sÍntesis, evaluaciÓn biolÓgica y estudios …
TRANSCRIPT

UNIVERSIDAD DE BUENOS AIRES
Facultad de Farmacia y Bioquímica
Departamento de Farmacología
Cátedra de Química Medicinal
Instituto de Química y Metabolismo del Fármaco
SÍNTESIS, EVALUACIÓN BIOLÓGICA Y
ESTUDIOS COMPUTACIONALES DE
TIOSEMICARBAZONAS DE 1-INDANONAS
N4-ARIL SUSTITUIDAS COMO POTENCIALES
ANTIVIRALES Y ANTIPARASITARIOS
Farm. María Cristina Soraires Santacruz
Director: Prof. Dra. Liliana Finkielsztein
Codirector: Dr. Esteban Bontempi
Tesis Doctoral | 2019


1
AGRADECIMIENTOS
A mi directora Lili y mi codirector Esteban por su orientación y apoyo para llevar a
cabo esta tesis. Por permitirme crecer profesionalmente.
Al grupo de investigación de la Dra. Cavallaro por los ensayos biológicos en virus y en
células MDBK.
A mis compañeros de medicinal por su apoyo y compañerismo. A Guido por compartir
las horas en medicinal. A Lucas por responder siempre mis inquietudes y su gran ayuda
en la realización de los espectros de RMN. A Gabriel por el intercambio de ideas.
A todas las personas del quinto y segundo piso del Fatala Chabén por hacer un ambiente
en el cual da gusto trabajar. A Mónica Esteva por su invalorable ayuda con el ensayo en
tripomastigotes. A Vilma Duschak por la asistencia con los geles de actividad y la
provisión de cruzipaína. A Octavio Fusco por siempre buscar la forma de ayudarme. A
Patricia Bustos por enseñarme a usar el citómetro. A Alina Perrone y Luz Piedad por
iniciarme en los cultivos de tripos y amastigotes.
A Nico por su amor y apoyo incondicional.
A mi familia, Tito y Teo, por estar siempre. Los amo.

2

3
RESUMEN
El objetivo del presente trabajo de tesis es contribuir en el hallazgo de compuestos
terapéuticos frente a la enfermedad de la diarrea viral bovina (DVB) y a las parasitosis
debidas a Trypanosoma cruzi y Trypanosoma brucei. Para cumplir con dicho objetivo,
se realizó un estudio exhaustivo y multidisciplinario que comprendió la síntesis,
evaluación biológica y estudios computacionales de tiosemicarbazonas de 1-indanonas
N4-aril sustituidas (N
4-TSCs).
Se sintetizaron y caracterizaron 30 N4-TSCs, de las cuales 24 son descriptas por primera
vez en el marco de esta tesis. Los compuestos preparados poseen diferentes
sustituyentes en el núcleo indánico y en la posición para del arilo unido al N4 con el fin
de evaluar la variación de la actividad según el patrón de sustitución. La síntesis fue
llevada a cabo mediante dos metodologías, una convencional y otra utilizando
tecnología de microondas, por lo cual se pudo determinar qué método resulta más
ventajoso según tiempos y rendimientos de reacción.
Se determinaron las actividades de las N4-TSCs frente al virus de la DVB (VDVB), a
partir de las cuales se realizó un estudio de la relación estructura-actividad en el que se
identificaron patrones estructurales claves para la actividad de las N4-TSCs como
agentes anti-VDVB. El compuesto más activo fue identificado como probable inhibidor
de la ARN polimerasa viral dependiente de ARN por lo cual se llevaron a cabo estudios
computacionales de docking y de dinámica molecular con esta enzima. Los resultados
computacionales obtenidos permitieron conocer en detalle las características de esta
serie de compuestos que serían determinantes para su actividad.
Por otro lado, se llevó a cabo la evaluación de las N4-TSCs frente a T. cruzi para las
formas epimastigote, tripomastigote y amastigote. Los resultados obtenidos mostraron
que cuatro compuestos fueron activos frente a los tres estadios del parásito. De acuerdo
a los antecedentes en literatura para este tipo de compuestos como inhibidores de la
enzima cruzipaína, varias N4-TSCs fueron evaluadas frente a esta enzima. Los
compuestos no mostraron ser inhibidores de la cruzipaína.
Se llevaron a cabo estudios in vitro frente a T. brucei, los cuales mostraron que varios
compuestos fueron activos frente al parásito. Uno de los derivados (N4-TSC 8) se

4
destacó por su potencia y se estudió su capacidad de inhibir las proteasas del parásito.
Los resultados indicaron que estas enzimas no serían el blanco de acción del compuesto.
Teniendo en cuenta los valores de actividad se realizó un estudio de relación estructura-
actividad cuantitativa (QSAR), el cual permitió establecer una posible conformación
activa y determinar las propiedades de las cuales dependería la actividad biológica
estudiada. Asimismo, mediante distintos ensayos se estudió por citometría de flujo el
tipo de muerte celular que induce el compuesto 8 en T. brucei. Los resultados indicaron
que el compuesto desencadena en los parásitos un proceso similar a la apoptosis.
Por otro lado, se evaluó la citotoxicidad de todos los compuestos sobre células de
mamífero, no evidenciándose efectos tóxicos a las máximas concentraciones que
pudieron ser investigadas las N4-TSCs.
A partir de los resultados de la presente tesis se identificaron compuestos activos como
antivirales y antiparasitarios, se estudió la relación estructura-actividad, se realizaron
estudios del mecanismo de acción y del tipo de muerte celular. Asimismo, se generaron
modelos teóricos de QSAR, y se llevaron a cabo estudios de docking y dinámica
molecular que permitieron conocer las características moleculares que estarían
implicadas en la acción de estos compuestos.
Finalmente, los resultados alcanzados permiten sentar bases para continuar con el
diseño de nuevas moléculas antivirales y antiparasitarias. La información obtenida
podrá ser aplicada en otras estrategias de la Química Medicinal como el cribado virtual
de librerías de compuestos y la posterior farmacomodulación de los candidatos más
prometedores para el tratamiento de la enfermedad de la DVB y las tripanosomiasis.

5
PUBLICACIONES
Parte de los resultados que se presentan en este trabajo de tesis doctoral dio lugar a la
redacción del siguiente artículo científico publicado:
M.C. Soraires Santacruz, M. Fabiani, E.F. Castro, L.C. Cavallaro, L.M. Finkielsztein.
“Synthesis, antiviral evaluation and molecular docking studies of N4-aryl
substituted/unsubstituted thiosemicarbazones derived from 1-indanones as potent anti-
bovine viral diarrhea virus agents”. Bioorganic & Medicinal Chemistry. 2017. 25, 15,
4055-4063.
La experiencia adquirida durante el desarrollo de la tesis permitió la siguiente
publicación:
A. Sólimo, M.C. Soraires Santacruz, A.I. Loaiza Perez, E. Bal de Kier Joffé, L.M.
Finkielsztein, M.A. Callero. “N4-aryl substituted thiosemicarbazones derived from 1-
indanones as potential anti-tumor agents for breast cancer treatment”. Journal of
Cellular Physiology. 2017. 233, 6, 4677-4687.

6

7
ABREVIATURAS Y SIGLAS
AM1: método semiempírico Austin Model 1
AV: anexina V
BHE: barrera hematoencefálica
BHT: medio líquido monofásico cerebro corazón triptosa
Bz: benznidazol
CC50: concentración citotóxica 50
CCD: cromatografía en capa delgada
CI50: concentración inhibitoria 50
CP: citopático
Cz: cruzipaína
3D: tridimensional
DMSO: dimetilsulfóxido
DMSO-d6: dimetilsulfóxido deuterado
DVB: diarrea viral bovina
E-64: trans-epoxisuccinil-L-leucilamido-(4-guanidino)-butano
ECP: efecto citopático
Efl: eflornitina
EM: espectrometría de masa
EtOH: etanol
F: estadístico de Fisher
FSC: dispersión frontal
FM: fase móvil
fs: femtosegundo
H2-DCF-DA: diacetato de 2’,7’-diclorodihidrofluoresceína
HF: método de Hartree-Fock para cálculos mecanocuánticos
HMBC: correlación heteronuclear a múltiples enlaces
HOMO: orbital molecular ocupado de mayor energía
HSQC: correlación heteronuclear a un enlace
IR: infrarrojo
IUPAC: unión internacional de química pura y aplicada
IS: índice de selectividad

8
J: constante de acoplamiento
LUMO: orbital molecular desocupado de menor energía
MCR: muerte celular regulada
MM: mecánica molecular
MM/PBSA: mecánica molecular/área de superficie de Poisson Boltzmann
MTT: bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazol
MTS/PES: 3-(4,5-dimetiltiazol-2-il)-5-(3-carboximetoxifenil)-2-(4-sulfofenil)-2H-
tetrazolio/etilsulfato de fenazina
MW: microondas
ns: nanosegundo
N4-TSC: tiosemicarbazona de 1-indanona N
4-aril sustituida
NOESY: espectroscopía de efecto nuclear Overhauser
OMS: Organización Mundial de la Salud
PBS: buffer fosfato salino
pCI50 calc: -logaritmo de la concentración inhibitoria 50 calculada o predicha
pCI50 exp: -logaritmo de la concentración inhibitoria 50 experimental
pCI50: -logaritmo de la concentración inhibitoria 50
PDB: banco de datos de proteínas
Pf: punto de fusión
PI: yoduro de propidio
ppm: partes por millón
PS: fosfatidilserina
QSAR: relación estructura-actividad cuantitativa
R2: coeficiente de determinación
RdRp: ARN polimerasa dependiente de ARN
RMN: resonancia magnética nuclear
13C-RMN: resonancia magnética nuclear de carbono
1H-RMN: resonancia magnética nuclear de hidrógeno
ROS: especies reactivas del oxígeno
rpm: revoluciones por minuto
SD: desvío estándar
SDS: dodecilsulfato sódico
SSC: dispersión lateral
PAGE: electroforesis en gel de poliacrilamida

9
Ptm: pentamidina
SFB: suero fetal bovino
TSC: tiosemicarbazona
TUNEL: marcado de corte terminal de dUTP mediado por la deoxinucleotidil
transferasa
VDVB: virus de la diarrea viral bovina
vdW: van der Waals
VHC: virus de la hepatitis C
VSG: variación antigénica de glicoproteínas
δ: desplazamiento químico
ΔG: diferencia de energía libre

10

11
ÍNDICE
Capítulo I. INTRODUCCIÓN 15
1. Desarrollo de fármacos 17
1.1. Desarrollo de un medicamento. Generalidades 17
1.2. Estrategias en el desarrollo de fármacos 18
1.3. Diseño de fármacos asistido por computadora 19
2. Tiosemicarbazonas 20
2.1. Síntesis de TSCs 20
2.2. Síntesis asistida por radiación de microondas 22
2.3. Actividades biológicas de las TSCs 22
2.4. Antecedentes en el grupo de investigación 24
3. Diarrea viral bovina 25
3.1. Generalidades 25
3.2. Virus de la diarrea viral bovina 26
3.3. Transmisión y replicación del virus 27
3.4. Control y prevención 28
4. Enfermedad de Chagas 29
4.1. Generalidades 29
4.2. Transmisión y ciclo de vida del parásito 29
4.3. Etapas de la enfermedad 31
4.4. Terapia antichagásica disponible 32
5. Enfermedad del sueño y nagana 33
5.1. Generalidades 33
5.2. Transmisión y ciclo de vida del parásito 33
5.3. Etapas de la enfermedad 35
5.4. Terapia anti Trypanosoma brucei disponible 36
Capítulo II. HIPÓTESIS Y OBJETIVOS 41
Capítulo III. RESULTADOS Y DISCUSIÓN 45
1. Síntesis de las N4-TSCs 47
1.1. Selección de los compuestos a sintetizar 47

12
1.2. Métodos de síntesis 48
1.3. Tiempos de reacción y rendimientos 49
1.4. Análisis de la isomería Z/E 52
2. Virus de la diarrea viral bovina 59
2.1. Actividad anti-VDVB de las N4-TSCs 59
2.2. Relación estructura-actividad 60
2.3. Estudios de resistencia cruzada 62
2.4. Docking molecular 64
2.5. Cálculo de energía libre de unión 68
3. Trypanosoma cruzi 73
3.1. Actividad anti-T. cruzi de las N4-TSCs 73
3.2. Relación estructura-actividad cualitativa 78
3.2.1. Resultados en epimastigotes 78
3.2.2. Resultados en tripomastigotes 79
3.3. Evaluación de las N4-TSCs frente a la cruzipaína 79
4. Trypanosoma brucei 83
4.1. Actividad anti-T. brucei de las N4-TSCs 83
4.2. Relación estructura-actividad cualitativa 85
4.3. Estudios de QSAR 86
4.4. Evaluación de la N4-TSC más activa frente a proteasas 96
4.5. Captación intracelular de las N4-TSCs 97
4.6. Mecanismo de muerte celular inducido por el compuesto 8 99
4.6.1. Determinación del tamaño celular 102
4.6.2. Determinación de especies reactivas del oxígeno 103
4.6.3. Determinación del potencial de membrana mitocondrial 105
4.6.4. Ensayo de doble marcación con anexina V e yoduro de propidio 106
4.6.5. Ensayo TUNEL 109
4.6.6. Microscopía electrónica de transmisión para ultraestructura 110
Capítulo IV. MATERIALES Y MÉTODOS 113
1. Síntesis de las N4-TSCs 115
1.1. Reactivos y solventes 115
1.2. Técnicas instrumentales 115
1.3. Procedimiento general para la síntesis de las N4-TSCs 116

13
1.4. Descripción de los compuestos nuevos 116
2. Estudios biológicos 131
2.1. Virus de la diarrea viral bovina 131
2.1.1. Cultivo celular y virus 131
2.1.2. Ensayo de reducción de placas 131
2.1.3. Estudios de resistencia cruzada 131
2.2. Trypanosoma cruzi 132
2.2.1. Cultivo celular de epimastigotes 132
2.2.2. Actividad anti-epimastigotes de T. cruzi 132
2.2.3. Obtención de tripomastigotes sanguíneos 133
2.2.4. Actividad anti-tripomastigotes sanguíneos de T. cruzi 133
2.2.5. Cultivo celular de tripomastigotes 133
2.2.6. Actividad anti-amastigotes de T. cruzi 133
2.2.7. Obtención de la cruzipaína 134
2.2.8. Zimografía 135
2.3. Trypanosoma brucei 135
2.3.1. Cultivo celular 135
2.3.2. Actividad anti-T. brucei 136
2.3.3. Lisado de T. brucei 136
2.3.4. Zimografía 136
2.3.5. Captación intracelular de las N4-TSCs 137
2.3.6. Determinación de tamaño celular 137
2.3.7. Determinación de especies reactivas del oxígeno 137
2.3.8. Determinación del potencial de membrana mitocondrial 138
2.3.9. Ensayo de doble marcación con anexina V e yoduro de propidio 138
2.3.10. Ensayo TUNEL 139
2.3.11. Microscopía electrónica de transmisión para ultraestructura 139
2.4. Ensayo de citotoxicidad 140
2.4.1. Ensayo de efecto citotóxico en células MDBK 140
2.4.2. Ensayo de efecto citotóxico en células Vero 140
3. Estudios computacionales 140
3.1. Modelado molecular 140
3.2. Docking molecular 141
3.3. Dinámica molecular y MM/PBSA 141

14
3.4. Cálculo de descriptores 142
3.5. QSAR 142
4. Análisis estadístico 143
Capítulo V. CONCLUSIONES Y PERSPECTIVAS 145
REFERENCIAS 151

.
Capítulo I
INTRODUCCIÓN


Introducción
17
Capítulo I. INTRODUCCIÓN
1. Desarrollo de fármacos
El fármaco es un compuesto químico de estructura definida que posee un efecto
farmacológico. Su combinación con otros componentes da lugar al medicamento, el
cual permite su administración para la prevención, diagnóstico, tratamiento de una
enfermedad o estado patológico y/o para modificar sistemas fisiológicos en beneficio de
la persona o animal a quien se le administre.
La Química Medicinal tiene como objetivo el estudio químico de los fármacos, sus
propiedades biológicas y el estudio de la relación estructura-actividad, con el fin último
de diseñar, descubrir, interpretar el modo de acción y optimizar compuestos
farmacológicamente activos.
1.1. Desarrollo de un medicamento. Generalidades
El desarrollo de un medicamento es un proceso multidisciplinario y complejo el cual se
estima que lleva entre 10 y 15 años, cuesta un promedio de 800 millones de dólares e
involucra un alto riesgo1,2
.
El proceso se inicia con la selección de las moléculas a estudiar mediante estrategias de
la Química Medicinal (Figura 1). Una vez definidos los compuestos se continúa con las
valoraciones biológicas y se optimiza el más promisorio, llamado compuesto líder. A
partir de este compuesto se avanza con los estudios pre-clínicos, los cuales engloban los
ensayos in vitro e in vivo en animales, se realizan estudios de estabilidad y se desarrolla
la formulación.
La siguiente etapa corresponde a los estudios clínicos en los cuales se diferencian
distintas fases, en la primera de ellas se evalúa la tolerabilidad en un grupo reducido de
personas. Si los resultados son los esperados se continúa con la fase II la cual
compromete a pacientes con la patología a estudiar, se observa la eficacia y se ajusta la
dosis. En la fase III el número de pacientes aumenta y es posible que los mismos
presenten no sólo la patología a estudiar sino también otras; en esta fase se confirma la

Introducción
18
dosis, se tienen en cuenta el régimen de administración, la tolerancia y los efectos
adversos3.
Finalmente, de superarse todas las etapas anteriores, el medicamento puede ser evaluado
por el organismo regulador, en nuestro país la ANMAT. En caso de aprobarse, el
medicamento podrá comercializarse. Sin embargo, el proceso no culmina aquí ya que
existe una última fase llamada farmacovigilancia, en la cual el medicamento y sus
efectos en la salud tienen un seguimiento continuo en la comunidad4.
Figura 1. Esquema del proceso de desarrollo de un medicamento.
1.2. Estrategias en el desarrollo de fármacos
Respecto al desarrollo de fármacos propiamente dicho, el proceso inicia con la
identificación de moléculas que muestren cierta actividad biológica deseada. Las vías de
hallazgo de estas moléculas pueden ser muy diversas, desde la observación fortuita de
los efectos biológicos, como fue el caso del descubrimiento de la penicilina, o bien de
los efectos adversos de fármacos ya existentes como las sulfas hipoglucemiantes, el
cribado sistemático de familias químicas en determinados ensayos
biológicos/bioquímicos, o el diseño racional basado principalmente en estudios
computacionales.
Una vez encontradas aquellas moléculas con la actividad biológica deseada por alguna
de las aproximaciones mencionadas, se selecciona el compuesto más promisorio. Para
esta selección, se tienen en cuenta los valores de actividad, toxicidad y propiedades
físico-químicas que permiten adelantar buenos perfiles farmacológicos y
farmacocinéticos. Sin embargo, a pesar de que el compuesto líder es prometedor, en
general es necesario introducirle ciertas modificaciones que mejoren su características,
es decir, es necesario realizar una farmacomodulación.
La farmacomodulación es una estrategia clave en Química Medicinal, la cual tiene tres
aproximaciones: la disyuntiva, la conjuntiva y la modulativa. En la disyuntiva el
objetivo es la simplificación molecular manteniendo la configuración mínima para que

Introducción
19
el producto final mantenga la actividad biológica (farmacóforo). En la aproximación
conjuntiva el fin es reunir en una única molécula dos fragmentos estructurales, en el
cual al menos uno posea actividad biológica, de esta forma se trata de solucionar un
problema terapéutico o farmacotécnico. En la tercer aproximación, la modulativa, la
complejidad de la molécula no se encuentra significativamente modificada, siendo de
gran utilidad para obtener copias terapéuticas (me too), explorar la importancia
conformacional y obtener resultados de relación estructura-actividad, entre otras. En
particular, dentro de la aproximación modulativa es posible encontrar varias
herramientas de modificación estructural como son: la formación, apertura o variación
del tamaño de anillos, la homología, la isomerización, la ramificación, saturación de
dobles enlaces, la isostería clásica y la bioisostería no clásica5.
1.3. Diseño de fármacos asistido por computadora
El diseño de fármacos asistido por computadora engloba una integración de métodos
mediados por software que tienen como objetivo facilitar el diseño o identificación de
nuevos compuestos, la selección de candidatos, o la optimización de compuestos
líderes. Estos objetivos conducen finalmente a la obtención de un compuesto con altas
probabilidades de ser un fármaco exitoso. Los métodos utilizados en esta tarea
dependerán en gran medida de la información experimental disponible y de los
objetivos específicos6.
En las últimas décadas, los métodos computacionales han permitido ahorrar tiempo,
dinero y aumentar las posibilidades para el hallazgo y optimización de fármacos. Casos
de fármacos exitosos en los cuales los métodos in silico han contribuido de manera
sustancial son: dorzolamida, para el tratamiento del glaucoma de ángulo abierto y la
hipertensión arterial; zanamivir y oseltamivir, utilizados para el tratamiento de algunos
tipos de influenza; saquinavir, para el tratamiento de la enfermedad por el virus de la
inmunodeficiencia humana; norfloxacina, como antibacteriano y donepezilo, para la
enfermedad de Alzheimer.
Por otro lado, más allá de las bondades de estos métodos, es importante desmitificar
ciertos preconceptos con respecto al uso de las computadoras en el proceso de
desarrollo de fármacos. Los métodos computacionales no diseñan por si solos los
fármacos sino que forman parte de una herramienta más, aunque muy valiosa, en la cual

Introducción
20
están en juego esfuerzos multidisciplinarios. El uso de métodos computacionales no
consiste en “apretar un botón” para obtener resultados (push-a-buttom drug discovery)
sino que demandan una gran preparación, análisis e interpretación de los datos
obtenidos. Finalmente, si bien estos métodos permiten el ahorro del tiempo, no por ello
se obtienen resultados instantáneos debido a la frecuente complejidad de los sistemas
analizados que deben ser calculados en las máquinas.
Existen varias estrategias para diseñar, seleccionar u optimizar fármacos por
computadora, las cuales se las puede clasificar en métodos directos y métodos
indirectos. Para el primer método es necesario conocer la diana biológica y contar con
su estructura tridimensional. Dicha estructura puede ser obtenida a partir de técnicas de
difracción de rayos X, resonancia magnética nuclear (RMN) o utilizando técnicas de
modelado por homología. De esta forma será posible llevar a cabo estudios de docking
en los cuales se estudia la complementariedad de un ligando con su sitio de acción, o de
dinámica molecular donde se pueden estudiar las interacciones de manera temporal.
Por el contrario, en los métodos indirectos no es necesario conocer la diana biológica ya
que el modelo se basa en conocer las propiedades de los compuestos para desarrollar
modelos de correlación con la actividad biológica. Dentro de este tipo de métodos se
encuentra la estrategia de QSAR (Quantitative Structure-Activity Relationship) la cual
busca una relación, expresada de forma matemática, entre las propiedades
fisicoquímicas de una molécula con una determinada actividad biológica.
2. Tiosemicarbazonas
2.1. Síntesis de TSCs
Entre las familias de compuestos que despiertan mayor interés en la búsqueda de nuevos
fármacos se encuentran las tiosemicarbazonas (TSCs), de considerable interés desde el
punto de vista farmacológico ya que presentan un amplio espectro de actividades. La
estructura química de las TSCs y la numeración de sus átomos, según las reglas de
nomenclatura IUPAC, se describe en la Figura 2.

Introducción
21
Figura 2. Estructura y numeración de TSCs.
Desde el punto de vista sintético presentan como característica principal su facilidad de
obtención. En general se obtienen por la reacción quimioselectiva de aldehídos y/o
cetonas con tiosemicarbazidas en medio alcohólico y en presencia de cantidades
catalíticas de ácido. Esta reacción se caracteriza por su rapidez y por los altos
rendimientos de obtención. La síntesis de estos compuestos tiene un bajo costo y una
gran economía de átomos ya que, a excepción de la molécula de agua que se libera en su
síntesis, todos los átomos de los reactivos están presentes en el producto final de la
reacción.
El mecanismo de reacción para la formación de TSCs transcurre a través de la
protonación del átomo de oxígeno del carbonilo para formar el intermediario ion
oxonio. Luego se produce el ataque nucleofílico del N1 de la tiosemicarbazida sobre el
carbono carbonílico para dar lugar a la formación de un hemiaminal protonado, el cual
pierde una molécula de agua, obteniéndose luego de la neutralización la TSC
correspondiente (Figura 3).
Figura 3. Mecanismo de reacción de TSCs
En este tipo de reacciones es importante trabajar en condiciones contraladas de pH
ácido, en general entre 4 y 5. Valores de pH menores a 4 no son recomendables ya que
conducen a la protonación del N1 de la tiosemicarbazida y, como consecuencia, a una
menor velocidad de reacción ya que pierde el carácter de nucléofilo por carecer de
electrones no compartidos. Asimismo, valores de pH mayores a 5 también

Introducción
22
comprometen la velocidad de reacción de formación de las TSCs ya que la protonación
del átomo de oxígeno del carbonilo no se encontraría favorecida, dejando el carbono
menos susceptible del ataque nucleofílico.
2.2. Síntesis asistida por radiación de microondas
Tradicionalmente el mecanismo de calentamiento en síntesis química ocurre utilizando
una fuente externa, la cual de forma conductiva calienta la pared del recipiente para
finalmente llegar al solvente y los reactivos. Este tipo de mecanismo se caracteriza por
una transferencia de calor ineficaz que vuelve lenta las reacciones químicas. Asimismo,
la formación de productos de descomposición se encuentra favorecida debido a los
tiempos de reacción y al gradiente de temperaturas existente entre el exterior, con
mayores temperaturas, y el seno de la reacción más frío.
El calentamiento mediante el uso de radiación de microondas ha demostrado ser de gran
valor en la síntesis química, ya que permite trabajar en condiciones controladas de
temperatura y disminuye los tiempos de reacción. Uno de los aspectos más apreciables
es la baja incidencia de reacciones secundarias debido a que los compuestos están poco
tiempo expuestos a altas temperaturas. Por otro lado, el uso de la tecnología de
microondas constituye un aspecto significativo en el concepto de química verde o eco-
compatible por el considerable ahorro de energía que permiten.
2.3. Actividades biológicas de las TSCs
Las TSCs presentan un abanico de actividades biológicas muy diversas, las cuales se
han venido estudiando desde 1946 cuando Domagk y col.7 describieron su actividad
antituberculosa. Desde entonces, se han reportado ésta y muchas otras actividades
biológicas tales como la antitumoral, antifúngica, antiparasitaria y antiviral8–12
.
En 1951, Hamre y col.13
reportaron la actividad antiviral de una serie de TSCs derivadas
de benzaldehídos, administradas en ratones de forma oral, frente a infecciones
producidas por el virus vaccinia. Posteriormente, Bauer y col.14
demostraron que el
derivado N-metilado de la TSC de la isatina (metisazona) (Figura 4) era efectivo en la
prevención de la enfermedad de personas expuestas al virus de la viruela. Estudios
posteriores demostraron la acción de TSCs frente al citomegalovirus15
, el VIH16
y el
herpes simple17
, entre otros.

Introducción
23
Figura 4. Metisazona
En cuanto a la actividad antitumoral de las TSCs, la primera observación fue realizada
por Brockman y col.18
quienes describieron la acción de la TSC derivada de la piridina-
2-carboxaldehído en un modelo de ratones con leucemia. A partir de ese momento,
muchos derivados de TSCs han sido descriptos con diversas acciones antitumorales. Sin
embargo, la más relevante debido a su potencial uso en la clínica es la TSC de la 3-
aminopiridina-2-carboxaldehído (triapina) (Figura 5), la cual se encuentra en la fase II
de estudios clínicos19
.
Figura 5. Triapina
En relación a las propiedades antibacterianas, la acción frente al Mycobacterium
tuberculosis fue la primera que se describió para esta familia química20
. Luego de ello
se han reportado numerosos derivados con acción frente a Staphylococcus aureus
Enterococcus faecalis, Salmonella typhimurium, Escherichia coli entre otros21,22
. Por
otro lado, varias TSCs mostraron tener una buena actividad antifúngica frente a distintas
especies como Aspergillus niger, Candida albicans, Amathia alternata y Fusarium
oxysporum23
.
Otra actividad biológica que presentan las TSCs es la antiparasitaria, en particular se ha
observado que tienen acción frente a Plasmodium falciparum, Plasmodium berghei,
Trypanosoma cruzi y Trypanosoma brucei. En 2002, Du y col.11
publicaron un estudio
exhaustivo de más de cien TSCs derivadas de aldehídos y cetonas y encontraron que
muchas de ellas resultaron activas frente a T. cruzi, destacándose la TSC de la 3’-
bromopropiofenona. En cuanto al mecanismo de acción se ha demostrado que la diana

Introducción
24
biológica de muchas TSCs activas frente a T. cruzi resulta ser la cruzipaína24
. Esta
enzima es una cisteína proteasa esencial en el ciclo de vida del parásito que participa en
la infección y la replicación de T. cruzi en las células huésped, por lo cual ha surgido
como un blanco válido para el desarrollo de nuevas drogas24,25
. En 2004, Greenbaum y
col.12
describieron la actividad de una librería de TSCs como inhibidores de las cisteína
proteasas de los parásitos P. falciparum, T. brucei y T. cruzi.
2.4. Antecedentes en el grupo de investigación
Nuestro grupo de investigación desarrolló la síntesis de una serie de TSCs derivadas de
1-indanonas, las cuales mostraron una excelente actividad frente al virus de la diarrea
viral bovina26
. Muchos de estos compuestos presentaron índices de selectividad
mayores que el de la ribavirina (droga de referencia), resultando la TSC derivada de la
5,6-dimetoxi-1-indanona (5,6-TSC) (Figura 6) la más activa de la serie estudiada (CE50
= 1,75µM). Asimismo, se dilucidó el mecanismo de acción de dicho compuesto
demostrándose que inhibe la ARN polimerasa dependiente de ARN viral (RdRp)27
, la
cual resulta una enzima imprescindible en la replicación del virus.
Figura 6. Tiosemicarbazona derivada de la 5,6-dimetoxi-1-indanona (5,6-TSC).
Estas TSCs derivadas de 1-indanonas a su vez fueron evaluadas en cuanto a su actividad
antiparasitaria frente a la forma epimastigote de T. cruzi (cepa Tulahuén), mostrando
varias de ellas valores de CI50 entre 1,8 y 7,5 µM, superiores al del nifurtimox (droga de
referencia: 7,7 µM)28
. La capacidad inhibitoria in vitro de las TSCs derivadas de 1-
indanonas sobre la cruzipaína mostró hasta un 60% de inhibición con respecto al
compuesto usado como referencia (TSC de 3’-bromopropiofenona: 100% de
inhibición).28
Por otro lado, los compuestos presentaron una limitada solubilidad tanto en solventes
acuosos como orgánicos. En tal sentido, su posterior utilización como fármacos se ve
comprometida debido a su posible baja biodisponibilidad. De esta manera, se

Introducción
25
prepararon nuevas TSCs derivadas de 1-indanonas que resultan de la incorporación de
un grupo arilo sustituido en la posición N4
de la función tiosemicarbazona (N4-TSCs)
(Figura 7), buscando así mejorar el balance lipo-hidrofílico de las TSCs. Se sintetizaron
seis nuevos compuestos, los cuales fueron evaluados frente a T. cruzi (cepa Tulahuén),
exhibiendo algunos de ellos una mejor actividad con respecto a las TSCs de las cuales
derivaron.28
Figura 7. Tiosemicarbazonas de 1-indanonas N4-aril sustituidas
3. Diarrea viral bovina
3.1. Generalidades
La diarrea viral bovina (DVB) es una enfermedad viral que afecta al ganado, así como
también a otras especies del orden artiodáctilo (cerdos, cabras, ovejas y otros
rumiantes)29,30
. Si bien en la actualidad la mayoría de las infecciones producidas por el
virus de la DVB (VDVB) son subclínicas, el impacto económico de los casos que se
manifiestan clínicamente es considerable. Las pérdidas económicas causadas por las
infecciones del VDVB se asocian con una reducción en la producción de leche, menores
tasas de concepción, trastornos respiratorios y muerte durante la infección aguda. La
infección fetal durante las etapas tempranas de la gestación puede causar aborto,
defectos congénitos, retraso del crecimiento y puede dar lugar al nacimiento de terneros
infectados persistentemente.31,32
Los porcentajes de animales infectados persistentemente alcanzan el 1-2% a nivel
mundial mientras que la sero-prevalencia del virus en áreas endémicas puede variar
entre el 40 y el 70%29–31,33–35
. En Argentina, la prevalencia de anticuerpos anti-VDVB
en el ganado adulto es aproximadamente del 70%, aunque este número varía
dependiendo la zona36–39
. Las diferencias de los porcentajes en cuanto a la sero-

Introducción
26
prevalencia se encuentran ligadas principalmente a factores como la densidad de
población de animales, sistemas de confinamiento y prácticas de manejo.
3.2. Virus de la diarrea viral bovina
El VDVB pertenece a la familia Flaviviridae, en la cual se incluyen los géneros
Flavivirus, Hepacivirus y Pestivirus. Dentro de este último género se encuentra el
VDVB con sus dos genotipos (VDVB1 y VDVB2), el virus de la peste porcina clásica y
el virus de la enfermedad de las fronteras.
En cuanto a la morfología, los virus que pertenecen a este género se caracterizan por su
pequeño tamaño, ya que tienen un diámetro entre 40 a 60 nm, la cápside es icosaédrica
y la misma se encuentra limitada por una envoltura conformada por tres glicoproteínas
de membrana: E1, E2 y Erns
. El VDVB puede ser inactivado por altas temperaturas y
detergentes. Asimismo, es sensible al éter etílico, cloroformo y tripsina. Con respecto al
pH, presenta resistencia en un amplio rango, encontrándose su mayor estabilidad entre
pH 5,7 y 9,3.
El genoma del VDVB está compuesto por ARN de polaridad positiva (ARNss+) por lo
cual el ARN genómico cumple la función de ARN mensajero. La traducción del
genoma genera una poliproteína que luego de ser procesada da lugar a las proteínas
estructurales y no estructurales del virus. Estas proteínas están ubicadas desde el
extremo N-terminal hacia el extremo C-terminal de la siguiente manera: Npro
-C-Erns
-E1-
E2-p7-NS2-NS3-NS4B-NS5A-NS5B, donde NS hace referencia a proteínas no
estructurales (Figura 8). NS5B es el gen que codifica para una proteína con actividad de
ARN polimerasa dependiente de ARN (RdRp) responsable de la replicación viral40,41
.
Según los efectos en cultivos celulares y la expresión de la proteína NS2-3, los
Pestivirus se pueden clasificar en dos biotipos: citopáticos (CP) y no citopáticos (NCP).
Los virus CP expresan las proteínas NS2 y NS3 de forma separada42
e inducen muerte
celular mediada por apoptosis, ocasionando la vacuolización del citoplasma. Por otro
lado, los virus NCP no ocasionan cambios visibles en el cultivo celular, manteniendo su
aspecto y crecimiento normal. Sin embargo, estas características no implican que dichos
virus no sean patogénicos. Este biotipo es el más común en la naturaleza y expresa NS2-
3 como proteína fusionada43
.

Introducción
27
Por otra parte, es interesante remarcar que la organización del genoma, la traducción, la
estrategia de replicación y las funciones proteicas de los Pestivirus guardan similitud
con el virus de la hepatitis C (VHC), el único miembro del género Hepacivirus, por lo
cual el VDVB se ha adoptado como modelo sustituto para los estudios de VHC dado
que este no puede replicar in vitro.44,45
Figura 8. Organización genómica del VDVB.
3.3. Transmisión y replicación del virus
La transmisión de los virus del género Pestivirus no depende de vectores sino que
ocurre de forma vertical (transplacentaria en hembras preñadas) o transmisión
horizontal por contacto directo o cercano indirecto con animales infectados ya que el
virus se propaga a través de las secreciones nasales y oculares y de las heces.
Adicionalmente, fluidos, gametas y otras células derivadas de algún animal enfermo
resultan fuentes de contaminación cuando son usados en la reproducción46
.
El virus ingresa a la célula por un mecanismo de endocitosis dependiente de clatrina
mediada por la unión del virus a receptores específicos del huésped47
. Luego de la
fusión de la membrana del virus con la del huésped, se desarma la nucleocápside y el
ARN se libera dentro del citoplasma celular. El ARNss+ es traducido a una poliproteína
la cual es clivada para dar origen a todas las proteínas estructurales y no estructurales.
La replicación viral se lleva a cabo en la superficie del retículo endoplasmático48
en un
complejo de replicación. Este complejo está compuesto por: ARN viral, polimerasas
virales y otras proteínas virales y celulares. Se sintetiza una cadena negativa de ARN a

Introducción
28
partir del ARNss+, dando como resultado intermedio ARNds los cuales se transcriben y
replican dando lugar a ARNm y nuevas ARNss+, respectivamente. Finalmente el virus
se ensambla y es liberado mediante exocitosis49,50
.
En la búsqueda de nuevos fármacos antivirales se tienen en cuenta las distintas etapas
del ciclo de replicación. Una estrategia prometedora es apuntar a la polimerasa viral,
como lo muestran los resultados obtenidos frente a la transcriptasa reversa del VIH, y
con los inhibidores de VHC NS5B, actualmente en uso clínico. La RdRp del VDVB
resulta ser una diana biológica validada como blanco de terapia antiviral por lo cual es
de interés en la búsqueda de compuestos que logren inhibir su actividad51
.
3.4. Control y prevención
Hasta el momento la enfermedad no cuenta con un tratamiento farmacológico por lo
cual se han centrado los esfuerzos en el control y prevención de la misma. El
diagnóstico de animales infectados persistentemente permite llevar a cabo la detección y
eliminación de dichos animales, ya que son la principal fuente de infección y reservorio
del virus. Debido al amplio tipo y severidad de lesiones inespecíficas, el diagnóstico se
basa únicamente en el aislamiento del virus o detección del antígeno viral específico.
Entre los métodos disponibles se encuentran: RT-PCR52,53
, ELISA de captura de
antígeno36
, aislamiento viral, o inmunohistoquímica en biopsia de la piel54
.
Por otro lado, es posible un control sistemático con vacunación. Entre las vacunas
disponibles se encuentran las inactivadas y las de virus vivo modificado55,56
. Las
vacunas inactivadas resultan seguras y se pueden administrar en cualquier momento de
la gestación pero requieren de una inmunización cada 6 meses para mantener el título de
anticuerpos. Estas vacunas no inducen inmunidad celular, pueden provocar reacciones
inflamatorias localizadas y son de alto costo. Por otro lado, las vacunas a virus vivo
modificados al contener cepas atenuadas del VDVB inducen la respuesta inmune en el
huésped de forma rápida, pudiéndose detectar anticuerpos dentro de las 4 semanas
postvacunal y manteniéndose estos por más de un año. Las desventajas de estas vacunas
son la inmunosupresión que provocan, predisponiendo al animal a infecciones con otros
patógenos, y su capacidad potencial de causar la infección fetal por atravesar la barrera
placentaria.

