
UNIVER!SlDADAUTONOMA METROPOLITANA
I Z T M ! A
Ingeniería de los Alimentos
Servicio Social
Asesor: M. Q. Salvador R. Tell0 Solís
Ing. Gabriel
oct / 1992

/ 9 'l E S P E C T R O S C O P I A I N F R A R R O J A . . . . . . . . . . . . . . . .I
-3.: INTRODUCCION. 1
VIBRACIONES MOLECUIARES. . . . . . . . . . . . . . . . . . . . . . . . .9 , C . 2 1: ABSORCION DE RADlAClON INFRARROJA POR
$:MOLECULAS DIATOMICAS. '. . . . . . . . . . . . . . . . . . . . . . . . . I I
2 \!
LA MOLECUIA COMO UN ROTOR RIGIDO. 12
OSCllADOR ARMONICO. 13
ABSORCION DE RADlAClON INFRARROJA POR MOLECUIAS POLIATOMICAS. . . . . . . . . . . . . . . . . . . . . 17
NATURALEZA DE LAS VIBRACIONES NORMALES. 17
CIASlFlCAClON DE IAS VIBRACIONES NORMALES. 18
REGIONES ESPECTRALES EN EL INFRARROJO. 23
INSTRUMENTACION. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
TIPOS DE ESPECTROFOTOMETROS INFRARROJO. 36
CARACTERISTICAS DE LOS INSTRUMENTOS COMERCIALES. 40
APLICACIONES DE IA ESPECTROSCOPIA INFRARROJA CUANTITATIVA. . . . . . . . . . . . . . . . . . . . . . . . . 42
IDENTIFICACION DE UN GRUPO FUNCIONAL: GRUPO CARBONILO. 42
APLICACION EXPERIMENTAL. . . . . . . . . . . . . . . . . . . . . . . . 48
RESULTADOS Y DISCUSION 52
CONCLUSION 55
PERSPECTIVAS 55
REFERENCIAS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56

E S P E C T R O S C O P I A I N F R A R R O J A .
INTRODUCCION.
ESPECTRO ELECTROMAGNETIC0 . La radiación electromagnetica es un tipo de energía que se transmite por el espacio a la
velocidad de la luz.
o Propiedades ondulatorias de la radiación electromagnética.
Para caracterizar muchas de las propiedades de la radiación electromagnetica, es
conveniente adjudicar una naturaleza ondulatoria a su propagaci6n y describir estas
ondas con parámetros como amplitud (A), y periodicidad; esta última puede ser descrita
en t6rminos de tres cantidades distintas : Longitud de onda (a) . Número de onda (3) . Frecuencia (w) .
FIGURA 1. Representación esquem&ica de un haz de radiación monocrom6tica de
longitud de onda (a) y amplitud mama (A). (5)
LBmina 1.
Tiompo o distancia ' ,.
-1 -

FIGURA 2. Regiones del espectro electromegn6tico. (5)
9.4 x 10'
9.4 A 105
9.4 z lol
9.4 y 10'
9.4 x 10"
9.4 X 10-3
9.4 x 10-6
9.4 x 10' '
4.1 X loe
4.1 X 10'
4.1 I( lo1
3.3 x 10'0
3.3 x l@
3.3 x los
4.1 X 3.3 X 10'
3 x 10'1
. .
3 X 10-9
3 x 10-7
. . . .
- 2 -

frecuencia ( 2) ) es el número de ondas que pasan por un punto determinado en una
unidad de tiempo o el número de oscilaciones del campo por segundo, y por consiguiente
tiene como unidad el inverso del segundo; la denominación de esta unidad es el hertz (
Hz ).
La frecuencia y la longitud de onda de l a s radiaciones electromagnéticas están
relacionadas a través de la ecuación
'*
donde c es la velocidad de la luz.
El número de onda ( Y ) es el número de ondas por centímetro por tanto su unidad es la
inversa del centímetro.
El número de onda y la frecuencia están directamente relacionadas por la velocidad de
la luz, y por esto se utiliza indistintamente cualquiera de las dos cantidades. La relaci6n
es (1 Y 5)
v ( cm" ) * velocidad de la luz ( cm/s ) = U ( s-l )
Lámina Ill.
* Propiedades corpusculares de la radiaci6n electromagn6tica.
El modelo ondulatorio para la radiaci6n no explica completamente los fen6menos
asociados con la absorción o la emisi6n de energía radiante; para estos procesos es
necesario considerar la radiaci6n electromagnética como un flujo de partículas discretas
de energía llamadas fotones. La energía de un fotón es proporcional a la frecuencia de la
radiación (5).
E = hm
donde h es la constante de Planck (6.63 x erg - S).
La radiaci6n absorbida por una molécula puede ser utilizada en tres formas diferentes :
- 3 -

1) Para causar una excitación electrónica.
2) Para causar cambios en el movimiento vibracional.
3) Para causar cambios en el movimiento rotacional.
Lámina N.
La radiación infrarroja es capaz de producir cambios en los estados vibracionales y
rotacionales de la molécula, cuando esta absorbe pequeñas cantidades de energía, sin
embargo, cuando absorbe cantidades de energía mayores, como en el caso de la luz
visible y ultravioleta, se producen cambios en los niveles de energía de los electrones.
IDENTIFICACION DE LA REGION INFRARROJA EN EL ESPECTRO ELECTROMAGNETIC0
La regiiin infrarroja abarca las regiones del espectro comprendidas entre los números de
onda de 12 800 a 1 O cm" aproximadamente, lo que corresponde a las longitudes de onda
de 0.78 a 1 O00 pm. El extremo de onda corta colinda con el limite de la percepción visual
en el rojo oscuro, mientras el extremo de onda larga se superpone con el espectro de
microonda en el .intervalo de onda de micrómetros.
Tanto desde el punto de vista de las aplicaciones, como de los instrumentos es con-
veniente subdividir la regi6n infrarroja del espectro en tres porciones denominadas:
infrarrojo cercano, medio y lejano. En la FIGURA 3 se indican los limites de cada una de
ellas, la gran mayoría de las aplicaciones analiticas se basan en el empleo de una parte
del infrarrojo medio comprendida entre los 4 O00 y los 670 cm" es decir, entre las
longitudes de onda de 2.5 y 15 pm. (3)

FIGURA 3. Regiones del espectro infrarrojo (5).
Umina V. ."
NATURALEZA DE LA RADlAClON INFRARROJA.
Las connotaciones espectroscópicas del infrarrojo fueron evidentes desde su des-
cubrimiento ( 1800 ) por Sir William Herschel quien, en realidad, repitib el experimento del
prisma de Newton, pero en búsqueda del efecto térmico en lugar de la distribución de
intensidad visible en el espectro. Parece que la palabra infrarrojo surgió por primera vez
en la literatura espectroscópica unos 75 años después, pero no se sabe con certeza a
quien se debe el origen del termino.
Hubo de transcurrir mucho tiempo para que se reconociera al infrarrojo como una forma
de radiacibn electromagn6tica y para descubrir luego sus otras formas en el sentido de
la mednica cubntica. Despues del descubrimiento de Herschel de la radiación calórica
en el espectro solar mas allb del rojo visible, no estaba claro si los rayos termicos y la luz
eran de la misma clase.
El propio Herschel estaba convencido de haber descubierto un nuevo tipo de radiación.
Solo después, que J. D. Forbes (1834) mostrb que la radiacibn térmica puede ser
polarizada de la misma manera que la luz, y despues que A. H. L. Fizeau y J. B. L. Foucault
(1837) determinaron realmente la longitud de onda de ondas del extremo cercano del
- 5 -

infrarrojo, por franjas de interferencia, se acepto ae modo genera! la identidad del
infrarrojo con la luz. !
J. C. Maxwell (1865) predijo teóricamente la existerda de ondas eWromagn6ticas y propuso el reconocimiento de la identidad de estas ondas con las ondZs.de luz. H. He*
(1887), produjo en el laboratorio ondas ele"-omagnéticas y confirmó que se
propagaban con la misma rapidez que la l u z y mia las mismas propiedades de
polarización que esta. E. F. Nichols y J. D. Tear (1B) lograron genarar ondas en el
extremo lejano del infrarrojo de 220 micras de 1ong.W de onda, por mecb de un oscilador
de chispa de tamaño miniatura.
M. Planck (1900) se di6 cuenta de que s610 era psible predecir b bstribuci6n de la
energía entre los osciladores elementales que cornpcnen el cuerpo de ndiacjbn termica,
' si se abandona el concepto de que la energía es GvMble de modo continuo. Planck
presentó su teoría ante la Sociedad Alemana de F i donde dijo que los estados de
energía de los osciladores elementales solo pu& variar por midIipks enteros de
cuantos elementales,
E = hv.
Ademas, Planck mostr6 que los cuantos de e m han de ser proporcionales a la frecuencia (v) y denomin6 correctamente la magnihd de la constante de proporcionalidad I>, - . ,
(h), que ahora se conoce como constante de Plan& . , -....F. ._. . ;". d&. \ ' ,. ,. ,..u,., . . -2"- .
Aunque este adelanto habría de tener consecuencb de largo alcance, pues finalmente
condujo al desarrollo de la mechica cuántica, su importancia no fue obvia inmediata-
mente. Cinco años despues del descubrimiento de f a n c k , A. Einstein fizo el primer uso
importante del concepto en su teoría del efecto hxléctrico, y esta;#ecb la idea de
"cuantos" de luz como forma moderna de los corpkdos dela l u z de Mewton. Aún mas
tarde, Bohr (1913) introdujo su teoría cuántica del sspectro atómico, que condujo a su
vez, a la fundación de la mecánica cuántica. Desde mtonces el concepto de l u z adquirid
el aspecto de una personalidad dividida. La radiaciór había de consldabatse unas veces
-6-

como onda y otras corno partícula, de acuerdo con la naturaleza de su interacción con la
materia. (2)
.i ORIGEN DEL ESPECTRO INFRARROJO
El primer espectro infrarrojo fue obtenido en 1840 por Sir John Herschel, hijo del
descubridor de la radiación infrarroja. Por un m6todo ingenioso, evaporación de alcohol
en papel ennegrecido (evapografía) obtuvo un registro de la parte infrarroja cercana del
espectro solar y mostró que estaba formada de tres regiones desconectadas entre sí.
Unos cuarenta años después, Langley publicó sus excelentes espectros Solares y mostr6
que habían bandas de absorción superpuestas al continuo solar. Gracias a los trabajos
de F.Paschen, E. Aschikinass y otros investigadores, en la década de 1890-1900, estas
bandas fueron identificadas como las de vapor de agua y dióxido de carbono. Cuando K.
J. Angstrom demostró que las bandas de diferentes gases formados por los mismos
&tomos (por ejemplo CO y Con) tienen diferentes espectros de absorción infrarrojos,
result6 evidente que los espectros infrarrojos están relacionados con las propiedades
moleculares mas que con las atómicas.
Esta evidencia se reforzó con las investigaciones en compuestos org6nicos (constituidos
principalmente por &tomos C y H ). Los iniciadores de la espectroscopia infrarroja en el campo de la química orgánica fueron W. de W. Abney y E. R. Festing, quienes usaron
placas fotogr&ficas especialmente sensibles a longitudes de onda hasta de 1.3m.
Por desgracia, en esta región sólo pudieron observar frecuencias armónicas y de
combinación de la vibraciones fundamentales C-H, que se encuentran mas all6 de 2.7m.
Esta limitación fue superada por W. H. Julius, quien extendió los espectros hasta unas
10m con el uso del prisma de sal gema y el barómetro. El estudio mas extenso de los
espectros infrarrojo de compuestos orgánicos en ese periodo fue hecho por W. W.
Coblentz, cuyo trabajo preparó el camino para el surgimiento de la espectroscopía
- 7 -
i

molecular estructural, con la utilización de espectros obtenidos del prisma de clorhidrato
de sodio. (2)
TABLA 1. Desarrollo del espectrofotómetro infrarrojo.
1908 Coblentz (1 8)
1910 Wood y Trowbridge (1 9)
191 8 Randall y Sleator (20)
1927 Pfund (21)
1931 -1 932 Randall (22)
1932 Firestone (23)
1942-1 945 Varios (24)
1945 Roess (25)
1955 Grubb Parsons ( 2 6 )
1961 Perkin- Elmer
1975 .
Diseño y aplicación de un espectro fotómetro de prisma de cloruro de sodio.
Introducción de las rejillas de difracción de echelette.
Diseño y uso del espectrofotómetro de rejillas de difracción de alta resolución.
Introducción de la parábola en foco.
Introducción del espejo condensador con formas parabólica y elipsoidal fuera de foco.
Primeros sistemas de detección.
Desarrollo de un espectrofotómetro Investigadores de doble haz.
Primer amplificador electrónico para usarlo con ter- mopares.
Introducción del primer prisma de rango amplio / espectrofotómetro de rejillas de doble haz.
Introducción del primer filtro de rango amplio / espectrofotómetro de rejillas de doble haz.
Espectrofotómetro basado en la transformada de Fourier.

