s4507431beaf003cb.jimcontent.coms4507431beaf003cb.jimcontent.com/download/version... · web viewen...
TRANSCRIPT

DEPARTAMENTO ACADÉMICO:
Morfología Humana
CURSO:
Genética Médica
TÍTULO:
Diagnóstico y tratamiento genético de las enfermedades
INTEGRANTES:
Pérez Ramírez, James
Polo Mejía, Juan Julio Joseph
Polo Saona, Christian
Poma González, Elka
Pretell Vargas, Crystel Yasmín
Pumamango Córdova, Jimmy Emerson
Quiroz Aldave, Juan Eduardo
Reyes Florián, Giuliana
Rivalles Alvarez, Renzo Renato
TRUJILLO – PERÚ
2010

Tabla de contenido
Introducción................................................................................................................................................3
Estrategias generales de diagnóstico de enfermedades..............................................................................4
Métodos de detección de mutaciones....................................................................................................4
Métodos de barrido.............................................................................................................................5
Métodos de comprobación...............................................................................................................35
Aplicación del ligamiento al diagnóstico (proceso de diagnóstico indirecto de las enfermedades genéticas)..............................................................................................................................................49
Tratamiento genético de las enfermedades..............................................................................................59
Proteínas recombinantes.......................................................................................................................69
Células madre......................................................................................................................................102
2

Introducción
Gran parte del conocimiento de la genética humana se funda en el estudio de las enfermedades
hereditarias (codificación genética que puede o no tener manifestaciones congénitas, como en la Corea
de Huntington, y la identificación de nuevos procedimientos clínicos ha estimulado el interés en su
estudio. El control en otro tipo de enfermedades ha propiciado que aumente la frecuencia de las de
etiología genética, por ejemplo, desde principios de siglo, los esfuerzos por preservar la salud han sido
dirigidos principalmente contra enfermedades infecciosas, pero debido al control epidemiológico
progresivo y uso de antibióticos, la esperanza de vida se ha incrementado en forma considerable, con la
consiguiente disminución de enfermedades.
En el presente trabajo ampliaremos a fondo respecto a las estrategias actualmente utilizadas para el
diagnóstico de enfermedades, los tratamientos genéticos seguidos como el uso de proteínas
recombinantes, y la aplicación de la tecnología de las células madre en el tratamiento y prevención de
enfermedades genéticas
Palabras clave: Métodos de detección de mutaciones, Aplicación del ligamiento al diagnóstico, Tratamiento
genético de las enfermedades, Proteínas recombinantes, Células madre
3

Estrategias generales de diagnóstico de enfermedades
Métodos de detección de mutaciones
Método de barrido
SSCP (single strand conformation polymorphism, Polimorfismo de la conformación de las
cadenas simples)
DGGE (Denaturing Gradient Gel Electrophoresis, Electroforesis en gel con gradiente
desnaturalizante)
DHPLC (Denaturing HPLC)
Rotura de desemparejamiento (Mismatch cleavage)
PPT (Protein Truncation Test, Prueba de truncamiento de proteínas)
Secuenciación
Métodos de comprobación
PCR – digestión
ASO (Allele Specific Oligonucleotide, Oligonucleótido específico de alelo)
ARMS (Amplification Refractory Mutation System)
OLA (Oligonucelotide Ligation Assay, Ensayo de ligación de oligonucleótidos)
4

Métodos de barrido
SSCP (single strand conformation polymorphism, Polimorfismo de la conformación de las cadenas
simples)1
En la búsqueda de mutaciones puntuales, se emplean métodos como el secuenciamiento directo,
RFLPs (Southern, 1975), clivaje químico,CCM (Cotton et al., 1988), clivaje por RNAsa (Goldrick et al.,
1996) u oligonucleótidos alelo específicos para hibridación (ASO) (Nollau & Wagener, 1997), entre
otros. Estos métodos presentan diferentes rangos de sensibilidad, además de requerir reactivos y
equipos sofisticados, que aumentan su complejidad y costo operativo.
Frente a ellos, el método de SSCP es fácil y económico, pues usa segmentos amplificados por PCR
visualizados en geles de acrilamida con adyuvantes (Orita et al., 1989).
El SSCP es un método capaz de identificar la variación de un sólo nucleótido en un segmento de
ADN, típicamente entre 150 a 200 nucleótidos de largo. En condiciones ideales el rango de
sensibilidad del SSCP varía entre 8090% para detectar mutaciones en fragmentos menores de 200
pares de bases (Sheffield et al., 1993). El método del SSCP se basa en que bajo condiciones no
desnaturalizantes una hebra individual de ADN adopta una conformación espacial que es específica
de la composición de su secuencia nucleotídica (Fig. 1).
Esta conformación sería dependiente de la hibridación entre distintas regiones de un segmento de
ADN replegado sobre sí mismo. La configuración diferente es provocada por el cambio de una sola
base, entonces podría ser detectada en algunas condiciones de migración electroforética en una
matriz de poliacrilamida (Orita et al., 1989, Humphries et al., 1997).
La técnica es simple, versátil y económica, pero exige la optimización de parámetros en la
composición del gel para cada fragmento a evaluar
1 Alejandro Estrada-Cuzcano, José Sandoval, María L. Guevara-Fujita y Ricardo Fujita. Uso de la técnica SSCP para detectar mutaciones puntuales del ADNmitocondrial humano
5

Figura 1. Representación esquemática del
fundamento de la técnica de SSCP durante la
electroforesis en un gel no desnaturalizante.
La movilidad electroforética de cada
fragmento depende del auto plegamiento de
acuerdo a la formación de estructuras
secundarias.
Procedimiento
El analisis de SSCP puede realizarse con la amplificacion por PCR para analizar pequeñas regiones
amplificadas directamente del DNA genomico o del cDNA.Para ello se sintetizan dos
oligonucleotidos cebadores que flanquean (con sentido y antisentido)la region de interes y el DNA
se amplifica por PCR en presencia de nucleotidos marcados radiactivamente.El producto de la PCR,
un DNA de doble hebra marcado, se desnaturaliza por calor en presencia de formamida al 80% e
inmediatamente se procede a sus analisis a traves de elctroforesis en un gel de poliacrilamida no
desnaturalizante .Durante esta los fragmentos de ADN de cadena sencilla se pliegan en una forma
tridimensional de acuerdo a su secuencia.La separacion depende,por tanto,de la forma de las
moleculas de cadena sencilla. Las movilidades de los productos de los pacientes se comparan con las
de los controles a partir de sujetos normales,pues si los productos normal y mutante difieren en
algun nucleotido adoptaran estructuras tridimensionales diferentes y mostraran distintas
movilidades electroforetica.
6

La conformación del ADN en el gel puede ser alterado por una serie de condiciones que tienen que
ser optimizado para detectar la mutación. Estas variables incluyen:
Propiedades y tamaño de los poros del gel, estos se determinan por el porcentaje de
poliacrilamida, la relación de acrilamida / bis (cross-linker), y la Temperatura en la que la
polimerización se lleva a cabo, incluyendo la Temperatura de los reactivos.
Presencia y porcentaje de glicerol,este tiene una accion desnaturalizante debil para abrir
parcialmente la estructura del DNA plegado en una mayor superficie para que el gel detecte
cambios conformacionales
7

Temperatura ala que se realiza la electroforesis
pH del buffer usado para hacer el gel
pH y fuerza ionica del sitema buffer.
Preparacion de muestras y electroforesis
Gel colorante cargado que contiene formamida impide la renaturalización del DNA de cadena simple
después de la desnaturalización. ADN de doble cadena (dsDNA) se ejecuta con las muestras para
actuar como un marcador y distinguir su banda de los creados por ssDNA
1. Las muestras serán preparadas como siguen en las etiquetas de los tubos
2. Los tubos tienen un límite y se centrifuga durante unos segundos a 4 ° C
3. Las muestras se desnaturalizaron a 95C durante 6 minutos. Esto se puede hacer en un bloque
caliente, pero es más conveniente utilizar un ciclador termal PCR.
4. Para evitar la renaturalización, los tubos son colocados inmediatamente en hielo durante 10
minutos.
5. Centrifugar los tubos de nuevo durante 1 min a 4 ° C, a continuación, inmediatamente vuelve a
poner en el hielo
6. Preparar el ADN de doble cadena simple en un tubo con la etiqueta:
7. Las muestras se cargan en el gel de poliacrilamida con un pico de pato punta de la pipeta
8. El gel se suele ejecutar a 25 W durante aproximadamente 18 horas en 1XTBE. Un simple DNA de
cadena simple de 200 pb tendrá una duración aproximada de tres cuartas partes del camino por el
gel. El poder puede ser modificado para variar el tiempo de ejecución y la separación.
8

Los patrones de bandas de SSCP se pueden detectar mediante el uso de la radiactividad, tinción de
plata o de fluorescencia.
Aplicaciones clinicas frecuentes del SSCP
Esta tecnica puede utilizarse para el diagnostico de gran numero de enfermedades y mutaciones
somaticas.
Aplicaciones clínicas más frecuentes del SSCP
1. Mutaciones somáticas en cáncer
RAS
P53
RB
2. Genes de enfermedades hereditarias
Neurofibromatosis tipo I
Poliposis colónica familiar (APC)
Síndrome de mujer XY
Síndrome de Marfan (gen de fibrilina)
Parálisis periódica hiperkalémica
Tumor de Wilms
9

Fibrosis quística
Hipertriglicerdemia
Tay Sachs tipo 1
Fenilcetonuria
Hemofilia B
Raquitismo vitamina D dependiente
Retinitis pigmentosa (gen de rhodopsina)
Peripherin – RDS gen
Enfermedad Pelizaeus – Merzbacher
Polimorfismos y alteraciones alélicas familiares
Desventajas de la técnica SSCP
Las desventajas del método PCR-SSPC son los falsos negativos, que como hemos indicado pueden ocurrir hasta en un 10% de las ocasiones y que no nos permiten estar seguros de que no haya ninguna mutacion en los casos negativos.
DGGE (Denaturing Gradient Gel Electrophoresis, Electroforesis en gel con gradiente desnaturalizante)
La electroforesis en gel con gradiente de desnaturalización, también referida como electroforesis en
gel desnaturalizante en gradiente, (en el ámbito científico comúnmente referida por su abreviación
en inglés: DGGE) es una técnica de huella, rastreo o trazado molecular (vulgarmente conocida en
Inglés como molecular fingerprinting) usada en diversas disciplinas de la biología y la química, que
consiste en la separación de cadenas de ADN de doble cadena dependiendo de su punto de
desnaturalización, siendo dicha desnaturalización definida en éste caso como la separación de
cadenas complementarias de ADN
10

Procedimiento
DGGE se basa en la moovilidad electroforetica de una molecula de doble cadena a traves de
concentraciones linealmente crecientes de un agente desnaturalizante.El gradiente desnaturalizante
en gel para electroforesis [DGGE] literalmente separa los fragmentos de acuerdo con sus
propiedades desnaturalizantes.En consecuencia,el éxito de DGGE depende de la eleccion cuidadosa
de las condiciones de desnaturalizacion. La foramida y la urea se utilizan sobretodo como agentes
desnaturalizantes,pero el aumento de gradiente de temperatura es usado con frecuencia por
muchos investigadores.
El punto de desnaturalización de tal fragmento de ADN aumentará con el tamaño de la secuencia de
nucleótidos que lo conforman, y también por la composición de nucleótidos de la secuencia, que
suele aumentar con altos contenidos de Guanina (G) y Citosina (C), aumentando la probabilidad en
el incremento de secuencias que lleven consecutivamente G o C. Hay relaciones derivadas de esta
11

relación: secuencias ricas en GC tienen, con sus secuencias de bases complementarias,
apareamientos más espontáneos (fenómeno conocido en inglés como Base Stacking) ya que en
estos casos, tienen energía de Gibbs más negativa y por tal motivo aumenta la temperatura mínima
de fundición (o desnaturalización en este caso, conocido en Inglés con la abreviatura Tm) de la
secuencia de ADN. Un DNA doble con un par mal unido se desnaturaliza con mayor rapidez que un
doble unido perfectamente. Si ocurre la desnaturalización, se acompañara por una disminución en la
migración sobre el gel y aparecerá como un alto peso molecular con respecto al control del doble
unido perfectamente. Generalmente se utilizan dos métodos para crear dicho gradiente
desnaturalizante en un gel: por incremento de concentraciones de sustancias desnaturalizantes
dependiendo de la distancia recorrida en el gel, o por incremento de la temperatura a través del
tiempo en que los fragmentos van migrando a través del gel
La tecnica de PCR-DGGE es muy poderosa cuando se aplica para la deteccion de variantes de
nucleotidos para heterocigotos.La desnaturalizacion continua y el realineamiento de moleculas de
cadena simple durante la PCR permiten la formacion de dobles homologos asi como de dobles
heterologos hibridados.La presencia de una simple union mal efectuada en dobles heterologos
disminuye en forma importante su temperatura de fusion y permite la separacion de los dobles
homologos y su facil visualizacion para la deteccion de mutaciones.Con los fragmentos de DNA de
mas de 500 pares de bases ,el DGGE ha sido altamente sensible y detecta el 99% de las secuencias
cambiadas si las condiciones del gel son optimas.La tecnica de exploracion detecta cerca del 50% de
toddos los cambios de bases simples.Aunque este es un campo de desarrollo rapido,la tecnica de
DGGE ha permanecido como la prueba del momento.
Esta tecnica presenta ventajas,pero tambien desventajas con respecto a otras tecnicas.Asi,una sw
las principales ventajas de la DGGE es la posibilidad de identificar los organismos presentes,
mediante el corte y secuenciacion de las bandas que aparecen despues de la elctroforesis,por
tecnicas de hibridacion con sondas especificas,o bien con tecnicas de clonacion,etc.Otra de las
ventajas es la posibilidad de obtener informacion sobre mutuaciones puntuales,ya que el cambio en
un solo nucleotido puedce ser detectado por esta tecnica.
La principal desventaja de esta tecnica es que distintas secuencias pueden migrar hasta el mismo
punto, esto sobre todo es importante cuando se recortan bandas para su secuenciacion ya que
pueden recogerse bandas situadas muy proximas entre si.Otra desventaja es que aunque es capaz
12

de identificar mutaciones en una secuencia no da informacion de a que nivel se encuentra la
mutacion ,aunque esta informacion puede obtenerse con la secuenciacion de fragmentos de ADN.
DHPLC (Denaturing HPLC)2
Antes de estudiar el fundamento del DHPLC tenemos que conocer cómo se procesan las muestras
previamente a ser analizadas. Así pues, una vez extraído el ADN, se amplifica el fragmento de gen a
estudiar por medio de una PCR convencional; posteriormente, se realiza una desnaturalización a 95
°C durante tres minutos, seguido de una disminución progresiva de la temperatura de 95 °C a 65 °C
durante 30 minutos, permitiendo así que las hebras renaturalicen lentamente.
Con este proceso se podrán detectar las mutaciones (mismatches) sobre la base de las diferentes
uniones formadas después de la renaturalización entre los fragmentos de gen amplificados, es decir,
al renaturalizar lentamente provocamos que se forme de nuevo la doble hebra de ADN, uniéndose
las hebras exactamente igual que las originales (la doble hebra de ADN wild type “normal”, por una
parte, y la mutada por otra), a las que llamaremos homodúplex, y además provocamos que se unan
combinaciones de las cadenas sentido y antisentido de cada homodúplex, para formar los llamados
heterodúplex.
Así pues, se puede decir que el DHPLC detecta las mutaciones según los diferentes homodúplex y
heterodúplex que se forman del fragmento de gen estudiado. Los heterodúplex, que son
2 A. Pons Castillo, A. Bennani, F. Cañizares Hernández. HPLC desnaturalizante (DHPLC): nuevo método descreening de enfermedades genéticas
13

térmicamente menos estables que su correspondiente homodúplex, se separan por cromatografía
líquida de fase reversa, que trabaja en un rango de 50-70. La clave del funcionamiento consiste en
una fase estacionaria inerte física y químicamente, compuesta por partículas de poliestireno-
divinilbenceno de 2-3 μm de diámetro. Dicha fase estacionaria es eléctricamente neutra e
hidrofóbica. El ADN, que posee grupos fosfato, está cargado negativamente y, por lo tanto, no
puede adsorberse a la matriz de la columna por sí mismo. Con el fin de permitir que el ADN se una a
la fase estacionaria, se usa una molécula conocida por el nombre de “reactivo de par-iónico”,
tratándose en este caso del acetato de trietilamonio (TEAA), que posee carga positiva, gracias a los
iones amonio. La carga positiva de los iones amonio del TEAA interacciona con la carga negativa de
los iones fosfato del ADN, mientras que las cadenas alquil del TEAA interaccionan con la superficie
hidrofóbica de la fase estacionaria. Por tanto, las moléculas de doble cadena de ADN se retienen
según su longitud. La elución de las moléculas de ADN se consigue haciendo pasar por la columna
acetonitrilo (solvente orgánico) que será la fase móvil, lo cual resulta en la desorción de los iones
anfifílicos y moléculas de ADN. Dicha elución se percibe en un detector de ultravioleta a una
absorbancia de 254 nm, de tal forma que se elimina el tedioso proceso de teñir geles.
El tiempo de retención de la doble cadena de ADN cambia con la temperatura de la columna. A
temperatura < 50 °C el orden de elución es estrictamente basado en la longitud de la doble cadena
de ADN, en cambio a temperatura > 50 °C las moléculas de DNA se desnaturalizan, separándose en
dos cadenas simples complementarias. La desnaturalización en los heterodúplex es más extensa
que la correspondiente a los homodúplex que tienen las bases perfectamente emparejadas, por
consiguiente, los heterodúplex se retienen menos, eluyendo delante de los homodúplex como uno
o más picos adicionales, dependiendo del número de mutaciones (mismatches) que existen.
14

La inestabilidad térmica de los heterodúplex dependerá de los puentes de hidrógeno entre las bases
que están causando la mutación (bases no opuestas), tales como GT y GA, y de la influencia de la
secuencia cercana. En la práctica cualquier cambio en el perfil de elución, incluyendo cambios
sutiles, como la apariencia del frente o un ensanchamiento de un pico, es diagnóstico y permite
establecer la localización y naturaleza de la base o bases alteradas.
Sensibilidad y especificidad en la detección de enfermedades
La temperatura es el parámetro más importante que afecta la sensibilidad del DHPLC en la
detección de mutaciones. Originariamente, la temperatura para el análisis de una secuencia
particular de ADN se determinaba empíricamente, realizando repetidas inyecciones de una muestra
y aumentando gradualmente la temperatura de la columna hasta que los picos empiezan a aparecer
a tiempos de retención más cortos. A partir de este punto, la presencia de mutación o mutaciones
(mismatches) se detecta por la aparición de uno o más picos que eluirán inmediatamente antes que
los homodúplex. Sin embargo, esta forma empírica para determinar la temperatura adecuada de
análisis, corre el riesgo de que los fragmentos ricos en dominios AT, que poseen baja temperatura
de meelting (desnaturalización), no se detecten debido a su completa desnaturalización. Por este
motivo se ha desarrollado un algoritmo, que calcula la temperatura en una secuencia conocida para
que el 50% del fragmento permanezca sin desnaturalizar. Este algoritmo está disponible en la web:
http://insertion.stanford.edu/melt.html.
El primer gen que se analizó por DHPLC fue el gen CACNL1A4, del canal de Calcio. Hasta la fecha se
han analizado más de 100 genes por DHPLC, obteniendo una sensibilidad y especificidad excelente,
15

para la detección de pequeñas mutaciones, estando todas ellas documentadas en la literatura
<http://insertion.stanford.edu/pub.html>.
En el análisis para detectar mutaciones en el exón H del gen del factor de coagulación IX (F9) y el
exón 16 del gen tipo 1 de la neurofibromatosis (NF1) se describe una sensibilidad y una especificidad
del cien por cien. Análisis previos del exón H del gen F9 por análisis conformacional de cadena
sencilla (SSCP) detectó únicamente el 50% de las mutaciones.
Se han estudiado más de 180 mutaciones diferentes en los genes de cáncer de mama BRCA1 y
BRCA2 por DHPLC, encontrando 179 perfiles de elución diferentes de las 180 mutaciones existentes.
El estudio del gen BRCA1 identificó en 41 casos, cuatro mutaciones causantes de enfermedad,
mientras que sólo tres de estas cuatro mutaciones se detectaron mediante electroforesis en gel
desnaturalizante (DGGE).
La sensibilidad del análisis conformacional de cadena sencilla (SSCP) disminuye enormemente
cuando se intentan analizar fragmentos más grandes de 300 pares de bases, o en el caso de
mutaciones localizadas dentro de estructuras conformacionales en horquilla.
La sensibilidad del DHPLC se mantiene con tamaños de fragmentos comprendidos entre 150 y 700
pares de bases y, además, no se ha identificado, hasta la fecha, ninguna estructura conformacional
que impida la detección de mutaciones por DHPLC.
En el ADN también nos podemos encontrar dominios en los que existen fragmentos que poseen
diferentes temperaturas meelting. El DHPLC demuestra estar menos afectado que el DGGE al
analizar este tipo de dominios, así pues, el análisis por DHPLC del exón 8 del gen HPRT detectó 20 de
20 mutaciones, independientemente de su localización en cualquiera de los dominios, con
temperaturas que diferían de 65 a 69 °C. En cambio, el análisis por DGGE no detectó mutaciones en
los dominios con mayores temperaturas meelting.
La secuenciación directa está considerada el “estándar oro” en el análisis de mutaciones.
Sin embargo, la secuenciación directa falla en la determinación de alelos mutantes poco frecuentes.
El DHPLC, en contraste, detecta fiablemente alelos mutantes presentes a frecuencias tan bajas como
el 10%. Por tanto, perfiles de heterodúplex que son reproducibles mediante el análisis por DHPLC no
deberían clasificarse de antemano como falsos positivos, si la secuenciación directa falla en
16

identificar los mismatch. Por consiguiente, los fragmentos amplificados deben ser clonados, y a
continuación realizar una secuenciación de un número suficiente de clones.
Análisis por desnaturalización completa
El análisis de fragmentos cortos de ADN (50-100 pb) se realiza en el DHPLC bajo condiciones
desnaturalizantes completas. El elevado poder de resolución del sistema de separación permite
discriminar entre dos ácidos nucleicos monocatenarios de idéntico tamaño, con unas diferencias en
la composición que pueden oscilar desde una base a 100 pares de bases.
Análisis multiplex por DHPLC
A diferencia de la electroforesis capilar, el HPLC convencional no se ha podido poner a punto para
analizar muestras en paralelo, usando una batería de columnas. Pero con la reciente introducción de
columnas capilares de poliestireno divinilbenceno, ahora es posible analizar varias muestras en
paralelo. El rendimiento aumenta con el número de columnas usadas en el sistema, y con el número
de muestras analizadas simultáneamente en la misma columna, lo cual es posible por la nueva
tecnología fluorescent color multiplexing.
Los diferentes amplificados por PCR se marcan con fluoroforos diferentes, usando primers
fluorescentes. Las reacciones de PCR se juntan después de haberse desnaturalizado y renaturalizado
por separado, analizándose simultáneamente en una misma columna. La elución de los diferentes
fragmentos se realiza usando un detector que registre las diferentes longitudes de onda
características de los fluoróforos empleados.
Puesto que los fluoróforos afectan al tiempo de retención de los ácidos nucleicos marcados, el
mismo amplificado marcado con diferentes fluoróforos dará diferentes perfiles.
Rotura de desemparejamiento (Mismatch cleavage)
Se produce los heterodúplex ADN:ADN por calor y reacoplamiento y se exponen a una endonucleasa
tras lo cual los fragmentos se analizan por electroforesis en gel. Se ha utilizado las nucleasas de la
haba mung y S1, pero este método es muy dependiente de la secuencia y no detecta muchas
mutaciones.
17

EMC con resolvasas de bacteriófagos:Es un método sensible para la detección de mutaciones en
heterodúlplexs de ADN ramificadas. Dos de estas enzimas, la T4 endonucleasa VII (T4E7) y la T7
endonucleasa I, (T7E1) rompen en ADN cerca de los sitios de discordancia de las bases. Los
productos de la rotura pueden ser analizados mediante electroforesis en gel y permite analizar
fragmentos de al menos 1500 bp. el método permite detectar un 95% de las mutaciones y parece
ser más sensible que SSCP en el cribado de algunos genes, como BRCA1215.
Heteroduplexes generados por la desnaturalización de calor de los productos PCR de ADN
polimórfico o de tipo salvaje y mutante alelos, respectivamente, se incuban y troceados por
cualquiera de la T4 YII bacteriófago T7 endonucleasa o fragmentos de ADN se analizan I. por
electroforesis. Tinción de plata deben ser factibles.
Tamaño del Fragmento: Las mutaciones son detectables en los productos de PCR entre 88 y
940 pb o de hasta 1,5 kb.
Límite de detección: No hay estudios sistemáticos sobre la menor proporción de mutantes
para detectar alelos de tipo salvaje se conocen
18

