0. anemias emuap (1)
TRANSCRIPT
ANEMIAS ANEMIAS

Anemia: DefiniciónAnemia: Definición
• Cifras de hemoglobina y hematocrito por debajo del límite inferior normal, según los valores de referencia para:
• Edad• Sexo• Altura sobre el nivel del mar.
• Cifras de hemoglobina y hematocrito por debajo del límite inferior normal, según los valores de referencia para:
• Edad• Sexo• Altura sobre el nivel del mar.

• Las cifras de hemoglobina y hematocrito tienen variables fisiológicas según:
–Edad–Sexo–Altura sobre nivel del mar–Embarazo
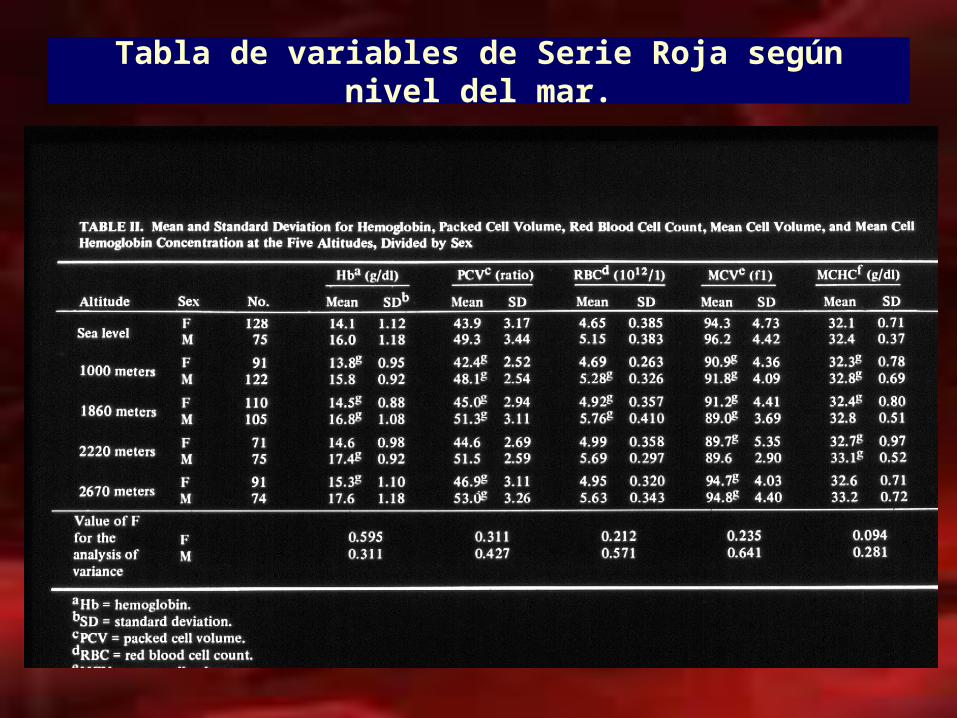
Tabla de variables de Serie Roja según nivel del mar.
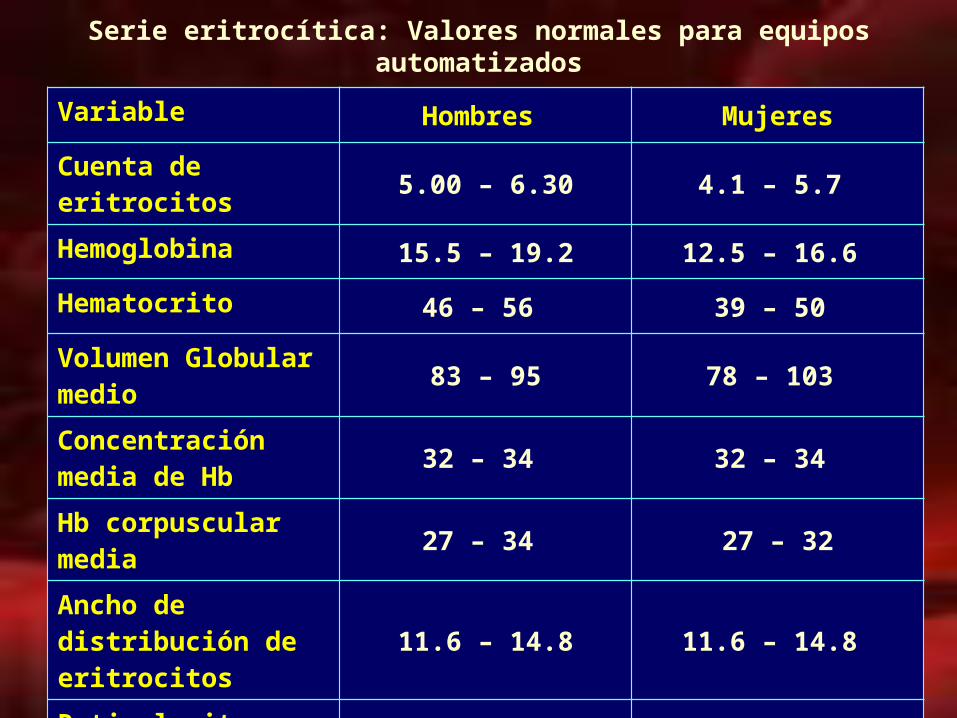
Serie eritrocítica: Valores normales para equipos automatizados
Variable Hombres Mujeres
Cuenta de eritrocitos 5.00 – 6.30 4.1 – 5.7
Hemoglobina 15.5 – 19.2 12.5 – 16.6
Hematocrito 46 – 56 39 – 50
Volumen Globular medio 83 – 95 78 – 103
Concentración media de Hb 32 – 34 32 – 34
Hb corpuscular media 27 – 34 27 – 32
Ancho de distribución de eritrocitos
11.6 – 14.8 11.6 – 14.8
Reticulocitos 0.5 – 1.5 0.5 – 1.5

Diagnóstico de anemia
• Hemoglobina: – < 12.5 g/dL en mujer– < 15.5 g/dL en hombre
• Hematocrito:– < 39 % en mujer– < 46 % en hombre

Clasificación de la anemia
• Por su evolución
• Por su grado
• Por su fisiopatología
• Por su etiología
• Por su morfología
• Por su evolución
• Por su grado
• Por su fisiopatología
• Por su etiología
• Por su morfología

Clasificación de anemias por su grado
Grado Hemoglobina
(g/dL)
I 12 – 9
II 9 – 6
III 6 – 3
IV < 3
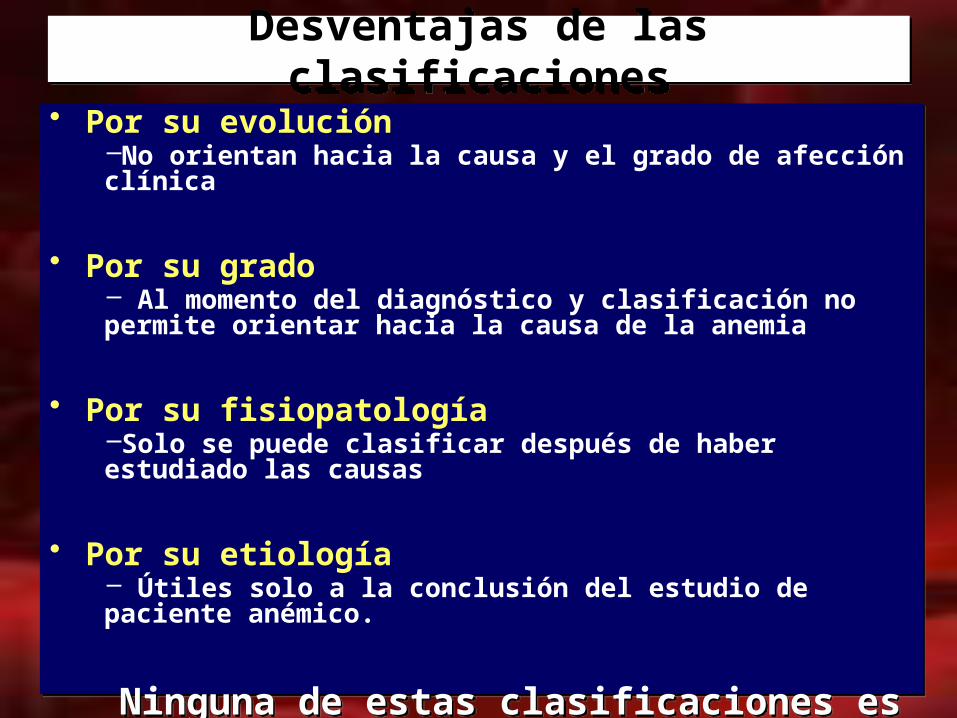
• Por su evolución–No orientan hacia la causa y el grado de afección clínica
• Por su grado– Al momento del diagnóstico y clasificación no permite orientar hacia la causa de la anemia
• Por su fisiopatología–Solo se puede clasificar después de haber estudiado las causas
• Por su etiología– Útiles solo a la conclusión del estudio de paciente anémico.
Ninguna de estas clasificaciones es útil, al momento del diagnóstico de anemia, para orientar la clínica y el laboratorio hacia la
causa de la anemia y al tratamiento específico.
• Por su evolución–No orientan hacia la causa y el grado de afección clínica
• Por su grado– Al momento del diagnóstico y clasificación no permite orientar hacia la causa de la anemia
• Por su fisiopatología–Solo se puede clasificar después de haber estudiado las causas
• Por su etiología– Útiles solo a la conclusión del estudio de paciente anémico.
Ninguna de estas clasificaciones es útil, al momento del diagnóstico de anemia, para orientar la clínica y el laboratorio hacia la
causa de la anemia y al tratamiento específico.
Desventajas de las clasificacionesDesventajas de las clasificaciones

CLASIFICACIÓN MORFOLÓGICA DE LAS ANEMIAS
CLASIFICACIÓN MORFOLÓGICA DE LAS ANEMIAS
• PERMITE ORIENTAR EL DIAGNOSTICO ETIOLÓGICO DE LA ANEMIA A PARTIR DE LOS SIGUIENTES ÍNDICES:
– VOLUMEN GLOBULAR MEDIO– HEMOGLOBINA CORPUSCULAR MEDIA– CONCENTRACIÓN MEDIA DE Hb
– RETICULOCITOS– ANCHO DE DISTRIBUCIÓN DE LOS ERITROCITOS
• PERMITE ORIENTAR EL DIAGNOSTICO ETIOLÓGICO DE LA ANEMIA A PARTIR DE LOS SIGUIENTES ÍNDICES:
– VOLUMEN GLOBULAR MEDIO– HEMOGLOBINA CORPUSCULAR MEDIA– CONCENTRACIÓN MEDIA DE Hb
– RETICULOCITOS– ANCHO DE DISTRIBUCIÓN DE LOS ERITROCITOS

Morfología de las anemiasMorfología Índices Estado
NORMOCITICASNORMOCROMICAS
VCMHCM
NORMALES
MICROCITICAS HIPOCROMICAS
VCMHCM
DISMINUIDOS
MACROCITICASNORMOCROMICAS
VCMHCM
AUMENTADONORMAL

Reticulocitos
• Precursores de eritrocitos.
• Valores normales: 0.5 – 1.5%
• Indican la intensidad de la
actividad de la eritropoyesis en
la médula ósea.
• Precursores de eritrocitos.
• Valores normales: 0.5 – 1.5%
• Indican la intensidad de la
actividad de la eritropoyesis en
la médula ósea.

