aseguramiento de la calidad en la producción de medicamentos … · 2019-10-17 · del plasma y el...
TRANSCRIPT

Aseguramiento de la Calidad en la Producción de Medicamentos Biológicos y Hemoderivados,
enfoque actual según las nuevas normativas.
Dra. Cecilia Sobrero [email protected]
Lic. Pablo Ponziani [email protected]
Comité de Expertos Aseguramiento de la Calidad

Sistema de Calidad Farmacéutico en la producción de medicamentos Biológicos y
Hemoderivados:
Similares Requerimientos aplicables a la implementación de un Sistema de Calidad Farmacéutico destinado a la producción de medicamentos denominados comúnmente “genéricos” o “convencionales”
Mismas Actividades del área de Aseguramiento de Calidad para demostrar una adecuada implementación del Sistema de Calidad
Introducción

Variabilidad intrínseca de los productos biológicos
consideraciones específicas que surgen de la naturaleza del producto biológico: materiales biológicos, tales como el cultivo de células o la extracción de organismos vivos.
consideraciones específicas asociadas a los procesos involucrados: fermentación; precipitación y digestión proteica; filtración tangencial . Pueden presentar una amplia variabilidad inherente (diferencia de los medicamentos convencionales, fabricados mediante tecnologías químicas y físicas con un alto grado de consistencia)
Donde radica la diferencia?

Aplicando los principios de Gestión de Riesgos para la Calidad (QRM) en el desarrollo de la estrategia del control a lo largo de las diferentes etapas del proceso de fabricación a fin de asegurar la trazabilidad, minimizar la variabilidad y reducir la oportunidad de contaminación y contaminación cruzada
Cómo mitigar esta variabilidad?

Los materiales y las condiciones de procesamiento utilizados diseñados para favorecer las condiciones para el crecimiento de células y microorganismos específicos, puede ocasionar crecimiento contaminantes microbianos extraños.
Productos con capacidad limitada para soportar un amplio rango de técnicas destinadas a inactivar o eliminar contaminantes virales adventicios
El diseño de los procesos, equipos, instalaciones, servicios, las condiciones de preparación y adición de buffers y reactivos, el muestreo y la formación de los operarios son puntos clave a tener en cuenta para minimizar el riesgo de contaminaciones
Cuáles son las características de los productos y de los procesos que debemos considerar?

Productos biológicos que no pueden ser esterilizados por filtración (ejemplo Vacunas), proceso aséptico para minimizar la introducción de contaminantes.
El control implica habitualmente técnicas analíticas biológicas, que normalmente presentan una mayor variabilidad que las determinaciones fisicoquímicas.
Los medicamentos biológicos que incorporan tejidos o células humanas, como algunos Medicamentos de Terapias Avanzadas, deben cumplir los requerimientos nacionales para las etapas de donación, obtención y análisis
Cuáles son las características de los productos y de los procesos que debemos considerar?

Los requisitos de trazabilidad son de aplicación desde el donante (mientras se mantenga la confidencialidad de este), hasta las fases aplicables a los centros de tejidos, y continuando hasta la institución donde el producto es usado, bajo la legislación sobre medicamentos.
Las sustancias biológicas activas y los medicamentos biológicos deben cumplir la última versión de los lineamientos nacionales sobre la reducción del riesgo de transmisión de agentes de encefalopatía espongiforme animal (EET) a través de medicamentos humanos.
Cuáles son las características de los productos y de los procesos que debemos considerar?

Disposición ANMAT 3827/18, Texto corregido y ordenado de la Disposición ANMAT 3602/18; Guía de Buenas Prácticas de Fabricación para Elaboradores, Importadores/Exportadores de Medicamentos para Uso Humano
Anexo 17 Fabricación de Productos Medicinales de Origen Biológico
Anexo 18 Fabricación de Medicamentos derivados de la Sangre o Plasma Humano
Estos Anexos son complementarios a los requerimientos generales de la Norma
Normas aplicables

Anexo 17 Fabricación de Productos Medicinales de Origen Biológico
• Parte A: requerimientos específicos y complementarios a las GMP aplicables a la fabricación de ingredientes farmacéuticos activos y medicamentos de origen biológico, (desde el control de los lotes semilla y los bancos de células hasta las actividades productivas finales y los ensayos)
• Parte B: establece lineamientos adicionales para determinados tipos de ingredientes farmacéuticos activos y medicamentos de origen biológico
Normas aplicables
De existir una potencial contraposición entre un requerimiento general y uno específico, prevalecerá el
requerimiento específico complementario