PPT (Protein Truncation Test, Prueba de truncamiento de proteínas)
El PTT (protein truncation test) sólo sirve para detectar mutaciones que implican la aparición de un
codón (secuencia de 3 nucleótidos) de fin de la traducción proteica (stop codon). Se realiza una PCR
para amplificación de un gen que codifica para una determinada proteína utilizando iniciadores a los
que se ha añadido secuencias de iniciación para células eucariotas y un promotor T7. Por lo tanto,
los productos de PCR (que contienen estas secuencias) se pueden someter a reacciones de
transcripción-traducción con lo que se consigue péptidos. El tamaño de los péptidos será menor en
el caso de que ADN contenga una mutación que determine la aparición de un codón de fin puesto
que la reacción se detendrá en este punto. El tamaño de los péptidos se determina mediante
electroforesis de los mismos para proteínas (Western blot).
La prueba de truncamiento de proteína (PTT) provee una excepción rara, apuntar a las mutaciones
que generan acortó proteínas, terminación de traducción principalmente prematura. La prueba de
truncamiento de la proteína (PTT) proporciona no un rara excepción, dirigido un las mutaciones que
19

generan proteínas acortado, terminación de la traducción, principalmente prematuros. PTT tiene
varias características atractivas, incluyendo precisando el sitio de una mutación, la buena
sensibilidad, una tasa del positivo falso bajo, y, más importante aún, el destacamiento de exclusiva
cercana de mutaciones que causan enfermedad. PTT tiene varias características atractivas,
ferrocarril elevado como señalando sitio de no un mutación, no un buena sensibilidad, no un baja
tasa de falsos positivos, y, más importante aún, la casi exclusiva destacando de mutaciones
causantes de enfermedad. Además, PTT facilitó la detección de un tipo nuevo de mutación,y, un
cambio de secuencia generando una región hipermutable saliendo a la superficie en la RNA.
Además, PTT facilitado la detección de tipo de mutación nueva, es decir, cambio de secuencia de
generación de no un ferrocarril elevado región hipermutables la superficie en ARN. Los problemas
técnicos principales son con los que se guardó relación el hecho que PTT generalmente usa un
blanco RNA, incluyendo las dificultades que provienen de la expresión diferencial potencial y la
estabilidad de los traslados derivativos del presente de dos alelos. Los problemas técnicos
principales se relacionan atraiga con engaño ferrocarril elevado hecho de que PTT generalmente
utiliza objetivo de ARN, incluyendo las dificultades que surgen de la expresión diferencial de
potencial y la estabilidad de los transcritos derivados de los dos alelos presentes. El PTT apenas ha
evolucionado del método originalmente descrito, con multiplexing y proteína terminal a N
etiquetando formando las únicas modificaciones que innova. El ferrocarril elevado del ferrocarril
elevado PTT apenas ha evolucionado desde método descrito originalmente, contra la proteína de la
multiplexación y de terminal a N marcado que forman las únicas modificaciones innovadoras.
Implementar rendimiento específico alto oculta usar a PTT, las mejoras principales del
procedimiento básico serán requeridas. La contra del rendimiento de la contralto Para Implementar
las pantallas de PTT, ferrocarril elevado las grandes mejoras de procedimiento básico se requiere.
Limitaciones
Detección de la mutación por PTT puede ser limitado de dos maneras: 1) el ferrocarril elevado falta
de amplificar alelo mutado y, 2) los deletions/insertions del marco falta de detección muy pequeño
en o mutaciones de mal-sentido supresiones marco inserciones o mutaciones pecan sentido. El
fracaso de amplificación puede tener varias explicaciones incluyendo una mutación de la cartilla
Error de amplificación puede tener varias explicaciones como no un sitio obligatorio mutación de la
cartilla (una secuencia alterada o suprimida), unas inserciones grandes, desplazamientos y unas
inversiones que traspasan largos del amplifiable, o sitio de unión (no un secuencia de alteración o
20

borrado), grandes inserciones, duplicaciones translocaciones e inversiones que del vehículo
remolque recubierto más allá de longitudes amplificable, o cuando único o sitios de ambos
imprimador de obligatorio consiste dentro del ferrocarril elevado duplicado del segmento
duplicaciones cuando uno o ambos sitios de unión cartilla se encuentran dentro de segmento
duplicado. Las mutaciones que afectan la cantidad de RNA Las mutaciones que afectan un la
cantidad de ARN producido o que entregue el mRNA mutado sensitivo también no puede ser
ferrocarril elevado detectado producidos o que hacen que ARNm mutado inestable tampoco
pueden ser detectados. PTT no podrá detectar muy parte pequeña hacia adentro no tender una
celada a PTT ferrocarril elevado detectar muy pequeñas dentro de marco deletions/insertions
porque los trajes rectos de movilidad son muy pequeños para detectar a . deleciones / inserciones,
hijo porque los cambios de la movilidad demasiado pequeños para detectarlos. Las mutaciones
Missense no saldrán de mutaciones de cambio de sentido no descubiertos detectada del mar.
Secuenciación
A finales de los años 70 se desarrollaron los métodos que permitieron de manera simple y rápida,
determinar la secuencia nucleotídica de cualquier fragmento de ADN. Estos primeros intentos de
secuenciar ácidos nucleicos siguieron los pasos empleados en la secuenciación de proteínas: romper
las moléculas en pequeños fragmentos, determinar su composición de bases y deducir la secuencia
21

a partir de fragmentos solapantes. Este método resulta relativamente sencillo para proteínas donde
estas resultan de la combinación de hasta 20 aminoácidos distintos, pero constituye un problema en
el caso de los ácidos nucleicos donde la secuencia resulta de la combinación de únicamente cuatro
nucleótidos diferentes.
Métodos clásicos de secuenciación
Los dos protocolos clásicos de secuenciación (método químico y enzimático) comparten etapas
comunes.
Marcado: Es necesario marcar las moléculas a secuenciar radiactiva o fluorescentemente
Separación: Cada protocolo genera una serie de cadenas sencillas de ADN marcadas cuyos
tamaños se diferencian en una única base. Estas cadenas de distintas longitudes pueden
separarse por electroforesis en geles desnaturalizantes de acrilamida-bisacrilamida-urea,
donde aparecen como una escalera de bandas cuya longitud varía en un único nucleótido.
Para obtener estas cadenas se emplean dos procedimientos:
Método químico de Maxam y Gilbert
Originalmente descrito por A. Maxam y W. Gilbert en 1977.
Esta técnica consiste en romper cadenas de ADN de cadena sencilla marcadas radiactivamente
con reacciones químicas específicas para cada una de las cuatro bases. Los productos de estas
cuatro reacciones se resuelven, por electroforésis, en función de su tamaño en geles de
poliacrilamida donde la secuencia puede leerse en base al patrón de bandas radiactivas
obtenidas.
Un fragmento de ADN se marca radiactivamente en sus extremos con gamma 32P ó gamma 32S
dATP por acción de la polinucleótido quinasa. La técnica consiste en romper estas moléculas
marcadas con reacciones químicas específicas para cada una de las cuatro bases. Cuatro
alícuotas de la misma muestra se tratan bajo condiciones distintas, posteriormente el
tratamiento con piperidina rompe la molécula de ADN a nivel de la base modificada. Los
productos de estas cuatro reacciones se resuelven en función de su tamaño en geles de
poliacrilamida donde la secuencia puede leerse en base al patrón de bandas radiactivas
obtenidas. Esta técnica permite la lectura de unas 100 bases de secuencia.
22

Método enzimático de Sanger
Este método de secuenciación de ADN fue diseñado por Sanger, Nicklen y Coulson también en
1977 y se conoce como método de los terminadores de cadena o dideoxi.
Para este método resulta esencial disponer de un ADN de cadena simple (molde) y un iniciador,
cebador o "primer" complementario de una región del ADN molde anterior a donde va a
iniciarse la secuencia. Este cebador se utiliza como sustrato de la enzima ADN polimerasa I que
va a extender la cadena copiando de forma complementaria el molde de ADN.
Se diseña un oligonucleótido sintético de unos 17-20 bases complementario de la cadena de
ADN que se quiere secuenciar y situado a unos 20-30 bases de distancia del comienzo de la
23

secuencia que se quiere leer. Si este oligo se diseña fuera de la región de clonaje de los
plásmidos podrá emplearse el mismo oligo para secuenciar distintos insertos.
El dúplex formado entre el oligo y el ADN complementario de cadena sencilla se convierte en
sustrato de la ADN polimerasa I que va a extender la cadena desde grupo OH libre del extremo 3
´ del oligo, incorporando dNTPs y copiando el molde de ADN al sintetizar la cadena
complementaria. Como enzima se emplea el fragmento Klenow de la ADN polimerasa I que
carece de actividad exonucleasa 5´- 3. También pueden emplearse otras polimerasas como la
Taq polimerasa o la polimerasa del fago T7.
Durante la secuenciación se llevan a cabo cuatro reacciones de síntesis separadas incluyendo en
cada una de ellas pequeñas cantidades de los dideoxinucleótidos (ddNTP: ddGTP; ddATP, ddCTP,
ddTTP) que carecen de extremo 3´ OH libre y que al incorporarse en la cadena de ADN que se
esta sintetizando acaban con la elongación de la misma.
La incorporación al azar de un ddNTP en competición con el dNTP correspondiente, implica la
formación de una mezcla de cadenas de distintas longitudes, todas ellas empezando en el
extremo 5´ y acabando en todas las diferentes posiciones posibles donde un ddNTP puede
incorporarse en lugar de un dNTP. El promedio de longitud de las cadenas puede alterarse
modificando la relación dNTP/ddNTP en la mezcla de reacción. Por ejemplo, aumentando la
concentración de ddNTP aumenta el número de cadenas de pequeña longitud al aumentar la
frecuencia de incorporación de este tipo de nucleótidos.
La sustitución de uno de los dNTPs por el mismo nucleótido marcado radiactivamente permite la
visualización de las bandas de distinta longitud en un gel de poliacrilamida donde cada una de
las reacciones se carga en un carril. En el gel la separación de las distintas bandas se produce en
función de su tamaño y la secuencia puede determinarse leyendo las bandas de los cuatro
carriles. En este tipo de geles pueden leerse hasta 300 bases. En carreras más largas la lectura
puede llegar a ser de hasta 500 bases.
24

Secuenciación automática empleando el metodo enzimático
Es una alternativa al método de Sanger. Consiste en marcar el oligo cebador o los terminadores
con un compuesto fluorescente y activar la reacción de secuencia. Los productos de la reacción
se detectan directamente durante la electroforésis al pasar por delante de un láser que al
excitar los fluoróforos permite detectar la fluorescencia emitida.
Secuenciación empleando cebadores fluorescentes
Se realizan cuatro reacciones de secuencia distintas en cada una de las cuales se añade el
oligonucleótido o cebador marcado con una sonda fluorescente distinta y un ddNTP diferente
en cada una de ellas. Al finalizar las cuatro reacciones se mezclan en un único tubo.
25

Se realizan cuatro reacciones de secuencia distintas en cada una de las cuales se añade el
oligonucleótido marcado en 5´ con una sonda fluorescente distinta, junto con la polimerasa, los
dNTPs y el ddNTP correspondiente. Las sondas empleadas son:
para A ---> JOE. Emisión: verde. Derivado de fluoresceína
para C ---> FAM. Emisión: azul. Derivado de fluoresceína
para G --> TAMRA. Emisión: amarillo. Derivado de rodamina
para T --> ROX. Emisión: rojo. Derivado de rodamina
Durante la electroforésis las bandas del ADN de cadena sencilla pasan a través de un láser de
argón que excita las sondas fluorescentes, la señal de emisión de fluorescencia, una vez
amplificada, es detectada a través de un filtro y asignada a la base correspondiente
Secuenciación empleando terminadores fluorescentes
Se realiza una única reacción de secuencia en presencia de los cuatro ddNTPs, cada uno de ellos
marcados con una sonda fluorescente distinta.
26

De modo alternativo, la secuenciación puede llevarse a cabo empleando terminadores marcados
cada uno con un fluoróforo diferente. Esta química es más sencilla porque permite llevar a cabo
la reacción en un solo tubo, en que se añaden los cuatro terminadores marcados.
Secuenciadores automáticos
Secuenciadores automáticos capilares
Existen instrumentos de electroforesis capilar que proporcionan una alternativa clara al sistema
basado en geles de acrilamida/bisacrilamida donde este soporte ha sido sustituido por un
polímero que se inyecta de forma automática en un capilar antes de cargar la muestra de
secuenciación; las muestras se van analizando una a una. Este tipo de secuenciadores se utiliza
para lecturas no superiores a unos 450pb.
El equipo funciona de forma completamente automática inyectando las muestras (que se
preparan exactamente igual que durante la secuenciación en geles y se colocan en una placa de
96 pocillos), en un capilar previamente cargado con un cierto polímero que funciona como lo
27

hace la matriz de acrilamida: bisacrilamida:urea de los geles de secuencia, permitiendo resolver
fragmentos de ADN de cadena sencilla que se diferencian en una única base.
A una altura determinada el laser detecta la fluorescencia emitida por cada cadena sencilla de
ADN fluorescente y traduce esta emisión de fluorescencia en la secuencia correspondiente. Una
vez desarrollada la electroforesis de la primera muestra, el capilar se vacía rellenándose
nuevamente con polímero fresco: Se inyecta a continuación una segunda muestra, se procede a
desarrollar nuevamente la electroforesis y así sucesivamente.
28

Las principales ventajas de estos nuevos equipos son:
Automatización completa de todo el proceso, no siendo necesaria siquiera la presencia
de un técnico que cargue las muestras en los diferentes capilares del equipo.
Rapidez en el analisis de cada muestra: dado el pequeño diámetro de los capilares, se
pueden aplicar voltajes mayores que en los geles de acrilamida:bisacrilamida:urea, por
lo que pueden leerse del orden de 450 nucleotidos en el plazo de una hora mientras que
esta misma longitud de secuencia necesita unas 2-4 horas en un secuenciador en gel.
El tiempo necesario del proceso es. para la polimerizacion del mismo, unas dos horas y
para la preelectroforesis, una hora aproximadamente.
Secuenciación automática en geles desnaturalizantes de acrilamida/bisacrilamida
La secuenciación automática mediante geles desnaturalizantes, se realiza polimerizando un gel
de acrilamida/bisacrilamida entre dos cristales montados adecuadamente sobre el cassette que
sirve de soporte para los mismos y que posteriormente se acoplará en el secuenciador
automático para proceder a la carga de la muestras
Secuenciación automática mediante
geles desnaturalizantes, se realiza
polimerizando un gel de acrilamida/bis
entre dos cristales montados
adecuadamente sobre el cassette que
sirve de soporte para los mismos y que
posteriormente se acopla en el
secuenciador automático para proceder
a la carga de la muestras.
El número de muestras que podemos cargar en cada gel, viene determinado por el número de
pocillos que posee el peine ( de dientes de tiburón) que utilicemos. Hay peines de cuatro
tamaños distintos: de 36, 48, 64 y 96 pocillos.
La electroforesis se desarrolla durante 7 horas, y se puede realizar una lectura de unos 700pb
aproximadamente, dependiendo siempre de la calidad del ADN, de la correcta cuantificación del
mismo, de la utilización del primer adecuado, y de la polimerización correcta del gel de
acrilamida/bis principalmente.
29

Cuando las muestras a secuenciar tienen una longitud no superior a 400pb, podemos disminuir
el tiempo de la electroforesis a 3.5 horas, aumentando la velocidad de barrido del laser, y el
resultado es igualmente optimo.
Otros métodos de secuenciación automática
Secuenciación de ADN empleando microarrays
Los microarrays constituyen la última línea de técnicas basadas en la interacción de cadenas
complementarias de ADN. Este tipo de técnicas introducen básicamente dos nuevas
innovaciones: el empleo de soportes sólidos no porosos tales como cristal que facilitan la
miniaturización y la detección basada en fluorescencia y el desarrollo de métodos de síntesis in
situ de oligonucleótidos a altas densidades sobre el soporte sólido.
Principio:
Existen muchas variedades de microarrays o ADN-chips como también se les denomina. Desde
los microarrays "artesanales" realizados en el laboratorio a los ofrecidos por compañías de
biotecnología, el principio que rige su diseño es el mismo: la complementariedad de las cadenas
aisladas de ADN: o propiedad de las citadas cadenas de hibridarse con otras cadenas aisladas de
ADN que posean una estructura complementaria.
Básicamente un microarray consiste en una matriz de "pocillos" microminiaturizados sobre un
substrato de vidrio en donde se implantan, utilizando diversas técnicas, cadenas simples de
oligonucleótidos. El poder adherir una cadena corta de oligo sobre una superficie plana es
decisivo en el diseño de los microarrays.
(Molecular interactions on microarrays)
En la figura se muestran esquemáticamente unas pocas celdas de un microarray. En cada celda,
adherida por uno de sus extremos, existe una determinada cadena de oligonucleótido. (Zona
inferior izquierda de la imagen) Si se pone el microarray en contacto con una mezcla de cadenas
30

simples de ADN a identificar, marcadas fluorescentemente, (zona superior izquierda) éstas se
hibridarán con su oligo complementario. (zona derecha).
Dado que conocemos la secuencia y distribución de los oligonucleótidos en el microarray
podemos, al excitar el microarrray con un laser y estar las cadenas hibridadas
fluorescentemente, conocer la ubicación de las cadenas de las mismas y su secuencia.
Visualización típica de una secuenciación
mediante microarrays
Fabricación:
En la fabricación de microarrays se emplean técnicas fotolitográficas análogas a las utilizadas en
microelectrónica. Un sustrato de vidrio se trata químicamente con determinados grupos
reactivos para permitir la implantación de los oligonucleótidos sobre el mismo; a continuación
se deposita sobre el substrato una película fotodegradable y mediante la utilización de una
plantilla y un haz luminoso, se crea la estructuura de celdas del microarray.
Fabricación de las celdas del microarray
Existen dos técnicas para para acoplar los oligos a cada una de las celdas.
31

Implantar en cada celda, mediante un brazo robotizado, el oligonucleótido
presintetizado que corresponda.
Sintetizar en las propias celdas, mediante ciclos sucesivos de síntesis, los
oligonucleótidos correspondientes.
El proceso de síntesis "in situ" de los nucleótidos en el microarray se realiza en las siguientes
fases:
Elaboración previa de un mapa de distribución del tipo de oligonucleótido
correspondiente a cada celda del microarray.
Preparación del sustrato y deposición de una película fotodegradable. (1)
Aplicación de una máscara que permita eliminar selectivamente la película protectora
en las zonas del microarray correspondiente a un determinado nucleótido. (2 y 3)
Incubación química y acoplamiento del nucleótido previsto (4).
Una nueva capa fotodegradable es aplicada sobre el microarray (5)
Se repiten los pasos anteriores para cada nucleótido hasta obtener la secuencia prevista
Se elimina definitivamente la película fotodegradable (10).
Pirosecuenciación
32

Se trata de un método simple y consistente, útil para la secuenciación de fragmentos de ADN de
tamaños pequeños o medianos
Etapas del proceso:
Un cebador específico se une a una cadena de ADN sencilla en presencia de dNTPS, ADN
polimerasa, ATP sulfurilasa, luciferasa y apirasa así como de los sustratos APS
(adenosina 5 fosfosulfato) y luciferina.
El primero de los cuatro dNTPs se añade a la reacción. La ADN polimerasa cataliza su
incorporación a la cadena; si este dNTP es complementario de la secuencia del ADN
molde se libera PPi en una cantidad equimolar a la cantidad de nucleótido incorporado.
La ATP sulfurilasa convierte cuantitativamente el Ppi en ATP en presencia de APS. Este
ATP media la conversión de luciferina en oxiluciferina por parte de la luciferasa,
produciéndose luz visible en intensidad proporcional a la cantidad de ATP. Esta luz es
detectada por una cámara CCD y se visualiza en el pirograma como un pico, siendo la
altura del pico proporcional al número de nucleótidos incorporados.
La apirasa degrada de forma continúa tanto el ATP como los dNTPs no incorporados
apagando de esta forma la emisión de luz y regenerando las condiciones para que pueda
ser añadido un nuevo nucleótido en la reacción.
33

La adición de nucleótidos ocurre de uno en uno. Se sustituye el dATP por dATPalfaS
porque este sustrato es reconocido eficientemente por la ADN polimerasa pero no por
la luciferasa.
Aplicaciones de la secuenciación de ADN
Detección de mutaciones
Secuenciación de ADNs fósiles
Diagnóstico de enfermedades genéticas
Identificación de especies y control de cruces entre animales
Proyecto genoma humano
34

Métodos de comprobación
PCR – digestión
REACCIÓN EN CADENA DE LA POLIMERASA (PCR)
En abril de 1983, Kary Mullis da a conocer la técnica de reacción en cadena de la polimerasa, o PCR.
Esta técnica ha invadido de tal forma la Biología Molecular, que hoy en día es muy difícil imaginar
esta ciencia sin ella. En 1989, Science seleccionó el PCR como el principal desarrollo científico, y la
Taq polimerasa como la molécula del año. En 1993, Kary Mullis recibió por este descubrimiento el
Premio Nobel de Química.
Gracias a la PCR, la "insuficiente cantidad de ADN" ya no es una limitación en la investigación en
Biología Molecular, ni en los procedimientos de diagnóstico basados en el estudio del ADN. Una de
sus posibles aplicaciones se centra en la secuenciación del ADN de muestras biológicas.
Procedimiento:
Las bases teóricas de la reacción de PCR son simples. En la reacción intervienen tres segmentos de
ADN: el segmento de doble cadena que queremos amplificar, y dos pequeños fragmentos de cadena
sencilla, los oligonucleótidos (primers), que tienen la misma secuencia que los extremos
flanqueantes del ADN molde. Además, participan en la reacción la enzima Taq polimerasa (Taq),
deoxinucleótidos-trifosfato (dNTPs), sales, y un tampón.
Los primers se añaden a la reacción en exceso con respecto al ADN que desea amplificarse. Los
primers hibridarán con las regiones complementarias del ADN, quedando orientados con sus
extremos 3´ enfrentados, de modo que la síntesis mediante la ADN polimerasa (que cataliza el
crecimiento de nuevas cadenas 5´-> 3´) se extiende a lo largo del segmento de ADN que queda entre
ellas.
El primer ciclo de síntesis producirá nuevas cadenas, de longitud indeterminada, las cuales, a su vez,
son capaces de hibridar con los primers. Este tipo de productos se irá acumulando de forma
geométrica, con cada ciclo de síntesis, utilizando como moldes las moléculas de ADN parental de la
reacción.
35

Durante el segundo ciclo de síntesis, estas cadenas hijas servirán a su vez como moldes,
generándose en este caso fragmentos cuyo tamaño será el del fragmento que engloba los extremos
de ambos primers. Estas nuevas cadenas hijas servirán de molde a su vez para sucesivos ciclos de
síntesis. La cantidad de estos productos se duplica con cada ciclo, por lo que se van acumulando de
forma exponencial. Al cabo de 30 ciclos, habrán aparecido 270 millones de estas moléculas, por cada
una de ADN original que hubiese.
El primer paso necesario en este proceso de la síntesis es la desnaturalización del ADN. Puesto que
el molde es una molécula de ADN de doble cadena, será necesaria la separación de estas para que
los primers puedan acceder a sus secuencias complementarias y unirse a ellas en el siguiente paso,
llamado de alineamiento ("annealing"). Una vez que los primers se han unido a las cadenas, sirven
como cebadores para la acción de la ADN polimerasa, la cual comienza a crear a partir de ellos
nuevas cadenas de ADN complementarias, en el proceso de elongación. Estos tres pasos
consecutivos (desnaturalización, alineamiento y elongación) constituyen un ciclo de síntesis.
Para las reacciones de PCR de secuenciación se ha diseñado una Taq polimerasa especifica, Taq FS
(fluorescente sequencing), a partir de la Taq polimerasa de Thermus aquaticus y modificada
genéticamente de forma que posee muy baja actividad exonucleasa 5´->3´ con lo que los resultados
son más limpios con menos ruido de fondo y apenas falsas terminaciones. Además este enzima
incorpora los ddNTPs marcados fluorescentemente más eficientemente, con lo cual son necesarios
menos ddNTPs-fluorescentes y menos ADN molde para conseguir la misma señal.
En los últimos años, también se ha avanzado notablemente en la obtención de sondas fluorescentes
cuyos espectros de emisión de fluorescencia presentan un mínimo solapamiento.
36

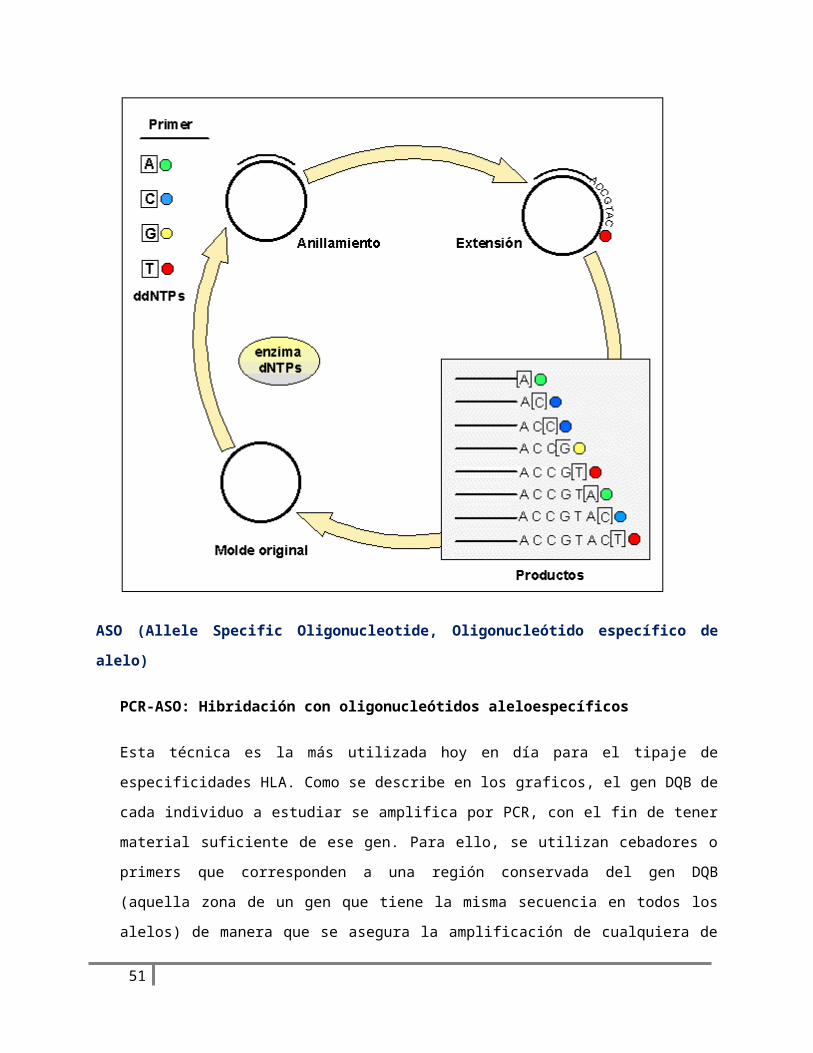
ASO (Allele Specific Oligonucleotide, Oligonucleótido específico de alelo)
PCR-ASO: Hibridación con oligonucleótidos aleloespecíficos
Esta técnica es la más utilizada hoy en día para el tipaje de especificidades HLA. Como se describe en
los graficos, el gen DQB de cada individuo a estudiar se amplifica por PCR, con el fin de tener
material suficiente de ese gen. Para ello, se utilizan cebadores o primers que corresponden a una
región conservada del gen DQB (aquella zona de un gen que tiene la misma secuencia en todos los
alelos) de manera que se asegura la amplificación de cualquiera de los alelos. El ADN amplificado se
deposita, en formato dot blot, sobre una membrana de nylon, para su hibridación con sondas
marcadas. En el caso del gen DQB, el ADN amplificado de cada individuo se reparte en 20
membranas (igual al número de sondas que se van a utilizar) . Cada una de estas membranas, en la
que se puede depositar ADN de hasta 96 individuos, se incuba con una de las sondas ASO
37

