
INSTITUTO POLITÉCNICO NACIONAL UNIDAD PROFESIONAL INTERDISCIPLINARIA
DE BIOTECNOLOGÍA
MANUAL PARA LABORATORIO DE
MÉTODOS CUANTITATIVOS
INSTITUTO POLITÉCNICO NACIONAL
SECRETARÍA ACADÉMICA
DIRECCIÓN DE EDUCACIÓN SUPERIOR
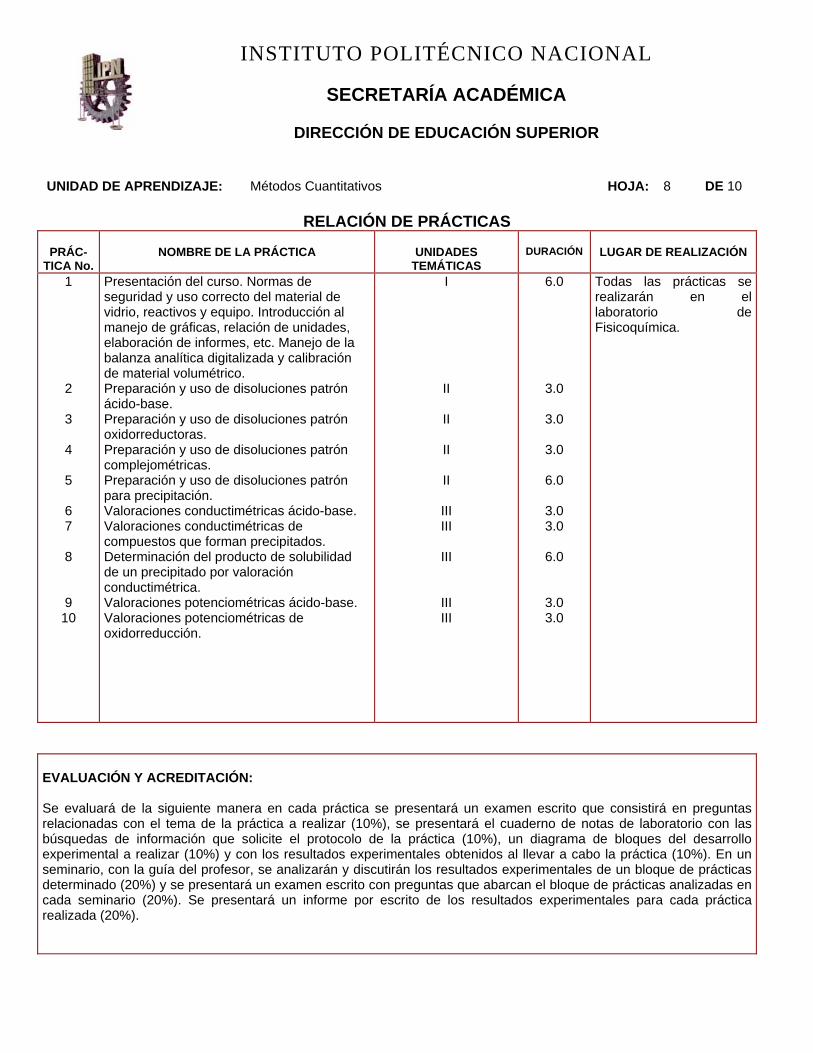
UNIDAD DE APRENDIZAJE: Métodos Cuantitativos HOJA: 8 DE 10
RELACIÓN DE PRÁCTICAS
PRÁC-TICA No.
NOMBRE DE LA PRÁCTICA
UNIDADES
TEMÁTICAS
DURACIÓN
LUGAR DE REALIZACIÓN
1
2
3
4
5
6 7
8
9 10
Presentación del curso. Normas de seguridad y uso correcto del material de vidrio, reactivos y equipo. Introducción al manejo de gráficas, relación de unidades, elaboración de informes, etc. Manejo de la balanza analítica digitalizada y calibración de material volumétrico. Preparación y uso de disoluciones patrón ácido-base. Preparación y uso de disoluciones patrón oxidorreductoras. Preparación y uso de disoluciones patrón complejométricas. Preparación y uso de disoluciones patrón para precipitación. Valoraciones conductimétricas ácido-base. Valoraciones conductimétricas de compuestos que forman precipitados. Determinación del producto de solubilidad de un precipitado por valoración conductimétrica. Valoraciones potenciométricas ácido-base. Valoraciones potenciométricas de oxidorreducción.
I II II II II
III III
III
III III
6.0
3.0
3.0
3.0
6.0
3.0 3.0
6.0
3.0 3.0
Todas las prácticas se realizarán en el laboratorio de Fisicoquímica.
EVALUACIÓN Y ACREDITACIÓN: Se evaluará de la siguiente manera en cada práctica se presentará un examen escrito que consistirá en preguntas relacionadas con el tema de la práctica a realizar (10%), se presentará el cuaderno de notas de laboratorio con las búsquedas de información que solicite el protocolo de la práctica (10%), un diagrama de bloques del desarrollo experimental a realizar (10%) y con los resultados experimentales obtenidos al llevar a cabo la práctica (10%). En un seminario, con la guía del profesor, se analizarán y discutirán los resultados experimentales de un bloque de prácticas determinado (20%) y se presentará un examen escrito con preguntas que abarcan el bloque de prácticas analizadas en cada seminario (20%). Se presentará un informe por escrito de los resultados experimentales para cada práctica realizada (20%).
INSTITUTO POLITÉCNICO NACIONAL
SECRETARÍA ACADÉMICA
DIRECCIÓN DE EDUCACIÓN SUPERIOR
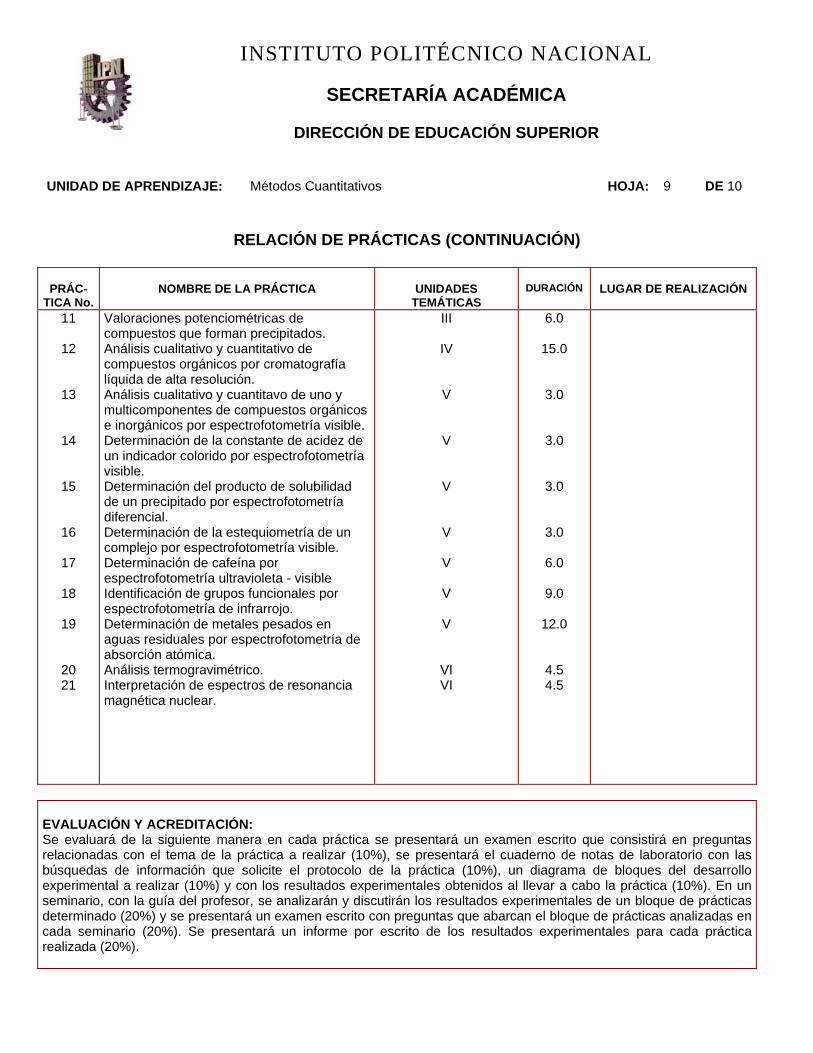
UNIDAD DE APRENDIZAJE: Métodos Cuantitativos HOJA: 9 DE 10
RELACIÓN DE PRÁCTICAS (CONTINUACIÓN)
PRÁC-
TICA No.
NOMBRE DE LA PRÁCTICA
UNIDADES
TEMÁTICAS
DURACIÓN
LUGAR DE REALIZACIÓN
11
12
13
14
15
16
17
18
19
20 21
Valoraciones potenciométricas de compuestos que forman precipitados. Análisis cualitativo y cuantitativo de compuestos orgánicos por cromatografía líquida de alta resolución. Análisis cualitativo y cuantitavo de uno y multicomponentes de compuestos orgánicos e inorgánicos por espectrofotometría visible. Determinación de la constante de acidez de un indicador colorido por espectrofotometría visible. Determinación del producto de solubilidad de un precipitado por espectrofotometría diferencial. Determinación de la estequiometría de un complejo por espectrofotometría visible. Determinación de cafeína por espectrofotometría ultravioleta - visible Identificación de grupos funcionales por espectrofotometría de infrarrojo. Determinación de metales pesados en aguas residuales por espectrofotometría de absorción atómica. Análisis termogravimétrico. Interpretación de espectros de resonancia magnética nuclear.
III
IV
V
V
V
V
V
V
V
VI VI
6.0
15.0
3.0
3.0
3.0
3.0
6.0
9.0
12.0
4.5 4.5
EVALUACIÓN Y ACREDITACIÓN: Se evaluará de la siguiente manera en cada práctica se presentará un examen escrito que consistirá en preguntas relacionadas con el tema de la práctica a realizar (10%), se presentará el cuaderno de notas de laboratorio con las búsquedas de información que solicite el protocolo de la práctica (10%), un diagrama de bloques del desarrollo experimental a realizar (10%) y con los resultados experimentales obtenidos al llevar a cabo la práctica (10%). En un seminario, con la guía del profesor, se analizarán y discutirán los resultados experimentales de un bloque de prácticas determinado (20%) y se presentará un examen escrito con preguntas que abarcan el bloque de prácticas analizadas en cada seminario (20%). Se presentará un informe por escrito de los resultados experimentales para cada práctica realizada (20%).

Métodos Cuantitativos Manual de Prácticas de Laboratorio
PARTE 1 VALORACIONES VOLUMÉTRICAS
Las valoraciones o titulaciones se basan en la reacción entre un analito y una disolución patrón conocida como reactivo valorante ó titulante. En una valoración volumétrica la disolución patrón ó reactivo titulante se añade desde una bureta y la reacción transcurre en un vaso de precipitados ó preferentemente en un matraz Erlenmeyer, como se muestra en la siguiente figura.
Las valoraciones volumétricas consisten en la medida del volumen de una disolución de concentración conocida necesario para reaccionar completamente con el analito. La reacción entre el reactivo titulante y el analito debe ser completa (que el valor de la constante de equilibrio sea grande) y rápida. Las valoraciones volumétricas más comunes se basan en las reacciones ácido-base, de oxidorreducción, de formación de complejos y de precipitación. En las valoraciones volumétricas el punto de equivalencia químico se detecta por el cambio de color de un indicador ó por el cambio de color de la disolución valorada. Las valoraciones volumétricas son ampliamente utilizadas para la determinación de ácidos, bases, oxidantes, reductores, iones metálicos, proteínas y otras especies más. TÉRMINOS EMPLEADOS EN EL ANÁLISIS VOLUMÉTRICO Un análisis volumétrico es cualquier procedimiento basado en la medida del volumen de reactivo necesario para que reaccione con el analito. La unidad principal de volumen en el sistema métrico es el litro (L). La milésima parte de un litro se denomina mililitro (mL). Esta magnitud corresponde en plena medida a un centímetro cúbico (cm3), es decir a la milésima parte del decímetro cúbico. Soluciones patrón o estándar Las disoluciones que contienen concentraciones conocidas de analito se llaman disoluciones patrón o estándar. Las disoluciones patrón desempeñan una función principal en todos los métodos de análisis por valoración. Por ello, es necesario considerar cuáles son las propiedades deseables para estas disoluciones, como se preparan y como se utilizan. Los reactivos utilizados como referencia se dividen en patrones primarios y patrones secundarios.
1

Métodos Cuantitativos Manual de Prácticas de Laboratorio
2
Patrones primarios Un patrón primario, es una sustancia suficientemente pura para ser pesada y ser usada directamente. Un patrón primario debe tener las siguientes características: a) Pureza mínima del 99.9% b) No descomponerse en condiciones normales de almacenamiento c) Debe ser estable al calor y al vacío, porque es preciso eliminar trazas de agua adsorbida de la
atmósfera d) Debe tener un peso molecular elevado para disminuir el porcentaje de error en la pesada de
los mismos. Los reactivos que no cumplen estos criterios reciben el nombre de patrones secundarios este es un reactivo cuya pureza hay que establecer por comparación con un patrón primario. Uso de patrones primarios Existen dos formas básicas para utilizar los patrones primarios: a) Método de pesadas separadas. Se pesa por triplicado la cantidad estequiométrica de patrón
primario, se disuelve en una medida de agua, se adiciona el indicador y se valora con la solución ácida o básica preparada, la cual se coloca en la bureta. El cálculo de la normalidad se hace en base al número de moles o peso equivalente.
b) Método de preparación de una disolución patrón. Se realiza el cálculo para preparar una
solución de normalidad conocida, se pesa, se disuelve el patrón primario y se afora a un volumen conocido. Se toma una alícuota de la sustancia ácido-base a estandarizar, se adiciona el indicador y se valora con el patrón primario que debe colocarse en la bureta.
Punto de equivalencia y punto final El punto de equivalencia de una valoración es aquel en el que la cantidad de reactivo titulante agregado es igual a la cantidad exactamente requerida para que reaccione estequiométricamente con el analito. Encontrar el punto de equivalencia es el fin ideal que se persigue en una titulación. En realidad, lo que se mide es el punto final. El punto final de una valoración se caracteriza por un cambio brusco en una propiedad física o química de la disolución. Indicadores Un indicador es un compuesto que posee una propiedad física (generalmente el color) que cambia bruscamente en las proximidades del punto de equivalencia. El cambio se debe a la rápida desaparición del analito o a la rápida aparición del reactivo titulante en el punto de equivalencia.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
PRÁCTICA No. 1 BALANZA ANALITICA DIGITAL Y CALIBRACIÓN DE MATERIAL
VOLUMÉTRICO 1. OBJETIVOS
1.1 El alumno utilizará la balanza analítica para determinar el peso de diferentes cuerpos. 1.2 El alumno conocerá y usará de manera adecuada el material volumétrico. 1.3 El alumno adquirirá los conocimientos necesarios para calibrar el material volumétrico. 1.4 El alumno determinará la precisión, en términos de desviación estándar, que puede
obtener con su material volumétrico. 2. INTRODUCCIÓN
2.1 Balanza electrónica Una balanza analítica (figura 1) es un instrumento para pesar diferentes cuerpos. La capacidad de la balanza analítica, generalmente, no debe ser mayor de 100-200 g, con una precisión de al menos 0.1 mg de su capacidad máxima. Muchas balanzas analíticas modernas superan la precisión de 0.001 mg de su capacidad total.
e por
Figura 1. Balanza analítica digital. La cantidad de materia que contiene una sustancia o un cuerpo equivale a su masa y es invariable. El peso de un objeto es la medida de la fuerza que la gravedad terrestre ejerce sobre él. La fuerza de la gravedad varía con la latitud y altitud terrestres, de acuerdo a ales variaciones, el peso de un objeto puede variar. La masa de un objeto se midtcomparación de su peso con el de una masa conocida. Las balanzas analíticas más comunes, microbalanzas, tienen una capacidad máxima que varía en un intervalo entre 160 y 200 g. Con éstas balanzas las mediciones se pueden hacer con una precisión de ± 0.1 mg. Las balanzas semi-microanalíticas tienen una carga máxima de 10 a 30 g con una precisión de ± 0.01 mg. Una balanza microanalítica típica tiene una capacidad de 1 a 3 g y una precisión de ± 0.001 mg. En la figura 2 se muestra un diagrama de la balanza analítica electrónica que sirve para explicar el fundamento de su operación. El platillo se encuentra sobre un cilindro metálico hueco rodeado por una bobina que está fija sobre el polo interior de un imán cilíndrico permanente. Una corriente eléctrica de la bobina crea un campo magnético que sostiene el cilindro, el platillo, el brazo indicador, así como cualquier carga que esté sobre el platillo. La corriente se ajusta de modo que el nivel del brazo indicador esté en la posición
3

Métodos Cuantitativos Manual de Prácticas de Laboratorio
4
obina, creando un campo magnético mayor, lo que ace regresar al platillo a su posición nula original. Un dispositivo como éste, en el cual
una pequeña corriente eléctrica hace que un sistema mecánico mantenga una posición nula, se llama sistema servo. La corriente necesaria para conservar el platillo y el objeto en la posición nula es directamente proporcional a la masa del objeto y es más fácilmente medida, digitalizada y mostrada en la pantalla de la balanza. Para calibrar una balanza electrónica se necesita emplear una masa patrón y ajustar la corriente de forma que la masa patrón aparezca en la pantalla.
nula cuando el platillo está vacío. Al colocar un objeto sobre el platillo éste y el brazo indicador se mueven hacia abajo, lo que aumenta la cantidad de la luz que choca en la foto celda del detector en la posición nula. El incremento de corriente de la foto celda es amplificado y sirve para alimentar la bh
Figura 2. Diagrama de una balanza analítica electrónica.
Para pe n recipiente limpio en el platillo de la baun botón
2.1.1 Cuidados básicos para la balanza analítica digital.
) Dejar siempre la balanza en el modo "stand by", evitando la necesidad de nuevo to ("warm up").
corrientes de
e) as durante la pesada.
quede sobre las partes móviles de la balanza.
medida y su contenido deben estar a la misma
sar una sustancia química se coloca primero ulanza. La masa del recipiente vacío se llama tara. En la mayoría de las balanzas hay
para ajustar la tara a cero.
a) Verificar siempre la nivelación de la balanza. b) Dejar siempre la balanza conectada a la toma y prendida para mantener el
equilibrio térmico de los circuitos electrónicos. c
tiempo de calentamiend) La balanza debe estar colocada en una mesa firme y fuera de las
aire y del polvo. Las puertas de la balanza deben permanecer cerrad
f) Emplear un pincel o una brocha pequeña para eliminar cualquier residuo de materiales o polvo que
2.1.2 Recipientes de medida.
a) Usar siempre el recipiente para pesar, de menor capacidad posible. b) La temperatura del recipiente de
temperatura del ambiente de la cámara de medida.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
5
ente con los dedos al ponerlos o sacarlos de
2.1.3 lytical Standard. pesar como máximo 250 g.
a) Encender con la tecla ON/Tare. 0
a tecla ON/Tare llevar a cero.
g) cipiente conteniendo el reactivo de interés.
2.1.4 za Kern ALS 220-4.
máximo 220 g con una precisión de
rque 0.0000 la balanza está lista para
) Colocar el material a pesar en el centro del platillo y esperar a que aparezca el
e) Colocar el material a pesar en el recipiente. el material con una precisión de 0.1 mg
g) Retirar el recipiente conteniendo el material de interés.
sando la tecla ON/OFF.
a no opera correctamente informe inmediatamente al instructor. Los e
2.2 Materi
Para mpipeta 2.2.1
as buretas (figura 3) se usan para las titulaciones y son tubos de vidrio de forma cilíndrica fabricados con precisión que tienen una graduación que permite medir el volumen de líquid que se encuentra en la parte
ferior. Las buretas, generalmente, se gradúan en mililitros (25 ó 50 mL) y sus
c) Nunca tocar los recipientes directamla cámara de medida.
Procedimiento de Operación de la balanza OHAUS AnaLa balanza OHAUS Analytical Standard permite
b) Esperar a que alcance el equilibrio y aparezca en la pantalla 0.000c) Colocar en el centro del platillo de pesada el recipiente donde va a pesar. d) Con le) Pesar el reactivo requerido. f) Anotar el peso del reactivo con precisión de 0.1 mg.
Retirar el reh) Ajustar a cero pulsando la tecla ON/ Tare.
Procedimiento de Operación de la balanLa balanza Kern ALS 220-4 permite pesar como0.1 mg. a) Conectar la balanza con la tecla ON/OFF. b) Luego que el indicador de peso ma
funcionar. c
peso. d) Apretar la tecla TARE para ajustar a cero.
f) Anotar el peso d
h) Ajustar a cero pulsando la tecla TARE. i) Apagar la balanza pul
Si la balanzstudiantes no deben intentar repararla por sí mismos.
al volumétrico
edir con precisión los volúmenes en el análisis cuantitativo se utilizan buretas, s y matraces aforados.
Buretas L
o vertido a través de una llaveindécimas; la división cero se halla en la parte superior de la bureta. Antes de llenar la bureta con la disolución, cuyo volumen se quiere medir, se debe lavar bien, el lavado de la bureta se puede terminar cuando el agua de lavado se escurra uniformemente por las paredes sin dejar alguna parte de gota.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
Figura 3. Diferentes tipos de buretas
Para no esperar a que la bureta lavada se seque, a fin de eliminar el agua, se le enjuaga dos veces con pequeñas cantidades de la disolución con la cual se piensa efectuar la titulación. La bureta se llena mediante un embudo que se introduce en su orificio superior y que luego se retira. Si durante la titulación el embudo no se retira, el líquido remante puede escurrir del embudo y la medición del volumen será inexacta. Es indispensable prestar una atención especial a que en el estrecho tubo inferior de la bureta no queden burbujas de aire. Para eliminarlas, se abre la llave y se deja salir de la bureta un fuerte chorro de líquido que se recoge en un vaso o un matraz.
fin posible, e p pios.
2.2.2
tudiada, son tubos estrechos y largos que se
A de que las buretas durante su almacenamiento, se ensucien lo menosueden llenar con agua hasta el borde y tapar con tubos de ensayo lims
Pipetas volumétricas Las pipetas volumétricas están destinadas para la medición precisa de volúmenes
inidos de la disolución esdefensanchan en el centro (figura 4a). En la parte superior estrecha de la pipeta hay una marca anular (aforo) hasta la cual se debe llenar de líquido. Las pipetas se constituyen, principalmente, para 100, 50, 25, 20, 5 y 1 mL.
6

Métodos Cuantitativos Manual de Prácticas de Laboratorio
a) b) c)
Figura 4. a) Pipeta volumétrica, b) Pipeta graduada y c) soporte para pipetas
Antes de llenar la pipeta con la solución estudiada, se lava a fondo para eliminar la
y otras impurezas y se enjuaga dos veces con la solución estudiada a fin de
uego, sosteniendo la parte superior de la pipeta entre el pulgar y el dedo índice de la mano derecha y sumergiendo profundamente su extremo inferior en el líquido, se
rga por succión m por arriba de la marca del aforo
plado de la última gota, uno no
retas y tienen la misma
2.2.3 Matraces volumétricos
grasaque no queden gotas de agua. L
ca(us
hasta, aproximadamente, 2 car preferentemente, perilla de succión). En caso de que no se este usando
perilla de succión, se cierra rápidamente el orificio superior de la pipeta con el dedo índice ligeramente húmedo (pero no mojado) y, entre abriéndolo, el líquido se deja escurrir muy lentamente hasta que el borde inferior del menisco llegue a la marca del aforo (los ojos deben hallarse al nivel de la marca). La pipeta se pasa a un recipiente preparado previamente para ese fin y, colocándola en posición vertical se deja escurrir el líquido. Luego con la punta de la pipeta se toca la pared del recipiente y se esperan 15 segundos. A continuación, la pipeta se retira del recipiente sin prestar atención a la gota remanente en ella. De ningún modo se debe soplar para sacar esa gota, lo importante es que su cantidad en todos los casos sean igual. Esto se obtiene, empleando siempre el método descrito de vaciar la pipeta. Si se recurre al sopuede, evidentemente, crear tales condiciones constantes, porque la fuerza del soplado será variable en diferentes casos. Además de pipetas volumétricas, a veces se emplean las llamadas pipetas graduadas (figura 4b), que su forma recuerdan las bugraduación. Una vez terminado el trabajo, las pipetas se lavan y se colocan en un soporte especial (figura 4c). Para preservarlas contra el polvo se cubren con tubos de ensayo invertidos o con tapones de algodón.
7

Métodos Cuantitativos Manual de Prácticas de Laboratorio
8
icos (figura 5) son recipientes de fondo plano con un cuello rgo y delgado, alrededor del cual está trazada una marca anular (aforo).
Estos recipientes se usan para diluir la disolución estudiada a un volumen definido y para preparar disoluciones valoradas. De esta manera, a diferencia de las buretas y de las pipetas, los matraces volumétricos no se destinan, generalmente, para emitir un volumen determinado de líquido. Los matraces volumétrla
Figura 5. Ma es volumétricos
Como todo recipiente graduado, el matraz volumétrico se debe lavar a fondo antes
races volumétricos están destinados para diluir una studia, no se deben enjuagar con esta
se hace en el caso de las pipetas y buretas.
el m uele midiendo la masa de agua vertida por el cipiente ontenid l, y ut a d e o ertir
masa en volumen. Se debe conocer la temperatura del agua en el momento de la ió ra realiz correc r temperatura para que los resultados se
orten grados Celsius. El material que se va a calibrar debe estar
trac
de utilizarlo. Puesto que los matcantidad definida de disolución que se edisolución como El matraz se llena primero usando un embudo y al final, mediante un cuentagotas, se agrega líquido gota a gota hasta que el borde inferior del menisco llegue a la marca del aforo. En este caso los ojos del observador deben hallarse al nivel de la marca del matraz. Una vez llevando el volumen de la disolución a la marca, el matraz se debe cerrar con un tapón y agitar bien la disolución. Hay que tener presente que no se permite calentar las disoluciones en los matraces volumétricos.
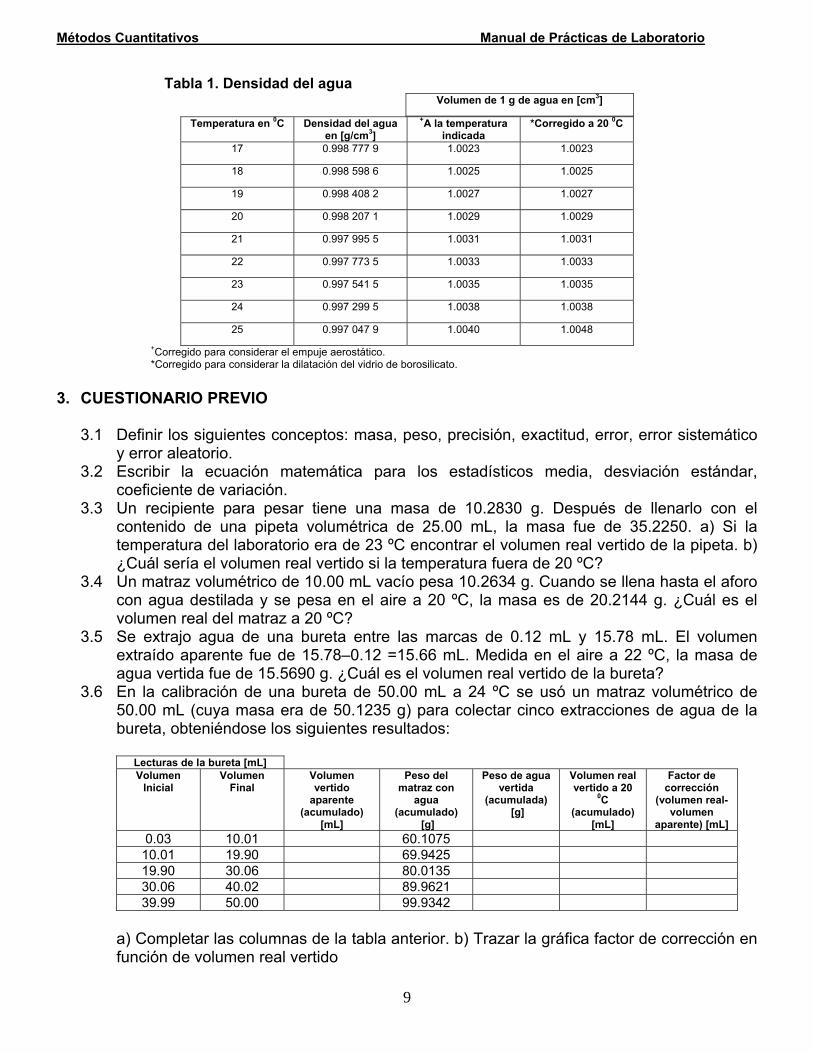
2.2.4 Calibración de material volumétrico La calibración del material volumétrico que se emplea para las determinaciones analíticas que se realizan en el Laboratorio de Métodos Cuantitativos, consiste en determinar la precisión que se puede alcanzar con dicho material a fin de medir exactamente los volúmenes vertidos ó contenidos en él. En los trabajos de gran exactitud se debe considerar la dilatación del vidrio y la dependencia de la concentración de las disoluciones con la temperatura. En la tabla 1 se muestra la dependencia del volumen de agua con la temperatura y se muestran las correcciones por considerando el empuje aerostático y la dilatación del vidrio.
calibLa re
ración d ó c
aterial sa en é
ar la
hacerseilizando l ensidad d ese líquid para conv
calibrac n pa ci poón repperfectamente limpio.
a 20
Métodos Cuantitativos Manual de Prácticas de Laboratorio
9
bla 1. ad del ag a
Ta Densid u
Temperatura en 0C Densidad del agua en [g/cm3]
+A la temperatura indicada
*Corregido a 20 0C
17 0.998 777 9 1.0023 1.0023
18 0.998 598 6 1.0025 1.0025
19 0.998 408 2 1.0027 1.0027
20 0.998 207 1 1.0029 1.0029
21 0.997 995 5 1.0031 1.0031
22 0.997 773 5 1.0033 1.0033
23 0.997 541 5 1.0035 1.0035
24 0.997 299 5 1.0038 1.0038
25 0.997 047 9 1.0040 1.0048 +Corregido para considerar el empuje aerostático.
l vidrio de bor*Corregido para considerar la dilatación de osilicato.
3. CUE
3.1 a, peso, precisión, exactitud, error, error sistemático
3.2 n matemática para los estadísticos media, desviación estándar, ión.
3.3 pesar tiene una masa de 10.2830 g. Después de llenarlo con el ipeta volumétrica de 25.00 mL, la masa fue de 35.2250. a) Si la
laboratorio era de 23 ºC encontrar el volumen real vertido de la pipeta. b) lumen real vertido si la temperatura fuera de 20 ºC?
.00 mL vacío pesa 10.2634 g. Cuando se llena hasta el aforo sa en el aire a 20 ºC, la masa es de 20.2144 g. ¿Cuál es el
3.5 extraídagua verti
3.6 En la cali50.00 mLbureta, ob
Lecturas de la bureta
REVIO STIONARIO P
Definir los siguientes conceptos: masy error aleatorio.
la ecuacióEscribir coeficiente de variacUn recipiente para ontenido de una pc
temperatura del Cuál sería el vo¿
3.4 Un matraz volumétrico de 10con agua destilada y se pevolumen real del matraz a 20 ºC? Se ex otraj agua de una bureta entre las marcas de 0.12 mL y 15.78 mL. El volumen
o aparente fue de 15.78–0.12 =15.66 mL. Medida en el aire a 22 ºC, la masa de da fue de 15.5690 g. ¿Cuál es el volumen real vertido de la bureta? bración de una bureta de 50.00 mL a 24 ºC se usó un matraz volumétrico de (cuya masa era de 50.1235 g) para colectar cinco extracciones de agua de la teniéndose los siguientes resultados:
[mL] Volumen
Inicial Vol
Final vertido
]
el matraz con
Peso de agua vertida
Volumen real vertido a 20
0C (acumulado)
[mL]
Factor de corrección
(volumen real-volumen
aparente) [mL]
umen Volumen Peso d
aparente (acumulado)
agua (acumulado)
(acumulada) [g]
[mL [g] 0.03 10.01 60.10 75 10.01 19.90 69.9425 19.90 30.06 80.01 35 30.06 40.02 89.96 21 39.99 50.00 99.9342
a) Completar las columnas de la tabla anterior. b) Trazar la gráfica factor de corrección en función
men de 1 g ua en [cm3Volu de ag ]
de volumen real vertido

