
SEMINARIO Amiloidosis hereditaria sistémica:
nuevos conocimientos y tratamiento
Carlos R. Mejía Chew
R3 Medicina Interna

Agenda
1. Nuevos conocimientos
a) Conceptos básicos de amiloidosis
b) Generalidades de Amiloidosis hereditaria
c) Presentación clínica de Amiloidosis hereditaria
2. Nuevos tratamientos
a) Dianas terapéuticas
b) Fármacos aprobados y en estudio

Agenda
1. Nuevos conocimientos
a) Conceptos básicos de amiloidosis
b) Generalidades de amiloidosis hereditaria
c) Presentación clínica de amiloidosis hereditaria
2. Nuevos tratamientos
a) Dianas terapéuticas
b) Fármacos aprobados y en estudio

Conceptos básicos de amiloidosis
• Rudolf Virchow 18511
• Nomenclatura: “AX”2
• Clasificación
– Sistémica o localizada
– Adquirida o hereditaria
1. Critical Reviews in Clinical Laboratory Sciences, 3 1 (4):325-354 (1994) 2. Amyloid, 2012; Early Online: 1–4 © 2012 Informa UK, Ltd. ISSN 1350-6129 print/ISSN
1744-2818 online. DOI: 10.3109/13506129.2012.734345
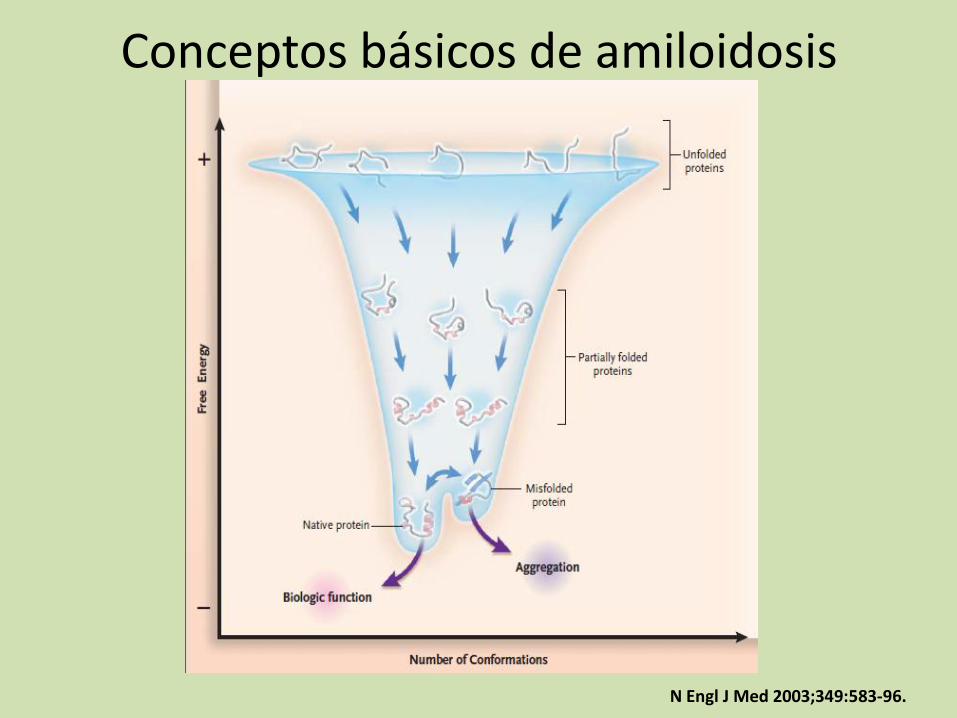
Conceptos básicos de amiloidosis
N Engl J Med 2003;349:583-96.