(oligonucleótidos correspondientes a regiones polimórficas -variables-) del gen DQB, de forma que
se hibridarán al ADN depositado en la membrana solo si esa variante polimórfica está presente. Una
vez reveladas todas las pruebas, se analizan los resultados; la combinación de sondas para las que
una muestra hibrida positivamente nos dará los dos alelos presentes en ese ADN genómico. La
mayor ventaja de esta técnica estriba en la posibilidad de tipar en poco tiempo un gran número de
muestras. No obstante, debido a un cierto nivel de reacción cruzada que existe entre las diferentes
sondas ASO, la interpretación de los resultados no es siempre sencilla, y se precisa de personal
experimentado en el HLA. Se están identificando nuevos tipos y subtipos continuamente, de forma
que la cantidad de sondas a utilizar y la complejidad del análisis es cada vez mayor, hasta el punto
de que existen equipos informáticos capaces de asignar los “subtipos más probables” a partir de los
resultados del dot blot.
38

ARMS (Amplification Refractory Mutation System)
Introducción
La técnica de PCR se puede adaptar de muchas maneras para facilitar el análisis de ADN. Diferentes
tipos de estrategias se utilizan para la detección de mutaciones puntuales. Los métodos actuales se
basan en la amplificación de destino, por lo general por PCR, seguida de la identificación de
variantes de ADN con sondas, enzimas de restricción, ligasas y polimerasas. En ARMS, el par de
primer está diseñado para que uno de los extremos 3’ coincida con una variante de nucleótidos en la
secuencia de destino.3
Cuando el primer no corresponde a la plantilla de la frecuencia, la extensión es muy baja y por lo
tanto el número efectivo de copias disponibles para la amplificación de la secuencia se reduce
considerablemente. ARMAS-PCR aprovecha una polimerasa termoestable que carece de actividad
exonucleasa 3’ (por lo general Taq polimerasa). Estas enzimas extienden primers obligados a su
objetivo de secuencias, que es muy ineficiente cuando la base 3 'no coincide. Debido a la actividad 3
'exonucleasa, necesario para la reparación de errores no está presente, la extensión de los
cebadores en la PCR es un evento raro y la amplificación se retrasa. Sin embargo, una vez que una
extensión de desajuste se ha producido, la amplificación procede, normalmente, de la línea de meta
de nueva síntesis.
El sistema de amplificación de mutación-refractario (ARMS) es un método simple para detectar
cualquier mutación que impliquen cambios en una sola base o pequeñas deleciones. ARMS se basa
en el uso secuencias específicas de cebadores de PCR que permite la amplificación de ADN de
prueba sólo cuando el alelo de destino se encuentra dentro de la muestra. Después de una reacción
ARMS, la presencia o ausencia de un producto de PCR es diagnóstico para detectar la presencia o
ausencia del alelo de destino.
El protocolo básico describe el desarrollo y la aplicación de una prueba de ARMS para una sola
mutación, el protocolo alterno extiende esto un para el análisis de dos o más mutaciones. El
soporte de protocolo describe un método rápido de extracción de ADN de muestras de sangre o el
enjuague bucal que produce ADN compatibles con el tipo de pruebas del sistema descrito. ARMS es
un método simple para detectar cualquier mutación que implicara cambios en una sola base. Por
3 http://www.currentprotocols.com/protocol/hg0908
39

tanto ARMS es un método simple para detectar cualquier mutación que implicaran cambios en una
sola base
El sistema de amplificación mutación refractarios (ARMS), que también ha sido descrita como un
PCR alelo-específico (ASP) y la amplificación por PCR de alelos específicos (PASA), es un método
basado en PCR para detectar mutaciones de una sola base. ARMS se ha aplicado con éxito para el
análisis de una amplia gama de polimorfismos, mutaciones de línea germinal y mutaciones
somáticas. La técnica tiene la capacidad de discriminar bajos niveles de “secuencias mutante” en un
fondo de ADN de difícil manejo. En una PCR- ARMS el nucleótido ubicado en el extremo 3' terminal
de uno de los cebadores de PCR coincide con la mutación de destino. La mayoría de las aplicaciones
del método de análisis se basan en "punto final", utilizando el método clásico de gel de
electroforesis. Sin embargo, en este tipo de análisis sólo se puede evaluar la presencia o ausencia de
secuencias mutantes o de tipo poco manejable y no da una indicación de la proporción de
secuencias mutantes de este tipo en una población mixta de ADN. Aquí se describe una adaptación
de la PCR en tiempo real de ARMS, ARMS cuantitativa, que permite la medición del tamaño de la
población de cada variante en una mezcla. Un método para la detección de mutaciones humanas
del virus de la hepatitis B que confiere resistencia a los antivirales.
Así, un importante principio de las ARMS es que no se emparejen (es decir, no se unan sólo en el
extremo 3 'de un cebador) plantilla puede ser amplificado. Los factores que contribuyen a este
aparente «fracaso» de ARMS es que cuando un número muy elevado de moléculas de plantilla se
añaden a la reacción, la amplificación es probable que continúe porque la pequeña proporción de
las extensiones de primer no coincidentes, alcanzan el umbral de sensibilidad de la reacción. En
segundo lugar, ciertos no coincidentes estén extendidos más eficientemente que los otros, por tanto
esas extensiones extendidas en cada ciclo de PCR varia.
40

Ilustración esquemática de ARMS
ARMS para el diagnóstico prenatal y la Fibrosis Quística
ARMS se ha aplicado al diagnóstico prenatal y la detección de portadores de fibrosis quística. Se ha
determinado la secuencia de nucleótidos de los dos alelos del polimorfismo de la restricción de
longitud de los fragmentos en el locus KM19, que muestra el desequilibrio de ligamiento con fibrosis
quística. ARMS permite el análisis directo de los alelos de este polimorfismo en el ADN aislado de la
biopsia de vellosidades coriónicas o células blancas de la sangre.
HFE genotipificación por ARMS y desnaturalización HPLC
La Hemocromatosis hereditaria (HC) es una de las enfermedades genéticas más frecuentes en
poblaciones caucásicas [prevalencia de hasta 1 en 300 en el norte de Europa, con una frecuencia
portadora estimada de 1 de cada 10]. Causado por un aumento progresivo de hierro, se caracteriza
por complicaciones graves y una progresión potencialmente letal que puede ser totalmente
prevenida con la remoción regular del hierro por medio de la sangre. La disponibilidad de un
tratamiento preventivo eficaz destaca el valor de la detección temprana. El descubrimiento del gen
HFE por Feder y otros, ha dado una prueba genética para el riesgo de la enfermedad. Dos
mutaciones C282Y y H63D, situadas respectivamente en los exones 4 y 2, la alteración de la función
41

HFE probados para, junto con una tercera variante que se encuentra en el exón 2. Dos genotipos
(homocigotos C282Y y heterocigotos compuestos C282Y/H63D) se asocian con el riesgo de HC
Debido a que muchos podrían beneficiarse de las pruebas, numerosos métodos han sido descritos,
además de los ensayos convencionales de PCR de restricción. Métodos descritos incluyen PCR en
tiempo real con sondas fluorescentes, ensayo convencional de restricción de PCR con el uso de HPLC
o la electroforesis capilar para resolver los fragmentos de restricción, los productos de PCR alelo-
específicas resueltos en geles de losa o por electroforesis capilar, y seguido por un paso de
extensión de un solo nucleótido la detección de electro quimioluminiscencia, o ensayo de
hibridación reversa.
Desnaturalización HPLC (DHPLC) es un nuevo método para la detección de polimorfismo de
nucleótido único, que ha sido eficaz en términos de costo por análisis, sensibilidad y especificidad.
Sin embargo, una limitación de (DHPLC es que en la mayoría de los casos apenas se diferencia de los
genotipos homocigotos mutantes homocigotos de tipo poco manejable, y por lo tanto, no se
recomienda para enfermedades que se caracterizan por un alta prevalencia de algunas mutaciones.
DHPLC fue utilizado con éxito para detectar pacientes con heterocigosis a C282Y y H63D, pero la
detección de los pacientes homocigotos requiere un doble análisis: el primero utilizando con la
muestra sola y luego un segunda PCR en el que se mezcla la muestra con un control de tipo poco
manejable para revelar pacientes homocigotos con mutaciones. Se ha diseñado un método que
utiliza un ARMS múltiple PCR y DHPLC (ARMS-DHPLC) para la determinación del genotipo HFE en un
procedimiento de dos pasos
Se utilizaron muestras de EDTA de sangre obtenidas de pacientes con exceso de hierro o familiares
que se refiere a nosotros para la determinación del genotipo HFE. El ADN se extrajo con fenol-
cloroformo, y los genotipos HFE se determinaron mediante ensayos de PCR de restricción Rsa I
(C282Y), HPH I (H63D), y HinfI (S65C).
Para cada (C282Y y (H63D), se diseñó un conjunto de primers, dos específicos para los alelos de tipo
poco manejable y con mutaciones y una tercera común. Para cada conjunto, la cartilla específica
para el alelo con mutaciones portada a 12 pb 5‘ de la cola. Para aumentar la especificidad de la
cartilla r-Y282, se introdujo se diseñó un desfase cercano al extremo 3'. Cebadores fueron
seleccionados y se calcula la compatibilidad en la técnica de PCR utilizando un software. Igualdad de
42

eficiencia de amplificación de cada conjunto logró mediante el ajuste de la concentración de cada
cebador.
El método de ARMS DHPLC combina el tamaño. Las propiedades de detección de la conformación
del ADN y la detección de las propiedades de DHPLC, lo que permite genotipo HFE rápido y fiable.
Las características principales de este método son que hace uso de una sola reacción básica de PCR
con patrón estándar de pH y la ADN polimerasa, primers regulares (es decir, sin marcaje
fluorescente o modificación de terminales), tubos estándar de PCR, sin etapas de reacción más o
manipulación, y 260 nm ADN de medida de absorción. Esto debería compararse con las limitaciones
técnicas en tiempo real de métodos de fluorescencia de PCR y otros métodos
Los ARMS DHPLC ensayos son altamente automatizados, de dos pasos método de determinación del
genotipo. Aplicada a la determinación del genotipo HFE, que da resultados sin ambigüedades de las
dos mutaciones más frecuentes asociadas HC (C282Y y (H63D), y es capaz de detectar otras
mutaciones en los productos de amplificado sin pérdida de precisión para las dos principales
mutaciones, como se muestra para la menos frecuente (S65C y la rara mutación E277K).
Una característica de HC es que en número sustancial de pacientes el típico fenotipo de HC,
genotipo HFE convencionales es decir, la rara mutación de detección de la conformación del de las
tres mutaciones más frecuentes, detecta una sola o ninguna mutación. Esta peculiaridad le pide
revisión exhaustiva de la secuencia del gen HFE para identificar mutaciones. Esto permitiría la
identificación de pacientes con exceso de hierro en los que la HFE no está implicada en la
enfermedad.
43

Determinación de haplotipos usando doble ARMS. La figura ilustra el doble haplotipo
de un individuo que es heterocigotos en ambos sitios polimórficos 1 y 2. a, b, c, y d son
las bases complementarias que corresponde a A, B, C y D, respectivamente. El objeto del
doble análisis de ARMS es determinar si el alelo A está relacionado con el alelo B o el
alelo A está relacionado con el alelo D. Este problema se puede resolver con relativa
facilidad utilizando ARMAS por llevar a cabo la amplificación mediante ARMS ARMS-A y-
B. Si un producto se observa ampliado, a continuación, el alelo A está relacionado con el
alelo B en el mismo cromosoma. La PCR confirmatoria segundo puede llevarse a cabo
utilizando armas A y ARMS-d, que debe ser negativa. Para un análisis más detallado del
número de reacciones de ARMS para resolver los haplotipos
44

OLA (Oligonucelotide Ligation Assay, Ensayo de ligación de oligonucleótidos)
Es un método rápido, sensible y específico para la detección de polimorfismos conocido un solo
nucleótido (SNPs). Este método se basa en la unión de dos sondas de oligonucleótidos adyacentes
utilizando una ligasa de ADN.4
En esta técnica se utilizan dos pares de iniciadores ,complementarios a lugares donde puede haber
mutaciones o polimorfismos en el ADN a estudio, de modo que estos iniciadores se acoplen con el
ADN quedando consecutivos ,es decir, que el extremo 3’ del primero se continúe con el extremo 5’
del segundo.
Cuando no hay mutación los iniciadores se colocan consecutivos y se puede ligar por la acción de
una ligasa tras lo cual es posible amplificar el ADN con PCR, pero si hay mutación los fragmentos no
se acoplan 100% al ADN muestra y no están consecutivos, por lo tanto la ligasa no puede unirlos.
La ligasa sella el corte creado entre 2 sondas adyacentes si hay una perfecta unión alrededor de él.
La ligación de bases mal apareadas es fuertemente desfavorable. La ligasa T4 puede hacer una
discriminación 5 veces mayor cuando el mal emparejamiento esta en el extremo 3’ de las sonda
aguas arriba que cuando está en la base 5’ terminal aguas abajo. El análisis de mutaciones
puntuales normalmente se lleva a cabo estableciendo la reacción OLA con 2 sondas alélicas
competidoras que ligadan en una sonda común solo si son perfectamente complementarias al
DNA blanco.
Esta técnica se ha usado para la discriminación alélica de mutaciones puntuales en la fibrosis
cística, b-globina, a-antitripsina, y genes de la 21-hidroxilasa.
4 Ana Alonso Pacho, José Sabán Ruiz. Control global del riesgo cardiometabólico. Ediciones Díaz de Santos, 2009. Pág. 205
45

Esquema del procedimiento del Ensayo de Ligación de Oligonucleótidos (OLA)
Un avance posterior, la reacción OLA múltiple, requiere balances de Tm's de todas las sondas para
que todas las reacciones puedan producirse bajo las mismas condiciones de tampón y temperatura.
Se consigue balancear la señal gracias al ajuste de la concentración relativa de cada set de sondas.
Sometiendo las sondas a ciclos que varían entre una gran temperatura de desnaturalización y una
baja temperatura a la que puedan anillar y ligar se promueven múltiples oportunidades para que
cada una pueda encontrar su complementaria perfecta. Esta técnica produce amplificación lineal de
la señal OLA y se llama reacción de detección de la ligasa (LDR). Tanto la OLA múltiple como la LDR
múltiple, necesita un ensayo para diferenciar unos productos OLA de otros. Este puede realizarse
utilizando diferentes marcajes para cada set de sondas OLA o usando diferentes ganchos. Si se
utiliza una captura en fase sólida para análisis de productos OLA suele emplearse el mismo gancho
46

para capturar los productos y detectar marcas diferentes. Esas marcas pueden ser diferentes
haptenos que se unan a diferentes anticuerpos o pueden ser moléculas directamente detectables
como tintes fluorescentes. La variación de los ganchos se suele usar cuando el análisis de productos
de OLA se realiza con electroforesis. En este caso se sintetiza una sonda de cada par con una cola de
nucleótidos no complementarios (tales como poli-A de diferentes tamaños), sin nucleótidos, o con
colas modificadoras de movilidad (Como colas de varias longitudes de oxido de hexaetileno). Cada
producto de OLA tendrá un único tamaño. Mediante combinaciones de ganchos y etiquetas se ha
creado un OLA múltiple con 60 alelos para mutaciones en el gen regulador transmembranal de la
fibrosis cística.5
Sondas de DNA: Pueden sintetizarse utilizando la química usual de síntesis de DNA. Las sondas del
ensayo de ligación aguas abajo deben ser fosforiladas en su extremo 5’ (el extremo que se enfrenta
al corte). Esto puede hacerse mediante fosforilación enzimática o mediante la adicción de fosfato
durante las síntesis de la sonda. El OLA puede hacerse con sondas que varían en su temperatura
media desde 45ºC a 70ºC. Para hacer la discriminación alélica, la Tm de sondas alélicas competentes
deben ser cuidadosamente ajustadas para evitar la preferencia por un alelo en la producción del
OLA, mientras que las sondas comunes deben tener una alta temperatura. Para LCR, es importante
que las 4 sondas tegan la Tm ajustada próxima para minimizar eficientemente las diferencias entre
las 2 cadenas. Cuando se usa la gap-LCR, es importante bloquear la extensión por DNA polimerasa
mediante modificación de los extremos exteriores de las sondas con los ganchos y etiquetas
requeridos para la posterior detección del producto o con un grupo no extensible como una amina.
Ligasa: Los primeros trabajos con OLA usaban ligasa de E. Coli o T4. Sin embargo, para muchas
aplicaciones se usan ligasas térmicamente estables como las de Thermus aquaticus (Taq) o
Pyrococcus furiosus (Pfu). Estas fuentes de enzima difieren en el cofactor que requieren como
proveedor de energía para la formación del enlace químico entre las sondas. Las ligasas T4 y Pfu
usan ATP, mientras que las de E. coli y Taq usan NAD.
Detección del producto.
Los productos de la reacción de la ligasa pueden ser detectados utilizando ensayos de
microtitulación, electroforesis en gel o un analizador clínico automático. Para la detección de
microtitulación en plato se modifica una sonda con un hapteno como la biotina que puede ser
5 http://eureka.ya.com/jaleo/7_detecc5.htm
47

capturada en una placa recubierta con avidina o estreptoavidina. La captura también puede hacerse
gracias a la adicción de colas de poli-A como hapteno para una de las sondas y capturando el
producto de ligación en bolas magnéticas de oligo dT. La segunda sonda contiene la etiqueta que
puede o detectarse directamente o unirse a un anticuerpo conjugado con una enzima para una
detección por amplificación enzimática.
48

Aplicación del ligamiento al diagnóstico (proceso de diagnóstico indirecto de
las enfermedades genéticas)
El estudio de ligamiento genético es un método indirecto que permite establecer la relación de una
condición o enfermedad genética entre distintos miembros de una familia. Para establecer esta relación
se utilizan marcadores genéticos localizados en la región cromosómica de interés, tratando de acotar el
gen o la mutación asociada a la enfermedad o a la alteración investigada.
En la práctica clínica, las principales limitaciones de este estudio son:
Es imprescindible que exista un diagnóstico clínico
Es necesaria la colaboración en el estudio del máximo número posible de miembros de la familia
El resultado se indica en términos de una probabilidad dependiente de la informatividad
genética de cada familia y de posibles sucesos de recombinación.
Aplicaciones al diagnóstico:
Segregación de una condición genética entre los individuos de una familia
- Diagnóstico genético de portadores
- Diagnóstico genético presintomático
- Diagnóstico genético de exclusión
- Diagnóstico genético de informatividad
- Diagnóstico genético prenatal
- Diagnóstico genético preimplantación
Pérdida de heterocigosidad (LOH)
Disomía uniparental (UPD)
Detección de deleciones o duplicaciones
Estudios de discriminación entre varios genes o loci asociados a una misma enfermedad
El diagnóstico indirecto
El diagnóstico indirecto es independiente del conocimiento previo de la naturaleza molecular de la
mutación a diagnosticar y para llevarlo a cabo solo es preciso conocer la localización cromosómica del
locus implicado. Dicha estrategia consiste en estudiar, en la familia del propositus, la segregación
49

conjunta de la enfermedad y secuencias polimórficas físicamente próximas (ligadas) al locus de la
misma.
El ADN es una molécula lineal en la que las secuencias de nucleótidos se hallan contiguas y distribuidas a
lo largo de la doble hélice en un mundo de una dimensión. Existe pues una relación física de proximidad
entre secuencias de ADN. Dicha relación de continuidad puede alterarse durante el proceso de la
división meiótica que se presenta durante la formación de las células germinales. En la profase de la
primera división meiótica, los cromosomas homólogos (uno proveniente de la línea paterna y el otro de
la línea materna) intercambian material por el proceso de entrecruzamiento. El resultado final es tal que
los cromosomas de la célula germinal madura (espermatozoide u óvulo) presentan nuevas
combinaciones de las secuencias existentes en la célula original (nuevos recombinantes). Dada la
relación física existente entre secuencias adyacentes en un mismo cromosoma, la frecuencia en que dos
de estas secuencias se transmitan (segregan) independientemente de una generación a la siguiente es
directamente proporcional a la distancia que las separa. Es decir, cuanto menor es la distancia que
separa a dos secuencias de ADN, menos probable es el entrecruzamiento entre ellas y por lo tanto
segregarn independientemente con una menor frecuencia. Cuando se da esta situación decimos que
ambas secuencias están ligadas.
El ligamiento entre secuencias de ADN se establece mediante el estudio de la segregación de los alelos
de dichas secuencias en genealogías. Este tipo de estudios permiten la obtención del mapa genético de
una determinada región cromosómica. Dicho mapa contendrá puntos de referencia al estilo de un mapa
de carreteras. Así, mientras que el mapa de carreteras nos informa de la posición de pueblos y ciudades,
el mapa genético contendrá información sobre la posición de los genes. Además de pueblos y ciudades
el mapa de carreteras contiene otros puntos de referencia orientativos como pueden ser un cruce de
carreteras, un puerto de montaña o una vista panorámica. Dichos puntos de referencia son
fundamentales para orientarse a lo largo de la carretera. Su equivalente en el mapa genético serán por
ejemplo los puntos de rotura de una determinada translocación o las secuencias polimórficas repartidas
a lo largo de la molécula de ADN.
Secuencias polimórficas
Como su nombre indica, las secuencias polimórficas (también conocidas como secuencias anónimas, loci
anónimos, loci polimórficos, marcadores anónimos o marcadores polimórficos) son secuencias de ADN
que, normalmente, no codifican para un producto génico, se distribuyen de forma más o menos
50
aleatoria a lo largo del genoma y presentan como característica singular el hecho de ser polimórficas.
Este último hecho es de suma importancia pues confiere a este tipo de secuencias la característica
primordial del análisis genético, la variabilidad.
Las variantes de un locus es lo que conocemos como alelos. Ya hemos utilizado este concepto
anteriormente al hablar de los alelos de loci implicados en enfermedades (el alelo 508 de la fibrosis
quística o el alelo S de la anemia falciforme). Sin embargo, este tipo de alelos son, afortunadamente,
muy poco frecuentes y no nos serían útiles en los estudios de ligamiento. Por el contrario las secuencias
anónimas o loci polimórficos presentan por definición varios alelos con una frecuencia
significativamente alta en la población (superior al 1% para el menos frecuente).
Supongamos que queremos saber si el locus a, que está implicado en una enfermedad hereditaria, esta
ligado al locus b, que es una secuencia anónima de ADN. Lo primero que necesitaremos será identificar
las variantes de la secuencia anónima en cada uno de los miembros de la familia y estudiar su
segregación juntamente con la de la enfermedad. La aplicación de diversos métodos estadísticos nos
permitirá establecer la existencia o no de ligamiento entre los dos loci investigados y estimar la distancia
que los separa.
En los estudios de ligamiento la distancia que separa a dos loci se mide en función de la frecuencia en
que ambos recombinan. La unidad de distancia es el centimorgan (cM). Si dos loci recombinan en el 1%
de las meiosis, decimos que les separa una distancia de 1 cM. La frecuencia de recombinación entre dos
loci puede variar desde 0 (decimos entonces que dichos loci están íntimamente ligados) hasta el 50% y
decimos entonces que son independientes. Aunque no nos podemos extender aquí sobre este aspecto,
no existe siempre una relación lineal entre distancia genética (frecuencia de recombinación medida en
cM) y distancia física (número de nucleótidos que las separan, medida en pares de bases). Para
frecuencias de recombinación inferiores al 20% se puede establecer una relación de 106 pares de bases
por cada 1% de recombinación.
Tipos de variación polimórfica en secuencias anónimas
Hemos indicado que la característica primordial de las secuencias anónimas es su variabilidad, veamos
ahora en que consiste dicha variabilidad. Básicamente existen dos tipos de polimorfismos, los que
derivan de la substitución de un nucleótido por otro y los que derivan de la inserción o deleción de
51

secuencias de ADN. En estos últimos distinguimos dos subtipos: las inserciones o deleciones de
fragmentos de ADN y las repeticiones de secuencias de entre dos y cientos de nucleótidos.
Polimorfismos del tamaño de los fragmentos de restricción (RFLP del inglés "restriction fragment
length polymorphism").- Cuando el cambio de un nucleótido altera la diana para un determinado ER o la
inserción o deleción de un fragmento de ADN se localiza entre dos dianas de un ER, podemos detectar el
polimorfismo en función de la variación que éste genera en el patrón de restricción. Los RFLP son
polimorfismos que presentan normalmente un número bajo de alelos y por lo tanto su utilización en el
diagnóstico indirecto es limitada.
Polimorfismos del tamaño de fragmentos de restricción (RFLP). La pérdida o ganancia de una
diana de restricción por efecto de la substitución de un nucleótido (A), o la inserción o deleción
de un fragmento de ADN (B), pueden ser detectados por las variaciones en los tamaños de los
fragmentos de restricción. En la figura se presenta el resultado esperado de este tipo de
polimorfismos en individuos homozigotos para la presencia de la diana o la deleción (+ +), los
homozigotos para la ausencia de diana o la inserción (- -) y los heterozigotos (+ -). El método de
detección puede ser tanto hibridación y southern (esquematizado en la figura) como mediante
PCR.
Polimorfismos de repetición.- Existen dos tipos de polimorfismos de repetición de uso en diagnóstico
genético, los VNTR-minisatélites y los VNTR-microsatélites. En ambos casos presentan un número
variable de repeticiones en tandem (VNTR del inglés "variable number of tandem repeats").
Los minisatélites son loci que corresponden a secuencias de ADN de unas pocas decenas de nucleótidos
repetidas en tandem. El número de dichas repeticiones varia de cromosoma a cromosoma, de forma
que en un cromosoma el número de repeticiones en tandem puede ser de 10, en otro de 15 en otro de
52