Métodos Cuantitativos Manual de Prácticas de Laboratorio
10
3.7 Dependiea) ¿Que para ser cy los matraces volumétricos para ser considerados clase B?
3.8 Realiz 4. PARTE EXPER
4.1 Material
2 matraces volumétricos de 25.00 mL 1 p lum e 5.02 pipetas graduadas de 5 1 termómetro 1 frasco lavador con agua destilada 1 embudo 1 soporte universal 1 pinzas 1 balanza analítica papel adsorbente
d
4.2 Desarrollo
4.2.1 Calibración a) Medir la temperatura del agua destilada que se va a usar para la
experimentación. Llenar la bureta con agua destilada. Expulsar cualquier burbuja de aire retenida en la punta. Ajustar el nivel del menisco en 0.00 mL. Dejar la bureta en reposo durante 5 minutos. Mientras, se pesa un matraz volumétrico vacío y seco de 25.00 mL con su tapón (repetir la pesada del matraz tres veces distintas). Verificar que no existan fugas en la bureta.
ndo de la precisión, el material volumétrico se clasifica como clase A o clase B. precisión deben alcanzar las pipetas, las buretas y los matraces volumétricos onsiderados clase A? b) ¿Que precisión deben alcanzar las pipetas, las buretas
ar un diagrama de bloques que esquematice la parte experimental de la práctica.
IMENTAL
y reactivos 1 bureta de 25.00 mL
ipeta vo étrica d 0 mL mL
Agua estilada
experimental
de una b reta de 2u 5.00 mL
Temperatura del agua en 0C
Peso del matraz vacío y seco con su tapón
Peso en gramos
1
2
3
Promedio
agua en el matraz previamente
raz y se determina la masa
b) Se vierten aproximadamente 5.00 mL depesado. Tapar el matraz para evitar perdidas por evaporación. Se deja durante 30 segundos, que la película del líquido adherida a la pared escurra antes de efectuar la lectura de la bureta. Todas las lecturas se efectúan hasta el centésimo de mL más cercano. Se pesa de nuevo el mat
transferida de agua por diferencia de peso.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
11
c) Ahora se extraen de la bureta 5.00 a 10.00 mL) en el mismo matraz volumétrico, se determina la masa de agua desalojada. Se repite el procedimiento para 15.00, 20.00 y 25.00 mL.
Datos experimental de de e 25Lecturas de la bureta [m
otros 5.00 mL (de
es la calibración la bureta d .00 mL L]
Volumen Inicial
Volumen Fina
Volumen vertido
aparente (acumulado)
[mL]
Peso del matraz con
agua mulado) [g]
Peso de agua rtida
(ac mulada) [g]
Volumen real vertido a 20
0C (acumulado)
[mL]
Factor de corrección
(volumen real-volumen
aparente) [mL]
l
(acu
veu
4.2.2 Calibración de un matraz volumétrico de 25.00 mL
a) Pesar e o y seco con su tapón (efectuar tres pes das indep
Peso del matraz vacío y seco con su tapón.
en os
l matraz volumétrico de 25.00 mL vacía endientes).
Peso gram
1
2
3
Promedio
b) Con un embudo llenar el matraz con agua destilada hasta que alcance un nivel
inferior al de la marca del aforo. Retirar el embudo procurando no mojar el
sorbente. Después de efectuar este ajuste se coloca el
tapón sobre el matraz y se controla lo siguiente: •• El exterior del matraz d• No deben existir burbu eridas a la pared del interior del
matraz. c) Pesar e mat ond) Posteriormente, se extraen con precaución unos mililitros del agua contenida en
el matraz procurando no mojar el cuello del mismo. Repetir el ajuste hasta la marca del aforo y pesar nuevamente el matraz con agua.
to experimentale de la calibrac n del matraz volumétrico
Temperatura del agua 0C
cuello del matraz. Completar con agua destilada hasta la marca del aforo con una pipeta hasta que el fondo del menisco coincida con la marca. En caso de pasarse ligeramente de la marca, es posible retirar un poco de líquido medianteun pedazo de papel ad
El cuello del matraz arriba de la marca debe estar seco. ebe estar seco.
jas de aire adh
l raz aforado c agua.
Da s s ió
Capacidad del matraz

Métodos Cuantitativos Manual de Prácticas de Laboratorio
12
(mL)
Peso del matraz con agua (g)
Peso de agua vertida (g)
Volumen real vertido a 20 0C
1 2 3
Promedio
4.2.3 Calibración de una pipeta volumétrica de 5.00 mL
a) Pesar un matraz volumétrico de 25 mL vacío y seco con su tapón. El interior del cuello esmerilado, el tapón y el exterior del
ar tres pesadas independientes).
Peso del matraz vacío y seco con su tapón. Peso en gramos
matraz no necesita estar seco, elmatraz deben estar secos (efectu
1
2
3
Promedio
b) Llenar la pipeta con agua destilada y ajustar el nivel de manera que el fondo del
exterior de la punta de la
rico a unos milímetros abajo de
n antes mencionada.
Datos experimentales de la calibración de la pipeta volumétrica Temperatura del agua 0C
menisco coincida con la marca del aforo. Verificar que no existan burbujas de aire adheridas a las paredes. Durante el ajuste, mantener la pipeta en forma vertical a la altura de los ojos. Después del ajuste, secar la partepipeta con un pedazo de papel adsorbente.
c) Introducir la pipeta en el cuello del matraz volumétla parte esmerilada. La pipeta debe estar en forma vertical y el matraz inclinado. Vaciar el agua contenida en la pipeta manteniendo la posició
d) Pesar el matraz con agua.
Capacidad del matraz
Peso del matraz
con agua (g) Peso de agua
vertida (g) Volumen real
vertido a 20 0C (mL)
1 2 3
Promedio
5. ANÁLISIS DE RESULTADOS
5.1 Con los resultados de la calibración de la bureta trazar la gráfica factor de corrección en función de volumen real vertido
5.2 Determinar la precisión de la bureta, del matraz volumétrico y de la pipeta volumétrica en términos de desviación estándar.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
13
5.3 Clasificar el material volumétric tomando en cuenta la precisión que
. CONCLUSIONES
6.1 os objetivos de la práctica?
7. BIBL
. Teoría y Problemas de Química Analítica
o en clase A o Bobtuviste al calibrarlo.
6
¿Se lograron l6.2 Obtener las conclusiones pertinentes.
IOGRAFÍA
7.1 Gordus, A. A . Primera Edición. Editorial éxico, S. A. de C. V. México, D. F. 1991. 255 págs. ISBN McGraw-Hill/Interamericana de M
968-422-942-9. 7.2 Hadjiioannou, T. P.; Christian, G. D.; Efstathiou, C. E.; Nikolelis, D. P. Problem Solving in
Analytical Chemistry. Primera Edición. Editorial Pergamon Press. Printed in Great Britain. 1988. 437 págs. ISBN 0-08-036968-5 Hardcover. ISBN 0-08-036967-7 Flexicover.
7.3 Hadjiioannou, T. P.; Christian, G. D.; Efstathiou, C. E.; Nikolelis, D. P. Problem Solving in Analytical Chemistry. Solutions Manual. Primera Edición. Editorial Pergamon Press. Printed in Great Britain. 1988. 164 págs. ISBN 0-08-036972-3.
7.4 Harris, D. C. Análisis Químico Cuantitativo. Segunda Edición. Editorial Reverté, S. A. Barcelona, España. 2001. 981 págs. ISBN 84-291-722-X.
7.5 Harris, D. C. Análisis Químico Cuantitativo. Primera Edición. Grupo Editorial Iberoamérica, S. A. de C. V. México, D. F. 1992. 886 págs. ISBN 970-625-003-4.
7.6 Skoog, D. A.; West, D. M.; Holler, F. J.; Crouch, S. R. Fundamentos de Química Analítica. Octava Edición. Editorial Thompson. México. 2005. 1065 págs. ISBN 970-686-369-9.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
14
CIONES PATRÓN ÁCIDO-BASE. 1. OBJ
1.1.
1.2. El alumno realizará la estandarización de las soluciones ácido-base preparadas anter
1.3. El alumno pract ra preparar n la estandarización d
2. INTRODUCCIÓN
La preparación y estandarización de soluciones son dos técnicas importantes en el análisis químico. Una disolución es una mezcla homogénea de un soluto y un solvente. El soluto es la susta lvente es aquel que está en mayor proporción. Existen soluciones sólidas, líquidas y gaseosas; algunos ejemplos de éstas son ede latón (cmolaridad (etc. En Quí Una vez qsoluto respecello se utilizestandarizar cuantitativo. 2.1 Sust
En Químla disolución sea ácida ó básica. Los iones que dan origen al comportamiento ácido son los protones y los iones hidróxido provocan el comportamiento alcalino. Por lo tanto, ácido es un electrolito que en disolución acuosa cede un protón y genera una base conjugada:
á conjugada
Una ba
H+ ⇄ HB
De acuer er protones al medio se le denomina fuerte o débil. Si el ácido está disociado más del 90% ó cede sus protones con suma facilidad al medio, se dice que es fuerte y si se disocia en un porcentaje ínfimo se
PRÁCTICA No. 2 PREPARACIÓN Y USO DE DISOLU
ETIVOS
El alumno preparará soluciones patrón ácido-base de HCl, ácido acético, amoniaco y NaOH 0.1 N.
iormente. icará las diferentes formas pa un patrón primario a utilizarse e
e soluciones ácido-base.
ncia que se encuentra en menor proporción, mientras que el so
l aire limpio (mezcla de nitrógeno y oxígeno), agua endulzada y algunas aleaciones obre y zinc). La forma de expresar la concentración para las soluciones es: M), normalidad (N), formalidad (F), ppm, molalidad (m), soluciones porcentuales, mica Analítica es común utilizar soluciones molares y normales.
ue las soluciones son preparadas se debe conocer con exactitud la concentración del to a la cantidad de disolvente, a éste proceso se le llama estandarización y para an sustancias llamadas patrones primarios y secundarios. Es importante las soluciones preparadas porque sólo así pueden ser utilizadas en el análisis
ancias ácido-base
ica Analítica son de gran interés aquellos electrolitos cuyos iones provocan que
HA ⇄ H+ + A-
cido base
se es una especie química que acepta un protón y genera un ácido conjugado:
B + base ácido
conjugado
do con la capacidad que tenga un ácido para ced

Métodos Cuantitativos Manual de Prácticas de Laboratorio
15
dice qhidróxidos Para estandarizar sustancias ácidas se emplean patrones primarios alcalinos y para estandarizque las sustancias ácidas o básicas se han comparado con un patrón primario se les puede usar como patrones secundarios, por ejemplo NaOH, HCl, H2SO4, EDTA, etc.
T es prim raciones á 1
s primario ma
ue es débil. Este mismo criterio se utiliza para una base pero la misma cede al medio.
ar sustancias básicas es necesario emplear patrones primarios ácidos. Una vez
abla 1. Principales patron arios para valos ácidos
cido-basePatrones priPatrone rios básicos
Ftalato ácido Tris-hidroximetilaminometano de potasio Yodato ácido de p xido mercotasio Ó úrico
Ácido sulfamíl rbonato deico Ca sodio Sal doble de ácido sulfosalicílico Bórax
2.1.1 Indicadores ácido-base
La estandarización de sustancias ácido-base requiere de un método para identificar
base ha reaccionado estequiométricamente con la sustancia por valorar.
étodo potenciométrico. Consiste en el monitoreo del pH de la solución que se n problema se estandariza el
o para la medición del pH y una posterior gráfica de pH = f (vol. de
an como
ste color va a depender del pH.
ara un indicador se puede escribir así:
HIn ⇄ H+ + In-
Forma disociada
Color 2
ender de la concentración de H+, es decir, del pH. Para decuado, en un caso específico se debe tomar en cuenta
iones:
ner un intervalo de vire que coincida con el pH del punto estequiométrico de la valoración. Si el indicador elegido se aparta demasiado
a condición se obtendrá un error importante.
el punto final de dicha reacción, es decir, el punto donde la especie valorante sea ácido o Algunos métodos para identificar el punto final en una valoración son:
a) Mestá estandarizando, ya que una vez que la soluciópH cambia drásticamente. Este método requiere de un potenciómetro y un electrodvalorante).
b) Utilización de un indicador químico. Las sustancias que se us
indicadores son sustancias orgánicas de carácter ácido-base muy débil, cuyos iones tienen un color diferente del de la forma sin disociar, y é
E l equilibrio p
Forma no disociada
Color 1
El color observado va a depseleccionar el indicador alas siguientes condic a) Debe te
de ést

Métodos Cuantitativos Manual de Prácticas de Laboratorio
16
or. Los colores de los indicadores os, que para 100 mL de disolución bastan dos gotas de indicador,
s se emplean en concentraciones muy diluidas (0.01-0.1 %). c) El primer cambio de color detectable del indicador debe ser tomado como punto
las valoraciones ácido-base con sus intervalos de
b) Debe usarse una cantidad pequeña de indicadson tan intenslos cuale
final.
Tabla 2. Indicadores más usuales para transición respectivos.1
Indicador Intervalo de transición pH
Color del ácido Color de la base
Anaranjado de metilo 3.1-4.4 Rojo Amarillo Rojo de metilo 4.8-6.0 Rojo Amarillo Fenolftaleína 8.0-9.6 Incoloro Rosa mexicano
Azul de bromotimol 6.0-7.6 Amarillo Azul
IO PREVIO
concepto de molaridad (M), normalidad (N), formalidad (F), ppm y soluciones les. concepto de peso equivalente en un sistema ácido-base y ejemplificar el en ácidos de fórmula general HA, H
3. CUESTIONAR
3.1 Definir el
porcentua3.2 Definir el
concepto M(OH)2.
3.3 ¿Qué 3.4 Buscar en
cada indic3.5 Realizar l
(pureza 3 N, NaOH 0.1N, ácido acético (pureza 99%, densid
3.6 Buscar enrealizar lo
4. PARTE EXPER
4.1 Materia
3 vasos de 2 vasos de 1 matraz vo4 matraces 2 pipetas vo1 pipeta1 bureta de1 probet
HCl concentrado NDisolución alcohólica de fenolftaleína al 0.1% (W/V)
2A y bases de fórmula general MOH,
es el punto final o estequiométrico de una valoración? la literatura una lista de indicadores ácido-base e indicar el intervalo de vire de ador. os cálculos para preparar 1L de cada una de las siguientes disoluciones de HCl 6%, densidad 1.21 g/mL) al 0.1
ad 1.05 g/mL) 0.1N y amoniaco ( pureza 28%, densidad 0.9 g/mL) 0.1N. la literatura la forma de preparar una disolución del indicador fenolfaleína y
s cálculos para preparar 10 mL de este indicador al 0.1% (W/V).
IMENTAL
l y reactivos
precipitados de 250 mL precipitados de 30 mL lumétrico de 1000 mL Erlenmeyer de 250 mL lumétricas de 10 mL
graduada de 10 mL 25 mL
a de 10 mL
aOH

Métodos Cuantitativos Manual de Prácticas de Laboratorio
17
DisoluciFtalato ácidCarbona
4.2 Desar
4.2.1 Pre
n lentejas, pesando con una precisión de 0.1mg. El o con la balanza, sino que debe
iamente pesado. Cualquier partícula de sólido que accidentalmente se vierta, debe
e el frasco que contiene el hidróxido de sodio quede perfectamente tapado después de utilizarlo. Transferir el hidróxido de sodio a un vaso de 50 mL limpio, adicionarle aproximadamente 10 mL de agua destilada, enseguida agitar con una varilla de vidrio hasta que el sólido se disuelva totalmente. Pasar cuantitativamente esta solución en un matraz volumétrico de 100 mL y llevar al aforo con agua
ar la so ción por inversiones y agitaciones repetidas. a solu
de u contenido, la fec malidad des
b) Dis 83 mL de HCl concentrado, llevar
cha, el
c) ac 0.5 H concentrado, pasarlo a un matraz volumétrico de 100 mL que contenga 10 mL de H2O destilada, agitar ligeramente para que se disuelva y posteriormente llevar al aforo en un matraz volumétrico de 100 mL con agua destilada y guardar en un frasco limpio. Etiquetar el frasco haciendo constar su contenido, la fecha, el nombre del alumno y dejando espacio para reseñar la normalidad después de
) atraz volumétrico de 100 mL que contenga
e disuelva y de 100 mL con agua
n exactitud. )
Envasar. ) ). Pesar 0.1 g de anaranjado de
4.2.2 dio 0.1 N
ón acuosa de anaranjado de metilo al 0.1% (W/V) o de potasio
to de sodio
rollo experimental
paración de una disoluciones a) Disolución de hidróxido de sodio 0.1 N. En una balanza analítica pesar 0.4 g
de hidróxido de sodio eproducto sólido no debe estar en contacto directutilizarse un vidrio de reloj ó un vaso de precipitados de 30 mL, prev
desecharse inmediatamente. Asegurarse de qu
destilada. HomogenizPasa
lur est ción a un frasco de un litro limpio y seco y taparlo con un tapón
bakelita o de goma. Etiquetar el frasco haciendo constar sha, el nombre del alumno y dejando espacio para reseñar la norpués de que se determine con exactitud. olución de ácido clorhídrico 0.1 N. Medir 0.
al aforo en un matraz volumétrico de 100 mL con agua destilada y guardar en un frasco limpio. Etiquetar el frasco haciendo constar su contenido, la fenombre del alumno y dejando espacio para reseñar la normalidad después de que se determine con exactitud. Disolución de ácido ético 0.1 N. Medir 7 mL de CH3COO
que se determine con exactitud. d Disolución de amoniaco (hidróxido de amonio) 0.1 N. Medir 0.67 mL de
hidróxido de amonio, pasarlo a un m10 mL de H2O destilada, agitar ligeramente para que sposteriormente llevar al aforo en un matraz volumétricodestilada y guardar en un frasco limpio. Etiquetar el frasco haciendo constar su contenido, la fecha, el nombre del alumno y dejando espacio para reseñar la normalidad después de que se determine co
e Indicador de fenolftaleína al 0.1% (W/V). Pesar 0.1 g de fenolftaleína y disolver en 100 mL de etanol.
f Indicador de anaranjado de metilo al 0.1% (W/Vmetilo y disolver con 100 mL de agua destilada. Envasar y etiquetar.
Normalización de la disolución de hidróxido de so

Métodos Cuantitativos Manual de Prácticas de Laboratorio
18
a) Pesar exactamente en una balanza analítica 0.2040 g de biftalato de potasio, previamente desecado a 105–110 ºC durante una hora.
ver el biftalato de potasio en un matraz Erlenmeyer de 250 mL, con un vol
c) A c icador fenolftaleína. d) Co r cada uno
de a que aparezca un ligero color rosa persistente por 30 segundos por lo menos.
alidad de la solución de NaOH.
g a d no La ecuación que deberá utilizar para este cálculo es:
b) Disolumen de agua destilada de 20 a 30 mL. ada uno de los matraces se le adiciona tres gotas de indlocar la solución de NaOH preparada en una bureta limpia, titulalos tres matraces con esta solución, hast
e) Anotar el volumen de hidróxido de sodio agregado y determinar la norm
f) Realizar el procedimiento anterior por triplicado ) La desviación medi e estos tres resultados debe exceder de 2%.
NaOHbiftalato
biftalato
mLxVPEN
)(=
mgw )(
ido de sodio gastado.
ltados.
Tabla 3. Resultados experimentales para la estandarización de NaOH. Por triplicado.
4.2.3 N rmali
a mente
bcd) d bur el Na2CO3
con esta disolución hasta qu olor amarillo vire rojo canela persistente por 30 segundos por lo menos.
e) Anotar el volumen de ácido clorhídrico gastado y determinar la normalidad de la disolución de HCl.
f) Repetir el procedimiento anterior por triplicado.
Peso del biftalato (mg)
Vol. gastado de NaOH (mL)
Normalidad del NaOH
Donde:
w(mg)biftalado es el peso en miligramos de biftalato de potasio. PEbiftalato es el peso equivalente del biftalato de potasio. V(mL)NaOH es el volumen en mililitros de hidróxN es la normalidad del hidróxido de sodio.
En la siguiente tabla puede vaciar los datos que se indican y los resu
o zación de la disolución de HCl 0.1 N ) Pesar exactamente en una balanza analítica 0.05 g de Na2CO3 , previa
desecado a 200 ºC por 30 minutos. ) Disolver en un matraz Erlenmeyer con 50 mL de agua destilada. ) d e me
Adicionar tres gotasColocar la solución
el indicador anaranjado de HCl preparada en una
tilo. eta limpia, titular
a u colore el c n
g) La desviación media de estos tres resultados no debe exceder de 2%.
La ecuación que deberá utilizar para este cálculo es:

Métodos Cuantitativos Manual de Prácticas de Laboratorio
HClCONa 32
CONa
mLxVPEmgw
N)(
)(32=
Donde:
w(mg) Na2CO3 es el peso en miligramos de carbonato de sodio. PENa2CO3 es el peso equivalente del carbonato de sodio. V(mL)HCl es el volumen en mililitros de ácido clorhídrico gastado.
es la normalidad del ácido clorhídrico.
En la siguiente tabla puede vaciar los datos que se indican y los resultados: Tabla 4 ado.
Peso del carbonato
N
. Resultados experimentales para la estandarización de HCl. Por triplicVol. gastado de HCl Normalidad del HCl
de sodio (g) (mL)
4.2.4 Normalización de la disolución de ácido acético 0.1 N
a) En una bureta colocar el NaOH valorado en el experimento 4.2.2 b) En un matraz Erlenmeyer colocar 10.00 mL de CH3COOH, medido con precisión. c Adicionar tres gotas del indicador fenolftaleína y titular con la disolución de NaOH
hasta el vire del indicador de incoloro a rosa y que sea persistente por 30 enos.
d) lizarlo por triplicado.
)
segundos por lo mEl procedimiento anterior rea
e) La desviación media de estos tres resultados no debe exceder de 2%.
La ecuación que deberá utilizar para el cálculo de la normalidad es:
221
VNN = 1
Donde: N
V
del NaOH V1 es el volumen de la alícuota de ácido acético
datos que se indican y los resultados:
ultados experimentales para la valoración de CH3COOH. Por triplicado.
1 es la normalidad del ácido acético N2 es la normalidad
V2 es el volumen gastado de NaOH en el punto de equivalencia
En la siguiente tabla puede vaciar los Tabla 5. Res
Número de matraz Vol. gastado de NaOH (mL)
Normalidad del CH3COOH
ción de la disolución de amoniaco (hidróxido de amonio) 0.1 N a) En una bureta colocar la disolución de HCl estandarizada en el punto 4.2.3.
4.2.5 Normaliza
19

Métodos Cuantitativos Manual de Prácticas de Laboratorio
20
10.00 mL de amoniaco medido con precisión. metilo y titular con la disolución
or de amarillo a rojo canela. d) El procedimiento anterior realizarlo por triplicado. e) La desviación media de estos tres resultados no debe exceder de 2%.
La ecuación que deberá utilizar para el cálculo de la normalidad es:
b) En un matraz Erlenmeyer colocar c) Adicionar tres gotas del indicador anaranjado de
de HCl hasta el vire del indicad
1
221 V
N = VN
Tabla 6. Resultados experimentales para la valoración de NH4OH. Por triplicado. Número de matraz Vol. gastado de HCl
(mL) Normalidad del
NH4OH
Donde: N1 es la normalidad del hidróxido de amonio N2 es la normalidad del HCl V1 es el volumen de la alícuota de hidróxido de amonio
l gastado en el punto de equivalencia V2 es el volumen de HC En la siguiente tabla puede vaciar los datos que se indican y los resultados:
5. ANÁLISIS DE RESULTADOS
5.1 Establecer la reacción química que se verifica entre biftalato de potasio e hidróxido de sodio
5.2 Establecer la reacción química que se verifica entre carbonato de sodio y ácido clorhídrico
5.3 Reportar la normalidad de las soluciones preparadas, indicando los cálculos realizados. Hacer el análisis dimensional pertinente.
5.4 Realizar el análisis estadístico demostrando que sus resultados no exceden el 2% de coeficiente de variación (CV). Llenar la siguiente tabla con los datos obtenidos para la valoración de NaOH y la de HCl.
Promedio de normalidad
Desviación estándar %C V
5.5 Calcular el error relativo y el error absoluto en la valoración de cada uno de las soluciones
valoradas. 5.6 Justificar ¿Por qué? Se utilizaron indicadores diferentes para las valoraciones anteriores,
usar para ello la bibliografía.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
21
6. CONCLUSIONES
6.1 ¿Se lograron los objetivos de la práctica? one para mejorar los resultados de la práctica?
6.3 Obtener las conclusiones pertinentes.
. BIBLIOGRAFÍA
.1. Ayres, G. H. “Análisis Químico Cuantitativo” Editorial Oxford University Press, Madrid
ica Analìtica”, 8ava edición. Ed. Thomson, Mèxico (2006), 1065 pàg
Kapelusz 2da. Edición, Buenos Aires 1960,
6.2 ¿Qué prop
7
7(1990), 740 pàgs:
7.2. Harris D.C. “Análisis Químico Cuantitativo”. Grupo Editorial Iberoamerica (1991), México, 981 págs
7.3. Orozco, D. Fernando “Análisis Químico Cuantitativo”, Porrúa, S.A. México (1987), 447 págs.
7.4. Skoog, D.A. y Leary J.J. “Análisis Instrumental”. 4ta edición. Ed. Mc Graw Hill. (1994) 7.5 Skoog, D.A. y West D. A. “Fundamentos de Quìm
7.5. Vogel, A.I. “Química Analítica Cuantitativa” 812 pàgs

Métodos Cuantitativos Manual de Prácticas de Laboratorio
22
PRÁCTICA No. 3 PREPARACIÓN Y USO DE DISOLUCIONES PATRÓN OXIDOREDUCTORAS
1.
1.1 p1.2 p
2. INTR U
na especie a otra. Un agente oxidante toma electrones de otra sustancia y se reduce. Un agente reductor cede electrones a otra sustancia y se oxida. La mayoría de los agentes oxidantes pueden utilizarse como titulantes, entre los que se encuentran el MnO4
- en medio ácido, el Cr2O7= en medio
ácido. Con lo que respecta a los titulantes reductores, estos son que suelen ser inestables en presencia del oxígeno atmosférico y,
2.1 potasio impurezas de productos de reducción, por ejemplo
ilmente por la acción de los reductores: amoniaco, cen con el agua, con el polvo, etc. Debido a ello, la
lución de KMnO4 disminuye una vez preparada. De aquí se deduce solución valorada de permanganato a partir de una porción pensable determinar su concentración exacta.
KMnO4 sea suficientemente estable y su concentración no se pensable eliminar el precipitado de MnO2, puesto que acelera scomposición de KMnO4. Hay que tener presente también que el
oma, tapones de corcho, papel y otras sustancias, por eso es tacto de la solución con estos materiales. Así, no se puede
MnO4 con filtros de papel, sino que se deben utilizar crisoles de olución de permanganato se debe conservar al abrigo de la luz o en
que la luz acelera la descomposición de KMnO4.
es de KMnO4 se han propuesto varias sustancias patrón Na2C2O4, As2O3, el hierro metálico, etc. Las
H2C2O4 . 2H2O y Na2C2O4, que deben ser s y corresponder rigurosamente a sus fórmulas.
2.2 sulfato de sodio l tiosulfato de sodio es el titulante reductor casi universal para el yodo. En soluciones
to (oxidándose a tetrationato) reduce el yodo a yoduro. La 2O no es lo suficientemente pura para ser patrón
prepar
3. CUESTIONAR3.1 Balancear
a) MnO4 ⇄ Mn
OBJETIVOS
Pre arar y estandarizar una disolución de KMnO4. zPre arar y estandari ar una disolución de Na2S2O3.
OD CCIÓN Una e educción implica la transferencia de electrones de ureacción d oxidorr
ácido y el Ce(IV) en mediomenos frecuentes debido a por tanto, deben conservarse en una atmósfera inerte.
Oxidación con permanganato deEl permanganato contiene siempreMnO2. Además, se descompone fác
s, que se introdusustancias orgánicaconcentración de la suque no se puede preparar una
spesada con precisión. Es indi
solución de A fin de que lamodifique, es indis
ecatalíticamente la dpermanganato oxida a la g
el conindispensable evitar filtrar la solución de Kidrio sinterizado. La sv
frasco de vidrio oscuro, puesto
ionPara estandarizar las solucprimario, por ejemplo, H2C2O4.2H2O,
onvenientes son: sustancias más caquímicamente pur
educción con tioR
Eneutras o ácidas, el tiosulfaforma usual del tiosulfato, Na2S2O3.5Hprimar Eio. l tiosulfato suele estandarizarse haciéndolo reaccionar con una solución recién
ada de I2 a partir de KIO3 más KI ó con una solución de I2 estandarizada con As4O6.
IO PREVIO las siguientes semirreacciones en medio ácido. – 2+

Métodos Cuantitativos Manual de Prácticas de Laboratorio
23
b) C2O4
c) S
3.2 Realizar loa) 100 mb) 100 mc) 1d) 50
3.3 Buscar el a) MnOb) Cc) S4 6 2 3
4. PART
4.1 Materi1 vaso1 vaso4 vasos d2 matr1 bureta de 25 mL
1 pipe1 pipe1 termó1 pinza pa1 sopo1 parri1 placa1 agitador magnético
2 2 3Disolución de H2SDisolución de almidó enYoduro de potasio Oxalato de sodio Yodato de potasio
4ransferirla a un
erior con agua destilada hasta 130 mL
2– ⇄ CO2 (g) 2– 2–
4O6 ⇄ S2O3
s cálculos para preparar las siguientes disoluciones. L de KMnO4 (pureza 99.2%) 0.02 M L de H2SO4 (pureza 96%, densidad 1.84 g/mL) 2.5 M
000 mL de Na2S2O3 (pureza 99.9%) 0.07 M mL almidón al 0.1% en peso
potencial estándar de los siguientes pares oxidorreductores. 4
– / Mn2+
2–2O4 / CO2(g) O 2– / S O 2–
E EXPERIMENTAL
al y reactivos de precipitados de 250 mL de precipitados de 150 mL
e precipitados de 100 mL aces volumétricos de 100 mL
1 probeta de 100 mL 1 pipeta volumétrica de 5 mL
ta graduada de 10 mL ta graduada de 1 mL
metro ra bureta
rte universal lla de calentamiento de agitación
Disolución de KMnO4 0.02 M Disolución de Na S O 0.07 M
O4 2.5 M n al 0.1% peso
4.2 Desarrollo experimental
4.2.1 Preparación de disoluciones a) Disolución de KMnO 0.02 M. En un vaso de precipitados de 250 mL poner a
ebullición 200 mL de agua destilada. Pesar 0.319 g de KMnO4 y tvaso de precipitados de 150 mL. Adicionar al KMnO4 50 mL de agua destilada en ebullición poco a poco, hasta que el sólido se disuelva completamente. Completar el volumen de la solución ant