VIBRACIONES MOLECULARES.
Los espectros moleculares pueden ser de tres clases : espectros de rotación, de vibración
y electrónicos. Los espectros de rotación se originan por la absorción de fotones por parte
de las moléculas, con la conversión completa de la energía del fotón en energía de rotación
molecular. Los espectros de vibración aparecen cuando la absorción de energía radiante
produce cambios en la energía de la vibración molecular. Solo están permitidas ciertas
energías discretas en las moléculas, y la absorción de luz corresponde a una transición
entre dos niveles de energía.
Una molécula puede considerarse como un conjunto de bolas y muelles, representando
las bolas los núcleos y los muelles los enlaces químicos. Tal sistema puede vibrar de
acuerdo con un amplio número de esquemas complejos. Puede esperarse un espectro
de absorción bastante complejo para la mayoría de los compuestos, porque una molécula
que contenga 'In" &tomos tiene 3n - 6 vibraciones normales (3n - 5 para una molécula
lineal). Una frecuencia fundamental característica estará asociada a cada una de estas
vibraciones normales. (6),
Las moléculas poliatómicas pueden manifestar mas de una banda de absorci6n
vibracional fundamental. El número de estas bandas fundamentales se encue,ntra
relacionado con los grados de libertad en una mol6cula. El número de grados de libertad
es igual a la suma de las coordenadas necesarias para localizar todos los 6tomos en la
molécula considerada en el espacio.La cantidad 3n - 6 se obtiene de la siguiente manera.
Con el fin de describir por completo el movimiento de los núcleos de una mol6cula, tienen
que especificarse tres coordenadas para cada núcleo, de este modo, para una mol6cula
con n átomos, se requieren en total 3n coordenadas, y se dice que tiene 3n grados de
libertad (1).
Los átomos tienen tres grados de libertad, cuando estos se combinan para formar las
moléculas, no se pierden grados de libertad. Los 3n grados de libertad de la mol6cula

están constituidos por los grados de libertad rotacional, vibracional y traslacionales. Los grados de libertad rotacionales resultan de la rotación de la molécula alrededor de un eje
que pasa a traves del centro de gravedad. Estas rotaciones resuttan de un grado de
libertad solamente cuando la posición de los átomos en el espacio cambia durante la
rotación.
Todos los grados de libertad que no estén representados por los grados de libertad
traslacionales y rotacionales, pertenecen a los grados de libertad vibracionales (5).
3n grados de libertad = traslacionales + rotacional + vibracional.
Para moléculas lineales :
Grados de libertad vibracionales = 3n - 5
Para moleculas no lineales :
Grados de libertad vibracionales = 3n - 6
LBmina VI.
El número de bandas en un espectro infrarrojo se puede ver aumentado por bandas que
no son fundamentales, como son : tonos de combinación, armónicos (sobretonos) y
tonos de diferencia. Un tono de combinación es la suma de dos o mas frecuencias
diferentes, tales como ~l y ~2 (esto es, el fotón absorbido excita las vibraciones 1 y 2 al mismo tiempo). Un arm6nico es un múltiplo de una frecuencia dada, como 2~ (primer
arm6nico), 3v (segundo armónico), etc. Un tono de diferencia es la diferencia entre dos
frecuencias como ~l y ~2. La molécula esta ya en un estado de vibración excitado (~2) y
absorbe suficiente energía radiante adicional para alcanzar otro estado excitado de
vibración ( ~ 1 ) (6, 3)
Lámina Vll.

Para que se verifique la absorción infrarroja deben cumplirse las siguientes condiciones:
1) La energía de la radiación debe coincidir con la diferencia de energía entre los estados excitado y el normal de la molécula. La molécula absorberá entonces la energía radiante aumentado su vibración natural.
2) La vibración debe ir acompañada de un cambio en el momento dipolar eléctrico (3).
Lámina Vlll.
ABSORCION DE RADlAClON INFRARROJA POR MOLECULAS DIATOMICAS.
Para iniciar el estudio de espectros moleculares es conveniente empezar por una molécula
diatómica, ya que esta es el caso mas sencillo.Las molkulas homopolares como H2, N2 , y 0 2 no absorben la radiación infrarroja, mientras que moléculas como HCI y CO
presentan absorción en el infrarrojo, ya que estas moléculas están asociadas a un cambio
del momento dipolar. En algunos casos, la magnitud del cambio del momento dipolar
puede ser muy pequeña, produciéndose únicamente bandas de absorción d6biles tal
como sucede en el grupo C= N, que es relativamente no polar. En contraste al alto
momento dipolar del grupo C =O que prod& bandas de absorción fuertes, que suele
ser la característica mas distintiva de un espectro infrarrojo.
Si no se crea un momento dipolar como sucede en el enlace C=C (estando situado
simétricamente en una molécula) cuando presenta una vibración de estiramiento, no se
absorbe radiación, y se dice que la forma de vibraci6n es inactiva en el infrarrojo. (3)
Lámina IX.
- 11 -

LA MOLECULA COMO UN ROTOR RlGlDO.
El modelo más simple posible de una molécula en rotación puede representarse con-
siderando que los dos átomos de masas m1 y m2 son puntuales y están unidos a una
distkcia y desde los extremos de una varilla rígida sin peso.En la mecánica clásica, la
energía de rotación E de un cuerpo rígido esta dada por (6)
E=1/2 I w 2
Aquí, w es la velocidad angular de rotación e I es el momento de inercia del sistema
alrededor del eje de rotación. La velocidad angular esta relacionada con el número de
rotaciones por segundo, (frecuencia rotacional) por
w = 2 S urot
el momento de inercia esta definido por
I = mi ?i
y el momento angular del sistema esta dado por
P = I w .
La energía de rotaci6n depende en escencia del momento de inercia. Para el modelo del
rotor rígido modelo, este vale :
donde
r l = m2/m1 + m2.r y r 2 = ml/m1+m2.r
y r l y r 2son las distancias entre las masas m1 y m2 al centro de gravedad, y r la distancia
entre las dos masas puntuales. AI sustituir
I = m1 m2/ml +m2. ?
- 1 2 -

el momento de inercia es el mismo que el de una masa
a una distancia y del eje; p se denomina masa reducida. Por tanto se puede considerar
la rotación de una masa sencilla alrededor de un punto a una disiancia fija y del eje de
rotación. Tal sistema se conoce como rotor rígido simple.
Lámina X.
De acuerdo con la electrodinámica clásica, un movimiento intramolecular conduce a la
radiación de luz solo si existe un momento dipolar variable asociado con él. Para un rotor
rígido, esto puede originarse por una masa puntual rotatoria con una carga o asociada a
un momento dipolar permanente. Esto Úkimo se aplica a todas las moléculas diatómicas
constituidas por átomos distintos, por que para estas los centros de las cargas positivas
y negativas no coinciden. Durante la rotación, el componente del dipolo en una dirección
fijada cambia periódicamente, lo que según la teoría clásica significa que debe emitirse
luz de una frecuencia v rot.
Para moléculas constituidas por dos átomos iguales no aparece un dipolo permanente,
y, en consecuencia, no se emite luz. Solo si existe un dipolo permanente pude absorberse
una frecuencia infrarroja, y , por esta razdn, se produce una rotación del sistema o se
incrementa otra ya existente. Según la teoría clbica el espectro absorbido o emitido del
rotor es continuo, puesto que v rot puede tomar cualquier valor (6).
OSCILADOR ARMONICO.
El concepto mas sencillo sobre la forma de vibración de una molécula diatómica es que
cada uno de los átomos se acerca o se aleja del otro con movimiento armónico, esto es,
el desplazamiento de la posición de equilibrio es una función sinusoidal del tiempo. Tal
movimiento de los dos átomos puede reducirse con facilidad a la vibración armónica de
-13-

una Sola masa puntual alrededor de una posición de equilibrio, es decir, al modelo del oscilador armónico (6,l).
Lgmina XI.
En mecánica clásica, un oscilador armónico puede definirse como un punto de masa m,
que esta sometido a una fuerza F, proporcional a la distancia x y dirigida hacia la ;)osición
de equilibrio.
Puesto que la fuerza = masa x aceleración.
F = k x = m(d% /d t2 )
Lh ina X/¡.
El factor de proporcionalidad k se llama constante de fuerza. La solución de la ecuación
diferencial es
x = xg sen ( 2 ~ vosc t + +)
Aquí la frecuencia de vibración uosc esta dada por:
osc = 1/2 X d k/m
La amplitud de la vibración es xg y # es una constante de fase dependiente de las
condiciones iniciales. Como la fuerza es una derivada negativa de la energía potencial V,
sededuceque
para el oscilador armónico
F = kx,
v = 1/2loi! = a2 m v 2 0 s d
Se puede también definir al oscilador armónico como un sistema cuya energía potencial
es proporcional al cuadrado de la distancia de su posición de equi1ibrio.k fuerza mutua
ejercida por dos átomos de una molécula cuando se desplazan de su posición de equilibrio
-14-

es, por lo menos, aproximadamente proporcional a la variación de la distancia inter-
nuclear. Si se supone que esta relación se mantiene con exactitud, se deduce que los &tomos en la molécula ejecutaran vibraciones armbnicas, cuando queden sueltos
después de haber sido desplazados de sus posiciones de equilibrio.Para el primer &tomo
de masa m1 ,
m1 (d 2r1 /d? ) = -k (r-r e)
y para el segundo átomo de masa m2
m2 (d2 r ddt 2, = -k (r-re )
donde rl y r2 son las distancias de los dos 6tomos al centro de gravedad, y es la distancia
entre los dos &tomos y r e es la distancia de equilibrio. Por sustituci6n de la ecuación del
momento de inercia del sistema alrededor del eje de rotación, mencionado anteriormente,
obtenemos apartir de las dos ecuaciones
(r-re ) (r e = constante), resulta
p . d2 (r-re )/d = -k (r-fe )
Por tanto, las vibraciones de los átomos de la molécula se han reducido a la vibración de
una sola masa puntual p , cuya amplitud es igual al cambio de la distancia internuclear
en la molécula. De la ecuacidn de la frecuencia de vibración con la ecuación anterior, se deduce que la frecuencia de vibración clásica de la molécula es
Si la molécula en su posición de equilibrio tiene un momento dipolar, como es siempre el caso de mol6culas constituidas por átomos distintos, este momento dipolar variara, en
- 1 5 -

general, si la distancia internuclear es lineal. Por consiguiente, el momento dipolar cambia
con la frecuencia de la vibración mecánica. Según la electrodinámica clbsica, esto
conducirá a la emisión de luz de frecuencia V . Por el contrario el oscilador se pondrb a
vibrar por absorción de luz de frecuencia VOSC. (6)
- 16-

