departamento de quimica organica - dialnet
TRANSCRIPT

DEPARTAMENTO DE QUIMICA ORGANICA
SÍNTESIS Y ESTUDIO
DE LAS REACCIONES DE CICLOADICIÓN DE
BIS-DITIOLO-1,4-TIAZINAS Y BIS-DITIOLOPIRROLES
Susana Barriga Falcón
Cáceres, 30 de octubre de 2001

Edita: Universidad de Extremadura Servicio de Publicaciones C / Pizarro, 8 10071 Cáceres E-Mail: [email protected] http://www.pcid.es/public.htm

UNIVERSIDAD DE EXTREMADURA
DEPARTAMENTO DE QUIMICA ORGANICA
SÍNTESIS Y ESTUDIO DE LAS REACCIONES DE CICLOADICIÓN
DE BIS-DITIOLO-1,4-TIAZINAS Y BIS-DITIOLOPIRROLES
Memoria que para optar al Título de
Doctora por la Universidad de Extremadura
presenta:
Susana Barriga Falcón
Cáceres, 30 de octubre de 2001


D. Tomás Torroba Pérez, catedrático del Departamento de Química de la Universidad
de Burgos y D. Carlos Fernádez Marcos, profesor asociado de la Universidad de
Extremadura
CERTIFICAN:
Que la memoria adjunta titulada “Síntesis y estudio de las reacciones
de cicloadición de bis-ditiolo-1,4-tiazinas y bis-ditiolopirroles”, para
optar al grado de Doctora en Ciencias Químicas presentada por
Susana Barriga Falcón, ha sido realizada bajo nuestra dirección en
los laboratorios del Departamento de Química Orgánica de la
Universidad de Extremadura.
Considerando que constituyen un trabajo de Tesis Doctoral,
autorizamos su presentación en la Universidad de Extremadura.
Y para que así conste, expedimos el presente certificado en Cáceres a
1 de Septiembre de 2001
Fdo.: Tomás Torroba Pérez Fdo.: Carlos Fernández Marcos


AGRADECIMIENTOS
Al Profesor Tomás Torroba Pérez, director de este trabajo, por darme la oportunidad de formar parte de
su grupo de investigación, por sus valiosas enseñanzas y consejos así como por haberme introducido en la
química del azufre y por haberme hecho madurar científicamente. Al Dr. Carlos Fernández Marcos,
director de este trabajo, por facilitarme los primeros contactos con el laboratorio, por estar siempre que lo
he necesitado al pie del cañón y por su enorme hospitalidad cada vez que le he pedido asilo en Cáceres.
Al Profesor Olivier Riant por acogerme en su grupo de investigación en dos ocasiones, Francia y Bélgica,
por el excelente trato recibido y por hacerme participe de sus grandes conocimientos científicos.
Al Dr. Gabriel García Herbosa, por introducirme en la voltametría cíclica y el EPR.
A mis compañeros de laboratorio, especialmente a Ana, Sara y Pedro, porque sé que sin sus ánimos,
consejos y apoyo quizás habría tirado la toalla y no habría llegado hasta el final. Además, es imposible
olvidar los buenos momentos vividos dentro y fuera del laboratorio, las múltiples risas y las aventuras
vividas en la SIBERIA. A Jose, porque siempre ha estado dispuesto a ayudarme en el laboratorio y sin él
las cosas son más complicadas.
A mi familia, especialmente a mi madre y mi hermana, por darme todo su apoyo y confiar ciegamente en
mí. A Jorge, por ser el receptor universal de mis agobios y tener la paciencia infinita de soportarlo sin
quejas. A mis amigos, porque sin ellos todo sería diferente, gracias por escucharme, sosegarme y hacer
que me divierta como una loca cuando estamos juntos. A Mercedes, mi madre científica, por escucharme
y animarme siempre, por aconsejarme científicamente y hacerlo tan bien.
A mis compañeros de laboratorio en el extranjero, sobre todo a Victor y a Ahlem, en Paris, y a James y a
Naouëlle, en Lovaina, por haber facilitado tanto mi integración en los laboratorios como en las ciudades
correspondientes.
Al Ministerio de Educación y Cultura, por la concesión de una beca F. P. I. mediante la cual ha sido
posible realizar este trabajo.
Al Imperial College de Londres por permitirnos usar sus servicios de masas, RMN, análisis y difracción
de rayos X. A la Universidad de Salamanca, por los servicios de masas y RMN. A la Universidad de
Valladolid, por la difracción de rayos X del compuesto 274c. A la Universidad de Santiago, por los
servicios de masas y RMN, especialmente a las Dras. Ana Fernández Gacio y Ana Gómez Neo, porque sé
que en más de una ocasión les he quitado su tiempo personal de utilización de los aparatos y además mis
espectros les han causado algún que otro quebradero de cabeza. A la Universidad de Burgos, por sus
servicios de RMN y análisis, y en especial a Jacinto, por soportar todas mis exigencias y ser tan generoso
con su tiempo.
En general, y espero no haberme olvidado de nadie, GRACIAS A TODOS LOS QUE HABEIS HECHO
QUE ESTO SEA POSIBLE.


ÍNDICE
1. INTRODUCCIÓN 1
2. OBJETIVOS 95
3. EXPOSICIÓN Y DISCUSIÓN DE RESULTADOS 99
4. PARTE EXPERIMENTAL 203
5. CONCLUSIONES 473
RESUMEN I
SUMMARY XV
ÍNDICE DE ESTRUCTURAS XXIX


ABREVIATURAS UTILIZADAS EN ESTA MEMORIA
A Amperios
Ar Aromático
BAIB Bis(acetoxi)iodobenceno
c Cuartete
ccf Cromatografía de capa fina
d Doblete
DABCO 1,4-Diazobiciclo[2.2.2]octano
dc Doble cuartete
DBA Dibenzoilacetileno
DCA Dicianoacetileno
DMAD Acetilendicarboxilato de dimetilo
E0 Potencial de semionda
EM Espectrometría de masas
Epox Potencial de pico de la onda de
oxidación
Epred Potencial de pico de la onda de
reducción
FAB Técnica de bombardeo con
átomos rápidos
IE Impacto electrónico
IR Infrarrojo
J Julios
K Kelvin
M Concentración molar
m Multiplete
M+· Ión molecular
m/z Masa por unidad de carga
νmáx Máximo de número de ondas
NLO Óptica no lineal
p.f. Punto de fusión
Ph Fenilo
RMN Resonancia magnética nuclear
s Singulete
sa Singulete ancho
st Stretching
S Siemens
δ Desplazamiento químico
t Triplete
TBDMS Terc-butildimetilsilano
TC Transferencia de carga
Tc Temperatura crítica
Θ Temperatura de Curie-Weiss
TEMPO 2,2,6,6-Tetrametilpiperidin-1-
oxilo
THF Tetrahidrofurano
TTF Tetratiafulvaleno
V Voltios


1. INTRODUCCIÓN


Introducción 3
QUÍMICA DE MATERIALES: INTRODUCCIÓN Y CONCEPTOS
Los materiales ejercen una gran influencia en el desarrollo cultural, socioeconómico,
demográfico y geográfico de la sociedad. La definición de material como “una sustancia
que presenta propiedades que pueden ser útiles en la construcción de maquinaria,
estructuras, aparatos y productos” 1
conecta significativamente material con función y,
a la vez, función con utilidad. La Química, a su vez, se define como “el estudio de la
composición, estructura y propiedades de las sustancias y de las transformaciones que
pueden sufrir dichas sustancias para dar lugar a otras sustancias”.2 Por tanto, la ciencia
de materiales, como subdisciplina de la Química, nos permite comprender y controlar
las conexiones fundamentales entre estructura y función desde el nivel molecular hasta
la escala macroscópica. Esta comprensión conduce a mejorar la composición, la
estructura y los métodos sintéticos, y permite el desarrollo de nuevos tipos de materiales
avanzados que presentan mejores propiedades.
1 Bever, M. B.; Ed. “Encyclopedia of Materials Science and Engineering”. Vol. 1, Pergamon Press,
Oxford, 1986. 2 Gillespie, R. J.; Humphreys, D. A.; Baird, N. C.; Robinson, E. A. “Chemistry”. 2nd ed. Prentice Hall,
Upper Saddle, NJ, 1989.

4 Tesis Doctoral Susana Barriga Falcón
Clasificación de materiales
Los materiales pueden ser clasificados en diversas categorías basándonos en su
constitución química y en sus propiedades físicas.3 Los materiales sólidos se agrupan
generalmente en tres categorías básicas: metales, cerámicas y polímeros. Además, hay
otras dos categorías importantes: composites, constituidos por combinación de dos o
más materiales diferentes, y semiconductores, que se distinguen por sus características
eléctricas inusuales. Además, los materiales pueden ser clasificados por su función en
materiales electrónicos, biomédicos, estructurales, ópticos (lineales o no lineales), etc.
Los materiales metálicos están normalmente constituidos por uno o más elementos
metálicos; son generalmente buenos conductores de la electricidad y del calor, y no son
transparentes a la luz visible. Estos materiales suelen ser muy maleables, por lo que son
muy usados en aplicaciones estructurales.
Las cerámicas son compuestos formados por elementos metálicos y no metálicos; no
conducen la luz ni la electricidad y son muy resistentes a las degradaciones térmicas o
ambientales. Son muy usados en aplicaciones estructurales, ópticas y electrónicas.
Los polímeros son moléculas de gran tamaño, formadas por una misma unidad química
que se repite numerosas veces, enlazándose covalentemente unas unidades con otras
para formar una cadena. Los polímeros tienen conductividades eléctricas y térmicas
muy bajas, y normalmente no soportan las altas temperaturas. Se usan en diversas
aplicaciones, como en aparatos electrónicos, pantallas digitales, litografías, soportes de
grabación, fibras para vestir, conservación de alimentos, etc.
El desarrollo de la ciencia y tecnología de materiales al comienzo del siglo veintiuno se
orienta hacia materiales con funciones específicas.4 Se buscan materiales que trabajen a
gran temperatura y que presenten estructuras resistentes, a la vez que una baja
dimensionalidad. Además, la investigación básica en ciencia de materiales persigue la
3 (a) Callister, W. D. Jr. “Materials Science and Engineering: An Introduction”. 3rd ed. Wiley, New
York, 1994. (b) Askeland, D. R. “The Science and Engineering of Materials”. 3rd ed. PWS, Boston, 1994.
4 Shi, C. X. “Highlights of Materials Science and Technology at the Tuning of the 21st century”.
Progress in Natural Science 1999, 9, 2-14.

Introducción 5
mejora de materiales convencionales, así como la síntesis de nuevos materiales
avanzados. La investigación más actual está orientada al descubrimiento de materiales
inteligentes, capaces de adaptar sus propiedades a necesidades más específicas en
función de la señal detectada.5 La química supramolecular ha dado lugar al desarrollo de
sustancias con propiedades electrónicas, magnéticas, ópticas, estructurales, mecánicas y
químicas de gran interés en el desarrollo de nuevos materiales. 6
Esto ha dado lugar a un
nuevo campo en la ciencia de materiales, el de los materiales moleculares o covalentes,
que incluyen cualquier tipo de material que esté constituido por moléculas discretas.
La manifestación de propiedades de gran interés tecnológico, como el ferromagnetismo
y la superconductividad, no depende de las moléculas individuales, sino del conjunto de
moléculas en estado sólido, y por lo tanto, estas propiedades son una consecuencia de
las interacciones moleculares. La comprensión de la estructura en estado sólido y los
métodos para predecir, controlar y modular la estructura son esenciales para el
conocimiento y la manipulación de dichas características.
1. Materiales orgánicos conductores, superconductores y magnéticos
El fenómeno de la electricidad y del magnetismo ha cautivado la imaginación y ha
fascinado a la humanidad desde su descubrimiento. La sociedad moderna sería
inconcebible sin los productos derivados de la explotación de estos fenómenos y, por
ello, la búsqueda de nuevos materiales que contengan propiedades inusuales es un
campo muy activo de investigación en la actualidad. Así, el descubrimiento de
materiales moleculares que exhiben ciertas propiedades vinculadas tradicionalmente con
la fase metálica, tales como la superconductividad7 y el ferromagnetismo,
8 ha impulsado
la investigación en este campo. Además, los materiales moleculares, en comparación
5 Mulhaupt, R. “From Alchemy to Modern Advanced Materials”. Chimia 1997, 51, 76-81.
6 Lehn, J. M. “Supramolecular Chemistry: Concepts and Perspectives”. VCH, Weinheim, 1995.
7 Williams, J. M.; Ferraro, J. R.; Thorn, R. J.; Carlson, K. D.; Geiser, U.; Wang, H. H.; Kini, A. M.;
Whangbo, M. H. “Organic Superconductors. Synthesis, Structure, Properties, and Theory”. Prentice Hall, Englewood Cliffs, New Jersey, 1992.
8 Gatteschi, D.; Kahn, O.; Miller, J. S.; Palacio, F. Ed. “Magnetic Molecular Materials”. NATO ASI
Series, Series E: Applied Sciences, Vol. 198, Kluwer, Dordrecht, 1991.

6 Tesis Doctoral Susana Barriga Falcón
con los metales, son generalmente más ligeros, solubles, transparentes y pueden tener
propiedades ópticas particulares. Por lo tanto, los materiales moleculares abren nuevas
perspectivas, en particular, para la industria electrónica, que conducen hacia el
desarrollo de aparatos a escala molecular y nanoescala.9
La conductividad eléctrica para los metales está comprendida entre 104 y 10
5 S/cm a
temperatura ambiente. La mayoría de los materiales presentan conductividades
menores, que oscilan entre 10-9
S/cm para los materiales aislantes, hasta los valores
intermedios de conductividad que presentan los semiconductores, y los elevados que
tienen los metales.
La superconductividad fue descubierta por Kammerlingh-Onnes estudiando el
mercurio.10
Por debajo de la temperatura crítica, Tc, un material superconductor tiene
resistividad cero. Además, si este material se sitúa en un campo magnético a
temperaturas inferiores a Tc, se vuelve perfectamente diamagnético (efecto Meissner).11
Sin embargo, si se aplica un campo magnético suficientemente elevado, se restaura el
estado metálico normal. Además, una densidad de corriente eléctrica elevada también
destruye la superconductividad.
1.1. Conductores y superconductores basados en sales de transferencia de carga
Los complejos de transferencia de carga (TC) están formados por un dador (D) (Figura
1.1) y un aceptor (A) (Figura 1.2) capaces de formar, respectivamente, catión-radicales
y anión-radicales estables y de presentar una estructura apilada en el estado sólido.
9 Crandall, B. C.; Lewis, J. Ed. “Nanotechnology. Research and Perspectives”. The MIT Press,
Cambridge, Massachusetts, 1992. 10
Kammerlingh-Onnes, H. Akad. Wetenschappen 1911, 14, 811. 11
Meissner, W.; Ochsenfeld, P. Naturwissenhasften 1933, 21, 787.

Introducción 7
S
S
S
S
Se
Se
Se
SeMe
MeMe
Me
S
S
S
S
S
SS
S
S
S
Se
Se
Me
MeS
S S
S
S
SS
S
S
S
S
S
O
OO
O Se
Se
Se
Se
S
SS
S
TTF, 1 TMTSF, 2 DMTE, 3 MDT-TTF, 4
BEDT-TTF, 5 BEDO-TTF, 6 BEDS-TTF, 7
Figura 1.1: Algunos dadores de electrones orgánicos usados en la formación de complejos de
transferencia de carga
Estos complejos presentan propiedades eléctricas conductoras con carácter
potencialmente metálico, o pueden tener propiedades magnéticas particulares,
dependiendo del tipo de apilamiento que presenten. Los complejos TC, debido a su
estructura, son sólidos de baja dimensionalidad y, además, sus propiedades físicas
muestran una anisotropía considerable.
Los primeros descubrimientos en el campo de los complejos de transferencia de carga
fueron realizados por Akamatu,12
que observó por primera vez una elevada
conductividad en una sal de bromuro de perileno. Además, en la compañía DuPont13
se
observó un fenómeno similar en varias sales de trasferencia de carga que contenían
TCNQ (7,7,8,8-tetraciano-p-quinodimetano). Sin embargo, la primera molécula
conductora no metálica fue preparada fortuitamente hace más de 150 años en el grupo
de Knop por oxidación del tetracianoplatinato de potasio, K2[Pt(CN)4], con cloro o
12
Akamatu, H.; Inokuchi, H.; Matsunaga, Y. Nature 1954, 173, 168. 13
(a) Acker, D. S.; Harder, R. J.; Hertler, W. R.; Mahler, W.; Melby, L. R.; Benson, R. E.; Mochel, W.
E. “7,7,8,8,-Tetracyanoquinodimethene and its Electrically Conducting Anion-radical Derivates”.
J. Am. Chem. Soc. 1960, 82, 6408-6409. (b) Melby, L. R.; Harder, R. J.; Hertler, W. R.; Mahler, W.;
Benson, R. E.; Mochel, W. E. “Substituted Quinodimethans. II. Anion-Radical Derivatives and
Complexes of 7,7,8,8-tetracyanoquinodimethane”. J. Am. Chem. Soc. 1962, 84, 3374-3387.
(c) Hertler, W. R.; Mahler, W.; Melby, L. R.; Miller, J. S.; Putscher, R. E.; Webster, O. W. “Cyanocarbons - Their History from Conducting to Magnetic Organic Charge-Transfer Salts”. Mol.
Cryst. Liq. Cryst. 1989, 171, 205-216.

8 Tesis Doctoral Susana Barriga Falcón
bromo.14
La conductividad de esta sustancia, conocida posteriormente como KCP, fue
medida hace tan sólo 25 años.
NC CN
CN
CN
CN
NCCN
CN
CN
CN
CN
NC CN
CN
CN
NC
CN
NC CN
NC CN
NC CN
NC CN
F
FF
F
NC CN
NC CN
I
IO
O
CN
CNCl
Cl
NCN
NNC
Me
Me
NNC
NNC
N
NCN
CN
NNC
O
O
NCN
S
S
NNC
NNC
N
NCN
CN
S
PtS
S
S CN
CN
NC
NC S
NiS
S
S
S
SS
S
SS
S
NiS
S
S
C3(CN)5, 8 C4(CN)6, 9 C3[C(CN)2], 10
TCNQ, 11 TCNQF4, 12 TCNQI2, 13 DDQ, 14 Me2DCNQI, 15
PTCI, 16 PDCI, 17 TCLDBT, 18
Pt[C2S2(CN)2]2, 19 Ni(dmit)2, 20 Ni(bds)2, 21
Figura 1.2: Algunos aceptores de electrones usados en la formación de complejos de transferencia de
carga
Desde el descubrimiento en 1973 del complejo de transferencia de carga
monodimensional, formado por el dador tetratiafulvaleno (TTF) y por el aceptor
TCNQ,15
que presenta una elevada conductividad a temperatura ambiente (500 S/cm) y
14
(a) Knop, W. Justus Liebigs Ann. Chem. 1842, 43, 111. (b) Knop, W.; Schnedermann, G. J. Prakt.
Chem. 1846, 37, 461. 15
(a) Ferraris, J.; Cowan, D. O.; Walatka, V. V.; Perlstein, J. H. “Electron Transfer in a New Highly
Conducting Donor-Acceptor Complex”. J. Am. Chem. Soc. 1973, 95, 948-949. (b) Coleman, L. B.; Cohen, M. J.; Sandman, D. J.; Yagamashi, F. G.; Garito, A. F.; Heeger, A. J. Solid State Commun.
1973, 12, 1125.

Introducción 9
comportamiento metálico, la investigación en este campo ha experimentado un gran
auge.
A partir del descubrimiento del [TTF][TCNQ] se han preparado y estudiado un gran
número de sales de transferencia de carga, y el primer superconductor basado en un
complejo TC se obtuvo en 1980 usando las sales de tetrametiltetraselenofulvaleno
(TMTSF, Figura 1.1-2). Así, la primera sal de transferencia de carga superconductora
bajo presión fue (TMTSF)2(PF6),16
y (TMTSF)2(ClO4) fue el primer superconductor
basado en una sal de transferencia de carga a temperatura ambiente.17
Otras
modificaciones del TTF conducen también a sustancias superconductoras
(Tabla 1.1).18
Complejo TC Conductividad (S/cm) Condiciones
[TTF][TCNQ] 500 Temperatura ambiente
(TMTSF)2(ClO4) Superconductor Tc = 1.4 K Presión atmosférica
[TTF][Ni(dmit)2]2 Superconductor Tc = 1.6 K P = 7 Kbar
Cu(Me2DCNQI)2 5·105 Temperatura ambiente
(BEDT-TTF)2Cu[N(CN)2]Br Superconductor Tc = 11.6 K Presión atmosférica
Tabla 1.1: Ejemplos seleccionados de complejos TC conductores y superconductores
El uso de sales de transferencia de carga basadas en compuestos de coordinación
inorgánicos para obtener superconductores, especialmente en complejos de metales de
transición, ha sido ampliamente estudiado. Existen varios superconductores basados en
16
(a) Jerome, D.; Mazaud, A.; Ribault, M.; Bechgaard, K. J. Phys. Lett. 1980, 41, 95. (b) Parkin, S. S.
P.; Ribault, M.; Jerome, D.; Bechgaard, K. “Superconductivity in the Family of Organic Salts Based on the Tetramethyltetraselenafulvalene (TMTSF) Molecule - (TMTSF)2X (X = ClO4, PF6, AsF6,
SbF6, TaF6) J. Phys. C - Solid State Physics 1981, 14, 5305-5326. 17
Bechgaard, K.; Carneiro, K.; Rasmussen, F. B.; Olsen, M.; Rindorf, G.; Jacobsen, C. S.; Pedersen, H.
J.; Scott, J. C. “Superconductivity in an Organic-Solid - Synthesis, Structure, and Conductivity of
Bis(Tetramethyltetraselenafulvalenium) Perchlorate, (TMTSF)2ClO4”. J. Am. Chem. Soc. 1981, 103,
2440-2442. 18
Williams, J. M.; Ferraro, J. R.; Thorn, R. J.; Carlson, K. D.; Geiser, U.; Wang, H. H.; Kini, A. M.;
Whangbo, M. H. “Organic Superconductors (Including Fullerenes)”. Prentice Hall, Upper Saddle
River, New Jersey, 1992.

10 Tesis Doctoral Susana Barriga Falcón
sales de transferencia de carga constituidos por complejos M(dmit)2
(dmit2-
= 2-tioxo-1,3-ditiol-4,5-ditiolato) (Figura 1.2 y Tabla 1.2) .19
Complejo CT Tc (K) P (Kbar)
[TTF][Ni(dmit)2]2 1.6 7
[Me4N]0.5[Ni(dmit)2]2 5 7
α’-[TTF][Pd(dmit)2]2 5.9 24
α-[TTF][Pd(dmit)2]2 1.7 22
β-[Me4N]0.5[Pd(dmit)2]2 6.2 6.5
[Me2Et2N]0.5[Pd(dmit)2]2 4 2.4
α-[BEDT-TTF][Ni(dmit)2]21.3 Atmosférica
Tabla 1.2: Algunos superconductores basados en aceptores M(dmit)2
Recientemente, el grupo de Haddon ha descubierto una nueva clase de superconductores
basados en sales de transferencia de carga, los fulleruros, AxC60
(A = K, Rb, Cs).20
El valor más elevado de Tc encontrado en estos compuestos ha sido
de 33 K, en la molécula RbCs2C60.21
1.1.1. Conductores basados en TTF y derivados
Tras el descubrimiento del primer metal orgánico [TTF][TCNQ] y de las primeras sales
superconductoras [TMTSF]2X se ha realizado un enorme trabajo para poder aumentar la
capacidad dadora de electrones de los derivados de TTF, con el fin de mejorar la
conductividad de las sales y de los complejos de transferencia de carga derivados de
ellos. Por otra parte, los derivados de TTF han sido ampliamente usados como bloques
19
Cassoux, P.; Valade, L. en “Inorganic Materials”. Bruce, D. W.; O’Hare, D. Ed. Wiley, Chichester,
West Sussex, England, 1992, pag. 1-58. 20
Hebard, A. F.; Rosseinsky, M. J.; Haddon, R. C.; Murphy, D. W.; Glarum, S. H.; Palstra, T. T. M.;
Ramirez, A. P.; Kortan, A. R. “Superconductivity at 18 K in Potassium-Doped C-60”. Nature 1991, 350, 600-601.
21 Hebard, A. F. “Doped Fullerenes - a Soot to Superconductivity Story”. Physica B 1994, 197, 544-
550.

Introducción 11
estructurales básicos en la construcción de estructuras macromoleculares y
supramoleculares.22
La química del TTF es muy rica y en la actualidad es muy fácil obtener una gran
variedad de derivados monosustituidos de TTF.23
Las modificaciones químicas clásicas realizadas sobre el esqueleto del TTF han estado
dirigidas a aumentar la conductividad eléctrica24
mediante el ajuste de la capacidad
dadora a través de la modificación de los sustituyentes presentes en el TTF, o bien
mediante la sustitución del azufre por otros calcógenos, o por la introducción de
calcógenos en posiciones periféricas del TTF.
Se observó que los conductores de baja dimensionalidad son inestables y que se produce
la pérdida del estado metálico al disminuir la temperatura. Por ello, se ha intentado
aumentar la dimensionalidad en la química del TTF. Bryce publicó en 1995 una
revisión en la que se recogían algunas de las estrategias más comunes para aumentar la
dimensionalidad, que incluían la síntesis de derivados de TTF funcionalizados, 22 y 23,
que pueden formar enlaces por puente de hidrógeno intermoleculares, así como la
síntesis de análogos de TTF con conjugación π-extendida, 24-28.25
La incorporación de
grupos espaciadores cíclicos entre dos anillos de 1,3-ditiafulvenilo se ha usado también
para extender la conjugación π en sistemas basados en TTF.
22
Segura, J. L.; Martín, N. “New Concepts in Tetrathiafulvalene Chemistry”. Angew. Chem. Int. Ed.
2001, 40, 1372-1409. 23
Ver por ejemplo: Garín, J. “Reactivity of TTF and TSeF”. Adv. Heterocycl. Chem. 1995, 62, 249-
304. 24
Bryce, M. R. “Recent progress on conducting CT salts”. Chem. Soc. Rev. 1991, 20, 355-390. 25
Bryce, M. R. “Current Trends in Tetrathiafulvalene Chemistry – Towards Increased Dimensionality”.
J. Mat. Adv. 1995, 5, 1481-1496.

12 Tesis Doctoral Susana Barriga Falcón
S
S
S
S OHS
SS
S
S
S NHR
S
S
S
S
SX
SS S
S
SS S
S
S S
S
SS
SS
SMeMeS
SS
SMeMeS
OMe
OMe
S
SS
S
24 25: X =O, S, NMe 26
27 28
22 23
También se han sintetizado dímeros de TTF, que presentan un comportamiento redox
multietapa provocado por las interacciones intramoleculares entre las unidades de
TTF.26
Las interacciones moleculares entre las dos zonas redox, así como entre los
apilamientos o eslabones, pueden modificarse de forma controlada mediante
modificación del grupo que actúa de puente entre las unidades de TTF. La figura 1.3
muestra los diferentes tipos de derivados “bis-TTF”.
26
(a) Otsubo, T.; Aso, Y.; Takimiya, K. “Dimeric Tetrathiafulvalenes: New Electron Donors”. Adv.
Mater. 1996, 8, 203-211. (b) Becher, J.; Lau. J.; Mørk, P. en “Electronic Materials: The Oligomer
Approach”. Müllen, K.; Wegner, G. Ed. Wiley-VCH, Weinheim, 1998, pag. 198-233.

Introducción 13
Dímeros con conectores no conjugados Dímeros con conectores conjugados
TTFσ
TTF
TTFπ
TTF
Un conector
TTFσ
TTFσ
TTFπ
TTFπ
Dos conectores
TTF
σTTF
σ
TTF
πTTF
π
Cíclicos
Figura 1.3: Diferentes tipos de derivados “bis-TTF”
Una nueva aproximación que busca aumentar la dimensionalidad se ha orientado hacia
la síntesis de derivados de TTF con estructuras no planares.27
Las sales de transferencia
de carga con moléculas que no son planas muestran una conductividad comparable a la
que presentan las sales de transferencia de carga de moléculas planas y presentan
estados metálicos más estables.
En la Figura 1.4 se muestran algunos ejemplos de derivados de TTF que no son planos,
29-33.
S
S
S
S
S
S
S
S
R
RR
R
S
S
S
S
S
SR
R S
S
S
S
S
M
S R
R
S
S
S
S
SS
S S
SMe
SMe
SMeMeS
SMeMeS
SS
S S
RR
RR
SS
S S
RR
RR
S
S
S
S
29 30
31 32 33: R = H, SMe
o R R = SCH2CH2S
Figura 1.4: Algunos ejemplos representativos de TTF que no son planos
27
Kato, H.; Kobayashi, T. “Nonplanar Tetrathiafulvalene Vinylogues”. Adv. Mater. 1993, 5, 750-751.

14 Tesis Doctoral Susana Barriga Falcón
Un grupo a priori muy interesante para modificar las propiedades de los derivados de
TTF es el ferroceno, debido a sus particulares propiedades estructurales y
electroquímicas. En los últimos años se han sintetizado diversos complejos de
transferencia de carga sustituidos con ferrocenos; sin embargo, la mayoría ha mostrado
conductividades moderadas.28
Fe
S
S
S
S
S
S
S
S
Fe Fe Fe
34, trans 34, cis
Fe S
S
S
S
S
S
S
S
Fe S
S
S
S
35 36
FeMe
Me
S
S
S
S
FeMe
Me
S
S
S
S
Me
Me
Me
Me
37 38
Se han diseñado también muchos sistemas dadores-aceptores intramoleculares basados
en C60, intentando explotar las propiedades de transferencia electrónica de los
28
(a) Ueno, Y.; Sano, H.; Okawara, M. J. Soc. Chem. Chem. Commun. 1980, 28-30. (b) Togni, A.;
Hobi, M.; Rihs, G.; Rist, G.; Albinati, A.; Zanello, P. Zech, D.; Keller, H. “1,1'-Disubstituted
Ferrocenes as Donors for Charge-Transfer Complexes - Synthesis, Structure, Conductivity, and
Magnetic-Properties”. Organometallics 1994, 13, 1224-1234. (c) Moore, A. J.; Skabara, P. J.; Bryce, M. R.; Batsanov, A. S.; Howard, J. A. K.; Daley, S. T. A. K. “Covalently Attached Ferrocene and
Tetrathiafulvalene Redox Systems”. J. Chem. Soc. Chem. Commun. 1993, 417-419.

Introducción 15
fullerenos. En estas moléculas, las unidadades de C60 –aceptor– y de TTF –dador– están
unidas mediante un espaciador (Figura 1.5).29
S
S
S
Sespaciador
NCH3
S
S
S
S R
RNCH3
CH2 S S
S
S
S SCH3
SCH3
H3CS
S
S
S
S
R
R
S
S R
R
S
S
n n
39 40
41 42
Figura 1.5: Esquema general para el diseño de moléculas de transferencia electrónica entre C60 y TTF y
algunos ejemplos representativos
1.1.2. Sales de transferencia de carga con heterociclos con azufre y nitrógeno
Aunque los derivados de TTF son los complejos de transferencia de carga conductores
más conocidos, existen varios ejemplos de materiales conductores basados en
heterociclos que contienen azufre y nitrógeno. Por ejemplo, el radical 43 forma un
complejo de transferencia de carga conductor, cuya conductividad en el monocristal es
29
(a) Martín, N.; Sánchez, L.; Seoane, C.; Andreu, R.; Garín, J.; Orduna, J. “Semiconducting Charge
Transfer Complexes from [60]Fullerene-Tetrathiafulvalene (C-60-TTF) Systems”. Tetrahedron Lett.
1996, 37, 5979-5982. (b) Llacay, J.; Mas, M.; Molins, E.; Veciana, J.; Powell, D.; Rovira, C. “The
First Diels-Alder Adduct of [60]Fullerene with a Tetrathiafulvalene”. Chem. Commun. 1997, 659-
660. (c) Herranz, M. A.; Martín, N. “A New Building Block for Diels-Alder Reactions in pi-
Extended Tetrathiafulvalenes: Synthesis of Novel Electroactive C-60-based Dyad” Org. Lett. 1999,
1, 2005-2007. (d) Guldi, D. M.; González, S.; Martín N.; Antón, A.; Garín, J.; Orduna, J. “Efficient Charge Separation in C-60-Based Dyads: Triazolino[4 ',5 ': 1,2][60]Fullerenes”. J. Org. Chem. 2000,
65, 1978-1983.

16 Tesis Doctoral Susana Barriga Falcón
de 460 S/cm a temperatura ambiente. Otros ejemplos son los complejos de transferencia
de carga 1:1 que se forman entre los dirradicales 44 y 45 y el yodo, que llegan a
alcanzar una conductividad de 100 S/cm a temperatura ambiente.30
S
SN
N
NS
SN N
S
SN N
S
SN
S
NSe
SeN N
Se
SeN
43 44 45
1.1.3. Películas de Langmuir-Blodgett
Las películas de Langmuir-Blodgett están constituidas por capas moleculares
depositadas sobre una superficie, generalmente metálica. Estás capas están formadas por
moléculas anfifílicas, que presentan una zona polar y una apolar, empaquetadas muy
juntas. Sobre la superficie se puede depositar una única capa, monocapa, o el proceso
puede repetirse varias veces para dar lugar a una estructura en multicapas. Las películas
de Langmuir-Blodgett son típicamente densas y tienen un orden muy elevado, aunque
no cristalino. El espesor de cada capa es uniforme y está determinado por la longitud de
la molécula y por su inclinación en el empaquetamiento de las moléculas en la capa.
Las películas de Langmuir-Blodgett constituyen superficies anisotrópicas que
encuentran aplicación en un amplio rango de aparatos, tales como aparatos
bioelectrónicos, transistores de efecto de campo (FET), interruptores de memoria,
detectores piroeléctricos, y aparatos diseñados para óptica no lineal. Las aplicaciones
industriales de estas películas incluyen desde la obtención de conductores
anisotrópicos31
hasta la obtención de materiales para prevenir la corrosión.32
Se han sintetizado derivados de TTF anfifílicos mediante la incorporación de cadenas
hidrofóbicas, 46-48, o de sustituyentes aromáticos cargados, 49, al núcleo de TTF, y se
30
Sunandana, C. S. “Techniques and Applications of Electron Spin Resonance”. Bulletin of Materials
Science 1998, 21, 1-70. 31
Bryce, M. R.; Petty, M. C. “Electrically Conductive Langmuir-Blodgett Films of Charge-Transfer
Materials”. Nature, 1995, 374, 771-776. 32
Roberts, G. G. “An Applied Science Perspective of Langmuir-Blodgett Films”. Adv. Phys. 1985, 34,
475-512.

Introducción 17
han usado para hacer películas de Langmuir-Blodgett de complejos de transferencia de
carga y de sales de transferencia de carga.33
S
S
S
SS
S S
S C18H37
S
S
S
S
N N
CH3H3C
H3C CH3
C18H37C18H37
46 47
S S
S S
C10H21S SC10H21
H3C S(CH2)4NHCOCH2S
S S
S S
C10H21S SC10H21
S(CH2)4NHCOCH2S
SS
SS
SC10H21C10H21S
CH3
S
S
S
SS
S
CH3
CH3
NH3C
I
48 49
Las películas de Langmuir-Blodgett no tienen aplicación únicamente en la obtención de
materiales conductores, sino que se han diseñado sistemas de Langmuir-Blodgett con
muy diversos fines. Un ejemplo de membrana luminiscente se ha obtenido mediante la
formación de monocapas del derivado antracénico 50.
COOH
50
Otro ejemplo de las diversas utilidades de las películas de Langmuir-Blodgett lo
constituyen los complejos de 5,10,15,20-tetraquis(4-hidroxifenil)porfirina con Fe(III),
Co(II), Ni(II), Cu(II) y Pt(II) que forman películas de Langmuir-Blodgett que pueden
ser usadas como sensores para detectar gases, como Cl2, HCl y O2.34
33
Nakamura, T. en “Handbook of Organic Conductive Molecules and Polymers” Vol. 1 Nalwa, H. S.
Ed. Wiley, 1997, pag 727-780. 34
“Molecular Electronic Devices”. Carter, F. L. Ed. Marcel Dekker, New York, 1987.

18 Tesis Doctoral Susana Barriga Falcón
1.2. Moléculas orgánicas magnéticas
El comportamiento magnético se detecta por la respuesta de repulsión o atracción de un
material frente a un campo magnético. Este comportamiento se debe al espín
mecanocuántico de cada electrón y a cómo éste interacciona con los demás espines
vecinos. En función de ésto, los materiales magnéticos se clasifican en paramagnéticos,
que presentan los espines desordenados, ferromagnéticos, con espines ordenados
paralelos, antiferromagnéticos, con espines ordenados y opuestos, y ferrimagnéticos,
que resultan de acoplamientos del mismo tipo que los antiferromagnéticos pero en los
que no se produce la cancelación total de los espines (Figura 1.6). Esta ordenación de
estado ferro-, antiferro- o ferrimagnético sólo ocurre por debajo de la temperatura crítica
o temperatura de ordenación magnética Tc.
Xxx xxx
Xxx
Paramagnético Ferromagnético Antiferromagnético Ferrimagnético
Figura 1.6: Comportamiento común de los espines emparejados
1.2.1. Moléculas ferromagnéticas basadas en sales de transferencia electrónica
En muchos casos la conductividad y el magnetismo están unidos. Así, en 1979 Miller
observó que existía ordenación ferromagnética en la sal de transferencia de carga
[FeCp*2][TCNQ] (Cp* = η5-C5Me5).
35 En 1985 se reemplazó el TCNQ por un aceptor
más pequeño, el TCNE (tetracianoetileno), y el complejo [FeCp*2][TCNE] mostró un
ferromagnetismo elevado.36
35
Candela, G. A.; Swartzendruber, L.; Miller, J. S.; Rice, M. J. “Metamagnetic Properties
of One-dimensional Decamethylferrocenium 7,7,8,8-Tetracyano-p-quinodimethamide (1:1):
[Fe(η5-C5Me5)2]+·[TCNQ]-·”. J. Am. Chem. Soc. 1979, 101, 2755-2756.
36 Miller, J. S.; Calabrese, J. C.; Rommelmann, H.; Chittapeddi, S. R; Zhang, J. H.; Reiff, W. M.;
Epstein, A. J. “Ferromagnetic Behavior of [Fe(C5Me5)2]9+·[TCNE]-·. Structural and Magnetic
Characterization of Decamethylferrocenium Tetracyanoethenide, [Fe(C5Me5)2]+·[TCNE]+·MeCN,
and Decamethylferrocenium Pentacyanopropenide, [Fe(C5Me5)2]+·[C3(CN)5]
-”. J. Am. Chem. Soc.
1987, 109, 769-781.

Introducción 19
1.2.2. Moléculas ferromagnéticas basadas en radicales nitroxilo
Hasta 1991 sólo se conocían cinco radicales orgánicos estables que mostraran un
comportamiento ferromagnético. Por el contrario, la mayoría de los radicales
sintetizados tenían favorecidas las interacciones antiferromagnéticas frente a las
ferromagnéticas.
Sin embargo, en la actualidad existen numerosos radicales orgánicos ferromagnéticos,
que han sido sintetizados introduciendo en una molécula aceptora o dadora de
electrones un radical estable como sustituyente.37
Los radicales más usados con este fin
son de tipo pirrolidin-1-oxilo, 51, y piperidin-1-oxilo, 52.
N O N O
51 52
El primer radical orgánico magnético estudiado fue 53,38
y el mejor caracterizado hasta
el momento es 54.39
N
O
O
O
O NO
N
N
N
O
O
O
O
53 54
O
Con la estrategia anteriormente descrita es posible preparar multitud de aceptores y
dadores susceptibles de formar complejos de carga conductores. De esta forma, se han
unido radicales tipo 51 y 52 a dadores como el TTF. Esta unión puede hacerse
37
Togashi, K.; Imachi, R.; Tomioka, K.; Tsuboi, H.; Ishida, T.; Nogami, T; Takeda, N.; Ishikawa, T.
“Organic Radicals Exhibiting Intermolecular Ferromagnetic Interactions with High Probability:
4-Arylmethyleneamino-2,2,6,6-tetramethylpiperidin-1-yloxyls and Related Compounds”.
Bull. Chem. Soc. Jpn. 1996, 69, 2821-2830. 38
Chouteau, G.; Veyret-Jeandey, C. “Metamagnetism in Tanol Suberate”. J. Phys. 1981, 42,
1441-1444. 39
Kinoshita, M. “Ferromagnetism of Organic Radical Crystal”. Jpn. J. Appl. Phys. 1994, 33,
5718-5733.

20 Tesis Doctoral Susana Barriga Falcón
directamente o mediante grupos que actúan como puente entre ambas estructuras40
(Figura 1.7). También se ha realizado la unión con aceptores quinónicos.
Así, por ejemplo, 56 y 57 pueden formar complejos de transferencia de carga con
tetrafluorotetracianoquinodimetano (TCNQF4), presentando una elevada conductividad
además de magnetismo.40b
NS
S
S
S
N O
S
S
S
S
N
N
O
O
5655
NO
NS
S
S
S N
N
O
O
O
O
57 58
Figura 1.7: Algunos ejemplos de dadores y aceptores de electrones con radicales
1.2.3. Moléculas ferromagnéticas basadas en metalocenos
Los metalocenos son candidatos excelentes para poder acoplar unidades magnéticas, no
sólo por su extensa química sino también porque son especies electroactivas cuyo
estado de oxidación puede ser controlado mediante estímulos químicos o
electroquímicos.
Existen varias estrategias para obtener moléculas ferromagnéticas con metalocenos. La
primera, como ya se dijo, consiste en la formación de sales de transferencia de carga
40
(a) Kumai, R.; Matsuhita, M. M.; Izuoka, A.; Sugawara, T. “Intramolecular Exchange Interaction in a
Novel Cross-Conjugated Spin System Composed of π-Ion Radical and Nitronyl Nitroxide”. J. Am. Chem. Soc. 1994, 116, 4523-4524. (b) Sugimoto, T.; Yamaga, S.; Nakai, M.; Tsujii, M,
Nakatsuji, H.; Hosoito, N. “Different Magnetic Properties of Change-Transfer Complexes and Cation
Radical Salts of Tetratiafulvalene Derivatives Susbstituted with Imino Pyrolidine- and
Piperidine-1-oxyls”. Chem. Lett. 1993, 1917-1820. (c) Nakazaki, J.; Matsushita, M. M.; Izuoka, A.;
Sugawara, T. “Novel Spin-Polarized TTF Donors Affording Ground State Triplet Cation Diradicals”.
Tetrahedron Lett. 1999, 40, 5027-5030. (d) Fijiwara, H.; Kobayashi, H. “New pi-extended Organic Donor Containing a Stable TEMPO Radical as a Candidate for Conducting Magnetic Multifunctional
Materials”. Chem. Commun. 1999, 2417-2417.

Introducción 21
entre metalocenos y aceptores de electrones. En la tabla 1.3 se muestran algunos
ejemplos, también se incluyen los valores de Curie-Weiss, Θ, que indican la magnitud
del carácter ferromagnético, así como la temperatura crítica, Tc.
Complejo CT
ΘΘΘΘ
(K)�
Tc (K)
[MnCp*2][TCNE] 22.6 8.8
[CrCp*2][TCNE] 22.2 3.65
[FeCp*2][TCNE] 16.8 4.8
[MnCp*2][TCNQ] 10.5 6.5
[CrCp*2][TCNQ] 12.8 3.5
[FeCp*2][TCNQ] 12.3 2.6
Tabla 1.3: Algunos ejemplos de complejos de TC con metalocenos. Se muestran los valores de Curie-
Weiss y la temperatura crítica
También se puede combinar un metaloceno con radicales orgánicos estables con el fin
de formar sales de transferencia de carga. Así, el radical 59 forma un complejo de
transferencia de carga con DDQ que presenta un comportamiento magnético
dependiente de la temperatura, aunque es predominantemente antiferromagnético.41
N
N
O
O
Fe
59
O
O
CN
CNCl
Cl
DDQ, 14
También se han utilizado los metalocenos como puente entre radicales para poder
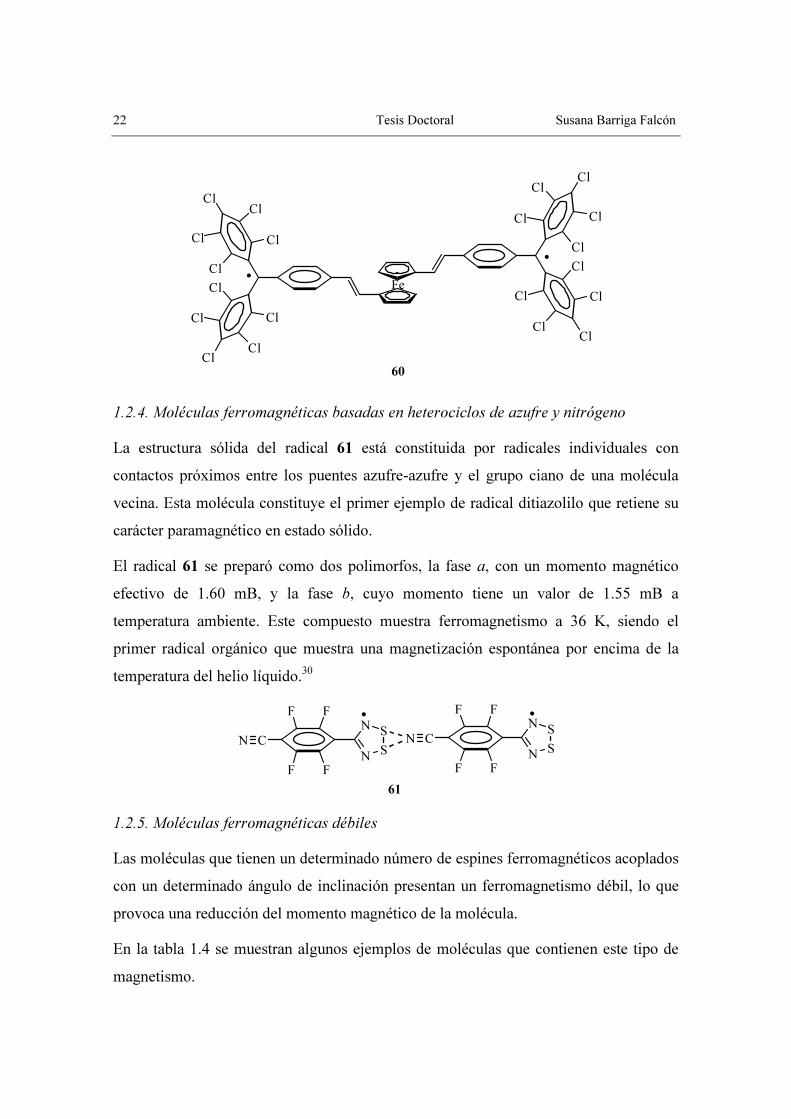
obtener nuevas moléculas ferromagnéticas, como por ejemplo 60.42
41
Nakamura, Y.; Koga, N.; Iwamura, H. “Synthesis and Characterization of 2-Ferrocenyl-4,4,5,5-
tetramethyl-2-imidazolin-1-oxyl 3-oxide and its CT-Complex with DDQ”. Chem. Lett. 1991, 69-72. 42
Elsner, O.; Ruiz-Molina, D.; Vidal-Gancedo, J.; Rovira, C.; Veciana, J. “Ferromagnetic interactions
between triphenylmethyl radicals through an organometallic coupler”. Chem. Commun. 1999,
579-580.
22 Tesis Doctoral Susana Barriga Falcón
Fe
Cl
ClCl
Cl
Cl
Cl
Cl
ClCl
Cl
Cl
ClCl
Cl
Cl
Cl
Cl
ClCl
Cl
60
1.2.4. Moléculas ferromagnéticas basadas en heterociclos de azufre y nitrógeno
La estructura sólida del radical 61 está constituida por radicales individuales con
contactos próximos entre los puentes azufre-azufre y el grupo ciano de una molécula
vecina. Esta molécula constituye el primer ejemplo de radical ditiazolilo que retiene su
carácter paramagnético en estado sólido.
El radical 61 se preparó como dos polimorfos, la fase a, con un momento magnético
efectivo de 1.60 mB, y la fase b, cuyo momento tiene un valor de 1.55 mB a
temperatura ambiente. Este compuesto muestra ferromagnetismo a 36 K, siendo el
primer radical orgánico que muestra una magnetización espontánea por encima de la
temperatura del helio líquido.30
NS
SN
FF
F F
C
NS
SN
FF
F F
CN N
61
1.2.5. Moléculas ferromagnéticas débiles
Las moléculas que tienen un determinado número de espines ferromagnéticos acoplados
con un determinado ángulo de inclinación presentan un ferromagnetismo débil, lo que
provoca una reducción del momento magnético de la molécula.
En la tabla 1.4 se muestran algunos ejemplos de moléculas que contienen este tipo de
magnetismo.

Introducción 23
Ferromagnetismo débil Tc (K)
Ftalocianina de manganeso xxxxxxx 8.3
Octaetiltetraazaporfirina de hierro 5.6
[κ-(BEDT-TTF)]2Cu [N(CN)2]Cl 22
Li[TCNQF4] 12
Cu(Me2DCNI)2-d16 8
TDAE(C60) (TDAE = Tetradimetilaminoetileno) 16.1
Tabla 1.4: Ejemplos representativos de ferromagnetismo débil
2. Materiales ópticos no lineales
La habilidad de manipular la frecuencia, la fase, la polarización o la trayectoria de la luz
tiene aplicaciones tecnológicas muy importantes en áreas tales como la producción de
luz láser sintonizada y el almacenamiento de datos ópticos.
En la actualidad, existe un enorme interés en la búsqueda de materiales orgánicos e
inorgánicos que presenten efectos de óptica no lineal (NLO), especialmente materiales
que producen armónicos de segunda generación.
2.1. Orígenes de la óptica no lineal
Cuando la radiación electromagnética interacciona con la materia provoca una
oscilación en la distribución de la densidad electrónica de la molécula en la misma
frecuencia que la luz incidente. La presencia de una carga oscilante provoca una
separación de cargas y la aparición de un dipolo inducido oscilante, con momento µ,
que es una función de la frecuencia de la luz incidente, ω, y que es también
proporcional a la magnitud del campo eléctrico, E. La magnitud del dipolo inducido
para un determinado E está determinada por la facilidad en la deformación de la
densidad electrónica en un átomo o molécula en particular. La facilidad de deformación
de la densidad electrónica es la polarizabilidad. Debido a que puede ser diferente según

24 Tesis Doctoral Susana Barriga Falcón
las diferentes direcciones, la polarizabilidad es un tensor, y la polarizabilidad molecular
total se expresa como:
Polarización = µ(ω) = αij(ω)E(ω)
Donde, αij(ω) es el tensor de polarizabilidad lineal a la frecuencia ω y describe la
variación lineal del dipolo inducido con el campo eléctrico. La oscilación resultante en
la polarización (dipolo oscilante) es, en efecto, una carga móvil, y por lo tanto emitirá
radiación de la misma frecuencia que la oscilación. Esto produce efectos de óptica lineal
como la birrefringencia y la refracción. Sin embargo, cuando una molécula se somete a
campos muy intensos, como los producidos por la luz láser, el material puede ser tan
polarizado que su polarizabilidad puede cambiar. En este caso, la polarización inducida
es una función no lineal de la fuerza del campo. De nuevo esta función es un tensor,
porque la luz incidente puede provenir de cualquier dirección e inducir una polarización
de respuesta en cualquier dirección.
La expresión matemática del tensor tiene un número infinito de términos, aunque a
campos muy altos el segundo y el tercer término son los realmente importantes. El
tensor puede expresarse como:
µ = µ0 + (αij)E + 1/2(βijk)E·E + 1/6(γijk)E·E·E + ...
El componente fundamental αE es lineal con respecto a E y representa las propiedades
de óptica lineal. El segundo, el tercero y los siguientes términos armónicos son no
lineales respecto a E y producen efectos de óptica no lineal. Los valores de β,
responsable de la generación de armónicos de segundo orden, y γ, que conduce a la
formación de armónicos de tercer orden, se refieren a la primera y segunda
hiperpolarizabilidad, respectivamente.
2.2. Materiales ópticos no lineales de segundo orden
Los criterios electrónicos generales para generar armónicos de segundo orden son los
siguientes:
� Gran variación en el momento dipolar bajo excitación

Introducción 25
� Grandes momentos dipolares transitorios
� Pequeñas diferencias energéticas entre el estado excitado y el estado normal
Las moléculas constituidas por un aceptor de electrones y un dador de electrones
conectados entre sí por un cable molecular son altamente polarizables y presentan
comportamiento de óptica no lineal.
En base a esto, Lehn ha desarrollado varios sistemas “push-pull” usando como dadores
grupos benzo-1,3-ditiol o p-dimetilaminofenilo, y una gran variedad de aceptores
(Figura 1.8).43
43
Blancharddesce, M.; Ledoux, I.; Lehn, J. M.; Malthete, J.; Zyss, J. “Push-Pull Polyenes and
Carotenoids - Synthesis and Non-Linear Optical-Properties”. J. Chem. Soc. Chem. Commun. 1988,
737-739.

26 Tesis Doctoral Susana Barriga Falcón
D A
(a)
S
S
A
N
A62 63
(b)
S
S
A
S
S
A
S
S
A
N
A
N
AN
A
64 65
66 67
68 69
O
HA =
CN
CN
N NCN NO2
"Cable molecular"
Figura 1.8 Polienos “push-pull” en los que un dador de electrones orgánicos y un aceptor están
conectados mediante un “cable molecular”: (a) diagrama esquemático; (b) ejemplos
Para conseguir una ordenación no centrosimétrica se han incorporado estos materiales
en monocapas de Langmuir-Blodgett formadas con ácidos grasos. Los ácidos grasos se
eligen de la misma longitud que el material aceptor-dador. Las monocapas así
producidas presentan unas marcadas propiedades ópticas no lineales (Figura 1.9).

Introducción 27
HO2C HO2C HO2C
N
S
S
N
S
S
N
S
S
HO2C HO2C HO2C
Figura 1.9: Organización de Langmuir-Blodgett formada por moleculas NLO entre ácidos grasos
La presencia de unidades organometálicas, tales como carbonilos de Fischer y
metalocenos, ofrece una amplia variedad de perspectivas sintéticas y electrónicas para la
obtención de materiales organometálicos de metales de transición. Los derivados con
metalocenos como 70 han sido ampliamente estudiados y presentan propiedades de
óptica no lineal. Los valores de β son comparables a los de las especies orgánicas del
mismo tipo, pero muestran además propiedades electrónicas y redox propias del
metaloceno.44
X
X
X
X
X
M
Y
n
X = H, Men = 1, 2Y = CN, CHO, NO2
M = Fe, Ru
70
44
Long, N. J. “Organometallic Compounds for Nonlinear Optics-The Search for En-light-enment!”.
Angew. Chem. Int. Ed. 1995, 34, 21-38.

28 Tesis Doctoral Susana Barriga Falcón
Las propiedades de óptica no lineal aumentan cuando aumenta la longitud de la cadena
y además la presencia de grupos metilo en los anillos de ciclopentadieno también
aumenta significativamente las propiedades de óptica no lineal. Del mismo modo, los
compuestos trans tienen mejores propiedades de óptica no lineal que los cis. En cuanto
al metal, los compuestos de hierro presentan mejores propiedades de óptica no lineal
que los que tienen rutenio y esto indica que el rutenio es un dador peor.
Coe obtuvo el primer interruptor molecular basado en propiedades de óptica no lineal.45
El complejo de rutenio 71 presenta un elevado valor de β. Cuando el rutenio (III) es
oxidado, el comportamiento de óptica no lineal del sistema se reduce drásticamente,
siendo restaurado tras una nueva reducción. Este proceso es completamente reversible.
N N
Me
O
Ru
NH3
H3N
H3N
H3N
NH3
3+
71
En la actualidad, existe también un gran interés en introducir dadores de electrones
como el TTF en materiales diseñados para óptica no lineal.46
Algunos ejemplos se
muestran en la figura 1.10.
45
Coe, B. J. “Molecular Materials Possessing Switchable Quadratic Nonlinear Optical Properties”.
Chem. Eur. J. 1999, 5, 2464-2471. 46
Bryce, M. R.; Green, A.; Moore, A. J.; Perepichka, D. F.; Batsanov, A. S.; Howard, J. A. K.; Ledoux-
Rak, I.; González, M.; Martín, N.; Segura, J. L.; Garín, J.; Orduna, J.; Alcalá, R.; Villacampa, B.
“Synthesis of Conjugated Tetreathiafulvalene (TTF)- π-Acceptor Molecules – Intramolecular Charge Transfer and Nonlinear Optical Properties”. Eur. J. Org. Chem. 2001, 1927-1937.

Introducción 29
S
S
S
S
R
R
CHO
S
S
S
S
R
R
CN
CNS
S
S
S
R
R
NO2
S
S
S
S
R
R
O
NCCN
S
S
S
S
R
RNH
NH
O
O O
S
S
S
S
R
RN
N
O
O O
Me
Me
S
S
S
S
R
RN
N
O
O S
Et
Et
74
75 76
7778
72 73
Figura 1.10: Ejemplos seleccionados de materiales NLO que contienen TTF
2.3. Materiales ópticos no lineales de tercer orden
En un principio, el diseño de materiales de tercer orden se centró en sistemas
π-conjugados extendidos.47
Se han estudiado ampliamente oligómeros y polímeros
como, por ejemplo, poliacetilenos, polidiacetilenos, poliarilenos y poli(arilenvinilenos).
En la actualidad se está también investigando la extensión del sistema π en dos y tres
direcciones, utilizando sistemas tales como ftalocianinas y fullerenos, respectivamente.
Las relaciones entre estructura y propiedades para los efectos de tercer orden son más
complejas que las existentes en los efectos de segundo orden. En principio, para generar
efectos de tercer orden no es necesaria la existencia de asimetría molecular.
Al igual que ocurría con los efectos de segundo orden, la introducción de metalocenos
puede ayudar a producir efectos de tercer orden. Se ha estudiado la óptica no lineal de
metalocenos tales como ferrocenos, hafnocenos, rutenocenos y zirconocenos. Sin
embargo, los ferrocenos son los compuestos que presentan mayor interés, debido a su
47
Bredas, J. L.; Adant, C.; Tackx, P.; Persoons, A.; Pierce, B. M. “3rd-Order Nonlinear-Optical
Response in Organic Materials - Theoretical and Experimental Aspects”. Chem. Rev. 1994, 94, 243-
278.

30 Tesis Doctoral Susana Barriga Falcón
estabilidad y a su versatilidad sintética, y los que han dado lugar a más ejemplos (Figura
1.11).48
Fe
CHO
Fe
Fe
Fe
Ph
Ph
Ph
Fe
OHC
n
79 80
81 82
Figura 1.11: Algunos derivados de ferrocenos estudiados para óptica no lineal de tercer orden
Otra buena aproximación para obtener este tipo de materiales implica el uso de
polialquinos organometálicos conjugados que tienen deslocalización extendida a través
de la cadena polimérica (Figura 1.12).49
Pt
PBu3
PBu3
H3C
CH3
Pt
PBu3
PBu3
Pd
PBu3
PBu3
Pt
PBu3
PBu3
H3C
CH3
H3C
CH3
Pt
PBu3
PBu3
n n n
n n
83 84 85
86 87
Figura 1.12: Algunos palatino- y paladapolienos que presentan propiedades NLO de tercer orden
Algunos ditiolanos con metales de transición, como por ejemplo 88, también presentan
efectos de tercer orden.50
48
Ghoshal, S.; Samoe, M.; Prasad, P. N.; Tufariello, J. J. “Optical Nonlinearities of Organometallic
Structures - Aryl and Vinyl Derivatives of Ferrocene”. J. Phys. Chem. 1990, 94, 2847-2851. 49
Guha, S.; Frazier, C. C.; Porter, P. L.; Kang, K.; Finberg, S. E. “Measurement of the 3rd-orden
Hyperpolarizability of Platinum Poly-ynes”. Optics Lett. 1989, 14, 952-954. 50
Oliver, S. N.; Winter, C. S.; Rush, J. D.; Underhill, A. E.; Hill, C. Proc. SPIE Int. Soc. Opt. Eng.
1990, 1337, 81.

Introducción 31
S
M
S
S
S
R
R
R
R
88, M = Ni, Pt R = CH3, Ph
Varias clases de materiales orgánicos están siendo estudiadas en la actualidad como
materiales de tercer orden: líquidos, sólidos moleculares, complejos de transferencia de
carga, polímeros π-conjugados, composites orgánicos y cristales líquidos.47
Una de las
principales aplicaciones de los materiales ópticos no lineales de tercer orden se
encuentra en el área del procesamiento de señales ópticas.
3. Cristales líquidos
Los cristales líquidos se descubrieron a finales del siglo XIX, pero ha sido en las
últimas dos décadas cuando sus propiedades han sido más ampliamente usadas.
Como su nombre indica, los cristales líquidos tienen algunas propiedades características
de los líquidos (fluyen) y algunas propiedades de los cristales (tienen propiedades
diferentes en las distintas direcciones).
Los cristales líquidos también reciben el nombre de líquidos cristalinos, mesofases o
fases mesomórficas. Los compuestos con propiedades mesomórficas pueden ser
llamados mesogénicos.51
3.1. Tipos de cristales líquidos
Los cristales líquidos pueden ser clasificados atendiendo a diversos criterios. Así, si se
realiza una clasificación general, debemos hablar de cristales líquidos termotrópicos,52
51
(a) Collings, P. J.; Hird, M.; “Introduction to Liquid Crystals Chemistry and Physics”. Taylor and
Francis, London, 1997. (b) Collings, P. J.; Patel, J. S. “Handbook of Liquid Crystal Research”.
Oxford University Press, Oxford, 1997. (c) “Handbook of Liquid Crystals”. Demus, D.; Goodby, J.
W.; Gray, G. W.; Spiess, H. W.; Vill, V. Ed. Wiley-VCH, Weinheim, Chichester, y New York, 1998.
(d) Percec, V. “Liquid Crystals 100 Years Later, What Are the New Concepts Used in the Design of
Molecular, Macromolecular and Supramolecular Liquid Crystals?”. Macromol. Symp. 1997, 117,
267-273. 52
(a) Gray, G. W. “Low-molar-mass Thermotropic Liquid Crystals”. Philos. Trans. R.. Soc. Lond. Ser.
A Math. Phys. Eng. Sci. 1990, 330, 73-94. (b) Gray, G. W. “The Chemistry of Liquid Crystals”
Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 1983, 309, 77-92. (c) Vertogen, G.; De Jeu,

32 Tesis Doctoral Susana Barriga Falcón
en los que la fase líquida cristalina existe en ciertas regiones de temperatura. Este tipo
de fases puede presentarse tanto en compuestos puros como en mezclas.
El otro tipo de cristal líquido genérico sólo existe en mezclas constituidas por
compuestos con elevada polaridad (compuestos anfifílicos) y ciertos disolventes;
reciben el nombre de cristales líquidos liotrópicos y la condición necesaria para su
existencia es la interacción fuerte entre los compuestos polares con las moléculas del
disolvente.53
Existen compuestos, llamados anfotrópicos, que son capaces de formar cristales
líquidos termotrópicos y liotrópicos.
Si atendemos a la estructura molecular podemos clasificar los cristales líquidos en:
� Cristales líquidos calamíticos: constituidos por moléculas alargadas, en forma de
varilla. Se consideran los cristales líquidos clásicos.
� Cristales líquidos discóticos: formados por moléculas circulares, en forma de disco.
Fueron descubiertos en 1977.54
� Cristales líquidos sanídicos: formados por moléculas en forma de ladrillos. Los
primeros se descubrieron en 1986 en cristales líquidos poliméricos.
También podemos hacer una clasificación atendiendo a la estructura de la fase. Las
estructuras de fase más importantes que pueden presentar son las siguientes:
� Nemáticas: la mayoría de los cristales líquidos presentan esta estructura. Al
contrario que en los líquidos isotrópicos, uno o dos de los ejes moleculares están
orientados paralelamente respecto a otro; esto origina un orden de orientación de
largo alcance. Dentro de este tipo se encuentran los cristales líquidos colestéricos,
W. H. “Thermotropic Liquid Crystals, Fundamentals”. Springer, Heidelberg, 1987. (d)
“Thermotropic Liquid Crystals, Structures and Phase Transitions”. Gray, G. W. Ed. Vol. 22, Wiley,
New York, 1987. 53
(a) Charvolin, J. “Lyotropic Liquid Crystals, Structures and Phase Transitions”. NATO ASI Ser B
1989, 211, 95-111. (b) Gelbart, W. M.; Ben-Shaul, A.; Masters, A.; McMullen, W. E. “Effects of
Interaggregate Forces on Micellar Size in Isotropic and Aligned Phases, Physics of Amphiphiles: Miceles, Vesicles and Microemulsions”. Elsevier, Amsterdam, 1985.
54 Chandrasekhar, S. “Liquid Crystals of Disc-like Molecules”. Philos. Trans. R. Soc. Lond. Ser. A
Math. Phys. Eng. Sci. 1983, 309, 93-103.

Introducción 33
que son una variante del estado nemático, que se da en moléculas quirales. La
denominación de colestérico proviene del hecho de que la primera vez que se
observó este tipo de fases fue en un derivado del colesterol. Suelen estar
organizados en forma de hélice y esta estructura en hélice les confiere propiedades
ópticas muy especiales.
� Esmécticas: estructuras en capas con muchas posibilidades de orden en las capas y
con posibilidades de diferentes interacciones mutuas entre las capas. Tienen una
orientación de largo alcance y un orden posicional más o menos establecido.
� Cúbicas: estructuras formadas por unidades micelares colocadas entre rejas o por
complicadas redes entretejidas.
� Columnares: estructuras organizadas en columnas formadas a través de
interacciones paralelas entre moléculas que tienen forma circular o de disco.
Figura 1.13: Representación de los tipos de mesofases más comunes que presentan los cristales líquidos

34 Tesis Doctoral Susana Barriga Falcón
3.2. Materiales basados en cristales líquidos calamíticos
Vorländer demostró que la mayoría de los cristales líquidos están constituidos por
moléculas alargadas, que habitualmente se construyen uniendo una o dos cadenas
alifáticas a un núcleo rígido formado por unidades planas ricas en electrones, como
anillos aromáticos, separadas por uniones cortas que contienen generalmente dobles
enlaces (Figura 1.14).
N
N
OC9H19
O
O
O
C8H17
O
C8H17
C10H21
N
C6H13
O
O
O
O
O
C6H13
O
O
C6H13
89 90
91 92
Figura 1.14: Algunos ejemplos típicos de cristales líquidos calamíticos con núcleo rígido y cadenas
flexibles
El problema que presenta este tipo de estructuras es que la temperatura a la que
manifiestan sus propiedades como cristales líquidos es elevada, debido a las
interacciones existentes entre las moléculas.
Se pueden controlar los puntos de fusión de estos compuestos modificando la longitud
de las cadenas unidas al núcleo rígido. A mayor longitud, menores puntos de fusión.
Pero además, estas cadenas largas tienden a interaccionar entre ellas formando capas en
las que las cadenas se sitúan en una región y el núcleo aromático en otra. Se promueve
así una fase esmética frente a una fase nemática.
En 1972, Gray y colaboradores sintetizaron dos moléculas orgánicas alargadas basadas
en cianobifenilos (Figura 1.15).55
Estas moléculas tienen tres características esenciales:
55
Gray, G. W.; Hird, M.; Ifill, A. D.; Smith, W. E.; Toyne, K. J. “The Synthesis and Transition-
Temperatures of Some 4'-Alkyl-4-Cyano-3-Fluorobiphenyls and 4'-Alkoxy-4-Cyano-3-
Fluorobiphenyls”. Liquid Crystals 1995, 19, 77-83.

Introducción 35
un grupo bifenilo, molécula químicamente estable; cadenas alquílicas con las que poder
controlar los puntos de fusión; y un grupo ciano que nos asegura la respuesta correcta al
someter estas moléculas a un campo eléctrico. Estas moléculas por sí solas no presentan
un rango de temperatura adecuado para su utilización como cristal líquido. Sin embargo,
mezclas de 93 y 94 proporcionan amplios rangos de temperatura variable que oscilan
entre los -3 ºC y los 52 ºC. En 1973, Gray sintetizó también el compuesto 95 que al ser
mezclado con 93 y 94 permite trabajar con en un rango de temperatura de -9 ºC a 59 ºC.
CNC5H11 CNO
C5H11
C5H11 CN
93 94 95
Figura 1.15: Algunos cristales líquidos que han revolucionado la industria de las pantallas digitales
La principal utilidad de este tipo de cristales líquidos está orientada a la fabricación de
pantallas digitales.
Recientemente, Fergason y Doane han desarrollado una nueva aplicación para las fases
nemáticas. Han diseñado un cristal líquido que está disperso en una matriz polimérica
transparente; se construye disolviendo el cristal líquido en los monómeros precursores
del polímero y realizando a continuación la polimerización. Al formarse el polímero, los
cristales líquidos se ordenan al azar, y cuando la luz pasa a través de la película formada
se produce su dispersión, por lo que la película aparece opaca. Si se aplica un campo
eléctrico los cristales líquidos se ordenan, y la película se vuelve transparente; cuando el
campo eléctrico cesa las moléculas de cristal líquido vuelven a su desorden inicial, y la
película vuelve a aparecer opaca. Esto constituye un interruptor eléctrico para la
obtención de láminas opacas o transparentes.

36 Tesis Doctoral Susana Barriga Falcón
Opaco Xxxxxxxx Transparente
Figura 1.16: Interruptor eléctrico basado en cristales líquidos dispersos en una matriz polimérica
3.3. Materiales basados en cristales líquidos discóticos
El primer cristal líquido discótico 96 fue sintetizado por Chandrasekhar en 1977. Desde
entonces, se ha descubierto un elevado número de cristales líquidos discóticos.56
Normalmente, estos cristales discóticos se componen de un núcleo aromático, que lleva
seis u ocho cadenas alifáticas unidas.57
O
C5H11
C5H11 O
C5H11
O
O
C5H11
O C5H11
C5H11
O
96
Las fases más simples que se suelen presentar son las fases nemáticas discóticas.
56
Vögtle, F. “Supramolecular Chemistry”. Wiley, Chichester y New York, 1993, pag. 231-281. 57
(a) Manickam, M.; Kumar, S. “New Mixed Tail Triphenylene Discotic Liquid Crystals”. Mol. Cryst.
Liq. Cryst. 1999, 326, 165-176. (b) Kumar, S.; Kumar Varshney, S. “A Room-temperature Discotic
Nematic Liquid Crystal”. Angew. Chem. Int. Ed. 2000, 39, 3140-3142.

Introducción 37
Los cristales líquidos discóticos han sido ampliamente explotados a nivel comercial,
pero en la actualidad existe un gran interés en su uso como cables moleculares para
transportar electrones en aparatos electrónicos a nanoescala.58
Si las moléculas de una misma columna están muy próximas entre ellas, el tránsito de
electrones será más fácil dentro de una misma columna que entre las columnas
adyacentes. Se ha encontrado que existen varios tipos de fases discóticas en las que las
moléculas se organizan en columnas (Figura 1.17).
Figura 1.17: Organización molecular entre cristales líquidos con fases columnares
Una de las posibles aplicaciones de estos cables moleculares se encuentra en la
fabricación de sensores medioambientales de gases o en aparatos electrónicos, debido a
que su conductividad depende fuertemente de la distancia entre las moléculas que varía,
por ejemplo, cuando las moléculas de gas son absorbidas por el material.
58
(a) Boden, N.; Borner, R. C.; Bushby, R. J.; Clements, J. “First Observation of an n-doped quasi-one-
dimensional Discotic Liquid Crystals”. J. Am. Chem. Soc. 1994, 116, 10807-10808. (b) Sikharulidze,
D.; Chilaya, G.; Praefcke, K.; Blunk, D. “Liquid Crystalline Compounds. 112. First Observation of
an Optically Controlled Electro-optic Effect in Nematic-discotic Liquid Crystals”. Liq. Cryst. 1997, 23, 439-442. (c) Kleppinger, R.; Lillya, C. P.; Yang, C. Q. “Discotic Liquid Crystals through
Molecular Self-assembly”. J. Am. Chem. Soc. 1997, 119, 4097-4102. (d) Boden, N; Bissell, R.;
Clements, J.; Movaghar, B. “Discotic Liquid Crystals Self-organizing Molecular Wires”. Curr. Sci.
1996, 71, 599-601. (e) Adam, D.; Schuhmacher, P.; Simmener, J.; Haussling, L.; Siemensmeyer, K.;
Etzbach, K. H.; Ringsdorf, H.; Haarer, D. “Fast Photoconduction in the Highly Ordered Columnar
Phase of a Discotic Liquid Crystal”. Nature 1994, 371, 141-143. (f) Chandrasekhar, S. Krishna Prasad, S. “Recent Developments in Discotic Liquid Crystals”. Contemporary Physics 1999, 40, 237-
245.

38 Tesis Doctoral Susana Barriga Falcón
3.4. Materiales basados en cristales líquidos colestéricos
Los cristales líquidos colestéricos han encontrado uso como sensores de temperatura.
En la organización en hélice, estos cristales líquidos pueden reflejar un determinado
color, que varía con la temperatura; normalmente varían del azul al rojo.
Para construir sensores de temperatura, el compuesto colestérico es encapsulado en una
matriz polimérica que se sitúa entre una superficie transparente y un soporte negro, que
absorbe cualquier luz extraña.
Además, los cristales líquidos colestéricos han sido utilizados recientemente en la
fabricación de pantallas digitales.59
La fase colestérica muestra dos texturas ópticas, una
que refleja fuertemente la luz, mientras que la otra no. Las dos texturas son estables y
pueden ser interconvertidas usando un campo eléctrico. Esto permite almacenar
información que podrá ser leída posteriormente.
Los cristales líquidos colestéricos que presentan fase esméctica quiral de tipo C, son
ferroeléctricos, es decir, presentan un momento dipolar permanente en ausencia de un
campo eléctrico externo. Esta propiedad puede ser usada en la construcción de pantallas
que pueden ser apagadas o encendidas muy rápidamente.
Más recientemente, Fukuda ha descubierto una fase esméctica quiral C que es
antiferroeléctrica, y en la que la dirección de la inclinación se alterna entre las capas. Su
uso en la fabricación de pantallas ofrece varias ventajas respecto al uso de fases
convencionales ferroeléctricas esmécticas quirales C, y permite reducir enormemente el
número de conexiones eléctricas necesarias en la fabricación de pantallas. 60
3.5. Materiales basados en cristales líquidos poliméricos
Los primeros cristales líquidos poliméricos sintéticos se observaron en disoluciones
concentradas de los polímeros poli(γ-metilglutamato) y poli(γ-benzilglutamato) 97 en
59
Yang, D. K.; Huang, X. Y.; Zhu, Y. M. “Bistable Cholesteric Reflective Displays: Materials and
Drive Schemes”. Annual Review of Materials Science 1997, 27, 117-146. 60
Takezoe, H.; Fukuda, A. “Application of Antiferroelectric Liquid-Crystals for Display and
Photoaddressed Spatial Light-Modulator”. Molecular Crystals and Liquid Crystals Science and
Technology Section A-Molecular Crystals and Liquid Crystals 1994, 255, 253-259.

Introducción 39
disolventes tales como dioxano o diclorometano (Figura 1.18). Los polímeros disueltos
adoptan una estructura en hélices extendidas que pueden comportarse como largas
varillas rígidas que pueden empaquetarse entre sí eficientemente.
Kwolek ha encontrado que los cristales líquidos poliméricos pueden constituir fibras
que presentan una elevada resistencia. Estas fibras están constituidas por poliamidas
aromáticas relativamente rígidas llamadas aramidas. El primer producto, basado en una
poli-benzamida fue la fibra B, pero fue sustituida rápidamente por poli(p-
fenilentereftalamida) 98 que es conocida comercialmente como Kevlar®.61
Otras aramidas de relevancia industrial son los poli(p-fenilenbenzobistiazoles) o PBZT
99,62
y sus análogos oxazoles, PBO, que debido a su elevada fortaleza y su excelente
estabilidad térmica pueden ser usados en la industria aeroespacial.
HN
PhOC O
O
NH
O
OHNN
S N
S
n nn
97 98, Kevlar 99, PBZT
Figura 1.18: Estructura química de algunos polímeros que presentan comportamiento de cristal líquido en
disolución
También existen cristales líquidos poliméricos termotrópicos, que se pueden dividir en
dos clases: rígidos y semiflexibles.
En 1980 se comercializaron varios copoliésteres aromáticos termoplásticos, incluyendo
el Vectra, 100, que ha sido el que más aplicaciones ha encontrado.63
61
Chatzi, E. G.; Koenig, J. L. “Morphology and Structure of Kevlar Fibers - A Review”. Polymer-
Plastics Technology and Engineering 1987, 26, 229-270. 62
Mehta, V.R.; Kumar, S. “Structure, morphology, and properties of PBZT and methyl pendant PBZT
fibers”. Journal of Applied Polymer Science 1999, 73, 305-314. 63
Brees, D. E; Ramírez, J. E. “Vectra a High-Performance Fiber”. Journal of the Textile Institute 1990,
81, 561-574.

40 Tesis Doctoral Susana Barriga Falcón
OO
OO
100, Vectra
0.750.25 n
Las aplicaciones más importantes de este tipo de cristales líquidos poliméricos se han
encontrado en componentes ópticos, discos compactos, conectores de fibras ópticas, etc.
Sin embargo, estos materiales son aún caros con lo que su comercialización está muy
limitada.
Los cristales líquidos poliméricos semi-flexibles se forman al unir los grupos de átomos
responsables del comportamiento como cristal líquido con uniones flexibles o
espaciadores. Al aumentar la flexibilidad se reducen las temperaturas de transición.
Grupo mesogénico Espaciador flexible
Figura 1.19: Esquema de cristal líquido polimérico semiflexible
Otro tipo de cristales poliméricos flexibles son los cristales líquidos poliméricos con
grupos laterales, en los que las unidades de cristales líquidos cuelgan de un polímero
guía a través de espaciadores flexibles (Figura 1.20). Estos materiales tienen
aplicaciones potenciales en tecnología electro-óptica avanzada.

Introducción 41
Figura 1.20: Cristales líquidos poliméricos unidos a un polímero guía mediante espaciadores flexibles
Wendorff ha demostrado que los polímeros que responden a la luz pueden ser usados
para almacenar información. Los materiales fotosensibles se construyen incorporando
unidades moleculares que cambian de estructura cuando son irradiadas por luz de una
determinada longitud de onda. Los materiales así formados permiten almacenar
información que se “escribe” mediante irradiación y puede ser borrada por simple
calentamiento, generalmente un láser de infrarrojos (Figura 1.21).64
Algunos cristales
líquidos que incluyen unidades de azobenceno presentan este comportamiento.
64
(a) Stracke, A.; Wendorff, J. H.; Janietz, D.; Mahlstedt, S. “Functionalized liquid-crystalline donor-
acceptor triple compounds containing azobenzene for optical storage” Advanced Materials 1999, 11,
667. (b) Stracke, A.; Wendorff, J. H.; Goldmann, D.; Janietz, D. “Optical storage in a smectic mesophase: thermal amplification of light-induced chromophore orientations and surface relief
gratings”. Liquid Crystals 2000, 27, 1049-1057.

42 Tesis Doctoral Susana Barriga Falcón
Figura 1.21: Sistema de almacenamiento de información mediante cristales líquidos que contienen
azobencenos
4. Información y señales: Semioquímica y Sensores Químicos
La semioquímica es un área de la química supramolecular. El término proviene de
“semiótica” que significa el estudio de las señales o los signos y su uso o interpretación.
El área más obvia de aplicación para la semioquímica es la producción de sensores
moleculares: compuestos que pueden llevar a cabo paralelamente las tareas de
reconocimiento molecular y señalización. Pero la semioquímica también se ocupa de
otros aspectos tales como la generación de señales, procesado, transferencia,
amplificación, conversión y detección.
4.1. Sensores Fotoquímicos
El concepto básico de sensor se ilustra en la figura 1.22. El sustrato (huésped, analito)
es atraído por una parte del sensor, que hace las veces de receptor, y puede estar
constituido por diversos sistemas, tales como éteres corona, criptandos, cavitandos, etc.
La unión debe ser selectiva para el sustrato en cuestión en presencia de otros sustratos
potenciales. El receptor debe estar además en comunicación con un señalizador que
genere una señal cuando se produzca la unión con el sustrato. Esta señal puede ser una
radiación electromagnética (señal fotoquímica), una corriente (señal electroquímica) o
cualquier otro cambio externo que pueda ser medido u observado (por ejemplo, color o
pH). Esto implica que el espaciador que une el receptor con el detector debe permitir la

Introducción 43
comunicación entre ellos y que la unión debe provocar intrínsecamente un cambio en
las propiedades con respecto a las que presentan el receptor y el sustrato libres.65
UNIDAD DE SEÑALIZACIÓN ESPACIADOR RECEPTOR SUSTRATO
Figura 1.22: Representación figurada de un sensor químico, reproducida la cita 65
Una de las aplicaciones potenciales de estos sistemas se encuentra en la química
analítica, en que el receptor se usa para unir y reconocer pequeñas cantidades de analito.
Si la concentración de analito es muy baja se requiere una elevada selectividad y
afinidad entre el receptor y el sustrato, aunque a veces esto es difícil de conseguir.
Un ejemplo simple de un sensor fluorescente modulable es el sistema preparado por de
Silva, que consiste en un antraceno unido a un aza-18-corona-6 que actúa como receptor
selectivo para el ión K+ frente a otros cationes alcalinos (Esquema 1.1). En general, el
antraceno libre presenta una elevada fluorescencia; sin embargo, en el compuesto 101
esta fluorescencia se pierde completamente como resultado de la transferencia
electrónica desde el par de electrones desapareados del nitrógeno. En presencia de K+,
este par de electrones interacciona con el metal como parte de la unión con el sistema
macrocíclico. De esta forma, al unirse el K+, la fluorescencia del antraceno vuelve a
manifestarse y puede detectarse y medirse (Esquema 1.1). Este tipo de sensores recibe
el nombre de sensores PET (transferencia de electrones fotoinducida).66
65
Fabbrizzi, L.; Poggi, A. “Sensors and Switches from Supramolecular Chemistry”. Chem. Soc. Rev.
1995, 24, 197-202. 66
de Silva, A. P.; Fox, D. B.; Moody, T. S.; Weir, S. M. “Luminescent Sensors and Photonic
Switches”. Pure and Applied Chemistry 2001, 73, 503-511.

44 Tesis Doctoral Susana Barriga Falcón
O O
O
O
N
O
X
K+
O O
O
O
N
O
K+
hνabs
hνfluohνabs
hνfluo
101 102
Esquema 1.1: Sensor de iones K+ basado en un azacoronando sustituido con antraceno
Existen otros sensores con antracenos, como por ejemplo el macro-triciclo 103, que
tienen un funcionamiento similar al anterior, y que sirven para reconocer cationes
diamonio lineales. Los grupos antraceno son utilizados como espaciadores al mismo
tiempo que como grupos fluorescentes. En función del tipo de espaciador utilizado se
podrán detectar cationes diamonio de diferente longitud.67
O O
N
O
N
O
O O
OO O
N
O
N
O
O
103
67
Fagues, F.; Desvergne, J. P.; Kampke, K.; Bouaslaurent, H.; Lehn, J. M.; Meyer, M.; Albrechtgary,
A. M. “Linear molecular recognition – spectroscopic, photophysical and complexation studies on alpha, omega-alkanediyldiammonium ions binding to a bisanthracenyl macrotricyclic receptor”.
J. Am. Chem. Soc. 1993, 115, 3658-3664.

Introducción 45
Las propiedades fluorescentes del catión [Ru(bpy)3]2+
han sido ampliamente usadas en
la fabricación de sensores para el reconocimiento de aniones. En 1995 Beer y Szemes68
diseñaron un sensor para iones cloruro usando bipiridinas, 104. El receptor puede
complejar iones cloruro mediante cuatro enlaces por puente de hidrógeno, lo que
produce cambios en la fluorescencia de la molécula. La presencia de dos centros
luminiscentes en 104 complica la señal de fluorescencia y por ello sintetizaron el sensor
mixto 105, que contiene una unidad luminiscente y una unidad electroactiva. Este
sensor tiene la misma afinidad que 104 por los iones cloruro, y en presencia de un
equivalente de iones cloruro aumenta significativamente la fluorescencia del sensor 105.
N
N
N
N
N
N
N
O
N
O
N
N
O
O
N
N N
N
N
N
Ru Ru
H H
H H
2+ 2+
104
N
O
N
O
N
N
O
O
N
N N
N
N
N
Ru
H H
H H
M2+
105, M = Fe, Co+
El desarrollo de medios para medir la distribución y la concentración de iones de
importancia biológica, tales como el Zn2+
, Ca2+
y Mg2+
, es un tema de máximo interés
en el diseño de sensores. Resulta especialmente interesante poder detectar uno de estos
68
Beer, P.D.; Szemes, F.; “Remarkable Chloride over Dihydrogen Phosphate Anion Selectivity
Exhibited by Novel Macrocyclic Bis[ruthenium(II) bipyridyl] and Ruthenium Bipyridyl-metallocene
Receptors”. J. Chem. Soc. Chem. Commun. 1995, 2245-2247.

