
Origen de los cetáceos
Un ejercicio práctico


¿Qué quiere decir esto?
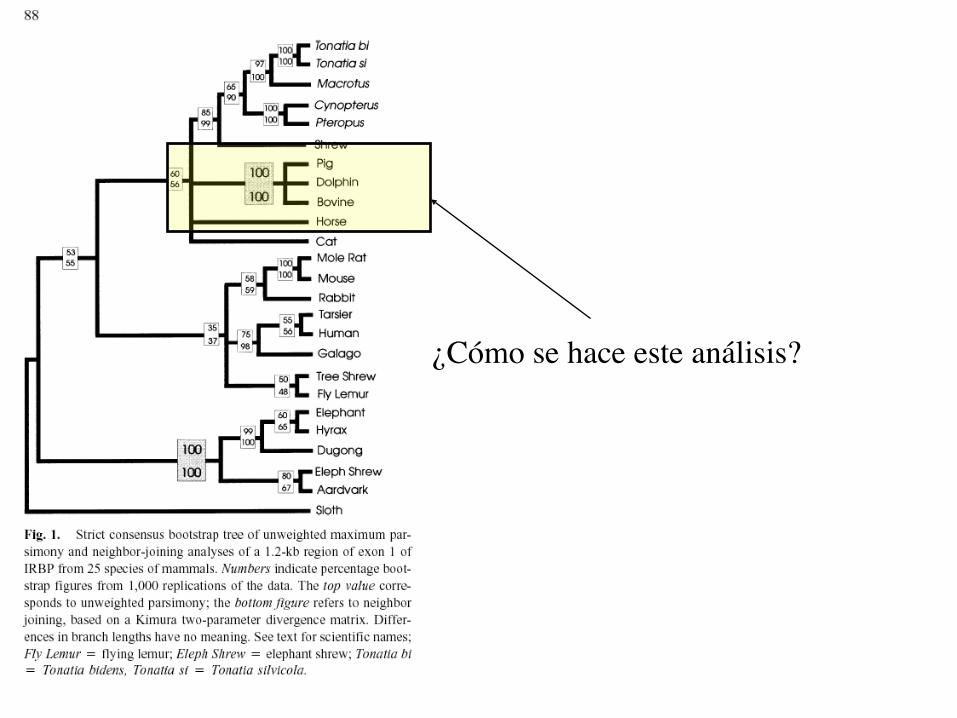
¿Cómo se hace este análisis?
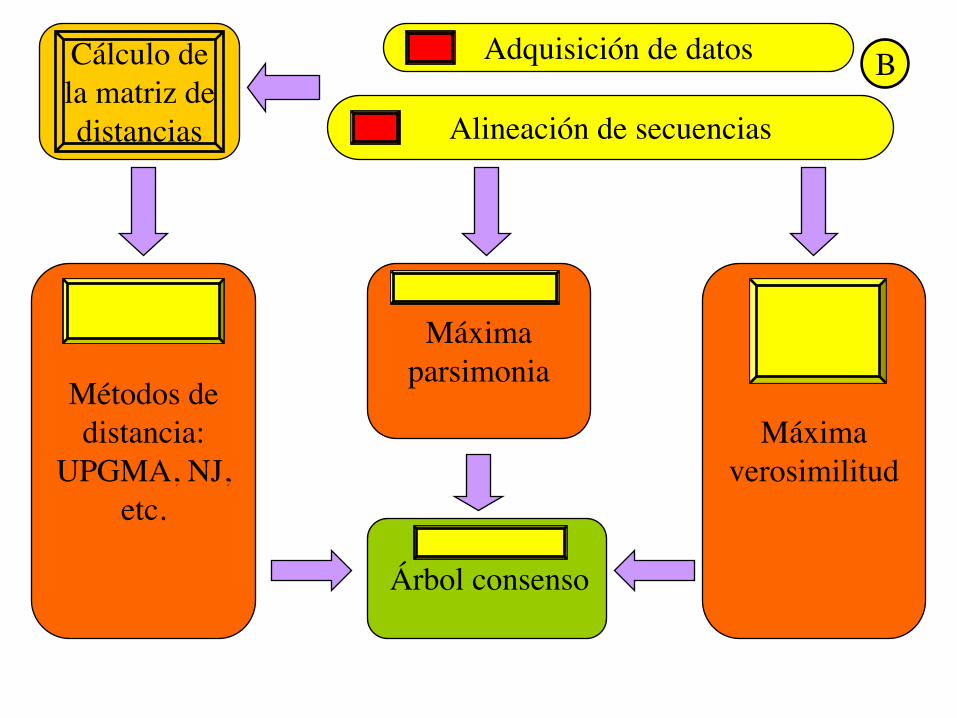
Adquisición de datos
Alineación de secuencias
Cálculo de la matriz de distancias
Métodos de distancia:
UPGMA, NJ, etc.
Máxima verosimilitud
Máxima parsimonia
Árbol consenso
B