Conceptos básicos de amiloidosis
Annals of Medicine. 2007; 39: 200–207
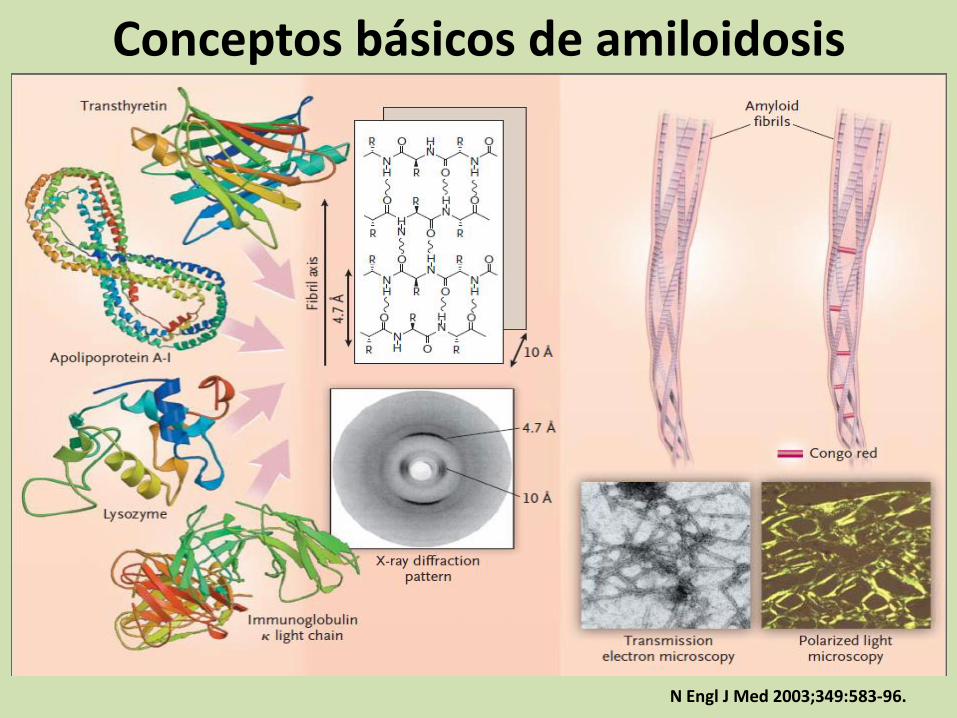
Conceptos básicos de amiloidosis
N Engl J Med 2003;349:583-96.

Agenda
1. Nuevos conocimientos
a) Conceptos básicos de amiloidosis
b) Generalidades de Amiloidosis hereditaria
c) Presentación clínica de Amiloidosis hereditaria
2. Nuevos tratamientos
a) Dianas terapéuticas
b) Fármacos aprobados y en estudio

Amiloidosis hereditaria: generalidades
• Enfermedades raras: incidencia < 1 por 100,0001
• Limitada a grupos poblacionales: – Portugal, Suecia, Japón y Finlandia
• Trastornos autosómico dominante
• Mutaciones puntuales (estructura primaria)
• Afección del SNP, corazón y riñón2
1. Amyloid: Int. J Exp. Clin. Invest. 7, 15-16 (2000) 2. N Engl J Med 2002, Vol. 346, No. 23

Amiloidosis hereditaria: generalidades
• Manifestación en la edad adulta
• Mutaciones principales: 1. Trantiretina (ATTR)
2. Fibrinogeno Aα (AFib)
3. Lisozima (ALys)
4. Apolipoproteína AI (AApoAI)
5. Apolipoproteína AII (AApoAII)
6. Gelsolina (AGel)
Amyloid: Int. J Exp. Clin. Invest. 7, 15-16 (2000)

Amiloidosis hereditaria: generalidades
• Evaluación diagnóstica:
– Biopsia
• Aspirado de grasa subcutánea abdominal
• Órgano afectado
• Mucosa gingival o rectal
• Nervio sural
– Tipificación del amiloide
• Inmunohistoquímica
• Secuenciación de ADN
Boston University. Amyloidosis Center. Abdominal Fat Tissue Aspirate Procedure 2007. Disponible en: http://www.youtube.com/watch?v=tctYTmxd9gQ

Agenda
1. Nuevos conocimientos
a) Conceptos básicos de amiloidosis
b) Generalidades de Amiloidosis hereditaria
c) Presentación clínica de Amiloidosis hereditaria
2. Nuevos tratamientos
a) Dianas terapéuticas
b) Fármacos aprobados y en estudio

