tema 5 separaciones tÉrmicas · consideraciones generales. ... en el diagrama de la figura se ha...
TRANSCRIPT

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 1 de 80
Muestreo y preparación de la muestra
TEMA 5
SEPARACIONES TÉRMICAS
Ciclo Formativo Análisis Químico y Control de Calidad

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 2 de 80
DESTILACIÓN
1. CONSIDERACIONES GENERALES.
Las operaciones básicas llamadas separaciones térmicas utilizan el calor como medio de separación de la mezcla de sustancias.
En la DESTILACIÓN esta separación se consigue debido a la diferencia en el punto de ebullición de las dos sustancias a separar.
En la EVAPORACIÓN se consigue separar el componente más volátil.
En el SECADO el componente a separar es más volátil que el sólido.
En la CRISTALIZACIÓN se separa por enfriamiento el componente menos soluble en la disolución.
Llamamos DESTILACIÓN a la separación de los componentes de una mezcla líquida por vaporización parcial de la misma. La concentración de componentes volátiles es mayor en el vapor obtenido que en la mezcla inicial, mientras que en el residuo aumenta la concentración de los componentes menos volátiles.
Las aplicaciones industriales más numerosas se encuentran en el campo de los compuestos de carbono; p. ej., la separación de mezclas de hidrocarburos o la obtención de compuestos puros. Sin embargo, no hay que olvidar la importancia de la destilación en industrias como la de separación del aire líquido en sus componentes, la recuperación del amoniaco o la obtención del H2O2 a partir de las disoluciones acuosas.
La destilación fraccionada también será estudiada en este tema.
2. EQUILIBRIOS DE VAPORIZACIÓN Y CONDENSACIÓN.
La separación de los componentes de una mezcla líquida por destilación sólo puede hacerse cuando el vapor producido en la ebullición tiene diferente composición que el líquido de que procede, y será tanto más fácil cuanto mayor sea la diferencia entre las dos composiciones.
Por esto, el fundamento básico de la destilación se encuentra en el estudio de los equilibrios vapor-líquido para las distintas mezclas. En general, y por ser de carácter más sencillo, nos limitaremos a las mezclas binarias; pero la mayoría de los conceptos estudiados pueden aplicarse también a las de tres o más componentes volátiles.
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 3 de 80
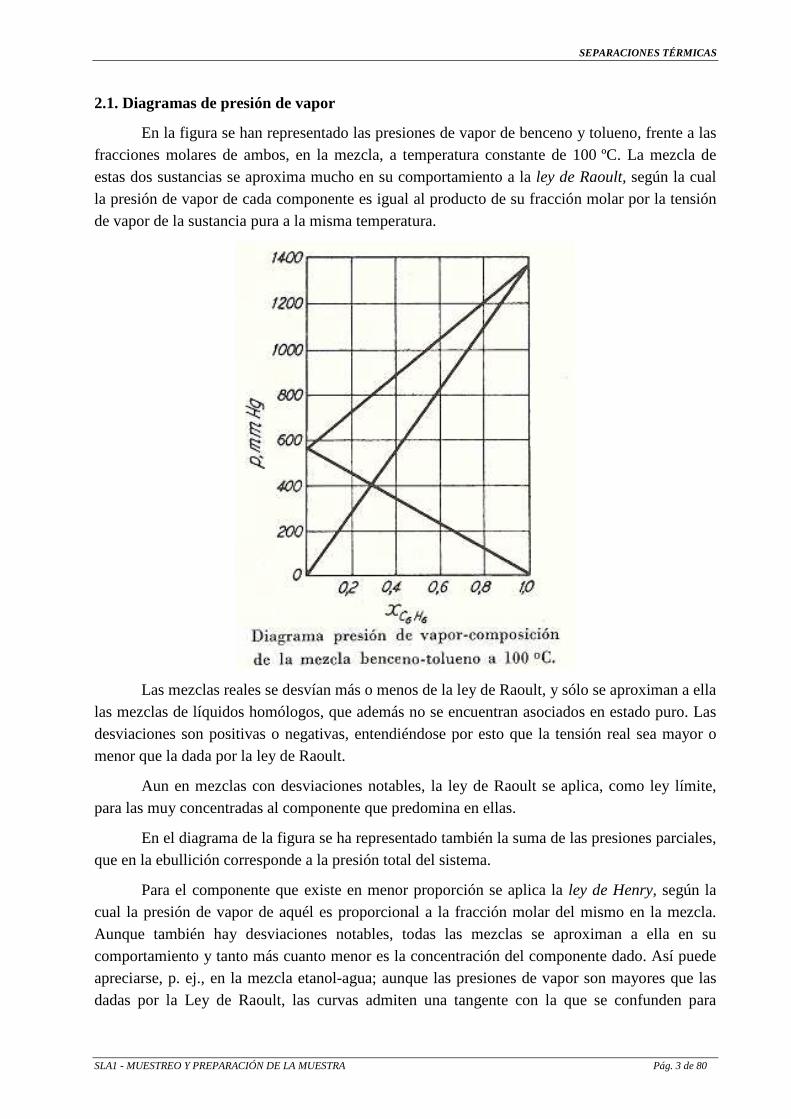
2.1. Diagramas de presión de vapor
En la figura se han representado las presiones de vapor de benceno y tolueno, frente a las fracciones molares de ambos, en la mezcla, a temperatura constante de 100 ºC. La mezcla de estas dos sustancias se aproxima mucho en su comportamiento a la ley de Raoult, según la cual la presión de vapor de cada componente es igual al producto de su fracción molar por la tensión de vapor de la sustancia pura a la misma temperatura.
Las mezclas reales se desvían más o menos de la ley de Raoult, y sólo se aproximan a ella las mezclas de líquidos homólogos, que además no se encuentran asociados en estado puro. Las desviaciones son positivas o negativas, entendiéndose por esto que la tensión real sea mayor o menor que la dada por la ley de Raoult.
Aun en mezclas con desviaciones notables, la ley de Raoult se aplica, como ley límite, para las muy concentradas al componente que predomina en ellas.
En el diagrama de la figura se ha representado también la suma de las presiones parciales, que en la ebullición corresponde a la presión total del sistema.
Para el componente que existe en menor proporción se aplica la ley de Henry, según la cual la presión de vapor de aquél es proporcional a la fracción molar del mismo en la mezcla. Aunque también hay desviaciones notables, todas las mezclas se aproximan a ella en su comportamiento y tanto más cuanto menor es la concentración del componente dado. Así puede apreciarse, p. ej., en la mezcla etanol-agua; aunque las presiones de vapor son mayores que las dadas por la Ley de Raoult, las curvas admiten una tangente con la que se confunden para

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 4 de 80
concentraciones pequeñas de componente (a para el etanol, b para el agua).
2.2. Diagramas de ebullición.
En la práctica de la destilación son más útiles los diagramas de ebullición, en los que se representa la temperatura de ebullición en función de la composición de la mezcla líquida, a presión constante.
El diagrama de la figura corresponde al de ebullición de mezclas binarias de sulfuro de carbono y tetracloruro de carbono a 760 mm de Hg.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 5 de 80
La curva inferior es la del líquido, y la superior la del vapor. Cada punto de esta última representa el vapor en equilibrio con el líquido dado por el punto que se halla en la curva inferior sobre la misma horizontal.
Una mezcla líquida de composición x = 0,30, empieza a hervir al alcanzar la temperatura de 62,00 C (punto A), dando un vapor de composición y = 0,55. Si los vapores producidos en la ebullición se mantienen en equilibrio con el líquido, el final de la ebullición tendrá lugar a la temperatura 69,5 ºC del punto C. Esto motiva la denominación de curvas de principio y fin de ebullición para las curvas del líquido y del vapor.
Al enfriar una mezcla vaporizada de composición y = 0,55, empieza a condensarse a 62ºC (punto B), dando en un principio un líquido de composición x1 = 0,30 (punto A).
El final de la condensación de la condensación de equilibrio, tendría lugar a la temperatura de 54,8 ºC del punto D. Esto justifica las denominaciones de curvas del principio y fin de condensación aplicadas también a las curvas del vapor y del líquido.
El diagrama estudiado anteriormente corresponde a una pareja de líquidos de tipo normal (I), en que las volatilidades de una mezcla de ambos componentes en proporciones cualesquiera es menor que la del líquido puro más volátil y mayor que la del menos volátil.
En la figura representamos una mezcla de tipo diferente (II), en la que existe un mínimo para la temperatura de ebullición.
La mezcla que hierve a esta temperatura se conoce con el nombre de mezcla azeotrópica, y tiene la propiedad de vaporizarse a temperatura constante, dando vapores de una y la misma composición. Esta mezcla, que a este respecto se comporta como un líquido puro, es más volátil que cualquiera de los dos componentes.
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 6 de 80
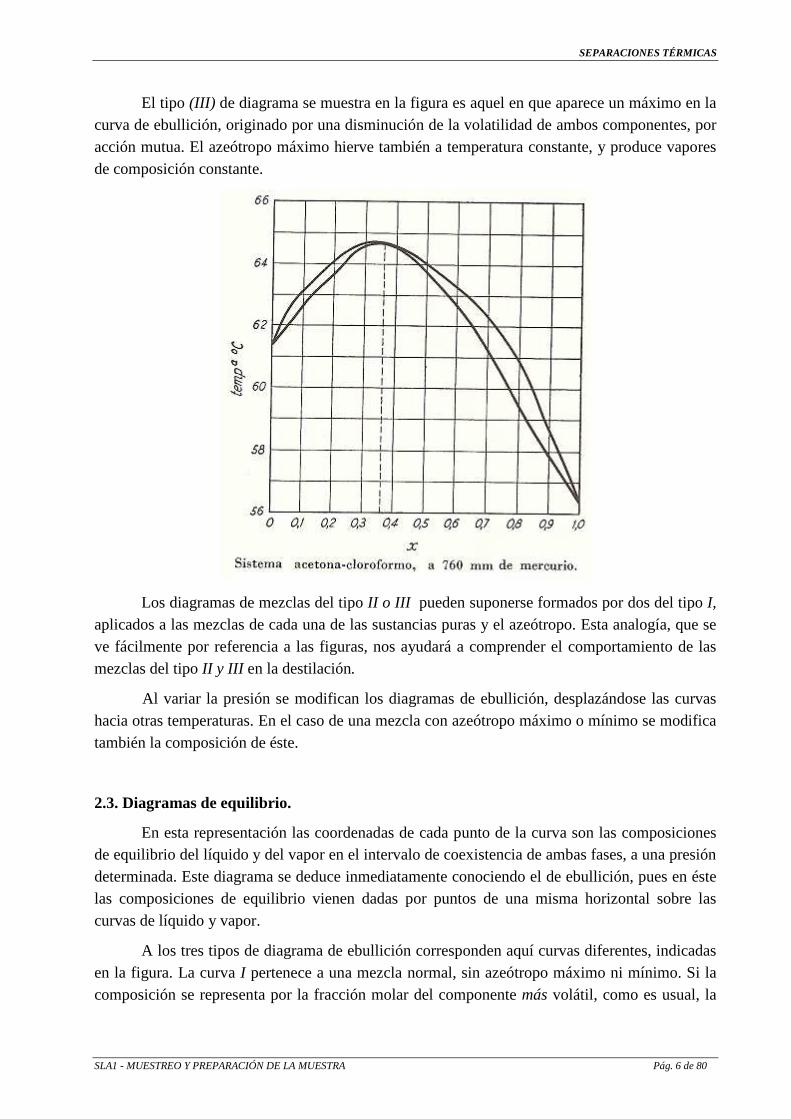
El tipo (III) de diagrama se muestra en la figura es aquel en que aparece un máximo en la curva de ebullición, originado por una disminución de la volatilidad de ambos componentes, por acción mutua. El azeótropo máximo hierve también a temperatura constante, y produce vapores de composición constante.
Los diagramas de mezclas del tipo II o III pueden suponerse formados por dos del tipo I,
aplicados a las mezclas de cada una de las sustancias puras y el azeótropo. Esta analogía, que se ve fácilmente por referencia a las figuras, nos ayudará a comprender el comportamiento de las mezclas del tipo II y III en la destilación.
Al variar la presión se modifican los diagramas de ebullición, desplazándose las curvas hacia otras temperaturas. En el caso de una mezcla con azeótropo máximo o mínimo se modifica también la composición de éste.
2.3. Diagramas de equilibrio.
En esta representación las coordenadas de cada punto de la curva son las composiciones de equilibrio del líquido y del vapor en el intervalo de coexistencia de ambas fases, a una presión determinada. Este diagrama se deduce inmediatamente conociendo el de ebullición, pues en éste las composiciones de equilibrio vienen dadas por puntos de una misma horizontal sobre las curvas de líquido y vapor.
A los tres tipos de diagrama de ebullición corresponden aquí curvas diferentes, indicadas en la figura. La curva I pertenece a una mezcla normal, sin azeótropo máximo ni mínimo. Si la composición se representa por la fracción molar del componente más volátil, como es usual, la

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 7 de 80
curva irá siempre por encima de la diagonal. Sin embargo, las mezclas con azeótropo mínimo o máximo nos dan las curvas del tipo II y III, con un punto de intersección con la diagonal para la composición de aquél.
La zona en que curva de equilibrio va por debajo de la diagonal, corresponde a una inversión de la volatilidad. En efecto: acudiendo al diagrama de ebullición vemos que en esta zona disminuye la volatilidad (aumenta el punto de ebullición) al aumentar la proporción del componente más volátil.
3. CÁLCULO DE LOS DATOS DE EQUILIBRIO.
En general los datos de equilibrio se determinan; experimentalmente, en aparatos que para cada temperatura permiten aislar muestras del vapor y del líquido en las condiciones de equilibrio.
Otras veces se parte de datos incompletos (p. ej., los relativos a la curva de ebullición) y se complementan por métodos de cálculo aproximados.
Por último, para mezclas cuyo comportamiento se aproxima al ideal, pueden construirse los diagramas a partir de los datos de tensión de vapor de ambos componentes. En efecto: siendo PA y PB las tensiones de vapor a una temperatura dada, xA y xB las fracciones molares de ambos componentes en el líquido, aplicando la ley de Raoult, tenemos que la presión necesaria para que

