Download - Demencias Trabajo Final

DEMENCIAS
Dr. Oscar Heredia.Cátedra de Neurología clínica.
Docente FMH-UNPRG.2013.

DEMENCIA
Síndrome progresivo de deterioro global de lasfunciones intelectuales (memoria y al menos otra comolenguaje, gnosias, praxias o función ejecutiva) conpreservación del nivel de vigilancia, que interfiere en elrendimiento laboral o social del individuo y le haceperder su autonomía personal.
El concepto de demencia exige que la causa sea unalesión o disfunción orgánica cerebral para excluir losdefectos cognitivos debidos a otros trastornospsicológicos.

DECLIVE COGNITIVO DEL ENVEJECIMIENTO Y
DETERIORO COGNITIVO LEVE.
Con el envejecimiento se produce un declive de las funcionescognitivas como de otras funciones motoras y sensoriales del sistemanervioso, pero hay una resistencia en aceptar que el envejecimiento porsí solo sea capaz de producir demencia.
� En biopsias cerebrales de algunos ancianos centenarios no se encuentra unaumento proporcional de las lesiones atribuidas al envejecimiento.
Afecta a la memoria, al lenguaje, habilidades visuoespaciales y funciones ejecutivas.
DETERIORO COGNITIVO LEVE (MCI): Es el estado incierto, zona depenumbra entre el estado normal y la demencia.
El MCI es un factor de riesgo para le desarrollo de la demencia (12% progresade MCI a demencia en un año), sin embargo no todos los pacientes con MCIevolucionan con demencia. Mejoran aquellos pacientes cuyas causas eranreversibles: Depresión, alteraciones metabólicas o efectos secundarios defármacos.
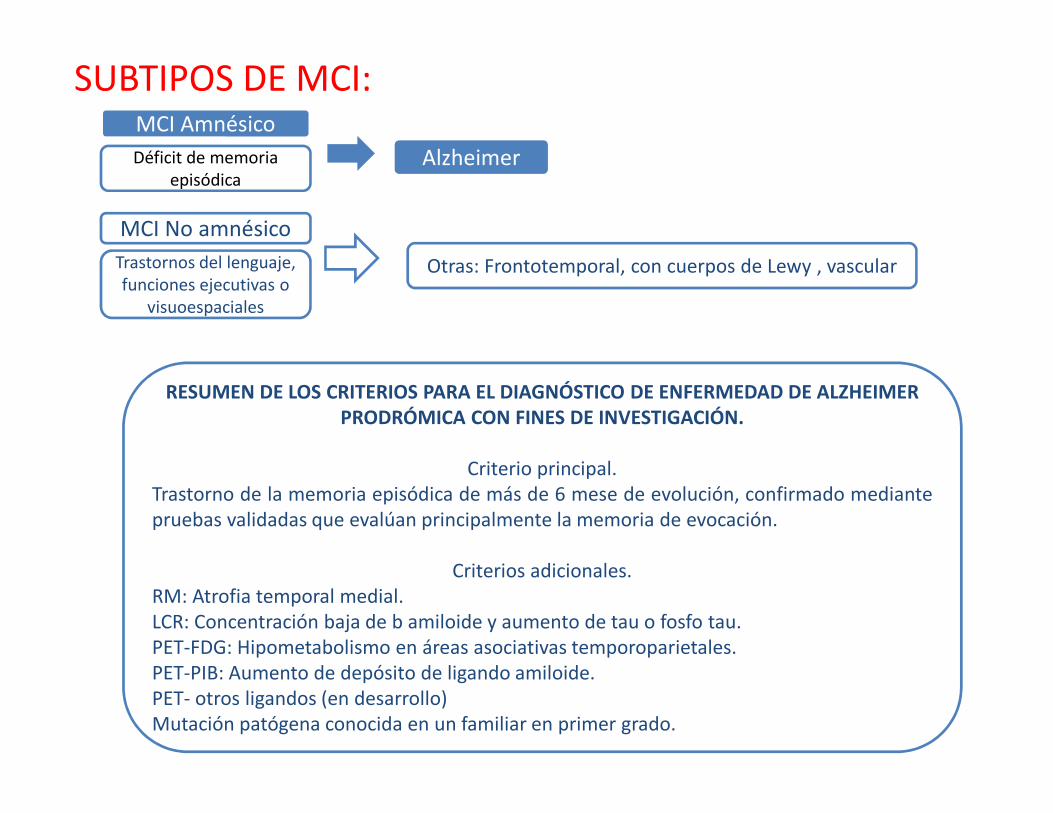
SUBTIPOS DE MCI: MCI Amnésico
AlzheimerDéficit de memoria episódica
MCI No amnésico
Otras: Frontotemporal, con cuerpos de Lewy , vascularTrastornos del lenguaje, funciones ejecutivas o
visuoespaciales
RESUMEN DE LOS CRITERIOS PARA EL DIAGNÓSTICO DE ENFERMEDAD DE ALZHEIMER PRODRÓMICA CON FINES DE INVESTIGACIÓN.
Criterio principal.Trastorno de la memoria episódica de más de 6 mese de evolución, confirmado mediantepruebas validadas que evalúan principalmente la memoria de evocación.
Criterios adicionales.RM: Atrofia temporal medial.LCR: Concentración baja de b amiloide y aumento de tau o fosfo tau.PET-FDG: Hipometabolismo en áreas asociativas temporoparietales.PET-PIB: Aumento de depósito de ligando amiloide.PET- otros ligandos (en desarrollo)Mutación patógena conocida en un familiar en primer grado.

EPIDEMIOLOGÍA
• Incidencia y prevalencia de las demencias se incrementa con la edad.
• Menos del 1% de las personas menores de 50 años tiene demencia, pero afecta a un 5% de las personas mayores de 65.– Entre 65-69 a: 5% demencia.– Entre 85-89a: 22% demencia.– Entre 85-99a: 35% demencia.
• Para el año 2050 la prevalencia de personas con demencia será al menos tres veces mayor que la actual.

ETIOLOGÍA
o Todas las agresiones del cerebro: degenerativa, tóxica, metabólica, traumática, infecciosa,tumoral o vascular pueden producir demencia.
o Las causas más frecuentes (80-90%) son: Degenerativa, vascular y degenerativa-vascular.
o En niños y jóvenes las principales causas son metabolopatías (leucodistrofias,neurolipoidosis, aminoacidurias).
o Los diagnósticos clínicos categóricos no se ajustan a la realidad neuropatológica:
� Un buen porcentaje de pacientes diagnosticados con Alzheimer tuvieron otrapatología concomitante demostrada por neuropatología: Demencia de etiologíamixta. La patología vascular está siempre como un cofactor cuando las lesionestipo Alzheimer son menores.
o Un porcentaje elevado de la población, (en especial los ancianos con deficiencias nutritivas)tienen niveles bajos de vitaminas (B12, ácido fólico o vitamina D), y también hipotiroidismosclínicos o subclínicos. La corrección de esos parámetros puede mejorar el estado general delos pacientes, pero no tienen influencia sobre su estado cognitivo.

Enfermedades degenerativas.� Enfermedad de Alzheimer.*�Enfermedad de Huntintong.�Enfermedad de los cuerpos de Lewy difusos.� Enfermedad de Parkinson idiopática.� Demencia “de tipo frontal” (taupatías y otras).� Complejo de la isla de Guam (parkinsonimos-demencia-esclerosis lateral amiotrófica).�Asociada a enfermedades (Wilson, Hellervorden-Spatz, ELA, degeneración espinocerebelosa).�Enfermedad de Creutzfeldt-Jakob y otras prionpatías.
Enfermedades vasculares� Infartos múltiples corticosubcorticales.*�Leucoencefalopatía subcortical (Binswanger).*�Arteritis (de células gigantes y otras).� Malformaciones arteriovenosas y otras.� Infartos selectivos bilaterales (tálamo, cerebral anterior).�Vasculitis (LES).�Estados de hipoperfusión por obstrucciones bicarotídeas.
Trastornos metabólicos adquiridos y tóxicos.*� Fármacos.�Anoxia.� Hipo e hipertiroidismo.� Hipoparatiroidismo primario o secundario.� Panhipopituitarismo.�Uremia y demencia dialítica.�Degeneración hepatocerebral adquirida.� Carencia de B1, B12, ácido fólico, pelagra.�Alcoholismo y otras drogas.