46 Tesis Doctoral Susana Barriga Falcón
cationes en presencia de los otros dos en muestras de tejido. El primer receptor usado
con este fin fue el TSQ, 106, que actúa selectivamente produciendo fluorescencia con el
Zn2+
en presencia de los otros dos cationes. Desafortunadamente la fluorescencia varía
con el medio y con la estequiometría de los complejos, por lo que este sistema no es
ideal. Se consiguen mejoras usando un tipo de aceptor denominado Zinquin, que debido
a su carácter lipófilo difunde a través de las membranas celulares, y una vez en la célula
es hidrolizado por esterasas y compleja el Zn2+
. Este método no sirve para detectar el
Zn2+
presente en metaloenzimas o en proteínas con dedos de Zn2+
.69
SO2
HN
N
MeO
106
Kimura ha diseñado un fluoróforo de Zn2+
muy efectivo, el dansilamidoetilencicleno
107,70
que presenta la doble ventaja de la alta afinidad del grupo dansilamida
desprotonado por el Zn2+
y el gran aumento de la intensidad de fluorescencia debido a la
quelatación del metal por el nitrógeno del cromóforo. Este último factor es conocido
como efecto CHEF (aumento de la fluorescencia por quelatación). Este fluoróforo es
capaz de detectar Zn2+
en concentraciones del rango de 0.1 a 5 µM y existe una
dependencia lineal entre la concentración y la intensidad de fluorescencia. La
69
Kimber, M. C.; Mahadevan, I. B.; Lincoln, S. F.; Ward, A. D.; Tiekink, E. R. T. “The synthesis and
fluorescent properties of analogues of the zinc(II) specific fluorophore zinquin ester”. J. Org. Chem.
2000, 65, 8204-8209. 70
Koike, T.; Watanabe, T.; Aoki, S.; Kimura, E.; Shiro, M. “A novel biomimetic zinc(II)-fluorophore,
dansylamidoethyl- pendant macrocyclic tetraamine 1,4,7,10-tetraazocyclododecane (cyclen)”.
J. Am. Chem. Soc. 1996, 118, 12696-12703.

Introducción 47
selectividad no se ve afectada en presencia de iones alcalinos o alcalinotérreos, aunque
el Cu2+
compite efectivamente con el Zn2+
.71
N
SO2HN
N
NH
HN
HN
N
O2S
N
N
NN
N
Zn2+
Zn2+
107 108
Esquema 1.2: Sensor detector de Zn2+ basado en un derivado de la dansilamida
4.2. Sensores electroquímicos
Los sensores electroquímicos incorporan un centro redox, por ejemplo un derivado de
ferroceno, que puede permitir la detección electroquímica de la unión del sustrato al
receptor.
Beer72
ha diseñado un sensor para moléculas neutras basado en receptores de tipo
calixareno. El receptor esta constituido por un calix[5]areno, 109, con tres derivados de
ferroceno unidos, y puede reconocer moléculas de disolvente dipolares como
acetonitrilo, dimetilsulfóxido y dimetilformamida, que se unen al receptor mediante
interacciones por puentes de hidrógeno con los dos grupos fenólicos restantes que
existen en el calixareno.
71
Kimura, E.; Koike, T. “Recent Development of Zinc Fluorophores”. Chem. Soc. Rev. 1998, 27,
179-184. 72
Beer, P. D.; Gale, P. A.; Chen, Z. Drew, M. G. B. “New bis- and tris-ferrocenyl and tris-benzoyl
lower-rim substituted calyx[5]arene esters: Synthesis, electrochemistry and X-ray crystal structures”.
Supramolecular Chemistry 1996, 7, 241-255.

48 Tesis Doctoral Susana Barriga Falcón
OFc FcO
OH
OFc
HOFe
O
O
OFc =
109
La presencia de dipolos adyacentes provoca un pequeño, pero significativo,
desplazamiento en el potencial redox de los grupos ferroceno, que no ocurre en
presencia de moléculas apolares como el diclorometano o el tolueno.
La unión del sustrato a este receptor es débil, como consecuencia de la debilidad de los
sitios de unión que se utilizan.
Tucker ha conseguido diseñar un receptor que tiene una respuesta electroquímica mayor
que el diseñado por Beer para moléculas neutras. Este receptor, 110, incorpora zonas
para el reconocimiento mediante enlaces de hidrógeno,73
y es capaz de detectar ácido
glutárico en cloroformo. Cada grupo carboxilato forma dos enlaces de hidrógeno, el
hidroxilo con el nitrógeno de la piridina y el carbonilo con la amida presente en el
receptor.
Fe
NH
NH3C
O
HNNH3C
O
HO
HO
O
O
110 111, Ácido Glutárico
73
Carr, J. D.; Lambert, L.; Hibbs, D. E.; Hursthouse, M. B.; Malik, K. M. A.; Tucker, J. H. R. “Novel
electrochemical sensors for neutral molecules”. Chem. Commun. 1997, 1649-1650.

Introducción 49
5. Aparatos moleculares electrónicos: Interruptores, cables y rectificadores
Una máquina molecular puede definirse como el conjunto de un número de
componentes moleculares discretos diseñado para realizar una función específica. Cada
componente molecular realiza un acto simple, mientras que el conjunto entero realiza
una función más compleja, que es el resultado de la cooperación de varios componentes
moleculares.
Las máquinas moleculares operan a través de impulsos electrónicos y/o nucleares y se
caracterizan por:
� El tipo de energía suministrada para realizar el trabajo
� El tipo de detección de la función que realizan
� La posibilidad de repetir la operación (proceso cíclico)
� El tiempo necesario para completar un ciclo
� La función realizada
5.1. Cables moleculares
El término de cable molecular se reserva para aquellos sistemas que están totalmente
conjugados, tienen una longitud precisa, una constitución determinada y una o dos
funcionalizaciones terminales.
Las propiedades básicas de un cable molecular consisten en poder ser conectado entre
dos componentes (generalmente un aceptor de electrones y un dador de electrones) y en
poder conducir una señal eléctrica o impulso entre ellos. La señal eléctrica debe poder
ser transportada a una distancia espacial significativa.
El grupo de Lehn ha preparado varios compuestos de tipo carotenoide, 112-114,
llamados caroviológenos.74
Mediante dicroísmo lineal, Lehn ha demostrado que estos
caroviológenos son capaces de atravesar membranas moleculares de dihexadecilfosfato,
74
Arrhenius, T. S.; Blanchard-Desce, M.; Dvolaitzky, M.; Lehn, J. M.; Malthete, J. “Molecular devices
– caroviologens as an approach to molecular wires synthesis and incorporation into vesicle
membranes”. Proc. Natl. Acad. Sci. USA, 1986, 83, 5355-5359.

50 Tesis Doctoral Susana Barriga Falcón
de espesor similar a la longitud de un cable molecular. En el compuesto 112 los grupos
piridinio hidrofílicos interactúan con las fases acuosas situadas en ambas caras de la
pared vesicular, actuando también como grupos electroactivos, aunque no modifican
significativamente la conductividad transmembranar.
N
N
112
N
N
-O3S
SO3
-
113
NN
N NN
N
N
N
114
Los mejores resultados se obtienen con el zwitterion 113, en presencia del cual se
observa un aumento de la conductividad de cuatro a ocho veces, entre una fase oxidante
interna que contiene ferricianuro potásico y una fase reductora externa de ditionito
sódico (Figura 1.23).
N
O3S
N
SO3
e - e -
Figura 1.23: Conductividad molecular a través de una vesícula de fosfolípido

Introducción 51
En máquinas moleculares electrónicas reales se requiere un grado de seguridad en caso
de daño, por lo que se ha diseñado un cable molecular triple 114. Del mismo modo,
Lehn ha desarrollado un aislante de cables moleculares, que consiste en hacer pasar a
los caroviológenos a través de la cavidad de una ciclodextrina, que actúa como “funda
protectora” (Figura 1.24).
N
N
Figura 1.24: Aislante molecular
Una aproximación alternativa hacia conductores moleculares fue realizada por Fox75
usando copolímeros de bloque que contienen unidades dadoras de electrones y unidades
aceptoras de electrones separadas por relés conductores, como por ejemplo 115.
Ph
O O
D
O O
R
O O
A
Ph
NMe
MeNC
CN
x y z
115
D = R = A =
Es posible realizar el control de la longitud del polímero y también del número de
monómeros que constituyen cada bloque polimérico. Los polímeros que se obtienen son
75
Fox, M. A. “Fundamentals in the design of molecular electronic devices: Long-range charge carrier
transport and electronic coupling”. Acc. Chem. Res. 1999, 32, 201-207.

52 Tesis Doctoral Susana Barriga Falcón
rígidos, y por lo tanto presentan características eléctricas bien definidas y muestran
transferencia electrónica vectorial desde el dador al aceptor.
Por otra parte, los materiales basados en polipéptidos tienen velocidades de
transferencia electrónica diferentes, dependiendo de la orientación del dipolo en la
hélice peptídica. Por lo tanto, esta transferencia electrónica puede ser controlada
mediante la aplicación de potenciales externos. Estos resultados representan un inicio
del control de las características de transferencia electrónica necesarias para diseñar
máquinas optoelectrónicas funcionales a nanoescala.75
El diseño de cables moleculares no está limitado únicamente a conductores orgánicos.
La incorporación de iones metálicos con sus diferentes estados de oxidación y actividad
fotoquímica proporciona nuevas perspectivas para la síntesis de conductores
moleculares con metales.6 En la figura 1.25 se muestran algunos ejemplos.
Re
Re
NOPh3P
PPh3ON
Re
PPh3ON
ReNOPh3P
ReNOPh3P
Re
PPh3ON RhHL
LL
n
116 117 118 119
Figura 1.25: Ejemplos de cables moleculares con metales de transición
Estas ideas, desarrolladas en moléculas pequeñas, se han usado también en la formación
de polímeros acetilénicos tales como 119, que son interesantes tanto por sus
propiedades conductoras como por sus propiedades ópticas no lineales.
5.2. Rectificadores moleculares
Un rectificador es un aparato que permite el flujo de electrones únicamente en una
dirección. En aparatos electrónicos voluminosos se utiliza para transformar corriente

Introducción 53
alterna en corriente continua. Los rectificadores convencionales se construyen con
semiconductores de tipo p (pobre en electrones) y de tipo n (rico en electrones) en
contacto. El rectificador causa un aumento de la capa de aislamiento en la zona de
contacto. Cuando la unión tipo n y tipo p se forma, los electrones fluyen del dador al
aceptor hasta que la carga se neutraliza dentro de la zona de contacto y, a partir de
entonces, el flujo de corriente queda imposibilitado. Si se aplica un potencial externo al
rectificador, dependiendo de la polaridad de dicho potencial, se producirá el cese o la
restauración de la corriente. Si se aplica una corriente alterna habrá flujo de corriente
sólo cuando el ciclo de alternancia sea de la polaridad adecuada.
En el ámbito molecular, se requieren las mismas características que en los componentes
supramoleculares y, por lo tanto, un rectificador molecular está formado por un dador
de electrones y un aceptor de electrones separados por una zona aislante.56
Los compuestos 120 y 121 tienen fragmentos aislantes de tipo σ, mientras que en
derivados de tipo carotenoide tales como 122 el dador y el aceptor están separados por
un sistema conductor π.
S
S
S
S
NC CN
NC CN
O
O
OMe
OMe
OMe
S
S
N
120 121
122

54 Tesis Doctoral Susana Barriga Falcón
5.3. Interruptores moleculares
La idea del uso de moléculas en la interrupción de señales eléctricas, fue sugerido por
primera vez por Aviram y Ratner en 1974.76
Los tipos más frecuentes de interruptores son aquellos en los que hay un cambio
estructural producido por una fotoexcitación, o bien se produce un cambio electrónico
drástico causado por una perturbación externa.
5.3.1. El sistema 1,2-ditienileteno como interruptor
Un excelente componente para los aparatos moleculares es el sistema 1,2-ditienileteno,
que ha sido ampliamente usado por el grupo de Lehn en varios sistemas de
interruptores.77
La base del interruptor es la transformación de las unidades de
ditienileteno entre dos estados estables en función de la longitud de onda de la radiación
incidente (Esquema 1.3).
S
F
F
F FF
F
SR2
R1
S SR1
R2
F
F
FFF
F
N
S
S
CN
CN
Abierto
365 nm
> 600 nm
Cerrado
123a: R1 = R
2 =
124a: R1 = R
2 =
N
123b: R1 = R
2 =
S
S
CN
CN
124b: R1 = R
2 =
Esquema 1.3: Estados del 1,2-ditienileteno biestable
Esta capacidad de interrupción es altamente reversible por simple alternancia entre
irradiación con luz ultravioleta y luz visible. Además los dos estados, “abierto” y
76
Aviram, A.; Ratner, M. A. Chem. Phys. Lett. 1974, 29, 277. 77
Kawai, S. H.; Gilant, S. L.; Lehn, J. M. “Dual-mode optical-electrical molecular switching device”.
J. Chem. Soc. Chem. Commun. 1994, 1011-1013.

Introducción 55
“cerrado”, tienen propiedades electrónicas muy diferentes. El estado cerrado contiene
un sistema π totalmente conjugado, permitiendo una comunicación electrónica efectiva
desde el sustituyente R1 hasta R
2.
En 123 los dos sustituyentes unidos al interruptor tienen grupos atractores de electrones
idénticos. La forma cerrada se reduce fácilmente mientras que la forma abierta no lo
hace. Por lo tanto, constituye el prototipo de un cable molecular que puede actuar como
un interruptor.
En 124, R1 es un grupo dador de electrones, mientras que R
2 es un aceptor de
electrones. La molécula resultante en la forma cerrada es un sistema de tipo push-pull
con marcadas propiedades de óptica no lineal. Si se abre el interruptor con luz visible,
se destruye la comunicación entre los dos grupos finales de la molécula y se pierden las
propiedades de óptica no lineal.
El sistema fotointerrumpible más destacado basado en este concepto es 125. La forma
abierta 125 es electroquímicamente inerte. Cuando se cierra fotoquímicamente, la forma
cerrada 126 es oxidada fácilmente a la bis-quinona 127. La forma quinónica es
fotoquímicamente inerte y no puede volver a la forma abierta hasta que se vuelve a
reducir. El sistema puede ser utilizado en sistemas de almacenaje de información. El
1,2-ditienileteno 125 representa una aproximación química para la creación de un
EDRAW (borrado directo de lectura tras escritura) que es un modo de almacenaje de
información en el que ésta se escribe ópticamente mediante irradiación con luz
ultravioleta y es salvaguardada por oxidación. La información puede ser leída muchas
veces sin destrucción antes de ser finalmente destruida mediante reducción.

56 Tesis Doctoral Susana Barriga Falcón
S
F
F
F FF
F
SS S
F
F
FFF
F
OHHOHO OH
[O]
S S
F
F
FFF
F
O O
365 nm
> 600 nm
> 600 nm
Fotoquímicamenteinerte
Electroquímicamente inerte
125 126
127
Esquema1.4: Interruptor foto y electroquímico 125
5.3.2. Luminiscencia electrointerrumpible
Otro ejemplo de un aparato molecular electro-óptico combinado lo constituye la pareja
de estructuras 128 y 129, que son electroquímicamente interconvertibles de forma
rápida. En la forma hidroquinónica reducida 129, el complejo Ru(bpy)32+
es altamente
luminiscente. La oxidación electroquímica hasta la forma quinónica 128 ocurre antes
que la oxidación del centro metálico de Ru(II) y provoca la interrupción de la
luminiscencia. Tanto la forma reducida como la oxidada de este compuesto pueden ser
aisladas, y la pareja redox es totalmente reversible y estable.78
78
Goulle, V.; Harriman, A.; Lehn, J. M. “An electro-photoswitch – redox switching of the
luminescence of a bipyridine metal-complex”. J. Chem. Soc. Chem. Commun. 1993, 1034-1036.

Introducción 57
N
N
N
N
N
N
Ru
O
O
N
N
N
N
N
N
Ru
HO
OH
2+
+2e-, -2H
+
-2e-, +2H
+
2+
128 129
Esquema1.5
5.3.3. Complejación fotointerrumpible
Las reacciones inducidas fotoquímicamente pueden ser usadas para generar un
interruptor en materiales que, cuando absorben luz, pasan desde un estado a otro,
provocando un cambio en sus propiedades.
Por ejemplo, un interruptor fotoquímico entre un receptor que puede unir fuertemente a
los cationes de los metales alcalinos y uno que los une mucho más débilmente puede
usarse para generar un pulso de luz cuando los cationes son liberados: una señal
fotoiónica. Este comportamiento se observa en un nitrofenil derivado del
[2.2.2]criptando, 130. Los criptandos se unen fuertemente a cationes tales como el K+,
incluso en agua; sin embargo, la irradiación con una longitud de onda de 330 nm
provoca la apertura del anillo conduciendo a 131. Esta especie tiene menor afinidad por
los cationes metálicos, y por lo tanto cualquier ion metálico unido es inmediatamente
liberado.

58 Tesis Doctoral Susana Barriga Falcón
O O
N
O
N
O
NO2
O O
O O
N
O
N
O
NO2
O ON
O
N
O
O O
O
O
OH
O2N
n+
n+
n+
330 nm
130 131
Esquema 1.6
Otro componente fotointerrumpible muy usado es el azobenceno, que puede sufrir
isomerización cis/trans bajo condiciones térmicas o fotoquímicas (Esquema 1.7).79
N
N
N NT
132, trans 132, cis
hν
Esquema 1.7
Esta funcionalidad reversible del azobenceno se ha incorporado en varios sistemas de
receptores moleculares basados en éteres corona (azocoronas), ciclofanos (azofanos) y
compuestos tales como azocriptandos y azociclodextrinas. Un azocoronando como 133
actúa como interruptor frente a la unión de metales alcalinos. La forma trans no se une a
los cationes alcalinos como consecuencia de la conformación que adopta el
azocoronando, mientras que la forma cis compleja a los metales y muestra un
comportamiento similar a los análogos [15]corona-5, [18]corona-6 y [21]corona-7.
79
Rau, H. en “Photochromism, Molecules and Systems”. Dürr, H.; Bouas-Laurent, H. Ed. Elsevier,
Amsterdam, 1990.

Introducción 59
N N
O
O
OO
O
O
ON
N
OO
O
O OO
O
∆T
hν
133-trans 133-cis
Esquema 1.8
La estabilidad relativa de los isómeros cis y trans en muchos azoderivados depende de
la naturaleza del sustrato, y la velocidad de la isomerización, tanto térmica como
fotoquímica, puede ser influenciada por este hecho.
5.3.4. Interruptores alostéricos
Un sistema alostérico es aquel cuya conformación depende del estado de complejación
de dos o más sitios de unión. Estos tipos de sistemas son muy comunes entre las
macromoléculas biológicas, por ejemplo, el caso de la hemoglobina. Esta proteína
transportadora puede modificar su afinidad por el oxígeno debido a la unión de
moléculas pequeñas, como el monóxido de carbono, a un centro alostérico alejado del
centro activo. La unión a un sitio produce un cambio conformacional que influye en la
capacidad de unión del segundo sitio. Uno de los primeros ejemplos de interruptor
alostérico artificial lo constituye el macrociclo 134 diseñado por Rebek.80
En el estado libre, la molécula tiene una conformación flexible y no compleja bien a los
cationes alcalinos. La complejación del derivado bipiridina con un metal de transición
que presente un estado de oxidación bajo, como el Ru(II), provoca un cambio
conformacional que induce una rotación a través del eje piridilo-piridilo, situando el éter
corona en una conformación más preorganizada que permite la unión selectiva de Na+.
De forma similar, la unión de metales alcalinos con el éter corona del ligando organiza
80
Rebek, J. Jr. “Binding forces, equilibria, and rates – new models for enzymic catalysis”. Acc. Chem.
Res. 1984, 17, 258-264.

60 Tesis Doctoral Susana Barriga Falcón
la parte que contiene la bipiridina para la unión con Ru(II) u otros metales de transición
(Esquema 1.9).
NN
O
O
OO
O
Na+
N
NOO
O
O ORu
2+
N
NOO
O
O O
134 135 136
Esquema 1.9
5.3.5. Interruptores para moléculas magnéticas
El magnetismo molecular puede ser fotocontrolado mediante la incorporación de
unidades fotocrómicas en el sistema. Es decir, cuando dos electrones desapareados se
sitúan en ambos extremos de una cadena π-conjugada, los dos espines de los electrones
desapareados interaccionan magnéticamente. Si puede controlarse la formación o
interrupción de un sistema π-conjugado usando una unidad fotocrómica, la interacción
magnética puede ser controlada mediante irradiación de luz.
Con este fin se han diseñado moléculas que tienen radicales nitroxilo colocados en los
extremos de sistemas que contienen la unidad fotocrómica diarileteno.81
Cuando las moléculas 137a y 138a se irradian a 313 nm, tiene lugar una ciclación
fotoquímica y la molécula pierde su carácter magnético inicial. Del mismo modo,
cuando 137b y 138b se irradian a 578 nm el anillo central vuelve a abrirse y la molécula
recupera su magnetismo inicial. Estos dos sistemas constituyen los primeros ejemplos
de interrupción reversible del magnetismo intramolecular.
81
(a) Matsuda, K.; Irie, M. “A Diarylethene with Two Nitronyl Nitroxides: Photoswitching of
Intramolecular Magnetic Interactions”. J. Am. Chem. Soc. 2000, 122, 7195-7201. (b) Matsuda, K.; Irie, M. “Photoswitching of Intramolecular Magnetic Interaction Using a Photochromic Spin
Coupler: An ESR Study”. J. Am. Chem. Soc. 2000, 122, 8309-8310.

Introducción 61
S
F
F
F FF
F
SS S
F
F
FFF
F
N
NN
N
O
OO
ON
N
O
O
N
N
O
O
S
F
F
F FF
F
S
N
N
O
O
N
N
O
O
S S
F
F
FFF
F
N
N
O
O
N
N
O
O
313 nm
578 nm
137a 137b
313 nm
578 nm
138a 138b
Esquema 1.10
6. Máquinas moleculares basadas en catenanos y rotaxanos
Una máquina molecular es un tipo particular de sistema supramolecular en el que las
partes que lo componen pueden presentar cambios en sus posiciones relativas como
resultado de estímulos externos.82
Este término sólo se aplica a sistemas que presentan
una gran amplitud de movimiento de sus componentes moleculares.
Es importante indicar que los movimientos moleculares están acompañados por
cambios de algunas de las propiedades físicas o químicas del sistema, lo que constituye
una señal externa que puede ser utilizada para detectar la operación que realiza la
máquina. La reversibilidad del movimiento es un requisito necesario para construir una
máquina molecular.
El diseño de máquinas moleculares se basa, en la actualidad, en la construcción de
pseudorrotaxanos, rotaxanos y catenanos.83
Un catenano es un compuesto que consiste en dos o más anillos entrelazados
mecánicamente sin que exista ninguna interacción química entre ellos. Generalmente,
los anillos no pueden separarse sin que se produzca la ruptura de un enlace químico. Los
82
Balzani, V.; Credi, A.; Raymo, F. M.; Stoddart, J. F. “Artificial Molecular Machines”. Angew. Chem.
Int. Ed. 2000, 39, 3348-3391. 83
Balzani, V.; Gómez-López, M.; Stoddart, J. F. “Molecular Machines”. Acc. Chem. Res. 1998, 31,
405-414.

62 Tesis Doctoral Susana Barriga Falcón
catenanos se nombran de acuerdo con el número de anillos entrelazados, así, por
ejemplo, un [2]catenano está constituido por dos anillos (Figura 1.26 a).
Un rotaxano consiste en un componente de geometría lineal de gran longitud que se
inserta en un anillo macrocíclico. De nuevo, para separar ambos componentes es
necesario la ruptura de algún enlace químico. Además la parte lineal acaba con dos
grupos voluminosos que son demasiado grandes como para pasar a través del
macrociclo (Figura 1.26 b).
Un pseudorrotaxano está constituido de la misma forma que un rotaxano pero no
presenta los grupos voluminosos al final de la parte lineal (Figura 1.26c), lo que permite
el ensamblaje o desensamblaje de las partes que constituyen el pseudorrotaxano,
modificándose de esta forma las propiedades del sistema. Los pseudorrotaxanos son a
menudo precursores necesarios en la síntesis de catenanos y rotaxanos.
(a) [2]Catenano (b) [2]Rotaxano (c) [2]Pseudorrotaxano
Figura 1.26: Representación esquemática de (a) un [2]catenano, (b) un [2]rotaxano y (c) un
[2]pseudorrotaxano
Recientemente, se ha conseguido una aproximación sintética eficiente y racional para la
preparación de sistemas moleculares tales como los pseudorrotaxanos, rotaxanos y
catenanos.84
Las estrategias elegidas por el grupo de Stoddart85
se basan en: (a)
transferencia de carga en interacciones π-π entre un aceptor y un dador de electrones y/o
84
Sauvage, J. P.; Dietrich-Buckecher, C. O. Ed. “Molecular Catenanes, Rotaxanes and Knots”. VCH-
Wiley, Weinheim, 1999. 85
Raymo, F. M.; Stoddart, J. F. “Interlocked Macromolecules”. Chem. Rev. 1999, 99, 1643-1663.

Introducción 63
(b) interacciones mediante puentes de hidrógeno entre iones amonio secundarios y
éteres corona.
La aplicación de estos sistemas incluye la formación de interruptores moleculares
sofisticados, el desarrollo de materiales con nuevas propiedades físicas y la
inmovilización superficial de especies catalíticas foto o redox activas sin la necesidad de
alterar químicamente sus propiedades.
6.1. Máquinas moleculares basadas en pseudorrotaxanos
Un pseudorrotaxano presenta un movimiento que recuerda a un pistón en un cilindro. El
primer intento de diseñar una máquina molecular86
basada en un pseudorrotaxano hizo
uso de un componente macrocíclico o lineal fotorreducible. Así, en el pseudorrotaxano
139 la irradiación con luz visible provoca la reducción de una de las unidades de
bipiridina del anillo, lo que destruye parcialmente la interacción responsable del
acoplamiento de las distintas unidades, y produce su separación. La oxidación del
sistema bipiridina restaura la interacción y produce de nuevo el acoplamiento.
N
N
N
N
O
O
O
O
OH
OO
HO
N
N
N
N
O O O OH
OOOHO[O]
hν
+
139
Esquema 1.11
Una segunda generación de máquinas que funcionan fotoquímicamente ha sido diseñada
de forma que el pseudorrotaxano incorpora el motor “canalizador de la luz” (por
ejemplo un componente fotosensible) en la zona lineal87
o en el anillo macrocíclico.88
86
Ballardini, R.; Balzani, V.; Gandolfi, M. T.; Prodi, L.; Venturi, M.; Philp, D.; Ricketts, H. G.;
Stoddart, J. F. “A Photochemically-Driven Molecular Machine”. Angew. Chem. Int. Ed. 1993, 32, 1301-1303.
87 Ashton, P. R.; Ballardini, R.; Balzani, V.; Constable, E. C.; Credi, A.; Kocian, O.; Langford, S. J.;
Perece, J. A.; Prodi, L.; Schofield, E. R.; Spencer, N.; Stoddart, J. F.; Wenger, S. “Ru(II)-

64 Tesis Doctoral Susana Barriga Falcón
En ambos casos, la excitación del componente fotosensible con luz visible, en presencia
de un dador, provoca la reducción de la unidad aceptora de electrones y, como
consecuencia, el desensamblaje del pseudorrotaxano. En presencia de oxígeno se
produce de nuevo el enlace del pseudorrotaxano. (Esquema 1.12)
N
N
N
N
N
N
Ru
O
O
O
O
O
O
O
O
O
O
N
N
O
O
O
O
O
O
O
O
O
O
N
N
N
N
N
N
RuN
N
2+2+
+
O2
hν
140
N
N
N
N
O
O
O
O
OH
OO
HO
N
N
N
N
O O O OH
OOOHO
O2
N N
Re ClOC
OC CO
Re ClOC
OC CO
hν
+
141
Esquema 1.12
Un sistema que actúa como interruptor entre dos colores diferentes (interruptor
crómico) resulta de la formación de dos pseudorrotaxanos diferentes. El color del
complejo, que cambia de rojo a violeta, depende del componente que intervenga en la
formación del pseudorrotaxano, 142 o 143. El interruptor en estado de reposo es de
color violeta, ya que el grupo más rico en electrones, el 1,5-dioxinaftaleno de 143, se
compleja con el ciclofano 144. Pero 143 también puede comportarse como un receptor
de cationes alcalinos, especialmente de iones K+, por la presencia del anillo de
[18]corona-6. Con la unión de los iones K+ se produce una repulsión inmediata entre el
componente lineal 143 y el ciclofano catiónico 144. Esta repulsión provoca la ruptura
Polypyrimidine Complexes Covalently Linked to Electron Acceptors as Wires for Light-Driven Pseudorotaxane-Type Molecular Machines”. Chem. Eur. J. 1998, 4, 2411-2422.
88 Ashton, P. R.; Balzani, V.; Kocian, O.; Prodi, L.; Spencer, N.; Stoddart, J. F. “A Light-Fueled
“Piston-Cylinder” Molecular-Level Machine”. J. Am. Chem. Soc. 1998, 120, 11190-11191.

Introducción 65
del pseudorrotaxano, y conduce a la complejación con el otro componente lineal 142, lo
que confiere color rojo a la disolución. De esta forma se puede detectar, mediante un
sencillo examen visual, la presencia de cantidades muy pequeñas de iones K+ en
disolución (Esquema 1.13).
O O
O
O
O
O O O O
O
HO O O
O
N
N
N
N N
N
N
N
O O
O
O
O
O O O O
O
+
+ 142
K+
N
N
N
N
O O
O
O
O
O O O O
O
K+
N
N
N
N
HO O O
O
+ (143)K+
Rojo
142
143
144
Violeta
Esquema 1.13: Interruptor cromóforo basado en un pseudorrotaxano
6.2. Máquinas moleculares basadas en rotaxanos
Los rotaxanos que contienen dos sitios de reconocimiento en el componente lineal,
susceptibles de unión con el macrociclo, pueden ser usados como interruptores en
función del sitio donde se produce la unión. Un ejemplo lo constituye el rotaxano 145,
formado por el macrociclo dibenzo[24]corona-8 y un componente lineal que contiene
un centro de dialquilamonio y una unidad dicatiónica 4,4’-bipiridina. Se incluye un

66 Tesis Doctoral Susana Barriga Falcón
grupo antraceno como grupo voluminoso, y se utilizan sus propiedades luminiscentes y
redox para detectar el estado en el que se encuentra el sistema. El anillo de
dibenzo[24]corona-8 presenta una efectividad del 100% para reconocer el sitio que
contiene el catión amonio. Cuando se desprotona el ión amonio, el anillo se desplaza
hacia la unidad bipiridina. La protonación provoca de nuevo el desplazamiento hacia el
centro amónico.89
O
O
O
O
O
O
O
O
NH2
N
N O
O
O
O
O
O
O
O
NH N
N- H
+
+ H+
145a 145b
Figura 1.27: Máquina molecular basada en un rotaxano
En la actualidad se están diseñando rotaxanos cuyo movimiento puede estar causado por
dos fuerzas motrices diferentes. Por ejemplo, en 146 el desplazamiento puede estar
provocado por un cambio del pH o por un proceso redox, que es la oxidación de la
unidad benzidínica. Ambos procesos son totalmente reversibles y el sistema puede ser
utilizado como un interruptor molecular.
89
Ashton, P. R.; Ballardini, R.; Balzani, V.; Baxter, I.; Credi, A.; Fyfe, M. C. T.; Gandolfi, M. T.;
Gómez-López, M.; Martínez-Díaz, M. V.; Piersanti, A; Spencer, N.; Stoddart, J. F.; Venturi, M.; White, A. J. P.; Williams, D. J. “Acid-base Controllable Molecular Shuttles”. J. Am. Chem. Soc.
1998, 120, 11932-11942.

Introducción 67
Si O O O HN
N
N
N
N
NH O O O O O O O Si
N
N
N
N
Si O O O N
H
HN O O O O O O O Si
H
H
- 2H+
+ 2H+
146a
146b
N
N
N
N
Si O O O N N O O O O O O O Si
146c
Figura 1.28: Interruptor molecular que responde a cambios redox y de pH basados en un [2]rotaxano
En este sentido, se han diseñado varios rotaxanos que actúan como interruptores redox o
fotoactivos. Para ello, se usan grupos finales fototoactivos como el antraceno o grupos
redox activos, como el grupo ferrocenilo. Estos grupos permiten controlar la velocidad
de recombinación de carga bajo fotoexcitación en los rotaxanos 147 y 148.
N
N
N
N
O
O
OOO
O
O O O
O
147

68 Tesis Doctoral Susana Barriga Falcón
N
N
N
N
O
O
OOO
O
O O O
O
Fe
Fe148
6.3. Máquinas moleculares basadas en catenanos
En un catenano, los cambios estructurales causados por cincunrotación de un anillo
respecto a otro son evidentes cuando uno de los anillos contiene dos unidades no
equivalentes.
En el catenano 149a-c de la figura 1.29, el anillo que contiene el aceptor de electrones
es simétrico, mientras que el otro anillo no lo es, ya que contiene dos unidades dadoras
de electrones diferentes, un TTF y un 1,5-dioxinaftaleno.90
En la estructura del
catenano, el dador de electrones situado en el interior experimenta el efecto de dos
unidades aceptoras de electrones, mientras que el que se encuentra fuera experimenta el
efecto de una sola.
En el catenano 149a, el mejor dador de electrones es el TTF y por lo tanto está dentro
del aceptor, mientras que el 1,5-dioxinaftaleno se encuentra en el exterior. Si se produce
una oxidación, la primera unidad que se oxida es el TTF, con lo que pierde sus
propiedades como dador de electrones. La oxidación provoca la repulsión entre el TTF+
y el macrociclo tetracatiónico lo que produce la cincunrotación de un anillo para dar
lugar a los isómeros traslacionales 149b o 149c, que contienen al 1,5-dioxinaftaleno en
el interior del aceptor. Si se produce la reducción, el sistema vuelve a la posición inicial,
149a, ya que el interruptor es totalmente reversible.
90
Asakawa, M.; Ashton, P. R.; Balzani, V.; Credi, A.; Hamers, C.; Mattersteig, G.; Montalti, M.;
Shipway, A. N.; Spencer, N.; Stoddart, J. F.; Tolley, M. S. ; Venturi, M.; White, A. J. P.; Williams, D. J. “A Chemically and Electrochemically Switchable [2]Catenane Incorporating a
Tetrathiafulvalene Unit”. Angew. Chem. Int. Ed. 1998, 37, 333-337.

Introducción 69
SS
SS
OO
O
O
O
O
OO
O
O
OO
O
O
O
S S
S S
149a 149b
- e-
+ e-
N
N
N
N
N
N
N
N
OO
O
O
O
S S
S S
149c
- e-
+ e-
N
N
N
N
Figura 1.29: Movimientos moleculares controlados electroquímicamente del catenano 149
Lo mismo ocurre con el catenano formado entre 150, que contiene dos sitios diferentes
de reconocimiento, una unidad bipiridina y una unidad bis(piridina)etileno, y 151, bis(p-
fenilen)[34]corona-10. Al formarse el catenano, la unidad de bipiridina se encuentra
dentro de la corona, tal como aparece en el esquema. Si se produjera la reducción de
150 para dar lugar a un tricatión radical, se produciría la circunrotación y la bipiridina
quedaría fuera del éter corona, ya que la reducción disminuye drásticamente sus
características de aceptor de electrones. De esta forma, la unidad que se encontraría
entonces dentro de la corona sería la de bis(piridina)etileno que sería el mejor aceptor de
electrones existente en el sistema. La oxidación conduciría al isómero inicial 150,
puesto que el sistema es reversible.
O
O
O
OO
O
O
O
O O
N
N
N
N
151
150

70 Tesis Doctoral Susana Barriga Falcón
7. Dendrímeros
Una de las clases más sorprendentes de compuestos macromoleculares, que han
adquirido un gran uso en los últimos años desde un punto de vista sintético, estructural
y funcional, han sido los dendrímeros. Los dendrímeros, o “moléculas en cascada”, son,
idealmente, macromoléculas monodispersas que presentan una estructura con gran
ramificación y arquitectura tridimensional. Un dendrímero puede ser considerado como
un compuesto que crece desde un núcleo central en forma de árbol. Por lo tanto,
estructuralmente, y a veces sintéticamente, un dendrímero comienza con un núcleo o
raíz multifuncional. Desde cada grupo funcional del núcleo se genera una nueva unidad
multifuncional para dar lugar a un oligómero con un núcleo central y numerosos grupos
funcionales en las ramas. De esta forma, se genera la primera generación del
dendrímero. Si se realiza una segunda iteración, se produce la unión de nuevas unidades
monoméricas a los grupos funcionales existentes en la primera generación del
dendrímero, de forma que se obtiene la segunda generación, mayor y mucho más
ramificada, del dendrímero. En principio, este proceso iterativo puede realizarse
indefinidamente para conducir a una macromolécula tridimensional bien definida
(Figura 1.30)
A A
A
A A
A
B
B
B
B
BB
A A
A
B
B
B
B
B B C C
C
C
C
CC
C
CC
C
C
Núcleo Primera generación Segunda generación
Tercera generación...
Figura 1.30: Diagrama esquemático de la preparación iterativa de dendrímeros

Introducción 71
Los dendrímeros han sido utilizados en el diseño de materiales funcionales y en la
construcción de materiales estructurales de aplicación en diversos campos.91
7.1. Preparación y propiedades de los dendrímeros
Existen dos estrategias para la síntesis de dendrímeros: la aproximación divergente, en
la que el dendrímero se construye desde el núcleo hacia la periferia, y la aproximación
convergente, en la que se construyen primero las ramas dendríticas y en el paso final se
unen a una molécula o átomo que constituye el núcleo del dendrímero. En la
aproximación divergente se realizan numerosas reacciones sobre la misma molécula;
por lo tanto, cada reacción debe hacerse con una gran selectividad y con un elevado
rendimiento, para evitar la introducción de errores o defectos. En la aproximación
convergente el número de reacciones individuales a realizar en cada etapa sintética es
menor, lo que minimiza la aparición de defectos estructurales.
La caracterización de dendrímeros, especialmente la determinación de la pureza
dendrítica, es difícil debido al gran tamaño y la gran simetría que presentan.
Generalmente se emplea la espectrometría de masas con técnicas de ionización como el
electro-spray.
En términos de propiedades físicas los dendrímeros son muy variables, dependiendo de
la naturaleza de los grupos que los componen y de las interacciones entre ellos. La
mayoría de los dendrímeros pueden sufrir cierto replegamiento, lo que reduce su
densidad de superficie y el número de grupos funcionales disponibles en la superficie.
Debido a su estructura tridimensional, las propiedades de los dendrímeros difieren
significativamente de las macromoléculas lineales en disolución. Esto presenta ciertas
ventajas, como por ejemplo que la viscosidad no aumenta con el peso molecular, y que
la forma adoptada depende en gran medida del disolvente.92
91
Fisher, M.; Vögtle, F. “Dendrimers: From Design to Application”. Angew. Chem. Int. Ed. 1999, 38,
884-905. 92
Bosman, A. W.; Janssen, H. M.; Meijer, E. W. “About Dendrimers: Structure, Physical Properties
and Applications”. Chem. Rev. 1999, 99, 1665-1669.

72 Tesis Doctoral Susana Barriga Falcón
7.2. Dendrímeros fotoactivos con metales de transición
La conectividad metal-ligando-metal de los complejos bipiridina-rutenio y compuestos
de coordinación relacionados se ha usado para construir metalodendrímeros. Los
dendrímeros resultantes son interesantes tanto por sus propiedades electroquímicas
como luminiscentes. El primer dendrímero que contenía un complejo rutenio-bipiridina
como núcleo fue descrito por el grupo de Balzani en 1994.93
N N
N
N
N
N
N
N
N
N
N
N
N N
N
N
N
N
N
N
N N
N
N
N
N
NN
N N
N
N
N
N
N
N
N N
N
N
N
N
NN
N N
N
N
N
N
N
N
N N
N
N
N
N
N
N
Ru
Ru Ru
Ru
Ru
Ru
Ru Ru
Ru
Ru
20+
152
En el dendrímero 153 el tiempo de vida del estado fotoexcitado del ion rutenio es
significativamente mayor que el que presentan complejos no dendríticos.94
Se preveen
aplicaciones en diagnósticos médicos para este tipo de dendrímeros supramoleculares.
93
Serroni, S.; Campagna, S.; Juris, A.; Venturi, M.; Balzani, V. “Polyether Arborols Mounted on a
Luminescent and Redox-Active Ruthenium(II) Polypyridine Core”. Gazz. Chim. Ital. 1994, 124, 423-
427. 94
Issberner, J.; Vögtle, F.; De Cola, L.; Balzani, V. “Dendritic Bipyridine Ligands and their
Tris(bipyridine)ruthenium(II) Chelates - Syntheses, Absorption Spectra, and Photophysical
Properties”. Chem. Eur. J. 1997, 3, 706-712.

Introducción 73
N N
O
HN
O
HNO
O
RO O
O
RO
O
O
OR
O
O
HN
O
O
ROO
O
RO
O
O
RO
O
O
NH
O
O
OR
O
O
OR
O
O
RO O
O
NH
O
NHO
O
ORO
O
OR
O
O
RO
O
O
NH
O
O
ORO
O
OR
O
O
OR
O
O
HN
O
O
RO
O
O
RO
O
O
ORO
N
N
O
NH
O
NH
O
O
OR
O
O
OR
O
O
RO O
O
NH
O
O
OR
O
O
ORO
O
ROO
O
HNO
O
RO O
O
RO
O
O
OR
O
O
NH
O
HN
O
ORO
O
O
RO
O
ORO
O
O
NH
O
O
RO
O
O
RO
O
ORO
O
O
NH
O
ORO
O
O
RO
O
ORO
O
N
N
O
HN
O
HN
O
O
RO
O
O
RO
O
O
ORO
O
HN
O
O
RO
O
O
ROO
O
ORO
O
NHO
O
ORO
O
OR
O
O
RO
O
O
HN
O
HN
O
O OR
O
O
OR
O
OOR
O
O
HN
O
O
OR
O
O
OR
O
OOR
O
O
HN
O
OOR
O
O
OR
O
O OR
O
Ru2+
153
Los dendrímeros con unidades de porfirina en su núcleo se han utilizado como modelos
biomiméticos de proteínas que contienen grupos hemo, y también como catalizadores
de oxidación impedidos estéricamente. El grupo de Inoue fue el primero en describir
dendrímeros que contenían metaloporfirinas en su núcleo, como por ejemplo 154.95
95
Jin, R. H.; Aida, T.; Inoue, S. “Caged Porphyrin - The 1st Dendritic Molecule Having a Core
Photochemical Functionality”. J. Chem. Soc. Chem. Commun. 1993, 1260-1262.

74 Tesis Doctoral Susana Barriga Falcón
NH N
N NH
O
Ar
O
OO
Ar
ArO
O
Ar
Ar
O O
O
OO
O
ArAr
OO
O
O
O
O
Ar
Ar
OO
Ar
Ar O
O
OO
O
O
Ar
Ar
O
O
O
O
O
Ar
O
O
O
Ar
Ar
O
OAr
Ar
O
O
OO
O
O
Ar
Ar
O
O
O
O
O
O
Ar
ArO
O
Ar
Ar
O
O
O
O
O
OAr
Ar O
O
O
O
O
Ar
O
OO
Ar
ArO
O
Ar
Ar
O O
O
O O O
ArAr
O O
O
O
ArO
O
Ar
Ar
OO
Ar
ArO
O
O O
O
O
Ar
Ar
O
O
O
O
O
Ar
O
O
O
Ar
Ar
O
OAr
Ar
O
O
OO
O
O
Ar
Ar
O
O
O
O
O
O
Ar
ArO
O
Ar
Ar
O
O
O
O
O
OAr
ArO
O
O
O
154
Zn2+
7.3. Sistemas fotoactivos no convencionales
La luz se utiliza habitualmente para manipular sistemas moleculares porque sus efectos
son rápidos, suaves, y a menudo reversibles. Así, el azobenceno se ha usado
ampliamente en la construcción de sistemas supramoleculares.
El primer dendrímero fotosensible fue sintetizado por el grupo de Vögtle en 1993.96
Este dendrímero, 155, contiene seis unidades de azobenceno en su periferia.
96
Mekelburger, H. B.; Rissanen, K.; Vögtle, F. “Repetitive Synthesis of Bulky Dendrimers - A
Reversibly Photoactive Dendrimer with 6 Azobenzene Side-Chains”. Chem. Ber. 1993, 126,
1161-1169.

Introducción 75
N
SO2 NO2S
NN
NSO2
NN
NSO2
NSO2
N
N
N
SO2
N
N
NO2S
N
SO2
NN
NO2S
NN
155
Recientemente, Junge y McGrath han sintetizado un dendrímero (156) que contiene un
grupo azobenceno en su núcleo, y han investigado su isomerización E/Z inducida
mediante luz ultravioleta (Figura 1.31).97
97
Junge, D. M.; McGrath, D. V. “Photoresponsive Dendrimers” Chem. Commun. 1997, 857-858.

76 Tesis Doctoral Susana Barriga Falcón
N N
O
O
O
O
O
O
O
O
O
O
O
O
O
O
N N
O
O
O
O
O
O
O
O
O
O
O
O
O
O
hν
hν
156a
156b
Figura 1.31: Dendrímero con núcleo de azobenceno capaz de sufrir isomerización fotoquímica
Uno de los últimos ejemplos de la utilización de azobencenos para construir
dendrímeros ha sido dado por Vögtle. El dendrímero 157 contiene 32 unidades de
azobenceno en la periferia y se ha investigado su comportamiento como interruptor
fotoquímico.98
98
Archut, A.; Vögtle, F.; De Cola, L.; Azzellini, G. A.; Balzani, V.; Ramanujam, P. S.; Berg, R. H.
“Azobenzene-Functionalized Cascade Molecules: Photoswitchable Supramolecular Systems”. Chem.
Eur. J. 1998, 4, 699-706.

Introducción 77
NN
NH
O
NN
NH
O
NN
N
HN
O
NN
HN
O
NN
N
NHN
ON
N HN
ON
N
N
HN
O
N
N
HNO
N
N
N
N
NHO
N
N
NHO
N
N
N
NHO
N
N
NHO
N
N
N
N
NH
O
NN
NH
O
NN
N
NH
O
NN
NH
O
NN
N
N
N
N
N NN
HN
O
N N
HN
O
N N
N
NH
O
NN
NH
O
NN
N
NNH
O NNNH
O N
N
N
NH
O
N
N
NHO
N
N
N
N
N
HNO
N
N
HN O
N
N
N
HNO
N
N
HNO
N
N
N
N
HN
O
NN
HN
O
NN
N
HN
O
NN
HN
O
N N
157
Además de las aplicaciones mencionadas anteriormente, estos dendrímeros
funcionalizados en la periferia han sido ensayados como materiales para el almacenaje
de datos holográficos.
7.4. Antenas moleculares dendríticas
En los dendrímeros, se puede introducir prácticamente cualquier grupo funcional
orgánico. En especial se busca introducir grupos foto- y electroquímicamente activos.
La introducción de grupos fotoactivos tiene como objetivo conseguir un efecto antena,
que consiste en que todas la unidades fotoactivas situadas en la periferia transfieran la
energía absorbida al núcleo del dendrímero, a través de enlaces covalentes.

78 Tesis Doctoral Susana Barriga Falcón
Xu y Moore han diseñado una antena molecular que consiste en un luminóforo
electroluminiscente sustituido con residuos dendríticos capaces de conducir electrones
hacia el punto focal de la molécula (Figura 1.32).99
Transferencia
de
energía
Absorción
Luz
Figura 1.32: Representación esquemática de una antena molecular
Se ha investigado también la transferencia de electrones fotoinducida en dendrímeros
que contienen fluoróforos (Figura 1.33).100
Estos compuestos pueden ser usados para
diseñar diodos electroluminiscentes.101
99
Xu, Z. F.; Moore, J. S. “Stiff Dendritic Macromolecules. 4. Design and Synthesis of a Convergent
and Directional Molecular Antenna”. Acta Polymer. 1994, 45, 83-87. 100
Devadoss, C.; Bharathi, P.; Moore, J. S. “Anomalous Shift in the Fluorescence Spectra of a High-
Generation Dendrimer in Nonpolar Solvents”. Angew. Chem. Int. Ed. 1997, 36, 1633-1635. 101
Wang, P. W.; Liu, Y. J.; Devadoos, C.; Bharathi, P.; Moore, J. S. “Electroluminescent Diodes from a
Single-Component Emitting Layer of Dendritic Macromolecules”. Adv. Mater. 1996, 8, 237.

Introducción 79
158
Figura 1.33: Ejemplo de antena molecular real
7.5. Dendrímeros que contienen TTF
En la actualidad existe un gran interés por la síntesis de dendrímeros que contengan
TTF, para incorporar las propiedades de este dador de electrones a estructuras
supramoleculares.102
Recientemente se ha diseñado un dendrímero que contiene 21
unidades de TTF, que están repartidas en la periferia de la molécula y en las ramas del
dendrímero.103
Como resultado de las múltiples unidades redox activas presentes en la
molécula, ésta se puede utilizar, por ejemplo, como catalizador redox.
102
Bryce, M. R.; Devonport, W.; Moore, A. J. “Dendritic Macromolecules Incorporating
Tetrathiafulvalene Units”. Angew. Chem. Int. Ed. 1994, 33, 1761-1763. 103
Christensen, C. A.; Goldenberg, L. M.; Bryce, M. R.; Becher, J. “Synthesis and Electrochemistry of a
Tetrathiafulvalene (TTF)(21)-glycol Dendrimer: Intradendrimer Aggregation of TTF Cation
Radicals”. Chem. Commun. 1998, 509-510.

80 Tesis Doctoral Susana Barriga Falcón
Me
MeMe
S
SS
SMe
SS
S
SMeS
S
S
S
S
S
S
MeS
S
S
S
O
O
SO
O
S
O
O
SO
O
S
SS
SS
S
S
S
S
SMe
MeS
SS
SS
MeS
SMe
SMe
SMe
S
S
O
O
S
MeS
O
O
S
S
SS
SS
S
S
S
S
MeS
SMe
SS
SS
SMe
MeS
MeS
MeS
S
S
O
O
S
SMe
O
O
S
S
S
S
S
SS
SS
SMe
MeS
S
S
S
S
MeS
SMe
SMe
SMe
S
S
O
O
S
MeS
O
O
S
SS
S
S
S
SS
SS
MeS
SMe
S
S
S
S
SMe
MeS
MeS
MeS
S
S
O
O
S
SMe
O
O
S
S
S
S
S
S
S
S
S
S
SMe
SMe
S
S
S
S
SMe
MeS
SMe
MeS
S
S
O
O
S
MeS
O
O
S
S
O O
S
O
O
S
S
S
S
S
S
S
S
S
SMe
SMe
S
S
S
S
SMe
MeS
SMe
MeS
S
S
O
O
S
SMe
O
O
S
S
159
8. Nuevos materiales heterocíclicos con azufre y nitrógeno
En los últimos años, el interés por desarrollar nuevos materiales orgánicos se ha centrado
en gran medida en la síntesis de heterociclos polinucleares de azufre y nitrógeno. Estos

Introducción 81
compuestos poseen unas características electrónicas especiales que los hacen idóneos para
su utilización en aplicaciones ópticas y electrónicas. Buenos ejemplos de ello lo
constituyen los interruptores moleculares104
y los complejos de transferencia de carga
superconductores105
basados en tetratiafulvalenos (TTFs), los cristales líquidos con
estructura tiazólica o ditiazólica106
y los tiofenos con propiedades ópticas no lineales.107
Los materiales orgánicos, y en concreto los compuestos heterocíclicos mencionados,
poseen grandes ventajas con respecto a los materiales utilizados tradicionalmente en la
industria electrónica y de computación. Así, es posible diseñar y sintetizar materiales
orgánicos a medida, que reúnan las propiedades físico-químicas deseadas.
Una de las mayores limitaciones de la investigación en el campo de la química
heterocíclica es la escasez de rutas sintéticas que conduzcan a sistemas de anillos
complejos o poco habituales. Los métodos clásicos de síntesis de compuestos
heterocíclicos se han utilizado ampliamente, pero se buscan incesantemente métodos que
conduzcan hacia nuevos derivados, que sean altamente selectivos y que puedan llevarse a
cabo bajo condiciones de reacción muy suaves.
Se han explorado tantos sistemas heterocíclicos que no es fácil encontrar estructuras
totalmente nuevas a partir de las cuales se puedan diseñar nuevos materiales útiles. Sin
embargo, existe una familia de estructuras que están en la línea intermedia entre la
104
(a) Jørgensen, T.; Hansen, T. K.; Becher, J. “Tetrathiafulvalenes as building-blocks in
supramolecular chemistry”. Chem. Soc. Rev. 1994, 23, 41-51. (b) Jager, W. F.; de Jong, J. C.; de
Lange, B.; Huck, N. P. M.; Meetsma, A.; Feringa, B. L. “A high stereoselective optical switching
process based on donor-acceptor substituted disymmetric alkenes”. Angew. Chem. Int. Ed. 1995, 34,
348-350. (c) Tsivgoulis, G. M.; Lehn, J. M. “Photonic molecular devices: reversibly photoswitchable
fluorophoros for nondestructive readout for optical memory”. Angew. Chem. Int. Ed. 1995, 34,
1119-1122. 105
(a) Williams, J. M.; Schultz, A. J.; Geiser, U.; Carlson, K. D.; Kini, A. M.; Wang, H. H.; Kwok, W.
K.; Whangbo, M. H.; Schirber, J. E. “Organic Superconductors – New Benchmarks”. Science, 1991,
252, 1501 -1508. (b) Misaki, Y.; Higuchi, N.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka,
S. “ (DTEDT)[Au(CN)2]0.4 - An organic superconductor based on the novel pi-electron framework of
vinylogous bis-fused tetrathiafulvalene”. Angew. Chem, Int. Ed. 1995, 34, 1222-1225. 106
(a) Day, P.; Bradley, D. C.; Bloor, D. Molecular Chemistry for Electronics, The Royal Society:
London 1990. (b) Gray, G. W. “Low-molar-mass thermotropic liquid-crystals”. Phil. Trans. R. Soc. London Series A-Mathematical Physical and Engineering Sciences. 1990, 330, 73-94.
107 Jen, A. K. Y.; Rao, V. P.; Wong, K. Y.; Drost, K. J. “Functionalized thiophenes - 2nd - order
nonlinear optical-materials”. J. Chem. Soc. Chem. Commun. 1993, 90-92.

82 Tesis Doctoral Susana Barriga Falcón
Química Orgánica y la Inorgánica y que se caracterizan por tener un gran porcentaje de
heteroátomos respecto del carbono. Los compuestos heterocíclicos con gran proporción de
heteroátomos son raros. Normalmente son difícilmente accesibles y, cuando se forman, en
muchos casos son inestables. Una vía de acceso a estructuras heterocíclicas de este tipo
parte de compuestos inorgánicos estables y derivados carbonados.108
Esta idea ha sido
especialmente fructífera en la producción de nuevos sistemas heterocíclicos con azufre y
nitrógeno, los heterociclotiazenos.109
Los sistemas heterocíclicos obtenidos con la filosofía anterior han mostrado tener una
química enormemente rica e inusual, que ha dado lugar a nuevas reacciones y nuevos tipos
de compuestos desconocidos hasta el momento. Las propiedades de algunos heterociclos
con azufre han demostrado que la unión covalente no es necesaria para generar
conductividad, sino que basta la interacción entre moléculas donadoras y aceptoras de
electrones para formar un complejo de transferencia de carga capaz de conducir la
corriente eléctrica.
8.1. Síntesis de heterociclos polinucleares con azufre y nitrógeno
En esta línea, en nuestro grupo de investigación se ha conseguido sintetizar una gran
variedad de heterociclos, que poseen un elevado número de átomos de azufre y que pueden
ser considerados como bloques de construcción para el diseño y la síntesis de nuevos
materiales. La investigación se ha centrado en la utilización de reactivos muy simples de
azufre, principalmente dicloruro de diazufre o esporádicamente dicloruro de azufre, sobre
compuestos nitrogenados, básicamente oximas, nitrilos y aminas.
108
Torroba, T. “Poly-Sulfur-Nitrogen Heterocycles via Sulfur Chlorides and Nitrogen Reagents”.
Journal für Praktische Chemie-Chemikerzeitung. 1999, 341, 99-113. 109
Los heterociclotiazenos, su filosofía de síntesis y sus aportaciones científicas aparecen revisadas en:
(a) Dunn, P. J.; Rees, C. W. “Novel Polysulfur-Polynitrogen Chemistry”. Reviews on Heteroatom
Chemistry. Volume 1, S. Oae, Editor, MYU, Tokio, 1988, 204-217. (b) Morris, J. L.; Rees, C. W.
“Organic Poly(sulfur-nitrogen) Chemistry,” Chem. Soc. Rev. 1986, 15, 1-15. (c) Morris, J. L.; Rees,
C. W. “Electron rich 10π and 14π heteroaromatic systems”. Pure & Appl. Chem. 1986, 58, 197-210. (d) Rees, C. W. “Polysulfur-nitrogen Heterocyclic Chemistry”. J. Heterocyclic Chem. 1992, 29, 639-651. (e) Duan, X-G.; Duan, X-L.; Rees C. W.; Yue T-Y. “Organic Chemistry of Trithiazyl
Trichloride”. J. Heterocyclic Chem. 1996, 33, 1419-1427.

Introducción 83
En nuestro grupo de investigación se pudo comprobar que la N-etildiisopropilamina, o
base de Hünig, considerada habitualmente como una base inerte, reacciona en
determinadas condiciones con dicloruro de diazufre para proporcionar distintos
heterociclos, mediante una reacción que implica múltiples etapas secuenciales en cascada
(Esquema 1.14).110
N
CHCl3S
N
S
SS
S
S SS2Cl2, DABCO
160 161
Esquema 1.14
Un estudio sistemático y el aislamiento de algunos intermedios de reacción permitió
formular un mecanismo constituido por quince etapas de reacción.111
En el mecanismo
propuesto, la reacción comienza con la oxidación de uno de los grupos isopropilo de la
base de Hünig por el dicloruro de diazufre, o más probablemente por su complejo con
DABCO, que es mucho más activo, formando un ion imonio 162.
N
HCl S
S Cl
DABCO
N
Cl
S2 S8
N
N
H Cl
160 162 163
Esquema 1.15
Esta sal de imonio 162 puede convertirse posteriormente en una enamina reactiva 164, que
es capturada por una nueva molécula de dicloruro de diazufre. La desprotonación del
nuevo ion imonio formado, 165, conduce a la enamina 166, que puede a su vez ciclar para
formar el primer anillo de ditiol 168.
110
Marcos, C. F.; Polo, C.; Rakitin, O. A.; Rees, C. W.; Torroba, T. “From Hünig’s Base to
Bis[1,2]dithiolo[1,4]thiazines in One Pot”: The Fast Route to Highly Sulfurated Heterocycles”. Angew. Chem. Int. Ed. 1997, 36, 281-283.
111 Rees, C. W.; White, A. J. P.; Williams, D. J.; Rakitin, O. A.; Marcos, C. F.; Polo, C.; Torroba, T.
“Selective synthesis of bis[1,2]dithiolo[1,4]thiazines and bis[1,2]dithiolopyrroles from Hünig’s
base”. J. Org. Chem. 1998, 63, 2189-2196.

84 Tesis Doctoral Susana Barriga Falcón
N
Cl H
H
H
DABCO
NN
S
SClH
HH
DABCON
S
SCl
N
S
S
HH
DABCO
N
S
S
H
S S
Cl
Cl
Cl
- HCl
- HCl - HCl
Cl
162 164 165
166 167 168
Esquema 1.16
La cloración y deshidrohalogenación del anillo de ditiol conduce a la sal estable 172, tal
como se propone en el esquema 1.17.
N
S
S
H
Cl S
S Cl
N
S
S
HCl
N
S
S
Cl
Cl S
S Cl
Cl
- HCl
168 169 170
DABCO
N
S
S
ClCl
HH
- HClN
S
S
Cl
H
Cl
Cl
171 172
DABCO
Esquema 1.17
La repetición de la reacción sobre el otro grupo isopropilo transforma la sal 172 en la
disal 173, que es el intermedio crucial de la reacción (Esquema 1.18).
N
S
S
Cl
H Cl
N
S
S
Cl
Cl S
S
ClS2Cl2
172 173
DABCO Cl
Esquema 1.18

Introducción 85
La disal 173 es muy sensible a las condiciones de reacción. En presencia de S2Cl2 y
DABCO en proporción equimolecular, se produce una nueva ciclación, seguida por la
extrusión de azufre, que da lugar a una nueva disal 174; sin embargo, cuando el DABCO
se encuentra en menor proporción que el dicloruro de diazufre, se favorece la cloración,
obteniéndose la disal clorada 175.
N
S
S
Cl
S
S
Cl
175
Cl Cl
N
S
S
Cl
S
S
Cl
173
Cl Cl
Cl Cl
S2Cl2
DABCO
S2Cl2
N
S
S
Cl
S
S
Cl
S S
Cl Cl
-[S]N
S
S
Cl
S
S
Cl
S Cl Cl
174
Esquema 1.19
Estas disales reactivas 174-175 (incluso 172) reaccionan con nucleófilos, que pueden estar
ya presentes en el medio de reacción, o pueden ser añadidos durante el último período de la
reacción. Cada una de estas reacciones multicomponente tiene lugar con gran eficacia,
permitiendo la preparación selectiva de cada producto final.
A partir de la disal 174 pueden obtenerse varios derivados de bis[1,2]ditiolo[1,4]tiazina,
tal y como se indica en el esquema 1.20.110,111

86 Tesis Doctoral Susana Barriga Falcón
N
S
S
Cl
S
S
Cl
S
174
Cl
CO2H
HCO2H
Cl
S8, S-2
N
S
S
S
S
S
S
S
161
N
S
S
S
S
S
O
S
176
N
S
S
O
S
S
O
S
177
Esquema 1.20
En esta transformación en una sola etapa experimental, los catorce enlaces carbono-
hidrógeno de los dos grupos isopropilo de la base de Hünig han sido reemplazados por
diez enlaces carbono-azufre y por dos dobles enlaces carbono-carbono, permaneciendo el
grupo etilo inalterado.
Todos los derivados de [1,2]ditiolo[1,4]tiazina adoptan una estructura plegada en estado
sólido, con el anillo de 1,4-tiazina que tiene una estructura tipo barca con el grupo
N-alquilo sobre el átomo de azufre de la tiazina (Figura 1.34).
Figura 1.34: Estructura obtenida por difracción de rayos X de la 4-etilbis[1,2]ditiolo
[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona (161)

Introducción 87
Estas reacciones son generales para diisopropilaminas terciarias. Las diisopropilaminas
con diversos sustituyentes sobre el nitrógeno conducen a ditiolotiazinas con estructura
de carpeta semi-plegada, similares a las obtenidas con la base de Hünig.
N
R
DABCO
S
N
S
SS
S
RS S
R = Me, Ph, CH2Cl, CH2SPh, CH2NPhth
S2Cl2
-NPhth =
N OO
Esquema 1.21
Hay una excepción a este hábito molecular. Así, el sistema de bis[1,2]ditiolo[1,4]tiazina
que posee como sustituyente un hidrógeno en el nitrógeno (180) es un compuesto plano
en estado sólido, como se puede ver en la Figura 1.35. Este compuesto cabeza de serie,
180, se obtiene por la desbencilación del N-bencilderivado de la
bis[1,2]ditiolo[1,4]tiazina (179).112
Este compuesto ha mostrado una actividad
inhibidora completa frente a diversas líneas de células tumorales a una concentración
baja (10-4
M) en pruebas preliminares in vitro.113
N
2) HCO2H
Ph
S
N
S
SS
S
O OPh
H2SO4
S
N
S
SS
S
O OH1) S2Cl2, DABCO
178 180179
Esquema 1.22
112
Marcos, C. F.; Rakitin, O. A.; Rees, C. W.; Torroba, T.; White, A. J. P.; Williams, D. J.
“Bis[1,2]dithiolo[3,4-b][4´,3´-e][1,4]thiazine-3,5-dione, a planar 1,4-thiazine”, Chem. Commun.
1999, 29-30. 113
Datos obtenidos de los ensayos realizados por el NCI de USA. Sin publicar.

88 Tesis Doctoral Susana Barriga Falcón
Figura 1.35: Estructura de difracción de rayos X de la bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-diona
(180)
Las bis[1,2]ditiolo[1,4]tiazinas son excelentes sustratos para reacciones de
desulfuración, ya que el átomo de azufre del anillo de 1,4-tiazina se extruye fácilmente
si el compuesto se calienta en disolventes de alto punto de ebullición, por ejemplo
clorobenceno, conduciendo a bis[1,2]ditiolopirroles (Esquema 1.23).114
N
R
S
S
X
S
S
Y
S
N
SSS
S
R
XY
X = O, SY = O, S
PhCl
∆
R = Me, Ph, CH2Cl, CH2SPh, CH2NPhth
Esquema 1.23
Este proceso está favorecido porque transcurre a través de un tiirano intermedio, muy
estabilizado por la cesión electrónica del nitrógeno y el efecto atractor de los grupos
carbonilo y/o tiocarbonilo, que genera una gran deslocalización electrónica del sistema
(Esquema 1.24).111
Asimismo, la formación de un compuesto plano y aromático resulta
favorable termodinámicamente.
114
Marcos, C. F.; Polo, C.; Rakitin, O. A.; Rees, C. W.; Torroba, T. “One-pot Synthesis and Chemistry
of Bis[1,2]dithiolopyrroles”. Chem. Commun. 1997, 879-880.

Introducción 89
S
N
S
SS
S
YX
S
N
S
SS
S
YXN
SSS
S
YX∆
181; X, Y = S
182; X = S, Y = O
183; X, Y = O
161; X, Y = S
176; X = S, Y = O
177; X, Y = O
Esquema 1.24
Estos nuevos derivados pirrólicos también pueden obtenerse mediante reacciones en una
sola etapa, si se hacen reaccionar las correspondientes diisopropilaminas en disolventes
de alto punto de ebullición, como el clorobenceno (Esquema 1.25).114
N
DABCO
HCO2H
N
SSS
S
OO
N
SSS
S
SODABCO
1/2 HCO2H
DABCO
N
SSS
S
SS
PhCl, ∆
PhCl, ∆
PhCl, ∆
S2Cl2
S2Cl2
S2Cl2182
181
183
160
Esquema 1.25
Las reacciones de la N-(2-cloroetil)diisopropilamina (184) constituyen un caso especial.
En principio, la reacción de esta amina con dicloruro de diazufre proporciona los derivados
tricíclicos de bis[1,2]ditiolo[1,4]tiazina esperados 185 y 186.115,116
115
Marcos, C. F.; Rakitin, O. A.; Rees, C. W.; Souvorova, L. I.; Torroba, T.; White, A. J. P.; Williams,
D. J. “Tertiary amine-S2Cl2 chemistry: interception of reaction intermediates”. Chem. Commun. 1998, 453-454.
116 Rees, C. W.; White, A. J. P.; Williams, D. J.; Rakitin, O. A.; Marcos, C. F.; Torroba, T. “Synthesis of
Bis[1,2]Dithiolo[1,4]thiazine from Terteary Diisopropylamines”. J. Org. Chem. 1999, 64,
5010-5016.

90 Tesis Doctoral Susana Barriga Falcón
N
DABCO
Cl
N
S
S
S
S
S
S
S
Cl
S2Cl2
184 186
DABCO
N
S
S
O
S
S
O
S
Cl
S2Cl2
185
HCO2H
Esquema 1.26
El átomo de cloro de la ditiona 185 resultó ser inerte frente a nucleófilos en reacciones
de sustitución. Además, a diferencia de lo que ocurría con su análogo N-etilo, este
compuesto extruye azufre de forma muy lenta para dar lugar al correspondiente
derivado pirrólico.
N
S
S
O
S
S
O
S
Cl
PhCl
N
SSS
S
Cl
OO
185 187
∆
Esquema 1.27
La difracción de rayos X de 185 muestra una estructura similar a la observada para el
análogo N-etilo, en la cual el sistema tricíclico presenta forma de carpeta, y el nitrógeno
tiene una geometría piramidal, pero la cadena cloroetilo está ligeramente desviada de la
posición simétrica por encima del anillo de la tiazina (Figura 1.36). Se observa, además,
una interacción débil entre el átomo de cloro y el azufre del anillo tiazínico.
Figura 1.36: Estructura de difracción de rayos X de la 4-(2-cloroetil)-bis[1,2]ditiolo
[3,4-b:4’,3’-e][1,4]tiazina-3,5-diona (185).

Introducción 91
Alternativamente, si en la última fase de la reacción de 2-cloroetildiisopropilamina con
dicloruro de diazufre se añade pentasulfuro de fósforo, el curso de la reacción cambia
totalmente. En este caso, el átomo de cloro es reemplazado por un átomo azufre en la sal
intermedia 188, para formar el tiolato 189, el cual evoluciona dando lugar a un nuevo
sistema heterocíclico de [1,2]ditiolo[1,4]tiazina 190.
N
Cl
S
N
S
S
S S
SS
H
2) P4S10
1) S2Cl2
190184
N
S
S
Cl
S
S
Cl
S
189
N
S
S
Cl
S
S
Cl
Cl
188
Esquema 1.28
La cristalografía de rayos X muestra que el anillo de 1,4-tiazina de 190 se encuentra en
una conformación en semi-silla, el nitrógeno presenta geometría piramidal y los anillos
de ditiol están orientados casi ortogonalmente para minimizar las interacciones estéricas
entre ellos.
Figura 1.37: Estructura de difracción de rayos X de la 5,6-dihidro-4-(3-tiono[1,2]ditiol-4-il)-[1,2]ditiolo-
[3,4-b][1,4]tiazina-3-tiona (190)
Además del nitrógeno, también el azufre puede activar grupos isopropilo. En este caso,
sólo uno de los dos grupos isopropilo es activado por el azufre en la reacción frente a
dicloruro de diazufre.117
De este modo, la reacción de sulfuro de diisopropilo con
dicloruro de diazufre en clorobenceno, en presencia de DABCO, proporciona
117
Rees, C. W.; Rakitin, O. A.; Marcos, C. F.; Torroba, T. “Synthesis of Sulfur Rich 1,2- and
1,3-Dithiolodisulfides and Thiodesaurines, from Diisopropyl Sulfide”. J. Org. Chem. 1999, 64,
4376-4380.

92 Tesis Doctoral Susana Barriga Falcón
4-isopropiltio-5-isopropilditio-1,2-ditiolo-3-tiona (192) y un bisditioldisulfuro dimérico
193.
S
S
SS
S
SS
SS
S
S
S
S
S
S
S
S
PhCl
191 192 193
+
DABCO
S2Cl2
Esquema 1.29
Cuando la reacción se realizó añadiendo disulfuro de diisopropilo en la última fase de la
reacción, se obtuvo selectivamente el compuesto monocíclico 192, ya que de esta forma
el equilibrio de formación del dímero 193 resulta inhibido.
S
S
SSS
S
S
191 192
S
S
PhCl, ∆
1) S2Cl2, DABCO
2)
Esquema 1.30
El compuesto monomérico 192 y el dimérico 193 pueden interconvertirse bajo
irradiación ultravioleta. Además, si se trata el compuesto 192 con dos equivalentes de
trifenilfosfina se obtiene la tiodesaurina 194, como mezcla de isómeros geométricos.
S
SS
S
S
S
S
S
S
S
S
S
S
S
Ph3P
CH2Cl2
192 194
Esquema 1.31

Introducción 93
Sin embargo, el oxígeno no es capaz de activar suficientemente los grupos isopropilo en
este tipo de reacciones, y así el diisopropil éter no reacciona con dicloruro de
diazufre.117
OS2Cl2
DABCO
No hay reacción
195
Esquema 1.32
Todas las estructuras heterocíclicas anteriores se caracterizan por poseer una gran
deslocalización electrónica, debido a la presencia de un gran número de átomos de
azufre en sus estructuras. La mayoría son compuestos sólidos muy coloreados y de gran
estabilidad, pero poseen una reactividad muy rica, que hace posible su transformación
en estructuras más complejas.
Todo ello convierte a estos poliheterociclos sulfurados en buenos puntos de partida para
la preparación de diversos tipos de nuevos materiales orgánicos.


2. OBJETIVOS


Objetivos 97
El objetivo general de esta Tesis Doctoral es el desarrollo de nuevas rutas sintéticas para
la preparación de poliheterociclos que contienen azufre y nitrógeno, con un doble
propósito: por una parte, el estudio de la estructura y propiedades de sistemas
heterocíclicos hasta el momento desconocidos, y por otra parte, la preparación de
compuestos potencialmente útiles como nuevos materiales orgánicos.
Para lograr estos propósitos se tratará de desarrollar el potencial sintético de una nueva
reacción descubierta recientemente en el grupo de trabajo en que se realiza esta Tesis
Doctoral, y que convierte diisopropilaminas terciarias en sistemas polinucleares del tipo
[1,2]ditiolo[1,4]tiazinas y [1,2]ditiolopirroles.
Los objetivos concretos del presente trabajo pueden resumirse en los siguientes puntos:
1. Aplicación de la metodología desarrollada anteriormente en nuestro grupo de
investigación para sintetizar heterociclos polinucleares con núcleo tiazínico o pirrólico
con distintos sustituyentes a partir de aminas terciarias.
2. Estudio de la reactividad de ditioltionas con núcleos tiazínicos y pirrólicos como
dipolarófilos frente a reactivos 1,3-dipolares como azidas y nitriliminas.
3. Estudio de la reactividad de las bisditioltionas sintetizadas con la metodología propia
del grupo de investigación frente a la adición de reactivos nitrogenados de tipo
cloraminas B y T.
4. Estudio de la reactividad de las ditioltionas sintetizadas con la metodología propia del
grupo de investigación como reactivos 1,3-dipolares frente a dipolarófilos. Estos
dipolarófilos serán, principalmente, acetilenos activados mediante grupos atractores de
electrones, alquenos y bencinos.
5. Aplicación de las nuevas rutas sintéticas desarrolladas para la preparación de nuevos
materiales con propiedades magnéticas o electrónicas específicas.
6. Estudio de las propiedades electroquímicas de los nuevos sistemas heterocíclicos
obtenidos mediante reacciones de cicloadición.
7. Estudio conformacional, mediante modelización molecular, de las estructuras
previsiblemente obtenibles y su comparación con los datos teóricos, experimentales y

98 Tesis Doctoral Susana Barriga Falcón
espectroscópicos que puedan obtenerse, con el fin de obtener conclusiones respecto a
la posible utilidad de dichos compuestos con vistas a su aplicación como nuevos
materiales.
8. El objetivo a medio plazo de la Tesis Doctoral es el estudio de las aplicaciones
potenciales de los nuevos compuestos sintetizados, basadas bien en sus propiedades
electrónicas, como referencia para la preparación de materiales de óptica no lineal y
conductores electrónicos, bien en su actividad biológica, con vistas a su posible uso
farmacológico o agroquímico.

3. EXPOSICIÓN Y DISCUSIÓN DE RESULTADOS


Exposición y discusión de resultados 101
En los últimos años, la investigación de nuestro grupo se ha centrado en la síntesis de
nuevos sistemas heterocíclicos polinucleares de azufre y nitrógeno, con vistas al
desarrollo de nuevos materiales orgánicos.
La metodología propia del grupo para obtener dichos heterociclos se basa en la
utilización de reactivos inorgánicos de azufre muy simples, principalmente dicloruro de
diazufre, o esporádicamente dicloruro de azufre, sobre compuestos nitrogenados,
básicamente oximas, nitrilos y aminas. Esto permite obtener de forma muy sencilla
diversos sistemas heterocíclicos y, entre ellos, 1,2-ditiol-3-tionas fusionadas a
1,4-tiazinas y pirroles.
La investigación de la química de estos compuestos es importante, puesto que permite
diseñar estrategias encaminadas a modificar de forma controlada las estructuras
moleculares de partida. En concreto, las reacciones de cicloadición, por ejemplo,
ofrecen un método adecuado para introducir modificaciones estructurales y funcionales
en las 1,2-ditiol-3-tionas.
Con esta idea, en este trabajo de Tesis Doctoral se ha realizado el estudio de la reactividad
frente a reacciones de cicloadición de las 1,2-ditiol-3-tionas poliheterocíclicas, obtenidas

102 Tesis Doctoral Susana Barriga Falcón
con anterioridad en nuestro grupo de investigación. Este estudio se describirá
detalladamente a continuación, pero se ha creído interesante explicar, en primer lugar y
muy brevemente, algunos de los antecedentes bibliográficos más importantes que existen
sobre reacciones de cicloadición con ditioltionas.
A. ANTEDECENTES BIBLIOGRÁFICOS: REACCIONES DE
CICLOADICIÓN DE 1,2-DITIOL-3-TIONAS
La reactividad de las ditioltionas ha sido estudiada con bastante intensidad, sobre todo a
partir de los años 60.
Las ditioltionas pueden ser atacadas por nucleófilos, reaccionar con carbenos y nitrenos,
sufrir reacciones de oxidación y de reducción. Pero las reacciones con más aplicaciones
sintéticas son, sin duda, las reacciones de cicloadición, que pueden tener lugar en
condiciones térmicas o fotoquímicas.
Las reacciones de cicloadición de 1,2-ditiol-3-tionas frente a reactivos 1,3-dipolares y
frente a filodienos se conocen desde hace mucho tiempo, apareciendo los primeros
estudios publicados en los años 1965 y 1966. La mayor parte de la bibliografía sobre
este tema hasta el año 1980 está recogida en una revisión sobre la química de 1,2-ditiol-
3-tionas y 1,2-ditiol-3-onas.118
El grupo ditioltiona puede participar en dos tipos diferentes de reacciones de
cicloadición: reacciones de cicloadición [3+2] con reactivos 1,3-dipolares involucrando
únicamente al grupo tiona, y cicloadiciones con reactivos dipolarófilos involucrando al
grupo tiona y al anillo de ditiol.
1. Reacciones de cicloadición 1,3-dipolares del grupo tiona
Las reacciones de cicloadición 1,3-dipolares son reacciones de [4π+2π] electrones, que
transcurren a través de un estado de transición electrónico “aromático” 6π en el que el
118 Pedersen C. Th. “1,2-Dithiole-3-thiones and 1,2-dithiol-3-ones”. Adv. Heterocycl. Chem. 1982, 31,
63-112.

Exposición y discusión de resultados 103
componente 4π contiene al menos un heteroátomo. En este caso el componente 4π
recibe el nombre de 1,3-dipolo y el componente 2π es llamado dipolarófilo.119
El grupo tiona presente en las 1,2-ditiol-3-tionas (dipolarófilo) puede reaccionar con
reactivos 1,3-dipolares dando lugar a un cicloaducto espiránico, que normalmente sufre
una ciclorreversión, u otro tipo de transformaciones, para conducir a sistemas estables.
S
S
S
XY
Z
S
S
Z
YX
S
+
S
S
Z
YX
SSISTEMA
ESTABLE
196 197 198
Esquema 3.1
Existe un considerable número de reactivos 1,3-dipolares que pueden dar lugar a este
tipo de reacciones. Huisgen ha clasificado los reactivos 1,3-dipolares que contienen
centros con carbono, nitrógeno y oxígeno en dos tipos: reactivos alenilpropargílicos y
reactivos alílicos.120
X N Y
X = C, N Y = C, N, O
C N
Y
O X
O
Y = C, N, O
Tipo alenilpropargílico Tipo alilo
C O
Y
Y = C, N, O
N X
Y
X = N, O Y = N, O X = N, O
Figura 3.1 Representación genérica de reactivos 1,3-dipolares alenilpropargílicos y alílicos.
En estos antecedentes bibliográficos se hablará únicamente de reactivos 1,3-dipolares de
tipo alenilpropargílico, y concretamente sólo de azidas y nitriliminas, que han sido
objeto de estudio en este trabajo, y además, se hablará de reacciones con óxidos de
nitrilo, ya que en nuestro grupo se había estudiado previamente la transformación del
grupo tiona en cetona mediante reacciones de cicloadición y ciclorreversión de
119 Carruthers, W. en “Cycloaddition reactions in organic synthesis”. Tetrahedron Organic Chemistry
Series. Ed. Baldwin, J. E.; Magnus, P.D. Pergamon Press, London, 1990, Vol. 8. 120 Huisgen, R. en “1,3-Dipolar Cycloaddition Chemistry”. Ed. Padwa, A. Wiley-Interscience. 1984,
Vol. 1, pag. 1.

104 Tesis Doctoral Susana Barriga Falcón
cloroximidoacetato de etilo con algunos de los sistemas heterocíclicos obtenidos con
anterioridad en nuestro grupo.
1.1. Estudios previos de la cicloadición de ditioltionas. Reacción con óxidos de nitrilo
Los óxidos de nitrilo son reactivos 1,3-dipolares que reaccionan con dipolarófilos
etilénicos para formar ∆2-isoxazolinas y con dipolarófilos acetilénicos para dar lugar a
isoxazoles.121
C N OR C N OR
R'R'
O
N
R'
R
O
N
R'
R
199 199'
200 201
202 203
Esquema 3.2
Por otra parte, la gran reactividad de los óxidos de nitrilo con el doble enlace carbono-
azufre de las tiocetonas ha sido también ampliamente estudiada (Esquema 3.3).122
S
R2
R1
RCNOS
O
N
R2
R1
R
+
204 199 205
Esquema 3.3
121 (a) Grundmann, C.; Grünanger, P. “The nitrile oxides”. Springer, Berlin, 1971. (b) Christl, M.;
Huisgen, R. “1,3-Dipolar Cycloadditions. 72. Reactions of Fulminic Acid with Unsaturated Compounds”. Chem Ber. 1973, 106, 3291-3311. (c) Huisgen, R. “1,3-Dipolar Cycloadditions. 76. Concerted Nature of 1,3-Dipolar Cycloadditions and the Question of Diradical Intermediates”. J. Org.Chem. 1976, 41, 403-419. (d) Lang, S. A. Jr.; Lin, Y. en “Comprehensive Heterocyclic Chemistry”. Ed. Katritzky, A. R.; Rees, C. W. Vol. 6, Pergamon, 1984, pag. 1. (e) Padwa, A. en “1,3-Dipolar Cycloaddition Chemistry”. Ed. Padwa, A. Wiley-Interscience. 1984, Vol. 2, pag. 368.
122 Por ejemplo, Schaumann, E.; Ruhter, G. “Fragmentation of 1,3-dithiolane-derived sulfur ylides – a convenient thioaldehydes synthesis” Tetrahedron Lett. 1985, 26, 5265-5268.

Exposición y discusión de resultados 105
Los óxidos de nitrilo se preparan normalmente in situ y no se aíslan. Son generados en
presencia del dipolarófilo y forman el cicloaducto directamente. Se usan dos métodos
generales para preparar óxidos de nitrilo, la deshidrocloración de cloruros de
hidroximoílo con trietilamina123 y la deshidratación de nitrocompuestos primarios con
un isocianato de arilo (Esquema 3.4).124
N
R
Cl OH
C N OR C N OR
Et3N
RCH2NO2
Et3N
PhNCO
206 199 199' 207
Esquema 3.4
El uso de óxidos de nitrilo para convertir grupos tiocarbonilo en carbonilos fue descrito
por Huisgen en 1961,125 y posteriormente fue utilizado de forma muy puntual por otros
investigadores.126
Previamente al inicio del presente trabajo, en nuestro grupo de investigación se llevó a
cabo un estudio de las reacciones de cicloadición de ditioltionas con cloroximidoacetato
de etilo. Como comparación con otras reacciones de cicloadición 1,3-dipolares
estudiadas en la presente Tesis, se incluye a continuación un breve resumen de los
resultados obtenidos con dicho óxido de nitrilo. En nuestro grupo de investigación se
observó que el cloroximidoacetato de etilo convertía 1,2-ditiol-3-tionas en
1,2-ditiol-3-cetonas.111 De esta forma, la dicetona 177 es obtenida con un rendimiento
del 90% a partir de la bisditioltiona 161, y con un rendimiento del 95% a partir de la
monoditioltiona 176.
123 Huisgen, R. “1,3-Dipolar Cycloadditions” Angew. Chem. 1963, 75, 604-637. 124 Mukaiyama, T.; Hoshino, T. “The Reactions of Primary Nitroparaffins with Isocyanates”. J. Am.
Chem. Soc. 1960, 82, 5339-5342. 125 Huisgen, R.; Mack, W.; Anneser, E. Angew. Chem. 1961, 73, 656. 126 (a) Boberg, F.; Knoop, J. Liebigs Ann. Chem. 1967, 708, 148. (b) Kim, J. N.; Ryu, E. K. “Convenient
Synthesis of Isothiocyanates from Nitrile Oxides”. Tetrahedron Lett. 1993, 34, 8283-8284.