¿Cómo conseguir los datos?
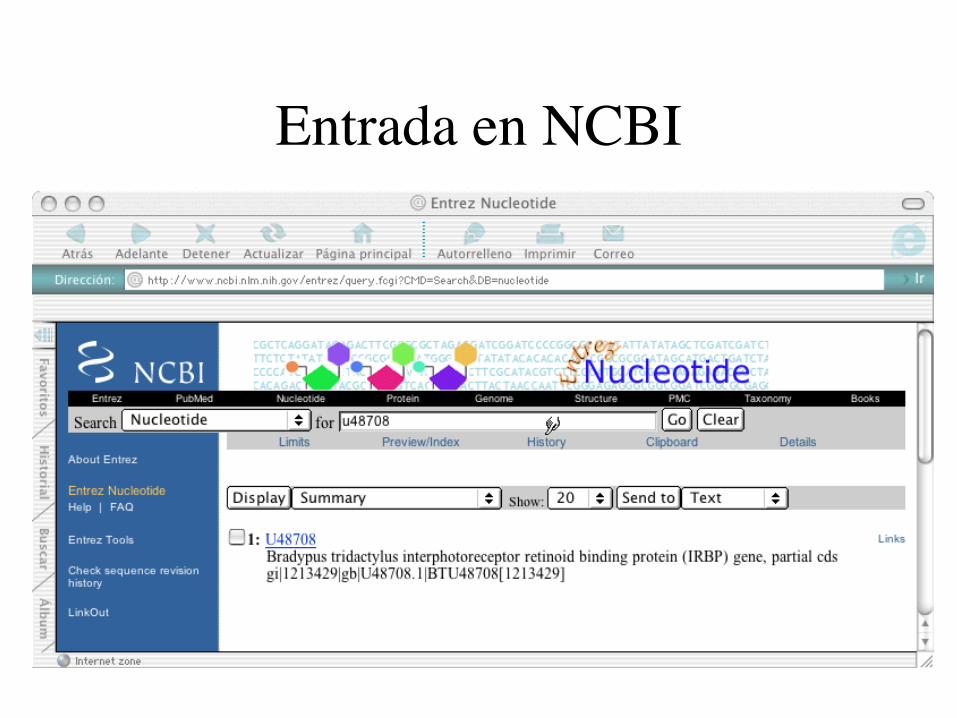
Entrada en NCBI
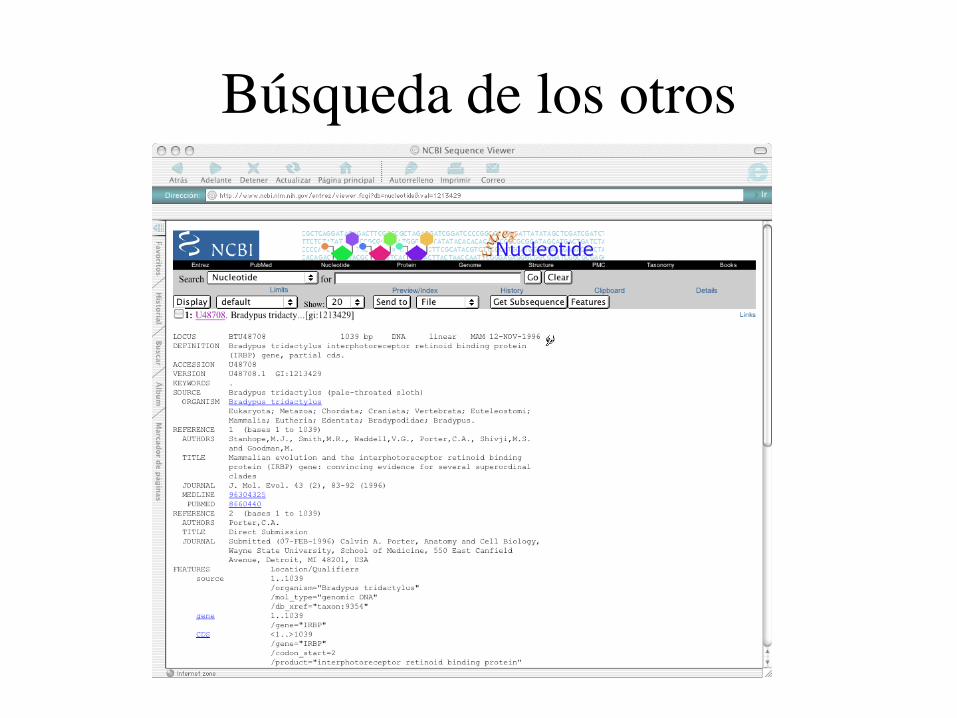
Búsqueda de los otros