Métodos Cuantitativos Manual de Prácticas de Laboratorio
24
imadamente y dejar ebullir suavemente durante 30 minutos. Enfriar la solución, filtrarla con lana de vidrio y envasarla en un frasco ámbar.
2S2O3 y transferirlos a un 0 mL. Disolver el Na2S2O3 con 30 mL de agua
destilada previamente hervida y fría. Transferir el contenido del vaso a un traz volumétrico de 100 mL y llevar hasta la marca con agua destilada
recipitados de 100 mL adicionar 50
poco a poco y por las paredes de
destilada.
4.2.2 Estandarización de una disolución de KMnO4 0.02 M
a) Pesar con exactitud 50 mg de oxalato de sodio y colocarlos en un vaso de precipitados de 100 mL.
b) Adicionar 40 mL de agua destilada y 10 mL de H2SO4 2.5 M. c) Calentar a una temperatura de 55-60 °C y con agitación constante adicionar con
una bureta la disolución de KMnO4 gota a gota hasta el cambio de color de transparente a ligeramente rosa.
d) Realizar por triplicado la estandarización.
4.2.3 Estandarización de una disolución de Na2S2O3 0.07 M a) En un vaso de precipitados de 100 mL pesar con exactitud 30 mg de KIO3. b) Adicionar 40 mL de agua destilada y 10 mL de H2SO4 2.5 M. c) Agregar a la disolución anterior 2 g de KI sólido y con agitación constante
valorarla con Na2S2O3 hasta que el color de la solución sea ligeramente amarilla.
d) Adicionar 1 mL de almidón y continuar la valoración hasta el vire del color azul al incoloro.
e) Realizar por triplicado la estandarización.
. ANÁLISIS DE RESULADOS 5.1 Llenar la siguiente tabla con los datos experimentales obtenidos.
5.2 Establecer la reacción que se verifica entre el oxalato de sodio y el permanganato de potasio.
5.3 Establecer la reacción que se verifica entre el yodato de potasio y el yoduro de potasio en medio ácido.
5.4 Establecer la reacción que se verifica entre el yodo y el tiosulfato de sodio. 5.5 Con los datos experimentales obtenidos, determinar la concentración exacta del
permanganato de potasio y del tiosulfato de sodio.
Repetición Peso en mg de Na2C2O4
Vol. en mL de KMnO4 gastados
Mg de KIO3 Vol. en mL de Na2S2O3
gastados
aprox
b) Disolución de Na2S2O3 0.07 M. Pesar 1.74 g de Navaso de precipitados de 10
mahervida y fría. Envasar en un frasco ámbar.
c) Disolución de H2SO4 2.5 M. En un vaso de pmL de agua destilada. Colocar el vaso en un baño de hielo. Medir 14 mL de H2SO4 y adicionarlos al vaso de precipitadoseste, transferir el contenido del vaso a un matraz volumétrico de 100 mL y llevar a la marca del aforo con agua
d) Indicador de almidón al 0.1% (W/V).
5
1 2 3

Métodos Cuantitativos Manual de Prácticas de Laboratorio
25
6. CONCLUSIONES
6.1 ¿Se lograron los objetivos de la práctica? as conclusiones pertinentes.
7.
.1 Charlot G., “Curso de Química Analítica General”
6.2 Obtener l
BIBLIOGRAFÍA 7 , 1ª edición, Tomo I, Editorial Toray-
1977, Barcelona, España, 282 páginas. Masson, S.A., 7.2 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo II, Editorial Toray-
Masson, S.A., 1977, Barcelona, España, 200 páginas. 7.3 Harris D.C., “Análisis Químico Cuantitativo”, 3ª edición, Editorial Iberoamerica S. A. de
C.V., 1992, México, D.F., 886 páginas. 7.4 Skoog D.A., West D.M., Holler F.J., “Química Analítica”, 6ª edición, Editorial McGraw-
Hill/Interamericana de México, 1995, México, D.F., 612 páginas

Métodos Cuantitativos Manual de Prácticas de Laboratorio
26
PREP 1. OBJ
1.1. Preparar y estandarizar una diso1.2. Determinar Ca2+ y Mg2+ (dureza total) en agua natural
2. INTRODUCCIÓN Las reacciones de formación de complejos son importantes en muchas áreas científicas y de la vida cotidiana. Estas reacciones, se emplean mucho en química analítica. Una de las aplicaciones principales de estas reacciones es la valoración volumétrica de cationes. Para que
plear en volumetría, debe ser rápida, esteq cos reaccionan con dadores (ligandos) de pares ddeben teneenlace. Sonhaluro. En acuosa perescribir al iopueden meácido etilen
En la industria alimenticia y particularmente la que se dedica a la fabricación de jugos y refrescos, el agua usada para la preparación de estas bebidas debe tener un estricto control de calidad. Uno de los con enido de sales de calcio y magnesio, es decir, la precisamente un ejemplo de import omplejos M–EDTA. 2.1 Equilibrios de formación de complejos
En lasligandocaractcompledisociasiguien
n
PRÁCTICA No. 4 ARACIÓN Y USO DE DISOLUCIONES PATRÓN COMPLEJOMÉTRICAS
ETIVOS lución de EDTA
la reacción de formación de complejos se pueda emuiométrica y cuantitativa. Muchos cationes metáli
e electrones para formar compuestos de coordinación o complejos. Los ligandos r por lo menos un par de electrones sin compartir disponible para la formación del ejemplos de ligandos inorgánicos comunes, el agua, el amoniaco y los iones
realidad, muchos iones metálicos existen como complejos hidratados en disolución o, en las ecuaciones químicas, habitualmente se simplifican estos complejos al n metálico como si no formara parte de un complejo. De los ligandos orgánicos se
ncionar a la etilendiamina, el trifosfato de adenosina (ATP) y el más importante es el diaminotetraacético (EDTA) y su sal disódica.
troles que se le realizan al agua, es el contde la dureza total del agua. Esta determinación es determinación
ancia práctica en la que se usa la formación de c
reacciones de formación de complejos, un ion metálico, Ma+, reacciona con un , nLb–, para formar el complejo MLn
a – nb. La etapa de formación del complejo esta erizada por una constante de equilibrio llamada constante de formación del jo (Kf). La inversa de la constante de formación del complejo es la constante de ción (Kd). De manera general, la formación de un complejo se representa por el te equilibrio:
Ma+ + b– ⇄ ML a – nb nL[ ]
[ ][ ] dnba
nban
f KLM
MLK 1==
−+
−
2.2 Formac n de comp
l ácid etilendiaminotetraacético (abreviado EDTA ) tiene la siguiente fórmula estructural: ióo
lejos M–EDTA E

Métodos Cuantitativos Manual de Prácticas de Laboratorio
27
dades ácido-base se puede representar como H4Y cuyos valores e pKa son: pKa1=2.00, pKa2=2.66, pKa3=6.16 y pKa4=10.24; por tanto las múltiples
sentan por: H4Y, H3Y–, H2Y2–, HY3– y Y4–. La sal disódica del
El EDTAmetálicoformaciósiguiente
Por lecuación
El EDTA por sus propiedespecies del EDTA se repreEDTA se representa por Na2H2Y•2H2O.
forma complejos estables de estequiometría 1:1 con la mayoría de los iones s, independientemente de la carga del catión. La reacción general para la n de complejos entre el ion metálico Mn+ y el EDTA esta representada por el equilibrio:
Mn+ + Y4– ⇄ MYn – 4
o tanto, la constante de formación del ion complejo MYn – 4 esta dada por la siguiente :
[ ] [ ][ ]−+= 4LM
MLK nMY −4n
2.2.1Lo
mcc
Indicadores para valoraciones con EDTA os indicadores de iones metálicos para valoraciones con EDTA son colorantes rgánicos que forman quelatos coloreados con iones metálicos en un intervalo de
pM que es característico de cada catión y colorante. Es habitual que los complejos tengan un color intenso y sean discernibles a simple vista en concentraciones
olares que van de 10–6 a 10–7 M. El eriocromo negro T es un indicador aracterístico de iones metálicos que se utiliza en la valoración de diversos ationes comunes. Su fórmula estructural se muestra en la siguiente figura y su
comportamiento como ácido débil se describe con los siguientes equilibrios:
H2In – ⇄ HIn 2– + H+ pKa = 6.3 rojo azul HIn 2– ⇄ In 3– + H+ pKa = 11.6 azul anaranjado
Los complejos metálicos del eriocromo negro T por lo general son rojos, como en el caso de H2In –. Por lo tanto, para la detección de iones metálicos es necesario ajustar el pH a un vp
alor mayor a 7, de modo que la forma azul de la especie HIn 2–, una valoración el indicador compleja
como consecuencia del siguiente equilibrio:
redomine en ausencia de un ion metálico. En el exceso de ión metálico de modo que la disolución es roja hasta el punto de equivalencia, ante el primer leve exceso de EDTA, la disolución se torna azul

Métodos Cuantitativos Manual de Prácticas de Laboratorio
28
3. CUESTIONAR3.1 Realiz 0.01
M, b) 23.2 Investigar3.3 Investigar mplejos de EDTA con Ca2+ y Mg2+.
4. PARTE EX4.1 Materi
4.2
4.2.1 a
trdemezclar bien hasta integración completa. Envasar.
b activos pleta. Envasar.
c e NH4Cl y disolverlos ezcla a un matraz
volumétrico de 250.00 mL y diluir hasta la marca del aforo con agua destilada. L pesar 24 g de
étrico
e) D1uE
f) te una so de precipitados de 100 mL
disolver con agua destilada. la marca
con agua destilada. Envasar. g) Disolución acuosa de HCl 1:1. Añadir 10 mL de HCl concentrado a 10 mL de
agua destilada, mezclar y envasar en un frasco gotero.
Enjuagar con una pequeña porción de EDTA disódico una bureta previaL.
ecador.
0.1 mg, adicionar 30 mL de agua destilada. Posteriormente agregar una gota de HCl 1:1 y observar el burbujeo originado por la disolución del CaCO , adicionar una gota mas de HCl 1:1 y obse C ya no
rbujea Nota: no excederse en la adici . Adicionar 10 mL de disolución cantidad (20–30 mg) del indicador
MIn – + HY3– ⇄ HIn 2– + MY2–
rojo azul
IO PREVIO ar los cálculos y describir la forma para preparar a) 1000 mL de EDTA disódico0 mL de ENT al 0.1% en (w/v) en etanol y c) 50 mL de HCl 1:1.
como se prepara una disolución reguladora de NH4+/NH3 (pH=10).
los valores de las K de los cof
PERIMENTAL al y reactivos
Desarrollo experimental Preparación de disoluciones ) Indicador de eriocromo negro T (ENT). Disolver 0.2 g de ENT en 15 mL de
ietanolamina más 5 mL de etanol absoluto. Envasar. Debido a la inestabilidad e la disolución líquida, el indicador se puede preparar en disolución sólida; para llo, pesar 0.5 g de ENT y 100 g de KCl, colocar los reactivos en un mortero y
) Indicador murexida. Pesar 20 mg de murexida y 5 g de KCl, colocar los reen un mortero y mezclar bien hasta integración com
) Disolución reguladora de NH4+/NH (pH=10). Pesar 17.5 g d3
en 142 mL de NH3 acuoso al 28% w/w. Transferir la m
d) Disolución de NaOH 6 M. En un vaso de precipitados de 100 mNaOH, disolver con agua destilada. Transferir la mezcla a un matraz volumde 100 mL y diluir hasta la marca con agua destilada. Envasar.
isolución de oxalato de amonio al 10% (w/v) . En un vaso de precipitados de 00 mL pesar 10 g de la sal, disolver con agua destilada. Transferir la mezcla a n matraz volumétrico de 100 mL y diluir hasta la marca con agua destilada. nvasar.
Disolución de EDTA disódico 0.01 M. Secar 4 g de Na2H2Y•2H2O duranhora a 80 oC y enfriar en un desecador. En un vapesar 3.75 g de la sal con precisión del 0.1 mg, Transferir la mezcla a un matraz volumétrico de 1000 mL y diluir hasta
4.2.2 Estandarización de una disolución de EDTA disódico 0.01 M
a) mente limpia. Llenar la bureta con la disolución de EDTA disódico y ajustarla a 0.00 m
b) Secar 500 mg de CaCO durante una hora a 100 3En un matraz erlenmeyer de 125 mL pesar 10-20 mg de CaCO
oC y enfriar en un des3 con precisión del
3rvar. No agregar mas H
ón de HCll si la disolución
bureguladora de pH=10. Agregar una pequeña
.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
29
T só o. Titular con la disolu ón de EDTA disódico hasta el cambio de color rojo v no a azul turquesa.
c) Anotar el volumen gastado de EDTA disódico hasta la centésima de mL.
ón exacta del EDTA disódico y etiquetar el envase con este dato.
4.2.3 Determinación de Mg2+ en agua natural
a) Llenar la bureta con la disolución de DTA disódico y ajus rla a 0.00 mL. b) Medir 50.00 mL de agua de la llave con una pipeta volumétrica y transferirlos a
un vaso de precipitados de 100 mL. Adicionar 10 mL de disolución reguladora de pH=10 y 10 mL de disolución de oxalato de amonio al 10% w/w.
c) Dejar reposar la mezcla durante 30 minutos y posteriormente filtrar. Recoger el filtrado en un matraz erlenmeyer de 250 mL.
L de agua destilada. e) A las aguas de filtrado, agregar una pequeña cantidad (20–30 mg) del indicador
sólido. Titular con la disolución de EDTA disódico hasta el cambio de color
EDTA disódico hasta la centésima de mL. g) Realizar por triplicado el procedimiento anterior.
cular el contenido de Mg2+ en la muestra*.
+ (dureza total) en agua natural
de la llave y transferirlos a un matraz erlenmeyer de 125 mL. Adicionar 10 mL de disolución reguladora de pH=10. Agregar una pequeña cantidad (20–30 mg) del indicador ENT sólido. Titular con la disolución de EDTA disódico hasta el cambio de color de rojo vino a azul turquesa.
c) Anotar el volumen gastado de EDTA disódico hasta la centésima de mL. d) Realizar por triplicado el procedimiento anterior. e) Calcular el contenido total de Ca2+ y Mg2+ en la muestra*. f) Calcular por diferencia el contenido de Ca2+ en la muestra*.
4.2.5 Determinación de Ca2+ en presencia de Mg2+ en agua natural
a) Llenar la bureta con la disolución de EDTA disódico y ajustarla a 0.00 mL b) Medir con una pipeta volumétrica de 50.00 mL, agua de la llave y transferirlos a
un matraz erlenmeyer de 125 mL. Adicionar 3 mL de NaOH 6 M, agitar y en caso de ser necesario ajustar el pH de la disolución entre 12 y 13 con la disolución de NaOH 6 M. Agregar 100 mg del indicador murexida sólido. Titular con la disolución de EDTA disódico hasta el cambio de color de rojo a violeta.
c) Anotar el volumen gas dico hasta la centésima de mL. d) Realizar por triplicado el procedimiento anterior.
5
RESULTADOS
EN lid cide i
d) Realizar por triplicado el procedimiento anterior. e) Calcular la concentraci
E ta
d) Lavar el precipitado con dos o tres porciones de 10 m
ENT de rojo vino a azul turquesa.
f) Anotar el volumen gastado de
h) Cal
4.2.4 Determinación de Ca2+ y Mg2
a) Llenar la bureta con la disolución de EDTA disódico y ajustarla a 0.00 mL b) Medir con una pipeta volumétrica de 50.00 mL, agua
tado de EDTA disó
e) Calcular el contenido total de Ca2+ en la muestra*. f) Comparar el contenido de calcio obtenido en los puntos 4.2.4 y 4.2.
. ANÁLISIS DE 5

Métodos Cuantitativos Manual de Prácticas de Laboratorio
30
5.3 el EDTA.
Peso en mg de CaCO3 Volumen en mL de EDTA gastado
N (eq/L) del EDTA
5.1 Establecer las reacciones que se llevan a cabo entre el indicador ENT y los iones Ca2+ y Mg2+.
5.2 Establecer las reacciones que se llevan a cabo entre el EDTA y los iones Ca2+ y Mg2+. Establecer las reacciones que se llevan a cabo entre el indicador ENT y
5.4 Llenar la siguiente tabla con los datos experimentales obtenidos en la valoración del EDTA:
1 2 3
5
.5 Llenar la siguiente tabla con los datos experimentales obtenidos en la determinación de Ca2+ y Mg2+ en el agua natural:
Dureza total (Ca2+ y Mg2+)*
Mg2+ * Ca2+ * por diferencia Ca2+ * directo
*
6. CONCLUSIONES 6.1 ¿Se lograron los objetiv6.2 Obtener las conclusiones pertinentes.
7. BIBLIOGRAFÍA 7.1 Harr
Reportar los valores en términos de carbonato de calcio (CaCO3)
os de la práctica?
is, D. C. Análisis Químico Cuantitativo. Segunda Edición. Editelona España. 2001. 981 págs.
orial Reverté, S. A. Barc ,
7.2 Skoog, D. A.; West, D. M.; Holler, F. J.; Crouch, S. R. Fundamentos de Química Analítica. Octava Edición. Editorial Thompson. México. 2005. 1065 págs.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
31
PRÁCTICA No. 5
P
1. O
1
1 a muestra
1 rlí todo de Mohr.
En considera una titu i tulante, nitrato de activo precipitante para que la reacción sea completa, permitiendo saber cuánto analito existe en la muestra.
REPARACIÓN Y APLICACIÓN DE UNA DISOLUCION PATRÓN MEDIANTE TITULACIÓN POR PRECIPITACIÓN (MÉTODO DE MOHR)
BJETIVOS
.1 El alumno aprenderá a valorar una disolución titulante de AgNO3 utilizando un estándar primario.
.2 Determinar el porcentaje de cloruros (cloruro de sodio y cloruro cúprico) en unsólida por medio de una titulación por precipitación (método de Mohr).
.3 Determinar el porcentaje de clo uros, cloruro de sodio y cloruro cúprico en una muestra quida por medio del mé
2. INTRODUCCIÓN
el método de Mohr se lleva a cabo una reacción de precipitación que se fácil de realizar, en la que se mide el volumen del tilac ón directa precisa y
plata, y a la vez es el re
En t mo indicador K2CrO4. La solución titulante es AgNO3, y el pH es a reacción se utiliza coóptimo es de 6 a 10. A pH menor de 6 el precipitado de cromato de plata formado en el punto de u edio, da lugar a la formación de dicromato de plata eq ivalencia debido a la acidez del mque es ble que el cormato de plata, por lo tanto se consume más ion plata lo mucho mas solucual daría resultados erróneos, no permitiendo observar el color rojizo característico de este pre to final. A pH mayor de 10, alcalino, la plata podría forma cipitado y por lo tanto el punhid e 8-8.3 es adecuado para la determinación. róxido de plata. Un pH d
Las reacciones que ocurren en la determinación de iones cloruro son:
Cl – + Ag+ → AgCl↓ (precipitado blanco)
El cloruro de plata se forma después de que todos los cloruros han reaccionado.
CrO4-2 + 2Ag+ → Ag2CrO4↓ (Precipitado rojo ladrillo en el punto final)
(amarillo)
El cromato de plata es resultado del primer exceso de titulante.
L l nitrato de a solución patrón de AgNO3 se puede preparar por el método directo dado que eplata es un reactivo tipo primario; con el objeto de compensar los errores en la precipitación del punto final se prefiere el método indirecto donde la solución se valora con NaCl q s uímicamente puro. Cuando la solución tipo se prepara por el método indirecto no e

Métodos Cuantitativos Manual de Prácticas de Laboratorio
32
necesario el ensayo en blanco, porque el exceso empleado en la valoración de la sustancia p
Este método se emplea satisfactoriamente en la determinación de Cl , Br – y CN –; El ion cloruro (Cl –), es uno de los aniones inorgánicos principales en el agua natural y residual. No e estros iones sobre el precipitado de plata.
3. CUESTIONARIO PREVIO
3.1 ¿Cuál es la característica de los compuestos iónicos que se generan en las titulaciones
isten? Describa brevemente el cada uno de ésto.
3 jar con el método potenciométrico para la determinación
3 nal requiere 20 mL de
4. PARTE EXPERIMENTAL
4.3 Material y reactivos
• nitrato de plata (preferentemente seco previemente por 2 hrs. a 100oC) • cromato de potasio • cloruro de sodio • fenolftaleína • papel indicado• bureta de 25 m• 6 matraces erl• 1 frasco ámbar matraz de aforo de 500 ml
• s•
roblema se compensa con el empleado en la valoración del AgNO3.
–
s eficiente para determinar yoduros o tiocinato por la adsorción de
de precipitación? 3.2 ¿Qué otros métodos de presipitación ex
3.3 investigar cual es el limite de cloruros permitido en agua naturales. 3.4 investigue la reacción de valoración y la reacción indicadora. .5 ¿cuando es recomendable traba
de cloruros? 3.6 Sugiera un método de titulación para determinar Cu2+ en una solución. 3.7 Para muestras o soluciones problema ácidas, ¿qué método debe ser usado? .8 Calcule la concentración de Cl – en 25 ml de solución si el punto fi
nitrato de plata 0.1 M.
r de pH l
enmeyer de 250 ml
•• matraz de aforo de 10 ml
oporte universal pinzas para bureta

Métodos Cuantitativos Manual de Prácticas de Laboratorio
33
4.2 Desarrollo experimental
4.2.1 Preparación de la solución estándar e indicador 1
a) Calcular los gramos necesarios para preparar 500 ml de una solución 0.02N. b) Pesar los gramos a contenida en un vaso deprecipitado y aforar
2. Preparar el indicador, solución de K2CrO4 al 0.5% (W/V) a) Cacular los miligramos necesarios para preparar 10 ml de solución de K2CrO4 al 0.5% b) Pesar la sal de K2CrO4, disolver y aforar a
4.2.2 Valoración de la solución de AgNO3. (consultar esquema 1) Precaución: el nitrato de plata tiñe ropa y piel, Cualquier salpicadura debe de enjuagarse con agua inmediatamente. a) Pesar a 0.25 g de NaCl. NOTA: Se utiliza sal grado reactivo, secada toda la noche y llevada a temperatura ambiente. b) Disolver en 50 ml de aces erlenmeyer de 250 ml. c) Ajustar el pH de las soluciones adicionando pequeñas cantidades de NaHCO3 solo hasta
que la efervescencia desaparezca. r de 3 a 5 gotas de indicador al 0.5%. La solución se pondrá amarilla.
r valorar. ador hasta
Donde:
lúmen gastado de nitrato (ml) Eq = peso equivalente del NaCl
NOTAS:
. Preparación del estándar de AgNO3.
calculados, disolverlos en agua destilad en un matraz volumétrico. Guardar la solución en un frasco ámbar.
10 ml con agua destilada.
con exactitud y por triplicado 0.2
agua destilada contenida en tres matr
d) Agregae) Llenar y aforar una bureta de 25 o 50 ml con solución estándar de AgNO3 pof) Titular cada uno de los matraces que contienen el estándar primario y el indic
que se presente el primer vire permanente de amarillo a color rojizo. g) Registrar los gasto de para calcular la normalidad del AgNO3.
e NaCl = e AgNO3
(a/Eq) NaCl = (V * N) AgNO3
N = (a/V*Eq) = (TNaCl/AgNO3/ Eq)
N = normalidad del Nitrato a = mg del NaCl (estándar primario) V = vo
T = título en términos de mg de NaCl/ ml de AgNO3

Métodos Cuantitativos Manual de Prácticas de Laboratorio
34
hasta centésimas.
on aquellos resultados que coincidan hasta la tercera cifra
numero de replica muestra de NaCl (g) (a)
volumen de AgNO3 (ml) gastados (V)
concentración N de AgNO3 (N)
a. Este cálculo de efectuará por triplicado y al menos dos resultados deben coincidir
b. En caso de haber más de tres integrantes por equipo, cada uno realizará una determinación.
c. Calcular un promedio cdecimal. Colocar los resultados obtenidos en la siguiente tabla:
Tabla 1. Estandarización del AgNO3
b ---------- lanco ---------- 1 2 3
Promedio Nprom=
Esquema 1. Valoración del AgNO 0.02 N
4.2 a) Secar tras a 110 C durante 1 hora y enfriar en el desecador b) Pe
Matraces de 250 ml.
a. el color del
indicador desaparezca.
3
.3 Determinación de cloruros en la muestra problema (muestra sólida)
por triplicado las mues O
sar de 0.3-0.5 g del material seco y disuelva en 50 ml de agua destilada contenida en
Con papel indicador verificar el pH de cada solución, el pH debe ser neutro. si la solución es básica agregar a cada matraz 1 gota de fenolftaleína y después agregue gota a gota de ácido nítrico diluido (1 ml en 150 de agua) hasta que
0.2-0.25 g de NaCl en 50 ml de agua destilada y 2-3 gotas de K2CrO4
AgNO
3hasta el vire de color Rosa salmón.
3
Titular con la solución de AgNO

Métodos Cuantitativos Manual de Prácticas de Laboratorio
35
. si la solución es ácida agregar a cada matraz 1 gota de fenolftaleína, adicione NaOH 0.1N hasta que la ente añada 1 a 2 gotas del
c) Agregue 5 gotas del indicador K2CrO4 al 0.5%, la soluciónes se pondrán amarillas. d) Titule con nitrato de plata estándar hasta el primer vire de amarillo a rosa salmón. Tratar cada
muestra de la mima manera.
a es de color rojizo, pero con el color amarillo del cromato de potasio, la tonalidad cambia a rosa salmón .
e AgNO3 gastado en cada uno de los matraces. Complete la
f) Con la epara c os, cloruro de sodio y cloruro cúpri
g) Repe 3h) Calc r
% NaCl ={[(N * V) AgNO3 * EqX ] / b} * 100
uestra (ml) menos gasto en el blanco b = masa (en gramos) o volumen (en ml) de la muestra problema
equivalente de la sustancia por analizar, ej. cloruros, cloruro de calcio, uro de sodio, etc.
bsolución este ligeramente rosa. Posteriorm
ácido nítrico dilu
ido hasta que el color del indicador desaparezca.
El precipitado de cromato de plat
) Efectuar la lectura del volumen detabla 2.
stequiometría de la reacción y el número de moles de la solución estándar necesarios al anzar el punto final, calcular el porcentaje de clorurco en la muestra problema. tir el procedimiento desde el punto b de esta sección usando CaCO como blanco.
ula el % de NaCl:
Donde: N = normalidad del nitrato de plata V = gasto del nitrato de plata en la m
EqX = peso clor
Tabla 2. Titulación de muestras sólidas. -Replica de la
muestra problema peso en g de la muestra (0.2- 0.25g)
ml de titulante gastados
% Cl en cada muestra
1 2 3
Promedio
4.2.4 Determinación de cloruro lema (muestra liquida) Este m tre 1.5 y 100
g/l. Para aguas cuya concentración de cloruros sea inferior a 30 ppm no utilizar este método.
sigue: b) Pipetee 10 ml de muestra en un matraz de 250 ml con 40 ml de agua destilada
s en la muestra prob
étodo es recomendado para aguas con concentraciones de cloruros enm Nota: si se determinará cloruros en aguas potables o superficiales es importante que no tengan excesico color o turbidez.
a) Eliminar la maetria orgánica mediante filtración. Proceda por triplicado como

Métodos Cuantitativos Manual de Prácticas de Laboratorio
36
K2Crn.
Efectuar la lectura del volumen de AgNO3 gastado en cada uno de los matraces.
aCl en el agua diluida y no diluida.
e muestras liquidas. rm
- -
c) Agregue 5 gotas del indicador O4 al 0.5%, d) Titule con nitrato de plata estándar hasta el primer vire de amarillo a rosa salmóe)
Complete la tabla 3. f) Calcular la concentración en mol/litro del en Cl- y de N
g) Repetir el procedimiento desde el punto b de esta sección usando CaCO3 como blanco. esta muestra trabaja como se describe en el apartado 4.2.2.2. Se recomienda realizar una prueba de titulación para familiarizarse con la coloración del punto final.
Tabla 3. Titula ión dc
eplica de la volumen en ml muestra problema de la muestra
l de titulante gastados
%Cl en cada muestra diliuda
%Cl en cada muestra no diliuda
1 2 3
promedio
5. ANÁLISIS DE RESULTADOS • realice la deducción de la ecuación con la que se realiza el % de NaCl en muestras
sólidas y líquidas, así como su analisis dimensional
• Discuta sus resultados, comparelos con los de otos equipos, y si es posible con los resultados téoricos reportados
• práctica
6.3 ¿Se lograron los objetivos de la práctica? inentes.
LIOGRAFÍA
7.7 edition. 7.8 r the Determination of Silver and
. Soc.; 1994; 46(12); 2707 7.9 R.A. Day J.R. and A.L. Enderwood. Química analítica cuantitativa. Ed. pHH. 5a, edición.
mination,
• Desarolle las reacciones que ocurren en la valoración
recomedaciones y/o propuestas para el desarollo de la
6. CONCLUSIONES
6.4 Desarolle las conclusiones pert
7. BIB
Gary D. Christian. Analytical Chemistry. Ed. Wiley. fifth Kraemer E. O. and Stamm A. J. Mohr’s Method fo
Halogens in other than Neutral Solutions. J. Am. Chem
7.10 Skoog D. A.; West D. M.; Holler F. J. Fundamentals of Analytical Chemistry, 7th Edition. 1996.
7.11 Sheen R.T. and Kahler H. L. Effects of Ions on Mohr Method for Chloride DeterInd. Eng. Chem. Anal. Ed.; 1938; 10(11); 628-629.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
37
VALORACIONES CONDUCTIMÉTRICAS n para permitir el paso
s disoluciones dtencial. Al igual que un conductor m, por lo tanto, el recíproco de la
en mhos o siemens.
PARTE 2 La conductancia electrolítica es una medida de la facilidad de una disolucióe corriente eléctrica. La e electrolitos conducen la corriente eléctrica por la d
migración de los iones bajo la influencia de un gradiente de poetálico las disoluciones de electrolitos obedecen la ley de Ohm
resistencia electrolítica se denomina conductancia y se expresa
RL 1
= (1)
Donde: L es la conductancia expresada en mhos o siemens. R es la resistencia de la solución electrolítica en Ohms.
La conductancia observada de una disolución que se encuentra entre dos electrodos de láminas de platino paralelos uno a otro, depende inversamente de la distancia entre electrodos y irectamente de su área. d
⎟⎠⎞
⎜⎝⎛=
dAkL (2)
Donde: d es la distancia entre electrodos en cm. A es el área de los electrodos en cm2. k es la conductancia específica denominada conductividad. Se define como el recíproco de
la resistividad en mhos·cm-1. Para una celda conductimétrica dada con electrodos fijos, la relación d/A es una constante llamada constante de celda y se representa por θ . La ecuación (2) queda entonces:
θ=
kL (3)
aciones analíticas dependen de la relación entre la conductividad y la oncentración de los diversos iones presentes en la disolución, resultando como consecuencia la
conductividad equivalente:
En conductimetría, las aplicc
Nk
=Λ (4)
Donde:
Λ es la conductividad equivalente en mhos·L·cm-1·eq-1
N es la concentración en eq/L. La
conductividad equivalente también se puede expresar según:
N
k⋅=Λ
1000 (5)

