
1
DESTILACIÓN
BLOQUE 5

2
DISOLUCIONES BINARIAS IDEALES
(1+2)
(1+2)
L
V
Disoluciónideal
Ley deRaoult: *
2L22
*1
L11
PxPPxP
=
=
En el vapor, de acuerdo con la ley de Dalton:
TOTV22TOT
V1121TOT PxP;PxP;PPP ==+=
Equilibrio L ↔ V
• Cumplen la ley de Raoult en toda la gama de concentraciones.
• Las interacciones A-A y B-B son similares a las interacciones A-B.
A una Temperatura constante

3
*2
L1
*2
*1
*2
L1
*2
L1
*121TOT
*2
L1
*2
*2
L1
*2
L22
L1
*1
*1
L11
Px)PP(PxPxPPPPPxPP)x1(PxP
xPPxP
+−=+−=+=
+−=−==
==
PTOT
P1
P1*
P2
P2*
Si P1* > P2
* P
x1L0 1
Diagrama Presión – Composición (P-x)
Curva de Presión frente a la composición del liquido (x1L)
T: cte

4
V1
*2
*1
*1
*2
*1
TOT*2TOT*
1
V1
*2
*1
*1
*2TOT*
1
V1
*2
*1*
2*1
TOTV1*
2*1TOT
TOT
*1
L1
TOT
1V1
x)PP(PPPP;PPP
x)PP(P
PPPx)PP(1;PP
Px)PP(P
PPx
PPx
−−==⋅−−=
−−+−=
==
Curva de Presión frente a lacomposición del vapor (x1
V)
P
x1V0 1
PTOT
P1*
P2*

5
Podemos representar ambas curvas (líneas) en un mismo diagrama:
Diagrama de fases Presión – Composición (P-x)(T = cte)
Por encima de la línea superior solo existe líquido; por debajo de la línea inferior solo vapor. En la zona comprendida entre las dos líneas coexisten líquido y vapor en equilibrio

6
P
x10 1
Disminuimos P a T cte de A hasta E
A: disolución líquidaB: empieza a producirse vaporC: líq + vapor en equilibrioD: Se evapora la última gota de líquidoE: Todo vapor
x1V
B
x1Vx1L
CD
x1L
A
E
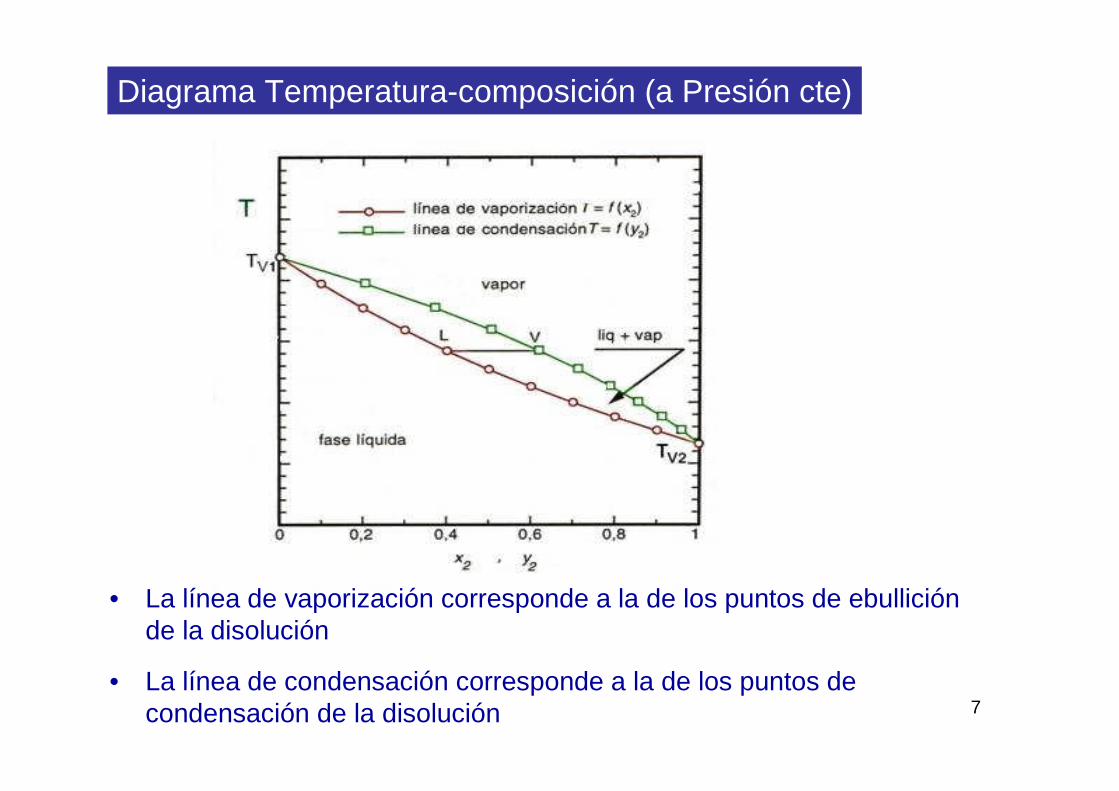
7
Diagrama Temperatura-composición (a Presión cte)
• La línea de vaporización corresponde a la de los puntos de ebullición de la disolución
• La línea de condensación corresponde a la de los puntos de condensación de la disolución

8
Diagrama Temperatura-composición (a P cte)
Para una misma temperatura, el vapor es mas rico en el componente mas volátil.

9
Aplicación: Destilación
Como el vapor es mas rico en el componente mas volátil que el líquido original, es posible separar los 2 componentes de una disolución ideal por destilaciones sucesivas.

10
DESTILACION
• Operación física que se utiliza para purificar líquidos, para separar un líquido de sus impurezas no volátiles o bien para separar dos líquidos.
• Se basa en la diferencia de los puntos de ebullición de los componentes de la mezcla.
• La destilación como proceso consta de dos fases:
Métodos de destilación :• Destilación simple o sencilla• Destilación fraccionada• Destilación a vacío
• El líquido pasa a vapor
• El vapor se condensa pasando de nuevo a líquido en un matraz distinto al de destilación

11
Aparato de destilación fraccionada
Termómetro
Refrigerante
AguaAguaColumna de fraccionamiento
Adaptador
Matraz de destilación
Matraz derecepción

12
Destilación fraccionada
Se construye una columna de destilación donde se producen un grannúmero de condensaciones y revaporizaciones sucesivas.
Destilado(vapor condensado,rico en componentemás volátil)
Residuo(líquido residual,rico en componentemenos volátil)

13
PT es mayor que la quepredice la ley de Raoult
PT es menor que la quepredice la ley de Raoult
FuerzaA-B
FuerzaA-A
FuerzaB-B< &
FuerzaA-B
FuerzaA-A
FuerzaB-B> &
DISOLUCIONES NO IDEALES
• Presentan desviaciones respecto a la ley de Raoult
• Las interacciones A-A e B-B son mas o menos fuertes que las A-B

14
Si las desviaciones de la ley de Raoult son grandes, puede aparecer un máximo o un mínimo en los diagramas P-x y T-x.
• En el máximo o mínimo, la composición del líquido es igual a la del vapor.
• Las mezclas que presentan este comportamiento se llaman azeotropos.
• La composición del máximo o mínimo, se denominan composición azeotrópica.

15
• Un azeótropo de punto de ebullición máximo (a) tiene un punto de ebullición superior al de cualquier componente.
• Un azeótropo de punto de ebullición mínimo (b) tiene un punto de ebullición inferior al de cualquier componente.
(a) (b)

16

1
BLOQUE 5
DISOLUCIONES

2
DEFINICIONES
• Disolución: mezcla homogénea de dos o más sustancias.
•
• El soluto es la sustancia presente en menor cantidad.
• El disolvente es la sustancia presente en mayor cantidad.
• Una de las sustancias es el disolvente y la/s otra/s el soluto.