ABSORCION DE RADlAClON INFRARROJA POR MOLECULAS POLIATOMICAS.
NATURALEZA DE LAS VIBRACIONES NORMALES.
Es evidente que una mol6cula poliatómica puede tener muchos niveles de energía de
vibraci6n, y por consiguiente, los modos de vibración son activos desde el punto de vista
espectroscópico. En la práctica la situación es complicada, por el hecho de que no todas
las frecuencias son activas, por la presencia de líneas espectrales debidas a armónicos
y por formación de tonos de combinación. La naturaleza de las vibraciones normales de
una molécula poliatómica puede aclararse por referencia al caso de la molécula triangular
sim6trica , que aparece en la FIGURA 5. Los desplazamientos de los tres &tomos
pueden representarse por medio de las coordenadas x, y y q; x es el desplazamineto
relativo desde el eje de simetría de átomo Y en el plano de la molécula; y es el
desplazamiento relativo de Y con respecto al centro de masa de los dos átomos X; y q
es el desplazamiento relativo de los átomos X a lo largo de la línea que los une. (6)
FIGURA 5. VIBRACIONES NORMALES DE UNA MOLECULA POLIATOMICA EN ELCASO
DE UNA MOLECULA TRIANGULAR SlMETRlCA YX2 (6).
Y
X X
Lhina XI¡¡.
En la FIGURA 6. se representan los diagramas que pueden considerarse como una
ilustración aproximada de la naturaleza de las vibraciones normales de una molécula
17

triangular simétrica tal como H 20, H 2S, SO 2. El movimiento de vibración real de la
molécula yx;! será complicado, pero, prescindiendo de la complejidad, puede tratarse
como equivalente a la superposición de los tres tipos de oscilación reflejados en esta
figura. +
En cada una de las vibraciones normales, los núcleos de los tres átomos se moverán
en fase en las direcciones de la flecha, auque las amplitudes de vibración pueden ser
diferentes para los tres núcleos.
FIGURA 6. VIBRACIONES AISLADAS DE LA MOLECUIA yx2 (6).
Lámina XN.
CLASlFlCAClON .DE LAS VIBRACIONES NORMALES.
En general cuando una molécula grande contiene un sistema de tres htomos, es posible
que existan vibraciones de flexión o deformaci6n. Estas son vibraciones que implican
movimientos de átomos que salen del eje de enlace. Se pueden considerar 4 tipos :
1. Deformación o tijereteo. Los dos átomos conectados a un átomo central se mueven acerdndose y alejándose uno del otro con deformación del ángulo de Valencia.
2. Balanceo o flexión plana. La unidad estructual se inclina alternativamente de un lado hacia el otro en el plano de simetría de la molécula.
3. Oscilaci6n o flexión espacial. La unidad estructural se inclina alternativamente de un lado hacia el otro en el plano perpendicular al plano de simetría de la mothla. (3)
18

Lámina XV.
Se han propuesto varios métodos de clasificación de los modos de vibración normales.
Un metodo de clasificación para moléculas que posean un eje de simetría depende de la
dirección de la variación del momento dipolar eléctrico de la molécula que acompana a
la vibración.
Esta clasificación suministra información sobre si la frecuencia particular es activa o
inactiva en el espectro infrarrojo de vibración de la molécula. Si no hay ningún cambio en
el momento dipolar que acompaña a la vibración, entonces esta vibración particular ser&
inactiva en el espectro infrarrojo. Si varía el momento dipolar, el cambio puede estar en
las direcciones paralela o perpendicular al eje de simetría.
En el primer caso, se dice que es una vibración paralela, y en el último caso se le d& el
nombre de vibración perpendicular. Ambas vibraciones, perpendicular y paralela, Serb
activas en el infrarrojo. En el caso de la molécula angular simétrica YX2, las tres vibraciones
estan acompañadas de cambios del momento dipolar elétrico de la molécula. Las vibraciones asociadas con las frecuencias U 1y U 2 cambian el momento en una dirección
paralela al eje de simetría, que en este caso es bisectriz del ángulo XYX; éstas son
vibraciones paralelas, y las correspondientes bandas infrarrojas tendrhn estructura similar.
La tercera frecuencia de vibración, v 3, conduce a una alteración del momento dipolar en
una dirección perpendicular al eje de simetría, y 6sta se denomina vibración perpendicular.
Con el propósito de identificar varios tipos de vibraciones normales, Mecke ha sugerido
clasificarlas como vibraciones de Valencia (tensión), para las que se utiliza el símbolo V, y
vibraciones de deformacibn (flexión), indicadas por el símbolo 6. De los 3n-6 modos de
vibración de una molecula que contiene n &tomos, n-1 son vibraciones de Valencia y 2n-5
son vibraciones de deformación. Las vibraciones U implican movimientos en la dirección
de los enlaces de Valencia, mientras que las vibraciones 6 están acompañadas por
movimientos en águlos rectos a estos enlaces. La naturaleza paralela o perpendicular de
las vibraciones está indicada por el uso de los símbolos y U, respectivamente. Por ello
19

U (X) representaría una vibraci6n de tensión paralela y 6 (O) sería una vibraci6n de
deformación perpendicular. Refiriéndose a la molécula yx2 , el movimiento de los &tomos
X en las vibraciones de frecuencias VI y u3 estan en la direccidn de los enlaces X-Y y se
consideran vibraciones de Valencia. La primera es paralda, y la ú!tima perpendicular, de
modo que son U (X) y U (S), respectivamente. En la vibracibn u2 , los &tomos X vibran en
ángulos rectos a los enlaces X-Y; de este modo la forma general de vibracibn se ve que
consiste en una flexión de la molecula como un todo. Es posible considerar esto como
un modo de deformaci6n y, como es paralelo, simbolizarse por 6 (X). Las notaciones para
moléculas angulares yx2 pueden resumirse como sigue :
Símbolo convencional : U 1 u 2 u3
Un metodo para clasificar las vibraciones normales para mol6culas relativamente com-
plejas, es el que emplea letras A y B para representar vibraciones no degeneradas; las
de la clase A son sim6tricas, esto es, su signo no cambia por rotación n-aria, mientras
que las de la clase B son antisimétricas para esta operación. Un subíndice da el valor de
"ni' en cada caso; por ejemplo A1 , A 2 ,B1 ,B 2,etc. Las vibraciones binarias degeneradas
se indican con la letra E, y la degeneracih temaria se designa con la letra F.
Si la molécula tiene un centro de simetría, se emplean l a s letras g y u como subindices
para indicar que las vibraciones son simétricas y antisimétricas, respectivamente, con
respecto a la inversi6n en el centro de simetría (6).
En algunos casos se emplea (') para indicar que la vibración es simetrica por flexión en
un plano perpendicular al eje principal. Un signo (") indica una vibraci6n que es
antisim6trica para esta operación.
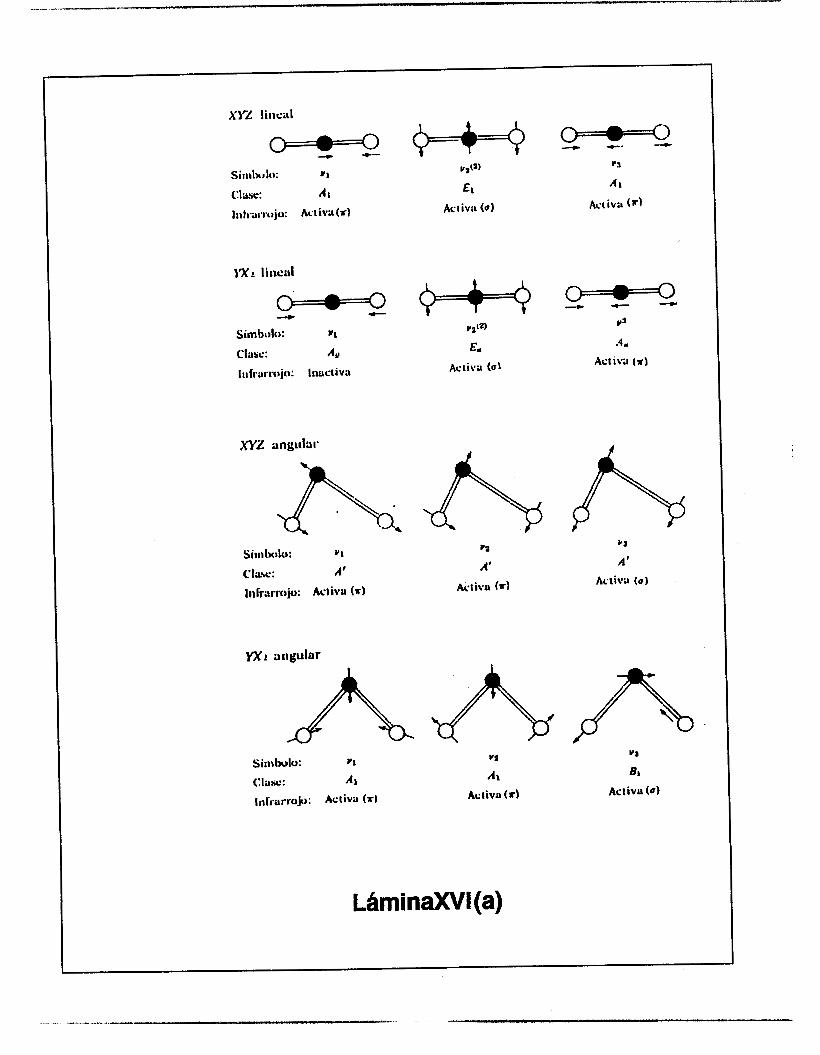
Los modos normales de vibraci6n de algunas mol6culas sencillas aparecen en la FIGURA
7. Un círculo de línea discontinua significa que el movimiento es isbtropo, es decir, no es
restringido en una direccibn, en el plano del círculo. La actividad o inactividad de las
20

características varias de las vibraciones están indicadas en cada caso. Las letras p y d
indican líneas Raman polarizadas y despolarizadas, respectivamente.
FlGURA7.MODOS NORMALES DEVIBRACION DEALGUNAS MOLECULAS SENCILLAS (6)
A

sinll~lo: y,
C lax : Al'
lnfrarr.: 111nctioa
Síalbtrlo: v i
Clnsc: A l
Inkan'.: lnactiva
Ltimina XVI

REGIONES ESPECTRALES EN EL INFRARROJO.
Las regiones espectrales mas importantes en el infrarrojo son (8) :
e Región de vibración por extensión del hidrógeno, 3 700 a 2 700 cm" (2.7
e Región de triple enlace entre 2 700 y I 850 cm -' (3.7 a 5.4 pm).
4
a 3.7 pm).
o Región de doble enlace entre 1 950 y 1 550 cm -' (5.1 a 6.5 pm).
o Región de "huella digital" entre 1 500 y 700 cm" (6.7 a 14 pm).
Lámina WII.
Regi6n de vibraci6n por extensl6n del hidr6geno.
L a aparición de fuertes picos de absorción en éSta región generalmente es resultado de
una vibración por extensibn entre un &tomo de hidrógeno y algún otro 6tomo. El movimmiento es en gran parte el del átomo de hidrógeno, por que es mucho mas ligero
que el átomo con el que se enlaza; como consecuencia la absorción no es muy afectada
por el resto de la molécula. Además, la frecuencia'de extensión del hidrógeno es mucho
mayor que la de otros enlaces químicos, con el resultado de que la interacci6n de esta
vibración con otras es generalmente pequeña.
Los picos de absorción en la región de 3 700 a 3 100 cm"(2.7 a 3.2 pm) se deben
ordinariamente a varias vibraciones de extensión O-H y N-H; la primera tiende a aparecer
en números de onda m6s altos. Las vibraciones de los C-H aliiticos se encuentran en la
región comprendida entre 3 O00 y 2 850 cm" (3.3 a 3.5 pm).