Métodos Cuantitativos Manual de Prácticas de Laboratorio
38
onde:
A dcon
Dk es la conductividad en mhos·cm-1. N es la concentración de los iones en solución en eq/L.
Λ es la conductividad equivalente en mhos·cm2·eq-1.
ilución infinita los iones teóricamente son independientes entre sí y cada ión contribuye a la ductividad equivalente total; por lo tanto:
∑ ∑ −+∞ λ+λ=Λ 00 (6) Do
y son las co es y aniones a dilución infinita, respectivamente.
Si e
ustituyendo (3) y (7) en (5) y reordenando se tiene finalmente:
nde:
∞Λ es la conductividad equivalente a dilución infinita. 0+λ 0
−λ nductividades equivalentes iónicas de cation
xperimentalmente trabajamos a concentraciones diluidas entonces:
∑ −+∞ λ+λ=Λ )( 00 (7)
s
θ⋅
= −+
1000L (8)
λλ +⋅∑ )( 00N
LAS VA La det nme nácidos y bases cuya detección es difícil por medi pH pueden efectuarse por Lascon les requieren condiciones de operación más estrictas.
En e eterminar por nductimetría ya que los medios de reacción necesarios para asegurar una buena cuantitatividad
e acabo en presencia de medios ácidos concentrados. En estos la movilidad del ión H+, no permite diferenciar los cambios en la
on terés durante la valoración.
LORACIONES
medición de la conductividad de una disolución en el transcurso de una valoración permite ar el punto final de la valoración. En particuermi lar la determinación del punto final por
dició conductimétrica es usado en disoluciones muy diluidas. Por ejemplo las valoraciones de ción potenciométrica del
el m déto o conductimétrico.
va solubles e insolubles pueden efectuarse ductimétricamente y así evitar el uso de electrodos selectivos de iones los cua
loraciones de formación de complejos
gen ral el punto final de las valoraciones de oxidorreducción no se puede dcorequieren que la reacción se llevcasos la alta concentración y c ductividad de los iones de in

Métodos Cuantitativos Manual de Prácticas de Laboratorio
39
PRÁCTICA No. 6 IONES CONDUCTIMÉTRICAS ÁCIDO-BASE
1. OBJ
1.1 D ductimétricas de un ácido fuerte con una base fuerte y de una mezcla de dos ácidos (fuerte y débil) con una base fuerte.
1.2 Determinar con precisión el punto final de una valoración conductimétrica de un ácido
fuerte con una base fuerte y de una mezcla de dos ácidos (fuerte y débil) con una base f
2. INTRODUCCIÓN
La concent midiendo la variación de la conductancia que se observa cuando se le agrega una base de concentración conocida, pues a partir de las medidas conductimétricas se deduce fácilmente el punto final de la reacción de neutralización.
Este tipo de valoraciones conductimétricas se ve muy favorecido en el caso de reacciones ácido-base por el hecho de que las conductancias equivalentes iónicas a dilución infinita del ion H+ y el ion OH – son muy grandes comparadas con las de los demás iones.
La valoración conductimétrica correspondiente a la reacción de un ácido y una base fuerte, por ejemplo el HCl y NaOH, se basa en la siguiente reacción iónica:
2–
En la valoración del HCl, al adicionar el NaOH se consumirán los iones H+ y se acumularan en la disolución iones Na+ y por consiguiente disminuirá la conductancia de la disolución hasta llegar al punto de equivalencia. En este punto, la conductancia sólo se debe a los iones Cl – y Na+ presentes en el medio. P diendo más cantidad de base fuerte los iones OH – aparecerán en la disoluc ente aumento de la conductancia de la misma.
3. CUESTIONARIO PREVIO
3.1 Realiza) 100b) 100 ncentración 0.1M (densidad
del ácido acético = 1.05 g/mL, pureza = 99%). c) 500
3.2 Buscarsiguientes especies: H 3
3.3 Esqueuna ba
3.4 Escriba) El áb) El á
3.5 Establea) HCl con Nab) Una mezcla de HCl y CH3COOH con NaOH.
VALORAC
ETIVOS eterminar las curvas de valoración con
uerte.
ración de una disolución de un ácido se puede valorar
H+Cl – + Na+OH – → H O + Na+Cl
ero si se sigue añaión, con el consigui
ar los cálculos para preparar las siguientes disoluciones: mL de HCl 0.1 M (Pureza 36%, densidad = 1.21 g/mL).
ambos en co mL de una mezcla de HCl y CH3COOH,
m de NaOH 0.1M los valores de las conductividades equivalentes iónicas a dilución infinita de las
+
L
, Na+, OH – y CH COO –. matizar la forma de la curva de valoración conductimétrica de un ácido fuerte con se fuerte.
ir la reacción que se verifica para los siguientes casos: cido clorhídrico y el hidróxido de sodio. cido acético y el hidróxido de sodio. cer el cuadro de variación de concentraciones para la valoración de:
OH.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
40
3.6 . 3.7 Busca3.8 Elabor
. 4. PARTE EXPERIMENTAL
1 probeta de 50 mL 11 ica 1 bureta de 25 mL 1 itador magnético1 placa de agitación 1 pinza para bureta 1 celda conductimétrica 1 conductímetro papel absorbente para secar la celda. 1 jeringa 1 soporte universal 1 piceta
c n H ac ). Disol ión estandariz
4.2 Desarrollo experimental 4.2.1 Valoración de un ácido fuerte con una base fuerte.
Bureta con NaOH
celda
Conductímetro probeta
Placa de agitación
ra 1.
Describir el principio de funcionamiento de una celda conductimétricar que tipo de valoraciones ácido-base son difíciles de realizar por medición del pH. ar un diagrama de flujo donde se indique la secuencia experimental.
4.1 Material y reactivos
probeta de 25 mL pipeta volumétr de 10 mL
ag
Disolución de HCl de concentración 0.1 M Mez la de ácidos e disolución (HCl y CH3COO en concentr ión ≅ 0.1 M c/u
uc ada de NaOH 0.1M
Fig. 1 Sistema para medir conductancia.
a) Montar el dispositivo de la figu

Métodos Cuantitativos Manual de Prácticas de Laboratorio
41
ar una alícuota de 10.00 mL de disolución de HCl de concentración desconocida y transferirla a una probeta de 50 mL.
ración (0.0 mL de reactivo titulante
de NaOH 0.1 M y
1.00 mL.
ión graficar la conductancia en función del
4.2.2 Valoración de una mezcla de ácidos con una base fuerte. a) Montar el dispositivo de la figura 1. b) Tomar una alícuota de 10.00 mL de la mezcla de ácidos y transferirla a una
probeta de 50 mL. c) Repetir los pasos c) a i)
. ANÁLISIS DE RESULTADOS 5.1 Reportar los resultados obtenidos en una tabla que contenga la siguiente información.
Valoración conductimétrica de HCl con NaOH 0.1M. mL de NaOH Conductancia mL de NaOH Conductancia
b) Tom
c) Adicionar 10 mL de agua desionizada. d) Conectar el conductímetro. e) Introducir a la probeta, el agitador magnético y la celda conductimétrica. Tomar
la lectura de conductancia al inicio de la valoagregado).
f) Llenar una bureta de 25.00 mL con la disolución estándarajustar la marca a 0.00 mL.
g) Valorar la disolución de HCl con adiciones de alícuota de h) Registrar el volumen agregado de titulante y la conductancia en cada punto. i) Conforme transcurra la valorac
volumen agregado de titulante. j)
5
Valoración conductimétrica de una mezcla de ácidos (HCl y CH3COOH) con NaOH 0.1 M.
H Conductancia mL de NaOH Conductancia mL de NaOH Conductancia mL de NaO

Métodos Cuantitativos Manual de Prácticas de Laboratorio
42
ón de la mezcla de ácidos y la concentración de cada uno de los componentes de la mezcla
6.
6.2 Obtener las conclusiones pertinentes.
. BIBLIOGRAFÍA
7.1 ca Analítica General”
5.2 Construir las gráficas de conductancia en función del volumen agregado de titulante. 5.3 Localizar con ayuda del diagrama conductancia en función del volumen de hidróxido de
sodio los puntos finales de cada valoración. 5.4 Con los puntos finales de la valoración calcular la concentración del HCl, la concentraci
de ácidos. CONCLUSIONES 6.1 ¿Se lograron los objetivos de la práctica?
7
Charlot G., “Curso de Quími , 1ª edición, Tomo I, Editorial Toray-, España, 282 páginas. Masson, S.A., 1977, Barcelona
7.2 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo II, Editorial Toray-Masson, S.A., 1977, Barcelona, España, 200 páginas.
7.3 Meloan C.E y Kiser R.M., “Problemas y Experimentos en Análisis Instrumental”, 1ª edición, Editorial Reverté Mexicana, 1973, México, D.F., 560 páginas.
7.4 Skoog D.A. y Leary J.J., “Análisis Instrumental”, 4ª edición, Editorial Mc Graw
7.5 “Electroquímica Analítica”Hill/Interamarcana de España, 1994, Madrid España, 935 páginas. Vassos B.H., Ewing G. W., , 1ª edición, Editorial Limusa, S. A.
7.6 Willard H.H., Merrit L.L., Dean J.A. y Settle F.A., “Métodos Instrumentales de Análisis, 1ª edición, Editorial Iberoamérica S.A. de C.V., 1991, México, D.F., 884 páginas.
de C.V., 1987, México, D. F., 303 páginas.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
43
PRÁCTICA No. 7 VALORACIONES CONDUCTIMÉTRICAS DE COMPUESTOS QUE FORMAN
. OBJETIVOS
conductimétrica de cloruro de sodio con nitrato de plata. 1.2 Determinar con precisión el onductimétrica de cloruro de
sodio
. INTRODUCCIÓN
Las valoraciones por precipitac se producen entre compuestos iónicos de limitada solubilida itante más utilizado es el nitrato de plata, el cual se emplea para la determinación de ha haluros (SCN –, CN –, CNO –), mercaptanos, ác ivalentes y trivalentes. El método más común para determinar la cacurea ó va de valoración para este método, consiste en una gráfica de pAg en función del volu
3. CU
3.1
3.2s , Na+, Cl – y NO3
–. ración conductimétrica de cloruro de sodio con
3.5 Establecer loración de cloruro de s
3.6 Elaborar un diagrama de flujo donde se. 4. PARTE
4.1 Material y reactivos 1 matraz volumétrico de 100 mL 1 matraz volumétrico de 250 mL 2 probetas de 100 mL 1 obeta de 250 mL1 bureta de 25 mL 1 agitador magnético
PRECIPITADOS 1
1.1 Determinar la curva de valoración
punto final de una valoración c con nitrato de plata.
2
ión se basan en reacciones que d. El reactivo precip
luros, aniones del tipo de losidos grasos y diversos aniones inorgánicos b
oncentración de iones haluro en disoluciones osas es la valoración con una disolución patrón de nitrato de plata. El producto de la
n es el haluro de plata sólido. Una curccimen de nitrato de plata añadido.
TIONARIO PREVIO ES
Realizar los cálculos para preparar las siguientes disoluciones: a) 100 mL de NaCl 0.002 N. b) 100 mL de AgNO 0.05 N. 3
Buscar los valores de las conductividades equivalentes iónicas a dilución infinita de las iguientes especies: Ag+
3.3 Esquematizar la forma de la curva de valonitrato de plata.
3.4 Escribir la reacción que se verifica entre el cloruro de sodio y el nitrato de plata. el cuadro de variación de concentraciones para la va
o ato de pdio con nitr lata. indique la secuencia experimental.
EXPERIMENTAL
pr
1 placa de agitación

Métodos Cuantitativos Manual de Prácticas de Laboratorio
1 pinza para bureta 1 celda conductimétrica
44
papel absorbente para secar la celda.
1 soporte universal
Disolución de NaCl 0.002 N. AgNO3 0.05 N
Conductímetro probeta
Placa de agitación
Fig. 1 Sistema para medir conductancia.
a) Montar el dispositivo de la figura 1. b) Tomar una alícuota de 250.00 mL de disolución de NaCl de concentración desconocida
y transferirla a una probeta de 250 mL. c) Conectar el conductímetro. d) Introducir a la probeta, el agita celda conductimétrica. Tomar la lectura
3 0.00 mL.
a disolución de NaCl con adiciones de alícuota de 1.00 mL. ante y la conductancia en cada punto.
en
5. ANÁLISIS sultados obtenidos en una tabla que contenga la siguiente información.
Valoración conductimétrica de NaCl con AgNO3. mL de AgNO3 Conductancia mL de AgNO3 Conductancia
1 conductímetro
1 jeringa
1 piceta
Disolución de
4.2 Desarrollo experimental 4.2.1 Valoración de un NaCl con AgNO3.
Bureta con AgNO3
celda
dor magnético y la de conductancia al inicio de la valoración (0.0 mL de reactivo titulante agregado).
e) Llenar una bureta de 25.00 mL con la disolución estándar de AgNO y ajustar la marca a
f) Valorar lg) Registrar el volumen agregado de titulh) Conforme transcurra la valoración graficar la conductancia en función del volum
agregado de titulante.
DE RESULTADOS 5.1 Reportar los re

Métodos Cuantitativos Manual de Prácticas de Laboratorio
45
ductancia en función del volumen agregado de titulante.
tancia en función del volumen de AgNO3 el
6.
7.
Curso de Química Analí
5.2 Construir la gráfica de con5.3 Localizar con ayuda del diagrama conduc
punto final de la valoración. 5.4 Con el punto final de la valoración calcular la concentración del NaCl.
CONCLUSIONES
6.3 ¿Se lograron los objetivos de la práctica? 6.4 Obtener las conclusiones pertinentes. BIBLIOGRAFÍA
7.1 Charlot G., “ tica General”, 1ª edición, Tomo I, Editorial Toray-
7.2 Charlot G., “Curso de Química Analítica General”Masson, S.A., 1977, Barcelona, España, 282 páginas.
, 1ª edición, Tomo II, Editorial Toray-r lona, España, 200 páginas. .M., “Problemas y Experimentos en Análisis Instrumental”
Masson, S.A., 1977, Ba7.3 Meloan C.E y Kiser R
ce, 1ª
inas. edición, Editorial Reverté Mexicana, 1973, México, D.F., 560 pág7.4 Skoog D.A. y Leary J.J., “Análisis Instrumental”, 4ª edición, Editorial Mc Graw
Hil /Interamarcana de España, l 1994, Madrid España, 935 páginas. 7.5 Vassos B.H., Ewing G. W., “Electroquímica Analítica”, 1ª edición, Editorial Limusa, S. A.
de C.V., 1987, México, D. F., 303 páginas. 7.6 Willard H.H., Merrit L.L., Dean J.A. y Settle F.A., “Métodos Instrumentales de Análisis, 1ª
edición, Editorial Iberoamérica S.A. de C.V., 1991, México, D.F., 884 páginas.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
46
DETERMINACIÓN DEL PRODUCTO DE SOLUBILIDAD DE UN PRECIPITADO ALORACIÓN CONDUCTIMÉTRICA
1.
1.1 ductimétrica de cloruro de bario con sulfato de sodio. 1.2 d del sulfato de bario. 1.3 te del producto de solubilidad (Ks) del sulfato de bario.
2. INTR El equilibrio para un compuesto poco soluble de fórmula AB, se puede representar por la sigui B –
y la constante de equilibrio termodinámico esta dada por:
PRÁCTICA No. 8
POR V
OBJETIVOS
Trazar la curva de valoración conDeterminar la solubilidaDeterminar la constan
ODUCCIÓN
ente reacción AB↓ ⇄ A+ +
)]([]][[
sABBAKeq
−+
=
si [AB
donde Ks se llama el producto de solubilidad.
Si en el equsolubilidad del do que la solución es ideal, que los iones que la forman no sufren másdel precipitado, entonces la solubilidad se puede relacionar con el producto de solubilidad. Así, si se disuelsiguiente equili
s s entonces, queda:
. CUESTIONARIO PREVIO
3.1 Realizar los cálculos para preparar las siguientes disoluciones: i) 100 mL de BaCl2 0.002 N j) 100 mL de Na SO 0.05 N
uctividades equivalentes iónicas a dilución infinita de las siguientes especies: Ba2+, Na+, Cl – y SO4
2–. 3.3 En un experimento se valoraron 250 mL de BaCl con Na SO4 0.05 N y se obtuvieron los
sigui
↓] = 1 ]][[][ −+==↓ BAKsABKeq
]][[ −+= BAKs
ilibrio se han disuelto s moles por litro del precipitado, entonces s se define como la precipitado. Asumien
reacción y que la solución no contiene iones comunes con aquellos provenientes
ven s moles/L de AB↓ para dar s moles/L de A+ y s moles/L de B –, de acuerdo al brio:
AB↓ ⇄ A+ + B –
2))((]][[ sssBAKs === −+
3
2 4 3.2 Buscar los valores de las cond
2 2entes resultados

Métodos Cuantitativos Manual de Prácticas de Laboratorio
47
V [mL] de Na2SO4 0.0 1.0 2.0 3.0 4.0 5.0 5.5 6.0 6.5 7.0 8.0 9.0 10 L [µS] 469 468 467 461 459 457 455 452 451 448 447 451 465
V [mL] de Na2SO4 11 12 13 14 15 16 17 18 L [µS] 498 1 677 711 747 530 566 601 64
a) Escribir la reacc 4. b) Tra ar la gráfica regado de titulante. c) Ded) Cae) Caf) Cag) Ca ul
jo donde se indique la secuencia experimental.
4
4.4 M1 matraz volumétrico de 100 mL 1 matraz volumétrico de 250 mL 2 probetas de 100 mL 1 obeta de 250 mL1 bureta de 25 mL 1 itador magnético1 placa de agitación 1 pinza para bureta 1 celda conductimétrica 1 conductímetro papel absorbente para secar la celda.
erimental
4.
tar el dispositivo de la figura 1. ar una alícuota de 250.00 mL de disolución de BaCl2 y transferirla a una
ión que se verifica entre el BaCl2 y el Na2SO de conductancia en función del volumen agz
terminar el volumen y la conductancia en el punto de equivalencia. lcular la concentración inicial del BaCl2.
2+ 2–lcular la concentración de los iones Ba y SO4 en el punto de equivalencia. lcular la solubilidad del BaSO4.
ar el pKs del BaSOlc 4. 3.4 Elaborar un diagrama de flu
ARTE EXPERIMENTAL P
aterial y reactivos
pr
ag
1 jeringa 1 soporte universal 1 piceta Disolución de BaCl2 0.002 N.
5 Disolución de Na2SO4 0.0 N
4.5 Desarrollo exp
4.5.1 Valoración de un BaCl2 con Na
2SO
a) Monb) Tom
probeta de 250 mL. c) Conectar el conductímetro. Prender el aparato 15 minutos antes de iniciar la
medición. d) Introducir a la probeta, el agitador magnético y la celda conductimétrica. Tomar
la lectura de conductancia al inicio de la valoración (0.0 mL de reactivo titulante agregado).
e) Llenar una bureta de 25.00 mL con la disolución estándar de Na2SO4 y ajustar la marca a 0.00 mL.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
48
celda
Conductímetro probeta
Placa de agitación
Fig. 1 Sistema para medir conductancia. f) Valorar la disolución de BaCl2 con adiciones de alícuota de 1.00 mL. g) Registrar el volumen agregado de titulante y la conductancia en cada punto. h) Conforme transcurra la valoración graficar la conductancia en función del
volumen agregado de titulante.
ANÁLISIS DE RESULTADOS 5.4 Reportar los resultados obtenidos en una tabla que contenga la siguiente información.
Valoración conductimétrica de BaCl2 con Na2SO4. mL de Na2SO4 Conductancia mL de Na2SO4 Conductancia
Bureta con Na2SO4
5
6.5 ¿Se lograron los objetivos de la práctica?
5.5 Construir la gráfica de conductancia en función del volumen agregado de titulante. 5.6 Determinar el volumen y la conductancia en el punto de equivalencia. 5.7 Calcular la concentración inicial del BaCl2. 5.8 Calcular la concentración de los iones Ba2+ y SO4
2– en el punto de equivalencia. 5.9 Calcular la solubilidad del BaSO4. 5.10 Calcular el pKs del BaSO4.
CONCLUSIONES 6

Métodos Cuantitativos Manual de Prácticas de Laboratorio
49
6.6 Obtener las conclusiones pertinentes.
7 BIBLIOGRAFÍA
7.1 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo I, Editorial Toray-Masson, S.A., 1977, Barcelona, España, 282 páginas.
7.2 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo II, Editorial Toray-Masson, S.A., 1977, Barcelona, España, 200 páginas.
7.3 Meloan C.E y Kiser R.M., “Problemas y Experimentos en Análisis Instrumental”, 1ª edición, Editorial Reverté Mexicana, 1973, México, D.F., 560 páginas.
7.4 Skoog D.A. y Leary J.J., “Análisis Instrumental”, 4ª edición, Editorial Mc Graw Hill/Interamarcana de España, 1994, Madrid España, 935 páginas.
7.5 Vassos B.H., Ewing G. W., “Electroquímica Analítica”, 1ª edición, Editorial Limusa, S. A. de C.V., 1987, México, D. F., 303 páginas.
7.6 Willard H.H., Merrit L.L., Dean J.A. y Settle F.A., “Métodos Instrumentales de Análisis, 1ª edición, Editorial Iberoamérica S.A. de C.V., 1991, México, D.F., 884 páginas.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
50
mica con la que entra en contacto. Dicha iones separadas, una con un electrodo indicador y la
tra con un electrodo de referencia, que son unidas por la muestra en disolución, de tal manera ión que compara un valor desconocido con otro conocido
ara cuantificar el parámetro problema, regido por la ecuación de Nernst.
l electrodo indicador es específico para cada parámetro a determinar, mientras que el de ferencia es común a todos ellos. Estos electrodos específicos pueden ser electrodos de embrana (como el electrodo para medir pH), la cual hace el papel de intercambiador de iones
on la solución que contiene a la muestra, lo que genera un cambio valorable en el potencial de embrana. La cadena galvánica de medición que está dentro del electrodo, determina la iferencia de los potenciales de ambos lados de la membrana.
l objetivo de una medición potenciométrica es obtener información acerca de la composición de na disolución mediante el potencial que aparece entre dos electrodos. La medición del potencial e determina bajo condiciones reversibles, en forma termodinámica, y esto implica que se debe ejar pasar el tiempo suficiente para llegar al equilibrio, extrayendo la mínima cantidad de tensidad para no influir sobre el equilibrio que se establece entre la membrana y la disolución uestra.
ara obtener mediciones analíticas válidas en potenciometría, uno de los electrodos deberá ser de otencial constante y que no sufra cambios entre uno y otro experimento. El electrodo que cumple sta condición se conoce como electrodo de referencia. Debido a la estabilidad del electrodo de ferencia, cualquier cambio en el potencial del sistema se deberá a la contribución del otro
lectrodo, llamado electrodo indicador o de trabajo.
l potencial registrado es en realidad la suma de todos los potenciales individuales, con su signo orrespondiente, producidos por los electrodos indicador y referencia.
e han podido diseñar electrodos que responden de manera específica a cierto analito. El uso de stos electrodos para que midan las diferencias de po nadas por la diferente
concentración las medidas potenciométrica
nciométricos comprenden dos tipos fundamentales de análisis. Uno implica la me n la con e la medición de los cambios del potencial originados por la adición de un titulante a la muestra (valoraciones
Me
PARTE 3 VALORACIONES POTENCIOMÉTRICAS
La potenciometría es una técnica analítica que permite cuantificar la concentración de una sustancia en disolución. Esto se logra relacionando la actividad iónica de la disolución con la
erza electromotriz existente en una celda electroquífucelda electroquímica consta de dos disolucoque permite formar un sistema de medicp Eremcmd Eusdinm Pperee Ec Se tencial eléctrico origi
ímica, constituyende una especie qu el fundamento de s.
Los métodos pote
dició directa de un potencial de electrodo a partir del cual se puede determinar centración de un ión activo (potenciometría directa). El otro tipo comprend
potenciométricas).
didas Potenciométricas Directas.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
51
En su forma más simple se puede utilizar la celda galvánica constituida; de una parte por la solparquecircmede
VaLaselede me en el recipiente de valoración se detecta por medio del electrodo indicador. El potencial de la solución varrecen punabs
ución de la especie a la cual se determinar su actividad, en contacto con un metal que sirve a conducir las cargas que producirán el cambio en el estado de oxidación, constituyendo así los se denomina un electrodo indicador ya que es el que causará la señal correspondiente en el uito eléctrico de medida. De otra parte, el cambio en el estado de oxidación de la especie ncionada arriba se promueve mediante el cambio en el estado de oxidación de otra que sirve referencia.
loraciones potenciométricas. valoraciones potenciométricas consisten en la medición de la diferencia de potencial entre un ctrodo indicador y un electrodo de referencia durante el transcurso de una titulación. El objetivo la medición potenciométrica es obtener información acerca de la composición de una solución diante el potencial que aparece entre los dos electrodos. El potencial de la solución
ía a medida que se agrega el reactivo titulante. El problema más crítico de una valoración es el onocimiento del punto en el cual las cantidades de las especies reaccionantes están presentes cantidades equivalentes (el punto final). La curva de valoración puede seguirse punto por to, localizando como ordenadas de una gráfica a los valores del potencial de la celda y como cisas a los valores de volumen de titulante.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
52
PRÁCTICA No. 9 VALORACIONES POTENCIOMÉTRICAS ÁCIDO-BASE
1. OBJETIVOS
1.1 e urva de valora n otenciométrica de una disolución problema de Na2CO3. 1.2 Determinar la concentración de una disolución problema de Na2CO3.
2. INTRODUCCIÓN Los ácidos y las bas tivos o catalizadores en diversos procesos industriales. e el de am bricar fertilizantes y explosivos, se prepara a partir de la neutralización de ácido nítrico y amoniaco; y el ácido
l etílico.
En muchos procesos biológicos participan también ácidos y bases; por ejemplo, la acumulación de ácido láctico en el tejido muscular durante una sesión de ejercicio pesado produce dolor muscular; este mismo ácido es el responsable del olor y sabor característico de la leche agria. El ácido clorhídrico es un componente de los jugos digestivos estomacales; si se encuentra en cantidad excesiva provoca úlceras y si la cantidad es demasiado pequeña, en ocasiones se produce anemia. Diversos medicamentos como la aspirina y la vitamina C son ácidos. El vinagre es una disolución diluida de ácido acético, la limonada contiene los ácidos cítrico y ascórbico. La leche de magnesia es una base, al igual que el carbonato de sodio. Los blanqueadores, los limpiadores de horno y la mayor parte de los destapacaños son bases. Las reacciones entre los ácidos y las bases constituyen uno de los tipos más importantes de reacciones químicas y es fundamental comprender su funcionamiento en las diversas áreas del conocimiento. Ácidos tales como el H2CO3 y el H3PO4 que contienen más de un hidrógeno intercambiable se llaman polipróticos. El ión hidrógeno que se intercambia más fácilmente da origen a la constante Ka de mayor valor, los siguientes valores de Ka son progresivamente más pequeños. En la valoración de ácidos polipróticos, si la diferencia entre los valores sucesivos de pKa es por lo menos de cuatro unidades, se observan puntos de equivalencia separados, mostrando ada uno un amplio cambio de pH.
gran
– +
El cu mplificadas que imponen el pH en el transcurso de la valoración se encuentran en la siguiente tabla.
Obten r la c ció p
es se emplean como reac Por jemplo, nitrato onio que se emplea para fa
fosfórico se usa como catalizador en la producción de alcoho
c La valoración de ácidos polipróticos o bases polipróticas tiene gran importancia y es de
–1interés práctico; por ejemplo, si se valora una disolución de Na2CO3 10 –1 M con HCl 10 M, en el transcurso de la valoración se presentan las siguientes reacciones sucesivas:
CO32– + H+ ⇄ HCO3
–
HCO3 + H ⇄ H2CO3
adro de variación de concentraciones y las ecuaciones si

Métodos Cuantitativos Manual de Prácticas de Laboratorio
53
→ HCO3
– Ecuaciones
CO32– + H+
X=0 i Co
CopKapH
21
217 2 ++=
0<X<1
APE (1-x)Co
εCo≅0
xCo [ ][ ]−
−
+=3
23
2 logHCOCO
pKapH
⎟⎠⎞
⎜⎝⎛ −
+=x
xpKapH 1log2
2=x 1 PE
21
Co21 Co
21
2pKapH = εCo ≅0
X=1 PE
εCo ≅0
Co εCo ≅0 2
12 pKapKapH
+=
+ → H2CO3 HCO3– + H
X´=0 i
2
12 pKapKapH
+= Co
[ ]
0<X´<1
(1-x)Co
≅
APE εCo 0 xCo
[ ]321 COH
3logHCO
pKapH−
+=
⎟⎠⎞
⎜⎝⎛ −
+=´
´1log1 xxpKapH
21´=x PE
21 Co
21 εCo ≅0 Co
21
1pKapH =
X´=1 PE εCo ≅0 εCo ≅0 Co
CopKapH log
21
2 1=1
−
DPE
εCo ≅0
(x´-1)Co
Co
pH=-log[HX´>1 +]=-log{(x´-1)Co}
La curva d
e valoración se representa en la siguiente figura.
Curva teórica de valoración deNa2CO3 0.1M con HCl 0.1M
0
2
8
10pH
4
6
12
0 1 2 3x (fracción de la especie titulada)

Métodos Cuantitativos Manual de Prácticas de Laboratorio
54
. CUESTIONARIO PREVIO
3.1 Busca cuales son las partes de las que esta constituido un electrodo para medir pH. 3.2 Describe el principio de funcionamiento del electrodo combinado para medir pH. 3.3 Con ayuda de las ecuaciones que imponen el pH en el transcurso de la valoración de
Na2CO3 0.1 M con HCl 0.1 M. Trazar la curva teórica de valoración (pH=f(x) de esta titulación.
3.4 Describe en que consiste cada uno de los siguientes métodos para la determinación del punto de equivalencia en una valoración potenciométrica. k) Método del paralelogramo l) Método de la primera derivada m) Método de la segunda derivada n) Método de la gráfica de Gran
3.5 Realizar los cálculos para preparar las siguientes disoluciones: a) 100 mL de HCl 0.1 ). b) 100 mL de Na2CO3 0.1 N
4. PARTE EXPERIMENTAL
4.1 Material y reactivos vasos de precipitados de 250 mL pipeta graduada de 10 mL pipeta volumétrica de 10.00 mL matráz volumétrico de 100.00 mL bureta de 50.00 mL soporte universal
1 pinzas para bureta electrodo para medir pH agitador magnético placa de agitación potenciómetro
isolución de HCl 0.1 N. isolución de Na CO 0.1N.
4.2 Desa
4.2.1 Pa)
frasco haciendo constar su contenido, la fecha, el nombre del alumno
4.2.2
y puede romperse. b) Conectar el potenciómetro a la corriente eléctrica. Conectar el electrodo
trada correspondiente del aparato. ”
para sacar, introducir, cambiar o enjuagar el electrodo. El electrodo se debe secar a que no se raye.
3
N (Pureza 36%, densidad = 1.21 g/mL
211111
1111 DD 2 3
rrollo experimental reparación de disoluciones Disolución de HCl 0.1 N. Medir 0.83 mL de HCl concentrado, llevar al aforo en un matraz volumétrico de 100 mL con agua destilada y guardar en un frasco limpio. Etiquetar el y dejando espacio para reseñar la normalidad después de que se determine con exactitud.
b) Disolución de Na2CO3. Calibración del potenciómetro Fisher Scientific a) Manejar en forma cuidadosa el electrodo combinado para medir pH debido a que
es muy frágil
combinado a la enc) Siempre colocar el botón de “function” del potenciómetro en la posición “STD BY
con un papel suave y absorbente par

Métodos Cuantitativos Manual de Prácticas de Laboratorio
55
d) Introducir el electrodo en la disolución buffer de pH = 7.0. Colocar el botón de “function” en la posición pH y ajustar el valor a 7.0 usando la perilla marcada como “standarize”.
e) Enjuagar el electrodo con agua destilada, se arlo e introducirlo en la disolución buffer de pH = 4.0. Ajustar el valor de pH en el potenciómetro con la perilla marcada como “slope”. Enjuagar el electrodo y secarlo.
f) Repetir los pasos d) y e) tantas veces como sea necesario, hasta que al introducir el electrodo de pH en las soluciones buffer, el potenciómetro marque el valor de pH de la disolución sin realizar ningún ajuste.
c
Conección para el electrodo
Fig. 2 Potenciómetro Fisher Scientific.
4.2.3 Valoración de Na2CO3 10 –1 M con HCl 10 –1 N
function
standardize
slope
temperature