22, etc, etc. La singularidad más especial de este tipo de polimorfismos está en que cada loci puede
presentar muchos alelos distintos (tantos como repeticiones), sin embargo presentan el inconveniente
que no están distribuidos por todo el genoma y por lo tanto solo pueden ser utilizados en el diagnostico
de un número muy reducido de enfermedades. Los VNTR-minisatélites han encontrado su máxima
aplicación en la determinación de la paternidad y en los protocolos de identificación genética en el
ámbito judicial. Cuando se habla de huellas dactilares del ADN se está hablando de este tipo de
polimorfismo.
Los VNTR-microsatélites son por excelencia los polimorfismos anónimos utilizados en el diagnóstico
genético. Corresponden a la repetición en tandem de secuencias de entre 2 y 5 nucleótidos. Los
microsatélites presentan dos características que los hacen ideales para su uso. En primer lugar, están
distribuidos de forma casi homogénea por todo el genoma y en segundo lugar, presentan un número
elevado de alelos con frecuencias similares entre sí, de forma que la probabilidad de que un individuo
sea heterozigoto es muy elevada (presentan una alta heterozigosidad). Su detección se realiza
normalmente mediante la técnica de PCR.
Detección mediante PCR de distintos alelos de un polimorfismo microsatélite. En el ejemplo se
presentan dos individuos heterozigotos para los alelos de 6 y 7 repeticiones y de 4 y 8
repeticiones del dinucleótido (CA)n. La detección se realiza mediante PCR utilizando como
53

cebadores oligonucleótidos que flanquean a la región que contiene la repetición (flechas). De
cada individuo se amplifican fragmentos de ADN que difieren entre si por el número de
repeticiones. El resultado de la amplificación se fracciona en un gel de acrilamida-urea para
determinar el genotipo de cada individuo.
Aplicación en el diagnóstico indirecto de enfermedades hereditarias
El ejemplo más sencillo de aplicación de los polimorfismos anónimos en el diagnóstico indirecto lo
tenemos en el caso de enfermedades ligadas al cromosoma X. En la familia en cuestión ha nacido un
varón afectado de una enfermedad hereditaria ligada al cromosoma X. De acuerdo con el patrón de
herencia, su padre presentará el alelo normal de la enfermedad, mientras que su madre será portadora,
es decir heterozigota. El varón afectado habrá heredado de su madre el cromosoma con el alelo de la
enfermedad y dada su condición de hemizigoto, manifestará el fenotipo correspondiente a la misma. Sin
embargo no podemos predecir, más allá del modelo mendeliano, el genotipo de la hermana ni el del
feto que se está desarrollando.
Supongamos que existe un locus polimórfico, tipo microsatélite, próximo al locus de la enfermedad,
íntimamente ligado (frecuencia de recombinación igual a 0). Si identificamos los alelos de dicho
polimorfismo en cada uno de los miembros de la familia podremos inferir el alelo del locus de la
enfermedad que porta cada individuo.
En la parte inferior de la figura se presenta, de forma simplificada, el resultado obtenido al caracterizar
mediante PCR el locus polimórfico. En el margen izquierdo se presenta un patrón correspondiente al
número de repeticiones del dinucleótido CA (de 1 a 6) y perpendicularmente a cada individuo el alelo
correspondiente a cada uno de ellos. Podemos ver que el varón afectado ha heredado de su madre el
alelo de 2 repeticiones, por lo que podemos inferir que dicho alelo está junto al alelo de la enfermedad.
Según ello, la hermana habrá heredado de su padre el alelo 4 del polimorfismo junto con el alelo normal
de la enfermedad y de su madre el alelo 6 que también en este caso irá junto al alelo normal. Por lo
tanto la hermana será homozigota para el alelo normal de la enfermedad. Siguiendo un razonamiento
similar podemos inferir que el feto será una niña (presenta dos alelos del polimorfismo) y heterozigota
para la enfermedad, pues ha heredado de su madre el alelo 2 del polimorfismo que como hemos
indicado anteriormente está acompañado por el alelo que causa la enfermedad. La Figura 20 es una
interpretación de lo anteriormente indicado.
54

Genealogía de una familia en la
que se hereda una enfermedad
ligada al cromosoma X. Véase el
texto.
En el ejemplo anterior hemos utilizado como marcador del locus de la enfermedad a un locus altamente
polimórfico, los cromosomas paternos presentan alelos distintos y por lo tanto los podemos identificar
sin ambigüedad alguna, y íntimamente ligado, es decir no existe recombinación entre ambos loci y por lo
tanto el alelo del polimorfismo que en la madre va junto al alelo de la enfermedad continuará junto a
éste en los cromosomas de la descendencia. Estas dos características son fundamentales en el momento
de escoger un determinado polimorfismo en el desarrollo de una estrategia de diagnóstico indirecto.
Representación gráfica del razonamiento
aplicado para determinar los genotipos
de los miembros de la familia de la figura
anterior. Los varones presentan dos
cromosomas distintos (un cromosoma X y
un cromosoma Y). La estrella vacía indica
el alelo normal y la estrella llena el alelo
de la enfermedad. Cada alelo del locus
del polimorfismo está indicado de
acuerdo con el número de repeticiones
del dinucleótido CA.
55

Si el marcador utilizado no presenta muchos alelos entonces pueden darse situaciones de ambigüedad
cuando uno o ambos padres sean homozigotos para un determinado alelo del marcador. En este caso
diremos que la familia es "no informativa" para dicho marcador y por lo tanto deberemos utilizar otro
polimorfismo en el diagnóstico de dicha familia. Por otra parte, si el marcador no está íntimamente
ligado, es decir existe recombinación entre el locus de la enfermedad y el locus del marcador, el
diagnóstico deberá tener en cuenta dicha posibilidad.
Veamos el siguiente ejemplo. En la siguiente figura se presenta la genealogía correspondiente a una
familia en la que se hereda una enfermedad con una herencia autosómica dominante. Con una
probabilidad del 50% la madre afectada transmitirá a sus descendientes el carácter. La utilización de un
locus polimórfico ligado al carácter en cuestión nos permitirá establecer un diagnóstico más ajustado
para el feto en desarrollo. En dicha figura, A se muestra el resultado obtenido al analizar un
polimorfismo microsatélite (repetición del dinucleótido CA). Vemos que la madre presenta los alelos 6 y
2 y el padre los alelos 4 y 7. Por su parte el hijo normal es portador de los alelos 2 y 7, el primero
procedente de la madre y el segundo del padre. Si el carácter es penetrante, podemos considerar que el
alelo 2 en la madre está junto al alelo normal del locus de la enfermedad. Por lo tanto, si el feto
presenta el alelo 2 del polimorfismo podremos inferir que también ha heredado el alelo normal del otro
locus.
Sin embargo, tal y como se presenta en la figura ya mencionada, B, si la distancia que separa a ambos
loci es tal que permite la recombinación entre ellos, es posible que el feto haya heredado de su madre el
alelo 2 del polimorfismo junto con el alelo de la enfermedad, debido a la recombinación de ambos loci
en la meiosis de la madre. La frecuencia con la que se presentará este segundo caso será la frecuencia
de recombinación entre los loci implicados. Así, si la distancia que les separa es de 5 cM, es decir su
frecuencia de recombinación es del 5%, el diagnóstico que daremos en el caso presentado será de un
95% de probabilidad de que el feto sea normal (apartado A) y de un 5% de que el feto haya heredado el
alelo de la enfermedad junto con el alelo 2 del polimorfismo (apartado B).
56

Diagnóstico indirecto de una enfermedad autosómica dominante. En A el locus del
polimorfismo y el de la enfermedad no presentan recombinación. En B, el feto en desarrollo
presenta un cromosoma materno en el cual se ha dado un evento de recombinación (flecha)
durante la meiosis de la madre. La simbología es la misma que la utilizada en la figura anterior.
Para obtener una mayor fiabilidad en el diagnóstico se utilizan varios loci polimórficos localizados a
ambos lados del locus de la enfermedad. Con ello se consiguen evitar las ambigüedades generadas por
el entrecruzamiento.
La dimensión social del diagnóstico genético
Si bien este aspecto será tratado con una mayor extensión y detalle en otros capítulos del libro, no
quisiera finalizar este capítulo sin abordar algunos de los aspectos ético – sociales que acompañan al
diagnostico genético. En la introducción se enumeraron algunas de las características diferenciadoras de
este tipo de diagnóstico respecto al diagnóstico convencional. Las diferencias no son de índole
metodológica o tecnológica, dado que en los distintos ámbitos de la biomedicina la complejidad
tecnológica es similar, sino que estas radican en el ámbito de sus implicaciones ético-sociales. Desearía
destacar algunos de los aspectos más significativos de esta dimensión ético – social.
El primero se refiere a la relación que se establece entre el paciente y su familia. Todo diagnóstico
genera en el paciente una tensión que se resuelve en un sentido positivo o negativo según sea el
57

resultado. El diagnóstico genético no solo genera tensión en el propositus sino que toda su familia se ve
implicada en él. Esta implicación no es solo de índole solidaria, la tensión en la familia obedece a una
reacción primaria al verse directamente implicados en la patología como potencialmente afectados. Es
por ello que el genetista debe considerar a la familia como la unidad de diagnóstico y aliviar mediante la
adecuada información, la angustia y la tensión que el diagnóstico pueda generar.
El segundo aspecto hace referencia a la componente predictiva del diagnóstico genético. Sin duda
alguna la fatalidad que acompaña al diagnostico positivo de una enfermedad hereditaria no tiene
parangón en otras disciplinas médicas. Ello es así, porque en la actualidad la mayoría de las
enfermedades hereditarias carecen de una terapia curativa. Cuando en una familia se diagnóstica un
hijo con fibrosis quística no solo se está dando una predicción sobre la progresión clínica del paciente en
un futuro próximo, sino que además se está asignando a la familia una predicción sobre futuros
embarazos. Así mismo, cuando uno de los miembros de una familia es diagnosticado como afecto de
una enfermedad hereditaria de manifestación tardía, los miembros jóvenes de la familia reciben una
carga de angustia y tensión. Un ejemplo de esta situación lo tenemos en el diagnóstico de la
enfermedad de Huntington. ¿Querrán los miembros de la familia saber cuál es el diagnóstico del
propositus? ¿Desearán los familiares asintomáticos ser sometidos a un diagnóstico? Estas y otras
cuestiones se podrán plantear y la respuesta no es obvia.
La determinación de los factores de riesgo y el estudio de la predisposición o susceptibilidad genética es
uno de los campos de futura aplicación del diagnostico genético. Sin duda alguna esto comportará
grandes beneficios medico-sociales, al facilitar la medicina preventiva en enfermedades como la
diabetes, la hipertensión, las enfermedades cardiovasculares u otras de carácter multifactorial, que
tienen una gran prevalencia en nuestra población. Sin embargo, todo lo positivo que comporta este tipo
de estudios puede verse truncado por una mala utilización de la información obtenida.
No se debe asignar rango de infalibilidad a una información de tipo probabilístico. La probabilidad de
padecer la enfermedad de Alzheimer se incrementa en un factor de 8 en los individuos homozigotos
para el alelo APO – 4, pero ello no implica que un individuo homozigoto para dichos alelos vaya a
manifestar inexorablemente la enfermedad. Los factores ambientales, la historia natural de cada
individuo, y el fondo genético tienen un papel muy importante en la manifestación fenotípica de un
carácter. Resulta curioso constatar que en la actualidad los más acérrimos defensores del papel del
ambiente en la ecuación: fenotipo = genotipo + ambiente, sean los genetistas.
58

Tratamiento genético de las enfermedades
Terapias génicas. Enfermedades tratables y tratamientos posibles6
Las enfermedades hereditarias provocadas por la carencia de una enzima o proteína son las más idóneas
para estos tratamientos. Pero también aquellas en las que no importa demasiado el control preciso y
riguroso de los niveles de la proteína cuya producción se pretende inducir mediante manipulación
genética (ej., el factor VIII de la sangre). Se trata, normalmente, de enfermedades monogénicas,
originadas por la alteración de un único gen recesivo anómalo y en las que basta la mera presencia del
producto génico para corregir el defecto.
A finales de los 80 se consideraban alteraciones idóneas para ser objeto de tratamiento génico la
enfermedad de Lesch-Nyhan (provocada por la ausencia de la enzima HPRT -hipoxantina-guanina
fosforribosil transferasa-, que provoca grave deficiencia mental y tendencia compulsiva al
automutilamiento) e inmunodeficiencias como PNP (muy grave, provocada por la carencia de una purina
nucleósido fosforilasa) y ADA (la que padecen los "niños burbuja", en los que la falta del enzima
adenosina desaminasa les deja absolutamente indefensos contra cualquier agente patógeno,
obligándoles a vivir en un ambiente absolutamente estéril). En estos casos se conocían y habían sido
clonados los genes implicados. Bastaría una pequeña presencia de los productos génicos necesarios para
corregir la alteración, y unos niveles ligeramente superiores de los mismos no parece tener
consecuencias negativas. Las células implicadas en la producción de estos enzimas se hallan en la
médula ósea, lo cual plantea el problema adicional de hallar donantes compatibles para iniciar el
tratamiento. Los experimentos indican que el tratamiento génico de algunas células extraídas a los
enfermos y su posterior reinserción está siendo eficaz -al menos en ADA-, y que, tras dos o más años de
tratamiento se ha conseguido la relativa normalización del sistema inmune y la restauración de la
inmunidad celular y humoral, gracias en parte a ventajas de crecimiento que las células tratadas
genéticamente parecen tener sobre las anómalas
Aparte de la médula ósea, los tejidos humanos mejor conocidos eran hasta hace muy poco la piel y la
sangre. En otras células se plantean múltiples problemas para extraer las células necesarias, cultivarlas
durante el tiempo requerido para manipularlas genéticamente y ser reimplantadas al paciente. El bajo
número de células madre en la médula ósea, no diferenciadas todavía -antes de constituir las células 6 http://www.biotech.bioetica.org/ap47.htm
59

específicas de la sangre- y problemas en su reconocimiento han constituido un importante obstáculo
para la terapia génica. Además, ninguna de las técnicas disponibles a comienzos de los 90 permitía
insertar un gen en su locus o lugar cromosómico específico dentro de la célula diana.
Cuadro 1. Algunas enfermedades hereditarias tratables mediante terapia génica
Enfermedad Incidencia Producto normal del
gen defectuoso
Células a modificar por
la terapia génica
Inmunodeficiencia
combinada severa
(SCID) (“niños
burbuja”)
Rara Enzima adenosin
desaminasa (ADA)
Células de la médula
ósea o linfocitos T
Hemoglobinopatías
(talasemias)
1 cada 600 personas en
ciertos grupos étnicos
β - globina de la
hemoglobina
Células de la médula
ósea
Hemofilia A 1/10.000 varones Factor VIII de
coagulación
Células del hígado o
fibroblastos
Hemofilia B 1/30.000 varones Factor IX de
coagulación
Células del hígado o
fibroblastos
Hipercolesterolemia
familiar
1/500 personas Receptor del hígado
para lipoproteínas de
baja densidad (LDL)
Células del hígado
Enfisema hereditario 1/3.500 personas α-1-antitripsina
(producto hepático que
protege los pulmones
de la degradación
enzimática)
Células del pulmón o
del hígado
Fibrosis quística 1/2.500 personas Producto del gen CFTR
que mantiene libre de
mucus los tubos aéreos
de los pulmones
Células del pulmón
Distrofia muscular de
Duchenne
1/10.000 varones Distrofina
(componente
Células musculares
60

estructural del
músculo)
Fuente: Lacadena Calero, R. Genética. Universidad Complutense (Madrid-España) 1999.
A fines del siglo XX, las posibilidades y aplicaciones de los tratamientos génicos se ampliaron
notablemente, con diferentes resultados según las líneas celulares manipuladas:
Tratamiento génico de enfermedades hepáticas
Los experimentos con ratones transgénicos, bioquímica y fenotípicamente modificados, han permitido
evaluar la eficacia terapéutica de la transferencia génica somática de vectores adenovirales con
replicación alterada que codificaban el ADNc de la ornitina transcarbamilasa (OTC). La duración de su
expresión está por determinar todavía, creo, pero los primeros resultados parecen esperanzadores. Algo
parecido puede decirse sobre la expresión de beta-galactosidasa y a 1-antitripsina humana en
hepatocitos aislados en modelos transgénicos. A pesar de los problemas técnicos que persisten en
relación con la transferencia ex vivo de células y de la duración limitada de la expresión terapéutica, hay
al menos dos protocolos clínicos aprobados para la transferencia ex vivo de genes al hígado. Los
tratamientos van destinados a pacientes con insuficiencia hepática aguda y al tratamiento de la
hipercolesterolemia familiar. De manera global, la precaución y el estudio detenido de los protocolos
clínicos presentados seguirán siendo la norma, mientras persistan los problemas de expresión
transitoria y grado de eficacia insuficiente en el tratamiento genético hepático tanto in como ex vivo.
Hemoglobinopatías y tratamiento génico de células hematopoyéticas
El dominio adquirido en los trasplantes de médula ósea y la capacidad que tienen las células
primordiales hematopoyéticas (CPH) para reconstituir totalmente la médula ósea, hacen del sistema
hematopoyético un candidato idóneo para el tratamiento génico. Las inmunodeficiencias combinadas
severas (ADA), las hemoglobinopatías (talasemias), las deficiencias de adhesión leucocitaria y las
enfermedades de depósito lisosomal (enfermedad de Gaucher) han acaparado la mayor parte de las
investigaciones en tratamiento génico. Recientemente se han identificado los genes responsables de
tres enfermedades inmunitarias ligadas al cromosoma X, y diversos tipos de leucemia, el cáncer y el
SIDA están siendo objeto de intensos estudios que incluyen aproximaciones terapéuticas de tipo
genético. En las CPH los vectores retrovirales parecen ser, de momento, los más eficaces para la
transferencia.
61

ADA (trastorno autosómico recesivo; frecuencia: 25% de las ICS)
Su deficiencia origina una disfunción intensa en las células T y B, que provoca la muerte de los pacientes
antes de los 2 años de edad por infección masiva. Actualmente, el tratamiento preferido es el trasplante
de médula ósea procedente de un donante HLA idéntico, que puede producir una curación completa
aunque conlleva una alta morbilidad. Pero menos de un 30% de los pacientes tienen un hermano HLA
idéntico. La sustitución enzimática no consigue una restitución inmunitaria completa y produce otros
efectos tóxicos. Un tratamiento sustitutorio, consistente en inyectar el enzima ADA combinada con
polietilenoglicol (PEG) permite en ciertos casos frenar los efectos de la enfermedad. Pero es un
tratamiento muy caro, ininterrumpido y de eficacia inconstante.
Tras numerosos experimentos en organismos modelo, se iniciaron a mediados de los años ‘90 ensayos
clínicos de transferencia génica de ADA hacia las células T periféricas, previamente tratadas in vitro.
Aunque algunos pacientes experimentaron una notable mejoría clínica e inmunitaria, los resultados
difieren considerablemente de unos a otros y no llegan a normalizarse todos los índices de la función
inmunitaria. En opinión de algunos, ni en este ensayo pionero ni en otros existen evidencias inequívocas
de que el tratamiento genético ha producido beneficios terapéuticos. Los riesgos de posible
mutagénesis insertiva de genes relacionados con el cáncer, después de repetidas transferencias
retrovirales, no se han visto confirmados hasta el momento. Pero los modestos resultados han
estimulado otras propuestas de ensayos clínicos en Italia y Países Bajos. Según sus autores, en uno de
estos últimos ensayos la transferencia genética había funcionado y se había conseguido la reconstitución
inmunitaria. En todo caso, es preciso tener en cuenta que los pacientes estuvieron recibiendo también
inyecciones rutinarias de ADA sintética, y estos tratamientos convencionales podrían ser responsables
en buena parte de su buena salud.
Enfermedad de Gaucher
Autosómica recesiva, es producida por el derivado proteico de un gen que codifica la enzima
glucocerebrosidasa. El trasplante alogénico de médula ósea ha corregido la enfermedad en algunos
pacientes, pero ya se ha conseguido la trasferencia génica retroviral de un gen recombinante normal de
la glucocerebrosidasa en células madre de ratón, seguida de la expresión proteica en macrófagos
diferenciados a partir de células madre transducidas.
Las hemoglobinopatías representan el trastorno genético más frecuente en humanos, y la mayor parte
de los experimentos con tratamiento génico persiguen la expresión regulada de altos valores del gen de
62

la globina utilizando vectores retrovirales. Pero a mediados de 1994 no se había conseguido la expresión
regulada de los genes de la globina utilizando vectores retrovirales.
Tratamiento genético del cáncer
Los procesos cancerígenos hereditarios como el retinoblastoma, poliposis adenomatosa familiar, cáncer
de mama y melanoma están relacionados con un gen único que predispone al paciente a presentar
neoplasia. Leucemia y linfomas no hereditarios parecen provocados por nuevas mutaciones
(translocaciones, a menudo) que confieren un nuevo rasgo genético a la célula maligna. La corrección
génica de todas las células malignas implicadas en procesos cancerígenos constituye un desafío
sorprendente. Muchos estudios han intentado incrementar la cantidad y citotoxicidad especifica de los
linfocitos que reaccionan con las células tumorales. Los primeros intentos incluían el marcaje de unas
células inmunes llamadas linfocitos de infiltración tumoral (TILs, tumor-infiltrating lymphocytes) para
seguir el progreso del tratamiento contra el melanoma maligno.
Steven Rosenberg, uno de los más conocidos investigadores sobre el cáncer de los NIH, consiguió en
1990 la aprobación definitiva para una segunda aplicación de la terapia génica a pacientes con casos
avanzados de melanoma, cáncer de piel que anualmente mata a unos 28.000 ciudadanos en EE.UU. El
enfoque adoptado por Rosenberg reconoce las limitaciones del recurso a fuerzas externas (radiación,
quimioterapia y cirugía) con los pacientes cancerosos y propone una estrategia basada en los propios
mecanismos internos del cuerpo, como la terapia génica, para conseguir que el propio cuerpo rechace la
enfermedad. Rosenberg y su equipo extrajeron linfocitos de infiltración tumoral procedentes de los
tumores de pacientes con melanoma. Los TILs fueron introducidos en una solución de interleuquina-2,
una sustancia natural que potencia su efecto destructor, y posteriormente expuestos a retrovirus de
leucemia de ratón manipulados. El sistema de transporte y distribución de Rosenberg había sido
neutralizado y dotado mediante técnicas de ADN recombinante con un gen humano. Este gen codifica
un factor de necrosis tumoral (TNF), una proteína que interfiere con el suministro de sangre al tumor y
debilita las células tumorales. Los virus alterados se insertan ellos mismos junto con su gen polizón
dentro del material genético de los TILs, y estos son inyectados en la sangre de los pacientes con
melanoma. Conforme a las previsiones, los TILs activados se hospedarían en los tumores como si fuesen
misiles teledirigidos, atacando las células cancerosas y a la vez liberando el factor antitumoral (tóxico)
para ayudar a exterminarlos.
63

Un año después, sin embargo, el comité de asesores científicos del National Cancer Institute's Division of
Cancer Treatment cuestionó la fiabilidad y algunos elementos cruciales de los experimentos de
Rosenberg, negándole un contrato de 3,9 millones de dólares por 3 años para desarrollar células TIL en
un laboratorio independiente. Han fallado varios aspectos importantes en los dos años de
experimentación. Aunque los TILs sí se dirigían al tumor, no lo hacen los modificados con el TNF. Parece
que algo relacionado con la inserción del TNF interfiere con su capacidad para hospedarse en el tumor.
El resultado es que la mayor parte de los linfocitos modificados quedan atrapados en el hígado, bazo y
pulmones, donde probablemente son destruidos. Y la expresión no regulada en estos órganos del TNF
puede originar procesos tóxicos secundarios.
Otro método basado en la inmunoterapia ("vacunación genética" o inmunoterapia activa) trata de
aumentar el carácter "extraño" de las células tumorales para estimular la acción antitumoral de las
células asesinas (linfocitos T y macrófagos) del sistema inmunitario. Las células tumorales producen en
su superficie unas proteínas anómalas capaces de activar las células asesinas. Algunos tumores incluso
son portadores de antígenos propios ("antígenos asociados a los tumores") que permanecen
"silenciosos" en las células normales.
Incremento de la inmunogenicidad de las células tumorales
Mediante modificación genética se intenta potenciar la respuesta inmunitaria dirigida contra el tumor.
Se utilizan diversas proteínas específicas, especialmente citoquinas y moléculas de adhesión celular
(interleuquina-2, interleuquina-4, el TNF, etc.) que no producen ningún efecto sobre el crecimiento de
las células tumorales in vitro pero inhiben el crecimiento del tumor in vivo. En ratón, las células
tumorales son eficazmente rechazadas cuando han sido manipuladas mediante técnicas de ingeniería
genética para expresar diversas citoquinas o bien el complejo principal de histocompatibilidad. Esto
hace pensar que en humanos, el protocolo debería incluir la eliminación de una parte del tumor, la
transducción de las células in vitro con las formas de expresión adecuadas y el reimplante de estas
células tumorales al paciente. Se han aprobado diversos protocolos clínicos en todo el mundo para la
transferencia in vitro a células tumorales de genes de citoquinas (IL-2, IL-4, el interferón g , el GM-CSF o
factor estimulante de colonias de granulocitos-macrófagos, etc.) para diferentes tipos de cáncer:
colonrrectal, de mama, melanoma maligno, neuroblastoma, carcinoma de pulmón, de riñón, etc.
Genes protectores y destructores
64

