antiagregantes plaquetarios - abril 2015
TRANSCRIPT

ANTIAGREGANTESANTIAGREGANTES
PLAQUETARIOSPLAQUETARIOSZappa Santiago AndresZappa Santiago Andres
BioquímicoBioquímico
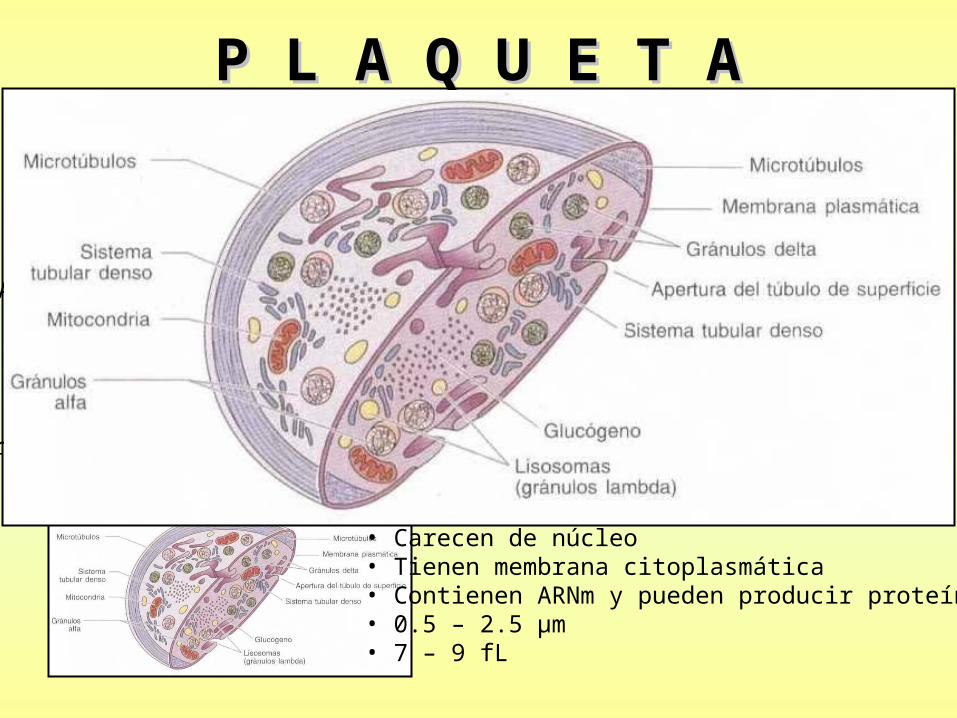
P L A Q U E T AP L A Q U E T A
Al salir de la Medula Oseaingresan a la
circulación sanguínea
Son fragmentos celulares derivados de Megacariocitos ubicados en la Medula Osea
Permanecen allí por 8 – 10 días
• Carecen de núcleo• Tienen membrana citoplasmática• Contienen ARNm y pueden producir proteínas• 0.5 – 2.5 µm• 7 – 9 fL
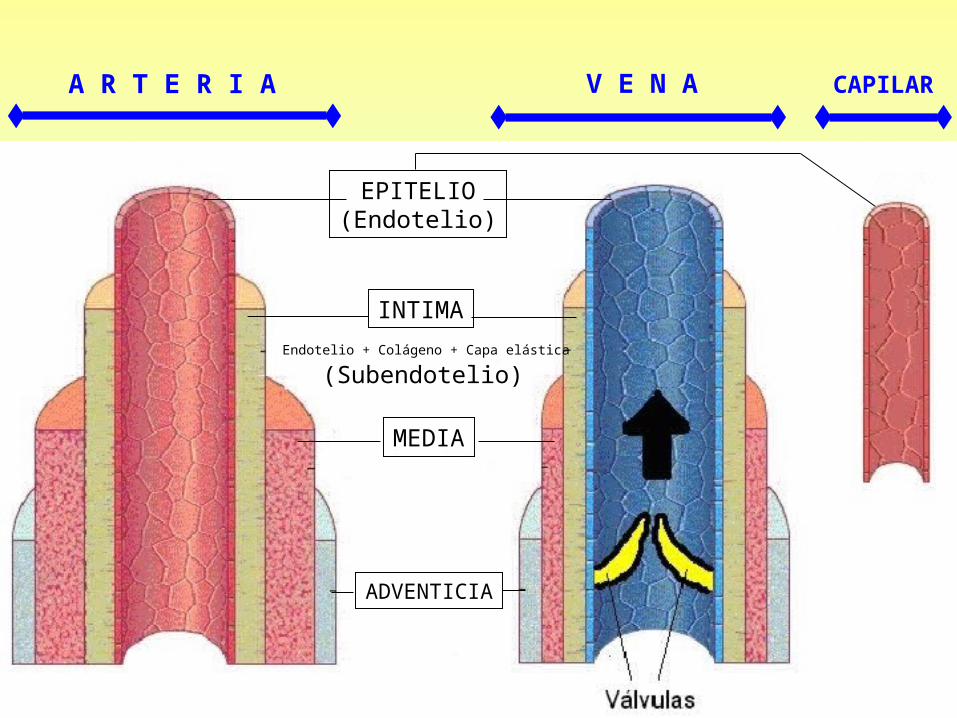
EPITELIO(Endotelio)
V E N A CAPILAR
INTIMA
MEDIA
ADVENTICIA
Endotelio + Colágeno + Capa elástica
A R T E R I A
(Subendotelio)

¿Cómo funcionan las plaquetas?¿Cómo funcionan las plaquetas?Lesión de la pared del Vaso Sanguíneo
Se expone el SUBENDOTELIO• Colágeno• Factor de Von Willebrand• Fibronectina• + Factor Tisular = activa la coagulación
Se adhiere la PLAQUETAPLAQUETA circulante
SUBENDOTELIOSUBENDOTELIO PLAQUETAPLAQUETA
COLÁGENO ↔ GP Ia/IIa + GP VI
FvW ↔ GP Ib/IX/V + GP IIIa/IIb
Fibronectina ↔ GP Ic/IIa
Laminina ↔ GP Ic
Trombospondina ↔ GP IV
Depende de la velocidaddel flujo sanguíneo RECEPTORES
TRANSMEMBRANA
UNIÓN LIGANDO - RECEPTORUNIÓN LIGANDO - RECEPTOR

¿Cómo funcionan las plaquetas?¿Cómo funcionan las plaquetas?La unión ligando-receptor induce un
cambio conformacional en el ReceptorSe desencadena una
respuesta intraplaquetaria
• Se contrae el Citoesqueleto → Cambio conformacional de la Plaqueta
• Exocitosis de los gránulos internos → Se liberan mediadores químicos
Gránulos densosATP/ADPCa2+/Mg2+
Serotonina
Gránulos α
Proteínas proagregantesFvWF. CrecimientoProteasasCitoquinas
Lisosomas Hidrolasas
Digieren lamatriz subendotelial

