333 presentación de casos - acta médica...
TRANSCRIPT

333 Presentación de Casos
INHIBIDOR HEPARINOIDE EN UN PACIENTE CON MIELOMA MULTIPLE Se describe una paciente con mieloma múltiple tipo IgA
con un inhibidor de la coagulación de tipo heparinoide, no inmune ni relacionado con la paraproteína. Fue identi-ficado por pruebas de neutralización con sulfato de pro-tamina, con una concentración plasmática menor de 0.3 U/mL. Se discute la naturaleza de este tipo de inhibidores y su relación con las neoplasias.
INTRODUCCION Las enfermedades neoplásicas raramente se asocian con
anormalidades de la coagulación, por lo común mediadas por inhibidores circulantes como inmunoglobulina, que se unen específicamente ya sea a factores individuales de la coagula-ción o a los fosfolípidos, o que interfieren en forma inespecí-fica con la polimerización de la fibrina o con la agregación plaquetaria (1, 2). Las disproteinemias y entre ellas específi-camente el mieloma múltiple, se asocian con defectos de los mecanismos hemostáticos mediados por la interacción direc-ta de la paraproteína con un factor de la coagulopatía, o indi-rectamente por interferencia con la polimerización de los monómeros de fibrina (3,4); además, se describe una coagu-lación en el mieloma múltiple, no directamente relacionada con la paraproteína, sino caracterizada por la presencia en la circulación de proteoglicanos con propiedades similares a la heparina (5, 6).
En este artículo describimos una paciente con mieloma múltiple tipo IgA, asociado con alteración del tiempo de trombina (T.T.) mediada por un inhibidor heparinoide no inmune, no relacionado con la paraproteína, fácilmente neu-tralizado por la protamina y sin repercusiones clínicas.
Presentación del caso H.J.G., paciente de sexo femenino, de 58 años de edad,
natural del área rural de Cocorná (Antioquia), procedente del área rural de Puerto Triunfo (Antioquia), casada, 17 hijos, ocupación oficios domésticos, con historia de cinco meses de evolución de su enfermedad, con acentuación del cuadro clí-nico dos meses antes del ingreso, consistente inicialmente en dolor en región lumbar, incapacitante, intensificado con los cambios de posición. Sin historia de fenómenos hemorrági-cos, trombóticos ni de tratamiento con heparina o algún otro tipo de anticoagulante.
Al examen físico, paciente consciente, orientada, normo-tensa, hidratada, pulso regular 100/min, TA 120/70. Existía dolor a la presión de arcos costales inferoanteriores izquierdos. La rotación del tronco despierta dolor intenso en la región lumbar. La percusión del cráneo y de la columna lumbar no causa dolor. Hiperreflexia osteotendinosa simétrica en miembros inferiores. Discreta distensión abdominal. El resto del examen físico es satisfactorio.
Estudio diagnóstico conclusivo de mieloma múltiple tipo
Acta Médica Colombiana Vol 14 N°5 - Septiembre-Octubre - 1989
inmunoglobulina A, estado clínico II A, según protocolo Col-ciencias No. 1115-05-070-86.
Hemograma con hemoglobina de 12.6 g/L, hematocrito 0.37; reticulocitos corregidos de 1.7%; volumen corpuscular medio 100 fL; concentración de hemoglobulina corpuscular media de 340 g/L. Leucocitos de 3.8 109/L, con 0.62 de gra-nulocitos, 0.09 de eosinófilos; 0.01 de basófilos, linfocitos 0.22 y monocitos 0.06. Sedimentación 91 mm/h. Química sanguí-nea normal. Parcial de orina normal y proteína de Bence Jo-nes negativa.
La electroforesis de proteínas séricas muestra pico mono-clonal que migra en las bandas Alfa 2-Beta de 4.78 g/L. Al-búmina sérica normal (46.8 g/L). Gammaglobulinas dismi-nuidas (0.7), Cuantificación de inmunoglobulinas: IgG 5.77 g/L (vn: 6.00- 18.00 g/L); IgA 0.28 g/L (vn: 1.00-4.60 g/L); IgM 0.54 g/L(vn: 0.50 - 3.50 g/L). Viscosidad sérica relativa al agua 2.55 (N: 1.7-2.14).
Mielograma esternal, conclusivo de mieloma múltiple con iñfiltración de 52% de plasmocitos maduros, y disminución de las demás líneas celulares. Ausencia de mastocitos. Estu-dio radiográfico óseo: deformidad en cuña de los cuerpos vertebrales de DIV, DVI, DVIII, LI-II-III con severa desmi-neralización ósea.
Gammagrafía ósea total con 99 mTc. muestra acúmulos anormales del radiotrazador, situados a nivel de arcos anteriores de las últimas costillas derechas, así como del arco anterior de la cuarta y sexta costillas izquierdas. Además zonas fotopénicas diseminadas en estas costillas. A nivel de columna, hipercaptación en T6, 7 y 8 y L2.