Parte B
Disposición
3827/18
Requisitos básicos para Ingredientes
Farmacéuticos Activos (IFA’s)
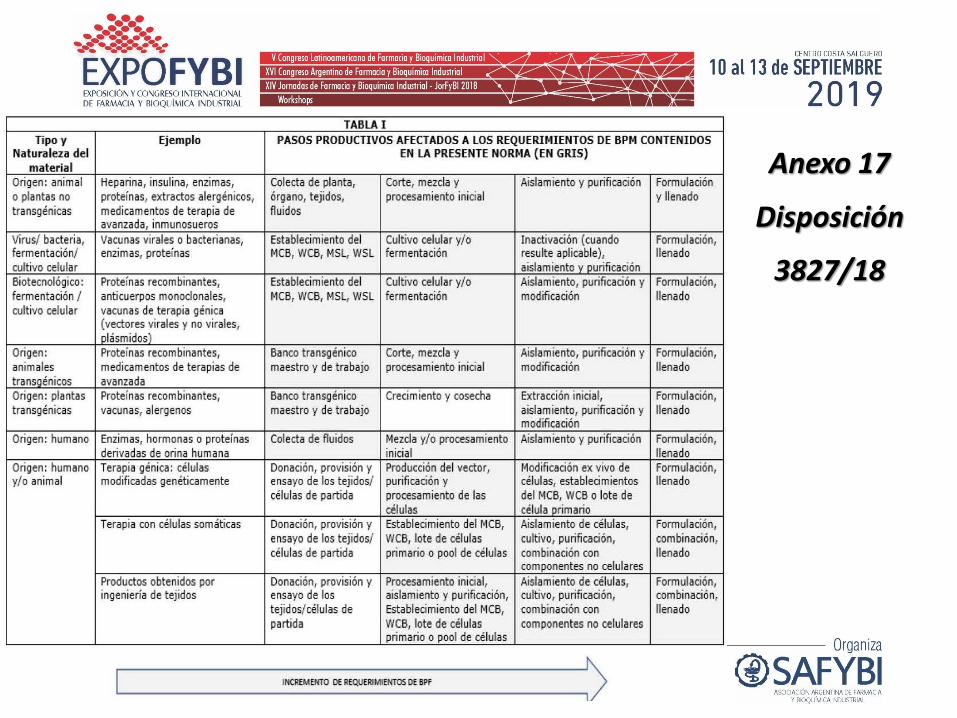
Anexo 17
Disposición
3827/18

Anexo 17 Disposición 3827/18. Requerimientos Específicos
• Personal:
Vacunación específicas adecuadas y controles médicos regulares.
El control del estado de salud del personal debe ser acorde con el riesgo. Incluir recomendaciones médicas para el personal que trabaje con organismos peligrosos.
Restricción del flujo de personal (incluyendo control de calidad, mantenimiento y personal de limpieza) de manera de minimizar el riesgo de contaminación cruzada.
El personal no debe pasar de zonas de exposición a microorganismos vivos, organismos modificados genéticamente, toxinas o animales, a zonas donde se manejen otros productos, productos inactivados u organismos diferentes; a menos que se evalué adecuadamente el Riesgo y se tomen las medidas adecuadas

Anexo 17 Disposición 3827/18. Requerimientos Específicos
• Instalaciones y Equipos:
Monitoreo ambiental con detección de la presencia de microorganismos específicos (es decir, organismos hospedadores, hongos, levaduras, anaerobios, etc.)
Áreas de producción dedicadas para el manejo de células vivas persistentes hasta su inactivación. También para la fabricación de organismos patógenos capaces de causar enfermedades humanas severas (nivel de bioseguridad 3 ó 4)
Sistemas de contención primarios diseñados y revisados periódicamente para prevenir escape de agentes biológicos en el entorno inmediato de trabajo
Sistemas de drenaje deben diseñados para neutralizar o descontaminar los efluentes (contaminación cruzada); cumpliendo regulaciones locales (contaminación del medioambiente)

Anexo 17 Disposición 3827/18. Requerimientos Específicos
• Animales:
Monitorear agentes adventicios relevantes (zoonosis, enfermedades de origen animal); con la participación de especialistas.
Poner especial atención en la prevención y monitoreo de infecciones en los animales de origen o donantes.
Deben tenerse en cuenta los requisitos las leyes, reglamentos y disposiciones en relación a la protección de los animales usados para propósitos experimentales. (Regla de las 3 R)
Los animales (reactivos biológicos) y los ensayos realizados deben ser objeto de un sistema de identificación y trazabilidad para prevenir cualquier
riesgo de confusión

