nacional de la - unlp
TRANSCRIPT

UNIVERSIDAD NACIONAL DE LA PLATAFACULTAD DE CIENCIAS EXACTAS
FOTOLUMINISCENCIA DEL INDOL Y DERIVADOS POSIBILIDADES ANALITICAS
Trabajo de Tesis presentado por
LILIANA BRUZZONEPara optar al grado de Doctor en Ciencias Químicas
(Orientación Analítica)

UNIVERSIDAD NACIONAL DE LA PLATAFACULTAD DE CIENCIAS EXACTAS
FOTOLUMINISCENCIA DEL INDOL Y DERIVADOS POSIBILIDADES ANALITICAS
Trabajo de Tesis presentado por
LILIANA BRUZZONEPara optar al grado de Doctor en Ciencias Químicas
(Orientación Analítica)
1981

A mis padres

INDICE
INTRODUCCION.................................................................................................1
PROPOSITO .................................................................................................. 4
1 PROCESOS FOTOFISICOS GENERALES. MECANISMOS ............................ 5
2 INSTRUMENTAL Y REACTIVOS.............................................................. 17
2.1 Absorción ultravioleta.............................................................. 17
2.2 Excitación y emisión.......................................................................17
2.3 Reactivos...........................................................................................19
2.4 Purificación de los reactivos.................................................. 19
3 CORRECCION DE ESPECTROS.............................................................. 21
4 METODOS DE CALCULO...........................................................................24
4.1 Determinación de rendimientos de fluorescencia................. 24
4.2 Determinación de rendimientos a baja temperatura....................... .. .25/c ia.
4.3 Determinación del tiempo de decaimiento de fosforescen-26
4.4 Cálculo del tiempo de vida media natural y del tiempo
de decaimiento de fluorescencia.............................................26
5 PARTE EXPERIMENTAL........................................................................... 31
5.1 Espectros de absorción..................... ............................................31
5.1.1 Fuerza del oscilador por resolución gráfica de las
bandas................................................................................................42
5.1.2 Análisis de los espectros.......................................................4b
5.2 Espectros de fluorescencia a temperatura ambiente . .53
5.2.1 Análisis de los espectros en ciclohexano..........................59
5.2.2 Efecto de la tempera terra..........................................................62
5.2.3 Efecto de átomos pesados..........................................................66
5.3 Espectros de luminiscencia a 77°K........................................69
5.3.1 Análisis de los espectros.......................................................74
5.3.2 Efecto de temperatura sobre la emisión de fosforescencia...............75
6 INTERPRETACION .......................................................................... 76
CONCLUSIONES ........................................................................... 84
APENDICE......................................................................................... 89
BIBLIOGRAFIA ................................................................................ 98

1
INTRODUCCION
El indol es una molécula de gran importan^
cía analítica ya que es el grupo cromóforo del aminoácido
triptofano. Estudiando sus propiedades espectroscópicas y las
de sus derivados en medios de diferente polaridad se obtienen
parámetros que pueden correlacionarse con las propiedades de
las proteínas y a los cambios que éstas sufren asociadas con
el medio que las rodea.
Existe gran número de trabajos dedicados
al estudio de esta molécula y la variedad de teorías que ex
plican sus distintas propiedades es peetroscópicas indica la
naturaleza compleja del problema.
La mayoría de los estudios realizados se
basan en el comportamiento observado cuando esta molécula se
disuelve en solventes de diferente polaridad.
Strickland, Horwitz y Billups identi
ficaron varias de las transiciones vibracionales del indol y
del 3-metilindol provocando perturbación por acción de solver^
tes en los espectros de absorción yf^poder así diferenciar los
corrimientos en las transiciones y , según la termino
logía de Platt 2 \
Zimmermann y Joop midieron los espec
tros de polarización de absorción y de fluorescencia. La tran
sición ^L, está polarizada en la dirección del eje molecular o
más largo y la ^L^ perpendicular a la a lo largo del eje
molecular corto. Los mismos autores dedujeron que la emisión
de fluorescencia se originaba fundamentalmente desde el esta
do ?l^ pero que una fracción lo hacía también desde el
Song y Kurtin efectuaron medidas de
polarización de luminiscencia y cálculos de orbitales molecu-

¿
lares llegando a la misma conclusión; o sea que la emisión
era dual, proveniente de los estados 1y JL() tanto en sol
ventes polares como en otros relativamente no polares.( 5)Van Duuren ' ' observó cambios pronuncia
dos en los espectros de emisión de fluorescencia del indol y
derivados con diferentes solventes, no así en los espectros
de absorción o de excitación de fluorescencia. Este comporta
miento se debería a la existencia de estados mesoméricos po
lares que generarían mayores contribuciones en la molécula a£
tivada que en el estado fundamental. Tales estados serían más
sensibles a los cambios de las constantes dieléctricas de los
solventes, produciendo el corrimiento en fluorescencia y no
en absorción.
El corrimiento hacia mayores longitudes
de onda de la banda de fluorescencia del indol cuando se exa
mina en diferentes solventes, al pasar de no polares a los po^
lares, ha sido atribuido a un incremento en el mohiento dipo
lar del estado excitado y a una mayor estabilización causada( 6 - 81por interacciones soluto-solvente ' .
También ha sido explicado el corrimiento
en los espectros de fluorescencia por la formación de uniones
hidrógeno entre el indol y las moléculas del solvente(o \
Konev determino que la emisión de
fluorescencia del indol involucraba dos transiciones electró
nicas ya que al ir de un solvente no polar a uno polar,
además del corrimiento, se producía la desaparición de la ban_
da de fluorescencia de menor longitud de onda, J, y un co
rrimiento mayor y ensanchamiento en la banda de longitud de
onda más larga, Además postuló que existen procesos de
relajación de solventes y como resultado de ello la banda a

3
mayor longitud de onda estaría formada por la luminiscencia
de moléculas con tiempos de vida mayores.
Walker, Bednar, Lumry y Hum--phries(13) sugieren la existencia de un complejo excitado so
luto-solvente (exciplex) el cual sería el responsable del
gran corrimiento y pérdida de estructura vibracional de los
espectros de fluorescencia del indol y derivados en solven
tes polares. Este complejo sería del tipo de transferencia de
carga, en el cual el indol excitado actuaría como dador y el
solvente polar como aceptor.
Varios autores postulan que el
proceso de desactivación no radiativo del estado singlete ex
citado puede producirse por dos caminos: uno independiente de
la temperatura y otro dependiente de la misma.( 14)Freed y Salmre v 7 obtuvieron espectros
de emisión de fosforescencia del indol y derivados, los cua
les tenían mayores diferencias entre sí que las halladas en
los espectros de fluorescencia.
La escasa literatura existente referente
a la fosforescencia (4.9) puntualiza que la misma se produci
ría desde el estado 3L .a.
Las teorías desarrolladas para la expli
cación del comportamiento espectroscópico del indol y deriva
dos son varias. Los diferentes datos bibliográficos e inter
pretaciones demuestran que el comportamiento de estas molécu
las en estado excitado es complejo.

4
El presente estudio está orientado a:
verificar los diversos datos experimentales, no siempre con
cordantes, establecidos por diferentes autores relativos al
proceso de absorción del indol (o indoles) en el ultraviole
ta, a hacer uso de valores que no son accesibles por expe
riencias en nuestro laboratorio -como los que surgen de pro
cesos que ocurren en la escala de los nanosegundos-. y com
plementarlos con el estudio de fluorescencia y fosforescencia
bajo efectos perturbadores: solventes, metilación, temperatu
ra , átomos pesados.
Correlacionando los fenómenos de absor
ción y emisión será posible intentar interpretar, sobre la
base de los valores encontrados, el proceso en sus aspectos
y dinámico^.
La interpretación, junto con los datos
espectroscópieos, son específicamente importantes en relación
con el comportamiento del triptofano en estado excitado y la
fluorescencia de proteínas.
Además, desde el punto de vista analíti
co, se pueden adecuar las condiciones para mejorar la sensi
bilidad y selectividad de las determinaciones que involucren
residuos de proteínas que contengan el grupo cromóforo indó-
1 i co .

5
1 PROCESOS FOTOFISICOS GENERALES. ME CAN ISM O S.
La absorción de luz por una molécula en
fase condensada produce transiciones a estados excitados de
diferentes configuraciones nucleares y electrónicas. EstosJe, los ¡es
estados exhiben distinta reactividad química^y—represen ttm
y'^los caminos por los cuales la energía absorbida es disipada.
Esta serie de procesos tiene como conse-
cuencia la emisión de la energía absorbida en forma parcial
o total (rendimiento ). La emisión parcial es debida a los
procesos competitivos en el estado excitado, que dependen de
los parámetros de estructura mo 1ecu1ar , ínti mamen te asociados
al medio que rodea a la molécula, el solvente, el cual afec
ta profundamente a los procesos actuando como perturbador,
por lo que el sistema debe ser considerado como una entidad
sol uto-sol ven te.
Todos los mec-a-ni sinos de—l-o-s procesos de
emisión o transferencia de energía pueden ocurrir entre nive
les excitados de la misma o diferente multiplicidad. El fenó
meno puede representarse por una serie de caminos que parten
del proceso inicial de la absorción de radiación que lleva a
la molécula (o sistema) a un estado cualquiera singlete exci
tado vi o rae iona1 mente (vibrónico) para luego ocurrir, como es
el caso casi general, la degradación de la energíadíl nivel
vibracional fundamental del primer estado singlete -e x-c-i-1 a d o .Li/GA'O de')Ln—4st-e proceso de degradación puede ocurrir la emisión de la
radiación como fluorescencia o f osf ores cenci a , según lo condi_
ciona una serie de constantes de velocidad que regulan estos
procesos fotofísicos, algunos dependientes y otros indepen
dientes de la temperatura.
En general, para una molécula en estado

6
electrónico y vibracional fundamental SQ, los procesos pueden
representarse según el diagrama de Jablonski:
Luego de .alcanzar un estado estacionario,
los procesos fotofísicos que pueden ocurrir se expresan me
diante las ecuaciones cinéticas siguientes:
diferente multiplicidad.
i. So + fu I , absorción a un estado elec- atrónico singlete excitado. Esta do vibróni co.
ii.★
S1 + calor Relajación vibracional.
ni. S J★
— So + h^f kf[Sj3» fluorescencia.
IV.★
— So kn[sj]» conversión interna.
V.★
So ^So + calor Relajación vibracional.
VI . s/★
“>Tj kg[S/l» cruce entre sistemas de

té rmi c a .
VII .*
b ’ b Relajación vibracional.
VIII. b* *
- b v b k^fSjlexpí- E/RT), activación
térmi ca .
IX. b + hb kp[Tj]» fosforescencia.
X. b*
— So k [T J , conversión interna.
XI . b* *
-*b —b k£[T7]exp(- E/RT), activación
El proceso fotofísico I es la excitación
electrónica de una molécula que involucra la transición de un
electrón de un orbital molecular en el estado S a un orbital o de un estado electrónico superior excitado vibracionalmente★
Sp transición -vibróniea. En el caso presente la energía de
excitación proviene de radiación óptica, no ionizante, dando
como resultado la absorción de fotones.
El proceso II corresponde a la disipación
de la energía, generalmente por colisión con el medio, condu
ciendo a la molécula al estado vibracional fundamental del
estado electrónico excitado S., se denomina relajación vibrado
cional (Rv). Se trata de una transición que involucra un es- - 13 -11tado electrónico. El proceso es del orden de 10 a 10 se
g undos.
El proceso III se denomina fluorescencia
y es la desactivación con emisión de radiación desde el nivel
vibracional fundamental del estado singlete excitado electró
nicamente Sj a cualquier nivel vibracional del estado funda
mental SQ. El proceso está determinado por una constante de- 9 - Rvelocidad k^ y es del orden de 10 a 10 segundos.
El proceso IV representa una relajación e^
lectrÓnica, transición isoenergética, en este caso entre el
nivel vibracional fundamental del estado electrónico excitado

3
a cualquier nivel vibracional excitado del estado electrónico
fundamental. Se denomina conversión interna (CI) y está deter
minado por una constante k .nEl proceso V es relajación vibracional
del estado fundamental.
El proceso VI, también es una relajación
electrónica; es una conversión que está prohibida por spin
desde el estado singlete al estado triplete cercano excitado
vibraciona1 men te , transición isoenergética. Se produce por un
proceso de acoplamiento vibracional, igual que en conversión
interna y su constante de velocidad k^es del orden de la de Ib
fluorescencia. Se denomina cruce entre sistemas y puede o no
ser activada por átomos pesados.
El proceso VII es relajación vibracional
del estado triplete.
El proceso VIII corresponde a la activa
ción térmica de niveles vibracionales del estado singlete ex
citado que aumenta la probabilidad de acoplamiento con nive
les vibraciona1 es de cualquier otro estado, k^ es el pre-expo/
nencial para este proceso dependiente de la temperatura.
El proceso IX es la desactivación con ra
diación del nivel triplete fundamental a cualquier nivel vi
bracional excitado del estado singlete fundamental. El proce
so se den omi na fosforescencia, es regido por una constante k p-4 y es del orden de 10 a varios segundos.
El proceso X es de conversión interna re
gido por una constante de velocidad km y es seguido por el
proceso V de relajación vibracional.
El proceso XI es de activación térmica
de la molécula en el estado triplete más bajo, que puede, en

J
algunos casos, ser suficiente para poblar el nivel singlete
excitado y producir emisión de radiación denominada fluores
cencia retardada, k? es el factor pre-exponenci a 1.
La relación entre el número de fotones
emitidos a fotones absorbidos define el rendimiento de la emi_
siÓn(^, fluorescencia ((^) o fosforescencia (^p) •
Si se quiere determinar la eficiencia de
la emisión de fotones, se debe medir la pérdida de población
del estado excitado en función del tiempo y la cantidad de fo
tones absorbidos por la sustancia irradiada. Esta última, 11 a_
mada intensidad de la radiación absorbida, se expresa como el
número de fotones absorbidos por segundo y está relacionada
con la ley de Beer por intermedio de la intensidad de la luz
incidente, IQ, mediante la ecuación Ifl = IQ(l-10 ) donde
£ es la absortividad molar de la sustancia, c la concentración
molar y 1 la longitud del camino Óptico.
En el caso menos general en que el fenóme_
no esté representado por los procesos I a III, toda la luz
absorbida es emitida, o sea (|)f = 1 ; se tiene entonces que:
De la ecuación (1) se define:
y la integración de la ecuación (2):
Lsj1 = Lsol exp (4)