Síndrome anémico
• Palidez de piel y mucosas• Taquicardia• Soplo cardiaco funcional• Disnea de esfuerzo• Cefalea• Hipodinamia• Astenia• Anorexia• Amenorrea

Anemia por hemorragia aguda
• Datos clínicos de hipovolemia– Taquicardia– Taquicardia– Hipotensión arterial– Disminución de PVC– Lipotimia
• Tratamiento– Detener la hemorrágia (hemostasia)– Sustitución de volumen – Transfusión (Hb: < 8- 10 g/dL)
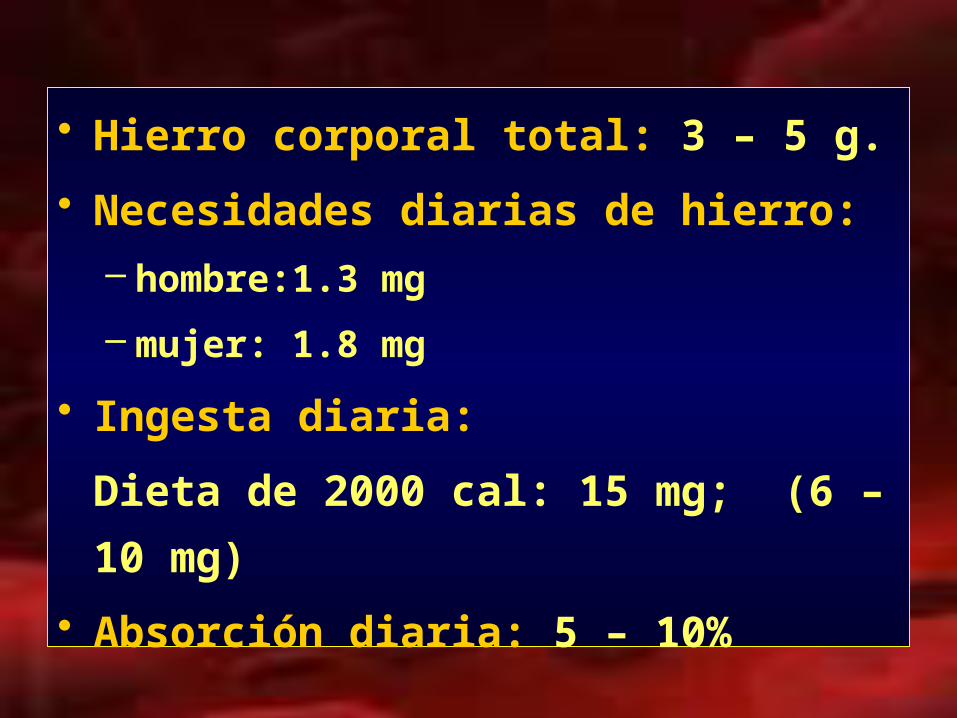
• Hierro corporal total: 3 – 5 g.
• Necesidades diarias de hierro:
– hombre:1.3 mg
– mujer: 1.8 mg
• Ingesta diaria:
Dieta de 2000 cal: 15 mg; (6 – 10 mg)
• Absorción diaria: 5 – 10%
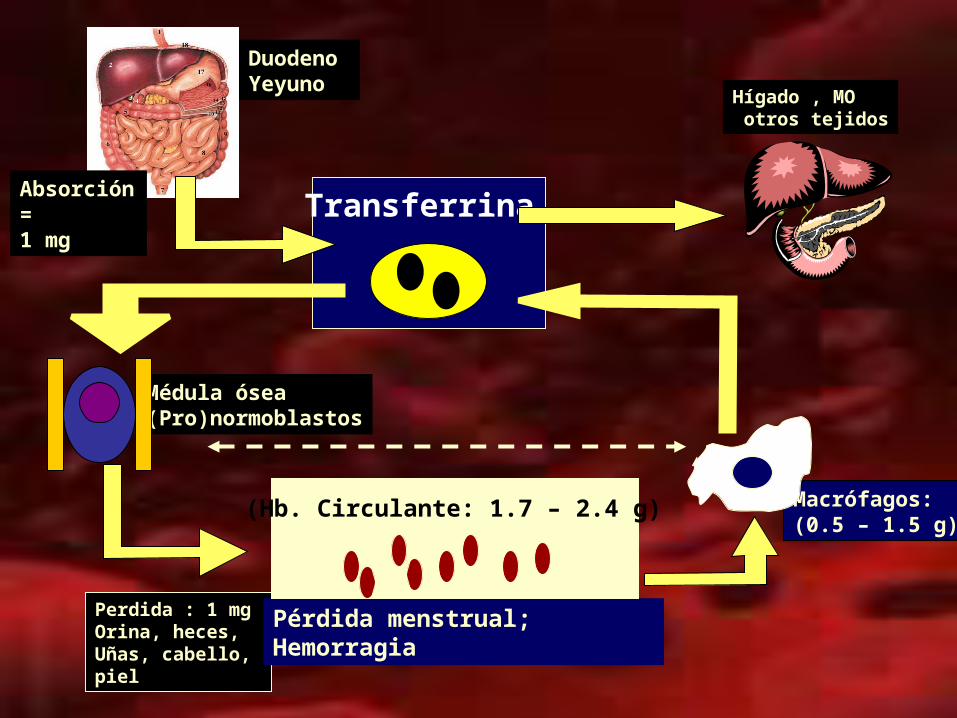
Perdida : 1 mg Orina, heces, Uñas, cabello,piel
Pérdida menstrual; Hemorragia
Transferrina
Duodeno Yeyuno
Absorción=1 mg
Médula ósea(Pro)normoblastos
Macrófagos:(0.5 – 1.5 g)
(Hb. Circulante: 1.7 – 2.4 g)
Hígado , MO otros tejidos
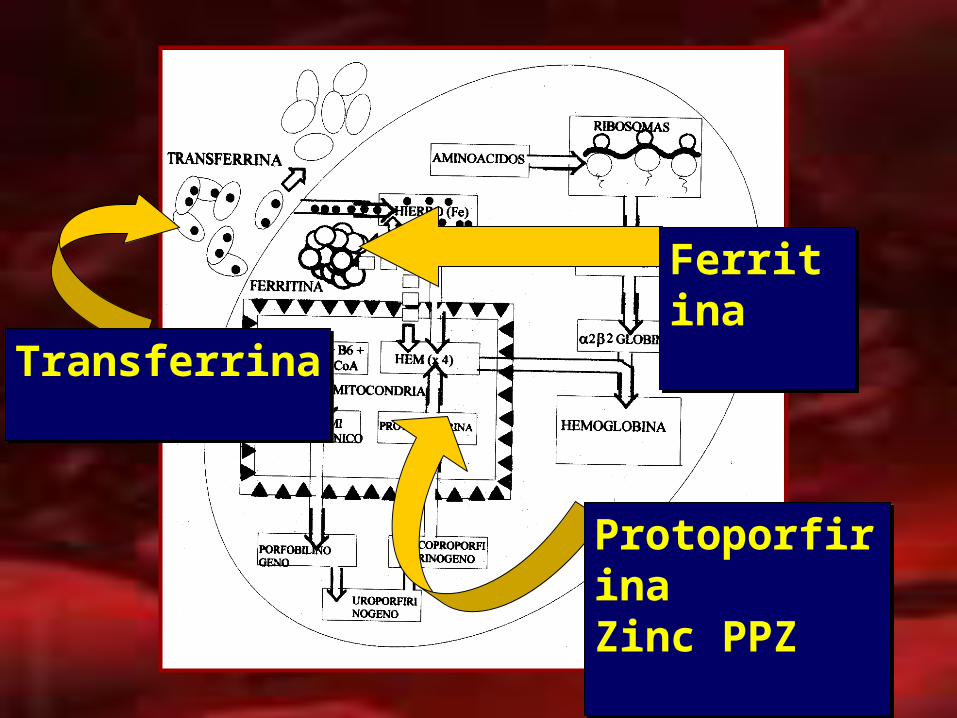
TransferrinaTransferrina
ProtoporfirinaZinc PPZProtoporfirinaZinc PPZ
FerritinaFerritina
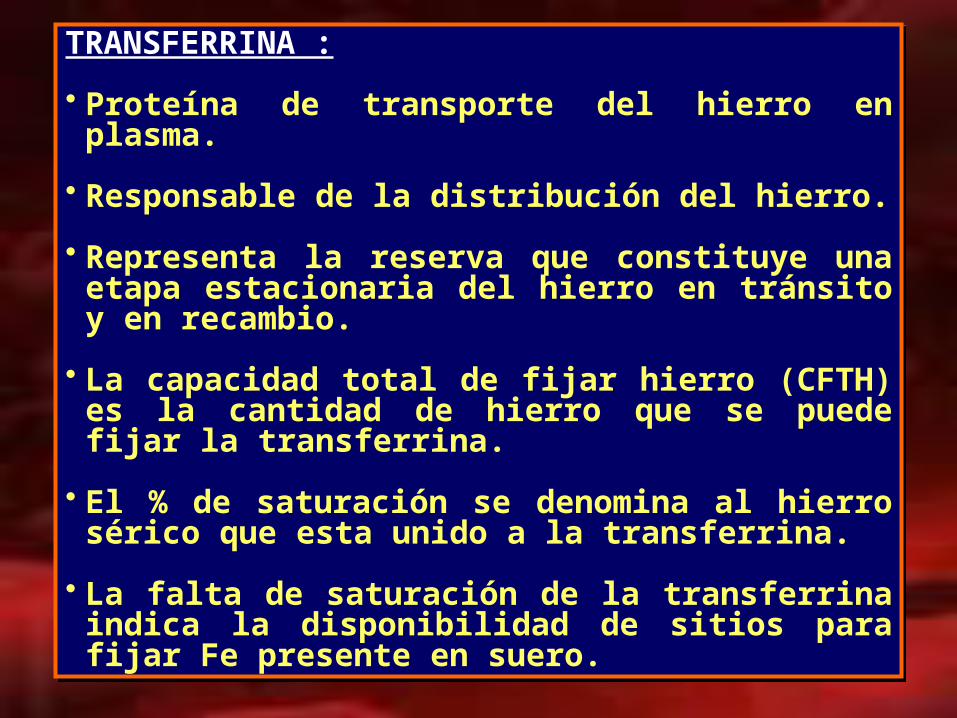
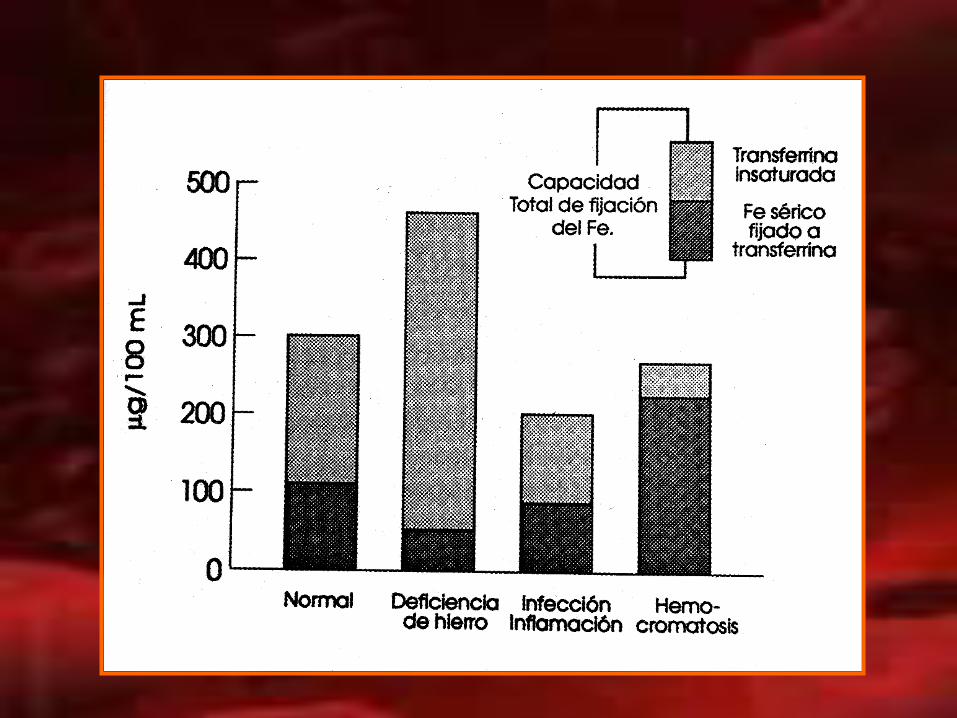
TRANSFERRINA :
• Proteína de transporte del hierro en plasma.
• Responsable de la distribución del hierro.
• Representa la reserva que constituye una etapa estacionaria del hierro en tránsito y en recambio.
• La capacidad total de fijar hierro (CFTH) es la cantidad de hierro que se puede fijar la transferrina.
• El % de saturación se denomina al hierro sérico que
esta unido a la transferrina.
• La falta de saturación de la transferrina indica la disponibilidad de sitios para fijar Fe presente en suero.
TRANSFERRINA :
• Proteína de transporte del hierro en plasma.
• Responsable de la distribución del hierro.
• Representa la reserva que constituye una etapa estacionaria del hierro en tránsito y en recambio.
• La capacidad total de fijar hierro (CFTH) es la cantidad de hierro que se puede fijar la transferrina.
• El % de saturación se denomina al hierro sérico que
esta unido a la transferrina.
• La falta de saturación de la transferrina indica la disponibilidad de sitios para fijar Fe presente en suero.

FERRITINA :
• Grupo heterogéneo de proteínas hidrosolubles que almacenan Fe .
• Se halla en casi todas las células orgánicas, pero principalmente en hígado, bazo y medula ósea.
• Agrupa y aísla el exceso de moléculas de hierro, lo que impide cualquier acción tóxica sobres las células tisulares.
• Es una fuente rápida de reserva de hierro.
• A pesar de sus variaciones con la edad y el sexo , la ferritina sérica es menos variable que las concentraciones de Fe sérico.
FERRITINA :
• Grupo heterogéneo de proteínas hidrosolubles que almacenan Fe .
• Se halla en casi todas las células orgánicas, pero principalmente en hígado, bazo y medula ósea.
• Agrupa y aísla el exceso de moléculas de hierro, lo que impide cualquier acción tóxica sobres las células tisulares.
• Es una fuente rápida de reserva de hierro.
• A pesar de sus variaciones con la edad y el sexo , la ferritina sérica es menos variable que las concentraciones de Fe sérico.

