1 5 3 2 2359
L,(A) 466,26 530,43 439,61 383,14 384,25 386,84
llv O (cm·') 15619 1561 7 1567 O 1568 J
Z llv O (cm·') 1349 O 1310,7 1336.5 1343 9 < � FWJlM,,(cm·', 10,75 1 1 .74 1 1.38 20,26 < �
lo/In 0,05 0,01 0,01 0,10 A An+An xlOO 1 1 94 1.85 2 " 1404
e � < T ..... (I('C) 77l 6" 766 741 67]

AlIeración criSlalinidad del grafilo por procesos cizalla 147
mayor cristalinidad de las mismas. Los resultados de FWHMtxJ1 reflejan el mismo comportamiento, traduciéndose en unas variaciones del tamaño de cristalito L desde ",,25 Á hasta =150 Á entre ambos grupos de muestras. En cambio, la relación de intensidades ItxJ/fOO4 no presenta ninguna tendencia clara. Según Landis (1971), el aumento del grado metamórfico produce sobre el grafito una evolución en su estruclUra que se manifiesta en la agudeza y simelrfa de la reflexión (002) y en la aparición de otras reflexiones en los diagramas de difracción de rayos X que se refleja en e[ aumento del valor de ItxJ/fro.. El mayor valor de esta relación, que por lo tanto se correspondería con una mayor cristalinidad, pertenece a una de las muestras de grafito diseminado en las cuarcitas y el menor valor (menor cristalinidad) a una de las muestras de grafito de la mineralización, pero el resto de valores se distribuyen de un modo aleatorio en [os dos grupos de muestras.
La espectroscopía Raman no permite apreciar ningún comportamiento representativo, excepto el visible aumento de la relación lilo en la muestra HORC-7. El resto de los parámetros no proporcionan una tendencia definida por 10 que, probablemente, sería necesario el estudio de un mayor número de mueSlras para conseguir un patrón más fiable. Respecto a los análisis térmicos, nuevamente el grafito de las cuarcitas presenta el máximo exotérmico a mayor temperatura. pero las variaciones respecto al grafito de la mineralización no son muy grandes. Solamente en una de las muestras la variación es más evidente llegando a una diferencia de casi 100 oC.
Discusión
Los efectos que la presión ejerce sobre el proceso de grafitización se han estudiado en diversos trabajos, tanto experimentalmente (Bonijoli et 01.1.. 1982; Bustin et 01.1., 1995 a.b), como en materiales naturales (Barrenechea et 01.1., 1992; Arkai et aL, 2002). En todos ellos
se confirma que los esfuerzos de cizalla aplicados sobre materiales carbonosos poco ordenados favorecen el aumento de su cristalinidad. En cambio. parece claro que el efecto de la cizalla sobre grafitos de elevada cristalinidad no es el mismo. Mediante el análisis experimental se ha puesto de manifiesto que existe una modificación de la cristalinidad del grafito debido a los efectos de una molienda prolongada. Cada una de las técnicas utilizadas muestra una sensibilidad distinta frente a esta alteración por lo que se puede elaborar un intervalo de uso recomendado. dentro del cual los valores calculados son fiables.
Los resultados obtenidos permiten que las muestras sean definidas como grafitos de 01.1101. cristalinidad. independientemente del tiempo de molienda empleado. Sin embargo. la XRO demuestra tener una gran capacidad para captar las transformaciones producidas en la estructura del mineral. La rotura de la estructura Iriperiódica y de la microtextura laminar del mineral se conseguiría con tiempos de molienda mucho mayores de los que se han aplicado en este trabajo (Salver-Dis.ma et 01.1.. 199901. y b), y de los que se utilizan habitualmente en la preparación de muestras geológicas. Parece que el dw' (y, por tanlo. el parámetro e) se ve afectado
· ligeramente por
el tipo de esfuerzo que ejerce el mortero sobre las mue·stras. provocando un aumento de este valor con respecto al espaciado original del grafito. Esta alteración artificial se debe tener en consideración yaque supone una fuente de error a la hora de estimar la temperatura del metamorfismo sufrido por las rocas. El intervalo de variación es pequeño. pero su reflejo en las estim"aciones geotermométricas es bastante significativo. El resto de los parámetros (anchura a media altura para la reflexión (002). FWHMO()!; relación de intensidades, IO()!I_; y tamaño de cristalito. L<) parecen más sensibles que el anterior a las variaciones en la cristalinidad. Se observa una clara diferencia entre los resultados obtenidos para los dos molinos utilizados. surgiendo la pregunta. de por qué los datos correspondientes al molino de

148 CRESPO FEO. E. el al.
aspas reflejan una menor cristalinidad frente a sus equivalentes del mortero de ágata. Este fenómeno se explicaría debido a los diferentes tipos de esfuerzos que intervienen en la disgregación del material en cada uno de los moli· nos. La deslaminaci6n de la rnueSlra es más efectiva en el mortero de ágata por lo que a tiempos menores muestra de forma más evidente el grado de cristalinidad real del material, pero su agresividad provoca abrasión sobre el plano de las láminas (plano a.b) como se observa en la disminución de los valores de la relación de áreas de los picos D y O de Raman y en las imágenes de HRTEM 0, incluso. la destrucción parcial de la estruclUra en la dirección de apilamiento como ocurre en los tiempos largos de molienda y que se correlaciona con el descenso en los valores de L,.
Las modificaciones a lo largo de la dirección de apilamiento (eje e) quedan muy bien definidas al relacionar los parámetros L� vs. dOOl• Se produce un salto evidente en las muestras de 60 y 120 minutos del mortero de ágata debido a la disminución del tamaño de cristalito L,. asi que este parámetro parece ser un marcador extremadamente sensible a las variaciones de cristalinidad del grafito.
En los parámetros medidos a partir de la espectroscopia Raman las variaciones son muy ligeras y no tienen apenas repercusión al utilizarlas para determinar las condiciones de temperatura. Solamente se produce un error significativo en el rango metamórfico al que corresponde la muestra con tiempo de molienda de 120 minutos en mortero de ágata.
La homogeneidad de estos valores definiría al Raman como una buena herramienta para realizar estimaciones geotermométricas. En ninguna de las muestras de ambos grupos se modifica el pico de orden que aparece en el espectro de segundo orden ya que las alteraciones estructurales no afectan al ordenamien-10 tridimensional (Lespade et al., 1982). Estas se producen a un nivel más superficial quedando reflejadas únicamente en el espectro de primer orden. En este rango de frecuencias se
miden los cambios en el grado de orden del plano a-b (tamaño de cristalito L). Observando los resultados, es evidente que con el mortero de ágata la transformación de la estructura es mayor, aunque con ambos molinos se produce. El aumento de los valores de FWHMo' 1110' y de la relación entre áreas se deben a la ondulación provocada en la superficie de la partfcula. como se ha comprobado mediante HRTEM. También afeclaría a FWHMoo2 (Klug y Alexander, 1974) y podría ser responsable de la aparente disminución de L�.
Comparando los resullados obtenidos en este estudio con los determinados por Wopenka y Pasteris ( 1993) para partículas de grafito de Sri Lanka sin tratamiento vemos que la posición de la banda O coincide con los 1578 cm·1 calculados para el mortero de ágata. Sólo a partir de 120 minutos de molienda con el mortero de ágata. el valor obtenido es mayor que el que aportan estos autores. interpretándosecomo variaciones importantes en LQ'
Con la ayuda del HRTEM se han confirmado todas las interpretaciones realizadas a partir de las técnicas anteriores. El mortero de ágata es mucho más agresivo para la estructura del grafito que el molino de aspas. Con media hora de molienda, las partículas más delgadas se flexionan y aunque en su interior siguen estando ordenadas. la pérdida de cristalinidad es obvia. Tras 120 minutos, el aspecto se asemeja a una estructura turbostrática, se pierde continuidad en el plano a-b y. por tanto, el tamaño de L. disminuye. Los dominios paralelos que se desorientan son de espesores cercanos a los 7.5 nm, y eSlán mucho más curvados que los previos. En este sentido. Sal verDisma el al. ( l999b) describen la tendencia de la materia orgánica grafitizable a formar agregados de morfología redondeada, en torno a 10 nm de diámetro. con un desarrollo de microtextura concéntrica por encima de las 300 horas de molienda. En el caso del molino de aspas la alteración sobre la superficie del plano a·b de las partfculas es mínima, debido a que la desagregación de la muestra se realiza a favor

Alteración cristalinidad del grafito por procesos cizalla 149
de esos planos. Los fragmentos separados mantienen su paralelismo y su continuidad horizontal, por eso se conservan los valores de L y L .
Con respecto a las propiedades tér;;' ica�. las modificaciones producidas en 1;1 cristalinidad del grafito no son lo suficientemente importantes como para variar. de forma evidente. la temperatura de la reacción exotérmica. Los valores aportados por el DTA se mantienen muy estables y además son muy similares para ambos tipos de molinos. La fiabilidad de los datos obtenidos a partir de ella queda limitada a las muestras con menos de 20 minutos en el mortero de ágata, y dc menos de 60 minutos en el molino de aspas. debido a la reducción de tamano de partícula de la muestra, más que por la pérdida de cristalinidad. La temperatura del máximo exotérmico en DT A es muy próxima a la dada por diversos autores (Dissanayake, 198 1 . 1994; Katz. 1987) para el grafito de Sri Lanka considerando evidencias geológicas y geotermometrfa de intercambio (T::750-800 oC). Los datos termogravimétricos no sufren ninguna modificación ya que describen el con· tenido en carbono de las muestras. Esta característica química, lógicamente. no varia por los efectos de esfuerzos mecánicos.
En las muestras naturales estudiadas también se reflejan modificaciones en la cristalinidad debidas a los efectos de la cizalla. Como ha ocurrido en el caso experimental. la XRD se revela como la técnica más sensible a las variaciones estructurales del mineral. La diferencia en los valores entre los grafitos dispersos en las cuarcitas y los que aparecen en la mineralización resulta evidente y se puede correlacionar con los efectos de los esfuerzos tectónicos sufridos. El valor de L. sufre una importante reducción y si se analiza junto con la variación en las temperaturas del máximo exotérmico obtenidas por DTA. que también disminuyen. se puede interpretar como un proceso de disminución del tamaño de partícula debido a la cizalla. En el estudio petrográfico de las muestras de la mineralización se aprecia que las concentra-
ciones del mineral están formadas por partículas de tamaño menor (entre 60 y 25 J.Lm) a las que aparecen en las muestras pertenecientes a las cuarcitas con grafito disperso (tamaños medios de 100 J.Lm). De nuevo 1<1 espectroscopía Raman parece no reflejar las modificaciones que se efectúan en la estructura. indicando que éstas se producen principalmente a lo largo del eje c. Únicamente la muestra Horc-7 refleja modificaciones en la continuidad de los plano a-b (disminución del tamaño de cristal ita L) representadas por un aumento de la relación 1,)10 con respecto al resto de las muestras estudiadas. El aumento de este valor se relnciona con un incremento del desorden del grafito debido a [a disminución en la continuidad de los planos a-b. Según Tu¡nstTa y Koenig (1970), la aparición de la banda de desorden se debe a un efecto del tamaño de partícula. Cuando el análisis Raman se realiza sobre un monocristal de grafito (longitud en el plano a-b suficientemente grande para que se le considere como un cristal infinito bajo los rangos de resolución de esta técnica). el único modo vibracional activo es el E, cuyo reflejo es la banda de orden del -. espectro Raman. en cambio. cuando la mues-tra es poli cristalina, la presencia de pequeños cristalitos generan un nuevo modo activo, el A, . cuyo reflejo es la banda de desorden. . -Esta banda será más intensa cuanta mayor cantidad de contactOS entre partículas haya, O sea. cuanto menor sea el tamaño de cristalito Lo' Parece evidente, por lo tanto, que cuando el grafito ha logrado alcanznr un elevado grado de ordenamiento estructural. la presión de cizalla ejercida sobre él provoca una disminución en su cristalinidad (Salver-Disma et al. 1999a; Wnkayama et ni, 1999; Ong y Yang. 2000)_
Implicaciones geotermométricas
Una de las ventajas del grafito como indicador geotermométrico es que este mineml aparece en muchos tipos de rocas: en rocas

150 CRESPO FEO. E. et al.
sedimentarias como mineral autigénico (Kucha y Wieczorek, 1988), en rocas ígneas desde ácidas a ultrabásicas (Luque el al., 1998), pero sobre todo en rocas metamórficas silleeas y carbonatadas (Kwiecinska, 1980; entre otros).
La disgregación mecánica producida de forma natural o durante la preparación de las muestras puede modificar sensiblemente los resultados del cálculo geotermométrico. En la figura 5, se han representado los valores de e correspondientes a Hore-2. Horc-7, I y 120 minutos para el molino de aspas, y de 1 y 60 minutos para el mortero de ágata. Estos valores son los que mejor reflejan la tendencia de evolución de este parámetro y los que permitirán establecer las limitaciones del geotermómetro. Los datos experimentales aparecen concentrados en un pequeño área, cercanos al valor de e determinado para el grafi
to de Sri Lanka (e = 6,714 Á) correspondiéndose con unos grafitos de elevada cristalinidad. tal y como demuestran los valores de FWHMOO! y L •. Con 1 minuto de molienda los valores para el mortero de ágata y el molino de aspas son semejantes, indicando que la estructura no se ve modificada y, por tanto, los valores de temperatura estimados pueden ser fiables. Al aumentar el tiempo de molienda, el mortero de ágata produce alteraciones en la estructura reflejadas en un mayor valor del parámetro c, que en el caso de los 60 minutos se traduce en una variación de temperatura de aproximadamente 30 oC con respecto al de I minuto. El molino de aspas provoca el efecto contrario, el parámetro e disminuye con el tiempo de molienda y la variación de temperatura llega a los 50 oc. Por tanto, la utilización del parámetro e como geotermómetro quedaría limitada a tiempos de molienda inferiores a 5 minutos. Para tiempos mayores las temperaturas estimadas se pueden considerar como temperaturas mfnimas cuando la preparación de la muestra se ha efectuado con un mortero de ágata, y por encima de la real cuando se realice con un molino de aspas.
Cuando se analiza la proyección del valor del parámetro e de las muestras Horc-2 (grafito diseminado en las cuarcitas) y Horc-7 (grafito concentrado en la zona de fractura) en el diagrama de Shengelia et al. ( 1979) se aprecia una diferencia de temperatura de unos 150 oC (figura 5). Esto supone una gran variación para un grafito que genéticamente es el mismo y cuya única diferencia radica en los esfuerzos de cizalla sufridos para su concentración. Es evidenle la necesidad del conocimiento de la geología de la zona de la que proviene el grafito para la posterior interpretación de los resultados geotermométricos obtenidos mediante esta técnica, los cuales podrán ser considerados como temperaturas mínimas en zonas en las que el grafito ha sido sometido a esfuerzos tectónicos posteriores a su formación.
La estimación de las condiciones de temperatura a partir del grafito por medio de otras técnicas se presenta como una alternativa al uso del parámetro e obtenido por XRD. La espectroscopfa Raman ha demostrado ser menos sensible a las variaciones en la cristalinidad del grafito provocadas por la molienda. En el gráfico de Wopenka y Pasteris ( 1993) correspondiente a la figura 6, se han
'.H
•. 11
'!: " •• 70
.. 11 ...... 1 • 11 ...... 1 tJ. '·Mo ....... ". ,Il",. m 60' MorI .... d. ';p'. ñ I · M.liu olt o""" O IlU·MoI¡ ... " ••• po.
100 lOO �OO �OD 400 100 100 900
Trq Figura 5: Estimaciones geotermométricas a 'partir de las muestras estudiadas, de acuerdo a la calibración de Shengelia el al. ( 1 979).

Alteración cristalinidad del grafito por procesos cizalla 151
representado los datos correspondientes a I
y 120 minutos de molienda en ambos molinos y los de Horc-2 y Horc-7. El valor de L. obtenido, coincide perfectamente con la facies metamórfica estimada para las rocas (granulitas) en las que encajnn las mineralizaciones de grafito de Sri Lanka (Dissanayake. 1994), y para las rocas (esquistos con sill imanita) del gr:lfito de Horcajuelo (Crespo el al.. 2002). Sólo a p:lrtir de 120 minutos de molienda con el mortero de ágata. las condiciones de metamorfismo estimadas son inferiores a las reales. Este gráfico permite delimitar el rango de valores al que pertenece la roca. aunque no proporciona un valor de temperatura cuantitativo. Este valor podría obtenerse a partir del DT A. ya que la temperatura del máximo exotérmico se considera muy cercana a la de formación y siempre está dentro del rango de temperatura de la facies metamórfica correspondiente (Kwiecinska, 1980). De esta forma. se puede precisar el intervalo de temperatura
con una mayor aproximación que mediante el uso exclusivo de la espectroscopfa Raman.
Las temperaturas estimadas mediante DT Á pueden considerarse muy fiables para muestras con menos de 30 minutos de moliend:l. A partir de este tiempo se asume que se introduce un error metodológico debido a la disminución del tamaño de las partfculas. obteniendo temperaturas inferiores a las reales. En el caso de muestras nuturales en zonas de cizalla ocurre lo mismo. por lo que la temperaturu obtenida también debe de considerarse como mínima. aunque es mucho más cercana a la real que la estimada mediante el parámetro c.
Recomendaciones para la preparación de las muestras
Durante la separación del grafito de la roca que lo contiene. la cristalinidad de éste puede ser modificada por la presencia de otras fases minerales que suelen ser más
M¡-------,
'" '" �
;:: o
:;< c
,------ " .-,-�
60 __ Clori"
40 6Iadta-4Gr.uak
"
"
,.
" E.!itauroUIa� o "'_Ia
� ,� J., (Al
3- 20 SIW_ 'b ..., GranuHIa
;;;,... O
O 1000 2000 L, (Ál
", -
O 120' Molino Aspas
e l ' Molino Aspas
* 120'Mortcro Ágata
O l ' Mortero Ág.1tll • I lore-7
• l'lore':!
Figora 6: Relación de :\reas de la banda de desorden (D) frenle a la banda de orden (O) y el tamai'lo de eristalito L. (Wopenka y Pasteris. 1993). y proyeeeión de los resultados de las muestras estudiadas.