106 Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
SS EtO2C C N O
S
N
S
SS
S
OO
S
N
S
SS
S
SO
THF, 0 ºC
161 177 176
EtO2C C N O
THF, 0 ºC
Esquema 3.5
La reacción tiene lugar de forma prácticamente instantánea en THF a 0 ºC. Se usa
exceso del óxido de nitrilo 209, que es generado in situ al tratar cloroximidoacetato de
etilo con trietilamina.
En la cicloadición probablemente se genera inicialmente un cicloaducto con estructura
oxotiazolínica (210), que no se aísla, y sobre el cual se produce una ciclorreversión que
conduce, tras una transposición con pérdida de etoxicarbonilisotiocianato de etilo, a la
ditiolcetona 177 (Esquema 3.6).127
N
Cl CO2Et
O
H
Et3N N
S
N
S
S S
S
S O
S
N
S
S S
S
O
O
NS
CO2Et
EtO2C
O
208 209 176 210
S
N
S
S S
S
O
O
NS
CO2Et
210
S
N
S
S S
S
O O
S
N
S
S S
S
O O
+
CO2Et
N
C
S
177177 211
Esquema 3.6
Igualmente pueden sufrir esta reacción los bis[1,2]ditiolopirroles, y así la dicetona 183
puede obtenerse con un rendimiento del 70% a partir de 181 y con un 75% a partir de
182.
127 Torssell, K. B. G. en “Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis. Novel Strategies
in Synthesis”, de la serie “Organic Nitro Chemistry Series”. Ed. Feuer, H. VCH Publishers, Inc. 1988, Indiana (USA).
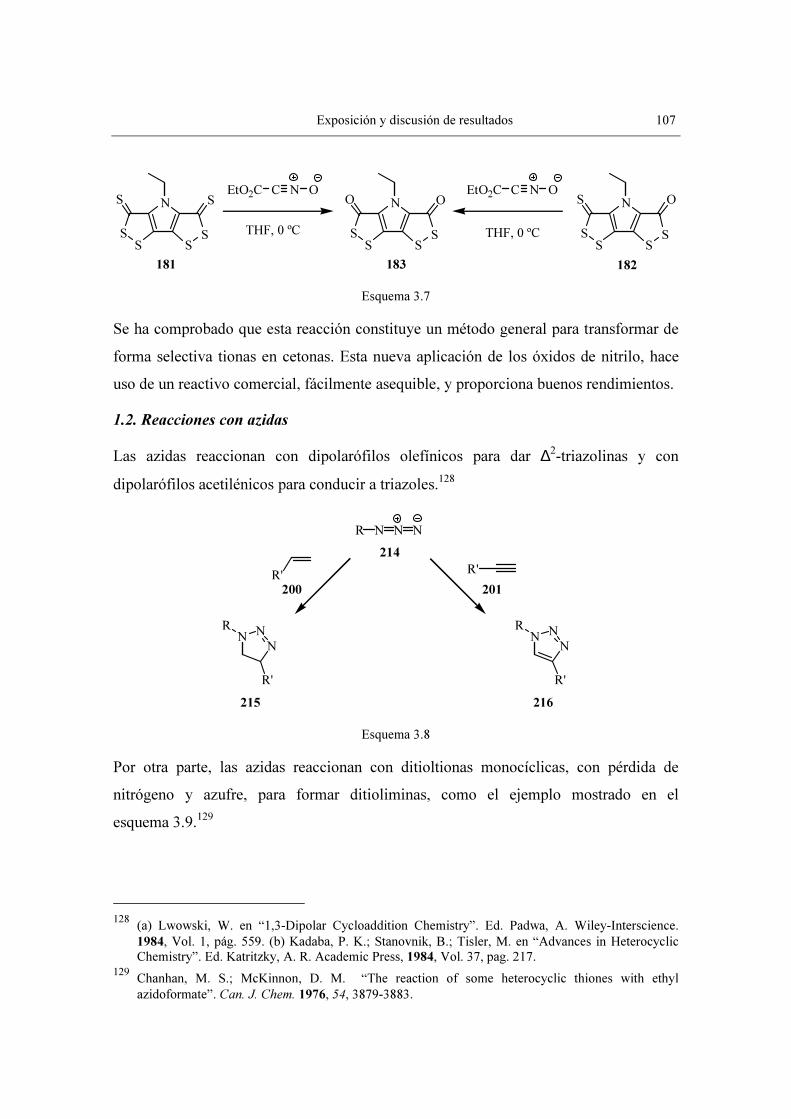
Exposición y discusión de resultados 107
EtO2C C N O EtO2C C N ON
SSS
S
S S N
SSS
S
O O N
SSS
S
S O
THF, 0 ºC
181 183 182
THF, 0 ºC
Esquema 3.7
Se ha comprobado que esta reacción constituye un método general para transformar de
forma selectiva tionas en cetonas. Esta nueva aplicación de los óxidos de nitrilo, hace
uso de un reactivo comercial, fácilmente asequible, y proporciona buenos rendimientos.
1.2. Reacciones con azidas
Las azidas reaccionan con dipolarófilos olefínicos para dar ∆2-triazolinas y con
dipolarófilos acetilénicos para conducir a triazoles.128
N N NR
R'R'
NN
N
R'
RN
N
N
R'
R
214
201200
215 216
Esquema 3.8
Por otra parte, las azidas reaccionan con ditioltionas monocíclicas, con pérdida de
nitrógeno y azufre, para formar ditioliminas, como el ejemplo mostrado en el
esquema 3.9.129
128 (a) Lwowski, W. en “1,3-Dipolar Cycloaddition Chemistry”. Ed. Padwa, A. Wiley-Interscience.
1984, Vol. 1, pág. 559. (b) Kadaba, P. K.; Stanovnik, B.; Tisler, M. en “Advances in Heterocyclic Chemistry”. Ed. Katritzky, A. R. Academic Press, 1984, Vol. 37, pag. 217.
129 Chanhan, M. S.; McKinnon, D. M. “The reaction of some heterocyclic thiones with ethyl azidoformate”. Can. J. Chem. 1976, 54, 3879-3883.

108 Tesis Doctoral Susana Barriga Falcón
S
S
S
PhS
SPh
NCO2C2H5N3CO2C2H5
217 218
Esquema 3.9
1.3. Reacciones con nitriliminas
Se conoce desde hace tiempo que las reacciones de ditioltionas con reactivos
1,3-dipolares del tipo nitrilimina proporcionan 1,3,4-tiadiazoles.130 Existen, sin
embargo, muy pocos ejemplos, y en todos ellos la asignación de la geometría del doble
enlace del producto final de la reacción es incierta. En el esquema 3.10 se muestra un
ejemplo.
S
S
S
Ph
Cl N
HN
Ph
PhS
N
N
Ph
PhPh
S+
Xilenos
217 219 220
Base
Esquema 3.10
1.4. Adición de nitrenos
Las reacciones de 1,2-ditioltionas monocíclicas con nitrenos fueron ya descritas por
McKinnon a principio de los años 80. Los nitrenos reaccionan con el grupo tioxo de las
ditioltionas dando lugar a tiaziridinas (222), que pueden, a su vez, extruir azufre para
formar 3-imino-1,2-ditioles (223).131
130
Maguet, M.; Poirier, Y.; Teste, J. “Heterocyclic sulfur compounds VI. Synthesis of derivatives of 1,3,4-thiadiazoline”. Bull. Soc. Chim. Fr. 1970, 1503-1509.
131 (a) McKinnon, D. M. “1,2-dithioles” en Comprehensive Heterocyclic Chemistry. Katrizky, A. R.; Rees, C. W., editores. Pergamon. Oxford, 1984. Vol 4, cap. 31, pag. 783-811. (b) McKinnon, D. M. en Comprehensive Heterocyclic Chemistry II. Katrizky, A. R.; Rees, C. W.; Scriven, E. F. V., Editores. Elsevier, Oxford, 1996. Vol 3, cap. 3.11.

Exposición y discusión de resultados 109
S
S
S
R2
R1
NR
S
S
R2
R1 N
S R
S
S
N
R2
R1
R
221 222 223
Esquema 3.11
2. Reacciones de cicloadición 1,3-dipolares que involucran a todo el anillo de ditiol
Este tipo de reacciones se centra fundamentalmente en las reacciones de los derivados
de 1,2-ditiol-3-tionas con alquenos o alquinos bajo condiciones fotoquímicas o
térmicas. El trabajo de un gran número de investigadores sobre las reacciones de
ditioltionas con alquinos se resume en el esquema 3.12.131 Las tionas reaccionan
térmicamente con alquinos para dar lugar a 2-tioacilmetilen-1,3-ditioles (225), que
pueden ser aislados o reaccionar con una nueva molécula de alquino para formar el
diaducto correspondiente, cuando se usan alquinos suficientemente activados.
Por otra parte, si tras la primera cicloadición se forma un tioaldehído (225, R2 = H),
éste puede, en muchas ocasiones, dimerizar para formar el correspondiente
bis-1,3-ditiol-2-ilidenbuteno (227).
A veces, también pueden obtenerse 1,6,6aλ4-tritiapentalenos (226). Esto puede ser
debido a una transposición de la tiona (225) en una reacción catalizada por azufre o por
reactivos que contienen azufre, o mediante la reacción directa de la tiona (221) con
azufre y el alquino.

110 Tesis Doctoral Susana Barriga Falcón
SS
S
R2
R1 R
3
R4
S
S
R1
R2
S
R4(R
3)
R3(R
4)
S SS
R1
R2
R3(R
4)
R4(R
3)
R3
R4
S
SR3
R4 R
1
H
S
S
S R3
R4
R1
R2
(R4)R
3R
4(R
3)
221 226
+
R2
= H
2
225
227 228
224
224
Esquema 3.12
La reacción con alquenos es similar. Las tionas pueden reaccionar térmicamente con
alquenos para formar 3-tioacilmetilen-1,3-ditiolanos (229), pero fotoquímicamente
forman 1,2-ditiolespirotietos (230). Estos compuestos de tipo espiránico también
pueden formarse con alquinos en reacciones activadas por la luz.
S
S
S SS S
229 230
A continuación se explicará con más detalle el uso de alquinos en reacciones de
cicloadición con ditioltionas, ya que gran parte del trabajo realizado en esta Tesis
Doctoral se ha centrado en ellos.
Los acetilenos activados con sustituyentes atractores de electrones son reactivos
dienófilos muy activos. Los sustituyentes activantes usados más frecuentemente son
COR, CO2R, CN y NO2, y los dienófilos que contienen uno o más de estos grupos en
conjugación con un doble o triple enlace reaccionan fácilmente con dienos. La reacción
de cicloadición transcurre mayoritariamente mediante interacciones entre el orbital
HOMO del dieno y el LUMO del dienófilo, y la presencia de grupos atractores de
electrones en el dienófilo facilita la reacción de cicloadición porque disminuye la

Exposición y discusión de resultados 111
energía del LUMO y, por tanto, disminuye la separación energética entre el LUMO del
dienófilo y el HOMO del dieno.119
La reactividad de los compuestos acetilénicos en estas reacciones disminuye, al
disminuir la capacidad atractora de electrones de los sustituyentes, en el orden:
ROCC≡CCOR > HC≡CCOR > ArC≡CCOR > HC≡CAr > ArC≡CAr
En la tabla 3.1 se recogen algunos de los grupos atractores de electrones que pueden ser
utilizados para activar triples enlaces, así como el valor de sus constantes de Hammett,
que dan una medida de su capacidad atractora.132,133
Sustituyente σσσσp Sustituyente σσσσp
H 0 CF3 0.54
CHO 0.42 CN 0.66
COPh 0.43 SO2Ph 0.68
COOMe 0.45 NO2 0.78
Tabla 3.1: Valores de las constantes de Hammett (σp) para algunos grupos atractores de electrones.132,133
Entre los acetilenos activados más comunes se encuentran el bis(trifluorometil)acetileno
(231), el dicianoacetileno (232), los acetilendiésteres (233) y los bis-acilacetilenos
(234).
CF3F3C CO2RRO2C CORROC
231 233 234
CNNC
232
En bibliografía existen numerosos ejemplos de reacciones de cicloadición de
ditioltionas con acetilenos activados. A continuación, se destacarán aquellos que hacen
132 Perrin, D. D.; Dempsey, B.; Serjeant, E. P. “Prediction for organic acids and bases”. Chapman &
Hall; London, 1981. 133 Hansch, C.; Leo, A.; Taft, R. W. “A survey of Hammett substituents constants and resonance and
field parameters”. Chem. Rev. 1991, 91, 165-195.

112 Tesis Doctoral Susana Barriga Falcón
referencia a acetilenos activados simétricamente tales como el acetilendicarboxilato de
dimetilo (235) y el dibenzoilacetileno (236), ya que estos han sido ampliamente usados.
La reacción de cicloadición 1,3-dipolar de 5-aril-1,2-ditiol-3-tionas, como 237, con
acetilendicarboxilato de dimetilo transcurre fácilmente, incluso a temperatura ambiente,
proporcionando exclusivamente los derivados de 1,3-ditiol, como por ejemplo 238, en
rendimiento alto.134
S
S
H3COC6H4
S
CH3OC6H4S
SS
R
CO2CH3
CO2CH3
CO2CH3H3CO2CR
237 238; 90%
acetona, temp. amb.
Esquema 3.13
La reacción de cicloadición de 1,2-ditiol-3-tionas de estructura más complicada, frente a
acetilendicarboxilato de dimetilo (DMAD), ha sido estudiada con bastante detalle por
varios grupos. Así, la ditioltiona polimérica 239 adiciona dos o tres moléculas de
DMAD por monómero, para dar mayoritariamente 240 y en pequeña proporción 241.135
SS
S S
S DMAD
S
S
S
S
S
CO2CH3
CO2CH3
H3CO2C
H3CO2C
S
S SS
H3CO2C
H3CO2CCO2CH3
CO2CH3
CO2CH3
H3CO2C
n
+
239 240 (69 %) 241 (12 %)
Esquema 3.14
El producto 241 proviene, muy probablemente, de la cicloadición de una tercera
molécula de DMAD sobre el compuesto 240, seguida de extrusión de azufre.135
134 Davy, H.; Decroven, J. M. “Composés organiques sulfurés. LIII. (Aryl-2-thioxo-2-ethylidine)-2
dithioles-1,3: synthèse, oxydation et transformation en tritia-1,6,6a SIV pentalenes. Oxydation des trithiapentalenes ainsi obtenus”. Bull. Soc. Chim. Fr. 1976, 115-119.
135 Doxsee, D. D.; Gallaway, C. P.; Rauchfuss, T. B.; Wilson, S. R.; Yang, X. “New carbon sulfides based on 4,5-dimercapto-1,2-dithiole-3-thione”. Inorg. Chem. 1993, 32, 5467-5471.
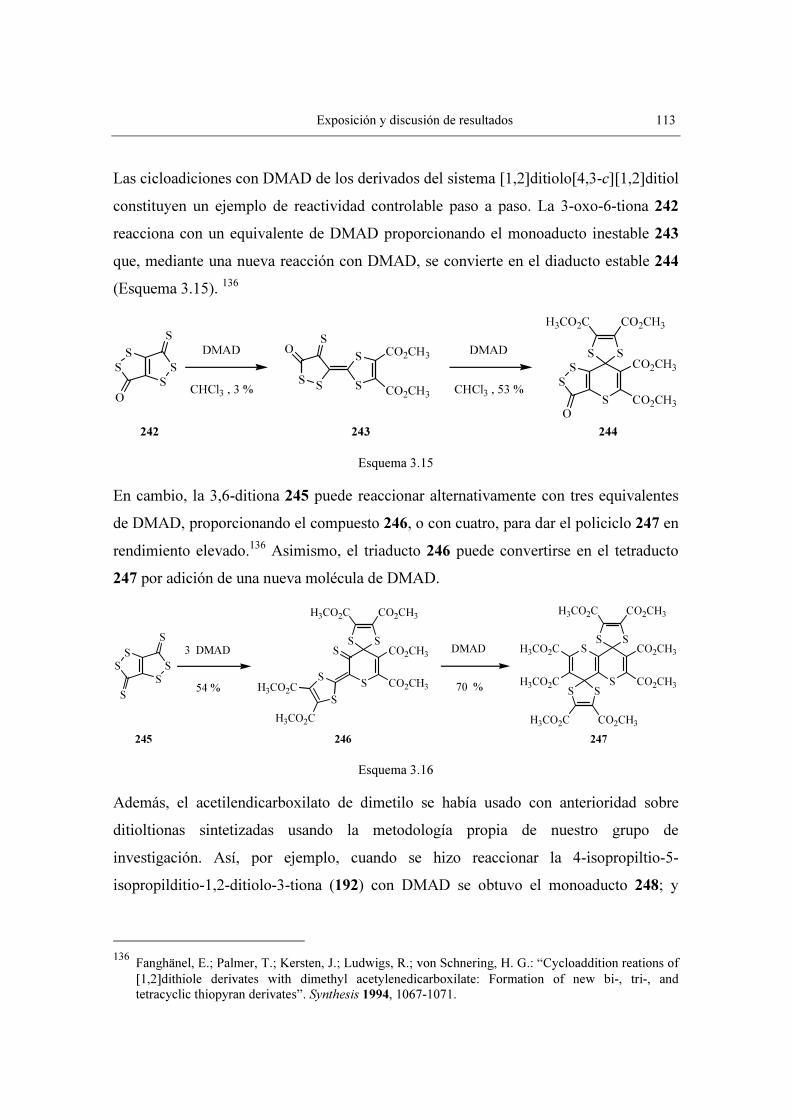
Exposición y discusión de resultados 113
Las cicloadiciones con DMAD de los derivados del sistema [1,2]ditiolo[4,3-c][1,2]ditiol
constituyen un ejemplo de reactividad controlable paso a paso. La 3-oxo-6-tiona 242
reacciona con un equivalente de DMAD proporcionando el monoaducto inestable 243
que, mediante una nueva reacción con DMAD, se convierte en el diaducto estable 244
(Esquema 3.15). 136
S
S
S
S
S
O
DMAD
SS S
S
SO CO2CH3
CO2CH3
DMAD
S
S
S
SS
CO2CH3H3CO2C
CO2CH3
CO2CH3
O
CHCl3 , 3 % CHCl3 , 53 %
242 243 244
Esquema 3.15
En cambio, la 3,6-ditiona 245 puede reaccionar alternativamente con tres equivalentes
de DMAD, proporcionando el compuesto 246, o con cuatro, para dar el policiclo 247 en
rendimiento elevado.136 Asimismo, el triaducto 246 puede convertirse en el tetraducto
247 por adición de una nueva molécula de DMAD.
S
S
S
S
S
S
S
SSS
S
S
H3CO2C
H3CO2C
CO2CH3
CO2CH3
CO2CH3H3CO2C
DMAD S
SS S
SSH3CO2C
H3CO2C
CO2CH3
CO2CH3
H3CO2C CO2CH3
H3CO2C CO2CH3
3 DMAD
54 % 70 %
245 246 247
Esquema 3.16
Además, el acetilendicarboxilato de dimetilo se había usado con anterioridad sobre
ditioltionas sintetizadas usando la metodología propia de nuestro grupo de
investigación. Así, por ejemplo, cuando se hizo reaccionar la 4-isopropiltio-5-
isopropilditio-1,2-ditiolo-3-tiona (192) con DMAD se obtuvo el monoaducto 248; y
136 Fanghänel, E.; Palmer, T.; Kersten, J.; Ludwigs, R.; von Schnering, H. G.: “Cycloaddition reations of
[1,2]dithiole derivates with dimethyl acetylenedicarboxilate: Formation of new bi-, tri-, and tetracyclic thiopyran derivates”. Synthesis 1994, 1067-1071.

114 Tesis Doctoral Susana Barriga Falcón
cuando se hizo la misma reacción con el dímero 193 se obtuvo 249 como resultado de
dos reacciones 1,3-dipolares sobre ambos grupos ditioltionas (Esquema 3.17). 117
SS
SS
S
S
S
S
S
S
SS
CO2CH3
CO2CH3
DMAD
192248
S
SS
S
S
S
S
S
S
S
S
S
S
S
S
S
CH3O2C
CH3O2C
CO2CH3
CO2CH3
S
S
S
S2 DMAD
193 249
Esquema 3.17
De forma análoga, las 1,2-ditiol-3-tionas reaccionan con un equivalente de
dibenzoilacetileno para dar monoaductos con estructura de dibenzoil-1,3-ditiol. Sin
embargo, cuando son tratadas con dos equivalentes de dibenzoilacetileno, se obtienen
diaductos provenientes de la reacción de hetero-Diels-Alder de los monoaductos. Así, la
5-fenil-1,2-ditiol-3-tiona 217 proporciona el monoaducto 250 o el diaducto 251,
mediante reacción con uno o dos equivalentes de dibenzoilacetileno, respectivamente.137
S
S
S
Ph
C C COPhPhOC
S
S
Ph
S COPh
COPh
S
S
SCOPh
COPh
Ph
PhOC COPh
250 251
83 %
C C COPhPhOC
84 %
217
Esquema 3.18
137 Ahmed, M.; Buchschriber, J. M.; McKinnon, D. M.: “Studies on some 4,5-diacyl-1,3-dithioles”. Can.
J. Chem. 1970, 48, 1991-1995.

Exposición y discusión de resultados 115
El dicianoacetileno, que también es un dienófilo muy reactivo, proporciona aductos de
Diels-Alder con una amplia variedad de dienos,138 y aductos 1,3-dipolares con diversas
tionas,139 aunque no ha sido descrita su utilización con ditioltionas.
Las 1,2-ditiol-3-tionas pueden reaccionar también, mediante reacciones de cicloadición
1,3-dipolares, con bencinos. Esta reacción constituye un método simple para obtener
benzoditiolanos.
La reacción de bencino con 1,2-ditiol-3-tionas proporciona generalmente mezcla de dos
productos, un 1,3-ditiol (254) y un tritiapentaleno (255). Los porcentajes de cada
producto dependen del método de obtención de bencino, pero en general el producto
mayoritario es el 1,3-ditiol, que en casos contados puede aislarse como producto
único.140
S
S
R
R
S
R
S
R
S
S
S
SS
R
R
253
252 254 255
+
Esquema 3.19
B. DESCRIPCIÓN DE LOS RESULTADOS OBTENIDOS EN ESTE
TRABAJO
Con los precedentes bibliográficos anteriormente descritos decidimos realizar el estudio
de la reactividad de las ditioltionas sintetizadas con anterioridad en nuestro grupo de
138 (a) Cookson, R. C.; Dance, J.; Godfrey, M. “Diels-Alder adducts of dicyanoacetylene and their
electronic spectra”. Tetrahedron 1968, 24, 1529-1535. (b) Weis, C. D. “Reactions of dicyanoacetylene”. J. Org. Chem. 1963, 28, 74-78.
139 (a) Huisgen, R; Rapp, J. “Thiocarbonyl S-Sulfides, a New Class of 1,3-Dipoles”. J. Am. Chem. Soc. 1987, 109, 902-903. (b) Jenny, C.; Heimgartner, H. “1,4-Dithiafulvalenes, Products of the Reaction of 4,4-Disubstituted 1,3-Thiazol-5(4H)-thiones and Acetylenic Compounds”. Helv. Chim. Acta 1986, 69, 419-429.
140 Paquer, D.; Pon, R. “ Action du benzyne sur differents hétérocycles sulfurés”. Bull. Soc. Chim. Fr.
1976, 120-124.

116 Tesis Doctoral Susana Barriga Falcón
trabajo frente a reacciones de cicloadición. Este estudio se ha realizado con reactivos
1,3-dipolares y con dienófilos, mayoritariamente con acetilenos activados. A
continuación, se discutirán detalladamente los resultados obtenidos.
1. Reacciones de cicloadición con reactivos 1,3-dipolares
Los reactivos 1,3-dipolares elegidos en este trabajo son las azidas y las nitriliminas.
Dentro de este apartado se incluirán también las reacciones de adición de nitrenos, pues
su mecanismo de acción involucra únicamente al grupo tiona.
1.1. Reacción de bisditiolotiazinas y bisditiolopirroles con azidas
Según los antecedentes bibliográficos antes descritos, las 1,2-ditiol-3-tionas reaccionan
con azidas para dar lugar a ditioliminas. Con el fin de obtener ditioliminas fusionadas a
núcleos tiazínicos y pirrólicos se realizó el estudio de las reacciones con azidas de
algunas de las ditioltionas y ditiolpirroles sintetizados en nuestro grupo.
En primer lugar, decidimos usar azidas que tienen grupos atractores de electrones sobre
el nitrógeno, ya que, según los antecedentes bibliográficos, parecen tener una buena
reactividad frente a ditioltionas.
La reacción a reflujo de benceno durante seis horas de la monoditioltiona 176 con
exceso de etoxicarbonilazida (256), preparada de acuerdo con el método de Froeyen,141
condujo al derivado 257, un sólido de color naranja, en un rendimiento del 36%.
S
N
S
SS
S
SO
S
N
S
SS
S
NO CO2Et
176 257256
+
O N
O
N N
Esquema 3.20
Basándonos en la bibliografía existente sobre este tipo de reacciones, hemos propuesto
el mecanismo que se indica en el esquema 3.21. La reacción de la etoxicarbonilazida
141 Froeyen, P. “A particulary convenient one-pot synthesis of N-alkoxycarbonyl, N-acyl and N-aroyl
substituted imino phosphoranes; improved preparations of azidoformates, aroyl and alkanoyl azides; an alternative route to complex amides”. Phosphorus, Sulfur Silicon Relat. Elem. 1993, 78, 161-172.

Exposición y discusión de resultados 117
(256) con el grupo tioxo de la bisditioltiona 176 daría lugar a un sistema espiránico (no
aislado) que contiene un anillo de tiatriazol. Una ciclorreversión in situ, produce la
eliminación de nitrógeno, dando lugar a la formación de una tiaziridina tampoco aislada
259, que a continuación extruye azufre, conduciendo al producto final 257.
N
N
NS
N
S
SS
S
SO
S
N
S
SS
S
ON
NN
S
EtO2C
S
N
S
SS
S
O NS
CO2Et
EtO2C
S
N
S
SS
S
O N CO2Et
176 258
259257
256
- N2
- [S]
Esquema 3.21
Esta reacción ofrece, en principio, la posibilidad de introducir diferentes sustituyentes
en los sistemas bisditiolotiazínicos, y, con este fin, se utilizaron azidas de
alcoxicarbonilos con diferentes funcionalidades. Por ejemplo, se utilizó la azida 260,
sustituida con una cadena hidrocarbonada que contiene un buen grupo saliente,142 con el
fin de poder realizar transformaciones posteriores en las ditioliminas obtenidas. Esto
permitiría aumentar la complejidad estructural, así como modificar algunas propiedades
respecto al cicloaducto original. Del mismo modo, se prepararon azidas con
sustituyentes quirales (261)143 para preparar derivados quirales, y también con
sustituyentes que contienen un radical estable (262)144 para formar sustancias orgánicas
ferromagnéticas que pudieran tener interés en el desarrollo de nuevos materiales
142 Delaby et al. C. R. Hebd. Seances Acad. Sci. 1958, 246, 3353. 143 Banks, M. R.; Blake, A. J.; Brown, A. R.; Cadogan, J. I. G.; Gaur, S.; Gosney, I.; Hodgson, P. K. G.;
Thorburn, P. “Exploiting Steric Shielding – Tuning Terpenoid-Derived Oxazolidin-2-ones as Chiral Auxiliaries for the Diels-Alder Reaction”. Tetrahedron Lett. 1994, 35, 489-492.
144 Hankovszky, H. O.; Hideg, K.; Lex, L.; Tigyi, J. “Nitroxyls; IV. Synthesis of Spin-Labelled N-(4-piperidinyloxycarbonyl)-imidazoles and 4-Piperidinyloxycarbonyl Azides and their Reaction with Amino Acid Derivatives”. Synthesis 1979, 530-531.

118 Tesis Doctoral Susana Barriga Falcón
magnéticos. Desafortunadamente, las reacciones con estas azidas dieron lugar a
productos inestables que se descompusieron durante su aislamiento, no pudiendo ser
caracterizados.
S
N
S
S S
S
S O
+
176 260
DescomposiciónOCl
S
N
S
S
O
O
176
+
261
DescomposiciónN
O
S
S
S
N N
S
N
S
S S
S
S O
176
+N
O
262
O
DescomposiciónN
O
N N
N N N
Esquema 3.22
Por otra parte, azidas de alquilo o arilsulfonilo (1-azidoadamantano o
4-carboxibencenosulfonilazida), todas ellas comerciales, tal como la mostrada en el
esquema 3.23, no reaccionaron con la monoditioltiona 176.
N
N
NS
N
S
S S
S
S O
176
+ No hay reacción
263
Esquema 3.23
En cuanto al comportamiento de otras ditioltionas, la reacción de la bisditioltiona 161
con etoxicarbonilazida (256) no dio lugar a la formación de ningún producto
caracterizable. En lugar de ello, se observó por cromatografía en capa fina la
desaparición del producto de partida y la aparición de línea base exclusivamente, por lo
que no se siguió investigando.

Exposición y discusión de resultados 119
S
N
S
S S
S
S S
+
161 256
DescomposiciónO N N N
O
Esquema 3.24
Sin embargo, la etoxicarbonilazida (256) sí reaccionó con los bisditiolpirroles 182 y 181
para dar lugar respectivamente a la monoimina 264 y a la diimina 265.
Estas reacciones se realizaron en tolueno, y para completar la reacción, en ambos casos
fue necesario calentar a reflujo durante cuatro horas. Tras la evaporación del disolvente
y purificación cromatográfica, se obtuvieron los compuestos 264 (67%), y 265 (94%),
como sólidos amarillos estables.
N
SSS
S
SO N
SSS
S
NO
+
182 264
CO2Et
256
O N N N
O
N
SSS
S
SS N
SSS
S
NN
+
181 265
CO2EtEtO2C
256
O N N N
O
Esquema 3.25
Así pues, las reacciones de los compuestos 176, 181 y 182 con azidas, sólo tuvieron
éxito en el caso de la etoxicarbonilazida, por lo que no permitieron introducir
sustituyentes complejos. Sin embargo, a través de estas reacciones de cicloadición, se
han podido obtener heterociclos del tipo bis[1,2]ditiolo[1,4]tiazina imina o
bis[1,2]ditiolopirrol iminas difícilmente obtenibles mediante otro tipo de reacciones.
1.2. Reacciones de cicloadición con nitriliminas
Como ya se dijo, las reacciones de ditioltionas con nitriliminas constituyen una buena
vía para obtener 1,3,4-tiadiazoles aunque no se conoce su geometría. Los
1,3,4-tiadiazoles y sus derivados son compuestos usados ampliamente en medicina,

120 Tesis Doctoral Susana Barriga Falcón
donde existen fármacos con esta estructura que han encontrado uso en enfermedades
tales como el glaucoma, epilepsia, fallos cardíacos, etc. Estos compuestos tienen uso
también en agricultura, donde pueden actuar como herbicidas, insecticidas, funguicidas
y bactericidas, y en diversas aplicaciones tecnológicas, usándose como tintes,
lubricantes, cristales líquidos ópticamente activos, materiales fotográficos, etc.
Por todo esto, se decidió estudiar con profundidad las reacciones de cicloadición de
nitriliminas con nuestros sistemas heterocíclicos, y asimismo, intentar elucidar la
geometría de los productos obtenidos.
Con este fin se han preparado precursores de varias nitriliminas aromáticas en dos
etapas de reacción, de acuerdo con el método de Huisgen y colaboradores.145 Primero se
prepararon las N-benzoil-N’-arilhidrazinas (268a-f), que se obtienen haciendo
reaccionar cloruros de aroílo (266a-f) con la correspondiente arilhidrazina (267a-f) en
presencia de piridina, calentando a reflujo de benceno durante 30 minutos.
O
Ar ClH2N
HN
Ar Ar NH
HN
Ar
O
+
266a-f 267a-f 268a-f
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
C5H5N
C6H6, ∆
Esquema 3.26
El rendimiento de esta etapa fue excelente, entre 90% y 100%, y los productos así
obtenidos se utilizaron sin purificación en la siguiente etapa, que es la formación de
N-(α-cloroariliden)-N’-arilhidrazonas. Para ello, se hicieron reaccionar los compuestos
268a-f con pentacloruro de fósforo en 1,2-dicloroetano, calentando a reflujo durante
toda la noche.
145 Huisgen, R.; Seidel, M.; Wallbillich, G.; Knupher, H. “1,3-Dipolar addition I. Diphenylnitrileimine
and its 1,3-dipolar additions to alkenes and alkynes.” Tetrahedron 1962, 17, 3-29.

Exposición y discusión de resultados 121
Ar NH
HN
Ar
O
PCl5 Ar N
HN
Ar
Cl
268a-f
+
270a-f269
ClCl
∆
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
Esquema 3.27
Los derivados 270a-f se purificaron por cromatografía rápida en gel de sílice y se
obtuvieron en muy bajo rendimiento. Sólo 270c fue obtenido con un rendimiento
moderado del 30%. A pesar de ello, la reacción puede hacerse en una escala apropiada
para obtener los reactivos en cantidad suficiente para llevar a cabo los ensayos
posteriores.
El esquema 3.28 muestra el mecanismo propuesto145 para la formación de las
N-(α-cloroariliden)-N’-arilhidrazonas.
Ar NH
HN
Ar
O
Cl
PCl4
Ar NH
HN
Ar
OCl4P
Cl+ +
268a-f 269 271a-f
Ar N
HN
Ar
Cl
O PCl4+
Ar N
HN
Ar
Cl+ HO PCl4
H
272a-f 270a-f
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
Esquema 3.28
Las clorohidrazonas 270a-c y 270f son compuestos conocidos que habían sido
sintetizados anteriormente por otros investigadores de acuerdo con el método de
Huisgen,145 mientras que los derivados 270d-e, que hemos preparado de forma análoga,
no habían sido descritos con anterioridad.

122 Tesis Doctoral Susana Barriga Falcón
Las nitriliminas reactivas se obtienen in situ por tratamiento de las N-(α-cloroariliden)-
N’-arilhidrazonas con trietilamina, en presencia de la ditioltiona. Las reacciones de
cicloadición con la tiazina 176 se llevaron a cabo con exceso de nitrilimina, en benceno
a reflujo durante una hora.
Se propone para estas reacciones un mecanismo concertado130 en el que, a la vez que se
adiciona la nitrilimina al grupo tiona para formar un nuevo ciclo de [1,3,4]tiadiazol, se
rompe el anillo de ditiol por el enlace azufre-azufre, produciéndose la pérdida de un
átomo de azufre. Esto puede conducir, en principio, a la formación de dos regio-
isómeros diferentes, 274a-f y 275a-f (Esquema 3.29).
S
N
S
SS
S
O S
Ar N
HN
Ar
Cl Et3N
S
N
S
SS
S
ON
NS
Ar
Ar
+
176 270a-f 273a-f
S
N
S
S
S
O
NN
S
Ar
Ar
275a-f
S
N
S
S
S
O
S
NNAr
Ar
274a-f
y/o
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
Esquema 3.29
Con el fin de determinar la estructura del producto obtenido, hemos utilizado dos
técnicas diferentes: la realización de difracción de rayos X de uno de los derivados y,
por otra parte, la minimización de energías mediante cálculos teóricos de las dos
estructuras posibles. Ambas técnicas condujeron a resultados idénticos que se
expondrán a continuación.
Experimentalmente, por cromatografía en capa fina hemos observado siempre la
aparición de sólo uno de los dos posibles regioisómeros. A este regioisómero se le ha
asignado la estructura 274a-f debido a que se han podido obtener monocristales de 274c

Exposición y discusión de resultados 123
y la estructura de este compuesto ha podido ser determinada mediante difracción de
rayos X. Dicha estructura posee configuración E en el doble enlace exocíclico.
S
N
S
SS
S
O Et3N
S
N
S
S
S
O
S
NNSAr
Ar
Ar N
HN
Ar
Cl
176 274a-f
+
270a-f
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
Esquema 3.30
En la tabla 3.2 se recogen los datos de los diferentes productos de reacción de la
monoditioltiona 176 con distintas nitriliminas, así como el rendimiento con el que han
sido obtenidos.
Precursor del reactivo 1,3-dipolar Producto obtenido
(Rendimiento)
N-(α-clorobenciliden)-N´-fenilhidrazona (270a) 274a (67%)
N-(α-cloro-p-clorobenciliden)-N´-(p-clorofenil)hidrazona (270b) 274b (77%)
N-(α-cloro-p-bromobenciliden)-N´-(p-bromofenil)hidrazona (270c) 274c (78%)
N-(α-cloro-p-iodobenciliden)-N´-(p-iodofenil)hidrazona (270d) 274d (87%)
N-(α-cloro-p-cianobenciliden)-N´-(p-cianofenil)hidrazona (270e) 274e (95%)
N-(α-cloro-2,4-diclorobenciliden)-N´-(2,4-diclorofenil)hidrazona (270f) 274f (35%)
Tabla 3.2: Productos obtenidos entre la monoditioltiona 176 y los precursores de nitriliminas 270a-f
Los compuestos 274a-f son muy coloreados. El color varía de rojo a violeta,
dependiendo de los sustituyentes en los anillos aromáticos. Los derivados 274a-f son
sólidos cristalinos y 274c-d presentan brillo metálico.

124 Tesis Doctoral Susana Barriga Falcón
Los cicloaductos obtenidos mediante la reacción de las diferentes nitriliminas, obtenidas
a partir de las correspondientes N-(α-cloroariliden)-N’-arilhidrazonas 270a-f, presentan
en RMN-1H y RMN-13C una serie de características peculiares, que son resumidas a
continuación.
El espectro de RMN-1H muestra entre 7 y 8 ppm los protones correspondientes a los dos
anillos aromáticos, el grupo metilo a 1 ppm, y dos protones metilénicos diastereotópicos
que aparecen entre 2.5 y 3 ppm. Cada protón del grupo metileno existe en un entorno
químico diferente y se acopla con el protón geminal con una constante de acoplamiento
de 14.2 Hz y con los protones del grupo metilo con una constante de acoplamiento de
7.1 Hz. Así pues, los dos protones metilénicos aparecen como sendas señales con
multiplicidad aparente de sextete, aunque son en realidad dobles cuartetes en los que la
constante geminal es el doble que la constante de acoplamiento con el metilo, tal como
se muestra en la figura 3.2 para el caso de 274c.
Figura 3.2: Ampliación de la zona del espectro de RMN-1H del derivado 274c en la que aparecen los
protones metilénicos diastereotópicos.
En el espectro de RMN-13C de los derivados 274a-f aparece el grupo tiocarbonilo a una
frecuencia entre 188 y 185 ppm, anormalmente baja, ya que el grupo tiona aparece
normalmente entre 195 y 192 ppm y en el caso de un anillo de ditiol entre 205 y 200
ppm. La señal correspondiente al grupo carbonilo aparece aproximadamente a un
desplazamiento de 182 ppm. La presencia de ambos grupos funcionales también fue
confirmada mediante espectroscopia infrarroja (C=O 1630-1650 cm-1 y C=S
1300-1320 cm-1).
Un caso especial es el del derivado 274f, que presenta un espectro de RMN-1H que
muestra un complejo patrón de señales para el grupo etilo. El espectro muestra dos
señales para el grupo metilo y cuatro grupos de seis señales (cada uno está constituido
por dos cuartetos) para el grupo metileno. Esto evidencia la existencia de dos

Exposición y discusión de resultados 125
atropisómeros conformacionales en cada uno de los cuales los protones metilénicos son
distereotópicos y aparecen como un sistema de tipo AB.
Al igual que ocurre con el espectro de RMN-1H, el espectro de RMN-13C de 274f,
muestra cuatro señales que corresponden al grupo etilo, que, por situarse en diferentes
entornos químicos en cada uno de los atropoisómeros, origina dos grupos de señales
distintas. Una explicación más detallada de esta situación, basada en cálculos
semiempíricos, se dará más adelante.
Como hemos mencionado anteriormente, en el caso del tiadiazol 274c hemos podido
obtener monocristales de calidad suficiente como para poder realizar la difracción de
rayos X (Figura 3.3).
Figura 3.3: Estructura de rayos X de la (E)-3-oxo-4-etil-5-{3,5-di(4-bromofenil)[1,3,4]tiadiazol-2-
ylideno}[1,2]ditiolo[3,4-b][1,4]tiazin-6-tiona (274c)
Los datos de rayos X se resumen en la tabla 3.3. La distancia entre el azufre de la tiona
y el azufre del [1,3,4]tiadiazol es 2.96 Å, lo que confirma la existencia de una
interacción entre los dos azufres (la suma de radios de Van der Waals es de 3.7 Å), sin
llegar a existir un enlace neto (la distancia típica del enlace azufre-azufre es de 2.05 Å).
Esta interacción podría explicar la frecuencia anormalmente baja a la que aparece la
señal del grupo tiocarbonilo en RMN-13C.

126 Tesis Doctoral Susana Barriga Falcón
Sistema cristalino Monoclínico
Grupo espacial P2(1)/c
Dimensiones de la celdilla unidad
a = 14.5880 Å α = 90º
b = 10.7454 Å β = 93.046º
c = 15.2356 Å γ = 90º
Número de moléculas por celdilla unidad 4
Tabla 3.3: Datos cristalográficos de 274c
Para justificar la obtención de un único isómero geométrico de los dos posibles, 274a-f
y 275a-f, se ha realizado el estudio conformacional asistido por ordenador de los
isómeros geométricos del sistema heterocíclico correspondiente al producto obtenido en
la reacción de la ditioltiona 176 y el precursor de la difenil nitrilimina 274a. La
conformación de mínima energía se ha obtenido, en cada caso, utilizando el método
semiempírico PM3, desarrollado por Stewart146 como una optimización del método
AM1, desarrollado por el grupo de Dewar en la Universidad de Texas en Austin. Se ha
utilizado la condición restringida de Hartree-Fock (RHF).
El algoritmo utilizado para la optimización semi-empírica es el de gradiente conjugado
de Polak-Rivière tal y como aparece implementado en el programa HyperChem ,147
que se ha usado sin adiciones ni modificaciones.
Los cálculos de energía de 274a y 275a por el método PM3 indican que 274a tiene una
entalpía de formación de 20 Kcal/mol más baja que 275a, y además existe una
interacción entre el átomo de azufre del grupo tiona y el átomo de azufre del
[1,3,4]tiadiazol. Esta interacción mantiene a ambos átomos de azufre interpenetrando
146 (a) Stewart, J. J. P. “Optimization of parameters for semiempirical methods 1. Method”. J. Comput.
Chem. 1989, 10, 209-220. (b) Stewart, J. J. P. “Optimization of parameters for semiempirical methods II. Aplications” J. Comput. Chem. 1989, 10, 221-264.
147 HyperChem 5.11, Hypercube, Inc. 419 Phillip Street. Waterloo. Ontario. N2L3X2, Canada 1999.

Exposición y discusión de resultados 127
cada uno en la esfera de Van der Waals del otro y esto puede provocar una mayor
estabilidad de 274a frente a 275a.
Los cálculos obtenidos para cada isómero se resumen a continuación en la tabla 3.4 y
las estructuras minimizadas para los isómeros 274a y 275a se muestran en la figura 3.4.
Isómero 274a Isómero 275a
Energía total -102032.0640528 (kcal/mol) -102052.5428171 (kcal/mol)
Energía de enlace -4913.4529338 (kcal/mol) -4933.9316981 (kcal/mol)
Energía atómica aislada -97118.6111190 (kcal/mol) -97118.6111190 (kcal/mol)
Energía atómica -890750.0825347 (kcal/mol) -900121.7609766 (kcal/mol)
Interacción núcleo-núcleo 788718.0184820 (kcal/mol) 798069.2181595 (kcal/mol)
Calor de formación 187.3260662 (kcal/mol) 166.8473019 (kcal/mol)
Gradiente 0.0108918 (kcal/mol/Ang) 0.0265153 (kcal/mol/Ang)
Tabla 3.4: Cálculos semiempíricos obtenidos para los isómeros 274a y 275a
274a 275a
Figura 3.4: Estructura minimizada para los isómeros geométricos 274a y 275a

128 Tesis Doctoral Susana Barriga Falcón
Como puede apreciarse 275a debe existir en una estructura minimizada muy torsionada
debido al impedimento estérico entre el grupo tiona y el anillo del N-fenilo, lo cual
explicaría su mayor energía.
De acuerdo con estos cálculos semiempíricos y teniendo en cuenta la estructura de 274c,
determinada mediante cristalografía de rayos X, se ha asignado la estructura del
regioisómero más estable, (274a-f), a todos los cicloaductos obtenidos.
Del mismo modo se hicieron cálculos semiempíricos para poder explicar el espectro de
RMN-1H que presenta el derivado 274f. En este caso la presencia de un átomo
voluminoso en la posición 2 del grupo N-arilo puede explicar la presencia de una
barrera rotacional que dé lugar a la existencia de dos atropisómeros por imposibilidad
de rotación del grupo N-arilo.
Efectivamente, los cálculos semiempíricos realizados parecen confirmar que 274f puede
existir como dos atropisómeros, tal y como se muestra en la figura 3.5.
Atropisómero S Atropisómero R
Figura 3.5: Atropisómeros S y R del derivado 274f
Puede verse que el entorno químico del grupo etilo es distinto en cada caso, lo que
explica la multiplicidad encontrada en el espectro de 1H-RMN del compuesto.
El atropisómero S tiene una energía de formación ligeramente inferior a la del
atropisómero R. Esto podría explicar el hecho de que los dos estereoisómeros estén en
distinta proporción, aunque este dato no es suficientemente significativo.

Exposición y discusión de resultados 129
En la tabla 3.5 se resumen los cálculos obtenidos para cada atropisómero.
Atropisómero S Atropisómero R
Energía total -129851.1332229 (kcal/mol) -129850.6008980 (kcal/mol)
Energía de enlace -4863.5803359 (kcal/mol) -4863.0480110 (kcal/mol)
Energía atómica aislada -124987.5528870 (kcal/mol) -124987.5528870 (kcal/mol)
Energía atómica -1141317.1958942 (kcal/mol) -1144270.8345235 (kcal/mol)
Interacción núcleo-núcleo 1011466.0626713 (kcal/mol) 1014420.2336255 (kcal/mol)
Calor de formación 144.7506641 (kcal/mol) 145.2829890 (kcal/mol)
Gradiente 0.0623883 (kcal/mol/Ang) 0.0644378 (kcal/mol/Ang)
Tabla 3.5: Cálculos semiempíricos obtenidos para los atropisómeros S y R correspondientes a la rotación
restringida del grupo N-arilo.
Así pues, los espectros de RMN de 274f pueden justificarse mediante los cálculos
teóricos por la presencia de una mezcla de dos isómeros rotacionales, en cada uno de los
cuales el grupo etilo se encuentra en un entorno químico diferente, y por lo tanto las
señales aparecen duplicadas.
También se realizaron reacciones de cicloadición entre la tiazina 176 y nitriliminas que
tienen sustituyentes diferentes en los dos grupos arilo. Las reacciones se realizaron de
manera análoga a las anteriores, es decir, generación de la nitrilimina in situ por
tratamiento con trietilamina, y calentamiento a reflujo de benceno durante una hora.
Desafortunadamente, en los casos ensayados (Esquema 3.31) los productos obtenidos
no son estables y se descomponen en presencia de gel de sílice durante la etapa de
purificación. La purificación cromatográfica en alumina o Florisil® condujo a resultados
similares.

130 Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
SO
Ar1 N
HN
Cl Et3N
+
176 270g-h
Ar2
270g, Ar1 = Ph, Ar
2 = p-NO2C6H4
270h, Ar1 = p-MeOC6H4, Ar
2 = p-NO2C6H4
Descomposición
Esquema 3.31
También se realizó la reacción de cicloadición de la monoditioltiona 176 con
nitriliminas cuyos sustituyentes fueran un grupo aromático y un grupo alifático
(Esquema 3.32). Al realizar la reacción en las condiciones habituales no se observó
producto de reacción, observándose en cromatografía en capa fina la monoditioltiona
176 inalterada.
S
N
S
S S
S
O S
+
176
Ph N
HN
Cl
O
O
276
No hay reacción.
Esquema 3.32
También se intentó realizar reacciones de cicloadición entre el monoditiolopirrol 182 y
algunas de las nitriliminas utilizadas con la tiazina 176, en condiciones similares, pero
en ningún caso se obtuvo un producto estable que pudiera ser caracterizado,
observándose en cromatografía por capa fina únicamente la descomposición de 182.
N
SSS
S
O S
N
ClHN
Et3N+ Descomposición
182 270a, Ar1 = Ar
2 = Ph
270c, Ar1 = Ar
2 = p-BrC6H4
270g, Ar1 = Ph; Ar
2 = p-NO2C6H4
C6H6, ∆Ar
1Ar
2
Esquema 3.33

Exposición y discusión de resultados 131
1.3. Adición de nitrenos
De acuerdo con los precedentes bibliográficos, la adición de algunos nitrenos sobre
1,2-ditiol-3-tionas conduce a la formación de 3-imino-1,2-ditioles.131
Conociendo estos precedentes, decidimos ensayar reacciones de adición de
bisditiolotiazinas con las cloraminas B y T (277a-b). Dichas cloraminas son una fuente
práctica de nitrógeno,148 y se postulan como precursores de nitrenos, aunque la
existencia de estos nitrenos no ha sido probada. Además, ambas cloraminas son
reactivos comerciales, relativamente baratos, estables y fáciles de manipular. Se han
ensayado estas reacciones de adición, tanto con bis[1,2]ditiolo[1,4]tiazina-3,5-ditiona
161, como con bis[1,2]ditiolo[1,4]tiazina-3-oxo-5-tiona 176. Las reacciones se
realizaron en condiciones térmicas con catálisis ácida.
En el caso del la bisditioltiona 161 y la monoditioltiona 176, la reacción con las
cloraminas B o T se realizó siempre en exceso de las mismas, a reflujo de benceno,
durante dos horas en el caso de 161 y tres horas para la tiazina 176, y se usó ácido
acético para catalizar la reacción (Esquemas 3.34 y 3.35).
S
N
S
SS
S
S S
ArS
NClNa
O O
S
N
SSS
S
N NSO2ArArO2S
+
161277a-b278a (48%)
278b (57%) a, Ar = Ph
b, Ar = C6H4CH3
CH3COOH
C6H6, ∆+
N
SSS
S
S S
181
Esquema 3.34
148 Jeong, J. U.; Tao, B.; Sagasser, I.; Henniges, H.; Sharpless, K. B. “Bromine-Catalyzed Aziridination
of Olefins. A Rare Example of Atom-Transfer Redox Catalysis by a Main Group Element”. J. Am.
Chem. Soc. 1998, 120, 6844-6845.

132 Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
O S
ArS
NClNa
O O
S
N
S
SS
S
O NSO2Ar
+
176277a-b
a, Ar = Ph
b, Ar = C6H4CH3
279a = 30%
279b = 36%
CH3COOH
C6H6, ∆
N
SSS
S
O S
182
+
Esquema 3.35
Las ditioliminas obtenidas, 278a-b y 279a-b son sólidos estables de color naranja.
Los rendimientos obtenidos con la monoditioltiona 176, tanto con la cloramina B como
con la cloramina T, son muy bajos. Además, es necesario calentar durante varias horas,
lo que provoca la extrusión de azufre del anillo tiazínico conduciendo a la formación del
monoditiolpirrol 182. Por ello, se buscaron nuevas condiciones de reacción más suaves
con el fin de intentar mejorar el rendimiento.
Con este objeto se pensó sustituir el ácido acético por un ácido de Lewis que, al mismo
tiempo, pudiera actuar activando al grupo tiona. Las bases de Lewis, particularmente los
grupos carbonilo y tiocarbonilo, pueden ser activadas mediante coordinación con el
centro metálico de los ácidos de Lewis, provocando grandes cambios en su reactividad y
en la estereoquímica del proceso.149
Recientemente han aparecido publicaciones que muestran buenos resultados
proporcionados por las sales de tierras raras y las sales trivalentes de escandio, tales
como el triflato de escandio, en la catálisis de reacciones, tales como cicloadiciones,150
condensaciones aldólicas, 151 reacciones de Friedel-Crafts,152 y otras.153 El triflato de
149 Yamamoto, H. en “Lewis Acid Reagents. A Practical Approach” Ed. Yamamoto, H. Oxford
University Press. 1999, Cap. 1. pag 1. 150 Kobayashi, S.; Hachiya, I.; Araki, M.; Ishitani, H. “Scandium Trifluoromethanesulfonate (Sc(OTf)3)
- A Novel Reusable Catalyst in the Diels-Alder Reaction”. Tetrahedron Lett. 1993, 34, 3755-3758. 151 Kobayashi, S.; Hachiya, I.; Ishitani, H.; Araki, M. “Scandium Trifluoromethanesulfonate (Sc(OTf)3)
as a Novel Reusable Lewis-Acid Catalyst in Aldol and Michael Reactions”. Synlett 1993, 472-474. 152 Kawada, A.; Mitamura, S.; Kobayashi, S. “Scandium Trifluoromethanesulfonate – A Novel Catalyst
for Friedel-Crafts Acylation”. Synlett 1994, 545-546. 153 Kobayashi, S. “Scandium triflate in organic synthesis”. Eur. J. Org. Chem. 1999, 1, 15-27.

Exposición y discusión de resultados 133
escandio presenta una acidez de Lewis mayor que otros ácidos de Lewis de las tierras
raras, debido probablemente a que tiene menor radio iónico que los lantánidos.154
Asimismo, existen ejemplos en la bibliografía en los que se favorece la coordinación del
grupo tiona con ácidos de Lewis frente al grupo carbonilo, si se utilizan ácidos de Lewis
fuertes.155 Tomando como base todos estos precedentes se decidió ensayar las
reacciones de ditioltionas y cloraminas en presencia de triflato de escandio.
La reacción de adición se realizó inicialmente usando exceso de cloramina B o T y una
proporción molar de triflato de escandio del 50% respecto a la tiazina 176. La reacción
transcurre de forma prácticamente inmediata en diclorometano a temperatura ambiente,
mejorando considerablemente los rendimientos respecto a los obtenidos en las
reacciones con ácido acético. Así, 279a se obtuvo con un rendimiento del 73% y 279b
con un rendimiento del 89%.
Las ventajas de esta metodología son evidentes, ya que además de aumentar de forma
eficaz los rendimientos (de un 30% a un 73% en el caso de 279a y de un 36% a un 89%
para 279b), se consigue reducir el tiempo de reacción considerablemente (de tres horas
a quince minutos), y las reacciones transcurren a temperatura ambiente en lugar de a
reflujo de benceno. Se evita además la formación del producto secundario de extrusión
de azufre 182.
Con el fin de optimizar las condiciones de reacción, se realizó un estudio sobre la
influencia de la concentración del catalizador en el rendimiento y en el tiempo de
reacción. Se eligió para ello como modelo la reacción de adición de la cloramina T
sobre la tiazina 176. Los resultados obtenidos se resumen en la tabla 3.6.
154 Fukuzawa, S. en “Lewis Acid Reagents. A Practical Approach” Ed. Yamamoto, H. Oxford
University Press 1999, Cap. 11. pag 185. 155 Braddock, D. C.; Brown, J. M.; Guiry, P. J. “Stereochemistry of the Catalyzed Diels-Alder Reaction
between Cyclopentadiene and Dimethyl Monothionofumarate – Soft versus Hard Lewis-Acids”. J. Chem. Soc. Chem. Commun. 1993, 1244-1246.

134 Tesis Doctoral Susana Barriga Falcón
Experimento Concentración molar de Sc(OTf)3 Tiempo de reacción Rendimiento
1 1% 15 min 48%
2 5% 12 min 52%
3 10% 10 min 80%
4 25% 7 min 93%
5 50% 5 min 89%
6 100% 5 min 85%
7 5% 12 h 80%
Tabla 3.6: Resumen de los resultados obtenidos de la reacción entre la monoditioltiona 176 y la cloramina
T con catálisis de Sc(OTf)3
Las reacciones se siguieron por cromatografía en capa fina hasta observar la completa
desaparición de la sustancia de partida.
A medida que aumenta la concentración del catalizador se produce una disminución en
el tiempo de reacción, y la mejor relación rendimiento-tiempo se consigue para
proporciones de catalizador comprendidas entre el 25% y el 50% (Tabla 3.6,
experimentos 4 y 5).
Para poder rebajar la cantidad del catalizador se hizo un nuevo experimento en el que se
aumentó el tiempo de reacción. Se usó una concentración de cinco por ciento de triflato
de escandio, y la reacción se mantuvo agitando durante doce horas. Con esto se
consiguió aumentar el rendimiento hasta el 80% (Tabla 3.6, experimento 7). De esta
forma queda demostrado que el triflato de escandio es un catalizador eficiente con el
que se puede trabajar en condiciones catalíticas consiguiendo rendimientos elevados en
tiempos de reacción relativamente cortos y en condiciones muy suaves.

Exposición y discusión de resultados 135
2. Reacciones que involucran a todo el anillo de ditioltiona
Dentro de este tipo de reacciones se han estudiado fundamentalmente las reacciones
frente a acetilenos. En este apartado incluiremos además el inicio de los estudios de
reactividad de las ditioltionas, sintetizadas con la metodología propia de nuestro grupo,
frente al bencino.
2.1. Reacciones de cicloadición 1,3-dipolares con acetilenos activados
En primer lugar se estudió la reactividad de algunas de las ditioltionas sintetizadas en
nuestro grupo de investigación frente a acetilenos simétricos sencillos, y una vez
conocidas las condiciones de reacción, se aplicó esta metodología para introducir grupos
más complejos, que serán descritos también en detalle.
2.1.1. Elección y síntesis de acetilenos simétricos sencillos
Como ya se dijo, el bis-(trifluorometil)acetileno es un alquino muy activado; sin
embargo, a pesar de ser un reactivo comercial, no se ha usado en este trabajo debido a
que se trata de un gas tóxico y no presenta, en principio, grandes ventajas frente a otros
reactivos de cicloadición.
Los primeros acetilenos elegidos en este estudio fueron, por tanto, los siguientes:
acetilendicarboxilato de dimetilo, (DMAD), dicianoacetileno (DCA) y
dibenzoilacetileno (DBA), compuestos muy reactivos y, en principio, fácilmente
accesibles y de manipulación sencilla.
La primera elección, por razones prácticas, se dirigió hacia el acetilendicarboxilato de
dimetilo (DMAD), un reactivo comercial que ha sido usado en ocasiones en reacciones
de cicloadición con ditioltionas.134,135,136,117
El segundo acetileno utilizado en el presente trabajo fue el dicianoacetileno, cuyos
grupos ciano activan más al triple enlace que los grupos éster del acetilendicarboxilato
de dimetilo. Este reactivo se prepara a partir de un precursor estable (280), que se
obtiene, a su vez, a partir de DMAD (Esquema 3.36). Se realiza, en primer lugar, una
sencilla amonolisis del acetilendiéster, que conduce a la formación de la
correspondiente diamida (280). El producto así formado se deshidrata a continuación

136 Tesis Doctoral Susana Barriga Falcón
con pentóxido de fósforo. La deshidratación se produce calentando a elevada
temperatura y aplicando vacío (2-20 mm Hg). Esto provoca la sublimación del
dicianoacetileno, que es recogido en un tubo colector enfriado en un baño de acetona-
hielo seco. Esta reacción entraña una dificultad experimental considerable, y
proporciona un rendimiento bajo.
CC
OCH3
OO
H3CO
NH3
CC
NH2
OO
H2N
235 280
CNNC
232
P2O5
Esquema 3.36
El dicianoacetileno se preparó independientemente por dos procedimientos alternativos,
que son, respectivamente, adaptación del método de Blomquist156 y del descrito por
Hopf y Witulski.157 La diferencia entre ambos métodos se encuentra en el paso de
deshidratación, siendo la formación de la acetilendicarboxamida (280) común a ambos.
En el método de Blomquist, se utiliza un gran exceso de pentóxido de fósforo y es
necesario calentar a 215 ºC. En cambio, de acuerdo con la metodología de Hopf y
Witulski, la deshidratación se realiza en una suspensión de sulfolano y se usa sólo un
ligero exceso de pentóxido de fósforo. Además, en este método la temperatura de la
reacción es considerablemente menor que la requerida en el método anterior. Por ambos
métodos hemos obtenido rendimientos similares, entre un 15 y un 18%.
El dicianoacetileno se descompone rápidamente y polimeriza formando un sólido de
color oscuro en las paredes del tubo colector. Esto puede ser evitado, durante un breve
espacio de tiempo, si se conserva el montaje a -78 ºC en atmósfera de nitrógeno. Por
esta razón, la reacción de deshidratación de la acetilendicarboxamida se ha realizado
siempre en una escala pequeña, únicamente para proporcionar la cantidad de
dicianoacetileno necesaria para la reacción en que va a emplearse. Así pues, el
156 Blomquist, A. C.; Winslow, E. C. “Unsaturated nitriles as dienophiles in the diene synthesis”. J. Org.
Chem. 1945, 10, 149-158. 157 Hopf, H.; Witulski, B. en “Modern acetylene chemistry”. Ed. Stang, P. J.; Diederich, F. VCH;
Weinheim, 1995, Cap 2, pag. 60-61.

Exposición y discusión de resultados 137
dicianoacetileno se ha preparado siempre inmediatamente antes de las correspondientes
reacciones de cicloadición con las ditioltionas.
El tercer dipolarófilo elegido fue el dibezoilacetileno (236), que es un reactivo de
cicloadición muy conveniente porque, además de ser muy reactivo, es de fácil manejo y
puede prepararse in situ a partir de un precursor muy estable y fácilmente obtenible a
partir de reactivos comerciales.
COPh
H
H
PhOC
Ph C C
O Br
H
Br
H
O
PhC
O
PhC
O
Ph
Br2 N(CH2CH3)3
281 282 236
Esquema 3.37
El dibenzoilacetileno se preparó de acuerdo con el método propuesto por Lutz y
colaboradores (Esquema 3.37).158 Para ello, se parte del trans-1,2-dibenzoiletileno (281)
comercial, que se transforma en el derivado dibromado correspondiente 282, sólido
estable, que se puede preparar en grandes cantidades y ser almacenado durante mucho
tiempo sin ningún tipo de cuidado especial.159 A continuación, se realiza una doble
deshidrobromación para formar el acetileno. De acuerdo con Lutz, los mejores
resultados se obtienen usando trietilamina en benceno o acetona a reflujo. Este método
permite la posterior utilización del dibenzoilacetileno sin purificación previa; basta con
filtrar las sales que se forman, y el reactivo está listo para realizar las reacciones de
cicloadición.158
Al igual que con el dicianoacetileno, la deshidrobromación se realiza in situ en el
momento de realizar la reacción de cicloadición y de esta forma se minimiza la posible
descomposición del reactivo.
Se ensayó además la reacción de bisditioltionas con bis(arilsulfonil)acetilenos, pues las
sulfonas son grupos muy activantes, lo que les confiere una gran reactividad en
158 Lutz, R.; Smithey, W. R. Jr. “The reactions of the dibromides and bromo derivatives of
dibenzoylethylene with amines”. J. Org. Chem. 1951, 16, 51-56. 159 Lutz, R. E. “Studies on unsaturated 1,4-diketones. I. Synthesis and structure of 1,2-di(2,4,6-
trimethylbenzoyl)ethenol”. J. Am. Chem. Soc. 1926, 48, 2905-2915.

138 Tesis Doctoral Susana Barriga Falcón
reacciones de cicloadición.160 Los estudios preliminares realizados sobre la
monoditioltiona 176 no dieron ningún resultado positivo. Tras realizar la cicloadición,
por cromatografía en capa fina, se observó la aparición de al menos siete productos
diferentes que no fueron aislados ni caracterizados.
Por otra parte, la síntesis de estos arilsulfonilacetilenos es bastante larga y laboriosa, por
lo cual estas reacciones fueron descartadas y no se tendrán en cuenta en este estudio.
Una vez elegidos los acetilenos, se procedió a estudiar las mejores condiciones de
reacción para llevar a cabo estas cicloadiciones. La elección del disolvente de reacción
fue un factor clave en estudio de las reacciones. Se realizaron ensayos con diversos
disolventes, pero las posibilidades eran bastante limitadas, porque las ditioltionas de
partida tienen una solubilidad muy baja en la mayoría de los disolventes más comunes.
Finalmente, el benceno resultó ser el disolvente más adecuado, puesto que no sólo
disuelve bien todos los sustratos, sino que tiene una temperatura de ebullición idónea
para realizar la reacción a reflujo. Algunas cicloadiciones menos favorecidas requieren
temperaturas de reacción más elevadas, por lo que fue necesario utilizar tolueno, que
tiene un punto de ebullición más alto. Las reacciones de cicloadición realizadas usando
triflato de escandio, que transcurren a temperatura ambiente, se realizan en
diclorometano, ya que las ditioltionas utilizadas son bastantes solubles en este
disolvente.
2.1.2. Reacciones de cicloadición de la monoditioltiona 176 con acetilenos activados
S
N
S
SS
S
OS EE
176
Las reacciones de cicloadición de la tiazina 176 con acetilenos activados se realizaron
siempre calentando en benceno a reflujo durante un breve tiempo (entre 5 y 30 minutos)
160
Pasquato, L; de Luchi, O.; Krotz, L. “Bis(arylsulphonyl)acetylenes”. Tetrahedron Lett. 1991, 32, 2177-2178.

Exposición y discusión de resultados 139
y usando, por lo general, un ligero exceso del alquino correspondiente. De esta forma se
obtuvieron los monoaductos representados en el esquema 3.38.
S
N
S
SS
S
OS EE
S
N
S
S
O
S
S
S
E
E
176 283, E = CO2Me
284, E = CN
285, E = COPh
C6H6, ∆
Esquema 3.38
Esta estructura ha sido confirmada mediante técnicas espectroscópicas y analíticas. La
presencia del grupo tiona ha sido confirmada mediante RMN-13C (δ: 192-195 ppm) y
mediante espectroscopia infrarroja (ν: 1250-1300 cm-1).
Los productos así formados con los diferentes alquinos son todos de color rojo y
perfectamente estables.
La adición de una única molécula de alquino a la ditioltiona 176 conduce, de acuerdo
con la bibliografía existente, a 2-tioacilmetilen-1,3-ditioles, compuestos que presentan
la estructura tricíclica que se muestra el esquema 3.38. Esta estructura está de acuerdo
con los antecedentes bibliográficos y con las consideraciones mecanísticas más
probables, ya que de acuerdo con Drozd y colaboradores los compuestos acetilénicos,
especialmente si tienen unidos grupos aceptores de electrones, reaccionan con
1,2-ditiol-3-tionas por un mecanismo de cicloadición con apertura simultánea del anillo
de 1,2-ditiol con formación de 2-(α-tioacilalquiliden)-1,3-ditioles (Esquema 3.39).161
161
Drozd, V. N.; Udachin, Y. M.; Bogomolova, G. S.; Sergeichuk, V. V. “Cycloaddition of 4-methyl-1,2- thiolene-3-thione to acetylenes”. Zh. Organ. Khim. 1980, 16, 883-886.

140 Tesis Doctoral Susana Barriga Falcón
S
S
S
S
S
S
R
RS
S
S
R
RR R+
[1,3]
196 286 287
Esquema 3.39
Los antecedentes bibliográficos existentes sobre este tipo de reacciones nos hicieron
albergar la esperanza de poder adicionar una segunda molécula de alquino mediante una
reacción de hetero-Diels-Alder. Así, forzando ligeramente las condiciones de reacción,
usando exceso de alquino y aumentando ligeramente el tiempo de calentamiento a
reflujo, se podrían obtener nuevos heterociclos con estructura tetracíclica.
Sin embargo, sólo se pudo obtener este tipo de heterociclo cuando se usó
dibenzoilacetileno, mientras que las reacciones con DMAD y DCA dieron
exclusivamente el producto de la monoadición.
Para obtener selectivamente el diaducto de la tiazina 176 con dibenzoilacetileno, fue
necesario añadir dos equivalentes y medio de dibenzoilacetileno a una disolución de la
monoditioltiona 176 en benceno y calentar la mezcla a reflujo durante treinta minutos,
mientras que la obtención exclusiva del monoaducto requiere usar la cantidad
estequiométrica de dibenzoilacetileno y calentar a reflujo de benceno durante tan sólo
cinco minutos. El diaducto tiene una gran tendencia a formarse y el mínimo exceso de
dibenzoilacetileno provoca rápidamente la formación de trazas de 288.
S
N
S
SS
S
OS COPhPhOC
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
176 288
2
S
N
S
S
O
S
S
S
PhOC
PhOC
287
Esquema 3.40
El tetraciclo 288 es un sólido de color amarillo, también muy estable.

Exposición y discusión de resultados 141
La formación del diaducto confirma también la estructura de los monoaductos, ya que
evidencia la formación de un nuevo sistema heterodiénico susceptible de sufrir una
nueva reacción de cicloadición con algunos acetilenos si éstos son suficientemente
reactivos.
La formación de este diaducto puede explicarse de acuerdo con el mecanismo propuesto
en el esquema 3.41. Recordemos que los compuestos 283, 284 y 285 contienen una
tiona α,β-insaturada, que constituye un heterodieno susceptible de sufrir una nueva
reacción de cicloadición. Así, en algunos casos se produce una reacción hetero-Diels-
Alder, y se forma un nuevo anillo heterocíclico de seis miembros (Esquema 3.41).
S
S
S
R
R
S
R R
SS R
RS
SSR
R
RRR R
D-A
+
287 286 289
Esquema 3.41
Los resultados obtenidos de la cicloadición de 176 con estos tres acetilenos se resumen
en la tabla 3.7.
Acetileno Número de equivalentes Tiempo de reacción Producto obtenido Rendimiento
DMAD 2.5 15 minutos 283 81%
DCA 2.5 15 minutos 284 68%
DBA 1 5 minutos 285 48%
DBA 2.5 30 minutos 288 73%
Tabla 3.7: Resultados obtenidos de la reacción de la tiazina 176 y diversos acetilenos en benceno a reflujo
Hay que resaltar que los cicloaductos descritos, obtenidos con dipolarófilos acetilénicos,
poseen estructuras electrónicas muy deslocalizadas, en las que los átomos de azufre
actúan como dadores de electrones, mientras que los grupos activantes de los acetilenos

142 Tesis Doctoral Susana Barriga Falcón
actúan como atractores, con lo que constituyen sistemas push-pull con potencial
aplicación como materiales para óptica no-lineal.
Por otra parte, tanto los monoaductos 283, 284 y 285 como el diaducto 288 son
asimétricos, debido a la existencia de un nitrógeno piramidal, que da lugar a dos
enantiómeros interconvertibles con dificultad. Esto es fácilmente apreciable en los
espectros de resonancia magnética nuclear de protón, en los que la señal
correspondiente al grupo metileno unido al nitrógeno aparece desdoblada, indicando la
existencia de un entorno químico diferente en cada uno de los hidrógenos de dicho
grupo metileno, como consecuencia de la inversión restringida del nitrógeno (Figura 3.6).
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57 .07.58.0
3.003 .103.203 .303.403.503.603.70
Figura 3.6: Espectro de RMN-1H de 283 y ampliación de la zona en la que aparececen los protones metilénicos
Se realizaron estudios de RMN dinámico sobre los monoaductos 283, 285 y 288. Para
ello, se efectuaron los espectros de RMN-1H en clorobenceno deuterado desde
temperatura ambiente hasta 125 ºC. En ningún caso, se llegó a la temperatura de
coalescencia del sistema. En la figura 3.7 se muestran los espectros obtenidos para el
S
N
S
S
O
S
S
S
MeO2C
MeO2C
283

Exposición y discusión de resultados 143
monoaducto 283 a distintas temperaturas. Como puede observarse, y tal como
explicamos anteriormente, a temperatura ambiente los protones metilénicos aparecen
como dos grupos de seis señales que corresponden a dos dobles cuartetes que están
separados entre sí 0.40 ppm. A medida que aumenta la temperatura se observa que las
dos señales disminuyen en intensidad y en resolución, de forma que a 125 ºC se
aprecian únicamente dos pequeños singuletes anchos. La separación de estos dos
singuletes es de 0.31 ppm.
Aunque no se ha llegado a alcanzar la temperatura de coalescencia, la aplicación de la
ecuación de Eyring,162 considerando que la temperatura de coalescencia es mayor de
125 ºC, permite deducir que la barrera de rotación debe ser mayor que 79.68 kJ/mol.
2.903.003.103.203 .303.403 .503.60
T= 398 K
2.903.003.103.203 .303.403 .503.60
T= 378 K
2.903.003.103.203 .303.403 .503.60
T= 353 K
2.903.003.103.203 .303.403 .503.60
T= 333 K
2.903.003.103.203 .303.403 .503.60
T= 313 K
2.903.003.103.203 .303.403 .503.60
T= 293 K
Figura 3.7: Espectros de RMN-1H del monoaducto 283 registrados entre 20 ºC y 125 ºC
Se realizó asimismo el estudio de las propiedades electroquímicas del aducto 283
mediante voltametría cíclica. Para ello, se utilizó una concentración molar de 5·10-4 M
de 283 en diclorometano a 20 ºC. Se utilizó como electrolito soporte Bu4NPF6 en
concentración aproximada 0.1 M, como electrodo de trabajo el de bola de platino, como
162 Hesse, M.; Meier, H.; Zeeh, B. en “Métodos espectroscópicos en Química Orgánica”. Síntesis,
Madrid, 1995.

144 Tesis Doctoral Susana Barriga Falcón
electrodo auxiliar un hilo de platino y como electrodo de referencia el de calomelanos
saturado.
El voltamograma se registró a diferentes velocidades de barrido, observándose siempre
que el proceso era irreversible. Se observa que existe una transferencia electrónica en la
que aparentemente están implicados el mismo número de electrones en la oxidación y
en la reducción, comparando la intensidad de las ondas existentes en ambos procesos,
aunque no se ha usado ni la técnica del electrodo rotatorio ni la columbimetría para
determinar el número de electrones implicados. En la figura 3.8 se representa el
voltamograma registrado a 100 mV/s para el compuesto 283. En él puede observarse
que el potencial de pico en la onda oxidativa aparece a 1.25 V (Epox = 1.25 V) y el
potencial de pico de la onda en las reducciones aparece a –1.00 V (Epred = -1.00 V).
Figura 3.8: Voltamograma del monoaducto 283 registrado en diclorometano a temperatura ambiente
S
N
S
S
O
S
S
S
MeO2C
MeO2C
283

Exposición y discusión de resultados 145
2.1.3. Ensayos de doble cicloadición de la tiazina 176 con diferentes reactivos de
cicloadición
S
N
S
SS
S
OS XX
176
YY
Basándonos en los resultados anteriormente descritos, se pensó que podría ser factible la
adición sucesiva de dos dipolarófilos distintos sobre la tiazina 176, con lo que podrían
obtenerse policiclos con diferentes sustituyentes. Así, se llevaría a cabo una reacción
1,3-dipolar en la que se adicionase un primer acetileno (como DMAD o DCA) y, a
continuación, se introduciría un segundo alquino, distinto, a través de una reacción de
hetero-Diels-Alder (Esquema 3.42).
S
N
S
SS
S
S O XX
S
N
S
S
O
S
S
S
X
X
YY
S
N
S
S
O
S
SS
XX
Y
Y
176 290 291
Esquema 3.42
En primer lugar, se pensó utilizar como segundo acetileno DBA porque con este
acetileno habíamos conseguido obtener el diaducto, lo que nos hizo pensar que era el
acetileno más reactivo. Así, se realizaron dos experimentos independientes, en los que
se añadió DBA a las disoluciones en benceno de los cicloaductos formados con
acetilendicarboxilato de dimetilo (283) y con dicianoacetileno (284). Después de
calentar la mezcla de reacción durante unas dos horas se observó que no había habido
reacción, y que el cicloaducto inicial permanecía inalterado. Se intentaron entonces
condiciones de reacción más enérgicas, para lo que se utilizó gran exceso de
dibenzoilacetileno y un disolvente de alto punto de ebullición, como es el clorobenceno,
y se calentó a reflujo durante cuatro horas. En este caso tampoco pudo apreciarse
ninguna reacción.

146 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S
H3CO2C
H3CO2C
∆
DBA
283
S S
N
S
S
SS O
PhOC
PhOC
CO2CH3H3CO2C
293
Esquema 3.43
S
N
S
S
O
S
S
S
NC
NC
284
S S
N
S
S
SS O
PhOC
PhOC
CNNC
294
∆
DBA
Esquema 3.44
A pesar de los resultados negativos obtenidos, estos experimentos sirvieron, al menos,
para corroborar la gran estabilidad térmica de los aductos 283 y 284, una propiedad muy
valiosa en su posible utilización futura como nuevos materiales.
A continuación, decidimos volver a realizar la reacción de cicloadición mixta entre la
tiazina 176 y los acetilenos simétricos sencillos utilizados en esta Tesis Doctoral,
realizando la secuencia en el sentido inverso al realizado anteriormente, es decir,
utilizando como monoaducto inicial el dibenzoilderivado 285, puesto que los grupos
benzoílos, presentes en el mismo, son grupos menos atractores de electrones que los
grupos ciano o ésteres (ver Tabla 3.1). Según esto, el heterodieno del monoaducto 285
debería ser más rico en electrones, puesto que los grupos benzoílos retiran menos carga
del mismo. Se supone que, por este motivo, el monoaducto 285 presenta una gran
tendencia a adicionar una segunda molécula de dibenzoilacetileno si éste se encuentra
en el medio de reacción. Por lo tanto, debería reaccionar también con otros dipolarófilos
presentes. Se eligió como segundo acetileno DMAD y la reacción se realizó en
clorobenceno. Tras calentar a reflujo durante 4 horas se observó por cromatografía en
capa fina la completa desaparición de 285 y la aparición de un nuevo producto de color
amarillo. Este producto se caracterizó como el correspondiente a la adición sobre la
ditioltiona 176 de una molécula de DBA, mediante una reacción 1,3-dipolar, sobre la

Exposición y discusión de resultados 147
que cual se adiciona una molécula de DMAD, mediante una reacción hetero-Diels-
Alder que conduce al producto final 295. Este aducto mixto es un sólido estable de
color amarillo que se obtiene en un rendimiento del 83% (Esquema 3.45).
S
N
S
S
O
S
S
S
COPh
COPh
DMAD
PhCl, ∆
S
N
S
S
S
SSO
CO2Me
CO2Me
COPhPhOC
287 295
Esquema 3.45
Para concluir este apartado, y dado que con dibenzoilacetileno no se consiguió obtener
el cicloaducto mixto con el monoaducto 283, decidimos probar con un dienófilo más
activo para poder lograrlo. Se eligió la 4-fenil-1,2,4-triazolin-3,5-diona (296), que es un
poderoso reactivo de cicloadición que reacciona instantáneamente con ciclopentadieno,
butadieno y cicloheptatrieno a temperatura muy baja.163
Como acabamos de mencionar, se eligió como cicloaducto de partida 283, que se hizo
reaccionar a reflujo de clorobenceno con 296 en exceso. Tras la purificación se obtuvo
un producto único de color anaranjado, estable, con un rendimiento del 48%.
La estructura esperable para este compuesto es 297, de acuerdo con el mecanismo
propuesto, mediante el cual se espera una reacción hetero-Diels-Alder sobre 283, que
conduzca al producto final.
163 Cookson, R. C.; Giliani, S. S. H.; Stevens, I. D. R. “4-Phenyl-1,2,4-triazolin-3,5-dione: a powerful
dienophile”. Tetrahedron Lett. 1962, 615-618.

148 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S
CO2CH3
CO2CH3
N
NN
O
O
Ph
S
N
S
S
O
SN
N
SS
N
CO2CH3H3CO2C
O
O
Ph
283
297
+
296
S
N
S
S N
HN
S
SO CO2CH3
CO2CH3
O
298
Ph
Esquema 3.46
Sin embargo, el espectro de masas del compuesto obtenido mostró un pico de masa de
567 uma, 73 uma menos de la esperada, que corresponde a una molécula de fórmula
C21H17N3O6S5. Un compuesto con esta fórmula podría formarse mediante la adición del
grupo tiona de 283 sobre la triazolina 296, produciéndose a continuación la apertura del
intermedio policíclico 300 para dar lugar a la imina 301, que posteriomente se hidroliza
con pérdida neta de un grupo SNCO y formación del producto final estable 298, una
imina de la difenilurea. Un posible mecanismo se muestra en el esquema 3.47.
S N
N N
O
O
Ph
N N NS
O
Ph
O H2OHN
ON
CO2
SN
NN
O
O
Ph
Ph
S
N
NN
O
O
Ph
[NS]
299
+
300 301
302 303 304298
+
Esquema 3.47
Los demás datos espectroscópicos y analíticos de 298 son compatibles con la estructura
propuesta.
En la actualidad se sigue trabajando sobre esta reacción modificando la triazolindiona
por introducción de diferentes grupos en el anillo aromático y también eligiendo

Exposición y discusión de resultados 149
monoaductos de partida diferentes que conduzcan a productos más cristalinos que
permitan obtener monocristales para poder usar la difracción de rayos X como medio
para determinar de forma inequívoca la estructura de este tipo de diaductos.
2.1.4. Reacciones de cicloadición de la bisditiolotiazina 161 con acetilenos activados
S
N
S
SS
S
SS EE
161
Se han realizado varios experimentos, que serán descritos a continuación, para estudiar
la reactividad de la bisditiolotiazina 161 frente al acetilendicarboxilato de dimetilo
(DMAD).
En primer lugar, la reacción se realizó en condiciones análogas a las utilizadas con la
monoditioltiona 176, es decir, se usaron cinco equivalentes de DMAD y la mezcla se
calentó a reflujo de benceno durante 15 minutos. Se observó la aparición de tres
productos diferentes por cromatografía de capa fina, que pudieron ser separados
cromatográficamente. Estos productos mostraron en sus espectros de masas picos del
ion molecular de masas correspondientes, respectivamente, a las de los productos de
adición de dos, tres, y cuatro moléculas de DMAD sobre la bisditiolotiazina 161. En
cambio, no se observó la formación del producto correspondiente a la adición de una
única molécula de DMAD sobre la bisditiolotiazina 161.
Las estructuras de los productos obtenidos contienen algunas peculiaridades que es
necesario resaltar. Así, en el caso en que dos moléculas de dipolarófilo cicloadicionan
sucesivamente mediante dos reacciones 1,3-dipolares consecutivas sobre cada uno de
los grupos ditioltiona de la bisditiolotiazina 161, se obtiene un compuesto 306 con una
estructura que puede considerarse un tetratiafulvaleno extendido,164 estructura muy
164 (a) Moore, A. J.; Bryce, M. R. “New vinylogous tetrathiafulvelene (TTF) pi-electron donors”.
Tetrahedron Lett. 1992, 33, 1373-1376. (b) Bryce, M. R.; Moore, A. J.; Lorcy, D.; Dhindsa, A. S.; Robert, A. “Unsymmetrical and highly-conjugated tetrathiafulvelene and selenatrithiafulvalene derivatives”. Chem. Commun. 1990, 470-472.
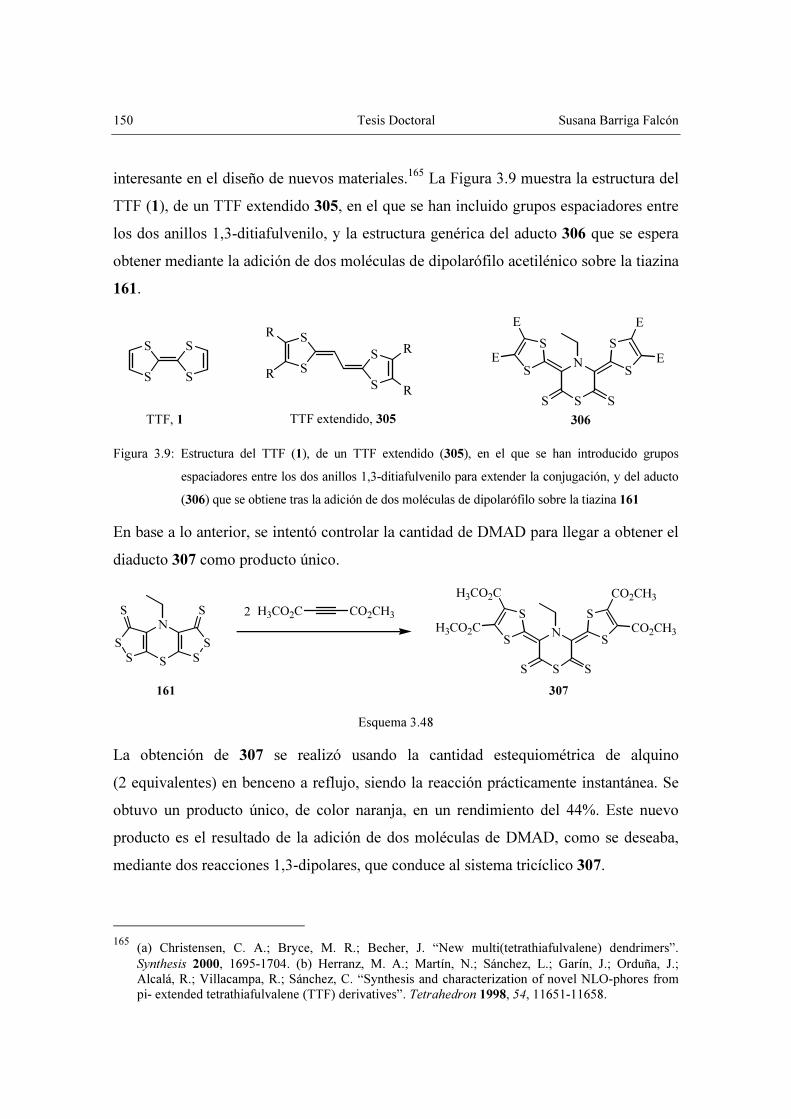
150 Tesis Doctoral Susana Barriga Falcón
interesante en el diseño de nuevos materiales.165 La Figura 3.9 muestra la estructura del
TTF (1), de un TTF extendido 305, en el que se han incluido grupos espaciadores entre
los dos anillos 1,3-ditiafulvenilo, y la estructura genérica del aducto 306 que se espera
obtener mediante la adición de dos moléculas de dipolarófilo acetilénico sobre la tiazina
161.
S
S
S
S
S
S
S
S
R
R
R
R
TTF, 1 TTF extendido, 305
S
N
SS
S
SS
SE
EE
E
306
Figura 3.9: Estructura del TTF (1), de un TTF extendido (305), en el que se han introducido grupos
espaciadores entre los dos anillos 1,3-ditiafulvenilo para extender la conjugación, y del aducto
(306) que se obtiene tras la adición de dos moléculas de dipolarófilo sobre la tiazina 161
En base a lo anterior, se intentó controlar la cantidad de DMAD para llegar a obtener el
diaducto 307 como producto único.
S
N
S
SS
S
S S H3CO2C CO2CH3
S
N
SS
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
161 307
2
Esquema 3.48
La obtención de 307 se realizó usando la cantidad estequiométrica de alquino
(2 equivalentes) en benceno a reflujo, siendo la reacción prácticamente instantánea. Se
obtuvo un producto único, de color naranja, en un rendimiento del 44%. Este nuevo
producto es el resultado de la adición de dos moléculas de DMAD, como se deseaba,
mediante dos reacciones 1,3-dipolares, que conduce al sistema tricíclico 307.
165 (a) Christensen, C. A.; Bryce, M. R.; Becher, J. “New multi(tetrathiafulvalene) dendrimers”.
Synthesis 2000, 1695-1704. (b) Herranz, M. A.; Martín, N.; Sánchez, L.; Garín, J.; Orduña, J.; Alcalá, R.; Villacampa, R.; Sánchez, C. “Synthesis and characterization of novel NLO-phores from pi- extended tetrathiafulvalene (TTF) derivatives”. Tetrahedron 1998, 54, 11651-11658.

