cardio-lipidología · lipoproteínas es la digestión, absorción, resíntesis y transporte de los...
TRANSCRIPT

Cardio-LipidologíaLipidología con enfoque cardiovascular
Metabolismo, Dislipidemias, Aterogenesis-Regresión, Estratificación, Metas y Tratamiento
Autor y Editor. Dr. Enrique C. Morales Villegas
Prólogo. Antonio M. Gotto Jr. MD, DPhil
Portada. Dr. Jorge Oseguera Moguel
Editorial.. Atheros-CIC. Aguascalientes, México
Atlas Ilustrado 2012

Cardio-LipidologíaLipidología con enfoque cardiovascular
Metabolismo, Dislipidemias, Aterogenesis-Regresión, Estratificación, Metas y Tratamiento
Autor y Editor. Dr. Enrique C. Morales Villegas
Prólogo. Antonio M. Gotto Jr. MD, DPhil
Portada. Dr. Jorge Oseguera Moguel
Editorial.. Atheros-CIC. Aguascalientes, México
Atlas Ilustrado 2012


Dedicatoria
Dedico este libro a los estudiantes de Medicina de pregrado y de postgrado así como a losMédicos Generales. A los primeros por ser ellos en quienes la semilla de la Cardio-Lipidología encuentra suterreno más fértil, y a los segundos por ser ellos quienes tienen el 75% de la responsabilidad para lograr quelos individuos en riesgo de o con enfermedad cardiovascular tengan una mejor expectativa y calidad de vida.
La enseñanza, el aprendizaje y la práctica de una Medicina basada en la Prevención sonimpostergables, siendo la Cardio-Lipidología el pivote de esta visión.

ndiceIPrólogo por Antonio M. Gotto Jr. MD, DPhil
Introducción por el Autor
Capítulo 1. Metabolismo de lípidos y lipoproteínas-Revisitando las bases de la fisiología-
. Generalidades
. Metabolismo exógeno de lípidos
. Metabolismo endógeno de lípidos
. Metabolismo reverso de lípidos
Capítulo 2. Dislipidemias-Trastornos primarios, secundarios y mixtos-
. Generalidades
. Incremento de LDL
. Reducción de LDL
. Incremento de triglicéridos
. Incremento de LDL y triglicéridos. Dislipidemias en México
. Incremento de HDL
. Reducción de HDL
Capítulo 3. Aterotrombogénesis y Ateroregresión-La liga entre la Lipidología y la Cardiología-
003-034
001-002
035-064
065-086

Cardio-Lipidología
. Generalidades
. LDL oxidado. Hipótesis fundamental
. Fases de la aterotrombogénesis
. Clasificación de la A.H.A para la aterosclerosis
. Ateroregresión
Capítulo 4. Salud, Riesgo, Estratificación y Metas-Información esencial para el Médico actual-
. Generalidades
. Factores de riesgo, biomarcadores y bioimágenes
. Estratificación básica del riesgo cardiovascular
. Cálculo del riesgo a mediano plazo -10 años-
. Cálculo del riesgo a largo plazo
. Estratificación avanzada del riesgo cardiovascular. Biomarcadores. Bioimágenes. Otros métodos de estratificación
. Metas terapéuticas en lípidos
Capítulo 5. Estatinas, Fibratos, Niacina y más-Evidencias, controversias e investigaciones-
. Generalidades
. Estatinas
. Ezetimibe
. Fibratos
. Acidos grasos Omega-3
. Niacina
. Estrategias en investigación:. Reductores de LDL. Incrementadores de HDL
087-122
123-189

Over the past several decades, our understanding of the relationship
between lipoprotein metabolism, atherosclerosis, and cardiovascular disease
has increased exponentially. It is now clear that elevated levels of serum
cholesterol increase risk for cardiovascular disease. Reduction of low-density
lipoprotein cholesterol levels with drug therapy has been shown in multiple
clinical trials to decrease cardiovascular risk. These advances have led to
improvements in the management of dyslipidemia and have contributed to an
age-adjusted decline in cardiovascular morbidity and mortality. Yet increases
in other risk factors, especially obesity and diabetes, threaten to reverse this
trend. In Mexico in particular, the prevalence of diabetes, obesity, and the
metabolic syndrome is rising dramatically. For example, between 1993 and
2006, the prevalence of diabetes in adults in Mexico increased from 6.7% to
14.4%, while the prevalence of metabolic syndrome increased from 27% to 37%.
These changes are likely to increase the burden of cardiovascular disease greatly
within the next 10-15 years.
Cardio-Lipidología, written by Dr. Enrique C. Morales Villegas, is a
comprehensive text on dyslipidemia and cardiovascular prevention with a
focus on Mexico. It provides the clinician with a clear guide to the management
of lipid disorders at a time when such texts are sorely needed. An authoritative
guide to lipidology, it offers a thorough overview of lipid metabolism, different
Foreword to Cardio-Lipidología

forms of dyslipidemia, the development and progression of the atherosclerotic
plaque, cardiovascular risk stratification, and treatment options for
dyslipidemia. It contains a wealth of helpful illustrations, diagrams,
photographs, and tables to accompany the text, and the information is
presented in a way that is both accessible and easy to understand. I am sure that
Cardio-Lipidología will prove to be a valuable resource for the busy clinician.
Antonio M. Gotto, Jr., MD, DPhil
Weill Cornell Medical College
New York, NY
Spring 2012

Introducción
Este libro nació con la idea de conjuntar 5 temas íntimamente vinculados y de grantrascendencia en la Medicina moderna. El conocimiento aislado de estos temas no se justifica, por elcontrario, su dominio e integración le permiten al Médico llegar al momento crítico de la prescripción conun alto grado de certeza.
El primer capítulo revisa el metabolismo de lípidos y lipoproteínas. Esta revisión de lafisiología es crucial para el entendimiento de los trastornos en lípidos y lipoproteínas y especialmente para lacomprensión de la farmacología de las estrategias terapéuticas orientadas al tratamiento de lasdislipidemias. El segundo capítulo, sobre dislipidemias, está enfocado a la revisión de las causas primarias ogenéticas, secundarias y mixtas de los trastornos en colesterol total, colesterol-LDL, triglicéridos ycolesterol-HDL. Si bien en Endocrinología, el termino dislipidemia implica cualquier trastorno en loslípidos, en Cardiología, el termino dislipidemia lleva intrínseca una connotación de aterogenicidad. Por estarazón, este capítulo destaca aquellas dislipidemias pro-aterogénicas, con énfasis en la dislipidemia mixta tancomún en nuestro País; esta es la razón por la cual este libro se intitula“Cardio-Lipidología. Lipidología conorientación cardiovascular”. El tercer capítulo sobre Aterogénesis y Ateroregresión es la liga natural entre laLipidología y la Cardiología. Como referí previamente, en la Medicina de nuestros días, los trastornos enlípidos y lipoproteínas tendrían otro peso específico si no estuvieran vinculados con la Aterosclerosis y lasEnfermedades Cardiovasculares. Partiendo de la revisión de los conceptos de nivel fisiológico de LDL yLDL oxidado como “patógeno mimetizado”, en este capítulo se revisan los fenómenos moleculares,celulares, tisulares y estructurales que caracterizan a la Aterogénesis y a su contraparte la Ateroregresión. Elcuarto capítulo trata los conceptos de salud, riesgo, estratificación cardiovascular y metas en lípidos. Lasalud cardiovascular ideal en el adulto es casi una utopía, en la mejor de las estadísticas sólo 1 de cada 100adultos posee un estado ideal de salud cardiovascular, por lo tanto para orientar al 99% de la poblaciónadulta carente de este ideal, el dominio de los conceptos de factor de riesgo, riesgo cardiovascular absoluto amediano y largo plazo y estratificación del riesgo cardiovascular general, es la mejor táctica que el MédicoClínico tiene para plantear estrategias científicas orientadas a incrementar la expectativa y calidad de vida de
1

2
Cardio-Lipidología
sus consultantes. Puesto que los niveles sanguíneos supra-fisiológicos de colesterol total, LDL y colesterolno-HDL son los factores de riesgo con mayor evidencia básica, epidemiológica, clínica y terapéutica deasociación con Aterosclerosis y Enfermedades Cardiovasculares, en este libro se analizan las metasactuales, las controversias y las posibles propuestas futuras para el logro de dichas metas. Finalmente paracerrar el ciclo de fisiología, fisiopatología, enfermedad, diagnóstico y tratamiento, el quinto capítulo estádedicado al tratamiento farmacológico del riesgo cardiovascular a través de la modificación de lípidos ylipoproteínas. En palabras del Dr. Alcocer Diaz-Barreiro “comprar salud artificial” es la mejor y quizáúnica estrategia que tiene la mayoría de los adultos para alcanzar un estado “ideal” de salud. Por orden denivel de evidencia se revisan los siguientes grupos farmacológicos: estatinas y ezetimibe como fármacospara reducir principalmente colesterol-LDL; fibratos y ácidos omega-3 como fármacos para reducirpreferencialmente colesterol no-HDL y triglicéridos, y niacina como el mejor fármaco disponible paraincrementar colesterol-HDL. Para todos los grupos farmacológicos se revisan mecanismos de acción yefecto en lípidos, biomarcadores y bioimágenes -ateroregresión- así como los efectos en la reducción deeventos clínicos en los diferentes escenarios estudiados, efectos adversos y controversias. Destacan lascontroversias sobre estatinas e incremento de glucosa y A1c, y las indicaciones actuales para ezetimibe,fibratos y niacina. Se revisan también las moléculas en investigación clínica con potencial para eltratamiento de las dislipidemias, especialmente los inhibidores-moduladores de la CEPT, como el grupomás avanzado en esta arena de la Cardio-Lipidología.
Así, este libro es una puesta al día sobre lípidos, lipoproteínas, dislipidemias, estratificacióncardiovascular, metas en lípidos y estrategias farmacológicas orientadas a reducir el riesgo cardiovascular através de la modificación eficiente de lípidos y lipoproteínas. La estructura del libro se caracteriza por untexto con conceptos claros basados en evidencia, con referencias bibliográficas clásicas, de “punta” y/o deconsenso integradas al texto y una profusa ilustración a color. Todas las ilustraciones pueden serdescargadas sin costo en Sea pues esta obra, una contribución más al desempeñowww.cicags.com.mx.científico y humanístico de nosotros los Médicos.
Enrique C Morales-VillegasAguascalientes, MéxicoPrimavera 2012

Metabolismo de lípidos y lipoproteínasRevisitando las bases de la fisiología
Capítulo 1

GeneralidadesEste libro inicia con una sección frecuentemente omitida en los libros de Medicina Clínica, la
Fisiología. La fisiología del metabolismo de lípidos y lipoproteínas es la piedra de toque para poder entenderla detección, el diagnóstico y el tratamiento de los trastornos de esta área del metabolismo intermedio taníntimamente ligada a la enfermedad cardiovascular.
El metabolismo de lípidos y lipoproteínas se ha dividido en tres apartados: metabolismoexógeno, metabolismo endógeno y metabolismo reverso. El metabolismo exógeno estudia la digestión,absorción, resíntesis y transporte de los lípidos ingeridos en la dieta y los que provienen de la bilis y losdetritus celulares del tracto gastrointestinal, del intestino -enterocito- hacia los tejidos periféricos en medioshídricos como la linfa y el plasma. El metabolismo endógeno trata la síntesis de novo y el transporte de loslípidos, del hígado -hepatocito- hacia los tejidos periféricos en un medio hídrico como el plasma. Elmetabolismo reverso analiza la transportación del colesterol acumulado en las células, especialmente en losmacrófagos, de los tejidos periféricos hacia el hígado para su catabolismo y eliminación hepatobiliar,igualmente en un medio hídrico como el plasma.
De esta forma, existen dos mecanismos -exógeno y endógeno- que proveen lípidoshidrofóbicos o neutros como el colesterol esterificado y los triglicéridos, y lípidos anfipáticos o polarescomo los fosfolípidos a todas las células de la economía. En contraparte, existe un mecanismo -reverso- quepermite la remoción, catabolismo y eliminación del exceso de colesterol acumulado en los tejidosperiféricos, y con ello mantiene un fino equilibrio entre metabolismo y catabolismo, especialmente decolesterol. Estos objetivos fisiológicos requieren de mecanismos especiales o “pivote” que permitan quelos lípidos, moléculas no solubles en la linfa y el plasma puedan viajar desde sus células de origen-enterocito, hepatocito y macrófago- hacia las células blanco. Esos mecanismos especiales comprenden lasíntesis de macromoléculas transportadoras de lípidos denominadas lipoproteínas; estas macromóleculascontienen los lípidos a transportar y diversas proteínas -apoproteínas y proteínas asociadas-. Estas dosúltimas dan a la lipoproteína: estructura terciaria, hidrosolubilidad, sustrato de unión o protección aenzimas hidrolíticas, sitio de reconocimiento por receptores de apoproteínas, estructura de unión contransferidores de lípidos, etc.
Una vez armadas las lipoproteínas, existen una serie de procesos fisiológicos que permiten laentrega o la captura de los lípidos, estos procesos fisiológicos al igual que los objetivos y los mecanismosfisiológicos de cada apartado del metabolismo de los lípidos y lipoproteínas son revisados en este capítulo.
4
Cardio-Lipidología

Metabolismo normal de lípidos y lipoproteínas
5
Al final del día el propósito último de los metabolismos exógeno y endógeno es transportarhacia los tejidos triglicéridos, colesterol y fosfolípidos. Los triglicéridos al hidrolizarse en la periferia donanácidos grasos y glicerol; los primeros, son sustratos de la betaoxidación para la generación de energía o bienpara la resíntesis de triglicéridos como fuente energética potencial y el segundo es sustrato para lagluconeogénesis hepática; el colesterol es molécula fundamental para la formación de membranascelulares, así como para la síntesis de hormonas esteroides, vitaminas liposolubles y sales biliares, encontraparte su exceso y especialmente su acumulación y oxidación es el factor etiopatogénico másimportante de la aterosclerosis, por ende la gran importancia del metabolismo reverso del mismo;finalmente los fosfolípidos al igual que los triglicéridos son donadores de ácidos grasos y como tales sonsustrato para la síntesis de múltiples moléculas de señalización intracelular y al igual que el colesterol sonmoléculas constitutivas de las membranas celulares.
Así este capítulo centrado en los objetivos, mecanismos y procesos fisiológicos de losmetabolismos exógeno, endógeno y reverso de lípidos y lipoproteínas, es una puesta al día con orientaciónclínica sobre este tema crucial en la Medicina moderna, la cual es sólo la base del conocimiento modernosobre lípidos, conocimiento en rápida y amplia expansión gracias al progreso en la conformación del Mapadel Lipidoma Humano que en un futuro nos permitirá comprender mejor entre otras, la liga entre losdiferentes tipos de lípidos en el plasma humano -esteroles, glicerofosfolípidos, glicerolípidos,esfingolípidos y ácidos grasos- y el estado de salud-enfermedad.
Quehenberger O, Dennis EA. The Human Plasma Lipidome. Review Article. New Engl JMed 2011; 365:1812-1823.

Metabolismo exógeno de lípidosObjetivo fisiológico. El objetivo fisiológico del metabolismo exógeno de lípidos y
lipoproteínas es la digestión, absorción, resíntesis y transporte de los lípidos ingeridos en la dieta, tantohidrofóbicos o neutros -colesterol esterificado y triglicéridos-, como anfipáticos o polares -colesterol libre yfosfolípidos-, del intestino hacia los tejidos periféricos y el hígado, en medios hídricos como la linfa y elplasma. También se incluye el metabolismo de los lípidos contenidos en la bilis y en los detritus de célulasexfaceladas del tracto gastrointestinal.
Mecanismo fisiológico pivote. El mecanismo fisiológico, eje del metabolismo
exógeno de lípidos y lipoproteínas es la síntesis intestinal de macromoléculas transportadoras de lípidosdenominadas Quilomicrones -QM-, equivalentes intestinales de las VLDL hepáticas. Los QM estánconstituidos por un “core” que al igual que las VLDL contienen los lípidos hidrofóbicos o neutros atransportar -colesterol esterificado y triglicéridos- y un recubrimiento integrado por los lípidos anfipáticoso polares -fosfolípidos, colesterol libre o no esterificado- y apoproteínas. Estas últimas le proporcionan alos QM: estructura, hidrosolubilidad plasmática, sustrato para la acción o inhibición de enzimas hidrolíticasy sitios de reconocimiento por receptores de lipoproteínas.
Procesos fisiológicos. Con fines didácticos, los procesos fisiológicos del metabolismo
exógeno de lípidos y lipoproteínas pueden segmentarse en: digestión, absorción y resíntesis intestinal delípidos; síntesis intestinal de QM; metabolismo linfático y plasmático de QM. A continuación se describecada uno de dichos procesos.
Digestión y absorción intestinal de grasas. Los triglicéridos, fosfolípidos y esteres decolesterol ingeridos en la dieta, así como los contenidos en la bilis y en los detritus de célulasgastrointestinales exfaceladas hacia la luz intestinal, son eficientemente hidrolizados por lipasas salivales,gástricas y pancreáticas así como por fosfolipasas y esterasas de colesterol. Los productos de dichahidrólisis, especialmente ácidos grasos y colesterol no esterificado son emulsificados por los ácidos biliaresy transportados distalmente para su absorción. Los ácidos grasos son incluidos al enterocito portransportación transmembranal facilitada y el colesterol no esterificado por transportacióntransmembranal activa y selectiva, ambos a nivel del “borde en cepillo” del aspecto luminal recubierto demucina de los enterocitos en duodeno y yeyuno proximal. La proteína ligadora de ácidos grasos -FABP-“fatty acid binding protein”, facilita en el enterocito el ingreso y la migración intracelular de los ácidos
Cardio-Lipidología
6

grasos y monoacilgliceroles. Los transportadores de colesterol de la luz intestinal hacia el enterocito son laproteína 1 similar a la proteína Nieman Pick C1 -NPC1L1- y el receptor depredador SR-B1; así mismo sehan identificado a los cassettes ABCG5 y ABCG8 como transportadores de colesterol del enterocito haciala luz intestinal. Se han propuesto a los cassettes ABCA1como transferidores de colesterol del enterocitohacia las HDL circulantes en la linfa. La fuente mayor de colesterol en el metabolismo exógeno es la bilis-800-1200mg/día-, seguida del colesterol ingerido en la dieta -300-500mg/día- y el colesterol provenientede detritus celulares exfacelados hacia el tracto gastrointestinal -300mg/día-. Dentro del enterocito a niveldel retículo endoplásmico liso, ácidos grasos, monoacilgliceroles, fosfatidilcolina y colesterol noesterificado son reconstituidos como triglicéridos, fosfolípidos y colesterol esterificado por diversosprocesos enzimáticos.
Síntesis intestinal de Quilomicrones. El proceso fisiológico que inicia la síntesis de los QMes la lipidación de la apoproteína B48 -apoB48-. La apoB48 es una proteína constitutiva derivada de unproceso de edición post-transcripcional exclusivo del enterocito, en el cual un codón de paro trunca pordesaminación enzimática al RNA mensajero de la apoB100, resultando una proteína con solo 48% delmaterial transcripcional de la apoB100. Una vez sintetizada, la apoB48 es trasladada por proteínaschaperonas de los ribosomas hacia el retículo endoplásmico liso. En el retículo endoplásmico liso, con lacolaboración de la MTP “microsomal transport protein”, la apoB48 es transformada en QM ricos entriglicéridos, 85 a 92% de los lípidos contenidos en los QM son triglicéridos. Secuencialmente los QM sonlipidados con fosfolípidos -6% a 12%- y colesterol esterificado -1 a 3%-. Los QM formados en el retículoendoplásmico liso son trasladados hacia el retículo endoplásmico rugoso y al aparato de Golghi en formade vesículas para su secreción hacia la circulación linfática que drena hacia el conducto torácico.
Metabolismo linfo-plasmático de QM. Los QM son secretados por el enterocito hacia lacirculación entero-linfática y de ahí acceden a través del conducto torácico hacia la circulación sistémica. Enla circulación sistémica los QM intercambian con las HDL, apoAI y apoAIV por apoCII, apoCIII y apoE.Gracias a esta remodelación apoprotéica los QM son retenidos e hidrolizados especialmente en loscapilares de los tejidos adiposo y muscular. Al igual que para las VLDL, este proceso es mediado por lainteracción entre la apoE de los QM y los proteoglicanos de la membrana luminal de las células endoteliales,sitio de anclamiento de la LPL. La interacción entre el sitio catalítico de la LPL y su sustrato, la apoCII de losQM, promueve la hidrólisis de los triglicéridos contenidos en los QM. Los ácidos grasos liberados de los
Metabolismo normal de lípidos y lipoproteínas
7

B48B48
QuilomicrónSíntesis. Estructura primaria con apoB48
1
B48B48
QuilomicrónSíntesis. Acoplamiento de triglicéridos por MTP
2
B48B48
QuilomicrónSíntesis. Acoplamiento de fosfolípidos
3
B48B48
AIVAIV
AIAI
TriglicéridosTriglicéridos
ColesterolColesterol
FosfolípidosFosfolípidos
Triglicéridos
FosfolípidosFosfolípidos
ColesterolColesterol
ColesterolColesterol
FosfolípidosFosfolípidos
Triglicéridos
ColesterolColesterol
FosfolípidosFosfolípidos
Triglicéridos
QuilomicrónSíntesis. Acoplamiento y esterificación de CE y otras apo
4
ACAT2
Síntesis de un QuilomicrónImágenes que muestran la estructura primaria de unQM con apoB48 y un QM en formación con laincorporación de triglicéridos por acción de la MTP
Síntesis de un QuilomicrónImágenes que muestran un QM ya con triglicéridosy con la incorporación de fosfolípidos y un QM yacon triglicéridos y fosfolípidos y con laincorporación de colesterol esterificado por acciónde la ACAT2
Cardio-Lipidología
8

B48B48
QuilomicrónIntercambio periférico de apoproteínas
AIVAIV
AIAI
5
CIIICIII
CIICII
EE
Metabolismo normal de lípidos y lipoproteínas
Quilomicrón remanenteInteracción apo CII - lipasa hepática
1
B48E
CII
B48B48EE
CIICII
7
1.- Proteoglicanos Ácidos grasos libres
Glicerol
Síntesis de VLDL
Gluconeog
Síntesis de VLDL
Gluconeog
HLHL
énesisénesis
Quilomicrón remanenteReconocimiento en apoE por LRP1
8
Hepatocito
B48B48B48
EEE
QuilomicrónInteracción apo CII - lipoproteínlipasa
1.- Proteoglicanos
2.- ApoA-V
GPI-HDL-BP1
1
B48
CIII
E
CII
B48B48
CIIICIII
EE
CIICII
6
Ácidos grasos libres
Glicerol
Betaoxidación-ATP
TGS-Lipogénesis
Betaoxidación-ATP
TGS-Lipogénesis
2 2
Metabolismo de un QuilomicrónImágenes que muestran un QM secretado hacia la
circulación, intercambiando apoproteínas; gananciade apoE, apoCII y apoCIII y pérdida de apo AI y
apo AIV y un QM anclado al endotelio por laatracción entre proteoglicanos y apoE y por la
acción de apoAV y GPIHDLBPI. Este anclamientopermite la unión en apoCII de la LPL y su acción
hidrolítica sobre TG con liberación de AGL yglicerol
Metabolismo de un QuilomicrónImágenes que muestran un QM anclado al
endotelio de los capilares hepáticos por la atracciónentre proteoglicanos y apoE. Este anclamiento
permite la unión de la HL y su acción hidrolíticasobre TG con liberación de AGL y glicerol y un
QM remanente reconocido en apoE y removido dela circulación por los receptores tipo LDLR y/o
LRP
LpLLpL
9

QM hacia la circulación, migran hacia el subendotelio y son incorporados por difusión transmembranal o através de transportadores específicos -CD36- a las células adiposas para la síntesis de triglicéridos comofuente energética potencial y/o a las células musculares esqueléticas y/o cardiacas para la formación deATP a través de la vía metabólica de la betaoxidación; un porcentaje variable de ácidos grasos se une aalbumina y es transportado a otros tejidos. De esta forma se cumple el principal objetivo fisiológico delmetabolismo exógeno de los QM, dotar en el estado prandial de ácidos grasos a las células de alto consumoenergético -miocitos esquelético y cardiaco- y a las células de almacenamiento energético -adipocitos-. LosQM depletados de triglicéridos son denominados QM remanentes; en su recirculación hepática, los QMremanentes son rehidrolizados por la lipasa hepática -HL- “hepatic lipase” y eficientemente sonreconocidos en apoE por los receptores LRP1 y LDLR y removidos por el hepatocito para su catabolismohepatobiliar. A diferencia de la apoB100, la apoB48 de los QM no es reconocida por receptores demembrana.
Mathews CK, Van Holde KE y Ahern KG. Metabolismo lipídico 1: ácidos grasos,triacilgliceroles y lipoproteínas. En Bioquímica. 3ª edición 2002; capítulo 18. Editores Chistopher KMathews, K E Van Holde y Kevin G Ahern. Molecular biology of lipoproteinsTsimikas S and Mooser V.and dyslipidemias. In Molecular Basis of Cardiovascular Diseases. Edition 2004; Chapter 21. EditorKenneth R Chien. Lipoprotein metabolism and vascular biology. InChoi BG, Badimon JJ and Fuster V.Therapeutic Lipidology. Edition 2007; chapter 1. Editors Michael H Davidson, Peter P Toth and Kavin CMaki. Definición de una dislipidemia. En Dislipidemias.Aguilar-Salinas CA, Gomez RA y Gómez FJ.De lo clínico a lo molecular. Edición 2008; capítulo 1. Editores Carlos A Aguilar, Rita A Gomez y FranciscoJ Gómez. Absorption and excretion of cholesterol and other sterols. InWang DHQ and Cohen DE.Clinical Lipidology. Edition 2009; chapter 3. Editor Christie M Ballantyne.
Cardio-Lipidología
10

Metabolismo endógeno de lípidosObjetivo fisiológico. El objetivo fisiológico del metabolismo endógeno de lípidos y
lipoproteínas es el transporte de los lípidos hidrofóbicos o neutros -colesterol esterificado y triglicéridos:constituidos por 3 ácidos grasos y glicerol- y de los lípidos anfipáticos o polares -colesterol libre yfosfolípidos: fosfatidilcolina y esfingomielina- del hígado hacia los tejidos periféricos en un medio hídricocomo el plasma.
Mecanismo fisiológico pivote. El mecanismo fisiológico eje del metabolismo
endógeno de lípidos y lipoproteínas, es la síntesis hepática de macromoléculas transportadoras de lípidosdenominadas VLDL “very low density lipoprotein”. Las VLDL están constituidas por un “core” quecontiene los lípidos hidrofóbicos o neutros a transportar -colesterol esterificado y triglicéridos- y unrecubrimiento integrado por los lípidos anfipáticos o polares -fosfolípidos, colesterol libre o noesterificado- y apoproteínas. Estas últimas le proporcionan a la VLDL: estructura, hidrosolubilidadplasmática, sustrato para la acción o inhibición de enzimas hidrolíticas y sitios de reconocimiento porreceptores de lipoproteínas.
Procesos fisiológicos. Con fines didácticos, los procesos fisiológicos del metabolismo
endógeno de lípidos y lipoproteínas pueden segmentarse en: síntesis hepática de VLDL; metabolismoplasmático de lipoproteínas con apoB100; metabolismo celular de LDL vía LDLR; metabolismo celular deLDL-modificado vía receptores depredadores. A continuación se describe cada uno de dichos procesos.
Síntesis hepática de VLDL. El proceso fisiológico que inicia la síntesis de las VLDL es lalipidación de la apoproteína B100 -apoB100-. La apoB100 es una proteína constitutiva de 550 kDa, nointercambiable, codificada por el gen aPOB localizado en el cromosoma 2, formada por 4,536 aminoácidosdispuestos en 4 dominios amfipáticos con 2 hojas β lipofílicas y 2 hélices α. Una vez sintetizada, la apoB100es trasladada por proteí nas chaperonas -heat shock protein- de los ribososmas hacia el retí culoendoplásmico liso. En el retículo endoplásmico liso, con la colaboración de la MTP “microsomaltriglyceride transfer protein”, la apoB100 es lipidada y transformada secuencialmente en: pre-VLDLo VLDL primordial, VLDL2 o VLDL pobre en triglicéridos y VLDL1 o VLDL rica en triglicéridos. LaapoB100 no lipidada por la MTP es proteolizada. El pool hepático de ácidos grasos no esterificados es elprincipal activador de la síntesis de triglicéridos y por ende de la actividad de la MTP, la lipidación de laapoB100 y la síntesis de VLDL. La adición de fosfatidilcolina y de colesterol esterificado -por acción de la
Metabolismo normal de lípidos y lipoproteínas
11

enzima esterificadora de colesterol ACAT2 “acylCoA cholesterol acyl transferasa 2”-, concluye lalipidación de las VLDL. Las VLDL2 y especialmente las VLDL1 ricas en triglicéridos y formadas en elretículo endoplásmico liso son trasladadas para su secreción hacia el retículo endoplásmico rugoso y alaparato de Golghi en forma de vesículas. Este último proceso es un transporte activo dependiente de unaGTPasa -Sar1- y de una proteína recubridora COPII “coatamer protein II”.
Metabolismo plasmático de lipoproteínas apoB100. Las VLDL son secretadas por elhepatocito hacia el espacio de Disse y de ahí hacia la circulación sistémica. Las VLDL no son hidrolizadasen el espacio de Disse gracias a que: la apoE -ligando de los receptores LRP1- “LDLR related protein-1” seencuentra oculta en la estructura terciaria de las VLDL recién secretadas; las VLDL recién secretadas sonricas en apoCIII -apoproteína inhibidora de lipoproteínlipasa-; los capilares hepáticos tienen una bajaconcentración de lipoproteínlipasa -LPL-. En la circulación periférica las VLDL son retenidas ehidrolizadas especialmente en los capilares de los tejidos adiposo y muscular -tejidos productores de LPL-.Este proceso es mediado por la interacción entre la apoE de la VLDL y los proteoglicanos de la membranaluminal de las células endoteliales, sitio de anclamiento de la LPL. La interacción entre el sitio catalítico de laLPL y su sustrato, la apoCII de las VLDL, promueve la hidrólisis de los triglicéridos contenidos en lasVLDL. Los ácidos grasos liberados de las VLDL hacia la circulación, migran hacia el subendotelio y sonincorporados por difusión transmembranal o a través de transportadores específicos -CD36- a las célulasadiposas para la síntesis de triglicéridos como fuente energética potencial y/o a las células muscularesesqueléticas y/o cardiacas para la formación de ATP a través de la vía metabólica de la betaoxidación. Deesta forma se cumple el primer objetivo fisiológico del metabolismo endógeno de las VLDL, dotar deácidos grasos a las células de alto consumo energético -miocitos esquelético y cardiaco- y a las células dealmacenamiento energético -adipocitos-. Recientemente se han identificado otras moléculas que modulanla interacción entre la LPL y la apoCII, entre ellas están la apoAV y la GPIHBP-1 “glycosyl phosphatidylinositol anchored HDL binding protein 1”, ambas con acción sinérgica a la LPL; por el contrario lasangiopoietin-like proteins 3 y 4 “angptl3” y “angptl4”, tienen una acción antagónica a la LPL.
Las VLDL depletadas parcialmente de triglicéridos son denominadas IDL “intermediatedensity lipoprotein”. La remodelación periférica de las IDL favorece la exposición de las apoE y apoB100.En su recirculación hepática, la atracción entre la apoE y los proteoglicanos de la membrana luminal de lossinusoides hepáticos permite la acción hidrolítica de la HL “hepatic lipase” sobre triglicéridos y
Cardio-Lipidología
12

fosfolípidos; secuencialmente, las IDL son reconocidas en apoE y apoB100 por los receptores LRP1 yLDLR y captadas por el hepatocito para su catabolismo hepatobiliar.
Las lipoproteínas apoB100 que escapan al fenómeno hidrolítico y catalítico de “red de pescarhepático”, se caracterizan por un contenido muy bajo de triglicéridos y alto de colesterol esterificado, y sondenominadas LDL “low density lipoprotein”. Las LDL recirculan sistémicamente y son reconocidas porlos receptores LDLR periféricos y/o hepáticos para la incorporación del colesterol esterificado almetabolismo celular. De esta forma se cumple el segundo objetivo fisiológico del metabolismo endógenode las VLDL, dotar de colesterol esterificado a las células para la formación de membranas celulares y/o lasíntesis de hormonas esteroides o ácidos biliares.
Mathews CK, Van Holde KE y Ahern KG. Metabolismo lipídico 1: ácidos grasos,triacilgliceroles y lipoproteínas. En Bioquímica. 3ª edición 2002; capítulo 18. Editores Chistopher KMathews, K E Van Holde y Kevin G Ahern. Molecular biology ofTsimikas S and Mooser V.lipoproteins and dyslipidemias. In Molecular Basis of Cardiovascular Diseases. Edition 2004; Chapter 21.Editor Kenneth R Chien. Lipoprotein metabolism and vascularChoi BG, Badimon JJ and Fuster V.biology. In Therapeutic Lipidology. Edition 2007; chapter 1. Editors Michael H Davidson, Peter P Tothand Kavin C Maki. Definición de una dislipidemia. EnAguilar-Salinas CA, Gomez RA y Gómez FJ.Dislipidemias. De lo clínico a lo molecular. Edición 2008; capítulo 1. Editores Carlos A Aguilar, Rita AGomez y Francisco J Gómez. Human plasma lipoprotein metabolism.Pownall HJ and Gotto Jr AM.Regulation and clearance of apolipoprotein B-containing lipoproteins. In Clinical Lipidology. Edition2009; chapter 1 and chapter 2. Editor Christie M Ballantyne.
Metabolismo celular de colesterol vía LDLR. Antes de iniciar este apartado vale la penarecordar que fueron Joseph Goldstein y Michael Brown los descubridores de gran parte de los conceptosaquí expresados.
El descubrimiento de Goldstein y Brown del receptor para LDL o LDLR y las implicacionesclínicas de dicho descubrimiento, los hicieron merecedores del premio Nobel en Medicina y Fisiología en1985. El colesterol esterificado transportado en las LDL es incorporado al metabolismo celular a partir delreconocimiento de la apoB100 por los receptores LDL de las membranas celulares. La unión apoB100-LDLR es un proceso altamente selectivo que explica 75% de la captación de las LDL; el otro 25% dependede la captación por receptores depredadores de LDL modificado -ver siguiente apartado-.
Metabolismo normal de lípidos y lipoproteínas
13

TriglicéridosTriglicéridos
ColesterolColesterol
FosfolípidosFosfolípidos
ColesterolColesterol
FosfolípidosFosfolípidos
Triglicéridos
B100B100
Very Low Density LipoproteinSíntesis. Acoplamiento de triglicéridos por MTP
2
B100B100
Very Low Density LipoproteinSíntesis. Acoplamiento de fosfolípidos
3
B100B100
Very Low Density LipoproteinSíntesis. Acoplamiento y esterificación de CE y otras apo
CIICII
CIIICIII
EE
4
ACAT2
B100B100
Very Low Density LipoproteinSíntesis. Estructura primaria con apoB100
1
Cardio-Lipidología
Síntesis de una VLDLImágenes que muestran la estructura primaria deuna VLDL con apoB100 y una VLDL enformación con la incorporación de triglicéridos poracción de la MTP
Síntesis de una VLDLImágenes que muestran una VLDL ya contriglicéridos y con la incorporación de fosfolípidos,y una VLDL ya con triglicéridos y fosfolípidos ycon la incorporación de colesterol esterificado poracción de la ACAT2
14
Triglicéridos
FosfolípidosFosfolípidos
ColesterolColesterol
ColesterolColesterol
FosfolípidosFosfolípidos
Triglicéridos

CIICIICIILpLLpL
Very Low Density LipoproteinInteracción apo CII - lipoproteínlipasa
1.- Proteoglicanos
2.- ApoA-V
GPI-HDL-BP1
1 22
B100
CIII
E
5
LpL
Ácidos grasos libres
Glicerol
Betaoxidación-ATP
TGS-Lipogénesis
Betaoxidación-ATP
TGS-Lipogénesis
CIICIICIILpLLpL1
B100E
HL
Intermediate Density LipoproteinInteracción apoCII - lipasa hepática
6
1.- Proteoglicanos Ácidos grasos libres
Glicerol
Síntesis VLDL
Gluconeogénesis
Síntesis VLDL
Gluconeogénesis
B100B100EE
Low Density LipoproteinDepleción de triglicéridos
7 Low Density Lipoprotein
B100
E
8
Células
Metabolismo normal de lípidos y lipoproteínas
Metabolismo de una VLDL a IDLImágenes que muestran una VLDL secretada haciala circulación, anclada al endotelio de los capilarespor la atracción entre proteoglicanos y apoE y por
la acción de apoAV y GPIHDLBPI. Esteanclamiento permite la unión en apoCII de la LPLy su acción hidrolítica sobre TG con liberación deAGL y glicerol y una IDL anclada al endotelio de
los capilares hepáticos por la atracción entreproteoglicanos y apoE. Este anclamiento permite la
unión de la HL y su acción hidrolítica sobre TGcon liberación de AGL y glicerol
Metabolismo de una IDL a LDLImágenes que muestran una IDL depletada de TG
o LDL con un centro lipídico rico en colesterolesterificado, reconocida en apoB100 y removida de
la circulación por los receptores tipo LDLR.
Reconocimiento por LDLR
15

Los receptores para LDL o LDLR son proteínas de 5 dominios ancladas en estructurasmembranales denominadas hoyos de clatrina, constituidas por la proteína clatrina. Las LDL con un “core”conteniendo un promedio de 1,600 moléculas de colesterol esterificado y un recubrimiento de fosfolípidos,colesterol libre y una molécula de apoproteína B100, en conjunto con su receptor y con fragmentos delhoyo de clatrina son incluidas en las células por endocitosis. En el endosoma así formado, el cual contieneLDL, LDLR y moléculas de clatrina, una reducción en el pH permite la disociación del LDLR, el cual serecicla cada 10 minutos durante su vida media de 20 horas o bien es proteolizado por acción de la PCSK9“proprotein convertase subtilisin kexin type 9”. Por fusión membranal, el contenido de LDL de losendosomas es vertido hacia lisosomas. En estos organelos la apoB100 y la clatrina son hidrolizadas haciaaminoácidos y el colesterol esterificado es desterificado. El colesterol libre liberado de los lisosomas esresterificado en el citoplasma por acción de la ACAT2 o bien es transferido hacia el sistema retículoendoplásmico a través de un proceso de reciente caracterización denominado transporte hidrofóbico, en elcual participan 2 proteínas de unión a colesterol, las proteínas de Nieman Pick C2 y C1. La concentración decolesterol no esterificado en las membranas del sistema retículo endoplásmico es la variable que modula amanera de “feed-back” la síntesis y la captación celulares de colesterol. El factor de transcripción SREBP2“sterol regulatory element binding protein 2”, es una proteína unida a la membrana del retículoendoplásmico. Esta proteína es transportada con su proteína ancla o SCAP “SREBP cleavage activatingprotein” hacia el aparato de Golghi. En este organelo por acción de 2 proteasas, S1P y S2P “site 1 protease”y site 2 protease”, el fragmento transcripcional del SREBP2 es liberado y migra hacia el núcleo dondecodifica para la producción de enzimas relacionadas con la síntesis de colesterol, especialmente laHMGCoAR y para la síntesis de LDLR. De esta forma una concentración infra-fisiológica de colesterol enlas membranas del retículo endoplásmico dispara la síntesis y la captación celulares de colesterol. Ensentido opuesto, una concentración fisio o suprafisiológica de colesterol en las membranas del retículoendoplásmico activa a la ACAT2 para la formación y almacenamiento citoplasmático de esteres decolesterol e inhibe la ruptura del binomio SCAP-SREBP2, suprimiendo de esta forma la síntesis y lacaptación celulares de colesterol.
Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425-430.Goldstein JL, Brown MS. History of Discovery. The LDL receptor. Arterioscler Thromb Vasc Biol 2009;29:431-438. Hidrophobic Handoff mechanism. Nobel Laureate Lecture.Goldstein JL, Brown MS.
Cardio-Lipidología
16

American Heart Association Meeting. Chicago Ill. Novembre 15 2010.Nivel fisiológico de LDL. Este concepto encierra gran relevancia, especialmente porqué su
entendimiento le da lógica a la idea de que los niveles supra-fisiológicos de LDL son condición “sin-equanon” para la aterogénesis. En palabras de Goldstein y Brown “es muy difícil producir aterosclerosispor cualquier medio experimental a menos que el nivel plasmático de colesterol este elevado”. Desde 1978los autores referidos demostraron que una concentración de 25mg/dl de LDL en el plasma es suficientepara lograr una concentración intersticial de LDL -10% de la concentración en plasma- que sature a losreceptores celulares de LDL e inhiba la síntesis y la captación de colesterol al bloquear la síntesis de laHMGCoAR y de los receptores de LDL.
En otras palabras 25mg/dl de LDL es una concentración biológicamente activa para cubrir lasfunciones celulares dependientes del colesterol contenido en las LDL. Especies animales como los
roedores, ovejas, bovinos, conejos, perros y gatos tienen valores de LDL 25mg/dl; los camellos, puercos,�
leones y los humanos recién nacidos o cazadores-recolectores tienen valores de LDL entre 25 y 50mg/dl.En condiciones naturales, ninguna de dichas especies desarrolla aterosclerosis. Como se analizará en elapartado de aterogénesis, 25mg/dl de LDL es una concentración 5 veces menor a la concentración de LDLconsiderada poblacionalmente “normal” -125mg/dl-. Estudios de la cinética de LDL han demostrado quela brecha entre el valor fisiológico y el valor poblacional “normal” de LDL no obedece a un incremento enla síntesis de LDL, dicha brecha se explica principalmente por una reducción en el catabolismo de lalipoproteína por represión de los receptores de LDL secundaria a la sobreingesta de colesterol y otrasvariables en investigación; por lo tanto el incremento en el catabolismo de LDL por sobre-expresión de losLDLR es una de las estrategias más eficientes para acercar hacia su valor fisiológico la cifra de LDL.
D Reichl, NB Myant, Brown MS, Goldstein JL. Biologically active Low DensityLipoprotein in human peripheral lymph. J Clin Invest 1978; 61:64-71. .Goldstein JL, Brown MSLipoprotein Receptors: Genetic defense against atherosclerosis. Clinical Research 1982; 30:417-426.
Metabolismo celular de colesterol vía receptores depredadores. En 1979 JosephGoldstein y Michael Brown demostraron in-vitro que el LDL nativo modificado por acetilación erarápidamente captado por macrófagos peritoneales; sin embargo la acetilación de LDL in-vivo nunca hapodido ser demostrada. En los 80´s diversos grupos de investigadores, especialmente el equipo de trabajode Daniel Steiberg y Joseph Wiztum en la Jolla, California, demostraron que la oxidación del LDL durante
Metabolismo normal de lípidos y lipoproteínas
17

B100
Ce
Endosoma
Reciclamiento de LDLR
Hepatocito
3
Hepatocito
Endosoma
2
B100
Ce
LDLRHoyos de Clatrina
Hepatocito
1Ce
LDL
B100
Lisosoma
Hepatocito
4
Endosoma
B100
Ce
B100
Ce
Cardio-Lipidología
Reconocimiento y endocitosis de LDLImágenes que muestran una LDL reconocida enapoB100 por el receptor tipo LDLR anclado en unhoyo de clatrina y una LDL incluida a la célula porendocitosis junto con su receptor y fragmentos delhoyo de clatrina
Reciclamiento de LDLRImágenes que muestran al LDLR abandonando elendosoma para su reciclamiento hacia la membranadel hepatocito y la fusión membranal entre unendosoma y un lisosoma con transferencia delcontenido de LDL hacia este último
18

Metabolismo normal de lípidos y lipoproteínas
NP2 - NP1
Hepatocito
7
C
Retículo EndoplásmicoAparato de Golghi
Transporte Hidrofóbico
C
«Acorazamiento» por NP2-1
Hepatocito
6
NP2 - NP1
C
Movilización intracelular de colesterolImágenes que muestran la proteolísis de la apoB100y la desesterificación del colesterol esterificado de laLDL. Una fracción del colesterol desesterificado esresterificado por acción de la ACAT2 y la fracción
no esterificada es acorazada por las proteínas NP2 yNP1
Inhibición de la síntesis de LDLRImágenes que muestran la transferencia del
colesterol desesterificado del centro hidrofóbico delbinomio NP2-NP1 hacia las membranas del
sistema retículo endoplásmico-aparato de Golghi ycomo la concentración fisiológica de colesterol
desterificado en dichos organelos es la señal quemantiene secuestrado al factor de transcripción
SREBP2 e inhibida la síntesis de LDLR eHMGCoAR
C
SREBP
Gen R-LDL
Gen HMGCoAR
Hepatocito
8
Retículo EndoplásmicoAparato de Golghi C
SCAP
Ce
Proteolisis
Resterificación
ACAT2
Hepatocito
5
Lisosoma
Desterificación
Ce Hidrolasa
B100
AA
Ce
C
19

su exposición a células endoteliales en cultivo generaba LDL oxidado -OxLDL-, el cual era ávidamentecaptado por macrófagos. A partir de este descubrimiento, el grupo de Steinberg-Witzum y otros,especialmente en Oslo, Cleveland, Nueva York y Dallas han demostrado que el OxLDL es unamacromolécula con homología a la pared celular de bacterias gram positivas y a la membrana de célulasapoptóticas, las cuales al igual que el OxLDL, tienen la capacidad de activar en los macrófagos la expresiónde receptores depredadores -SR-A, CD-36, SR-B1, CD-68, SR-PSOX y LOX-1. En condicionesfisiológicas, la acción depredadora de OxLDL del macrófago permite captar en promedio 25% del LDLcirculante y eliminarlo vía el metabolismo reverso de colesterol -ver más adelante-. Así mismo losmacrófagos con participación de moléculas del sistema mayor de histocompatibilidad son células“presentadoras” de OxLDL a los linfocitos; ante este estímulo antigénico los linfocitos producenanticuerpos tipo IgM, IgG y diversas linfocinas dirigidas contra OxLDL.
El OxLDL se genera a partir del LDL nativo por acción de diversos sistemas enzimáticosoxidativos, entre ellos lipoxigenasas, mieloperoxidasas, NADPH-oxidasa y óxido nítrico sintasadesacoplada. Tanto los receptores depredadores como los anticuerpos IgM e IgG e incluso pentraxinascomo la PCR, reconocen al OxLDL en su componente fosfocolina oxidada. Esta misma molécula es elligando de la pared celular bacteriana y de las membranas de células apoptóticas. A la fecha, la hipótesis deque el OxLDL, específicamente su componente fosfocolina oxidada, es un epítope de patronesmoleculares de reconocimento de patógenos o PAMPs y de patrones de reconocimiento de dañomembranal celular o DAMPs, con capacidad de activar una respuesta de inmunidad innata y adaptativa, esuna hipótesis ampliamente validada y que explica otra vía de degradación de LDL.
El desbalance entre la generación de OxLDL y la capacidad de eliminarlo vía el metabolismoreverso de colesterol ocasiona la formación de macrófagos pletóricos de OxLDL -células espumosas-. Laasociación entre la formación de células espumosas y la aterogénesis será tratada ampliamente en el capítulo3 de este libro que trata sobre aterotrombogénesis.
Steinberg D and Witztum J. History of Discovery.Oxidized low-density lipoprotein andatherosclerosis. Arterioscler Thromb Vasc Biol 2010; 30:2311-2316.
Cardio-Lipidología
20
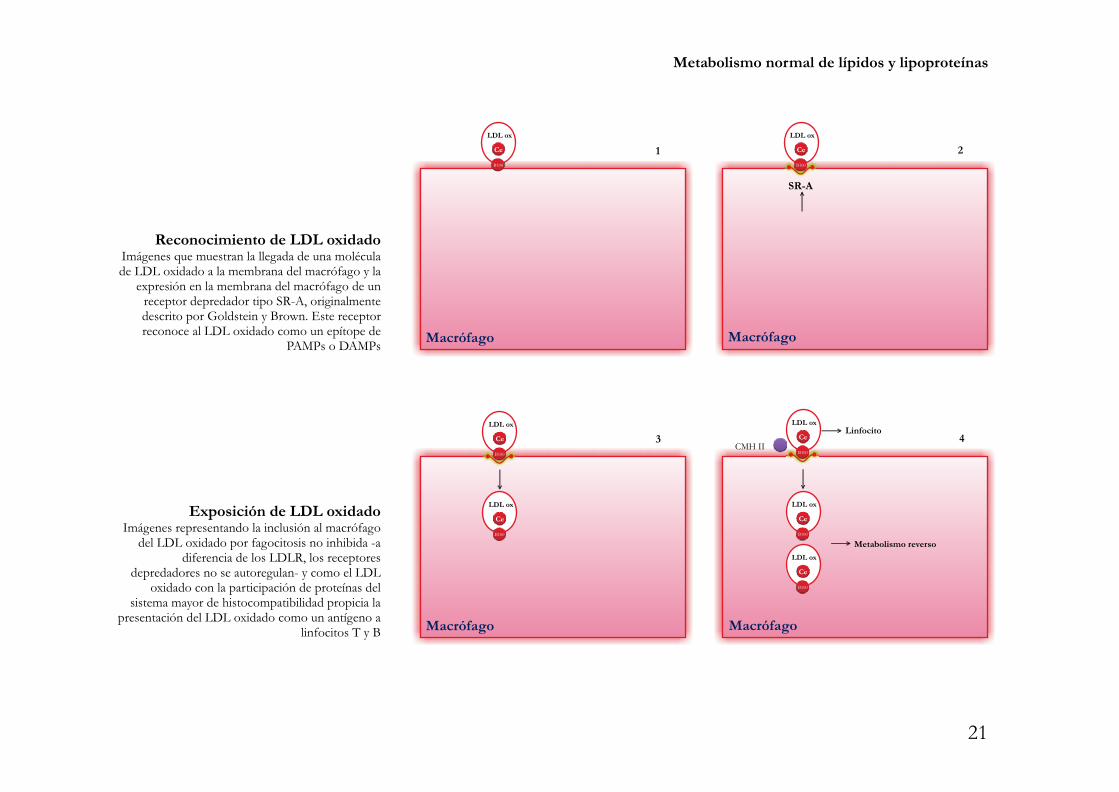
Metabolismo normal de lípidos y lipoproteínas
1
Macrófago
B100
Ce
LDL ox
Macrófago
2
SR-A
Ce
LDL ox
B100
Macrófago
3
B100
Ce
LDL ox
Ce
LDL ox
B100
Reconocimiento de LDL oxidadoImágenes que muestran la llegada de una moléculade LDL oxidado a la membrana del macrófago y la
expresión en la membrana del macrófago de unreceptor depredador tipo SR-A, originalmentedescrito por Goldstein y Brown. Este receptorreconoce al LDL oxidado como un epítope de
PAMPs o DAMPs
Exposición de LDL oxidadoImágenes representando la inclusión al macrófago
del LDL oxidado por fagocitosis no inhibida -adiferencia de los LDLR, los receptores
depredadores no se autoregulan- y como el LDLoxidado con la participación de proteínas del
sistema mayor de histocompatibilidad propicia lapresentación del LDL oxidado como un antígeno a
linfocitos T y B
CMH II
Macrófago
4
Metabolismo reverso
B100
Ce
LDL ox
B100
Ce
LDL ox
Ce
LDL ox
B100
Linfocito
21

Metabolismo reverso de lípidosObjetivo fisiológico. El objetivo fisiológico del metabolismo reverso de lípidos es la
transportación del colesterol acumulado en las células, especialmente en los macrófagos, desde los tejidosperiféricos hacia el hígado para su metabolismo y eliminación hepatobiliar, en un medio hídrico como elplasma. Este proceso fue denominado transporte reverso de colesterol en 1968 por Glomset. Esimportante recordar que el colesterol es una molécula que si bien es sintetizada por las células, no puede serdegradada por las mismas. Por lo tanto el metabolismo reverso de colesterol es uno de los mecanismos másimportantes en los seres vivos para eliminar el exceso de este lípido.
Mathews CK, Van Holde KE y Ahern KG. Metabolismo lipídico 1: ácidos grasos,triacilgliceroles y lipoproteínas. En Bioquímica. 3ª edición 2002; capítulo 18. Editores Chistopher KMathews, K E Van Holde y Kevin G Ahern.
Recientemente se han descrito otros mecanismos para la eliminación del colesterol vía enteral,sin la participación hepatobiliar. El denominado TICE “trans intestinal cholesterol excretion” es unmecanismo alterno al transporte reverso de colesterol “clásico”. Este mecanismo fue recientementerevisado por Brufau y Cols y no es tratado in extenso en este apartado.
Brufau G, Groen AK and Kuipers F. Reverse cholesterol transpor revisited. Contribution ofbiliary versus intestinal cholesterol excretion. Arterioscler Thromb Vasc Biol 2011; 31:1726-1733.
Mecanismo fisiológico pivote. El mecanismo fisiológico eje del metabolismo reverso
de colesterol es la síntesis hepática e intestinal de moléculas aceptoras, transportadoras y eliminadoras decolesterol denominadas HDL “high density lipoprotein”. Las HDL se originan a partir de pro-apoAI,primordio constituido fundamentalmente por la apoproteína AI o apoAI. La apoAI una vez secretada porel hepatocito y/o enterocito, desarrolla su potencial aceptor, transportador y eliminador de colesterol pormedio de los procesos fisiológicos que se describen adelante.
Tsimikas S and Mooser V. Molecular biology of lipoproteins and dyslipidemias. InMolecular Basis of Cardiovascular Diseases. Edition 2004; chapter 21. Editor Kenneth R Chien. Choi BG,Badimon JJ and Fuster V. Lipoprotein metabolism and vascular biology. In Therapeutic Lipidology.Edition 2007; chapter 1. Editors Michael H Davidson, Peter P Toth and Kavin C Maki. Brewer Jr HB.High Density Lipoprotein metabolism. In Clinical Lipidology. Edition 2009; chapter 4. Editor Christie MBallantyne.
Procesos fisiológicos. Con fines didácticos, los tres procesos fisiológicos del
Cardio-Lipidología
22

metabolismo reverso de colesterol pueden segmentarse en: captación de LDL oxidado por los macrófagosvía receptores depredadores; síntesis y expresión de cassettes para la transferencia de colesterol de losmacrófagos hacia las HDL; metabolismo plasmático de las HDL que incluye: eliminación de colesterol víadirecta a través de SR-B1 hepáticos; eliminación de colesterol vía directa a través de LRP1 hepáticos;eliminación de colesterol vía indirecta con mediación de la CEPT. A continuación se describe cada uno dedichos procesos y como corolario se tratarán algunos aspectos relevantes de las HDL más allá de losrelacionados con el transporte reverso de colesterol.
Captación de LDL oxidado por los macrófagos vía receptores depredadores. Como yafue referido en el apartado de metabolismo endógeno, en condiciones fisiológicas, aproximadamente 25%de las LDL son captadas en su forma de LDL oxidadas por los receptores SR-A, CD-36 y otros receptoresdepredadores de los macrófagos.
Steinberg D and Witztum J. History of Discovery. Oxidized low-density lipoprotein andatherosclerosis. Arterioscler Thromb Vasc Biol 2010; 30:2311-2316.
Síntesis y expresión de cassettes transferidores de colesterol. En el macrófago unafracción del colesterol libre derivado del catabolismo de las LDL oxidadas es esterificado por acción de laenzima ACAT1 “acylCoA cholesterol acyl transferasa 1” y almacenado como “gotas lipídicas” en elcitoplasma, la fracción no esterificada es transferida hacia las HDL. El acúmulo intracelular de colesterollibre y oxisterol -colesterol oxidado- en el macrófago es el “gatillo” para la activación de los receptoresnucleares LXR “liver X receptors” y su heterodimerización con los RXR “retinoid X receptors”. Estebinomio de factores de transcripción nuclear codifica para la síntesis de los cassettes ABCA1 “ATP bindingcassette A1” y ABCG1 “ATP binding cassette G1”. Ambos cassettes propician 75% del flujo centrífugo decolesterol del macrófago hacia las HDL. El ABCA1 es el principal cassette transferidor de colesterol; elmecanismo de este proceso involucra diversas moléculas que regulan el tráfico de colesterol libre desde losendosomas hacia la membrana celular a través del sistema retículo endoplásmico-aparato de Golghi yconstituyen un verdadero “reóstato celular” de colesterol. La unión de la apoA1 al ABCA1 y a la ATP-sintasa de la membrana celular, con consumo de ATP, promueve el flujo centrífugo de colesterol libre de lasmembranas del macrófago hacia la apoA1. Este proceso puede llevarse a cabo en la membrana celular o enlas membranas de los endosomas por internalización del complejo ABCA1-apoA1. En este último, elABCA1 es reciclado y la apoA1 con su cargo de colesterol libre es retroendocitada.
Metabolismo normal de lípidos y lipoproteínas
23

B100
Ce
1
SR CD-36 Macrófago
Lisosoma
3
C
Retículo EndoplásmicoAparato de Golghi
C
NP2 - NP1
C
SR CD-36 Macrófago
Lisosoma
4
Retículo EndoplásmicoAparato de Golghi
LXR-RXR
Gen ABCA1-ABCG1
ABCA1SR CD-36 Macrófago
CC
C
Metabolismo reverso de colesterolImágenes que muestran la inclusión del LDLoxidado al macrófago y su inclusión a un lisosomacon la proteolísis de la apoB100 y ladesesterificación del colesterol esterificado del LDL.Una fracción del colesterol desesterificado esresterificado por acción de la ACAT2
Metabolismo reverso de colesterolImágenes que muestran el acorazamiento delcolesterol libre o desesterificado por las proteínasNP2 y NP1 y su transferencia del centrohidrofóbico hacia las membranas del sistemaretículo endoplásmico-aparato de Golghi. En elmacrófago esta es la señal induce la actividad de losfactores de transcripción LXR-RXR para la síntesisde cassettes transferidores de colesterol
Cardio-Lipidología
SR CD-36 Macrófago
AA
CProteolisis
Resterificación
ACAT2
Ce
2
Lisosoma
Desterificación
Ce Hidrolasa
LDL ox
B100
Ce
24
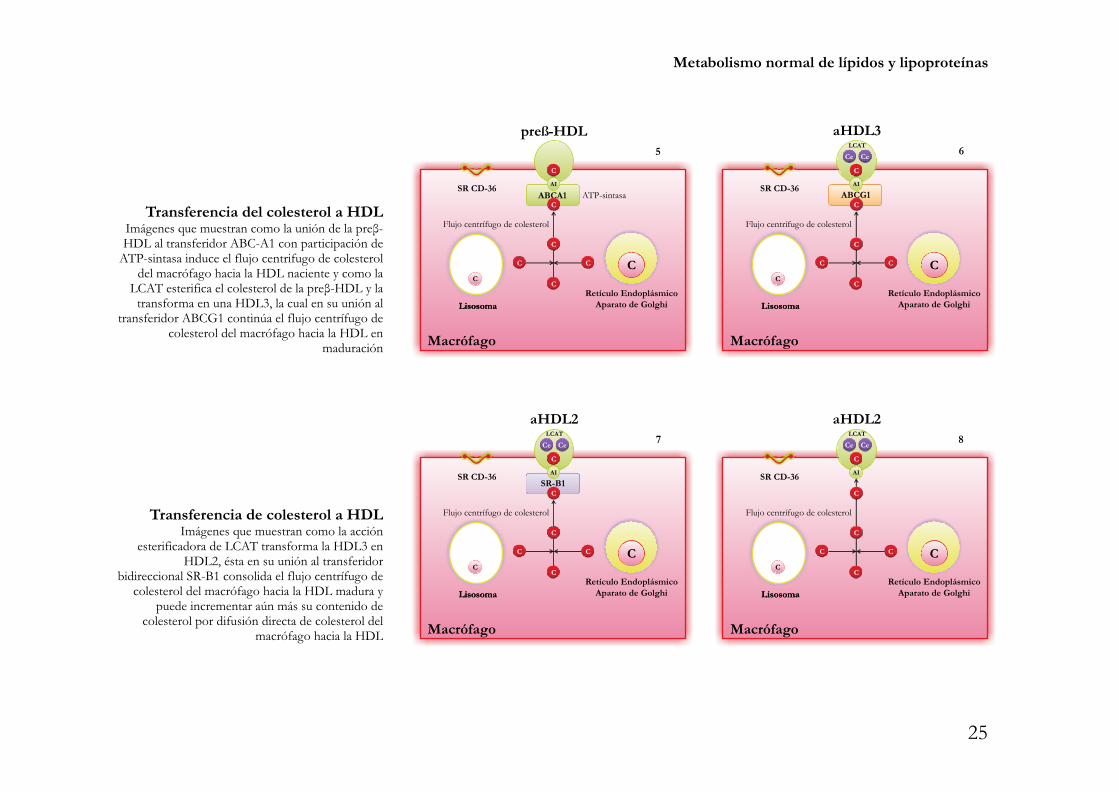
Metabolismo normal de lípidos y lipoproteínas
LisosomaLisosoma
Macrófago
5
C
Retículo EndoplásmicoAparato de Golghi
C
preß-HDL
Flujo centrífugo de colesterol
ATP-sintasaSR CD-36
ABCA1
C C
C
C
AI
C
C
6
aHDL3
CeCe
LisosomaLisosoma
Macrófago
C
Retículo EndoplásmicoAparato de Golghi
C
Flujo centrífugo de colesterol
SR CD-36
C C
C
C
AI
C
C
LCAT
ABCG1
7
aHDL2
CeCe
LisosomaLisosoma
Macrófago
C
Retículo EndoplásmicoAparato de Golghi
C
Flujo centrífugo de colesterol
SR CD-36
C C
C
C
AI
C
C
LCAT
SR-B1
8
aHDL2
CeCe
LisosomaLisosoma
Macrófago
C
Retículo EndoplásmicoAparato de Golghi
C
Flujo centrífugo de colesterol
SR CD-36
C C
C
C
AI
C
C
LCAT
Transferencia del colesterol a HDLImágenes que muestran como la unión de la preβ-HDL al transferidor ABC-A1 con participación de
ATP-sintasa induce el flujo centrifugo de colesteroldel macrófago hacia la HDL naciente y como la
LCAT esterifica el colesterol de la preβ-HDL y latransforma en una HDL3, la cual en su unión al
transferidor ABCG1 continúa el flujo centrífugo decolesterol del macrófago hacia la HDL en
maduración
Transferencia de colesterol a HDLImágenes que muestran como la acción
esterificadora de LCAT transforma la HDL3 enHDL2, ésta en su unión al transferidor
bidireccional SR-B1 consolida el flujo centrífugo decolesterol del macrófago hacia la HDL madura y
puede incrementar aún más su contenido decolesterol por difusión directa de colesterol del
macrófago hacia la HDL
25