Tumores.� Gliomas o linfomas del cuerpo calloso.� Meningiomas frontales.� Gliomatosis celebri.� Linfoma endovascular.�Síndromes paraneoplásicos (“encefalitis límbica”).
Traumatismo �Hematoma subdural crónico.� Secuela de contusiones cerebrales múltiples.� Demencia pugilística.
Infecciones y encefalopatías inflamatorias (microencefalitis crónica).� Bacterias (Brucella, Listeria, sífilis o micobaterias).� Hongos (criptococos).� Virus de acción lenta (panencefalisits esclerosante subaguda, leucoencefalopatía multifocal progresiva, VIH)� Encefalitis autoinmune.�Síndrome de Sjogren y otros estados disinmunes.� Enfermedad de Whipple.
Enfermedades desmielinizantes.�Esclerosis múltiple y sus variedades.� Leucodistrofias.
Enfermedades de depósito y metabólicas congénitas. �Enfermedad de Lafora.�Enfermedades de los cuerpos poliglucosados (poliglucosan-body)� Enfermedades con cuerpos de inclusión intranucleares.� Lipoidosis.� Mucopolisacaridosis� Eminoacidurias.
Hidrocefalia (“a presíón normal”)*
EA: 60% de todas las demencias. La suma de los (*) = 95% del total
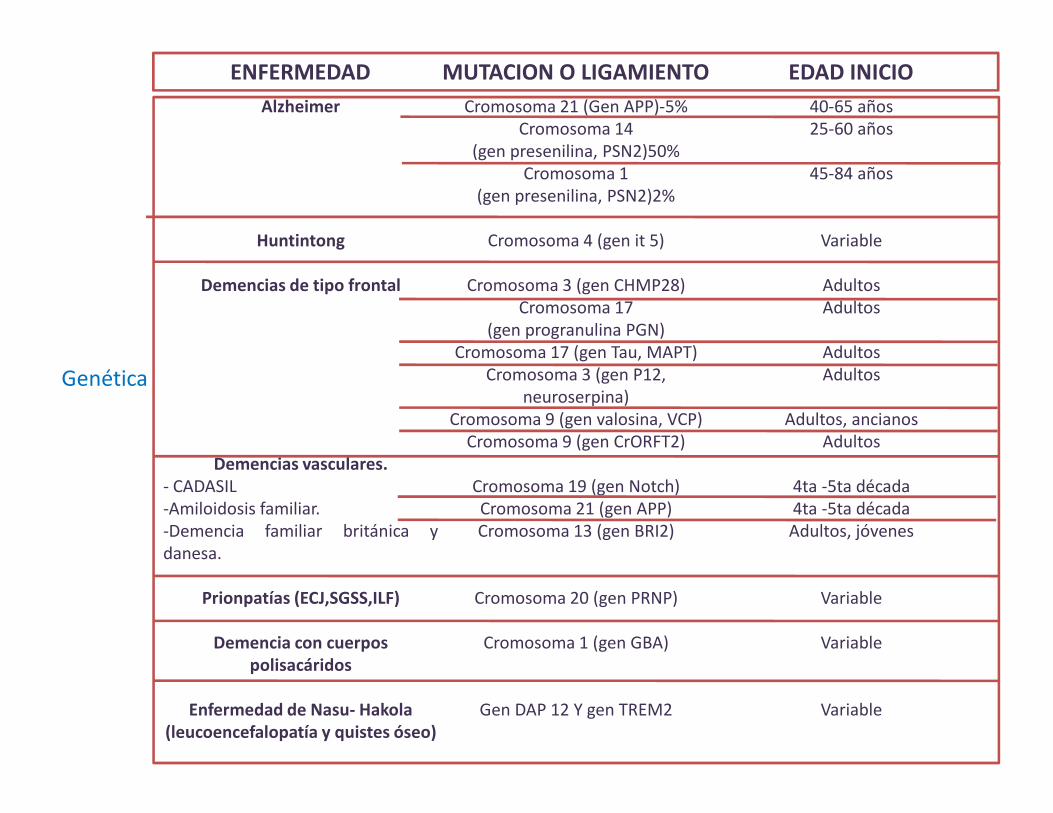
Genética
Alzheimer
Huntintong
Demencias de tipo frontal
Demencias vasculares.- CADASIL-Amiloidosis familiar.-Demencia familiar británica ydanesa.
Prionpatías (ECJ,SGSS,ILF)
Demencia con cuerpos polisacáridos
Enfermedad de Nasu- Hakola(leucoencefalopatía y quistes óseo)
Cromosoma 21 (Gen APP)-5%Cromosoma 14
(gen presenilina, PSN2)50%Cromosoma 1
(gen presenilina, PSN2)2%
Cromosoma 4 (gen it 5)
Cromosoma 3 (gen CHMP28)Cromosoma 17
(gen progranulina PGN)Cromosoma 17 (gen Tau, MAPT)
Cromosoma 3 (gen P12, neuroserpina)
Cromosoma 9 (gen valosina, VCP)Cromosoma 9 (gen CrORFT2)
Cromosoma 19 (gen Notch)Cromosoma 21 (gen APP)Cromosoma 13 (gen BRI2)
Cromosoma 20 (gen PRNP)
Cromosoma 1 (gen GBA)
Gen DAP 12 Y gen TREM2
40-65 años25-60 años
45-84 años
Variable
AdultosAdultos
AdultosAdultos
Adultos, ancianosAdultos
4ta -5ta década4ta -5ta décadaAdultos, jóvenes
Variable
Variable
Variable
ENFERMEDAD MUTACION O LIGAMIENTO EDAD INICIO