En individuos normales hay una correlación
directa importante entre la ferritina y la
concentración de hierro almacenado por lo
tanto es un indicador sensible de los depósitos
de hierro
Se ha demostrado que presenta una buena
correlación con el hierro de médula ósea
detectable por tinción por lo tanto representa
un método simple y no invasivo de exploración
del metabolismo del hierro
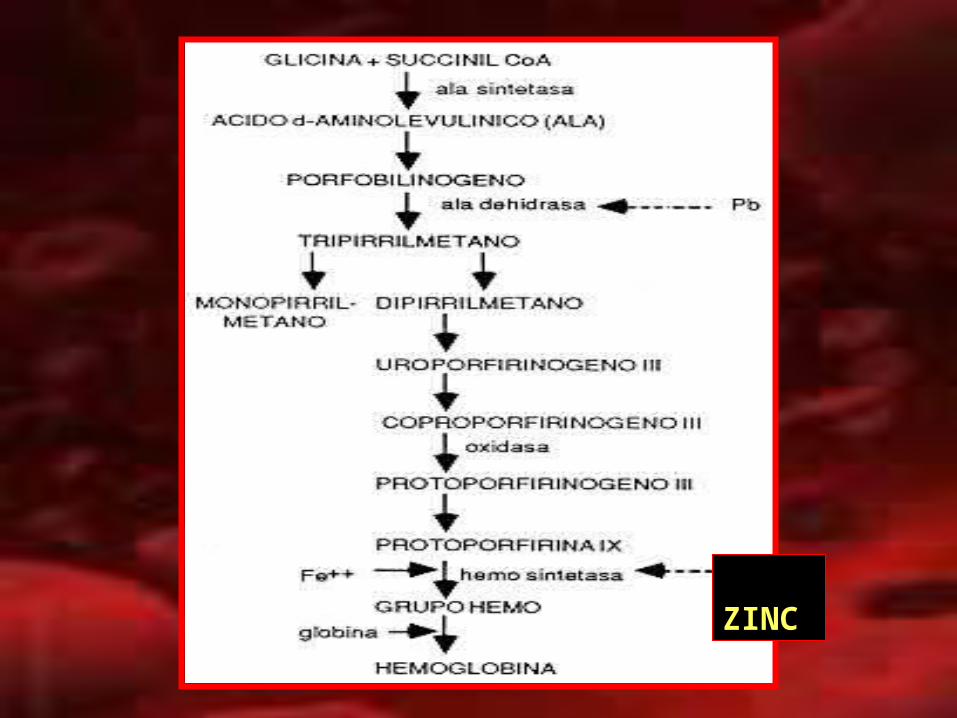
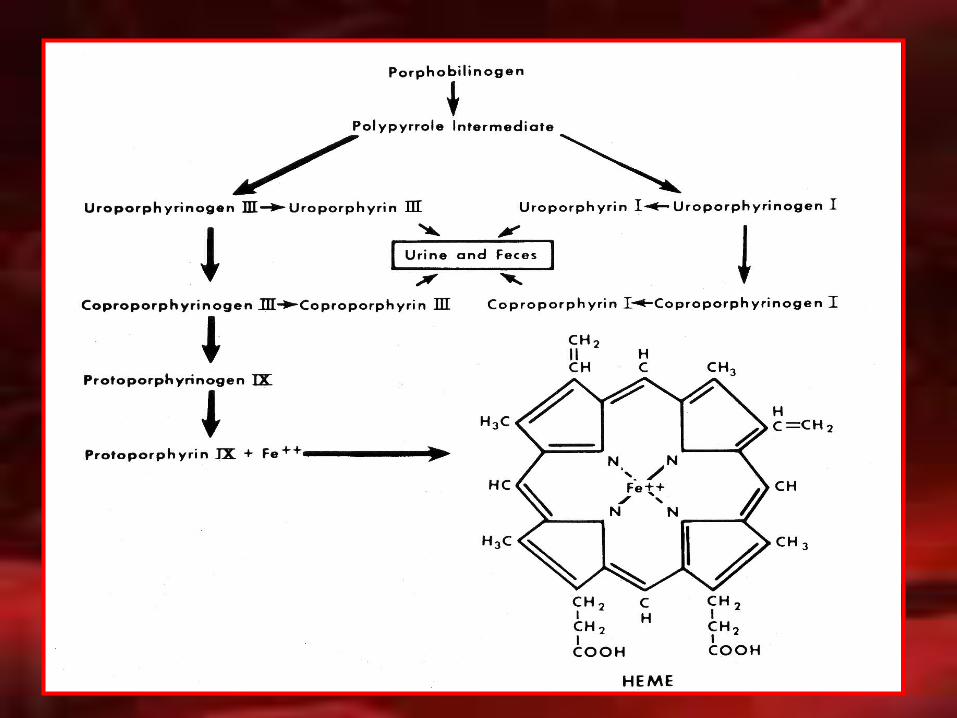
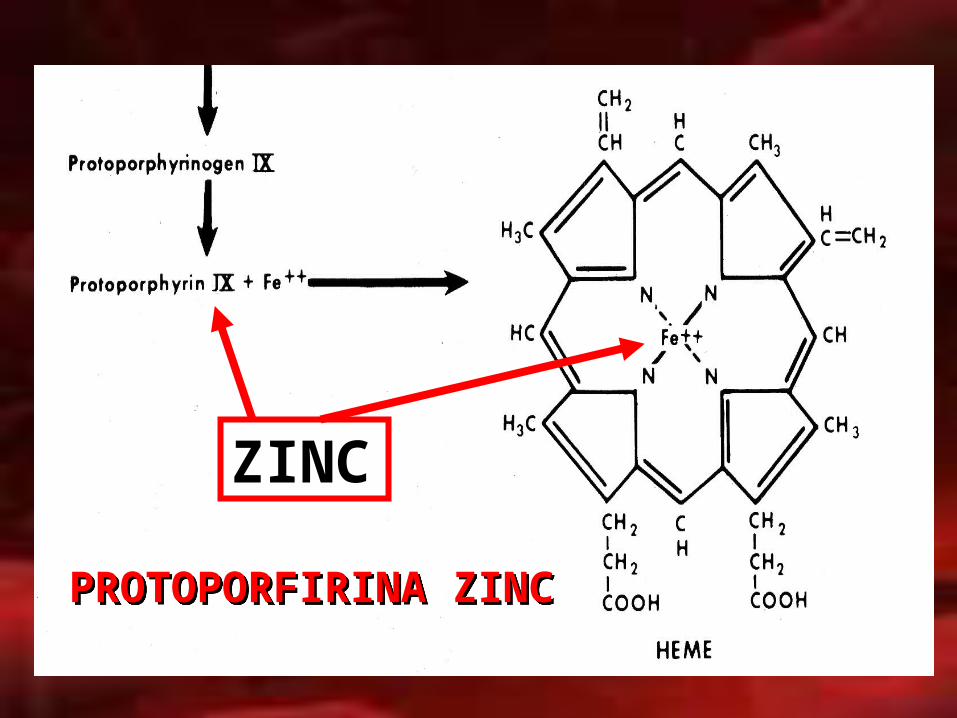
ZINC
PROTOPORFIRINA ZINCPROTOPORFIRINA ZINC
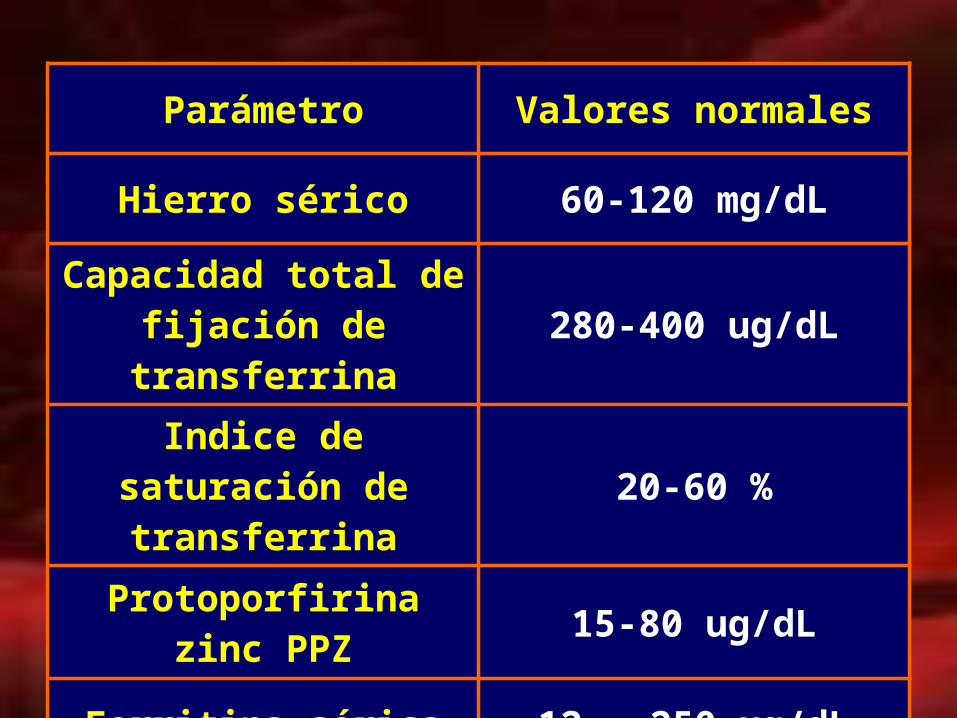
Parámetro Valores normales
Hierro sérico 60-120 mg/dL
Capacidad total de fijación de transferrina 280-400 ug/dL
Indice de saturación de transferrina 20-60 %
Protoporfirina zinc PPZ 15-80 ug/dL
Ferritina sérica 12 – 250 ug/dL

Anemia de sangrado crónico
• Varices esofágicas• Gastritis erosiva• Ulcera gsatroduodenal• Hernia hiatal• Angiodisplasia• Hemorroides• Cáncer • Telangiectasia• Cáncer renal /vejiga• Tumores ginecológicos• Litiasis• STV anormales
• Varices esofágicas• Gastritis erosiva• Ulcera gsatroduodenal• Hernia hiatal• Angiodisplasia• Hemorroides• Cáncer • Telangiectasia• Cáncer renal /vejiga• Tumores ginecológicos• Litiasis• STV anormales
Deficiencia de hierro

Anemia por deficiencia de hierro• Pérdida de hierro mayor a capacidad de
absorción.
• Hombres: 1.6%
• Mujeres: 14.3 a 25%
• Manifestaciones clínicas:– Pica– Platoniquia, coiloniquia– Caída de cabello– Irritabilidad– Síndrome anémico
• Pérdida de hierro mayor a capacidad de absorción.
• Hombres: 1.6%
• Mujeres: 14.3 a 25%
• Manifestaciones clínicas:– Pica– Platoniquia, coiloniquia– Caída de cabello– Irritabilidad– Síndrome anémico

Historia natural de la ferropenia
FERRITINA
HIERRO ; % SAT. TRANSFERRINA
HCM: HIPOCROMIA
VGM: MICROCITOSIS
HB ; HTO: ANEMIA
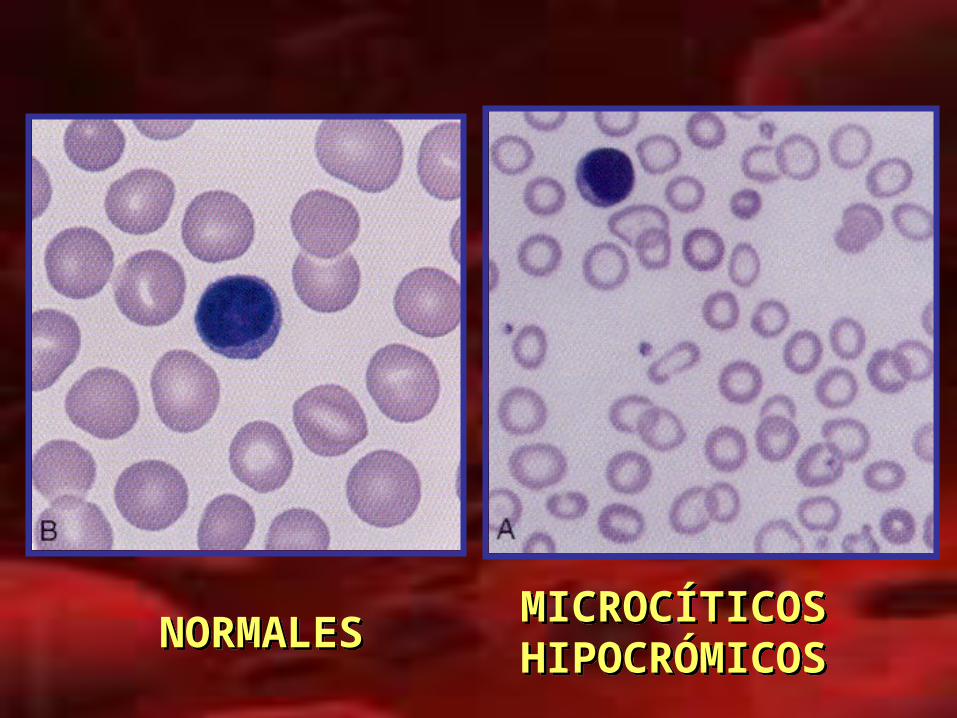
NORMALESNORMALES MICROCÍTICOS HIPOCRÓMICOSMICROCÍTICOS HIPOCRÓMICOS