El ABCG1 es un cassette menos estudiado que el ABCA1, el cual, al igual que el ABCA1promueve el flujo centrífugo de colesterol libre y también de fosfolípidos del macrófago hacia las αHDL.La función del reóstato de colesterol en el macrófago y el balance entre flujo y eflujo mantieneconcentraciones fisiológicas de colesterol en las membranas celulares e inhibe la exposición y activación delos receptores TLR “toll like receptors” localizados en “rafts” de la membrana celular y endosomal-lisosomal. Este mecanismo mantiene inactivas a las cadenas enzimáticas de activación de los factores detranscripción pro-inflamatorios como el NFkB. En resumen, el acúmulo de colesterol y oxisterol en elmacrófago activa la heterodimerización de los factores de transcripción LXR-RXR y la síntesis de lostransferidores ABCA1 y ABCG1. Ambos cassettes facilitan el flujo centrífugo de colesterol y limitan en elmacrófago la exposición de los receptores TLR a sus ligandos, impidiendo la activación de factores detranscripción pro-inflamatorios como el NFkB. De esta forma el papel depredador de colesterol de lasHDL evita la formación de células espumosas; cuando este mecanismo disfunciona -por causas genéticascomo la enfermedad de Tangier o por causas ambientales como el exceso en el consumo de colesterol-,inicia la actividad inflamatoria a partir del macrófago convertido en célula espumosa, como se verá en elcapítulo 3 que concierne a la aterogénesis.
Schmitz G and Grandl M. History of Discovery. The molecular mechanisms of HDL andassociated vesicular trafficking mechanisms to mediate cellular lipid homeostasis. Arterioscler ThrombVasc Biol 2009; 29:1718-1722. . Role of HDL, ABCA1, andYvan-Charbet L, Wang N and Tall ARABCG1 transporters in cholesterol efflux and inmune responses. Arterioscler Thromb Vasc Biol 2010;30:139-143.
Existen dos mecanismos más que contribuyen al flujo reverso de colesterol. La difusióntransmembranal pasiva de colesterol por gradiente de concentración y la transferencia de colesterol delmacrófago hacia las HDL a través del receptor depredador SR-B1 “scavenger receptor B type 1”. Esteúltimo receptor tiene la capacidad de transferir colesterol de las células hacia las HDL y visceversa; estacapacidad depende de la localización del SR-B1. En los macrófagos, el SR-B1 promueve el flujo centrífugode colesterol y en los hepatocitos promueve la captación hepática y la eliminación hepatobiliar del lípido.Amén de su participación en el flujo reverso de colesterol, el SR-B1 y su interacción con las HDL regulavarios de los mecanismos cardioprotectores de la lipoproteína; por ejemplo en la célula endotelial la uniónapoAI-SR-B1 promueve la producción de óxido nítrico.
Cardio-Lipidología
26

Saddar S, Mineo C and Shaul PW. Signaling by the high affinity HDL receptor scavengerreceptor B type 1. Arterioscler Thromb Vasc Biol 2010; 30:144-150.
Metabólismo plasmático de las HDL.Una vez analizados los procesos que permiten lacaptación del exceso de colesterol-LDL circulante por los macrófagos y la síntesis de transferidores decolesterol de los macrófagos hacia las HDL circulantes, analizaremos como estas lipoproteínas de altadensidad concluyen el proceso de transporte reverso de colesterol a través de los siguientes 3 mecanismos.
. Eliminación de colesterol vía directa a través de SR-B1 hepáticos. Como se refirió alprincipio de este apartado, las HDL nacientes o pro-apoAI están constituidas fundamentalmente por laapoproteína AI. La apoAI, a diferencia de la apoB100, es una proteína intercambiable, codificada en elcromosoma 11, constituida por 243 aminoácidos dispuestos en una región globular y 11 hélices α lipofílicas.La apoAI una vez secretada por el hepatocito y/o enterocito, desarrolla secuencialmente su potencialaceptor, transportador y eliminador de colesterol. En su circulación sistémica, la interacción de la apoA1con el transferidor ABCA1 y la ATP-sintasa de membrana de los macrófagos, promueve por el mecanismoya analizado más del 50% del flujo centrífugo de colesterol hacia la apoA1 y la transforma en una preβ-HDL. La apoA1 de la preβ-HDL activa a la enzima LCAT “lecithin cholesterol acyl transferase”-proteí naasociada a la lipoproteína- y con su participación, el colesterol libre contenido en la preβ-HDL esesterificado y concentrado en el “core” hidrofóbico de la lipoproteí na, transformándola en una αHDLinmadura o αHDL3 con capacidad para captar más colesterol libre de los macrófagos. Así la αHDL3 alinteractuar de nuevo con los macrófagos y aceptar colesterol libre y fosfolí pidosa través de los cassettesABCG1 y/o los receptores SR-B1, promueve al máximo su capacidad aceptora de colesterol. Cabemencionar que el papel de los ABCG1 en el humano aún es controversial. Las αHDL maduras o αHDL2,en su circulación sistémica son remodeladas por acción de las proteí nas asociadas PLTP “phospholipidtransfer protein” y CEPT “cholesterol ester transfer protein” -ver adelante- y en su circulación hepáticason hidrolizadas por la HL “hepatic lipase” y reconocidas por los SR-B1, localizados en la membrana basal-lateral de los hepatocitos, los cuales promueven el flujo selectivo del colesterol esterificado del “core” de laαHDL2 hacia el hepatocito para su metabolismo y/o eliminación hepatobiliar en forma de ácidos biliares.A diferencia de los receptores LRP1 o LDLR que captan por endocitosis a la lipoproteína completa, los SR-B1 sólo captan el colesterol esterificado contenido en las αHDL2, facilitando de esta forma la recirculaciónde la lipoproteína. Por este mecanismo denominado vía directa del flujo reverso de colesterol se elimina
Metabolismo normal de lípidos y lipoproteínas
27

1
aHDL2
CeCe
AI
LCAT
SR-B1
Ce
Hepatocito
SR-B
1
2
AI
SR B1
Hepatocito
SR-B
1
Ce
-
Ce Ce Ce
2
CeCe
Ce
Hepatocito
E
LRP1
Endocitosis
Eliminación hepática de colesterolImágenes que muestran como la HDL2 transfiereselectivamente hacia el hepatocito su contenido decolesterol gracias a la participación del SR-B1, ycomo este mismo receptor localizado en lamembrana hepato-biliar elimina el colesterol haciael aspecto biliar del hepatocito
Cardio-Lipidología
Eliminación hepática de colesterolImágenes que muestran como la HDL2 esreconocida en apoE y endocitada por el hepatocito,y como una vez endocitadala lipoproteína escatabolizada en el interior del hepatocito
1
aHDL2
CeCe
Ce
Hepatocito
E
LRP1
28
Absorción selectiva de colesterol
Endocitosis de HDL
Ce
Catabolismo de HDL
Metabolismo y/o Eliminación

aproximadamente el 75% del colesterol acumulado en los tejidos periféricos.. Eliminación de colesterol vía directa a través de LRP1 hepáticos. En forma similar al
proceso descrito previamente, un pequeño porcentaje de colesterol se elimina por HDL que contienenapoA1 y apoE. Estas apoproteínas apoA1-E son reconocidas en apoE por los receptores hepáticos LRP1de igual forma que lo son las IDL con apoE -ver apartado de metabolismo endógeno-.
. Eliminación de colesterol vía indirecta con mediación de la CEPT. En promedio un20% de colesterol acumulado en los tejidos periféricos se elimina por esta vía. Las αHDL2, a través de suproteína asociada CEPT “cholesterol ester transfer protein”, transfieren colesterol esterificado hacia lasapoproteínas B100 -en condiciones fisiológicas preferencialmente a las LDL-, a cambio, las αHDL2pueden ser cargadas con triglicéridos . De esta forma el colesterol esterificado transferido de las αHDL2hacia las lipoproteinas apoB100 es eliminado con mayor rapidez al ingresar al metabolismo endógenorevisado en el apartado previo. Las HDL pobres en colesterol y ricas en triglicéridos son hidrolizadas por laslipasas LPL, EL “endothelial lipase” y HL y recicladas y/o catabolizadas por vía hepática y/o renal.
Aguilar-Salinas CA, Gomez RA y Gómez FJ. Definición de una dislipidemia. EnDislipidemias. De lo clínico a lo molecular. Edición 2008; capítulo 1. Editores Carlos A Aguilar, Rita AGomez y Francisco J Gómez. . . In ClinicalBrewer Jr HB High Density Lipoprotein metabolismLipidology. Edition 2009; chapter 4. Editor Christie M Ballantyne.
Recientemente se ha descrito que la CEPT es una proteína asociada a las αHDL que notransfiere únicamente colesterol esterificado de las αHDL2 hacia las lipoproteínas con apoB100-transporte heterotípico-. La CEPT también transfiere colesterol de las αHDL3 “inmaduras” hacia lasαHDL2 “maduras”. Este transporte denominado homotí picofavorece la maduración de una fracción delas αHDL3 hacia αHDL2 y la transformación de otra fracción de las αHDL3 hacia preβ-HDL. En suconjunto el transporte homotí picosinergiza al transporte heterotí picoe incrementa el flujo de colesteroldel macrófago hacia las HDL. Como se verá en la sección de tratamiento, la inhibición completa de ambostipos de transporte vía CEPT -homotípico y heterotípico- podría ser una estrategia menos eficiente que lainhibición parcial o modulación, con inhibición específica del transporte heterotípico sin afección delhomotípico.
Niesor EJ. Different effects of compounds decreasing cholesteryl ester transfer proteinactivity on lipoprotein meabolism. Current Opinion in Lipidology 2011; 22:000-000. Khera AV, Cuchel
Metabolismo normal de lípidos y lipoproteínas
29

CEPT. Transferencia homotípicaImágenes que muestran la acción de la CEPT en latransferencia homotípica de colesterol de las HDL3hacia las HDL2 y como la acción de la CEPT en latransferencia homotipica permite que una fracciónde HDL3 por acción de la proteínatransferidoramadure y se transforme en una HDL2y otra fracción “involucione” hacia preβ-HDL conalta capacidad aceptora de colesterol. .
CEPT. Transferencia heterotípicaImágenes que muestranla acción de la CEPT en latransferencia heterotípica de colesterol de las HDL2hacia las LDL -condición fisiológica- y como laHDL2 que transfirió colesterol hacia las LDL puedeadoptar de nuevo su roll de HDL3 aceptora decolesterol del macrófago
Cardio-Lipidología
1
AI
Ce
B100E
Ce
Ce
Ce
HeterotípicoCETP Ce
Ce
HDL2
Ce
CETP Ce Homotípico2
AI
HDL3
Ce Ce
AI
Ce Ce
Ce
CETP
HDL2
Ce Homotípico1
AI
HDL3
Ce Ce
AI
HDL3
LDLR
AI
Ce
B100E
Ce
Ce
Ce
HeterotípicoCETP Ce
Ce
HDL2
Ce
Ce Ce
2
30AI
Ce Ce
CeHDL2
AI
pre HDL
Ce
LDL LDL

M, de la Llera-Mora M et al. Cholesterol efflux capacity, high-density lipoprotein function, andatherosclerosis. N Engl J Med 2011; 364:127-35.
HDL más allá del transporte reverso. Las lipoproteínas denominadas como HDL
no sólo participan en el transporte reverso de colesterol, conservar esta visión de las HDL sería un granerror conceptual. Por estudios proteómicos con espectrometría de masas, se ha descrito queindependientemente de las apoproteínas: apoA1 con 6 isoformas, apoAII, apoAIV con 6 isoformas, apoEcon 6 isoformas, apo M con 2 isoformas, ApoCII, ApoCIII con 3 isoformas; las HDL contienen variasdecenas de proteínas denominadas asociadas. En un proteoma normal, se han descrito entre las másimportantes a las siguientes proteínas asociadas: PAFAH “platelet activating factor acetyl hidrolase”,sPLA2 “secretory phospho lipase A2”, MPO “mielo peroxidase”, SAA1 “serum amiloid A1”, PLTP“phospholipid transfer protein”, PON1 “paroxonase 1”, CEPT “cholesterol ester transfer protein”, LCAT“lecithin cholesterol acyl transferase”, etc. A la fecha se propone que la identidad de las HDL sea clasificadapor el contenido de sus proteínas asociadas y que estas sean agrupadas en “clusters funcionales”: proteínasasociadas a metabolismo y transporte de lípidos, proteínas asociadas a inmunidad innata y sistema decomplemento, proteínas-factores de crecimiento, proteínas de unión a hormonas, proteínas asociadas conla hemostasis, etc. De esta forma, más allá de la visión clásica de las HDL, el conocimiento extenso yprofundo del proteoma de las HDL en condiciones de salud y enfermedad en conjunto la aplicación deestudios funcionales como el flujo de colesterol del macrófago hacia las HDL, nos permitirá en un futuroentender mejor, entre otras, la paradoja actual, aún no resuelta entre el efecto cardioprotector en losestudios epidemiológicos de los niveles espontáneamente altos de HDL vs el potencial y esperado efectocardioprotector de los niveles farmacológicamente elevados de HDL.
Davidsson P, Hulthe J, Fagerberg B and Camejo G. Proteomics of apolipoproteins andassociated proteins from plasma High-Density Lipoproteins. Arterioscler Thromb Vasc Biol 2010; 30:156-163.
Metabolismo normal de lípidos y lipoproteínas
31
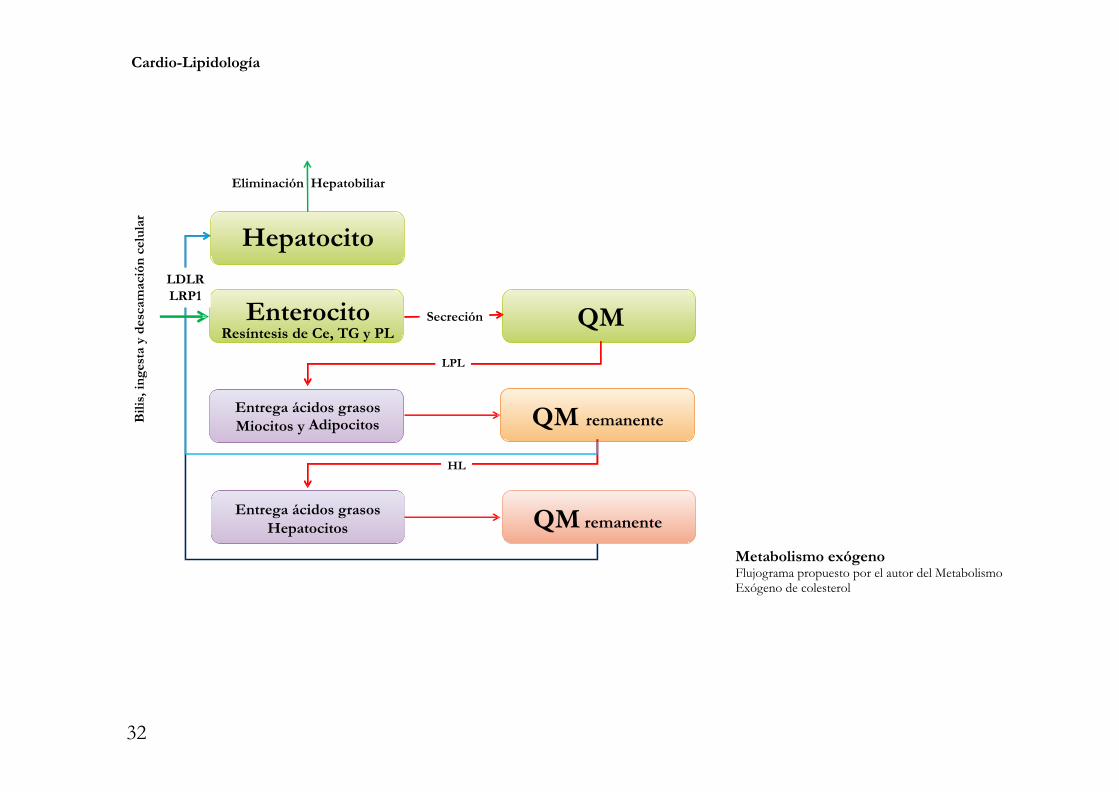
Cardio-Lipidología
EnterocitoResíntesis de Ce, TG y PL
Entrega ácidos grasosMiocitos y Adipocitos
Entrega ácidos grasosHepatocitos
QM remanente
QM remanente
QM
LPLLPL
HLHL
SecreciónSecreción
LDLRLRP1
Bili
s,in
ges
tay
des
cam
ació
nce
lula
r
Hepatocito
Eliminación Hepatobiliar
Metabolismo exógenoFlujograma propuesto por el autor del MetabolismoExógeno de colesterol
32

Metabolismo normal de lípidos y lipoproteínas
Hígado
Entrega ácidos grasosMiocitos y Adipocitos
Entrega colesterolCélulas: membranas y hormonas
Entrega ácidos grasosHepatocitos
IDL
LDL
OxLDLMacrófago
SR-A CD36
VLDLSR-B1LRP1
LDLR
TRC vía HDL
LPLLPL
HLHL
SecreciónSecreción
LDLRLRP1
Eliminación Hepatobiliar
Metabolismo endógenoFlujograma propuesto por el autor del Metabolismo
Endógeno de colesterol
33

Cardio-Lipidología
HígadoIntestino
Recoge colesterolMacrófago
Recoge colesterolMacrófagos
Recoge colesterolMacrófagos
preß-HDL
HDL3
HDL2
apoA1SR-B1LRP1
SR-B1-Difusión
ABCA1ABCA1
ABCG1ABCG1
SecreciónSecreción
Eliminación Hepatobiliar
LCAT
LCAT
LCAT-PLTP
LDL
CEPT hetero
CEPT homo
LDLRLRP1
Metabolismo reversoFlujograma propuesto por el autor del MetabolismoReverso de colesterol
34

DislipidemiasTrastornos primarios, secundarios y mixtos
Capítulo 2

GeneralidadesLiteralmente hablando Dislipidemia es toda alteración en los lípidos, sea esta pro-aterogénica,
anti-aterogénica o neutra. Sin embargo la liga contemporánea entre lípidos, aterosclerosis y enfermedadcardiovascular le ha dado al termino dislipidemia un sentido aterogénico; en otras palabras en el contextoactual, dislipidemia se conceptualiza como toda aquella alteración de los lípidos sanguíneos que incrementael riesgo de aterosclerosis y enfermedad cardiovascular.
En este apartado abordaremos la clasificación de las dislipidemias con un enfoque etiológico.Por su importancia, en primer orden se enuncian las principales características de la clasificación deFredrickson, Levy y Less; a continuación abordaremos las dislipidemias asociadas a incremento y reducciónde LDL, las dislipidemias asociadas a incremento y reducción de triglicéridos, las dislipidemias asociadas aincremento de LDL y triglicéridos y finalmente se analizarán las dislipidemias asociadas a incremento yreducción de HDL. Por su alta frecuencia y gran impacto aterogénico, las dislipidemias en México,especialmente las relacionadas a obesidad central y síndrome metabólico son tratadas con amplitud en unapartado especial.
Este capítulo será fácilmente comprensible en tanto se tengan las bases fisiológicas revisadasen el capítulo previo, por lo que recomendamos estudiar y comprender los aspectos relacionados con elmetabolismo normal de los lípidos en sus apartados de metabolismo exógeno, endógeno y reverso, esteejercicio permitirá una óptima integración entre el conocimiento de la fisiología y la comprensión de lafisiopatología de los lípidos sanguíneos.
Clasificación de Fredrickson. Esta clasificación propuesta en el año 1967 por
Fredrickson, Levy y Less y adoptada por la Organización Mundial de la Salud, se basó en la descripción delaspecto visual de la muestra de plasma tomada en ayuno, después de 18 a 24 horas de refrigeración; lainspección 100% descriptiva permite suponer un incremento de QM por la presencia de una capa superior“cremosa” o bien un incremento de VLDL por un plasma de aspecto turbio; dicho análisis escomplementado con ultracentrifugación del plasma y electroforesis de lipoproteínas.
Con esta metodología las Dislipidemias fueron clasificadas por Fredrickson y la OrganizaciónMundial de la Salud en los siguientes fenotipos.
Tipo I. Aspecto cremoso en la capa superior con aspecto claro de las capas inferiores. Implicaincremento de QM con colesterol total normal y triglicéridos incrementados.
Tipo IIA. Aspecto claro. Implica incremento de LDL con VLDL normal con colesterol total
Cardio-Lipidología
36

incrementado y triglicéridos normales.Tipo IIB. Aspecto claro o discretamente turbio. Implica incremento de LDL y VLDL con
colesterol total y triglicéridos incrementados.Tipo III. Aspecto cremoso en la capa superior delgada con capas inferiores turbias. Implica
incremento de IDL con colesterol total y triglicéridos incrementados.Tipo IV. Aspecto turbio. Implica incremento de VLDL con colesterol total normal o
incrementado y triglicéridos incrementados.Tipo V. Aspecto cremoso en la capa superior con aspecto turbio de las capas inferiores.
Implica incremento de QM y VLDL con colesterol total y triglicéridos incrementados.Esta clasificación es relevante ya que sin tener un carácter diagnóstico etiológico y sin incluir a
las HDL, fue el punto de partida para la estructuración en el diagnóstico de los trastornos en lípidos ylipoproteínas.
Fredrickson DS, Levy RI, Less RS. Fat transport in lipoproteins; an integrated approach tomechanisms and disorders. N Engl J Med 1967; 276:34-43. Evaluación del riesgo y reducciónGotto Jr AM.de los factores de riesgo. En diagnóstico y tratamiento actual de los trastornos lipídicos. 3ª edición; capítulo3. Editor Antonio M. Gotto. Dyslipidemia. In Atlas of Cardiovascular RiskGornik HL and Plutzky J.Factors. Edition 2006; chapter 8. Editor J Michael Gaziano.
Dislipidemias
37

Incremento de LDLDislipidemias primarias y secundarias. Efecto aterogénico alto.Estos trastornos tienen un fenotipo IIA de la clasificación de Fredrickson, Levy y Less con
colesterol total 200mg/dl y LDL 130mg/dl, con triglicéridos 150mg/dl y HDL 60mg/dl. Se� � � �
subclasifica como hipercolesterolemia grave aquella con colesterol total 300mg/dl y LDL 160mg. Los� �
trastornos primarios del metabolismo de los lípidos que ocasionan estas dislipidemias son los siguientes.
Alteración en el receptor de LDLHipercolesterolemia Familiar Homocigota. Es un trastorno autosómico dominante que
afecta ambos alelos del gen del receptor LDL -LDLR-, determina alteraciones cuantitativas y/o cualitativasen la totalidad de dichos receptores, su prevalencia es 1/1,000,000 individuos y su fenotipo lipídico secaracteriza por cifras de LDL entre 500mg/dl y 1,000mg/dl, asociadas a la presencia de arco corneal,xantomas tendinosos y aterosclerosis precoz manifestada por eventos cardiovasculares en la infancia -sehan reportado casos de infarto miocárdico a los 18 meses de edad- y adolescencia.
Hipercolesterolemia Familiar Heterocigota. Al igual que la previa es un trastornoautosómico dominante que afecta sólo un alelo del gen del receptor LDL, determina alteracionescuantitativas y/o cualitativas en aproximadamente 50% de dichos receptores, su prevalencia es 1/500individuos y su fenotipo lipídico se caracteriza por cifras de LDL entre 250mg/dl y 500mg/dl, asociadas a lapresencia de arco corneal, xantomas tendinosos y aterosclerosis precoz manifestada por eventoscardiovasculares en la etapa adulta temprana. En promedio 5% de los individuos con infarto miocárdicoantes de los 60 años de edad tienen este trastorno.
Se han descrito más de 600 mutaciones en el gen del receptor LDL, las cuales han sidoagrupadas en 5 clases. Clase 1. Síntesis nula del receptor. Clase 2. Trastornos en la traslación del receptor delretículo endoplásmico hacia el aparato de Golghi y/o la membrana celular. Clase 3. Trastornos en la unióndel receptor con apoB100 y reconocimiento normal de apoE. Clase 4. Trastornos en la internalización delreceptor por defectos en su unión a los hoyos de clatrina. Clase 5. Trastornos en la disociación entre elreceptor y el endosoma. En México Robles-Osorio y Cols., publicaron 46 casos de HipercolesterolemiaFamiliar.
Alteración en la proteína adaptadora -ARH-Hipercolesterolemia Autosómica Recesiva. Es un trastorno autosómico recesivo que
afecta al -los- gen -es- de la proteína adaptadora de unión entre el receptor LDL y la proteína clatrina o
Cardio-Lipidología
38

proteína ARH-l, determina alteraciones en la unión de LDL con su receptor, su prevalencia es muy baja y sufenotipo lipídico depende de su expresión homo o heterocigota, con cifras de LDL entre 250mg/dl y1,000mg/dl, asociadas a la presencia de arco corneal, xantomas tendinosos y aterosclerosis precoz,manifestada por eventos cardiovasculares en la adolescencia o en la edad adulta. En México Canizales yCols., publicaron 1 familia con esta alteración.
Alteración en la apoproteína B100Es un trastorno que afecta al gen de la apoB100, determina la síntesis anormal de apoB100, la
cual es disfuncional para su unión con el receptor LDL, siendo normal la unión entre dicho receptor yapoE, su prevalencia es variable, en promedio 1/750 individuos, su fenotipo lipídico es similar al de laHipercolesterolemia Familiar Heterocigota, asociado también a la presencia de arco corneal, xantomastendinosos y aterosclerosis precoz. Se han descrito 3 tipos de mutaciones en este trastorno. En MéxicoRobles-Osorio y Cols., publicaron 1 caso de esta alteración.
Alteración -ganancia de función- en la PCSK9Hipercolesterolemia Autosómica Dominante. Es un trastorno que afecta el gen de la
PCSK9 -proprotein convertase subtilisin kexin type 9-, determina la expresión a la alta en la función de laPCSK9 -proteasa que proteoliza al receptor LDL-, por lo tanto, el incremento de función de la PCSK9provoca un mayor catabolismo y reducción de receptores LDL con incremento de LDL circulante, suprevalencia es muy baja y su fenotipo lipídico se caracteriza por cifras de LDL entre 250mg/dl y1,000mg/dl, asociadas a la presencia de arco corneal, xantomas tendinosos y aterosclerosis precoz,manifestada por eventos cardiovasculares en la adolescencia o en la edad adulta. En México Canizales yCols., publicaron 1 familia con la mutación de sobre-expresión de la PCSK9.
SitoesterolemiaEs un trastorno que afecta los genes de los cassettes ABCG5 y ABCG8 -cassettes
transportadores de esteroles-, determina alteraciones en la absorción intestinal y la excreción biliar deesteroles neutros; específicamente en la sitoesterolemia se incrementa la absorción intestinal y se reduce laexcreción biliar de esteroles neutros derivados de plantas con incremento en su concentración plasmática,lo cual ocasiona la regulación a la baja de los receptores LDL e incremento secundario en el nivel de LDL en
el plasma, su prevalencia es muy baja y su fenotipo lipídico se caracteriza por cifras de LDL 250mg/dl e�
incremento de sitoesteroles, asociados a la presencia de arco corneal, xantomas tendinosos y aterosclerosis
Dislipidemias
39

precoz, manifestada por eventos cardiovasculares en la adolescencia y edad adulta.
Hiperlipidemia familiar combinadaOriginalmente descrita por Goldstein y Brown, es un trastorno oligogénico -varios genes-, con
influencia ambiental, se ha propuesto como mecanismo un incremento en la expresión de los genesreguladores de la síntesis de VLDL y de los genes moduladores del catabolismo de dichas lipoproteínas,específicamente de apoB100 y apoCIII, su prevalencia es 1/100-200 individuos en la población general y
� 1/10-15 individuos con enfermedad cardiovascular precoz, su fenotipo lipídico se caracteriza por
elevaciones percentila 90 en las cifras de apoB100 - 110mg/dl- y elevaciones variables en las cifras de� �
colesterol - 200mg/dl- y triglicéridos - 150mg-; la elevación de la apoB100 y el incremento de la� �
fracción densa y pequeña de las LDL -patrón β- son hallazgos característicos de esta dislipidemia, la cual nose asocia a la presencia de arco corneal ni xantomas tendinosos. Existe una coexistencia no causal condisfunción metabólico-hemodinámica secundaria a resistencia a la insulina e incremento en el riesgo deaterosclerosis precoz, manifestada por eventos cardiovasculares en la edad adulta con agregación familiar.
Hipercolesterolemia familiar poligénicaEs un trastorno con agregación familiar sin una alteración genética identificada, su fenotipo
lipídico se caracteriza por cifras de LDL 200mg/dl en varios miembros de una familia e incremento en el�
riesgo de aterosclerosis precoz, manifestada por eventos cardiovasculares en la edad adulta con agregaciónfamiliar.
Patologías con dislipidemia secundaria e incremento de las LDLSíndrome nefróticoHipotiroidismoColestasisSíndrome metabólicoIngesta alta de grasas saturadas y/o colesterolDiabetes mellitus tipo 1 en descontrolFármacos: Tiazidas, isotretinoina, corticoesteroides, ciclosporina y esteroides anabólicos.
Cardio-Lipidología
40

Incremento primario de LDLLa tabla enuncia las Dislipidemias de origen
genético determinantes de incremento de LDL. Seenuncia denominación, alteración genética, cifras de
LDL, signos clínicos y grado de asociación aAterosclerosis. Para mayor información ver texto
Dislipidemias
Denominación Alteración LDL mg/dl SignosHipercolesterolemia
Familiar Homocigota
Gen R-LDL 500-1000 Arco corneal y xantomas
tendinosos.
Aterosclerosis muy precoz
Hipercolesterolemia
Familiar Heterocigota
Gen R-LDL 250-500 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Hipercolesterolemia
Autosómica Recesiva
Gen prot. ARH 250-1000 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Alteración apoB100 Gen apoB100 250-500 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Hipercolesterolemia
Autosómica Dominante
Gen PCSK9. Ganancia de función
250-1000 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Sitoesterolemia Gen ABCG5 y G8 =250
? Sitoesteroles
Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Hiperlipidemia Familiar
Combinada
Oligogénico Patrón beta
? apoB100
Asociación a Sx.M.
Aterosclerosis precoz
Hipercolesterolemia
Familiar Poligénica
Poligénica =200 Aterosclerosis precoz
Denominación Alteración LDL mg/dl SignosHipercolesterolemia
Familiar Homocigota
Gen R-LDL 500-1,000 Arco corneal y xantomas
tendinosos.
Aterosclerosis muy precoz
Hipercolesterolemia
Familiar Heterocigota
Gen R-LDL 250-500 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Hipercolesterolemia
Autosómica Recesiva
Gen prot. ARH 250-1,000 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Alteración apoB100 Gen apoB100 250-500 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Hipercolesterolemia
Autosómica Dominante
Gen PCSK9. Ganancia de función
250-1,000 Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Sitoesterolemia Gen ABCG5 y G8 250
Sitoesteroles
Arco corneal y xantomas
tendinosos.
Aterosclerosis precoz
Hiperlipidemia Familiar
Combinada
Oligogénico Patrón beta
apoB100
Asociación a Sx.M.
Aterosclerosis precoz
Hipercolesterolemia
Familiar Poligénica
Poligénica 200 Aterosclerosis precoz
41
Causas primarias de incremento de LDL

Reducción de LDLDislipidemias primarias y secundarias. Efecto anti-aterogénico.Estos trastornos tienen un fenotipo lipídico caracterizado por colesterol total 150mg con�
disminución de LDL, VLDL e IDL y con HDL variable. Los trastornos primarios del metabolismo de loslípidos que ocasionan estas dislipidemias son los siguientes.
AbetalipoproteinemiaEs un trastorno autosómico recesivo que afecta al gen de la MTP -mycrosomal transfer
protein-, proteína encargada de la lipidación de QM y VLDL, determina alteraciones cuantitativas ycualitativas en la síntesis de QM y VLDL, su prevalencia es muy baja y su fenotipo lipídico está en funcióndel genotipo; el genotipo heterocigoto tiene cifras normales de colesterol, triglicéridos y lipoproteínas, elgenotipo homocigoto tiene cifras muy bajas de colesterol y triglicéridos e indetectables de QM, VLDL,IDL y LDL, asociadas a protección aterogénica aunque con deficiencia de vitaminas liposolubles -lasvitaminas E, A y K normalmente son transportadas por QMs y VLDL-, acantocitosis, retinitis pigmentosay degeneración neuromuscular precoz.
Hipobetalipoproteinemia FamiliarEs un trastorno autosómico dominante que afecta en forma heterocigota u homocigota al gen
de la apoB100, determina alteraciones cuantitativas y/o cualitativas en la síntesis y/o catabolismo deapoB100 con incapacidad para el ensamble de VLDL a partir de apoB100 e incremento en la concentraciónintra-hepatocito de triglicéridos, fosfolípidos y colesterol; este acúmulo de lípidos ocasiona en el hígado:represión de la síntesis de colesterol y del receptor LDL, incremento en la síntesis de cassettes ABCG5 yABCG8 con incremento en la excreción biliar de colesterol, y en el enterocito reducción en la absorciónintestinal de colesterol; su prevalencia es muy baja y su fenotipo lipídico se caracteriza por cifras de apoB100
y LDL percentila 50, asociadas a protección aterogénica; el genotipo homocigoto se caracteriza por�
cifras prácticamente indetectables de apoB100 y LDL, asociadas a deficiencia de vitaminas liposolubles,acantocitosis, retinitis pigmentosa y degeneración neuromuscular similares a las observadas en laAbetalipoproteinemia. Se han descrito varias decenas de mutaciones en el gen de la apoB100.
Alteración -pérdida de función- en la PCSK9Es un trastorno que afecta el gen de la PCSK9 -proprotein convertase subtilisin kexin type 9-,
determina la expresión a la baja en la función de la PCSK9 -proteasa que proteoliza al receptor LDL-, por lo
Cardio-Lipidología
42

tanto la reducción de función de la PCSK9 provoca reducción del catabolismo e incremento de receptoresLDL con disminución de LDL circulante, su prevalencia es de 2% en población afro-americana y sufenotipo lipídico se caracteriza por cifras de LDL <80mg/dl, asociadas a una reducción de 80% en el riesgorelativo de eventos cardiovasculares.
Patologías con dislipidemia secundaria y reducción de las LDLDesnutriciónVegetarianismo estrictoSíndromes de malabsorción intestinalInsuficiencia hepáticaInmunoglobulinopatíasSíndromes mieloproliferativosEnfermedades infiltrativas de sistema retículo endotelialHipertiroidismoSepsis
Aguilar-Salinas CA, Gomez RA y Gómez FJ. Abordaje diagnóstico de las dislipidemias. EnDislipidemias. De lo clínico a lo molecular. Edición 2008; capítulo 3. Editores Carlos A Aguilar, Rita AGomez y Francisco J Gómez. Molecular biology of lipoproteins andTsimikas S and Mooser V.dyslipidemias. In Molecular Basis of Cardiovascular Diseases. Edition 2004; chapter 21. Editor Kenneth RChien. Genetic disorders of lipoprotein metabolism. In TherapeuticCuchel M, Qasim A and Rader D.Lipidology. Edition 2007; chapter 2. Editors Michael H Davidson, Peter P Toth and Kavin C Maki.Brunzell JD. Genetic dyslipidemia. In Clinical Lipidology. Edition 2009; chapter 6. Editor Christie MBallantyne. Atlas of Cardiovascular Risk Factors. EditionGornik HL and Plutzky J. Dyslipidemia. In2006; chapter 8. Editor J Michael Gaziano.
Dislipidemias
43
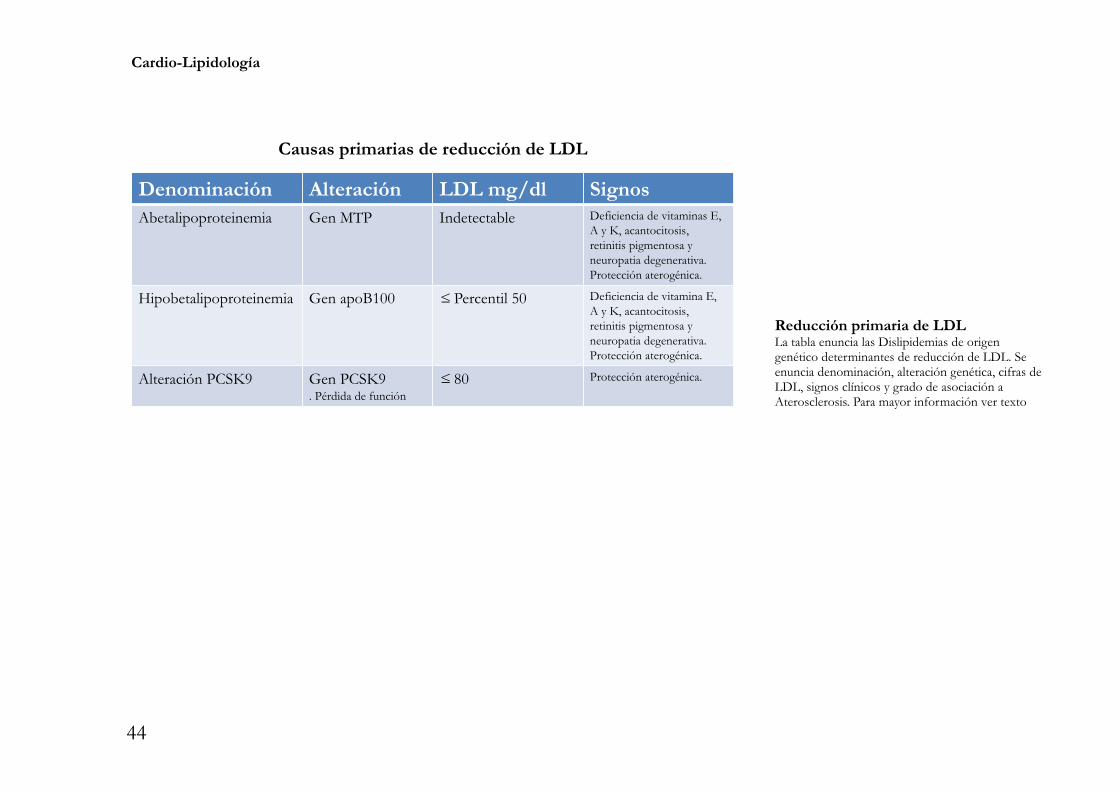
Cardio-Lipidología
Reducción primaria de LDLLa tabla enuncia las Dislipidemias de origengenético determinantes de reducción de LDL. Seenuncia denominación, alteración genética, cifras deLDL, signos clínicos y grado de asociación aAterosclerosis. Para mayor información ver texto
Denominación Alteración LDL mg/dl SignosAbetalipoproteinémia Gen MTP Indetectable Deficiencia de vitaminas E,
A y K, acantocitosis,
retinitis pigmentosa y
neuropatia degenerativa.
Protección aterogénica.
Hipobetalipoproteinemia Gen apoB100 = Percentil 50 Deficiencia de vitamina E,
A y K, acantocitosis,
retinitis pigmentosa y
neuropatia degenerativa.
Protección aterogénica.
Alteración PCSK9 Gen PCSK9. Perdida de función
= 80 Protección aterogénica.
Denominación Alteración LDL mg/dl SignosAbetalipoproteinemia Gen MTP Indetectable Deficiencia de vitaminas E,
A y K, acantocitosis,
retinitis pigmentosa y
neuropatia degenerativa.
Protección aterogénica.
Hipobetalipoproteinemia Gen apoB100 Percentil 50 Deficiencia de vitamina E,
A y K, acantocitosis,
retinitis pigmentosa y
neuropatia degenerativa.
Protección aterogénica.
Alteración PCSK9 Gen PCSK9. Pérdida de función
80 Protección aterogénica.
44
Causas primarias de reducción de LDL

Incremento de triglicéridosDislipidemias primarias y secundarias. Efecto aterogénico variable.Estos trastornos tienen un fenotipo variable de la clasificación de Fredrickson, Levy y Less:
tipo I o V en la deficiencia de LPL y apoCII por incremento de QM, VLDL y TG 500mg/dl y tipo IV en�
las hipertrigliceridemia familiar, hiperlipidemia familiar combinada e hipertrigliceridemias secundarias por
incremento de VLDL con TG 200mg/dl. Se subclasifica como hipertrigliceridemia grave aquella con�
triglicéridos 500mg/dl y colesterol <200mg/dl con una relación 5:1 entre triglicéridos y colesterol total,�
puesto que las lipoproteínas de muy baja densidad -VLDL- contienen 5 moléculas de triglicéridos por cadamolécula de colesterol. Los trastornos primarios del metabolismo de los lípidos que ocasionan estasdislipidemias son los siguientes.
Deficiencia de lipoproteínlipasa -LPL-Es un trastorno autosómico recesivo -rara vez dominante- que afecta al gen de la LPL -lipasa
que hidroliza los TG contenidos en QM, VLDL e IDL-, sin afectación de la lipasa hepática -HL-, determinadeficiencia en la hidrólisis de los triglicéridos contenidos en las lipoproteínas referidas, su prevalencia esmuy baja, aproximadamente 1/1,000,000 individuos y su fenotipo lipídico se caracteriza por
quilomicronemia y cifras de TG 1,000mg/dl desde la infancia, asociadas a la presencia de xantomas�
eruptivos, lipemia retiniana, hepatoesplenomegalia, incremento en el riesgo de pancreatitis recurrente en lainfancia -aparentemente por conglomerados de ácidos grasos, lisolecitina y albumina en los capilarespancreáticos- y asociación controversial a eventos cardiovasculares.
Se han descrito más de 60 mutaciones en el gen de la LPL, asociadas con alteraciones en lasíntesis celular, secreción hacia el espacio endotelial, anclamiento a los proteoglicanos y/o acciónhidrolítica de la LPL sobre la apoCII de VLDL y QM; se agrupan como mutaciones clase I las asociadas a laausencia de LPL y clase II o III las asociadas a alteraciones en su función.
Deficiencias de apoCII y apoAVLa deficiencia de apoCII es un trastorno autosómico dominante o recesivo que afecta al gen de
la apoCII -apoproteína sustrato para la acción de la LPL-, la variante homocigota determina un patrónsimilar a la deficiencia de LPL con deficiencia en la hidrólisis de triglicéridos contenidos en QM y VLDL, suprevalencia es muy baja -se ha reportado menor a la de la deficiencia de LPL- y su fenotipo lipídico se
caracteriza por quilomicronémia y cifras de TG 1,000mg/dl de aparición más tardía que en la deficiencia�
Dislipidemias
45

de LPL, generalmente en la adolescencia, asociadas a la presencia de xantomas eruptivos, lipemia retiniana,hepatoesplenomegalia, incremento en el riesgo de pancreatitis recurrente en la adolescencia o la edadadulta temprana y asociación controversial a eventos cardiovasculares. Recientemente se ha descrito ladeficiencia de apoAV -sustrato accesorio para la acción de la LPL sobre apoCII- como un trastorno con unfenotipo similar a la deficiencia de apoCII.
Hipertrigliceridemia familiarEs un trastorno familiar de etiología no determinada -probable sobreproducción hepática de
TG y VLDL ricas en triglicéridos-, autosómico dominante, con prevalencia variable, en promedio 1/500-1,000 individuos y fenotipo lipídico caracterizado por elevación de VLDL con cifras de TG entre 250mg/dly 1,000mg/dl con colesterol total y HDL normales, aunque el primero puede estar elevado y las segundasdisminuidas en forma directamente proporcional al incremento de TG; existe una asociación con síndromemetabólico y DM2 que incrementan el riesgo de eventos cardiovasculares.
Hiperlipidemia familiar combinada.Ya descrita con anterioridad.
Patologías con dislipidemia secundaria e incremento de TGDM en descontrolAlcoholUremiaDiálisis peritonealSepsisLipodistrofiasMieloma múltipleLupus eritematoso sistémicoGlucogenosisAcromegaliaFármacos: Diuréticos, bloqueadores-beta, anticonceptivos, estrógenos, isotretinoina,
esteroides anabólicos, inhibidores de proteasas.
Cardio-Lipidología
46
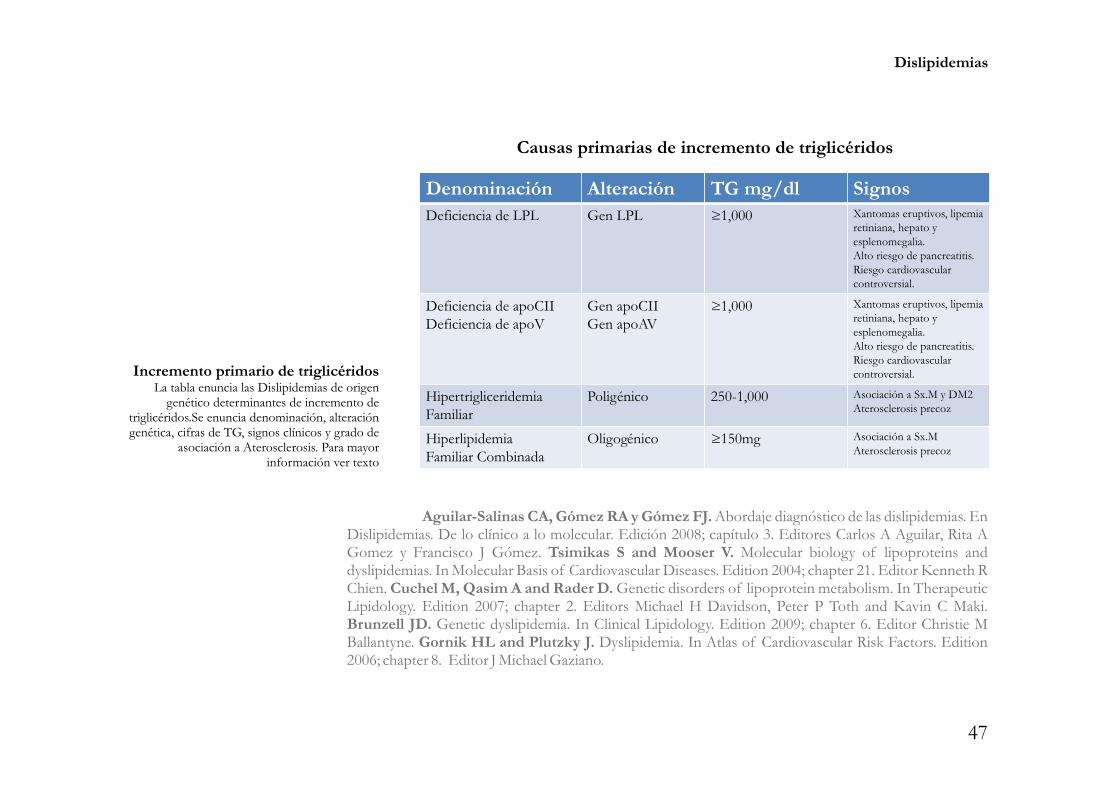
Aguilar-Salinas CA, Gómez RA y Gómez FJ. Abordaje diagnóstico de las dislipidemias. EnDislipidemias. De lo clínico a lo molecular. Edición 2008; capítulo 3. Editores Carlos A Aguilar, Rita AGomez y Francisco J Gómez. Molecular biology of lipoproteins andTsimikas S and Mooser V.dyslipidemias. In Molecular Basis of Cardiovascular Diseases. Edition 2004; chapter 21. Editor Kenneth RChien. Genetic disorders of lipoprotein metabolism. In TherapeuticCuchel M, Qasim A and Rader D.Lipidology. Edition 2007; chapter 2. Editors Michael H Davidson, Peter P Toth and Kavin C Maki.Brunzell JD. Genetic dyslipidemia. In Clinical Lipidology. Edition 2009; chapter 6. Editor Christie MBallantyne. Dyslipidemia. In Atlas of Cardiovascular Risk Factors. EditionGornik HL and Plutzky J.2006; chapter 8. Editor J Michael Gaziano.
Dislipidemias
47
Incremento primario de triglicéridosLa tabla enuncia las Dislipidemias de origen
genético determinantes de incremento detriglicéridos.Se enuncia denominación, alteracióngenética, cifras de TG, signos clínicos y grado de
asociación a Aterosclerosis. Para mayorinformación ver texto
Denominación Alteración TG mg/dl SignosDeficiencia de LPL Gen LPL =1000 Xantomas eruptivos, lipemia
retiniana, hepato y
esplenomegalia.
Alto riesgo de pancreatitis.
Riesgo cardiovascular
controversial.
Deficiencia de apoCII
Deficiencia de apoV
Gen apoCII
Gen apoAV
=1000 Xantomas eruptivos, lipemia
retiniana, hepato y
esplenomegalia.
Alto riesgo de pancreatitis.
Riesgo cardiovascular
controversial.
Hipertrigliceridemia
Familiar
Poligénico 250-1000 Asociación a Sx.M y DM2
Aterosclerosis precoz
Hiperlipidemia
Familiar Combinada
Oligogénico =150mg Asociación a Sx.M
Aterosclerosis precoz
Denominación Alteración TG mg/dl SignosDeficiencia de LPL Gen LPL 1,000 Xantomas eruptivos, lipemia
retiniana, hepato y
esplenomegalia.
Alto riesgo de pancreatitis.
Riesgo cardiovascular
controversial.
Deficiencia de apoCII
Deficiencia de apoV
Gen apoCII
Gen apoAV
1,000 Xantomas eruptivos, lipemia
retiniana, hepato y
esplenomegalia.
Alto riesgo de pancreatitis.
Riesgo cardiovascular
controversial.
Hipertrigliceridemia
Familiar
Poligénico 250-1,000 Asociación a Sx.M y DM2
Aterosclerosis precoz
Hiperlipidemia
Familiar Combinada
Oligogénico 150mg Asociación a Sx.M
Aterosclerosis precoz
Causas primarias de incremento de triglicéridos

Incremento de LDL y triglicéridosDislipidemias primarias y secundarias. Efecto aterogénico alto.Estos trastornos tienen un fenotipo III -disbetalipoproteinemia- y IIB de la clasificación de
Fredrickson, Levy y Less con colesterol total 200mg/dl, triglicéridos 150mg/dl y HDL 60mg/dl.� � �
Los trastornos primarios del metabolismo de los lípidos que ocasionan estas dislipidemias son lossiguientes.
DisbetalipoproteinemiaEs un trastorno autosómico generalmente recesivo -existen casos dominantes- que determina
la ausencia o defectos de la síntesis de apoE -la isoforma disfuncional más frecuente es la apoE2- y por lotanto existe ausencia o alteración en el reconocimiento en apoE de los QM, VLDL e IDL por susreceptores específicos -LDLR y LRP- con incremento en el nivel y tiempo de circulación de dichaslipoproteínas, su prevalencia es baja con 0.5% de prevalencia en individuos con historia de infartomiocárdico y su fenotipo lipídico se caracteriza por cifras de colesterol y triglicéridos “igualadas”entre450mg/dl y 700mg/dl con LDL normal; dicha equalización se explica porque tanto remanentes de QMcomo IDL contienen concentraciones iguales de colesterol y triglicéridos, asociadas“patognomónicamente” a la presencia de xantomas estriados palmares, también pueden existir xantomastuberosos en codos, nudillos y rodillas y xantomas eruptivos con aterosclerosis precoz manifestada poreventos cardiovasculares tempranos por compromiso de cualquier territorio arterial -supra o infra-diafragmático-.
Las isoformas anómalas de apoE además de modificar el reconocimiento de QM remanentes,VLDL e IDL por los receptores LDL y LRP, también incrementan la síntesis hepática de VLDL y reducenla actividad de la LPL, ambos mecanismos contribuyen al incremento en el tiempo de circulación de VLDL.Así mismo cualquier condición que comprometa la función de los receptores de LDL -obesidad, DM2,hipotiroidismo etc- sinergiza la expresión de este trastorno.
Hiperlipidemia familiar combinadaYa descrita con anterioridad.
Patologías con dislipidemia secundaria e incremento de LDL y TGObesidad, síndrome metabólico, diabetes mellitus tipo 2. Ver siguiente apartado de
dislipidemias en México.
Cardio-Lipidología
48

Dislipidemias en MéxicoPor la alta prevalencia de obesidad en nuestro país y por el gran impacto aterogénico de la
dislipidemia asociada a ella, en este apartado se tratará en detalle este común y grave problema de saludpública.
Prevalencia de obesidad en México. En México de acuerdo al reporte de ENSANUT 2006,62.9% de los hombres y 83.8% de las mujeres >20 años de edad cumplen el criterio de la IDF -International
Diabetes Federation- de obesidad visceral o central -perímetro abdominal 90cm y 80cm en hombres y� �
mujeres respectivamente-. La prevalencia de obesidad central en adultos es sólo el continuo del mismofenómeno en la infancia y adolescencia. En un estudio longitudinal reciente, individuos Israelies evaluadosinicialmente a los 17 años de edad y seguidos en promedio hasta los 45 años de edad, se observó que endicho lapso de tiempo hubo un incremento promedio de 15kg o 4 unidades de IMC. Al comparar el quintil1 vs el quintil 5 de incremento ponderal se estableció una asociación directamente proporcional con elincremento en las cifras de presión arterial, glucosa, triglicéridos, colesterol LDL, TGO, TGP y tabaquismoy con la reducción en las cifras de colesterol HDL y actividad física y aumentos de 2.7 y 5.4 en el radio deriesgo para DM2 y enfermedad aterosclerosa coronaria confirmada por angiografía coronaria.
Así la obesidad central en el 74.2% de la población mexicana adulta representa el sustrato másfrecuente para el desarrollo de resistencia a la insulina y sus manifestaciones hemodinámicas -disfunciónendotelial e HTA- y metabólicas -dislipidemia y disglucemia-, procesos interactuantes conocidos comofactores de riesgo cardiovascular mayores, ya que ocasionan daño endotelio-vascular, enfermedadcardiovascular, invalidez y muerte.
Epidemiología de las dislipidemias en México. En México en la muestra ENSANUT2006 -encuesta más reciente en nuestro país- de 4040 individuos entre 20 y 69 años de edad, estudiados trasun ayuno de 9 a 12 horas, las concentraciones medias de los lípidos sanguíneos fueron colesterol 198.5mg/dl, triglicéridos 139.6 mg/dl, colesterol HDL 39.0 mg/dl, colesterol no-HDL 159.5 mg/dl y colesterolLDL 131.5 mg/dl.
La anormalidad aislada más común fue la hipoalfalipoproteinemia -colesterol HDL < 40
mg/dl-; su prevalencia fue 60.5% -IC 95% 58.2-62.8%-. La hipercolesterolemia -colesterol 200 mg/dl-�
fue la segunda anormalidad en frecuencia, con 43.6% -IC 95% 41.4-46.0%-. La hipertrigliceridemia - 150�
Dislipidemias
49

mg/dl- fue observada en 31.5% -IC 95% 29.3-33.9%-. Cifras de colesterol LDL >130 mg/dl fueronreportados en 41.4% -37.6-45.3%- en hombres y 50.0% -46.7-53.3%- en mujeres con un promedio de46.0 % -43.5-48.6%-. Valores de colesterol no-HDL >160 mg/dl fueron reportados en 39.1% -35.3-43.0%- en hombres y 45.3% -42.0-48.6%- en mujeres con un promedio de 42.4% -39.9-45.0%-.
La dislipidemia mixta con elevación simultánea de colesterol y triglicéridos -dislipidemiaaltamente aterogénica- fue reportada en 18.2% -IC 95% 16.3-20.2%- de la población, con una prevalenciamáxima en la población entre 40-49 años. La combinación de hipertrigliceridemia ehipoalfalipoproteinemia fue reportada en 18.3% de la población con una prevalencia mayor en los hombres-p < 0.01-. La hipercolesterolemia aislada fue reportada en 25.3% y la hipertrigliceridemia aislada en 13.3%de la población.
De la muestra referida 1,534 individuos tenían sobrepeso y 1,240 obesidad; las anormalidadesen los lípidos no difirieron entre ambos grupos, siendo la prevalencia de hipertrigliceridemia,hipoalfalipoproteinemia y dislipidemia mixta significativamente mayor en los individuos con sobrepeso uobesidad en comparación a individuos con IMC <25kg/m2. Lo anterior indica que en México ladislipidemia aparece aún con bajos índices de adiposidad, muy probablemente por una susceptibilidadgenética propia de nuestra etnia, en la cual se han reportado una serie de genotipos “precipitadores” dedichos trastornos metabólicos.
En resumen se estima que en México, 20.5, 14.8, 10.6 y 6.1 millones de individuos tienenhipoalfalipoproteinemia, hipercolesterolemia, hipertrigliceridemia y dislipidemia mixta respectivamente; lagran mayoría de ellos ->90%- sin diagnóstico ni tratamiento, realmente éllo constituye un gran retoepidemiológico cuya consecuencia es la epidemia de enfermedades cardiovasculares. En el año 2010 elINEGI en su reporte de mortalidad 2009 informó 78,000 muertes asociadas a Diabetes Mellitus y 111,000muertes asociadas a Enfermedades Cardiovasculares; en total 189,000 fallecimientos secundarios albinomio Diabetes Mellitus-Enfermedades Cardiovasculares -un fallecimiento en promedio cada 2.5minutos-.
Obesidad como disparador de resistencia a la insulina, disfunción metabólica yhemodinámica. Los tejidos de reserva energética y de respuesta inmune están íntimamente relacionados ytienen un origen común. En la mosca Drosophila ambos tejidos están contenidos en un órganodenominado cuerpo graso, en los seres superiores dichos sistemas están representados por el tejido adiposo
Cardio-Lipidología
50