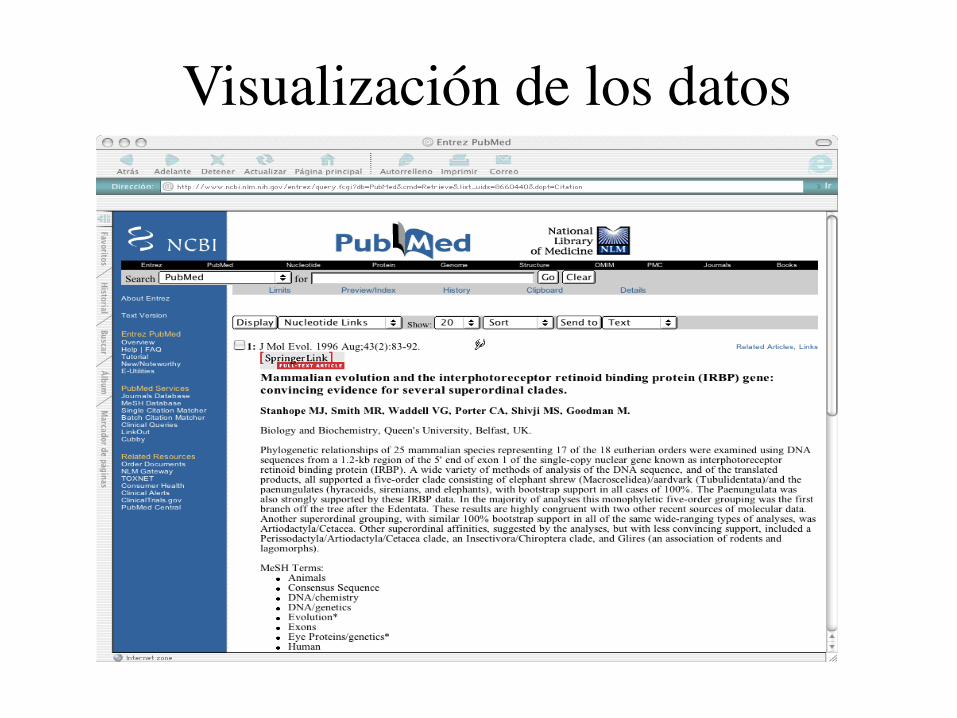
Visualización de los datos
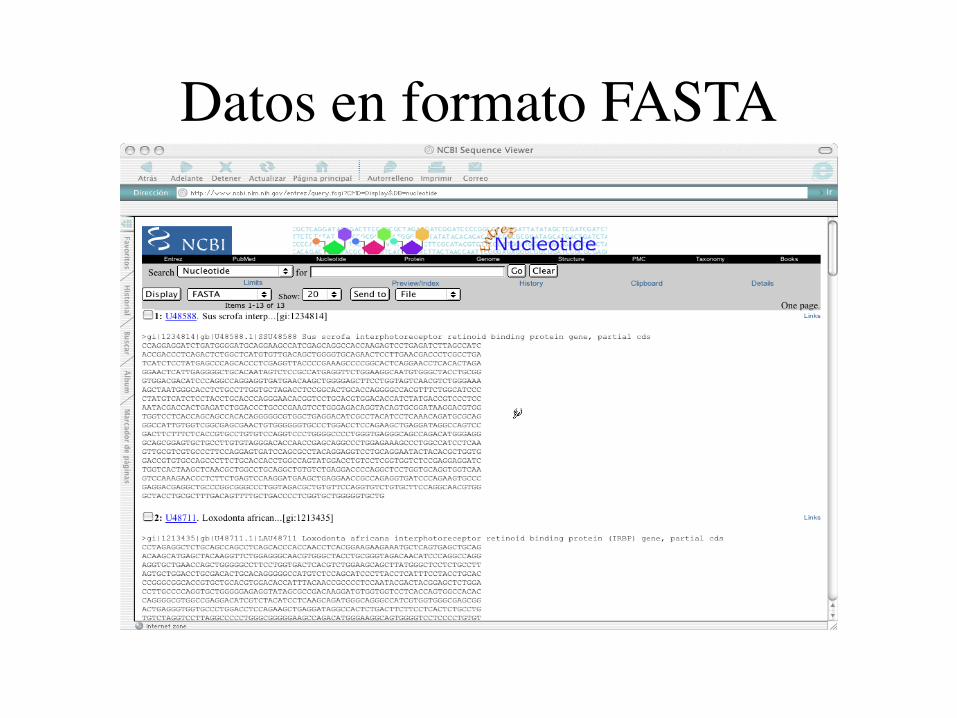
Datos en formato FASTA

