desarrollo de un método de reducción dimensional no … · los perfiles de expresión génica a...
TRANSCRIPT

Desarrollo de un método de reducción
dimensional no lineal y clustering para
la visualización e interpretación de
single cell RNA-seq data
Estudiante: Miguel Juliá Molina
MÁSTER EN BIOINFORMÁTICA Y BIOLOGÍA COMPUTACIONAL
ESCUELA NACIONAL DE SALUD- INSTITUTO DE SALUD CARLOS III
2013-2014
Swiss Institute of Bioinformatics (SIB)
University Hospital of Lausanne (CHUV)
Antonio Rausell, Amalio Telenti, Ioannis Xenarios
David González
Septiembre de 2014

1
Índice
1. Introducción …………………………………………………………… 2
2. Objetivos ……………………………………………………………….. 8
3. Resultados ……………………………………………………………... 9
4. Discusión y Conclusiones ………………………………………… 27
5. Materiales y Métodos ……………………………………………… 29
6. Bibliografía …………………………………………………………… 33

2
1. Introducción
1. El transcriptoma y los métodos tradicionales usados para su análisis
Hablamos de transcriptómica para referirnos al estudio del RNA (RNAr, RNAt, RNAm, RNAi, miRNA), que existe en una célula, tejido u órgano. La transcriptómica permite cuantificar el nivel de expresión de genes, empleando técnicas que permiten analizar miles de moléculas de RNA al mismo tiempo. Las dos principales tecnologías para el estudio del transcriptoma a gran escala son los microarrays y el RNA-seq (ver [1]):
Microarrays: es una superficie sólida a la cual se une una colección de fragmentos de RNA. Las superficies empleadas para fijar el RNA son muy variables y pueden ser de vidrio, plástico e incluso de silicio. Los chips de RNA se usan para analizar la expresión diferencial de genes, y se monitorean de manera simultánea los niveles de miles de ellos. Su funcionamiento consiste, básicamente, en medir el nivel de hibridación entre la sonda específica, y la molécula diana, y se detectan generalmente mediante fluorescencia y a través de un análisis de imagen, lo cual indica el nivel de expresión del gen.
RNA-seq: es una tecnología que utiliza los métodos de secuenciación de próxima generación (NGS) para revelar una instantánea de la presencia y la cantidad de RNA sintetizado a partir de un genoma en un momento dado en el tiempo. Su funcionamiento se basa en la fragmentación del material genético, amplificación mediante el uso de PCR, y secuenciación de short reads.
2. Single cell RNA-seq Recientes avances técnicos han permitido la secuenciación del RNA en células individuales (single cell RNA-seq). Estos métodos consisten en el aislamiento de las células individuales previo a la secuenciación, donde la falta de cantidad de material genético se suple mediante el uso de PCR en pocillos independientes para cada célula. Los estudios exploratorios ya han dado lugar a ideas sobre la dinámica de la diferenciación, las respuestas celulares a la estimulación y la naturaleza estocástica de la transcripción, y con la ayuda de los recientes métodos de análisis de single cell RNA-seq se han podido además identificar isoformas alternativas de los tránscritos, biomarcadores candidatos para identificación de tumores y

3
localización de tránscritos con escaso número de copias (ver [2] para una revisión reciente). Estamos entrando en una era de transcriptómica unicelulares que promete impactar sustancialmente la biología y la medicina. Los perfiles de expresión génica a resolución de single cell ofrecen un extraordinario nivel de detalle nunca visto anteriormente, añadiendo la posibilidad de distinguir la heterogeneidad de los estados celulares. En este estudio contaremos con los análisis de datos de células CD4 T aisladas mediante el sistema C1 Single-Cell AutoProp System de Fluidigm [6], seguido de la secuenciación del transcriptoma completo de hasta 96 librerías single cell por muestra biológica, mediante Illumina HiSeq paired-end 50-bp (ver [7]). El sistema de Fluidigm consiste en un workflow completo que aísla las células individuales y preamplifica su transcriptoma previo paso a su secuenciación por Next Generation Sequencing (NGS).
3. Limitaciones técnicas No obstante, esta nueva técnica también arrastra ciertas limitaciones técnicas (ver [3]) y biológicas (ver [4] y [5]): 1. Técnicas:
a) Límite de detección: incluso un transcrito altamente expresado puede ser
ignorado, ocultado por el ruido técnico.
b) Sesgo introducido por la PCR: puede no existir proporcionalidad entre el número de moléculas antes y después de la amplificación.
2. Biológicas:
a) Estocasticidad biológica de la transcripción: la cantidad y tipo de tránscritos
que produce una célula varía dependiendo de muchos factores, hasta el punto que dos células hermanas (provenientes de la división de una misma célula) en el mismo instante de tiempo no tienen los mismos niveles de expresión.
b) Correspondencia entre los niveles de RNA y proteínas: no todos los tránscritos se traducen y la cantidad de proteína presente en la célula no tiene por qué ser proporcional a la cantidad del tránscrito que la codifica, ya que depende además depende de otros factores como es el tiempo de vida de la proteína o si la proteína traducida es intracelular o extracelular.
4. Métodos para el análisis de jerarquías de estados celulares a partir de single
cell RNA-seq data Existen actualmente un conjunto reducido de métodos diseñados para analizar datos provenientes de experimentos de single cell. Estos métodos se basan en la premisa de que,

4
en el contexto de un proceso de diferenciación o activación celular, existe un gradiente de estados celulares continuo entre los estados estables de partida e inicio. Así, los métodos tratan de determinar estos gradientes y el estado de cada célula de la muestra respecto a ellos mediante el cálculo de estructuras jerárquicas en forma de grafos (Minimum Spanning Tree) y clústeres o mediante el examen visual directo en un gráfico en 2D (de nuevo hablamos de un grafo o un Scatter Plot) basados en las relaciones de parecido entre las células. Es común entre estos métodos, incluso en aquellos que no devuelven sus resultados como un simple Scatter Plot, realizar una reducción dimensional de los datos para facilitar el cálculo de las distancias entre las células a la hora de calcular la estructura jerárquica de los datos. Hay que tener en cuenta que cuando hablemos de “árbol” de ahora en adelante nos estaremos refiriendo a un subtipo de grafo que nada tiene que ver con los árboles filogenéticos. En los árboles filogenéticos solo las hojas hacen referencia a los datos analizados, mientras que los nodos intermedios son añadidos para la construcción de la jerarquía. En los árboles y grafos jerárquicos de los que hablaremos en este documento todos y cada uno de los nodos corresponderá a una célula de la muestra. Por último, cabe mencionar que, aunque todos los resultados de experimentos de single cell pueden ser codificados como una matriz numérica y que ésta es la forma interna con la que operan todos los métodos descritos, los métodos basados en datos de citometría utilizan como entrada archivos binarios. Presentamos a continuación cada uno de los métodos existentes [ver Fig. 1] para pasar después a explicar las limitaciones que nos motivaron a desarrollar un nuevo método de análisis.
Tipo de datos de Single Cell
Papers / Métodos publicados
Distancia y Reducción Dimensional
Representación Gráfica
Citometría SPADE Qiu P, Nature America, 2011
2 dim PCA + distancia Euclídea
Minimum Spanning Tree
ViSNE Amir ED, Nature Biotechnology, 2013
2 dim t-SNE + distancia Euclídea
Scatter Plot
Wanderlust Bendall SC, Cell, 2014
2 dim t-SNE + distancia Euclídea
-
RNA-seq MAP-CAP Jaitin DA, Science, 2014
Mixture a Posteriori Probability
Circular a Posteriori Projection
Monocle Trapnell C, Nature Biotechnology, 2014
2 dim ICA + distancia Euclídea
Minimum Spanning Tree