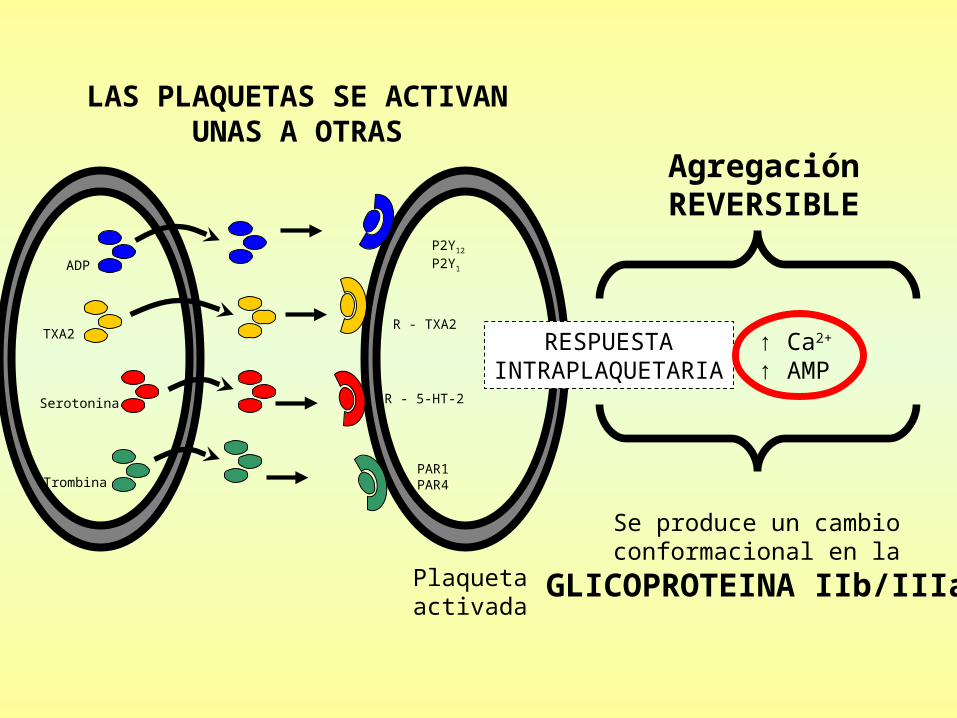
ADPADP
TXATXA22
SerotoninaSerotonina
TrombinaTrombina
P2YP2Y1212
P2YP2Y11
R - TXAR - TXA22
R - 5-HT-2R - 5-HT-2
PAR1PAR1PAR4PAR4
+ Trombina (proveniente de la cascada de coagulación)
ADP
TXA2
Serotonina
Trombina
P2Y12
P2Y1
R - TXA2
R - 5-HT-2
PAR1PAR4
Plaquetaactivada
LAS PLAQUETAS SE ACTIVANUNAS A OTRAS
RESPUESTAINTRAPLAQUETARIA
↑ Ca2+
↑ AMP
AgregaciónREVERSIBLE
Se produce un cambioconformacional en la
GLICOPROTEINA IIb/IIIa

Se produce un cambioconformacional en la
GLICOPROTEINA IIb/IIIa
AUMENTA MUCHOla AFINIDAD por
Fibrinogeno FvW
Se generan verdaderospuentes entre las plaquetas
AgregaciónIRREVERSIBLE

REGULACIÓN PLAQUETARIAREGULACIÓN PLAQUETARIAEL ENDOTELIO NATURALMENTE
LIBERA SUSTANCIAS ANTIAGREGANTES
GENERA UNA SUPERFICIE ANTITROMBOTICA
OXIDO NITRICO
Difunde dentro de la plaqueta
↑↑↑ GMPc
↓↓↓ Ca2+
Inhibe el cambio conformacional de la
GP IIb/IIIa
Prostaglandina I2
PROSTACICLINA
R – PGI2 en la plaqueta
↑ AC
↓↓↓ Ca2+
ATP ↑↑↑ AMPc
Inhibe la agregación
ECTO – ADPasa(en la superficie endotelial)
Degradan ADP y ATP circulantes