Anexo 17 Disposición 3827/18. Requerimientos Específicos
• Documentación:
Documentación adicional sobre fuente, origen, cadena de distribución, método de fabricación y controles para las especificaciones de materiales de partida y materias primas de origen biológico.
Se requiere trazabilidad completa de los materiales de partida y materias primas de donantes de células o tejidos humanos. Registros de trazabilidad conservados durante 30 años a partir de la fecha de caducidad del medicamento.
Para los componentes de la sangre, cuando sean utilizados como material de partida o como materias primas en el proceso productivo, deben cumplimentarse los requerimientos nacionales; incluyendo los relativos a los requisitos de trazabilidad, notificación de reacciones adversas serias y eventos.

Anexo 17 Disposición 3827/18. Requerimientos Específicos • Producción:
Las revisiones de calidad del producto (RAP) deberían reevaluar los pasos para aumentar la robustez del proceso y por tanto reducir su variabilidad y asegurar su reproducibilidad en las diferentes fases del ciclo de vida del producto.
• Materiales de partida y materias primas:
Debe definirse claramente procedencia, origen e idoneidad de los materiales de partida y materias primas biológicas.
Cambios en el origen y/o fabricante de la materia prima activa debe ser evaluado e informado a la autoridad regulatoria.

Anexo 17 Disposición 3827/18. Requerimientos Específicos
• Principios de operación:
Confinar los procesos de centrifugación y mezcla de productos que pueden dar lugar a la formación de aerosol (contaminación cruzada)
Disponer de medidas de descontaminación validadas para cada organismo o grupo de organismos relacionados.
En el caso de que se lleve a cabo durante la fabricación un proceso de inactivación o eliminación de virus, se deberán tomar medidas para evitar el riesgo de recontaminación de productos tratados con productos no tratados.
Para los productos inactivados por la adición de un reactivo (ej. microorganismos en el curso de fabricación de una vacuna) el proceso debe asegurar la inactivación completa del organismo vivo.

Anexo 17 Disposición 3827/18. Requerimientos Específicos
• Control de Calidad:
Relevancia de los controles en proceso para garantizar la consistencia de la calidad de las sustancias biológicas activas y los medicamentos biológicos.
El programa de estabilidad on-going debe incluir los productos terminados fabricados con intermedios almacenados durante el periodo máximo.
La liberación de ciertos productos biológicos (vacunas)requiere de la intervención previa de la Autoridad Regulatoria mediante la presentación del Protocolo Resumido de Producción y Control correspondiente (Anexo I- Ítem 4 de la Disposición ANMAT 705/05).

Anexo 17 Disposición 3827/18. Parte B - LINEAMIENTOS ESPECÍFICOS APLICABLES A TIPOS DE PRODUCTOS SELECCIONADOS
• Productos derivados de inmunosueros de origen animal
Importancia en la preparación de materiales usados en la inmunización de los animales (ej. Antígenos, adyuvantes, agentes estabilizadores) y el almacenamiento de esos materiales inmediatamente antes de la inmunización.
Ajustar los Calendarios de inmunización, test sanguíneo y extracción de sangre a los aprobados en la en la Autorización del estudio de farmacología clínica o en la Autorización de Comercialización.
Condiciones de fabricación para la preparación de sub-fragmentos de anticuerpos (Ej. Fab o F(ab’)2) y cualquier modificación adicional deben ser conformes con los parámetros validados y aprobados.

Análisis sanitario de los animales previo al ingreso. Vacunación contra tétanos, influenza y anti- encefalomielitis.
Identificación y caracterización del antígeno a ser
utilizado.
Seguimiento “in vitro” de la respuesta inmune.
Utilización de antisépticos.
Sangrado del animal en un ambiente acorde a los
lineamientos de la “WHO guideline for the
Production, Control and Regulation of snake
antivenom immunoglobulins”.
Procedimientos y registros específicos.
Entrenamiento del personal en sangría; separación
del plasma y el paquete globular.
Control del título de anticuerpos en el pool de
plasma previo a su uso.
Control diario de la temperatura de la
heladera. Calibración del sensor de temperatura de
la heladera/Calificación equipo.
Productos derivados de inmunosueros de origen animal: Etapa Obtención del Plasma