Métodos Cuantitativos Manual de Prácticas de Laboratorio
a) Montar el dispositivo de la figura 1. una alícuota de 10 mL de Na CO . Agregar 40 mL de agua destilada e
la valoración (0.00 mL de reactivo titulante agregado).
l y ajustar la marca a 0.00 mL.
disolución de Na2CO con adiciones de alícuota de 0.50 mL. l volumen agregado e titulante y el pH en cada punto.
f) Conforme transcurra la valoración graficar el pH en función del volumen agregado de titulante.
5. AN5 contenga la siguiente información.
de HCl pH
b) Tomar 2 3introducir el electrodo lenta y cuidadosamente en la disolución de Na2CO3 cuyo pH se va a determinar. Evitar que la barra magnética al girar, golpee al electrodo. Tomar la lectura de pH al inicio de
c) Llenar una bureta de 25.00 mL con la disolución estándar de HC
d) Valorar la 3e) Registrar e d
ÁLISIS DE RESULTADOS .1 Reportar los resultados obtenidos en una tabla que
Valoración potenciométrica de Na2CO3 con HCl. mL mL de HCl pH
56

Métodos Cuantitativos Manual de Prácticas de Laboratorio
57
encia de la valoración potenciométrica por los siguientes métodos.
6 CO
6.7 ¿Se lograron los objetivos de la práctica?
7 BIBLIOGRAFÍA
.1 Ayres, G. H. “Análisis Químico Cuantitativo” Ed. Harper and Row, Madrid (1990). 7.2 Ga ía-Avila J Apun Ana a". U IB7.3 is Químico Cuantitativo. Editorial Rev rté (2001). 7.4 itativo”, Porrúa, S. A. México (1987). 7.5 Skoog, D. A. , West,D.M. Holler, F.J. y Crouch, S.R. "Fundamentos de Química Analítica"
8a. edición Ed. Thompson. (2005). 7.6 S og D. A. y Lear J J.. "A lisis nstru tal".4ta e ición. Ed. Mc Graw Hill. (1994). 7.7 g . I. “Química Analítica” Kapeluz 5ª. Edición
5.2 Construir la gráfica de pH en función del volumen agregado de titulante. 5.3 Localizar con ayuda del diagrama de pH en función del volumen de HCl el punto de
equivala) Método del paralelogramo b) Método de la primera derivada c) Método de la segunda derivada d) Método de la gráfica de Gran
5.4 Con el punto de equivalencia de la valoración calcular la concentración del Na2CO3.
NCLUSIONES
6.8 Obtener las conclusiones pertinentes.
7 rc . " tes de Química lític P I. IPN.
Harris D.C. AnálisOrozco, D. “Análisis Químico Cuant
e
ko , y . ná I men d Vo el, A

Métodos Cuantitativos Manual de Prácticas de Laboratorio
58
1.
1.1 Determinar con precisión el punto final de la valoración potenciométrica de I2 con
te la curva de valoración potenciométrica de I2 con Na2S2O3. 1.3 a disolución de I2 cuando se valora con Na2S2O3 por el
2. INTR
En la ciométricas de oxidorreducción habitualmente se usa un electrodo indic tectar el punto de equivalencia. En ocasiones se recurre a otros y el mercurio. Una s usadas en potenciometría es la ecuación de Nernst.
PRÁCTICA No. 10 VALORACIONES POTENCIOMÉTRICAS DE OXIDORREDUCCIÓN
OBJETIVOS.
Na2S2O3. 1.2 Trazar experimentalmen
Determinar la concentración de unmétodo potenciométrico.
ODUCCIÓN. s valoraciones poten
ador inerte de platino para de metales, como la plata, el oro
de las ecuaciones má
][Re
][ln0E +d
OxnFRT
dond
E encial observado) E uctor dado. R T condiciones normales. F Faraday. n ectrones que intervienen en la reacción de oxidorreducción.
constantes y pasando de logaritmos naturales a
logar d
E =
e:
es el potencial del s stemai (potº es el potencial normal del par oxidorred
iversal de los gases. es la constante un a es la temperatura
es la constante dees el número de el
Sustituyendo los valores numéricos de lasitmos iec males, obtenemos:
][Re
][log06.00
dOx
nEE +=
Una valoracióconcentración a) La mezcla r e en todas las etapas de
la valorab) La constan
que, en el pproductos.
c) Si se están s valores del potencial, Eº, para las reacciones oxidorreductoras individuales deben tener una d esario para la distinción de los puntos d
n potenciométrica de oxidorreducción se puede utilizar para calcular la de un oxidante o de un reductor siempre que:
eaccionante alcance el equilibrio casi instantáneamentción.
te de equilibrio para la reacción de valoración sea suficientemente grande para unto de equivalencia el 99.9% (mínimo) de los reactivos se hayan convertido en
determinando conjuntamente la concentración de más de una especie, lo
iferencia entre si de por lo menos 0.2 V, lo que es nece equivalencia en la curva de valoración.
Métodos Cuantitativos Manual de Prácticas de Laboratorio
59
d) La celdoxidorreducdebe ser un electrodo de potencial constante.
El cuadro de variación de concentraciones y las ecuaciones simplificadas que imponen el potencial, E con tiosulfato se encuentran en la siguiente tabla.
+ S4O6 Ecuaciones
a electroquímica debe estar dispuesta de tal manera que la reacción de ción ocurra solamente en la mitad del compartimiento, la otra mitad de la celda
, en el transcurso de la valoración de yodo
I2 + 2S2O3 2– → 2I– 2–
x=0
i
Co
][Re][log06.00
dOx
nEE +=
0log
206.054.0 CoE +=
0<x<2
APE
Cox )
211( −
≅0
xCo
xCo21 2
2
][[log
206.054.0 −+=
IIE ]
2)(
)2
1(log03.054.0
CoxE
−+=
1
xCo
X=2
PE
≅0
≅0
2Co
Co 2)1.0(2)54.0(2 +
=E
x>2
DPE
≅0
(x-2)Co
2Co
Co 22
32
264
][][
log206.010.0 −
−
+=OSOS
E
2])2[(log03.010.0
CoxCoE
−+=
La curva de valoración se representa en la siguiente figura.
Curva teórica de valoración de I2 0.1M con Na2S2O .1 M
0,1
0,3
0,4
0,5
0,6
0,7
2 3x (fracción de la es ecie titulada)
E(V)
3. CUESTIONARIO PREVIO.
3.1 Realizar los cálculos para preparar las siguientes disoluciones:
3 0
0,2
0 1p

Métodos Cuantitativos Manual de Prácticas de Laboratorio
60
a) 100 mL de una disolución de H2SO4 3 M. b) 100 mL de I2 0.05 M. c) 100 mL de Na2S2O3.
3.3 el potencial estándar de los siguientes pares oxidorreductores.
3.4 e reunir un electrodo indicador. 3.5 debe reunir un electrodo de referencia. Da algunos
3.6 stigar el principio de funcionamiento de los siguientes electrodos de referencia Ag+/AgCl y E.C.S.
as ecuaciones que imponen el potencial en el transcurso de la valoración a curva teórica de valoración (E=f(x) de esta
.8 Elaborar un diagrama de flujo donde se indique la secuencia experimental.
4. 4.1
1 parrilla de calentamiento 1 placa de agitación 1 agitador magnético Disolución de I2 0.05 M Disolución de Na S O 0.05 M
Disolución de almidón al 0.1% Yoduro de potasio
sodio
o de precipitados de 100 mL. Disolver el KI con L de agua destilada. Transferir el contenido del vaso a un matraz
3.2 Escribir la reacción que se verifica entre I2 y Na2S2O3. a) I2 / I –. b) S4O6
2– / S2O3 2–.
Investigar a) I2 / I – b) S4O6
2– / S2O3 2–.
Investigar las características que debInvestigar las características queejemplos de este tipo de electrodos. Inve
3.7 Con ayuda de lde I2 0.1 M con S2O3
2– 0.1 M. Trazar ltitulación.
3
PARTE EXPERIMENTAL. Material y reactivos.
1 vaso de precipitados de 250 mL 1 vaso de precipitados de 150 mL 4 vasos de precipitados de 100 mL 1 bureta de 25 mL 1 probeta de 100 mL 1 pipeta volumétrica de 5 mL 1 pipeta graduada de 10 mL 1 pipeta graduada de 1 mL 1 pinza para bureta 1 soporte universal
2 2 3Disolución de H2SO4 3 M
en peso
Oxalato deYodato de potasio
4.2 Desarrollo experimental. 4.2.1 Preparación de disoluciones
a) Disolución de KI 0.01 M. Pesar la cantidad necesaria para preparar 100 mL de KI 0.01 M y transferirla a un vas30 mvolumétrico de 100 mL y llevar hasta la marca con agua destilada. Envasar.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
61
ámbar y proteger de la luz. c) Disolución de Na2S2O3 0.05 M. Pesar la cantidad necesaria para preparar 100
4.2.2 Estandarización de una disolución de Na2S2O3.
gregar a la solución anterior 2 g de KI sólido y con agitación constante valorarla con Na2S O3 hasta que el color de la solución sea ligeramente amarilla.
d) Adicionar 1 mL de almidón y continuar la valoración hasta el vire del color azul al i loro.
e) Realizar por triplicado la estandarización.
. alorac ón potenciométrica de I2 2S2O3. ) Montar el esquema de la figura 1.
i) Con una pipeta volumétrica, medir 10.00 mL de la disolución de I2 y transferirla a u aso d p o de 1 mL. A icionar con una pipeta graduada 1 mL de H2SO4 3 M. Agregar con una probeta 40 mL de agua destilada. Introducir a la disolución la barra de agitación y los electrodos de referencia e indicador.
ión de Na2S2O3 estandarizada. k) Valorar la disolución de I2 con adiciones 0.5 mL de Na2S2O3.Registrar el
volumen de titulante agregado y el potencial en cada punto. l) Conforme transcurra la valoración graficar el potencial en función del volumen
del reactivo titulante agregado.
b) Disolución de I2 0.05 M. Pesar la cantidad necesaria para preparar 50 mL de I2 0.05 M y transferirla a un vaso de precipitados de 50 mL. Disolver el I2 con 30 mL de una disolución de KI 0.01 M. Transferir el contenido del vaso a un matraz volumétrico de 50 mL y llevar hasta la marca con la disolución de KI 0.01 M. Envasar en un frasco color
mL de Na2S2O3 0.07M y transferirla a un vaso de precipitados de 100 mL.Disolver el Na2S2O3 con 30 mL de agua destilada previamente hervida y fría.Transferir el contenido del vaso a un matraz volumétrico de 100 mL y llevar hasta la marca con agua destilada hervida y fría. Envasar en un frasco ámbar.
a) En un vaso de precipitados de 100 mL pesar con exactitud 30 mg de KIO3. b) Adicionar 40 mL de agua destilada y 10 mL de H2SO4 2.5 M. c) A
2
nco
3 V4.2 i con Na
h
n v e recipitad s 00 d
j) Llenar una bureta de 25.00 mL con la disoluc

Métodos Cuantitativos Manual de Prácticas de Laboratorio
62
ra 1
5. te información:
de titulante Potencial (mV) mL de titulante Potencial (mV)
Figu
ANÁLISIS DE RESULTADOS. 5.1 Construir una tabla que contenga la siguien
mL
5.2 en función del volumen agregado de titulante. 5.3 del diagrama de potencial en función del volumen de titulante
equivalencia de la valoración potenciométrica por los siguientes
e Gran 5.4 de la valoración calcular la concentración de la disolución de
6. 6.1 6.2 Obten
7. BIBLIOGRAFÍ7.1 Charlot
Graficar el potencial Localizar con ayuda(Na2S2O3) el punto demétodos. a) Método del paralelogramo b) Método de la primera derivada ) Método de la segunda derivada c
d) Método de la gráfica dCon el punto de equivalenciaI2.
CON ICLUS ONES.
¿Se lo rgra on los objetivos de la práctica? er las conclusiones pertinentes.
A. G., “Curso de Química Analítica General”, 1ª edición, Tomo I, Editorial Toray
, S.A., 1977, Barcelona, España, 282 páginas. Masson7.2 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo II, Editorial Toray-
A., 1977, Barcelona, España, 200 páginas. y Kiser R.M.,
Masson, S.7.3 Meloan C.E “Problemas y Experimentos en Análisis Instrumental”, 1ª edición,
verté Mexicana, 1973, México, D.F., 560 páginas. Editorial e7.4 S
Rkoog D.A. y Leary J.J., “Análisis Instrumental”, ill/Inter a
4ª edición, Editorial McGraw H
7.5 Vassos am rcana de España, 1994, Madrid España, 935 páginas. B.H., Ewing G. W., “Electroquímica Analítica”, 1ª edición, Editorial Limusa, S. A. de 87, México, D. F., 303 páginas.
C.V., 19
7.6 Willard H.H., Merrit L.L., Dean J.A. y Settle F.A., “Métodos Instrumentales de Análisis”, 1ª Editorial Iberoamérica S.A. de C.V., 1991, México, D.F., 884 páginas. edición,

Métodos Cuantitativos Manual de Prácticas de Laboratorio
63
VALORACIO
1. OBJETIVOS.
1.1 Determcon AgNO
1.2 Trazar1.3 Determina ción de KCl cuando se valora con AgNO3 por el
método potenciométrico. 2. INTRODUCCIÓN.
La concentración de los iones que forman p s puede determinarse por medio de una valoración potenciométrica. Para este tipo de valoraciones, lo que se hace es diseñar una
de salida sea proporcional a pX= – log [X ], donde X es el ión e precipitación son prácticas
olamente si el producto de la reacción es muy insoluble. Los electrodos indicadores que se requieren pa n tión o del anión que ha de seguirse durante la titulación. Resulta útil deducir una curva teórica de valoración para entender lo que ocurre durante la valoración por precipitación. La curva de valoración es una gráfica que muestra como varía la concentración de uno de los reactivos a medida que se agrega un titulante. Para las curvas teóricas de valoración por precipitación se traza la gráfica pX en función del volumen agregado de titulante ó la gráfica pX=f(x). El cuadro de variación de concentraciones y las e s que imponen el otencial, E, en el transcurso de la valoración de yodo con tiosulfato se encuentran en la iguiente tabla.
PRÁCTICA No. 11 NES POTENCIOMÉTRICAS DE COMPUESTOS QUE FORMAN
PRECIPITADOS
inar con precisión el punto de equivalencia de la valoración potenciométrica de KCl . 3
experimentalmente la curva de valoración potenciométrica de KCl con AgNO3. r la concentración de una disolu
recipitado
celda electroquímica cuyo voltajeque será determinado o el ión valorante. Las valoraciones ds
ra una valoració potenciométrica de precipitación dependen del ca
cuaciones simplificadaps
– + Cl + Ag → AgCl↓ Ecuaciones
x=0 i
Co
]log[ −−= ClpCl
CopCl log−= (1-x)Co ≅0 0<
1
])1log[( CoxpCl −−=
x<1 APE
X
1 pKspCl21
= =1 PE ≅0 ≅0
x>1
DPE
])1log[( CoxpKspCl −+=
≅0 (x-1)Co 1
uLa c rva de valoración se representa en la siguiente figura.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
Curva teórica de valoración de KCl 0.1 M con
10pCl
6
8
4
64
AgNO3 0.1 M
0
2
0 1 2 (fracción de la especie titulada)
. CUESTIONA
indique la secuencia experimental.
rial y reactivos.
dicador de Ag
ntal.
x
3 RIO PREVIO. 3.1 Realizar los cálculos para preparar las siguientes disoluciones:
a) 100 mL de una disolución de KCl 0.1 M b) 100 mL de AgNO3 0.1 M
3.2 Escribir la reacción que se verifica entre el KCl y el AgNO3. 3.3 Buscar el producto de solubilidad del AgCl 3.4 Buscar las características que debe reunir un electrodo indicador metálico de primera
especie. 3.5 Buscar las características que debe reunir un electrodo indicador metálico de segunda
especie. 3.6 Con ayuda de las ecuaciones que imponen el pCl en el transcurso de la valoración de KCl
0.1 M con AgNO3 0.1 M. Trazar la curva teórica de valoración (pCl=f(x) de esta titulación. 3.7 Elaborar un diagrama de flujo donde se
4. PARTE EXPERIMENTAL.
4.1 Mate2 vasos de precipitados de 250 mL 1 bureta de 25.00 mL 2 pipetas volumétrica de 20 mL 1 pinza para bureta 1 soporte universal 1 placa de agitación 1 agitador magnético 1 potenciómetro 1 electrodo in1 electrodo de referencia de Ag/AgCl Disolución de KCl 0.1 M Disolución de AgNO3 0.1 M
4.2 Desarrollo experime

Métodos Cuantitativos Manual de Prácticas de Laboratorio
65
olución de KCl 0.1 M. Pesar la cantidad necesaria para preparar 100 mL de KCl 0.1 M y transferirla a un vaso de precipitados de 100 mL. Disolver el KCl
a. Transferir el contenido del vaso a un matraz volumétrico de 100 mL y llevar hasta la marca con agua destilada. Envasar.
M. Pesar la cantidad necesaria para preparar 100 mL de AgNO3 0.1 M y transferirla a un vaso de precipitados de 50 mL. Disolver el
arca con agua desionizada. Envasar en un frasco color ámbar y proteger de la luz.
rización de una disolución de AgNO3.
dicador K2CrO4 al 0.5%.
ión potenciométrica de KCl con AgNO3.
a) Montar el esquema de la figura 1.
40 mL de agua desionizada. Introducir a la disolución la barra de agitación y los
ia e indicador. ndarizada.
d) Valorar la disolución de I2 con adiciones 0.5 mL de AgNO3.Registrar el volumen encial en cada punto.
e) Conforme transcurra la valoración graficar el potencial en función del volumen
4.2.1 Preparación de disoluciones a) Dis
con 30 mL de agua destilad
b) Disolución de AgNO3 0.1
AgNO3 con 50 mL de agua desionizada. Transferir el contenido del vaso a un matraz volumétrico de 100 mL y llevar hasta la m
4.2.2 Estanda
a) En un matráz erlenmeyer de 125 mL pesar con exactitud 10 mg de NaCl. b) Adicionar 40 mL de agua desionizada y dos o tres gotas del in
c) Con agitación constante valorarla con AgNO3 hasta el cambio de color de amarillo a rojo ladrillo.
d) Realizar por triplicado la estandarización.
4.2.3 Valorac
b) Con una pipeta volumétrica, medir 10.00 mL de la disolución de KCl y transferirla a un vaso de precipitados de 100 mL. Agregar con una probeta
electrodos de referencc) Llenar una bureta de 25.00 mL con la disolución de AgNO3 esta
de titulante agregado y el pot
del reactivo titulante agregado.
Figura 1

Métodos Cuantitativos Manual de Prácticas de Laboratorio
66
mL de titulante Potencial (mV) mL de titulante Potencial (mV)
5. ANÁLISIS DE RESULTADOS. 5.1 Construir una tabla que contenga la siguiente información:
5.2 Graficar el potencial en función del volumen agregado de titulante.
imera derivada gunda derivada
5.3 Localizar con ayuda del diagrama de potencial en función del volumen de titulante (Na2S2O3) el punto de equivalencia de la valoración potenciométrica por los siguientes métodos. a) Método del paralelogramo b) Método de la prc) Método de la sed) Método de la gráfica de Gran
5.4 Con el punto de equivalencia de la valoración calcular la concentración de la disolución de I2.
6. CONCLUSIONES.
6.1 ¿Se lograron los objetivos de la práctica? 6.2 Obtener las conclusiones pertinentes.
. BIBLIOGRAFÍA. 77.1 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo I, Editorial Toray
Masson, S.A., 1977, Barcelona, España, 282 páginas. 7.2 Charlot G., “Curso de Química Analítica General”, 1ª edición, Tomo II, Editorial Toray-
sson, S.A., 1977, Barcelona, España, 200 páginas. loan C.E y Kiser R.M., “Problemas y Experimentos en Análisis Instrumental”
Ma7.3 Me , 1ª edición,
itorial Reverté Mexicana, 1973, México, D.F., 560 páginas. og D.A. y Leary J.J., “Análisis Instrumental”
Ed7.4 Sko , 4ª edición, Editorial McGraw
, 935 páginas. ”
Hill/Interamarcana de España, 1994, Madrid España7.5 Vassos B.H., Ewing G. W., “Electroquímica Analítica , 1ª edición, Editorial Limusa, S. A. de
C.V., 1987, México, D. F., 303 páginas. 7.6 Willard H.H., Merrit L.L., Dean J.A. y Settle F.A., “Métodos Instrumentales de Análisis”, 1ª
edición, Editorial Iberoamérica S.A. de C.V., 1991, México, D.F., 884 páginas.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
67
ARTE 4 S DE CROMATOGRÁFICOS
La cromatografía es un procedimiento de separación de los constituyentes de una mezcla. Es un
ra identificar y cuantificar los componentes de una fase líquida o gaseosa homogénea. La característica que distingue a la cromatografía de la mayoría de los
fisicoquímicos de separación, es que se ponen en contacto dos fases mutuamente miscibles. Una fase estacionaria y la otra móvil. Una muestra que se introduce en la fase móvil
ase estacionaria. Las especies (repartos) entre la fase móvil y
se e tacionaria. Los componentes de la muestra se separan gradualmente en bandas en la
do
La cromatografía es una técnica analítica de separación con un doble fundamento: termodinámico cinético.
os aspectos termodinámicos rigen el equilibrio de distribución o reparto de los solutos entre las aria; y por lo tanto, son responsables de características
ble papel: por una parte, debe considerarse el tiempo en el ue se alcanza el equilibrio de distribución de cada plato teórico; por otra parte, la velocidad de
LASIFICACIONES Para clasificar globalmente los procesos cromatográficos es necesario atender a dos criterios básicos: (a) fundamento del proceso cromatográfico, lo que conduce a los tipos de romatografías, (b) forma de realizar el proceso cromatográfico, es decir, lo que constituye las
estacionaria y móvil. Según la naturaleza de la fase estacionaria.
a) Si es un sólido, cabe distinguir entre:
-Cromatografía de adsorción
PMÉTODO
método analítico que se usa pa
métodosines transportada a lo largo de una columna que contiene a la f
etidascomponentes de la muestra experimentan interacciones repfa sla
fase móvil. Al final del proceso los componentes separados emergen en orden creciente de interacción con la fase estacionaria. El componente menos retardado emerge primero, el retenimás fuertemente, eluye al último.
y Ldos fases, móvil y estacioncromatográficas tan importantes como la retención y la selectividad. Los aspectos cinéticos juegan un doqdesplazamiento diferencial de la mezcla de solutos en el lecho cromatográfico. C
cdistintas técnicas cromatográficas. Tipos de cromatografías Los distintos tipos de cromatografías pueden clasificarse de acuerdo a la naturaleza de las fases
. El sólido ad orbe al componente que inicialmente estaba en la fase móvil (líquida o gaseosa) (fuerzas de Van der Waals). -Cromatografía de cambio iónico
s
. El sólido es un cambiador de iones (fuerzas electrostáticas). -Cromatografía de exclusión (o de geles). El sólido es un gel formado por polímeros no iónicos porosos que retienen a las moléculas de soluto según su tamaño.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
68
-Cromatografía de afinidad. Es un tipo e ografía de adsorción, utilizada especialmente en bioquímica, en la que un sólido tiene enlazado a un llamado ligando de afinidad que puede ser, por ejemplo, un inhibidor enzimático o un anticuerpo.
b) Si es un líquido
-Cromatografía de partición. El líquido (fase estacionaria) se encuentra soportado en un sólido inerte. El soluto se parte entre la fase móvil (líquido o gas) y la fase estacionaria.
Según la naturaleza de la fase móvil. La técnica cromatográfica correspondiente recibe el nombre de dicha fase móvil.
a) Su es un líquido. Cromatografía líquida.
-Cromatografía líquido-líquido
special de cromat
. En la que ambas fases son líquidas y, por lo tanto, se trata de una cromatografía de partición.
-Cromatografía líquid
o-sólido. En la que la fase estacionaria esidad).
sólida (adsorción, cambio iónico, exclusión, afin
b) Si es un gas. Cromatografía de gases.
-Cromatografía gas-líquido. Es un tipo de cromatografía de partición. -Cromatografía gas-sólido. Es una cromatografía de adsorción.
c) Si es un fluido supercrítico (fluido calentado a una temperatura superior a su temperatura su presión crítica).
fluidos supercríticos
crítica, pero simultáneamente comprimido a una presión mayor que
-cromatografía de . Puede ser de absorción y partición.
e consigue: 1) por
rom
Técnicas cromatográficas Según la forma de llevar a acabo la separación cromatografica, es decir, según el dispositivo utilizado para conseguir el contacto entre la fase móvil y la estacionaria, cabe distinguir dos grandes tipos de técnicas cromatográficas: en columna y plana. Cromatografía en columna. Se utiliza un tubo cilíndrico, en cuyo interior se coloca la fase estacionaria y a su través se hace asar la fase móvil. El flujo de la fase móvil (líquido o gas) a través de la estacionaria sp
( presión, (2) por capilaridad, (3) por gravedad. Cromatografía plana. La fase estacionaria esta colocada en una superficie plana que en realidad es tridimensional, aunque una sola de sus dimensiones es muy reducida, por lo que puede considerarse bidimensional. Se divide en dos tipos generales. -C atografía en papel. En la que el papel actúa como soporte de la fase estacionaria (cromatografía de partición).

Métodos Cuantitativos Manual de Prácticas de Laboratorio
-Cromatografía en capa fina. En la que un sólido actúa como fase estacionaria (adsorbente, cambiador), o como soporte de la fase estacionaria (cromatografía de partición), se extiende en una capa delgada sobre una placa, generalmente de vidrio. CONCEPTOS BÁSICOS. Cromatograma. Es la representación de la respuesta o señal del sistema de detección, en función del
canza el detector. Tiempo de retención, tiempo de retención absoluto (tr). El tiempo de retención de un soluto es el tiempo que transcurre desde su inserción en la columna hasta que alcanza el detector. Tiempo n el que el soluto recorre la columna.
Tiemsolu tr =
Cau Volumen de retención (Vr). Es el volumen de fase móvil que se requiere para eluir un soluto dado e la columna cromatográfica.
r = tr F
onstante de distribución, razón de distribución, coeficiente de distribución, coeficiente de eparto (K). Todas las separaciones cromatográficas se basan en el grado en que los solutos se istribuyen entre la fase móvil y la fase estacionaria. Para el soluto A, el equilibrio implicado es:
AM ⇄ AE
a constante de distribución es:
tiempo, volumen de eluyente o distancia en el lecho cromatográfico. Tiempo muerto (t0). Es el tiempo que transcurre desde que se introduce a la columna cromatográfica una sustancia no retenida hasta que al
e
po de retención corregido, tiempo de retención ajustado (tr’). Es el tiempo que pasa el to en la fase estacionaria.
t0 + tr’
tr’ = tr – t0
dal (F). Gasto volumétrico de fase móvil en [mL/min].
d V Crd
L
M
E
AAK
][][
=
que frecuentemente se escribe:
M
E
CCK =
donde:
]E = CE = concentración molar del soluto en la fase estacionaria. ]M = CM = concentración molar del soluto en la fase móvil.
actor de capacidad, factor de retención, relación de retención, relación de reparto (k’). Se sa con frecuencia para describir las velocidades de migración de los analitos en las columnas.
[A[A Fu 69

Métodos Cuantitativos Manual de Prácticas de Laboratorio
MMM VVCEEE VKVCk ==´
00 tt0´ ttt
k r =−
=´r
EPARACIÓN DE MEZCLAS.
do aborda la separación y determinación de los comdefi
selectividad, factor de separación (α). Es un parámetro referido a la sep
SLa cromatografía adquiere su total sentido cuan
ponentes de una mezcla. A continuación se expondrán los conceptos que son utilizados para nir la separación cromatográfica
Retención relativa, factor de
aración entre si de dos analitos.
1
222
´´
´´ r
tt
kk
KK
===
70
112/1
r
α
sepanc
Resolución (Rs). Es un factor que se refiere a la capacidad de un sistema cromatográfico para
arar dos sustancias, teniendo en cuenta no solo la separación de picos; sino también los hos de zona o banda en el momento de la detección.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
71
TICA No. 12
PRÁC
Evaluación de la calidad de los alimentos mediante la identificación y cuantificación de HMF por HPLC.
1. OBJE
1.1. Realizar las curvas de calibración para un estándar de HMF 1.2. Separar os
1.3. Cuantificar los niveles de HMF en diversas muestras de alimentos
1.4. Ev
. INTRO UCCIÓN
Los alimentos ricos en azucares reductores que son sometidos a altas temperaturas esarrollan un oscurecimiento café no enzimático denominado reacción de Maillard. Esta reacción
causando alteraciones en el color (melanoidinas), ab cetonas) y valor nutricional (bloqueo o destrucción de lisina) de los alimentos.
alimentos, los factores que favorecen esta reacción son temper rpm y contenidoalto conte transporte a C y pHs de entre 4 a 7.
as de la reacción de Maillard pueden ser evaluadas mediante la detlact o
lucos de la reacción de oscurecimiento o z or la degradación de los azucares a altas temperaturas son el
furf metil furfural (HMF).
De ta monitoreada mediante munoensay feros, pruebas microbiológicas, y ediante métodos químicos en los que se mide el contenido de diversos compuestos furánicos,
métodos colorimétricos están siendo remplazados por las técnicas ayor especificidad y precisión. La más común es la cromatografía
qu da para la e temente se a n jugos y concentrados de frutas, leche, café, vino y
cer ontenidos de humedad, principalmente.
na fas ión delDic
TIVOS
e identificar el HMF en aliment
aluar la calidad de los alimentos en base al contenido de HMF
2 D
docurre entre azucares reductores y aminoácidos sDurante la preparación de los
or (aldehídos y
aturas mayores a 180oC en combinación con agitaciones superiores a 100 s de humedad menores del 15%. En alimentos con contenido de humedad intermedio y nido de proteínas y carbohidratos, la reacción es favorecida durante su almacenamiento
temperaturas mayores de 50oy
Las etapas tempranerminación de furosina, un aminoácido formado durante la hidrólisis ácida de fructosil-lisina, ul sil-lisina y maltulosil-lisina, mediante la reacción de los grupos ε-amino de la lisina con
a, lactosa y maltosa. Otros de los productos intermediarios gn en imáticos que se forman p
ral, metilfurfural y el 5-hidroxiu
l manera que la reacción de Maillard pueos, bioensayos de crecimiento de células de mamí
de ser inmcomo el HMF. Actualmente los romatográficas debido a su mc
lí ida de alta resolución (HPLC), que debido a su amplia versatilidad es empleantificación y cuantificación de diversos compuestos en alimentos y bebidas. Recienid
h empleado en la determi ación de HMF en tos ceales para bebés, y en alimentos con al
Para lograr lo anterior, la muestra del alimento a analizar será llevada, mediante el flujo de uumna. La separace móvil liquida, hacia una fase estacionaria localizada dentro de una col
HMF ocurrirá por su interacción con cada una de las dos fases del equipo de cromatografía. ho equipo deberá de estar integrado de (figura 1):

