choque cirugia
TRANSCRIPT

ChoqueSCHWARTZ. PRINCIPIOS DE CIRUGÍA - 10° ED.

“El choque es la manifestación del trastorno grave de la maquinariade la vida.”1—Samuel V. Gross, 1872.
En su definición más rudimentaria y sin importar su causa, el choque es la incapacidad para cubrir las necesidades metabólicas de la célula y las consecuencias de esto.
La lesión celular inicial es reversible, pero la lesión se vuelve irreversible si la hipoperfusión hística se prolonga o es lo bastante grave para que ya no sea posible la compensación a nivel celular.

Por lo general, las manifestaciones clínicas de estas respuestas fisiológicas son las que llevan a los médicos al diagnóstico de choque, además de servir de guía para el tratamiento de pacientesen choque. Sin embargo, los parámetros hemodinámicos, como la presión sanguínea y la frecuencia cardiaca, son mediciones más bien insensibles del choque, debe considerarse algún otro parámetro para ayudar al diagnóstico temprano y tratamiento de pacientes en choque.

Claude Bernard sugirió que el organismo intenta mantener la constancia del ambiente internocontra las fuerzas externas que alteran el medio interior. 2 Walter B. Cannon complementó estas observaciones e introdujo el término “homeostasis”; él enfatizó que la capacidad de un organismo para sobrevivir se relacionaba con la conservación de la homeostasis.3
El fracaso de los sistemas fisiológicos para proteger al organismo contra fuerzas externas tiene como resultado la disfunción orgánica y celular, que se conoce en términos clínicos como choque.
Primero describió la “respuesta de pelea o huida” generada por las concentraciones altas de catecolaminas en la sangre.

El choque hipovolémico, el tipo más común, es consecuencia de la pérdida de volumen sanguíneo circulante. Puede deberse a la pérdida de sangre entera (choque hemorrágico), plasma, líquidointersticial (obstrucción intestinal) o una combinación de estos elementos.
El choque vasógeno es efecto de una atenuación de la resistencia dentro de los vasos de capacitancia, que se observa a menudo en la septicemia.
El choque neurógeno es una forma del choque vasógeno en la que la lesión de la médula espinal o la
anestesia raquídea causan vasodilatación por pérdida aguda del tono vascular simpático.
El choque cardiógeno se atribuye a la falla del corazón como bomba, tal y como sucede en las arritmias o en el infarto agudo del miocardio.

En la práctica clínica reciente se describen seis tipos de choque:
1. Hipovolémico.
2. Séptico (vasodilatador)
3. Neurógeno.
4. Cardiógeno.
5. Obstructivo.
6. Traumático.

El choque obstructivo, causado por embolia pulmonar o neumotórax a tensión, origina una depresión del gasto cardiaco, consecuencia del impedimento mecánico en la circulación, no de una insuficiencia cardiaca primaria.
choque por traumatismo, la lesión del tejido blando y óseo inducen la activación de célulasinflamatorias y liberación de factores circulantes, como citocinas y moléculas intracelulares que modulan la respuesta inmunitaria.
Las investigaciones recientes revelaron que los mediadores inflamatorios liberados como respuesta a la lesión hística [patrones moleculares relacionados con la lesión (DAMP, damage-associated molecular patterns)] son reconocidos por muchos de los mismos receptores celulares [receptores de reconocimiento de patrones (PRR, pattern recognition receptors)] y activan vías de señalización similares a las que activan productos bacterianos producidos en la septicemia (patrones moleculares relacionados con patógenos), como el lipopolisacárido.

El fenómeno de redistribución de líquido después de un traumatismo mayor que implica pérdida sanguínea se denominó formación de tercer espacio y describe el desplazamiento de líquido intravascular al peritoneo, intestino, tejidos quemados o sitios de lesión por aplastamiento.
apareció un nuevo proceso patológico, la insuficiencia pulmonar fulminante aguda, como una causa temprana de muerte después de una operación al parecer satisfactoria para controlar la hemorragia.
Este problema clínico, llamado al inicio pulmón de DaNang o pulmón de choque, se reconoció más adelante como síndrome de insuficiencia respiratoria aguda (ARDS, acute respiratory distress syndrome).

principios centrales
Los principios centrales del tratamiento inicial del paciente muy grave o lesionado incluyen:
a) asegurar el control definitivo de las vías respiratorias;
b) controlar la hemorragia activa a la brevedad (el retraso en el control de la hemorragia aumenta la mortalidad y los datos recientes de campos de batalla sugieren que en la población joven, y por lo demás saludable que a menudo resulta herida en combate, el control de la hemorragia es prioritario)
c) reanimar con suministro de eritrocitos y soluciones cristaloides en tanto se lleva a cabo el control quirúrgico de la hemorragia;
d) la hipoperfusión no identificada o corregida de modo inadecuado aumenta la morbilidad y la mortalidad (es decir, la reanimación incorrecta causa muertes tempranas por choque que pueden evitarse), y
e) la reanimación excesiva con líquidos puede exacerbar la hemorragia (la reanimación no regulada es peligrosa). Por consiguiente, la reanimación inadecuada y el volumen no controlado son perjudiciales.

Definiciones y desafíos actuales
Estado de choque éste consiste en:
1. perfusión inadecuada de los tejidos,
2. marcada por descenso en el aporte de los sustratos metabólicos requeridos y
3. eliminación insuficiente de los productos de desecho celular.
Esto implica falla del metabolismo oxidativo que puede incluir defectos en el aporte, transporte y utilización del oxígeno (O2).

FISIOPATOLOGÍA DEL CHOQUE.
Sin importar la causa, las respuestas fisiológicas iniciales en el choque están impulsadas por la hipoperfusiónhística y el desarrollo de déficit de energía celular.
Muchas de las respuestas específicas de los órganos están dirigidas a mantener la perfusión en la circulación cerebral y la coronaria. Éstas se encuentran reguladas en múltiples niveles, incluidos
a) receptores de estiramiento y barorreceptores en el corazón y vasculatura (seno carotídeo y cayado aórtico);
b)quimiorreceptores;
c) respuestas a la isquemia cerebral;
d) liberación de vasoconstrictores endógenos;
e) desplazamiento de líquido al espacio intravascular, y
f) reabsorción y conservación renales de sal y agua.