CUADRO CLÍNICO.
• Heterogéneo.
• Combinación de síntomas y signos: Deterioro cognitivo y afectivos, conductuales, de sueño y hasta alteraciones motoras (movimientos anormales, signos piramidales y extrapiramidales, ataxia, crisis epiléptica o mioclonías).
• Los trastornos no cognitivos (conducta) suelen aparecer en fases graves del deterioro cognitivo, pero pueden aparecer precozmente (demencias frontotemporales).
o Inicio con amnesia episódica y se sigue de otros defectos práxicos: EA (lesiones temporolímbicas).o Inicio con trastornos precoces de la personalidad y conducta: Demencias frontotemporales.
� Demencias corticales: EA� Demencias subcorticales: El resto de demencias.
• Rasgos depresivos: tristeza, llanto, angustia, deseos de muerte y de suicidio.• Rasgos de Apatía y aplanamiento de afectos• Rasgos psicóticos : obsesivos, suspicaces y paranoides, fabulan amenazas y deliran o tienen alucinaciones.
ETAPAS:� 1ERA:Síntomas de deterioro cognitivo y cambios de la personalidad y conducta. Mantiene su autonomía
personal y requieren poca supervisión familiar.� 2DA: Pierde progresivamente la capacidad de llevar a cabo tareas instrumentales de la vida diaria y su
autonomía personal, y requiere supervición y ayuda progresiva hasta ser constante.� 3ERA: Completamente dependiente para las actividades básicas de la vida diaria como comida e higiene.
Dificultades motoras para comer y tragar, andar y sostenerse de pie. Silla de ruedas, y finalmente confinado a la cama.
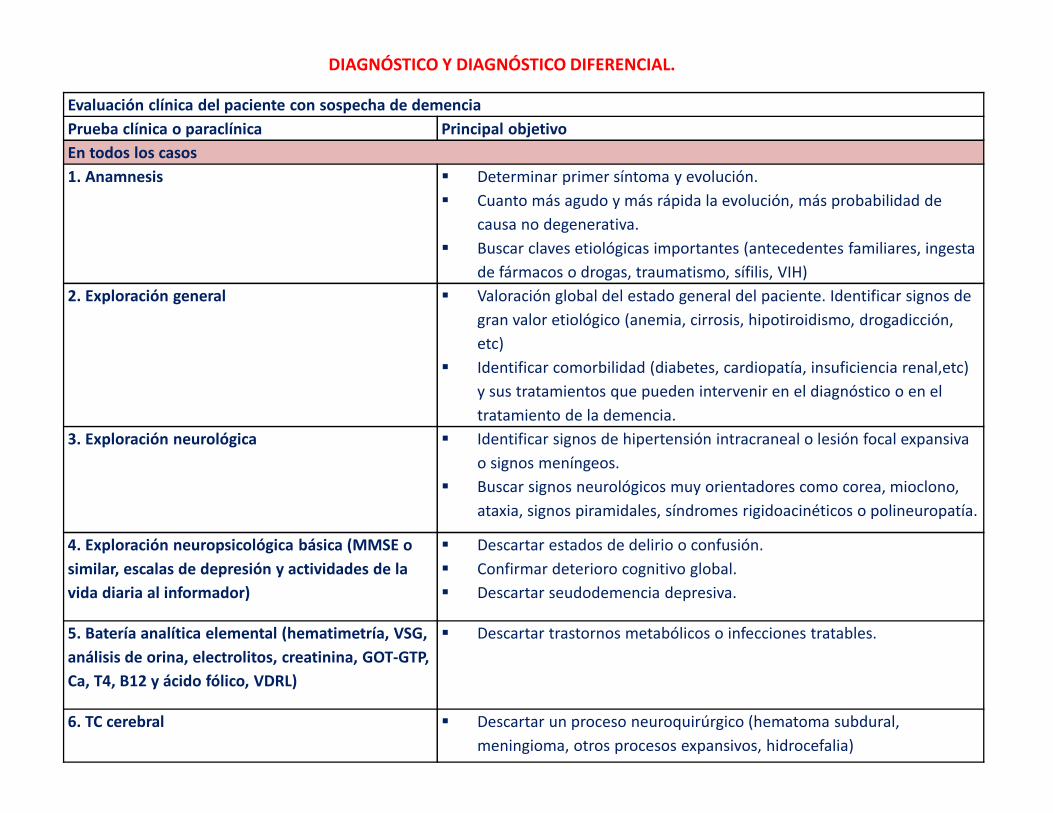
DIAGNÓSTICO Y DIAGNÓSTICO DIFERENCIAL.
Evaluación clínica del paciente con sospecha de demencia
Prueba clínica o paraclínica Principal objetivo
En todos los casos
1. Anamnesis � Determinar primer síntoma y evolución.
� Cuanto más agudo y más rápida la evolución, más probabilidad de
causa no degenerativa.
� Buscar claves etiológicas importantes (antecedentes familiares, ingesta
de fármacos o drogas, traumatismo, sífilis, VIH)
2. Exploración general � Valoración global del estado general del paciente. Identificar signos de
gran valor etiológico (anemia, cirrosis, hipotiroidismo, drogadicción,
etc)
� Identificar comorbilidad (diabetes, cardiopatía, insuficiencia renal,etc)
y sus tratamientos que pueden intervenir en el diagnóstico o en el
tratamiento de la demencia.
3. Exploración neurológica � Identificar signos de hipertensión intracraneal o lesión focal expansiva
o signos meníngeos.
� Buscar signos neurológicos muy orientadores como corea, mioclono,
ataxia, signos piramidales, síndromes rigidoacinéticos o polineuropatía.
4. Exploración neuropsicológica básica (MMSE o
similar, escalas de depresión y actividades de la
vida diaria al informador)
� Descartar estados de delirio o confusión.
� Confirmar deterioro cognitivo global.
� Descartar seudodemencia depresiva.
5. Batería analítica elemental (hematimetría, VSG,
análisis de orina, electrolitos, creatinina, GOT-GTP,
Ca, T4, B12 y ácido fólico, VDRL)
� Descartar trastornos metabólicos o infecciones tratables.
6. TC cerebral � Descartar un proceso neuroquirúrgico (hematoma subdural,
meningioma, otros procesos expansivos, hidrocefalia)
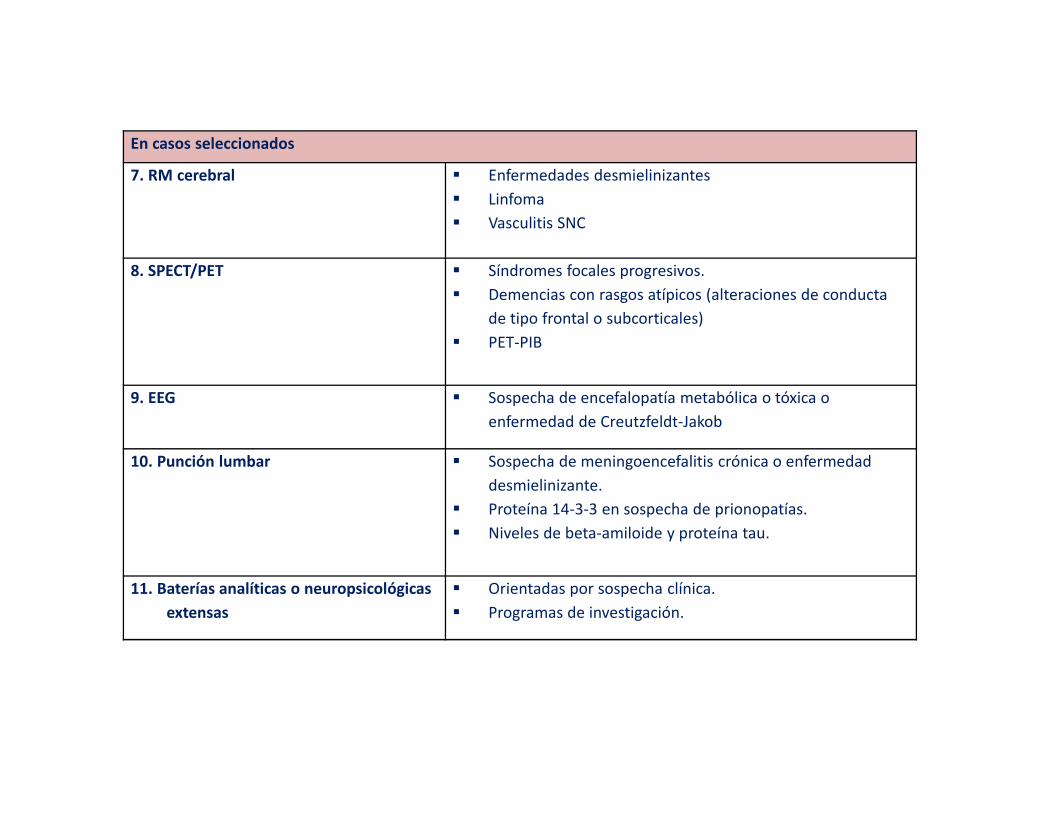
En casos seleccionados
7. RM cerebral � Enfermedades desmielinizantes
� Linfoma
� Vasculitis SNC
8. SPECT/PET � Síndromes focales progresivos.
� Demencias con rasgos atípicos (alteraciones de conducta
de tipo frontal o subcorticales)
� PET-PIB
9. EEG � Sospecha de encefalopatía metabólica o tóxica o
enfermedad de Creutzfeldt-Jakob
10. Punción lumbar � Sospecha de meningoencefalitis crónica o enfermedad
desmielinizante.
� Proteína 14-3-3 en sospecha de prionopatías.
� Niveles de beta-amiloide y proteína tau.
11. Baterías analíticas o neuropsicológicas
extensas
� Orientadas por sospecha clínica.
� Programas de investigación.


Tratamiento general del paciente demente.
Apoyo farmacológico eficiente, psicológico y apoyo a la familia del
paciente.
Primera fase: Ansiolíticos, hipnoticosAntidepresivos
Transtorno de personalidad: Neuroleptico
Hablar con familiares sobre cuidados higienicosde los pacientes

Enfermedades que cursan con demencia

Enfermedad de Alzheimer
Familiar
• 3 genes: Presenilina 1 o PS1, PS2 y prot precurso del amiloide o APP
Esporadico
• Factores ambientales, edad, Apo E, e historia familiar.
• Factor protector: Alelo e2

Enfermedad de Alzheimer y sindrome de Down
• Sufren casi invariablemente las lesiones neuropatologícas de enf deAlzheimer .
• Se debe descartar con otras causas como depresión, aislamiento,privacion sensorial.
• Se ha atribuido a la copia extra de APP en el cromosoma 21
Patogenia
Formacion del beta amiloide
Acumulacion del amiloide
Disfuncion de tau y degeneracion neurofibrilar
Reaccion inflamatoria microglial y glia
Perdida sinaptica y neuronal

Cuadro Clínico
• Son muy variables, algunos cuadro clinico por síndromes focales progresivos, otros sintomasparkinsonianos, otros predominio psicotico, etc.

Alzheimer familiar
De comienzo tardio.
• Pasado los 65 años y hasta ahora no hay base genetica conocida. El cuadro clinico es indistingible de las esporadicas.
Por mutaciones del gen APP
• Cromosoma 21. Edad de inicio 45 y 65 años.Incremento de B amiloide
Mutación del gen PS1
• Cromosoma 14. Representan el 50% de los casos de enfermedade de Alzheimer familiar. Edad de inicio 35 y 55 años. Frecuentes las crisis epilepticas y sintomas extrapiramidales
Alzheimer esporádica.
Cuadro clínico
• Insidiosamente por la perdida de memoria reciente y dificultad para incorporar nueva información. Ligeramente alteraciones de conducta, afectivo y sueño
Deterioro cognitivo
• Desorientación temporal � Síntoma precoz Estas avanzados afasia progresiva con anomia y dificultad para expresar conceptos. La alexia y la agrafia siguen un curso de deterioro.
Trastorno de personalidad
• Depresion interna, ansiedad, alucinaciones, delirios --> Manifestaciones frecuentes. Extension de las lesiones hacia los lobulos frontales.
• Apatia transtorno mas comun, mas que la depresion