5
Fig. 1: Tabla resumen de los métodos publicados para el análisis de jerarquías de single-cell en la que se describen las características principales de cada uno, así como su autor y tipo de datos para los que han sido diseñados.
SPADE:
Este método es capaz de agrupar las células en montones de diminutos clústeres para luego construir un árbol por Minimum Spanning Tree (ver Métodos) usándolos como nodos, de forma que ordena los estados celulares dentro del proceso de diferenciación. Este árbol se extrae a partir del grafo completo generado con la matriz de distancias euclídeas, calculada a su vez sobre la matriz de datos previamente reducida por PCA. Ver [8].
ViSNE: Versión mejorada del anterior. Ahora además puede calcular una trayectoria que estima la ruta del proceso de diferenciación de las células. Esta trayectoria la obtiene como el mínimo camino que recorre todas las células sobre un grafo I-Nearest Neighbours. Más adelante explotaremos la idea subyacente de este método para el desarrollo del algoritmo K-Nearest Neighbout (ver Resultados). Este camino se proyecta sobre un plano y se suaviza para obtener una curva que se ajuste a los datos. Ver [11]. Wanderlust: Versión mejorada del anterior. Ahora además puede calcular una trayectoria que estima la ruta del proceso de diferenciación de las células. Esta trayectoria la obtiene como el mínimo camino que recorre todas las células sobre un grafo I-Nearest Neighbours, cuya idea subyacente es muy parecida a la modificación que veremos más adelante del K-Nearest Neighbout (ver Resultados). Este camino se proyecta sobre el plano y suaviza para obtener una curva que se ajuste a los datos. Ver [11]. MAP-CAP: Obtiene clústeres parciales de la población de células (es decir, puede dejar individuos sin pertenencia a ninguno de los clústeres) que utiliza posteriormente para calcular la probabilidad de incluir a cada una de las células sin grupo en cada uno de ellos. Finalmente, utiliza estas probabilidades para hacer un plot circular en el que cada clúster se encuentra en un punto distante de la circunferencia y las células se distribuyen por el interior según sus probabilidades de pertenencia a cada uno de ellos. Ver [12]. Monocle: Realiza un fuerte filtrado de datos y posterior reducción de la dimensión para ordenar las células en un mínimum spanning tree en dos dimensiones. Luego calcula la posible ruta de diferenciación como el camino más largo dentro del árbol

6
y define cada nodo de ese camino como un pseudo-tiempo y cada rama lateral como un estado sobre el que posteriormente calcular enriquecimiento funcional. Ver [13].
Otros métodos:
También hemos tenido en cuenta varios métodos de reducción dimensional diseñados para él análisis de datos multivariables de otros campos distintos a la biología (ver [10] y [14]), pero en muchos aspectos resultaban inapropiados e inadaptables a nuestro caso de estudio.
Si bien los métodos anteriores han demostrado ser capaces de reconstruir el progreso de diferenciación o la heterogeneidad subyacente en poblaciones celulares, su diseño es limitante en diversos aspectos que desarrollamos a continuación:
a) Necesidad de adaptar los algoritmos de los métodos de citometría de flujo o masas al análisis de datos de RNA-seq:
Estos programas se han desarrollado y optimizado para citometría de flujo y citometría de masas. Diversas limitaciones excluyen la posibilidad de ser generalmente aplicable a los datos de RNA-seq de single cell en su estado actual. Por ejemplo, en la citometría de flujo se evalúa solo un pequeño número de marcadores para un gran número de células, mientras que en el caso de RNA-seq permite la cuantificación de un gran número de genes para un reducido grupo de células (<96 células y >53000 genes en nuestro caso). Las diferencias en la naturaleza de los datos también requerirían adaptar el proceso de normalización, la transformación y filtrado y la evaluación del ratio ruido-señal.
b) Reducción de la dimensionalidad Todos los métodos expuestos realizan una reducción de la dimensionalidad de los datos previa al cálculo de distancias entre ellas, que podría no ser apropiada. Esto puede deberse a que la heterogeneidad de los datos sumada al enorme número de variables puede distribuir la variabilidad de la muestra en un número mucho mayor de dimensiones de las que se pretenden capturar [ver Fig. 2].

7
Fig. 2: Scatter plot de las dos primeras componentes de un análisis de componentes principales
(PCA, izquierda) realizado sobre una matriz de expresión de RNA-seq junto al porcentaje de variación en los datos explicados por cada variable de la misma PCA (derecha). En este ejemplo
puede apreciarse claramente cómo una reducción dimensional no captura más que una ínfima parte (~10%) de la variabilidad de los datos.
c) Uso de distancias lineales versus no lineales Todos los métodos utilizan una distancia pre-implementada que no puede ser sustituida por otra, por ejemplo funcional o no lineal, para realizar distintos tipos de análisis. Lo mismo ocurre con los tipos de normalización de datos y filtros de ruido que aceptan cada uno de las pipelines. La capacidad para variar la distancia puede ser de gran ayuda a la hora de comparar la estructura de los datos analizándolos desde diferentes puntos de vista, por ejemplo alternando entre una distancia de información funcional no lineal y otra de correlación de expresión.
d) Necesidad de dar soporte estadístico a los árboles calculados Ningún método realiza tests para comprobar si la jerarquía celular o su representación de baja dimensión obtenida son fiables estadísticamente ante pequeñas variaciones de los datos de partida. Carecen por tanto de un soporte estadístico que permita distinguir una estructura de estados celulares determinada por una señal biológica de un artefacto determinado por una señal ruidosa.
e) Necesidad de disponer de un entorno informático integrado donde se puedan combinar los diferentes abordajes
Como ya hemos comentado anteriormente, la elección de un método condiciona al usuario a utilizar unas distancias y reducciones dimensionales específicas, además de un tipo de datos y una representación gráfica. No hay posibilidad de combinar diferentes partes de las pipelines para hacer un análisis personalizado. Es por todas estas razones que creímos conveniente desarrollar un entorno de software que incluyera nuevos algoritmos de análisis con el objetivo de complementar a los métodos ya

8
existentes y aportara soluciones a algunas de las limitaciones señaladas. Como resultado, se ha desarrollado un paquete de R llamado sincell que será publicado en Bioconductor en breve. Este paquete permite al usuario introducir una matriz de datos proveniente de cualquier tipo de experimento y normalización y reconstruir la jerarquía de los estados celulares presentes en una muestra en forma de árbol o grafo con clústeres calculados de forma independiente, y posteriormente dotarlo de un soporte estadístico que de pruebas de la robustez del método y testar los niveles de enriquecimiento funcional de sets de genes de interés.

9
2. Objetivos
Las prácticas se realizaron orientadas a cumplir tres objetivos: Las prácticas se realizaron orientadas a cumplir tres objetivos:
1. Desarrollo e implementación de un algoritmo similar a SPADE o ViSNE orientado al análisis de jerarquías de estados celulares a partir de single cell RNA-seq data
Basándonos en los métodos ya publicados para citometría SPADE y ViSNE, desarrollamos un nuevo algoritmo para el análisis de single cell RNA-seq data. Este algoritmo se vale de un set de distancias lineales y no lineales (basadas en la teoría de la información, rank-based o correlación) combinadas con estrategias de reducción dimensional como PCA, ICA, t-SNE o MDS. También incluye herramientas de clustering tanto jerárquicas como no jerárquicas, y puede trabajar sin límites de dimensión sobre el conjunto de células o genes. La implementación de métodos de reducción dimensional se complementa con la capacidad de representar gráficamente los datos. Se ofrecen estrategias para la interpretación funcional de las jerarquías obtenidas a través del análisis de los niveles de expresión de ontologías como GO terms, pathways o sets de marcadores celulares.
2. Desarrollo de algoritmos para dotar de soporte estadístico a los ordenamientos o jerarquías de estados celulares derivadas a partir de single cell RNA-seq data.
Hasta ahora, ningún método de ordenamiento celular para análisis de single cell permite dotar a la jerarquía resultante de un valor de significación estadística. Teniendo en cuenta la gran cantidad de ruido que arrastran estos métodos y la estocasticidad de la transcripción celular, entre otros muchos factores (ver [4] y [5]), resulta de gran interés desarrollar un algoritmo que permita determinar la influencia del ruido en la estructura y así determinar si esas jerarquías obtenidas representan una heterogeneidad confiable de estados celulares.
3. Aplicación del software para la interpretación de los datos obtenidos a partir de la secuenciación de las células CD4 T activadas en condiciones ex-vivo procedentes de dos donantes, uno de ellos permisivo y el otro resistente a la infección del VIH
El algoritmo es aplicado sobre los datos de experimentos de sigle cell que se están realizando en el laboratorio y varias hipótesis obtenidas de análisis previos. Partiendo de los datos de células T CD4 de dos pacientes sanos, uno precaracterizado como resistente a la infección y el otro como permisivo, se han realizado varios tipos de análisis. Primero, si el algoritmo era capaz de reconstruir los diferentes estados de diferenciación celular, considerando los resultados del clustering y expresión de marcadores de superficie.