10
define el tiempo de vida medio como el tiempo requerido para que bJ = TsJ/e , de aquí que:
En este caso particular, en que toda la
energía absorbida es emitida, el tiempo de vida medio es el
tiempo de vida medio natural o radiativo,; el proceso de
emisión no está afectado por otros procesos competitivos,
siendo la constante de velocidad la radiativa, k^.
En realidad, ocurre en forma general que
la energía es distribuida entre todos los procesos fotofísi-
cos regulados por los siguientes parámetros de decaimiento
desde S j :
donde e corresponde a datos experimentales.
La luz absorbida:
Reemplazando en la ecuación (3):
De las ecuaciones (5), (6) y (7) se tiene:
Es de interés, para la interpretación de
los procesos y para distinguir la morfología molecular en el
estado excitado respecto del fundamental, hacer una considera
ción según la forma en que se obtiene el valor del tiempo de
vida natural. Cuando^0 se calcula mediante la medida experi-

11
Otra manera de obtenerlo es mediante la integración del es
pectro de absorción ( •
Los dos valores pueden diferir, porgue
es obtenido de la fuerza de la absorción y está íntegra
mente asociado a la molécula en estado fundamental y'Co es
calculado sobre la base de las propiedades de la molécula en
estado excitado (ecuación 9). Cualquier diferencia en sus va_
lores demostraría algún cambio de configuración en el estado
exc i tado.
El proceso VI representa el fenómeno de
transferencia de energía a un estado de diferente multiplicó
dad, regulado por una constante de velocidad k^, mediante el
cual se consigue poblar el estado triplete excitado cuando
su valor compite con la constante de fluorescencia. Este es
tado excitado, de tiempo de vida largo y altamente reactivo,
es de fundamental importancia en las reacciones fotoquímicas
dentro de la química orgánica. Es desactivado en estado con-
densado, generalmente por colisión, llegando al estado funda
mental por un proceso de conversión interna regido por una
constante de velocidad k .mSe define la vida media radiativa de fo£
forescencia como:
(10)
y teniendo en cuenta el proceso de desactivación, la constan^
te del proceso total:
mental de y del rendimiento c uán t i co (J)f, se lo indica co-
(9)

12
(ID
y el tiempo de decaimiento:
(12)
Como en el caso de fluorescencia, la ecua
ción del rendimiento queda expresada por:
(13)
además de una ecuación que expresa el rendimiento en la p o ó 1 a_
ción del estado triplete , que es directamente proporcional
al producto del tiempo de decaimiento de fluorescencia
por la constante de velocidad del proceso de transferen ci a
de energía entre ambos sistemas de diferente multiplicidad
(14)
En un medio rígido (de un vidrio transpa
rente), la constante kn puede despreciarse:
(15)
(16)
Utilizando las ecuaciones (13), (10),
(16) y (12) se obtienen:
(17)
(18)

13
A bajas temperaturas la emisión de fluo
rescencia ocurre desdo a niveles vibraci on a 1 es del estado
electrónico fundamental. El espectro de fluorescencia puede
consistir en una serie de bandas cuyas diferencias en ener
gía caracterizan los diferentes modos vibraci on a 1 es de la mo
lécula sin excitar, generalmente vibraciones C - C y C - H.
Cuando se aumenta la temperatura se produce un ensanchamien
to del espectro con pérdida de estructura. Este efecto pro
duciría la población de niveles vibrae iona1 es excitados del★
nivel electrónico superior Sj con una energía E^ en exceso
respecto a la correspondiente al nivel vibracional fundamen
tal del estado electrónico excitado Sj.
Si Ej es la energía de activación del es^
tado singlete excitado, la nueva ecuación que define al esta_
do estacionario es:
Operando matemáticamente se obtiene:
(22)
= kf + kp + + k^ exp(- Ej/RT)
kf + kn + kg + k? exp(' E ;/RT)

14
donde:
Kj es una constante que representa a los procesos independien
tes de la temperatura en el intervalo estudiado.
A temperaturas del nitrógeno líquido (77°K)
se eliminan ciertos caminos no radiativos y la emisión desde
el nivel singlete aumenta en intensidad al mismo tiempo que
ocurre la emisión desde el nivel triplete fundamental T.»
dando lugar al fenómeno de fosforescencia.
Un efecto de elevación de temperatura a
partir de estas condiciones produce la población de niveles
vibracional es del estado triplete. Estos niveles pueden con
tribuir a la emisión fosforescente o puede ocurrir que la e-
nergía de activación E£ sea suficiente para alcanzar a poblar
el estado singlete, dando como resultado una emisión de la
misma energía que la de fluorescencia con un valor del pará
metro de decaimiento del orden del de fosforescencia, denomi-(15) nada fluorescencia retardada ' .
La constante de velocidad del decaimien
to de la fosforescencia está representada por:
(23)
Reemplazando la ecuación (11) en la (23)
y tomando la inversa:
(24)
Operando se obtiene, por ecuación (12):

(25)
La medida de la eficiencia de fluorescen
cia y tiempos de decaimiento de la fosforescencia en función
de la temperatura permitiría obtener valores de constantes de
velocidades y energías de activación de los procesos no radia_
ti vos .
Cuando en el sistema existe una molécula
extraña, el proceso que puede ocurrir se representa mediante:
Este proceso está regulado por la constan^
te kq que puede ser calculada mediante la ley de Stern - Vol-
mer (16).
La intensidad de la luz absorbida cuando
existe una molécula extraña:
la que combinada con al ecuación (3) resulta:
(26)
rescenci a
Si se considera^ la eficiencia de fluo-
en ausencia de la sustancia extraña, de las ecuacio
nes (7) y (26) se obtiene:
(27)
siendo K$v 1 a cons tan te de Stern Volmer, la cual, cuando se

ib
conoce el tiempo de decaimiento de la fluorescencia,
calcular el valor de la constante k^:
permi te
(28)

17
2 INSTRUMENTAL Y REACTIVOS
2 . 1 Absorción ultravioleta
Los espectros de absorción de las molécu
las estudiadas fueron registrados en un espectrofo tóme tro
Bausch Si Lomb 600 E y las longitudes de onda medidas con una
aproximación de 1 nm. Ancho de banda: 0,5 nm.
2.2 Excitación y emisión
Los espectros de excitación y emisión
fueron estudiados con un es pectrofotof1uorómetro Aminco Bow
man SPF convencional equipado con fuente de xenón de
150 vatios, espejo condensador elipsoidal y fotómetro propor
cional con fototubo multiplicador RCA 4837.
El sistema condensador elipsoidal aumenta
la sensibilidad del equipo convencional y la resolución de
los espectros de excitación y emisión.
El sistema consiste en una fuente de xenón
y dos espejos: uno es el condensador elipsoidal y el otro es
uno plano que dirige la radiación condensada hacia el equipo
a través de una ranura variable. Por medio de este sistema se
disminuyen las fluctuaciones debidas a la oscilación del arco
de la lámpara de xenón ya que los dos espejos rotan la imagen
del arco 90° con respecto a su eje original. La oscilación
del arco se mueve así a lo largo de la ranura en lugar de ha
cerlo a lo ancho de la misma manteniendo aproximadamente cons
tante las condiciones de la excitación.
El fotómetro proporcional varía la sensi
bilidad del fototubo multiplicador en forma inversa a las va
riaciones en la intensidad de la radiación de excitación debi_

Id
das a las fluctuaciones del arco de la lámpara.
La radiación de excitación pasa a través
de un divisor de haces: parte penetra al compartimiento de la
muestra y la otra porción es desviada, sin atravesar la celda,
alternándose ambos haces mecánicamente en su incidencia sobre
el fototubo multiplicador.
Durante la fase I la radiación de fluores_
cencía de la muestra pasa por el monocromador de emisión y el
fototubo produce una señal. Al continuar girando el alterna
dor, la radiación de fluorescencia se bloquea y la porción de
radiación de excitación que había sido desviada llega al foto
tubo (fase II). La señal resultante es retenida momentáneamen
te y llevada a un amplificador diferencial de referencia don
de se compara con un potencial fijo. Si las dos señales son
iguales, no hay variación en la salida del amplificador dife
rencial y no es necesaria ninguna corrección en el potencial
del fototubo. Si la intensidad de la lámpara de xenón varía,
se producirá una variación en la señal de salida del amplifi
cador diferencial de referencia. Esta señal modificará el po
tencial del fototubo, o sea la sensibilidad del mismo, hasta
que la señal en el amplificador diferencial sea igual a la de
referencia fija, manteniéndose sin cambio la salida del ampli_
fi ca dor di feren ci a 1.
Esta corrección requiere aproximadamente
150 mi 1 isegundos, lo que es considerablemente mucho menor que
la constante de tiempo del registrador X-Y, por lo que no se
observan cambios.
La escala de longitudes de onda del mono
cromador de emisión se calibra con una lámpara de mercurio de
baja presión tipo lapicera. La calibración de la escala de

19
longitudes de onda del monocroma dor de excitación se realiza
después de calibrado el de emisión, colocando en la celda una
suspensión de sulfato de bario en agua y encontrando el máxi
mo de dispersión para distintas posiciones del monocromador
de emisión.
2.3 Reactivos
La pureza de las especies usadas para la
investigación de su luminiscencia debe ser muy alta ya que
cualquier sustancia extraña podría atenuarla.
Los solventes empleados también deben ser
muy puros. En las medidas absorciométricas un criterio de pu
reza suficiente es que no absorba en la región espectral de
estudio.
En las medidas fosforimétricas, además de
las condiciones anteriores, el solvente debe formar vidrios
transparentes a la temperatura del nitrógeno líquido y poseer
la capacidad de disolver los compuestos de interés.
Los solventes empleados en el presente es_
tudio fueron de diferente polaridad y constante dieléctrica,
cicloñexano (yu= 0, £ = 2,0) y alcohol etílico (yU-= 1,66 ,
E = 24,5). Para efectuar la calibración del equipo Arninco
Bowman SPF se empleó etilenglicol como disolvente de la roda-
mina B.
2.4 Purificación de los reactivos
Alcohol etílico 8
El alcohol de 95% se mantuvo a reflujo
durante varias horas con 25 ml/1 de I^SO^ 12N y luego se des_
tilo. El destilado se trató con 20 g de KOH y 10 g de AgJO^
por litro y fue destilado nuevamente. Este destilado se man

20
tuvo durante una semana sobre amalgama de aluminio activada,
se filtró y destiló.
Ci elohexano
Reactivo Matheson, Coleman & Bell calidad
es pectroscópica ; se utilizó sin tratamiento adicional de puri
f i c a c i ó n .
E t i lenglicol
Reactivo Merck, se utilizó sin tratamien
to de purificación.
S u1 fa to de quinina
Reactivo Merck, se recristalizó como d i h i_
drato en H^SÜ^ 0,05 M seguido por dos recristalizaciones en
agua. Se secó a 100°C.
Rodamina B
Reactivo BDH, se recristalizó en una solu
ción de éter-benceno (1+1).
Indol (Fluka) (p.f. 52 ,0 °C - 52,4oC) y 2-meti1indol (Aldrich)
(p.f. 58,0oC-58,2°C) fueron recristalizados en éter de petró
leo y luego sublimados al vacío.
3-meti 1 i ndol
Reactivo Fluka, fue recrista1 izado en he£
taño y luego sublimado al vacío (p.f. 9 5 ,0° C-9 5,3° C) .
L(_) triptofano
Reactivo Carlo Erba, fue recristalizado
en una solución de etanol-agua (1+1) (p.f.292,5°C d.)
Yoduro de sodio
Reactivo Cario Erba, fue utilizado sin
purificación adicional.

¿1
3 _de_ e_sp_ectros
Los espectros de excitación y emisión re
gistrados por la mayoría de los espectrofotof1uorómetros son
aparentes.
A fin de obtener el espectro verdadero de
un compuesto es necesario calibrar el equipo, ya que el espe£
tro obtenido es una resultante de la geometría óptica, de la
intensidad de la fuente de excitación en función de la longi
tud de onda y de la sensibilidad del fotodetector.
El método utilizado para la corrección de(19) ambos espectros fue el desarrollado por Meluish x .
Se utiliza una solución concentrada de ro
damina B (3 g/1 en eti 1 eng1 i col ) como pantalla fluorescente
empleando el accesorio frontal de fluorescencia.
Se registra el espectro de excitación de
fluorescencia con el monocromador de emisión colocado a 640 nm.
Este espectro es la respuesta combinada de la lámpara y del
monocromador: Q(X).
Para calibrar el sistema de detección se
reemplaza el contador cuántico por un espejo recientemente
aluminizado y se mide la energía reflejada: R(A).
El factor de calibración para obtener los
espectros de emisión verdaderos es:

¿¿
Q(A): respuesta a la energía del contador cuántico.
M(X): transmisión de la ventana de la celda en que se mide la
respuesta del contador.
R(X): respuesta del monocromador-fototubo a la energía refle
jada.
B(X): ancho de banda del monocromador de emisión.
W(A): reflectancia del espejo a 1umi nizado.
Para longitudes de onda mayores de 400 nm,
se interpone una placa de vidrio a la salida del monocromador
de excitación para absorber el segundo orden de radiación.
Cuando se determina R(X) se debe fijar
exactamente el espejo aluminizado, cualquier movimiento intro
ducirá error en R(^). Se registra la emisión para cada valor
de longitud de onda de excitación, prácticamente cada 10 nm,
obteniéndose una distribución en forma de pico agudo. La al
tura del pico es el valor de R(A) y el ancho medio el de B (A ) .
Dado que se trabaja con red de difracción, B(X) es constante.
Los espectros de excitación se corrigie
ron multiplicando cada valor de intensidad registrada a una
determinada longitud de onda por el factor correspondiente
en 1 a curva de Q(A ).
Las curvas de S(X) y Q(A) se muestran en
la figura 1.