Otros síntomas:� 10% presentan crisis convulsiva� La polimioclonia (fases avanzadas)� Crisis epiléptica y mioclonias� Fases avanzadas:
• Disfagia• Rigidez muscular• Reflejos primitivos• Alteraciones de la marcha y del equilibrio• Estado próximo al vegetativo
Correlación clinicopatológica:� DNF� Placas neríticas
Mejor marcador de la evolución cuantitativa de la demencia
Casos atípicos:Sd. de afasia no fluida progresiva o de degeneración corticalbasal (50%)Demencia de tipo frontal con alteraciones conductuales o de afasia semántica (10%)
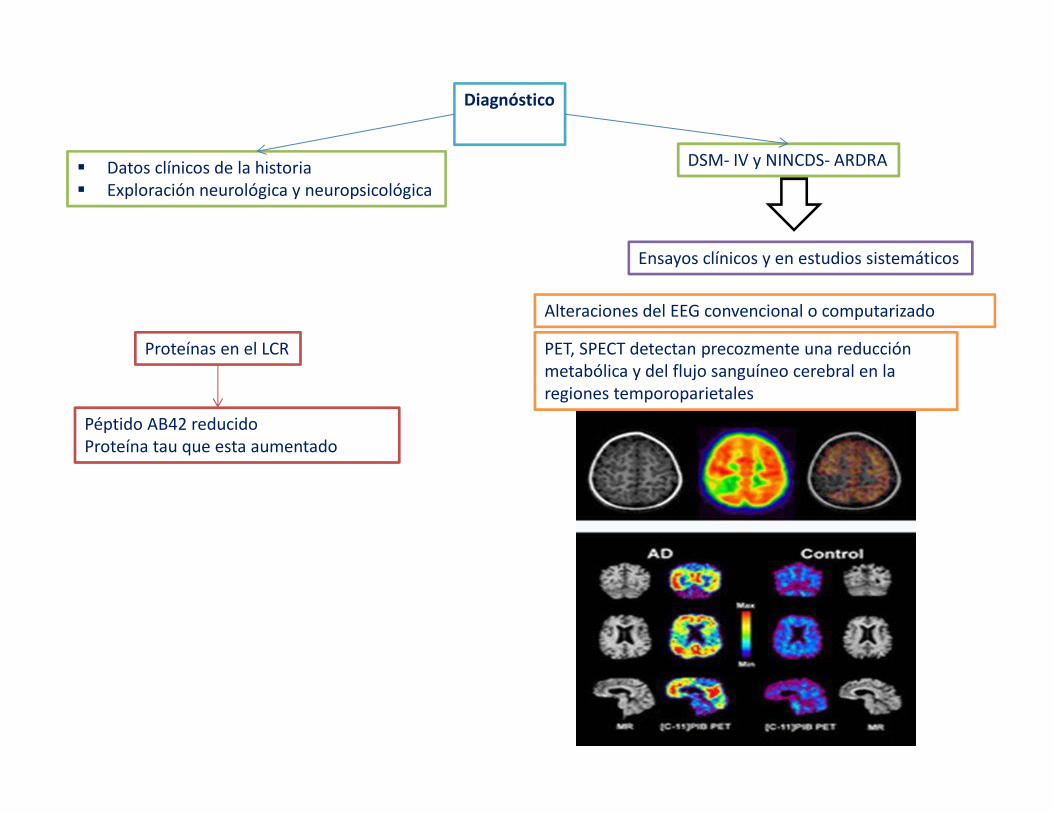
Diagnóstico
� Datos clínicos de la historia� Exploración neurológica y neuropsicológica
DSM- IV y NINCDS- ARDRA
Ensayos clínicos y en estudios sistemáticos
Proteínas en el LCR
Péptido AB42 reducido Proteína tau que esta aumentado
Alteraciones del EEG convencional o computarizado
PET, SPECT detectan precozmente una reducción metabólica y del flujo sanguíneo cerebral en la regiones temporoparietales
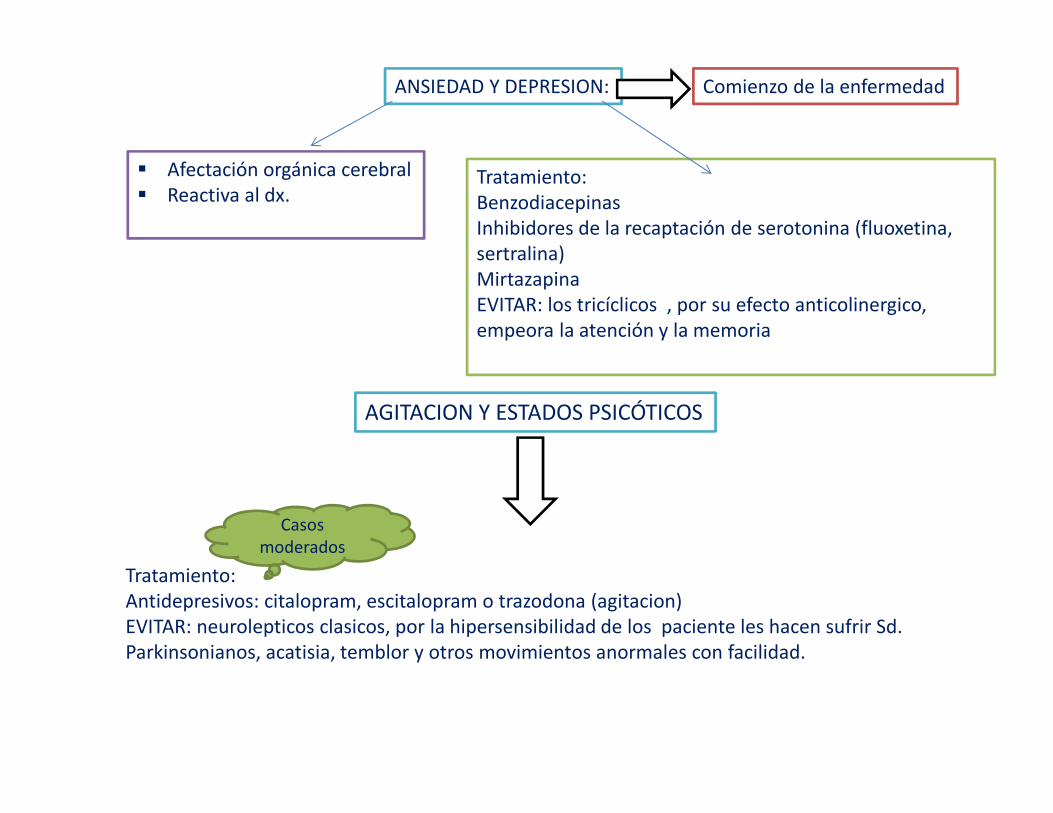
ANSIEDAD Y DEPRESION: Comienzo de la enfermedad
� Afectación orgánica cerebral� Reactiva al dx.
Tratamiento:Benzodiacepinas Inhibidores de la recaptación de serotonina (fluoxetina, sertralina)MirtazapinaEVITAR: los tricíclicos , por su efecto anticolinergico, empeora la atención y la memoria
AGITACION Y ESTADOS PSICÓTICOS
Tratamiento:Antidepresivos: citalopram, escitalopram o trazodona (agitacion)EVITAR: neurolepticos clasicos, por la hipersensibilidad de los paciente les hacen sufrir Sd. Parkinsonianos, acatisia, temblor y otros movimientos anormales con facilidad.
Casos moderados
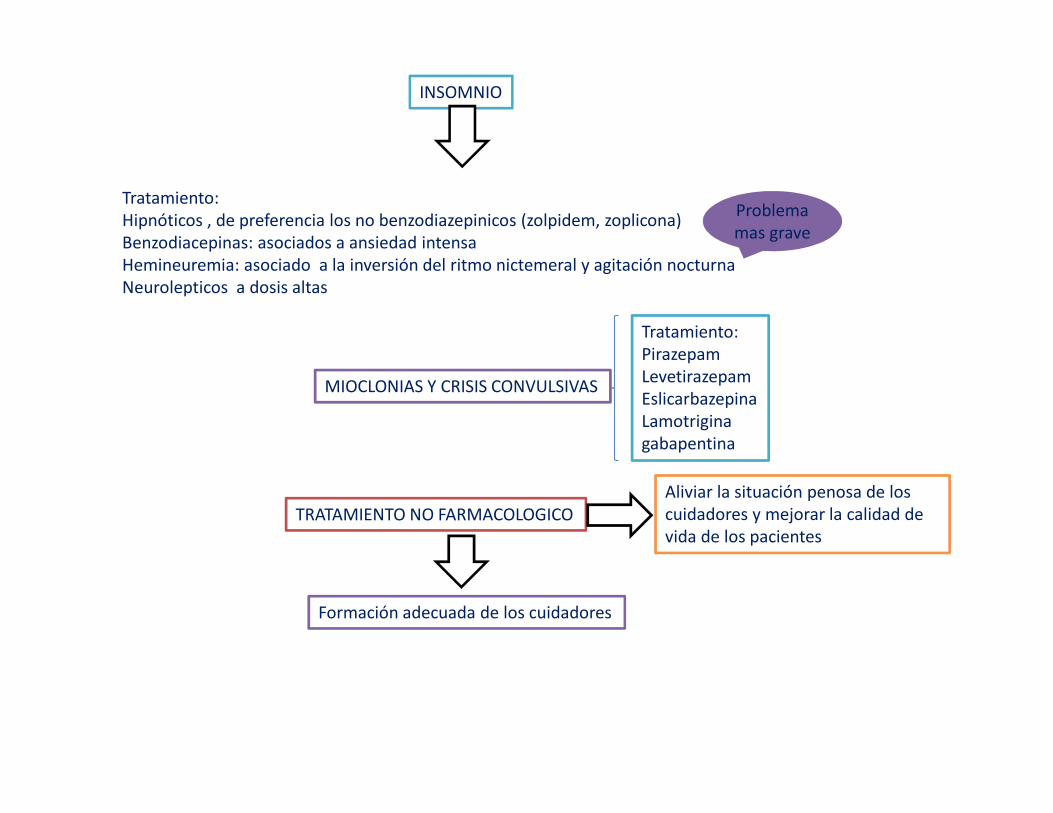
INSOMNIO
Tratamiento:Hipnóticos , de preferencia los no benzodiazepinicos (zolpidem, zoplicona)Benzodiacepinas: asociados a ansiedad intensaHemineuremia: asociado a la inversión del ritmo nictemeral y agitación nocturnaNeurolepticos a dosis altas
Problema mas grave
MIOCLONIAS Y CRISIS CONVULSIVAS
Tratamiento:PirazepamLevetirazepamEslicarbazepinaLamotriginagabapentina
TRATAMIENTO NO FARMACOLOGICOAliviar la situación penosa de los cuidadores y mejorar la calidad de vida de los pacientes
Formación adecuada de los cuidadores
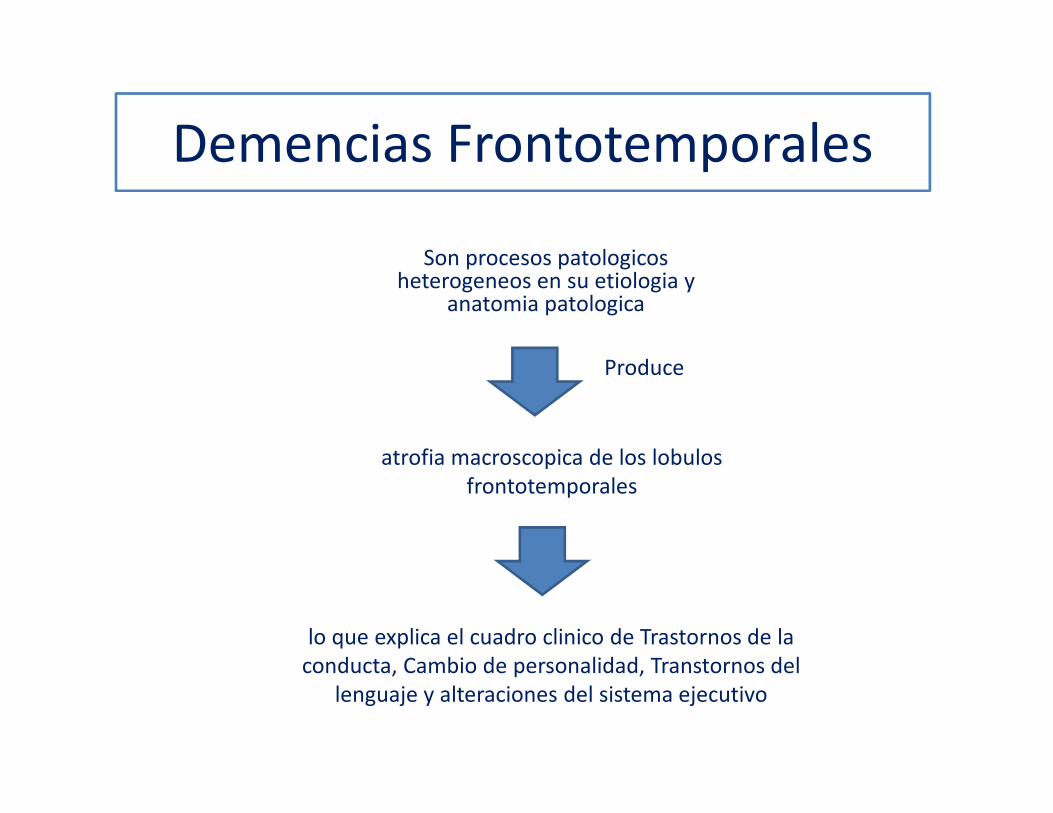
Demencias Frontotemporales
atrofia macroscopica de los lobulosfrontotemporales
lo que explica el cuadro clinico de Trastornos de la conducta, Cambio de personalidad, Transtornos del
lenguaje y alteraciones del sistema ejecutivo
Son procesos patologicosheterogeneos en su etiologia y
anatomia patologica
Produce