Los estudios de coagulación, excepto T.T., se pueden ver en las Tablas 1 y 2.
METODOS El estudio de tiempos de protrombina (T.P.) y trombo-
plastina parcial (T-P-T.) se llevó a cabo por métodos manua-les y de observación del coágulo en tubo de vidrio, según descripción hecha en revisión anterior (7). Para el tiempo de trombina (T.T.) se empleó trombina humana no comercial, con actividad de 1000 W/mL, diluida en solución salina 0.15M para producir un T .T. de plasma normal entre 17 y 20 segundos.

Presentación de casos
Se obtuvo plasma pobre en plaquetas mediante centrifu-gación de sangre completa citratada (una parte de citrato de sodio al 3.8%, por nueve partes de sangre completa) a 3600 rpm durante 15 minutos. Plasma pobre en plaquetas de la paciente y de un grupo normal para correcciones del T.T. Los métodos de corrección del T.T. con plasma normal (P.N.) fueron descritos por Pizzuto y col (8), con una única modifi-cación respecto a la adición de plasma pobre en plaquetas, con la finalidad de evitar en lo posible efectos antiheparínicos del factor plaquetario 4 (F.P4) que se libera al ser activada la plaqueta por acción de la trombina (9), con la consecuente normalización del T.T. en un plasma con sospecha de anti-coagulante héparinoide.
Prueba de tamizaje para inhibidores de las proteínas de la coagulación según método descrito por Godwin y Roberts (14). Para la prueba de neutralización con protamina, se empleó sulfato de protamina comercial según método descrito por Peener, Bowie y Owen (10, 11).
RESULTADOS La adición de solución salina en diferentes proporciones
al plasma del paciente, corrige el T.T. en la dilución 1:4; esto indica la presencia de un inhibidor circulante que al ser di-luido pierde actividad y que, al agregar protamina en la prue-ba basal, se neutraliza en 50%, completándose su inactiva-ción con la adición de protamina a las diluciones de solución salina (Tabla 3). Este efecto habla de un inhibidor de tipo heparinoide y no es el efecto esperado en casos de disfibrino-genemia, paraproteinemia o interferencia en la polimerización de la fibrina por productos de degradación de la fibrina (12,
13); se descarta además déficit asociado de fibrinógeno dado el comportamiento que sigue el T.T. tras las diluciones seria-das con solución salina (7).
La corrección significativa del T.T. con la adición de plas-ma normal en proporciones de 1:2 y 1:4 (Tabla 4), descarta un inhibidor del tipo inmunoglobulina tanto para la trombina como para la formación de fibrina (14) y corrobora aún más la presencia de un heparinoide circulante que, al unirse ines-pecíficamente con otros polianiones del plasma adicionado (fibrinógeno, B-lipoproteínas, fibronectina, gammaglobulinas) disminuye su efecto enzimàtico hasta su neutralización (15); además, aunque sería de esperar la potenciación del efecto al agregar plasma normal (por la adición de antitrombina III), los heparinoides en sistemas in vitro no forman complejos con actividad antitrombínica (15). In vivo, su efecto se supone diferente por la interacción con estructuras vasculares de las que proceden, con plaquetas, con productos derivados del metabolismo del ácido araquidónico, heparinasas, etc. (16,17).
La corrección del T.T. al agregar plasma normal en la pro-porción 1:4, habla de una concentración de heparinoide por debajo de 0.3 U/mL (8), que explica la falta de complicacio-nes clínicas. Otros resultados obtenidos, una ligera prolonga-ción del tiempo de sangría con un recuento de plaquetas nor-mal, se podría explicar por efecto de la paraproteína mielo-matosa.
DISCUSION La actividad anticoagulante presente en el plasma de esta
paciente habla de un anticoagulante heparinoide asociado al mieloma múltiple inmunoglobulina A, que se caracterizó como no inmune y no relacionado con la paraproteína; probable-mente dependiente de proteoglicanos libres en la circulación.
El ácido hialurónico, condroitín sulfato y heparitín sulfato son polisacáridos compuestos de aminoazúcares y de ácidos urónicos y han sido clasificados junto con la heparina como mucopolisacáridos o glicosaminoglicanos, formando la mo-lécula carbohidratada de los proteoglicanos (15). Los proteo-glicanos son componentes estructurales importantes de las arterias elásticas y junto con su función de mantener la inte-gridad estructural, las propiedades viscoelásticas y la per-meabilidad macromolecular, varios proteoglicanos inhiben la

Presentación de casos 335
agregación plaquetaria inducida por trombina y las funciones del factor Xa en la coagulación (6).