En el choque hemorrágico, el cuerpo puede compensar la pérdida sanguínea inicial, sobre todomediante una respuesta neuroendocrina para mantener el estado hemodinámico. Esto representa la fase
compensada del estado de choque.
Con la hipoperfusión sostenida, que puede pasar desapercibida, continúan la muerte y lesión celulares, y sobreviene la fase de descompensación del estado de choque.
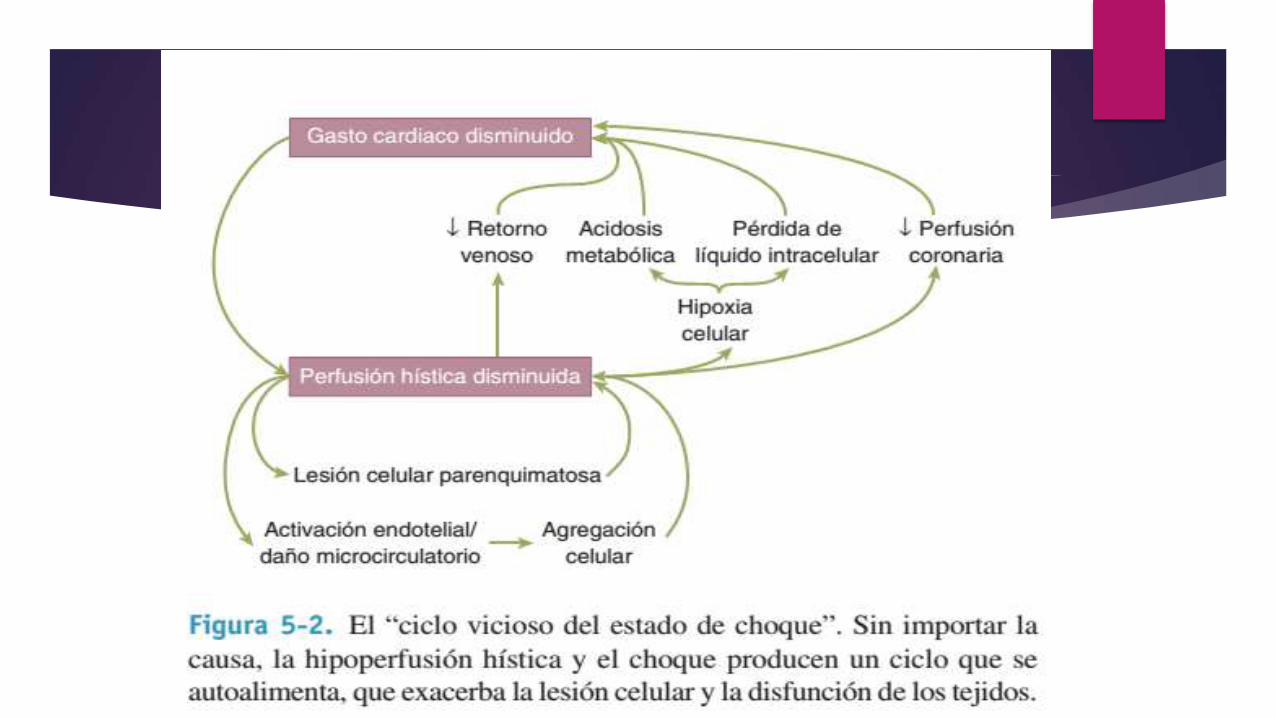
La disfunción microcirculatoria, el daño del tejido parenquimatoso y la activación de células inflamatorias perpetúan la hipoperfusión. La lesión por isquemia y reperfusión a menudo exacerba la lesión inicial. Si no se tratan estos efectos a nivel celular, conducen al compromiso de la función al nivel de sistema orgánico, lo que genera el “círculo vicioso” del estado de choque.
La hipoperfusión persistente causa trastornos hemodinámicos adicionales y colapso cardiovascular. A esto se le denomina fase irreversible del estado de choque y puede desarrollarse en formamuy insidiosa, a veces sólo es evidente en retrospectiva.

Respuestas neuroendocrinas específicos a la hemorragia
La finalidad de la respuesta neuroendocrina a la hemorragia es conservar la perfusión al corazón y cerebro, incluso a expensas de otros sistemas y órganos.
El estímulo inicial en el choque hemorrágico es la pérdida del volumen sanguíneo circulante. La magnitud de la reacción neuroendocrina se basa en el volumen de la pérdida de sangre y el ritmo al que se pierde.

Señales aferentes
Los impulsos aferentes transmitidos desde la periferia se procesan en el sistema nervioso central (SNC) y activan las respuestas efectoras reflejas o impulsos eferentes.
El acontecimiento desencadenante inicial es la pérdida del volumen sanguíneo circulante. Otros estímulos que pueden activar la reacción neuroendocrina incluyen dolor, hipoxemia, hipercapnia, acidosis, infección, cambios de la temperatura, excitación emocional o hipoglucemia.
La sensación de dolor proveniente del tejido lesionado se transmite a través de las vías espinotalámicas y tiene como resultado la activación del eje hipotálamo-hipófisis-suprarrenales y asimismo del sistema nervioso autónomo (SNA) para inducir la estimulación simpática directa dela médula suprarrenal para liberar catecolaminas.

Los barorreceptores también son una vía aferente de importancia en el inicio de la respuesta de adaptación al choque.
En las aurículas del corazón se encuentran receptores de volumen, sensibles a cambios de la presión de la cámara como al estiramiento de la pared. Se activan con una hemorragia de volumen bajo o
disminuciones menores de la presión de la aurícula derecha.
Los receptores en el cayado aórtico y los cuerpos carotídeos reaccionan a alteraciones de la presión o al estiramiento de la pared arterial y responden a reducciones mayores del volumen o la presión
intravasculares.
En condiciones normales, estos receptores anulan la inducción del sistema nervioso autónomo. Cuando se activan, tales barorreceptores disminuyen sus impulsos eferentes y desinhiben enconsecuencia el efecto del sistema nervioso autónomo.

Los quimiorreceptores de la aorta y los cuerpos carotídeos son sensibles a cambios de la tensión de oxígeno, la concentración del ion H+ y las concentraciones de dióxido de carbono (CO2).
La estimulación de los quimiorreceptores causa vasodilatación de las arterias coronarias, disminución de la frecuencia cardiaca y vasoconstricción de la circulación esplácnica y esquelética.

Señales eferentes.
Respuesta cardiovascular. Los cambios de la función cardiovascular son efectos de las respuestas neuroendocrina y del SNA al estado de choque y constituyen una característica prominentedel mecanismo de respuesta de adaptación del cuerpo y los signos y síntomas clínicos del paciente en choque.
La hemorragia causa reducción del retorno venoso al corazón y disminución del gastocardiaco. Esto se compensa al incrementar la frecuencia y contractilidad del corazón y asimismo por vasoconstricción venosa y arterial.
La estimulación de las fibras simpáticas que inervan el corazón conduce a activación de receptores adrenérgicos β1 que aumentan la frecuencia y contractilidad cardiacas como intento de incrementar el gasto cardiaco.
La estimulación simpática directa de la circulación periférica a través de la activación de receptores adrenérgicos α1 en arteriolas ocasiona vasoconstricción e incremento compensador de la resistencia vascular y la presión arterial sistémicas.

La vasoconstricción arterial no es uniforme y su consecuencia es la redistribución notable del flujo sanguíneo.
Se establece entonces una perfusión selectiva de los tejidos por variaciones regionales de la resistencia arteriolar con derivación de sangre de los lechos orgánicos menos esenciales, como intestino, riñones y piel. En cambio, el cerebro y el corazón tienen mecanismos autorreguladores que preservan su flujo sanguíneo a pesar de la reducción global del gasto cardiaco.
El incremento de los impulsos simpáticos da lugar a la liberación de catecolaminas de la médula suprarrenal,cuya concentración llega al máximo en el transcurso de 24 a 48 h tras la lesióny a continuación regresa a la basal.

