examen de problemas (solucionado) · en resumen, la relacion´ de intensidades entre las l´ıneas...
TRANSCRIPT

Aplicaciones de la Quımica Cuantica3◦ de Quımicas Convocatoria de Febrero Curso: 2004-05
Examen de problemas (SOLUCIONADO) (Version: 28 de septiembre de 2005)
1. [3.0 puntos]
a) Deduce la expresion que permite obtener el valor de Jmax para el cual la intensidad de la lınea del espectro de microondas sea
maxima (utiliza el modelo del rotor rıgido para una molecula diatomica).
b) Calcula el valor de Jmax para los espectros de microondas del 12C16O y 13C16O, a 300 K, sabiendo que la distancia de enlace es
1.1282 A.
c) Sabiendo que la abundancia natural de los isotopos de 12C y 13C es del 98.91 % y del 1.09 %, respectivamente, ¿cual sera la
intensidad relativa entre las lıneas espectrales de ambas moleculas en una mezcla con esa abundancia?
a) Obtencion de la expresion de Jmax
De acuerdo con la estadıstica de Maxwell-Boltzmann la poblacion de un estado J con respecto al estado
J = 0 viene dada por,NJ
N0= (2J + 1) · exp
(
−BJ(J + 1)
kT
)
El maximo de intensidad se dara desde el estado que tenga mayor poblacion,
maximo⇒ d(NJ/N0)
dJ= 0
d(NJ/N0)
dJ= 2 · exp
(
−BJ(J + 1)
kT
)
+ (2J + 1) · exp(
−BJ(J + 1)
kT
) (
− B
kT(2J + 1)
)
= 0
2 − (2J + 1)2B
kT= 0 ⇒ (2J + 1)2 =
2kT
B⇒ J =
√
kT
2B− 1
2
Anadimos el subindice max por ser el valor de J correspondiente al de maxima intensidad,
Jmax =
√
kT
2B− 1
2=
√
kT
2hcB− 1
2
En la expresion anterior, B es la constante rotacional que, para una molecula diatomica, viene dada por la
expresion:
B =~2
2I=
h2
8π2I=
h2
8π2µr2
o bien en numero de onda:
B =h
8π2cµr2
En ambas expresiones vemos que al aumentar la masa reducida (µ) disminuye la constante rotacional B
y por tanto aumenta el valor de Jmax, es decir la masa y el valor de Jmax son proporcionales:
Jmax ∝ √µ
b) Para el caso concreto de las moleculas 12C16O y 13C16O, tenemos:
µ(12C16O) =15.9949 · 12.000015.9949 + 12.0000
g mol−1 kg
1000 g
1
6.0221 · 1023 mol−1= 1.1385 · 10−26 kg
µ(13C16O) =15.9949 · 13.003415.9949 + 13.0034
g mol−1 kg
1000 g
1
6.0221 · 1023 mol−1= 1.1910 · 10−26 kg