Grabar en un archivo
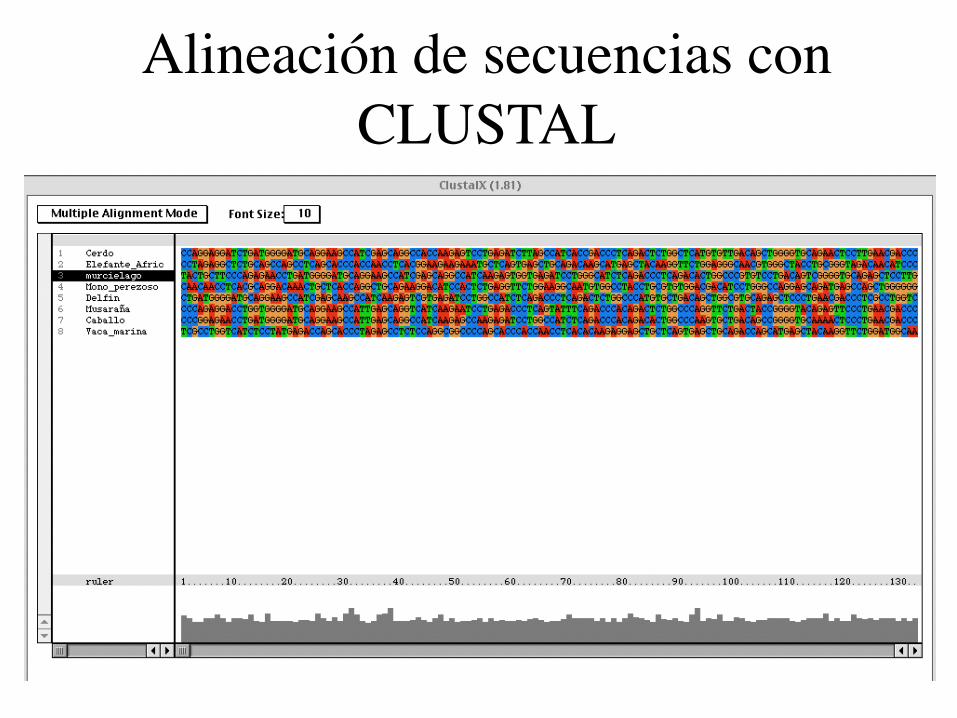
Alineación de secuencias con CLUSTAL
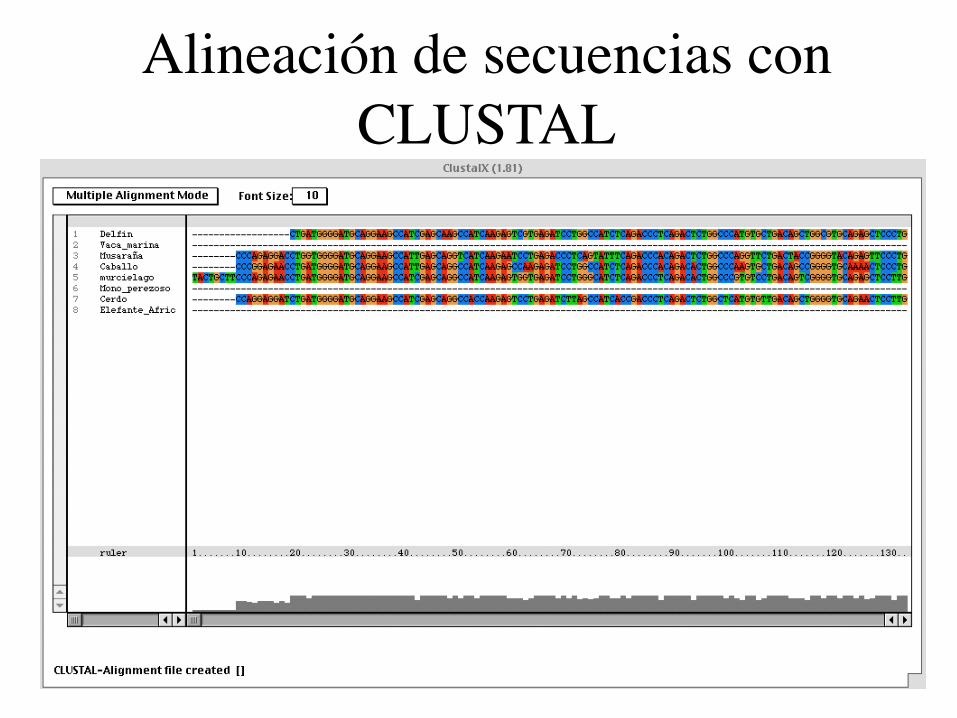
Alineación de secuencias con CLUSTAL