10
Segundo, se han usado sets de genes con niveles de expresión característicos de diferentes subtipos celulares para dar sentido a la ruta de diferenciación, con la ayuda de las ontologías y pathways antes mencionadas. Y tercero, detectando patrones de diferenciación en la expresión de genes entre los dos donantes.

11
3. Resultados
1.1. El paquete de bioconductor en R “sincell” Inspirados en los métodos ya publicados para citometría SPADE (ver [8]) y ViSNE (ver [9]), desarrollamos un paquete informático para el cálculo e interpretación funcional de jerarquías de estados celulares obtenidas a partir de single cell RNA-seq data. El paquete se ha implementado en el lenguaje R para la plataforma Bioconductor. La Figura 3 esquematiza el diseño del paquete y los diferentes workflows de análisis que permite. Este software provee de un entorno de análisis en el que se integran los abordajes ya existentes (señalados en la tabla de la Fig. 1 y destacados en letra negra en la Fig. 3) y se complementan con novedades algorítmicas que se detallan a continuación (destacadas en letra negra en la figura Fig. 3). El paquete tiene las siguientes funcionalidades:
I. Cálculo de jerarquías de estados celulares a partir de sus perfiles de expresión derivados de single-cell RNA-seq
i. Reducción de la dimensión y cálculo de distancias entre células: se provee de un set de distancias lineales y no lineales (p.ej basadas en la teoría de la información o correlación de rango), combinadas con métodos de reducción dimensional como PCA, ICA, t-SNE o MDS (ver Métodos).
ii. Clustering de células: se incluyen herramientas de clustering tanto jerárquicas como no jerárquicas, y puede trabajar sin límites de dimensión sobre el conjunto de células o genes.
iii. Estructuración en forma de grafo: algoritmos para el cálculo de jerarquías
de estados celulares.

12
II. Dar un soporte estadístico: sincell permite dotar de soporte estadístico a la estructura obtenida mediante dos algoritmos distintos que se presentan como novelad:
iv. Soporte estadístico por sampleo de subsets de genes de las céulas de partida.
v. Soporte estadístico por generación in-silicio de pseudo-réplicas de las células de partida seguida de bootstrapping.
III. Dar una interpretación funcional
vi. Cálculo de correlación entre el grafo obtenido con el grafo obtenido cuando
se restringe el análisis a los niveles de expresión de un set de genes basados en ontologías como GO terms, pathways o sets de marcadores celulares.
IV. Representación gráfica: el paquete se complementa con la capacidad de representar
gráficamente los datos. vii. Plotting en 2D de las reducciones dimensionales y los grafos obtenidos
viii. Coloreado del grafo por niveles de expresión
Fig. 3: Workflow propuesto por sincell en el que se pueden ver diferenciadas todas sus funciones y las relaciones entre estas. Los nombres en azul hacen referencia a los nuevos algoritmos propuestos en este

13
trabajo y los negros a los ya presentados en otros métodos. Los números romanos identifican cada etapa del workflow con cada una de las funcionalidades antes descritas.
Antes de empezar a utilizar sincell el usuario debe llevar a cabo el preproceso de los datos, el único paso para el cual el paquete no trae funciones preimplementadas, que consiste en la cuantificación y normalización de los niveles de expresión por gen a partir de los datos de RNA-seq. Puede aplicarse cualquier combinación de métodos que se desee, con la única condición de que este proceso de como resultado una matriz numérica cuyas columnas corresponden a las células secuenciadas y las filas a cada uno de los genes cuantificados, como puede verse en el ejemplo de la Fig. 4.
Fig. 4: Ejemplo de formato que acepta sincel. Se trata de una matriz numérica donde cada columna corresponde a una célula de la muestra y cada fila a los valores log-normalizados de las RPKM de los genes en cada una de las células.
I. Cálculo de jerarquías de estados celulares a partir de sus perfiles de expresión derivados de single-cell RNA-seq
i. Reducción de la dimensión y cálculo de distancias entre células Una vez tenemos los datos listos para ser usados por sincell, lo primero que debemos hacer es calcular la matriz de distancias entre células individuales asociada, que servirá de base para las siguientes etapas. Para ello, opcionalmente se puede realizar una reducción dimensional previa al cálculo de distancias, mediante diferentes estrategias: PCA, ICA, t-SNE o MDS (ver Métodos), La reducción de la dimensión puede ser útil como forma de retener la parte más informativa de la varianza de los datos y eliminar la parte que puede considerarse desestructurada o ruidosa. Sin embargo, dependiendo de los datos de partida, puede ocurrir que su estructura no se recoja bien en unas pocas dimensiones, por lo que una reducción severa de la dimensión descartaría información relevante. En sincell se permite al usuario explorar estas alternativas (p.ej. a través de la inspección de los autovalores de un PCA) y decidir cuánta información conservar para el cálculo de las distancias entre las células. Además, el paquete permite explorar distancias tanto lineales o no lineales, como continuas o discretas. En Métodos se describen sucintamente estas distancias. El paquete permite así cualquier combinación de métodos de reducción dimensional y distancias posible, dando flexibilidad al usuario sin afectar al funcionamiento del resto del workflow.

14
ii. Clustering A continuación, el paquete implementa diferentes algoritmos para la detección de conglomerados de células especialmente similares entre ellas y diferentes del resto de la muestra. Los algoritmos se describen a continuación:
Fig. 5: Ilustración de un clustering de células individuales producido por el método K-NN Mutual con K=3. Los nodos en azul representan cada una de las células, las aristas representan la pertenencia de dos células a un mismo agrupamiento y su longitud representa distancia entre cada para de células. En la figura se muestran tres conglomerados detectados en los que sólo se mantienen las aristas entre pares de nodos que cumplen una condición recíproca de similitud (ver texto). Las posiciones de las células corresponden a las generadas aleatoriamente por un modelo de repulsión física entre nodos no unidos.
Métodos de clustering:
a) Subgraphs
Partiendo de un grafo completo (cada nodo conectado con todos los demás, en el que los nodos representan cada una de las células, las aristas representan la pertenencia de dos células a un mismo agrupamiento y su longitud representa distancia entre cada para de células) la forma más intuitiva de formar clústeres es eliminando todas las aristas menos las de menor distancia. Esto puede hacerse o bien fijando una distancia máxima y eliminando todas las mayores o bien fijando un número de aristas mínimas que se quieren conservar.
b) K-NN No confundir con el método tradicional de aprendizaje supervisado k nearest neighbours. Este ha sido modificado para poder operar de forma no supervisada, uniendo cada nodo con sus k vecinos más cercanos y devolviendo un grafo donde las componentes conexas corresponden a cada uno de los clústeres encontrados. El clustering pueden ser parcial (células quedan sin pertenecer a ningún grupo) y las células pertenecientes a grupos pueden estar unidas por más de una arista. Una ilustración del output producido por este método se representa en la Fig. 5.