En resumen…En resumen…
E N D O T E L I O
Lesión del EndotelioExposición del Subendotelio
Activa la coagulación
Adhesión plaquetaria
↑ Trombina
Fibrina Fibrinogeno
Activación plaquetaria
Agregación plaquetaria
FORMACIÓN FORMACIÓN DEL TROMBODEL TROMBO
ONPGI2
ADPTXA2
SHT
Cambioconformacional
GP IIb/IIIa
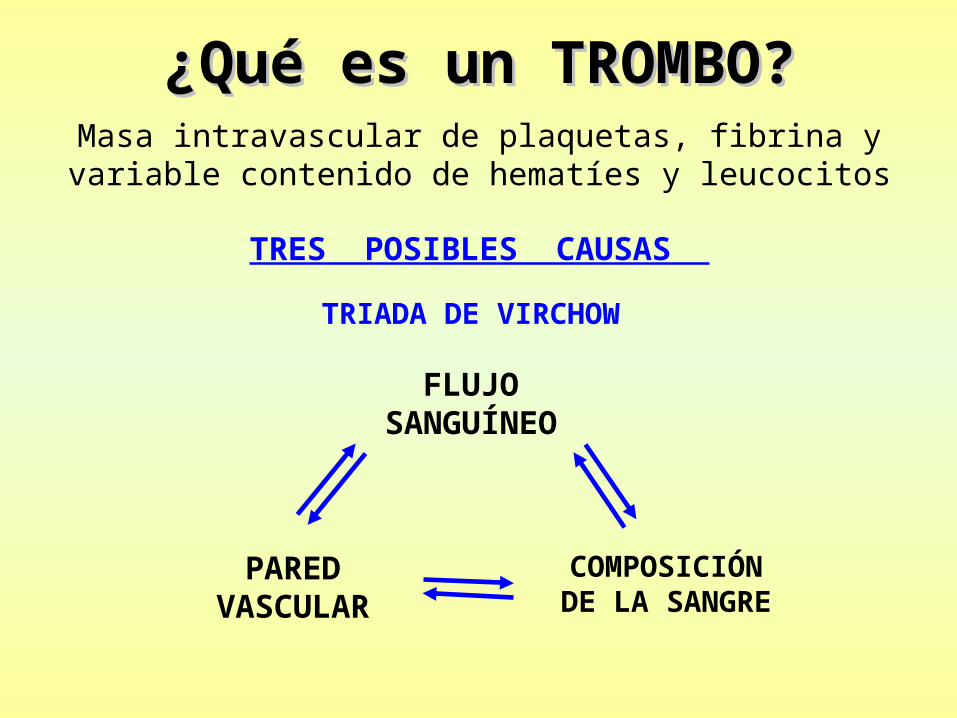
¿Qué es un TROMBO?¿Qué es un TROMBO?Masa intravascular de plaquetas, fibrina y variable
contenido de hematíes y leucocitos
TRES POSIBLES CAUSAS
TRIADA DE VIRCHOW
FLUJO SANGUÍNEO
PARED VASCULAR
COMPOSICIÓN DE LA SANGRE
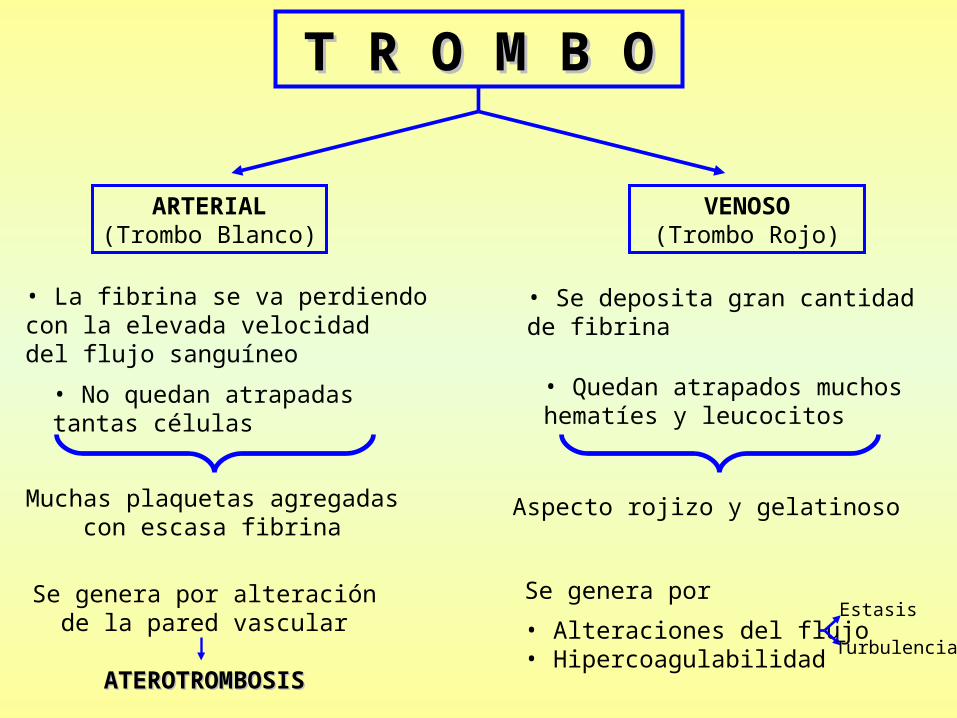
T R O M B OT R O M B O
ARTERIAL(Trombo Blanco)
VENOSO (Trombo Rojo)
Se genera por alteraciónde la pared vascular
ATEROTROMBOSISATEROTROMBOSIS
• La fibrina se va perdiendocon la elevada velocidaddel flujo sanguíneo
• No quedan atrapadastantas células
Muchas plaquetas agregadascon escasa fibrina
Se genera por
• Alteraciones del flujo• Hipercoagulabilidad
• Se deposita gran cantidadde fibrina
• Quedan atrapados muchoshematíes y leucocitos
Aspecto rojizo y gelatinoso
Estasis
Turbulencia

ARTERIOSCLEROSISARTERIOSCLEROSISEngrosamiento y perdida de elasticidad de las paredes arteriales
ATEROSCLEROSIS ESCLEROSIS CALCIFICADA ARTERIOLOSCLEROSIS
Afecta a las Arterias más grandes y
elásticas
Engrosamiento de la INTIMA y
depósito de lípidos
Afecta a Arterias de mediano tamaño
Calcificación y endurecimiento de
la MEDIA
Afecta a las Arterias más pequeñas
(Arteriolas)
Engrosamiento de las paredes por depósito
de material hialino(Asociado a HTA y DBT)
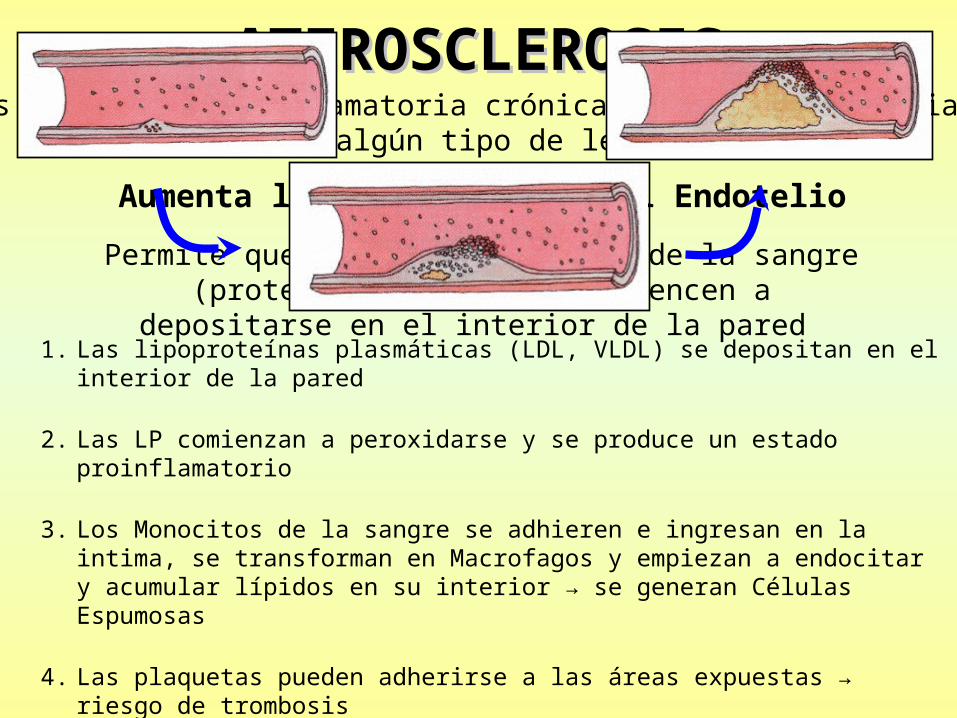
ATEROSCLEROSISATEROSCLEROSISEs una reacción inflamatoria crónica de la pared arterial
que se inicia por algún tipo de lesión del Endotelio
Aumenta la permeabilidad del Endotelio
Permite que los constituyentes de la sangre (proteínas y lípidos) comiencen a depositarse en el interior de la pared
1. Las lipoproteínas plasmáticas (LDL, VLDL) se depositan en el interior de la pared
2. Las LP comienzan a peroxidarse y se produce un estado proinflamatorio
3. Los Monocitos de la sangre se adhieren e ingresan en la intima, se transforman en Macrofagos y empiezan a endocitar y acumular lípidos en su interior → se generan Células Espumosas
4. Las plaquetas pueden adherirse a las áreas expuestas → riesgo de trombosis
5. Migran células musculares lisas desde la media para regenerar colágeno y proteoglicanos