Alineación de secuencias con CLUSTAL
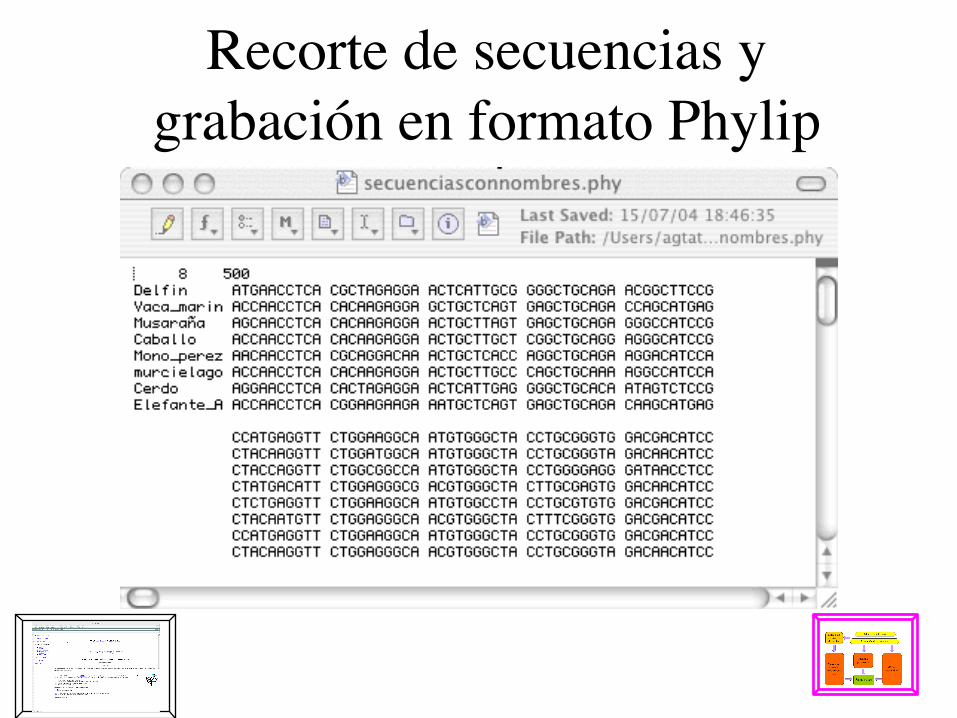
Recorte de secuencias y grabación en formato Phylip
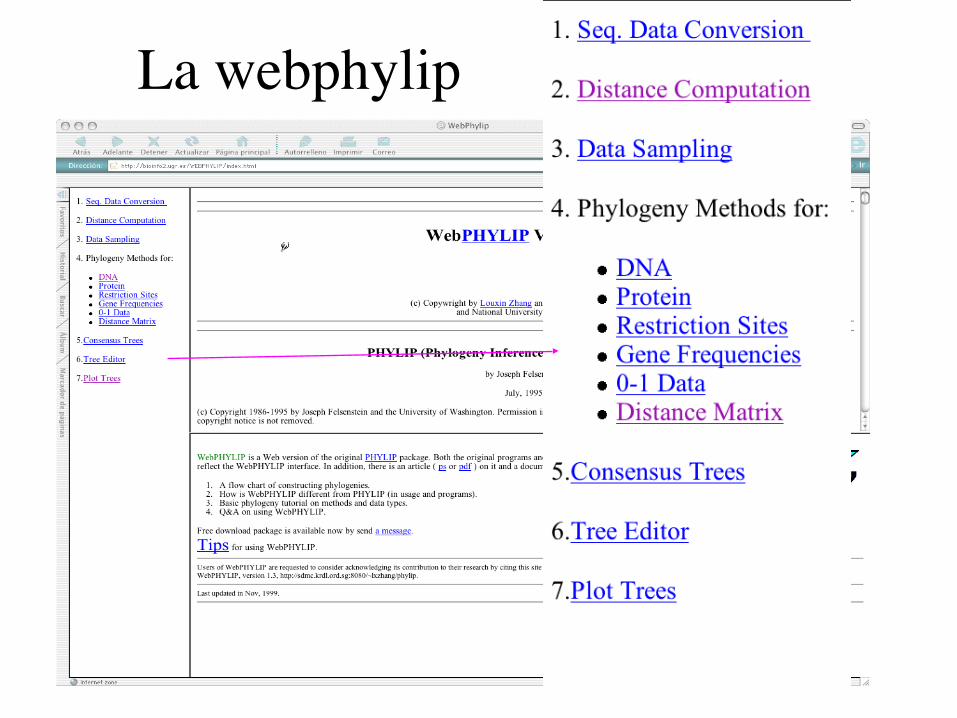
La webphylip
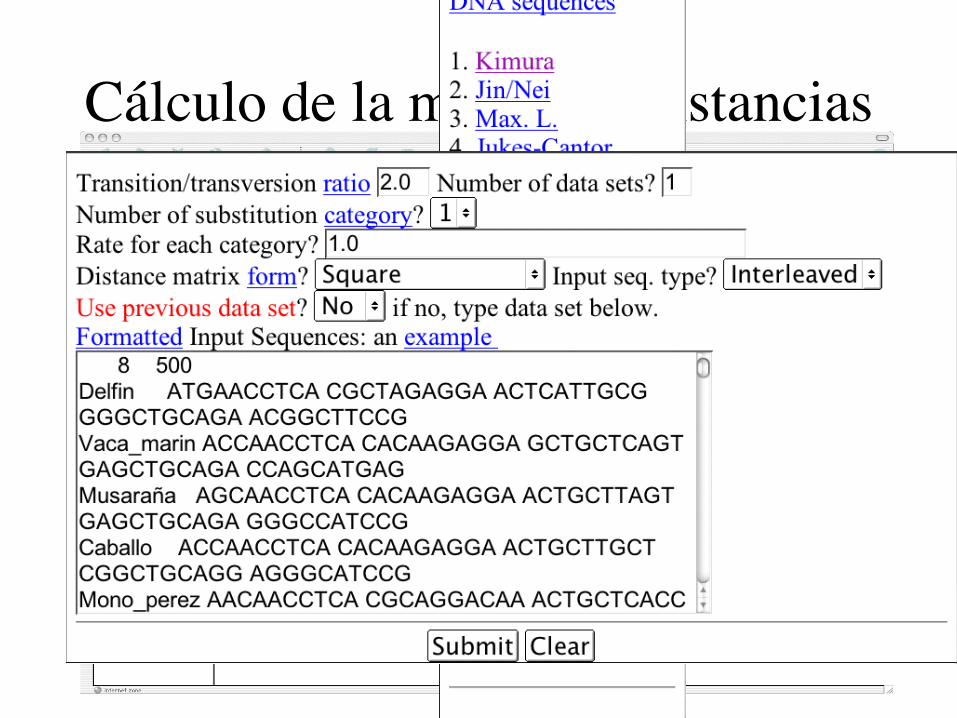
Cálculo de la matriz de distancias