62.9
Hombres
83.8
Mujeres
20%
40%
60%
80%
Obesidad y Dislipidemias en MéxicoLas gráficas muestran la prevalencia de obesidad
central por perímetro abdominal >80cm en mujeresy >90cm en hombres -criterio IDF- y la prevalenciade dislipidemias aisladas por orden de frecuencia de
acuerdo a ENSANUT 2006
Dislipidemias en MéxicoLas gráficas muestran las prevalencias de
hipercolesterolemia total, hipercolesterolemia-LDL,hipercolesterolemia no-HDL y dislipidemias mixtas
de acuerdo a ENSANUT 2006
Dislipidemias
43.6
HDLCTTG
60.520%
40%
60%
80%
31.5
<40mg200mg150mg<<
20%
40%
60%
80%
43.643.6
CTLDLno-HDL
41.441.4 39.139.1
200mg130mg160mg
<<<
18.218.2
CT-TGTG-HDL
18.318.320%
40%
60%
80%
200mg- 150mg150mg-<40mg<<
<
51

y el tejido linfohemático. Ante un reto infeccioso, la respuesta inmune -de alto consumo energético- seinicia gracias a la liberación de energía -ácidos grasos- acumulada en forma de triglicéridos en el tejidoadiposo; en caso requerido dicha respuesta inmune puede sostenerse debido al establecimiento de unestado transitorio de resistencia a la insulina mediado por moléculas secretadas por monocitos y linfocitos-vg TNFα, IL6 etc.-.
Por dicho mecanismo se propicia un “switch energético” en el cual, los sustratos energéticos-ácidos grasos- no se acumulan como triglicéridos en los adipocitos y son derivados directamente para elsostén de la respuesta inmune. Este estado fisiológico y transitorio de resistencia a la insulina cede una vezque la respuesta y la agresión inmunes también ceden. Así en su origen el tejido adiposo fue un sistemaconcebido para almacenar -lipogénesis- y en su momento liberar -lipolisis- sustratos energéticos en unmedio ambiente que ofrecía escasas fuentes de energía y un alto grado de estrés ambiental. A mayoreficiencia para almacenar-liberar sustratos energéticos, mayor posibilidad de sobrevivencia -hipótesis delgen ahorrador-.
En nuestros días la premisa anterior ha cambiado. El medio ambiente actual se caracteriza porun exceso de fuentes hiperenergéticas y un bajo nivel de estrés ambiental condicionantes de obesidad-hiperplasia e hipertrofia de tejido adiposo- y detonante de un estado de inflamación subclínica y crónicadenominada Metainflamación -MI-. La obesidad, especialmente la visceral genera MI en el momento en elcual las adipocitos rebasan su límite fisiológico para la captación de ácidos grasos y la síntesis detriglicéridos. En esta condición de estrés celular adipocitario, mitocondrias y retículo endoplásmico, a travésde la producción de radicales libres de O2 activan vías enzimáticas de señalización intracelular quemodifican el fenotipo del adipocito. Con fines didácticos, la MI puede dividirse en tres etapas.
Primera etapa. El estrés oxidativo adipocitario, es el detonante que activa cascadasenzimáticas inflamatorias como: IKK “Inhibitor NFkB Kinase”, JNK “Janus Kinase” y AP1 “ActivatedProtein 1”, las cuales provocan la regulación a la alta de factores de transcripción “primitivos”. En eladipocito se regula a la alta el NFkB -factor de transcripción maestro inflamatorio-, revirtiendo su genotipoal de un preadipocito con un fenotipo inflamatorio, similar al de los monocitos. Esta desdiferenciación deladipocito hacia su fenotipo inflamatorio es factible dado el origen común de los tejidos adiposo ylinfohemático. Así el adipocito “reacciona” ante la agresión metabólica convirtiéndose en una célulainflamatoria con capacidad para la producción de moléculas como la MCP1 -proteína 1 quimiotáctica de
Cardio-Lipidología
52

Cuerpo graso
Drosophila
Leucocitos Adipocitos
Reserva EnergéticaTejido adiposo
Adipocitos
Reserva EnergéticaTejido adiposo
Adipocitos
Respuesta InmuneTejido linfohemático
Leucocitos
Respuesta InmuneTejido linfohemático
Leucocitos
Ingesta Liberación AGL
Reserva EnergéticaTejido adiposo
Adipocitos
Reserva EnergéticaTejido adiposo
Adipocitos
Respuesta InmuneTejido linfohemático
Leucocitos
Respuesta InmuneTejido linfohemático
Leucocitos
Switch Energético
Reserva EnergéticaTejido adiposo
Adipocitos
Reserva EnergéticaTejido adiposo
Adipocitos
Respuesta InmuneTejido linfohemático
Leucocitos
Respuesta InmuneTejido linfohemático
Leucocitos
MamíferosEnergía e Inmunidad
Imágenes que muestran el cuerpo graso -órganoprimitivo de regulación energética e inmunológica-
en la mosca Drosophila y la diferenciación delrudimentario cuerpo graso en los sistemas de
regulación energética -tejido adiposo- y derespuesta inmunológica -tejido linfohemático- en
los mamíferos
Energía e InmunidadImágenes que muestran la colaboración entre el
tejido adiposo como donador de ácidos grasos y eltejido linfohématico como receptor de dicho
sustrato energético para el inicio de la respuestainmune y el switch energético con resistencia a la
insulina fisiológica y transitoria durante la respuestainmune. En el switch, los ácidos grasos se derivandirectamente hacia el tejido linfohemático sin ser
procesados por el tejido adiposo
Dislipidemias
RI transitoriaSeñal inhibitoria
RI transitoriaSeñal inhibitoria
53
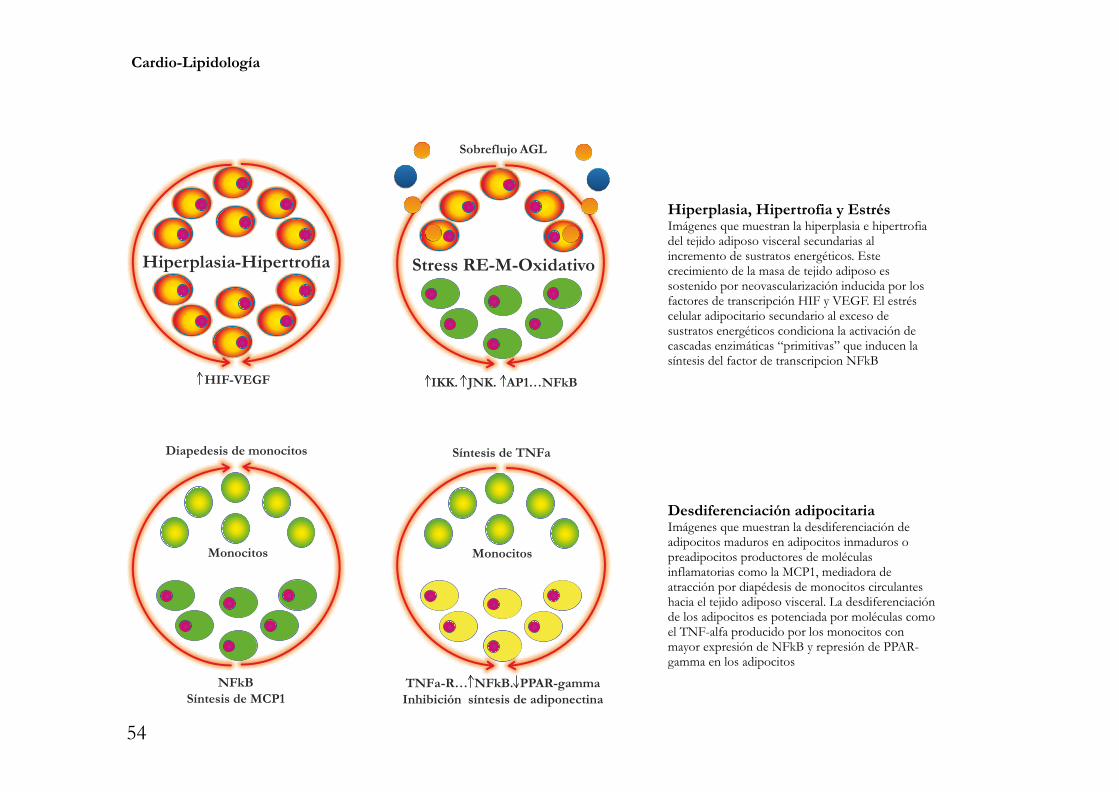
NFkBSíntesis de MCP1
Diapedesis de monocitos
Monocitos
Cardio-Lipidología
Hiperplasia, Hipertrofia y EstrésImágenes que muestran la hiperplasia e hipertrofiadel tejido adiposo visceral secundarias alincremento de sustratos energéticos. Estecrecimiento de la masa de tejido adiposo essostenido por neovascularización inducida por losfactores de transcripción HIF y VEGF. El estréscelular adipocitario secundario al exceso desustratos energéticos condiciona la activación decascadas enzimáticas “primitivas” que inducen lasíntesis del factor de transcripcion NFkB
Desdiferenciación adipocitariaImágenes que muestran la desdiferenciación deadipocitos maduros en adipocitos inmaduros opreadipocitos productores de moléculasinflamatorias como la MCP1, mediadora deatracción por diapédesis de monocitos circulanteshacia el tejido adiposo visceral. La desdiferenciaciónde los adipocitos es potenciada por moléculas comoel TNF-alfa producido por los monocitos conmayor expresión de NFkB y represión de PPAR-gamma en los adipocitos
Hiperplasia-Hipertrofia
HIF-VEGF IKK. JNK. AP1…NFkB
Sobreflujo AGL
Stress RE-M-Oxidativo
Inhibición síntesis de adiponectina
Síntesis de TNFa
Monocitos
TNFa-R… NFkB. PPAR-gamma
54
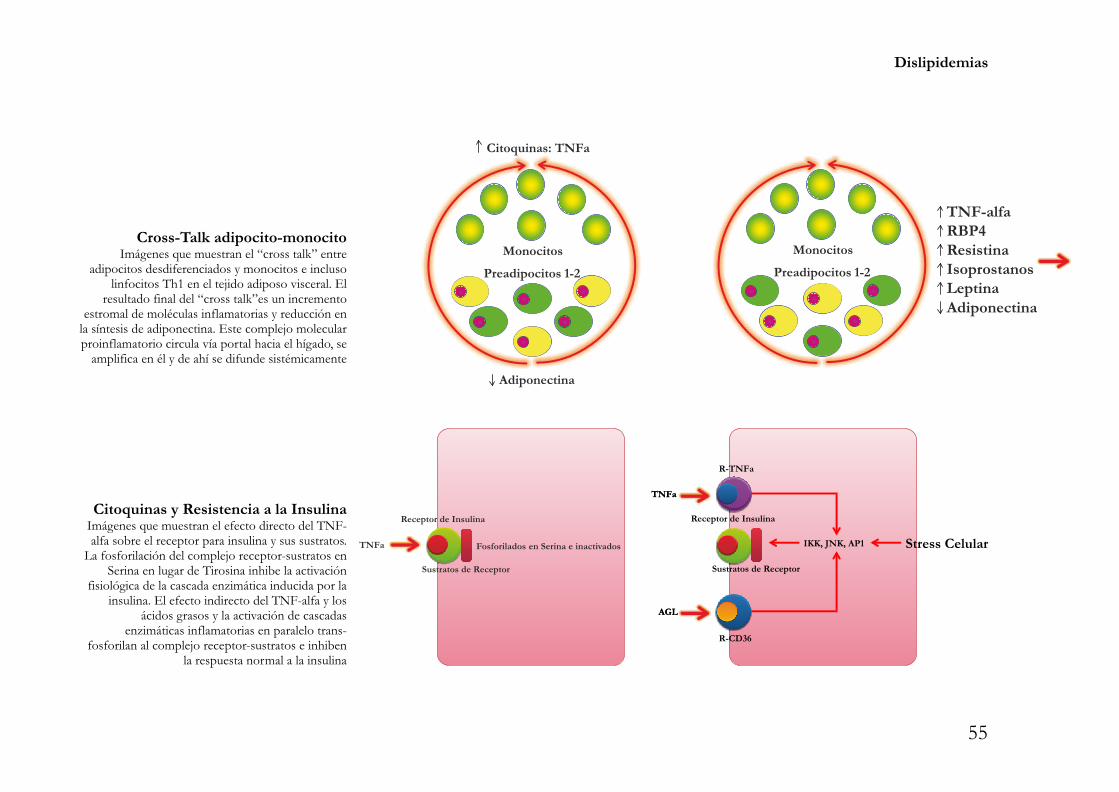
Cross-Talk adipocito-monocitoImágenes que muestran el “cross talk” entre
adipocitos desdiferenciados y monocitos e inclusolinfocitos Th1 en el tejido adiposo visceral. El
resultado final del “cross talk”es un incrementoestromal de moléculas inflamatorias y reducción en
la síntesis de adiponectina. Este complejo molecularproinflamatorio circula vía portal hacia el hígado, se
amplifica en él y de ahí se difunde sistémicamente
Citoquinas y Resistencia a la InsulinaImágenes que muestran el efecto directo del TNF-alfa sobre el receptor para insulina y sus sustratos.
La fosforilación del complejo receptor-sustratos enSerina en lugar de Tirosina inhibe la activación
fisiológica de la cascada enzimática inducida por lainsulina. El efecto indirecto del TNF-alfa y los
ácidos grasos y la activación de cascadasenzimáticas inflamatorias en paralelo trans-
fosforilan al complejo receptor-sustratos e inhibenla respuesta normal a la insulina
Dislipidemias
Preadipocitos 1-2
Monocitos
TNF-alfaRBP4ResistinaIsoprostanosLeptinaAdiponectina
TNFa
Sustratos de Receptor
Receptor de Insulina
Fosforilados en Serina e inactivados
TNFaTNFa
R-TNFa
IKK, JNK, AP1 Stress Celular
AGLAGL
R-CD36
Receptor de Insulina
Sustratos de Receptor
Adiponectina
Preadipocitos 1-2
Citoquinas: TNFa
Monocitos
55

monocitos- y otras citoquinas. Por acción de la MCP1, monocitos y otras células inflamatorias como loslinfocitos Th1migran por diapédesis desde la circulación sanguínea hacia el estroma adiposo visceral con elobjetivo de reforzar la respuesta inflamatoria iniciada por el preadipocito. Este fenómeno es favorecido porla síntesis de factores de transcripción angiogénicos como: HIF “Hypoxia Induced Factor” y VEGF“Vascular Endothelial Growth Factor” con neovascularización del tejido adiposo hiperplásico ehipertrófico.
Segunda etapa. La interacción celular o “cross talk” entre preadipocitos, monocitos ylinfocitos Th1, sostiene un estado inflamatorio parácrino, centralizado al estroma adiposo visceral. Entrelas moléculas inflamatorias producidas dentro del estroma adiposo visceral inflamado destacan: RBP4“retinoid binding protein 4”, Resistina, Isoprostanos, IL6, TNF-alfa etc.-, de ellas, la más importante yoriginalmente descrita por Hotamisligil, es el TNF-alfa “tumor necrosis factor-alfa”. Esta potentecitoquina es ligando de sus receptores específicos TNF-alfa-R1 y TNF-alfa-R2, expresados en lamembrana celular de los adipocitos desdiferenciados o preadipocitos; la activación de estos receptoressostiene la estimulación de las cascadas enzimáticas que retroalimentan el fenotipo inflamatorio oinmaduro de los adipocitos e inhiben el fenotipo metabólico o maduro del adipocito a través de laregulación a la baja de los receptores de membrana para insulina y de los receptores nucleares tipo PPAR-gamma que codifican entre otras, la síntesis de adiponectina -molécula con múltiples accioneseumetabólicas y eucardiovasculares-. Así el estroma adiposo visceral se transforma en un “adiposomainflamatorio”.
Tercera etapa. El ambiente inflamatorio que en un inicio se limitó al “adiposomainflamatorio” se torna sistémico gracias a la comunicación venosa portal del estroma adiposo visceral conel flujo sanguíneo sistémico. A su paso por el hígado, el fenómeno inflamatorio se amplifica por laparticipación de las células de Küppfer. El resultado final es un exceso sistémico de ácidos grasos ymoléculas inflamatorias más una deficiencia de moléculas antinflamatorias, iniciado por trastornos en elmetabolismo intermedio y condicionante de resistencia a la insulina, en una palabra Metainflamación. Laresistencia a la insulina tanto en las células del eje metabólico -adipocito, hepatocito, miocito- como en lascélulas endoteliales, obedece esencialmente a la transfosforilación en serina y no en tirosina de los sustratosdel receptor para insulina -IRS-. Esta fosforilación anómala puede ser directa sobre el receptor de insulinay/o sus sustratos por acción de moléculas como el TNF-alfa y/o indirecta a través de cascadas enzimáticas
Cardio-Lipidología
56

inflamatorias activadas por la unión de diversos ligandos a sus receptores específicos -vg ácidos grasossobre CD36, angiotensina II sobre AT1, AGEs sobre RAGEs e incluso el propio TNF-alfa sobre TNF-alfaR1 o R2, etc.-.
Fisiopatología de la dislipidemia mixta asociada a obesidad central. La dislipidemiamixta generada por el continuo obesidad visceral-MI-RI se caracteriza por incremento de triglicéridos masreducción de colesterol-HDL mas colesterol total normal con incremento “oculto” de la fracción decolesterol-LDL” denso y pequeño; este patrón lipídico es altamente aterogénico por lo que también ha sidodenominado Dislipidemia Aterogénica -DLA-. El origen de esta DLA tiene un fondo genético de nivelvariable, sin embargo la modulación por el medio ambiente juega un papel etiopatogénico fundamental. Elincremento en la síntesis y la reducción en el catabolismo de las VLDL son los ejes de la DLA tal como seanaliza a continuación.
Incremento en la síntesis de VLDL vouyant. El continuo ya revisado de obesidad central-MI-RI propicia la proliferación de adipocitos viscerales desdiferenciados, incapaces de captar ácidos grasosy sintetizar triglicéridos a partir de ellos; en consecuencia el exceso de ácidos grasos circulantes es un gran
Exceso de EnergíaSobreingesta + Sedentarismo
Hipertrofia-HiperplasiaEstrés adipocitario
HipoxiaNeovascularización
InflamaciónMetainflamación
Resistencia a la InsulinaDisfunción hemodinámico-metabólica
Adiposoma Inflamatorio
1ª etapa
2ª etapa
3ª etapa
1ª etapa
2ª etapa
3ª etapa
Metainflamación y RI. AlgoritmoDiagrama que resume las tres etapas de la
Metainflamación. Etapa 1. Estrés celular cuando lahipertrofia e hiperplasia adipocitaria dejan de sersuficientes para compensar el exceso de sustratos
energéticos. Etapa 2. Actividad celular inflamatoriay neovascularización reactiva con la constitución deun “adiposoma inflamatorio”. Etapa 3. Resistencia
sistémica a la insulina inducida por el exceso demoléculas inflamatorias y la deficiencia deadiponectina proveniente del “adiposoma
inflamatorio”
Dislipidemias
57

sustrato para la síntesis hepática de VLDL ricas en triglicéridos o vouyant. En el hepatocito del individuocon RI coexisten: sobre-expresión del factor de transcripción SREBP-1c el cual codifica la producción deenzimas determinantes de la síntesis de triglicéridos, reducción en el catabolismo de la apoB100 -esqueletopara la lipidación de las VLDL- e incremento en la síntesis de apoCIII -apoproteína protectora de la acciónhidrolítica de la LPL-. De esta forma, el exceso de ácidos grasos circulantes -sustrato- en conjunto con lasreferidas tres modificaciones hepáticas que incrementan el metabolismo y disminuyen el catabolismo de lasVLDL favorecen la síntesis, secreción y concentración plasmática de VLDL vouyant o VLDL enriquecidasde triglicéridos.
Reducción en el catabolismo de VLDL vouyant. Colateralmente a lo descrito en el párrafoanterior, en el estado de RI coexiste subexpresión de LPL y sobrexpresión de CETP, lo primero en sinergiacon la sobrexpresión de apoCIII disminuye el catabolismo de las VLDL circulantes, incrementando sutiempo de circulación, y lo segundo incrementa el intercambio de triglicéridos por colesterol con las HDL.Las VLDL ricas en triglicéridos “donan” triglicéridos a las HDL y “reciben” colesterol de ellas. Así lasVLDL vouyant por la acción hidrolítica de LPL y especialmente de HL se transforman secuencialmente enIDL y lipoproteínas de pequeño tamaño y alta densidad, denominadas LDL densas y pequeñas o de patrónbeta -altamente aterogénicas-; las HDL ricas en triglicéridos, también por la acción hidrolítica de LPL y deHL se transforman en HDL inmaduras; este fenómeno que afecta la calidad de las HDL se potencia con lareducción en la síntesis y por ende en la cantidad de apoAI y HDL secundarias a la deficiencia deadiponectina y de ABC-A1 en el adipocito, propias del estado de RI.
En resumen, el incremento en la producción de VLDL rica en triglicéridos, si bien, compensatransitoriamente el exceso de ácidos grasos circulantes, a la postre determina al menos tres mecanismosaterotrombogénicos, a saber: exceso en la concentración de ácidos grasos circulantes, los cuales por simismos retroalimentan la RI -teoría tóxico lipolítica de Wang-; incremento en la concentración delipoproteínas ricas en TG y especialmente de la fracción más aterogénica de las lipoproteínas apoB100, lasLDL densas y pequeñas -teoría infiltrativa de Zilversmit-; deficiencia en la cantidad y calidad de las HDL.
Así, como se analizará en el apartado de Aterogénesis, la DLA “per se” en sinergia con ladisfunción endotelial y la disglucemia son los tres factores de lesión endotelio-vascular que explican la altaprevalencia e incidencia de enfermedad cardiovascular en los individuos con sobrepeso y obesidad,subgrupo que es mayoritario en nuestro país. Si en eras paleolíticas se cumplía la premisa “a mayor
Cardio-Lipidología
58
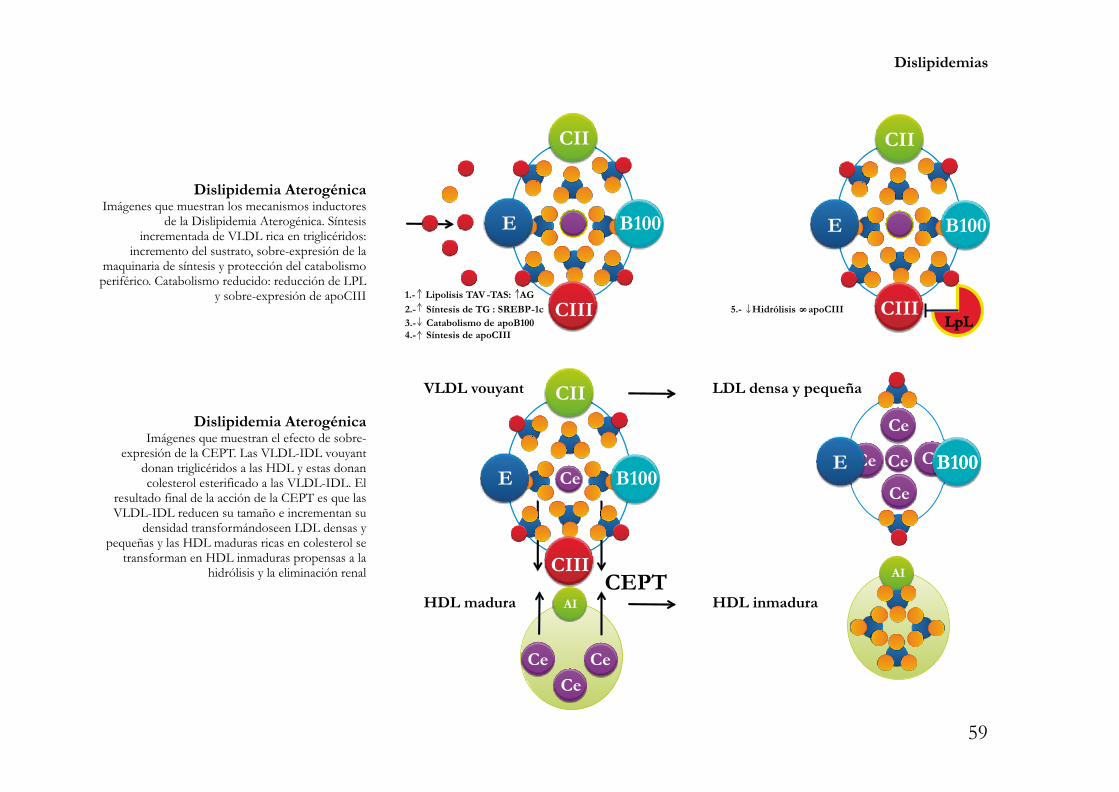
Ce
Ce
B100B100Ce
Ce
Ce
AI
E
AI
Ce
CII
B100E
CIIICEPT
Ce
Ce Ce
Dislipidemia AterogénicaImágenes que muestran los mecanismos inductores
de la Dislipidemia Aterogénica. Síntesisincrementada de VLDL rica en triglicéridos:
incremento del sustrato, sobre-expresión de lamaquinaria de síntesis y protección del catabolismo
periférico. Catabolismo reducido: reducción de LPLy sobre-expresión de apoCIII
Dislipidemias
CII
1.- Lipolisis TAV -TAS: AG
1
2.- Síntesis de TG : SREBP-1c3.- Catabolismo de apoB1004.- Síntesis de apoCIII
B100E
CIII
CII
LpL5.- Hidrólisis 8 apoCIII
LpL
B100E
CIII
Dislipidemia AterogénicaImágenes que muestran el efecto de sobre-
expresión de la CEPT. Las VLDL-IDL vouyantdonan triglicéridos a las HDL y estas donancolesterol esterificado a las VLDL-IDL. El
resultado final de la acción de la CEPT es que lasVLDL-IDL reducen su tamaño e incrementan su
densidad transformándoseen LDL densas ypequeñas y las HDL maduras ricas en colesterol se
transforman en HDL inmaduras propensas a lahidrólisis y la eliminación renal
59
VLDL vouyant LDL densa y pequeña
HDL madura HDL inmadura

eficiencia para almacenar-liberar sustratos energéticos, mayor posibilidad de sobrevivencia”, ennuestra era esa premisa se ha invertido “a mayor restricción para almacenar-liberar sustratosenergéticos, mayor posibilidad de sobrevivencia libre de eventos cardiovasculares”.
Aguilar-Salinas CA, Gómez RA y Gómez FJ. Abordaje diagnóstico de las dislipidemias. EnDislipidemias. De lo clínico a lo molecular. Edición 2008; capítulo 3. Editores Carlos A Aguilar, Rita AGomez y Francisco J Gómez. Molecular biology of lipoproteins andTsimikas S and Mooser V.dyslipidemias. In Molecular Basis of Cardiovascular Diseases. Edition 2004; chapter 21. Editor Kenneth RChien. Genetic disorders of lipoprotein metabolism. In TherapeuticCuchel M, Qasim A and Rader D.Lipidology. Edition 2007; chapter 2. Editors Michael H Davidson, Peter P Toth and Kavin C Maki.Brunzell JD. Genetic dyslipidemia. In Clinical Lipidology. Edition 2009; chapter 6. Editor Christie MBallantyne. Dyslipidemia. In Atlas of Cardiovascular Risk Factors. EditionGornik HL and Plutzky J.2006; chapter 8. Editor J Michael Gaziano. Aguilar-Salinas CA, Gómez-Pérez FJ, Rull J, VillalpandoS, Barquera S, Rojas R. Prevalence of dyslipidemias in the Mexican National Health and NutritionSurvey. Salud Publica Mex 2010; 52:S1:544-553. Adolescent BMI trajectoryTirosh A, Shai I, Afek A et al.and risk of Diabetes versus Coronary Disease. N Engl J Med 2011; 364: 1315-25. .Hotamisligil GSInflammation and Metabolic Disease. Nature 2006; 444: 860-866. Gustafson B, Hammarstedt A,Anderson CX et al. Inflammed adipose tissue. A culprit underlying the metabolic syndrome andatherosclerosis. Atheroscler Thromb Vasc Biol 2007; 27:2276-2283. Kinstcher U, Hartge M, Hess K etal. T-lymphocyte infiltration in visceral adipose tissue. A primary event in adipose tissue inflammation andthe development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 2008; 28:1304-1310. Hypoxia and Inflammation. N Engl J Med 2011; 364:656-665.Eltzschig HK and Carmeliet P.Taskinen MR, Adiels M, Westerbacka J et al. Dual metabolic defects are required to producehypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol 2011; 31:2144-2150. Chung SK,Sawyer JK, Gebre AK et al. Adipose tissue ABC-A1 contributes to HDL biogenesis in vivo. Circulation2011; 124:1663-1672. Adipose modulation of HDLMc Gilliducay FC, Reylly MP, Rader D.cholesterol. Implications for obesity, HDL metabolism and cardiovascular disease. Circulation 2011;124:1602-1605.
Cardio-Lipidología
60

Incremento de HDLDislipidemias primarias y secundarias. Efecto anti-aterogénico variableEstos trastornos no fueron incluidos en la clasificación de Fredrickson y se caracterizan por
HDL >60mg/dl. Los trastornos primarios del metabolismo de los lípidos que ocasionan estasdislipidemias son los siguientes.
Deficiencias de Cholesterol Ester Transfer Protein -CEPT-Hiperalfalipoproteinémia familiar. Es un trastorno autosómico recesivo o dominante que
afecta al gen de la CEPT -proteína transferidora de esteres de colesterol por triglicéridos entre las HDL y lasVLDL-IDL-LDL-, determina un menor catabolismo de las HDL con incremento en todas sus fraccionescirculantes y reducción en la síntesis de LDL con incremento en la expresión de receptores LDL ydisminución variable de LDL circulante; en población japonesa su prevalencia es relativamente alta -2 a27%- y su fenotipo lipídico se caracteriza por cifras de HDL que oscilan de 40 a 120mg/dl, asociadas a unacontroversial protección cardiovascular, la cual en el estudio Honolulu fue más probable con cifras de HDLentre 60 y 80mg/dl y menos probable con cifras <40mg/dl o >80mg/dl -fenómeno de curva U-.
Se han identificado al menos 17 mutaciones en el gen de la CEPT, las cuales determinandiferentes estructuras moleculares de la proteína y probablemente efectos biológicos también variables; losindividuos homocigotos carecen de actividad de la CEPT en tanto los heterocigotos conservan entre 50 y75% de la actividad de la transferasa.
Condiciones con incremento secundario de HDLEstrógenosEjercicio aeróbicoDosis bajas de alcohol -1 ración al día-.ColestasisFármacos: corticoesteroides, fenitoina, insulina.Interrupción de tabaquismoFármacos inhibidores de CEPTFármacos moduladores de CEPTOtros fármacos incrementadores de HDL -niacina-
Dislipidemias
61

Reducción de HDLDislipidemias primarias y secundarias. Efecto aterogénico variableEstos trastornos al igual que el incremento de HDL no fueron incluidos en la clasificación de
Fredrickson y se caracterizan principalmente por HDL bajo. Los trastornos primarios del metabolismo delos lípidos que ocasionan estas dislipidemias son los siguientes.
Alteración en el transferidor ATP Binding Cassette -ABC-A1-Hipoalfalipoproteinemia familiar. Es un trastorno autosómico dominante que afecta en
forma heterocigota al gen del ABC-A1, determina una disminución en la transferencia de colesterol delmacrófago hacia la apoA1 con disminución en la síntesis de HDL y reducción en todas sus fraccionescirculantes, su fenotipo lipídico se caracteriza por cifras de HDL por abajo del percentil 10 -en México<23mg/dl en hombre y <26mg/dl en mujeres-, no necesariamente asociadas a un incremento en el riesgode eventos cardiovasculares. En vías a establecer el diagnóstico de hipoalfalipoproteinemia aislada oprimaria deben descartarse causas secundarias de HDL bajo -ver adelante- e investigar el fenotipo lipídicofamiliar en busca de otros casos asociados.
Enfermedad de Tangier. Es un trastorno autosómico dominante que afecta en formahomocigota al gen del ABC-A1, determina una incapacidad para la transferencia de colesterol delmacrófago hacia la apoAI la cual es catabolizada en la circulación sin evolucionar a formas maduras deHDL -preβ-HDL, α-HDL, HDL3 etc- con proliferación subendotelial de macrófagos “espumosos”, sufenotipo lipídico se caracteriza por cifras de HDL por abajo del percentil 5 -<10mg/dl-, LDL bajo ytriglicéridos altos, asociadas a infiltración por macrófagos espumosos en amígdalas -orange tonsils-,hígado, bazo, ganglios linfáticos y mucosa intestinal con alteraciones hematológicas, incremento en elriesgo de eventos cardiovasculares y neuropatía de presentación variable -mono o polineuropatía, simétricao asimétrica, intermitente o progresiva-.
Se han identificado al menos 100 mutaciones en el gen del ABC-A1 con expresiones biológicasdiversas que van desde el estado asintomático con reducción de HDL -20 a 40mg/dl- hasta la enfermedadde Tangier.
Alteración en la Lecitin Cholesterol Acyl Transferase -LCAT-Enfermedad de “ojo de pescado” y enfermedad de Norum. Son trastornos autosómicos,
recesivos que afectan al gen de la LCAT, determinan disminución en la esterificación del colesterolcontenido en las formas inmaduras de HDL -preβ-HDL y α-HDL-, por deficiencia de αLCAT en la
Cardio-Lipidología
62

enfermedad de “ojo de pescado” o deficiencia para la esterificación del colesterol en todo tipo delipoproteínas -HDL, VLDL, IDL y LDL-, por deficiencia de α y β-LCAT en la enfermedad de Norum condisminución en la síntesis de HDL y reducción en todas sus fracciones circulantes con un fenotipo lipídicoque se caracteriza por cifras de HDL <10mg/dl, no asociadas según Norum y Cols. a incremento en elriesgo de eventos cardiovasculares aunque con opacidades corneales en la enfermedad de “ojo de pescado”y con dicho signo más anemia hemolítica, hematuria, proteinuria e insuficiencia renal en la enfermedad deNorum. Esta última complicación se atribuye al depósito renal de lipoproteínas ricas en colesterol yfosfolípidos no esterificados.
Se han descrito al menos 45 mutaciones en el gen de la LCAT con fenotipos variables enfunción del grado de deficiencia de la α y/o β actividad de la LCAT.
Alteraciones en apoAISon trastornos autosómicos, que afectan al gen de la apoAI, determinan un espectro que va
desde la ausencia en la síntesis de apoAI con ausencia de síntesis de HDL hasta mutaciones en la estructurade la apoproteína con tipos especiales de apoAI como la apoAI Milán o la apoAI París con HDLfuncionales, su fenotipo lipídico se caracteriza por reducción de HDL en las variedades como apoAI Milano París hasta ausencia completa de apoAI y HDL, esta última asociada a incremento en el riesgo de eventoscardiovasculares, xantomas planos y opacidades corneales; los tipos apoAI Milán y París tienengeneralmente un efecto cardioprotector.
Se han descrito múltiples mutaciones en el gen de la apoAI con fenotipos variables en funcióndel grado de deficiencia de la síntesis de la apoproteína.
Condiciones con reducción secundaria de HDLTabaquismoObesidad-síndrome metabólicoEstrésInfeccionesEnfermedades sistémicas: neoplasias, hepatopatíasFármacos: andrógenos, progestágenos, probucol, corticoesteroides, bloqueadores-beta,
diuréticos especialmente tiazídicos -hidroclorotiazida-.Aguilar-Salinas CA, Gómez RA y Gómez FJ. Abordaje diagnóstico de las dislipidemias. En
Dislipidemias
63

Denominación Alteración HDL mg/dl SignosHiperalfalipoproteinémia
Familiar
Gen CEPT 40-120 Protección cardiovascular
controversial (60-80mg).
Hipoalfalipoproteinémia
Familiar
Gen ABC-A1. Heterocigoto
< Percentil 10 Riesgo cardiovascular
controversial
Enfermedad de Tangier Gen ABC-A1. Homocigoto
< Percentil 5 Infiltración de células
espumosas: amígdalas,
hígado, bazo, ganglios,
mucosas intestinales;
alteraciones hematológicas,
neuropatía etc.
Aterosclerosis precoz.
Enfermedad de «ojo de
pescado».
Enfermedad de Norum
Gen LCAT
. a-LCAT
. aß-LCAT
<10 Opacidades corneales,
anemia hemolítica,
hematuria, proteinuria e
IRC.
Riesgo cardiovascular
controversial.
Ausencia apoAI Gen apoAI Ausencia Opacidades corneales y
xantomas planos.
Aterosclerosis precoz
Alteraciones apoAI Gen apoAI. Milán
. París
10-40 Protección cardiovascular
Denominación Alteración HDL mg/dl SignosHiperalfalipoproteinemia
Familiar
Gen CEPT 40-120 Protección cardiovascular
controversial -60-80mg-.
Hipoalfalipoproteinemia
Familiar
Gen ABC-A1. Heterocigoto
< Percentil 10 Riesgo cardiovascular
controversial
Enfermedad de Tangier Gen ABC-A1. Homocigoto
< Percentil 5 Infiltración de células
espumosas: amígdalas,
hígado, bazo, ganglios,
mucosas intestinales;
alteraciones hematológicas,
neuropatía etc.
Aterosclerosis precoz.
Enfermedad de «ojo de
pescado».
Enfermedad de Norum
Gen LCAT
. α-LCAT
. αß-LCAT
<10 Opacidades corneales,
anemia hemolítica,
hematuria, proteinuria e
IRC.
Riesgo cardiovascular
controversial.
Ausencia apoAI Gen apoAI Ausencia Opacidades corneales y
xantomas planos.
Aterosclerosis precoz
Alteraciones apoAI Gen apoAI. Milán
. París
10-40 Protección cardiovascular
Cardio-Lipidología
Modificaciones primarias de HDLLa tabla muestra las Dislipidemias de origengenético determinantes de incremento, reducción ovariaciones en las HDL. Se enuncia denominación,alteración genética, cifras de HDL, signos clínicos ygrado de asociación a Aterosclerosis. Para mayorinformación ver texto
64
Dislipidemias. De lo clínico a lo molecular. Edición 2008; capítulo 3. Editores Carlos A Aguilar, Rita AGomez y Francisco J Gómez. Genetic disorders of lipoproteinCuchel M, Qasim A and Rader D.metabolism. In Therapeutic Lipidology. Edition 2007; chapter 2. Editors Michael H Davidson, Peter PToth and Kavin C Maki. ClinicalAssmann G and Seedorf U. High-density lipoprotein mutations. InLipidology. Edition 2009; chapter 7. Editor Christie M Ballantyne.
Causas primarias de modificaciones de HDL

A Aterotrombogénesis y teroregresiónLa liga entre la Lipidología y la Cardiología
Capítulo 3

GeneralidadesLa Aterosclerosis ha sido calificada como una enfermedad creada por el hombre -man-made
disease-, paradójicamente esta enfermedad se ha convertido en la “asesina número 1” de las generacioneshumanas que la hemos creado. La evolución de aterosis hacia aterotrombosis es un continuo biológico queinicia desde la concepción, evoluciona y puede complicarse en algún momento de la vida de cualquierindividuo. Su espectro histopatológico va desde el hallazgo microscópico de un macrófago en elsubendotelio fagocitando LDL oxidado, hasta aquel visible en la sala de autopsia con una placaaterosclerosa rota que irrumpe trombosada en la luz arterial del tronco coronario izquierdo.
A partir de la aterosis, el proceso evoluciona, desde las fases iniciales de la aterogénesis comolesiones tipos I, II y III, hasta la aterosclerosis “madura” con lesiones tipos IV, V, VII y VIII, o las másgraves, las lesiones tipo VI, constituidas por placas aterosclerosas complicadas por trombosis in situ. Laevolución de la Aterosclerosis es variable y directamente asociada a la precocidad, intensidad y cronicidadde los factores de lesión endotelio-vascular o factores de riesgo cardiovascular. De ellos, el desbalance entreel depósito de LDL -especialmente su fracción oxidada- en el subendotelio y el transporte reverso decolesterol del subendotelio hacia el hígado a favor del primero, es el factor más importante en la génesis yevolución de la enfermedad.
En este apartado se revisará con cierta amplitud la hipótesis aterogénica fundamental,originalmente propuesta por Joseph Goldstein y Michael Brown y consolidada por Daniel Steinberg,Joseph Witztum y colaboradores sobre la modificación por oxidación de las LDL. Esta hipótesis es hoy porhoy la que mejor explica el mecanismo etiopatogénico “pivote” de los cambios citológicos e histológicosque caracterizan al continuo que inicia con la aterosis y eventualmente concluye con la ruptura y trombosisde una placa aterosclerosa. Para algunos autores como Roberts no existen diez ni más factores de riesgocardiovascular, existe sólo uno, y ese es la hipercolesterolemia; este concepto que podría parecer radical oLDL-céntrico se sustenta entre otras, en tres observaciones con sólidos fundamentos básicos y clínicos: a)en el ser humano, la hipercolesterolemia como factor único -vg niños con Hipercolesterolemia Familiar-, essuficiente para generar Aterosclerosis y causar infartos miocárdicos a edades tan tempranas como los 18meses, por el contrario individuos con hipocolesterolemia a lo largo de toda su vida -vg individuos con lamutación de pérdida de función de la PCSK9- tienen un riesgo muy bajo de Aterosclerosis y de eventoscardiovasculares; b) la hipercolesterolemia incrementa proporcionalmente la fracción oxidada de LDL; c) elLDL oxidado es un epítope de patrones moleculares asociados a patógenos o PAMPs y de patrones
66
Cardio-Lipidología

moleculares asociados a daño celular o DAMPs, y como tal es capaz de invocar una respuesta inflamatoriaendotelio-vascular, la cual a diferencia de la iniciada por PAMPs y DAMPs no es autolimitada ygeneralmente es precoz, crónica e intensa.
A continuación de la revisión de la hipótesis fundamental del LDL oxidado, se analizarán losaspectos más importantes sobre las respuestas celulares inflamatorias que caracterizan a la aterogénesis y ala aterotrombogénesis. La activación de la inmunidad innata por epítopes como el OxLDL y suamplificación por la inmunidad adquirida es un verdadero engaño a la biología endotelio-vascular por un“patógeno mimetizado” que es el LDL oxidado, cuya consecuencia histológica, la formación de una placaaterosclerosa -para algunos un granuloma con lípidos- sería irrelevante e incluso inocente; sin embargo suubicación subendotelial y su tendencia a la ruptura con exposición de su contenido a la luz vascular,trombosis y oclusión arterial en los órganos vitales, es lo que erradica la inocencia de las placasaterosclerosas, y al mismo tiempo en ello radica su importancia en la Medicina actual. Finalmente en estecapítulo se presentará la clasificación anatomopatológica de la Aterosclerosis propuesta por Herbert CStary y evidencias clásicas y recientes sobre Ateroregresión.
De esta forma en este capítulo continuaremos la secuencia didáctica que inició en el capítulo 1con la revisión del metabolismo de los lípidos y las lipoproteínas y continuó en el capítulo 2 con el análisis delos trastornos primarios y secundarios en dicha área del metabolismo intermedio, cuya consecuencia másimportante, la que liga a la Lipidología con la Cardiología, es la aterotrombogénesis que aquí nos ocupa.
Stary HC. Hystologic classification of human atherosclerotic lesions. Atherotrombosis andCoronary Heart Disease: pp 441-449. Editors Fuster V, Topol E and Nabel E. Edition 2004, Elsevier.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis. State of the art paper. J Am CollCardiol 2009; 54:2129-2138. History of Discovery. Oxidized low-densitySteinberg D and Witztum J.lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol 2010; 30:2311-2316. O´Donnel CJ andNabel EG. Genomics of Cardiovascular Medicine. Review Article. New Engl J Med 2011; 365:2098-2109.Artensein AW and Opal SM. Proprotein Convertases in health and disease. Mechanisms of disease. NEngl J Med 2011; 365:2507-2518. A tale of coronary artery disease andNabel EG and Braunwald E.myocardial infarction. 200 NEJM Anniversary article. N Engl J Med 2012; 366:54-63.
67
Aterotrombogénesis y Ateroregresión

LDL oxidado. Hipótesis fundamentalEn la Naturaleza el colesterol y las lipoproteínas transportadoras del mismo, especialmente las
LDL son moléculas vitales y no fueron diseñadas para inducir Aterosclerosis. Por ello antes de abordar lahipótesis de la aterogénesis a partir de la modificación por oxidación de las LDL, vale la pena recordar lossiguientes conceptos revisados en el capítulo 1.
Nivel fisiológico de LDL nativo. Este concepto encierra gran relevancia, especialmenteporque su entendimiento da lógica a la idea de que los niveles suprafisiológicos de LDL son condición “sin-equanon” para la aterogénesis. En palabras de Goldstein y Brown “es muy difícil producir aterosclerosispor cualquier medio experimental a menos que el nivel plasmático de colesterol este elevado”. Desde 1978los autores referidos demostraron que una concentración en el plasma de 25mg/dl de LDL es suficientepara lograr una concentración intersticial de LDL de 2.5mg/dl, la cual satura los receptores celulares deLDL e inhibe la síntesis y la captación de colesterol al bloquear la síntesis de la HMGCoAR y de losreceptores de LDL.
En otras palabras 25mg/dl de LDL en el plasma es una concentración biológicamente activapara cubrir las funciones celulares dependientes del colesterol contenido en las LDL. Especies animales
como los roedores, ovejas, bovinos, conejos, perros y gatos tienen valores de LDL 25mg/dl; los�
camellos, puercos, leones y los humanos recién nacidos o cazadores-recolectores tienen valores de LDLentre 25 y 50mg/dl. En condiciones naturales, ninguna de dichas especies desarrolla aterosclerosis. Es claroque una concentración de LDL de 25mg/dl es 500% menor a la concentración de LDL consideradapoblacionalmente “normal” -131.5mg/dl en México-. Estudios de la cinética de las LDL han demostradoque la brecha entre el valor fisiológico y el valor poblacional “normal” de LDL no obedece a un incrementoen la síntesis de LDL, dicha brecha se explica principalmente por una reducción en el catabolismo de lalipoproteína por represión de los receptores de LDL secundaria a la sobreingesta de colesterol y otrasvariables en investigación. De esta forma la brecha entre la cifra fisiológica de LDL -25 a 50mg/dl- y la cifrareal de LDL, en promedio 130mg/dl, -en nuestro país 131.5mg/dl- es linear y directamente proporcional alriesgo de Aterosclerosis y Enfermedad Cardiovascular.
D Reichl, NB Myant, Brown MS, Goldstein JL. Biologically active Low DensityLipoprotein in human peripheral lymph. J Clin Invest 1978; 61:64-71. Goldstein JL, Brown MS.Lipoprotein Receptors: Genetic defense against atherosclerosis. Clinical Research 1982; 30:417-426.
LDL oxidado “un patógeno mimetizado”. En 1979 Joseph Goldstein y Michael Brown
68
Cardio-Lipidología
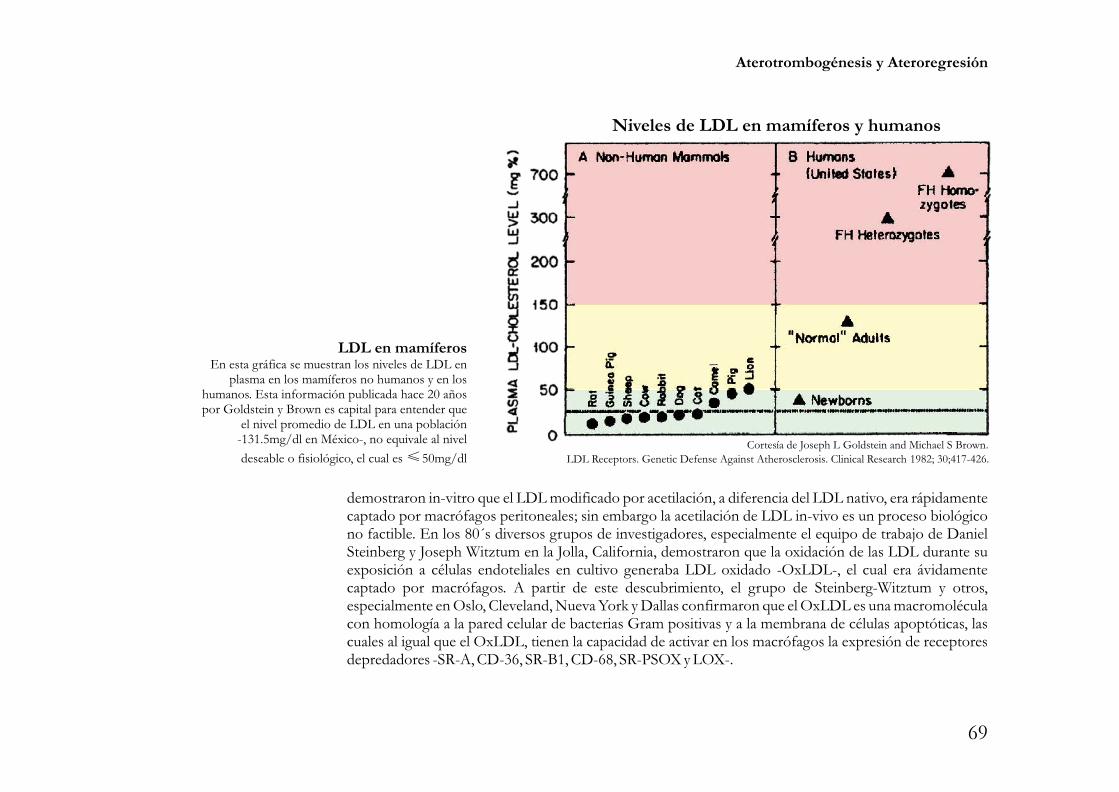
demostraron in-vitro que el LDL modificado por acetilación, a diferencia del LDL nativo, era rápidamentecaptado por macrófagos peritoneales; sin embargo la acetilación de LDL in-vivo es un proceso biológicono factible. En los 80´s diversos grupos de investigadores, especialmente el equipo de trabajo de DanielSteinberg y Joseph Witztum en la Jolla, California, demostraron que la oxidación de las LDL durante suexposición a células endoteliales en cultivo generaba LDL oxidado -OxLDL-, el cual era ávidamentecaptado por macrófagos. A partir de este descubrimiento, el grupo de Steinberg-Witztum y otros,especialmente en Oslo, Cleveland, Nueva York y Dallas confirmaron que el OxLDL es una macromoléculacon homología a la pared celular de bacterias Gram positivas y a la membrana de células apoptóticas, lascuales al igual que el OxLDL, tienen la capacidad de activar en los macrófagos la expresión de receptoresdepredadores -SR-A, CD-36, SR-B1, CD-68, SR-PSOX y LOX-.
LDL en mamíferosEn esta gráfica se muestran los niveles de LDL en
plasma en los mamíferos no humanos y en loshumanos. Esta información publicada hace 20 añospor Goldstein y Brown es capital para entender que
el nivel promedio de LDL en una población-131.5mg/dl en México-, no equivale al nivel
deseable o fisiológico, el cual es 50mg/dl�
Cortesía de Joseph L Goldstein and Michael S Brown.
LDL Receptors. Genetic Defense Against Atherosclerosis. Clinical Research 1982; 30;417-426.
69
Aterotrombogénesis y Ateroregresión
Niveles de LDL en mamíferos y humanos

En condiciones fisiológicas, la acción depredadora de OxLDL del macrófago permite captaren promedio 25% del LDL circulante y eliminarlo vía metabolismo reverso de colesterol -ver capítulo 1-.Así mismo, un subtipo de macrófagos -células dendríticas-, con participación de moléculas del sistemamayor de histocompatibilidad son células “presentadoras” de OxLDL a los linfocitos T y B; ante esteestímulo antigénico los linfocitos producen diversas linfocinas y anticuerpos “naturales” tipo IgM e IgGdirigidos contra OxLDL. El OxLDL in-vivo se genera a partir del LDL nativo por acción de diversossistemas enzimáticos oxidativos, entre ellos lipoxigenasas, mieloperoxidasas, NADPH-oxidasa y óxidonítrico sintasa desacoplada. Tanto los receptores depredadores como los anticuerpos IgM e IgG e inclusopentraxinas como la PCR, reconocen al OxLDL en su componente fosfocolina. Esta misma molécula es elligando común de la pared celular bacteriana y de las membranas de células apoptóticas. A la fecha, lahipótesis de que el OxLDL, específicamente su componente fosfocolina, es un epítope de patronesmoleculares de reconocimiento de patógenos o PAMPs y de daño membranal celular o DAMPs, concapacidad de activar una respuesta de inmunidad innata y adquirida, es una hipótesis ampliamente validaday explica la fisiología del catabolismo del OxLDL y la fisiopatología de la génesis de la Aterosclerosis.
Tsimikas S, Glass CK, Steinberg D, Witztum J. Lipoprotein oxidation, macrophages,immunity and atherogenesis. In Molecular Basis of Cardiovascular Diseases. Edition 2004, Saunders;Chapter 22. Editor Kenneth R Chien.
De macrófago “removedor” a macrófago “inflamatorio”. Hasta cierto límite fisiológicoel colesterol contenido en el OxLDL es removido y eliminado vía transporte reverso de colesterol -vercapítulo 1-. Más allá de dicho límite, el macrófago modifica su fenotipo de un macrófago “removedor” decolesterol hacia el de un macrófago “inflamatorio”. Esta transformación o selección fenotípica delmacrófago se genera cuando existe desequilibrio entre la concentración citoplasmática de colesterol en elmacrófago y la capacidad de las HDL para promover el flujo centrífugo de colesterol. Ello ocasionaincremento en la concentración intracelular de colesterol, reducción en la fluidez de las membranascelulares y formación de “rafts” en la membrana celular y en la membrana de los lisosomas de losmacrófagos. Estos “rafts” o balsas membranales son estructuras que facilitan el anclamiento de múltiplesreceptores primitivos o Tool Like Receptors -TLR-. La exposición de los TLR y la unión con ligandos comoLPS -lipopolysacharids-, AGEs, apoCIII, etc., inicia cascadas enzimáticas que concluyen con la proteolisisde las proteínas inhibidoras de los más importantes factores de transcripción inflamatorios; por ejemplo la
70
Cardio-Lipidología

proteolisis del IkB -Inhibitor of NFkB- y la liberación del NFkB, dicta al núcleo del macrófago la síntesis demás de un centenar de moléculas inflamatorias como MCP1, MCSF, selectinas P, E y L, e inmunoglobulinasde adhesión celular -VCAM, ICAM, PECAM-, PCR, etc.
De esta forma la esencia de la aterogénesis puede explicarse por un exceso suprafisiológico deLDL que origina un incremento de OxLDL y otras moléculas en estudio -apoBDS-1-, con sobrecargafuncional al macrófago; o bien una deficiencia cuali o cuantitativa en los procesos celulares -macrófago-dependientes- o moleculares -HDL-dependientes- involucrados en el flujo centrífugo o reverso decolesterol. Uno de ellos o la combinación de ambos mecanismos determinan procesos citológicos de tipoinflamatorio que cuando se gestan a nivel endotelio-vascular dan pie a cambios histológicos denominadosAterosclerosis.
Yvan-Charbet L, Wang N and Tall AR. Role of HDL, ABCA1, and ABCG1 transporters incholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol 2010; 30:139-143. KetelhuntDF, Rios FJO, Wang Y et al. Identification of a Danger-Associated Peptide from apoB100 -apo BDS-1-that triggers innate proatherogenic responses. Circulation 2011; 124:2433-2443.
Macrófago removedor vs inflamatorioImagen que muestra como un macrófago confenotipo removedor, ante una concentración
fisiológica de colesterol, activa factores detranscripción tipo LXR-RXR que favorecen el flujo
reverso de colesterol. El macrófago ante unaconcentración suprafisiológica de colesterol, vira a
su fenotipo inflamatorio con expresión membranalde TLR y activación de factores de transcripción
tipo NFkB que retroalimentan la respuestainflamatoria
Retículo EndoplásmicoAparato de Golghi
Tooll Like Receptors
Lisosoma
Macrófago «inflamatorio»
Retículo EndoplásmicoAparato de Golghi
Tooll Like Receptors
NFkB
B100
CeC C
C
C
C
RetAparato de Golghi
LXR-RXR
Macrófago «removedor»
Lisosoma
C
C
Retículo EndoplásmicoAparato de Golghi
LXR-RXR
Gen ABCA1-ABCG1
C
ABCA1SR CD-36 AI
C
preß-HDL
Macrófago «removedor»
71
Aterotrombogénesis y Ateroregresión

Fases de la aterotrombogénesisFase de infiltración monocitaria -inmunidad innata-. La teoría inflamatoria de la
Aterosclerosis originalmente propuesta por Rusell Ross en los 70´s y corroborada por múltiplesinvestigadores, ahora es incontrovertible y de ella se conocen la mayoría de los procesos moleculares,celulares e histológicos, mismos que revisaremos a continuación.
El gradiente de LDL entre la luz vascular y el subendotelio y las características anatómicas delos cojinetes endocárdicos son las dos principales determinantes de la migración de las LDL de la luzvascular hacia regiones arteriales ateropronas. Las LDL son “atrapadas” preferencialmente en el aspectoexterno de las bifurcaciones arteriales por los siguientes mecanismos: atracción eléctrica entre la cargapositiva de la apoB100 y la carga negativa del grupo sulfato de los abundantes proteoglicanos de la matrizsubendotelial de los cojinetes endocárdicos, ausencia de vasa vasorum en el subendotelio que impide el“barrido” de las LDL del subendotelio hacia la circulación, secreción de moléculas como LPL,esfingomielasa secretoria -sSMase-, fosfolipasa A2 secretoria -sPLA2- y otras que propician oxidación,proteolisis y lipolisis de las LDL .
Las LDL retenidas en el subendotelio, generalmente son modificadas “in-situ”, y al igual queactivan el fenotipo inflamatorio del macrófago nativo, activan el fenotipo inflamatorio de la célulaendotelial. La célula endotelial expuesta a OxLDL reprime su fenotipo fisiológico y expresa su fenotipoinflamatorio -de respuesta al daño- con síntesis y expresión de moléculas de adhesión celular tipo selectinaP, E y L; con ello se propicia el rodamiento lento de leucocitos y plaquetas sobre la membrana luminal delendotelio. Esta acción de rodamiento se logra por la “atracción” entre el ligando celular SLe -sialina Lewis-y las selectinas endoteliales; el efecto adhesivo de las selectinas es relativamente débil y breve; moléculas deadhesión tipo inmunoglobulina como VCAM, ICAM y PECAM, otorgan una acción de adhesión intensa yprolongada gracias a la atracción entre dichas inmunoglobulinas y sus ligandos celulares tipo integrina de lafamilia VLA, especialmente el VLA4 expresado en monocitos y linfocitos, no así en neutrófilos. Una vezadheridos al endotelio, leucocitos, plaquetas y mastocitos liberan citoquinas y autacoides que fosforilan a lasproteínas contráctiles del citoesqueleto endotelial incluyendo a la proteína de unión caderina, con ello, lascélulas endoteliales se contraen en sentido centrípeto, sus uniones intercelulares “apretadas” se tornan laxasy se “abre paso” para la migración de macromoléculas y células sanguíneas inflamatorias -monocitos,linfocitos, etc.-, de la luz vascular hacia el subendotelio.
La migración de monocitos o diapédesis del aspecto luminal del endotelio hacia el
72
Cardio-Lipidología

subendotelio, es inducida por moléculas quimiotácticas como la MCP1 -Monocyte ChemoatractantProtein 1-; en el subendotelio, los monocitos proliferan por efecto de moléculas tipo citoquina con afectomitogénico como el MCSF -Monocyte Colony Stimulant Factor-, ambas moléculas MCP1 y MCSF sonsintetizadas por monocitos y células endoteliales activadas. Los monocitos migrados y multiplicados en elsubendotelio, especialmente la subpoblación de fenotipo proinflamatorio con el marcador de membranaPSGL -P Selectin Glycoprotein Ligand-, retroalimentan los procesos de inmunidad innata originalmenteactivados por OxLDL. Esta subpoblación de monocitos a diferencia de la subpoblación sin el fenotipoPSGL, por acción de factores de transcripción como el NFkB sintetizan y expresan múltiples receptoresprimitivos tipo TLR -CD14 para LPS, TLR4, TLR2 para apoCIII, SR-A y LOX para OxLDL, RAGES paraAGEs entre los más importantes- y sintetizan moléculas como proteasas, RLO2, IL1, TNF-alfa etc., y asíretroalimentan la conducta celular inflamatoria innata que se complementa con la participación de larespuesta celular inflamatoria inmunomodulada o adquirida que se comenta enseguida.
Fase de infiltración linfocitaria -inmunidad adquirida-. La subpoblación de monocitosdenominados células dendríticas, es reconocida como una estirpe de células “presentadoras” de antígenos alos linfocitos T. Las células dendríticas tienen la capacidad de presentar a los linfocitos T complejosantigénicos formados como PAMPs, DAMPs o bien OxLDL; el tipo de respuesta celular está en función dela población de linfocitos T estimulada. Las subpoblaciones de linfocitos CTL -Cytolitic T Cell- son células“asesinas” de la célula que hospeda al antígeno; los linfocitos T “helper” Th1 y Th17 sintetizan citoquinascomo interferon-gamma y TNF-alfa que sinergizan la respuesta inflamatoria; los linfocitos T “helper” Th2sintetizan citoquinas como IL4 y si bien aún es controversial, antagonizan la respuesta inflamatoria de loslinfocitos Th1 y Th17; la acción antinflamatoria de los linfocitos Th2 es favorecida por la subpoblación delinfocitos T “regulators” que sintetizan citoquinas como IL10 y otras moléculas inhibidoras demetaloproteinasas, PDGF o factor de crecimiento derivado de plaquetas que propicia la atracción decélulas musculares lisas, su transformación en fibroblastos y la producción de tejido fibroso, TGF-beta ofactor beta de crecimiento y transformación, el cual induce una respuesta fibroproliferativa y decalcificación al codificar la síntesis de la proteína morfogenética y el factor básico de crecimientofibroblástico -bFGF-, que induce una reacción proliferativa de fibroblastos. Finalmente los linfocitos Bcuando son activados por la presentación de OxLDL sintetizan anticuerpos contra este epítope, con unefecto final anti-aterogénico.
73
Aterotrombogénesis y Ateroregresión