La aparición de actividad antitrombínica, dependiente de proteoglicanos (actividad heparinoide) en sangre, debe dife-renciarse de los inhibidores inmunes que pueden aparecer en las disproteinemias, mediante las pruebas de tamizaje para inhibidores (14). Las razones para la liberación de heparinoi-des a la circulación en cantidades que alteren los mecanismos de coagulación, siguen siendo oscuras, pero por su presenta-ción en entidades neoplásicas acompañadas de algún tipo de invasión tumoral (daño vascular relacionado con la enferme-dad), sugiere que el heparinoide provenga de los proteoglica-nos constituyentes de las paredes vasculares. Otra posibilidad es que el inhibidor heparinoide represente una sustancia fetal o embrionaria que en el adulto sea expresada en ciertos des-órdenes malignos (18).
Aunque en nuestra paciente no hubo clínicamente sangra-do relacionado con la actividad heparinoide del plasma, pro-bablemente por la baja concentración del inhibidor (menos de 0.3 U/mL), hay descritos casos de sangrado fatal (6).
SUMMARY A 58 years-old woman with IgA multiple myeloma and
heparin-like non-immune anticoagulant is reported. The anti-coagulant was not related with the paraprotein and was iden-tified by a heparin neutralizing protamine sulfate assay; its plasma concentration of less than 0.3 U/ml did not have clini-cal manifestations. The relation of this anticoagulant with malignant tumors is discussed.
O. MARTINEZ F. CUELLAR L. ALVAREZ
REFERENCIAS 1. Margol ius J R , Jackson DP, Ra tnoff OD. Circulating anticoagulants:
a study of 40 cases and a review of the literature. Medicine (Baltimore) 1961; 40:145-201.
2. Harris EN, Phil M, Asherson RA, Hughes GRV. Antiphospholipid antibodies - autoantibodies with a difference. Ann Rev Med 1988; 39: 261-271.
3. Perkins HA, Machenz ie M R , Fudenberg H. Hemostatic defects in dysproteinemias. Blood 1970; 35:695-707.
4 . Coleman M, V igl iano EM, Weksler ME, Nackman RL. Inhibition of fibrin monomer polymerization by lambda myeloma globulins. Blood 1972; 39 :210 .
5 . Khoory MS, Meshe im ME, Bowie W, Mann K. Circulating heparin sulfate proteoglycan anticoagulant from a patient with a plasma cell disorder. J Clin Invest 1980; 65: 666-674.
6 . P a l m e r RN, Rick M E , Rick P, Zeller J A , Gra lnick HR. Circulating heparin sulfate anticoagulant in a patient with a fatal bleeding disorder. N Eng J Med 1984; 26:1696-1699.
7. Cuéllar F, Lozano J, Sarmiento JJ, et al. Protocolo para el estudio de las hemofilias A y B en Medellín. Acta Med Col 1985; 10: 192-196.
8. Pizzuto J , García-Méndez S, De la Paz Reina M, et a l . Thrombin time dilution test: a simple method for the control of heparin therapy. Thromb Haemost (Stuttgart) 1972; 42:1276-1285.
9. Harr i son R, Binnote R. The thrombin clotting time. Am J Clin Pathol 1988; 89 :81-87.
10. Peener J A. Experience with a thrombin clotting time assay for measur-ing heparin activity. Ann J Clin Pathol 1974; 61: 645- 653.
11. Bowie EJW, Owen ChA. The clinical and laboratory diagnosis of hem-orrhagic disorders. In: Ratnoff OD, Forbes ChD. Disorders of hemosta-sis. Orlando, Florida: Grune & Stratton Inc. 1984: 43-72.
12. Flute PT. Disorders of plasma fibrinogen synthesis. Br Med Bull 1977; 33 : 253-259.
13. Ratnoff OD, Forman WB.Criteria for the differentiation of dysfibrino-genemic states. Sem Hematol 1976; 13: 141-157.
14. Godwin J, Roberts R. Immunology of acquired inhitors to clotting pro-teins. In: Friedman H, Fahey JL. Immunology of acquired inhitors to clotting proteins. Washington: Eds. Noel Rose 1986: 635-649.
15. Jacques LB. Heparins-anionic polyelectrolyte drugs. Pharmac Rev 1980; 31 :99-166.
16. Ts'Ao Hh-H, Eisentein R, Schumacher B. Effect of an aortic proteogly-can on platelet aggregation an thrombin time: plasma requirement and active moieties. Proc Soc Exp Biol Med 1977; 156:162-167.
17. Kjel len L, Peterson I, Hook M. Cell surface heparin sulfate: an intaca-lated membrane proteoglycan. Proc Natl Acad Sci USA 1981; 78: 5371-5375.
18. Rodgers G M , Corash L. Acquires heparin like anticoagulant in a pa-tient with metasiasic breast carcinoma. West J Med 1985; 143: 672-675.