Cálculo de la matriz de distancias

Cálculo de la matriz de distancias
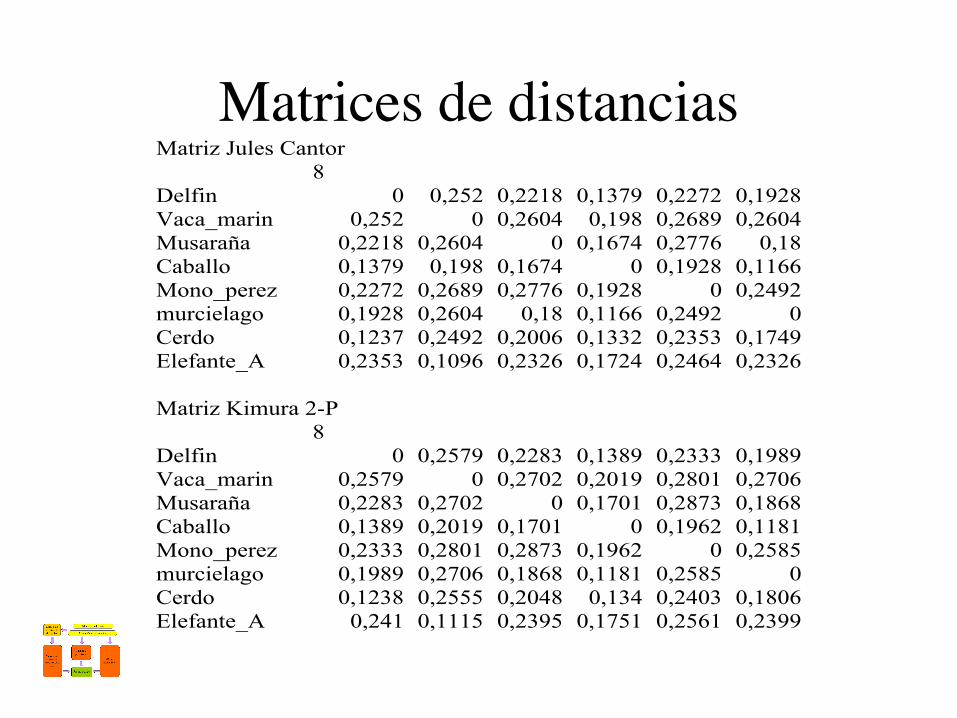
Matrices de distanciasMatriz Jules Cantor
8Delfin 0 0,252 0,2218 0,1379 0,2272 0,1928Vaca_marin 0,252 0 0,2604 0,198 0,2689 0,2604Musaraña 0,2218 0,2604 0 0,1674 0,2776 0,18Caballo 0,1379 0,198 0,1674 0 0,1928 0,1166Mono_perez 0,2272 0,2689 0,2776 0,1928 0 0,2492murcielago 0,1928 0,2604 0,18 0,1166 0,2492 0Cerdo 0,1237 0,2492 0,2006 0,1332 0,2353 0,1749Elefante_A 0,2353 0,1096 0,2326 0,1724 0,2464 0,2326
Matriz Kimura 2-P8
Delfin 0 0,2579 0,2283 0,1389 0,2333 0,1989Vaca_marin 0,2579 0 0,2702 0,2019 0,2801 0,2706Musaraña 0,2283 0,2702 0 0,1701 0,2873 0,1868Caballo 0,1389 0,2019 0,1701 0 0,1962 0,1181Mono_perez 0,2333 0,2801 0,2873 0,1962 0 0,2585murcielago 0,1989 0,2706 0,1868 0,1181 0,2585 0Cerdo 0,1238 0,2555 0,2048 0,134 0,2403 0,1806Elefante_A 0,241 0,1115 0,2395 0,1751 0,2561 0,2399