El aumento persistente de los valores de catecolaminas después de este tiempo sugiere la continuación de
estímulos nocivos aferentes.
Casi toda la epinefrina circulante la produce la médula suprarrenal, en tanto que la norepinefrina procede de las sinapsis del sistema nervioso simpático.
Los efectos de las catecolaminas en tejidos periféricos incluyen:
1. Estimulación de la glucogenólisis y gluconeogénesis hepáticas para incrementar la disponibilidad de glucosa circulante a los tejidos periféricos.
2. Incremento de la glucogenólisis en el músculo esquelético.
3. Supresión de la liberación de insulina y aumento de la liberación de glucagón.

Respuesta hormonal.
La respuesta a la lesión incluye la activación del sistema nervioso autónomo y la del eje hipotálamo-hipófisis-suprarrenales. El choque estimula al hipotálamo para producir hormona liberadorade corticotropina, que a su vez activa la secreción de la hormona adrenocorticotrópica (ACTH, adrenocorticotropic hormone) por la hipófisis.
El cortisol, que actúa de modo sinérgico con la adrenalina y el glucagón para inducir un estado catabólico.
El cortisol activa la gluconeogénesis y resistencia a la insulina y da lugar a la aparición de hiperglucemia y catabolismo de proteínas de las células musculares y lipólisis para proporcionar sustratos para la gluconeogénesis hepática.
El cortisol propicia la retención de sodio y agua por las nefronas del riñón.

Respuesta hormonal.
Si bien la angiotensina I no tiene una actividad funcional de importancia, la angiotensina II es un vasoconstrictor potente de lechos vasculares esplácnicos y periféricos y asimismo estimula la secreción de aldosterona, ACTH y hormona antidiurética (ADH).
La aldosterona, un mineralocorticoide, actúa en la nefrona para promover la resorción de sodio y, como consecuencia, de agua. Se pierden iones potasio e hidrógeno por la orina en unintercambio por sodio.
La epinefrina, la angiotensina II, el dolor y la hiperglucemia incrementan la producción de hormona antidiurética (ADH). Los valores de esta última permanecen elevados casi una semana después de laagresión inicial, de acuerdo con la gravedad y persistencia de las anomalías hemodinámicas.
La ADH actúa en el túbulo distal y el conducto colector de la nefrona para acentuar la permeabilidad al agua, disminuir las pérdidas de agua y sodio y preservar el volumen intravascular. La ADH(arginina vasopresina), vasoconstrictor mesentérico potente y deriva sangre circulante de los órganos esplácnicos durante la hipovolemia.

La vasopresina también incrementa la gluconeogénesis y glucólisis hepáticas.
En estados sépticos, la endotoxina estimula de manera directa la secreción de vasopresina arginina al margen de los cambios de la presión arterial, osmóticos o del volumen intravascular.


Homeostasis circulatoria.
Precarga. En reposo, la mayor parte del volumen sanguíneo se encuentra en el sistema venoso.
Casi todas las alteraciones del gasto cardiaco en el corazón normal se relacionan con cambios en la precarga.
Volumen sanguíneo esplácnico, que habitualmente representa el 20% del volumen de sangre.
Las respuestas agudas al volumen intravascular incluyen:
Cambios del tono venoso, resistencia vascular sistémica y presión intratorácica, con menos importancia de los cambios hormonales más lentos en la respuesta temprana a la pérdida de volumen.

Contracción ventricular.
La curva de Frank-Starling describe que la fuerza de la contracción ventricular guarda relación con suprecarga. Esta relación se basa en que la longitud inicial del músculo determina la fuerza de contracción.
Poscarga. La poscarga es la fuerza de resistencia al trabajo del miocardio durante la contracción. La presión arterial es el principal componente de la poscarga que influye en la fracción de expulsión.Esta resistencia vascular la determinan los esfínteres de músculo liso precapilares.
Microcirculación. El lecho microvascular está inervado por el sistema nervioso simpático y tiene un efecto profundo en las arteriolas más grandes. Después de una hemorragia, las arteriolas más grandes se constriñen; sin embargo, en caso de septicemia o choque neurógeno, los vasos se dilatan.
El flujo en el lecho capilar a menudo es heterogéneo en estados de choque, lo que tal vez se deba a múltiples mecanismos locales, como edema, disfunción y activación de células endotelialesmarcadas por la atracción de leucocitos. En conjunto, estos mecanismos disminuyen la perfusión capilar, y podrían persistir después de la reanimación.

Datos interesantes sugieren que en la septicemia, la respuesta para limitar el consumo de O2por parte de las células parenquimatosas del tejido es una respuesta de adaptación a la señalización inflamatoria y al descenso en la perfusión.
La disfunción capilar también es secundaria a la activación de las células endoteliales por mediadores inflamatorios circulantes generados en los estados de choque séptico o traumático. Esto exacerba el edema de las células endoteliales y la fuga capilar, además de aumentar la adhesión de leucocitos, lo cual produce oclusión capilar que puede persistir después de la reanimación y se denomina falta de reflujo.

EFECTOS METABÓLICOS.
El metabolismo celular se basa sobre todo en la hidrólisis del trifosfato de adenosina (ATP). La división del enlace fosfoanhidro del segmento terminal o el fosfato- γ del ATP es la fuente de energíapara la mayor parte de los procesos dentro de la célula en condiciones normales.
La mayor parte de ATP se genera en nuestros cuerpos mediante metabolismo aeróbico en el proceso de fosforilación oxidativa en las mitocondrias.
Este proceso depende de la disponibilidad de oxígeno como receptor final de electrones en lacadena de transporte de electrones.
Cuando el aporte de oxígeno se afecta tanto que es imposible sostener la fosforilación oxidativa,el estado se denomina disoxia. Cuando la fosforilación oxidativa es insuficiente, las células cambian a metabolismo anaerobio y glucólisis para generar ATP.