Dr. Octavio Martínez Betancur: Residente de Hematología del Hospital Militar Central, Bogotá; Dr. Francisco Cuéllar Ambrosi: Jefe Sección de He-matología, Departamento de Medicina Interna, Facultad de Medicina, H.U.S.V.P., Medellín; Dra. Leonor Alvarez Peláez: Profesora Hematología, Departamento de Medicina Interna, Facultad de Medicina, H.U.S.V.P., Medellín. Autor responsable: Francisco Cuéllar Ambrosi. A. A. 55953, Medellín, Colom-bia. Trabajo auspiciado por Colciencias a través del Programa Protocolizado de Hematología 1115 - 05 - 070 - 86.
HIPOVENTILACION ALVEOLAR PRIMARIA (¿Enfermedad de Monge en Bogotá?)
Se presentan dos casos de hipoventilación alveolar sin enfermedad pulmonar ni apneas de sueño, secundarias a trastornos no anatómicos del sistema de control de la res-piración. Se discuten los diagnósticos diferenciales y se plantea la hipótesis que su etiología es la hipoventilación crónica de la altura o enfermedad de Monge.
INTRODUCCION El término hipoventilación alveolar es sinónimo de hiper-
capnia y es frecuentemente identificado en la práctica clínica, acompañando a diversas enfermedades pulmonares, especial-mente la bronquitis crónica.
Sin embargo, hay un grupo de enfermedades que se carac-terizan por hipoventilación alveolar en personas con pulmo-nes normales. Estos pacientes se identifican por la ausencia de síntomas y signos de enfermedad pulmonar, acompañados
Acta Med Colomb Vol 14 N°5 - 1989
336 Presentación de casos
de hipercapnia e hipoxemia (que inicialmente se corresponde milímetro a milímetro de Hg con el aumento del CO 2 ) , dife-rencia (A-a) O2 normal, y espirometría normal. Como sus pulmones son normales, cuando hiperventilan voluntariamente son capaces de corregir la retención de C O 2 y normalizar el PaO 2 . Como consecuencia de la hipercapnia e hipoxemia aparecen eritrocitosis, hipertensión pulmonar y cor pulmona-res (1,2).
Caso 1 Mujer de 49 años, natural y procedente de Bogotá, con
historia de hipertensión arterial y disnea de varios años de evolución. Fue atendida en el Hospital Simón Bolívar por insuficiencia cardíaca derecha, hipertensión pulmonar y cor pulmonale de etiología no definida, por lo cual fue estudiada en la Fundación Santa Fe de Bogotá.
La radiografía de tórax mostró signos de hipertensión pul-monar arteriolar, con crecimiento ventricular derecho, sin anomalías del parénquima pulmonar ni de los huesos. Elec-trocardiograma: crecimiento y sobrecarga de cavidades dere-chas. La espirometría fue normal y los gases arteriales mos-traron acidosis respiratoria crónica con hipoxemia que se co-rrigen con hiperventilación voluntaria (Tabla 1). Química sanguínea, T3,T4,TSH: normales; hemoglobina 180 g/L; Hct:
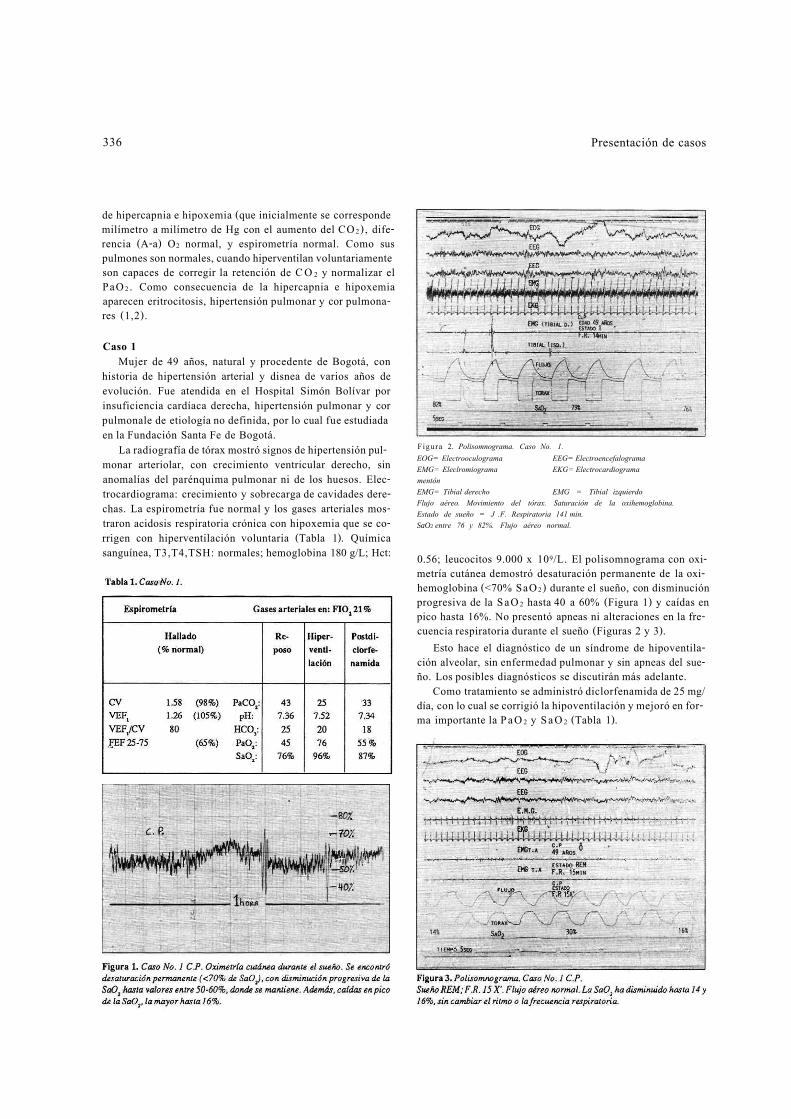
Figura 2. Polisomnograma. Caso No. 1. EOG= Electrooculograma EEG= Electroencefalograma EMG= Eleclromiograma EKG= Electrocardiograma mentón EMG= Tibial derecho EMG = Tibial izquierdo Flujo aéreo. Movimiento del tórax. Saturación de la oxihemoglobina. Estado de sueño = J .F. Respiratoria 141 min. SaO2 entre 76 y 82%. Flujo aéreo normal.