Exposición y discusión de resultados 151
Una reacción similar se llevó a cabo también con la bisditioltiona 161 y
dibenzoilacetileno como dipolarófilo en condiciones similares, de la que se obtuvo 308,
que presenta una estructura de tipo TTF extendido, análoga a la obtenida en la adición
de DMAD. Este diaducto se obtuvo en un rendimiento del 52% como un sólido estable
de color naranja.
S
N
S
SS
S
S S PhOC COPh
S
N
SS
S
SS
S
COPh
COPhPhOC
PhOC
161 308
Esquema 3.49
Por otra parte, se abordó la obtención del producto de adición de cuatro moléculas de
DMAD sobre la tiazina 161. Este producto se había formado en muy pequeña cantidad
en el ensayo inicial, siendo identificado únicamente mediante espectrometría de masas.
Con el fin de preparar 310, se realizó un nuevo experimento añadiendo un gran exceso
de DMAD y se calentó la reacción durante un tiempo considerablemente largo para
forzar la adición de cuatro moléculas de DMAD sobre la tiazina 161. Por cromatografía
en capa fina se observó la rápida formación del producto correspondiente a la doble
adición de DMAD, así como su completa desaparición tras unos minutos de reacción.
Sin embargo, la reacción no llegó a completarse para dar lugar, como se deseaba,
únicamente al tetraaducto, 310. Por el contrario, éste se obtiene como producto muy
minoritario de la reacción, siendo siempre el producto mayoritario 309, que corresponde
a la adición de tres moléculas de DMAD sobre la bisditiolotiazina 161.
S
N
S
SS
S
SS
S
N
SS
S
S
SSCO2Me
CO2Me
CO2MeMeO2CMeO2C
MeO2C
161 309, mayoritario
DMAD
S
N
SS
CO2Me
CO2MeMeO2C
MeO2CS
S
CO2Me
CO2Me
SS
MeO2C
CO2Me
310, trazas
+
Esquema 3.50

152 Tesis Doctoral Susana Barriga Falcón
Dado que el producto de adición de cuatro moléculas de DMAD (310) no pudo
obtenerse con un rendimiento aceptable decidimos, al menos, desarrollar unas
condiciones de reacción que permitieran obtener adecuadamente el producto de adición
de tres moléculas de DMAD.
H3CO2C CO2CH3
S
N
S
SS
S
SS
S
N
SS
S
S
SSCO2CH3
CO2CH3
CO2CH3H3CO2CH3CO2C
H3CO2C
161 309
3
Esquema 3.51
Para ello, se calentó a reflujo de benceno durante diez minutos una disolución de la
tiazina 161, que contenía cinco equivalentes de DMAD. La reacción se siguió por
cromatografía en capa fina hasta que se observó la formación del aducto deseado 309
como producto mayoritario, junto con una pequeña cantidad del producto de adición de
cuatro moléculas de DMAD (310).
Después de la separación cromatográfica en columna, se obtuvo el producto 309 en un
27% de rendimiento, como un sólido rojizo. Los datos analíticos y espectroscópicos de
este compuesto permitieron asignarle la estructura tetracíclica representada,
correspondiente, efectivamante, a la adición de tres moléculas de DMAD.
La tendencia de la bisditioltiona 161 a adicionar tres moléculas de acetileno es
sorprendente, pues inicialmente se supuso que tendría un comportamiento análogo al de
la tiazina 176, con lo cual se obtendría sólo el producto de la adición de dos moléculas
de DMAD, que tiene una estructura de tipo tetratiafulvaleno extendido. La formación
del triple aducto puede ser explicada si tenemos en cuenta las características
electrónicas del anhídrido de tioácido intermedio 307, formado al adicionarse las dos
primeras moléculas de DMAD (Esquema 3.53), que contrastan con las características
electrónicas del aducto 283 de la tiazina 176, que es un ditioéster (Esquema 3.52).
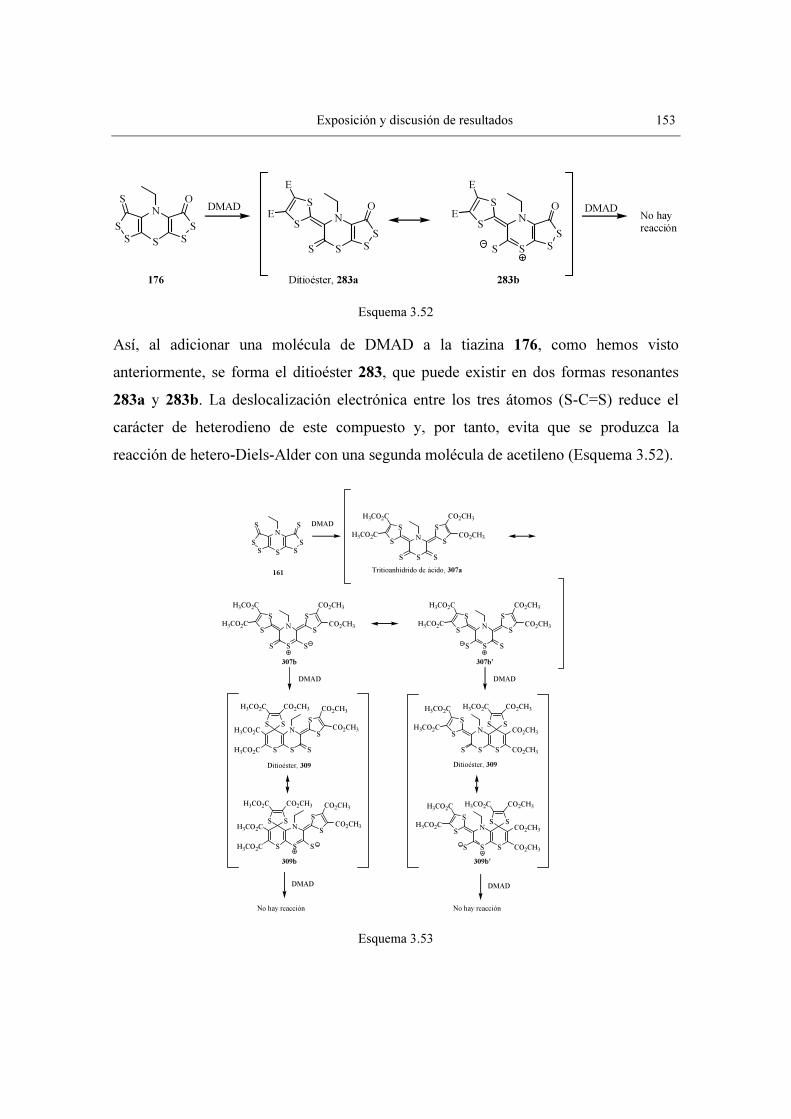
Exposición y discusión de resultados 153
S
N
S
SS
S
OS
S
N
SS
O
S
S
S
E
EDMAD
S
N
SS
O
S
S
S
E
EDMAD
176 Ditioéster, 283a 283b
No hay
reacción
Esquema 3.52
Así, al adicionar una molécula de DMAD a la tiazina 176, como hemos visto
anteriormente, se forma el ditioéster 283, que puede existir en dos formas resonantes
283a y 283b. La deslocalización electrónica entre los tres átomos (S-C=S) reduce el
carácter de heterodieno de este compuesto y, por tanto, evita que se produzca la
reacción de hetero-Diels-Alder con una segunda molécula de acetileno (Esquema 3.52).
S
N
SSS
S
SS DMAD
S
N
SS
S
SS
S
CO2CH3
CO2CH3
H3CO2C
H3CO2C
Tritioanhídrido de ácido, 307a
S
N
S
S
SS
S
CO2CH3
CO2CH
3
H3CO2C
H3CO2C
307b
S
N
SS
S
SS
S
CO2CH3
CO2CH
3
H3CO2C
H3CO2C
307b'
S
S
N
SS
S
S
SSCO2
CH3
CO2CH3
CO2CH
3H3CO
2CH
3CO
2C
H3CO2C
DMAD
S
N
S S
S
S
S SH3
CO2C
H3CO2C
H3CO
2C CO
2CH
3 CO2CH
3
CO2CH3
DMAD
S
N
S
S
S
S SH3CO2C
H3CO
2C
H3CO2C CO2CH3 CO2CH3
CO2CH3
S
N
SS
S
S
SSCO2CH3
CO2CH3
CO2CH3H3CO2CH3CO2C
H3CO2C
S
DMAD DMAD
161
Ditioéster, 309 Ditioéster, 309
309b
No hay reacción No hay reacción
309b'
Esquema 3.53

154 Tesis Doctoral Susana Barriga Falcón
El caso de la tiazina 161 es bastante diferente. En principio se adiciona una molécula de
acetileno a cada una de las dos funciones ditioltiona, con lo que se obtiene un
tritioanhídrido 307, que puede existir como tres formas resonantes. En este intermedio
(307) el enlace carbono-azufre exocíclico tiene mayor carácter de doble enlace que en el
caso del ditioéster 283 y, por tanto, existe un sistema heterodiénico claro, que puede dar
una adición hetero-Diels-Alder con una tercera molécula de alquino. El triple aducto
(309) formado es un ditioéster cíclico que, como en el caso de 283, debido a la
deslocalización electrónica del grupo S-C=S, no da lugar fácilmente a una segunda
reacción de Diels-Alder. (Esquema 3.53).
Al igual que ocurre con los productos de cicloadición de la monoditioltiona 176, el
espectro de RMN-1H del compuesto 309 muestra que la señal del metileno aparece
desdoblada en dos grupos de seis señales, correspondientes a dos dobles cuartetes, lo
que indica que existe una gran barrera de energía para la inversión del átomo de
nitrógeno (Figura 3.10).
Figura 3.10: Espectro de RMN-1H del compuesto 309
Ya que fue imposible preparar el tetraaducto con DMAD, decidimos utilizar el
dibenzoilacetileno con este fin, ya que este reactivo había mostrado dar fácilmente
productos de doble adición sobre la monoditioltiona 176.
S
N
S S
S
S
S SH3CO2C
H3CO2C
H3CO2C CO2CH3 CO2CH3
CO2CH3
309

Exposición y discusión de resultados 155
S
N
S
SS
S
SS
S
N
SS
COPh
COPhPhOC
PhOC S
S
COPh
PhOC
S
S
PhOC
COPh
S
N
SS
S
S
SSCOPh
COPh
COPhPhOCPhOC
PhOCDBA
161 312311
Esquema 3.54
Así, la reacción de la tiazina 161 con un exceso de dibenzoilacetileno (cinco
equivalentes), en disolución de benceno a reflujo durante quince minutos da lugar,
según se observa por cromatografía de capa fina, a la aparición de dos productos, uno
minoritario, 311, de color naranja, y un producto principal, 312, de color amarillo. Estos
compuestos, tras su purificación, fueron caracterizados provisionalmente, mediante
espectrometría de masas como los productos correspondientes a la adición de tres y
cuatro moléculas de dibenzoilacetileno, respectivamente.
Para obtener exclusivamente el producto correspondiente a la adición de cuatro
moléculas de dibenzoilacetileno sobre la tiazina 312, se forzaron las condiciones de
reacción. Para ello, se añadió dibenzoilacetileno en gran exceso (veinte equivalentes) y
se calentó a reflujo de benceno durante dos horas y media. Así se pudo aislar de esta
reacción el pentaciclo 312 como producto único, en un 95% de rendimiento
(Esquema 3.48). La estructura de 312 ha sido confirmada mediante las técnicas
analíticas y espectroscópicas habituales.
Así pues, en este caso, de forma similar a lo que ocurría con la tiazina 176, es posible la
doble cicloadición con dibenzoliacetileno sobre cada uno de los grupos ditioltiona.
Este procedimiento permite, a partir de productos comerciales sencillos y en tan sólo
dos pasos de reacción, obtener un compuesto pentaheterocíclico de elevado peso
molecular (1275 uma), que además posee grupos funcionales que podrían intervenir en
posteriores pasos de síntesis con el fin de modificar la estructura básica generada en esta
reacción. Esta metodología ofrece, por tanto, un potencial sintético considerable.
El espectro de RMN de protón del tetraaducto 312 muestra un singulete ancho
correspondiente al grupo metileno, indicando la presencia de isómeros

156 Tesis Doctoral Susana Barriga Falcón
conformacionales, de lo que se deduce que existe una asimetría inherente en la
molécula.
Para justificar este hecho se llevó a cabo una modelización molecular de un sistema
poliheterocíclico modelo, 313. En este modelo se prescindió de los sustituyentes
benzoílo, para reducir el tiempo de cálculo. El cálculo de la geometría más estable
proporcionó dos conformaciones de energía mínima prácticamente idéntica
(Figura 3.11, Modelo 1 y Modelo 2). Se aprecia que la primera conformación es
asimétrica, estando los grupos 1,3-ditiolano en disposición anti, y la segunda es
simétrica, con los grupos ditiolano en disposición syn.
S
N
SS
SS SS
HH
H
H
H
H
HH
313
Modelo 1 Modelo 2
Figura 3.11: Representación de las conformaciones de mínima energía obtenidas para 313
En la tabla 3.8 se muestran los valores obtenidos para cada una de las dos posibles
conformaciones de 313.

Exposición y discusión de resultados 157
Modelo 1 Modelo 2
Energía total -82021.8280517 (kcal/mol) -82022.1247289 (kcal/mol)
Energía de enlace -3812.2683707 (kcal/mol) -3812.5650479 (kcal/mol)
Energía atómica aislada -78209.5596810 (kcal/mol) -78209.5596810 (kcal/mol)
Energía atómica -682825.5193992 (kcal/mol) -685486.8176120 (kcal/mol)
Interacción núcleo-núcleo 600803.6913474 (kcal/mol) 603464.6928830 (kcal/mol)
Calor de formación 177.0976293 (kcal/mol) 176.8009521 (kcal/mol)
Gradiente 0.0619128 (kcal/mol/Ang) 0.0598000 (kcal/mol/Ang)
Tabla 3.8: Cálculos semiempíricos obtenidos para los Modelos 1 y 2
La realización de nuevos cálculos de minimización energética, usando como base tanto
el Modelo 1 como el Modelo 2 (Figura 3.11) a los que se les han añadido grupos
benzoílo a los 1,3-ditiolanos, dio lugar, en ambos casos, a una misma conformación de
mínima energía que está representada en la figura 3.12 (Modelo 3). Como puede
observarse en dicha figura, los ditiolanos están dispuestos en anti. En esta disposición
anti la repulsión estérica es menor.
La conformación del Modelo 3 (Figura 3.12) es asimétrica, pudiendo existir en dos
formas anti enantioméricas, interconvertibles a través de una conformación syn. Es
probable que la interconversión sea relativamente lenta a temperatura ambiente,
pudiéndose así obtener para el grupo metileno una señal ancha, en 1H-RMN. Esta señal
corresponde a un entorno químico distinto para cada protón en un espacio de rotación
lenta o restringida para el grupo etilo, debido al impedimento estérico para la inversión
de la conformación.

158 Tesis Doctoral Susana Barriga Falcón
Modelo 3
Figura 3.12: Conformación de mínima energía obtenida al adicionar cuatro grupos benzoílo
La tabla 3.9 muestra los valores obtenidos en la minimización correspondiente al
Modelo 3.
Modelo 3
Energía total -191209.8346504 (kcal/mol)
Energía de enlace -9664.4874334 (kcal/mol)
Energía atómica aislada -181545.3472170 (kcal/mol)
Energía atómica -2437807.2652330 (kcal/mol)
Interacción núcleo-núcleo 2246597.4305826 (kcal/mol)
Calor de formación 181.6665666 (kcal/mol)
Gradiente 0.0897522 (kcal/mol/Ang)
Tabla 3.9: Cálculos semiempíricos obtenidos para el Modelo 3 minimizado
Además de los cálculos teóricos, que nos dan la estructura en fase gaseosa, se hicieron
estudios de RMN dinámico del pentaciclo 312, para conocer la estructura existente en
disolución. En la figura 3.12 se muestran los espectros obtenidos en intervalos de 10 ºC,

Exposición y discusión de resultados 159
desde -80 ºC hasta 40 ºC. Recordemos que el tetraaducto 312 muestra, a temperatura
ambiente, un espectro de RMN-1H en el que los protones metilénicos aparecen como un
singulete ancho. Este singulete se resuelve completamente si el espectro es registrado a
40 ºC, obteniéndose un cuadruplete que presenta una constante de acoplamiento idéntica
a la del triplete correspondiente a los protones metílicos. A medida que disminuye la
temperatura se observa que el singulete disminuye en intensidad y se ensancha hasta
que aparentemente desaparece a –20 ºC. Si realizamos el espectro a –30 ºC, se observa
que comienzan a aparecer dos nuevos singuletes, que corresponden a cada uno de los
protones metilénicos. Estos singuletes van afinándose al continuar disminuyendo la
temperatura. Sin embargo, al llegar a –50 ºC el singulete que aparece sobre 4.30 ppm se
desdobla a su vez en dos siguletes anchos que continúan integrando como un único
protón.
Como puede observarse en la Figura 3.12, la temperatura de coalescencia es 283 K y, de
acuerdo con la ecuación de Eyring,162 el valor de la barrera de rotación es de 60.00 kJ/mol.
Ecuación de Eyring: ∆G≠ = 19.12 Tc (10.32 + log Tc – log kc) kc = [π(∆ν)]/√2 (Ec. 3.1)
Ecuación de Gibbs-Hemholtz: ∆G≠ = ∆H≠ – T ·∆S≠ (Ec. 3.2)
∆G≠ = Energía de activación libre de Gibbs; ∆H≠ = Entalpía de activación; ∆S≠ = Entropía de activación;
Tc = Temperatura de coalescencia; kc = velocidad de intercambio a la temperatura de coalescencia
Si calculamos las constantes de velocidad de intercambio k para cada una de las
temperaturas a las que se registró es espectro y aplicamos la ecuación de Gibbs-
Hemholtz, se puede determinar la entalpía y la entropía de activación, que son,
respectivamente, de 59.85 KJ/mol y –0.63 KJ/mol.

160 Tesis Doctoral Susana Barriga Falcón
3.23.43.63.84.04.24.44.6
T= 313 K
T= 303 K
T= 293 K
T= 283 K
T= 273 K
T= 263 K
T= 253 K
T= 243 K
T= 233 K
T= 223 K
T= 213 K
T= 203 K
T= 193 K
Figura 3.12: Espectros de RMN-1H del aducto 312 registrados entre –80 ºC y 40 ºC
Asimismo, se realizó el estudio de las propiedades electroquímicas del aducto 312
mediante voltametría cíclica. Para ello, se utilizó una concentración molar de 5·10-4 M
de 312 en diclorometano a 20 ºC. Se utilizó como electrolito soporte Bu4NPF6 en
concentración aproximada 0.1 M, como electrodo de trabajo el de bola de platino, como
electrodo auxiliar un hilo de platino y como electrodo de referencia el de calomelanos
saturado.
El voltamograma se registró a diferentes velocidades de barrido, observándose siempre
que el proceso era irreversible. Se observa que existe una transferencia electrónica en la
que aparentemente está implicado un número de electrones distinto en la oxidación y en
la reducción, de acuerdo con la intensidad de las ondas correspondientes a ambos
procesos, aunque no se ha usado ni la técnica del electrodo rotatorio ni la columbimetría
para determinar el número de electrones implicados. En la figura 3.14 se representa el
voltamograma registrado a 100 mV/s para el compuesto 312. Como puede observarse,

Exposición y discusión de resultados 161
el potencial de pico en la onda oxidativa aparece a 1.45 V (Epox = 1.45 V) y el potencial
de pico de la onda en las reducciones aparece a –1.10 V (Epred = -1.10 V).
Figura 3.14: Voltamograma del pentaciclo 312 registrado en diclorometano a temperatura ambiente
2.1.5. Síntesis y reacciones de cicloadición de tiazinas, con cadenas largas sobre el
nitrógeno, y acetilenos activados
N
R
DABCO
S
N
S
SS
S
R SSEES2Cl2
La generalización a otros sistemas tiazínicos de las reacciones antes descritas puede
permitir la obtención de estructuras con diversas funciones, susceptibles de sufrir
posteriores transformaciones. Con esta idea, y a la vista de los resultados obtenidos
mediante las reacciones de cicloadición de la tiazina 161 con acetilendicarboxilato de
dimetilo y con dibenzoilacetileno, nos planteamos obtener nuevas estructuras de tipo
tetratiafulvaleno extendido con otros derivados tiazínicos, y también nuevas estructuras
pentacíclicas altamente ramificadas.
Actualmente, el interés de los heterociclos con capacidad para formar complejos
conductores se ha extendido hacia la formación de películas de Langmuir-Blodgett, que
tienen un gran interés industrial para la formación de conductores anisotrópicos.31
S
N
SS
COPh
COPhPhOC
PhOC S
S
COPh
PhOC
S
S
PhOC
COPh
312

162 Tesis Doctoral Susana Barriga Falcón
Por ello, nos propusimos sintetizar heterociclos que contuvieran cadenas
hidrocarbonadas largas que podrían ser anclados sobre una superficie metálica y ser
usados, de este modo, en la preparación de películas de Langmuir-Blodgett. Debido a la
gran cantidad de átomos de azufre, con grandes nubes electrónicas fácilmente
polarizables, en el sistema heterocíclico, estos compuestos podrían tener la aplicación
antes mencionada.
Como ya se explicó en la introducción de esta memoria, la reacción de formación de
bisditiolotiazinas y bisditiolopirroles desarrollada en nuestro grupo de investigación
puede aplicarse de forma genérica a diisopropilaminas terciarias. Por ello, decidimos
utilizar la metodología propia de nuestro grupo de investigación111 con
disopropilaminas sustituidas con cadenas hidrocarbonadas largas, tales como el grupo
lauroílo o el estearoílo. Estas aminas terciarias se sintetizaron a partir de
diisopropilamina y el cloruro de ácido correspondiente, a través de la formación de una
amida166 y posterior reducción con hidruro de litio y aluminio (Esquema 3.55).167
HN
R
O
ClN
OR
N
RLiAlH4
+
314 315, R = C11H23
316, R = C17H35
317, R = C11H23
318, R = C17H35
319, R = C11H23
320, R = C17H35
Esquema 3.55
El método empleado para la obtención de los heterociclos fue el mismo que el utilizado
para la tiazina 161,111 empleando N-laurildiisopropilamina o N-estearildiisopropilamina
en lugar de N-etildiisopropilamina. El rendimiento de estas reacciones es muy bajo, 5%
en el caso de 321 y 20% para 322. Esto es debido, en gran medida, a la insolubilidad de
los productos, que hace extremadamente difícil su purificación cromatográfica.
166 Ghosez, L.; George-Koch, I.; Patiny, L.; Houtekie, M.; Bovy, P. “General and Practical Method of
Synthesis of 2-disubstituted-1-chloro- and 1-bromoenamines”. Tetrahedron 1998, 54, 9207-9222. 167 Anderson, J. E.; Casarini, D.; Lunazzi, L. “Stereodynamics of Inversion and Rotation in
Trialkylamines. N,N-Diisopropyl Primary Alkylamines Studied by Dynamic NMR Spectroscopy and Molecular Mechanics Calculations”. J. Chem. Soc. Perkin Trans. 2 1990, 1791-1796.

Exposición y discusión de resultados 163
N
R
2) Et3N S
N
S
SS
S
R SS1) S2Cl2, DABCO
319, R = C12H25
320, R = C18H37
321, R = C12H25
322, R = C18H37
Esquema 3.56
La difracción de rayos X de monocristal del compuesto 321 (Figura 3.15) reveló un
empaquetamiento molecular en el que las cadenas hidrofóbicas se sitúan juntas
linealmente, y los núcleos heterocíclicos, más polares, se empaquetan cara contra cara
mediante atracciones dipolo-dipolo. El ordenamiento molecular resultante recuerda al
encontrado en la doble capa lipídica de las membranas celulares, así como al de los
cristales líquidos calamíticos (Figura 3.15).
Figura 3.15: Estructura de rayos X y empaquetamiento de la tiazina 321
Los resultados obtenidos en las reacciones realizadas sobre la N-etilbisditiolotiazina 161
aconsejaron usar siempre DMAD para formar los derivados con estructura de tipo TTF
extendido, y DBA para obtener las estructuras pentacíclicas.
Para llegar a obtener el derivado pentacíclico de 321 se necesitó, como en el caso de la
tiazina 161, utilizar dibenzoilacetileno en exceso (20 equivalentes) y calentar durante un
período de tiempo largo (dos horas y media) a reflujo de benceno. Por cromatografía en
capa fina se observó la aparición de un producto único de color amarillo. Este producto

164 Tesis Doctoral Susana Barriga Falcón
corresponde, de acuerdo con los resultados espectroscópicos, a la adición de cuatro
moléculas de DBA sobre la tiazina 321 inicial, y posee una estructura análoga a la del
pentaciclo 312. Este nuevo derivado 323 se obtuvo como un sólido de color amarillo,
con un rendimiento del 93% (Esquema 3.57).
S
N
S
SS
S
SS
S
N
SS
COPh
COPhPhOC
PhOC
C12H25
SS
COPh
PhOC
SS
PhOC
COPh
DBA
321323
C12H25
C6H6, ∆
Esquema 3.57
Una reacción análoga se realizó también sobre la tiazina de cadena más larga 322. La
reacción se llevó a cabo en las mismas condiciones descritas anteriormente, y de esta
forma se obtuvo, en un rendimiento del 55%, el pentaciclo 324, que al igual que en los
casos anteriores es un sólido estable de color amarillo.
S
N
S
SS
S
SS
S
N
SS
COPh
COPhPhOC
PhOC
C18H37
SS
COPh
PhOC
SS
PhOC
COPh
DBA
322324
C18H37
C6H6, ∆
Esquema 3.58
Por otra parte, la reacción de la tiazina 322 con dos equivalentes de DMAD, para formar
el diaducto, fue prácticamente instantánea a reflujo de benceno, aunque por precaución
se prolongó el calentamiento durante veinte minutos y se usaron dos equivalentes
exactos del alquino para evitar la formación de otros productos. Tras la purificación
cromatográfica se obtuvo el aducto 325 como un sólido rojizo y estable, con un
rendimiento del 56%. Su estructura fue corroborada mediante las técnicas
espectroscópicas y analíticas habituales.

Exposición y discusión de resultados 165
S
N
S
SS
S
C18H37
S S H3CO2C CO2CH3
S
N
SS
C18H37
S
SS
SCO2CH3
CO2CH3H3CO2C
H3CO2C
322 325
2
C6H6, ∆
Esquema 3.59
Pensando en la posibilidad de obtener cicloaductos sobre los que se pudieran introducir
distintos grupos funcionales en la cadena lateral, se decidió preparar también la
estructura tipo TTF extendido, 306, usando como sustrato de cicloadición la tiaziana
186, que contiene un átomo de cloro en la posición terminal. Además, es importante
estudiar el comportamiento del grupo cloruro de alquilo en el cicloaducto formado, ya
que debemos recordar que en la tiazina 186 este cloruro es inerte en reacciones de
sustitución nucleófila (véase la introducción de esta memoria).
S
N
S
SS
S
S SH3CO2C CO2CH3
S
N
SS
S
SS
SCO2CH3
CO2CH3H3CO2C
H3CO2C
186 326
Cl Cl
2
C6H6, ∆
Esquema 3.60
Al igual que en los casos anteriores, la tiazina 186 reaccionó con dos equivalentes de
DMAD a reflujo de benceno durante 45 minutos. Tras la purificación cromatográfica se
obtuvo el diaducto 326 en un rendimiento del 68%, como un sólido anaranjado estable.
Para estudiar la reactividad del cloruro de alquilo en el diaducto 326, se hizo un ensayo
cualitativo con yoduro sódico, con el fin de producir la sustitución nucleofílica del
átomo de cloro por yodo. La reacción se siguió por cromatografía en capa fina y se
observó la aparición de un nuevo producto anaranjado de polaridad algo menor que la
del diaducto 326. Esta reacción se hizo a modo de ensayo, a muy pequeña escala, y el
producto no fue aislado ni, por lo tanto, caracterizado, pero hace albergar futuras
esperanzas acerca de la reactividad del átomo de cloro en 326.

166 Tesis Doctoral Susana Barriga Falcón
Se realizó el estudio de las propiedades electroquímicas de los aductos con estructura
tipo TTF extendido, 307, 308 y 326 mediante voltametría cíclica, con objeto de ver la
influencia de los sustituyentes de los anillos fulvenilo así como de la cadena alquílica
situada sobre el nitrógeno. Se utilizó en todos los casos una concentración molar de
5·10-4 M del aducto correspondiente en diclorometano a 20 ºC. Se utilizó como
electrolito soporte Bu4NPF6 en concentración aproximada 0.1 M, como electrodo de
trabajo el de bola de platino, como electrodo auxiliar un hilo de platino y como
electrodo de referencia el de calomelanos saturado.
Para los tres aductos, los voltamogramas se registraron a diferentes velocidades de
barrido, observándose siempre que los procesos eran irreversibles. Se observa que existe
una transferencia electrónica en la que aparentemente está implicado el doble de
electrones en la reducción que en la oxidación, comparando la intensidad de las ondas
de ambos procesos, aunque no se ha usado ni la técnica del electrodo rotatorio ni la
columbimetría para determinar el número de electrones implicados. En la figura 3.16 se
ha representado el voltamograma registrado a 100 mV/s para el compuesto 307. Como
podemos observar, el potencial de pico en la onda oxidativa aparece a 1.38 V
(Epox = 1.38 V) y el potencial de pico de la onda en las reducciones aparece a –1.09 V
(Epred = -1.09 V).
Figura 3.16: Voltamograma del diaducto 307 registrado en diclorometano a temperatura ambiente
S
N
SS
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
307
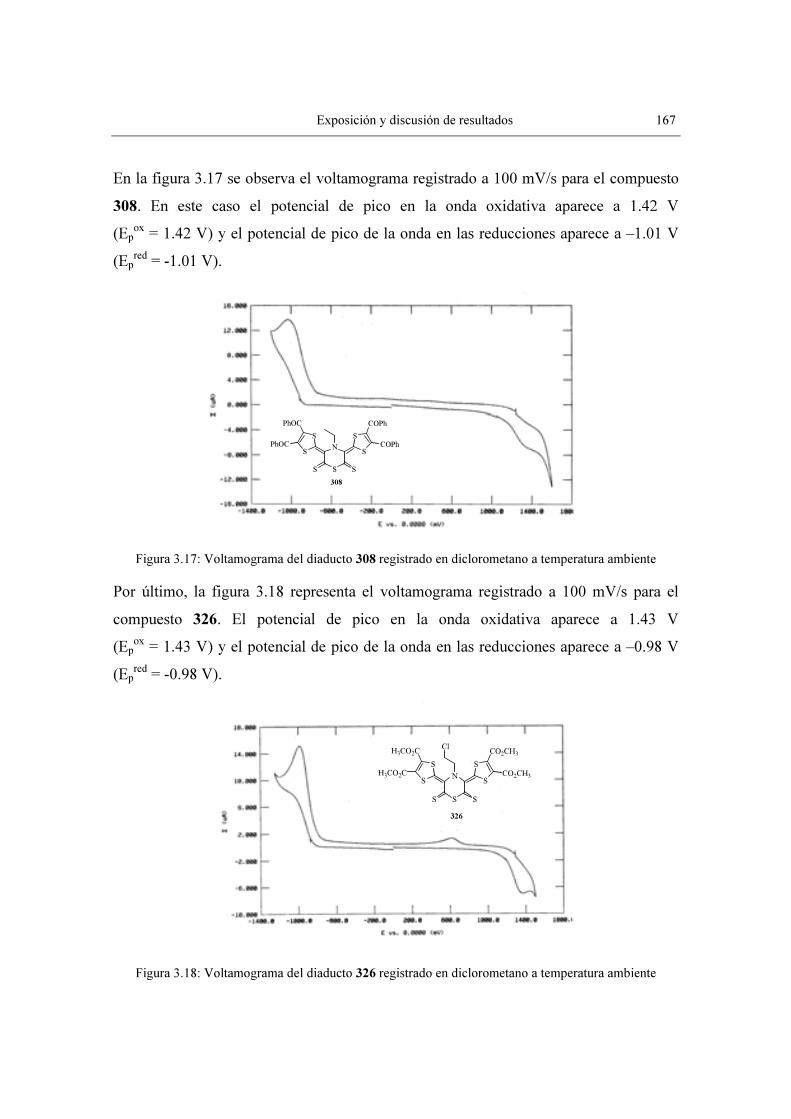
Exposición y discusión de resultados 167
En la figura 3.17 se observa el voltamograma registrado a 100 mV/s para el compuesto
308. En este caso el potencial de pico en la onda oxidativa aparece a 1.42 V
(Epox = 1.42 V) y el potencial de pico de la onda en las reducciones aparece a –1.01 V
(Epred = -1.01 V).
Figura 3.17: Voltamograma del diaducto 308 registrado en diclorometano a temperatura ambiente
Por último, la figura 3.18 representa el voltamograma registrado a 100 mV/s para el
compuesto 326. El potencial de pico en la onda oxidativa aparece a 1.43 V
(Epox = 1.43 V) y el potencial de pico de la onda en las reducciones aparece a –0.98 V
(Epred = -0.98 V).
Figura 3.18: Voltamograma del diaducto 326 registrado en diclorometano a temperatura ambiente
S
N
SS
S
SS
S
COPh
COPhPhOC
PhOC
308
S
N
SS
S
SS
SCO2CH3
CO2CH3H3CO2C
H3CO2C
326
Cl

168 Tesis Doctoral Susana Barriga Falcón
Como conclusión, podemos decir que los diferentes sustituyentes no modifican
significativamente las propiedades electroquímicas de los derivados tipo TTF,
obteniéndose siempre sistemas irreversibles con Epox comprendidos entre 1.43 y 1.38 V,
y Epred comprendidos entre –1.09 y –0.98 V.
2.1.6. Reacciones de cicloadición de la tiazina 190 con acetilenos activados
S
N
S
S
SS
SS
EE
190
La tiazina 190 contiene dos grupos ditioltiona, susceptibles de sufrir reacciones de
cicloadición análogas a las descritas anteriormente para la monoditioltiona 176 y para
las bisditioltionas 161, 321 y 322. Además, se pensó usar la tiazina 190 en reacciones de
cicloadición, debido a que los cicloaductos que se pueden obtener deberían tener
también estructura angular como 190. Estos nuevos derivados deberían ser más solubles
que 190, característica muy conveniente para facilitar el estudio posterior de sus
posibles aplicaciones. Igualmente, se buscaba obtener moléculas altamente ramificadas
con núcleo heterocíclico diferente al resto de las moléculas ramificadas obtenidas hasta
el momento, 312, 323 y 324.
Se realizó la reacción de cicloadición de la tiazina 190 con acetilendicarboxilato de
dimetilo en benceno a reflujo. Inmediatamente, la disolución cambió bruscamente de
color y tras calentar a reflujo durante quince minutos se observó por cromatografía en
capa fina que el producto de partida había desaparecido y se habían formado tres nuevos
productos, en proporción similar. Se intentó purificar dichos productos, pero se
descompusieron durante el proceso y no pudieron ser caracterizados.

Exposición y discusión de resultados 169
S
N
S
S
SS
SS DMAD
Descomposición
190
Esquema 3.61
A pesar de los resultados obtenidos con DMAD, se decidió volver a intentar la reacción
de cicloadición de la tiazina 190, en este caso con dibenzoilacetileno, esperando obtener
productos más estables que nos permitieran su aislamiento y caracterización.
En primer lugar, la reacción se hizo en las condiciones habituales, es decir, utilizando
cinco equivalentes de alquino y calentando a reflujo de benceno durante 15 minutos. Se
observó, por cromatografía en capa fina, la desaparición de la tiazina original y la
aparición de tres nuevos productos, que fueron separados cromatográficamente. Los dos
primeros productos, 327 y 328, son sólidos marrones que se obtienen con rendimientos
respectivos del 20% y del 30%. El tercer producto, 329, es un sólido de color rojo que
se obtiene con un rendimiento del 15%.
Por espectrometría de masas, el primero de estos productos (327) muestra un ion
molecular de masa 807, que corresponde a la adición de dos moléculas de acetileno. Los
otros dos productos (328 y 329) presentan picos moleculares idénticos, de masa 1041,
compatibles con la adición de tres moléculas de alquino sobre la tiazina inicial. Los
espectros de RMN-1H de 327 y 328 presentan una señal a aproximadamente 11 ppm,
que parece indicar la existencia de una función tioaldehído en estas moléculas. La
presencia de esta función fue corroborada también por los espectros de RMN-13C, en los
que aparece una señal alrededor de 200 ppm, que se asigna a un carbono terciario a
partir de la señal correspondiente en experimentos DEPT. A partir de los datos
espectroscópicos de estos compuestos, así como de las consideraciones del mecanismo
de la reacción, se han asignado a los compuestos 327, 328 y 329 las estructuras
mostradas en el esquema 3.62.
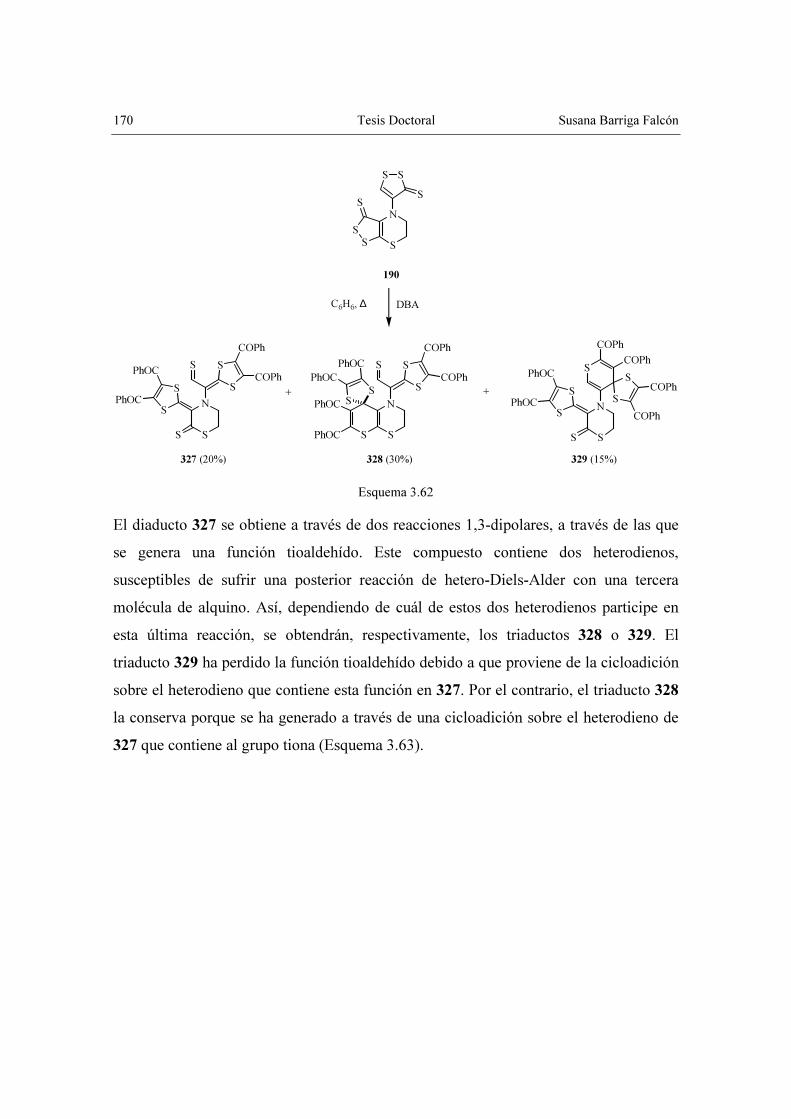
170 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
SS
SS
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC
S
N
SPhOC
PhOC
SS
PhOC
PhOCS
SS
COPh
COPh
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
190
DBA
+ +
327 (20%) 328 (30%) 329 (15%)
C6H6, ∆
Esquema 3.62
El diaducto 327 se obtiene a través de dos reacciones 1,3-dipolares, a través de las que
se genera una función tioaldehído. Este compuesto contiene dos heterodienos,
susceptibles de sufrir una posterior reacción de hetero-Diels-Alder con una tercera
molécula de alquino. Así, dependiendo de cuál de estos dos heterodienos participe en
esta última reacción, se obtendrán, respectivamente, los triaductos 328 o 329. El
triaducto 329 ha perdido la función tioaldehído debido a que proviene de la cicloadición
sobre el heterodieno que contiene esta función en 327. Por el contrario, el triaducto 328
la conserva porque se ha generado a través de una cicloadición sobre el heterodieno de
327 que contiene al grupo tiona (Esquema 3.63).

Exposición y discusión de resultados 171
S
N
S
S
SS
SS
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC
S
N
SPhOC
PhOC
S
S
PhOC
PhOCS
SS
COPh
COPh
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
190
2 DBA
327
328
329
DBA
DBA
Esquema 3.63
A continuación se intentó optimizar las condiciones de reacción para mejorar su
rendimiento, así como para la obtención selectiva de cada uno de los poliheterociclos
327-329. Así, la utilización de dos equivalente de DBA conduce a la formación
selectiva de 327, mientras que si se usan tres equivalentes de DBA se obtiene una
mezcla de los triaductos 328 y 329. Por otra parte, si se utiliza exceso de DBA y
condiciones de reacción más enérgicas, se obtiene un nuevo derivado pentacíclico 330.
S
N
S
S
SS
SS
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
S
S
PhOC
PhOC
190 330
DBA
Esquema 3.64
Las condiciones óptimas de reacción y los rendimientos obtenidos se resumen en la
tabla 3.10.

172 Tesis Doctoral Susana Barriga Falcón
Equivalentes DBA Condiciones de reacción Productos obtenidos (rendimiento)
2 20 minutos a t. ambiente 327 (36%)
3 45 minutos a t. ambiente 328 (40%) y 329 (20%)
10 60 minutos a reflujo en benceno 330 (68%)
Tabla 3.10: Condiciones de reacción y productos obtenidos en los diferentes ensayos de reacción de la
tiazina 190 con DBA
Los tioaldehídos 327 y 328 son moléculas muy reactivas y se descomponen a
temperatura ambiente en pocos días, pero son bastante estables si se conservan bajo
nitrógeno a -18 ºC. Son también muy inestables en disolución. La reactividad de estas
moléculas podría aprovecharse precisamente para realizar posteriores modificaciones a
las estructuras obtenidas, con el fin de preparar productos más complejos. La química
de estos tioaldehídos queda pues abierta a posteriores investigaciones. El tetraciclo 329,
en cambio, es más estable a temperatura ambiente, no observándose, en este sentido,
gran diferencia con el resto de los cicloaductos obtenidos a partir de otras ditioltionas.
Al igual que el resto de los sistemas pentacíclicos obtenidos hasta el momento, 330 es
un sólido de color amarillo y presenta una gran estabilidad.
2.1.7. Estudio de la reactividad de la ditiolopirroltiona 182 frente a acetilenos activados
N
SSS
S
OS
182
EE
A continuación, se expondrá el estudio realizado con los heterociclos que tienen un
núcleo central pirrólico, los bisditiolopirroles 181 y 182, en reacciones de cicloadición
frente a acetilenos activados. Los alquinos elegidos son los mismos que se han
empleado en el estudio realizado previamente con las tiazinas, es decir, DMAD, DCA y
DBA. Se expondrá en primer lugar del estudio de la reactividad de la ditiolopirroltiona
182.

Exposición y discusión de resultados 173
En principio, debe esperarse que la reactividad del sistema ditiolopirroltiona 182 sea
sustancialmente menor que la de la tiazina análoga 176, ya que la adición de una
molécula de acetileno provoca una pérdida de aromaticidad del anillo central y, por lo
tanto, juega en contra de la termodinámica del proceso. Efectivamente, la reacción de
cicloadición de 182 con alquinos activados requiere condiciones de reacción más
enérgicas que las empleadas con la tiazina 176. Sin embargo, paradójicamente, la
pirroloditioltiona siempre adiciona dos moléculas del reactivo de cicloadición.
N
SSS
S
OS
182
EE
N
SS
O
SE
E
331, E = CO2CH3
SS
EE
332, E = CN
333, E = COPh
Esquema 3.65
La tabla 3.11 resume las condiciones de reacción y los productos obtenidos a partir del
pirrol 182 y los diversos dipolarófilos sometidos a estudio.
Acetileno empleado Condiciones de reacción Producto obtenido (rendimiento)
DMAD 2 horas a reflujo en tolueno 331 (65%)
DCA 2 horas a reflujo en benceno 332 (38%)
DBA 15 minutos a reflujo en benceno 333 (73%)
Tabla 3.11: Condiciones de reacción y productos obtenidos con 182 y los acetilenos DMAD, DCA y DBA
Los aductos obtenidos, 331, 332, y 333, son sólidos de color amarillo. Los productos de
adición de DMAD (331) y DBA (333) son, además, sólidos estables; sin embargo, 332
se descompone lentamente a temperatura ambiente. Esta inestabilidad puede explicar el
bajo rendimiento obtenido en este caso.

174 Tesis Doctoral Susana Barriga Falcón
El hecho de que siempre se adicionen dos moléculas de acetileno es probablemente
debido a que, al producirse la primera cicloadición, la previsible inestabilidad del
monoaducto no aromático 334 motiva que se adicione a éste una segunda molécula de
acetileno, con lo que se recupera la aromaticidad del sistema. De hecho, en ningún caso
se han podido llegar a aislar monoaductos ya que, cuando se forman, evolucionan
rápidamente mediante una segunda reacción de cicloadición, que lleva a la restauración
del anillo aromático de pirrol (Esquema 3.66).
N
S
SS
S
OS E
E
N
S
S
O
S
S
SE
E
E
E
182 334 335
N
S
SS
EE
E
E
SS
O
Esquema 3.66
Por el contrario, como ya se ha explicado, las estructuras análogas a 334 con un anillo
central de tiazina pueden aislarse como productos estables, aunque en algunos casos son
también bastante reactivas frente a dipolarófilos. Dos ejemplos de 1,4-tiazinas de
estructura relacionada son 283 y 309, descritos anteriormente. Las diferencias
estructurales, en este caso, se traducen en diferencias de reactividad, no de estabilidad,
ya que, en el cálculo energético de cada uno de los modelos moleculares de los
monoaductos con núcleo tiazínico y pirrólico, no se aprecian diferencias
suficientemente grandes que impidan la existencia de ninguna de ellas.
Las estructuras 283, 309 y 334 participan en reacciones de cicloadición de hetero-Diels-
Alder frente a filodienos activados por los grupos atractores de electrones. En todos los
casos el sistema heterodiénico consiste en una tiona α,β-insaturada muy rica en
electrones, ya que está sustituida por dos átomos de azufre en la posición 4 y un
nitrógeno en la posición 3. La cicloadición estará, en todos los casos, conducida por el
orbital HOMO del heterodieno y el LUMO del dienófilo. Puesto que, los filodienos son
los mismos, las diferencias que dan lugar a distinta reactividad deberán encontrarse en

Exposición y discusión de resultados 175
los orbitales HOMO de los heterodienos. Se han calculado los orbitales HOMO de las
conformaciones de mínima energía de 283, 309 y 334, mediante modelos simplificados
(sin sustituyentes en los anillos de ditiolano y de dihidrotiano), 283s, 309s y 334s, y la
distribución de la densidad electrónica en cada uno de estos orbitales se muestra en la
figura 3.19.168 De la comparación de las tres representaciones se aprecia que el orbital
HOMO de 334s concentra la mayor parte de la densidad electrónica en las posiciones
1 y 4 del heterodieno, que son justamente las posiciones más reactivas frente al orbital
LUMO del filodieno. En cambio, los orbitales HOMO de 283 y 309 concentran la
mayor densidad electrónica en la agrupación S-C=S, desviando parte de la densidad
electrónica fuera del sistema diénico, y empobreciendo mucho la posición 4 del dieno.
Esto explica que el intermedio correspondiente a 334 sea mucho más reactivo frente a
filodienos que los compuestos 283 y 309, que son por ello aislables.
S
N
S
S
S
SS
S
H
H
H
H
HH
N
SS S
S
SO
H
H
S
N
S
S
S
H
H
S
S
O
309s 334s 283s
Figura 3.19: Representación de la densidad electrónica de los orbitales HOMO de modelos simplificados
de los heterociclos 309s, 334s y 283s como el cuadrado de la función de onda
168 Se usó para ello el programa HyperChem . Se utilizó el método semiempírico PM3 y la condición
restringida de Hartree-Fock (RFH).

176 Tesis Doctoral Susana Barriga Falcón
2.1.8. Estudio de la reactividad del bisditiolopirrol 181 frente a acetilenos activados
N
SSS
S
SS
181
EE
El comportamiento del bisditiolopirrol 181 es totalmente análogo al del
monoditiolopirrol 182. Así, el producto que se obtiene tras la reacción de cicloadición
con acetilenos es el que corresponde a la adición de cuatro moléculas de dipolarófilo
sobre el ditiolopirrol 181, tal como se muestra en el esquema 3.67.
N
SSS
S
SS
181 336, E = CO2CH3
EE
337, E = COPh
N
SS
SSS
S
E
EEE
E
EE
E
4
∆
Esquema 3.67
La tabla 3.12 resume las condiciones de reacción y los productos obtenidos entre el
pirrol 181 y los diversos acetilenos sometidos a estudio.
Acetileno empleado Condiciones de reacción Producto obtenido (rendimiento)
DMAD 3 horas a reflujo en tolueno 336 (61%)
DBA 15 minutos a reflujo en benceno 337 (38%)
DCA 30 minutos a reflujo en benceno Descomposición
Tabla 3.12: Condiciones de reacción y productos obtenidos con 181 y los acetilenos DMAD y DBA.
Los aductos 336 y 337 son sólidos estables de color amarillo. En el caso de la reacción
de cicloadición con dicianoacetileno, el producto que se obtiene resultó ser muy
inestable y no se llegó a aislar.

Exposición y discusión de resultados 177
Como puede observarse, no se encuentra diferencia de reactividad con relación a las
reacciones de cicloadición de los ditiolopirroles 181 y 182 con DMAD y DBA,
obteniéndose siempre el producto que corresponde a la adición de dos moléculas de
alquino por cada grupo tiona.
Se supone que la reacción comienza mediante una cicloadición del dipolarófilo sobre
uno de los grupos ditioltiona, para dar lugar al intermedio 338. Esta tiona intermedia
posee un sistema heterodiénico capaz de dar lugar a una reacción de hetero-Diels-Alder.
Si se repiten estas reacciones sobre la otra ditioltiona, se obtiene el producto de adición
de cuatro moléculas de alquino, 341 (Esquema 3.68).
N
S
SS
S
SS E
E
N
S
S
S
S
S
SE
E
E
E
E
E
N
S
S
S
E
E
S
SS
EE
E
E
E
E
N
S
SS
EE
E
E
S
SS
E
E
E
E
181 338 339
340341
N
S
SS
EE
E
E
SS
S
Esquema 3.68
En ningún caso hemos sido capaces de detectar ninguno de los intermedios, de lo que se
deduce que, en este caso, la reacción más lenta es la cicloadición 1,3-dipolar inicial, y
los intermedios no aromáticos se transforman rápidamente, mediante reacciones de
Diels-Alder, en el producto aromático final.

178 Tesis Doctoral Susana Barriga Falcón
Una vez realizado el estudio de la reactividad de las diferentes tiazinas y pirroles frente
a reacciones de cicloadición con los acetilenos activados elegidos, se decidió sintetizar
nuevos acetilenos buscando introducir grupos que fueran útiles para una posible
aplicación de los aductos obtenidos como nuevos materiales. Concretamente se buscaba
introducir precursores con reactividad posterior a la cicloadición, así como grupos
susceptibles de modificar las propiedades electroquímicas de los aductos.
2.1.9. Cicloadiciones con acetilenos activados por grupos aldehído
S
N
S
SS
S
O S
CHOOHC+
176
En primer lugar, se decidió introducir el grupo aldehído en nuestros heterociclos con el
fin de poder obtener estructuras de mayor complejidad. Esto podría llevarse a cabo
mediante reacciones de cicloadición con alquinos que contuvieran esta función. El
grupo aldehído es un grupo que activa fuertemente el filodieno y además, es muy
reactivo. Así, los aductos obtenidos contendrían al menos un grupo aldehído,
susceptible de sufrir reacciones con nucleófilos, y de esta forma obtener estructuras más
complejas.
Un ejemplo de posible utilización de la reactividad de grupos aldehido en la obtención
de nuevos materiales es la producción de radicales estables. Un radical estable utilizado
frecuentemente en los materiales orgánicos magnéticos es el óxido de imidazol 344.
Este heterociclo puede obtenerse fácilmente a partir de un grupo aldehído, por reacción
con tetrametiletilendihidroxilamina (343) y posterior oxidación (Esquema 3.69).169 En
la actualidad existe un gran interés en la fabricación de “imanes moleculares”. Por ello,
nos planteamos obtener heterociclos funcionalizados que pudieran ser transformados en
radicales estables de utilidad para la construcción de materiales magnéticos.
169 Kalai, T.; Jeko, J.; Szabo, L.; Parkanyi, L.; Hideg, K. “Synthesis and reactions of new nitronyl
nitroxides”. Synthesis 1997, 1049-1055.

Exposición y discusión de resultados 179
R CHO
NHOHHOHN
R
N
N
O
O
1)
2) PbO2
343
344342
Esquema 3.69
En primer lugar optamos por introducir un único aldehído. Por ello, se eligieron para
realizar las reacciones de cicloadición los alquinos 345 y 346, que contienen la función
aldehído protegida en forma de acetal, además de un segundo grupo atractor de
electrones. Los alquinos muy pobres en electrones tienen gran tendencia a polimerizar y
por ello es preferible desproteger el aldehído in situ, o después de haber realizado la
cicloadición.
CN
EtO
EtO
CO2Me
MeO
MeO
345 346
El propargilnitrilo 345 fue preparado, de acuerdo con el método propuesto por Murray y
Zweifel, en dos etapas de reacción.170 En primer lugar, se obtiene el cianato de fenilo
(348) y a continuación se hace reaccionar éste con el anión 349 para dar lugar al alquino
345 (Esquema 3.70)
OH BrCN Et3N O
CN
Et3N·HBr
EtO
EtO
O
CN EtO
EtO
CN
+ + +
+
348
348 345
347
349
Esquema 3.70
170 Murray, R. E.; Zweifel, G. “Preparation of phenyl cyanate and its utilization for the synthesis of
α,β-unsaturated nitriles”. Synthesis 1980, 150-151.

180 Tesis Doctoral Susana Barriga Falcón
El acetal 346 se sintetizó de acuerdo con Gorgues y colaboradores, por tratamiento de
propiolato de metilo con ortoformiato de trimetilo en presencia, de yoduro de zinc
(Esquema 3.71).171
H CO2Me MeO
OMe
OMe
H
ZnI2
CO2Me
MeO
MeO
+
350 351 346
Esquema 3.71
Con ambos acetales, la reacción de cicloadición con la monoditioltiona 176 requería
condiciones muy drásticas, como calentar a reflujo en ácido fórmico para desproteger el
aldehído, con lo que se producía parcialmente tanto la extrusión de azufre en la tiazina
176 como la descomposición del reactivo de cicloadición. Tanto en el caso de 345 como
en el de 346, al realizar la reacción de cicloadición con la monoditioltiona 176 se
obtuvieron productos que no pudieron ser caracterizados, pues se descomponían durante
el proceso de purificación, o rápidamente tras haber sido purificados.
S
N
S
SS
S
O S
CN
EtO
EtO
HCO2H+
345
Descomposición
176
Esquema 3.72
CO2Me
EtO
EtO
346
S
N
S
SS
S
O S
+
176
HCO2HDescomposición
Esquema 3.73
A la vista de los resultados obtenidos se descartaron estos acetilenos y se pensó en
introducir dos grupos aldehído sobre las ditioltionas, utilizando para ello acetilenos
171 Gorgues, A.; Simon, A.; Le Coq, A.; Hercouet, A.; Corre, F. “Versatilité de réactivité de l’acétylène
dicarbaldéhyde et des aldéhydes α-acétyléniques à l’égard des diènes conjugués cycliques et hétérocycliques en milieu acide”. Tetrahedron 1986, 42, 351-370.

Exposición y discusión de resultados 181
simétricos. Se decidió utilizar simultáneamente dos tipos de acetilenos activados
mediante grupos aldehído; por un lado, el acetilendicarbaldehído (352), que, de acuerdo
con la bibliografía existente, ya había sido ensayado en reacciones de cicloadición con
ditioltionas172 y, por otro lado, un alquino activado con grupos aldehído unidos al triple
enlace mediante grupos espaciadores, como por ejemplo, el grupo furoílo.
Se discutirá a continuación el estudio realizado con el acetilendicarbaldehído (352) y la
ditioltiona 176.
El acetilendicarbaldehído (352) es un compuesto muy interesante para la síntesis de
precursores de nuevos materiales, como por ejemplo el dirradical 353 (Esquema 3.74).
NHOHHOHN
OHC CHO
N
N
O
O
N
N
O
O
1)
2) PbO2
343
352 353
Esquema 3.74
Dado que el acetilendicarbaldehído (352) es muy reactivo, difícilmente aislable y muy
inestable,173 se decidió realizar la cicloadición con una de las dos funciones aldehído
protegida, por lo que se sintetizó el monoacetal dietílico del acetilendicarbaldehído
(356). Para ello el alquino 354, un reactivo comercial estable, se transformó en el
172 Frère, P.; Belyasmine, A.; Gouriou, Y.; Jubault, M.; Gorgues, A.; Duguay, G.; Wood, S.; Reynolds,
C. D.; Bryce, M. R. “4,5-Diformyl-1,3-dithiol-2-ylidene substituted ethanals or ethanones and vinylogs of tetraformyltetrathiafulvalene from acetylenedicarbaldehyde and 3-thioxo-1,2-dithioles”. Bull. Soc. Chim. Fr. 1995, 132, 975-984.
173 Gorgues, A.; Stephan, D.; Belyasmine, A.; Khanous, A.; Le Coq, A. “Acétylènedicarbaldéhyde: Isolement et diénophilie exclusive en milieu neutre”. Tetrahedron 1990, 46, 2817-2826.

182 Tesis Doctoral Susana Barriga Falcón
alcohol 355 por tratamiento sucesivo con butillitio y formaldehído.174 Este alcohol fue
posteriormente oxidado con óxido de manganeso para dar lugar a 356.175
H
O
O
BuLi
CH2O
CH2OH
O
O
MnO2
CHO
O
O
354 356355
Esquema 3.74
Para evitar la descomposición del reactivo se buscaron condiciones de reacción suaves,
y para compensar la menor activación del acetileno se pensó de nuevo en utilizar ácidos
de Lewis, cuyos usos en reacciones de cicloadición son bien conocidos.176 En vista de
los buenos resultados obtenidos con el triflato de escandio en las reacciones con las
cloraminas, se decidió usar este ácido de Lewis para catalizar la reacción de
cicloadición 1,3-dipolar, y de esta forma evitar calentar a temperatura elevada.
La reacción se realizó en diclorometano entre la tiazina 176 y el monoacetal dietílico del
acetilendicarbaldehído (356) y se usó una concentración molar de triflato de escandio
del 50%. La reacción sucede de forma prácticamente instantánea a temperatura
ambiente en diclorometano. Tras la purificación se obtuvo el aducto 357 como un sólido
rojo que es una mezcla de los isómeros Z y E.
CHO
O
OS
N
S
SS
S
O S
S
N
S
S
O
S
S
SCHO
CH(OEt)2
356
+
357 (Z + E)176
Sc(OTf)3
CH2Cl2
Esquema 3.76
174 Raffin, C.; Bernard, D.; Fournet, G.; Gore, J.; Chantepie, J.; Quash, G. “Synthèse de composés
acétyléniques α−α’-difuntionnels utilisables dans l’inhibition sélective de la croissance de cellules transformées”. J. Chem. Res. Miniprint 1995, 152-172.
175 Macrides, T. A.; Thaller, V. “Natural Acetylenes. Part 55. Acetylenic Aldehydes. Synthesis and Hydrate, Hemiacetal, and Oxime Formation”. J. Chem. Res. Miniprint 1981, 2001-2016.
176 Santelli, M.; Pons, J. M. en “Lewis acids and selectivity in organic síntesis” de la serie “New directions in organic and biological chemistry”. Editor Rees, C. W. CRC Press. Florida. 1996.

Exposición y discusión de resultados 183
A continuación se realizó la desprotección del acetal presente en el aducto 357. Esta
desprotección se realiza calentando en ácido fórmico a 60 ºC durante media hora y es
cuantitativa. Tras la purificación se obtuvo un único producto 358 sólido, de color rojo,
estable. El rendimiento global de estas dos reacciones fue del 83%.
S
N
S
S
O
S
S
S
CHO
CH(OEt)2
357 (Z + E)
HCO2H
S
N
S
S
O
S
S
S
CHO
CHO
358
Esquema 3.77
Se pensó obtener un nuevo derivado heterocíclico de tipo pirazina a partir del aducto
358, mediante reacción con hidrazina, una reacción clásica de los 1,3-ditiol-4,5-
carbaldehídos.172 Sin embargo, tras añadir la hidrazina sólo se observó la
descomposición del aducto 358, con lo que no pudimos obtener los derivados deseados.
S
N
S
S
O
S
S
SCHO
CHO
358
N2H4·H2O
Descomposición
Esquema 3.78
El aducto 358 tiene dos aldehídos y por lo tanto, es un sustrato adecuado para obtener
un derivado que contenga dos radicales libres mediante tratamiento con la
tetrametilendihidroxilamina (343), y posterior oxidación (Esquema 3.79). Esta reacción
será estudiada en un futuro próximo como continuación del tema de trabajo.
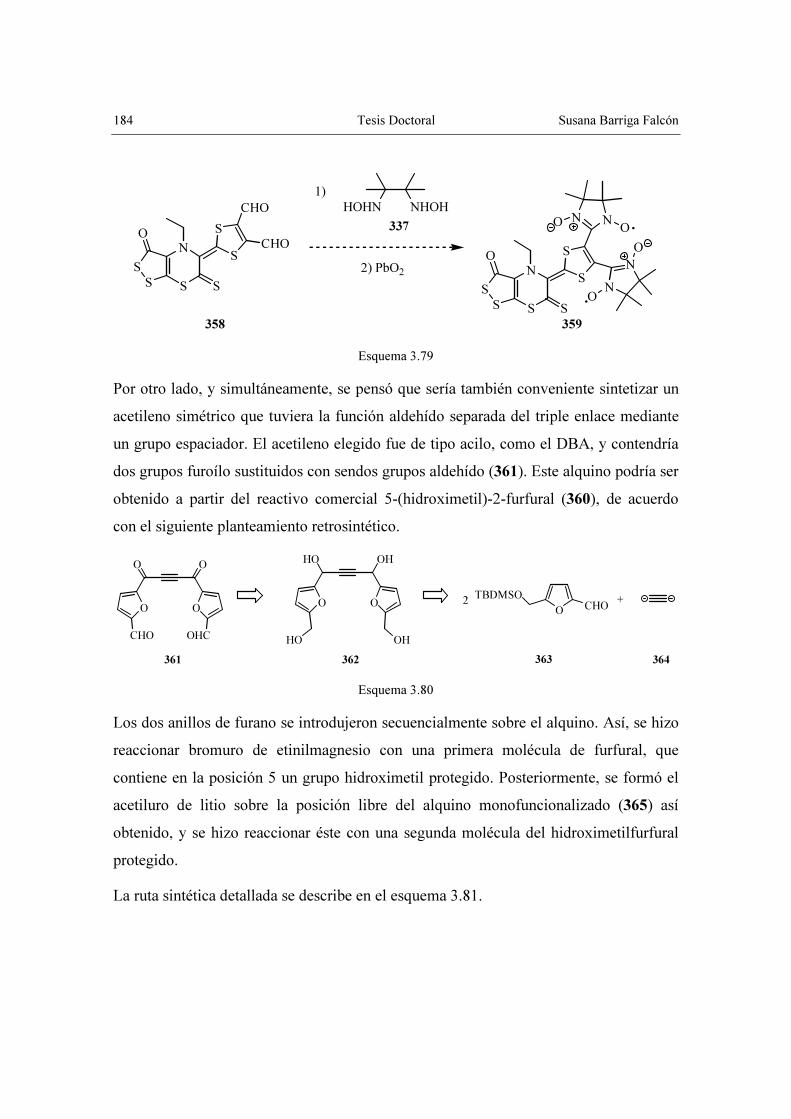
184 Tesis Doctoral Susana Barriga Falcón
NHOHHOHN
N
N
O
O
1)
2) PbO2
337
359
S
N
S
S
O
S
S
S
NNO O
S
N
S
S
O
S
S
S
CHO
CHO
358
Esquema 3.79
Por otro lado, y simultáneamente, se pensó que sería también conveniente sintetizar un
acetileno simétrico que tuviera la función aldehído separada del triple enlace mediante
un grupo espaciador. El acetileno elegido fue de tipo acilo, como el DBA, y contendría
dos grupos furoílo sustituidos con sendos grupos aldehído (361). Este alquino podría ser
obtenido a partir del reactivo comercial 5-(hidroximetil)-2-furfural (360), de acuerdo
con el siguiente planteamiento retrosintético.
O O
O
CHO
O
OHC
HO OH
O O
HO OH
O
TBDMSO
CHO2 +
361 362 364363
Esquema 3.80
Los dos anillos de furano se introdujeron secuencialmente sobre el alquino. Así, se hizo
reaccionar bromuro de etinilmagnesio con una primera molécula de furfural, que
contiene en la posición 5 un grupo hidroximetil protegido. Posteriormente, se formó el
acetiluro de litio sobre la posición libre del alquino monofuncionalizado (365) así
obtenido, y se hizo reaccionar éste con una segunda molécula del hidroximetilfurfural
protegido.
La ruta sintética detallada se describe en el esquema 3.81.

Exposición y discusión de resultados 185
HO
O
TBDMSO
OH
O
OSMDBT
O
O
TBDMSO
O
O
OSMDBT
AcOH1) BuLi
2) 363
367366
O
O
HO
O
O
OH
O
O
CHO
O
O
OHC
368 361
[O]
BAIB
TEMPO
O
HOCHO
TBDMSCl
O
TBDMSOCHO
MgBr
HO
O
TBDMSO360 363 365
THF-H2O
Esquema 3.81
En primer lugar se realizó la protección del grupo hidroxilo del 5-hidroximetilfurfural
de partida, 360, con cloruro de terc-butildimetilsililo.177 La adición de bromuro de
etinilmagnesio da lugar al alcohol propargílico 365. Este alcohol propargílico 365 se
trata con dos equivalentes de butillitio, y el anión formado se hace reaccionar con una
nueva molécula del furaldehído protegido 363, dando lugar al diol 366. Este diol se
oxida, a la correspondiente dicetona con bisacetoxiiodobenceno (BAIB) y
2,2,6,6-tetrametilpiperidin-1-oxilo (TEMPO).178 El intento de desprotección de los
sililéteres con fluoruro de tetrabutilamonio produjo instantáneamente la descomposición
de 367. Sin embargo, afortunadamente, el uso de ácido acético-THF-agua en proporción
3:1:1179 permitió obtener el diol 368 con un buen rendimiento, en unas condiciones de
reacción muy suaves.
177 McNelis, B.J.; Sternbach, D. D.; Macphail, A. T. “Synthetic and Kinetic-Studies of Substituent
Effects in the Furan Intramolecular Diels-Alder Reaction”. Tetrahedron 1994, 50, 6767-6782. 178 DeMico, A.; Margarite, R.; Parlati, L.; Vescovi, A.; Pancatelli, G. “A versatile and highly selective
hypervalent iodine (III)/ 2,2,6,6-tetramethyl-1-piperidenyloxyl-mediated oxidation of alcohols to carbonyl compounds”. J. Org. Chem. 1997, 62, 6974-6977.
179 Corey, E. J.; Venkateswarlu, A. “Protection of hydroxyl groups as tert-butyl-dimethylsilyl derivates”. J. Am. Chem. Soc. 1972, 94, 6190-6191.

186 Tesis Doctoral Susana Barriga Falcón
A continuación, se intentó realizar la oxidación del diol 368 para obtener el dialdehído
361, pero no se encontró ningún método que condujera al producto deseado. Los
agentes oxidantes ensayados fueron el óxido de manganeso, BAIB/TEMPO,
Magtrieve®,180 y N-óxido de N-metilmorfolina/perrutenato de tetrapropilamonio.181 Con
todos estos reactivos, en diversas condiciones de reacción, sólo se obtuvo la
descomposición del diol 368, no pudiéndose llegar a obtener el aldehído deseado. Se
pensó, por ello, llevar a cabo la oxidación tras realizar la reacción de cicloadición con la
monoditioltiona 176.
La reacción de cicloadición se realizó en diclorometano a temperatura ambiente, con
catálisis de triflato de escandio en una concentración molar del 50%. Se obtuvo de esta
manera un nuevo monoaducto 369 como un sólido estable de color rojo, en un
rendimiento del 84% (Esquema 3.82).
S
N
S
SS
S
SO
OO
OO
HO OH S
N
S
S
S
O
S
S O
O
OO
OH
HO
Sc(OTf)3
176
+
368 369
CH2Cl2
Esquema 3.82
Se intentó entonces oxidar el aducto 369 con los mismos métodos que los usados para el
diol 368, pero sólo se pudo constatar la descomposición prácticamente inmediata de 369
en todos los casos estudiados.
Con este sustrato no se ha podido conseguir la función aldehído deseada y, por lo tanto,
no han podido introducirse radicales estables, pero se ha llegado a introducir la función
180 Lee, R. A.; Donald, D. S. “MagtrieveTM and efficient, magnetically retrievable and recyclable
oxidant”. Tetrahedron Lett. 1997, 38, 3857-3860. 181 Ley, S. V.; Norman, J.; Grifffith, W. P.; Marsden, S. P. “Tetrapropylammonium Perruthenate,
Pr4N+RuO4
-, TPAP: A Catalytic Oxidant for Organic Synthesis”. Synthesis 1994, 639-666.

Exposición y discusión de resultados 187
alcohol, sobre la cual se pueden realizar también otras transformaciones posteriores. La
investigación en este sentido está en curso de realización.
2.1.10. Cicloadiciones con acetilenos activados por grupos ferrocenilo
S
N
S
SS
S
SX
161, X = S
176, X = 0
Fe
O O
FeR
R
+
Es sobradamente conocida la utilidad de los compuestos organometálicos en la
preparación de nuevos materiales.182 Los metales de transición tienen una serie de
características que permiten modificar las propiedades de las moléculas orgánicas a las
que se unen.
En nuestro grupo teníamos un gran interés en introducir grupos que pudieran modificar
las propiedades electroquímicas de las ditioltionas. El grupo elegido fue el ferroceno,
que es ampliamente utilizado en la ciencia de materiales ya que, además de modificar
las propiedades electroquímicas, puede formar complejos de transferencia de carga,
participar en la formación de radicales estables o formar parte de cristales líquidos
termotrópicos.183 Además, la utilización de ferrocenos con quiralidad planar es una
excelente vía para introducir quiralidad en la molécula.
Las reacciones con ferrocenos se realizaron en el laboratorio del Profesor Olivier Riant,
en la Université de Paris-Sud. Se han usado ferrocenos aquirales y también ferrocenos
quirales, con el fin de estudiar el efecto sobre los confórmeros posibles de la tiazina
176, que recordemos presenta asimetría en el nitrógeno.
En primer lugar el estudio se llevó a cabo con acetilenos activados únicamente
en una parte del triple enlace. Los acetilenos de este tipo elegidos fueron
182 Miller, J. S. “Organometallic- and Organic-based Magnets: New Chemistry and New Materials for
the New Millenium”. Inorg. Chem. 2000, 39, 4392-4408. 183 Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science. Editores: Togni, A.;
Hayashi, T. VCH and John Wiley & Sons, Weinheim and New York. 1995.

188 Tesis Doctoral Susana Barriga Falcón
1-ferrocenil-2-propin-1-ona (370), (R)-1-[α-(p-metoxifenil)ferrocenil]2-propin-1-ona
(371) y 1-[(S)-α-(trimetilsilil)ferrocenil]-2-propin-1-ona (372).
H
O
Fe
370
Fe
372
TMS
O
H
371
Fep-MeOC6H4
O
H
Se utilizó en primer lugar el ferroceno no quiral 370, que se preparó en dos etapas. En la
primera se hizo reaccionar el ferrocencabaldehído comercial (373) con bromuro de
etinilmagnesio, para obtener de esta forma el alcohol propargílico 374. En la segunda
etapa, 374 es oxidado con óxido de manganeso para dar lugar al correspondiente
ferrocenilacetileno 370.
BrMg H
Fe
CHOMnO2
Fe
HO
H
Fe
O
H
373 374 370
85% 92%
Esquema 3.83
Este método es muy sencillo y permite obtener el acetileno 370 en buen rendimiento.
Existen precedentes en la bibliografía de la obtención de 374 y 370, aunque los métodos
de obtención son experimentalmente más difíciles y los rendimientos son menores.184
Los primeros ensayos para llevar a cabo la reacción de cicloadición de la tiazina 176
con el acetileno 370 se realizaron en condiciones térmicas. Tras seis horas de reacción a
reflujo de benceno, la reacción de cicloadición no fue completa, y se observó la
formación de dos productos, uno que correspondía al producto de cicloadición de la
184 (a) Schloegl, K.; Mohar, A. “Über Ferrocen-acetylene, 2. Mitt.: Darstellung und Reaktionen von
Ferrocenyl-alkinen und -alkinyl-ketonen (11. Mitt. über Ferrocenderivate)”. Monatsh. Chem. 1962, 93, 861-876. (b) Schloegl, K.; Steyrer, W. “Ferrocen-acetylene, 5. Mitt.: Eine allgemeine Methode zur Darstellung von Ferrocenyl-acetylenen und -allenen aus Acylferrocenen”. Monatsh. Chem. 1965, 96, 1520-1535. (c) Akiyama, T.; Hattori, T.; Ito, K.; Uryu, T.; Kajitani, M.; Sugimori, A. “Formation of Ethynyl Metallocenyl Ketone from 1,1,1-Trichloro-3-alkoxy-3-metallocenylpropane”. Chem. Lett. 1980, 361-364.

Exposición y discusión de resultados 189
monoditioltiona 176 con el alquino 370, y otro que resultó ser el monoditiolopirrol 182,
que aparecía como consecuencia de la extrusión de azufre del anillo central de la tiazina
de partida.
Se probó entonces realizar esta misma reacción con catálisis de triflato de escandio.
Se usó una concentración molar de triflato de escandio del 50%, y la reacción se hizo en
diclorometano a temperatura ambiente. La reacción resultó ser prácticamente
instantánea, aunque se mantuvo la agitación durante 15 minutos para asegurar que la
reacción se completara.
S
N
S
SS
S
S
+
176
Sc(OTf)3
S
N
SS
S
S
S
O
H
Fe
O Fe
S
N
SS
S
S
S H
O
O
+
O
Fe
O
H
CH2Cl2
370 (E)-375 (Z)-375
Esquema 3.84
En estas condiciones se obtuvo el cicloaducto 375, mezcla de los correspondientes
isómeros Z y E, como un sólido rojo en un rendimiento del 91%.
De esta forma hemos introducido, de forma sencilla, un centro activo redox adicional
sobre la ditioltiona 176, que sin duda es capaz de modificar las propiedades
electroquímicas del sistema.
A continuación se sintetizaron los acetilenos quirales 371 y 372. Estos acetilenos son
especialmente útiles, pues además de permitir modificar las propiedades
electroquímicas de la tiazina 176 original, nos permiten introducir un segundo centro
quiral en la molécula.
Los ferrocenilacetilenos quirales se prepararon con la misma metodología que la
empleada para sintetizar el acetileno no quiral 370 (Esquema 3.85). Los aldehídos

190 Tesis Doctoral Susana Barriga Falcón
quirales de partida, 376 y 377, fueron sintetizados previamente en el grupo del profesor
Riant.185
BrMg H
Fe R
CHO
376, R = p-MeOC6H4
377, R = TMS378, R = p-MeOC6H4 (88%)
379, R = TMS (92%)
MnO2
371, R = p-MeOC6H4 (88%)
372, R = TMS (95%)
Fe R
HO
H
Fe R
O
H
Esquema 3.85
Las reacciones de cicloadición de la tiazina 176 y los acetilenos 371 y 372 se realizaron
usando condiciones análogas a las descritas para la reacción con el acetileno no quiral
370 (Esquema 3.86).
S
N
S
SS
S
S
+
176
Sc(OTf)3
371, R = p-C6H4OMe
372, R = TMS
S
N
SS
S
S
S
O
H
Fe
RO
S
N
SS
S
S
S H
O
O
+
(E)-380, R = p-C6H4OMe
(E)-381, R = TMS
(Z)-380, R = p-C6H4OMe
(Z)-381, R = TMS
O
Fe R
O
H
CH2Cl2
R
Esquema 3.86
La tabla 3.13 recoge los productos obtenidos de la cicloadición de la tiazina 176 con los
distintos ferrocenilacetilenos, así como los rendimientos con los que se han obtenido.
Acetileno Producto obtenido (Rendimiento)
364 375 (91%)
365 380 (87%)
366 381 (85%)
Tabla 3.13: Productos obtenidos entre los acetilenos 370, 371 y 372 y la tiazina 176
Todos estos cicloaductos son sólidos de color rojo muy estables.
185 Riant, O.; Samuel, O.; Flessner, T.; Taudien, S.; Kagan, H. B. “An Efficient Asymmetric Synthesis
of 2-Substituted Ferrocenecarboxaldehydes”. J. Org. Chem. 1997, 62, 6733-6745.

Exposición y discusión de resultados 191
En estos compuestos, al igual que en otros heterociclos obtenidos anteriormente, existe
restricción en la inversión del nitrógeno, lo que hace que los productos 380 y 381 se
obtengan como mezcla de dos diastereoisómeros, ya que se ha introducido un ferroceno
quiral en la molécula. Desgraciadamente, no se pudieron separar los diastereoisómeros
de 380 y 381.
Para evitar el problema de la generación de isómeros geométricos, y también para
introducir un mayor número de centros quirales en la molécula que pudieran facilitar la
separación de diastereoisómeros, se prepararon reactivos de cicloadición acetilénicos
sustituidos de forma simétrica. Así, se sintetizaron los diferrocenilacetilenos 382 y 383
en forma ópticamente pura.
Fe Fe
OO
382
Fe
O O
FeR
R
383, R = p-MeOC6H4
De forma análoga a la obtención de los furanos acetilénicos, se sintetizaron las
dicetonas 382 y 383 a partir de los alcoholes propargílicos 374 y 378. Se realizó el
tratamiento de los mismos con butillitio para formar los correspondientes acetiluros, que
se hacen reaccionar con el aldehído correspondiente. De esta forma, se forman los
dioles 384 y 385 con rendimientos moderados (56% y 62%, respectivamente). Estos
dioles fueron oxidados con óxido de manganeso para obtener las dicetonas 382 y 383 en
rendimientos muy buenos (98% y 82%, respectivamente). La síntesis de estos
compuestos se resume en el esquema 3.87.
374, R = H
378, R = p-MeOC6H4
1) BuLi
FeR
CHO
373, R = H
376, R = p-MeOC6H4
2)
MnO2
382, R = H (98%)
383, R = p-MeOC6H4 (82%)
Fe R
HO OH
384, R = H (56%)
385, R = p-MeOC6H4 (62%)
Fe R
HO
H
Fe
R
Fe R
O O
Fe
R
Esquema 3.87

192 Tesis Doctoral Susana Barriga Falcón
Las dicetonas 382 y 383 se hicieron reaccionar con la tiazina 176 mediante reacciones
de cicloadición. Las reacciones se llevaron a cabo con catálisis de triflato de escandio,
en condiciones análogas a las utilizadas con los acetilenos 370, 371 y 372, es decir, con
una concentración molar del 50% de triflato de escandio en diclorometano, y usando la
cantidad estequiométrica de las dicetonas 382 y 383. La reacción se completó en 15
minutos a temperatura ambiente.
S
N
SS
S
S
S
O
O
O
Fe
R
S
N
S
SS
S
O S
+
Sc(OTf)3
176 386, R = H
387, R = p-C6H4OMe
CH2Cl2
382, R = H (98%)
383, R = p-MeOC6H4 (82%)
Fe R
O O
Fe
R
Fe
R
Esquema 3.88
Al igual que ocurrió en las reacciones con ferrocenilacetilenos quirales
monofuncionalizados 371 y 372, la reacción del diferrocenilacetileno quiral 383 con la
tiazina 176 proporcionó el aducto 387 que fue obtenido como una mezcla inseparable de
diastereoisómeros.
De forma análoga se realizó también la reacción de cicloadición de la bisditioltiona 161
con la dicetona 382, que se completó igualmente en 15 minutos a temperatura ambiente,
proporcionando el aducto 388.
Fe
S
N
S
S
S
O
O
FeS
N
S
SS
S
SS
Fe Fe
OO Sc(OTf)3
382
+
161 388
S
S
S
O
O
Fe
Fe
CH2Cl2
Esquema 3.89
Los resultados obtenidos en estas reacciones de cicloadición se resumen en la
tabla 3.14.

Exposición y discusión de resultados 193
Tiazina Acetileno Producto obtenido (rendimiento)
176 382 386 (97%)
176 383 387 (78%)
161 382 388 (65%)
Tabla 3.14: Productos obtenidos en las reacciones de cicloadición de las dicetonas 382 y 383 con las
tiazinas 176 y 161
Los cicloaductos 386, 387 y 388 son sólidos estables de color rojo.
Se realizó el estudio de las propiedades electroquímicas del aducto 386 mediante
voltametría cíclica. Se utilizó una concentración molar de 5·10-4 M de 386 en
diclorometano a 20 ºC. Se utilizó como electrolito soporte Bu4NPF6 en concentración
aproximada 0.1 M; como electrodo de trabajo, el de bola de platino; como electrodo
auxiliar, un hilo de platino; y como electrodo de referencia, el de calomelanos saturado.
El voltamograma se registró a diferentes velocidades de barrido, observándose siempre
que el proceso era reversible. Se observa que la relación entre la intensidad de la onda
de oxidación y la onda de reducción es constante a las velocidades en las que se ha
realizado el estudio (50 mV/s, 100 mV/s, 200 mV/s y 500 mV/s), así como que la
intensidad es proporcional a la raíz cuadrada de la velocidad de barrido. En la
figura 3.21 se observa el voltamograma registrado a 100 mV/s para el compuesto 386.
Como podemos observar, el potencial de semionda se encuentra a 0.78 V (E0 = 0.78 V).
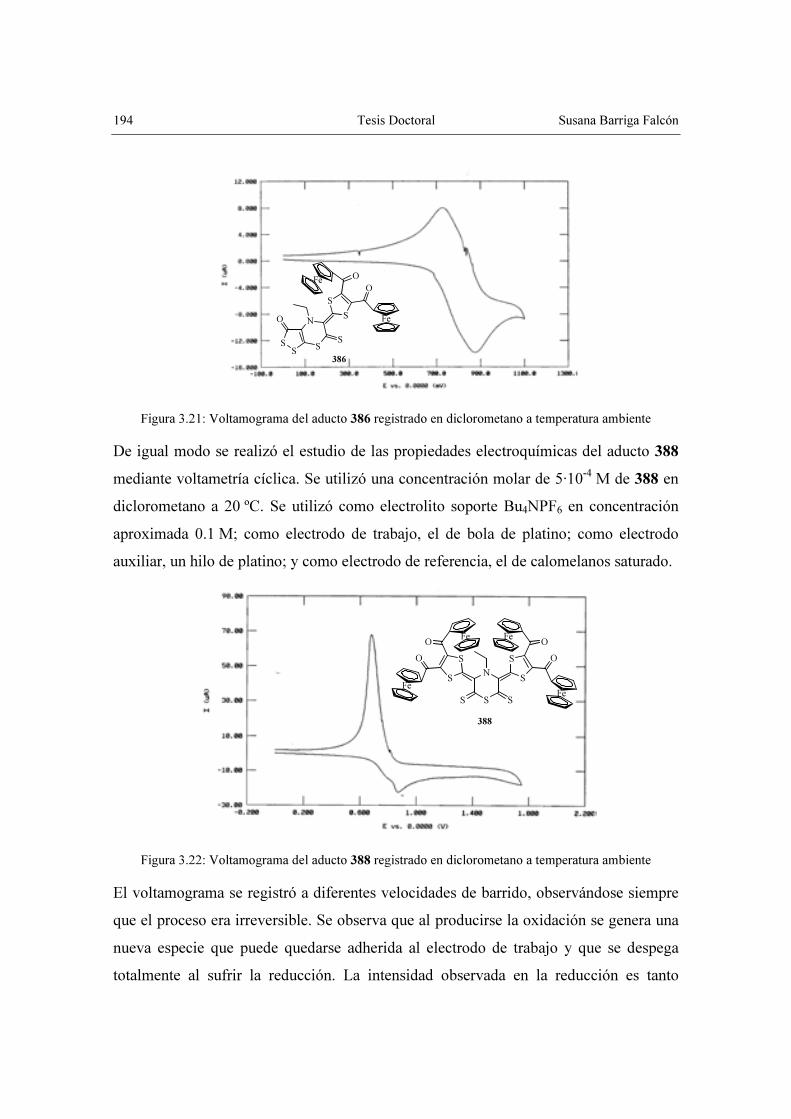
194 Tesis Doctoral Susana Barriga Falcón
Figura 3.21: Voltamograma del aducto 386 registrado en diclorometano a temperatura ambiente
De igual modo se realizó el estudio de las propiedades electroquímicas del aducto 388
mediante voltametría cíclica. Se utilizó una concentración molar de 5·10-4 M de 388 en
diclorometano a 20 ºC. Se utilizó como electrolito soporte Bu4NPF6 en concentración
aproximada 0.1 M; como electrodo de trabajo, el de bola de platino; como electrodo
auxiliar, un hilo de platino; y como electrodo de referencia, el de calomelanos saturado.
Figura 3.22: Voltamograma del aducto 388 registrado en diclorometano a temperatura ambiente
El voltamograma se registró a diferentes velocidades de barrido, observándose siempre
que el proceso era irreversible. Se observa que al producirse la oxidación se genera una
nueva especie que puede quedarse adherida al electrodo de trabajo y que se despega
totalmente al sufrir la reducción. La intensidad observada en la reducción es tanto
S
N
SS
S
S
S
O
O
O
Fe
386
Fe
Fe
S
N
S
S
S
O
O
Fe
388
S
S
S
O
O
Fe
Fe

Exposición y discusión de resultados 195
mayor cuanto mayor sea el tiempo que mantengamos la especie nueva formada en la
zona de oxidaciones. En la figura 3.22 se observa el voltamograma registrado a
100 mV/s para el compuesto 388. Como podemos observar el potencial de pico de la
onda de oxidación se encuentra a 0.84 V (Epox = 0.84 V).
Estos acetilenos, tanto los activados sólo en una parte del triple enlace, como los que
están activados en ambas partes, permiten introducir de forma muy sencilla en las
ditioltionas de partida, núcleos de ferroceno mediante reacciones de cicloadición. Estos
nuevos dipolarófilos sustituidos con ferrocenos deberían reaccionar fácilmente, no sólo
con ditioltionas, sino también frente a otros reactivos 1,3-dipolares y dienos.
2.1.11. Cicloadiciones con acetilenos activados por grupos tienilo
S
N
S
SS
S
OS
176
OO
S
R
S
R+
A continuación decidimos utilizar grupos tienilo sustituidos con cadenas
hidrocarbonadas largas para modificar también las propiedades electroquímicas de la
tiazina 176. Este tipo de sustratos es comúnmente utilizado para realizar procesos de
electropolimerización. Efectivamente, la electropolimerización de monómeros de
pirroles y tiofenos conduce a una deposición rápida y directa del polímero resultante
sobre la superficie del electrodo. Esta técnica permite el control total de las
características del polímero resultante incluyendo conductividad, permeabilidad,
propiedades redox, selectividad frente a otras especies y solubilidad. Además, la
electropolimerización permite obtener películas muy delgadas de polímeros conductores
de una forma controlada. Esto es muy útil para obtener electrodos reproducibles.186
Con esta idea, se pensó en sintetizar un alquino simétrico 394 que contuviera dos grupos
tiofeno sustituidos con cadenas hexilo, con el fin de utilizarlo en reacciones de
186 Genies, E. M.; Bidan, G. J. “Spectroelectrochemical Study of Polypyrrole films”. J. Electroanal.
Chem. 1983, 149, 101-113.