15
c) K-NN Mutual Este método se propone aquí como novedad. Se trata de una modificación del método K-NN ya descrito que solo considera cercanos a un nodo aquellos vecinos de orden menor que k que a su vez tengan a dicho nodo dentro de sus vecinos de orden menor que k. Esto permite localizar puntos de convergencia (estados estables) en los datos y obtener clústeres más robustos.
Además de estos 3 métodos de clustering, el paquete sincell implementa un intérprete para poder integrar en el workflow los clústeres generados por el paquete mclust (ver [16]). De esta forma el usuario puede optar por la multitud de algoritmos de clúster ofrecidos por este paquete especializado. En el caso de que el usuario quisiera realizar el clustering usando otro paquete o software externo a R, debería parsear manualmente al formato apropiado que se describe en la documentación del paquete sincell. Estructuración en forma de grafo El principal objetivo del paquete sincell es el cálculo de ordenamientos o jerarquías de estados celulares subyacentes a un experimento de single-cell RNA-seq. Estos ordenamientos pueden interpretarse como la reconstrucción del continuo de estados intermedios originados a lo largo de un proceso de diferenciación o activación celular. Para el cálculo de estos ordenamientos o jerarquías, sincell ofrece una variedad de opciones, como su representación en forma de Scatter Plot sobre las dimensiones reducidas, formas arborescentes como el Mínimum Spanning Tree (MST), o métodos más complejos como el Maximum Similarity Spanning Tree (SST) o el Iterative Clustering. Estos métodos se describen a continuación:
a) Mínimum Spanning Tree (MST) Dado un grafo conexo y no dirigido, un árbol recubridor (Spanning Tree) de ese grafo es un subgrafo que tiene que ser un árbol y contener todos los nodos del grafo inicial. Cada arista tiene asignado un peso proporcional entre ellos, que representa la distancia entre los nodos que une, y se usa para asignar un peso total al árbol recubridor computando la suma de todos los pesos de las aristas del árbol en cuestión. Un árbol recubridor mínimo es un árbol recubridor que pesa menos o igual que todos los posibles árboles recubridores del grafo. Éste es el método de representación más extendido, a pesar de que el árbol recubridor mínimo no es necesariamente el que une los nodos más cercanos entre sí, es decir, las células más parecidas. Para evitar que células muy similares acaben en puntos lejanos del árbol, hemos implementado la opción de añadir a posteriori al MST las aristas resultantes correspondientes a clústeres generados de la forma descrita en la sección anterior. De esta forma evitamos que la ordenación obtenida por el algoritmo MST esté escondiendo como artefacto relaciones de parecido que parecen robustas de acuerdo a un método de clustering.

16
b) Maximum Similarity Spanning Tree (SST) Presentamos como novedad aquí este nuevo algoritmo de construcción de ordenamientos jerárquicos. A diferencia del anterior, que minimiza la suma de distancias de todas las aristas del árbol, el árbol recubridor por máxima similitud minimiza las distancias entre los nodos, de forma que cada uno de ellos quede unido a su nodo más cercano. Este algoritmo ofrece un nuevo punto de vista no usado hasta ahora en ningún método de análisis de single cell, y que consideramos que ofrece un mejor modelo de aproximación a las relaciones reales que hay en la biología celular, ya que prioriza el parecido entre las células ante el peso total de las aristas del grafo a la hora de construirlo. Como característica relevante para nuestra pipeline de análisis, este algoritmo de construcción de grafos admite como entrada no solo una matriz de distancia, sino también información sobre clústeres que se deseen conservar en el grafo resultante. Esto se consigue haciendo que el algoritmo trabaje sobre clústeres en lugar de células independientes, considerando en la primera etapa cada célula como un clúster de un solo individuo. La distancia entre clústeres se define como la mínima de las distancias entre nodos de clústeres distintos. Partiendo de los clústeres de los datos y considerando las células no pertenecientes a ningún clúster como clústeres de una sola célula (en caso de no haberse realizado un clustering todas las células serían consideradas clústeres individuales), se unen iterativamente los dos clústeres más cercanos hasta alcanzar un grafo completamente conexo. Este grafo será un árbol si no se han tomado clústeres iniciales, y la distancia entre dos clústeres se calcula como la mínima de las distancias entre nodos de clústeres distintos. Puede verse un ejemplo del resultado que aporta este método en la Fig. 6.
c) Iterative Mutual Clustering (IMC) También como novedad presentamos aquí como método alternativo el que hemos llamado Método de Clustering Mutuo Iterativo (IMC) que sigue la idea del algoritmo anterior, para obtener un grafo que represente de forma óptima los estados de diferenciación celular y agrupe las células en clústeres en función de ellos. El IMC consiste en la iteración sucesiva del método de clustering K-NN Mutual descrito anteriormente, tomando como datos de entrada en cada paso la salida de la iteración anterior y trabajando sobre clústeres en lugar de hacerlo sobre células independientes (mismo razonamiento que en el SST). El método finaliza cuando el grafo de salida es conexo, es decir, cuando hay al menos un camino a lo largo de la jerarquía que une cada nodo con cada uno de los demás. Este proceso permite ir ampliando a cada paso los clústeres obtenidos en la primera iteración y relacionándolos entre sí mediante sus células más parecidas, al mismo tiempo que dota a los clústeres de una estructura interna de relaciones entre sus componentes. El algoritmo comienza considerando cada célula un clúster independiente, se repite iterativamente el proceso de clustering seleccionado hasta obtener un único grafo conexo. La distancia entre dos clústeres se calcula como la mínima de las distancias entre nodos de clústeres distintos.