Si el estimulo que generaba la inflamación desaparece
Se resuelve la lesión y se restituye la función endotelial
Si el estimulo permanece de manera CRÓNICA
La lesión progresa hasta generar una “Placa ateromatosa” o ATEROMA
ESTÍMULOESTÍMULO
HIPERCOLESTEROLEMIATrastornos hemodinámicosEndotoxinas (Homocisteína)HipoxiaHipertensión
FACTORES DE FACTORES DE RIESGORIESGO
HiperlipidemiaHipertensiónDiabetesTabaquismoSedentarismoObesidadEstrésFacotres genéticos+ Edad, Sexo masculino


HIPERCOLESTEROLEMIAHIPERCOLESTEROLEMIA• LDL es el más aterogenico, le siguen VLDL y remanentes de QM
LDL oxidada o glicosilada (diabéticos)Quimiotáctica para los MonocitosCitotóxica para las Células Endoteliales
Cuanto mayor es la cantidad de LDL en sangre, más fácilmente ingresa a la pared arterial
LA HIPERLIPIDEMIA CRÓNICA INDUCE UNA MAYOR PRODUCCIÓN DE SUSTANCIAS REACTIVAS DEL OXÍGENO
(Radicales libres, superoxidos, etc)
QUE INHIBEN LA PRODUCCIÓN DE OXIDO NITRICOPOR LAS CELULAS ENDOTELIALES(antitrombotico y protector endotelial)
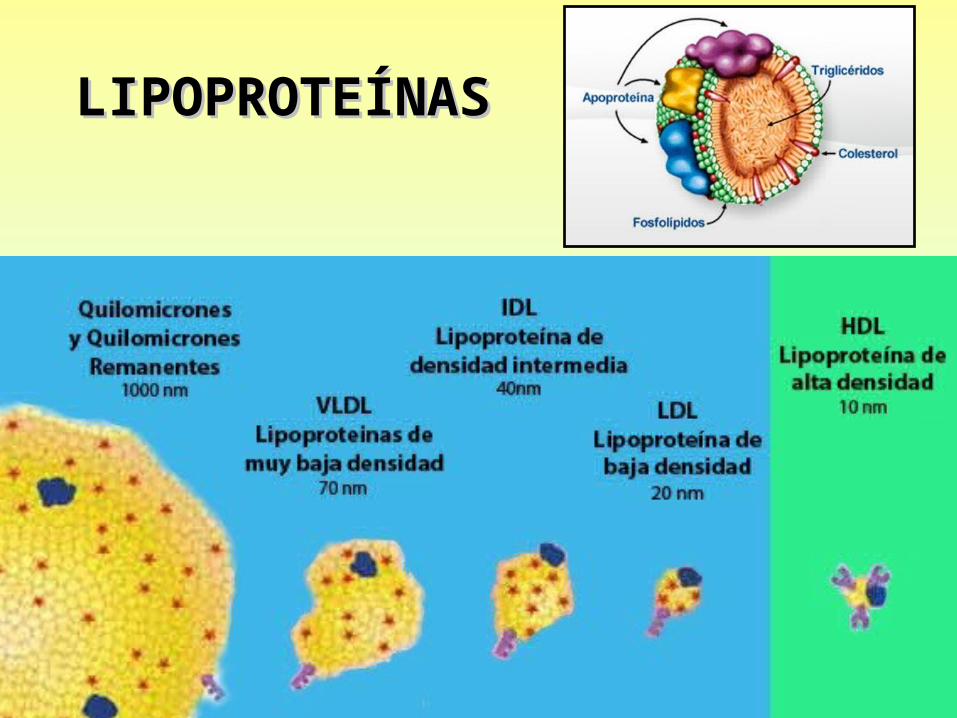
LIPOPROTEÍNASLIPOPROTEÍNAS


COMPLICACIONES del ATEROMACOMPLICACIONES del ATEROMA
Estrechamiento lento y progresivo de la luz
ISQUEMIAISQUEMIA
Ulceración del Endotelio
ATEROTROMBOSIS
Vasos pequeños
Oclusión súbita
INFARTOINFARTO
Vasos grandes
Desprendimiento de la placa
TROMBOEMBOLISMOTROMBOEMBOLISMO
Adelgazamiento progresivo de la media
ANEURISMAANEURISMA
Debilitamiento de la pared
Dilatación de la arteria
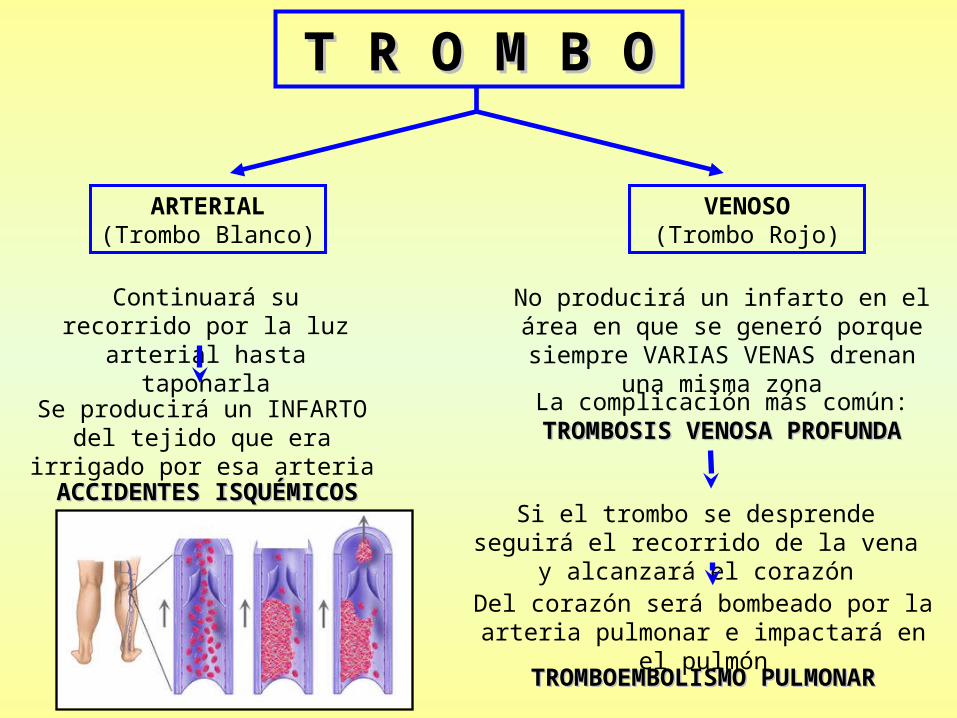
T R O M B OT R O M B O
ARTERIAL(Trombo Blanco)
VENOSO (Trombo Rojo)
Continuará su recorrido por la luz arterial hasta taponarla
Se producirá un INFARTO del tejido que era irrigado por esa arteria
No producirá un infarto en el área en que se generó porque siempre VARIAS
VENAS drenan una misma zona
La complicación más común:TROMBOSIS VENOSA PROFUNDATROMBOSIS VENOSA PROFUNDA
Si el trombo se desprende seguirá el recorrido de la vena y alcanzará el corazón
Del corazón será bombeado por la arteria pulmonar e impactará en el pulmón
TROMBOEMBOLISMO PULMONARTROMBOEMBOLISMO PULMONAR
ACCIDENTES ISQUÉMICOSACCIDENTES ISQUÉMICOS