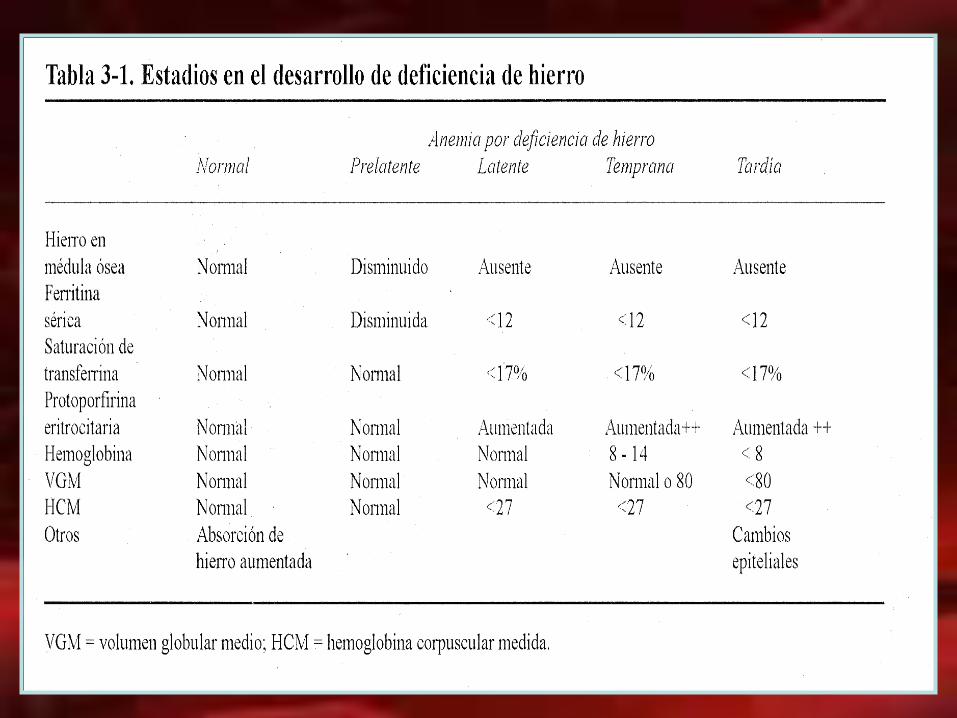
Laboratorio en el diagnóstico de anemia por deficiencia de hierro
examen
VGM
HCM
ADE (CV-VGM)
Fe SERICO
CTF TRANFERRINA
% SATURACION DE TRANSFERRINA
FERRITINA SERICA
PROTOPORFIRINA ZINC
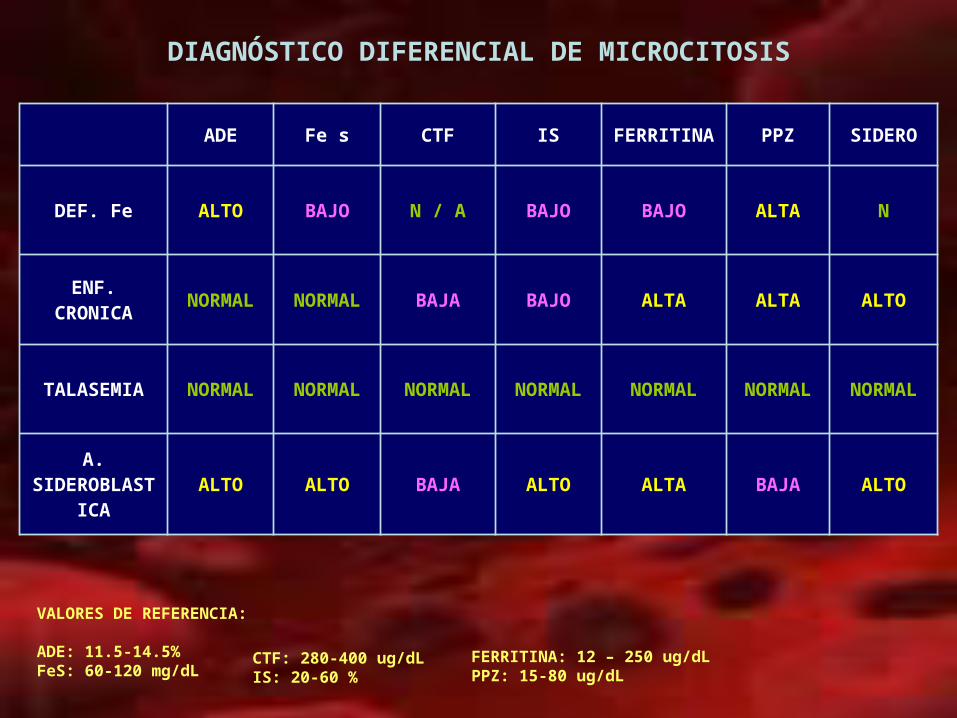
DIAGNÓSTICO DIFERENCIAL DE MICROCITOSIS
ADE Fe s CTF IS FERRITINA PPZ SIDERO
DEF. Fe ALTO BAJO N / A BAJO BAJO ALTA N
ENF. CRONICA
NORMAL NORMAL BAJA BAJO ALTA ALTA ALTO
TALASEMIA NORMAL NORMAL NORMAL NORMAL NORMAL NORMAL NORMAL
A. SIDEROBLAS
TICAALTO ALTO BAJA ALTO ALTA BAJA ALTO
VALORES DE REFERENCIA:
ADE: 11.5-14.5%FeS: 60-120 mg/dL
FERRITINA: 12 – 250 ug/dLPPZ: 15-80 ug/dL
CTF: 280-400 ug/dLIS: 20-60 %

Déficit de Fe: peso Kg x (50 – 2.4 x Hb)
(15-Hb) x peso x 3
Tratamiento de deficiencia de hierro
• Vía oral– Adulto: 200 mg de Fe elemental al día– Niño: 1.5 a 2.5 mg de Fe elemental / kg x 3
• Sulfato ferroso.• Fumarato ferroso.• Gluconato ferroso.• Complejo polimaltosado férrico
• Vía parenteral– Reservado a casos especiales
• Vía oral– Adulto: 200 mg de Fe elemental al día– Niño: 1.5 a 2.5 mg de Fe elemental / kg x 3
• Sulfato ferroso.• Fumarato ferroso.• Gluconato ferroso.• Complejo polimaltosado férrico
• Vía parenteral– Reservado a casos especiales

Anemias megaloblásticasAnemias megaloblásticas
• Deficiencia de B12• Deficiencia de folatos• Alteración en la maduración de los núcleos de
los normoblastos.• Maduración Megaloblástica• Eritropoyesis aumentada• Descontrol del crecimiento celular• Retardo en síntesis de ARN y ADN• Exceso en síntesis de componentes
citoplásmicos
• Deficiencia de B12• Deficiencia de folatos• Alteración en la maduración de los núcleos de
los normoblastos.• Maduración Megaloblástica• Eritropoyesis aumentada• Descontrol del crecimiento celular• Retardo en síntesis de ARN y ADN• Exceso en síntesis de componentes
citoplásmicos

Acido FólicoAcido Fólico
• 90% poliglutamatos– Vegetales de hojas verdes– Levaduras– Derivados de leche– Órganos parenquimatosos (hígado, riñón)
• Requerimiento diario: 200 – 800 ug• Absorción: Intestino delgado proximal• Folato corporal: 5-10 mg• Concentración sérica: 5-10 ng/dL
• 90% poliglutamatos– Vegetales de hojas verdes– Levaduras– Derivados de leche– Órganos parenquimatosos (hígado, riñón)
• Requerimiento diario: 200 – 800 ug• Absorción: Intestino delgado proximal• Folato corporal: 5-10 mg• Concentración sérica: 5-10 ng/dL

Deficiencia de ácido fólicoDeficiencia de ácido fólico• Dieta• Enf. Hepática: cirrosis / alcoholismo• Embarazo• Infancia• Mala absorción• Anticonvulsivantes• Anticonceptivos• QT, otros inhibidores de dihidrofolato
reductasa• Proliferación celular• Mutación de genes responsables de
absorción o metabolismo (Gen: MTHFR)
• Dieta• Enf. Hepática: cirrosis / alcoholismo• Embarazo• Infancia• Mala absorción• Anticonvulsivantes• Anticonceptivos• QT, otros inhibidores de dihidrofolato
reductasa• Proliferación celular• Mutación de genes responsables de
absorción o metabolismo (Gen: MTHFR)

Deficiencia de folatos durante el embarazo
• Perdidas fetales• Desprendimiento de placenta• Peso bajo en R.N.• Malformaciones esqueléticas• Defecto del tubo neural (espina bífida;
anencefalia)• Hiperhomocisteinémia (A.F – B12)
• Perdidas fetales• Desprendimiento de placenta• Peso bajo en R.N.• Malformaciones esqueléticas• Defecto del tubo neural (espina bífida;
anencefalia)• Hiperhomocisteinémia (A.F – B12)

Hiperhomocisteinemia (A.F – B12)Hiperhomocisteinemia (A.F – B12)
• Riesgo relativo para trombosis 40% mayor que por hipercolesterolemia.
• > 50% de riesgo relativo para Enfermedad Vascular Cerebral y Enfermedad arterial periférica.
• A. Fólico, B12 y B6. disminuyen los niveles de homocisteína y el riesgo de enfermedad arterial coronaria.
• Riesgo relativo para trombosis 40% mayor que por hipercolesterolemia.
• > 50% de riesgo relativo para Enfermedad Vascular Cerebral y Enfermedad arterial periférica.
• A. Fólico, B12 y B6. disminuyen los niveles de homocisteína y el riesgo de enfermedad arterial coronaria.
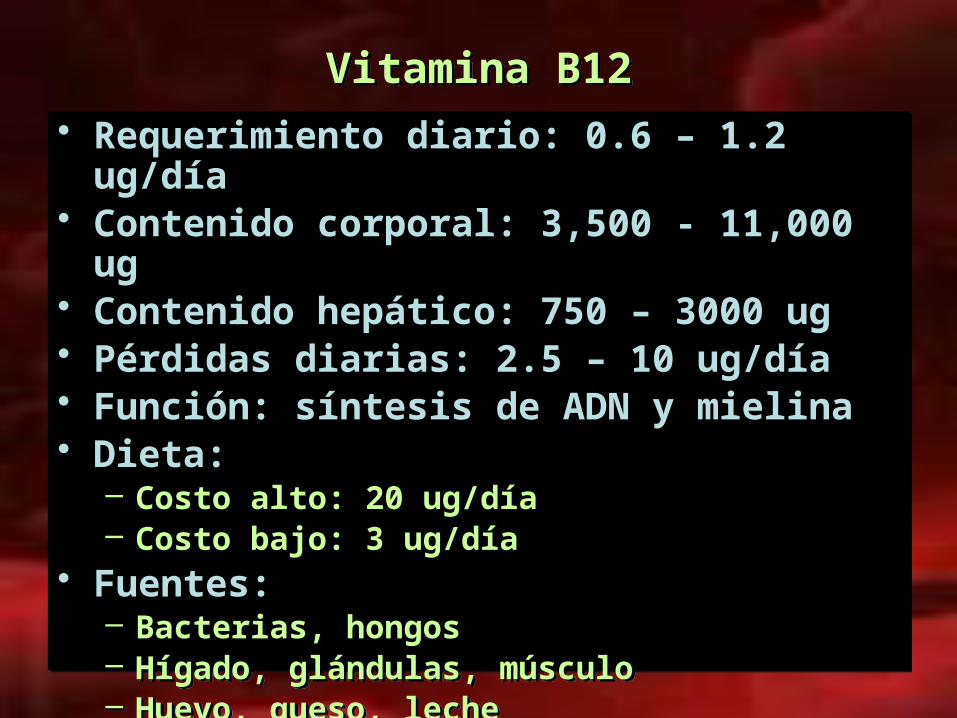
Vitamina B12Vitamina B12
• Requerimiento diario: 0.6 – 1.2 ug/día• Contenido corporal: 3,500 - 11,000 ug• Contenido hepático: 750 – 3000 ug • Pérdidas diarias: 2.5 – 10 ug/día• Función: síntesis de ADN y mielina • Dieta:
– Costo alto: 20 ug/día– Costo bajo: 3 ug/día
• Fuentes:– Bacterias, hongos– Hígado, glándulas, músculo– Huevo, queso, leche
• Requerimiento diario: 0.6 – 1.2 ug/día• Contenido corporal: 3,500 - 11,000 ug• Contenido hepático: 750 – 3000 ug • Pérdidas diarias: 2.5 – 10 ug/día• Función: síntesis de ADN y mielina • Dieta:
– Costo alto: 20 ug/día– Costo bajo: 3 ug/día
• Fuentes:– Bacterias, hongos– Hígado, glándulas, músculo– Huevo, queso, leche
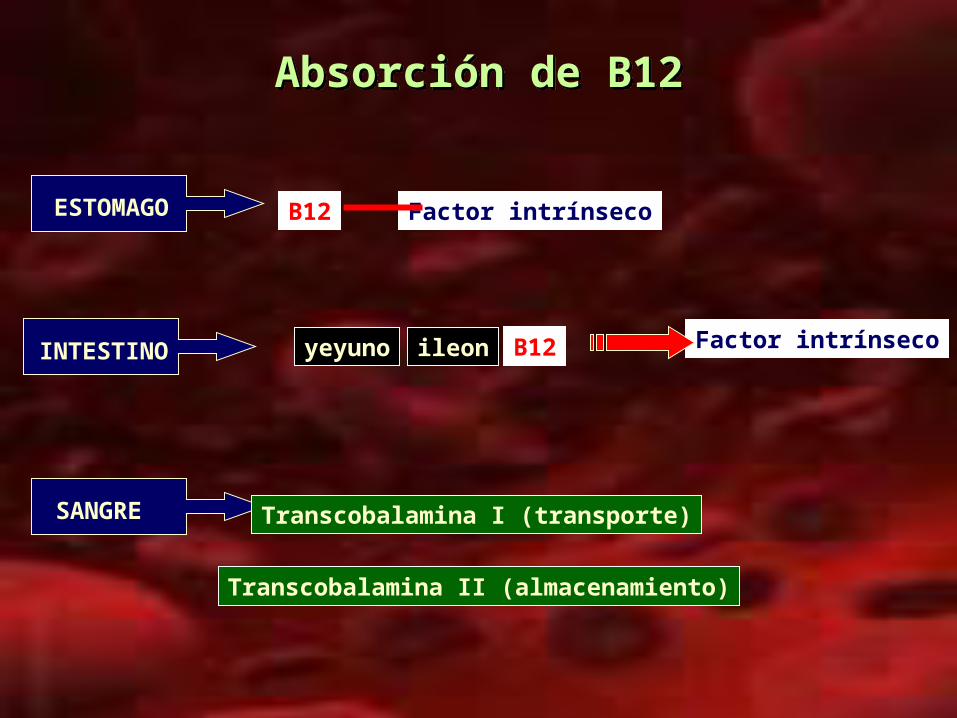
Absorción de B12Absorción de B12
B12 Factor intrínseco
yeyuno ileon
Transcobalamina I (transporte)
Transcobalamina II (almacenamiento)
Factor intrínsecoB12
ESTOMAGO
INTESTINO
SANGRE