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 8 de 80
se produzca la ebullición a esa temperatura será:
BBAABA PxPxppp ⋅+⋅=+=
Las fracciones molares de los componentes en el vapor, YA e YB, son proporcionales a sus presiones parciales, luego:
p
Px
p
py AAA
A
⋅==
p
Px
p
pyy BBB
BA
⋅===−1
Estas ecuaciones se aplican a la construcción de los diagramas de equilibrio a presión constante o a temperatura constante. Este último caso es más sencillo, porque dada la temperatura, y con ello las tensiones PA y PB, la presión de vapor para cada composición de líquido se obtiene de la primera ecuación, y las composiciones del vapor de la segunda, ambas directamente.
El diagrama a presión constante requiere un cálculo por tanteo si -dada la composición del líquido- se busca la temperatura de equilibrio, que es aquella para la cual las tensiones de vapor PA y PB satisfacen la primera ecuación. Cuando se trata de calcular el diagrama de ebullición, el valor de x para cada temperatura se halla por la ecuación:
BA
BA PP
Ppxx
−−
=≡
deducida de las anteriores.
Si tenemos en cuenta que BA xx −≡ 1 . En todos los casos la composición del vapor resulta
inmediatamente de las ecuaciones anteriores.
Para mezclas muy diluidas en uno de los componentes puede admitirse la Ley de Henry, ya mencionada, que expresa la relación directa entre las composiciones de vapor y el líquido en equilibrio:
Kxy =
K es la llamada constante de equilibrio. Esta última ecuación se aplica también a las mezclas de dos o más hidrocarburos en concentraciones cualesquiera; en este caso K es independiente de las concentraciones y de la naturaleza de los otros hidrocarburos; pero varía con la presión total y la temperatura.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 9 de 80
4. FORMACIÓN DE AZEÓTROPOS BINARIOS
EI hecho de que las sustancias que forman azeótropos no puedan separarse en una sola destilación y, por otra parte, el que su formación se aproveche para la separación de mezclas más complicadas, realza la importancia del estudio de las mezclas azeotrópicas.
En la actualidad se han descrito más de 3000 mezclas binarias con azeótropo mínimo, y sólo unas 250 con azeótropo máximo.
Se han hecho muchos intentos para predecir el fenómeno de la formación de mezclas azeotrópicas binarias en función de la naturaleza de los componentes. La causa determinante está en las desviaciones de la ley de Raoult: la probabilidad de formación de un azeótropo aumenta con la magnitud de las desviaciones respecto de la ley de Raoult y disminuye tanto más cuanto mayor es la diferencia entre los puntos de ebullición normales de ambos componentes (esta última puede tomarse como medida cualitativa del cociente de las tensiones de vapor a temperatura constante). Esto nos explica que mezclas con desviaciones muy pequeñas puedan dar azeótropo si los puntos de ebullición de los componentes son muy próximos; y también que en las mezclas binarias de una sustancia con los distintos términos de una serie homóloga sólo se produce azeótropo con los que hierven a temperatura próxima a la de aquélla.
Desplazamiento del azeótropo con la presión.-Un punto interesante en el estudio de la formación de azeótropos binarios es el desplazamiento de la composición del azeótropo con la presión. Según la regla de Wrewski, que se cumple en muchos casos, al aumentar la presión, y con ella la temperatura total del sistema, aumenta en el azeótropo mínimo la proporción del componente que tiene mayor calor latente de vaporización, y en el azeótropo máximo la del que tiene menor calor latente. Este hecho se ha aprovechado para separar la mezcla etanol-agua, pues al operar a presión reducida disminuye la proporción de agua en el azeótropo, pudiendo llegar a anularse prácticamente.
En muchos casos se ha demostrado la desaparición de la azeotropía por encima y por debajo de determinados valores de la presión total, y así parece suceder en el anteriormente mencionado.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 10 de 80
5. SISTEMAS INMISCIBLES.
En teoría, no puede hablarse de líquidos totalmente inmiscibles; la inmiscibilidad es un límite al que se acercan más o menos las parejas de líquidos, sin haber ninguna que lo alcance. Sin embargo, en la práctica hay líquidos de solubilidad mutua tan pequeña que se puede considerar nula. Así sucede en el caso clásico del mercurio y el agua.
La división entre líquidos inmiscibles y parcialmente miscibles no puede hacerse de un modo tajante, pues cualquier intento de clasificación ha de basarse en un límite totalmente arbitrario. Tengamos además en cuenta que la miscibilidad varía con la temperatura. Un sistema esencialmente inmiscible puede pasar a parcialmente miscible, y este último transformarse en miscible, al aumentar la temperatura. Otras veces puede suceder lo mismo al descender la temperatura, aunque el caso no sea tan corriente, porque antes de llegar a la miscibilidad puede tener lugar la congelación de los líquidos. En teoría, es posible la existencia de dos temperaturas críticas de solución, la superior y la inferior, y entre ellas pueden darse todos los grados de miscibilidad parcial.
Las mezclas de dos líquidos, A y B, parcialmente miscibles existen como sistemas monofásicos para pequeñas concentraciones de A o de B; los límites de estas concentraciones son sólo función de la temperatura. Para concentraciones medias aparecen dos fases: solución saturada de A en B y solución saturada de B en A. Las concentraciones de ambos quedan fijadas automáticamente en función de la temperatura, como puede deducirse por aplicación de la regla de las fases. Al variar la composición global dentro de los límites de inmiscibilidad sólo lo-graremos variar la proporción relativa de ambas fases, y no su composición. En el equilibrio del sistema líquido con el vapor quedan también determinadas la composición y la presión de ést'e; esta última será la suma de las tensiones parciales de los dos componentes. La tensión de vapor de cada componente es la misma en ambas fases.
Por su mayor aplicación nos interesan los sistemas considerados como totalmente inmiscibles. Las tensiones de vapor de ambos líquidos son independientes de sus proporciones relativas, o sea de la composición global de la mezcla. La ebullición se produce cuando la suma de las tensiones de vapor iguala a la presión que reina en el espacio gaseoso. La composición del vapor es independiente de la que tenga el conjunto líquido, y se calcula fácilmente con ayuda de las leyes de los gases perfectos (las desviaciones son mínimas a presiones ordinarias).
Al enfriar los vapores a presión y composición constantes, la condensación comienza cuando la presión de vapor de cualquiera de los componentes iguala a su tensión de vapor. La temperatura a que esto ocurre es la de rocío o comienzo de condensación (o bien, de final de ebullición); si continúa el enfriamiento se separa como líquido el componente puro que alcanzó la saturación, y el vapor se enriquece en el otro componente.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 11 de 80
Ejemplo: Calcular la temperatura de ebullición de la mezcla benceno-agua a 760 mm. de Hg y la composición de los vapores. Calcúlense también las temperaturas de rocío de los vapores de composición y(C6H6) = 0,60 e y(C6H6) = 0,80. Las tensiones de vapor de los componentes puros son:
Temperatura ºC 60 65 70 75 80
P C6H6 391,2 465,4 550,4 647,4 757,4
P H2O 149,4 187,5 233,7 289,1 355,1
760 – PH2O 610,6 572,5 526,3 470,9 404,9
SOLUCIÓN.
Hay que determinar la temperatura para la cual las tensiones de vapor suman 760 mm. Hg. En primera aproximación se ve que aquélla está comprendida entre 65 ºC y 70 ºC. Después puede recurrirse a una interpolación aritmética de la suma de las tensiones en función de la temperatura. Para mayor exactitud se representa gráficamente P frente a t, mediante una curva que une los cinco puntos dados, y en ella se determina el valor de t para P = 760 mm Hg. Otro procedimiento, equivalente a éste, consiste en buscar la intersección de las curvas que re-presentan P(C6H6) y (760 – PH2O) frente a la temperatura, tal y como se representa
TENSIONES VAPOR BENCENO-AGUA
300
400
500
600
700
800
60 65 70 75 80
TEMPERATURA
TE
NS
IÓN
VA
PO
R
P benceno
760 - P(H20)
La solución es: t = 69,15 ºC; P(C6H6) = 535 mm Hg; PH2O = 225 mm Hg.
La composición del vapor, referida al benceno, será:
704,0760
53566 ==HCy
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 12 de 80
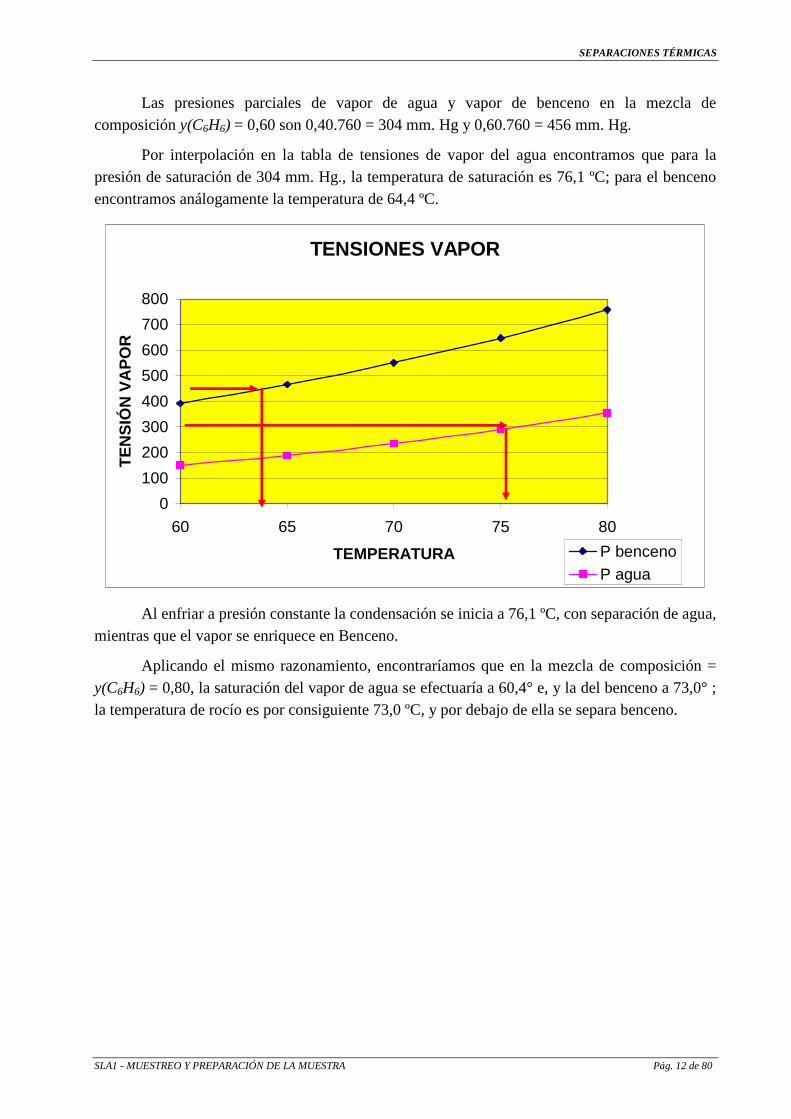
Las presiones parciales de vapor de agua y vapor de benceno en la mezcla de composición y(C6H6) = 0,60 son 0,40.760 = 304 mm. Hg y 0,60.760 = 456 mm. Hg.
Por interpolación en la tabla de tensiones de vapor del agua encontramos que para la presión de saturación de 304 mm. Hg., la temperatura de saturación es 76,1 ºC; para el benceno encontramos análogamente la temperatura de 64,4 ºC.
TENSIONES VAPOR
0
100
200
300
400
500
600
700
800
60 65 70 75 80
TEMPERATURA
TE
NS
IÓN
VA
PO
R
P bencenoP agua
Al enfriar a presión constante la condensación se inicia a 76,1 ºC, con separación de agua, mientras que el vapor se enriquece en Benceno.
Aplicando el mismo razonamiento, encontraríamos que en la mezcla de composición = y(C6H6) = 0,80, la saturación del vapor de agua se efectuaría a 60,4° e, y la del benceno a 73,0° ; la temperatura de rocío es por consiguiente 73,0 ºC, y por debajo de ella se separa benceno.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 13 de 80
En la figura siguiente se representa el diagrama de ebullición de un sistema inmiscible a presión constante. Cualquiera que sea la composición inicial, la ebullición se produce al alcanzarse la línea DE, y el vapor tiene la composición constante correspondiente. Los vapores que por su composición están a la izquierda del punto C condensan a lo largo de la línea BC,
dando como condensado el líquido B puro, y enriqueciéndose en el componente A. Al llegar a C, la condensación tiene lugar a temperatura constante, dando un líquido difásico de la misma composición global. Las características de una mezcla de composición C son en este sentido las de un azeótropo, que se designa con el nombre de heteroazeótropo para señalar la existencia de dos fases líquidas. Las diferencias con los homoazeótropos son tan notables como las analogías.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 14 de 80
6. DESTILACIÓN SIMPLE.
Aunque el método de destilación más importante es el de rectificación, la destilación simple tiene numerosas aplicaciones. En esencia, consiste en la vaporización parcial del líquido con producción de una cantidad de vapor, más rica en componentes volátiles que el líquido inicial, y un residuo liquido más concentrado en los componentes menos volátiles. La destilación simple puede efectuarse con arreglo a dos métodos diferentes, que estudiamos a continuación.
6.1. Destilación de equilibrio, o cerrada.
El líquido de una composición definida se lleva a una temperatura constante, intermedia entre la de principio y la de fin de ebullición, hasta el establecimiento del equilibrio. Las fracciones líquida y vapor que resultan al alcanzarse éste tienen composiciones definidas, que coinciden con las composiciones de equilibrio a la temperatura de destilación y la presión total de trabajo.
Para un sistema binario, la determinación de las composiciones puede efectuarse con ayuda del diagrama de ebullición a la presión dada, pues son las correspondientes a los puntos M y N que se encuentran en la intersección de la isoterma (horizontal) con las curvas del líquido y del vapor.
de aquí que las cantidades de líquido y de vapor son inversamente proporcionales a las longitudes de los segmentos MP y PN determinados en el diagrama por las composiciones respectivas y la del líquido inicial.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 15 de 80
Ejemplo: Una mezcla de 60 moles de benceno y 40 de tolueno se somete a destilación cerrada a presión de 800 mm Hg y temperatura constante de 95° C. Hallar:
a) Las composiciones de líquido y de vapor en equilibrio.
b) La cantidad (en moles) de benceno que pasa al estado de vapor.
SOLUCIÓN:
a) Las composiciones de equilibrio se determinan generalmente con ayuda del diagrama de ebullición. En este caso podemos calcularlas por aplicación de la ley de Raoult, en función de las tensiones de vapor de los componentes puros:
Temperatura ºC PB
mm. Hg PT
mm. Hg
80 757,4 290,7
85 881,6 344,6
90 1021 406,4
95 1177 476,7
100 1351 556,2
105 1545 646,0
110 1759 746,9
De la ecuación: TTBBTB PxPxppp ⋅+⋅=+=
( ) TBBB PxPxp ⋅−+⋅= 1
TBTBB PxPPxp ⋅−+⋅=
( ) TTBB PPPxp +−⋅=
462,07,4761177
7,476800 =−−=
−−
=TB
TB PP
Ppx
La composición del vapor al ser las fracciones molares de los componentes en el vapor By e Ty
proporcionales a sus presiones parciales:
p
Px
p
py BBB
B
⋅==
p
Px
p
pyy TTT
TB
⋅===−1
679,0800
1177462,0 =⋅=By