• Mujer de 61 años que acude por dificultad para hablar, adormecimiento de la lengua, disfagia y pérdida de peso de 20 Kg de 3 años de evolución
• Antecedentes personales
– Depresión
– Neuropatía periférica inespecífica idiopática
– Síndrome de Intestino Irritable
• Antecedentes familiares
– Madre fallecida de ICC a los 72 años
– Tía materna fallecida a los 70 años, tenía amiloidosis renal y gastrointestinal
Caso clínico No. 1

Caso clínico No. 1
• Exploración: adenopatías periféricas,
e hipoestesia nociceptiva en manos y pies
• Electroforesis de proteínas séricas y urinarias normal
• Biopsia de la adenopatía axilar: Rojo Congo +
• Inmunohistoquímica inconclusa

Amiloidosis TTR: generalidades
• TTR:
– Proteína plasmática tetramérica (T4, Retinol-BP/vitA)
– Cromosoma 18
– Sintetizada en hígado, plexo coroideo y epitelio pigmentado de la retina
• Destabilización y disociación del tetrámero
Cell 2005;121:73

Amiloidosis TTR: generalidades
• Oporto, Portugal 1952:
– Andrade C. “A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves.” 1
• Polineuropatía amiloide familiar (FAP)
• > 100 mutaciones del gen de la TTR2
1. Brain 75 (3): 408–27 2. Cell 2005;121:73

Amiloidosis TTR: generalidades
• Mutaciones más comunes1:
– V30M (neuropatía)
– V122I (cardiomiopatía)
• Supervivencia media: 10 años TTR-FAP
• Tratamiento clásico: Transplante hepático2
1. J Med Genet 2005;42:953 2. Transplantation 2004;77:64

Amiloidosis TTR: manifestaciones clínicas
• SNP: + común
• SNA: ortostatismo y alteraciones gastrointestinales
• Corazón: cardiomiopatía restrictiva
• Vascular:
– SNC
• Amiloidosis leptomeningea (raro)
• Infartos cerebrales
• Afección renal

TTR-FAP: fisiopatología
Amyloid, 2012; 19(S1): 28–29

TTR-FAP: cuándo lo sospecho? • Antecedente familiar de TTR-FAP
• Polineuropatía axonal progresiva idiopática:
– Disfunción autonómica – Cardiopatía – Síndrome de túnel del carpo – Opacidades vítreas
• Diagnóstico diferencial:
– Polineuropatía inflamatoria crónica desmielinizante – Neuropatía autonómica hereditaria tipo 1 – AL, AApoAI y AGel
Amyloid, 2012; 19(S1): 25–27. Imágen: N Engl J Med 1975 Oct 30;293(18):914-5.

TTR-FAP: estadios clínicos
0 • Difunción autonómica mínima
I • Neuropatía en MMII. Ambulante
II
• Neuropatía MMII y MMSS
• Deambula con ayuda
III
• Neuropatía severa, disfunción autonómica intensa
• No ambulante
Amyloid and amyloidosis. Amsterdam: Excepta Medica; 1980. pp 88–98.

TTR-FAP: temprano vs. tardío
Amyloid, 2012; 19(S1): 55–57

TTR-FAP: cardiopatía…TTR-FAC
• Manifestación principal en1: – Val122Ile (descendencia africana)
– Ile68Leu (Italianos)
– Leu111Met (Daneses)
• Cardiomiopatía restrictica en la 5-7a década de la vida
• Menor infiltración de cavidades derechas que en pacientes con AL (87 vs. 59%, p = 0.03)2
1. Amyloid, 2012; 19(S1): 16–21 2. Amyloid, 2012; 19(2): 99–105

TTR-FAC: manifestaciones clínicas
1. Amyloid, 2012; 19(2): 99–105 2. Amyloid, 2012; 19(S1): 16–21

TTR-FAC: tratamiento
1. Transplante hepático ortotópico (OLT) en pacientes con ATTR-V30M
– En pacientes con variantes no ATTR-V30M, el OLT acelera la cardiomiopatía
2. Estabilizadores de TTR: diflunisal y tafamidis.
– Estudio Fx1B-201 (tafamidis 20 mg/día por 12 meses en 35 pacientes)
Amyloid, 2012; 19(S1): 16–21