17
Fig. 6: Grafo final obtenido en un análisis de sincell usando el algoritmo SST combinado con un K-NN mutual de tres vecinos, y coloreado posteriormente con un gradiente de colores basado en la expresión de un marcador (desde verde: no expresado, a rojo intenso: altamente expresado). Pueden apreciarse pequeños clústeres que sirven como semilla para construir el grafo y representan grupos de células muy homogéneas entre sí.
II. Dar un soporte estadístico
La ausencia de soporte estadístico es una de las principales limitaciones de los abordajes existentes para el cálculo de jerarquías de estados celulares (ver Fig. 1). Así, estos métodos, dada una matriz de niveles de expresión génica siempre van a producir como output un ordenamiento o jerarquía. Sin embargo, puede ocurrir que la jerarquía calculada sea muy inestable frente a pequeñas variaciones de los datos de entrada. Esta posibilidad es especialmente relevante en el caso de datos de single-cell RNA-seq que, como se describió en la Introducción, son especialmente ruidosos tanto por razones técnicas como biológicas. Como principal utilidad del paquete sincell, se presentan dos abordajes para dotar de soporte estadístico a los ordenamientos o jerarquías de estados celulares obtenidas por los diferentes métodos descritos anteriormente. El primero se basa en la realización de múltiples submuestreos aleatorios (resampling) de los genes utilizados en el cálculo de las jerarquías. El segundo se basa en la generación in-silicio de pseudo-réplicas de las células de partida bajo un modelo de generación de variabilidad derivado de los mismos datos observados. Ambos abordajes se describen a continuación.
iii. Soporte estadístico por submuestreo de genes (subsampling) Se genera un conjunto de árboles a partir de sampleos aleatorios de un número fijo de genes para todos ellos y se comparan con el grafo obtenido con todos los genes. Realizando varios tests a la vez que se varía el número de genes con el que se genera la población de contraste se puede comprender cómo de robusta es la estructura final frente al ruido. El algoritmo parte fijando un número representativo de genes como tamaño de los subsets para el muestreo, y se generan los subsets como un submuestreo aleatorio sin reposición de los genes de la matriz original. Con cada uno de los subsets se construye un grafo siguiendo los mismos parámetros que para el árbol global. Finalmente se calcula la similitud de cada
18
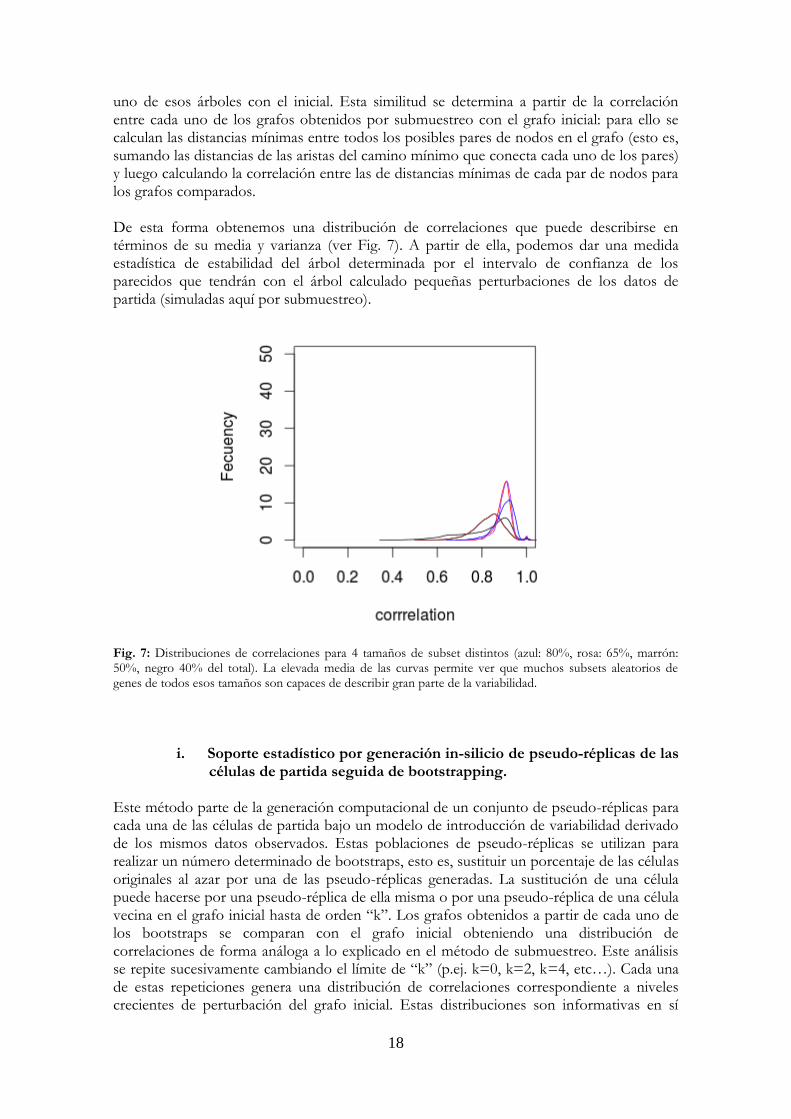
uno de esos árboles con el inicial. Esta similitud se determina a partir de la correlación entre cada uno de los grafos obtenidos por submuestreo con el grafo inicial: para ello se calculan las distancias mínimas entre todos los posibles pares de nodos en el grafo (esto es, sumando las distancias de las aristas del camino mínimo que conecta cada uno de los pares) y luego calculando la correlación entre las de distancias mínimas de cada par de nodos para los grafos comparados. De esta forma obtenemos una distribución de correlaciones que puede describirse en términos de su media y varianza (ver Fig. 7). A partir de ella, podemos dar una medida estadística de estabilidad del árbol determinada por el intervalo de confianza de los parecidos que tendrán con el árbol calculado pequeñas perturbaciones de los datos de partida (simuladas aquí por submuestreo).
Fig. 7: Distribuciones de correlaciones para 4 tamaños de subset distintos (azul: 80%, rosa: 65%, marrón: 50%, negro 40% del total). La elevada media de las curvas permite ver que muchos subsets aleatorios de genes de todos esos tamaños son capaces de describir gran parte de la variabilidad.
i. Soporte estadístico por generación in-silicio de pseudo-réplicas de las células de partida seguida de bootstrapping.
Este método parte de la generación computacional de un conjunto de pseudo-réplicas para cada una de las células de partida bajo un modelo de introducción de variabilidad derivado de los mismos datos observados. Estas poblaciones de pseudo-réplicas se utilizan para realizar un número determinado de bootstraps, esto es, sustituir un porcentaje de las células originales al azar por una de las pseudo-réplicas generadas. La sustitución de una célula puede hacerse por una pseudo-réplica de ella misma o por una pseudo-réplica de una célula vecina en el grafo inicial hasta de orden “k”. Los grafos obtenidos a partir de cada uno de los bootstraps se comparan con el grafo inicial obteniendo una distribución de correlaciones de forma análoga a lo explicado en el método de submuestreo. Este análisis se repite sucesivamente cambiando el límite de “k” (p.ej. k=0, k=2, k=4, etc…). Cada una de estas repeticiones genera una distribución de correlaciones correspondiente a niveles crecientes de perturbación del grafo inicial. Estas distribuciones son informativas en sí

19
mismas del grado de soporte estadístico que tienen los árboles obtenidos a cada nivel de perturbación. Adicionalmente, mediante un test de rango puede determinarse si estas distribuciones son significativamente diferentes entre sí, de modo que se evalúa el gradiente de desestructuración de un árbol ante grados crecientes de perturbación. De esta manera se permite detectar el grado de diferenciación o heterogeneidad jerárquica de una muestra biológica a partir del su gradiente de desestructuración: muestras con alta homogeneidad entre células van a tender a generar árboles con distancias pequeñas entre ellas y cuya distribución de correlación ante perturbaciones no va a empeorar significativamente a medida que se amplíe el grado de la perturbación (“k”). Por el contrario, muestras biológicas bien diferenciadas con una clara estructura jerárquica subyacente y distancias elevadas entre sus células mostrarán un gradiente de desestructuración evidente a medida que se sustituyen algunas de sus células por pseudo-réplicas de vecinos cada vez más lejanos. Los detalles de este abordaje se desarrollan a continuación
a) Adición de ruido
El algoritmo para la generación in silicio de la población de pseudo-réplicas correspondiente a cada una de las células originales es el siguiente: Sea M nuestra matriz de datos original, donde las columnas corresponden a las células y las filas a cada uno de los genes (Fig. 8):
Fig. 8: Cálculo de las varianzas de las filas (genes) de la matriz de expresión inicial.
Se calcula la varianza de cada uno de los genes y se ordenan los genes en deciles según el valor de su varianza (Fig. 9). La razón para agrupar los genes de esta forma es porque se ha observado que existe una relación entre la media y varianza de los genes en el RNA-seq (ver [19]), y ya que la razón de generar las pseudo-réplicas es tratar de simular todo el

20
continuo del proceso de diferenciación, en lugar de aplicar la varianza observada en la muestra utilizaremos una tomada al azar entre los genes que muestran varianzas similares.
Fig. 9: Deciles de los genes ordenados por sus varianzas.
Usando M como base, calculamos M´ sumando a cada gen gi un número aleatorio
perteneciente a la distribución uniforme de media 0 y varianza σi2´
, U(0, σi2´), donde σi
2´ es
una varianza escogida al azar entre las varianzas de los genes que pertenecen al mismo decíl
que gi .
ci = (gi,1,, gi,2, …, gi,m) = (gj)i ci
’ = (gj + U(0, σj2´))i
De esta forma, introduciremos un nivel de variación aleatoria en un gen de acuerdo a un valor aleatorio acotado entre los límites esperables de varianza en genes con niveles de expresión media similares. Con este procedimiento se obtienen nuevas muestras introduciendo niveles de ruido aleatorios en todas las variables que oscila entre límites derivados de la variabilidad observada en los mismos datos considerando además la relación media-varianza característica de la tecnología RNA-seq. Como resultado, se generan “células artificiales” o pseudo-réplicas in silicio intermedias que ayudan a poblar el espacio continuo de diferenciación (Figura 10).

21
Fig. 10: Scatter plot de las células originales (azul) y sus pseudo-réplicas (rojo) obtenidas computacionalmente.
b) Modelo de simulación celular multiparamétrico
Este modelo celular tiene en cuenta 3 parámetros para simular el comportamiento de cada
gen (gi): la media (μi), la varianza (σi2), y la proporción de células en las que el gen aparece
expresado (αi). La construcción está basada en el modelo de estocasticidad observada en la
expresión de un gen en single cells publicado por Shalek et al 2014 (ver [15]). Para construir cada una de las nuevas células, se calcula el valor de cada gen como un valor
de la distribución normal de media μi y varianza σi2, N(μi, σi
2), y se multiplica por el valor
obtenido de una distribución de Bernoulli de probabilidad de éxito αi, Be(αi).
ci = (gi,1,, gi,2, …, gi,m) = (gj)i ci
’ = ((gj + N(μj, σj2)) * Be(αi))i
Soporte estadístico por pseudo-réplicas de las células de partida seguida de bootstrapping. Partiendo de una población de pseudo-réplicas (explicadas anteriormente), podemos realizar sustituciones de células en la matriz de datos original de diversos modos para incluir distintos niveles de ruido. Una vez fijado el porcentaje de células (“nodos”) que vamos a sustituir (típicamente el 15 % de las células), los distintos modos de sustitución son:
a) Sustitución por una pseudo-réplica propia:
Equivalente a añadir ruido a los valores de cada gen proporcionalmente a su rango de expresión en la muestra original.
b) Sustitución por una pseudo-réplica de un vecino de hasta orden k:
Las células son sustituidas por una pseudo-réplica generada a partir de la misma célula o a partir de una que no esté a más de k aristas de distancia de ella sobre el grafo que se quiere evaluar.