Métodos de distancias

Métodos de distancias

Resultado con
Neighbor Joining y matriz de Kimura
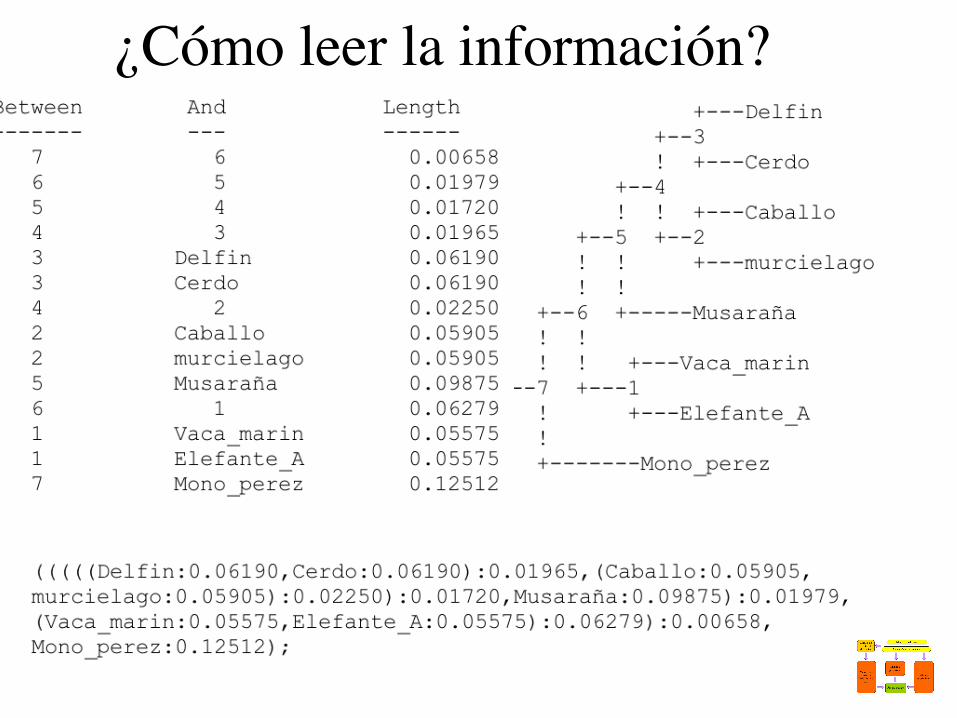
¿Cómo leer la información?