3
Clasificación de las disoluciones
• Dependiendo del estado de la disolución resultante:
Sólida Líquida Gaseosa
Las sustancias pueden ser sólidas, líquidas o gaseosas

4
TIPOS DE DISOLUCIONES

5
• Dependiendo del disolvente• Acuosas
• No acuosas
• Dependiendo de la naturaleza del soluto:
• Electrolíticas• El soluto se disocia en iones, p. ej. sal
• Conducen la corriente eléctrica
• No Electrolíticas• El soluto no se disocia en iones, p. ej. azucar
• No conducen la corriente eléctrica
Clasificación de las disoluciones líquidas

6
• Un electrolito es una sustancia que, cuando se disuelve en agua, forma una disolución que conduce la electricidad.
• Un no electrolito es una sustancia que, cuando se disuelve, forma una disolución que no conduce la electricidad.
no electrolito electrolito débil electrolito fuerte

7
• Fracción molar (x)
Tot
ii n
nx =
1xi
i =∑
• Representa el tanto por uno en moles de i• Adimensional• 0 ≤ xi ≤ 1 ;
• Molalidad (m)
disolvente kg
nm i
i =• Unidades: mol⋅kg-1 (molal,m)• Ventaja: No varía con T
Concentración: cantidad de soluto presente en una cantidad dada de disolvente.
UNIDADES DE CONCENTRACIÓN

8
• Molaridad (M)Unidades: mol/L (molar,M)
• Desventaja: Varía con T• Ventaja: Facilidad para medir Vdisolución L
nM i
i =
• Porcentaje en peso (% p/p)
100disolución masa
soluto masapeso% ⋅=
610disolución masa
soluto masappm ⋅=
• Partes por millón (ppm)µg/g
mg/L

9
• Una disolución está saturada cuando de ha disuelto la máxima cantidad de soluto en el disolvente, a una temperatura dada. La concentración del soluto en ese momento es igual a la solubilidad del soluto.
• Una disolución no saturada (disolución diluida) contiene menor cantidad de soluto que la que es capaz de disolver, a una temperatura dada.
• Una disolución sobresaturada contiene más soluto que el que puede haber en una disolución saturada.
SOLUBILIDAD
• Cantidad máxima de soluto que podemos disolver en una cantidad dada de disolvente

10
DISOLUCIONES SÓLIDO -LÍQUIDO
sólido + líquido disolución ∆Hdisolucón > 0
• La solubilidad de los sólidos aumenta con la temperatura

11
DISOLUCIONES GAS - LIQUIDO
• La solubilidad de un gas en un líquido depende de los siguientesfactores:
Temperatura (ºC)S
olub
ilida
d (m
ol/L
)
• Naturaleza del gas y del líquido
• Temperatura del líquido
• Presión parcial del gas
La solubilidad de un gas en un líquido disminuye al aumentar la temperatura del líquido:
gas + líquido disolución ∆Hdisolucón < 0

12
Presión y solubilidad de los gases
La solubilidad de un gas, a una temperatura determinada, en un líquido aumenta a medida que se incrementa la presión del gas sobre el disolvente.
c = kH ·P
c : concentración (M) del gas disuelto
P : presión del gas sobre la disolución (atm)
KH: constante de Henry (mol/L•atm) que dependesólo de la temperatura
baja P
Ley de Henry
alta c
baja cbaja P

13
• A estas presiones, la cantidad de CO2 que se disuelve en la bebida es superior a la que se disolvería en condiciones atmosféricas normales.
• Cuando abrimos la botella, escapa el gas a presión. La presión en la botella cae a la presión atmosférica y la cantidad de CO2 que permanece en la bebida está determinada por la presión parcial del CO2 (0,03 atm).
• El exceso de CO2 abandona la disolución creando la efervescencia.
Ejemplo: Bebidas carbonatadas
• Cuando abrimos una botella de Coca-Cola (bebida carbonatada) se observa la formación de burbujas.
• Las bebidas carbonatadas están presurizadas con CO2 a presiones superiores a 1 atm, generalmente de 3 a 5 atm.

14
Aplicación de la ley de Henry
¿Que pasa cuando se abre una Coca-Cola?
CO2(g) + H2O ←→ H2CO3
Cuando se destapa, la P de CO2
cae a 0,03 atm, ↓ la solubilidad y
por lo tanto el CO2 que sobre se
escapa de la solución.

15
PROPIEDADES COLIGATIVAS
1. Disminución de la presión de vapor2. Aumento de la temperatura de ebullición3. Descenso de la temperatura de fusión/congelación4. Presión osmótica
Las propiedades coligativas son propiedades que dependen sólo del número de partículas de soluto en la disolución y no de la naturaleza de las partículas del soluto.
DISOLUCIONES NO ELECTROLÍTICAS
Soluto no electrolítico: Sustancia que se disuelve en agua como moléculas neutras, su disolución no conduce la corriente eléctrica: metanol, glucosa, sacarosa.

16
DISMINUCIÓN DE LA PRESIÓN DE VAPOR
Ley de Raoult
Si la disolución contiene sólo un soluto:
X1 = 1 – X2
P 1o = presión de vapor del disolvente puro
X1 = fracción molar del disolvente
X2 = fracción molar del soluto
P1 = X1 P 1o
Si añadimos un soluto no volátil a un disolvente puro, la presión de vapor de la disolución es menor que la del disolvente puro.
P 1o - P1 = ∆P = X2 P 1
o
P 1 = presión de vapor de la disolución

17
• Las interacciones moleculares soluto - disolvente son de igual magnitud a las disolvente - disolvente y las soluto - soluto
DISOLUCIONES IDEALES
• Todos sus componentes (soluto y disolvente) cumplen la Ley de Raoult en todo el intervalo de concentraciones
Las disoluciones que obedecen a la Ley de Raoult se denominan disoluciones ideales.

18
AUMENTO EBULLOSCÓPICO
∆Te = Te - Teo = ke·m
Te : punto de ebullición de la disoluciónTe
o: punto de ebullición del disolvente puroKe : constante ebulloscópica (constante molar del punto de ebullición)m : molalidad de la disolución
Te > Teo ∆Te > 0
Consecuencia de ladisminución de la presión de
vapor
la temperatura de ebullición de la disolución es mayor que la del
disolvente puro.

19
DESCENSO CRIOSCÓPICO.
Consecuencia de ladisminución de la presión de
vapor
la temperatura de fusiónde la disolución es menor que
la del disolvente puro.
Tfo : punto de congelación del disolvente puro
Tf : punto de congelación de la disoluciónKf : constante crioscópica (constante molal del punto de congelación)M : molalidad de la disolución
∆Tf = Tfo - Tf = Kf ·m
Tfo > Tf ∆Tf > 0

20
kf > ke
El descenso crioscópico es más acusado que el aumento ebulloscópico
• Determinación de pesos moleculares• Anticongelantes, añadir sal a las carreteras, ...Aplicaciones

21
PRESIÓN OSMÓTICA.
La ósmosis es el paso selectivo de moléculas del disolvente a través de una membrana porosa desde una disolución diluida hacia una de mayor concentración.
Una membrana porosa permite el paso de moléculas del disolvente pero impide el paso de moléculas del soluto.
La presión osmótica (π) es la presión que se requiere para detener la ósmosis.

22

23
PRESIÓN OSMÓTICA
π = c R T Ecuación de van’t Hoff
Molaridad
• Determinación de pesos moleculares:(especialmente para moléculas con altos pesosmoleculares como, p.ej., macromoléculas biológicas).
• Ósmosis inversa desalinización(aplicar a la disolución una presión mayor que la π, provocando un flujo de salida del disolvente).
Aplicaciones

24
Osmosis inversa: desalinización

25
RESUMEN
• Las propiedades coligativas son propiedades que depende sólo del número de partículas de soluto en la disolución y no de la naturalezade las partículas de soluto.
• La concentración de partículas en disolución de compuestos No electrolitos (compuestos que no se disocian en disolución) es igual a la concentración del compuesto total adicionado.
• Disminución de la presión de vapor
• Elevación del punto de ebullición
• Disminución del punto de congelación
• Presión osmótica (π)
P1 = X1 P1o
∆Tb = Kb m
∆Tf = Kf m
Π = MRT

26
PROPIEDADES COLIGATIVAS
Las propiedades coligativas de las disoluciones dependen de la concentración total de partículas de soluto, sin importar si las partículas son iones o moléculas.
Disolución de electrolitos
• El soluto se disocia en iones
• Conducen la corriente eléctrica
Electrolitos
DISOLUCIONES ELECTROLITICAS
• Fuertes
• Débiles