Introducción
29
Por lo mencionado anteriormente, las vacunas convencionales (formulaciones con virus
atenuado o inactivado) pueden no ser óptimas para controlar las infecciones por
VDVB57,58
. En consecuencia, existe la necesidad de agentes antivirales como una
alternativa útil para este propósito51
.
4. Enfermedad de Chagas
4.1. Generalidades
La enfermedad de Chagas o tripanosomiasis americana es una enfermedad parasitaria
endémica de América Latina, causada por el protozoo Trypanosoma cruzi. Representa
una de las enfermedades más serias de la región debido a su impacto social y
económico59,60
. Pertenece al grupo de las denominadas enfermedades desatendidas u
olvidadas según la clasificación de la OMS.
Actualmente, se estima que existen 6 a 8 millones de personas infectadas con T. cruzi, y
cada año ocurren más de 10 mil muertes asociadas a esta parasitosis61
. La incidencia de
la enfermedad está relacionada con el nivel socioeconómico y, en particular, con las
viviendas precarias, ya que el vector de la enfermedad encuentra allí un ambiente
favorable para desarrollarse. Por otro lado, debido al creciente movimiento migratorio
de las últimas décadas, el patrón epidemiológico tradicional de la enfermedad se vio
modificado, ya que el número de pacientes infectados ha aumentado en las zonas
urbanas y en los países desarrollados62,63
.
4.2. Transmisión y ciclo de vida del parásito
La transmisión del T. cruzi puede darse a través del vector Triatoma infestans conocido
en nuestro país con el nombre de vinchuca. El vector es un insecto hematófago, el cual
transmite el parásito a través de las heces contaminadas que elimina al picar. Otras
formas de infección son mediante transfusión de sangre, transmisión congénita (el
parásito puede atravesar la placenta), trasplante de órganos y los accidentes de
laboratorio. A su vez, otra forma de infección que ha cobrado relevancia es la

Introducción
30
denominada vía digestiva, debida al consumo de alimentos contaminados con heces de
la vinchuca portadora de T. cruzi.
El parásito T. cruzi presenta un ciclo de vida complejo que involucra estadios
diferenciados para su adaptación tanto en el vector como en el mamífero. Según
criterios morfológicos y la posición relativa del kinetoplasto respecto del núcleo, se
distinguen tres formas principales del parásito: 1) Amastigote, replicativa e intracelular
en el mamífero. Es de forma redondeada con un flagelo de corta longitud que no
sobresale del bolsillo flagelar y con el kinetoplasto anterior al núcleo. 2)
Tripomastigote, forma infectiva no replicativa, que puede encontrarse tanto en el insecto
vector como en el mamífero. Es de forma ahusada con el kinetoplasto ubicado posterior
al núcleo. 3) Epimastigotes, es la forma replicativa que se encuentra en el insecto
vector. Es de forma ahusada con el kinetoplasto ubicado anterior al núcleo (Figura 9).
Figura 9. Principales estadios que adopta T. cruzi durante su ciclo de vida.
El ciclo de vida de T. cruzi se puede describir como se ilustra en la Figura 10. El vector
luego de picar libera tripomastigotes metacíclicos con sus defecaciones, los que
ingresan al torrente sanguíneo del nuevo huésped a través de la piel dañada, iniciando
de esta manera el ciclo de vida en el mamífero. Una vez que los tripomastigotes están en
la sangre, estos invaden células nucleadas para diferenciarse en amastigotes, donde
sufren varios ciclos de división y se diferencian en tripomastigotes sanguíneos. Cuando
los mismos alcanzan alta densidad en la célula, y debido al movimiento de los mismos,
se provoca la ruptura de la célula hospedadora, liberando los parásitos a la circulación
sanguínea donde pueden alcanzar otras células. Por otro lado, cuando un nuevo vector
se alimenta de un mamífero infectado, junto con la sangre estará ingiriendo
tripomastigotes, los cuales en el insecto se diferencian a la forma epimastigote. Éstos se

Introducción
31
dividen repetidas veces y finalmente se diferencian a tripomastigotes metacíclicos en el
intestino, cerrando así el ciclo de vida del parásito.
Figura 10. Ciclo de vida de T. cruzi.
4.3. Etapas de la enfermedad
La enfermedad de Chagas es una infección que persiste durante toda la vida del
paciente. Presenta dos fases claramente diferenciadas: la etapa aguda y la crónica64
.
La fase aguda suele durar unos dos meses después de contraerse la infección y se
caracteriza por la presencia de un gran número de parásitos en circulación. En la
mayoría de los casos no se presentan síntomas o son leves e inespecíficos: fiebre,
malestar general, agrandamiento de ganglios linfáticos, palidez, dolores musculares,
dolor abdominal y dificultad para respirar. En el caso de transmisión vectorial se
observa la presencia de reacciones cutáneas en el sitio de la picadura llamado chagoma,
o la reacción conjuntiva ocular llamado signo de Romaña. Es en esta fase aguda cuando
los parásitos invaden y se multiplican en diferentes células del mamífero.

Introducción
32
La fase crónica suele presentarse asintomática durante 10 a 20 años. Sin embargo, los
parásitos se encuentran en el músculo cardíaco y digestivo lo cual da lugar a tres formas
de la enfermedad: la cardíaca, la digestiva y la cardiodigestiva. La forma cardíaca se
caracteriza por daño miocárdico en la que pueden presentarse desde arritmias y
tromboembolismos hasta insuficiencia y falla cardíaca con muerte súbita, siendo esta
última la principal causa de muerte en los pacientes con enfermedad de Chagas. Por otro
lado, la forma digestiva se caracteriza por alteraciones funcionales y anatómicas en el
esófago y colon debido a la denervación intrínseca del sistema parasimpático. Las
alteraciones clínicas que se observan son las modificaciones en la movilidad peristáltica
y el agrandamiento de las vísceras. Por último, la forma cardiodigestiva es la asociación
de la enfermedad cardíaca con megaesófago y/o megacolon.
4.4. Terapia antichagásica disponible
A pesar de que la enfermedad de Chagas afecta a millones de personas, no es
considerada una patología rentable por la industria farmacéutica. No existen vacunas ni
tratamientos seguros, siendo la eliminación del insecto vector la forma de controlar la
aparición de nuevos casos.
Los únicos dos fármacos que se utilizan para el tratamiento de la enfermedad, desde
hace más de cuatro décadas, son el nifurtimox (1972) y el benznidazol (1974), (Figura
11)65
. Ambos presentan diversos efectos adversos que incluyen: anorexia, vómitos,
dermatitis alérgica, polineuropatía periférica, leucopenia, entre otros. Estos efectos
adversos pueden llevar a la falta de adhesión al tratamiento. Asimismo, la existencia de
varias cepas que han sido aisladas en distintas zonas geográficas, marca diferencias
significativas en cuanto a la resistencia y susceptibilidad a las drogas.
Figura 11. Quimioterápicos disponibles para la enfermedad de Chagas

Introducción
33
El mecanismo de acción más aceptado para el nifurtimox es la alteración del equilibrio
redox del parásito. Por otro lado, el efecto tripanocida del benznidazol probablemente se
debe a la unión covalente de los metabolitos reducidos del fármaco con
macromoléculas. A pesar de ser fármacos ampliamente utilizados, su modo de acción
todavía no es comprendido totalmente.
Considerando entonces las notables limitaciones de los quimioterápicos disponibles, se
presenta como un importante desafío el tratamiento de esta enfermedad olvidada. Es
necesario encontrar pronto un fármaco más efectivo y seguro65
.
5. Enfermedad del sueño y nagana
5.1. Generalidades
Trypanosoma brucei spp, es el agente etiológico de la tripanosomiasis africana, que
puede afectar tanto a humanos como al ganado. En el caso de los humanos la
enfermedad es conocida como la enfermedad del sueño y es producida por las
subespecies T. b. gambiense y T. b. rhodesiense. Por otro lado, la subespecie T. b.
brucei es la responsable de la enfermedad que afecta a los animales, la cual se conoce
como nagana.
La enfermedad del sueño amenaza a millones de personas en 36 países del África
subsahariana, las cuales en muchos casos viven en áreas rurales remotas con acceso
limitado a servicios de salud, lo que complica la vigilancia y, por lo tanto, el diagnóstico
y tratamiento. De acuerdo con el último informe de la OMS, aproximadamente 20.000
personas se infectan cada año y más de 65 millones de personas están en riesgo
de contraer la enfermedad 66
.
Por otro lado, la enfermedad veterinaria también reviste gran relevancia ya que
compromete la actividad ganadera, restringiendo así el desarrollo de la economía, lo que
a su vez contribuye a la pobreza de las zonas más afectadas.
5.2. Transmisión y ciclo de vida del parásito
La transmisión del T. brucei se da a través de la mosca tsé-tsé (transmisión vectorial),
aunque también es posible mediante transfusión de sangre, transmisión congénita (el

Introducción
34
parásito puede atravesar la placenta), trasplante de órganos y los accidentes de
laboratorio.
T. brucei presenta un ciclo de vida complejo el cual comprende a un vector, la mosca
tsé-tsé (Glossina spp), y a un hospedador mamífero (Figura 12). Durante cada etapa del
ciclo de vida el parásito sufre diferentes cambios bioquímicos y morfológicos que le
permite su adaptación al entorno.
La mosca tsé-tsé inocula tripomastigotes metacíclicos en el hospedador mamífero, los
cuales se diferencian a tripomastigotes sanguíneos de forma alargada (long slender) y se
multiplican en la sangre y linfa, pudiendo alcanzar también el sistema nervioso central
al traspasar la barrera hematoencefálica (BHE). A medida que se incrementa la
parasitemia algunos tripomastigotes se diferencian a la forma sanguínea redondeada
(stumpy), la cual no es proliferativa pero está preadaptada a las condiciones que
encontrará en el vector.
El sistema inmune del hospedador reconoce las glicoproteínas de la superficie celular de
los parásitos y comienza a producir anticuerpos. De esta manera se neutralizan los
tripanosomas y disminuye la parasitemia. Sin embargo, T. brucei tiene la capacidad de
cambiar las glicoproteínas de superficie por otras con una región antigénica diferente.
Dicho mecanismo conocido como variación antigénica de glicoproteínas (VSG) permite
al parásito no ser reconocido por los anticuerpos circulantes. De esta manera logra
evadir el sistema inmune del huésped y puede seguir proliferando alcanzando una
infección persistente.
La mosca al alimentarse ingiere los tripomastigotes redondeados, los cuales poseen la
capacidad de diferenciarse rápidamente en el tracto digestivo a tripomastigotes
procíclicos, la forma replicativa no infectiva. Los parásitos migran a las glándulas
salivales de la mosca donde se adhieren como formas epimastigotes, las cuales son
replicativas. Posteriormente se transforman a tripomastigotes metacíclicos que serán
transmitidos al hospedador mamífero, completándose así el ciclo.

Introducción
35
Figura 12. Ciclo de vida de T. brucei67
.
5.3. Etapas de la enfermedad
La enfermedad del sueño presenta dos fases patológicas principales. La primera etapa,
conocida como fase hemolinfática, se caracteriza por la presencia del parásito
únicamente en sangre y en linfa68
, describiéndose los siguientes síntomas: fiebre,
dolores musculares, debilidad, inflamación de los ganglios linfáticos, dolores de cabeza
y pérdida de peso. En la segunda etapa conocida con el nombre de fase neurológica, el
parásito logra atravesar la BHE invadiendo así el sistema nervioso central, causando
cambios en el comportamiento, confusión, mala coordinación, alteración de los ciclos
del sueño, coma y finalmente la muerte69
.
La progresión de la enfermedad depende tanto de las características del parásito, del
hospedador y de la interacción parásito-hospedador. En el caso de la infección sin
tratamiento con la subespecie T. b. gambiense la enfermedad se caracteriza en general
por una progresión lenta de aproximadamente 3 años, hasta la muerte de la persona
infectada, mientras que en el caso de una infección por T. b. rhodesiense, la muerte en
general sucede en el plazo de semanas o pocos meses.

Introducción
36
Los animales también pueden verse infectados con T. b. gambiense y T. b. rhodesiense,
aunque no desarrollan la enfermedad. Sin embargo, constituyen una reserva a partir de
las cuales las moscas tsé-tsé pueden adquirir la infección70
. Por otro lado, la infección
con T. b. brucei causa enfermedad en el ganado, la cual se caracteriza por fiebre
intermitente, anemia, diarrea, inflamación de tejidos con compromiso en muchos casos
del sistema nervioso central y debilitamiento del animal, que con frecuencia termina
causando la muerte. Hay que mencionar que esta última subespecie no es infectiva en
humanos por la presencia en sangre de apolipoproteina L1, conocida como “factor
tripanolítico” la cual produce la lisis del parásito.
5.4. Terapia anti Trypanosoma brucei disponible
El manejo de la enfermedad comprende en primer lugar la detección de la infección
mediante la determinación directa o indirecta del parásito, el control de signos y
síntomas clínicos y la evaluación del grado de avance de la enfermedad. En este sentido,
es importante realizar el diagnóstico lo antes posible para evitar la etapa neurológica de
la enfermedad.
En cuanto a la inmunización, no existe en la actualidad una vacuna disponible. Si bien
se han intentado diversos planes de inmunización en animales, todos ellos han fracasado
en gran parte debido a la asombrosa capacidad de los parásitos para cambiar la cubierta
antigénica o VSG. En este contexto, la principal alternativa para el tratamiento de la
enfermedad sigue siendo la quimioterapia.
Los fármacos disponibles para controlar la enfermedad del sueño son: pentamidina,
suramina, melarsoprol, eflornitina y nifurtimox en combinación con eflornitina. Estas
drogas tienen notables limitaciones que incluyen: toxicidad, ineficacia, alto costo y
largos períodos de administración.71,72
La pentamidina (Figura 13) es una diamidina aromática que se ha utilizado desde 1937
en la etapa temprana de la tripanosomiasis humana causada por T. b. gambiense. A pH
fisiológico se encuentra principalmente bajo la forma cargada positivamente, por lo cual
su paso a través de la BHE se encuentra desfavorecido, explicando así su eficiencia sólo
en la primera etapa de la enfermedad del sueño73
. Recientemente se descubrió que la
pentamidina tiene un paso a través de la BHE, pero es excluida por transportadores 74
.
Introducción
37
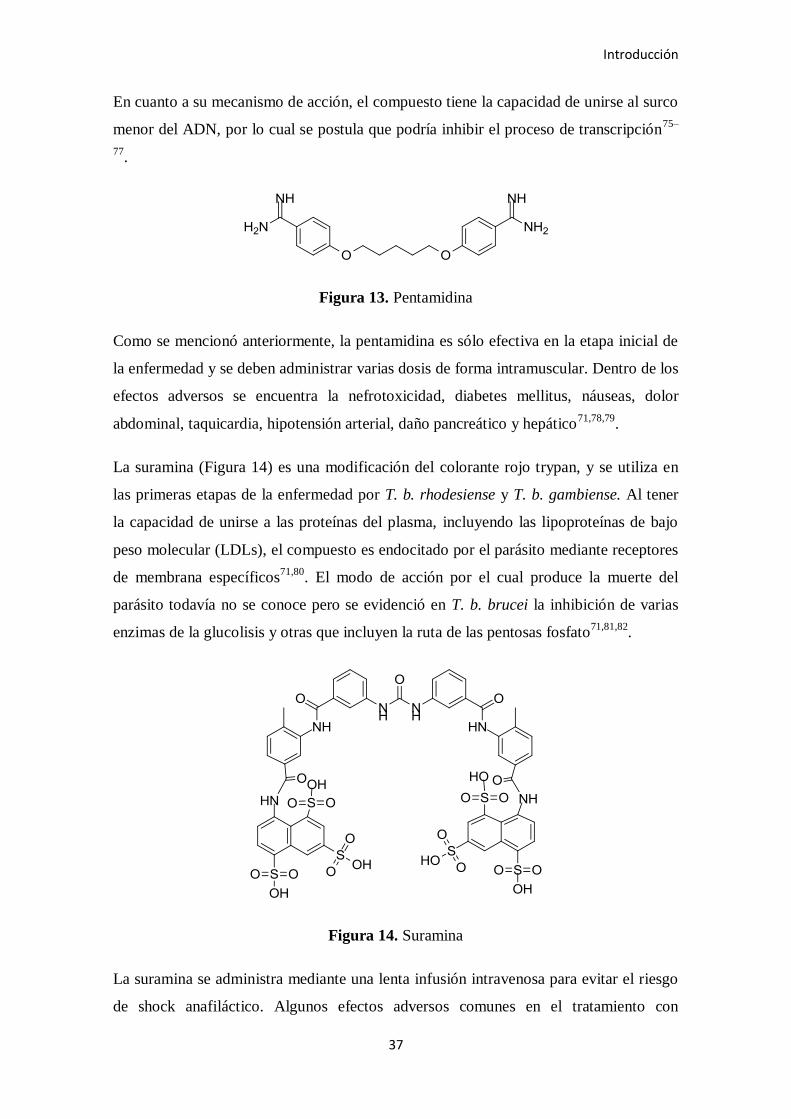
En cuanto a su mecanismo de acción, el compuesto tiene la capacidad de unirse al surco
menor del ADN, por lo cual se postula que podría inhibir el proceso de transcripción75–
77.
Figura 13. Pentamidina
Como se mencionó anteriormente, la pentamidina es sólo efectiva en la etapa inicial de
la enfermedad y se deben administrar varias dosis de forma intramuscular. Dentro de los
efectos adversos se encuentra la nefrotoxicidad, diabetes mellitus, náuseas, dolor
abdominal, taquicardia, hipotensión arterial, daño pancreático y hepático71,78,79
.
La suramina (Figura 14) es una modificación del colorante rojo trypan, y se utiliza en
las primeras etapas de la enfermedad por T. b. rhodesiense y T. b. gambiense. Al tener
la capacidad de unirse a las proteínas del plasma, incluyendo las lipoproteínas de bajo
peso molecular (LDLs), el compuesto es endocitado por el parásito mediante receptores
de membrana específicos71,80
. El modo de acción por el cual produce la muerte del
parásito todavía no se conoce pero se evidenció en T. b. brucei la inhibición de varias
enzimas de la glucolisis y otras que incluyen la ruta de las pentosas fosfato71,81,82
.
Figura 14. Suramina
La suramina se administra mediante una lenta infusión intravenosa para evitar el riesgo
de shock anafiláctico. Algunos efectos adversos comunes en el tratamiento con

Introducción
38
suramina incluyen: toxicidad renal, fiebre, nauseas y vómitos. Otros efectos menos
comunes son: ictericia, diarrea severa, anemia y anafilaxia que puede llevar a la
muerte81–83
.
El melarsoprol (Figura 15), un derivado orgánico del arsénico, es el fármaco de elección
en la segunda etapa de la enfermedad del sueño causada por T. b. gambiense o T. b.
rhodesiense debido a su capacidad de atravesar la BHE. Si bien también es activo en las
etapas tempranas de la infección, no se lo suele utilizar hasta no verse avanzada la
enfermedad debido a los peligrosos efectos adversos que puede ocasionar. Se administra
de forma intravenosa y el paciente requiere estar hospitalizado ya que el 10% sufre de
encefalopatías que resultan mortales en el 50% de los casos. Asimismo, en los últimos
años, se han registrado casos de falla terapéutica luego de utilizar melarsoprol,
especialmente frente a T. b. gambiense en áreas de alta transmisión84,85
.
Figura 15. Melarsoprol
La eflornitina (Figura 16) fue desarrollada originalmente como antitumoral, pero
actualmente es una alternativa al tratamiento para la enfermedad del sueño. El
mecanismo de acción comprende la inhibición específica e irreversible de la enzima
ornitina decarboxilasa, la cual actúa en el primer paso de la biosíntesis de
poliaminas73,84
.
Figura 16. Eflornitina
Muchas desventajas han limitado el uso de la eflornitina en el tratamiento, entre ellas
están el alto costo y la corta vida media en el plasma luego de una inyección intravenosa
(alrededor de 3 h), con la consiguiente eliminación por orina del 80% del fármaco luego
de 24 h. Asimismo, el tratamiento con eflornitina está relacionado a efectos adversos

Introducción
39
tales como: anemia, leucopenia, pancitopenia, dolores de cabeza y convulsiones86
.
Además, se ha demostrado que la resistencia a la eflornitina puede ser inducida
rápidamente ya que en un estudio con T. b. brucei cepa 427 se observó que luego de
exponer los parásitos a concentraciones crecientes de la droga, estos se volvían
resistentes luego de 60 días. Por este motivo, en un intento para prevenir la pérdida de
efectividad del tratamiento, se introdujo una terapia combinada de eflornitina con
nifurtimox.
El nifurtimox (Figura 11), se utiliza desde la década de 1960 para tratar la forma aguda
de la enfermedad de Chagas y recientemente en la etapa neurológica de la enfermedad
del sueño. Las ventajas de este fármaco son principalmente su administración oral y su
costo relativamente bajo. Sin embargo, presenta numerosos efectos adversos y la
duración de su tratamiento es muy prolongada por lo cual no puede ser empleada como
monoterapia, administrándose en conjunto con la eflornitina.
En cuanto a los fármacos de elección para el tratamiento de la tripanosomiasis animal
africana (nagana), uno de los más utilizados es el aceturato de diminazeno (Figura 17).
Dicho compuesto pertenece a la familia de las diamidinas aromáticas, resulta de bajo
costo relativo y es efectivo frente a varias infecciones animales. Sin embargo, en los
últimos años se han registrado casos de resistencia a la droga, lo cual ha complicado la
eficacia del tratamiento.
Figura 17. Aceturato de diminazeno
En conclusión, la falta de una vacuna contra esta enfermedad desatendida y las notables
limitaciones de los tratamientos quimioterápicos representan un importante desafío para
la química medicinal. Es necesario encontrar una terapia más efectiva, segura y menos
costosa.

40

Capítulo II.
HIPÓTESIS Y OBJETIVOS


Hipótesis y objetivos
43
Capítulo II. HIPÓTESIS Y OBJETIVOS
Visto y considerando los antecedentes anteriormente mencionados se propone la
siguiente hipótesis: “El estudio de compuestos pertenecientes a la familia de las
tiosemicarbazonas de 1-indanonas N4-aril sustituidas permitirá el hallazgo de
nuevos agentes líderes como antivirales y antiparasitarios”
Para demostrar esta hipótesis se propone como objetivo general de la presente tesis: la
síntesis de 30 TSCs N4-aril sustituidas derivadas de 1-indanona (N
4-TSCs) siendo 24 de
ellas derivados nuevos, las cuales serán evaluadas como probables agentes terapéuticos
frente a la enfermedad causada por el virus de la DVB y a las parasitosis debidas a T.
cruzi y T. brucei. Asimismo, se buscará para los compuestos más promisorios su
probable blanco de acción mediante la aplicación de métodos computaciones y/o
ensayos in vitro.
Con el fin de avanzar en la concreción del objetivo general formulado, se plantean los
siguientes objetivos específicos:
I. Sintetizar y caracterizar estructuralmente una serie de 30 N4-TSCs que posean
variabilidad estructural tanto en los sustituyentes del anillo indánico como en
los del arilo en posición N4. De esta forma se podrán llevar a cabo estudios
de relación estructura-actividad.
II. Evaluar la actividad biológica de las N4-TSCs:
a) Antiviral frente al VDVB.
b) Antiparasitaria frente a T. cruzi en sus formas epimastigote,
tripomastigote y amastigote y frente a T. brucei en su forma
tripomastigote sanguíneo.
c) Selectividad mediante estudios de citotoxicidad frente a células de
mamíferos.
III. Frente al virus de la diarrea viral bovina:
a) Estudiar la relación entre las estructuras de los compuestos con sus
actividades biológicas, relación estructura-actividad.
b) Determinar in silico interacciones ligando-diana biológica (N4-TSC-
RdRp) y definir los aspectos moleculares claves en la actividad del

Hipótesis y objetivos
44
compuesto más activo mediante estudios computacionales de docking y
de dinámica molecular.
IV. Frente a Trypanosoma cruzi y Trypanosoma brucei:
a) Estudiar la relación entre las estructuras de los compuestos con sus
actividades biológicas de forma cualitativa y cuantitativa, relación
estructura-actividad y relación estructura-actividad cuantificada (QSAR)
respectivamente. De esta manera se podrán encontrar patrones
fisicoquímicos claves que modulan la actividad.
b) Investigar para el compuesto de mayor actividad su probable mecanismo
de acción y tipo de muerte celular mediante ensayos in vitro específicos.

Capítulo III
RESULTADOS Y DISCUSIÓN


Resultados y discusión | Síntesis de las N4-TSCs
47
Capítulo III. RESULTADOS Y DISCUSIÓN
1. Síntesis de las N4-TSCs
Teniendo en cuenta los antecedentes de las actividades biológicas de las TSCs derivadas
de 1-indanonas y, en particular, los resultados obtenidos previamente en el grupo de
investigación, se propuso para el presente trabajo de tesis estudiar nuevas N4-TSCs
como antivirales y antiparasitarios. Para ello se sintetizó una serie de análogos para
evaluar su acción biológica con el fin de encontrar compuestos promisorios y realizar
estudios de relación estructura-actividad. En esta sección se describen los métodos de
síntesis, los tiempos y rendimientos de la reacción de formación de los compuestos y el
análisis de la isomería Z/E.
1.1. Selección de los compuestos a sintetizar
Antes de llevar a cabo la síntesis se hizo una selección de los sustituyentes del núcleo
indánico y el arilo unido al N4 (Figura 18). Para ello se tuvieron en cuenta los resultados
de actividad obtenidos previamente por el grupo de investigación. En este sentido, la
TSC derivada de la 5,6-dimetoxi-1-indanona sin sustituir en el N4 (5,6-TSC) demostró
ser la más activa frente al VDVB con un valor de CE50 de 1,75 µM.26
Por otro lado, en
los estudios frente a epimastigotes de T. cruzi cepa Tulahuén 2 las TSCs derivadas de la
4,5-dimetoxi-, 5-bromo- y 5-metil-1-indanona fueron las que resultaron más activas con
valores de CI50 de 1,8, 2,3 y 3,6 µM respectivamente28
. Asimismo, se observó que la
sustitución en el N4 por restos arilos favorecía la actividad de los compuestos, resultando
el compuesto derivado de la 5,6-dimetoxi-1-indanona con la sustitución p-cloro en el
arilo unido al N4
la más activa, con un valor de CI50 de 1,0 µM.
Figura 18. Tiosemicarbazonas de 1-indanonas N4-aril sustituidas (N
4-TSCs)

Resultados y discusión | Síntesis de las N4-TSCs
48
De acuerdo a estos antecedentes, se decidió sintetizar las N4-TSCs (Figura 18) que
tuvieran sustituyentes R1 = H; 5,6-dimetoxilo (5,6-diOCH3); 4,5-dimetoxilo (4,5-
diOCH3); 5-bromo (5-Br) y 5-metilo (5-CH3), seleccionados teniendo en cuenta la
actividad antiviral y anti T. cruzi observada en las TSCs y N4-TSCs previamente
evaluadas. Por otro lado, los sustituyentes R2 fueron los siguientes: H; CH3; F; Cl;
OCH3; NO2, los cuales fueron elegidos en función de las distintas calidades electrónicas
y/o estéricas de los mismos que permitieran así proporcionar variabilidad en cuanto a
propiedades fisicoquímicas de los compuestos para los posteriores ensayos biológicos y
análisis de la relación estructura-actividad.
1.2. Métodos de síntesis
Con respecto al método de síntesis, el grupo de investigación describió dos posibles
caminos alternativos, el Método A: a partir de las hidrazonas de 1-indanonas con
fenilisotiocianatos ambos convenientemente sustituidos en condiciones de reacción
convencionales; y el Método B: a partir de 1-indanonas convenientemente sustituidas,
hidrato de hidracina y fenilisotiocianatos, también sustituidos convenientemente, por
medio de una reacción one-pot multicomponente utilizando tecnología de microondas.
Se decidió entonces realizar la síntesis de cada compuesto mediante ambas
metodologías para comparar sus rendimientos y tiempos de reacción.
En el siguiente esquema se muestran las dos rutas sintéticas:
Ruta de síntesis método A:
Las hidrazonas se obtuvieron previamente por tratamiento de las correspondientes 1-
indanonas con hidrato de hidracina.

Resultados y discusión | Síntesis de las N4-TSCs
49
Ruta de síntesis método B:
R1= H; 5,6-diOCH3; 4,5-diOCH3; 5-Br; 5-CH3
R2= H; CH3; F; Cl; OCH3; NO2
La determinación de las estructuras se realizó en base a métodos espectroscópicos (IR,
1H,
13C-RMN mono y bidimensionales) y espectrométricos (masa de alta resolución).
1.3. Tiempos de reacción y rendimientos
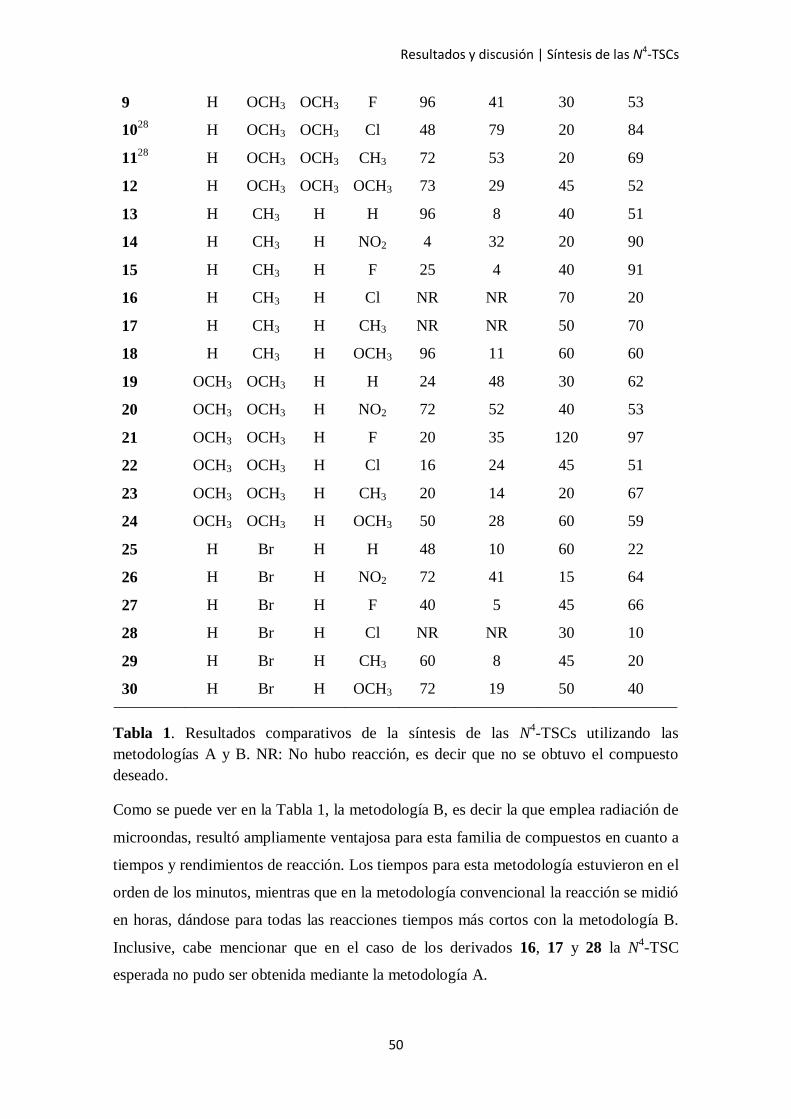
Se sintetizaron 30 N4-TSCs de las cuales 24 son compuestos nuevos, empleando ambas
metodologías (A y B). Los resultados de tiempo y rendimiento de las reacciones se
detallan en la Tabla 1.
N4-TSCs R1 R2 R3 R4
Método A Método B
Tiempo
(h)
Rendimiento
(%)
Tiempo
(min)
Rendimiento
(%)
128
H H H H 25 87 20 89
2 H H H NO2 20 46 45 70
3 H H H F 27 19 130 87
428
H H H Cl 4 79 30 73
528
H H H CH3 48 72 10 78
6 H H H OCH3 72 86 60 79
728
H OCH3 OCH3 H 25 77 15 74
8 H OCH3 OCH3 NO2 72 56 65 100
Resultados y discusión | Síntesis de las N4-TSCs
50
9 H OCH3 OCH3 F 96 41 30 53
1028
H OCH3 OCH3 Cl 48 79 20 84
1128
H OCH3 OCH3 CH3 72 53 20 69
12 H OCH3 OCH3 OCH3 73 29 45 52
13 H CH3 H H 96 8 40 51
14 H CH3 H NO2 4 32 20 90
15 H CH3 H F 25 4 40 91
16 H CH3 H Cl NR NR 70 20
17 H CH3 H CH3 NR NR 50 70
18 H CH3 H OCH3 96 11 60 60
19 OCH3 OCH3 H H 24 48 30 62
20 OCH3 OCH3 H NO2 72 52 40 53
21 OCH3 OCH3 H F 20 35 120 97
22 OCH3 OCH3 H Cl 16 24 45 51
23 OCH3 OCH3 H CH3 20 14 20 67
24 OCH3 OCH3 H OCH3 50 28 60 59
25 H Br H H 48 10 60 22
26 H Br H NO2 72 41 15 64
27 H Br H F 40 5 45 66
28 H Br H Cl NR NR 30 10
29 H Br H CH3 60 8 45 20
30 H Br H OCH3 72 19 50 40
Tabla 1. Resultados comparativos de la síntesis de las N4-TSCs utilizando las
metodologías A y B. NR: No hubo reacción, es decir que no se obtuvo el compuesto
deseado.
Como se puede ver en la Tabla 1, la metodología B, es decir la que emplea radiación de
microondas, resultó ampliamente ventajosa para esta familia de compuestos en cuanto a
tiempos y rendimientos de reacción. Los tiempos para esta metodología estuvieron en el
orden de los minutos, mientras que en la metodología convencional la reacción se midió
en horas, dándose para todas las reacciones tiempos más cortos con la metodología B.
Inclusive, cabe mencionar que en el caso de los derivados 16, 17 y 28 la N4-TSC
esperada no pudo ser obtenida mediante la metodología A.
Resultados y discusión | Síntesis de las N4-TSCs
51
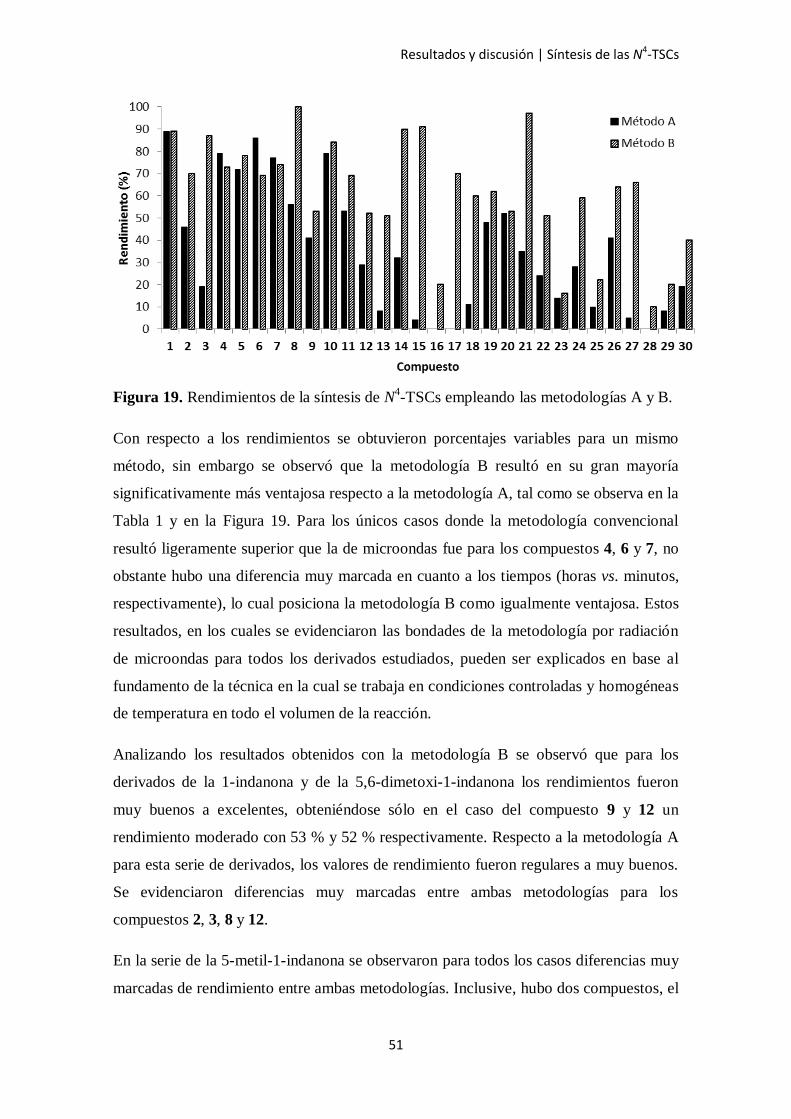
Figura 19. Rendimientos de la síntesis de N4-TSCs empleando las metodologías A y B.
Con respecto a los rendimientos se obtuvieron porcentajes variables para un mismo
método, sin embargo se observó que la metodología B resultó en su gran mayoría
significativamente más ventajosa respecto a la metodología A, tal como se observa en la
Tabla 1 y en la Figura 19. Para los únicos casos donde la metodología convencional
resultó ligeramente superior que la de microondas fue para los compuestos 4, 6 y 7, no
obstante hubo una diferencia muy marcada en cuanto a los tiempos (horas vs. minutos,
respectivamente), lo cual posiciona la metodología B como igualmente ventajosa. Estos
resultados, en los cuales se evidenciaron las bondades de la metodología por radiación
de microondas para todos los derivados estudiados, pueden ser explicados en base al
fundamento de la técnica en la cual se trabaja en condiciones controladas y homogéneas
de temperatura en todo el volumen de la reacción.
Analizando los resultados obtenidos con la metodología B se observó que para los
derivados de la 1-indanona y de la 5,6-dimetoxi-1-indanona los rendimientos fueron
muy buenos a excelentes, obteniéndose sólo en el caso del compuesto 9 y 12 un
rendimiento moderado con 53 % y 52 % respectivamente. Respecto a la metodología A
para esta serie de derivados, los valores de rendimiento fueron regulares a muy buenos.
Se evidenciaron diferencias muy marcadas entre ambas metodologías para los
compuestos 2, 3, 8 y 12.
En la serie de la 5-metil-1-indanona se observaron para todos los casos diferencias muy
marcadas de rendimiento entre ambas metodologías. Inclusive, hubo dos compuestos, el

Resultados y discusión | Síntesis de las N4-TSCs
52
16 y 17, que no pudieron ser obtenidos empleando la metodología A pero sí con la B.
En tanto, para los otros derivados los rendimientos alcanzados con el método A fueron
muy pobres.
En la serie de la 4,5-dimetoxi-1-indanona los rendimientos con el método B fueron
variables, con valores desde 51 % con R5 = Cl a 97 % con R5 = F. En el caso del método
A todas las reacciones tuvieron un rendimiento pobre, el máximo fue de 52 % para el
compuesto 20 y el mínimo de 14 % para el 23.
Por último, la serie de la 5-bromo-1-indanona fue la que presentó en general los
rendimientos más bajos con valores máximos de 41 % y 66 % para la metodología A y
B respectivamente. Al igual que con las otras series, para ésta se siguió observando las
ventajas del uso de microondas frente a la metodología convencional ya que en todos
los casos fue posible mejorar el rendimiento de la reacción con el método B e incluso
para el caso del derivado 28 se logró obtener el compuesto cuando de la otra forma no
pudo ser posible.
Respecto a los sustituyentes en el grupo arilo del N4 los rendimientos en general fueron
aleatorios según la serie que se trató, concluyéndose que no influyen en las variables
analizadas.
1.4. Análisis de la isomería Z/E.
Las TSCs tienen la capacidad de formar isómeros Z y E alrededor de la unión C=N1, por
lo cual en la síntesis es probable obtener uno de ellos o una mezcla de los mismos
(Figura 20). La formación y estabilidad de estas dos configuraciones depende de cada
TSC en particular.
Figura 20. Isómeros Z (izquierda) y E (derecha) de las N4-TSCs.