Otros ensayos persiguen la utilización de genes protectores, mediante la transferencia de genes que
incrementen la resistencia a varios fármacos en células madre de pacientes con tumores sólidos o
leucemia y permitan niveles superiores de quimioterapia para erradicar la enfermedad residual. Otros
estudios proponen utilizar genes destructores como los de la toxina diftérica y los que codifican el TNF
para eliminar las células cancerosas; o el empleo de "genes suicidas", que transforman un producto no
tóxico (por ejemplo, un antivírico como el aciclovir) en un veneno que provoca la muerte de las células.
Otros enfoques del tratamiento génico contra el cáncer pretenden el marcaje de las células malignas
con proteínas codificadas por genes de supresión tumoral como el p53 (alterado en el 50% de los casos)
o ras (alterado sólo en el 30%). In vitro, las células malignas a las que se ha transferido la forma natural
del p53 ya no tienen capacidad tumorigénica o la tienen más débil que las células originales.
Poco tiempo antes de concluir el siglo XX se ha desvelado el funcionamiento de otro gen muy
directamente implicado en la supresión de tumores, el p16. De momento, se están diseñando los
vectores para introducirlo adecuadamente en las células tumorales y conseguir su expresión con arreglo
a las previsiones. Pero el propio director de equipo, el español Manuel Serrano, reconoce las dificultades
inherentes todavía a las terapias génicas y deposita más esperanzas en el diseño de alguna molécula por
síntesis química capaz de imitar la acción del gen p16.
Todas estas alternativas presentan numerosos inconvenientes todavía. Sigue siendo difícil extraer y
modificar células tumorales. Sólo una fracción de ellas -poco controlable- resulta manipulable para una
posible fabricación de vacunas parciales. No todos los tumores son físicamente accesibles. El problema
más importante lo constituye la elevada eficacia necesaria en la transferencia de las células modificadas
a las células neoplásicas, de modo que no perjudiquen a las normales. Por último, la manipulación de
células tumorales con genes inhibidores de la proliferación celular como el p53 requiere la modificación
de todas las células tumorales, y como la mayoría de cánceres proceden de una cascada de anomalías
genéticas, la reversión de una sola de ellas no bastaría seguramente para detener la enfermedad.
Tratamiento genético de las células respiratorias
Para la fibrosis quística (enfermedad autosómica recesiva, que afecta a 1/2.500 recién nacidos blancos)
existe un tratamiento convencional (fluidificación de las secreciones del sistema respiratorio,
tratamiento de las infecciones y sustitución de las enzimas pancreáticas). Se ha descubierto
recientemente el gen implicado en la enfermedad, el regulador de la conductancia transmembrana
(CFTR), lo que ha supuesto un importante avance hacia su tratamiento génico. Se están siguiendo
65

estrategias in vivo para el tratamiento de la FQ, más adecuadas y viables que las ex vivo. Después de
algunos intentos con ratones (instilación traqueal de un adenovirus recombinante con replicación
defectuosa que codifica el CFTR) con buenos resultados -aunque la expresión del gen no ha durado más
de 42 días- se aprobaron varios ensayos clínicos en humanos utilizando un vector adenoviral con CFTR y
transferencia genética de un complejo ADN-liposoma. Los riesgos de toxicidad parecen descartados en
los primeros intentos y basta algo tan sencillo como un inhalador para conseguir la expresión del gen y
aliviar en un 30% los síntomas de la enfermedad.
Tratamiento genético muscular (Síndrome de Duchenne)
En ratones se ha conseguido la sustitución de las secuencias anómalas del gen mutante por secuencias
normales, corrigiendo así el defecto genético.
Cada día la literatura científica da cuenta de una nueva enfermedad sometida a esta terapia. El tumor de
páncreas –por ejemplo-, que carece de un tratamiento realmente efectivo y es de pronóstico tan
negativo, justifica la estrategia de la terapia genética. La técnica en desarrollo se basa en genes suicidas,
que por sí mismos carecen de actividad nociva; y que codifican para una proteína capaz de transformar
una pro-droga (fármaco con baja toxicidad) en un metabolito tóxico, que es el que causará la muerte de
la célula cancerosa. Este sistema tiene la ventaja de que el metabolito tóxico puede difundirse de una
célula a otra con lo que, modificando una célula mediante herramientas de terapia génica, se puede
conseguir la muerte de las células tumorales adyacentes.
Otro caso logrado por el equipo de investigadores dirigidos por Joseph Glorioso, del Departamento de
Genética Molecular y Bioquímica de la Universidad de Pittsburgh, en Estados Unidos, quienes han
obtenido la patente para un vector genético capaz de bloquear respuestas dolorosas en ratones. El
vector se basa en la utilización del herpes virus, uno de cuyos genes produce una enzima que bloquea el
dolor. Esta técnica –cuyos resultados en cobayos se han mantenido hasta por siete días- podrá utilizarse
para tratar el dolor asociado al cáncer, la artritis, la angina y las neuropatías periféricas, patologías cuya
medicación actual está basada en narcóticos que provocan, entre otros efectos secundarios, confusión
mental y letargo. En comparación con estos métodos convencionales, la terapia genética es muy
específica, mientras que la liberación de sustancias analgésicas se encuentra limitada a la
hiperestimulación de un grupo limitado de nervios.
En 1990 fue posible que una niña de cuatro años afectada de inmunodeficiencia combinada aguda
(IDCA) fuera inoculada con glóbulos blancos genéticamente modificados conteniendo una copia
66

funcional del gen de la enzima adenosina-desaminasa (ADA), proteína esencial para el desarrollo y
funcionamiento del sistema inmunitario humano. La niña había heredado de ambos padres las copias
defectuosas del gen de la ADA; ellos, a su vez, había heredado una variante defectuosa del gen de uno
de sus progenitores pero el mismo había quedado “compensado” por la copia normal heredada del otro
progenitor, y habían gozado siempre de perfecta salud. La niña podía haber heredado las copias
normales o al menos una normal, y hubiese sido saludable. La carencia de ADA es uno de los más de
7.000 “trastornos de un solo gen” que provocan enfermedades genéticas o hereditarias en los seres
humanos. La terapia somática a la que fue sometida no alteró en la niña su genoma (y las copias que de
ella heredarán sus hijos serán defectuosas), sino que modificó funcionalmente su sistema inmunológico
al introducir los nuevos glóbulos blancos alterados para producir la proteína deseada en las cantidades
necesarias, en el momento adecuado. Aún falta alterar las células-madre productoras de los glóbulos
blancos, con la instrucción correcta de producir la enzima ADA. La alteración del genoma -mediante
terapia germinal- podría ser intentada en el cigoto (embrión de una sola célula) de los hijos de la niña o,
quizás, en las gametas que una vez adulta se destinen a la procreación, la cual debería ser llevada a cabo
in vitro
Otros métodos alternativos que se están ensayando son, por ejemplo, el de inyectar directamente genes
normales que codifican para la distrofina para tratar de curar la distrofia muscular de Duchenne, o
inhalar mediante pulverización con aerosol virus o liposomas portadores de genes normales que, una
vez dentro de las células de los pulmones, permitan curar la fibrosis quística.
Un grupo importante de enfermedades que se originan por la alteración de multiples genes es el de los
cánceres. Dejando de lado los factores ambientales que en estas enfermedades –como en otras- tienen
una decisiva relevancia, y centrándose en el ámbito estrictamente genético se verifica que los genes
protagonistas de la cancerización pueden pertenecer a uno de dos grupos antagónicos: 1) los
procarcinogénicos denominados oncogenes y 2) los que suprimen la formación de tumores conocidos
como antioncogenes o emerogenes. De los cien mil genes promedio en cada célula de nuestro
organismo, se calcula que hay aproximadamente cien oncogenes y cien antioncogenes. Los primeros
pueden producir cáncer cuando su estructura se altera por mutaciones, traslocaciones y por pérdidas
parciales o totales de su secuencia. Pero además de los cambios en los oncogenes, la cancerización
también implica la pérdida de la función de uno o múltiples antioncogenes en prácticamente todos los
cánceres humanos. Los antioncogenes, al sufrir mutaciones o perdidas parciales o totales de su
secuencia, dejarán de suprimir algunos pasos que evitan la cancerización de las células. De esta forma, la
67

cancerización se conceptualiza actualmente en términos de activación de oncogenes asociada a la
inactivación de antioncogenes, y se inscriben en la lista de candidatos para las terapias génicas.
Esto ocurre con productos génicos descubiertos en algunos melanomas humanos, las proteínas MAGE
(melanoma antigen) y MART (melanoma antigen recognized by T-cells) descubiertas recientemente por
los equipos de T. Boon y Rosenberg, respectivamente. Normalmente, los antígenos son presentados a
las células inmunitarias en forma de fragmentos por las proteínas del complejo mayor de
histocompatibilidad (HLA) presentes en las superficies de las células. Pero una de las diferencias
fundamentales entre las células normales y las tumorales está en que estas últimas no presentan
correctamente los antígenos tumorales. Esta es una de las características que se pretenden modificar.
68

Proteínas recombinantes
Biotecnología de las proteínas recombinantes 7
La Biotecnología de proteínas está implicada en el aislamiento, producción y mejoramiento de las
propiedades biológicas de proteínas específicas a partir de diversas fuentes naturales tales como
plantas, animales o bien microorganismos, para su subsiguiente empleo en diversas aplicaciones. La
tecnología del DNA recombinante ha jugado un papel muy importante en el desarrollo de la
Biotecnología de proteínas ya que gracias a la manipulación de genes ha sido posible producir grandes
cantidades de proteínas, muchas de las cuales se encuentran en concentraciones muy bajas en su
ambiente natural. Se considera una proteína recombinante o proteína heteróloga aquella proteína cuya
síntesis se realiza en un organismo distinto al organismo nativo. Actualmente se ofrecen en el mercado
internacional una gran variedad de proteínas recombinantes para un amplio abanico de aplicaciones,
incrementándose día a día la lista de estos productos, lo que ha llegado a constituir toda una revolución
en el mercado biotecnológico y a la vez ha impulsado la investigación en este campo (Walsh &Headon
1995).
El desarrollo de la tecnología del DNA recombinante y el avance logrado en la optimación de
bioprocesos con organismos recombinantes ofrece una amplia gama de posibilidades para la producción
de proteínas nativas o modificadas con nuevas y mejores propiedades.
Para la producción de proteínas recombinantes se hace uso de uno de los múltiples sistemas de
expresión disponibles comercialmente o de aquellos desarrollados con fines de investigación, o bien se
pueden diseñar y construir sistemas de expresión según necesidades específicas. Un sistema de
expresión lo conforma un organismo hospedero y un vector de expresión o fragmentos de DNA que
poseen los elementos génicos necesarios para realizar los procesos de trascripción y traducción en dicho
organismo hospedero, además de recibir las moléculas de DNA recombinante.
Para la selección de un sistema de expresión adecuado para la síntesis de una proteína recombinante se
tienen que tener en consideración el origen biológico y las propiedades químicas y biológicas de la
proteína de interés, su aplicación posterior y el bioproceso que se empleará para su producción (Tabla
7 Guerrero - Olazarán, Cab - Barrera, Galán - Wong y Viader – Salvadó. Biotecnología de Proteínas Recombinantes para la Aplicación en Acuacultura. 2004. Avances en Nutrición Acuícola VII. Memorias del VII Simposium Internacional de Nutrición Acuícola. Hermosillo, Sonora, México. Págs. 419 - 427
69

1). El origen biológico de la proteína es importante ya que generalmente un sistema de expresión
eucariota es más eficiente para sintetizar una proteína de origen eucariota que un sistema de expresión
procariota. Respecto a las propiedades químicas y biológicas de la proteína es importante analizar su
secuencia nucleotídica codificante para evaluar el uso de codones preferenciales y la presencia de
secuencias de finalización prematura de la transcripción, la secuencia aminoacídica para evaluar el peso
molecular, punto isoeléctrico y tipo de modificaciones postraduccionales presentes, la estabilidad al pH,
temperatura y proteólisis, la posible toxicidad sobre el hospedero, el destino celular deseado
(extracelular o intracelular) y el grado de pureza deseado. Un tercer factor a considerar es la aplicación
que se le dará posteriormente a la proteína, si ésta se aplicará en las áreas biomédica (agentes
terapéuticos, vacunas y diagnóstico clínico) o de alimentos (procesos industriales, aditivos y
nutraceúticos) para emplearla en humanos, animales o plantas, o en el área de la Química de la
transformación como la industria del papel, textil y del cuero, farmacéutica y química, o bien como
reactivo en el área de la química analítica o en el medio ambiente como fuente de energía o tratamiento
de residuos. Por último se tiene que considerar el bioproceso que se empleará para su producción
específicamente el tipo y condiciones operacionales de cultivo, la escala de producción de la proteína de
interés según si sólo se requieren unos pocos miligramos para fines de investigación o en la escala de
gramos o kilogramos para fines industriales, además de evaluar el costo del bioproceso.
Tabla 1. Factores determinantes en la selección de un sistema de expresión
ORIGEN DE LA
PROTEÍNA
Procariota
Eucariota
PROPIEDADES DE
LA PROTEÍNA
Secuencia nucleotídica
Propiedades químicas
Estabilidad
Uso de colores
preferenciales
Tamaño
pI
Modificaciones
postraduccionales
Proteólisis
pH
70

Toxicidad
Destino celular
Grado de pureza
Temperatura
Extracelular
Intracelular
APLICACIÓN
Biomedicina
Alimentos
Química de la Trasformación
Reactivo químico
Medio ambiente
Agente terapéutico
Vacunas
Diagnóstico
Procesos Industriales
Aditivos
Nutracéuticos
Suplemento alimenticio
Industria del papel
Industria textil y del cuero
Industria farmacéutica
Industria química
Reactivo analítico
Energía y combustibles
Tratamiento de residuos
Humanos
Animales
Vegetales
Catálisis enzimática
Síntesis estereoselectiva
BIOPROCESO Tipo
Condiciones de cultivo
Escala
Costo
Actualmente se emplean una gran variedad de hospederos que van desde organismos unicelulares tales
como bacterias, hongos, levaduras y líneas celulares de mamíferos, insectos o plantas, hasta organismos
completos como animales y plantas transgénicas.
Dentro de las bacterias, Escherichia coli ha sido la más empleada para estos fines, aunque también se
han utilizado Bacillus subtilis, Caulobacter crescentus, Pseudomonas sp., Streptomyces lividans,
71

Lactobacillus lactis, etc. (Walsh & Headon 1995). Debido al nivel de conocimiento sobre su fisiología y
genética, E. coli ofrece varias ventajas (Tabla 2) respecto a otros hospederos, lo que ha permitido el
desarrollo de una gran variedad devectores de expresión y cepas mutantes. Sin embargo, el empleo de
E. coli tiene también desventajas, tales como su ineficiencia para secretar proteínas al medio de cultivo y
además éstas suelen precipitar como cuerpos de inclusión insolubles en el citoplasma celular.
Adicionalmente, la síntesis de las proteínas heterólogas en E. coli induce a un incremento de la
proteólisis celular, cuenta con un sistema ineficiente para realizar modificaciones postraduccionales y
genera endotoxinas dañinas para la salud.
Si se emplea Bacillus subtilis, se tiene la ventaja de la secreción de la proteína heteróloga, sin embargo
esta bacteria secreta una gran cantidad de proteasas, además se dispone de una menor cantidad de
vectores de expresión, elementos promotores para la regulación de la expresión de los transgenes y se
ha reportado menor estabilidad genética de los plásmidos incorporados en el hospedero.
Los sistemas de expresión de proteínas recombinantes basados en levaduras han demostrado ser una
fuente eficiente y económica de proteínas de eucariotas superiores de interés industrial y/o académico
(Buckholz & Gleeson 1991, Romanos et al. 1992). Las levaduras ofrecen un ambiente apto para el
plegamiento de proteínas eucarióticas, llevan a cabo algunas modificaciones postraduccionales y
pueden secretar las proteínas recombinantes al medio de cultivo. Además las levaduras mantienen las
ventajas de los microorganismos al ser unicelulares, de fácil manipulación y rápido crecimiento. Por otro
lado tienen una organización celular eucariótica que permite la realización de procesos de expresión y
maduración característicos de células animales y vegetales (Eckart & Bussineau 1996), lo cual les da una
ventaja para la producción de proteínas de origen Eucariota.
El empleo de Saccharomyces cerevisiae como hospedero en un sistema de expresión eucariota ofrece la
ventaja de ser uno de los organismos eucariotes mejor caracterizado, su genoma ha sido totalmente
secuenciado y es considerado como organismo GRAS (Generally Recognized as Safe) por la FDA (Food
and Drug Administration) americana.
Por estos motivos, muchos de los sistemas de expresión desarrollados para la producción de proteínas
heterólogas están basados en S. cerevisiae. Las limitaciones de este microorganismo se han puesto de
manifiesto al aumentar de manera significativa el número de sistemas de expresión basados en
levaduras no convencionales (no Saccharomyces), los cuales exhiben mejores prestaciones como
productores de ciertas proteínas heterólogas. Entre estos nuevos microorganismos hospederos se
72

incluyen Hansenula polymorpha, Pichia pastoris, Yarrowia lipolytica y Kluyveromyces lactis, los cuales
constituyen la base de diversos procesos desarrollados a escala industrial.
Tabla 2. Ventajas y desventajas de diferentes hospederos para la producción de proteínas
recombinantes
BACTERIAS
Escherichia coli
VENTAJAS DESVENTAJAS
Buen nivel de conocimiento sobre su fisiología y
genética
Se cuenta con una gran variedad de vectores de
expresión y cepas mutantes
Fácil manipulación
Cultivos baratos con altas densidades celulares
Niveles moderados de producción de la proteína
recombinante
Ineficiente secreción de la proteína al medio de
cultivo
Formación de cuerpos de inclusión insolubles en el
citoplasma celular
La síntesis de las proteína heterólogas induce a un
incremento de la proteólisis celular
Realiza pocas modificaciones postraduccionales
Generación de endotoxinas dañinas para la salud
humana y animal
Bacillus subtilis
Secreción de proteína heteróloga Secreta una gran cantidad de proteasas afectando el
rendimiento del producto recombinante
Dispone de pocos vectores de expresión y elementos
promotores para la regulación de la expresión de los
transgenes comparado con E. coli
Menor estabilidad genética de los plásmidos
incorporados en el hospedero
73

LEVADURAS
Saccharomyces cerevisiae
Sistema eucariota mejor caracterizado desde el punto de vista de la Biología
Molecular y la Fisiología
Genoma totalmente secuenciado
Vectores de expresión y cepas disponibles
Manipulación sencilla a nivel de laboratorio e industrial
Cultivos baratos
Se producen altos niveles de proteína recombinante
Realiza modificaciones postraduccionales
Es considerado como organismo GRAS
Produce
hiperglicosilaciones
Baja eficiencia en la
producción de proteína
heteróloga
Pichia pastoris
Emplea los promotores más fuertes y eficientemente regulados, de los conocidos
Vectores de expresión y cepas disponibles
Manipulación sencilla a nivel de laboratorio e industria
Secreción eficiente de proteína heteróloga
Medios de cultivo son baratos y formulados libre de toxinas y pirógenos
Los cultivos alcanzan altas densidades celulares
Altos niveles de producción
Bajos niveles de secreción de proteínas endógenas facilitando la purificación de la
proteína heteróloga
Produce patrones de
glicosilación no
deseables para
algunas proteínas
Actividad
proteolítica
Algunas proteínas
no se producen de
forma eficiente
74

El sistema de expresión basado en la levadura metilotrófica Pichia pastoris ha ganado aceptación como
hospedero para la producción de proteínas recombinantes (Romanos 1995, Escamilla-Treviño et al.
1999, Cereghino & Cregg 2000). Pichia pastoris comparte las ventajas de fácil manipulación genética y
bioquímica con S. cerevisiae pero supera a ésta de 10 a 100 veces en los niveles de producción de
proteínas heterólogas (Cregg et al. 1993, Romanos 1995, Guerrero-Olazarán & Viader-Salvadó 2003).
Ambas levaduras se han utilizado en la producción de muchas proteínas eucariotas y en la mayoría de
los casos la productividad de P. pastoris ha sido muy superior a la de S. cerevisiae (Romanos 1995,
Cereghino & Cregg 2000). Pichia pastoris comparada con S. cerevisiae, presenta importantes ventajas
para la expresión de proteínas heterólogas. En P. pastoris el promotor usado para transcribir genes
heterólogos proviene del gen de la enzima alcohol oxidasa 1 (AOX1) de P. pastoris cuya expresión es
regulada por metanol. Este promotor es uno de los más eficientes y fuertemente regulados de los
conocidos actualmente. Esta capacidad para mantener los cultivos con la expresión heteróloga
“apagada” en ausencia de metanol es importante para minimizar el efecto tóxico que presentan muchas
proteínas heterólogas sobre el hospedero (Cereghino & Cregg 1999). Por otro lado, mientras que S.
cerevisiae presenta dificultades para conseguir elevadas densidades celulares, P. pastoris es
relativamente fácil de cultivar a densidades celulares de ~100 g/L, en peso seco. Además, S. cerevisiae
puede presentar baja eficiencia en la secreción de proteínas heterólogas (Cereghino & Cregg 1999,
Guerrero-Olazarán & Viader-Salvadó 2003). Adicionalmente, la glicosilación en el proceso de post-
traducción en P. pastoris se lleva a cabo con oligosacáridos más pequeños que en S. cerevisiae,
reduciendo los fenómenos de hiperglicosilación de muchas proteínas de eucariotas expresadas en este
último sistema (Romanos 1995, Higgins & Cregg 1998). Cabe destacar los bajos niveles de secreción al
medio de cultivo de proteínas endógenas en P. pastoris, lo cual supone una gran ventaja para la
purificación de la proteína recombinante producida. Finalmente, el medio de cultivo empleado para el
cultivo de P. pastoris en un biorreactor es una mezcla definida de sales, oligoelementos, biotina y fuente
de carbono y nitrógeno que no resulta ser caro y puede ser formulado libre de toxinas y pirógenos
(Higgins & Cregg 1998, Viader-Salvadó & Guerrero-Olazarán 2003).
Aunque muchos metabolitos se obtienen a partir de microorganismos no modificados por ingeniería
genética, la incorporación de esta metodología permitió optimizar la eficiencia del proceso de
producción y/o la calidad del producto. Por un lado, fue posible modificar el control de vías metabólicas,
75

por ejemplo para la sobreproducción de metabolitos, y por otro, permitió fabricar proteínas bajo la
forma de proteínas recombinantes.8
Las ventajas que presenta la producción de una proteína bajo la forma de proteína recombinante son:
Permite obtener a partir de un microorganismo, cultivo de células, planta o animal una proteína
completamente ajena, tal es el caso de la producción de insulina en bacterias, anticuerpos
humanos en plantas y vacunas en levaduras.
Se obtienen grandes cantidades del producto, fácil de purificar y más barato, en comparación
con el purificado a partir de su fuente natural (en el caso de la insulina, se obtenía a partir de
páncreas de animales).
Se obtienen productos libres de patógenos y otros riesgos potenciales. Esto es particularmente
importante en el caso de los productos farmacéuticos, ya que mucha gente ha contraído
enfermedades como SIDA y hepatitis B o C por empleo de hormonas o factores derivados de
suero o sangre (por ejemplo, los factores de coagulación para el tratamiento de la hemofilia
ahora pueden administrarse como proteínas recombinantes, libres de contaminación).
Pueden producirse proteínas que no existen en la naturaleza, como ciertas estructuras de
anticuerpos denominadas “anticuerpos de cadena simple”, útiles en el diagnóstico y tratamiento
de algunas enfermedades.
En los últimos años han sido publicados diversos estudios focalizados a la optimización de la producción
y escalado de proteínas recombinantes (proteínas producidas en el laboratorio mediante ingeniería
genética producidas en células distintas a las que se producen en la naturaleza) para uso médico,
veterinario e industrial. Aunque se han producido en células de mamíferos, insectos y levaduras, la
bacteria Escherichia coli continúa siendo el microorganismo más empleado para la expresión de genes
heterólogos o recombinantes.
Muchas proteínas heterólogas expresadas intracelularmente en E. coli se producen rutinariamente con
niveles superiores al 15% del total de proteínas celulares producidas en esta bacteria, a menudo en
forma de agregados insolubles intracelulares, llamados cuerpos de inclusión. Los cuerpos de inclusión
están determinados por una combinación de factores relativos a la velocidad de plegamiento y
8 Concejo Argentino para la Información y el Desarrollo de la Biotecnología http://www.argenbio.org/h/biotecnologia/07.php
76

agregación, solubilidad de la proteína, estabilidad termodinámica y plegamiento intermediario así como
susceptibilidad a degradación proteolítica e interacción con chaperonas.
Al elegir un sistema de expresión de proteínas recombinantes para incrementar los niveles de
producción de las mismas deben tenerse en cuenta muchos factores. Estos incluyen características del
crecimiento celular, niveles de expresión, expresión intracelular y extracelular, modificaciones post-
traduccionales, actividad biológica de la proteína de interés, así como características especiales en el
caso de la producción de proteínas terapéuticas. La selección de un sistema particular de expresión
requiere además la valoración de costos del proceso así como de otras consideraciones económicas. Si
bien E. coli es el microorganismo más empleado para la producción de proteínas recombinantes, debido
en parte al conocimiento de la biología molecular del mismo, no todos los genes pueden expresarse
eficientemente en este microorganismo. Esto puede estar determinado por las características de la
secuencia génica, estabilidad y eficiencia traduccional del ARN mensajero (ARNm; molécula usada como
molde, formada a partir de la secuencia de ADN necesaria para la síntesis de una determinada proteína),
plegamiento proteico, degradación proteica por enzimas celulares llamadas proteasas, diferencias de
uso de determinados codones (código de ADN que indica un determinado aminoácido), así como la
toxicidad potencial de la proteína heteróloga en la célula donde se va a producir. Las mayores
desventajas de los sistemas de expresión en E.coli incluyen la incapacidad de realizar modificaciones
post-traduccionales (o sea modificaciones que se realizan en las proteínas inmediatamente después que
la misma ha sido sintetizada en la célula) en proteínas eucariotas (proteínas sintetizadas en células no
bacterianas), la carencia de mecanismos de secreción para la liberación eficiente de proteínas al medio
de cultivo, y limitada capacidad de formación de puentes disulfuro. Sin embargo, por otro lado, muchas
proteínas eucariotas retienen su actividad biológica en forma no glicosilada pudiendo ser producidas
eficientemente en E. coli.9
Factores antihemolíticos
Los factores de la coagulación, a excepción del 6, se enumeran con números romanos, y son:
I: Fibrinógeno
II: Protrombina
9 Este artículo está basado en parte del trabajo de tesis de la Maestría en Biotecnología: “Producción de Estreptolisina-O recombinante para uso diagnóstico” realizado por Blanca Velázquez (Facultad de Ciencias, Universidad de la República, Uruguay)
77