Métodos Cuantitativos Manual de Prácticas de Laboratorio
72
A) itos para la fase móvil (disolventes)
Los recipientes que se utilicen para almacenar la fase móvil deben de ser inertes. Suelen ser botellas de vidrio y tubos de teflón. Estarán provistos de unos filtros o aditamentos,
ontener la fase móvil. La fase móvil podrá ser un solvente puro o una mezcla de solventes.
B) onseguir y regular la presión y la velocidad del flujo de la fase móvil. Cuando
la fas programarse para que tome los solven s ción determinada y llevarlos hacia la cámara de mezclado
F r C) Sistema de inyección de muestras
Son v u e la muestra en volúmenes de 5 a 500 µl.
D) Columna cromatográfica de acero inoxidable
pre E) Detectores selectivos
Depós
indispensables para eliminar los gases disueltos y partículas que pudiera c
Sistema de bombeo Necesario para c
e móvil es una mezcla de solventes, la bomba podráte de las diferentes botellas en una propor
igu a 1. Esquema de los elementos de un equipo de HPLC. Referido en el texto.
álv las dosificadoras que permiten regular la aplicación d
Deberá contener las micropartículas de la fase estacionaria. Pudiera estar equipada con columnas y de termostatos reguladores de temperatura.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
73
flu ue responden a los ido a la presencia de la muestra; el más
F) Sistema para el tratamiento de datos y registrador
na propiedad en función del tiempo. La representación gráfica de estos registros se denomina cromatogramas. En estos registros
pos para la identificación cualitativamuestra. on la de un estándar primario. Mientras que en las determinaciones cualitativas se comparará la
. CUESTIONARIO PREVIO
3.1. Investigue qué otros compuestos pueden emplearse como parámetros para evaluar la
3.2. En que alimentos podemos encontrar furfuraldehídos. Ejemplos
3.3 s alimentos ¿qué indica?
3.4. investigu
3.5. Describa la reacción de Maillard.
4. PARTE EXPERIMENTAL 4.1. Materiales
rse las manos para evitar
contaminación por grasa.
• Vasos de precipitados de 10 ml • Pipetas volumétricas de 1 ml • Pipetas graduadas de 10 ml • Matraz kitazato de 1 lt
• Equipo de HPLC con todos los aditamentos y equipos requeridos. o bomba o columna C-18 o detector UV
• Sonicador
2-0.45 m
Responderán a una propiedad del soluto en la fase móvil. Pueden ser detectores UV/VIS, de orescencia y electroquímicos. Existen los detectores universales q
cambios en alguna propiedad de la fase móvil debcomún es el detector de índice de refracción.
Indispensable para registrar los cambios en algu
aparecerá una serie de picos simétricos sobre el eje de los tiem (posición en el eje) y cuantitativa (área bajo los picos) de los componentes de una Para las determinaciones cuantitativas se comparará la altura o área de un pico c
posición del pico (tiempo de retención) con cromatogramas estándares.
3
calidad de los alimentos durante su procesamiento y/o almacenamiento.
. La acumulación de furfuraldehídos, furfural y metilfurfural, en lo
e las estructuras del hidroximetilfurfural, furfural y metilfurfural
Todo el material debe estar libre de polvo. Es importante lava
• unidad de filtración millipore • Jeringa de vidrio de 10 µl
-VIS VARIAN
• Bomba de vacío • Jeringas con soporte para membranas de 0.2

Métodos Cuantitativos Manual de Prácticas de Laboratorio
74
• Filtros de nylon o de acetato de celulosa de 0.22-0.45 µm
4.2.
2. Para evitar las obstrucciones, todas las soluciones y muestras deben de filtrarse en
membranas de 0.22-0.45 µm
• A
ara preparar las soluciones patrón de 0.02-0.5 y 0.5-5.0 mg/L (en agua)
icación:
rocianuro de potasio 2. solución Carrez II: 30% (w/v) de acetato de zinc
.3 Desarrollo experimental
xperimentales. Referencia, García Villanova B. et al, 1993.
b) fc) c
µ
f) ep
a) Fb) C fía de líquidos
d) Presentación del equipo de HPLC y explicación para su operación (por los maestros)
Reactivos
NOTAS: 1. Todos los reactivos empleados en esta práctica deben ser de grado reactivo.
gua desionizada • Solución de HMF (Merck, Darmstandt, Germany) a 200 mg/L (en metanol) p
• Solución de clarif
1. solución Carrez I: 15% (w/v) de fer
4 .
4.3.1 Parámetros e
a) fase fija: Columna C18 en fase inversa. ase móvil: agua-acetonitrilo (95:5). ondiciones de flujo: 1 ml/min
d) cantidad de muestra a inyectar: 20 l de las muestras o soluciones previamente filtradas e) longitud de onda: realizar las lecturas en el detector UV a 280-285 nm
spectro experimental que se debe emplear como referencia para los resultados de esta ráctica. Tr= 6 – 8 minutos con un tiempo de corrimiento de 15 minutos.
Primera sesión 4.3.2 Explicación teórica de las bases de HPLC (seminario por los alumnos)
undamentos teóricos onceptos empleados en cromatogra
c) Aplicaciones • Comparación con otros métodos cromatográficos • Partes que integran el equipo de HPLC

Métodos Cuantitativos Manual de Prácticas de Laboratorio
75
Un día antes de la segunda sesión
a) Filtrar la fase móvil a través de membranas de 0.45 µm. b) Desgasificar la fase móvil que será empleada en la segunda y tercera sesión. Si el equipo
no cuenta con desgasificadores, este pro o debe realizarse mediante ultrasonido. Colocar la fase móvil en el sonicador durante 20 minutos y posteriormente en el cromatógrafo.
c) Purgar el equipo haciendo fluir a través de la columna 1.2 ml/min de por lo menos 60 ml de
fase móvil o hasta la desaparición de las burbujas de aire.
4.3.3 Curva de calibración a) Una vez seleccionadas rámetros experimentales de
va se deberá leer por triplicado.
d) Con una soluciones patrón empleada para la curva de calibración, realizar variaciones en la longitud de onda del detector. Discutir la longitud de onda de trabajo
en tubos de centrífuga de 10 ml. akes, pan casero o pan tostado comercial. La
c) Centrifugar durante 10 min a 5000 rpm. Repetir los paso anteriores una vez mas
cedimient
Segunda sesión
las condiciones de trabajo (pa
referencia), construir una curva de calibración para el HMF. b) Con siete puntos, construir una curva tipo de calibración empleando las disoluciones de
HMF de 0.02 a 0.5 y de 0.5 a 5 mg/L.
o Se debe de inyectar 1 ml de cada disolución patrón de modo de saturar y limpiar el loop del inyector (consultar el anexo del equipo).
o Cada punto de la cur o Graficar el área del pico en función de la concentración.
c) Mediante regresión lineal se deberán obtener dos ecuaciones, una para cada rango de concentración, del tipo: Y = m X + b, donde Y es la altura del pico (calculada por el alumno), y X es la concentración de HMF
Tercera sesión
4.3.4 Preparación y análisis de la (s) muestra (s)
a) Pesar 0.4 g de muestra molida La muestra puede ser cornfldeterminación de HMF se puede realizar en la superficie y/o en el cuerpo del pan.
b) Adicionar 7 ml de agua desionizada, agitar vigorosamente con el vortex durante 1 minuto

Métodos Cuantitativos Manual de Prácticas de Laboratorio
76
d) Recuperar el sobrenadante y clarificar agregando 1 ml de cada solución Carrez I y II.
ezclar y centrifugar 10 minutos a 5000 rpm. Recuperar el sobrenadante y didluirlo con agua desionizada hasta 25 ml.
grafo, filtrar 2 ml de la muestra anterior a través de un filtro de ulosa o de Nylon de 0.22-0.45 µm.
on una jeringa de 5 ml unida al filtro. filtrado y el resto se recoge en un recipiente
• Si es necesario, la (s) muestra (s) debe (n) de ser diluida (s) para obtener valores dentro del intervalo de la curva de calibración.
g)
Muestra Tr Área bajo la curva = Y
e) M
f) Antes de inyectar al cromató
celmembrana de acetato de
r a acabo c• El filtrado se puede llevaDescartar el primer mililitro delimpio.
• El volumen de muestra por inyectar debe ser lo más pequeño posible. ¿Para
que? Consultar los parámetros experimentales.
Esperar la aparición del pico y registrar el tiempo de retensión
1. 2. 3.
h) Con la ecuación obtenida en la primera sesión calcular la concentración de HMF en la
muestra problema y comparar este resultado con las lecturas del equipo.
Opcional para una cuarta sesión
4.3.4 Pueden realizarse variaciones en el flujo de análisis, a longitud de onda constante, para que el alumno discuta la eficiencia de la separación.
5. TRATAMIENTO Y ANÁLISIS DE LOS RESULTADOS
5.1. En el cromatograma del HMF represente la anchura del pico, el tiempo vacío o muerto, el tiempo de retención y el tiempo de retención corregido.
5.2. Compare el valor del HMF de la muestra analizada con los valores reportados. Qué puede
discutir respecto a la calidad, procesamiento y almacenaje del alimento analizado.
5.3. Calcule en número de platos teóricos de la columna para la separación del HMF y discuta
su relación con la eficiencia de la columna.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
77
5.4. Propuestas para mejorar las condiciones experimentales de esta práctica. 6. CONCLUSIONES
6.10 Obtener las conclu
7. IB
he Determination of 5-(Hydroxymethyl)-2-furaldehyde in Breackfast
and 5-methylfurfural and their relation with cork polysaccharides.
.3
w-Hill Interamericana.
methylfurfural content of selected bakery products. Food
.6. Ramírez-Jiménez A., Guerra.Hernández E. y García Villanova B. (2000). Browning
Indicators in ric. Food Chem. 48, 41764181
6.9 ¿Se lograron los objetivos de la práctica?
siones pertinentes.
LIOGRAFÍA B
7.1. García Villanova B., Guerra Hernández E., Martínez Gómez E. y Montilla J. (1993) Liquid Chromatography for tCereals. J.Agric.Food Chem 41, 1254-1255.
7.2. María Rocha S., A.Coimbra M. y Delgadillo I. (2004) Occurrence of furfuraldehydes during
the processing of Quercus suber L. cork. Simultaneous determination of furfural, 5-hydroxymethylfurfural Carbohydrate Polymers 56, 287–293
. Principios de Análisis Instrumental. 5ª ed (2001). Douglas A. Skoog, F.J. Holler,T.A. 7Nieman. Mc Gra
7.4. Química Analítica. 8ª ed. (2005). Skoog, West, Holler. Crouch. Thomson
7.5. Ramírez-Jiménez A., García Villanova B. y Guerra.Hernández E.. (2000). Hydroxymethylfurfural and Research International 33, 833-838
7 Bread. J. Ag

Métodos Cuantitativos Manual de Prácticas de Laboratorio
78
LasinteLosproméLasultr La La vel magnética en las regiones de UV-visible, y en ocasiones a la de región IR, si bien el sentido estricto del término abarca sólo a la radiación visible.
uede describirse como una onda con propiedades de longitud de ond
PARTE 5 ESPECTROSCOPIA
medidas basadas en la radiación electromagnética se utilizan mucho en química analítica. Las racciones de la radiación con la materia son el tema de la ciencia denominada espectroscopia. métodos analíticos espectroscópicos se fundamentan en medir la cantidad de radiación que ducen o absorben las especies moleculares o atómicas de interés. Es posible clasificar los todos espectroscópicos según la región del espectro electromagnético utilizado para la medida. regiones del espectro que se han utilizado abarcan los rayos gamma, rayos X, radiación violeta (UV), radiación infrarroja (IR), microondas y radiofrecuencias (RF). a
radiación electromagnética.
radiación electromagnética es una forma de energía que se transmite por el espacio a enorme ocidad. Se denomina luz a la radiación electro
La radiación electromagnética pa, frecuencia, velocidad y amplitud.
elo de onda no explica fenómenos relacionados con la absorción y emisión de la energía puede considerar a la radiación electromagnética
retos de energía o partícul
El modradiante. En relación con estos procesos, se como paquetes disc as, llamados fotones o cuantos. Estas dos con idera ulas y o , sino más bien comp Ley de Bee La ley de absorción, también llamada ley de Beer-Lambert o simplemente ley de Beer, relaciona las potencias de un haz monocromático incidente, Po y mitido la l del trayecto recorrido, de acuerdo con las siguientes ecuaciones.
s ciones de la radiación como partlementarias.
íc ndas no son excluyentes entre sí
r
tras P, con ongitud
bceP
T ε−==0
P
donde:

Métodos Cuantitativos Manual de Prácticas de Laboratorio
79
T = Transmitancia P = Potencia del haz transmitido Po = Pε = Constante propia del sistema componente-disolvente b = L gc = Concentración expresada en gramos por litro Como la a ritmo inverso de la transmitancia, se establece la siguienespectrofo
otencia del haz incidente
on itud del trayecto recorrido por la luz
bsorbancia A es por definición el logate ecuación que es la base en el desarrollo y operación de técnicas analíticas
tométricas.
bcT
A ε==1log
sistema componente – disolvente, denominado coeficiente de cuando la concentración se expresa en moles por litro.
ε = Constante propia del absortividad molar

Métodos Cuantitativos Manual de Prácticas de Laboratorio
80
ANÁLISIS CUADE COM ICOS MEDIANTE
1. OBJETIVOS.
1.1 Obtener el anaranja
1.2 Determinar
anaranja 1.3 Trazar la cu 1.4 Determin
anaranja 1.5 Realizar un
y anaranjado de metilo cuando estos componentes forman parte de una muestra problema.
2. INTRPara un siabsorción d l la concentración y la longitud del
onocromático, es idéntica a la ley de las aditividades y se
A la lo
A 2
ε max1 b C λmax1
A la longit de nda áxim del c pon te 2, max2, se tiene:
ε b 2
ε1
λmax1 y 1 o fi s b id o [ L 1] para los componentes 1 y 2, respectivamente, evaluados a la longitud de onda máxima del componente 1, λmax1.
onda máxima
1 y C2 son las concentraciones de los componentes 1 y 2, respectivamente, en [mol L–1]. Con los valores conocidos de λmax1, ε2
λmax1, ε1 se tie un sistema de dos ecuacio d ógnita se p ed resolv ci
PRÁCTICA No. 13 LITATIVO Y CUANTITATIVO DE UNO Y MULTICOMPONENTES
PUESTOS ORGÁNICOS E INORGÁNESPECTROFOTOMETRÍA VISIBLE
espectro de absorción del verde de bromocresol y el espectro de absorción del do de metilo.
la longitud de onda de máxima absorción del verde de bromocresol y del do de metilo.
rva de calibración del verde de bromocresol y del anaranjado de metilo.
ar el coeficiente de absortividad molar, ε, del verde de bromocresol y del do de metilo.
análisis cuantitativo para determinar la concentración de verde de bromocresol
ODUCCIÓN. stema de dos componentes la expresión matemática derivada que describe la
a radiación electromagnética en función deetrayecto recorrido por un haz mexpresa de la siguiente manera.
ngitud de onda máxima del componente 1, λmax1, se tiene:
A = 1 + A A = 1
λ1 + ε2 b C2
ud o m a om en λ
A = A1 + A2 A = 1
λmax2 C1 + ε2 λmax b C2
ε2 λmax son l s coe ciente de a sortiv ad m lar en cm–1 mol–
ε1λmax2 y ε2
λmax2 son los coeficientes de absortividad molar en [cm–1 L mol–1] para los componentes 1 y 2, respectivamente, evaluados a la longitud de del componente 2, λmax2.
C
ε1s qu
λmax2 λmax2 b , ε2 y lidad
ne ne ons c os inc e u e er c n fao .

Métodos Cuantitativos Manual de Prácticas de Laboratorio
81
La ley citada tiene diversas aplicaciones en el desarrollo y adecuación de técnicas analíticas espectrofotométricas, para demostrar que ε es una co tante verd dera dentro de un intervalo estrecho de longitudes de onda cercano a la λ máxima en el cálculo de la absorbancia o
ansmitancia de un haz monocromático a partir de resultados obtenidos con un haz
expe inámicas aparentes y condicionales como las de la acidez,
que rticipar en el equilibrio
tiene propias.
ntes disoluciones: g/mL, pureza 36%).
rde de bromocresol de concentración 1000 ppm en etanol-agua (50:50).
el
de una serie de dis 1 M y las absorbancias de una serie de disoluciones de anaranjado de metilo en HCl 0.1 M. Se leyo también la absorbancia de una disolución compuesta de una mezcla de verde de bromocresol y anaranjado de metilo de concentración desconocida. Los resultados experimentales se resumen en las siguientes tablas:
Absorbancias experimentales para una serie de disoluciones de verde de bromocresol en HCl 0.1 M.
Absorbancias experimentales para una serie de disoluciones de anaranjado de metilo en HCl 0.1 M.
[ppm] A λ = 513 A λ = 435 [ppm] A λ = 513 A λ = 435
ns a
trpolicromático con longitudes de onda cercano a la λ máxima y en la determinación
rimental de constantes termodcomplejación, hidratación y deshidratación de equilibrio y potencial de electrodo.
Los resultados experimentales esperados para un indicador ácido-base pueden predecirse ya estos indicadores son ácidos monopróticos débiles, capaces de pa
ácido-base consecutivo, generando dos especies, la ácida HIn y la básica In–, las cuales n propiedades químicas y físicas
3. SCUE TIONARIO PREVIO
iguie3.1 Realizar los cálculos para preparar las sa) 250 mL de HCl 0.1 M (densidad 1.21 b) 25 mL de vec) 25 mL anarajado de metilo de concentración 1000 ppm en etanol-agua (50:50).
3.2 Buscar en la bibliografía la fórmula condensada y estructura del verde de bromocresol y del anaranjado de metilo.
3.3 Buscar en la bibliografía el valor de la λmax para la forma ácida del verde de bromocresol y el valor de la λmax para la forma ácida del anaranjado de metilo.
3.4 En un experimento, se trazó el espectro de absorción de una disolución de verde de bromocresol en HCl 0.1 M y la λmax del indicador resultó de 435 nm. Se trazó tambiénespectro de absorción de una disolución de anaranjado de metilo en HCl 0.1 M y la λmax del indicador fué de 513 nm. A estas longitudes de onda máximas, se leyeron las absorbancias
oluciones de verde de bromocresol en HCl 0.
10 0.049 0.249 2.5 0.225 0.034 15 0.077 0.380 5.0 0.470 0.069 20 0.102 0.489 7.0 0.655 0.095 25 0.131 0.626 9.0 0.845 0.120 35 0.190 0.884 10.0 0.902 0.129
Absorbancias experimentales para una disolución que contiene verde de bromocresol y anaranjado de metilo en HCl 0.1 M.
A λ = 513 A λ = 4350.546 0.459

Métodos Cuantitativos Manual de Prácticas de Laboratorio
82
Con la información anterior: Transformar la concentración de las disoluciones de verde de bromocresol y anaranjado
de metilo de ppm a [mol L–1]. Trazar la curva de calibració romocresol y el anaranjado de metilo
usando la concentración en [mol L ].
brom as (435 nm y 51
la concentración de verde de bromocresol y anaranjado de metilo de la mezcla
4. PART
lumétricos de 10.00 mL
Disolución del indicador verde l de 1000 ppm en etanol-agua (50:50).
disoluciones.
a) Disolución de verde de bromocresol 1000 ppm. En un vaso de precipitados pesar 25 mg de verde de bromocresol y disolver con 12.5 mL de etanol. Transferir la disolución a un matraz volumétrico de 25.00 mL y llevar a la marca del aforo con agua destilada. Envasar y etiquetar.
b) Disolución de anaranjado de metilo 1000 ppm. En un vaso de precipitados pesar 25 mg de anaranjado de metilo y disolver con 12.5 mL etanol. Transferir la disolución a un matraz volumétrico de 25.00 mL y llevar a la marca del aforo con agua destilada. Envasar y etiquetar.
c) Disolución de HCl 0.1 M. d) Disolución de verde de bromocresol 100 ppm. Medir 2.5 mL de verde de
bromocresol de 1000 ppm y transferirlos a un matraz volumétrico de 25.00 mL y llevar a la marca del aforo con HCl 0.1 M. Envasar y etiquetar.
e) Disolución de anaranjado de metilo 100 ppm. Medir 2.5 mL de anaranjado de metilo y transferirlos a un matraz volumétrico de 25.00 mL y llevar a la marca del aforo con HCl 0.1 M. Envasar y etiquetar.
4.2.2
n para el verde de b–1
Calcular los coeficientes de absortividad molar en [cm–1 L mol–1] del verde de ocresol y del anaranjado de metilo a las dos longitudes de onda máxim
3 nm). Calcular
problema en [mol L–1] y en ppm.
E EXPERIMENTAL 4.1 Material y reactivos
6 vasos de precipitados de 30 mL 3 vasos de precipitados de 100 mL 1 matraz volumétrico de 250.00 mL 2 matraces volumétricos de 25.00 mL 2 matraces vo6 pipetas graduadas de 10 mL Balanza analítica Espectrofotómetro UV-VIS Un par de celdas cuadradas de vidrio o de cuarzo de 3 mL
de bromocresoDisolución del indicador anaranjado de metilo de 1000 ppm en etanol-agua (50:50). Disolución de HCl 0.1 M Etanol al 96% Agua destilada Mezcla problema (concentración desconocida)
4.2 Desarrollo experimental4.2.1 Preparación de
Obtención del espectro de absorción de los colorantes.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
83
a) Preparar una serie de disoluciones de 10.00 mL de verde de bromocresol cuyas de 10, 15, 20, 25 y 30 ppm a partir de la disolución de
ppm. Usar HCl 0.1 M como diluyente para llevar a la marca del aforo. c) Obtener el espectro de absorción de la disolución de 15 ppm de verde de
bromocr ión de la λ. Obtener la longitud
de onda máxima, max. d) Obtener el espectro de absorción de la disolución de 6 ppm de anaranjado de
metilo, realizando un barrido de longitud de onda en el intervalo de 460 a 560
a de calibración de cada uno de los colorantes. a) A las dos longitudes de onda máxima, leer la absorbancia para la serie de
e cada uno de los colorantes.
5. AN5.1 con la información que se solicita.
sol 5
concentraciones sean100 ppm. Usar HCl 0.1 M como diluyente para llevar a la marca del aforo.
b) Preparar una serie de disoluciones de 10.00 mL de anaranjado de metilo cuyas concentraciones sean de 2, 4, 6, 8 y 10 ppm a partir de la disolución de 100
esol, realizando un barrido de longitud de onda en el intervalo de 400 a500 nm. Trazar el diagrama absorbancia en func
λ
nm. Trazar el diagrama absorbancia en función de la λ. Obtener la longitud de onda máxima, λmax.
4.2.3 Obtención de la curv
disoluciones d
ÁLISIS DE RESULTADOS Llenar las siguientes tablas
Datos experimentales para la determinación del espectro de absorción del verde de bromocre
λ[nm] 400 405 410 415 420 425 430 435 440 445 450 45A
λ[nm] 460 465 470 475 480 485 490 495 500 A
Datos experimentales para la determinación del espectro de absorción del anaranjado de metilo.
λ[nm] 460 465 470 475 480 485 490 495 500 505 510 515 A
λ[nm] 520 525 530 535 540 545 550 555 560 A
5.2 Trazar el diagrama abs da uno de los colorantes. 5.3 Obtener la longitud de o de los colorantes. 5.4 Llenar la siguiente tabla con la información que se solicita.
Absorbancias experimental una serie de disoluciones de verde de bromocresol en HCl 0.1 M.
Ab experimentales para una seri luciones de anaranjado de metilo en HCl 0.1 M.
[ppm] A λ = A [ A λ = A λ =
orbancia en función de la λ para canda máxima, λmax, para cada uno
es para
sorbanciasoe de dis
λ = ppm] 10 2 15 4 20 6 25 8 30 10

Métodos Cuantitativos Manual de Prácticas de Laboratorio
84
5.5 ón de las disoluciones de verde de bromocresol y anaranjado
5.6 T anaranjado de metilo
.7 Calcular los coeficientes de absortividad molar en [cm–1 L mol–1] del verde de bromocresol
5.8 verde de bromocresol y anaranjado de metilo de la mezcla problema en [mol L–1] y en ppm.
6. CO
6.1 6.2 Obtener las conclusiones pertinentes.
7. BIB GR
7.1 Harris D.C. Análisis Quím o ona, España (2001).
7.2 amentos de Química Analítica” 8ava. Edición, Editoria
7.3 edición Ed. Mc Graw Hill (1994).
7.47.5 Meloan C.E. y R. W. Kiss Problems and Experiments". 1era.
edición. Charles E. Merrill Publishing Co. (1963).
Transformar la concentracide metilo de ppm a [mol L–1].
razar la curva de calibración para el verde de bromocresol y el usando la concentración en [mol L–1].
5y del anaranjado de metilo a las dos longitudes de onda máximas.
Calcular la concentración de
NCLUSIONES ¿Se lograron los objetivos de la práctica?
LIO AFÍA Cuantitativo. Editorial Reverté S.A., Barcelic
Skoog, D. A., Donald M.W, F James, H, Stanley R. C. “Fundl Thomson, México D.F. (2005).
Skoog, D. A. y Leary J.J.. "Análisis Químico Cuantitativo". 4ta.
Skoog, D. A. y Leary J.J.. "Análisis Instrumental".4ta edición. Ed. Mc Graw Hill. (1994). er. "Instrumental Analysis.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
85
PRÁCTICA No. 14
DETER ÉTRICA DE LA CONSTANTE DE ACIDEZ VERDE DE BROMOCRESOL
1. OBJE
1.1 Trazar el espectro de absorción de la forma ácida y de la forma básica del verde de bro
1.2 Determinar la longitud de onda
del 1.3 Determinar el valor de la constante de acidez del verde de bromocresol.
2. INTROEl indicador verde de bromocresol es un ácido monoprótico débil, capaz de participar en un
pecies, la ácida HIn y la básica In–, las cuales tienen icho equilibrio esta caracterizado por la siguiente
reacc
En el sistema H, y como los espectros deintersectan eniguales (εIn- = cumplir las leyeseleccionado
MINACIÓN ESPECTROFOTOMDEL INDICADOR QUÍMICO
TIVOS.
mocresol.
de máxima absorción de la forma ácida y de la forma básica verde de bromocresol.
DUCCIÓN.
equilibrio ácido-base, generando dos espropiedades químicas y físicas propias. D
ión: HIn ⇄ In– + H+
HIn/In , las concentracione–
– s de HIn y de In– se definen fijando el p absorción de HIn y de In en condiciones de predominio extremas e intermedias,
un punto donde los coeficientes de absortividad molar de las dos especies son εHIn), el resultado espectroscópico mostrará un punto isosbéstico y deberá s fundamentales de la espectrofotometría, si el intervalo de concentración
es el adecuado.
Espde b
Determinación Muchos compuestos orgánicos existen en más de una forma, según el pH del sistema. La determinación de la constante de ionización de estos compuestos proporciona datos útiles
ectros de absorción experimentales de una disolución de 20 ppm de verde romocresol a diferentes valores de pH.
de valores de pKa.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
86
cuando se trabase:
La ecuación de Henderson-Hasselbalch queda establecida por:
tan de realizar estudios cinéticos y de equilibrio. Para el siguiente equilibrio ácido-
HIn ⇄ In– + H+
[ ][ ]HInInpKpH a
−
+= log
Si se traza una gSi el sistema obuna gráfica de absorbancia en función del pH darconcentración en función del pH.
3. CUESTIONARIO PREVIO 3.1 Realizar los cálculos para preparar las siguientes disoluciones:
a) 250 mL de HCl 0.1 M (densidad 1.21 g/mL, pureza 36%). b) 100 mL de NaOH 0.1 M. c) 100 mL de KCl 2 M. d) 25 mL de verde de bromocres de con ración 1000 ppm en una mezcla etanol-agua
(50:50). 3.2 Buscar en la bibliografía la fórmula condensada y estructura del verde de bromocresol. 3.3 Buscar en
verde de brom3.4 Buscar en la 3.5 Buscar en la e del indicador verde de bromocresol. 3.6 En un ex
bromocresol espectro de absorción de una disolución de del indicaabsorbanciaspH. Los resultados experimentales se
una serie de disoluciones de verde bromocresol a diferentes valores
ráfica de pH en función de la razón de concentración puede obtenerse el pKa. edece la ley de Beer, la absorbancia resulta proporcional a la concentración y
a los mismos resultados que una gráfica de
ol cent
la bibliografía el valor de la λmax para la forma ácida y para la forma básica del ocresol.
bibliografía el valor del pKa del indicador verde de bromocresol. bibliografía el intervalo de vir
perimento, se trazó el espectro de absorción de una disolución de verde de en HCl 0.1 M y la λmax del indicador resultó de 442 nm. Se trazó también el
verde de bromocresol en NaOH 0.1 M y la λmax dor resultó de 615 nm. A estas longitudes de onda máximas, se leyeron las
de una serie de disoluciones de verde de bromocresol a diferentes valores de resumen en la siguientes tabla:
Absorbancias experimentales para
de de pH.
pH A λ = 442 A λ = 615ácido 0.535 ------- 2.92 0.530 0.034 3.47 . 0.480 0 106 4.00 0.391 0.345 4.50 . 0.278 0 604 5.00 0.186 0.881 5.50 0.115 1.051 6.04 0.087 1.174
básico 0.065 -------
Con la información anterior:

Métodos Cuantitativos Manual de Prácticas de Laboratorio
87
a) T a a b n e rm id e a forma bás a del verde de bromocresol en función del pH.
b Obtener el valor del pKa y del Ka del indicador verde de bromocresol.
3.7 P la forma ácida (λmax = 442 nm) del verde de bromocresol
razar en una mism gráfic , la a sorba cia d la fo a ác a y d l ic
)
ara la longitud de onda dededucir la siguiente ecuación:
442442log == −+= λλ
basema AA
pKpH442442 == λλ − mácido AA
Para la deducción tomar en cuenta las siguientes ecuaciones: [ ][ ]HInInpKpH a
−
+= log
Am
λ=442 = AHIn λ=442 + AIn-
λ=4 bso ia ob a 442 nm de una disolución que –.
AHIn λ=442 = εHIn
λ=442b[HIn] = absorbancia de la especie HIn a 442 nm.
AIn-λ=442 = εIn-
λ=442b[In–] = absorbancia de la especie In a 442 nm
Aácido λ=442 = εHIn
λ=442bC0 = bancia btenida a 442 nm de una disolución que solo e a la specie HI .
Abaseλ=442 = εIn-
λ=442bC0 = absorbancia obtenida a 442 nm de una disolución que solo –
42 = acontiene a la especie HIn y a la especie In
rba cn ten dai
–
absor o contien
e n
contiene a la especie In .
on la información del problema 3.6 3.8 Ca) Llenar la siguiente tabla:
Estudio logarítmico para la determinación experimental del pKa del indicador verde de bromocresol.
pH 442442
442442 == − λλ AAlog == − λλ
basem
mácido
AA
2.00 2.92 3.47 4.00 4.50 5.00 5.50 6.04
lb) Trazar la gráfica de pH en función de 442442 == − λλbasem AA
) Con la gráfica
442442
g== − λλ
mácido AA .
c del estudio logaritmico, obtener el valor del pKa y del Ka del indicador verde de bromocresol.
lo

Métodos Cuantitativos Manual de Prácticas de Laboratorio
88
4.
6 vasos de precipitados de 100 mL lumétrico de 250.00 mL
aces volumétricos de 10.00 mL
de celdas cuadradas de vidrio o de cuarzo de 3 mL
Disolución de HCl 2 M Disolución de NaOH 0.1 M Etanol al 96% Agua destilada
4.2 Desarrollo experimental 4.2.1 Preparación de disoluciones.
a) Disolución de HCl 0.1 M. Medir 2.1 mL de HCl concentrado, transferirlos a un matraz volumétrico de 250 mL y llevar a la marca del aforo con agua destilada. Mezclar adecuadamente. Etiquetar y envasar.
b) Disolución de HCl 2 M. c) Disolución de NaOH 0.1 M. En un vaso de precipitados de 50 mL pesar 0.4 g
de NaOH y disolver con agua destilada. Transferir la disolución a un matraz volumétrico de 100 mL y llevar a la marca del aforo con agua destilada. Mezclar adecuadamente. Etiquetar y envasar.
d) Disolución de KCl 2 M. En un vaso de precipitados de 50 mL pesar 7.82 g de KCl y disolver con agua destilada. Transferir la disolución a un matraz volumétrico de 100 mL y llevar a la marca del aforo con agua destilada. Mezclar adecuadamente. Etiquetar y envasar
e) Disolución de verde de bromocresol 1000 ppm. En un vaso de precipitados pesar 25 mg de verde de bromocresol y disolver con 12.5 mL de etanol. Transferir la disolución a un matraz volumétrico de 25.00 mL y llevar a la marca del aforo con agua destilada. Envasar y etiquetar.
f) Disolución de verde de bromocresol 100 ppm. Medir 2.5 mL de verde de bromocresol de 1000 ppm y transferirlos a un matraz volumétrico de 25.00 mL y llevar a la marca del aforo con agua destilada. Envasar y etiquetar.
4.2.2 Obtención del espectro de absorción de la forma ácida y de la forma básica
del verde de bromocresol. a) Preparar 10.00 mL de verde de bromocresol de concentración 20 ppm en HCl
0.1 M a partir de la disolución de 100 ppm. b) Preparar 10.00 mL de verde de bromocresol de concentración 20 ppm en
NaOH 0.1 M a partir de la disolución de 100 ppm. c) Obtener el espectro de absorción de la disolución de ácida de verde de
bromocresol, realizando un barrido de longitud de onda en el intervalo de 400 a 500 nm. Trazar ancia en función de la λ. Obtener la longitud de onda máxima, λmax.
PARTE EXPERIMENTAL 4.1 Material y reactivos
1 matraz vo2 matraces volumétricos de 25.00 mL 2 matr6 pipetas graduadas de 10 mL Balanza analítica Espectrofotómetro UV-VIS Un par Disolución del indicador verde de bromocresol de 1000 ppm en etanol-agua (50:50) Disolución de HCl 0.1 M
el diagrama absorb