27
Electrolitos fuertes
• Se disocian totalmente en iones
NaCl (s) Na+ (aq) + Cl- (aq)
• El cloruro de sodio se disocia en iones Na+ y Cl-.
• No hay unidades NaCl no disociadas en la disolución
Electrolitos débiles
• No se disocian totalmente. Se produce un equilibrio entre lo disociado y lo que no se disocia
Al2(SO4)3 (s) 2Al3+ (aq) + 3SO42- (aq)

28
• La disociación de electrolitos en iones tiene una influencia directa en las propiedades de las disoluciones que están determinadas por el número de partículas presentes.
• Para las disoluciones electrolíticas con validas las mismas fórmulas (propiedades coligativas) vistas anteriormente pero deben multiplicarse por el factor de van`t Hoft (i).
factor de van’t Hoff (i) =número real de partículas en la
disolución después de la disociaciónnúmero de unidades fórmula disueltas
inicialmente en la disolución

29
Factor de van’t Hoff ( i)
• Electrolitos fuertes
NaCl (s) Na+ (aq) + Cl- (aq) i = 2
CaCl2 (s) Ca2+(aq) + 2Cl- (aq) i = 3
• Electrolitos débiles
Al2(SO4)3 2Al3+ + 3SO42-
n 0 0
nα 2nα 3nα
n(1-α) 2nα 3nα
n(1-α) + 2nα + 3nαn
i =
(α : grado de disociación)

30

31
• Disminución de la presión de vapor
• Elevación del punto de ebullición
• Disminución del punto de congelación
• Presión osmótica (π)
P1 = i ·X1 P1o
∆Tb = i · Kb m
∆Tf = i · Kf m
Π = i ·MRT
PROPIEDADES COLIGATIVAS
DISOLUCIONES ELECTROLITICAS

1
DISOLUCIONES (II)
BLOQUE 5

2
DISOLUCIONES BINARIAS IDEALES
(1+2)
(1+2)
L
V
Disoluciónideal
Ley deRaoult: *
2L22
*1
L11
PxP
PxP
=
=
En el vapor, de acuerdo con la ley de Dalton:
TOTV22TOT
V1121TOT PxP;PxP;PPP ==+=
Equilibrio L ↔ V
• Cumplen la ley de Raoult en toda la gama de concentraciones.
A una Temperatura constante:

3
En una disolución ideal ambos componentes siguen la ley de Raoult
( ) ( )* * * * *1 2 1 1 2 1 2 1 2 11= + = + − = + −P P P P X P X P P P X
Diagrama Presión – Composición (P-X)
P: Presión Total de la disolución
Xi: Fracción molar del componente i en la fase líquida
Representación gráfica entre la Presión total y la fracción molar en la fase líquida (Xi) :
Temperatura constante

4
1 1=P y P
( )*
1 1 11 * * *
2 1 2 1
= =+ −
P P Xy
P P P P X
*1 1= X P
Relación entre la Presión total y la fracción molar en la fase gas (y1) :
( )* *
1 2* * *
1 1 2 1
P PP
P P P y=
− −
P: Presión Total de la disolución
y i: Fracción molar del componente i en la fase vapor
11 *
1
=y P
XP
Fase vapor: Ley de Dalton

5
Podemos representar ambas curvas (líneas) en un mismo diagrama:
Diagrama de fases Presión – Composición (P-X)(T = cte)
Por encima de la línea superior solo existe líquido; por debajo de la línea inferior solo vapor. En la zona comprendida entre las dos líneas coexisten líquido y vapor en equilibrio
Representación gráfica entre la Presión total y la fracción molar en la fase vapor (y i) :
Representación gráfica entre la Presión total y la fracción molar en la fase líquida (Xi) :

6
La composición del vapor, en unidades de fracción molar, no es la misma que la composición de la disolución líquida con la que está en equilibrio.
Ejemplo:
Mezcla benceno – tolueno:Xbenceno=0.33
Xtolueno=0.67
torrPobenceno 75=
torrPotolueno 22=
Calcular la presión total y la composición del vapor en unidades de fracción molar.

7
Se tiene una disolución de 5 moles de tolueno en 5 moles de benceno a 60 ºC que se comporta idealmente. Las presiones de vapor a esa temperatura de tolueno y benceno puros son 139 mmHg y 392 mmHg.Calcular:Las presiones parciales de tolueno y benceno y sus fracciones molares en la fase de vapor.
5.51

8
liquidaBencenoTotalBenceno
liquidaToluenoTotalTolueno
XPP
XPP
⋅=
⋅=
mmHgmmHgP
mmHgmmHgP
Benceno
Tolueno
19655
5392
5.6955
5139
=+
⋅=
=+
⋅=
mmHgPPP BencenoToluenoTotal 5.2651965.69 =+=+=
738.05.265
196
262.05.265
5.69
===
===
Total
BencenoBenceno
Total
ToluenoTolueno
P
Py
P
Py
RaoultdeLey
DaltondeLey vaporiTotali yPP ⋅=

9
A 85 ºC, la presión de vapor de C2H4Br2 puro es de 173 mmHg, y la del C3H6Br2 puro es de 127 mmHg. Se disuelven 10 g de C2H4Br2 en 80 g de C3H6Br2.Calcular:• La presión de vapor de la disolución a 85 ºC y la presión parcial de
cada componente.• b) Las fracciones molares de C2H4Br2 y C3H6Br2 en la fase de vapor
en equilibrio con la disolución.Datos: Br = 79.9
5.46
PTOT
PA
173
PB
127
P
x1L0 1

10
molgBrHCmolarMasa /8.201)( 263 =
molgBrHCmolarMasa /8.187)( 242 =
molesmolg
gn BrHC 0532.0
/8.187
10242
==
molesmolg
gn BrHC 396.0
/8.201
80263
==
molesnTotales 450.0396.00532.0 =+=
mmHgmmHgXPPBrHC ovaporvapor 5.20
450.0
0532.0173:242 =⋅=⋅=
mmHgmmHgXPPBrHC ovaporvapor 8.111
450.0
396.0127:263 =⋅=⋅=
mmHgPP iTotal 3.1328.1115.20 =+==∑
845.03.132
8.111
155.03.132
5.20
263
263
42
242
===
===
Total
BrHCBrHC
Total
BrHCBrHC
P
Py
P
Py

11
Diagrama Temperatura-composición (a Presión cte)
• La línea de vaporización corresponde a la de los puntos de ebullición de la disolución
• La línea de condensación corresponde a la de los puntos de condensación de la disolución

12
• La temperatura de ebullición de una disolución ideal varía con su composición.
• La curva de condensación coincide con la temperatura de ebullición referida a la composición del vapor y la curva inferior referida a la composición del líquido.
• A temperaturas elevadas la disolución estará en estado vapor, a temperaturas bajas se encontrará en estado líquido, y en la zona intermedia de las dos curvas coexistirán las fases vapor y líquido en equilibrio.
Diagrama T-χ para una disolución ideal líquido-líquido a presión constante
Diagrama Temperatura-composición (a Presión cte)

13
PT es mayor que la quepredice la ley de Raoult
PT es menor que la quepredice la ley de Raoult
FuerzaA-B
FuerzaA-A
FuerzaB-B< &
FuerzaA-B
FuerzaA-A
FuerzaB-B> &
DISOLUCIONES NO IDEALES
• Presentan desviaciones respecto a la ley de Raoult
• Las interacciones A-A e B-B son mas o menos fuertes que las A-B
ideali iP P>
ideali iP P<

14
Si las desviaciones de la ley de Raoult son grandes, puede aparecer un máximo o un mínimo en los diagramas P-x y T-x.
• En el máximo o mínimo, la composición del líquido es igual a la del vapor.

15
• Las mezclas que presentan este comportamiento se llaman azeotropos.
• La composición del máximo o mínimo, se denominan composición azeotrópica.
• Un azeótropo de punto de ebullición máximo (a) tiene un punto de ebullición superior al de cualquier componente.
• Un azeótropo de punto de ebullición mínimo (b) tiene un punto de ebullición inferior al de cualquier componente.
(a) (b)

16
Diagrama Temperatura-composición (a Presión cte)
Para una misma temperatura, el vapor es mas rico en el componente mas volátil.