152 CRESPO FEO. E. el al.
resistentes y no licnen morrología laminar. y que producen mayor abrasión sobre ¡,]S partfculas de grafilo durante el proceso de molienda. Además. los ataques ácidos neceSiJTios para aislar al grafilo del resto de minerales de la roca también pueden modificar su estructura (Gaweda y Ccbulllk, 1999; Cebulak el al.. 1999), así que parece evidente que hay que reducir en lo posible el efecto de estos tratamientos. La espectroscopia Raman permite la realización de análisis sobre láminas delgadas o probetas de la roca global (Wopenka y Pasteris, 1993). Con esta técnica, no hace falta ni separar ni concenIrar el grafito, de modo que parece ser la mejor opción para las estimaciones geolermomélricas, teniendo en cuenta las consideraciones sobre el pulido indicadas por Pasteris (1989).
Las recomendaciones que se pueden hacer tras este eslUdio para el tratamiento de materiales níllur.l.les con grafito en las estimaciones geotermomélricas. se dirigen a la ulilización de la especlroscopía Raman como método analítico.junto con DTA con el fin de obtener un dato cuantitativo. Si esto no es posible o se quieren efectuar medidas de comprobación medianle XRD. sería preferible realizar una molienda controlada. Deberla realizarse en intervalos de I o 1.5 minutos e ir tamizando enlre cada uno de ellos. De este modo el grafila separado no permanece en el molino. evitando la aplicación durante mayor tiempo de esfuerzos que alteren su cristalinidad. En cualquier caso. debe procurarse que la molienda no produzca parlículas con un lamaño inferior al original. por lo que es indispensable un estudio previo mediante microscopía óptica. De este modo. se puede eslablecer el liempo de molienda necesario para individualizar los granos minerales. reduciendo al máximo la posibilidad de provocar alteraciones en la estructura del grafito y su utiliUlción geotermométrica será más fiable.
Conclusiones
Los procesos de cizalla. ya sean generados mediante molienda mecánica o de origen
tectónico en medios geológicos. provocan alleraciones en la cristalinidad del grafito que se reflejan duran le la caracterización estruclural y térmica del mineral.
La XRD es la técnica más sensible a eSlos cambios por lo que. en los estudios de caracterización eslructural con esta técnica. se debe reducir en lo posible el proceso de molienda evitando que sea superior a los 10 minutos. Para moliendas más prolongadas o para muestras naturales procedentes de zonas de cizalla. la modificación del parámetro c supone la introducción de un error en las estimaciones geotermométricas realizadas a partir de él, y los valores calculados se deben de considerar como temperaturas mínimas.
La combinación de la espectroscopra Raman y del DT A resulta ser una allernativa muy fiable para el cálculo geolermomélrico. Los parámetros delerminados por Raman no reflejan variaciones significativas ni en las muestras naturales ni en las preparadas experimentalmente. La posibilidad de realizar los análisis directamente sobre las láminas delgadas elimina la introducción de modificaciones en la crislalinidad del grafito debidas a la molienda. En esta combinación de técnicas únicamenle deberá tenerse en cuenta que moliendas superiores a 30 minUIOS generan. en los análisis de DTA. temperaturas inferiores a las reales debido a la disminución del tamaño de partícula. al igual que ocurre en las muestras nalurales que han sufrido esfuerzos de cizalla.
Agradecimientos
El presente trabajo ha sido financiado por el proyecto DGICYT PB98-0836. Los aUlores quieren agradecer a la Dra. Rosa Rojas (lnslituto de Ciencia de Materiales de Madrid). Dr. J-P. Boudou (Université Pierre el Marie Curie. Parfs, Francia) y Dr. J.L. 8aldonedo (Centro Microscopfa Electrónica "Luis Bru". Univ. Complutense de Madrid) su colabora-

Alteración cristalinidad del grafito por procesos cizalla 153
ción en las técnicas térmicas. espectroscopra Raman y HRTEM respectivamente. Al Dr. José Fernández Barrenechea y al Dr. Fernando Nieto por sus sugerencias y comentarios que han contribuido a la mejora sustancial del presente trabajo.
Bibliografía
Árkai. P., Ferreiro-Mahlmann, R .• Suchy. V., Balogh, K., SYkorova, l.. Frey, M. (2002): Possible effects of tcclonic shear strain on phyllosilicates: a case study from Ihe Kaderslcg area, Helvclic domain. Central Alps, Switzerland. Se//lvei:.. Milleral. Petrogr. Mili. 82, 273-290.
Barrenechea, J. F., Rodas, M . • Arche. A. ( 1992): Relation belween graphitizalion of organic malter and clay mineralogy, Silurian black shales in Cenlral Spain. Mil/ual. Mag. 56. 477.485.
Bonijoly. M., Oberlin, M., Oberlin. A. (1982): A possible mechanism for natural graphite formation. /m. 1. Coa/ Cco/. 1 . 283-312.
Buseck, P. R. Y Bo-Jun. H. ( 1985): Conversion of carbonaceous material to graphite during metamorphism. Ceoclrim. Coslllochim. Aeta 49. 2003-2016.
Bustin, R. M . • Ross, J. V., Rouzaud, J. N. (1995a): Mechanisms of graphite formation from kerogen: experimenlal evidence. lIu. J. Coal. Cco/. 28. 1·36.
__ o Rouzaud. J.-N., Ross. J. V. ( 1995b): Natural graphitization of anthracite: Experimental considerations. CarbOI! 33, 679-691 .
Cebulak, S .• Gaweda, A., Hanak, B . ( 1999): Dispersed organic maller as an indieator of the metamorphic processes- the example of graphites from western Talra crystalline basamento Aeta MOlllal/istiea Slovaea 4. 197-198.
Crespo, E.. Rodas. M., Luque, F. J . • Barrenechea. J. F. (2002): Graphile mobilization as an alternative mechanism
for vein-type mineralizalion. Proeeetlillgs 18'� Imema/iolla/ Millera/ogieal AssociatiOll 830-4, 269.
Dissanayake. C. B. ( 1981) : The origin of graphite of Sri Lanka. Orgallje Ceoehem. 3, 1-7.
__ ( 1994): Origin of vein graphite in highgrade metamorphic terrains. Millera/. Deposita 29. 57-67.
Fernández-Caliani. J. C. y Galán, E. ( 1992): Innuence of leClonic faclors 00 illile crystallinity: a case study in the lberian Pyrite Bell. C/ay Millcrals 27, 385-388.
Gaweda. A. y Cebulak. S. ( 1999): The origio of graphite in the cryslalline b3sameot of the Western Tatra Mis. (Western Carpatians. S-Poland). Ccologiea Carpatlliea 50, 295-303.
Katz, M. B. ( 1987): Graphite deposits of Sri Lanka: a consequence of gr3nulite facies metamorphism. Milleral. Deposita 22. 1 8-25.
Klug, H. P. Y Alexander, L. E. ( 1974): X-ray diffraction procedures for polycryslallioe and amorphous materials. 2"" editioo. John Wiley ed., New York. 966.
Kucha. H. y Wieczorek, A. ( 1988): Graphite in Kupferschiefer (Poland) 3nd ils genetic meaning. Mil/eral. Deposita 23. 174-178.
Kwiecinska, B. ( 1980): Miner310gy of natural graphites. Polska Akad. Nauk .• Praee Mil1ualogiev1e 67. 5-79.
Landis. C. A. (1971) : Graphitization of dispersed c3rbonaceous material in metamorphic rocks. COlllrib. Milleral. Petrol. 30, 34-45.
Lesp3de, P .• Al-Jishi. R., Dresselhaus. M. S. ( 1982): Model for Raman scaltering from iocompletely graphitized carboos. Carbol! 20, 427-43 1.
Luque. F. J., Barrenechea. J. F., Rod3s. M. ( 1993): Graphite geothermometry io low and high temperature regimes: two case studies. Ceo/. Mag. 1 30. 501-5 1 1 .
_, Pasteris, J. O .. Wopenka, B . • Rodas. M .• Barrenechea. J. F. (1998): Natural

154 CRESPO FEO. E. el al.
f1uid-deposited graphile: mineralogical. characterislics and mechanisms of formation. Am. J. Sci. 298, 471-497.
Malisa, E. P. (1998): Application of graphite as a geothermometer in hydrolhermally altered metamorphic rocks of (he Merelani-Lelatema area, Mozambique Bell. norlheaslern Tanzania. J. African Earrll Se;. 26. 3 1 3-316.
Nakamizo, M., Honda. H., Inagaki, M. (1978): Raman speclra of ground natural graphite. CarbOIl 16, 281-283.
Ong, T. S. y Yang, H. (2000): Errecl of almosphere on the mechanical milling of natural graphite. Carboll 38. 2077-2085.
Pasteris, J. D. ( 1989): /11 siru analysis in geotagical Ihin-seclions by Laser Raman Microprobe Spectroscopy: a cautionary note. Appl. Spectrosc. 43, 567-570.
___ y Wopenka, B . (1991): Raman spectra of graphite as indicators of degree of metamorphism. Call. Mil/era/. 29. 1-9.
Pérez-Rodríguez, J. L., Sánchez del VilIar, L., Sánchez Soto. P. J. (1988): EffecIs of dry grinding on pyrophyllite. Clay Mil/erais 23, 399-410.
Salver-Disma, F., Du Pasquier, A., Tarascon, J.-M., Lassegues, J.-C., Rouzaud, J.-N. ( 1999a): PhysicaJ characterization of carbonaceous malerials prepared by mechanical grinding. J. Power SOI//'ces 81-82. 291-295.
____ -,- ' Tarascan, J.-M., Clinard, C., Rouzaud, J.-N. (l999b): Transmission electron microscopy studies on carbon malerials prepared by mechanical milling. Carbol! 37, 1941-1959.
Sánchez-Solo, P. J . • Jiménez de HaTO. M. c., Pérez Maqueda. L. A., Varona, l., PérezRodríguez, J. L. (2000): Effects of grinding on the structural changes of kaolinile powders. J. Am. Cera",. Soco 83, 1649-1657.
Shengelia, D. M., Akhvlediani, R. A., Ketskhoveli, D. N. ( 1979): The graphite geothermometer. Dokl. Akad. Nallk. SSSR. 235. 1 32-134.
_-::-_ ' Miko. O .. Bezák, V. ( 1978): Determination of ¡he degree of regional met;.morphism in Tocks of Ihe Veporidic cryslalline (Hron complex) using "graphite thermometer". Mil/eralia SloI'aca 10, 321-328.
Stepkowska, E. T., Pérez-Rodríguez, 1. L., Jiménez de Haro, M. e., Sánchez-Soto, P. J., Maqueda. C. (2001): Effecls of grinding and water vapour on the particle size of kaolinite and pyrophyllite. C/ay Mil/erais 36, 105- 1 14.
Tuinstra, F. y Koenig. J. L. ( 1970): Raman speclrum of graphite. J. Chem. Phys. 53, 1 126- 1 1 30.
Wada, H., Tomita, T., Matsuura, K., luchi, K., Ito, M., Morikiyo, T. ( 1994): Graphitization of carbonaceous matter during metamorphism with references to carbonate and pelitic rocks of COnlact and regional metamorphisms. Japan. COllfrib. Mil/eral. Pe/rol. 1 1 8, 2 17-228.
Wakayama, H., Mizuno, J. , Fukushima, Y., Nagano, K., Fukunaga, T., Mizulani, U. (1999): Slructural defects in mechanically ground graphite. CarbOl! 37, 947-952.
Wopenka, B . y Pasteris, J. D. (1993): Structural characterization of kerogens to granulile-facies graphite: Applicability of Raman micropobe speclroscopy. Am. Mineral. 78, 533-557.
Yui, T. F., Huang. E., Xu, J. (1996): Raman spectrum of carbonaceous material: a possible metamorphic grade indicator for Jow-grade metamorphic rocks. J. Metamorp/¡ic Geo/. 14, 1 15-124.
Recibido: Septiembre 2003 Aceptado: Diciembre 2003

Boletín de la Sociedad E$pmiola de Mil/ua/ogía. 26 (2003). /55-J65 ¡SS
Efectos de la molienda en la caracterización mineralógica de materiales sepiolíticos y esmectíticos
David RUIZ DE LEÓN, Jaime CUEVAS, Raúl FERNÁNOEZ. Laura SÁNCHEZ y Santiago LEGUEY Opto. Química Agrícola, Geología y Geoquímica, Universidad Autónoma de Madrid Campus de Cantoblanco. 28049 Madrid
Abstraet: Grinding eonditions can af{eel the minenllogical characterization o{ day minerals due to Ihe different hardness of layer silicates and acccssory minerals, inducing different effects depending on the lime. To evaluale the grinding erfects in these materials, a group ofsamples {rom the Neogen Basin of Madrid, containing high levels of Mg-smectites and sepiolite have been selccted. The accessory minerals found werc dolomite. quartz and feldspars. An agnte bnll-micromill hns been used with a frequency of 1/ 25 Hz, chnnging the time {rom 5 10 60 minutes. The products have been studied by X-Ray dirfraetion in random powder and N1 adsorption.
The delamination of smectite nnd illile minerals is observed when grinding time i5 above 10 minutes. This fael produces a slight underestimalion in the phyllosilieate eontent. Sepiolile bceame only modiried al longer times. In thi5 case, the ehange5 delermined in its quanlifieation are of greater signil"icance than in smeetitic-illitic samples.
Keywords: Grinding, Sepiolite, Smectite
Resumen: Las condiciones de molienda innuyen en la caracterización mineralógica de los materiales arcillosos debido a la diferencia de dureza enlre los filosilicatos y los minerales accesorios, pudiendo provocar distintos efectos en función del liempo. Con el fin de valorar los efectos de la molienda en eSIOs materiales se han estudiado muestras de la Cuenca Neógena de Madrid con un alto contenido en esmeetitas magnésicas y sepiolita ademb de minerales accesorios tales como dolomita, cuarzo y feldespatos. Se ha usado un micromolino de bolas de ágata a un:1 frecuencia de 1/25 S" , variando el tiempo ente 5 y 60 minutos. Los productos obtenidos se han estudiado mediante difracción de rayos X en polvo policrislalino y análisis de la porosidad y la superficie especffica por adsorción de N,.
Hemos comprobado que 11 tiempos próximos a 1 0 minutos de molienda empieza a producirse la delaminaeión de los filosiliclltos esmeclita e ilita, hecho que innuye en una ligera subestimación en la semieuantificaeión de los filosilicatos. La scpiolita no sufre modificaciones importantes en el mismo periodo. Sin embargo, la molienda prolongada de eSle material provoca una disminución imponante del porcentaje cuantificado.
Palabras clave: Molienda, Sepiolita. Esmeetita
Introducción y Objetivos
Las condiciones de molienda pueden influir en la caracterización mineralógica de los materiales arcillosos debido a la diferencia de dureza entre los filosilicalOS y los minerales
accesorios, pudiendo provocar diSlintos efectos en función del tiempo. Los minerales detríticos, de mayor tamaño cristalino que las esmectilas o la sepiolita, pueden quedar en tamaños relalivamente grandes despu�s de una molienda manual. Esto hace que los patrones