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 16 de 80
b) La relación entre las cantidades de líquido y de vapor en el estado final es:
572,0462,0600,0
600,0679,0 =−−=
V
L
por otra parte L + V = 100, luego 6,63572,1
100 ==V ; 4,36=L
La cantidad de benceno en el vapor será: 2,430679,06,63 =⋅ moles
A este tipo de destilación se le conoce también por vaporización súbita (flash), y se emplea mucho en la industria del petróleo.
6.2. Destilación diferencial o abierta
La destilación diferencial se emplea mucho, en particular en los laboratorios, para la separación de mezclas de compuestos cuyas volatilidades difieren apreciablemente.
En la destilación diferencial o abierta el líquido se lleva hasta que comienza la ebullición, y el vapor producido se retira continuamente del espacio gaseoso, llevándolo a un condensador separado. De acuerdo con el mecanismo ya estudiado, el líquido se empobrece en los componentes más volátiles, ascendiendo continuamente su temperatura de ebullición y produciendo un vapor que es también más pobre en componentes volátiles.
Supongamos una mezcla binaria de composición inicial: xo. La ebullición comienza a la temperatura to, con producción de un vapor de composición inicial yo, correspondiente al equilibrio a esa temperatura.
Al separarse este vapor la composición del líquido se desplaza hacia la izquierda, siguiendo la curva de equilibrio, y, en consecuencia, aumenta la temperatura de ebullición. Cuando la composición del líquido residual es x, la burbuja de vapor que se desprende de él tiene la composición y; si hemos recogido todo el destilado en el mismo recipiente su composición global será intermedia entre y e yo.
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 17 de 80
6.3. Diagramas de ebullición-destilación.
A partir de los diagramas de ebullición de componentes simples y mezclas, pueden conseguirse experimentalmente otros diagramas de equilibrio, que informan debidamente de cómo se desarrollará el proceso de una destilación cualquiera.
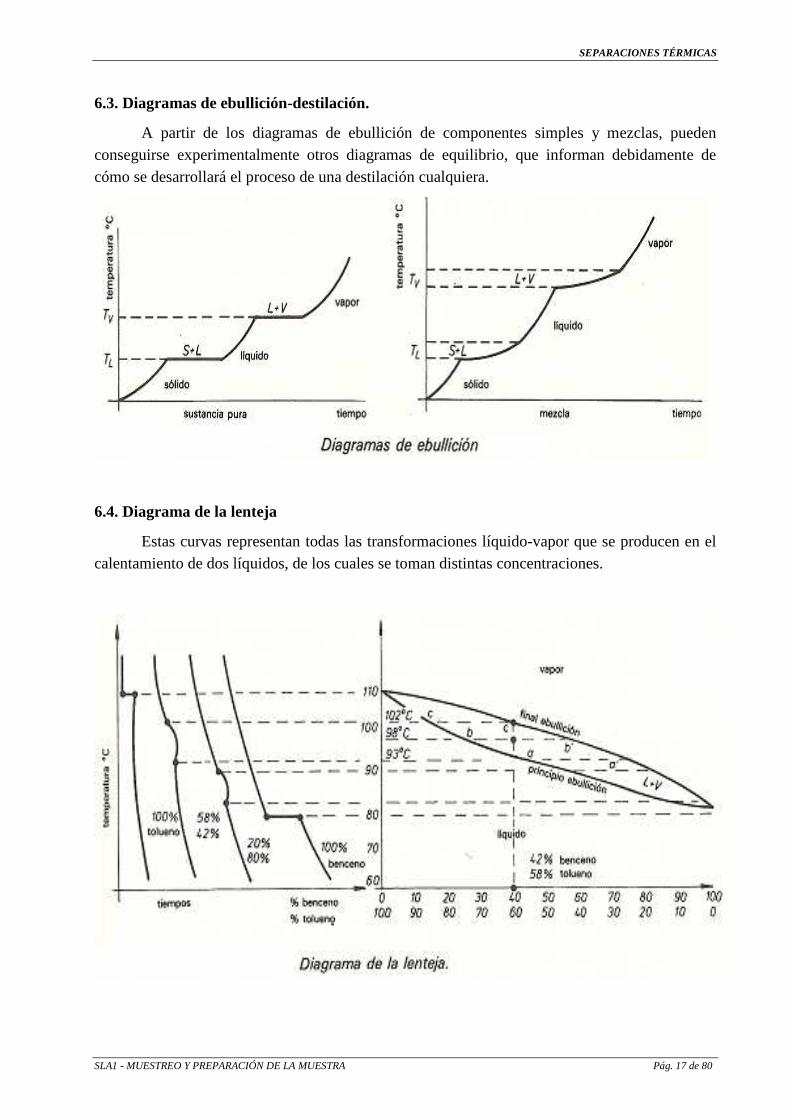
6.4. Diagrama de la lenteja
Estas curvas representan todas las transformaciones líquido-vapor que se producen en el calentamiento de dos líquidos, de los cuales se toman distintas concentraciones.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 18 de 80
El diagrama de la izquierda representa la evolución a cada concentración y el de la derecha, de la lenteja, el resultado transformado a unos ejes de coordenadas con temperaturas y concentraciones.
Tomando un punto como referencia, sea 42 % de benceno, se observa que la mezcla empieza a hervir a 93 °C y termina a 102 °C aproximadamente; y así, sucesivamente, para cada relación que se requiera.
Analícese la composición del vapor y del residuo líquido de la destilación citada:
Empieza a hervir a 93 ºC Vapor: 72 % benceno – 28 % tolueno Punto a`
Líquido. 42 % benceno – 58 % tolueno Punto a
A 98 ºC Vapor: 55 % benceno - 45 % tolueno Punto b`
Líquido: 25 % benceno - 75 % tolueno Punto b
Termina la ebullición a 102 ºC
Vapor: 42 % benceno - 58 % tolueno Punto c`
Líquido: 15 % benceno - 85 % tolueno Punto c
6.5. Diagrama de máximo y mínimo: mezclas azeotrópicas
Se construyen idénticamente al de la lenteja, ya que se consideran dos de éstos con el azeótropo superior o inferior como un componente puro.
Las mezclas azeotrópicas, de gran importancia en las destilaciones químicas, son mezclas de líquidos cuyo punto de ebullición a concentración determinada (máximo o mínimo) es único y destila toda la mezcla hasta terminarse. Por tanto, representa un inconveniente para obtener mezclas de gran pureza, puesto que al llegar al azeótropo no se consigue pasar de ella en cuanto a riqueza.
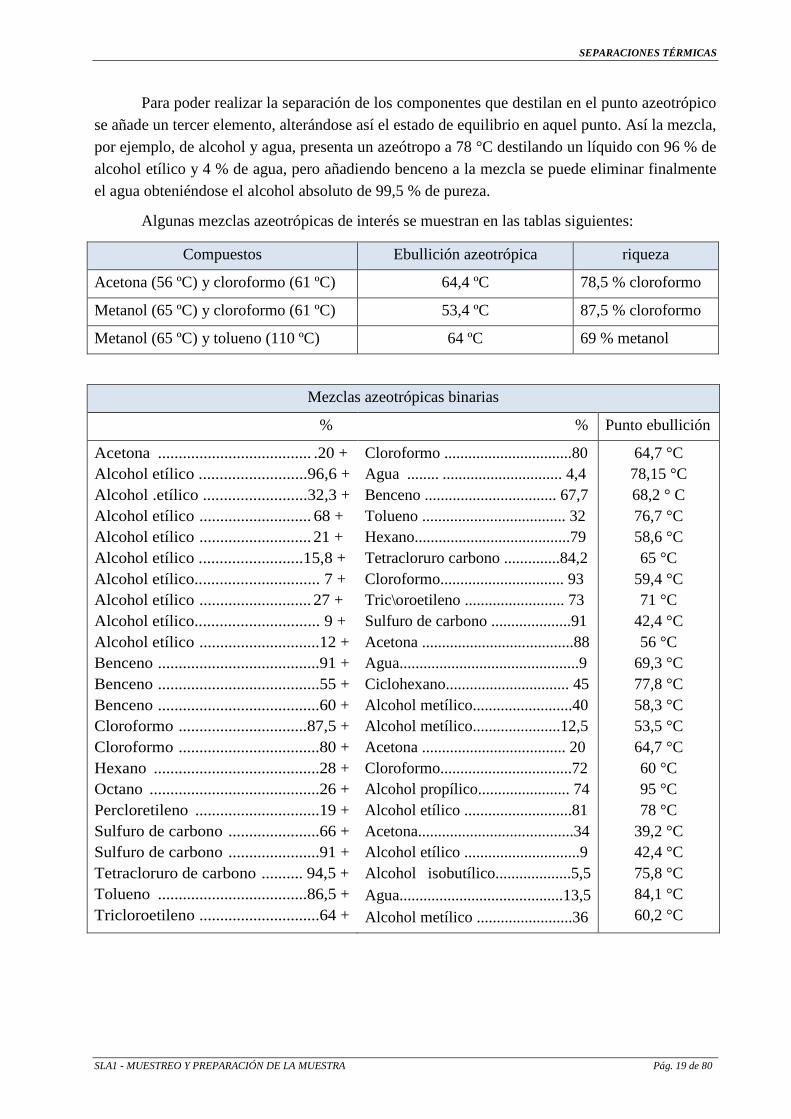
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 19 de 80
Para poder realizar la separación de los componentes que destilan en el punto azeotrópico se añade un tercer elemento, alterándose así el estado de equilibrio en aquel punto. Así la mezcla, por ejemplo, de alcohol y agua, presenta un azeótropo a 78 °C destilando un líquido con 96 % de alcohol etílico y 4 % de agua, pero añadiendo benceno a la mezcla se puede eliminar finalmente el agua obteniéndose el alcohol absoluto de 99,5 % de pureza.
Algunas mezclas azeotrópicas de interés se muestran en las tablas siguientes:
Compuestos Ebullición azeotrópica riqueza
Acetona (56 ºC) y cloroformo (61 ºC) 64,4 ºC 78,5 % cloroformo
Metanol (65 ºC) y cloroformo (61 ºC) 53,4 ºC 87,5 % cloroformo
Metanol (65 ºC) y tolueno (110 ºC) 64 ºC 69 % metanol
Mezclas azeotrópicas binarias
% % Punto ebullición
Acetona ..................................... .20 + Alcohol etílico ..........................96,6 + Alcohol .etílico .........................32,3 + Alcohol etílico ........................... 68 + Alcohol etílico ........................... 21 + Alcohol etílico .........................15,8 + Alcohol etílico.............................. 7 + Alcohol etílico ........................... 27 + Alcohol etílico.............................. 9 + Alcohol etílico .............................12 + Benceno .......................................91 + Benceno .......................................55 + Benceno .......................................60 + Cloroformo ...............................87,5 + Cloroformo ..................................80 + Hexano ........................................28 + Octano .........................................26 + Percloretileno ..............................19 + Sulfuro de carbono ......................66 + Sulfuro de carbono ......................91 + Tetracloruro de carbono .......... 94,5 + Tolueno ....................................86,5 + Tricloroetileno .............................64 +
Cloroformo ................................80 Agua ........ .............................. 4,4 Benceno ................................. 67,7 Tolueno .................................... 32 Hexano.......................................79 Tetracloruro carbono ..............84,2 Cloroformo............................... 93 Tric\oroetileno ......................... 73 Sulfuro de carbono ....................91 Acetona ......................................88 Agua.............................................9 Ciclohexano............................... 45 Alcohol metílico.........................40 Alcohol metílico......................12,5 Acetona .................................... 20 Cloroformo.................................72 Alcohol propílico....................... 74 Alcohol etílico ...........................81 Acetona.......................................34 Alcohol etílico .............................9 Alcohol isobutílico...................5,5 Agua.........................................13,5 Alcohol metílico ........................36
64,7 °C 78,15 °C 68,2 ° C 76,7 °C 58,6 °C 65 °C
59,4 °C 71 °C
42,4 °C 56 °C
69,3 °C 77,8 °C 58,3 °C 53,5 °C 64,7 °C 60 °C 95 °C 78 °C
39,2 °C 42,4 °C 75,8 °C 84,1 °C 60,2 °C

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 20 de 80
Mezclas azeotrópicas terciarias Punto ebullición
Alcohol ................18,5 + Benceno .............74,1 + Agua ……………7,4 64,9 ºC
Alcohol etílico ..... 4,0 + Cloroformo…….92,5 + Agua ……………3,5 55,5 ºC
Alcohol etílico …. 9,7 + Tet. Carbono….....86 + Agua ……………4,3 61,8 ºC
Alcohol propílico ..9,0 + Benceno……..82,4 + Agua ……………8,6 68,5 ºC
6.6. Utensilios para destilación en el laboratorio.
Es el sistema de destilación consistente en separar el líquido más volátil por destilación y posterior condensación, mientras el residuo o líquido de ebullición alta queda en el matraz destilador.
En el laboratorio se emplea para la obtención de agua destilada y otros componentes en los que exista gran proporción de líquido volátil en el conjunto a destilar.
Las partes esenciales de un destilador son: el matraz destilador, el refrigerante, la alargadera y el colector.
El matraz, con el líquido a destilar, puede estar calentado por gas y eléctricamente. Son de gran utilidad las mantas calefactoras de matraces que llevan una resistencia eléctrica incorporada y evitan así el contacto de vapores con la llama.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 21 de 80
El refrigerante se encarga de condensar los vapores procedentes de la destilación. La entrada del agua se efectúa por la parte inferior y su salida por a superior, con lo que se logra tener el refrigerante lleno de agua produciendo una refrigeración eficaz.
La alargadera y el colector conducen el condensado y lo depositan debidamente.
Deben emplearse asimismo, en la destilación, un termómetro para medida de la temperatura del vapor a la entrada del refrigerante; soportes, trípodes, tapones o juntas esmeriladas y otros accesorios.
6.7. Destilación de vino
El vino fundamentalmente es una mezcla de alcohol etílico-agua-azúcar y colarantes. Se destila considerándolo como una mezcla de alcohol y agua.
Se montará un aparato de destilación y se añadirán 150 c.c. de vino, calentándolo suavemente y tomando la temperatura cada minuto para obtener unos 100 c.c. de destilado; ya que se considera terminada la destilación del alcohol cuando se han obtenido los 2/3 de la mezcla. Comprobar por combustión del destilado si se trata de un compuesto inflamable como es el alcohol.
Completar la tabla y gráfica de tiempo y temperatura y estudiar qué conclusiones se san de la curva obtenida.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 22 de 80
6.8. Destilación de mezclas azeotrópicas
Como comprobación de mezclas azeotrópicas, se efectuará la destilación de acetona-c1oroformo.
Tómense 40 g de acetona (CH3COCH) y 160 g de cloroformo (CHCl3), introducidos en un matraz de destilación, como en el esquema práctico, y efectuar las conexiones necesarias. Se sustituirá el mechero Bunsen por una placa o manta calefactora eléctrica y se efectuará la destilación en vitrina de gases, ya que los vapores del destilado son inflamables.
Al cabo de unos minutos de ebullición observar la temperatura a que destila y la desviación respecto al azeótropo (64,7 °C).
Efectuar la tabla y gráfica de temperatura y tiempo.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 23 de 80
7. DESTILACIÓN A BAJA PRESIÓN.
Al vacío se destilan aquellas sustancias inestables a su temperatura de ebullición o bien de punto de ebullición muy alto.
El vacío puede llegar a menos de 1 mm de mercurio y se consigue descender la temperatura de 80 a 150 ºC, según el caso. Los equipos de destilación al vacío son algo especiales y debe procurarse tengan uniones esmeriladas para evitar cualquier entrada de aire.
7.1. Variación de la destilación con el vacío
En líneas generales, una caída de presión atmosférica de 10 mm de Hg rebaja el punto de ebullición de un líquido en 1 ºC.
Véase en la tabla la destilación de una sustancia hipotética, a varias presiones:
SISTEMA EMPLEADO Presión en mm. de Hg Punto de ebullición
Destilación simple 760 250 ºC
Trompa de agua Verano Invierno
25 15
144 ºC 134 ºC
Bomba de aceite Mala Buena Excelente
10 3 1
124 ºC 99 ºC 89 ºC
Bomba de vapor de mercurio 0,01 30 ºC
7.2. Utensilios para destilar al vacío en el laboratorio.
Los componentes de una instalación al vacío son semejantes a los de la figura:

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 24 de 80
1. Matraces, algo diferentes a los empleados en destilaciones simples, ya que deben poseer unas piezas para conectar al refrigerante, termómetro y tubo capilar. En general, para laboratorio se emplean los matraces Claisen.
2. Frasco de seguridad de Woulf y tubo capilar. El frasco de Woulf dispone de una llave para quitar lentamente el vacío y, a la vez, efectuar de colector en caso de verterse agua o gas, desde la trompa de agua o desde el frasco de destilado. El capilar penetra hasta el fondo del matraz destilador y, en el extremo opuesto, lleva una llave por donde penetran pequeñas burbujas de aire durante la destilación, a fin de evitar los saltos bruscos del líquido debido a la succión.
3. La alargadera conecta la entrada al colector con el refrigerante.
4. Los colectores especiales con separador son necesarios si se desea efectuar un fraccionamiento del destilado, recogiéndose en cada recipiente la parte que se requiera, con solo girar la llave de acoplamiento.
5. Bomba de vacío y vacuómetro que tienen la función de producir y medir el vacío deseado.
7.3. Aplicaciones de la destilación a presión reducida
Gran cantidad de productos orgánicos se impurifican en contacto con el aire o se descomponen a elevada temperatura, siendo necesario, en tales casos, destilarlos en vacío.
Se destilan en vacío el benzaldehído, butilmalonato de etilo, etc. Se concentran jugos vegetales, eliminando el agua a presión reducida a fin de evitar destrucciones de los compuestos vitamínicos y nutritivos.
7.4. Destilación de agua al vacío
Montar un equipo de destilación al vacío como se ha representado en la última figura; llenar la mitad del matraz Claisen con agua, e instalar en su interior el tubo capilar para que burbujee lentamente el aire. Comenzar la calefacción, conectar la trompa de agua para vacío en el frasco de seguridad y con el tubo central, con la llave cerrada.
Determinar a cada minuto la temperatura conseguida para construir la gráfica tiempo-temperatura. Una vez está hirviendo, variar la presión mediante la llave del frasco Woulf e ir anotando la nueva temperatura de ebulición para construir la gráfica presión-temperatura.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 25 de 80
7.5. Destilación de etilenglicol y agua al vacío
Tomar unos 30 cm3 de etilenglicol (CH20H -CH20H) o bien glicerina CH20H-CHxOH-CH20H) y mezclarlos con 100 cm3 de agua. Repetir enteramente las operaciones del caso anterior y trazar asimismo las gráficas correspondientes a tiempo-temperatura y a presión-temperatura.
Punto de ebullición a 1 atmósfera de presión
Agua Etilenglicol Glicerina
100 ºC 197 ºC 290 ºC
7.4. Destilación de zumos vegetales al vacío
En la instalación de vacío, empleada en los casos anteriores, se destilarán, mejor concentrarán, 100 c.c. de zumo de naranja, de limón o bien leche natural.
La ebullición debe llevarse a cabo de una forma suave, mediante el burbujeo constante y uniforme, a través de la punta del capilar. El agua destilará, mientras el líquido se va espesando; en cuanto se crea que se ha destilado toda el agua se abre la llave del frasco de Woulf y se da por terminada la operación cerrando la calefacción y parando la bomba de vacío.
Pesar el extracto obtenido y calcular el % sobre el inicial.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 26 de 80
9. DESTILACIÓN POR ARRASTRE DE VAPOR.
Se utiliza para la separación de sustancias insolubles en agua y de elevado punto de ebullición, destilándose a menor temperatura y evitando su descomposición. Los vapores del producto volátil son arrastrados por vapor de agua sobrecalentado o vapores de gas inerte.
El líquido hierve a menor temperatura de su punto de ebullición, ya que presión de sus vapores, junto con los del vapor de agua, sumados, vencen a la presión atmosférica y dan lugar a la destilación.
Presión atmosférica = Presión vapores del líquido + Presión vapor de agua
Así se tiene para el tolueno, la temperatura de ebullición a 1 atmósfera de presión a 110 °C, mientras que por arrastre de vapor de agua, 83 °C figura siguiente:

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 27 de 80
9.1. Instalación por arrastre de vapor
El equipo consta de:
- Generador de vapor: se efectúa en un calderín de vidrio resistente o metálico, con un indicador de nivel.
- Tubo de seguridad, que actúa de válvula de escape en caso de acumulación de vapor en su interior.
- Matraz de destilación, con el líquido a evaporar, en donde actúa el vapor de agua para arrastrar a los vapores volátiles hacia la condensación.
- Refrigeración, alargadera y colector, como en todos los destiladores.
Si se llega a condensar vapor de agua en el matraz del líquido, se calentará procurando que no llegue a hervir. Asimismo, si la sustancia solidificara en el refrigerante, se cortaría de vez en cuando el agua de refrigeración para que se fundiera el sólido.
Si los productos obtenidos son inmiscibles, se formarán capas en el matraz colector que se separarán por decantación.
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 28 de 80
9.2. Aplicaciones.
La destilación por arrastre de vapor suple, en muchos casos con ventaja, a la destilación en vacío.
Hay muchos compuestos orgánicos de punto de ebullición elevado que, con vapor de agua, destilan a menos de 100 °C.
Por ejemplo:
Mezclas de xileno y benceno.
Bromobenceno.
Anilinas.
Aceites minerales.
Tratamientos de grasas y petróleos.
Eliminación de disolventes en aceites vegetales y animales.
Separación de benceno yagua.
9.3. Destilación por arrastre de vapor de compuestos orgánicos
Efectuar una instalación, con un productor de vapor de agua, matraz de destilación, refrigerante y colector de sustancias.
Se tomarán 100 c c de líquido a vaporizar y se calentarán a unos 20 °C por debajo del punto de ebullición del componente más volátil y se pasará seguidamente una corriente de vapor
de agua. Enfriando convenientemente los vapores en el refrigerante se obtendrá en poco tiempo en el colector una separación de líquido destilado y gua (inmiscibles) que podrán decantarse para su separación final.
Sustancias para destilación por arrastre de vapor de compuestos orgánicos:
Compuesto Punto de ebullición a 1 atm. Ebullición por arrastre de
vapor a 100 ºC.
Yoduro de etilo 72 °C 64 ºC
Benceno 80 °C 69 ºC
Anilina 183 °C 96 ºC
Yodobenceno 188 ºC 98 ºC
Nitrobenceno 210 ºC 99 ºC
Naftaleno 218 ºC 99 ºC

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 29 de 80
DESTILACIÓN FRACCIONADA O RECTIFICACIÓN
1. DESTILACIÓN FRACCIONADA O RECTIFICACIÓN
El proceso de destilación más empleado en la práctica para separar entre sí líquidos volátiles es el de la rectificación; en él se hace circular el vapor en contracorriente con el líquido en un aparato, llamado columna de rectificación, que permite el contacto entre ambos.
En líneas generales, el dispositivo de rectificación consta de la columna, un calderín en su base en el que hierve continuamente una mezcla de los componentes a separar, para dar el vapor ascendente, y un condensador conectado en la parte superior, que suministra el líquido descendente. Este líquido se llama re/lujo, y resulta imprescindible para el funcionamiento de la columna. En la figura se representa esquemáticamente el dispositivo empleado en la rectificación. En lugar del condensador total podría emplearse un desflemador, obteniéndose entonces el destilado en forma de vapor.
El sistema recibe calor en el calderín y lo pierde en el condensador, mientras que la columna está aislada térmicamente del exterior.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 30 de 80
9.1. Columnas de platos.
Como ya hemos dicho, la columna de rectificación no es más que un dispositivo para lograr un contacto adecuado entre el líquido y el vapor.
Aunque preponderan las columnas de platos, descritas a continuación, va incrementándose el uso de las columnas de relleno, que estudiaremos más adelante en este capítulo.
La columna de platos consiste en una cámara cilíndrica vertical con platos horizontales separados a intervalos regulares. Cada plato está provisto de una serie de tubos cortos, de diseños variados, y cubiertos con campanas en la forma indicada en el esquema superior. El plato queda cubierto de líquido, que desborda hacia el plato inferior por uno o varios conductos. Éstos se sumergen en el líquido, que hace de cierre hidráulico. De este modo, el vapor, en su marcha ascendente por la columna, atraviesa cada plato por las cámaras formadas entre los tubos cortos y las correspondientes campanas, borboteando en el líquido.
Los numerosos diseños dados a estos dispositivos tienden a conseguir el mejor contacto entre el líquido y el vapor, y con ello la mayor aproximación al equilibrio de destilación, sin aumentar al mismo tiempo la pérdida de carga al pasar el vapor por cada plato.
En resumen: los platos de campanas suministran el contacto del líquido y el vapor y los obligan a seguir caminos determinados en su marcha a través de la columna. El mismo resultado puede conseguirse con platos de diseños diferentes

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 31 de 80
Evaporando y condensando tantas veces como sea preciso se obtiene, por la parte superior, el componente más volátil y por el inferior, el de punto de ebullición más alto.
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 32 de 80
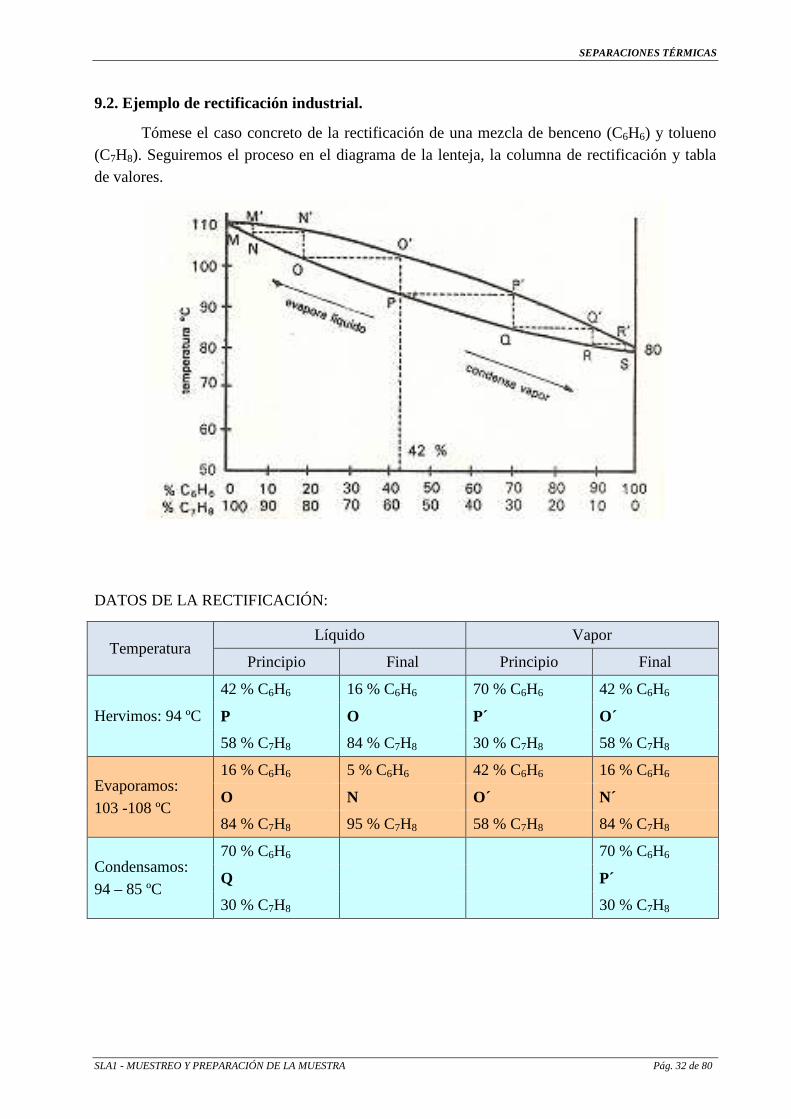
9.2. Ejemplo de rectificación industrial.
Tómese el caso concreto de la rectificación de una mezcla de benceno (C6H6) y tolueno (C7H8). Seguiremos el proceso en el diagrama de la lenteja, la columna de rectificación y tabla de valores.
DATOS DE LA RECTIFICACIÓN:
Temperatura Líquido Vapor
Principio Final Principio Final
Hervimos: 94 ºC
42 % C6H6 16 % C6H6 70 % C6H6 42 % C6H6
P O P´ O´
58 % C7H8 84 % C7H8 30 % C7H8 58 % C7H8
Evaporamos:
103 -108 ºC
16 % C6H6 5 % C6H6 42 % C6H6 16 % C6H6
O N O´ N´
84 % C7H8 95 % C7H8 58 % C7H8 84 % C7H8
Condensamos:
94 – 85 ºC
70 % C6H6 70 % C6H6
Q P´
30 % C7H8 30 % C7H8

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 33 de 80
Se procede a entrar en la columna una mezcla de 42 % de benceno y 58 % de tolueno, que empieza a hervir a 94 °C.
Al empezar a hervir, da una composición en vapor de 70 % de benceno y 30 % de tolueno (Punto P'); conforme avanza la destilación y se eleva la temperatura, el vapor recorre de P' a O' y el líquido de P a O´, por lo que, al final de esta primera evaporación se obtendrá un vapor con 42 % de benceno y 58 % de tolueno (Punto O') y un líquido con 16 % de benceno y 84 % de tolueno (Punto O).
Pero el vapor de esta primera evaporación se habrá condensado en el plato superior (al menos en parte), dando el condensado de P' el valor Q con un líquido de 70 % de benceno y 30 % de tolueno. Al destilar éste, arrastrado por los vapores ascendentes, se obtendrá un vapor Q' y así en casos sucesivos, hasta obtener por la parte superior un vapor final de aproximadamente 100 % de benceno y un líquido por la inferior de 100 % de tolueno. Todo ello gracias a las sucesivas evaporaciones y condensaciones que logran una eficacia y rendimiento muy grandes en cuanto a pureza del producto obtenido.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 34 de 80
9.3. Columnas de rectificación utilizadas en el laboratorio.
En las columnas de relleno y las Vigreux, el líquido no se mantiene en la columna, sino que condensa y cae goteando constantemente; por el contrario en las columnas Bruun de platos, el líquido llena los platos y no desciende hasta que el rebosadero está lleno.
9.4. Destilación fraccionada de vino
Efectuar un montaje de destilación fraccionada con empleo de cualquiera de las columnas reseñadas Vigreux, de platos o de relleno.
El mismo ensayo y con idénticas cantidades que en la destilación simple del vino reseñada en este tema se efectuará fraccionadamente a fin de observar su diferencia. Tomar las temperaturas cada minuto hasta recoger 100 c.c. de los 150 c.c. iniciales y construir la gráfica tiempo-temperatura.
Estudiar la curva obtenida y compararla con la curva de la destilación simple, explicando asimismo las ventajas del fraccionamiénto respecto a la destilación a presión normal.
Tampoco por este sistema puede obtenerse un alcohol de pureza superior a 96 º ya que se forma la mezcla azeotrópica de ebullición constante a unos 78 ºC.
Puede investigarse si los 100 c.c. primeros son alcohol etílico de 96º efectuando un ensayo del punto de ebullición.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 35 de 80
9.4. Destilación fraccionada de mezclas orgánicas.
Sabiendo que son factibles de destilación fraccionada cualesquiera mezclas que difieran más de 20 °C en sus puntos de ebullición, pueden ensayarse algunas sólo con consultar unas tablas de estas características.
Puede presentarse el inconveniente, como es el caso del alcohol etílico, de formarse mezcla azeotrópica, en cuyo caso destilará a concentración determinada constante, pero con pureza inferior al 100 %.
Como orientación se indican algunas mezclas susceptibles de ser rectificadas.
Tetracloruro de carbono (77 °C) - Tolueno (110°C)
Benceno (80°C) - Metanol (65 °C): Azeotropo a 58 °C.
Metanol (65°C) - Tolueno (110°C) : Azeotropo a 64 °C.
Metanol (65°C) - Agua (100 °C)
Benceno(80 °C) - Tolueno (110°C)
9.5. Fraccionamiento industrial.
Desde el punto de vista de su construcción interna, las torres de fraccionamiento industrial se clasifican en:
• Torre de platos de borboteo: Cada plato lleva un conducto para la entrada del vapor con un sombrerete para obligarle a borbotear en el líquido y un tubo para evacuar el líquido del rebosadero hacia el plato inferior.
• Torres de platos perforados: Constan de unas chapas perforadas que permiten el paso del vapor a través de sus orificios, pero no al líquido que forma un pequeño depósito sobre cada plato.
• Torres rellenas : Son cilindros verticales rellenos con material poroso para permitir que el líquido condensado escurra y se ponga en contacto con el vapor ascendente El relleno se produce con escorias, carbones, anillos Rasching de metal o de porcelana…