17
Aplicación: Destilación
Como el vapor es mas rico en el componente mas volátil que el líquido original, es posible separar los 2 componentes de una disolución ideal por destilaciones sucesivas.

18
1T
2T3T
4T
Destilación

19
DESTILACION
• Operación física que se utiliza para purificar líquidos, para separar un líquido de sus impurezas no volátiles o bien para separar dos líquidos.
• Se basa en la diferencia de los puntos de ebullición de los componentes de la mezcla.
• La destilación como proceso consta de dos fases:
Métodos de destilación :• Destilación simple o sencilla• Destilación fraccionada• Destilación a vacío
• El líquido pasa a vapor
• El vapor se condensa pasando de nuevo a líquido en un matraz distinto al de destilación

20
Aparato de destilación sencilla
El destilado queda enriquecido en el más volátil, pe ro no al 100%

21
Aparato de destilación fraccionada
Termómetro
Refrigerante
AguaAguaColumna de fraccionamiento
Adaptador
Matraz de destilación
Matraz derecepción

22
destilación fraccionada 1

23
destilación fraccionada 2

24
destilación fraccionada 3
Tº2
Tº3

25
Destilación fraccionada
Se construye una columna de destilación donde se producen un grannúmero de condensaciones y revaporizaciones sucesivas.
Destilado(vapor condensado,rico en componentemás volátil)
Residuo(líquido residual,rico en componentemenos volátil)

1
ESTADO LÍQUIDO
BLOQUE 5

2
La tensión superficial es la cantidad de energía por unidad de área, necesaria para estirar o aumentar la superficie de un líquido.
TpnAG
AW
,,
γ
∂∂==
dd
En ausencia de otras fuerzas (como la gravedad y las interacciones con el recipiente), los líquidos tienden a adoptar la forma de menor superficie: la esfera
Propiedades de los líquidos: tensión superficial

3
Las moléculas de la superficie son atraídas hacia el interior, dificultando el aumento de superficie:
A mayores fuerzas intermoleculares mayor tensión superficial
Propiedades de los líquidos: tensión superficial

4
Capilaridad: Interfase sólido-líquido
La cohesión es la atracción intermolecular entre moléculas semejantes
La adhesión es una atracción entre moléculas o materiales distintos
adhesión > cohesión
cohesión > adhesión
Meniscos y capilaridad

5
Propiedades de los líquidos: viscosidad
La viscosidad es una medida de la resistencia de un fluido a moverse ante una fuerza tangencial.
Se utiliza el coeficiente de viscosidad o viscosidad dinámica, η
Feje y
A .
v0
yvηA
F∂∂=
placa fija
fluido
placa móvil

6
Densidad máxima4 ºC
El hielo es menos denso que el agua
Densidad del agua
El agua es una sustancia única
Temperatura (ºC)
Den
sida
d (g
/mL)
Estructura y propiedades del agua

7
• La evaporación tiene lugar en la superficie del líquido.
• Cuando las moléculas de un líquido tienen suficiente energía cinética para escapar de la superficie, sucede un cambio de fase. La evaporación o vaporización es el proceso en el cual un líquido se transforma en un gas.
• El proceso inverso a la vaporización se llama condensación,, es el paso de gas a líquido
EVAPORACIÓN

8T2 > T1
Energía cinética E
Núm
ero
de m
oléc
ulas
Energía cinética E
Núm
ero
de m
oléc
ulas
Para evaporarse una partícula debe luchar contra la fuerza de atracción que ejercen otras partículas del líquido.
Las partículas que se evaporan, y abandonan la superficie del líquido, son las de mayor energía cinética.

9
Antes de laevaporación
En el equilibrio
vacío
Espaciovacío
Líquido Líquido
PRESIÓN DE VAPOR

10
H2O (l) H2O (g)
velocidad decondensación
velocidad deevaporación=
Equilibrio dinámico
La presión de vapor es la presión parcial de las moléculas del líquido en estado vapor sobre el líquido cuando se ha alcanzado el equilibrio condensación-evaporación.
Sistema cerrado
PRESIÓN DE VAPOR

11
• La presión de vapor de un líquido aumenta con la temperatura.
• La presión de vapor también depende de la naturaleza del líquido.
Pvapor baja a Tambiente
Sustancias
Volátiles: Pvapor alta a Tambiente
No volátiles:
PRESIÓN DE VAPOR

12
Variación de la presión de vapor con la temperatura
a) Éter dietílico, b) benceno, c) agua, d) tolueno, e) anilina
Curvas depresión de vapor
a) Éter dietílico , b) benceno, c) agua , d) tolueno , e) anilina

13
• La presión de vapor depende de la naturaleza del lí quido
a) Éter dietílico , b) benceno, c) agua , d) tolueno , e) anilina
• Líquidos con enlace por puentes de hidrógeno tienen una presión de vapor baja.
• A medida que aumenta el peso molecular disminuye la presión de vapor.

14
El calor molar de vaporización (∆Hvap) es la energía necesaria para evaporar 1 mol de un líquido.
CALOR MOLAR DE VAPORIZACIÓN

15
ln P = -∆Hvap
RT+ C
P = presión de vapor (equilibrio)
T = temperatura (K)
R = constante de los gases (8,314 J/K•mol)
Variación de Pv con la T: ecuación de Clausius-Clapeyron

16
PUNTO DE EBULLICIÓN DE UN LÍQUIDO
• El punto de ebullición es la temperatura a la cual la presión de vapor iguala a la presión externa que actúa sobre la superficie del líquido:
Pvapor = Pexterna
• Se define el punto de ebullición normal de un líquido, a la temperatura a la cual la presión de vapor es igual a la presión externa, cuando ésta sea igual a 1 atm.
¿Cuándo hierve un líquido?
• Para todo líquido existe una temperatura a la cual empieza a hervir.

17
El punto de ebullición de un líquido varía con la p resión
a) Éter dietílico , b) benceno, c) agua , d) tolueno , e) anilina

18
Siempre hay dos formas de que un líquido hierva:
� Aumentar su temperatura
� Disminuir la presión externa
Siempre hay dos formas de condensar un gas:
� Disminuir su temperatura suficientemente, y en cualquier caso por debajo de la temperatura crítica
� Aumentar la presión externa suficientemente, siempre que la temperatura sea inferior a la crítica

19
La temperatura crítica (Tc) es la temperatura por arriba de la cual la fase gaseosa no se puede licuar, independientemente de la magnitud de la presión que se aplique.
La presión crítica (Pc) es la mínima presión que se debe aplicar para llevar a cabo la licuefacción a la temperatura crítica.
TEMPERATURA Y PRESIÓN CRÍTICAS

20
Isotermas para los gases reales. Licuefacción
líquido + vapor

21
Monte Kilimanjaro (Tanzania)5895 m de altitud, P = 350 mmHg
Consecuencias
Teb (agua) = 79ºC

22
P » 2 atm
⇓Teb (agua) » 120ºC
Aplicaciones
Tiempos de cocción más rápidos
Olla a presión

23

24
• El punto de fusión de un sólido (o el punto de congelación de un líquido) es la temperatura a la cual la fase sólida y líquida están en equilibrio.
Sólido � Líquido
• El punto de fusión normal de un sólido (o el punto de congelación de un líquido) es la temperatura a la cual la fase sólida y líquida están en equilibrio a la presión de 1 atm.
• La transformación de un líquido a sólido se llama congelación y el proceso inverso se llama fusión .
EQUILIBRIO SÓLIDO-LIQUIDO
H2O (s) H2O (l)

25
El calor molar de fusión (∆Hfus) es la energía necesaria para fundir 1 mol de una sustancia sólida.
Tabla 11.8 Calor molar de fusión para sustancias selectasSustancia Punto de fusión* (ºC) ∆Hfus (kJ/mol)
* Medido a 1 atm.
Argón (Ar)
Benceno (C6H6)
Etanol (C2H5OH)
Éter dietílico (C2H5OC2H5)
Mercurio (Hg)
Metano (CH4)
Agua (H2O)
CALOR MOLAR DE FUSION

26
Curva de calentamiento
Tem
pera
tura Punto de ebullición
Líquido
Tiempo
Punto de ebullición
Punto de fusión
Sólido
Vapor
Líquido y vaporen equilibrio
Sólido y líquidoen equilibrio

27
• Los sólidos también sufren evaporaciones y en consecuencia tienen presión de vapor.
• La presión de vapor de un sólido es la presión que ejerce el vapor en el equilibrio sólido-vapor.
• El proceso en el cual las moléculas pasan directamente de sólido a vapor se llama sublimación, y el proceso inverso (de vapor a sólido directamente) se llama deposición.
EQUILIBRIO SÓLIDO-VAPOR
sublimación
deposición

28
Transición de fase: Conversión de una fase en otra
Gas
Líquido
Sólido
Sublimación
Vaporización
Fusión
Condensación
CongelaciónDeposición

29
Calentar o reducirpresión
Calentar o reducir presión
Enfriar o comprimir
ESTADOS DE AGREGACIÓN DE LA MATERIA
Enfriar ocomprimir

30
DIAGRAMAS DE FASES
• Los límites de fase muestran las condiciones en las que dos fases pueden coexistir en equilibrio dinámico
• En el punto triple (B) coexisten las tres fases en equilibrio.
Un diagrama de fases proporciona las condiciones de presión y temperatura en las cuales una sustancia existe como sólido, líquido o gas.