196 Tesis Doctoral Susana Barriga Falcón
cicloadición con ditioltionas. Para ello se partió del 3-hexiltiofeno (389), comercial. Se
introdujo un grupo aldehído en la posición 2 mediante dos etapas. En primer lugar se
introdujo un átomo de bromo en esa posición siguiendo el método de Wang.187 A
continuación se realizó la transmetalación con butillitio, y se usó DMF para introducir
la función aldehído en la posición deseada. Una vez sintetizado el aldehído 391, la ruta
sintética fue similar a la ya descrita anteriormente para otros alquinos. Primero se formó
el alcohol propargílico 392 por tratamiento con bromuro de etinilmagnesio. Este alcohol
393 se trató con dos equivalentes de butillitio y el anión acetiluro resultante se hizo
reaccionar de nuevo con el aldehído 391 para obtener el diol 393, que fue oxidado a
continuación para dar lugar a la dicetona 394.
S
NBS
S Br S CHO
MgBrH
H
HO
S
HO
S
OH
S
BAIB
O
S
O
S
389 390
1) BuLi
2) DMF
391 392
2) 391 TEMPO
393 394
1) BuLi
Esquema 3.90
El acetileno 394 se hizo reaccionar con la tiazina 176 en las condiciones habituales de
catálisis con triflato de escandio. Tras la reacción de cicloadición se obtuvo el
monoaducto 395 en un rendimiento del 50% como un sólido de color rojo estable.
187 Wang, F.; Rauh, R. D.; Rose, T. L. “An electrically conducting star polymer”. J. Am. Chem. Soc.
1997, 119, 11106-11107.

Exposición y discusión de resultados 197
S
N
S
S S
S
O S
176
OO
S S
Sc(OTf)3
S
N
S
S
O
S
S
S O
OS
S
+
394 395
Esquema 3.91
La estructura 395 se corresponde con los datos espectroscópicos obtenidos mediante las
técnicas habituales.
De igual modo se realizó el estudio de las propiedades electroquímicas del aducto 395
mediante voltametría cíclica. Se utilizó una concentración molar de 5·10-4 M de 395 en
diclorometano a 20 ºC. Se utilizó como electrolito soporte Bu4NPF6 en concentración
aproximada 0.1 M; como electrodo de trabajo, el de bola de platino; como electrodo
auxiliar, un hilo de platino; y como electrodo de referencia, el de calomelanos saturado.
En el voltamograma registrado a 100 mV/s se observaron dos ondas en la zona de
oxidaciones cuyos potenciales de pico son 0.81 V y 1.20 V, respectivamente. La
primera onda tiene una de onda de reducción de respuesta cuyo potencial de pico es de
0.55 V. Además, el proceso es irreversible (Figura 3.23).
Figura 3.23: Voltamograma del aducto 395 registrado en diclorometano a temperatura ambiente
S
N
S
S
O
S
S
S O
O
S
S
395

198 Tesis Doctoral Susana Barriga Falcón
Si se repite el proceso de oxidación sucesivas veces se observa que va paulatinamente
desapareciendo la onda que aparecía a 0.81 V, así como su onda respuesta hasta dar un
voltamograma similar al obtenido en el caso del aducto 283.
Figura 3.24: Voltamograma obtenido tras realizar sucesivas oxidaciones sobre el aducto 395
Cuando se registra el voltamograma a diferentes velocidades de barrido se observa
siempre que el proceso es irreversible. La comparación de las intensidades de las ondas
de oxidación y reducción muestra que existe una transferencia electrónica en la que
aparentemente está implicado el mismo número de electrones en ambos procesos,
aunque no se ha usado ni la técnica del electrodo rotatorio ni la columbimetría para
determinar el número de electrones implicados. En la figura 3.24 se observa el
voltamograma registrado a 100 mV/s para el compuesto 395. El potencial de pico en la
onda oxidativa aparece a 1.20 V (Epox = 1.20 V) y el potencial de pico de la onda en las
reducciones aparece a –1.05 V (Epred = -1.05 V).
Podemos hablar de la existencia de un proceso electroquímicamente catalizado, de
forma que cuando la especie existente en disolución, A, se oxida para dar A+, esta
especie oxidada se transforma en una nueva especie, B+, en cantidad catalítica. Esta
nueva especie B+ puede reaccionar con la especie inicial A provocando la generación de
más especie catalítica B+. Este proceso se repite hasta agotar totalmente la especie
inicial A y transformarla completamente en una nueva especie, B (Esquema 3.92).
S
N
S
S
O
S
S
S O
O
S
S
395

Exposición y discusión de resultados 199
A - 1e-
A+
B+
B+
+ A B + A+
Esquema 3.92
2.2. Estudios de reactividad del bencino sobre ditioltionas
S
N
S
SS
S
O S
+
176
Como complemento al estudio de las reacciones con acetilenos activados se ensayaron
dienófilos sin activar, como el ciclohexeno y otros alquenos, pero no resultaron
suficientemente reactivos para dar lugar a productos de cicloadición con las ditioltionas.
El bencino es un dienófilo muy reactivo,188 por lo que nos planteamos estudiar su
comportamiento frente a la tiazina 176.
En primer lugar pensamos en utilizar bencino clásico, sin sustituir, que debería
generarse in situ en presencia de la ditioltiona 176. Para ello pensamos en emplear
algunos de los métodos conocidos de generación de bencino, que se discutirán a
continuación.
El bencino se generó, en un primer intento, a partir del ácido antranílico189 en presencia
de la tiazina 176. En estas condiciones la reacción no dio lugar a la aparición de ningún
producto siguiendo la reacción mediante cromatografía en capa fina, sino que sólo se
observó la descomposición total de la tiazina 176 (Esquema 3.93).
188 “Dehydrobenzene and cycloalkanes” Hoffmann Ed. Academic Press, New York, 1967, pag. 200-239. 189 Kiyoshi, M.; Takane, U.; Kinuyo, A.; Masahiko, N.; Takeshi, K.; Tadashi, O. “Synthesis and
Reactions of 1,2-Fused 3-Cyanoindolizines”. J. Heterocycl. Chem. 1988, 25, 1793-1801.

200 Tesis Doctoral Susana Barriga Falcón
COOH
NH2H3C ONO
CH3
THF
COO
N2
S
N
S
S S
S
O S
ClCl
+
396 397 398
Descomposición176
Esquema 3.93
Seguidamente se intentó generar el bencino usando 1-aminobenzotriazol (400), que se
obtuvo por aminación del benzotriazol (399).190 El bencino se generó in situ mediante
oxidación del 1-aminobenzotriazol (400). Se ensayaron diversos oxidantes como
tetraacetato de plomo,191 N-iodosuccinimida192 y BAIB, pero en ningún caso se
consiguieron resultados satisfactorios en la reacción con la tiazina 176.
NH
N
N
N
N
N
NH2
399 400 253
[O]
Esquema 3.94
La generación de bencino por el método de Himeshima y colaboradores,193 por
tratamiento de triflato de o-trimetilsililfenilo (401) con fluoruro de cesio y paladio
tetraquis(trifenil)fosfina en presencia de la tiazina 176, también condujo a la total
descomposición de ésta (Esquema 3.95).
190 Campbell, C. D.; Rees, C. W. “Reactive Intermediates. Part I. Synthesis and Oxidation of 1- and 2-
Aminobenzotriazole”. J. Chem. Soc. (C) 1969, 742-747. 191 Campbell, C. D.; Rees, C. W. “Reactive Intermediates. Part II. Some Addition Reactions of
Benzyne”. J. Chem. Soc. (C) 1969, 748-751. 192 Birkett, M. A.; Knight, D. W.; Little, P. B.; Mitchell, M. B. “A New Approach to
Dihydrobenzofurans and Dihydrobenzopyrans (Chromans) Based on the Intramolecular Trapping by Alcohols of Benzyne Generated from 7-Substituted-1-aminobenzotriazoles”. Tetrahedron 2000, 56, 1013-1023.
193 Himeshima, Y.; Sonoda, T.; Kobayashi, H. “Fluoride-Induced 1,2-Elimination of Ortho-trimethylsilylphenyl Triflate to Benzine under Mild Conditions”. Chem. Lett. 1983, 1211-1214.

Exposición y discusión de resultados 201
TMS
OTf
Pd(PPh3)4
CsF
S
N
S
SS
S
O S
176Descomposición
401 253
Esquema 3.95
Por último se generó el bencino de acuerdo con el método de Kitamura.194 Este método
utiliza una sal aromática de yodo hipervalente, sustituida en la posición orto con un
grupo hidroxidimetilsilano, que por tratamiento con fluoruro de tetrabutilamonio en
diclorometano a 0 ºC da lugar al bencino en condiciones muy suaves (Esquema 3.96).
Si(OH)Me2
I(Ph)OTf
Bu4NF
402 253
CH2Cl2
Esquema 3.96
Cuando se genera de esta manera el bencino en presencia de la tiazina 176 se observa,
por cromatografía en capa fina, la formación inmediata de un producto de cicloadición.
La reacción es prácticamente instantánea a temperatura ambiente. Tras la purificación se
obtuvo el benzoditiolano 403 en un rendimiento del 71%, como un producto estable de
color rojo (Esquema 3.97).
194 Kitamura, T.; Meng, Z.; Fujiwara, Y. “Synthesis of a new hypervalent iodine compound,
[2-(hydroxydimethylsilyl)phenyl](phenyl)iodonium triflate as a convenient approach to benzyne”. Tetrahedron Lett. 2000, 41, 6611-6614.

202 Tesis Doctoral Susana Barriga Falcón
Si(OH)Me2
I(Ph)OTf
Bu4NFS
N
S
SS
S
O S
176
402 253
S
N
S
S S
O
S
S
403
CH2Cl2
Esquema 3.97
Este método ha abierto una nueva vía de trabajo en la que, en un futuro próximo, se
investigará esta reacción con otras ditioltionas policíclicas. Asimismo, se estudiará la
viabilidad de este método para la preparación de benzinos sustituidos más complejos.

4. PARTE EXPERIMENTAL


Parte experimental 205
1. Procedimientos generales
Las reacciones han sido realizadas bajo atmósfera de nitrógeno, en matraces
previamente flameados y purgados en una corriente de nitrógeno.
Los disolventes utilizados se purificaron siguiendo los procedimientos de D. Perrin195
y
se secaron inmediatamente antes de su uso por destilación en atmósfera inerte sobre un
agente desecante adecuado. Los agentes desecantes utilizados fueron: Na/benzofenona
para el THF, los éteres, el benceno, el tolueno y los xilenos; P2O5 para los disolventes
clorados; CaH2 para las aminas y para la acetona; Mg/I2 para los alcoholes; KOH
seguido de CaH2 para la piridina.
El butillitio fue valorado justo antes de su utilización de acuerdo con el método
propuesto por Kofron.196
Para las reacciones a baja temperatura se emplearon baños de hielo seco con acetona.
Las temperaturas indicadas se refieren a las del baño externo.
Las adiciones de líquidos se realizaron, generalmente, utilizando jeringa o cánula y bajo
atmósfera de nitrógeno. Todas las reacciones se llevaron a cabo con agitación
magnética.
Para el secado de las disoluciones procedentes de la elaboración de las diversas
reacciones se utilizó Na2SO4 anhidro. Las concentraciones se realizaron en un rotavapor
Heidolph modelo Laborota 4000.
Se utilizaron jeringas de plástico (Discardit) y de vidrio (Hamilton) con agujas Llorach-
Luer.
195 Perrin, D. D.; Armarego, W. L. F. “Purification of Laboratory Chemicals”. Ed. Pergamon Press;
Oxford, 1988. 196
Kofron, W. G.; Baclawski, L. M. “A Convenient Method for Estimation of Alkyllithium
Concentrations”. J. Org. Chem. 1976, 41, 1879-1880.

206 Tesis Doctoral Susana Barriga Falcón
La cromatografía de capa fina (c.c.f.) fue realizada en gel de sílice Merck 60 GF-254,
utilizando como reveladores luz ultravioleta de 254 nm, ácido fosfomolíbdico (5%) en
metanol, o bien una mezcla de p-anisaldehído (2.5%), ácido acético (1%) y ácido
sulfúrico (3.4%) en etanol del 95%. Para la cromatografía en columna rápida (flash) se
utilizó gel de sílice 60 (230-400 mesh, Merck). El dispositivo de purificación utilizado
se construyó según las indicaciones de Still.197
La cromatografía de columna de media
presión fue realizada en un aparato Gilson usando gel de sílice Merck 60 (menor de 230
mesh). Las cromatografías preparativas se desarrollaron sobre placas de 20 x 20 cm y 1
mm de espesor.
Los puntos de fusión se determinaron en un aparato Gallenkamp, y no están corregidos.
Los espectros de infrarrojo (IR) fueron realizados en pastilla de bromuro potásico o en
película líquida, en espectrofotómetros Bruker Vector 200 A y Nicolet Impact 410.
Los espectros de resonancia magnética nuclear de protón (RMN-1H) y de carbono
(RMN-13
C) fueron realizados en la disolución del disolvente deuterado que se indica en
cada caso en aparatos Bruker o Varian (200, 250, 300 y 400 MHz) en los respectivos
Servicios de Resonancia Magnética Nuclear de las universidades de Salamanca,
Valladolid, Santiago de Compostela, Burgos, Extremadura, Paris-Sud, Catholique de
Louvain y del Imperial College of Science, Technology and Medicine de Londres. Los
espectros fueron transformados en los propios equipos o con el programa MestRe-C
2.3.198
Los espectros de masas (EM) fueron realizados en un espectrómetro VG7070E y VG-
AutoSpec, con impacto electrónico a una energía de 70 eV.
Los análisis elementales fueron realizados con un Perkin Elmer 240-B.
197 Still, W. C.; Kahn, M.; Mitra, A. “Rapid Chromatographic Technique for Preparative Separations
with Moderate Resolution”. J. Org. Chem. 1978, 43, 2923-2925. 198
MestRe-C 2.3. Copyright 1995-2000. Departamento de Química Orgánica. Universidad de Santiago
de Compostela. Programadores: Sardina, F. J.; Cruces, J.; Cobas, C.

Parte experimental 207
Los estudios electroquímicos se realizaron en un equipo EG&G VersaStat, utilizando
una celda de tres electrodos. Como electrodo de trabajo se utilizó el de bola de platino;
como electrodo auxiliar, un hilo de platino; y como electrodo de referencia, el de
calomelanos saturado.
En todos los casos, se utilizó como referencia interna el ferroceno o el
decametilferroceno (dependiendo de la zona de potenciales objeto de estudio),
asignando a las referencias los potenciales que les corresponden según el disolvente
utilizado. Como electrolito soporte, se utilizó siempre la sal Bu4NPF6 en concentración
aproximada 0.1 M.
Las disoluciones de los compuestos en estudio se prepararon en diclorometano,
trabajando con concentraciones de 5·10-4
M.
Los cálculos moleculares se realizaron con el programa HyperChem 5.11 , utilizando
el método PM3.

208 Tesis Doctoral Susana Barriga Falcón
2. Reacción de cicloadición de la bisditiolotiazina 176 con etoxicarbonilazida
S
N
S
SS
S
OS
257176
S
N
S
SS
S
ONO N
O
N N
O
O
C6H6 , 37%
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(35 mg, 0.108 mmol) en benceno (5 mL) se añadió etoxicarbonilazida141
(31 mg, 0.270 mmol). La mezcla de reacción se calentó a reflujo durante 12 horas. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano), lo que permitió separar la ditiolimina 257 pura (15 mg, 0.040 mmol, 37%),
como un sólido anaranjado; p.f.: 191-192 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 1653 (C=O st), 1614 (C=N st), 1519, 1454, 1370, 1299, 1255.
RMN-1H (CDCl3, 400 MHz) δ: 4.33 (c, J = 7.2 Hz, 2 H, NCH2CH3), 3.97 (c, J = 7.2 Hz,
2 H, OCH2), 1.38 (t, J = 7.2 Hz, 3 H, NCH2CH3) y 1.28 (t, J = 7.2 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 182.45 (C=O), 174.82 (C=N), 165.44 (C=O éster),
148.39 (C sp2), 147.74 (C sp
2), 139.62 (C sp
2), 136.98 (C sp
2), 63.60 (OCH2),
44.16 (NCH2CH3), 14.38 (NCH2CH3) y 14.19 (OCH2CH3) ppm.
EM (IE) m/z (%): 378 (M+·, 100), 363 (M
+ - CH3, 78), 345 (20), 333 (M
+ - OCH2CH3, 5), 317
(M+ - CH2CH3 - S, 17), 291 (M
+ - NCO2CH2CH3, 20), 277 (25), 245 (43), 45 (OCH2CH3
+,43).
Masa exacta: calculada para C11H10N2O3S5: 377.9295; hallada: 377.9307
141 Froeyen, P. “A particulary convenient one-pot synthesis of N-alkoxycarbonyl, N-acyl and N-aroyl
substituted imino phosphoranes; improved preparations of azidoformates, aroyl and alkanoyl azides;
an alternative route to complex amides”. Phosphorus, Sulfur Silicon Relat. Elem. 1993, 78, 161-172.

Parte experimental 209
257
S
N
S
SS
S
ON
O
O

210 Tesis Doctoral Susana Barriga Falcón
257
S
N
S
SS
S
ON
O
O

Parte experimental 211
3. Reacción de cicloadición del bisditiolopirrol 182 con etoxicarbonilazida
N
SSS
S
SON
SSS
S
NOO N
O
N NO
OTolueno, 67%
182 264
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-5-tiona
(30 mg, 0.103 mmol) en tolueno (5 mL) se añadió etoxicarbonilazida141
(27 mg, 0.232 mmol). La mezcla de reacción se calentó a reflujo durante 4 horas. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, diclorometano - éter de petróleo
2:1, hasta diclorometano), lo que permitió separar el compuesto 264 puro
(24 mg, 0.069 mmol, 67%), como un sólido amarillo; p.f.: 123-124 ºC (CH2Cl2).
IR (KBr, cm-1
): 1656 (C=O st), 1649 (C=O st, heterociclo), 1630 (C=N st), 1529, 1426,
1275, 1263.
RMN-1H (CDCl3, 400 MHz) δ: 4.94 (c, J = 7.1 Hz, 2 H, NCH2CH3),
4.39 (c, J = 7.1 Hz, 2 H, OCH2) y 1.43 (t, J = 7.1 Hz, 6 H, NCH2CH3 y OCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 181.13 (C=O), 171.85 (C=N), 165.07 (C=O éster),
140.11 (C sp2), 135.34 (C sp
2), 130.78 (C sp
2), 130.02 (C sp
2), 63.69 (OCH2),
40.59 (NCH2CH3), 17.14 (NCH2CH3) y 14.43 (OCH2CH3) ppm.
EM (IE) m/z (%): 346 (M+·
, 40), 273 (M+ - CO2CH2CH3, 40),
258 (M+ + 1 - NCO2CH2CH3, 40), 245 (M
+ + 1 - CO2CH2CH3 - CH2CH3, 58),
241 (M+ -CO2 CH2CH3 - S, 56), 208 (50), 181 (43), 94 (80), 84 (100), 64 (85).
Masa exacta: calculada para C11H10N2O3S4: 345.9574; hallada: 345.9576.

212 Tesis Doctoral Susana Barriga Falcón
N
SSS
S
NO
O
O
264

Parte experimental 213
N
SSS
S
NO
O
O
264

214 Tesis Doctoral Susana Barriga Falcón
4. Reacción de cicloadición del bisditiolopirrol 181 con etoxicarboniloazida
N
SSS
S
SSN
SSS
S
NNO N
O
N NO
O
O
OTolueno, 93%
181 265
Sobre una disolución agitada de 4-etil-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-3,5-ditiona
(50 mg, 0.163 mmol) en tolueno (5 mL) se añadió etoxicarbonilazida141
(94 mg, 0.815 mmol). La mezcla de reacción se calentó a reflujo durante 4 horas. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano), lo que permitió separar el compuesto 265 puro
(63 mg, 0.151 mmol, 93%), como un sólido amarillo; p.f.: 145-146 ºC (CH2Cl2).
IR (KBr, cm-1
): 1626 (C=N st), 1433, 1426, 1315, 1284, 1269.
RMN-1H (CDCl3, 400 MHz) δ: 5.31 (c, J = 6.9 Hz, 2 H, NCH2CH3),
4.40 (c, J = 7.1 Hz, 4 H, 2 OCH2), 1.45 (t, J = 6.9 Hz, 3 H, NCH2CH3) y
1.44 (t, J = 7.1 Hz, 6 H, 2 CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 171.81 (C=N), 165.20 (C=O), 140.54 (C sp2),
130.93 (C sp2), 63.67 (OCH2), 41.63 (NCH2CH3), 17.10 (NCH2CH3) y
14.49 (OCH2CH3) ppm.
EM (IE) m/z (%): 417 (M+·
, 90), 372 (M+ - OCH2CH3, 10), 344 (M
+ - CO2CH2CH3, 20),
239 (M+ - 2 CO2CH2CH3 - S, 100), 211(60), 208 (50), 180 (43), 94 (75), 84 (73), 49 (75).
Masa exacta: calculada para C14H15N3O4S4: 416.9945; hallada: 416.9936.
Análisis: Calculado para C14H15N3O4S4: C, 40.27; H, 3.62; N, 10.06; hallado: C, 40.38;
H, 3.59; N, 9.84.

Parte experimental 215
N
SSS
S
NN
O
O
O
O
265

216 Tesis Doctoral Susana Barriga Falcón
N
SSS
S
NN
O
O
O
O
265

Parte experimental 217
5. Reacción de cicloadición de la bisditiolotiazina 176 con N-(αααα-cloro-benciliden)-
N’-fenilhidrazona (270a)
S
N
S
S S
S
S O
S
N
S
S
OEt3N
S
S
NN
Ph N
HN
Ph
Cl
Ph
Ph+
C6H6, 67%
270a 176 274a
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(50 mg, 0.155 mmol) en benceno (5 mL) se añadió N-(α-cloro-benciliden)-N’-
fenilhidrazona199
(89 mg, 0.387 mmol). Sobre esta mezcla se añadió lentamente trietilamina
(39 mg, 54 µL, 0.387 mmol). A continuación la mezcla resultante se calentó a reflujo
durante una hora y después se eliminó el disolvente en el rotavapor. El crudo obtenido se
purificó mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano 5:1), lo que
permitió separar el compuesto 274a puro (50 mg, 0.104 mmol, 67%), como un sólido rojo;
p.f.: 161-162 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2959, 2926, 2855, 1736 (C=O, st), 1262 (C=S, st).
RMN-1H (CDCl3, 400 MHz) δ: 7.90-7.87 (m, 5 H, C6H5), 7.55-7.47 (m, 5 H, C6H5),
3.04 (dc, J = 14.2 y 7.1 Hz, 1 H, CH2), 2.60 (dc, J = 14.2 y 7.1 Hz, 1 H, CH2) y
1.01 (t, J = 7.11 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 185.22 (C=S), 182.31 (C=O), 154.96 (C=N),
153.36 (C sp2), 152.10 (NC Ar), 140.44 (C sp
2), 131.87 (C sp
2), 131.69 (N=C-C Ar),
129.66 (C sp2), 129.31 (CH Ar), 129.23 (CH Ar), 128.60 (CH Ar), 127.37 (C-C=N),
123.85 (CH Ar), 121.32 (CH Ar), 47.75 (NCH2CH3) y 12.71 (NCH2CH3) ppm.
EM (IE) m/z (%): 485 (M+·
, 48), 456 (M+ - CH2CH3, 100).

218 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C21H15N3OS5: 484.9819 encontrada: 484.9815.
Análisis: Calculado para C21H15N3OS5: C, 51.93; H, 3.11; N, 8.65; hallado: C, 51.97;
H, 2.89; N, 8.42.
199
Sui, Z.; Guan, J.; Ferro, M. P.; McCoy, K.; Wachter, M. P.; Murray, W. V.; Singer, M.; Steber, M.;
Ritchie, D. M.; Argentieri, D. C. “1,3-Diarylcycloalkanopyrazoles and Diphenyl Hydrazides as
Selective Inhibitors of Cyclooxygenase-2”. Bioorg. Med. Chem. Lett. 2000, 10, 601-604.

Parte experimental 219
S
N
S
S
O
S
S
NN
Ph
Ph
274a

220 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NN
Ph
Ph
274a
Parte experimental 221
6. Cicloadición de la bisditiolotiazina 176 con N-(αααα-cloro-p-clorobenciliden)-
N’-(p-clorofenil)hidrazona (270b)
S
N
S
S S
S
S O
S
N
S
S
O
Et3N
S
N
HN
Cl
ClCl S
NN
Cl
Cl
+
C6H6, 77%
270b 176 274b
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(40 mg, 0.124 mmol) en benceno (5 mL) se añadió N-(α-cloro-p-clorobenciliden)-
N’-(p-clorofenil)hidrazona199
(93 mg, 0.31 mmol). Sobre esta mezcla se añadió lentamente
trietilamina (31 mg, 43 µL, 0.31 mmol). A continuación la mezcla resultante se calentó a
reflujo durante una hora y después se eliminó el disolvente en el rotavapor. El crudo
obtenido se purificó mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano
4:1), lo que permitió separar el compuesto 274b puro (53 mg, 0.096 mmol, 77%), como un
sólido rojo violáceo; p.f.: 229-230 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2961, 2924, 2853, 1638 (C=O, st), 1310, 1089.
RMN-1H (CDCl3, 400 MHz) δ: 7.81 (d, J = 7.2 Hz, 2 H, 2 CH Ar),
7.48 (d, J = 7.2 Hz, 2 H, 2 CH Ar), 7.44-7.40 (m, 4 H, 2 CH Ar),
3.02 (dc, J = 14.3 y 7.1 Hz, 1 H, CH2), 2.56 (dc, J = 14.3 y 7.1 Hz, 1 H, CH2) y
1.01 (t, J = 7.1 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 186.92 (C=S), 182.34 (C=O), 154.26 (C=N),
152.30 (C sp2), 151.88 (NC Ar), 138.61 (C sp
2), 138.01 (C sp
2), 135.64 (N=C-C Ar),
131.66 (C sp2), 129.62 (CH Ar), 129.37 (CH Ar), 128.47 (CH Ar), 127.04 (C-C=N),
125.03 (CH Ar), 121.29 (CH Ar), 47.96 (NCH2CH3) y 12.54 (NCH2CH3) ppm.
EM (IE) m/z (%): 557 (M+ + 4, 15), 555 (M
+ + 2, 43), 553 (M
+·, 46),
528 (M+ + 4 - CH2CH3, 30), 526 (M
+ + 2 - CH2CH3, 93), 524 (M
+- CH2CH3, 100).

222 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C21H13N3OS5Cl2: 552.9039 encontrada: 552.9047.

Parte experimental 223
S
N
S
S
O
S
S
NN
Cl
Cl
274b

224 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NN
Cl
Cl
274b

Parte experimental 225
7. Cicloadición de la bisditiolotiazina 176 con N-(αααα-cloro-p-bromobenciliden)-
N’-(p-bromofenil)hidrazona (270c)
S
N
S
S S
S
S O
S
N
S
S
O
Et3N
S
N
HN
Cl
BrBr S
NN
Br
Br
+
C6H6, 78%
270c 176 274c
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(45 mg, 0.139 mmol) en benceno (5 mL) se añadió N-(α-cloro-p-bromobenciliden)-
N’-(p-bromofenil)hidrazona200
(135 mg, 0.348 mmol). Sobre esta mezcla se añadió
lentamente trietilamina (35 mg, 49 µL, 0.348 mmol). A continuación la mezcla resultante se
calentó a reflujo durante una hora y después se eliminó el disolvente en el rotavapor. El
crudo obtenido se purificó mediante cromatografía rápida (SiO2, éter de petróleo-
diclorometano 1:1), lo que permitió separar el compuesto 274c puro (70 mg, 0.109 mmol,
78%), como un sólido morado; p.f.: 251-252 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2961, 2923, 2853, 1643 (C=O, st), 1352.
RMN-1H (CDCl3, 400 MHz) δ: 7.74 (d, J = 7.2 Hz, 2 H, 2 CH Ar),
7.64 (d, J = 7.2 Hz, 2 H, 2 CH Ar), 7.56 (d, J = 8.4 Hz, 2 H, 2 CH Ar),
7.33 (d, J = 8.4 Hz, 2 H, 2 CH Ar), 3.02 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3),
2.56 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3) y 1.01 (t, J = 7.1 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 187.06 (C=S), 182.28 (C=O), 154.14 (C=N),
152.22 (C sp2), 151.97 (NC Ar), 139.12 (C sp
2), 132.58 (CH Ar), 132.33 (CH Ar),
132.19 (N=CC Ar), 131.66 (C Ar), 128.60 (CH Ar), 127.48 (C sp2),
200 Rivett, D. E.; Rosevear, J.; Wilshire, J. F. K. “The Preparation and Spectroscopic Properties of Some
Di- and Tri-substituted 1,3,5-Triphenyl-2-pyrazolines and Related 2-Pyrazolines”. Aust. J. Chem.
1983, 36, 1649-1658.

226 Tesis Doctoral Susana Barriga Falcón
126.39 (CH Ar), 125.26 (C Ar), 123.70 (C sp2), 121.32 (C Ar), 47.96 (NCH2CH3) y
12.55 (NCH2CH3) ppm.
EM (IE) m/z (%): 645 (M+ + 4, 32), 643 (M
+ + 2, 53), 641 (M
+·, 22), 616 (M
+ + 4 - CH2CH3,
100), 614 (M+ + 2 - CH2CH3, 100), 612 (M
+ - CH2CH3, 50), 598 (M
+ + 2 - OCH2CH3).
Masa exacta: calculada para C21H13N3OS5Br2: 644.7988 encontrada: 644.7973.

Parte experimental 227
S
N
S
S
O
S
S
NN
Br
Br
274c

228 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NN
Br
Br
274c

Parte experimental 229
8. Síntesis de N-(p-iodobenzoil)-N’-(p-iodofenil)hidrazina (268d)
I I I
NH
HN
I
Cl
HN
H2N
O OC5H5N
+
C6H6, 93%
266d 267d 268d
Sobre una disolución agitada de p-iodofenilhidrazina (1.5 g, 6.41 mmol) y piridina
(4mL) en benceno (6 mL) se añadió lentamente cloruro de 4-iodobenzoilo
(1.0 g, 3.76 mmol) disuelto en benceno (5 mL). La mezcla de reacción se calentó a
reflujo durante 30 minutos. A continuación se vertió agua (100 mL) sobre la mezcla de
reacción; seguidamente se separaron las fases y la fase acuosa se extrajo con
diclorometano (3 x 20 mL). Los extractos orgánicos combinados se lavaron con una
disolución acuosa saturada de carbonato sódico (2 x 20 mL), y a continuación con una
disolución de ácido clorhídrico al 10% v/v (2 x 20 mL). Finalmente la fase orgánica se
secó sobre sulfato sódico y el disolvente se eliminó en el rotavapor. El crudo obtenido,
268d, sólido de color marrón, (1.62 g, 3.50 mmol, 93%) se utilizó sin purificación en la
siguiente etapa; p.f.: 210-211ºC (benceno).
IR (KBr, cm-1
): 3346 y 3238 (NH, st), 1660 (C=O, st), 1589, 1581 y 1532 (C6H4, st),
1061 (C-I, st).
RMN-1H (DMSO-d6, 200 MHz) δ: 7.89 (d, J = 8.3 Hz, 2 H, 2 CH Ar),
7.67 (d, J = 8.3 Hz, 2 H, 2 CH Ar), 7.44 (d, J = 8.6 Hz, 2 H, 2 CH Ar) y
6.61 (d, J = 8.6 Hz, 2 H, 2 CH Ar) ppm.
EM (IE) m/z (%): 464 (M+·
, 24), 247 (M+ - HNC6H4I, 2), 234 (M
+ - HNHNC6H4I, 100).

230 Tesis Doctoral Susana Barriga Falcón
I
NH
HN
I
O
268d

Parte experimental 231
I
NH
HN
I
O
268d

232 Tesis Doctoral Susana Barriga Falcón
9. Síntesis de N-(αααα-cloro-p-iodobenciliden)-N’-(p-iodofenil)hidrazona (270d)
NH
HN
I I
O
N
HN
I I
ClPCl5
ClCH2CH2Cl, 8%
268d 270d
Sobre una disolución agitada de N-(p-iodobenzoil)-N´-(p-iodofenil)hidrazina
(1.5 g, 3.23 mmol) en 1,2-dicloroetano (20 mL) se añadió pentacloruro de fósforo
(0.81 g, 3.88 mmol). La mezcla de reacción se mantuvo a reflujo durante toda la noche.
Después se dejo enfriar la disolución y se eliminó el disolvente en el rotavapor. El crudo
obtenido se purificó mediante cromatografía rápida (SiO2, desde éter de petróleo hasta
éter de petróleo-diclorometano 4:1), lo que permitió obtener la clorohidrazona 270d
pura (125 mg, 0.26 mmol) como un sólido amarillo; p.f.: 185-186ºC (eter de petróleo-
diclorometano).
IR (KBr, cm-1
): 3244 (N-H, st), 2956, 2924 y 2853 (C-H, st), 1655 (C=N, st), 1600 y
1500 (CH Ar, st).
RMN-1H (CDCl3, 200 MHz), δ: 7.77 (d, J = 8.2 Hz, 2 H, 2 CH Ar),
7.55 (d, J = 8.2 Hz, 2 H, 2 CH Ar), 7.42 (d, J = 8.6 Hz, 2 H, 2 CH Ar) y
7.33 (d, J = 8.6 Hz, 2 H, 2 CH Ar) ppm.
EM (IE) m/z (%): 482 (M+·
, 4), 280 (M+ - C6H4I, 100), 203 (M
+ - NHNCClC6H4I, 9).

Parte experimental 233
N
HN
I I
Cl
270d

234 Tesis Doctoral Susana Barriga Falcón
N
HN
I I
Cl
270d

Parte experimental 235
10. Cicloadición de la bisditiolotiazina 176 con N-(αααα-cloro-p-iodobenciliden)-
N’-(p-iodofenil)hidrazona (270d)
S
N
S
S S
S
S O
S
N
S
S
O
Et3N
S
N
HN
Cl
II S
NN
I
I
+
C6H6, 88%
270d 176 274d
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en benceno (5 mL) se añadió N-(α-cloro-p-iodobenciliden)-
N’-(p-iodofenil)hidrazona (112 mg, 0.232 mmol). Sobre esta mezcla se añadió lentamente
trietilamina (24 mg, 33 µL, 0.237 mmol). A continuación la mezcla resultante se calentó a
reflujo durante una hora y después se eliminó el disolvente en el rotavapor. El crudo
obtenido se purificó mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano
4:1), lo que permitió separar el compuesto 274d puro (60 mg, 0.082 mmol, 88%), como un
sólido violeta; p.f.: 235-236 ºC (CH2Cl2-éter de petróleo, descomposición).
IR (KBr, cm-1
): 2960, 2921, 2851, 1647 (C=O, st), 1350.
RMN-1H (CDCl3, 200 MHz) δ: 7.84 (d, J = 8.6 Hz, 2 H, 2 CH Ar),
7.77 (d, J = 8.6 Hz, 2 H, 2 CH Ar), 7.58 (d, J = 8.6 Hz, 2 H, 2 CH Ar),
7.29 (d, J = 8.6 Hz, 2 H, 2 CH Ar), 3.02 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3),
2.54 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3) y 1.00 (t, J = 7.1 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (C5D5N, 50 MHz) δ: 186.06 (C=S), 182.58 (C=O), 154.55 (C=N),
152.53 (C sp2), 152.29 (C Ar), 140.38 (C sp
2), 138.56 (CH Ar), 138.36 (CH Ar),
132.13 (CH Ar), 128.83 (C Ar), 128.22 (C sp2), 126.20 (C sp
2), 121.39 (CH Ar),
99.50 (CI Ar), 96.27 (CI Ar), 47.93 (NCH2CH3) y 12.25 (NCH2CH3) ppm.
EM (FAB+) m/z (%): 738 (M
+ + 1, 36), 737 (M
+·, 35), 708 (M
+ - CH2CH3, 24),
447 (M+ + 1 - C8H5NOS5, 92).

236 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C21H14I2N3OS5 (M+ + 1): 737.7830 encontrada: 737.7858.

Parte experimental 237
S
N
S
S
O
S
S
NN
I
I
274d

238 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NN
I
I
274d

Parte experimental 239
11. Síntesis de N-(p-cianobenzoil)-N’-(p-cianofenil)hidrazina (268e)
NC CN NC
NH
HN
CN
Cl
HN
H2N
O OC5H5N
+
266e 267e 268e
C6H6, 90%
Sobre una disolución agitada de p-cianofenilhidrazina (1.33 g, 10.00 mmol) y piridina
(4mL) en benceno (6 mL) se añadió lentamente cloruro de 4-cianobenzoilo
(1.0 g, 6.04 mmol) disuelto en benceno (5 mL). La mezcla de reacción se calentó a
reflujo durante 30 minutos. A continuación se vertió agua (100 mL) sobre la mezcla de
reacción; seguidamente se separaron las fases y la fase acuosa se extrajo con
diclorometano (3 x 20 mL). Los extractos orgánicos combinados se lavaron con una
disolución acuosa saturada de carbonato sódico (2 x 20 mL), y a continuación con una
disolución de ácido clorhídrico al 10% v/v (2 x 20 mL). Finalmente la fase orgánica se
secó sobre sulfato sódico y el disolvente se eliminó en el rotavapor. El crudo obtenido,
268e, sólido de color marrón, (1.43 g, 5.45 mmol, 90%) se utilizó sin purificación en la
siguiente etapa; p.f.: 205-206ºC (benceno).
IR (KBr, cm-1
): 3281 (NH, st), 2222 (CN, st), 1641 y 1605 (C=O, st), 1542, 1536 y
1509 (CH Ar, st).
RMN-1H (CD3OD, 200 MHz) δ: 8.13 (d, J = 8.5 Hz, 2 H, 2 CH Ar),
7.97 (d, J = 8.5 Hz, 2 H, 2 CH Ar), 7.88 (s, NH), 7.61 (d, J = 9.00 Hz, 2 H, 2 CH Ar) y
7.00 (d, J = 9.00 Hz, 2 H, 2 CH Ar) ppm.
EM (IE) m/z (%): 262 (M+·
, 26), 130 (NCC6H4CO+, 100), 102 (NCC6H4
+, 41).
240 Tesis Doctoral Susana Barriga Falcón
NC
NH
HN
CN
O
268e

Parte experimental 241
NC
NH
HN
CN
O
268e

242 Tesis Doctoral Susana Barriga Falcón
12. Síntesis de N-(αααα-cloro-p-cianobenciliden)-N’-(p-cianofenil)hidrazona (270e)
NH
HN
NC CN
O
N
HN
NC CN
ClPCl5
ClCH2CH2Cl, 2%
268e 270e
Sobre una disolución agitada de N-(p-cianobenzoil)-N’-(p-cianofenil)hidrazina
(1.43 g, 5.45 mmol) en 1,2-dicloroetano (20 mL) se añadió pentacloruro de fósforo
(1.37 g, 6.60 mmol). La mezcla de reacción se mantuvo a reflujo durante toda la noche.
Después se dejo enfriar la disolución y se eliminó el disolvente en el rotavapor. El crudo
obtenido se purificó mediante cromatografía rápida (SiO2, desde éter de petróleo hasta
diclorometano), lo que permitió obtener la clorohidrazona 270e pura
(33 mg, 0.12 mmol, 2%) como un sólido amarillo de p.f.: 234-235ºC (diclorometano).
IR (KBr, cm-1
): 3280 (N-H, st), 2220 (CN, st), 1610 (C=O, st), 1585, 1572 y
1552 (CH Ar, st).
RMN-1H (CDCl3, 200 MHz), δ: 8.40 (s ancho, 1H, NH),
8.04 (d, J = 8.8 Hz, 2 H, 2 CH Ar), 7.72 (d, J = 8.8 Hz, 2 H, 2 CH Ar),
7.62 (d, J = 8.9 Hz, 2 H, 2 CH Ar) y 7.25 (d, J = 8.9 Hz, 2 H, 2 CH Ar) ppm.
EM (IE) m/z (%): 282 (M++2, 16), 280 (M
+·, 46), 244 (M
+ - Cl, 24),
116 (NCC6H4N+, 100).

Parte experimental 243
N
HN
NC CN
Cl
270e

244 Tesis Doctoral Susana Barriga Falcón
N
HN
NC CN
Cl
270e

Parte experimental 245
13. Cicloadición de la bisditiolotiazina 176 con N-(αααα-cloro-p-cianobenciliden)-
N’-(p-cianofenil)hidrazona (270e)
S
N
S
S S
S
S O
S
N
S
S
O
Et3N
S
N
HN
Cl
CNNC S
NN
CN
NC
+
C6H6, 95%
270e 176 274e
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en benceno (5 mL) se añadió N-(α-cloro-p-cianobenciliden)-
N’-(p-cianofenil)hidrazona (65 mg, 0.232 mmol). Sobre esta mezcla se añadió lentamente
trietilamina (24 mg, 33 µL, 0.237 mmol). A continuación la mezcla resultante se calentó a
reflujo durante una hora y después se eliminó el disolvente en el rotavapor. El crudo
obtenido se purificó mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano
1:1), lo que permitió separar el compuesto 274e puro (47 mg, 0.088 mmol, 95%), como un
sólido rojo; p.f.: 279-280 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2956, 2923, 2852, 2227 (CN, st), 1654 (C=O, st).
RMN-1H (CDCl3, 200 MHz) δ: 7.98 (d, J = 9.0 Hz, 2 H, 2 CH Ar),
7.79 (d, J = 9.0 Hz, 2 H, 2 CH Ar), 7.75 (d, J = 8.4 Hz, 2 H, 2 CH Ar),
7.29 (d, J = 8.4 Hz, 2 H, 2 CH Ar), 2.96 (dc, J = 13.5 y 6.8 Hz, 1 H, NCH2CH3),
2.57 (dc, J = 13.5 y 6.8 Hz, 1 H, NCH2CH3) y 1.01 (t, J = 6.8 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (C5D5N, 50 MHz) δ: 188.19 (C=S), 183.08 (C=O), 154.20 (C=N), 151.29,
143.84, 133.94, 133.19, 132.65, 131.94, 129.05, 128.01, 126.39, 125.40, 118.86 y 118.37
(C sp2, CH Ar y C Ar) , 114.81 (CN), 113.37 (CN), 48.05 (CH2) y 12.17 (CH3) ppm.
EM (IE) m/z (%): 535 (M+·
, 10), 506 (M+ - CH2CH3, 23), 418 (M
+ - NC6H4CN, 93),
262 (M+ - C15H8N4O, 56).

246 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C23H13N5OS5: 534.9724 encontrada: 534.9716.

Parte experimental 247
S
N
S
S
O
S
S
NN
CN
NC
274e

248 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NN
CN
NC
274e

Parte experimental 249
14. Cicloadición de la bisditiolotiazina 176 con N-(αααα-cloro-2,4-diclorobenciliden)-
N’-(2,4-diclorofenil)-hidrazona (270f)
S
N
S
S S
S
S O
S
N
S
S
O
Et3N
S
N
HN
Cl
ClCl S
NN
Cl
Cl
ClClCl
Cl
+
C6H6, 35%
270f 176 274f
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en benceno (5 mL) se añadió N-(α-cloro-2,4-diclorobenciliden)-
N’-(2,4-diclorofenil)hidrazona201
(90 mg, 0.244 mmol). Sobre esta mezcla se añadió
lentamente trietilamina (25 mg, 34 µL, 0.244 mmol). A continuación la mezcla resultante se
calentó a reflujo durante una hora y después se eliminó el disolvente en el rotavapor. El
crudo obtenido se purificó mediante cromatografía rápida (SiO2, éter de petróleo-
diclorometano 17:3), lo que permitió separar el compuesto 274f puro
(20 mg, 0.033 mmol, 35%), como un sólido rojo; p.f.: 93-94 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2959, 2924, 2854, 1655 (C=O, st), 1346, 1326.
RMN-1H (CDCl3, 200 MHz) δ: 7.89 (d, J = 4.1 Hz, ½ H, ½ CH Ar),
7.85 (d, J = 4.1 Hz, ½ H, ½ CH Ar), 7.69-7.19 (m, 5 H, 5 CH Ar),
3.09 (dc, J = 14.2 y 7.1 Hz, ½ H, ¼ NCH2CH3), 2.90 (dc, J = 14.2 y 7.1 Hz, ½ H, ¼ NCH2CH3),
2.87 (dc, J = 14.2 y 7.1 Hz, ½ H, ¼ NCH2CH3), 2.56 (dc, J = 14.2 y 7.1, ½ H, ¼ NCH2CH3),
1.04 (t, J = 7.1 Hz, 1.5 H, ½ NCH2CH3) y 1.03 (t, J = 7.1 Hz, 1.5 H, ½ NCH2CH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 186.89 (C=S), 182.97 (C=O), 156.13 (C=N),
153.14, 149.43, 138.04, 136.46, 133.61, 132.43, 131.76, 131.00, 130.77, 130.35,
128.25, 127.89, 127.65, 126.22, 121.60 y 121.14 (C sp2, CH Ar y C Ar), 49.47 y 49.29
(CH2), 12.36 y 12.03 (CH3) ppm.

250 Tesis Doctoral Susana Barriga Falcón
EM (IE) m/z (%): 627 (M+ + 6, 3), 625 (M
+ + 4, 8), 623 (M
+ + 2, 15), 621 (M
+·, 9),
531 (M+ + 4 - CH2CH3, 21), 529 (M
+ + 2 - CH2CH3, 33), 527 (M
+ - CH2CH3, 22),
175 (M+ + 4 - C6H8N2OS5Cl2, 14), 173 (M
+ + 2 - C6H8N2OS5Cl2, 67), 171 (M
+- C6H8N2OS5Cl2,
100).
Masa exacta: calculada para C21H13N3OS5Br2: 620.8260 encontrada: 620.8236.
201
Roussel-Uclaf. Patent. Chem.Abstr. 1972, 77.

Parte experimental 251
S
N
S
S
O
S
S
NN
Cl
Cl
Cl
Cl
274f

252 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NN
Cl
Cl
Cl
Cl
274f

Parte experimental 253
15. Reacción de cicloadición de la bisditiolotiazina 176 con cloramina B
S
N
S
SS
S
OS
S
N
S
S
O
S
O
O
N(Cl)Na·xH2O
S
S
NS
O
O
CH2Cl2 , Sc(OTf)3, 75%
176 279a
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(35 mg, 0.108 mmol) en diclorometano (2 mL) se añadió cloramina B hidratada (58 mg) y
triflato de escandio (26 mg, 0.054 mmol). La mezcla de reacción se agitó a temperatura
ambiente durante 10 minutos. A continuación se filtraron las sales blancas, formadas en el
transcurso de la reacción, y se lavaron con diclorometano. El filtrado se concentró a presión
reducida y el sólido resultante se purificó mediante cromatografía en capa fina preparativa
(SiO2, eluyendo dos veces en diclorometano), lo que permitió separar el compuesto 279a
puro (36 mg, 0.081 mmol, 75%), como un sólido anaranjado; p.f.: 189-190 ºC (CH2Cl2).
IR (KBr, cm-1
): 1662 (C=O st), 1524 (C=N st), 1475, 1463, 1446, 1304 (S=O st),
1141 (S=O st), 1080, 743, 547.
RMN-1H (CDCl3, 400 MHz) δ: 7.96 (d, J = 7.2 Hz, 2 H, 2 CH Ar),
7.58 (t, J = 7.2 Hz, 1 H, CH Ar), 7.50 (t, J = 7.2 Hz, 2 H, 2 CH Ar),
3.76 (c, J = 7.2 Hz, 2 H, NCH2CH3) y 1.21 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 182.10 (C=0), 168.17 (C=N), 147.38 (C sp2),
147.30 (C sp2), 139.85 (C sp
2), 139.60 (C sp
2), 137.20 (C Ar), 133.17 (CH Ar),
128.96 (CH Ar), 126.86 (CH Ar), 43.54 (NCH2CH3) y 14.29 (NCH2CH3) ppm.
EM (IE) m/z (%): 446 (M+·
, 10), 414 (M+ - S, 15), 305 (M
+ - SO2Ph, 100),
277 (M+ + 1 - CH2CH3 - SO2Ph, 32), 262 (M
+ - NCH2CH3 - SO2Ph, 30), 77 (Ph
+)
Masa exacta: calculada para C14H10N2O3S6: 445.9016; hallada: 445.9025.

254 Tesis Doctoral Susana Barriga Falcón
Análisis: Calculado para C14H10N2O3S6: C, 37.65; H, 2.26; N, 6.27; hallado: C, 37.67;
H, 2.22; N, 6.24.

Parte experimental 255
S
N
S
S
O
S
S
NS
O
O
279a

256 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
NS
O
O
279a

Parte experimental 257
16. Reacción de cicloadición de la bisditiolotiazina 176 con cloramina T
S
N
S
S S
S
O S
S
N
S
S
O
S
O
O
N(Cl)Na·xH2O
S
S
N S
O
O
CH3H3C
CH2Cl2 , Sc(OTf)3, 93%
176 279b
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en diclorometano (2 mL) se añadió cloramina T hidratada (42 mg) y
triflato de escandio (11 mg, 0.022 mmol). La mezcla de reacción se agitó a temperatura
ambiente durante 7 minutos. A continuación se filtraron las sales blancas, formadas en el
transcurso de la reacción, y se lavaron con diclorometano. El filtrado se concentró a presión
reducida y el sólido resultante se purificó mediante cromatografía en capa fina preparativa
(SiO2, eluyendo dos veces en diclorometano), lo que permitió separar el compuesto 279b
puro (40 mg, 0.087 mmol, 93%), como un sólido anaranjado; p.f.: 164-165 ºC (CH2Cl2).
IR (KBr, cm-1
): 1660 (C=O st), 1520 (C=N st), 1456, 1302 (S=O st), 1284,
1142 (S=O st), 1080, 834
RMN-1H (CDCl3, 400 MHz) δ: 7.86 (d, J = 8.1 Hz, 2 H, 2 CH Ar),
7.31 (d, J = 8.1 Hz, 2 H, 2 CH Ar), 3.77 (c, J = 7.2 Hz, 2 H, NCH2CH3),
2.43 (s, 3 H, ArCH3) y 1.23 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 182.22 (C=O), 167.91 (C=N), 147.57 (C sp2),
147.14 (C sp2), 144.23 (C sp
2), 139.48 (C sp
2), 137.18 (C sp
2), 136.81 (C sp
2),
129.62 (CH Ar), 126.95 (CH Ar), 43.55 (NCH2CH3), 21.65 (ArCH3) y
14.34 (NCH2CH3) ppm.
EM (IE) m/z (%): 460 (M+·
, 1), 428 (M+ - S, 4), 305 (M
+ - SO2C6H4CH3, 25),
273 (M+ - SO2C6H4CH3 - S, 10), 155 (CH3C6H4SO2
+,35), 91 (PhCH2
·+, 100), 64 (75)

258 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C15H12N2O3S6: 459.9172; hallada: 459.9169.
Análisis: Calculado para C15H12N2O3S6: C, 39.11; H, 2.63; N, 6.08; hallado: C, 39.41;
H, 2.58; N, 5.86.
Parte experimental 259
S
N
S
S
O
S
S
N S
O
O
CH3
279b
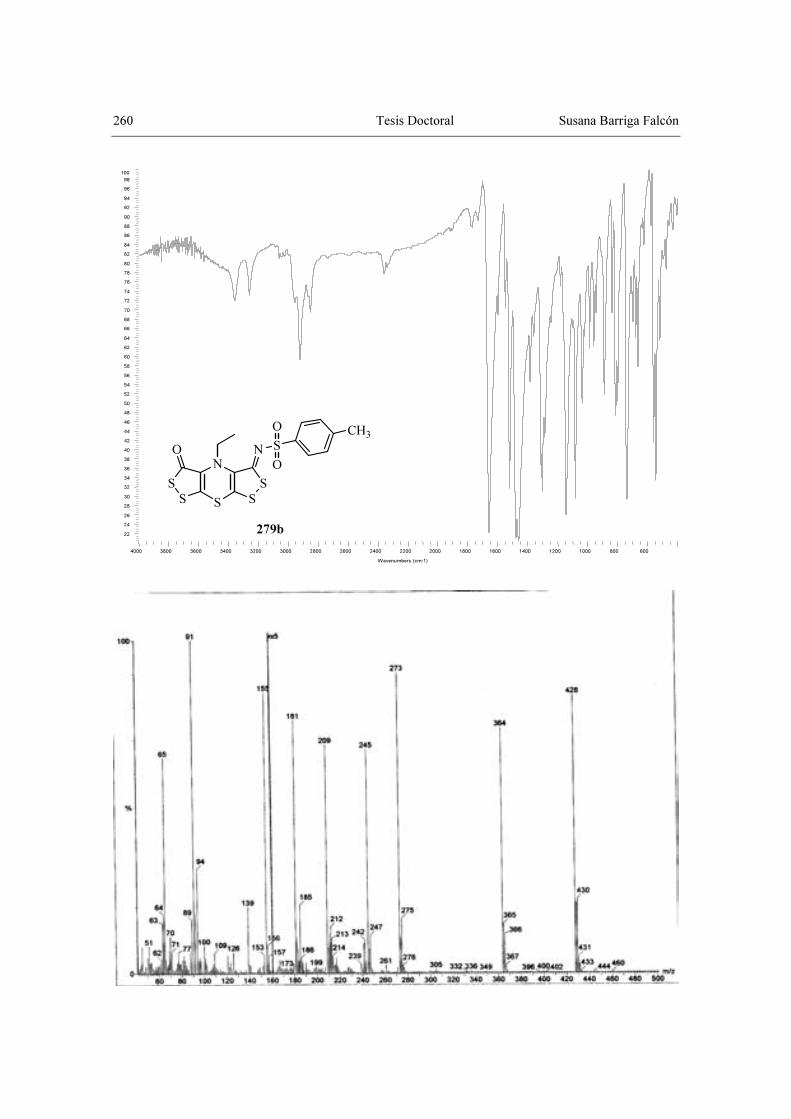
260 Tesis Doctoral Susana Barriga Falcón
22
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600
800
1000
1200
1400 1600 1800 2000 2200 2400 2600 2800
3000
3200
3400 3600
3800
4000
Wavenumbers (cm-1)
S
N
S
S
O
S
S
N S
O
O
CH3
279b

Parte experimental 261
17. Reacción de cicloadición de la bisditiolotiazina 176 con acetilendicarboxilato de
dimetilo (DMAD)
S
N
S
SS
S
OS CO2CH3H3CO2C
S
N
S
S
O
S
S
S
H3CO2C
H3CO2C
176
C6H6 , 81%
283
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(60 mg, 0.186 mmol) en benceno (5 mL) se añadió acetilendicarboxilato de dimetilo
(57 µL, 66 mg, 0.462 mmol). La mezcla de reacción se calentó a reflujo durante 10 minutos.
A continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano), lo que permitió separar el compuesto 283 puro (70 mg, 0.150 mmol, 81%),
como un sólido rojo; p.f.: 159-160 ºC (CH2Cl2-éter de petróleo).
IR (KBr cm-1
): 1753 (C=O st, éster), 1714 (C=O st, éster), 1668 (C=O st, heterociclo),
1436, 1295, 1235, 710.
RMN-1H (CDCl3, 400 MHz) δ: 3.94 (s, 3 H, CH3), 3.92 (s, 3 H, CH3),
3.51 (dc, J = 14.4 y 7.2 Hz, 1 H, CH2), 3.25 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3) y
1.22 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 192.44 (C=S), 184.77 (C=O), 161.15 (C=O éster),
159.53 (C=O éster), 159.48 (C sp2), 153.27 (C sp
2), 134.51 (C sp
2), 133.01 (C sp
2),
132.17 (C sp2), 132.06 (C sp
2), 53.91 (OCH3), 53.76 (OCH3), 47.87 (NCH2CH3) y
13.43 (NCH2CH3) ppm.
EM (IE) m/z (%): 465 (M+·
, 38), 436 (M+ - CH2CH3, 100), 376 (M
+ - CH2CH3 - CO2CH3,
5), 332 (M+ + 1 - CH2CH3 - CS - COS, 8), 274 (M
+ - CH2CH3 - 2 CO2CH3 - CS, 38),

262 Tesis Doctoral Susana Barriga Falcón
262 (M+ - DMAD - 2 S, 8), 218 (M
+ - DMAD - CS2 - S, 8), 174 (M
+ - DMAD - 2 CS2, 12),
158 (M+ - DMAD - CS2 - COS2, 15), 126 (M
+ - DMAD - CS2 - COS2- S, 40)
Masa exacta: calculada para C14H11NO5S6: 464.8962; hallada: 464.8961
Análisis: Calculado para C14H11NO5S6: C, 36.12; H, 2.38; N, 3.01; hallado: C, 36.12;
H, 2.15; N, 2.93.

Parte experimental 263
S
N
S
S
O
S
S
S
H3CO2C
H3CO2C
283

264 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S
H3CO2C
H3CO2C
283

Parte experimental 265
18. Reacción de cicloadición de la bisditiolotiazina 176 con dicianoacetileno (DCA)
S
N
S
SS
S
OS NC C C CN
S
N
S
S
O
S
S
S
NC
NC
176
C6H6 , 68 %
284
Sobre una disolución agitada de 4-etil-3-oxobis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(50 mg, 0.15 mmol) en benceno (10 mL) se añadió dicianoacetileno156,157
(30 mg, 0.39 mmol). La mezcla de reacción se calentó a reflujo durante 10 minutos. A
continuación se eliminó el disolvente en el rotavapor. El residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano), lo que permitió separar el compuesto 284 puro (42 mg, 0.10 mmol, 68%),
como un sólido rojo; p.f.: 173-175 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1): 2925, 2240 (CN st, débil), 1667 (C=O st), 1640 (C=O st), 1364, 1305, 1000, 710.
RMN-1H (CDCl3, 400 MHz) δ: 3.60 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3),
3.21 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3) y 1.23 (t, J = 7.1 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 196.59 (C=S), 184.43 (C=O), 154.28 (C=CS2),
151.89 (C=C-CO), 133.84 (NC-C=C-CN), 131.78 (NC-C=C-CN), 119.76 (CN),
118.92 (CN), 108.81 (C=C-S), 108.30 (C=C-S), 48.18 (NCH2CH3) y 13.42 (NCH2CH3) ppm.
EM (IE) m/z (%): 399 (M+·
, 40), 370 (M+ - CH2CH3, 100), 310 (M
+ - CH2CH3 - OCS, 5),
266 (M+ - CH2CH3 - OCS - CS, 17), 234 (M
+ - CH2CH3 - OCS - CS - S, 11),
208 (M+ - CH2CH3 - OCS - CS2 - CN, 27), 76 (NCCCCN
+, 14), 70 (SCCN
+, 18).
156 Blomquist, A. C.; Winslow, E. C. “Unsaturated nitriles as dienophiles in the diene synthesis”. J. Org.
Chem. 1945, 10, 149-158. 157
Hopf, H.; Witulski, B. en “Modern acetylene chemistry”. Ed. Stang, P. J.; Diederich, F. VCH;
Weinheim, 1995, Cap 2, pág 60-61

266 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C12H5N3OS6: 398.8757; hallada: 398.8750.
Análisis: Calculado para C12H5N3OS6: C, 36.07; H, 1.26; N, 10.52; hallado: C, 36.00;
H, 1.36; N, 10.48.

Parte experimental 267
S
N
S
S
O
S
S
S
NC
NC
284
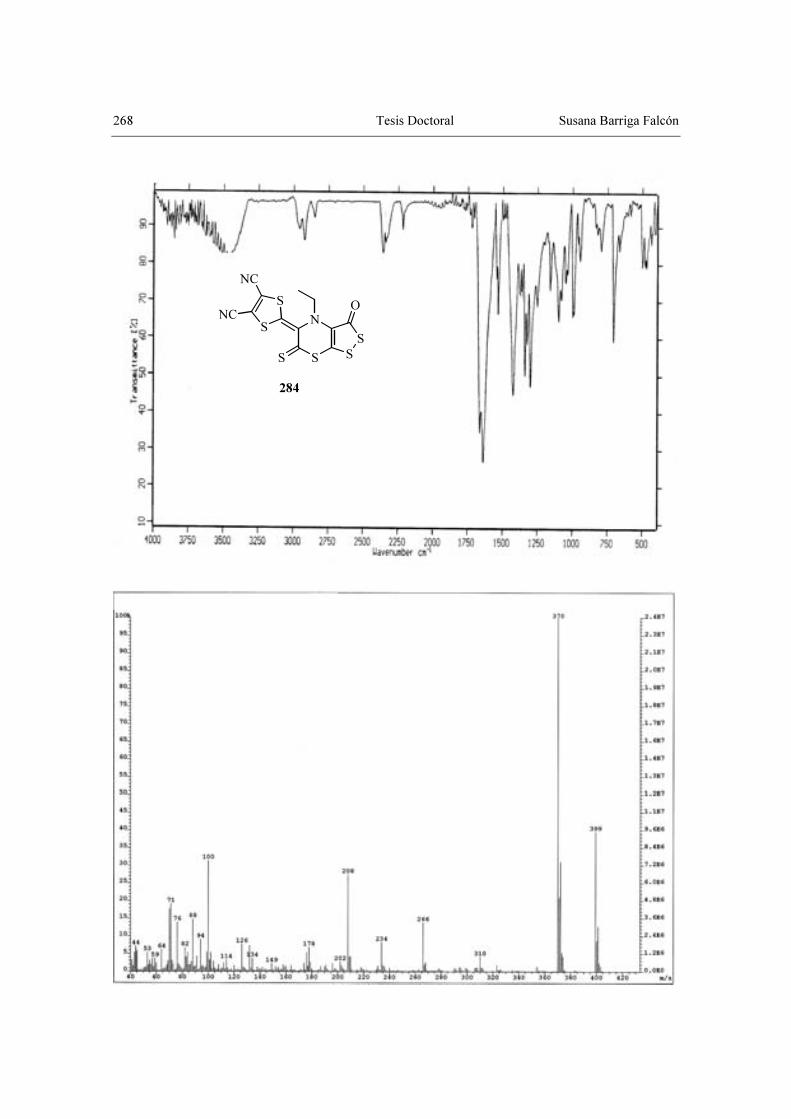
268 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S
NC
NC
284

Parte experimental 269
19. Reacción de cicloadición de la bisditiolotiazina 176 con un equivalente de
dibenzoilacetileno (DBA)
S
N
S
SS
S
OS COPhPhOC
S
N
S
S
O
S
S
S
PhOC
PhOC
176 285
C6H6, 48%
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.09 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(24 mg, 0.10 mmol). La mezcla de reacción se calentó a reflujo durante 5 minutos. A
continuación se eliminó el disolvente en frío, y el residuo resultante se purificó mediante
cromatografía líquida de media presión (SiO2, éter de petróleo hasta diclorometano-acetato
de etilo, 90: 10), lo que permitió separar el compuesto 285 puro (25 mg, 0.04 mmol, 48%),
como un sólido rojo; p.f.: 78-79 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2960, 2923, 1657 (C=O st), 1597, 1448, 1261 (C=S st), 1049, 695.
RMN-1H (CDCl3, 400 MHz) δ: 7.50-7.44 (m, 5 H, CH Ar), 7.29-7.22 (m, 5 H, CH Ar),
3.59 (dc, J = 14.2 y 7.1 Hz, 1H, NCH2CH3), 3.34 (dc, J = 14.2 y 7.1 Hz, 1H,
NCH2CH3), 1.27 (t, J = 7.1 Hz, 3H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 192.26 (C=S), 186.65 (COPh), 184.71 (C=O), 161.60 (C
sp2), 153.10 (C sp
2), 141.64 (C sp
2), 140.30 (C sp
2), 136.69 (C sp
2), 136.41(C sp
2), 134.33
(CH Ar), 134.20 (CH Ar), 134.02 (CH Ar), 133.94 (CH Ar), 132.23 (C sp2), 132.02 (C sp
2),
128.94 (CH Ar), 128.72 (CH Ar), 48.55 (NCH2CH3) y 13.48 (NCH2CH3) ppm.
158 Lutz, R.; Smithey, W. R. Jr. “The reactions of the dibromides and bromo derivatives of
dibenzoylethylene with amines”. J. Org. Chem. 1951, 16, 51-56. 159
Lutz, R. E. “Studies on unsaturated 1,4-diketones. I. Synthesis and structure of 1,2-di(2,4,6-
trimethylbenzoyl)ethenol”. J. Am. Chem. Soc. 1926, 48, 2905-2915. 202
Zhang, J. J.; Schuster, G. B. “Ylidions – A New Reactive Intermediate Prepared by One-electron
Oxidation of Phenacyl Sulfonium Ylides”. J. Am. Chem. Soc. 1989, 111, 7149-7155.

270 Tesis Doctoral Susana Barriga Falcón
EM (IE) m/z (%): 557 (M+·
, 12), 542 (M+ - CH3, 4), 528 (M
+ - CH2CH3, 33),
513 (M+ - CS, 8), 501 (M
+ - C2S, 3), 452 (M
+ - PhCO, 3), 351 (M
+ - CCOPh - CH2CH3 - SCO, 12),
162 (C3NOS3H+, 14), 105 (PhCO
+, 100), 77 (Ph
+, 58).
Masa exacta: calculada para C24H15NO3S6: 556.9282; hallada: 556.9376.
Análisis: Calculado para C24H15NO3S6: C, 51.68; H, 2.71; N, 2.51; hallado: C, 51.75;
H, 2.88; N, 2.74.

Parte experimental 271
S
N
S
S
O
S
S
S
PhOC
PhOC
285
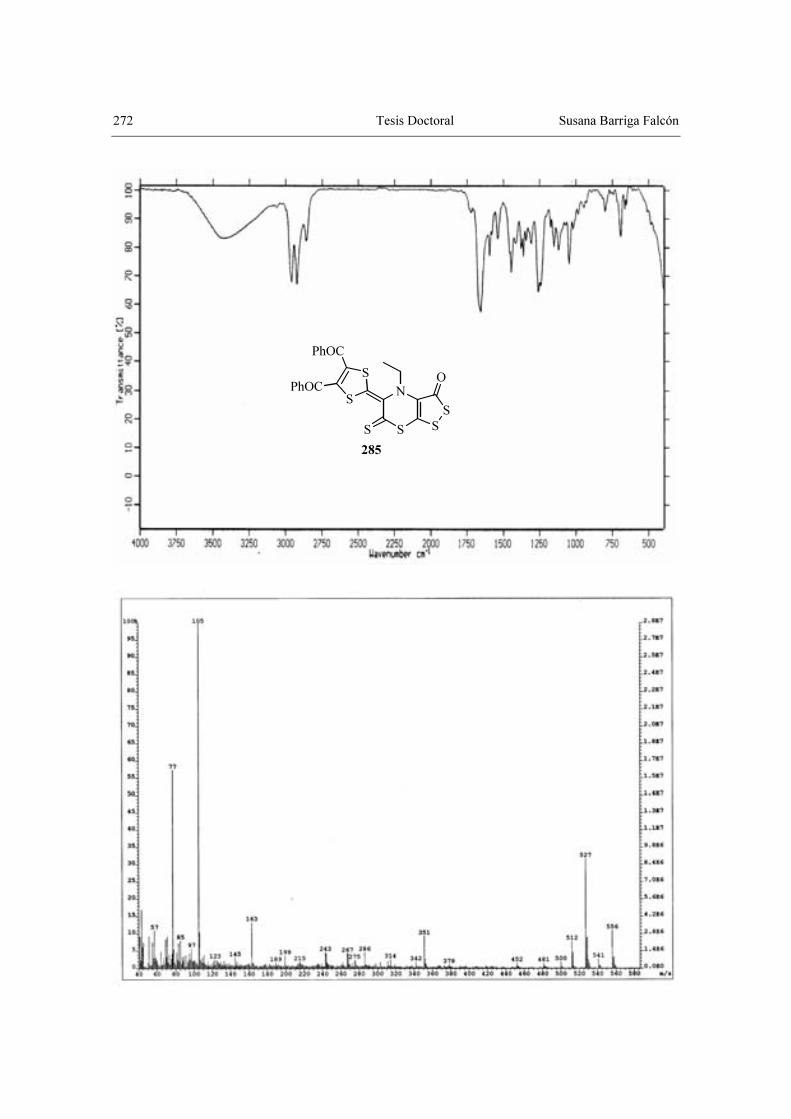
272 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S
PhOC
PhOC
285

Parte experimental 273
20. Reacción de cicloadición de la bisditiolotiazina 176 con dos equivalentes de
dibenzoilacetileno (DBA)
S
N
S
SS
S
OSCOPhPhOC
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
176 288
2
C6H6 , 73 %
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.09 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(54.4 mg, 0.23 mmol). La mezcla de reacción se calentó a reflujo durante 30 minutos. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta diclorometano),
lo que permitió separar el compuesto 288 puro (54 mg, 0.07 mmol, 73%), como un sólido
amarillo; p.f.: 104-106 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
):1660 (C=O st,), 1595, 1532, 1447, 1257, 689.
RMN-1H (CDCl3, 400 MHz) δ: 7.69-7.19 (m, 20 H, 4 x C6H5),
3.97 (dc, J = 14.4 y 7.2 Hz, 1 H, CH2), 3.82 (dc, J = 14.4 y 7.2 Hz, 1 H, CH2) y
1.44 (t, J =7.2 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 192.71 (C=O heterociclo), 189.59 (C=O),
187.65 (C=O), 186.45 (C=O), 182.58 (C=O), 155.68 (C sp2), 140.57 (C sp
2),
138.54 (C sp2), 137.72 (C sp
2), 137.38 (C sp
2), 137.30 (C sp
2), 136.78 (C sp
2),
136.69 (C sp2), 136.05 (C sp
2), 135.00 (C sp
2), 134.87 (C sp
2), 134.60 (CH Ar),
134.23 (CH Ar), 133.69 (CH Ar), 133.35 (CH Ar), 133.14 (CH Ar), 129.89 (CH Ar),
129.62 (CH Ar), 129.11 (CH Ar), 128.82 (CH Ar), 128.62 (C sp2), 128.41 (CH Ar),
128.23 (CH Ar), 121.50 (C sp2), 70.83 (C espirano), 50.23 (CH2) y 13.37 (CH3) ppm.

274 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 792 (M+·
+1, 2), 686 (M+ - COPh, 1), 549 (M
+ - 2 PhCO - S, 1),
521 (M+ - 2 PhCO - SCO, 1), 147 (25), 105 (PhCO
+, 33).
Análisis: Calculado para C40H25NO5S6: C, 60.66; H, 3.18; N, 1.77; hallado: C, 60.55;
H, 3.28; N, 1.64.
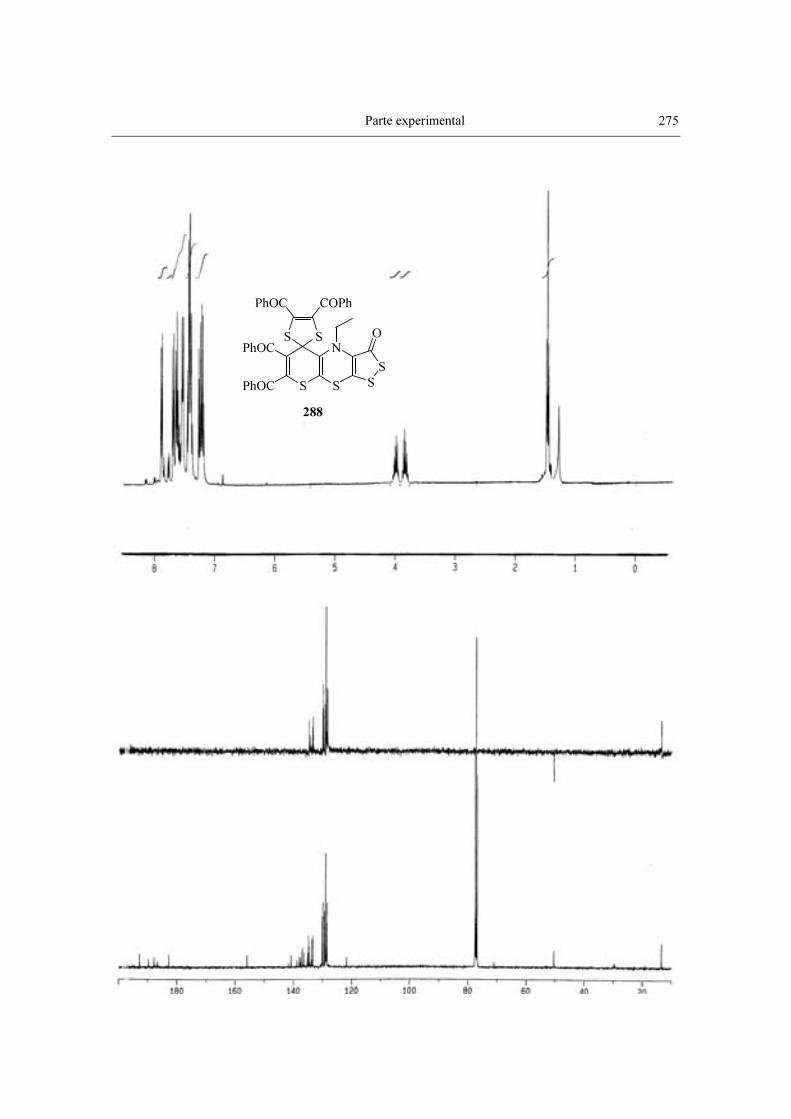
Parte experimental 275
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
288

276 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
288
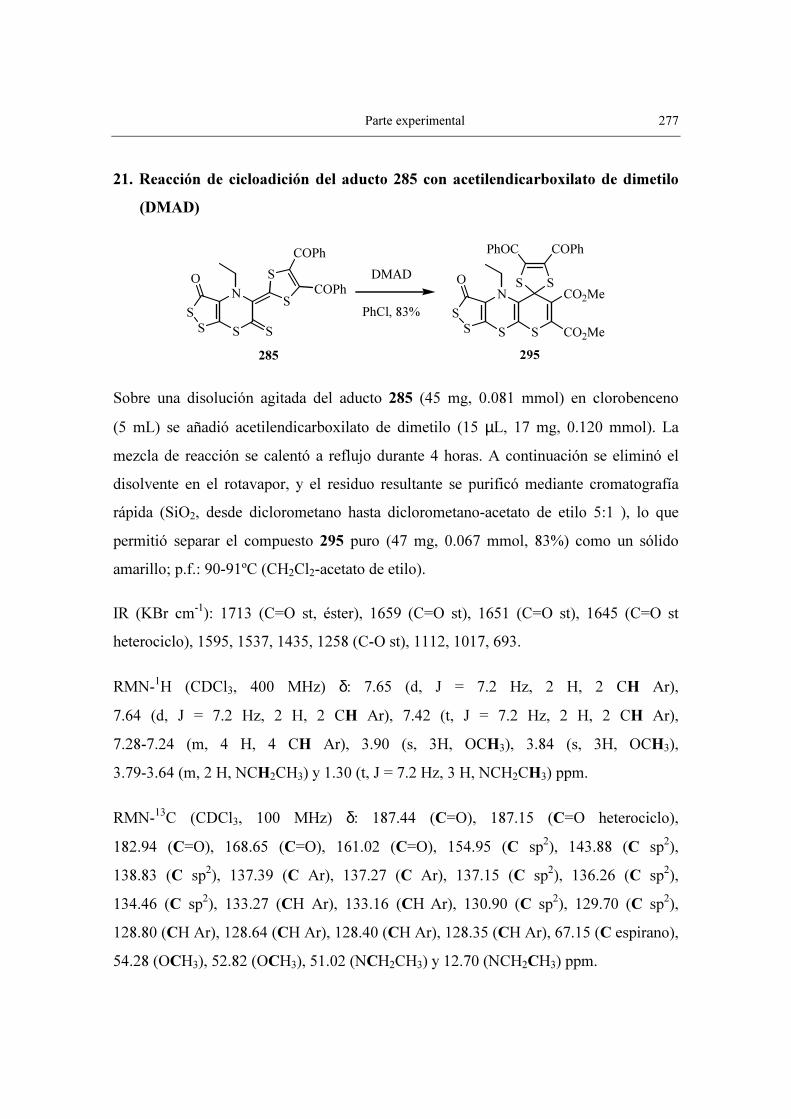
Parte experimental 277
21. Reacción de cicloadición del aducto 285 con acetilendicarboxilato de dimetilo
(DMAD)
S
N
S
S
O
S
S
S
COPh
COPh
DMAD
PhCl, 83%
S
N
S
S
S
SSO
CO2Me
CO2Me
COPhPhOC
285 295
Sobre una disolución agitada del aducto 285 (45 mg, 0.081 mmol) en clorobenceno
(5 mL) se añadió acetilendicarboxilato de dimetilo (15 µL, 17 mg, 0.120 mmol). La
mezcla de reacción se calentó a reflujo durante 4 horas. A continuación se eliminó el
disolvente en el rotavapor, y el residuo resultante se purificó mediante cromatografía
rápida (SiO2, desde diclorometano hasta diclorometano-acetato de etilo 5:1 ), lo que
permitió separar el compuesto 295 puro (47 mg, 0.067 mmol, 83%) como un sólido
amarillo; p.f.: 90-91ºC (CH2Cl2-acetato de etilo).
IR (KBr cm-1
): 1713 (C=O st, éster), 1659 (C=O st), 1651 (C=O st), 1645 (C=O st
heterociclo), 1595, 1537, 1435, 1258 (C-O st), 1112, 1017, 693.
RMN-1H (CDCl3, 400 MHz) δ: 7.65 (d, J = 7.2 Hz, 2 H, 2 CH Ar),
7.64 (d, J = 7.2 Hz, 2 H, 2 CH Ar), 7.42 (t, J = 7.2 Hz, 2 H, 2 CH Ar),
7.28-7.24 (m, 4 H, 4 CH Ar), 3.90 (s, 3H, OCH3), 3.84 (s, 3H, OCH3),
3.79-3.64 (m, 2 H, NCH2CH3) y 1.30 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 187.44 (C=O), 187.15 (C=O heterociclo),
182.94 (C=O), 168.65 (C=O), 161.02 (C=O), 154.95 (C sp2), 143.88 (C sp
2),
138.83 (C sp2), 137.39 (C Ar), 137.27 (C Ar), 137.15 (C sp
2), 136.26 (C sp
2),
134.46 (C sp2), 133.27 (CH Ar), 133.16 (CH Ar), 130.90 (C sp
2), 129.70 (C sp
2),
128.80 (CH Ar), 128.64 (CH Ar), 128.40 (CH Ar), 128.35 (CH Ar), 67.15 (C espirano),
54.28 (OCH3), 52.82 (OCH3), 51.02 (NCH2CH3) y 12.70 (NCH2CH3) ppm.

278 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 700 (M+ + 1, 10), 641 (M
+ + 1 – CO2CH3, 10), 154 (75), 105
(PhCO+, 87), 73 (100).
Análisis: Calculado para C30H21NO7S6: C, 51.48; H, 3.02; N, 2.00; hallado: C, 51.34; H,
2.96; N, 1.80.

Parte experimental 279
S
N
S
S
S
SSO
CO2Me
CO2Me
COPhPhOC
295
280 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
S
SSO
CO2Me
CO2Me
COPhPhOC
295

Parte experimental 281
22. Reacción de cicloadición del aducto 283 con 4-fenil-1,2,4-triazolin-3,5-diona (296)
S
N
S
S
O
S
S
S
CO2Me
CO2Me
N
NN
O
O
Ph
S
N
S
SN N
H
S
SO CO2CH3
CO2CH3
O+
283 296 298
PhCl, 48% Ph
Sobre una disolución agitada del aducto 283 (50 mg, 0.107 mmol) en clorobenceno (5 mL) se
añadió 4-fenil-1,2,4-triazolin-3,5-diona (296) (48 mg, 0.274 mmol). La mezcla de reacción se
calentó a reflujo durante 3 horas. A continuación se eliminó el disolvente en el rotavapor, y el
residuo resultante se purificó mediante cromatografía rápida (SiO2, éter de petróleo-
diclorometano 1:4), lo que permitió separar el compuesto 298 puro (29 mg, 0.051 mmol, 48%)
como un sólido anaranjado; p.f.: 81-82 ºC (CH2Cl2-éter de petróleo).
IR (KBr cm-1
): 3353 (N-H st), 2951, 2924, 2851, 1731 (C=O st carbamida), 1661 (C=O
st), 1515, 1429, 1258 (C-O st), 1200 (C-O st), 1099, 1017, 753.
RMN-1H (CDCl3, 400 MHz) δ: 7.62 (d, J = 7.7 Hz, 2 H, 2 CH Ar), 7.51 (s ancho, 1 H, NH),
7.37 (t, J = 7.7 Hz, 2 H, 2 CH Ar), 7.14 (t, J = 7.7 Hz, 1 H, 1 CH Ar), 3.93 (s, 3H, OCH3),
3.90 (s, 3H, OCH3), 3.59 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3),
3.22 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3) y 1.16 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 185.58 (C=O heterociclo), 162.21 (C=O),
160.26 (C=O), 159.62 (C=O), 157.60 (C=N), 151.91 (C sp2), 151.15 (C sp
2), 137.46 (C
Ar), 135.97 (C sp2), 133.02 (C sp
2), 129.09 (CH Ar), 124.45 (CH Ar), 120.58(C sp
2),
119.25 (CH Ar), 118.85 (C sp2), 53.69 (OCH3), 53.63 (OCH3), 46.53 (NCH2CH3) y
13.56 (NCH2CH3) ppm.
EM (FAB) m/z (%): 567 (M+·
, 38), 538 (M+ - CH2CH3, 32), 475 (M
+ - Ph - CH3, 38),
462 (M+ - CO2CH3 - OCH3 - CH3, 42), 448 (M
+ - CO2CH3 - OCH3 - CH2CH3, 12),

282 Tesis Doctoral Susana Barriga Falcón
419 (M+ - Ph - C2O2CH3, 55), 257 (M
+ - DMAD - CS2 - Ph - CH3, 23), 219 (40), 119
(85), 91 (PhN+, 100), 73 (92).
Masa exacta: calculada para C21H17N3O6S5: 566.9721 hallada: 566.9665.

Parte experimental 283
298
S
N
S
S NHN
S
SO
CO2CH3
CO2CH3
O
Ph

284 Tesis Doctoral Susana Barriga Falcón
298
S
N
S
S NHN
S
SO
CO2CH3
CO2CH3
O
Ph

Parte experimental 285
23. Reacción de cicloadición de la bisditiolotiazina 161 con dos equivalentes de
acetilendicarboxilato de dimetilo (DMAD)
S
N
S
SS
S
SS CO2CH3H3CO2C S
S
H3CO2C
H3CO2C
S
N
S S
S
S
CO2CH3
CO2CH3
C6H6, 44%
2
161 307
Sobre una disolución agitada de 4-etilbis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(55 mg, 0.162 mmol) en benceno (10 mL) se añadió acetilendicarboxilato de dimetilo
(50 µL, 58 mg, 0.408 mmol). La mezcla de reacción se calentó a reflujo durante 10
minutos. A continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se
purificó mediante cromatografía en capa fina preparativa (SiO2, eluyendo dos veces en
diclorometano), lo que permitió separar el compuesto 307 puro (45 mg, 0.072 mmol,
44%), como un sólido anaranjado; p.f.: 119-120 ºC (CH2Cl2).
IR (KBr, cm-1
): 1754 (C=O st, éster), 1731 (C=O st, éster), 1576, 1437, 1235 (C=S st),
1002, 966, 684.
RMN-1H (CDCl3, 400 MHz) δ: 3.98 (s, 3 H, OCH3), 3.93 (s, 3 H, OCH3),
3.38 (c, J = 7.2 Hz,2 H, NCH2CH3) y 1.27 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 195.55 (C=S), 160.87 (C sp2), 159.86 (C=O),
159.60 (C=O), 133.67 (C sp2), 133.14 (C sp
2), 132.80 (C sp
2), 53.98 (OCH3),
53.75 (OCH3), 49.84 (NCH2CH3) y 13.63 (NCH2CH3) ppm.
EM (FAB) m/z (%): 624 (M+ + 1, 45), 594 (M
+ - CH2CH3, 30), 562 (M
+ - 2 OCH3, 10),
533 (M+ - S - CO2CH3, 6), 91 (50), 77 (55), 69 (75), 55 (100).
Análisis: Calculado para C20H27NO8S7: C, 38.51; H, 2.75; N, 2.25; hallado: C, 38.52;
H, 2.78; N, 2.16.