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 36 de 80
9.6. Destilación fraccionada del petróleo.
Como complemento a la destilación fraccionada se describe aquí el proceso de mayor importancia industrial en esta técnica, como es el fraccionamiento del petróleo en sus componentes más elementales. La práctica tiene lugar en las grandes refinerías de petróleo.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 37 de 80
El petróleo crudo, sacado de los pozos petrolíferos, es calentado antes de entrar en la torre de rectificación y, una vez en ella, separa las distintas fracciones aprovechables. La fracción líquida depositada en cada plato es conducida al Striper (recolector de porciones), donde, por arrastre con vapor de agua, se elimnan aquellos vapores más volátiles haciéndolos regresar de nuevo hacia la torre de rectificado.
El líquido obtenido en cada depósito del Striper se hace circular por un serpentín que actúa, por un lado, como refrigerante de las fracciones del petróleo y por otro, como calefactor del crudo de entrada, presentándose el sistema como un verdadero recuperador de calor.
9.6.1. Componentes del fraccionamiento del petróleo.
1. Gas de refinería. Disuelto en el crudo de petróleo, este gas se emplea en las mismas refinerías para calentar; transformándolo convenientemente puede utilizarse también como gas ciudad. Se compone de vapor de agua, CO2, N2, H2, etcétera.
2. Gases licuados. Algunos disolventes como éter, pero en especial el propano y el butano que pueden licuarse fácilmente por presión, y reciben la denominación de gases líquidos del petróleo (GLP).
3. Bencina. El punto de ebullición de este componente oscila entre 50 y 180°C. Para emplearla directamente como carburante tiene un poder antidetonante demasiado reducido para los motores de alguna potencia, por lo que se le adiciona plomo tetraetílico, especialmente. A partir de esta fracción del petróleo, se obtienen los productos para la vasta industria petroquímica
4. Queroseno. Su punto de ebullición varía entre 180 y 230°C y se emplea para aviones a reacción, estufas, lámparas, etc.
5. Aceites ligeros y gas-oil. Estas fracciones, con un punto de ebullición, que varía entre 250 y 310°C, se usan para combustible en motores Diesel y para la calefacción en los hogares.
6. Fuel-oil pesado. El punto de ebullición de esta fracción del petróleo varía entre 330 y 360°C y se emplea como combustible industrial y para la generación de corriente. Su principal inconveniente es el elevado contenido en azufre 1,5 a 2 % y como consecuencia su contaminación atmosférica.
7. Parafinas, vaselinas y alquitranes. Son fracciones difícilmente destilables y cuya aplicación se halla como lubricantes, pinturas de protección, construcción de carreteras, fabricación de cartón alquitranado, etc.
Tratando del fraccionamiento del petróleo no pueden descuidarse los productos de la petroquímica. En la actualidad, más del 90 % de los productos de la química orgánica, proceden del petróleo y se encarga la denominada petroquímica de transformarlos en los materiales de base deseados por la industria química.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 38 de 80
9.6.2. Productos petroquímicos
En la figura se relacionan los productos petroquímicos de mayor interés y su fuente de procedencia, a partir de las fracciones del petróleo.
El procedimiento que emplea la petroquímica para obtener estos productos es el craqueo
(cracking) de la fracción del petróleo comprendida entre la bencina y los gases licuados. Estos componentes formados por hidrocarburos de cadena larga, se calientan y se someten a alta presión hasta lograr la ruptura de la cadena de las moléculas obteniendo otras más cortas que, naturalmente, tienen más bajo el punto de ebullición.
Los componentes obtenidos de esta manera pueden ser separados o transformados en los productos químicos deseados, con más facilidad, en el curso de diversos procesos físico-químicos.
Los productos primarios más importantes del craqueo son gas residual, etileno, propileno, fracción C4 y gasolina cracking.
Estos productos son transformados ulteriormente en productos petroquímicos y se usan, luego, como componentes de muchos productos químicos, como plásticos, caucho sintético, fibras, colorantes, detergentes, etc.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 39 de 80

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 40 de 80
EVAPORACIÓN
1. TEORÍA DE LA EVAPORACIÓN
La evaporación consiste en la eliminación de los componentes volátiles, no deseables, de una mezcla líquida, mediante calentamiento a temperatura inferior al punto de ebullición.
En general, la separación es parcial, con lo que se obtiene una mezcla de mayor concentración. El líquido indeseable suele ser el agua que acompaña a las disoluciones.
Aunque el calor necesario para la evaporación puede suministrarse por cualquier procedimiento, lo general de esta operación a nivel industrial, es el empleo del vapor de agua saturado, como fuente calorífica.
En algunos casos la evaporación persigue una segunda finalidad, que es la obtención del disolvente, o disolvente y soluto, a la vez.
La cristalización, secado y destilación, son también operaciones básicas de separación de sustancias líquidas mediante la evaporación total o parcial de los componentes volátiles. Se diferencian unas de otras en el grado de la eliminación y, si se recuperan o no los vapores producidos, por poseer un cierto valor como tales. Estas diferencias son las que determinan el método a seguir.
En el tema presente se estudiará la evaporación como operación de separación térmica, cuyas aplicaciones más importantes son las siguientes:
o Recuperación de sustancias sólidas disueltas: sal común
o Concentración de disoluciones: zumos vegetales
o Determinación de extractos secos: leche.
2. CÁLCULOS Y DIAGRAMA DE LA EVAPORACIÓN:
2.1. Presión de vapor de una disolución
Es conocido que, al disolver un soluto volátil en un disolvente disminuye la presión de vapor. La disminución relativa de la presión de vapor es igual a la fracción molar del soluto, según la ley de Raoult:
ds
s
o
o
nn
n
p
pp
+=−
p = presión de vapor del disolvente en la disolución
po = presión de vapor del disolvente puro
ns = número de moles de soluto
nd = número de moles de disolvente

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 41 de 80
Esta disminución de la presión de vapor del disolvente en una disolución aumenta gradualmente el punto de ebullición de ésta.
2.2. Aumento del punto de ebullición
Mientras en un líquido puro la temperatura se mantiene constante durante la evaporación (si no varía la presión), en el caso de una disolución el punto de ebullición aumenta gradualmente al concentrarse, dependiendo de la cantidad de soluto en la disolución.
La elevación del punto de ebullición es proporcional a la molaridad (m), según la fórmula de la ebulloscopía:
MG
gKmKT eee ⋅
⋅⋅=⋅=∆ 1000
Ke = cte. de ebulloscopía (para el agua 0,52 °C por mol)
g = gramos de soluto
G = gramos de disolvente
M = peso molecular del soluto
∆Te = incremento de temperatura de ebullición, °C
Si la disolución es iónica, la ecuación debe ir afectada de un coeficiente i, que representa el número de iones disociados del soluto:
MG
gKmiKT eee ⋅
⋅⋅=⋅⋅=∆ 1000
En los evaporadores, este aumento del punto de ebullición de la disolución una vez se concentra, requiere una aportación mayor de calor o bien provocar un vacío a la salida.
2.3. Regla de Dühring
Aunque, por aplicación de la ley de Raoult y de la ebulloscopía, se puede determinar la temperatura a que hervirá una disolución de concentración dada, es más cómodo y, en la práctica, así se hace: utilizar la regla de Dühring.
Esta regla es empírica y se basa en que la temperatura de ebullición de una disolución a
una presión dada, es proporcional a la temperatura de ebullición del disolvente a la misma
presión:
-

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 42 de 80
Kteconsdisoluciónebulliciónatemperaturdiferencia
líquidoebulliciónatemperaturdediferencia == tan
Ktt
tt =−−
12
12
´´ )´´( 1212 ttKtt −⋅=− ecuación lineal
Es decir, que llevados dichos valores a un sistema de ejes de coordenadas, los puntos representativos se encuentran en una misma recta para cada concentración.
El diagrama se construye, pues, sólo con conocer para cada concentración dos valores del punto de ebullición de la disolución a dos presiones dadas y uniendo ambos puntos con una recta. Evidentemente, cuando la concentración en soluto es 0 % (agua pura) esta recta tendrá una inclinación de 45º, si las escalas de los ejes son iguales.
2.4. Diagrama de Dühring
Constrúyase seguidamente el diagrama de Dühring para una disolución de NaCl, cuyo grado aparente de disociación lo fijaremos en un 80 %.
NaCl Na+ + Cl- 0,80 0,80
Algunos de los puntos del diagrama son:
- Agua pura: punto de ebullición a 760 mm Hg, 100 ºC
- Disolución al 20 % de Na CI:
CTe º36,35,5880
100020280,052,0 =
⋅⋅⋅⋅⋅=∆
Punto de ebullición: 103,36 °C
- Disolución al 40 % de Na CI:
CTe º12,95,5860
100040280,052,0 =
⋅⋅⋅⋅⋅=∆
Punto de ebullición: 109,12 °C y así sucesivamente.
Tabulando los resultados, se tendrá la tabla siguiente:
Composición Agua NaCl NaCl NaCl NaCl NaCl 20% 40% 60% 80% 90%
Punto de
ebullición 100 103,36 109,12 121 154,7 223,2

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 43 de 80
Estos valores no se cumplen en la práctica exactamente, puesto que el grado de disociación de la sal varía con la concentración, y en el ejemplo se ha supuesto constante.
Constrúyase la recta del agua y, paralelas a ellas, las rectas de ebullición de la disolución de NaCl a distintas concentraciones tal como indica el diagrama para las disoluciones acuosas de NaCl.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 44 de 80
En la figura siguiente se representa el diagrama de Dühring para disoluciones acuosas de NaOH.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 45 de 80
3. SISTEMAS DE EVAPORACIÓN EN EL LABORATORIO
3.1. Al aire libre
Se lleva a cabo en recipientes de gran superficie y poco fondo, como cristalizadores y cápsulas, ya que la evaporación depende directamente de la superficie:
Se denomina volatilización y tiene poca aplicación en el laboratorio, utilizándose en todo caso como operación de secado.
Industrialmente constituye el sistema de salinas para obtener y purificar la sal común, a partir del agua del mar.
La evaporación del agua destilada al aire libre sigue la gráfica que se ha representado en la figura:
3.2. Mediante calor
El sistema más empleado en el laboratorio para llevar a cabo las evaporaciones es por acción del calor, sin llegar a la ebullición del líquido. Se efectúa a fuego lento con mechero Bunsen y cápsulas de porcelana o en baños María o de arena, teniendo como finalidad la de concentrar llevando a sequedad un producto de análisis:
Las cápsulas o "cristalizadores empleados deben cubrirse con un vidrio de reloj o un embudo, a fin de que no penetre polvo en el interior de la disolución.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 46 de 80
3.3. En vacío.
Constituye el sistema más rápido de llevar a cabo las evaporaciones, ya que al disminuir la tensión de vapor de su superficie se logra un desprendimiento de moléculas más rápido y eficaz. Está ampliamente extendido para sustancias que no deben llegar a hervir, llamadas termolábiles, como son la evaporación de leche, jugos de frutas, extractos de carne, soluciones de nicotina, ácido láctico, etcétera. .
Existen, actualmente, varios dispositivos para ello, pudiéndose efectuar en una cámara de vacío conectada a la bomba de aceite, o a la trompa de agua.
Modernamente se utilizan los evaporadores rotativos de vacío (Rota-vapor), los cuales, además de evaporar intensamente, permiten recoger el producto de la evaporación. Consisten en un balón, que gira accionado por un motor eléctrico y un engranaje, en el cual se sitúa el líquido a evaporar y un refrigerante a su salida, donde se condensan los vapores, recogiéndose en un colector. La toma de vacío se efectúa desde la trompa de agua o a partir de una bomba de aceite.
En este sistema rotativo, el efecto de la rotación crea una superficie de evaporación que abarca todo el balón evaporador, lo cual triplica en todos los casos su eficacia.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 47 de 80
4. EVAPORACIÓN INDUSTRIAL:
Los evaporadores industriales funcionan generalmente por calefacción con vapor de agua, que cede su calor y se condensa, mientras la mezcla líquida eleva su temperatura, se evapora y concentra.
4.1. Clasificación
Los evaporadores se pueden clasificar según los distintos tipos de soluciones (grado de viscosidad) y según el método de calefacción pero, en general, todos los evaporadores tienen unas características comunes:
En cuanto al recinto de calefacción, generalmente el fluido calefactor es vapor de agua, a baja presión y saturado (se le llama vapor vivo). No tiene importancia el recalentamiento del vapor, puesto que la salida es de agua condensada, a la misma temperatura y presión que el vapor y lo que cede en la transmisión de calor, es el calor de condensación. Se utilizan otros fluidos, como sustancias orgánicas que funden a una determinada temperatura, que interesa en el proceso.
En la figura siguiente se representa el esquema básico de un evaporador y el esquema simbólico con las entradas y salidas de materia en el mismo. En la primera se trata del esquema de un evaporador industrial y la segunda es la representación esquemática del proceso.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 48 de 80

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 49 de 80
4.2. Evaporadores sencillos
Consisten en recipientes corrientemente semiesféricos provistos de un doble fondo para la circulación del vapor de calefacción y se utilizan en procesos en los que la evaporación es débil o cuando el consumo de vapor no influye en el costo del producto, ya que el rendimiento es muy bajo por la poca superficie de calefacción que presenta:
Pueden ser abiertos o cerrados, según que haya de trabajarse a presión atmosférica o al vacío. Se utilizan en la industria cervecera, preparación de ciertas proteínas, leche condensada, jugos de frutos, etc. En general, se trabaja al vacío para evitar la alteración de los productos delicados. En algunos casos, para acelerar la operación, van dotados de agitadores. El doble fondo puede ser sustituido por serpentines de calefacción.
4.3. Evaporador de tubos horizontales
La misión de los tubos es aumentar la superficie de calefacción y, con ello, la capacidad de evaporación. En el sistema de tubos horizontales la solución se concentra en el exterior de los tubos, mientras el vapor condensa dentro; se emplea para líquidos poco viscosos, que no formen espuma ni incrustaciones:

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 50 de 80
4.4. Evaporadores tubulares verticales (evaporador Kestner).
Los evaporadores verticales han desplazado a los de haz horizontal por su mayor rendimiento y facilidad de acceso para su limpieza. Uno de los de mayor aplicación es el evaporador Kestner.
Está formado por un haz de tubos de 3 a 7 m de longitud alejados verticalmente dentro del cuerpo del evaporador.
El líquido a evaporar entra por la parte inferior y al producirse una evaporación parcial del mismo, la mezcla líquido-vapor, que tiene una densidad menor que la de la solución de la alimentación, asciende por los tubos, en los cuales la evaporación progresa hasta desembocar en la parte superior. Mediante una pantalla reflectora, se rompen las espumas separando el líquido del vapor. Esta separación puede completarse por medio de un ciclón.
Este evaporador tiene el inconveniente de ser de difícil limpieza, por lo que sólo se utiliza para productos de poca viscosidad o que no depositen sales.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 51 de 80
4.5. Evaporadores de múltiple efecto
Los vapores que salen del evaporador llevan una cantidad importante de calor que puede emplearse en otro aparato similar, en el que se suele tratar también la misma disolución. Esta asociación es lo que se llama evaporación en doble, triple o múltiple efecto, según el número de aparatos reunidos. En la práctica no se suele pasar de 3 ó 5 evaporadores asociados.
En la figura se representa un triple efecto con circulación directa.
El líquido a concentrar penetra por el primer evaporador y pasa sucesivamente por los otros dos, aumentando su concentración, con lo que el punto de ebullición aumenta, disminuyendo el poder de calentamiento del vapor que recibe del efecto anterior. Para mantener la necesaria diferencia de temperaturas se reduce convenientemente la presión en cada uno de los efectos siguientes.
La operación se inicia abriendo la llave del vapor en el primer evaporador y se pone en marcha la bomba de vacío para mantener una temperatura constante en el último efecto. Sucesivamente, en cada aparato se calienta la solución, se evapora, y vapores y concentrado pasan al efecto siguiente.
En este tipo de alimentación directa el líquido fresco entra en el primer efecto, que es el que se encuentra a mayor temperatura y presión, y sale, ya concentrado, por el último. Tiene la ventaja de que no se precisan más que dos bombas: una, para la alimentación del primer evaporador y otra, para extraer el concentrado por el último. Se utilizan para concentración de productos de no muy elevada viscosidad y cuando se precisa alcanzar una fuerte concentración, como en la industria azucarera.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 52 de 80
En la figura se representa un triple efecto con circulación en contra corriente.
El régimen de temperaturas en un evaporador de triple efecto se establece al cabo de algún tiempo de funcionamiento, manteniéndose la temperatura y presión constantes en cada evaporador, tal como se representa en la figura siguiente.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 53 de 80
HUMIDIFICACIÓN Y SECADO
1. ESTADO HIGROMÉTRICO DEL AIRE.
1.1. Aire húmedo
El aire, a cada temperatura, disuelve una cantidad determinada de agua; cuando existe más de la que puede disolver, el exceso se precipita en forma de gotas.
El aire se llama saturado de humedad, cuando ya no admite más cantidad de agua.
1.2. Humedad relativa y estado higrométrico
Se denomina humedad relativa del aire la cantidad de agua en forma de vapor que contiene, respecto a la que podría tener, si estuviera saturado.
saturadodoescontendríaqueaguadevaporPeso
CtaairedekgcontienequeaguadevaporPesorelativaHumedad
tan
º1=
El aire hasta 50 % de humedad relativa se llama seco; de 50 a 80 % húmedo y más del 80 % fuertemente húmedo, hasta llegar al 100 % o saturado.
1.3. Psicrómetro
Consta, en esencia, de dos termómetros iguales: uno, cuyo depósito va envuelto por una tela constantemente mojada con agua destilada, termómetro húmedo; y el otro, denominado seco:
Cuanto más seca esté la atmósfera, mayor será la diferencia entre las dos temperaturas y, si el aire fuera saturado, marcarían igual.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 54 de 80
1.4. Presión actual
Mediante las lecturas de los dos termómetros se puede calcular la presión de vapor contenido en el aire, que se llama presión actual.
´)(´ ttHApp ma −⋅⋅−=
pa = presión actual en mm de mercurio. p m = presión de saturación del vapor de agua a t´ ºC (tabla). t = temperatura marcada por el termómetro seco. t´ = temperatura marcada por el termómetro húmedo.
A = constante. Para temperaturas entre 0 ºC y 40 ºC; A = 0,5/760. H = presión atmosférica en mm de mercurio.
Tabla presiones de vapor de agua
Temp. ºC
P. vapor mm Hg
Temp. ºC
P. vapor mm Hg
Temp. ºC
P. vapor mm Hg
Temp. ºC
P. vapor mm Hg
-20 -15 -10 -5 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
0,8 1,2 2,0 3,0 4,57 4,92 5,29 5,68 6,10 6,54 7,01 7,51 8,04 8,60 9,20 9,84 10,51 11,23 11,98 12,78 13,63 14,53 15,47 16,47 17,53 18,65 19,82 21,06 22,37
25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52
23,75 25,20 26,73 28,34 30,04 31,82 33,69 35,66 37,72 38,89 42,17 44,56 47,06 49,69 52,44 55,32 58,34 61,50 64,80 68,26 71,88 75,65 79,90 83,71 88,02 92,51 97,20 102,09
53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80
107,20 112,51 118,04 123,80 129,82 136,08 142,60 149,38 156,43 163,77 171,38 179,31 187,54 196,09 204,96 214,17 223,73 233,70 243,90 254,60 265,70 277,20 289,10 301,40 314,10 327,30 341,00 355,10
81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 102 104 106 108 110 112 114 116
369,70 384,90 400,60 416,80 433,60 450,90 468,70 487,10 506,10 525,76 546,05 567,00 588,60 610,90 633,90 657,62 682,07 707,27 733,24 760,00 815,86 875,06 937,92 1004,40 1074,60 1148,70 1227,20 1309,90

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 55 de 80
1.5. Humedad relativa
El psicrómetro da Pa, para calcular el estado higrométrico se efectúa:
Humedad relativa:
100×=m
a
p
pϕ
pa = presión actual en mm de mercurio. pm = presión de saturación a t ºC que nos da el termómetro seco. φ = % de humedad relativa.
1.6. Punto de rocío
Es aquella temperatura en la que el aire tiene una humedad relativa del 100 %. Matemáticamente es la temperatura a que se debería descender para que la presión actual, determinada por el psicrómetro, fuera idéntica a la de saturación.
La similitud del punto de rocío es bien patente con el llamado rocío de la mañana que aparece al amanecer en forma de gotitas de agua sobre las plantas. Ello se debe a que el aire, durante el día contenía una cierta cantidad de vapor, que durante la noche, al descender la temperatura, ha saturado el ambiente cayendo en forma de fina lluvia.
1.6. Humedad relativa y bienestar personal
La humedad relativa del aire, en un momento dado, está íntimamente ligada con nuestro bienestar personal como se muestra en la figura:

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 56 de 80
La evaporación de la humedad de la superficie de nuestro cuerpo es un proceso de enfriamiento; basta comprobarlo al salir de la ducha. En los días calurosos de verano el aire está casi saturado de humedad; entonces la evaporación es muy lenta y se experimenta la sensación de malestar, como de calor pegajoso. Si, en cambio, la humedad relativa es baja, la evaporación es rápida y nos sentimos frescos a pesar de la elevada temperatura.
La influencia decisiva de la humedad y temperatura en muchas producciones industriales, especialmente en el tratamiento de fibras textiles, hace que estos parámetros deban determinarse constantemente, empleando para ello el termohigrógrafo. Este aparato registra constantemente aquellos valores en una gráfica que suele cambiarse con periodicidad semanal.
1.7. Determinación práctica del estado higrométrico en el interior del laboratorio
Utilizando el psicrómetro y el barómetro tomar las condiciones ambientales del laboratorio: temperatura del termómetro seco, del termómetro húmedo y presión atmosférica.
Consultar la tabla de vapor de agua y a partir de ella y las fórmulas del psicrómetro hallar el % de humedad relativa:
´)(´ ttHApp ma −⋅⋅−=
Humedad relativa: 100×=m
a
p
pϕ
pa = presión actual en mm de mercurio. p m = presión de saturación del vapor de agua a t´ ºC (tabla). pm = presión de saturación a t ºC hallada por el termómetro seco. t = temperatura marcada por el termómetro seco. t´ = temperatura marcada por el termómetro húmedo.
A = constante. Para temperaturas entre 0 ºC y 40 ºC; A = 0,5/760. H = presión atmosférica en mm de mercurio.
Comprobar si los resultados se ajustan a lo dado por la tabla siguiente incluida en los psicrómetros. Pasar los datos a la gráfica de condiciones ambientales, justificando en qué zona se halla el ambiente atmosférico del laboratorio en aquellos momentos.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 57 de 80
2. HUMIDIFICACIÓN.
Es el proceso de la evaporación del agua dentro de un gas, que generalmente es el aire:
La humidificación se logra poniendo el aire en contacto con agua, en condiciones tales que alcance la saturación a una determinada Tª correspondiente a una determinada humedad.
2.1. Métodos de humidificación
- Calentar el agua hasta la temperatura para la cual la humedad del aire sea la requerida. Por ejemplo, a partir de agua a 8 ºC se calienta hasta 30 ºC para obtener una humedad del 60 %.
- Se calienta el aire hasta temperatura adecuada y luego se humidifica. Los sistemas de humidificación adquieren su máxima aplicación en el acondicionamiento de aire; necesitando de un proceso de humidificación en invierno, falto de humedad, y de deshumidificación en verano, exceso de vapor de agua en la atmósfera.
2.2. Deshumidificación
Es menos corriente que la humidificación y sólo se presenta cuando se trata de acondicionar el aire en países cálidos o húmedos, generalmente en verano.
Para la deshumidificación es necesario enfriar hasta la temperatura para la cual la humedad del aire sea la requerida. Se consigue por circulación del aire en contacto con un serpentín que contenga el refrigerante adecuado o por contacto con agua enfriada previamente.
2.3. Acondicionamiento de aire
Se entiende por acondicionamiento de aire en un determinado local o edificio, la conservación por medios artificiales de los factores atmosféricos ambientales: temperatura, humedad y pureza dentro de los límites del bienestar.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 58 de 80
Se trata de acondicionar un local, humidificándolo adecuadamente y calentándolo hasta condiciones de máximo bienestar.
La instalación consta de un aspirador de aire fresco, unos elementos calefactores y unos inyectores de agua pulverizada. La pulverización del agua arrastra polvo que, junto con las gotitas de aquélla, es precipitado en una serie de obstáculos colocados delante del pulverizador; así, el aire caliente y húmedo es posteriormente calentado hasta su temperatura final para ser enviado al local objeto del acondicionamiento.
2.4. Estufas de humidificación para laboratorios
Empleadas para aquellos materiales que deben ensayarse a temperaturas y humedad elevados, generalmente materiales de la construcción.
Debajo de la estufa se crea un ambiente saturado de humedad mediante unas resistencias que calientan y evaporan el agua que procede de un depósito contiguo. Este aire, cargado de humedad, recorre a través de las bandejas de la estufa, succionado por un aspirador instalado en la parte superior de la misma.
Se miden la temperatura y el estado higrométrico instalando en la parte superior de la estufa, dos termómetros, uno seco y otro húmedo, bañado en una cubeta de agua destilada con nivel constante.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 59 de 80
3. SECADO.
Consiste en eliminar el agua o líquidos volátiles de las sustancias sólidas y gaseosas. Se diferencia de la ebullición y evaporación en que el secado se efectúa en corriente de aire que arrastra el líquido evaporado. En los secaderos, el sólido húmedo desprende vapor hacia la atmósfera, saturando a ésta finalmente, lo que requiere una deshumidificación o renovación periódica del aire circulante.
El tipo de secado más rutinario es extender el material al aire libre y, con ayuda del calor solar, evaporar el agua que contenga; se emplea aún para secado de cereales, bacalao, madera, carnes, etc.
3.1. Ventajas y aplicaciones del secado
La aplicación de esta operación básica en gran cantidad de industrias se justifica por:
� Manejo más fácil del producto seco.
� Reducción del peso.
� Menor coste del transporte.
� Preservación de los productos durante el almacenamiento.
Algunas aplicaciones específicas del secado son:
� Industria de cerámica, papeleras, pinturas, productos alimenticios, forrajes, frutos, pescados, carnes, etc.
� En el laboratorio: humedades de carbón, yesos, productos químicos, deshidrataciones de sales químicas, secado de gases, etc.
3.2. Secado de sólidos en el laboratorio a temperatura ambiente:
De poca aplicación, sólo se utiliza en aquellas comprobaciones que deben reflejar el estado del material: secado de yeso, pastas, carbones, algunos productos químicos, etc. Se colocan sobre un papel o vidrio de reloj y se exponen al aire libre hasta no variar su peso.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 60 de 80
3.3. Secado de sólidos en el laboratorio en estufas de secado:
Se coloca el material pesado en un vidrio de reloj o pesasustancias y se introduce en las bandejas de la estufa. Esta debe tener control automático de temperatura por termostato para así ajustarla a las necesidades del material y durante el tiempo que convenga.
Se construyen estufas para vacío y con atmósfera de gases inertes para los casos de materiales que requieran estas condiciones.
3.3.1. Secado de sustancias sólidas.
Se efectuarán ensayos para sacar la humedad del carbón, tierra mojada, granos, sustancias verdes, etc., manteniéndolos en una estufa de secado a la temperatura indicada hasta constancia de peso según el esquema representado a la derecha de esta página.
Si se efectúan ensayos para hallar la humedad del yeso (CaSO4·2 H20) deberá mantenerse sólo unas 2 horas a 50°C, debido a la facilidad de perder el agua de cristalización, que luego se contabilizaría como humedad.
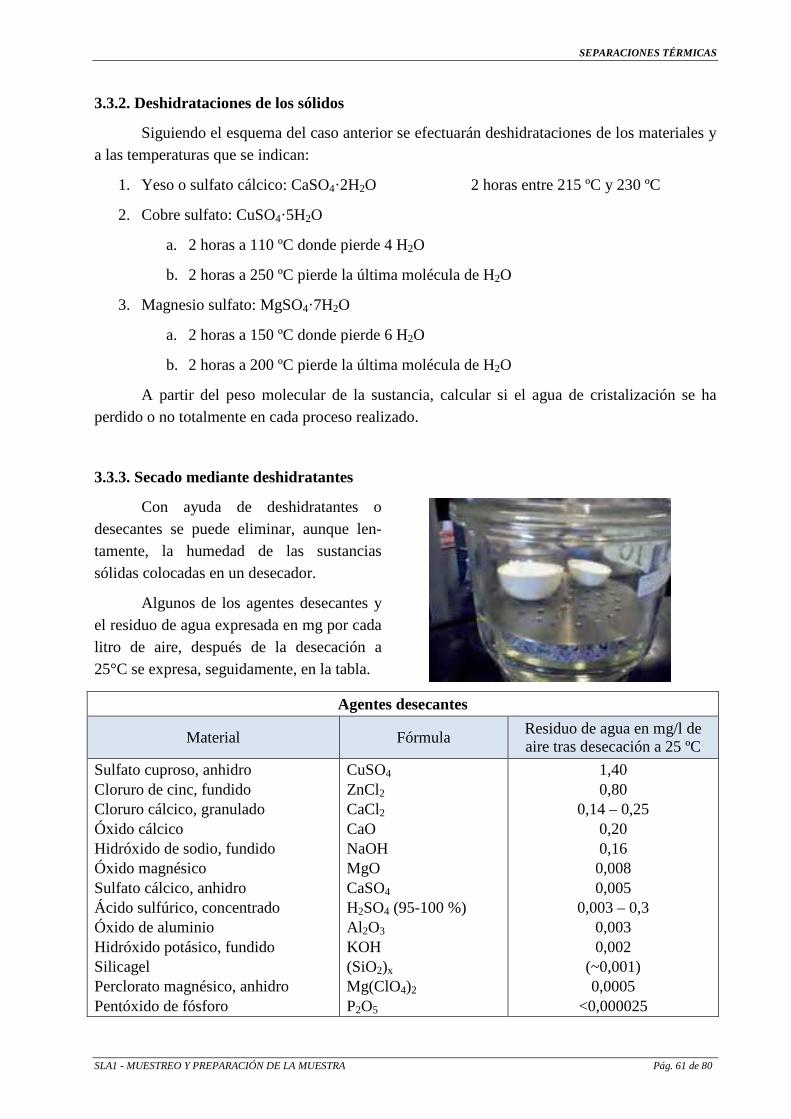
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 61 de 80
3.3.2. Deshidrataciones de los sólidos
Siguiendo el esquema del caso anterior se efectuarán deshidrataciones de los materiales y a las temperaturas que se indican:
1. Yeso o sulfato cálcico: CaSO4·2H2O 2 horas entre 215 ºC y 230 ºC
2. Cobre sulfato: CuSO4·5H2O
a. 2 horas a 110 ºC donde pierde 4 H2O
b. 2 horas a 250 ºC pierde la última molécula de H2O
3. Magnesio sulfato: MgSO4·7H2O
a. 2 horas a 150 ºC donde pierde 6 H2O
b. 2 horas a 200 ºC pierde la última molécula de H2O
A partir del peso molecular de la sustancia, calcular si el agua de cristalización se ha perdido o no totalmente en cada proceso realizado.
3.3.3. Secado mediante deshidratantes
Con ayuda de deshidratantes o desecantes se puede eliminar, aunque len-tamente, la humedad de las sustancias sólidas colocadas en un desecador.
Algunos de los agentes desecantes y el residuo de agua expresada en mg por cada litro de aire, después de la desecación a 25°C se expresa, seguidamente, en la tabla.
Agentes desecantes
Material Fórmula Residuo de agua en mg/l de aire tras desecación a 25 ºC
Sulfato cuproso, anhidro Cloruro de cinc, fundido Cloruro cálcico, granulado Óxido cálcico Hidróxido de sodio, fundido Óxido magnésico Sulfato cálcico, anhidro Ácido sulfúrico, concentrado Óxido de aluminio Hidróxido potásico, fundido Silicagel Perclorato magnésico, anhidro Pentóxido de fósforo
CuSO4 ZnCl2 CaCl2 CaO NaOH MgO CaSO4 H2SO4 (95-100 %) Al 2O3 KOH (SiO2)x Mg(ClO4)2 P2O5
1,40 0,80
0,14 – 0,25 0,20 0,16 0,008 0,005
0,003 – 0,3 0,003 0,002
(~0,001) 0,0005
<0,000025