31
Punto triple: coexisten
las tres fases
TEMPERATURA / K
PR
ES
ION
LIQUIDO
VAPOR
SOLIDO
GAS
Linea de presión de vapor del líquidoen función de la temperatura:Coexisten el vapor y el líquido
Línea de temperatura de fusión en función de la presión: coexisten el sólido y el líquido
Linea de presión de vapor del sólido en función de latemperatura: coexisten el sólido y el vapor
Punto crítico
DIAGRAMAS DE FASES

32
Curvas de Presión de VaporTemperatura de fusión a P
Temperatura de ebullición a P
Punto de ebullición normal:Temperatura a la que la presiónde vapor del líquido es igual a la presión de 1 bar ( o 1 atm).
Punto de fusión normal:Temperatura a la que funde el sólido si la presión es de 1 bar.
DIAGRAMAS DE FASES

33
0
100
200
300
400
500
600
700
800
900
1000
1100
0 10 20 30 40 50 60 70 80 90 100 110 120
T/ºC
P/m
mH
g
DIAGRAMAS DE FASES.
Curva de presión de vapor de un líquido
La curva nos informa desituaciones de equilibrio
líquido-gas.
Curva de equilibrio entre fases
Cada punto nos da una pareja de valores(P, T) para los cuales existe equilibrio
Si, a una P dada, T > Teb : La fase más estable es el gas.Si, a una P dada, T < Teb : La fase más estable es el líquido.
GasLíquido

34
• La curva de presión de vapor del líquido termina en el punto C (punto crítico ).
• Temperatura crítica (Tc): temperatura por encima de la cual no se puede licuar un gas, esto es, temperatura máxima a la cual la sustancia puede estar en fase líquida.
• Presión crítica (Pc): presión necesaria para provocar la licuefacción de un gas a la temperatura crítica. Es la presión de vapor mas elevada de un líquido.
DIAGRAMAS DE FASES.
LIQUIDO
VAPOR
GAS
PUNTO CRITICO
Tc
pc

35
Diagrama de fases del agua
Un aumento de favorece la fase más densa, produciendo fusión del agua: ρSOLIDO < ρLIQUIDO

36
Diagrama de fases del CO 2
Curva pto. fusión:Pendiente positiva
Como PPT > 1 atm
Sublima
CO2 (s):hielo seco
Utilidad: efectosde humo y nieblaTemperatura
Pre
sión
Líquido
Vapor
Sólido

37
Diagrama de fases del CO2 Diagrama de fases del agua

38
∆=∆
d P Hd T T V
H∆ > 0
V∆ > 0
Curva de pendiente positiva
dPdT
sólido → líquidoH∆ > 0
V∆ > 0
En general,curva de pendiente positiva
dPdT
Excepciones: H2O, Ga, Bi ∆V < 0 ⇒ curva de pendiente negativa
sólido → gasH∆ > 0
V∆ > 0
Curva de pendiente positiva
dPdT
Ecuación de Clapeyron
líquido → gas

39
Sublimación: Aplicaciones
Liofilización: deshidratación a baja presión
1) Congelar café molido2) Disminuir la presión3) El agua sólida pasa a agua gas, que se elimina.
Ventajas:* Evita secado por calentamiento (destruiría moléculas del sabor)* Evita que se estropee (en ausencia de agua no crecen bacterias)

40
ESTADO LÍQUIDO
PROBLEMAS

41
Construir un diagrama de fases para el xenon, a partir de la siguiente información: temperatura normal de ebullición, -108 ºC, temperatura normal de fusión,´-112 ºC, punto triple, -121 ºC (a una presión de 0,37 atm).
a) ¿En qué fase se encuentra el xenon a temperatura ambiente y a 1,0 atmde presión?.
b) Si la presión ejercida sobre una muestra de xenon es de 0,75 atm, y la temperatura es de -114 ºC. ¿en qué estado está el xenon?.
c) Si mides una presión de vapor de una muestra de xenon líquido y encuentras un valor de 0,5 atm. ¿cuál es la temperatura de la fase líquida?.
d) ¿Cuál es la presión de vapor del sólido a -122 ºC.
e) ¿Cua´es la fase mas densa, la sólida o la líquida?
5.38

42
Los puntos de ebullición y fusión normales del oxígeno son –183 ºC y –218 ºC respectivamente. Su punto triple está a –219 ºC y 1.14 torr y su punto crítico está a –119 ºC y 49.8 atm.
a) Dibuje el diagrama de fases mostrando los cuatro puntos correspondientes a cada fase.
b) Indique con flechas y nombres, los cambios de estado entre ellos.c) Al calentar oxígeno sólido, ¿sublima o funde a 1 atm de presión?.d) A temperaturas de –100 ºC puede licuarse aplicando una presión
adecuada.e) Sublima a una presión superior a 1.14 torr.f) Puede hervir a una presión superior a 1.14 torr, en el intervalo –218 ºC
a –100 ºC.g) La densidad del O2 (l) es mayor, igual o menor que la del O2 (s).

1
BLOQUE 5
GASES

2
GAS LÍQUIDO SÓLIDO

3
• Adoptan el volumen y la forma del recipiente que los contiene.
• Se consideran los más compresibles de los estados de la materia.
• Cuando se encuentran confinados en el mismo recipiente se mezclan completa y uniformemente.
• Cuentan con densidades mucho menores que los sólidos y líquidos.
Características físicas de los gases

4
Un gas queda definido por cuatro variables:
� Cantidad de sustancia
� Volumen
� Presión
� Temperatura
� moles
� L, m3, …
� atm, mm Hg o torr, Pa, bar
� ºC, K
Unidades:
� 1 atm = 760 mm Hg = 760 torr = 1,01325 bar = 101.325 Pa
� K = ºC + 273
� 1L = 1dm3
Medidas en gases

5
Unidades de presión
1 pascal (Pa) = 1 N/m2
1 atm = 760 mmHg = 760 torr
1 atm = 101,325 PaBarómetro
Presión = FuerzaÁrea
Presiónatmosférica

6
V α 1/P (a n y T ctes) P x V = constante
P1 x V1 = P2 x V2
Ley de Boyle
El volumen de una cantidad fija de un gas mantenido a temperatura constante es inversamente proporcional a la presión del gas.

7
V α T (a n y P ctes) V = constante x T
V1/T1 = V2/T2
Ley de Charles y de Gay-Lussac
El volumen de una cantidad fija de un gas mantenido a presión constantees directamente proporcional a la temperatura absoluta del gas del gas.

8
Ley de Avogadro
V α número de moles (a T y P ctes) V = constante x n
V1/n1 = V2/n2
A presión y temperatura constante, el volumen es directamente proporcional al número de moles del gas presente.