156 RUlZ DE LEÓN, D. el al.
de difracción muestren una escasa reproducibilidad. hecho que se ha observado en experimentos de difracción con cuarzo (Klug y Alexander. 1974). Los mélOdos de molienda automáticos aunque solventan en parte este problema homogeneizando el tamaño de partkula. pueden provocar la pérdida p3rcial de información al dañar, en cierta medida, las estructuras laminares (Bish. 1994).
Un exceso de molienda puede presentar otra serie de problemas. especialmente en aquellos minerales que son blandos en direcciones preferentes. como por ejemplo los filosilicatos. Las renexiones de difracción de rayos-X se ensanchan ya que se destruyen [os apilamientos coherentes. De este modo. la molienda en silic:l.los laminares da lugar a la delaminación de partlculas, disminuyendo su espesor y ocasionando cristales separados de mayor superficie específica (Mingelgrin et al., 1978). Por contra, las moliendas muy prolongadas (24-48 horas) dan lugar al colapso de las unidades laminares y a su aglomeración, disminuyendo la superficie específica. En estos casos también se h:l descrito la transferencia de protones ligados al agua interlaminar hacia posiciones de borde y, finalmente, la amorfización de la estructura (Cornejo y Hermosín, 1988, Stepkowska et 0.1., 2000).
Estudios similares en caolines revel:ln que la utilización de técnicas como la sonicación causan efectos análogos de reducción del tamaño de partícula y del ami nación de capas de filosilicatos, provoc:lndo un aumento de la superficie especffica de las muestras (Franco el 0.1., 2003).
La. sepiolita es un mineral industrial de especial importancia en nuestro país. La. zona este de Madrid concentra alrededor de 100 Mt de mineral, lo que constituye las mayores reservas mundiales conocidas (Murray, 2002). Los depósitos de la Cuenca Neógena de Madrid han sido descritos entre otros por Huertas et aL, (1971), Galán y Castillo (1984), Martín de Vidaleset al., (1988) y Leguey et al., (1995). Aun siendo un mineral poco común, se en-
cuentra ampl ¡amente representado en España y países de la Cuenca Mediterránea (Torres Ruiz el al., 1994, Callen, 1984). Estos hechos hacen que la investigación de estos m:lteriales naturales en cuanto a sus propiedades y usos sea de gran interés en nuestro entorno.
Nuestro objetivo en este trabajo es determinar las condiciones de molienda más adecuadas para la preparación de muestras :lrcillosas de cara a su caracterización mediante difracción de rayos-X en polvo policristalino. Para ello se ha usado un micro molino de bolas automático variando los tiempos de molienda. Las muestras estudiadas contienen ilita, esmectitas magnésicas, sepiolita y dolomita junto a otros minerales accesorios. La diferencia estructural y morfológica enlre la sepiolita y el resto de los filosil icatos hace que tengamos que buscar condiciones de molienda que, manteniendo una correcta homogeneización, no alteren las características de los perfiles de difracción de rayos-X. Este trabajo forma parte de la puesta a punto de un protocolo de caracterización de muestras arcillosas asociadas a sepiolita y materiales sepiolíticos situados al SE de Madrid en la zona de Barajas.
Materiales y métodos
Las muestras proceden de una serie de sondeos realizados en el Sudeste de Madrid (área de Barajas) a unas profundidades comprendidas entre los 32 y los 61 m. El muestreo inicial reunió un total de 1005 muestras en las que se realizó un análisis cualitativo por adición de Hel para determinar la existencia de carbonatos y realizar una estimación de su contenido. Por otra parte, se caracterizó la mineralogía de las capas de sepiolita por difracci6n de rayos X en polvo policristalino. De estos sondeos se ha escogido una secuencia de 7.35 m. de espesor situada a una profundidad de 32.55 m. En ella, la sepiolita (58 %) coexiste con dolomita (34 %) y está

Efectos de la molienda en la caractcrización m i neralógica . . . 157
nanqueada a muro y a techo por sedimentos arcillosos ricos en esmectita e i l i ta . Adcm.ís, presenta una intercalación decimélJ'ica de cuarzo criptocristalino (Figura 1 ) .
Para e l estudio de los efecLOs de las condiciones de molienda sobre estos materiales se han seleccionado dos muestras del tramo. La primera (4273) corresponde al lecho de la capa y no contiene sepiolita (muestra eSl11cctítica). La segunda muestra seteccionada (4282) est.í compuesta por sepiolita y dolomita en su mayoría (mueslra sepiolílica) (tabla 1 ) .
La preparación de muestras para cstos cnsayos consistió en el secado a 90nC en estufa y la posterior mol ienda « 1 mm. ) mediante un molino de mart i l los con t.¡miz incorporado. Después se tomaron 2 g. Y se molieron cn un micromolino de bolas de ágata. La frecuencia fue de 1 /25 s·, y los liempos de molienda fueron de 5 , 7, 1 0 , 1 5 , 20, 25, 30 Y 60 minutos respectivamente. Con el fin de eSludiar la reproducib i l idad del mélodo, para cada tiempo se realizaron 2 moliendas.
Los patrones de d i fracción en polvo de la muestra se realizaron en un cl i fractómetro
SO:IOEO
'" '"
'"
,JI
Siemens D-5000 (Cu-Ka). Las rendijas de d i vergencia-anti-scattering y de recepción fueron de 2 mm. y 0.6 m m . respectivamentc. El registró se obtuvo a una vclocid.íd de 2 s por paso, siendo éste de 0.04°, con unos valores de lensión y corrienle de 40 KV Y 40 mA.
Tabla l . Mi neralogía dc las muestras (muestras molidas 10 minutos). Porcentaje en peso.
4273 4282
Esmetítica Scpiolít ica
Filosil icatos 73' 58
Scpiolila - 58
Dolomita 1 1 34
Cuaílo 6 2
Albila 7 5
Microclin<l 3 I
, , *38 Yo Esmeellla, 35 Yo lIila
al Cuano tflllIOCnS::l400 0 ,. .. (113 • L1rno o Nena
I
SEcuEIICIA
539
538
'" 4273
03'
535
'"
4:182 533
El SNIl()!4"�:EI���t""",,",,, o QUtlS r'OSI.C¡;tl� � DOlcrn,:a El FckJe�p.:lto'
Figura l . Pc rril litoJógico del sondeo. La columnrl de la derecha corresponde al tramo estudiado (COlaS en metros).

158 RUIZ DE LEÓN, D. el al.
La determinación de los filosilicatos se realizó mediante agregados orientados solvatados con etilenglicol. Para ello se extrajo la fracción menor de 2 ¡J.m y se homoionizó en Ca2+, Posteriormente se extendió la suspensión en un portaobjetos de 5 cm! y. después de dejar secar, se solvaló con etilenglicoJ. Las condiciones de difracción fueron las mismas que en el caso de la difracción en polvo policrislalino y el juego de rendijas de focalizaci6n fue de 1 mm tanto para la rendija de abertura como para la de anli-scatlering.
La semicuantificación de estos materiales se ha realizado según la norma europea UNE 22-161-92, utilizando la relación de inlensidades entre Jos picos 4.45 A de los filosilicatos y 4.30 A de la sepiolita para la cuantificaci6n de este mineral. Se ha utilizado la relaci6n de áreas. considerando que la medida del área es más indicativa de la intensidad que la altura del pico.
La determinación del cuarzo se ha realizado en la reflexión 3.34 A. ya que la correspondiente a 1 .8 17 A (norma UNE 22- 161-92) es muy poco intensa en nuestros difractogramas. Para utilizar la reflexi6n a 3.34 A se ha seguido un método de deconvo1ución implementado en el programa DRXWIN 2.2.56 @ (Primo. 2002). Esta reflexión está afectada principalmente por la contribución de la ilita (d (003» y de la sepiolita (d (080», aunque esta última es poco intensa (30 %), y en general, muy ancha. Estas dos contribuciones se han tenido en cuenta mediante la localización de dos picos muy próximos. Sin embargo, existe gran dificultad en su asignación por lo que la cuantificación de cuarzo está sujeta a una incertidumbre importante. No obstante, la proporción de cuarzo en la muestra es pequeña en comparación con el conjunto de los filosilicatos.
La medida del área de los picos donde se cuantifican los filosilicatos (4.49 A) y la sepiolita (4.30 Á) se ha realizado también mediante una deconvolución en la región de 18.5 - 22.0 °28 (Figura 2). Esta deconvolución
es necesaria, dado que existe una contribuci6n de cuarzo a 4.26 Á.
Con el fin de conocer como afecta el incremento en el tiempo de molienda a la textura de los minerales que componen la muestra se realizaron medidas de superficie BET y distribución de diámetro de poros por adsorción de Nz con un porosímetro Micromeritics" ASAP 2010. Las muestras se tuvieron en una estufa a 90"C durante 24 horas y posteriormente se desgasificaron a 90"C durante otras 24 h. Este análisis se ha realizado sobre muestras molidas durante 10 y 60 minutos. tomándose como referencia la muestra sin moler y tamizada a 1 mm. Los cálculos de distribución de volúmenes de poro se han realizado según el método BJH (BarreuJoyner-Halenda; Webb y Orr, 1997).
Resultados y discusión
SemiCUOl/lificaci611 y reprodllcibilidad En las Figuras 3 y 4 puede verse el valor
medio y el error absoluto del contenido en filosilicatos y dolomita determinado en función del tiempo de molienda, para cada una de las muestras estudiadas. El error absoluto para los filosilicatos es menor de ± 5 % cuando el tiempo de molienda es menor de 20 minutos.
4.49
" lO'(CII I Ka,)
Figura 2 . DeconvoluciÓn de la región donde se cuantilica la scpiolila.

Efectos de la molienda en la caracterización mineralógica . . . 159
La reproducibilidad es mejor entre 10 y 15 minutos para la muestra esmectftica, mientras que para la muestra sepiolftica el error absoluto entre réplicas aumenta con el tiempo. No obstante, el tiempo a partir del cual se empiezan a observar cambios importantes oscila entre 20 y 30 minutos. En ambos casos, los filosilicatos tienden a disminuir con el tiempo, y por tanto el otro mineral mayoritario (dolomita) aumenta. Este efecto es mucho más acu· sado en la muestra sepiolftica, ya que la mues-
•
•
j -i .. o • " . "-l · : •
• " • •
tra constituida por ilita y esmectita presenta variaciones mfnimas en su cuantificación a tiempos prolongados de molienda. La disminución en la semicuantificación del contenido de filosilicatos comienza a partir de los 7 minutos en la muestra esmectftica y a partir de los 20 minutos en la muestra sepiolftica. Esto está de acuerdo con el carácter más lábil del enlace en la superficie basal de los filosilicatos laminares y lo atribuimos a que, en el caso de la muestra esmectftica, se produce una rápida
. . , ' .
• .. � • .. • .. • .. • • • " ..
" • • • •
• • • -
TIempo de monelldl ' mIli)
j'''fbiUc.lllo¡ --�� . ; ] Figura 3. Variación del contenido medio y de la reprodueibilidad eon el tiempo de filosilieatos y dolomita en la muestra esmectftiea (4273)
..
.. J , _1 ..
.. "
� L-__ � __ � __ �� ______________ � o 10 20 30 «1 " """",po" I'l\0l''''' '''')
• $tpIoIQ -OoIooriCI
Figura 4. Variación del contenido medio y de 13 reproducibilidad con el tiempo de scpiolita y dolomita en la muestra sepioHliea (4282)

160 RUIZ DE LEÓN, D. el al.
delaminación de los filosilicatos sin que se produzcan efectos posteriores de amorfiZilci6n. En la Tabla 1 se muestra el resultado de la cuantificación para un tiempo de molienda de 10 minutos.
EfeclOS de la Molienda (DRX y porosimelrfa de N1)
Las Figuras 5 y 6 muestran la variación que experimentan los difraclogramíls cuando la molienda se alarga en el tiempo (5 mino y 60 min.).
En la muestra esmectíticn, un tiempo de molienda de 60 minutos provoca la desaparición del pico correspondiente al espaciado basal a 15 Á Y la disminución en intensidad del pico de ilila. La reflexión a 15 Á empieza a verse afectada de forma notable a los JO minutos. No obstante. el resto del perfil permanece virtualmente inallerado por encima de este tiempo. Esto implic3 que 13 semicu3ntific::lción ::1 tiempos más I.trgos es muy similar.
En el caso de la muestra sepioHtica se produce una disminución del tam3ño de cristal (renexión ( 1 10» , pasando de una anchura a mitad de la altura de 0.58 "26 a 5 minutos de molienda a 1.03 "26 3 60 minutos de molienda (Figura 6). También destaca el aumento de
,,�, .. � ..... , h ....... " '. 11 ... o l· ··P"'·· I D , 1>., •• iI. • f •• N,· f......... ...... ¡
� ��.� .. ',:� ....... ... ".". ! F4·H f
lE ' F l ! " ';' , .c ·1 \ li E.I .. �: .. \.,,:, I�\.\ ; · � �/·J . ; /1 ,j ..... �..-. • ...... .....'"', �
J '" \
S ·,_..".... .... 14 ... • '4.1( .. �" .....
" .. 10 21 ,. " llh ... IG ••••• ,
..
Figura S. Muestra 4273 (csmectftica). Patrones de difracción de rayos-X. a: S minutos de molienda, b: 60 minutos de molienda.
la señal de fondo en el perfil de difracción al aument::lr el tiempo de molienda ::1 tiempos mayores de la minutos y la marc::lda asimetría del pico hacia ángulos más altos (7-9 o 26) a 60 minutos. Este hecho puede corresponderse con la existencia de láminas de tipo talco, si bien el efecto tiene que estudiarse con más detalle.
Para estudiar los cambios texturales que se han producido en las dos muestras consideradas se realizaron medidas de superficie BET y distribución del tamaño de poros. El comportamiento fue distinto para cada una de las muestras.
En la muestra esmectítica, las isotermas son de tipo rv con ciclos de histéresis que varían entre H2 y H3 (Sing et al.. 1985) (Figura 7). En cualquier caso, el tipo de isoterma se corresponde con un sólido mesoporoso. Los ciclos correspondientes a la muestra de referencia y la molida a 10 minutos se encuadran dentro del tipo H2. Este ciclo de histéresis es típico de las esmectitas (Ej. Huang et al.. 2002, Pernyeszi y Dékány, 2003). La rama de adsorción se caracteriza por un llenado paulatino de poros cada vez más grandes a medida que aumenta PIPO. Sin embargo. la desorción está relativamente impedida para PIPO < 0.9 Y aumenta bruscamente a PIpO < 0.5. Este hecho
, • • • 10 12 2'TI>oto�'1
..
Figura 6. Muestra 4282 (sepiolflica). Patrones de dirracción de rayos-X. Gris claro: 5 minutos de molienda. gris oscuro: 60 minutos de molienda.

Efectos de la molienda en la caracterización mineralógica . . . 1 6 1
se puede interpretar por l a dificultad que se le presenta al N1 para encontrar caminos conectados en la red de mesoporos (Huang el al.. 2002). Es evidente que en la muestra molida a 60 minutos se transforma la estructura porosa. El ciclo de histéresis se hace más estrecho y pasa a ser de tipo H3. Este ciclo es caracte· rístico de poros de tipo rendija con alta conectividad. lo que estaría de acuerdo con la delaminaci6n observada y el consiguiente aumento en la superficie BET (Tabla 2).
La distribución de meso y m¡¡croporos calculada a partir de las isotermas de la Figura 7 se muestra en la Figura 8. A 60 minutos. la distribución presenta un máximo entre 20 y 40 A. mientras que las muestras menos alteradas
. "
no
, ... � 1I .. ;; t .. • < • e " • �
" . . . . - . . . . � ... _� ... . _ .. . ... ... .. :: . . . . .. .. .
o o o .• o., 0.3 0.<
presentan Un3 distribución continua y decre· ciente desde el rango de microporos « 20 A). El incremento del tiempo de molienda aumenta la cantidad de meso poros entre 20 y 1000 A. Esto refuerza la idea de que se produce un cambio de textura. y de que existen aglomerados estables de mayor tamaño que al inicio. los cuales dejan entre sI poros más grandes.
La mayor verticalidad de las ramas de adsorción-desorción a partir de PIpO = 0.9 en la muestra molida a 60 minutos renejaría una tendencia hacia ciclos de histéresis de tipo HJ . los cuales son característicos de la existencia de aglomerados de tamaño uniforme . lo que hace que la distribución de tamaños presente máximos discretos (Daza el al. •
.... .... -. . . ..
o .• o .• 0.7 o .• O.> P .... 1ón Plrel ..
• � -10m1n -6O!rin
Figura 7. Isotermas de adsorción de NI para la muestra 4273 (esmectftica).
Tabla 2. Variación de la superficie BET con el tiempo.
Tiempo molienda (min) BET (m Ig) (esmectltica) BET (m Ig) (sepiolltica) O 66 91
10 52 133
60 82 11

162 RUIZ DE LEÓN, D. el al.
1989). Estos aglomerados han de ser porosos en la muestra esmeclílica dado el aumento de la superficie SET.
En el caso de la muestra sepiolftica los ciclos de histéresis son todos del tipo H3 y la forma es similar en los tres casos (Figura 9). En todos los casos el material se compone de poros rend�ja interconectados. De nuevo In verticalidad de las ramas de 3dsorcióndesoreión indicaría la presencia de aglomera-
dos de mayor tamaño cuando la muestra se ha molido prolongadamente (60 minutos).
Este caso podrfa ser un paso incipiente hacia la amorfización y aglomeración de la estructura de la sepiolita observada por Cornejo y Hermosfn (1988}) cuando los tiempos de molienda de la sepiolita son hasta de 48 horas. Estos autores, justifican con este pro· ceso el descenso detecl.:ldo en la superficie BET (tabla 2). La secuencia de variación de
a,ODla --_ •... .... _----
0,00111
0.0014
0,0012
• .OO, � .. o.oooa
.. - i , \.
......
0.0002
•
L--_ ....... ".-=:::. ..
. .. ..... :::.::::::; . . ....... ��
" ,oo '''' �molroIA)
E:7::.L�=-·1��-""1
"."
Figura 8. Distribución BJH de lamallo de poro p,ara la muestra 427) (esmecllticil). DV/dD: dcrivildil del volumen respeclo ill di6metro de poro.
'" . ------------
". '"
� 1.0 " � ".
1 ': < .. i .. >
m
•
. . . .
•
. .. •. . .
.. , ••• '.' • •• '.' .. , �nt.kllI PIl"'oI
r-.:.;:ímln-:..:.... l�=.�l • •• • ••
Figura 9. Isotermas de adsorción de N¡ para la muestra 4282 (scpioHtica).