Resultados y discusión | Síntesis de las N4-TSCs
53
Mediante el análisis espectroscópico de las N4-TSCs por
1H-RMN y
13C-RMN se
observó la presencia de señales únicas para cada protón y carbono respectivamente
(Figuras 21 y 22, Tabla 2), demostrando la formación preferencial de uno de los
isómeros frente al otro. Asimismo, se realizó la asignación de los protones y carbonos
por medio de espectros bidimensionales de HSQC y HMBC (Figuras 23 y 24, Tabla 3).
Mediante dichos análisis se asignó el H unido al N2, cuyo desplazamientos químico para
los compuestos sintetizados se encuentra entre 8,4 y 8,6 ppm, mientras que el H unido al
N4 tiene un desplazamiento químico entre 9,1 y 9,7 ppm.
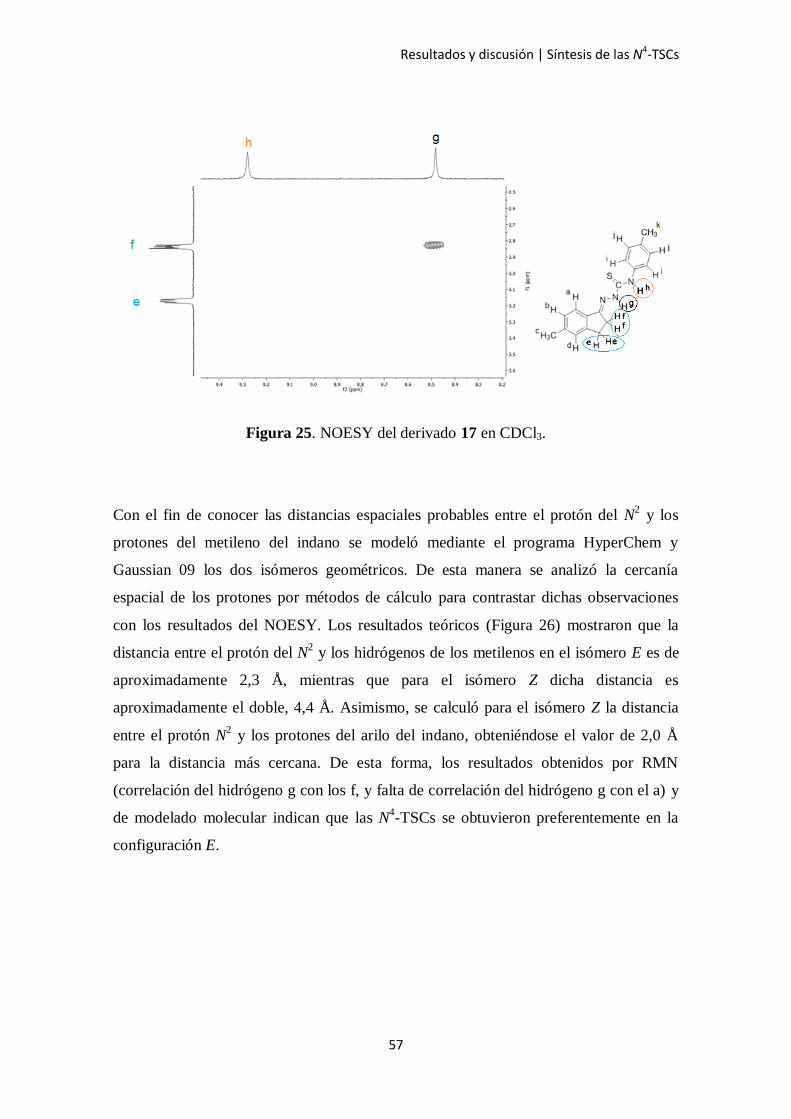
Luego de la asignación de las señales se analizó cuál de los isómeros Z ó E fue
obtenido. Para ello, se llevó a cabo un estudio de NOESY el cual permite determinar
qué protones se aproximan entre sí en el espacio independientemente del número de
enlaces que haya entre ellos. Luego del análisis se observó que el protón de
aproximadamente 8,4 a 8,6 ppm que corresponde al N2 se acopla con el protón de
aproximadamente 2,3 ppm que corresponde a los hidrógenos del metileno del núcleo
indánico más próximos a la función tiosemicarbazona (Figura 25). Cabe destacar que en
ningún espectro se observó correlación entre el protón del N2 con los protones del arilo
del indano.
Los espectros mostrados a continuación corresponden al compuesto 17, el cual fue
considerado como ejemplo. Las señales y correlaciones observadas y analizadas fueron
repetitivas para toda la serie de compuestos estudiada.

Resultados y discusión | Síntesis de las N4-TSCs
54
Figura 21. A) 1H-RMN del derivado 17 en CDCl3. B) Ampliación de espectro
correspondiente a la zona aromática.
A
B

Resultados y discusión | Síntesis de las N4-TSCs
55
H δ (ppm) Multiplicidad Integración J (Hz)
a 7,66 d 1H 7,88
b 7,15 d 1H 7,88
c 2,42 s 3H -
d 7,19 s 1H -
e 3,17 m 2H -
f 2,84 m 2H -
g 8,48 s 1H -
h 9,28 s 1H -
i 7,55 d 2H 8,27
j 7,22 d 2H 8,27
k 2,38 s 3H -
Tabla 2. Señales del espectro 1H-RMN del derivado 17 en CDCl3.
Figura 22. 13
C-RMN del derivado 17 en CDCl3

Resultados y discusión | Síntesis de las N4-TSCs
56
Figura 23. HSQC del derivado 17 en CDCl3.
Figura 24. HMBC del derivado 17 en CDCl3.
δ 1H (ppm) δ
13C (ppm) Correlación
H C
2,38 21,0 k o
2,42 22,0 c e
2,84 27,0 f i
3,17 28,6 e h
7,15 128,8 b c
7,19 126,4 d f
7,22 129,6 j n
7,55 124,8 i m
7,66 121,7 a b
Tabla 3. Correlaciones carbono-hidrógeno del derivado 17 en CDCl3 determinadas por
HSQC y HMBC.
Resultados y discusión | Síntesis de las N4-TSCs
57
Figura 25. NOESY del derivado 17 en CDCl3.
Con el fin de conocer las distancias espaciales probables entre el protón del N2 y los
protones del metileno del indano se modeló mediante el programa HyperChem y
Gaussian 09 los dos isómeros geométricos. De esta manera se analizó la cercanía
espacial de los protones por métodos de cálculo para contrastar dichas observaciones
con los resultados del NOESY. Los resultados teóricos (Figura 26) mostraron que la
distancia entre el protón del N2 y los hidrógenos de los metilenos en el isómero E es de
aproximadamente 2,3 Å, mientras que para el isómero Z dicha distancia es
aproximadamente el doble, 4,4 Å. Asimismo, se calculó para el isómero Z la distancia
entre el protón N2 y los protones del arilo del indano, obteniéndose el valor de 2,0 Å
para la distancia más cercana. De esta forma, los resultados obtenidos por RMN
(correlación del hidrógeno g con los f, y falta de correlación del hidrógeno g con el a) y
de modelado molecular indican que las N4-TSCs se obtuvieron preferentemente en la
configuración E.

Resultados y discusión | Síntesis de las N4-TSCs
58
Figura 26. Distancias interatómicas para el isómero A) E y B) Z
En resumen:
Se sintetizaron 30 N4-TSCs de las cuales 24 son nuevas.
Se evaluaron dos metodologías de síntesis, siendo la síntesis one-pot
mediante el uso de tecnología de microondas la mejor opción para preparar
estos derivados. Los compuestos se obtienen en tiempos cortos de reacción
y con buenos rendimientos.
Se estudió la isomería Z/E mediante diversas técnicas de RMN resultando el
isómero E el obtenido preferentemente en la síntesis de los compuestos.
A B

Resultados y discusión | Virus de la diarrea viral bovina
59
2. Virus de la diarrea viral bovina
Como se mencionó en la introducción, nuestro grupo de investigación sintetizó una
serie de TSCs las cuales fueron evaluadas frente al VDVB26
. De dicha serie, el
compuesto derivado de la 5,6-dimetoxi-1-indanona (5,6-TSC) resultó ser el más activo.
Por este motivo, se continuó en un principio con el estudio de N4-TSCs derivadas de
esta indanona con el objetivo de mejorar el perfil de liposolubilidad. Para completar
dicho estudio se evaluaron nuevas N4-TSCs obtenidas de otras indanonas que habían
resultado activas.
En esta sección se describen las actividades de las N4-TSCs frente al VDVB y se
presenta un estudio de la relación estructura-actividad con el fin de determinar las
propiedades que modulan la actividad observada. Se exponen resultados de resistencia
cruzada del compuesto más activo y la 5,6-TSC frente a la enzima RdRp (wild type y
mutada N264D/A392E) y estudios de docking molecular con el fin de conocer el
probable mecanismo de acción de los compuestos como inhibidores de la polimerasa
viral. Finalmente, se muestran los resultados del cálculo de energía libre por MM/PBSA
para conocer las contribuciones energéticas claves del complejo de la enzima con los
compuestos.
2.1. Actividad antiviral de las N4-TSCs
Se evaluaron 24 N4-TSCs frente al VDVB mediante el ensayo de reducción de unidades
formadoras de placas virales (UFP).
Dicha metodología permite titular las partículas virales que se producen en una célula
cuando es infectada. El fundamento de la técnica se centra en la infección viral de
células (en este caso MDBK), la cual genera una progenie viral que al liberarse inicia
nuevos ciclos de infección y replicación en células adyacentes dando lugar a zonas
localizadas de células muertas denominadas placas. Este tipo de estudio permite la
evaluación de compuestos como antivirales, ya que se determina la capacidad de los
mismos de disminuir el número de placas formadas. Dado que hasta el momento no
existe un fármaco para el tratamiento de la enfermedad de la DVB, para el ensayo se
tomaron como compuestos de referencia la 5,6-TSC y la ribavirina (análogo sintético de
guanosina de amplio espectro).

Resultados y discusión | Virus de la diarrea viral bovina
60
Los resultados de actividad antiviral mostraron que varias N4-TSCs fueron activas frente
al virus. Los compuestos 7 a 11 y el 14 presentaron valores de CE50 por debajo de 10
µM (Tabla 4). A su vez, se determinó la citotoxicidad de los compuestos frente a células
MDBK mediante el método MTS/PES y se determinó la selectividad de los mismos (IS)
como el cociente entre la máxima concentración evaluada para la citotoxicidad y la CI50.
Los resultados indicaron que los compuestos 17 y 23 presentaron una CC50 de 24,1 y
13,0 µM. Para el resto de las N4-TSCs no se evidenció citotoxicidad a la máxima
concentración soluble en el medio de cultivo (Tabla 4).
2.2. Relación estructura-actividad
Con el fin de encontrar las características estructurales de las N4-TSCs que modifican la
actividad anti-VDVB, se llevó a cabo un análisis de la relación estructura-actividad.
Los resultados indicaron que el compuesto más activo fue el derivado 8, con un valor de
CE50 de 0,7 µM. Dicho compuesto fue 11 veces más activo que la droga de referencia
ribavirina (CE50 = 7,7 µM) y 5 veces más que la TSC de la cual deriva (CE50 de la 5,6-
TSC = 3,8 µM).
Se observó que los derivados con la sustitución 5,6-dimetoxilo al igual que el
compuesto de referencia presentaron muy buenas actividades con valores de CE50 de 0,7
a 12,9 µM. Los compuestos con 5-metilo en el núcleo indánico presentaron buenos
valores de CE50 de 8,7 a 19,9 µM, con excepción del compuesto 13 para el cual el valor
de CE50 no fue definido y el derivado 17 al cual no se le determinó su actividad por
haber presentado citotoxicidad. En este análisis se pone en contraste la pobre actividad
presentada por todos los compuestos sin sustituir en el núcleo indánico y con la
sustitución 4,5-dimetoxilo, para los cuales no se pudo establecer un valor definido de
CE50 para ningún compuesto, encontrándose valores superiores a 10, 15 ó 20 µM. Se
evidencia así la importancia de los sustituyentes y su posición en el núcleo indánico
para que el compuesto resulte activo/inactivo frente al VDVB.

Resultados y discusión | Virus de la diarrea viral bovina
61
N4-TSC R1 R2 R3 R4 CE50 (µM) CC50 (µM) IS
1 H H H H >10 >10 -
2 H
hHH
H H NO2 >10 >10 -
3 H
H H F >20 >20 -
4 H H H Cl >20 >20 -
5 H H H CH3 >20 >20 -
6 H H H OCH3 >10 >10 -
7 H OCH3 OCH3 H 8,4 ± 0,4 >10 >1,2
8 H OCH3 OCH3 NO2 0,7 ± 0,1 >10 >14,3
9 H OCH3 OCH3 F 4,0 ± 0,8 >20 >5,0
10 H OCH3 OCH3 Cl 6,6 ± 0,2 >10 >1,5
11 H OCH3 OCH3 CH3 8,8 ± 0,1 >20 >2,3
12 H OCH3 OCH3 OCH3 12,9 ± 0,9 >20 >1,6
13 H CH3 H H >10 >10 -
14 H CH3 H NO2 8,7 ± 1,1 >20 >2,3
15 H CH3 H F 11,3 ± 1,7 >20 >1,8
16 H CH3 H Cl 13,1 ± 2,0 >20 >1,5
17 H CH3 H CH3 ND 24,1 ± 1,7 -
18 H CH3 H OCH3 19,9 ± 2,6 >20 >1,0
19 OCH3 OCH3 H H >15 >15 -
20 OCH3 OCH3 H NO2 >15 >15 -
21 OCH3 OCH3 H F >15 >15 -
22 OCH3 OCH3 H Cl >15 >15 -
23 OCH3 OCH3 H CH3 ND 13,0 ± 3,3 -
24 OCH3 OCH3 H OCH3 >15 >15 -
5,6-TSC - - - - 3,8 ± 0,4 >80 >21,1
Ribavirina - - - - 7,7 ± 1,5 54,4± 3,8 7,1
Tabla 4. Valores de actividad in vitro anti-VDVB de las N4-TSCs. ND: no determinado.
En cuanto a las variaciones en la posición para del arilo unido al N4, el compuesto más
activo de esta familia de N4-TSCs posee la sustitución nitro (derivado 8). Dentro de la
serie 5-metilo, el 14 que también presenta dicho sustituyente fue el más activo (CE50 =
8,7 µM). Cabe mencionar que el nitro es el grupo con mayor efecto atractor de
electrones estudiado en esta serie, y fue al mismo tiempo el que resultó asociado a una

Resultados y discusión | Virus de la diarrea viral bovina
62
mejor actividad antiviral. Asimismo, la incorporación de halógenos también produjo un
aumento de la actividad comparada con aquellos compuestos que están sin sustituir en
posición para. El compuesto 9 sustituido con fluor y el derivado 10 con cloro
presentaron una CE50 de 4,0 y 6,6 µM respectivamente. Para la serie 5-metilo dichos
sustituyentes presentaron actividad en el mismo orden, es decir más activo el sustituido
con fluor con una CE50 de 11,3 µM (derivado 15) y luego con el cloro con una CE50 de
13,1 µM (derivado 16). Estas observaciones pueden ser explicadas por el notable efecto
inductivo de los halógenos. En particular, el fluor presenta una mayor
electronegatividad que el átomo de cloro, lo cual lo posiciona como más activo. Por otro
lado, la sustitución del arilo con metoxilo (dador de electrones) fue la que dio lugar a
compuestos con la más baja actividad para la serie 5,6-dimetoxilo y 5-metilo con un
valor de CE50 de 12,9 y 19,9 µM para el compuesto 12 y 18 respectivamente. En el caso
de la serie 5,6-dimetoxilo, la sustitución con un grupo metilo con menor efecto dador de
electrones respecto al metoxilo presentó una mayor actividad (CE50 del compuesto 11 =
8,8 µM)
En resumen, teniendo en cuenta el análisis de las características de los sustituyentes en
la posición para del arilo unido al N4, se pudo observar que grupos atractores de
electrones favorecen la actividad mientras que grupos dadores la disminuyen. Se puede
inferir que el efecto electrónico en el R4 es el que modula la actividad antiviral y que la
sustitución en el indano juega un rol determinante para definir activos/inactivos frente al
VDVB para esta serie de compuestos.
2.3. Estudios de resistencia cruzada
La ARN polimerasa dependiente de ARN codificada por el virus (RdRp) lleva a cabo la
replicación del genoma viral. La estructura tridimensional de la enzima se parece a una
mano derecha compuesta de palma, pulgar y dedos (Figura 27) como se observa en
otras clases de polimerasas: ARN polimerasa dependiente de ADN, ADN polimerasa
dependiente de ADN y ADN polimerasa dependiente de ARN (transcriptasa reversa).
Sin embargo, los dedos y los dominios del pulgar varían significativamente entre las
diferentes clases de polimerasas. Una característica única de la estructura de la RdRp
son las ''yemas de los dedos'', que consisten en varias hebras de cadenas polipeptídicas
que conectan los dedos y el pulgar87
. Esta conexión restringe el gran movimiento que

Resultados y discusión | Virus de la diarrea viral bovina
63
estos dominios exhiben en otras clases de polimerasas. Las yemas de los dedos
contienen los motivos I y II de la RdRp los cuales se han propuesto que unen el
templado de ARN y los nucleótidos trifosfatos88,89
.
Figura 27. Estructura de la RdRp. La palma, dedos y pulgar se encuentran coloreados
en rojo, verde y azul respectivamente.
En trabajos previos del grupo de investigación, la 5,6-TSC fue caracterizada como un
inhibidor no nucleosídico de la RdRp viral27
. En dicho estudio fueron seleccionadas
cinco mutantes del virus resistentes al compuesto pero sensibles a ribavirina, cuatro de
las cuales llevan la mutación N264D, ubicada en la yema de los dedos de la RdRp y la
quinta mutante muestra además la mutación A392E, ubicada en el dominio de los
dedos.
Teniendo en cuenta estos resultados, se estudió mediante ensayos de resistencia cruzada
la nueva N4-TSC 8 frente al virus con la RdRp wild type y al virus con la doble mutante
N264D/A392E resistente a 5,6-TSC (de aquí en adelante RdRp mutada) con el fin de
dilucidar el mecanismo de acción del compuesto como probable inhibidor de la enzima.
Asimismo, se evaluó también la 5,6-TSC como compuesto de referencia para contrastar
los resultados encontrados. La resistencia cruzada fue evaluada mediante la reducción
del efecto citopático (ECP) por MTS/PES. Dicho ensayo se basa en los cambios
bioquímicos y moleculares que ocurren en las células infectadas, los cuales alteran la
viabilidad celular y pueden determinarse de forma espectroscópica.
Los resultados mostraron que el virus con la RdRp wild type fue sensible a la N4-TSC 8
y a la 5,6-TSC (CE50 = 0,45 y 1,95 µM respectivamente), es decir que dichos
compuestos fueron activos como anti-VDVB (Tabla 5). Estos resultados estuvieron en
concordancia con la actividad encontrada previamente para los compuestos con el

Resultados y discusión | Virus de la diarrea viral bovina
64
método de reducción de UFP. Por otro lado, los resultados del ensayo de ECP de los
compuestos frente al virus con la RdRp doble mutada mostraron valores de CE50
mayores a 10 y 80 µM para el derivado 8 y la 5,6-TSC respectivamente. De esta forma
se evidenció que el efecto de los compuestos es sensible a las mutaciones de la
polimerasa ya que para la 5,6-TSC y el derivado 8 se redujo la actividad más de 41 y 22
veces respectivamente. De los resultados obtenidos se concluye que la RdRp, al igual
que para la 5,6-TSC, sería el blanco de acción del compuesto 8.
CE50 (µM)
5,6-TSC Compuesto 8
VDVB con la RdRp wild type 1,95 ± 0,54 0,45 ± 0,08
VDVB con la RdRp N264D/A392E >80 >10
Tabla 5. Resultados de resistencia cruzada para la 5,6-TSC y la N4-TSC 8.
2.4. Docking molecular
Dados los resultados de resistencia cruzada y con el fin de explorar las interacciones
ligando-RdRp que determinan el modo de unión y la influencia de los aminoácidos
mutados en el desarrollo de resistencia, se realizó un estudio de docking molecular.
Dicho estudio es considerado una técnica computacional directa ya que se debe conocer
la estructura tridimensional de la diana biológica, la cual es utilizada para predecir la
conformación que adopta un ligando en el espacio explorado de la macromolécula. Sin
embargo, cabe aclarar que los estudios de docking no suelen ser adecuados para analizar
los valores teóricos de energía libre de unión debido a las numerosas aproximaciones
del método90,91
.
Para la realización de los cálculos de docking, el programa efectúa una búsqueda
conformacional del ligando dentro de una grilla tridimensional de puntos en la cual se
encuentra la macromolécula de interés. El programa arroja una serie de resultados que
corresponden a diferentes poses del ligando ordenadas en un ranking en función del
tamaño del cluster (conjunto de conformaciones similares) y de la energía del complejo.
Para el estudio de docking se realizó el procesamiento computacional de la estructura
cristalizada de la RdRp (PDB: 1S48) en la cual se reemplazaron las seleniometioninas

Resultados y discusión | Virus de la diarrea viral bovina
65
(las que provienen de la técnica utilizada para la cristalografía de rayos X) por
metioninas y se refinó el modelo teniendo en cuenta el diagrama de Ramachandran. En
paralelo se modeló y minimizó energéticamente el compuesto 8 y la 5,6-TSC.
En el proceso para realizar un docking resulta importante definir un espacio
tridimensional en la macromolécula en el cual se llevarán a cabo los cálculos. Este
espacio puede delimitarse a partir de conocer el sitio activo o de unión de ligandos.
Teniendo en cuenta que ningún ligando fue co-cristalizado con la enzima, se utilizó el
programa metaPocket 2.0 para identificar sitios putativos de unión en la superficie de la
enzima. Dicho programa utiliza ocho métodos distintos de predicción de sitios de unión
y los combina para mejorar los cálculos92
. Mediante este programa fue posible encontrar
tres sitios putativos de unión de los compuestos estudiados con la enzima, de los cuales
sólo uno se ubicó en la región cercana a los aminoácidos 264 y 392 donde se encuentran
las mutaciones de la RdRp resistente (Figura 28). Por lo tanto, éste fue el sitio donde se
llevó a cabo el estudio de docking.
Figura 28. Representación de la caja que delimita el sitio donde se llevó a cabo el
estudio de docking. Los aminoácidos 264 y 392 se encuentran ilustrados como esferas.
Los resultados mostraron las distintas conformaciones que puede adoptar cada ligando
en el sitio estudiado de la enzima. De todas ellas, se eligió la que tuvo el valor teórico de
energía libre de unión más favorable. Las interacciones observadas del compuesto 8 con
la RdRp wild type (Figura 29 A) fueron: i) dos puentes de hidrógeno entre el N2 y N
4 de
la función tiosemicarbazona con los aminoácidos E226 y F224, respectivamente; (ii)
puente de hidrógeno entre el oxígeno del grupo 6-metoxilo y el grupo hidroxilo de
Y289; (iii) interacción π-π stacking entre el arilo unido al N4 y la cadena lateral de
F224; (iv) interacción no polar entre el anillo indánico y A222.

Resultados y discusión | Virus de la diarrea viral bovina
66
Por otro lado, en la posición de mejor puntuación del complejo RdRp mutada-
compuesto 8 (Figura 29 B) se observó: i) puente de hidrógeno entre el N1 y el
aminoácido A222; ii) puente de hidrógeno entre el N4 y G220; iii) interacción no polar
entre el anillo indánico y A222. Teniendo en cuenta que la disposición espacial entre los
dos complejos estudiados fue muy distinta, se decidió analizar una segunda orientación
del ligando con la RdRp mutada que fuese similar a la adoptada en el complejo con
RdRp wild type (Figura 29 C). Se observó que a pesar de la orientación similar del
ligando en el sitio de unión, sólo se formó un puente de hidrógeno entre N2 y el
aminoácido E226. Además, cabe destacar que no se formaron puentes de hidrógeno
entre el grupo 5,6-dimetoxilo del indano y los aminoácidos de la enzima mutada.
Asimismo, con el fin de comparar si el modo de unión y las interacciones observadas
para el derivado 8 se mantenían en la 5,6-TSC con la enzima wild type, se realizó un
docking para este último compuesto. Los resultados indicaron que la conformación
adoptada en ambos casos con la enzima fue superponible y se encontraron, excepto las
correspondientes al arilo N4, el mismo tipo de interacciones con la RdRp (Figura 30).
En resumen, los resultados mostraron la diferente calidad y cantidad de interacciones
entre el compuesto 8 y la enzima wild type y mutada. Dicho estudio resaltó la
importancia de la sustitución 5,6-dimetoxilo en el núcleo indánico ya que a diferencia
de la wild type, no pudo establecerse ningún puente de hidrógeno con la enzima mutada.
Además, la superposición del complejo enzima-ligando para el compuesto 8 y la 5,6-
TSC en la enzima wild type permitió visualizar que las conformaciones adoptadas
fueron similares, por lo cual el mecanismo de acción de los compuestos sería similar.
Dichos resultados de cálculo están en concordancia con los obtenidos en el estudio de
resistencia cruzada, ya que ambos compuestos fueron activos en el virus con la enzima
wild type pero dicha actividad resultó afectada cuando el virus presentaba la doble
mutación. Por otro lado, los resultados también concuerdan con lo observado en la
relación estructura-actividad, dado que las N4-TSCs con la sustitución 5,6-dimetoxilo
resultaron las más activas, por lo tanto dicha sustitución es clave para la inhibición de la
enzima y por lo tanto para la acción anti-VDVB.
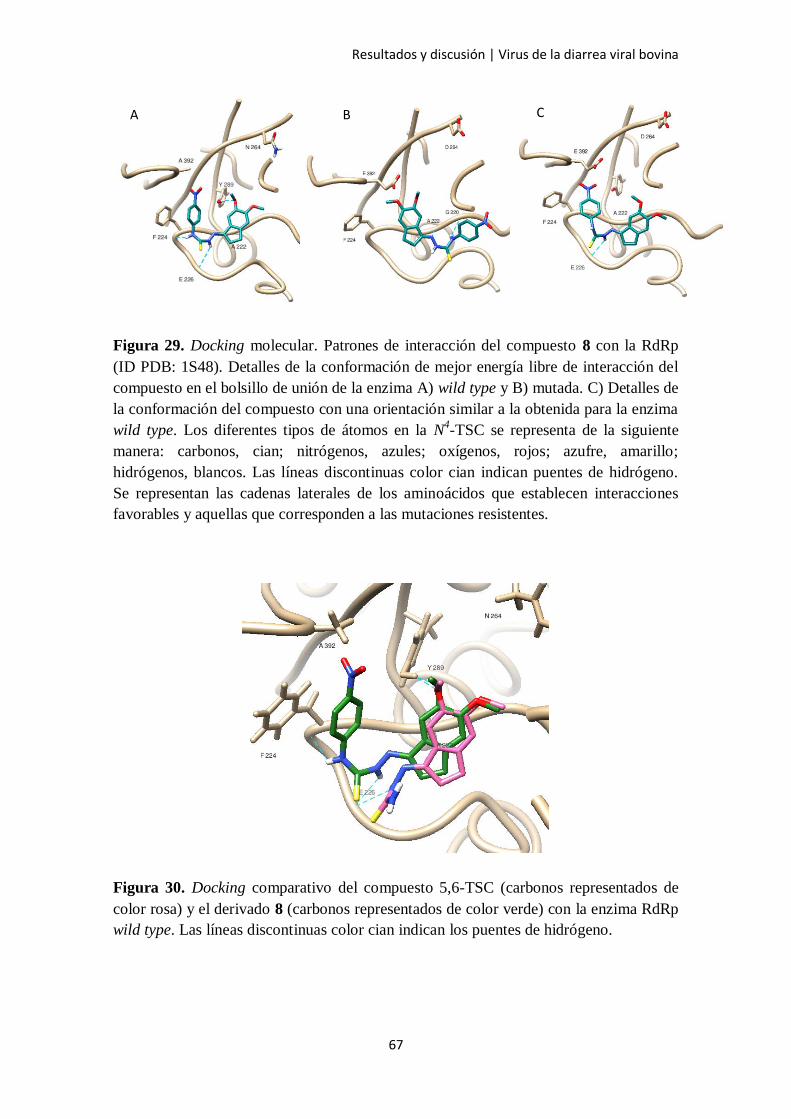
Resultados y discusión | Virus de la diarrea viral bovina
67
Figura 29. Docking molecular. Patrones de interacción del compuesto 8 con la RdRp
(ID PDB: 1S48). Detalles de la conformación de mejor energía libre de interacción del
compuesto en el bolsillo de unión de la enzima A) wild type y B) mutada. C) Detalles de
la conformación del compuesto con una orientación similar a la obtenida para la enzima
wild type. Los diferentes tipos de átomos en la N4-TSC se representa de la siguiente
manera: carbonos, cian; nitrógenos, azules; oxígenos, rojos; azufre, amarillo;
hidrógenos, blancos. Las líneas discontinuas color cian indican puentes de hidrógeno.
Se representan las cadenas laterales de los aminoácidos que establecen interacciones
favorables y aquellas que corresponden a las mutaciones resistentes.
Figura 30. Docking comparativo del compuesto 5,6-TSC (carbonos representados de
color rosa) y el derivado 8 (carbonos representados de color verde) con la enzima RdRp
wild type. Las líneas discontinuas color cian indican los puentes de hidrógeno.
A B C

Resultados y discusión | Virus de la diarrea viral bovina
68
2.5. Cálculo de la energía libre de unión
Las simulaciones de dinámica molecular son un complemento para analizar mecanismos
de acción ya que permiten describir un sistema de manera temporal. A partir de un
modelo inicial formado por N partículas que interaccionan, el cual puede provenir de
datos experimentales o teóricos, se determinan sus movimientos individuales en función
del tiempo. Dicha trayectoria sigue las ecuaciones correspondientes a las leyes de
movimiento de Newton, mediante las cuales se genera la secuencia temporal de la
evolución del sistema.
Debido a la complejidad de los cálculos, las ecuaciones de movimiento son integradas
en pequeños pasos sucesivos luego de un cierto tiempo denominado time step (dt), en el
cual en cada uno de ellos se obtiene una descripción del sistema (frame). Asimismo, en
cada paso se calculan las fuerzas de los átomos y luego se combinan con las posiciones
iniciales para generar la trayectoria. La elección del dt resulta de considerar la duración
de los fenómenos que se desean observar y el “costo” computacional. Para las
biomoléculas los movimientos de las vibraciones de los enlaces se encuentran en el
rango de los femtosegundos (fs), por lo cual el dt debería ser lo suficientemente
pequeño para reproducir estos movimientos.
Las interacciones entre macromoléculas y ligandos pueden ser estudiadas utilizando
herramientas de dinámica molecular, mediante las cuales es posible estimar la energía
libre de unión. Entre ellas, se encuentra el método de mecánica molecular/Poisson
Boltzmann surface area (MM/PBSA), el cual permite obtener tendencias comparativas
de gran utilidad para el estudio de interacciones ligando-proteína.
El método de MM/PBSA evalúa la energía libre de los estados iniciales y finales, por lo
cual se lo considera como un método de punto final. El cálculo de energía libre puede
dividirse en tres partes como se indica en la siguiente ecuación:
El primer término de ∆EMM es la energía de mecánica molecular, que incluye las
contribuciones de enlace, de van der Waals (∆EvdW) y electrostática (∆Eele). Cabe aclarar
que cuando no existen uniones covalentes entre el ligando y la macromolécula la
contribución de enlace es nula. El segundo término es la energía de solvatación (∆Gsol),

Resultados y discusión | Virus de la diarrea viral bovina
69
que incluye el componente polar (∆Gpol) y no polar (∆Gnopol). El último término es la
temperatura absoluta por la entropía (T∆S).
Con el fin de medir el ∆Gunión para el derivado 5,6-TSC y la N4-TSC 8 con la RdRp wild
type y mutada, se realizaron los cálculos de MM/PBSA de dichos compuestos con las
dos formas de la enzima. Como se indica en la Tabla 6, el complejo correspondiente al
compuesto 8 con la RdRp wild type presentó el valor de energía más favorable (∆Gunión
= -8,37 kcal/mol), seguido por el complejo de la 5,6-TSC con la RdRp wild type
(∆Gunión = -7,38 kcal/mol). En cambio, los complejos con la enzima mutada mostraron
un cambio desfavorable de energía con valores de ∆Gunión de -6,57 y -5,89 kcal/mol para
el derivado 8 y la 5,6-TSC respectivamente. De esta manera, se observó que el orden de
las energías estuvo en concordancia con los valores de actividad experimentales, es
decir las interacciones de ∆Gunión con valores más negativos se correspondieron con las
de menor CE50.
Con el fin de determinar el tipo de interacciones que más contribuyen en el ∆Gunión, se
realizaron comparaciones de los componentes de las energías libres (Tabla 6). En todos
los complejos la energía de van der Waals debida a las interacciones de tipo
hidrofóbicas fue la interacción más contribuyente para la unión. También se registraron
en todos los casos interacciones favorables debido a la energía electrostática y en menor
medida debida a la energía de solvatación no polar. Por otro lado, dichas contribuciones
favorables fueron contrarrestadas por la energía de solvatación polar y la entropía.
Al comparar las variaciones de cada componente de la energía entre los dos sistemas
correspondientes al derivado 8, se observó que en la contribución electrostática hubo
una diferencia de 1,60 kcal/mol a favor del complejo con la enzima wild type, siendo
este tipo de energía el que mostró la mayor variación entre los dos sistemas estudiados.
En el caso de las diferencias de energía de van der Waals el valor de diferencia
encontrado fue de 1,39 kcal/mol nuevamente a favor del sistema wild type. Para el
componente de la energía de solvatación polar, la variación fue de 0,94 kcal/mol a favor
del sistema mutado. Cabe señalar que la contribución de la solvatación no polar y
entrópica no mostraron diferencias entre los modelos estudiados. Para el compuesto 5,6-
TSC se encontraron resultados análogos, es decir, los cambios de energía entre los
complejos se centraron principalmente en la contribución electrostática (variación de
0,96 kcal/mol) y las interacciones de van der Waals (variación de 0,95 kcal/mol).

Resultados y discusión | Virus de la diarrea viral bovina
70
Teniendo en cuenta que las mayores variaciones de energía entre los complejos con la
RdRp wild type y mutada estuvieron centradas en las interacciones electrostáticas, se
analizó el número de puentes de hidrógenos a lo largo del tiempo (Figura 31). Los
resultados indicaron que el complejo entre la enzima wild type con el compuesto 8
mantuvo al menos un puente de hidrógeno el 35% del tiempo. Además, cabe destacar,
que en dicho complejo se observó la presencia de los 3 puentes de hidrógeno
encontrados previamente en el estudio de docking. En cuanto al complejo 5,6-TSC y la
enzima wild type, hubo al menos un puente de hidrógeno el 30 % del tiempo. Por el
contrario, los complejos con la enzima mutada mostraron que a lo largo del tiempo
estudiado los puentes de hidrógeno estuvieron presentes en menos del 15% de los
frames. De esta forma, se evidenció que la formación de puentes de hidrógeno sería
determinante para la interacción entre los compuestos y la enzima y por lo tanto para las
actividades observadas en el ensayo de resistencia cruzada.
Los valores de ∆Gunión calculados por MM/PBSA indicaron diferencias entre los cuatro
modelos estudiados, resultando los sistemas con la enzima wild type las de menor
energía y por lo tanto las más favorables termodinámicamente. Asimismo, se estableció
como factor principal de las diferencias observadas la contribución electrostática. Estos
resultados computacionales estuvieron en concordancia con el ensayo de resistencia
cruzada y proporcionaron información sobre la magnitud y calidad de interacciones
establecidas entre la polimerasa viral y el ligando. Por lo tanto, a partir de los resultados
teóricos se infiere una directa correlación entre la capacidad del ligando de interaccionar
con la enzima y la actividad anti-VDVB.
Compuesto 8 Compuesto 8 5,6-TSC 5,6-TSC
RdRp wild type RdRp mutada RdRp wild type RdRp mutada
ΔEvdW -37,08 -35,69 -36,10 -35,15
ΔEele -14,12 -12,52 -13,02 -12,06
ΔGNP -4,07 -4,27 -3,89 -3,92
ΔGPB 32,82 31,88 33,38 33,12
-TΔS 14,08 14,03 12,25 12,12
ΔGunión -8,37 -6,57 -7,38 -5,89
Tabla 6. Energías libre de unión y sus componentes para el compuesto 8 y 5,6-TSC con
la enzima RdRp wild type y mutada. Los valores de energía se expresan en kcal/mol.