I(12C16O) = µ · r2 = 1.1385 · 10−26 kg · (1.1282 · 10−10)2 m2 = 1.4494 · 10−46 kg m2
I(13C16O) = µ · r2 = 1.1910 · 10−26 kg · (1.1282 · 10−10)2 m2 = 1.5162 · 10−46 kg m2
B(12C16O) =h2
8π2I=
(6.6261 · 10−34)2 J2 s2
8π2 1.4494 · 10−46 kg m2= 3.8366 · 10−23 J (= 1.9314 cm−1)
B(12C16O) =h2
8π2I=
(6.6261 · 10−34)2 J2 s2
8π2 1.5162 · 10−46 kg m2= 3.6674 · 10−23 J (= 1.8462 cm−1)
Jmax(12C16O) =
√
1.3807 · 10−23 J K−1 · 300 K2 · 3.8366 · 10−23 J
− 1
2= 6.85 ≈ 7.0
Jmax(13C16O) =
√
1.3807 · 10−23 J K−1 · 300 K2 · 3.6674 · 10−23 J
− 1
2= 7.01 ≈ 7
El paso de la constante rotacional B de Julio a cm−1 se efectua dividiendo por h y c:
B(J) · 1
h (J · s) · 1
c (cm · s−1)= B(J) · 5.0341 · 1022
(
cm−1
J
)
= B (cm−1)
c) La intensidad de una lınea espectral es proporcional a la poblacion del nivel de partida, que viene dada por
la ley de distribucion de Maxwell-Boltzmann:
NJ = N0 · gJ · exp
(
−EJ
kT
)
o en terminos de la poblacion total,
NT =∑
J
NJ = N0
∑
J
gJ exp
(
−EJ
kT
)
= N0Zrot
donde Zrot es la funcion de particion rotacional. Ası podemos reescribir:
NJ =NT
ZrotgJ exp
(
−EJ
kT
)
Como hemos visto en el apartado anterior, las constantes rotacionales de las dos moleculas 12C16O,13C16O son similares, por lo que podemos considerar, en una primera aproximacion, que tanto Zrot como
el factor exp(
−EJ
kT
)
son iguales. Ası, la relacion de intensidades sera:
I12C16O
I13C16O
=NJ(12C16O)
NJ(13C16O)≈ NT (12C16O)
NT (13C16O)=
98.91
1.09= 90.74
En resumen, la relacion de intensidades entre las lıneas espectrales de ambas moleculas en una mezcla
con abundancia natural sera de 90.7 frente a 1.
p2-025 Examen Febrero 2005

2. [4.0 puntos]El espectro infrarrojo de la molecula 1H19F muestras cuatro bandas, cuyas frecuencias e intensidades relativas a dos
temperaturas se muestran en la siguiente tabla:
ν/cm−1 3779 3959 7737 11336
I (300 K) 6·10−9 1.0 0.4 0.1
I (1000 K) 3·10−3 1.0 0.4 0.1
a) Asigna estas bandas a las correspondientes transiciones vibracionales indicando los numeros cuanticos vibracionales correspondien-
tes a dichas transiciones.
b) Calcula la frecuencia fundamental de vibracion νe y la constante de anarmonicidad νexe.
c) Halla la energıa del punto cero y las energıas de disociacion De y D0 en cm−1 y en kcal/mol.
d) Obten la constante de fuerza del enlace.
a) Se presentan cuatro bandas, la primera (3779 cm−1) tiene una intensidad pequena y su intensidad relativa
cambia apreciablemente con la temperatura lo que indica que es una banda caliente, ademas, esta 180
cm−1 a la izquierda de la banda mas intensa que sera la banda Fundamental. Las bandas tercera y cuarta
estan aproximadamente a dos y tres veces la frecuencia de la banda fundamental y por tanto, corresponden
al primer y segundo armonico (o sobretono).
ν(v′′ → v′)/cm−1 Denominacion v′′ → v′ Ecuacion
3779 cm−1 Banda caliente v′′ = 1 → v′ = 2 νe − 4νexe
3959 cm−1 Banda Fundamental v′′ = 0 → v′ = 1 νe − 2νexe
7737 cm−1 1er Sobretono v′′ = 0 → v′ = 2 2νe − 6νexe
11336 cm−1 2o Sobretono v′′ = 0 → v′ = 3 3νe − 12νexe
b) La niveles de energıa vibracional en la aproximacion anarmonica con una constante de anarmonicidad son:
G(v) =Evib(v)
h c= νe
(
v +1
2
)
− νexe
(
v +1
2
)2
Para calcular la frecuencia fundamental y la constante de anarmonicidad podemos usar las transiciones
desde el nivel fundamental, es decir desde v′′ = 0 a v′:
ν0→v′ = νe
(
v′ +1
2
)
− νexe
(
v′ +1
2
)2
− νe
(
0 +1
2
)
+ νexe
(
0 +1
2
)2
ν0→v′ = v′νe − v′(v′ + 1)νexe
dividiendo todos los terminos por v′ obtenemos:
ν0→v′
v′= νe − (v′ + 1)νexe
representando ν0→v′
v′ frente a (v′ + 1) obtenemos del corte en ordenadas y de la pendiente los valores de
νe y νexe.
v’ ν0→v′
v′ /cm−1 (v′ + 1)
1 3959 2
2 3868.5 3
3 3778.7 4
Los valores que obtenemos son νe = 4139 cm−1 y νexe = 91 cm−1 (xe = 0.02199)