Efectos de la moliend:l en la c:lr:lcteriZ:lción minemI6gic:l . . . 163
la superficie SET es diferente :11 caso anterior, ya que en la' primer:l molienda ( 10 minutos) aumenta la superficie específica para luego disminuir considerablemente cuando la molienda se prolonga. El primer efecto es la desagregación de los haces de fibras y, por tanlo, el aumento de la superficie externa.
Las condiciones de molienda que se han empleado en este trabajo son de una agresividad moderada comp:lrada con los de otros autores ya que las modificaciones producidas afectan más a cambios texturales que estructurales. En el caso de la muestra esmectHica sus propiedades adsorbentes mejoran con esta molienda, ya que aumenta su superficie especrfic:l y la interconectividad en el sistema poroso. No es este el caso si consideramos una molienda prolongada en la muestra sepiolítica, donde los fenómenos de aglomeración de partículas conducen a una disminución drástica de la superficie específica. Este hecho, ha sido relacionado con la amorfización parcial de la estructura y la existencia de recubrimientos amorras en la superficie de las partrculas (Cornejo y Hermosfn, 1988).
0.0018
0.0018
o.oou
0,00'2
I! 0,001
� 0.0008
0,0008
0,000.
Conclusiones.
La moliend::a influye de forma importante en la caracterización de muestras arcillosas heterogéneas debido a la diferencia de dureza y texturas entre los distintos materiales. Un exceso de molienda genera la delaminación y fragmentación de los filosilicatos. asr como la formación de aglomerados.
Hemos comprobado que a tiempos próximos a lO minutos de molienda empieza a producirse la delaminación de los filosilicatos esmectita e ilita. hecho que influye en una ligera subestimación en la semicuantificación de los filosilicatos. La sepiolita no sufre modificaciones importantes en el mismo periodo. Sin embargo. la molienda prolongada de este material provoca una disminución importante del porcentaje cuantificado
Estos datos llevan a recomendar que se evite el uso de moliendas prolongadas a la hora de preparar muestras arcillosas para su caracterización mediante difracción de rayos-X.
La continuación de este trabajo pasa por estudiar. mediante t�cnicas microscópicas. los
0.000: L __________ -=::::::::�:;s:.... _____ � >O '00 >O"
DLtmeuo lA) ',::,::.1 iñm .. -l�',!!!..1t! -. . - !l.��ln !
>0'00
Figura 10, Distribución BJH dc tamafto de poro para la muestra 4282 (sepiOlftica), DV/dD: derivada del volumen respecto al diámetro de poro.

164 RUIZ DE LEÓN, D. el al.
cambios Icxlurales producidos y determinar su posible interés. tanto para la caracterización de los materiales como para la evolución de sus propiedades.
Bibliografía.
Bish. D.L. Quantitative X·ray diffraction analysis of soils. l.E. Amonellc y L.W. Zelazny (editors). SSSA Miscd/lllreolls PublicatiOlls. (1994): QUllmiraril1e Melhods ;11 so;/ Milleralogy Chapler 9 p. 267-277.
Bish. D.L. y Reynolds. R.e. Jr. ( 1989): Sample preparation for X-Ray Diffraction. D.L Bish y l.E. Post, Editors. WashinglOn D.C. Modutl Powder Diffractiol/ vol. 20 Chapler 4 p. 73-80
Callen. R. A. (1984): Clays of ¡he palygorskitesepiolitc group: depositional cnvironmenl, age, and distribution, in Singer, A., y Galán. E. (eds.), Pafygorsk;le-sepiolile: OCCllrre/lCes, genesis antl l/ses: tle�eloplllellls ;11 setlilllelltology. v. 37, Elsevier. New York, p. 1-38.
Cornejo, J. y Hermosfn, C. (1988): Slruclural alleralion of sepiolite by dry grinding. Clay Milluals 23. Number 4. p. 391-398.
Daza, L.. Mendioroz. S. y Pajares, J.A. (1989): Influence of texture and chemical composition on sulphur deposition onlo sepiolites. Applietl Clay Sciellce, 4, p. 389-402.
Franco.F, Pérez-Maqueda, L.A. y PérezRodríguez, J.L. (2003): The influence of ultrasound on the thermal behavior of a well ordered kaolinite. TI,ulllochimica acta 7 1247 p. 1-9.
Galán. E. y Castillo. A. ( 1984): "Sepiolitepalygorskite in spanish terliar)' basins: genelical panerns in continental environments" in: Palygorkire-sepiolire: occurre/lces. genesis al/d l/ses: developmellts ;/1 sedimelllo{ogy, v. 37, Elsevier. New York. p. 87-124.
Huang, F.C .. Lee, J.F.. Lee, C.K:, Tseng, W.N.
y Juang, L.C. (2002): Effects of exchange titanium catlons on Ihe pore struclure and adsorplion characterislics of montmorillonite. JOl/mal 01 colloitl and illterface sciellce, 256, p. 360-366.
Huertas, F., Linares, J. Y Martín Vivaldi, J.L. (1971): Minerales fibrosos de la arcilla en cuencas sedimentarias espai\olas. 1. Cuenca del Tajo. 801. Ceo/. y Min. 82. p. 534-542.
Klug, H.P., y Alexander. L.E. ( 1 974). X·Ray diffraclion procedures for polycryslalline and Ilmorphous materials. Wiley, New York.
Leguey, S., Martfn Rubl, J.A., Casas, J., Marta, l., Cuevas. J., Álvarez, A. y Medina, J.A. (1995): Diagenetic evolution and mineral fabric in sepiolitic material s from the Vidlvilro deposil (Madrid Basin). G.l. Churchman, R. W. Fitzpalrick and R.A. Eggleton (Editors), Clays: Colltrofli"g ,he EI/virol/lllellt. Proc. la· l. elay COI/f. Adeltlitle, AllStralia, /993. CSIRO Publishing, Melbourne, Auslralia, p. 383-392.
Martín de Vidales, J.L., Pozo. M., Medina, LA. y Leguey S. (1988): Formación de sepiolila-paligorskita en lilofacies lUIÍlicocarbonític:ls en el sector de BoroxEsquivias (cuenca de Madrid). Estlldios Geol., 44, p. 7-18.
Meckhemer. A.H.G. y Ismail. H.M. (2004).: Dispersion or ceria and lanthana on silica and alumina supposts X-ray diffractometry and nitrogen sorptometry studies. Colloitls ami SlIrfaces: Pllysicochelll. El/S. Aspecrs 235, p. 129-1 36.
Mingelgrin, U., Kliger. L., Gal, M. y Saltzman. S. ( 1978): The effect of grinding on the structure and beh:lviour of bentonile. Clays al/d Clay Millera/s. 26. p. 299-307.
Murr:ly, H. (2002). Industrial clays case sludy. Minning. minerals and sustainable development projecl. Report 110. 64. 11 Ed (Infemm;ollal itlstüllte for ellvirolllllellt

Efectos de hl molienda en la caracterización mineralógica . . . 165
alld developme"t. WBCSD (World Business Coullcil for Sustaillable Developmem J. p. 9.
Norma Espanola UNE 22-161-92 ( 1992): Cuantificación del contenido de sepiolita.
Pernyeszi, T. y Dékány, 1. (2003): Surface fractal and structural properties of layered clay minerals monitored by small-angle X-ray scauering and low-temperature nitrogen adsorption experiments. Colloid Polym. Sci. 281: p. 73-78.
Primo Martín V. (2002): DRXWin 2.2.56. [email protected] http://icmuv.uv.es/drx wi n
Sing. K.S.W.; Everelt. D.H.; Haul. R.A.W.; Moscou. L.; Pieroui. KA.; Rouquérol. J.; Siemieniewska. T. (lUPAC) ( 1985): Reporting physisorption data for gas/sol id systems with special reference to ¡he determination ofsurface area and porosity. Pure alld Appl. C¡'em., Vol. 57, No 4. p. 603-619.
Stepkowska. E.T., Pérez-Rodríguez, J.L.. Jiménez de Haro, M.C., Sánchez-Soto, P,j.
y Maqueda C. (2000): The influence of grinding on the particle size as measured by water sorption. En "SEA, IlItegración ciencia tecnología de las arcillas ell el colltexto tecllológico actllal del lluevo /IIilenio". J. Zapatero, A.J. Ramírez y M.V. Moya editores. Edita: Sociedad Espunola de Arcillas. P. 3 15-326
Torres-Ruiz, J., López-Galindo A., GonzálezLópez, and Delgado, A. ( 1 994): Geochemistry of Spanish sepiolitepalygorskite deposits: Genetic considerations based on trace elements and isolopes. Chemical Ge% gy, 1 12 p.221-245.
Webb, P.A. y Orr, C. ( 1 997): Analytical methods in fine particle technology. Micromeritics Instrument CorpoTtltion ed. Norcross, GA, 301p.
Recibido: Noviembre 2003 Aceptado: Diciembre 2003

Bof�l(n de fa Sociedad Espmiofa de Minerllfog(a. 26 (2003). /67·/77 167
Caracterización mediante microscopía electrónica de ba
rrido de partículas atmosféricas del área industrial de
Huelva (SW de España)
Characterization of atmospheric particles by scanning electron
microscopy (Huelva industrial area, SW Spain)
Jost M. BERNABÉ Y M. Isabel CARRETERO Opto. Cristalografía y Mi neralogía. Facultad de Química. Universidad de Sevilla. Apdo. 553. 41071 Sevilla
Abstrael: The study of :umospherie: partie:1cs from liuelva industrial arca (SW Srain) by me::lns of seanning e:lectron microscopy wilh EOX analysis have: bee:n done:. Results point out. hcside:s new morphologie:al and e:hemical data, the seasonal variability and probable origín of partides. Moreover. when possible ¡he: mineralogy has been establishcd. Partieles sampling are located al the Odid and Tinto rivers junction, within the industrial area of Huelva and jusI bdore Iheir oUllet in the Atlantic Oe:ean. According 10 their morphologies four groups ofpartieles have been differentiate:d: sphericals. irregulars, cristal1izalions and biologicals. Spherieal parlieles exhibit spongy, smooth and rough surfaces and are m3ioly e:omposed of S 3nd Si. They are of :lOlropie: origin aUribuled lO fonil fud combustion (ehemiul industries, vehides and thermic power plant). Irregular partic1es are maioly eompased of S, Si or Ca allhough Imees of other elements have been delected (Fe, Cu and Zn). The proposed origio is both natural (soils) and antropie (metallurgie and fertilizer industries). Partieles named crystalli1.ations are made up of S, Cl and P with minor eonlent in other elements (Na. Ca. Mg. etc.). BOlh, nalural (seo. water) and 3ntropie (SO,eminions. fertilizer and eh[orine industries) sources have been established.
The most abund3nt particles are antropie in origin and inelude: a) Spherieal particles (spongy surface) from fonil fud combustion are predominant. b) Irregular parlicles eomposed of S, Fe. Cu or Zn, sourced in melallurgieal industries. e) Cryslalli1.ations eomposed of P and Mg (fertili1.er industries) and S and Ca (SOl emissions). Natural parlicles are subordinated showing dirrerent sources: mineral (soils). biological (pollen) and marine (sallS).
V;lriation in the atmospheric particle content along the year is negligible beeause the conlinuous antropie emission$, only biological panicles (pollen) show a nOleworthy increase during spring season.
Key words: atmospheric panicles. scanning eleClron mieroseopy, polluting sources, Huclva (SW Spain)
Resumen: El empleo de la microscopia electrÓnica de barrido con sistema de análisis qulmico por dispersiÓn de energla de rayos X, para e[ estudio de I;lS partlculas atmosféricas del área industrial de Huelva, ha aponlldo nuevos datos morfológicos y qufmicos. asl como su posible variabilidad estacional y origen. deduciendo su mineralogra en los casos en [05 que ha sido posible. La captaciÓn de partlculas se ha realizado en la eonnuencia de los rfos Tinto y Odie!. dentro del área industrializada de Huelva y cerca del Oehno Atl:\ntico.
De acuerdo con su morfo[ogfa. las partlcu[as encontradas se han clasificado en cuatro grupos: esféricas. irregulares. cristalizaciones y biológicas. Las partlculas esrérieas presentan texturas

168 BERNABÉ. J. M. Y CARRETERO. M. 1.
espongiformes, lisas y rugosas. y están compuestas en su mayorla por S y Si . Estas partfculas son de origen 3n1rópico, y proceden de la combustión de combustibles fósiles utiliz:.dos principalmente en 13 eentral l�nnica, industrias pelroqufmicas y vehfculos de mOlor. US partlculas irregul:lrcs están compueslas principalmente por S, Si o Ca, junIo a otros elementos en menor proporción (Fe, Cu y Zn). Su origen es natural (polvo del tcrreno). y anlr6pico (industrias mct¡¡lúrgicils y de fertilizantes). Las cristalizaciones están compuestas predominantemente por S, el o P, junIo con aIras elementos (Na, Ca. Mg, etc.), y presentan tanto origen antrópico (industrias de fertiliznnlcs y de producción de cloro y derivados. y emisiones de 501) como natural (agua del mar).
Las partículas m:!.s abundantes son de origen antrópico, destacando: a) las partrculas esféricas espongiformes procedentes de gases de combustión de combustibles fósiles; b) las partículas irregulares formadas por S, Fe, Cu o Zn, procedentes de la industria metalúrgica; y e) las cristalizaciones compuestas por P y Mg, Y S Y Ca, procedentes de la industria de ferlilizantes y de las emisiones de SO,. Las partículas de origen natural son menos abundantes, con una procedencia principalmente mineral (Polvo del terreno), biológica (polen) y marina (sales).
Debido a la continuidad a lo largo del año tle las emisiones antrópicas. no se detectan variaciones en la cantidad de partículas atmosféricas encontradas. exceptuando las partículas biológicas (polcn) que abundan en primavera.
Palabras clave: partículas atmosftricas. microscopia electrónica de barrido. origen de contaminanles, Huelva.
Inlroducción
Dentro del ámbito de la contaminación atmosférica se definen como partfcuhls atmosféricas a todas aquellas sustancias en estado sólido o Ifquido. excepto el agua no combinada, dispersas en un gas, con tamaño entre 6xlO"Jlm y 500 Jlm (Umbría et al., 1999). Aunque las particulas no suponen un porcentaje muy elevado sobre el total de contaminantes atmosféricos. su riesgo potencial es muy grande, causando importantes daños en el patrimonio arquitectónico (Ausset et al., 1992; Sabbioni, 2002) y en los seres vivos (Bignon, 1990; Guthrie y Mossman, 1993).
Las partfculas atmosféricas pueden clasificarse según su origen, morfología o composición química. En función del origen, se clasifican en parHculas antropogénicas y partfculas na!orales. A escala global. entre el 15 y 20 % del total de las partículas presentan un origen antrópico, siendo el porcentaje restante d e origen natural (Amoroso y Fassina, 1983). A escala local. los porcentajes antes descritos pueden variar para ZOn:lS muy industrializadas, como seda el caso del área de Huelva (Querol el al., 2002) o de la provincia de Castellón (Álvarez Mota, 2001).
Considerando su morfologfa, las partículas se clasifican en esféricas (espongiformes, lisas o rugosas), irregulares, biológicas, amorfas y cristalizaciones (Umbria el al., 1999). En función de su composición qulmica, las partículas pueden agruparse en carbonáceas, aluminico-silicatadas, cálcicns, metálicas, elc. (Delaieux et al., 2001 ; Sabbioni, 2002). La caracterización quimica de las partículas aporta una información adicional muy útil a la hora de diferenciar y deducir el origen.
Los m6todos empleados para la toma de muestras de las partículas atmosféricas y sus t6cnicas de estudio son muy diversos (Van Grieken tI al, 1998). El muestreo puede realizarse por gravedad, precipitación electrostática, filtración, impaclación, etc., siendo los dos últimos los más utilizados. Asimismo, las diferentes técnicas analíticas que pueden emplearse son espectTOscopía de emisión y de absorción atómica, cromatografía iónica, nuorescencia de rayos X (FRX) (Chabas y Lefevre; 2000, Torfs y Van Grieken, 1997), espectroscopia de masas y emisión atómica con plasma acoplado inductivo (Brewer y Belzer, 2001; Queral el al., 2002), o emisión de rayos X inducida por protones (PIXE) (Bernabé tI al., 2003; Bogo et al., 2003). To-

Caracterización de partículas atmosféricas del área de Huelva 169
das estas téc nicas están enfocadas a la caracterización química de la muestra global, siendo la mayoría de e l las destructivas (excepto FRX y PIXE). Para el eSludio de las panículas desde el punto de v i sta morfológico, el método óptimo es la m icroscopía electrónica de barrido. Cuando esta técnica lleva acopIado un sistema de anál is is químico por dispersión de energía de rayos X (EDX) se pueden analizar las partículas desde un punto de viSla químico además de morfológico, pudiéndose deduci r en muchos casos su origen (Chabas y Lefévre. 2000; Esberl el a/ . , 1 996; Umbría el a/., 1 999).
El área i ndustrial de Huelva, situada próxima a la ciudad y cercana al Océano AtUntico, alberga un gran n(¡mero de actividades industriales contaminantes, por lo que es una zona muy interesante para el estudio de partículas atmosféricas. Los estudios previos sobre partículas atmosféricas rea l izados en el entorno de Huelva han estado enfocados a la caracterización química, aponando información sobre su composición y posible origen (Querol el a/
., 2002; Bernabé el a/ . , 2003). El presenle
trabajo se ha centrado en el estudio de las partículas atmosféricas de este (¡rea industrial , aponando nuevos datos morfológicos y químicos, su pos ib le vari abi l idad estacional y origen, y deduciendo su m i n eralogía en los casos en los que ha sido posible.
Área de estudio
El muestreo se ha realizado en la confluencia de los Ríos T i nto y Odiel . lugar conocido como la "Punta del Sebo" (SW España). Este punto se encuentra s i tuado entre la ciudad de I-Iuelva (al Norle) y el Océano AIlántico (al Sur), distando aproximadamente 5 Km de ambos (Fig. 1 ) . El área prese"Ia desde 1 960 una fuerte industrial ización, época durante la cual un gran número de industrias químicas básicas y petroquímicas comenzaron a desarrollar su actividad. Por su cercanía
al punto de muestreo, destacan el polo químico induslrial "Punra del Sebo" y e l polo químico industri a l "Nuevo Puerto", exist iendo un tercer polo industr ia l deno m i nado "Tanesas", localizado a 1 2 K m en dirección Noreste. También hay que destacar la presencia de una r;íbrica de cemento en la localidad de Niebla situada a 30 K m en esa misma d irección (Fig. 1 ) .
E l Polo Químico I ndustrial "Punta del Sebo" concentra la mayor parte de las i ndustrias dedicadas a l a fabri cación de ferti l izantes (Ferriberia Hue/va, F/WC Forel y Rhodia
(� .. . . t D �1 HU��A: � Mo,.o,
�\t/Gt ...... .
�.W' OCEANO AT�ANTI� Leyenda
I ��I I el Oi ," • "'" N " . . . , . * P ... nlo do m ... ollreo
[;J Pobl:.clonol
� DIII.:I. de r •• ld ... o. • Polos Ind .... lrl.'los:
1) Punl:' dol Sobo
W (' (;I()'\ . � � ... . 2) Nuovo PuOrlO
J) Tarlolol
'. ' . ':"' , ... ' I 1'1 • • '
s
a)
b)
Figura l . a) Localización del punto de muestreo y de las actividades industri.tles en el área dc Huelva. b) Representación de la dirección y frecuencia del viento en el Jño 2000 (datos facilitados por la Consejería de Medio Ambiente de la Junta de Andalucía).