III: Factor tisular (Tromboplastina)
IV: Calcio
V: Proacelerina (factor labil)
VII: Proconvertina (factor estable)
VIII: Factor antihemofílico A
IX: Factor antihemofílico B, Factor de Christmas
X: Factor de Stuart-Prower
XI: Factor antihemofílico C
XII: Factor de Hageman
XIII: Factor estabilizante de la fibrina
Son esenciales para que se produzca la coagulación, y su ausencia puede dar lugar a trastornos
hemorrágicos graves. Se destacan:
El factor VIII: Su ausencia produce hemofilia tipo A.
El factor IX: Su ausencia provoca hemofilia B.
El factor XI: Su ausencia provoca hemofilia C.
El factor antihemofílico (FAH), o proteína coagulante factor VIII, es una proteína que regula la activación
del factor X por las proteasas generadas en la vía intrínseca de la coagulación. Se sintetiza en el hígado y
circula formando un complejo con otra proteina, el factor de Von Willebrand (FvW). El factor VIII está
presente en concentraciones bajas (10ug/L) y el gen que codifica este factor esta en el cromosoma X.10
Uno de cada 10.000 varones nace con un déficit o defecto funcional de la molécula del factor VIII. El
proceso resultante, la Hemofilia A, se caracteriza por hemorragias en los tejidos blandos, los músculos y
las articulaciones que soportan peso. Los pacientes con manifestaciones clínicas suelen tener
concentraciones del factor VIII inferiores al 5%. Los pacientes que tienen menos de 1% de actividad del
factor VIII padecen una forma grave de hemofilia; sangran con frecuencia incluso sin traumatismos
perceptibles. Los pacientes con una concentración de 1 a 5% tienen una hemofilia moderada con
episodios hemorrágicos menos frecuentes; y los que tienen concentraciones de más del 5% presentan
10 HARRISON T.R. Principios de medicina interna. Vol 1. Edicion 15. New York, 2001; Mc Graw Hill Corp; pag 882-901.
78

un cuadro leve con hemorragias infrecuentes desencadenadas casi siempre por algún traumatismo. La
mayoría de los pacientes con hemofilia A tienen concentraciones del factor VIII inferiores al 5%.11
Las hemorragias de la hemofilia aparecen horas o días después de sufrir una lesión, pueden afectar a
cualquier órgano y si no se tratan pueden persistir durante días o semanas. Así, pueden formarse
grandes acúmulos de sangre parcialmente coagulada que comprimen los tejidos próximos y producir
necrosis musculares, congestión venosa y lesiones isquémicas de los nervios. Normalmente el
hemofílico consulta por dolor seguido de una hinchazón en la articulación que soporta peso como la
cadera, la rodilla o el tobillo. La acumulación de sangre en la articulación (hemartrosis) y la hematuria
sin afección genito urinaria son frecuentes en estos pacientes. Los pacientes con sospecha de hemofilia
deben estudiarse mediante recuento plaquetario, TH, TP y TPT. Generalmente el paciente tiene un TPT
prolongado con normalidad de las otras pruebas es necesario realizar análisis especifico para cuantificar
el factor VIII y X e identificar que factor es el afectado. El 10-20% de los pacientes presentan inhibidores
del factor VIII y el tratamiento médico depende de la presencia o ausencia de estos, en casos de no
presentarlos el manejo puede incluir la utilización de estimulantes transitorios del factor VIII como
(DDAVP), agentes antifibrinoliticos como el acido epsilon-aminocaproico (EACA) o el acido tranexamico
o la sustitución de factores con crioprecipitados, plasma fresco congelado, factor VIII purificado a alta
temperatura o utrapuro. En casos de cifras bajas o moderadas de los inhibidores de Factor VIII se
pueden utilzar dosis altas de factor VIII purificado, esteroides o concentraciones de factor VIII de porcino
y en los casos de niveles altos de inhibidores de factor VIII se utilizan concentrados de complejo
protrombina, factor IX activado, esteroides y concentraciones de factor VIII de porcino.
Los pacientes con hemofilia A deben ser manejados teniendo en cuenta la ínter consulta con el
hematólogo el cual establece el diagnostico, el grado de deficiencia del factor VIII y la presencia de algún
inhibidor de este. Posteriormente, selecciona el tipo de sustitución a realizar y determina la dosis.
DNAasa I (DNASA I - Desoxirribonucleasa I - Bovine Páncreas - Páncreas de Bovino)
Desoxirribonucleasa I (generalmente llamada DNAasa I), es una endonucleasa codificada por el gen
humano DNASE1. DNasa I es una nucleasa que corta el ADN preferentemente en los enlaces fosfodiéster
adyacente a nucleótidos de pirimidina, produciendo polinucleótidos terminados en 5'-fosfato- con un
grupo hidroxilo libre en posición 3', en tetranucleótidos de producción promedio. Actúa sobre cadena de
11 KASPER D, BRAUNWALD E, FAUCI A, HAUSER S. Harrison’s Principles of Internal Medicine. Vol 1. 2005; Mc Graw Hill; pag 681-689.
79

ADN simple, doble cadena de ADN y la cromatina. Se ha sugerido una de las desoxirribonucleasas como
responsable de la fragmentación del ADN durante la apoptosis.12
DNasa I se une a la proteína de citoesqueleto, actina. Se une monómeros de actina con muy alta
afinidad (sub-nanomolar) y polímeros de actina con menor afinidad. La función de esta interacción no
está clara. Sin embargo, la unión actina – DNasa I es enzimáticamente inactiva, el complejo DNasa -
actina podría ser una forma de almacenamiento de DNasa I que previene el daño de la información
genética.
Este gen codifica un miembro de la familia DNasa. Esta proteína se almacena en los gránulos de
zimógeno de la envoltura nuclear y funciona en el clivaje del ADN de una manera endonucleolítica. Al
menos seis alelos codominantes autosómica se han caracterizados, DNASE1*1 a DNASE1*6. Las
mutaciones en este gen, así como el factor de inactivación de su producto de enzima, se han asociado
con el lupus eritematoso sistémico (LES), una enfermedad autoinmune. Una forma recombinante de
esta proteína se utiliza para tratar uno de los síntomas de la fibrosis quística hidrolizando el DNA
extracelular en el esputo y la reducción de su viscosidad. Variantes del empalme transcripcional
alternativo de este gen se han observado, pero no han sido bien caracterizadas.
Riesgos
12 http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=1773
80

Regulación de la expresión génica a nivel de la cromatina13
Existen cuatro subniveles de regulación al nivel de la cromatina:
1. Condensación de la cromatina: sitios sensibles e hipersensibles a la DNasa I
2. Zonas superenrolladas
3. Metilación de las citosinas
4. Reordenamiento del genoma
Condensación de la cromatina: sitios sensibles e hipersensibles a la DNasa I
La estructura cromatiniana descondensada representa, al parecer, el primer y más elevado nivel de
regulación. Es a este nivel que se llevará a cabo la selección entre los genes que la célula está
autorizada a transcribir y aquellos que ella no debe transcribir. La cromatina está constituida por el
DNA enrollado alrededor de una serie de nucleosomas, empaquetada más relajada en las regiones
que contienen genes activos. Además de los cambios generales que ocurren en las regiones activas o
potencialmente activas, ocurren cambios estructurales en sitios específicos asociados con la
iniciación de la transcripción o con determinadas características estructurales del DNA. Estos
13 http://fbio.uh.cu/sites/genmol/confs/conf7/index_euc.htm
81

cambios se detectaron por primera vez gracias a los efectos de la digestión con concentraciones
muy débiles de la enzima DNAsa I.
Cuando la cromatina se digiere con DNAsa I, el primer efecto es la introducción de cortes en la doble
cadena en sitios hipersensibles específicos. Ya que la susceptibilidad a la DNAsa I refleja la
disponibilidad del DNA en la cromatina, consideramos que estos sitios representan regiones de la
cromatina en las cuales el DNA está específicamente expuesto porque no está organizado en la
estructura nucleosómica usual. Un sitio hipersensible típico en la cromatina es cien veces más
sensible al ataque de las enzimas. Estos sitios también son hipersensibles a otras nucleasas y a
agentes químicos. Ellos representan fragmentos de DNA desprovistos de nucleosomas.
Muchos de los sitios hipersensibles están relacionados con la expresión génica. Cada gen activo
tiene su sitio en la región del promotor y a veces más de un sitio. La mayoría de los sitios
hipersensibles se encuentran solamente en la cromatina de las células en las cuales se está
expresando el gen asociado; no se encuentran cuando el gen está inactivo. Se asume que un sitio
hipersensible es el resultado de la unión de proteínas reguladoras específicas que excluyen los
nucleosomas. Los factores de transcripción pueden generar sitios hipersensibles asociados a la
transcripción.
Zonas superenrolladas
El superenrollamiento negativo del DNA hace que las bases estén más accesibles a las proteínas.
Algunos resultados experimentales demuestran que la variación del grado de torsión del DNA se
utiliza como medio para modificar el acceso de las proteínas al promotor, lo mismo en eucariontes
que en procariontes, regulando así la expresión de los genes correspondientes. Las mutaciones en
los genes de las topoisomerasas, que son las enzimas que crean o eliminan los supergiros,
disminuyen su actividad y también disminuyen importantemente la transcripción de numerosos
genes. Este efecto también se obtiene por los inhibidores de las topoisomerasas. Sin embargo, este
resultado no es general, y sólo algunos genes están afectados. Las topoisomerasas implicadas en
esta regulación parecen fijarse a determinadas secuencias específicas del DNA situadas antes de los
promotores.
82

La expresión génica está asociada a la no-metilación
La metilación del DNA tiene lugar en sitios específicos. En bacterias está asociado a la identificación
de una determinada cepa bacteriana y también con la diferencia entre el DNA replicado y el no
replicado. En eucariontes, su función primordial conocida está asociada al control de la
transcripción. Entre el 2 y el 7% de las citosinas en el DNA de las células animales está metilado (el
valor varía con las especies). La mayoría de los grupos metilo se encuentran en los "dupletes" CG, y,
de hecho, la mayoría de las secuencias CG están metiladas. Generalmente, los residuos C en ambas
cadenas de este tipo de secuencia palindrómica están metiladas. Cuando un duplete está metilado
en una sola de las dos cadenas, se dice que está hemimetilado.
Muchos genes tienen un patrón de metilación que es constante en la mayoría de los sitios, pero
puede variar en otros. Una minoría de sitios está metilado en tejidos en los cuales no se expresa el
gen, pero no están metilados en los tejidos en los cuales el gen se encuentra activo. Por tanto, un
gen activo se puede describir como hipometilado. Un gen metilado es inactivo, pero el no metilado
es activo. Una región hipometilada coincide con una región de máxima sensibilidad a la DNAsa I. La
metilación en el extremo 5´ de un gen puede estar directamente relacionada con su expresión.
Muchos genes no están metilados en el extremo 5´ cuando se expresan, aunque permanecen
metilados en el extremo 3´. Al igual que como sucede con otros cambios en la cromatina, parece
probable que la ausencia de grupos metilo esté asociada con la posibilidad de transcripción y no con
el propio acto de la transcripción.
Reordenamiento del genoma
Entre los genes cuya expresión está condicionada por un reordenamiento genómico figuran los
genes de determinados antígenos de superficie en el tripanosoma, los genes de las proteínas del
sistema inmune y los genes que intervienen en la esporulación de la levadura (mating-type o
fenotipo sexual). Los reordenamientos del DNA generan diversidad, tanto en procariontes como en
eucariontes. Las consecuencias generales del reordenamiento pueden ser:
a) Crear nuevos genes como es el caso de las inmunoglobulinas, necesarias para la expresión en
determinadas circunstancias.
b) El reordenamiento puede ser responsable del cambio de la expresión de un gen ya existente en
otro. Esto ofrece un mecanismo para la regulación génica. Este es el caso del fenotipo sexual o
83

esporulación de las levaduras.
Eritropoyetina (EPO) 14
La eritropoyetina es una hormona glicoproteica de gran importancia para la formación de glóbulos rojos
durante la generación de sangre (hematopoyesis). La palabra deriva del griego erythros “rojo” y poiein
“hacer”, y también se la suele llamar EPO, epoetina y antiguamente hematopoyetina.
Tiene 30 400 d de peso molecular y regula la proliferación y diferenciación de los precursores eritroides
en la médula ósea, su gen se expresa en el cromosoma 7 (q11- q22) y codifica una proteína de 193
aminoácidos.
La EPO está entre los Agentes Estimulantes de la Eritropoyesis. Como medicamento, la eritropoyetina se
fabrica de forma biotecnológica para el tratamiento de la anemia en pacientes que hacen diálisis, en los
cuales la formación de sangre está alterada debido a un fallo en los riñones, y también después de ciclos
de quimioterapia agresivos. La EPO también es tristemente famosa por los numerosos escándalos de
dopaje en los que se ha visto implicada, en especial en el ciclismo, lo que ha hecho que algunos la
llamen “droga del ciclista”. El principal efecto adverso que produce es un incremento del riesgo de
complicaciones cardiovasculares si se usa para elevar los niveles de hemoglobina por encima de 13
gramos por decilitro de sangre.
Biosíntesis y función biológica
La eritropoyetina pertenece filogenéticamente a la familia de las citoquinas, entre las que se encuentran
también la somatotropina, la prolactina, las interleuquinas 2-7, y los llamados “Factores Estimulantes de
la Colonia” (G-CSF, M-CSF y GM-CSF). En el cuerpo humano, la EPO se forma en un 85-90% en el riñón
mediante el endotelio de los capilares situados alrededor de los canales nefríticos, y en un 10-15% en los
hepatocitos del hígado. Además, podría sintetizarse también en el cerebro, la matriz, los testículos y el
bazo.
El gen de la EPO en el hombre se encuentra en el cromosoma 7 (posición 7q21-7q22). La síntesis es
estimulada por la saturación de oxígeno (hipoxia) en la sangre. Esto lleva a la translocación de la
subunidad alfa del “Factor Inducido por Hipoxia” (brevemente FIH) desde el citoplasma en el núcleo de
las células productoras de EPO. Allí, el FIH se enlaza a la subunidad β (FIH-β) acompañante, de donde se
origina el heterodímero HIF-1. Este se enlaza otra vez con el factor de transcripción CREB y el p300. El 14 http://www.eritropoyetina.com/
84

complejo proteínico resultante consiste de tres elementos iniciales mediante una conexión a tres
bandas del gen de la EPO.
La concentración de la hormona en el suero de un hombre sano es de hasta 19 mU/mL. En la
eritropoyesis, la EPO se enlaza en la médula al receptor de la eritropoyetina en las células del precursor
tipo BFU-E (Unidad Formadora de Colonias Explosivas Eritroides), primero a las células más maduras del
tipo CFU-E (Unidad Formadora de Colonias Eritroides), y finalmente a los eritrocitos diferenciados.
EPO: ruta Jak – Stat (Receptores de la EPO y transducción de señales)
El receptor (EpoR) pertenece a la familia de los receptores de las citoquinas, cuyas características
estructurales comunes consisten en dos o más dominios similares a la inmunoglobulina, cuatro residuos
de cisteína y la secuencia extra-celular WSXWS (Triptófano-Serina-variable Aminoácido-Triptófano-
Serina). La conexión de la EPO produce un homodímero del receptor que activa la enzima quinasa
acoplada al receptor otra vez vía transfosforilación. Además, los restos de tirosina enlazados al receptor
85

se fosforilan y sirven como una estación de acoplamiento para la proteína de transducción de señales
STAT5 mediante la cual se producen diferentes cascadas de señalización. En conjunto hay 94 proteínas
implicadas.
Cada día se generan aproximadamente doscientos mil millones de eritrocitos. Además de en la
eritropoyesis, la EPO actúa en la diferenciación de las células de precursor y también estimula en
pequeña medida la formación de megacariocitos. La insuficiencia aguda y crónica debida a
enfermedades degenerativas del riñón, lleva a una menor formación de EPO y por eso se produce una
anemia renal.
La función de la EPO en el organismo no es sólo la formación de nuevos eritrocitos. El receptor de la EPO
se ha encontrado en diferentes tipos de células somáticas como las neuronas, los astrocitos, células de
la microglía y las células del músculo del corazón. Se han observado interacciones entre la EPO y su
receptor en tejidos que no forman sangre, como en la quimiotaxis, angiogénesis, activación intracelular
del calcio y la inhibición de la muerte celular. Se han encontrado lugares específicos para la EPO en las
neuronas, especialmente en el hipocampo, una región del cerebro muy afectada cuando se produce
falta de oxígeno.
Cualidades estructurales
El gen de la EPO (5,4 kb, 5 exones y 4 intrones) codifica una proteína pro-EPO con 193 aminoácidos.
Mediante una modificación posterior, el terminal N se convierte en un péptido con 27 aminoácidos
residuales así como el residuo de asparagina del terminal C restante mediante la acción de la
carboxipeptidasa intracelular.
Químicamente la EPO humana es un polipéptido ácido no ramificado a partir de 165 monómeros de
aminoácido y un peso molecular de aproximadamente 34kDa. La estructura terciaria consiste de cuatro
hélices α antiparalelas de lazos vecinos. La cantidad de hidratos de carbono es aproximadamente el 40%
de la masa molecular, consistente en un O-glicosídico (Ser. 126) y tres N-glicosídicos (Asn 24, Asn 38 y
Asn 83). Las cadenas laterales, por su parte, están formadas por los monosacáridos manosa, galactosa,
fucosa, N-acetilglucosamina, N-acetilgalactosamina y N-acetilneuramianoácido. El N-glicosídico ocupa
las cadenas laterales que poseen varias ramas exteriores que se llaman “antenas”.
86

Al contrario que los aminoácidos constantes de la molécula de EPO, las estructuras de azúcares son
variables. En este sentido se habla de una microheterogeneidad de la molécula de EPO, que se presenta
no sólo en estado natural sino también en la EPO recombinante. Ésta está marcado por una parte por
secuencias variables de monosacáridos en las cadenas laterales de azúcares, y por otra parte por una
cantidad variable de N-acetilneuramínico al final; éste último, también conocido por el nombre común
de ácido siálico, es decisivo para la actividad biológica de las glicoproteínas: cuanto más alto sea el nivel
de ácido siálico más alta será la actividad y la concentración en sangre de la hormona. Hay que destacar
que la isoforma de ácido siálico muestra una afinidad más pequeña al receptor de la EPO en
experimentos in vitro. Las isoformas funcionales son desmontadas poco a poco por las células del
cuerpo que llevan el receptor de EPO. En el desmontaje, las moléiculas de EPO son descompuestas por
endocitosis inducida por el receptor en la parte interna del lisosoma. Una particularidad de la molécula
de EPO es la sulfatación en las cadenas laterales de azúcar del N-glicosídico. Hasta el momento se
desconoce la función exacta de la sulfatación, que se produce tanto en la forma nativa como en la
recombinante.
Estructura de la EPO
87

Indicaciones para la terapia
De los factores de crecimiento empleados clínicamente en la actualidad, es la EPO la que posee el mayor
espectro de indicación. La terapia clásica con EPO está diseñada para poner en marcha la formación de
glóbulos rojos sanguíneos en pacientes con anemia de tipo renal, cancerosa o como consecuencia de
quimioterapias. Además, se considera como seguro que la tasa de reacción puede incrementarse
mediante hipoxia del tumor o quimioterapia del tumor después de la aplicación de EPO.
El mecanismo patológico de una anemia tumoral se produce por una perturbación en la utilización del
hierro que puede ser reparada con la adición de EPO. Dado que estos mecanismos se muestran también
en infecciones crónicas (como la enfermedad de Crohn o la colitis ulcerosa) o sepsis, se ha utilizado la
EPO como terapia de apoyo desde hace años en estudios clínicos. Se están evaluando otras formas de
terapia con EPO, en el tratamiento del síndrome de fatiga crónica, el síndrome mielodisplásico, la
anemia aplásica, la osteomielofibrosis y las infecciones por VIH. Sus cualidades citoprotectoras en
ensayos con cultivos celulares y con animales hacen a la EPO una candidata interesante para el
tratamiento de enfermedades neurológicas agudas como, por ejemplo, los ataques de apoplejía, y
también para enfermedades neurodegenerativas. Según un estudio piloto publicado en 2006, la EPO
puede producir, posiblemente, una ligera mejora de las capacidades cognitivas como medicamento de
apoyo en el tratamiento de pacientes esquizofrénicos. Los científicos suponen que el efecto observado
podría basarse en las cualidades protectoras de la EPO frente a mecanismos neurodegenerativos; sin
embargo, los resultados no han sido confirmados hasta ahora por otros grupos de investigación. En otro
estudio se ha observado que la EPO produce efectos neuropsicológicos que mejoran el estado de ánimo
en pacientes con miedo y depresión.
EPO y dopaje
El uso de la EPO como droga de dopaje en el deporte está prohibido. El efecto "positivo" de la EPO se
debe a que aumenta la masa eritrocitaria (elevando el hematocrito), lo que permite un mejor
rendimiento del deportista en actividades aeróbicas. De esta forma se aumenta la resistencia al ejercicio
físico.
Glucocerebrosidasa
La enfermedad de Gaucher es una rara enfermedad autosómica recesiva producida por un déficit de
glucocerebrosidasa (glucosilceramidasa), enzima que interviene en la degradación lisosómica de los
88

glucolípidos. En ausencia de dicha enzima se produce una acumulación secundaria de glucocerebrósidos
insolubles (glucosilceramida) en los lisosomas de los macrófagos. La enfermedad de Gaucher es más
frecuente en la población judía, de origen Ashkenazi , aunque también se presenta en otras poblaciones
no judías. La glucocerebrosidasa está codificada por un gen (GBA) situado en 21q en el cromosoma 1.15
Es una enzima lisosomal que degrada lípidos (grasas) complejos, glucoesfingolípidos, como la
glucosilceramida.
La imiglucerasa es una forma modificada de la β-glucosidasa ácida humana producida mediante
tecnología de ADN recombinante utilizando un cultivo celular de mamífero procedente de ovario de
hámster chino (CHO), con modificación en la manosa dirigida a macrófagos. Su utiliza en el tratamiento
de la enfermedad de Gaucher16
Hormona del crecimiento
La Hormona de Crecimiento Humana se sintetiza actualmente mediante tecnología de ADN
recombinante y su secuencia es similar a la hormona de crecimiento humana. Se utiliza en niños con
baja talla idiopática o por déficit de Hormona de Crecimiento, en pacientes con Insuficiencia Renal
Crónica, Síndrome de Turner o Síndrome de Prader Willy.
Existen en la actualidad, diversos preparados comerciales de hormona de crecimiento disponibles en el
mercado. En este informe se analizó la información sobre la composición de cada una de las
presentaciones. Se realizó una búsqueda bibliográfica con la intención de determinar si existe evidencia
científica publicada sobre la superioridad de un tipo de preparado o presentación sobre otro en cuanto a
su eficacia, efectividad o seguridad.17
En el país, todas las presentaciones de hormona crecimiento son realizadas mediante tecnología de ADN
recombinante. Existen 9 marcas comerciales disponibles en un total de 14 presentaciones. Las
principales diferencias entre las distintas presentaciones están relacionadas con el modo de aplicación
(frasco ampolla, jeringas prellenadas, etc.) y no con la droga en sí. Si bien las presentaciones existentes
15 http://www.iqb.es/hematologia/monografias/gaucher/gaucher01.htm16 http://www.iqb.es/cbasicas/farma/farma04/i029.htm17 Instituto de Efectividad Clínica y Sanitaria. Análisis sobre las diferentes presentaciones disponibles deHormona de Crecimiento Humana. Documentos de Evaluación de Tecnologías Sanitarias, Informe deRespuesta Rápida N° 12, Buenos Aires, Argentina. Octubre 2003.
89

pueden diferir en ciertos aspectos técnicos como el material al que se transfiere el material genético o
las técnicas de purificación, en todos los casos se trata del mismo principio activo: hormona de
crecimiento humana elaborada mediante tecnología de ADN recombinante. No se encontró evidencia
científica que haya demostrado la superioridad de alguna forma de hormona de crecimiento sobre otra
ni de una forma de aplicación o presentación sobre otra.
La hormona del crecimiento (HC) es un polipéptido natural producido por la glándula hipofisaria bajo el
control del hipotálamo. Es una hormona esencial para el crecimiento en niños y también tiene
importantes efectos en el metabolismo de proteínas, lípidos y carbohidratos. En los niños, la HC
promueve el crecimiento, estimulando la secreción de hormonas (somatomedinas) en el hígado. En los
adultos, la HC tiene efectos anabólicos estimulando la síntesis de proteínas en el músculo y la secreción
de ácidos grasos del tejido adiposo. Inhibe la captación de glucosa por el músculo y estimula la captación
de aminoácidos.
La deficiencia de la hipófisis da lugar a una producción y secreción insuficiente de la hormona.
Esta deficiencia conduce al retraso del crecimiento. El tratamiento se basaba en el empleo de hormona
del crecimiento obtenida de glándulas hipofisarias de cadáveres humanos. Sin embargo, se interrumpió
esta práctica al ser asociada con la enfermedad de Creutzfeld-Jakob, trastorno neurológico progresivo
producido por un virus lento. Actualmente su producción se realiza mediante tecnología de ADN
recombinante y su secuencia es idéntica a la de la hormona de crecimiento humana.
Se administra en forma subcutánea 6-7 veces por semana. Los efectos adversos son raros y pueden
incluir cefaleas, nauseas y vómitos, problemas visuales, artralgias, mialgias, parestesias, hipotiroidismo y
reacciones locales en el sitio de inyección.
Todavía hay, sin embargo, incertidumbre sobre su seguridad en el largo plazo. Ha generado
preocupación la asociación encontrada entre el uso de HC y cáncer colorrectal aunque no puede saberse
si esta asociación existiría también con las dosis que se emplean actualmente. El riesgo de desarrollo de
diabetes, sobre todo en la población predispuesta, también debe ser vigilado.
Por otro lado, si bien la HC aumenta en la mayor parte de los casos la velocidad de crecimiento, no en
todas las situaciones clínicas se ha demostrado claramente su capacidad para mejorar la talla final
adulta, que es finalmente el principal objetivo de tratamiento.
90