“Cuestionar continuamente, explorar nuevos caminos, no dar nunca nada por sentado y tener distintos puntos de vista requiere ser imaginativo, pero es la clave del avance científico”. Albert Einstein
Muchas Gracias por la Atención

DESAFÍOS EN EL ASEGURAMIENTO DE LA CALIDAD EN LA OBTENCIÓN DE LOS
HEMODERIVADOS PLASMÁTICOS
Comité de Expertos de
Aseguramiento de la Calidad

Desafíos en el aseguramiento de la calidad de los Hemoderivados REGULACIONES
GMP • DE MEDICAMENTOS. ANEXO 1: FABRICACIÓN DE MEDICAMENTOS ESTÉRILES,
ANEXOS 17 Y 18 : MEDICAMENTOS BIOLÓGICOS Y HEMODERIVADOS
MP
• LEY DE SANGRE
• NORMATIVAS DE LOS BANCOS DE SANGRE – FARMACOPEAS ARGENTINA, USP, EU.
PT • FARMACOPEA ARGENTINA E INTERNACIONALES RECONOCIDAS
Trazabilidad
• TRAZABILIDAD – ANMAT: DESDE EL DONANTE AL RECEPTOR, NO SOLO POR LOTE DE PRODUCCIÓN SINO, ENVASE A ENVASE, CON UN CÓDIGO DE SEGUIMIENTO ÚNIVOCO / INEQUÍVOCO, EL CUAL ES SEGUIDO POR LA AUTORIDAD SANIATRIA.
Disposición N° 963/2015 N° 10564/16, N° 1831/12, 247/13 y N° 10564/16.

Desafíos en el aseguramiento de la calidad: TRAZABILIDAD

Hemoderivados - Qué los hace diferentes a otros medicamentos?
OBTENCIÓN DE MATERIA PRIMA PLASMA MASTER FILE Recurso estratégico Variabilidad,Trazabilidad
R vs D?
FRACCIONAMIENTO purificación de los
distintos PA (fracciones proteicas)
FORMULACIÓN E INACTIVACIÓN VIRAL
ENVASADO ASÉPTICO DISTRIBUCIÓN – TRAZABILIDAD
DISPONIBILIDAD Y USOS FARMACOVIGILANCIA

Desafíos en el aseguramiento de la calidad de los Hemoderivados - Plasma Master File

Desafíos Algunos aspectos claves en la calificación de los proveedores:
REQUISITOS DEL SISTEMA DE CALIDAD (DOCUMENTACIÓN, DEFINICIÓN DE LAS
TAREAS Y LAS RESPONSABILIDADES)
CRITERIOS DE SELECCIÓN DE DONANTES Y
ANÁLISIS. MÉTODOS DE EXTRACCIÓN
REQUISITOS PARA LA SEPARACIÓN DE SANGRE
EN PLASMA Y COMP. SANGUÍNEOS;
CONGELAMIENTO DEL PLASMA (CRÍTICO PARA LA
RECUPERACIÓN DE PROTEÍNAS LÁBILES)
ALMACENAMIENTO Y TRANSPORTE DEL PLASMA.
RESULTADOS DE LOS ENSAYOS DE TODAS LAS
UNIDADES SUMINISTRADAS POR EL
ESTABLECIMIENTO DE SANGRE QUE DEBEN
ESTAR A DISPOSICIÓN DE LA PLANTA FABRICANTE
DEL MEDICAMENTO.

Algunos aspectos claves en la calificación de los proveedores:
Controles serológicos para los siguientes agentes infecciosos (realizados por el Banco de Sangre):
• HBs Ag • Anti - VHC •Anti - VIH 1 y 2
• Anti-core VHB. Se podrán recibir unidades de plasma Anti-core VHB reactivas para fraccionamiento plasmático, siempre que HBs Ag sea no reactivo y Anti - HBs Ag reactivo.
• Ag-VHC (*) Antígeno P24 de VIH (*) Cuando se realice tamizaje por biología molecular para VIH y VHC se exime de realizar los Ags correspondientes)
• Sífilis • Brucelosis • Chagas (empleando 2 métodos diferentes)
LOS REQUISITOS ANALÍTICOS DE VIRUS U OTROS AGENTES INFECCIOSOS HAN DE CONSIDERARSE A LA LUZ DEL CONOCIMIENTO EMERGENTE EN
AGENTES INFECCIOSOS Y EN FUNCIÓN DE LA DISPONIBILIDAD DE MÉTODOS ANALÍTICOS ADECUADOS Y VALIDADOS

Desafíos en el Aseguramiento de la Calidad de los Hemoderivados Plasma Master File
INFORMACIÓN LOOK-BACK
Los bancos de sangre deben notificar a la planta de fraccionamiento cualquier evento que pueda afectar la calidad o seguridad del producto, incluyendo las reacciones adversas
serias y otra información relevante hallada tras la liberación del plasma.