170 BERNABÉ. J. M. Y CARRETERO. M. 1.
Iberia S.A), y a las actividades metalúrgicas (Alfollfic Copper). Las primeras basan su actividad en la generación de ácido fosfórico. fosfato sódico y amónico. ácido sulfúrico y silicato sódico. Durante estas actividndes industriales pueden emitirse a la atmósfera Fundamentalmente PO/, F', SO/o y NH;. A su vez, estas actividades generan una gran canti· dad de residuos sólidos y líquidos (ricos en PO/o F', SO/o Fe'· y CuJo) que son acumulados en grandes balsas (aproximadamente de 3 Kml) situadas muy cercanas del Ifmilc Este del núcleo urbano de Huelva (Balivar. 1995). Las induSlrias metalúrgicas, muy importantes entre los años 60 y 70 debido al apogeo de la minería en la Faja Pirftica (Norte de Huelva), en la nctualidad se han reducido n las actividades desarrolladas por Atfamic Copper, dedicnda a la producción de cobre a partir de sulfuros importados. Las principales emisiones son S02' H1S04' As, Sb, Pb, Zn, Sn y otros metales. Además de estas dos principales actividades, existe una planta de generación de energra eléctrica (Celllral Térmica Co1611, ENDESA). que utiliza gns o fuel-oil como combustible.
El Polo Químico Industrial "Nuevo Puerto" agrupa a las industrias pelroqufmicas del área (CEPSA Pefroqll/",ica y CEPSA ERTISA) en las cuales se generan productos derivados del petróleo como fuel-oil. gas-oil. gasolina, acetonas. etc. las principales emisiones que pueden generarse de estas actividades industriales son hidrocarburos, SOl' NO,. Ni Y V. Otras actividades desarrolladas en esta zona son la generación de hidróxido sódico. cloro y derivados (hipoclorito sódico. ácido clorhídrico. clorometano. etc.) por Aragollesas; In producción de pigmentos de TiO! por Hlllltsmall Tioxide. y In obtención de nmoninco y urea por Ferliberia-Palos. Estns tres últimns actividndes pueden emitir principalmente a la atmósFera cloro. titanio y amoniaco (Bolivar. 1995).
En el Polo Qu{mico Industrial "Tartesos" se encuenlTa ubicnda ENCE Celtllosa, que pro-
duce pasta de papel mediante el método pasta de sulFato. emitiendo principalmente H1S y SO!, Como último foco de emisión importante en la zona está la fábrica de cemento de la cercana localidad de Niebla. que emite principnlmente S0l y partículas.
los vientos predominantes del área de Huelva son de componente NE y NW. circulando a través de los vnlles del Río Tinto y Río Odiel, y de componente SW, de procedencia Atlántica (Fig. l ) .
Metodología
El muestreo de las partlculas atmosfériCólS se hól renlizndo medinnte un caplador de partículns en suspensión, que constól de una unidad de filtmdo formada por un soporte de filtro, cnudalímetro y bomba de vacío de bajo volumen (Fig. 2). El equipo estaba diseñado de formól que se evitaba la recogida de partículas por gravedad o la acción directa del agua de lluvia. Se utilizaron filtros de membrnna de policarbonato (Nucleopore) con un diámetro de 47 mm y tamaño de poro de 0,4 J-lm. los muestreos se realizaron durante una semana completa, con una Frecuencia bimensual. n lo largo del año 2000.
los filtros obtenidos se han estudiado al microscopio electrónico de barrido utilizando un equipo lEOl lSM-5410 con sistema de análisis químico punlUal por dispersi6n de energra de rayos X. La metalizaci6n de las muestras se ha realizado con carbono. Para establecer el posible origen de las partrculas se ha tenido en cuenta la localización de las industrias dentro del Polo Industrial, los datos aportados por Querol et al., (2002), la clasificación que establece Umbrfn el af .• ( 1999) Y los datos meteorológicos fncilitados por la Consejería de Medio Ambiente de la Junta de Andalucía, correspondientes a la época en la que se realizó el muestreo.

Caracterización de p;'lrtículas atlllosféricas del .írea de I-Iuelva 1 7 1
Resultados y discusión
Las partículas atmosféricas encontradas se pueden c l as i ricar en cuatro grupos: panícu las esféricas , part ícu las i rregu lares, cristal izaciones y panículas biológicas. Estas úl t imas aparecen allamenre estructuradas y con texturas complejas. Se han el1conrrado granos de polen de d i ferentcs morfolog ías, con diámetro medio de 1 5 �lIn.
Parttcu/as e.\féricas En función de la textura que presentan.
las partículas esféricas pueden d i fcrenciarse en partículas espollgiformes, l i sas y rugosas . Las parttcu/as e!Jféricas espollgi/o/'lIles (Fi g .
3 ) presentan diámetros comprendidos entre 7
y 35 �l1l1, es tando compueslas principa lmen te por S y S i , Y en menor proporción por A l . Ca. p. Na. K. el . V e Fe. Las partículas esféricas espongi ronnes proceden genera lme nte de la
combustión incomp leta de los componentes orgánicos del fuel-o i l y/o fuel-gas. dando lugar a l a formación de panículas de gran superficie específica y baja densidad. La textura porosa se generaría por la e l imi nación de voI.hiles durante el proceso de combustión (UI11-
PARTicULAS ATMOSFERICAS
BOMBA DE VACIO
bría el al. 1 999). En el caso del área de I-Iuelva. estas partículas pueden proven i r de la u t i l i za
ción de combustibles fós i les para la generación de energía en la Central Térmica Colón,
a escasas decenas de metros del punto de Illuestreo, donde se u t i l i za fuel-oi l y/o fllelgas para su funcionamiento, así como las propias emisiones de los vehículos de motor, destacando el transporte de gran tonelaje por carretera y vía férrea. Otras posibles fuentes de emisión serían las industrias pctroquímicas
de "Nuevo Puerto", pero teniendo en cuenta las d irecciones preferencia les de viento (SE, NW y SW). el aporle d ircclO desde estas fuentes de emisión hac ia el punto de muest reo no es pos ible (Fig. 1 ) . aunque no hay que descar
tar un aporlc ind i recto por procesos de recirculación elel vienLo.
Las pal'ttcu/as e.\!érica.\· lisa.') presentan di .ímctros comprendidos entre 2,5 y 6,5 �m.
Tienen composición a lumin ico-s i l icatada, pre
sentando también en algunos casos T i (Fig. 4). Al igual que las anteriores, las partícu las esféricas lisas se generan por procesos ele combust ión . En este caso, su origen radica en l a fusión de l as impurezas que presenta el mate
rial combust ible, que a l fundirse da lugar a
CAUDALiMETRO
CAPTADOR OE
PARTicULAS
Figura 2. Equipo de captación de panículas atJllosféric�s.

172 BERNABÉ, 1. M. Y CARRETERO, M. 1 .
Figura 3 . Panícula esférica c spong i fonnc compuesta principalmenLe por S.
gotas l íquidas de forma esférica debido a s u tensión superfi c i a l . Esto suele ocurrir e n los procesos de combustión del carbón m i neral que pueden presentar i m purezas m i nerales (Umbría el (/1 . , 1 999). Si el proceso de enfriamiento es rápido, las part íc u l as pueden retener elementos mel:íl icos como T i , V, Fe, N i , ClI, Z n y Pb (Fassi n a , 1 992) . S i n embargo l a u t i l i zación d e l carbón m i neral e n el caso del [¡fea i ndustrial ele I-Iuel va es escasa, debido a l a i m p lantación ele refinerías petrol íferas que abastecen a las industrias con fucl-oil y otros combustibles. Al ser partículas de pequeño tamaño, pueden provenir de fuentes remotas del punto de muestreo, por ejemplo de l a cementera u b i cada a 30 K m en l a localidad de Nieb[a, donde durante el proceso de fabricaciÓn del cemento se produce [ a calcinación de materiales c a l i zos con s i l icatos y otros componentes (óxidos) a temperaturas de 1 500 oC. Durante este proceso de calci nación se produciría la fusión parcial de. los m a teriales y l a formación de las partículas sól idas esféricas. Estas partículas podrían l legar a l ¡írea de muestreo en los periodos donde el viento presenta u n a d i rección preferencial Noreste.
Las partícl/las esféricas ntgosns presentan diámetros comprendidos entre 1 ,5 a 2 �1 1l1 . Dentro d e e l l a s se pueden d i ferenciar partícu-
Figura 4. Partícula esférica l i s a compuesta por S i .
Al Y T i .
las compuestas principal mente por Ti, y partículas compuestas por S, Fe y S i . Las partículas esféricas rugosas compuestas principalmente por Ti son de pequeño tamaño con 1 ,5 )lm de cli¡í metro. Observadas a grandes aumentos, estas partículas aparecen como agregados de part ículas m¡ís pequeñas, s i n que se aprecien procesos de fusión en su generación. Su origen puede estar l i gado a la industria de p igmentos ubicada en el entorno del Polo Químico "Nuevo Puerto", basada en l a generación de Ti0l' El segundo tipo de partículas esféricas rugosas, compuestas por S, Fe y Si laJ11pOCO presentan signos evidentes de fusión. En general las partículas esféricas rugosas proceden principa[mente de hornos de fundición de metales y estún compuestas principal mente por metales en forma de óxidos ( U m bría el al. 1 999).
Por tanto las partículas encontradas pueden tener su origen en los procesos metalúrgicos que se [levan a cabo en el Polo Químico ·'Punta del Sebo", basados en l a transformación de la piri ta y otros s u l furos metálicos.
Todas las partículas esféricas encontradas son de origen f/Ilfrópico, siendo las partículas esféricas espongiformes, procedentes de l a central térmica, vehículos de motor de combustión e industrias petroquímicas. [as que aparecen en mayor cant idad.

Caracterización de partículas ,1Imosféricas del área de 1-luel v<1 173
Partíc/llas irregulares Las partíc ulas irregulares pueden d iferen
ciarse en tres tipos, en función de su composición química: partículas irregulares compuestas principalmente por S, partículas irregulares compuestas principalmente por S i , y partículas irregulares compuestas por Ca y otros elementos como P o Mg.
Las partículas irregulares COlllpIlCS/aS prillcipalmellte por S, contienen en menor proporción Fe-Cu-Zn o Ba. Las partículas compuestas por S , Fe, Cu o Zn, presentan un tamaño variable, oscilando entre los 3 y 30 ,tm de diúmetro medio. Pueden presentarse como partículas de contornos redondeados (Fig. 5), o en forma de partículas irregulares con contornos definidos. Las primeras partículas podrían ser sulfatos y pueden tener un origen secundario por reacción del Fe, Cl! o Zn con en S. El segundo tipo de partículas podrían ser sulfuros, tratándose probablemente de pirita (S, Fe), calcopirita (S, Fe y
Cu) y caleosina-covellina (S y Culo Todas estas partículas (sulfatos y sulfuros) pueden generarse por las industrias metalúrgicas ubicadas en el Polo Químico "Punta del Sebo". Las partículas i rregulares compuestas por S y Ba presentan un d iámetro medio entre 2-2,5 �lm y morfología poco den nida, pudiendo tratarse de sul rato b,írico (barita).
Figura 5. Partícula irregular compuesta por S. Fe y Cu.
Las panlcl/las irregulares compuestas pr;ncipalmellfe por Si pueden presentarse formando partículas monoelementales (Si) , o partículiJS de S i y Olros elementos. Entre los elementos más comunes con los que suele aparecer asociado el S i estún Al , Ca y Na (Fig. 6), Y Al, K, Fe y Mg. Por su morfología y composición química, este t ipo de partículas se han considerado partículas m i nerales (s i l icatos), probablemente cuarzo (Si) , plagioclasas (S i , Al , Ca. Na) y filosil icatos (S i , Al , K, Fe y Mg).
Las panículas irregulares compuestas por Ca y P, con 20 �l1n de d iámetro medio (Fig. 7), pueden tener su origen en las industrias de ferti l izantes ubicadas en el Polo Químico "Punta del Sebo", basadas en la transformación de rocas fosfatadas. para generar fosfatos cálcicos, sódicos y amónicos, entre otros. Las pan{elllas ¡rregl/lares compuestas por Ca y Mg presentan d iámetros medios de 10 pm. Estas partículas podrían ser c�lrbonatos (probablemente dolomita) por su composición química y morfología, y provenir del sustrato carbonatado existente en el entorno del punto de muestreo.
Dentro de todo el conjunto de partículas irregulares, las partículas de origen al/trópico son aquellas compuestas por S (sulfuros met¡íl icos tipo pir i ta, calcopir i ta , calcosina y
Figura 6. Partícula irregular compuesta por S i . Al . Ca y Nu (posiblcmcnte pl;¡gioclas�l).

174 BERNABÉ, J. M. Y CARRETERO, M . ! .
cove l l ina, entre otros, y sulfatos de origen secundario) y por Ca y P (posiblemente fosfatos cálcicos procedentes de la transformación de rocas ricas en fosfatos) . El resto de las partículas irregulares. compuestas por Si y otros elementos, así como por Ca y Mg, son de origen natural, c las ificándose de forma general como partículas minerales. A las partículas compuestas por S y Ba (que puede tratarse de barita) no se le puede atribuir UIl origen determinado (natural o anlrópico), ya que podría presentarse como impureza de las materias primas de las industrias, o bien proceder del sustrato C0l110 polvo mineral.
Considerando los d iferentes grupos de partículas irregulares encontrados, las panículas irregulares formaclas por S, Fe, el! o Zn, procedentes de I�I industria metalúrgica, son las más abundantes.
Cristal i:aciofles Las cristalizaciones son panículas debidas
a la cristalización de sales que previamente estaban disueltas en un medio acuoso. Estas partículas pueden l legar al filtro como partículas ya formadas O cristalizar directamente so· bre el propio filtro. Las cristalizaciones encontradas pueden clasificarse en seis tipos, según su composición química: a) compuestas por CI, S y Pb; b) compuestas principalmente por
Figura 7. Partícula irregular compuesta por Ca y P.
CI; c) compuesl<ls por P y Mg, d) compuestas por S y Ca; e) compuestas por CI y Na; )' f) compuestas por S, Mg Y K .
Las cristalizaciones compuestas por Cl, S y Pb son de h{tbito prismático y poseen un tamaño de cristal inferior a los 5 )lIll. Las cristalizaciones cOlI/pl/estas principalll/ente por CI aparecen tapizando di versas zonas del filtro (Fig. 8), l legando a abarcar superficies cercanas a los 200 pm2• Estos dos tipos de cristalizaciones pueden tener su origen en las reacciones secundarias de emisiones industriales ricas en S y Pb, con emisiones de CI , generadas por acti v idades industriales basa· das en la producción de cloro y derivados (hipoclorito sódico, ácido clorhídrico, cte.), o en la cristalización d i recta (en forma de cia· ruro amónico) de aerosoles ricos en cloro sobre la superficie del filtro. Las criswli:aciones eOll/pl/estas por p 'y Mg, cristalizan en forma de eflorescencias, dando lugar a agregados cristalinos de gran tamaño (20-30 �l!n de di ¡í· metro medio) (Fig. 9). Las cristalizaciones cOlI/pllestas por S y Ca (po.�·iblelllel1le yeso), cristalizan también en forma de eflorescencias, dando lugar agregados cristalinos de 1 5 �lm de diámetro medio. Estas crista l izaciones en algunas ocasioncs contienen además P. Estos dos úl t imos tipos de crista l izaciones encontradas pued�n tener su origen en las cmisio-
f-igura 8. Cristalizaciones compuestas por Cl.