Aunque la glucólisis es un proceso rápido, no es eficiente, sólo permite la producción de
dos moles de ATP a partir de un mol de glucosa, en comparación con la oxidación completa de uno de glucosa glucosa que produce 38 moles de ATP.
Además, en condiciones de hipoxia en el metabolismo anaeróbico, el piruvato se convierte a lactato, lo que produce acidosis metabólica intracelular.
Las catecolaminas incrementan la glucogenólisis y gluconeogénesis hepáticas, cetogénesis, catabolismo de las proteínas de músculos esqueléticos y lipólisis del tejido adiposo.
Durante el estado de choque, el cortisol, el glucagón y la ADH favorecen asimismo el catabolismo. La epinefrina induce la liberación adicional de glucagón, en tanto que suprime la de insulina por las células
pancreáticas β.
El resultado es un estado catabólico con desplazamiento de la glucosa, hiperglucemia, catabolismo proteínico, equilibrio negativo del nitrógeno, lipólisis y resistencia a la insulina durante el choque y la lesión. La utilización menor relativa de glucosa por tejidos periféricos la conserva para los órganos dependientes de la glucosa como el corazón y el cerebro.
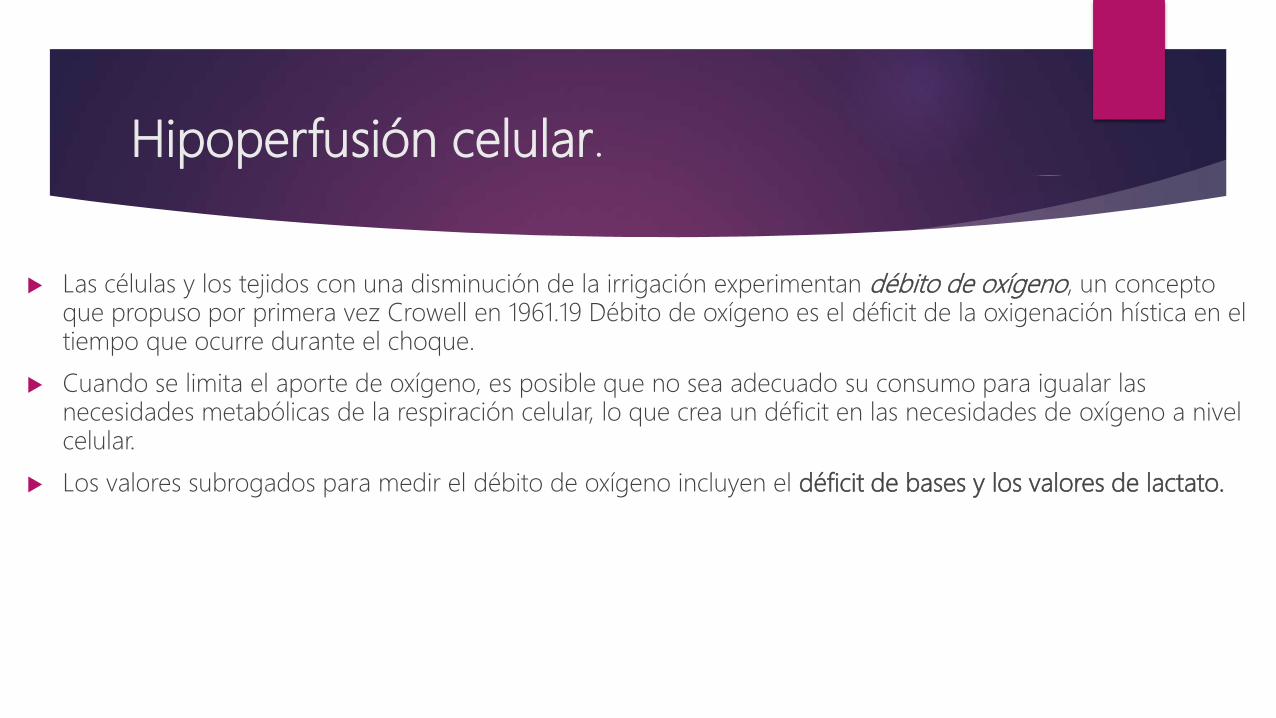
Hipoperfusión celular.
Las células y los tejidos con una disminución de la irrigación experimentan débito de oxígeno, un concepto que propuso por primera vez Crowell en 1961.19 Débito de oxígeno es el déficit de la oxigenación hística en el tiempo que ocurre durante el choque.
Cuando se limita el aporte de oxígeno, es posible que no sea adecuado su consumo para igualar las necesidades metabólicas de la respiración celular, lo que crea un déficit en las necesidades de oxígeno a nivel celular.
Los valores subrogados para medir el débito de oxígeno incluyen el déficit de bases y los valores de lactato.

RESPUESTAS INMUNITARIA E INFLAMATORIA
La falla en el control adecuado de la activación, incremento o supresión de la respuesta inflamatoriapuede originar un síndrome de respuesta inflamatoria sistémica y falla potencial de múltiples órganos.
Cuando estos mediadores de predominio paracrino poseen acceso a la circulación sistémica, pueden precipitar una diversidad de cambios metabólicos que se denominan en conjunto reacción inflamatoria del hospedador.
Sólo desde hace poco se reconoció que la liberación de productos intracelulares de las células lesionadas y dañadas puede tener efectos paracrinos y endocrinos en tejidos distantes para activar las respuestas inflamatorias e inmunitarias. Esta hipótesis, propuesta por primera vez por Matzinger, se conoce como señalización de peligro.
Según este novedoso paradigma de función inmunitaria, las moléculas endógenas son capaces de emitir señales sobre la presencia de peligro a las células y tejidos circundantes. Tales moléculas que se liberan de las células se conocen como patrones moleculares relacionados con la lesión (DAMP)

Los receptores de la superficie celular reconocen a los DAMP para iniciar la señalización que ceba y amplifica la respuesta inmunitaria.Estos receptores se conocen como receptores de reconocimiento de patrones (PRR, pattern recognition
receptors) e incluyen los receptores tipo Toll (TLR, Toll like receptors) y los receptores paraproductos finales de la glucosilación avanzada.

Citocinas/quimiocinas
Durante la respuesta a la lesión, el TNF α contribuye al catabolismo de proteí-nas musculares y caquexia.
La interleucina-1 (IL-1) ejerce acciones similares a las del TNF α. La IL-1 posee una semivida muy corta (6 min) y actúa sobre todo en forma paracrina para modular las respuestas celulares locales.
A nivel sistémico, la IL-1 genera una reacción febril a la lesión por activación de prostaglandinas en el hipotálamo posterior y causa anorexia porque estimula el centro de la saciedad.
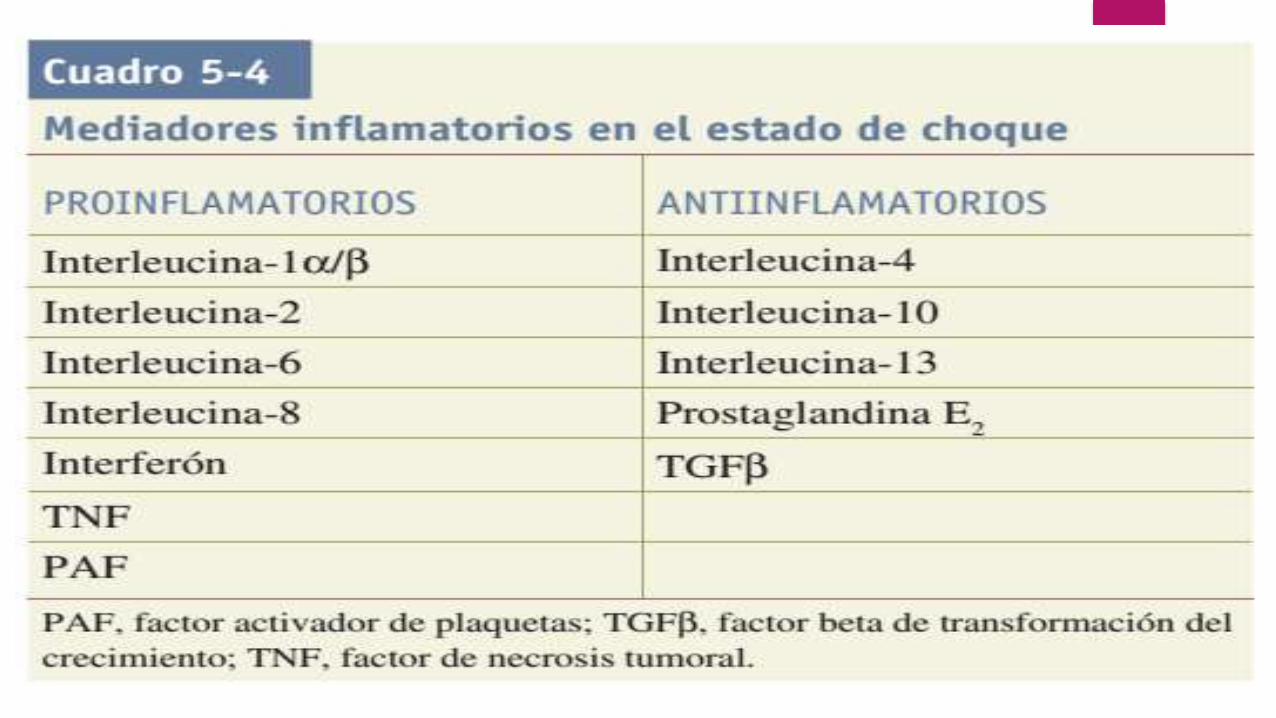