Cuadro ClínicoRasgos Clínicos de las demencias por regeneraciones frontotemporales
Trastornos de la conducta
Perdida de la autoconciencia o introspección (insight), Negación de la enfermedad,Negligencia del cuidado personal, Descuido de la formas sociales y desinhibición con comportamientos inadecuados, Irresponsabilidad, Abandono de la familia o del trabajo, Ausencia del sentido de culpabilidad, Tendencia al aislamiento social, falta de atención, rigidez mental, estereotipias mentales y de comportamiento, jocosidad, hiperfagia
Trastornos Afectivos
Apatía, indiferencia afectiva, retraimiento social, animo depresivo o eufórico, ansiedad exagerada
Alteraciones del lenguaje y el habla
Tendencia al mutismo reducción de la espontaneidad del lenguaje, estereotipias, ecolalia, disartria y disprosodia
Otros Signos
Aparición precoz de estereotipias gestuales, manipulaciones, reflejos de prensión forzada y de liberación frontal, incontinencia de esfínteres e indiferencia ante la suciedad, rigidez paranoica, signos parkinsonianos o de enfermedad de las motoneuronas

Variedades clinicopatologicas y geneticas de las demencias frontotemporales
Rasgos neuropatologicos comunes:
a) Dilatacion macroscopica de los surcos y de sistema ventricularb) La corteza esta atroficac) La transicion entre las circunvoluciones atroficas y las sanas
puede ser brusca y las adyacentes pueden ser muy variablesd) La sustancia blanca de los lobulos frontales puede tener un
aspecto grisaceo y rarefactadoe) Microespongiosis de tipo laminar de las capas superficiales de la
corteza cerebralf) Neuronas de perfil globoso y citoplasma palido que se tiñen
intensamente con anticuerpos antineurofilamentos fosforilados(neuronas de pick)

Variedades clinicopatologicas y geneticas de las demencias frontotemporales
Existes otros hallazgos histologicos, moleculares y geneticos que permiten individualizar un buen numero de identidades clinicopatologicas dentro de las DFT, como:
a) Demencia frontal sin rasgos histologicos especificosb) Demencia frontal con inclusiones positivas para ubicuitinac) Enfermedad de Pickd) DFT familiar ligada al cromosoma 17 (DFTP cr 17)e) DFT por enfermedad de alzehimer esporadica familiarf) Demencia con inclusiones de neurofilamentosg) Demencia frontal con inclusiones de neuroserpinah) DFT por distrofia miotonicai) Demencia frontal con miositis con cuepos de inclusion y
enfermedad de Paget

Demencia frontal sin rasgos histologicos especificos
Presentan un cuadro clinico de deterioro cognitivo y conductual de tipo frontal, sin historia familiar
En la autopsia presentan atrofia cortical con microespongiosislaminar, pero las tinciones de inmunohistoquimica son negativas

Demencia frontal con inclusiones positivas para la ubicuitina
DFT con inclusiones positivas para TDP-43
Mutaciones en CHMP2B
Mutaciones en PGRN
Mutaciones en CrORF72
DFT con inclusiones y FUS positivas

Enfermedad de Pick
Es la variedad clasica de DFT, pero no la mas frecuente.
Suele comenzar hacia los 60 años y la duracion media de la enfermedad es 9 años.
Casi siempre esporadica, pero hay familias con las mutaciones en el gen MAPT enCr17.
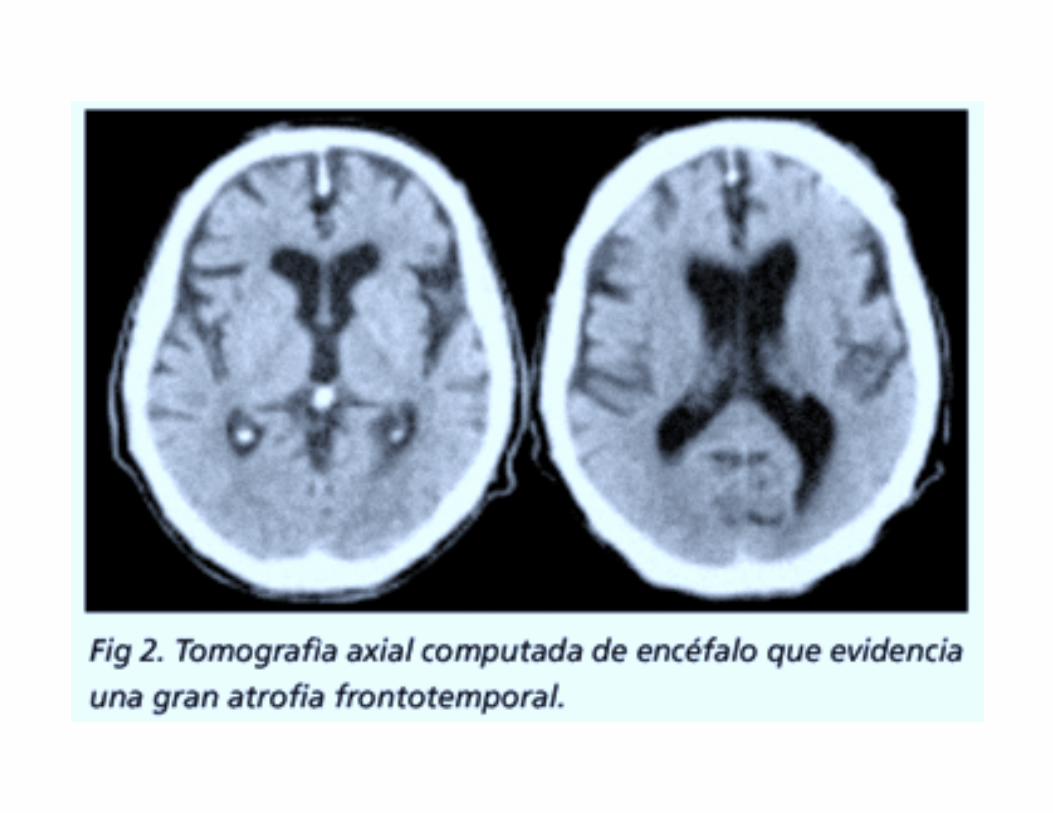
La atrofia frontotemporal suele ser muy aguda en las imágenes del cerebro.
El rasgo histologico distintivo es la presencia de cuerpos de inclusion redondeados,argirofilos en las tinciones de Bielschowsky y Bodian, pero negativos en Gallyas.
El cuadro clinico responde al esquema general anterior, pero con un predominio detrastornos conductuales sobre alteraciones del lenguaje.