Caracterización de panículas atmosféricas del área de Huelva 1 75
nes de industrias de fert i l izantes. En el caso de las cristalizaciones de yeso, también es muy probable que se generen por reacción del S02 <Itmosférico (emit ido por todas las industrias del úrea en cantidades importantes) con partículas cúlcicas sobre el propio filtro. Las cristalizaciones eOll/puestas por el y Na aparecen C0l110 crista l izaciones formadas sobre el propio filtro formando agregados cristalinos con morfología c llb ica característica (probablemente ha l i ta) (Fig. 1 0) , l legando al filtro en forma de aerosoles marinos. Por últ imo, las crisrali:aciones cOlI/puesra.\· por S. Mg y K están formadas por crista les que l legan al filtro en forma de partículas, presentando cierto habito prismático.
Dentro del grupo de las cristali zaciones, aquellas compuestas por a) CL S y Pb, b) C I , e ) P y Mg, Y d) S Y C a son cristal izaciones de origen al/lrópico, m ientras que las cristalizac iones compuestas por a) el y Na, y b) S , Mg Y K poseen un origen natural y proceden de la cristal ización de sales que provienen del agua del mar. Las cristal izaciones más abundantes son aquellas formadas por P y Mg, Y S Y Ca, procedentes de la industria de fert i l izantes y de las emis iones de SOr
En reSfll1/el/, los t ipos de partículas encontrados en el área industrial de l-Iuelva, su
Figura 9. Cristal izaciones compuestas por P y Mg.
composición química, origen natural o antrópico, posible procedencia y abundancia se recogen en la tabla l .
Las partículas y compuestos precipitados de origen antrópico no presentan muchas diferencias eSlflciollales, ya que las fuentes emisoras actúan prácticamente del mismo modo durante lodo el año. En el caso de las panículas de origen natural, su presencia se hace l11�ís evidente en la época estival (primavera-verano). Esto es debido al aumento de la actividad biológica de las plantas (polen) y a la mayor concentración de partículas por una menor abundancia de precipitaciones atmosféricas y mayor resuspensión de panículas del terreno.
Conclusiones
Las partículas encontradas en el úrea industrial de I-Iuelva se han clasificado en cuatro grupos: esféricas, i rregulares, cristal izaciones y biológicas. Las partículas esféricas, todas ellas de origen antrópico, muestran texturas espongiformes, lisas y rugosas, presentando diferencias en su composición química, aunque predomina la presencia de S y S i . Las partículas irregulares y cristalizaciones son de origen antrópico y natural. Las primeras se clas ifican
Figura 1 0. Cristalizaciones compuestas por el y Na (posiblcmcnte halita).

176 BERNABÉ. J. M. Y CARRETERO. M. 1.
Tabla 1.- Tipo. composición química, origen, posible procedencia y abundancia de las partículas atmosféricas del área industrial de Huelva.
TIpo do P&IIlcvlu eom_ltlOn Q�rmlc. Or""" PoIII>IfI 1',,, ...... ,,<1. Abundo"."
�""ell6rmic.o
• bpo"flllormol S. Si. Al. Ca. P. Na, K.O. V. Fa -- Vehiculoo do molO!' oSe oomo<.<ollOn + • 1 ... :".eI';"pe!roQ� � • U_ Si. AI {f� -- c. ....... wu" • • n -- _ ... <lo .,...._ lIug_
S. F •• Si -- Induslti& molw�
S. F •• Cu. ZtI AA_ • InOUSI:ia .... IalUrgQ + � . .. �IUI" '" • • NII .... j , • 51. ..... Ca. NI Nf, l ural O • Si. AI.K. F., MI;! "'1II'aI • •
'" _ .. � " . ... Ha ..... CI, S. Po AtllIÓpÓCO
• O --� O , .... --
� s. Ca (1') �-
• CI,NI ��, O S.�K �"
BlOI..ÓGICAS ��,
en función de su composición química en partículas compuestas principalmente por S, Si o Ca. junto a otros elememos en menor proporción. Las cristalizaciones presentan diferentes morfologías y composiciones químicas. predominando la presencia de S, CI o P.
Las partículas de origen antrópico aparecen en mayor cantidad que las de origen natural, debido a la gran influencia las actividades industriales en el entorno. Las más abundantes son las partículas esféricas espongiformes procedentes de gases de combustión de combustibles fósiles (tráfico, central eléctrica, etc.); las partfculas irregulares formadas por S, Fe. Cu o Zn, procedentes de la industria metalúrgica; y las cristalizaciones compuestas por P y Mg. Y S y Ca, procedentes de la industria de fertilizante$ y de las emisiones de SOr
Las partfculas de origen natural son principalmente de procedencia mineral (polvo del terreno). biológico (polen) y marino (sales). No se
SiIi<aIoo (cuatlO¡ Silicato. (fKe;glod ..... ) SiIC_ (OI�IQSI
InOUSIN lit I<I�I CatborIaIOS 1Il0l0 ..... ) ""'USItta do � do dOro 1 "" .... ".,. lnduStrla do p.oduocOlto do doto Y "'tiII_ 1�""I ... IIiz",,,," + __ c. r .... 'UtlIH + Etniaionel SO, "" ... .... ...... ... � -PIanl •• (pelan)
han detectado importantes variaciones en el tipo y composición de las partículas en las diferenles épocas del año, exceptuando las partículas biológicas (polen) que abundan en primavera.
Bibliografía
Álvarez Mota. C. (2001): Caracterización y modelización estadística de contaminantes atmosféricos en un área industrial. Tesis Doctoral, Universitat Jaume 1, Caslellón.
Amoroso, G.G. y Fassina, V. ( 1983): Stone decay and conservation. Atmospheric pollution, cleaning, consolidation and proleclion. Materials Science Monographs. 1 1 , Elsevier ed., 453 pp.
Ausset, P., Lefevre, R., Philippon. J .• Venel, C. (1992): Large scale distribution of flyash particles inside weathering crusts on calciurn carbonate substrates. Sorne

Caracterización de partículas atmosféricas del área de Huelva 177
examples on French monuments. in: "2001 Inl. Symp. on the Conservation of Monuments in the Mediterranean Bas;n". D. Decrouez. J. Chamay. F. Zezza eds .. Geneve (Switzerland). 121-139.
Bernabt, J.M., Carretero, M.I., García Orellana, 1. (2003): PIXE analys;s from aerosols collecled around Columbus Monument at Huelva (Spa;n). in AbstraclS Book of «Air Pollution and Cultural Heritage", CSIC ed., Sevilla. 65-66.
Bignon. J., (Ed.) (1 990): Heallh related effects ofphyllosílicates. NATO ASI Series. Vol. G 2 1 . Springer-Verlag ed., Berlin, Heidelberg, 447 pp.
Bolivar, J.P. ( 1995): AplicOlciones de las espectrometrías gamma y alfa al estudio del impacto radiactivo producido por industrias no nucleares. Tesis Doctoral, Universidad de Sevilla, 291 pp.
Bogo, H., Olero, M., Caslro, P., Ozafrán, M.J . • Kreiner, A., Calvo, EJ., Martín Negri, R. (2003): Sludy of almospheric parliculale maller in Buenos Aires cily. Armosplleric Ellvirolllllellt 37. 1 135-1 147.
Brewer, R. y Belzer, W. (2001): Assessmenl of metal concentrations in atmospheric particles from Burnab)' lake, Brilish Columbia, Canada. Atmosplleric Ellv;rOllmellt 35, 5223-5233.
Chabas, A. )' Lef�vre, R.A. (2000): Chemislry and microscopy of atmospheric parliculales at Delos (C)'clades·Greece). Atmosplleric Ellvirolllllellt 34, 225-238.
Delalieux, F., Cardell, C., Todorov, V., Dekov. V., Van Grieken, R. (2001): Environmental conditions controlling Ihe chemical weathering of the Madara Horseman monument. NE Bulgaria. Jo/ullal 01 Cultllral Heritage 2, 43-54.
Esberl, R.M., Draz Pache, F . . Alonso, F.L. Ordaz, J., Grossi, C.M. ( 1996): Solid particles of atmospheric pollution found on the Hontoria limeslone of Burgos CathedrOlI (SpOlin). in: "Proc. 8th Inl. Congress on Deterioration ,nd
Conservation of Slone". J. Riederer, ed., Berlín. Alemania. 393-399.
Fassina, V. (1992): The Slone deca)' of the porlal of the basílica of SS. Giovani e Paolo in Venice. in: "Proc. 7th Inl. Congress on Deterioration and ConservOltion of Slone". J. Delgado Rodrigues, F. Henriques, F.T. Jeremias eds., Lisboa (Portugal), 1 19-128.
Guthrie, G.D. )' Mossman, B.T . • (Eds.) ( 1993): Health effects of mineral dusls. Reviews in Mineralog)', Vol. 28. Mineralogical Societ)' of America ed., 584 pp.
Queral , X., Alastue)', A., de la Rosa, J., Sánchez de la Campa, A. Plana, F., Ruiz, C.R. (2002): Source apportionment anal)'sis of atmospheric particulates in an industrialised urban site in suthwestem Spain. Atmosferic EI/I'irol/l"em 36, 3 1 13-3 J 25.
Sabbioni. C. (2002): Multipollutants: evidence in stone and mortar deca)'. in: "Protection and Conservation of the Cultural Heritage of Ihe Mediterranean Cities", E. Galán )' F. Zezza eds., Balkema Publishers. Lisse (The Netherlands), 19-22.
Torfs, K. )' Van Grieken, R. (1997): Chemical relations belween almospheric aerosols, deposition and stone deca)' la)'ers on historie buildings al the Mediterranean coas!. A/llIosplleric E""irOIll1l(!IIr 31. 2179·2192.
Umbría, A .. Gervilla, J., Galán, M. )' Valdts, R. (1999): Caracterización de partículas. Consejería de Medio Ambiente, Junla de Andalucía ed., Sevilla, 163 pp.
Van Grieken, R., Cardell, C., Delalieux, F., Eyckmans, K. ( 1998): Anal)'lical, melhods to stud)' atmospheric pollution and weathering of materials. in: "Sciences and lechnologies of the materials and of Ihe environment for the protection of stained glass and slone monumenls", R.A. Lef�vre ed., Universitt Paris XII, Crtteil, 163-170.
Recibido: Noviembre 2003 Aceptado: Diciembre 2003

Boletín de la Sociedad Espa¡iola de Mineralogía, 26 (2003), 179-/86
NEW MINERALS APPROVED IN 2002 NOM ENCLATURE MODIFICA TIONS APPROVED 1998-2002
BYTHE COMMISSION ON NEW MINERALS AND MINERAL NAMES
INTERNATIONAl. MINERALOGICAL ASSOCIATION
Joel D. ORICE' (Chairman- Emeritus, CNMMN)
Giovanni FERRARIS" (Vice-Chainnan, CNMMN)
, Canadian Museum ofNature, P. Q. Box 3443, Slalion 'O', OUawa. Cnlario, Canada KIP 6P4 [email protected]
" Dipanimento di Scienze Mineralogiehe c Pctrologichc, Univcrsili di Torino, Via Valpcrga Caluso 35, 1-10125 Torino. Ital)' � [email protected]
NOTE: ncw mineral proposals should be senl to Ihc nc\\' Chairman: Ernst A, J. Burke Facllfly ofúwth (//Id Ufo Sciellces I fij/! U"iversi!/!;I, Ik lloeMlIlIn 1085 f081 111' ,I/IIsten/am. nI/! 1\·ethl!"'tlll(I.�
e-mail addreu: [email protected]
The ¡nfonnation gíven hcrc is provídcd by the Commission en Ncw Mincrals and Mineral Namcs, I.M.A. for comparative purposa and as a scrvice 10 mincralogiSlS working on new species.
Ench mineral is describcd in the following ronnal: IMA No. Chemica! Formula (any relationship to olher minerals; struc::ture analysis) Crys.lal syslem, space group
unít eetl pamnetcrs Colour; lustre; diophaneit)' Oplical propcrties Strongest lines in !hc X-ra)' powdcr difTractíon pattem
Thc names of these approved spccies are considcrcd confidential ¡nformalion until the authors havc publishcd Ihcir dcscriptiolls or relcascd informal ion themsclvcs.
NO OnlER lNFORMAT10N WILI. UE REI.EASED BY TUE COMMISSION
2002 PROPOSALS
lMA No. 2002-00 I IMA No. 2002-002
179
(Ce, La,Nd.Ba)( Pel' .AIM (As,Al)O.h< ml)6 Fe· (O.KJ.(Mg. Fe1'j¡Fcl'lISiuOJOI Milarilc dominanl analogue ofarscnoOon:ncitc-(Cc)
Tligooal: R3 ni a 1.260. e 16.11 A
Light-green 10 brownish; resinous; Iransparenl Uniaxial(-), mean refractive index D 1.91 !I.906(25), 3.636(40), 3.0!l2(100), 2,192(30), 2.239(35), 1.811(35)
group: structun: dctcrmincd Hexagonal: P6Imcc
a 10.OSO, e 14.338 A Dccp bluc 10 yeJlowish-green; vilrtous; translucent Uniaxial (-), CIl 1.589, t 1.586 8.70(97), 7.11(100), 5.535(96), 5.026(61), 4.352(53), 3.207(85)

180 GRICE. J. D. Y FERRARIS. G.
11\1/\ No. 2001-003 N:!.SrKZn(Ti.NbMSi�OllMO.ml)�·nllO
Labu1llsovilc group: 5truclUrc tktcnnincd Monoclinic: Cm
0 14.495. b 13.945. e 1.838 A. II 1 17.150 WhilC. p.1lc-brown: \'itrcous; InUlslUCCnl lO lrunsparenl Bia.xial (+). a 1.680. j1 1.6H7. Y 1.787. 2V(mC¡lS.) 25·, 2V(Cillc.) 31· 6.96( 100), 3.21(80). 3. 1 1(90), 2.6O(lS), 2.50(40), 1.74(30). 1.10(40)
IMA No. 1002-004 CoSO�+hO Kicscritc group Monoclinic: C21c
a 6.980. b 7.588, e 7.639 A. 1l 1 18.6S· Pink; powdcl}'; transportnl Binxinl (+), n - 1.65 (CIlJe.) 4.83(33), 3.405(100), 3.339(34), 3.291 (32), 3.062(56), 2.561(30), 2.513(49)
IMA No. 2002--005 (K.On.Na)iTi,Nb)I(Si�On){OH.Oh']B10
Labunlsoyite group: struclurc dctermincd Monoclinic: CIII
o 14.327. b 13.802. e 7.783 A. 11 1 16.95· l.ight brown. while. amI colourlc$l: \'ilrcOIIS: lr.II1Spllrenl lliu.-.:illl (+). a 1.689. Jl 1.700. '( 1.775, 2V(mC¡L�.)35°. 2V(cnlc.) 43° 6.87( 100), 4.85(50), 3.95(50), 3.20(60), 3.05(80), ).00(60). 2.SO(90)
IMA No. 2001-006 (lla.Na,Kh",Cn,Nbh<Si.0Il)(OH,Oh·4H10
Labllnlsovilc group: slruclure dClcrmiocd Monoclinic: C2Jm
a 14.551, b 14.001, e 15.702 A, fJ 1 17.58° [)rown; vilrcous; Imnsparent Ilia:�ial (+), a 1.667, p 1.674, Y 1.770, 2V(mcas.) 30", 2V(calc.) 31° 7. 1 1(100), 4.08(80), 3.95(100), 3.24(90), 3.1 1(80), 2.403(80), 1.914(90)
IMA No. 1001·007 1IIat\J¡:c(Ti .Nb ).(Si�Onh(O.OII ),'61'120
Lnbllntsovile gmllp: SlnlClun: detcnnilk!d Monoclinic: Cm
a 14.450, b 13.910. d.836 A. P 1 17.42° 1'lIlc·bro\\'n: vitrcous: Iransluccnt 10 Imnsplll\:nl Uinxial (+), a 1.677, p 1.684, Y 1.790, 2V(rneas.) 25°, 2V(calc.) 30° 6.93( 1 00), 4.93(80), 3.21 ( 100), 3 . 1 1(90), 2.62(60), 2.49(50), 1.687(40)
IMA No. 1001-008 NalH(PO�)· 81-110 Ncw Slructure Iypc Onhorhombic: loca
0 1 1.488. b 1 1.647. e 16,435 A C!lloudess: \'ilrCtlIlS lo resinolls: Iransparcnl l3iaxial (-). CJ. 1 .'1-13. fJ 1.457. Y IA5K. 2V(mCIlS.) 29". 2V(cnlc.) 30" 5.78(40). 4.90(43). 4.73(62). 3.75(81). 2.K76(77). 2.782(100), 2.744(74)
IMA No. 1001-010 NnNaJ(AI1Mg,)(Si,AI)On(F.OB)1 Amphibolc
group; slruclure detcrmined Monoclinic: C2Jm
0 9.666, b 17.799, c 5.31 1 A,p 104.100 D1uish-grey; luSlrc 001 givcn; transluccnt I3inxilll (+), Q 1.633. fJ 1 .624, Y 1.626, 2V mcdium; calculaled from chemical composilion 8.31(64),4.45(26), 3.38(42), 3.079(58), 2.691( 100), 2.571(32), 2.532(47)
IMA No. 2001-01 1 GaO(OJ-1) lsostructural wilh gocthilc Onhorhombic: I'bl/III
(1 4.512. b 9.772. e 2.967 A Palc gn."\:lIish )'ellol\' 111 beige: Jk!"rly; lrilllslucenl Bia-.:i,,1. ¡rlcnle.) 1 .96. 4.09( 1 00). 2.632(33). 2.530(22), 2A04{ 100), 1.690(26). U3K(21)
IMA No. 1001·012
Na!(Na,Ca)�Ca..(Mn,CahZr2 TitCS ilO,MO,F)� F 4 Roscnbuschile group; slructure- detcnnincd
Triclinic: P I o 10.032, b 1 1.333, e 7.202 A, a 90.19. �
100.33, Y 1 1 1.55° Colourtess lo pate shadc of brown; vilrcous; transparenl Biaxial (+), a 1.684, p 1.695, y 1.718, 2V(mcas.)
·
73°, 2V{calc.) 70" 3.951 (30), 3.028(60), 2.908(100), 2.600(80), 1.868(60), 1.670(50)
IMA Nu. 1001-013 Uil,N"C�'{\'O�,,(F.CI) Ha·dominnnt :maloguc of
belovite-(Cc); slnlclurc dClcnnined
Trigonal: l' 3 a 9.909. e 7.402 A Lighl rose; vitr�'()us: Imnsluccnl Uninxial H ro 1 .694. e 1.669 4.078(40), 3.693(40), 2.969(100), 2.867(60), 1.965(80), 10863(60)