¿Cómo tratamos la TROMBOSIS?¿Cómo tratamos la TROMBOSIS?
Si el TROMBO ya se generóTratamiento quirúrgico
Tratamiento fibrinolítico
Para prevenir una RE-TROMBOSISTrombo venoso = anticoagulantes
Trombo arterial = antiagregantes
A N T I A G R E G A N T E S
Prevención de accidentes isquémicos
• Coronarios: Angina de pecho + Infarto agudo de miocardio + Reoclusión tras bypass
• Cerebrales: Accidente cerebrovascular + Ictus cerebral
• Periféricos: Claudicación intermitente

ANTIAGREGANTES PLAQUETARIOSANTIAGREGANTES PLAQUETARIOS
Interferencia con la vía del ácido araquidonico
• Inhibidores de Ciclooxigenasa → Aspirina, Triflusal, Indobufeno, otros
• Inhibidores de Tromboxano Sintasa
• Antagonistas de Recptores de TXA2 → Vapiprost, Ifetrobán
• Inhibidores de Fosfodiesterasa → Dipiridamol
Modulación de mecanísmos mediados por AMPc/GMPc
• Moduladores de ciclasas → Prostaciclina, iloprost
• Inhibidores de Receptores de ADP → Ticlopidina, Clopidogrel, Prasugrel
Interferencia con la función del complejo GP IIb/IIIa
• Antagonistas de Receptores de GP IIb/IIIa → Eptifibatide, tirofibán, abciximab
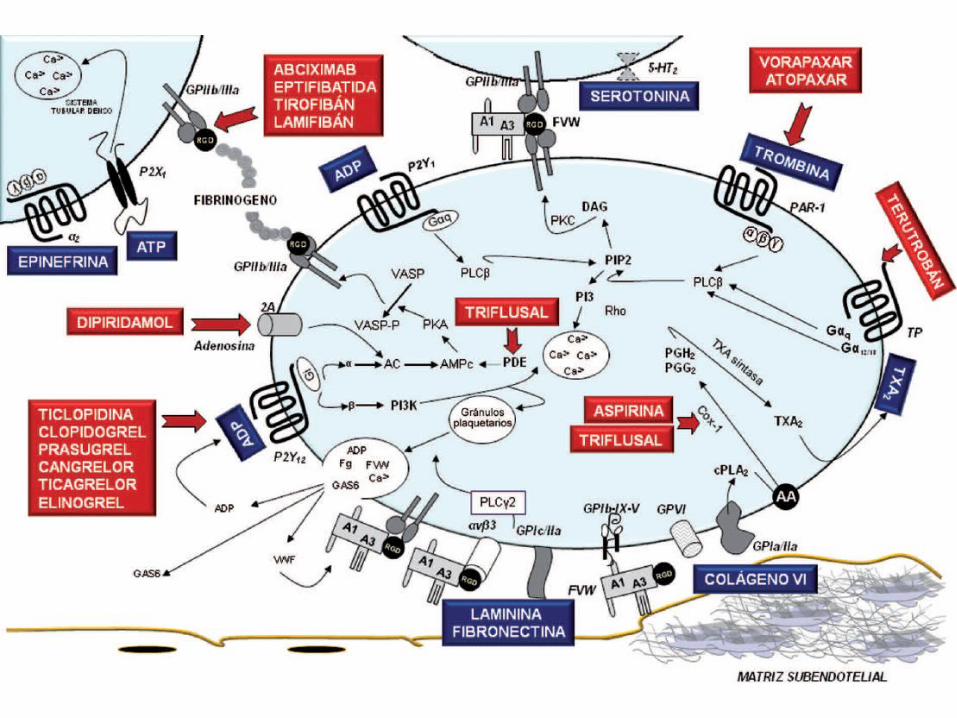

Activación de Plaqueta
Generación de AA
Síntesis deEndoperoxidos
cíclicos
Síntesis de TXA2
Expresión deReceptores GP IIb/IIIa
Unión entre plaquetasadyacentes mediante la unión
de fibrinogeno a losreceptores GP IIb/IIIa
Acidoacetilsalicilico
Antagonistasdel receptor
de TXA2
Antagonistas de losreceptores GP IIb/IIIa(abciximab, tirofiban)
Epoprostenol, ON
Epoprostenol
TiclopidinaClopidogrel
Inhibidoresde la síntesis
de TXA2
Síntesis de TXA2
A G R E G A C I Ó NP L A Q U E T A R I A
Exposición afosfolipidos
ácidos
Procesos decoagulación
Trombina

1. Interferencia con la vía del ácido araquidonico1. Interferencia con la vía del ácido araquidonico
INHIBIDORES DE CICLOOXIGENASA (COX-1 – COX-2)INHIBIDORES DE CICLOOXIGENASA (COX-1 – COX-2)
Acido araquidonico
Endoperoxido PGG2
Endoperoxido PGH2
Tromboxano A2 (TXA2)
• Agregante• Vasoconstrictor
COXCOX
En la plaqueta
Inhibición IRREVERSIBLE
AAS
Triflusal
Inhiben la síntesis de TXA2
Prostaglandina I2 (PGI2)
• Antiagregante• Vasodilatador
En el endotelio
A dosis terapéuticas prevalece el efecto inhibidor sobre TXA2
más que sobre PGI2
El efecto permanece duranteTODA LA VIDA DE LA PLAQUETA
(Los demás AINES la inhiben sólo reversiblemente)

ACIDO ACETIL SALICILICO (AAS – ASPIRINA)ACIDO ACETIL SALICILICO (AAS – ASPIRINA)
• ↓ Secreción de Granulos Densos →↓ Sust. proagregantes durante
la activación plaquetaria
• ↓ Prostaciclina, IL-6, inhibidores de la ONsintasa → Efecto antiinflamatorio
Ocurre a nivel endotelial al inhibir la COX-2 endotelial
La inhibición no es completa → El endotelio SI puede regenerar la COX
(SÓLO A ALTAS DOSIS)
• ↓ Producción plaquetaria de DAG → Inhibe la agregación mediada por Trombina, ADP y Colágeno(SÓLO A ALTAS DOSIS y Efecto menos duradero)
DOSIS REQUERIDAS
Muy inferiores a las analgésicas
Entre 80 y 100 mg por día se inhibe casi el 100% de la síntesis de TXA2
Dosis de carga: 200 – 300 mg + efecto máximo a los 30 min.100 – 150 mg/día previene accidentes isquémicos en pacientes de ALTO riesgo
Reduce eventos clínicos (IAM) en un 30-40% → ANTIPLAQUETARIO DE REFERENCIA
EFECTOS ADVERSOSTrastornos Gastrointestinales (raro a bajas dosis)Hemorragias a nivel del tracto digestivo altoEn prevención primaria: ↑54% hemorragias extracraneales

