membranopaties de l’eritrÒcit€¦ · imatges cedides per j. carrillo farga. citoesquelet de...
TRANSCRIPT

MEMBRANOPATIES DE L’ERITRÒCIT
Ines RodríguezInstitut Català d’Oncologia
Hospital Germans Trias i Pujol14 de novembre de 2013

MEMBRANOPATIES
• Esferocitosi hereditària (EH)
• Eliptocitosi congènita (EC)
• Ovalocitosi del Sud‐Est Asiàtic
• Transtorns congènits de la permeabilitat iònica

MEMBRANA PLASMÀTICA
Característiques:
‐ antigèniques
‐ de transport
‐mecàniques
Imatges cedides per J. Carrillo Farga

CITOESQUELET DE L’ERITRÒCIT
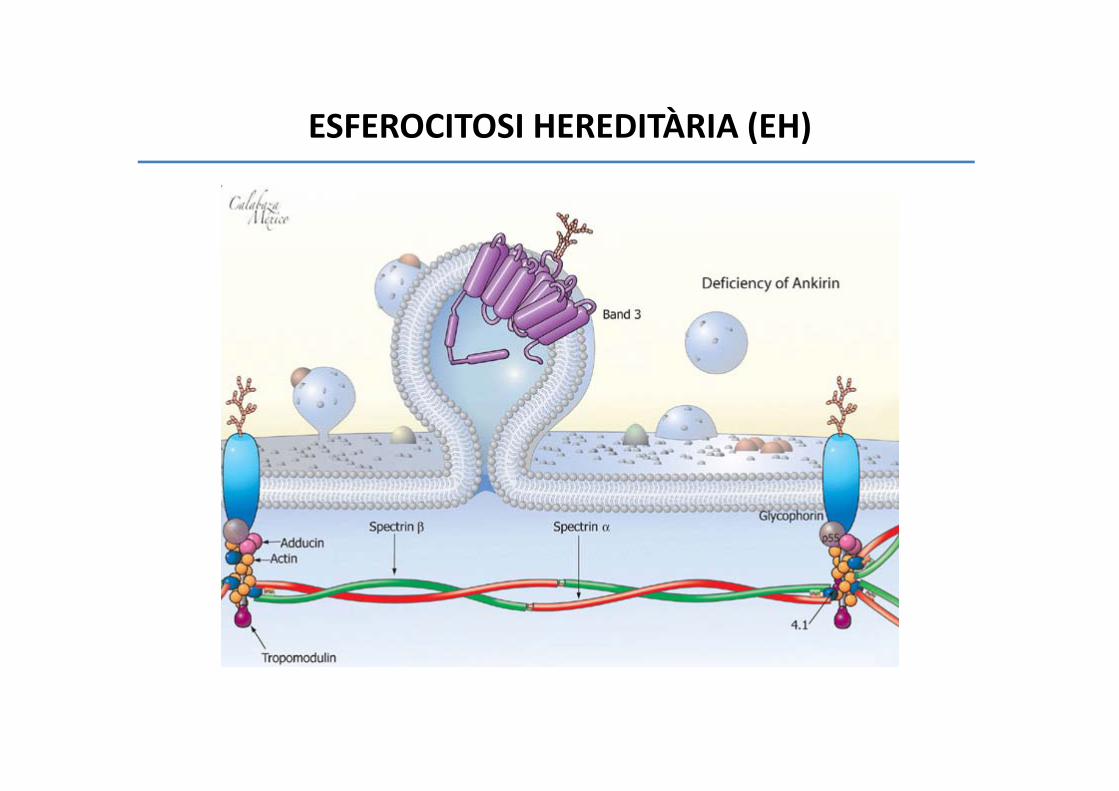
ESFEROCITOSI HEREDITÀRIA (EH)

• 75% herència autosòmica dominant
• Origen caucàsic
• 1/1000‐ 1/5000 casos
• Anemia hemolítica moderada‐greu
• Icterícia variable
• Esplenomegàlia
• Colelitiasi
EH: característiques

Mariani M. et al. Haematologica 2008
EH: característiques
N= 300 pacients amb EHClassificats segons SDS‐PAGE