Resultados y discusión | Virus de la diarrea viral bovina
71
Figura 31. Número de puentes de hidrógeno en función del tiempo. Complejo entre el
compuesto 8 y la: A) RdRp wild type; B) RdRp mutada. Complejo entre el compuesto
5,6-TSC y la: C) RdRp wild type; D) RdRp mutada.
A
B
C
D

Resultados y discusión | Virus de la diarrea viral bovina
72
En resumen:
La N4-TSC 8 es la más activa de la serie.
Grupos atractores de electrones en R4 y la sustitución 5,6-dimetoxilo en el
indano potencian la actividad antiviral.
Para la mayoría de los compuestos no se evidencian efectos citotóxicos en
células MDBK.
Los estudios de resistencia cruzada, docking y dinámica molecular indican
que la diana biológica del derivado 8 sería la RdRp viral.
.

Resultados y discusión | Trypanosoma cruzi
73
3. Trypanosoma cruzi
Con el fin de investigar la acción anti-tripanosomátida de las 30 N4-TSCs sintetizadas,
se determinaron de forma paralela las actividades biológicas de las mismas frente a T.
cruzi y T. brucei. En esta sección se describen y discuten los resultados de evaluación
antichagásica frente a las tres formas del parásito: epimastigote, tripomastigote y
amastigote y se presenta el análisis del estudio de la relación estructura-actividad. A la
vez, se exponen los resultados de citotoxicidad en células Vero. Por último, se muestran
los resultados de actividad de los compuestos más activos frente a la enzima cruzipaína,
llevada a cabo para dilucidar su probable mecanismo de acción.
3.1. Actividad anti T. cruzi de las N4-TSCs
Las 30 N4-TSCs fueron evaluadas frente a la forma epimastigote (clon CL Brener) y
tripomastigote (cepa Tulahuén, stock Tul2). Se realizó un screening a la concentración
final de 20 µM para conocer la potencia de los compuestos, de tal forma que se pudiese
identificar las N4-TSCs prometedoras. Asimismo, cabe mencionar que los valores de
CI50 determinados previamente frente a epimastigotes para esta familia de compuestos,
habían presentado valores desde 1,0 µM a mayores de 50 µM28
.
En los estudios frente a la forma epimastigote los resultados fueron expresados como
porcentaje de inhibición a la concentración de 20µM. En este primer experimento
ninguno de los compuestos resultó más activo que la droga de referencia benznidazol
(inhibición del 67,1 %). No obstante, los compuestos 7 y 10 mostraron porcentajes de
inhibición superiores al 50 % (Tabla 7). Cabe destacar que los compuestos 1, 4, 5, 7, 10
y 11 habían sido previamente estudiados frente a una cepa distinta de epimastigotes
(cepa Tulahuén), para la cual se había medido la actividad por un método
espectrofotométrico28
. En aquella experiencia, el compuesto 4 había presentado una CI50
de 25 µM y los otros cuatro compuestos habían mostrado una CI50 por debajo de 10
µM. Sin embargo, en esta oportunidad no se evidenciaron actividades similares, a
excepción del compuesto 4. Dichas diferencias pueden ser explicadas por la distinta
cepa estudiada, la variación inter-laboratorio y de operador, y el distinto método
empleado para la determinación de la actividad anti-chagásica.

Resultados y discusión | Trypanosoma cruzi
74
En una siguiente etapa, se continuó con los estudios para la forma tripomastigote (forma
infectiva en el mamífero) cuyos resultados fueron expresados como porcentaje de lisis.
Cuatro compuestos (1, 2, 4 y 7) mostraron valores de lisis superiores al del benznidazol
para esta forma del parásito (Tabla 7). Debido a estos resultados alentadores, se
ensayaron concentraciones crecientes de dichas N4-TSCs con el fin de determinar el
valor de CE50. Sin embargo, dada la solubilidad limitada de los compuestos en el medio
de cultivo no se pudo determinar dicho valor.
La comparación de los resultados entre las dos formas del parásito mostró para los
compuestos 1, 2, 4 a 7, 12, 13, 15 a 17, 21 y 28 una misma tendencia de actividad. Sin
embargo, para el resto de los compuestos hubo divergencias en los resultados obtenidos
entre ambas formas del parásito. En particular, hubo notables diferencias de actividad
para los compuestos 11 y 29 para los cuales el porcentaje de inhibición alcanzó casi el
50% y el 38% respectivamente en parásitos epimastigotes. No obstante, cuando se
estudió el efecto de dichas N4-TSCs para la forma tripomastigote la actividad fue nula
para ambos compuestos. El caso inverso ocurrió para los compuestos 14 y 20, es decir,
nula o muy pobre actividad fue observada en la forma epimastigote, pero valores de lisis
cercanos al 50 % fueron observados para la forma tripomastigote. De esta forma, según
el compuesto fue posible evidenciar o no una correlación de actividad medida entre los
dos estadios de T. cruzi.
En lo que respecta a la citotoxicidad de las N4-TSCs, se evaluó su efecto en células de
mamífero Vero mediante la reducción metabólica de MTT. Ninguno de los compuestos
evidenció citotoxicidad a la máxima concentración soluble en el medio de cultivo (entre
20 y 100 µM según el compuesto). Por otro lado, el benznidazol tampoco evidenció
efecto citotóxico para el valor de concentración final de 100 µM. Asimismo, para dicho
compuesto de referencia se pudo determinar la CC50 obteniéndose un valor de 291 µM.

Resultados y discusión | Trypanosoma cruzi
75
N4-TSC R1 R2 R3 R4 Epimastigotes Tripomastigotes
Células
Vero
% inhibición
(a 20 µM)
% lisis
(a 20 µM)
CC50
(µM)
1 H H H H 42,6 ± 1.9 56,7 ± 5,9 >20
2 H H H NO2 39,6 ± 3.4 56,0 ± 4,1 >50
3 H H H F 22,7 ± 2.1 9,1 ± 0,7 >80
4 H H H Cl 40,3 ± 3.4 67,9 ± 3,4 >40
5 H H H CH3 31,4 ± 2,0 27,3 ± 1,8 >50
6 H H H OCH3 16,3 ± 1,2 11,4 ± 1,1 >70
7 H OCH3 OCH3 H 55,0 ± 2,9 56,0 ± 5,5 >25
8 H OCH3 OCH3 NO2 27,7 ± 2,6 46,7 ± 3,7 >30
9 H OCH3 OCH3 F 41,2 ± 3,1 23,3 ± 2,6 >30
10 H OCH3 OCH3 Cl 65,4 ± 2,4 44,3 ± 0,8 >30
11 H OCH3 OCH3 CH3 49,2 ± 1,0 0,0 ± 0,0 >50
12 H OCH3 OCH3 OCH3 25,7 ± 2,3 18,2 ± 0,9 >30
13 H CH3 H H 39,7 ± 4,1 31,8 ± 2,7 >35
14 H CH3 H NO2 1,5 ± 1,2 51,1 ± 1,4 >60
15 H CH3 H F 42,7 ± 1,9 38,7 ± 1,8 >80
16 H CH3 H Cl 34,6 ± 2,6 46,0 ± 2,9 >50
17 H CH3 H CH3 15, 7 ± 2,5 27,0 ± 2,2 >50
18 H CH3 H OCH3 29,1 ± 1,8 36,5 ± 1,2 >50
19 OCH3 OCH3 H H 30,2 ± 2,6 40,9 ± 1,6 >25
20 OCH3 OCH3 H NO2 10,6 ± 1,3 42,9 ± 2,5 >30
21 OCH3 OCH3 H F 30,3 ± 1,7 36,4 ± 3,0 >30
22 OCH3 OCH3 H Cl 29,3 ± 1,9 4,5 ± 0,6 >30
23 OCH3 OCH3 H CH3 20,3 ± 1,7 41,3 ± 1,9 >50
24 OCH3 OCH3 H OCH3 8,6 ± 1,0 31,7 ± 2,8 >30
25 H Br H H 30,4 ± 4,2 50,7 ± 2,1 >20
26 H Br H NO2 25,5 ± 2,3 44,4 ± 3,9 >100

Resultados y discusión | Trypanosoma cruzi
76
27 H Br H F 45,4 ± 3,0 23,8 ± 1,5 >40
28 H Br H Cl 41,1 ± 2,7 36,2 ± 3,4 >30
29 H Br H CH3 38,0 ± 2,4 0,0 ± 0,0 >40
30 H Br H OCH3 30,1 ± 2,9 16,0 ± 1,2 >100
Bz - - - - 67,1 ± 2,1 52,4 ± 2,6 291±16,8
Tabla 7. Porcentajes de inhibición de T. cruzi en su forma epimastigote y de lisis para
la forma tripomastigote para una concentración 20 µM de compuesto. Resultados de
CC50 en células Vero. Bz: benznidazol.
En una etapa posterior, se ensayaron en amastigotes los cuatro compuestos que
presentaron la mayor actividad frente a los tripomastigotes, los cuales también habían
resultado activos frente a la forma epimastigote (1, 2, 4 y 7).
Los resultados obtenidos mostraron que los cuatro compuestos fueron activos frente a
los amastigotes de T. cruzi. En particular los compuestos 1 y 7 fueron los de mayor
actividad, con porcentajes de inhibición del 86 y 82 % (a la concentración final de 20
µM), respectivamente (Tabla 8 y Figuras 32 y 33). Por otro lado, los compuestos 2 y 4
también presentaron actividad pero con valores de inhibición del parásito menores que
los compuestos anteriores, de 54 y 64 % respectivamente para la misma concentración
(Tabla 8 y Figura 32). Teniendo en cuenta dichos resultados, se determinó la CI50 de las
N4-TSCs 1 y 7 para los cuales se obtuvieron los valores de 7,54 y 7,40 µM,
respectivamente (Tabla 8). El control positivo utilizado fue el benznidazol para el cual
se obtuvo una CI50 de 1,41 µM. Si bien las N4-TSCs no superaron la actividad de la
droga de referencia para esta forma del parásito, su actividad no resulta despreciable y
resultan de interés para ser utilizadas como punto de partida de futuras
farmacomodulaciones.
N4-TSC % de inhibición para: CI50 (µM)
20 µM 10 µM 1 µM
1 86,4 ± 13,0 61,6 ± 9,1 4,1 ± 1,1 7,54 ± 1,5
2 54,2 ± 8,4 31,1 ± 3,5 2,8 ± 0,9 ND
4 64,4 ± 12,6 17,5 ± 5,3 5,0 ± 1,7 ND
7 81,8 ± 9,3 71,7 ± 6,6 15,0 ± 2,8 7,40 ± 1,1
Tabla 8. Porcentajes de inhibición de T. cruzi en su forma amastigote luego del
tratamiento con N4-TSCs a las concentraciones de 20, 10 y 1 µM. ND: no determinado.
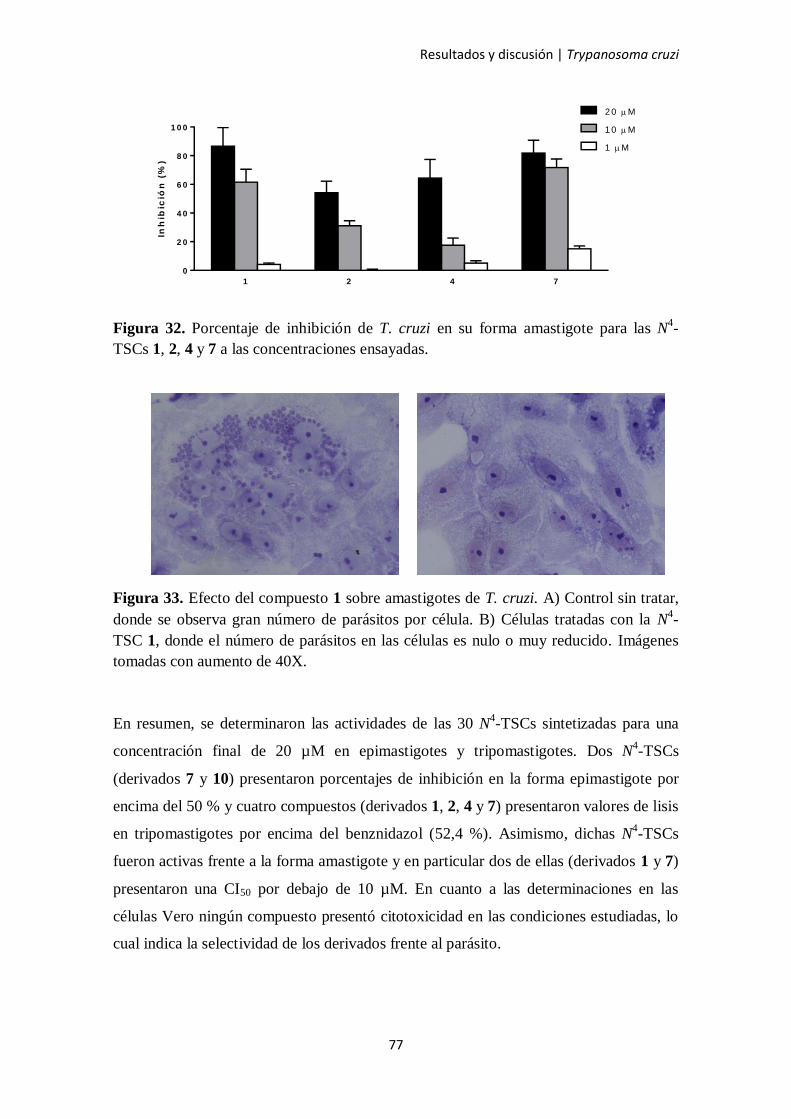
Resultados y discusión | Trypanosoma cruzi
77
Inh
ibic
ión
(%
)
1 2 4 7
0
2 0
4 0
6 0
8 0
1 0 0
2 0 M
1 0 M
1 M
Figura 32. Porcentaje de inhibición de T. cruzi en su forma amastigote para las N4-
TSCs 1, 2, 4 y 7 a las concentraciones ensayadas.
Figura 33. Efecto del compuesto 1 sobre amastigotes de T. cruzi. A) Control sin tratar,
donde se observa gran número de parásitos por célula. B) Células tratadas con la N4-
TSC 1, donde el número de parásitos en las células es nulo o muy reducido. Imágenes
tomadas con aumento de 40X.
En resumen, se determinaron las actividades de las 30 N4-TSCs sintetizadas para una
concentración final de 20 µM en epimastigotes y tripomastigotes. Dos N4-TSCs
(derivados 7 y 10) presentaron porcentajes de inhibición en la forma epimastigote por
encima del 50 % y cuatro compuestos (derivados 1, 2, 4 y 7) presentaron valores de lisis
en tripomastigotes por encima del benznidazol (52,4 %). Asimismo, dichas N4-TSCs
fueron activas frente a la forma amastigote y en particular dos de ellas (derivados 1 y 7)
presentaron una CI50 por debajo de 10 µM. En cuanto a las determinaciones en las
células Vero ningún compuesto presentó citotoxicidad en las condiciones estudiadas, lo
cual indica la selectividad de los derivados frente al parásito.
A B

Resultados y discusión | Trypanosoma cruzi
78
3.2. Relación estructura-actividad cualitativa
Con el fin de encontrar las características estructurales de las N4-TSCs que modifican la
actividad anti-T. cruzi se analizaron los resultados en la forma epimastigote y
tripomastigote.
3.2.1. Resultados en epimastigotes
Los resultados en epimastigotes mostraron que las variaciones en el arilo unido al N4
modifican la actividad. Compuestos con los sustituyentes: H, F y Cl fueron los que
presentaron los mejores valores de inhibición con valores de 30 a 55 %, 22 a 45 % y 29
a 65 % respectivamente. Cabe destacar que el hidrógeno y el fluor comparten la
característica de tener un reducido radio de van der Waals por lo cual el reemplazo de H
por F se lo considera desde la farmacomodulación como un cambio de isostería no
clásico. Asimismo, el reemplazo de F por Cl es un reemplazo isostérico clásico ya que
se conserva el mismo número de electrones de valencia. De esta forma, se evidenció que
las N4-TSCs que presentan dichos cambios isostéricos conservan la actividad evaluada.
Por otro lado, los sustituyentes nitro y metoxilo en el arilo unido al N4 presentaron los
porcentajes de inhibición más bajos, con valores de 1 a 39 % y 8 a 30 %
respectivamente. En cuanto a la sustitución con un grupo metilo, los valores de
actividad fueron variables, obteniéndose un rango de valor de inhibición entre 15 % y
49 %.
Respecto a la sustitución en el anillo indánico los derivados de 5,6-dimetoxilo fueron
los más activos, presentando cuatro compuestos (7, 9, 10 y 11) porcentajes de inhibición
entre 41 y 65 %. Los derivados con H y Br en posición 5 presentaron buenos valores de
actividad con porcentajes de inhibición que llegaron a 42 y 45 % respectivamente. Por
otro lado, la sustitución con 5-metilo presentó actividades anti-epimastigote variables,
con un valor máximo de inhibición del 42 % para el compuesto 15 y un valor mínimo
del 1 % para el derivado 14. La sustitución 4,5-dimetoxilo fue la menos favorable para
la actividad ya que ningún compuesto superó el 30 % de inhibición.
En resumen, teniendo en cuenta el análisis de las características de los sustituyentes en
el arilo unido al N4 se pudo observar que F y sus isósteros H y Cl presentaron los
mejores valores de actividad. Asimismo, se observó que la sustitución en el indano

Resultados y discusión | Trypanosoma cruzi
79
modula también la actividad, resultando la sustitución 5,6-dimetoxilo la más
conveniente para las N4-TSCs estudiadas.
3.2.2 Resultados en tripomastigotes
Los resultados en tripomastigotes también mostraron que las variaciones en el arilo
unido al N4 modifican la actividad. En este análisis, al igual que lo observado para
epimastigotes, la sustitución con R4 = Cl fue la que presentó el compuesto más activo
(N4-TSC 4) con el 68% de lisis. En cuanto a los compuestos con R4 = NO2 e H se
obtuvieron buenos valores de actividad para todos sus derivados, con porcentajes de
lisis de 42 a 56 % y de 31 a 56% respectivamente. En este sentido, hubo diferencia con
lo observado para la sustitución con nitro respecto a los epimastigotes. Asimismo, otra
diferencia con lo observado en epimastigotes, fue la actividad para los derivados con
fluor ya que en tripomastigotes no presentaron los mejores valores de actividad,
observándose el máximo porcentaje de lisis del 38 % y el mínimo del 9 %. Por otro
lado, los derivados con metilo y metoxilo presentaron pobres valores de lisis, a
excepción de los derivados 18 y 23 con 36 y 41 % respectivamente.
En cuanto a la sustitución en el núcleo indánico no se observó una tendencia definida,
todas las series de indanonas evaluadas presentaron actividades variables.
En síntesis, la actividad observada para las N4-TSCs en tripomastigotes fue buena para
los compuestos con la sustitución nitro e hidrógeno en R4. Sin embargo, más allá de esta
tendencia no se evidenció una relación directa de las actividades con las propiedades
estructurales de los compuestos.
3.3. Evaluación de las N4-TSCs frente a la cruzipaína
La cruzipaína (Cz) es la principal cisteína proteasa de T. cruzi. Dicha enzima es
codificada por una extensa familia de genes polimórficos y se expresa en los distintos
estadíos del ciclo de vida del parásito aunque su mayor concentración se presenta en los
epimastigotes25,93
. Respecto a su localización subcelular, se halla principalmente en los
lisosomas pero también se ha observado algunas isoformas minoritarias en la membrana
plasmática del parásito94
.

Resultados y discusión | Trypanosoma cruzi
80
La Cz juega un rol fundamental en la invasión de las células huésped ya que degrada los
tejidos, facilita la penetración del parásito y participa en la evasión del sistema inmune.
Asimismo, participa en la nutrición y proliferación de epimastigotes y amastigotes y en
la metaciclogénesis (transformación de epimastigotes a tripomastigotes metacíclicos)
por lo cual resulta esencial en el ciclo de vida del parásito93,95
. Por otro lado, la
inhibición de la Cz altera su paso desde el complejo de Golgi a los lisosomas
induciendo concomitantemente su acumulación en las vesículas de Golgi. Dicha
acumulación provoca alteraciones ultraestructurales que llevan finalmente a la muerte
del parásito. Por estos motivos la Cz ha resultado un blanco quimioterapéutico de gran
interés en la búsqueda de antichagásicos.
Desde el trabajo de Du y col.11
en 2002, numerosas TSCs han sido descriptas como
inhibidoras tanto de la enzima recombinante (cruzaína) como la nativa extraída del
parásito (Cz). De esta forma, se decidió investigar si los compuestos 1, 2, 4 y 7, los
cuales habían resultado ser los más activos sobre las tres formas del parásito, son
capaces de inhibir la enzima. Para determinar dicha inhibición se procedió a detectar la
actividad proteolítica de la Cz mediante una zimografía en presencia y ausencia de los
compuestos a ensayar. La técnica consiste en una electroforesis de la enzima en gel de
poliacrilamida, dicho gel tiene en su composición gelatina la cual sirve como sustrato de
la Cz. Luego de la incubación y tinción con Coomassie blue se puede detectar una zona
sin coloración la cual se corresponde con la degradación del gel debido a la actividad
proteolítica de la enzima.
Se realizaron dos zimografías debido a las limitaciones de tamaño del gel. En el primer
gel se incubaron las N4-TSCs 1 y 2. En el segundo gel se incubaron los compuestos 4 y
7. En ambos ensayos se corrió un estándar comercial de sustancias con pesos
moleculares conocidos, el cual permite identificar la posición correspondiente a la
enzima en la corrida, y los siguientes controles: Cz sin tratar (control sin tratar), Cz
incubada con 5% de DMSO (control de vehículo) y Cz incubada con E-64, un epóxido
que inhibe irreversiblemente una amplia gama de cisteína peptidasas96,97
(control
positivo de inhibición).
Los resultados en ambos geles mostraron la acción proteolítica de la Cz al encontrarse
degradado en el gel una región en forma de banda ancha correspondiente a un peso
molecular aparente cercano a los 45 kDa. Se observó que en el control de vehículo, el

Resultados y discusión | Trypanosoma cruzi
81
gel fue degradado de igual manera que en el control sin tratar, poniendo así en evidencia
que el DMSO, a la concentración evaluada, no tuvo efecto sobre la actividad
enzimática. Cabe aclarar que la banda de intensidad más tenue que se observó cercana a
los 55 kDa corresponde a una impureza que no se logra separar de la Cz con el método
de extracción utilizado98
. La calle correspondiente al control de inhibición de la enzima,
mostró que el gel se mantuvo integro en la región de la Cz luego de la incubación,
demostrando así la ausencia de actividad proteolítica debida a la presencia del inhibidor
de referencia.
Los resultados de las dos zimografías mostraron que luego de la incubación con los
compuestos 1, 2, 4 y 7 el gel fue degradado de igual forma que en el control sin tratar y
que el control de vehículo (Figuras 34 y 35). De esta forma se determinó que dichos
compuestos estudiados no tuvieron la propiedad de inhibir la Cz en las condiciones
evaluadas.
Teniendo en cuenta los resultados de la zimografía para los cuatro compuestos
ensayados, se concluye que ninguno de ellos logró inhibir a la cruzipaína. Por lo tanto,
esta enzima no sería el blanco de las N4-TSCs ensayadas y su efecto en el parásito sería
debido a otros mecanismos aún no estudiados.
Figura 34. Primera zimografía. a: estándar de pesos moleculares; b: Cz (control sin
tratar); c: Cz incubada con E-64 (control positivo de inhibición); d: Cz incubada con el
compuesto 1; e: Cz incubada con el compuesto 2; f: Cz incubada con DMSO (control de
vehículo). Las zonas más claras se deben a la degradación de la gelatina por parte de las
proteasas.
a b c d f
e
220 kDa
91 kDa
50 kDa
35 kDa

Resultados y discusión | Trypanosoma cruzi
82
Figura 35. Segunda zimografía. a: estándar de pesos moleculares; b: Cz (control sin
tratar); c: Cz incubada con E-64 (control positivo de inhibición); d: Cz incubada con el
compuesto 4; e: Cz incubada con el compuesto 7; f: Cz incubada con DMSO (control de
vehículo). Las zonas más claras se deben a la degradación de la gelatina por parte de las
proteasas.
En resumen:
Los derivados 1, 2, 4 y 7 son activos frente a las tres formas de T. cruzi y su
diana biológica no sería la cruzipaína.
Los átomos de H, F y Cl en R4 y la sustitución 5,6-dimetoxilo en el indano
potencian la actividad frente a epimastigotes.
No se estableció una relación directa entre las propiedades estructurales de
las N4-TSCs y la actividad frente a tripomastigotes.
Ningún compuesto resultó citotóxico en células Vero a la máxima
concentración evaluada
a b c d f
e
220 kDa
91 kDa
50 kDa
35 kDa

Resultados y discusión | Trypanosoma brucei
83
4. Trypanosoma brucei
Como se mencionó en la sección anterior, las 30 N4-TSCs sintetizadas fueron evaluadas
también como antiparasitarios frente a T. brucei. En esta sección se describen las
actividades de estos compuestos y se presenta y discute un análisis de la relación
estructura-actividad y QSAR llevado a cabo para determinar las propiedades que
modulan la actividad observada. Se exponen los resultados de inhibición de proteasas
por parte del compuesto más activo, realizada con el fin de conocer el probable
mecanismo de acción. Finalmente se muestran resultados de la captación intracelular del
compuesto y diversos estudios por citometría de flujo y microscopía electrónica para
evaluar el tipo de muerte celular que induce el compuesto más activo.
4.1. Actividad anti-T. brucei de las N4-TSCs
Las 30 N4-TSCs fueron evaluadas in vitro frente a T. brucei brucei en su forma
sanguínea (bloodstream) cepa 427. Se determinó para todos los compuestos la CI50
excepto para seis derivados (18, 20, 21, 22, 23 y 24) ya que la misma se encontraría por
encima de 30 µM, concentración en la cual los compuestos tienen una solubilidad
limitada.
Los resultados indicaron que seis compuestos (2, 8, 10, 14, 15 y 25) presentaron valores
de CI50 por debajo de 10 µM (Tabla 9). En particular, el compuesto 8 fue el más activo
de toda la serie estudiada con un valor de CI50 de 2,51 µM y resultó ocho veces más
activo que una de las drogas de referencia utilizada (CI50 de la eflornitina = 21,55 µM).
Teniendo en cuenta los valores de citotoxicidad frente a células Vero, se calculó el
índice de selectividad (IS) para las N4-TSCs como el cociente entre la máxima
concentración evaluada para la citotoxicidad y la CI50 (Tabla 9). Dentro de los 30
compuestos, aquel que presentó mayor selectividad fue el derivado 8 (IS > 11,95). Por
este motivo, los estudios posteriores de mecanismo de acción y de tipo de muerte
celular se llevaron a cabo con dicho derivado.

Resultados y discusión | Trypanosoma brucei
84
N4-TSC R1 R2 R3 R4 CI50 (µM)
CC50
(µM) IS
1 H H H H 12,07 ± 1,43 >20 >1,66
2 H H H NO2 6,96 ± 0,59 >50 >7,18
3 H H H F 11,22 ± 0,73 >80 >7,13
4 H H H Cl 14,25 ± 1,74 >40 >2,81
5 H H H CH3 35,23 ± 3,50 >50 >1,42
6 H H H OCH3 19,99 ± 1,27 >70 >3,50
7 H OCH3 OCH3 H 14,55 ± 1,37 >25 >1,72
8 H OCH3 OCH3 NO2 2,51 ± 0,21 >30 >11,95
9 H OCH3 OCH3 F 17,10 ± 1,21 >30 >1,75
10 H OCH3 OCH3 Cl 7,25 ± 0,49 >30 >4,14
11 H OCH3 OCH3 CH3 12,97 ± 1,40 >50 >3,86
12 H OCH3 OCH3 OCH3 18,29 ± 1,86 >30 >1,64
13 H CH3 H H 13,10 ± 0,74 >35 >2,67
14 H CH3 H NO2 8,41 ± 0,54 >60 >7,13
15 H CH3 H F 9,48 ± 0,42 >80 >8,44
16 H CH3 H Cl 25,71 ± 3,10 >50 >1,94
17 H CH3 H CH3 33,30 ± 2,99 >50 >1,50
18 H CH3 H OCH3 >30 >50 -
19 OCH3 OCH3 H H 12,34 ± 1,42 >25 >2,03
20 OCH3 OCH3 H NO2 >30 >30 -
21 OCH3 OCH3 H F >30 >30 -
22 OCH3 OCH3 H Cl >30 >30 -
23 OCH3 OCH3 H CH3 >30 >50 -
24 OCH3 OCH3 H OCH3 >30 >30 -
25 H Br H H 8,70 ± 0,58 >20 >2,30
26 H Br H NO2 11,52 ± 1,50 >100 >8,68
27 H Br H F 18,45 ± 0,83 >40 >2,17

Resultados y discusión | Trypanosoma brucei
85
28 H Br H Cl 20,86 ± 1,18 >30 >1,44
29 H Br H CH3 23,91 ± 0,95 >40 >1,67
30 H Br H OCH3 24,54 ± 0,79 >100 >4,07
Ptm - - - - (9,1 ± 0,12)x10-4
ND -
Efl - - - - 21,55 ± 2,29 580 ± 61 26,91
Tabla 9. Resultados de CI50 en T. brucei brucei en su forma sanguínea (bloodstream)
cepa 427 y CC50 en células Vero. ND: no determinado. Ptm: pentamidina. Efl:
eflornitina.
8
lo g [c o n c e n tra c ió n (M )]
% i
nh
ibic
ión
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0
0
2 0
4 0
6 0
8 0
1 0 0
1 0
lo g [c o n c e n tra c ió n (M )]
% i
nh
ibic
ión
0 .0 0 .5 1 .0 1 .5 2 .0
0
2 0
4 0
6 0
8 0
1 0 0
2
lo g [c o n c e n tra c ió n (M )]
% i
nh
ibic
ión
0 .0 0 .5 1 .0 1 .5
0
2 0
4 0
6 0
8 0
1 0 0
Figura 36. Curvas de porcentaje de inhibición vs. log de la concentración para los
derivados: A) 2; B) 8 y C) 10.
4.2. Relación estructura-actividad cualitativa
La variabilidad de los sustituyentes en el indano y en la posición para del arilo unido al
N4, junto con la determinación de los valores de CI50, permitieron estudiar de forma
cualitativa aquellas propiedades estructurales que se encuentran relacionadas con la
actividad medida.
Se observó que la naturaleza del sustituyente en posición para del grupo N4-arilo tiene
un efecto importante en la actividad biológica ensayada. Las N4-TSCs con un grupo
nitro en la posición R4 presentaron las mejores actividades dentro de la serie derivada de
1-indanona (CI50 2 = 6,96 µM), 5,6-dimetoxi-1-indanona (CI50 8 = 2,51 µM), y 5-metil-
1-indanona (CI50 14 = 8,41 µM). En el caso de los derivados de la 5-bromo-1-indanona
el compuesto sustituido con el grupo nitro (26) fue el segundo más activo de dicha serie
con un valor de CI50 de 11,52 µM. Asimismo, los compuestos sin sustituir (R4 = H)
mostraron en general buenos valores de actividad, aunque por debajo de aquellos
A B C

Resultados y discusión | Trypanosoma brucei
86
sustituidos con el grupo nitro. Por otro lado, los grupos metilo y metoxilo en R4 se
correspondieron con bajas actividades para cada una de las series de 1-indanonas. La
sustitución con los átomos de F y Cl en R4 mostraron valores de actividad variables.
El análisis de la sustitución del núcleo indánico mostró que la presencia de grupos
metoxilos en las posiciones 4 y 5 conllevó a la perdida de actividad, con excepción del
compuesto 19 con un valor de CI50 de 12,34 µM. Sin embargo, cuando en la indanona
los metoxilos se encuentran en posición 5 y 6 la actividad alcanzó los mejores valores
(entre 2,51 y 18,29 µM), demostrando así la importancia de la posición de estos
sustituyentes en la indanona. Por otro lado, las N4-TSCs derivadas de la 5-bromo y 5-
metil presentaron en general una reducción en su actividad respecto de sus análogos
derivados de la 1-indanona (sin sustituyentes en el núcleo indánico).
En síntesis, la actividad observada fue buena para los compuestos con la sustitución
nitro e hidrógeno en R4. Asimismo, se evidenció que el patrón y la sustitución en el
núcleo indánico influyen en la actividad, siendo los derivados de la 1-indanona y la 5,6-
dimetoxi-1-indanona los de mejor efecto anti-T. brucei.
4.3. Estudios de QSAR
Teniendo en cuenta los resultados encontrados previamente de relación estructura-
actividad cualitativa se decidió llevar a cabo un estudio en el mismo sentido pero de
forma cuantitativa (QSAR) con el objeto de describir matemáticamente la actividad
anti-T. brucei para esta serie de compuestos.
Como se mencionó en la introducción, QSAR es un método que contribuye en el diseño
de fármacos. Es una estrategia para la cual no se necesita conocer la diana biológica ya
que el modelo se basa en conocer las propiedades de los compuestos para desarrollar
ecuaciones de correlación con la actividad biológica medida. Para llevar a cabo el
estudio se necesita básicamente tres tipos de información: estructura molecular, datos de
actividad biológica y propiedades fisicoquímicas de los compuestos (las cuales se
expresan por medio de descriptores numéricos). Los datos de actividad biológica son
experimentales y es recomendable que provengan de un solo laboratorio de
investigación para asegurar que los resultados fueron obtenidos bajo las mismas
condiciones y bajo los mismos errores sistemáticos, operativos y de manipulación

Resultados y discusión | Trypanosoma brucei
87
humana. En cuanto a los descriptores moleculares, estos pueden ser obtenidos de forma
experimental o calculados computacionalmente.
Cuando se cuenta con la información previamente mencionada se puede utilizar el
método de regresión lineal múltiple para obtener la ecuación de correlación. Para ello se
toma como variable dependiente los valores de actividad biológica y como variables
independientes los descriptores. Las expresiones más sencillas de estos modelos
normalmente adoptan la siguiente forma:
Donde “y” representa la actividad biológica medida, “x” son los descriptores y “a” los
coeficientes para el ajuste de la ecuación.
La ecuación de QSAR permite dos tipos de aplicaciones: uno prospectivo y otro
retrospectivo. El primero de ellos permite predecir la actividad biológica de compuestos
aun no sintetizados, los cuales deben compartir características estructurales con los
compuestos incluidos en el estudio para no salir del patrón químico tenido en cuenta,
por lo cual es conveniente una amplia variabilidad estructural. El otro tipo de aplicación
(el retrospectivo) permite analizar las moléculas ya sintetizadas y evaluadas para
entender la correlación no siempre evidente entre estructuras y actividades.
Todeschini y Consonni definen a un descriptor molecular como el resultado final de un
procedimiento lógico y matemático, el cual transforma la información química
codificada dentro de una representación simbólica de una molécula en un número útil o
como el resultado de algún experimento estandarizado99
. Los descriptores moleculares
se pueden subclasificar en:
-Descriptores 0D: obtenidos partir de contar átomos.
-Descriptores 1D: obtenidos a partir de contar fragmentos de la molécula.
-Descriptores 2D: obtenidos a partir de incorporar información topológica
-Descriptores 3D: obtenidos a partir de tener en cuenta la estructura tridimensional de la
molécula.
El número de descriptores a considerar está en función de las herramientas
computacionales y del número de moléculas incluidas en el estudio. Un error frecuente

Resultados y discusión | Trypanosoma brucei
88
consiste en agregar un espacio numérico sobredimensionado para describir cada
molécula, es decir cuando el número de descriptores es elevado respecto al número de
las moléculas lo cual conduce a un overfitting.
El modelo debe ser validado de forma interna y externa. La validación interna tiene
como fin corroborar la correlación entre la actividad biológica y los descriptores
seleccionados. Para ello se tienen en cuenta distintos estadísticos: R2 (coeficiente de
determinación), SD (desvío estándar), F (estadístico de Fisher) y el test de student para
cada variable del modelo. Por otro lado, la validación externa permite estimar la
incertidumbre asociada a las predicciones del modelo. Para ello se dividen los datos en
dos grupos: el denominado training set y el test set. Los modelos se generan con los
datos del training set y esos mismos modelos se ponen a prueba aplicándolos sobre el
test set para corroborar si predicen la actividad biológica conocida.
Con el objetivo de llevar a cabo un estudio de QSAR que explicite la relación entre las
N4-TSCs y la actividad anti-T. brucei, se modelaron en primer lugar cada uno de los
compuestos que presentaron un valor definido de CI50. Para ello, se utilizó el programa
HyperChem teniendo en cuenta la configuración E de las N4-TSCs previamente
identificada mediante el análisis de los espectros de RMN.
En el primer estudio de QSAR se generó un pool de confórmeros para cada una de las
N4-TSCs con configuración E mediante el programa Ballon, a partir del cual se
calcularon diversos descriptores 0D, 1D, 2D y 3D con el programa PaDEL Descriptor.
Luego, teniendo en cuenta los valores de actividad anti-T. brucei expresados como
pCI50 (log 1/CI50) y los descriptores calculados anteriormente se generaron diferentes
modelos mediante algoritmo genético utilizando el programa McQSAR. Si bien en este
estudio no fue posible encontrar un modelo matemático que describiese la actividad
biológica, se observó la inclusión de descriptores 3D en la mayoría de las ecuaciones
generadas.
Como se mencionó anteriormente, los descriptores tridimensionales dependen de las
coordenadas x, y, z de los átomos de la molécula. Por este motivo, se decidió realizar un
estudio de QSAR que mantuviera fija, no sólo la configuración E, sino también las
conformaciones más estables de los compuestos. Considerando el ángulo diedro (ϕ)
formado por los átomos N-N-C-N (Figura 37 A) se describen en literatura dos