Z3
Figura 1. Curvas de calibración
............ Q(X)-----------s(A)

24
En el método seguido se debe tener cuida-
4 METODOS DE CALCULO
4 • 1 Determinación de rendimientos de fluorescencia
Se utilizó el método propuesto por Parker(20)y Rees k . Para ello se determinan los espectros de emisión
de fluorescencia de soluciones de la sustancia en estudio y
de una sustancia de rendimiento cuántico conocido, bajo las
mismas condiciones instrumentales.
Como la emisión de fluorescencia es pro
porcional a IQc1 y, por consiguiente, al área integrada del
espectro de fluorescencia corregido, se cumple:
donde:
2 y 1 identifican muestra y referencia respectivamente.
£: absortividad molar a la longitud de onda de excitación,
(p: rendimiento de fluorescencia.
c: concentración.
1: camino óptico.
Como sustancia de referencia se utilizó
sulfato de quinina disuelto en N. Se tomó como va
lor del rendimiento, para una longitud de onda de excitación
de 366 nm, el dado por Meluish de 0,55.
Cuando se utilizan solventes distintos al
agua se debe corregir por la diferencia en los índices de re
fracción. El valor del rendimiento debe ser multiplicado por
n2(so 1 vente)/ n2(agua) ^22’.
Precauci ones

do de no cometer errores debidos a: efecto de filtro interno,
uso de luz de excitación no monocromática, apagado por oxíge
no. La ecuación mencionada sólo es aplicable en condiciones
de absorción muy pequeña de luz. Se utilizaron soluciones muy
diluidas, con absorbancias menores de 0,05. Para evitar erro
res en la medida de la absorbancia se usaron celdas de 100 mm
de camino óptico. Antes de cada ensayo se eliminó el oxígeno
disuelto mediante el burbujeo con nitrógeno (99,998%).
4.2 Determinación de rendimientos a baja temperatura
Los espectros de emisión a baja temperate
ra y las curvas de decaimiento de fosforescencia se obtuvie
ron haciendo uso del accesorio de fosforescencia El fun.
damento del método empleado para la determinación es el mismo
que el descripto en la sección anterior operando a la temper^
tura del nitrógeno líquido (77°K) y midiendo primero a esa
temperatura y después a temperatura ambiente. El valor de ab
sorbancia empleado posteriormente en el cálculo es el medido
a temperatura ambiente.
En el caso especial de los indoles los
espectros de fluorescencia y fosforescencia no se solapan y
por consiguiente es posible calcular las áreas correspondiera
tes a cada proceso en forma individual.
Precauci ones
Además de las requeridas para el caso de
la fluorescencia se debe tener cuidado de evitar la formación
de burbujas en el nitrógeno líquido, por cuanto podrían pro
vocar la difusión de la radiación de excitación.
Una vez registrado el espectro de emisión
a baja temperatura, se deja equilibrar el sistema a la tempe-

Zb
ratura ambiente, y sin mover la celda, a fin de mantener inal
terada su posición, se mide nuevamente el espectro de emisión.
4 . 3 Determinación del tiempo de decaimiento de fosforescencia
Se establecieron las curvas de decaimien
to de fosforescencia intercalando en el camino óptico de la
radiación de excitación un interruptor mecánico.
No es necesario usar el cilindro rotato
rio ya que los tiempos de decaimiento son largos y los espec
tros de fluorescencia y fosforescencia están separados entre
ellos.
Temperaturas comprendidas entre 77°K y la
ambiente fueron alcanzadas permitiendo que la temperatura de
la muestra se elevase lentamente por evaporación del nitróge
no líquido. Las temperaturas fueron medidas insertando una
termocupla en la celda.
La intensidad de la emisión de fosfores
cencia se gráfico versus tiempo en un registrador con base de
tiempo (velocidad: 0,2 pulg/s).
4.4 Cálculo del tiempo de vida media natural y del tiempo
de decaimiento de fluorescencia
Se utilizó el método propuesto por Strick
ler y Berg ^3) empleando la fórmula:
donde:
X°a : tiempo de vida media natural,
g^ : estado degenerado inferior,
g^ : estado degenerado superior.

27
en transiciones s i n g 1 e te - s i n gl e te .
n : índice de refracción a la longitud de onda media de la
banda de fluorescencia.(é d 1 n >7 : integral bajo la banda de absorción electrónica.
Vf Zprom
(I (3)d3= ---- =----------- — : valor recíproco del término medio
lU)ds _3Sf en el espectro de fluorescencia.
Los espectros de absorción se integraron
en forma gráfica por el método de Simpson utilizando una com
putadora Monroe 1656.
Para facilitar los cálculos en las medi
das de rendimientos y tiempos de decaimiento se utilizó un
programa por computación. El programa, denominado CINEF
fue escrito en lenguaje FORTRAN IV para una computadora
IBM 360.
En la figura 2 se representa el diagrama
de flujo con la siguiente secuencia: hasta el punto (1) son
leídos y controlados todos los datos referentes a la curva de
calibración. Estos son introducidos según un orden determina
do y cualquier alteración será detectada e indicada, luego de
lo cual el procesamiento se detendrá. Los pasos siguientes
hasta el punto (2) son semejantes a la secuencia anterior pe
ro referidos al espectro que se desea corregir.
Si hasta aquí no ha habido inconvenientes,
los datos introducidos son listados y comienza la ejecución
de la corrección propiamente dicha.
La subrutina INTERP es una interpolación
parabólica progresiva que puede alcanzar hasta el grado cua
tro; en ella se fija el error deseado y durante el procesa-

¿ó
miento éste progresará hasta el grado necesario (siempre que
no sea mayor que cuatro) para que el valor obtenido tenga un
error menor al indicado. En esta parte, dicha subrutina inter
pola el valor del factor de corrección para cada longitud de
onda introducido como dato en el espectro problema.
A continuación se lleva a cabo la correc
ción de la intensidad de la emisión para cada punto y se tra
baja en número de onda, nm~^, en lugar de longitud de onda.
Con el fin de precisar el trazado del es
pectro se vuelve a llamar a la subrutina INTERP para obtener
valores cada 0,01 nm”^.
El espectro así obtenido se regulariza( 2 5 por cuadrados mínimos según el concepto aplicado por Golayv
En las secuencias siguientes se llevan a
cabo dos integraciones gráficas por el método de Simpson usan_
do la subrutina que ya posee la biblioteca de programas de la
máquina.
La primera integración es I (s ) d donde
I es la intensidad de la fluorescencia corregida: la segunda
es \ (ñ ). d Ñ . El cociente de ambas da el valor de
que interviene en la fórmula de Strickler y Berg para
la obtención del tiempo de vida natural de un estado excitado
electrónicamente por integración del espectro de absorción.
El espectro corregido es impreso gráfica
mente luego de normalizado, lo que permite obtener como resul_
tado el factor de normalización y el área del espectro.
Conjuntamente con el espectro se imprime
un listado de los valores interpolados y los grados utiliza
dos, esto hace posible un examen rápido de la efectividad del
proceso y del espaciamiento entre puntos. Si el grado es cero

significa que la longitud de onda de la curva de calibración
y del espectro coinciden y no se lleva a cabo la interpola
ción; si el grado es cuatro, los puntos a tomar deben estar
más próximos. Esta subrutina permite elegir los puntos toma
dos como datos en el espectro con entera libertad, sin que
sea necesario mantener un AAconstante, ni que haya coinciden^
cia con la longitud de onda de calibración.
29

30
Figura 2. Diagrama de flujo correspondiente al programa CINEF.

31
5 P A R T F EXPERIMENTAL
5 . 1 Es pe ctr o_s_ de absorción
Se registration los espectros de absorción
en ciclohexano y etanol en el intervalo
33000 y 40000 cm'1.
co in prendido entre
En las figuras 3, 4 y 5 se muestran los
espectros en ambos solventes del indol, 3-metil indol y 2 - m e t i 1_
indol normalizados en los respectivos máximos de absorción.
En las figuras 7 y 8 se observan los es
pectros de los tres indoles estudiados en ciclohexano y eta-
nol respectivamente. En la figura 8 se incluye el espectro
del L( jtriptofano. Se han
de ordenadas para apreciar
desplazado los valores
las frecuencias de las
absolutos
distintas
bandas.
Todos los espectros se barrieron con soli¿ _ 5
ciones aproximadamente 5.10 M en celdas de 100 mm de camino.
El indol en fase vapor muestra un espec
tro con estructura fina lo cual ha permitido a Strickland y
colaboradores determinar la secuencia vibrónica de las
bandas.
Comparando las posiciones de las bandas
de los compuestos disueltos en ciclohexano con las obtenidas
en fase vapor se trató de identificar las transiciones a las
cuales corresponden.
Todos los espectros demuestran alguna ana
logia en lo que respecta a la energía de las transiciones, p£
ro se observan ensanchamientos de las bandas y variaciones en
los contornos por efecto del solvente.
Las transiciones correspondien tes al indol
se dan en la tab la 1.

32
Luego de identificadas las bandas en ci
clohexano es posible compararlas con las obtenidas en etanol
(tabla 2) .
Tabla 1. Comparación de las posiciones de las bandas del indol di suelto en ciclohexano con las correspondientes en fase vapor.
NP: no resueltas, HP: no nuras
Ciclohexano v a no r (Strickland)
Designación (cm -1) Designación ( cm -1)
0 -0 34750 0-0 '4, 3 5 248
í°+ 907 36155
0 + 950 '4, 35700 >9 + 968 'L6 362 16
Io + 989 36237
0 + 1100 '4 ? 35850 0 + 1120 ?o 36368
0 + 1300 '4 36050 0 + 1313 36561
Q .-0 JLa 36050 0-0 36 56 1
0 + 700 a 36750 NR 0 + 750 3 7 3 1 1 HR
0 + 1000 ’la 37050 NR 0 + 1000 'l
a 3 7 56 1 HP
0 + 1250 'la 37300 HP 0 + 1700 33261 Kip
9 + 2100 38250 HP 0 + 2450 3)011 HP
Tabla 2. Comparación de las posiciones de las bandas del indol disuelto en etanol con las corresnondi en tes en ciclohexano.
Etanol Ciclohexano
Designación (cm”1) Des i g nac i ón \) (cm"1) Av(cm”1)
9-0 ’l,5 34650 0-0 >L„ 347 50 100
0 + 930 35600 0 + 950 '4, 35700 100
0-0 ’la. 35900 0-0 ’l
a 36050 150
0 + 700 ’la 36600 0 + 700 JL
a 367 50 150
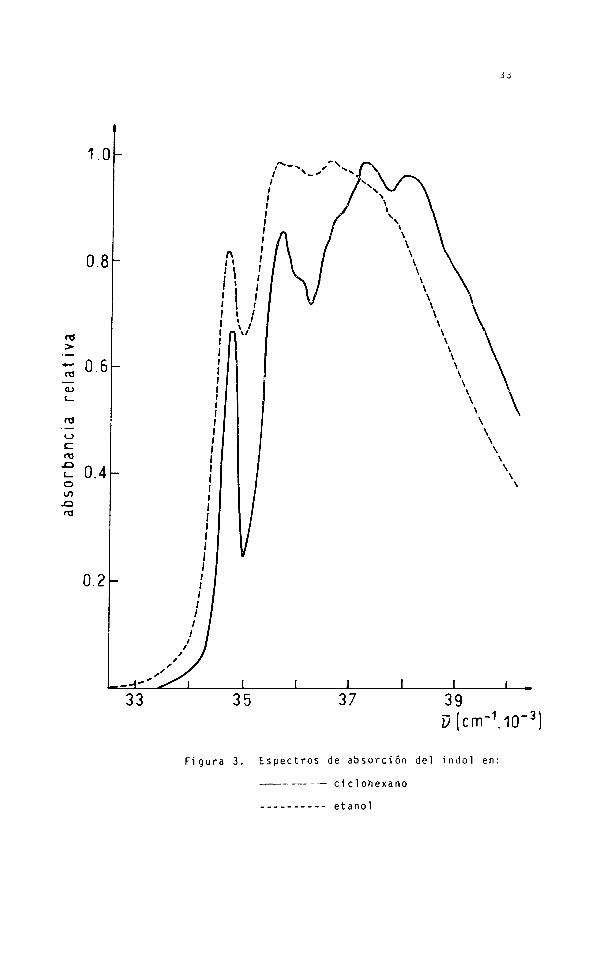
Figura 3. Espectros de absorción del indol en:
------------------ ciclohexano
------------------ etanol
33

34
El espectro del 3-meti 1 indol es más difí
cil de interpretar que el del indol debido a que las bandas a
mayores longitudes de onda son el resultado de un solapamien-
to de las dos transiciones. No obstante, efectuando el mismo
tipo de análisis que el practicado para el caso del indol, se
determinaron las transiciones correspondientes a las bandas
más características (tabla 3).
Tabla 3. Comparación de las posiciones de las bandas del
3-metilindol disuelto en ciclohexano con las corres_
pondientes en fase vapor.
Ciclohexano Vapor (Strickland)
Designación *9 (cm-1) Designación ^(cm"1)
0-0 34500 0-0 34831
0-0 'l 34500 0-0 'l 35510a a0 + 950 35450 0 + 980 3 581 1
0 + 1400 35900 0 + 1340 36171
0 + 2100 36600 0 + 2100 36931
0 + 27 50 37250 0 + 2800 'l.0
376 31
Las tres últimas bandas pueden ser solapami en tos de 'La
La única banda posible de identificar en
etanol es la de 34450 cm"1 que correspondería a la de
34500 cm"1 en ciclohexano.
El 2-meti 1 indol no fue estudiado por
Strickland. En la tabla 4 se dan las transiciones correspon
dientes a las distintas bandas observadas considerando los mo
délos del indol y del 3-metilindol.

35ab
sorb
anci
a rela
tiva
Figura 4. Espectros de absorción del 3-metilindol
en :
-----------------ci clohexano
---------------- etanol

36
El espectro del 2-meti 1 indo 1 (figura 5)
sufre variaciones similares a las producidas en el indol cuan
do se cambia de solvente. La banda 0-0 se puede determi
nar considerando el aumento de su intensidad relativa en eta
nol respecto a ciclohexano, por analogía con lo ocurrido en
el caso del indol (tabla 5).
Tabla 4. Posiciones de las bandas del 2-metilindol disuelto
en ciclohexano.
Designación (cm"1)
0-0 34800
0 + 900 1 35700
0 + 1050 35850
0 + 1400 36200
0-0 ’la 36200
0 + 800 'la 37000
0 + 2200 37000
0 + 2800 37600
0 + 3150 379 50
Las dos últimas bandas pueden ser solapamientos de \ y ^L,el O
Tabla 5. Comparación de las posiciones de las bandas del
2-metilindol disuelto en etanol con las correspon
dientes en ciclohexano.
Etanol Ciclohexano
Designación ^ (c m 1) Designación (cm 1) A^ (cm-1)
o-o ’l,o0-0 ’l
a
34700
36100
0-0 TLb
0-0 JLa
34800
36200
100
100

37
Figura 5. Espectros de absorción del 2-metilindol en:
-----------------ci clohexano
---------------- etanol

38
Se registró el espectro de absorción del
L(_)triptofano sólo en etanol (figura 6) debido a su insolubi
lidad en ciclohexano. Se observó que dicho espectro coincide
aproximadamente con el del 3-metilindol, lo que permite supo
ner que las transiciones son las mismas para las dos sustan
cias.

39ab
sorb
anci
a rela
tiva
Figura 6. Espectro de absorción del tri p tofano
en etanol.

40ab
sorb
anci
a
Fi gura 7. Espectros de absorción del (1) indol, (2) 2-inetil-
indol y (3) 3-metilindol en ciclohexano.