La IL-6 se eleva como respuesta al choque hemorrágico, procedimientos quirúrgicos mayores o traumatismos. Los valores elevados de IL-6 se correlacionan con la mortalidad en estados dechoque. La IL-6 contribuye a la lesión pulmonar, hepática e intestinal después del estado de choque hemorrágico.
La IL-10 se considera una citocina antiinflamatoria que puede tener propiedades inmunodepresoras. Su producción aumenta después de estados de choque y traumatismos y se presenta con depresión inmunitaria clínica y mayor susceptibilidad a las infecciones.

Complemento.
La lesión, estado de choque e infecciones graves suelen activar la cascada del complemento, la cual contribuye a la defensa del hospedador y la activación proinflamatoria. Después de un choque hemorrágico ocurre un consumo considerable del complemento.
Los factores del complemento activados C3a, C4a y C5a son mediadores potentes delincremento de la permeabilidad vascular, la contracción de la célula de músculo liso, la liberación de histamina y el producto accesorio ácido araquidónico y de la adhesión de neutrófilos al endoteliovascular.

Neutrófilos.
La activación de neutrófilos es un acontecimiento temprano en el aumento de la respuesta inflamatoria; son las primeras células que se incorporan al sitio de la lesión.
Los leucocitos polimorfonucleares (PMN) eliminan agentes infecciosos, sustancias extrañas quepenetraron las defensas de barrera del hospedador y tejido no viable mediante fagocitosis. Sin embargo, los PMN activados y sus productos también pueden infligir lesión celular y disfunción orgánica; generan y liberan diversas sustancias que pueden ocasionar lesión celular o hística, como especies de oxígeno reactivo, productos de peroxidación lipídica, enzimas proteolíticas (elastasa, catepsina G) y mediadores vasoactivos(leucotrienos, eicosanoides y factor activador de plaquetas).

FORMAS DE CHOQUE
Choque hipovolémico o hemorrágico
La causa más común de choque en el paciente quirúrgico o traumatizado es la pérdida de volumen circulante por una hemorragia.
la falta de efectos simpáticos en los vasos cerebrales y coronarios y la autorregulación local promueven la conservación del flujo sanguíneo cardiaco y del sistema nervioso central (SNC).
Diagnóstico.
Pueden ser obvios los signos clínicos de choque en un paciente agitado e incluyen extremidades frías y pegajosas, taquicardia, ausencia o debilidad de pulsos periféricos e hipotensión. Este choqueclínico aparente resulta de una pérdida cuando menos de 25 a 30% del volumen sanguíneo.

Las respuestas clínica y fisiológica a la hemorragia se clasifican según sea la magnitud de la pérdida de volumen.
Una pérdida hasta de 15% del volumen circulante (700 a 750 ml en un paciente de 70 kg) puede ocasionar pocas alteraciones en términos de síntomas obvios, en tanto que la pérdida hastade 30% del volumen circulante (1.5 L) produce taquicardia leve, taquipnea y ansiedad.
Es posible que no sean obvias la hipotensión, taquicardia intensa (es decir, pulso > 110 a 120 latidos por minuto [lpm]) y confusión hasta que se pierda más de 30% del volumen sanguíneo; la pérdida de 40% del volumen circulante (2 L) pone en peligro la vida de inmediato y debe realizarse control quirúrgico de la hemorragia


Datos recientes en pacientes traumatizados sugieren que una presión sanguínea sistólica (SBP, systolic bloodpressure) menor de 110 mmHg es una definición de hipotensión e hipoperfusión quetiene relevancia clínica, basada en el índice creciente de mortalidad con cifras menores a esta presión.
De igual manera, los valores de deficiencia de base derivados del análisis de gases sanguíneos arteriales brindan a los médicos un cálculo indirecto de la acidosis hística causada porhipoperfusión.
Davis et al. estratificaron la magnitud del déficit de base en:
1. leve (−3 a −5 mmol/L),
2. moderada (−6 a −9 mmol/L) y
3. grave (< −10 mmol/L),
con lo que establecieron una relación entre el déficit de base al momento del ingreso con las necesidades de transfusión, desarrollo de insuficiencia de múltiples órganos y muerte

Aunque es probable que los cambios en el hematócrito no reflejen con rapidez el volumen total de la pérdida sanguínea, se demostró que al momento de la hospitalización el hematócrito serelaciona con las necesidades de líquidos y transfusión en 24 h, y tiene una relación más marcada con la
transfusión de concentrados eritrocíticos que la taquicardia, hipotensión o acidosis.
Cuando no es visible de inmediato una pérdida de sangre mayor, debe sospecharse hemorragia interna (intracavitaria).
Cada cavidad pleural puede alojar 2 a 3 L de sangre y, por consiguiente,ser un punto de pérdida notable de sangre.
La hemorragia retroperitoneal mayor se relaciona de manera característica con fracturas pélvicas, que se confirman mediante radiografía de la pelvis en la unidad de reanimación.

Tratamiento.
reanimación con control de daños.
Dicha estrategia inicia en la sala de urgencias, continúa en el quirófano y en la unidad de cuidados intensivos (ICU).
La reanimación inicial se limita a mantener la SBP en alrededor de 90 mmHg. Esto impide una hemorragia renovada en los vasos recién coagulados.
La reanimación y la reanimación del volumen intravascular se hace con hemoderivados y soluciones cristaloides limitadas; Es peligroso que el volumen sea demasiado pequeño y permita la hipotensión e hipoperfusión persistentes, pero una reanimación con volumen muy vigoroso puede ser igual de nociva.