RESISTENCIA A LA RESISTENCIA A LA ASPIRINAASPIRINALa respuesta a las Aspirina presenta gran variabilidad interindividual
Hasta un 45% de los pacientes no responden al tratamiento
Falla la↓TXA2
↑ RENOVACIÓN DE PLAQUETAS (Transfusión, CRM)
↑ SINTESIS EXTRAPLAQUETARIA DE TXA2
INTERACCIONES DEL AAS CON OTROS AINES
PRESENCIA DE POLIMORFISMOS GENETICOSCOX
TXA2 sintasa
+ ↓Absorción, ↑Metabolismo, ↓Adherencia al tratamiento
LA RESISTENCIA A LA ASPIRINA ES UN MARCADOR POTENCIAL DE RIESGO CARDIOVASCULAR
NITRO-ASPIRINA → ↑ONVasodilatador
Antitrombotico

OTROS INHIBIDORES DE COXOTROS INHIBIDORES DE COXT R I F L U S A LT R I F L U S A L
Además de inhibir irreversiblemente a la COX, inhibe la FOSFODIESTERASA de AMPc y GMPc
FOSFODIESTERASA
AMPcGMPc
AMPGMP
Limita la movilización de Ca2+ →→ ↓Agregación plaquetaria
• Inhibe solamente a COX-1 → no afecta la función del endotelio vascular
• Estimula la ON sintasa → ↑ON
Ventajas:Similar potencia antiagregante que el AASMenor riesgo de hemorragia
Administraciónvía oral
Degradación hepáticat1/2 = 30min
Genera metabolito activot1/2 = 40hs
Eliminación porvía renal
+

1. Interferencia con la vía del ácido araquidonico1. Interferencia con la vía del ácido araquidonico
ANTAGONISTAS DE RECEPTORES DE TXAANTAGONISTAS DE RECEPTORES DE TXA22 (R- (R-αα y R- y R-ββ))
TXA2 – RTXA2
PGq
PG12
↑PLC↑DAG y ↑IP3 ↑Ca2+ y ↑PKC
↑Rho
AGREGACIÓNPLAQUETARIA
ACTIVACIÓNPLAQUETARIA
Ventaja respecto al AAS →→Si hay aumentada síntesis extraplaquetaria
de TXA2 (o existen otros agonistas delR-TXA2) el AAS no es eficaz
Previene aterogenesis, disminuye placa ateromatosa y mejora la función endotelial
RECEPTORES DE TXA22
+ + +
T E R U T R O B Á NT E R U T R O B Á N

2. 2. Moduladores del AMPc/GMPcModuladores del AMPc/GMPcAMPc → Inhibidor de la agregación plaquetaria
↑ AMPc → ↓ Ca2+ intracitoplasmático (se secuestra en los gránulos densos)
DOS FORMAS:
AUMENTAR LA PRODUCCIÓN
DE AMPc
DISMINUÍR LA DEGRADACIÓN
DE AMPc
A T P
A M P c
A M P
ADENILATOCICLASA
FOSFODIESTERASA
↑↑↑
↑↑↑
↓↓↓ DIPIRIDAMOL
PROSTACICLINA

2. 2. Moduladores del AMPc/GMPcModuladores del AMPc/GMPc
D I P I R I D A M O LD I P I R I D A M O L
Inhibe la Fosfodiesterasa
Menos eficaz que la Aspirina, aunque potencia
su función
Administración vía oral
Degradación vía hepática
Efectos adversos:Cefalea y diarrea (16%)
P R O S T A C I C L I N AP R O S T A C I C L I N A
Activa la Adenilato Ciclasa+ Vasodilatador
Tiempo de vida media: 2-3minExisten análogos con mayor t1/2
ILPROST (EV), CIPROSTENO (EV),CICAPROST (ORAL)
Mayor uso:Perfusión intravenosa durante la Dialisis Renal
Efectos adversos:Rubor facial, cefalea, bradicardia, hipotensión