• Esferòcits a frotis: 97% pacients
– Fragilitat osmòtica augmentada
– Disminució de la deformabilitat
EH: característiques

EH: frotis de sang perifèrica

EH: diagnòstic
‐ Test del glicerol acidificat‐ Resistència Osmòtica Eritrocitària (NaCl) immediata/ incubada 37º‐ Test de la criohemòlisi
‐ EMA‐binding test ‐ Ectacitometria
Bianchi P. et al. Haematologica 2012

ELIPTOCITOSI CONGÈNITA (EC)

FORMACIÓ D’ELIPTÒCITS

• Herència autosòmica dominant
• Distribució mundial
• Més freqüent a àrees endèmiques de malària. Incidència desconeguda
• Heterozigots EC comú(mutació espectrina): assimptomàtics. Només es
manifesta si coexisteix amb polimorfisme (α LELY)
• Homozigots: anemia hemolítica moderada‐greu, severitat variable entre
mateix pacient i entre familiars
• Mutació proteïna 4.1: hemòlisi lleu en estat heterozigot, severa si
homozigot.
• Icterícia variable, esplenomegàlia, colelitiasi
EC: característiques

EC HETEROZIGOT / EC AMB ALFA LELY

• EC esferocítica: ROE augmentada, esplenomegàlia
• Poiquilocitosi infantil: evolució a EC en el primer any de vida
– Piropoiquilocitosi hereditària: més frequent a raça negra
• Herència autosòmica recessiva
• Intensa hemòlisi neonatal
• Fragmentació a 44‐46ºC (N 49‐50º)
EC: tipus

OVALOCITOSI DEL SUD‐EST ASIÀTIC
J. Carrillo Farga

• Herència autosòmica dominant. Elevada prevalència a algunes
ètnies (5‐25%)
•Deleció de 9 aa a banda 3
• Eliptòcits estomatocítics característics (macrocítics
hipocròmics) i esferoeliptòcits (normocítics hipercròmics)
• No anèmia ni hemòlisi
• Estat homozigot incompatible amb la vida
• Histograma característic
OVALOCITOSI DEL SUD‐EST ASIÀTIC

OVALOCITOSI DEL SUD‐EST ASIÀTIC

OVALOCITOSI DEL SUD‐EST ASIÀTIC

TRASTORNS CONGÈNITS DE LA PERMEABILITAT IÒNICA
• Herència autosòmica dominant
• Augment de la permeabilitat passiva a Na i K
• D’hidrocitosi congènita a xerocitosi congènita. Formes intermitges.
• Morfologia no característica.
Hidrocitosi congènita: 2 formes. Hemòlisi compensada sense anèmia
/ hemòlisi crònica intensa. Excepcional. ROE disminuïda, CHCM <280
g/L, reticulocitosi marcada, baixa deformabilitat.
Xerocitosi congènita: hemòlisi compensada o lleu anèmia amb
reticulocitosi intensa (>200x10e9/L). Freqüent. Fragmentació a >
51ºC

ESTOMATOCITOSI HEREDITÀRIA

Membranopaties: tractament
Hemòlisi lleu: dietes riques en folats (vísceres/ verdures de fulla verda/ llegums/ fruits secs)
Anèmia crònica: afegir àcid folínic
Formes greus (especialment transfusió‐ dependents): esplenectomia (disminueix hemòlisi, augmenta supervivència)
esplenectomia a anèmia moderada
Nens/ adults joves: només si litiasi biliar/ altres complicaciones d’hemòlisi crònica. Només en > 5 anys per risc de sepsis
Contraindicada a trastorns congènits de la permeabilitat iònica pel risc posterior de trombosi venosa.