Hemoderivados - Qué los hace diferentes a otros medicamentos?

Hemoderivados - Qué los hace diferentes a otros medicamentos?
Obtención de materia prima Plasma Master
File.
FRACCIONAMIENTO purificación de los
distintos PA (fracciones proteicas)
FORMULACIÓN E INACTIVACIÓN VIRAL
Envasado aséptico Distribución – trazabilidad
Disponibilidad de los mismos y usos.
Farmacovigilancia

Hemoderivados - Qué los hace diferentes a otros medicamentos?
FORMULACIÓN E INACTIVACIÓN VIRAL: Métodos validados
Segregación de lo inactivado / no
(instalaciones y equipos dedicados y distintos para las etapas de fabricación pre y post inactivación viral)
PROCESAMIENTO ASÉPTICO: clase A entorno B
Áreas clasificadas y calificadas, equipos validados, validación de limpieza química y microbiológica. Minimizar la
contaminación microbiológica y la contaminación cruzada

TIEMPOS ( t) MÁXIMOS Y MÍNIMOS DETERMINADOS Y VALIDADOS MÁS CARACTERIZACIONES DE PROCESO
ENTRE LA LIMPIEZA Y LA ESTERILIZACIÓN ,Y ENTRE ÉSTA Y SU UTILIZACIÓN. DE ENSAMBLE DE TUBULADURAS EQUIPOS Y ACCESORIOS.
t DE FORMULACIÓN PRE FILTRACIÓN. BIOBURDEN EN CADA LOTE.
FILTRACIÓN: DEFINIDO EL t MAX Y MIN. IDEM INACTIVACIÓN VIRAL.
t ENTRE FILTRACIÓN Y DISPENSACIÓN EN LOS ENVASES, t DE EXPOSICIÓN DEL PRODUCTO EN EL LLENADO HASTA EL TAPONADO (MAN O AUT).
MEDIA FILL (PRUEBA SIMUL. PROCESO) : 3 INICIALES Y LUEGO DEBEN REPETIRSE DOS VECES AL AÑO POR TURNO Y POR PROCESO. LA FRECUENCIA CADA 6 MESES JUSTIFICANDO UN ANALISIS DE RIESGO (productos per se). PERSONAL 1 PSP EXITOSO.

Desafíos en el aseguramiento… INSTALACIONES Y EQUIPOS - LAY OUT ADECUADOS - CUIDADO!!!
DE LIMPIEZA Y MOVIMIENTO DE LOS MATERIALES
DE PROCESOS (PRE Y POST INACTIVADO)
DEL PERSONAL (DE VESTIMENTA, DE MUESTREOS, DE INSPECTORES)
DE INSUMOS Y MATERIALES
DE RESIDUOS (HIGIENE Y SEGURIDAD ATENDIENDO A LA BIOSEGURIDAD)
DE MANTENIMIENTO, DE LOS SERVICIOS AUXILIARES (WFI, SISTEMA DE AIRE)
CIRCUITOS, MOVIMIENTOS, DURACIONES, CRUCES, SEGREGADOS,
Que las operaciones se vean desde afuera, alarmas en las esclusas y vestuarios, alarma del sistema de aire, superficies lisas de fácil limpieza, evitar repisas, estantes, sumideros, cielo rasos herméticos!

Preparación aséptica - Media Fill

Desafíos en el aseguramiento de la calidad de los Hemoderivados
ARCHIVO MAESTRO DE PLASMA MASTER FILE
FORMULA MAESTRA, REVISIÓN ANUAL DE PRODUCTO, ANÁLISIS DE RIESGO
Si se procesa plasma / intermedios de orígenes distintos, se debe hacer un RIGUROSO ANÁLISIS DE GESTIÓN DE RIESGOS (atendiendo a posibles diferencias epidemiológicas); con el resultado, se diseña
una producción por campaña que incluya una clara segregación y procedimientos de limpieza validados

Desafíos FUTUROS de los Hemoderivados
PREGUNTAS? GRACIAS POR SU ATENCION!