Amiloidosis renal no-neuropática
• Benno Ostertag 19321
• Incluye el resto de genes causantes de amiloidosis hereditaria2:
– La cadena α A del Fibrinógeno (AFib)
– Apolipoproteina AI
– Apolipoproteina AII
– Lisozima
– Gelsolina
1. Z Menschl Vererbungs Konstit Lehre 1950;30:105–115. 2. Amyloid, 2012; 19(S1): 81–84

Amiloidosis Apolipoproteina AI
• 12 mutaciones descritas (Gly26Arg)
• Mutaciones en la porción N-terminal de Apo AI: renal, hepatica y ocasionalmente cardiaca
• Mutaciones en la porción C-terminal de Apo AI (a partir del residuo 90): cardiaca, cutánea y frecuentemente depositos laringeos
Amyloid, June 2005; 12(2): 75–87

• Enfermedad lentamente progresiva
• No hay tratamiento
– Apo AI se sintetiza en hígado y tracto intestinal
– El OLT disminuye un 50% la concetración plasmática de Apo AI
Amiloidosis Apolipoproteina AI
Amyloid, June 2005; 12(2): 75–87

Amiloidosis Fibrinógeno (AFib) • Se debe a una mutación en la proteina de la cadena Aα
del fibrinógeno (1993)
• 9 mutaciones (Glu526Val)
• Depósitos amiloides en los glomérulos: – Hipertensión – Proteinuria – Hiperazoemia
• Puede afectar a hígado y bazo
• No hay tratamiento específico
Amyloid, June 2005; 12(2): 75–87

Pregunta No.1 Varón de 60 años con un tipo de
amiloidosis hereditaria que tipicamente produce una facies de “mascara flácida colgante”. Usted sospecha el siguiente tipo de amiloidosis hereditaria: a) AA
b) ALys
c) AApoAII
d) AGel
e) ATTR

Amiloidosis Gelsolina (AGel): generalidades
• Jouko Meretoja en 1969
• FAP tipo IV o amiloidosis hereditaria tipo Finlandes
• 2 mutaciones en el cromosoma 9: – G654A
– G654T
• Penetrancia cercana del 100%
Critical Reviews in Biochemistry and Molecular Biology, 2012; 47(3): 282–296

AGel: fisiopatología
Critical Reviews in Biochemistry and Molecular Biology, 2012; 47(3): 282–296

AGel: manifestaciones clínicas
• Distrofia corneal reticular
• Paralisis facial periférica
• Cutis laxa
• Nefropatía
1. Amyloid, 2012; 19(S1): 30–33 2. Amyloid: Int. .I.E xp. Clin. Invest. 5, 55-66( 1998)

AGel: tratamiento
Disponible en: www.cartoonstock.com

Resumen: Amiloidosis hereditaria
Amyloid, June 2005; 12(2): 75–87

Agenda
1. Nuevos conocimientos
a) Conceptos básicos de amiloidosis
b) Generalidades de Amiloidosis hereditaria
c) Presentación clínica de Amiloidosis hereditaria
2. Nuevos tratamientos
a) Dianas terapéuticas
b) Fármacos aprobados y en estudio

Dianas terapéuticas
US Neurology, 2012;8(1):24–32

Dianas terapéuticas
Amyloid 2006;13:236–249.
“Datos in vitro indican que la disociación del tetrámero de transtiretina parece ser el paso limitante para la formación de fibrilla amiloide”

Agenda
1. Nuevos conocimientos
a) Conceptos básicos de amiloidosis
b) Generalidades de Amiloidosis hereditaria
c) Presentación clínica de Amiloidosis hereditaria
2. Nuevos tratamientos
a) Dianas terapéuticas
b) Fármacos aprobados y en estudio

Fármacos aprobados: Tafamidis
• Estabilizador cinético de la transtiretina (TKS)
• Aprobado por la EMA: 21/07/2011
• Polineuropatía amiloidótica familiar estadio 1
Neurology® 2012;79:785–792

Tafamidis: mecanismo de acción
PNAS | June 12, 2012 | vol. 109 | no. 24 | 9631

Tafamidis: la evidencia
• Objectivo: Eficacia y seguridad de tafamidis en pacientes con TTR-FAP V30M
• Objetivo primario (ITT y PP): Mejoría scores de NIS-LL y TQOL
• Objetivos secundarios: – Cambios en la función neurológica (fibras nerviosas)
– Estado nutricional (IMC modificado)
– Estabilización de la TTR (ensayos inmunoturbidimétrico)
Neurology® 2012;79:785–792