New minerals approved in 2002 181
IMA No. 1001-014 Pbl( UO�>.O.<OH)2J(�hO)�; x - 3 New
Slructurc type Monodinic: C21e
a 28.355, b 1 1.990, e 13.998A. P 104.248° Brighl orangc; vitreous; Imnsparcnl Biaxial.lI_ 1.807, "mu 1.891 6.92(60). 6.02(30), 3.46(80), 3.10( 100). 2.74(30). 2.01(30). 1.918(6O)
IMA No. 2002·015 IJalk¡ShOJ
Monoclinic: I'm
DilllOll'hllUS wilh bal)·lit..:: struclur..: d":lcnllincd
ti 1 1.637. b 4.918. c 4.668 A. P 89.800 Colourless: vilrcous: InUlspan.:nl Iliaxial (+). a 1.698. 11 1.700. Y 1.705, 2V(mcas.) 70", 2V(calc.) 650 3.39(84). 3.25(45), 3.04(40), 2.926(55), 2.458(100), 2.335(48). 2.076(38)
IMA No. 1001-016 CaFcl·P'c'·(Mn,Fc1')(ShOl)O(OI·I) Mn-
dominnnl analogue orilvailc Monoclinic: P2¡/a
(1 13.0246, b 8.851 1, e 5.8485 A. P 90.11" Block; vilrcous; opaque In reneclcd ¡¡ghl (in air): grey to bluish grey; intemal rcncctions: red: anisolropy: slrong in blucgTl:yish. R .... Ilnd R .... ,: 8.3-10% (460 nm). 7.5-9.8% (540 nm). 7.9.7%{580 nm), 6.1·9.5% (640 11m) 2.875(85). 2.848(90). 2.718(100). 2.61:7(70). 2.180(48). 2. 1 1 1 (47). 1 .475(48)
IMA No. 2002-0 17 MnV¡O.·41·hO New SlructUfC Iype Munoclinic: ale
a 13.171. b 10.128, e 6.983 A. P I I UJO Carmine red: adamantine; Iransparent Uiaxial, "_ 1.797, "m .. 1.856 7.82(100), 5.69(20). 5.06(20), 4.51(30), 3.91(JO), 3.029(10)
IMA No. 1001-018 {Mg.Fe)(Ta,Nbh06
Onhorhombic: Pbc"
Columbilc-Ialllal ile group
(1 14.355, b 5.735. e 5.058 A Black: scmi-mctallic to melallic: opaque Lighl-gn:)� inlcmal renections (in oir): brownish-rcd: nnisotropism: wCllk: bircncctancc: ver)' weak. R ..... aud It ... ,: \3.97-12.82% (460 n01). IJ.33-\3.20% (540 nm). 14.25·1].94% (S8n nrn). 15.61·15.31% (640 11m)
3.61(60), 2.96( 1 00), 1.174(60), 1.728(70), 1 .'62(90), 1 . 196(60), 1 . 105(60)
IMA No. 1001-019 l3a:¡{La, Th,Ce)(COJ)JF La-dominant analogue
orkukharcnkoitc-(Cc); structurc delcnnined Monotlinic: P21/m
/1 13.396, b 5. I I I . c 6.672 A, p 106.6Jo Pale l«k-green. colourlcss. white; vilreous: lrunsparent 10 Irnnsl\lccnt Uin.xial H u 1.581. P 1.715. Y 1.715. 2V(mcns.) S°, lV(cnlc.)OO 4.01( 1001. J.27( 100). 2.5<1(50). 2.3K(20), 2.14(80). 1.99K(80). 1.636(20)
IMA No. 2.002-010 (Cn.K,Nnh .• ("n,NbMSi�O¡!XOI-l,Oh·4H!O
Lnbunlsovi!c group: struclurc dclcrmincd Monotlinic: C21nr
(1 14.484, b 14.191,c7.907Á, P 1 1 7.26° White, pale brownish; vitrcous; Iransparcnt l3iaxial (+), a 1.666, p 1.676, Y 1.780, 2V(mcns.) 300, 2V(calc.) 36° 1.02(60), 6.38('0), 3.SJ(45), 3.16(100), 2.62(45), 2.51(85), 1 .718(50)
IMA No. 2002-021 (Nn,K,Ca)�,SiW\I}601.w[(SO�hCIJI"JH10
Cancrinitc-sodalilc group; structurc discusscd
I'kxagolllll or trigonal: P 6 2e or 1'3 le (1 12.880. b J 1.761 A
Colourless: \"ilreul1s: transparcnl Uniaxinl{ +). t: 1 .497. 1') 1 .495 '1.20(42). 3.72S( I (0). 3.513(8U). 3.296(35). J.OK9{<lO). 2.555(35). 2.150(40)
[MA No.
2002-022 Hgl'Hg�'OI
Monoclinic: ClIe
Rclatcd 10 Icrlinguailc; new structurc t)'pe
a 17.580, b 6.979, e 6.693 A. P 101.71° D:u-k grey-block: mClnllic; opaquc Calculalcd index or rcrrnction: 2.35-2.38 8.55(70), 3.275(100), 2.993(80), 2.873(80), 2.404(50), 1.878(50)
IMA No. 2001-013 Ce1Si101 lsoslructural with Ln1Si10J Tctrngonnl: P41
. (I 6.781. c 24.689 A Whilc to colourlcss: resinous: Imnsp;¡rcnt Unia,xial (+). (1) 1.840. e UI46 3.27(31). 3 . 14(27). ).12(2-1), 3.0K( IOO). ).01 1( 18). 2.846(22). 2.034( 19)

182 GRICE, 1, D. Y FERRARIS, G.
1!IoIA No. 2002·024 {CU� lAg1 1h::.GCS6
-Cubic: F4 3m
a 10.201 A
J\r�c1lli:m \'ariel)' 01" Q-CUIGCS�
lron-black; vilreous lo mcla11ic; opaque In rcncclcd lighl (¡¡ir): pale rose-brownish; inlemal rcnctlions: no; R ... and R .... : 29.4% (460 nml, 2J.6% (560 oml, 26.0% (580 nm), 25.3% (640 nm) 5.90(30),3.07(60), 2.94l( 100), 1.962(50), 1.805(70)
IMA No. 2002·025 CcJCaMg¡AllSi¡OI9l:0Hhf Rclatcd lo
cpidolc group: struclun: dClcrnlincd MonDe!inic: 1>'2,/m
(J 8.939, b 5.706. e 15.1155 A. fl 94.S8 Dark brown: vilrcous Bmsial (+). a I .1RI . 1I 1 .192(c¡,Ic.). " 1.810. 2V{mcas.1 7S0, 2V(calc.) 7!1° 4.64( 10), 3.50(20). 2.979( 100). 2.IIH( 10). 2.682( I J), 2.622( 1 9}. 2. 1 85( I S)
1M" No. 2002-026 (Na,CaMCa,Na)JSiI60ll(f,OHh'31'hO Rcycritc
_ group: slructure dctcrmined Triclinic: p I
Q 9.613, b 12.I IS, e 9.S89 A, a 92.9S, fJ 1 19.81, Y 96.6� Colourlc5s; pearly Dia. .. illl (-), a I.S22. p 1 .528, Y I.S29. 2V{mcas.) 48°, 2V{c:alc.) 44° 1 1 .99(100), S.97{8S), 3.97(40), 2.967{SO), 2.888( 100), 1.820(SO)
IM/\ No. 2002-027 Ila01Si!O.
Onhorhombic:: 1'/11110
Ua-dominanl analogue of rJnnburitc: Slruclur� d�lamined
a 8. 1 4 1 . 8.176.c 9.0311 A "'hile: vitreQus: lronsparcnl tliaxial (-). a 1.649. p 1 .6S6. y 1.6S6. 2V(mc:a.'I.) So. 2V(calc.) O" 6.07(60), 4.86(30), 3.62(100), 3.39(60). 2.83(SO), 2.481(40). 2.021(70)
IMA No. 2002-028 Cau(f'cl+,Mg,Fc'+MSi,AI)40.o(O¡·lh·4Hl0
Smc:c:tilc group MOlloclinic: probably C-cel!
a S.363, b 9.306, e 1 4.64 A, fJ 94.98°
Dark-grccn. bmwnish-grc..:n: \'iln:ou�. lfllllsluccnl. 13ia. .. ial (-).a 1 .<I<lH (calc.), 1I 1.6<1 1 . 'f 1.6<12: 2V(mcas.) So, 2V(calc.) 7.So. 7.37(90), <1.72(90), 3.80(80), 3.03(1 00), 2.S8S(90), 2.429(90), I.S49(90)
IMA No. 2002-029 N�MnTi�Si.OD·4H10
Orthorhombie: PCCII
Mn-dominant analoguc of kukisY\Jmitc
Q 29.0S, b 8.612, e S.220 Á Colourlcss; vitrcous; lransparcnt l3iaxiaJ (. l,a (e:llc.) \.6S7, p 1.744, Y 1.792,2V(mctlS.) 700,2V(cnlc.)70" 14.4 7( 100), 6.43(20). 4.83( 1 O), 3.02S(40). 2.881 (20)
IM/\ No, 2002-030 Mg�(UOJW
Orthorhornbic: 1'11//2.
ISOSlmclural wjlh M¡h(BO¡)f: Slruelur..: d":lcrmincd
" 20.490, h <I.S71. ,. I I.IIIJO A Colourlcss: \'ilrcouS: lransparcnl Bia. .. ial (+), a 1.609. 1"1 1.620, y 1.642. 2V(mcas.) 6So. 2V(ealc.) 71-2.743(77), 2.474(49), 2A 14(46), 2.241 ( 1 00), 2,234(49), 1.708(92), 1 .705(44)
IM/\ No. 2001-031 N:llK(V,R��} ¡Si60'SJ K and REE analoguc of
Na, Y (Si60nJ; struelurc dctcrmincd Orthorhombie: Ibml/l
Q 10.623, b 14.970, e 8.5S2 A "'hile; vitrcous; transparenl Ili:utial (+), C1 USS. P I.5S8, y 1 .566, 2V (mcas.) 64°. 2V (c:llc.) 63-S.32(3S), 4.98(100), 3.4S(SO), 3.26(8S), 3.0S(7S), 2.7S3(42). 2.490(4S)
1M" No. 2002-0l3 Na •. l{TLFc¡·}.¡(Si,AI)On(OI·IMI-l20j Rd¡lIcd 10
\'inllgrild()I'itc: slrUClUrc dClcrmincd Triclinic:l't
(, l.2S33. b 8.741 1 . e 12.9480 A. Q 70.4 7. P 78.47, y 89.93· Whilej Yilrcous; transluccnl lo Iransparenl Di:utial (-), a 1 .707, p 1.741. Y 1.7SS, 2V(mcas.) 64°, 2V(calc.) 64° 1 1 .9{S8), S.98(3S), S.88(6S), 4.3S(38), 3.182(100), 3.08S(29), 2.73S(2\)
IM/\ No. 2002-034 CdS04'4H10 Rozenitc group Monoclinie: P2,/n
Q 6.S8S9, b 14.329, e 8.S7I2 A, P 91.51°

New minerals approved in 2002 183
Colour1css lO I¡ght bloc: \'i¡rcous, lransparcnt Unio..'I:ial H ti 1.4]0. P 1.454. Y 1.470. 2V(ml!iIS.) -700. 2V(ca1c.} 77.3° 5.98(85), 4,84(10). ].1 46(8,5). 2.967(85). 2.70S(75). 2.6$4(100)
IMA No. 1001-0]5 (O.Cu!'.VI'), Al. (1)O�). I:, (1'110hl Ncll'
SlrUClurc I)'pc Onhorhombic: Pmlllll
Q 12.I23.b 18.999, c 4.%1 A, Pale grttn 10 turquoise; vitrcous; transluccnt Siaxial H. Q 1.S40. P 1.548. Y L553. 2V(mclIs.) 76°. 2V(calc.l 76° 9.54(80), 6.08(100), '.62(90), 3.430(40), 2.983(60), 2.661(40)
I¡\IA No. 2002-0]6 (l3l1.Co.)lAh(Si,I\I).¡OloCCO)(OH),¡-nH10
Suntc series Monodinic: CU/ll. C2 or CII/
a .1.176, b 8.989. e 16.166 A, 1l 96.44° White wilh light-gl'l:l!nish linl; �nr1)� translucent Uinxial (-). o 1 .580. P 1.625, Y 1.62S, 2V(mcas.1 0-1 0". 2V(cnlc.) O" 4.49(90). 3.68(60). 2.S8S( 100). 2.230<90). 2J)69(80}. 1.692(6Ol
lMA No. 2002-0]7 (Ca.NII)(BlI.K)(fc1' ,Mn)�Til(Si4014)Ol(f,OH,Oh
Bafcrtisite series; slrUClure detennined Monodinic: C2
Q 10.723, b 13.826, e 20.791 A,1l 95.00" l3rownish red; vitrcous; transparent 10 lranslueelll Bia.xilll (-). a 1.790(calc.), p 1.8.18, Y 1.888, 2V(meas.) 65" 10.39(20), 3.4S4( 100), 3.186( 1 S). 2.862( 1 5), 2 . .192(70), 2.074(40), l . 728( I S)
IMA No. 2001-0]8 M&l(A11.l.Mg�Sn.)(l30l)Ol
Monoclinie: J'2Jm
Hulsile group; structurc detennincd
II .1.3344. b 3.0300, e 10 . .106) A. O 94.'16° Ilrown to bluc-gn.-cn in trnnsmincd light; lusll'l: not obso.:rved: trnnspal'l:nt. I3in.',inl (+), o' 1.78. y' 1 .805. 2V(ml!ilS.) 33°. 2V(enlc.} 39D. 10.47(29), 5.24(49). 4.90(32), 2.61 8(.10).
2.532( 100). 2.3 18(30), 2.001(54), I.SI 5(28)
IMA No. 1001-0]9 I lgI' 4AI(P04), 14(OH)tn Monoclinic: C21c
New SlruCluTC t)'pc
(1 17.022, b 9.014, e 7.015 A, fl 101.20" Colorlcss lO white; vitreous: transparcnt 10 transluecnt Uiaxial (+). I"calc.) 1.94 8.3](100). 4.74(50). 2.979(80). 2.952(50). 2.784(80). 2.660(75)
IMA No. 2002-041 Kl'bL ,ZnCu.O�ScO) lCl IO Ncw structure I)'pe Orthorhombic: P"'"II
o: 9.132, b 19.41 S, e 13.213 A Olive grtt:n; vitrcous, lransparcnl Biaxial (-l, no rcfrnctivc indices given , 8.26(70), 7.63(60), 4.1 1 (90), 3.660(100), 2.996(40), 2.887(50), 2.642(40)
IMA No. 2002-04] Naz(Ba,K),CezFcl'ri)Sill0l6(0,"I)I(OH,H10)o
New structure typc
Trigonal: R 3 a 10.7 1 3 . c 60.67 A
Yellowish orange: vitrcous: transparcnl Unia:.;ial (+), 01 1 .705, e 1 .708 10.12(27). 3.236( 100), 3.094(21). 2.6S/I(38). 2.642(44). 2.2J.1( 19). 2.026(6 1 )
IMA No. 2{]02-Q47 lnife)O. Relaled to spiroffitc Monoclinic: C21c
o 12.676, b 5.198, e 1 1.781 A. 11 99.6( 1 t Gre)'; vilreous; trunslucent. In rcnecled light (air): gre)'; inlernal rcncetions nOI obscrved. anisolrop)' wcak. R .... and R_.: 6.7 -7.3% (460 nm). 7.4·7.80.4 (540 nm) 4.76(w), ].240(11'), 2.928(m), 2.820(w), 2.IS5(w), 1.985(1'0'), 1.599(11') .
IMA No. 2002-048 K(O,Nah(Mn,fc,Mgh(Bc,AIMSilz0lOl
Milarite group; slrueturc detcnnincd Hexagonal: P6Imcc
o 9.997. e 14.090 11. Yellow lO ornngc: vitrcous; transparent Uniaxial (-). 6) 1.560. e 1.5S9 7.0.1(40). 5.00("0). ,1.08(80). 3 . 1 87(901. 2.882{ 100). 2.732(SO). 1 .826(40)
IMA No. 2002-049 (Mnl' ,Ca)(Cc,REl::)AIMnl'Mnl'SizOJ SiO� 0(01-1)
Epidote group; struclurc delennined Monoclinic: nI/m
08.901, b5.738, e 10.068 A, P 1 1 3.42S· Dark brolVn; vitreous 10 adamanline; transpnrent