3750
3800
3850
3900
3950
4000
2 3 4 ν
v / v
’ (v’ + 1)
Cort. ord. = 4139.2(6) cm-1
Pendiente = -91.1(2) cm-1
σ = 0.286
c) La energıa1 del punto cero viene dada por la expresion,
G(0) =Evib(0)
h c= νe
(
1
2
)
− νexe
(
1
2
)2
= 4139/2 − 91/4 = 2092 cm−1 ≡ 5.98kcal/mol
A partir de los valores de νe y νexe podemos calcular la energıa de disociacion.
Cuando la consideracion de una sola constante de anarmonicidad proporciona una buena aproximacion al
potencial real, la energıa de disociacion, De, puede evaluarse de forma aproximada mediante la expresion
De =ν2
e
4νexe=
41392
4 · 91 = 47064 cm−1 ≡ 134.50 kcal/mol
que difiere de D0 en la energıa en el punto cero: De = D0 +G(0).
D0 = De −G(0) = 47064 − 2092 = 44972 cm−1 ≡ 128.53 kcal/mol
La expresion anterior se obtiene calculando el valor de G(vmax), siendo vmax el valor de v en el que la derivada
dG(v)/dv se hace cero.dG(v)
dv= νe − 2νexe(vmax + 1/2) = 0;
(vmax + 12 ) = 1
2xe
; vmax = 12xe
− 12
sustituyendo vmax en la ecuacion de G(v) obtenemos,
G(vmax) =νe
2xe
− νexe
4x2e
=νe
4xe
=ν2
e
4νexe
= De
Otra forma de calcular el valor de De consiste en busca el valor de vmax como aquel en el que la diferencia de
energıa entre dos estados se hace cero.
G(vmax + 1) −G(vmax) = νe − 2νexe(vmax + 1/2) = 0
vmax + 1 =1
2xe
; vmax =1
2xe
− 1
Vemos que el valor de vmax es ligeramente distinto del anterior. Sustituyendo vmax en la Ec. (??) obtenemos:
G(vmax) = νe(1
2xe
− 1 + 12 ) − νexe(
12xe
− 1 + 12 )2 = νe(
12xe
− 12 ) − νexe(
12xe
− 12 )2
= νe
2xe
− νe
2 − νexe
4x2e
− νexe
4 + νexe
2xe
= νe
2xe
− νe
4xe
− νexe
4
= νe
4xe
− νexe
4 = De
1Para cambiar de unidades,
Energıa(Kcal/mol) = Energıa(cm−1) · c(cm · s−1) · h(J · s) · 1 cal4.1858 J
·1 kcal
1000 cal· NA(mol−1)
= Energıa(cm−1) · 2.9979 · 1010· 6.6261 · 10−34
·1
4.1858·
11000
· 6.0221 · 1023
= Energıa(cm−1) · 2.8579 · 10−3

(d) La constante de fuerza se obtiene con la ecuacion de la frecuencia clasica:
νe =1
2π
√
k
µ; k = µ · (2πcνe)
2
µ(1H19F ) =18.9984 · 1.007818.9984 + 1.0078
g mol−1 kg
1000 g
1
6.0221 · 1023 mol−1= 1.5892 · 10−27 kg
k = 1.5892 · 10−27 kg · (2 π 2.9979 · 1010 cm · s−1 4139 cm−1)2 = 965.97 kg · s−2 ≡ 965.97 N ·m−1
p3-035 Examen Febrero 2005