Disfunción y activación endotelialImagen que muestra como el LDL oxidado reprimeen la célula endotelial la síntesis de ON, siendo elloel principal marcador de disfunción endotelial. ElLDL oxidado a través de receptores como LOX,activa en la célula endotelial su fenotipoinflamatorio con síntesis y expresión de moléculasde adhesión
Monocitos. Diapedesis y mitogénesisImagen que muestra la adhesión de monocitoscirculantes a los segmentos de endotelio activados yla acción citoquinética de moléculas como la MCP1que induce la migración o diapédesis de monocitoshacia el subendotelio. Los monocitos infiltrados enel subendotelio, se multiplican por la accióncitomitogénica de moléculas como el MCSF
74
Disfunción endotelial
Reducción en la síntesis de ON
Activación endotelial
Moléculas de adhesión
Diapédesis de monocitos
MCP-1 MCP-1
Infiltración y reproducción monocitaria
M-CSF
-
Cardio-Lipidología
OxLDL
OxLDL OxLDL

Linfocitos y NeovascularizaciónImagen que muestra como al igual que para los
monocitos, la adhesión de linfocitos circulantes alos segmentos de endotelio activados, es
complementada por la acción citoquinética demacrófagos activados “presentadores” de antígenoscomo OxLDL. La acción de interferón-gamma y el
incremento de volumen con hipoxia intraplacaactivan la síntesis de moléculas angiogénicas comoHIF y VEGF que inducen la angiogénesis de vasa-
vasorum 2ª a partir de la vasa-vasorum 1ª oadventicial
Hemorragia intraplaca y rupturaImagen que muestra la ruptura de la vasa-vasorum
2ª con hemorragia intraplaca, acúmulo de colesterollibre y hemoglobina que producen crecimiento e
inflamación acelerados. La preponderancia demacrófagos y linfocitos con fenotipo inflamatorio
Th1 determina la proteolisis de la cubierta fibrosa yla ruptura de la placa con trombosis secundaria. La
repercusión de la trombosis está en función defactores intrínsecos y sistémicos
75
Infiltración linfocitaria
-
Linfocitos Th1
Neovascularización intraplaca
-
HIF-VEGF
Hemorragia Intraplaca
-
Eritrocitos
Ruptura de placa y trombosis
ProteinasasF. Tisular
-
Aterotrombogénesis y Ateroregresión

El balance entre la actividad celular de monocitos proinflamatorios, linfocitos T tipo CTL,Th1, Th17 versus la actividad celular de monocitos antiinflamatorios, linfocitos Th2, T “regulators” ylinfocitos B, dictará el resultado entre la susceptibilidad a la ruptura vs la estabilidad intrínseca de la placaaterosclerosa. Extrínsecamente el estrés hemodinámico, la inflamación sistémica condicionada por otrosfactores de lesión endotelio-vascular como hipertensión arterial, hiperglucemia, etc., y las infecciones sonentre otros, gatillos para precipitar la ruptura de la placa aterosclerosa.
Fase de ruptura de placa. El centro de una placa aterosclerosa contiene una mezcla defragmentos celulares resultantes de la muerte celular por apoptosis, especialmente de macrófagos, condetritus conteniendo colesterol libre, colesterol esterificado, colesterol cristalizado y factor tisularproducido por el macrófago y liberado tras su apoptosis. La ruptura de la capa fibrosa de la placaaterosclerosa, la exposición de los componentes de la matriz subendotelial y la liberación del contenido dela placa aterosclerosa inician los procesos de trombosis patológica.
Los mecanismos reconocidos para explicar la ruptura de la capa fibrosa de una placaaterosclerosa son la fractura por factores extrínsecos “de fuera hacia dentro”, la erosión del aspecto luminalde la placa aterosclerosa y la fractura por hemorragia intraplaca “de dentro hacia fuera”. Los determinantesde la vulnerabilidad para la ruptura de una placa aterosclerosa son los siguientes. LaEstructurales.apoptosis de las células endoteliales con denudación endotelial es una causa frecuente de trombosis enregiones con flujo sanguíneo no laminar. Por otra parte, el tamaño del centro necrótico de la placaaterosclerosa se relaciona directamente con la fragilidad de la misma, principalmente si su localización esexcéntrica. El grosor de la capa fibrosa se asocia inversamente con la fragilidad de la placa aterosclerosa. Encontra del sentido común, la mayor probabilidad de un evento de trombosis arterial, no está en relacióndirecta con el grado de estenosis luminal, las placas con mayor probabilidad de ruptura y trombosis sonaquellas con una remodelación positiva o centrífuga, con poca pérdida de la luz vascular y por ende con unamayor tensión radial. Un centro necrótico que ocupa más de 40% del volumen de la placa y es excéntricohacia el “hombro” de la misma, en donde la capa fibrosa generalmente es delgada, es un marcador de altaprobabilidad de ruptura. La acumulación de macrófagos conteniendo OxLDL y linfocitosInflamatorios.CTL, Th1 y Th17 hacia las zonas excéntricas de la placa aterosclerosa, favorece su ruptura en esta regióndadas las características inflamatorias y proteolíticas que se han referido. . Como fueAngiogénicosanalizado con anterioridad, en procesos como la aterosclerosis, el incremento progresivo del grosor de la
76
Cardio-Lipidología

placa ocasiona hipoxia tisular intraplaca que incita la síntesis de factores de transcripción angiogénicoscomo HIF -Hipoxia Induced Factor- y VEGF -Vascular Endothelial Growth Factor-. La vasa-vasorumintraplaca, generada a partir de la vasa-vasorum primaria o adventicial es una estructura vascular con unendotelio activado, de elevada permeabilidad y susceptible a la ruptura, lo cual favorece la acumulaciónrápida de elementos formes de la sangre -hemorragia intraplaca-. Especialmente los eritrocitos contribuyenal crecimiento acelerado y eventualmente a la ruptura de la placa dado su alto contenido de colesterol libreen la membrana eritrocitaria que a la postre será oxidado y la liberación de hemoglobina con formación decomplejos hem-haptoglobina-2, que al igual que el OxLDL son proinflamatorios. Dichos procesos hacende la angiogénesis intraplaca un mecanismo cada vez más reconocido como factor de ruptura de la placaaterosclerosa de “dentro hacia fuera”.
Inci
den
cia
en%
de
EC
V
0
5
10
15
20
4.9
1.3
10.2
1.7 1.7 1.9
16.418.2
H.RValor PPrevalencia
3.9<0.001
46.7
6.5<0.001
15.9
10.8<0.001
10.1
11.0<0.001
4.2
Capa fibrosa delgada
Característica presenteCaracterística ausente
Tipo de placas por USG y Eventos CardiovascularesTipo de placa y eventos
Greg Stone con el estudio PROSPECT demostróin-vivo que la carga de ateroma, el diámetro luminal
mínimo y el grosor de la capa fibrosa de la placaaterosclerosa son los 3 principales determinantes deeventos cardiovasculares subsecuentes. Estos datos
prospectivos confirman los hallazgosanatomopatológicos retrospectivos generalmente
“post-mortem” y anticipan el gran potencial de losmétodos de imagen invasivos y no invasivos en la
predicción de eventos cardiovasculares
77
Aterotrombogénesis y Ateroregresión
Capa fibrosa delgada Capa fibrosa delgada Capa fibrosa delgadaDiámetro luz < 4 mm2 Cor ateroma <70% Diámetro luz < 4 mm2
Cor ateroma <70%

Los datos anatomopatológicos referidos con anterioridad, generalmente retrospectivos ypost-mortem, recientemente han sido confirmados por estudios prospectivos in-vivo con el empleo deultrasonido intracoronario en escala de grises y radiofrecuencia. En el estudio PROSPECT, en individuospost-SICA, hasta un 50% de eventos cardiovasculares subsecuentes se originaron en segmentos arterialescoronarios diferentes al segmento “índice”, con placas aterosclerosas que en promedio tenían 30% de
estenosis angiográfica, siendo: un centro ateromatoso de 70% del volumen de la placa, un área luminal�
mínima 4mm2 o una capa fibroateromatosa delgada, los principales predictores de eventos�
cardiovasculares en individuos con aterosclerosis establecida e historia de un SICA.Fase de trombosis. La ruptura de la placa aterosclerosa con exposición de la matriz
subendotelial y el contenido del corazón necrótico de la misma, activa rápida e intensamente latrombogénesis. En un endotelio activado por la acción de las selectinas e inmunoglobulinas de adhesión sefavorece la adhesión plaquetaria; así mismo se pierde el flujo sanguíneo laminar y la electronegatividad delendotelio y por lo tanto existe una atracción electrostática entre la plaqueta y el subendotelio favorecida porla falta de síntesis de acción de óxido nítrico, prostaciclina y ecto-ADPasa y el exceso de endotelina 1 ytromboxano A2. La adhesión y activación plaquetaria se consolida por la exposición de moléculas de lamatriz subendotelial como colágena, factor de VW y otras proteínas, y su unión a los receptoresplaquetarios tipo integrina como el Ia-IIa para la colágena o el Ib-IX-X para el factor de VW. Las plaquetasasí activadas liberan sus gránulos intracitoplasmáticos con tromboxano A2, adenosina y trombina. Estasmoléculas retroalimentan la activación plaquetaria por su unión a los receptores plaquetarios de membranaasociados a proteínas G: receptores TP-alfa para el tromboxano A2; P2Y1, P2Y12 y P2X-1 para laadenosina y PAR 1-3-4 para la trombina. Finalmente la activación por fosforilación de cascadasenzimáticas dependientes de fosfolipasas C con incremento de calcio en el citoplasma plaquetario yexpresión del receptor tipo integrina o glucoproteína IIb-IIIa consolida la agregación plaquetaria por laformación de uniones plaqueta-plaqueta, plaqueta-fibrinógeno y plaqueta-factor de VW.
Una vez formado el trombo plaquetario, su estabilización por una red de fibrina estádeterminada por la pérdida de la capacidad de síntesis endotelial del inhibidor del factor tisular y por la granliberación de este factor contenido en la placa aterosclerosa. El factor tisular tras su unión al factor VIIa,acelera la formación del factor Xa y este transforma al factor II o protrombina en factor IIa o trombina.Finalmente la trombina convierte al fibrinógeno en fibrina, macromolécula que a manera de red estabiliza al
78
Cardio-Lipidología

trombo plaquetario. En forma inversa al proceso activado de la coagulación, la fibrinolisis fisiológica estáinhibida, existiendo deficiencia de AHPT y sobreproducción del inhibidor 1 de activador de plasminógeno-PAI1-. Así la ruptura de la capa fibrosa y la exposición del contenido de la placa aterosclerosa al torrentesanguíneo, se caracteriza por adhesión, activación y agregación plaquetaria aceleradas, coagulación exaltaday fibrinolisis inhibida, resultado un estado trombogénico causante de la gran mayoría de muertes súbitasy/o síndromes isquémicos agudos.
La repercusión funcional de un trombo depende de su localización, la velocidad de formación,su tamaño y otras variables extrínsecas al trombo como: vasoreactividad del segmento arterialcomprometido, características de la circulación colateral, preacondicionamiento miocárdico a la isquemia,eficiencia de los mecanismos fisiológicos y/o farmacológicos antiagregantes plaquetarios, anticoagulantesy trombolíticos, entre otras condiciones. En base a lo anterior, el concepto de placa vulnerable se hatrasladado de una visión centrada al tipo de placa aterosclerosa hacia el concepto de paciente vulnerable, enel cual el ambiente sistémico de cada individuo modula la trombogenicidad y repercusión funcional de unao varias placas aterosclerosas.
VWF
GP Ibß-IX-V GP Ia-IIa ( a2ß1)
Colagena TxA2 ADP Trombina
TxA2-R P2Y12 PAR1
Fibrinógeno
GP IIb-IIIa (aIIbß3)
Activación-lesión endoteilalPerdida de flujo laminar
Perdida de electronegatividad endotelialReducción de síntesis de ON, PGI y ADP-asa
Incremento de síntesis de E1 y TxA2
Ruptura de placa y exposición-liberación de ligandos a receptores plaquetarios
Activación vía extrínseca de la coagulación+
Inhibición de la fibrinolisis
Trombosis
+
+
TrombogénesisLa ruptura de la placa aterosclerosa es el eventofinal que dispara el gatillo para la trombosis. Sin
embargo existen diversos componentes pro-trombogénicos que preceden y/o acompañan a la
ruptura de la placa aterosclerosa, entre ellos:activación y lesión endotelial, expresión de
múltiples ligandos de receptores plaquetarios,activación extrínseca de la coagulación e inhibición
de la fibrinolisis son los más importantes
79
Aterotrombogénesis y Ateroregresión

Ross R, Glomset JA. The pathogenesis of atherosclerosis. N Engl J Med 1976; 295:369-77.Ross R. Herrman J, LermanAtherosclerosis an inflammatory disease. N Engl J Med 1999; 340:115-126.L, Rodríguez-Porcel M. Coronary vasa vasorum neovascularization precedes epicardial endothelialdisfunction in experimental hypercholesterolemia. Cardiovasc Res 2001; 51:762-766. Kolodgie FD, GoldHK, Burke AP. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med 2003;349:2316-2325. Pathogenesis of atherothrombosis. In Atherothrombosis and CoronaryFalk E, Salk PK.Heart Disease: pp 451-465. Editors Fuster V, Topol E and Nabel E. Edition 2004, Elsevier. Corti R,Badimon L, Fuster V, Badimon JJ. Coronary thrombosis. Local and systemic factors. InAtherothrombosis and Coronary Heart Disease: pp 475-485. Editors Fuster V, Topol E and Nabel E.Edition 2004, Elsevier. Inflammation and immunity in atherogenesis.Libby P, Hansson GK, Pober JS.In Molecular Basis of Cardiovascular Diseases. Chapter 20. Editor Kenneth R Chien. Edition 2004,Saunders. Inflammation, atherosclerosis, and coronary heart disease. N Engl J Med 2005;Hanson GK.352:1685-1695. . Endothelial activation and theDe Caterina R, Zampolli A, Lazzerini G, Libby Pinitiation of atherosclerosis. In endothelial disfunctions and vascular disease. Chapter 2. Editors RaffaeleDe Caterina, Peter Libby. First edition 2007, Blackwell Futura. De Caterina R, Zampolli A, Del Turco S,Libby P. Mechanisms of plaque progression and complications. In endothelial disfunctions and vasculardisease Chapter 3. Editors De Caterina R, Libby P. First edition 2007, Blackwell Futura. Libby P, RidkerPM, Hansson GK. Inflammation in atherosclerosis. State of the art paper. J Am Coll Cardiol 2009;54:2129-2138. . Aterotrombogenesis. En Atlas deMorales-Villegas E, Di Sciascio G, Briguori CEndotelio, Aterotrombosis y Estatinas. Capítulo 3. Editor Morales . Edición 2010-11. EditorialEnriqueAtheros. . PROSPECT trial. A prospective natural-history studyStone GW, Maehara A, Lansky AJ et alof coronary atherosclerosis. N Engl J Med 2011; 364:226-235. Adventitia vasa vasorum andVirmani R.inflammation.A war in progress of defense vs betrayal. In 44th Annual New York CardiovascularSymposium; 2011. Book Program: 40-42. What fans the fire. Insights intoMajor AS, Harrison DG.mechanisms of inflammation in Atherosclerosis and Diabetes Mellitus. Circulation 2011; 124:2809-2811.Keaney JF. Eriksson EE.Immune modulation in Atherosclerosis. Circulation 2011; 124:e559-e560.Intravital microscopy on Atherosclerosis in apoE-deficient mice establishes microvessels as mayor entrypathway for leukocytes to advanced lesions. Circulation 2011; 124:2129-2138.
80
Cardio-Lipidología

Clasificación de la A.H.A para la aterosclerosisInicialmente propuesta por Stary, la clasificación micro y macroscópica de la aterosclerosis que
aquí se presenta es la clasificación avalada por la American Heart Association.Tipos de lesiones:
Caracterizada por macrófagos aislados en el subendotelio, con lípidos fagocitados, sinlípidos extracelulares ni distorsión de la anatomía arterial.Caracterizada por macrófagos agrupados en el subendotelio, con lípidos fagocitados, sinlípidos extracelulares ni distorsión de la anatomía arterial.Caracterizada por macrófagos infiltrando estructuras para y subendoteliales, con lípidosfagocitados, lípidos extracelulares y distorsión incipiente de la anatomía arterial.Ateroma clásico.Ateroma con fibrosis incipiente.Ateroma complicado con erosión, hemorragia, ruptura y/o trombo.Ateroma con calcificación predominante.Ateroma con fibrosis predominante.
Evolución. Las lesiones tipo I, II, III y IV evolucionan en forma progresiva, sin embargo unavez constituida una lesión tipo IV, esta puede hacer la transición hacia otros tipos de lesiones más avanzadassin una secuencia cronológica. Las lesiones tipo I, II y III no tienen manifestaciones clínicas por sí mismas.Las lesiones tipo IV y V pueden transformarse en lesiones tipo VI que son características de los síndromesisquémicos agudos. Las lesiones tipo VII y VIII pueden ser clínicamente silentes o causar síndromesisquémicos, generalmente estables y en raras ocasiones agudos.
Lesiones tempranas tipo I y II. Macroscópicamente descritas como puntos y/o estríasgrasas, las lesiones tipo I y II están formadas por macrófagos aislados o acúmulos de macrófagos cargadosde lípidos. Pueden coexistir también células musculares lisas, linfocitos y mastocitos nativos del espaciosubendotelial cargados de lípidos, no existen lípidos extracelulares y la estructura endotelial, subendotelial,media y adventicia arteriales están conservadas. Estas lesiones no provocan estenosis de la luz arterial y sedetectan a lo largo de toda la vida desde la infancia hasta la edad adulta. Su prevalencia en lactantes yadolescentes es de 45% y 70% respectivamente. La coexistencia de factores de riesgo cardiovascular acelerasu progresión hacia lesiones de tipo III y IV.
Lesiones intermedias tipo III. Macroscópicamente similares a las estrías grasas, están
81
I
II
III
IVV
VIVII
VIII
Aterotrombogénesis y Ateroregresión

formadas por macrófagos cargados de lípidos y restos de macrófagos apoptóticos con lípidos y detrituscelulares en el espacio subendotelial, la estructura subendotelial esta distorsionada y en forma incipiente seve afectada la capa media y la adventicia arteriales. Estas lesiones no provocan estenosis de la luz arterial yson el preámbulo de lesiones tipio IV.
Lesiones intermedias tipo IV y V. Denominadas ateromas clásicos, macroscópicamenteestán formadas por macrófagos cargados de lípidos, linfocitos y restos de macrófagos apoptóticos conlípidos y detritus celulares en el espacio subendotelial formando un corazón necrótico, limitado por unadelgada capa constituida por proteoglicanos de la matriz intercelular y células musculares lisas. Laestructura del subendotelio está francamente distorsionada al igual que la media y adventicia arteriales.Estas lesiones son proclives a la ruptura y se han descrito dos tipos de proceso adaptativo que reducen suvulnerabilidad a la ruptura. El primero es la elongación centrífuga con ruptura de la lámina elástica externa.Esta remodelación denominada positiva o Glagowiana, limita la potencial obstrucción de la luz arterial y secomplementa con el segundo proceso adaptativo que es la formación de una capa de tejido fibrosoalrededor del centro necrótico, la cual se desarrolla gracias a la transformación fibroblástica de las célulasmusculares lisas productoras de colágena. Por su estructura, grado de inflamación y contenido de vasa-vasorum secundaria, las lesiones tipo IV-V pueden romperse y/o erosionarse, y propiciar la transformaciónen una lesión tipo VI.
Lesiones complicadas tipo VI. La erosión y/o ruptura de una placa aterosclerosa, induce latrombosis mural del segmento comprometido y con ello la obstrucción súbita de una arteria. Dicho trombopuede lisarse por los mecanismos trombolíticos ya analizados o bien reorganizarse y contribuir alcrecimiento intermitente de la placa aterosclerosa.
Lesiones tardías tipo VII-VIII. Cuando una placa aterosclerosa contiene más de 50% decálcio se denomina tipo VII o calcificada. Si el contenido es predominantemente tejido fibroso, ladenominación será de una lesión tipo VIII o fibrótica.
Stary HC. Atlas de progresión y regresión de la aterosclerosis. Segunda edición 2003. Ed.Parthenon Publishing.
82
Cardio-Lipidología

Tipos de placa aterosclerosaBifurcación de arteria descendente anterior con
cojinete o engrosamiento endocárdico fisiológico-sitio ateroprono-. Arteria descendente anterior
proximal con lesión aterosclerosa tipo IV o ateromaclásico, estadísticamente presente en 1 de cada 4
individuos mayores de 25 años de edad
Tipos de placa aterosclerosaArteria descendente anterior proximal con lesión
aterotrombótica tipo VI causante de muerte súbita.Arteria descendente anterior proximal con lesión
aterosclerosa tipo VIII con estenosis significativa ycausante de angor pectoris estable
83
Arteria normal Arteria con lesión tipo IV
Arteria con lesión tipo VI
Arteria con lesión tipo VII-VIII
Aterotrombogénesis y Ateroregresión

AteroregresiónAl igual que los cambios anatomopatológicos del proceso aterotrombogénico, también están
descritos aquellos que caracterizan a la regresión de una placa aterosclerosa y le dan racional a las estrategiasfarmacológicas para lograr tal fin. En estudios con monos Macacus Rhesus se han descrito dos etapas de laateroregresión, una fase temprana y una tardía. Con el advenimiento del ultrasonido intravascular se hapodido confirmar que los cambios en animales son reproducibles en el ser humano.
Fase temprana. Esta fase se observa en Macacus Rhesus al mantener la concentración decolesterol total en suero por debajo de 180 mg/dl durante un lapso de 3 a 6 meses. Histológicamente secaracteriza por la degradación de los lípidos de la matriz subendotelial, lo cual induce la involución delesiones tipo I y II. En lesiones tipo III y IV amén de la reducción de lípidos en la matriz subendotelial,también se ha documentado su reducción en el interior de los macrófagos y células musculares lisas conapoptosis de los mismos y cese de su proliferación.
Fase tardía. Descrita en Macacus Rhesus al sostener la concentración de colesterol total ensuero por debajo de 180 mg/dl por más de 6 meses. Se caracteriza por la transformación de lesiones tipo IVy V en lesiones tipo VII y VIII con escasa o nula celularidad y un corazón necrótico escaso o nulo, sustituidopor tejido fibroso y/o cálcio. Este cambio morfológico reduce la vulnerabilidad para la ruptura de una placaaterosclerosa, por lo cual es una involución benéfica y factible de lograr en un escenario clínico, y explicapor ejemplo, el incremento en la densidad de cálcio en placas aterosclerosas meses después del tratamientocon estatinas.
Ateroregresión en humanos. Tras varios decenios de evaluar la aterosclerosis coronaria enforma semi o cuantitativa por medios angiográficos, el advenimiento del USG intracoronario ha permitidouna aproximación más precisa en el estudio de la Aterosclerosis in-vivo. Por USG intracoronario yradiofrecuencia es posible evaluar el diámetro luminal mínimo, el porcentaje de ateroma, el grosor de lacapa fibrosa e incluso los componentes del ateroma; esta información facilita la calificación y lacuantificación de las placas aterosclerosas, siendo factible demostrar cambios en la calidad de la placa oplacas aterosclerosas así como cambios en el volumen de la mismas.
El primer estudio con USG intra-coronario que hizo evidente la estabilización en el volumende la placa aterosclerosa e incluso la regresión del mismo fue el estudio REVERSAL. En este estudio seconfirmó que la regresión en el volumen de la placa aterosclerosa se mueve en función directa a lareducción del nivel circulante de LDL y de PCR -a mayor reducción del binomio, mayor posibilidad de
84
Cardio-Lipidología
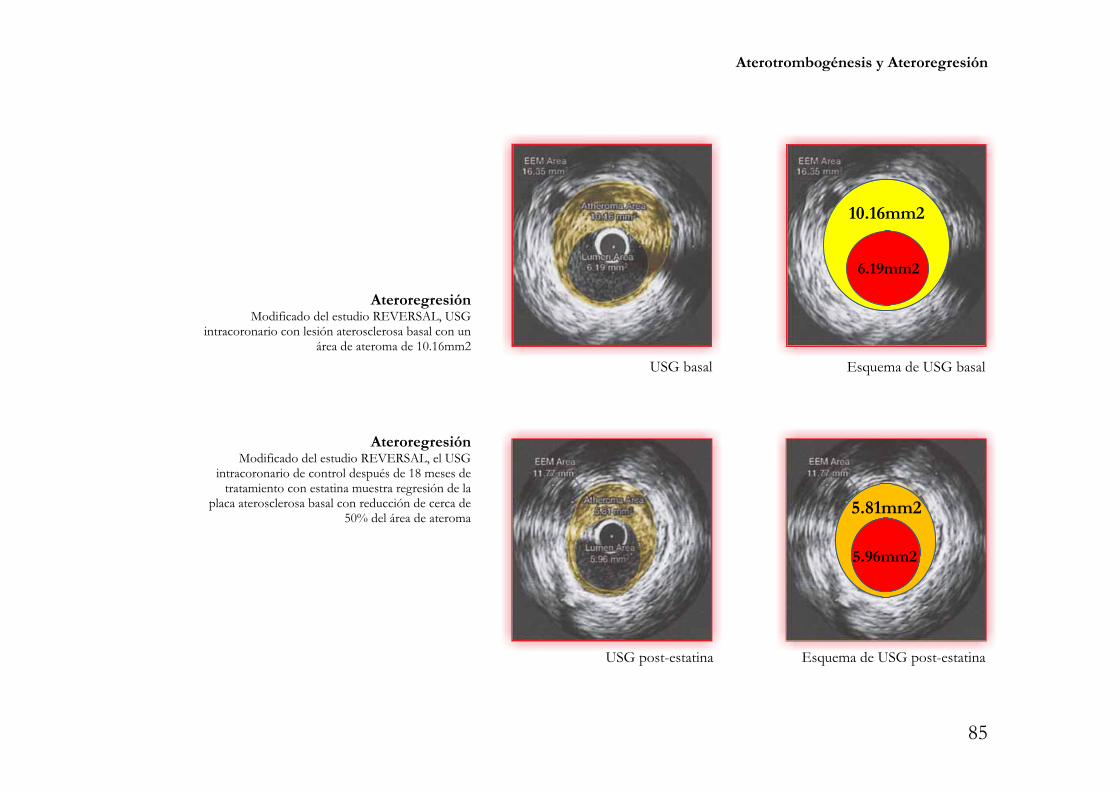
Aterotrombogénesis y Ateroregresión
AteroregresiónModificado del estudio REVERSAL, el USG
intracoronario de control después de 18 meses detratamiento con estatina muestra regresión de la
placa aterosclerosa basal con reducción de cerca de50% del área de ateroma
AteroregresiónModificado del estudio REVERSAL, USG
intracoronario con lesión aterosclerosa basal con unárea de ateroma de 10.16mm2
USG basal
10.16mm210.16mm2
6.19mm2
Esquema de USG basal
5.81mm25.81mm2
5.96mm25.96mm2
Esquema de USG post-estatinaUSG post-estatina
85

Cardio-Lipidología
86
ateroregresión-. Posteriormente los estudios METEOR con USG carotideo para evaluar el grosor intima-media carotideo, ASTEROIID, ERASE y JAPAN-ACS, entre otros estudios con USG intracoronario hanconfirmado la factibilidad de la ateroregresión. Como fue comentado previamente, el estudio PROSPECTcon USG-intracoronario y radiofrecuencia permitió clasificar la proclividad a la complicación de la placaaterosclerosa y a eventos cardiovasculares basado en tres características: área luminal mínima, carga deateroma y grosor de la capa fibrosa.
El estudio más reciente sobre ateroregresión en el humano es el estudio SATURN. Esteestudio reportó que en individuos con aterosclerosis coronaria y enfermedad cardiovascular, en un tiempopromedio de 2 años, en dos terceras partes de los individuos es factible inducir ateroregresión
estadísticamente significativa. Esta meta se alcanzó al lograr niveles de LDL 70mg/dl con el empleo de�
estatinas -atorvastatina 80mg y rosuvastatina 40mg- , el porciento de volumen de ateroma se redujo enpromedio 1%, equivalente a 5mm3 de ateroma, sin diferencias significativas entre ambas estatinas.
De esta forma, la ateroestabilización y la ateroregresiónson dos procesos clínicamentedeseables que se mueven en función de la reducción de LDL y de los marcadores de inflamación endotelio-vascular. Nuevas tecnologías como la MRI -magnetic resonance imaging- y la PET -positron emissiontomography- permiten en forma no invasiva el estudio de la anatomía de la placa aterosclerosa y suactividad inflamatoria, siendo ambos métodos útiles en el análisis de la eficiencia de diversas estrategiasterapéuticas anti-Aterosclerosis -ver capítulo 5-.
Herbert C Stary. Atlas de progresión y regresión de la aterosclerosis. Segunda edición 2003.Ed. Parthenon Publishing. Stephen J Nicholls, Christie M Ballantine, Phillip J Barter et al. SATURNtrial. N Engl J Med 2011; published on line 15 november 2011.

Salud, Riesgo, Estratificación y MetasInformación esencial para el Médico actual
Capítulo 4

GeneralidadesLa tendencia de este siglo en Medicina Cardiovascular está evolucionando de la Medicina
enfocada al individuo de alto riesgo y/o con enfermedad cardiovascular, hacia la Medicina Preventiva. LaMedicina Cardiovascular de hoy es un abanico que inicia con la Cardiología Preventiva Primordial yconcluye con la Rehabilitación Cardiovascular. Así mismo existen dos grandes líneas de acción, la línea deacción individual propia del Médico Clínico frente a su consultante, y la creciente y en rápidatransformación línea de acción poblacional, en la cual la participación multisectorial complementa elenfoque individual con el poblacional y tiene como objetivo reducir significativamente la epidémica morbi-mortalidad por enfermedades cardiovasculares.
En este capítulo daremos prioridad a la Estratificación del Riesgo Cardiovascular -ERCV-como la mejor táctica que tiene el Médico Clínico para construir una estrategia orientada a reducir en susconsultantes la probabilidad de un evento cardiovascular.
Antes de iniciar nuestro capítulo sobre ERCV y como sustento al mismo, vale la pena repasaralgunas ideas sobre el concepto de Salud Cardiovascular Ideal. En el año 2010 la Asociación Americana deCorazón planteó el concepto de salud cardiovascular ideal. Esta idea de tipo motivacional tiene el propósitode comunicar a la población cual es el perfil ideal de salud cardiovascular. La iniciativa intenta reorientar enuna forma propositiva la idea del riesgo cardiovascular. Ambos enfoques persiguen el mismo fin, motivar alas poblaciones civil y médica sobre la imperiosa necesidad de ir en búsqueda de una mejor saludcardiovascular alrededor del orbe. El perfil ideal de salud cardiovascular propuesto por la AHA estáintegrado por siete características, cuatro de ellas son conductas de vida y tres son variables, dos metabólicasy una hemodinámica.
Específicamente, un individuo con una salud cardiovascular ideal es aquel sin historiade hábito tabáquico al menos durante el año previo a la evaluación, con un IMC <25 kg/m2, quepractica actividad física moderada al menos 30 minutos o intensa al menos 15 minutos cinco días ala semana y consume una dieta sana. Lo anterior en conjunto con un nivel de colesterol total<200mg/dl sin tratamiento; glucosa de ayuno <100mg/dl sin tratamiento; y tensión arterial<120/80mmHg sin tratamiento.
De acuerdo a una reciente publicación de Claudia Bambs y colaboradores, en población adultacaucásica con edad promedio de 59 años, 0.1%, sólo 1 de cada 1000 individuos adultos reúne las sietecaracterísticas del perfil de salud cardiovascular ideal y menos de 10% reúnen de 5 a 7 características. En
Cardio-Lipidología
88

otras palabras, el 90% de los individuos adultos tienen estados de salud cardiovascular regulares o malos. En
el mismo orden de ideas, a principios del 2012 se publicó el análisis de 14,515 individuos 20 años de edad�
incluidos en la cohorte NHANES 2003-2008, divididos por estratos de 20-39 años, 40-64 años y 65 años�
de edad. Utilizando el constructo de Salud Cardiovascular Ideal de la AHA se demostró que menos de 1%de los individuos tenía una Salud Cardiovascular Ideal, con una reducción en el número de conductas devida y variables ideales en relación directa con el incremento de edad. En orden de frecuencia las conductasde vida ideales fueron: tabaquismo negativo, actividad física, IMC <25kg/m2 y dieta sana; y en el mismoorden las variables fueron: glucosa <100mg/dl, colesterol total <200mg/dl y presión arterial
<120/80mmHg. Ford a finales de enero del 2012, en el análisis de 7,622 individuos 20 años de edad�
incluidos en la cohorte NHANES 1999-2002 correlacionada con la mortalidad de dicha cohorte en el 2006,demostró la relación inversa y significativa entre el número de conductas de vida y variables ideales y la
mortalidad cardiovascular; comparando individuos con 5 variables óptimas, aquellos con 0 variables�
óptimas tuvieron un incremento en el riesgo de mortalidad cardiovascular de 88% y 78% de mortalidadtotal.
Siendo evidente y universal el pobre estado de salud cardiovascular de la población adulta,desde un enfoque clínico, el cálculo individualizado del riesgo cardiovascular y su manejo conmodificaciones conductuales más el empleo científico -basado en evidencia de eficacia, seguridad,tolerancia, adherencia y costo/eficiencia- de fármacos para la optimización de lípidos, glucosa y presiónarterial se plantean como la mejor táctica y estrategia, que como Médicos Clínicos, podemos adoptar paraatenuar el grave impacto de nuestro ominoso perfil de riesgo cardiovascular. Estas iniciativas son lajustificación de este capítulo.
Bambs C, Kip KE, Dinga A, Mulukutla SR, Aiyer AN, Reis SE. Low Prevalence of “IdealCardiovascular Health” in a Community-Based Population. The Heart Strategies Concentrating on RiskEvaluation -Heart SCORE- Study. Circulation 2011; 123:850:857. Is Ideal CardiovascularYancy CW.Health Attainable?. Editorial. Circulation 2011; 123:835-837. Shay CH,Ning H, Allen NB, CarnethonMR, Chiuve SE, Greenlund KJ et al. Status of Cardiovascular Health in US Adults. Prevalence Estimatesfrom the NHANES 2003-2008. Circulation 2012; 125:45-56. IdealFord ES, Greenlund KJ, Hong Y.Cardiovascular Health and Mortality from All Causes and Diseases of the Circulatory System amongAdults in the United States. Circulation 2012 online January 30.
Salud, Riesgo, Estratificación y Metas
89

Factores de riesgo, biomarcadores y bioimágenesEn ausencia de un perfil ideal de salud cardiovascular en el 99% de los individuos adultos, la
ERCV es una tarea obligada para cualquier Médico Clínico. Antes de presentar los métodos para cumplircon dicha tarea, es importante revisar los conceptos vigentes sobre los factores de riesgo cardiovascular,biomarcadores, bioimágenes y el valor predictor de enfermedad cardiovascular de todos ellos.
Factores de riesgo cardiovascular -FRCV-. Se considera a la hipercolesterolemia, lahipertensión arterial y la hiperglucemia -Hx3- como la triada de los principales factores de riesgo para eldesarrollo de enfermedad arterial aterosclerosa, siendo la hipercolesterolemia-LDL el eje etiopatogénico ylas otras dos moduladores sinérgicos -ver capítulo 3-. La activación y disfunción endotelio-vascular, puedenser documentadas por la positividad de los biomarcadores de inflamación y/o disfunción endotelial, reflejodel “switch off-on” en el status endotelio-vascular, de uno de reposo hacia otro activado por los FRCV. Laevolución de daño funcional hacía daño endotelio-vascular estructural, caracteriza a las diferentes etapas dela aterosclerosis. Esta enfermedad podría cursar subclínica durante toda la vida de un individuo, perotambién podría detectarse por métodos de imagen o bioimágenes antes de alcanzar el horizonte clínico, obien alcanzar este y dar alguna manifestación clínica aguda como un SICA por aterotrombosis, o crónicacomo angor pectoris por estenosis arterial hemodinámicamente significativa.
Dado que la edad de un individuo es un indicador del tiempo de exposición a los FRCV conuna susceptibilidad modulada por la herencia, el género y el medio ambiente, resulta lógico que estasúltimas y el tabaquismo sean junto con la triada Hx3 los siete FRCV principales o clásicos. El algoritmoFramingham y su versión Framingham General 2008 basado en los siete FRCV referidos, será analizadocon detalle en este capítulo.
D ´Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro J, Kannel WB.General Cardiovascular Risk Profile for Use in Primary Care. The Framingham Heart Study. Circulation2008; 117:743-753.
Biomarcadores y bioimágenes. Se define a un biomarcador o bioimagen, como unacaracterística que es posible medir objetivamente y que representa un proceso fisiológico o patológico, obien la respuesta a una intervención terapéutica farmacológica o de otra índole. En la predicción del riesgocardiovascular, los biomarcadores y las bioimágenes son complemento, nunca sustituto de los FRCV
clásicos. El poder de discriminación 0.75 de los métodos de predicción que incluyen en un puntaje total la�
suma del valor predictor de: edad, sexo, tabaquismo, cifras de colesterol, tensión arterial y glucosa, hasta hoy
Cardio-Lipidología
90

no ha sido mejorado por algún biomarcador o bioimagen aislados, ni por métodos multimarcador basadosen ellos. En el año 2009, el “US Preventive Services Task Force” y la “National Academy of ClinicalBiochemestry” concluyeron que la evidencia actual es insuficiente para sustentar que el balancebeneficio/riesgo de los nuevos biomarcadores-bioimágenes pueda justificar su empleo sobre el uso de losmétodos fundados en los FRCV clásicos. La indicación actual de los biomarcadores y/o bioimágenes en laERCV será tratada en este capítulo, con énfasis en su utilización para la ERCV en el individuo asintomático,basada en la última guía publicada por la AHA en conjunto con la ACCF en el 2010 para la valoración delriesgo cardiovascular en el individuo asintomático.
U.S. Preventive Services Task Force. Recommendation Statement. UsingNontraditional Risk Factors in Coronary Heart Disease Risk Assessment. Ann Intern Med 2009; 151:474-482. Emerging Biomarkers for Primary Prevention ofNational Academy of Clinical Biochemistry.Cardiovascular Disease and Stroke. Washington, DC. National Academy of Clinical Biochemistry; 2009.Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fallad Z et al. 2010 ACCF/AHAGuideline for Assessment of Cardiovascular Risk in Asymptomatic Adults: Executive Summary. A reportof the American College of Cardiology Foundation/American Heart Association Task Force on PracticeGuidelines. Circulation 2010; 122:2748-2764.
Valor predictivo de FRCV, biomarcadores y bioimágenes. La estadística médica actualaplicada a la predicción del riesgo cardiovascular tiene una alta exigencia para la validación de un nuevobiomarcador, bioimagen o método -algoritmo- para la predicción de enfermedad cardiovascular. Dado quemétodos como Framingham, iniciado en 1961 por William Kannel, validado y perfeccionado a los largo de50 años y tres generaciones, son hasta hoy la “regla de oro” en la predicción del riesgo de enfermedadescardiovasculares, los nuevos biomarcadores, bioimágenes y/o métodos de predicción tienen que demostrarsuperioridad o al menos optimización sobre el perfil predictor de los primeros. Para cumplir lo anterior losbiomarcadores y/o métodos candidatos deben reunir los siguientes requisitos: prueba de concepto;validación prospectiva; valor incremental basado en su poder de discriminación, calibración yreclasificación sobre el método a comparar -en la mayoría de los estudios, el método de Framingham-;utilidad clínico-terapéutica y balance costo/eficiencia.
De las características previas, aquellas que definen el valor incremental: discriminación,calibración y reclasificación son las más importantes. La discriminación se refiere a la capacidad de
Salud, Riesgo, Estratificación y Metas
91

un biomarcador-bioimagen-algoritmo para predecir dentro de una población con niveles de riesgodiferentes, quienes desarrollaran una enfermedad cardiovascular -sensibilidad- y quienes no lo harán-especificidad-. Un valor de 1.0 en el modelo estadístico C, corresponde a una prueba con unadiscriminación perfecta o de 100%; un valor de 0.5 corresponde a una prueba con una discriminaciónequivalente a un “volado” es decir 50%. El método de Framingham basado en los siete FRCV clásicosposee un valor de discriminación promedio de 0.75, es decir discrimina correctamente en un 75% de lasocasiones quien, en una población con diferentes niveles de riesgo, cursará con la enfermedad y quién no.Por lo tanto un nuevo biomarcador-bioimagen-algoritmo de predicción, al menos debe tener unadiscriminación de 0.75 o bien incrementar significativamente ->0.05-, la discriminación del método dereferencia. La calibración se refiere a la capacidad de un biomarcador-bioimagen-algoritmo paracomprobar que el número de casos observados de una enfermedad en una población con diferentes nivelesde riesgo, es igual al número de casos pronosticados. Una calibración es adecuada cuando el valor de la P enel modelo estadístico de Hosmer-Lomeshow es >0.05; este valor implica que no existe una diferencia
Características requeridas para la adopción de un nuevo Biomarcador
Biomarcadores. Validación clínicaEn esta tabla se enumeran las 5 característicasbásicas propuestas por Hlatky que debe reunir unnuevo biomarcador para ser considerado en laclínica; la mayoria de los nuevos biomarcadoressólo reúnen las 2 primeras
1.- Prueba de concepto
2.- Validaci ón prospectiva
3.- Valor incremental
a) Discriminaci ón
b) Calibraci ón
c) Reclasificaci ón
4.- Utilidad cl ínico-terapéutica
5.- Balance costo/eficiencia
1.- Prueba de concepto
2.- Validaci
3.- Valor incremental
a) Discriminación
b) Calibración
c) Reclasificación
4.- Utilidad clínico-terapéutica
5.- Balance costo/eficiencia
ón prospectiva
Correlaci ón etio, fisio y anatomopatol ógica
Correlaci ón entre el biomarcador y los eventos cl ínicos
Capacidad de incrementar el valor predictor
Predecir casos verdaderos positivos y verdaderos
negativosPredecir casos a observar
Mover con certeza casos de un nivel de riesgo a otro
La aplicaci ón impacta en la incidencia de eventos cl ínicos
Balance favorable incluyendo los costos derivados
Correlación etio, fisio y anatomopatológica
Correlación entre el biomarcador y los eventos clínicos
Capacidad de incrementar el valor predictor
Predecir casos verdaderos positivos y verdaderos negativos
Predecir casos a observar
Mover con certeza casos de un nivel de riesgo a otro
La aplicación impacta en la incidencia de eventos clínicos
Balance favorable incluyendo los costos derivados
Característica Concepto
Cardio-Lipidología
92

significativa entre los casos de la enfermedad pronosticados y los casos observados. Un demodelopredicción puede ser recalibrado para una población diferente a la que le dio origen; para ello es necesarioconocer la prevalencia de los FRCV y de enfermedad cardiovascular en la población índice. Lareclasificación se refiere a la capacidad de un biomarcador-bioimagen-algoritmo para mover a un individuoo una población de un nivel de riesgo dado hacia otro nivel; la reclasificación puede ser hacia arriba o haciaabajo, es decir hacia niveles de riesgo superior o inferior. Puesto que es importante comprobar que el casoreclasificado efectivamente tuvo el comportamiento del nivel de riesgo reclasificado, Michael Pencina hapropuesto el empleo del Indice de Reclasificación Neta o NRI, el cual indica el valor de los casos bienreclasificados menos los casos mal reclasificados; en este sentido un valor de 0 equivale a una reclasificaciónmala, en la cual el número de casos bien reclasificados es igual al número de casos mal reclasificados. Amayor numerador sobre denominador el NRI será mejor, un valor mínimo aceptable es de 10.
De esta forma un nuevo biomarcador, bioimagen o algoritmo de predicción de enfermedadcardiovascular debe tener con la enfermedad en cuestión: asociación etiopatogénica -prueba de concepto-;asociación epidemiológica -validación prospectiva-; valor incremental basado en su poder dediscriminación, calibración y reclasificación e idealmente demostración de modificar favorablemente elcurso clínico-terapéutico así como el balance costo/eficiencia en el diagnóstico y/o tratamiento de laenfermedad.
Hlatky MA, Greenland P, Arnett DK, Ballantyne CM, Criqui MH, Elkind MSV et al.Criteria for Evaluation of Novel Markers of Cardiovascular Risk. A Scientific Statement from theAmerican Heart Association. Circulation 2009; 119:2408-2419. . Cardiovascular RiskLloyd-Jones DMPrediction. Basic Concepts, Current Status and Future Directions. Circulation 2010; 121:1768-1777. deLemos JA, Rohatgi A. Separating the Contenders from the Pretenders. Competitive High-ThroughputBiomarker Screening in Large Population-Based Studies. Editorial. Circulation 2010; 121:2381-2383.Cooney MT, Dudina A, D´Agostino R, Graham IM. Cardiovascular Risk-Estimation Systems inPrimary Prevention. Do they differ?. Do they make a difference?. Can we see the future?. Circulation 2010;122:300-310. Assessing the Role of Circulating, Genetic, and Imaging Biomarkers inWang TJ.Cardiovascular Risk Prediction. Circulation 2011; 123:551-565. Evaluating thePletcher MJ, Pignone M.Clinical Utility of a Biomarker. A Review of Methods for Estimating Health Impact. Circulation 2011;123:1116-1124.
Salud, Riesgo, Estratificación y Metas
93

Estratificación básica de riesgo cardiovascularLa elaboración de sistemas para predecir eventos cardiovasculares, como el de Framingham
nacido en 1961 y basado en la incorporación y ponderación del valor predictivo de variables inmutablescomo edad, género y otras mutables susceptibles de tratamiento como hipercolesterolemia, hipertensiónarterial, hiperglucemia -Hx3- y tabaquismo, nos han dado el potencial de calcular el riesgo cardiovascular“per se”, tratar a los factores que lo determinan y disminuir la probabilidad estadística de un eventocardiovascular fatal y/o invalidante.
Kannel WB, Dawber TR, Kagan A, Revotskie N, Stokes JI. Factors of Risk in theDevelopment of Coronary Heart Disease -Six Year Follow-Up Experience-: the Framingham Study. AnnIntern Med 1961; 55:33-50. Validation of theD´Agostino RB, Grundy S, Sullivan LM, Wilson P.Framingham Coronary Heart Disease Prediction Scores: Results of a Multiple Ethnic GroupInvestigation. JAMA 2001; 286:180-187. D ´Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M,Massaro J, Kannel WB. General Cardiovascular Risk Profile for Use in Primary Care. The FraminghamHeart Study. Circulation 2008; 117:743-753.
Antes de revisar los diferentes métodos de Estratificación básica del Riesgo Cardiovascular-EbRCV-, es importante resaltar que en un escenario como el consultorio médico, se han de tener en cuentalas siguientes premisas.
Premisa 1.- Independientemente de su condición clínica y/o motivo de consulta médica, laprimera posibilidad de enfermedad y/o muerte en cualquier individuo adulto es la secundaria a patologíacardiovascular. Por ejemplo, en México en el año 2009 murieron 189, 000 mexicanos, 1 cada 180 segundospor diabetes mellitus y/o enfermedad cardiovascular. Muchas de esas muertes pudieron haberse prevenidosi el riesgo cardiovascular se hubiera evaluado y tratado 10, 20 o probablemente 30 años antes deconvertirse en una enfermedad cardiovascular.
Premisa 2.- El individuo con una enfermedad cardiovascular conocida y/o síntomas o signoscardiovasculares, es un sujeto de riesgo alto con una probabilidad estadística superior al 2% anual de unnuevo evento y/o muerte cardiovascular. En este escenario más allá de estratificar, importa tratarintensamente todos los FRCV modificables y la enfermedad cardiovascular “per se” de acuerdo a las guíasterapéuticas vigentes.
Premisa 3.- El individuo 20 años de edad, hombre o mujer, sin conocimiento de una�
enfermedad cardiovascular, asintomático y/o asignológico cardiovascular es el prototipo en quien la
Cardio-Lipidología
94
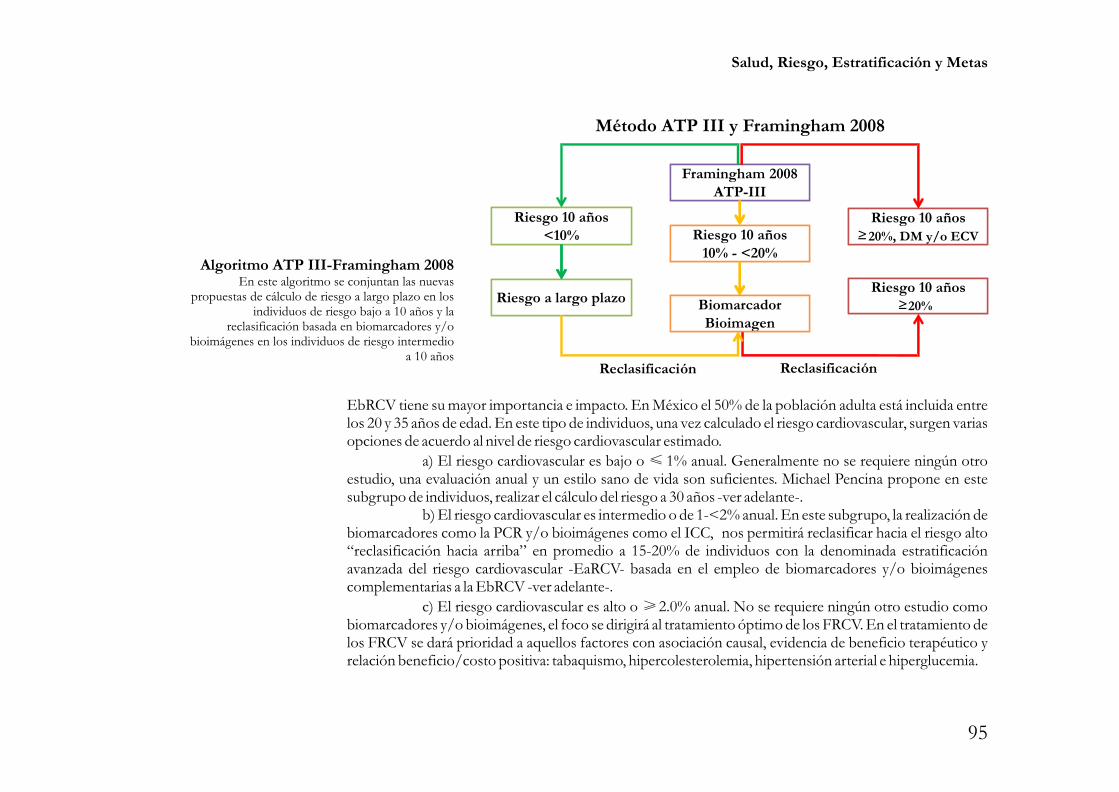
EbRCV tiene su mayor importancia e impacto. En México el 50% de la población adulta está incluida entrelos 20 y 35 años de edad. En este tipo de individuos, una vez calculado el riesgo cardiovascular, surgen variasopciones de acuerdo al nivel de riesgo cardiovascular estimado.
a) El riesgo cardiovascular es bajo o 1% anual. Generalmente no se requiere ningún otro�
estudio, una evaluación anual y un estilo sano de vida son suficientes. Michael Pencina propone en estesubgrupo de individuos, realizar el cálculo del riesgo a 30 años -ver adelante-.
b) El riesgo cardiovascular es intermedio o de 1-<2% anual. En este subgrupo, la realización debiomarcadores como la PCR y/o bioimágenes como el ICC, nos permitirá reclasificar hacia el riesgo alto“reclasificación hacia arriba” en promedio a 15-20% de individuos con la denominada estratificaciónavanzada del riesgo cardiovascular -EaRCV- basada en el empleo de biomarcadores y/o bioimágenescomplementarias a la EbRCV -ver adelante-.
c) El riesgo cardiovascular es alto o 2.0% anual. No se requiere ningún otro estudio como�
biomarcadores y/o bioimágenes, el foco se dirigirá al tratamiento óptimo de los FRCV. En el tratamiento delos FRCV se dará prioridad a aquellos factores con asociación causal, evidencia de beneficio terapéutico yrelación beneficio/costo positiva: tabaquismo, hipercolesterolemia, hipertensión arterial e hiperglucemia.
Método ATP III y Framingham 2008
Algoritmo ATP III-Framingham 2008En este algoritmo se conjuntan las nuevas
propuestas de cálculo de riesgo a largo plazo en losindividuos de riesgo bajo a 10 años y la
reclasificación basada en biomarcadores y/obioimágenes en los individuos de riesgo intermedio
a 10 años
Salud, Riesgo, Estratificación y Metas
Framingham 2008ATP-III
Riesgo 10 años10% - <20%
Riesgo 10 años=20%, DM y/o ECV
Riesgo 10 años<10%
Riesgo a largo plazo BiomarcadorBioimágen
Riesgo 10 años>20%
Reclasificación Reclasificación
Framingham 2008ATP-III
Riesgo 10 años10% - <20%
Riesgo 10 años20%, DM y/o ECV
Riesgo 10 años<10%
Riesgo a largo plazo BiomarcadorBioimagen
Riesgo 10 años20%
95
>
>

Cálculo del riesgo a mediano plazo -10 años-Framingham Clásico. Sin negar la relevancia de diversos algoritmos para el cálculo del riesgo
cardiovascular, entre ellos el PROCAM, la guía Británica-Neo Zelanda, los del Reino Unido, QRISK yASSIGN y el SCORE ampliamente validado en Europa y recomendado por las Guías ESC-EAS para laERCV, el método Framingham en sus diferentes versiones, incluyendo la más reciente del 2008, integra lacohorte con mayor seguimiento, validación, “tecnología” estadística y calibración para la etnia hispana, porello se ha seleccionado para su revisión y ejemplificación en este libro y es sugerido por el autor para suaplicación en el consultorio.
Basado en modelos estadísticos de regresión multivariable, los investigadores del proyectoFramingham, originalmente seleccionaron entre más de dos centenas, a las variables que tenían una mayorasociación positiva con la incidencia de un primer evento cardiovascular coronario: angor pectoris, infartomiocárdico y/o muerte cardiovascular. Las variables seleccionadas por su “peso predictor” fueron género yedad como variables inmutables o no susceptibles de tratamiento, y como variables mutables, por endesusceptibles de tratamiento: colesterol total, colesterol-HDL, tensión arterial sistólica, diabetes mellitus ytabaquismo. Este esquema de predicción de un primer evento clínico cardiovascular, en individuos sinevidencia de enfermedad cardiovascular clínica, es susceptible de calibración de acuerdo a la prevalencia defactores de riesgo y enfermedad cardiovascular en una población dada.
Algunos aspectos son importantes para entender la trascendencia de un sistema de predicciónde eventos cardiovasculares como el de Framingham.
a) Este sistema predice un evento cardiovascular, no la magnitud de la aterosclerosis. Lapredicción de aterosclerosis es denominada predicción diagnóstica y debe diferenciarse de la predicción deeventos cardiovasculares “Aterosclerosis vemos, eventos no sabemos”.
b) El nivel de riesgo cardiovascular, indica la magnitud necesaria de la intervención sobre losFRCV con el fin de evitar un evento cardiovascular clínico. En su esencia la ERCV está enfocada altratamiento óptimo de los FRCV, no a la indicación de estudios intervencionistas.
c) El tratamiento y la optimización y/o erradicación de las variables mutables como: colesteroltotal y/o colesterol-LDL, colesterol-HDL, tensión arterial, glucemia y tabaco, es hasta hoy nuestra mejorestrategia para reducir la carga global de enfermedad cardiovascular y brindar una vida más larga y de mayorcalidad a los individuos.
Dawber TR, Kannel WB. The Framingham Study. An Epidemiological Approach to
Cardio-Lipidología
96

Coronary Heart Disease. Circulation 1966; 34:553-555. Screening Tests. In Atlas ofMark DB.Cardiovascular Risk Factors; pp 39-55. Editors Gaziano JM, Braunwald E. Editoria. Current Medicine LLC2006. An Updated Coronary Risk Profile: AAnderson KM, Wilson PW, Odell PM, Kannel WB.Statement for Health Professionals. Circulation 1991; 83:356-362. Greenland P, Smith SC, Grundy SM.Improving Coronary Heart Disease Risk Assessment in Asymptomatic People: Role of Traditional RiskFactors and Noninvasive Cardiovascular Tests. Circulation 2001; 104:1863-1867. Wilson PW,D´Agostino RB, Levy D, Belanger AM, Silbershatz H, Kanel WB. Prediction of Coronary HeartDisease Using Risk Factor Categories. Circulation 1998; 97:1837-1847. .Morales-Villegas EEstratificación del Riesgo Cardiovascular. No todos tenemos el mismo riesgo, estratificar es la clave. EnEndocardiocrinología 1ª Edición 2005. Capítulo 7. Editor Enrique Morales-Villegas. Editorial. Rupestre.
Framingham General 2008. Ralph D´ Agostino a la cabeza del equipo Framingham, publicóen febrero del 2008, el método llamado Framingham General. Este método está validado en hombres ymujeres entre 30 y 75 años de edad, sin evidencia de enfermedad cardiovascular clínica y utiliza las mismassiete variables del Framingham Clásico. Las ventajas de la versión 2008 sobre la 1998 son las siguientes.
1.-A diferencia de la versión Framingham 1998, la cual permite predecir sólo eventoscoronarios, este método permite calcular el riesgo general de eventos cardiovasculares, incluyendo:arteriales coronarios, arteriales carotideo-vertebrales, arteriales periféricos e insuficiencia cardiaca. Sinduda este es un avance, ya que un Médico Clínico y su consultante desean saber el riesgo de cualquier eventocardiovascular mayor, no sólo el riesgo de un evento coronario.
2.-En conjunto con el cálculo del nivel de riesgo absoluto, también es posible calcular la edadcardiovascular de un individuo. El concepto de edad cardiovascular tiene gran implicación en lasensibilización del consultante ante su nivel de riesgo cardiovascular. En otras palabras, comentarle a unamujer de 60 años de edad que su riesgo cardiovascular a 10 años es de 10% o 1% por año, puede sersubjetivo y poco intuitivo. Por el contrario decirle que la edad calculada de su sistema cardiovascular es de 75años, cuando su edad cronológica es 15 años menor, es más objetivo, tiene un mayor impacto emocional, ypor lo tanto será más sencillo para el Médico Clínico inducir cambios conductuales favorables, incluyendola adherencia al tratamiento.
3.-Es factible calcular el riesgo cardiovascular general sin parámetros de laboratorio,sustituyendo colesterol-total y el colesterol-HDL por el IMC. Esta última aproximación en el cálculo del
Salud, Riesgo, Estratificación y Metas
97

riesgo cardiovascular no se recomienda de rutina, sin embargo puede ser útil en algunos escenarios médicosque no cuentan con un laboratorio clínico.
D ´Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro J, Kannel WB.General Cardiovascular Risk Profile for Use in Primary Care. The Framingham Heart Study. Circulation2008; 117:743-753.
El ABC para la aplicación del Framingham. Tanto para la aplicación del FraminghamClásico como la versión 2008, el autor propone el siguiente A B C ejemplificado en este capítulo con uncaso clínico de un hombre fumador de 61 años de edad.
Análisis. Consiste en adquirir la información de los siete FRCV.Balance. Una vez adquirida la información de los siete FRCV, se otorga un puntaje a cada uno
de ellos de acuerdo a la tabla de puntuación para el género correspondiente.Cálculo. En función de la puntuación de cada FRCV, se procede al cálculo de la puntuación
total y con ella es posible obtener el riesgo cardiovascular general absoluto a 10 años, así como la edadcardiovascular. El primero nos sirve de guía para establecer las metas terapéuticas para LDL, colesterol no-HDL y tensión arterial e incluso para sustentar la prescripción profiláctica de Aspirina. El HDL no tienemetas terapéuticas en base a la ERCV, sin embargo en bases epidemiológicas y observacionales, existenniveles deseables y como ya se comentó el cálculo de la edad cardiovascular es un concepto intuitivo quefavorece la comunicación entre el Médico Clínico y su consultante.
Mark DB. Screening Tests. In Atlas of Cardiovascular Risk Factors; pp 39-55. EditorsGaziano JM, Braunwald E. Editorial Current Medicine LLC 2006. Estratificación delMorales-Villegas E.Riesgo Cardiovascular. Del síntoma al genoma. En Atlas de Endotelio, Aterotrombosis y Estatinas. 1ªEdición 2010-11. Capítulo 4. Editor Enrique Morales-Villegas. Editorial Atheros.
Cardio-Lipidología
98