Resultados y discusión | Trypanosoma brucei
89
conformaciones relevantes por su estabilidad, la syn y la anti (Figura 37 B)100–103
. La
anti corresponde a la conformación en la cual el N1 y el N
4 se encuentran en forma
opuesta, mientras que la syn corresponde a la conformación donde dichos átomos se
encuentran del mismo lado del plano considerado. Resultó entonces necesario en el
estudio de 3D-QSAR tener en cuenta los isómeros E syn y E anti, que serán
denominados como “Syn” y “Anti”. Se generaron dos estudios de QSAR separados
para cada isómero, considerando sólo un conjunto de compuestos elegidos en función
de asegurar la diversidad estructural y el rango de actividades biológicas para la
construcción del modelo (training set de 17 compuestos, 1, 2, 5, 6, 7, 8, 10, 11, 13, 14,
15, 16, 19, 27, 28, 29 y 30) y reservando el resto (test set) para la futura validación
externa.
Figura 37. A) Ángulo diedro considerado para el estudio de 3D-QSAR. B)
Conformaciones de mínima energía del compuesto 1 correspondientes al isómero E anti
(izquierda) y E syn (derecha).
Por otro lado, se comparó gráficamente mediante el mapa de potencial electrostático la
distribución de la densidad electrónica para los compuestos más activos frente a los
menos activos y además entre el confórmero “Syn” y el “Anti”. En la Figura 38 se
representan los dos confórmeros estudiados del compuesto 8 (el más activo de la serie)
y los dos confórmeros estudiados del compuesto 5 (el menos activo con valor conocido
de CI50). A partir de dichas imágenes se pudo visualizar que los distintos sustituyentes
A

Resultados y discusión | Trypanosoma brucei
90
en el indano y en el arilo unido al N4 tienen un efecto diferencial sobre la distribución
electrónica en la función tiosemicarbazona. Se observó en general que la densidad
electrónica de dicha función es menor para los compuestos más activos y para la
conformación “Syn” (Figura 38).
Figura 38. Superficies de potencial electrostático para los confórmeros “Syn” y “Anti”
del compuesto 8 y 5. El color rojo de la escala cromática representa la mayor densidad
electrónica mientras que el color azul indica que la densidad electrónica es la de menor
valor.
Teniendo en cuenta los resultados diferenciales observados en los mapas de potencial
electrostático, se calcularon una serie de descriptores electrónicos para los estudios de
QSAR. Mediante el programa Gaussian 09, se obtuvieron: las energías de HOMO y
LUMO, dureza química, momento dipolar, electrofilicidad, refractividad, y distintos
esquemas de cargas (RESP y Mulliken) sobre distintos átomos (Tabla 10).
N4-TSCs 8 “Syn”
N4-TSCs 8 “Anti”
N4-TSCs 5 “Syn”
N4-TSCs 5 “Anti”

Resultados y discusión | Trypanosoma brucei
91
N4-TSC CI50 EHOMO ELUMO qN1 qN2 qC(=S) qS(=C) qN4
1 12,07 -8,5376 -0,7745 -0,3605 -0,5726 -0,2176 -0,3402 -0,8478
2 6,96 -9,0324 -2,2537 -0,3789 -0,5697 0,1996 -0,3068 -0,8518
3 11,22 -8,6218 -0,8837 -0,3644 -0,5737 0,4515 -0,3367 -0,8536
4 14,25 -8,6388 -0,8959 -0,3657 -0,5730 -0,1527 -0,3328 -0,8520
5 35,23 -8,4945 -0,7565 -0,3595 -0,5732 0,0251 -0,3437 -0,8478
6 19,99 -8,4267 -0,7578 -0,3591 -0,5748 0,4198 -0,3449 -0,8490
7 14,55 -8,4869 -0,7752 -0,3750 -0,5718 -0,2205 -0,3437 -0,8437
8 2,51 -8,7492 -2,3193 -0,3945 -0,5682 0,1947 -0,3136 -0,8569
9 17,10 -8,5563 -0,8848 -0,3792 -0,5728 0,4490 -0,3405 -0,8503
10 7,25 -8,5590 -0,8993 -0,3807 -0,5721 -0,1561 -0,3376 -0,8558
11 12,97 -8,4614 -0,7601 -0,3739 -0,5726 0,0215 -0,3480 -0,8540
12 18,29 -8,4336 -0,7650 -0,3736 -0,5742 0,0215 -0,3504 -0,8538
13 13,10 -8,5145 -0,7536 -0,3655 -0,5725 -0,2178 -0,3429 -0,8481
14 8,41 -8,9952 -2,2419 -0,3844 -0,5694 0,1994 -0,3096 -0,8499
15 9,48 -8,5982 -0,8618 -0,3698 -0,5735 0,4509 -0,3399 -0,8502
16 25,71 -8,6145 -0,8742 -0,3712 -0,5729 -0,1532 -0,3360 -0,8377
17 33,30 -8,4732 -0,7362 -0,3646 -0,5731 0,0250 -0,3463 -0,8323
19 12,34 -8,5236 -0,5656 -0,3699 -0,5720 -0,2182 -0,3426 -0,8432
25 8,70 -8,6258 -0,7722 -0,3579 -0,5730 -0,2177 -0,3322 -0,8487
26 11,52 -9,1191 -0,9825 -0,3711 -0,5693 0,2018 -0,3010 -0,8518
27 18,45 -8,7082 -2,3075 -0,3622 -0,5745 0,4532 -0,3288 -0,8478
28 20,86 -8,7249 -1,0724 -0,3632 -0,5736 -0,1521 -0,3252 -0,8479
29 23,91 -8,5800 -0,9658 -0,3571 -0,5736 0,0238 -0,3355 -0,8484
30 24,54 -8,5058 -0,9731 -0,3569 -0,5752 0,4207 -0,3370 0,8488
Tabla 10. Propiedades electrónicas calculadas para el isómero “Syn”. Los valores de
CI50 están expresados en µM y los de energía de HOMO (EHOMO) y energía de LUMO
(ELUMO) en eV. qN1: carga Mulliken en el nitrógeno 1. qN2: carga Mulliken en el
nitrógeno 2. qC(=S): carga Mulliken en el carbono unido al azufre. qS(=C): carga
Mulliken en el azufre de la función tiosemicarbazona. qN4: carga Mulliken en el
nitrógeno 4.
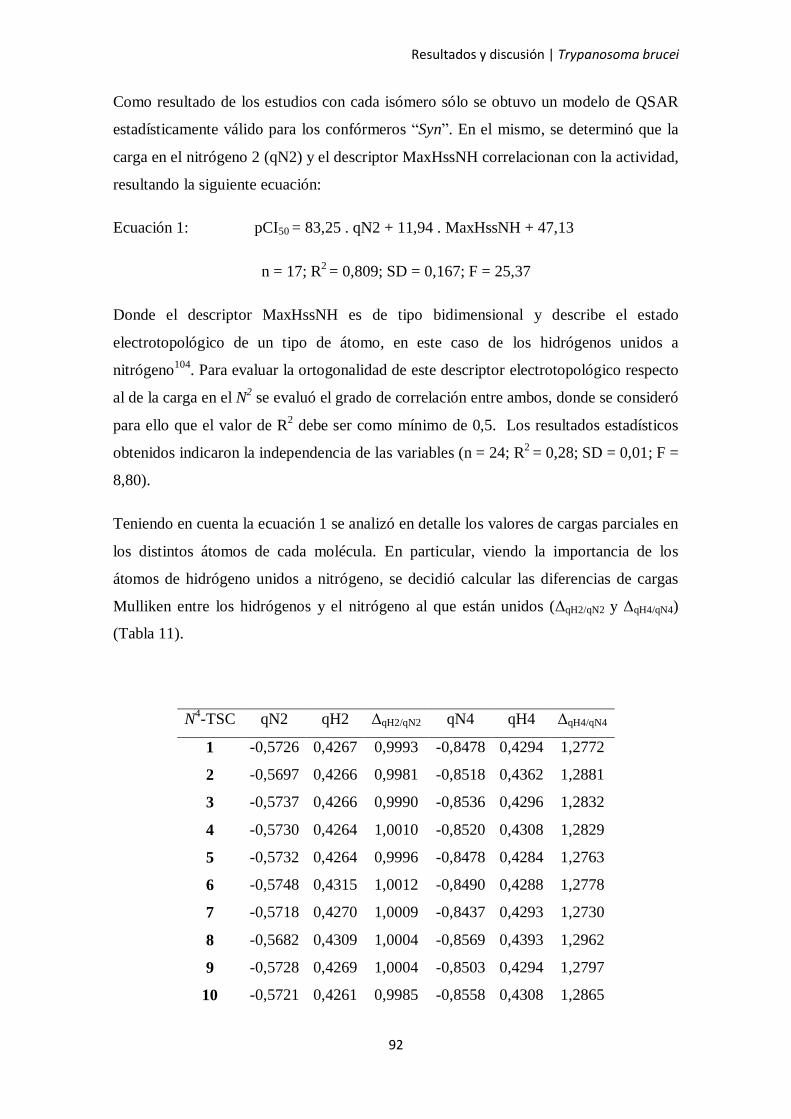
Resultados y discusión | Trypanosoma brucei
92
Como resultado de los estudios con cada isómero sólo se obtuvo un modelo de QSAR
estadísticamente válido para los confórmeros “Syn”. En el mismo, se determinó que la
carga en el nitrógeno 2 (qN2) y el descriptor MaxHssNH correlacionan con la actividad,
resultando la siguiente ecuación:
Ecuación 1: pCI50 = 83,25 . qN2 + 11,94 . MaxHssNH + 47,13
n = 17; R2 = 0,809; SD = 0,167; F = 25,37
Donde el descriptor MaxHssNH es de tipo bidimensional y describe el estado
electrotopológico de un tipo de átomo, en este caso de los hidrógenos unidos a
nitrógeno104
. Para evaluar la ortogonalidad de este descriptor electrotopológico respecto
al de la carga en el N2 se evaluó el grado de correlación entre ambos, donde se consideró
para ello que el valor de R2 debe ser como mínimo de 0,5. Los resultados estadísticos
obtenidos indicaron la independencia de las variables (n = 24; R2
= 0,28; SD = 0,01; F =
8,80).
Teniendo en cuenta la ecuación 1 se analizó en detalle los valores de cargas parciales en
los distintos átomos de cada molécula. En particular, viendo la importancia de los
átomos de hidrógeno unidos a nitrógeno, se decidió calcular las diferencias de cargas
Mulliken entre los hidrógenos y el nitrógeno al que están unidos (ΔqH2/qN2 y ΔqH4/qN4)
(Tabla 11).
N4-TSC qN2 qH2 ΔqH2/qN2 qN4 qH4 ΔqH4/qN4
1 -0,5726 0,4267 0,9993 -0,8478 0,4294 1,2772
2 -0,5697 0,4266 0,9981 -0,8518 0,4362 1,2881
3 -0,5737 0,4266 0,9990 -0,8536 0,4296 1,2832
4 -0,5730 0,4264 1,0010 -0,8520 0,4308 1,2829
5 -0,5732 0,4264 0,9996 -0,8478 0,4284 1,2763
6 -0,5748 0,4315 1,0012 -0,8490 0,4288 1,2778
7 -0,5718 0,4270 1,0009 -0,8437 0,4293 1,2730
8 -0,5682 0,4309 1,0004 -0,8569 0,4393 1,2962
9 -0,5728 0,4269 1,0004 -0,8503 0,4294 1,2797
10 -0,5721 0,4261 0,9985 -0,8558 0,4308 1,2865

Resultados y discusión | Trypanosoma brucei
93
11 -0,5726 0,4322 1,0004 -0,8540 0,4289 1,2829
12 -0,5742 0,4274 1,0002 -0,8538 0,4280 1,2818
13 -0,5725 0,4273 1,0007 -0,8481 0,4295 1,2776
14 -0,5694 0,4284 1,0023 -0,8499 0,4369 1,2868
15 -0,5735 0,4263 0,9981 -0,8502 0,4310 1,2812
16 -0,5729 0,4273 1,0020 -0,8377 0,4305 1,2682
17 -0,5731 0,4289 1,0020 -0,8323 0,4281 1,2604
19 -0,5720 0,4323 1,0023 -0,8432 0,4290 1,2722
25 -0,5730 0,4280 1,0009 -0,8487 0,4338 1,2825
26 -0,5693 0,4258 0,9989 -0,8518 0,4290 1,2808
27 -0,5745 0,4286 1,0011 -0,8478 0,4315 1,2793
28 -0,5736 0,4282 1,0001 -0,8479 0,4317 1,2796
29 -0,5736 0,4276 1,0011 -0,8484 0,4284 1,2768
30 -0,5752 0,4274 0,9999 0,8488 0,4288 1,2776
Tabla 11. Cargas parciales Mulliken para el confórmero “Syn”. qN2: carga en el
nitrógeno 2. qH2: carga en el hidrógeno unido directamente al nitrógeno 2. ΔqH2/qN2:
diferencia entre la carga en el hidrógeno y el nitrógeno 2. qN4: carga en el nitrógeno 4.
qH4: carga en el hidrógeno unido directamente al nitrógeno 4. ΔqH4/qN4: diferencia entre
la carga en el hidrógeno y el nitrógeno 4.
A partir de estos nuevos datos de cargas parciales y sus diferencias se propuso generar
nuevos modelos de QSAR superadores, donde el significado fisicoquímico de sus
descriptores fuera más simple de interpretar. Para ello, se mantuvieron los mismos
compuestos del training set anterior y se incluyeron los descriptores que se muestran en
la Tabla 11. Se obtuvieron varias ecuaciones de correlación estructura-actividad para el
confórmero de tipo “Syn”, mientras que para el confórmero “Anti” no se encontró
ninguna ecuación de correlación válida estadísticamente. A continuación se muestra el
modelo de QSAR con el mayor nivel de significación generado en esta etapa (Ecuación
2) y el gráfico de correlación entre los valores de actividad calculados y los
determinados de forma experimental (Figura 39).
Ecuación 2: pCI50 = 71,40 . qN2 + 20,54. ΔqH4/qN4 + 19,46
n = 17; R2 = 0,800; SD = 0,132; F = 28,04

Resultados y discusión | Trypanosoma brucei
94
Figura 39. Correlación entre los valores de actividad calculados vs. los experimentales.
Para esta última ecuación se evaluó la ortogonalidad de las variables y se comprobó la
independencia de las mismas a través de sus estadísticos (n = 24; R2
= 0,22; SD = 0,01;
F=6,15). Además, se calcularon los estadísticos para los coeficientes de cada descriptor
donde para qN2 se obtuvo un valor de t de Student de 3,16 y un p-valor de 0,007; y en
el caso de ΔqH4/qN4 un valor de t de Student de 3,22 y un p-valor de 0,006. Se distinguió
así para ambos descriptores valores estadísticamente significativos (p-valor < 0,05).
Las ecuaciones 1 y 2 permiten explicar la relación estructura-actividad, siendo sus
valores estadísticos calculados similares. En ambos casos se encuentra incluido el
descriptor qN2, en la primera ecuación el coeficiente que lo acompaña tiene un valor de
83,25 mientras que en la segunda ecuación es de 71,40, su valor positivo indica que para
lograr una buena actividad es necesario que qN2 adquiera los mayores valores posibles.
Por otro lado en la ecuación 1 el otro descriptor presente fue MaxHssNH el cual como
se mencionó anteriormente brinda información sobre el estado electrónico y topológico
de los hidrógenos unidos a N, considerando para ello las distancias con los átomos
vecinos y el estado intrínseco del átomo. En este caso mayores valores de MaxHssNH
se correlacionan con una mejor actividad biológica. Respecto a la ecuación 2, la
segunda variable es ΔqH4/qN4, es decir el valor dado por la diferencia de carga entre el
hidrógeno unido al nitrógeno 4 y este átomo. Considerando esta variable, su pendiente
positiva de 20,54 indica que valores más positivos del descriptor (Tabla 11) se
corresponden con las mejores actividades, lo cual es coincidente con lo observado para
los compuestos 2, 8, 14 y 26 (con R4 = NO2). Analizando en conjunto los valores de los

Resultados y discusión | Trypanosoma brucei
95
estadísticos obtenidos y el grado de abstracción e interpretación desde el punto de vista
fisicoquímico la ecuación 2 resultó superadora.
La validación externa de la ecuación 2 se realizó con los compuestos que no habían sido
empleados en la generación del modelo. Se tomaron las N4-TSCs 3, 4, 9, 12, 17, 25 y 26
como test set para evaluar el poder predictivo de la ecuación. Se calculó el valor de
r2
PRESS el cual es un estadístico que permite evaluar si el modelo es robusto. Cuando su
valor es mayor a 0,5 se puede indicar una buena predictibilidad externa105
. Dicho
estadístico se define matemáticamente como:
Donde SD = ∑ (pCI50 experimental del test set - pCI50 promedio del training set)2 y
PRESS = ∑ (pCI50 experimental del test set - pCI50 predicha del test set)2
Los resultados encontrados mostraron que para los compuestos evaluados en esta
instancia, el modelo resultó adecuado para la predicción de los valores de pCI50 ya que
el valor de r2
PRESS fue de 0,63. En la Tabla 12 se presentan los resultados de actividad
predichos y experimentales, así como los residuos respecto al valor experimental para el
test set.
N4-TSC pCI50exp pCI50calc Residuo
3 4,950 4,854 -0,096
4 4,846 4,896 0,049
9 4,767 4,846 0,079
12 4,738 4,789 0,052
17 4,478 4,428 -0,049
25 5,060 4,889 -0,171
26 4,939 5,119 0,180
Tabla 12. Valores de actividad biológica experimentales y calculados. pCI50exp: valor
de pCI50 experimental. pCI50calc: valor de pCI50 calculado a partir de la ecuación 2.

Resultados y discusión | Trypanosoma brucei
96
En resumen, según los resultados de este estudio de QSAR la actividad anti-T. brucei de
las N4-TSCs dependería de la carga sobre el N
2 y de la diferencia de cargas entre el
nitrógeno 4 y el hidrógeno unido directamente a este átomo. Además, se resalta la
importancia de las conformaciones adoptadas por la molécula, siendo las “Syn” las que
estarían implicadas en la actividad biológica evaluada.
4.4. Evaluación de la N4-TSC más activa frente a proteasas
Las proteasas de T. brucei juegan un rol fundamental en el ciclo de vida del parásito y
en la patogénesis de la enfermedad del sueño y nagana. Las proteasas descriptas en T.
brucei incluyen aquellas pertenecientes a la familia de las cisteína, serina, treonina y
metalo peptidasas. Estudios in vitro e in vivo han demostrado que estas enzimas son
blancos validados para el desarrollo de drogas frente a dicha tripanosomiasis95,106,107
.
Teniendo en cuenta que numerosas TSCs han sido reportadas como inhibidoras de
cisteína proteasas de T. brucei 108,109
, se decidió investigar si el compuesto más activo de
la serie de N4-TSCs ensayada (derivado 8) manifiesta la propiedad de inhibir alguna
proteasa del parásito. Con este objetivo, se realizó una zimografía a partir de un lisado
de parásitos en donde el sustrato fue la gelatina previamente agregada a un gel de
poliacrilamida. Cabe mencionar la conveniencia de aplicar este método separativo ya
que permite investigar varias proteasas al mismo tiempo a partir de un lisado celular sin
necesidad de purificar las enzimas110,111
.
En el ensayo se corrió un estándar comercial con sustancias de pesos moleculares
conocidos, un lisado de parásitos sin tratar (control sin tratar), un lisado de parásitos
incubado con 5% de DMSO (control de vehículo), un lisado de parásitos con E-64
(control positivo de inhibición) y un lisado de parásitos con el compuesto 8.
Los resultados mostraron la presencia de varias proteasas en el lisado de parásito
sembrado. La incubación con el compuesto 8 no logró la inhibición de ninguna de las
proteasas de T. brucei reveladas en el gel, ya que el patrón de bandas observado fue el
mismo que el del control sin tratar y el control de vehículo (Figura 40). Esto indica que
el compuesto más activo no estaría ejerciendo su efecto a través de la inhibición de estas
enzimas, por lo cual sería otro su mecanismo de acción.

Resultados y discusión | Trypanosoma brucei
97
Figura 40. Zimografía de un lisado de T. brucei. a: estándar de pesos moleculares; b:
lisado de parásitos (control sin tratar); c: lisado de parásitos incubado con E-64 (control
positivo de inhibición); d: lisado de parásitos incubado con el compuesto 8; e: lisado
incubado con DMSO (control de vehículo). Las zonas más claras se deben al consumo
de la gelatina por parte de las proteasas.
4.5. Captación intracelular de las N4-TSCs
Se observó que las N4-TSCs tienen la capacidad de emitir fluorescencia luego de ser
excitadas con una luz de longitud de onda de 405 nm. En la Figura 41 se representa la
fluorescencia emitida a distintas longitudes de onda para seis de los compuestos
evaluados. Se evidenció que el compuesto 14 es el que emite mayor intensidad de
fluorescencia. En el caso del compuesto más activo frente a T. brucei (compuesto 8), se
observó un pico a 415 nm y otro de similar intensidad a 468 nm. Estas propiedades de
emisión de fluorescencia de las N4-TSCs se encuentran relacionadas con su estructura
química debido a la presencia de dobles enlaces conjugados que producen una elevada
deslocalización electrónica.
Aprovechando la cualidad de las N4-TSCs estudiadas de emitir fluorescencia, se
procedió a evaluar si el compuesto 8 (el más activo) y el compuesto 14 (por ser el que
fluoresce con mayor intensidad) eran internalizados por los parásitos luego de su
tratamiento. Para ello, los parásitos fueron tratados durante 4 y 24 h con el compuesto
correspondiente y luego se visualizaron mediante microscopio confocal (laser de
a b c d e
220 kDa
91 kDa
50 kDa
35 kDa

Resultados y discusión | Trypanosoma brucei
98
excitación de 405 nm). En las imágenes obtenidas se pudo observar la localización de la
fluorescencia en el interior del parásito, lo cual demuestra que ambos compuestos
fueron internalizados por la célula tanto a las 4 h como 24 h de tratamiento (Figura 42).
Además, cabe destacar que en las imágenes se visualizó la fluorescencia de los
compuestos en todo el espacio intracelular del parásito por lo cual las N4-TSCs
estudiadas no presentaron una localización subcelular específica para los tiempos
ensayados.
Por otro lado, se estudió el perfil de fluorescencia de las N4-TSCs luego de ser excitadas
a 488 nm, por ser esta una longitud de onda muy utilizada en los laser de microscopía y
citometría de flujo. En dicho estudio se determinó que no hay emisión de fluorescencia
luego de la excitación a esta longitud de onda.
Figura 41. Espectro de emisión para distintas N
4-TSCs luego de ser excitadas a 405 nm.

Resultados y discusión | Trypanosoma brucei
99
Figura 42. Microscopía confocal. Parásitos sin tratar (A, a, B, b); tratados con el
compuesto 8 durante 4 h (C, c) y 24 h (D, d); y con el compuesto 14 durante 4 h (E, e) y
24 h (F, f).
4.6. Mecanismo de muerte celular inducido por el compuesto 8
La apoptosis es un tipo de muerte celular regulada (MCR) dependiente de energía en la
cual se reconocen determinados cambios morfológicos y bioquímicos. Las alteraciones
celulares que ocurren durante este proceso en los metazoos incluyen: la activación de
caspasas, disminución del volumen celular, exposición de fosfatidilserina (PS) en la
superficie externa celular, alteraciones mitocondriales, condensación de la cromatina,
fragmentación nuclear, compactación del citoplasma, membrana con aspecto de burbuja
y formación de cuerpos apoptóticos112
.
Si bien varias de las características clásicas de apoptosis descriptas para organismos
multicelulares han sido observadas en organismos unicelulares como los protozoarios,
aún no han sido detectadas las caspasas, consideradas fundamentales para la apoptosis
Co
ntr
ol
l
N4-T
SC 8
8
N4-T
SC 1
4
4 h 24 h
A
C
E
a
c
e
b
d
f
B
D
F

Resultados y discusión | Trypanosoma brucei
100
en metazoos113–115
. Por este motivo, se considera que en los protozoos el evento que
ocurre puede considerarse similar a la apoptosis116
.
La necrosis es otro tipo de muerte celular que se caracteriza por edema celular, aumento
de la permeabilidad de la membrana plasmática y finalmente la ruptura de la misma. De
esta forma se libera el contenido celular al medio circundante pudiendo desencadenar un
proceso inflamatorio en el huésped. Por este motivo, en la búsqueda de un tratamiento
quimioterapéutico, resulta de interés conocer el tipo de muerte en el caso que esta haya
sido desencadenada por la acción del fármaco, ya que gatillar un proceso necrótico no
resulta conveniente. Diferentes marcadores, que pueden ser analizados por citometría de
flujo, permiten poner en evidencia el tipo de muerte celular que ocurre luego de la
exposición a un agente químico o físico.
La citometría de flujo es una técnica de análisis celular multiparamétrica que permite
ser utilizada para diversas aplicaciones en la investigación básica y aplicada, ensayos
clínicos, diagnóstico y seguimiento de pacientes entre otras. El principio en el que se
basa esta tecnología radica en hacer pasar las células suspendidas de forma alineada y
de a una por delante de una luz láser (Figura 43). El impacto del haz de luz en cada
célula produce señales que corresponden a diferentes características de la misma, las
cuales son recogidas por distintos detectores. La información obtenida puede ser
clasificada en aquellas generadas por la dispersión de la luz incidente y la emisión de
luz por los fluoróforos que son utilizados como marcadores. Cabe destacar que los datos
obtenidos corresponden a un gran número de células (habitualmente 10 mil), por lo cual
posiciona a esta metodología sobre otras donde el tamaño de muestra suele ser bastante
más pequeño.
Los citómetros de flujo están formados por un complejo sistema de fluídica el cual
permite el enfoque hidrodinámico del flujo celular que asegura el alineamiento de las
células y su paso una por una. El equipo cuenta con un sistema óptico el cual
comprende la óptica de excitación y de lectura. El primero de ellos está constituido por
el láser y las lentes que permiten enfocar y colimar la luz. La óptica de lectura está
compuesta por las lentes de recolección y filtros ópticos de la luz emitida luego de la
interacción entre el láser y las células. De esta manera, se seleccionan las longitudes de
onda que se dejan pasar hacia el sistema de detección. Finalmente, el análisis

Resultados y discusión | Trypanosoma brucei
101
electrónico de la luz dispersada y la fluorescencia emitida se cuantifica con
herramientas computacionales.
Figura 43. Esquema de disposición de elementos en un citómetro de flujo.
Como se mencionó previamente, cierta información proviene de las señales de
dispersión de la luz. Dicha dispersión está en función de las características morfológicas
de las células como son el tamaño celular y su granulosidad. En el equipo se miden dos
fracciones de dispersión mediante un detector en línea con el rayo de luz denominado
FSC (por Forward Scatter) y una segunda fracción mediante el detector SSC (por Side
Scatter) ubicado en ángulo recto con respecto al láser.
El segundo tipo de información que se puede obtener es el originado por las señales de
fluorescencia. Para ello se emplean diversos fluoróforos o marcadores fluorescentes.
Cada uno posee un pico de excitación y una longitud de onda de emisión característica
que son detectados por diversos detectores conocidos como FL1-3.
Teniendo en cuenta la importancia de determinar el tipo de muerte celular y
aprovechando la gran utilidad de la citometría de flujo para este análisis, se utilizó dicha
tecnología en los parásitos tratados con el derivado 8. De esta manera, se evaluaron
varias de las características de las células y los procesos moleculares vinculados a la
apoptosis luego de la exposición de los parásitos a la droga. Se determinó: variación del
tamaño celular, presencia de especies reactivas del oxígeno (ROS), cambios en el
potencial de membrana mitocondrial, exposición de PS en la cara extracelular de la
membrana plasmática y fragmentación del ADN.

Resultados y discusión | Trypanosoma brucei
102
4.6.1 Determinación del tamaño celular
Los gráficos de FSC resultan útiles para detectar cambios en el tamaño celular y
pueden, por lo tanto, ser utilizados para evaluar la disminución de este parámetro
relacionado con la muerte celular117,118
.
Para determinar si el tratamiento con el compuesto 8 afecta el volumen celular de T.
brucei se analizó mediante citometría de flujo los valores de FSC y se compararon con
los valores del control sin tratar. Los parásitos se incubaron durante 24 h con una
concentración final de 5 y 10 µM de compuesto. Los resultados mostraron que a la
máxima concentración evaluada se evidenciaron células con una reducción en el tamaño
celular con respecto al control (Figuras 44 y 45), las cuales fueron definidas en el
gráfico de FSC vs SSC como una población de “tamaño pequeño”. Por otro lado,
cuando los parásitos se trataron con una concentración final de 5 µM se observó
también la disminución del tamaño pero en menor proporción. En conclusión se pudo
inferir que el tratamiento con el compuesto 8 induce una disminución del tamaño
celular, la cual es concentración dependiente. Estos resultados están en concordancia
con una de las características de los procesos definidos como similares a la apoptosis
que pueden ocurrir en los tripanosomátidos.
Figura 44. Tamaño celular en T. brucei luego de 24 h de tratamiento con el derivado 8.
A) control de parásitos. B) Incubados con el compuesto 8 en una concentración final de
5 µM y C) 10 µM. En forma ovalada se marca a la derecha la población de parásitos
que corresponden a un tamaño normal y a la izquierda la población de menor tamaño. El
gráfico es un típico dot plot de al menos tres experimentos independientes.
A B C
SSC
FSC

Resultados y discusión | Trypanosoma brucei
103
Figura 45. Variaciones en el tamaño celular en T. brucei luego de 24 h de tratamiento
con el derivado 8. Los histogramas de relleno gris representan el control sin tratar y los
histogramas de línea gris sin relleno corresponden a los parásitos tratados. A) Parásitos
totales incubados con 5 µM y B) 10 µM de compuesto. El gráfico es un típico
histograma de al menos tres experimentos independientes. C) Gráfico de barras de las
poblaciones de tamaño normal y pequeño. Los resultados representan la media ± SD (n
= 3). p-valor < 0,05 (*) y < 0,01 (**) fue considerado significativo con respecto al
control.
4.6.2. Determinación de especies reactivas del oxígeno
El equilibrio redox intracelular resulta esencial para la homeostasis de toda célula. En
los tripanosomátidos dicho equilibrio es regulado por mecanismos distintos al de las
células de mamíferos ya que emplean el tripanotión (bis-glutationilespermidina) como
principal cofactor redox, y carecen de enzimas tales como: glutatión reductasa,
tiorredoxina reductasa, catalasa, y glutatión peroxidasas selenio dependientes119–121
.
Además, se ha evidenciado la relación directa entre el aumento intracelular de las
especies reactivas del oxígeno (ROS) y la muerte celular regulada en T. brucei, por lo
cual el mantenimiento del equilibrio redox tiene un rol esencial en las decisiones de
vida/muerte114,122,123
. Por estos motivos, alterar el equilibrio redox de los parásitos
puede representar una estrategia útil para el desarrollo de antitripanosomátidos124–126
.
Por otro lado, se ha reportado que algunas TSCs tienen la capacidad de mediar sus
efectos biológicos a través de la inducción de ROS127,128
. Para evaluar si el tratamiento
de los parásitos con el compuesto 8 conlleva a un aumento en los niveles de ROS, se
recurrió a la tecnología de citometría de flujo utilizando la sonda diacetato de 2’,7’-
diclorodihidrofluoresceína (H2-DCF-DA). Este reactivo no fluorescente atraviesa la
Even
tos
FSC
A B C
Ev
en
tos
(%
)
C o n tr o l 5 M 1 0 M
0
2 0
4 0
6 0
8 0
1 0 0
C é lu la s n o rm a le s
C é lu la s p e q u e ñ a s
***
**
*

Resultados y discusión | Trypanosoma brucei
104
membrana plasmática e intracelularmente es hidrolizado a diclorodihidrofluoresceína
(H2-DCF), la cual ante la presencia de ROS es rápidamente oxidada a
diclorofluoresceina (DCF), altamente fluorescente (Figura 46).
Figura 46. Activación de la sonda diacetato de 2’,7’-diclorodihidrofluoresceína
(H2DCF-DA) para detectar ROS.
La N4-TSC 8 fue evaluada a la concentración final de 10 µM durante 2 h y 4 h y los
controles considerados fueron: los parásitos tratados con DMSO (control) y los
parásitos tratados con H2O2 (control positivo). Los resultados mostraron que la
intensidad de fluorescencia de la sonda aumentó significativamente luego de las 4 h, la
señal de DCF se incrementó ocho veces respecto al control. Por otro lado, no se
encontraron diferencias significativas entre el tratamiento a las 2 h con respecto al
control de células sin tratar (Figuras 47 y 48). De esta manera, se evidenció la inducción
de especies oxidantes luego de 4 h en presencia de la N4-TSC, lo cual pone de
manifiesto un probable mecanismo por el cual ejerce su acción el compuesto
relacionado con el tipo de muerte del parásito.
Figura 47. Estrés oxidativo en T. brucei. Parásitos incubados con A) DMSO (control);
luego del tratamiento con el compuesto 8 durante B) 2h y C) 4h y D) con peróxido de
hidrógeno. Los gráficos son un típico histograma de al menos tres experimentos
independientes.
A B D C
Even
tos
DCF
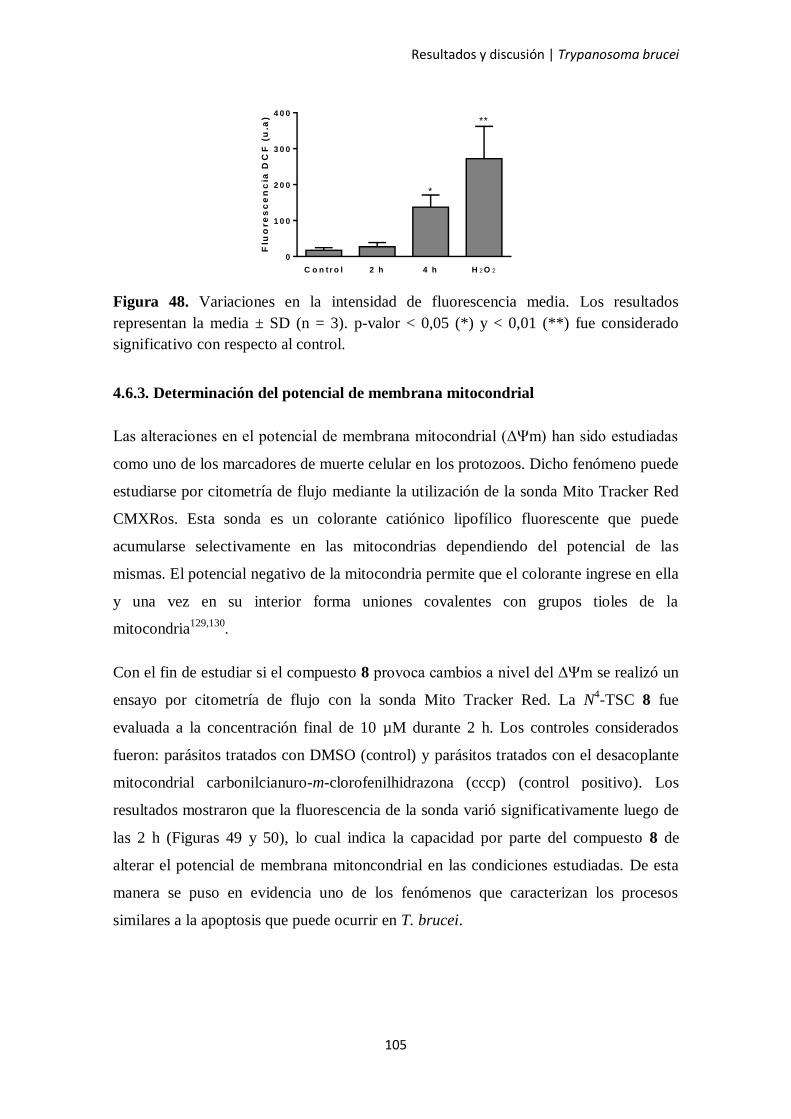
Resultados y discusión | Trypanosoma brucei
105
Flu
ore
sc
en
cia
DC
F (
u.a
)C o n tr o l 2 h 4 h H 2 O 2
0
1 0 0
2 0 0
3 0 0
4 0 0
*
**
Figura 48. Variaciones en la intensidad de fluorescencia media. Los resultados
representan la media ± SD (n = 3). p-valor < 0,05 (*) y < 0,01 (**) fue considerado
significativo con respecto al control.
4.6.3. Determinación del potencial de membrana mitocondrial
Las alteraciones en el potencial de membrana mitocondrial (ΔΨm) han sido estudiadas
como uno de los marcadores de muerte celular en los protozoos. Dicho fenómeno puede
estudiarse por citometría de flujo mediante la utilización de la sonda Mito Tracker Red
CMXRos. Esta sonda es un colorante catiónico lipofílico fluorescente que puede
acumularse selectivamente en las mitocondrias dependiendo del potencial de las
mismas. El potencial negativo de la mitocondria permite que el colorante ingrese en ella
y una vez en su interior forma uniones covalentes con grupos tioles de la
mitocondria129,130
.
Con el fin de estudiar si el compuesto 8 provoca cambios a nivel del ΔΨm se realizó un
ensayo por citometría de flujo con la sonda Mito Tracker Red. La N4-TSC 8 fue
evaluada a la concentración final de 10 µM durante 2 h. Los controles considerados
fueron: parásitos tratados con DMSO (control) y parásitos tratados con el desacoplante
mitocondrial carbonilcianuro-m-clorofenilhidrazona (cccp) (control positivo). Los
resultados mostraron que la fluorescencia de la sonda varió significativamente luego de
las 2 h (Figuras 49 y 50), lo cual indica la capacidad por parte del compuesto 8 de
alterar el potencial de membrana mitoncondrial en las condiciones estudiadas. De esta
manera se puso en evidencia uno de los fenómenos que caracterizan los procesos
similares a la apoptosis que puede ocurrir en T. brucei.