Deficiencia de B12Deficiencia de B12
• Gastrectomía• Corrosivos• Factor Intrínseco anormal• Crecimiento bacteriano• Enfermedad pancreática crónica• Hemodiálisis• Medicamentos• Mala absorción intestinal
• Gastrectomía• Corrosivos• Factor Intrínseco anormal• Crecimiento bacteriano• Enfermedad pancreática crónica• Hemodiálisis• Medicamentos• Mala absorción intestinal
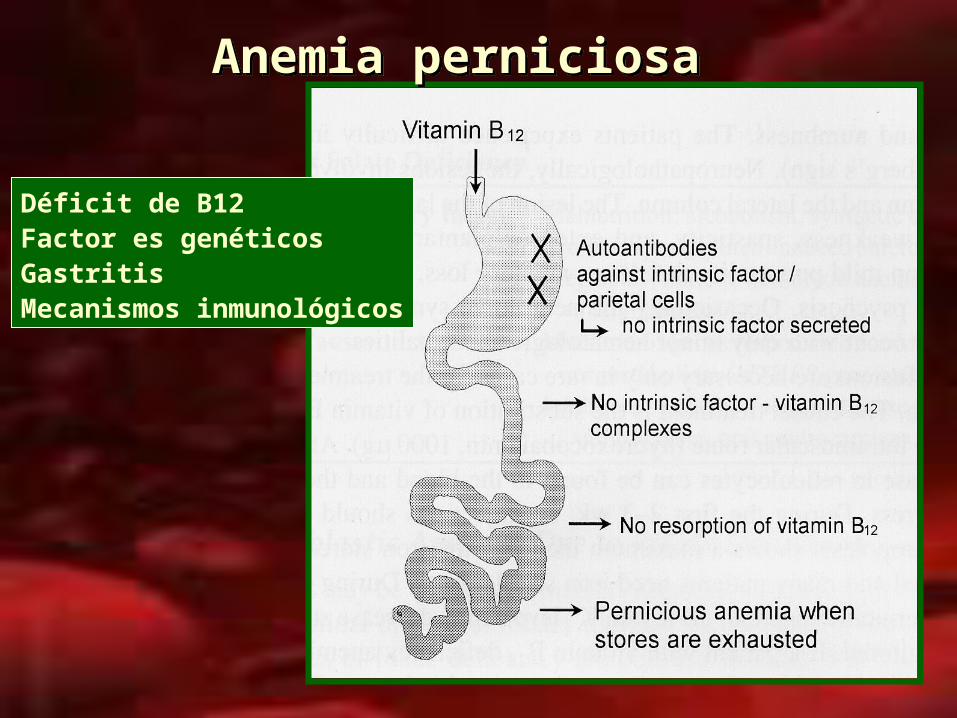
Anemia perniciosaAnemia perniciosa
Déficit de B12Factor es genéticosGastritisMecanismos inmunológicos

Anemia megaloblásticaAnemia megaloblástica
• Manifestaciones clínicas:– Glositis– Ictericia– Pérdida de peso– Trastornos de tubo digestivo– Pancitopenia– Fiebre– Púrpura– Neuropatía

Laboratorio en el diagnóstico de anemia megaloblástica
Laboratorio en el diagnóstico de anemia megaloblástica
• Macrocitosis• ADE alto• DHL alta• Prueba de supresión de la deoxiuridina• Excreción urinaria de metilmalonato• Folato sérico disminuido• B 12 sérica: disminuida• Prueba de Shilling positiva• Anticuerpos antifactor intrínseco• Anticuerpos anti- mucosa gástrica (células parietales)
• Macrocitosis• ADE alto• DHL alta• Prueba de supresión de la deoxiuridina• Excreción urinaria de metilmalonato• Folato sérico disminuido• B 12 sérica: disminuida• Prueba de Shilling positiva• Anticuerpos antifactor intrínseco• Anticuerpos anti- mucosa gástrica (células parietales)
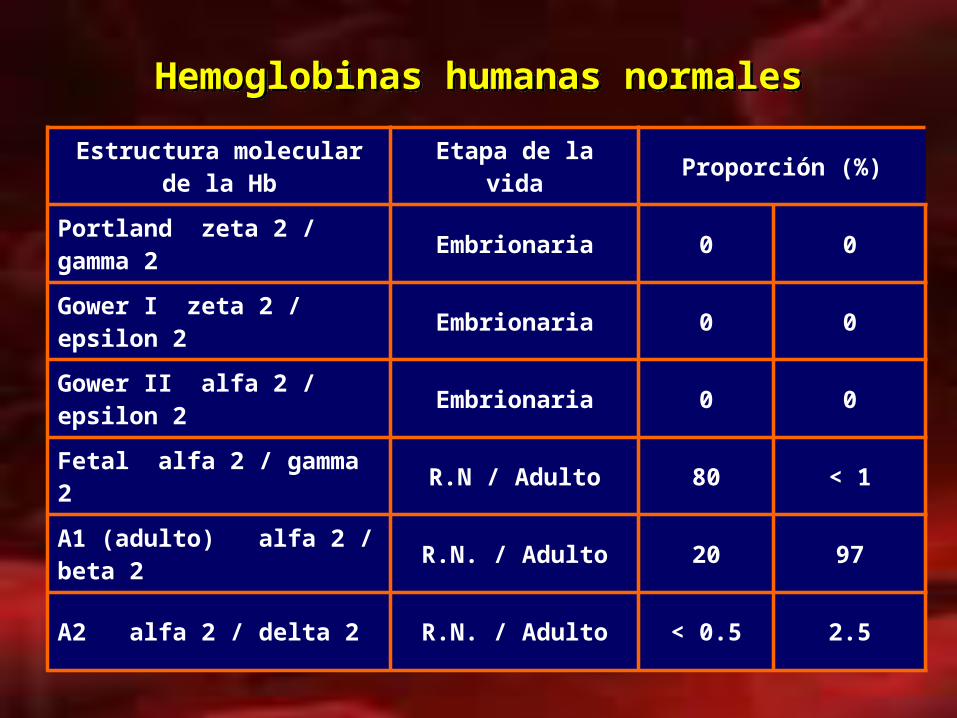
Hemoglobinas humanas normalesHemoglobinas humanas normales
Estructura molecular de la Hb
Etapa de la vida Proporción (%)
Portland zeta 2 / gamma 2 Embrionaria 0 0
Gower I zeta 2 / epsilon 2 Embrionaria 0 0
Gower II alfa 2 / epsilon 2 Embrionaria 0 0
Fetal alfa 2 / gamma 2 R.N / Adulto 80 < 1
A1 (adulto) alfa 2 / beta 2 R.N. / Adulto 20 97
A2 alfa 2 / delta 2 R.N. / Adulto < 0.5 2.5
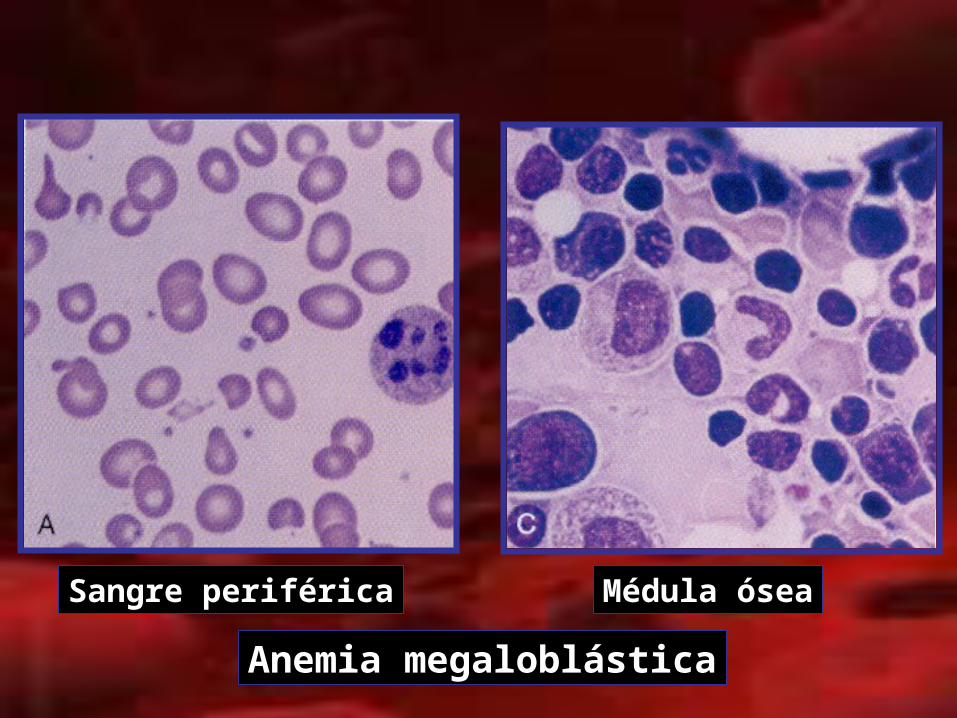
Sangre periférica Médula ósea
Anemia megaloblástica

Estudios de gabinete en anemia megaloblástica
• ENDOSCOPIA DE TUBO DIGESTIVO
• BIOPSIAS DE MUCOSA GÁSTRICA
• SERIE E.G.D. Y TRANSITO INTESTINAL
• PRUEBA DE SHILLING
• ENDOSCOPIA DE TUBO DIGESTIVO
• BIOPSIAS DE MUCOSA GÁSTRICA
• SERIE E.G.D. Y TRANSITO INTESTINAL
• PRUEBA DE SHILLING

Tratamiento de anemia megaloblásticaTratamiento de anemia megaloblástica
• Def. folatos
– Ácido fólico por vía oral: 5 a 15 mg/día
– Ácido folínico: 6 – 25 mg c/ 8 hs vía
parenteral
– Ácido folínico: 15 mg/ semana VO
• Def. folatos
– Ácido fólico por vía oral: 5 a 15 mg/día
– Ácido folínico: 6 – 25 mg c/ 8 hs vía
parenteral
– Ácido folínico: 15 mg/ semana VO
• Deficiencia B12:
– 2 – 5 ug.
– 100 ug / día dos a tres semanas + 1000 ug /
semana x 4 + 1000 ug/mes
– Intramuscular
• Deficiencia B12:
– 2 – 5 ug.
– 100 ug / día dos a tres semanas + 1000 ug /
semana x 4 + 1000 ug/mes
– Intramuscular