286 Tesis Doctoral Susana Barriga Falcón
S
S
H3CO2C
H3CO2C
S
N
S S
S
S
CO2CH3
CO2CH3
307

Parte experimental 287
S
S
H3CO2C
H3CO2C
S
N
S S
S
S
CO2CH3
CO2CH3
307

288 Tesis Doctoral Susana Barriga Falcón
24. Reacción de cicloadición de la bisditiolotiazina 161 con dos equivalentes de
dibenzoilacetileno (DBA)
S
N
S
SS
S
SS COPhPhOC
S
N
SS
S
SS
SCOPh
COPhPhOC
PhOC
161
C6H6, 61%
308
Sobre una disolución agitada de 4-etil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(20 mg, 0.059 mmol) en benceno (5 mL) se añadió dibenzoilacetileno158,159,202
(35 mg, 0.150 mmol). La mezcla de reacción se calentó a reflujo durante 5 minutos. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano 2:3), lo que permitió
separar el compuesto 308 puro (29 mg, 0.036 mmol, 61%), como un sólido anaranjado;
p.f.: 115-116 ºC (CH2Cl2-éter de petróleo).
IR (KBr cm-1
): 1650 (C=O st), 1595, 1537, 1446, 1436, 1261 (C=S st), 974, 693.
RMN-1H (CDCl3, 400 MHz) δ: 7.50-7.42 (m, 12 H, 12 CH Ar), 7.29-7.22 (m, 8 H, 8 CH Ar),
3.48 (c, J = 7.2 Hz, 2 H, NCH2CH3) y 1.36 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 195.40 (C=S), 186.77 (C=O), 186.56 (C=O),
161.54 (C sp2), 140.56 (C sp
2), 140.27 (C sp
2), 136.63 (C Ar), 136.45 (C Ar),
134.14 (CH Ar), 134.03 (CH Ar), 133.04 (C sp2), 128.93 (CH Ar), 128.88 (CH Ar),
128.73 (CH Ar), 49.84 (NCH2CH3) y 13.76 (NCH2CH3) ppm.
EM (FAB) m/z (%): 808 (M+ + 1, 5%), 778 (M
+ - C2H5, 3), 776 (M
+ + 1 - S, 1%),
371 (13), 309 (14), 278 (12), 263 (13), 231 (64), 154 (96), 137 (100),109 (27).
Análisis: Calculado para C40H25NO4S7: C, 59.45; H, 3.12; N, 1.73; hallado: C, 59.54;
H, 3.20; N, 1.58.

Parte experimental 289
S
N
SS
S
SS
SCOPh
COPhPhOC
PhOC
308

290 Tesis Doctoral Susana Barriga Falcón
S
N
SS
S
SS
SCOPh
COPhPhOC
PhOC
308

Parte experimental 291
25. Reacción de cicloadición de la bisdiolotiazina 161 con tres equivalentes de
acetilendicarboxilato de dimetilo (DMAD)
S
N
S
SS
S
SS CO2CH3H3CO2C
S
N
S S
S
S
S SH3CO2C
H3CO2C
H3CO2C CO2CH3 CO2CH3
CO2CH3
C6H6, 27%
3
161 309
Sobre una disolución agitada de 4-etilbis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(40 mg, 0.12 mmol) en benceno (10 mL) se añadió acetilendicarboxilato de dimetilo
(73 µL, 84 mg, 0.59 mmol). La mezcla de reacción se calentó a reflujo durante
20 minutos. A continuación se eliminó el disolvente en el rotavapor, y el residuo resultante
se purificó mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano-acetato de etilo, 90:10), lo que permitió separar el compuesto 309 puro
(24 mg, 0.31 mmol, 27%), como un sólido anaranjado; p.f.: 87-88 ºC (CH2Cl2-éter de
petróleo).
IR (KBr cm-1
): 1735 (C=O st, éster), 1580, 1432, 1250.
RMN-1H (CDCl3, 400 MHz) δ: 3.95 (s, 3 H, OCH3), 3.93 (s, 3 H, OCH3),
3.92 (s, 3 H, OCH3), 3.84 (s, 6 H, 2 OCH3), 3.73 (s, 3 H, OCH3),
3.64 (dc, J = 14.2 Hz y 7.1 Hz, 1 H, NCH2CH3), 3.18 (dc, J = 14.2 Hz y 7.1 Hz, 1 H, NCH2CH3)
y 1.19 (t, J = 7.1 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 195.85 (C=S), 165 .09 (C=O), 164.84 (C=O),
161.29 (C=O), 160.35 (C=O), 160.11 (C=O), 159.51 (C=O), 137.31 (C sp2),
134.89 (C sp2), 132.22 (C sp
2), 131.76 (C sp
2), 128.71 (C sp
2), 128.33 (C sp
2),
127.16 (C sp2), 126.80 (C sp
2), 124.72 (C sp
2), 115.45 (C sp
2), 71.42 (C espirano),
53.80 (OCH3), 53.65 (OCH3), 53.20 (OCH3), 53.07 (OCH3), 49.98 (NCH2CH3) y
12.87 (NCH2CH3) ppm.

292 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 766 (M+·
+ 1, 5), 577 (M+ + 1 - DMAD - CH3 - S, 3),
459 (M+ - DMAD - C3S4, 2), 401 (M
+ + 1 - DMAD - C3S4 - CO2CH3, 2), 355 (2),
327 (5), 281 (5), 221 (5), 207 (DMAD + 2S + 1, 7).
Análisis: Calculado para C26H23NO12S7: C, 40.77; H, 3.03; N, 1.83; hallado: C, 40.84;
H, 3.14; N, 1.84.

Parte experimental 293
S
N
S S
S
S
S S
H3CO2C
H3CO2C
H3CO2C CO2CH3 CO2CH3
CO2CH3
309

294 Tesis Doctoral Susana Barriga Falcón
S
N
S S
S
S
S S
H3CO2C
H3CO2C
H3CO2C CO2CH3 CO2CH3
CO2CH3
309

Parte experimental 295
26. Cicloadición de la bisditiolotiazina 161 con tres equivalentes de
dibenzoilacetileno (DBA)
S
N
S
SS
S
S S DBA
S
N
SS
S
S
SS
PhOC
PhOC
COPhPhOC
COPh
COPh
S
N
SS
COPh
COPhPhOC
PhOCS
S
COPh
COPh
SS
PhOC
COPh
312
+
161 311
Sobre una disolución agitada de 4-etil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(30 mg, 0.088 mmol) en benceno (5 mL) se añadió dibenzoilacetileno158,159,200
(103 mg, 0.440 mmol). La mezcla de reacción se calentó a reflujo durante 15 minutos. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano 2:3 hasta
diclorometano), lo que permitió separar los compuestos 311 (21 mg, 0.020 mmol, 23%) y
312203
(61 mg, 0.048 mmol, 55%) puros como un sólido rojizo; p.f.: 115-116 ºC (CH2Cl2-
éter de petróleo) y un sólido anaranjado; p.f.: 135-136 ºC (CH2Cl2-éter de petróleo)
respectivamente.
Datos espestroscópicos de 311:
IR (KBr cm-1
): 2924, 2853, 1661 (C=O st), 1595, 1538, 1448, 1260 (C=S st), 691.
RMN-1H (CDCl3, 400 MHz) δ: 7.93 (d, J = 7.4 Hz, 2 H, 2 CH Ar),
7.72 (d, J = 7.4 Hz, 2 H, 2 CH Ar), 7.62-7.06 (m, 26 H, 26 CH Ar),
3.76 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3), 3.40 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3)
y 1.36 (t, J = 7.1 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 196.49 (C=S), 193.03 (C=O), 189.90 (C=O),
187.04 (C=O), 186.81 (C=O), 186.62 (C=O), 186.34 (C=O), 164.01 (C sp2),
203
La descripción espectroscópica y analítica de 312 se realizará posteriormente cuando se describa la
obtención de éste como producto único de reacción.

296 Tesis Doctoral Susana Barriga Falcón
142.20 (C sp2), 141.14 (C sp
2), 140.28 (C sp
2), 138.06 (C sp
2), 137.23 (C sp
2),
137.12 (C sp2), 137.03 (C sp
2), 136.69 (C sp
2), 136.52 (C sp
2), 136.13 (C sp
2),
135.13 (C sp2), 134.55 (CH Ar), 134.03 (CH Ar), 133.99 (CH Ar), 133.93 (CH Ar),
133.23 (CH Ar), 133.08 (CH Ar), 131.75 (C sp2), 131.37 (C sp
2), 129.80 (CH Ar),
129.02 (CH Ar), 128.97 (CH Ar), 128.83 (CH Ar), 128.78 (CH Ar), 128.72 (CH Ar),
128.69 (CH Ar), 128.65 (CH Ar), 128.38 (CH Ar), 128.69 (CH Ar), 128.27 (CH Ar),
71.14 (C espirano), 50.56 (NCH2CH3) y 13.10 (NCH2CH3) ppm.
EM (FAB) m/z (%): 1042 (M+ + 1, 1%), 744 (M
+ + 1 - DBA - 2 S, 1%),
550 (M+ + 1 - DBA - CS2 - PhCO - Ph, 60%), 522 (M
+ + 1 - DBA - CS2 - 2 PhCO, 40%),
105 (PhCO+, 35), 81 (40), 69 (75), 55 (100).
Análisis: Calculado para C56H35NO6S7: C, 64.53; H, 3.38; N, 1.34; hallado: C, 64.64;
H, 3.31; N, 1.25.

Parte experimental 297
S
N
SS
S
S
SS
PhOC
PhOC
COPhPhOC
COPh
COPh
311

298 Tesis Doctoral Susana Barriga Falcón
S
N
SS
S
S
SS
PhOC
PhOC
COPhPhOC
COPh
COPh
311

Parte experimental 299
27. Reacción de cicloadición de la bisditiolotiazina 161 con cuatro equivalentes de
dibenzoilacetileno (DBA)
S
N
S
SS
S
SS COPhPhOC
S
N
SS
COPh
COPhPhOC
PhOCS
S
COPh
COPh
SS
PhOCCOPh
161 312
4
C6H6, 95%
Sobre una disolución agitada de 4-etil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(50 mg, 0.15 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(206 mg, 0.88 mmol). La mezcla de reacción se calentó a reflujo durante 60 minutos. Se
añadió una porción adicional de dibenzoilacetileno (69 mg, 0.29 mmol) y se calentó
durante 30 minutos. Este proceso se repitió dos veces más. Finalmente se eliminó el
disolvente en el rotavapor, y el residuo resultante se purificó mediante cromatografía
líquida de media presión (SiO2, éter de petróleo hasta diclorometano-acetato de etilo 98:2),
lo que permitió separar el compuesto 312 puro (177 mg, 0.14 mmol, 95%), como un
sólido anaranjado; p.f.: 135-136 ºC (CH2Cl2-éter de petróleo).
IR (KBr cm-1
): 3060, 2925, 1664 (C=O st), 1595, 1578, 1536, 1448, 1258, 691.
RMN-1H (CDCl3, 400 MHz) δ: 7.94-7.04 (m, 40 H, 8 C6H5), 3.88 (s ancho, 2 H, CH2),
1.49 (t, J = 7.0 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 193.13 (C=O), 189.80 (C=O), 187.15 (C=O), 186.68 (C=O),
143.37 (C sp2), 137.29 (C sp
2), 137.12 (C sp
2), 136.95 (C sp
2), 135.21 (C sp
2),
134.51 (CH Ar), 134.00 (CH Ar), 133.04 (C sp2), 132.92 (CH Ar), 132.88 (C sp
2),
132.60 (C sp2), 129.80 (CH Ar), 129.65 (CH Ar), 128.78 (CH Ar), 128.65 (CH Ar),
128.23 (CH Ar), 71.24 (C espirano), 52.55 (NCH2CH3) y 13.16 (NCH2CH3) ppm.
EM (FAB) m/z (%): 1275 (M+·, 1), 1170 (M
+ - PhCO, 1), 1011 (M
+ + 1 - CH2CH3 - DBA, 1),
977 (M+ + 2 - DBA - 2 S, 1), 914 (M
+ + 1 - DBA - 4 S, 1), 884 (M
+ + 1 - DBA - 4 S - CH2CH3, 1),

300 Tesis Doctoral Susana Barriga Falcón
732 (M+ + 1 - 2 DBA - CS2, 1), 716 (M
+ - 2 DBA - CS2 - CH3, 1), 670 (M
+ - 2 DBA - CS3 - CH2CH3, 1),
550 (6), 522 (M+ + 1 - 2 DBA - CS3 - CH2CH3 - PhCOCS, 4), 105 (PhCO
+, 42).
Análisis: Calculado para C72H45NO8S7: C, 67.74; H, 3.55; N, 1.10; hallado: C, 67.52;
H, 3.18; N, 1.33.

Parte experimental 301
S
N
SS
COPh
COPhPhOC
PhOCS
S
COPh
COPh
SS
PhOC
COPh
312

302 Tesis Doctoral Susana Barriga Falcón
S
N
SS
COPh
COPhPhOC
PhOCS
S
COPh
COPh
SS
PhOC
COPh
312

Parte experimental 303
28. Síntesis de diisopropillaurilamida (317)
HN
C11H23 Cl
O
N
C11H23 O
+
CH2Cl2, 93%
314 315 317
Sobre una disolución agitada de diisopropilamina (6.40 mL, 4.65 g, 45.95 mmol) en
diclorometano (100 mL) se añadió lentamente cloruro de lauroilo (5.00 g, 22.85 mmol).
Tras completar la adición, la mezcla de reacción se agitó a temperatura ambiente
durante 4 horas. A continuación se vertió sobre agua (50 mL) y se separaron la fases. La
fase acuosa se extrajo con diclorometano (3 x 20 mL) y los extractos orgánicos
combinados se lavaron con una disolución acuosa saturado de NaHCO3 (3 x 50 mL) y se
secaron sobre sulfato sódico. A continuación se eliminó el disolvente en el rotavapor y
se obtuvo el compuesto 317 (6.02 g, 21.25 mmol, 93%) como un aceite de color
amarillo claro que se utilizó sin purificación en la siguiente etapa.
IR (KBr cm-1
): 2925 (C-H st), 2854 (C-H st), 1642 (C=O st), 1439, 1370, 1216, 1135,
1044, 607.
RMN-1H (CDCl3, 400 MHz) δ: 3.87-3.80 (m, 1 H, CH(CH3)2),
3.36-3.33 (m, 1 H, CH(CH3)2), 2.13 (t, J = 7.5 Hz, 2 H, NCOCH2),
1.51-1.43 (m, 2 H, NCOCH2CH2), 1.24 (d, J = 7.0 Hz, 6 H, CH(CH3)2),
1.20-1.12 (m, 18 H, 9 CH2), 1.06 (d, J = 7.0 Hz, 6 H, CH(CH3)2) y
0.74 (t, J = 7.0 Hz, 3 H, CH2) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 171.64 (C=O), 47.94 (CH(CH3)2), 45.16 (NCH2),
35.09 (CH2), 31.62 (CH2), 29.23 (CH2), 29.06 (CH2), 25.17 (CH2), 22.38 (CH2),
20.68 (CH2), 20.40 (CH(CH3)2) y 13.76 (CH3) ppm.

304 Tesis Doctoral Susana Barriga Falcón
0.0
0.0
0.5
0.5
1.0
1.0
1.51.52.02.02.52.53.03.03.53.54.04.04.54.55.0
5.0
5.5
5.5
6.0
6.0
6.5
6.5
7.0
7.0
7.5
7.5
8.0
8.0
-25 -25 0 0 252550507575100100125 125 150 150 175 175 200 200

Parte experimental 305
N
C11H23 O
317

306 Tesis Doctoral Susana Barriga Falcón
29. Síntesis de diisopropillaurilamina (319)
N
C11H23 O
317
LiAlH4
THF, 97%
N
C11H23
319
Sobre una disolución agitada de diisopropillaurilamida (5.00 g, 17.64 mmol) en
tetrahidrofurano (100 mL) se añadió lentamente hidruro de litio y aluminio
(1.34 g, 35.28 mmol). Tras completar la adición la mezcla de reacción se calentó a
reflujo durante 96 horas. A continuación la mezcla de reacción se vertió sobre una
disolución THF-H2O 1:1 (60 mL). Seguidamente se filtraron las sales y éstas se lavaron
con agua y con éter dietílico. El filtrado se extrajo con éter dietílico (6 x 50 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico. Tras eliminar el
disolvente en el rotavapor se obtuvo compuesto 319 (4.61 g, 17.11 mmol, 97%) como
un aceite de color amarillo que se utilizó sin ningún tipo de purificación posterior.
IR (KBr cm-1
): 2961 (C-H st), 2925 (C-H st), 2854 (C-H st), 1465, 1383, 1360, 1206.
RMN-1H (CDCl3, 200 MHz) δ: 3.01-2.91 (m, 2 H, 2 CH(CH3)2), 2.34-2.25 (m, 2 H, NCH2),
1.38-1.11 (m, 20 H, 10 CH2), 0.96-0.92 (m, 12 H, 2 CH(CH3)2) y 0.87-0.78 (m, 3 H, CH3)
ppm.
RMN-13
C (CDCl3, 500 MHz) δ: 48.46 (CH(CH3)2), 45.40 (NCH2), 34.78 (CH2),
31.84 (CH2), 31.52 (CH2), 29.57 (CH2), 29.28 (CH2), 27.41 (CH2), 25.47 (CH2),
22.59 (CH2), 20.43 (CH(CH3)2) y 13.97 (CH3) ppm.

Parte experimental 307
-0.2-0.20.00.00.20.20.40.40.60.60.80.81.01.01.21.21.41.41.61.61.81.82.02.02.22.22.42.42.62.62.82.83.03.03.23.2
252550507575100100125125150150175175200200
N
C11H23
319

308 Tesis Doctoral Susana Barriga Falcón
N
C11H23
319

Parte experimental 309
30. Síntesis de 4-lauril-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona (321).
N
C12H25
S
N
S
SS
S
C12H25 SS2) Et3N
CHCl3, 5%
319 321
1) S2Cl2, DABCO
Se adicionó lentamente dicloruro de diazufre (3.0 mL, 37.5 mmol) a una disolución de
N-lauroildiisopropilamina (1.0 g, 3.71 mmol) y 1,4-diazabiciclo[2.2.2]octano
(DABCO, 3.32 g, 29.6 mmol) en cloroformo (100 mL) mantenida bajo nitrógeno y
enfriada a -40 ºC. La mezcla se agitó bajo nitrógeno durante 15 minutos a -40 ºC, y
después durante tres días a temperatura ambiente. A continuación la mezcla de reacción
se enfrió a 0 ºC y se añadió lentamente trietilamina (6.7 mL, 48.1 mmol). Tras
completar la adición la mezcla de reacción se agitó a temperatura ambiente durante
2 horas. Seguidamente la mezcla de reacción se filtró a través de celita y se eliminó el
disolvente en el rotavapor. El residuo se disolvió en diclorometano (100 mL), se filtró y
se lavó con ácido sulfúrico al 70% v/v (15 mL). La fase orgánica se extrajo con ácido
sulfúrico concentrado (95-98%, 20 mL). La fase de ácido sulfúrico concentrado se vertió
sobre hielo (200 g) y se extrajo con diclorometano (3 x 50 mL). La fase orgánica se lavó
con agua (3 x 30 mL), se secó sobre sulfato magnésico anhidro y se concentró en el
rotavapor. El crudo se purificó por cromatografía líquida de media presión (SiO2, éter de
petróleo hasta diclorometano). Esto permitió obtener el compuesto 321 puro
(89 mg, 0.19 mmol, 5%) como un sólido negro; p.f.: 147-149 ºC (CH2Cl2-éter de
petróleo).
IR (KBr, cm-1
): 2920, 2848, 1551, 1434, 1308 (C=S st), 1063.
RMN-1H (CDCl3, 400 MHz) δ: 4.09 (t, J = 7.9 Hz, 2 H, NCH2), 1.76-1.72 (m, 2 H, NCH2CH2),
1.37-1.25 (m, 18 H, 9 CH2) y 0.88 (t, J = 7.0 Hz, CH3) ppm.

310 Tesis Doctoral Susana Barriga Falcón
RMN-13
C (CDCl3, 100 MHz) δ: 201.94 (C=S), 155.38 (C=C-CS), 149.07 (C=C-CS),
47.97 (NCH2), 31.84 (CH2), 29.56 (CH2), 29.50 (CH2), 29.41 (CH2), 29.26 (CH2),
26.77 (CH2), 22.62 (CH2) y 14.06 (CH3) ppm.
EM (IE) m/z (%): 479 (M+·, 12), 447 (M+ - S, 100), 414 (25), 280 (20), 192 (16),
100 (25), 64 (82), 55 (56), 43 (78).

Parte experimental 311
S
N
S
SS
S
C12H25 SS
321

312 Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
C12H25 SS
321

Parte experimental 313
31. Reacción de cicloadición de la bisditiolotiazina 321 con cuatro equivalentes de
dibenzoilacetileno (DBA)
S
N
S
SS
S
SS COPhPhOC
S
N
SS
COPh
COPhPhOC
PhOC
C12H25
SS
COPh
COPh
SS
PhOCCOPh
321 323
4
C6H6, 93%
C12H25
Sobre una disolución agitada de 4-lauril-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(20 mg, 0.042 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(59 mg, 0.252 mmol). La mezcla de reacción se calentó a reflujo durante 60 minutos. Se
añadió una porción adicional de dibenzoilacetileno (38 mg, 0.162 mmol) y se calentó
durante 30 minutos. Finalmente se eliminó el disolvente en el rotavapor, y el residuo
resultante se purificó mediante cromatografía líquida de media presión (SiO2, éter de
petróleo-diclorometano 1:1, hasta diclorometano), lo que permitió separar el compuesto 323
puro (55 mg, 0.039 mmol, 93%), como un sólido anaranjado; p.f.: 65-66 ºC (CH2Cl2-éter de
petróleo).
IR (KBr, cm-1
): 2923, 2852, 1663 (C=O st), 1596, 1578, 1539, 1448, 1258, 1242.
RMN-1H (CDCl3, 400 MHz) δ: 7.92-7.06 (m, 40 H, 8 C6H5), 3.78 (m, 2 H, NCH2),
1.26 (m, 20 H, 10 CH2) y 0.88 (t, J = 7.0 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 193.06 (C=O), 189.83 (C=O), 187.09 (C=O),
186.61 (C=O), 143.04 (C sp2), 137.36 (C sp
2), 137.26 (C sp
2), 137.16 (C sp
2),
137.04 (C sp2), 135.27 (C sp
2), 134.45 (CH Ar), 133.89 (CH Ar), 133.52 (C sp
2),
132.85 (CH Ar),132.79 (C sp2), 132.49 (C sp
2), 129.85 (CH Ar), 129.63 (CH Ar),
128.76 (CH Ar),128.73 (CH Ar), 128.65 (CH Ar), 128.24 (CH Ar), 128.21 (CH Ar),
71.11 (C espirano), 58.04 (CH2), 53.34 (CH2), 31.88 (CH2), 29.72 (CH2), 29.62 (CH2),
29.59 (CH2), 29.30 (CH2), 28.03 (CH2), 27.34 (CH2), 22.64 (CH2) y 14.06 (CH3) ppm.

314 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 1416 (M+·
+ 1, 9), 1309 (M+ - PhCO, 1), 1118 (M
+ - DBA - 2 S, 2),
531 (2), 207 (15), 147 (25), 133 (15), 105 (PhCO+, 100), 91 (40), 73 (95) y 55 (80).
Análisis: Calculado para C82H65NO8S7: C, 69.51; H, 4.62; N, 0.99; hallado: C, 69.38;
H, 4.66; N, 0.97.
Parte experimental 315
S
N
SS
COPh
COPhPhOC
PhOC
C12H25
SS
COPh
COPh
SS
PhOC
COPh
323
316 Tesis Doctoral Susana Barriga Falcón
S
N
SS
COPh
COPhPhOC
PhOC
C12H25
SS
COPh
COPh
SS
PhOC
COPh
323

Parte experimental 317
32. Síntesis de diisopropilestearilamida (318)
HN
C17H35 Cl
O
N
C17H35 O
+
CH2Cl2, 88%
314 316 318
Sobre una disolución agitada de diisopropilamina (4.60 mL, 3.34 g, 33.00 mmol) en
diclorometano (100 mL) se añadió lentamente cloruro de estearoilo (5.00 g, 16.50
mmol). Tras completar la adición, la mezcla de reacción se agitó a temperatura ambiente
durante 4 horas. A continuación se vertió sobre agua (50 mL) y se separaron la fases. La
fase acuosa se extrajo con diclorometano (3 x 20 mL) y los extractos orgánicos
combinados se lavaron con una disolución acuosa saturado de NaHCO3 (3 x 50 mL) y se
secaron sobre sulfato sódico. A continuación se eliminó el disolvente en el rotavapor y
se obtuvo el compuesto 318 (5.34 g, 14.52 mmol, 88%) como un sólido de color beige,
p. f.: 33-34 ºC, que se utilizó sin purificación en la siguiente etapa.
IR (KBr cm-1
): 2917 (C-H st), 2814 (C-H st), 1641 (C=O st), 1467, 1368, 1217, 1134,
1042, 721, 606.
RMN-1H (CDCl3, 400 MHz) δ: 3.95-3.90 (m, 1 H, CH(CH3)2),
3.48-3.423 (m, 1 H, CH(CH3)2), 2.21 (t, J = 7.1 Hz, 2 H, NCOCH2),
1.57-1.53 (m, 2 H, NCOCH2CH2), 1.32 (d, J = 7.1 Hz, 6 H, CH(CH3)2),
1.24-1.20 (m, 30 H, 15 CH2), 1.13 (d, J = 7.1 Hz, 6 H, CH(CH3)2) y
0.82 (t, J = 7.1 Hz, 3 H, CH2) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 171.91 (C=O), 48.11 (CH(CH3)2), 45.33 (NCH2),
35.30 (CH2), 31.81 (CH2), 29.57 (CH2), 29.38 (CH2), 25.35 (CH2), 22.57 (CH2),
20.90 (CH2), 20.59 (CH(CH3)2) y 13.98 (CH3) ppm.

318 Tesis Doctoral Susana Barriga Falcón
0.0
0.0
0.5
0.5
1.0
1.0
1.51.52.02.02.52.53.03.03.53.54.04.04.54.55.0
5.0
5.5
5.5
6.0
6.0
6.5
6.5
7.0
7.0
7.5
7.5
8.0
8.0
25 25 50507575100100125125150 150 175 175 200 200

Parte experimental 319
N
C17H35 O
318

320 Tesis Doctoral Susana Barriga Falcón
33. Síntesis de diisopropilestearilamina (320)
N
C17H35 O
318
LiAlH4
THF, 91%
N
C17H35
320
Sobre una disolución agitada de diisopropilestearilamida (5.00 g, 13.60 mmol) en
tetrahidrofurano (100 mL) se añadió lentamente hidruro de litio y aluminio
(1.03 g, 27.20 mmol). Tras completar la adición la mezcla de reacción se calentó a
reflujo durante 96 horas. A continuación la mezcla de reacción se vertió sobre una
disolución THF-H2O 1:1 (60 mL). Seguidamente se filtraron las sales y éstas se lavaron
con agua y con éter dietílico. El filtrado se extrajo con éter dietílico (6 x 50 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico. Tras eliminar el
disolvente en el rotavapor se obtuvo compuesto 320 (4.38 g, 12.38 mmol, 91%) como
un aceite de color amarillo que se utilizó sin ningún tipo de purificación posterior.
IR (KBr cm-1
): 2924 (C-H st), 2853 (C-H st), 1466, 1360, 1206, 721.
RMN-1H (CDCl3, 200 MHz) δ: 3.62-3.52 (m, 2 H, 2 CH(CH3)2), 2.92-2.79 (m, 2 H, NCH2),
1.92-1.90 (m, 2 H, CH2), 1.43-1.39 (m, 12 H, 2 CH(CH3)2), 1.27-1.21 (m, 30 H, 15 CH2) y
0.84 (t, J = 6.5 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 500 MHz) δ: 53.72 (NCH2), 47.21 (CH(CH3)2), 32.71 (CH2),
31.81 (CH2), 29.58 (CH2), 29.38 (CH2), 29.25 (CH2), 28.82 (CH2), 27.29 (CH2),
26.78 (CH2), 25.67 (CH2), 22.57 (CH(CH3)2), 18.00 (CH2) y 14.01 (CH3) ppm.

Parte experimental 321
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.5
00252550507575100100125125150150175175200200
N
C17H35
320

322 Tesis Doctoral Susana Barriga Falcón
N
C17H35
320

Parte experimental 323
34. Síntesis de 4-estearil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona (322)
N
C18H37
S
N
S
SS
S
C18H37 SS
1) S2Cl2, DABCO
2) Et3N
CHCl3, 20%
320 322
Se adicionó lentamente dicloruro de diazufre (2.26 mL, 28.3 mmol) a una disolución de
N-estearildiisopropilamina (1.0 g, 2.83 mmol) y 1,4-diazabiciclo[2.2.2]octano
(DABCO, 2.54 g, 22.7 mmol) en cloroformo (100 mL) mantenida bajo nitrógeno y
enfriada a -40 ºC. La mezcla se agitó bajo nitrógeno durante 15 minutos a -40 ºC, y
después durante tres días a temperatura ambiente. A continuación la mezcla de reacción
se enfrió a 0 ºC y se añadió lentamente trietilamina (5.1 mL, 36.6 mmol). Tras
completar la adición la mezcla de reacción se agitó a temperatura ambiente durante
2 horas. Seguidamente la mezcla de reacción se filtró a través de celita y se eliminó el
disolvente en el rotavapor. El residuo se disolvió en diclorometano (100 mL), se filtró y
se lavó con ácido sulfúrico al 70% v/v (15 mL). La fase orgánica se extrajo con ácido
sulfúrico concentrado (95-98%, 20 mL). La fase de ácido sulfúrico concentrado se vertió
sobre hielo (200 g) y se extrajo con diclorometano (3 x 50 mL). La fase orgánica se lavó
con agua (3 x 30 mL), se secó sobre sulfato magnésico anhidro y se concentró en el
rotavapor. El crudo se purificó por cromatografía líquida de media presión (SiO2, éter de
petróleo hasta diclorometano). Esto permitió obtener el compuesto 322 puro
(319 mg, 0.57 mmol, 20%) como un sólido negro; p.f.: 134-135 ºC (CH2Cl2-éter de
petróleo).
IR (KBr, cm-1
): 2919, 2847, 1451, 1310 (C=S st), 1065.
RMN-1H (CDCl3, 400 MHz) δ: 4.10 (t, J = 7.9 Hz, 2 H, NCH2), 1.76-1.72 (m, 2 H, NCH2CH2),
1.37-1.25 (m, 30 H, 15 CH2) y 0.88 (t, J = 7.0 Hz, CH3) ppm.

324 Tesis Doctoral Susana Barriga Falcón
RMN-13
C (C5D5N, 50 MHz) δ: 203.75 (C=S), 162.15 (C=C-CS), 149.64 (C=C-CS),
48.42 (NCH2), 32.01 (CH2), 29.96 (CH2), 29.88 (CH2), 29.81 (CH2), 29.76 (CH2), 29.73 (CH2),
29.62 (CH2), 29.54 (CH2), 29.50 (CH2), 27.15 (CH2), 22.83 (CH2) y 14.18 (CH3) ppm.
EM (FAB) m/z (%): 564 (M+ + 1, 2), 532 (M
+ + 1 - S, 4), 500 (M
+ + 1 - 2 S, 3),
312 (M+ + 2 - C18H37, 7), 280 (3), 248 (4), 216 (4), 192 (3), 69 (52), 55 (100).
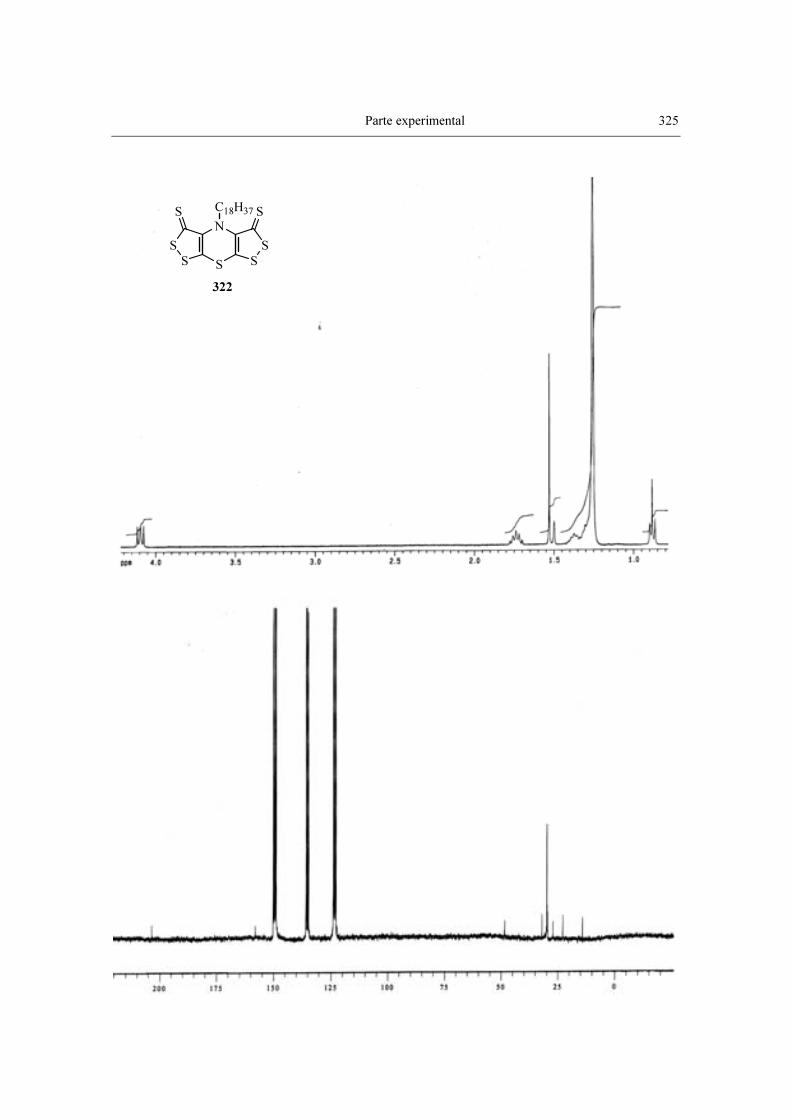
Parte experimental 325
S
N
S
SS
S
C18H37 SS
322

326 Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
C18H37 SS
322

Parte experimental 327
35. Reacción de cicloadición de la bisditiolotiazina 322 con cuatro equivalentes de
dibenzoilacetileno (DBA)
S
N
S
SS
S
SS COPhPhOC
S
N
SS
COPh
COPhPhOC
PhOC
C18H37
SS
COPh
COPh
SS
PhOCCOPh
322 324
4
C6H6, 55%
C18H37
Sobre una disolución agitada de 4-estearil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(50 mg, 0.089 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(125 mg, 0.534 mmol). La mezcla de reacción se calentó a reflujo durante 60 minutos. Se
añadió una porción adicional de dibenzoilacetileno (63 mg, 0.269 mmol) y se calentó durante
30 minutos. Este proceso se repitió dos veces más. Finalmente se eliminó el disolvente en el
rotavapor, y el residuo resultante se purificó mediante cromatografía líquida de media presión
(SiO2, éter de petróleo hasta diclorometano-acetato de etilo 95:5), lo que permitió separar el
compuesto 324 puro (70 mg, 0.049 mmol, 55%), como un sólido anaranjado; p.f.: 76-77 ºC
(CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2923, 2852, 1664 (C=O st), 1596, 1578, 1536, 1448, 1314, 1258, 691.
RMN-1H (CDCl3, 400 MHz) δ: 7.92-7.04 (m, 40 H, 8 C6H5), 3.88 (s ancho, 2 H, NCH2),
1.26 (m, 32 H, 16 CH2) y 0.89 (t, J = 7.0 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 193.20 (C=O), 189.88 (C=O), 187.19 (C=O),
186.70 (C=O), 143.14 (C sp2), 137.39 (C sp
2), 137.27 (C sp
2), 137.21 (C sp
2),
137.14 (C sp2), 135.26 (C sp
2), 134.55 (CH Ar), 133.97 (CH Ar), 133.44 (C sp
2),
132.94 (CH Ar), 132.87 (C sp2), 132.49 (C sp
2), 129.88 (CH Ar), 129.64 (CH Ar),
128.81 (CH Ar), 128.68 (CH Ar), 128.30 (CH Ar), 128.28 (CH Ar), 126.87 (CH Ar),
71.03 (C espirano), 32.28 (CH2), 31.92 (CH2), 29.78 (CH2), 29.72 (CH2), 29.66 (CH2),
29.32 (CH2), 28.02 (CH2), 27.36 (CH2), 22.85 (CH2), 22.69 (CH2) y 14.14 (CH3) ppm.

328 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 1500 (M+ + 1, 100), 1396 (M
+ + 2 - PhCO, 30),
1202 (M+ + 1 - DBA - 2 S, 78), 1096 (M
+ - PhCO - DBA - 2 S, 20), 904 (M
+ + 1 - 2 DBA - 4 S, 22),
545 (M+ + 1 - 3 DBA - C18H37, 45), 439 (M
+ - 3 DBA - C18H37 - PhCO, 72).
Análisis: Calculado para C88H77NO8S7: C, 70.42; H, 5.17; N, 0.93; hallado: C, 70.66;
H, 5.16; N, 0.80.

Parte experimental 329
S
N
SS
COPh
COPhPhOC
PhOC
C18H37
SS
COPh
COPh
SS
PhOC
COPh
324

330 Tesis Doctoral Susana Barriga Falcón
S
N
SS
COPh
COPhPhOC
PhOC
C18H37
SS
COPh
COPh
SS
PhOC
COPh
324

Parte experimental 331
36. Reacción de cicloadición de la bisditiolotiazina 322 con dos equivalentes de
acetilendicarboxilato de dimetilo (DMAD)
S
N
S
SS
S
SSC18H37
S
N
S S
S
SS
SCO2CH3
CO2CH3H3CO2C
H3CO2C
H3CO2C CO2CH3
325
C6H6, 56%
322
C18H37
2
Sobre una disolución agitada de 4-estearil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(35 mg, 0.062 mmol) en benceno (5 mL) se añadió acetilendicarboxilato de dimetilo
(19 µL, 22 mg, 0.155 mmol). La mezcla de reacción se calentó a reflujo durante 20 minutos. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano, 3:2), lo que permitió
separar el compuesto 325 puro (30 mg, 0.035 mmol, 56%), como un sólido aceitoso de color
rojo.
IR (KBr, cm-1): 2956, 2924, 2852, 1736 (C=O st), 1729 (C=O st), 1260 (C=S st).
RMN-1H (CDCl3, 200 MHz) δ: 3.98 (s, 6H, 2 OCH3), 3.93 (s, 6H, 2 OCH3),
3.29-3.21(m, 2 H, NCH2), 1.24-1.21 (m, 32 H, 16 CH2) y 0.87 (t, J = 6.4 Hz, 3 H, CH3)
ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 195.58 (C=S), 160.46 (C=O), 158.82 (C=O),
159.58 (C sp2), 133.57 (C sp
2), 133.47 (C sp
2), 132.83 (C sp
2), 55.42 (NCH2),
53.92 (OCH3), 53.79 (OCH3), 53.75 (CH2), 53.70 (CH2), 53.63 (CH2), 53.41 (CH2),
53.16 (CH2), 53.04(CH2), 31.89 (CH2), 29.66 (CH2), 29.56 (CH2), 29.48 (CH2),
29.35 (CH2), 29.32 (CH2), 29.26 (CH2), 28.18 (CH2), 27.27 (CH2), 22.66 (CH2) y
14.08 (CH3) ppm.
EM (FAB) m/z (%): 848 (M+ + 1, 4), 816 (M
+ + 1 - S, 1), 594 (M
+ - C18H37, 5),
562 (M+ - C18H37 - S, 3), 275 (4), 69 (62), 55 (85).

332 Tesis Doctoral Susana Barriga Falcón
Análisis: Calculado para C36H49NO8S7: C, 50.97; H, 5.82; N, 1.65; hallado: C, 51.37;
H, 5.79; N, 1.76.

Parte experimental 333
252550507575100100125125150150175175200200
S
N
S S
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
325
C18H37

334 Tesis Doctoral Susana Barriga Falcón
S
N
S S
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
325
C18H37

Parte experimental 335
37. Reacción de cicloadición de la bisditiolotiazina 186 con dos equivalentes de
acetilendicarboxilato de dimetilo (DMAD)
S
N
S
SS
S
SS
S
N
S S
S
SS
SCO2CH3
CO2CH3H3CO2C
H3CO2C
H3CO2C CO2CH3
ClCl
C6H6, 68%
2
326186
Sobre una disolución agitada de 4-cloroetil-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(15 mg, 0.040 mmol) en benceno (5 mL) se añadió acetilendicarboxilato de dimetilo
(12 µL, 14 mg, 0.100 mmol). La mezcla de reacción se calentó a reflujo durante 45 minutos. A
continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se purificó
mediante cromatografía en capa fina preparativa (SiO2, diclorometano dos veces), lo que
permitió separar el compuesto 326 puro (18 mg, 0.027 mmol, 68%), como un sólido rojo;
p.f.: 178-179 ºC (CH2Cl2).
IR (KBr, cm-1): 1732 (C=O st), 1712 (C=O st), 1574, 1453, 1428, 1448, 1268 (C=S),
1019, 978.
RMN-1H (CDCl3, 250 MHz) δ: 3.96 (s, 6 H, 2 OCH3), 3.91 (s, 6 H, 2 OCH3),
3.77-3.71 (m, 2 H, CH2) y 3.66-3.59 (m, 2 H, CH2) ppm.
RMN-13
C (CDCl3, 62.5 MHz) δ: 195.42 (C=S), 160.81 (C sp2), 159.60 (C=O),
159.46 (C=O), 133.53 (C sp2), 133.10 (C sp
2), 132.24 (C sp
2), 55.94 (CH2Cl),
54.04 (OCH3), 53.80 (OCH3) y 40.77 (NCH2) ppm.
EM (FAB) m/z (%): 658 (M+ + 1, 5), 626 (M
+ + 1 - S, 1), 594 (M
+ - CH2CH2Cl , 6),
563 (M+ + 1 - 2 CO2CH3 - CH2CH2Cl - CH3, 2), 399 (M
+ + 1 - DMAD - CH2CH2Cl - CS, 3)
147 (25), 91 (45), 81 (32), 73 (100), 55 (60).

336 Tesis Doctoral Susana Barriga Falcón
Análisis: Calculado para C20H16ClNO8S7: C, 36.50; H, 2.45; N, 2.13; hallado: C, 36.55;
H, 2.35; N, 1.91.
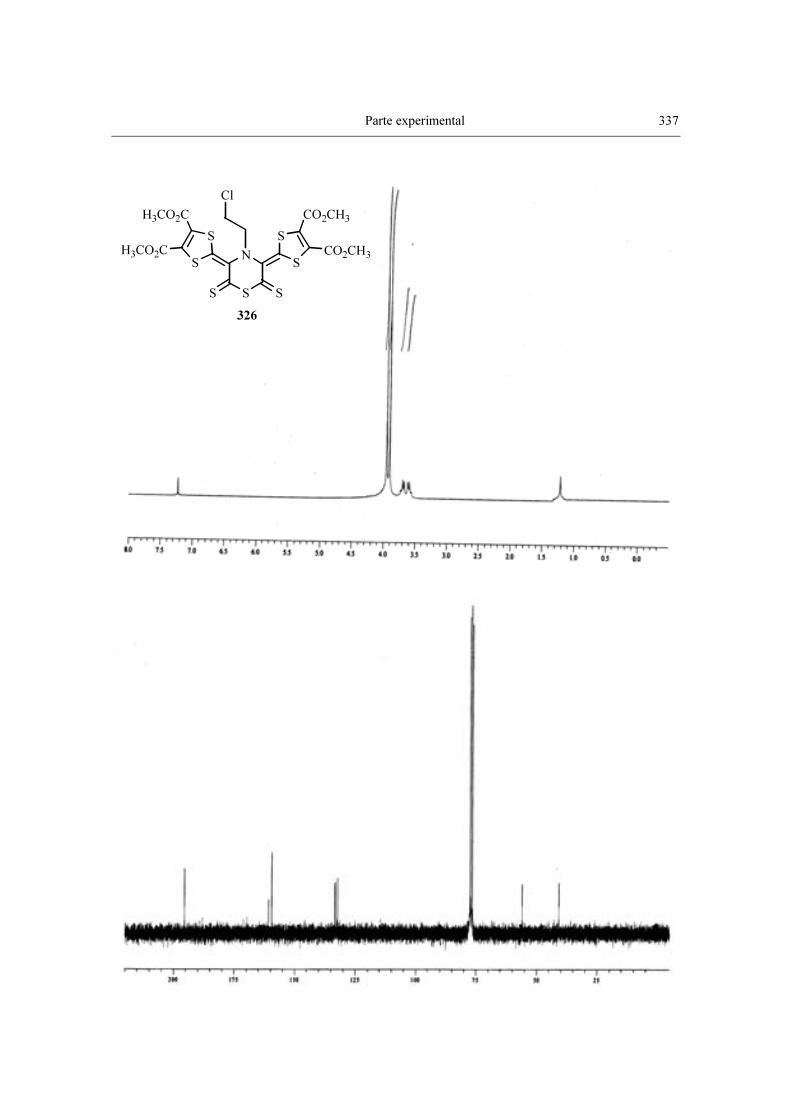
Parte experimental 337
S
N
S S
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
Cl
326

338 Tesis Doctoral Susana Barriga Falcón
S
N
S S
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
Cl
326

Parte experimental 339
38. Reacción de cicloadición de la tiazina 190 con dos equivalentes de
dibenzoilacetileno (DBA)
S
N
S
S
SS
SS PhOC COPh
C6H6, 36%
190 327
2
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC
Sobre una disolución agitada de 5,6-dihidro-4-(3-tiono[1,2]ditiol-4-il)-[1,2]ditiolo-
[3,4-b][1,4]tiazina-3-tiona (20 mg, 0.059 mmol) en benceno (5 mL) se añadió
dibenzoilacetileno158,159,202
(35 mg, 0.149 mmol). La mezcla de reacción se agitó a
temperatura ambiente durante 20 minutos. A continuación se eliminó el disolvente en frío, y el
residuo resultante se purificó mediante cromatografía rápida (SiO2, éter de petróleo-
diclorometano 1:1), lo que permitió separar el compuesto 327 puro (17 mg, 0.021 mmol, 36%),
como un sólido marrón; p.f.: 118-119 ºC (CH2Cl2-éter de petróleo).
IR (KBr, cm-1
): 2923, 1662, 1654 (C=O st), 1647, 1595, 1447, 1261 (C=S st),
1231 (C=S st), 692.
RMN-1H (CDCl3, 400 MHz) δ: 10.74 (s, 1 H, CSH), 7.50-7.43 (m, 10 H, 2 C6H5),
7.30-7.24 (m, 10 H, 2 C6H5),4.14-4.10 (m, 1 H, ½ CH2), 3.63-3.42 (m, 1 H, CH2) y
3.12-3.02 (m, 1 H, ½ CH2) ppm.
RMN-13
C (CD2Cl2, 75 MHz) δ: 199.20 (CSH), 197.69 (C=S), 187.59 (C=O),
187.23 (C=O), 187.13 (C=O), 159.88 (C sp2), 155.79 (C sp
2), 142.89 (C sp
2),
141.33 (C sp2), 140.61 (C sp
2), 140.16 (C sp
2), 137.34 (CH Ar), 137.19 (CH Ar),
136.01 (C sp2), 134.73 (CH Ar),134.64 (CH Ar), 134.51 (CH Ar), 133.83 (C sp
2),
129.46 (CH Ar), 129.40 (CH Ar),129.31 (CH Ar), 129.25 (CH Ar), 46.24 (CH2) y
33.04 (CH2) ppm.

340 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 808 (M+ + 1, 2), 105 (PhCO
+, 50), 77 (Ph
+, 45), 69 (70), 55 (100).

Parte experimental 341
327
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC

342 Tesis Doctoral Susana Barriga Falcón
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
327
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC

Parte experimental 343
39. Reacción de cicloadición de la tiazina 190 con tres equivalentes de
dibenzoilacetileno (DBA)
S
N
S
S
SS
SS
S
N
SPhOC
PhOC
SS
PhOC
PhOCS
SS
COPh
COPh
C6H6
190 328 (40%)
3 DBA
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
329 (20%)
+
Sobre una disolución agitada de 5,6-dihidro-4-(3-tiono[1,2]ditiol-4-il)-[1,2]ditiolo-
[3,4-b][1,4]tiazina-3-tiona (25 mg, 0.074 mmol) en benceno (5 mL) se añadió
dibenzoilacetileno158,159,202
(60 mg, 0.256 mmol). La mezcla de reacción se agitó a
temperatura ambiente durante 45 min. A continuación se eliminó el disolvente en el
rotavapor, y el residuo resultante se purificó mediante cromatografía rápida (SiO2, éter
de petróleo-diclorometano 1:1), lo que permitió separar el compuesto 328 puro
(31 mg, 0.030 mmol, 40%), como un sólido marrón; p.f.: 109-110 ºC (CH2Cl2-éter de
petróleo) y el compuesto 329 puro (16 mg, 0.015 mmol, 20%) como un sólido
anaranjado; p.f.: 116-117 ºC (CH2Cl2-éter de petróleo).
Datos espectroscópicos de 328:
IR (KBr, cm-1
): 2923, 1655 (C=O st), 1649, 1595, 1447, 1260, 1177, 692.
RMN-1H (CDCl3, 400 MHz) δ: 10.87 (s, 1 H, CSH), 8.06-6.95 (m, 30 H, 6 C6H5),
4.00 (d, J = 13.6 Hz, 2 H, CH2), 3.39-3.23 (m, 2 H, CH2) y
3.11 (d, J = 13.6 Hz, 2 H, CH2) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 199.77 (CSH), 193.03 (C=S), 189.79 (C=O),
187.31 (C=O), 187.09 (C=O), 186.45 (C=O), 160.51 (C sp2), 142.77 (C sp
2),
138.64 (C sp2), 137.01 (C sp
2), 134.26 (CH Ar), 133.91 (C sp
2), 133.88 (CH Ar),
133.81 (CH Ar), 133.04 (CH Ar),132.98 (CH Ar), 130.06 (CH Ar), 129.92 (CH Ar),

344 Tesis Doctoral Susana Barriga Falcón
129.70 (CH Ar), 129.20 (CH Ar), 128.87 (CH Ar), 128.82 (CH Ar), 128.66 (CH Ar),
128.46 (CH Ar), 128.27 (CH Ar), 128.22 (CH Ar), 128.15 (CH Ar), 119.99 (C sp2),
68.13 (C espirano), 49.21 (CH2) y 26.73 (CH2) ppm.
EM (FAB) m/z (%): 1042 (M+ + 1, 2), 711 (M
+ - DBA - 3 S, 3), 689 (M
+ - DBA - 2 S - CSH, 2),
638 (M+ - DBA - 2 S- PhCO, 3), 105 (PhCO
+, 70), 69 (80), 55 (100).
Análisis: Calculado para C56H35NO6S7: C, 64.53; H, 3.38; N, 1.34; hallado: C, 64.36;
H, 3.54; N, 1.23.
Datos espectroscópicos de 329:
IR (KBr, cm-1
): 2924, 1655 (C=O st), 1648, 1595, 1448, 1260, 1230 (C=S st), 1177,
690.
RMN-1H (CDCl3, 200 MHz) δ: 7.86-7.19 (m, 30 H, 6 C6H5), 6.43 (HC=C) y
4.28-3.14 (m, 4 H, 2 CH2) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 197.17 (C=S), 193.38 (C=O), 190.77 (C=O),
187.31 (C=O), 186.94 (C=O), 186.25 (C=O), 159.16 (C sp2), 141.45 (C sp
2),
136.71 (C sp2), 134.58 (CH Ar), 134.07 (CH Ar), 134.02 (CH Ar), 133.41 (CH Ar),
130.33 (CH Ar), 130.11 (CH Ar), 129.20 (CH Ar), 129.08 (CH Ar), 128.98 (CH Ar),
128.87 (CH Ar), 128.72 (CH Ar), 128.58 (CH Ar), 128.38 (CH Ar),128.28 (CH Ar),
128.23 (CH Ar), 128.15 (CH Ar), 110.47 (HC=C), 67.51 (C espirano), 48.75 (CH2) y
26.66 (CH2) ppm.
EM (FAB) m/z (%): 1042 (M+ + 1, 2), 711 (M
+ - DBA - 3 S, 3), 105 (PhCO
+, 50),
69 (80), 55 (100).

Parte experimental 345
S
N
SPhOC
PhOC
S
S
PhOC
PhOCS
SS
COPh
COPh
328

346 Tesis Doctoral Susana Barriga Falcón
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
SPhOC
PhOC
S
S
PhOC
PhOCS
SS
COPh
COPh
328

Parte experimental 347
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
329

348 Tesis Doctoral Susana Barriga Falcón
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
329

Parte experimental 349
40. Reacción de cicloadición de la tiazina 190 con cuatro equivalentes de
dibenzoilacetileno (DBA)
S
N
S
S
SS
SS PhOC COPh
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
SS
PhOC
PhOC
C6H6, 68%
190 330
4
Sobre una disolución agitada de 5,6-dihidro-4-(3-tiono[1,2]ditiol-4-il)-[1,2]ditiolo-
[3,4-b][1,4]tiazina-3-tiona (20 mg, 0.059 mmol) en benceno (5 mL) se añadió
dibenzoilacetileno158,159,202
(69 mg, 0.295 mmol). La mezcla de reacción se calentó a
reflujo durante 60 minutos. Se añadió una porción adicional de dibenzoilacetileno
(20 mg, 0.085 mmol) y se calentó durante 30 minutos. Este proceso se repitió dos veces
más. Finalmente se eliminó el disolvente en el rotavapor, y el residuo resultante se
purificó mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano 1:1, hasta
diclorometano), lo que permitió separar el compuesto 330 puro
(51 mg, 0.040 mmol, 68%), como un sólido amarillo; p.f.: 209-210 ºC (CH2Cl2).
IR (KBr, cm-1
): 1660 (C=O st), 1595, 1579, 1534, 1448, 1258, 1173, 691.
RMN-1H (CDCl3, 400 MHz) δ: 8.00-6.88 (m, 40 H, 8 C6H5), 6.31 (s, 1 H, CH),
5.34 (d, J = 13 Hz, 1 H, CH2), 3.64-3.48 (m, 2 H, CH2) y 3.32 (d, J = 13 Hz, 1 H, CH2) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 193.02 (C=O), 191.08 (C=O), 189.57 (C=O),
188.04 (C=O), 187.17 (C=O), 186.82 (C=O), 139.52 (C sp2), 137.40 (C sp
2),
137.28 (C sp2), 133.20 (CH Ar), 132.99 (CH Ar), 130.40 (C sp
2), 130.11 (C sp
2),
129.62 (CH Ar), 129.49 (CH Ar), 129.01 (CH Ar), 128.95 (CH Ar), 128.80 (CH Ar),
128.54 (CH Ar), 128.40 (CH Ar), 128.36 (CH Ar), 128.32 (CH Ar), 128.27 (CH Ar),
128.20 (CH Ar), 128.11 (CH Ar), 69.61 (C espirano), 67.71 (C espirano), 49.47 (CH2)
y 25.77 (CH2) ppm.

350 Tesis Doctoral Susana Barriga Falcón
EM (FAB) m/z (%): 1275 (M+·
, 9), 1169 (M+ + 1 - PhCO, 1), 1009 (M
+- DBA - S, 2),
711 (M+ - 2 DBA - 3 S, 2), 679 (M
+ - 2 DBA - 4 S, 2), 105 (PhCO
+, 100).
Análisis: Calculado para C72H45NO8S7: C, 67.74; H, 3.55; N, 1.10; hallado: C, 67.56;
H, 3.47; N, 1.18.

Parte experimental 351
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
S
S
PhOC
PhOC
330
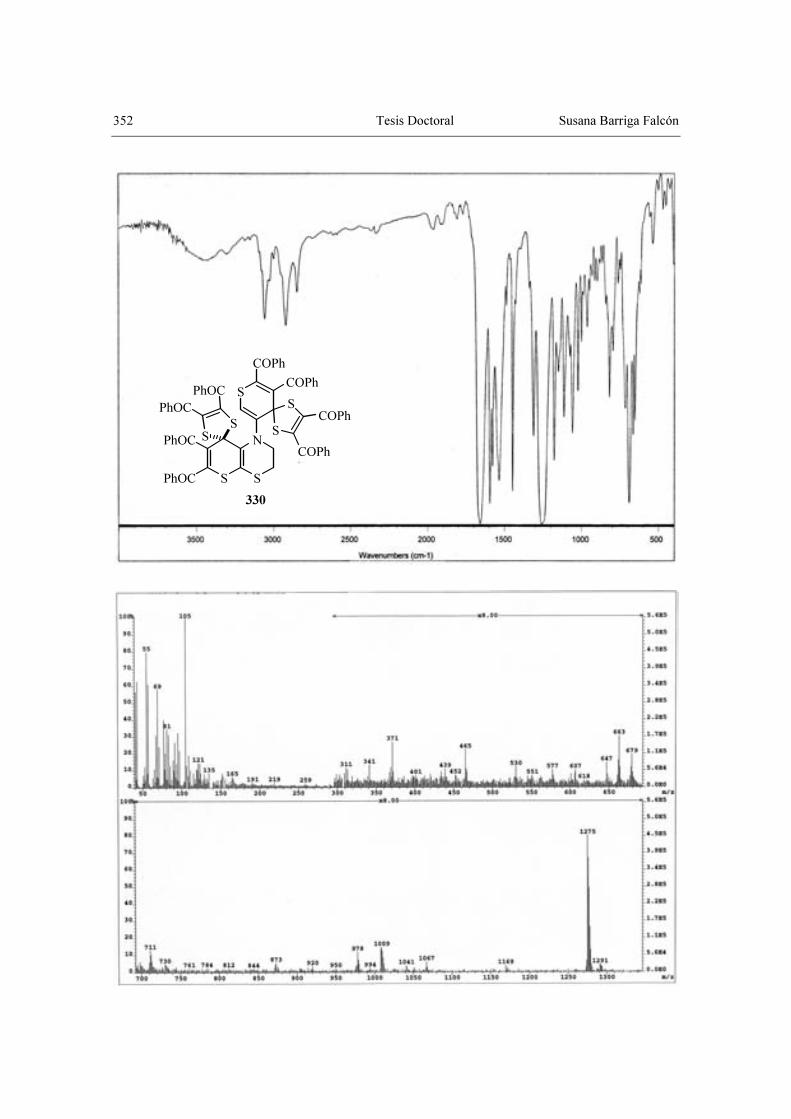
352 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
S
S
PhOC
PhOC
330

Parte experimental 353
41. Reacción de cicloadición del bisditiolopirrol 182 con acetilendicarboxilato de
dimetilo (DMAD)
N
SSS
S
OS
182
CO2CH3H3CO2C
N
SS
O
SH3CO2C
H3CO2C
331
SS
CO2CH3
H3CO2C2
Tolueno, 65%
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-5-tiona
(116 mg, 0.40 mmol) en tolueno (15 mL) se añadió acetilendicarboxilato de dimetilo
(123 µL, 142 mg, 1.00 mmol). La mezcla de reacción se agitó y se calentó a reflujo
durante dos horas. A continuación se eliminó el disolvente en el rotavapor, y el residuo
resultante se purificó mediante cromatografía líquida de media presión (SiO2, éter de
petróleo hasta diclorometano-acetato de etilo, 20:1), lo que permitió separar el
compuesto 331 puro (150 mg, 0.26 mmol, 65%), como un sólido amarillo;
p.f.: 218-220 ºC (CH2Cl2-éter de petróleo).
IR (KBr cm-1
): 2955, 2196, 1739 (C=O st, éster), 1703 (C=O st, éster),
1667 (C=O st, heterociclo), 1573, 1430, 1249, 1208.
RMN-1H (CDCl3, 400 MHz) δ: 4.76 (c, J = 7.0 Hz, 2 H, NCH2CH3), 3.90 (s, 3 H, CH3),
3.88 (s, 3 H, CH3), 3.82 (s, 6 H, 2 CH3) y 1.58 (t, J = 7.0 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 179.30 (C=O), 165.00 (C=O), 162.84 (C=O),
160.09 (C=O), 135.59 (C sp2), 129.88 (C sp
2), 129.00 (C sp
2), 127.17 (C sp
2),
125.78 (C sp2), 104.71 (C sp
2), 63.97 (C espirano), 54.00 (OCH3), 53.42 (OCH3),
53.33 (OCH3), 52.89 (OCH3), 42.80 (NCH2CH3) y 15.87 (NCH2CH3) ppm.
EM (IE) m/z (%): 575 (M+·
, 13), 516 (M+ - CO2CH3, 35),
456 (M+ - CH2CH3 - CO2CH3 - OCH3, 5), 400 (M
+ + 1 - DMAD - S, 25),
341 (M+ - DMAD - 2 S - OCH3, 13), 313 (M
+ - DMAD - 2 S - CO2CH3, 5),

354 Tesis Doctoral Susana Barriga Falcón
283 (M+ + 1 - DMAD - 2 S - CO2CH3 - OCH3, 8), 254 (M
+ - DMAD - 2 S - 2 CO2CH3,
4), 206 (C2S2(CO2CH3)2+,4), 103 (CSCO2CH3
+, 6), 59 (CO2CH3
+, 100).
Masa exacta: calculada para C20H17NO9S5: 574.9507; hallada: 574.9505.
Análisis: Calculado para C20H17NO9S5: C, 41.73; H, 2.98; N, 2.43; hallado: C, 41.90; H, 3.00; N, 2.41.

Parte experimental 355
N
SS
O
SH3CO2C
H3CO2C
331
SS
CO2CH3
H3CO2C
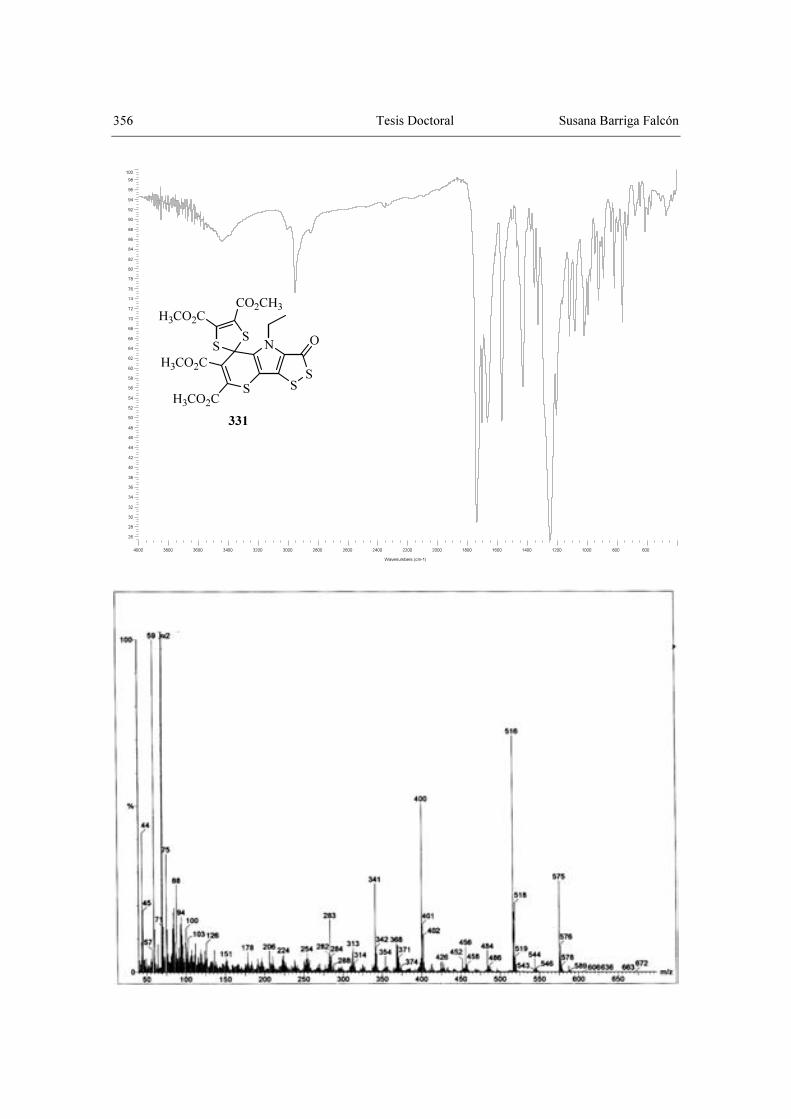
356 Tesis Doctoral Susana Barriga Falcón
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
N
SS
O
SH3CO2C
H3CO2C
331
SS
CO2CH3
H3CO2C

Parte experimental 357
42. Reacción de cicloadición del bisditiolopirrol 182 con dicianoacetileno (DCA)
N
SSS
S
OS
182
CNNC
N
SS
O
SNC
NC
332
SS
CNNC
2
C6H6, 38%
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-5-tiona
(55 mg, 0.189 mmol) en benceno (10 mL) se añadió dicianoacetileno156,157
(36 mg, 0.474 mmol). La mezcla de reacción se calentó a reflujo durante 2 horas. A
continuación se eliminó el disolvente en el rotavapor. El residuo resultante se purificó
mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano-acetato de etilo, 90:10), lo que permitió separar el compuesto 332204
puro
(31 mg, 0.070 mmol, 38%), como un sólido amarillo; p.f.: 228-229 ºC (CH2Cl2-éter de
petróleo).
IR (KBr, cm-1
): 2222 (CN st, débil), 1662 (CO st), 1530, 1063, 914.
RMN-1H (CDCl3, 400 MHz) δ: 4.71 (c, J = 7.0 Hz, 2 H, CH2) y 1.60 (t, J = 7.0 Hz, 3 H, CH3)
ppm.
EM (IE) m/z (%): 443 (M+·
, 32), 428 (M+ - CH3, 14), 414 (M
+ - CH2CH3, 12),
383 (M+ - SCO, 11), 353 (M
+ +1 - SCO - CH2CH3, 12), 335 (M
+ - NCCCCN - S, 90),
320 (M+ - NCCCCN - S - CH3, 27), 307 (M
+ + 1 - NCCCCN - S - CH2CH3, 38),
291 (M+ - DCA - CS2, 12), 276 (M
+ - DCA - CS2 - CH3, 35),
261 (M+ - DCA - CS2 - CH2CH3, 5), 140 (NCC2S2CN
+, 22), 76 (NCCCCN
+, 14),
70 (SCCN+, 100).
204 No se pudo realizar el RMN-13C de 332 ya que el producto es muy insoluble y además, se
descompone en disolución.

358 Tesis Doctoral Susana Barriga Falcón
Masa exacta: calculada para C16H5N5OS5: 442.9083; hallada: 442.9098.
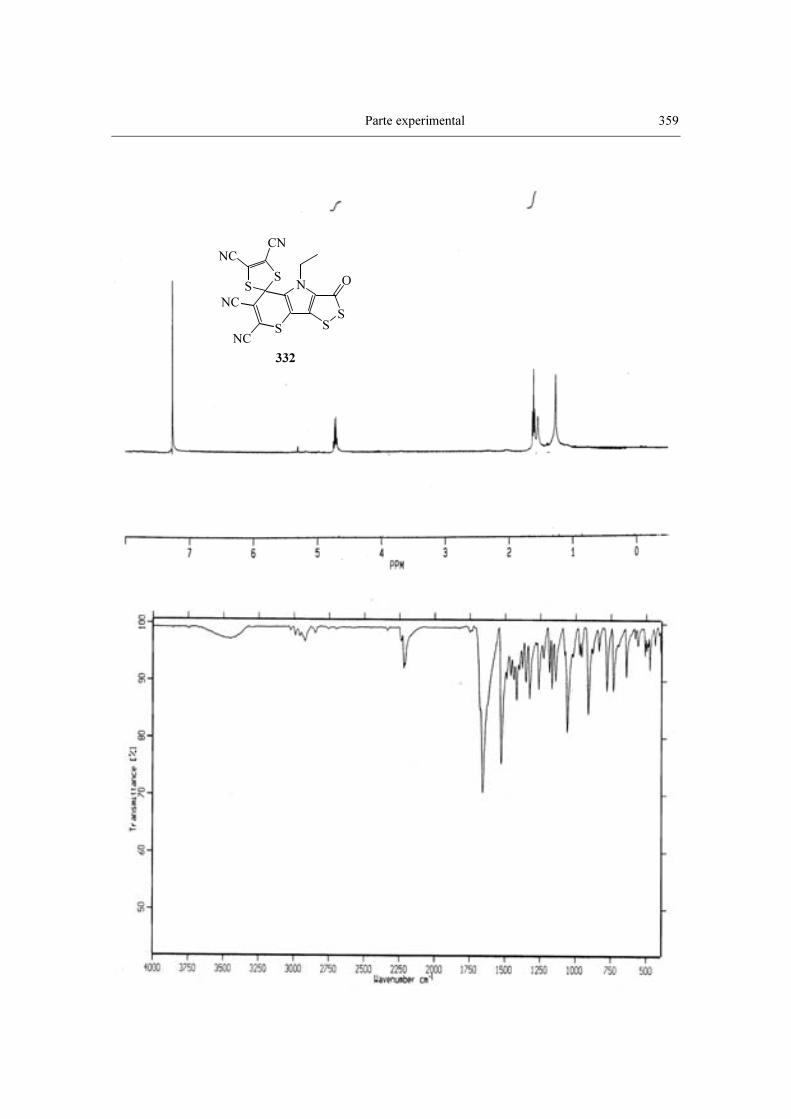
Parte experimental 359
N
SS
O
SNC
NC
332
SS
CN
NC

360 Tesis Doctoral Susana Barriga Falcón
43. Reacción de cicloadición del bisditiolopirrol 182 con dibenzoilacetileno (DBA)
N
SSS
S
OS
182
N
SS
O
SPhOC
PhOC
333
SS
COPh
PhOC
COPhPhOC
C6H6 , 73%
2
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-5-tiona
(100 mg, 0.344 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(201 mg, 0.860 mmol). La mezcla de reacción se agitó y se calentó a reflujo durante 30
minutos. A continuación se eliminó el disolvente en el rotavapor, el residuo resultante se
purificó mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano) lo que permitió separar el compuesto 333 puro
(191 mg, 0.251 mmol, 73%), como un sólido amarillo; p.f.: 242-244 ºC (CH2Cl2-éter de
petróleo).
IR (KBr, cm-1
): 1660 (C=O st), 1594, 1529, 1446, 1244, 689.
RMN-1H (CDCl3, 400 MHz) δ: 7.71-7.19 (m, 20 H, 4 C6H5), 5.00 (c, J = 6.9 Hz, 2 H, CH2)
y 1.73 (t, J = 6.9 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 193.17 (C=O), 191.19 (C=O), 186.55 (C=O),
179.31 (C=O), 138.81 (C sp2), 136.98 (C sp
2), 136.05 (C sp
2), 135.66 (C sp
2),
135.17 (C sp2), 134.70 (CH Ar), 133.84 (CH Ar), 133.47 (CH Ar), 132.24 (C sp
2),
131.05 (C sp2), 130.50 (CH Ar), 130.33 (CH Ar), 129.74 (C sp
2), 129.26 (C sp
2),
128.90 (CH Ar), 128.47 (CH Ar), 128.41 (CH Ar), 104.72 (C sp2), 63.26 (C espirano),
43.03 (CH2) y 16.06 (CH3) ppm.

Parte experimental 361
EM (FAB) m/z (%): 760 (M+ + 1, 10), 654 (M
+ - COPh, 1), 494 (M
+ - DBA- S, 2),
462 (M+ + 1 - DBA- 2 S, 3), 434 (M
+ - DBA- S2CO, 3), 290 (M
+ - 1 - 2 DBA, 38),
105 (PhCO+, 43).
Análisis: Calculado para C40H25NO5S5: C, 63.22; H, 3.32; N, 1.84; hallado: C, 63.33;
H, 3.39; N, 1.82.
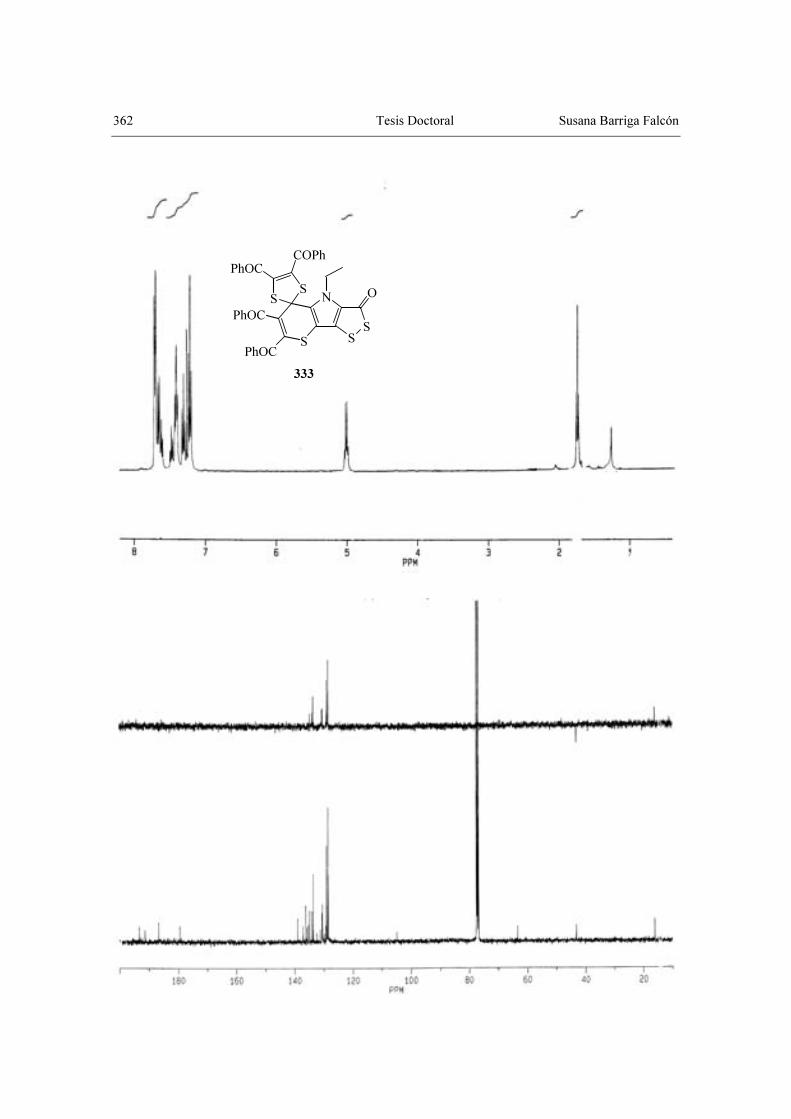
362 Tesis Doctoral Susana Barriga Falcón
N
SS
O
SPhOC
PhOC
333
SS
COPh
PhOC
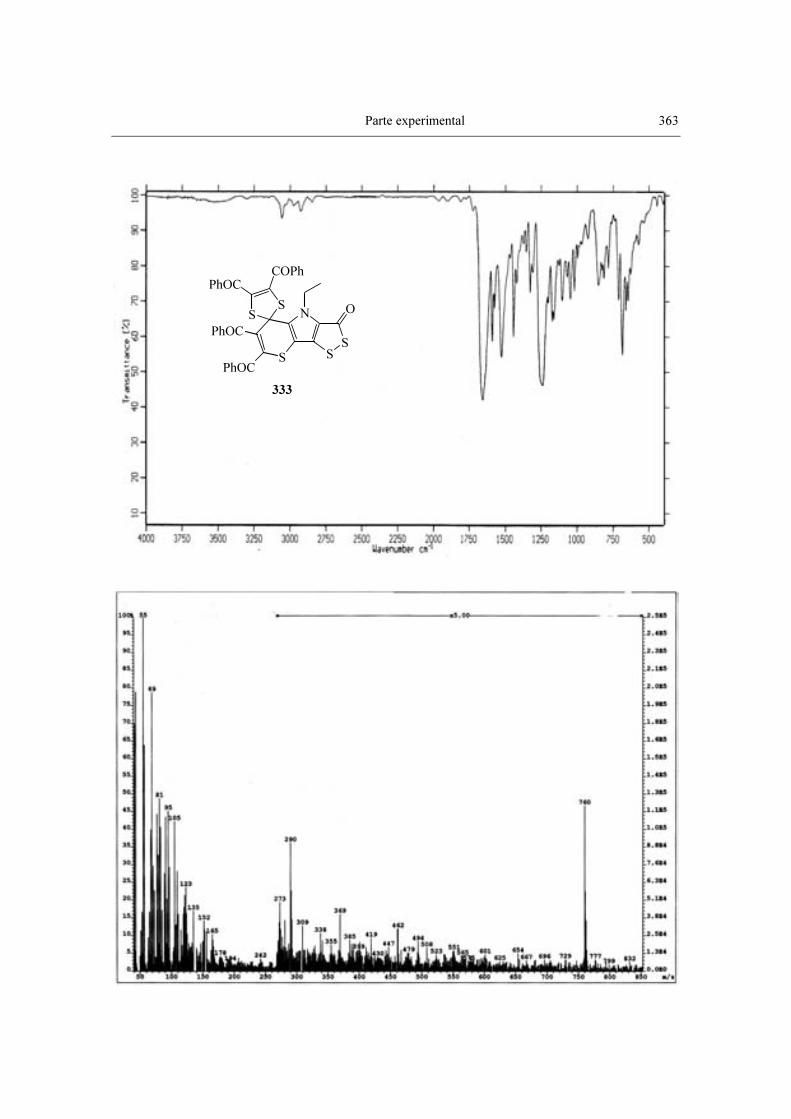
Parte experimental 363
N
SS
O
SPhOC
PhOC
333
SS
COPh
PhOC

364 Tesis Doctoral Susana Barriga Falcón
44. Reacción de cicloadición del bisditiolopirrol 181 con acetilendicarboxilato de
dimetilo (DMAD)
N
SSS
S
SS
181
CO2CH3H3CO2C
N
SH3CO2C
H3CO2C
336
S
SS
CO2CH3
CO2CH3
CO2CH3
CO2CH3
SS
H3CO2C
CO2CH3
4
Tolueno, 61%
Sobre una disolución agitada de 4-etil-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-3,5-ditiona
(100 mg, 0.326 mmol) en tolueno (30 mL) se añadió acetilendicarboxilato de dimetilo
(200 µL, 231 mg, 1.627 mmol). La mezcla de reacción se calentó a reflujo durante
3 horas. A continuación se eliminó el disolvente en el rotavapor, el residuo resultante se
purificó mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano-acetato de etilo, 85:15), lo que permitió separar el compuesto 336 puro
(175 mg, 0.20 mmol, 61%), como un sólido amarillo; p.f.: 226-227 ºC (CH2Cl2-éter de
petróleo).
IR (KBr cm-1
): 1730 (C=O st), 1572, 1433, 1256, 1042, 783.
RMN-1H (CDCl3, 500 MHz) δ: 5.22 (c, J = 8 Hz, 2 H, NCH2CH3),
3.91 (s, 6 H, 2 OCH3), 3.87 (s, 6 H, 2 OCH3), 3.83 (s, 12 H, 4 OCH3) y
1.85 (t, J = 8 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 125 MHz) δ: 165.32 (C=O), 162.83 (C=O), 160.29 (C=O),
131.04 (C sp2), 127.52 (C sp
2), 125.18 (C sp
2), 122.82 (C sp
2), 108.13 (C sp
2),
64.7 (C espirano), 53.84 (OCH3), 53.28 (OCH3), 53.18 (OCH3), 43.30 (NCH2CH3) y
16.56 (NCH2CH3) ppm.

Parte experimental 365
EM (FAB) m/z (%): 876 (M+ + 1, 100), 844 (M
+ - OCH3, 10), 816 (M
+ - CO2CH3, 20),
702 (M+ - DMAD - OCH3, 5), 670 (M
+ + 1 - DMAD- 2 S, 6),
551 (M+ + 1 - DMAD- 2 S -2 CO2CH3, 4), 495 (M
+ - 2 DMAD- 3 S, 5).
Análisis: Calculado para C32H29NO16S6: C, 43.88; H, 3.34; N, 1.60; hallado: C, 43.62;
H, 3.31; N, 1.49.

366 Tesis Doctoral Susana Barriga Falcón
N
SH3CO2C
H3CO2C
336
S
SS
CO2CH3
CO2CH3
CO2CH3
CO2CH3
SS
H3CO2C
CO2CH3

Parte experimental 367
N
SH3CO2C
H3CO2C
336
S
SS
CO2CH3
CO2CH3
CO2CH3
CO2CH3
SS
H3CO2C
CO2CH3

368 Tesis Doctoral Susana Barriga Falcón
45. Reacción de cicloadición del bisditiolopirrol 181 con dibenzoilacetileno (DBA)
N
SSS
S
SS
181
COPhPhOC
N
SPhOC
PhOC
337
SS
COPhPhOC
S
SS
COPh
COPh
COPh
COPh
4
C6H6, 38%
Sobre una disolución agitada de 4-etil-bis[1,2]ditiolo[3,4-b:4’,3’-e]pirrol-3,5-ditiona
(125 mg, 0.407 mmol) en benceno (10 mL) se añadió dibenzoilacetileno158,159,202
(476 mg, 2.035 mmol). La mezcla de reacción se calentó a reflujo durante 15 minutos.
A continuación se eliminó el disolvente en el rotavapor, y el residuo resultante se
purificó mediante cromatografía líquida de media presión (SiO2, éter de petróleo hasta
diclorometano-acetato de etilo, 97:3) lo que permitió separar el compuesto 337 puro
(193 mg, 0.155 mmol, 38%), como un sólido anaranjado; p.f.: 140-142 ºC (CH2Cl2-éter
de petróleo).
IR (KBr, cm-1
): 3059, 1662 (C=O st), 1595, 1531, 1448, 1255, 817, 689.
RMN-1H (CDCl3, 400 MHz) δ: 7.80-7.20 (m, 40 H, 8 C6H5),
5.69 (c, J = 6.7 Hz, 2 H, NCH2CH3) y 2.10 (t, J = 6.7 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 193.30 (C=O), 191.27 (C=O), 186.84 (C=O),
138.38 (C sp2), 137.28 (C sp
2), 136.78 (C sp
2), 135.19 (C sp
2), 134.55 (CH Ar),
133.76 (CH Ar), 133.34 (CH Ar), 132.91 (C sp2), 131.88 (C sp2), 130.46 (CH Ar),
130.31 (CH Ar), 128.94 (CH Ar), 128.45 (CH Ar), 128.39 (C sp2), 123.57 (C sp
2),
108.77 (C sp2), 64.18 (C espirano), 43.93 (NCH2CH3) y 17.14 (NCH2CH3) ppm.
EM (FAB) m/z (%): 1243 (M+·
, 15), 1138 (M+ - COPh, 1), 1034 (M
+ + 1 - 2 PhCO, 1),
978 (M+ + 1 - DBA - S, 2), 681 (M
+ + 2 - 2 DBA - 3 S, 1), 105 (PhCO
+, 65).

Parte experimental 369
Análisis: Calculado para C72H45NO8S6: C, 69.49; H, 3.64; N, 1.13; hallado: C, 69.62;
H, 3.78; N, 1.12.

370 Tesis Doctoral Susana Barriga Falcón
N
SPhOC
PhOC
337
SS
COPh
PhOC
S
SS
COPh
COPh
COPh
COPh

Parte experimental 371
N
SPhOC
PhOC
337
SS
COPh
PhOC
S
SS
COPh
COPh
COPh
COPh

372 Tesis Doctoral Susana Barriga Falcón
46. Reacción de cicloadición de la bisditiolotiazina 176 con 4,4-dietoxi-2-butinal
176
S
N
S
SS
S
O S OHC CH(OEt)2
S
N
S
S S
O
S
SCHO
CH(OEt)2
HCO2H
S
N
S
S S
O
S
SCHO
CHO
(Z)-357
Sc(OTf)3, CH2Cl2, 83%
358
+
S
N
S
S S
O
S
SCH(OEt)2
CHO
(E)-357
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(75 mg, 0.232 mmol) en diclorometano (2 mL) se añadió 4,4-dietoxi-2-butinal174,175
(72 mg, 0.462 mmol) y triflato de escandio (57 mg, 0.116 mmol). La mezcla de reacción se
agitó a temperatura ambiente durante 10 minutos. A continuación se eliminó el disolvente a
presión reducida y el sólido resultante se purificó mediante cromatografía rápida
(SiO2, desde CH2Cl2-éter de petróleo 3:1 hasta diclorometano), lo que permitió separar un
producto sólido rojo, mezcla de los isómeros (Z)-357 y (E)-357 (92 mg, 0.192 mmol, 83%).
A continuación este sólido fue disuelto en ácido fórmico y se calentó a 60º C durante
30 minutos. Tras llevar la mezcla de reacción a temperatura ambiente, se vertió sobre agua
(30 mL) y se separaron las fases. La fase acuosa fue extraída con diclorometano (3 x 30 mL).
Los extractos orgánicos combinados se secaron sobre Na2SO4 y se eliminó el disolvente en
el rotavapor. El sólido obtenido se purificó por cromatografía rápida (SiO2, CH2Cl2) lo que
174 Raffin, C.; Bernard, D.; Fournet, G.; Gore, J.; Chantepie, J.; Quash, G. “Synthese de composes
acetyleniques α−α’-difuntionnels utilisables dans l’inhibition selective de la croissance de cellules
transformees”. J. Chem. Res. Miniprint 1995, 152-172. 175
Macrides, T. A.; Thaller, V. “Natural Acetylenes. Part 55. Acetylenic Aldehydes. Synthesis and
Hydrate, Hemiacetal, and Oxime Formation”. J. Chem. Res. Miniprint 1981, 2001-2016.