3. [3.0 puntos]En la primera banda de vibracion del espectro electronico del 12C16O se observa que la separacion entre el origen de
banda y la cabeza de banda es de 31.6 cm−1. La banda esta degradada al rojo y su cabeza o canto aparece para J ′′ = 16.
a) Determinar las constantes rotacionales del estado fundamental B ′′ y del excitado B′.
b) Determinar la longitud del enlace 12C16O en su estado electronico fundamental y en el excitado.
c) Dibujar cualitativamente el correspondiente diagrama de energıas, incluyendo en el diagrama las energıas de disociacion D′
0, D′
e,
D′′
0 , D′′
e , ν00, νlim y νatomica.
d) Dibujar cualitativamente la estructura y posicion respecto al origen de banda de las lıneas de las ramas R y P para la banda v′′ =
0 → v′ = 0.
a) Si la banda degrada al rojo entonces es la rama R la que nos dara el canto o cabeza de banda.
Rama R νR(J) = νe,v + B′(J + 1)(J + 2) − B′′J(J + 1)
= νe,v + B′[J2 + 3J + 2] − B′′[J2 + J ]
= νe,v + J2(B′ − B′′) + J(3B′ − B′′) + 2B′
Sustituyendo en valor de JCB = 16 en la expresion anterior podemos obtener la frecuencia de la cabeza
de bandaνR(16) = νe,v + J2(B′ − B′′) + J(3B′ − B′′) + 2B′
= νe,v + 162(B′ − B′′) + 16(3B′ − B′′) + 2B′
La diferencia entre el centro u origen de la banda y el canto de banda es:
νR(16) − νe,v = 162(B′ − B′′) + 16(3B′ − B′′) + 2B′ = 306B′ − 272B′′ = 31.6 cm−1
Por otro lado, el valor de J que nos da el canto de banda lo obtenemos a partir de la derivada de la rama R
respecto a J e igualando a cero,(
δνR
δJ
)
= 2J(B′ − B′′) + (3B′ − B′′) = 0
Despejando el valor de J
JCB =−(3B′ − B′′)
2(B′ − B′′)= 16 ⇒ 35B′ − 33B′′ = 0
Obtenemos de esta manera dos ecuaciones con dos incognitas
306B′ − 272B′′ = 31.6 cm−1
35B′ − 33B′′ = 0 cm−1
de cuya solucion obtenemos las constantes rotacionales, B′ = 1.8042 cm−1 y B′′ = 1.9135 cm−1.
b) Los valores de las distancias interatomicas pueden obtenerse a partir de las constantes rotacionales,
B =h
8 π2 c I=
h
8 π2 c µ r2⇒ r =
√
h
8 π2 c µ B
µ(12C16O) =15.9949 · 12.000015.9949 + 12.0000
g mol−1 kg
1000 g
1
6.0221 · 1023 mol−1= 1.1385 · 10−26 kg
r′(est. excitado) =√
6.6261·10−34J s8 π2 2.9979·1010 cm s−1 1.1385·10−26 kg1.8042 cm−1
= 1.1674 · 10−10 m ≡ 1.1674 A
r′′(est. fundament.) =√
6.6261·10−34J s8 π2 2.9979·1010 cm s−1 1.1385·10−26 kg1.9135 cm−1
= 1.1336 · 10−10 m ≡ 1.1336 A

Figura 1: Diagrama de energıas para una molecula diatomica.
c) y d) Ver Figuras 1 y 2
p4-014 Examen Febrero 2005
Nota: NA = 6.0221·1023 mol−1, c = 2.9979·108 m s−1, h = 6.6261·10−34 J s,
k = 1.3807·10−23 J K−1. m(1H) = 1.0078 uma, m(19F ) = 18.9984 uma,
m(16O) = 15.9949 uma, m (12C) = 12.0000 uma, m (13C) = 13.0034 uma.