0.56; leucocitos 9.000 x 109/L. El polisomnograma con oxi-metría cutánea demostró desaturación permanente de la oxi-hemoglobina (<70% SaO 2 ) durante el sueño, con disminución progresiva de la S a O 2 hasta 40 a 60% (Figura 1) y caídas en pico hasta 16%. No presentó apneas ni alteraciones en la fre-cuencia respiratoria durante el sueño (Figuras 2 y 3).
Esto hace el diagnóstico de un síndrome de hipoventila-ción alveolar, sin enfermedad pulmonar y sin apneas del sue-ño. Los posibles diagnósticos se discutirán más adelante.
Como tratamiento se administró diclorfenamida de 25 mg/ día, con lo cual se corrigió la hipoventilación y mejoró en for-ma importante la P a O 2 y S a O 2 (Tabla 1).

337
Caso 2 Hombre de 52 años, natural y procedente de Bogotá, sin
historia de enfermedad cardiopulmonar, en quien se encon-traron en un examen de rutina leucocitos 7.580 x 109 /L, hemo-globina de 180 g/L, que había aumentado en los últimos tres años desde una hemoglobina de 140 g/L, El examen clínico, la radiografía del tórax y electrocardiograma son normales. La espirometría fue normal. Los gases arteriales mostraron acidosis respiratoria e hipoxemia moderada, que se corrigieron con la hiperventilación voluntaria (Tabla 2).
Tabla 2. Caso No. 2.
La oximetría cutánea durante el sueño muestra desatura-ción constante de la oxihemoglobina entre 60-70%. En este paciente no se realizó polisomnograma, pero la ausencia de caídas de la SaO2 durante el sueño descarta la presencia de apneas (Figura 4).
El cuadro corresponde igualmente a un síndrome de hipo-ventilación alveolar sin enfermedad pulmonar y sin apneas de sueño. Se formuló diclorfenamida de 25 mg/día, con me-joría de la hipoventilación y corrección de la hipoxemia (Ta-bla 2).
DISCUSION Uno de los objetivos básicos del sistema respiratorio es la
eliminación de CO2 mediante la ventilación alveolar. Siem-pre que se encuentra la presión arterial de CO2 (PaCO2) ele-vada, hay hipoventilación alveolar. Como consecuencia de esto se produce acidosis respiratoria, aumento del bicarbonato sérico, hipoxemia, desaturación de la oxihemoglobina y eri-trocitosis. La hipoxemia potenciada por la acidosis respirato-ria, lleva a hipertensión pulmonar y cor pulmonale (1,2).
Las causas de hipoventilación alveolar se clasifican así (1, 2, 3,4):
1. Hipoventilación alveolar por enfermedad pulmonar. a) EPOC. b) Asma. c) Fibrosis pulmonar avanzada. 2. Alteraciones mecánicas del aparato respiratorio. a) Enfermedades neuromusculares. b) Deformidad de la pared del tórax. c) Obstrucción mecánica de las vías aéreas superiores. 3. Hipoventilación alveolar por trastorno en el sistema de
control de la ventilación. a) Alteración funcional de los centros respiratorios. a.l.) Trastornos del sueño. a.2.) Trastornos metabólicos. a.3.) Drogas. a.4.) Disminución en la sensibilidad de los quimiorrecep-
tores. b) Alteración anatómica de los centros respiratorios. 4. Mixtas.