Resultados y discusión | Trypanosoma brucei
106
Figura 49. Determinación de potencial de membrana mitocondrial. Parásitos incubados
con A) DMSO (control); B) luego del tratamiento con el compuesto 8 durante 2h y C)
con cccp. Los gráficos son un típico histograma de al menos tres experimentos
independientes.
Flu
ore
sc
en
cia
(u
.a)
C o n tr o l 8 c c c p
0
5 0 0
1 0 0 0
1 5 0 0
2 0 0 0
**
**
Figura 50. Determinación de potencial de membrana mitocondrial. Los resultados
representan la media ± SD (n = 3). p-valor < 0,01 (**) fue considerado significativo con
respecto al control.
4.6.4. Ensayo de doble marcación con anexina V e yoduro de propidio
La anexina V (AV) tiene la capacidad de unirse a la PS, la cual desde las etapas
tempranas de apoptosis transloca desde el citosol hacia el lado externo de la membrana
plasmática. En estas etapas la membrana conserva su capacidad de excluir colorantes
vitales como el yoduro de propidio (IP). Cuando la membrana plasmática pierde
integridad dicha sonda ingresa y se intercala entre las bases de los ácidos nucleicos de
doble cadena. Por lo tanto, la detección de fenotipos apoptóticos se realiza mediante la
doble tinción con AV (marcada con FITC) e IP. De esta forma, la posibilidad de una
Even
tos
Mito-Tracker Red CMXRos

Resultados y discusión | Trypanosoma brucei
107
doble tinción permite distinguir cuatro tipos de poblaciones celulares: células viables
(AV- IP
-), células apoptóticas tempranas (AV
+ IP
-), células apoptóticas
tardías/necróticas (AV+ IP
+) y células necróticas (AV
- IP
+).
Un aspecto importante a tener en cuenta en este tipo de ensayo es la degradación del
ADN, que conlleva a falsos negativos de IP. Por este motivo se debe realizar el ensayo a
diferentes tiempos luego de la inducción de la muerte celular, para observar los cambios
de la población de parásitos entre los cuatro tipos posibles.
Con el fin de determinar si el compuesto 8 tiene la capacidad de inducir la exposición de
PS, se evaluó mediante citometría de flujo la fluorescencia de las sondas AV e IP luego
de incubar los parásitos con el compuesto a una concentración final de 10 µM durante
12h, 24h y 48h. Se utilizaron como controles: parásitos sin tratar (control) y parásitos
sometidos a un shock térmico de 60°C durante 3 min (ST) (control positivo).
Como se observa en las Figuras 51 y 52, a las 12 h de incubación con el compuesto no
hubo cambios significativos en la distribución de la población de parásitos. Sin
embargo, cuando el tratamiento duró 24 h un porcentaje significativo ocupó el
cuadrante AV+ IP
- indicando la presencia de células que conservan la integridad de su
membrana pero exponen PS, siendo este fenómeno un marcador de apoptosis temprana.
Por otro lado, a las 48h de tratamiento la mayor parte de la población se ubicó en los
cuadrantes de IP+ lo cual indica apoptosis tardía y necrosis.
De esta manera, los resultados indicaron que el compuesto estudiado desencadena en T.
brucei uno de los procesos (exposición de PS) característicos de un fenotipo apoptótico.

Resultados y discusión | Trypanosoma brucei
108
Figura 51. Determinación de la exposición de PS. Parásitos incubados con A) DMSO
(control); B) sometidos a shock térmico (ST) (control positivo); tratados con el
compuesto 8 durante C) 12 h, D) 24h y E) 48h.
Ev
en
tos
(%
)
C o n tr o l 1 2 h 2 4 h 4 8 h S T
0
2 0
4 0
6 0
8 0
1 0 0
A V - IP -
A V + IP -
A V + IP +
A V - IP +
**
**
**
Figura 52. Poblaciones celulares luego del tratamiento. Los resultados representan la
media ± SD (n = 3). p-valor < 0,01 (**) fue considerado significativo con respecto al
control.
IP
Anexina V

Resultados y discusión | Trypanosoma brucei
109
4.6.5. Ensayo TUNEL
La técnica de TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling)
permite la detección de ADN fragmentado. Se basa en la adición in situ de dUTPs en las
regiones cortadas del ADN catalizado por la enzima deoxinucleotidil transferasa.
Dichos dUTPs se encuentran marcados fluorescentemente por lo cual pueden
identificarse de forma simple y selectiva mediante citometría de flujo.
Se evaluó la capacidad del compuesto 8 de inducir cortes en el ADN mediante el kit “in
situ cell death” el cual se basa en la técnica de TUNEL. Para el ensayo se testeó el
compuesto en una concentración final de 5 y 10 µM durante 24h. Como controles se
tuvieron en cuenta: parásitos incubados con DMSO (control) y parásitos tratados con
ADNasa I (control positivo).
Los resultados mostraron que luego de las 24 h con el tratamiento de la N4-TSC la
intensidad de fluorescencia detectada fue mayor respecto al control (Figuras 53 y 54).
En el caso del tratamiento con 5 µM se obtuvo una media de células apoptóticas del
33%. Asimismo, cuando la concentración de compuesto fue el doble, 10 µM final, el
porcentaje de células apoptóticas tuvo una media del 81%. De esta forma, se evidenció
la capacidad del compuesto de inducir cortes en el ADN para los tiempos y
concentraciones estudiadas.
Figura 53. Efecto del compuesto 8 en la fragmentación del ADN de T. brucei. Los
cortes en el ADN fueron analizados mediante la técnica de TUNEL. Los parásitos se
incubaron con A) DMSO durante 24 h (control), B) ADNasa I (control positivo), y con
la N4-TSC a la concentración final de C) 5 µM y D) 10 µM.
dUTP-fluoresceína
Even
tos

Resultados y discusión | Trypanosoma brucei
110
Cé
lula
s p
os
itiv
as
%C o n tr o l 5 M 1 0 M AD N a s a I
0
2 5
5 0
7 5
1 0 0
**
**
**
Figura 54. Poblaciones celulares positivas para TUNEL luego del tratamiento
correspondiente. Los resultados representan la media ± SD (n = 3). p-valor < 0,01 (**)
fue considerado significativo con respecto al control.
En resumen, con las técnicas empleadas de citometría de flujo para el compuesto 8 se
observaron las siguientes características en los parásitos: reducción del volumen,
inducción de ROS, despolarización de ΔΨm, exposición de PS al exterior de la
membrana plasmática y fragmentación del ADN. Como conclusión de dichos resultados
se infiere que el compuesto 8 desencadena procesos característicos del fenotipo
apoptótico en T. brucei.
4.6.6. Microscopía electrónica de transmisión para ultraestructura
Para determinar el efecto del compuesto 8 sobre la morfología ultraestructural de T.
brucei se trataron los parásitos en fase exponencial con dicho derivado a la
concentración final de 7,5 µM durante 24 h y se observaron los cortes al microscopio
electrónico de transmisión. Asimismo, se observaron parásitos sin tratar como control.
Los cambios más frecuentes observados en los aislamientos tratados con el compuesto
fueron: i) alteraciones en la forma de la membrana conservando la integridad (Figura 55
d, e, h); bolsillo flagelar dilatado (Figura 55 f); apariencia de dos o más flagelos por
parásito (Figura 55 d, g) y alteraciones en el kinetoplasto (Figura 55 e). Dichas
características ultraestructurales en conjunto con los resultados de citometría de flujo
permiten afirmar la muerte celular de manera regulada. Asimismo, alteraciones en el
bolsillo flagelar, flagelos y kinetoplasto son descriptas como típicos cambios
morfológicos de la MCR.

Resultados y discusión | Trypanosoma brucei
111
Figura 55. Cambios ultraestructurales en T. brucei tratados con el compuesto 8. a, b y
c) Células control sin tratar. d, e, f, g, h) Células tratadas con la concentración final 7,5
µM del derivado 8 durante 24 h. Nótese en: d) el número de flagelos y la alteración de
la membrana plasmática; e) la alteración del kinetoplasto y la alteración de la
membrana plasmática; f) el bolsillo flagelar dilatado; g) el número de flagelos; h)
alteración de la membrana plasmática. La barra de escala representa 0,5 µm.
Abreviaciones y símbolos utilizados: N: núcleo; F: flagelo; FP: bolsillo flagelar; K:
kinetoplasto; A: acidocalcisoma; flecha blanca: glicosoma; fecha negra: alteraciones de
membrana.

Resultados y discusión | Trypanosoma brucei
112
En resumen:
El derivado 8 es el más activo de la serie y su diana biológica no serían las
proteasas del parásito.
La sustitución con nitro en R4 potencia la actividad frente al parásito. Los
derivados de la 1-indanona sin sustituyentes en el núcleo indánico y de la
5,6-dimetoxi-1-indanona fueron los de mejor efecto anti-T. brucei.
Se establece una correlación cuantitativa de la actividad biológica con la
carga del nitrógeno 2 y la diferencia de carga entre el nitrógeno 4 y su
hidrógeno.
El compuesto 8 ejerce su acción de forma selectiva sobre T. brucei
mediante un mecanismo de muerte regulado, el cual concuerda con
características del fenotipo apoptótico.

.
CAPÍTULO IV
MATERIALES Y MÉTODOS


Materiales y métodos
115
CAPÍTULO IV. MATERIALES Y MÉTODOS
1. Síntesis de las N4-TSCs
1.1. Reactivos y solventes
Todos los reactivos utilizados en las síntesis, cuya preparación no haya sido descrita,
fueron adquiridos comercialmente en Sigma-Aldrich.
Los solventes empleados fueron de calidad analítica y fueron suministrados por
Sintorgan.
1.2. Técnicas instrumentales
Las síntesis llevadas a cabo utilizando tecnología de microondas fueron realizadas en un
equipo Anton Parr Monowave 300, de potencia regulable entre 1 y 600 W, equipado
con sensor de presión y temperatura.
Los puntos de fusión se determinaron en un equipo Thomas Hoover, las unidades están
expresadas en grados centígrados (°C) y no están corregidas.
Los espectros de RMN fueron realizados en un equipo Bruker Avance II de 600 Mhz o
en un Bruker Avance II de 300 Mhz. Los solventes usados fueron CDCl3 o DMSO-d6
según se indica en cada caso. Los desplazamientos (δ) se expresan en ppm, tomando
como referencia interna la señal del tetrametilsilano (TMS) en CDCl3 o DMSO-d6. Las
constantes de acoplamiento (J) se expresan en hertz (Hz). Las multiplicidades de las
señales en los espectros de RMN están abreviadas de la siguiente forma: s, singlete; d,
doblete; dd doble doblete; t, triplete; y m, multiplete. La transformación y visualización
de los espectros se realizó con el software MestreNova.
Los espectros de IR se obtuvieron a partir de pastillas de KBr utilizando un equipo de
FT-IR Perkin Elmer Spectrum One o en un equipo FT-IR Shimadzu IRAffinity.
Los espectros de masa de alta resolución se obtuvieron en un equipo Bruker micrOTOF-
Q II con fuente de ionización a presión atmosférica por electrospray (ESI). Los
espectros fueron tomados en modo positivo y los datos se expresan en unidades de
masa/carga (m/z).

Materiales y métodos
116
1.3. Procedimiento general para la síntesis de las N4-TSCs
Método A: 1) Una mezcla de la correspondiente 1-indanona (3,8 mmol) e hidrato de
hidrazina al 85% (0,9 ml; 18,0 mmol) en isopropanol (5 ml) se calentó a reflujo. El
avance de la reacción se monitoreó por CCD usando CHCl3:EtOH (1:0,1) como FM. El
solvente se evaporó y el residuo se disolvió en CHCl3 (20 ml), se secó sobre Na2SO4
anhidro, se filtró y se evaporó nuevamente para obtener el derivado bruto. Las
hidrazonas obtenidas fueron recristalizadas en metanol. Las características físicas de las
hidrazonas derivadas de la 1-indanona, 5,6-dimetoxi-1-indanona y 5-metil-1-indanona
se correspondieron con las descriptas en literatura131–133
. 2) Una solución del
correspondiente isotiocianato (3,0 mmol) en benceno (5 ml) se añadió gota a gota a una
solución de la hidrazona (2,5 mmol) en benceno (10 ml). La mezcla resultante se agitó a
temperatura ambiente hasta desaparición de la hidrazona, monitoreado por CCD usando
CHCl3:EtOH (1:0,1) como FM. El sólido obtenido se filtró y se lavó con benceno (5 ml)
y con etanol (2 ml). Las N4-TSCs obtenidas se recristalizaron de etanol.
Método B: una mezcla de la correspondiente 1-indanona (0,378 mmol), hidrato de
hidrazina al 85% (21 µl; 0,435 mmol), el correspondiente isotiocianato (0,430 mmol),
ácido acético (20 µl) e isopropanol (0.5 mL) contenidos en un tubo de vidrio con tapón,
se colocó en un microondas de síntesis a 90°C, 30 W, 2,5 bar con agitación magnética.
El avance de la reacción se monitoreó por CCD usando CHCl3:EtOH (1:0,1) como FM.
La mezcla obtenida se suspendió en agua (5 ml), se filtró y lavó con etanol (2 ml). Las
N4-TSCs obtenidas se recristalizaron de etanol.
1.4. Descripción de los compuestos nuevos
4,5-dimetoxiindan-1-ona hidrazona
Rendimiento: 68%
Pf: 149-155°C.

Materiales y métodos
117
IR v/cm-1
(KBr): 3333 y 3187 (estiramiento N-H), 1633 (estiramiento C=N), 1268 y
1060 (estiramiento C-O).
1H-RMN (CDCl3) δ ppm: 3,05 (m, 4H, 2CH2), 3,90 (s, 3H, OCH3), 3,92 (s, 3H, OCH3),
5,10 (s, 2H, NH2), 6,93 (d, J = 8,2 Hz, 1H, H-Ar), 7,63 (d, J = 8,2 Hz, 1H, H-Ar).
13C-RMN (CDCl3) δ ppm: 25,7 (CH2), 28,8 (CH2), 56,4 (OCH3), 60,4 (OCH3), 112,5,
118,4, 132,3, 142,8, 145,4, 155,0, 169,4 (C=N).
EM de alta resolución (ESI) m/z: [M+H]+ C11H15N2O2 calculado: 207,1134; encontrado:
207,1153.
5-bromoindan-1-ona hidrazona
Rendimiento: 70%
Pf: 184-185°C
IR v/cm-1
(KBr): 3344 y 3198 (estiramiento N-H), 1639 (estiramiento C=N).
1H-RMN (CDCl3) δ ppm: 3,05 (m, 2H, CH2), 3,10 (m, 2H, CH2), 5,22 (s, 2H, NH2),
7,46 (d, J = 8,2 Hz, 1H, H-Ar), 7,54 (s, 1H, H-Ar), 7,78 (d, J = 8,2 Hz, 1H, H-Ar).
13C-RMN (CDCl3) δ ppm: 28,3 (CH2), 28,4 (CH2), 123,8, 125,6, 128,9, 130,4, 137,3,
151,7, 169,6 (C=N).
EM de alta resolución (ESI) m/z: [M+H]+ C9H9BrN2 calculado: 225,0069; encontrado:
225,0027.
Indan-1-ona N-(4-nitrofenil)tiosemicarbazona (2)
Pf: 171-172°C.
IR v/cm-1
(KBr): 3362 y 3218 (estiramiento N-H), 1597 (estiramiento C=N), 1523 y
1337 (vibración NO2), 1192 (estiramiento C=S).

Materiales y métodos
118
1H-RMN (CDCl3) δ ppm: 2,87 (m, 2H, CH2), 3,24 (m, 2H, CH2), 7,36 (m, J = 7,5 Hz,
1H, H-Ar), 7,40 (d, J = 7,5 Hz, 1H, H-Ar), 7,46 (m, J = 7,5 Hz, 1H, H-Ar), 7,80 (d, J =
7,5 Hz, 1H, H-Ar), 8,08 (d, J = 9,0Hz, 2H, H-Ar), 8,28 (d, J = 9,0 Hz, 2H, H-Ar), 8,61
(s, 1H, NNH), 9,71 (s, 1H, NH-Ar).
13C-RMN (CDCl3) δ ppm: 27,0 (CH2), 28,7 (CH2), 122,0, 122,4, 124,8, 126,0, 126,5,
127,7, 132,0, 136,5, 144,1, 144,5, 149,3, 158,2 (C=N), 175,1 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+
C16H15N4O2S calculado: 327,09102;
encontrado: 327,09136.
Indan-1-ona N-(4-fluorofenil)tiosemicarbazona (3)
Pf: 170-172°C.
IR v/cm-1
(KBr): 3279 y 3199 (estiramiento N-H), 1606 (estiramiento C=N), 1230
(estiramiento C-F), 1181 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,85 (m, 2H, CH2), 3,22 (m, 2H, CH2), 7,10 (m, 2H, H-Ar),
7,33 (m, J = 7,5 Hz, 1H, H-Ar), 7,38 (d, J = 7,5 Hz, 1H, H-Ar), 7,42 (m, J = 7,5 Hz, 1H,
H-Ar), 7,62 (dd, J1 = 8,8 Hz, J2 = 4,8 Hz, 2H, H-Ar), 7,77 (d, J = 7,5 Hz, 1H, H-Ar),
8,56 (s, 1H, NNH), 9,26 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 26,8 (CH2), 28,7 (CH2), 115,7 y 115,8 (d, J = 22,7 Hz),
121,9, 125,9, 126,7 y 126,7 (d, J = 8,3 Hz), 127,8, 131,6, 134,1, 136,8, 149,0, 157,4
(C=N), 160,1 y 161,7 (d, J = 245,9 Hz) (C-F), 176,6 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C16H15FN3S calculado: 300,09652;
encontrado: 300,09646.
Indan-1-ona N-(4-metoxifenil)tiosemicarbazona (6)
Pf: 172-173°C.

Materiales y métodos
119
IR v/cm-1
(KBr): 3308 y 3210 (estiramiento N-H), 1594 (estiramiento C=N), 1247 y
1037 (estiramiento C-O), 1187 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,83 (m, 2H, CH2), 3,21 (m, 2H, CH2), 3,83 (s, 3H, OCH3),
6,94 (d, J = 8,2 Hz, 2H, H-Ar), 7,32 (m, J = 7,5 Hz, 1H, H-Ar), 7,38 (m, J = 7,5 Hz, 1H,
H-Ar), 7,41 (m, J = 7,5 Hz, 1H, H-Ar), 7,53 (d, J = 8,2 Hz, 2H, H-Ar), 7,77 (d, 7,5 Hz,
1H, H-Ar), 8,47 (s, 1H, NNH), 9,20 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 26,7 (CH2), 28,7 (CH2), 55,6 (OCH3), 114,2, 121,9, 125,9,
126,6, 126,7, 127,5, 131,1, 131,4, 137,0, 148,9, 156,9, 157,0 (C=N), 158,1, 176,8
(C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C17H18N3OS calculado: 312,11651;
encontrado: 312,11651.
5,6-Dimetoxiindan-1-ona N-(4-nitrofenil)tiosemicarbazona (8)
Pf: 214-216 °C.
IR v/cm-1
(KBr): 3273 y 3191 (estiramiento N-H), 1598 (estiramiento C=N), 1528 y
1344 (vibración NO2), 1261 y 1075 (estiramiento C-O), 1197 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,86 (m, 2H, CH2), 3,14 (m, 2H, CH2), 3,94 (s, 3H, OCH3),
3,96 (s, 3H, OCH3), 6,84 (s, 1H, H-Ar), 7,16 (s, 1H, H-Ar), 8,04 (d, J = 8,9 Hz, 2H, H-
Ar), 8,26 (d, J = 8,9 Hz, 2H, H-Ar), 8,56 (s, 1H, NNH), 9,58 (s, 1H, NH).
13C-RMN (DMSO-d6) δ ppm: 28,6 (CH2), 28,7 (CH2), 56,1 (OCH3), 56,3 (OCH3),
105,1, 108,3, 112,9, 124,7, 126,9, 129,5, 143,7, 146,1, 149,3, 152,9 (C=N), 160,6, 175,7
(C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C18H19N4O4S calculado: 387,11215;
encontrado: 387,11081.

Materiales y métodos
120
5,6-Dimetoxiindan-1-ona N-(4-fluorofenil)tiosemicarbazona (9)
Pf: 185-186°C.
IR v/cm-1
(KBr): 3300 y 3196 (estiramiento N-H), 1605 (estiramiento C=N), 1255 y
1079 (estiramiento C-O), 1219 (estiramiento C-F), 1198 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,86 (m, 2H, CH2), 3,15 (m, 2H, CH2), 3,95 (s, 3H, OCH3),
3,96 (s, 3H, OCH3), 6,85 (s, 1H, H-Ar), 7,12 (d, J1 = 8,6 Hz, 2H, H-Ar), 7,18 (s, 1H, H-
Ar), 7,61 (dd, J1 = 8,6 Hz, J2 = 4,8 Hz, 2H, H-Ar), 8,52 (s, 1H, NNH), 9,19 (s, 1H, NH).
13C-RMN (DMSO-d6) δ ppm: 28,0 (CH2), 28,1 (CH2), 55,6 (OCH3), 55,8 (OCH3),
104,4, 107,8, 114,8 y 115,0 (d, J = 22,4 Hz), 128,4 y 128,4 (d, J = 8,1 Hz), 129,4, 135,7,
142,6, 148,8, 152,1 (C=N), 158,5, 158,9, 159,7 y 161,3 (d, J = 242,0 Hz) (C-F), 177,0
(C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C18H19FN3O2S calculado: 360,11765;
encontrado: 360,11645.
5,6-Dimetoxiindan-1-ona N-(4-metoxifenil)tiosemicarbazona (12)
Pf: 198-199 °C.
IR v/cm-1
(KBr): 3312 y 3216 (estiramiento N-H), 1603 (estiramiento C=N), 1249, 1238
y 1075, 1023 (estiramiento C-O), 1196 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,82 (m, 2H, CH2), 3,12 (m, 2H, CH2), 3,82 (s, 3H, OCH3),
3,92 (s, 3H, OCH3), 3,93 (s, 3H, OCH3), 6,83 (s, 1H, H-Ar), 6,93 (d, J = 9,0 Hz, 2H, H-
Ar), 7,15 (s, 1H, H-Ar), 7,49 (d, J = 9,0 Hz, 2H, H-Ar), 8,47 (s, 1H, NNH), 9,11 (s, 1H,
NH).

Materiales y métodos
121
13C-RMN (DMSO-d6) δ ppm: 28,3 (CH2), 28,6 (CH2), 55,8 (OCH3), 56,1 (OCH3), 56,3
(OCH3), 104,9, 108,3, 113,8, 128,3, 130,0, 132,7, 142,9, 149,3, 152,5 (C=N), 157,5,
158,6, 177,1 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C19H22N3O3S calculado: 372,13764;
encontrado: 372,13836.
5-Metilindan-1-ona N-feniltiosemicarbazona (13)
Pf: 184-186 °C.
IR v/cm-1
(KBr): 3262 y 3195 (estiramiento N-H), 1594 (estiramiento C=N), 1190
(estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,41 (s, 3H, CH3), 2,83 (m, 2H, CH2), 3,16 (m, 2H, CH2),
7,14 (d, J = 7,9 Hz, 1H, H-Ar), 7,18 (s, 1H, H-Ar), 7,24 (t, J = 7,5 Hz, 1H, H-Ar), 7,41
(t, J = 7,5 Hz, 2H, H-Ar), 7,65 (d, J = 7,9 Hz, 1H, H-Ar), 7,70 (d, J = 7,5 Hz, 2H, H-
Ar), 8,49 (s, 1H, NNH), 9,36 (s, 1H, NH).
13C-RMN (DMSO-d6) δ ppm: 22,2 (CH3), 27,2 (CH2), 28,7 (CH2), 121,8, 124,6, 126,4,
126,6, 128,9, 129,1, 134,5, 138,4, 142,4, 149,6, 157,4 (C=N), 176,2 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C17H18N3S calculado: 296,12159; encontrado:
296,12260.
5-Metilindan-1-ona N-(4-nitrofenil)tiosemicarbazona (14)
Pf: 203-204 °C.
IR v/cm-1
(KBr): 3209 y 3143 (estiramiento N-H), 1595 (estiramiento C=N), 1558 y
1329 (vibración NO2), 1199 (estiramiento C=S).

Materiales y métodos
122
1H-RMN (CDCl3) δ ppm: 2,42 (s, 3H, CH3), 2,85 (m, 2H, CH2), 3,18 (m, 2H, CH2),
7,17 (d, J = 8,1 Hz, 1H, H-Ar), 7,67 (d, J = 8,1 Hz, 1H, H-Ar), 8,07 (d, 2H, J = 9,1 Hz,
H-Ar), 8,27 (d, J = 9,1 Hz, 2H, H-Ar), 8,59 (s, 1H, NNH), 9,70 (s, 1H, NH).
13C-RMN (DMSO-d6) δ ppm: 22,0 (CH3), 27,2 (CH2) ,28,5 (CH2), 104,9, 121,7, 122,3,
124,7, 126,5, 128,3, 126,5, 142,1, 142,8, 144,16, 144,5, 149,7, 168,8, 175,0 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C17H17N4O2S calculado: 341,10667;
encontrado: 341,10601.
5-Metilindan-1-ona N-(4-fluorofenil)tiosemicarbazona (15)
Pf: 173-175 °C.
IR v/cm−1
(KBr): 3183 y 3111 (estiramiento N-H), 1602 (estiramiento C=N), 1215
(estiramiento C-F), 1182 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,41 (s, 3H, CH3), 2,83 (m, 2H, CH2), 3,16 (m, 2H, CH2),
7,09 (d, J = 8,9 Hz, 2H, H-Ar), 7,14 (d, J = 7,9 Hz, 1H, H-Ar), 7,18 (s, 1H, H-Ar), 7,62
(dd, J1 = 8,9 Hz, J2 = 4,8 Hz, 2H, H-Ar), 7,65 (d, J = 7,9 Hz, 1H, H-Ar), 8,51 (s, 1H,
NNH), 9,25 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 22,2 (CH3), 27,2 (CH2), 28,7 (CH2), 115,8 y 116,0 (d, J =
22,7 Hz), 121,8, 126,6, 126.9 y 126,9 (d, J = 8,25 Hz), 129,0, 134,4, 142,5, 149,6, 157,7
(C=N), 160,2 y 161,9 (d, J = 245,7 Hz) (C-F), 176,7 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C17H17FN3S calculado: 314,11217;
encontrado: 314,11265.
5-Metilindan-1-ona N-(4-clorofenil)tiosemicarbazona (16)
Pf: 169–170°C.

Materiales y métodos
123
IR v/cm−1
(KBr): 3264 y 3162 (estiramiento N-H), 1586 (estiramiento C=N), 1190
(estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,40 (s, 3H, CH3), 2,82 (m, 2H, CH2), 3,15 (m, 2H, CH2),
7,14 (d, J = 8,3 Hz, 1H, H-Ar), 7,17 (s, 1H, H-Ar), 7,35 (d, J = 8,9 Hz, 2H, H-Ar), 7,64
(d, J = 8,9 Hz, 2H, H-Ar), 7,66 (d, J = 8,3 Hz, 1H, H-Ar), 8,51 (s, 1H, NNH), 9,31 (s,
1H, NH).
13C-RMN (CDCl3) δ ppm: 22,2 (CH3), 27,2 (CH2), 28,7 (CH2), 121,9, 125,8, 126,6,
128,9, 129,2, 131,6, 134,3, 137,0, 142,6, 149,7, 157,8 (C=N), 176,1 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C17H17ClN3S calculado: 330,08262;
encontrado: 330,08262.
5-Metilindan-1-ona N-(4-metilfenil)tiosemicarbazona (17)
Pf: 179–180°C.
IR v/cm−1
(KBr): 3345 y 3302 (estiramiento N-H), 1591 (estiramiento C=N), 1171
(estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,37 (s, 3H, CH3), 2,41 (s, 3H, CH3), 2,82 (m, 2H, CH2), 3,16
(m, 2H, CH2), 7,14 (d, J = 7,9 Hz, 1H, H-Ar), 7,18 (s, 1H, H-Ar), 7,21 (d, J = 8,3 Hz,
2H, H-Ar), 7,54 (d, J = 8,3 Hz, 2H, H-Ar), 7,65 (d, J = 7,9 Hz, 1H, H-Ar), 8,47 (s, 1H,
NNH), 9,26 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 21,4 (CH3), 22,2 (CH3), 27,2 (CH2), 28,7 (CH2), 121,7,
124,8, 126,5, 128,9, 129,7, 134,4, 136,2, 136,3, 142,3, 149,4, 157,2 (C=N), 174,7
(C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C18H20N3S calculado: 310,1378; encontrado:
310,1384

Materiales y métodos
124
5-Metilindan-1-ona N-(4-metoxifenil)tiosemicarbazona (18)
Pf: 178–180°C.
IR v/cm−1
(KBr): 3323 y 3141 (estiramiento N-H), 1608 (estiramiento C=N), 1239 y
1060 (estiramiento C-O), 1164 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,40 (s, 3H, CH3), 2,82 (m, 2H, CH2), 3,15 (m, 2H, CH2),
3,83 (s, 3H, OCH3), 6,94 (d, J = 8,7 Hz, 2H, H-Ar), 7,13 (d, J = 7,9 Hz, 1H, H-Ar), 7,18
(s, 1H, H-Ar), 7,52 (d, J = 8,7 Hz, 2H, H-Ar), 7,65 (d, J = 7,9 Hz, 1H, H-Ar), 8,49 (s,
1H, NNH), 9,19 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 21,9 (CH3), 26,9 (CH2), 28,5 (CH2), 55,6 (OCH3), 114,2,
121,6, 126,4, 126,7, 128,7, 131,1, 134,3, 142,1, 149,3, 157,1, 158,1 (C=N), 176,7
(C=S).
EM de alta resolución (ESI) m/z: [M+H]+C18H20N3OS calculado: 326,1322; encontrado:
326,1326.
4,5-Dimetoxiindan-1-ona N-feniltiosemicarbazona (19)
Pf: 197-199 °C.
IR v/cm−1
(KBr): 3259 y 3161(estiramiento N-H), 1594 (estiramiento C=N), 1256 y
1060 (estiramiento C-O), 1186 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,86 (m, 2H, CH2), 3,21 (m, 2H, CH2), 3,93 (s, 3H, OCH3),
3,95 (s, 3H, OCH3), 6,93 (d, J = 8,5 Hz, 1H, H-Ar), 7,24 (t, J = 7,3 Hz, 1H, H-Ar), 7,41
(t, J = 7,3 Hz, 2H, H-Ar), 7,50 (d, J = 8,5 Hz, 1H, H-Ar), 7,72 (d, J = 7,3 Hz, 2H, H-
Ar), 8,49 (s, 1H, NNH), 9,35 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 25,9 (CH2), 27,5 (CH2), 56,6 (OCH3), 60,7 (OCH3), 113,1,
117,8, 124,6, 126,4, 129,1, 130,9, 138,4, 142,3, 150,5, 156,9 (C=N), 162,2, 176,8
(C=S).

Materiales y métodos
125
EM de alta resolución (ESI) m/z: [M+H]+ C18H20N3O2S calculado: 342,1271;
encontrado: 342,1261.
4,5-Dimetoxiindan-1-ona N-(4-nitrofenil)tiosemicarbazona (20)
Pf: 223-225 °C.
IR v/cm−1
(KBr): 3227 y 3154 (estiramiento N-H), 1595 (estiramiento C=N), 1545 y
1324 (vibración NO2), 1271 y 1058 (estiramiento C-O), 1199 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,86 (m, 2H, CH2), 3,20 (m, 2H, CH2), 3,91 (s, 3H, OCH3),
3,94 (s, 3H, OCH3), 6,95 (d, J = 8,5 Hz, 1H, H-Ar), 7,50 (d, J = 8,5 Hz, 1H, H-Ar), 8,07
(d, J = 9,1 Hz, 2H, H-Ar), 8,27 (d, J = 9,1 Hz, 2H, H-Ar), 8,58 (s, 1H, NNH), 9,67 (s,
1H, NH).
13C-RMN (CDCl3) δ ppm: 25,9 (CH2), 27,2 (CH2), 58,8 (OCH3), 60,3 (OCH3), 111,1,
117,2, 122,1, 124,0, 130,9, 141,8, 145,1, 155,2, 157,2 (C=N), 177,2 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C18H19N4O4S calculado: 387,1122;
encontrado: 387,1125.
4,5-Dimetoxiindan-1-ona N-(4-fluorofenil)tiosemicarbazona (21)
Pf: 182–183°C.
IR v/cm−1
(KBr): 3290 y 3119 (estiramiento N-H), 1601 (estiramiento C=N), 1268 y
1076 (estiramiento C-O), 1220 (estiramiento C-F), 1202 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,84 (m, 2H, CH2), 3,19 (m, 2H, CH2), 3,90 (s, 3H, OCH3),
3,93 (s, 3H, OCH3), 6,93 (d, J = 8,5 Hz, 1H, H-Ar), 7,09 (d, J = 8,9 Hz, 2H, H-Ar), 7,48
(d, J = 8,5 Hz, 1H, H-Ar), 7,61 (dd, J1 = 8,9 Hz, J2 = 4,8 Hz, 2H, H-Ar), 8,49 (s, 1H,
NNH), 9,22 (s, 1H, NH).

Materiales y métodos
126
13C-RMN (CDCl3) δ ppm: 25,9 (CH2), 27,5 (CH2), 56,6 (OCH3), 60,7 (OCH3), 113,1,
115,9 y 116,0 (d, J = 22,1 Hz), 117,8, 126,9 y 126,9 (d, J = 8,27 Hz), 130,8, 134,4,
142,4, 145,9, 155,3, 157,2 (C=N), 160,2 y 161,9 (d, J = 246 Hz) (C-F), 176,6 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C18H19FN3O2S calculado: 360,1177;
encontrado: 360,1183.
4,5-Dimetoxiindan-1-ona N-(4clorolfenil)tiosemicarbazona (22)
Pf: 179-180 °C.
IR v/cm−1
(KBr): 3288 y 3142 (estiraminto N-H), 1583 (estiramiento C=N), 1271 y
1076 (estiramiento C-O), 1199 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,84 (m, 2H, CH2), 3,19 (m, 2H, CH2), 3,90 (s, 3H, OCH3),
3,93 (s, 3H, OCH3), 6,93 (d, J = 8,4 Hz, 1H, H-Ar), 7,36 (d, J = 8,7 Hz, 2H, H-Ar), 7,48
(d, J = 8,4 Hz, 1H, H-Ar), 7,66 (d, J = 8,7 Hz, 2H, H-Ar), 8,50 (s, 1H, NNH), 9,31 (s,
1H, NH).
13C-RMN (CDCl3) δ ppm: 25,9 (CH2), 27,5 (CH2), 56,6 (OCH3), 60,7 (OCH3), 113,1,
117,8, 125,8, 129,2, 130,7, 131,6, 137,0, 142,4, 145,9, 155,4, 157,3 (C=N), 176,0
(C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C18H19ClN3O2S calculado: 376,0881;
encontrado: 376,0880.
4,5-Dimetoxiindan-1-ona N-(4-metilfenil)tiosemicarbazona (23)
Pf: 179-181°C.
IR v/cm−1
(KBr): 3298 y 3145 (estiramiento N-H), 1590 (estiramiento C=N), 1265 y
1064 (estiramiento C-O), 1185 (estiramiento C=S).

Materiales y métodos
127
1H-RMN (CDCl3) δ ppm: 2,39 (s, 3H, CH3), 2,85 (m, 2H, CH2), 3,21 (m, 2H, CH2),
3,92 (s, 3H, OCH3), 3,95 (s, 3H, OCH3), 6,95 (d, J = 8,5 Hz, 1H, H-Ar), 7,23 (d, J = 8,3
Hz, 2H, H-Ar), 7,50 (d, J = 8,5 Hz, 1H, H-Ar), 7,55 (d, J = 8,3 Hz, 2H, H-Ar), 8,47 (s,
1H, NNH), 9,26 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 21,2 (CH3), 25,6 (CH2), 27,2 (CH2), 56,4 (OCH3), 60,5
(OCH3), 112,9, 117,6, 123,0, 124,7, 129,5, 129,5, 130,7, 135,6, 136,1, 142,1, 155,0
(C=N), 173,3 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C19H22N3O2S calculado: 356,1433;
encontrado: 356.1456.
4,5-Dimetoxiindan-1-ona N-(4-metoxilfenil)tiosemicarbazona (24)
Pf: 176-177°C.
IR v/cm−1
(KBr): 3301 y 3154 (estiramiento N-H), 1598 (estiramiento C=N), 1262,
1243 y 1073, 1058 (estiramiento C-O), 1180 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2.85 (m, 2H, CH2), 3.21 (m, 2H, CH2), 3,85 (s, 3H, OCH3),
3,92 (s, 3H, OCH3), 3,95 (s, 3H, OCH3), 6,95 (d, J = 8,4 Hz, 1H, H-Ar), 6,96 (d, J = 8,9
Hz, 2H, H-Ar), 7,50 (d, J = 8,4 Hz, 1H, H-Ar), 7,54 (d, J = 8,9 Hz, 2H, H-Ar), 8,48 (s,
1H, NNH), 9,18 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 25,5 (CH2), 27,1 (CH2), 55,5 (OCH3), 56,3 (OCH3), 60,3
(OCH3), 112,7, 114,0, 117,4, 126,6, 130,6, 131,0, 141,9, 145,5, 154,9, 157,9 (C=N),
177,8 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C19H22N3O3S calculado: 372,1376;
encontrado: 372,1383.
5-Bromoindan-1-ona N-feniltiosemicarbazona (25)

Materiales y métodos
128
Pf: 196-198°C.
IR v/cm−1
(KBr): 3286 y 3185 (estiramiento N-H), 1590 (estiramiento C=N), 1167
(estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,85 (m, 2H, CH2), 3,20 (m, 2H, CH2), 7,26 (t, J = 7,5 Hz,
1H, H-Ar), 7,41 (t, J = 7,9 Hz, 2H, H-Ar), 7,46 (d, J = 8,3 Hz, 1H, H-Ar), 7,53 (d, 1H,
H-Ar), 7,63 (d, J = 8,3 Hz, 1H, H-Ar), 7,69 (d, J = 7,9 Hz, 2H, H-Ar), 8,52 (s, 1H,
NNH), 9,31 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 26,7 (CH2), 28,3 (CH2), 122,8, 124,2, 125,7, 126,2, 128,8,
129,0, 130,8, 135,87, 137,9, 145,3, 150,4 (C=N), 175,9 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C16H15BrN3S calculado: 360,01646;
encontrado: 360,1629.
5-Bromoindan-1-ona N-(4-nitrofenil)tiosemicarbazona (26)
Pf: 191-193°C.
IR v/cm−1
(KBr): 3287 y 3214 (estiramiento N-H), 1595 (estiramiento C=N), 1508 y
1329 (vibración NO2), 1167 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,88 (m, 2H, CH2), 3,23 (m, 2H, CH2), 7,44 (d, J = 9,1 Hz,
2H, H-Ar), 8,02 (s, 1H, H-Ar), 8,06 (d, J = 9,1 Hz, 1H, H-Ar), 8,24 (d, J = 9,1 Hz, 2H,
H-Ar), 8,28 (d, J = 9,1 Hz, 1H, H-Ar), 8,61 (s, 1H, NH), 9,64 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 27,8 (CH2), 29,3 (CH2), 122,0, 124,0, 129,0, 130,35, 142,1,
149,5 (C=N), 157,3, 176,9 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C16H14BrN4O2S calculado: 405,00154;
encontrado: 405,00247.