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 62 de 80
3.4. Secado de líquidos en el laboratorio
3.4.1. Mediante deshidratantes
Se mantiene el líquido varias horas, o incluso días, en contacto con el producto secante agitándolo, de vez en cuando, y separándolos final-mente por filtración y destilación. Con ello se habrá perdido gran parte del agua que contenía. Los deshidratantes son cloruro cálcico, pentóxido de fósforo y sulfato magnésico.
3.4.2. Empleo de tamices moleculares
Los tamices moleculares son ceolitas sintéticas cristalinas, con numerosas oquedades que comunican entre sí mediante poros de radio constante (3, 4, 5 Á, etc.). Si se elimina calentando el agua contenida en las oquedades o poros, se obtiene un material muy empleado como adsorbente. Adsorben las moléculas de sustancia cuyos diámetros son inferiores a los poros, quedando separadas del resto que constituyen el material a separar.
Su principal aplicación es en desecación de disolventes orgánicos haciéndolos pasar por columnas rellenas con el tamiz en forma de perlas o barritas. El agua queda retenida y pasan las moléculas del disolvente con un % de humedad prácticamente despreciable.
Los tamices moleculares se pueden regenerar tantas veces como se quiera, sin perder su capacidad de adsorción. Se realiza a unos 300-350 °C y en corriente de gas seco o mejor en vacío. Luego se conservan en sitio totalmente deshumidificado.
Algunos disolventes orgánicos en que es corriente el empleo de tamices moleculares para su deshidratación, se relacionan en la tabla siguiente:

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 63 de 80
Disolventes orgánicos
Disolvente Tamiz requerido
Acetona Benzol Carbono tetracloruro Ciclohexano Cloroformo Dioxano Etanol Éter etílico Metanol Xilol
3 Å 4 Å 4 Å 4 Å 4 Å 4 Å 3 Å 4 Å 3 Å 4 Å
3.5. Secado de gases en el laboratorio
El gas circula a través de la torre, rellena del producto deshidratante y, finalmente, se hace burbujear con ácido sulfúrico.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 64 de 80
3.5.1. Práctica del secado de gases.
Efectúese una instalación como indica el esquema de la figura de la página siguiente:
- Carga de caliza, mármol u otra variedad de carbonato cálcico (CaC03) en la bola inferior del Kipp.
- Agua destilada en la botella de lavado.
- Pentóxido de fósforo (P20S) en la torre de secado.
- Gel de sílice (Si02) en la torre de comprobación de humedad.
El aparato de Kipp funciona del siguiente modo:
Se añade ácido clorhídrico 1:1 por el tubo de seguridad, manteniendo la llave del gas cerrada y hasta que cubra al mineral. Cuando el desprendimiento de gas carbónico (C02) sea abundante abrir la llave que dará paso al borboteo del gas en el frasco de lavado y posterior circulación a las torres de secado de gel de sílice.
El gas saldrá purificado y seco mientras la torre con gel de sílice mantenga su color azul debiéndose cambiar el pentóxido de fósforo cuando aquella cambie a rosado.
El gel de sílice rosado se secará en estufa a unos 80 °C y podrá utilizarse de nuevo una vez haya tomado su vivo color azul.
El aparato de Kipp tiene la ventaja de que, cerrando la llave de salida del gas se para el desprendimiento del mismo, puesto que la misma presión obliga a descender el reactivo del mineral en contacto y ascender por el tubo central.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 65 de 80
4. SECADO EN LA INDUSTRIA
Los aparatos industriales de secado se proyectan teniendo en cuenta:
- Las sustancias a tratar:
• Granular: cereales.
• Pastosa: pinturas.
• Fibrosas: carnes. - El sistema de calefacción
• Aire precalentado.
• Vapor de agua.
• Gases de combustión de hornos.
• Rayos infrarrojos.
• Inducción eléctrica. Construcción de los secaderos
Teniendo en cuenta las anteriores particularidades, los secaderos se construyen:
- Para sólidos:
• De túnel.
• De bandejas.
• Rotativos.
- Para líquidos.
• Por pulverización. - Para gases
• Cilindros con regeneración.
4.1 Secaderos de aire caliente
4.1.1. De túnel para sólidos
Es un secadero de tipo continuo en el que el material a secar se pone en contacto con el aire caliente que circula en contracorriente. Las vagonetas, en su movimiento, ponen en contacto el material con el aire seco de entrada. Son muy utilizados para deshidratar frutos, legumbres y productos químicos.
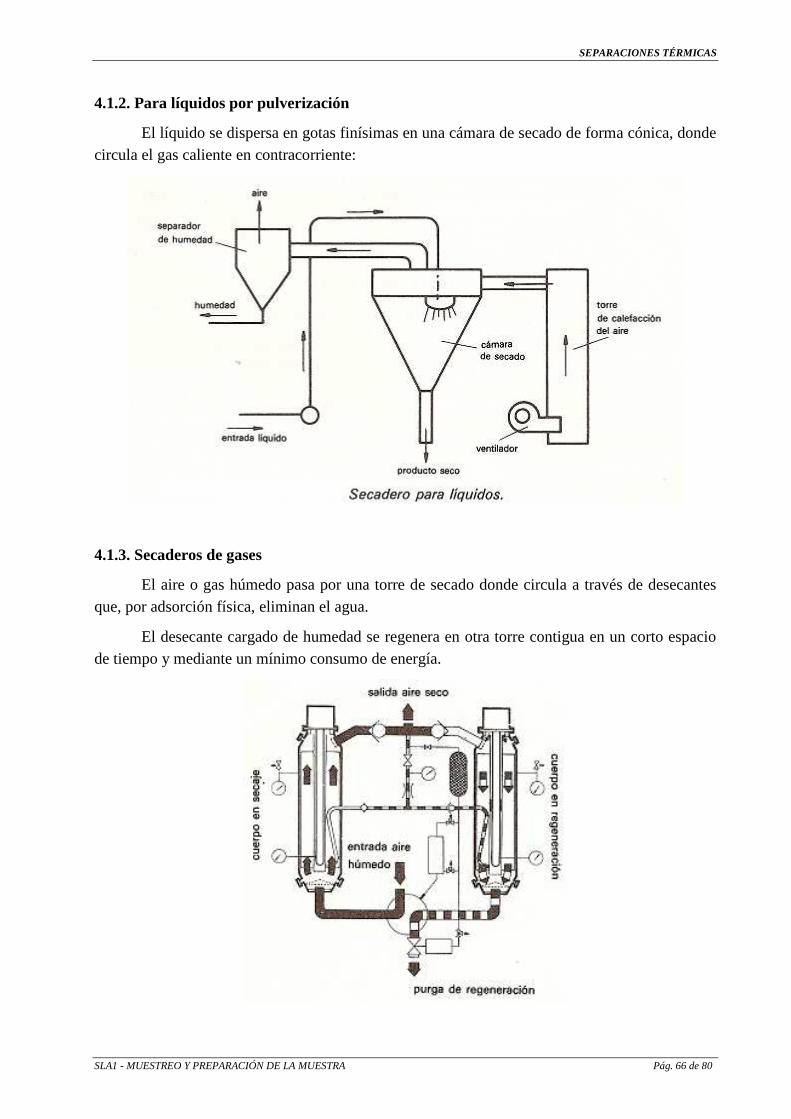
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 66 de 80
4.1.2. Para líquidos por pulverización
El líquido se dispersa en gotas finísimas en una cámara de secado de forma cónica, donde circula el gas caliente en contracorriente:
4.1.3. Secaderos de gases
El aire o gas húmedo pasa por una torre de secado donde circula a través de desecantes que, por adsorción física, eliminan el agua.
El desecante cargado de humedad se regenera en otra torre contigua en un corto espacio de tiempo y mediante un mínimo consumo de energía.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 67 de 80
4.1.4. Secado por infrarrojos
Emplean generalmente el sistema de túnel con desplazamiento de los cuerpos a desecar, arrastrándolos por una banda deslizante de forma que vayan pasando bajo las lámparas de incandescencia productoras de las radiaciones infrarrojas según el esquema de la figura. Estas lámparas van equipadas con reflectores para dirigir la radiación sobre los cuerpos húmedos.
El sistema posee mayor rapidez que la desecación por aire caliente. Las radiaciones infrarrojas empleadas son ondas de gran poder de penetración y van desde 8.000 Á a 20,000 Á de longitud de onda (1 Á = 10-8 cm).
4.2 Empleo
Se emplean en secado y tostación de harinas, galletas, frutos, pescados, carnes, pinturas de automóviles y bicicletas, torrefacción del café y otras.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 68 de 80
CRISTALIZACIÓN
1. CRISTALIZACIÓN EN EL LABORATORIO
En líneas generales, la cristalización tiene por objeto obtener un compuesto en la forma de sólido cristalino partiendo de la sustancia en disolución, fundida o en fase de vapor. Prácticamente tiene interés la cristalización a partir de disoluciones químicas.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 69 de 80
La condición principal para que se realice la cristalización de una sustancia es que su solubilidad, en un determinado disolvente, varíe considerablemente con la temperatura, tal y como se ve en la gráfica siguiente. Si se trata de cristalizar una sustancia pura se logra enfriándola por debajo de su punto de fusión:
AGUA -----------------� HIELO (enfriando)
La segunda condición para que se realice la cristalización es que la disolución se halle saturada del compuesto a cristalizar. Por eso, cuando se parte de una disolución, debe saturarse o mejor sobresaturarse y posteriormente enfriarse
Na2SO4 saturado -------------� Na SO4 •. 10 H20 cristalizado
En la práctica industrial, además de enfriar una disolución, es conveniente evaporar parte del disolvente.
2. Cuerpos cristalinos y amorfos
Un cristal es un sólido que presenta una estructura poliédrica bien definida; sus átomos, iones o moléculas se ordenan en el espacio dando lugar a una red cristalina.
Tanto en un mineral como en una sal química es difícil, a veces, observar exteriormente su estructura cristalina, ya que resulta alterada por las impurezas, agregados cristalinos, formación de maclas (asociación de cristales), etc. Hay 32 clases de cristales agrupados en 7 sistemas cristalinos, que se caracterizan por tener constantes sus ángulos y caras.
Las variedades cristalinas se estudian en Cristalografía y en este tema se representan las variedades fundamentales ordenadas en sistemas de cristalización:
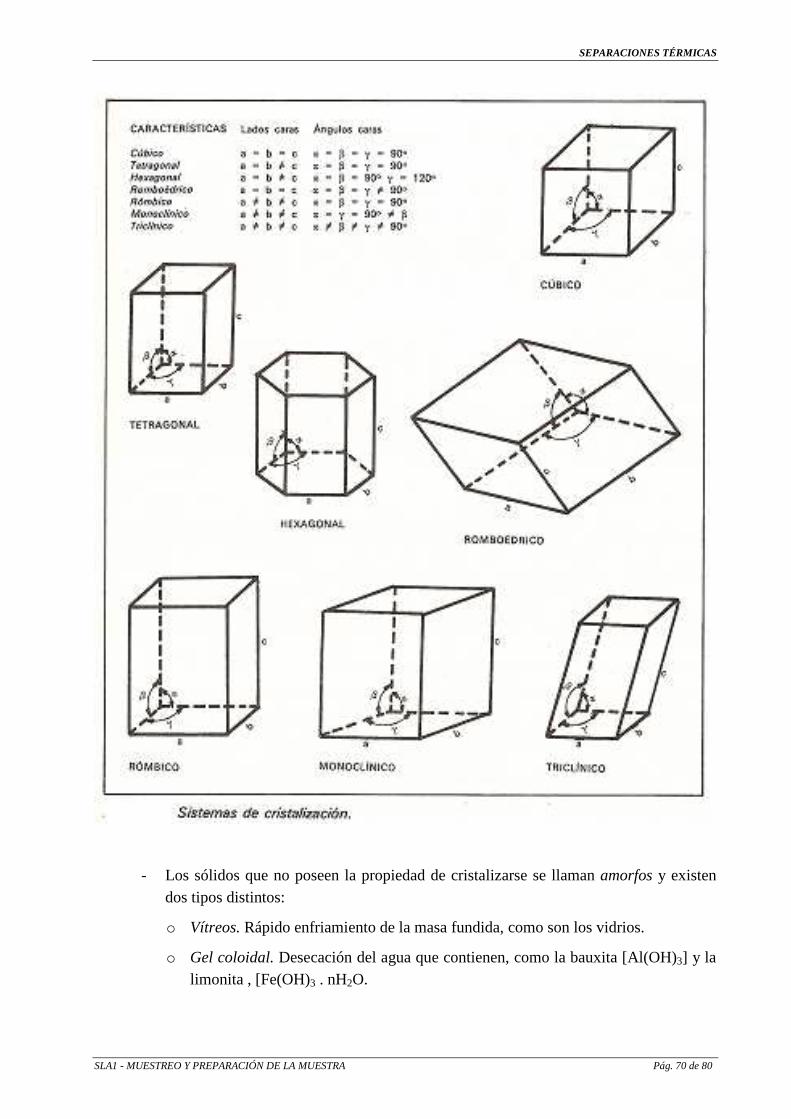
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 70 de 80
- Los sólidos que no poseen la propiedad de cristalizarse se llaman amorfos y existen dos tipos distintos:
o Vítreos. Rápido enfriamiento de la masa fundida, como son los vidrios.
o Gel coloidal. Desecación del agua que contienen, como la bauxita [Al(OH)3] y la limonita , [Fe(OH)3 . nH2O.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 71 de 80
3. FORMACIÓN DE NÚCLEOS CRISTALINOS Y CRECIMIENTO D E LOS CRISTALES.
Seguir el proceso en la figura:
Una vez enfriada la sustancia sobresaturada, el proceso de cristalización se realiza en dos etapas:
3.1. Formación de núcleos o gérmenes cristalinos
Es una agrupación de átomos, iones o moléculas (en número de 10 a 1000 que, al enfriarse, da lugar al primer núcleo de cristalización. A partir de él se adjuntarán otros, dando lugar a la cristalización de toda la masa. Esta primera agrupación se logra por adición de otras sustancias, intervención del polvo, enfriamientos localizados, etc.
3.2. Crecimiento de los cristales
Una vez formados los núcleos de cristalización empieza el crecimiento de las -caras paralelamente a las iniciales y, en general, no de una forma regular sino deformada.
Para la obtención de cristales grandes se procede al sistema de siembra que consiste en añadir al producto en cristalización, cristales de la misma sustancia. No es conveniente que los cristales sean excesivamente grandes ya que entonces, incluyen excesivas impurezas.
3. CRISTALIZACIÓN FRACCIONADA.
Se conoce por cristalización fraccionada aquélla que consigue separar o más productos de una mezcla. Se fundamenta en la distinta solubilidad de componentes; según ella, cada producto alcanza la saturación a una temperatura determinada, depositándose como producto cristalino.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 72 de 80
Tiene gran aplicación en la industria química, al poder separar varios prodructos disueltos en un mismo disolvente. Como ejemplo concreto, puede citarse separación de la silvinita (KCl) de la sal común (Na CI), que se extraen juntos.
4. HIDRATACIÓN, EFLORESCENCIA Y DELICUESCENCIA.
Un hidrato es una sal cristalizada con moléculas de agua:
Cu SO . 5 H20. . . . . . sulfato de cobre pentahidratado
Ba CI2 . 2 H20 . . . . . . . cloruro bárico dihidratado
Na2C03 . 10 H20 . . . . . carbonato sódico decahidratado
Al . K . (SO4)2 . 24 H20 sulfato alumínico-potásico (alumbre), con veinticuatro moléculas de agua .
Ca SO4 . 2 H20 . . . . . . sulfato cálcico dihidratado (yeso).
El agua que contienen los hidratos se denomina de hidratación o de cristalización y, si la pierden, se tornan sales anhidras.
El fenómeno de la eflorescencia ocurre cuando la presión del vapor de agua de cristalización de las sales anteriores es superior a la del ambiente, por lo que el vapor pasa del cristal a la atmósfera. En tales casos, las sales pierden su estructura cristalina primitiva pasando al hidrato inferior o a sal anhidra:
Cuando ocurre lo contrario, tomar humedad una sal anhidra o hidrato para pasar a un estado de mayor hidratación, se denomina delicuescencia: en este caso se pierde la estructura cristalina por disolverse el cristal en el agua absorbida de la atmósfera.
5. FINALIDAD DE LA CRISTALIZACIÓN.
Además de facilitar la expedición del producto en forma sólida en vez de líquida, cuyo manejo, transporte y dosificación son mucho más fáciles, la cristalización persigue tres finalidades principales:
- Preparar un producto puro a partir de una muestra de menor pureza: cristalización de sales inorgánicas.
- Fraccionamiento de una mezcla compleja: mezclas de sales de distinta solubilidad como el NaCl y KCl.
- Aislamiento de una sustancia contenida en una mezcla: mezclas de productos en química orgánica.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 73 de 80
6. CRISTALIZACIONES POR DISOLUCIÓN Y PRECIPITACIÓN.
Seguir el proceso de la figura:
Se partirá de 100 g de sulfato sódico (Na2SO4 . H20) que se disolverán en 150 c.c. de agua entre 30 y 35°C (máxima solubilidad de la sal). Consúltense las tablas de las páginas siguientes:
Filtrar en caliente y recoger el filtrado en dos pequeños cristalizadores; taparlos con un vidrio de reloj y dejarlos enfriar en completo reposo.
En uno de ellos se producirá la cristalización por siembra, adicionando un pequeño cristalito de la sal hidratada. En el otro, si no cristaliza por sí solo, se acelerará golpeando la pared del cristalizador para provocar la aparición del primer foco cristalino o se rascará con la varilla de vidrio el fondo del cristalizador, a fin de producir partículas de vidrio que actúen de foco.
Filtrar al vacío y secar los cristales sobre papel de filtro en el desecador.
Secar igualmente algunos cristalitos en la estufa, a unos 100 °C, con lo que se volverá anhidra la sal inicial (Na2S04).
Calcular:
- cantidad de sal solubilizada a 35 °C, aproximadamente;
- pérdida de peso al pasar de sal decahidratada a sal anhidra.
Idénticamente al caso del sulfato sódico, pueden efectuarse ensayos de cristalización con otros productos, como se muestra en la tabla de ensayo de cristalización de los productos más comunes en el laboratorio.
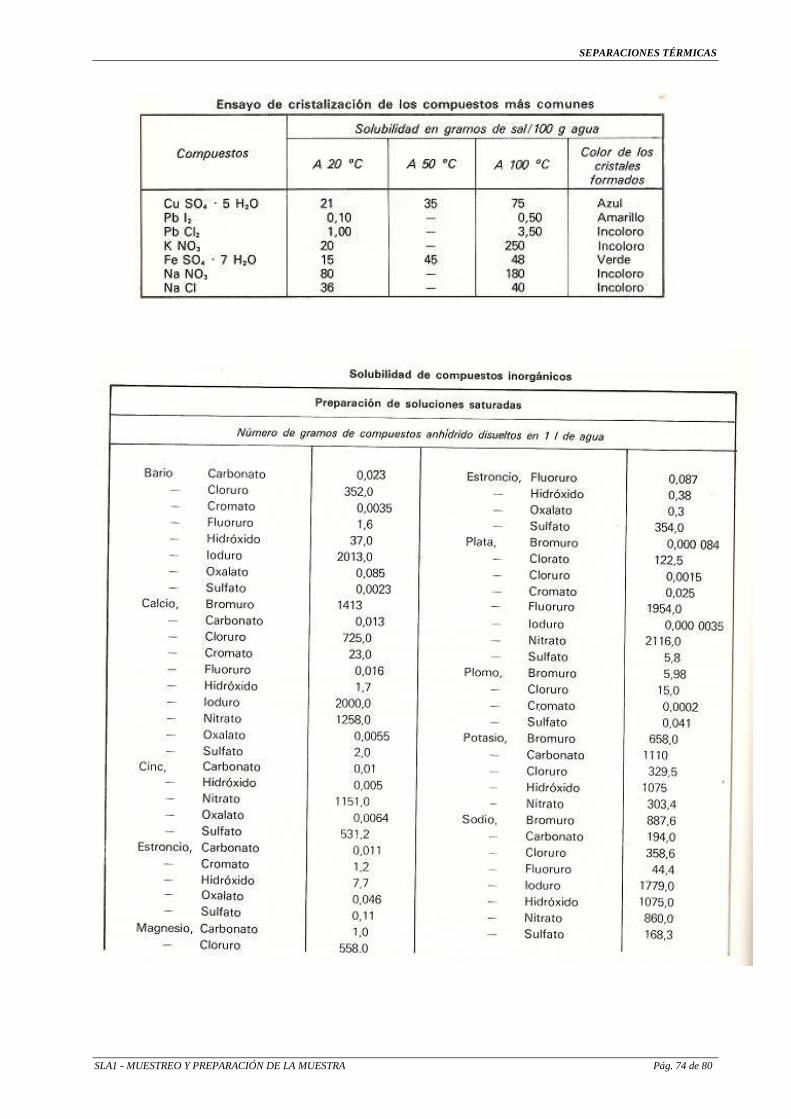
SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 74 de 80