Parte experimental 373
permitió obtener el compuesto 358 puro (78 mg, 0.192 mmol, 83% del total de ambos
pasos), como un sólido rojo; p.f.: 76-77 ºC (CH2Cl2).
IR (KBr, cm-1
): 2956, 2923, 2851, 1668 (C=O st), 1653 (C=O st),
1636 (C=O st, heterociclo), 1261, 1103, 1075, 1047, 800.
RMN-1H (CDCl3, 200 MHz) δ: 10.42 (s, 2 H, 2 CHO), 3.59 (dc, J = 7.2 y 14.4 Hz, 1 H,
NCH2CH3), 3.26 (dc, J = 7.2 y 14.4 Hz, 1 H, NCH2CH3) y 1.21 (t, J = 7.2 Hz, 3 H,
NCH2CH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 196.06 (C=S), 184.50 (C=O heterociclo),
180.02 (CHO), 179.16 (CHO), 156.28 (C sp2), 151.95 (C sp
2), 147.53 (C sp
2),
146.08 (C sp2), 132.81 (C sp
2), 132.00 (C sp
2), 47.90 (NCH2CH3) y 13.43 (NCH2CH3)
ppm.
EM (IE) m/z (%): 405 (M+·
, 65), 376 (M+ - CHO, 100), 348 (M
+ - 2 CHO, 60), 186 (25),
149 (35), 71 (30).
Masa exacta: calculada para C12H7NO3S6: 404.8750; hallada: 404.8746.
Análisis: Calculado para C12H7NO3S6: C, 35.54; H, 1.74; N, 3.45; hallado: C, 35.44;
H, 1.88; N, 3.62.
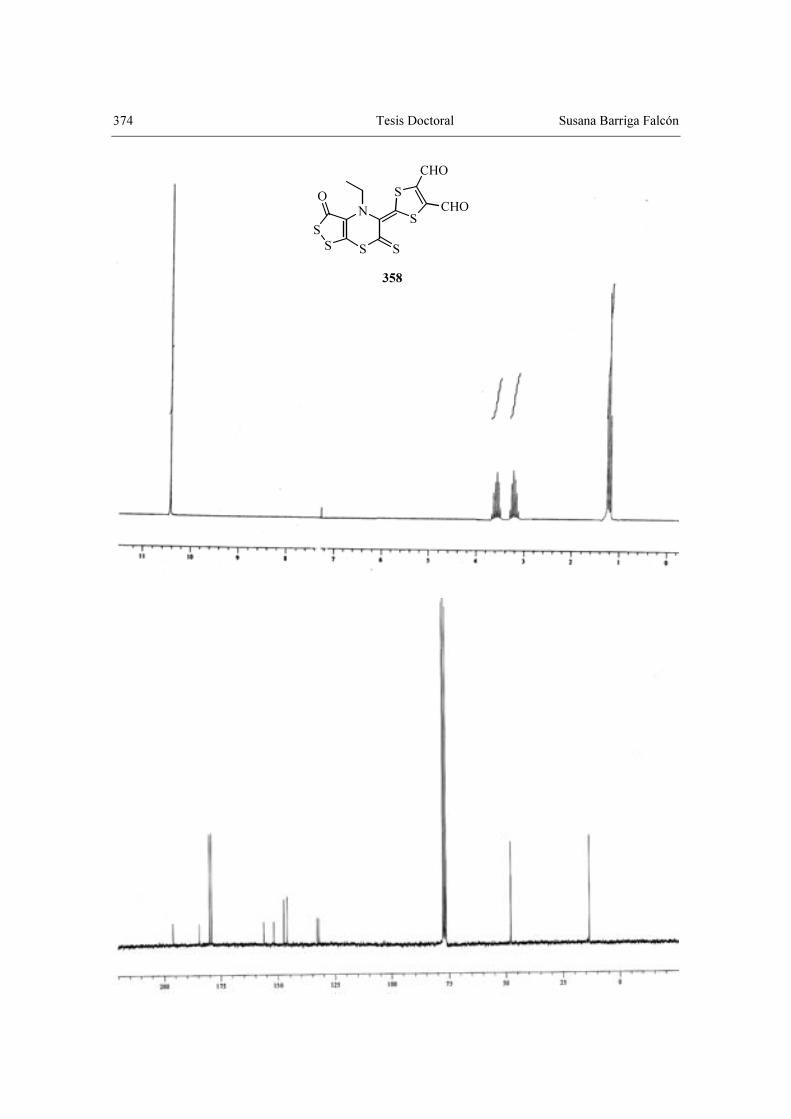
374 Tesis Doctoral Susana Barriga Falcón
S
N
S
S S
O
S
S
CHO
CHO
358

Parte experimental 375
22
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
S
S S
O
S
S
CHO
CHO
358

376 Tesis Doctoral Susana Barriga Falcón
47. Obtención de 1-[5-terbutildimetilsililoximetil-2-furil]-2-propin-1-ol (365)
O CHOO
SiH MgBr
O
OSi
OH
H
THF, 77%
363 365
Sobre una disolución agitada de 5-terbutildimetilsililoximetilfurfural (363)177
(1.80 g, 7.50 mmol) en THF (10 mL) se añadió lentamente bromuro de etinilmagnesio
(20.0 mL, 9.00 mmol, 0.45 M en THF). Tras completar la adición la mezcla de reacción
se agitó a temperatura ambiente durante 60 minutos. Seguidamente se añadió sobre la
mezcla de reacción una disolución acuosa saturada de NH4Cl (5 mL). A continuación se
separaron las fases y la fase acuosa fue extraída con éter dietílico (3 x 10 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico y el disolvente fue
eliminado en el rotavapor. El crudo obtenido se purificó mediante cromatografía rápida
(SiO2, éter de petróleo-diclorometano 1:1), lo que permitió separar el compuesto 365
puro (1.54 g, 5.79 mmol, 77%), como un aceite de color amarillo.
IR (película líquida, cm-1
): 3311 (OH st), 2954, 2929, 2885, 2857, 1472, 1464, 1257,
1079, 838 (Si-C st), 779 (Si-C st).
RMN-1H (CDCl3, 200 MHz) δ: 6.39 (d, J = 3.0 Hz, 1 H, CH Ar),
6.18 (d, J = 3.0 Hz, 1 H, CH Ar), 5.42 (s ancho, 1 H, CHOH), 4.62 (s, 2 H, OCH2),
2.59 (d, J = 2.0 Hz, 1 H, C≡CH), 0.89 (s, 9 H, 3 CH3) y 0.08 (s, 6 H, 2 CH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 154.92 (C Ar), 151.86 (C Ar), 108.55 (CH Ar),
107.90 (CH Ar), 81.01 (C≡CH), 73.92 (C≡CH), 58.15 (CHOH), 57.87 (OCH2),
25.81 (CH3), 18.33 (CSi) y - 5.30 (CH3) ppm.
177
McNelis, B.J.; Sternbach, D. D.; Macphail, A. T. “Synthetic and Kinetic-Studies of Substituent Effects in the Furan Intramolecular Diels-Alder Reaction”. Tetrahedron 1994, 50, 6767-6782.

Parte experimental 377
O
OSi
OH
H
365
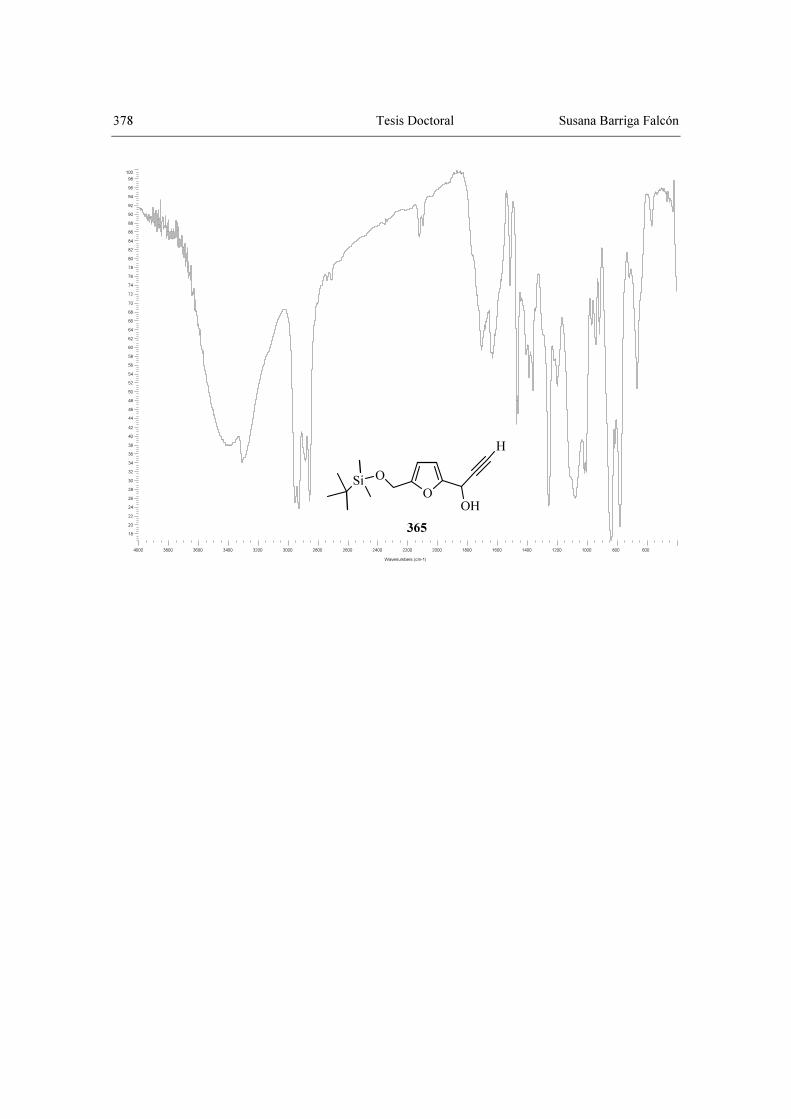
378 Tesis Doctoral Susana Barriga Falcón
18
20
22
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
O
OSi
OH
H
365

Parte experimental 379
48. Obtención de 1,4-bis[5-terbutildimetilsilioximetil-2-furil]-2-butin-1,4-diol (366)
O CHO
O
TBDMSO
OH
H
TBDSOOHHO
OO
OTBDMSTBDMSO
363
365
2)
THF, 56%
366
1) BuLi
Sobre una disolución agitada del alcohol propargílico 365 (996 mg, 3.74 mmol) en THF
(5 mL), enfriada a -78 ºC se añadió lentamente butillitio (5.2 mL, 8.32 mmol, 1.6 M en
hexanos). Tras completar la adición la mezcla de reacción se agitó a esta temperatura
durante 15 minutos. A continuación se añadió lentamente el aldehido 363
(898 mg, 3.74 mmol). La mezcla de reacción se agitó hasta alcanzar la temperatura
ambiente y se continuó la agitación a esta temperatura durante 30 minutos más.
Seguidamente se añadió sobre la mezcla de reacción una disolución acuosa saturada de
NH4Cl (5 mL). A continuación se separaron las fases y la fase acuosa fue extraída con
éter dietílico (3 x 10 mL). Los extractos orgánicos combinados se secaron sobre sulfato
sódico y el disolvente fue eliminado en el rotavapor. El crudo obtenido se purificó
mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano 1:1 hasta
diclorometano-acetato de etilo 17:3), lo que permitió separar el compuesto 366 puro
(1.058 g, 2.09 mmol, 56%), como un aceite de color marrón.
IR (película líquida, cm-1
): 3433 (OH st), 2955, 2930, 2858, 1257, 1101, 838 (Si-C st),
779 (Si-C st).
RMN-1H (CDCl3, 200 MHz) δ: 6.38 (d, J = 3.0 Hz, 2 H, 2 CH Ar),
6.18 (d, J = 3.0 Hz, 2 H, 2 CH Ar), 5.49 (s ancho, 2 H, 2 CHOH),
4.62 (s, 4 H, 2 OCH2), 0.89 (s, 9 H, C(CH3)3) y 0.07 (s, 6 H, 2 SiCH3) ppm.

380 Tesis Doctoral Susana Barriga Falcón
RMN-13
C (CDCl3, 50 MHz) δ: 154.92 (C Ar), 151.86 (C Ar), 108.65 (CH Ar),
107.92 (CH Ar), 83.02 (C≡C), 58.20 (CHOH), 58.09 (OCH2), 25.82 (C(CH3)3),
18.35 (CSi) y - 5.27 (SiCH3) ppm.

Parte experimental 381
OHHO
OO
OTBDMSTBDMSO
366

382 Tesis Doctoral Susana Barriga Falcón
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
OHHO
OO
OTBDMSTBDMSO
366

Parte experimental 383
49. Obtención de 1,4-bis[5-terbutildimetilsililoximetil-2-furil]-2-butin-1,4-diona (367)
OHHO
OO
OTBDMSTBDMSO
366
OO
OO
OTBDMSTBDMSO
367
BAIB, TEMPO
CH2Cl2, 72%
Sobre una disolución agitada del diol 366 (1.025 g, 2.03 mmol) en diclorometano
(5 mL) se añadió bisacetoxiiodobenceno (BAIB) (1.440 g, 4.47 mmol) y
2,2,6,6-tetrametilpiperidin-1-oxilo (TEMPO) (64 mg, 0.41 mmol).178
La mezcla de
reacción se agitó a temperatura ambiente durante 2 horas. A continuación la mezcla de
reacción se diluyó con diclorometano (5 mL) y se añadió una disolución acuosa saturada
de Na2S2O3 (5 mL). Después se separaron las fases y la fase acuosa fue extraída con
diclorometano (4 x 10 mL). Los extractos orgánicos combinados se lavaron con una
disolución saturada de NaHCO3 y posteriormente con una disolución saturada de NaCl.
La fase orgánica se secó sobre sulfato sódico y el disolvente fue eliminado en el
rotavapor. El crudo obtenido se purificó mediante cromatografía rápida (SiO2, éter de
petróleo-diclorometano, 1:1), lo que permitió separar la dicetona 367 pura
(734 mg, 1.46 mmol, 72%), como un sólido de color naranja; p.f.: 97-98 ºC (CH2Cl2-
éter de petróleo).
IR (KBr, cm-1
): 2956, 2930, 2857, 1643 (C=O st), 1633 (C=O st), 1513, 1372, 1312,
1207, 1123, 1084, 1022, 848, 777.
RMN-1H (CDCl3, 200 MHz) δ: 7.48 (d, J = 3.5 Hz, 2 H, 2 CH Ar),
6.53 (d, J = 3.5 Hz, 2 H, 2 CH Ar), 4.77 (s, 4 H, 2 OCH2), 0.94 (s, 18 H, 2 C(CH3)3) y
0.13 (s, 12 H, 4 SiCH3) ppm.
178
DeMico, A.; Margarite, R.; Parlati, L.; Vescovi, A.; Pancatelli, G. “A versatile and highly selective hypervalent iodine (III)/ 2,2,6,6-tetramethyl-1-piperidinyloxyl-mediated oxidation of alcohols to carbonyl compounds”. J. Org. Chem. 1997, 62, 6974-6977.

384 Tesis Doctoral Susana Barriga Falcón
RMN-13
C (CDCl3, 50 MHz) δ: 162.80 (C=O), 161.93 (C Ar), 151.42 (C Ar),
124.16 (CH Ar), 109.68 (CH Ar), 82.91 (C≡C), 58.42 (OCH2), 25.47 (C(CH3)3),
18.02 (CSi) y - 5.68 (SiCH3) ppm.
EM (IE) m/z (%): 445 (M+ - C(CH3)3, 5), 387 (M
+ - TBDMS, 2).

Parte experimental 385
OO
OO
OTBDMSTBDMSO
367

386 Tesis Doctoral Susana Barriga Falcón
OO
OO
OTBDMSTBDMSO
367

Parte experimental 387
50. Desprotección de la 1,4-bis[5-terbutildimetilsililoximetil-2-furil]-2-butin-1,4-
diona (367)
OO
OO
OTBDMSTBDMSO
367
OO
OO
OHHO
368
AcOH, THF, H2O
82%
Una disolución agitada de la dicetona 367 (105 mg, 0.209 mmol) en una mezcla de
ácido acético-THF-agua en proporción 3:1:1 (10 mL) se calentó a reflujo durante
2.5 horas.179
Tras enfriar la disolución se vertió sobre agua (20 mL) y se extrajo con
diclorometano (4 x 20 mL). Los extractos orgánicos combinados se lavaron con
disolución saturada de NaHCO3 (10 mL) y a continuación con agua (10 mL). La fase
orgánica se secó sobre sulfato de sodio. Seguidamente se eliminó el disolvente en el
rotavapor y el crudo resultante se purificó mediante cromatografía rápida (SiO2, acetato
de etilo) lo que permitió separar la dihidroxidicetona 368 pura (47 mg, 0.172 mmol,
82%), como un sólido de color naranja; p.f.: 86-87 ºC (acetato de etilo).
IR (KBr, cm-1
): 3426 (OH st), 1626 (C=O st), 1511, 1203, 1126, 421.
RMN-1H (CD3OCD3, 200 MHz) δ: 7.68 (d, J = 2.6 Hz, 2 H, 2 CH Ar),
6.68 (d, J = 2.6 Hz, 2 H, 2 CH Ar) y 4.73 (t, J = 5.0 Hz, 4 H, 2 CH2OH) ppm.
RMN-13
C (CD3OCD3, 50 MHz) δ: 165.82 (C=O), 153.56 (C Ar), 151.42 (C Ar),
126.50 (CH Ar), 112.05 (CH Ar), 84.24 (C≡C) y 58.57 (OCH2) ppm.
EM (IE) m/z (%): 256 (M+ - H2O, 10).
179
Corey, E. J.; Venkateswarlu, A. “Protection of hydroxyl groups as tert-butyl-dimethylsilyl derivates”. J. Am. Chem. Soc. 1972, 94, 6190-6191.
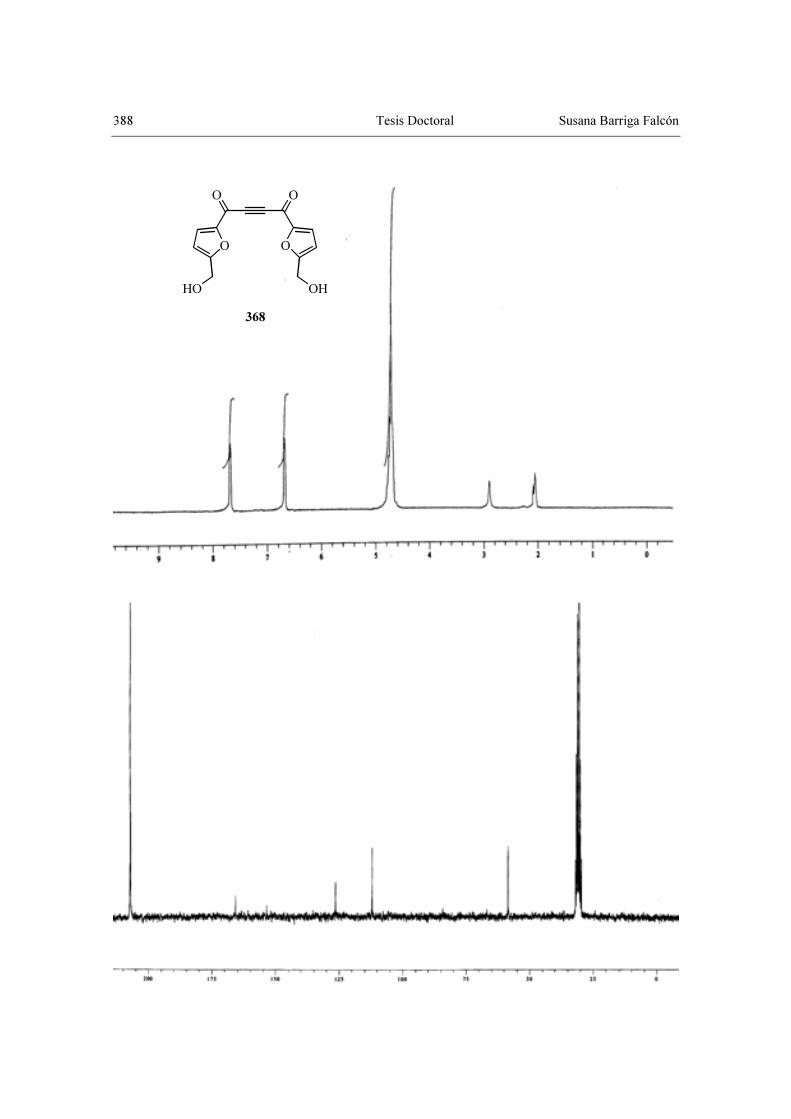
388 Tesis Doctoral Susana Barriga Falcón
OO
OO
OHHO
368

Parte experimental 389
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
OO
OO
OHHO
368

390 Tesis Doctoral Susana Barriga Falcón
51. Reacción de cicloadición de la bisditiolotiazina 176 con la dihidroxidicetona 368
S
N
S
SS
S
SO
OO
OO
HO OH S
N
S
S
S
O
S
S O
O
OO
OH
HO
Sc(OTf)3
CH2Cl2, 84%
176
+
368 369
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(40 mg, 0.124 mmol) en diclorometano (2 mL) se añadió el diol 368 (41 mg, 0.150 mmol) y
triflato de escandio (30 mg, 0.060 mmol). La mezcla de reacción se agitó durante 10 minutos
a temperatura ambiente. Tras eliminar el disolvente en el rotavapor, el crudo obtenido se
purificó mediante cromatografía rápida (SiO2, diclorometano-acetato de etilo 1:1), lo que
permitió separar el compuesto 369 puro (62 mg, 0.104 mmol, 84%), como un sólido de color
rojo; p.f.: 134-135 ºC (diclorometano-acetato de etilo).
IR (KBr cm-1
): 3426 (OH st), 2924, 1656 (C=O st heterociclo), 1639 (C=O st), 1631
(C=O st), 1513, 1414, 1285, 1261, 1046 (C-O, st), 1036 (C-O, st).
RMN-1H (CD3COCD3, 400 MHz) δ: 7.42 (d, J = 3.5 Hz, 1 H, CH Ar),
7.36 (d, J = 3.5 Hz, 1 H, CH Ar), 6.54-6.52 (m, 2 H, 2 CH Ar), 4.71-4.67 (m, 2 H, 2 OH),
4.52 (t, J = 5.9 Hz, 4 H, 2 CH2OH), 3.54 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3),
3.37 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3), 0.94 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CD3COCD3, 100 MHz) δ: 193.31 (C=S), 186.14 (C=O heterociclo),
173.60 (C=O), 173.46 (C=O), 164.99 (C Ar), 164.82 (C Ar), 164.39 (C sp2),
155.12 (C sp2), 151.97 (C Ar), 151.84 (C Ar), 142.29 (C sp
2), 141.19 (C sp
2),
133.96 (C sp2), 133.50 (C sp
2), 124.94 (CH Ar), 124.66 (CH Ar), 112.00 (CH Ar),
111.96 (CH Ar), 58.37 (CH2OH), 49.47 (NCH2CH3) y 14.68 (NCH2CH3) ppm.

Parte experimental 391
EM (IE) m/z (%): 582 (M+ - CH3, 5).
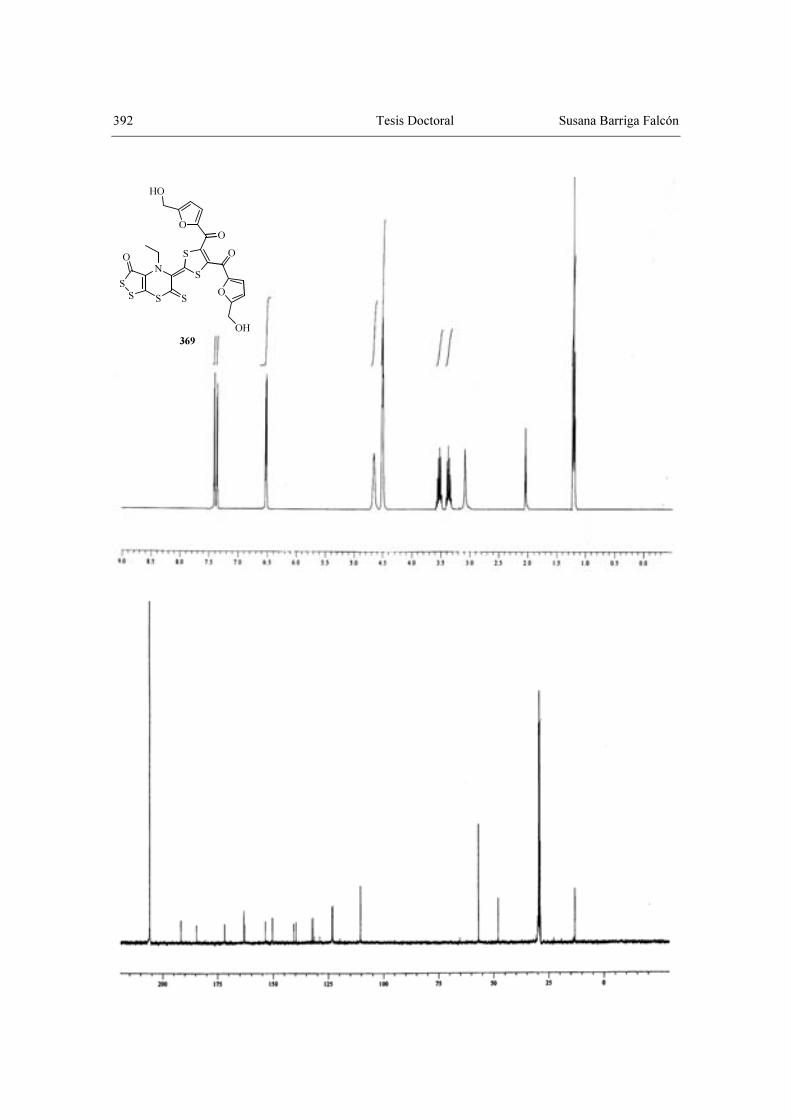
392 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
S
O
S
S O
O
O
O
OH
HO
369

Parte experimental 393
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
S
S
S
O
S
S O
O
O
O
OH
HO
369

394 Tesis Doctoral Susana Barriga Falcón
52. Obtención de 1-ferrocenil-2-propin-1-ol (374)184
FeCHO
BrMg H H
HO
FeTHF, 85%
373 374
Sobre una disolución agitada de ferrocenilcarbaldehido (373) (535 mg, 2.50 mmol) en
THF (5 mL) se añadió lentamente bromuro de etinilmagnesio (8.33 mL, 3.75 mmol,
0.45 M en THF). Tras completar la adición la mezcla de reacción se agitó a temperatura
ambiente durante 60 minutos. Seguidamente se añadió sobre la mezcla de reacción una
disolución acuosa saturada de NH4Cl (5 mL). A continuación se separaron las fases y la
fase acuosa fue extraída con éter dietílico (3 x 10 mL). Los extractos orgánicos
combinados se secaron sobre sulfato sódico y el disolvente fue eliminado en el
rotavapor. El crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietilico, 8:2), lo que permitió separar el compuesto 374 puro
(510 mg, 2.12 mmol, 88%), como un sólido de color amarillo, p.f.: 82-83 ºC
(ciclohexano-éter dietílico).
IR (KBr cm-1
): 3289 (OH st), 3266 (OH st), 1389, 1294, 1227, 1105, 1044, 1015, 997,
988, 810, 508, 489.
RMN-1H (CDCl3, 200 MHz) δ: 5.18-5.16 (m, 1H, CHOH), 4.39-4.35 (m, 2 H, 2 CH Ar),
4.23 (s, 5 H, 5 CH Ar), 4.21-4.20 (m, 2H, 2 CH Ar ), 2.62-2.61 (m, 1 H, C≡CH) y
2.51-2.48 (m, 1 H, OH) ppm.
184
(a) Schloegl, K.; Mohar, A. “Über ferrocen-acetylene, 2. Mitt.: Darstellung und Reaktionen von ferrocenyl-alkinen und –alkinyl-ketonen (11. Mitt. über ferrocenderivate)”. Monatsh. Chem. 1962, 93, 861-876. (b) Schloegl, K.; Steyrer, W. “Ferrocen-Acetylene, 5. Mitt.: Eine allgemeine Methode zur Darstellung von ferrocenyl-acetylenen und –allenen aus acylferrocenen”. Monatsh. Chem. 1965, 96, 1520-1535. (c) Akiyama, T.; Hattori, T.; Ito, K.; Uryu, T.; Kajitani, M.; Sugimori, A. Chem. Lett. 1980, 361-364.

Parte experimental 395
RMN-13
C (CDCl3, 50 MHz) δ: 89.14 (C Ar), 83.51 (C≡CH), 72.66 (C≡CH),
68.78 (CH Ar), 68.35 (CH Ar), 68.13 (CH Ar), 66.40 (CH Ar) y 60.61 (CHOH) ppm.
EM (IE) m/z (%): 240 (M+·
, 85), 214 (M+ - C2H2, 15), 186 (Fc
+, 20), 138 (CpFeOH
+,
100), 121 (CpFe+, 25), 56 (Fe
+, 20).
Masa exacta: calculada para C13H12OFe: 240.0238 encontrada: 240.0216.

396 Tesis Doctoral Susana Barriga Falcón
H
HO
Fe
374
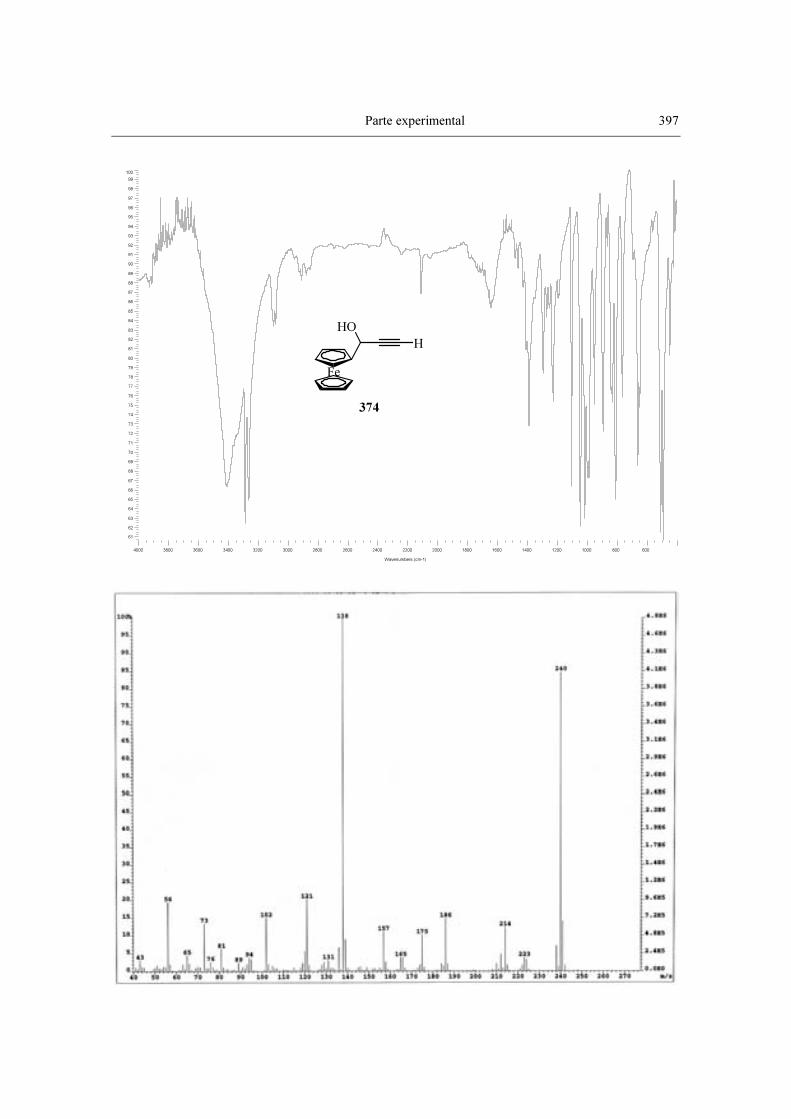
Parte experimental 397
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
H
HO
Fe
374

398 Tesis Doctoral Susana Barriga Falcón
53. Obtención de 1-ferrocenil-2-propin-1-ona (ferrocenil etinil cetona) (370)184
H
O
Fe
370
MnO2
CH2Cl2, 92%
H
HO
Fe
374
Sobre una disolución agitada del alcohol propargílico 374 (200 mg, 0.83 mmol) en
diclorometano (5 mL) se añadió dióxido de manganeso (2.17 g, 24.94 mmol). La mezcla
de reacción se agitó a temperatura ambiente durante 15 minutos. A continuación se filtró
sobre celita y se lavó con diclorometano hasta que el filtrado dejó de salir coloreado.
Tras eliminar el disolvente en el rotavapor, el crudo obtenido se purificó mediante
cromatografía rápida (SiO2, ciclohexano-éter dietílico, 8:2), lo que permitió separar el
compuesto 370 puro (181 g, 0.76 mmol, 92%), como un sólido de color rojo;
p.f.: 77-78ºC (ciclohexano-éter dietílico).
IR (KBr cm-1
): 1618 (C=O st), 1446, 1273, 1115, 824, 508, 489, 470.
RMN-1H (CDCl3, 200 MHz) δ: 4.93-4.91 (m, 2 H, 2 CH Ar ), 4.62-4.60 (m, 2 H, 2 CH Ar),
4.25 (s, 5 H, 5 CH Ar) y 3.29 (s, 1 H, C≡CH) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 180.12 (C=O), 81.31 (C≡CH), 79.62 (C≡CH), 77.14 (C Ar),
73.53 (CH Ar), 73.43 (CH Ar) y 70.47 (CH Ar) ppm.
EM (EI) m/z (%): 238 (M+·
, 100), 210 (25), 184 (M+ - C3H2O, 13), 121 (CpFe
+, 10),
56 (Fe+, 30).
Masa exacta: calculada para C13H10OFe: 238.0081 encontrada: 238.0061.

Parte experimental 399
H
O
Fe
370

400 Tesis Doctoral Susana Barriga Falcón
H
O
Fe
370

Parte experimental 401
54. Reacción de cicloadición de la bisditiolotiazina 176 con ferrocenil etinil cetona (370)
S
N
S
SS
S
O S
+
176
H
O
Fe
370
Sc(OTf)3
CH2Cl2, 91%
S
N
SS
S
S
S
O
H
Fe
O Fe
S
N
SS
S
S
S H
O
O
+
(E)-375 (Z)-375
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(50 mg, 0.155 mmol) en diclorometano (2 mL) se añadió la cetona propargílica 370
(84 mg, 0.187 mmol) y triflato de escandio (38 mg, 0.077 mmol). La mezcla de reacción se
agitó durante 10 minutos a temperatura ambiente. Tras eliminar el disolvente en el rotavapor,
el crudo obtenido se purificó mediante cromatografía rápida (SiO2, ciclohexano-éter dietílico
8:2), lo que permitió separar el cicloaducto puro, mezcla de isómeros (Z)-375 y (E)-375
(79 mg, 0.141 mmol, 91%), como un sólido rojo; p. f.: 114-115 ºC (ciclohexano-éter
dietílico).
IR (KBr cm-1
): 1652 (C=O st), 1645 (C=O st), 1622 (C=O st cetona), 1616 (C=O st
cetona), 1346, 1284 (C=S st), 1119.
RMN-1H (CDCl3, 400 MHz) δ: 8.03 (s, 1H, C=CH), 7.86 (s, 1H, C=CH), 4.96 (s, 2 H, 2 CH Ar),
4.93 (s, 2 H, 2 CH Ar), 4.91 (s, 4 H, 4 CH Ar), 4.70 (s, 10 H, 10 CH Ar),
3.63-3.52 (m, 2 H, NCH2CH3), 3.36-3.26 (m, 2 H, NCH2CH3) y 1.27-1.24 (m, 6 H, 2 NCH2CH3)
ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 191.02 (C=S), 190.05 (C=S), 187.25 (C=O),
186.81 (C=O), 184.88 (C=O cetona), 184.58 (C=O cetona), 166.14 (C sp2),
165.68 (C sp2), 154.18 (C sp
2), 153.22 (C sp
2), 141.23 (C sp
2), 140.71 (C sp
2),
132.26 (C sp2), 131.90 (C sp
2), 131.81 (C sp
2), 129.98 (C sp
2), 129.81 (C sp
2),
73.66 (CH Ar), 73.57 (CH Ar), 73.54 (CH Ar), 71.12 (CH Ar), 70.81 (CH Ar),
70.77 (CH Ar), 70.68 (CH Ar), 70.56 (CH Ar), 48.08 (NCH2CH3), 47.43 (NCH2CH3),
13.52 (NCH2CH3) y 13.42 (NCH2CH3) ppm.
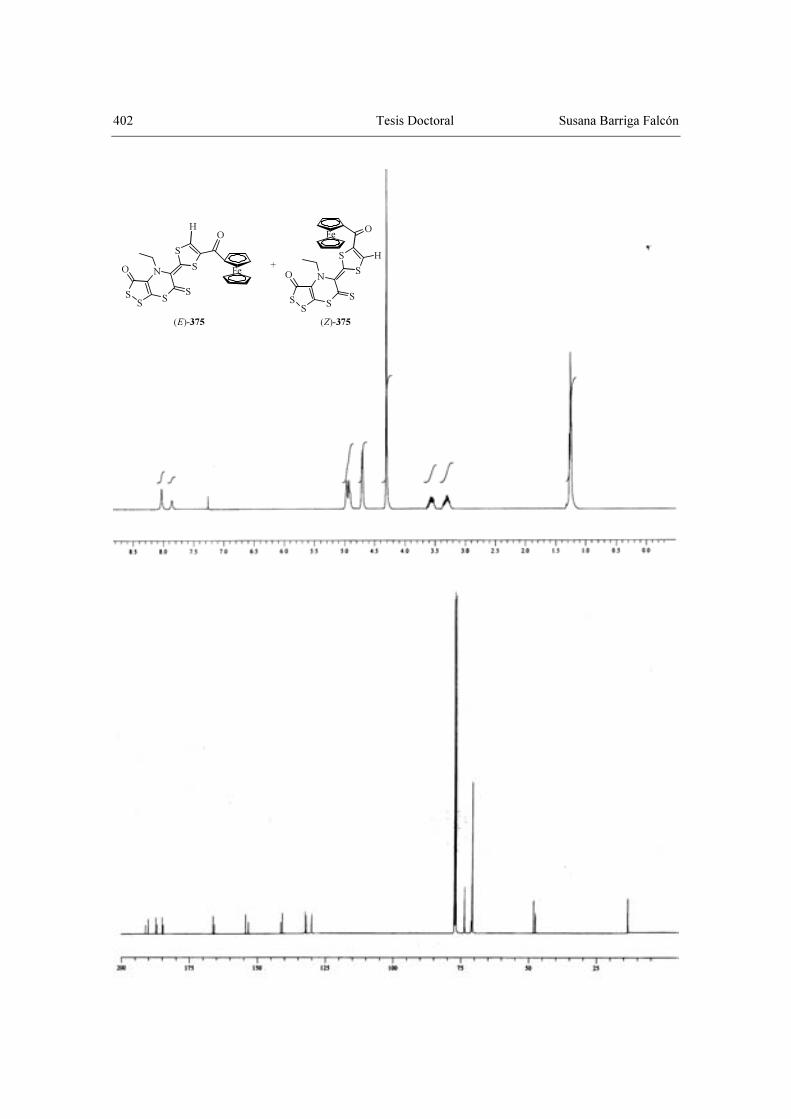
402 Tesis Doctoral Susana Barriga Falcón
S
N
SS
S
S
S
O
H
Fe
O Fe
S
N
SS
S
S
S H
O
O
+
(E)-375 (Z)-375

Parte experimental 403
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
SS
S
S
S
O
H
Fe
O Fe
S
N
SS
S
S
S H
O
O
+
(E)-375 (Z)-375

404 Tesis Doctoral Susana Barriga Falcón
55. Obtención de (R)-1-[αααα-(p-metoxifenil)ferrocenil]-2-propin-1-ol (378).
BrMg H
Fe p-MeOC6H4
CHO
THF, 88%
376
Fe
378
p-MeOC6H4
HO
H
Sobre una disolución agitada de (R)-α-(p-metoxifenil)ferrocenocarboxaldehído (376)185
(500 mg, 1.56 mmol) en THF (5 mL) se añadió lentamente bromuro de etinilmagnesio
(4.15 mL, 1.87 mmol, 0.45 M. en THF). Tras completar la adición la mezcla de reacción
se agitó a temperatura ambiente durante 60 minutos. Seguidamente se añadió sobre la
mezcla de reacción una disolución acuosa saturada de NH4Cl (5 mL). A continuación se
separaron las fases y la fase acuosa fue extraída con éter dietílico (3 x 10 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico y el disolvente fue
eliminado en el rotavapor. El crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietílico, 8:2), lo que permitió separar el alcohol 378 puro
(475 mg, 1.37 mmol, 88%), como un sólido aceitoso de color amarillo.
IR (película líquida, cm-1
): 3282 (OH st), 2956, 2925, 2851, 2836, 1610, 1522, 1453,
1438, 1248 (C-O st), 1179, 1031, 831.
RMN-1H (CDCl3, 200 MHz) δ: 7.53 (d, J = 8.8 Hz, 2 H, 2 CH Ar),
6.89 (d, J = 8.8 Hz, 2 H, 2 CH Ar), 5.28 (s ancho, 1 H, CHOH), 4.55 (m, 1 H, CH Ar),
4.48 (m, 1 H, CH Ar), 4.29 (m, 1 H, CH Ar), 4.23 (s, 5 H, 5 CH Ar),
3.83 (s, 3 H, OCH3) y 2.57 (m, 2 H, C≡CH y OH) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 158.34 (C Ar), 130.09 (CH Ar), 129.12 (C Ar),
113.49 (CH Ar), 89.14 (C Ar), 87.27 (C Ar ), 83.76 (C≡CH), 73.17 (C≡CH),
185
Riant, O.; Samuel, O.; Flessner, T.; Taudien, S.; Kagan, H. B. “An Efficient Asymmetric Synthesis of 2-Substituted Ferrocenecarboxaldehydes”. J. Org. Chem. 1997, 62, 6733-6745.

Parte experimental 405
70.18 (CH Ar), 69.86 (CH Ar), 67.10 (CH Ar), 66.98 (CH Ar), 58.98 (CHOH) y
55.19 (OCH3) ppm.
EM (IE) m/z (%): 346 (M+·
, 69), 320 (M+ - C2H2, 52), 292 (M
+ + 1- C2H3O, 10),
208 (M+ - CpFe - OH, 75), 149 (100), 121 (CpFe
+, 20).
Masa exacta: calculada para C20H18O2Fe: 346.0656 encontrada: 346.0681.

406 Tesis Doctoral Susana Barriga Falcón
Fe
378
p-MeOC6H4
HO
H
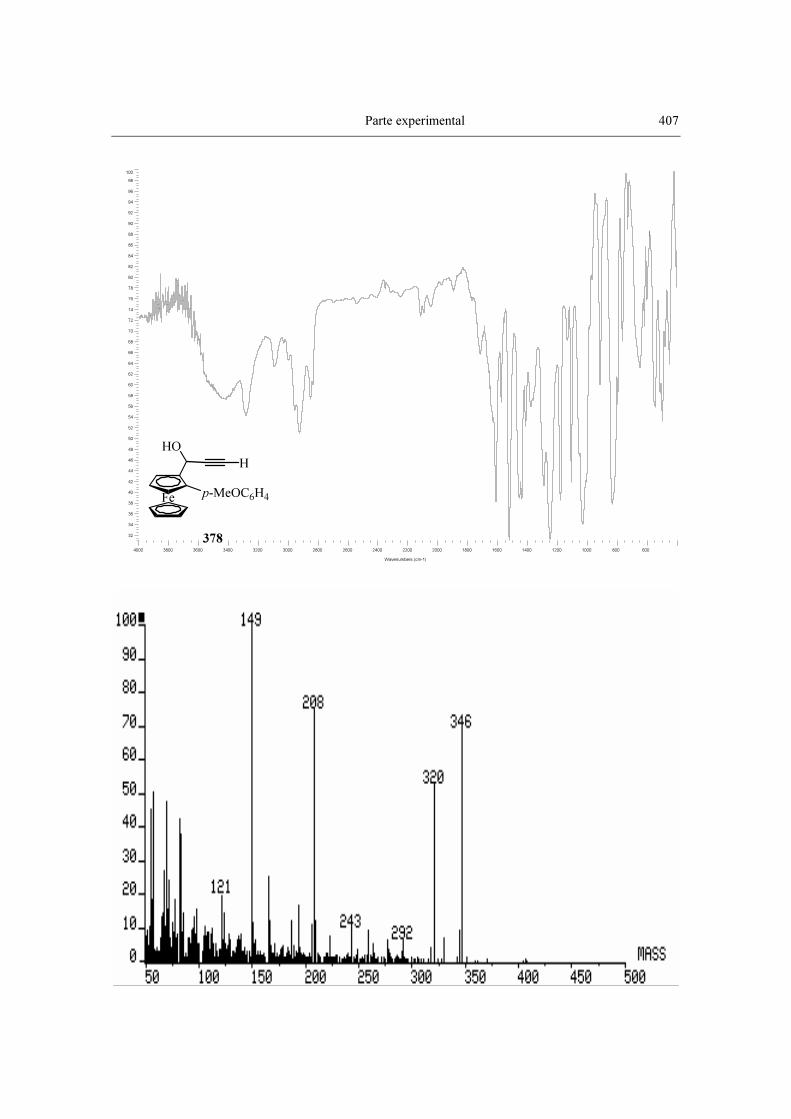
Parte experimental 407
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
Fe
378
p-MeOC6H4
HO
H

408 Tesis Doctoral Susana Barriga Falcón
56. Obtención de (R)-1-[αααα-(p-metoxifenil)ferrocenil]-2-propin-1-ona (371)
MnO2
CH2Cl2, 88%
371378
Fep-MeOC6H4
HO
H
Fep-MeOC6H4
O
H
Sobre una disolución agitada del alcohol 378 (250 mg, 0.72 mmol) en diclorometano
(5 mL) se añadió dióxido de manganeso (1.88 g, 21.68 mmol). La mezcla de reacción se
agitó a temperatura ambiente durante 15 minutos. A continuación se filtró sobre celita y
se lavó con diclorometano hasta que el filtrado dejó de salir coloreado. Tras eliminar el
disolvente en el rotavapor, el crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietílico, 8:2), lo que permitió separar el compuesto 371 puro
(218 g, 0.63 mmol, 88%), como un sólido de color rojo; p.f.: 149-150 ºC ciclohexano-
éter dietílico).
IR (KBr, cm-1
): 3250, 1629 (C=O st), 1520, 1247, 1233, 1183, 1029.
RMN-1H (CDCl3, 200 MHz) δ: 7.52 (d, J = 8.6 Hz, 2 H, 2 CH Ar),
6.86 (d, J = 8.6 Hz, 2 H, 2 CH Ar), 5.06 (m, 1 H, CH Ar), 4.80 (m, 1 H, CH Ar),
4.66 (m, 1 H, CH Ar), 4.27 (s, 5 H, 5 CH Ar), 3.83 (s, 3 H, OCH3) y
3.18 (s, 1 H, C≡CH) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 180.02 (C=O), 158.72 (C Ar), 131.15 (CH Ar),
127.98 (C Ar), 112.85 (CH Ar), 91.12 (C≡CH), 82.10 (C≡CH), 76.02 (C Ar),
73.68 (CH Ar), 71.98 (CH Ar), 71.68 (CH Ar) y 55.20 (OCH3) ppm.
EM (IE) m/z (%): 344 (M+·
, 100), 320 (M+ + 1 - C2H, 10), 198 (M
+ - CpFe - C2H, 50),
183 (M+ - CpFe - C2H CH3, 20), 121 (CpFe
+, 25), 56 (Fe
+, 13).
Masa exacta: calculada para C20H16O2Fe: 344.0500 encontrada: 344.0490.

Parte experimental 409
371
Fep-MeOC6H4
O
H

410 Tesis Doctoral Susana Barriga Falcón
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
371
Fep-MeOC6H4
O
H

Parte experimental 411
57. Reacción de cicloadición de la bisditiolotiazina 176 con
(R)-1-[αααα-(p-metoxifenil)ferrocenil]-2-propin-1-ona (371)
S
N
S
SS
S
S
+
176
Sc(OTf)3
CH2Cl
2, 87%
S
N
SS
S
S
S
O
H
Fe
PMPO
S
N
S
S S
S
SHO
+
(E)-380, PMP = p-MeOC6H4
O
371
Fep-MeOC
6H4
O
HO
Fe
PMP
(Z)-380, PMP = p-MeOC6H4
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en diclorometano (2 mL) se añadió la cetona propargílica 371
(39 mg, 0.113 mmol) y triflato de escandio (23 mg, 0.047 mmol). La mezcla de reacción se
agitó durante 10 minutos a temperatura ambiente. Tras eliminar el disolvente en el rotavapor,
el crudo obtenido se purificó mediante cromatografía rápida (SiO2, ciclohexano-éter dietílico
8:2), lo que permitió separar el cicloaducto puro, mezcla de isómeros (Z)-380 y (E)-380
(54 mg, 0.081 mmol, 87%), como un sólido rojo; p. f.: 133-134 ºC (ciclohexano-éter dietílico).
IR (KBr cm-1
): 1667 (C=O st), 1651 (C=O st), 1633 (C=O st, cetona), 1616 (C=O st,
cetona), 1519, 1403, 1342, 1246 (C=S st), 1097, 826.
RMN-1H (CDCl3, 400 MHz) δ: 7.72 (s, 1 H, C=CH), 7.70 (s, 1 H, C=CH),
7.62 (s, 1 H, C=CH), 7.56 (s, 1 H, C=CH), 7.41 7.30 (m, 4 H, 4 CH Ar),
6.83-6.72 (m, 4 H, 4 CH Ar), 4.80 (s, 4 H, 4 CH Ar), 4.64 (s, 2 H, 2 CH Ar),
4.34 (s, 2 H, 2 CH Ar), 4.30 (s, 2 H, 2 CH Ar), 4.28 (s, 6 H, 6 CH Ar),
3.80 (s, 3 H, OCH3), 3.78 (s, 3 H, OCH3), 3.56-3.46 (m, 2 H, NCH2CH3),
3.26-3.18 (m, 2 H, NCH2CH3) y 1.37-1.21 (m, 6 H, 2 NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 190.17 (C=S), 189.86 (C=S), 188.58 (C=O),
188.50 (C=O), 184.81 (C=O cetona), 184.45 (C=O cetona), 165.76 (C sp2),
165.70 (C sp2), 158.66 (C sp
2), 158.57 (C sp
2), 154.14 (C sp
2), 153.07 (C sp
2),
142.16 (C sp2), 141.93 (C sp
2), 141.70 (C sp
2), 141.66 (C sp
2), 132.17 (C=CH),
132.11 (C=CH), 132.04 (C Ar), 131.80 (C Ar), 130.80 (CH Ar), 130.75 (CH Ar),

412 Tesis Doctoral Susana Barriga Falcón
128.19 (CH Ar), 128.16 (CH Ar), 113.33 (CH Ar), 113.25 (CH Ar), 90.95 (C Ar),
90.68 (C Ar), 74.29 (CH Ar), 73.99 (CH Ar), 73.82 (CH Ar), 73.66 (CH Ar),
72.09 (CH Ar), 72.06 (CH Ar), 71.93 (CH Ar), 71.87 (CH Ar), 71.83 (CH Ar),
71.57 (CH Ar), 70.79 (CH Ar), 70.71 (CH Ar), 70.60 (CH Ar), 55.20 (OCH3),
48.06 (NCH2CH3), 47.32 (NCH2CH3), 13.45 (NCH2CH3) y 13.30 (NCH2CH3) ppm.
EM (FAB) m/z (%): 668 (M+·
, 3).

Parte experimental 413
S
N
SS
S
S
S
O
H
Fe
PMPO
S
N
S
S S
S
SHO
+
(E)-380, PMP = p-MeOC6H4
O
Fe
PMP
(Z)-380, PMP = p-MeOC6H4

414 Tesis Doctoral Susana Barriga Falcón
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
SS
S
S
S
O
H
Fe
PMPO
S
N
S
S S
S
SHO
+
(E)-380, PMP = p-MeOC6H4
OFe
PMP
(Z)-380, PMP = p-MeOC6H4

Parte experimental 415
58. Obtención de 1-[(S)-αααα-(trimetilsilil)ferrocenil]-2-propin-1-ol (379)
BrMg H
Fe TMS
CHO
THF, 92%
377
Fe
379
TMS
HO
H
Sobre una disolución agitada de (S)-α-(trimetilsilil)ferrocenocarboxaldehído (377)185
(200 mg, 0.699 mmol) en THF (5 mL) se añadió lentamente bromuro de etinilmagnesio
(2.33 mL, 1.049 mmol, 0.45 M en THF). Tras completar la adición la mezcla de
reacción se agitó a temperatura ambiente durante 60 minutos. Seguidamente se añadió
sobre la mezcla de reacción una disolución acuosa saturada de NH4Cl (5 mL). A
continuación se separaron las fases y la fase acuosa fue extraída con éter dietílico
(3 x 10 mL). Los extractos orgánicos combinados se secaron sobre sulfato sódico y el
disolvente fue eliminado en el rotavapor. El crudo obtenido se purificó mediante
cromatografía rápida (SiO2, ciclohexano-éter dietílico, 8:2), lo que permitió separar el
compuesto 379 puro (201 mg, 0.644 mmol, 88%), como un sólido aceitoso de color
amarillo.
RMN-1H (CDCl3, 200 MHz) δ: 5.12 (m, 1 H, CHOH), 4.62 (m, 1 H, CH Ar),
4.36 (m, 1 H, CH Ar), 4.23 (s, 5 H, 5 CH Ar), 4.16 (m, 1 H, C≡CH Ar),
2.52 (m, 1 H, CH), 2.32 (m, 1 H, OH), y 0.28 (s, 9 H, 3 SiCH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 96.30 (C Ar), 83.91 (C≡CH), 74.88 (C≡CH),
72.92 (CH Ar), 70.45 (C Ar), 70.04 (CH Ar), 69.30 (CH Ar), 68.66 (CH Ar),
60.00 (CHOH) y 0.33 (SiCH3) ppm.

416 Tesis Doctoral Susana Barriga Falcón
Fe
379
TMS
HO
H

Parte experimental 417
59. Obtención de 1-[(S)-αααα-(trimetilsilil)ferrocenil]-2-propin-1-ona (372)
MnO2
CH2Cl2, 95%Fe
379
TMS
HO
H
Fe
372
TMS
O
H
Sobre una disolución agitada del alcohol 379 (150 mg, 0.481 mmol) en diclorometano
(5 mL) se añadió óxido de manganeso (1.25 g, 14.43 mmol). La mezcla de reacción se
agitó a temperatura ambiente durante 15 minutos. A continuación se filtró sobre celita y
se lavó con diclorometano hasta que el filtrado dejó de salir coloreado. Tras eliminar el
disolvente en el rotavapor, el crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietílico 9:1), lo que permitió separar el compuesto 372 puro
(142 mg, 0.458 mmol, 95%), como un sólido de color rojo; p.f.: 136-137 ºC
(ciclohexano-éter dietílico).
IR (KBr, cm-1
): 3289 (OH st), 3266, (OH st), 1407, 1105, 1044, 1015, 997, 988,
810 (Si-C st), 508, 489.
RMN-1H (CDCl3, 200 MHz) δ: 5.14 (m, 1 H, CH Ar), 4.71 (m, 1 H, CH Ar),
4.58 (m, 1 H, CH Ar), 4.27 (s, 5 H, 5 CH Ar), 3.23 (m, 1 H, C≡CH) y
0.29 (s, 9 H, 3 SiCH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 181.04 (C=O), 83.91 (C≡CH), 81.76 (C≡CH),
80.41 (CH Ar), 76.51 (CH Ar), 74.86 (CH Ar), 70.46 (CH Ar) y -0.08 (SiCH3) ppm.
EM (IE) m/z (%): 311 (M+ + 1, 16), 310 (M
+·, 72), 295 (M
+ - CH3, 100),
267 (M+ - SiCH3, 9), 121 (CpFe
+, 22), 56 (Fe
+, 15).
Masa exacta: calculada para C16H18FeOSi: 310.0476 encontrada: 310.0512.

418 Tesis Doctoral Susana Barriga Falcón
Fe
372
TMS
O
H

Parte experimental 419
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
Fe
372
TMS
O
H

420 Tesis Doctoral Susana Barriga Falcón
60. Reacción de cicloadición de la bisditiolotiazina 176 con la
1-[(S)-αααα-(trimetilsilil)ferrocenil]-2-propin-1-ona (372)
S
N
S
SS
S
S
+
176
Sc(OTf)3
CH2Cl2, 87%
S
N
SS
S
S
S
O
H
Fe
TMSO
S
N
SS S
S
SHO
+
(E)-381
O
372
Fe TMS
O
HO
Fe
TMS
(Z)-381
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en diclorometano (2 mL) se añadió la dicetona 372
(35 mg, 0.113 mmol) y triflato de escandio (23mg, 0.047 mmol). La mezcla de reacción se
agitó durante 10 minutos a temperatura ambiente. Tras eliminar el disolvente en el rotavapor,
el crudo obtenido se purificó mediante cromatografía rápida (SiO2, ciclohexano-éter dietílico
9:1), lo que permitió separar el cicloaducto puro, mezcla de isómeros (Z)-381 y (E)-381
(50 mg, 0.079 mmol, 85%), como un sólido rojo; p.f.: 105-106 ºC (ciclohexano-éter
dietílico).
IR (KBr cm-1
): 1667 (C=O st), 1652 (C=O st), 1633 (C=O st, cetona), 1622 (C=O st,
cetona), 1616, 1408, 1346, 1311, 1246 (C=S st), 1143, 1118, 1107, 1046, 837,
822 (Si-C st).
RMN-1H (CDCl3, 400 MHz) δ: 8.13 (s, 1 H, C=CH), 8.10 (s, 1 H, C=CH),
4.99 (s, 2 H, 2 CH Ar), 4.78 (s, 2 H, 2 CH Ar), 4.66 (s, 2 H, 2 CH Ar),
4.34 (s, 5 H, 5 CH Ar), 4.35 (s, 5 H, 5 CH Ar), 3.62-3.51 (m, 2 H, NCH2CH3),
3.38-3.24 (m, 2 H, NCH2CH3), 0.98-0.87 (m, 6 H, 2 NCH2CH3), 0.32 (s, 9H, Si(CH3)3)
y 0.07 (s, 9H, Si(CH3)3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 190.09 (C=S), 189.83 (C=S), 188.32 (C=O),
188.22 (C=O), 184.97 (C=O cetona), 184.78 (C=O cetona), 166.51 (C sp2),
166.24 (C sp2), 165.92 (C sp
2), 154.38 (C sp
2), 154.23 (C sp
2), 153.28 (C sp
2),

Parte experimental 421
141.50 (C sp2), 141.35 (C sp
2), 141.02 (C sp
2), 132.35 (C=CH), 132.30 (C=CH),
131.98 (C sp2), 131.85 (C sp
2), 130.88 (C sp
2), 130.22 (C sp
2), 129.84 (C sp
2),
128.78 (C sp2), 82.41 (C Ar), 82.28 (C Ar), 79.95 (CH Ar), 79.85 (CH Ar),
75.02 (CH Ar), 70.66 (CH Ar), 70.56 (CH Ar), 47.51 (NCH2CH3), 47.44 (NCH2CH3),
14.10 (NCH2CH3), 14.03 (NCH2CH3), 0.98 (Si(CH3)3) y 0.19 (Si(CH3)3) ppm.
EM (IE) m/z (%): 633 (M+·, 10), 604 (M
+ - CH2CH3, 10), 601 (M
+ - S, 5), 285 (23), 149 (87).

422 Tesis Doctoral Susana Barriga Falcón
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.08.58.5
252550507575100100125125150150175175200200
S
N
SS
S
S
S
O
H
Fe
TMSO
S
N
SS S
S
SHO
+
(E)-381
O
Fe
TMS
(Z)-381

Parte experimental 423
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
SS
S
S
S
O
H
Fe
TMSO
S
N
SS S
S
SHO
+
(E)-381
O
Fe
TMS
(Z)-381

424 Tesis Doctoral Susana Barriga Falcón
61. Obtención de 1,4-bisferrocenil-2-butin-1,4-diol (384)
Fe CHO
THF, 56%Fe Fe
OHHO
374
1) BuLi
2)
373
384
Fe
H
HO
Sobre una disolución agitada del alcohol propargílico ferrocénico 374
(240 mg, 1.00 mmol) en THF (5 mL) y enfriada a -78 ºC se añadió lentamente butillitio
(1.54 mL, 2.20 mmol, 1.42 M en hexanos). Tras completar la adición la mezcla de
reacción se agitó a -78 ºC durante 15 minutos. A continuación se añadió lentamente el
ferrocenilcarbaldehído (373) (214 mg, 1.00 mmol). La mezcla de reacción se mantuvo
bajo agitación hasta alcanzar la temperatura ambiente y después se prolongó la agitación
a temperatura ambiente durante 30 minutos más. Seguidamente se añadió sobre la
mezcla de reacción una disolución acuosa saturada de NH4Cl (5 mL). A continuación se
separaron las fases y la fase acuosa fue extraída con éter dietílico (3 x 10 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico y el disolvente fue
eliminado en el rotavapor. El crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietílico, 1:1), lo que permitió separar el compuesto 384 puro
(254 g, 0.56 mmol, 56%), como un sólido de color amarillo claro;
p.f.: 128-129 ºC (ciclohexano-éter dietílico).
IR (KBr, cm-1
): 3095 (OH st), 1291, 1234, 1125, 1105, 1039, 1022, 998, 972, 821, 505, 486.
RMN-1H (CD3COCD3, 400 MHz) δ: 5.38-5.36 (m, 2 H, 2 CHOH), 4.55-4.53 (m, 2 H, 2 CH Ar),
4.48-4.46 (m, 2 H, 2 CH Ar), 4.36-4.35 (m, 2 H, 2 CH Ar), 4.24 (s, 10 H, 10 CH Ar), y
4.18-4.16 (m, 4H, 2 CH Ar y 2 OH) ppm.

Parte experimental 425
RMN-13
C (CD3COCD3, 100 MHz) δ: 86.38 (C≡C), 70.79 (C Ar), 70.68 (CH Ar),
69.73 (CH Ar), 69.46 (CH Ar), 68.78 (CH Ar) y 62.31 (CHOH) ppm.
EM (IE) m/z (%): 454 (M+·
, 100), 436 (M+ - OH - H, 11), 420 (M
+ - 2 OH, 16),
316 (M+ - Cp - Fe - OH, 87), 214 (M
+ - Fc - C3H2O, 20), 186 (Fc
+, 35), 121 (CpFe
+, 40),
56 (Fe+, 27).
Masa exacta: calculada para C24H22O2Fe2: 454.0319 encontrada: 454.0297.

426 Tesis Doctoral Susana Barriga Falcón
Fe Fe
OHHO
384

Parte experimental 427
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
Fe Fe
OHHO
384

428 Tesis Doctoral Susana Barriga Falcón
62. Obtención de 1,4-bisferrocenil-2-butin-1,4-diona (382)
Fe Fe
OHHO
384
Fe Fe
OO
382
MnO2
CH2Cl2, 98%
Sobre una disolución agitada del diol 384 (227 mg, 0.50 mmol) en diclorometano
(5 mL) se añadió óxido de manganeso (2.61 g, 30.00 mmol). La mezcla de reacción se
agitó a temperatura ambiente durante 15 minutos. A continuación se filtró sobre celita y
se lavó con diclorometano hasta que dejó de salir coloreado. Tras eliminar el disolvente
en el rotavapor, el crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietílico, 8:2), lo que permitió separar el compuesto 382 puro
(220 g, 0.49 mmol, 98%), como un sólido de color morado; p.f.: 161-162 ºC
(ciclohexano-éter dietílico).
IR (KBr, cm-1
): 1618 (C=O st), 1446, 1273, 1115, 824, 508, 489, 470.
RMN-1H (CDCl3, 200 MHz) δ: 5.00 (dd, J = 3.8 y 1.9 Hz, 4 H, 4 CH Ar),
4.71 (dd, J = 3.8 y 1.9 Hz, 4 H, 4 CH Ar) y 4.34 (s, 10 H, 10 CH Ar) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 179.56 (C=O), 83.68 (C≡C), 79.72 (C Ar),
74.12 (CH Ar), 70.80 (CH Ar) y 70.58 (CH Ar) ppm.
EM (IE) m/z (%): 450 (M+·
, 100), 186 (Fc+, 20), 149 (CpFeCO
+, 60), 121 (CpFe
+, 25).
Masa exacta: calculada para C24H18O2Fe2: 450.0006 encontrada: 450.0008.
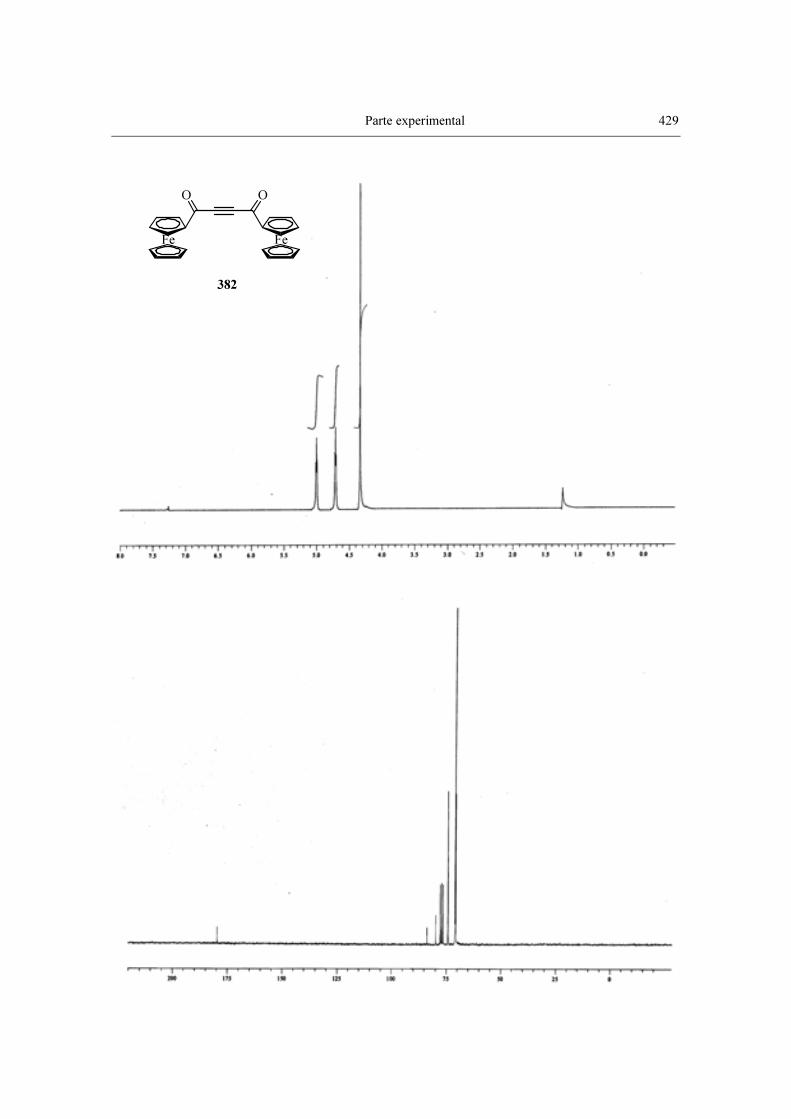
Parte experimental 429
Fe Fe
OO
382

430 Tesis Doctoral Susana Barriga Falcón
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
Fe Fe
OO
382

Parte experimental 431
63. Reacción de cicloadición de la bisditiolotiazina 176 con la 1,4-bisferrocenil-2-
butin-1,4-diona (382)
Fe
S
N
SS
S
S
S
O
O
O
Fe
S
N
S
SS
S
SO
Fe Fe
OOSc(OTf)3
382
+
CH2Cl2, 97%
176 386
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(40 mg, 0.124 mmol) en diclorometano (2 mL) se añadió la dicetona 382
(67 mg, 0.149 mmol) y triflato de escandio (30mg, 0.060 mmol). La mezcla de reacción se
agitó durante 10 minutos a temperatura ambiente. Tras eliminar el disolvente en el rotavapor,
el crudo obtenido se purificó mediante cromatografía rápida (SiO2, ciclohexano-éter dietílico
1:1), lo que permitió separar el compuesto 386205
puro (93 mg, 0.120 mmol, 97%), como un
sólido de color rojo; p.f.: >300ºC (ciclohexano-éter dietílico).
IR (KBr cm-1
): 1664 (C=O st, heterociclo), 1612 (C=O st), 1348, 1272 (C=S st), 1085,
1049, 489.
RMN-1H (CDCl3, 400 MHz) δ: 4.89 (m, 1 H, CH Ar), 4.86 (m, 1 H, CH Ar),
4.72-4.59 (m, 6 H, 6 CH Ar), 4.37 (s, 5 H, 5 CH Ar), 5.35 (s, 5 H, 5 CH Ar),
3.64 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3), 3.39 (dc, J = 14.4 y 7.2 Hz, 1 H, NCH2CH3)
y 1.32 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
EM (FAB) m/z (%): 774 (M+ + 1, 3), 663 (M
+ + 1 - C5H4 - S - CH3, 2),
633 (M+ - Cp - S - NCH2CH3, 1), 577 (M
+ - Cp - Fe - S - NCH2CH3, 20), 95 (38),
81 (45), 69 (75), 55 (100).
Masa exacta: calculada para C32H24Fe2NO3S6 (M+ + 1): 773.8779 encontrada: 773.8791.
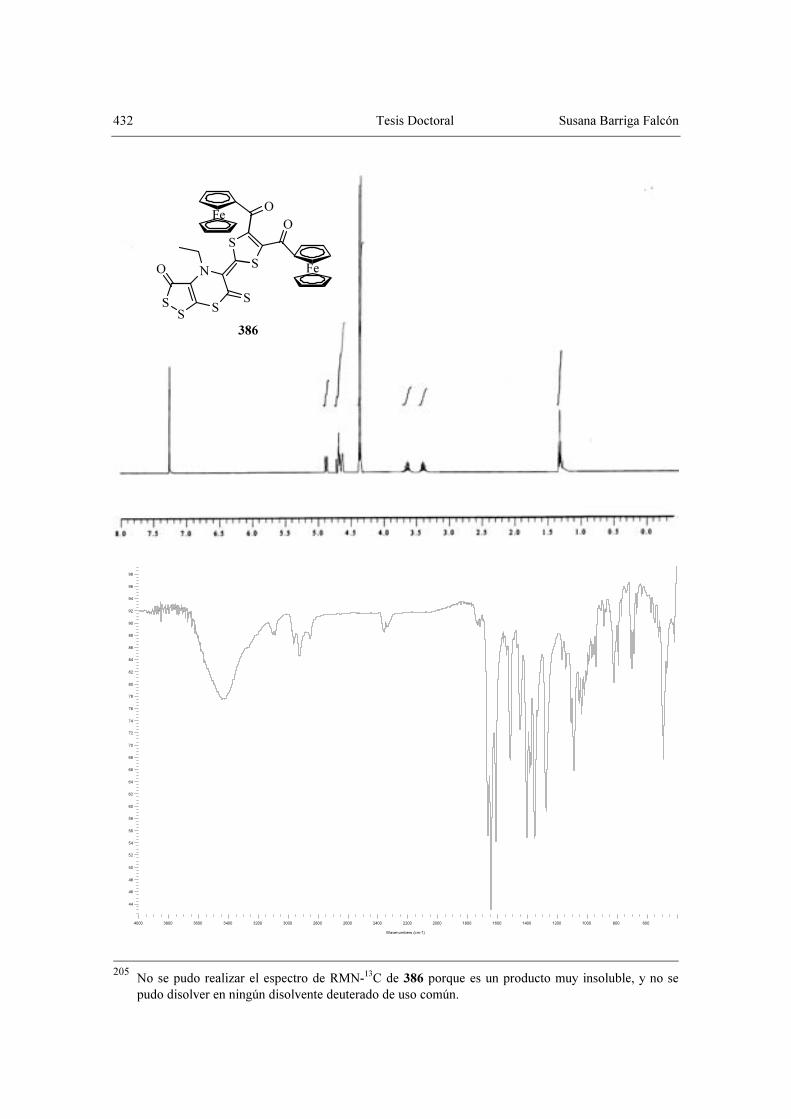
432 Tesis Doctoral Susana Barriga Falcón
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
205
No se pudo realizar el espectro de RMN-13C de 386 porque es un producto muy insoluble, y no se
pudo disolver en ningún disolvente deuterado de uso común.
Fe
S
N
S
S
S
S
S
O
O
O
Fe
386

Parte experimental 433
Fe
S
N
S
S
S
S
S
O
O
O
Fe
386

434 Tesis Doctoral Susana Barriga Falcón
64. Obtención de 1,4-bis[(R)-αααα-(p-metoxifenil)ferrocenil]-2-butin-1,4-diol (385)
THF, 62%
Fe p-C6H4OMe
CHO
1) BuLi
2)
376
378
Fe p-MeOC6H4
HO
H
385, PMP = p-MeOC6H4
Fe
HO OH
Fe
PMP
PMP
Sobre una disolución agitada del alquino 378 (200 mg, 0.578 mmol) en THF (5 mL),
enfriada a -78 ºC, se añadió lentamente butillitio (0.90 mL, 1.272 mmol, 1.42 M en
hexanos). Tras completar la adición la mezcla de reacción se agitó a esta temperatura
durante 15 minutos. A continuación se añadió lentamente el aldehido 376
(185 mg, 0.578 mmol). La mezcla de reacción se mantuvo bajo agitación hasta alcanzar
la temperatura ambiente y después se prolongó la agitación a temperatura ambiente
durante 30 minutos más. Seguidamente se añadió sobre la mezcla de reacción una
disolución acuosa saturada de NH4Cl (5 mL). A continuación se separaron las fases y la
fase acuosa fue extraída con éter dietílico (3 x 10 mL). Los extractos orgánicos
combinados se secaron sobre sulfato sódico y el disolvente fue eliminado en el
rotavapor. El crudo obtenido se purificó mediante cromatografía rápida (SiO2,
ciclohexano-éter dietílico 1:1), lo que permitió separar el compuesto 385 puro (238 g,
0.358 mmol, 62%), como un sólido de color amarillo; p.f.: 71-72 ºC (ciclohexano-éter
dietílico).
IR (KBr, cm-1
): 3427 (OH st), 2923, 1522, 1247, 1033, 831.
RMN-1H (CDCl3, 200 MHz) δ: 7.48 (d, J = 8.8 Hz, 4 H, 4 CH Ar), 6.83 (d, J = 8.8 Hz, 4 H, 4 CH Ar),
5.28 (m, 2 H, 2 CHOH), 4.41 (m, 2 H, 2 CH Ar), 4.24 (m, 2 H, 2 CH Ar),
4.23 (m, 2 H, 2 CH Ar), 4.17 (s, 10 H, 10 CH Ar) y 3.81 (s, 3 H, OCH3) ppm.

Parte experimental 435
RMN-13
C (CDCl3, 50 MHz) δ: 158.28 (C Ar), 130.24 (CH Ar), 129.39 (C Ar), 113.42
(CH Ar), 88.75 (C≡CH), 87.20 (C Ar), 85.25 (C Ar), 70.28 (CH Ar), 69.92 (CH Ar),
67.62 (CH Ar), 66.82 (CH Ar), 59.50 (CHOH) y 55.22 (OCH3) ppm.
EM (FAB) m/z (%): 667 (M+ + 1, 6).
Masa exacta: calculada para C38H35O4Fe2: 667.1234 encontrada: 667.1277.

436 Tesis Doctoral Susana Barriga Falcón
385, PMP = p-MeOC6H4
Fe
HO OH
Fe
PMP
PMP
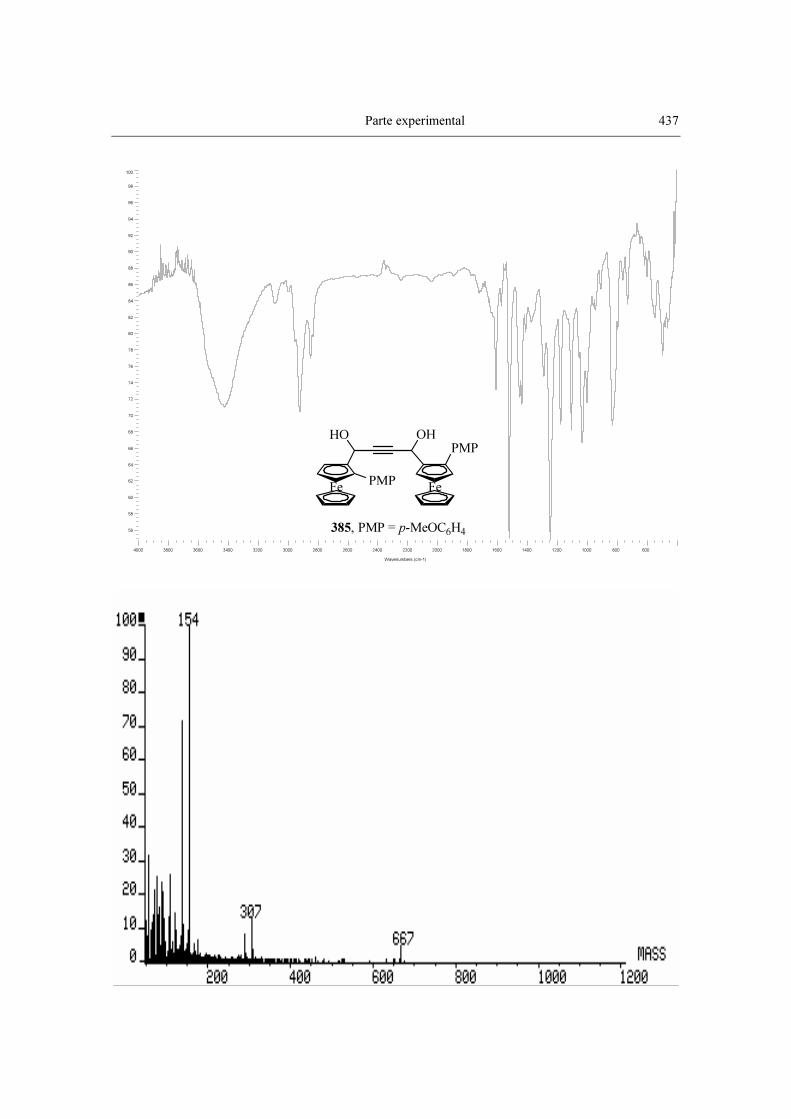
Parte experimental 437
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
385, PMP = p-MeOC6H4
Fe
HO OH
Fe
PMP
PMP

438 Tesis Doctoral Susana Barriga Falcón
65. Obtención de 1,4-bis[(R)-αααα-(p-metoxifenil)ferrocenil]-2-butin-1,4-diona (383)
MnO2
CH2Cl2, 82%
385, PMP = p-MeOC6H4
Fe
HO OH
Fe
PMP
PMPFe
O O
Fe
PMP
PMP
383, PMP = p-MeOC6H4
Sobre una disolución agitada del diol 385 (200 mg, 0.30 mmol) en diclorometano
(5 mL) se añadió óxido de manganeso (1.57 g, 18.00 mmol). La mezcla de reacción se
agitó a temperatura ambiente durante 15 minutos. A continuación se filtró sobre celita y
se lavó con diclorometano hasta que el filtrado dejó de salir coloreado. Tras eliminar el
disolvente en el rotavapor, el crudo obtenido se purificó mediante cromatografía rápida
(SiO2, ciclohexano-éter dietílico 8:2), lo que permitió separar el compuesto 383 puro
(165 mg, 0.25 mmol, 82%), como un sólido de color rojo; p.f.: 65-66 ºC (ciclohexano-
éter dietílico).
IR (KBr, cm-1
): 2924, 1630 (C=O st), 1609 (C=O st), 1521, 1245 (ArC-O-C st), 1107,
826.
RMN-1H (CDCl3, 200 MHz) δ: 7.55 (d, J = 8.8 Hz, 4 H, 4 CH Ar),
6.88 (d, J = 8.8 Hz, 4 H, 4 CH Ar), 5.02 (m, 2 H, 2 CH Ar), 4.85 (m, 2 H, 2 CH Ar),
4.71 (m, 2 H, 2 CH Ar), 4.30 (s, 10 H, 10 CH Ar) y 3.82 (s, 6 H, 2 OCH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 179.39 (C=O), 158.82 (C Ar), 131.12 (CH Ar),
127.69 (C Ar), 113.05 (CH Ar), 91.24 (C Ar), 84.76 (C≡C), 76.35 (CH Ar),
76.03 (C Ar), 73.61 (CH Ar), 72.15 (CH Ar) y 55.23 (OCH3) ppm.
EM (EI) m/z (%): 663 (M+ + 1, 10).
Masa exacta: calculada para C38H31O4Fe2 (M+ + 1): 663.0921 encontrada: 663.0866.

Parte experimental 439
Fe
O O
Fe
PMP
PMP
383, PMP = p-MeOC6H4

440 Tesis Doctoral Susana Barriga Falcón
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
Fe
O O
Fe
PMP
PMP
383, PMP = p-MeOC6H4

Parte experimental 441
66. Reacción de cicloadición de la bisditiolotiazina 176 con la dicetona 383
S
N
S
S S
S
SO
O
O
S
N
S
SS
S
O S
+
Sc(OTf)3
CH2Cl2, 78%
176 383, PMP = p-MeOC6H4
Fe
O O
Fe
PMP
PMP
Fe
PMP
Fe
PMP
387, PMP = p-MeOC6H4
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(35 mg, 0.108 mmol) en diclorometano (2 mL) se añadió la dicetona 383
(86 mg, 0.130 mmol) y triflato de escandio (26 mg, 0.053 mmol). La mezcla de reacción se
agitó durante 10 minutos a tempratura ambiente. Tras eliminar el disolvente en el rotavapor,
el crudo obtenido se purificó mediante cromatografía rápida (SiO2, ciclohexano-éter dietílico
7:3), lo que permitió separar el compuesto 387 puro (83 mg, 0.084 mmol, 78%), como un
sólido de color rojo; p.f.: 103-104 ºC (ciclohexano-éter dietílico).
IR (KBr cm-1
): 2955, 2924, 2852, 1667 (C=O st heterociclo), 1632 (C=O st), 1520,
1405, 1342, 1244 (C=S st), 1036, 828.
RMN-1H (CDCl3, 400 MHz) δ: 7.48-7.13 (m, 4 H, 4 CH Ar ), 6.72-6.64 (m, 3 H, 3 CH Ar),
6.44 (d, J = 8.4 Hz, 1 H, CH Ar), 5.04 (m, 1 H, CH Ar), 4.90 (m, 1 H, CH Ar),
4.79-4.65 (m, 4 H, 4 CH Ar), 4.50 (s, 3 H, 3 CH Ar), 4.46 (s, 3 H, 3 CH Ar),
4.40 (s, 2 H, 2 CH Ar), 4.37 (s, 2 H, 2 CH Ar), 3.72 (s, 3 H, OCH3),
3.68 (s, 1 H, OCH3), 3.54 (s, 2 H, OCH3), 3.40 (dc, J = 14.0 y 7.0 Hz, ½ H, ½ NCH2CH3),
3.39 (dc, J = 14.0 y 7.0 Hz, ½ H, ½ NCH2CH3), 3.00 (dc, J = 13.8 y 6.9 Hz, H, NCH2CH3) y
1.15 (t, J = 7.2 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 189.68 (C=S), 189.63 (C=O), 188.01 (C=O),
184.66 (C=O), 163.98, 158.72, 158.59, 158.54, 141.12, 140.53, 132.26, 131.50, 131.41,
131.24, 131.07, 130.23, 128.072, 127.94, 127.69, 126.89, 113.01, 112.86, 112.83,
112.59, 93.75, 93.61, 93.03, 92.54, 77.16, 76.36, 76.31, 76.15, 76.11, 72.08, 71.96,

442 Tesis Doctoral Susana Barriga Falcón
71.92, 71.60, 71.52, 71.48, 71.42, 70.51, 70.34, 55.22 (OCH3), 55.14 (OCH3),
55.12 (OCH3), 55.10 (OCH3), 47.68 (NCH2CH3), 13.42 (NCH2CH3) y
13.34 (NCH2CH3) ppm.
EM (FAB) m/z (%): 985 (M+·
, 15), 920 (M+ - Cp, 1), 865 (M
+ +1 - CpFe, 1),
694 (M+ - CpFe - C5H3 - C6H4OCH3, 1), 662 (M
+ - CpFe - C5H3 - C6H4OCH3 -S, 1),
95 (45), 81 (50), 69 (85), 55 (100).
Masa exacta: calculada para C46H35Fe2NO5S6: 984.9538 encontrada: 984.9563.

Parte experimental 443
S
N
S
S S
S
SO
O
O
Fe
PMP
Fe
PMP
387, PMP = p-MeOC6H4

444 Tesis Doctoral Susana Barriga Falcón
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
S
S S
S
SO
O
O
Fe
PMP
Fe
PMP
387, PMP = p-MeOC6H4

Parte experimental 445
67. Reacción de cicloadición de la bisditioltiazina 161 con la 1,4-bisferrocenil-2-
butin-1,4-diona (382)
Fe
S
N
S
S
S
O
O
FeS
N
S
SS
S
SS
Fe Fe
OO Sc(OTf)3
382
+
CH2Cl2, 65%
161 388
S
S
S
O
O
Fe
Fe
Sobre una disolución agitada de 4-etilbis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-3,5-ditiona
(25 mg, 0.074 mmol) en diclorometano (2 mL) se añadió la dicetona 382
(83 mg, 0.184 mmol) y triflato de escandio (18 mg, 0.037 mmol). La mezcla de reacción se
agitó durante 30 minutos a temperatura ambiente. Tras eliminar el disolvente en el rotavapor,
el crudo obtenido se purificó mediante cromatografía rápida (SiO2, ciclohexano-acetato de
etilo 8:2), lo que permitió separar el compuesto 388 puro (59 mg, 0.048 mmol, 65%), como
un sólido de color rojo; p.f.: 152-153 ºC (ciclohexano-acetato de etilo).
IR (KBr, cm-1
): 2922, 2851, 1630 (C=O st), 1441, 1270 (C=S st), 1081, 486.
RMN-1H (CDCl3, 200 MHz) δ: 4.88-4.61 (m, 16 H, 16 CH Ar),
4.37 (s, 10 H, 10 CH Ar), 4.25 (s, 10 H, 10 CH Ar), 3.60 (c, J = 7.1 Hz, 2 H,
NCH2CH3) y 1.46 (t, J = 7.1 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 194.36 (C=S), 188.97 (C=O), 188.84 (C=O),
162.57 (C sp2), 139.08 (C sp
2), 138.71 (C sp
2), 133.73 (C sp
2), 78.00 (C Ar),
77.73 (C Ar), 74.05 (CH Ar), 73.91 (CH Ar), 73.82 (CH Ar), 70.83 (CH Ar),
70.75 (CH Ar), 70.70 (CH Ar), 70.62 (CH Ar), 70.56 (CH Ar), 70.43 (CH Ar),
49.69 (NCH2CH3) y 13.97 (NCH2CH3) ppm.
EM (FAB) m/z (%): 1239 (M+·
, 6), 663 (M+ - C24H18Fe2O2 - Cp - CH2CH3 - S, 7),
186 (Fc+, 6), 121 (CpFe
+, 15), 95 (38), 81 (50), 69 (80), 55 (100).