Framingham 2008Tabla Framingham 2008 para mujeres. Puntuación
para cada factor de riesgo cardiovascular
Puntos Edad HDL Colesterol TAS noTx TAS Tx Tabaco DM
-03 <120
-02 60+
-01 50-59 <120
00 30-34 45-49 <160 120-129 No No
01 35-44 160-199 130-139
02 35-39 <35 140-149 120-129
03 200-239 130-139 Si
04 40-44 240-279 150-159 Si
05 45-49 280+ 160+ 140-149
06 150-159
07 50-54 160+
08 55-59
09 60-64
10 65-69
11 70-74
12 75+
Puntos Edad HDL Colesterol TAS noTx TAS Tx Tabaco DM
-03 <120
-02 60+
-01 50-59 <120
00 30-34 45-49 <160 120-129 No No
01 35-44 160-199 130-139
02 35-39 <35 140-149 120-129
03 200-239 130-139 Si
04 40-44 240-279 150-159 Si
05 45-49 280+ 160+ 140-149
06 150-159
07 50-54 160+
08 55-59
09 60-64
10 65-69
11 70-74
12 75+
Salud, Riesgo, Estratificación y Metas
Framingham General 2008. Tabla para género femenino
99

Puntos Grupo riesgo
-02- -01
-01 1.0
00 1.2
01 1.5
02 1.7
03 2.0
04 2.4
05 2.8
06 3.3
07 3.9
08 4.5
09 5.3
10 6.3
11 7.3
12 8.6
13 10.0
14 11.7
15 13.7
16 15.9
17 18.5
18 21.5
19 24.8
20 28.5
21+ 30+
Puntos Grupo riesgo
-02 -01
-01 1.0
00 1.2
01 1.5
02 1.7
03 2.0
04 2.4
05 2.8
06 3.3
07 3.9
08 4.5
09 5.3
10 6.3
11 7.3
12 8.6
13 10.0
14 11.7
15 13.7
16 15.9
17 18.5
18 21.5
19 24.8
20 28.5
21+ 30+
Puntos Edad vascular
-01 -30
01 31
02 34
03 36
04 39
05 42
06 45
07 48
08 51
09 55
10 59
11 64
12 68
13 73
14 79
15+ 80+
Puntos Edad vascular
-01 -30
01 31
02 34
03 36
04 39
05 42
06 45
07 48
08 51
09 55
10 59
11 64
12 68
13 73
14 79
15+ 80+
Framingham 2008Tablas Framingham 2008 para mujeres. A laizquierda la tabla para el cálculo del riesgo absolutoa 10 años y sus categorías de riesgo bajo -verde-,intermedio -ambar- y alto -rojo-. A la derecha latabla para el cálculo de la edad vascular
Cardio-Lipidología
Framingham General 2008. Riesgo Absoluto y Edad Vascular
100

Puntos Edad HDL Colesterol TAS noTx TAS Tx Tabaco DM
-02 60+ <120
-01 50-59
00 30-34 45-49 <160 120-129 <120 No No
01 35-44 160-199 130-139
02 35-39 <35 200-239 140-159 120-129
03 240- 160+ 130-139 Si
04 280+ 140-159 Si
05 40-44 160+
06 45-49
07
08 50-54
09
10 55-59
11 60-64
12 65-69
13
14 70-74
15 75+
Puntos Edad Colesterol TAS noTx TAS Tx Tabaco DM
-02 60+ <120
-01 50-59
00 30-34 45-49 <160 120-129 <120 No No
01 35-44 160-199 130-139
02 35-39 <35 200-239 140-159 120-129
03 240-279 160+ 130-139 Si
04 280+ 140-159 Si
05 40-44 160+
06 45-49
07
08 50-54
09
10 55-59
11 60-64
12 65-69
13
14 70-74
15 75+Framingham 2008
Tabla Framingham 2008 para hombres. Puntuaciónpara cada factor de riesgo cardiovascular
Salud, Riesgo, Estratificación y Metas
Framingham General 2008. Tabla para género masculino
101
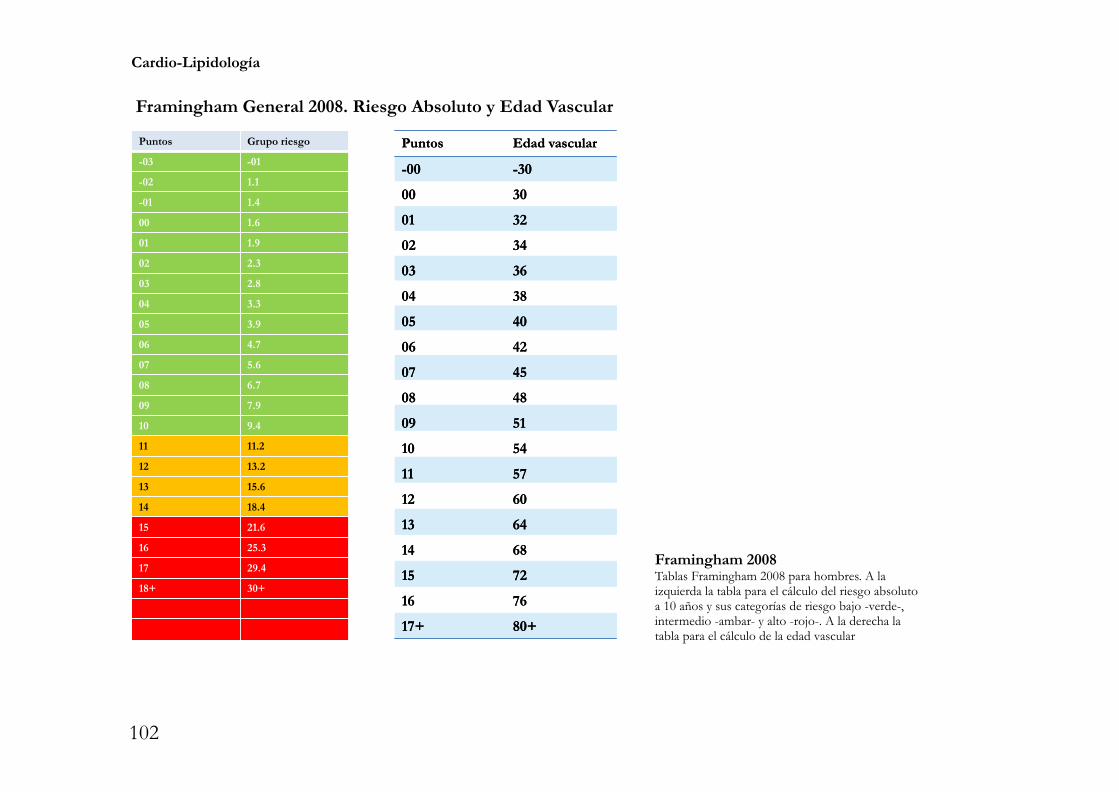
Puntos Grupo riesgo
-03- -01
-02 1.1
-01 1.4
00 1.6
01 1.9
02 2.3
03 2.8
04 3.3
05 3.9
06 4.7
07 5.6
08 6.7
09 7.9
10 9.4
11 11.2
12 13.2
13 15.6
14 18.4
15 21.6
16 25.3
17 29.4
18+ 30+
Puntos Grupo riesgo
-03 -01
-02 1.1
-01 1.4
00 1.6
01 1.9
02 2.3
03 2.8
04 3.3
05 3.9
06 4.7
07 5.6
08 6.7
09 7.9
10 9.4
11 11.2
12 13.2
13 15.6
14 18.4
15 21.6
16 25.3
17 29.4
18+ 30+
Puntos Edad vascular
-00 -30
00 30
01 32
02 34
03 36
04 38
05 40
06 42
07 45
08 48
09 51
10 54
11 57
12 60
13 64
14 68
15 72
16 76
17+ 80+
Puntos Edad vascular
-00 -30
00 30
01 32
02 34
03 36
04 38
05 40
06 42
07 45
08 48
09 51
10 54
11 57
12 60
13 64
14 68
15 72
16 76
17+ 80+
Framingham 2008Tablas Framingham 2008 para hombres. A laizquierda la tabla para el cálculo del riesgo absolutoa 10 años y sus categorías de riesgo bajo -verde-,intermedio -ambar- y alto -rojo-. A la derecha latabla para el cálculo de la edad vascular
Cardio-Lipidología
Framingham General 2008. Riesgo Absoluto y Edad Vascular
102
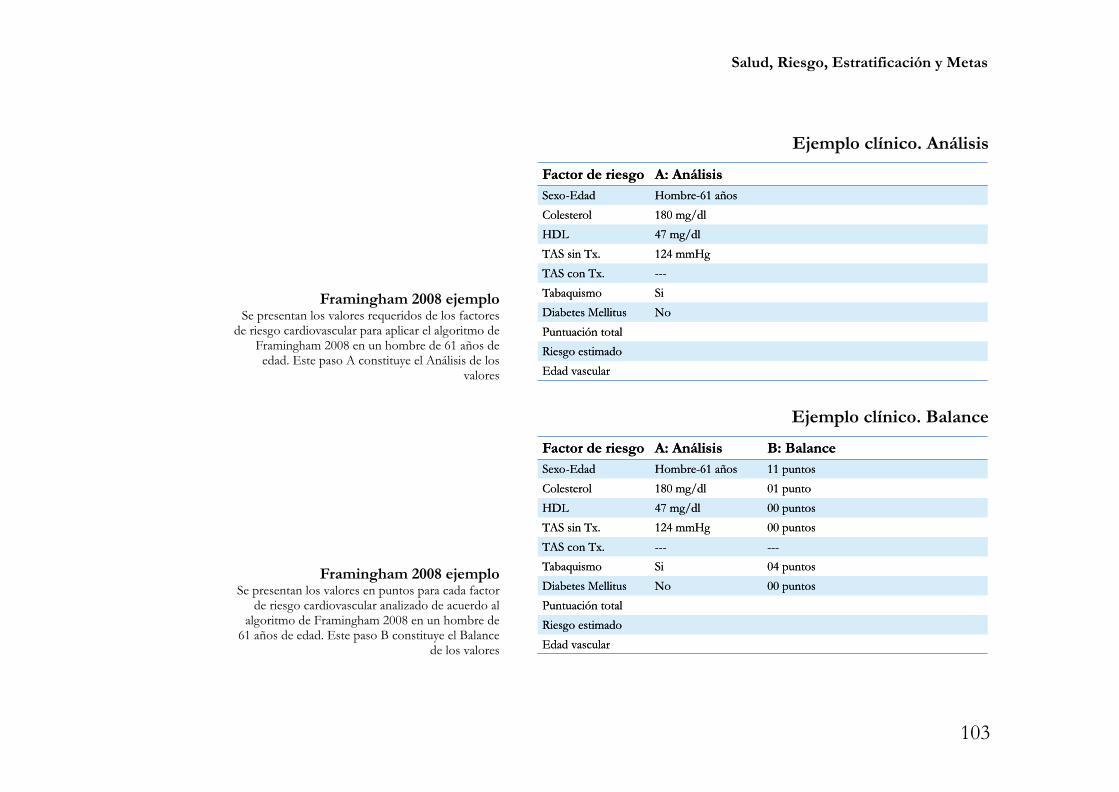
Factor de riesgo A: Análisis B: BalanceSexo-Edad Hombre-61 años 11 puntos
Colesterol 180 mg/dl 01 punto
HDL 47 mg/dl 00 puntos
TAS sin Tx. 124 mmHg 00 puntos
TAS con Tx. --- ---
Tabaquismo Si 04 puntos
Diabetes Mellitus No 00 puntos
Puntuación total
Riesgo estimado
Edad vascular
Factor de riesgo A: Análisis B: BalanceSexo-Edad Hombre-61 años 11 puntos
Colesterol 180 mg/dl 01 punto
HDL 47 mg/dl 00 puntos
TAS sin Tx. 124 mmHg 00 puntos
TAS con Tx. --- ---
Tabaquismo Si 04 puntos
Diabetes Mellitus No 00 puntos
Puntuación total
Riesgo estimado
Edad vascular
Framingham 2008 ejemploSe presentan los valores en puntos para cada factor
de riesgo cardiovascular analizado de acuerdo alalgoritmo de Framingham 2008 en un hombre de
61 años de edad. Este paso B constituye el Balancede los valores
Factor de riesgo A: AnálisisSexo-Edad Hombre-61 años
Colesterol 180 mg/dl
HDL 47 mg/dl
TAS sin Tx. 124 mmHg
TAS con Tx. ---
Tabaquismo Si
Diabetes Mellitus No
Puntuación total
Riesgo estimado
Edad vascular
Factor de riesgo A: AnálisisSexo-Edad Hombre-61 años
Colesterol 180 mg/dl
HDL 47 mg/dl
TAS sin Tx. 124 mmHg
TAS con Tx. ---
Tabaquismo Si
Diabetes Mellitus No
Puntuación total
Riesgo estimado
Edad vascular
Ejemplo clínico. Análisis
Framingham 2008 ejemploSe presentan los valores requeridos de los factores
de riesgo cardiovascular para aplicar el algoritmo deFramingham 2008 en un hombre de 61 años deedad. Este paso A constituye el Análisis de los
valores
Salud, Riesgo, Estratificación y Metas
Ejemplo clínico. Balance
103

Factor de riesgo A: Análisis B: Balance C: CálculoSexo-Edad Hombre-61 años 11 puntos
Colesterol 180 mg/dl 01 punto
HDL 47 mg/dl 00 puntos
TAS sin Tx. 124 mmHg 00 puntos
TAS con Tx. --- ---
Tabaquismo Si 04 puntos
Diabetes Mellitus No 00 puntos
Puntuación total 16 puntos
Riesgo estimado 25.3% a 10 años
Edad vascular 76 años
Factor de riesgo A: Análisis B: Balance C: CálculoSexo-Edad Hombre-61 años 11 puntos
Colesterol 180 mg/dl 01 punto
HDL 47 mg/dl 00 puntos
TAS sin Tx. 124 mmHg 00 puntos
TAS con Tx. --- ---
Tabaquismo Si 04 puntos
Diabetes Mellitus No 00 puntos
Puntuación total 16 puntos
Riesgo estimado 25.3% a 10 años
Edad vascular 76 años
Factor de riesgo A: Análisis B: Balance C: CálculoSexo-Edad Hombre-61 años 11 puntos
Colesterol 180 mg/dl 01 punto
HDL 47 mg/dl 00 puntos
TAS sin Tx. 124 mmHg 00 puntos
TAS con Tx. --- ---
Tabaquismo
Diabetes Mellitus No 00 puntos
Puntuación total 12 puntos
Riesgo estimado 13.2% a 10 años
Edad vascular 60 años
Factor de riesgo A: Análisis B: Balance C: CálculoSexo-Edad Hombre-61 años 11 puntos
Colesterol 180 mg/dl 01 punto
HDL 47 mg/dl 00 puntos
TAS sin Tx. 124 mmHg 00 puntos
TAS con Tx. --- ---
Tabaquismo No 00 puntos
Diabetes Mellitus No 00 puntos
Puntuación total 12 puntos
Riesgo estimado 13.2% a 10 años
Edad vascular 60 años
Framingham 2008 ejemploSe presentan los valores de la puntuación total, elriesgo absoluto estimado a 10 años y la edadvascular de acuerdo a los pasos A y B del algoritmode Framingham 2008 en un hombre de 61 años deedad. Este paso C constituye el Cálculo de lasresultantes de los valores
Framingham 2008 ejemploSe presentan los valores de la puntuación total, elriesgo absoluto estimado a 10 años y la edadvascular de acuerdo a los pasos A y B del algoritmode Framingham 2008 en un hombre de 61 años deedad después de haber eliminado el tabaquismo.En este paso C se puede apreciar la modificaciónfavorable tanto en el riesgo estimado a 10 añoscomo en la edad vascular
Cardio-Lipidología
Ejemplo clínico. Cálculo
Ejemplo clínico. Cálculo modificado sin tabaquismo
104

Cálculo del riesgo a largo plazoPartiendo del concepto de Namby y Ballantyne de que “10 años no es una vida” y que
especialmente en hombres menores de 50 años o mujeres menores de 60 años de edad, el riesgocardiovascular calculado a 10 años frecuentemente luce bajo o insignificante, se han propuesto dos nuevasvisiones para minimizar esta situación de “miopía” en la ERCV.
Nambi V, Ballantyne CM. Risky Business. Ten years is not a life. Editorial. Circulation2009; 119:362-364.
Cálculo del riesgo a los 50 años de edad. Esta propuesta fue publicada originalmente en el2006 por Donald Lloyd-Jones y Colaboradores y es denominada cálculo de riesgo cardiovascular “de porvida”. Lloyd-Jones consideró cuatro factores de riesgo cardiovascular clásicos: tensión arterial sistólica,tensión arterial diastólica, colesterol total y tabaquismo; no incluyó a la diabetes mellitus por considerarseun factor de riesgo a plazo intermedio. Para el diseño de este método se evaluaron individuos que a los 50años de edad permanecían sin evidencia de enfermedad cardiovascular clínica. En ellos, el perfil de riesgocardiovascular estimado a los 50 años, fue correlacionado con la probabilidad de sobrevivir libre de eventoscardiovasculares hasta los 95 años. De acuerdo a este método, el perfil de riesgo se divide en 5 estratosmutuamente excluyentes:
1.-Perfil con las cuatro variables en nivel ideal. Tensión arterial sistólica <120mmHg sintratamiento, diastólica<80mmHg sin tratamiento, colesterol total <180mg/dl sin tratamiento, no fumadory sin diabetes mellitus.
2.-Perfil con una o más variables subóptimas. Tensión arterial sistólica 120-139mmHg sintratamiento, diastólica 80-89mmHg sin tratamiento, colesterol total 180-199mg/dl sin tratamiento, nofumador y sin diabetes mellitus.
3.-Perfil con una o más variables altas. Tensión arterial sistólica 140-159mmHg sin tratamiento,diastólica 90-99mmHg sin tratamiento, colesterol total 200-239mg/dl sin tratamiento, no fumador y sindiabetes mellitus.
4.-Perfil con un FRCV presente. Tensión arterial sistólica 160mmHg virgen o tratada�
independientemente de la cifra, diastólica 100mmHg virgen o tratada independientemente de la cifra,�
colesterol total 240mg/dl virgen a tratamiento o tratado independientemente de la cifra, tabaquismo y/o�
diabetes mellitus.5.-Perfil con dos o más FRCV presentes.
Salud, Riesgo, Estratificación y Metas
105

Lloyd-Jones DM, Leip EP, Larson MG, D´Agostino RB, Beiser A, Wilson PWF et al.Prediction of Lifetime Risk for Cardiovascular Disease by Risk Factor Burden at 50 Years of Age.Circulation 2006; 113:791-798.
Basado en este método a finales de enero del 2012 fueron publicados los resultados delProyecto Global de Riesgo Cardiovascular “de por vida”. En este proyecto meta-analítico se incluyeron 18estudios con 257,384 individuos hombres y mujeres, blancos y negros evaluados por decenios entre los 45 ylos 75 años de edad. Los resultados principales de este estudio son los siguientes.
a) De la población analizada, sólo 5% tuvo el perfil ideal -grupo 1- y dos terceras partestuvieron perfiles con 1 o más FRCV -grupos 4 y 5-. Existió incremento en la prevalencia de DM e HTA yreducción de tabaquismo, sin cambio en el nivel de colesterol, en relación directa a la edad. Sin embargo enigualdad de variables, el impacto de los FRCV es similar independientemente de la edad. En otras palabras,tener un perfil ideal a los 45 años o a los 75 años de edad implica un mejor pronóstico que tener un perfil convariables subóptimas, altas o 1 o más FRCV. En generaciones nacidas más recientemente, existe unatendencia a menor número de variables subóptimas, altas y/o FRCV relacionados con tabaquismo, lípidose HTA y mayor número de DM.
b) Los FRCV clásicos conservan su efecto de riesgo cardiovascular en forma directamenteproporcional al número de FRCV y al tiempo de exposición a ellos. Por ejemplo, un hombre de 55 años de
Riesgo de “por vida”En esta tabla se muestran las 5 variables propuestaspor Lloyd-Jones para estratificar el riesgocardiovascular de “por vida”. Se muestran losvalores para clasificar las variables como óptima,subóptima, alta o como FRCV
n de las variables según LloydClasificació -Jones
Variable1.- T.A.S. mmHg
2.- T.A.D. mmHg
3.- C.T. mg/dl
4.- Tabaquismo
5.- D.M.
Óptima<120
<080
<180
Negativo
Negativo
Subóptima120-139
080-089
180-199
Negativo
Negativo
Alta140-159
090-099
200-239
Negativo
Negativo
FRCV160 o Tx
100 o Tx
240 o Tx
Positivo
Positivo
>
>
>
Cardio-Lipidología
106

edad con un perfil óptimo tendría un riesgo de muerte cardiovascular a los 80 años de edad de 4.7%,comparado con un riesgo de 29.6% si el perfil fuera de 2 o más FRCV; de muerte cardiovascular o infartomiocárdico no fatal de 3.6% vs 37.5% y de enfermedad vascular cerebral fatal o no fatal de 2.3% vs 8.3%; enuna mujer de 55 años de edad los riesgos de los eventos referidos serían de 6.4% vs 20.5%; 1% vs 18.3% y5.3% vs 10.7%.
c) El porcentaje de variables subóptimas, altas y/o de FRCV es mayor en individuos negrosque en blancos. Sin embargo en igualdad de variables, el impacto de los FRCV es similar en individuosnegros que en blancos.
Berry JD, Dyer A, Cai X, Garside DB, Ning H et al. Lifetime Risk of CardiovascularDisease. N Engl J Med 2012; 366:321-329.
Riesgo de “por vida”En esta tabla se muestran para hombres y mujeres
los riesgos cardiovasculares de 45 y 55 hacia 85años y de 65 y 75 hacia 90 años en relación a la
clasificación de las variables propuestas por Lloyd-Jones y evaluadas 257,384 individuos del Proyecto
Global de Riesgo Cardiovascular de “por vida”
Salud, Riesgo, Estratificación y Metas
Hombres TodasÓptimas
1Subóptima
1Alta
1FRCV
2FRCV
45 hacia 80 años 1.4 31.2 39.6 39.6 49.5
55 hacia 80 años 14.6 19.7 33.9 32.2 46.8
65 hacia 90 años 29.5 29.4 38.2 37.2 49.5
75 hacia 90 años 17.5 22.8 28.9 36.1 38.5
Hombres TodasÓptimas
1Subóptima
1Alta
1FRCV
2FRCV
45 hacia 80 años 1.4 31.2 39.6 39.6 49.5
55 hacia 80 años 14.6 19.7 33.9 32.2 46.8
65 hacia 90 años 29.5 29.4 38.2 37.2 49.5
75 hacia 90 años 17.5 22.8 28.9 36.1 38.5
Mujeres TodasÓptimas
1Subóptima
1Alta
1FRCV
2FRCV
45 hacia 80 años 4.1 12.2 15.6 20.2 30.7
55 hacia 80 años 10.1 13.3 15.3 16.7 29.2
65 hacia 90 años 12.4 25 29.3 31.9 38.7
75 hacia 90 años 12.4 19.9 21.8 29.4 36.3
Mujeres TodasÓptimas
1Subóptima
1Alta
1FRCV
2FRCV
45 hacia 80 años 4.1 12.2 15.6 20.2 30.7
55 hacia 80 años 10.1 13.3 15.3 16.7 29.2
65 hacia 90 años 12.4 25 29.3 31.9 38.7
75 hacia 90 años 12.4 19.9 21.8 29.4 36.3
>>>
>>>
Riesgo de Eventos Cardiovasculares Totales asociados a AterosclerosisModelo Time Risk Pooling Proyect » para hombre y mujer« Life
107

El mensaje de estos estudios es simple, el nivel de riesgo cardiovascular con que se llega a los 45o 55 años de edad implicará un premio o un costo a pagar en el mediano y largo plazo. Por lo tanto, el cálculode riesgo “de por vida”, permite plantear a cualquier individuo de 20 años de edad la necesidad de laprevención, detección y/o tratamiento de las variables de riesgo cardiovascular lo más precoz posible,mucho antes de los 45 o 55 años de edad, con el objetivo de llegar a dicha edad con una buena perspectivacardiovascular para la edad adulta avanzada y en la vejez.
Cálculo del riesgo a 30 años. Michael Pencina como primer autor, publicó en el 2009 elmétodo para el cálculo del riesgo cardiovascular a 30 años. El sentido de esta visión, también está enfocadoa predecir el riesgo cardiovascular a largo plazo. La población estudiada fueron individuos entre 20 y 59años de edad sin historia de enfermedad cardiovascular clínica. Las variables analizadas fueron las clásicasde Framingham: edad, sexo, tensión arterial sistólica virgen a tratamiento o en tratamiento, colesterol total,colesterol-HDL, diabetes mellitus y tabaquismo; también se incluyó el índice de masa corporal como unmarcador de adiposidad. Las conclusiones más importantes de esta publicación son las siguientes:
a) A mayor número de FRCV, mayor probabilidad de un evento cardiovascular en laproyección de 10 años hacia 30 años.
b) A menor edad y mayor número de FRCV, mayor amplificación en la probabilidad de unevento cardiovascular en la proyección de 10 años hacia 30 años; por ejemplo un individuo de 25 años deedad con múltiples FRCV, tiene un riesgo cardiovascular calculado a 10 años de 5%, el cual daría una falsaimpresión de salud cardiovascular, sin embargo proyectado a 30 años, cuando tenga 55 años su riesgo seráde 40%, una amplificación del riesgo de 800% que elimina la falsa imagen de salud cardiovascular y sustentala relevancia de la estratificación y el control temprano de los FRCV. Es deseable que el cálculo del riesgocardiovascular y el tratamiento de los FRCV sea lo más temprano posible, en el “Statement” AHA-ACCFpublicado la última semana del 2010, se propuso que la estratificación y en su caso el tratamiento preventivoinicien a partir de los 20 años de edad en hombres y mujeres.
c) El índice de masa corporal tiene valor únicamente como una variable integrada al resto de losFRCV, como variable aislada, el IMC tiene un poder predictor pobre y es posible calcular el riesgocardiovascular en ausencia de parámetros de laboratorio, sin embargo al igual que lo planteó D´Agostino sibien es factible, no es ideal.
Recientes reportes de los estudios ARIC, CARDYA y MESA, han mostrado que en individuos
Cardio-Lipidología
108

Dislipidemia
Hipertensión
Tabaquismo
Diabetes
No
No
No
No
Si
No
No
No
Si
Si
No
No
Si
Si
Si
No
Si
Si
Si
Si
%es
tim
ado
de
ries
gode
EC
V
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Riesgo estimado a 10 años
Riesgo estimado a 30 años
Amplificación del riesgo de 10 a 30 años
Modelo de Michael Pencina
<50 años de edad, la prevalencia de aterosclerosis subclínica fue significativamente mayor en individuoscon riesgo bajo a corto plazo y riesgo alto a largo plazo, comparado con individuos con riesgo bajo a corto ylargo plazo, en conjunto con los ya analizados reportes NHANES 1999-2002, 2003-2008 y el ProyectoConjunto del Riesgo Cardiovascular “de por vida”, está justificado incorporar el cálculo del riesgocardiovascular a largo plazo en individuos con riesgo bajo a 10 años.
Pencina MJ, D´Agostino RB, Larson MG, Massaro JM, Vasan RS. Predicting the 30-YearRisk of Cardiovascular Disease. The Framingham Heart Study. Circulation 2009; 119:3078-3084. BerryJD, Liu K, Folsom AR, Lewis CE, Carr JJ, Polak JF et al. Prevalence and Progression of SubclinicalAtherosclerosis in Younger Adults with Low Short-Term but High Lifetime Estimated Risk forCardiovascular Disease. The Coronary Artery Risk Development in Young Adults Study -CARDYA Study-and Multi-Ethnic Study of Atherosclerosis -MESA Study-. Circulation 2009; 119:382-389.
Riesgo a 30 añosEn esta tabla se muestra para un hombre de 45
años el cálculo de riesgo a 10 años- -barras verdes-y a 30 años -barras rojas- de acuerdo a la ausencia opresencia de dislipidemia, HTA, tabaquismo y DM.
Las barras negras representan la amplificación delriesgo de 10 hacia 30 años, destacando la
importancia de calcular el riesgo en un horizontemás allá de los 10 años
Salud, Riesgo, Estratificación y Metas
109

Estratificación avanzada de riesgo cardiovascularEn diciembre del 2010 fue publicada la Guía Conjunta AHA-ACCF para la estratificación del
riesgo cardiovascular en el individuo asintomático y sin historia de enfermedad cardiovascular. Estedocumento recopiló, analizó y sintetizó en forma de Guías la mejor bibliografía sobre biomarcadores ybioimágenes en la estratificación del riesgo cardiovascular publicada hasta el año 2010, clasificando en laforma convencional AHA-ACCF, el nivel de indicación como I, II y III y el tipo de evidencia como A, B yC. En forma sencilla podríamos considerar que las indicaciones clase I son ampliamente recomendadas, lasindicaciones clase II son recomendadas a la luz del juicio clínico individualizado y las indicaciones clase IIINO son recomendadas. El nivel de evidencia A es aquel basado en bibliografía sólida, un nivel C enbibliografía débil y un nivel B en bibliografía aceptable.
Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fallad Z et al. 2010ACCF/AHA Guideline for Assessment of Cardiovascular Risk in Asymptomatic Adults: ExecutiveSummary. A Report of the American College of Cardiology Foundation/American Heart AssociationTask Force on Practice Guidelines. Circulation 2010; 122:2748-2764.
De esta Guía se desprende que la única recomendación con indicación clase I es la siguiente:1.- Estratificación del riesgo cardiovascular con métodos como Framingham, SCORE,
PROCAM o Reynolds. Los autores recomiendan su realización en todo individuo, hombre o mujer de 20años de edad o mayor. La integración de la historia de enfermedad cardiovascular precoz en familiares deprimer orden -padres e hijos- debe ser parte de este proceso.
A continuación revisaremos de acuerdo a la Guía referida, el tipo de indicación y el nivel deevidencia de los 6 biomarcadores y las 10 bioimágenes con mayor evidencia:
Biomarcadores1.- Lípidos, lipoproteínas y apoproteínasa) La determinación de lípidos, lipoproteínas, apoproteínas más allá de colesterol total, HDL,
LDL y no-HDL y triglicéridos tiene indicación Clase III con nivel de evidencia C.2.- Proteína C reactivaa) La determinación de PCR en hombres de 50 años de edad o más, o en mujeres de 60 años de
edad o más con LDL <130mg/dl, sin terapia modificadora de lípidos, sin criterios de alto riesgocardiovascular -incluye DM- y sin causas no cardiovasculares para el incremento de PCR tiene indicaciónClase IIa con nivel de evidencia B. Su utilidad es la indicación potencial de estatinas en individuos que por
Cardio-Lipidología
110

EbRCV podrían no tenerla.b) La determinación de PCR en hombres de menos de 50 años o en mujeres de menos de 60
años y riesgo intermedio en la EbRCV tiene indicación Clase IIb con nivel de evidencia B.c) La determinación de PCR en individuos de alto riesgo tiene indicación Clase III con nivel de
evidencia B.d) La determinación de PCR en hombres de menos de 50 años o en mujeres de menos de 60
años y riesgo bajo en la EbRCV tiene indicación Clase III con nivel de evidencia B.3.- Microalbuminuriaa) La determinación de microalbuminuria en individuos con HTA y/o DM tiene indicación
Clase IIa con nivel de evidencia B.b) La determinación de microalbuminuria en individuos sin HTA ni DM con riesgo
intermedio en la EbRCV tiene indicación Clase IIb con nivel de evidencia B.4.- Hemoglobina Glicosilada -A1c-a) La determinación de A1c en individuos sin DM tiene indicación Clase IIb con nivel de
evidencia C.b) La determinación de A1c en individuos con DM tiene indicación Clase IIb con nivel de
evidencia B.5.- Lipoproteína Fosfolipasa Asociada A2 -Lp-PLA2-a) La determinación de Lp-PLA2 en individuos con riesgo intermedio en la EbRCV tiene
indicación Clase IIb con nivel de evidencia B.6.- Péptido natriurético atrial -BNP-a) La determinación de BNP tiene indicación Clase III con nivel de evidencia B.7.- Genotipoa) La determinación del genotipo tiene indicación Clase III con nivel de evidencia B.En base a este análisis sobre la evidencia en biomarcadores se podría concluir que como
complemento a la EbRCV las opciones con mejor perfil son la PCR -inciso 2a y 2b- y la Microalbuminuria,especialmente en individuos con HTA o DM- inciso 3a-, considerando que el individuo con DM, entérminos generales en EbRCV se considera de riesgo alto.
Salud, Riesgo, Estratificación y Metas
111

Biomarcadores. Utilidad en la clínica. ACCF-AHA 2010
Bioimágenes1.- ECG en reposo:a) La realización de ECG de reposo en individuos con HTA y/o DM tiene indicación Clase IIa
con nivel de evidencia C.b) La realización de ECG de reposo en individuos sin HTA ni DM tiene indicación Clase IIb
con nivel de evidencia C.2.- ECG de esfuerzoa) La realización de ECG de esfuerzo en individuos sedentarios de riesgo intermedio en la
Estratificación básica I B 20 años
Hx familiar de ECV I B 20 años
PCR IIa B 50-60-LDL <130
PCR IIb B <50-60-RCV 10-
PCR III B RCV 20%-DM y/o ECV
PCR III B <50-60-RCV <10%
Microalbumina IIa B Con HTA y/o DM
Microalbumina IIb B Sin HTA ni DM
A1c IIb C Sin DM
A1c IIb B Con DM
Lp-PLA2 IIb B RCV 10%-<20%
BNP III B No indicado
Genotipo III B No indicado
Estratificación básica I B 20 años
Hx familiar de ECV I B 20 años
PCR IIa B 50-60-LDL <130
PCR IIb B <50-60-
PCR III B RCV 20%-DM y/o ECV
PCR III B <50-60-RCV <10%
Microalbumina IIa B Con HTA y/o DM
Microalbumina IIb B Sin HTA ni DM
A1c IIb C Sin DM
A1c IIb B Con DM
Lp-PLA2 IIb B RCV 10%-<20%
BNP III B No indicado
Genotipo III B No indicado
Biomarcador Indicación Evidencia Observaciones
Biomarcadores en asintomáticosEn esta tabla se muestra la propuesta AHA-ACCF2010 para el empleo de biomarcadores en laestratificación del riesgo del individuo asintomático.Nótese que sólo la estratificación básica y elinterrogatorio de ECV precoz tienen indicaciónclase I
Cardio-Lipidología
RCV 10%-<20%
112

EbRCV para la evaluación de su capacidad física tiene indicación Clase IIb con nivel de evidencia B.3.- Ecocardiograma trans-torácico en reposoa) La realización de ecocardiograma para determinar HVI en individuos con HTA tiene
indicación Clase IIb con nivel de evidencia B.b) La realización de ecocardiograma para determinar HVI en individuos sin HTA tiene
indicación Clase III con nivel de evidencia C.4.- Ecocardiograma trans-torácico de estrésa) La realización de ecocardiograma de estrés en individuos de riesgo bajo o intermedio tiene
indicación Clase III con nivel de evidencia C.4.- Grosor íntima-media carotidea -IMT-a) La determinación de IMT en individuos de riesgo intermedio en la EbRCV tiene indicación
Clase IIa con nivel de evidencia B. Deben cumplirse los requisitos de operador-equipo determinados porlas guías respectivas.
5.- Indice tobillo-brazo -ITB-a) La determinación de ITB en individuos de riesgo intermedio en la EbRCV tiene indicación
Clase IIa con nivel de evidencia B.6.- Indice de calcio coronario -ICC-a) La determinación de ICC en individuos de riesgo intermedio 10%-<20% tiene indicación
Clase IIa con nivel de evidencia B.b) La determinación de ICC en individuos de riesgo intermedio “bajo” -6%-<10%- tiene
indicación Clase IIb con nivel de evidencia B.c) La determinación de ICC en individuos de riesgo bajo -<6%- tiene indicación Clase III con
nivel de evidencia B.
d)La determinación de ICC en individuos 40 años de edad con DM tiene indicación Clase�
IIa con nivel de evidencia B.7.- Reserva vasodilatadora braquial -RVB-a) La determinación de RVB tiene indicación Clase III con nivel de evidencia B.8.- Rigidez arteriala) La determinación de rigidez arterial tiene indicación Clase III con nivel de evidencia C.
Salud, Riesgo, Estratificación y Metas
113

Bioimágenes. Utilidad en la clínica. ACCF-AHA 2010
ECG en reposo IIa C Con HTA y/o DM
ECG en reposo IIb C Sin HTA ni DM
ECG de esfuerzo IIb B RCV 10- <20% sedentario
ECO en reposo HVI IIb B Con HTA
ECO en reposo HVI III C Sin HTA
ECO de estrés III C RCV <20%
IMT IIa B RCV 10%-<20%
ITB IIa B RCV 10%-<20%
ICC IIa B RCV 10%-<20%
ICC IIb B RCV 6%-<10%
ICC III B RCV <6%
ICC IIa B DM=40 años
RVB III B No indicado
ECG en reposo IIa C Con HTA y/o DM
ECG en reposo IIb C Sin HTA ni DM
ECG de esfuerzo IIb B RCV 10%-<20% sedentario
ECO en reposo HVI IIb B Con HTA
ECO en reposo HVI III C Sin HTA
ECO de estrés III C RCV <20%
IMT IIa B RCV 10%-<20%
ITB IIa B RCV 10%-<20%
ICC IIa B RCV 10%-<20%
ICC IIb B RCV 6%-<10%
ICC III B RCV <6%
ICC IIa B 40 años con DM
RVB III B No indicado
Bioimagen Indicación Evidencia Observaciones
Rigidez arterial III C No indicado
Gamagrafía de estrés IIb C RCV =20%, DM, ICC
=400
Gamagrafía de estrés III C RCV <20%
Angiotomografía III C No indicado
RMN III C No indicado
Rigidez arterial III C No indicado
Gamagrafía de estrés IIb C RCV 20%, DM, ICC 400
Gamagrafía de estrés III C RCV <20%
Angiotomografía III C No indicado
RMN III C No indicado
Bioimágenes en asintomáticosEn esta tabla se muestra la propuesta AHA-ACCF2010 para el empleo de bioimágenes en laestratificación del riesgo del individuo asintomático.Nótese que ninguna de ellas tiene indicación clase I,la mayoría son clase II y III
Cardio-Lipidología
114

9.- Gamagrafía miocárdica de estrésa) La realización de gamagrafía de estrés en individuos de riesgo alto con historia familiar de
enfermedad cardiovascular precoz, DM y/o ICC 400 UA tiene indicación Clase IIb con nivel de�
evidencia C.b) La realización de gamagrafía de estrés en individuos de riesgo bajo o intermedio tiene
indicación Clase III con nivel de evidencia C.10.- Angiotomografía coronariaa) La realización de angiotomografía tiene indicación Clase III con nivel de evidencia C.11.- Resonancia magnética nuclear -RMN-b) La realización de RMN tiene indicación Clase III con nivel de evidencia C.
Otros métodos de estratificación.Método de Reynolds-Ridker. El grupo de la Universidad de Harvard, encabezado por el
Dr. Paul Ridker y con el patrocinio de la Fundación Donald W. Reynolds y el NHLBI, propuso laincorporación de la PCR de alta sensibilidad y la historia familiar de eventos cardiovasculares tempranos,para optimizar la valoración del riesgo cardiovascular en población sin evidencia de enfermedadcardiovascular clínica y/o diabetes mellitus. El denominado índice de Reynolds fue validado originalmenteen población femenina y en noviembre del 2008 apareció publicada su validación en población masculina.Evidencias sólidas han confirmado que un valor de PCR > 3 mg/lt, de 1-3 mg/lt y < 1 mg/lt, confieren demanera significativa e independiente, un riesgo cardiovascular alto, intermedio y promedio.
Ridker integrando a los factores de riesgo cardiovascular “clásicos” el valor de PCR y la historiafamiliar de eventos cardiovasculares antes de los 60 años en familiares de primer orden, demostró que esfactible reclasificar entre 15% y 20% de individuos a un subgrupo de riesgo mayor. Sin entrar en detallessobre las características requeridas para un nuevo algoritmo, ya analizados al principio de este capítulo, elmétodo de Reynolds es especialmente útil en los individuos que por el método de Framingham tienen unaprobabilidad de un evento cardiovascular de 6%-<10% ó 10%-<20%. En el primer subgrupo, 9% sonreclasificados hacia el subgrupo de riesgo intermedio y del subgrupo de riesgo intermedio, 8% sonreclasificados hacia el subgrupo de riesgo alto. Esta reclasificación “hacia arriba” de 17% de un subgrupode la población tiene implicaciones terapéuticas significativas, ya que el umbral terapéutico para LDL,colesterol no-HDL y presión arterial se modifican “hacia abajo” con la reclasificación, es decir se hacen más
Salud, Riesgo, Estratificación y Metas
115

estrictos. Un aspecto muy importante sobre la reclasificación es la reclasificación “hacia abajo”, es decirreclasificar a individuos del subgrupo de riesgo alto hacia un riesgo intermedio o del subgrupo de riesgointermedio hacia un riesgo bajo. Esta reclasificación al contrario de la reclasificación “hacia arriba”, implicaun riesgo potencial, ya que al “bajar” a un individuo a un subgrupo de riesgo más bajo, su meta terapéuticase eleva, es decir se hace menos estricta. El principal cuestionamiento a la adopción de esta conducta es queal incrementar el umbral terapéutico a valores menos exigentes, el individuo podría quedar desprotegido alargo plazo, especialmente porque el seguimiento en el que se fundamenta el método de Reynolds es de sólo7 años, no comparable con el seguimiento de varios decenios de métodos como el de Framingham.
En base a lo anterior, en la clínica no se recomienda adoptar la conducta de reclasificar“hacia abajo”. En la reclasificación “hacia arriba” no existe dicho riesgo, ya que en esta conducta elincremento en la exigencia terapéutica es congruente con la tendencia actual de establecer niveles “meta”más estrictos o fisiológicos para todos los factores de riesgo cardiovascular modificables.
Framingham 2008ATP-III
Riesgo10% - <20%
Riesgo=20%, DM y/o ECV
Riesgo<6%
Reclasificaciones hacia arriba con impacto clínico
Riesgo6%-<10%
Método de Reynolds-RidkerBasada en PCR + historia familiar de ECV precoz
9.2% 8.0%
Framingham 2008ATP-III
Riesgo10% - <20%
Riesgo20%, DM y/o ECV
Riesgo<6%
Reclasificaciones hacia arriba con impacto clínico
Riesgo6%-<10%
- . Reclasificación en hombres
9.2% 8.0%
Reynolds-Ridker en hombresEn esta tabla se observa como a partir de los 4grupos de riesgo propuestos por Ridker, lasreclasificaciones con validez clínica son aquellas delsubgrupo de riesgo de 6% a <10% hacia riesgointermedio y del subgrupo de riesgo intermediohacia riesgo alto; en total 17.2%. Lasreclasificaciones entre los subgrupos de bajo riesgono tienen impacto y la reclasificación hacia abajo nose recomienda en la clínica
Cardio-Lipidología
116

Ridker PM, Buring JE, Rifai N, Cook RN. Development and Validation of ImprovedAlgorithms for the Assessment of Global Cardiovascular Risk in Women: the Reynolds Risk Score. JAMA2007; 297:611-619. C-Reactive Protein andRidker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR.Parenteral History Improve Global Cardiovascular Risk Prediction. The Reynolds Risk Score for Men.Circulation 2008; 118:2243-2251.
Método de Laval-Després. El grupo del Instituto de Corazón de Quebec, encabezado por elDr. Jean Pierre Després, ha propuesto la incorporación de la adiposidad central o visceral, en su expresiónde cintura abdominal >102cm en hombres y >88cm en mujeres ->90cm y >84cm en hombres y mujereslatinos-, para una mejor valoración del riesgo cardiovascular. Para este grupo de trabajo, la coexistencia deadiposidad central más triglicéridos >150 mg/dl o “cintura hipertrigliceridémica”, es un binomio clínico,sencillo, económico y con un alto valor predictor para inferir la existencia de un riesgo cardiovascularincrementado por factores metabólicos no considerados dentro de los clásicos.
En su propuesta el grupo de Quebec conserva el resto de FRCV“clásicos”: colesterol total y/ocolesterol-LDL, colesterol-HDL, hipertensión arterial, diabetes mellitus, edad, sexo masculino ytabaquismo, dejando incluso cabida a otros factores de riesgo cardiovascular en validación. Esta propuestabrinda al clínico una buena herramienta para la predicción de eventos cardiovasculares. Efectivamente lacoexistencia de adiposidad central más triglicéridos >150mg/dl, es un signo dual, sensible y específico paraintuir que la adiposidad visceral ha condicionado metainflamación, resistencia a la insulina, disfunciónendotelial y metabólica, y por tanto un riesgo cardiovascular mayor al calculado tradicionalmente. En estecontexto, la “cintura-hipertrigliceridémica”, cuando se acompaña de otros factores de riesgo cardiovascular“clásicos”, indicará una probabilidad de un evento cardiovascular mayor a la calculada sólo por estosúltimos. La propuesta de Després, quizá fue la primera propuesta formal para integrar en uno, dosconceptos hasta hace poco tiempo separados e incluso antagónicos, el método de Framingham y laclasificación de Síndrome Metabólico. Actualmente está fuera de toda controversia el que ambos conceptosse vinculan sinérgicamente entre sí. Por ejemplo, un individuo de riesgo alto por enfermedadcardiovascular, si desarrollará síndrome metabólico se transformaría en un individuo de riesgo muy alto.
Després JP, Lemieux I. Abdominal Obesity and Metabolic Syndrome. Nature 2006;444:881-887.
Salud, Riesgo, Estratificación y Metas
117

Metas terapéuticas de lípidosEn espera de los resultados de los mega-ensayos clínicos que investigan el tratamiento
poblacional del riesgo cardiovascular con estrategias como la poly-pill o la poli-cap -ver capítulo 5-, en laMedicina Clínica, la EbRCV y la EaRCV son las tácticas clínicas de mayor utilidad en un consultoriomédico, ellas nos permite establecer las estrategias terapéuticas para cumplir con nuestra meta másimportante como Médicos, la cual consiste en mostrarle a todo individuo una vía para que su vida sea largay de calidad.
En este apartado se hará una integración clínico-práctica de los conceptos expresados en lostres documentos más importantes en esta arena terapéutica: las Guías aún vigentes del Panel deTratamiento del Adulto número III -ATP-III modificado-, las Guías Conjuntas EAS-ESC 2011 y las GuíasConjuntas ACCF-AHA2011. En su esencia las tres Guías están basadas en la mejor evidencia científica yaunque emplean diferentes algoritmos de ERCV, conceptualmente son similares con diferencias sutiles enla interpretación del peso específico de la evidencia.
Third Report of the National Cholesterol Education program -NCEP- Expert Panelon Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Adult TreatmentPanel III. Grundy SM, Cleeman JI, Merz CN, BrewerFinal Report. Circulation 2002; 106:3141-3421.HB Jr, Clark LT, Huninghake DB et al. Implications of Recent Clinical Trials for the NationalCholesterol Education Program Adult Treatment Panel III Guidelines. Circulation 2004; 110:227-239.Reiner Z, Catapano AL, De Becker G, Graham I, Taskinen M, Wiklund O, Agewall S et al. The TaskForce for the Management of Dyslipidemias of the ESC-EAS. Eur Heart J 2011; 32:1769-1818. Smith SC,Benjamin EJ, Bonow RO, Braun LT, Creager MA, Franklin BA et al. AHA/ACCF SecondaryPrevention and Risk Reduction Therapy for Patients with Coronary and Other Atherosclerotic VascularDisease: 2011 Update. Circulation 2011; 124: published on line 29 November 2011.
Guías y LípidosAnte la reciente publicación de las Guías EAS-ESC -junio 2011- y las Guías AHA-ACCF
-noviembre 2011- y en vísperas de la publicación en “combo” del ATP IV en conjunto con el JNC8 y lasGuías para el manejo del sobrepeso y la obesidad, son válidas las siguientes consideraciones en relación a lasmetas lipídicas:
1.- La Aterosclerosis es una enfermedad que inicia con la concepción y es progresiva a lo largode la vida de un individuo. Por lo tanto la predicción de los eventos cardiovasculares derivados de la
Cardio-Lipidología
118

Aterosclerosis y la prevención y/o control de los FRCV, debería ser precoz, óptima e indefinida.2.- La brecha entre el nivel fisiológico de LDL -<50mg/dl- y el nivel promedio en una
población -130mg/dl- es el factor de riesgo más importante para la génesis de la Aterosclerosis. Por lotanto, en prevención primordial, primaria y secundaria se debe privilegiar el concepto de que el niveldeseable de dicha lipoproteína es el más cercano al valor fisiológico.
3.- La adopción o modificación del estilo de vida hacia uno saludable debe ser una indicaciónmédica universal; el empleo clínicamente juicioso -basado en evidencia- de fármacos como las estatinas esla estrategia terapéutica que hasta hoy tiene sustento en los escenarios de prevención primaria y secundaria.
Considerando a la herencia, el género y la edad como variables no modificables, el ideal enprevención cardiovascular sería que todo individuo tuviera a lo largo de su vida el nivel óptimo de lasvariables modificables de riesgo cardiovascular más importantes: colesterol total <180mg/dl, tensiónarterial sistólica y diastólica <120mmHg y <80mmHg en ausencia de hábito tabáquico; concepto esencialde la Prevención Primordial. Dado que como ya se presentó, este perfil ideal es escaso en la poblaciónadulta, se ha establecido un nivel mínimo deseable para cada variable en función del riesgo cardiovascularestimado para un individuo; concepto esencial de la Prevención Primaria.
Considero importante destacar que, la aplicación individual del concepto de meta terapéuticatiene cierta artificialidad, especialmente por el establecimiento de puntos de corte “arbitrarios” paravariables que son continuas y por su vinculación a la influencia de análisis de costo/eficiencia. En otraspalabras, el riesgo cardiovascular inicia con niveles inferiores a los establecidos como “metas” para todoslos FRCV y las metas se han establecido en función del cociente: costo del tratamiento/costo e incidenciadel evento cardiovascular potencialmente evitado. Una estrategia terapéutica o el logro de una meta serácosto/eficiente en la medida que el costo del tratamiento sea menor. Considerando lo anterior es oportunomencionar que las metas ATP III para lípidos se establecieron en la época que las estatinas eficientes noeran genéricas y por lo tanto tenían un costo “alto”.
Así, por ejemplo, la meta terapéutica para LDL en un análisis de costo/eficiencia se establecióen <160mg/dl para la población de riesgo cardiovascular bajo. Esta decisión estuvo influida por laincidencia baja de eventos cardiovasculares en esta población y el costo alto que tendría tratar a todos losindividuos en riesgo bajo con estatinas “caras” si se estableciera, por ejemplo una meta terapéutica<130 mg/dl o <100mg/dl. Sin embargo, que pasaría si una estatina eficiente -eficaz, segura, tolerada y con
Salud, Riesgo, Estratificación y Metas
119

alta adherencia- fuera gratis o de bajo costo, lógicamente el análisis de costo/eficiencia permitiría reducir elnivel de la meta terapéutica de LDL hacia los valores más cercanos a los fisiológicos, independientementedel nivel de riesgo cardiovascular -concepto poly-cap de Prevención Primaria-.
Los aspectos referidos: aterosclerosis enfermedad pediátrica como inductor de tácticas para laprevención, detección y control precoz de los FRCV; nivel fisiológico de LDL como inductor de estrategiasconductuales-farmacológicas que eviten el desarrollo de aterosclerosis o bien que induzcan suestabilización y/o regresión; costo/eficiencia de la estatinas como inductor de estrategias para el uso deestatinas eficientes, están en juego en la elaboración entre otras muchas iniciativas, de las nuevas Guías aproponer por el 4º Panel para el Tratamiento del Adulto o ATP IV cuya meta global es la reducción en laincidencia de eventos cardiovasculares en el transcurso de este decenio. Dicho lo anterior, a continuación serevisan las metas lipídicas de acuerdo a las Guías vigentes.
Black HR, Wilson PWF. Should We Still be LDL-Centric. Medscape Cardiology;www.medscape.com; posted 27 December 2011.
Meta primaria -LDL-Este apartado se complementa con el capítulo 5 de terapéutica farmacológica. Existe consenso
universal en las tres Guías referidas de que la reducción de las LDL dado su roll etiopatogénico y opcionesterapéuticas eficientes, debe ser la estrategia primaria del tratamiento. Las metas terapéuticas, hasta ahoraestán determinadas por el perfil de riesgo cardiovascular; a mayor riesgo, menor nivel de LDL.
El ATP-III y las Guías AHA-ACCF establecen como meta terapéutica de LDL: <160 mg/dl
en riesgo bajo; <130mg/dl en riesgo intermedio; <100mg/dl o reducción 30% en riesgo alto y opcional�
<70 mg/dl o reducción 50% en riesgo muy alto: enfermedad cardiovascular con Diabetes Mellitus,�
síndrome metabólico, tabaquismo y/o SICA.
La Guía EAS-ESC establecen como meta terapéutica de LDL: <70mg/dl o reducción 50%�
en individuos de riesgo muy alto que incluye: enfermedad cardiovascular documentada, DM2 o DM1 condaño a órgano blanco, individuos con depuración de creatinina <60ml/min/1.73m2 o un valor en el
algoritmo SCORE 10%; <100mg/dl en individuos de riesgo alto que incluye: dislipidemia familiar, HTA�
“severa” o un valor en el algoritmo SCORE 5% a <10%; <115mg en individuos de riesgo intermedio�
que incluye: un valor en el algoritmo SCORE 1% a <5%. En promedio el valor del algoritmo SCORE�
Cardio-Lipidología
120
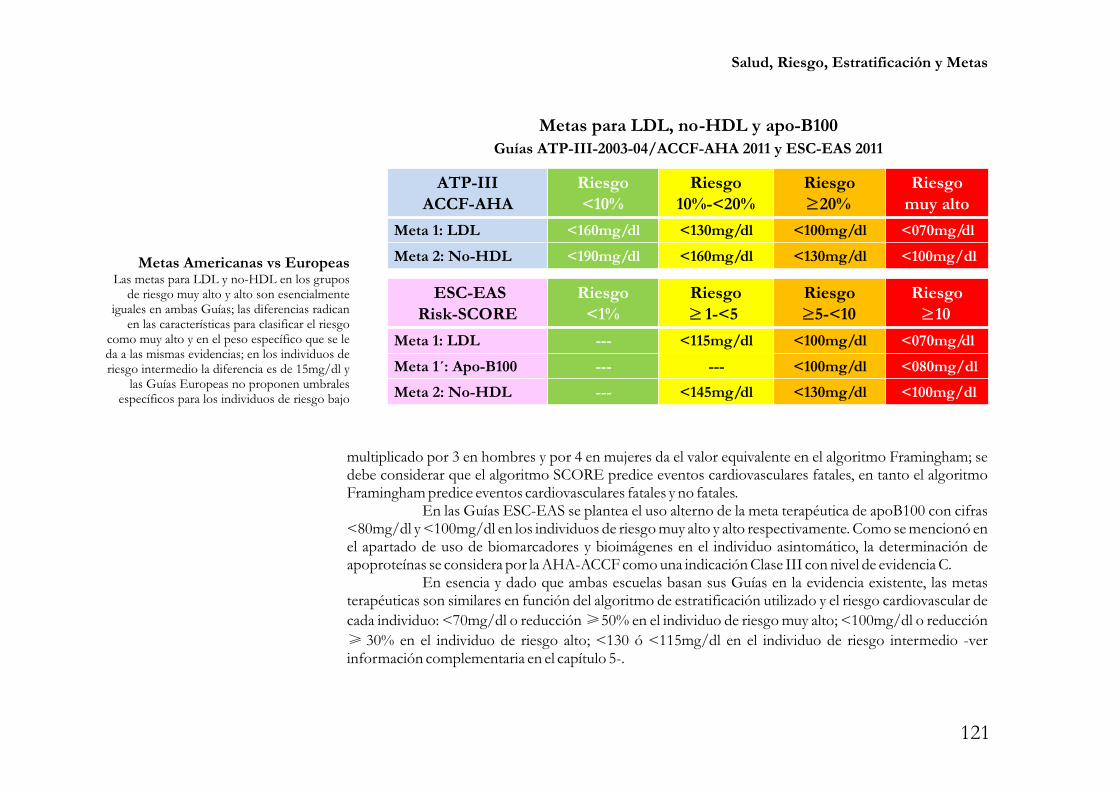
multiplicado por 3 en hombres y por 4 en mujeres da el valor equivalente en el algoritmo Framingham; sedebe considerar que el algoritmo SCORE predice eventos cardiovasculares fatales, en tanto el algoritmoFramingham predice eventos cardiovasculares fatales y no fatales.
En las Guías ESC-EAS se plantea el uso alterno de la meta terapéutica de apoB100 con cifras<80mg/dl y <100mg/dl en los individuos de riesgo muy alto y alto respectivamente. Como se mencionó enel apartado de uso de biomarcadores y bioimágenes en el individuo asintomático, la determinación deapoproteínas se considera por la AHA-ACCF como una indicación Clase III con nivel de evidencia C.
En esencia y dado que ambas escuelas basan sus Guías en la evidencia existente, las metasterapéuticas son similares en función del algoritmo de estratificación utilizado y el riesgo cardiovascular de
cada individuo: <70mg/dl o reducción 50% en el individuo de riesgo muy alto; <100mg/dl o reducción�
� 30% en el individuo de riesgo alto; <130 ó <115mg/dl en el individuo de riesgo intermedio -verinformación complementaria en el capítulo 5-.
Metas para LDL, no-HDL y apo-B100Guías ATP-III-2003-04/ACCF-AHA 2011 y ESC-EAS 2011
ATP-IIIACCF-AHA
Riesgo<10%
Riesgo10%-<20%
Riesgo20%
Riesgomuy alto
Meta 1: LDL <160mg/dl <130mg/dl <100mg/dl <070mg/dl
Meta 2: No-HDL <190mg/dl <160mg/dl <130mg/dl <100mg/dl
ATP-IIIACCF-AHA
Riesgo<10%
Riesgo10%-<20%
Riesgo20%
Riesgomuy alto
Meta 1: LDL <160mg/dl <130mg/dl <100mg/dl <070mg/dl
Meta 2: No-HDL <190mg/dl <160mg/dl <130mg/dl <100mg/dl
ESC-EASRisk-SCORE
Riesgo<1%
Riesgo1-<5
Riesgo5-<10
Riesgo10
Meta 1: LDL --- <115mg/dl <100mg/dl <070mg/dl
Meta 1´: Apo-B100 --- --- <100mg/dl <080mg/dl
Meta 2: No-HDL --- <145mg/dl <130mg/dl <100mg/dl
ESC-EASRisk-SCORE
Riesgo<1%
Riesgo1-<5
Riesgo5-<10
Riesgo10
Meta 1: LDL --- <115mg/dl <100mg/dl <070mg/dl
Meta 1´: Apo-B100 --- --- <100mg/dl <080mg/dl
Meta 2: No-HDL --- <145mg/dl <130mg/dl <100mg/dl
Metas Americanas vs EuropeasLas metas para LDL y no-HDL en los grupos
de riesgo muy alto y alto son esencialmenteiguales en ambas Guías; las diferencias radican
en las características para clasificar el riesgocomo muy alto y en el peso específico que se leda a las mismas evidencias; en los individuos deriesgo intermedio la diferencia es de 15mg/dl y
las Guías Europeas no proponen umbralesespecíficos para los individuos de riesgo bajo
Salud, Riesgo, Estratificación y Metas
121