1. Hipoventilación alveolar con enfermedad pulmonar En este grupo se encuentra la enfermedad pulmonar obs-
tructiva crónica y los estados avanzados de fibrosis pulmo-nar. Son pacientes fáciles de identificar con los hallazgos clí-nicos y con la espirometría.
La aparición de hipoventilación alveolar en estos pacien-tes se relaciona con la severidad de la obstrucción y la inte-gridad del sistema del control de la ventilación. Así, si los mecanismos de control de la ventilación son normales no se presentará hipoventilación alveolar hasta tanto el VEF1 no sea menos de un litro, o menos de 40% de lo normal. Además, si la causa de la hipoventilación es la enfermedad pulmonar intrínseca o alteración mecánica, el paciente es incapaz de disminuir más de 20% de la PaCO2 durante la hiperventilación voluntaria (1, 2, 5).
La historia clínica, el examen físico, los resultados de es-pirometría y los gases arteriales, permiten excluir de este gru-po de enfermedades a nuestros dos pacientes. 2. Alteraciones mecánicas del aparato respiratorio
En este grupo se incluyen las enfermedades neuromuscu-lares (poliomielitis, miastenia, esclerosis lateral, Guillain-Barré, distrofias musculares, etc.), las enfermedades de la pared del tórax (cifoescoliosis) y la obstrucción mecánica de las vías aéreas superiores (estenosis de tráquea, etc.).
La historia clínica y el examen físico son generalmente
Acta Med Colomb Vol 14 N°5 ~ 1989

338 Presentación de casos
suficientes para sospechar estos diagnósticos. Además, antes de causar hipoventilación crónica, se encuentran alteraciones en la espirometría, los volúmenes pulmonares o disminución en la fuerza muscular (6). En los casos presentados, la historia clínica, el examen físico y la espirometría con VVM normal prácticamente excluyen estas posibilidades. En ambos casos la presión inspiratoria máxima fue normal. 3. Hipoventilación alveolar por trastorno en el sistema de control de la ventilación
a) Alteración funcional de los centros respiratorios: en este grupo se incluyen las siguientes enfermedades:
a.l.) Trastornos del sueño: apneas obstructivas, centrales o mixtas; hipopneas y trastornos del ritmo y la frecuencia res-piratoria.
Estos pacientes se estudian con polisomnograma que re-gistra durante el sueño los movimientos del tórax, el abdo-men y movimientos oculares; electromiografía del mentón; electroencefalograma; flujo aéreo y saturación de O2 transcu-táneo. Es característico que durante el sueño se encuentren disminuciones en la SaO 2 mayores de 4%, que aparecen y se corrigen en forma intermitente (3, 7, 8).
Este valor de 4% es aceptado y creo que puede ser útil a nivel del mar, donde debido a la mayor PaO 2 , pequeños cam-bios en la PaO 2 durante el sueño no causan variaciones im-portantes en la saturación arterial de la oxihemoglobina (S aO2). En Bogotá, donde la P aO 2 normal es 60 mmHg, las variacio-nes normales durante el sueño (3 a 10 mmHg) pueden produ-cir mayor disminución en la SaO 2 sin ser patológicas; esto no ha sido estudiado en la altura (9). En el segundo caso presen-tado, no se encuentran alteraciones en la SaO 2 durante el sue-ño, lo cual excluye a este paciente de este grupo de enferme-dades.
El primer caso presenta diferentes episodios de desatura-ción durante el sueño; sin embargo, en el polisomnograma no se demostraron apneas ni trastornos en la frecuencia y el rit-mo respiratorio. La única explicación para la variación de la S aO 2 durante el sueño en esta paciente es una disminución en la ventilación minuto, que produciría hipoventilación alveo-lar, retención de CO 2 e hipoxemia secundaria (8, 11).
a.2.) Trastornos metabólicos: alcalosis metabólica, hipo-tiroidismo. En los dos casos las pruebas de función tiroidea fueron normales y los gases arteriales no muestran alcalosis metabólica sino acidosis respiratoria crónica, con aumento secundario del bicarbonato.
a.-3.) Drogas que deprimen los centros respiratorios: se-dantes, narcóticos, etc.; ninguno de los pacientes recibía estas drogas.