Máxima parsimonia

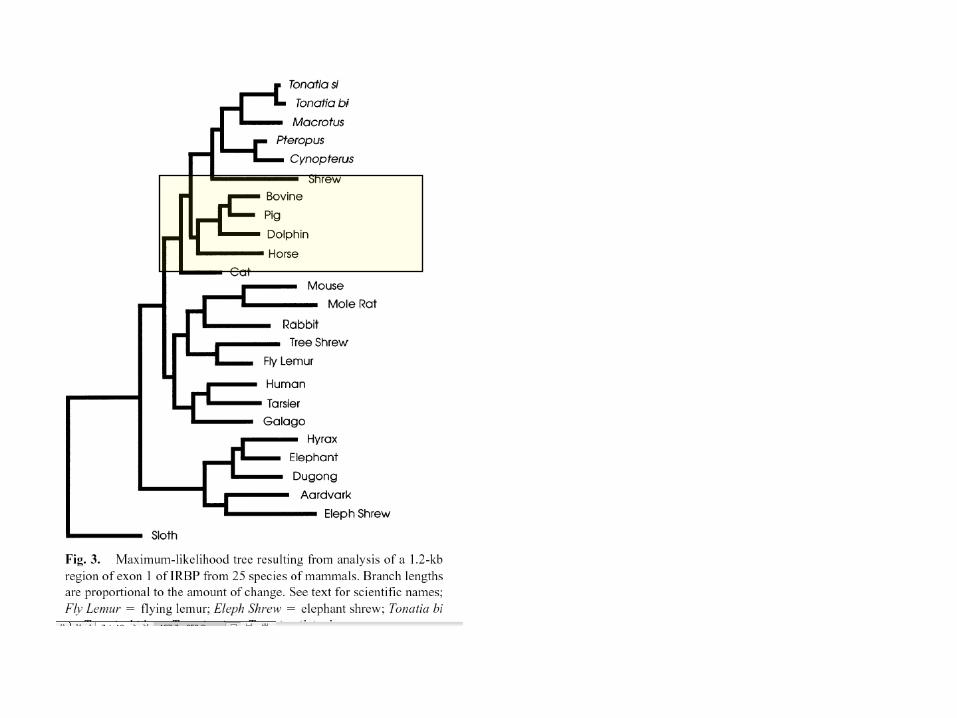
Máxima verosimilitud

Resultados Máx. Verosimilitud

Árbol consensoGeneralmente se obtienen resultados dispares según los métodos usadosEl uso de genes distintos o incluso, dentro del mismo gen, zonas distintas, pueden dar lugar a resultados diferentes. Se impone por lo tanto estipular un método para integrar toda esa información en un único árbol
El método más usado es el del consenso.
Veámoslo sobre nuestros resultados

¿Son iguales los arboles?
NJ
MP
MV

Nuestro consenso

Bootstrap y JackknifeSon técnicas de remuestreo para mejorar la estimación.
En el caso del bootstrap consiste en elegir tantas columnas del conjunto de datos como las que hay originalmente, pero con reemplazamiento, es decir, las columnas se pueden repetir.
En el caso del jacknife, se trata de elegir aleatóriamente un subconjunto, generalmente la mitad.
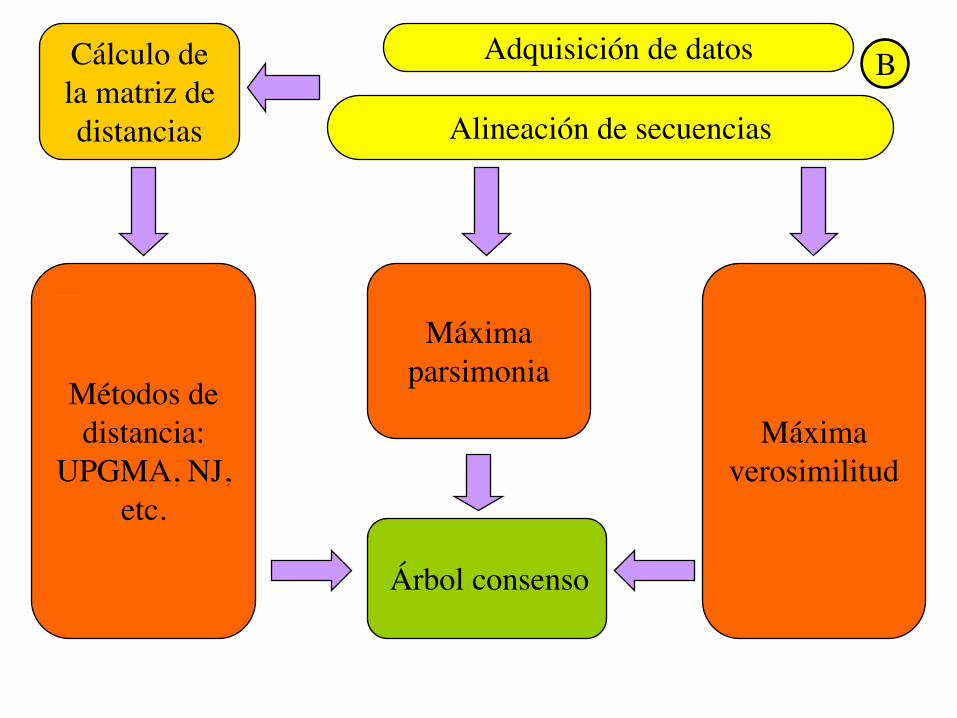
Adquisición de datos
Alineación de secuencias
Cálculo de la matriz de distancias
Métodos de distancia:
UPGMA, NJ, etc.
Máxima verosimilitud
Máxima parsimonia
Árbol consenso
B