Figura 2: Estructura y posicion respecto al origen de banda de las lıneas de las ramas R y P

Aplicaciones de la Quımica Cuantica3◦ de Quımicas Convocatoria de Febrero Curso: 2004-05
Examen de teorıa. (SOLUCIONADO) (Version: 28 de septiembre de 2005)
1. [2.0 puntos]Indica que tipos de espectroscopias pueden usarse para obtener la geometrıa molecular de las siguientes moleculas:
N2, NO, CO2, CF4
Justifica brevemente.
En todos los casos solo podra resolverse la estructura rotacional si los compuestos estan en fase gaseosos.
N2 La molecula N2, diatomica homonuclear, tiene momento dipolar cero y, por tanto, no dara espectro de
microondas. Sin embargo, su geometrıa, es decir, la distancia de enlace N − N se puede obtener por
espectroscopıa Raman rotacional ya que la polarizabilidad si cambia con la rotacion.
NO La molecula NO, diatomica heteronuclear, si tiene momento dipolar y, por tanto ,podemos obtener el
espectro de microondas y de el la distancia de enlace rN−O.
CO2 La molecula CO2, triatomica lineal, no presenta momento dipolar y, por tanto, no dara espectro de mi-
croondas. Sin embargo, al igual que la molecula N2, su geometrıa, es decir, la distancia de enlace rC=O
se puede obtener por espectroscopıa Raman rotacional ya que la polarizabilidad si cambia con la rotacion.
CF4 La molecula CF4, trompo esferica, no presenta momento dipolar, por tanto, no podemos estudiarla por
microondas. Por otro lado, su elipsoide de polarizabilidad es una esfera (polarizabilidad isotropica), es
decir no cambia con la rotacion y tampoco puede estudarse por Raman rotacional. Una posibilidad es
estudiar la estructura fina (rotacion-vibracion) de los modes de vibracion que den lugar a un cambio en el
momento dipolar. La geometrıa en equilibrio de esta molecula viene determinada por una unica distancia
(rC−F ) y podrıa obtenerse a partir de una constante rotacional que obtendrıamos analizando la estructura
de rotacion-vibracion de algun modo de vibracion asimetrico.
p3-032s Examen de Febrero, curso 2004-2005.

2. [2.0 puntos]Dibuja un diagrama de niveles energeticos vibracionales para una molecula triatomica cuyos modos normales tienen
energıa de 500, 1200 y 1600 cm−1. Calcula la energıa del punto cero e indica la energıa del estado (1,2,1). Supon siempre la aproxi-
macion armonica.
En al aproximacion armonica la energıa de vibracion de una molecula poliatomica viene dada por la expresion:
Evib =
nvib∑
j=1
Evj= h · c
nvib∑
j=1
νj
(
vj +1
2
)
donde nvib son los grados de libertad vibracional (3N−6 o 3N−5), vj son los numeros cuanticos de vibracion
para el modo normal Qj . Estos numeros toman los valores vj = 0, 1, 2, ....
Para una molecula no lineal tendriamos tres modos de vibracion (3N − 6 = 3 ) cuyas frecuencias nos da el
enunciado del problema.
En base a la ecuacion anterior y dando diferentes valores a los numeros cuanticos obtendrıamos el siguiente
diagrama de energıas:
1000
2000
3000
4000
5000
Evi
b/(h
c)
(cm
-1)
(0,0,0) 1650
(0,0,1) 3250
(0,0,2) 4850
(0,1,0) 2850
(0,1,1) 4450
(0,2,0) 4050
(1,0,0) 2150
(1,0,1) 3750
(1,1,0) 3350
(1,1,1) 4950
(1,2,0) 4550
(2,0,0) 2650
(2,0,1) 4250
(2,1,0) 3850
(3,0,0) 3150
(3,0,1) 4750
(3,1,0) 4350
Figura 3: Diagramas para los primeros niveles de energıa de una molecula triatomica no lineal con ν1 = 500, ν2 = 1200 y ν3 = 1600.
La energıa del punto cero corresponde a la energıa para el estado (0, 0, 0, . . .), es decir con los numeros cuanticos
vj = 0.
Evib(0, 0, 0, . . .) =1
2h · c ·
nvib∑
j=1
νj
En nuestro caso y asumiendo una molecula no lineal, la energıa del punto cero es:
Evib(0, 0, 0) = 12 h · c · (500 + 1200 + 1600)
= 0.5 · 6.6261 · 10−34J s · 2.9979 · 1010cm s−1 · (500 + 1200 + 1600)cm−1
= 3.2776 · 10−20 J (≡ 1650 cm−1)
La energıa en el estado (1, 2, 1) se calcula de modo similar:
Evib(1, 2, 1) = h · c · (500(1 + 0.5) + 1200(2 + 0.5) + 1600(1 + 0.5))
= h · c · (1.5 · 500 + 2.5 · 1200 + 1.5 · 1600)= 6.6261 · 10−34J s · 2.9979 · 1010cm s−1 · 6150cm−1
= 1.2217 · 10−19 J (≡ 6150 cm−1)