5.1.1 Fuerza del oscilador por resolu c i ó n g r áfiea de las
bandas .
Es posible determinar la intensidad de u-
na transición sobre la base de la fuerza del oscilador, f (26),
el cual puede alcanzar el valor unitario para la máxima proba
bilidad de la transición.
La fuerza del oscilador se calcula median
te la expresión conocida:
f = 4,319. 10’9 Jí di
en que /£ di es la intensidad integrada de la banda correspon
diente del espectro de absorción y es la frecuencia (cm~^) .
El cálculo de las fuerzas de los oscilado
res de las bandas y para el indol y sus derivados, a
causa de su solapamien to, requieren ser resueltas descontando
sobre el espectro, la absorción correspondiente a cada una de
ellas.
Como ejemplo del procedimiento se dan en
la figura 9 las bandas y halladas para el indol disuel
to en ciclohexano.
En las tablas 6 y 7 figuran los valores
obtenidos en ambos solventes para las fuerzas de los oscila
dores y los momentos de transición de las bandas ^L^ y ^L^
de las sustancias en estudio suponiendo el mismo tipo de sola^
pami en to.
En la tabla 8 se indican las relaciones
de las fuerzas de los osciladores de las bandas y L^ en
los mismos solventes.
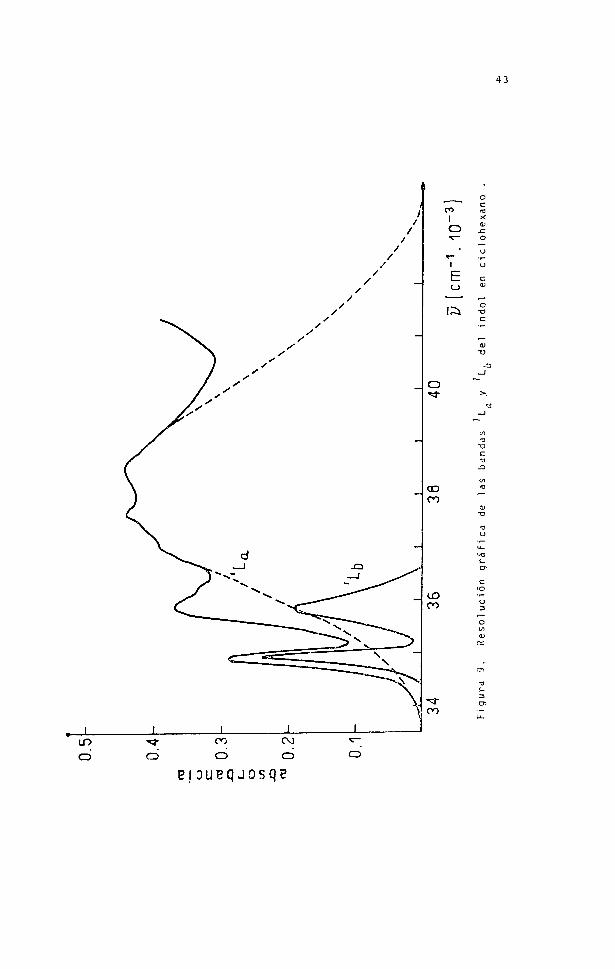
43
LOO O O
Figu
ra 9. R
esol
ució
n grá
fica d
e las b
anda
s L y L,
del ind
ol en
cicl
ohex
ano

44
«3- en o •—<
oj cm en cj
lo en eno >—•
o o
en -3- m•-< o
o o
ena)
caj
-ac oe endJU í-o o
enO</)
Tabl
a 6. Fuerz
as de
l osci
lado
r oara
la ba
nda L
del ind
ol y s
us de
rivad
os.
en o
■cro >
O O O LO loLO LO co en
o o o o en • LO LO en en
S- cu
•U
en o en
>
oT9c
2- m
etilin
dol
3480
0 0,
013
0,9
350
3460
0 0,
009
3- m
etilin
dol
3459
0 0,
021
1,1
750
3435
0 0,
012
L, -tri
ntof
ano
3425
0 0,
012

45
Tabla 8. Relación entre las fuerzas del oscilador de las
bandas y del indol y sus derivados en fun
ción del solvente.
^^Sol vente
Compuest o^\^^
Ci el oh exano
f 'L? f \
Etanol
f ’>V f ’Lfa
Indol 11.1 12,0
2-meti1indol 11,6 16,6
3-meti1indol 4,4 13,2
L( jtriptofano 6,2

46
5.1.2 Análisis de los espec tros
En general se observa que las transicio
nes son más sensibles a las perturbaciones por efecto del
sol vente que las ^L^.
Los espectros ultravioleta de las sustan
cias estudiadas en un determinado solvente y de cada uno de
los compuestos en los dos solventes usados, exhiben una carac
terística fundamental, la banda de mayor longitud de onda su
fre muy pequeños cambios respecto a la energía e intensidad
y las mayores variaciones aparecen a partir de los 36000 cm"^
en el sentido de las frecuencias decrecientes.
De los solventes estudiados el ciclohexa
no es el que produce menos alteraciones en los espectros de
absorción lo que cabe atribuir a su no polaridad.(9)
Según Konev ' cuando se pasa de solu
ciones en hidrocarburos a solventes más polares el máximo co
rrespondiente a bajas longitudes de onda sufre un gran corri
miento (700 cm"l) hacia el rojo; en cambio, el máximo a lon
gitudes de onda mayores se corre muy poco (150 cm”*). La in
terpretación de Konev es que se producen uniones hidrógeno en_
tre las moléculas del indol y las del solvente.
Observando la tabla 2 se advierte que en
los sistemas estudiados no se produce el gran corrimiento po£
tulado por Konev; el corrimiento se encuentra dentro de los
150 cm”l. La discrepancia surge del hecho de que en el cálcu
lo de los valores encontrados, dicho autor solo computó los
máximos de absorción sin tener en cuenta el estado vibracio
nal al cual pertenecían.
Si se consideran los máximos para el in
dol disuelto en ciclohexano (37300 cm *) y en etanol

47
(36600 cm el corrimiento da el mismo valor que encontró Ko
nev ( 700 cm’1) .
El corrimiento de los máximos (únicos va
lores susceptibles de ser comparados) del 3-metilindol en ci
clohexano (35900 cm y en etanol (35500 cm’h es de 400 cm~^,
menor que en el caso indol, lo que estaría indicando que se
produce un gran solapamiento de las bandas y *.
En cambio, para el 2-metilindol, el corri_
miento de los máximos en ciclohexano (37600 cm’^) y en etanol
(36750 cm es de 850 cm comparable al del indol.
Los estudios sobre polarización de los es_
pectros de absorción 9) han permjtido llegar a esta
blecer que la banda a mayor longitud de onda del indol en ci
clohexano es una transición con una leve superposi
ción de la banda en la zona de mayor longitud de onda.
Si suponemos que los dos primeros máximos
corresponden a la misma banda, determina por resolución
gráfica la frecuencia vibracional, la que da un valor de apro
ximadamente 900 cm”^ para las sustancias disueltas en ciclo
hexano y muy próximo a este valor para las disueltas en eta
nol (tab 1 a 9 ) .
Los espectros fueron medidos con una reso
lución de 0,5 nm que corresponde a + 50 cm en la región ba
rrida.

48
Tabla 9. Frecuencias vibraciona1 es de la banda L, . b
^^\So^ven teCiclohexano Etanol
Compuest (cm"1) *9 (cm"1)
Indol 0 + 940 0 + 975
2-meti1indol 0 + 820 0 + 900
3 - m e t i 1 i n d o 1 0 + 830 0 + 890
L(_)tri ptofano 0 + 760
Esta vibración de intensidad fuerte co
rrespondería a la de 980 cm 1 dada por Strickland en metilci_
clohexano y a la que Hollas caracterizó a 907 cm’1 en fa_‘
se vapor.
Yamamoto y Tanaka en sus estudios en_
cuentran que el cristal de indol exhibe prácticamente el mis
mo espectro que el del compuesto en solución y una frecuencia
vibracional de 850 cm"1 para la banda Esto está de acuer
do con lo anterior y si se tiene en cuenta que Kaneda y Tana-(29)ka k 7 han establecido que la molécula de indol es muy plana,
se puede concluir que la conformación del estado fundamental
en solución es la misma que la del cristal, lo que además es
taría de acuerdo con el hecho probado y conocido de que cuan
to más plano es el estado fundamental más estructurado resul
ta el espectro de absorción.
Se observa también que las dos frecuen
cias vibraci ona 1es de menor energía de cada sustancia en los
dos solventes ensayados, permanecen aproximadamente constan
tes (tab 1 a >).
Analizando las relaciones de las fuerzas
de los osciladores (tabla 8) en ciclohexano se observa que

49
los valores son parecidos para el indol y el 2-metilindol y
mucho menor para el 3-meti1indol , lo cual corroboraría que en
este solvente la banda a 34500 cm es un solapamiento muy
grande de ^L y ^L^, en concordancia, por lo demás, con lo ob
servado anteriormente.
La relación de las fuerzas de los oscila
dores de las dos bandas en el L(-) triptofano es mucho menor
que los valores alcanzados en los otros derivados; cabe atri-
buir este efecto al aumento de la fuerza del oscilador de . oEl solapamiento también determina un au
mento en el ancho medio de la banda, en el caso del espectro
del indol cuando se pasa de ciclohexano a etanol como solven
te, y en el del 3-metilindol respecto al del indol ambos en
ciclohexano.
Valeur y Weber en un trabajo recien
te, midieron los espectros de excitación polarizados, selec
cionando, por medio de filtros, el nivel desde el cual se on
gina la transición. Consiguen de esta manera resolver el es
pectro en las dos bandas y de las moléculas de tripto
fano y de indol disueltos en propi 1 eng1 i co 1. La figura 10 re
presenta en forma comparativa la resolución del espectro estu_
diado por el método simple usado en nuestro laboratorio y el
que usaron los autores.
Ambos espectros están en plena correspon
dencia y dan una frecuencia vibracional de 0 + 760 cm Lo
mismo sucede en el caso del indol en ciclohexano; para éste
la frecuencia vibracional hallada en nuestro estudio es de
0 + 940 cm~l. Los valores siempre resultan muy próximos a las
dos frecuencias dadas por Strickland que son de 0 + 730
cm”1 y 0 + 980 cm .

50
Figura 10. Resolución gráfica de las bandas y
Jl_b para el Ljtriptofan o .
a) Método de Valeur y Weber.
b) Método usado en el laboratorio.

bl
En general se puede decir qirc los espec
tros de absorción del L_jtripto fa no y del indol en solución
están constituidos por dos transiciones bien características
en la zona estudiada; una banda *a mayor longitud de onda,
con cierto solapamiento de que es posible resolver grá
ficamente con cierta seguridad y una transición para la
cual es muy aventurado establecer una analogía con la fase
vapor.
En solventes diferentes, la banda so a - lapa con la en un grado que depende de la polaridad de
aquellos.
Puede observarse que los dos estados cum
píen, aun en solución, lo que está demostrado para los espec
tros respectivos en fase gaseosa, a saber: que el primer es
tado da una banda aguda y que el segundo es mucho más ancho,
con el agregado de que en este caSo, el ensanchamiento vibró
nico no alcanza a anular la estructura vibracional.
La conservación, en lo sustancial, de la
forma y contorno de las bandas demuestra que la sustitución
afecta poco la geometría molecular y no hay prácticamente e-
fee to i n d u c t i v o del grupo metilo sobre la frecuencia v i b r a c i o_
nal de 1 a banda y solamente con alcance limitado sobre
1 a s de 1 a banda ’l .a
Se puede decir también que en el indol
y derivados estudiados la interacción se produce entre dos
estados del mismo tipo orbital, es decir, los dos electrones
del heteroátomo contribuyen a la estructura electrónica Tí,
la transición es Tí - TT como en el pirrol, dando como resul
tado que se conserve el modelo vibrónico del estado excitado.
En los espectros del Lj_jtriptofa no y del

52
3-metilindol en etanol se observa que las bandas -i mayor lon
gitud de onda sufren un corrimiento hacia el rojo con respec
to al indol o al 2-metilindol conservando básicamente su c o n_
torno.
Esta estabilización del estado excitado
no puede ser atribuida a una transferencia de carga con el
grupo ácido aceptor del triptofano ya que el 3-meti 1 indo1 ex
hibe un espectro análogo sin tal posibilidad; en todo caso
esto demuestra la relevancia de la posición 3 a la estabili-(31)zación cuando el H es sustituido v .
Esta propiedad y la planaridad del grupo
cromóforo quedan demostradas en el estudio de Rivero y cola
boradores sobre la estructura cristalina del complejo
de transferencia de carga entre escatol y tetracianoeti1eno.

53
5 • 2 Espectros de fluorescencia a temperatura ambiente.
Los espectros de emisión de fluorescencia
a temperatura ambiente se registraron termostatizando a 25°C
el compart i mi en to donde se aloja la celda.
Las soluciones empleadas para correr los
espectros de fluorescencia fueron las mismas usadas para los
espectros de absorción.
Todos los espectros de fluorescencia co
rregidos se han representado, en forma normalizada, conjunta
mente con los espectros de absorción. Es importante destacar
que el espectro de excitación corregido se extiende entre los
límites correspondien tes al de absorción y coincide en su as
pecto vibracional con éste.
En ciclohexano se observa una cierta es
tructura en el contorno de la banda de fluorescencia además
de tener una imagen especular parcial respecto al de absor
ción; por lo tanto se ha tratado de establecer la concordan
cia entre las frecuencias vibraciona1 es de ambos espectros.
En las figuras 11, 12 y 13 se representan
los espectros del indol, 3-metilindol y 2-metilindol disuel
tos en ciclohexano. Las flechas indican las frecuencias vi b ra_
cionales de la banda de menor energía asignada a la transi
ción ?Lfa <—4A del cristal de indol.
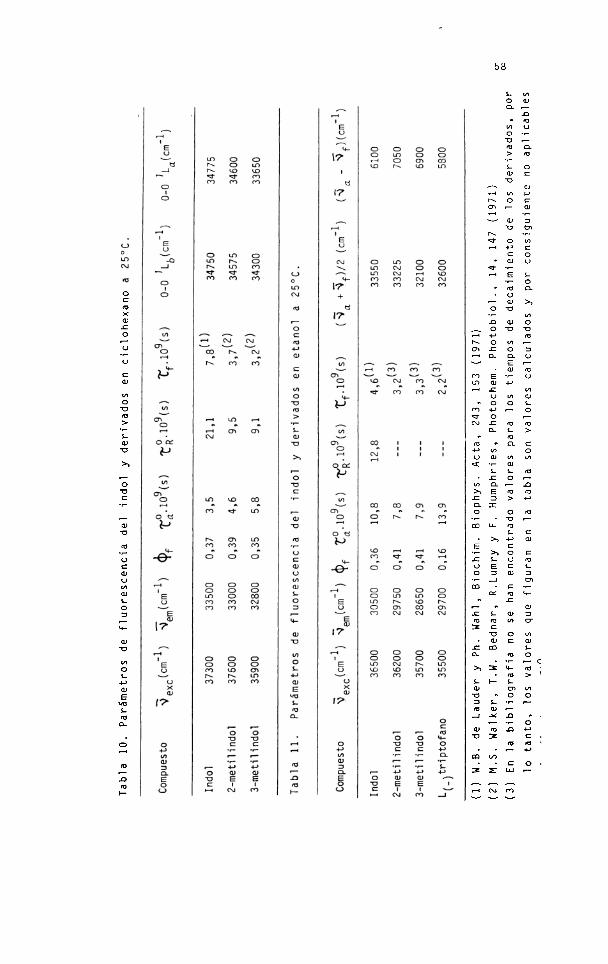
En la tabla 10 figuran los valores de los
parámetros de fluorescencia de los compuestos citados disuel
tos en ciclohexano.
Cuando se utiliza etanol como solvente
se observa que los espectros de emisión de fluorescencia ca
recen completamente de estructura y sus máximos se desplazan
hacia frecuencias menores.