ANTAGONISTAS DEL RECEPTOR DE TROMBINAANTAGONISTAS DEL RECEPTOR DE TROMBINA
PAR-1
PAR-4
(PRINCIPAL)
(COMPLEMENTA)
Receptores acoplados
a Proteínas G
Se inicia laactivación plaquetaria
ANTAGONISTAS DE PAR-1 → V O R A P A X A R + A T O P A X A R
• Son proteínas que compiten con la Trombina• Administración vía oral• Metabolización hepática y eliminación por materia fecal
P E P D U C I N A S → Peptidos que inhiben la unión PAR – ProteínaG intraplaquetaria
A P R O T I N I N A → Inhibidor de Serino-proteasas (inhibe la Trombina)
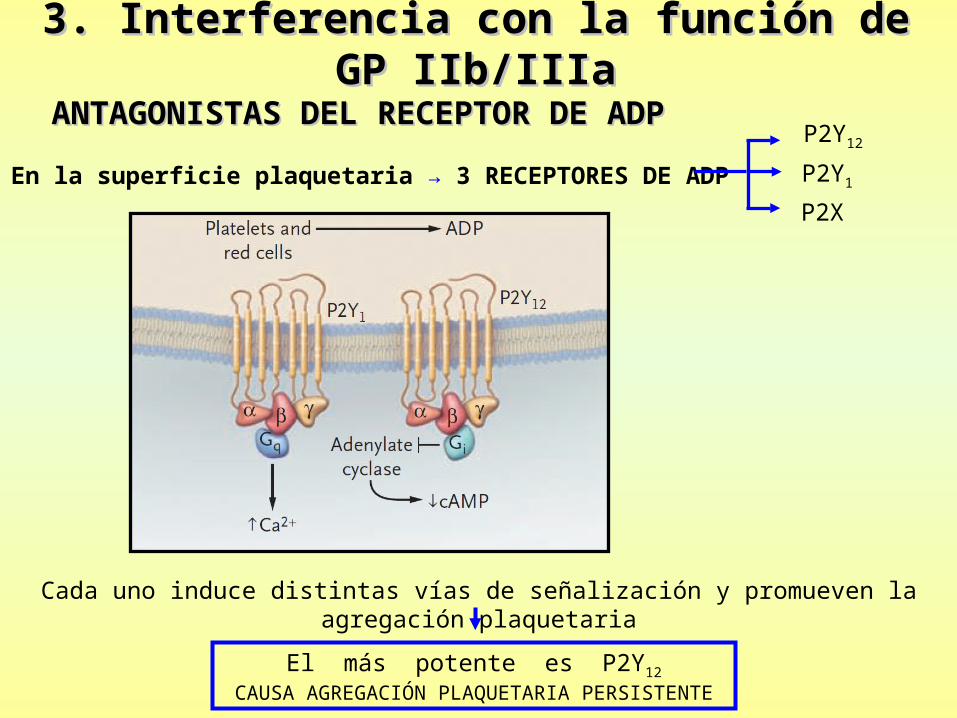
3. Interferencia con la función de GP IIb/IIIa3. Interferencia con la función de GP IIb/IIIa
ANTAGONISTAS DEL RECEPTOR DE ADPANTAGONISTAS DEL RECEPTOR DE ADP
• En la superficie plaquetaria → 3 RECEPTORES DE ADP
P2Y12
P2Y1
P2X
Cada uno induce distintas vías de señalización y promueven la agregación plaquetaria
El más potente es P2Y12
CAUSA AGREGACIÓN PLAQUETARIA PERSISTENTE
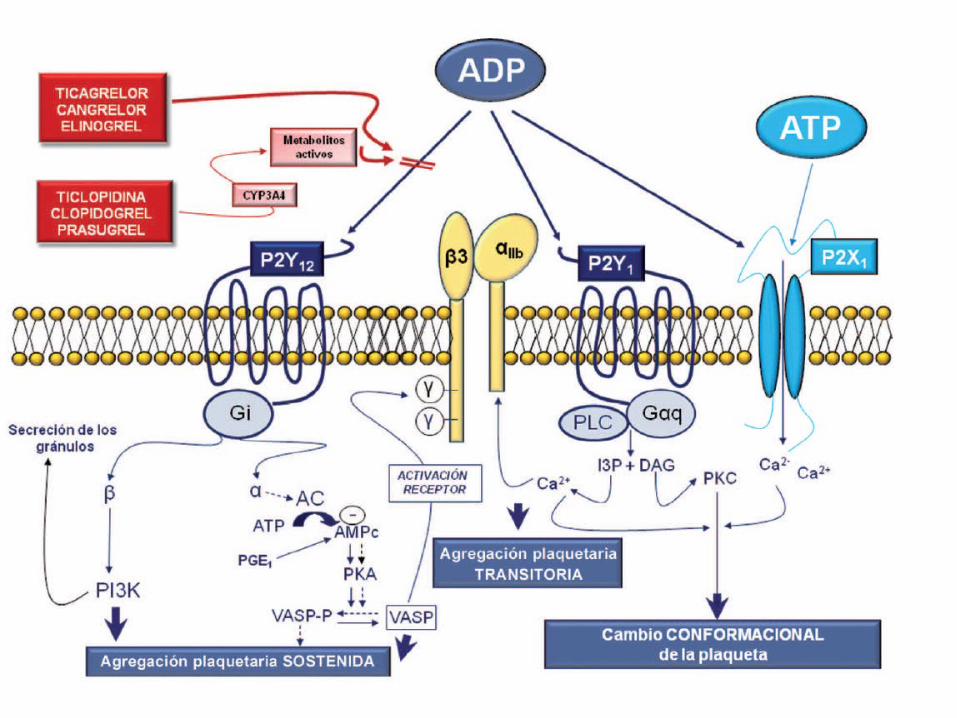

TIENOPIRIDINASTIENOPIRIDINAS• Primeros antagonistas del R-ADP:
TICLOPIDINA
CLOPIDOGREL
PLASUGREL
ANTAGONISTAS DEL RECEPTOR DE ADPANTAGONISTAS DEL RECEPTOR DE ADP
• Inhiben la unión del ADP a su Receptor• Inhiben la unión del fibrinogeno a la GP IIb/IIIa• Suprimen la acción inhibitoria del ADP sobre el AMPc
REDUCEN
Agregación plaquetariaSecreción plaquetariaInteracción leucocito – plaquetaSíntesis de TXA2
• Son PROFÁRMACOS →→se metabolizan a nivel hepático
generando metabolitos ACTIVOS más potentes

ANTAGONISTAS DEL R-ADPANTAGONISTAS DEL R-ADP
T I C L O P I D I N AT I C L O P I D I N A
Inhibidor IRREVERSIBLE del R-ADP P2Y12
• Ventaja: Alternativa terapéutica para pacientes con Resistencia a Aspirina
• Desventajas: Tarda mucho tiempo en aparecer su efecto (3 a 5 días)
Efectos adversos:
Limita su uso en pacientes con SCA
Neutropenia marcada (8%, desaparece al suspenderlo)
Trombocitopenia grave (reversible)
PTT y anemia aplásica
• Vía de administración: vía oral• Tiempo de vida media: 25-40hs

TROMBOCITOPENIA INDUCIDATROMBOCITOPENIA INDUCIDAPOR DROGASPOR DROGAS
• Los síntomas aparecen 1 semana después de haber iniciado el tratamiento
• Se generarían por una reacción autoinmune (aparecen nuevos epitopes)
• Si ya preexistían auto-ac los síntomas aparecen en 1 a 2 días
• Al suspenderse la medicación los síntomas desaparecen en 1 a 2 días
• Incidencia: 10 casos por millón al año
• Drogas asociadas: Heparina Quininas Antibióticos Antiagregantes Analgésicos Diuréticos Sedantes

ANTAGONISTAS DEL R-ADPANTAGONISTAS DEL R-ADPC L O P I D O G R E LC L O P I D O G R E L
Inhibidor IRREVERSIBLE del R-ADP P2Y12
• Ventaja: No tiene tantos efectos adversos como Ticlopidina y es económico
• Desventajas: Tarda mucho tiempo en aparecer su efecto (4 a 5 días)
Efectos adversos:
Limita su uso en pacientes con SCA Neutropenia marcada (0.5%)
• Vía de administración: vía oral• Tiempo de vida media: 8hs• Pico de inhibición: 4-6hs postadministración
Recomendado para prevenir TROMBOSIS LUEGO DE SCA o SHOCK ISQUÉMICO
Dosis inicial de 300mg + dosis diaria de 75mg

ANTAGONISTAS DEL R-ADPANTAGONISTAS DEL R-ADPTICLOPIDINA Y CLOPIDOGRELTICLOPIDINA Y CLOPIDOGREL
Al inhibir irreversiblemente al R-ADP, el efecto tardará en desaparecer lo que tarda en renovarse la plaqueta (5-10 días)
En caso de cirugía → Suspender UNA semana antes
Presentan gran VARIABILIDAD INTRA E INTERINDIVIDUAL ya que dependen de enzimas hepáticas para su metabolización
• Gran heterogenicidad en la ACTIVIDAD de las isoenzimas del Cit. P450
• Compiten con otros fármacos que utilizan esta vía de activación (Ej.: Estatinas)
↓ Biodisponibilidad → Falla del tratamiento “Resistencia al Clopidogrel”
VENTAJA DE LAS TIENOPIRIDINASNo interfieren en el metabolismo del Acido Araquidonico
Pueden COMBINARSECOMBINARSE con ASPIRINA y POTENCIAR EL EFECTO
El beneficio de la terapia combinada sólo se comprueba en pacientes sintomáticos de ALTO RIESGO CARDIOVASCULAR
SCA
Colocaciónde STENT