• Hereditarias– Defecto de membrana
• Esferocitosis• Eliptocitosis
– Defectos enzimáticos• Deficiencia G6PD• Deficiencia de piruvato
cinasa– Hemoglobinopatías
• Drepanocitosis– Talasemias
• Beta-Talasemias• Alfa-Talasemias• Talasemia intermedia
• Hereditarias– Defecto de membrana
• Esferocitosis• Eliptocitosis
– Defectos enzimáticos• Deficiencia G6PD• Deficiencia de piruvato
cinasa– Hemoglobinopatías
• Drepanocitosis– Talasemias
• Beta-Talasemias• Alfa-Talasemias• Talasemia intermedia
• Adquiridas– Autoanticuerpos
• Ac. Calientes• Ac. Frios
– Hemoglobinuria Paroxistica Nocturna
– Enfermedad hemolítica del RN
• Anti-ABO• Anti Rh
– Infección parasitaria• Paludismo• Babesiosis
– Microangiopática– CID
• Adquiridas– Autoanticuerpos
• Ac. Calientes• Ac. Frios
– Hemoglobinuria Paroxistica Nocturna
– Enfermedad hemolítica del RN
• Anti-ABO• Anti Rh
– Infección parasitaria• Paludismo• Babesiosis
– Microangiopática– CID
Anemias hemolíticasAnemias hemolíticas

Anemias hemolíticas hereditarias por defectos de membrana
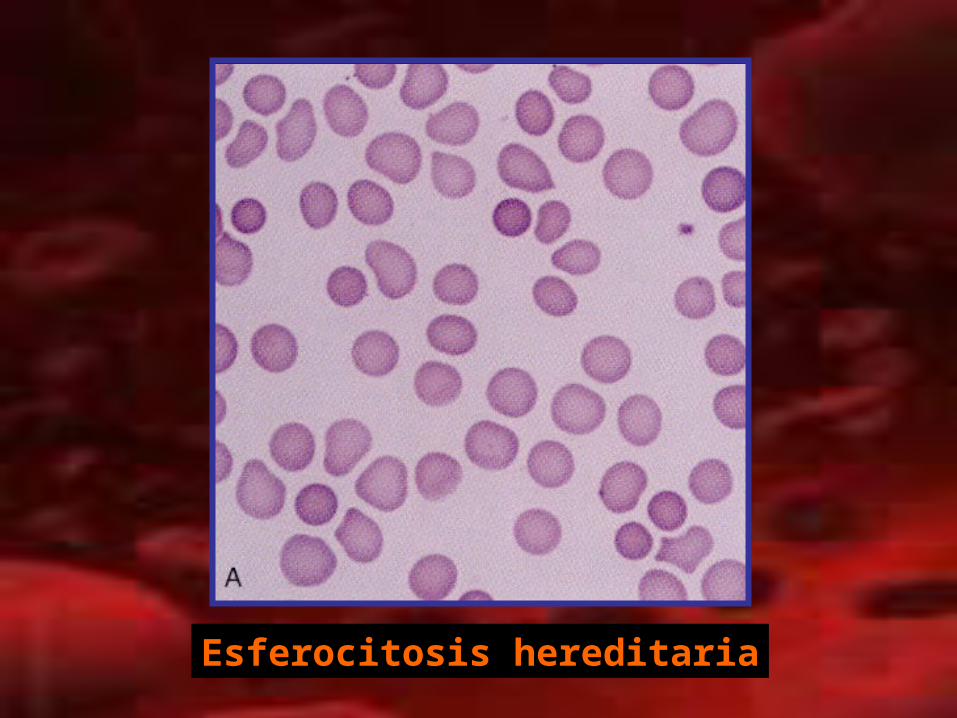
• Esferocitosis hereditaria:– Autosómica dominante– Defecto en proteínas estructurales de la membrana del
eritrocito (espectrina)– Disminución de lipidos de membrana– Disminución de la resistencia osmótica– Genes: ANK1, EPB3, ELB42, SPTA1, SPTB– Anemia crónica, exacerbación con infección– Anemia, ictericia, dolor abdominal (litiasis), esplenomegalia.– Crisis aplástica: parvovirus B19. – Esferocitos, Fragilidad osmótica.– DHL +; BI+; haptoglobina disminuida.– Esplenectomía.
• Esferocitosis hereditaria:– Autosómica dominante– Defecto en proteínas estructurales de la membrana del
eritrocito (espectrina)– Disminución de lipidos de membrana– Disminución de la resistencia osmótica– Genes: ANK1, EPB3, ELB42, SPTA1, SPTB– Anemia crónica, exacerbación con infección– Anemia, ictericia, dolor abdominal (litiasis), esplenomegalia.– Crisis aplástica: parvovirus B19. – Esferocitos, Fragilidad osmótica.– DHL +; BI+; haptoglobina disminuida.– Esplenectomía.

• Eliptocitosis hereditaria– Eritrocitos ovales o elípticos– Autosómica dominante– Defecto molecular: espectrina, proteína 4.1,
glicoferina C.– Sintomática en homocigotos– Se exacerba con infección– Esplenectomía
• Eliptocitosis hereditaria– Eritrocitos ovales o elípticos– Autosómica dominante– Defecto molecular: espectrina, proteína 4.1,
glicoferina C.– Sintomática en homocigotos– Se exacerba con infección– Esplenectomía
Anemias hemolíticas hereditarias por defectos de membrana

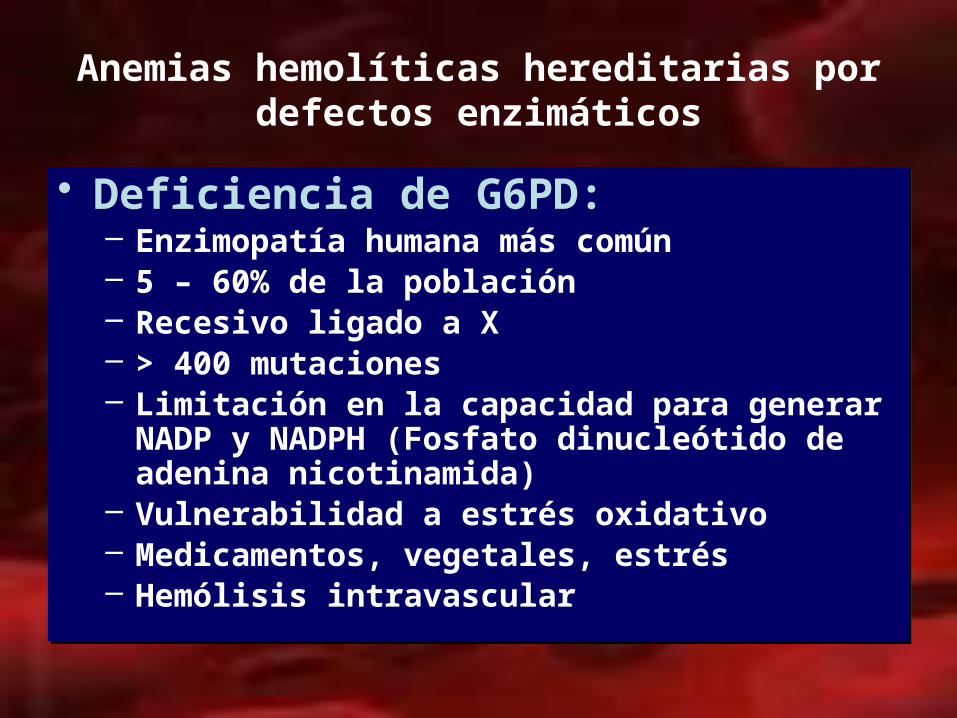
Anemias hemolíticas hereditarias por defectos enzimáticos
• Deficiencia de G6PD:– Enzimopatía humana más común– 5 – 60% de la población– Recesivo ligado a X– > 400 mutaciones – Limitación en la capacidad para generar NADP y
NADPH (Fosfato dinucleótido de adenina nicotinamida)
– Vulnerabilidad a estrés oxidativo– Medicamentos, vegetales, estrés– Hemólisis intravascular
• Deficiencia de G6PD:– Enzimopatía humana más común– 5 – 60% de la población– Recesivo ligado a X– > 400 mutaciones – Limitación en la capacidad para generar NADP y
NADPH (Fosfato dinucleótido de adenina nicotinamida)
– Vulnerabilidad a estrés oxidativo– Medicamentos, vegetales, estrés– Hemólisis intravascular

• Deficiencia de G6PD:– Hemólisis intravascular– Hemoglobinuria– Fiebre– Dolor lumbar– Ictericia– Medición de actividad enzimática– Cuerpos de Heinz (Hb oxidada)– Episodio hemolítico: medidas de apoyo (Tf)– Medida profiláctica: Evitar agentes hemolíticos
• Deficiencia de G6PD:– Hemólisis intravascular– Hemoglobinuria– Fiebre– Dolor lumbar– Ictericia– Medición de actividad enzimática– Cuerpos de Heinz (Hb oxidada)– Episodio hemolítico: medidas de apoyo (Tf)– Medida profiláctica: Evitar agentes hemolíticos
Anemias hemolíticas hereditarias por defectos enzimáticos

• Deficiencia de piruvato cinasa:– Autosómica recesiva– Alteración en la síntesis de dinucleótido adenina-
nicotinamida (NAD) y disminución de adenosin trifosfato (ATP)
– Hemólisis: homocigotos– Acantocitos, equinocitos, esferocitos– Esplenomegalia, litiasis vesicular, ictericia– Exacerbación con infección– Esplenectomía: alta demanda transfusional
• Deficiencia de piruvato cinasa:– Autosómica recesiva– Alteración en la síntesis de dinucleótido adenina-
nicotinamida (NAD) y disminución de adenosin trifosfato (ATP)
– Hemólisis: homocigotos– Acantocitos, equinocitos, esferocitos– Esplenomegalia, litiasis vesicular, ictericia– Exacerbación con infección– Esplenectomía: alta demanda transfusional
Anemias hemolíticas hereditarias por defectos enzimáticos

Hemoglobinopatías
• Anormalidades estructurales de la Hb– Drepanocitosis Hb S– Hb C– Hb E
• Anormalidades por sintesis reducida
de cadenas α o β–Talasemias
• Anormalidades estructurales de la Hb– Drepanocitosis Hb S– Hb C– Hb E
• Anormalidades por sintesis reducida
de cadenas α o β–Talasemias

Anemia drepanocíticaAnemia drepanocítica• Mayor frecuencia: África• Hemoglobina S• Mutación: sustitución de timina por
adenina en sexto codón del gen de glonina β ocasionando la codificación de valina por glutamina.
• Hb S: insoluble y se precipita en cristales con baja presión de oxigeno.
• Deformación del eritrocito en forma de hoz• Bloqueo de microcirculación
• Mayor frecuencia: África• Hemoglobina S• Mutación: sustitución de timina por
adenina en sexto codón del gen de glonina β ocasionando la codificación de valina por glutamina.
• Hb S: insoluble y se precipita en cristales con baja presión de oxigeno.
• Deformación del eritrocito en forma de hoz• Bloqueo de microcirculación
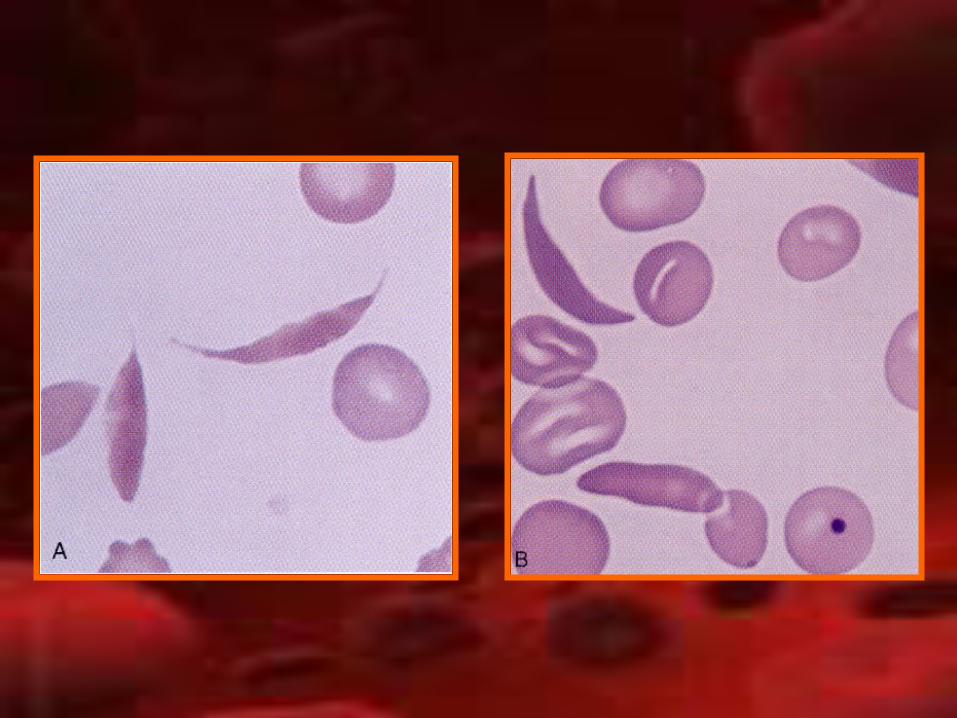