DFT familiar ligada al Cr17
Se trasmite por herencia autosomica dominante.
Se debe a la mutacion del gen que codifica proteina tau en el Cr17.
Ademas de los rasgos clinicos de la DFT, los pacientes tienen signos parkinsonianos y de enfermedad de las motoneuronas.
En el estudio histologico, ademas de la degeneracion cortical frontotemporal, de la amigdala y del conjunto del sistema limbivo, existen una degeneracion nigrica sin CL.
El hallazgo histologico mas caracteristico son las inclusiones que contienen proteina tau fosforilada

DFT por enfermedad de Alzheimer esporadica y familiar
Entre el 10 y 30% de los pacientes con un sindrome de DFT tienen enfermedad deAlzheimerLa presentacion atipica de Alzheimer es habitual en las mutaciones del gen depreselina (PS1)
Demencia con inclusiones de neurofilamentos
Se caracteriza por las inclusiones intraneuronales de neurofilamentos; sonpositivas para ubicuitina pero negaticas para tau y a-sinucleina
Ademas de signos de demencia de tipo frontal, pueden aparecer signosparkinsonianos y de enfermedad de motoneuronas

Demencia frontal con inclusiones de neuroserpinas
Enfermedad familiar con mutaciones en el gen PI12 en el Cr3q26 que codifica neuroserpina.
Los portadores de la mutacion presentan signos precoces de disfuncion frontal.
Se caracteriza histologicamente por inclusiones intraneuronales denominadas CUERPOS DE COLLINS, eosinofilia y PAS positivo.
DFT por distrodia miotonica
En las dos variables de esta enf. se describen trastornos neuropsicologicos en las formas tardias, pero no llegan a tener un deterioro cognitivo grave.

Demencia frontal con miositis con cuepos de inclusion y enfermedad de Paget
Enfermedad de herencia autosomica dominante y se debe a mutaciones en el gen de valosina en el Cr9.
En la neuropatologia en rasgo caracteristico es la presencia de abundantes neuritas e inclusiones TDP-43 positivas intranucleares.

Diagnostico y Tratamiento
El Dx es neuropatologico, salvo en aquellas variedades geneticasen las que el Dx se pueda hacer por analisis del ADN
Algunas correlaciones clinicopatologicas que orientan al Dx:
Demencia que comienza con afasia semantica
Demencia que comienza con Afasia no fluida
La DFT con asociacion con ELA
La DFT con asociacion con parkinsonismo
Inclusiones ubi-TDP-43 (+)
Inclusiones tau
Inclusiones ubi-TDP-43
Inclusiones tau (taupatia)

Diagnostico y Tratamiento
El TRATAMIENTO es puramente SINTOMATICO, similar al de todas las demencias en general
Al igual que en otras demencias degeneraticas, es preciso utilizar los farmacos con menos capacidad de inducir un sindrome parkinsoniano

DEMENCIAS CON CUERPOS DE LEWY

1.ETIOPATOGENIA
• La mayoria de los casos de DCL son esporadicos.
• Existen familias en las que se transmite por herenciadominante, pero en las que se desconoce su base genetica.
• En la mutacion E46K en el gen de la a-sinucleina todos los casos conocidos desarrollan demencia y enfermedad de Parkinson.
• En las otras mutaciones en ese mismo gen,la incidencia de la demencia es variable.

2.ANATOMIA PATOLOGICA
El hallazgo histologicocaracteristico son los CL y lasneuritasdistroficas(NL),repartidos porla corteza cerebral.
Ambos inmunorreactivospara ubicuitina y a-sinucleina, e identicos a los que se encuentran en la enfermedad de Parkinson en la sustancianegra y en otros nucleosdel tronco cerebral y ganglios simpaticos.
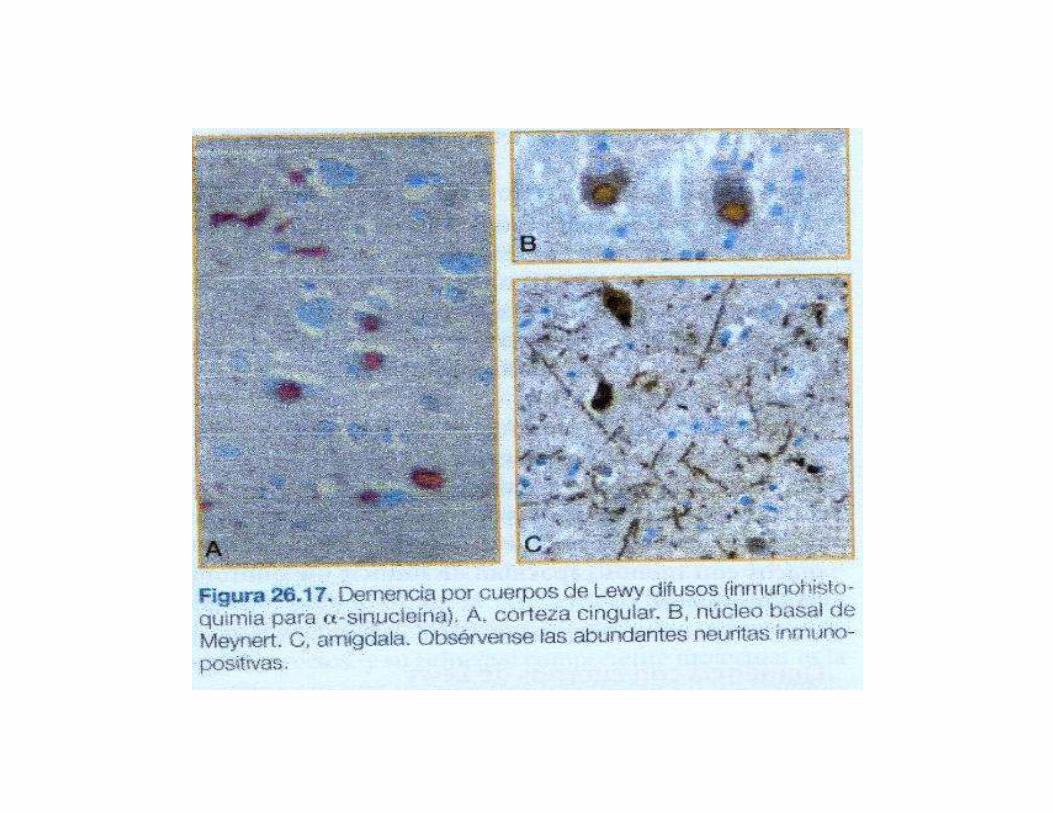