ANTAGONISTAS DEL R-ADPANTAGONISTAS DEL R-ADPP R A S U G R E LP R A S U G R E L
Inhibidor IRREVERSIBLE del R-ADP P2Y12
• Ventaja: Su ACTIVACIÓN no depende de tantos intermediarios
• Desventaja: Muy costoso
• Vía de administración: vía oral• Tiempo de vida media: 3-4hs• Pico de inhibición: 2-4hs postadministración
Recomendado en pacientes con bajo riesgo de sangrado que van a someterse a INTERVENCIÓN CORONARIA PERCUTÁNEA
MENOR VARIABILIDAD INTERINDIVIDUAL+ Es más potente → requiere menores dosis+ Es más rápido → útil en casos que requieren inhibición plaquetaria rápida

NUEVOS ANTAGONISTAS DEL R-ADPNUEVOS ANTAGONISTAS DEL R-ADPT I C A G R E L O RT I C A G R E L O R
Inhibidor REVERSIBLE del R-ADP P2Y12
• Vía de administración: vía oral• Tiempo de vida media: 12hs• Pico de inhibición: 1,5hs postadministración
• Ventajas:
• Desventaja: Se asocia a tiempos de sangrado más prolongados
Recomendado para PREVENCIÓN de EVENTOS TROMBOTICOS en pacientes con SCA
Efecto más rápido y más potente que ClopidogrelMenor variabilidad biológica que Clopidogrel
El efecto se REVIERTE 24hs después de la última dosisVía de excreción: Bilis/Heces (independiente del riñón!)
Asociado a disnea (dosis dependiente)

NUEVOS ANTAGONISTAS DEL R-ADPNUEVOS ANTAGONISTAS DEL R-ADPC A N G R E L O RC A N G R E L O R
Inhibidor REVERSIBLE del R-ADP P2Y12
• Vía de administración: endovenosa• Tiempo de vida media: < 10 minutos (2 a 5 min)
• Ventajas:
• Desventaja: Se asocia a tiempos de sangrado menor más prolongados
Inhibe >90% la agregación plaquetaria a pocos minutos de su administración
Menor variabilidad biológica que Clopidogrel
El efecto se REVIERTE CASI TOTALMENTE 1-2 hs postinfusiónExcelente perfil de seguridad
RÁPIDAMENTE INACTIVADO POR ECTONUCLEASAS
Ideal para pacientes que requieren ser sometidos a procedimientos invasivos
CUIDADO con el SHIFT entre CANGRELOR y OTRO ANTIAGREGANTE
Cangrelor compite con Clopidogrel y PrasugrelPueden aparecer períodos de baja inhibición plaquetaria durante el shift
↑↑↑ riesgo de trombosis!

3. Interferencia con la función de GP IIb/IIIa3. Interferencia con la función de GP IIb/IIIa
ANTAGONISTAS LA GLICOPROTEÍNA IIb/IIIaANTAGONISTAS LA GLICOPROTEÍNA IIb/IIIa
GP IIb/IIIa reconoce una misma secuencia de aminoácidos que comparten varias proteínas que
actúan por distintas vías
• FIBRINOGENO• VITRONECTINA• F. VON WILLEBRAND• FIBRONECTINA
Principal (mayor cantidad de secuencias de aa)
Al inhibir la GP IIb/IIIa se bloquea la ETAPA FINAL del proceso trombótico
INDEPENDIENTEMENTE DE CUÁL ES EL ACTIVADOR INICIAL DEL PROCESO

3. Interferencia con la función de GP IIb/IIIa3. Interferencia con la función de GP IIb/IIIa
ANTAGONISTAS LA GLICOPROTEÍNA IIb/IIIaANTAGONISTAS LA GLICOPROTEÍNA IIb/IIIa
• A B C I X I M A B → Es un anticuerpo monoclonal anti-GPIIb/IIIa
Inhibe IRREVERSIBLEMENTE la GP IIb/IIIa
• E P T I F I B A T I D E → Es un Peptido ciclico
Inhibe REVERSIBLEMENTE la GP IIb/IIIa
• T I R O F I B A N → Es una molécula no peptídica
Inhibe REVERSIBLEMENTE la GP IIb/IIIa
Administración: siempre por vía endovenosa Efectos adversos: asociados a eventos hemorrágicos + trombocitopenia (1-2%)
Abciximab tiene capacidad inmunogénica (2-5%)
Se utilizan para TRATAR SCA combinados con ASPIRINA+ En pacientes que se someten a Intervención coronaria percutánea (↓ riesgo trombótico)


M U C H A SM U C H A S
G R A C I A S ! ! G R A C I A S ! ! !!

BibliografíaBibliografía• Jackson S. The growing complexity of platelet aggregation. The American Society of Hematology,
Blood, Volume 109, number 12, Junio 2007: 5087-5095.• Blanco A, Kordich L. Fundamentos para el manejo práctico en el Laboratorio de Hemostasia.
Grupo CAHT (Grupo Cooperativo Argentino de Hemostasia y Trombosis). Segunda Edición.• Badimon L, Vilahur G. Mecanísmos de acción de los diferentes agentes antiplaquetarios.
Revista Española de Cardiología, 2013: 8-15.• Palomo I, Torres C. Antiagregantes plaquetarios: mecanísmos de acción y riesgos asociados al
uso. Vitae, revista de la facultad de química farmacéutica, Volumen 16, número 1, 2009.• Raju N, Eikelboom J. Platelet ADP-receptor antagonists for cardiovascular disease: past,
present and future. Nature clinical practice, Volumen 5, número 12, Diciembre 2008.• Dullewe Y. Anti-platelet therapy: ADP receptor antagonists. British Journal of clinical
Pharmacology. 72:4, 647-657, Abril 2011.• Sánchez D. Protocolos Tratamiento Antiagregante. Sociedad Española de Medicina Interna.• Giovanni D, Patrono C. Platelet activation and atherothrombosis. The New England Journal of
Medicine. 357:24. Diciembre 2007.• Aster R, Bougie D. Drug induced immune thrombocytopenia. The New England Journal of
Medicine. 357:6. Agosto 2007.• Baron T, Kamath P. Management of Antithrombotic therapy in patients undergoing invasive
procedures. The New England Journal of Medicine. 368:22. Mayo 2013.