9
La ecuación del gas ideal
Ley de Charles: V α T (a n y P constantes)
Ley de Avogadro: V α n (a P y T constantes)
V αnT
P
V = constante x = RnT
P
nT
PR : constantede los gases
P·V = n·R·T
V α 1/P (a n y T ctes)Ley de Boyle:

10
Ecuación de estado ( p = f (V,T,n)Ley del Gas Ideal (gas perfecto)

11
TEORIA CINÉTICO-MOLECULAR DE LOS GASES
• Relaciona las propiedades de los gases (P,V,T) con las propiedades de las moléculas de los gases (masa y velocidad).
• La teoría cinético-molecular explica la ecuación de estado de los gases ideales.
Modelo molecular:
• Un gas está formado por un gran número de partículas muy pequeñas llamadas moléculas. El volumen total ocupado por las moléculas es despreciable en comparación con el del recipiente que las contienen por lo que se pueden considerar como masas puntiformes.
• Maxwell, Clausius y Boltzmann desarrollaron esta teoría, basada en la idea de que todos los gases se comportan de forma similar en cuanto al movimiento de partículas se refiere.

12
• Las moléculas de un gas no ejercen entre sí ni fuerzas de atracción ni de repulsión.
• Las moléculas que constituyen el gas están en continúo movimiento.
• La energía cinética media de las moléculas de un gas es proporcional a la temperatura absoluta del gas.
• Dos gases cualesquiera a la misma temperatura tienen la misma energía cinética.
Se deduce:
• La presión que ejerce un gas se produce por las colisiones de las moléculas con las paredes del recipiente que lo contiene.
• Las moléculas de un gas chocan entre si y con las paredes del recipiente que lo contiene. Estos choque se suponen elásticos, es decir, las partículas no ganan ni pierden energía cinética en ellos.

13
Representación gráfica de los postulados de la teoría cinética de los gases. Las moléculas que constituyen el gas están en continúo movimiento y sus colisiones son elásticas; al producirse contra las paredes originan la presión del gas.

14
La teoría cinético-molecular se basa en el movimiento independiente de las moléculas del gas.
V
cNmP
3
2
=
Aplicando las leyes de la física al modelo molecular, se deduce que la presión que ejerce un gas en un recipiente de volumen V viene dada por:
N = Número de moléculas
m = masa de la nolécula
moléculaslastodasdesvelocidadelasdecuadradoslosdemediac =2

15
nRTPV =
3
2cNmnRT=
N
nRTcm
32=
2
2cmEc =
N
nRTEc 2
3=
n
NNA = )( AvogadrodeNúmeroNA =
AN
RTmoléculaporEc
2
3=
V
cNmP
3
2
=
La energía cinética de una molécula gaseosa viene dada por:

16
1231038.1 −−×=== JKBoltzamndectekN
R
A
kTN
RTmoléculaporEc
A 2
3
2
3==
)/31.8(2
3
2
3
2
3molKJRRTTN
N
RkTNmolporEc A
AA ====

17
kTcm
Ec 2
3
2
2
==
M
RT
mN
RT
m
kTc
A
3332===
)/(2 smeficazvelocidadc =)/( molKggasdelmolarmasaM =
• La velocidad eficaz es directamente proporcional a la temperatura absoluta.
• La velocidad eficaz es inversamente proporcional a la masa molar.

18
• La velocidad eficaz es directamente proporcional a la temperatura absoluta.
Distribución de velocidades de las moléculas de N2 a tres temperaturas diferentes
( )( ) 1
2
12
22
T
T
c
c=
Para un mismo gas, a dos temperaturas distintas, se verifica:

19
• La velocidad eficaz es inversamente proporcional a la masa molar.
( )( ) A
B
B
A
M
M
c
c=
2
2
Para dos gases A y B que se encuentran a la misma temperatura:
Distribución de velocidades de tres gases a la misma temperatura

20
LEY DE GRAHAM
Las velocidades eficaces de dos gases, que se encuentran a la misma temperatura, son inversamente proporcionales a las raíces cuadradas de sus pesos moleculares.
( )( ) A
B
B
A
M
M
c
c==
2
2
Aplicación de la Ley de Graham:
• Efusión• Difusión

21
Efusión
• La efusión es el proceso mediante el cual un gas bajo presión escapa de un compartimento a otro pasando a través de una pequeña abertura (la velocidad de paso a través de la abertura es la velocidad de efusión).
Orificio pequeño
Gas Vacío
Ley d e Graham de la efusión:
( )( ) A
B
B
A
efusión
efusión
M
M
c
c
BV
AV==
2
2
)(
)(

22
La efusión también se define como el flujo de moléculas de un gas a través de orificios estrechos o poros.
t
VQ
tiempo
VolumenCaudal =⇔=
)(
)(
)(
)(
)(
)(
BV
AV
t
BVt
AV
BQ
AQ
efusión
efusión
B
A==
La velocidad de efusión es inversamente proporcional al tiempo que tarda un volumen de gas en pasar a través de un orificio.
B
A
efusión
efusión
B
A
M
M
AV
BV
t
t==
)(
)(

23
vefusion (CH4) = 1,9· vefusion (hidrocarburo)
Masa molar del CH4 = 16 g/mol
Butano (C4H10): Masa molar =12·4 + 10·1 = 58 g/mol
El metano (CH4) efunde 1,9 veces más rápido que cierto hidrocarburo. Hallar la masa molar del hidrocarburo y decir de quien se puede tratar
( ) molgmolgM
M
M
CHV
rohidrocarbuV
rohidrocarbu
rohidrocarbu
CH
efusión
efusión
/8.57/169.1
9.1)(
)(
2
4
4
=×=
==

24
Difusión
• La difusión es el proceso mediante el cual las moléculas de un gas se mezclan con las moléculas de otro u otros gases.
• Si en un recipiente introducimos dos gases, la mezcla de ambos gases no será instantánea ya que las velocidades a la que fluyen ambos gases no es la misma.
NH317 g/mol
HCl36 g/mol
NH4Cl
1
2
2
1
M
M
v
v
d
d=
,
,
Ley de Graham de la difusión:relaciona la velocidad de difusión para dos gases puros.

25
Modelo Molecular para la Ley de Boyle.
El aumento de presión exterior origina una disminución del volumen,
que supone el aumento de choques de las partículas con las paredes
del recipiente, aumentando así la presión del gas.
Teoría cinética de los gases
V α 1/P (a n y T ctes)

26
Modelo Molecular para la Ley Gay-Lussac
Al aumentar la temperatura aumenta la velocidad media de las partículas, y con ello el número de choques con las paredes. Eso provoca un aumento de la presión interior que desplaza el émbolo hasta que se iguala con la presión exterior, lo que supone un aumento del volumen del gas.
Teoría cinética de los gases
V α T (a n y P ctes)

27
Modelo Molecular para la Ley de Avogadro
La adición de más partículas provoca un aumento de los choques contra las paredes, lo que conduce a un aumento de presión, que desplaza el émbolo hasta que se iguala con la presión externa. El proceso global supone un aumento del volumen del gas.
Teoría cinética de los gases
V α número de moles (a T y P ctes)

28
GASES REALES
• La ecuación de estado de los gases ideales, PV=nRT, solo es valida cuando los gases se encuentran a presiones bajas (no muy superiores a 1 atmósfera) y temperaturas elevadas (muy superiores a sus puntos de congelación).
• Los gases se desvían del comportamiento ideal ya que las moléculasreales tienen un volumen finito y porque se atraen entre sí. El efecto de estos dos factores depende de la temperatura y presión del gas.
• Al aumentar la presión de un gas y/o disminuir su temperatura la conducta de un gas se desvía progresivamente del comportamiento que predice la ecuación de los gases ideales.

29
realPVZ
RT=
Si el gas real se comporta idealmente:
Desviación del comportamiento ideal
Para medir la desviación que un gas real puede presentar respecto a un gas ideal, se define el factor de compresibilidad, Z
real
ideal
VZ
V=
1=⇒= ZVV idealreal

30
T cte
Comportamiento de un mol de diferentes gases a la misma temperatura. La desviación del comportamiento ideal se acentúa al aumentar la presión.