Si la molecula fuera lineal tendrıamos cuatro modos normales de vibracion (3N −5), dos de ellos degenerados
que posiblemente corresponderan a la vibracion de flexion que suele ser la de menor numero de onda (en este
caso 500 cm−1). En este caso, que no vamos a ver en detalle, en los sumatorios de las ecuaciones anteriores
habrıa que considerar dos veces el modo de vibracion degenerado, por ejemplo, la energıa del punto cero serıa:
Evib(0, 0, 0, 0) = 12 h · c · (500 + 500 + 1200 + 1600)
= 0.5 · 6.6261 · 10−34J s · 2.9979 · 1010cm s−1 · 3800 cm−1
= 3.7742 · 10−20 J (≡ 1900 cm−1)
p3-033 Examen de Febrero de 2005

3. [2.0 puntos]
a) ¿Por que entre lıneas Stokes y las Anti-Stokes del espectro Raman vibracional existe una marcada diferencia de intensidades?
b) Justifıcalo para el caso de la molecula N2 a 110 K (Dato: ν0 = 2358 cm−1).
c) ¿Sucede algo similar en el espectro Raman rotacional puro?, razonalo.
Figura 4: Espectro de vibracion Raman. La linea intensa de la dispersion Rayleigh correspondiente a la frecuencia de excitacion (ν0) se ha
eliminado del espectro para mayor claridad.
a) En vibracion Raman, las lıneas de Stokes tienen una intensidad mucho mayor que las de anti-Stokes. Las
transiciones de Stokes son ”equivalentes”2 a transiciones desde el estado fundamental (v′′ = 0) a un estado
excitado (generalmente v′ = 1), por tanto, su intensidad depende de la poblacion del estado fundamental
vibracional que a temperatura ambiente es muy alta.
Las transiciones anti-Stokes son ”equivalentes” a transiciones desde un estado excitado (generalmente
v′′ = 1) al estado fundamental (v′ = 0). Estas transiciones son dificiles de detectar (por su menor inten-
sidad) porque se necesita tener suficientemente poblados los estados excitados de vibracion y a tempera-
tura ambiente, en general, unicamente tiene una poblacion suficiente el estado vibracional fundamental
(v = 0).
b) Para justificarlo en el caso de la molecula N2 podemos comprobar la relacion de poblaciones entre el estado
vibracional fundamental (v = 0) y el primer estado excitado (v = 1)
N(v=1)
N(v=0)= exp
(
−Ev=1−Ev=0k·T
)
= exp(
−h·c·ν0k·T
)
= exp(
−6.6261·10−34 J s 2.9979·1010 cm s−1 2357 cm−1
1.3807·10−23 J K−1 110 K
)
= 4.09 · 10−14
Vemos que la relacion de poblaciones es muy pequena, es decir la poblacion en el estado fundamental es
del orden 1014 veces la poblacion del estado excitado.
c) En rotacion Raman, el mecanismo es similar, es decir, las transiciones de Stokes son ”equivalentes” a
transiciones desde un estado rotacional (J ′′) a un estado excitado superior (J ′ > J ′′). Las transiciones
2Decimos ”equivalentes” ya que en realidad, tanto las transiciones de Stokes como las de anti-Stokes, las hemos estudiado como transiciones
de un estado a otro pasando por un hipotetico estado vitutual.