22
c) Sustitución aleatoria:
Las células son sustituidas por una pseudo-réplica cualquiera al azar.
Los modos de sustitución a) y c) son equivalentes a k=0 y k=máximo(k) en la notación del apartado b). Una vez se han llevado a cabo las sustituciones, se procede a construir nuevamente el grafo con los datos modificados. Repitiendo el proceso un número determinado de veces (generalmente construyendo hasta 100 grafos), se calcula la similitud entre ellos con el grafo original, de forma que obtenemos una distribución de correlaciones. Estas correlaciones se calculan a partir de las distancias entre todos los pares de células siguiendo el método de los caminos mínimos entre nodos descrito anteriormente. Si superponemos distintas distribuciones de correlaciones correspondientes a distintos métodos de sustitución (k=0, k=2, k=4, etc…), podremos comprobar si nuestro grafo es estable (si las distribuciones de correlación para un k=0 tiene una media de correlación cercana a 1 y poca varianza) y cómo niveles crecientes de perturbación (representados por las curvas correspondientes a vecinos de orden “k” mayores) se traducen en distribuciones de correlación significativamente desplazadas hacia correlaciones menores (determinadas mediante un test de Wilcoxon). Como se comentó anteriormente, esto permite observar el gradiente de desestructuración de un árbol ante grados crecientes de perturbación e interpretar este gradiente como reflejo del grado de homogeneidad/heterogeneidad en la jerarquía inicial. A continuación pueden verse unos ejemplos (Fig. 11 y Fig. 12):
Fig. 11: Distribuciones de correlaciones para dos métodos de reposición por pseudo-réplicas distintos. La curva negra corresponde a sustituir las células por pseudo-réplicas propias (es decir, añadir ruido blanco en función de su variabilidad a los genes) y la curva roja a una sustitución al azar. Las altas medias de correlación con el árbol original indican que los datos tienden a converger a esa estructura a pesar de gran cantidad de ruido, al tiempo que las diferencias de medias y varianza de las dos distribuciones confirman que no se trata de una estructura aleatoria sino claramente definida.

23
Fig. 12: Distribuciones de correlaciones correspondientes a distintos métodos de sustitución (de derecha a izquierda las curvas corresponden a k=0, k=4, k=8, k=16, k=all). La baja media ante la menor introducción de ruido revela una estructura poco fiable, que degenera rápidamente al incrementar ligeramente el ruido hasta ser prácticamente aleatoria. III. Dar una interpretación funcional: test de asociación funcional con la
jerarquía de estados celulares
ii. Correlación del grafo con los niveles de expresión de un set de genes con una función común
Una vez determinada la existencia de una jerarquía fiable de estados de diferenciación celular, podemos evaluar si la estructura de grafo obtenida está asociada a los niveles de expresión de un determinado set de genes implicados en una determina función. Para ello se construye el árbol utilizando únicamente los valores de expresión de un set “S” de genes, como puede ser una ruta KEGG (ver [17]) o GO terms (ver [18]). A continuación, se evalúa el parecido de este nuevo grafo “S” con el original “O” mediante el cálculo de correlación entre dos grafos, esto es: calculando las distancias mínimas entre todos los pares de nodos dentro del grafo (esto es, sumando el peso de las aristas del camino mínimo que conecta dos nodos) y luego calculando la correlación entre las dos listas de distancias mínimas.
En un segundo paso, se genera una población de 1000 grafos “Ai, i=1…1000”a partir de
sets de genes aleatorios del mismo tamaño del set de interés, generando así la distribución de parecidos de fondo sobre la que atribuir un p-valor al parecido observado entre “S” y “O”. Si este proceso se realiza a gran escala para un conjunto elevado de sets de genes, el p-valor se corrige por testeo múltiple.

24
Representación gráfica
iii. Plotting en 2D
La información generada sobre las relaciones de parecido y la estructura jerárquica de los estados celulares, incluyendo la reducción dimensional si esta se ha llevado a cabo, puede representarse gráficamente en plots de dos dimensiones. Las reducciones dimensionales se representan como un Scatter Plot, y las coordenadas de los puntos visualizados pueden usarse posteriormente como layout para construir el grafo jerárquico sobre ellas. De no usarse reducción dimensional u otra matriz de coordenadas introducida por el usuario, la representación de las posiciones de los nodos del grafo se realiza por un modelo físico de repulsión entre nodos manteniendo fija la distancia de las aristas.
iv. Coloreado del grafo por niveles de expresión Tanto los nodos del grafo como los puntos de un Scatter Plot pueden colorearse fácilmente siguiendo un gradiente de colores basado en los niveles de expresión de marcadores (genes de interés) mediante una herramienta incluida en el paquete. Esto resulta extremadamente útil para seguir la variación de un gen a lo largo de las diferentes etapas de la ruta de diferenciación y encontrar genes clave que condicionen ese comportamiento.
3.2. Evaluación de la metodología de soporte estadístico desarrollada mediante su aplicación a jerarquías celulares previamente caracterizadas de mioblastos esqueléticos
Para ilustrar la utilidad de los algoritmos de soporte estadístico implementados en el paquete sincell , escogimos un set de datos publicados recientemente en Trapnell et al 2014 (ver [13] y Fig. 13). En este trabajo los autores analizan un conjunto de 4 experimentos de single cell RNA-seq realizados sobre muestras de mioblastos primarios tomadas a 4 tiempos diferentes (0, 24, 48 y 72 horas) tras su exposición a un agente de diferenciación celular: Para ello colocaron células de mioblasto de músculo esquelético humano en estado de mitosis avanzada e indujeron la diferenciación cambiando las células a un medio low-serum. En el mismo trabajo los autores presentan el algoritmo de determinación de jerarquías de estados celulares “monocle” descrito en la Introducción. Básicamente, monocle analiza conjuntamente las células de todos los tiempos y las ordena en un Minimum Spanning Tree, usando la mayor distancia entre nodos de ese árbol para definir una ordenación en el proceso de diferenciación a la que denominan pseudotiempos. Este ordenamiento se hace siguiendo la expresión de los genes diferencialmente expresados entre dos grupos de tiempos: 0 horas contra el conjunto formado por la unión de 24, 48 y 72 horas. Los autores muestran que el proceso de diferenciación se va alcanzando paulatinamente desde el tiempo 0 (célula primaria no diferenciada) hasta el tiempo 72 a través de estados celulares continuos. De forma importante para nuestro trabajo, los autores muestran también que,

25
una vez comenzada la diferenciación, dentro de un determinado tiempo existe un cierto nivel de heterogeneidad celular que puede llegar a recapitular todo el rango de estados intermedios de diferenciación.
Fig. 13: Ejemplo de árbol generado por monocle tomada de la página 7 de la vignette del paquete de R del mismo nombre. Este conjunto de datos nos permite evaluar nuestros algoritmos de soporte estadístico para jerarquías de estados celulares sobre la siguiente hipótesis: las jerarquías obtenidas a partir de las células de tiempo “0” en estado indiferenciado, deberían ser menos fiables estadísticamente que aquellas obtenidas a tiempos de diferenciación mayores. Así, a tiempo “0” la estructura de datos inicial debería ser más homogénea que la mostrada a tiempos mayores que contienen una heterogeneidad de pseudotiempos más elevada. Utilizando nuestro paquete, hemos realizado pruebas de soporte estadístico para los árboles construidos utilizando las células de cada tiempo por separado. Para ello se ha calculado las distribuciones de correlación de los parecidos con el grafo inicial obtenidos al realizar boostrap de pseudo-réplicas celulares a grados crecientes de vecindario “k”: k=0, k<=2, k<=4, k<=8, k<=16 y k<=max(k). Estas distribuciones se representan en la Fig. 12. Como se observa en la Figura 14, para los tiempos 0 (indiferenciado) y 24 (diferenciación temprana de los mioblastos), las distribuciones de correlación obtenidas presentan una media baja y una varianza elevada, mostrando que los ordenamientos obtenidos no son estables estadísticamente ante pequeñas introducciones de ruido. Adicionalmente en estos tiempos, las perturbaciones a grados “k” crecientes no se traducen en diferencias significativas entre sus correspondientes distribuciones de correlación, reflejando la ausencia de un gradiente significativo de desestructuración progresiva propio de muestras con escasa heterogeneidad. En conjunto, ambos resultados muestran la ausencia en estos tiempos de una heterogeneidad marcada que permita establecer jerarquías creíbles.