Regi6n de triple enlace.
Un número limitado de grupos absorbe en esta región espectral por lo que su presencia
suele ser fácilmente evidente. La vibración por extensib de los enlaces triples produce
un pico de 2 250 a 2 225 cm" (4.44 a 4.49 pm) para el -C N, en 2 180a 2 120 cm -' (4.59
a 4.72 pm) para el -N C , y en 2 260 a 2 190 cm -' (4.42 a 4.57 mm), para el -C C-.
Se encuentran presentes también en esta región picos para S-H en 2 600 a 2 550 cm" (3.85
a 3.92 pm), P-H en 2 440 a 2 350 cm" (4.10 a 4.26 pm), y Si-H en 2 260 a 2 O90 cm" (4.42
a 4.78 pm).
**
Regi6n de doble enlace
La vibración por extensi6n del carbonilo - C = O - se caracteriza por absorción en toda
esta región; cetonas, aldehidos, ácidos, aminas y carbonatos tienen picos de absorción
alrededor de 1 700 cm " (5.9 pm). Estos, cloruros ácidos y anhídridos de ácidos tienden
a absorber en número de onda ligeramente superiores, es decir, 1 770 a 1725 cm"(5.65
a 5.80 pm). Los picos de absorcibn producidos por la vibraciones de extensión C = C y
C = N estan situados en la regibn de 1 690 a l 1 600 cm(5.9 a 6.2pm) se obtiene valiosa
informaci6n referente a la estructura de las olefinas de la posición exacta de este.pico. La
regi6n comprendida entre 1 650 y 1 450 cm " (6.1 a 6.9 pm) proporciona importante
información sobre los anillos arom6ticos. Los compuestos aromáticos con un bajo grado
de sustitución exhiben cuatro picos cerca de 1 600, 1 580, 1 500 y 1 460 cm'' (6.25,6.33,
6.67 y 6.85 pm). La variación de los espectros en esta región de acuerdo al número y a
la disposición de los grupos sustituyentes son generalmente constantes, pero inde-
pendientes del tipo de sustituyente, puede recogerse a s í información estructural del
estudio de la absorción arom6tica en la región I.R.
24
_-"_."- .. - . . ""

Regi6n de "huella digital".
En esta región se encuentran algunas importantes frecuencias de grupo. Entre ellas figuran la vibración por extensión C-O-C de los éteres y ésteres a 1 200 cm" (8.3 pm) y la
vibración por extensión C-CI a 700 a 800 cm-'(14.3 a 12.5 pm). Algunos grupos
inorgánicos como sulfato, fosfato, nitrato y carbonato absorben también en números de
onda inferiores a 1 200 cm".
I
4 I
FIGURA 8. ABSORCION DE GRUPOS FUNCIONALES.
-1 cm
4000 m m m
Lámina XVIII.

INSTRUMENTACION.
El espectrofotómetro infrarrojo común, es un instrumento que transmite luz policromhtica
en dos longitudes de onda distintas y mide la absorbancia de las muestras en los intervalos
de 4 O00 a 650 cm" (2.5 a 15.4 pm). Consta de tres sistemas separados que operan en
conjunci6n como se observa en la FIGURA 9, estos son :
1) Una conexión de radiación indicada por líneas de trazos.
2) Una conexión eléctrica indicada por líneas estrechas.
3) Una conexión mecánica indicada por líneas obscuras y gruesas.(5)
FIGURA 9. DIAGRAMA ESQUEMATICO DE UN ESPECTROFOTOMETRO DE DOBLE
HAZ (5).
r hcc.on0
- Grófica
I Atenuador
... ! S
t
Detector ""_
I Filtrador, P r r
amp' modulo&r, amplificador Rectificodor
si ncrono
Linea obscura gruesa enlace mednico; línea delgada enlace el6ctrico; línea de trazo trayectoria de la radiaci6n.
-26-
" I_ x__ - - _II

Ldmina NX.
Los componentes básicos que conforman los espectrofot6metros infrarrojos son (3,7) :
o Fuente de radiación.
o Selector de longitud de onda (monocromador).
o Cor,ipartimiento para la muestra.
o Detector.
a Atenuador.
o Sistema de Registro.
LBmina XX.
Fuente de radiaci6n infrarroja.
La radiaci6n infrarroja continua se produce calentando un sólido inerte. Una varilla de
carburo de silicio, denominada Globar, emite energía radiante en un intervalo compren-
dido entre 1 y 40 pm (IO O00 -250 cm") cuando se calienta eléctricamente a 1 500°C,-
aproximadamente. Un emisor de Nemst es una varilla de 6xidos de Circonio, Itrio, Torio,
o Cerio que se calienta tambih a 1 500 OC, aproximadamente, por el paso de corriente;
produce radiaci6n en la regi6n comprendida entre 0.4 y 20 pm (25 OOO-500 cm-'). Un
filamento en espiral de Nicromo calentado es otra fuente útil de radiaci6n infrarroja desde
0.7 a 20 pm (14 200-500 cm"), aproximadamente (54).
Todas estas fuentes de radiaci6n se aplican para el caso del infrarrojo medio, en el caso del infrarrojo cercano se utilizan lbparas incandescentes de Tungsteno , y para el
infrarrojo lejano Ibmparas de arco de mercurio de alta presión con recubrimiento de
cuarzo para reducir las pérdidas térmicas, esto aumenta el rendimiento con longitudes de
onda largas (3).

Lámina n//.
Selector de longitud de onda (monocromador).
El monocrornador consta principalmente de lentes para enfocar la radiación, rendijas de
entrada y de salida para restringir la radiación indeseable y ayudar a controlar la pureza
del espectro de la radiación emitida por el monocromador y un medio dispersante para
separar las longitudes de onda de la radiación policromática de la fuente.(8)
La selección de la longitud de onda en la región infrarroja puede realizarse por medio de
filtros de interferencia, prismas o rejillas de difracción.
Lámina XX///.
Filtros de interferencia.
Estos filtros se basan en un fenómeno de la interferencia para producir bandas relatíva-
mente estrechas de radiación. Un filtro de interferencia consiste en un diel6ctrico
transparente (frecuentemente fluoruro de calcio o fluoruro de magnesio) que ocupa el- espacio comprendido entre dos peliculas m&icas semitransparentes que revisten l a s
superficies interiores de dos placas de vidrio. Existen filtros de interferencia para anhlisis
cuantitativo en el infrarrojo de compuestos específicos, por ejemplo el filtro con una
transmisibn de 9.Opm (1 100 cm") para la cuantificacibn del acetaldehido. Por lo general,
estos filtros tienen anchos de banda efectivos de aproximadamente 1.5 % del valor de la
longitud de onda de su pico.
FIGURA IO. FILTROS DE INTERFERENCIA

Filtros de interferencia en cuña.
Un filtro de interferencia en forma de cuña, consiste en un par de placas metalizadas en
espejo y parcialmente transparentes, separadas por una capa de material diel6ctrico en
forma de cuña, que proporciona bandas variables estrechas y continuas de radiación
infrarroja. Los intervalos cubiertos por este dispositivo son de 2.5 a 4.5, de 4.5 a 8 y de 8
a14.5~(4OOOa2220,de2220a1250yde1250a690cm").
Prismas.
Se han utilizado distintos materiales para la construcci6n de prismas. El cuarzo se puede
usar para la regi6n del infrarrojo cercano (0.8 a 3 pm), aunque sus características de
dispersibn para esta regidn del espectro estan muy lejos de ser las ideales. Este material
absorbe intensamente alrededor de los 4pm (2 500 cm"). El material mbs común para la
construccidn de prismas para el infrarrojo, 8s el Cloruro de Sodio cristalizado; su
dispersi6n es elevada en la regi6n entre 5 y 15 pm (2 O00 a 670 cm") y es adecuada hasta
los 2.5 pm (4 O00 cm"). M6s all6 de los 20 pm (so0 cm"), el Cloruro de Sodio absorbe
intensamente y no puede utilizarse. Los prismas de cristales de Bromuro de Potasio y de
Bromuro de Cesio son adecuados para la regidn del infrarrojo lejano 15 a 40 mm (670 a
250 cm"), mientras que el Fluoruro de Litio es m& adecuado en la región del infrarrojo
cercano de 1 a 5 pm (10 O00 a 2 O00 cm"). Muchos espectrofot6metros han sido
diseñados en base al intercambio de prismas. Desgraciadamente todos los materiales
utilizados comúnmente como materiales de transmisidn para infrarrojo, con la única
excepci6n del cuarzo, se deterioran fbcilmente, son hidrosolubles y es necesario usar
agentes desecadores o calor para protegerlos de la condensación de humedad. (5)
-29-
- ." "" -

FIGURA 11. PRISMA CON MEDIO DE DISPERSION (3)
Angulo de! dpia
Luz incidente
Luz dispersa
i es el Bngulo de incidencia, r el hngulo de refracci6n, t es la anchura de la base del prisma y el hngulo 6ptico es de 30 grados.

Compartimiento para la muestra.
El compartimiento depende del tipo de muestra que se va a analizar ya sea líquida, s6lida
o gaseosa, y para esto existen accesorios dependiendo del caso.
En los espectrofotómetros de difracción, donde se emplea un sistema de doble haz, se tienen dos posibles compartimientos para la muestra. En los espectrofot6metros moder-
nos que emplean la transformada de fourier se tiene s610 un compartimiento para la
muestra, ya que no existen interferencias como son la humedad y el CO2 en el sistema
óptico.
FIGURA 12. CELDAS QUE SE EMPLEAN COMUNMENTE EN EL INFRARROJO (8).
-31 -