anti-Stokes son ”equivalentes” a transiciones desde un estado excitado (J ′′) a un estado inferior energıa
(J ′ < J ′′). La diferencia, con respecto a vibracion, estriba en que la poblacion de los primeros estados
excitados rotacionales no es depreciable e incluso, como ya hemos visto en numerosos ejemplos, en algu-
nos estados excitados su poblacion es superior a la del estado fundamental. Por tanto, se observa tanto la
parte de Stokes como las de anti-Stokes con intensidades parecidas.
p3-034 Examen de Febrero curso 2004-2005.

4. [2.0 puntos]Define fluorescencia y fosforescencia e indica sus diferencias.
Fluorescencia y fosforescencia
La fluorescencia y fosforescencia, que se denominan genericamente como luminiscencia, corresponden a la
emision espontanea de radiacion de especies excitadas.
Se denomina fluorescencia si la emision de radiacion corresponde un transito electronico en el que ∆S = 0
(entre dos estados con el mismo numero cuantico de espın) y fosforescencia si la emision se debe a la transicion
es entre dos estados con numero cuantico de espın distinto, y, por tanto, es prohibida por espın.
Las principales diferencias fenomenologicas entre fluorescencia y fosforescencia son que la primera es muy
rapida y cesa al cesar la radiacion de excitacion, mientras que la segunda persiste durante algun tiempo, conse-
cuencia de las reglas de seleccion.
Estado fundamental singlete
Estado excitado triplete
Estado excitado singlete
Figura 5: Curvas de energıa potencial para varios estados electronicos de la molecula C2. (Barrow Fig. 10-20)
p4-013 Examen de Febrero de 2005
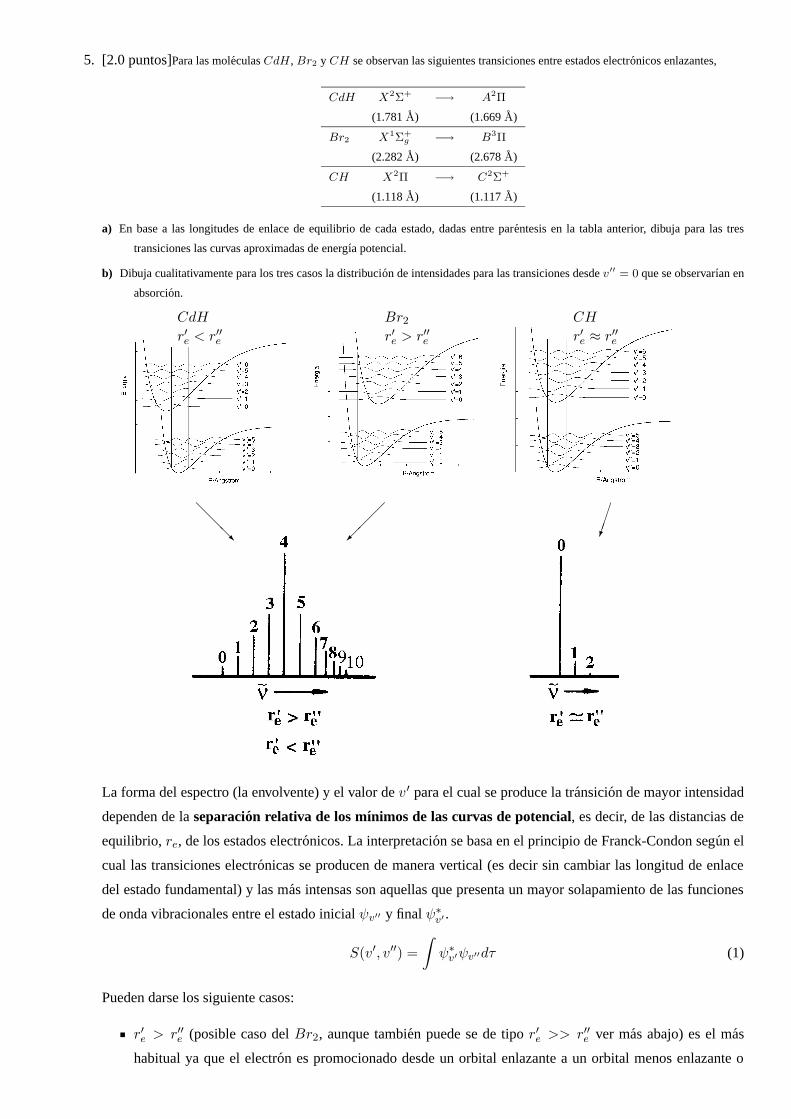
5. [2.0 puntos]Para las moleculas CdH , Br2 y CH se observan las siguientes transiciones entre estados electronicos enlazantes,
CdH X2Σ+−→ A2Π
(1.781 A) (1.669 A)
Br2 X1Σ+g −→ B3Π
(2.282 A) (2.678 A)
CH X2Π −→ C2Σ+
(1.118 A) (1.117 A)
a) En base a las longitudes de enlace de equilibrio de cada estado, dadas entre parentesis en la tabla anterior, dibuja para las tres
transiciones las curvas aproximadas de energıa potencial.
b) Dibuja cualitativamente para los tres casos la distribucion de intensidades para las transiciones desde v′′ = 0 que se observarıan en
absorcion.
CdH Br2 CH
r′e < r′′e r′e > r′′e r′e ≈ r′′e
@@
@R
��
�
���
La forma del espectro (la envolvente) y el valor de v′ para el cual se produce la transicion de mayor intensidad
dependen de la separacion relativa de los mınimos de las curvas de potencial, es decir, de las distancias de
equilibrio, re, de los estados electronicos. La interpretacion se basa en el principio de Franck-Condon segun el
cual las transiciones electronicas se producen de manera vertical (es decir sin cambiar las longitud de enlace
del estado fundamental) y las mas intensas son aquellas que presenta un mayor solapamiento de las funciones
de onda vibracionales entre el estado inicial ψv′′ y final ψ∗
v′ .
S(v′, v′′) =
∫
ψ∗
v′ψv′′dτ (1)
Pueden darse los siguiente casos:
r′e > r′′e (posible caso del Br2, aunque tambien puede se de tipo r′e >> r′′e ver mas abajo) es el mas
habitual ya que el electron es promocionado desde un orbital enlazante a un orbital menos enlazante o