31
Mismo gas
Al aumentar la temperatura, el comportamiento se acerca al de una gas ideal.

32
• Los gases reales se desvían del comportamiento ideal porque las moléculas reales ocupan un determinado volumen e interaccionan entre sí.
Ecuación de van der Waals
• Se han propuesto varias ecuaciones de estado para los gases reales.
• van der Waals modificó la ley de los gases ideales para tener en cuenta los dos factores anteriores.
( ) nRTnbVV
anp =−
+
2
2

33
Efecto de las fuerzas intermoleculares en la presión ejercida por un gas
Pared del recipiente
Partícula frenada
En un gas real, las partículas se atraen y se repelen.
El resultado de estas interacciones es una presión menor a la presión que predice la ecuación del gas ideal.
ideal realP P>
ideal realP P p= +
Efecto del volumen ocupado por las moléculas del gas
• El volumen real para el movimiento molecular es menor que el volumen del recipiente
Vreal = V – Vexcluido

34
2
2
V
anpp realideal +=
• Donde a (interacción entre moléculas) es una constante y n y Vson el número de moles y el volumen del recipiente.
• El volumen real para el movimiento molecular es menor al volumen del recipiente: Vef= V – nb
( ) nRTnbVV
anp =−
+
2
2
Corrige las fuerzas de interacción molecular
La constante b está relacionada con el volumen ocupado por las moléculas. Sus unidades son (L / mol).

35

36
Isotermas para los gases reales. Licuefacción
líquido + vapor
líquido
gas
vapor
Vm / L mol -1
p/ a
tm
Licuefacción del N 2

37
GASES
PROBLEMAS

38
¿Cuántos mL de CO2 saldrían por un capilar por el que salen 47,2 mL de O2 en las mismas condiciones y en el mismo tiempo?
5.29
En una botella de helio a presión hay un pequeño escape por donde fluye a razón de 3,4 milimoles/hora. ¿Qué tiempo tardaría en fluir por el mismo orificio y bajo la misma presión 10 milimoles de CO?
5.8
La efusión de 10 cm3 de H2 a través de una membrana porosa tarda 26 segundos. Si la misma cantidad de otro gas tarda 130 segundos en las mismas condiciones de presión y temperatura. ¿Cuál es el peso molecular de este gas?
5.60

39
Dos recipientes contienen nitrógeno. Uno de los recipientes se calienta a 125 ºC y el otro permanece a 25 ºC. Si cada uno de los recipientes tieneun orificio de dimensiones similares. ¿Cuántas veces será mas rápida la efusión del nitrógeno, en el recipiente caliente o en el frío?
16 Kg de O2 están contenidos en una botella cuya capacidad es de 20,0 L. Sabiendo que la presión máxima que pueden soportar las paredes de la botella es de 150 atm, calcular la temperatura máxima a la que se puede calentar la botella, utilizando:
a) La aproximación de los gases perfectos
b) La aproximación de Van der Waals.
Datos: a = 1,360 atm·L2·mol-2 b = 0,03183 L·mol-1
5.40

DETERMINACIÓN DEL GRADO ALCOHÓLICO DE UN VINO
PRACTICA 6

2
DESTILACION• Operación física basa en la diferencia de los puntos de ebullición de los
componentes de la mezcla.
• Se utiliza para separar un líquido de sus componentes no volátiles o bien para separar los componentes líquidos de la muestra.
• La destilación como proceso consta de dos fases:• El líquido se calienta hasta su punto de ebullición, en un matraz de destilación produciéndose su paso a la fase de vapor.
• El vapor se condensa en contacto con un refrigerante o condensador, formando el destilado que se en un matraz distinto al de destilación, el matraz colector.

Métodos de destilación:• Destilación simple o sencilla• Destilación fraccionada• Destilación a vacío
DESTILACIÓN SIMPLE O SENCILLA
Se utiliza, generalmente, cuando la muestra tiene un único componente volátil que se desea separar. También permite la separación de dos componentes líquidos de la mezcla cuando la diferencia en sus puntos de ebullición es mayor de 30 ºC, o cuando un líquido es casi puro.
El proceso se lleva acabo en una sola etapa, es decir que se evapora el líquido de punto de ebullición mas bajo y se condensa por medio de un refrigerante (Ver figura).

DESTILACIÓN SIMPLE O SENCILLAFundamentalmente, consiste en calentar la mezcla en un matraz de destilación hasta que su presión de vapor iguale a la presión exterior, hierva. El vapor sale por la tubuladura lateral hasta que el condensador o refrigerante donde se condensan los vapores producidos, cayendo por gravedad al matraz colector.Para evitar un sobrecalentamiento del líquido y una ebullición tumultuosa, se introduce en el matraz de destilación algunos trocitos de plato poroso.La correcta posición del termómetro es la que deja a la misma altura el extremo superior del depósito de mercurio y la parte inferior de la tubuladura lateral que comunica con el refrigerante.

Plato Poroso
El bulbo del termómetro debe estar a la altura de la tubuladura lateral.
El agua de refrigeración debe circular en sentido contrario al vapor.
DESTILACIÓN SIMPLE O SENCILLA

DESTILACIÓN FRACCIONADAPermite la separación de los componentes de una muestra cuando la diferencia en sus puntos de ebullición es inferior a 30 ºC (puntos de ebullición muy próximos).
El proceso tiene lugar en múltiples etapas por medio de una columna de destilación o fraccionamiento en la cual se llevan acabo numerosas evaporaciones y condensaciones.
En la destilación fraccionada la mezcla se calienta y el vapor ascendente pasa a través de la columna de fraccionamiento. La columna de fraccionamiento puede estar llena de bolitas de vidrio o tener diversas geometrías con el fin de proporcionar una gran superficie para el intercambio de calor entre el vapor que asciende y el condensado que desciende, de manera que se efectúe un número mas o menos elevado de vaporizaciones y condensaciones a lo largo de la columna. El vapor se condensa sobre las bolitas y luego pasa nuevamente al estado vapor.
Al ir avanzando a lo largo de la columna (hay un gradiente de temperatura), la composición del vapor es mas rica en el componente mas volátil y la concentración del líquido que condensa es mas rica en el componente menos volátil.

7
DESTILACIÓN FRACCIONADA

destilación fraccionada

DESTILACIÓN A VACIO
Este método se utiliza cuando se necesita separa compuestos que tengan un punto de ebullición elevado (> 200 ºC) y que al calentar a temperaturas elevadas se puedan descomponer antes de llegar a hervir.
La destilación al vacío consiste en generar un vacío parcial por dentro del sistema de destilación para destilar sustancias por debajo de su punto de ebullición normal. Este tipo de destilación se utiliza para purificar sustancias inestables.Lo importante en esta destilación es que al crear un vacío en el sistema se puede reducir el punto de ebullición de la sustancia casi a la mitad.
Adaptador para destilación a vacío

DESTILACIÓN A VACIO

Un VINO es una mezcla muy compleja; contiene agua, etanol, azúcares, ácidos orgánicos, pigmentos (que le dan color) y otros ingredientes. Los componentes volátiles que se encuentran en cantidad considerable son precisamente el agua y el etanol, cuyos puntos de ebullición son, respectivamente, 100,0 °C y 78,3 °C. Ambos forman un azeótropo que hierve a 78,2 °C y cuya composición es 96 % de masa de etanol y 4% en masa de agua. En el vino, el contenido en alcohol se expresa en porcentaje de volumen y es algo mayor del 10 %. En la destilación de vino no se puede obtener ninguna fracción que contenga alcohol al 100 %, debido a que el “componente” más volátil es precisamente el azeótropo.
PRÁCTICA 6

PRÁCTICA 6
La mezcla etanol-agua forma un azeótropo de punto de ebullición mínimo que hierve a 78.2 ºC. El punto de ebullición del agua y del etanol son 100 ºC y 78.3 ºC.Respectivamente.La composición del azeótropo es del 96% en masa de etanol y del 4% en masa de agua. En el punto azeotrópico la fase líquida y vapor tienen la misma composición y por tanto tienen la misma temperatura de ebullición, por lo que no es posible separar los dos componentes por destilación.En la destilación no se puede obtener ninguna fracción que contenga alcohol al 100% debido a que el componente mas volátil es el azeótropo . Obtenemos en el destilado una mezcla hidroalcohólica denominada mezcla azeotrópica.
DETERMINACIÓN DEL GRADO ALCOHÓLICO DE UN VINO COMERCIAL

GRADO ALCOHOLICO:Porcentaje en volumen de alcohol presente en una muestra.• Se determina midiendo la densidad.• El vino es una mezcla compleja: agua, etanol, azucares, ácidos, pigmentos, etc. Como consecuencia de ello, no se puede medir directamente el grado alcohólico midiendo su densidad.• Es necesario someter al vino a un proceso de destilación.
OBJETIVO DE LA PRÁCTICA:Destilar todo el alcohol contenido en una muestra de vino comercial para determinar su grado alcohólico, para ello utilizaremos una destilación sencilla en una sola etapa.
PRÁCTICA 6“La finalidad de la práctica no es conseguir la mezcla ezeotrópica, para ello tendríamos que realizar una destilación fraccionada.”