Métodos Cuantitativos Manual de Prácticas de Laboratorio
89
b el intervalo de 560 a 660 nm. Trazar el diagrama λ. Obtener la longitud de onda máxima, λmax.
3 3 5 mL de KCl 2 M y
r al pH ta a
d) Obtener el espectro de absorción de la disolución de básica de verde deromocresol, realizando un barrido de longitud de onda en
absorbancia en función de la
4.2.3 Obtención del pKa del verde de bromocresol.
a) Preparar una serie de disoluciones reguladoras de ácido acético/acetato (CH COOH/CH COO–) a diferentes valores de pH. Pesar en un vaso de precipitados de 100 mL 2 g de acetato de sodio, adicionar25 mL de agua desionizada. Introducir el electrodo combinado y ajusta
ionar goindicado en la siguiente tabla con la disolución de HCl 0.1 M (adicgota y agitar) Envasar y etiquetar.
g de
CH3COONamL de
KCl 2 M mL de
H2O x mL de
HCl 0.1 MpH
2 5 25 6.00 2 5 25 5.50 2 5 25 5.00 2 5 25 4.50 2 5 25 4.00 2 5 25 3.50 2 5 25 3.00
b) Apartir de la disolución de 100 ppm de verde de bromocresol, preparar una
l, preparar una ión 20 ppm usando como
5. 5.1
serie de disoluciones de verde de bromocresol de concentración 20 ppm usando como diluyente las disoluciones reguladoras de ácido acético/acetato preparadas anteriormente.
c) Apartir de la disolución de 100 ppm de verde de bromocresol, preparar una disolución de verde de bromocresol de concentración 20 ppm usando como diluyente HCl 0.1 M.
d) Apartir de la disolución de 100 ppm de verde de bromocresodisolución de verde de bromocresol de concentracdiluyente NaOH 0.1 M.
e) A las dos longitudes de onda máxima, leer la absorbancia para las disoluciones preparadas en los incisos b) al d) 4.2.3.
ANÁLISIS DE RESULTADOS
Llenar las siguientes tablas con la información que se solicita. Datos experimentales para la determinación del espectro de absorción de la forma ácida del verde de bromocresol.
λ[nm] 400 405 410 415 420 425 430 435 440 445 450 455 A
λ[nm] 460 465 470 475 480 485 490 495 500 A
rción de la forma
λ[nm] 560 565 570 575 580 585 590 595 600 505 610 615
Datos experimentales para la determinación del espectro de absobásica del verde de bromocresol.
A

Métodos Cuantitativos Manual de Prácticas de Laboratorio
90
λ[nm] 620 625 630 635 640 645 650 655 660 A
5.2 Trazar el diagrama absorbancia en función de la λ para la forma ácida y la forma básica
del verde de bromocres5.3 btener la longitud de onda máxima, λ , para la forma ácida y la forma básica del verde
5.4
una serie de disoluciones de verde l a diferentes valores
ol. O maxde bromocresol. Llenar la siguiente tabla con la información que se solicita.
Absorbancias experimentales para
de bromocresode pH.
pH A λ = A λ = ácido 3.00 3.50 4.00 4.50 5.00 5.50 6.00
básico 5.5 Trazar en una misma gráfica, la absorbancia de la forma ácida y de la forma básica del
5.6 5.7
verde de bromocresol en función del pH. Obtener el valor del pKa y del Ka del indicador verde de bromocresol. Realizar el estudio logarítmico para el cálculo del pKa y llenar la siguiente tabla:
Estudio logarítmico para la determinación experimental del pKa del indicador verde de bromocresol.
pH 442442
442442
log ==
==
−−
λλ
λλ
basem
mácido
AAAA
5.8 razar la gráfica de pH en f n del 442442
442442
log=λ
ácidoAT unció ==
=
−−
λλ
λm
AAA .
5.9 Cde br
basem
on la gráfica del estudio logaritmico, obtener el valor del pKa y del Ka del indicador verde omocresol

Métodos Cuantitativos Manual de Prácticas de Laboratorio
91
6. CONCLU
6.1 ¿6.2 btener las conclusiones pertinentes.
. BIBLIOGRAFÍA
uímico Cuantitativo. Editorial Reverté S.A., Barcelona, España
7.2 M.W, F James, H, Stanley R. C. “Fundamentos de Química ón, Editorial Thomson, México D.F. (2005).
7.3 s Químico Cuantitativo". 4ta. edición Ed. Mc Graw Hill
7.4 J.J. "Análisis Instrumental".4ta edición. Ed. Mc Graw Hill. (1994). 7.5 l Analysis. Problems and Experiments". 1era.
s E. Merrill Publishing Co. (1963).
SIONES Se lograron los objetivos de la práctica?
O
77.1 Harris D.C. Análisis Q
(2001). Skoog, D. A., DonaldAnalítica” 8ava. EdiciSkoog, D. A. y Leary J.J. "Análisi(1994). Skoog, D. A. y LearyMeloan C.E. y R. W. Kisser. "Instrumentaedición. Charle

Métodos Cuantitativos Manual de Prácticas de Laboratorio
92
DETERMINAC O DE SOLUBLILIDAD DE UN PRECIPITADO AL
1. .1 Identificar experimentalmente un oxalato insoluble desconocido por el método de
espectrofotometría diferencia1.2 Preparar el blanco adecuado que permita realizar la determinación del oxalato
desconocido. 1.3 Determinar experimentalmente la solubilidad del oxalato desconocido. 1.4 Determinar experimentalmente el producto de solubilidad del oxalato desconocido. 1.5 eterminar experimentalme el nombre y fórmula del oxalato desconocido.
2.
lque sea la técnica más
a p or la sensibilidad de su . En estos instrumentos la incertidumbre en la medida de la transmitancia,
típica de un instrumento que tiene un pequeño i duda, importantes errores analíticos cuando la
enor de 10% (A=1.0) o mayor de 70% (A=0.15).
. STIONARIO PREVIO 3.1 Realizar los cálculos para preparar las siguientes disoluciones:
a. 100 mL de permanganato de potasio 0.01 M. b. 100 mL de H2SO4 3 M.
3.2 ¿Por qué es necesario utilizar un blanco para realizar cualquier determinación espectrofotométrica y cómo se efectúa el ajuste del instrumento con el blanco?
3.3 Si por medio de un método espectrofotométrico ordinario se desea determinar una especie química X (en forma directa o indirecta) presente en una matriz M. ¿Qué compuesto deberá contener el blanco?
3.4 ¿Cuál es la diferencia fundamental entre un blanco preparado para efectuar una medida espectrofotométrica por un método ordinario y el que se utilizaría para hacer la lectura en forma diferencial?
3.5 Escribe la reacción iónica balanceada entre el ion oxalato y el ion permanganato en medio ácido. Datos:
Sistema Eº (V/ENH) a pH=0 MnO4
–/Mn2+ 1.51 CO2/C2O4
2– – 0.49 3.6 Buscar en la bibliografía los valo guientes oxalatos.
PRÁCTICA No. 15 IÓN DEL PRODUCT
POR ESPECTROFOTOMETRÍA DIFERENCI
O1
BJETIVOS
l.
D nte
INTRODUCCIÓN La espectrofotoscopía molecular basada en la radiación ultravioleta, visible e infrarrojo se emp ea mucho en la identificación y determinación de muchas especies inorgánicas, orgánicas y bioquímicas. Se emplea en el análisis cuantitativo y es probable utilizada en los laboratorios químicos y clínicos de todo el mundo.
recisión de muchos fotómetros y espectrofotómetros está limitada pLdispositivo de lecturaT, es constante. Esta incertidumbre puede ser
sinmed dor de tipo D´Arsonval. Se encuentranconcentración del analito es tal que %T es m Los métodos diferenciales proporcionan un medio de expandir la escala de estos instrumentos
ia, de reducir en forma significativa este tipo de error. Dichos métodos y en consecuencemplean soluciones de analito para ajustar los valores de transmitancia cero, transmitancia 100% o ambos, ajustando el fotómetro o el espectrofotómetro y no el obturador o el disolvente. CUE3
res de pKs de los si

Métodos Cuantitativos Manual de Prácticas de Laboratorio
93
Compuesto Valor de pKsAg2C2O4 CuC O 2 4BaC2O4 CdC2O4 ZnC2O4
.7 En un experimento, se trazó el espectro de absorción de una disolución de permanganato
x10–4 M y la λmax resultó de 530 nm. A esta longitud de onda máxima, se leyeron las absorbancias de una serie de disoluciones de permanganato de potasio. Los resultados experimentales se resumen en la siguientes tabla:
3de potasio 2.8
Absorbancias experimentales para una serie de disoluciones de permanganato de potasio.
[M] A 5.5x10–5 2 0.251.1x10–4 0.482 1.7x10–4 0.743 2.2x10 0.992 –4
2.8x10–4 1.234 3.3x10–4 1.482
a. Trazar la curva de absorbancia en función de la concentración molar de permanganato de potasio.
b. Determinar el coeficiente de absortividad molar del permanganato de potasio.
3.8 En un experimento, con una disolución saturada de oxalato de bario se preparó una dilución de oxalato de bario 1:1 con agua destilada. Se tomaron 10.00 mL de la disolución diluida y se colocaron en un matraz volumétrico de 25.00 mL (se etiqueto como M1), se adicionaron 10 mL de H2SO4 3 M, se agregaron 400 µL de permanganato de potasio 0.0138 M, se llevó a la marca del aforo con agua destilada y el matraz se calentó a baño maria durante 2 minutos a 50 °C (este procedimiento se realizó por triplicado). En otro
experimentales.
matraz volumétrico de 25.00 mL (se etiqueto como B) se adicionaron 10 mL de H2SO4 3 M, se agregaron 400 µL de permanganato de potasio 0.0138 M, se llevó a la marca del aforo con agua destilada y el matraz se calentó a baño maria durante 2 minutos a 50 °C. Se esperó a que las disoluciones estuvieran a temperatura ambiente y se leyeron las absorbancias a 530 nm de acuerdo al siguiente procedimiento: se ajustó el espectrofotómetro a 0 de absorbancia con la disolución M1 y posteriormente se leyó la absorbancia de la disolución B, se repitió éste procedimiento para las disoluciones M2 y M3. Los resultados se resumen en la siguiente tabla.
Absorbancias
ABM1 0.120 M2 0.127 M3 0.120

Métodos Cuantitativos Manual de Prácticas de Laboratorio
94
a. Con la información anterior, determinar la concentración de la disolución diluida (1:1)
b. Determinar la solubilidad (concentración de la disolución saturada del oxalato de bario) para cada una de las repeticiones realizadas.
c. Determinar el valor del Ks y el valor del pKs del oxalato de bario.
3.9 Indica. 4. PARTE EXPERIM
4.1 Material y reactivos
ipeta de volumétrica 10.00 mL 5 matraces volumétricos de 25.00 mL
1 agitador magnético con barra magnética 1 termómetro 1 perilla de succión 1 piceta 1 espectrofotómetro visible
Disolución de H2SO4 3 M DisolucióDisolución saturada de oxalato insoluble desconocido Agua destilada
4.2
4.2.1 Preparación de disoluciones
oluble desconocido. En un vaso de precipitados de 250 mL colocar una cantidad del oxalato desconocido, agregar 150 mL de agua de nstante, dejar que alcance el equilibrio, a presión y temperatura constante durante media hora.
4.2.2 Secuencia Exp
a. Tomar una alícuota del sobrante de la disolución saturada y diluir con un volumen igual de agua. Esta disolución, X, es la que se usará en los siguientes
b. En un matraz volumétrico de 25.00 mL, mezclar en el siguiente orden 10.00 mL de disolución X, 10 mL de H2 3 M, más 1.00 mL de la disolución de KMnO4 0.01 M y el volumen adecuado de agua destilada para completar el aforo. Realizar este procedimiento por triplicado. Etiquetar estas disoluciones como M1, M2 y M3.
c. En otro matraz volumétrico de 25.00 mL, mezclar en el siguiente orden 10 mL de H2SO4 3 M, más 1.00 mL de la disolución de KMnO4 0.01 M y el volumen adecuado de agua destilada para completar el aforo. Etiquetar esta disolución como B.
d. Calentar en baño maría (entre 40 y 50 ºC durante 2 minutos) las disoluciones preparadas. Dejar que las disoluciones alcancen la temperatura ambiente y leer
del oxalato de bario para cada una de las repeticiones realizadas.
ENTAL
1 bureta de 25.00 mL 1 pipeta de 1.00 mL 1 p
1 probeta de 25 mL
n de KMnO4 0.01 M
Desarrollo experimental
a) Disolución de H2SO4 3 M. b) Disolución de KMnO4 0.01 M. c) Disolución saturada del oxalato ins
stilada y con agitación co
erimental
pasos del procedimiento.
SO4

Métodos Cuantitativos Manual de Prácticas de Laboratorio
95
las absorbancias a 530 nm de acuerdo al siguiente procedimiento: ajustar el espectrofotómetro a 0 de absorbancia con la disolución M1 y posteriormente
5. ANÁLISIS DE RESULTADOS
5.1
leer la absorbancia de la disolución B, repitir éste procedimiento para las disoluciones M2 y M3.
Indica los valores de absorbancia de las disoluciones medidas.
Absorbancias experimentales.
ABM1 M2 M3
Determinar la concentración de la disolución diluida (1:1) del oxalato de bario para cada 5.2 una de las repeticiones realizadas.
5.3 Determinar la solubilida da del oxalato de bario) para cada una de las repeticiones realizadas.
5.4 to de bario. 5.5 Comparar el pKs que se obtuvo experimentalmente con los valores de pKs buscados en
.
6. CO6.16.2 Obtener las conclusiones pertinentes.
7. BIBLIOGRAFÍA.
7.1 Douglas A. Skoog, y Donald M. West “Análisis Instrumental”. 2da. edición. Ed. Mc. Graw Hill. (1994) 7.2 Hobart H. Willard, Lynnel L. Merrit, Jr. “Métodos Instrumentales de Análisis”. 2da. Edición
d (concentración de la disolución satura
Determinar el valor del Ks y el valor del pKs del oxala
la bibliografía y dar el nombre y fórmula del compuesto MX(C2O4)y
NCLUSIONES ¿Se lograron los objetivos de la práctica?
Ed. Compañía Editorial Continental (1988).

Métodos Cuantitativos Manual de Prácticas de Laboratorio
96
PRÁCTICA No.16 DETERMINACIÓN DE LA ESTEQUIOMETRÍA DE UN COMPLEJO POR
1. OB
1.5
2. INT
trofotometría es el conjunto de procedimientos que utilizan la luz para medir concentraciones químicas y es una herramienta valiosa para determinar la composición de ionrad consideración.
h) El método de relación de moles.
conreactivos en cada mezcla son constantes, pero la relación de moles de los reactivos varía de forma sistemática. Así que cada solución se mide a una longitud de onda apropiada y se corrige con respecto a la absorbancia que pudiera mostrar la mezcla si no hubiera reacción.
En el método de proporción de moles, se prepara una serie de soluciones en las que se mant a de un reactivo, mientras que se varía la del otro. Se elabora una gráfica de la absorbancia frente a la proporción molar de los reactivos. Si la constante de formación es favorable, se obtienen dos rectas de pendiente distinta, que se cortan en un valor de proporción de moles correspondientes a la proporción de combinación en el complejo.
L entes es útil en particular para complejos débiles, pero
sólo e ser forma un complejo. En este método la reacción de formación del complejo, se pue iante un exceso cuantitativo de cualquiera de los reactivos; que en estas circunstancias se cumple por la Ley de Beer, y que sólo el complejo absorbe a la lo el experimento.
de con
ESPECTROFOTOMETRÍA VISIBLE
JETIVOS.
El alumno determinará espectrofotométricamente una constante de equilibrio.
RODUCCIÓN
La espec
es complejos en solución y sus constantes de formación. La capacidad de esta técnica ica en el hecho de que se pueden llevar a cabo, sin alterar los equilibrios en
g) El método de variaciones continúas.
i) El método de la relación de pendientes. En el método de variaciones continúas, se mezclan soluciones de catión y ligando en centraciones analíticas idénticas, de manera que el volumen total y los moles de los
iene constante la concentración analític
a técnica de proporción de pendies aplicable a sistemas en los qu
de forzar para que se complete med
ngitud de onda elegida para
Tanto I2 como I2* piridina absorben radiación visible, pero la piridina es incolora. El análisis los cambios espectrales asociados con la variación de concentración de piridina con una centración total constante de yodo permite evaluar la K de relación.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
2.1 Determinación de la Constante de Equilibrio (Gráfico de Scatchard)
Para medir una constante de equilibrio se deben medir las concentraciones de las especies químicas que intervienen en el equilibrio. Se puede usar la espectrofotometría con para este fin.
Primero se examina el equilibrio en el que las ies P y X reaccionan para formar PX:
Puesto que bancia es proporcional a la c ión ctividad nen que convertir las centracio en activid para obtenerve ras cons de equilib
espec
(1)
Despreciando los coeficientes de actividad se puede escribir,
Consideremos una serie de disuna cantidad constante de P. Sentonces se puede escribir la s
Se puedbalance
la absor(no a la aoncentrac ) se tie
con nes ades las rdade tantes rio.
Ahora bien, la expresión del eq
97
(2)
oluciones en las que se va añadiendo incrementos de X a ea Po la concentración total de P (en forma de Po de PX), iguiente ecuación:
[P] = P0 - [PX] (3)
e comprobar que la ecuación es un de masas.
uilibrio, ecuación 2 se puede cambiar como sigue:
(4)

Métodos Cuantitativos Manual de Prácticas de Laboratorio
98
La representación de [PX]/[X] e igual a - K, y se llama Gráfico de Scatchard ntes de equilibrio, especial-mente en bioquímica.
Si se conoce [PX], se puede hallar [X] utilizando el balance de masas:
trofotométrica. Se hace suponer que gitud de onda,
frente a [PX] tendrá una pendient. Se emplea mucho para medir consta
X0 = [total X] = [PX] + [X] (5)
Para medir [PX], se podría usar la absorbancia espec P y PX absorben a la lon , y que X no presenta absorbancia a esta longitud
de onda. Por simplicidad, se suponen que las medidas se hacen en una celda de paso óptico 1,000 cm. Esta r scribir la ley de Beer. La absorbancia a una longitud de onda es la suma de las absorbancias de PX y P.
A = ε ε (6)
A = εPX [PX] + ε P [PX] (7)
Ao
Un gráfico de Scatchard es una representación de [PX]/[X] nte a [PX] cuya pendiente es -K.
condición pe mite suprimir b (= 1,000 cm) al e
PX [PX] + P [P]
Haciendo [P] = Po – [PX], se puede escribir:
P o
fre
Pero P adir nada de X. Reordenando la ecua n
ε Po es Ao, la absorbancia inicial antes de añció 7, resulta:
A = [PX] (εPX - εP) + Ao ⇒ [PX] = ε∆∆
=APX][ (8)
Donde ∆ε es igual a εpx - εp y A (= a – a 0) es la absorbancia observada menos la ción. Aplicando la expresión de [PX] de la
ión 6 se obtiene: absorbancia inicial en cualquier punto de la valoraecuación 8 en la ecuac
Ecuación de Scatchard:
AKPKXA
o ∆−∆=∆ ε
][ (9)

Métodos Cuantitativos Manual de Prácticas de Laboratorio
99
[X] frente a ∆A es una línea recta de pendiente - K. La absorban-cia medida mientras se valora P con X se puede utilizar para hallar la constante de
En io se presentan dos casos. Si la constante de
foseind
En la práctica, los errores inherentes a un gráfico de Scatchard pueden ser importantes y a veces se pasan p o:
La representación de ∆A/
equilibrio de la reacción de X con P.
la aplicación de la ecuación 9, de ordinarequilibrio es pequeña, se necesitan grandes concentraciones de X para observar la
rmación de PX. Por consiguiente, Xo » Po, y la concentración de X libre en la ecuación 9 e puede considerar igual a la concentración inicial, Xo. O bien, si K no es pequeña, ntonces [X] no es igual a Xo, y se puede medir [X]. El mejor procedimiento es una medida dependiente de [X], ya sea a otra longitud de onda o midiendo una propiedad física istinta.
or alto. Si se define la fracción de saturación de P com
Fracción de saturación = oP 0)
Se puede hacer ver que se obtienen los datos más exactos para 0,2 < S < 0,8.6. Para verificar que se cum quilibrio deben ob datos en un intervalo que represente aproximad el 75% d rva de sat . Algunos cometen errores explorando un trozo muy pequeño de la curva de reacción sin incluir la región 0,2 < S < 0,8.
2.2 EL MÉTODO DE LAS VARIACIONES CONTINUAS
Se considera el siguiente el equilibrio:
PXS ][= (1
ple el eamen
(1) se e la c
tener te u uración
(11)
Y si se forman varios complejos:
P + 2X 2 (12)
P + 3X
Si plo, PXllamado método de Job) nos permite predominante. El procedimiento clásico coequ emente sforma que la concentración total (formal) de
s de partida de la de
PX
predomina un complejo (por ejem
imoleculares de P y X (probabl
podrían mezclar disolucione tabla 1, originando varias relaciones
3 (13) PX
2), el método de variaciones continuas (también identificar la estequiometría del complejo nsiste en mezclar alícuotas de disoluciones eguida de dilución a un volumen constante) de P + X permanezca constante. Por ejemplo, se P 2,50 mM y X 2,50 mM, como se muestra en X:P pero una concentración total constante de

Métodos Cuantitativos Manual de Prácticas de Laboratorio
100
1,00 mm. A continuación se mide la absorbancia de cada disolución a una longitud de onda a
Tabla 1. Disoluciones para el m iones continuas.
mL de P
2.50 mM
Relación
(X:
Fracción molar de X
decuada.
étodo de variac
mL de X
2.50 mM
molar
P) ⎟⎠⎞
⎜⎝⎛
+ molPmolXmolX
1.00 9.00 9.00:1 0.900 2.00 8.00 4.00:1 0.800 2.50 7.50 3.00:1 0.750 3.33 6.67 2.00:1 0.667 4.00 6.00 1.50:1 0.600 5.00 5.00 1.00:1 0.500 6.00 4.00 1:1.50 0.400 6.67 3.33 1:2.00 0.333 7.50 2.50 1:3.00 0.250 8.00 2.00 1:4.00 0.200 9.00 1.00 1:9.00 0.100
Nota: Todas las disolucion a un volumen total de 25,0 mL con un tampón.
Se reprfracción moa la estequiometría del complejo predominante. La absorbancia corregida se define como la absorbancia medida menos la absorbancia que se produciría por P0X solos:
es se diluyen
Absorbancia corregida = Absorbancia medida - εPbPT - εXbXT (14)
esentan las absorbancias corregidas (definidas en la ecuación 14) frente a la lar de X. Se alcanza la absorbancia máxima a la composición que corresponde
Fig. 1. Repres mportami ráficos a formaci plejos P3X, PX y PX2
Donde εP y εX absortividades molares de P X puros, respectivamente, b es la lon-gitud de camin muestra, XT son las concentraciones totales (formales) de P y de X en la dis Para la p disolución de la tabla 1, PT = (1,00/25,0)(2,5 mM) = 0,100 mM y X 0/25,0)(2, ) = 0,900 mM Si P y X no absorben a la longitud de onda de interés, no es necesaria la corrección de absorbancia.
enta el co ento ideal de los g de Job para l ón de com
son laso de
yla
olucióy PT y rimern.
T = (9a
50 m,0 M .

Métodos Cuantitativos Manual de Prácticas de Laboratorio
101
Amortigua ing) es el roceso de debilitamie emisión de un molécula excitada ferencia energé ca a otra molécula ( uador).
ción (quenchnto de
pa
por trans tiel amortig
El máximo de absorbancia se presenta a la fracción molar de X que corresponde a la stequiometría del compuesto (figura 1). Si el complejo predominante es PXe
presenta a la fracción molar de X = 2/(2 + 1) = 0,667.
F
2, el máximo se
racción molar de X en:
abbabXP+
= (=0.667 cuando b=2 y a=1)
Mé
P + nX n
todo de variaciones contínuas:
PX
El máximo de absorbancia se presenta cuan
=X
Si la especie predominante fuera P3X, el má
(11+
=X
Al aplicar este método de variaciones contin
3 Verificar que el complejo cumple la 4 Usar una fuerza iónica y un pH cons5 Hacer lecturas a más de una longit
misma fracción molar para todas las
4. Hacer experiencias a diferentes cootro conjunto de disoluciones en lpartiendo de disoluciones 5,00 mMmisma fracción molar.
do la fracción molar de:
( )1+n n
ximo se presentaría a una fracción molar de:
) 250.03
=
uas hay que tomar algunas precauciones:
ley de Beer. tantes, cuando sea necesario. ud de onda; el máximo se debe presentar a la longitudes de onda.
ncentraciones totales de P + X. Si se prepara as proporciones dadas en la tabla 20.2, pero , el máximo debe continuar presentándose a la

Métodos Cuantitativos Manual de Prácticas de Laboratorio
102
3. CUESTIONARIO PREVIO.
s cálculos para preparar las siguientes disoluciones
2
ciclohexano y la max resultó de 523 nm. Se trazó también el espectro de absorción de una x10– 4 M y piridina 2x10– 4 M en ciclohaxano y se obsevaron dos longitudes
3.1 Realizar lo
a. 25 mL I2 0.01 M en ciclohexano b. 25 mL piridina 0.01 M en ciclohexano
3.2 En un experimento, se trazó el espectro de absorción de una disolución de I 5x10– 4 M en λ
mezlcla de I2 5de onda máximas una a λmax =523 nm y otra a λmax = 422 nm. A la longitud de onda máxima de 422, se leyeron las absorbancias de una serie de disoluciones preparadas mezclando I2 0.01 M en ciclohexano y piridina 0.01 M en ciclohexano. Los resultados experimentales se resumen en la siguientes tabla:
Absorbancias experimentales para una serie de disoluciones preparadas mezclando I2 0.01 M en ciclohexano y piridina 0.01 M en ciclohexano, para obtener la estequiometría y la constante de formación del complejo yodo-piridina.
I [M] Piridina [M] A 2 λ = 4225x10–4 1x10–4 0.167 5x10–4 2x10–4 0.221 5x10–4 3x10–4 0.263 5x10–4 4x10–4 0.315 5x10–4 5x10–4 0.338 5x10 6x10 0.370 –4 –4
5x10–4 7x10–4 0.383 5x10–4 8x10–4 0.401 5x10–4 9x10–4 0.410 5x10–4 1x10–3 0.415
a. Con la información anterior, determinar la estequiometría del complejo yodo-piridina
por el método de la razón de moles, para ello trazar el diagrama razón molar de piridina/yodo en función de la absorbancia.
b. Determinar el valor de la constante de formación del complejo, usar el diagrama razón molar de piridina/yodo en función de la absorbancia.
3.3 ridina? y ¿cuál es su grado toxic
4. PARTE EXPERIMENTAL.
4.1
4.2
Buscar en la bibliografía ¿qué es la pi ológico?
Material y reactivos. 6 matraces aforados de 10.00 mL 1 pipeta volumétrica de 1.0 mL I2 0.01 M en ciclohexano Piridina 0.01 M en ciclohexano
Desarrollo experimental 4.2.1 Preparación de disoluciones

Métodos Cuantitativos Manual de Prácticas de Laboratorio
103
4.2.2 Secuencia experimental
a) Preparar 10 mL de una serie de disoluciones mezclando I2 0.01 M en ciclohexano y piridina 0.01 M en ciclohexano de acuerdo a la siguiente tabla.
I2 [M] Piridina [M]
5x10–4 ----- 5x10–4 1x10–4
5x10–4 2x10–4
5x10 3x10–4 –4
5x10–4 4x10–4
5x10–4 5x10–4
5x10–4 6x10–4
5x10 7x10–4 –4
5x10–4 8x10–4
5x10–4 9x10–4
5x10–4 1x10–3
5x10–4 4x10–3
5x10–4 6x10–3
5x10–4 8x10–3
c) línea base de todas absorbancias que se midan. Si
odo más conveniente de hacer esta práctica es con un espectrofotómetro de barrido provisto de registrador, pero también se puede utilizar uno de
las de llenado y vaciado de la celda del espectrofotómetro. • Para hacer las medidas, hay que sacar cerradas las cubetas de la vitrina. • Las soluciones usadas se deben recoger en un recipiente de residuos, colocado
en la vitrina, y no tirar a la pila.
5. ANÁLISIS DE RESULTADOS. 5.1 Llenar la siguiente tabla con las absorbancias leidas a 422 y 523 nm.
I [M] Piridina [M] A λ = 422 A λ = 523
b) Leer las absorbancias de las disoluciones a 422 y 523 nm. Usar celdas de vidrio o de cuarzo. Restar la absorbancia de laes posible, todos los espectros, incluyendo el de la línea de base, en una hoja de papel registrador.
OBSERVACIONES • El m
medidas puntuales. • Todas las operaciones que se describen a continuación se deben hacer en
vitrina, incluyendo
25x10–4 ----- 5x10–4 1x10–4 5x10–4 2x10–4 5x10–4 3x10–4 5x10–4 4x10–4 5x10–4 5x10–4 5x10–4 6x10–4 5x10–4 7x10–4

Métodos Cuantitativos Manual de Prácticas de Laboratorio
104
5x10–4 8x10–4 5x10–4 9x10–4 5x10–4 1x10–3 5x10–4 4x10–3 5x10–4 6x10–3 5x10–4 8x10–3
5.2 Con la información anterior, determinar la estequiometría del complejo yodo-piridina por el
método de la razón de moles, para ello trazar el diagrama razón molar de piridina/yodo en función de la absorbancia.
5.3 Determinar el valor de la constante de formación del complejo, usar el diagrama razón molar de piridina/yodo en función de la absorbancia.
6. CONCLUSIONES.
1. ¿Se lograron los objetivos de la práctica? 2. Obtener las conclusiones pertinentes con respecto al análisis de resultados. 3. Comparar los resultados y conclusiones con la referencia teórica.
, D.A. y Leary J.J “Análisis Instrumental” 4ª. Edición Ed. Mc Graw Hill (1994) Voguel, A.I. “Química analítica” Kapelutz 5ª Edición.
7. BIBLIOGRAFÍA
Skoog “Fundamentos de Química Analítica” 8ª. Edición THOMSON. Harris Daniel C. “Análisis Químico Cuantitativo” 2ª. Edición Editorial Reverté S.A. Skoog