Meta secundaria -no-HDL-Para ambas escuelas, el nivel de colesterol no-HDL es la segunda meta terapéutica en lípidos.
Puesto que 150 a 200mg/dl de triglicéridos representan en promedio 30mg/dl de colesterol contenido enlas lipoproteínas apoB100 -VLDL e IDL-, ambas potencialmente aterogénicas -ver capítulo 2 y 3-, las
Guías ESC-EAS recomiendan el cálculo del colesterol no-HDL con triglicéridos 150mg/dl en tanto el�
ATP-III y la Guía AHA-ACCF con triglicéridos 200mg/dl. En ambas Guias, las metas terapéuticas para�
colesterol no-HDL son las metas para LDL más un valor de 30: <100mg/dl en el individuo de riesgo muyalto; <130mg/dl en el individuo de riesgo alto; <160mg/dl en el individuo de riesgo intermedio.
Nivel deseable de HDLEl incremento farmacológico del nivel de HDL es hasta hoy una estrategia controversial y no
existen niveles de meta terapéutica. Si bien en bases epidemiológicas y observacionales un nivel alto y
espontáneo de HDL - 40mg/dl en hombres y 50mg/dl en mujeres- lleva implícita una menor� �
prevalencia de enfermedad cardiovascular, la evidencia sobre el beneficio de incrementarfarmacológicamente el colesterol-HDL aún es inconsistente. Esta aseveración tiene al menos 2 ejemplosrecientes y paradójicos. El programa ILLUMINATE con torcetrapib vs placebo mostró en su fase III queel inhibidor de CEPT, incrementó significativamente el nivel basal de HDL, sin embargo lamorbimortalidad cardiovascular también fue mayor. El estudio AIM-HIGH con niacina de liberaciónprolongada vs placebo -con dosis de 50mg de niacina- mostró que en individuos con enfermedadcardiovascular y terapia óptima con estatinas -LDL <70mg/dl-, la adición de niacina no es superior aplacebo en la reducción de morbimortalidad cardiovascular.
El consenso actual en relación a HDL es intensificar las conductas de vida sana y esperar losresultados de estudios en marcha con diversas estrategias farmacológicas incrementadoras de HDL-Niacina + Laropiprant, Anacetrapib, Dalcetrapib, Evacetrapib, agonistas PPAR-delta, péptidos miméticosde apoAI etc-. Así mismo es muy relevante considerar que la clasificación de las HDL a través del estudio desu proteoma y su calidad funcional -vg capacidad de flujo de colesterol macrófago-HDL, efectoantinflamatorio, efecto antioxidante etc. - nos permitirá conocer las características cuali y cuantitativas pro yanti-aterogénicas de esta compleja lipoproteína -ver capítulo 5-.
Cardio-Lipidología
122

Estatinas, Fibratos, Niacina y másEvidencias, controversias e investigaciones
Capítulo 5

GeneralidadesEste capítulo de terapéutica clínica está orientado a la reducción del riesgo cardiovascular a
través del tratamiento farmacológico de las Dislipidemias. Como fue presentado en el capítulo 4 sobreestratificación del riesgo cardiovascular, el Médico Clínico debe basar la prescripción de fármacos en laestratificación individual del riesgo cardiovascular; así mismo debe establecer una estrategia que contemplela optimización secuencial de los niveles de LDL, colesterol no-HDL e idealmente colesterol HDL. En estecapítulo se revisarán los siguientes grupos terapéuticos.
Fármacos reductores de LDL. Las estatinas son los fármacos de aplicación clínica máseficientes para la reducción de LDL. Se revisarán sus mecanismos de acción para la reducción de LDL,reducción de biomarcadores de inflamación endotelio-vascular y la inducción de citoprotección; así mismose revisarán las evidencias sobre ateroregresión y reducción de eventos cardiovasculares en prevenciónprimaria y prevención secundaria en síndromes coronarios estables, síndromes coronarios inestables,intervención coronaria percutánea y enfermedad vascular cerebral. Los datos con aplicación clínica sobreezetimibe también serán revisados.
Fármacos reductores de triglicéridos y colesterol no-HDL. Los fibratos, si bien conindicaciones aún controversiales, son los fármacos de aplicación clínica más eficientes para la reducción detriglicéridos y colesterol no-HDL. Se revisarán sus mecanismos de acción para la reducción de triglicéridos,colesterol no-HDL y biomarcadores de inflamación endotelio-vascular; así mismo se revisarán lasevidencias sobre reducción de eventos cardiovasculares en prevención primaria y prevención secundaria,con foco en su papel como fármacos adyuvantes a las estatinas en individuos con triglicéridos elevados yHDL bajo residuales al tratamiento óptimo con estatinas. Los datos más importantes con aplicación clínicasobre ácidos omega-3 también serán revisados.
Fármacos incrementadores de HDL. La niacina, aún después de 56 años de uso clínico,también tiene indicaciones aún controversiales, sin embargo es el fármaco de aplicación clínica máseficiente para incrementar HDL. Se revisará su mecanismo de acción para incrementar el HDL; así mismose revisarán las evidencias sobre eventos cardiovasculares en prevención primaria y secundaria con énfasisen su papel como fármaco adyuvante a las estatinas en individuos de alto riesgo con HDL bajo persistente.
Fármacos en investigación. Este es un campo en gran actividad en los tres rubrosterapéuticos. Se enunciarán las moléculas, estudios y resultados publicados de las diferentes moléculas parala reducción de LDL, reducción de colesterol no-HDL y triglicéridos e incremento de HDL.
124
Cardio-Lipidología

EstatinasMecanismo de acción y efecto en lípidosLa primera estatina fue extraída del Penicillum citrinum por el Dr. Akira Endo en los años 70s.
Este investigador basado en el conocimiento sobre la síntesis de colesterol a partir deAcyl y Acetyl CoA,demostró que la compactina bloquea la síntesis de colesterol al competir con la Hidroxi Metil Glutaril CoApor el sitio catalítico de su reductasa. Ahora se sabe que todas las estatinas compiten eficientemente con laHMGCoA por el sitio catalítico de la HMGCoAR y que este mecanismo inhibe el paso metabólico deHMGCoA hacia mevalonato, molécula precursora de la síntesis de colesterol, isoprenoides y otrasmoléculas. El descubrimiento de Akira Endo se complementó y tomó sentido clínico con los estudios deJoseph Goldstein y Michael Brown; ambos Investigadores descubrieron al receptor para LDL ydescribieron los conceptos de endocitosis mediada por receptor, autoregulación del receptor y reciclaje delreceptor. Estos conceptos que son tratados in extenso en el capítulo 1 sobre metabolismo de lípidos ylipoproteínas, revolucionaron la Medicina y les hicieron merecedores del premio Nobel de Medicina yFisiología en 1985.
Al inhibir la HMGCoAR, las estatinas reducen la concentración de LDL por los siguientesmecanismos: reducción de la síntesis hepática de colesterol, la lipidación de apoB100 y por ende de lasíntesis de VLDL, precursora de IDL y LDL; reducción en la concentración de colesterol libre en lasmembranas del hepatocito con activación del factor de transcripción SREBP2 e incremento en la síntesis yexpresión del receptor de LDL; incremento en la captación hepática de LDL por LDLR con aumento en laeliminación hepatobiliar de colesterol esterificado.
Endo A. The discovery and development of HMGCoA reductase inhibitors. J Lipid Res 1992;33:1569-1582. A tribute to Akira Endo, discoverer of a “Penicillin” for theBrown MS, Goldstein JL.cholesterol. Atherosclerosis 2004 Supp -5-:13-16. . A receptor-mediatedBrown MS, Goldstein JLpathway for cholesterol homeostasis. Nobel Lecture 9 December 1985. Downloadedwww.nobelprices.com. . Regulation of the mevalonate pathway. Nature 1990;Goldstein JL, Brown MS343:425-430. . The LDL receptor. History of discovery. Arterioscler ThrombGoldstein JL, Brown MSVasc Biol 2009; 29:431-438.
La eficacia reductora de LDL de las estatinas está en relación directa con el número de enlacesque estas moléculas establecen con el sitio catalítico de la HMGCoAR, esta homología entre la estructuratridimensional de las estatinas y la región catalítica de la HMGCoAR, se traduce en coeficientes de 50% de
125
Estatinas, Fibratos, Niacina y más

inhibición de la acción de la HMGCoAR o IC50 de 6.8, 12 y 15.2nM/L para pitavastatina, rosuvastatina yatorvastatina respectivamente. En la clínica, la comparación de la eficacia de las estatinas para reducir LDLdebe basarse en la bioequivalencia de sus dosis. En términos generales 4mg de pitavastatina -dosisterapéutica máxima- tiene la misma eficacia que 10mg de rosuvastatina o 20mg de atorvastatina, siendo lasdosis terapéuticas máximas 40mg para rosuvastatina y 80mg para atorvastatina.
La reducción media de LDL, está en función de la estatina y su dosis. Dosis de 10mg deatorvastatina, 5mg de rosuvastatina o 2mg de pitavastatina logran una reducción media de 37.5%, en tantoque dosis de 20mg, 10mg y 4mg producen una reducción media de 42.5%; cada duplicación de la dosis decualquier estatina, agrega un 6% a 8% de reducción de LDL y 4% de reducción media de colesterol no-HDL. Este efecto de las estatinas es independiente de la hidrofilicidad por difusión facilitada porpolipéptidos transportadores de aniones orgánicos u OATP o lipofilicidad por difusión directatransmembrana y permite calcular desde un inicio cual será la dosis óptima de una estatina en función delnivel basal de LDL, la meta de acuerdo al nivel de riesgo y la brecha terapéutica. Por ejemplo, un individuocon DM2 y colesterol LDL de 150mg/dl con una meta de LDL <100mg/dl y una brecha de 50mg/dl,requerirá una dosis de alguna estatina que reduzca al menos 33.3% -150mg/dl = 100% por lo tanto50mg/dl x 100 / 150 = 33.3- el nivel basal de LDL; 20mg de atorvastatina, 10mg de rosuvastatina o 4mg depitavastatina serían buenas opciones con un excelente margen terapéutico.
Jones PH, Davidson MH, Stein EA et al. Comparison of the efficacy and safety ofrosuvastatin versus atorvastatin, simvastatin and pravastatin across doses -STELLAR Trial-. Am J Cardiol2003; 92:152-160. Comparison of the efficacy ofMcKenney JM, Jones PH, Adamczyk MA et al.rosuvastatin versus atorvastatin, simvastatin and pravastatin in achieving lipid goals. Results from theSTELLAR Trial. Curr Med Res Opin 2003; 19:689-698. Nicholls SJ, Brandrup-Wognsen G, Palmer M,Barter PJ. Meta-analysis of comparative efficacy of increasing dose of atorvastatin versus rosuvastatinversus simvastatin on lowering levels of atherogenic lipids -from VOYAGER-. Am J Cardiol 2010; 105:69-67. Critical appraissal of the role of pitavastatin in treating dyslipidemias and achieving lipid goals.Saito Y.Vasc Health and Risk Managment 2009; 5:921-936. Pitavastatin calcium: ClinicalYee LL, Wright EA.Review of a new antihyperlipidemic medication. Download Clin Ther 2011 July 07 2011. . TheTeramoto Tclinical impact of pitavastatin: comparative studies with other statins on LDL-C and HDL-C. Expert OpinPharmacother 2012, February 15, PubMed.gov, published ahead of print.
126
Cardio-Lipidología

127
Estatinas, Fibratos, Niacina y más
Hepatocito
ColesterolHMG-CoA-R E
HMGCoA
Mevalonato
E
Hepatocito
ColesterolHMG-CoA-R B100 VLDL
HMGCoA
Mevalonato
E
Retículo EndoplásmicoAparato de Golghi
SREBP
Gen R-LDL
Hepatocito
Colesterol Col
R-LDL
R-LDLR-LDL
B100
LDL
B100B100B100
CeCe
LDL
Hepatocito
Retículo EndoplásmicoAparato de Golghi
Col
SREBP2
Gen R-LDL
Estatinas. Mecanismo de acciónSe esquematiza como la estatina al ocupar el
sitio catalítico de la HMGCoAR inhibe lasíntesis de mevalonato y con ello la síntesis de
colesterol. La inhibición de la síntesis decolesterol, disminuye la lipidación de apoB100 y
la síntesis de VLDL
Estatinas. Mecanismo de acciónSe esquematiza como la estatina al inhibir la
síntesis de colesterol, reduce la concentracióndel lípido en las membranas del retículo
endoplásmico-aparato de Golghi, siendo esta laseñal para la desrepresión del factor de
transcripción SREBP2 y la síntesis dereceptores para LDL. Este mecanismo
incrementa el catabolismo de LDL circulante
Lipidación
3
2
4
1
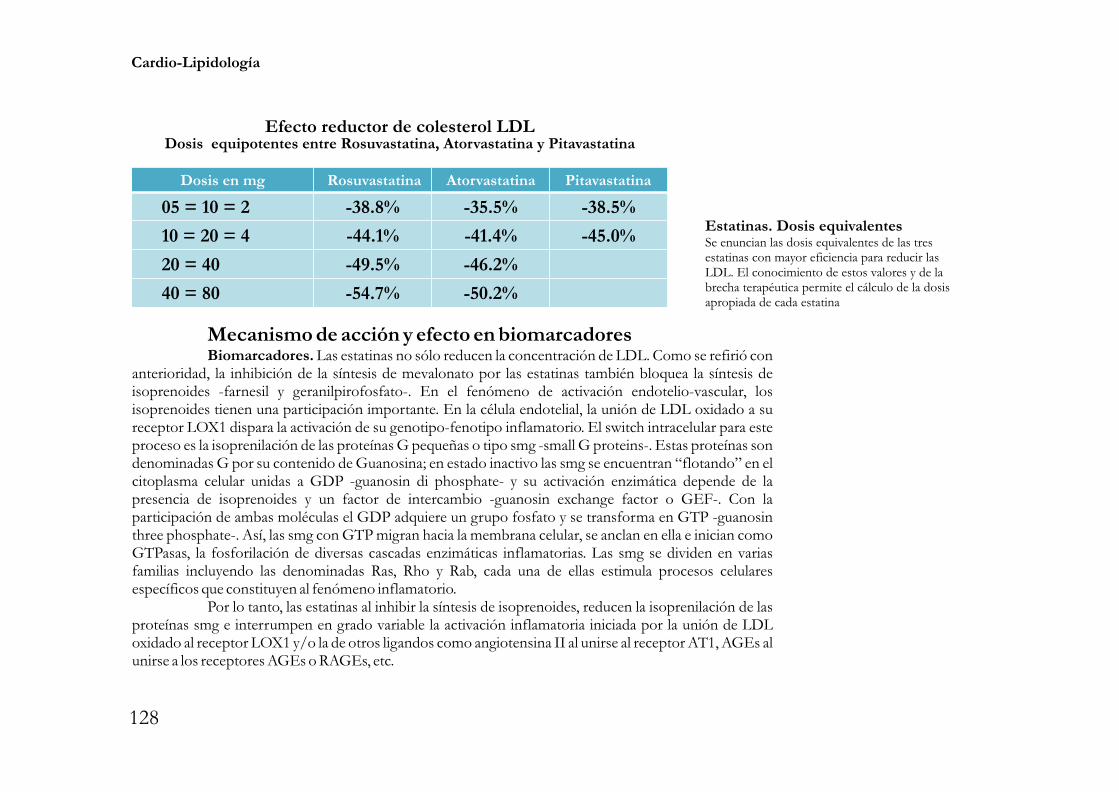
Mecanismo de acción y efecto en biomarcadoresBiomarcadores. Las estatinas no sólo reducen la concentración de LDL. Como se refirió con
anterioridad, la inhibición de la síntesis de mevalonato por las estatinas también bloquea la síntesis deisoprenoides -farnesil y geranilpirofosfato-. En el fenómeno de activación endotelio-vascular, losisoprenoides tienen una participación importante. En la célula endotelial, la unión de LDL oxidado a sureceptor LOX1 dispara la activación de su genotipo-fenotipo inflamatorio. El switch intracelular para esteproceso es la isoprenilación de las proteínas G pequeñas o tipo smg -small G proteins-. Estas proteínas sondenominadas G por su contenido de Guanosina; en estado inactivo las smg se encuentran “flotando” en elcitoplasma celular unidas a GDP -guanosin di phosphate- y su activación enzimática depende de lapresencia de isoprenoides y un factor de intercambio -guanosin exchange factor o GEF-. Con laparticipación de ambas moléculas el GDP adquiere un grupo fosfato y se transforma en GTP -guanosinthree phosphate-. Así, las smg con GTP migran hacia la membrana celular, se anclan en ella e inician comoGTPasas, la fosforilación de diversas cascadas enzimáticas inflamatorias. Las smg se dividen en variasfamilias incluyendo las denominadas Ras, Rho y Rab, cada una de ellas estimula procesos celularesespecíficos que constituyen al fenómeno inflamatorio.
Por lo tanto, las estatinas al inhibir la síntesis de isoprenoides, reducen la isoprenilación de lasproteínas smg e interrumpen en grado variable la activación inflamatoria iniciada por la unión de LDLoxidado al receptor LOX1 y/o la de otros ligandos como angiotensina II al unirse al receptor AT1, AGEs alunirse a los receptores AGEs o RAGEs, etc.
Efecto reductor de colesterol LDLDosis equipotentes entre Rosuvastatina, Atorvastatina y Pitavastatina
Dosis en mg Rosuvastatina Atorvastatina Pitavastatina
05 = 10 = 2 -38.8% -35.5% -38.5%
10 = 20 = 4 -44.1% -41.4% -45.0%
20 = 40 -49.5% -46.2%
40 = 80 -54.7% -50.2%
Dosis en mg Rosuvastatina Atorvastatina Pitavastatina
05 = 10 = 2 -38.8% -35.5% -38.5%
10 = 20 = 4 -44.1% -41.4% -45.0%
20 = 40 -49.5% -46.2%
40 = 80 -54.7% -50.2%
128
Cardio-Lipidología
Estatinas. Dosis equivalentesSe enuncian las dosis equivalentes de las tresestatinas con mayor eficiencia para reducir lasLDL. El conocimiento de estos valores y de labrecha terapéutica permite el cálculo de la dosisapropiada de cada estatina

129
Estatinas, Fibratos, Niacina y más
Estatinas. Mecanismo de acciónSe esquematiza la cascada enzimática inflamatoria
de la célula endotelial, la cual es similar a la activadaen otras estirpes celulares inflamatorias. Especial
importancia tiene la isoprenilación de las proteínassmg o proteínas G pequeñas. Al igual que la
estatinas inhiben la síntesis de colesterol, inhiben lasíntesis de isoprenoides
Estatinas. Mecanismo de acciónSe esquematiza como la estatina al inhibir la síntesis
de isoprenoides, inhibe la isoprenilación de lasproteínas smg y con ello bloquea en grado variable
la activación de las cascadas enzimáticasinflamatorias. El resultado final es la reducción en
los denominados biomarcadores de inflamación
Célula endotelial
IsoprenoidesHMG-CoA-R
HMGCoA
Mevalonato
E
Célula endotelial
Isoprenilación
IsoprenoidesHMG-CoA-R E
HMGCoA
Mevalonato
MCP1, MCSFICAM, VCAM, PCR, etc
Célula endotelial
Isoprenilación
MCP1, MCSFICAM, VCAM, PCR, etc
Lox
K
smg
LDL
NFkB
Célula endotelial
Isoprenilación
IsoprenoidesHMG-CoA-R E
HMGCoA
Mevalonato
Lox
LDL
Lox
smg
LDL
K
NFkB
3
2
4
1

En este mismo escenario el grupo de Jain demostró in-vitro que las estatinas favorecen en lacélula endotelial la expresión del factor de transcripción KLF2 y con ello la síntesis de eNOS y la actividadde la cascada enzimática PI3Kinasa-Akt2-eNOS. De esta forma, las estatinas incrementan la síntesisendotelial de óxido nítrico y su acción como molécula que impide la proteolisis del IkB, favoreciendo el“secuestro” intracitoplasmático del factor de transcripción proinflamatorio NFkB.
Casey PJ. Zhang FL, CaseyProtein lipidation in cell signaling. Science 1995; 268:221-225.PJ. Protein prenylation: molecular mechanism and functional consequences. Annu Rev Biochem 1996;65:241-249. Isoprenoids as mediators of the biological effects of statins. J Clin Invest 2002;Liao JK.110:285-288. Dichtl W, Dulak J, Frick H, Schwarzacher SP, Ares MPS, Nilsson J, Pachinger O,Weidinger F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in humanendothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2003; 23:58-66. McFarlaneSI, Muniyappa R, Francisco R, Sowers JR. Pleiotropic effects of statins: Lipid reduction and beyond. JClin Endocrinol Metab 2002; 87:1451-1458. Pleiotropic effects of statins. Annu RevLiao JK, Laufs U.Pharmacol Toxicol 2005; 45:89-118. Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, BanerjeeP, Kumar A, Jain MK. Kruppel-Like Factor 2 as a novel mediator of statin effects in endothelial cell.Circulation 2005; 112:720-726. 1a. Edición 2005.Atlas de Estatinas, Aterotrombosis y Pleiotropismo.Autor y Editor Morales Enrique. Editorial Atheros-CIC.
Estos mecanismos explican por qué el efecto antinflamatorio de las estatinas va más allá de lareducción de LDL y/o LDL oxidado, su acción antinflamatoria está en función de su potencial para inhibirprocesos fundamentales de la activación celular inflamatoria. En el animal de experimentación y en elindividuo con aterosclerosis, se han publicado múltiples evidencias que demuestran que las estatinasatenúan el proceso inflamatorio endotelio-vascular propio de la aterosclerosis en forma independiente,aunque sinérgica con la reducción de LDL. Las evidencias van desde la multi-referida reducción debiomarcadores inflamatorios como la PCR, hasta la demostración experimental y en el ser humano de laatenuación del proceso inflamatorio en placas aterosclerosas. En el animal de experimentación, en elmodelo animal de aterosclerosis aortocoronaria precoz en conejos, las estatinas al compararse en formaciega con placebo, logran disminuir en forma aguda la infiltración monocitaria en zonas de aterosclerosisaortocoronaria; este fenómeno experimental equivale a la reducción aguda de la señal de actividadinflamatoria intraplaca en aortas de individuos con Aterosclerosis tratados con estatinas. Rudd empleó
130
Cardio-Lipidología

como marcador de actividad inflamatoria a la 18 fluoro deoxiglucosa y como método de imagen latomografía por emisión de positrones y publicó que la administración aguda de estatinas reducesignificativamente la señal del trazador de inflamación en las placas aterosclerosas; esta atenuación en laactividad inflamatoria sucede en días y no se explica por modificaciones significativas en la concentraciónde LDL.
Kircher MF, Grimm J, Swirski FK, Libby P, Gerszten RE, Allport JR et al. Noninvasive invivo imaging of monocyte trafficking to atherosclerotic lesions. Circulation 2008; 117:388-395. RuddJHF, Hyafil F, Fayad ZA. Inflammation imaging in atherosclerosis. Arterioscler Thromb Vasc Biol 2009;29:1009-1016.
Mecanismo de acción y efecto en citoprotecciónLas estatinas han demostrado en animales de experimentación un efecto cardioprotector,
específicamente miocitoprotector, el cual se sustenta en mecanismos que van más allá de su acciónreductora de LDL y antinflamatoria. Este efecto se relaciona con la activación de dos sistemas enzimáticosde protección celular, la denominada RISK pathway “reperfusion ischemic salvage kinases pathway” y laAMPK “AMP activated kinase”.
131
Estatinas, Fibratos, Niacina y más
PET -18-FDG-/CT pre y post estatina
18-FDG positivobasal
18-FDG negativo24 hr post-estatina18-FDG negativo
24 hr post-estatinaEstatinas. Inhibición de inflamaciónEsta imagen de PET con 18-FDG en aorta de unser humano con aterosclerosis muestra claramentela desaparición de la señal de inflamación -medialuna amarilla en aorta- 24 horas después de laadministración de una carga de estatina -efectoLDL-independiente-

RISK pathway. La producción de óxido nítrico por activación de la cascada enzimáticaPI3Kinasa-Akt2-eNOS es un proceso fisiológico exaltado en los tejidos sometidos a un incremento enla demanda de O2 -preacondicionamiento isquémico-. Existe evidencia sobre la activación indirecta dedicha cascada enzimática tras la administración aguda de estatinas. Bell y Yellon publicaron en el 2003 queen ratas de experimentación sometidas a isquemia-reperfusión miocárdicas, el cociente del tejidomiocárdico necrótico/isquémico post-reperfusión se reduce con la administración pre-isquemia de unacarga de atorvastatina. Sin embargo dicho efecto cardioprotector se atenúa significativamente cuando laestatina se administra en forma crónica pre-isquemia. Esta reducción del efecto cardioprotector secorrelacionó con incremento significativo en la síntesis y expresión de PTEN “phosphatase tensin”,fosfatasa con efecto antagónico a PI3k. El hallazgo más importante de esta serie de experimentos fue lademostración de que la recarga con estatina logró reducir significativamente el cociente de tejidonecrótico/isquémico, hallazgo asociado a represión de PTEN y re-expresión de PI3K con incremento en laproducción de óxido nítrico. Como se verá más adelante, estas evidencias dieron racional y nombre alestudio ARMYDA-RECAPTURE.
Bell RM, Yellon DM. Atorvastatin administered at the onset of reperfusion and independentof lipid lowering protects the myocardium by up-regulating a pro-survival pathway. J Am Coll Cardiol 2003;41:508-515. . Failure to protect the myocardium againstMensah K, Mocanu MM, Yellon DMischemia/reperfusion injury after chronic atorvastatin treatment is recaptured by acute atorvastatintreatment: a potential role for phosphatase and tensin homolog deleted onchromosome ten? J Am CollCardiol 2005; 45:1287-1291. . Statins andLudman A, Venugopal V, Yellon DM, Hausenloy DJcardioprotection. More than just lipid lowering. Pharmacology and Therapeutics 2009; 122:30-43. EllisSG, Anwaruddin S. Recapturing the magic. Revisiting the pleiotropic effects of statins in percutaneouscoronary revascularization. Editorial Comment. J Am Coll Cardiol 2009; 54:558-560. Hauseloy DJ,Yellon DM. Cell membrane repair as a mechanism for ischemic preconditioning?. Editorial. Circulation2010; 121:2547-2549.
AMPK o kinasa activada por AMP. Esta enzima es considerada el switch energético de lacélula isquémica. En la célula isquémica existe una reducción en el cociente ATP/AMP con incremento enla concentración citoplasmática de AMP. Este sustrato es la señal que activa a la subunidad gamma de laAMPK y con ello esta kinasa inicia la fosforilación de los transportadores de glucosa -GLUT4- y de ácidos
132
Cardio-Lipidología

grasos -CD36- así como de las enzimas clave de las vías de la glucolisis y la betaoxidación. Por estemecanismo, en la célula isquémica, la activación fisiológica de la AMPK incrementa el ingreso y lautilización de glucosa y ácidos grasos libres para la producción de ATP.
Existe evidencia de que moléculas como la adiponectina, la metformina y las estatinas sonactivadores indirectos de la AMPK, ejerciendo con ello un efecto cardioprotector, especialmente enescenarios de isquemia miocárdica.
Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra.Basic Science for Clinicians. Circulation 2008; 117:832-840.
Resumiendo, la RISK pathway y la AMPK son activadas in-vitro por las estatinas en una formaindependiente a su efecto reductor de colesterol e isoprenoides y propician la miocito-protección,especialmente ante la isquemia post-reperfusión. La RISK pathway incrementa la producción de óxidonítrico e induce vasodilatación en los tejidos isquémicos, la AMPK al activar transportadores de glucosa y
E
133
Estatinas, Fibratos, Niacina y más
Estatinas. CitoprotecciónSe esquematiza la activación indirecta por la
estatina de la cascada enzimática PI3K-eNOS-ON en la célula endotelial. Por acción
vasodilatadora del ON se incrementa el aportede sustratos energéticos al miocito y por
activación de AMPK, el miocito ve favorecida laincorporación y utilización de glucosa y ácidos
grasos para la formación de ATP
Cardiomiocito
AMPKinasa
AMPK
Glucosa
GLUT4
BetaoxidaciónGlucolisis
Ácidos grasos
CD36
Célula endotelial
RISK pathway
PI3k-Akt2
eNOS
EE
ON ATP

ácidos grasos así como enzimas claves de la glucolisis y betaoxidación, incrementa el ingreso y la utilizacióncelular de glucosa y ácidos grasos, con lo cual se optimiza la eficiencia energética de la célula isquémica.
Mecanismo de acción y efecto en ateroregresiónEl fenómeno de la ateroregresión es un proceso factible, no solamente en el animal de
experimentación, también en el ser humano con aterosclerosis coronaria -ver capítulo 3-. Este hechodepende del grado de reducción de LDL, pero también del grado de reducción de la inflamación endotelio-vascular.
Como evidencias representativas de este concepto basta revisar las gráficas con los resultadosde los estudios REVERSAL, ASTEROID, ERASE, JAPAN-ACS y SATURN. En estos estudios clínicos sedemostró que la ateroregresión coronaria estudiada por ultrasonido intra-arterial, está directa ysinérgicamente relacionada con la reducción de LDL e inflamación endotelio-vascular; a mayor reducciónde ambos parámetros, mayor ateroregresión. Los índices más altos de ateroregresión y la rapidez de lamisma, se mueven en función del tiempo de inicio, dosis, lapso de administración y estatina empleada.Hasta ahora atorvastatina 80mg, rosuvastatina 40mg y pitavastatina 4mg son las estatinas con evidenciapara sostener el concepto y la indicación de ateroregresión.
Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA et al.REVERSAL Investigators. Effect of intensive compared with moderate lipid-lowering therapy onprogression of coronary atherosclerosis. A randomized controlled trial. JAMA 2004; 291:1071-1080.Nissen SE, Tuscu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J et al. REVERSALInvestigators. Statin therapy, LDL cholesterol, C-Reactive Protein and coronary artery disease. N Engl JMed 2005; 352:29-38. Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM et al.ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronaryatherosclerosis: the ASTEROID trial. J Am Med Assoc 2006; 295:1556-1565. Rodés-Cabau J, Tardif JC,Cossette M, Bertrand OF, Ibrahim R, Larose E et al. ERASE trial.Acute effects of statin therapy oncoronary atherosclerosis following acute coronary syndromes. Am J Cardiol 2009; 104:750-757. Hibi K,Kimura K, Morimoto T, Hiro T, Miyauchi K et al. JAPAN-ACS trial. Clinically evident polyvasculardisease and regression of coronary atherosclerosis after intensive statin therapy in patients with ACS: Serialintravascular ultrasound from the Japanese assessment of pitavastatin and atorvastatin in JAPAN-ACStrial. Atherosclerosis 2011 .www.elsevier.com/locate/atherosclerosis Nicholls SJ, Ballantyne CM,
134
Cardio-Lipidología

Barter PJ, Chapman J, Erbel RM, Libby P et al. SATURN trial. Effect of two intensive statinregimens on progression of coronary disease. N Engl J Med 2011 www.nejm.org.
Efecto en reducción de eventos clínicos.Prevención primaria. La reducción de colesterol y de biomarcadores, la citoprotección
miocárdica e incluso la ateroregresión serían hechos irrelevantes en ausencia de reducción de eventoscardiovasculares. En individuos con diversos niveles de riesgo cardiovascular -en su mayoría intermedio yalto-, sin antecedente de enfermedad cardiovascular, la historia de las estatinas ha sido documentada por losmeta-análisis 2005 y 2010 del grupo colaborativo en colesterol. En esta arena clínica los tres estudiosclínicos de mayor relevancia son los estudios ASCOT-LLA, CARDS y JUPITER. El estudio ASCOT-LLAdemostró que en población con hipertensión arterial y al menos otro factor de riesgo cardiovascular,atorvastatina a dosis de 10mg reduce el riesgo de un primer evento cardiovascular en 36% con una
LDL Progresión vs Regresión
135
Estatinas, Fibratos, Niacina y más
Estatinas. AteroregresiónEsta gráfica resume los estudios con USG
intracoronario enfocados al estudio de laateroregresión coronaria. El último estudio de lasaga, SATURNO corroboró la factibilidad de la
ateroregresión con la utilización de rosuvastatina40mg o atorvastatina 80mg, sin existir diferencias
entre ambas estatinas. Lograr un LDL <70mg/dl esel punto donde la ateroregresión es factible
120100806040
2
1
0
-1
-2
PravastatinaREVERSAL
PlaceboCAMELOT
PlaceboSTRADIVARIUS
PlaceboA-plus
Atorvastatina-PlaceboILLUSTRATE
AtorvastatinaREVERSAL
Atorvastatina 80mgSATURNO
Rosuvastatina 40mgSATURNO
RosuvastatinaASTEROID
LDL promedio mg/dl
%ca
mb
iod
evo
lum
end
eat
erom
a
p ns
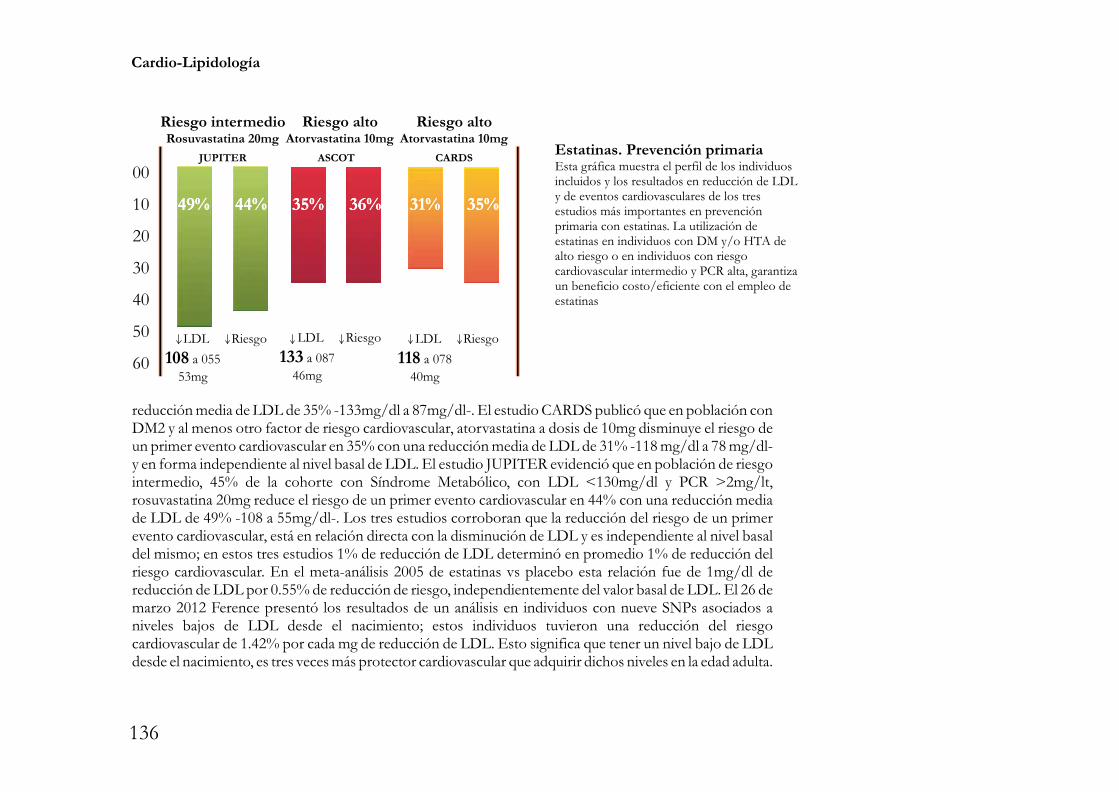
reducción media de LDL de 35% -133mg/dl a 87mg/dl-. El estudio CARDS publicó que en población conDM2 y al menos otro factor de riesgo cardiovascular, atorvastatina a dosis de 10mg disminuye el riesgo deun primer evento cardiovascular en 35% con una reducción media de LDL de 31% -118 mg/dl a 78 mg/dl-y en forma independiente al nivel basal de LDL. El estudio JUPITER evidenció que en población de riesgointermedio, 45% de la cohorte con Síndrome Metabólico, con LDL <130mg/dl y PCR >2mg/lt,rosuvastatina 20mg reduce el riesgo de un primer evento cardiovascular en 44% con una reducción mediade LDL de 49% -108 a 55mg/dl-. Los tres estudios corroboran que la reducción del riesgo de un primerevento cardiovascular, está en relación directa con la disminución de LDL y es independiente al nivel basaldel mismo; en estos tres estudios 1% de reducción de LDL determinó en promedio 1% de reducción delriesgo cardiovascular. En el meta-análisis 2005 de estatinas vs placebo esta relación fue de 1mg/dl dereducción de LDL por 0.55% de reducción de riesgo, independientemente del valor basal de LDL. El 26 demarzo 2012 Ference presentó los resultados de un análisis en individuos con nueve SNPs asociados aniveles bajos de LDL desde el nacimiento; estos individuos tuvieron una reducción del riesgocardiovascular de 1.42% por cada mg de reducción de LDL. Esto significa que tener un nivel bajo de LDLdesde el nacimiento, es tres veces más protector cardiovascular que adquirir dichos niveles en la edad adulta.
136
Cardio-Lipidología
Estatinas. Prevención primariaEsta gráfica muestra el perfil de los individuosincluidos y los resultados en reducción de LDLy de eventos cardiovasculares de los tresestudios más importantes en prevenciónprimaria con estatinas. La utilización deestatinas en individuos con DM y/o HTA dealto riesgo o en individuos con riesgocardiovascular intermedio y PCR alta, garantizaun beneficio costo/eficiente con el empleo deestatinas
Riesgo intermedioRosuvastatina 20mg
Riesgo altoAtorvastatina 10mg
Riesgo altoAtorvastatina 10mg
00
10
20
30
40
50
60
44%44%49%49%
LDL
108 a 055
53mg
Riesgo
31%31%
LDL
118 a 078
40mg
Riesgo
36%36%35%35%
LDL
133 a 087
46mg
Riesgo
JUPITER ASCOT CARDS
35%35%

Este trabajo abre nuevas interrogantes sobre las tácticas y estrategias idoneas para alcanzar una reducciónmáxima del riesgo cardiovascular.
Cholesterol Treatment Trialist -CTT- Collaboration. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomized trials ofstatins. Lancet 2005; 366:1267-78. Sever PS, Dahlof B, Poulter NR on behalf ASCOT-LLAInvestigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients whohave average or lower-than average cholesterol concentration, in the Anglo-Scandinavian CardiacOutcomes Trial-Lipid Lowering Arm -ASCOT-LLA-. A multicentre randomized controlled trial. Lancet2003; 361:1149-1158. Colhoun HM, Betteridge DJ, Durrington PN on behalf CARDSInvestigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in theCollaborative Atorvastatin Diabetes Study -CARDS-. Multicentre randomized placebo-controlled trial.Lancet 2004; 364:665-696. Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, KasteleinJJP et al. JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women withelevated C-Reactive Protein. N Engl J Med 2008; 359:2195-2207. Cholesterol Treatment Trialist -CTT-Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of datafrom 170,000 participants in 26 randomized trials. Lancet 2010; 376:1670-1681. StartFerence B.Lowering LDL in Childhood. American College of Cardiology 61st Annual Scientific Sesion.
Al igual que lo comentado para la ateroregresión, en prevención primaria la reducción dual deLDL y PCR, también se asocia a un mayor beneficio que aquel alcanzado por la reducción aislada de LDL.En este sentido, el subanálisis LDL-PCR del estudio JUPITER reportó una reducción del riesgocardiovascular de 65% en la sub-población que logró una reducción de LDL <70mg/dl + PCR <1mg/ltdurante el tratamiento con estatinas.
Ridker PM, Danielson E, Fonseca FAH, Gotto AM, Kastelein JJP et al. JUPITERStudy Group. Reduction in C-Reactive Protein and LDL cholesterol and cardiovascular event rates afterinitiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet 2009; 373:1175-1182.
Prevención secundaria. La historia de éxito terapéutico de las estatinas, también incluye a losindividuos con historia de un evento cardiovascular. El uso óptimo de estatinas reduce en formasignificativa el riesgo de un evento cardiovascular subsecuente. En este apartado la evidencia abarca lossiguientes escenarios clínicos.
137
Estatinas, Fibratos, Niacina y más

Síndromes coronarios estables. En individuos con antecedente de enfermedadcardiovascular y un curso clínico estable, el beneficio de las estatinas también quedó plasmado en los meta-análisis 2005 y 2010 del grupo colaborativo en colesterol. Los estudios HPS, IDEAL y TNT han sido losmás importantes en este escenario clínico; de ellos, el estudio TNT por ser el más reciente, con seguimientoa largo plazo y con resultados estadísticamente significativos es quizá el más relevante en esta arenaterapéutica. Este estudio comparó atorvastatina 80mg contra atorvastatina 10mg en individuos conenfermedad coronaria estable y demostró que el tratamiento con atorvastatina 80mg vs atorvastatina 10mgreducía significativamente la probabilidad de un evento cardiovascular subsecuente. Este logro terapéuticofue especialmente significativo en el subgrupo de individuos que alcanzó un nivel de LDL <70mg/dldurante el tratamiento con estatinas. En su más reciente reporte de eventos cardiovasculares, el mismogrupo de investigadores TNT comunicó que el beneficio de la dosis de 80mg de atorvastatina se mantiene alargo plazo con reducción de 19% -estadísticamente significativa- en la incidencia de eventoscardiovasculares subsecuentes, sin incremento significativo en la incidencia de eventos adversos. En elmeta-análisis 2010 de estatinas a dosis “bajas” vs estatinas a dosis “altas”, la conclusión fue similar a la del2005, en promedio cada reducción de 1mg/dl de LDL, reduce 0.58% -22% por 38mg/dl- el riesgo de unevento cardiovascular, independientemente del nivel basal de LDL. En otras palabras y basado en unejemplo de la vida real, un individuo con DM2 con LDL de 150mg/dl, al ser tratado por ejemplo conatorvastatina 80mg o rosuvastatina 40mg y reducir a 75mg/dl -50%- su nivel de LDL, logra una reducciónde 43.5% -75mg/dl x 0.58- en su riesgo de un evento cardiovascular, sin incremento en el riesgo de otroseventos adversos serios, especialmente cáncer o hemorragia cerebral.
La Rosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, Gotto AM, GretenH, Kastelein JJ, Sheperd J, Wenger NK. Intensive lipid lowering with atorvastatin in patients with stablecoronary disease. N Engl J Med 2005; 352:1425-1435. Pedersen TR, Faergerman O, Kastelein JJ,Olson AG, Tiskkanen MJ, Holme I et al. IDEAL Study Group. High-dose atorvastatin vs usual-dosesimvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomizedcontrolled trial. JAMA 2005; 294:2437-2445. La Rosa JC, Grundy SM, Kastelein JJ, Kostis JB, GretenH. Treating to New Targets -TNT- Steering Committee and Investigators. Safety and efficacy ofatorvastatin induced very low density lipoprotein cholesterol levels in patients with coronary heart disease -a post hoc analysis of the Treating to New Targets Study-. Am J Cardiol 2007; 100:747-752. La Rosa JC,
138
Cardio-Lipidología

Deedwania PC, Shepherd J, Wagner NK, Greten H, DeMicco DA et al. Comparison of 80mg versus10mg of atorvastatin on occurrence of cardiovascular events after the first event -from the TNT trial-. AmJ Cardiol 2010; 105:283-287.
A la fecha y como se comentó en el capítulo 4, en individuos con antecedente de enfermedadcardiovascular existe cierta discrepancia en relación a las metas de LDL entre las Guías AHA-ACCF dePrevención Secundaria, publicadas en noviembre del año 2011 y las Guías Europeas ESC-EAS publicadasen junio del 2011. En el fondo esta controversia es sólo conceptual y vale la pena analizar este punto conmás detalle.
Las Guías Americanas consideran como individuos de riesgo alto a aquellas personas conantecedente de enfermedad cardiovascular, en ausencia de diabetes mellitus, tabaquismo, síndromemetabólico, especialmente con dislipidemia aterogénica y/o síndrome coronario agudo,en este subgrupo de individuos se propone una meta de LDL <100mg/dl como indicación Clase I connivel de evidencia C. Esta recomendación está sustentada en los estudios HPS, IDEAL, TNT, el meta-análisis 2010 del CTT y las guías ATP-III. Se consideran como individuos de riesgo muy alto a aquellaspersonas con antecedente de enfermedad cardiovascular, en presencia de diabetes mellitus,
139
Estatinas, Fibratos, Niacina y más
Estatinas vs GenéticaIndependientemente del escenario clínico y delvalor basal de LDL, cada 38mg -1mmol- dereducción de LDL con estatinas, equivale a 22% dereducción del riesgo relativo de un eventocardiovascular; en otras palabras cada mg/dl dereducción de LDL, disminuye 0.57% el riesgorelativo de un evento cardiovascular. Ferencedocumentó que este beneficio casi se triplica enindividuos con patrones genéticos determinantes deLDL bajo desde el nacimiento
Meta-análisis Ference B2010-CTT
00
10
20
30
40
50 38mg
22%Riesgo
LDL
54%Riesgo
SNP- LDL

tabaquismo, síndrome metabólico, especialmente con dislipidemia aterogénica y/o síndromecoronario agudo; en este sub-grupo se propone una meta de LDL <70mg/dl o una reducción 50%�
como una indicación Clase IIa con nivel de evidencia B. Esta recomendación está sustentada básicamenteen los estudios TNT, IDEAL, el meta-análisis 2010 del CTT, PROVE-IT, el meta-análisis 2006 de Cannon yel sub-análisis LDL-PCR de PROVE-IT.
Las Guías Europeas consideran como individuos de riesgo muy alto a todo individuo conantecedente de enfermedad cardiovascular, independientemente de la ausencia o presencia deotro factor de riesgo; se incluyen también en este grupo individuos con DM2, DM1 con daño a órgano
blanco, insuficiencia renal moderada-severa y/o un SCORE 10%; en este grupo de individuos se�
propone una meta de LDL <70mg/dl o una reducción 50% como una indicación Clase I con nivel de�
evidencia A. Esta recomendación está sustentada fundamentalmente en los estudios IDEAL, TNT y elmeta-análisis 2010 del CTT.
Dado que los estudios en los que se basan ambas Guías son los mismos, siendo mayor laevidencia conjuntada por las Guías Americanas, la diferencia en metas es conceptual ya que en el fondo lamayor diferencia es el hecho de que las Guías Europeas consideran siempre como de muy alto riesgo a unindividuo con antecedente de enfermedad cardiovascular. La otra diferencia entre ambas Guías es menor yconsiste en el “peso específico” otorgado a las mismas evidencias; para los proponentes de las GuíasAmericanas los estudios referidos sólo son suficientes para dar una indicación de Clase IIa con nivel deevidencia B, en tanto que para los de las Guías Europeas son suficientes para otorgar una indicación Clase I
con nivel de evidencia A, a la meta de LDL <70mg/dl o reducción 50%. Al final del día e�
independientemente de complejas evaluaciones de costo-eficiencia, la gran mayoría de los individuos conantecedente de enfermedad cardiovascular tendrán alguna otra variable de riesgo y será deseable una meta
de LDL <70mg/dl o al menos una reducción 50% en el nivel basal de LDL.�
Para los individuos de riesgo intermedio las Guías Americanas recomiendan una meta de LDL<130mg/dl -potencialmente <100mg/dl en el ATP IV-, en tanto que las Guías Europeas recomiendan unameta de LDL <115mg/dl; nuevamente diferencias limítrofes.
Reiner Z, Catapano AL, De Becker G, Graham I, Taskinen M, Wiklund O, Agewall S etal. The Task Force for the Management of Dyslipidemias of the ESC-EAS. Eur Heart J 2011; 32:1769-
140
Cardio-Lipidología

1818. AHA/ACCFSmith SC, Benjamin EJ, Bonow RO, Braun LT, Creager MA, Franklin BA et al.Secondary Prevention and Risk Reduction Therapy for Patients with Coronary and Other AtheroscleroticVascular Disease: 2011 Update. Circulation 2011; 124: published on line 29 November 2011.
Síndromes coronarios agudos. Al igual que el estudio TNT en síndromes coronariosestables, el estudio PROVE-IT, inspirado por el resultado del estudio MIRACL, es el icono del beneficioterapéutico de las estatinas en síndromes coronarios agudos. El estudio PROVE-IT comparó atorvastatina80mg contra pravastatina 40mg en individuos después de un síndrome coronario agudo y demostró que eltratamiento con atorvastatina 80mg vs pravastatina 40mg reducía significativamente la probabilidad de unevento cardiovascular post-SICA. Los subanálisis del estudio PROVE-IT en función de LDL, PCR yneopterina -molécula soluble de activación monocitaria-, arrojaron datos que modificaron en el año 2007las guías de tratamiento para este tipo de individuos de riesgo muy alto. En PROVE-IT el subgrupo deindividuos que alcanzó en el día 30 después del evento clínico índice un nivel de LDL <70mg/dl + PCR<2mg/lt tuvo la incidencia más baja de infarto miocárdico o muerte cardiovascular post-SICA;2.4 eventos por cada 100 individuos tratados por año, comparada con 4.6 en el subgrupo cuyo LDL
� �70mg/dl + PCR 2mg/lt. Un subgrupo ideal resultó aquel cuyo LDL fue <70mg/dl + PCR <1mg/lt;en este subgrupo, la incidencia de infarto miocárdico o muerte cardiovascular fue de 1.9 eventos por cada100 individuos tratados por año. El análisis de la relación entre el régimen terapéutico y el logro de la metadual de colesterol LDL <70 mg/dl + PCR <2 mg/lt, demostró que en el grupo que alcanzó la doble meta-20% del total-, el subgrupo de atorvastatina 80mg llegó a esta doble meta en 8 de cada 10 casos en tanto quepravastatina 40mg en 2 de cada 10 casos.
Shwartz G, Olson A, Ezekowitz M et al. Atorvastatin effects on recurrent acute coronarysyndromes. J Am Med Assoc 2001; 285:1711-1718. Cannon CP, Braunwald E, McCabe CH, Rader DJ,Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM. Intensive versus moderate lipidlowering with statins after acute coronary syndrome. N Engl J Med 2004; 350:1495-1504. Ridker PM,Cannon CP, Morrow D, Rifai N, Rose LM, Mc Cabe CH et al. PROVE-IT. TIMI 22 Investigators.C-Ractive Protein levels and outcomes after statin therapy. N Engl J Med. 2005; 352:20-28. Ray KK,Morrow DA, Sabatine MS, Shui A, Rifai N, Cannon CP, Braunwald E. Long term prognostic value ofneopterin: a novel marker of monocyte activation in patients with acute coronary syndromes. Circulation2007; 115:3071-3078.
141
Estatinas, Fibratos, Niacina y más

ARMYDA OriginalN 153 naive (76/77)
ACS Non ACS Atorvastatina vs Placebo40mg/7 days pre-PCI
NAPLES I
N 451 naive (226/225)
ACS 8% Non ACS 92% Varias estatinas vs Control
Diferentes dosis >72hr pre-PCI
NAPLES II
N 668 naive (338/330)
ACS 2% Non ACS 98% Atorvastatin
80mg/24hr pre-PCI
ARMYDA-ACS
N 171 naive (85/86)
Non STEMI ACS Atorvastatina vs Placebo
120mg/12hr pre-PCI
ARMYDA-RECAPTURE
N 383 preTx (192/191)
Non STEMI ACS 47% Non ACS 53% Atorvastatin Placebo
120mg/12hr pre-PCI
Korean Non STEMI
N 445 naive (220/225)
Non STEMI ACS Rosuvastatina vs Control
40mg/16hr pre-PCI
Korean STEMI
N 171 naive (86/85)
STEMI ACS Atorvastatin a dosis alta vs baja
Vaselka
N 200 naive (100/100)
Non ACS Atorvastatina vs Control
80mg/48hr pre-PCI
Bozbas
N 93 naive
Non ACS Pravastatina 2 dosis vs Placebo
10mg vs 40mg/7days pre PCI
Konoshita
N 42 naive
Non ACS Atorvastatina a dosis baja vs intermedia
5-20mg/14 días pre PCI
Jia
N 228 naive
STEMI ACS 29%
Non STEMI ACS 71%
Simvastatina a dosis baja vs alta
20mg vs 80mg/7 días pre-PCI
Hara
N 37 naive y pre-Tx
Non STEMI ACS Atorvastatina vs Placebo
20mg/24hr pre-PCI
Cay
N 299 naive
Non ACS Rosuvastatina vs Placebo
40mg/24 hr pre-PCI
ARMYDA OriginalN 153 naive (76/77)
ACS Non ACS Atorvastatina vs Placebo40mg/7 days pre-PCI
NAPLES I
N 451 naive (226/225)
ACS 8% Non ACS 92% Varias estatinas vs Control
Diferentes dosis >72hr pre-PCI
NAPLES II
N 668 naive (338/330)
ACS 2% Non ACS 98% Atorvastatina vs Control
80mg/24hr pre-PCI
ARMYDA-ACS
N 171 naive (85/86)
Non STEMI ACS Atorvastatina vs Placebo
120mg/12hr pre-PCI
ARMYDA-RECAPTURE
N 383 preTx (192/191)
Non STEMI ACS 47% Non ACS 53% Atorvastatina vs Placebo
120mg/12hr pre-PCI
Korean Non STEMI
N 445 naive (220/225)
Non STEMI ACS Rosuvastatina vs Control
40mg/16hr pre-PCI
Korean STEMI
N 171 naive (86/85)
STEMI ACS Atorvastatin a dosis alta vs baja
80mg vs 10mg en PCI primaria
Vaselka
N 200 naive (100/100)
Non ACS Atorvastatina vs Control
80mg/48hr pre-PCI
Bozbas
N 93 naive
Non ACS Pravastatina 2 dosis vs Placebo
10mg vs 40mg/7days pre PCI
Konoshita
N 42 naive
Non ACS Atorvastatina a dosis baja vs intermedia
5-20mg/14 días pre PCI
Jia
N 228 naive
STEMI ACS 29%
Non STEMI ACS 71%
Simvastatina a dosis baja vs alta
20mg vs 80mg/7 días pre-PCI
Hara
N 37 naive y pre-Tx
Non STEMI ACS Atorvastatina vs Placebo
20mg/24hr pre-PCI
Cay
N 299 naive
Non ACS Rosuvastatina vs Placebo
40mg/24 hr pre-PCI
13 Estudios, 3,341 Pacientes con Estatinas en ICP
Estudio Escenario Intervencionista Tratamiento con Estatina
142
Cardio-Lipidología