446 Tesis Doctoral Susana Barriga Falcón
Fe
S
N
S
S
S
O
O
Fe
388
S
S
S
O
O
Fe
Fe
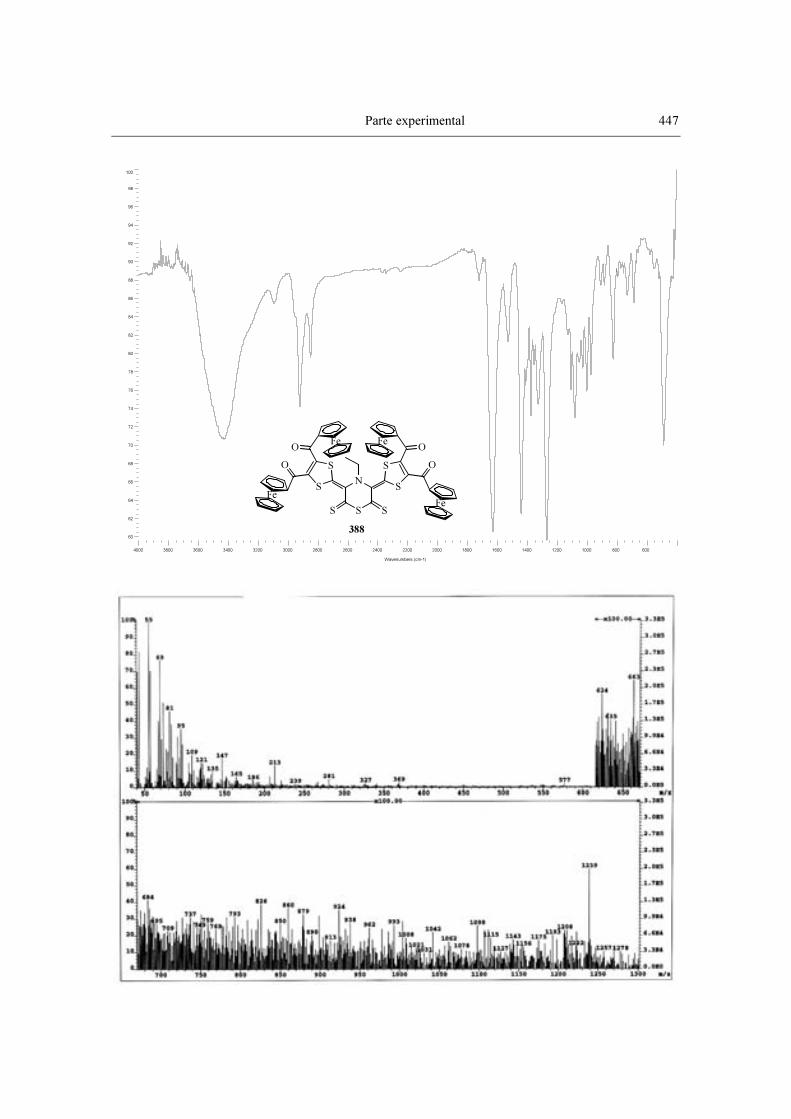
Parte experimental 447
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
Fe
S
N
S
S
S
O
O
Fe
388
S
S
S
O
O
Fe
Fe

448 Tesis Doctoral Susana Barriga Falcón
68. Obtención de 3-hexiltien-2-carbaldehído (391)
S Br S CHO
1) BuLi
2) DMF
THF, 89%
390 391
Sobre una disolución agitada de 2-bromo-3-hexiltiofeno187
(1.20 g, 4.86 mmol) en THF
(5 mL), enfriada a -78 ºC, se añadió lentamente butillitio (3.64 mL, 5.83 mmol, 1.6 M
en hexanos). Tras completar la adición la mezcla de reacción se agitó a esta temperatura
durante 15 minutos. A continuación se añadió lentamente dimetilformamida
(710 mg, 0.75 mL, 9.72 mmol). Se dejó alcanzar la temperatura ambiente a la mezcla de
reacción, bajo agitación, y después se mantuvo bajo agitación durante otros 30 minutos.
Seguidamente se añadió sobre la mezcla de reacción una disolución acuosa saturada de
NH4Cl (5 mL). A continuación se separaron las fases y la fase acuosa fue extraída con
éter dietílico (3 x 10 mL). Los extractos orgánicos combinados se secaron sobre sulfato
sódico y el disolvente fue eliminado en el rotavapor. El crudo obtenido se purificó
mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano, 3:1), lo que
permitió separar el compuesto 391 puro (847 mg, 4.32 mmol, 89%), como un aceite de
color amarillo.
IR (película líquida, cm-1
): 2958, 2925, 2849, 1657 (C=O st), 1432, 1237, 672.
RMN-1H (CDCl3, 200 MHz) δ: 10.04 (s, 1 H, CHO), 7.64 (d, J = 5.0 Hz, 1 H, CH Ar),
7.00 (d, J = 5.0 Hz, 1 H, CH Ar), 2.96 (t, J = 7.6 Hz, 2 H, CH2), 1.66 (m, 2 H, CH2),
1.31 (m, 6 H, 3 CH2) y 0.87 (t, J = 6.5 Hz, 3 H, CH3) ppm.
187
Wang, F.; Rauh, R. D.; Rose, T. L. “An electrically conducting star polymer”. J. Am. Chem. Soc.
1997, 119, 11106-11107.

Parte experimental 449
RMN-13
C (CDCl3, 50 MHz) δ: 181.66 (CHO), 152.42 (C Ar), 137.25 (C Ar),
133.98 (CH Ar), 130.36 (CH Ar), 31.18 (CH2), 31.01 (CH2), 28.55 (CH2), 28.05 (CH2),
22.16 (CH2) y 13.67 (CH3) ppm.
EM (IE) m/z (%): 196 (M+·
, 60), 139 (M+ - C4H9, 100), 126 (70), 97 (35).
Masa exacta: calculada para C11H16OS: 196.0922 encontrada: 196.0914.

450 Tesis Doctoral Susana Barriga Falcón
SCHO
391

Parte experimental 451
16
18
20
22
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
SCHO
391

452 Tesis Doctoral Susana Barriga Falcón
69. Obtención de 1-(3-hexiltien-2-il)-2-propin-1-ol (392)
S CHO
H MgBr
S
H
OH
391
THF, 91%
392
Sobre una disolución agitada de 3-hexil-2-tien-2-carbaldehído (490 mg, 2.50 mmol)
en THF (5 mL) se añadió lentamente bromuro de etinilmagnesio
(8.3 mL, 3.74 mmol, 0.45 M en THF). Tras completar la adición la mezcla de reacción
se agitó a temperatura ambiente durante 60 minutos. Seguidamente se añadió sobre la
mezcla de reacción una disolución acuosa saturada de NH4Cl (5 mL). A continuación se
separaron las fases y la fase acuosa fue extraída con éter dietílico (3 x 10 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico y el disolvente fue
eliminado en el rotavapor. El crudo obtenido se purificó mediante cromatografía rápida
(SiO2, éter de petróleo-diclorometano 1:1), lo que permitió separar el compuesto 392
puro (504 mg, 2.27 mmol, 91%), como un aceite de color amarillo intenso.
IR (película líquida, cm-1
): 3307 (OH st), 3289 (OH st), 2954, 2924, 2855, 1674, 1661,
1464, 1028, 659.
RMN-1H (CDCl3, 200 MHz) δ: 7.20 (d, J = 5.0 Hz, 1 H, CH Ar),
6.86 (d, J = 5.0 Hz, 1 H, CH Ar), 5.68 (s ancho, 1 H, CHOH),
2.64 (m, 3 H, CH2 y C≡CH), 1.61 (m, 2 H, CH2), 1.31 (m, 6 H, 3 CH2) y
0.90 (t, J = 7.0 Hz, 3 H, CH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 140.59 (C Ar), 136.52 (C Ar), 129.10 (CH Ar),
124.26 (CH Ar), 84.17 (C≡CH), 73.94 (C≡CH), 58.00 (CHOH), 31.57 (CH2),
30.58 (CH2), 29.07 (CH2), 28.22 (CH2), 22.51 (CH2) y 14.00 (CH3) ppm.

Parte experimental 453
EM (IE) m/z (%): 222 (M+·
, 25), 196 (M+ - C2H2, 20), 137 (M
+ - C6H13, 100), 123 (30),
97 (25), 77 (20).

454 Tesis Doctoral Susana Barriga Falcón
S
H
OH
392

Parte experimental 455
16
18
20
22
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
H
OH
392

456 Tesis Doctoral Susana Barriga Falcón
70. Obtención de 1,4-bis(3-hexiltien-2-il)-2-butin-1,4-diol (393)
S
H
OH
S CHO
OHHO
S S
392
1) BuLi
391
2)
THF, 56%
393
Sobre una disolución agitada del alcohol propargílico 392 (500 mg, 2.25 mmol) en THF
(5 mL), enfriada a -78 ºC se añadió lentamente butillitio (3.1 mL, 4.96 mmol, 1.6 M en
hexanos). Tras completar la adición la mezcla de reacción se agitó a esta temperatura
durante 15 minutos. A continuación se añadió lentamente el aldehido 391
(441 mg, 2.25 mmol). La mezcla de reacción se agitó hasta alcanzar la temperatura
ambiente y después se mantuvo la agitación a temperatura ambiente durante 30 minutos
más. Seguidamente se añadió sobre la mezcla de reacción una disolución acuosa
saturada de NH4Cl (5 mL). A continuación se separaron las fases y la fase acuosa fue
extraída con éter dietílico (3 x 10 mL). Los extractos orgánicos combinados se secaron
sobre sulfato sódico y el disolvente fue eliminado en el rotavapor. El crudo obtenido se
purificó mediante cromatografía rápida (SiO2, éter de petróleo-diclorometano 1:1 hasta
diclorometano-acetato de etilo 19:1), lo que permitió separar el compuesto 393 puro
(531 mg, 1.27 mmol, 56%), como un aceite de color marrón.
IR (película líquida, cm-1
): 3425 (OH st), 3352 (OH st), 2955, 2926, 2856, 1633, 1465,
1264, 1003, 738, 704.
RMN-1H (CDCl3, 200 MHz) δ: 7.21 (d, J = 5.0 Hz, 2 H, 2 CH Ar),
6.87 (d, J = 5.0 Hz, 2 H, 2 CH Ar), 5.70 (m, 2 H, 2 CHOH), 2.65 (t, J = 6.0 Hz, 4 H, 2 CH2),
1.61 (m, 4 H, 2 CH2), 1.31 (m, 12 H, 6 CH2) y 0.90 (t, J = 6.5 Hz, 6 H, 2 CH3) ppm.

Parte experimental 457
RMN-13
C (CDCl3, 50 MHz) δ: 140.71 (C Ar), 136.54 (C Ar), 129.20 (CH Ar),
124.34 (CH Ar), 83.16 (C≡C), 58.09 (CHOH), 31.62 (CH2), 30.64 (CH2), 29.11 (CH2),
28.28 (CH2), 22.54 (CH2) y 14.03 (CH3) ppm.

458 Tesis Doctoral Susana Barriga Falcón
OHHO
S S
393
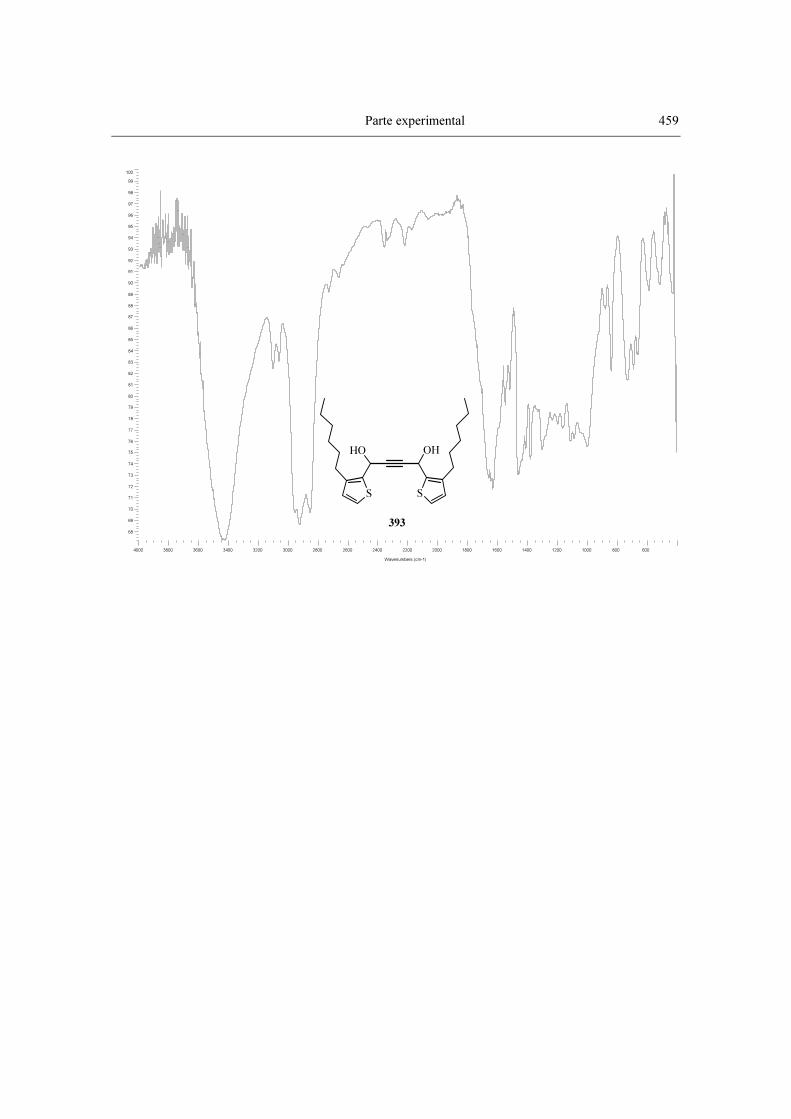
Parte experimental 459
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
OHHO
S S
393

460 Tesis Doctoral Susana Barriga Falcón
71. Obtención de 1,4-bis(3-hexiltien-2-il)-2-butin-1,4-diona (394)
OHHO
S S
OO
S S
393 394
BAIB, TEMPO
CH2Cl2, 80%
Sobre una disolución agitada del diol 393 (209 mg, 0.50 mmol) en diclorometano
(2 mL) se añadió bisacetoxiiodobenceno (BAIB) (355mg, 1.10 mmol) y
2,2,6,6-tetrametilpiperidin-1-oxilo (TEMPO) (16 mg, 0.10 mmol). La mezcla de
reacción se agitó a temperatura ambiente durante 2 horas. A continuación la mezcla de
reacción se diluyó con diclorometano (5 mL) y se añadió una disolución acuosa saturada
de Na2S2O3 (5 mL). Después se separaron las fases y la fase acuosa fue extraída con
diclorometano (4 x 10 mL). Los extractos orgánicos se lavaron con una disolución
acuosa saturada de NaHCO3 y posteriormente con una disolución saturada de NaCl. La
fase orgánica se secó sobre sulfato sódico y el disolvente fue eliminado en el rotavapor.
El crudo obtenido se purificó mediante cromatografía rápida (SiO2, éter de petróleo-
diclorometano, 1:1), lo que permitió separar el compuesto 394 puro
(167 mg, 0.40 mmol, 80%), como un aceite de color amarillo oscuro.
IR (película líquida, cm-1): 3102 (OH st), 2955, 2926, 2856, 1627 (C=O st), 1515, 1409, 1279, 1242.
RMN-1H (CDCl3, 200 MHz) δ: 7.57 (d, J = 5.0 Hz, 2 H, 2 CH Ar),
7.01 (d, J = 5.0 Hz, 2 H, 2 CH Ar), 3.05 (t, J = 7.4 Hz, 4 H, 2 CH2),
1.61 (m, 4 H, 2 CH2), 1.30 (m, 12 H, 6 CH2) y 0.87 (t, J = 6.6 Hz, 6 H, 2 CH3) ppm.
RMN-13
C (CDCl3, 50 MHz) δ: 168. 78 (C=O), 152.44 (C Ar), 136.48 (C Ar),
133.91 (CH Ar), 131.84 (CH Ar), 81.45 (C≡C), 31.57 (CH2), 30.17 (CH2), 30.12
(CH2), 29.14 (CH2), 22.53 (CH2) y 14.03 (CH3) ppm.

Parte experimental 461
EM (IE) m/z (%): 414 (M+·
, 3), 344 (M+ - C5H10, 13), 273 (M
+ - C6H13 - C4H8, 8),
247 (M+ - C6H13 - C4H2S, 14), 219 (M
+ - C6H13 - C4H2S - CO, 45),
176 (M+ - C6H13 - C4H2S - CO - C3H7, 100), 163 (M
+ - C6H13 - C4H2S - CO - C4H8, 29),
151 (M+ - C6H13 - C4H2S - CO - C - C4H8, 54), 97 (25).
Masa exacta: calculada para C24H30O2S2: 414.1687 encontrada: 414.1691.
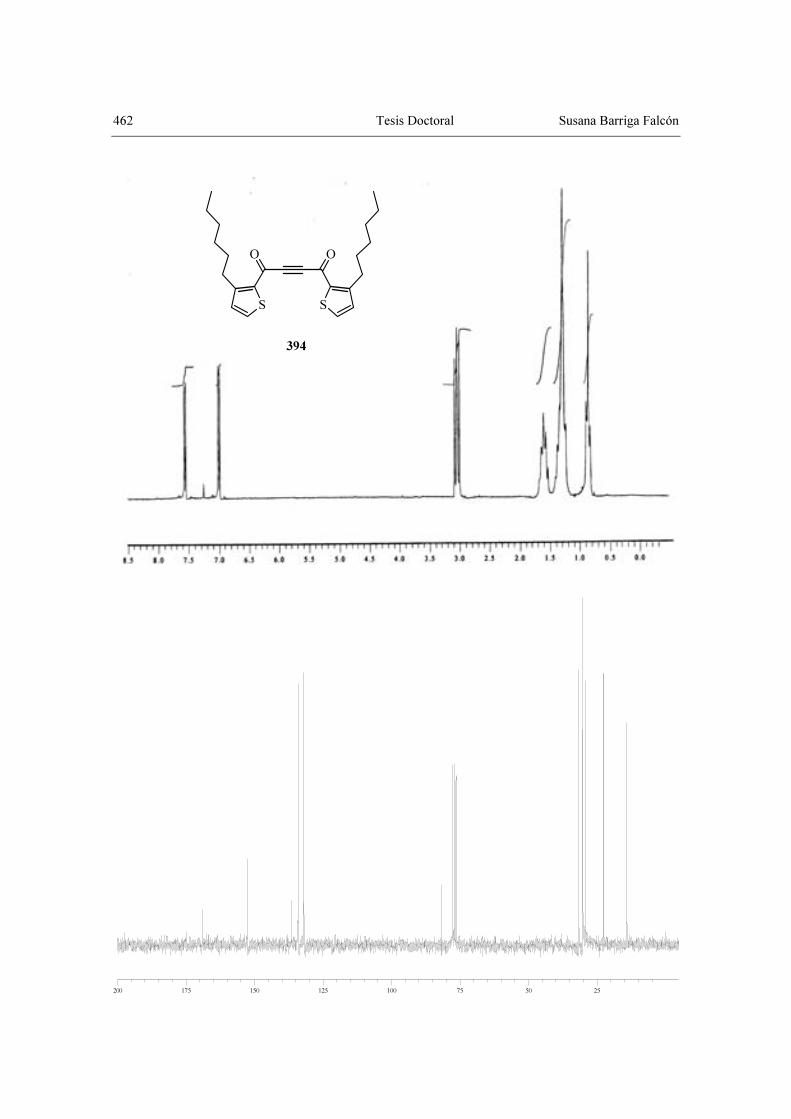
462 Tesis Doctoral Susana Barriga Falcón
252550507575100100125125150150175175200200
OO
S S
394

Parte experimental 463
18
20
22
24
26
28
30
32
34
36
38
40
42
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
OO
S S
394

464 Tesis Doctoral Susana Barriga Falcón
72. Reacción de cicloadición de la bisditiolotiazina 176 con
1,4-bis(3-hexiltien-2-il)-2-butin-1,4-diona (394)
S
N
S
S S
S
O S
176
OO
S S
Sc(OTf)3
S
N
S
S
O
S
S
S O
OS
S
+
CH2Cl2, 50%
394 395
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) en diclorometano (1.5 mL) se añadió 1,4-bis(3-hexiltien-2-il)-2-butin-
1,4-diona (394) (77 mg, 0.186 mmol) y triflato de escandio (23 mg, 0.047 mmol). La mezcla
de reacción se agitó a temperatura ambiente durante 10 minutos. A continuación se eliminó
el disolvente en el rotavapor, y el residuo resultante se purificó mediante cromatografía
rápida (SiO2, éter de petróleo-diclorometano 1:1), lo que permitió separar el compuesto 395
puro (34 mg, 0.046 mmol, 50%), como un sólido rojo; p.f.: 58-59 ºC (CH2Cl2-éter de
petróleo).
IR (KBr cm-1
): 2952, 2924, 2852, 1672 (C=O st), 1666 (C=O st), 1630 (C=O st,
heterociclo), 1407, 1237.
RMN-1H (CDCl3, 400 MHz) δ: 7.46 (d, J = 5.0 Hz, 2 H, 2 CH Ar),
6.92 (dd, J = 5.0 y 2.5 Hz, 2 H, 2 CH Ar), 3.57 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3),
3.31 (dc, J = 14.2 y 7.1 Hz, 1 H, NCH2CH3), 2.72 (c, J = 7.2 Hz, 4 H, 2 CH2),
1.43 (m, 4 H, 2 CH2), 1.25 (m, 12 H, 6 CH2) y 0.87 (m, 9 H, NCH2CH3 y 2 CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 191.47 (C=S), 184.63 (C=O heterociclo),
177.94 (C=O), 177.79 (C=O), 161.97, 153.61, 153.28, 153.24, 142.42, 141.32, 134.03,
133.82, 133.45, 133.18, 132.19, 131.79, 131.61 y 131.48 (C sp2, CH Ar y C Ar),
47.84 (NCH2CH3), 31.59 (CH2), 30.13 (CH2), 30.07 (CH2), 29.14 (CH2), 22.55 (CH2),
14.07 (CH3) y 13.43 (NCH2CH3) ppm.

Parte experimental 465
EM (FAB) m/z (%): 738 (M+ + 1, 10).
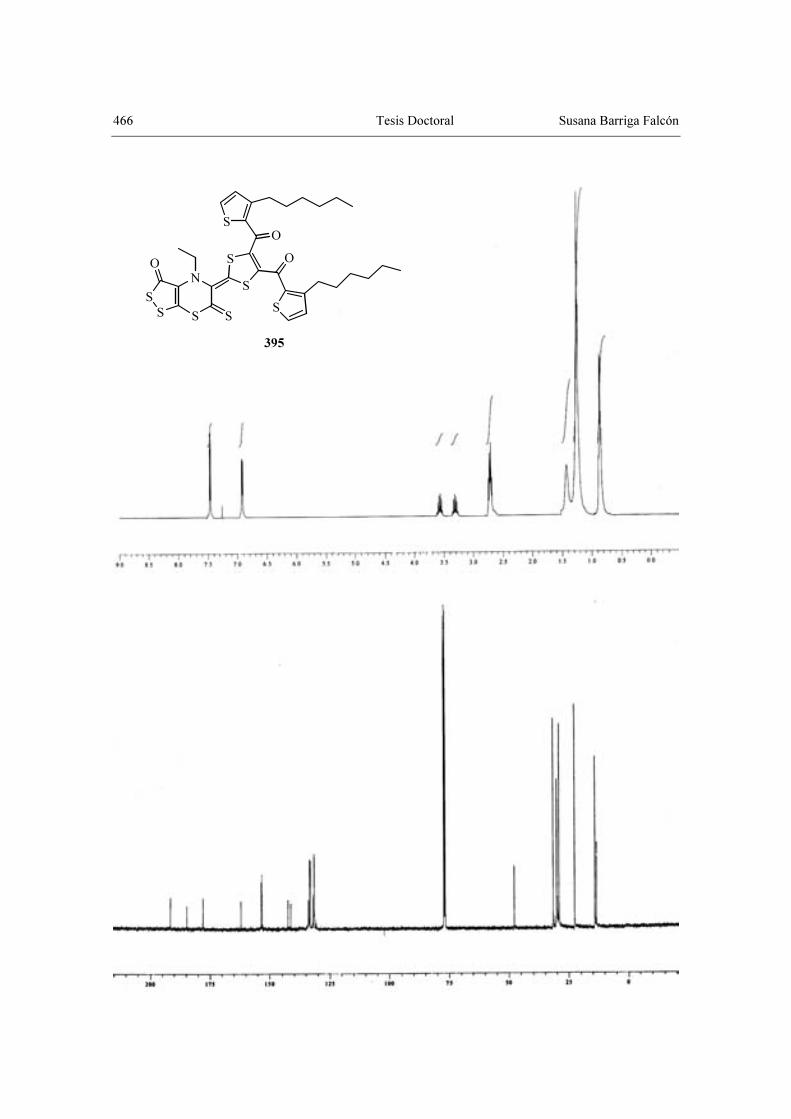
466 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S O
O
S
S
395
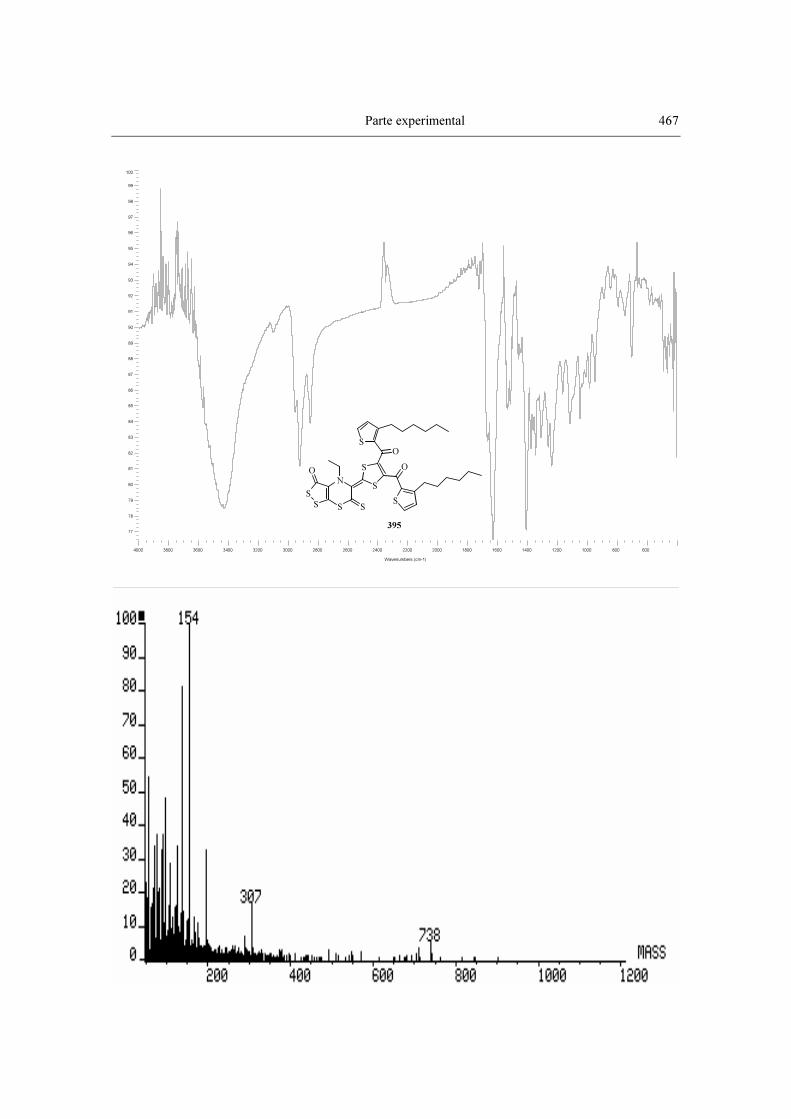
Parte experimental 467
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
S
S
O
S
S
S O
O
S
S
395

468 Tesis Doctoral Susana Barriga Falcón
73. Reacción de cicloadición de la bisditiolotiazina 176 con bencino
S
N
S
SS
S
O S Bu4NF
S
N
S
S
O
S
S
S
+
176
CH2Cl2, 70%
403402
Si(OH)Me2
I(Ph)OTf
Sobre una disolución agitada de 4-etil-3-oxo-bis[1,2]ditiolo[3,4-b:4’,3’-e][1,4]tiazina-5-tiona
(30 mg, 0.093 mmol) y triflato de [2-(hidroxidimetilsilil)fenil](fenil)iodonio194
402
(117 mg, 0.232 mmol) en diclorometano (2 mL) enfriada a 0 ºC se añadió lentamente una
disolución de fluoruro de tetrabutilamonio (0.232 mL, 0.232 mmol, 1.0 M en THF). Tras
completar la adición la mezcla de reacción se agitó durante 30 minutos a temperatura
ambiente. A continuación se vertió agua (10 mL) sobre la mezcla de reacción. Seguidamente
se separaron las fases y la fase acuosa se extrajo con diclorometano (3 x 10 mL). Los
extractos orgánicos combinados se secaron sobre sulfato sódico y el disolvente se eliminó en
el rotavapor. El crudo obtenido se purificó mediante cromatografía rápida (SiO2, éter de
petroleo-diclorometano 9:1) lo que permitió separar el compuesto 403 puro
(26 mg, 0.065 mmol, 70%), como un sólido rojo; p.f.: 72-73 ºC (éter de petroleo-
diclorometano).
IR (KBr cm-1
): 1646 (C=O st), 1635, 1398, 1337, 1308, 1290, 1262, 1105, 1044.
RMN-1H (CDCl3, 400 MHz) δ: 7.68-7.65 (m, 2H, 2 CH Ar), 7.45-7.43 (m, 2H, 2 CH Ar),
3.56 (dc, J = 14.0 y 7.0 Hz, 1 H, NCH2CH3), 3.35 (dc, J = 14.0 y 7.0 Hz, 1 H, NCH2CH3)
y 1.25 (t, J = 7.0 Hz, 3 H, NCH2CH3) ppm.
RMN-13
C (CDCl3, 100 MHz) δ: 192.23 (C=S), 185.14 (C=O), 165.38 (C sp2),
153.96 (C sp2), 137.54 (C sp
2), 134.78 (C sp
2), 132.52 (C Ar), 131.84 (C Ar),
194
Kitamura, T.; Meng, Z.; Fujiwara, Y. “Synthesis of a new hypervalent iodine compound,
[2-(hydroxydimethylsilyl)phenyl](phenyl)iodonium triflate as a convenient approach to benzyne”.
Tetrahedron Lett. 2000, 41, 6611-6614.

Parte experimental 469
127.57 (CH Ar), 127.31 (CH Ar), 123.87 (CH Ar), 122.08 (CH Ar), 48.18 (NCH2CH3)
y 13.48 (NCH2CH3) ppm.
EM (EI) m/z (%): 399 (M+·
, 40), 370 (M+ - CH2CH3, 100), 208 (45), 178 (30), 108 (25),
69 (25).
Masa exacta: calculada para C14H9NOS6: 398.9008 encontrada: 398.9006.

470 Tesis Doctoral Susana Barriga Falcón
S
N
S
S
O
S
S
S
403

Parte experimental 471
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000 3200 3400 3600 3800 4000
Wavenumbers (cm-1)
S
N
S
S
O
S
S
S
403


5. CONCLUSIONES


Conclusiones 475
1. En el presente trabajo se han sintetizado nuevas bisditioltionas con núcleo tiazínico
y cadenas largas sobre el nitrógeno, a partir de aminas terciarias y dicloruro de
diazufre, utilizando la metodología desarrollada en nuestro grupo de investigación.
2. Se ha comprobado que las bisditioltionas con núcleo tiazínico y pirrólico,
sintetizadas previamente por el grupo de investigación, reaccionan con azidas
sencillas, permitiendo obtener heterociclos del tipo bis[1,2]ditiolo[1,4]tiazina imina
o bis[1,2]ditiolopirrol imina. Esta reacción constituye un método óptimo para la
transformación de ditioltionas en ditioliminas.
3. Asimismo, se ha desarrollado un método alternativo para llevar a cabo esta
transformación, que consiste en el tratamiento de las ditioltionas con cloraminas B o
T en medio ácido. Además, se han encontrado nuevas condiciones de reacción, que
utilizan catálisis de triflato de escandio, y permiten obtener
bis[1,2]ditiolo[1,4]tiazina iminas en elevados rendimientos, en tiempos de reacción
muy cortos y en condiciones de reacción muy suaves. Esta metodología es
probablemente generalizable a otras reacciones con cloraminas.
4. Se han sintetizado precursores de nitrilimina del tipo N-(α-cloroariliden)-N’-
arilhidrazona y se ha comprobado que reaccionan con la ditioltiona 176
proporcionando nuevos sistemas heterocíclicos del tipo
[1,3,4]tiadiazoliliden[1,2]ditiolo[1,4]tiazina. Los estudios teóricos, espectroscópicos
y de difracción de rayos X han permitido determinar la configuración E del doble
enlace exocíclico de estos cicloaductos.
5. Se ha comprobado que las ditioltionas reaccionan con diversos acetilenos activados,
dando lugar a los productos de mono o doble adición, dependiendo de la naturaleza
de la ditioltiona y de la reactividad del alquino, pudiéndose controlar selectivamente
la obtención de los monoaductos, diaductos, triaductos o tetraaductos
correspondientes. La adición de la primera molécula de alquino sobre el grupo tiona
del anillo de ditiol transcurre mediante un mecanismo 1,3-dipolar, mientras que la
segunda molécula se adiciona (en su caso) a través de una reacción de hetero-Diels-
Alder. La reacción con acetilenos ha permitido introducir en los heterociclos

476 Tesis Doctoral Susana Barriga Falcón
iniciales grupos que modifican las propiedades electroquímicas de los mismos, y
otros que permiten posteriores funcionalizaciones. Se ha comprobado también en
este caso que el triflato de escandio cataliza eficientemente estas reacciones.
6. Se ha comprobado que las ditioltionas con núcleo pirrólico requieren condiciones de
reacción más enérgicas que sus análogos 1,4-tiazínicos; sin embargo dan lugar en
todos los casos a los productos de adición doble sobre cada anillo de ditiol. Esto se
explica si tenemos en cuenta que la adición de la primera molécula de acetileno
conlleva una pérdida de aromaticidad del anillo central, que sin embargo se recupera
en la segunda adición hetero-Diels-Alder. Los cálculos teóricos realizados están de
acuerdo con esta conclusión.
7. La modelización molecular asistida por ordenador de los cicloaductos sintetizados ha
permitido predecir la estructura conformacional más estable en cada uno de los casos
en los que existen varios confórmeros posibles. Las conclusiones acerca de la
estructura de los confórmeros más estables están de acuerdo con los datos obtenidos
mediante técnicas espectroscópicas.
8. Se ha comprobado que las bisditiolotiazinas reaccionan frente a bencino generado en
las condiciones de Kitamura, para obtener fácilmente sistemas benzoditiolanos.
9. Tras el estudio electroquímico mediante voltametría cíclica de algunos de los aductos
obtenidos, se pudo comprobar que todos ellos constituyen sistemas irreversibles, a
excepción del compuesto 386, que incluye grupos ferroceno en su estructura. En este
caso, el sistema heterocíclico modifica de forma considerable el potencial redox del
sistema organometálico, constituyendo una forma sencilla de regular las propiedades
electroquímicas del ferroceno.

RESUMEN


Resumen III
1. Nuevos materiales basados en moléculas orgánicas
La ciencia de materiales, subdisciplina de la Química, permite desarrollar compuestos
que presentan propiedades avanzadas que los hacen útiles como nuevos materiales en
muy diversos campos. Los materiales pueden ser clasificados en diversas categorías,
basándonos en su constitución química y en sus propiedades físicas. En la actualidad la
búsqueda de nuevos materiales se orienta hacia materiales con funciones específicas;
además, la química supramolecular ha dado lugar al desarrollo de sustancias con
propiedades electrónicas, magnéticas, ópticas, estructurales, mecánicas y químicas de
gran interés en el desarrollo de nuevos materiales.
Los materiales que más interés están suscitando en este momento son:
� Los materiales orgánicos conductores, superconductores y magnéticos. Estos
materiales conductores y superconductores están basados fundamentalmente en
complejos de trasferencia de carga formados entre un aceptor y un dador de
electrones existiendo un especial interés en el desarrollo de los derivados de TTF.
En cuanto a los materiales orgánicos magnéticos, pueden obtenerse usando diversas
estrategias tales como la formación de sales de transferencia de carga, introducción
de radicales nitroxilo, uso de metalocenos, etc.
� Los materiales ópticos no lineales. Existe un gran interés en poder manipular la
frecuencia, la fase, la polarización o la trayectoria de la luz. Si una molécula es
sometida a campos eléctricos muy intensos, el material es polarizado y esta
polarización es una función no lineal de la fuerza del campo. Esto permite obtener
materiales con propiedades ópticas especiales. En especial existe un gran interés en
el desarrollo de compuestos que presenten armónicos de segundo y tercer orden.
� Cristales líquidos. Estos materiales presentan propiedades que se sitúan entre las
propiedades de los sólidos y de los líquidos. Existen diversos tipos de cristales
líquidos de acuerdo con la estructura molecular y la estructura de las fases. La
aplicación más frecuente se encuentra en la fabricación de pantallas digitales,
aunque pueden ser utilizados como sensores, interruptores o para fabricar fibras muy
resistentes como el Kevlar®.

IV Tesis Doctoral Susana Barriga Falcón
� Sensores químicos. Los sensores son capaces de detectar selectivamente un sustrato
en presencia de otros y generar una señal cuando esto ocurre. Existen diversos tipos
de sensores y normalmente se clasifican en función del tipo de señal que generan.
� Aparatos moleculares electrónicos: interruptores, cables y rectificadores. Un
aparato molecular puede ser definido como el conjunto de un número de
componentes moleculares discretos diseñado para realizar una función específica.
Existe un especial interés en el desarrollo de aparatos moleculares que actúen como
cables moleculares, interruptores de muy diversos tipos y rectificadores.
� Máquinas moleculares basadas en catenanos y rotaxanos. Un máquina molecular es
un tipo particular de sistema supramolecular en el que las partes que lo componen
pueden presentar cambios en sus posiciones relativas como resultado de estímulos
externos.
� Dendrímeros. Los dendrímeros son macromoléculas monodispersas que presentan
una estructura con gran ramificación y arquitectura tridimensional. Los dendrímeros
son interesantes en una gran variedad de contextos, siendo muy importantes en el
diseño de materiales funcionales y en la construcción de materiales estructurales de
aplicación en diversos campos.
Particularmente, los nuevos materiales heterocíclicos con azufre y nitrógeno poseen
características electrónicas especiales que los hacen idóneos para su utilización en
aplicaciones ópticas y electrónicas.
En esta línea, en nuestro grupo de investigación se ha conseguido sintetizar una gran
variedad de heterociclos, que poseen un elevado número de átomos de azufre, y que
pueden ser considerados como bloques sintéticos para el diseño y la preparación de
nuevos materiales. Para ello, se utilizan reactivos muy simples de azufre, como por
ejemplo dicloruro de diazufre, con compuestos nitrogenados también muy sencillos,
como aminas, oximas y nitrilos. En el esquema 1 se muestra un ejemplo.

Resumen V
N
CHCl3S
N
S
SS
S
S SS2Cl2, DABCO
160 161
Esquema 1
Las ditioltionas como 161 son compuestos que tienen una elevada reactividad frente a
numerosos reactivos, aunque las reacciones con más aplicaciones sintéticas son
probablemente las reacciones de cicloadición.
El propósito general de esta Tesis Doctoral es la síntesis de nuevos sistemas
poliheterocíclicos que contengan azufre y nitrógeno, tengan un elevado interés teórico y
estructural, y, además, sean potencialmente útiles como nuevos materiales. Así, se ha
estudiado la reactividad de estos sistemas heterocíclicos obtenidos en nuestro grupo,
frente a reacciones de cicloadición para obtener nuevos derivados poliheterocíclicos con
azufre y nitrógeno, con estructuras que puedan tener aplicaciones potenciales como
nuevos materiales.
2. Estudio de la reactividad de bisditioltionas frente a reactivos 1,3-dipolares
Las bisditioltionas reaccionan frente a reactivos 1,3-dipolares involucrando unicamente
al grupo tiona. Se genera de esta forma un cicloaducto de estructura espiránica, que
normalmente sufre una ciclorreversión, u otro tipo de transformaciones, para conducir a
sistemas estables.
En esta Tesis Doctoral se ha abordado únicamente el estudio de los sistemas
heterocíclicos sintetizados con anterioridad en el grupo de investigación, frente a
reactivos 1,3 dipolares de tipo azida y nitrilimina.
Las reacciones con azidas conducen a derivados del tipo bis[1,2]ditiolo[1,4]tiazinimina
y bis[1,2]ditiolopirroliminas (Esquema 2). En nuestro caso sólo se consiguieron
resultados satisfactorios frente a azidas sencillas, tales como la etoxicarbonilazida.

VI Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
SO
S
N
S
SS
S
NO CO2Et
176 257256
+
O N
O
N N
N
SSS
S
SO N
SSS
S
NO+
182 264
CO2Et
256
O N N N
O
N
SSS
S
SS N
SSS
S
NN+
181 265
CO2EtEtO2C
256
O N N N
O
Esquema 2
Las ditioltionas reaccionan con nitriliminas para dar lugar a 1,3,4-tiadiazoles. Se
sintetizaron diversas diarilnitriliminas y se estudió su reactividad frente a la
bisditioltiona 176. Se obtuvieron de esta forma (E)-3-oxo-4-etil-5-(3,5-
diaril[1,3,4]tiadizol-2-ilideno)[1,2]ditiolo[3,4-b][1,4]tiazin-6-tionas (Esquema 3) en
rendimiento moderados a buenos. La estructura del cicloaducto ha podido ser
confirmada mediante difracción de rayos X de monocristal, así como mediante cálculos
teóricos y las técnicas espectroscópicas habituales, siendo la primera vez que ha podido
determinarse inequívocamente la configuración geométrica de este tipo de compuestos.
S
N
S
SS
S
O Et3N
S
N
S
S
S
O
S
NNSAr
Ar
Ar N
HN
Ar
Cl
176 274a-f
+
270a-f
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
Esquema 3

Resumen VII
Se decidió abordar también la adición de las cloraminas B y T sobre las
bisditiolotiazinas 161 y 176 para obtener de esta forma nuevas bisditioliminas
(Esquema 4). Estas reacciones se realizaron térmicamente.
S
N
S
SS
S
S S
ArS
NClNa
O O
S
N
SSS
S
N NSO2Ar
ArO2S
+
161277a-b278a (48%)
278b (57%)a, Ar = Ph
b, Ar = C6H4CH3
CH3COOH
C6H6, ∆
S
N
S
SS
S
O S
ArS
NClNa
O O
S
N
S
SS
S
Ac NSO2Ar
+
176277a-b
a, Ar = Ph
b, Ar = C6H4CH3
279a = 30%
279b = 36%
CH3COOH
C6H6, ∆
Esquema 4
Dado que los rendimientos obtenidos no son muy elevados se decidió buscar un
catalizador para mejorar los rendimientos y suavizar las condiciones de reacción. Se
obtuvieron excelentes resultados utilizando cantidades catalíticas de triflato de escandio.
La reacción con catálisis de triflato de escandio transcurre a temperatura ambiente en
condiciones suaves de forma prácticamente instantánea.
3. Estudio de la reactividad de bisditioltionas frente a reactivos dipolarófilos
Este tipo de reacciones involucran a todo el anillo de ditiol y se han estudiado
fundamentalmente las reacciones de las bisditioltionas con alquinos bajo condiciones
térmicas, con o sin catálisis de triflato de escandio.
En primer lugar se eligieron como reactivos de cicloadición acetilenos sencillos
activados mediante grupos atractores de electrones. Los acetilenos elegidos fueron
acetilendicarboxilato de dimetilo, dicianoacetileno y dibenzoilacetileno. Se estudió la
reactividad de diversas ditioltionas frente a los citados acetilenos. Así en el caso de la
tiazina 176 se obtienen 2-tioacilmetilen-1,3-ditioles (Esquema 5).
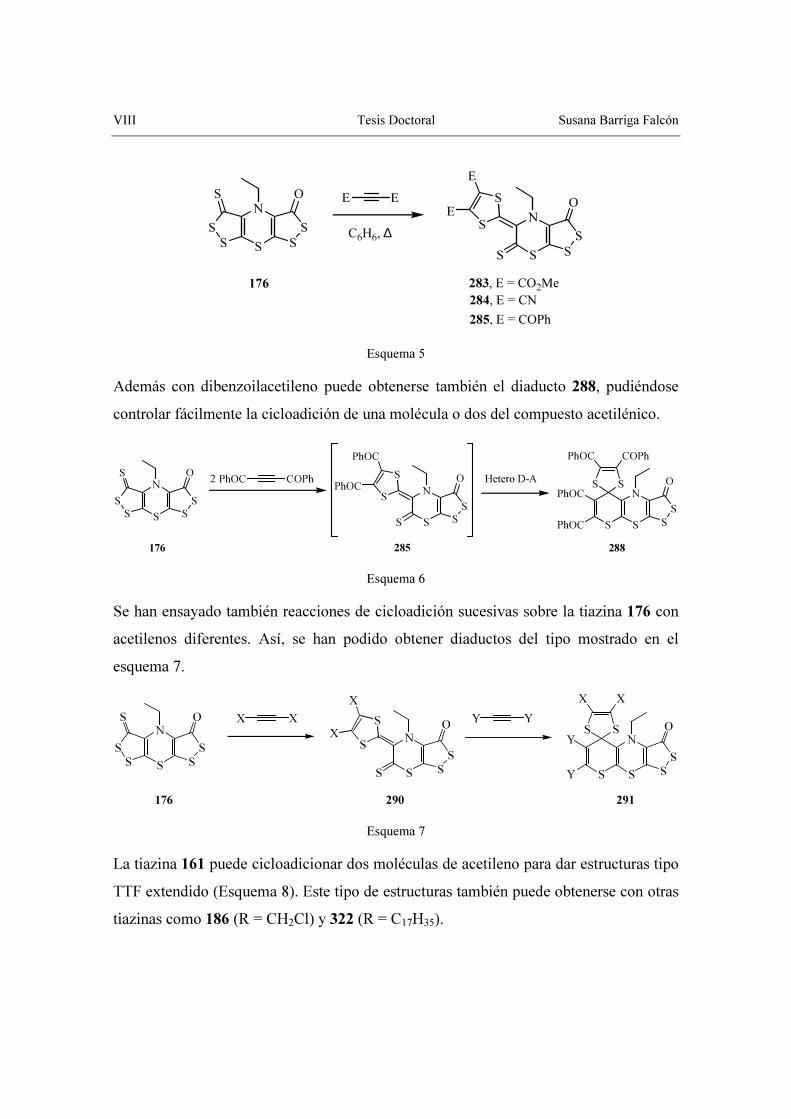
VIII Tesis Doctoral Susana Barriga Falcón
S
N
S
SS
S
OS EE
S
N
S
S
O
S
S
S
E
E
176 283, E = CO2Me
284, E = CN
285, E = COPh
C6H6, ∆
Esquema 5
Además con dibenzoilacetileno puede obtenerse también el diaducto 288, pudiéndose
controlar fácilmente la cicloadición de una molécula o dos del compuesto acetilénico.
S
N
S
SS
S
OSCOPhPhOC
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
176 288
2
S
N
S
S
O
S
S
S
PhOC
PhOC
285
Hetero D-A
Esquema 6
Se han ensayado también reacciones de cicloadición sucesivas sobre la tiazina 176 con
acetilenos diferentes. Así, se han podido obtener diaductos del tipo mostrado en el
esquema 7.
S
N
S
SS
S
S O XX
S
N
S
S
O
S
S
S
X
X
YY
S
N
S
S
O
S
SS
XX
Y
Y
176 290 291
Esquema 7
La tiazina 161 puede cicloadicionar dos moléculas de acetileno para dar estructuras tipo
TTF extendido (Esquema 8). Este tipo de estructuras también puede obtenerse con otras
tiazinas como 186 (R = CH2Cl) y 322 (R = C17H35).

Resumen IX
S
N
S
SS
S
RS S E E
S
N
SS
R
S
SS
SE
EE
E
R = CH3, CH2Cl, C17H35 R = CH3, CH2Cl, C17H35
E = CO2CH3, COPh
2
Esquema 8
Además la tiazina 161 puede adicionar tres moléculas de DMAD o DBA
(Esquema 9).
E E
S
N
S
SS
S
SS
S
N
SS
S
S
SSE
E
EEE
E
161 E = CO2CH3, COPh
3
Esquema 9
Con dibenzoilacetileno puede conseguirse la adición de cuatro moléculas de acetileno
sobre la tiazina 161 obteniéndose un derivado pentacíclico. Este tipo de derivados
también puede conseguirse con las tiazinas 321 (R = C11H23) y 322 (R = C17H35).
S
N
S
SS
S
SSR
S
N
SS
COPh
COPhPhOC
PhOC
R
SS
COPh
PhOC
SS
PhOC
COPh
DBA
R = CH3, C11H23, C17H35R = CH3, C11H23, C17H35
Esquema 10
En cuanto a la tiazina angular 190, ésta reacciona con dibenzoilacetileno pudiéndose
controlar las condiciones de reacción para obtener selectivamente el diaducto 327, la
mezcla de los dos posibles triaductos 328 y 329, o el tetraaducto 330 (Esquema 11).
Mediante estas reacciones de cicloadición se generan heterociclos que contienen la
función tioaldehído, un grupo poco común en Química Orgánica.

X Tesis Doctoral Susana Barriga Falcón
S
N
S
S
SS
SS
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
SS
PhOC
PhOC
190
330
DBA
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC
327
S
N
SPhOC
PhOC
SS
PhOC
PhOCS
SS
COPh
COPh
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
+
328 329
DBA
DBA
Esquema 11
Además, se estudió la reactividad de los bisditiolopirroles 181 y 182 frente a acetilenos
activados y, aunque las condiciones de reacción requeridas son más enérgicas que para
las tiazinas anteriormente descritas, siempre se produce la adición de dos moléculas de
acetileno por cada grupo tiona, no observándose diferencias apreciables de reactividad
entre los distintos acetilenos.

Resumen XI
N
SSS
S
OS
182
N
SS
O
SE
E
SS
EE
N
SSS
S
SS
181
EEN
SS
SSS
S
E
EEE
E
EE
E
4
341
335
EE
Esquema 12
Las reacciones con alquinos son efectivamente catalizadas con triflato de escandio, lo
que permitió llevar a cabo estas reacciones en condiciones muy suaves y con elevados
rendimientos.
A continuación, se han sintetizado alquinos más complejos y se han hecho reaccionar
con la tiazina 176. Los acetilenos elegidos han sido acetilendicarbaldehído, acetilenos
activados mediante grupos furoílo, mediante grupos ferrocenilo (aquirales y quirales), y
mediante grupos tienilo.
El método de síntesis empleado para la síntesis de estos acetilenos activados fue similar
en todos los casos. La molécula de partida fue siempre un aldehído y se introdujo el
triple enlace mediante el acoplamiento con bromuro de etinilmagnesio. Se obtiene así el
alcohol propargílico correspondiente. Sobre este alcohol se genera un acetiluro de litio,
que a continuación se hace reaccionar con una nueva molécula del aldehído inicial.
Finalmente el diol obtenido se oxida para dar lugar a la correspondiente dicetona
(Esquema 13).
R CHO
MgBrH
H
HO
R R CHO
HO
R R
OH [O] O
R R
O1) BuLi
2)
Esquema 13
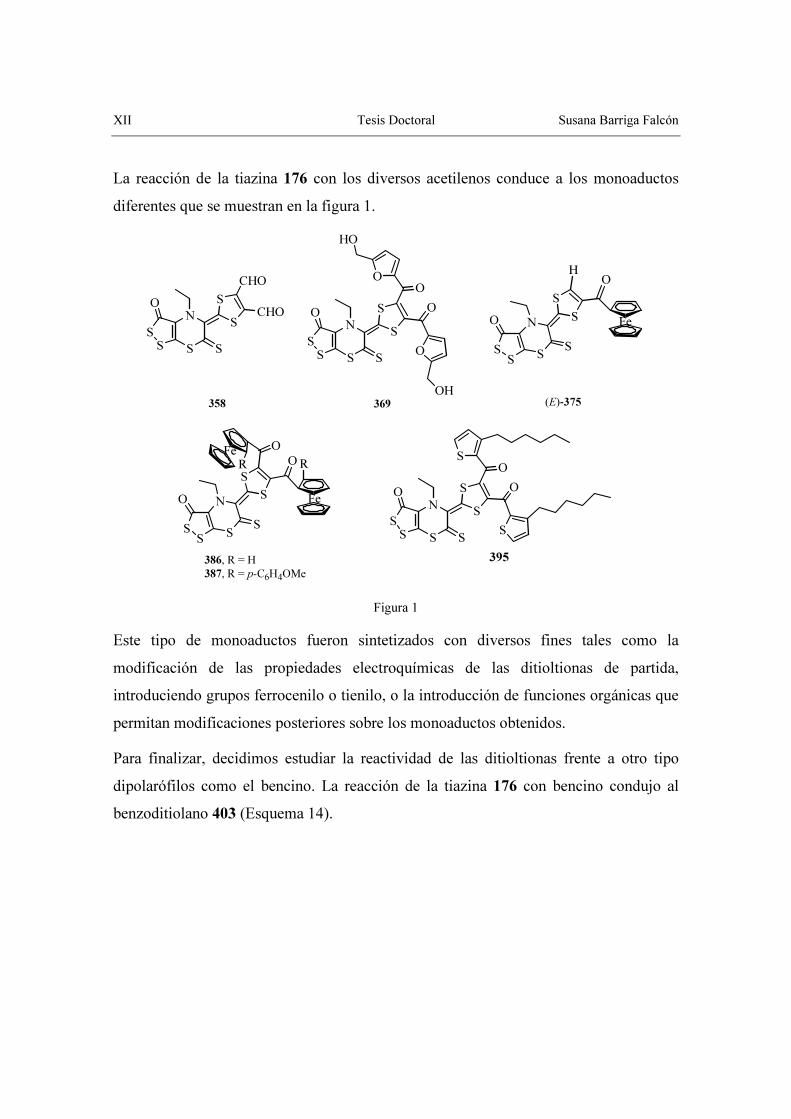
XII Tesis Doctoral Susana Barriga Falcón
La reacción de la tiazina 176 con los diversos acetilenos conduce a los monoaductos
diferentes que se muestran en la figura 1.
S
N
S
S
O
S
S
S
CHO
CHO
S
N
S
S
S
O
S
S O
O
O
O
OH
HO
S
N
SS
S
S
S
O
H
Fe
O
S
N
SS
S
S
S
O
O
O
Fe
R
Fe
R
S
N
S
S
O
S
S
S O
O
S
S
395
358 369 (E)-375
386, R = H
387, R = p-C6H4OMe
Figura 1
Este tipo de monoaductos fueron sintetizados con diversos fines tales como la
modificación de las propiedades electroquímicas de las ditioltionas de partida,
introduciendo grupos ferrocenilo o tienilo, o la introducción de funciones orgánicas que
permitan modificaciones posteriores sobre los monoaductos obtenidos.
Para finalizar, decidimos estudiar la reactividad de las ditioltionas frente a otro tipo
dipolarófilos como el bencino. La reacción de la tiazina 176 con bencino condujo al
benzoditiolano 403 (Esquema 14).
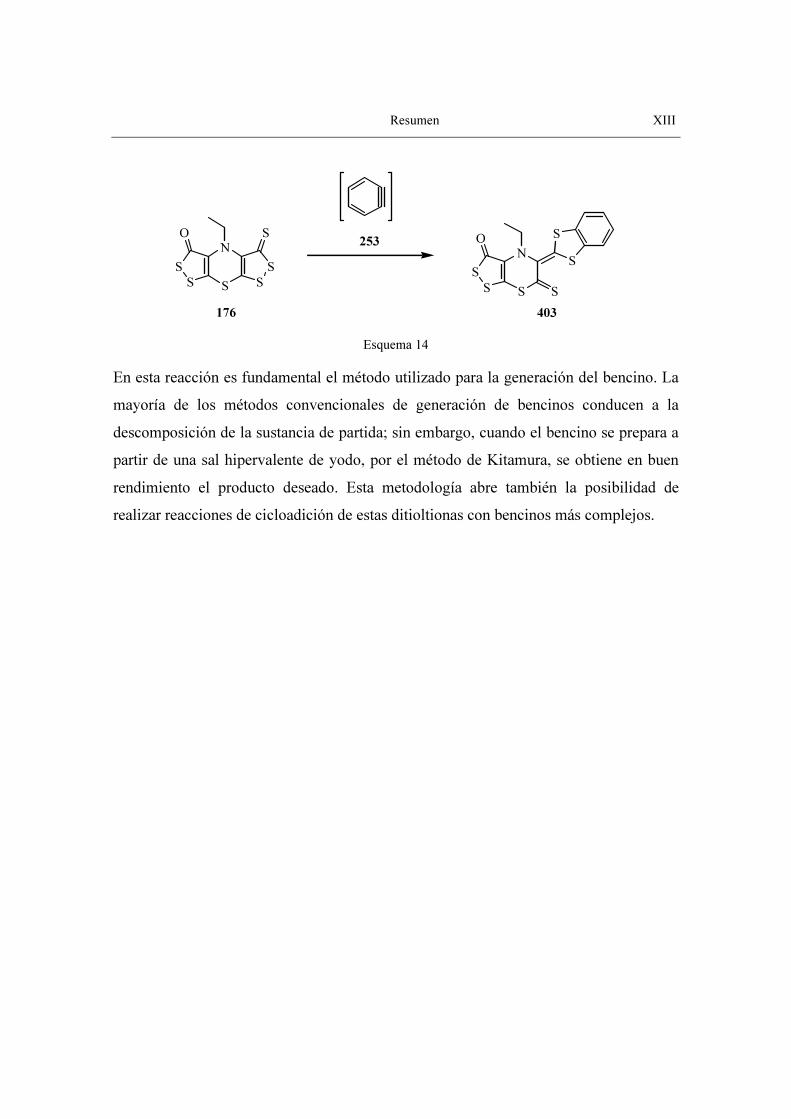
Resumen XIII
S
N
S
SS
S
O S
176
253
S
N
S
S S
O
S
S
403
Esquema 14
En esta reacción es fundamental el método utilizado para la generación del bencino. La
mayoría de los métodos convencionales de generación de bencinos conducen a la
descomposición de la sustancia de partida; sin embargo, cuando el bencino se prepara a
partir de una sal hipervalente de yodo, por el método de Kitamura, se obtiene en buen
rendimiento el producto deseado. Esta metodología abre también la posibilidad de
realizar reacciones de cicloadición de estas ditioltionas con bencinos más complejos.


SUMMARY


Summary XVII
1. New materials based on organic molecules
Materials science allows developing compounds that present advanced properties and
can be used as new materials in very diverse fields. Materials can be classified in
several categories, depending on their chemical constitution and physical properties.
The search of new materials is currently oriented towards the preparation of materials
with very specific functions; in this sense, supramolecular chemistry has made possible
the development of substances with electronic, magnetic, optical, structural, mechanical
and chemical properties very useful in the preparation of new materials.
In the last few years a big interest has raised for the finding of several types of
materials, including the following:
� Conductors, superconductors and magnetic organic materials. Conductors and
superconductors are constituted basically by transference-charge complexes made
up of an acceptor and a donor. TTF derivatives are between the most useful donors.
As far as the magnetic organic materials are concerned, they can be obtained by
using diverse strategies, such as the formation of transference-charge salts,
introduction of nitroxyl radicals, use of metallocenes, etc.
� Non-linear optical materials. There is a high interest in manipulating the frequency,
the phase, the polarization, or the trajectory of light. If molecules are subjected to
very intense electric fields, this results in polarization of the material, and this
polarization is a non-linear function of the field strength. This basic property of
matter allows the preparation of materials with special optical response. The main
aim in the field of non-linear optics is the development of compounds that present
second and third order harmonics.
� Liquid crystals. The properties of these materials are in between the properties of
solids and liquids. Liquid crystals are usually classified in different types according
to their molecular and phase structures. The most frequent application of liquid
crystals is the manufacture of digital screens, though they can also be used in the
production of sensors, switches, and even very resistant fibres, such as Kevlar®.

XVIII PhD Thesis Susana Barriga Falcón
� Chemical sensors. Sensors are able to detect selectively a substrate in the presence
of others, and to generate a signal when this happens. Diverse types of sensors exist,
and they are usually classified according to the type of signal they generate.
� Molecular electronic devices: switches, wires and rectifiers. A molecular electronic
device can be defined as the assembly of a number of discreet molecular
components designed to make a specific function. Today is especially interesting the
existence of molecular devices that function as molecular wires, switches of very
diverse types and rectifiers.
� Molecular machines based on catenanes and rotaxanes. A molecular machine is a
particular type of supramolecular system in which the parts that compose it can
show changes in their relative positions as a result of external stimuli.
� Dendrimers. Dendrimers are monodisperse macromolecules showing extensively
branched structures and three-dimensional architecture. Dendrimers are interesting
in a wide variety of contexts, being very important both in the design of functional
materials and in the construction of structural materials with application in diverse
fields.
Particularly, new materials based on sulfur and nitrogen heterocycles have special
electronic characteristics that make them suitable for their use in optical and electronic
applications.
In this line, our research group has synthesized a wide variety of heterocycles
containing a high number of sulfur atoms, and which can be considered as synthetic
blocks for the design and the preparation of new materials. Very simple sulfur reagents,
such as sulfur dichloride, and nitrogen compounds, as amines, oximes and nitriles, have
been used for this purpose. Scheme 1 shows an example.

Summary XIX
N
CHCl3S
N
S
SS
S
S SS2Cl2, DABCO
160 161
Scheme 1
Dithiolethiones as 161 are compounds that show high reactivity with numerous
reagents, though cycloaddition reactions are probably the reactions with more synthetic
applications.
The general object of this PhD Thesis is the synthesis of new polyheterocyclic systems
containing sulfur and nitrogen, with the aim of developing new molecules with high
theoretical and structural interest, and potentially useful as new materials. Thus, some of
the heterocyclic systems previously obtained in our group have been studied, regarding
their reactivity in cycloaddition reactions that lead to new sulfur and nitrogen
polyheterocyclic derivatives.
2. Study of the reactivity of bisdithiolethiones versus 1,3-dipole reagents
The reaction of bisdithiolethiones with 1,3-dipolar reagents involves only to the thione
group. In this way, a spiranic cycloadduct is generated, and usually this undergoes
cycloreversion, or other type of transformation, leading to a stable system.
In the present work the study 1,3-dipolar reagents has been limited to azides and
nitrilimines.
Reaction of bis[1,2]dithiolo[1,4]thiazine-3-thiones and bis[1,2]dithiolopirrolo-3-thiones
with azides leads to the corresponding imine derivatives (Scheme 2). In our case, we
have only obtained satisfactory results in reactions with simple azides, such as
ethoxycarbonylazide.

XX PhD Thesis Susana Barriga Falcón
S
N
S
SS
S
SO
S
N
S
SS
S
NO CO2Et
176 257256
+
O N
O
N N
N
SSS
S
SO N
SSS
S
NO+
182 264
CO2Et
256
O N N N
O
N
SSS
S
SS N
SSS
S
NN+
181 265
CO2EtEtO2C
256
O N N N
O
Scheme 2
On the other hand, dithiolethiones react with nitrilimines giving 1,3,4-thiadiazoles.
Diverse diarylnitrilimines have been synthesised and its reactivity has been studied
versus bisdithiolethione 176. In this way, we have obtained (E)-3-oxo-4-ethyl-5-
(3,5-diaryl[1,3,4]thiadiazole-2-yliden)[1,2]dithiolo[3,4-b][1,4]thiazin-6-thiones (Scheme 3)
in yields ranging from moderate to good. The structure of these compounds has been
confirmed by the monocrystal X ray diffraction of one of the cycloadducts, theoretical
calculations and the usual spectroscopic techniques. In this way we have been able to
unequivocally determine, for the first time, the geometric configuration of this type of
compounds.
S
N
S
SS
S
O Et3N
S
N
S
S
S
O
S
NNSAr
Ar
Ar N
HN
Ar
Cl
176 274a-f
+
270a-f
a, Ar = C6H5
b, Ar = p-ClC6H4
c, Ar = p-BrC6H4
d, Ar = p-IC6H4
e, Ar = p-CNC6H4
f, Ar = 2,4-Cl2C6H3
Scheme 3

Summary XXI
We have also studied the thermal addition of chloramines B and T on
bisdithiolothiazines 161 and 176 to obtain new bisdithioleimines (Scheme 4).
S
N
S
SS
S
S S
ArS
NClNa
O O
S
N
SSS
S
N NSO2Ar
ArO2S
+
161277a-b278a (48%)
278b (57%)a, Ar = Ph
b, Ar = C6H4CH3
CH3COOH
C6H6, ∆
S
N
S
SS
S
O S
ArS
NClNa
O O
S
N
S
SS
S
Ac NSO2Ar
+
176277a-b
a, Ar = Ph
b, Ar = C6H4CH3
279a = 30%
279b = 36%
CH3COOH
C6H6, ∆
Scheme 4
The yields of these reactions were not very high, and we decided to do some further
research to try to find a catalyst to improve the yields and to make the reaction
conditions milder. The use of catalytic amounts of scandium triflate resulted in an
excellent outcome of the reaction. The catalysed reaction takes place at room
temperature, in smooth conditions, and is practically instantaneous.
3. Study of the reactivity of bisdithiolethiones with dipolarophiles
Reactions of bisdithiolethiones with dipolarophiles are characterised because they
involve the whole dithiole ring. We have basically studied the reactions of
bisdithiolethiones with alkynes, under thermal conditions, both with and without
catalysis of scandium triflate.
In the first place, we chose as cycloaddition reagents simple acetylenes activated by
electron-withdrawing groups, as dimethylacetylenedicarboxylate, dicyaneacetylene and
dibenzoylacetylene. The reactivity of diverse dithiolethiones with the referred acetylenes
has been studied. Thus, for example, in the case of thiazine 176, we have got
2-thioacylmethylen-1,3-dithioles (Scheme 5).
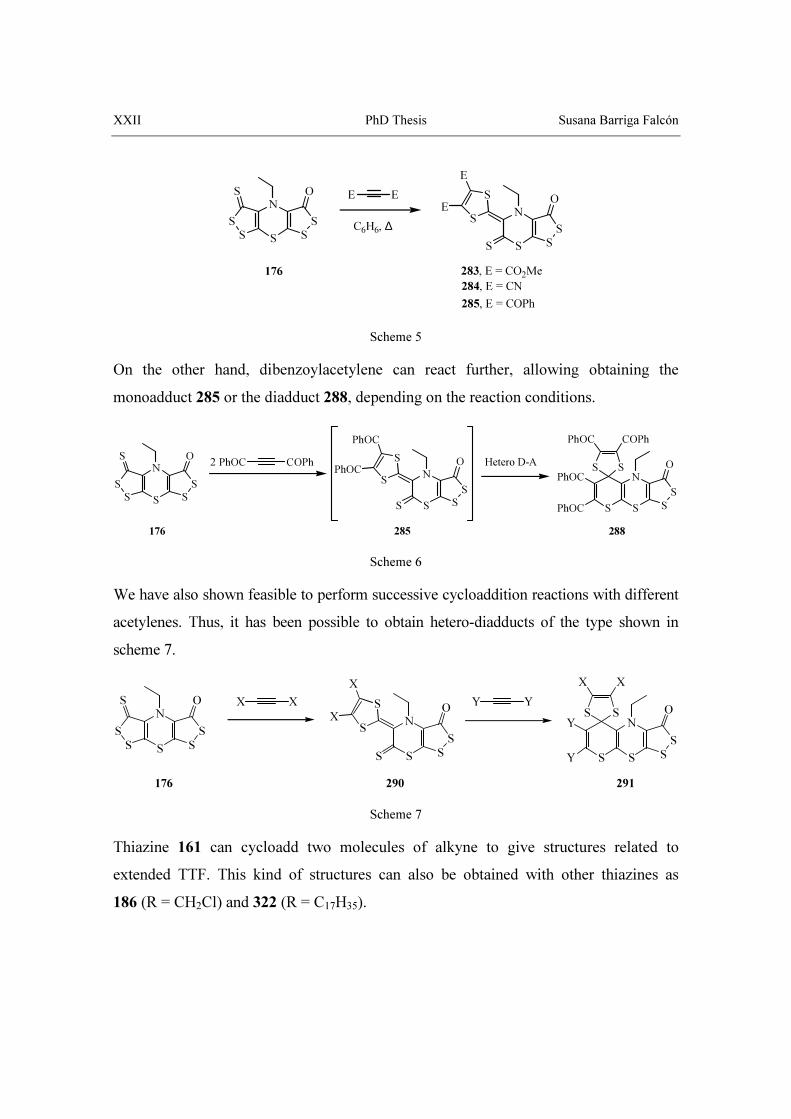
XXII PhD Thesis Susana Barriga Falcón
S
N
S
SS
S
OS EE
S
N
S
S
O
S
S
S
E
E
176 283, E = CO2Me
284, E = CN
285, E = COPh
C6H6, ∆
Scheme 5
On the other hand, dibenzoylacetylene can react further, allowing obtaining the
monoadduct 285 or the diadduct 288, depending on the reaction conditions.
S
N
S
SS
S
OSCOPhPhOC
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
176 288
2
S
N
S
S
O
S
S
S
PhOC
PhOC
285
Hetero D-A
Scheme 6
We have also shown feasible to perform successive cycloaddition reactions with different
acetylenes. Thus, it has been possible to obtain hetero-diadducts of the type shown in
scheme 7.
S
N
S
SS
S
S O XX
S
N
S
S
O
S
S
S
X
X
YY
S
N
S
S
O
S
SS
XX
Y
Y
176 290 291
Scheme 7
Thiazine 161 can cycloadd two molecules of alkyne to give structures related to
extended TTF. This kind of structures can also be obtained with other thiazines as
186 (R = CH2Cl) and 322 (R = C17H35).

Summary XXIII
S
N
S
SS
S
RS S E E
S
N
SS
R
S
SS
SE
EE
E
R = CH3, CH2Cl, C17H35 R = CH3, CH2Cl, C17H35
E = CO2CH3, COPh
2
Scheme 8
Thiazine 161 can also add three molecules of DMAD or DBA, as shown in scheme 9.
E E
S
N
S
SS
S
SS
S
N
SS
S
S
SSE
E
EEE
E
161 E = CO2CH3, COPh
3
Scheme 9
The addition of four molecules of alkyne on thiazines 161, 321 (R = C11H23) and 322
(R = C17H35), in order to get pentacyclic derivatives, could be achieved with the use of
dibenzoylacetylene.
S
N
S
SS
S
SSR
S
N
SS
COPh
COPhPhOC
PhOC
R
SS
COPh
PhOC
SS
PhOC
COPh
DBA
R = CH3, C11H23, C17H35R = CH3, C11H23, C17H35
Scheme 10
On the other hand, the angular thiazine 190 reacts with dibenzoylacetylene to get the
diadduct 327, the mixture of the two possible triadducts 328 and 329, or the tetraadduct
330, depending on the particular reaction conditions (Scheme 11). These cycloaddition
reactions give rise to heterocycles containing a thioaldehyde function, which a quite
uncommon group.

XXIV PhD Thesis Susana Barriga Falcón
S
N
S
S
SS
SS
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
SS
PhOC
PhOC
190
330
DBA
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC
327
S
N
SPhOC
PhOC
SS
PhOC
PhOCS
SS
COPh
COPh
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
+
328 329
DBA
DBA
Scheme 11
We have as well studied the reactivity of bisdithiolopyrroles 181 and 182 with activated
acetylenes. Although these reactions require more harsh conditions than those
previously used for the thiazines, the addition of two acetylene molecules on each
thione group always takes place. Furthermore, no significant differences of reactivity
have been observed between the different acetylenes.
N
SSS
S
OS
182
N
SS
O
SE
E
SS
EE
N
SSS
S
SS
181
EEN
SS
SSS
S
E
EEE
E
EE
E
4
341
335
EE
Scheme 12

Summary XXV
Scandium triflate catalyses effectively these reactions, allowing to get the desired
products in very smooth conditions and high yields.
Complex alkynes have also been synthesised and tested in reactions with thiazine 176.
Between these more complex acetylenes we have prepared acetylenedicarbaldehyde,
and acetylenes containing furanyl, thienyl, and chiral and non-chiral ferrocenyl groups.
The method used for the synthesis of these activated acetylenes was similar in all the
cases. The starting material was always an aldehyde, and the triple bond was introduced
by reaction with ethynylmagnesium bromide. In this way, the corresponding
propargylic alcohol is obtained. A lithium acetylide is generated from this alcohol by
the addition of two equivalents of a base. This anion then reacts with a new molecule of
the initial aldehyde, and the diol obtained is finally oxidized to get the corresponding
diketone (Scheme 13).
R CHO
MgBrH
H
HO
R R CHO
HO
R R
OH [O] O
R R
O1) BuLi
2)
Scheme 13
The reaction of thiazine 176 with diverse acetylenes leads to the different monoadducts
that are showed in figure 1.

XXVI PhD Thesis Susana Barriga Falcón
S
N
S
S
O
S
S
S
CHO
CHO
S
N
S
S
S
O
S
S O
O
O
O
OH
HO
S
N
SS
S
S
S
O
H
Fe
O
S
N
SS
S
S
S
O
O
O
Fe
R
Fe
R
S
N
S
S
O
S
S
S O
O
S
S
395
358 369 (E)-375
386, R = H
387, R = p-C6H4OMe
Figure 1
These monoadducts were synthesized with diverse aims. For example modification of
the electrochemical properties of the initial dithiolethiones was achieved by introducing
in the molecule ferrocenyl or thienyl groups. The use of cycloadditions to introduce
organic functions that would allow further modifications of the adducts, was other of
the strategies used in this context.
In the last term, we studied the reactivity of dithiolethiones with other types of
dipolarophiles, as benzyne. Accordingly, the reaction of thiazine 176 with benzyne led
to benzodithiolane 403 (Scheme 14).
S
N
S
SS
S
O S
176
253
S
N
S
S S
O
S
S
403
Scheme 14

Summary XXVII
In this reaction, the method used for the generation of benzyne is essential. Most of the
conventional methods to generate benzyne led to the decomposition of the starting
dithiolethione; however, fortunately, when benzyne was prepared from a hypervalent
iodine salt, according to Kitamura’s method, the desired product was obtained in good
yield. This methodology will hopefully allow successfully performing cycloaddition
reactions of dithiolethiones with complex benzynes.


ÍNDICE DE ESTRUCTURAS

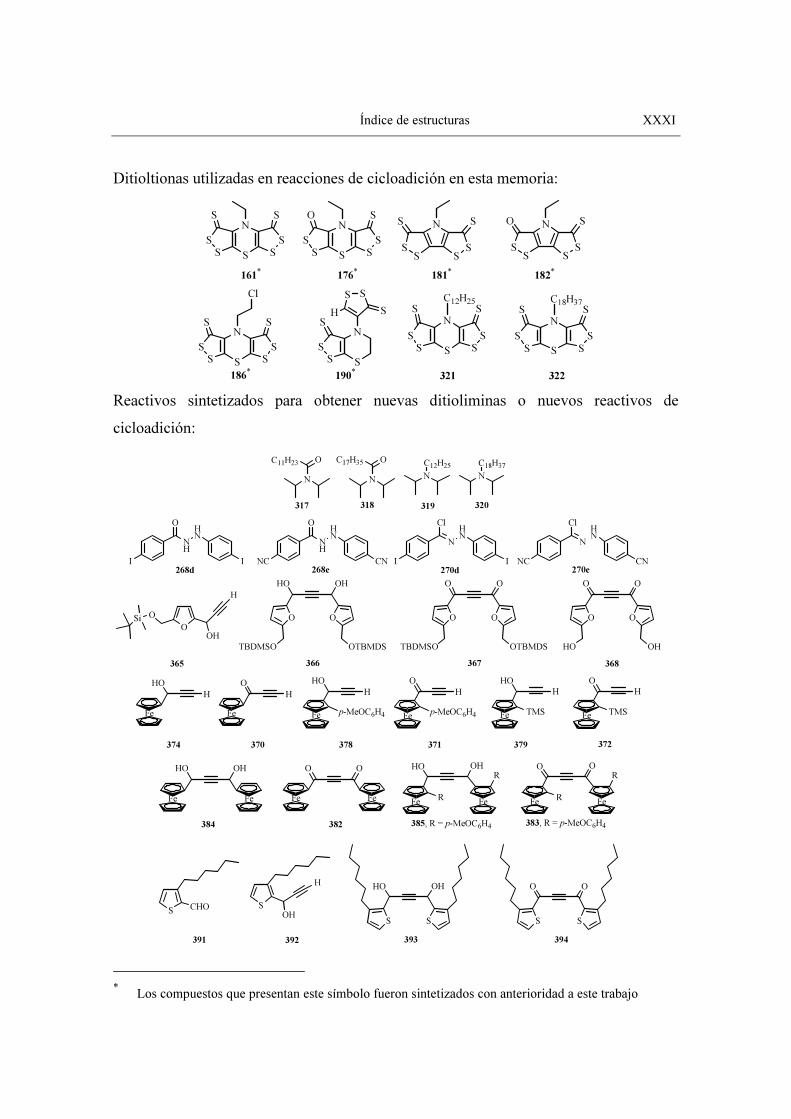
Índice de estructuras XXXI
Ditioltionas utilizadas en reacciones de cicloadición en esta memoria: *
N
S
S
S
S
S
S
S
N
S
S
S
S
S
O
S
N
S
S
S
S
S
S
S
Cl
S
N
S
S
S S
S
SH
N
SSS
S
SS
161*
176*
182*
186*
190*
181*
S
N
S
SS
S
C12H25
SS
S
N
S
SS
S
C18H37
SS
321 322
N
SSS
S
SO
Reactivos sintetizados para obtener nuevas ditioliminas o nuevos reactivos de
cicloadición:
N
C12H25
N
C18H37
N
OC11H23
N
OC17H35
319 320317 318
NH
HN
O
CNNC
NH
HN
O
II
N
HN
Cl
II
N
HN
Cl
CNNC268e268d 270d 270e
O
OSi
OH
H
OHHO
OO
OTBMDSTBDMSO
OO
OO
OTBMDSTBDMSO
OO
OO
OHHO
365 366 367 368
H
HO
Fe
H
O
Fe
374 370
Fe p-MeOC6H4
HO
H
Fep-MeOC6H4
O
H
Fe TMS
HO
H
Fe TMS
O
H
378 371 379 372
Fe Fe
OHHO
Fe Fe
OO
Fe
HO OH
Fe
R
RFe
O O
Fe
R
R
384 382 385, R = p-MeOC6H4383, R = p-MeOC6H4
OHHO
S S
OO
S S
S CHO S
H
OH
393 394391 392
* Los compuestos que presentan este símbolo fueron sintetizados con anterioridad a este trabajo
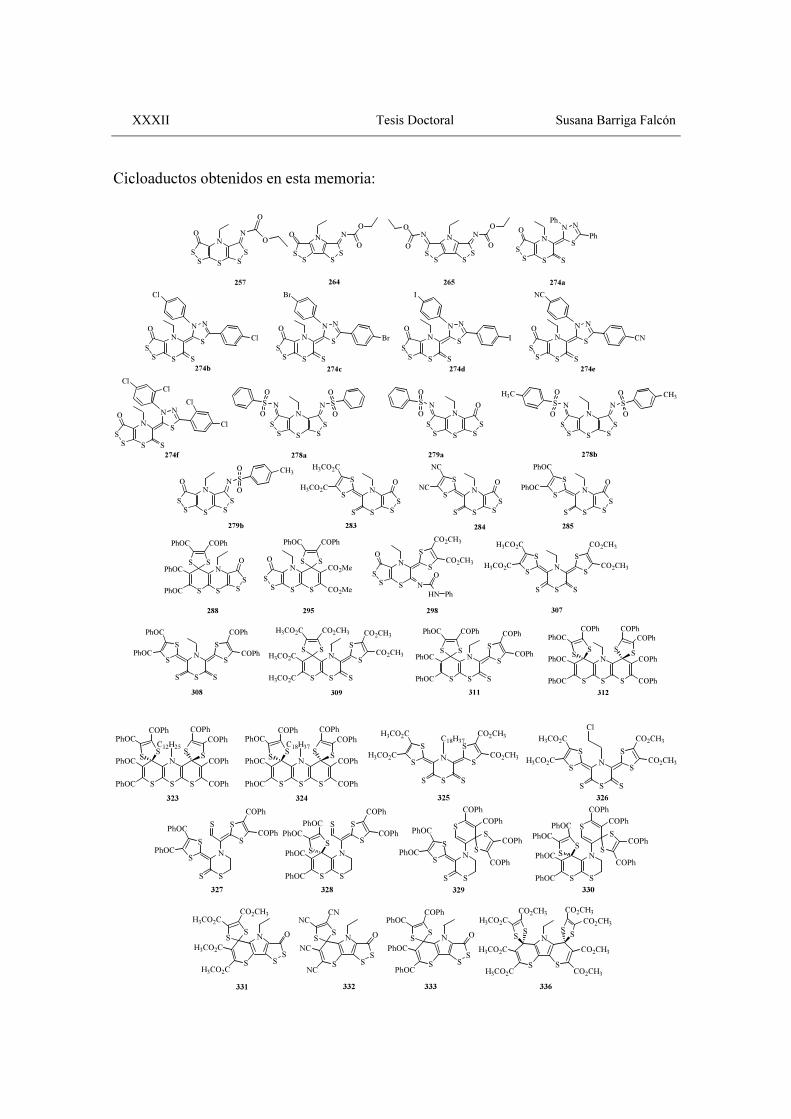
XXXII Tesis Doctoral Susana Barriga Falcón
Cicloaductos obtenidos en esta memoria:
S
N
S
S S
S
O N
O
O N
SSS
S
NO
O
O
N
SSS
S
NN
O
O
O
O
S
N
S
S
O
S
S
NN
Ph
Ph
257 264 265 274a
S
N
S
S
O
S
S
NN
Cl
Cl
S
N
S
S
O
S
S
NN
Br
Br
S
N
S
S
O
S
S
NN
I
I
S
N
S
S
O
S
S
NN
CN
NC
274b 274c 274d 274e
S
N
S
S
O
S
S
NN
Cl
Cl
Cl
Cl
274f
S
N
S
SS
S
N N SS
O
O
O
O
278a
S
N
S
S
O
S
S
NS
O
O
279a
S
N
S
SS
S
N N SS
O
O
O
O
CH3H3C
278b
S
N
S
S
O
S
S
N S
O
O
CH3
279b
S
N
S
S
O
S
S
S
H3CO
2C
H3CO2C
283
S
N
S
S
O
S
S
S
NC
NC
S
N
S
S
O
S
S
S
PhOC
PhOC
285284
S
N
S
S
O
S
SS
COPhPhOC
PhOC
PhOC
288
S
N
S
S
S
SSO
CO2Me
CO2Me
COPhPhOC
295
S
N
S
S N
O S
S
H3CO2C
H3CO2C
S
N
S S
S
S
CO2CH3
CO2CH3
307
S
S
HN
O
Ph
CO2CH3
CO2CH3
298
S
N
SS
S
SS
SCOPh
COPhPhOC
PhOC
308
S
N
S S
S
S
S SH3CO2C
H3CO2C
H3CO2C CO2CH3 CO2CH3
CO2CH3
309
S
N
S S
S
S
S S
COPh
COPh
PhOC COPh
PhOC
PhOC
S
N
SS
COPh
COPhPhOC
PhOCS
S
COPh
COPh
SS
PhOC
COPh
312311
S
N
SS
COPh
COPhPhOC
PhOC
C12H25
SS
COPh
COPh
SS
PhOC
COPh
S
N
SS
COPh
COPhPhOC
PhOC
C18H37
SS
COPh
COPh
SS
PhOC
COPh
S
N
S S
S
SS
SCO2CH3
CO2CH3H3CO2C
H3CO2C
C18H37
S
N
S S
S
SS
S
CO2CH3
CO2CH3H3CO2C
H3CO2C
Cl
S
N
S
S
SS
COPh
COPh
S
S
PhOC
PhOC
S
N
SPhOC
PhOC
SS
PhOC
PhOCS
SS
COPh
COPh
S
N
S
S
S
PhOC
PhOC
S
S
SCOPh
COPh
COPh
COPh
S
N
S
S
S
S
PhOC
PhOC
COPh
COPh
COPh
COPh
S
S
PhOC
PhOC
N
SS
O
SH3CO2C
H3CO2C
331
SS
CO2CH3
H3CO2C
N
SS
O
SNC
NC
332
SS
CN
NC
N
SS
O
SPhOC
PhOC
333
SS
COPh
PhOC
N
SH3CO2C
H3CO2C
336
S
SS
CO2CH3
CO2CH3
CO2CH3
CO2CH3
SS
H3CO2C
CO2CH3
323 324 325 326
327 328 329 330

Índice de estructuras XXXIII
S
N
SS
S
S
S
O
H
Fe
RO
S
N
SS
S
S
S
O
H
Fe
TMSO
380, R = p-MeOC6H4 381
Fe
S
N
SS
S
S
S
O
O
O
Fe
S
N
S
S S
S
SO
O
O
Fe
R
Fe
R
392 387, R = p-MeOC6H4
Fe
S
N
S
S
S
O
O
FeS
S
S
O
O
Fe
Fe
S
N
S
S
O
S
S
S O
OS
S
395
S
N
S
S
O
S
S
S
388 403
N
SPhOC
PhOC
337
SS
COPhPhOC
S
SS
COPh
COPh
COPh
COPh
S
N
S
S S
O
S
SCHO
CHO
S
N
S
S
S
O
S
S O
O
OO
OH
HO
S
N
SS
S
S
S
O
H
Fe
O
358 370 375