a.4.) Disminución en la sensibilidad de los quimiorrecep-tores: puede ser por disfunción de los cuerpos carotídeos (hipoxemia crónica o cirugía) o por disminución en la res-puesta al CO 2 . En este grupo se incluye la hipoventilación alveolar primaria y la hipoventilación crónica de la altura (enfermedad de Monge). En los dos casos presentados no se midió la respuesta ventilatoria a la hipoxia o a la hipercapnia,
pero se podrían incluir en este grupo. b) Depresión de los centros respiratorios por alteración
anatómica; en este grupo se encuentran las siguientes enfer-medades: encefalitis, poliomielitis bulbar, infartos o hemo-rragias del SNC, enfermedades desmielinizantes, tumores del SNC y médula. Los dos casos presentados no tienen alteraciones al examen neurológico, por lo cual se pueden excluir de este grupo de enfermedades. 4. Causas mixtas de hipoventilación alveolar
a) Hipoventilación alveolar secundaria a alteraciones en el sistema de control de la ventilación y a enfermedades pul-monares (algunos casos de bronquitis crónica).
b) Hipoventilación alveolar secundaria a alteraciones en el control de la ventilación y alteraciones mecánicas del apa-rato respiratorio (apnea obstructiva durante el sueño, síndro-me de obesidad-hipoventilación).
Los casos presentados no corresponden a bronquitis cró-nicas, no tienen apneas durante el sueño, ni son obesos; por lo tanto, se pueden excluir de este grupo.
Revisando los diagnósticos diferenciales, los casos pre-sentados se podrían clasificar dentro del grupo de hipoventi-lación alveolar secundaria a trastornos en el control de la ven-tilación por alteración funcional, por disminución en la sensi-bilidad de los quimiorreceptores; y deben corresponder a hipoventilación alveolar primaria o hipoventilación crónica de la altura (enfermedad de Monge).
El diagnóstico de hipoventilación primaria se ha reservado para aquellos pacientes con hipercapnia e hipoxemia sin en-fermedades neurológicas, musculares, pulmonares, ni de la pared del tórax (1,2,5,10). Estos pacientes corresponden a la definición de hipoventilación alveolar primaria; sin embargo, la frecuencia con que estamos identificando este tipo de pa-cientes nos hace pensar en otro diagnóstico diferencial que sería la enfermedad de Monge o hipoventilación crónica de la altura (12-15). La forma de aclarar esto sería llevando estos pacientes a vivir a nivel del mar y observando su evolución, que debe demostrar normalización de la hipercapnia y de la hipoxemia con disminución de la hemoglobina. Sin embargo, esto no ha sido posible en ninguno de nuestros pacientes.
En otros casos no publicados, al igual que en estos ejem-plos presentados, se pueden identificar dos tipos de pacien-tes: uno, con cuadro clínico severo (mayor hipecapnia, mayor hipoxemia, más hipertensión pulmonar en las radiografías del tórax y electrocardiogramas), y que presentan severa dismi-nución de la S aO 2 durante el sueño, pero sin apneas. El otro grupo tiene menos hipoventilación, menos hipoxemia y no tiene signos clínicos, radiológicos, ni electrocardiográficos de hipertensión pulmonar; además, la S aO 2 durante el sueño permanece estable, aunque en valores menores de lo normal.
El primer tipo de pacientes podría corresponder a una for-ma de hipopneas durante el sueño que cause hipoventilación e hipoxemia secundaria (8,16), sin cambiar la frecuencia res-piratoria, por disminución en el volumen comente. Sin em-bargo, con la tecnología con que contamos actualmente no

Presentación de casos 339
podemos medir el volumen durante el sueño para confirmar esta hipótesis.
Los pacientes que no presentan disminución en la S a O 2
durante el sueño, pienso que pueden corresponder a casos de hipoventilación alveolar crónica de la altura o enfermedad de Monge.
Los pacientes con alteración en la S aO 2 durante el sueño, sin apneas y en este caso sin cambios en la frecuencia y ritmo respiratorio, podrían corresponder simplemente a pacientes con hipopneas que con la tecnología actual no podemos de-tectar; o también ser el estado más avanzado de la enferme-dad de Monge. En este caso, una posible secuencia fisiopato-lógica sería: hipoventilación crónica en la altura por disminu-ción en la respuesta ventilatoria a la hipoxemia, y secundario a esto hipercapnia, aumento del HCO3 y mayor hipoxemia. Con el tiempo se produciría mayor disminución en la sensibilidad de los mecanismos de control de la ventilación con lo cual se perdería el control durante el sueño apareciendo mayor hipoventilación nocturna e hipoxemia con eritrocitosis, hipertensión pulmonar y cor pulmonale (9, 17,18).
Como tratamiento se dio diclorfenamida, un diurético in-hibidor de la anhidrosa carbónica con el fin de aumentar la excreción del bicarbonato y producir acidosis metabólica es-timulando los centros respiratorios. El control de gases arte-riales después de iniciado el tratamiento muestra disminución de la PaCO 2 hasta valores normales y mejoría importante en la P a O 2 (9,19,20).