26
Por el contrario, los tiempo 48 y 72 tienen medias más altas y sus varianzas mucho menores, lo que indica que lo árboles expuestos al ruido son similares al árbol obtenido y por tanto están soportados estadísticamente. Así mismo, los tests de Wilcoxon indican un gradiente significativo de desestructuración ante niveles crecientes de “k”, lo que indica un nivel de heterogeneidad relevante que se traduce en ordenamientos jerárquicos estables. En resumen, nuestros resultados muestran que para los tiempos 0 y 24 el ruido de los datos es indiferenciable de la variabilidad de las células, obteniendo estructuras aleatorias que varían drásticamente con cualquier nivel de ruido y guardan escaso parecido con el árbol original, mientras que para los tiempos 48 y 72 el nivel de diferenciación de las células es mayor que el ruido base y se obtiene una estructura estable que se va deteriorando gradualmente al incrementar los niveles de ruido.
Fig. 14: Diferentes distribuciones de correlación obtenidas por generación de pseudo-réplicas a diferentes límites de k-vecinos (ver texto; k=0: azul oscuro, k=4: azul claro, k=8: verde, k=16: rojo, k=all: morado) a partir de los 4 subsets de tiempo de los datos de monocle junto a sus resultados en el test de Wilcoxson (significancia positiva o negativa) Estos resultados muestra la capacidad de sincell para dotar de soporte estadístico relevante a las jerarquías de estados celulares derivadas de muestras tomadas a tiempos aislados. Como principal resultado de este trabajo concluimos que el test de soporte estadístico basado en boostrap con pseudo-réplicas in silicio es capaz de inferir la existencia de estados heterogéneos de diferenciación a nivel de célula individual dentro de una única muestra biológica.

27
3.3. Aplicación al estudio de la heterogeneidad celular en la activación de CD4 T cells
Hemos utilizado las herramientas del paquete sincell para analizar las muestras secuenciadas de dos pacientes sanos en condiciones comparables (ver Fig. 15). Células del primer donante habían sido caracterizadas como permisivas a la infección del virus mientras que células del segundo donante eran altamente resistentes. Secuenciado las células T CD4 tras 72 horas de activación con TCR de ambos donantes esperábamos encontrar un gradiente de diferenciación en las células del sistema inmute que permitiera explicar los distintos niveles de permisividad entre los individuos. Los datos crudos de ambos donantes fueron normalizados mediante RPKM log10 transformado. Aunque esta técnica no se considera en general la más apropiada para analizar datos de NGS, se ha comprobado internamente mediante réplicas técnicas que sí lo es a la hora de trabajar con datos de single cell. Inicialmente comenzamos analizando solo al primer donante, ya que los datos del segundo no llegaron hasta pasado el segundo mes de las prácticas. Los métodos de reducción dimensional ordinarios no permitían diferenciar ningún patrón o ruta de diferenciación entre las células, ya que el porcentaje de variabilidad explicado por cada componente reducida era muy escaso (Fig. 2). Por ello, nos vimos en la necesidad de empezar a trabajar con distintas combinaciones de distancias y reducciones dimensionales para tratar de encontrar un sentido a los datos. Dado que la representación en dos dimensiones no arrojaba ninguna luz, probamos con una representación en forma de grafo, donde además de la posición relativa de las células en un plano podíamos estudiar a la vez las relaciones entre las células más semejantes y los clústeres internos. Sin embargo, la representación gráfica nos mostraba siempre una nube de nodos entre las que apenas había diferenciación alguna, y ningún marcador genético o grupo funcional parecía imponer un orden en el proceso de diferenciación. Los datos del segundo paciente eran bastante diferentes a los del anterior. Inicialmente, debido al bajo número medio de reads por librería, descartamos muchas células achacándolo a un error de PCR, al igual que había ocurrido con unas pocas células del primer donante. Más adelante veríamos que no, que efectivamente esas células estaban bien secuenciadas pero que el número de reads era menor simplemente porque las células estaban en un estado de activación más inmaduro, y por ello tuvimos que reconsiderar las células que se incluirían en el estudio. Las células del segundo donante se comportaban de manera muy distinta, formando multitud de pequeños clústeres que resultaron fácilmente ordenables aplicando los métodos que no habían arrojado luz sobre el primero. También formaba estructuras mucho más robustas y que tendían a converger entre ellas, cosa que tampoco pasaba con los datos del primer donante. No fue hasta la llegada de los datos del segundo paciente y el análisis combinado de ambos donantes junto con algunas células a nivel de muestra completa previamente caracterizadas que pudo verse que el continuo de diferenciación debido a la activación por TCR realmente existía en ambos pacientes. Analizando esta nueva ruta obtuvimos una serie de marcadores que correlacionaban y extrajimos los que mayor relación parecían tener con el proceso de diferenciación para tratar de analizar las muestras por separado.

28
Fig. 15: Grafos obtenidos mediante sincell a partir de los datos de los dos donantes (izquierda primer donante, derecha el segundo) y coloreados posteriormente por los niveles de expresión del marcador IFITM, el mejor candidato de los genes correlacionados con la ruta de diferenciación. El color rojo indica mayor nivel de expresión, decreciendo según avanza en el gradiente hacia el color verde.
Al representar las células de ambos pacientes superpuestas sobre un eje que seguía la ruta de diferenciación pudo comprobarse que las células del primer donante eran efectivamente indiferenciables porque quedaban prácticamente todas agrupadas en el extremo inicial del proceso, mientras que las del segundo donante se repartían en pequeñas islas a lo largo de todo el eje.

29
4. Discusión y conclusiones
En resumen, sincell es un paquete de R que permite realizar análisis muy flexibles sobre matriz de datos de gran dimensión especialmente desarrollado para el análisis de datos de expresión de single-cell RNA-seq. Este análisis se basa en las siguientes etapas: 1. Extracción de los niveles de similitud de las células individuales junto con una
reducción dimensional que permite la representación bidimensional de los datos.
2. Búsqueda de clústeres y generación de una estructura jerárquica que mantenga la información sobre la relación entre las células.
3. Dotar a dicha estructura jerárquica de un soporte estadístico que permita asegurar la
existencia de heterogeneidad de estados de diferenciación o activación a nivel de célula individual.
4. Realizar tests de asociación funcional de sets de genes con la estructura jerárquica. 5. Finalmente, obtenemos una representación en dos dimensiones que muestra las células
bajo una estructura jerárquica probada robusta y donde las aristas contienen la información de la distancia real entre las células, información que mantenemos en la representación final a pesar de haber reducido los datos a dos dimensiones, añadiendo así la información que permite reconstruir las rutas de diferenciación celular y, en caso de los clústeres, distinguir los estados celulares intermedios de las células en proceso de diferenciación.
De esta forma permitimos que el grafo represente procesos continuos de diferenciación celular en los que pueden diferenciarse los estados celulares o etapas intermedias entre estados en las que se encuentra cada una de las células.
Como característica relevante de nuestra plataforma de software, el paquete es capaz de dotar de soporte estadístico a los ordenamientos o jerarquías obtenidos con los demás métodos existentes para el estudio de datos de single cell, complementándolos. Además, nuestro paquete provee la posibilidad de portar sus algoritmos a otros tipos de datos, combinar distintas pipelines de análisis o modificarlas incluyendo código propio para un análisis personalizado, y obtener una idea sobre la idoneidad del modelo de análisis usado en cada set de datos particular. Además, su estructura modular y de objetos simples permite