• Clínica:– Crisis dolorosas– Crisis vaso-oclusivas (infección, deshidratación,
cirugía)– Bazo atrófico y susceptibilidad a infección– Infartos óseos– Úlceras en piernas– Retinopatía proliferativa– Litiasis vesicular– Priapismo– Promedio de vida: 40 – 50 años
Anemia drepanocíticaAnemia drepanocítica

• Diagnóstico:– Drepanocitos (inducción)– Electroforesis de Hb– Reacción de polimerasa en cadena (PCR)
• Tratamiento:– Evitar factores que precipitan crisis– Ácido fólico– Analgesia, hidratación, Transfusión.– Exanguineo transfusión– Infarto esplénico grave: esplenectomía.– Hidroxiurea (induce Hb F y disminuye inflamación)– Futuro: Clotrimazol, magnesio, oxido nítrico,
ácidos grasos de cadenas cortas.
• Diagnóstico:– Drepanocitos (inducción)– Electroforesis de Hb– Reacción de polimerasa en cadena (PCR)
• Tratamiento:– Evitar factores que precipitan crisis– Ácido fólico– Analgesia, hidratación, Transfusión.– Exanguineo transfusión– Infarto esplénico grave: esplenectomía.– Hidroxiurea (induce Hb F y disminuye inflamación)– Futuro: Clotrimazol, magnesio, oxido nítrico,
ácidos grasos de cadenas cortas.
Anemia drepanocíticaAnemia drepanocítica

Talasemias
• Hemoglobina del adulto: – hemoglobina A : 2 α y 2 β
• Talasemia: Síntesis reducida o ausente de cadenas de globina.
• Talasemia α: – reducción o ausencia de cadenas α.– África, Mediterráneo, Asia
• Talasemia β: – reducción a ausencia de cadenas β– Asia.
• Hemoglobina del adulto: – hemoglobina A : 2 α y 2 β
• Talasemia: Síntesis reducida o ausente de cadenas de globina.
• Talasemia α: – reducción o ausencia de cadenas α.– África, Mediterráneo, Asia
• Talasemia β: – reducción a ausencia de cadenas β– Asia.

Talasemia α
• Deleción de los 4 genes de α: – incompatible con la vida.
• Ausencia de 3 cadenas: – Hb H, microcitosis e hipocromia grave.
• Ausencia de 2 cadenas: – hemólisis compensada, concentración de
Hb normal con microcitosis e hipocromia.
• Deleción de los 4 genes de α: – incompatible con la vida.
• Ausencia de 3 cadenas: – Hb H, microcitosis e hipocromia grave.
• Ausencia de 2 cadenas: – hemólisis compensada, concentración de
Hb normal con microcitosis e hipocromia.

Talasemia β
• Ausencia total de cadenas β: β0
– HbF aumentada; HbA ausente; HbA2 variable.
• Producción reducida de cadenas β (10-20%) : β+
– HbF aumentada; HbA variable; HbA2 normal
• Se han descrito más de 150 mutaciones. • Talasemia mayor (Enf. de Cooley):
Homocigotos• Síntomas a los 4-6 meses de edad.• Cadenas alfa: efecto tóxico, eritropoyesis
ineficaz.
• Ausencia total de cadenas β: β0
– HbF aumentada; HbA ausente; HbA2 variable.
• Producción reducida de cadenas β (10-20%) : β+
– HbF aumentada; HbA variable; HbA2 normal
• Se han descrito más de 150 mutaciones. • Talasemia mayor (Enf. de Cooley):
Homocigotos• Síntomas a los 4-6 meses de edad.• Cadenas alfa: efecto tóxico, eritropoyesis
ineficaz.

Talasemia β• Homocigotos:
– Hemólisis severa– Ictericia– Hepatoesplenomegalia– Cambios óseos por hiperplasia eritroide– Fracturas por adelgazamiento de corteza
ósea– Sobrecarga de hierro– Hemosiderosis– Infecciones frecuentes.
• Homocigotos:– Hemólisis severa– Ictericia– Hepatoesplenomegalia– Cambios óseos por hiperplasia eritroide– Fracturas por adelgazamiento de corteza
ósea– Sobrecarga de hierro– Hemosiderosis– Infecciones frecuentes.


• Diagnóstico– Anemia microcítica hipocrómica– Reticulocitosis– Normoblastos– Células en diana, dianocitos o blanco de tiro– Sobrecarga de hierro, Electroforesis de Hb con ausencia o
banda tenue de HbA y HbF aumentada– PCR: defecto molecular
• Tratamiento– Transfusiones– Desferoxamina (ped: 20-40 mg/k; ad: 2 g )– Ácido fólico– Esplenectomía– Transplante de células hematopoyéticas totipotenciales
• Diagnóstico– Anemia microcítica hipocrómica– Reticulocitosis– Normoblastos– Células en diana, dianocitos o blanco de tiro– Sobrecarga de hierro, Electroforesis de Hb con ausencia o
banda tenue de HbA y HbF aumentada– PCR: defecto molecular
• Tratamiento– Transfusiones– Desferoxamina (ped: 20-40 mg/k; ad: 2 g )– Ácido fólico– Esplenectomía– Transplante de células hematopoyéticas totipotenciales
Talasemia β

Anemias hemolíticas adquiridas
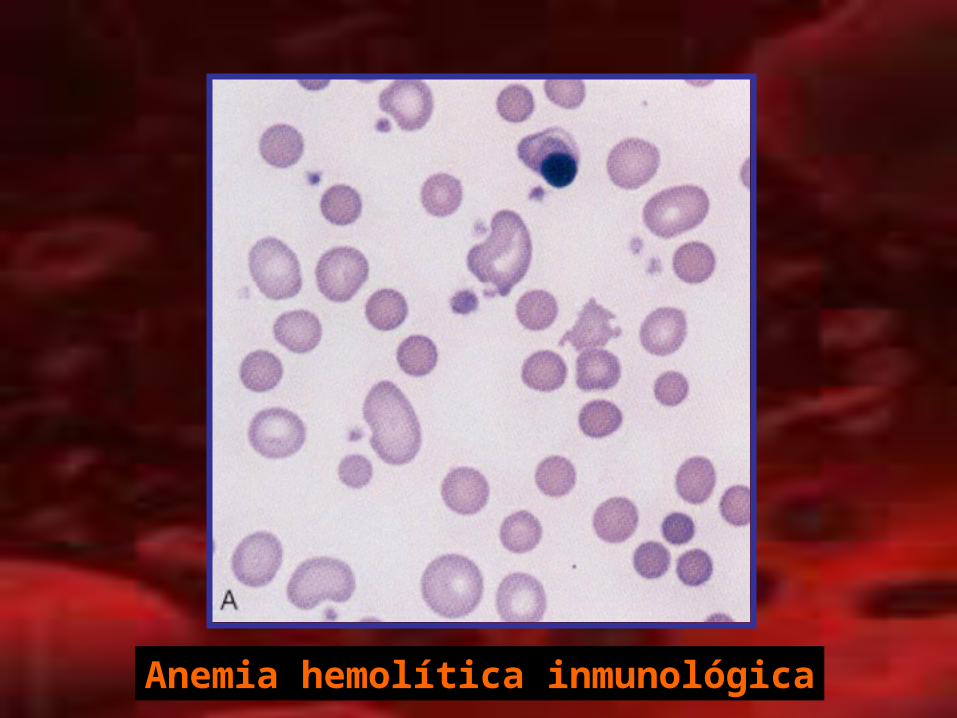
• Anemia hemolítica autoinmune– De anticuerpos calientes (reaccionan a 37°C)
– De anticuerpos fríos (4°C)
– Prueba de antiglobulina Directa (Coombs Directo)
positiva (negativo: no excluye)
– Anticuerpos IgG, IgA o IgM y C3, C3d.
– Presentación aguda o crónica
– Idiopática o Secundaria
– Hemólisis: Sistema reticuloendotelial del bazo
• Anemia hemolítica autoinmune– De anticuerpos calientes (reaccionan a 37°C)
– De anticuerpos fríos (4°C)
– Prueba de antiglobulina Directa (Coombs Directo)
positiva (negativo: no excluye)
– Anticuerpos IgG, IgA o IgM y C3, C3d.
– Presentación aguda o crónica
– Idiopática o Secundaria
– Hemólisis: Sistema reticuloendotelial del bazo
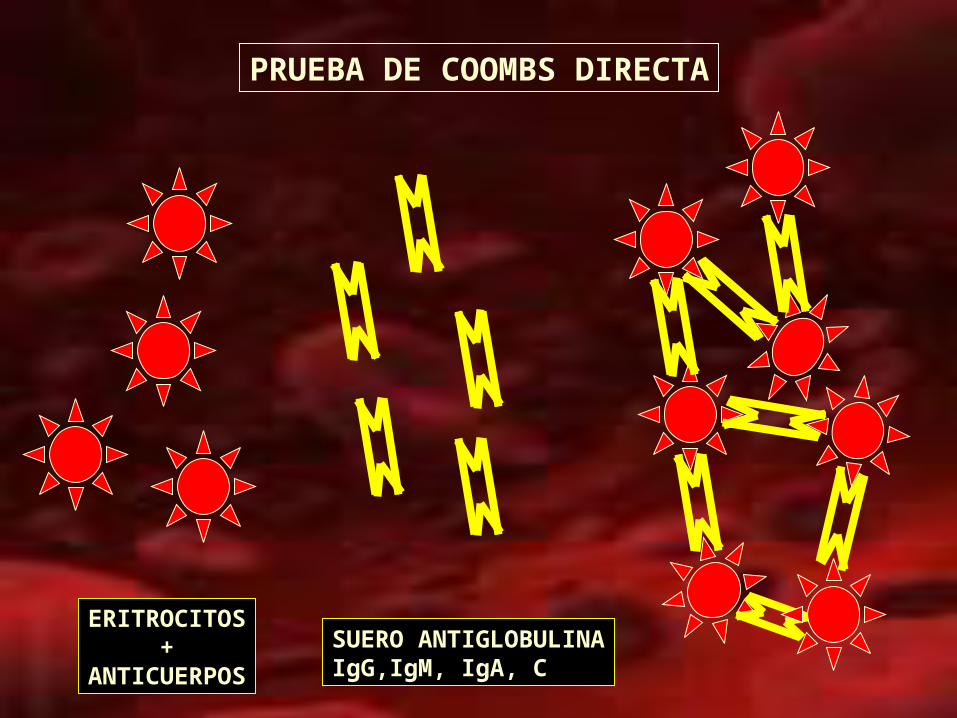
PRUEBA DE COOMBS DIRECTA
ERITROCITOS+
ANTICUERPOS
SUERO ANTIGLOBULINAIgG,IgM, IgA, C

Anemia hemolítica autoinmune anticuerpos calientes
• Idiopática; Sec: LES, linfoma, LLC, Metildopa, Procainamida, Diclofenaco, etc
• Clínica:– Fatiga, disnea, ictericia, fiebre.– Esplenomegalia.– Trombocitopenia (S. Evans)
• Laboratorio:– Microesferocitos– Antiglobulina +– Autoanticuerpos: dirigidos a grupo sanguíneo
específico: Rh, Kidd, LW.
• Idiopática; Sec: LES, linfoma, LLC, Metildopa, Procainamida, Diclofenaco, etc
• Clínica:– Fatiga, disnea, ictericia, fiebre.– Esplenomegalia.– Trombocitopenia (S. Evans)
• Laboratorio:– Microesferocitos– Antiglobulina +– Autoanticuerpos: dirigidos a grupo sanguíneo
específico: Rh, Kidd, LW.