Esto demuestra que no existe una causa en el pulmón ni en las vías neuromusculares para la hipoventilación ya que el solo estímulo químico la corrige. Esto está de acuerdo con la hipó-tesis de hipoventilación sin enfermedad pulmonar secundaria a trastorno funcional y no anatómico en el control de la venti-lación; es decir, hipoventilación alveolar primaria, o hipo-ventilación crónica de la altura (enfermedad de Monge).
La publicación de estos dos casos tiene como objeto pre-sentar la hipótesis de que en Bogotá, a 2.640 m sobre el nivel del mar existe la enfermedad de Monge, hasta ahora no reco-nocida en nuestro medio, y plantear que quizás la presencia de hipopneas con disminución de la SaO 2 durante el sueño, tengan un papel importante en la aparición de eritrocitosis y cor pulmonale en este tipo de pacientes.
SUMMARY Two cases of alveolar hypoventilation without lung dis-
ease nor sleep apnea caused by non anatomic disturbances of the breathing control system are reported. Its possible differ-ential diagnosis is discussed. It is postulated that its etiology is high-altitude chronic hypoventilation or Monge's disease.
R. ACERO
REFERENCIAS 1. Bradley TD, Day A, Hyland R, Webster P., et al. Chronic ventilatory
failure caused by anormal respiratory pattern generation during sleep. Am Rev Respir Dis 1984; 130: 678-680.
2. Braun N M T , Arora NS, Rochester DF. Respiratory muscle and pul-monary function in polymyositis and other proximal myophaties. Thorax 1983; 38 :616-623.
3. Fletcher E, Gray B, Levin C. Nonapneic mechanisms of anerial oxy-gen desaturation during rapid-eye-movement sleep. J Appl Physiol 1983; 54(3): 632-639.
4. Gould G A, White KF, Rhind GB, Airlie A A, et al.The sleep hypopnea syndrome. Am Rev Respir Dis 1988; 137: 895-898.
5. Gronbeck III Ch. Chronic mountain sickness at an elevation of 2.000 meters. Chest 1984; 85: 577-578.
6. Hecht H. A sea level view of altitude problems. Am J Med 1971; 50: 703-708.
7. Hurtado A. Some clinical aspects of life at high altitude. Ann Intern Med 1960; 53: 247-258.
8. Kryger M, McCullough R, Collins D, Scogginch, et al. Treatment of excessive polycythemia of high altitude with respiratory stimulant drugs. Am Rev Respir Dis 1978; 117 :455-454.
9. Kryger M, McCullough R, Doekel R, Collins D, et al. Excessive pol-ycythemia of high altitude: role of ventilatory drive and lung disease. Am Rev Respir Dis 1978; 118: 659-665.
10. Kryger M, Weil J, Grover R. Chronic mountain polycythemia: a disor-der of the regulation of breathing during sleep? Chest 1978; 73: 303305.
11. Lopata M, Lourenso RV. Evaluation of respiratory control. Clinics in Chest Medicine 1980; 1: 33-45.
12. Lopata M, Onale. Mass loading, sleep an apneas, and the pathogenesis of obesity hipoventilation. Am Rev Respir Dis 1982; 126: 640-645.
13. Millaman R, Fishman AP. Disorders of alveolar ventilation. En: Fish-man AP Edit. Pulmonary diseases and disorders. McGraw-Hill Book Company. New York 1988: 1335-1345.
14. Peñaloza D, Sime F. Chronic cor pulmonale due to loss of altitude ac-climatization (chronic mountain sickness). Am J Med 1971; 50:728-743.
15. Phillipson EA. Hypoventilation syndrome. En: Murray JF and Nadel JA eds. Textbook of respiratory medicine. Edit WB Sounders Company, Philadelphia 1988: 1831-1440.
16. Phillipson EA. Sleep disorders. En: Textbook of respiratory medicine. Murray/Nadel Editores. Edit. WB Saunder Company, Philadelphia 1988: 1841-1860.
17. Reichel J. Primary alveolar hypoventilation. Clinics in Chest Medicine 1980; 1:119-123.
18. Sullivan CE, Issa FG. Obstructive sleep apnea. Clinics in Chest Medicine 1985; 6 :633-650.
19. Sutton JR, Houston Ch, Mansell A, McFadden M, et al . Effect of acetazolamide on hypoxemia during sleep at high altitude. N Engl J Med 1979; 301 : 1329-1331.
20. Weil J V. Sleep at high altitude. Clinics in Chest Medicine 1985; 6: 615-622.
Rafael Acero Colmenares: Neumólogo, Laboratorio de Fisiología Pulmo-nar, Fundación Santa Fe de Bogotá.
Acta Med Colomb Vol 14 N°5 - 1989