Si bien diversos análisis post-hoc y meta-análisis sobre el efecto terapéutico de las estatinas enindividuos con SICA han comunicado resultados controversiales, la solidez de los resultados de losestudios controlados MIRACL y PROVE-IT dieron fundamento como una indicación Clase I con un nivelde evidencia A, a la administración precoz de una estatina a dosis óptima con la meta doble de LDL<70mg/dl + PCR <2mg/lt. En su reporte de eventos cardiovasculares subsecuentes, los investigadores dePROVE-IT reportaron el mismo hallazgo ya comentado para TNT, el beneficio de atorvastatina 80mg semantuvo a largo plazo con una reducción estadísticamente significativa en la incidencia de eventoscardiovasculares subsecuentes, sin incremento clínicamente significativo de eventos adversos.
In the AHA Guidelines and Scientific Statements Handbook. Editor Fuster V. Edition2009. Chapter 2; pp 25-45 and 3; pp 46-90. Murphy SA, Cannon CP, Wiviott SD, McCabe CH,Braunwald E. Reduction in recurrent cardiovascular events with intensive-lowering statin therapycompared with moderate-lipid lowering statin therapy after acute coronary syndromes from the PROVE-IT. TIMI 22 trial. J Am Coll Cardiol 2009; 54:2363-2365.
Intervención coronaria percutánea e infarto peri-procedimiento. En el año 2011 en lasGuías AHA-ACCF-SCAI sobre Intervención Coronaria Percutánea -ICP- quedó plasmada como unaindicación Clase IIa con nivel de evidencia A, el uso de estatinas a dosis altas -carga o recarga- antes de unprocedimiento de ICP en individuos vírgenes a estatinas, y IIa-B en individuos en tratamiento con ellas.
Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, Cercek B et al. 2011 ACCF-AHA-SCAI Guideline for Percutaneous Coronary Intervention. Executive Summary. Circulation 2011;124:000-000 November 11 2011.http://circ.ahajournals.org
Esta nueva indicación para el uso de estatinas nació con estudios observacionales como el deChang, maduró con estudios aleatorizados, especialmente con los proyectos Italianos ARMYDA yNAPLES y los estudios Coreanos, y se consolidó con el análisis paciente por paciente de los 3,341individuos incluidos en los trece estudios publicados hasta el año 2010 en esta arena terapéutica, publicadopor Patti y Colaboradores en abril 2011.
Chang SM, Yazbeck N, Lakkis NM. Use of statins prior to percutaneous coronaryintervention reduces myonecrosis and improves clinical outcome. Cathether Cardiovasc Interv 2004;62:193-197. Pasceri V, Patti G, Nusca A, Pristipino C, Richichi G, Di Sciascio G. ARMYDAInvestigators. Randomized trial of atorvastatin for reduction of myocardial damage during coronary
143
Estatinas, Fibratos, Niacina y más

intervention: results from the ARMYDA -Atorvastatin for Reduction of Myocardial Damage duringAngioplasty- study. Circulation 2004; 110:674-678. Patti G, Chello M, Pasceri V, Colonna D, Nusca A,Miglinico M et al. Protection from procedural myocardial injury by atorvastatin is associated with lowerlevels of adhesion molecules after percutaneous coronary intervention. Results from the ARMYDA-CAMs -Atorvastatin for Reduction of Myocardial Damage during Angioplasty-Cell Adhesion Molecules-.J Am Coll Cardiol 2004; 48:1560-1566. Statin administration beforeBriguori C, Colombo A, Airoldi F.percutaneous coronary intervention: impact on periprocedural myocardial infarction. Eur Heart J 2004;25:1822-1828. NovelBriguori C, Visconti G, Focaccio A, Golia B, Chieffo A, Castelli A et al.Approaches for Preventing or Limiting Events -Naples II trial-. Impact of a single high loading dose ofatorvastatin on periprocedural myocardial infarction. J Am Coll Cardiol 2009; 54:00-00. Patti G, PasceriV, Colonna G, Miglionico M, Fischetti D, Sardella G et al. Atorvastatin pretreatment improvesoutcomes in patients with acute coronary syndromes undergoing early percutaneous coronaryintervention. Results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol 2007; 49:1272-1278. DiSciascio G, Patti G, Pasceri V, Gaspardone A, Colonna G, Montinaro A. Efficacy of atorvastatinreload in patients on chronic statin therapy undergoing percutaneous coronary intervention. Results of theARMYDA-RECAPTURE randomized trial. J Am Coll Cardiol 2009; 54:558-565. Yun KH, Jeong MH,Oh SK, Rhee SJ, Park EM, Lee EM et al. The beneficial effect of high loading dose of rosuvastatinbefore percutaneous coronary intervention in patients with acute coronary syndromes. Int J Cardiol 2009;137:246-251. Efficacy of high-dose atorvastatinKim JS, Kim J, Choi D, Lee CJ, Lee SH, Ko YG et al.loading before primary percutaneous coronary intervention in ST-segment elevation myocardialinfarction. J Am Coll Cardiol Intv 2010; 3:332-339. Patti G, Cannon CP, Murphy SA, Mega S, Pasceri V,Briguori C et al. Clinical benefit of statin pretreatment in patients undergoing percutaneous coronaryintervention. A collaborative patient-level meta-analysis of 13 randomized studies. Circulation 2011;123:1622-1632.
El grupo italiano encabezado por Germano DiSciascio inició con su primer estudioARMYDA en el año 2004 -primer autor Pasceri- la saga de 13 estudios clínicos que hoy sustentan elconcepto de preacondicionamiento miocárdico por estatinas ante la isquemia advertida. Es importantedestacar que el infarto miocárdico peri-procedimiento es frecuente, su incidencia se ha reportado tan altacomo 70% -media 35%-, en su etiopatogenia participan entre otros factores el estado endotelio-vascular
144
Cardio-Lipidología

del territorio comprometido, la microembolización espontánea e inducida por el trinomio catéter-balón-Stent con compromiso de la microcirculación distal, complicaciones mecánicas trans-intervención y lacapacidad del miocardio para responder ante la isquemia trans y post-reperfusión. El infarto miocárdicoperi-procedimiento, aun con elevación aislada de CPK-MB o troponinas, se asocia a deterioro en elpronóstico. La morbimortalidad reportada a mediano y largo plazo tiene una asociación directamenteproporcional con la magnitud de la elevación de los marcadores de daño miocárdico peri-intervencióncoronaria percutánea. A partir del 2007 el criterio diagnóstico de infarto miocárdico asociado aintervención coronaria percutánea o tipo 4A, es la elevación mayor a 3 veces sobre el valor máximo normalde CPK-MB y/o troponinas -antes del 2007 el criterio era la elevación mayor a 2 veces-.
Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHFTask Force for the Redifinition of Myocardial Infarction. Universal definition of myocardialinfarction. Eur Heart J 2007; 28:2535-2538. Myocardial Infarction due toPrasad A, Herrmann J.Percutaneous Coronary Intervention. Review Article. New Engl J Med 2011; 364:453-464.
En el meta-análisis referido se incluyeron 3,341 individuos de los trece estudios publicados
05
10
15
7.07.0
11.911.9
IMP CPK-MB >3
20
p 0.00001-RR 44%
NNT 20
7.47.4
12.612.6
MACE-30
05
10
15
20
p 0.00001-RR 44%
NNT 19
EstatinaEstatina PlaceboPlacebo
145
Estatinas, Fibratos, Niacina y más
Estatinas. Reducción de IMPEn el meta-análisis de Patti y colaboradores, la
conclusión es sencilla. El empleo de estatinas antesde una ICP en forma de carga o recarga evita un
infarto miocárdico peri-procedimiento 1 de cada 20ocasiones y evita una muerte, revascularización no
programada y/o IMP 1 de cada 19 ocasiones
Meta-Análisis. Estatinas e IMP-MACES

sobre el efecto de las estatinas administradas antes de una ICP en cualquier tipo de escenario clínico; lasconclusiones del mismo fueron las siguientes: la delta de PCR -pre y post-ICP-, la incidencia de mionecrosispost-ICP, la incidencia de infarto miocárdico asociado a la ICP y los eventos de revascularización y muertecardiovascular fueron significativamente menores en el grupo de individuos tratados con estatinascomparados con el grupo de individuos control o tratados con placebo. El beneficio de las estatinas fuemayor en individuos con PCR elevada pre-ICP así como en individuos con síndromes coronariosinestables. El NNT para evitar un infarto miocárdico peri-ICP es de 20 y de 19 el NNT para evitar unevento cardiovascular mayor peri-ICP. Sin lugar a duda el beneficio de la administración de una carga orecarga de estatinas en esta arena clínica de riesgo muy alto, está demostrado y como fue referido, estáreflejado en las Guías 2011 AHA-ACCF-SCAI de ICP con una indicación Clase IIa con nivel de evidenciaB en individuos vírgenes a estatinas y IIa-B en individuos en tratamiento con estatinas, siendo la carga o larecarga con atorvastatina 120mg la dosis más estudiada.
Morales-Villegas E, Di Sciascio G, Briguori C. Statins: Cardiovascular risk reduction inPercutaneous Coronary Intervention-basic and clinical evidence of hyperacute use of statins-. ReviewArticle. Int J of Hypertension Volume 2011, article ID 904742.
Enfermedad vascular cerebral. El estudio SPARCL -Stroke Prevention by AggressiveReduction in Cholesterol Levels- publicado en el 2006, demostró que en individuos sin enfermedadcoronaria clínica, con enfermedad vascular cerebral o isquemia cerebral transitoria en los 6 meses previos asu inclusión al estudio y LDL entre 100mg/dl y 190 mg/dl -medio 137mg/dl-, la administración deatorvastatina 80mg al día comparada con placebo, redujo significativamente la incidencia subsecuente deeventos cerebrovasculares fatales y no fatales así como de eventos cardiovasculares totales yrevascularización arterial coronaria. La incidencia de eventos cerebrovasculares fatales y no fatales fue de11.2% en el brazo atorvastatina contra 13.1% en el brazo placebo, lo que equivalió a una reducción relativade 16%, reducción absoluta de 2.2% y p de 0.03. Los eventos cerebrovasculares de origen isquémico -infarto cerebral- mostraron una reducción especialmente significativa del riesgo relativo de 22%. Loseventos cardiovasculares totales -incluyendo eventos coronarios y revascularización arterial coronaria-tuvieron una reducción absoluta de 3.5%. El NNT fue de 46 para evitar en 5 años un eventocerebrovascular y 29 y 32 para evitar en el mismo lapso de tiempo un evento cardiovascular y unarevascularización arterial coronaria respectivamente. No existió diferencia significativa en mortalidad.
146
Cardio-Lipidología

El nivel de LDL durante el estudio en el grupo tratado con atorvastatina fue de 73mg/dl contra129mg/dl en el grupo placebo. Esta reducción de colesterol LDL mostró una correlación significativa conla reducción de eventos cerebrovasculares de origen isquémico, eventos coronarios y procedimientos derevascularización arterial coronaria. La asociación entre eventos cerebrovasculares hemorrágicos yestatinas no fue concluyente, sin embargo se recomienda precaución en el uso de estos fármacos enindividuos con historia y/o un evento cerebrovascular hemorrágico. No existieron diferencias clínicamentesignificativas en los niveles de enzimas hepáticas y musculares, así como tampoco en eventos adversosmusculoesqueléticos. El meta-análisis 2010 del CTT y el meta-análisis colaborativo publicado porHackman en 2011, corroboraron los resultados del estudio SPARCL con una reducción promedio de 16%en el riesgo de enfermedad vascular cerebral, estadísticamente significativa a favor de los subgrupos deindividuos tratados con dosis “altas” de estatinas, sin incremento en el riesgo de hemorragia cerebral.
Stroke Prevention by Aggressive Reduction in Cholesterol Levels -SPARCLInvestigators-. High dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 2006; 355:549-559. Cholesterol Treatment Trialist -CTT- Collaboration. Efficacy and safety of more intensivelowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomized trials.Lancet 2010; 376:1670-1681. Hackman DG, Woorward M, Newby K, Bath DL, Shao M, Smith EE etal. Statins and Intracerebral Hemorrhage. Collaborative Systematic Review and Meta-Analysis. Circulation2011; 124: 2233-2242.
Artritis reumatoide. Siendo la AR una enfermedad crónica, incapacitante, autoimmune y conun riesgo cardiovascular incrementado, el empleo de estatinas con el objetivo de reducir la actividadinflamatoria de la enfermedad ha mostrado resultados controversiales. Sin embargo, recientemente fuepublicado el primer estudio observacional sobre el efecto de estatinas en prevención cardiovascularprimaria y secundaria en individuos con AR. La exposición a estatinas en individuos con AR sinenfermedad cardiovascular, redujo la cifra de colesterol total en 16% y el riesgo cardiovascular en 55%; esteefecto no se observó en individuos con AR y enfermedad cardiovascular. Estos hallazgos abren unapotencial nueva indicación al uso de estatinas en enfermedades inflamatorias.
Sheng X, Murphy MJ, MacDonald TM, Wei L. Effectiveness of statins on total cholesteroland cardiovascular disease and all-cause mortality in osteoarthritis and rheumatoid arthritis. J Rheumatol2012 39:32-40.
147
Estatinas, Fibratos, Niacina y más

148
Cardio-Lipidología
Efectos adversosLas estatinas como todo grupo farmacológico tienen efectos adversos.En relación a los más importantes, en este capítulo se han referido las evidencias que descartan
el incremento en el riesgo de cáncer y de enfermedad vascular cerebral hemorrágica.
En cuanto al incremento de enzimas hepáticas 3 ULN, están bien identificados los factores�
que incrementan el riesgo de dicho evento adverso: dosis altas especialmente simvastatina 80mg, bajo peso,sexo femenino, insuficiencia hepática, insuficiencia renal, hipotiroidismo e interacción farmacológica. Labaja incidencia de dicho evento adverso -1.4% vs 1.1% al comparar con placebo y 1% a 3% al comparardosis “bajas vs dosis “altas”-, su transitoriedad y la infima asociación a insuficiencia hepática grave -1 en 1millón personas-año de tratamiento-, cifra similar a la incidencia de insuficiencia hepática grave en lapoblación general, han motivado la reciente recomendación de la FDA que sugiere realizar determinaciónde enzimas hepáticas antes de iniciar tratamiento con estatinas, sin necesidad de un monitoreo rutinarioposterior, a menos que surjan signos y/o síntomas de lesión hepática.
La miopatía o elevación de CPK 10ULN asociada a síntomas tiene una incidencia de 5 por�
100,000 personas-año de tratamiento y la rabdomiolisis de 1.6 por 100,000 personas-año tratamiento, esteevento adverso está asociado a ciertos polimorfismos -SLCO1B1- que determinan subexpresión de lostransportadores hepáticos de estatinas o OATP1B1.
En resumen, el daño hepático y el daño muscular asociados a estatinas son muy pocofrecuentes, son previsibles y generalmente no se asocian a desenlaces fatales.
McKenney JM, Ganz P, Wiggins BS, Saseen JS. Statins. In Clinical Lipidology. Chapter22. Editor Christie M Ballantine. Editorial Saunders Elsevier. Edition 2009.
Incremento de glucosa. Sin lugar a duda, en la actualidad, este es el principal “talón deAquiles” de las estatinas. Con el antecedente de estudios como HPS, ASCOT-LLA y JUPITER quemostraron una tendencia a mayor incidencia de DM2 de novo en los grupos tratados con estatinas, el meta-análisis más balanceado en este campo, es el realizado por Naveed Sattar, publicado en el 2010. Este meta-análisis mostró un incremento de 9% en el riesgo relativo de Diabetes Mellitus de novo en los grupos deestudio tratados con estatinas vs los grupos placebo con un NNH de 255 y un NNT de 5, lo anterior implicaque se requiere tratar a 255 individuos para inducir un caso de Diabetes Mellitus de novo contra 5

individuos para evitar un evento cardiovascular mayor. Una relación 50 a 1 entre el beneficio cardiovasculary el riesgo metabólico. Kausik Ray en su meta-análisis de estudios de dosis “altas” vs dosis “bajas” deestatinas, informó que el riesgo relativo de Diabetes Mellitus de novo es dosis dependiente, sin embargoesta conclusión no fue confirmada por el meta-análisis de David Waters de los estudios TNT, IDEAL ySPARCL. Más recientemente Annie Culver, informó un desproporcionado incremento de 48% en el riesgorelativo de DM2 de novo en mujeres post-menopausicas tratadas con estatinas vs las no tratadas con dichosfármacos. Este estudio ha sido “de-construido” y se considera un estudio con múltiples sesgos intrínsecos asu diseño observacional, en una población muy selectiva -mujeres de 50 a 75 años de edad- entre las cualessólo un 7% recibió tratamiento con estatinas -probable sesgo de selección-.
Partiendo del meta-análisis de Sattar, así como de otras evidencias acerca del “potencialdiabetogénico” de las estatinas, vale la pena hacer las siguientes consideraciones.
1.- La reducción del riesgo relativo de eventos cardiovasculares con el empleo científico deestatinas es de 0.58% por cada mg/dl de reducción de LDL, siendo esta relación linear e independiente delnivel basal de LDL. Basados en el ejemplo de un individuo con riesgo cardiovascular alto y un nivel basal deLDL de 150mg/dl, la reducción de 75mg/dl -50%- de LDL, le reduce su riesgo relativo de un eventocardiovascular en 43.5%.
2.- Asumiendo como valido el incremento de 9% en el riesgo relativo de DM2, el individuo denuestro ejemplo, al soslayar la prescripción de una estatina, tendría la oportunidad de evitar un riesgorelativo de 9% para DM2, sin embargo, también perdería la oportunidad de reducir 43.5% su riesgorelativo de un evento cardiovascular.
3.- Si bien existe evidencia experimental de incremento de A1c relacionada con reducción en lasecreción de insulina a incremento en la resistencia a la insulina secundaria al empleo de estatinas,especialmente lipofílicas, dicho incremento es de 2% a 5% -dosis dependiente- sobre el nivel basal de A1c.Los mecanismos propuestos para este fenómeno son una reducción Ca-dependiente en la secreción deinsulina y/o inhibición en la fosforilación de los GLUT4. Lo anterior implica que un individuo con una A1cde 6.4% podría incrementar dicho valor a 6.72% y cruzar el umbral diagnóstico de DM2.
4.- En la historia de la terapéutica orientada a la reducción del riesgo cardiovascular, en ordende importancia, las estrategias que tienen mayor impacto positivo son: la reducción de LDL, la reducción dela tensión arterial y aún con un valor controversial, la reducción de la A1c.
149
Estatinas, Fibratos, Niacina y más

5.- Por lo anterior, la recomendación de los expertos es no modificar nuestraprescripción de estatinas siempre y cuando ella se base en la evidencia científica reflejada en lasGuías. Entre las recomendaciones dadas en febrero del 2012 por la FDA también se encuentra laadición de una advertencia sobre la posibilidad de incremento de glucosa y/o A1c con el empleode estatinas, la cual se tiene que ponderar en bases individuales.
Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulinresistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardio 2010;55:1209-1216. Predictors of new-onset diabetes in patientsWaters DD, Ho JE, DeMicco DA et al.treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535-1545- Statins and risk of incident diabetes: a collaborative meta-Sattar N, Preiss D, Murray HM et al.analysis of randomized statin trials. Lancet 2010; 375:735-742. Culver AL, Ockene IS, BalasubramarianR et al. Statin use and risk of diabetes mellitus in postmenopausal women in the Women Health Initiative.Arch Intern Med 2012; 172:144-152. FDA communication on line 28 February 2012. Minder CM, BlahaMJ, Horne A, Michos ED, Kaul S, Blumenthal RS. Evidence-based use of statins for prymary preventionof cardiovascular disease. Am J Med 2012; 125:440-446.
Esta gráfica resume el meta-análisis de Sattar. Eldocumentó en una población de 91,140 individuosun incremento del riesgo relativo para DM2 denovo de 9% con un NNH o Número Necesario deTratar para producir 1 nuevo caso de DM2 de 255 yun NNT o Número Necesario de Tratar para evitar1 caso de ECV -fatal o no fatal- de 5.4. En otraspalabras un beneficio cardiovascular 50 veces mayorque el riesgo metabólico.
Cardio-Lipidología
150
Estatinas e Hiperglucemia
N RR NNH NNT
5.4255+9%91,140

EzetimibeMecanismo de acción y efecto en lípidosEl ezetimibe es un inhibidor de la absorción de colesterol a nivel intestinal. Como
monoterapia, el ezetimibe reduce en forma modesta las LDL -10% a 15%-. En combinación con estatinas,el efecto reductor de LDL se sinergiza en grado significativo en función de la estatina y de la dosis empleada.El ezetimibe es una molécula con alta afinidad por la permeasa de colesterol Nieman-Pick-C1-L1. Estaproteína de membrana es la encargada de permear el colesterol de la luz intestinal hacia el enterocito; de estaforma el ezetimibe al competir con el colesterol exógeno por la permeasa NP-C1-L1, disminuye la tasa deabsorción de colesterol y el sustrato para la síntesis de lipoproteínas apo-B100.
Mecanismo de acción y efecto en biomarcadores-bioimágenesEl efecto de ezetimibe sobre biomarcadores tiene evidencia negativa y limitada. En relación a
ateroregresión, en el estudio ENHANCE en individuos con hipercolesterolemia familiar, la combinaciónde ezetimibe 10mg más simvastatina 40mg vs placebo más simvastatina 40mg, no fue estadísticamentesuperior en la modificación del grosor íntima-media carotídeo,-objetivo primario-.
Kastelein JJP, Akdim F, Stroes ESG, Zwinderman AH, Bots ML, Stalenhoef AFH et alfor the ENHANCE Investigators. Simvastatin with ezetimibe in familiar hypercholesterolemia. N EnglJ Med. 2008; 358:1431-1443.
Efecto en reducción de eventos clínicosEn combinación con simvastatina los dos estudios relevantes son los siguientes.Estudio SEAS. Este protocolo fue el primero en evaluar el efecto de la combinación
ezetimibe más simvastatina sobre eventos cardiovasculares. En este estudio, en individuos con estenosisvalvular aórtica, la combinación de ezetimibe 10mg más simvastatina 40mg vs placebo más simvastatina 40mg, no fue estadísticamente superior en la reducción del objetivo primario: combo de muertecardiovascular, cirugía de remplazo valvular aórtico, infarto miocárdico no fatal, hospitalización por angorinestable, angioplastía coronaria, cirugía de revascularización coronaria y/o enfermedad cerebrovascularno hemorrágica, -35.5% vs 38.2% con p ns-. En el análisis por objetivos secundarios, la única diferenciasignificativa a favor de la terapia combinada fue una menor tasa de revascularización coronaria. Sinembargo, dicha diferencia se vio ensombrecida por un incremento significativo -p 0.01- en la incidencia decáncer en el grupo de terapia combinada -105 vs 70 casos-.
151
Estatinas, Fibratos, Niacina y más

Estudio SHARP. Este estudio evaluó el efecto de la combinación ezetimibe 10mg más
simvastatina 20mg vs placebo sobre eventos cardiovasculares en individuos 40 años de edad con�
creatinina 1.7mg/dl en hombres y 1.5mg/dl en mujeres. En el diseño de este estudio, durante el primer� �
año de seguimiento existieron 3 brazos de tratamiento: ezetimibe 10mg más simvastatina 20mg, placebomás simvastatina 20mg y placebo más placebo; al final del primer año, el brazo placebo más simvastatina20mg fue “switcheado” 50% y 50% a los brazos ezetimibe 10mg más simvastatina 20mg y placebo másplacebo. El objetivo primario del estudio: combo de muerte cardiovascular, infarto miocárdico no fatal,enfermedad vascular cerebral no hemorrágica no fatal y revascularización arterial, tuvo una incidencia de11.3% en el grupo de terapia combinada vs 13.4% en el grupo placebo con una reducción del riesgo relativode 17% con p 0.0021. En relación a la seguridad de la combinación, en este estudio no se documentóexceso en la incidencia de cáncer -hallazgo concordante con el meta-análisis de los estudios con ezetimibe-,así como tampoco en la incidencia de elevación de enzimas hepáticas, miopatía y/o rabdomiolisis. Laconclusión de este estudio es que la combinación referida es una alternativa al empleo de estatina sola. Aligual que lo mostró este estudio, el meta-análisis 2010 del CTT demostró una reducción de 20% en el riesgorelativo de eventos cardiovasculares por cada 38mg/dl de reducción de LDL con estatinas en individuoscon insuficiencia renal. En espera de los resultados del estudio IMPROVE-IT, el consenso actual para eluso de ezetimibe en combinación con estatinas es el siguiente. Ezetimibe puede asociarse a una estatinacuando el inhibidor de la HMGCoAR a su dosis óptima no alcance la meta de LDL o bien no sea tolerado;una tercera indicación de la combinación sería en individuos con el perfil SHARP.
González CA, Rubio-Guerra AF, Pavia A, Redding FJ, Cervantes JL, Zacarias JL et al.Effectiveness and safety of ezetimibe added to statin therapy in patients with primary dyslipidemia notachieving the LDL-C treatment goal on statin monotherapy. Clin Drug Invest 2007; 27:333-337. RosseboAB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K et al for the SEAS Investigators.Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008; 359:1343-1356. Baignet C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C et al for the SHARPInvestigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients withchronic kidney disease -Study of Heart and Renal Protection-; a randomized placebo-controlled trial.Lancet; published on line June 9 2011.
152
Cardio-Lipidología

FibratosMecanismo de acción y efecto en lípidosLos fibratos son moléculas agonistas de los receptores nucleares PPAR-alfa. Issemann y Green
en 1990 describieron la proliferación de peroxisomas en hepatocitos de ratas por un efecto activador deciertas moléculas derivadas del ácido fíbrico, sobre un subgrupo de receptores nucleares pertenecientes a la“súper-familia” de los receptores para hormonas esteroides. Estos receptores a la postre fuerondenominados PPAR-alfa o Receptores-alfa Activadores de Proliferación de Peroxisomas.
Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamilyby peroxisome proliferators. Nature 1990; 347:645-650.
Los peroxisomas son organelos rudimentarios, primordios de las mitocondrias, cuyasfunciones básicas en especies inferiores son: la peroxidación de sustancias citotóxicas, la betaoxidación delos ácidos grasos para su transformación en acetil-CoA sin incorporación a la fosforilación oxidativa y lainactivación del exceso de especies reactivas de oxígeno. En el ser humano, las funciones de losperoxisomas son desarrolladas con mayor eficiencia por las mitocondrias, en las cuales, la betaoxidación delos ácidos grasos es seguida por la incorporación de acetil-CoA al ciclo del ácido tricarboxilico para lageneración de moléculas donadoras de electrones y la síntesis de fosfatos de alta energía -ATP- en elproceso bioquímico de la fosforilación oxidativa.
Purdue PE, Lazarow PB. Peroxisome biogénesis. Annu Rev Cell Dev Biol 2001; 17:701-752.Tabak HF, Brakkman I, Distel B. Peroxisomes: simple in function but complex in manteinance. TrendsCell Biol 1999; 9:447-453.
En el tejido muscular, la fuente principal de energía son los ácidos grasos; el miocito formaATP primariamente a partir de ácidos grasos. Las VLDL, IDL y QM ricas en triglicéridos son hidrolizadaspor acción de la lipoproteínlipasa -LPL-; los ácidos grasos derivados de dicha hidrólisis difunden portransporte transmembrana hacia el citoplasma del miocito y actúan como ligandos naturales y activadoresde los receptores nucleares PPAR-alfa. La activación de los PPAR-alfa y su heterodimerización con losreceptores nucleares RXR propicia la unión del heterodimero PPAR-RXR con elementos nucleares derespuesta de proliferación peroxisómica -PPER- e induce o reprime la transcripción de múltiples genes. Enel miocito induce los genes para la síntesis y la expresión de las enzimas que participan en la β-oxidación. Las3 enzimas más importantes en dicho proceso son la acyl sintasa, que transforma a los ácidosgrasos en acyl-CoA; la carnitin acyl-CoA tranferasa I que transfiere la acyl-CoA hacia la matriz mitocondrial
153
Estatinas, Fibratos, Niacina y más

y la carnitin acyl-CoA transferasa II que transforma a la acyl-CoA en acetil-CoA en la matriz mitocondrial.La acetil-CoA se incorpora al ciclo del ácido cítrico y posteriormente a la fosforilación oxidativa, con laproducción de 8.2 moléculas de ATP por cada molécula de ácido graso.
Plutzky J. Peroxisome Proliferator-Activated Receptors.In Hypertension Primer.ThirdEdition.USA. Editorial Lippincott Williams and Wilkins Editors Izzo JL, Black HR 2003:60-62. Alberts B,Johnson A, Lewis J. Cell chemistry and biosynthesis In Molecular Biology of The Cell. 4 Edition USA.
th
Editorial Garland Science. Editors Alberts B, Johnson A, Lewis J et al. 2002:47-128.Este proceso naturalmente activado por los ácidos grasos, puede ser activado por agonistas
sintéticos de los PPAR-alfa como los fibratos. Los efectos clínicos de los fibratos se explican a través de lossiguientes mecanismos -ver capítulo 1 y 2, apartado de dislipidemia aterogénica-.
Reducción de triglicéridos. Los fibratos, en los hepatocitos inhiben la síntesis de apoCIII-apoproteína inhibidora de la LPL-; en los adipocitos, miocitos y hepatocitos incrementan la síntesis deLPL, de los transportadores de ácidos grasos -CD36 y FABP- y de apoAV -apoproteína facilitadora de lahidrólisis de triglicéridos- y en los hepatocitos y miocitos incrementan la síntesis de acyl-CoA sintasa ycarnitin acyl-CoA sintasa I y II -enzimas claves en la síntesis de acetyl-CoA- e inhiben la de acyl-CoA-carboxilasa y sintasa de ácidos grasos -enzimas clave en la síntesis de ácidos grasos a partir de acetyl-CoA .De esta forma los fibratos incrementan la lipolisis de lipoproteínas ricas en triglicéridos -VLDL, IDL yQM- y el pool de ácidos grasos para la síntesis de acyl-CoA, la transformación de acyl-CoA en acetil-CoA y
154
Cardio-Lipidología
PeroxisomasMicrofotografía de peroxisomas en hígado derata. Isseman y Green demostraron que estosorganelos rudimentarios proliferan por efecto
de derivados del ácido fíbrico. Este hallazgopublicado en 1990 dio nombre a una amplia
familia de receptores nucleares antes“huérfanos” y ahora conocidos como PPAR
con tres subfamilias alfa, beta/delta y gamma
Peroxisomas hepáticos

Miocito
A
A
CD36
Miocito
A
CD36
RXR
APPARa
155
Estatinas, Fibratos, Niacina y más
Miocito
PPERActivador
GenAcyl-sintasaCarnitil-acyl-Coa-transferasa ICarnitin-acyl-CoA-transferasa II
Betaoxidación
Miocito
PPERActivador
Gen
Acyl-sintasaCarnitil-acyl-Coa-transferasa ICarnitin-acyl-CoA-transferasa IICD36FABP
CD36CD36 CD36
Betaoxidación
Acidos grasos. Agonistas PPARSe esquematiza como las ácidos grasos portransporte transmembrana en el citoplasma
agonizan al receptor PPAR-alfa y este seheterodimeriza con el receptor RXR, integrando
el heterodimero PPAR-RXR
Acidos grasos. Agonistas PPAREl heterodimero PPAR-RXR migra al núcleo y se
fusiona con fragmentos de DNA conocidos comoelementos de respuesta a PPAR o PPRE. En el
miocito la activación de los PPRE induce la síntesisde diversas proteínas como los transportadores
CD36 y FABP y enzimas clave de la betaoxidación.El objetivo de este proceso es la utilización de losácidos grasos en la formación de fosfatos de alta
energía o ATP
A
A
PPARa
RXR
APPARa
RXR
APPARa
A A A
A A A
A
3
2
4
1

la incorporación de esta a la fosforilación oxidativa para la formación de ATP; en paralelo inhiben la síntesisde ácidos grasos a partir de acetyl-CoA. Como resultado final se disminuye la concentración delipoproteínas ricas en triglicéridos y el sustrato de ácidos grasos para la síntesis hepática de triglicéridos, lalipidación de apoB100 y la formación de VLDL. En estudios clínicos randomizados, el fenofibrato y sumetabolito activo, el ácido fenofíbrico -agonista PPAR-alfa- en comparación con placebo reducetriglicéridos de -26.7% a -35.5% vs +1.3% a +13.6%; colesterol total -5.2% a -26.9% vs -3.7% a +1.6% yLDL +1.2% a -32.5% vs -5.6% a +0.5%; así mismo reduce significativamente -p <0.05- lasconcentraciones de apoB100, VLDL, no-HDL y apo CIII con incremento en el tamaño de las LDL. Lareducción de triglicéridos y el incremento de HDL -ver adelante- son proporcionales a los niveles basales deambos; a mayor nivel de triglicéridos, mayor reducción de triglicéridos e incremento de HDL.
Incremento de HDL. En el hepatocito, los fibratos incrementan la síntesis de apoAI,apoAII, PLTP y receptor SR-B1 e inhiben la actividad de CEPT; en el macrófago, incrementan la síntesisdel transferidor ABC-A1. En su conjunto estos efectos favorecen el incremento de HDL y su acción detransporte reverso de colesterol. En estudios clínicos randomizados, el fenofibrato y su metabolito activo,el ácido fenofíbrico -agonista PPAR-alfa- en comparación con placebo incrementa HDL de +8% a +13%vs -3.7% a +1.3% e incrementa significativamente -p <0.05- las concentraciones de apo AI y apoAI.
En base a estas evidencias en 1993, el fenofibrato fue aprobado en Estados Unidos para eltratamiento de la hipertrigliceridemia severa, en 1999 fue aprobado para su uso conjunto con estatinas parael tratamiento de la hipercolesterolemia y para el control de la hipoalfalipoproteinemia y en el 2008, el ácidofenofíbrico fue aprobado para su uso clínico con las mismas indicaciones que el fenofibrato. Estasindicaciones fueron autorizadas en base a la modificación favorable del perfil lipídico, en ausencia deevidencias sobre eventos cardiovasculares.
Chapman MJ. FruchartFibrates: therapeutic Review. Br J Diabetes Vasc Dis 2006; 6:11-19.JC, Duriez P. Mode of action of fibrates in the regulation of trygliceride and HDL-cholesterolmetabolism. Drugs today -Barc- 2006; 42:39-63. Is there a role for fibrates in theBarter PJ, Rye KA.management of dyslipidemia in the metabolic syndrome?. Arterioscler Thromb Vasc Biol 2008; 28:39-46.Goldfine AB, Kaul S, Hiatt WR. Fibrates in the treatment of dyslipidemias. Time for a reassessment.New Engl J Med 2011; 365:481-484. A Review of its lipid-modifying effects inKeating GM. Fenofibrate.dyslipidemia and its vascular effects in type 2 Diabetes Mellitus. Am J Cardiovasc Drugs 2011; 11:227-247.
156
Cardio-Lipidología

Mecanismo de acción y efecto en biomarcadores-bioimágenesBiomarcadores. Los fibratos independientemente de sus efectos lipídicos tienen otras
acciones que potencialmente podrían contribuir a reducir el riesgo cardiovascular. A continuación seenumeran las acciones “pleiotrópicas” de los fibratos. Reducción en la concentración deDislipidemia.fibrinógeno, PAI-1, PCR, IL6, IL1-beta, TNF-alfa, MCP1, ICAM, LpPLA2 e incremento de PON1 yadiponectina; también se ha documentado reducción de ácido úrico, incremento de homocisteina eincremento en la vasodilatación arterial dependiente de endotelio. Reducción deSíndrome metabólico.fibrinógeno, PAI-1, PCR, IL6, IL1-beta, TNF-alfa, MCP1, ICAM, VCAM, LpPLA2, viscosidad sanguínea
157
Estatinas, Fibratos, Niacina y más
? SíntesisAcidos Grasos
? LipolisisVLDL-IDL-QM
? BetaoxidaciónAcidos grasos
? Acyl-CoA-carboxilasa ? Lipoproteínlipasa ? CD36 y FABP
? Sintasa de ácidos grasos ? Apoproteína CIII ? Acyl-sintasa
? Apoproteína AV ? Carnitin-acyl-CoA-trans I
? Carnitin-acyl-CoA-trans II
? SustratoSíntesis de VLDL
? Hidrólisis periféricaVLDL-IDL-QM
? MetabolísmoAcidos grasos
SíntesisAcidos Grasos
LipolisisVLDL-IDL-QM
BetaoxidaciónAcidos grasos
Acyl-CoA carboxilasa Lipoproteínlipasa CD36 y FABP
Sintasa de ácidos grasos Apoproteína CIII Acyl sintasa
Apoproteína AV Carnitin acyl-CoA trans I
Carnitin acyl-CoA trans II
SustratoSíntesis de VLDL
Hidrólisis periféricaVLDL-IDL-QM
MetabolismoAcidos grasos
Mecanismos que explican el incremento de HDL
? SíntesisApoproteinas
RemodelaciónPLTP-CEPT
MovilizaciónABCA1-SRB1
? Apoproteína AI ? PLTP ? ABCA1
? Apoproteina AII ? CEPT ? ? SRB1
? ApoproteínasSíntesis de HDL
? MaduraciónHDL3-HDL2
? TRCMacrófago-Hepatocito
SíntesisApoproteínas
RemodelaciónPLTP-CEPT
MovilizaciónABCA1-SRB1
Apoproteína AI PLTP ABCA1
Apoproteína AII CEPT SRB1
ApoproteínasSíntesis de HDL
MaduraciónHDL3-HDL2
TRCMacrófago-Hepatocito
Fibratos. Efectos en lípidosLos fibratos como agonistas de los receptores
PPAR-alfa a través de los mecanismosenunciados en las tablas, provocan respuestasbiológicas cuyo resultado final es la reducción
de triglicéridos y VLDL y el incremento deHDL
Mecanismos que explican la reducción de VLDL y TG

postprandial e incremento de adiponectina. Reducción de fibrinógeno, PCR eDiabetes Mellitus.incremento en homocisteina y en vasodilatación arterial dependiente de endotelio. La mayoría de lasacciones extra-lipídicas de los fibratos se relacionan con el agonismo PPAR-alfa en estirpes celularesinvolucradas en los procesos inflamatorios y aterotrombogénicos. En dichas estirpes celulares, incluyendoadipocitos desdiferenciados, monocitos y linfocitos del estroma adiposo visceral del individuo conobesidad central, el agonismo PPAR-alfa estimula PPER con efecto represor de los genes que dictan lasíntesis de moléculas inflamatorias; excepción a esta regla sería el incremento en la síntesis de adiponectinay PON1. Sin embargo, la disección de estas acciones de aquellas secundarias a la reducción de las fraccioneslipídicas inflamatorias y aterogénicas -VLDL-IDL-LDL- y/o al incremento de HDL, así como sutrascendencia clínica, es difícil de establecer.
Al igual que drogas como metformina, adiponectina y estatinas, los fibratos tienen un efectoactivador indirecto de la AMPK. Este efecto no está relacionado con su agonismo PPAR-alfa y se traduceen efectos antinflamatorios y antiapoptóticos especialmente relacionados con la activación de cascadasenzimáticas celulares pro-supervivencia como la PI3K-Akt-eNOS con incremento en la producción deóxido nítrico con sus múltiples acciones autócrinas, parácrinas y endócrinas.
Bioimágenes. El efecto de los fibratos en ateroregresión evaluado por angiografía coronariacuantitativa y USG carotideo es neutro -estudios LOCAT, BECAIT y DAIS-
Fruchart JC. Peroxisome proliferator-activated receptor-alpha -PPAR-alfa-: at the crossroadsof obesity, diabetes and cardiovascular disease. Atherosclerosis 2009; 201:1-8. Tomizawa A, Hattori Y,Inoue T et al. Fenofibrate suppresses microvascular inflammation and apoptosis through adenosinemonophosphate-activated protein kinase activation. Metabolism 2011; 60:513-522.
Efecto en reducción de eventos clínicosEn la introducción de este capítulo se comentó que la información actual sobre fibratos es
controversial y ello obedece al siguiente hecho. Los dos estudios más recientes sobre el uso de fibratos enindividuos con DM2 -FIELD y ACCORD-, en su cohorte general no demostraron que el fibrato -FIELD-y la adición del fibrato a estatina -ACCORD-, al compararse con placebo, disminuyera significativamente elobjetivo primario de ambos estudios -incidencia de eventos cardiovasculares-.
Dichos resultados han suscitado la controversia en relación a la autorización para el uso clínicode fibratos, basada sólo en la modificación favorable del perfil lipídico. Las conclusiones y las racionales de
158
Cardio-Lipidología

dicha controversia son presentadas en este apartado.Más allá de los estudios con fibratos de la era pre-estatinas -World Health Organization
Clofibrate Study, Helsinki Heart Study con gemfibrozil, Veterans Affairs High-Density LipoproteinIntervention Trial con gemfibrozil y Bezafibrate Infarction Prevention Study, cuyo diseño se apartasustancialmente de la práctica actual del tratamiento en el individuo con riesgo cardiovascular alto enprevención primaria y/o del individuo en prevención secundaria, a continuación se hará un análisis sobre laestatus actual y las recomendaciones de las organizaciones regulatorias en salud sobre el uso clínico de losfibratos.
Estudio FIELD -Fenofibrate Intervention and Event Lowering in Diabetes-. Esteestudio incluyó entre los años 1998 y 2000 a 9,765 individuos con DM2 con y sin enfermedadcardiovascular. En forma doble-ciega y aleatorizada se asignó un grupo a tratamiento con placebo y otrocon fenofibrato 200mg; los valores de triglicéridos de la cohorte general oscilaron entre 90 y 450mg/dl conuna media de 156mg/dl. El objetivo primario del estudio fue mostrar una reducción significativa en laincidencia de infarto miocárdico y/o muerte cardiovascular. En un seguimiento promedio de 5 años, elobjetivo primario sucedió en 288 de 4,900 individuos en el grupo placebo y en 256 de 4,895 individuos en elgrupo fenofibrato -5.9% vs 5.2%- con un HR de 0.89 -IC 95% 0.75 a 1.05- con valor de p de 0.89. En otraspalabras una reducción relativa del riesgo de infarto miocárdico y/o muerte cardiovascular de 11%,estadísticamente no significativa.
159
Estatinas, Fibratos, Niacina y más
Fibratos. Efectos “pleitrópicos”Los fibratos en forma paralela a sus efectos
lipídicos producen ciertos efectosantinflamatorios. Los mecanismos para
explicarlos son la activación indirecta de laAMPK y la activación en estirpes celulares
inflamatorias de PPRE represores de los genesde moléculas o biomarcadores de inflamación Célula inflamatoria
RXR
PPERRepresor
Gen
FibrinógenoTNF-alfaIL6-PCRIL1-betaMCP1ICAMVCAMLpPLA2
AMPK PI3k-eNOS-ON
FPPARa
F
F

Estudio ACCORD -Action to Control Cardiovascular Risk in Diabetes-. Este estudio ensu brazo de intervención sobre lípidos, incluyo entre los años 2001 y 2005 a 5,518 individuos con DM con ysin enfermedad cardiovascular y tratamiento óptimo con estatinas. En forma doble-ciega y aleatorizada seasignó un grupo a tratamiento con placebo y otro con ácido fenofíbrico 160mg; los valores de triglicéridosde la cohorte general fueron <400mg/dl -criterio de inclusión- con una media de 162mg/dl. El objetivoprimario del estudio fue mostrar una reducción significativa en la incidencia de infarto miocárdico no fatal,enfermedad vascular cerebral no fatal y/o muerte cardiovascular. En un seguimiento promedio de 4.7 años,el objetivo primario sucedió en 310 de 2,765 individuos en el grupo placebo y en 291 de 2,753 individuos enel grupo de ácido fenofíbrico -11.2% vs 10.6%- con un HR de 0.92 -IC 95% 0.79 a 1.08- con p de 0.32. Loanterior equivale a una reducción relativa del riesgo de infarto, “stroke” y/o muerte cardiovascular de 8%,estadísticamente no significativa.
Dado el reporte de los estudios “antiguos” con fibratos sobre un mayor beneficio en lossubgrupos de individuos con dislipidemia mixta, en ambos estudios se pre-estableció un sub-análisis de lapoblación con triglicéridos altos y HDL bajo; los resultados de ambos subanálisis son los siguientes.
Estudio FIELD -sub-análisis en dislipidemia mixta-. Incluyo 21% de la cohorte general
-2,050 individuos- con triglicéridos 204mg/dl y HDL <50mg/dl y <40mg/dl en mujeres y hombres�
respectivamente. El objetivo primario sucedió en 173 de 970 individuos en el grupo placebo y en 141 de1,044 individuos en el grupo fenofibrato -17.8% vs 13.5%- con un HR de 0.73 -IC 95% 0.58 a 0.91- convalor de p de 0.053. En otras palabras una reducción relativa de 27% en el riesgo de infarto miocárdico y/omuerte cardiovascular, estadísticamente en el margen de la significancia.
Estudio ACCORD -sub-análisis en dislipidemia mixta-. Incluyó 17% de la cohorte
general -9,380 individuos- con triglicéridos 204mg/dl y HDL 34mg en mujeres y hombres. El� �
objetivo primario sucedió en 79 de 456 individuos en el grupo placebo y en 60 de 485 individuos en el grupofenofibrato -17.3% vs 12.4%- con un HR de 0.69 -IC 95% 0.49 a 0.97- con valor de p de 0.057. Las cifrasreferidas equivalen a una reducción relativa de 31% en el riesgo de infarto miocárdico y/o muertecardiovascular, también estadísticamente en el margen de la significancia.
Keech A, Simes RJ, Barter PJ, Best J, Scott R, Taskinen MR et al. Effects of long-termfenofibrate therapy on cardiovascular events in 9,795 people with type 2 diabetes mellitus -the FIELD trial-: randomized controlled trial. Lancet 2005; 366:1849-1861. Effects ofThe ACCORD Study Group.
160
Cardio-Lipidología

combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563-1574. Goldfine AB,Kaul S, Hiatt WR. Fibrates in the treatment of dyslipidemias. Time for a reassessment. New Engl J Med2011; 365:481-484.
Con esta información, los analistas de la Food and Drug Administration han concluido lossiguientes puntos en relación al uso clínico de fibratos.
1.- En individuos con DM el beneficio de la adición de fibratos al tratamiento óptimocon estatinas aún no está confirmado por la evidencias.
2.- En individuos con DM y dislipidemia mixta -triglicéridos >200mg/dl + HDL<40mg/dl en hombres o <50mg/dl en mujeres- el beneficio de la adición de fibratos altratamiento óptimo con estatinas no puede descartarse en base a las evidencias.
3.- Se sugiere el empleo de fibratos en individuos de alto riesgo en prevención primariao bien en prevención secundaria con dislipidemia mixta residual al tratamiento óptimo conestatinas. En individuos con hipertrigliceridemia grave - 500mg/dl- los fibratos tienen�
indicación Clase I en la prevención de pancreatitis, independientemente del riesgocardiovascular.
De esta forma, en un escenario clínico la recomendación basada en las evidencias revisadas, laspropuestas de las agencias regulatorias y las Guías vigentes serían las siguientes:
27%27%
-7.5%
-15%
-22.5%
-30%
RR
Rd
eE
ven
tos
Car
dio
vasc
ula
res
Generalns
11%
Estudio FIELD Estudio ACCORD
31%31%General
p ns
8%
161
Estatinas, Fibratos, Niacina y más
Fibratos. Beneficio potencialLos subanálisis prestablecidos de los estudiosFIELD y ACCORD muestran claramente elbeneficio de los fibratos en la población conDM2 sin dislipidemia, a diferencia de aquella
subpoblación con triglicéridos altos más HDLbajo. A la fecha se acepta que los fibratos son
fármacos indicados en individuos condislipidemia mixta residual al uso óptimo de
estatinas
TG 204+HDL 40-50p 0.053 p 0.057
TG 204+HDL 34

a) Cumplir la meta de LDL.
b) Medir colesterol no-HDL cuando persista un valor de triglicéridos 150mg/dl -Guía�
Europea- o 200mg/dl -Guía Americana-. Si el valor de colesterol no-HDL está por arriba de la meta de�
acuerdo al nivel del riesgo cardiovascular, las opciones terapéuticas son incrementar la dosis de estatina siaún existe margen terapéutico -recordar que la duplicación en la dosis de estatina reduce 4% el valor decolesterol no-HDL-, si ello no fuese suficiente para lograr la meta, la adición de fibrato tienen indicación IIcon nivel de evidencia B, otras opciones serían niacina o ácidos omega 3.
c) En individuos con triglicéridos 500mg, los fibratos tienen indicación Clase I con nivel de�
evidencia C.Reiner Z, Catapano AL, De Becker G, Graham I, Taskinen M, Wiklund O, Agewall S et
al.The Task Force for the Management of Dyslipidemias of the ESC-EAS. Eur Heart J 2011; 32:1769-1818. AHA/ACCFSmith SC, Benjamin EJ, Bonow RO, Braun LT, Creager MA, Franklin BA et al.Secondary Prevention and Risk Reduction Therapy for Patients with Coronary and Other AtheroscleroticVascular Disease: 2011 Update. Circulation 2011; 124: published on line 29 November 2011.
Efectos adversosLos principales efectos adversos reportados son: incremento transitorio y reversible de
creatinina y homocisteina, incremento de INR en individuos anticoagulados, incremento en el riesgo deelevación de enzimas hepáticas y CPK en terapia combinada con estatinas e incremento en el riesgo decoleslitiasis. Se recomienda la determinación de GFR y el ajuste de la dosis de fibrato -50% con GFR 30-60ml/min, interrupción con <30ml/min-, monitoreo de INR en individuos anticoagulados y enzimashepáticas y CPK en individuos en tratamiento combinado con estatinas.
Jones PH. Fibrates. In Clinical Lipidology. Chapter 26. Editor Christie M Ballantine. EditorialSaunders Elsevier. Edition 2009.
162
Cardio-Lipidología

Acidos grasos omega-3Mecanismo de acción y efecto en lípidosLa ubicación de la doble ligadura en la estructura terciaria de un ácido graso, define la
denominación del mismo. Omega-3 si ella está a 3 posiciones del carbono Omega o terminal u Omega-6 silo está a 6 posiciones del carbono terminal. Los ácidos grasos Omega-3 son de origen vegetal -ácidolinoleico- o marino -eicosapentanoico o EPA, docosapentaenoico o DPA y docohexaenoico o DHA-.Dosis diarias de 3 a 4 gramos de EPA o DHA producen reducciones entre 25% y 35% en el nivel detriglicéridos; esta disminución es directamente proporcional al nivel basal de triglicéridos. Los ácidos grasosOmega-3 no modifican directamente el nivel de LDL, la reducción de LDL en individuos con un nivel altode VLDL “vouyant” es el reflejo de la reducción en la síntesis y el incremento en el catabolismo de dichalipoproteína; en otras palabras, la reducción en el nivel de triglicéridos ocasiona disminución en la síntesis deVLDL y por lo tanto reducción secundaria de LDL determinada por la reducción en la síntesis de VLDL.En individuos con un nivel muy alto de triglicéridos la administración de ácidos grasos Omega-3 puedeoriginar un incremento “paradójico” de LDL “vouyant”. El efecto de los ácidos grasos Omega-3 en el nivelde HDL es neutro.
Tras las publicaciones clásicas de Bang y Dyerberg en los 70´s sobre el perfil anti-aterogénico yanti-trombogénico de la tribu esquimal Inuit y tras 30 años de investigación básica y clínica, ahoraconocemos diversos mecanismos de acción con múltiples efectos biológicos de los ácidos grasos Omega-3,entre ellos, los mecanismos más importantes que explican la modificación del perfil de lípidos son lossiguientes. Los ácidos grasos Omega-3 inhiben la transcripción de las enzimas que regulan la síntesishepática de triglicéridos DAGAT1-2 -di acyl glicerol acyl transferasa 1-2- y PAPH -phosphatidic acidphospho hidrolasa- e inhiben la lipasa adipocitaria sensible a hormonas. Ambos efectos se explican por lainhibición del factor de transcripción SREBP -sterol regulatory element binding protein-. Así mismo se hadescrito que los ácidos grasos Omega-3 activan la transcripción de las enzimas que regulan la beta-oxidación al actuar como agonistas de los PPAR-alfa -efecto fibrato-like- e incrementan la actividad de lalipoproteinlipasa; esta enzima hidroliza los triglicéridos contenidos en las VLDL e IDL. A través de estos 4mecanismos los ácidos grasos Omega-3 producen su efecto terapéutico al disminuir la síntesis hepática detriglicéridos y fosfolípidos, principales lípidos en la síntesis de VLDL; inhibir de la hidrólisis adipocitaria detriglicéridos dependiente de catecolaminas e incrementar la hidrólisis periférica de VLDL “vouyant”.
Dyerberg J, Bang HO, Stoffersen B, Moncada S, Vane JR. Eicosapentanoic acid and
163
Estatinas, Fibratos, Niacina y más

prevention of thrombosis and atherosclerosis?. Lancet 1978; 2:117-119. n-3 fatty acids inDe Caterina R.cardiovascular disease. Review Article. N Engl J Med 2011; Published on line June 23 2011.
Mecanismo de acción y efecto en biomarcadores-bioimágenesEs relevante referir que más importante que la modificación a los biomarcadores inducida por
los Omega-3, ellos mismos son marcadores de riesgo cardiovascular. El nivel de EPA y DHA en suero y enla membrana del eritrocito tienen una asociación inversa al riesgo cardiovascular. En bioimágenes deateroregresión la información es escasa.
Harris WS, Jacobson TA. Omega-3 fatty acids. In Clinical Lipidology. Chapter 27; pp 326-338. Editor Ballantyne C. Edition Saunders 2009.
Efectos sobre eventos cardiovascularesSe han publicado múltiples estudios sobre el efecto clínico de los ácidos grasos Omega-3, gran
parte de ellos han sido estudios observacionales, abiertos, no aleatorizados y/o controlados; entre losestudios con mejor diseño y mayor trascendencia están los siguientes:
Estudio GISSI-Prevenzione. Este ensayo comparó en forma abierta la terapia de EPA yDHA 1 gramo contra un grupo control; también existió un brazo con vitamina E y otro con Omega-3 másvitamina E. La población estudiada fueron individuos con infarto miocárdico en los tres meses previos a laselección. El objetivo primario fue analizar la incidencia del combinado: muerte, infarto miocárdico no fataly/o enfermedad vascular cerebral no fatal, entre los grupos de tratamiento. En la comparación entre losgrupos Omega-3 vs control, el resultado del objetivo primario mostró una reducción del riesgo relativo de15% a favor del grupo Omega-3, estadísticamente significativa -p <0.02-, con una llamativa reducción de21% del riesgo de la mortalidad total temprana y de 44% en muerte súbita hacia el 3º y 4º mes detratamiento. Estos resultados abrieron la puerta al concepto de “acción antiarrítmica” de los Omega-3.Este concepto no ha sido confirmado en estudios “ad-hoc”. Por otra parte, los resultados no se asociaron auna diferencia en el perfil de lípidos, entre ambos grupos. El resto de los objetivos secundarios no tuvierondiferencia significativa entre ambos grupos. La principal crítica a este ensayo fue su diseño abierto y el altoporcentaje de abandono ->25%-. El grupo de vitamina E no mostró beneficio al compararse con el grupocontrol.
Estudio JELIS. Este estudio de prevención primaria y secundaria, comparó en forma abiertala terapia de EPA 1.8 gramos más estatinas contra “dieta japonesa habitual” más estatinas, en 14,981
164
Cardio-Lipidología

individuos sin y 3,664 con enfermedad cardiovascular e hipercolesterolemia -colesterol total >250mg/dl-.El objetivo primario fue analizar la incidencia del combinado: muerte cardiovascular, infarto miocárdico nofatal, angina inestable y/o revascularización, entre el grupo de Omega-3 y el grupo “dieta japonesahabitual”. El resultado mostró en el subgrupo de prevención secundaria, no así en el de prevenciónprimaria, una reducción del riesgo del objetivo primario de 19% a favor del grupo Omega-3,estadísticamente significativo -p 0.01-. Entre los objetivos secundarios, la reducción de 19% del riesgo deinfarto miocárdico no fatal y de 14% de angina inestable fueron estadísticamente significativos. En base aeste perfil de protección cardiovascular, surgió el concepto de “estabilización de placa aterosclerosa” paralos Omega-3. La principal crítica a este ensayo fue su diseño abierto, la administración no controlada deestatinas y el que la “dieta habitual japonesa” es por si misma alta en Omega-3, con un contenido entre 800 y1,000 mg de Omega-3, lo cual probablemente explica la baja incidencia de eventos cardiovasculares en elgrupo control -8 veces menor al observado en grupos control de dieta “occidental”- y atenúe la diferenciaen el objetivo primario entre ambos grupos de tratamiento.
Estudio GISSI-HF. Este protocolo comparó en forma aleatoria la terapia de EPA y DHA 1gramo contra placebo en individuos con insuficiencia cardiaca clase II-IV de la NYHA. El objetivoprimario fue analizar la incidencia de muerte total y el combinado: muerte cardiovascular y/ohospitalización por indicación cardiovascular, entre en grupo de Omega-3 y el grupo placebo. El resultadomostró una reducción del riesgo de mortalidad total de 9%, estadísticamente significativa aunque limítrofe-0.04-, así como una reducción del riesgo de muerte cardiovascular y/o hospitalización por indicacióncardiovascular de 8% a favor del grupo Omega-3, estadísticamente significativo -p 0.009-. La etiología delas muertes cardiovasculares fueron “falla eléctrica” presumida o falla cardiaca progresiva. Así como, paralos estudios GISSI-Prevenzione y JELIS, en este estudio se plantearon varias hipótesis del efecto“cardiomiocito protector” de los Omega-3.
Estudio Alfa-Omega. Este protocolo comparó en forma aleatoria, doble-ciego, la terapiasuplementaria de EPA mas DHA 400mg, ALA -ácido alfa-linoleico- 2 gramos, EPA mas DHA y ALA yplacebo en individuos entre 60 y 80 años con historia de infarto miocárdico. El objetivo primario fueanalizar la incidencia de eventos cardiovasculares totales y revascularización, entre los diferentes grupos desuplementación y el grupo placebo. El resultado de este estudio fue negativo, los tres grupos desuplementación no mostraron reducción del objetivo primario al compararse con placebo. La principal
165
Estatinas, Fibratos, Niacina y más

crítica a este estudio fue las dosis bajas de EPA y DHA y altas de ALA en la suplementaciónMarchioli R, Barzi F, Bomba E. Early protection against sudden death by 3-n polyunsatured
fatty acids after myocardial infarction. Time-course analysis of the results of the Gruppo Italiano per loStudio della Sopravvivenza nell´´Infarto Miocardico -GISSI-Prevenzione-. Circulation 2002; 105:1897-1903. Effects of eicosapentaenoic acid on major coronaryYokoyama M, Origasa H, Matsuzaki M.events in hypercholesteroleamic patients -JELIS-. A randomized open-label, blinded end point analysis.Lancet 2007; 369:1090-1098. Effect of n-3 polyinsaturatedTavazzi I, Maggioni AP, Marchioli R, et al.fatty acids in patients with chronic heart failure -the GISSI-HF trial-: a randomized, double-blind, placebo-controlled trial. Lancet 2008; 372:1223-1230. n-3 fatty acids andKromhout D, Giltay EJ, Geleijnse JM.cardiovascular events after myocardial infarction. N Engl J Med 2010; 363:2015-2026.
En base a las evidencias referidas, las indicaciones aprobadas por la AHA para el uso de losácidos grasos Omega-3, son las siguientes:
1.- En el tratamiento de la hipertrigliceridemia grave con >500mg/dl con una dosissugerida de 4 gramos de EPA y DHA.
2.- En individuos con insuficiencia cardiaca, se recomienda el consumo de 1 gramo deEPA y DHA; en individuos sin insuficiencia cardiaca y sin hipertrigliceridemia, se recomienda elconsumo de comidas ricas en Omega-3 al menos 2 veces a la semana.
3.- Como alternativa a fibratos en individuos con elevación de colesterol no-HDLresidual a estatinas a dosis óptima.
Kris-Etherton PM, Harris WS, Appel LJ. Omega-3 fatty acids and cardiovascular disease:new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol 2003;23:151-151. Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN et al.Tryglicerides and cardiovascular disease. A Scientific Statement from the American Heart Association.Circulation 2011; 123:2292-2333.
166
Cardio-Lipidología