Por esto motivos, falta de evidencia clara acerca de sus beneficios en determinadas situaciones e
incertidumbre acerca de su seguridad a largo plazo, se debe ser cauto a la hora de decidir su indicación
Se ha postulado su uso en los siguientes escenarios clínicos:
Niños con baja estatura idiopática
Niños con déficit de hormona de crecimiento
Pacientes con insuficiencia renal crónica
Pacientes con síndrome de Turner
Pacientes con síndrome de Prader-Willi
Insulina
La hormona insulina, involucrada en la regulación y metabolismo de la glucosa, fue hasta la década de
los 80 obtenida de páncreas de cerdo o bovino. Fue a partir de 1982, cuando se introdujo en el mercado
insulina humana producida por ingeniería genética, empleando como organismo productor la bacteria
Escherichia coli. Actualmente se emplean además de E. coli otros microorganismos, tales como algunos
tipos de levaduras.
Es de destacar que la insulina humana obtenida por ingeniería genética fue la primera molécula
producida con fines terapéuticos que se introdujo en el mercado. Desde su introducción en el mercado,
ha sustituido paulatinamente a la insulina obtenida de páncreas de cerdos y bovinos. En la década de los
90 la “insulina humana recombinante” representaba el 70% de la insulina disponible en el mercado,
siendo actualmente el 93% de la insulina producida.18
La producción de “insulina recombinante” posee las ventajas de tener protocolos de producción más
sencillos, con altos rendimientos de producto, por lo tanto, a medida que los mismos son introducidos
en el mercado, dan como resultado una sensible reducción en los costos de los mismos.
Las nuevas técnicas de ingeniería genética empleando plantas o animales transgénicos, permitirían aún
mayores rendimientos de producción, sin necesidad de contar con grandes reactores de cultivo en el
laboratorio. Estas nuevas tecnologías aplicadas a la producción en gran escala de insulina actualmente
están en distintas fases de desarrollo y restan realizar en algunos casos pruebas de seguridad y eficacia
del producto; pero de ser los resultados positivos, se estima que en poco tiempo, quizás en unos 3 años,
18 http://www.bioero.com/index.php?tag=insulina-recombinante
91

puedan estar disponibles para el tratamiento de pacientes con diabetes 1 o diabetes tipo 2 con
requerimientos de insulina.
La producción de la molécula de insulina ha sido pionera en el advenimiento de la “era de la ingeniería
genética” , y es sin lugar a dudas una de las moléculas de gran importancia en el mundo de las moléculas
de uso terapéutico; ya que el número de personas, en especial de los países desarrollados que padecen
diabetes es importante, considerando la enfermedad como una pandemia.
Existen en el mundo cerca de 200 millones de personas que padecen diabetes y las proyecciones
epidemiológicas dicen que esta cifra tenderá a duplicarse en los próximos quince años. La producción de
insulina tiene una importante demanda en el mercado mundial alcanzando los 5000 millones de dólares.
Si tomamos el caso de Argentina, existen poco más de un millón y medio de pacientes diabéticos; con
una demanda de insulina de 50 millones de dólares anuales. El tratamiento genera un gran impacto
económico en los pacientes y en los sistemas de salud por tratarse de una terapia crónica con un
producto de considerable valor.
Por tal motivo la posibilidad de producir insulina humana a través de bovinos transgénicos,
obteniéndose a partir de leche vacuna, significará un cambio radical; ya que con sólo 25 vacas se
obtendría los 200 kilos de insulina humana que se necesitan por año en la Argentina, y que actualmente
se importan. En tal sentido, la empresa Bio Sidus ha apostado, al producir insulina en bovinos
transgénicos, llegar a la comunidad con este nuevo desarrollo tendiente a disminuir los costos de
producción, que según los expertos sería del 30 – 40%; conjuntamente con las ventajas que conlleva la
independencia de productores extranjeros. Es de elogiar este nuevo avance de Bio Sidus, pues es la
primera insulina humana recombinante en el mundo obtenida por bovinos transgénicos. Sólo hay 2
multinacionales que producen insulina humana empleando cabras transgénicas, purificándose la misma
a través de la leche; y una empresa canadiense que obtiene insulina humana en plantas. Si bien estos
desarrollos no están aún en el mercado, se estima que en poco tiempo estarán disponibles.
Es de destacar que este desarrollo científico-tecnológico posiciona a la empresa Bio Sidus, y a la
Argentina a un nivel privilegiado en lo que respecta a nivel biotecnológico.
Más allá de la inversión monetaria; que sin lugar a dudas fue muy importante, ya que se invirtieron 4
millones de dólares para la realización de este proyecto; así como el trabajo conjunto de más de 50
científicos; lo que se transluce es el sentido de visión estratégica, el alto compromiso, dedicación y
92

profesionalidad que se ha apostado en el área biotecnológica en Argentina. En países como Argentina y
Brasil, esta área ha adquirido un importante desarrollo, contando con carreras universitarias de grado y
postgrados, con maestrías y doctorados específicos en el área, y con polos tecnológicos altamente
desarrollados, con una fuerte y seria interrelación industria-academia. El hecho que la Argentina
mediante la empresa Bio Sidus haya logrado este importante éxito, sumado a los que ya están puestos
en marcha, así como otros emprendimientos vinculados a la producción de insulina humana mediante
ingeniería genética, por distintos laboratorios argentinos, nos demuestran de la pujanza, dinamismo,
capacidad emprendedora y compromiso que tiene este país en lo que a biotecnología respecta.
Interferon alfa
ACCIÓN TERAPÉUTICA: Antineoplásico, modulador de la respuesta biológica (inmunomodulador).
INDICACIONES
Leucemia, célula vellosa (tratamiento): Interferón alfa-2b recombinante está indicado para tratamientos
de leucemia de célula vellosa en pacientes esplenectomizados y no esplenectomizados.
Condiloma acuminado (tratamiento): Interferón alfa-2b está indicado para inyección intralesional para
tratamientos de condiloma acuminado refractarioso externo recurrente (verrugas genitales).
Hepatitis, crónica, activa (tratamiento): Interferón alfa-2b está indicado para tratamientos de hepatitis
no-A, no-B/C en pacientes de 18 años de edad o mayores con enfermedades hepáticas compensadas
que posean historia de exposición de sangre o productos de la sangre y/o que tengan anticuerpos
positivos.
Hepatitis B crónica (tratamiento): Interferón alfa-2b está indicado para tratamiento de hepatitis B
crónica en pacientes de 18 años de edad o mayores con enfermedad hepática compensada y replicación
del virus de hepatitis B (HBV). Se realizará a los pacientes exámenes positivos para el antígeno sérico de
la hepatitis B por lo menos durante 6 meses y deberán tener una replicación del HBV con
aminotransferasa alanina sérica elevada.
Sarcoma de Kaposi, relacionado con el SIDA (tratamiento): Interferón alfa-2b recombinante indicado
para el tratamiento de sarcoma de Kaposi relacionado con el SIDA en pacientes seleccionados de 18
años de edad o mayores.
93

Carcinoma, renal (tratamiento): Interferón alfa-2b recombinante, indicado para el tratamiento del
carcinoma renal y la leucemia mielocítica crónica.
Papilomatosis, de laringe (tratamiento): Interferón alfa-2b recombinante, indicado para tratamiento de
papillomatosis de laringe, incluyendo papilloma de laringe juvenil.
Micosis fungoides (tratamiento): Interferón alfa-2b recombinante, especialmente pequeños linfomas
foliculares de célula partida (tipos nodulares deficientemente diferenciados).
Carcinoma, ovárico, epitelial (tratamiento) o carcinoma, piel (tratamiento): Interferón alfa-2b
recombinante.
Policitemia vera (tratamiento): Interferón alfa-2b recombinante como tratamiento médico razonable en
algún punto en el manejo de la policitemia vera.
A pesar de que la eficacia de todos los interferones alfa parece ser similar para varias indicaciones,
pueden existir diferencias en la eficacia relativa para una indicación particular.
Se ha estudiado el interferón alfa-2b recombinante, en tratamiento en combinación, para uso en el
tratamiento de carcinoma cervical. Algunos expertos médicos consideran que este agente es un
tratamiento médico razonable en algún punto del manejo del carcinoma cervical avanzado (a pesar de
que no es recomendado como un tratamiento de primera-línea).
Se ha demostrado en diferentes estudios que el interferón alfa-2b recombinante, en tratamiento en
combinación, es inefectivo en el tratamiento del carcinoma colorrectal.
CONTRAINDICACIONES:
Las consideraciones/contraindicaciones médicas relacionadas han sido seleccionadas sobre la base
desde su potencial importancia clínica.
• Historia de enfermedad autoimmune (se recomienda tener precaución debido a que el interferón alfa
puede aumentar la actividad del sistema inmunológico.
• Depresión de médula ósea.
» Enfermedad cardíaca, severa, incluyendo infarto de miocardio reciente o
» Diabetes melitus tendiente a cetoacidosis o
94

» Alteraciones isquémicas
» Enfermedades pulmonares (pueden verse agravadas como resultado de la tensión de la fiebre y los
escalofríos que se presentan en la mayoría de los pacientes que reciben interferón alfa).
(El riesgo de cardiotoxicidad del interferón alfa puede aumentar en pacientes con una historia de
enfermedad cardíaca; muy rara vez se ha informado sobre infarto de miocardio).
» Varicela, existente o reciente, incluyendo exposición reciente o
» Herpes zóster (riesgo de enfermedad severa generalizada).
» Función CNS, comprometida o
» Historia de condición psiquiátrica severa, o
» Trastornos de ataques.
• Enfermedad hepática, severa.
» Trastornos infecciosos.
• Enfermedad renal, severa.
» Sensibilidad al interferón alfa.
» Disfunción de la tiroides (se ha informado que el interferón alfa-2b recombinante causa anomalías en
la función de la tiroides.
• Trastornos de coagulación (se recomienda tomar precauciones; el interferón alfa-2b recombinante
puede prolongar el PT y el PTT).19
La infección con el virus de la hepatitis C puede causar enfermedad hepática crónica para la cual no hay
tratamiento efectivo está disponible. Se estudiaron los efectos del interferón alfa recombinante humano
en un estudio prospectivo, aleatorizado, doble ciego, controlado con placebo en pacientes con bien
documentada crónica de la hepatitis C. Cuarenta y un pacientes fueron reclutados en el ensayo, 37 de
los cuales fueron encontrados más tarde a tiene anticuerpos contra virus de la hepatitis C. Veinte y un
pacientes recibieron interferón alfa (2 millones de unidades) por vía subcutánea tres veces por semana
19 http://www.minsa.gob.pe/portalbiblioteca2/biblio/plm/PLM/productos/52823.htm
95

durante seis meses, y 20 recibieron placebo. Los niveles de aminotransferasa sérica media y las
características histológicas del hígado mejoraron significativamente en los pacientes tratados con
interferón, pero no en los pacientes que recibieron placebo. Diez pacientes tratados con interferón (48
por ciento) presentaron una respuesta completa, definida como un descenso en la media de los niveles
de aminotransferasa sérica a niveles normales durante el tratamiento, otros tres tuvieron una
disminución en los niveles de aminotransferasa media de más del 50 por ciento. Después de finalizado el
tratamiento, sin embargo, el suero aminotransferasas por lo general volvió a los niveles pre-
tratamiento, 6 a 12 meses después de la interrupción del tratamiento con interferón, sólo dos pacientes
(10 por ciento) todavía tenía los valores normales.20
Interferon gamma – 1b
El interferon gamma ha sido utilizado con resultados alentadores en ensayos clinicos para el tratamiento
de la Artritis Reumatoide y de la Artritis Reumatoide Juvenil, por lo que se emplea en este trabajo. El
hecho de haber obtenido mejoria en pacientes resistentes a otros tratamientos y ser posible la
suspension de los esteroides, hace que los resultados sean muy alentadores y justifican un ensayo
donde se emplee el IFN gamma como primer inductor de remision.
Interleucina 2, 3, 4
La interleucina-2 es una glicoproteína humana de 133 aminoácidos que resulta vital para obtener una
respuesta inmunológica efectiva. En el Centro de Ingeniería Genética y Biotecnología ha podido
obtenerse por vía recombinante a partir de una cepa de Escherichia coli. Para el control de la calidad de
cada lote producido se hizo necesaria la elaboración de un material de referencia de trabajo. En esta
publicación se describen los pasos seguidos en la obtención del lote candidato y la preparación del
material de referencia como tal. Se demostró que el patrón de trabajo preparado es homogéneo para el
uso en las técnicas analíticas de control de calidad de la interleucina-2 y se predijo una pérdida de
actividad inferior al 5 % anual mediante un estudio de estabilidad acelerada. Se obtuvieron valores
adecuados para la pureza del material de referencia, evaluada por electroforesis y para la actividad
biológica, que se estableció con trazabilidad al material de referencia internacional para este producto.21
20 http://www.nejm.org/doi/full/10.1056/NEJM19891130321220421 Maribel Vega, Kelly Gorrín, Gerardo García, Haydeé Gerónimo, Galina Moya, Marisel Quintana y Mirta Castiñeira. Establecimiento de un material de referencia de trabajo para interleucina-2 recombinante
96

Las proteínas denominadas citoquinas, dentro de las cuales se encuentran las interleucinas, están
involucradas en la patogénesis de muchas enfermedades y pueden también ser utilizadas en su
tratamiento. La primera interleucina descubierta y caracterizada fue la interleucina-2 (IL-2),
glicoproteína de 133 aminoácidos, con masa molecular entre 13 y 17,5 kDa y puntos isoeléctricos entre
6,6 y 8,2; se conoce que resulta vital para la generación de una respuesta inmunológica efectiva. En
1985 se reportó que la IL-2 desempeñaba una función importante en el tratamiento de tumores sólidos,
con lo cual se amplió su utilización en la clínica. También puede ser empleada para aumentar la
respuesta inmunológica, reparar las deficiencias celulares y/o humorales, en pacientes con
enfermedades infecciosas u otros trastornos inmunológicos. Esta proteína se ha obtenido por técnicas
de ingeniería genética, aunque la IL-2 recombinante, a diferencia de la proteína humana natural, no es
glicosilada; pero esta diferencia no tiene un efecto significativo sobre las funciones biológicas de la
proteína.
Activador tisular del plasminógeno
Es una proteína proteolítica implicada en la disolución de coágulos de sangre. Específicamente, es una
serina proteasa que se encuentra en las células endoteliales, las células que recubren el interior de los
vasos sanguíneos. Como una enzima, cataliza la conversión de plasminógeno a plasmina, que es la
enzima principal para la disolución de coágulos de sangre. El t-PA es empleado en medicina para el
tratamiento del ictus o isquemia cerebral provocado por un coágulo de sangre. El t-PA es segregado por
el endotelio vascular después de sufrir una lesión y su función es activar el plasminógeno
transformandolo en plasmina.22
Activadores del plasminógeno tisular recombinante (r-TPA) incluyen alteplasa, reteplasa y tenecteplasa
(TNKase).
La alteplasa es aprobado por la FDA para el tratamiento del infarto de miocardio con elevación del ST
(STEMI), accidente cerebrovascular isquémico agudo (AIS), embolia pulmonar aguda masiva, y el centro
de dispositivos de acceso venoso (CVAD).
22 Rijken DC (1988). «Relationships between structure and function of tissue-type plasminogen activator». Klin. Wochenschr. 66 Suppl 12: pp. 33–9
97

Reteplasa es aprobado por la FDA para el infarto agudo de miocardio, donde cuenta con una
administración más conveniente y más rápido que con alteplasa trombólisis.
La tenecteplasa también está indicada en el infarto agudo de miocardio, mostrando menos
complicaciones hemorrágicas, pero las tasas de mortalidad similares de alguna manera después de un
año en comparación con alteplasa. R-TPA adicionales, tales como desmoteplase, están en fase de
desarrollo clínico.
Anticuerpos para inmunoterapia celular
En los últimos años se observó que los anticuerpos (Ac) monoclonales han crecido en importancia como
drogas potenciales para el tratamiento contra el cáncer. Esto se basa en los efectos de la terapia con Ac
nativos que van desde el aumento de los mecanismos efectores inmunes, tales como la citotoxicidad
dependiente del complemento (CDC) y la citotoxicidad celular dependiente de anticuerpos (CCDA),
hasta el efecto antiproliferativo y la inducción de la apoptosis inducida por el bloqueo de la interacción
receptor/ligando. Por lo tanto, para llevar a cabo dichas acciones, el Ac debe unirse a los receptores de
la fracción Fc de la inmunoglobulina (Ig).
Por un lado, la CCDA depende de los monocitos/macrófagos y de las células natural killer (NK). Además,
los autores han demostrado que existe un Ac IgG humano anti-antígeno carcinoembrionario (CEA) que
parece presentar una importante CCDA contra las células tumorales que expresan dicho antígeno.23
Se ha demostrado que la fracción FcalfaRI representa el receptor Fc de neutrófilos más potente para
desencadenar la CCDA y que los Ac IgA humanos han podido provocar este tipo de respuesta contra
células tumorales de linfoma y tumores sólidos.
Los autores sostienen que en el presente estudio han formulado un Ac recombinante humano de tipo
IgA anti CEA, y evaluaron su eficacia para mediar CCDA con los neutrófilos como células efectoras contra
células que expresen CEA.
Anticuerpos monoclonales
23 http://www.bago.com/bago/bagoarg/biblio/oncoweb286.htm. Zhao J, Kuroki M, Kuroki M y colaboradores, Recombinant Human Monoclonal IgA Antibody Against CEA to Recruit Neutrophils to CEA-Expressing Cells. Oncology Research 17(5):217-222, 2008
98

Los autores comentan que el Ac humano se obtuvo a partir de la infección de las larvas de gusano de
seda con el virus que contenía la secuencia genética del Ac. Luego de que se recolectara la hemolinfa de
las larvas, se le agregó una sustancia que provocó la precipitación de la proteína (Ac) recombinante.
Posteriormente se procedió al proceso de purificación y, de esta forma, se separó el Ac IgA
recombinante humano.
En este trabajo, se evaluó la capacidad del Ac IgA recombinante de inducir una CCDA, para lo cual se
utilizaron neutrófilos contra células que expresaban CEA. Con el objetivo de potenciar esta respuesta,
los autores trataron a los neutrófilos con interferón gamma, que se sabe aumenta la expresión del
receptor del Ac IgA en esta estirpe celular. Se observó que, ante la presencia de la IgA recombinante
humana, los neutrófilos activados ejercieron una respuesta citotóxica contra las células COH que
expresaban CEA; por lo tanto, se asume que los neutrófilos mataron a dichas células ya que
reconocieron la porción Fc del Ac recombinante que se había unido previamente a las células que
expresaban el antígeno.
Vacunas (en mascotas)
El avance en el estudio de las vacunas, ha ofrecido las herramientas necesarias para la prevención de las
enfermedades infecciosas.24
El uso de vacunas muertas o vivas atenuadas en el siglo 20 ha resultado en espectaculares avances en el
control y erradicación de algunas enfermedades mortales para humanos y los animales. Sin embargo, en
el caso de las primeras si bien eran seguras no daban protección prolongada; mientras que las segundas
podían llegar a tener algunos problemas post - vacunales, principalmente en aquellos animales
parasitados, afectados por algunas enfermedades preexistentes, desnutridos, o incluso estresados.
Gracias al constante trabajo de los científicos se pudieron lograr que desaparezcan las desventajas
propias de las vacunas convencionales como las nombradas anteriormente.
Hoy y después de más de 20 años de investigación, estamos en presencia de una nueva era de la
medicina veterinaria preventiva, a través de la ingeniería genética por medio de la Tecnología
24 http://www.foyel.com/cartillas/5/vacunas_recombinantes.html
99

Recombinante. Esta agrega armas nuevas, más seguras y efectivas para la prevención de las
enfermedades infecciosas en los animales de compañía.
Que es un Recombinante
Puede ser cualquier microorganismo, entre ellos una bacteria, un virus, o una levadura en el cual su
información interna, lo que nosotros llamamos material genético, ha sido modificado en forma artificial
o recombinado.
Porqué debemos utilizar un Recombinante
Porque es 100 % seguro, porque no contiene el virus completo ( de ésta forma se elimina el riesgo de
otras complicaciones post - vacunales), sino sólo las partes necesarias para proteger al animal.
Las vacunas Recombinantes se utilizan en otras partes del mundo. En el mercado hay varias vacunas
disponibles; incluso en Estados Unidos y Europa se utiliza una vacuna contra la Rabia vía oral que se
administra mediante cebos los que se distribuyen por medio de helicópteros en las cuevas de los
animales salvajes. Gracias a éste tipo de tecnología segura y efectiva se está eliminando el gran
problema que existe de rabia en esos países.
Hay otros tipos de vacunas Recombinantes para otras especies y en el caso de animales de compañía,
podemos mencionar la de Rabia para gatos , la de Moquillo canino; gracias a éstas se han evitado todos
los problemas post - vacunales que solían ocasionar las tradicionalmente utilizadas hasta ahora.
En el caso del virus del Moquillo, los científicos pudieron estudiar su información interna y determinar
que parte del virus era la responsable de dar protección contra esa enfermedad. Luego que pudieron
determinar esto, por medio de lo que llamamos estudio de su código genético, se extrajeron esas partes
específicas que llevan la información y que más tarde van a dar la respuesta protectora en el animal.
Buscaron un virus que sea totalmente inocuo para el animal, y para todas las especies, incluso en
medicina humana se está utilizando éste virus para la fabricación de la vacuna del H.I.V. (Sida)
justamente por ser totalmente seguro, otras de las características de éste virus es ser lo suficientemente
grande para permitir incorporar más información, el virus que se utiliza es el de la viruela del canario.
Por esta razón lleva el nombre de Recombinante, porque se utiliza éste virus que cuando ingresa al
animal por medio de la vacunación ayuda a expresar esa respuesta protectora contra la enfermedad del
100

Moquillo. Por lo tanto, tenemos protegido al animal sin tener que suministrarle todo el virus entero,
sino la parte específica que estimula el sistema de defensa del animal.
Cuando le damos éste tipo de vacunas Recombinantes el sistema de defensa del animal no se distrae
con un montón de respuestas, sino únicamente con la específica de protección. Con esto nos evitamos
cualquier problema postvacunal asociado a la administración de todo el virus.
Por suerte hoy ésta tecnología está también disponible en la Argentina , al alcance de todos y es lo que
vamos a utilizar de ahora en más.
Para resumir podemos decir que: las vacunas Recombinantes usan sólo las partes específicas del virus
que necesita el animal para quedar protegido y se eliminan todas las partes patógenas o sea toda
aquello que pueda causar algún daño al animal incluso llegar a producirle la enfermedad.
Por lo tanto para mantener a nuestras mascotas con buen estado de salud, es muy importante que se
vacune con el mejor producto disponible, la Tecnología Recombinante nos da esa posibilidad, debido a
su Excelente Eficacia y su Seguridad sin igual.
101

Células madre
Las células madre son células que tienen la capacidad de dividirse indefinidamente y que pueden dar
origen a células más especializadas. Cuando se dividen, las células madre producen un tipo de célula
más especializada y además generan más células madre.25
Concepto de célula madre. A. Vista generalizada de la cascada a partir de la célula madre
pluripotente pasando por la célula madre comprometida, luego por la célula progenitora,
hasta la célula diferenciada. Cuando una célula madre se divide forma una célula más
comprometida y otra célula madre. B. Ejemplo de formación de células sanguíneas basado
en el esquema superior. (Según National Institutes of Health 2001.)
Algunas células madre individuales son capaces de generar todas las estructuras del embrión. Estas
células, denominadas células madre pluripotentes (que pueden formar al embrión, pero no a los tejidos
extraembrionarios), pueden generar ectodermo, mesodermo y células germinales.
Así como dan origen a más células madre pluripotentes, también generan células madre
comprometidas. Estas células madre comprometidas pueden dar origen a una pequeña población de
25 Scott Gilbert. Biología del desarrollo. Ed. Panamericana, 2005. Págs. 73 – 74
102

células. Por ejemplo, un tipo de célula madre comprometida (hemangioblasto) da origen a todos los
vasos sanguíneos, células sanguíneas y linfocitos. Otro tipo de célula madre comprometida, la célula
madre mesenquimática, puede dar origen a la totalidad de los diferentes tejidos conectivos (cartílago,
músculo, adiposo, etc.)
Las células madre comprometidas pueden originar a células madre comprometidas más
específicamente, como las células madre que generan sólo células sanguíneas y linfocitos, mas no vasos
sanguíneos, o éstas pueden generar lo que se conoce como células progenitoras.
Las células progenitoras, también denominadas células precursoras, no son más células madre debido a
que sus divisiones no crean otra célula progenitora similar. En su lugar, la célula progenitora se divide
para formar uno o pocos tipos celulares relacionados, dependiendo del ambiente celular en que se
encuentre. Por ejemplo, hay un precursor celular sanguíneo (la célula progenitora mieloide) que puede
generar todos los tipos diferentes de células sanguíneas. Generalmente, la célula progenitora muestra
alguna evidencia de diferenciación, pero el proceso no se completa hasta que se ha formado la célula
diferenciada.
La restricción del potencial de la célula madre es gradual, y los potenciales de estas células están
determinados por su entorno. Sin embargo, generalmente una vez comprometidas no cambian su
compromiso. Cuando se colocan en un nuevo ambiente, no cambiarán el tipo de células que generan.
Las células madre son críticas para mantener poblaciones celulares que sobreviven períodos
prolongados y que deben ser renovadas. Debido a esto, son importantes para nuestra población
continua de células de la sangre, del pelo, de la epidermis y del epitelio intestinal.
Células madre musculares y medicina regeneradora del aparato locomotor
Investigadores del Instituto de Tecnología Biomédica de la Universidad de Twente en Enschede (Países
Bajos) han desarrollado una nueva técnica de ingeniería molecular que podría permitir utilizar células
madre humanas para reconstruir huesos en pacientes con huesos dañados o desaparecidos. Los
resultados de su trabajo se publican esta semana en la edición digital de la revista 'Proceedings of the
National Academy of Sciences' (PNAS).26
26 http://www.granadadigital.com/gd/amplia.php?id=87074&parte=Sociedad
103

Hasta el momento no ha sido posible utilizar las aplicaciones con células madre mesenquimales (CMM)
en pacientes humanos, pero los investigadores sugieren que han descubierto cómo conseguir producir
cantidades suficientes de tejido óseo a partir de estas células madre.
Los investigadores, dirigidos por Ramakrishnaiah Siddappa, informan de que la activación de la proteína
quinasa A, o PKA, en un modelo de ratón desencadena la capacidad de formar hueso que poseen las
células madre mesenquimales. Añaden que la activación de PKA induce una secreción continua de
factores de crecimiento asociados a los huesos, que conduce a una formación ósea controlada y
sustancial.
Los científicos utilizaron células madre mesenquimales derivadas de más de una docena de pacientes
ortopédicos de edades comprendidas entre los 23 y los 82 años y muestran que al tratarlas estas células
produjeron la matriz necesaria para dar lugar a la formación de hueso. Al estar generadas a partir de las
propias células del paciente las convierten en células personalizadas para cada individuo, añaden los
autores.
Los investigadores sugieren, además, que su método podría facilitar la introducción de tales técnicas de
ingeniería de tejidos óseos en aplicaciones clínicas.
Curación de lesiones musculares con células madre
El uso de células madre para reparar tejido muscular está en fase de investigación, pero en el momento
en el que se llegue a concretar la forma adecuada de hacerlo en humanos será una alternativa para
diversas enfermedades, entre ellas la distrofia muscular.27
Por otra parte, cabe pensar que mas de un laboratorio buscará la forma de extender su uso para
promover el crecimiento de tejido muscular, tal como hoy dia lo hacen los esteroides, con lo que
podrían ser utilizados por deportistas y atletas de diversas especialidades en su afán de ser cada vez
mejores o superiores.
Investigadores de la Universidad de Colorado en Boulder Estados Unidos, han identificado un tipo de
célula madre del músculo esqueletal que contribuye a la reparación de los músculos dañados de los
ratones.
27 http://www.puntofape.com/curacion-de-lesiones-musculares-con-celulas-madre-1403/
104