Materiales y métodos
129
5-Bromoindan-1-ona N-(4-fluorofenil)tiosemicarbazona (27)
Pf: 198-199°C.
IR v/cm−1
(KBr): 3273 y 3168 (estiramiento N-H), 1593 (estiramiento C=N), 1192
(estiramiento C-F), 1165 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,88 (m, 2H, CH2), 3,23 (m, 2H, CH2), 7,13 (d, J = 8,9 Hz,
2H, H-Ar), 7,47 (d, J = 8,3 Hz, 1H, H-Ar), 7,56 (s, 1H, H-Ar), 7,63 (dd, J1 = 8,9 Hz, J2
= 4,8 Hz, 2H, H-Ar), 7,64 (d, J = 8,3 Hz, 1H, H-Ar), 8,56 (s, 1H, NNH), 9,23 (s, 1H,
NH).
13C-RMN (CDCl3) δ ppm: 27,1 (CH2), 28,7 (CH2), 116,0 y 116,1 (d, J = 22,8 Hz),
123,2, 126,1, 126,9 y 127,0 (d, J = 8,3 Hz), 129,4, 131,2, 134,2, 136,1, 150,9 (C=N),
156,1, 159,9 y 161,6 (d, J = 264,17 Hz) (C-F), 176,9 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C16H14BrFN3S calculado: 378,00704;
encontrado: 378,00699.
5-Bromoindan-1-ona N-(4-clorofenil)tiosemicarbazona (28)
Pf: 199-201°C.
IR v/cm−1
(KBr): 3273 y 3133 (estiramiento N-H), 1586 (estiramiento C=N), 1166
(estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,85 (m, 2H, CH2), 3,20 (m, 2H, CH2), 7,37 (d, J = 8,8 Hz,
2H, H-Ar), 7,46 (d, J = 8,3 Hz 1H, H-Ar), 7,54 (s, 1H, H-Ar), 7,63 (d, J = 8,3 Hz, 1H,
H-Ar), 7,66 (d, J = 8,8 Hz, 2H, H-Ar), 8,52 (s, 1H, NNH), 9,26 (s, 1H, NH).
13C-RMN (CDCl3) δ ppm: 26,7 (CH2), 28,3 (CH2), 122,9, 125,4, 125,8, 128,9, 129,0,
130,9, 131,4, 135,7, 136,5, 150,5 (C=N), 155,7, 176,0 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C16H14BrClN3S calculado: 393,97748;
encontrado: 393,97843.

Materiales y métodos
130
5-Bromoindan-1-ona N-(4-metilfenil)tiosemicarbazona (29)
Pf: 182-184°C.
IR v/cm−1
(KBr): 3288 y 3138 (estiramiento N-H), 1591 (estiramiento C=N), 1164
(estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,37 (s, 3H, CH3), 2,84 (m, 2H, CH2), 3,20 (m, 2H, CH2),
7,21 (d, J = 8,2 Hz, 2H, H-Ar), 7,45 (d, J = 8,3, 1H, H-Ar), 7.52 (d, J = 8,2, 2H, H-Ar),
7,53 (s, 1H, H-Ar), 7,62 (d, J = 8,3 Hz, 1H, H-Ar), 8,50 (s, 1H, NNH), 9,22 (s, 1H,
NH).
13C-RMN (CDCl3) δ ppm: 21,4 (CH3), 27.0 (CH2), 28,7 (CH2), 123,2, 124.9, 126,0,
129,4, 129,7, 131,2, 135,7, 136,3, 136,5, 150,8 (C=N), 155,7, 176,6 (C=S).
EM de alta resolución (ESI) m/z: [M+H]+ C17H17BrN3S calculado: 374,0327;
encontrado: 374,0331.
5-Bromoindan-1-ona N-(4-metoxifenil)tiosemicarbazona (30)
Pf: 184-185°C.
IR v/cm−1
(KBr): 3288 y 3129 (estiramiento N-H), 1592 (estiramiento C=N), 1247 y
1056 (estiramiento C-O), 1168 (estiramiento C=S).
1H-RMN (CDCl3) δ ppm: 2,84 (m, 2H, CH2), 3,20 (m, 2H, CH2), 3,83 (s, 3H, OCH3),
6,94 (d, J = 8,9 Hz, 2H, H-Ar), 7,45 (d, J = 8,3 Hz, 1H, H-Ar), 7,51 (d, J = 8,8 Hz, 2H,
H-Ar), 7,53 (s, 1H, H-Ar), 7,62 (d, J = 8,3 Hz, 1H, H-Ar), 8,50 (s, 1H, NNH), 9,14 (s,
1H, NH).
13C-RMN (CDCl3) δ ppm: 26,6 (CH2), 28,3 (CH2), 55,5 (OCH3), 105,8, 114,1, 122,8,
126,5, 127,6, 129,0, 130,9, 135,2, 150,8 (C=N), 155,8, 158,7, 174,7 (C=S).

Materiales y métodos
131
EM de alta resolución (ESI) m/z: [M+H]+ C17H17BrN3OS calculado: 390,0270;
encontrado: 390,0287.
2. Estudios biológicos
2.1. Virus de la diarrea viral bovina
2.1.1. Cultivo celular y virus
Células de riñón bovino Madin Darby (MDBK) se hicieron crecer en el medio E-MEM
(medio esencial mínimo Eagle) suplementado con 10% de suero fetal bovino (SFB,
Internegocios), 2mM de L-glutamina, 100 µM de aminoácidos no esenciales, 100 µg/ml
de estreptomicina y 100 UI/ml de penicilina (medio de crecimiento). Se utilizó el
VDVB cepa tipo 1 NADL, biotipo CP (VDVB-1, ATCC VR 534).
2.1.2. Ensayo de reducción de placas
Se sembraron en una placa de cultivo de 24 pocillos 1,3x105 células MDBK por pocillo.
Después de incubar durante 24 h con 5% de CO2 a 37°C se removió el medio de
crecimiento, se lavaron las células con buffer fosfato salino (PBS) y se infectaron con
100 UFP del VDVB. Después de 1 h de incubación a 37°C se lavaron las células con
PBS y se agregaron los compuestos a evaluar en cada pocillo. Se utilizó ribavirina y
5,6-TSC como controles positivos. Asimismo, se incluyeron como controles células sin
tratar y células sin infectar. Después de incubar durante 72 h con 5% de CO2 a 37°C, las
células se fijaron con 10% de formaldehído, se tiñeron con 0,75% de cristal violeta y se
contaron las placas virales. La concentración efectiva 50 (CE50) fue definida como la
concentración del compuesto que reduce el número de placas virales en un 50% con
respecto al control infectado sin tratar y fue determinada a partir de curvas de
concentración-respuesta utilizando GraphPad Prism. Se llevaron a cabo tres
experimentos independientes por triplicado.
2.1.3. Estudios de resistencia cruzada
El compuesto 8 y la 5,6-TSC fueron evaluados mediante el ensayo de reducción del
ECP frente al virus con la enzima RdRp wild type y con las mutaciones N264D y

Materiales y métodos
132
A392E27
. En el ensayo de reducción del ECP se sembraron en una placa de cultivo de
96 pocillos 1x104 células MDBK por pocillo. Después de incubar durante 24 h en una
estufa con 5% de CO2 a 37°C, las células se infectaron con el VDVB a una
multiplicidad de infección de 0,01 UFP/célula. Se utilizó ribavirina y 5,6-TSC como
controles positivos. Asimismo, se incluyeron células sin tratar y células sin infectar.
Después de incubar durante 72 h a 37ºC, se determinó el ECP midiendo la viabilidad
celular por MTS/PES (Cell Titer 96 Aqueous One Solution Cell Proliferation Assay,
Promega). La concentración efectiva 50 (CE50) fue definida como la concentración del
compuesto que reduce el ECP en un 50% con respecto al control infectado sin tratar y
fue determinada a partir de curvas de concentración-respuesta utilizando GraphPad
Prism. Se llevaron a cabo dos experimentos independientes por cuadruplicado.
2.2. Trypanosoma cruzi
2.2.1. Cultivo celular de epimastigotes
Se cultivaron epimastigotes de T. cruzi (clon CL Brener) a 28°C durante 5 a 7 días (fase
exponencial de crecimiento) en medio líquido monofásico Brain Heart Triptose (BHT:
infusión cerebro-corazón 33 g/l, triptosa 3 g/l, Na2HPO4 4 g/l, KCl 0,4 g/l, glucosa 0,3
g/l y hemina 20 mg/l) suplementado con 10% de SFB (Natocor), 100 UI/ml de
penicilina y 100 µg/ml de estreptomicina.
2.2.2. Actividad anti-epimastigotes de T. cruzi
Las células se cosecharon mediante centrifugación y se resuspendieron en medio fresco,
realizando el recuento en un contador de células Coulter Counter (Beckman). Se
sembraron en placas de 96 pocillos 1x106 parásitos/pocillo. Se agregaron los
compuestos a evaluar en una concentración final de 20 µM a partir de una solución
madre en DMSO. La concentración final de DMSO en el medio de cultivo nunca superó
el 1% y el control se realizó en presencia de 1% de DMSO y en ausencia de cualquier
compuesto. Se utilizó benznidazol (Bz) como control positivo. Los parásitos se
incubaron a 28°C, durante 120 h. Luego de este tiempo se procedió a realizar el
recuento de los parásitos vivos mediante el contador de células. Se determinó el
porcentaje de inhibición para una concentración 20 µM de compuesto. Se llevaron a
cabo tres experimentos independientes por triplicado.

Materiales y métodos
133
2.2.3. Obtención de tripomastigotes sanguíneos
Los parásitos fueron tomados de ratones BALB/c infectados previamente con la cepa
Tulahuén, stock Tul2. Dicha cepa es mantenida de rutina en el estadio de tripomastigote
sanguíneo por infecciones sucesivas en el bioterio del Instituto Nacional de
Parasitología “Dr. Mario Fatala Chabén”.
2.2.4. Actividad anti-tripomastigotes sanguíneos de T. cruzi
Se cuantificaron los parásitos mediante recuento en cámara de Malassez y se sembraron
en placas de 96 pocillos fondo en U 5x104
parásitos/pocillo, utilizando el medio BHT
sin suplementar con SFB. Se trataron las células con una concentración final de 20 µM
de compuesto a partir de una solución madre en DMSO. La concentración final de
DMSO en el medio de cultivo nunca superó el 1% y el control se realizó en presencia de
1% de DMSO y en ausencia de cualquier compuesto. Se utilizó Bz como control
positivo. Los parásitos se incubaron durante 24 h a 4°C. Luego de ese tiempo se
procedió a realizar el recuento de los parásitos vivos mediante el método de Pizzi-
Brener. Se determinó el porcentaje de lisis para una concentración final de 20 µM de
compuesto. Se llevaron a cabo al menos tres experimentos independientes por
duplicado.
2.2.5. Cultivo celular de tripomastigotes
El mantenimiento de tripomastigotes (clon CL Brener) se realizó en cultivos de células
Vero. Se cultivaron células Vero (células epiteliales de riñón derivadas de mono verde
africano) a 37°C, con 5% de CO2 en medio RPMI suplementado con 10% de SFB
(Natocor), 100 UI/ml de penicilina y 100 µg/ml estreptomicina. Los parásitos fueron
cosechados del sobrenadante de cultivo, centrifugados a 1800 rpm durante 10 min, re-
suspendidos en medio RPMI suplementado con 10% SFB y mantenidas a 4°C durante
todo el procedimiento.
2.2.6. Actividad anti-amastigotes de T. cruzi
Se sembraron un total de 1x104 células Vero/pocillo sobre cubreobjetos circulares de 12
mm introducidos en placas de 24 pocillos e incubados a 37°C con 5% de CO2 durante 2
h. A continuación se infectaron con 1x105 tripomastigotes/pocillo obtenidos

Materiales y métodos
134
previamente de cultivos de células Vero. Las células fueron incubadas durante 24 h a
37°C con 5% de CO2. Luego de dicho tiempo, se lavaron los cultivos para eliminar los
parásitos no internalizados y las células fueron tratadas con distintas concentraciones de
los compuestos a evaluar diluidas en DMSO. La concentración final de DMSO en el
medio de cultivo nunca superó el 0,25% y el control se realizó en presencia de 0,25% de
DMSO y en ausencia de cualquier compuesto. Se utilizó Bz como control positivo. Las
células se incubaron durante 72 h adicionales. Los cubreobjetos con las células
adheridas se retiraron de la placa y las células se fijaron con metanol y se tiñeron con
Giemsa (Merck) para su observación al microscopio de campo claro. Se exploraron los
cubreobjetos con aumentos de 40X fotografiando un mínimo de 10 campos distintos al
azar (Laica CTR Mic, Alemania). Se cuantificó el número de amastigotes/célula
realizando un recuento mínimo de 200 células/cubreobjeto. Se llevaron a cabo tres
experimentos independientes por duplicado.
2.2.7. Obtención de la cruzipaína
La purificación de la Cz se llevó a cabo a partir de epimastigotes de T. cruzi. La primera
parte de la purificación incluyó la obtención del extracto libre de células. Para ello, se
lisaron los parásitos mediante dos ciclos consecutivos de congelado y descongelado y se
re-suspendieron en una solución de sacarosa 0,25 M y KCl 5 mM (3 ml/gr de parásitos).
El homogenato resultante se centrifugó a 7000 rpm durante 10 min a 5 °C, el
sobrenadante se recuperó y el precipitado se volvió a resuspender en la misma solución
y se centrifugó a 12000 rpm durante 15 min a 5°C. Los sobrenadantes se clarificaron
por centrifugación a 19000 rpm durante 30 min a 4°C. A continuación se realizó la
precipitación con solución saturada de sulfato de amonio pH 7,5. La suspensión se
centrifugó a 12000 rpm durante 15 min a 5 °C. Se recuperó el pellet y se re-suspendió
en buffer TBS (Tris-HCl 50 mM, pH 7,5, NaCl 150 mM). Se dializó 2 veces y se dejó
durante toda la noche a 4 °C.
La segunda parte de la purificación de la Cz se realizó por medio de una cromatografía
de afinidad con Concanavalina A (Con-A). Para ello, al extracto libre de células
dializado se le agregó CaCl2, MgCl2 y MnCl2 hasta alcanzar una concentración final 3
mM de cada uno, y el mismo se centrifugó a 10000 rpm durante 12 min a 5°C. El
sobrenadante resultante se eluyó utilizando α-metil-manopiranósido 10% en una
columna de ConA-Sepharosa. La columna se lavó hasta absorbancia nula a 280 nm en

Materiales y métodos
135
el eluato. Se recuperaron las fracciones con actividad enzimática sobre sustrato sintético
cromogénico Bz-Pro-Phe-Arg-pNA, y se dializaron contra Tris-HCl 50 mM pH 7.6 a
4°C. Los pasos de la purificación fueron controlados mediante determinación de
proteínas y actividad enzimática frente a sustratos sintéticos cromogénicos, SDS PAGE
y Western Blot con suero policlonal específico para Cz.
2.2.8. Zimografía
En un gel de poliacrilamida 10% con dodecilsulfato sódico (SDS-PAGE) y 0,15% de
gelatina incorporada como sustrato, se sembraron 10 µL de una dilución 1:100 de la
enzima purificada (20 U/ml) en PBS 50 mM pH 7,3 y DTT 5 mM por calle. Asimismo,
se corrió un estándar comercial de sustancias con pesos moleculares conocidos
(Prestained Broad Range Protein Marker 2-220 kDa, Genbiotech). La corrida se realizó
a 120 V durante 2 h a 4°C. Una vez finalizada se lavó el gel dos veces durante 15 min
con 0,1% Tritón X-100 y se incubó cada calle con el compuesto correspondiente
durante 24 h a 37°C en buffer MES 0,1 M con agregado de DTT 0,5 mM. Se incluyeron
los siguientes controles: la Cz sin tratar, la Cz incubada con 5% de DMSO y la Cz
incubada con E-64. Luego de la incubación se procedió a teñir el gel con Coomassie
brilliant blue R-250. Los resultados entre las distintas condiciones las bandas fueron
analizados mediante el programa Fiji134
. Se llevaron a cabo tres experimentos
independientes.
2.3. Trypanosoma brucei
2.3.1. Cultivo celular
Se cultivó la forma sanguínea (bloodstream) long slender de T. brucei brucei (cepa 427)
a 37°C, 5% de CO2, en medio CMM (Creek’s minimal médium)135
suplementado con
10% SFB (Natocor), 100 UI/ml de penicilina y 100 µg/ml de estreptomicina. Para la
conservación y preservación del cultivo anterior, se cosecharon los parásitos en la mitad
de la fase exponencial de crecimiento (1x106 parásitos/ml), se les agregó 10% de
glicerol estéril y se los colocó en criotubos, los cuales fueron congelados lentamente
hasta -80°C. A las 24 h los criotubos fueron colocados en N2 líquido hasta el momento
de su utilización. Para su uso se descongelaron rápidamente en estufa de 37°C. Se los

Materiales y métodos
136
colocó en 5 ml de medio de cultivo CMM suplementado con 10% SFB y se centrifugó a
3000 rpm durante 3 minutos. El pellet se re-suspendió en 3 ml de medio de cultivo y se
lo trasvasó a una botella de cultivo celular.
2.3.2. Actividad anti-T. brucei
Las células se cosecharon en fase exponencial y se re-suspendieron en medio CMM
fresco, haciendo el recuento en el contador de células Coulter Counter (Beckman). Se
sembraron en placas de 96 pocillos 5x105 parásitos/pocillo. Se agregó la cantidad
indicada del compuesto estudiado a partir de una solución madre en DMSO. La
concentración final de DMSO en el medio de cultivo nunca superó el 0,8%. El control
se realizó en presencia de 0,8% de DMSO y en ausencia de cualquier compuesto y se
utilizó eflornitina y pentamidina como controles positivos. Se incubaron los parásitos a
37°C, 5% de CO2 durante 72 h. Luego de este tiempo se procedió a realizar el recuento
de los parásitos vivos mediante el contador de células. La concentración inhibitoria 50
(CI50) definida como la concentración de compuesto que inhibe en un 50% el
crecimiento con respecto al control sin tratar, se determinó a partir de curvas
concentración-respuesta usando GraphPad Prism. Se llevaron a cabo al menos tres
experimentos independientes por triplicado.
2.3.3. Lisado de T. brucei
A partir de un cultivo celular de T. brucei se realizó el recuento de parásitos mediante el
contador de células, se cosecharon mediante centrifugación a 3000 rpm durante 3 min y
se lavaron con PBS glucosa 3%. Se preparó luego el lisado de T. brucei mediante tres
ciclos consecutivos de congelado a -20°C y descongelado en hielo.
2.3.4. Zimografía
En un gel 10% SDS-PAGE con 0,15% de gelatina incorporada como sustrato se sembró
15 µL del lisado de T. brucei por calle (equivalente a 2x106 parásitos) y se corrió un
estándar comercial de sustancias con pesos moleculares conocidos (Prestained Broad
Range Protein Marker 2-220 kDa, Genbiotech). La corrida se realizó a 120 V durante 2
h a 4°C. Una vez finalizada se lavó el gel dos veces durante 15 min con 0,1% Tritón X-
100 y se incubó cada calle con el compuesto correspondiente durante 24 h a 37°C en

Materiales y métodos
137
buffer MES 0,1 M con agregado de DTT 0,5 mM. Se incluyeron los siguientes
controles: lisado de parásitos sin tratar, lisado de parásitos incubados con 5% de DMSO
y lisado de parásitos incubados con E-64. Luego de la incubación se procedió a teñir el
gel con Coomassie brilliant blue R-250. Se llevaron a cabo tres experimentos
independientes.
2.3.5. Captación intracelular de las N4-TSCs
Se trataron 5x106 T. brucei con el compuesto 8 ó 14 durante 24 h a una concentración
final de 5 µM y durante 4 h a una concentración final de 20 µM a 37°C y 5% de CO2.
Se utilizó como control parásitos sin tratar. Posteriormente se los lavó con PBS y se los
fijó con 2% de solución de paraformaldehído durante 15 min a temperatura ambiente.
Se los lavó dos veces con PBS y se los montó sobre un portaobjeto con glicerol. Las
muestras fueron visualizadas mediante microscopio confocal Leica TCS SP5 excitando
a 405 nm. Las imágenes fueron procesadas con el programa Fiji134
.
2.3.6. Determinación del tamaño celular
Se trataron 1x105 T. brucei con el compuesto 8 a una concentración final de 5 µM y 10
µM durante 24h a 37°C y 5% de CO2, luego se lavaron dos veces en PBS y se
detectaron en un equipo de citometría de flujo FACSCalibur (Becton-Dickinson, USA).
Se utilizó un control de células sin tratar y tratadas con el vehículo de DMSO. Un total
de 10 mil eventos fueron adquiridos en la región correspondiente a los parásitos. El
análisis y los histogramas del parámetro FSC fueron realizados utilizando el programa
FlowJo Versión X.0.7. Se llevaron a cabo tres experimentos independientes por
triplicado.
2.3.7. Determinación de especies reactivas del oxígeno
Se trataron 3x105 parásitos con el compuesto 8 a una concentración final de 10 µM
durante 2 y 4 h a 37°C y 5% de CO2. Se utilizó como control positivo 1 mM de H2O2
durante 10 min. Luego del tratamiento correspondiente se lavaron las células dos veces
con PBS. A continuación se agregó la sonda 2’,7’-diclorodihidrofluoresceina diacetato
(H2-DCF-DA) en una concentración final de 30 µM, se incubó 30 min a 37°C. Se
utilizaron como controles: i) células sin tratar con agregado de sonda; ii) células tratadas

Materiales y métodos
138
con el vehículo de DMSO y con sonda; iii) células sin tratar y sin agregado de sonda y;
iv) células tratadas y sin agregado de sonda. La fluorescencia fue medida en el
citómetro de flujo FACSCalibur (Becton-Dickinson, USA). Un total de 10 mil eventos
fueron adquiridos en la región correspondiente a los parásitos. Para el análisis y las
representaciones gráficas se usó el programa FlowJo Versión X.0.7. Se llevaron a cabo
tres experimentos independientes por triplicado.
2.3.8. Determinación del potencial de membrana mitocondrial
Se sembraron en una placa de 96 pocillos 4x105 parásitos/pocillo. Se trataron las células
con el compuesto 8 a una concentración final de 10 µM durante 2 h a 37°C y 5% de
CO2. Se utilizó como control positivo parásitos tratados por 1 h con 300 µM de
carbonilcianato m-clorofenil-hidrazona (cccp). Se lavaron los parásitos dos veces con
PBS, se agregó la sonda Mito Tracker® Red CMXRos y se incubó 30 min a 37°C. Se
utilizaron como controles: i) células sin tratar con agregado de sonda; ii) células tratadas
con el vehículo de DMSO y con sonda; iii) células sin tratar y sin agregado de sonda y
iv) células tratadas y sin agregado de sonda. La fluorescencia fue medida mediante
citometría de flujo. El equipo y el software utilizado fue el mismo que en las
determinaciones anteriores por citometría de flujo. Se llevaron a cabo tres experimentos
independientes por triplicado.
2.3.9. Ensayo de doble marcación con anexina V e yoduro de propidio
Se siguió el procedimiento descripto por el fabricante del kit I de FITC Annexin V
Apoptosis Detection (BD) con algunas modificaciones. Se sembraron en una placa de
96 pocillos 4x105 parásitos/pocillo. Se trataron las células con el compuesto 8 a una
concentración final de 10 µM durante 12, 24 y 48 h a 37°C y 5% de CO2. Se utilizó
como control positivo células sometidas a shock térmico de 60°C durante 3 min. Se
lavaron los parásitos con PBS, se agregó 5 µl de la sonda FITC anexina V y 6 µl de IP y
se dejó incubando 30 min. Se utilizaron como controles: i) células sin tratar con
agregado de sonda; ii) células tratadas con el vehículo de DMSO y con sonda; iii)
células sin tratar y sin agregado de sonda y; iv) células tratadas y sin agregado de sonda.
La fluorescencia fue medida mediante citometría de flujo. El equipo y el software
utilizado fue el mismo que en las anteriores determinaciones por citometría de flujo. Se
llevaron a cabo tres experimentos independientes por triplicado.

Materiales y métodos
139
2.3.10. Ensayo TUNEL
Se siguió el procedimiento descripto por el fabricante del kit In situ cell death detection
kit, fluorescein (Roche). Se sembraron en una placa de 96 pocillos 1x105
parásitos/pocillo. Se trataron las células con el compuesto 8 a una concentración final de
5 y 10 µM durante 24 h a 37°C y 5% de CO2. Se lavaron los parásitos con PBS, se
fijaron con 2% de paraformaldehído durante 20 min y se volvieron a lavar con PBS. Se
permeabilizaron con 0,1% de Tritón X-100 en 0,1% de citrato de sodio durante 2 min en
hielo y se lavaron con PBS. Se utilizó como control positivo parásitos permeabilizados
y tratados con 1 µl de ADNasa I (Promega) durante 10 min. Se agregó 5 µl de la enzima
deoxinucleotidil transferasa, 45 µl de dUTP-fluoresceína y se incubó durante 1 h a
37°C. Se utilizaron como controles: i) células sin tratar con agregado de enzima y dUTP
marcados; ii) células tratadas con el vehículo de DMSO con agregado de enzima y
dUTP marcados; iii) células tratadas con agregado de dUTP marcados y sin el agregado
de enzima. El equipo y el software utilizado fue el mismo que en las determinaciones
anteriores por citometría de flujo. Se llevaron a cabo tres experimentos independientes
por triplicado.
2.3.11. Microscopia electrónica de transmisión para ultraestructura
Se trataron 3x108
parásitos con 7,5 µM de concentración final del derivado 8 durante 24
h a 37°C y 5% de CO2. Luego de dicho tiempo se cosecharon y se lavaron dos veces
con PBS. En paralelo se trabajó con un control de 1x108 parásitos sin tratar. Las células
se fijaron con una solución de glutaraldehído al 2,5% en buffer fosfato 0,1 M pH 7.4
durante 4 h a 4 °C, se lavaron dos veces y se fijaron posteriormente con 1% de tetróxido
de osmio en el mismo buffer durante 60 min a 4 °C. Las células se lavaron con agua
doblemente destilada y el material se deshidrató gradualmente en concentraciones
crecientes de etanol. Las muestras fueron embebidas en resina epoxi “Durcupan”. Una
vez polimerizada la resina se realizaron cortes ultrafinos de 70-90 nm de espesor. Las
secciones se montaron en grillas de cobre y se contrastaron con acetato de uranilo y
citrato de plomo. Se observó en un microscopio electrónico de transmisión MET Zeiss
109 y se obtuvieron imágenes con la cámara Gatan W10000.

Materiales y métodos
140
2.4. Ensayo de Citotoxicidad
2.4.1. Ensayo de efecto citotóxico en células MDBK
Se sembraron en una placa de cultivo celular de 96 pocillos 1x104 células/pocillo.
Después de 24 h de incubación a 37°C, 5% de CO2, se agregaron diluciones de las N4-
TSCs en DMSO. Se utilizó como control células sin tratar. Las células proliferaron
durante 72 h a 37°C. Luego de dicho tiempo se determinó la viabilidad celular mediante
el método MTS/PES del kit Cell Titer 96 Aqueous One Solution Cell Proliferation Assay
(Promega) siguiendo el procedimiento del fabricante. Se incubó durante 2 h 30 min a
37°C y se leyeron las absorbancias a 490 nm. Se llevaron a cabo tres experimentos
independientes por triplicado.
2.4.2. Ensayo de efecto citotóxico en células Vero
Se sembraron en una placa de cultivo celular de 96 pocillos 1x104 células/pocillo en
medio de cultivo RPMI. Después de 24 h de incubación a 37°C, 5% de CO2, se
agregaron diluciones de las N4-TSCs en DMSO. La concentración final de DMSO en el
medio de cultivo nunca superó el 0,6 % y el control se realizó en presencia de 0,6 % de
DMSO y en ausencia de cualquier compuesto. Las células proliferaron durante 72 h a
37°C con 5% de CO2. Luego de dicho tiempo se lavaron con PBS y se les agregó 100 µl
de medio fresco y 15 µl de una solución madre de MTT (5mg/ml). Se incubó durante 1
h 30 min en oscuridad, 37°C y 5% de CO2. Se lavaron las células y se disolvió el
colorante con 100 µl de DMSO. Se leyeron las absorbancias a 578 nm con referencia de
630 nm. Se llevaron a cabo tres experimentos independientes por triplicado.
3. Estudios computacionales
3.1 Modelado molecular
Las N4-TSCs fueron modeladas con el programa HyperChem 8.0.7, pre-optimizadas con
el campo de fuerza MM+ y posteriormente con el método semi-empírico AM1,
utilizando el algoritmo Polak-Ribiere con criterio de convergencia inferior a 0.001
Kcal/(Å.mol) ó máximo de 9000 iteraciones. Las geometrías fueron finalmente

Materiales y métodos
141
optimizadas utilizando el programa Gaussian 09 mediante el método de HF con la base
6-31G(d).
3.2. Docking molecular
La estructura de rayos X de la enzima RdRp del VDVB se obtuvo de la base de datos
PDB (http://www.rcsb.org) con el código 1S48136
. Los residuos de selenometionina se
modificaron de nuevo a residuos de metionina, se añadieron hidrógenos polares y las
moléculas de agua fueron removidas. La enzima fue optimizada con el programa
NAMD 2.9137
utilizando el campo de fuerza CHARMM. El sitio de unión putativo para
los compuestos seleccionados en la RdRp se determinó utilizando el servidor
Metapocket 2.092
. Mediante dicho procedimiento se encontraron tres sitios de unión
putativos en la superficie de la enzima; sin embargo, sólo uno se encontró en la región
cercana a los aminoácidos 264 y 392 de la RdRp mutada utilizada en el ensayo de
resistencia cruzada. Por lo tanto, este sitio fue elegido para llevar a cabo el
procedimiento de docking molecular. Se utilizó el software AutodockTools 1.5.6 para
obtener los archivos inputs de la macromolécula y de los ligandos con el formato pdbqt.
Los cálculos fueron llevados a cabo mediante el programa Autodock 4.2138
. Para los
cálculos se definió un espacio de 60 x 60 x 60 puntos en el plano xyz y una distancia de
0,375 Å y centrado en el sitio elegido. Los ligandos fueron completamente flexibles
durante el procedimiento, mientras que la proteína se mantuvo rígida. El docking se
realizó con 1000 corridas utilizando el algoritmo genético lamarckiano. La selección
final del modo de unión se realizó teniendo en cuenta la energía de unión libre estimada
y el tamaño del cluster. Las interacciones de unión fueron visualizadas en el programa
Chimera 1.8139
.
3.3. Dinámica molecular y MM/PBSA
Las mejores conformaciones del docking molecular fueron utilizadas como estructuras
de partida para el estudio de dinámica molecular. Las simulaciones se realizaron con el
programa NAMD 2.9 utilizando el campo de fuerza Amber. Cada complejo fue
solvatado en un caja de agua explícita descripta con el potencial TIP3P con la distancia
mínima de 10 Å desde la superficie de la proteína hasta el borde de la caja. Se agregaron
iones cloruro para neutralizar el sistema. La primera minimización de energía del

Materiales y métodos
142
sistema se realizó con la proteína restringida conformacionalmente, seguida de una
segunda minimización sin restricciones. Luego, la temperatura se incrementó
gradualmente hasta 300 K durante 300 ps usando un ensamble canónico (NVT) y se
equilibró durante un período de 3 ns a 1 atm y una temperatura de 300 K. Las
producciones se llevaron a cabo durante 6 ns en condiciones de número de partículas,
presión y temperatura constantes (NPT) y con un paso de tiempo de integración de 2 fs.
La malla de partículas Ewald se utilizó para tratar interacciones electrostáticas de largo
alcance; el algoritmo SHAKE se aplicó a todos los átomos de hidrógeno y la distancia
límite de van der Waals se estableció en 10 Å. Las trayectorias se analizaron utilizando
el programa VMD. Las energías libres de unión (ΔGunión) se estimaron mediante el
método MM/PBSA a partir de 1000 frames obtenidos en la producción. Para el cálculo
de entropía se llevó a cabo el método de modos normales en la zona de la interacción
proteína-ligando. La formación de puente de hidrógeno fue considerada para una
distancia menor a 2,5 Å y un ángulo mayor a 120°.
3.4. Cálculo de descriptores
A cada una de las N4-TSCs se le calcularon distintos descriptores moleculares (0D, 1D,
2D y 3D), utilizando el programa PaDEL Descriptor 2.15140
. El cálculo de las
propiedades electrónicas (HOMO, LUMO, momento dipolar, potencial electrostático y
distintos esquemas de carga: Mulliken, RESP, al igual que los mapas de potencial
electrostático) se realizó con el programa Gaussian 09 mediante el método de HF con la
base 6-31G(d).
Las imágenes de los mapas de potencial electrostático fueron obtenidas con el programa
GaussView 4.1, representados en una escala de colores que va del rojo (densidad de
carga negativa) hasta el azul (densidad de carga positiva).
3.5. QSAR
Los primeros modelos de QSAR se llevaron a cabo con el programa McQSAR 1.2141
a
partir de los conformeros generados con el programa Balloon142
. Se tomaron en cuenta
descriptores moleculares calculados con el programa PaDEL Descriptor y los datos de
actividad biológica sobre T. brucei expresados como pCI50. El algoritmo genético

Materiales y métodos
143
empleado en el programa McQSAR fue configurado con el valor de 0,5 como máxima
colinealidad entre descriptores y con 3 descriptores como máximo para cada ecuación.
La segunda generación de modelos de QSAR se calculó a partir de las propiedades
electrónicas determinadas mediante Gaussian 09. Las ecuaciones matemáticas y el
análisis estadístico de las mismas se realizaron con el módulo de análisis de datos
incluido en el programa Microsoft Excel 2007.
4. Análisis estadístico
Las pruebas estadísticas se realizaron utilizando el programa GraphPad Prism versión
6.00 ó el módulo de análisis de datos incluido en el programa Microsoft Excel 2007. La
comparación entre las medias de dos grupos se realizó mediante la prueba t-Student
(para muestras con varianzas iguales). Para las comparaciones entre múltiples grupos se
utilizó el análisis de varianza (ANOVA), seguido de los post tests de Bonferrony o
Dunnett. Los p-valor < 0,05 (*) y p-valor < 0,01 (**) fueron considerados
estadísticamente significativos. Los modelos de QSAR se realizaron a partir de
regresiones lineales múltiples y se tomaron en cuenta para las ecuaciones el coeficiente
de determinación, el estadístico de Fisher, el desvío estándar y el test de Student para
cada variable del modelo.

144

Capítulo V.
CONCLUSIONES Y PERSPECTIVAS


Conclusiones y perspectivas
147
Capítulo V. CONCLUSIONES Y PERSPECTIVAS
Se han sintetizado y caracterizado estructuralmente 30 tiosemicarbazonas de 1-
indanonas N4-aril sustituidas con distintos sustituyentes en el anillo indánico así como
en la posición para del arilo unido al N4. La variedad de los sustituyentes en cuanto a las
distintas calidades electrónicas y/o estéricas permitieron, luego de los ensayos
biológicos, el análisis de la relación estructura-actividad.
Se compararon los rendimientos y tiempos de reacción de la síntesis de las N4-TSCs y
se llegó a la conclusión que la metodología one-pot que utiliza la radiación de
microondas es más ventajosa en tiempos y rendimientos de reacción que la metodología
convencional.
Los compuestos fueron evaluados como antivirales frente al virus de la diarrea viral
bovina. Se concluyó en el estudio de relación estructura-actividad que sustituyentes
atractores de electrones en el arilo unido al N4 son importantes para la actividad
antiviral, mientras que dadores de electrones en dicha posición no se relacionan con
buenos valores de actividad. Asimismo se concluyó que los compuestos con la
sustitución 5,6-dimetoxilo o 5-metilo son activos frente al VDVB, mientras que la
actividad se pierde con las N4-TSCs sin sustituir en el indano o con la sustitución 4,5-
dimetoxilo. De esta manera se concluyó que el tipo y patrón de sustituciones en el
indano juegan un rol determinante para definir N4-TSCs activas/inactivas frente al
VDVB.
Por otro lado, a partir del ensayo de resistencia cruzada, el cual sugiere que el
compuesto 8 actuaría inhibiendo la ARN polimerasa viral dependiente de ARN, se
llevaron a cabo estudios computacionales. En los estudios de docking molecular con la
enzima wild type se evidenció para el compuesto estudiado interacciones de puente de
hidrógeno, π-π stacking y no polar entre el grupo 5,6-dimetoxi, el indano, y la función
tiosemicarbazona con la enzima. En el modelo de la RdRp mutada se perdieron
interacciones de puente de hidrógeno y de π-π stacking. De esta forma, en concordancia
con el ensayo de resistencia cruzada, se concluyó la importancia de los aminoácidos
mutados en la interacción dada entre la molécula y el sitio de la enzima donde se llevó a
cabo la simulación. Por otro lado, en las dinámicas moleculares realizadas se evidenció
que la componente electrostática y de van der Waals son las de mayor incidencia en la

Conclusiones y perspectivas
148
energía del complejo compuesto-enzima. Asimismo, se encontró que la formación de
puentes de hidrógeno resultaría un factor determinante para la interacción del
compuesto con la enzima y por ende para su actividad. Estos datos, juntos con los de
resistencia cruzada, dan sustento a la hipótesis de establecer esta enzima y este sitio
como potencial blanco de la N4-TSC estudiada.
Por otro lado, en la evaluación de los compuestos frente a la forma epimastigote,
tripomastigote y amastigote de T. cruzi se identificaron derivados activos frente a las
tres formas. Asimismo, se evidenció que su mecanismo de acción no resulta ser la
inhibición de la cruzipaína. De esta forma surge como perspectiva explorar otros
posibles blancos de acción para los compuestos. En los análisis de relación estructura-
actividad para la forma epimastigote se concluyó que el H, F y Cl en la posición R4 y la
sustitución 5,6-dimetoxilo en el indano potencian la actividad. En cuanto a la forma
tripomastigote, no se estableció una relación directa entre las propiedades estructurales
de los compuestos y su actividad.
En los estudios frente a T. brucei seis N4-TSCs presentaron una CI50 por debajo de 10
µM, siendo el compuesto 8 el más activo de la serie. El análisis de estructura-actividad
en T. brucei permitió concluir que la sustitución con nitro en el arilo unido al N4
potencia la actividad. Por otro lado se concluyó que los derivados sin sustituir en el
indano y los que poseen la sustitución 5,6-dimetoxilo se vinculan con los mejores
valores de actividad. En los estudios de QSAR se pudo observar que la actividad está
condicionada a descriptores cuánticos, la carga parcial sobre el átomo de N2
y la
diferencia de cargas entre el N4 y el hidrógeno unido a este. Además, a partir del modelo
de QSAR obtenido se concluyó que la conformación relacionada con la actividad
correspondería a la “Syn” ya que sólo para dicha conformación se encontró correlación
con la actividad.
Por otro lado, en el estudio del compuesto más activo frente a las proteasas presentes en
el lisado de parásitos se observó que éstas no resultan ser su blanco. Se determinaron los
eventos que contribuyen a la muerte del parásito mediante estudios de citometría de
flujo y microscopía. Los parásitos tratados con la N4-TSC más activa evidenciaron la
disminución del tamaño celular, generación de especies reactivas del oxígeno,
despolarización de la membrana mitocondrial, exposición de PS en la superficie externa
de la membrana plasmática, degradación del ADN y alteraciones ultraestructurales.