antienlazante. En estas transiciones el transito vibronico mas intenso se corresponde con un ∆v tanto
mayor cuanto mayor sea la separacion relativa de los mınimos. La intensidad de los transitos, respecto al
maximo, disminuye lentamente dando lugar a la presencia de progresiones largas en el espectro.
r′e < r′′e , (caso del CdH), es un caso poco comun, se da ejemplo cuando un electron es promocionado
desde un orbital antienlazante a uno enlazante o no-enlazante. La forma del espectro es similar a la de
r′e > r′′e . Aunque hay que tener en cuenta como se comporta el oscilador anarmonico y que la pendiente
de la curva de potencial no es la misma a la derecha que a la izquierda de la distancia de equilibrio.
r′e u r′′e (caso de CH) el transito mas intenso se corresponde con ∆v = 0, disminuyendo rapidamente la
intensidad para transitos con ∆v 6= 0, y observandose en el espectro progresiones muy cortas.
Si r′e >> r′′e (posible caso del Br2) habra una intensidad apreciable cerca de los niveles de vibracion del
continuo o encima del lımite de disociacion (como se indica en la figura siguiente).
En el caso del Br2 aunque la transicion esta prohibida si se puede detectar en determinadas circunstacias.
p4-015 Examen Febrero 2005
Nota: c = 2.9979·108 m s−1, h = 6.6261·10−34 J s, k = 1.3807·10−23 J K−1.