54
En la tabla 11 figuran los valores de los
parámetros de fluorescencia de los compuestos estudiados di
sueltos en etanol, cuyos espectros se dan, conjuntamente con
los de baja temperatura, en las figuras 18, 19, 20 y 21.

55
Figu
ra 11.
Esoect
ros de
abso
rció
n y de
emis
ión a
25°C
del ind
ol en
cicl
ohex
ano.
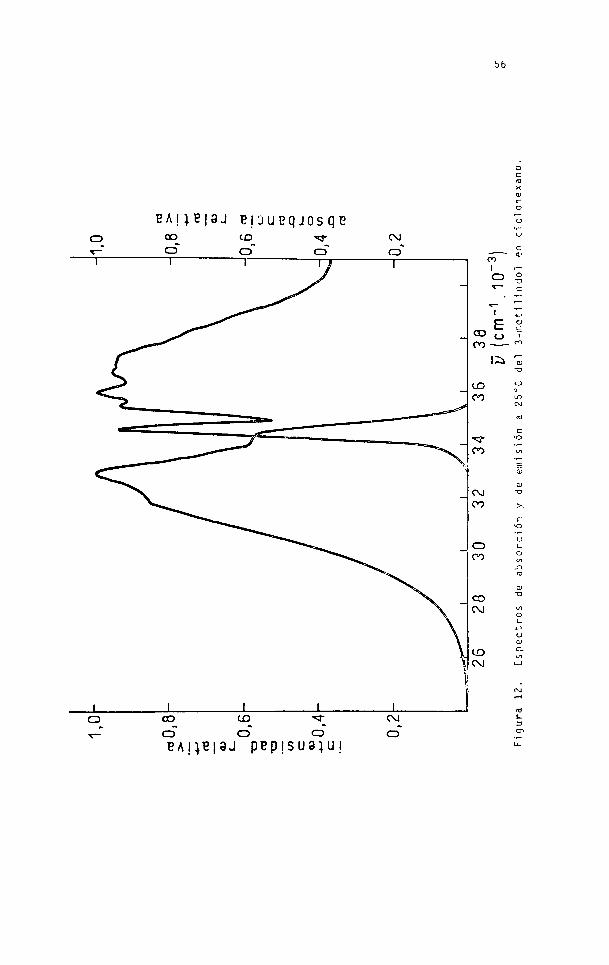
56
rdU
cn
CM

57
ea i )E|9j eioueqjosqe
Figu
ra 13.
Espect
ros de
abso
rció
n y de
emis
ión d
el 2-m
etilin
dol en
cicl
ohex
ano.
o oLO CXI
<T3
58
Tabl
a 10. Pa
rám
etro
s de flu
ores
cenc
ia de
l indol
y de
rivad
os en
cicl
ohex
ano
(1)
W.B
. de La
uder
y Ph
. Wahl
, Bio
chim
. Biop
hys.
Acta
, 243
, 153 ( 19
7 1)
(2)
M.S
. Wal
ker, T
.W. Be
dnar
, R.Lu
mry
y F.
Hum
phrie
s, Ph
otoc
hem
. Phot
obio
l., 14,
147 (19
71)
(3)
En la
biblio
graf
ía no
se han
enco
ntra
do va
lore
s para
los tiem
pos de
deca
imie
nto d
e los d
eriv
ados
, por
lo tan
to, los
valo
res qu
e figu
ran e
n la tab
la son
valo
res c
alcu
lado
s y po
r cons
igui
ente
no ap
licab
les

59
5.2.1 Análisis de los espectros en ciclohexano.
La imagen especular parcial de la figura
11 se observa en la figura 14,donde se hacen coincidir las
alturas de las bandas 0-0 de absorción y de fluorescencia,
proyectando luego el espectro sobre la línea de simetría.
Los picos a 34750 cm'1 correspondientes
a la transición 0-0 ^L^ en absorción y en fluorescencia son
coincidentes y el pico a 35700 cm 1 en absorción corresponde^
ría a la inflexión observada a 33750 cm'1 en fluorescencia.
La frecuencia vibracional de esta banda arroja un valor de
1000 cm 1 para el estado fundamental, en correspondencia con
la frecuencia vibracional en absorción, que daba un valor de
940 cm’l por resolución del espectro (Tabla 9).
Las frecuencias del máximo de fluorescen^
cia a 33500 cm’^ y la inflexión a 32500 cm"1 pueden ser com
paradas con las de absorción a 36050 cm'1 y 37050 cm’1 respe£
tivamente correspondientes a la transición \ . Estas ener:
gías difieren en los espectros de emisión y absorción en el
mismo intervalo: 1000 cm'1.
La transición correspondiente a 36750 cm'-
en absorción no se observa en el espectro de fluorescencia,
supuesto que deba existir a 32800 cm 1.
Considerando la simetría parcial de los
espectros, el origen de la transición JL^ se encontraría a
34775 cm"1, muy cercana a la transición 0-0 1L^ (34750 cm b.
El corrimiento de Stokes para la banda
0-0 ^L es de 2550 cm'1.d
Se estudiaron los otros compuestos efec
tuando el mismo tipo de consideraciones.

Figura 14. Proyección del espectro de absorción
sobre el de fluorescencia del indol
en c i elonexano .

61
Para el 3-metilindol la energía a 34100
cm 1 en el espectro de fluorescencia correspondería a la de
34500 cm 1 en absorción (el origen de la transición esta
ría en 34300 cm'1).
Si el máximo de la emisión de fluorescen^
cia corresponde a la transición \^(3^800 cm’1) y de la absor_
ción a 34500 cm 1 a 1 a misma transición, el origen de la ban
da estaría en 33650 cm-1 o sea a mayor longitud de onda
que la 0-0 1 .
El corrimiento de Stokes para la banda
0-0 es de 400 cm"1, y para la banda 0-0 es de 1 700 cm"1
Para el 2-metilindol, el pico a 34300 cm 1
en absorción se corresponde con el hombro a 34350 cm"1 en
fluorescencia, por lo cual el origen de la transición es
taría en 3457b cm"1.
El hombro a 33400 cm"1 en fluorescencia
se halla a 950 cm"1 del de 34350 cm"1 y se corresponde con
el pico a 35700 cm”1 en absorción, que se halla a 900 cm 1 del
de 34300 cm"1, lo cual significa que corresponden a la misma
transición 1 .
El máximo a 33000 cm"1 en fluorescencia
correspondería al pico a 36200 cm”1 en absorción y la infle-
ción a 32200 cm"1 en fluorescencia con la de 37000 cm"1 en
absorción, serían transiciones \ .
El origen de la banda 1La estaría en
34600 cm"1, con lo cual el origen de esta transición sería el
mismo que el de la 1.
El corrimiento de Stokes para la banda
0-0 es de 450 cm’1 y para la banda 0-0 JL es de 3200 cm 1L) a

62
5.2.2 Efecto de la temperatura
El efecto de la temperatura sobre la emi
sión de fluorescencia fue estudiado en el intervalo de 2 0 °C
a 60°C. Con la medida experimental del rendimiento de flúores
cencia se determinó la energía de activación.
En la figura 15 se muestra la variación
del rendimiento de fluorescencia con la temperatura para el
indol y el L(_jtriptofano disueltos en etanol. El aumento de
la temperatura afecta las velocidades de los procesos de des
activación en solventes polares. En ciclohexano no se ha en
contrado variación con la temperatura en el rendimiento de
fluorescencia.
Con la hipótesis de la existencia de dos
caminos de desactivación del estado excitado es posible calcu
lar la energía de activación para el proceso dependiente de
la temperatura, mediante la ecuación (22) que expresada en
forma logarítmica resulta:
La
valores en la ecuación
a 1 representar
constante Kse
hasta conseguir
obtiene introduciendo, . (33,34)la mejor recta
debe ser considerado como aproximado ya que
El valor de K
existe la posibi
1 idad de incurrir en errores sistemáticos en su evaluación.
En la figura 16 se muestran las rectas ob
teñidas para el indol y el L_jtriptofano disueltos
El trazado de la recta obliga a
en etanol.
despreciar
el valor de Kj, lo que indica que el proceso de desactivación
es únicamente dependiente de la temperatura.
(31)

63
2
Figura 15. Variación del rendimiento de
con la temoeratura del (1)
°C
fluorescencia
indol v
trintofano en etanol.

64
En la tabla 12 se muestran los valores ha
liados para las energía de activación y las constantes corres
pondi entes .
En el caso del ciclohexano (solvente no
polar), al ser el oroceso de desactivación independiente de
la temperatura, la ecuación (22) se simplifica ya que
= Kj , lo que conduce a valores de > 1 para los
compues tos estudiados, lo que significa que k < k n + k .
Tabla 12. Resultados experimentales correspondientes a la
ecuación (22) para el apagado de la fluorescencia
en etanol por efecto de la temperatura, = 0.
Compuesto K2(cm
Indol 52,4 700
2-me ti 1 i n do 1 25,0 600
3-met i 1indol 22,9 600
L( jtriptofano 3568,8 1400
ka.1C'9 (sJ11(1)
k¿.exn( -E,/RT)
(25°C)
4.1 1,4. 108
3,2 1,8. 108
2,9 1,9. 108
256 .9 3.5 108
(1) Valores calculados con 7^ de tabla 11

Figura 16. Representación gráfica de la aplicación
de la ecuación (31) para el
( 1) indol y
(2) L( jtriptofano en etanol, Kj 0

66
5.2.3 Efecto de átomos pesados
La eficiencia de fluorescencia en un sis
tema puede disminuir debido a la presencia de ciertas sustan
cias que producen apagado.
La cinética del apagado en soluciones se
describe mediante la ley de S tern-Volmer (ecuación 27). Grafi_
cando la relación de eficiencias de fluorescencia en función
de la concentración de átomo pesado que produce el apagado,
se obtiene una recta cuya pendiente es la constante de Stern-
Volmer, X $ v , con la cual se calcula la constante de apagado,
kq, (ecuación 28). El valor de esta constante determina si el
proceso está controlado di fus i ona linen te, para lo cual es necc
sario que el tiempo de vida del estado excitado sea mayor de
10 1 segundos. En este caso, las constantes de apagado deoen-
den de la viscosidad del solvente y de la temperatura.
La constante de velocidad de un proceso (35)controlado di fusi on a 1 men te es v .
Aj : viscosidad del solvente en poises.
Básicamente pueden existir dos mecanismos
mediante los cuales se pierde la energía en forma colisional:
aumento del cruce entre sistemas o transferencia electrónica.
En el presente trabajo se estudió el efec
to producido por el agregado de yoduro de sodio a soluciones
de un indol en etanol. Se observó que la forma del espectro
de emisión de fluorescencia no variaba, pero sí disminuía su
intensidad. La disminución de la fluorescencia es debida al
I" que actúa como átomo pesado; el Na + no produce apagado.
Las concentraciones de yoduro de sodio se

67
mantuvieron lo suficientemente bajas como para que no exis
tiera absorción de la luz de excitación.
En la figura 17 se adopta el indol para
ejemplificar la representación gráfica de la ley de Stern-Vol
me r.
En la tabla 13 se dan los valores halla
dos para las constantes de apagado del indol y derivados di
sueltos en etanol a 25°C. El apagado del
I" en etanol es despreciable.
L(-) tri pto fan o por
Se puede observar que los valores encon
trados son comparables con el de la constante de control di-
fusional calculada mediante la ecuación (32):
Tabla 13. Resultados experimentales del apagado de la
fluorescencia por efecto del Jal en etanol a 25°C.
(1) Valores calculados con de tabla 11.
Com pues to K (M1)S V 7
k .10'9(M'1 s'1) (1)
Indol 19.1 4,1
2-meti 1 i ndo 1 11.2 3,5
3-meti1 indol 6.1 1,9
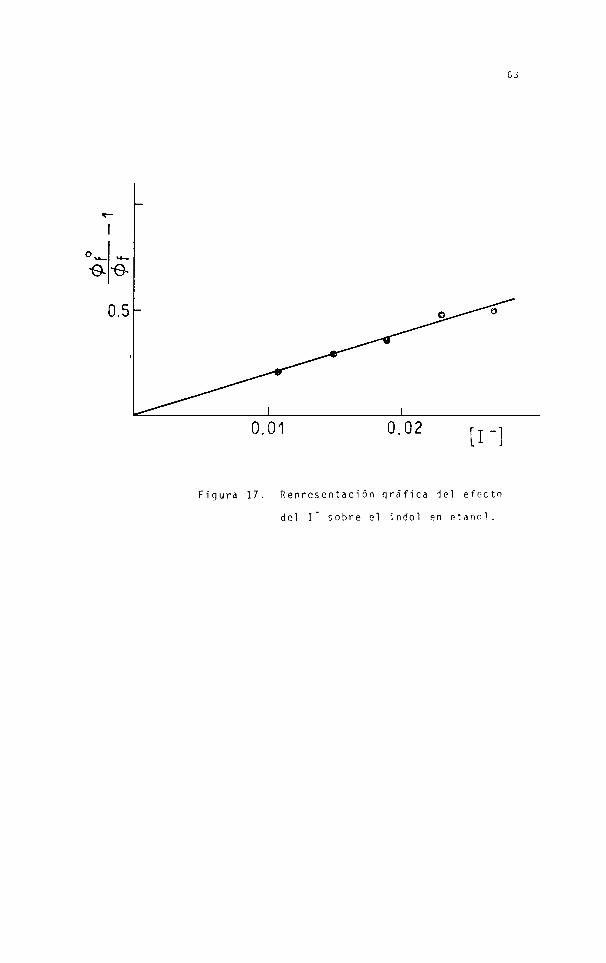
Figura 17. Representación qráfica del efecto
del I” sobre el indol en etanol.