Pacientes con lesiones cerradas, en los que la principal causa de muerte es un traumatismo cefálico cerrado, debe evitarse el aumento en la mortalidad con hipotensión en presencia de lesión cerebral. En esta circunstancia, parece más apropiada una cifra de 110 mmHg para la presión sanguínea sistólica.
Existe un subgrupo de personas que no responde a los esfuerzos de reanimación a pesar del control adecuado de la hemorragia. Estos enfermos tienen necesidades constantes de líquidos aunqueel control de la hemorragia sea adecuado, presentan hipotensión persistente no obstante la restitución del volumen intravascular que requirió apoyo vasopresor, y pueden presentar un ciclo fútilde hipotermia no corregible, hipoperfusión, acidosis y coagulopatía que no es posible interrumpir pese al tratamiento máximo.
Estos individuos se deterioran hasta choque descompensado o irreversible con vasodilatación periférica y resistencia a la infusión de vasopresores. Una vez que el enfermo se encuentra en estado de choque en sus etapas terminales es inevitable la muerte. Por desgracia,muchas veces esto se diagnostica en forma retrospectiva.

el tipo ideal de líquidos que deben administrarse a pacientes en choque. Sin embargo, las soluciones cristaloides se mantienen como los líquidos de elección.
Varios estudios demostraron el aumento de riesgo en pacientes con traumatismos con hemorragia tratados con coloides, en comparación con los que recibieron cristaloides. En caso de hemorragia grave, la restauración del volumen intravascular debe hacerse con hemoderivados.
Hay estudios en proceso que valoran el uso de solución salina hipertónica como auxiliar en la reanimación de pacientes con hemorragia.
El beneficio de las soluciones salinas hipertónicas podría ser inmunomodulador.

La transfusión de concentrados de eritrocitos y otros hemoderivados es esencial para el tratamiento de pacientes en estado de choque hemorrágico. Las recomendaciones actuales en sujetosestables de la ICU incluyen el logro de un nivel de hemoglobina de 7 a 9 g/100 ml.
El estándar actual en pacientes con lesiones graves se denomina reanimación con control de daños y consiste en transfusión con eritrocitos, plasma fresco congelado (FFP, fresh frozen plasma) y unidades de plaquetas administradas en cantidades iguales.
Las plaquetas deben transfundirse al paciente con hemorragia para mantener cifras superiores a 50 × 109/L.
Existe una aplicación potencial para otros productos de factores de coagulación, como concentrados de fibrinógeno y concentrados de complejo de protrombina.
El uso de estos agentes puede guiarse con la caída en la concentración de fibrinógeno a < 1 g/L o,de manera menos específica, por los datos en el tromboelastograma sugestivos de hiperfibrinólisis.

Desde hace poco se usa la tromboelastografía (TEG) como una herramienta de valoraciónmás rápida y completa de la coagulopatía y la fibrinólisis en el paciente lesionado.
Holcomb et al. publicaron en fecha reciente que la TEG es un mejor factor pronóstico de los pacientes con hemorragia sustancial y transfusión de eritrocitos mejor que las pruebas convencionales para coagulopatía, anticipa la necesidad de transfusión plaquetaria mejor que el recuento plaquetario y anticipa la necesidad de transfusión de plasma mejor que la concentración de fibrinógeno.

Estado de choque por traumatismo.
Los ejemplos de estado de choque por traumatismo incluyen hemorragia de volumen pequeño acompañada de lesión de tejidos blandos (fractura del fémur, lesión por aplastamiento) o cualquier combinación de choque hipovolémico, neurógeno, cardiógeno y obstructivo que precipita la activación proinflamatoria rápidamente progresiva.
El tratamiento del estado de choque por traumatismo se dirige a corregir los elementosindividuales para atenuar la cascada de activación proinflamatoria e incluye control rápido de la hemorragia, reanimación adecuada de volumen para corregir el déficit de oxígeno, desbridamiento de tejido no viable, estabilización de lesiones óseas y tratamiento apropiado del daño de tejidos blandos.

Estado de choque septicémico (vasodilatador)
En la circulación periférica, la vasoconstricción profunda es la respuesta fisiológica típica a la disminución de la presión arterial y la perfusión hística secundaria a hemorragia, hipovolemia o insuficiencia cardiaca aguda. Ésta no es la respuesta característica en el choque por vasodilatación.
Es resultado de la disfunción del endotelio y la vasculatura secundaria a mediadores y células inflamatorias circulantes, o como respuesta a la hipoperfusión prolongada y grave.
En el choque vasodilatador, la hipotensión resulta de la falta de contracción apropiada del músculo liso vascular.
El choque vasodilatador se caracteriza por vasodilatación periférica con hipotensión resultante y resistencia al tratamiento con vasopresores.

A pesar de la hipotensión, se encuentran aumentadas las concentraciones de catecolaminas en plasma y activado el sistema renina-angiotensina.
La forma de choque vasodilatador que se encuentra con mayor frecuencia es el choque septicémico.
Otras causas de choque vasodilatador incluyen acidosis láctica hipóxica, envenenamiento por monóxido de carbono, choque hemorrágico descompensado e irreversible, choque cardiógeno terminal y poscardiotomía.
Por consiguiente, al parecer el choque vasodilatador representa la vía común final del estado de choque intenso y prolongado de cualquier causa. A pesar de los adelantos en cuidados intensivos, la mortalidad por septicemia grave es aún de 30 a 50%.
El estado de choque septicémico es un producto secundario de la respuesta corporal a la alteración en el equilibrio entre el microbio y el hospedador, que resulta en una infección localizada invasiva o grave.


Cuando esta reacción es muy exagerada o se torna sistémica en lugar de localizada, casi siempre son notorias las manifestaciones de septicemia.
Tales datos incluyen mayor gasto cardiaco, vasodilatación periférica, fiebre, leucocitosis, hiperglucemia y taquicardia.
En el choque septicémico, los efectos vasodilatadores se deben en parte al aumento de la isoforma inducible de la óxido nítrico sintasa (iNOS o NOS 2) en la pared de los vasos.
La iNOS produce grandes cantidades de óxido nítrico por periodos constantes. Este potente vasodilatador suprime el tono vascular y torna resistente la vasculatura a los efectos de fármacos vasoconstrictores

Diagnóstico.
Se emplean los términos septicemia, septicemia grave y choque septicémico para cuantificar la magnitud de la reacción inflamatoria sistémica.
Los pacientes con septicemia tienen datos de infección y asimismo signos sistémicos de inflamación (p. ej., fiebre, leucocitosis y taquicardia).
La hipoperfusión con signos de disfunción orgánica se denomina septicemia grave.
El choque septicémico requiere la presencia de los anteriores, junto con datos más notables de hipoperfusión hística e hipotensión sistémica. Además de la hipotensión, la distribución deficiente del flujo sanguíneo y la derivación en la microcirculación alteran de forma adicional el aporte de nutrimentos a los lechos hísticos.

Tratamiento.
Esta reanimación debe ser de al menos 30 ml/kg en las primeras 4 a 6 h. Deben continuarse los bolos crecientes de líquido con base en el criterio de valoración de la reanimación, que incluye la eliminación de lactato.
Deben evitarse las soluciones coloides con almidón, ya que la evidencia reciente sugiere que estos líquidos pueden ser nocivos en presencia de septicemia.
Los antibióticos intravenosos no son suficientes para tratar de manera apropiada el episodio infeccioso cuando existen acumulaciones de líquidos infectadas, cuerpos extraños infectados ytejido desvitalizado.
Esta situación se denomina control del origen e incluye drenaje percutáneo y manejo quirúrgico para dirigirse al foco de infección. Tales casos podrían requerir múltiples operaciones paraasegurar una higiene y cicatrización adecuadas de la herida.