Tafamidis: metodología
n = 128
n =65
tafamidis 20mg/día n = 52
n = 63
placebo n = 50
n =162
Neurology® 2012;79:785–792
OLT
Seguimiento: semanas 2,4,8 y 12 mes 6,12 y 18

Tafamidis: objetivo primario
Neurology® 2012;79:785–792

Tafamidis: objetivos secundarios
Neurology® 2012;79:785–792
Menor progresión en fibras nerviosas pequeñas
Mejoría del estado nutricional

Tafamidis: limitaciones
• Score menos severo del NIS-LL en el brazo de tafamidis
• Un solo sitio proporcionó al 58% de los pacientes
– No evidencia de eficacia al excluir este sitio
• No significativo en análisis ITT

Tafamidis: conclusiones
• Estabiliza la molécular de TTR
• Inhibe la formación de amiloide y la progresión de la enfermedad
Neurology® 2012;79:785–792

Fármacos en estudio: Diflunisal
• Aleatorizado, doble ciego, controlado con placebo, multicéntrico e internacional
• Objetivo primario: Mejoría de la neuropatía
Amyloid, 2012; 19(S1): 37–38
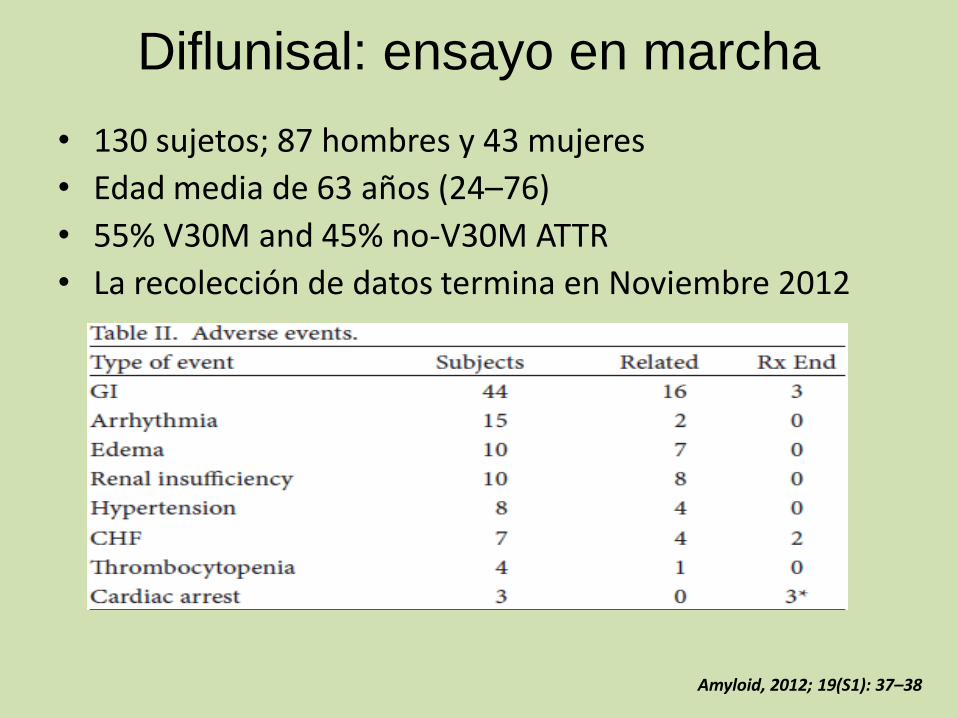
Diflunisal: ensayo en marcha
• 130 sujetos; 87 hombres y 43 mujeres
• Edad media de 63 años (24–76)
• 55% V30M and 45% no-V30M ATTR
• La recolección de datos termina en Noviembre 2012
Amyloid, 2012; 19(S1): 37–38

Fármacos en estudio: otros
Amyloid, 2012; 19(S1): 34–36; 34–44;-45–46

Ciudad de Antigua Guatemala, Sacatepéquez, Guatemala