6 9
5.3 Espectros de luminiscencia a 7 7°K.
Los espectros de emisión y las curvas de
decaimiento se barrieron usando el accesorio de fosforescen
cia y enfriando con nitrógeno líquido.
Las curvas de decaimiento de la fosfores
cencia se obtuvieron interrumpiendo la radiación de excitación
(280 nm) y graficando la intensidad versus tiempo para la lon
gitud de onda de emisión corres pond i en te a la banda 0-0 en el
espectro de fosforescencia.
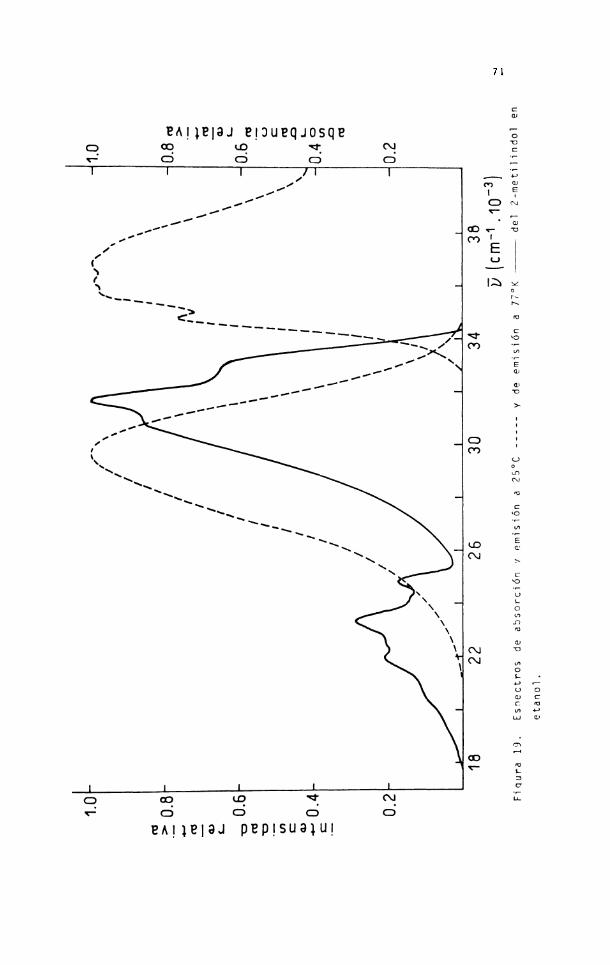
En las figuras 13, 19, 20 y 21 se repre
sentan los espectros de absorción y emisión a temperatura am
biente y el espectro de emisión total en etanol a 77°K de los
compuestos estudiados.
Los parámetros de emisión a 77°K se mues
tran en la tabla 14.
Tabla 14. Parámetros de la luminiscencia del indol y derivados a 7 7 ° K en etanol (excitación: 280 nm).
Fluorescenci a
Compuesto^em (cm’1)
' i
í i i
1 i
1 i
(^77°K‘^293°K)
Indol 31100 32600 33850 0,79 2100
2-meti1 i ndol 30800 31650 32800 0,68 1900
3-meti1indol 30500 31550 33300 0,69 2900
Lj jtriptofano 30700 31750 0,60 2050
Fosforescencia
Compuesto <pp p( s
Indol 24700 23900 23100 22200 21800 20600 0,12 6,6
2-metilindol
25050 24250 23450 22600 22050 20550 0,17 6,2
3 - m e t i 1 - indol
24000 22700 21400 20100 0,13 5,1
L( _) trinto í a n o
24600 23900 23200 22200 21800 20600 0,15 6,6
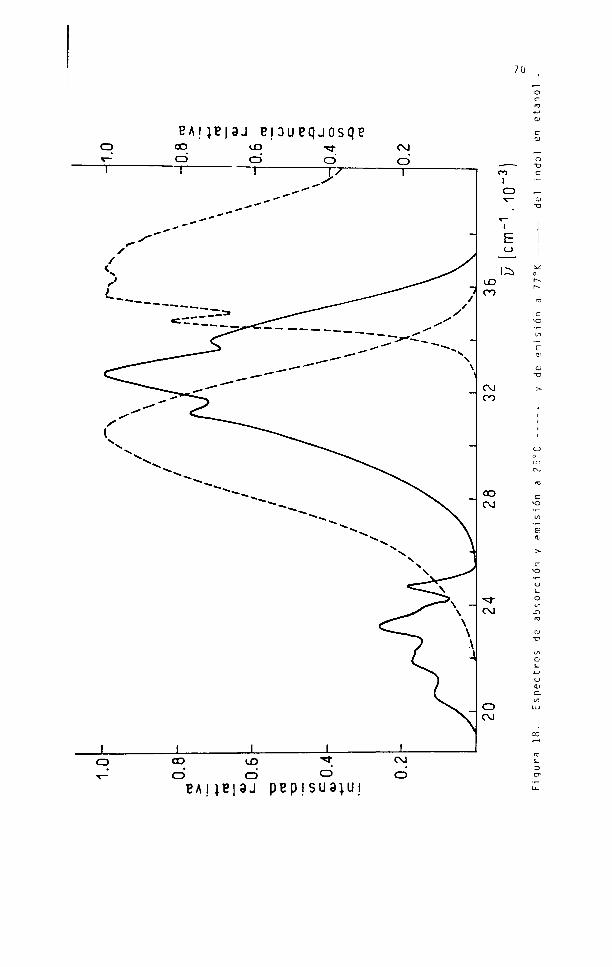
-H.0
J_________ I__________I----------------LCO UDCD CD ° CD
EAHB|3J PEPISU3|UJ Figu
ra 18.
Espect
ros de
abso
rció
n y em
isió
n a 25
°C ----
---- y d
e emis
ión a
77°K
-------
-- del
indo
l en et
anol
71

72
-M.0
cm o Fi
qura
2 0. Esp
ectro
s de ab
sorc
ión y
emis
ión a
25°C
-------
-y de
emis
ión a
7 7 °
K--
-----
----
- del 3-
met
ilindo
l en
EA I |e|9J PEPISU9JUI

7 3-H
.0
I CD
J__________ I------------------ L-oo coCD O O
I CXJ o Fi
qura
21. Esp
ectro
s de ab
sorc
ión y
emis
ión a
25'C --
-----
- V de
emis
ión a
77°K
-------
-- de
l L,_v
tript
ofan
o
BAI)E|9J pEpiSU9|UI

74
5.3.1 A.nálisis de los espectros
Enfriando a 77°K?los máximos de emisión
de fluorescencia de todos los compuestos experimentan un efec
to batocrómico muy grande.
Analizando el espectro del indol, aparece
un máximo a 32600 cm 1 con un corrimiento de 2100 cm"1 resoe£
to al de 25°C. A su vez, aparecen dos vibraciones bastante
bien resueltas a ambos lados del mismo: una a 33850 cm"1 y o-
tra a 31100 cm"1.
Para los derivados, los espectros de fluo
rescencia a 77°K también demuestran estructuras parecidas a
la del indol, sufriendo corrimientos de 2000 cm'1 resoecto a
los de 25°C, excepto en el caso del 3-metilindol cuyo máximo
se desolaza unos 3000 cm”1 (tabla 14).
Los espectros de fosforescencia muestran
varios picos bien definidos (tabla 14) con energías e intensi_
dades del mismo orden. Lo mismo ocurre con resoecto a la dis
tribución vibracional si se excita a otra longitud de onda.
La orimer vibración corresponde a la t r a £
sición 0-0 3l .d
Cuando se agregó yoduro de sodio, los es
pectros mantuvieron sus aspectos vibraciona1 es y no hubo va
riación en las intensidades.
Los espectros corregidos de excitación a
77°K presentan enerqías iguales que las de los mismos a 25°C,
lo que indica que el efecto de temneratura sobre el espectro
de absorción es nrácticamente nulo.

7 5
5.3.2 Efecto de temperatura sóbre la emisión de fosforescencia.
En la figura 22 se muestra la recta obte
nida para el indol disuelto en etanol cuando se representa la
ecuación (25) en forma logarítmica en función de la inversa
de la temperatura.
El valor de la energía de activación encon
trado fue de 0,29 kcal/mol y el del nreexnonenci a 1 (k^) de
Figura 22. Representación gráfica de la aplicación de
la ecuación (25) para el indol en etanol.

76
6 INTE RP RETAC IOM
Por el análisis efectuado del espectro de
emisión del indol en ciclohexano, se asigna la transición 0-0
a la banda de mayor energía por coincidir con la oanda 0-0 en
absorción; este so 1apamien to es una confirmación de que los
estado de equilibrio fundamental y excitado tienen la misma
conformaci ón.
Si consideramos la transición 0-0 de ab
sorción en el cristal a 34250 cm \ se observa que está des
plazada 500 cm 1 hacia el rojo respecto a la absorción corres^
pondiente en solución.
Está bien establecido por estudios de sol
vación, que las transiciones en solventes no polares están co
rridas hacia mayores frecuencias respecto a transiciones en
el cristal cuando las moléculas de soluto tienen la misma con
formación de equilibrio que el estado cristalino.
El efecto de solvente no polar es prácti
camente nulo y el nivel de emisión puede considerarse como un
nivel Franck-Condon , indicando también que el momento dipolari 4
de la transición L. <— A - es el mismo.o f
Además, el espectro de excitación corregí
do coincide en sus rasgos característicos con las dos transi
ciones <— y <—4Ay y existe una relación especular
parcial con el espectro de emisión, el cual no carnoia en su
contorno con la longitud de onda de la excitación, lo que in
dica la población de los dos niveles !La y
El desol azaini en to de la banda \ muestra a.
un corrimiento de 2500 cm’1, lo que está de acuerdo con lo es
tablecido por varios autores (3¿,37) respect0 a que e] moaen-

77
to dipolar de la transición es mayor que el de la ?L,a !o
De lo expuesto, se puede tomar como origen
de la banda L, a 34750 cm y de la banda a 34775 cm’^.o a
El origen de la banda es un valor ra
zonable comparado con el dado por Mani y Lombardi
3b232 cm en su estudio del espectro electrónico del indol
en fase gaseosa, por lo que el solvente tendría el efecto de
correr la banda en aproximadamente 500 cm~^.
La banda \ de 36050 cm en absorción a
está en franca coincidencia con la banda ^L dada por Kadiri, a r
(30)Martinaud y Cazeau-Dubroca los cuales recientemente hi
cieron un estudio de las bandas del indol a distintas tempera
turas.
El cromóforo metilado en posición 2 ó 3
demuestra pérdida de estructura en los espectros de emisión
en ciclohexano.
La banda 0-0 en indol se transforma en un
hombro con frecuencia muy cercana a la transición 0-0 en ab
sorción, siempre corrida unos cientos de cm'^ hacia el azul
con respecto al cristal del indol; esto hace suponer que no
hay diferencias entre la conformación de equilibrio en el es
tado cristalino del indol y estos derivados.
Las frecuencias vibraciona1 es correspon
den aproximadamente a las del indol, el rendimiento de fluo
rescencia es prácticamente el mismo y sólo se nota un ensan
chamiento de la banda de emisión dando una indicación del so
lapamiento de niveles superiores y de que existe un cambio en
la relación de los niveles que emiten.
El 2-metilindol presenta un corrimiento
de Stokes mayor que el 3-metilindol, lo que puede atribuirse,

78
además del efecto inductivo, a la posible interacción del
grupo metilo con los hidrógenos en posición 1 y 3, lo que da-
ría un balance de repulsión estérica y estabilización por re-
sonancia.
Para el 3-meti1indol . el origen de la banda :fL^ estaría en 34300 cm" 1 y e1 déla 'l
a en 33650 cm -1
Para el 2 -me ti 1 i n do 1 , el origen de la ban
da 1i L^ estaría en 34575 cm" 1 y e1 déla 'la en 34600 cm -1
Los espectros de emisión en etanol a tern-
pe ra tura amb i en te no poseen estructura ; por lo tanto es d i f í -
ci 1 extraer conclusiones de ellos.
Los rendimientos de fluorescencia a 77°K
se acercan a un valor parecido para todos ellos, lo que indi
ca la eliminación del mismo tipo de proceso no radiativo.
El corrimiento hacia el azul puede deber
se a que la matriz de vidrio bloquea parcialmente la rotación
del estado excitado y la emisión puede considerarse desde el
estado Franck-Condon.
La diferencia de 1250 cm"1 para el indol,
es una vibración del estado fundamental. La banda de mayor e-
nergía a 33850 cm"1 está separada 800 cm 1 de la banda a
34650 cm”1 del espectro de absorción en solución. El promedio
da un valor de 34250 cm"1, en coincidencia con la transición
0-0 del cristal, lo que indicaría una pérdida de la energía
por desactivación vibracional representada por 400 cm
El gran corrimiento de los máximos a las dos
temperaturas extremas estudiadas indica que la reorientación
del solvente a baja temperatura es parcialmente impedida.
Si se toma el origen de las bandas como
el promedio de los máximos de absorción y emisión da un valor

79
nara el indol a 25°C de 33550 cm’*, muy próximo a la vibración
de mayor energía del espectro de emisiÓrr a 77°K; nara el 2-me
tilindol, 3-metilindol y el triptofa no el origen correspon
dería a 33225, 33050 y 32600 cm respectivamente.
En general, los espectros a baja tempera
tura tienen una imagen especular con el espectro de absorción.
El espectro de emisión del trintofano a
77°K tiene una característica: pierde la estructura en la zo
na de mayor energía de la banda, la frecuencia vibracional
que existe en los otros compuestos desaparece y el efecto de
temperatura es mucho mayor. Esto se manifiesta sobreponiendo
los espectros de emisión a las dos temperaturas, oudiendo así
observarse que a 77°K hay una contracción de la banda, lo que
no ocurre en los demás compuestos. Además, el espectro es la
réplica especular de la región identificada como en el es
pectro de absorción, indicando que este estado es el mayor e-
mi s or .
Salvo esta característica del trintofano,
los demás compuestos tienen espectros de emisión muy semejan
tes en solventes no polares a 25°C y polares a 77°K.
Comparando los tiempos de vida radiativa
y los obtenidos por absorción (tabla 10 y 11) se observa que
sólo guarda relación con la polaridad del solvente: aumen
ta en etanol y es del mismo orden para los diferentes comoues^
tos en cada solvente; mientras que'Up, por el contrario, mués
tra una disminución cuando se nasa de ciclohexano a etanol .
En ciclohexano se observa que Tp es mayor
quet° : unas seis veces en indol y aproximadamente el doble en
I os otros deri vados.
En cambio, en etanol y son anro-