3.CUADRO CLINICO
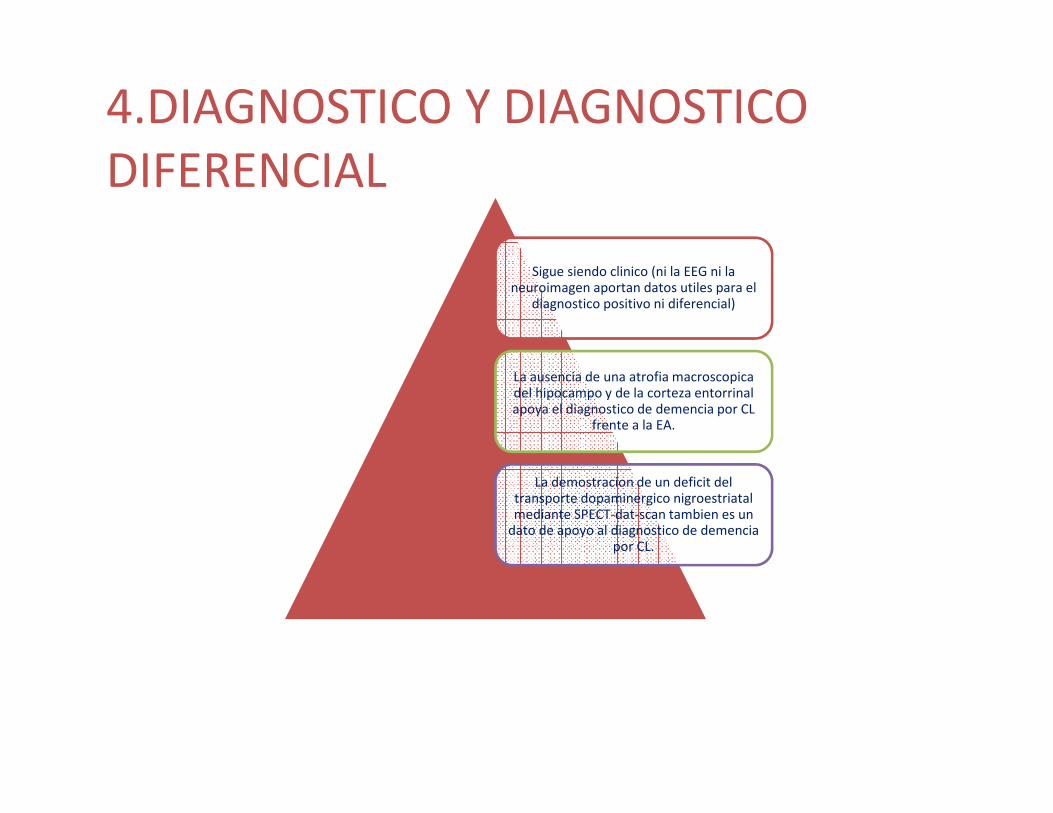
4.DIAGNOSTICO Y DIAGNOSTICO DIFERENCIAL
Sigue siendo clinico (ni la EEG ni la neuroimagen aportan datos utiles para el
diagnostico positivo ni diferencial)
La ausencia de una atrofia macroscopicadel hipocampo y de la corteza entorrinalapoya el diagnostico de demencia por CL
frente a la EA.
La demostracion de un deficit del transporte dopaminergico nigroestriatalmediante SPECT-dat-scan tambien es un
dato de apoyo al diagnostico de demenciapor CL.

5.TRATAMIENTO
• Inhibidores de la acetilcolinesterasa(rivastigmina)
• Levodopa en dosis muy bajas y fragmentadas
• Neurolepticos modernos en dosis muy bajas e intermitentes (quetiapina)
• Otras opciones: ziprasidona,aripiprazol y clozapina(con precauciones de control hematologico)

DEMENCIA VASCULAR(DVA)Y DETERIORO COGNITIVO DE ORIGEN
VASCULAR(DCVA)

1.ETIOPATOGENIA
• El DCVA es multifactorial, el daño cerebral deriva de la isquemia y anoxia, pero tambien de factores de predisposicion de enfermedadaterotrombotica y de la arterioesclerosis.(HTA,DM, hipercolesterolemia, tabaquismo,obesidad,etc)
• Otros factores: hiperhomocistinemia, descenso de folato y vit.B12
• La esclerosis de arteriolas perforantes en la sustancia blanca y en los ganglios basales, puede dar lugar al daño del parenquima cerebra; con perdida neuronal y desmielinizacion a traves de edema y anoxia, sin llegar a producir verdaderos infartos.
• ICC,crisis hipotensoras, arritmias, apneas obstructivas del sueño, tambien añaden lesiones vasculares sin la presencia de infarto.
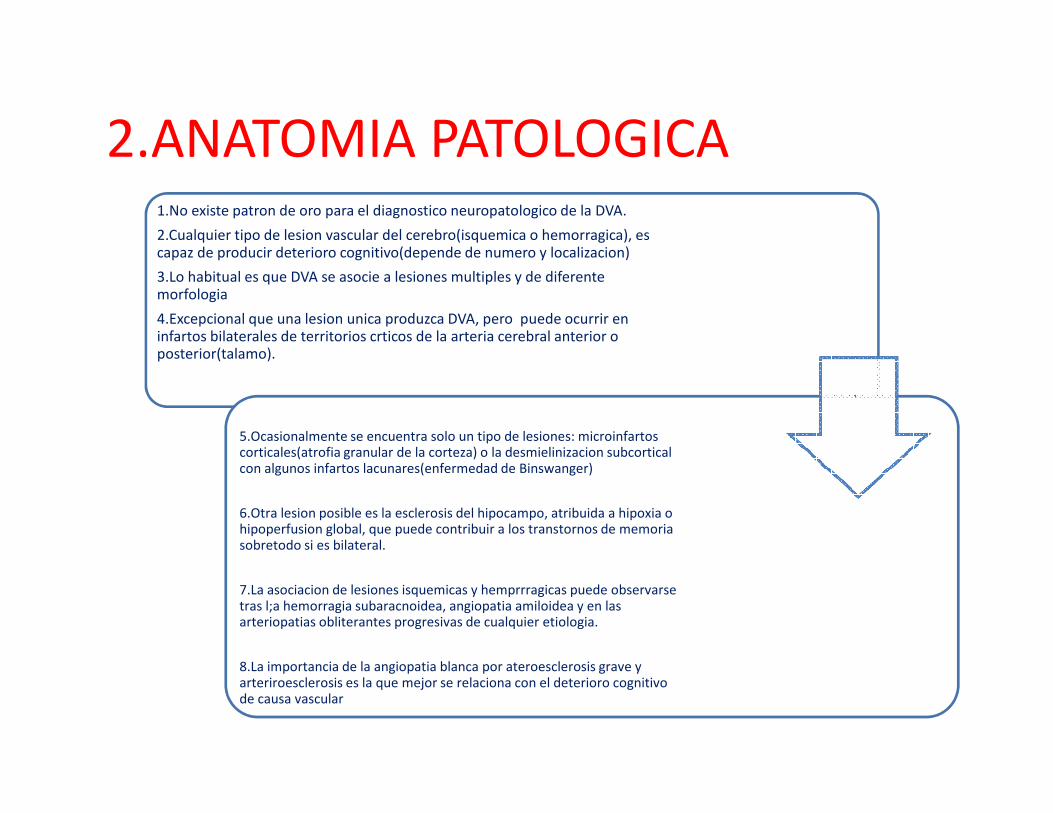
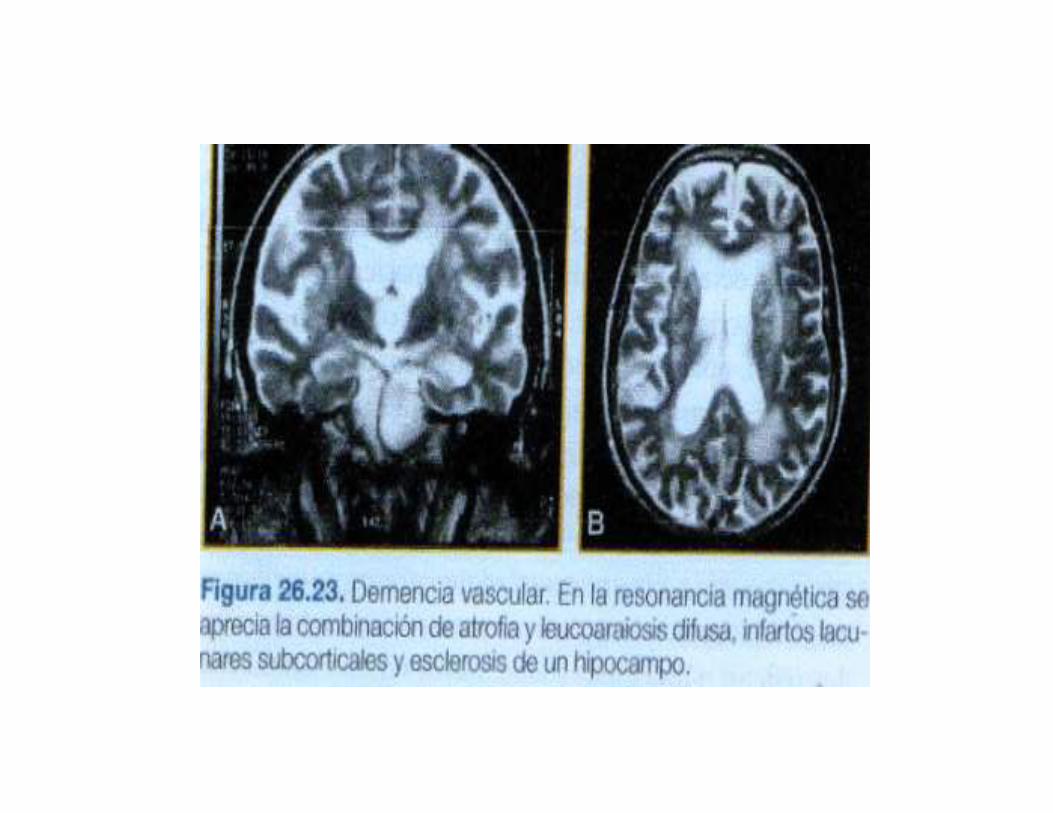
2.ANATOMIA PATOLOGICA1.No existe patron de oro para el diagnostico neuropatologico de la DVA.
2.Cualquier tipo de lesion vascular del cerebro(isquemica o hemorragica), escapaz de producir deterioro cognitivo(depende de numero y localizacion)
3.Lo habitual es que DVA se asocie a lesiones multiples y de diferentemorfologia
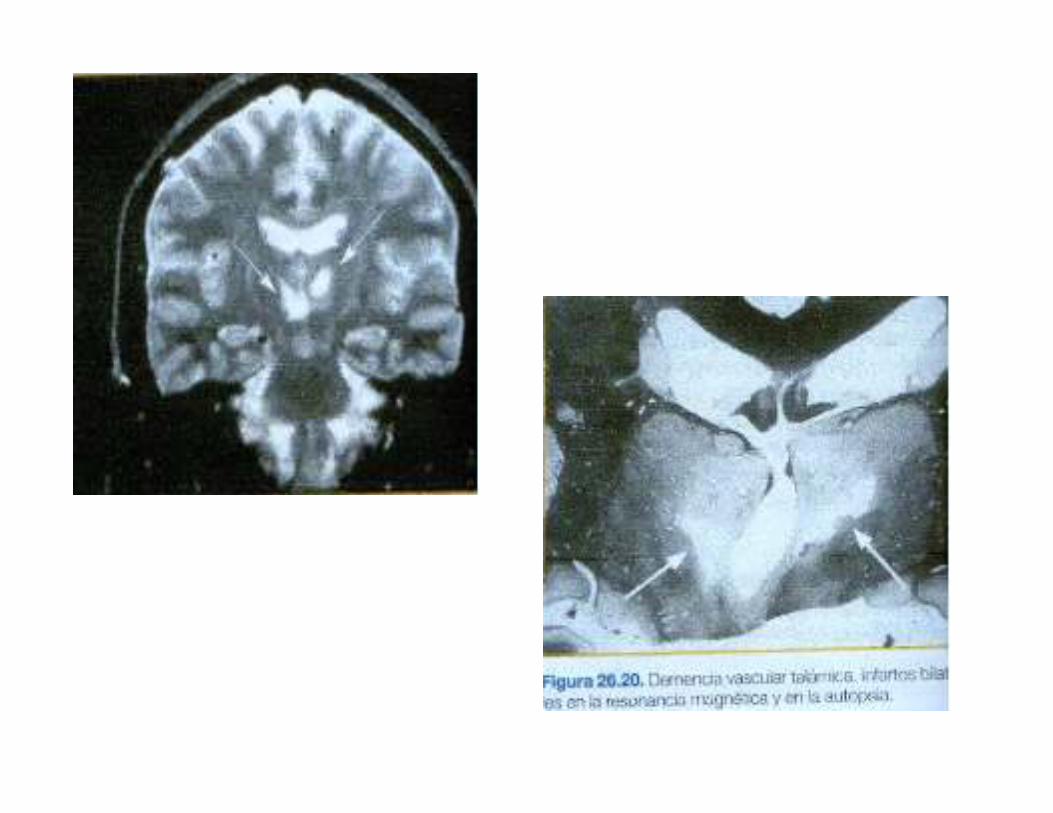
4.Excepcional que una lesion unica produzca DVA, pero puede ocurrir en infartos bilaterales de territorios crticos de la arteria cerebral anterior o posterior(talamo).
5.Ocasionalmente se encuentra solo un tipo de lesiones: microinfartoscorticales(atrofia granular de la corteza) o la desmielinizacion subcorticalcon algunos infartos lacunares(enfermedad de Binswanger)
6.Otra lesion posible es la esclerosis del hipocampo, atribuida a hipoxia o hipoperfusion global, que puede contribuir a los transtornos de memoriasobretodo si es bilateral.
7.La asociacion de lesiones isquemicas y hemprrragicas puede observarsetras l;a hemorragia subaracnoidea, angiopatia amiloidea y en lasarteriopatias obliterantes progresivas de cualquier etiologia.
8.La importancia de la angiopatia blanca por ateroesclerosis grave y arteriroesclerosis es la que mejor se relaciona con el deterioro cognitivode causa vascular