Conclusiones y perspectivas
149
Todas ellas indicaron que el compuesto produce muerte celular regulada con fenotipo
apoptótico.
Respecto a los ensayos en células MDBK y Vero ninguna N4-TSC presentó
citotoxicidad, lo cual resulta una característica relevante en la búsqueda de fármacos y
apoya la continuación de los estudios.
En conclusión, los modelos teóricos indirectos permitieron conocer las características
estructurales que son determinantes en la actividad anti-T. brucei. Los modelos teóricos
directos permitieron conocer y entender el mecanismo de acción del compuesto más
activo frente al VDVB. El compuesto 8 surge como una molécula líder que permitirá su
optimización para un futuro uso como antiviral y antiparasitario. La relación entre el
patrón estructural y las características biológicas estudiadas en el presente trabajo
proporcionan valiosas bases para continuar con el diseño de nuevas moléculas
antivirales y antiparasitarias.

150

Capítulo V.
REFERENCIAS


Referencias
153
REFERENCIAS
1. Dickson, M., Gagnon, J. P. Key factors in the rising cost of new drug discovery
and development. Nat. Rev. Drug Discov., 2004, 3(5), 417-429.
2. Saldívar-González, F., Prieto-Martínez, F. D., Medina-Franco, J. L.
Descubrimiento y desarrollo de fármacos: un enfoque computacional. Educ.
Quim., 2017, 28(1), 51-58.
3. Lombardino, J. G., Lowe III, J. A. The role of the medicinal chemist in drug
discovery-then and now. Nat. Rev. Drug Discov., 2004, 3(10), 853.
4. http://www.anmat.gov.ar/farmaco/farmacovigilancia.asp.
5. Patani, G. A., LaVoie, E. J. Bioisosterism: A rational approach in drug design.
Chem. Rev., 1996, 96(8), 3147-3176.
6. Baig, M. H., Ahmad, K., Roy, S., Ashraf, J. M., Adil, M., Siddiqui, M. H., Khan,
S., Kamal, M. A., Choi, I. P. Computer aided drug design: success and
limitations. Curr. Pharm. Des., 2016, 22(5), 572-581.
7. Domagk, G., Behnish, R., Mietzsch, F., Schmidt, H. New compound active
against tuberculosis bacilli in vitro. Naturwissenschaften, 1946, 33, 315.
8. Finch, R. A., Liu, M.-C., Cory, A. H., Cory, J. G., Sartorelli, A. C. Triapine (3-
aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of
ribonucleotide reductase with antineoplastic activity. Adv. Enzyme Regul., 1999,
39, 3-12.
9. Mishra, V., Pandeya, S. N., Pannecouque, C., Witvrouw, M., De Clercq, E. Anti-
HIV activity of thiosemicarbazone and semicarbazone derivatives of (±)-3-
menthone. Arch. Pharm. (Weinheim), 2002, 335(5), 183-186.
10. Varma, R. S., Nobles, W. L. Synthesis and antiviral and antibacterial activity of
certain N-dialkylaminomethylisatin β-thiosemicarbazones. J. Med. Chem., 1967,
10(5), 972-974.
11. Du, X., Guo, C., Hansell, E., Doyle, P. S., Caffrey, C. R., Holler, T. P.,
McKerrow, J. H., Cohen, F. E. Synthesis and structure-activity relationship study
of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine
protease cruzain. J. Med. Chem., 2002, 45(13), 2695-2707.
12. Greenbaum, D. C., Mackey, Z., Hansell, E., Doyle, P., Gut, J., Caffrey, C. R.,
Lehrman, J., Rosenthal, P. J., McKerrow, J. H., Chibale, K. Synthesis and
structure−activity relationships of parasiticidal thiosemicarbazone cysteine
protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and
Trypanosoma cruzi. J. Med. Chem., 2004, 47(12), 3212-3219.
13. Hamre, D., Brownlee, K. A., Donovick, R. Studies on the chemotherapy of
vaccinia virus. J. Immunol., 1951, 67(4), 305-312.
14. Bauer, D. J. The antiviral and synergic actions of isatin thiosemicarbazone and
certain phenoxypyrimidines in vaccinia infection in mice. Br. J. Exp. Pathol.,

Referencias
154
1955, 36(1), 105-114.
15. Sidwell, R. W., Arnett, G., Dixon, G. J., Schabel, F. M. Purine analogs as
potential anticytomegalovirus agents. Proc. Soc. Exp. Biol. Med., 1969, 131(4),
1223-1230.
16. Bal, T. R., Anand, B., Yogeeswari, P., Sriram, D. Synthesis and evaluation of
anti-HIV activity of isatin β-thiosemicarbazone derivatives. Bioorg. Med. Chem.
Lett., 2005, 15(20), 4451-4455.
17. Shipman, C., Smith, S. H., Drach, J. C., Klayman, D. L. Antiviral activity of 2-
acetylpyridine thiosemicarbazones against herpes simplex virus. Antimicrob.
Agents Chemother., 1981, 19(4), 682-685.
18. Brockman, R. W., Thomson, J. R., Bell, M. J., Skipper, H. E. Observations on the
antileukemic activity of pyridine-2-carboxaldehyde thiosemicarbazone and
thiocarbohydrazone. Cancer Res., 1956, 16(2), 167-170.
19. https://clinicaltrials.gov/ct2/results?cond=&term=triapine.
20. Hinshaw, H. C., McDermott, W. Thiosemicarbazone therapy of tuberculosis in
humans. Am. Rev. Tuberc. Pulm. Dis., 1950, 61(1), 145-157.
21. As, Z., Kaplanc, J., Dilek, M., Alt, J., Sever, B., Cantürk, Z., Özdemir, A.
Synthesis and in vitro evaluation of new thiosemicarbazone derivatives as
potential antimicrobial agents. J. Chem., 2016, 2016, 1-7.
22. Khan, S. A., Yusuf, M. Synthesis and biological evaluation of some
thiazolidinone derivatives of steroid as antibacterial agents. Eur. J. Med. Chem.,
2009, 44(6), 2597-2600.
23. Gingras, B. A., Colin, G., Bayley, C. H. Antifungal activity of
thiosemicarbazones. J. Pharm. Sci., 2018, 54(11), 1674-1675.
24. Duschak, V. G., Couto, A. S. Cruzipain, the major cysteine protease of
Trypanosoma cruzi: a sulfated glycoprotein antigen as relevant candidate for
vaccine development and drug target. A review. Curr. Med. Chem., 2009, 16(24),
3174-3202.
25. Ndao, M., Beaulieu, C., Black, W. C., Isabel, E., Vasquez-Camargo, F., Nath-
Chowdhury, M., Massé, F., Mellon, C., Methot, N., Nicoll-Griffith, D. A.
Reversible cysteine protease inhibitors show promise for a Chagas disease cure.
Antimicrob. Agents Chemother., 2014, 58(2), 1167-1178.
26. Finkielsztein, L. M., Castro, E. F., Fabián, L. E., Moltrasio, G. Y., Campos, R.
H., Cavallaro, L. V., Moglioni, A. G. New 1-indanone thiosemicarbazone
derivatives active against BVDV. Eur. J. Med. Chem., 2008, 43(8), 1767-1773.
27. Castro, E. F., Fabián, L. E., Caputto, M. E., Gagey, D., Finkielsztein, L. M.,
Moltrasio, G. Y., Moglioni, A. G., Campos, R. H., Cavallaro, L. V. Bovine viral
diarrhea virus RNA synthesis inhibition by the Thiosemicarbazone derived from
5, 6-dimethoxy-1-indanone. J. Virol., 2011, 85(11), 5436-5445.
28. Caputto, M. E., Fabian, L. E., Benítez, D., Merlino, A., Ríos, N., Cerecetto, H.,

Referencias
155
Moltrasio, G. Y., Moglioni, A. G., González, M., Finkielsztein, L. M.
Thiosemicarbazones derived from 1-indanones as new anti-Trypanosoma cruzi
agents. Bioorg. Med. Chem., 2011, 19(22), 6818-6826.
29. Paton, D. J., Christiansen, K. H., Alenius, S., Cranwell, M. P., Pritchard, G. C.,
Drew, T. W. Prevalence of antibodies to bovine virus diarrhoea virus and other
viruses in bulk tank milk in England and Wales. Vet. Rec., 1998, 142(15), 385-
391.
30. Houe, H. Epidemiological features and economical importance of bovine virus
diarrhoea virus (BVDV) infections. Vet. Microbiol., 1999, 64(2-3), 89-107.
31. Nelson, D. D., Duprau, J. L., Wolff, P. L., Evermann, J. F. Persistent bovine viral
diarrhea virus infection in domestic and wild small ruminants and camelids
including the mountain goat (Oreamnos americanus). Front. Microbiol., 2016, 6,
1415.
32. Lanyon, S. R., Hill, F. I., Reichel, M. P., Brownlie, J. Bovine viral diarrhoea:
pathogenesis and diagnosis. Vet. J., 2014, 199(2), 201-209.
33. Lindberg, A., Brownlie, J., Gunn, G. J., Houe, H., Moennig, V., Saatkamp, H.
W., Sandvik, T., Valle, P. S. The control of bovine viral diarrhoea virus in
Europe: today and in the future. Rev. Sci. Tech., 2006, 25(3), 961-979.
34. Khodakaram-Tafti, A., Farjanikish, G. H. Persistent bovine viral diarrhea virus
(BVDV) infection in cattle herds. Iran. J. Vet. Res., 2017, 18(3), 154-163.
35. Ståhl, K., Alenius, S. BVDV control and eradication in Europe -an update. Jpn. J.
Vet. Res., 2012, 60, 31-39.
36. Pacheco, J. M., Lager, I. Indirect method ELISA for the detection of antibodies
against bovine diarrhea virus in bovine serum. Rev. Argent. Microbiol., 2003,
35(1), 19-23.
37. Altamiranda, E. A. G., Kaiser, G. G., Weber, N., Leunda, M. R., Pecora, A.,
Malacari, D. A., Morán, O., Campero, C. M., Odeón, A. C. Clinical and
reproductive consequences of using BVDV-contaminated semen in artificial
insemination in a beef herd in Argentina. Anim. Reprod. Sci., 2012, 133(3-4),
146-152.
38. Fernandez Sainz, I. Seroprevalencia de la diarrea viral bovina, herpesvirus
bovino y virus sincicial respiratorio en Argentina. Vet. Arg., 2001, 82(4), 216-
220.
39. Streitenberger, N., Ferella, A., Aguirreburualde, P., Sammarruco, A., Dus Santos,
M. J., Mozgovoj, M. V, Maidana, S., Romera, S. A., Pecora, A., Quiroga, M. A.
Complejo respiratorio bovino: evidencia de circulación viral múltiple en un
establecimiento de cría. Rev. Investig. Agropecu., 2017, 43(2), 149-155.
40. Collett, M. S., Larson, R., Belzer, S. K., Retzel, E. Proteins encoded by bovine
viral diarrhea virus: The genomic organization of a pestivirus. Virology, 1988,
165(1), 200-208.
41. Collett, M. S., Wiskerchen, M. A., Welniak, E., Belzer, S. K. Bovine viral

Referencias
156
diarrhea virus genomic organization. Arch. Virol. Suppl., 1991, 3, 19-27.
42. Donis, R. O., Dubovi, E. J. Differences in virus-induced polypeptides in cells
infected by cytopathic and noncytopathic biotypes of bovine virus diarrhea-
mucosal disease virus. Virology, 1987, 158(1), 168-173.
43. Bolin, S. R., Grooms, D. L. Origination and consequences of bovine viral
diarrhea virus diversity. Vet. Clin. Food Anim. Pract., 2004, 20(1), 51-68.
44. Moon, S. L., Blackinton, J. G., Anderson, J. R., Dozier, M. K., Dodd, B. J. T.,
Keene, J. D., Wilusz, C. J., Bradrick, S. S., Wilusz, J. XRN1 Stalling in the 5’
UTR of hepatitis C virus and bovine viral diarrhea virus is associated with
dysregulated host mRNA stability. PLOS Pathog., 2015, 11(3), e1004708.
45. Buckwold, V. E., Beer, B. E., Donis, R. O. Bovine viral diarrhea virus as a
surrogate model of hepatitis C virus for the evaluation of antiviral agents.
Antiviral Res., 2003, 60(1), 1-15.
46. Gard, J. A., Givens, M. D., Stringfellow, D. A. Bovine viral diarrhea virus
(BVDV): Epidemiologic concerns relative to semen and embryos.
Theriogenology, 2007, 68(3), 434-442.
47. Lecot, S., Belouzard, S., Dubuisson, J., Rouillé, Y. Bovine viral diarrhea virus
entry is dependent on clathrin-mediated endocytosis. J. Virol., 2005, 79(16),
10826-10829.
48. Zhang, G., Flick-Smith, H., McCauley, J. W. Differences in membrane
association and sub-cellular distribution between NS2-3 and NS3 of bovine viral
diarrhoea virus. Virus Res., 2003, 97(2), 89-102.
49. Ohmann, H. B., Bloch, B. Electron microscopic studies of bovine viral diarrhea
virus in tissues of diseased calves and in cell cultures. Arch. Virol., 1982, 71(1),
57-74.
50. Grummer, B., Beer, M., Liebler-Tenorio, E., Greiser-Wilke, I. Localization of
viral proteins in cells infected with bovine viral diarrhoea virus. J. Gen. Virol.,
2001, 82(11), 2597-2605.
51. Finkielsztein, L. M., Moltrasio, G. Y., Caputto, M. E., Castro, E. F., Moglioni, L.
V. C. and A. G. What is known about the antiviral agents active against bovine
viral diarrhea virus (BVDV)? Curr. Med. Chem., 2010, 17(26), 2933-2955.
52. Zhang, N., Liu, Z., Han, Q., Qiu, J., Chen, J., Zhang, G., Li, Z., Lou, S., Li, N.
Development of one-step SYBR Green real-time RT-PCR for quantifying bovine
viral diarrhea virus type-1 and its comparison with conventional RT-PCR. Virol.
J., 2011, 8, 374.
53. van Iddekinge, B. J. L. H., van Wamel, J. L. B., van Gennip, H. G. P.,
Moormann, R. J. M. Application of the polymerase chain reaction to the
detection of bovine viral diarrhea virus infections in cattle. Vet. Microbiol., 1992,
30(1), 21-34.
54. Thür, B., Zlinszky, K., Ehrensperger, F. Immunohistochemical detection of
bovine viral diarrhea virus in skin biopsies: a reliable and fast diagnostic tool. J.

Referencias
157
Vet. Med. Ser. B., 2018, 43(3), 163-166.
55. Newcomer, B. W., Chamorro, M. F., Walz, P. H. Vaccination of cattle against
bovine viral diarrhea virus. Vet. Microbiol., 2017, 206, 78-83.
56. Oirschot, J. T., Bruschke, C. J., Rijn, P. A. Vaccination of cattle against bovine
viral diarrhoea. Vet. Microbiol., 1999, 64(2-3), 169-183.
57. Bolin, S. R. Control of bovine viral diarrhea infection by use of vaccination. Vet.
Clin. Food Anim. Pract., 1995, 11(3), 615-625.
58. Pinior, B., Firth, C. L., Richter, V., Lebl, K., Trauffler, M., Dzieciol, M., Hutter,
S. E., Burgstaller, J., Obritzhauser, W., Winter, P., Käsbohrer, A. A systematic
review of financial and economic assessments of bovine viral diarrhea virus
(BVDV) prevention and mitigation activities worldwide. Prev. Vet. Med., 2017,
137, 77-92.
59. Pereira, P. C. M., Navarro, E. C. Challenges and perspectives of Chagas disease:
A review. J. Venom. Anim. Toxins Incl. Trop. Dis., 2013, 19(1), 1-8.
60. Pinheiro, E., Brum-Soares, L., Reis, R., Cubides, J. C. Chagas disease: Review of
needs, neglect, and obstacles to treatment access in Latin America. Rev. Soc.
Bras. Med. Trop., 2017, 50(3), 296-300.
61. http://www.who.int/chagas/epidemiology/en/.
62. Pinto Dias, J. C. Human chagas disease and migration in the context of
globalization: some particular aspects. J. Trop. Med., 2013, 2013, 789758.
63. https://www.dndi.org/diseases-projects/chagas/.
64. http://www.who.int/es/news-room/fact-sheets/detail/chagas-disease-(american-
trypanosomiasis).
65. Coura, J. R., De Castro, S. L. A critical review on Chagas disease chemotherapy.
Mem. Inst. Oswaldo Cruz, 2002, 97(1), 3-24.
66. http://www.who.int/es/news-room/fact-sheets/detail/trypanosomiasis-human-
african-(sleeping-sickness).
67. https://www.cdc.gov/parasites/sleepingsickness/biology.html.
68. Matthews, K. R. The developmental cell biology of Trypanosoma brucei. J. Cell
Sci., 2005, 118(2), 283-290.
69. Brun, R., Blum, J., Chappuis, F., Burri, C. Human African trypanosomiasis.
Lancet, 2010, 375(9709), 148-159.
70. Njiokou, F., Laveissière, C., Simo, G., Nkinin, S., Grébaut, P., Cuny, G., Herder,
S. Wild fauna as a probable animal reservoir for Trypanosoma brucei gambiense
in Cameroon. Infect. Genet. Evol., 2006, 6(2), 147-153.
71. Barrett, M. P., Boykin, D. W., Brun, R., Tidwell, R. R. Human African
trypanosomiasis: pharmacological re-engagement with a neglected disease. Br. J.
Pharmacol., 2009, 152(8), 1155-1171.

Referencias
158
72. Babokhov, P., Sanyaolu, A. O., Oyibo, W. A., Fagbenro-Beyioku, A. F.,
Iriemenam, N. C. A current analysis of chemotherapy strategies for the treatment
of human African trypanosomiasis. Pathog. Glob. Health, 2013, 107(5), 242-252.
73. Denise, H., Barrett, M. P. Uptake and mode of action of drugs used against
sleeping sickness. Biochem. Pharmacol., 2001, 61(1), 1-5.
74. Sanderson, L., Dogruel, M., Rodgers, J., De Koning, H. P., Thomas, S. A.
Pentamidine movement across the murine blood-brain and blood-cerebrospinal
fluid barriers: effect of trypanosome infection, combination therapy, p-
glycoprotein, and multidrug resistance-associated protein. J. Pharmacol. Exp.
Ther., 2009, 329(3), 967-977.
75. Wilson, W. D., Nguyen, B., Tanious, F. A., Mathis, A., Hall, J. E., Boykin, C. E.
S. and D. W. Dications that target the DNA minor groove: compound design and
preparation, DNA interactions, cellular distribution and biological activity. Curr.
Med. Chem. Anticancer Agents, 2005, 5(4), 389-408.
76. Millan, C. R., Acosta-Reyes, F. J., Lagartera, L., Ebiloma, G. U., Lemgruber, L.,
Martínez, J. J. N., Saperas, N., Dardonville, C., De Koning, H. P., Campos, J. L.
Functional and structural analysis of AT-specific minor groove binders that
disrupt DNA-protein interactions and cause disintegration of the Trypanosoma
brucei kinetoplast. Nucleic Acids Res., 2017, 45(14), 8378-8391.
77. Gillingwater, K., Kumar, A., Ismail, M. A., Arafa, R. K., Stephens, C. E.,
Boykin, D. W., Tidwell, R. R., Brun, R. In vitro activity and preliminary toxicity
of various diamidine compounds against Trypanosoma evansi. Vet. Parasitol.,
2010, 169(3-4), 264-272.
78. Legros, D., Ollivier, G., Gastellu-Etchegorry, M., Paquet, C., Burri, C., Jannin, J.,
Büscher, P. Treatment of human African trypanosomiasis-present situation and
needs for research and development. Lancet Infect. Dis., 2002, 2(7), 437-440.
79. Kennedy, P. G. Clinical features, diagnosis, and treatment of human African
trypanosomiasis (sleeping sickness). Lancet Neurol., 2013, 12(2), 186-194.
80. Bacchi, C. J. Chemotherapy of human african trypanosomiasis. Interdiscip.
Perspect. Infect. Dis., 2009, 2009, 1-5.
81. De Koning, H. P. Uptake of pentamidine in Trypanosoma brucei brucei is
mediated by three distinct transporters: implications for cross-resistance with
arsenicals. Mol. Pharmacol., 2001, 59(3), 586-592.
82. Fairlamb, A. H. Chemotherapy of human African trypanosomiasis: current and
future prospects. Trends Parasitol., 2003, 19(11), 488-494.
83. Pepin, J. The treatment of human African trypanosomiasis. Adv Parasitol, 1994,
33, 1-47.
84. Delespaux, V., de Koning, H. P. Drugs and drug resistance in African
trypanosomiasis. Drug Resist. Updat., 2007, 10(1-2), 30-50.
85. Pépin, J., Milord, F., Khonde, A., Niyonsenga, T., Loko, L., Mpia, B. Gambiense
trypanosomiasis: frequency of, and risk factors for, failure of melarsoprol

Referencias
159
therapy. Trans. R. Soc. Trop. Med. Hyg., 1994, 88(4), 447-452.
86. Lutje, V., Seixas, J., Kennedy, A. Chemotherapy for second-stage Human
African trypanosomiasis. Cochrane Database Syst. Rev., 2013, 2013, 6201.
87. Choi, K. H., Gallei, A., Becher, P., Rossmann, M. G. The structure of bovine
viral diarrhea virus RNA-dependent RNA polymerase and its amino-terminal
domain. Structure, 2006, 14(7), 1107-1113.
88. Butcher, S. J., Grimes, J. M., Makeyev, E. V, Bamford, D. H., Stuart, D. I. A
mechanism for initiating RNA-dependent RNA polymerization. Nature, 2001,
410(6825), 235-240.
89. Ferrer-Orta, C., Arias, A., Perez-Luque, R., Escarmís, C., Domingo, E.,
Verdaguer, N. Structure of foot-and-mouth disease virus RNA-dependent RNA
polymerase and its complex with a template-primer RNA. J. Biol. Chem., 2004,
279(45), 47212-47221.
90. Warren, G. L., Andrews, C. W., Capelli, A.-M., Clarke, B., LaLonde, J.,
Lambert, M. H., Lindvall, M., Nevins, N., Semus, S. F., Senger, S., Tedesco, G.,
Wall, I. D., Woolven, J. M., Peishoff, C. E., Head, M. S. A critical assessment of
docking programs and scoring functions. J. Med. Chem., 2006, 49(20), 5912-
5931.
91. Enyedy, I. J., Egan, W. J. Can we use docking and scoring for hit-to-lead
optimization? J. Comput. Aided. Mol. Des., 2008, 22(3-4), 161-168.
92. Zhang, Z., Li, Y., Lin, B., Schroeder, M., Huang, B. Identification of cavities on
protein surface using multiple computational approaches for drug binding site
prediction. Bioinformatics, 2011, 27(15), 2083-2088.
93. Cazzulo, J. Proteinases of Trypanosoma cruzi: potential targets for the
chemotherapy of Chagas disease. Curr. Top. Med. Chem., 2002, 2(11), 1261-
1271.
94. Cazzulo, J. J., Stoka, V., Turk, V. Cruzipain, the major cysteine proteinase from
the protozoan parasite Trypanosoma cruzi. Biol. Chem., 1997, 378(1), 1-10.
95. Caffrey, C. R., Steverding, S. S., D. Cysteine proteinases of trypanosome
parasites novel targets for chemotherapy. Curr. Drug Targets, 2000, 1(2), 155-
162.
96. Barrett, A. J., Kembhavi, A. A., Brown, M. A., Kirschke, H., Knight, C. G.,
Tamai, M., Hanada, K. L- trans -Epoxysuccinyl-leucylamido(4-guanidino)butane
(E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins
B, H and L. Biochem. J., 1982, 201(1), 189-198.
97. Cazzulo, J. J., Cazzulo Franke, M. C., Martínez, J., Franke de Cazzulo, B. M.
Some kinetic properties of a cysteine proteinase (cruzipain) from Trypanosoma
cruzi. Biochim. Biophys. Acta, 1990, 1037(2), 186-191.
98. Labriola, C., Sousa, M., Cazzulo, J. J. Purification of the major cysteine
proteinase (cruzipain) from Trypanosoma cruzi by affinity chromatography. Biol.
Res.,1993, 26(1-2), 101-107.

Referencias
160
99. Todeschini, R., Consonni, V., Handbook of molecular descriptors. Wiley-VCH:
2000.
100. Krasowska, M., Kochel, A., Filarowski, A. The conformational analysis of 2-
hydroxyaryl Schiff thiosemicarbazones. Cryst. Eng. Comm., 2010, 12(6), 1955-
1962.
101. Marković, V., Joksović, M. D., Marković, S., Jakovljević, I. Influence of
anthraquinone scaffold on E/Z isomer distribution of two thiosemicarbazone
derivatives. 2D NMR and DFT studies. J. Mol. Struct., 2014, 1058, 291-297.
102. Soares, M. A., Lessa, J. A., Mendes, I. C., Da Silva, J. G., Dos Santos, R. G.,
Salum, L. B., Daghestani, H., Andricopulo, A. D., Day, B. W., Vogt, A.,
Pesquero, J. L., Rocha, W. R., Beraldo, H. N4-Phenyl-substituted 2-
acetylpyridine thiosemicarbazones: Cytotoxicity against human tumor cells,
structure-activity relationship studies and investigation on the mechanism of
action. Bioorg. Med. Chem., 2012, 20(11), 3396-3409.
103. Nuwan De Silva, N. W. S. V., Albu, T. V. A theoretical investigation on the
isomerism and the NMR properties of thiosemicarbazones. Cent. Eur. J. Chem. 5,
396-419 (2007).
104. Hall, L. H. & Kier, L. B. Electrotopological state indices for atom types: a novel
combination of electronic, topological, and valence state information. J. Chem.
Inf. Comput. Sci., 1995, 35(6), 1039-1045.
105. Veerasamy, R., Rajak, H., Jain, A., Sivadasan, S., van Iddekinge, B. J. L. H.,
Agrawal, and R. K. Validation of QSAR models - strategies and importance. Int.
J. Drug Des. Discov., 2011, 2, 511-519.
106. Steverding, D., Sexton, D. W., Wang, X., Gehrke, S. S., Wagner, G. K., Caffrey,
C. R. Trypanosoma brucei: Chemical evidence that cathepsin L is essential for
survival and a relevant drug target. Int. J. Parasitol., 2012, 42(5), 481-488.
107. O’Brien, T. C., Mackey, Z. B., Fetter, R. D., Choe, Y., O’Donoghue, A. J., Zhou,
M., Craik, C. S., Caffrey, C. R., McKerrow, J. H. A parasite cysteine protease is
key to host protein degradation and iron acquisition. J. Biol. Chem., 2008,
283(43), 28934-28943.
108. Mallari, J. P., Shelat, A., Kosinski, A., Caffrey, C. R., Connelly, M., Zhu, F.,
McKerrow, J. H., Guy, R. K. Discovery of trypanocidal thiosemicarbazone
inhibitors of rhodesain and TbcatB. Bioorg. Med. Chem. Lett., 2008, 18(9), 2883-
2885.
109. Fonseca, N. C., Da Cruz, L. F., Da Silva Villela, F., Do Nascimento Pereira, G.
A., De Siqueira-Neto, J. L., Kellar, D., Suzuki, B. M., Ray, D., De Souza, T. B.,
Alves, R. J., Júnior, P. A. S., Romanha, A. J., Murta, S. M. F., McKerrow, J. H.,
Caffrey, C. R., De Oliveira, R. B., Ferreira, R. S. Synthesis of a sugar-based
thiosemicarbazone series and structure-activity relationship versus the parasite
cysteine proteases rhodesain, cruzain, and Schistosoma mansoni cathepsin B1.
Antimicrob. Agents Chemother., 2015, 59(5), 2666-2677.
110. Robertson, C. D., North, M. J., Lockwood, B. C., Coombs, G. H. Analysis of the

Referencias
161
proteinases of Trypanosoma brucei. J. Gen. Microbiol., 1990, 136(5), 921-925.
111. Lima, L., Ortiz, P. A., da Silva, F. M., Alves, J. M. P., Serrano, M. G., Cortez, A.
P., Alfieri, S. C., Buck, G. A., Teixeira, M. M. G. Repertoire, genealogy and
genomic organization of cruzipain and homologous genes in Trypanosoma cruzi,
T. cruzi-like and other Trypanosome species. PLoS One, 2012, 7(6), e38385.
112. Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P.,
Alnemri, E. S., Altucci, L., Amelio, I., Andrews, D. W., Annicchiarico-
Petruzzelli, M., Antonov, A. V., Arama, E., Baehrecke, E. H., Barlev, N. A.,
Bazan, N. G., …, Kroemer, G. Molecular mechanisms of cell death:
Recommendations of the nomenclature committee on cell death 2018. Cell Death
Differ., 2018, 25(3), 486-541.
113. Jiménez-Ruiz, A., Alzate, J. F., MacLeod, E. T., Lüder, C. G. K., Fasel, N., Hurd,
H. Apoptotic markers in protozoan parasites. Parasit.Vectors, 2010, 3(104), 1-15.
114. Duszenko, M., Figarella, K., Macleod, E. T., Welburn, S. C. Death of a
trypanosome: a selfish altruism. Trends Parasitol., 2006, 22(11), 536-542.
115. Jimenez, V., Paredes, R., Sosa, M. A., Galanti, N. Natural programmed cell death
in T. cruzi epimastigotes maintained in axenic cultures. J. Cell. Biochem., 2008,
105(3), 688-698.
116. Kaczanowski, S., Sajid, M., Reece, S. E. Evolution of apoptosis-like programmed
cell death in unicellular protozoan parasites. Parasit.Vectors, 2011, 4(44), 2-9.
117. Alzate, J. F., Álvarez-Barrientos, A., González, V. M., Jiménez-Ruiz, A. Heat-
induced programmed cell death in Leishmania infantum is reverted by Bcl-XL
expression. Apoptosis, 2006, 11(2), 161-171.
118. de Silva Rodrigues, J. H., Stein, J., Strauss, M., Rivarola, H. W., Ueda-
Nakamura, T., Nakamura, C. V., Duszenko, M. Clomipramine kills Trypanosoma
brucei by apoptosis. Int. J. Med. Microbiol., 2016, 306(4), 196-205.
119. Jaeger, T., Flohé, L. The thiol-based redox networks of pathogens: Unexploited
targets in the search for new drugs. BioFactors, 2006, 27(1-4), 109-120.
120. Krauth-Siegel, R. L., Meiering, S. K., Schmidt, H. The parasite-specific
trypanothione metabolism of trypanosoma and leishmania. Biol. Chem., 2003,
384(4), 539-549.
121. Krauth-Siegel, R. L., Comini, M. A. Redox control in trypanosomatids, parasitic
protozoa with trypanothione-based thiol metabolism. Biochim. Biophys. Acta,
2008, 1780(11), 1236-1248.
122. Figarella, K., Rawer, M., Uzcategui, N. L., Kubata, B. K., Lauber, K., Madeo, F.,
Wesselborg, S., Duszenko, M. Prostaglandin D2 induces programmed cell death
in Trypanosoma brucei bloodstream form. Cell Death Differ., 2005, 12(4), 335-
346.
123. Barth, T., Bruges, G., Meiwes, A., Mogk, S., Mudogo, C. N., Duszenko, M.
Staurosporine-induced cell death in Trypanosoma brucei and the role of
endonuclease G during apoptosis. Open J. Apoptosis, 2014, 3(2), 1-16.

Referencias
162
124. Schmidt, A., Krauth-Siegel, R. L. Enzymes of the trypanothione metabolism as
targets for antitrypanosomal drug development. Curr. Top. Med. Chem., 2002,
2(11), 1239-1259.
125. Turrens, J. F. Oxidative stress and antioxidant defenses: A target for the
treatment of diseases caused by parasitic protozoa. Mol. Aspects Med., 2004,
25(1-2), 211-220.
126. Maya, J. D., Cassels, B. K., Iturriaga-Vásquez, P., Ferreira, J., Faúndez, M.,
Galanti, N., Ferreira, A., Morello, A. Mode of action of natural and synthetic
drugs against Trypanosoma cruzi and their interaction with the mammalian host.
Comp. Biochem. Physiol. A Mol. Integr. Physiol., 2007, 146(4), 601-620.
127. Martins, S. C., Lazarin-Bidóia, D., Desoti, V. C., Falzirolli, H., da Silva, C. C.,
Ueda-Nakamura, T., Silva, S. de O., Nakamura, C. V. 1,3,4-Thiadiazole
derivatives of R-(+)-limonene benzaldehyde-thiosemicarbazones cause death in
Trypanosoma cruzi through oxidative stress. Microbes Infect., 2016, 18(12), 787-
797.
128. Rodríguez Arce, E., Machado, I., Rodríguez, B., Lapier, M., Zúñiga, M. C.,
Maya, J. D., Olea Azar, C., Otero, L., Gambino, D. Rhenium(I) tricarbonyl
compounds of bioactive thiosemicarbazones: Synthesis, characterization and
activity against Trypanosoma cruzi. J. Inorg. Biochem., 2017, 170, 125-133.
129. Kholmukhamedov, A., Schwartz, J. M., Lemasters, J. J. Mitotracker probes and
mitochondrial membrane potential. Shock, 2013, 39(6), 543.
130. Cottet-Rousselle, C., Ronot, X., Leverve, X., Mayol, J. F. Cytometric assessment
of mitochondria using fluorescent probes. Cytom. Part A, 2011, 79(6), 405-425.
131. Newkome, G. R., Fishel, D. L. Synthesis of simple hydrazones of carbonyl
compounds by an exchange reaction. J. Org. Chem., 1966, 31(3), 677-681.
132. Lansbury, P. T., Mancuso, N. R. Further studies of bond insertion and
nonstereospecific beckmann rearrangements in 7-Alkyl-1-indanone oximes1. J.
Am. Chem. Soc., 1966, 88(6), 1205-1212.
133. Friedrichsen, W., Betz, M., Büldt, E., Jürgens, H. J., Schmidt, R., Schwarz, I.,
Visser, K. Diels-Alder-reaktionen von o-benzochinonen mit fulvenen. Justus
Liebigs Ann. Chem.,1978, 1978(3), 440-472.
134. Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch,
T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D.
J., Hartenstein, V., Eliceiri, K., Tomancak, P., Cardona, A. Fiji: an open-source
platform for biological-image analysis. Nat. Methods, 2012, 9(7), 676.
135. Creek, D. J., Nijagal, B., Kim, D. H., Rojas, F., Matthews, K. R., Barrett, M. P.
Metabolomics guides rational development of a simplified cell culture medium
for drug screening against trypanosoma brucei. Antimicrob. Agents Chemother.,
2013, 57(6), 2768-2779.
136. Choi, K. H., Groarke, J. M., Young, D. C., Kuhn, R. J., Smith, J. L., Pevear, D.
C., Rossmann, M. G. The structure of the RNA-dependent RNA polymerase
from bovine viral diarrhea virus establishes the role of GTP in de novo initiation.

Referencias
163
Proc. Natl. Acad. Sci., 2004, 101(13), 4425-4430.
137. Zhang, J. X., Meng, F. C., Yu, S. Y. Two contrasting HP/LT and UHP
metamorphic belts: Constraint on early paleozoic orogeny in qilian-altun orogen.
Acta Petrol. Sin., 2010, 26(7), 1967-1992.
138. Morris, G., Huey, R. AutoDock4 and AutoDockTools4: Automated docking with
selective receptor felxibility. J. comput. Chem., 2009, 30(16), 2785-2791.
139. Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M.,
Meng, E. C., Ferrin, T. E. UCSF Chimera - a visualization system for exploratory
research and analysis. J. Comput. Chem., 2004, 25(13), 1605-1612.
140. Yap, C. W. PaDEL-descriptor: An open source software to calculate molecular
descriptors and fingerprints. J. Comput. Chem., 2010, 32(7), 1466-1474.
141. Vainio, M. J., Johnson, M. S. McQSAR: A multiconformational quantitative
structure-activity relationship engine driven by genetic algorithms. J. Chem. Inf.
Model., 2005, 45(6), 1953-1961.
142. Vainio, M. J., Johnson, M. S. Generating conformer ensembles using a
multiobjective genetic algorithm. J. Chem. Inf. Model., 2007, 47(6), 2462-2474.