NiacinaMecanismo de acción y efecto en lípidosLa niacina es un derivado de la nicotinamida -vitamina B3-. Es un agonista de los receptores de
membrana asociados a proteínas G denominados HM74A, también conocidos como GPR109A. Estosreceptores de membrana se expresan principalmente en adipocitos, hepatocitos y macrófagos, incluyendolas células de Langerhans que son macrófagos de la dermis. Las acciones en las diferentes estirpes celularesson las siguientes. La niacina en el adipocito activa al receptor HM74A asociado a una proteínaAdipocito.Gi o inhibidora la cual regula a la baja la síntesis de AMPc e inhibe a la lipasa adipocitaria. La inhibición deesta enzima bloquea la hidrólisis de triglicéridos inducida por hormonas contra-reguladoras y por lo tantodisminuye el sustrato de ácidos grasos libres para la síntesis de VLDL “vouyant”. La niacinaHepatocito.en el hepatocito reduce la tasa de catabolismo de la apo-A1 y HDL2, el mecanismo de este proceso no estábien definido. En forma similar a la inhibición de la lipasa adipocitaria, la niacina en el hepatocito inhibe a laDAGAT1, enzima participante en la síntesis de triglicéridos y VLDL. La disminución en la síntesis deVLDL “vouyant” tiene como reflejo una reducción en la actividad de la CEPT. En su conjunto, la reducciónen la liberación de ácidos grasos, la inhibición de la DAGAT1 y la regulación a la baja de la CEPTpromueven la reducción de la concentración en suero de VLDL “vouyant” y un efecto positivo en laremodelación de las HDL. La niacina en el macrófago activa al receptor HM74A asociado aMacrófago.una proteína Gi que transactiva a proteínas Gq o quelantes. Estas proteínas inducen la actividad de lafosfolipasa A2 o PLA-2, enzima que facilita la síntesis de ácido araquidónico. Se ha publicado que uno de losmetabólitos de esta molécula, específicamente el 15PGJ2 es un agonista selectivo y potente de los PPAR-gamma del macrófago, siendo este factor de transcripción el que induce en el macrófago la síntesis de CD-36 y ABC-A1, depredador y transferidor de colesterol respectivamente y explicación plausible del efectopositivo de la niacina sobre el trasporte reverso de colesterol. Macrófago dérmico -célula deLangerhans-. La niacina al agonizar al receptor HM74A en el macrófago de la dermis, como ya se comentóestimula a la PLA-2 y con ello la producción de ácido araquidónico que por acción de la COX-1 setransforma en prostaglandina E2 y D2. En forma parácrina, la prostaglandina-D2 migra a la capa muscularmedia arteriolar y estimula al receptor D1 de la célula muscular lisa ocasionando vasodilatación y elcaracterístico flushing asociado a niacina.Lorenzen A, Stannek C, Lang H, Andrianov I, Kalvinsh U, Schwabe U. Characterization of a Gprotein-coupled receptor for nicotinic acid. Mol Pharmacol 2001; 59:349-357. Zhang Y, Robert S,
167
Estatinas, Fibratos, Niacina y más
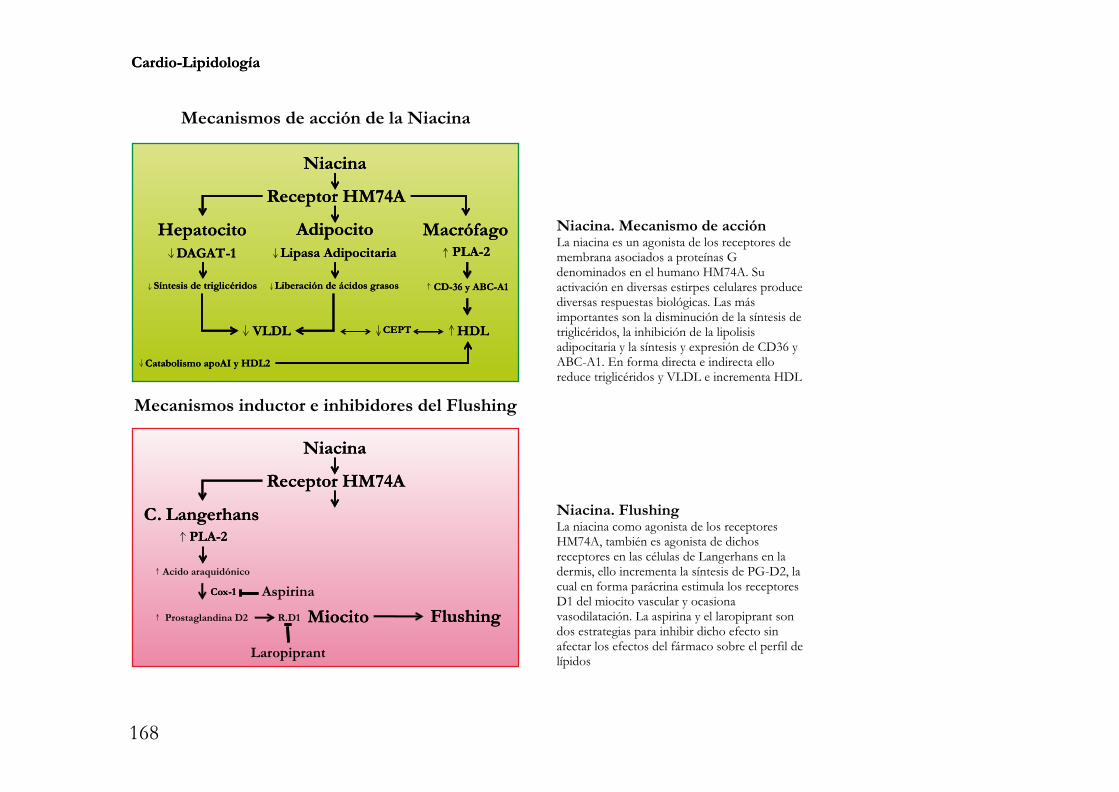
168
Cardio-LipidologíaCardio-Lipidología
Niacina
Receptor HM74A
AdipocitoLipasa Adipocitaria
HepatocitoDAGAT-1
MacrófagoPLA-2
Liberación de ácidos grasosSíntesis de triglicéridos CD-36 y ABC-A1
VLDL
Catabolismo apoAI y HDL2
HDLCEPT
Niacina
Receptor HM74A
AdipocitoLipasa Adipocitaria
HepatocitoDAGAT-1
MacrófagoPLA-2
Liberación de ácidos grasosSíntesis de triglicéridos CD-36 y ABC-A1
VLDL
Catabolismo apoAI y HDL2
HDLCEPT
Niacina
Receptor HM74A
Miocito
C. Langerhans
Flushing
PLA-2
Cox-1
Niacina
Receptor HM74A
Miocito
C. Langerhans
Flushing
PLA-2
Cox-1
Acido araquidónico
Prostaglandina D2 R.D1
Aspirina
Laropiprant
Niacina. Mecanismo de acciónLa niacina es un agonista de los receptores demembrana asociados a proteínas Gdenominados en el humano HM74A. Suactivación en diversas estirpes celulares producediversas respuestas biológicas. Las másimportantes son la disminución de la síntesis detriglicéridos, la inhibición de la lipolisisadipocitaria y la síntesis y expresión de CD36 yABC-A1. En forma directa e indirecta elloreduce triglicéridos y VLDL e incrementa HDL
Niacina. FlushingLa niacina como agonista de los receptoresHM74A, también es agonista de dichosreceptores en las células de Langerhans en ladermis, ello incrementa la síntesis de PG-D2, lacual en forma parácrina estimula los receptoresD1 del miocito vascular y ocasionavasodilatación. La aspirina y el laropiprant sondos estrategias para inhibir dicho efecto sinafectar los efectos del fármaco sobre el perfil delípidos
Mecanismos de acción de la Niacina
Mecanismos inductor e inhibidores del Flushing

Foxworthy P, Emkey R, Oler K, Jennifer O, Thomas HL, Wang H, Erik WS, Marion KM, PatrickIE, Cao G. Niacin mediates lipolysis in adipose tissue through its G-protein coupled receptor HM74A.Biochem Biophys Res Commun 2005; 334:729-732. Guanine nucleotide binding proteins.Garrison GC.Chapter A29, pp: 80-83. In Hypertension Premier. Edition 2003. AHA. Editors Izzo JL, Black HR. TangY, Zhou L, Gunnet JW, Wines PG, Cryan EV, Demarest KT. Enhancement of arachidonic acidsignaling pathway by nicotinic acid receptor HM74A. Biochem Byophis Res Commun 2006; 345:29-37.Liewer SA, Lenhard JM, Wilson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocytesdifferentiation. Cell 1995; 83:813-819. Inositol phospholipids and inositol phosphates. ChapterBalla T.A31, pp: 88-91. In Hypertensión Premier. Edition 2003. AHA. Editors. Izzo JL, Black HR. Nasjletti A,McGiff JC. Prostaglandins and P450 metabolits. Chapter A20, pp 55-57. In Hypertensión Premier.Edition 2003. AHA. Editors Izzo JL, Black HR. Lipoxigenase products. Chapter A21, pp 58-Nadler JL.59. In Hypertensión Premier. Edition 2003. AHA. Editors Izzo JL, Black HR.
La principal acción lipídica de la niacina es el incremento de HDL. El incremento de HDL conuna dosis promedio de 2 gramos de niacina oscila entre 25% y 35%. Al día de hoy está catalogada como elfármaco de uso clínico que logra el mayor incremento de HDL. La niacina reduce entre 10% y 25% lostriglicéridos y las diferentes fracciones de las lipoproteínas apo-B-100, incluyendo a la lipoproteína -a-.
Brown BG, Canner PL, McGovern ME, Guyton JR, Carlson LA. Nicotinic Acid. InClinical Lipidology. Chapter 25; pp 298-314. Editor Ballantyne Ch. Edition Saunders 2009.
Mecanismo de acción y efecto en biomarcadores-bioimágenesLa niacina tiene una larga e importante historia en la investigación sobre ateroregresión. El
estudio CLAS, combinando niacina y colestipol fue el primer estudio que utilizó la angiografía coronariacuantitativa para evaluar la factibilidad de ateroregresión coronaria. Posteriormente se han publicado almenos una decena de estudios clínicos con metodología diferente pero con el mismo objetivo, demostrar laateroregresión con niacina sola o en combinación con otros fármacos como colestipol y estatinas de“primera generación” -estudios CLAS, CLAS-II, UC-SCOR, FATS, SCRIP, HARP, FATS-TRx10, HATS,AFREGS y ARBITER-2, 3 y 6-.
Estos estudios fundaron conceptos que ahora son pilares en la terapéutica cardiovascular,entre ellos los más relevantes son los siguientes. El nivel de LDL, HDL y presión arterial sistólica son
169
Estatinas, Fibratos, Niacina y más

variables con asociación significativa a la evolución de la aterosclerosis; la ateroregresión es un fenómenofactible; la incidencia de eventos cardiovasculares está en relación directa con la intensidad del tratamientofarmacológico. Si bien estos conceptos ahora suenan elementales, en los 90s sentaron las bases de la terapiaactual de la aterosclerosis. Más allá del análisis individual de los estudios referidos, el meta-análisis publicadopor Greg Brown en el 2007, permite integrar la información de 50 años de investigación clínica en esta área.En este meta-análisis Brown evaluó la factibilidad de progresión o regresión de la aterosclerosis coronaria yanalizó los 12 estudios con mejor diseño. Incluyó estudios con cinco estrategias terapéuticas -placebo,fibratos, estatinas, estatinas más resina y niacina más otros fármacos-. Las dos estrategias que se asociaron aregresión de la aterosclerosis coronaria fueron las estatinas más resina y la niacina más estatinas o resina. Enforma opuesta, el placebo favoreció la progresión más importante de la aterosclerosis coronaria, seguido dela monoterapia con fibratos y estatinas de primera generación. El corolario de este meta-análisis fue que elpredictor con mayor potencia estadística para la regresión de la aterosclerosis coronaria, es la suma dereducción de LDL más incremento HDL, logrado por una estrategia farmacológica. Para confirmar estaobservación fue diseñado el estudio ARBITER-6.
Brown BG, Zhao XQ, Cheung MC. Should both HDL cholesterol and LDL cholesterol betargets for lipid therapy?. A review of current evidence. J Clin Lipidology 2007; 1:88-94.
El estudio ARBITER-6 partió de un cuestionamiento de gran importancia clínica: ¿enindividuos con enfermedad cardiovascular clínica o riesgo cardiovascular alto y tratamiento óptimo conestatinas y LDL en meta <100 mg/dl, la estrategia de reducir aún más el LDL con ezetimibe será superior ala estrategia de incrementar el HDL con niacina?. Resultado, la estrategia de reducir el nivel de LDL conezetimibe fue inferior a incrementar el nivel de HDL con niacina. La aterosclerosis en este estudio fueevaluada por ultrasonido carotideo de alta resolución. El grupo tratado con niacina 2 gramos fuecomparado con el grupo tratado con ezetimibe 10mg. En el primero el HDL incrementó 18.4% en tanto enel segundo el LDL disminuyó 19.2%. El grosor medio y máximo del binomio íntima-media carotídea tuvoregresión estadísticamente significativa en el grupo niacina, en tanto que el grupoezetimibe tuvo tendencia a la progresión. Estos cambios correlacionaron con una mayor incidencia deeventos cardiovasculares en el grupo ezetimibe -análisis no pre-establecido-. Si bien este estudio ha sidocontrovertido, su resultado está bien fundamentado y apoyado por los resultados de otros estudiosrecientes como el estudio de Oxford, en el cual la evaluación del grosor carotideo con resonancia magnética
170
Cardio-Lipidología

nuclear, también demuestra regresión de la aterosclerosis carotidea con niacina.Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M et al. Extended-
realease niacin or ezetimibe and carotid intima-media thicknes. N Engl J Med 2009; 361:3113-2122. LeeJMS, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I et al. Effects of high-dosemodified-release nicotinic acid on atherosclerosis and vascular function. A randomized placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol 2009; 54:1787-1794.
Efecto en reducción de eventos clínicosEstudio Coronary Drug Proyect -CDP-. En la historia de la niacina, la evidencia positiva
sobre eventos cardiovasculares surgió anticipada a la evidencia sobre los estudios de imagen. El estudioCDP, iniciado en 1966 y concluido en 1974 es el ensayo que mantiene el interés en este fármaco. Esteestudio incluyó 8,341 hombres con historia de infarto miocárdico documentado por ECG, clínicamenteestables y en clase funcional NYHA I-II al momento de su randomización. Las estrategias terapéuticasoriginalmente comparadas contra placebo -2,789 individuos- fueron: estrógenos 2.5 mg, estrógenos 5.0
Estudio ARBITER-6Cambio en el grosor íntima-media carotideo
-0.014
00
Cam
bio
enel
GIM
Cen
mm
148Meses de seguimiento
Ezetimibe 10mg
Niacina 2 gramos
0
-0.014
00
Cam
bio
enel
GIM
Cen
mm
148Meses de seguimiento
Ezetimibe 10mg
Niacina 2 gramos
0
171
Estatinas, Fibratos, Niacina y más
Estudio ARBITER-6Utilizando el USG carotídeo y la medición delgrosor íntima-media como subrrogado deAterosclerosis, los Investigadores de esteestudio demostraron que en individuos tratadoscon estatina, reducir más el LDL con ezetimibeno modificó el grosor íntima-media a diferenciade la estrategia de incrementar el HDL conniacina, la cual si disminuyó en formasignificativa el valor de dicho subrogado

mg, clofibrato 1.8 gramos, dextrotiroxina 6mg y niacina de liberación inmediata 3 gramos -1,119individuos-. Las estrategias de estrógenos y dextrotiroxina fueron interrumpidas prematuramente por laalta incidencia de eventos adversos. La comparación de niacina con placebo mostró los siguientesresultados: a) mortalidad total sin diferencia significativa; b) infarto miocárdico no fatal 10.7% contra14.8% con p 0.001; c) revascularización coronaria 3.3% contra 7.2% con p <0.0001. La incidencia deinfarto miocárdico no fatal y de revascularización coronaria fue de 13.8% y 6.3% respectivamente en elgrupo clofibrato, cifras similares a las del grupo placebo. Esta observación es clínicamente relevante ya quela niacina y el clofibrato provocaron reducciones similares de lípidos y sólo la niacina redujosignificativamente la incidencia de infarto miocárdico no fatal y revascularización coronaria. La incidenciade eventos adversos en el grupo niacina fue alta, especialmente algún tipo de “flushing” en 91% de losindividuos, lo cual fue motivo estadísticamente significativo de abandono del tratamiento con p < 0.0001.Estos resultados especialmente la baja tolerancia a la niacina de liberación inmediata “enterraron”temporalmente el interés por la niacina. Sin embargo la iniciativa del NIHLB de analizar en 1975 el estatusvital de los 6,088 sobrevivientes de la cohorte original del estudio modificó el panorama. En un análisis deun promedio de 15 años de seguimiento, el grupo original de niacina comparado con el grupo originalplacebo mostró una mortalidad total de 52.0% en el grupo niacina contra 58.2% en el grupo placebo con p0.0004. Esta diferencia en mortalidad estuvo originada principalmente por una reducción en la mortalidadcardiovascular 36.5% en el grupo niacina contra 41.3% en el grupo placebo con p 0.005.
Dichos hallazgos renovaron el interés por este interesante fármaco y han motivado una saga deestudios clínicos cuyo último ejemplo el ensayo AIM-HIGH con niacina de liberación prolongada -veradelante- ha creado una gran controversia y estamos en espera de la publicación del protocolo HPS-2 conniacina más laropiprant.
The Coronary Drug Proyect Research Group. Clofibrate and niacin in coronary heartdisease. JAMA 1975; 231:360-381. Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L,Prineas RJ, Friedwald W. Fifteen year mortality in Coronary Drug Proyect patients: long term benefitwith niacin. J Am Coll Cardiol 1986; 8:1245-1255. Reduction of mortalityCarlson LA, Rosenhamer G.in the Stockholm Ischaemic Heart Disease Secondary Prevention Study by combined treatment withclofibrate and nicotinic acid. Acta Med Scand 1988; 223:405-418. Brown G, Albers JJ, Fisher L, SchaeferSM, Lin JT, Kaplan C et al. Regression of coronary artery disease as a result of intensive lipid-lowering
172
Cardio-Lipidología

therapy in men with high levels of apolipoprotein B. N Engl J Med 1990; 323:1289-1298. Brown B, ZhaoX, Chait A, Fischer L, Cheung M, Morse J et al. Simvastatin and niacin, antioxidant vitamins, or thecombination for the prevention of coronary disease. N Engl J Med 2001; 345:1583-1592. Taylor AJ,Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the TreatmentEffects of Reducing Cholesterol -ARBITER-2-: a double-blind, placebo-controlled study of extendedrelease niacin on atherosclerosis progression in secondary prevention patients treated with statins.Circulation 2004; 110:3512-3517. Whitney EJ, Krasuski RA, Personius BE, Michalek JE, MaranianAM, Kolasa MW, Monick E, Brown BG, Gotto AM Jr. A randomized trial of a strategy for increasinghigh-density lipoprotein cholesterol levels: effects on progression of coronary heart disease and clinicalevents. Ann Intern Med 2005; 142:95-104. El LDL no lo es todo. Redefiniendo elMorales-Villegas EC.uso terapéutico de la Niacina. Rev. Pharmalife México. Agosto 2008:22-28.
Estudio AIM-HIGH. Por sus implicaciones y lo reciente de su publicación analizaremos esteestudio con cierto detalle. Publicado en noviembre del 2011, este estudio ha iniciado un compás de esperaen relación al uso clínico de niacina.
Niacina
Placebo
15
Eve
nto
sca
rdio
vasc
ula
res
IAM no fatal Cirugía de RVC
05
10
20
00
14.8
10.7
7.2
3.3
p .001
p .0001
173
Estatinas, Fibratos, Niacina y más
Estudio Coronary Drug ProyectEste estudio “pivote” en la historia de la niacinademostró que la administración de 3 gramos de
niacina de liberación inmediata, redujo en formasignificativa eventos coronarios, especialmente
el infarto miocárdico y la revascularizacióncoronaria. Estos hallazgos han mantenido viable
la hipótesis del incremento farmacológico deHDL
Estudio Coronary Drug Proyect

El estudio fue patrocinado por el NHLBI con donación de las drogas en estudio. Después deun periodo abierto de 4 a 8 semanas para evaluar la tolerancia de simvastatina 40mg y niacina en dosisescalada de 500 a 1500mg, el grupo de estudio “tolerante” se aleatorizó 1:1 a un brazo simvastatina másniacina y otro simvastatina más placebo -conteniendo 50mg de niacina en cada tableta de placebo de niacina
de 500 o 1000mg-. La población de estudio fueron individuos 45 años de edad con enfermedad�
cardiovascular estable -coronaria, carotideo-vertebral o periférica-, HDL <40mg/dl en hombres o<50mg/dl en mujeres, triglicéridos 150mg/dl a 400mg/dl y LDL <180mg/dl en el subgrupo de individuossin tratamiento con estatinas. Durante el tratamiento la dosis de estatina y/o la adición de ezetimibe fuepermitida para lograr una meta de LDL entre 40mg/dl y 80mg/dl. El objetivo primario del estudio fueevaluado por la incidencia del combo de muerte cardiovascular, infarto miocárdico no fatal, enfermedadvascular cerebral isquémica no fatal, hospitalización por SICA y/o revascularización indicada por síntomasen territorio coronario o carotideo. El estudio fue interrumpido por cumplir los criterios pre-establecidosde futilidad -no eficacia- amén de una tendencia a mayor incidencia de enfermedad vascular cerebral en elgrupo simvastatina más niacina. De los 8,162 individuos seleccionados inicialmente, 4,273 -52.4%-reunieron los criterios de inclusión-exclusión para ingresar a la fase abierta de tolerancia, de esta cohorte3,414 -79.9%- fueron aleatorizados -este dato implica que 20.1% de los individuos no tuvieron unatolerancia aceptable de los fármacos de estudio-. La edad media de los individuos fue de 64 años, 85.2%hombres, 92.2% blancos, 81% con síndrome metabólico, 71.4% con hipertensión arterial y 33.9% conDM2. El 93.6% de los individuos al momento de su aleatorización estaban en tratamiento con estatinas conun LDL promedio de 71mg/dl, HDL 35mg/dl y triglicéridos 161mg/dl; los 210 individuos “naives” aestatinas tuvieron valores de 124mg/dl, 33mg/dl y 215mg/dl respectivamente. A los 2 años de tratamientoel HDL incremento 25% en el grupo niacina y 9.8% en el grupo placebo -42mg/dl vs 38mg/dl- p <0.001;los triglicéridos descendieron 28.6% en el grupo niacina y 8.1% en el grupo placebo y el LDL descendió12% en el grupo niacina y 5.5% en el grupo placebo. La reducción y descontinuación del tratamiento fuemás frecuente en el grupo niacina, en la mayoría de los individuos la dosis de simvastatina fue 40mg con unamayor frecuencia de simvastatina 80mg y/o ezetimibe en el grupo placebo sin otras diferenciassignificativas en el resto del tratamiento farmacológico.
La incidencia del objetivo primario fue de 16.4% -282 de 1,718- en el grupo niacina contra16.2% -274 de 1,696- en el grupo placebo -HR con niacina 1.02 con IC 95% 0.87 a 1.21 y p de 0.80-, con un
174
Cardio-Lipidología

desbalance no estadísticamente significativo en la incidencia de enfermedad vascular cerebral isquémica1.6% en el grupo niacina contra 0.9% en el grupo placebo; no existió diferencia significativa en los objetivossecundarios y no se identificó alguna interacción significativa en relación a las características basales de lacohorte.
En la discusión verbal e impresa del artículo, así como en la Editorial de NEJM al mismo y otraspublicaciones relacionadas, se concluye que en individuos con enfermedad cardiovascular estable, entratamiento óptimo con estatinas y cifras de LDL <70mg/dl -promedio 60mg/dl- más terapia adyuvantetambién óptima -antiagregantes plaquetarios, inhibidores del SRAA, bloqueadores beta, etc-, este estudiocon niacina al igual que otros con diferentes estrategias por ejemplo fibratos -ACCORD-, no han logradoun beneficio terapéutico adicional basado en la modificación del perfil de lípidos. Amén de las potencialeslimitaciones de este estudio, como se comentó en el capítulo 4, antes de descartar la hipótesis del beneficioterapéutico de incrementar farmacológicamente el HDL habrá que valorar los resultados de estudios condiferentes estrategias incrementadoras de HDL -ver adelante-.
The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels
Niacina
Placebo
15
Eve
nto
sca
rdio
vasc
ula
res
en% 16.4
05
10
20
00
16.2
p ns
1.6 0.9
p ns
En
ferm
edad
vasc
ula
rce
reb
rale
n%
15
05
10
20
00
175
Estatinas, Fibratos, Niacina y más
Estudio AIM-HIGHParadójicamente el estudio AIM-HIGH no
confirmó la hipótesis del beneficio delincremento farmacológico de HDL. En una
población con enfermedad cardiovascular,tratamiento óptimo y LDL promedio de
60mg/dl, no existió diferencia entre niacina yplacebo en la incidencia de nuevos eventos
cardiovasculares. Entre muchas otrascontroversias, este estudio pone de manifiesto
las grandes diferencias en el perfil de laspoblaciones de los estudios CDP y AIM-HIGH
Estudio AIM-HIGH

receiving intensive statin therapy. N Engl J med. Published on-line November 17 2011. Giugliano RP.Niacin at 56 years of age. Time for an early retirement. N Engl J Med. Published on line November 17 2011.
Estrategias para incrementar la tolerancia a la niacina. Es un hecho bien conocido que eluso de la niacina está limitado por la incidencia de flushing o sensación de rubicundez y calor de gradovariable en la cara y el torso. El 90% de los individuos que toman niacina experimentan algún grado deflushing, siendo la causa del 90% de los casos de abandono terapéutico y/o dosis subterapéuticas de niacinaque restringen el beneficio terapéutico del fármaco. Las opciones actuales para limitar el flushing son lassiguientes: Como ya fue referido, en la génesis del flushing participa la síntesisaspirina a dosis de 325 mg.de diversas prostaglandinas, especialmente la PGD2. La aspirina como inhibidor parcial de la síntesis deprostaglandinas produce una reducción también parcial de 10% a 25% en el flushing asociado a niacina. Ladosis recomendada es de 325mg, treinta minutos previos a la ingesta de la niacina y por tiempo indefinido.La dosis y el tiempo de administración de la aspirina limitan su uso en pacientes con intolerancia gástrica a lamisma, ello en conjunto con su efecto parcial la hacen sólo una medida complementaria para incrementar latolerabilidad a la niacina; Dado que la intensidad delniacina de liberación extendida reformulada.flushing está determinada en gran parte por la velocidad de absorción de la niacina, se han investigadovarias alternativas para extender la absorción del fármaco. La niacina de liberación extendida, aprobada porla FDA en 1999, mostró modificar el perfil de lípidos igual que la niacina de liberación rápida, disminuir laintensidad y frecuencia del flushing, y carecer de efectos hepatotóxicos. Recientemente la niacina deliberación extendida fue reformulada para incrementar aún más su tolerabilidad. En un estudio doble ciegoy controlado en población sana, diseñado para inducir flushing con una dosis de 2 gramos de niacinareformulada en una sola toma y sin premedicación con aspirina, se demostró una menor incidencia deflushing en comparación con la niacina original -89% vs 98% con p 0.0027- y reducción en la duración eintensidad del flushing -42% y 43% con p <0.0001-. El perfil de tolerabilidad global fue superior y el deseguridad fue similar entre la nueva formulación y la fórmula original de niacina de liberación extendida.Los autores concluyen que la niacina de liberación extendida reformulada es una buena opción paraincrementar el apego al tratamiento. Se enfatiza que es necesaria una buena educación del paciente,orientada al conocimiento del fármaco y sus beneficios cardiovasculares así como el empleo correcto decorrecto de aspirina para abatir el abandono del tratamiento a cifras inferiores a 10% en el largo plazo;laropiprant -bloqueador selectivo del receptor D1 de la prostaglandina D2-. El laropiprant es un
176
Cardio-Lipidología

bloqueador selectivo de los receptores D1 para PGD2, responsable del 80% del flushing asociado a niacina.En estudios de investigación fase II y III, el laropiprant ha demostrado las siguientes características:Reducción significativa de flushing en forma aguda; reducción significativa de flushing en forma crónica;ausencia de interferencia con el efecto favorable de la niacina sobre el perfil lipídico. Se propone para usoclínico el uso de 1 gramo de niacina más 20 mg de laropiprant en una sola tableta como dosis de nivel I oinicial durante 4 semanas y duplicar la dosis a nivel II como dosis de mantenimiento con 2 gramos deniacina más 40 mg de laropiprant. El laropiprant o MK0524 bloquea selectivamente al receptor D1 dePGD2 y reduce la respuesta vasodilatadora post-niacina en un 65%. El laropiprant no bloquea al receptorHM74A, por lo tanto no interfiere con las acciones de la niacina en adipocitos, macrófagos o hepatocitos.El programa de investigación de fase III comprobó que la combinación niacina más laropiprant lograincrementar significativamente la tolerancia a la niacina, conservando los efectos favorables de esta últimasobre la concentración de lipoproteínas. Finalmente, lo relevante será confirmar a mediano y largo plazo,que la combinación de niacina con laropiprant, impacte favorablemente en la incidencia de eventoscardiovasculares -estudioHPS-2-.
Cefali EA, Simmons PD, Stanek EJ, Shamp TR. Improved control of niacin-inducedflushing using an optimized once-daily extended-release niacin formulation. Int J Clin Pharmacol Ther2006; 44:633-640. Lai E, DeLepeleire I, Crumley TM, Liu F, Wenning LA, Michiels N, Vets E,O´Neill GO, Wagner JA, Gottesdiener K. Suppression of niacin-induced vasodilatation with anantagonist to prostaglandin D2 receptor subtype 1. Clin Pharmacol Ther 2007; 81:849-857. Paolini JF,Mitchel YB, Reyes R, Kher U, Lai E, Watson DJ, Norquist JM, Meehan AG, Bays HE, DavidsonM, Ballantyne CM. Effects of laropiprant on nicotinic acid-induced flushing in patients withdislipidemia. Am J Cardiol 2008; 101:625-630. Maccubbin D, Sirah W, Batteridge A, Kuznetsova O, YuQ, McCrary Sisk C, Bays HE, Olsson AG, Pasternak RC, Mitchel Y, Paolini JF. Flushing profile ofER niacin/laropiprant in patients with primary hypercholesterolemia or mixed dislipidemia. Abstractpresented at AHA Meeting 2007.
177
Estatinas, Fibratos, Niacina y más

178
Cardio-Lipidología
Estrategias farmacológicas en investigaciónLas dos grandes líneas de investigación sobre estrategias para modificar el perfil de lípidos son
las orientadas a potenciar el efecto reductor de LDL de las estatinas y las dirigidas a incrementar la cantidady/o calidad de las HDL.
Estrategias en investigación para reducir LDLEn este rubro existen cuatro estrategias principales, la inhibición de la escualeno sintasa, la
inhibición de la MTP “microsomal transfer protein”, la inhibición de la apoproteína B y la más reciente, lainhibición de la PCSK9 “proprotein convertase subtilisyn kexin type 9”.
Inhibidores de la escualeno sintasa. La inhibición distal de la síntesis de colesterol,bloqueando el paso de farnesil pirofosfato a escualeno con el empleo de inhibidores de la enzima escualenosintasa, tiene su racional más importante en la no inhibición de la síntesis de isoprenoides y por ende lafacilitación de la isoprenilación de diversas moléculas, entre ellas no-esteroles, grupo hem A, ubiquinona,dolicol, isopentanil tRNA etc. La inhibición de la isoprenilación de dichas moléculas por las estatinas se haasociado a su miotoxicidad y se plantea que un bloqueo distal a nivel de escualeno sintasa podría limitardicho evento adverso. El lapaquistat o TAK-475 ha sido la molécula con mayor desarrollo en este campo,sin embargo su programa de investigación fue interrumpido en fase III por incremento significativo enenzimas hepáticas a la dosis de 100mg; se espera un probable ajuste a la dosis de la molécula.
Inhibidores de la MTP. La inhibición de la síntesis de VLDL y QM, bloqueando la lipidaciónde la apoB con inhibidores de la proteína microsomal transferidora de lípidos o MTP, tiene su modelogenético humano en individuos con abetalipoproteinemia e hipobetalipoproteinemia familiar -ver capítulo2-. Se han desarrollado varias moléculas inhibidoras de la MTP, la más avanzada en su desarrollodenominada implitapide o AEGR-427 ha mostrado una gran eficacia para reducir las cifras de lipoproteínascon apoB, sin embargo la dosis estudiada de 80mg se ha asociado a esteatosis hepática con elevaciónenzimática y malabsorción intestinal por lo cual se exploran opciones de dosis <80mg, aplicables a gruposseleccionados de individuos con intolerancia y/o contraindicación para el uso de estatinas.
Otra estrategia para inhibir la síntesis de MTP es la denominada terapia anti-sense. Estaconsiste en el diseño de moléculas con acción RNA-asa “like activity” que interfieren a nivel del RNAmensajero con la codificación de una proteína determinada. La mayoría de las moléculas anti-sense sonoligonucleótidos denominados ASO “anti sense oligonucleotides”. El mipomersen o ISIS-301012 es unASO contra apoB100 de aplicación intramuscular y semanal, el cual a dosis de 400mg en estudios de fase IImostró una gran eficacia para reducir LDL -12% a 70%-, sin embargo las reacciones cutáneas en el sitio de

aplicación y especialmente la elevación de enzimas hepáticas han sido eventos adversos limitantes en elprograma de investigación; a la fecha se plantea su utilización selectiva en subgrupos de individuos conhipercolesterolemia grave, especialmente hipercolesterolemia familiar homocigota -ver capítulo 2-.
Inhibidores de la PCSK9. La inhibición de la acción proteolítica de la PCSK9 sobre elreceptor para LDL y con ello el incremento de la vida media y el reciclamiento del LDLR, tiene también sumodelo natural humano en los individuos con la mutación de “perdida de función” de la PCSK9, los cualestienen cifras <80mg/dl de LDL durante toda su vida y una baja incidencia de enfermedad cardiovascular-RRR 80%-. Recientemente se publicó el primer estudio fase I con terapia anti-sense contra PCSK9. En 20voluntarios sanos la administración intravenosa de 0.25mg/kg de ALN-PCS comparada con placeboprovocó una reducción hasta de 60% en la concentración de PCSK9 -p <0.001- y hasta de 50% -promedio39%- en el nivel de LDL -p <0.05-. Se espera la continuación del programa de investigación de ALN-PCS,el cual se clasifica como una molécula de interferencia al RNA basada en una nano-partícula lipídica. El 22de marzo de este año Stein publicó en NEJM el primer estudio con un anticuerpo monoclonal -REGN727-dirigido contra PCSK9, el cual interfiere con la unión de la proteasa al LDLR y por lo tanto incrementa lavida media de este. En un diseño de tres etapas, el estudio de fase I en voluntarios sanos, individuos conhipercolesterolemia no familiar e individuos con hipercolesterolemia familiar heterocigota, demostró unbuen perfil de seguridad y tolerancia al anticuerpo vs PCSK9 y reducciones de LDL -sobre estatinas- de39.2% a 61% en forma dosis dependiente, con reducción de colesterol no-HDL, apoB100 y lipoproteína-a-, así como un llamativo incremento de HDL -18%- y apoAI -13%-. Este promisorio y el reporte deMcKenney -en prensa- nagrega evidencia sólida en este terreno terapéutico aditivo a la acción de lasestatinas.
Bays H. Investigational agents affecting atherogenic lipoproteins. In Clinical Lipidology.Chapter 44; pp 530-543. Editor Ballantyne C. Edition Saunders 2007. Stiles S. Novel LDL-gene-targetedagent promising. Alnylam reports positive preliminary clinical results for ALN-PCS, an RNAi therapeutictargeting PCSK9 for the treatment of severe hypercholesterolemia. January 4 2012.www.medscape.comStein EA, Mellis S, Yancopoulus GD, Stahl N, Logan D, Smith WB et al. Effect of a monoclonalantibody to PCSK9 on LDL cholesterol. N Engl J Med 2012; 366:1108-1118. Mc Kenney JM, Koren MJ,Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to PCSK9,in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am CollCardiol 2012;59:000-000 -in press-
Estatinas, Fibratos, Niacina y más
179

Estrategias en investigación para incrementar el HDLEste es un campo en amplia investigación especialmente por tres razones; la evidencia básica y
clínica sobre el papel del HDL en la ateroregresión y en otras acciones biológicas cardiovascular ymetabólicamente favorables; la evidencia epidemiológica sobre la relación inversa entre el nivelespontáneo de HDL y la incidencia de eventos cardiovasculares -mayor HDL menos eventos- yfinalmente la evidencia del riesgo residual a la terapia óptima con estatinas, aún en individuos que logran unLDL <70mg/dl. Estas tres líneas de evidencia mantienen el interés por la investigación sobre estrategiasfarmacológicas para incrementar el HDL.
Sin embargo, desde el punto de vista del autor, es justo referir que la solidez de dichasevidencias ha sido retada por otras recientes, siendo la evidencia epidemiológica la única incontrovertibleen el momento actual. En relación al papel del HDL en la ateroregresión, los programas ILLUMINATE,ILLUSTRATE y RADIANCE con torcetrapib pusieron en tela de juicio la solidez de la hipótesis, ya queaun con elevaciones muy significativas de HDL ->50%- no existió ateroregresión a nivel coronario y/ocarotídeo y la morbi-mortalidad cardiovascular fue 25% mayor con torcetrapib que con placebo; si bienestos efectos se han atribuido a una acción activadora del SRAA del torcetrapib, los datos generaroncontroversia y extrema precaución. En relación al riesgo residual a estatinas en individuos con LDL <70mg,varios subanálisis, especialmente subanálisis TNT mostró que el riesgo residual después de estatina y LDLóptimos es inversamente proporcional al valor de HDL trans-estatina, sin embargo el estudio AIM-HIGHfue incapaz de demostrar que el incremento farmacológico de HDL con niacina, sea superior a placebo enla reducción de eventos cardiovasculares en individuos con enfermedad cardiovascular, terapia óptima conestatinas, LDL promedio de 60mg/dl y tratamiento adyuvante óptimo; nuevamente datos que han creadocontroversia y escepticismo.
Los puntos anteriores hacen de este rubro de la terapéutica cardiovascular uno, si no es que elmás interesante, siendo el objetivo universal demostrar que después de estatinas a dosis óptimas y LDL<70mg/dl, la adición de un incrementador de HDL reduce significativamente el riesgo residual a losinhibidores de la HMGCoAR.
A continuación se enunciarán los datos más relevantes de los programas de investigación,dando prioridad a aquellos que han alcanzado fases II y III en humanos y han sido publicados.
Rader D. Therapeutic targeting of high-density lipoprotein Metabolism. In Clinical
Cardio-Lipidología
180

Lipidology. Chapter 45; pp 544-552. Editor Ballantyne C. Edition Saunders 2007. Natarajan P, Ray KK,Cannon CP. High-density lipoprotein and coronary heart disease. Current and future therapies. J Am CollCardiol 2010; 55:1283-1299.
Inhibidores de la CETP- Cholesterol Ester Transfer Protein-. La inhibición de la proteínatransportadora de esteres de colesterol de HDL hacia LDL-IDL-VLDL y de triglicéridos en forma inversa,tiene su racional y modelo natural humano en los individuos con deficiencia de esta proteína, en quienes elnivel de HDL es significativamente mayor y la incidencia de enfermedad cardiovascular es de acuerdo aVasan, significativamente menor a sus contrapartes sin la mutación -ver capítulo 2-.
Vasan RS, Pencina MJ, Robins SJ et al. Association of circulating CEPT activity withincidence of cardiovascular disease in the community. Circulation 2009; 120:2414-2420.
Excluyendo al torcetrapib por ser una molécula cuyo programa de investigación clínica estácerrado, existen otros tres fármacos que inhiben o modulan a la CETP. Los dos inhibidores de la CETP sonel anacetrapib y el evacetrapib y como modulador el dalcetrapib.
Dalcetrapib. Esta molécula fue la primera de su clase en publicar sus resultados en fase II en elaño 2002. En estudios de fase II dalcetrapib mostró un incremento promedio de HDL de 35% con un buenperfil de seguridad. Estos reportes impulsaron el desarrollo del programa de investigación denominadodal-HEART que incluye 5 estudios, a la fecha se han publicado los estudios dal-PLAQUE y dal-VESSEL.
. Estudio dal-VESSEL. Este fue un estudio de seguridad endotelio-vascular, cuyo objetivoprimario fue la evaluación de la reserva vasodilatadora braquial dependiente de flujo y la presión arterialambulatoria en un periodo de 24 horas; los objetivos secundarios fueron el cambio en el perfil de lípidos,biomarcadores de inflamación, insulina, electrolitos séricos, concentración-actividad de la CETP y elregistro de eventos cardiovasculares dentro del programa dal-HEART.
En 476 individuos randomizados con riesgo de o con enfermedad arterial coronaria, LDL<100mg/dl en tratamiento con estatinas y/o ezetimibe y HDL <50mg/dl, se comparó en forma doble-ciego, dalcetrapib 600mg contra placebo. En un diseño de no-inferioridad de dalcetrapib contra placebo elobjetivo primario fue positivo. El valor basal promedio de la reserva vasodilatadora braquial fue 4.1% en elgrupo placebo y 4% en el grupo dalcetrapib, sin cambio significativo a las 12 y 36 semanas de tratamiento.La presión arterial ambulatoria promedio basal fue de 125/74mmHg en el grupo placebo y 128/75mmHgen el grupo dalcetrapib, sin cambio significativo a las 36 semanas de tratamiento; para ambas evaluaciones
Estatinas, Fibratos, Niacina y más
181

no existieron diferencias significativas entre los grupos pre-especificados. Dalcetrapib corregido contraplacebo, incremento HDL y apoA1 hasta 31% y 10% respectivamente, redujo la actividad de la CETP, hasta56%, triglicéridos, LDL y apo B100 hasta 14%, 4% y 5%; todos estos valores fueron estadísticamentesignificativos. Los electrolitos séricos, la creatinina y la relación glucosa/insulina no tuvieron diferenciassignificativas entre ambos grupos. Los biomarcadores de inflamación, excepto la Lp-PLA2 tampocotuvieron diferencias significativas entre ambos grupos; la Lp-PLA2 tuvo un incremento significativo en elgrupo dalcetrapib -cambio asociado al incremento de HDL-. Los eventos adversos y los eventoscardiovasculares pre-especificados fueron similares, sin diferencia significativa entre ambos grupos.
La conclusión de este estudio es que dalcetrapib no afecta la función endotelio-vascular,conclusión basada en la ausencia de modificación en la respuesta vasodilatadora braquial dependiente deendotelio y la ausencia de incremento en la presión arterial ambulatoria. Una observación a este estudio fueque, si bien se cumplió el objetivo primario de no inferioridad, es intrigante la ausencia de beneficio dedalcetrapib en los parámetros evaluados, incluyendo los biomarcadores de inflamación. Los autoresproponen como la explicación más viable, la elevación de sólo 31% en las cifras de HDL.
Luscher TF, Taddei S, Kaski JC, Jukema W, Kallend D, Munzel T et al. Vascular effectsand safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomizedclinical trial. Eur Heart J. Advance acces published February 16, 2012.
Estudio dal-PLAQUE. Este fue un estudio de seguridad sobre estructura e inflamaciónarteriales cuyo objetivo primario fue la evaluación por MRI “magnetic resonance imaging” y PET“positron emission tomography” con 18-FDG “18-fluoro deoxi glucose” de los cambios estructurales y/oinflamatorios en la pared arterial de carótidas y aorta ascendente; como parte del programa dal-HEART, losobjetivos secundarios fueron similares al estudio dal-VESSEL.
En 130 individuos con riesgo de o con enfermedad arterial coronaria, LDL <100mg/dl en
tratamiento con estatinas, triglicéridos 400mg/dl y una relación arteria/sangre 1.6 en la captación de� �
18-FDG, se comparó en forma doble-ciego, dalcetrapib 600mg contra placebo. En un diseño de no-inferioridad de dalcetrapib contra placebo, el objetivo primario fue positivo. Por RMN los valores de áreavascular total, área de pared, grosor de pared y área de pared/área vascular total a los 24 meses detratamiento no fueron estadísticamente diferentes entre el grupo dalcetrapib y el grupo placebo y sereportó una tendencia a un menor incremento de área vascular total en el grupo dalcetrapib. Por PET
Cardio-Lipidología
182

8
Cam
bio
enm
mH
gen
laA
BP
Msi
stól
ica
-6
1
15
-13
5
Cam
bio
enm
mH
gen
laA
BP
Md
iastólica
-5
1
10
-10
Reserva vasodilatadoraSe ilustra en el estudio dal-VESSEL la ausencia de
deterioro de la RVB con la administración de600mg de dalcetrapib; las mediciones basal, a las
12 y 36 semanas de tratamiento fueron similares enel grupo placebo y en el grupo dalcetrapib
1.5
Cam
bio
por
cen
tual
enla
rese
rva
vaso
dila
tad
ora
0.5
1.0
2.0
00
p nsp ns
p ns
Basal Semana 12
Dalcetrapib 600mg
Placebo
Semana 36
Dalcetrapib 600mg
Placebo
Dalcetrapib 600mg
Placebo
Presión arterial ambulatoriaSe ilustra en el estudio dal-VESSEL la ausencia de
deterioro de la presión arterial ambulatoria de 24horas con la administración de 600mg de
dalcetrapib; las mediciones a las 12 y 36 semanasde tratamiento fueron similares en el grupo
placebo y en el grupo dalcetrapib
Estatinas, Fibratos, Niacina y más
Estudio dal-VESSEL
Estudio dal-VESSEL
4 12 36
Semana
4 12 36
Semana
Dalcetrapib 600mg
Placebo
Dalcetrapib 600mg
Placebo
p ns p ns
183

tampoco existió diferencia estadísticamente significativa en el grado de inflamación vascular cuantificadopor captación de 18-FDG en los segmentos índice a los 6 meses de tratamiento. La presión arterial enconsultorio no difirió entre ambos grupos. Dalcetrapib corregido contra placebo incremento HDL yapoA1 26.9% y 6.8% respectivamente, incremento la masa y redujo la actividad de la CETP, hasta 55.5%;triglicéridos, LDL y apo B100 no tuvieron diferencias significativas entre ambos grupos y paradójicamentela PCR incremento 33.3% en el grupo dalcetrapib sin cambio en el grupo placebo. Al igual que en el estudiodal-VESSEL, la Lp-PLA2 tuvo un incremento significativo en el grupo dalcetrapib -cambio asociado alincremento de HDL-. Los eventos adversos y los eventos cardiovasculares pre-especificados fueronsimilares, sin diferencia significativa entre ambos grupos.
La conclusión de este estudio es que dalcetrapib no favorece la ateroprogresión ni lainflamación vascular carotideo-aórtica, existiendo una tendencia al beneficio vascular en ambosparámetros correlacionada con un incremento temprano y sostenido de HDL. Al igual que para dal-VESSEL, se comprueba la hipótesis de no-daño vascular por dalcetrapib, si bien con ausencia de beneficiode dalcetrapib en los parámetros evaluados y una paradógica elevación de PCR. Los autores proponencomo la explicación más viable a estos hallazgos el seguimiento limitado a 24 meses y la disociación entre lainflamación intra-arterial y los biomarcadores sistémicos.
Fayad ZA, Mani V, Woodward M, Kallend D, Abt M, Burgess T, Fuster V et al. Safetyand efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging -dal-PLAQUE-: a randomized clinical trial. Lancet . Published online September 11, 2011.www.thelancet.com
El estudio dal-PLAQUE 2 evaluará en 900 individuos con enfermedad arterial coronaria elefecto de dalcetrapib 600mg contra placebo en la aterosclerosis carotideo-coronaria con el empleo de USGcarotideo e intra-coronario. El estudio dal-ACUTE evalúa en 300 individuos con síndromes isquémicoscoronarios agudos, la seguridad y eficacia de la administración precoz de dalcetrapib 600mg contra placebo.El estudio dal-OUTCOMES 1 evalúa en 15,600 individuos con síndrome isquémico coronario reciente, elefecto en eventos cardiovasculares de dalcetrapib 600mg contra placebo. Finalmente el estudio dal-OUTCOMES 2 evalúa en 20,000 individuos con enfermedad cardiovascular establecida, DM de alto riesgoe individuos sin ECV ni DM pero con alto riesgo cardiovascular -en fase de inclusión- el efecto en eventoscardiovasculares de dalcetrapib 600mg contra placebo. Sin duda el programa dal-HEART es un ambiciosoprograma de investigación.
Cardio-Lipidología
184

Una característica de dalcetrapib que lo diferencia de anacetrapib y evacetrapib, es suselectividad para inhibir sólo el transporte heterotípico de colesterol -de HDL hacia LDL-IDL-LDL-, sininterferir en el transporte homotípico de colesterol entre las diferentes subclase de HDL. Esta característicaque se ha denominado modulación de la CETP, se ha asociado a una mayor maduración de HDL3 haciaHDL2, así como una mayor generación de preβ-HDL -ver capítulo 1-, factores que plausiblementeexplican el incremento del flujo de colesterol macrófago-HDL-bilis-excremento demostrado paradalcetrapib. Potencialmente esta es la principal razón para plantear un potencial beneficio clínico en losestudios referidos con esta molécula.
Eric J Niesor. Different effects of compounds decreasing cholesteryl ester transfer proteinactivity on lipoprotein meabolism. Current Opinion in Lipidology 2011; 22:000-000. Amith V Khera,Marina Cuchel, Margarita de la Llera-Mora et al. Cholesterol efflux capacity, high-density lipoproteinfunction, and atherosclerosis. N Engl J Med 2011; 364:127-35.
Anacetrapib. Esta molécula ha sido estudiada en fases I y II y a finales del año 2010 se reportóel estudio fase III DEFINE. Este fue un estudio mediano de eficacia y seguridad en individuos con riesgocardiovascular alto ->20% por Framingham o equivalente- o enfermedad arterial coronaria documentadamás de tres meses antes de la inclusión, con LDL de 50mg/dl a 100mg/dl en terapia con estatinas, HDL
<60mg/dl y TG 400mg/dl. El objetivo primario del estudio fue el cambio en el perfil de lípidos -LDL�
objetivo primario y HDL objetivo secundario- y en parámetros de seguridad: signos vitales con mediciónautomatizada de la tensión arterial, electrolitos séricos, aldosterona, enzimas hepáticas y CPK entre los másimportantes; así mismo se evaluó el combo de: muerte cardiovascular, infarto miocárdico no fatal,enfermedad vascular cerebral no fatal y hospitalización por angina inestable. Se incluyeron a la fase doble-ciego con anacetrapib 100mg vs placebo a 1623 individuos; 55% con enfermedad cardiovascular y 45% conriesgo cardiovascular alto.
Los valores basales promedio de LDL y HDL fueron 81mg/dl y 41mg/dl respectivamente. Laevaluación del perfil de lípidos a las 24 semanas de tratamiento, mostró una reducción de LDL de 81mg/dl a45mg/dl en el grupo anacetrapib vs 82mg/dl a 77mg/dl en el grupo placebo -reducción de 39.8% con p<0.001-; el HDL incremento de 41mg/dl a 101mg/dl en el grupo anacetrapib vs 40mg/dl a 46mg/dl en elgrupo placebo -incremento de 138.1% con p <0.001-. Apoproteína B, colesterol no-HDL, Lp -a- ytriglicéridos mostraron disminución significativa, en tanto que apoAI tuvo incremento también
Estatinas, Fibratos, Niacina y más
185

significativo. Estos cambios en el perfil lipídico se mantuvieron durante las 76 semanas de tratamiento. Lasevaluaciones de seguridad, entre ellas: eventos adversos, signos vitales, electrolitos séricos, enzimashepáticas, CPK, aldosterona etc., no mostraron diferencias entre los grupos anacetrapib y placebo. Elcombo pre-especificado de eventos cardiovasculares tuvo una incidencia de 2% en el grupo anacetrapib yde 2.6% en el grupo placebo -HR 0.76 con IC 95% 0.39 a 1.45 y p 0.04-.
Se concluye que los resultados del estudio DEFINE dan una certeza de 94% en la predicción
de que anacetrapib no incrementará la incidencia de eventos cardiovasculares en forma parecida a
torcetrapib. Con estos resultados se fortalece la investigación clínica con inhibidores de la CET , si bienP
quedan las interrogantes sobre la funcionalidad de la HDL y la repercusión en eventos cardiovasculares en
un estudio “ad-hoc”. Para contestar esta última pregunta se diseñó y recién inicio el estudio REVEAL
HPS3 TIMI-55 en el cual en forma doble-ciego, randomizada y comparada con placebo, la estrategia de
anacetrapib 100mg será investigada en 30,0000 individuos con enfermedad cardiovascular o diabetes
mellitus con aterosclerosis coronaria y terapia óptima con atorvastatina -LDL <70mg/dl-, siendo el
objetivo primario la evaluación del combo: muerte cardiovascular, infarto miocárdico no fatal y
revascularización coronaria.Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM et al. DEFINE
Investigators. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med2010; 363:2406-15.
Evacetrapib. Esta molécula ha sido estudiada en fases I y II y a finales del año 2011 se reportópor Nicholls el estudio doble ciego, comparado con placebo sobre eficacia y tolerancia de evacetrapib adosis de 30mg, 100mg y 500mg como monoterapia y 100mg en combinación con estatinas -simvastatina40mg, atorvastatina 20mg o rosuvastatina 10mg-. El objetivo primario del estudio fue evaluar el cambio enLDL y HDL a las 12 semanas de tratamiento.
En individuos con un LDL promedio de 144.3mg/dl y HDL promedio de 55.1mg/dl,evacetrapib como monoterapia incrementó en forma dosis dependiente de 53.6% a 128.8% el nivel deHDL -30mg/dl a 60mg/dl sobre el basal- comparado con una reducción de -3% en el grupo placebo -p<0.001- y redujo de 13.6% a 35.9% el nivel de LDL -20.5mg/dl a 51.4mg/dl abajo del basal- comparadocon un incremento de 3.9% en el grupo placebo -p < 0.001-. En combinación con estatinas, evacetrapib100mg incremento el nivel de HDL de 78.5% a 88.5% comparado con estatina sola -p <0.001- y redujo el
Cardio-Lipidología
186

-4.01mm2p 0.04
MRIArea vascular total
-2.2mm2p 0.12
MRIArea de pared
-0.03mmp 0.45
MRIGrosor de pared
0.6p 0.57
MRIIndice Vascular
0.07p 0.51
PETIndice TBR
-1
-3
-2
0
-4
Valores absolutos de Dalcetrapib 600mg - Placebo
Placa e inflamaciónSe ilustra en el estudio dal-PLAQUE la ausencia de
incremento en los parámetros morfológicos y deinflamación vasculares. Los valores de área de
pared, grosor de pared e índice vascular medidospor MRI no tuvieron diferencia estadística entre el
grupo placebo y el grupo dalcetrapib, el áreavascular total tuvo una diferencia significativa a
favor del grupo dalcetrapib; el índice deinflamación TBR medido por PET fue similar en
ambos grupos
Comparación entre I-CEPTEsta tabla muestra los porcentajes de inhibición deCEPT, los cambios en HDL y LDL y el efecto enpresión arterial y nivel de aldosterona in-vitro con
los inhibidores de la CEPT -torcetrapib,anacetrapib y evacetrapib y el modulador
dalcetrapib
Estatinas, Fibratos, Niacina y más
Estudio dal-PLAQUE
Comparación entre los inhibidores y modulador de CEPT
CETP
HDL
LDL
Presión arterial
Aldosterona
Variable Dalcetrapib600mg
Torcetrapib60mg
Anacetrapib150 mg
Evacetrapib100mg
37.2% 90% 70%>80%
26.5% 129% 95%62%
-5% -40% -22%-11%
No
No
No
No
No
No
Si
Si
187

nivel de LDL de -11.2% a 13.9% comparado con estatinas -p <0.001-. No se reportaron eventos adversosobservados. La conclusión de este estudio de eficacia fue que evacetrapib comparado con placebo, comomonoterapia o en combinación con estatinas incrementa significativamente el nivel de HDL y reduce elnivel de LDL.
Nicholls SJ, Brewer HB, Kastelein JJP, Krueger KA, Wang MD, Shao M et al. Effects ofthe CEPT inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL andLDL cholesterol. JAMA 2011; 306:2099-2109.
ApoAI y apoAI-miméticos. Dada la gran capacidad aceptora de colesterol de la apoAI, se haplanteado la estrategia que parte de la demostración de que la infusión de apoAI nativa, apoAI tipo Milán,péptidos miméticos o análogos de apoAI y más recientemente la inducción de la síntesis de apoAI, tienenpotencial para inducir ateroregresión en el humano. Esta es una hipótesis en evolución en la cual, el estudioERASE con apoAI más fosfatidilcolina mostró un resultado neutro en volumen de placa con cambiofavorable en la constitución de la placa y elevación de enzimas hepáticas que motivo la interrupción delprograma; el estudio clásico con apoAI Milán más fosfolípidos mostró un resultado positivo en volumen yconstitución de la placa; aún en fase de experimentación animal se encuentra la investigación con péptidosmiméticos orales con resultado positivo en volumen y constitución de placa en ratones y finalmente laestimulación de la transcripción de apoAI con la molécula RVX-208 aun esta en fases preliminares deestudio.
En el conocimiento de que la hidrólisis de HDL por la HL “hepatic lipase” y la EL “endotheliallipase” genera HDL pobres en lípidos, cuyo contenido de apoAI se cataboliza en un tercio por la proteínacubilina, localizada en las células epiteliales del túbulo proximal y dos tercios a nivel hepático, se haplanteado la estrategia de inhibir dichas enzimas para limitar la hidrólisis de las HDL y el catabolismo de laapoAI. Dado que la HL también participa en la hidrólisis de lipoproteínas apoB100 a diferencia de la ELque interviene principalmente en la hidrólisis de fosfolípidos de HDL, esta última se plantea como unblanco terapéutico, cuyo modelo genético se caracteriza por pérdida de función de la EL e incremento deHDL y apoAI.
Agonistas LXR. La hipótesis de esta estrategia parte de la factibilidad de agonizar a losreceptores nucleares LXR del macrófago con el objetivo de incrementar la síntesis y expresión de lostransferidores ABCA1 y ABCG1 -ver capítulo 1-. La prueba de esta hipótesis en animales de
Cardio-Lipidología
188

experimentación ha sido positiva con incremento del TRC y enlentecimiento de la aterosclerosis; sinembargo, el agonismo no selectivo LXR-alfa/beta se asocia a esteatosis hepática, hipertrigliceridemia ehipercolesterolemia por agonismo paralelo de los LXR-alfa -hepáticos- y el factor de transcripciónSREBP1c con incremento en la síntesis de ácidos grasos, sustrato para la síntesis de triglicéridos y VLDL.Esta estrategia continúa con la investigación de agonistas selectivos de los LXR-beta de predominio en elmacrófago con el objetivo de maximizar la síntesis de los cassettes ABCAI y GI sin incremento en la síntesisde ácidos grasos.
Agonistas PPAR. La evidencia con fibratos ya fue revisada en este capítulo; el desarrollo deagonistas de los receptores nucleares PPAR-delta/beta es una estrategia en investigación animal y fase I conresultados, aún no concluyentes en relación a su efecto sobre HDL, TRC y aterosclerosis.
Inhibición de la ACAT. Siendo la ACAT1 la enzima responsable de la esterificación delcolesterol en los macrófagos, se ha propuesto la estrategia de inhibición de la ACAT1 basada en la hipótesisde que la inhibición de la ACAT, al inhibir la formación de macrófagos espumosos, podría inducirateroregresión en el ser humano. Sin embargo tres estudios en humanos han hecho nula dicha hipótesis; elestudio A-Plus con avasimive y los estudios ACTIVATE y CAPTIVATE con pactimibe mostraron unefecto nulo en la aterosclerosis coronaria cuantificada por IVUS, incluso con progresión de la aterosclerosiscarotidea cuantificada por el GIMC en el estudio CAPTIVATE. La hipótesis para explicar este resultadoparadójico es que la inhibición de la esterificación del colesterol en el macrófago, inhibe la expresión deltransportador ABCA1 y con ello se limita el TRC.
Rader D. Therapeutic targeting of high-density lipoprotein Metabolism. In ClinicalLipidology. Chapter 45; pp 544-552. Editor Ballantyne C. Edition Saunders 2007. Natarajan P, Ray KK,Cannon CP. High-density lipoprotein and coronary heart disease. Current and future therapies. J Am CollCardiol 2010; 55:1283-1299.
Estatinas, Fibratos, Niacina y más
189

Cardio-Lipidología es la saga de una serie de 7 libros escritos y editados por Enrique C.
Morales Villegas. Esta saga inició en el año 1992 con la pequeña obra “Manual de HemodinámicaDiagnostica”, este pequeño manual sustentó la práctica protocolizada de la CardiologíaIntervencionista en Aguascalientes. Después de un lapso de 13 años dedicado al desarrollo delIntervencionismo Cardiovascular en su estado adoptivo y a la asesoría de diversos grupos médicos deintervencionismo cardiovascular en otros estados de nuestro país, en el año 2005 Morales Villegaspresentó su obra “Endocardiocrinología”, la cual marca su evolución de Cardiólogo Intervencionistahacia Cardiólogo enfocado a la Investigación, Enseñanza y Asistencia centradas en los factores deriesgo cardiovascular. Con dicha obra Enrique Morales arrancó la escritura y edición -editorial Atheros-CIC- de una sería de Atlas Ilustrados que han tenido gran aceptación e influencia en la enseñanza de laCardiología Preventiva en nuestro país, centro y sudamérica.
Las obras nacidas en el actual Centro de Investigación Cardiometabólica de Aguascalientes-CIC- son las siguientes. Año 2008 “Atlas de Aterotrombosis, Pleiotropismo y Estatinas”, 2009 “Atlasde Riesgo Cardio Metabólico Total”, 2010 “Atlas de Endotelio, Aterotrombosis y Estatinas” en co-autoría con los Profesores GermanoDi Sciascio y Carlos Briguori, y prologado por los Maestros Valentin Fuster y Marco Antonio Martínez-Rios, 2011 “Atlas deHipertensión Arterial” en co-autoría con el Maestro Luis Alcocer Diaz-Barreiro y en este año “Cardio-Lipidología. Lipidología conenfoque cardiovascular”, prologado por el Maestro Antonio M. Gotto.
Cardio-Lipidología es un recorrido actualizado a marzo del 2012 sobre metabolismo de lípidos y lipoproteínas;dislipidemias primarias, secundarias y mixtas; aterotrombogénesis y ateroregresión; salud, riesgo, estratificación y metas en lípidos yfarmacología clínica sobre estatinas, ezetimibe, fibratos, ácidos omega-3, niacina y estrategias en investigación. Un continuo deconocimientos con alto impacto clínico, enmarcado por la portada ilustrada con la obra “La Verónica” del Cordiólogo Jorge OsegueraMoguel, compañero de formación cardiológica del Autor y ahora destacado impulsor de la fusión entre la ciencia y el arte centrados enel órgano vital.
De esta forma en el CIC, Morales-Villegas como Director del mismo, impulsa la integración y formación de postgrado denuevos recursos humanos en Investigación Clínica y armoniza su actividad asistencial cotidiana como Cardiólogo Preventivo con las deInvestigador y Educador, a la fecha con la participación en 67 estudios clínicos en las áreas de dislipidemias, hipertensión arterial ydiabetes mellitus e innumerables invitaciones como conferencista a foros nacionales y extranjeros.
Finalmente, es de especial interés mencionar la sinergia naciente en entre el CIC y el grupo médico Mac “Médica AvanzadaContigo”, la cual entre otros, permitirá ampliar y reproducir en otros estados de nuestro país los proyectos médicos hasta ahoradesarrollados en Aguascalientes por el CIC.