80
ximadamente del mismo orden para el indol suponiéndose que lo
mismo ha de suceder para los otros derivados.
La diferencia encontrada para el indol en
ciclohexano puede interpretarse que las transiciones que invo
lucra la fluorescencia emiten en una relación distinta a la
que corresponde a las fuerzas de los osciladores en absorción.
Además, esto está de acuerdo con la imagen
especular parcial que se ha encontrado entre los dos espectros
a que se ha hecho referencia en la sección 5.2.
En solvente polar como etanol, el despla
zamiento masivo de la banda hacia mayor longitud de onda
colocaría a esta banda a menor energía que la ^L^» de ahí que
la emisión podría considerarse proveniente principalmente de
esta transi ci ón .
La energía de activación del triptofano
en alcohol respecto del proceso de apagado de la emisión, en
el orden de temperatura estudiado, indica una constante de ve_
locidad (tabla 12) mayor que la de los otros compuestos, pero
dentro del orden de la constante de velocidad de la fluores
cencia .
El valor del factor de frecuencia es de
1011 para el triptofano y 109 para los otros compuestos. El
valor de 1011 se aproxima más a los procesos permitidos que
a los prohibidos; por lo tanto, esta energía de activación co
rrespondería a la desactivación por conversión interna (kp)
involucrando algún modo vibracional del estado excitado, cami_
no que lleva a una disminución del rendimiento de fluorescen
cia.
El triptofano presenta el menor valor de
en un medio como etanol y conjuntamente el menor valor de

di
4>f (tabla 11).
Teniendo en cuenta que el rendimiento y
decaimiento de la fosforescencia de los compuestos son prácti
camente iguales, los dos caminos posibles de desactivación se
rían k y k ; si se aplica la fórmula
de <$>t.
La diferencia de energía entre el primer
estado singlete excitado y el primer estado triplete da un va_
lor de aproximadamente 8000 cm"^ para todos los compuestos.
La energía de activación calculada dió
100 cm 1 (0,29 kcal/mol) con un factor de frecuencia muy pe
queño. La separación entre los niveles singlete (sp y triple-
te (T|) es un valor grande para que el proceso de transferen
cia de energía s ea di recto al nivel T^; por lo tanto debe exis
tir un nivel superior con modos vibraciona1es solapados con
los de o , lo cual facilita este tipo de transferencia,
explicándose así que el rendimiento de fosforescencia sea
igual en todos los compuestos. Asimismo, el bajo valor de la
energía de activación indica una desactivación independiente
de la temperatura —>SQ.
Los valores obtenidos de los picos e in
flexiones en los espectros de estos compuestos se han compara
do con el espectro Raman del indol dado por Heckman y
con el espectro del cristal dado por Zuclich (tabla 15).
Las frecuencias vibraciona1es que corresponderían al estado
fundamental son las mismas, lo que indicaría que el efecto de
la sustitución no afecta la conformación de este estado. La
ello indicaría un incremento

82
frecuencia más fuerte, de 1600 cm‘^, para indol corresponder
a la vibración OC del ciclo bencénico dado por Bellamy
aunque la vibración de deformación del grupo >.*iH tiene la
misma energía.

Tabl
a 15. Com
para
ción
de la
s frec
uenc
ias v
ibra
cion
a1 e
s obte
nida
s para
el in
dol y
deriv
ados
con las
de
l cris
tal de
indol
y de
l espe
ctro
Raman
(40).

d4
CONCLUSIONES
La investigación sobre la polarización d
la absorción y la emisión del indol, realizada por varios au( 3 4 9 )tores * ’ , y el estudio efectuado por nosotros sobre 1
espectros de excitación correqidos (absorción) que can una i
magen especular respecto al de emisión, es un argumento más
que confirma que los dos niveles están emitiendo simultánea
mente.
El cambio de la frecuencia de excitación
no altera la distribución de intensidades de los esnectros y
además las medidas experimentales realizadas por varios auto
res que han encontrado una curva exponencial de decaimiento
de la fluorescencia, todo lo cual concurre a concluir que e-
xiste un estado de equilibrio térmico entre los niveles
y en todos los compuestos estudiados.
La absorción de luz en estos derivados
conduce a la población de dos estados muy cercanos energetic
mente de una dada multiplicidad ñor lo que presentan nroniec
des luminiscentes que no son comunes a todas las moléculas.
Los fenómenos de emisión pueden ser renr
sentados por medio de dos procesos: uno desde un estado Frar
Condon, que se produciría en solventes no polares o polares
estado vitreo, y otro que se produciría en solvente nolar a
temperatura ambiente, donde hay una fuerte reorientación del
mismo con el estado ^L .a
Si se supone que se alcanza el equilibri
a través de una coordenada de relajación vibracional y una c
ordenada función de la reorientación del solvente, los procc
sos se pueden representar, sin hacer distinción entre los es

85
tados, de la siguiente manera:
en ciclohexano o en solventes polares en estado vitreo y
en solventes polares a temperatura ambiente.
r representa la coordenada de reorientación por el solvente
y v la de relajación vibracional.
En sol ven tes no polares, la banda de absor
ción a mayor energía está compuesta por
po r
dos frecuencias viora-
850 cm 1. Esta bandacionales bien definidas y separadas
asignada a la transición es bi
po cromóforo y resiste cualquier perturbación por efecto del
en es pee i fi ca del gr u-
solvente o temperatura (según se lo na comprobado por medio
del espectro de excitación). La modificación de esta banda en
el solvente polar es debida al corrimiento que sufre la tran
sición IL^ <— 7A al ser perturbada por solvente o sustitución,
lo que la hace solapar en mayor o menor medida hasta un punto
tal que esta transición puede llegar a ser la de menor energía.
Las características de los espectros y los
parámetros derivados de la fluorescencia son propiedades úti
les para controlar los cambios que ocurren en el medio que ro
dea a las proteínas, principio de aplicación en el campo bio
lógico, ya que ellas presentan las propiedades luminiscentes
del aminoácido del cual derivan.
De los resultados obtenidos se puede infe

86
rir que el mejor solvente de los dos usados para el estudio
de estos derivados es el etanol cuando se trabaja a 77°K. A
esta temperatura la sensibilidad de la emisión es diez veces
superior a la de la temperatura ambiente y cien veces respec
to a la que se alcanza en absorción.
Los espectros de fluorescencia a 77°K son
característicos de cada derivado, con máximos de emisión d i f £
rentes para cada uno de ellos y son análogos a los que se ob
tienen en solventes no polares a tempera tura ambiente.
Los máximos de emisión en etanol a tempe
ratura ambiente son también carae terísti eos de cada derivado.
El efecto de apagado por I sobre el ren
dimiento de la fluorescencia a temperatura ambiente, con una
constante de Stern-Vo 1 mer determinada, es un elemento más de
juicio para la identificación de los compuestos.
La característica que iiiani fiesta el
de yoduro, a diferencia de los
Ltriptofano de mantener constante el valor del rendimiento
de fluorescencia en presencia
otros derivados considerados, permitiría desarrollar un méto-
do analítico de de te rmi n ac i ón de aquél en presencia de alguno
de los otros.
Los espectros luminiscentes han permitido
obtener una serie de parámetros asociados con el estado exci
tado de los compuestos estudiados (tabla 16). Sobre la oase
de ellos, se representan en la figura siguiente los valores
obtenidos para la molécula del L_jtriptofano en etanol.


88

89
APENDICE
Programa CINEF
C INEF CEFIMCICN DE VAR IABLES
NPUNT : NUMERO OE PINTOS DE LA CURVA CE CALIERACICNFOTCT : TIPO DE FOTOTUBOFC : FACTORES DE COFRECCICNRLC : LONG. DE ONDA DE LCS FACT. DE CCRRECCICNNPROBL : NUMERO DE PROBLEMA (IDENTIF ICATCRIC)ITERM : NUMERO DE PUNTOS DEL ESPECTRO PRCBUERAFUI : INTENSIDAD DE FLUORESCENCIARUF : UONG. DE ONDA DE FLUORESCENCIANOTA : UOS VAUORES DE UAS VARIABUES DEBEN SER INTPOCUCIDOS EN ORDEN DECRECIENTE DE LONGITUDES DE ONDA

90
F1R
TRA
N IV
G LEV
EL 21
FAIN
O
ATE
= 772
46
10/1
4/05
'J)l
INTF
GER
-2 R E
NK
1 rl
I / • ,
<? *
*,9í«
•,»
. *,9
0« «,«
. • ,9
4:»
»,1*
. ’.9
**
• ,S*'
»,*.
»,9*
» * ♦
’. ’»*.
• ,9*«
»t»
. »,
2 9 ¿
,•r ’ /,
3FEN
2(1
0 1 )
/ ’ O
',’
1 »,
»2
»,9*
'. ’,'3
’,9*’.
',’
4 »,49
*'w ’»’
5 ’,9*’.
• ,
• 6. «t9
*'.
',’7 ',9
*».
','8 ’
,9*»
. ’.’
9 ».59
* ' . ',
'O '/,
IAST
EP /
• * ' /
» ' J2
CIH
FNSI
ON
FC ( 1
00 ) ,
WLC
( l'»
¿)) t
F L I
( 109
) ,H
F(in
i , FR
EC (1
O )
) ,C I
( 100
),1 C
HA
D(3
) ,D
CU
AD
(2) .F
üTC
TÍ 5
)i )
> 3 D
IMEN
SIO
N PQL
( lD
rG ) ,
FE E
CL
(loC
*l ) ,
V» A
VE ( 1
J V’) )
, A F
E A ( 1
006 )
, C
UeC
FP ( 1
0 310
),SC
ATA
(V')O
)•
J>4
CO
MM
ON
OFD ( 5
) ,A
BC
(5) ,
PGLI,
INTG
C C
CO
NTR
OL CE
L FAN
GO
Y CFD
FNA
MIE
NTO
DE LC
S PAFA
FETR
CS CE
LA C
UR
VA CE
C
CO
RR
ECC
ION
C)J
..)5
NPA
SC=3
)J >ó
FEA
C (5,
1)
NPU
NT ,
(FO
TOT(
I ) ,1
=1,
5)) )
)7
1 FOE m
at ( I 3
, 5A
4)i)J
3 FE
AC
(5,91
(FC( I
) ,WLC
(I 1 , I
=1 ,N
PUN
T)U
)9
9 FOF w
AT(
8 ( F
6.3,
F4.0
) )U
IJ
1=1
.'11
2 IF ( I.
EO.l)
GO TC
3JJ
12
IF (UL
C< I
• 1 ) .
LE.V
IC( I
) ) GC TC
4) 11
3 IF
( I.NE.
NPU
NT)
GO TC
3j j
_ 4
GO
ID 6
3)15
3 1 =
1 + 1
))16
GO
K 2
i.*17
4 WR
I T E
( 6 , 5
)
JJlrt
5 FJ
FMA
T ( lh ,'L
OS VA
LOR
ES CE
LCN
G. DE
CN
CA
DE C
DFF
ECC
ICN
ESTA
N MAL
70!- D
ENA
DO
S’ )
>319
ST
OP
C C
CO
NTR
OL DE
L FAN
GO
Y CR
OEN
DF IO
S DATO
S DEL PR
CB
LEM
AC
.) ) 2 >
6 FEA
T (5,F,
ENC
= 2
0C ) NP
RO
RL,
ITFR
M) )2
i 8 FOR
WAT ( 2
13)
> *22
FEA
C (5,
888)
(FL I
(J 1 ,
VLF(J
) , J =
1 , I I
FRM
)) 2 3
888 FO
FMA
T (8(F
5.1,
F5.1
)).» >2
4 J=
1•
>2 5
7 IF (J
.EC
.l) GO
Tr 12
IF (RL
P(J 1 )
. LE
. W L
p ( J
) ) GC TC
13Jd
.>7
IF (J.N
F.IT
ERM
) QC TC
14j >2
8
IF ( RL
F ( J
) . G
E.W
L C( N
PLN
T 1 1 G-
n To 2
0»>
2 9
GO
TC 15
U3)
12
IF
< VL C < 1
) • G
E . V
mL F
( 1 ) >
GO
TC
14
031
1 5
UF I
TE ( 6
, 17 )
0)32
17
FO
RM
AT (IF
,«HA
Y VALC
FFS
CE
LCN
G. DE
CN
CA
CUE CAE
N FUF
FA CEL
FANC
GX DE
LA C
UFV
A DE
CA
I I B
F AC
I CN
• )))3
3 ST
OP

91
3 3 34
1 3
V F I I
F ( 6 , 1
8 )
”35 7 8
XF8r
CEN
- ) 1H
’’Lr,S
''4LC
PES CF
LCNG
- nE C
NC
A DEL
PECB
LEPA
NC ES
TAN
EN>¡
36
^rIIE
<6,99
9) UL
F(J)
tJ3 3
37
999 FO
R MA
T <F 1
2.2
, I 8
I>J
38
STO
P>3
39
14 J =
J + 1
3343
G
O TC
70 C C
CO
MIE
NZA
LA IN
TER
POLA
CIO
NC
3 341
2)
IF( N
PASC
.NE.
OG
O TO
1633
42
R R- I
T E (
6 , 1
9 )Ía ^
"u:2%
g^f;
í^l%
7h^:
í/ncp8
.t>'
’5x’'lc
ng
- ^dí,)
•J45
26
FOEV
AT(
T42,
I5,lO
X.Fe
.3,8
>,F7
.O)
J3**
7 27
FOR m
A T
( / / T
5 7 ,
’ I N
T. F L
LO
F . * ,
5 X
, * L C
NG
. CNCA
»)>
w I T
E ( 6
,28
) ( J ,
FL I (
J ) ,
ULF
( J ) ,
J =
1 , I
TER
m )
*349
28
FOR w
A T
( T 4
2 , I
5 ,10
X ,F
8.1
, 8 X
,F 7
.0 )
.) J *) I
1=9
JJ!?
1 V.
PÍTE
(6,132
>)J
>2
1a'tO
Í2xII
vAL.
5,IÑ
kfP!
Í)S DC
IATF
FpC
LAC
IGN
-//T5
.,-LC
NG
. GNC
A-.5
X.-G
F’3
53
CO
E?*\ =
i ,I T
ERM
J]?*
* l
= L il
LF<K
) WLC
{LH
21,22
,23
3 3 9
6 ‘ G J ’ IC
433
97
22 INT
C=i
^jn^
[fi,K
’"LF
(K,’i
Ms’
Fc,L
’JJ
'j.'
GO
TC n2
J )6L
?3
INT =
L 23 3
62
Cl 2 5
1 = 1
,5' >6
3 IT
( I NT+
5.G
T.N
PUN
T)G
0 TO 3(
’ ’64
Ok í)
( I ) =
F C ( I
N T
♦ I )
) J 6
5 A
BC
(I)=k
lC(lN
T+I)
t J c>
6 G
2 TO 2 5
’ )67
3 ' OH
) ( I )
=FC
(I • I +
l)' 3
68
ABC
( I ) =
v>LC
( L 1 + 1
))j
j9
25 C IN
I IN
U-
' ’ 7 j
APR
C x=
IR D ( 2
) / 2 )
•) 1
71
CA
IL INT
ER n
( RLF
( K ),
AP
R ilX
)’ '7
2 w
RIT
l(6,
29)k
,ViL
F(K)
,INTG
,F0L
I3J
7j
29 E OR
w A
T( Tf
;3, I
4 ,F1
1 . 1
, I 1 )
, F 1
2. 3)
C