• Tratamiento:
– Transfusión en crisis graves
– Esteroides: • PDN: 1-2 mg/k por día,
• MPS: IV
– Esplenectomía
– Inmunosupresores: CFM; CyA; AZAT
– Dosis altas de Igs
Anemia hemolítica autoinmune anticuerpos calientes

• Idiopática; Sec: Infecciones (Mycoplasma, VEB), linfomas, síndrome de aglutininas frías.
• Anticuerpos IgM• Complemento• Fagocitosis en SER de hígado.• Síntomas que se agravan con el frío• Acrocianosis, Ictericia y Esplenomegalia. • Coombs +• Anti-I• Anti- sistemas ABO, MNS o LW• Anticuerpos policlonales: Linfoma• Anticuerpos monoclonales: S. Aglutininas frías.
• Idiopática; Sec: Infecciones (Mycoplasma, VEB), linfomas, síndrome de aglutininas frías.
• Anticuerpos IgM• Complemento• Fagocitosis en SER de hígado.• Síntomas que se agravan con el frío• Acrocianosis, Ictericia y Esplenomegalia. • Coombs +• Anti-I• Anti- sistemas ABO, MNS o LW• Anticuerpos policlonales: Linfoma• Anticuerpos monoclonales: S. Aglutininas frías.
Anemia hemolítica autoinmune anticuerpos fríos

• Tratamiento:– Evitar el frío– Tratar la causa– Corticosteroides y esplenectomía: poco efectivos– Plasmaféresis– Inmunosupresores: CLB, CFM– Transfusión con eritrocitos lavados (evitar C) a
alta t°C– Hemoglobinuria paroxística fría; Ac. IgG bifásico
(Ac Donath Landsteiner) (Sífilis terciaria)
• Tratamiento:– Evitar el frío– Tratar la causa– Corticosteroides y esplenectomía: poco efectivos– Plasmaféresis– Inmunosupresores: CLB, CFM– Transfusión con eritrocitos lavados (evitar C) a
alta t°C– Hemoglobinuria paroxística fría; Ac. IgG bifásico
(Ac Donath Landsteiner) (Sífilis terciaria)
Anemia hemolítica autoinmune anticuerpos fríos

Hemoglobinuria Paroxística Nocturna (HPN)
• Alteración clonal de la célula totipotencial• Adquirida• Defecto en la molécula ancla de Fosfatidil-inositol-
glicano (PIG)• Mutación: gene de PIG-A en brazo corto de
cromosoma X• El defecto se observa en una o en todas las líneas
celulares hematopoyéticas• El glicolípido regula las moléculas de complemento
a través de la unión con proteínas de la membrana celular (CD59, CD55 C8)
• Defecto de estas moléculas: Incremento de actividad de lisis celular mediada por C y trombosis.

• Clínica:– Anemia hemolítica crónica o pancitopenia– Crisis hemolíticas nocturnas y hemoglobinuria
matutina– Complicación mayor: trombosis (porta. SNC)– Deficiencia de hierro– Anemia N-N, neutropenia, trombocitopenia– Pruebas de Sucrosa-Inulina y HAM +– Disminución en la expresión de CD55 y CD59– Heparina, Fe, A.Fólico, Esteroides, Danazol,
Andrógenos– TCTH
• Clínica:– Anemia hemolítica crónica o pancitopenia– Crisis hemolíticas nocturnas y hemoglobinuria
matutina– Complicación mayor: trombosis (porta. SNC)– Deficiencia de hierro– Anemia N-N, neutropenia, trombocitopenia– Pruebas de Sucrosa-Inulina y HAM +– Disminución en la expresión de CD55 y CD59– Heparina, Fe, A.Fólico, Esteroides, Danazol,
Andrógenos– TCTH
Hemoglobinuria Paroxística Nocturna (HPN)
Anemia de las enfermedades crónicas
• Anemia que acompaña a:– Infección crónica - A. sideroblástica– Enfermedad reumática - Enfermedad hepática– Neoplasia - Embarazo– Desnutrición - Insuf. Renal– Endocrinopatías - Intoxicación por plomo
• Hierro sérico normal o disminuido• Receptores de transferrina normales• Ferritina alta• Eritropoyetina y eritropoyesis disminuida
• Anemia que acompaña a:– Infección crónica - A. sideroblástica– Enfermedad reumática - Enfermedad hepática– Neoplasia - Embarazo– Desnutrición - Insuf. Renal– Endocrinopatías - Intoxicación por plomo
• Hierro sérico normal o disminuido• Receptores de transferrina normales• Ferritina alta• Eritropoyetina y eritropoyesis disminuida

Mecanismos para el desarrollo de anemia por enfermedad crónica.
Mecanismos para el desarrollo de anemia por enfermedad crónica.
• Desviación del transito del hierro del suero hacia las reservas del SER
• Eritropoyesis disminuida• Pobre respuesta a eritropoyetina• Disminución de la vida media
eritrocítica• Eritrofagocitosis
• Desviación del transito del hierro del suero hacia las reservas del SER
• Eritropoyesis disminuida• Pobre respuesta a eritropoyetina• Disminución de la vida media
eritrocítica• Eritrofagocitosis

Desviación del transito del hierro del suero hacia las reservas del SER
• Citocinas y metabolitos• Interleucina-1• Factor de necrosis tumoral• Citocinas pro-inflamatorias• Citocinas anti-inflamatorias: IL: 4, 10. 13
ANEMIA MEDIADA POR CITOCINASANEMIA MEDIADA POR CITOCINAS
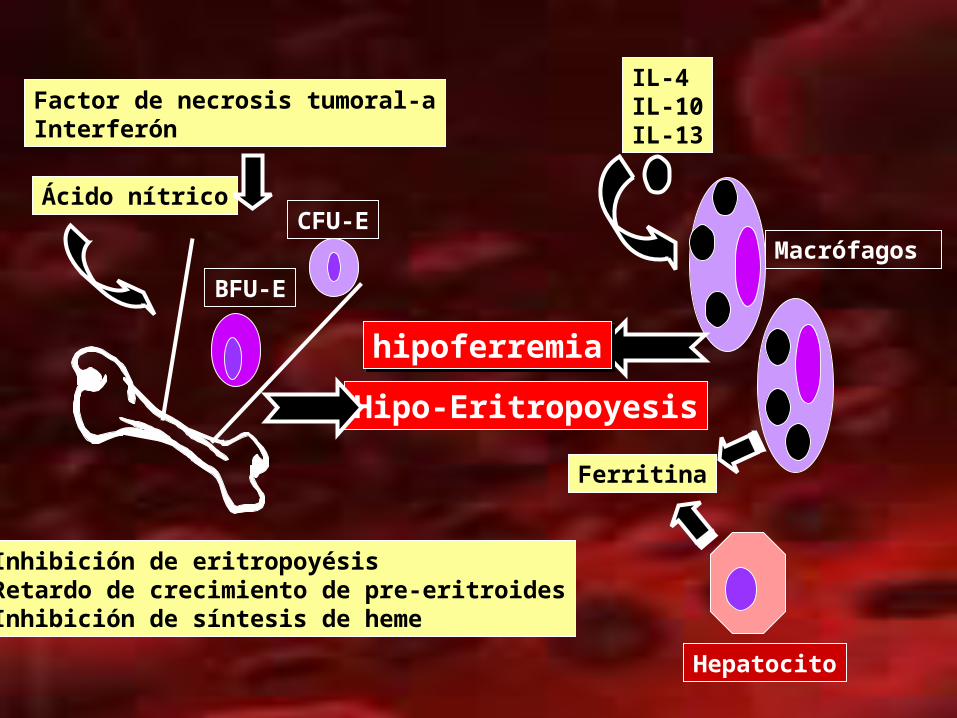
IL-4IL-10IL-13
Ferritina
Hepatocito
Macrófagos
hipoferremiahipoferremia
Hipo-Eritropoyesis
Factor de necrosis tumoral-aInterferón
BFU-E
CFU-EÁcido nítrico
Inhibición de eritropoyésisRetardo de crecimiento de pre-eritroidesInhibición de síntesis de heme

Tratamiento de la anemia por enfermedad crónica
Tratamiento de la anemia por enfermedad crónica
• Mejorar o eliminar la enfermedad de base• Anticuerpos anti-Factor de necrosis
tisular• Evitar el uso de hierro VO o parenteral
– Favorece crecimiento bacteriano– Exacerba la sobrecarga de hierro– Controversia con enfermedades autoinmunes
o reumáticas.• Eritropoyetina (?)
• Mejorar o eliminar la enfermedad de base• Anticuerpos anti-Factor de necrosis
tisular• Evitar el uso de hierro VO o parenteral
– Favorece crecimiento bacteriano– Exacerba la sobrecarga de hierro– Controversia con enfermedades autoinmunes
o reumáticas.• Eritropoyetina (?)

Anemia asociada a enfermedad hepática
• Macrocítica; dianocitos (colesterol)• Deficiencia de folatos• Sangrado crónico• Toxicidad del alcohol• Hemólisis• Hiperesplenísmo• Síndrome de Zieve: hemólisis
aguda, hiperlipidemia y hepatitis alcohólica
• Macrocítica; dianocitos (colesterol)• Deficiencia de folatos• Sangrado crónico• Toxicidad del alcohol• Hemólisis• Hiperesplenísmo• Síndrome de Zieve: hemólisis
aguda, hiperlipidemia y hepatitis alcohólica

Anemia en Insuficiencia RenalAnemia en Insuficiencia Renal
• Normocítica-normocrómica• Reticulocitopenia• Inadecuada producción de eritropoyetina• Disminución de sobrevida eritrocitaria• Efecto tóxico sobre eritropoyesis• Pérdida sanguínea durante la diálisis• Infección• Incremento de niveles de aluminio• Eritropoyetina: 50 150 U/K (3 veces por
semana, IV, SC)• Efectos colaterales: HTA, Trombosis.
• Normocítica-normocrómica• Reticulocitopenia• Inadecuada producción de eritropoyetina• Disminución de sobrevida eritrocitaria• Efecto tóxico sobre eritropoyesis• Pérdida sanguínea durante la diálisis• Infección• Incremento de niveles de aluminio• Eritropoyetina: 50 150 U/K (3 veces por
semana, IV, SC)• Efectos colaterales: HTA, Trombosis.

Abordaje Diagnóstico de AnemiaAbordaje Diagnóstico de Anemia
• Cifras de Hb y Hto por abajo del valor normal
según altitud, edad y sexo.
• Clasificación morfológica.
• Cuenta de reticulocitos
• Ancho de distribución es eritrocitos (ADE,
RDW, CV-VGM)
• Clínica
• Estudios especiales
• Cifras de Hb y Hto por abajo del valor normal
según altitud, edad y sexo.
• Clasificación morfológica.
• Cuenta de reticulocitos
• Ancho de distribución es eritrocitos (ADE,
RDW, CV-VGM)
• Clínica
• Estudios especiales
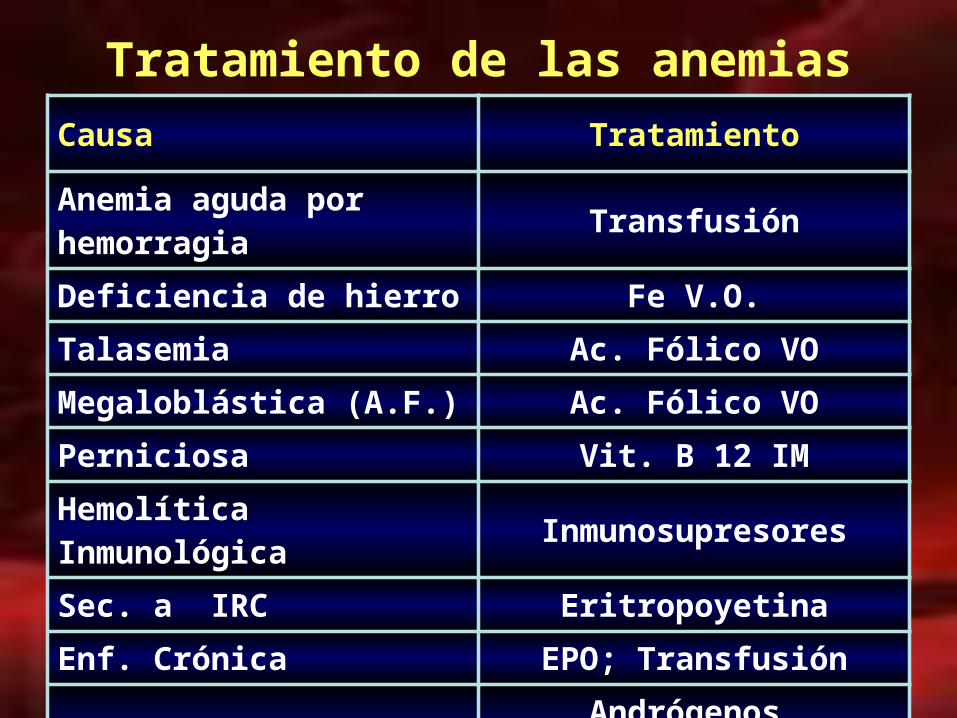
Tratamiento de las anemias
Causa Tratamiento
Anemia aguda por hemorragia Transfusión
Deficiencia de hierro Fe V.O.
Talasemia Ac. Fólico VO
Megaloblástica (A.F.) Ac. Fólico VO
Perniciosa Vit. B 12 IM
Hemolítica Inmunológica Inmunosupresores
Sec. a IRC Eritropoyetina
Enf. Crónica EPO; Transfusión
H.P.N. Andrógenos, esteroides