184 GRICE, J. D. Y FERRARIS. G.
Biaxial (+), a > 1.74, 2V(meas.) 8 \ o 3.S1(17), 2.896(100), 2.71 3(34), 2.707(43), 2.622('8), 2.'91 (32), 2. 18'(3 1 )
IMA No. 2002·050 Ca.A1Si(S04)Fu' I 21-hO
Cubic: Fd3 a 16.722 A
Rclnlcd lO chukhrovite-(Cc:l
Whill! 10 y.:llowish: vilrcous: trnnspnrcnt lsolropic; lI(calc.) 1.430 9.63(100), S.91(46). S.04(27), 4.17(19). 3.219(32). 2.235(28), 2. 178()3)
IMA No. 1002·051 (Na,K)Co1(MSlAh)SisAI10U(OI'I)1 Amphibolc
group; slruclurc dClcrmincd Monoclinic: e Um
a 9.905. bI S.OO, c S.J22 A. P 10S.47" Brownish bJack; vitrcous; transluccnt Biuial (+), a 1 .674, � (cale.) 1.683. 1 1.694, 2V(mcas.) 8S· 8.47(70), 3.38(60), 3.1 3(70), 2.70(100), 2.19(70), 2.!i7(100), 2.1 6(60), 1.447(60)
IMA No. 2002·051
K\(AI.Znh(As.SihOaI Monoc:linic: elJc
Fddspnr group; slruclurc dClcrmincd
a 1J.416.h 13.370, c 8.772 A, p 100.067" Colorlcss: vitrcous: Imn$p.1renl Biaxial (-)_ o. 1.532, P 1.535, '1 1 .537, 2V(meas.) 60": 2V(caJc.) 78" 4.33(70), 3.90(70). 1.164( 100). 3.300(50). 3.066(40), 2.981(60), 2.646(40)
IMA No. 2002-053
T1.A&lCu.As.¡SJI
Triclinic: pI
Related 10 imhofitc; slructurc delennined
Q 12.138, b 12.196, e 15.944 A. a 78.537, ji 84.715, Y 60.470" l3Iack; melallic; lranslucenl In rtOeeted ¡¡gIll (air): while; inlemal rcO«lion5 rreqlJCnl, anisolropy weak. R: 30.7% (460 nm), 29.4% (540 nml. 28.2% (580 nml, 26.8% (640 nm) 15.63(100), 3.53 1(80), 3.263(50), 3.143(90), 2.978(60), 2.911 (70), 2.520(60)
IMA No. 2002-054
\.a(C03)(OI-I) Ancylile gruup Unhorhomhic: I'IIICII
n 4.986. b 8.:513. C 7.227 A
Pale pinkish purple lO whitc; vitrcous; diaphaneity nOI given No optical data 4.31{ 100), 3.69(72), 2.93(57), 2.64(30), 2.49(29), 2.33(�0), 2.06(48), 1.994(3')
IMA No. 2002·055
NtlllSrIC",r-cIZr,NbSiJSOJI(O,OH,¡'¡�O)ICI1 Eudialyte group: 51fUClurt delennincd
Trigonal: R3m tI 14.286. (" 29.99 A
Clove brown to )'ellowi511 brown: \'jlreOIlS: transpllrenl Uniaxial (.); fIl 1 .649, & 1.638 1 1 .49(:50), 9.51(90), 3.43(90), 3.19(80), 2.98(1 00), 2.86(100)
IMA No. 2002·056
(Na,\.I)ll(Na,CehCa.Mnlü¡Nb(SiuOu)(OHMCOI) 'H10 Eudialyle group; struelore delermined Trigonal: R3m
a 1-4.239, e 30.039 A Yellow; vilreous; transparenl Uniaxial (-1; ro 1.645. t 1.635 6.39(25), 4.30(24), 3.204(38), 3.1 :55(35), 3.01 9(34), 2.970(83), 2.849( 100), 2. 134(23)
IMA No. 2002-057
(Na,' :)ll(Ce,Nn)ICi16MnIZrINb(SiJs071)(0¡'Ij¡(CO¡) -1-110 Eudialytc group: slnu:lurc determined Trigonal: R3111
a 1 4.248. c 30.076 A Crcllm: \'itreous: transparcnl Uniaxilll(-): (al 1 .648. e 1 .637 4.32(�1), 3.975(37). 3.:536(33). 3.220(1 00). 3. 1 66(56), 2.979(95), 2.857(88)
IMA No. 2002-058 Cu�AgPbll3itSl'
Moneel;nie: e2/m
Related lo makoviekyite; struClurt detennincd
a 13.396, b 4.013, e 29.93 A, JI 100.07" Grey; melallie; opaque In renected ligllt (air): grcyish white; ¡nlemal reneelions not observed, ani50tropy moderate. R ... and R.o.: 42.3 - 48.5% (460 nm), 4 1 . 1 - 47.1% (�40 nm), 40.0 - 46.0"/0 (580 nm). 39.8 - 45.2% (640 nm) 3.645(56), 3.486(40), 3.478(100), 3.345(32). 2.964(33), 2.885(29), 2.842(95), 2.282(3 \ )
lMA No. 2002·059
(Ni,Co.Cu)�AslO,)U Moneelinic: e2
Ncl\' struclurc Iypc

New m i nerals approved in 2002 185
a 33.256. b 8.<182 A. e 14.191 A. P 104. 145° [)"rk violel-red lo dark brownish red: vilrcous; lranslucenl In reOeeled lighl (nir): dark grey; internal rcneelions amnge, anisolropy nol obvious. R: 9.63% (460 nm), 9.33% (540 nm), 9.27% (580 nm), 9.33% (640 nm) 4.23(30), 3 . 1 18( 1 00), 3.005(60), 2.567(50), 1.637(50), 1.507(30)
IMA No. 1001-060 Cu2Pd¡Se�
Monoclinie: P2¡/c
Chrisstanleyite series; strueture detennined
a 5.672. b 9.910. e 6.264 A. P 1 15.40(2)° Silvery grey: mclallie: opaque In renceled lighl (air): bulr w grC)'-l!reen: inlernal relleelions 1101 obscr"ed. anisolrop)' moderole. It",;� and R .... ,: 40.'1 - 48.4% t460 nm). 44.2 - 50.7% (540 nm), 44.7 - 50.6% (580 nm). 45.1 - 50.6% (640 nm) 2.776(22), 2.759(23). 2.676(1 00), 2.630(64). 2.508(3 1), 2.269(27)
IMA NO. lOOl-06. Na(UIO){UO!)¡(SeOJ)lOl·4¡'hO Relaled 10
haynesile; slruelure detennined Monoclinie: PI I m
a 6.9806, b 17.249, e 7.6460 A. � 90.039° Yellow; vilreOlJs; transparcnl Bia.xial (-). Cl 1.597, � 1.770, Y 1 .775, 2V(meas.) 20"; 2V(ealc.) 18° 8.63(43), 7.67( 100), 7.02(33), 3.85(40), 3. 107(77), 2.874(53). 1 .41 1(30)
IMA No. 2002-062
CUlHgPbllSbllS�S s New slruelure Iypc
Monoclinic: C2 or C2/m
a 43.1 13. b 4.059. e 37.1174 A. P 1 1 7.35° Hlack: melallic, opaque In rcneeled ligh! (air): while: inlernnl reOcelions red, nnisotropy distinel. R: 39.0% (460 nm). 36.4% (540 nm), 35.2% (580 nm), 33.4% (640 nm) 3.114(31 l, 3.402( I 00), 3.369(74), 2.81 5(70), 2.756(36), 2.251 (JI " 2.1 16(31 " 1.955(30)
IMA No. 2002-063 (Ni,Zn)AI.(V01)z(OH)ll(H10hJ Ni-dominnn!
analogue of nlvanitc; slruelure delermined Monoelinic: nl/II
a 17.8098,b 5.1228,c 8.86651\, � 92. 141° Colorless lO white, lighl green 10 lighl blue; I'ilrcous: diaphaneity 001 given lliaxial (-), Cl 1 .653, P 1.680. Y 1 .106, 2V(mcas.) 86°, 2V(enle.) 88°
8.89( I 00), 7.83( 1 00), 3.266(50). 1 .970(80), 1 .904(70), 1.605(50), 1.481 (80)
IMA No. 2002-064 (K,Na,CJ) (MnZ+,fel+ ,Li)l (AI,Si). Si4 011(01'1). (F,OH)� Carpholilegroup Orthorhombie: Ceca
a 13.715, b 20.302, e S. 138 A White lo slraw-ycllow; si lky; diaphaneity nOI givcn J)i�ial (-), a. 1 .578, 13 1.592 Y 1.598, 2V(meas.) 57°, 2V(eale.) 66° S.70(100). 3.819(80), 3.43(80). 3.048(90), 2.744(80). 2.613( I 00). 2.050(80). 1 .467(80)
IMA No. 2002-065 (Na,K.Sr)nC¡lllFcJZroTiSi,IOIH{O.OI I.I I!O}�CI¡
Emlinlylc group: Slruclure delcrmincd Trigonal: In
(1 14.239. e 60.733 A Pink; vilreous: lransparent Uniaxial (+). ro 1 .597. t 1.601 6.45(33), 5.70(34). 4.32(68), 3.55(39), 3.230(44), 3.049(36), 2.977( 100), 2.853(88)
IMA No. 1002-066 (H¡O}¡(Na,K,Sr)sC%Zr¡Si16066(OI'I)9CI
Eudialyte group; struelure detennincd Trigonnl: R3
a 14.078, e 3 1 .24 A Pink; vilreous; translueenl Uni�¡al (+), 00 1.569, t 1 .571 1 1 .43 (39), 10.50(44), 7.06(42). 6.63(43), 4.39( 100), 3.624(4 1 " 2.987( 100), 2.850(79)
IMA No. 2002-067 NalSCn¡Fc¡{Nn.7.r)¡Zr¡{SLNb)(Si!,\07JXOJ·I.H!O}¡ (CI.OII) EudialYle group:slruclurc dctermined Trigonal: RJ
(/ 14.229. (' 30.019 A Red: vilrcous: transparcnl Unia;t.;inl (+), 00 1 .608, t 1.61 1 1 1 .48(33), 5.72{35). 4.31(66). 4.09{37), 3.209{58). 3.023(40), 2.974(86), 2.853{1 00)
PROPOSALS FROM PREVIOUS YEARS
APPROVED IN 2002
lMA No. 2000-010 (Na,H¡O)1 s(Ca,Mn,REEk,Fc" ¡Zr)(: .,Zr)(i_ ',S i)Sil�066 (O.OHl6CI·oH10 EudialYle group;
Trigonal: R3m a J4.167, c 30.081 A
Yellow; vilrcous; lranslueent
slruelure determined

186 GRICE. J. D. Y FERRARIS, G.
Unin.xial (+), ro 1.612, e 1.615 6.41(41), 4.30(91),3.521(57), 3.205(44), 2.963(92), 2.841(100), 2.588(37)
IMA No. 2000·018 Nal1K,Ca,:FclZr¡,Sis!O,H(OI·I.04Ch Elldial)' ... !
group: SlmClun: dctcnn;ncd Trigunal: lB",
(1 14.249, C' 60.969 A Pink; V;II'I:OUS: Irnnsparcnl Uniaxial (+), el) 1.598, e 1.600 6.48(47), 4.34(81), 3,565(41), 2.987(100), 2.861(73), 2.695(40)
1M" No. 2001·069
3.249(57),
Na(Nal.o.l ,LilU.1.oh(Fc )·1 Mg z Li)SJ .Ol1(OHh Amphibolc graup; slructure dctcrmincd
Monoclinic: el/m 0 9.712, b 17.851, c S.297 A, P
103.6](21 Bluish black; yjtrtous; transluccnt No oplical dala could be givcn 3.392(3]), 3.098(37). 2.701(100), 2.576(14), 2.S24( 100). 2.1 57(20), 1.646(20), ¡.S81( I 5)
1M" No. lOOI·070 CO)(1'04) Rclmcd lo whillockilc Trigonal: U3m
n S.2SII. e 18.727 A While to )'ello\\;sh grey: vill\:ous: dinplmncilY not given Uniaxial (�), ro 1.706 1.701 2.891(80), 2.628( 1 00), 2.214(20), 2.078( 12),
2.047(16), 1 .945(47), 1.730(2S)
Nomentlature modifications 1998-2002
IMA Code 98-0 - Monsrnedite discredited ... \'ollailc.
IMA Cooc 98-E - Arsenobismite diseredilrd -mixture of preisingerite, minor atc1estitc and minor bcudantitc/scgn ite
IMA COOe 99-A - Plalynile discredited - mixture or laitllkarite Ilnd sdcnian galena.
IMA Cadc 99-0 - Peprossiitl!-(Cc) retlelincd as
(19S5) el labuntsovite-Mn; labuntsovite of Millon el al. (I9S8) · paralabunlsovite-Mg. IMA Code 00-13 - Kurgllnlaite revlllidllted.
IMA Code OO·C - Baiyutlcboitc-(Cc) d iseredited CE eordylilc-(Ce).
IMA Codc OO-D - Nomo.:nclalurc of jnaquinilc grollp rrddinetl 10 etlnfllrm wilh lhe Lcvin�H1 s)'stcm. Tito.: memhcl'll 01' tho.: g.roup are: onhojoaquinilc-(I.a): jO!1<luinilt.iCc). onho-joaquinitc-{Ce), onhojoaquiniIC-(La), slronlio-joaquinite, strontio-onhojoaquinite, bario-orthojoaquinite, byclorussite-(Ce).
IMA Code OO·E. Oestinezite redefined as trielinic Fel(P04)(S04)(OH).6H20.
IMA COOc OO-F. - Redefinition (the new name is Ihe seeond one): hellandile = hellandite-(Y); tadzhikile - tadzhikite-(Ce).
IMA Code 00-0 - Neotypc approved and magnesium - zippcite ndefined as monocJinie Mg (U01h (S04) (OI·lk 1.5H20 IMA Code OI_A - Redefinition (Ihe new namo.: is Ihe sccond one): htsgl.xnnitl!-Rlf magnesi(lhüg.h(lmile-2:\'2S: hUghomill!- I01' mngncsiohiíg.bomitc-2:\·lS·; hllgomn ile-24 U nmgnl!siohl't¡;homill!-6.\'6S; zincohilg.bomill!-811 ... zincohOgoomitl!-2N2.\': zincohügbornill!- 1611 7.incohügbomile-2:\'6S: nigcrito.:-61' '" Icrronigl!rile-2NIS; nigerile-24R fcrronigeritc-6N6S: pcngzhizhongitc-67' ... lllagncsionigerilc-2NI.S� pcnv. hizhongite-24R .. magncsionigerite-6N6S: tufTe;te '" magnesiot8affeitc-2N'2S; musgmvile -magnesiolaafTeitc-6N'3S; pchrmanile -ferrolaaffe1te·6N' 3S;
IMA Code 01-13 • Duhamelilc disereditcd -mottramite.
IMA COOe 02-A - Tripuh)'itc is redefined as FeSbO� and squawereekile or Ford el al. (1991) is disereditrd.
IMA Code 02-11 - Arhb.1rite is retlefinrtl as trielinic Cu:Mg(A�O�)(OI-I)I.
(Ce.IJl)(AIJOh·Jn�OIn- IMA Codo.: 02-1) - 'IlII! minl!r.l1 rmml! nmhlr\loodilc
IMA Codc OO-A - Retlefinhion (lhl! ne\\' name is Ihe sceond one): vuoriyarvill! - vuori)'llrvilc-K: kuzmcnkoile - kuzlllcnkoilc-Mn; IcmmlcinilC -Icmmlcinilc-K; l!lbuntsovite or Semeno\' & Burova
is correcled in nl¡llhmuodile.
Name ehan¡;c npprovcd - "magnocolumbilc" is 1l0W magnesio<:olumbilc