9 2
c C
CA
LCLL
O OF
LA CO
RR
ECC
ION
C C
O7‘f
82
C I ( K
) = P
OL I
*F L
I ( K
) *( V»
L F
( K ) /
40C
. ) *
4 2
JJ7
5 FT
EC
(K ) =
IOD
O./W
LF(K
)O
7o
86 CO
NTI
NU
EJ»
77
WR
ITE (6
,111
) NPFO
BL,
FOTC
T07
8 11
1 FOR
MA
T ( 1H
1 , 4 8
X , ’
E S p
EC
TP C C
OR
R E
G I
CO
•/17H
PRCPL
EMA
NUMER
O , I 3
♦60X
,X9
HFC
T0TU
B0 , 5
A4)
C C C
INTE
RPO
LAC
ION
DE PU
NTO
S CCR
REG
IDC
SC C
.079
LI
NE=
jo8
1 K
=203
1 L=
108
2 12
• FF
ECl(L
) = F
FRC
(l)+e
.Cl*(
L-ll
033
121
IF(F
REC
U(L
)-FR
EC(K
)) 12
5,12
6,12
71)
84
125
INT=
K 2
0 3 5
CO
115 1=1
,5O
Bo
IF( IN
T +
5.G
T. IT
ER N
)GO
TO
11608
7 O
R D
( I
) = C
I(INT
+ I )
) *88
AB
C (
I ) =
FF E
C ( I
NT+
I )
; H9
CO
TC 11
5J >
9)
116 O
RD
( I ) =
C I
(K 1 + 1
)• )
9 1
AB
C ( I
)=FF
EC ( K
I + 1 )
092
Uc CO
NT I
NU
E09
3 A
P R G
X =
A H
S ( (
3 R D
( 5 )
O
R C
( 1) )
/O'C
. )J O
4
A 8
S I =
F F
E C U
( L )
1199
C
ALL
INTER
P( A
BSI
, APR
CX
I) *9
o p IL
( L ) =
PO
LI09
7 IF
( PC
L ( L
) .L T
.'J. )
POL (
L ) =
»•, I 9
8 L
= L +
109
9 G
O TO
12C
»1M
12
6 P IL
( L )=
0R D
( K )
j 1 • 1
IF ( K
.EQ
. I T
ER *
)G0 TO
12 9•1
12
127 IF
(K.E
Q.IT
ERM
) GO TC
128I
1j i
K =
K 4
]J1
J4
GO
TC 12
1C C
F -G
IL A
R I I
AC
I O
NC.
•i'5
12« L=
L 1JO
O
126 CA
LL SMO
OTH
( L ,P
OL ,
M t S
DA
TA )
> 1 )7
0 1 I C
O
I = O
m0'
8 FF
CC
K
I ) =
FF E
CU
I +4)
II >9
C
UB
O FF
(
I ) = S
D A
T A ( I
)/F
F EC
U ( I
) ¿ *
3lil
i F A
VE ( I
I
= 1'0
'1. /
FF E
CU
( I )
• 111
1 O
CON T
INU
E

93
C C
INTE
GR
AC
ION
C>1
12
CA
LL QSF
( r. )1
,SD
ATA
,AFE
A ,M
) 1 1
3 AR E
A 1 =
AR
EA( M
)'1
14
CA
LL OS
F(C
.O 1
.CU
BC
FF ,A
FEA
,M)
*115
A
F EA
2 =
AF E
A ( M
)C C
NO
RM
ALI
ZAC
ION
C>1
16
OFD
MA
X=r.
1117
DC) 1 5 •
? k = 1
.m>1
18
IF(S
CAT
A(k)
.GT.
C)F
DM
AX)O
I-DM
AX
= SD
ATA(
K)•1
19
150 CO
NTI
NU
E>1
20
FAC
NC
F= 1
./OF
CM
AX
J 121
C
O 14)
J=1,
M>1
22
SDA
TA(J
)=SD
ATA
(J)/O
FDM
AX
>123
PO
L I=S
DA
TA( J
)>1
24
NPA
Sn=l
>125
11
10 IND
= PO
L I *
1DD
.*1. 5
>126
IF
(IND
.LT.
1) IND
= 1
•127
IF
(IND
.GT.
1j1)
IND=1
C1
J128
IF
(LINE.
EQ.0
) GC TQ
1112
3 129
LI
NE=
l.IN
F 1’1
31
IG=R
EN
K IN
D )
•131
R
FNK
IND
) = IA
STEF
’I?2
,, , , I 3E( 6
, 111
1 )PO
L I ,F
EM
, V A
VE ( J
)>1
33
1111
FIRM
AT (8
X,F6
.4,2
X, 1C
1A1 ,
F7. 1
)>1
94
F EN
1 ( IN
C ) =
1G
>135
G
O TC
140
•13b
11
12 IG=
PEN
?(IN
D)
■> 1
3 7
L IN
E =
9>1
38
FEN
KIN
D ) =
I AST
EF'1
?9
FF I T
E ( 6
,1 I
1 3 ) F
F ^fU
i J ) ,
PO
L I , F
FM
, F A
VE ( J
)nt
; 11
13 ^
“nnn
,rA-2
’Fq-4 -
2x’14
2 14
) c )N
T IN
UE
>14
3 W
't-IT
E (6,1
114)
F rN2
'144
11
14F)
FMA
T(16
X,1)
1A1)
''1
45
PR PR
WQ =
AF F
A1/
AR
F A
2‘1
n Wk I TE(
6, 13
• ) A
PEA
1 ,F
F PF
M3 t
F AC
NC
PJ1
13
\ XG
CO
GC
DC
nPC
Oni
. Z:’^
12-5
-Tfl5
,.INVE
PSA
CE <F
FEC
*“ T>
*14
8 G ) TC
a '
’ " ’
* ’ '
'• 1
49
? * ) M'
)P•1
5 ;
END

J4
c C C
SUB
RO
UTI
NE SM
OO
TH 9 P
OIN
TC
j i il
SUB
RO
UTI
NE SM
OO
TH ( N
, ND
A T
A ,
M , m
0 A
T ¿ )
CJ_
)o2
F-FA
L ndat
a,mc
ata ,
np.n
sum
) J 0
3 D
IMEN
SIO
N ND
A T
A( Id
JO ) ,
MQ
A T
A ( 1
b r) )
,NP(
9)C C
SEG
MEN
TO DE
INI C
I A
L I Z
AC
I C N
CjJ
>4
M =
N- E
) J j
5 D
O 1G
I = 2
,9JO
>6
J=I 1
j 007
1 ) N
P( I
)=N
CA
TA(J
)C C
SMO
CTI
NG
LOOP
Ci J
)8
CO
20
0 1 = 1
,MJ J
J 9
J = I
4 6
J J 1
J D
O
1 1 K =
1,8
j)ll
KA
=K«1
) il2
11 NP
(K )=
NP(
KA
)) )i
3 N
D( c
) = N
CA
TA(J
)01
4 N
SU M
= 5
9''N
P ( 5
)*54
KN
P(4)
4NP(
6H4-
39*(
NP(
3)4N
P(7)
)414
«(N
P(2)
*NP
(8))~
121*
(Np(
1 I +
NP(
9) )
>015
m
oat
A( I
)=N
SUM
/231
)J16
2 )0 CO
NT I
NU
E00
17
RET
UR
N00
18
END

9 5
MJ1
SU
BR
OU
TIN
E INTE
R P(
VAF ,
EFFC
P )30
'2
CO
MM
ON
Y(5),X
(5),P
OLI
,INTG
J J J
3 M
- 1J )
J4
mm
= 1
J 3 ) 5
M
MM
=
1) 0
I 6
X X =
> ( 3
)33
J7
DEL
P1=
X ( 2
) X( 1 )
• > »8
4 )
DEL V1
= XX
-X( 1
)3
>9
POL I
= Y (
1)*
(Y(2
)-Y(1
> )/D
ELPl
*DEL
Vl33
13
Gn TO
( 500,
510
,520
) ,MU
li 5n
) A2=
\ (3)
PCL I
3)12
I F
( A P
S ( A
2 ) •
L T •
Ec F
OR ) GO
TC 5C
13)
13
x X
= X (
4 ))3
14
M=3
)J15
C
O TC
400
U16
53
1 XX=V
AF
)317
m
=233
18
INTG
=13)
19
GO
TC 40
C3 3
23
520
CEL
P2=(
X(3)
X(
l) )♦
( X(3
) X(
2)
)3)
21
DEL
V2=
DEL
V1*(
XX-X
(? ) )
3 >22
PO
LI =
PO
LI*A
2/0E
LP2*
DEL
V2) >
23
GO
TO ( 5
3C,5
10,5
40) ,M
MM
24
531
A3=
Y(4)
POLI
))25
I F ( A
E S (
A 3 )
• L T
• ER
F O
F ) GO
TC 53
1.» ) 2
6 m
m= t
3327
X X
= X( 5
)3)
2 8
CO
TC 43
0M
29
531
XX=V
AF
)3 3O
M
M=?
J > 3 1
IN
T G
= 2
J)32
G
O TC
4?n
M 3
3 54
3 rE
LP3=
(X(4
) X (
1 )
) ♦ ( X
( 4 )
X ( 2
) ) *
( X ( 4
) X(3
))J)
34
OEL
V? =
DEL
V2*(X
X•X(
3) )
3*35
PO
LI=P
OLl
4A3/
DEL
P3*D
ELV3
))36
GO
TO ( 5
5C,51
C,B
6C)
’ » 37
55 A 4= Y
(5 > •
PCI I
33.3
8 I F
( A P
S ( A
4 ) .
L T. E
R F
OR ) GO
TO 55
1)J
39
X x= V
AR
) )4 >
))4
1 IN
TG=4
J ’42
GJ TO
4
DC
3)43
55
1 XX=V
AR
3344
m
mm
=2> >
45
INTG
= 3
)3 4
6 G
’1 TO
4< C
3347
56
) DELP
4=( X
( 5) ■
X(1 )
)★( x
(5) X ( 2
) ) ♦(
X ( 5
) - X
( 3 ) )
* ( X
( 5 )
- X U
) )D
48
CEL
V4
= D
ELV3
*(XX
X(4)
))J
4 9
P'JL
I = P
OL
I 4A
4/D
EL P
4-'D
ELV4
J)5j
51
■ RET
LFN
M51
EN
D

96
BIBLIOGRAFIA
1- E.H. Strickland, J. Horwitz y C. Billups, Biochemistry,
9, 4914 (1970).
2- J.R. Platt, J. Chem. Phys., 19, 101 (1951).
3- H. Zimmermann y N. Joop, J. E1ektrochem . , 65, 61 (1961).
4- P.S. Song y W.E. Kurtin, J. Am. Chem. Soc., JI, 469 2 (1969)
5- B.L. Van Duuren, J. Org. Chem., 26, 2954 (1961).
6- N. Mataga, Y. Torihashi y K. Ezumi, Theoret. Chim. Acta,
2 , 158 ( 19 64 ) .
7- E. Vander Donckt, Bull. Soc. Chim. Beiges, 78, 69 (1969).
8- A. Kawski y J. Czajko, Z. Jaturforsch, 29a, 84 (1974).
9- S.V. Konev, Fluorescence and Phosphorescence of Proteins
and Nucleic Acids, S. Udenfriend, Tr., Plenum
Press, N. Y. , ( 1967) .
10- M.S. Walker, T.W. Bednar y R. Lumry, J . Chein. Phy s . ,
45, 3455 (1966)
11- M.S. Walker, T.W. Bednar y R. Lumry, J. Chem. Phys.,
47, 1020 (1967).
12- M.S. Walker, T.W. Bednar y R. Lumry, Mol. Lumin., I nt.Conf
Ed., Benj ami n ,N.Y. ,
135 (1969).
13- M.S. Walker, T.W. Bednar, R. Lumry y F. Humphries, Pnoto-
che m. Pnotobiol., 14, 147 (1971).
14- S. Freed y W. Sal in re, Science, 128, 1341 (1958).
15- C.A. Parker y C.G. Hatchard, Trans. Faraday Soc.,
59, 284 (1963).
16- Q. Stern y M. Volmer, Phys. Z., 20, 183 (1919).
17- American Instrument Co., Inc. (U.S.A.), Manual de instruc
ción 768 G, 959-Al y 904-A.

97
18- P.A. Leighton, R.W. Crary y L.T. Schipp, J. Am. Chem. Soc.,
53 , 301 7 ( 1931).
19- W.H. Meluish, J. Opt. Soc. Am., 52, 1256 (1962).
20- C.A. Parker y W.T. Rees, The Analyst, 85,587 (1960).
21- W.H. Meluish, New Zealand J. Sci. Technol., 37, 142 (1955).
22- J.J. Hermans y S. Levinson, J. Opt. Soc. Am., 41, 460 (1951)
23- S.J. Strickler y R.A. Berg, J. Chem. Phys., 37, 814 (1962).
24- J.F. Magallanes, L. Bruzzone y M.E. Roselli, Anales Asoc.Qui
Argén tina, 65,23
( 19 77 ) .
25- A. Savitzky y M. Golay, Anal.Chem., 36, 1627 (1964).
26- H. Suzuki, Electronic Absorption Spectra and Geometry of
Organic Molecules, Academic Press, N.Y., (1967).
27- J.M. Hollas, Spectrochim. Acta, 19, 753 (1963).
28- Y. Yamamoto y J. Tanaka, Bull. Chem. Soc. of Japan,
45 , 1362 ( 1 972 ) .
29- T. Kane da y J. Tanaka, Bull. Chem. Soc. of Japan,
49, 1799 (1976).
30- B. Valeur y G. Weber, Photochem. Photobiol., 25, 441 (1977)
31- L. Bruzzone y M.E. Roselli, Anales Asoc. Quim. Argentina,
65 , 107 ( 19 77 ) .
32- B.E. Rivero, A.D. Podjarny, E.E. Castellano y M.E. Roselli,
Acta Cryst. , B 36 , 1 726 (1980).
33- K.K. Turoverov, Opt. Spectrosc., 26, 310 (1969).
34- E.P. Kirby y R.F. Steiner, J. Phys. Chem., 74, 4480 (1970).
35- C.A. Parker, Photo 1uminiscence of Solutions, E1sevier , ( 1968)
36- G. Jacyno y A. Kawski, Bull. Acad. Polon. Sci., Ser. Sci.,
Math. Astr. e Phys., 21, 289 (1973)
37- M. Sung y P.S. Song, Photochem. Photobiol., 25, 3 (1977).
38- A. Mani y J.R. Lombardi, J. Mol. Spect., 31, 308 (1969).

98
39- A. Kadiri, M. Martinaud y C. Cazeau-Dubroca , Chem. Phys.
Letters, 65, 484 (1979).
40- R.C. Heckman, J. Mol. Spect., ¿ , 27 (1958).
41- J. Zuclich, J.V. von Schutz y A.H. Maki, J. Am. Chem. Soc.
96 , 710 ( 1974).
42- L.J. Bellamy, The Infrared Spectra of Complex Molecules,
J. Wiley & Sons , Inc. , J.Y. , ( 1956 ) .