Lámina xxv/.
Las celdas utilizada en el espectrofotdmetro infrarrojo deben de ser transparentes en l a s
regiones de 2.5 a 17 pm (4 O00 a 600 cm"), y de longitud variable en espesor de
aproximadamente 0.002 - 3mm (6,7). * I
Tknicas de manipuiacibn de la muestra.
Cuando las muestras son liquidos puros, suelen correrse sin diluir en la regidn del
infrarrojo, para esto la longitud de la celda debe ser corta, con el objeto de mantener la
absorbancia dentro de la región dptima. Si se prepara una solucidn de la muestra debe
correrse una concentracidn bastante elevada, porque ningún disolvente es totalmente
transparente en la regidn de infrarrojo y así podrh reducirse la absorbancia del disolvente
al mínimo (17).
El espectro de gases o líquidos de bajo punto de ebu¡lición puede lograrse mediante la
expansión de la muestra en el interior de una celda al vacío.
La determinación de los espectros infrarrojos de compuestos volátiles,es posible ahora
mediante el uso del espectrofotómetro infrarrojo de rastreo repido (1 1).
Los líquidos puros se examinan entre placas de sal y normalmente sin un separador. El
prensado de una muestra líquida entre placas planas produce una pellcula de 0.01 mm o
menos de espesor; l a s placas se mantienen unidas por capilaridad.
Los líquidos vol&iles se examinan en celdas selladas con separadores sumamente
delgados. Las placas KRS-5 o de CLORURO DE PUTA se utilizan para las muestras que
disuelven l a s placas de Cloruro de Sodio (7).
Los sdlidos no suelen ser suficientemente solubles en los disolventes disponibles, para
que la concentracidn sea bastante elevada y pueda efectuarse la medición en el infrarrojo;
pueden correse polvos en forma de suspensión en un líquido viscoso que tenga
-32-

aproximadamente el mismo índice de refracción, para reducir la dispersibn de la luz. La
muestra se muele con el líquido que generalmente es Nujol. Las muestras tambi6n pueden
molerse con KBr (que es transparente en la regibn del infrarrojo) y comprimirse formando
una pastilla con la cual puede efectuarse la medicibn.
La t6cnica del disco prensado depende del hecho de que el Bromuro de Potasio
pulverizado y seco ( u otros haluros de metal alcalino ) se puede prensar bajo presión al
vacío para formar discos transparentes (3,8).
Detector.
En el extremo de longitudes de onda cortas, aproximadamente debajo de 1.2 pm (8 330
cm"), los m6todos de deteccibn son iguales a los empleados para la radiacibn visible o
ultravioleta. Los detectores que se usan en las longitudes de onda más largas se clasifican
en dos grupos generales :
1) Detectores cuánticos, que dependen de los efectos fotoconductores internos, que resultan de la transicibn de un electrbn de una banda de Valencia a una banda de conduccibn del semiconductor del receptor.
2) Detectores t6rmicos, en los que la radiacibn produce un efecto de calentamiento que afecta a una propiedad física del detector.
Los detectores cu&nticos son m& rbpidos y sensibles, pero estan muy limitados con respecto al intervalo de longitudes de onda en el que producen respuestas. Los detectores
t6rmicos se pueden emplear en un amplio intervalo de longitudes de onda, de hecho en
la totalidad de la regibn espectral en la que se puede considerar al elemento absorbente
como cuerpo negro, pero tiene sensibilidades relativamente bajas y una respuesta lenta.
Normalmente se utilizan detectores t6rmicos en los espectrofotbmetros comerciales
(3,141.

Las formas bhsicas de detectores de radiación termicas son :
o Termopar de radiaci6n.
o Detector Golay.
o Bolómetro. *
El termopar, que es el mhs común de los detectores infrarrojos, generalmente se fabrica
con una pequeña pieza de Ih ina de oro ennegrecida para absorber la radiacibn, soldada
a las puntas de dos alambres conductores hechos con materiales diferentes (3,7).
FIGURA 13. TERMOPAR.
UNION DE MElALES
TERMOPAR
El detector Golay, opera con principios neum&cos. La unidad consiste de un pequeño
cilindro metblico, cerrado con una placa rígida de metal ennegrecido en un extremo y un
diafragma flexible plateado en el otro. La &mara se llena con Xen6n. La radiacibn pasa
através de una pequeña ventana de transmisi6n infrarroja y es absorbida por la placa
ennegrecida. El calor es conducido al gas y ocaciona que este se dilate y deforme el diafragma que puede ser flexible o rígido. Se mide el grado de deformacibn con un mecanismo óptico donde una fuente de luz independiente estb enfocada mediante la

membrana de espejo sobre una fotocelda. Entre la fuente y la membrana se coloca una
rejilla de tal forma que cuando el espejo de membrana est6 sin distorsidn la imagen de
las líneas cae en las aberturas de la rejilla. Cuando el espejo se distorciona las imagenes
reflejadas de las líneas de la rejilla están obstruidas parcialmente por la rejilla misma y por
eso cambia la cantidad de luz que cae sobre la fotocelda.
El detector Golay tiene una sensibilidad parecida al termopar, y es mucho mas eficiente
para el infrarrojo lejano de más de 50 pm (20 cm").
El boldmetro es un termómetro de resistencia en miniatura que generalmente se con-
struye con un metal o un semiconductor. Una pequeña escama de Silicio o Germanio un
poco impuro, enfriado con Helio líquido, constituye un bolómetro muy efectivo. Otro de
los detectores que se usan en el infrarrojo lejano consiste en un trozo de lnSb muy puro.
Generalmente se considera como un bolómetro eléctrico, donde el haz de electrones
puede calentarse durante un tiempo corto sin que haya acoplamiento reticular (3).
Atenuador y Sistema de Registro.
En los espectrofot6metros de difraccidn se logra la atenuacidn imponiendo un artificio que
elimina una fracci6n variable del haz de referencia. El atenuador comúnmente adopta la forma de un peine de dientes finos, los cuales se adelgazan gradualmente hacia la punta,
de modo que existe una relacidn lineal entre el movimiento lateral del peine y la reduccibn
de la potencia del haz. Se produce movimiento del peine cuando el detector percibe una
diferencia en la potencia de los haces. Este movimiento se sincroniza con la pluma
registradora, de modo que su posicidn de una medida de la potencia relativa de los dos
haces y, por lo tanto, de la transmitancia de la muestra (5).

DE ESPECTROFOTOMETROS INFRARROJO.
I
Espectrofotbmetros de doble haz.
Para el trabajo cualitativo en el infrarrojo se emplean espectrofotdmetros de doble haz ya
que el diseño es menos exigente en cuanto a rendimiento de la fuente y del detector que
el de los instrumentos de un solo haz. Estos instrumentos son por lo general del tipo de
compensaddn a cero, en l o s que el haz se atenúa con un peine o una cuña de absorción (3).
El funcionamiento general de este tipo de instrumentos se describe a continuación :
La radiación procedente de la fuente se divide en dos haces, la mitad pasa por la celda
de la muestra y la otra mitad por la celda de referencia. El haz de referencia pasa luego
por el atenuador y se dirige hacia el divisor periódico. Este consiste en un disco impulsado
por un motor que refleja en forma atenuada el haz de referencia o transmite el haz de
muestra al monocromador. Despues de la dispersión por el prisma o la rejilla, los haces
alternativos caen sobre el detector y son convertidos en una señal elbctrica. La señal se
amplifica y pasa al rectificador sincrónico, aparato acoplado macánica o el6ctricamante
al divisor periódico de forma que el interruptor del rectificador y el haz que abandona el
divisor cambien simultheamente.
Si los dos haces son id6nticos en potencia, la señal procedente del rectificador es una
corriente continua no fluctuante. Si los dos haces difieren en potencia, se produce una
corriente fluctuante o alterna, cuya polaridad es determinada por el haz que es m&
intenso. La corrinte procedente del rectificador es fittrada y amplificada aún mhs para
impulsar el motor sincrbnico en una direccidn u otra, según la polaridad de la corriente
de entrada. El motor sincrónico está conectado mecánicamente al atenuador y a la pluma
del registrador, y hace que ambos se muevan hasta que se alcanza el punto nulo. Un
segundo motor sincrónico impulsa el papel y varía simultáneamente la longitud de onda.
Existe por lo general, un acoplamiento mecánico entre los sistemas que accionan el
control de la longitud de onda y la ranura, de modo que la potencia radiante que llega al
-36-

detector se conserve aproximadamente constante gracias a la modificación del ancho de
la ranura.
En la FIGURA 14. Se muestra la óptica de un espectrofotómetro comercial simple, donde
la radiación que proviene del alambre caliente de Nicromo que constituye la fuente, se
divide y pasa através del compartimiento de la muestra. Ambos haces poseen
atenuadores, uno para el ajuste de T (transmitancia) a loo%, y el otro para reducir el haz
de referencia a cero para la medida del porcentaje de T. Los haces se recombinan por
medio de un divisor periódico segmentado, se dispersan por la acción del monocromador
y la radiación se detecta por medio de un termopar. La salida de corriente alterna de éste
último, después de amplificarse, acciona el atenuador de la referencia y una posición cero;
este mecanismo controla el movimiento de la pluma del registrador que está acoplado
mechicamente (5,3)
FIGURA 14. DIAGRAMA DEL ESPECTROFOTOMETRO INFRARROJO DE DOBLE HAZ (3)
Fwnh de n h m a
I , .. . o loo% “””
_ .
. C... .. - . -

EspectrofOt6metrO interferom&rico, o de transformada de Fourier
La configuraci6n bhsica de la porción del interferómetro de un espectr6metro de
transformación de Fourier, tal como el que se muestra en la FIGURA 15. Incluye dos
espejos planos formando un ángulo recto entre si y un separador de haces a 45' con los
espejos. La luz modulada de la fuente se colima y pasa al separador de haces que la
divide en dos haces iguales para los dos espejos, se coloca un espesor igual de material
de soporte llamado compensador, en uno de los brazos del interferómetro, para equilibrar
la longitud de trayecto óptico en ambos brazos.
Cuando éstos espejos se colocan de tal manera que las longitudes de los trayectos
ópticos de los haces reflejado y transmitido son iguales, los dos haces estarán en fase al
regrasar al separador de haces y se interferirán constructivamente. Desplazando el espejo
móvil un cuarto de longitud de onda provocar6 un desfazamiento de 180' de los dos haces
y se interferirh destructivamente. Al continuar el movimiento del espejo en cualquier
dirección, se hará que el campo oscile de la luz a la oscuridad por cada movimiento de
un cuarto de longitud de onda del espejo, lo que corresponde a A/2 cambios. Cuando el
interferómetro se ilumina con luz monocromgfica de longitud de onda A y el espejo se
mueve a una velocida U, la señal del detector tiene une frecuencia f = 2~b. Una grbfica
de la señal en funci6n de la distancia del espejo es una onda consenusoidal pura. Con
luz policromática, la señal de salida es la suma de todas las ondas consenusoidales, que
es la transformación de Fourier del espectro. En lugar de dispersar la radiación
policrom&ica como lo haría un espectrómetro dispersante convensional, el espectrómetro
de transformación de Fourier efectúa una transformacibn de frecuencia (3,15).
Ademas de procesar la radiación espectral de entrada, se usa una línea de una fuente
laser para producir una señal discreta con acoplamiento de tiempo con el movimiento del
espejo y, por lo tanto, con el interograma. La posición del espejo pude determinarse
midiendo el interferograma de la línea laser, contando la franjas a medida que se mueve
el espejo desde la posici6n inicial denotada por un destello de luz de una fuente
incandescente.
-38-
I