Métodos Cuantitativos Manual de Prácticas de Laboratorio
105
PRÁCTICA No. 18 ID E GRUPOS FUNCIONALES POR ESPECTROFOTOMETRÍA
DE INFRARROJO 1.
presentes en distintas sustancias de interés ría infrarrojo como una técnica de caracterización
2. INTRODUC
2.3 La porcióncercano, medio y lejano, así nombrados por su relación con el espectro visible. El infrarrojo lejano (aproximadamente 400-10 cm
ENTIFICACIÓN D
OBJETIVOS 1.6 El alumno identificará los grupos funcionales 1.7 El alumno reconocerá la espectrofotomet
y de cuantificación.
CIÓN infrarroja del espectro electromagnético se divide en tres regiones; el infrarrojo
-1) se encuentra adyacente a la región de dio (aproximadamente 4000-400 cm-microondas, posee una baja energía, el infrarrojo me
1) puede ser usado para estudiar las vibraciones fundamentales y la estructura rotacional vibracionall mientras que el infrarrojo cercano (14000-4000 cm-1) puede excitar
onossobret o vibraciones armónicas. De estas tres regiones la más empleada para car o elucidar estructuras es la zona del infrarrojo medio, ya que la mayoría de los identifi
grupos funcionales orgánicos absorben en dicha zona.
Esta técnica funciona exclusivamente con enlaces covalentes, y como tal es de gran utilidad en química orgánica. Espectros nítidos se obtenlaces activos al IR y altos niveles de pureza. Es
ienen de muestras con pocos tructuras moleculares más complejas
evan técnica
El infradigitale
ll a más bandas de absorción y a un espectro más complejo. Sin embargo esta se ha podido utilizar para la caracterización de mezclas complejas
jo medio a su vez se divide en zona de grupos funcionales y zrro ona de huellas de s:
G R U
Lás mMolécuconjugpuedecomún
P O S F U N C I O N A L E S HUELLAS DIGITALES
oléculas diatómicas simples tienen solamente un enlace, el cual se puede estirar. las más complejas pueden tener muchos enlaces, y las vibraciones pueden ser
adas, llevando a absorciones en el infrarrojo a frecuencias características que n relacionarse a grupos químicos. Los átomos en un grupo CH2, encontrado mente en compuestos orgánicos pueden vibrar de seis formas distintas,
tos simétricos y asimétricos, flexiones simétricas y asimétricas en el plano estiramien(scissplano (
oring y rocking, respectivamente), y flexiones simétricas y asimétricas fuera del wagging y twisting, respectivamente); como se muestra a continuación

Métodos Cuantitativos Manual de Prácticas de Laboratorio
PR
Las muestras gaseosas requieren poca preparación más allá de su purificación, pero se usa uestra con una larga longitud de celda
EPARACIÓN DE LAS MUESTRAS:
una celda de m (usualmente 5-10 cm) pues los gases muestran absorbancias relativamente débiles.
Las muestras líquidas se pueden disponer entre dos placas de una sal de alta pureza (comúnmente cloruro de sodio, o sal común, aunque también se utilizan otras sales tales como bromuro de potasio o fluoruro de calcio. Las placas son transparentes a la luz infrarroja y no introducirán líneas en el espectro. Algunas placas de sal son altamente solubles en agua, y así la muestra, completamente anhidros
agentes de lavado y similares deben estar (sin agua).
Las muestras sólidas se pueden preparar principalmente de dos maneras. La primera es moler la muestra con un agente aglomerante para la suspensión (usualmente nujol) en un mortero de mármol o ágate. Una fina película del agente aglomerante se aplica en las placas de sal y se realiza la medición.
El segundo método es triturar una cantidad de la mezcla con una sal especialmente purificada (usualmente bromuro de potasio) finamente (para remover efectos dispersores de los cristales grandes). Esta mezcla en polvo se comprime en una prensa de troquel mecánica para formar un pellet translúcido a través del cual puede pasar el rayo del espectrómetro.
Es importante destacar que el espectro obtenido a partir de preparaciones distintas de la muestra se verán ligeramente distintas entre sí debido a los diferentes estados físicos en los que se encuentra la muestra.
106
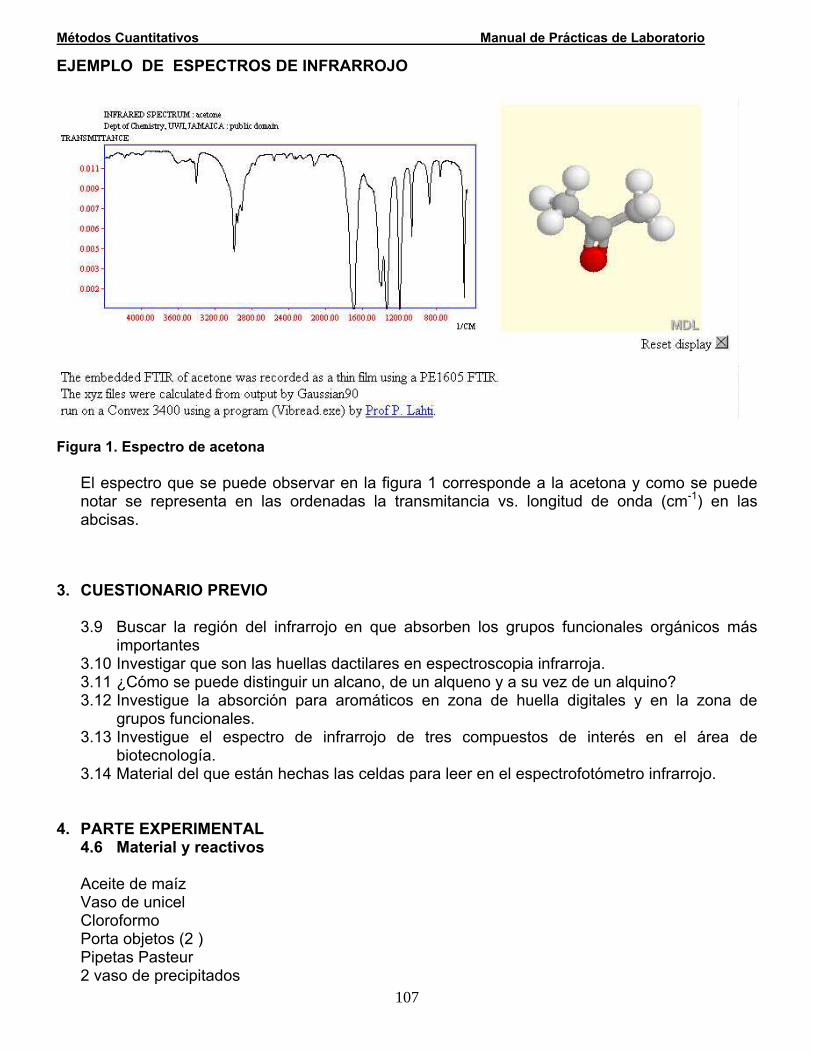
Métodos Cuantitativos Manual de Prácticas de Laboratorio
107
EJEMPLO DE ESPECTROS DE INFRARROJO
Figura 1. Espectro de acetona
snotarabcis
3. CU
3.9 l región del infrarrojo en que absorben los grupos funcionales orgánicos más importantes
3.10 Investigar que son las huellas dactilares en espectroscopia infrarroja. 3.11 ¿Cómo se puede distinguir un alcano, de un alqueno y a su vez de un alquino? 3.1 ue la absorción para aromáticos en zona de huella digitales y en la zona de
grupos funcionales. 3.13 Investigue el espectro de infrarrojo de tres compuestos de interés en el área de
ra leer en el espectrofotómetro infrarrojo.
4. PAR4.6 AceitVasoCloroPortaPipe2 vaso de precipitados
El e pectro que se puede observar en la figura 1 corresponde a la acetona y como se puede se representa en las ordenadas la transmitancia vs. longitud de onda (cm-1) en las as.
STIONARIO PREVIO E
Buscar a
2 Investig
biotecnología. 3.14 Material del que están hechas las celdas pa
TE EXPERIMENTAL Material y reactivos
e de maíz de unicel formo objetos (2 )
tas Pasteur

Métodos Cuantitativos Manual de Prácticas de Laboratorio
108
Bolsa de plástico CaucÁcidoAcetAcetCeldParri 4.7
se distribuye uniformemente en la celda, se introduce al equipo y se adquiere el espectro indicando la longitud de absorción de cada banda observada
etanilida y ácido salicílico
ispositivo para la adquisición de muestras sólidas (ATR) en el equipo de Infrarrojo; con la ayuda de una espátula se toma un poco de acetanilida, se introduce al equipo y se adquiere el espectro.
f) De la misma forma que el inciso a) se adquiere el espectro del ácido salicílico. g) Indicar la frecuencia de absorción de cada banda en el espectro.
Se recomienda limpiar el dispositivo para sólidos antes de usarlo con un papel ue contenga un poco de cloroformo a fin de limpiar completamente dicho ispositivo.
4.7.4 Espectro de muestra poliméricas
te de la experimentación se puede trabajar usando una muestra de unicel, de caucho, etc.
abaja colocándola en un portaobjetos, con la ayuda una pipeta Pasteur se le adiciona 5 ó más gotas de cloroformo, hasta que el
unicel se disuelva bien. b) Una vez que se disolvió el unicel, se coloca encima otro portaobjetos y se
desliza con suavidad. c) Se deja evaporar el disolvente y una vez evaporado, se levanta la película se
coloca en un portaceldas y se adquiere el espectro. d) En el caso de la muestra de caucho ó plástico duro, se puede disolver un poco
en un vaso de precipitados usando ciclohexano en el caso del caucho y cloroformo en el caso
ho salicílico
ato de isoamilo anilida as lla de agitación.
Desarrollo experimental 4.7.1 Película de acetato de isoamilo
a) En una celda para espectroscopia infrarrojo, que puede ser de NaCl, KBr o ZnSe hacer una película de acetato de isoamilo, y adquirir el espectro de infrarrojo.
4.7.2 Película de aceite de maíz a) En una celda de infrarrojo se coloca con la ayuda de una pipeta Pasteur una gota de aceite de maíz y
4.7.3 Espectro de ac
e) Se coloca el d
qd
Esta paruna bolsa de plástico, una muestraa) La muestra de unicel se tr
de
de plástico duro

Métodos Cuantitativos Manual de Prácticas de Laboratorio
109
e) Una vez disuelto, se coloca una película sobre una celda de infrarrojo y se traza el espectro.
f) En el caso de la bolsa de plástico, se corta un rectángulo y se coloca sobre un rectamente el espectro.
5.
.11 Reportar cada espectro obtenido indicando los valores de absorción para cada banda. rupos funcionales principales en cada espectro y las huellas dactilares
.13 De los espectros investigados de tres compuestos de interés en el área biotecnológica (investigado desde el cuestionario previo), identifíque los grupos funcionales de cada
6.
6.11 ¿Se lograr6.1
7. BIBLIOGRAFÍA
7.12 Silverstein Robert M., Bassler Clayton y Morril Terenc ctrométrica de compuestos
7.13 Harris D.C. “Análisis Químico Cuantitat 991). 7.3 Skoog, D.A. y Leary J.J. “Analysis Instrumental”. 4ta ed w Hill. (1994).
con una pipeta Pasteur
portaceldas y se traza di
ANÁLISIS DE RESULTADOS
55.12 Identificar los g5
espectro y las huellas dactilares. 5.14 Señale aplicaciones de ésta práctica en su carrera.
CONCLUSIONES
on los objetivos de la práctica? 2 Obtener las conclusiones pertinentes.
e. Identificación espeorgánicos.
ivo”. Grupo Editorial Iberoamerica (1ición. Ed. Mc Gra

Métodos Cuantitativos Manual de Prácticas de Laboratorio
110
PRÁCTICA No. 19 DE SIDUALES POR
ABSORCIÓN ATÓMICA 1. OB
1.1 1.2
2. INTLas cas se pueden clasificar en general como basadas en procesos de emisión atómica y La espectroscopia de emisión atómica implica, en el caso normal, la emisión de fotones cuando los electrones regresan de est
Las técnicas espectroscópicas en cauantificar la energía (midiendo la longitud de onda y la intensidad) absorbida de una fuente de radiación incidente, para exitar los electrones elementales respecto al estado fundamental. La longitud de onda y la absorción se pueden medir y registrar en un espectro. Los espectros atómicos se originan de las transiciones electrónicas entre orbitales atómicos y proden líneas de absorción extremadamente delgadas, con anchos de banda de longitud de onda de 0.1 nm, más o menos. 2.1
mica de flama es la forma más usada de espectroscopia atómica. Se pueden determinar con facilidad concentraciones de ppm de muchos iones metálicos. En la práctica, la técnica se basa en suministrar átomos o iones electrónicamente excitados
ica; la absorción que se produce se mide entonces en el instrumento. Su desventaja principal radica en su naturaleza de técnica unielemental, impuesta por la necesidad de una lámpara distinta para cada elemento.
.1
l) .1.2
a)
c) 2.2
grafito, en forma de un tubo hueco. Entonces, las muestras se separan en átomos por una atomización electrotérmica. El
TERMINACIÓN DE METALES PESADOS EN AGUAS REESPECTROFOTOMETRÍA DE
JETIVOS
RODUCCIÓN técn as espe troscópica atómi
procesos de absorción atómica.ic c s
ados exitados a estados fundamentales. En contraste, las técnicas de absorción atómica se basan en la captura de fotones, cuando los electrones son promovidos o a veces hasta se pierden en la formación de un ion.
de absorción atómica consisten
Absorción atómica de flama La absorción ató
por luz monocromát
2 .1
j) k)
2
b)
Absorción atómica con horno de grafito Los hornos de grafito eliminan totalmente la necesidad de una llama porque usan un elemento de calentamiento eléctrico resistivo, de
método del horno de grafito se ha vuelto la forma más adoptada de atomización electrotérmica. Estos métodos se acercan a una eficiencia de atomización cercana al 100%. El tubo de grafito rodea a la muestra con un volumen muy pequeño, y (cuando se combina con calentamiento eléctrico) permite un control mucho mayor de la temperatura que el que se obtiene con una llama. Además, no hay fluctuaciones en la longitud de trayectoria óptica debidas a convección térmica en el interior de la llama. También se

Métodos Cuantitativos Manual de Prácticas de Laboratorio
111
elimina, claro está, la generación no deseada de luz por combustión de los gases dentro
Las muestras se inyectan por una ventana en el techo del horno de tubo de grafito con una micropipeta. Los cm de longitud y sus diámetros aproximados son de 1 cm. Los tubos son diseñados para que se intercambien, se limpien y se reemplacen con facilidad. Los contactos eléctricos para el elemento calentador se disponen en ambos lados del tubo. En el caso normal, todo el tubo esta rodeado por una chaqueta de enfriamiento con agua. Una corriente externa de gas noble (argón normalmente) evita el ingreso de oxígeno de la atmósfera, que causaria la incineración del tubo. También se pasa gas inerte por ambos extremos del tubo del horno de grafito que sale por la ventana de inyección de la muestra. Este gas no solo sirve para apartar el oxígeno, sino también ayuda a arrastrar los vapores generados durante las etapas iniciales de calentamiento. La temperatura suele aumentar suavemente para evaporar el solvente. Una vez seca por completo la muestra, se puede aumentar rápidamente para vaporizar el analito. La fuente de luz incidente de la lámpara de cátodo hueco, entra por un extremo del tubo de grafito y por la trayectoria de la muestra evaporada. En el caso típico, la muestra evaporada atraviesa la trayectoria de la luz durante aproximadamente 1 s, lo cual también ayuda a aumentar la sensibilidad de este método. La luz transmitida se puede medir, entonces, después de atravesar y salir del otro extremo del tubo de grafito. 2.2.1
3. CUESTIONARIO PREVIO 3.10 3.11
4. PARTE EXPERIMENTAL 4.3 Material y reactivos 4.4 Desarrollo experimental
4.4.1 d) e)
h)
j) 5.
5.6 5.7
6. CO6.3 ¿Se lograron los objetivos de la práctica? 6.4 Obtener las conclusiones pertinentes.
7. BIBLIOGRAFÍA 7.8 7.9
de la llama.
tubos de horno de grafito suelen tener 5
f) 4.4.2
i)
ANÁLISIS DE RESULTADOS
NCLUSIONES

Métodos Cuantitativos Manual de Prácticas de Laboratorio
112
ANÁLISIS TÉRMICO 1. OBJETIVOS.
1.8 El alumno identificará los diferentes métodos de análisis térmico. 1.9 El alumno aplicará el análisis termogravimétrico en la caracterización de oxalato de calcio.
2. INTRODUCCIÓN
Un análisis térmico comprende el estudio de la evolución de las propiedades de una muestra o compuesto cuando es sometida a un calentamiento a altas temperaturas. También el análisis térmico es definido como una serie de técnicas donde se mide la dependencia de una propiedad física con respecto a la variación de la temperatura para una substancia específica o un producto de reacción y bajo un programa dado. Las propiedades físicas incluyen: masa, temperatura, entalpía, dimensión características dinámicas.
De acuerdo a las propiedades físicas que se miden es como se clasifican las técnicas de
análisis térmico:
Propiedad Física
Técnica
PRÁCTICA No. 20
Masa Termogravimetría (TG) Temperatura Análisis Térmicos diferencial (DTA)
Entalpía Calorimetría Diferencial de Barrido (DSC) Dimensiones Termodilatometría
Propiedades mecánicas Análisis Termomecánico Propiedades ópticas Termomicroscopía
Propiedades magnéticas Termomagnetometría Propiedades eléctricas Termoelectrometría Propiedades acústicas Termosonometría
Evolución de gas radiactivo Análisis Térmico de Emanación Evolución de partículas Análisis de Termopartículas
Aspectos prácticos de la medición incluyen:
• Evaporación • Sublimación • Descomposición • Oxidación • Reducción Adsorción • Desorción de gas

Métodos Cuantitativos Manual de Prácticas de Laboratorio
Varias reacciones pueden ocurrir cuando una sustancia se calienta:
3 2(g) F Dentro de este contexto, la termogravimetría ambios de la masa
en u n una termobalanza. A conti
érmicos
A (S1) A(cristal) A(goma)
A(s) + B(g) A(s) + B(g) gases A(s) + (gases (s) + (gases)2 A(s) + B(s) (s)
2.1 L
La termobalanza es un instrumen
temperade temp so (ga
La b
que la mcambioselectro-óptico con un obturador uniintensidaposiciónmasa. L
BaCl2 * 2H2O(s) BaCl(s) + 2 H2O(g) CaCO CaO(s) + CO
AAttmmóóssffeerraa IInneerrttee
NH4Cl NH3(g) + HCl(g)
2 (s) OAg + ½ 2(s) Ag2O(g) CuO(s) + H2(g) Cu(s) + H2O(g) C(s) + O2(g) CO2(g)e2O3(s) + MgO(s) Mg(s) + Fe2O4(s)
es la técnica que mide los cna muestra manteniendo una temperatura específica y se realiza enuación en la Tabla 2 se muestra algunos de los fenómenos térmicos:
Tabla 1. Fenómenos T
C(s)
)1 A
AB
AB(s) + CD(s) AD(s) + CB(s)
a Termobalanza
turas en diversos procesos eratura, cambio de pe
alanza de modo nulo es la muestra permanezca siempr
de masa. La desviación de
d de luz que llega a la foto del brazo, en su punto nula sensibilidad de pesada de
AAttmmóóssffeerraa AAmmbbiieennttee
A(S2) A(l) A(g) B(s) + gases gases
Fase de TransiciónFusión Sublimación
Descomposición
n
Cristal de Transició113
to de medición utilizado para determinar el equilibrio de las
nancia o pérdida).
a un dispositivo do al externo del brazo. El movimiento del brazo altera la
ada con su tara máxima.
y/o reacciones industriales y químicas. Esto es, a cambios
ás utilizada en Termogravimetría (TG). En ella se asegura e en la misma zona del horno independientemente de los l brazo de la balanza del punto nulo se utiliz
celda, y esta señal amplificada se utiliza para restaurar la o, al mismo tiempo que sirve como medida del cambio de la balanza está relacion
Heterogéneo Catálisis Adición
ble descomposición
Combustión Volatilización
Oxidación Corrosión
Do

Métodos Cuantitativos Manual de Prácticas de Laboratorio
114
Así,
eléctrica
Fig. 1 Partes de una microbalanza electrónica
.1.2. Ventajas del uso de la Termobalanza:
avacío (<10-4 Pa) a alta presión (3000 kPA).
Se puede trabajar en atmósferas de gases inertes, oxidantes, reductores o corrosivos.
A presiones atmosféricas se puede trabajar con un flujo dinámico, con las ventajas: Reducir
la condensación de los productos de reacción en las partes más frías del mecanismo de pesada. Limpiar los productos corrosivos. Minimizar reacciones secundarias. Actuar como refrigerante para el mecanismo de la balanza.
para valores máximos de carga de 1 g se obtienen sensibilidades de 1 mg. La señal de salida se transforma en una curva derivada termogravimétricamente.
2 L s termobalanza permiten realizar medidas a diferentes presiones atmosféricas, desde el

Métodos Cuantitativos Manual de Prácticas de Laboratorio
115
Fig. 2 Termobalanza Mettler TA 3000
2.2. Preparación de la muestra, atmósfera de medida y co Muestras de igual composición exhiben diferentes comporta
dependencia de la preparación de las muestras. Existe diferenciade cristales individuales, como polvo o en masa. No es convcantidades de masa debido a la temperatura en la misma no rescantidades pequeñas de masa protege al aparato de explosionesmuestra, siempre que sea posible, se prepara de forma dispersacon lo que facilita el desprendimiento de gases de la misma.
La temperatura de la muestra, TM, normalmente ocurre
temperatura del horno, TH, y por lo tanto no puede ser medida ráel proceso de pesada. La medida de la temperatura se suele hacy a veces se utilizan dos, para controlar de manera independietemperatura se regula mediante programadores especiales quevelocidades de calentamiento, desde fracciones de grado a 1000
a
a
G
muestra
o
o
o
Anillo de la tap
R
Gas de purgo
Horn
Microbalanza
p
o
Cámara del hornEntrada del Aire frí
Tapón de hule
Salida del Aire frí
Platillo pesado uía de conducto
Anillo sellad
Tornillo centrado eflectores de calor
Salida de Gas de purga
Cargador de muestra
ntrol de temperatura.
mientos térmicos debido a la al calentar un sólido en forma eniente trabajar con grandes ulta homogénea. Trabajar con o deflagraciones fortuitas. La y uniforme en el contenedor,
con retraso respecto a la idamente sin que se interfiera
er por un termopar (de platino) nte TH y TM. El control de la
permiten un amplio rango de ºC por minuto.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
116
Disposiciones de la muestra con relación al horno:
Ser capaz de alcanzar una temperatura superior en 100 o 200 ºC a la deseada de trabajo.
Disponer de una amplia zona de calentamiento homogéneo. Alcanzar la temperatura deseada de inicio tan rápido como sea. No afectar al mecanismo de la balanza por radiación o convección. A temperaturas < 500 ºC las muestras se suelen contener en porta muestras de hoja de aluminio.
En ellos se puede encerrar y sellar muestras líquidas y volátiles. A partir de 500 ºC se utiliza porta muestras de oro o de grafito. El material de referencia para las aplicaciones de Calorimetría de Diferencial de Barrido (CDB) es simplemente un porta muestras vacío.
Fig. 3 Preparación de muestras para CDB
2.3 Interpretación de las curvas
Tipo (i): La muestra no sufre descomposición con pérdida de productos volátiles en el rango de temperatura mostrado. Pudiera ocurrir reacciones tipo: transición de fase, fundido, polimerización. Tipo (ii): Una rápida pérdida de masa inicial es característica de procesos de
desorción o secado. Tipo (iii): Esta curva representa la descomposición de la muestra en un proceso
simple. La curva se puede utiliz
Métodos Cuantitativos Manual de Prácticas de Laboratorio
116
Disposiciones de la muestra con relación al horno:
Ser capaz de alcanzar una temperatura superior en 100 o 200 ºC a la deseada de trabajo.
Disponer de una amplia zona de calentamiento homogéneo. Alcanzar la temperatura deseada de inicio tan rápido como sea. No afectar al mecanismo de la balanza por radiación o convección. A temperaturas < 500 ºC las muestras se suelen contener en porta muestras de hoja de aluminio.
En ellos se puede encerrar y sellar muestras líquidas y volátiles. A partir de 500 ºC se utiliza porta muestras de oro o de grafito. El material de referencia para las aplicaciones de Calorimetría de Diferencial de Barrido (CDB) es simplemente un porta muestras vacío.
Fig. 3 Preparación de muestras para CDB
2.3 Interpretación de las curvas
Tipo (i): La muestra no sufre descomposición con pérdida de productos volátiles en el rango de temperatura mostrado. Pudiera ocurrir reacciones tipo: transición de fase, fundido, polimerización. Tipo (ii): Una rápida pérdida de masa inicial es característica de procesos de
desorción o secado. Tipo (iii): Esta curva representa la descomposición de la muestra en un proceso
simple. La curva se puede utilizar para definir los límites de estabilidad del reactante, determinar la estequiometría e investigar la cinética de las reacciones. Tipo (iv): Se indica una descomposición multietapa con intermedios relativamente
estables. Se puede definir los límites de estabilidad del reactante e intermedios, y de forma más compleja la estequiometría la reacción.
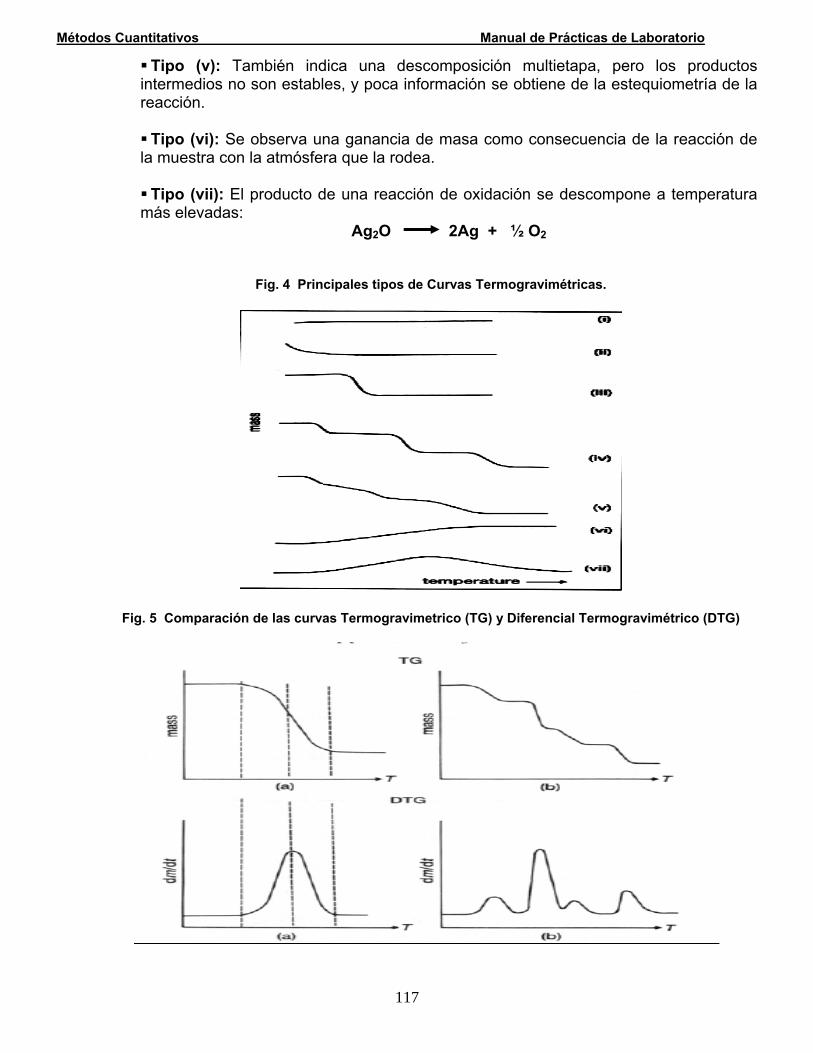
Métodos Cuantitativos Manual de Prácticas de Laboratorio
117
Tipo (v): También indica una descomposición multietapa, pero los productos intermedios no son estables, y poca información se obtiene de la estequiometría de la reacción.
Tipo (vi): Se observa una ganancia de masa como consecuencia de la reacción de
la muestra con la atmósfera que la rodea. Tipo (vii): El producto de una reacción de oxidación se descompone a temperatura
más elevadas: AgB2 BO 2Ag + ½ OB2 B
Fig. 4 Principales tipos de Curvas Termogravimétricas.
Fig. 5 Comparación de las curvas Termogravimetrico (TG) y Diferencial Termogravimétrico (DTG)

Métodos Cuantitativos Manual de Prácticas de Laboratorio
118
2.4 Ventajas del Análisis Térmico:
El análisis térmico se ha usado en control de calidad.
Obtención de la información respecto a la comparación y estructura detallada de los
diferentes fases de una muestra dada. Mediante las temperaturas de los cambios de fase y de las reacciones al igual que
los calores de reacción sirven para determinar la pureza de los materiales.
En el campo de la producción, control de procesos y en inspección de materias primas y productos de síntesis.
Tiene aplicaciones en varios campos como en polímeros, vidrio, cerámicos, metales,
explosivos, semiconductores, medicinas y comidas.
3. CUESTIONARIO PREVIO.
2) Investigar, ¿qué es el análisis termogravimétrico? 3) Investigar, ¿cómo está constituido el equipo para el análisis termogravimétrico? 4) Investigar algunas aplicaciones del análisis termogravimétrico (TG) y calorímetría
Diferencial de Barrido (DSC) en diferentes áreas. 5) Investigar algunas aplicaciones del análisis termogravimétrico (TG) y calorímetría
Diferencial de Barrido (DSC) en relación a su carrera. 6) Elaborar un diagrama de flujo con el procedimiento a seguir durante la práctica.
4. PARTE EXPERIMENTAL.
4.3 Material y reactivos. • Equipo TGA-50 Shimadzu • Oxalato de calcio • Muestra de Poliestireno • Charolas de aluminio para colocar las muestras 4.4 Desarrollo experimental
d) Pesar las charolas donde se colocorá la muestra. e) Pesar 10 mg de oxalato de calcio. f) Cerrar con la prensa las charolas conteniendo la muestra. g) Transferir las charolas preparadas con la muestra al equipo para realizar el análisis
TG. Las charolas deberán estar previamente pesadas y preparadas con la muestra, procurar no tocarlas con los dedos, todo hacerlo con pinzas.

Métodos Cuantitativos Manual de Prácticas de Laboratorio
119
h) Establecer las condiciones de trabajos del equipo: Vel. de Calentamiento= 20ºC/seg,
TBinicioB=0 ºC, TBfinalB=1000 ºC. i) Registrar lecturas gráficas. j) Interpretar los resultados obtenidos.
5. ANÁLISIS DE RESULTADOS.
1. Obtener la curva del análisis térmico para el Oxalato de Calcio. 2. Indique, ¿cuántos pasos de descomposición se observan para dicha sustancia y en
que temperatura?
3. Con el Peso Molecular, calcule las pérdidas de peso en cada paso.
4. E Indique los productos tras cada pérdida de peso.
5. Consulte la bibliografía e indique si era lo esperado.
6. De acuerdo al valor obtenido en el Análisis por Calorimetría Diferencial de Barrido (DSC), ¿qué sustancia es probable que contenga la muestra?
6. CONCLUSIONES.
4. ¿Pudo familiarizarse con al menos dos técnicas del análisis térmico? 5. El empleo del análisis térmico es adecuado en la caracterización, ¿de qué tipo de
muestras? 6. Obtener las conclusiones pertinentes con respecto al análisis de resultados. 7. Comparar los resultados y conclusiones con la referencia teórica. 8. Sugerencias para obtener resultados más confiables.
7. BIBLIOGRAFÍA
Shimadzu, Corporation. Introduction to Termal Análisis.