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 75 de 80
7. CRISTALIZACIÓN DE ALUMBRES.
Alumbre alumínico-potásico:
En dos vasos de precipitado de 200 c.c. disolver por separado: 30 g de Al2(SO4)3 . 18 H20 en 50 c.c. de agua a unos 50 °C; 10 g de K2SO4 en 50 c.c. de agua a 50 °C
Diluir con 100 c c de agua cada uno de los anteriores y verterlos juntos a un cristalizador, esperando a que se lleve a cabo la cristalización; ésta puede surgir en pocos minutos o al cabo de horas. Si se observa una masa espesa excesivamente cuajada, deberá ensayarse diluyéndola con agua o calentándola. Se filtran los cristales en vacío y se secan:
La reacción de formación del alumbre es la siguiente:

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 76 de 80
Alumbre crómico-potásico:
Partir de 20 g de dicromato potásico finamente pulverizados y disolver en 100 c.c. de agua en un erlenmeyer, añadir mientras se agita, 20 c.c. de ácido sulfúrico concentrado y dejarlo enfriar.
Luego se añadirán, poco a poco y agitando, 10 c.c. de alcohol etílico (CH3 CH2 OH); se echa en un cristalizador y se deja evaporar depositándose cristales de color violeta de forma octaédrica.
Observar el cambio de color que experimentan al calentar en tubo de ensayo unos cristalitos del alumbre formado.
Escribir la reacción que da por resultado la formación del alumbre Cr . K (SO4)2 . 12 H20.
8. CRISTALIZACIÓN DE COMPUESTOS ORGÁNICOS.
Solubilidad a 20 °C = 0,3 g/100 g de agua.
Solubilidad a 100 °C = 7 g/100 g de agua.
Preparar una disolución saturada a 100 °C y filtrarla en caliente con fitro de pliegues. Las aguas madres recogidas se dejan enfriar hasta cristalización y luego se filtran los cristales a presión reducida, secándolos en el desecador para obtener el ácido benzoico puro.
Idénticamente al ácido benzoico, pueden efectuarse:
Solubilidad a: 20 °C 100 °C
Acetato de plata: 1 g/100 g agua 3 g/100 g agua
Si alguna de las disoluciones es excesivamente coloreada o sucia, cosa frecuente en compuestos orgánicos, debe decolorarse con carbón activo u otro decolorante.
9. CRISTALIZACIÓN POR FUSIÓN.
Se emplea para el caso del azufre y del cloruro sódico fundido. Seguir el proceso en la figura:
9.1. Azufre.
En un crisol de porcelana o de barro se calentará hasta fluidez un poco de azufre y se dejará enfriar parcialmente; verter una parte de este azufre sobre un cristalizador con agua fría y se obtendrá azufre plástico de color oscuro y elástico como la goma.
En el mismo crisol se espera a que se forme una costra, se rompe ésta con una varilla y se vierte la masa fundida. Quedarán en las paredes del crisol adheridas unas agujas de azufre cristalizado, dando lugar al azufre mono clínico.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 77 de 80
Si los cristales monoclínicos se dejan a la temperatura ambiente se volverán opacos por pasar a la variedad rómbica (ver figura).
El líquido viscoso obtenido después del calentamiento puede echarse sobre un vidrio de reloj y dejarlo enfriar y cristalizar para observar al microscopio.
9.2. Tiosulfato sódico
En una cápsula de porcelana se calientan hasta su total fusión (56 °C) 10 g. de tiosulfato sódico (Na2S2O3). Se tapa con un vidrio de reloj y se mantiene varias horas fundido. Si se añade un cristalito de tiosulfato en el interior de la cápsula se formarán cristales grandes de bonita estructura.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 78 de 80
10. CRISTALIZACIÓN POR SUBLIMACIÓN.
Se emplea en la cristalización de azufre, yodo y sulfuro de cadmio.
Los vapores se condensan en las paredes del recipiente frío en forma de cristales de pequeño tamaño:
En el interior de un matraz de 1 litro se calienta suavemente 1 g de yodo. El I2 sublima y se deposita sobre las paredes del matraz en forma de pequeños cristales.
11. CRISTALIZACIÓN DE COMPUESTOS ORGÁNICOS E INORGÁ NICOS.
- En química inorgánica no resulta difícil la obtención de cristales y se emplea el agua casi como único disolvente. Se preparan con gran facilidad:
- En química orgánica, la cristalización es más difícil y tiene gran importancia la elección del disolvente adecuado en el cual sea soluble la sustancia a cristalizar. La tabla resume los puntos de ebullición de disolventes orgánicos para los productos mayormente empleados.
Si la sustancia a cristalizar posee coloración inicial, caso frecuente en compuestos orgánicos, es aconsejable decolorarla inicialmente con carbón activo y filtrarla posteriormente.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 79 de 80
12. CRISTALIZACIÓN INDUSTRIAL.
Las cristalizaciones industriales pueden c1asificarse atendiendo a varias particularidades:
- Según la sobresaturación: por enfriamiento, evaporación y mixto.
- Según el funcionamiento: continuo o discontinuo.
- Por la separación de cristales: en clasificadores de tamaño y sin clasificación.
- Según la agitación: con agitación y sin ella.
12.1. Cuna de cristalización
Consiste en una gran cuba metálica con fondo bombeado y montada sobre rodillos en unos carriles para facilitar su movimiento de oscilación o balanceo:
Interiormente está dividida en compartimientos por tabiques transversales incompletos que permiten un recorrido en zig-zag de la disolución, la cual penetra por la parte superior y desciende lentamente hasta la inferior. Entra la disolución saturada y caliente y, debido a la gran superficie de evaporación, se provoca la cristalización lenta con producción de cristales grandes y bien formados.

SEPARACIONES TÉRMICAS
SLA1 - MUESTREO Y PREPARACIÓN DE LA MUESTRA Pág. 80 de 80
12.2. Cristalizador con recirculación
Se consigue un rápido crecimiento de los cristales y su clasificación por tamaños. Se logra la cristalización por enfriamiento de la disolución saturada entrante:
13. APLICACIONES DE LA CRISTALIZACIÓN.
13.1. En la naturaleza:
Se obtienen grandes masas rocosas y de minerales en forma cristalizada:
Cuarzo (Si02) Calcita (CaCO3)
Galena (Pb S) Hielo (H2O)
Blenda (ZnS) Oro (Au)
Pirita (Fe S) Azufre (S)
13.2. En el laboratorio
Obtención de sales cristalizadas:
compuestos anhidros: KClO3, NaCl, S, etc.
sales hidratadas: Na2SO4.10 H20, CuSO4.5 H20, etc.
compuestos orgánicos: ácido benzoico, antraceno, resorcina.
13.3. En la industria
Productos para laboratorios, margarina, helados, parafinas, aceites, grasas productos farmacéuticos, alumbres, minerales, etc.