No se requiere dispersión o filtros, por lo que no se necesitan rendija, que desperdician
energía, y esto representa una gran ventaja. Puesto que la energía es costosa en el infrarrojo lejano, el gran poder colector de luz del espectrómetro de interferencia es una
gran ayuda para esta regi6n espectral. 1
En el infrarrojo cercano y medio, el material separador de haces mas común es el recubrimiento de Germanio sobre una sal transparente, tal como NaCI, Kbr o Csl. En los espectrómetros para infrarrojo lejano, el separador de haces es una película delgada de
Mylar, cuyo espesor debe seleccionarse de acuerdo con la región espectral de
interés. (3,9)
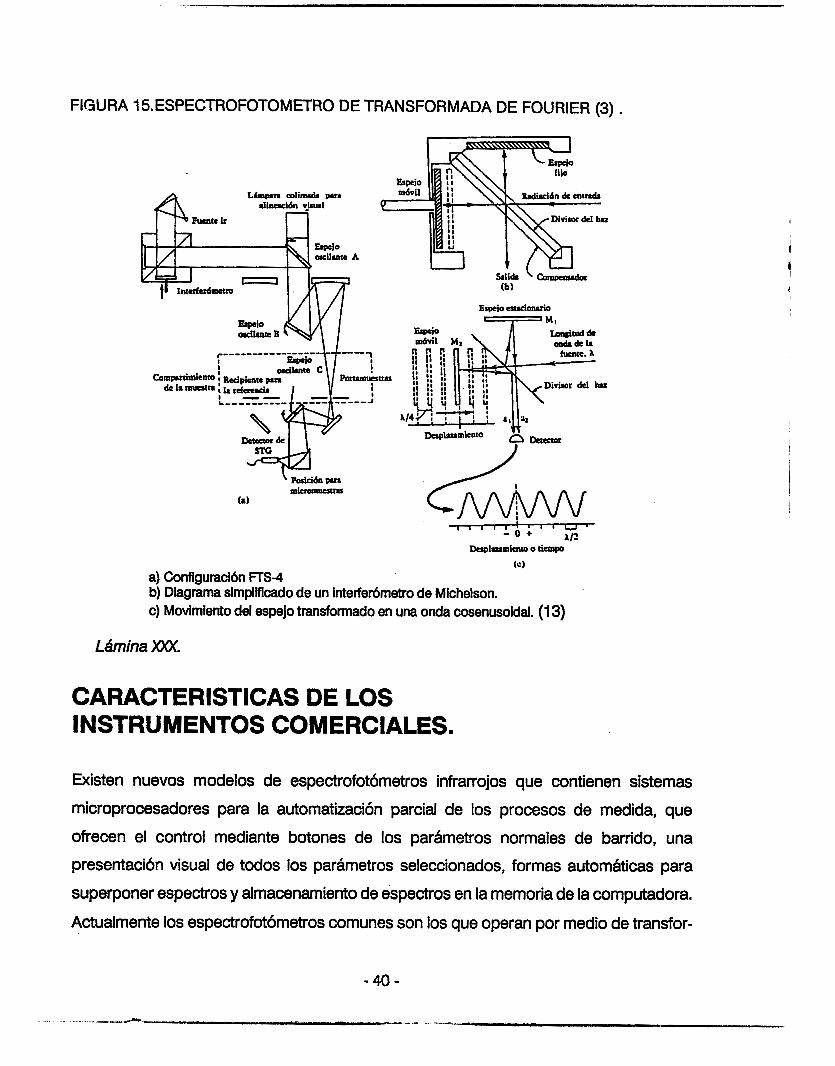
FIGURA 15ESPECTROFOTOMETRO DE TRANSFORMADA DE FOURIER (3) .
L4mp.n mlinuds ~ . n dbmcl6n r i w l
L""""..
(S) mioomucnnr
Derplaaaticnm o ti-
(c) a) Configuracibn nS-4 b) Diagrama simplificado de un interferbrnetro de Michelson. c) Movimiento del espejo transformado en una onda cosenusoidal. (1 3)
CARACTERISTICAS DE LOS INSTRUMENTOS COMERCIALES.
Existen nuevos modelos de espectrofotómetros infrarrojos que contienen sistemas
microprocesadores para la automatización parcial de los procesos de medida, que
ofrecen el control mediante botones de los parámetros normales de barrido, una
presentación visual de todos los parámetros seleccionados, formas automáticas para
superponer espectros y almacenamiento de espectros en la memoria de la computadora.
Actualmente los espectrofotómetros comunes son los que operan por medio de transfor-
-40-

mada de Fourier, estos equipos son fáciles de manejar, y de gran resoluciijn, además de
ser muy rápidos (16).
Se fabrican varios tipos de instrumentos para la región del infrarrojo medio; la mayoría
posee fuentes y divisores de haz intercambiables que permiten cubrir varios intervalos de
número de onda.
En el comercio se encuentran espectrofotbmetros que se basan en la transformada de
Fourier, su precio varia entre 90 y 120 millones de pesos, incluyendo la computadora
dedicada para calcular la transformada de Fourier y el almacenamiento de datos. (9, IO) .
Entre los sistemas m& novedosos que se encuentran en el mercado, está el
espectrofotómetro FTIR-8101 de la compañía Shimadzu, este equipo se puede manejaren
conjunciijn con un microscopio, utilizando un compartimiento para muestras secundario.
Para este tipode equipo se utilizan normalmente detectores de LTaO de alta estabilidad
que no requieren de un sistema de control de temperatura. (9)
En el caso de equipos como el espectrofotómetro 1605 de la compañía Perkin-Elmer, se
emplean detectores de Lira03 o bien de DTGS en un rango de longitud de onda de 4
400 a 450 cm” con una resolucidn de 2 cm’la 16 cm-‘, este tipo de equipo adembs tiene
banco de datos donde se encuentran los espectros comúnmente utilizados tanto en la
investigacibn como en la industria. (IO).

APLICACIONES DE LA ESPECTROSCOPIA INFRARROJA CUANTITATIVA.
La espectroscopía infrarroja tiene dderentes aplicaciones, como son :
1) El conocimiento de la estructura molecular.
2) Identificación de grupos funcionales.
3) El análisis de muestras de compuestos químicos.
4)EI seguimiento de una reacción química, tomando como base la aparición o
desaparición de diferentes grupos funcionales.
5) La versatilidad de poder obtener espectros de muestras, tanto en forma líquida, sólida,
gaseosa, como soportada en películas.
ldentificacibn de un grupo funcional: grupo carbonilo.
La banda de absorción de vibración longitudinal del carbonilo es un excelente parhmetro
para la determinación de estructuras moleculares. La intensidad de la banda es muy
elevada y su mhximo de longitud de onda se puede determinar con mucha presici6n.
La posici6n exacta de la banda de absorción del carbonilo varía con :
1) El estado físico de la molécula.
2) Las interacciones electrostáticas con grupos vecinos.
3) La conjugación.
4) Los puentes de hidrógeno.
5) La tensión en el anillo.
- 42 - ... . .~.. "_"" -

La posición de la banda es prácticamente la misma para aldehidos alifáticos, 5.75 a 5.82
(1 740 a 1 720 cm"), y cetonas, 5.80 a 5.87~ (1 725 a 1 705 cm"), pero es muy diferente
para sistemas insaturados conjugados, acidos carboxílicos, ésteres, anhídridos y
halogenuros de acilo. Su posición exacta en la región, que va desde 1 825 a 1 575 cm " (5.48 a 6.35 rm), depende del caracter de enlace doble del grupo carbonilo como se
muestr a en la FIGURA 16.
FIGURA 16. ABSORCIONES DE CARBONILOS (14).
Tlpica ¿e
~~
Número de Longitud de ondas cm" onda pm
-~
hnhfdridos de dcidos carbonílicos: llifdticos
aromdticos
Cloruros de kidos carboxílicos Aados carbaxff icos ( mon6muas ) Esteres fenílim Btuw vinílicos de dcidos carboxilicos &teres vinilidCnicoa de Pcidos carboxílicos Carbonatos tipo vinflico c8rboMtoo nomde8
Estaes de &idos urborllicor &teres del dado f&mia, mehldos otonrs:
Esteres mctni dc kidos c8tboxllicos
8IifAtifXS uomiticn
Ad& catboxllicos (dimeros) orhmrcos Amidas ( 16) de dcidos Cruboxnims
Amidos (20)- de &idos' carboxflicos., Amidas (30) dc dcidos urboxílicos Sales de icidos carboxflicos
182Sb 1754
. 180Zb 1754 1812 *
1776 1770 1770 1764 1761 175 1 1748 1736 1733 1736
1724 1680-1645 1720-1700
1689 1718 e y d a 1684 1669 1667 1575
5.48 5.70 5.59 5.70 5.52 5.63 5.65 5.65 5.67 5.68 5.71 5.72 5.76 5.77 5.76
5.80 5.95-6.08 5.81-5.88
5.92 5.84 aguda 5.94 5.99 6.00 6.39
A continuación se presenta el seguimiento de un grupo funcional característico: grupo
c = o, en diferentes compuestos.
~ ~.
-43-

FIGURA 17. ESPECTRO EN EL INFRAROJO DE LA ACETOFENONA (15).
La posición de la vibración longitudinal de C =O de los Bsteres dependen tambibn de la
conjugacidn y del tamaño del anillo; como por ejemplo, el acetato de n- butilo ( ester alifbtico), tiene una vibración longitudinal de C = O en 5.75 p (1 739 cm-'), ver la FIGURA
18., mientras que un Bste arombtico la tienen a 5.80 p (1 724 cm"). En el acetato de
n-butilo se encuentra ademhs, una banda ancha de vibración longitudinal de GO,
centrada a 8.05 p (1 242 cm"), característica del grupo acetato.
FIGURA 18.ESPECTRO EN EL INFRARROJO DEL ACETATO DE N-BUTILO (15)
1 .O(
Go(
-44-

Los ácidos carboxílicos generalmente se encuentran dimerizados en el estado sólido y
líquido. Las unidades del monómero están unidas por puentes de hidrógeno entre los dos grupos carboxilo, medianamente fuerte. En el espectro del ácido n-hexanoic0 (Figura
23), se puede apreciar la vibración longitudinal característica de C-H alifático a 3.43 p (2
915 cm”). La banda de vibraci6n longitudinal de carbonilo se encuentra a 5.89 p (1 698
cm”’) y es m& intensa que la banda corespondiente de éster.
FIGURA 19. ESPECTRO EN EL INFRARROJO DEL ACID0 N-HEXANOIC0 (15).
1 .M
OOC
Otros ejemplos de espectros infrarrojos siguiendo el grupo carbonilo, en diferentes
compuestos son :

FIGURA 20. ESPECTRO DEL ACID0 ACETICO.
. - 1000 1 ' so00 2boo I 3 600 1200 600
A C I D 0 A C E T I C O ( F S T F R
F E N I L I C O )
2<
0.0
t>.O!,
O. 1
0.2
O.?,
1 .o 7.0
>O
FIGURA 21. ESPECTRO DEL ACID0 BENZOICO.
0.0
, ' 0.05
. 0 .1
I
. -0.6
. l . O
4 O00 3000 2000 1600 1 500 ROO 400 200 1-
F R E c u E N c I A ( e r n " )
A C I D 0 B E N Z O I C O ( S A L D E A M O N I O )

o I .
. .. -
GURA 22. ESPECTRO DEL ANHlDRlDO PROPIONICO.
A D S 0 B A N C I A
I 4000 3000
- "" C..-
2000 1600 1 O00 600 b
20c
0.0
0.05
o. 1
0.2
0.1
2.0 1 .o
1
-47 -

Este es el primero de una serie de experimentos para poder estudiar, posteriormente, la
cinética de una reacción fotoquímida por espectroscopía infrarroja.
El anhlisis se enfoca en el seguimiento de la desaparición de la banda de absorción del
grupo ciano (2500 cm"), para la reacción.
P- Rosanilinu 1
+ + CN-
Con la finalidad de obtener el espectro de infrarrojo de la muestra se utilizaron dos
m6todos de preparaci6n: impregnación en película y pastilla en Bromuro de Potasio.
METODO EXPERIMENTAL
El compuesto Cianuro de Pararosanilina fué sintetizado por el Dr. Manuel Barceló et al
(comunicación personal, patente en trhmite) de la Universidad de Yucatb.
1) El compuesto fu6 impregnado en una película plástica para la obtencidn de los espectos infrarrojos y aislada de la luz visible-uttravioleta, ya que el compuesto es sumamente
.sensible.
Se obtuvo el espectro infrarrojo de la película impregnada: film en un I.R. de transformada
de Fourier marca Nicolet MXI.
En la FIGURA 23, se muestra el espectro del compuesto sin irradiar impregnado en una
película. En este espectro, no se observa la banda característica del grupo ciano, ya que
hay un enmascaramiento por parte de la película en la que se soporta el compuesto.