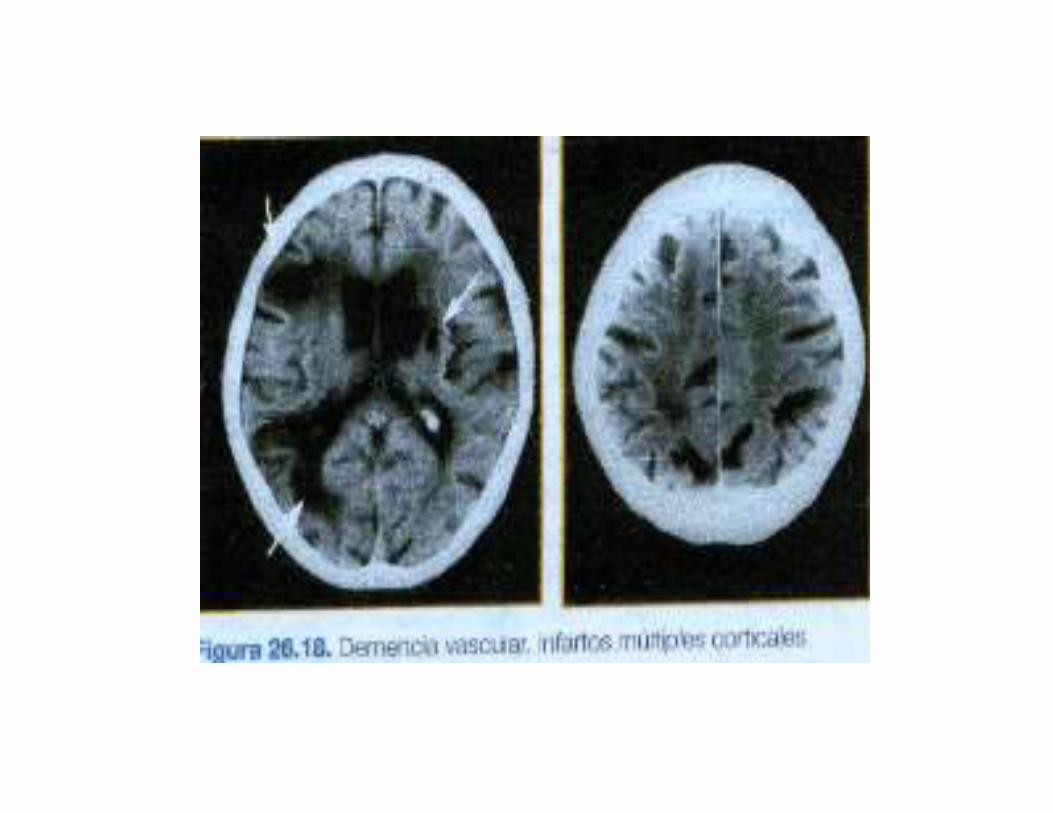

3.CUADRO CLINICO


4.TRATAMIENTO
• Prevencion primaria: Control de la P.A(P.max=135-150 mmHg y Pmin<90 mmHg)
• Profilaxis secundaria con antiagregantes plaquetarios, anticoagulacion,cirugia en pacientes con ictus previos.
• El DCVA es un sindrome heterogeneo, por lo que tratamiento farmacologico debe ser individualizado.
• Medidas terapeuticas: calcioantagonistas,antiagregantes.
• En el estado de demencia de causaisquemica(sobretodo por lesiones subcorticales) pueden estar indicados galantamina,donepezilo u otrosanticolinesterasicos.

5.DEMENCIAS VASCULARES DE ETIOLOGIA ESPECIFICARasgos Clínicos de las demencias por regeneraciones frontotemporales
CADASIL(ARTERIOPATIA CEREBRAL AUTOSOMICA DOMINANTE CON INFARTOS SUBCORTICALES Y LEUCOENCEFALOPATIA)
Debida a mutaciones en el gen notch 3 en el cromosoma 19(19q2). Comienzo de los sintomas entre los 30-50 años: cursan con ictus repetidos y leucoaraiosisintensa hasta la demencia; en algunas familias variantes fenotipicas con asociacion de migraña, ictus y depresion psicotica. Las lesiones vasculares consisten en engrosamientos de la capa media, en la que se deposita material granular basofilo que reemplaza las fibras musculares, ademas de fragmentacion y reduplicacion de la elastica interna.
CARASIL
Casos raros de pacientes(mayoria japoneses), herencia autosomicarecesiva, base genética desconocida, a menudo hay consanguinidad en los padres. Ademas de las lesiones cerebrales subcorticales por la arteriolopatia(hay degeneracion hialina pero sin los granos PAS+ caracteristicos del CADASIL), los pacientes cursan con lumbago, dolor de espalda, cifoescoliosis y alopecia.