30
combinarlo fácilmente con otros paquetes de R o incluso programas de análisis para incrementar sus funcionalidades según las necesidades del usuario. En el caso de que la tecnología de secuenciación de RNA-seq siga mejorando y abaratándose, llegará un punto en que los experimentos de single cell podrán realizarse sobre un gran número de células al mismo tiempo. Si ese número fuera lo suficientemente grande es posible que la mecánica de generar pseudo-réplicas para simular los estados de transición entre estados celulares pierda parte de su importancia en el análisis, pero todas herramientas proporcionadas por el paquete seguirán siendo plenamente funcionales a pesar del aumento del tamaño de las matrices de datos, ya que los límites de procesamiento vienen impuestos por el hardware donde se ejecuta y no el software. Un detalle a tener en cuenta, es que el modelo de ruido generado por el método de soporte estadístico basado en la generación de pseudo-réplicas puede no resultar óptimo para determinados sets de datos. En tal caso, al tratarse de un software de código abierto y documentado, el usuario puede modificar la generación de ruido por el método que crea óptimo para su experimento. A pesar de ello, el ruido genérico implementado ha dado resultados positivos en los dos sets de datos analizados hasta ahora. La parte de análisis funcional del paquete está aún incompleta, y actualmente seguimos explorando posibilidades para dotar a sincell de todas las herramientas necesarias para convertirlo en una herramienta completamente independiente de las demás incluso para análisis funcionales complejos.

31
5. Materiales y Métodos
Paquetes / librerías de R utilizadas La versión de R utilizada para realizar los experimentos ha sido la 3.0.2. Todos los paquetes de R son públicos y pueden descargarse desde CRAN, excepto sincell que es suministrado junto a este documento y pronto será público a través de Bioconductor: igraph 0.7.1 entropy 1.2.0 scatterplot3d 0.3-35 MASS 7.3-33 TSP 1.0-9 ggplot2 1.0.0 reshape 1.4 fields 7.1 spam 0.41-0 grid (R-base) maps 2.3-7 proxy 0.4-12 parallel (R-base) fastICA 1.2-0

32
Métodos de reducción dimensional:
a) PCA El análisis de componentes principales es una técnica utilizada para reducir la dimensionalidad de un conjunto de datos. Intuitivamente la técnica sirve para hallar las causas de la variabilidad de un conjunto de datos y ordenarlas por importancia. Técnicamente, el ACP busca la proyección según la cual los datos queden mejor representados en términos de mínimos cuadrados. El PCA comporta el cálculo de la descomposición en autovalores de la matriz de covarianza, normalmente tras centrar los datos en la media de cada atributo.
b) MDS El escalamiento multidimensional es un medio de visualizar el nivel de similitud de los casos individuales de un conjunto de datos. Se refiere a un conjunto de técnicas de ordenación relacionados utilizados en la visualización de la información, en particular, para visualizar la información contenida en una matriz de distancia, por lo que puede usarse para reducir la dimensionalidad de los datos de los cuales proviene dicha matriz. En algoritmo MDS tiene como objetivo colocar cada objeto en el espacio N-dimensional de tal manera que las distancias entre objetos se conservan, así como sea posible. Cada objeto se le asigna coordenadas en cada uno de los N dimensiones. El número de dimensiones de una parcela MDS N puede ser superior a 2 y se especifica a priori. La elección de N = 2 optimiza las ubicaciones de los objetos de un gráfico de dispersión bidimensional.
c) t-SNE t-Distributed Stochastic Neighbor Embedding es una técnica para la reducción de dimensionalidad que es especialmente adecuado para la visualización de conjuntos de datos de alta dimensión. La técnica se puede implementar a través de aproximaciones Barnes-Hut, lo que le permite ser aplicado en grandes conjuntos de datos del mundo real.
d) ICA El Análisis del Componente Independiente es un método computacional que sirve para separar una señal multivariante en subcomponentes aditivos suponiendo que la señal de origen tiene una independencia estadística y es no-Gausiana. Distancias:
a) Euclidean La distancia euclidiana o euclídea es la distancia "ordinaria" (que se mediría con una regla) entre dos puntos de un espacio euclídeo, la cual se deduce a partir del teorema de Pitágoras.

33
b) Mutual Information por bines La información mutua o transinformación de dos variables aleatorias es una cantidad que mide la dependencia mutua de las dos variables, es decir, mide la reducción de la incertidumbre (entropía) de una variable aleatoria, X, debido al conocimiento del valor de otra variable aleatoria Y. En este caso particular, los datos se discretizan previamente en bines y se calcula la dependencia de las variables sobre estos bines.
c) Dissimilarities La medida de disimilitud viene definida como una transformación lineal de la correlación (Pearson o Spearman) entre las variables que le otorgan las características matemáticas de distancia. Esta medida indica la fuerza y la dirección de una relación lineal y proporcionalidad entre dos variable. Se considera que dos variables cuantitativas están correlacionadas cuando los valores de una de ellas varían sistemáticamente con respecto a los valores homónimos de la otra. Datos de Monocle Los datos usados pertenecientes al paper de monocle son publicamente accesibles a través de GEO: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52529 Datos de RNA-seq HIV Los datos de los dos donantes secuenciados por single cell RNA-seq no son públicos y no pueden ser facilitados ya que actualmente están siendo analizados por el equipo de Amalio Telenti.

34
6. Bibliografía
1. Wang Z, Gerstein Z, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Genetics 10, 57-63. doi:10.1038/nrg2484. 2. Tsioris K, Torres AJ, Douce TB, Love JC (2014) A New Toolbox for Assessing Single Cells. Annu. Rev. Chem. Biomol.. 5, 455–77. doi:10.1146/annurev-chembioeng-060713-035958 3. Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, et al. (2013) Accounting for technical noise in single-cell RNA-seq experiments. Nature Methods 10, 1093–1095. doi:10.1038/nmeth.2645.
4. Rand U, Rinas M, Schwerk J, Nöhren G, Linnes M, et al. (2012) Multi‐layered
stochasticity and paracrine signal propagation shape the type‐I interferon response. Molecular Systems Biology 8, 584. doi: 10.1038/msb.2012.17 5. Vogel C, Marcotte EM (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature reviews Genetics 13(4):227-32. doi:10.1038/nrg3185. 6. Fluidigm C1™ Single-Cell Auto Prep System: http://www.fluidigm.com/c1-single-cell-auto-prep-system.html 7. Illumina HiSeq Performance Parameters: http://systems.illumina.com/systems/hiseq_2500_1500/performance_specifications.ilmn 8. Qiu P, Simonds EF, Bendall SC, Gibbs KD Jr, Bruggner RV, et al. (2011) Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol 29: 886–891. doi:10.1038/nbt.1991. 9. Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, et al. (2013) viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31: 545–552. doi:10.1038/nbt.2594. 10. Roweis ST, Saul LK (2000) Nonlinear dimensionality reduction by locally linear embedding. Science 290: 2323–2326. doi:10.1126/science.290.5500.2323.

35
11. Bendall SC, Davis KL, Amir ED, Tadmor MD, Simonds EF, et al. (2014) Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B Cell Development. Cell 157: 714-725. doi:10.1016/j.cell.2014.04.005. 12. Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, et al. (2014) Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition into Cell Types. Science 343: 776-779. doi:10.1126/science.1247951. 13. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, et al.(2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology 32: 381-356. doi:10.1038/nbt.2859. 14. Kaski S, Peltonen J (2011) Dimensionality Reduction for Data Visualization. IEEE Signal Processing Magazine 100-104. doi: 10.1109/MSP.2010.940003 15. Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, et al. (2014) Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature aop. doi:10.1038/nature13437 16. Fraley C, Raftery AE (1999) MCLUST: Software for Model-Based Cluster Analysis. Journal of Classification, Volume 16, Issue 2 , 297-306. doi: 10.1007/s003579900058 17. Kanehisa M, Goto S (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl. Acids Res. 28 : 27-30. doi: 10.1093/nar/28.1.27 18. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25 – 29. doi:10.1038/75556 19. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biology 2010, 11:R106. doi:10.1186/gb-2010-11-10-r106.