FIGURA 23. ESPECTRO DEL COMPUESTO SIN IRRADIAR EN UNA PELICUIA.
: F R E c u E N c I A rem-')
Se impregnaron varias películas para ser irradiados con l u z ultravioleta a diferentes
tiempos.
FIGURA 24. IRRADIACION CON LUZ ULTRAVIOLETA.
Se obtuvieron espectros de películas irradiadas durante 10, 20, 30, 40, 50 y 60 minutos.
Se muestra el espectro a 40 minutos en la FIGURA 25.
- 49 - _... ". ~. _-. _yu - """_

FIGURA 25. ESPECTRO DEL COMPUESTO IRRADIADO DURANTE 40 MINUTOS.
HUESTRA. 40 MIN.
‘ m r
2) Se prepararon pastillas del compuesto en Bromuro de Potasio las cuales se irradiaron
al igual que l a s películas. En esta ocasión se obtuvo el espectro infrarrojo en un equipo
Perkin-Elmer 283.(FIGURA 26).
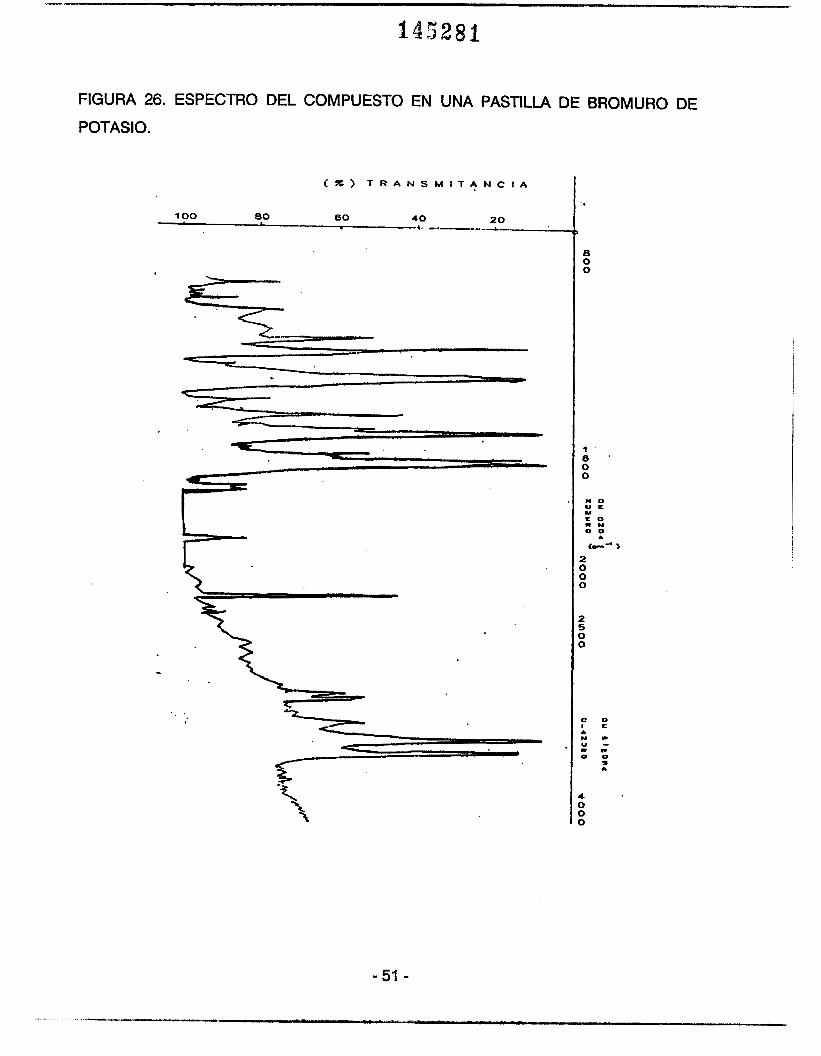
FIGURA 26. ESPECTRO DEL COMPUESTO EN UNA PASTILLA DE BROMURO DE
POTASIO.
1 O0 SO 60 40 20 a- ".""" .
1
8 O O
8 ' 1
O O
Y O U L Y C Q R N O 0
A
<" >
O L
D
R O S A
-
RESULTADOS Y DISCUSION
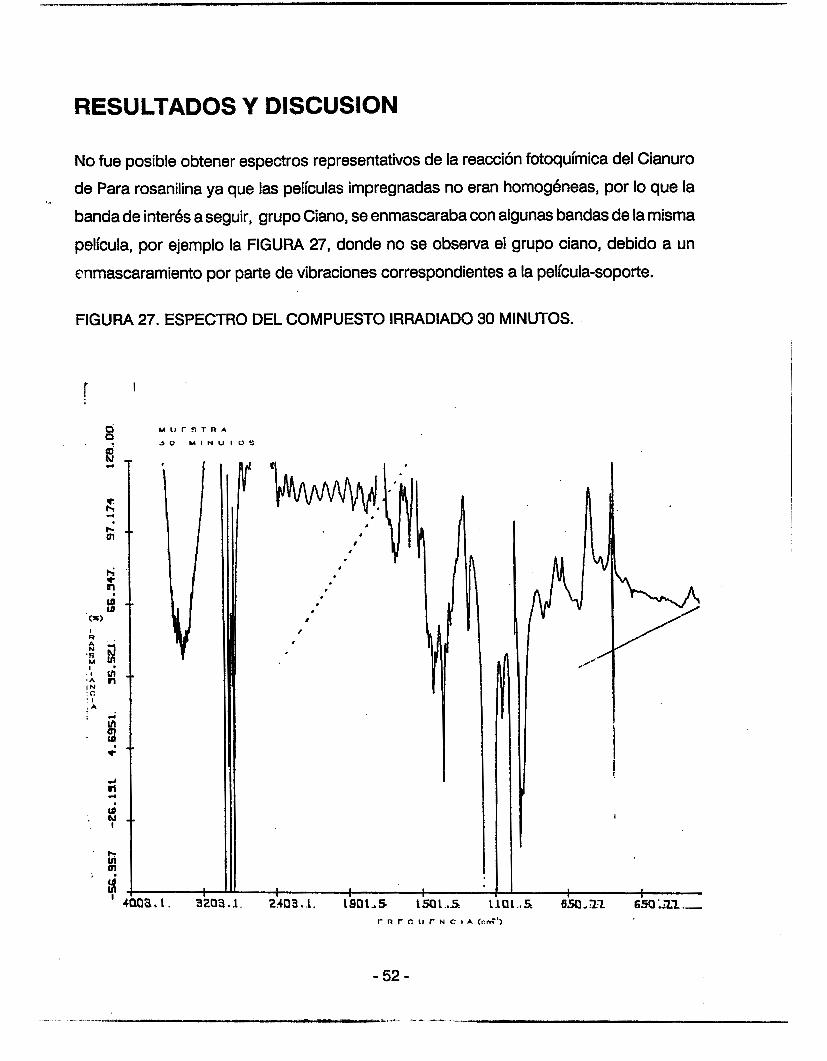
No fue posible obtener espectros representativos de la reacción fotoquímica del Cianuro
de Para rosanilina ya que las películas impregnadas no eran homogéneas, por lo que la
banda de interés a seguir, grupo Ciano, se enmascaraba con algunas bandas de la misma
película, por ejemplo la FIGURA 27, donde no se observa el grupo ciano, debido a un
cnmascararniento por parte de vibraciones correspondientes a la película-soporte.
FIGURA 27. ESPECTRO DEL COMPUESTO IRRADIADO 30 MINUTOS.
, I
I
I
#
,/'
r R r c 11 r N c I A (cm")
- 52 - _. . - - "_ - - ".".._
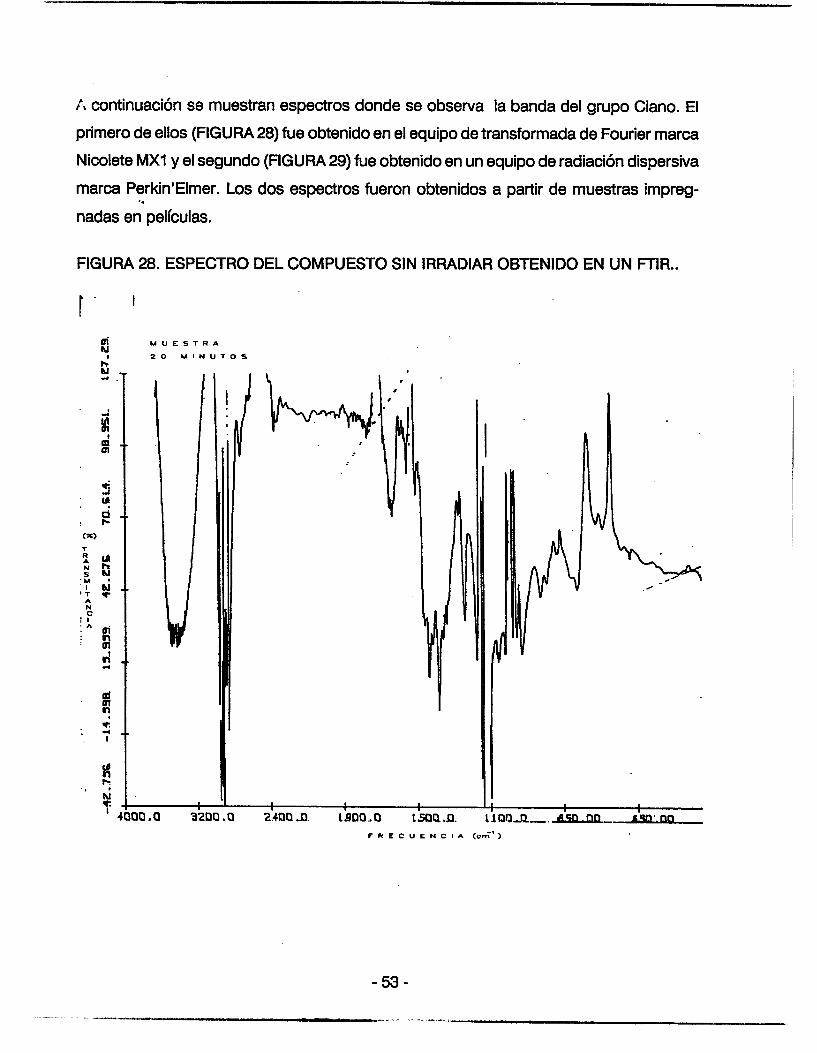
A continuación se muestran espectros donde se observa la banda del grupo Ciano. El
primero de ellos (FIGURA 28) fue obtenido en el equipo de transformada de Fourier marca
Nicolete MX1 y el segundo (FIGURA 29) fue obtenido en un equipo de radiación dispersiva
marca Perkin'Elmer. Los dos espectros fueron obtenidos a partir de muestras impreg-
nadas en películas. ' I
FIGURA 28. ESPECTRO DEL COMPUESTO SIN IRRADIAR OBTENIDO EN UN FTIR..
m Y
M U E S T R A
2 0 M I N U T O S
bl i L .
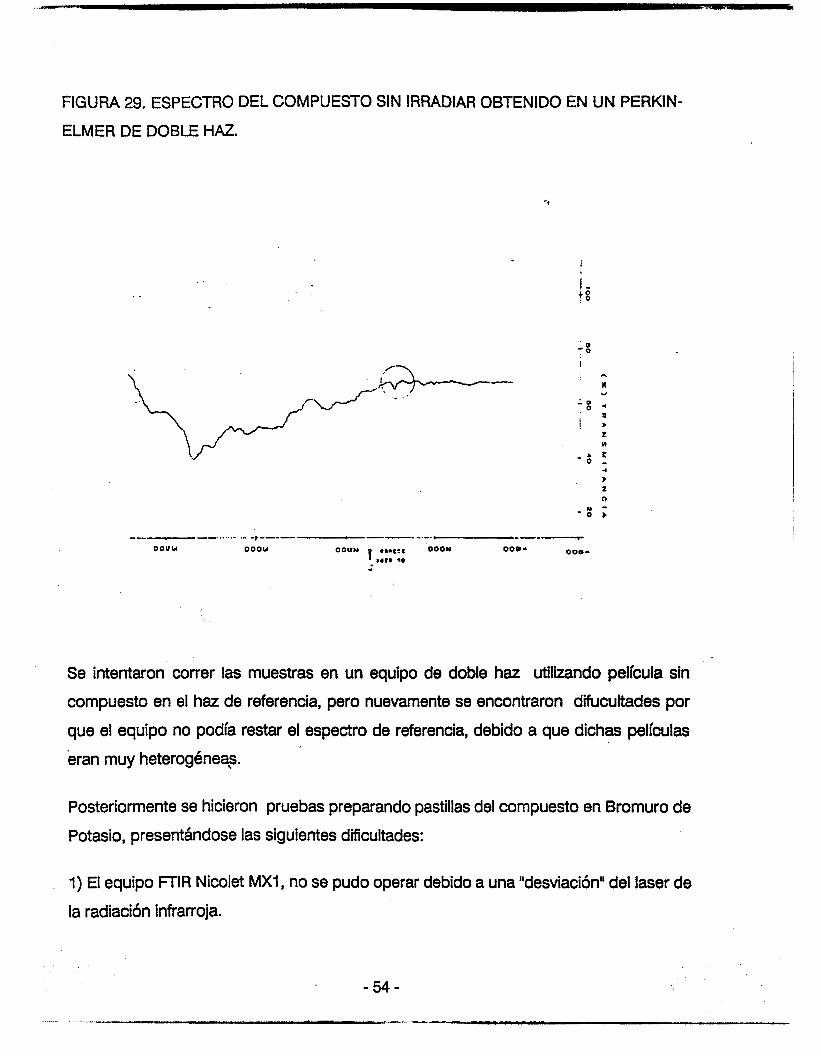
FIGURA 29. ESPECTRO DEL COMPUESTO SIN IRRADIAR OBTENIDO EN UN PERKIN-
ELMER DE DOBLE HAZ.
. .
Se intentaron correr las muestras en un equipo de doble haz utilizando película sin
compuesto en el haz de referencia, pero nuevamente se encontraron difucultades por
que el equipo no podía restar el espectro de referencia, debido a que dichas películas
eran muy heterogéneq.
Posteriormente se hicieron pruebas preparando pastillas del compuesto en Bromuro de
Potasio, presentándose las siguientes dificultades:
1) El equipo FTlR Nicolet MX1, no se pudo operar debido a una "desviación" del laser de
la radiación infrarroja.
-54-