Las catecolaminas son los vasopresores más usuales, la norepinefrina es el compuesto de primera línea, seguido por la epinefrina.
En ocasiones, los pacientes con choque septicémico desarrollan resistencia a las catecolaminas. La arginina
vasopresina, un potente vasoconstrictor, a menudo es eficaz en estas situaciones y con frecuencia se agrega a a la norepinefrina.
Casi todos los sujetos con septicemia tienen una fisiología hiperdinámica con gasto cardiaco supranormal y resistencia vascular sistémica baja. En ocasiones, estos enfermos pueden tener ungasto cardiaco bajo a pesar de la reanimación de volumen e incluso el apoyo vasopresor.
Se recomienda el tratamiento con dobutamina para los pacientes con disfunción cardiaca demostrada por presiones de llenado elevadas y gasto cardiaco bajo o por signos de hipoperfusión después de restaurar la presión sanguínea mediante la reanimación con líquidos.


Estado de choque cardiógeno.
Estado de choque cardiógenoEl choque cardiógeno se define desde el punto de vista clínico como una falla de la bomba circulatoria que conduce a reducción del flujo anterógrado e hipoxia hística subsecuente, con un volumen intravascularadecuado.
Los criterios hemodinámicos incluyen1. hipotensión sostenida (SBP < 90 mmHg cuando menos durante 30 min),
2. índice cardiaco reducido (< 2.2 L/min/m2) y
3. presión en cuña de la arteria pulmonar alta (> 15 mmHg).

Las tasas de mortalidad del estado de choque cardiógeno son de 50 a 80%.
La causa más común de este trastorno es infarto del miocardio (MI) agudo yextenso; un infarto más pequeño en un paciente con disfunción delventrículo izquierdo también puede precipitar choque. El choquecardiógeno es una complicación de 5 a 10% de los MI agudos;por el contrario, es la causa más común de muerte en personashospitalizadas con MI agudo. Aunque el choque puede presentarseen un momento temprano después de un infarto del miocardio, demanera característica no se reconoce al ingresar al hospital. El 75%de los pacientes con choque cardiógeno como complicación de unMI agudo desarrolla signos de este trastorno en el transcurso de24 h tras el inicio del infarto (promedio, 7 h).

El grado de este últimodespués de una angioplastia coronaria transluminal percutánea(PTCA) se correlaciona con la mortalidad en el hospital (33% conla reanudación de la perfusión completa, 50% con la reanudaciónincompleta del riego y 85% de mortalidad cuando no se reanuda). 96Una función cardiaca inadecuada puede ser el resultado directo deuna lesión cardiaca, incluidos la contusión profunda del miocardio, lesión valvular cardiaca contusa o daño directo del miocardio


Diagnóstico.
Los signos dechoque circulatorio comprenden hipotensión, piel fría y marmó-rea, depresión del estado mental, taquicardia y disminución de lospulsos. La exploración cardiaca puede incluir arritmias, levantamiento precordial o tonos cardiacos distantes. La confirmacióndel estado de choque de origen cardiaco requiere un electrocardiograma y ecocardiografía urgentes.
Otras pruebas diagnósticas útilesincluyen radiografía de tórax, gases en sangre arterial, electrólitos,biometría hemática y enzimas cardiacas. Por lo general es útil lavigilancia cardiaca con penetración corporal, que casi nunca esnecesaria, para excluir infarto del ventrículo derecho, hipovolemiay posibles complicaciones mecánicas.

Tratamiento.
Deben corregirse las anomalías eletrolíticas, las más de las veceshipopotasemia e hipomagnesemia.
Cuando existe disfunción cardiaca grave puede estar indicadala administración de fármacos inotrópicos para mejorar la contractilidad y gasto cardiacos. La dobutamina estimula sobre todo a losreceptores cardiacos β1 para incrementar el gasto cardiaco, perotambién puede causar vasodilatación de los lechos vasculares periféricos y reducir la resistencia periférica total y la presión arterialsistémica por efecto sobre los receptores β2. En consecuencia, antesde instituir el tratamiento con dobutamina se debe asegurar unaprecarga y volumen intravascular adecuados. La dopamina estimulaa los receptores α (vasoconstricción), receptores β1 (estimulacióncardiaca) y receptores β2 (vasodilatación), con efectos en los receptores β en particular a dosis más bajas. En el tratamiento de ladisfunción cardiaca en personas hipotensas puede ser preferiblela dopamina a la dobutamina.

La epinefrina estimula a los receptores α y β y aumenta lacontractilidad y frecuencia cardiacas; no obstante, también puedetener efectos vasoconstrictores periféricos intensos que deterioran de modo adicional la función del corazón.
Los líneamientos actuales de la American Heart Associationrecomiendan la angiografía coronaria transluminal percutánea enpersonas con choque cardiógeno, elevación de ST, bloqueo de ramaizquierda y edad menor de 75 años.101,102 El paso más importanteen el tratamiento de los individuos con choque cardiógeno por MIagudo es la definición de la anatomía coronaria y revascularizacióntemprana. 103 Cuando es factible, el tratamiento de elección es laPTCA (por lo general con la colocación de una prótesis).

Estado de choque obstructivo
los pacientes traumatizados, por lo regular la obstrucción se debe a la presencia de neumotórax a tensión. Se identificataponamiento cardiaco cuando se acumula suficiente líquido en elsaco pericárdico para obstruir el flujo sanguíneo a los ventrículos.Las anomalías hemodinámicas en el taponamiento pericárdico sedeben a la elevación de las presiones intracardiacas con limitacióndel llenado ventricular en la diástole y disminución consiguiente delgasto cardiaco. El pericardio no se distiende de manera aguda;en consecuencia, volúmenes pequeños de sangre pueden causartaponamiento cardiaco. Si se acumula lentamente el derrame (p. ej.,en casos de uremia, insuficiencia cardiaca o derrame maligno),la cantidad de líquido que ocasiona taponamiento cardiaco puedellegar a 2 000 ml. El principal determinante del grado de hipotensión es la presión pericárdica.


Diagnóstico y tratamiento. El diagnóstico de neumotórax a tensión debe establecerse en la exploración clínica. Los datos comunesincluyen insuficiencia respiratoria (en un paciente despierto), hipotensión, disminución de los ruidos respiratorios en un hemitórax,hiperresonancia a la percusión, distensión venosa yugular y desviación de las estructuras mediastínicas hacia el lado no afectado condesviación traqueal.