El descubrimiento, que se publica en la revista Cell Stem Cell, podría tener implicaciones en el
tratamiento de los músculos humanos dañados, enfermos o envejecidos.
Estas células madre se encuentran en poblaciones de células satélite localizadas entre las fibras
musculares y en los alrededores del tejido conectivo responsable de la reparación y mantenimiento de
los músculos esqueletales.
Cuando las fibras musculares están estresadas o traumatizadas, las células satélite se dividen para hacer
más células musculares especializadas y reparar el músculo. Las células madre identificadas entre las
células satélite, denominadas células satélite-SP, renovaban la población de células satélite después de
una inyección en las células musculares dañadas, lo que contribuía a la recuperación del tejido muscular
en los ratones de laboratorio.
Según Bradley Olwin, coautor del estudio, “esta investigación muestra cómo las células satélite
mantienen sus poblaciones en los tejidos dañados. La esperanza es que este nuevo método nos permita
reparar el tejido muscular esqueletal dañado o enfermo”.
Las células madre se distinguen por su habilidad para renovarse a si mismas mediante la división y
diferenciación celular en tipos de células especializadas. En el tejido muscular esqueletal sano, se
mantiene de forma constante la población de células satélite, lo que llega a los autores a pensar que
alguna de estas poblaciones de células satélite incluían células madre.
Los investigadores inyectaron 2.500 células satélite-SP en una población de células satélite del tejido
muscular de un ratón herido. Descubrieron que el 75 por ciento de las células satélite que se
reproducían derivaban de aquellas inyectadas en el tejido. Los resultados demostraron que las células
inyectadas renovaban la cantidad de células satélite.
“El punto clave es si sólo reparamos el tejido. Inyectamos una población autorenovadora permanente
de células madre. Una ventaja de utilizar esta tecnología es que podemos utilizar un número pequeño
de células madre y realizar el trabajo con un número pequeño de inyecciones, en este caso sólo una”,
explica Olwin.
La investigación tiene implicaciones para una variedad de enfermedades humanas. En la distrofia
muscular, la pérdida de una proteína llamada distrofina produce que el músculo se desgaste, un proceso
que no se puede reparar sin un tratamiento celular. Aunque las células inyectadas reparen las fibras
musculares, su mantenimiento requiere inyecciones celulares adicionales.
105

Células madre para el infarto al miocardio
VALLADOLID.- Un hombre de 65 años ha sido el primer paciente del mundo al que se le ha implantado,
directamente en el corazón, quince inyecciones de células mesenquimales a través de un catéter que,
conectado a una máquina llamada NOGA, es capaz de identificar aquellas zonas enfermas donde hay
que colocar las células que, previamente, fueron extraídas de su propia médula ósea.28
Esta intervención pionera la realizaron los doctores Francisco Fernández Avilés, Ricardo Sanz y Javier
López, el 21 de febrero de este año. El enfermo, que había sufrido varios infartos crónicos y contaba con
zonas del corazón absolutamente muertas, experimentó una mejoría sensible.
Hasta el momento, tan novedosa terapia ha sido aplicada en el Servicio de Cardiología del Hospital
Clínico de Valladolid a dos pacientes, con resultados positivos. Su máximo responsable, el doctor José
Alberto San Román, se muestra muy satisfecho con los efectos, "clínicamente ha evolucionado muy
bien, pero con un número tan pequeño de pruebas, nada se puede decir, hay que continuar la
investigación".
El objetivo de este proyecto de terapia celular es conseguir curar a pacientes a los que es imposible
aplicar ninguna otra acción terapéutica, porque tienen el corazón muy estropeado, con las arterias
coronarias "hechas un desastre", por lo que ni tan siquiera se las puede abrir con un catéter y, por
supuesto, evitar el trasplante cardiaco, única solución a la que están abocados alguna de estas personas.
El doctor San Román es un convencido de que la terapia celular "va a ayudar a que cada vez sea menos
necesario el trasplante cardiaco". Según asegura, "sería fantástico, porque el trasplante necesita una
cantidad de recursos humanos y económicos brutales".
Ahora, concluida la primera fase, aún "quedan al menos dos años hasta obtener resultados definitivos",
comenta el también director del Instituto de Ciencias del Corazón. Pero, por lo obtenido hasta ahora, las
conclusiones que ofrecen son prometedoras porque se ha "comprobado una mejoría en la función
ventricular, es decir, en la función de bomba del corazón".
El Hospital Clínico de Valladolid adquirió hace dos meses el sistema de navegación cardiaca NOGA, que
es la tecnología más avanzada disponible en la actualidad en el mercado, y sirve para crear imágenes
altamente precisas en tres dimensiones del corazón. Costó unos 250.000 euros y, para su financiación,
contó con ayudas, tanto del Instituto Carlos III, como de la Consejería de Sanidad.28 http://www.elmundo.es/elmundo/2008/05/18/castillayleon/1211135370.html
106

Hasta el momento, sólo existen tres máquinas de estas características en España. El doctor López
aprendió su funcionamiento durante una estancia de formación en Estados Unidos y es el encargado del
estudio, el sistema NOGA "proporciona un mapa claro, que ayuda a identificar los tejidos enfermos",
apunta este joven cardiólogo. "Es un sistema de análisis muy sofisticado, que ofrece dos mapas del
corazón. En uno puedes ver las zonas que generan electricidad, es decir, que tienen vida, y la partes que
ya no la producen", aclara el doctor.
Corazón bioartificial
Cómo se desarrolla un ser vivo, ya sea un ratón de laboratorio o una persona, a partir de una sola célula
es uno de los grandes misterios de la biología. A pesar de los avances de la genómica y de la
investigación con células madres, los biólogos están muy lejos de entenderlo. Ni siquiera saben cómo
hacer un solo órgano. Las células madre pueden ayudar a regenerar un tejido, pero fabricar en el
laboratorio un órgano completo, pongamos un corazón, es hoy por hoy ciencia-ficción. Si al menos se
tuviera un molde, un andamiaje para que las células madre prendieran y por sí solas se pusieran manos
a la obra de crear un músculo cardiaco que empezara a latir y a bombear.29
Ésta es la ingeniosa idea que han puesto en práctica con éxito científicos de la Universidad de Minnesota
en EE UU y que ayer anunciaron en la edición digital de revista Nature Medicine. El logro ha sido un
primer corazón bioartificial de ratón desarrollado a partir del esqueleto fibroso de un corazón de
cadáver de ratón al que se le inyectaron células madre cardiacas. Y significa un paso adelante hacia los
futuros bancos de órganos bioartificiales para trasplante.
"El objetivo sería poder desarrollar vasos sanguíneos u órganos completos, listos para trasplante, que se
generarían a partir de las células del propio paciente", explica Doris Taylor, autora principal del trabajo y
directora del Centro de Reparación Cardiaca de la Universidad de Minnesota. Hoy, los trasplantes de
corazón de donate sólo alcanzan para el 5% de los pacientes que los necesitan. En España, la mitad de
los enfermos seleccionados para trasplante cardiaco mueren mientras lo esperan.
El logro del grupo de Minnesota es todavía experimental y muy inicial, pero "demuestra que la terapia
celular es una apuesta firme", destaca Francisco Fernández-Avilés, jefe de servicio de Cardiología del
hospital Gregorio Marañón de Madrid, que ya conocía los trabajos de Taylor. Ahora hay que
perfeccionar el modelo en animales más próximos (cerdos) antes de probarlo en humanos. Lo primero
29 http://www.elpais.com/articulo/salud/Creado/corazon/bioartificial/celulas/madre/elpepusocsal/20080115elpepisal_5/Tes?print=1
107

podría demorarse de tres a cinco años, y lo segundo, una década y media, aventura Fernández-Avilés,
que es coordinador nacional de la Red Temática de Investigación Cooperativa en Enfermedades
Cardiovasculares.
"Lo importante de este trabajo es que representa un salto conceptual, que permite seguir una línea de
trabajo y ver el final del túnel, aunque sea lejano", destaca Fernández-Avilés, que se reunirá mañana con
Taylor en Nueva York.
La clave del avance ha sido lo que han llamado "descelularización de órgano completo", un
procedimiento que consiste en eliminar todas las células del corazón del animal muerto dejando intacta
la matriz extracelular. Tras obtener corazones descelularizados de ratón, los investigadores les
inyectaron células madre obtenidas de los corazones de ratas recién nacidas.
El desarrollo de los acontecimientos fue espectacular: a los cuatro días, el esqueleto fibroso sembrado
de células madre empezó a contraerse. "Al ver las primeras contracciones nos quedamos sin palabras",
recuerda uno de los investigadores, Harald C. Ott, que trabaja en el hospital General de Massachusetts
en Boston (EE UU). A los ocho días, el corazón empezó a bombear y podía considerarse un esbozo de
órgano bioartificial.
El grupo de Doris Taylor es uno de los seis equipos que han confirmado que el corazón puede
regenerarse, un hito logrado por el investigador italiano afincado en EE UU Piero Anversa en 2001. Esto
contradecía el dogma científico vigente y resultó polémico. Taylor no sólo ha confirmado que el corazón
tiene células madre cardiacas residentes, capaces de dividirse y producir tejido muscular, vascular o de
conducción, sino que ha sido capaz de aislarlas, de cultivarlas en el laboratorio y de regenerar con ellas
todo un órgano funcional. Este modelo conceptual y experimental también podría ser válido con otros
órganos.
Células madre y sordera
108

Los investigadores lograron recrear en un laboratorio las células de las microscópicas vellosidades que
recogen las ondas de sonido en el oído interno y las transmiten al cerebro humano.30
Según el equipo de la Universidad de Sheffield, Inglaterra, este avance podría eventualmente ayudar a
quienes han perdido esa células, conocidas como ciliadas, por el daño causado por sonidos fuertes o a
las personas que nacieron con problemas de audición hereditarios.
Sin embargo, todavía faltan más investigaciones para que la nueva cura esté disponible, dicen los
científicos en la revista Stem Cell .
"El problema con la sordera es que las células ciliadas sólo se producen durante las etapas de
desarrollo", explicó a la BBC el doctor Marcello Rivolta, quien dirigió el estudio.
"Si estas células se pierden durante la adultez, es imposible regenerarlas. Así que lo que hicimos fue
volver a esas etapas iniciales de desarrollo en el embrión para identificarlas, aislarlas y multiplicarlas en
el laboratorio", dijo el científico.
Los expertos afirmaron que hasta el momento los resultados han sido exitosos y que el siguiente paso
será comprobar con animales si las células creadas logran recuperar las funciones auditivas.
Daño irreversible
Hasta ahora, el daño a las ciliadas es irreversible y causa problemas del oído en un 10% de la población
mundial.
Las células madre pueden solucionar este problema porque tienen la capacidad única de convertirse en
cualquier tipo de célula humana.
Y tal como señaló el doctor Rivolta, no sólo podrían utilizarse para reemplazar las células auditivas
perdidas, sino también cualquier neurona dañada involucrada en las señales que el oído genera y
transmite al cerebro.
"Lo que hemos logrado es contar con un sistema en el laboratorio que hará posible generar ciliadas y
neuronas humanas", dijo el científico.
Y explicó que, en teoría, este procedimiento podría funcionar con cualquier tipo de sordera, incluso la
vinculada al envejecimiento.30 http://news.bbc.co.uk/hi/spanish/science/newsid_7978000/7978673.stm
109

"Todavía falta mucho tiempo para eso, pero creemos que todo lo que está relacionado con pérdida de
células auditivas o neuronas podría ser restaurado con la nueva terapia", explica el doctor Rivolta.
El avance, afirmaron los investigadores, servirá también para probar nuevos tratamientos para la
sordera y entender mejor la función de los genes en el oído.
"Este sistema es lo más cercano que hemos llegado a poder disponer de un oído humano real en el
laboratorio", aseguró el investigador.
"Esperamos que en el futuro nos sirva como un tratamiento contra la sordera, pero a un plazo más
inmediato lo usaremos como un modelo para estudiar la diferenciación de las células humanas", añadió.
Células madre y córnea
La grasa, el tejido con peor fama del organismo, también es el de la reparación. Las células madre
obtenidas de la grasa pueden restaurar fístulas que no cierran y tratar corazones dañados. Ahora el
Instituto Oftalmológico de Alicante ha demostrado que también podría reparar los ojos dañados por
traumatismos, quemaduras o infecciones y convertirse en una alternativa eficaz al trasplante de córnea
de cadáver.31
Bastaría con hacer una liposucción al paciente para tener un material inagotable y sin riesgo de rechazo
para recuperar los ojos dañados. El equipo del profesor Jorge Alió, en colaboración con el Hospital La
Paz, utilizó grasa sobrante de liposucciones para obtener células madre que pudieran reparar la córnea,
la principal lente del ojo. Una vez aisladas y purificadas, las células derivadas de tejido adiposo se
cultivaron y expandieron en el laboratorio para obtener el concentrado celular necesario. Después las
células no se depositaron directamente sobre la superficie ocular. Se realizó una incisión con láser,
similar a la que se realiza en la cirugía de la miopía para llegar al estroma, una de las capas de la córnea.
En ese lecho se colocaron las células madre a la espera de que adquirieran características específicas del
entorno y pudieran reparar la córnea. En el experimento, realizado con conejos y grasa sobrante de
pacientes de liposucción, no hubo rechazo alguno. Se constató que las células humanas trasplantadas se
transformaban en células corneales (queratocitos).
El nuevo tejido mantenía una transparencia funcional, sin ninguna opacidad, característica necesaria
para recuperar la visión. La córnea es una membrana transparente que enfoca las imágenes de la retina,
31 http://www.madrimasd.net/noticias/Regeneran-corneas-danadas-celulas-madre-grasa/34503
110

y cuando se daña pierde su transparencia. Los resultados de este experimento, publicados en la revista
científica «Stem Cells», abre nuevas perspectivas al tratamiento de las dolencias de la córnea. Pero,
sobre todo, sortea el problema de la falta de donaciones y el riesgo de rechazo del organismo. Además,
el trasplante de cadáver obliga a tomar fármacos inmunosupresores para combatir el rechazo del
sistema inmune.
Prótesis humanizadas
Para aplicarlo en pacientes, los oftalmólogos buscan otras fórmulas para que las células de la grasa se
transformen en células corneales en el laboratorio. «De esa forma, se evitaría la agresión con láser y se
controlaría mejor el proceso de diferenciación de las células», explicó Silvia Pastor, responsable del
laboratorio de cultivos celulares.
Las nuevas células se colocarían en un material biocompatible para obtener una prótesis fabricada a
partir de células del propio paciente. Esas prótesis «humanizadas» serían útiles cuando fracasen los
trasplantes clásicos de córnea. La terapia con grasa también podría ser una opción para tratar úlceras
superficiales y el queratocono, un trastorno que deforma la córnea.
Células madre y cáncer
El cultivo de células madre cancerígenas marcará el desarrollo de la neurocirugía y de la oncología
cerebral en los próximos años, según se ha puesto de manifiesto en el décimo tercer Congreso de la
Sociedad Española de Neurocirugía, que se celebra en Valencia.32
En el encuentro participan alrededor de trescientos especialistas en Neurocirugía, que han coincidido en
destacar que su especialidad médica camina hacia el desarrollo de técnicas menos invasivas con el
apoyo de las nuevas tecnologías y la biología molecular, según han informado en un comunicado
fuentes de la organización.
El secretario del congreso y jefe del Servicio de Neurocirugía del Hospital de la Ribera, en Alzira
(Valencia), José Piquer, ha señalado que el desarrollo de la biología molecular ha permitido aislar células
tumorales y conseguir a partir de ellas células madre tumorales.
32 http://actualidad.terra.es/sociedad/articulo/neurocirugia_cultivo_celulas_madre_cancerigenas_2513210.htm
111

'De esta manera podemos entender mucho mejor el comportamiento de los tumores y diferenciar
diferentes vías del origen de los mismos y con ello encontrar nuevas alternativas de tratamiento' de los
tumores cerebrales, que desarrollan diez de cada cien mil personas.
Otro de los grandes avances neuroquirúrgicos es la llamada 'realidad virtual', y en este sentido el doctor
Piquer ha señalado que la resonancia y la navegación 'permiten introducir esta tecnología con una
precisión milimétrica para ubicarnos dentro del cerebro y localizar cualquier lesión con una precisión
exacta'.
Según el experto, el avance de la neurocirugía se sustenta también en nuevas técnicas desarrolladas a
partir de viejas prácticas, como el caso de la psicocirugía y la neurología funcional.
Así, La neuroestimulación de determinados centros cerebrales 'está dando buenos resultados en la lucha
contra enfermedades como el parkinson o la distonía' y también hay buenas experiencias en pacientes
obsesivos.
Células madre en enfermedades hereditarias
La Universidad Autónoma de Madrid albergará el próximo 22 de mayo la Jornada “Terapia celular en la
medicina del siglo XXI”, enmarcada dentro del Foro de Ciencia y Tecnología madri+d dedicado a
Biomedicina y Ciencias de la Salud. La iniciativa, destinada tanto a empresas del sector como a
investigadores, consultores y gestores de este ámbito, ofrecerá una amplia perspectiva sobre los nuevos
avances en terapia celular y medicina regenerativa.33
Actualmente, las enfermedades degenerativas constituyen uno de los principales problemas sanitarios
de los países desarrollados. Estas enfermedades se deben, por lo general, a defectos génicos, lesiones
traumáticas, estilo de vida o al progresivo envejecimiento de la población.
Una característica común en este tipo de enfermedades es la alteración de tejidos y/o la desaparición de
ciertos tipos celulares, provocando una disfunción en distintos órganos. Los fármacos utilizados en el
tratamiento de estas enfermedades frecuentemente palian únicamente los síntomas, en vez de reparar
el daño tisular. Precisamente, el objetivo de la terapia celular es restaurar esos componentes dañados o
ausentes para así intentar recuperar la función del órgano en este tipo de enfermedades.
33 http://www.madridiario.es/2008/Mayo/ciencia-tecnologia/75928/foro-ciencia-tecnologia-salud-terapia-celular-medicina-regenerativa.html
112

Las aproximaciones de la terapia celular son muy variadas: el uso de células autólogas (del propio
paciente), alogénicas (de un donante), xenotransplantadas (procedentes de animales generalmente), el
uso de células madre adultas o embrionarias, células humanas modificadas genéticamente para producir
determinadas sustancias o células transdiferenciadas que ejercen funciones celulares que antes le eran
ajenas (por ejemplo, la producción de insulina por hepatocitos transdiferenciados en células beta), entre
otras.
Todas estas técnicas presentan frente a otras la ventaja de su versatilidad, por lo que las patologías a las
que pueden dirigirse son muy variadas. Han sido aplicadas con mayor o menor éxito a patologías tan
diversas como disfunciones hormonales (diabetes o déficit en la hormona del crecimiento), lesiones
cardiovasculares (isquemia, infarto de miocardio), enfermedades neurodegenerativas (Parkinson,
Alzheimer, Corea de Huntington), lesiones osteoarticulares o simplemente en lesiones en las que haya
que regenerar un tejido (como una fístula).
Parece claro el interés que tiene en el tratamiento de patologías hasta ahora incurables y como la
investigación en este área permitirá identificar los aspectos claves para hacer de la terapia celular una
técnica segura y eficaz que constituya un avance sin precedentes en el campo de la medicina.
La Jornada “Terapia Celular, la medicina del siglo XXI” ofrecerá varias perspectivas sobre la aplicación y
transferencia de conocimiento en este campo teniendo en cuenta sus aplicaciones clínicas en el área de
la medicina regenerativa. La Jornada comenzará con una conferencia magistral impartida por Damián
García Olmo, profesor del Departamento de Cirugía de la Universidad Autónoma de Madrid-Hospital
Universitario La Paz, quien ofrecerá información sobre los ensayos clínicos en terapia celular que se
están desarrollando en España. Posteriormente, tendrá lugar una mesa redonda donde se debatirá
acerca de la transferencia del conocimiento en terapia celular, conducida por Antonio Verde, director de
la OTRI de la Universidad Autónoma de Madrid y que contará con la participación de Jorge Alemany,
director de Estrategia y Operaciones de Cellerix; José Luis Jorcano, director General de la Fundación
Genoma España; y Víctor González, de la Subdirección de Terapia Celular y Medicina Regenerativa del
Instituto de Salud Carlos III.
La importancia de la terapia celular en la aportación de nuevos enfoques para el tratamiento de
enfermedades está reflejada en el interés creciente que la investigación en el área está suscitando. Tan
sólo en el ámbito nacional, podemos encontrar más de 50 grupos dedicados a la investigación en éste
área, existiendo varios centros de investigación dedicados casi exclusivamente a la medicina
113

regenerativa y terapia celular como por ejemplo el Centro Andaluz de Biología Molecular y Medicina
Regenerativa de Sevilla, el Banco de Células Madre de Granada y el Centro de Medicina Regenerativa de
Barcelona.
En la Comunidad de Madrid existen numerosos investigadores que realizan estudios en este campo. Los
intereses en la investigación de un 39 por ciento de los grupos se centran en el estudio de las células
madre, tanto embrionarias como adultas, mientras que un 22 por ciento investigan en las áreas de
regeneración tisular y medicina regenerativa.
El Consejo Superior de Investigaciones Científicas con centros como el Centro de Investigaciones
Biológicas (CIB), el Centro Nacional de Biotecnología (CNB) y el Centro de Biología Molecular (CBM),
ambos centros mixtos UAM-CSIC, concentra una importante actividad investigadora en terapia celular.
El Centro Nacional de Investigaciones Cardiovasculares (CNIC) y el Centro Nacional de Investigaciones
Oncológicas (CNIO) son instituciones de referencia en el área, así como centros hospitalarios de
excelencia como La Paz y Gregorio Marañón. La Universidad Complutense y la Universidad Autónoma de
Madrid albergan programas de investigación cooperativa en terapia celular.
Para hacernos una idea de la importancia que se otorga a la investigación en el área de terapia celular,
basta con hacer una revisión del número de ayudas y programas que incluyen esta línea de investigación
dentro de las acciones prioritarias a nivel europeo (el VII Programa Marco), nacional (por ejemplo,
acciones dentro del Plan Nacional o del Instituto de Salud Carlos III a través de las ayudas RECTIS y las
ayudas de apoyo a la investigación Biomédica para Consorcios en red CIBER) y regional.
Consciente de la importancia que la terapia celular tiene para la sociedad y de las posibles aplicaciones
en el ámbito clínico de los resultados de estas investigaciones, la Comunidad de Madrid ha puesto en
marcha varias iniciativas encaminadas a promocionar la investigación de excelencia y la transferencia de
conocimiento en este ámbito.
La Dirección General de Universidades e Investigación ha promovido y financiado programas de
investigación dedicados al estudio de células madre, centrando, por ejemplo, sus investigaciones en
determinar las variables que permitirán aplicar con total seguridad el uso de la células madre para el
tratamiento de diversas enfermedades; o determinando las propiedades biológicas y posibles
aplicaciones clínicas de las células madre mesenquimales. Además, otros grupos identificados en la
Comunidad de Madrid realizan estudios relacionados con la biología de precursores hematopoyéticos,
terapia génica y estudios sobre la estabilidad, biología y aplicación clínica de células madre.
114

El estudio de las células madre nos permitirá conocer los mecanismos de especialización celulares. Qué
mecanismos hacen que un gen sea activo y haga su trabajo y qué mecanismos inhiben la expresión de
ese gen. El cáncer, por ejemplo, es un caso de especialización celular anormal.
Las células madre pueden servir para probar nuevos medicamentos en todo tipo de tejidos antes de
hacer las pruebas reales en animales o en humanos.
Las células madre tendrán aplicaciones en terapias celulares, medicina regenerativa o ingeniería tisular.
Muchas enfermedades son consecuencia de malfunciones celulares o destrucción de tejidos. Uno de los
remedios, en casos muy graves, es el transplante. Las células madre pluripotentes estimuladas a
desarrollarse como células especializadas ofrecen frecuentemente la posibilidad de reemplazar células y
tejidos dañados. Así se podrán emplear para casos de Parkinson y Alzheimer, lesiones medulares,
quemaduras, lesiones de corazón o cerebrales, diabetes, osteoporosis y artritis reumatoide.
Veamos ejemplos de aplicaciones: 34
Según publicó Science Abril de 2000, a dos bebés que nacieron con un defecto genético que les
ocasionaba una severa inmunodeficiencia, les extrajeron células madre de médula ósea. Se cultivaron
las células, se reemplazó el gen defectuoso y se transfirieron de nuevo a los niños. Este experimento, en
el que se emplearon células madre de los propios bebés, constituyó el primer éxito de curación
mediante terapia genética.
Por primera vez en España en la Clínica Universitaria de Navarra se ha curado (PDF) un corazón infartado
implantándo células madre del propio paciente. El paciente tenía una parte del músculo cardíaco
muerta a acusa de varios infartos. Se le extrajeron células del muslo se seleccionaron y purificaron las
células madre. Después de cultivarlas durante tres semanas se inyectaron en el músculo infartado. La
recuperación fue prodigiosa.
Un trabajo de la Universidad Johns Hopkins, en Baltimore, presentado durante el encuentro anual de la
Sociedad Americana de Neurociencia explicaba que la inyección de células madre en el líquido
cefalorraquídeo de los animales lograba devolver el movimiento a unos roedores con parálisis. Los
expertos introdujeron células madre neuronales en los roedores paralizados por un virus que ataca
34 http://www.ecojoven.com/uno/05/celulasm.html
115

específicamente a las neuronas motoras y comprobaron que el 50 por ciento recuperaba la habilidad de
apoyar las plantas de una o de dos de sus patas traseras.
Las investigaciones son muy prometedoras y avanzan muy rápidamente, pero queda mucho por hacer
para llegar a aplicaciones clínicas reales. Todavía falta por conocer los mecanismos que permiten la
especialización de las células madre humanas para obtener tejidos especializados válidos para el
transplante.
116