DESTILACIÓN SENCILLA EN UNA SOLA ETAPA
• Se destila la mitad del volumen inicialmente utilizado (100 mL). (Nos aseguramos que todo el alcohol contenido en el vino ha pasado al matraz colector)• En el destilado se obtiene todo el alcohol contenido en el vino y agua.• Añadimos agua al destilado hasta completar los 100 mL para que el alcohol se encuentre disuelto en el mismo volumen que en la muestra de vino inicial.• Se sumerge un alcohómetro en esta disolución y se lee directamente el grado alcohólico del vino.
PRÁCTICA 6

1
ESTADO SÓLIDO CRISTALINO
BLOQUE 5

2
Un sólido cristalino está constituido por el apilamiento de bloques idénticos que constituyen el sólido.
SÓLIDOS CRISTALINOS
Los átomos, iones o moléculas (elementos constituyentes) ocupan posiciones específicas, es decir tienen una ordenación regular en el espacio.
Tipos de sólidos cristalinos : Sólidos iónicos, sólidos covalentes, sólidos metálicos, sólidos moleculares.

3
SÓLIDOS CRISTALINOS
• La unidad básica de un sólido cristalino se llama celda unitaria .
• La unidad de volumen mas pequeña de un cristal que reproduce por repetición la red cristalina.
• Mínima unidad que da toda la información acerca de la estructura del sólido.
Los puntos reticulares representan:• Átomos• Moléculas• Iones

4
Celda unidad
Translación eje y
Translación eje X
Translación eje Z
La estructura del sólido cristalino se representa mediante la repetición de la celda unidad en las tres direcciones del espacio
SÓLIDOS CRISTALINOS

5
Sistemascúbico a = b = c α = β = γ =90ºtetragonal a = b ≠ c α = β = γ =90ºortorrómbicoa ≠ b ≠ c α = β = γ =90ºmonoclínicoa ≠ b ≠ c α = γ =90ºβ≠90ºtriclínico a ≠ b ≠ c α ≠ β ≠ γ ≠90ºhexagonal a = b ≠c α = β =90º γ =120ºromboédricoa = b = c α=β= γ ≠90º
TIPOS DE CELDAS UNITARIAS
En función de los parámetros de la celda unitaria, es decir, de las longitudes de los lados, o ejes, del paralelepípedo elemental y de los ángulos que forman, se distinguen siete sistemas cristalinos

6
Los siete tipos de celdas unitarias
Cúbica simple Tetragonal Ortorrómbica Romboédrica
Monoclínica Triclínica Hexagonal

7
REDES DE BRAVAIS
En función de las posibles localizaciones de los átomos en la celda unitaria se establecen 14 estructuras básicas, denominadas Redes de Bravais.

8
CELDA UNITARIA CÚBICA
• Cada punto reticular está ocupado por un átomo (una esfera).• Todos los lados y todos los ángulos de la celda son iguales.• Según como estén ubicados los átomos en la celda:
• Celda cúbica simple• Celda cúbica centrada en el cuerpo• Celda cúbica centrada en las caras

9
Celda unitaria cúbica
Cúbica simple Cúbica centrada en el cuerpo Cúbica centrada en las caras

10
Cada celda unitaria en un sólido cristalino es adyacente a otras celdas unitarias la mayoría de los átomos son compartidos con las celdas unitarias vecinas.- Cada átomo en un vértice es compartido por ocho celdas unitarias.- Un átomo centrado en las caras es compartido por dos celdas unitarias
Compartido por 8 celdas unitaria
Compartido por 2 celdas unitaria
Un átomo del vértice y unátomo centrado en las caras

11
1 átomo/celda unitaria
(8 x 1/8 = 1)
2 átomos/celda unitaria
(8 x 1/8 + 1 = 2)
4 átomos/celda unitaria
(8 x 1/8 + 6 x 1/2 = 4)
Cúbica simple Cúbica centrada en el cuerpo Cúbica centrada en las caras

12
INDICE O NÚMERO DE COORDINACIÓN:
Número de átomos (esferas) que rodean a un átomo (esfera) en una red cristalina.
Cúbica simple:I.C = 6
Cúbica centrada en el cuerpo:I.C = 8
Ejemplos:

13
EFICACIA DE EMPAQUETAMIENTO: porcentaje de espacio de la celda ocupado por las esferas.
100×=celdaladevolumen
celdaladedentroesferaslasdeVolumenientoempaquetamdeEficacia
3
3
23
4
3
4
== aresferaladeVolumen π
3aunitariaceldaladeVolumen =
%5210023
4
3
3
=×
=
a
a
ientoempaquetamdeEficacia
π
Celda cúbica simple

14
Nº de coordinación:6
Átomos por celda: 8 vértices*1/8 =1
Relación entre la longitud de arista y el
radio del átomo: 2r = a
Eficacia del empaquetamiento: 52%
( ) ( )52.0
6)r2(
r34
a
r34
V
V3
3
3
3
celda
ocupado====πππ
Celda cúbica simple

15
Celda cúbica centrada en el cuerpo
Nº de coordinación:8
Átomos por celda: 8 aristas*1/8 + 1centro =2
Relación entre la longitud de arista y el radio del átomo:
4
a 3r =
Eficacia del empaquetamiento: 68%
( ) ( )68.0
83
)3
r4(
r342
a
r342V
V3
3
3
3
celda
ocupado=
π=
π=
π=

16
Celda cúbica centrada en las caras
Nº de coordinación:12
Átomos por celda: 8 aristas*1/8 + 6caras*1/2=4
Relación entre la longitud de arista y el
radio del átomo: (4r)2=a2+a2
Eficacia del empaquetamiento: 74%
( ) ( )74.0
2
r4r34
a
r344V
V
2/1
3
3
3
celda
ocupado=
π=
π⋅=

17
Relación entre la longitud de la arista y el radiode los átomos en tres celdas unitarias diferentes

18
PROBLEMAS
ESTADO SÓLIDO CRISTALINO

19
El plomo metal cristaliza en una red cúbica centrada en las caras. Si el radio atómico del plomo es de 175,0 pm. Calcular:
a) Longitud de la arista de la celdilla unidad.
b) Volumen de la celdilla unidad
c) Porcentaje de ocupación de la celdilla unitaria.
d) Densidad del metal.
Datos: Pb = 207,19
4.50

20
3aunitariaceldaladeVolumen =
4 átomos/celda unitaria
(8 x 1/8 + 6 x 1/2 = 4)
( )a
r344
V
V3
3
celda
ocupado π⋅=
d = mV
m = 4 átomos Pb207,19 g
mol Pbx
1 mol Pb
6,022 x 1023 átomosx = 1,37· 10-21g
d = mV
1,37· 10-21g
1,21· 10-22 cm3= = 11,32 g/cm3

21
El wolframio cristaliza con una estructura cúbica centrada en el cuerpo. Su densidad es 19.3 g/cm3. ¿Cuál será el radio atómico del wolframio?Dato: W = 183.85
4.25

22
El radio metálico del cobre es 128 pm, y cristaliza con una estructura cúbica centrada en las caras. Calcular la densidad del metal.Dato: Cu=63.54
4.48
a
4r

23
El polonio es un elemento que cristaliza en el sistema cúbico y su celda unidad es simple. Sabiendo que la densidad del polonio es 9.4 g·cm-3, calcular su radio atómico suponiendo que en la red cristalina los átomos contiguos son tangentes y que los átomos son esféricos.Dato: Po=210
4.47
a
r

24
Cuando la plata cristaliza, forma celdas cúbicas centradas en las caras. La longitud de la arista de la celda unitaria es de 409 pm. Calcule la densidad de la plata.
d = mV
V = a3 = (409 pm)3 = 6,83 x 10-23 cm3
4 átomos/celda unitaria
m = 4 átomos Ag107,9 g
mol Agx
1 mol Ag
6,022 x 1023 átomosx = 7,17 x 10-22 g
d = mV
7,17 x 10-22 g
6,83 x 10-23 cm3= = 10,5 g/cm3
a
4r