Son suficientes tres datos paraestablecer el diagnóstico de neumotórax a tensión: insuficienciarespiratoria o hipotensión, disminución de los ruidos pulmonarese hipertimpanismo a la percusión. Los datos de las radiografías detórax que pueden observarse incluyen desviación de las estructurasmediastínicas, depresión del hemidiafragma y menor opacificacióncon ausencia de marcas pulmonares. Como se comentó, el tratamiento definitivo del neumotórax a tensión es una toracostomía consonda inmediata. La sonda torácica debe insertarse a la brevedad,pero con cuidado, y ser lo bastante grande para evacuar cualquiercantidad de sangre que pueda haber en el espacio pleural. La ubicación más recomendada es el cuarto espacio intercostal (al niveldel pezón) en la línea axilar anterior.

El taponamiento cardiaco también puede acompañarse de disnea, ortopnea, tos, edema periférico,dolor torácico, taquicardia, tonos cardiacos amortiguados, distensión venosa yugular y elevación de la presión venosa central. Latríada de Beck consiste en hipotensión, tonos cardiacos amortiguados y distensión de las venas del cuello.
La ecocardiografía es laprueba preferida para el diagnóstico del taponamiento cardiaco

La ventana pericárdica diagnóstica es un método más directopara determinar la presencia de sangre en el pericardio. El procedimiento se practica mejor en el quirófano bajo anestesia general.Puede efectuarse a través de una vía subxifoidea o transdiafragmática.
Es posible exponerel corazón si se extiende la incisión hasta una esternotomía medialo a través de una toracotomía anterior izquierda o toracotomíasanteriores bilaterales (“en concha de almeja”).

Estado de choque neurógenoEl choque neurógeno se refiere a una disminución en la perfusiónhística como efecto de la pérdida del tono vasomotor en lechos arteriales periféricos.
Por lo general, el choque neurógenoes secundario a lesiones de la médula espinal por fracturas de loscuerpos vertebrales de la región cervical o torácica alta que alteranla regulación simpática del tono vascular periférico (


mulación de neurotransmisores y liberación de radicaleslibres. Como hecho importante, la hipotensión contribuye al empeoramiento de la lesión aguda de la médula espinal como resultado dela reducción adicional del flujo sanguíneo a la médula espinal.

Diagnóstico.
Lossujetos con lesiones motoras completas tienen probabilidad cincoveces mayor de requerir vasopresores por choque neurógeno, encomparación con aquellos que sufren lesiones incompletas.104 Ladescripción típica del estado de choque neurógeno incluye disminución de la presión arterial acompañada de bradicardia (ausencia de taquicardia refleja por alteración de la descarga simpática),extremidades calientes (pérdida de la vasoconstricción periférica),déficit motores y sensoriales que indican una lesión de la médulaespinal y prueba radiológica de una fractura de la columna vertebral.

Tratamiento.
El suministrode vasoconstrictores mejora el tono vascular periférico, atenúa lacapacitancia vascular e incrementa el retorno venoso, pero sólodebe considerarse tras excluir hipovolemia como causa de la hipotensión y establecer el diagnóstico de choque neurógeno. Si la presiónarterial del enfermo no responde a una reanimación de volumenadecuada, puede emplearse primero dopamina. Por lo regular seutiliza un agonista alfa puro, como fenilefrina, o en personas queCuadro 5-9Causas del choque neurógenoTraumatismo de médula espinalNeoplasia de médula espinalAnestesia espinal/epiduralBiblioteca Médica Virtual130ParTe iConsideraCiones BásiCasno responden a la dopamina. El tratamiento específico de la hipotensión casi siempre es breve, ya que de manera característica sóloes necesario administrar vasoconstrictores por 24 a 48 h

CRITERIOS DE VALORACIÓN EN LA REANIMACIÓNEl choque se define como una perfusión inadecuada para conservarla función normal de los órganos. Con el metabolismo anaerobioprolongado, se acumulan la acidosis hística y el déficit de oxígeno.Por consiguiente, el objetivo en el tratamiento del estado de choquees restablecer la perfusión adecuada de los órganos y la oxigenación de los tejidos. La reanimación es completa cuando secorrige el déficit de oxígeno y la acidosis hística y se restablece el metabolismo aerobio. Aún es un desafío confirmar enclínica este punto final.


Los criterios de valoración en la reanimación pueden dividirse en parámetros sistémicos o globales, parámetros específicosde tejido y parámetros celulares. Los criterios de valoración globales incluyen signos vitales, gasto cardiaco, presión en cuña de laarteria pulmonar, aporte y consumo de oxígeno, lactato y déficit debases

Aspectos que se valoran en la reanimación
Lactato. El lactato se produce por conversión del piruvato a lactato por acción de la lactato deshidrogenasa en casos de insuficiencia de oxígeno. El lactato llega a la circulación y se capta ymetaboliza de manera predominante por el hígado y los riñones.El hígado recoge alrededor de 50% del lactato total del cuerpo y elriñón cerca de 30%. El aumento del lactato sérico es una medidaindirecta del débito de oxígeno y por consiguiente una aproximación de la magnitud y duración de la gravedad del estado de choque.

Déficit de bases. El déficit de bases es la cantidad de bases enmilimoles necesaria para ajustar 1 L de sangre entera a un pH de7.40 con la muestra saturada en su totalidad con oxígeno a 37°C yCuadro 5-10Criterios de valoración de la reanimaciónSistémicos/globalesLactatoDéficit de basesGasto cardiacoAporte y consumo de oxígenoEspecíficos de tejidoTonometría gástricaValores hísticos de pH, oxígeno, dióxido de carbonoEspectroscopia cuasi infrarrojaCelularesPotencial de membranaTrifosfato de adenosina (ATP)7Biblioteca Médica Virtual131CAPÍTuLo 5Choqueuna Paco2 de 40 mmHg
El déficit de bases puede dividirse en las categorías leve (3a 5 mmol/L), moderada (6 a 14 mmol/L) y grave (15 mmol/L),con tendencia a una mortalidad más alta con el empeoramientodel déficit de bases en pacientes con traumatismos. La magnitud del déficit de la perfusión indicada por el déficit de bases yel tiempo necesario para corregirlo son factores importantes queestablecen el resultado final en el choque.De hecho, cuando persiste elevado el déficit de bases (o acidosis láctica) en un enfermo traumatizado, la causa es casi siempreuna hemorragia en curso.

Los factores que pueden alterar la utilidad del déficit de basesen la estimación del débito de oxígeno son la administración debicarbonato, hipotermia, hipocapnia (ventilación excesiva), heparina, alcohol y cetoacidosis.
ástrica con una sonda nasogástrica diseñada especialmentepara ello. Con el principio de que el bicarbonato gástrico es igual alas concentraciones séricas, se calcula el pH de la mucosa gástrica(pHi) aplicando la ecuación de Henderson-Hasselbalch. El pHdebe ser > 7.3; es bajo en casos de menor oxigenación de los tejidos.El pHi es un buen indicador pronóstico; los pacientes con pHinormal tienen mejores resultados que aquellos que tienen un valor< 7.3.

Espectroscopia cuasi infrarroja. La espectroscopia cuasi infraroja (NIR) puede medir la oxigenación de los tejidos y el estado deoxidorreducción del citocromo a,a3 en una forma continua que noimplica penetración corporal. La sonda de NIR emite múltiples lo
ngitudes de onda de luz en el espectro de NIR (650 a 1 100 nm