MICROANGIOPATIES TROMBÒTIQUES

MICROANGIOPATIES TROMBÒTIQUES
• Púrpura trombòtica trombocitopènica (PTT)
• Síndrome hemolítica‐urèmica (SHU)
SHUa
• Síndrome antifosfolipídic
• Crisi renal esclerodèrmica
• Hipertensió maligna
• Associada a fàrmacs: mitomicina, ciclosporina, tacrolimus,
quinina, TPH, ICT o combinacions QT

MAT: diagnòstic
Trombocitopènia perifèrica
Hemòlisi intravascular
Absència de causa que justifiqui les troballes
• Pràctica clínica: trombocitopènia, esquistòcits i augment LDH (necrosi)
• Severitat segons l’extensió de l’agregació microvascular (intrarenal o
sistèmica)
CRITERIS DIAGNÒSTICS PRIMARIS
CRITERIS DIAGNÒSTICS SECUNDARIS: Alteracions de la funció renal/ neurològiques/ debilitat/ símptomes
abdominals/ febre

MAT: frotis de sang perifèrica

PÚRPURA TROMBÒTICA TROMBOCITOPÈNICA
• Agregació plaquetar microvascular sistèmica: afectació cerebral,
altres òrgans
• < 20 000 plaquetes/µL
• PTT familiar
• PTT post‐ciclopidina, clopidogrel.
• PTT durant gestació/ post‐part

PTT: fisiopatologia

• Agregació plaquetar: mediada per multímers de factor Von Willebrand
• Diagnòstic:
Demostració d’anticossos anti‐ADAMTS‐13.
Demostració d’activitat ADAMTS‐13 reduïda <5%
Pacients amb activitat normal: mecanismes?
• Tractament:
Infància: infusió plasma fresc pobre en plaquetes (o crioprecipitat/ plasma rentat)
Adults: recanvi plasmàtic diari (plasmafèresi + infusió plasma fresc). Supervivència > 90% sense dany orgànic permanent
Paper de corticoides, esplenectomia, vincristina, rituximab.
PTT: diagnòstic, tractament

SÍNDROME HEMOLÍTICA‐URÈMICA
• Agregació plaquetar microvascular sistèmica: afectació
predominantment renal
• 90% relació amb infecció per BGN: gastroenteritis hemorràgica per E.
coli productora de toxina Shiga
• Zones endèmiques: Buenos Aires, Calgary
• Habitualment episodi únic
• No associat a pèrdua d’activitat ADAMTS‐13

SHU: fisiopatologia

• Diagnòstic: clínic
Activitat ADAMTS‐13 normal
• Tractament: suport
Infància, oligoanúria < 24h: aport hidroelectrolític. Si > 24h HD urgent
Adults: afectació renal més greu. Tractament com a MRC terminal.
Paper de plasmafèresi/ recanvi plasmàtic: resultats dubtosos
Paper d’antidiarreics, antimicrobians. Immunització.
SHU: diagnòstic, tractament

SÍNDROME HEMOLÍTICA‐URÈMICA ATÍPICA
• 10% de SHU: no precedit d’infecció bacteriana.
• Mal pronòstic: 25% de mortalitat al moment agut.
‐ 50% evolució a IRC terminal
• Fisiopatologia: alteració de la via alternativa del complement
‐ 50% casos: mutacions amb alteració de les proteïnes
reguladores del complement

• Qualsevol edat
• Manifestacions clíniques conseqüència de trombosi microvascular lesió
isquèmica hemòlisi microangiopàtica
‐ anèmia (Hb < 100 g/L), hemòlisi intravascular i esquistòcits a SP
‐ fracàs renal agut greu : azotèmia
‐ HTA
‐ proteinuria (rang nefròtic)
‐ hematuria
• 20% afectació extrarenal
• 10% manifestacions neurològiques
• 5% debut en forma de fracàs multiorgànic
SHU ATÍPICA

SHU ATÍPICA: histologia
Nayer and Asif. IJKD. 2013

SHU ATÍPICA: fisiopatologia
Nayer and Asif. IJKD. 2013

SHU ATÍPICA: fisiopatologia
Nayer and Asif. IJKD. 2013

SHU ATÍPICA: tractament
• Transfusió de plasma: restitució de proteïnes reguladores del C
• Recanvi plasmàtic: eliminació d’inhibidors de proteïnes reguladores del C
• Transplant renal: mals resultats. Recurrències > 80% (amb pèrdua
d’injert)
• Eculizumab: bloqueig escisió C5 en C5a/C5b
‐ No formació de CAM
‐ efecte inhibidor de la coagulació i fibrinolisi

SHU ATÍPICA: tractament

Hospital Universitari Germans Trias i Pujol