2) El equipo de doble haz Perkin Elmer, modelo 283 tuvo problemas en el cambio de
prismas por lo que no se pudo barrer el espectro en la zona donde aparece la banda de
interés.
Adembs, comparando ambos mbtodos para la preparación de la muestra, encontramos
que la banda en estudio fué mbs intensa cuando se utiliza una pastilla de Bromuro de
Potasio como soporte de la muestra; que cuando se utilizó la película.
CONCLUSION
En los experimentos realizados durante el desarrollo de este estudio, se observó que la
banda de inter& : gropo ciano, presentaba mayor intensidad en los espectros de la
muestra preparada en pastilla de Bromuro de Potasio con respecto a las impregnadas
en película, por lo tanto resulta de gran importancia la elección de la preparación de la
muestra para la obtención de los espectros infrarrojos; ya que de esta manera se pueden
obtener espectros mejor definidos.
Se observa adembs un problema de elección en el material de las peliculas sobre las que
se impregna el compuesto, ya que estas no son homogéneas.
En el presente trabajo, se utilizc5 la espectroscopía infrarroja, para el seguimiento de una
reacción fotoquímica, y no solo como un estudio estructural, que es la aplicación mbs
frecuente que se db a la espectroscopía infrarroja.
PERSPECTIVAS
Para la obtención de mejores resultados , se sugiere cambiar la calidad de las películas
utilizando'dferentes tipos de Polímeros para soportar el compuesto de para-rosanilina y
lograr de esta manera películas homogéneas, y tratar de observar una banda m& intensa
del grupo ciano.

(1) Milton Orchin y H. H. Jaff6. Simetria orbitales y Espectros. Ediciones Bellaterra,
(2) Simon Ivan. Radiaci6n Infrarroja. Ed. Revert6 Mexicana, S. A.. M6xico,1968. (3) Willard H. H. Merritt L. L., M6todos Instrumentales de An&s¡s. CECSA. M6xico,
(4) Daves. Infrared Espectroscopy and Molecular Structure. Etsevier Publishing
(5) Skoog Douglas. AnAlisis Instrumental. Segunda Edicih. Ed. Interamericana.
(6) Conley R. T. Espectroscopía Infrarroja. Ed. Alhambra. Espah, 1979. PP. 10-26,
(7) Robert M., Silverstein G. Clayton Bassler. ldentificacibn Espectrom6trica de
(8) Christian G. D.. Química Analítica. Ed. Limusa Noriega. Universidad de
(9) Comunicaci6n con Shimadzu Corporation. International Marketing Division.
(10) Comunicaci6n con Perkin Elmer Corporation. International Marketing Division.
(1 1) Comunicaci6n con Beckman Instruments, Inc., Fullerton, Caliomia, Modelo
(1 2) Comunicaci6n con Wilks Scientific Corporation, w'como Barnes Engeneering
(13) Comunicaci6n con Block Engeneering /M=.. 1980.
(14) M. Gianturco en S. K. Freeman, Ed. Interpretive Spectroscopy, Van Nostrand
(1 5) Comunicaci6n con Sadtler Research Laboratories. 1980.
(16) Comunicaci6n con Sadtler Research Laboratories. 1981.
S. A., España, 1975, PP. 2,3, 239, 247.
D. F. 1981. PP. 175-217.
Company. London, 1963. PP. 22-28.
M6xi~0, D. F., 1987. PP. 98-1 13.
218-235.
Compuestos Orgbicos. Ed. Diana. M6xico, 1981. PP. 85-97.
Washington, 1990.
1992.
1992.
IR-102. 1980.
Corporation. 1981.
Reinhold, New York, New York, 1965. Pbg. 86.

3 0
TI O
L.
3
O b ti'
C
.

m r )
D
O
I CD -T rt- N
m m O U m O
U 9 n
.c W
r- O
G) P
U m O z U
n L w
3 3

9.4 X 107
9.4 A 105
9.4 z 10'
9.4 10'
9.4 x 10"
9.4 X 10-3
9.4 x 10-5
9.4 x 10' '
4.1 X 106
4.1 x 104
4.1 r( 102
4.1 X le
4.1 X IOez
4.1 x 104
4.1 X
4.1 X 10''
3.3 x 10'0
3.3 x 10'
3.3 x 106
3.3 X 10'
3.3 x 102
3.3 x 1oO
3.3 x 10-2
3.3 x 10-4
3 x 10"'
. .
3 X 10-9
3 x 10-7
3 x 10-5
i X 10-3
3 x 10-1
.. *
3 x 10' +"

LA RADlAClON ABSORBIDA POR UNA MOLECULA PUEDE SER UTILIZADA EN TRES FORMAS:
1 ) PARA CAUSAR UNA EXITACION ELECTRONICA (U.V.)
2) PARA CAUSAR CAMBIOS EN EL MOVIMIENTO VIBRACIONAL (I.R.)
3) PARA CAUSAR CAMBIOS EN EL MOVIMIENTO ROTACIONAL (I.R.)

P , P W C
!"Lk . x x x > C " C oooc " L
X X X )
F
O H

-0 O W
ó 9 7. a . . u I w I I LJ4
O II o II O
n
r= 7 m 9 r I
P O v, 0 m E W m 7u
o 4
51
" 7 m > r" J
I\
I /O
x O 2
9 o O -
'm v, II w
b o O 7
-
9 r m v,
I I w
E: 2 r II LJ x u
I I (9
-z- O c 7 m 9 r I
W
E o O 7 > r
-
W O
2 - O 7 > r <
-0 O X J
'?
ó
m c 73
. .

L
EL NUMERO DE BANDAS DE UN ESPECTRO INFRARROJO SE PUEDE VER AUMENTADO POR :
TONOS DE COMBINACION-SUMA DE DOS O ,MAS FRECUENCIAS DIFERENTES.
ARMONICOS ( SOBRETONOS )-ES UN MULTIPLO DE UNA FRECUENCIA DADA.
TONOS DE DIFERENCIA-ES LA DIFERENCIA ENTRE DOS FRECUENCIAS.
I

PARA QUE SE VERIFIQUE LA ABSORUON INFRARROJA DEBEN CUMPLIRSE LAS SIGUIENTES CONDICIONES :
1) LAENERGIA DE LA RADlAClON DEBE COINCIDIR CON LA DIFERENCIA DE ENERGIA ENTRE LOS ESTADOS EXITADOS Y EL NORMAL DE LA MOLECULA, ABSORBERA ENTONCES LA ENERGIA RADIANTE AUMENTANDO SU VIBRACION NATURAL.
2) LA VIBRACION DEBE DE IR ACOMPANADA DE UN CAMBIO EN EL MOMENTO DIPOLAR ELECTRICO.

XI O
w-
"rl
W c) m 23 O
n z O


D U m cn c -U O
c) O z W m
U O __I m z c>
U c m W m u m 7
D u x > U O
c> c "< D
c, O
W
I
m c> 'D U O
m m
I ' I ._II /u m o m
O D


r
<
x

Z P x ¿ W

CLASlFlCAClON DE LAS VIBRACIONES NORMALES.
SE PUEDEN CONSIDERAR CUATRO TIPOS :
1) DEFORMACION O TIJERETEO.
2) BALANCEO O FLEXION PLANA.
3) OSCll
4) TORS
ACION O FLEXION ESPACIAL.
ON.
angular
A LgminaXVl (a)

y2
Acliva (u)
,¡
y4
E' Activa (a)
/-., & j & .- ".
r , ,. ". * "". , , 'L.. . * ""e

1
O z m cn D 0 m 7J
m m ;rr 3 3
+ < O m o 9 E
1 O CQ I" m
I D "-
W m I n
PO U 'D c> 'D c) m
m m m z m
4 O O
m I
m O O I m
O G) m z O
- 00 cn O o
CT CT O o
4 O O o

ABSORCION DE GRUPOS FUNCIONALES -1
cm
4000 m m m

rincrono Gráfica
5 Motor rincrono
1 Atenuador 1'
Fuente I
4?. h. !
T
Muestra I
Monocromador
" "9 Detector
I
I Fiitrodor, P r o
amp' modulodot, - amplificador
Rectificador sincrono
. . , . I ... .

FUENTE DE RADlAClON .
SELECTOR DE LONGITUD DE ONDA (MONOCROMADOR).
LA M U ESTRA.
COMPONENTES DE LOS ESPECTROFOTOMETROS INFRARROJOS.
COMPARTIMIENTO PARA
DETECTOR.
ATENUADOR.
SISTEMA DE REGISTRO.

‘D c U D 7J D.
o n
W -
I m c > z O W
U m
U ZT O
c m U O - W
c> r O UJ D 7J
I
c,
c,
cm c 7J O U m
I c) O -
I I
W O -
Y
U m
m c> m 533 c, D z O W
”I U O m

””“ _I_
I
I
I I
I
I
I I
m
U

LA SELECCION DE LA LONGITUD DE ONDA EN EL INFRARROJO PUEDE REALIZARSE POR MEDIO DE :
O FILTROS DE INTERFERENCIA.
O PRISMAS.
O REJILLAS DE DIFRACCION.

Rendija ‘y
Pellcula transparente separadora (media longitud de onda de espesor)
Pelfculas de plata semitransparente

Y
LáminaXXV
aluminizada


u3 O 'O z m + m O n
'D "<
O z n
O "<
D z O W
c m W O - W

UNION METAL
TERMOPAR

Fuente de nicromo
I""" Tornocupla
"""
.. - . " . "I.
-. . .. +. Colimador
LBminaXXlX








